USRE47142E1 - Compounds and methods for treating inflammatory and fibrotic disorders - Google Patents
Compounds and methods for treating inflammatory and fibrotic disorders Download PDFInfo
- Publication number
- USRE47142E1 USRE47142E1 US14/534,450 US201414534450A USRE47142E US RE47142 E1 USRE47142 E1 US RE47142E1 US 201414534450 A US201414534450 A US 201414534450A US RE47142 E USRE47142 E US RE47142E
- Authority
- US
- United States
- Prior art keywords
- compound
- yield
- synthesis
- general procedure
- prepared
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related, expires
Links
- 0 C=C1b**cN1C1=C(C)C=CC=[K]1 Chemical compound C=C1b**cN1C1=C(C)C=CC=[K]1 0.000 description 46
- SOJCMDOKMPOJFI-UHFFFAOYSA-N CC1=CN(C2=CC=C(OC(C)C)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(OC(C)C)C=C2)C(=O)C=C1 SOJCMDOKMPOJFI-UHFFFAOYSA-N 0.000 description 3
- AGOWXBWUPJERCU-UHFFFAOYSA-N BrC1=CC=CC(Br)=N1.O=C1C=CC=C(Br)N1.O=C1C=CC=C(Br)N1C1=CC=CC=C1 Chemical compound BrC1=CC=CC(Br)=N1.O=C1C=CC=C(Br)N1.O=C1C=CC=C(Br)N1C1=CC=CC=C1 AGOWXBWUPJERCU-UHFFFAOYSA-N 0.000 description 2
- GYCPLYCTMDTEPU-UHFFFAOYSA-N Brc1cncnc1 Chemical compound Brc1cncnc1 GYCPLYCTMDTEPU-UHFFFAOYSA-N 0.000 description 2
- NYZGKZONOQPOAJ-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC(S(C)(=O)=O)=CC=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC(S(C)(=O)=O)=CC=C3)C=CC2=O)=CC=C1 NYZGKZONOQPOAJ-UHFFFAOYSA-N 0.000 description 2
- GYDIDNCRLFJFQM-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 GYDIDNCRLFJFQM-UHFFFAOYSA-N 0.000 description 2
- WHOWUBQXNXNNAG-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CN(C)C=N3)C(C)=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CN(C)C=N3)C(C)=CC2=O)=CC=C1 WHOWUBQXNXNNAG-UHFFFAOYSA-N 0.000 description 2
- MWUKCGQGXKWNKI-UHFFFAOYSA-N CC(C)N(C)C(=O)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound CC(C)N(C)C(=O)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 MWUKCGQGXKWNKI-UHFFFAOYSA-N 0.000 description 2
- IPUMKYZFANUYIS-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(Br)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(Br)=C3)C=C1 IPUMKYZFANUYIS-UHFFFAOYSA-N 0.000 description 2
- NEDKQIXGIXATOQ-UHFFFAOYSA-N CC(NC(=O)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2)C1=CC=CC=C1 Chemical compound CC(NC(=O)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2)C1=CC=CC=C1 NEDKQIXGIXATOQ-UHFFFAOYSA-N 0.000 description 2
- YDNFSZKYWAHWKZ-UHFFFAOYSA-N CC1=CC(=O)N(C2=CC=NC=C2)C=C1C1=CN(C)N=C1 Chemical compound CC1=CC(=O)N(C2=CC=NC=C2)C=C1C1=CN(C)N=C1 YDNFSZKYWAHWKZ-UHFFFAOYSA-N 0.000 description 2
- IKTCWTVJVXLKRU-UHFFFAOYSA-N CC1=CN(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C1 IKTCWTVJVXLKRU-UHFFFAOYSA-N 0.000 description 2
- PLMQFHAKZKHJAT-UHFFFAOYSA-N CC1=CN(C2=CC=CC(C(F)(F)F)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(C(F)(F)F)=C2)C(=O)C=C1 PLMQFHAKZKHJAT-UHFFFAOYSA-N 0.000 description 2
- RKQHBDKKVCVPEC-UHFFFAOYSA-N CC1=CN(C2=CC=CN=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CN=C2)C(=O)C=C1 RKQHBDKKVCVPEC-UHFFFAOYSA-N 0.000 description 2
- UKLKPKCNLIEFED-UHFFFAOYSA-N CCC1=CC=C(N2C=C(C)C=CC2=O)C=C1 Chemical compound CCC1=CC=C(N2C=C(C)C=CC2=O)C=C1 UKLKPKCNLIEFED-UHFFFAOYSA-N 0.000 description 2
- XIRVTPPVJPYHPN-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C3=CN(C)C=N3)C=CC2=O)C(C)=C1 Chemical compound CCOC1=CC=C(N2C=C(C3=CN(C)C=N3)C=CC2=O)C(C)=C1 XIRVTPPVJPYHPN-UHFFFAOYSA-N 0.000 description 2
- PMXJCTZQIGEMBK-UHFFFAOYSA-N CN1CCN(C(=O)C2=CC3=C(N2)N(C2=CC=C(Cl)C=C2)C(=O)C=C3)CC1 Chemical compound CN1CCN(C(=O)C2=CC3=C(N2)N(C2=CC=C(Cl)C=C2)C(=O)C=C3)CC1 PMXJCTZQIGEMBK-UHFFFAOYSA-N 0.000 description 2
- ZXQSASNHGXUJSP-UHFFFAOYSA-N CNC(=O)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound CNC(=O)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 ZXQSASNHGXUJSP-UHFFFAOYSA-N 0.000 description 2
- NULUCMSYOIHTEO-UHFFFAOYSA-N COC1=CC=C(N2C=C(C)C=CC2=O)C=C1F Chemical compound COC1=CC=C(N2C=C(C)C=CC2=O)C=C1F NULUCMSYOIHTEO-UHFFFAOYSA-N 0.000 description 2
- UZVULEVWLYCJKL-UHFFFAOYSA-N COC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(Cl)C=C2)C(=O)C=C3)=C1 Chemical compound COC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(Cl)C=C2)C(=O)C=C3)=C1 UZVULEVWLYCJKL-UHFFFAOYSA-N 0.000 description 2
- SRRDNTHCYIOQKM-UHFFFAOYSA-N COC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(F)C=C2)C(=O)C=C3)=C1 Chemical compound COC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(F)C=C2)C(=O)C=C3)=C1 SRRDNTHCYIOQKM-UHFFFAOYSA-N 0.000 description 2
- FJKKZIVNCVXCBP-UHFFFAOYSA-N COC1=CC=CC=C1C1=CN(C2=CN=CN=C2)C(=O)C=C1 Chemical compound COC1=CC=CC=C1C1=CN(C2=CN=CN=C2)C(=O)C=C1 FJKKZIVNCVXCBP-UHFFFAOYSA-N 0.000 description 2
- GWZRFTSIZBPMAM-UHFFFAOYSA-N COCC1=CC=CC=C1N1C=C(C)C=CC1=O Chemical compound COCC1=CC=CC=C1N1C=C(C)C=CC1=O GWZRFTSIZBPMAM-UHFFFAOYSA-N 0.000 description 2
- CLIQWJWCDSHTKH-UHFFFAOYSA-N COc(cc1)ncc1-c1cncnc1 Chemical compound COc(cc1)ncc1-c1cncnc1 CLIQWJWCDSHTKH-UHFFFAOYSA-N 0.000 description 2
- XADICJHFELMBGX-UHFFFAOYSA-N COc(cc1)ncc1Br Chemical compound COc(cc1)ncc1Br XADICJHFELMBGX-UHFFFAOYSA-N 0.000 description 2
- DHADXDMPEUWEAS-UHFFFAOYSA-N COc1ccc(B(O)O)cn1 Chemical compound COc1ccc(B(O)O)cn1 DHADXDMPEUWEAS-UHFFFAOYSA-N 0.000 description 2
- JFIMMEZRPIQGIN-UHFFFAOYSA-N CS(=O)(=O)C1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 Chemical compound CS(=O)(=O)C1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 JFIMMEZRPIQGIN-UHFFFAOYSA-N 0.000 description 2
- GXRBHQILPYQRQV-UHFFFAOYSA-N CSC1=CC=C(N2C=C(C)C=CC2=O)C=C1 Chemical compound CSC1=CC=C(N2C=C(C)C=CC2=O)C=C1 GXRBHQILPYQRQV-UHFFFAOYSA-N 0.000 description 2
- PTZDUZGYOYTRMZ-UHFFFAOYSA-N O=C(C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2)N1CCOCC1 Chemical compound O=C(C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2)N1CCOCC1 PTZDUZGYOYTRMZ-UHFFFAOYSA-N 0.000 description 2
- DZTTUUIMXGKDCR-UHFFFAOYSA-N O=C(NC1=CC(OC2=CC=CC=C2)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC(OC2=CC=CC=C2)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 DZTTUUIMXGKDCR-UHFFFAOYSA-N 0.000 description 2
- JRQNTNNHIVDJQD-UHFFFAOYSA-N O=C(NCC1=CC=CC=C1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1=CC=CC=C1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 JRQNTNNHIVDJQD-UHFFFAOYSA-N 0.000 description 2
- MERSMXLHLMMLGR-UHFFFAOYSA-N O=C(NCC1CCCO1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1CCCO1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 MERSMXLHLMMLGR-UHFFFAOYSA-N 0.000 description 2
- TZAOGQXEQXSLLV-UHFFFAOYSA-N O=C(NCCN1CCCC1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound O=C(NCCN1CCCC1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 TZAOGQXEQXSLLV-UHFFFAOYSA-N 0.000 description 2
- DFZINVABUWBJEQ-UHFFFAOYSA-N O=C1C=C(O)C=CN1C1=CC=CC=C1 Chemical compound O=C1C=C(O)C=CN1C1=CC=CC=C1 DFZINVABUWBJEQ-UHFFFAOYSA-N 0.000 description 2
- CDAYHNIXCXDSOF-UHFFFAOYSA-N O=C1C=CC(C(F)F)=CN1C1=CC=C(C(F)(F)F)C=C1 Chemical compound O=C1C=CC(C(F)F)=CN1C1=CC=C(C(F)(F)F)C=C1 CDAYHNIXCXDSOF-UHFFFAOYSA-N 0.000 description 2
- SQZHFSVQICMBRF-UHFFFAOYSA-N O=C1C=CC(C2=NC=CS2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=NC=CS2)=CN1C1=CC=C(OC(F)(F)F)C=C1 SQZHFSVQICMBRF-UHFFFAOYSA-N 0.000 description 2
- GACPZPNDWHHNAR-UHFFFAOYSA-N O=C1C=CC(N2CCOCC2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(N2CCOCC2)=CN1C1=CC=C(OC(F)(F)F)C=C1 GACPZPNDWHHNAR-UHFFFAOYSA-N 0.000 description 2
- LDZIHHDRHPNEPB-UHFFFAOYSA-N O=C1C=CC=C(Cl)N1C1=CC=CC=C1 Chemical compound O=C1C=CC=C(Cl)N1C1=CC=CC=C1 LDZIHHDRHPNEPB-UHFFFAOYSA-N 0.000 description 2
- NMBKCFKNHMYQRB-UHFFFAOYSA-N BC(=O)N1C=C(B(O)O)C=N1.CC.CC.O=C1C=CC(C2=CNN=C2)=CN1C1=CC=CC=C1.O=C1C=CC(I)=CN1C1=CC=CC=C1 Chemical compound BC(=O)N1C=C(B(O)O)C=N1.CC.CC.O=C1C=CC(C2=CNN=C2)=CN1C1=CC=CC=C1.O=C1C=CC(I)=CN1C1=CC=CC=C1 NMBKCFKNHMYQRB-UHFFFAOYSA-N 0.000 description 1
- QLJMKWOQEWXCCZ-UHFFFAOYSA-M Br.BrC1=CN=CN=C1.C.CCO.COC1=NC=C(B(O)O)C=C1.COC1=NC=C(C2=CN=CN=C2)C=C1.O=C1C=CC(C2=CN=CN=C2)=CN1.O=COO[K].[KH] Chemical compound Br.BrC1=CN=CN=C1.C.CCO.COC1=NC=C(B(O)O)C=C1.COC1=NC=C(C2=CN=CN=C2)C=C1.O=C1C=CC(C2=CN=CN=C2)=CN1.O=COO[K].[KH] QLJMKWOQEWXCCZ-UHFFFAOYSA-M 0.000 description 1
- FSQYNHYRPTYBKW-UHFFFAOYSA-M Br.BrC1=CN=CN=C1.CCO.COC1=NC=C(B(O)O)C=C1.COC1=NC=C(C2=CN=CN=C2)C=C1.O=C1C=CC(C2=CN=CN=C2)=CN1.O=COO[K].[KH] Chemical compound Br.BrC1=CN=CN=C1.CCO.COC1=NC=C(B(O)O)C=C1.COC1=NC=C(C2=CN=CN=C2)C=C1.O=C1C=CC(C2=CN=CN=C2)=CN1.O=COO[K].[KH] FSQYNHYRPTYBKW-UHFFFAOYSA-M 0.000 description 1
- FCULZYPRQFFQKO-UHFFFAOYSA-N Br.BrC1=NC=CS1.CCO.COC1=NC=C(B(O)O)C=C1.COC1=NC=C(C2=CN=C(OC)C=C2)C=C1.COC1=NC=C(C2=NC=CS2)C=C1.O=C1C=CC(C2=NC=CS2)=CN1 Chemical compound Br.BrC1=NC=CS1.CCO.COC1=NC=C(B(O)O)C=C1.COC1=NC=C(C2=CN=C(OC)C=C2)C=C1.COC1=NC=C(C2=NC=CS2)C=C1.O=C1C=CC(C2=NC=CS2)=CN1 FCULZYPRQFFQKO-UHFFFAOYSA-N 0.000 description 1
- KGCRTPSXAJWCKF-UHFFFAOYSA-N Br.C.CC1=CC(=O)N([Ar])C=C1C1=CN(C)N=C1.CC1=CC(=O)NC=C1C1=CN(C)N=C1.CCO.CN1C=C(B2OC(C)(C)C(C)(C)O2)C=N1.COC1=NC=C(Br)C(C)=C1.COC1C=C(C)C(C2=CN(C)N=C2)=CN1 Chemical compound Br.C.CC1=CC(=O)N([Ar])C=C1C1=CN(C)N=C1.CC1=CC(=O)NC=C1C1=CN(C)N=C1.CCO.CN1C=C(B2OC(C)(C)C(C)(C)O2)C=N1.COC1=NC=C(Br)C(C)=C1.COC1C=C(C)C(C2=CN(C)N=C2)=CN1 KGCRTPSXAJWCKF-UHFFFAOYSA-N 0.000 description 1
- HJPBDTCQWNQULM-WNYJDVODSA-N Br.C.CCO.N.O=C1C=CC(C2=NC=CN2)=CN1C1=CC=CC=C1.[2H]C([2H])(C)C(F)OO.[H]C(=O)C([H])=O.[H]C(=O)C1=CN(C2=CC=CC=C2)C(=O)C=C1.[H]C(=O)C1=CN=C(OC)C=C1.[H]C(=O)C1=CNC(=O)C=C1 Chemical compound Br.C.CCO.N.O=C1C=CC(C2=NC=CN2)=CN1C1=CC=CC=C1.[2H]C([2H])(C)C(F)OO.[H]C(=O)C([H])=O.[H]C(=O)C1=CN(C2=CC=CC=C2)C(=O)C=C1.[H]C(=O)C1=CN=C(OC)C=C1.[H]C(=O)C1=CNC(=O)C=C1 HJPBDTCQWNQULM-WNYJDVODSA-N 0.000 description 1
- IRSUZJHRAKFBSB-UHFFFAOYSA-M Br.C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CN=CC=C2)C=C1.O=C1C=CC(C2=CN=CC=C2)=CN1.O=COO[K].OB(O)C1=CC=CN=C1.[KH] Chemical compound Br.C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CN=CC=C2)C=C1.O=C1C=CC(C2=CN=CC=C2)=CN1.O=COO[K].OB(O)C1=CC=CN=C1.[KH] IRSUZJHRAKFBSB-UHFFFAOYSA-M 0.000 description 1
- BSUQITAPYQGUIW-UHFFFAOYSA-N Br.CC(=O)NC1=CC(N2C=C(C3=NC=CN3)C=CC2=O)=CC=C1.CCO.N.O=CCCC=O.[H]C(=O)C1=CN(C2=CC=CC(NC(C)=O)=C2)C(=O)C=C1.[H]C(=O)C1=CN=C(OC)C=C1.[H]C(=O)C1=CNC(=O)C=C1 Chemical compound Br.CC(=O)NC1=CC(N2C=C(C3=NC=CN3)C=CC2=O)=CC=C1.CCO.N.O=CCCC=O.[H]C(=O)C1=CN(C2=CC=CC(NC(C)=O)=C2)C(=O)C=C1.[H]C(=O)C1=CN=C(OC)C=C1.[H]C(=O)C1=CNC(=O)C=C1 BSUQITAPYQGUIW-UHFFFAOYSA-N 0.000 description 1
- PFSCOHSGTVEXNG-UHFFFAOYSA-N Br.CCO.COC1=NC=C(N)C=C1.COC1=NC=C(S(=O)(=O)Cl)C=C1.COC1=NC=C(S(N)(=O)=O)C=C1.N.NS(=O)(=O)C1=CNC(=O)C=C1 Chemical compound Br.CCO.COC1=NC=C(N)C=C1.COC1=NC=C(S(=O)(=O)Cl)C=C1.COC1=NC=C(S(N)(=O)=O)C=C1.N.NS(=O)(=O)C1=CNC(=O)C=C1 PFSCOHSGTVEXNG-UHFFFAOYSA-N 0.000 description 1
- ZZZYZLVZPBWRIW-UHFFFAOYSA-N BrC1=CC2=CC=CN=C2N1.C.C.C.C.C.C.C.C1=CN=C2NC=CC2=C1.CC(C)(C)OC(=O)N1C(Br)=CC2=CC=C(O)N=C21.CC(C)(C)OC(=O)OC1=CC=C2C=C(Br)N(C(=O)OC(C)(C)C)C2=N1.CC(C)OC1=CC=C(B(O)O)C=C1.CC(C)OC1=CC=C(N2C(=O)C=CC3=C2N(C(=O)OC(C)(C)C)C(Br)=C3)C=C1.CC1=CC=C(S(=O)(=O)Cl)C=C1.CC1=CC=C(S(=O)(=O)N2C(Br)=CC3=CC=CN=C32)C=C1.CC1=CC=C(S(=O)(=O)N2C=CC3=CC=CN=C32)C=C1.OC1=CC=C2C=C(Br)NC2=N1.ON1=C2NC(Br)=CC2=CC=C1 Chemical compound BrC1=CC2=CC=CN=C2N1.C.C.C.C.C.C.C.C1=CN=C2NC=CC2=C1.CC(C)(C)OC(=O)N1C(Br)=CC2=CC=C(O)N=C21.CC(C)(C)OC(=O)OC1=CC=C2C=C(Br)N(C(=O)OC(C)(C)C)C2=N1.CC(C)OC1=CC=C(B(O)O)C=C1.CC(C)OC1=CC=C(N2C(=O)C=CC3=C2N(C(=O)OC(C)(C)C)C(Br)=C3)C=C1.CC1=CC=C(S(=O)(=O)Cl)C=C1.CC1=CC=C(S(=O)(=O)N2C(Br)=CC3=CC=CN=C32)C=C1.CC1=CC=C(S(=O)(=O)N2C=CC3=CC=CN=C32)C=C1.OC1=CC=C2C=C(Br)NC2=N1.ON1=C2NC(Br)=CC2=CC=C1 ZZZYZLVZPBWRIW-UHFFFAOYSA-N 0.000 description 1
- RKUPSDNNRVDASH-UHFFFAOYSA-N BrC1=CC=NC=C1.C.O=C1C=CC=CN1.O=C1C=CC=CN1C1=CC=NC=C1 Chemical compound BrC1=CC=NC=C1.C.O=C1C=CC=CN1.O=C1C=CC=CN1C1=CC=NC=C1 RKUPSDNNRVDASH-UHFFFAOYSA-N 0.000 description 1
- HLEXQDSKYXTIML-UHFFFAOYSA-M BrC1=CN=CN=C1.COC1=NC=C(B(O)O)C=C1.COC1=NC=C(C2=CN=CN=C2)C=C1.O=COO[K].[KH] Chemical compound BrC1=CN=CN=C1.COC1=NC=C(B(O)O)C=C1.COC1=NC=C(C2=CN=CN=C2)C=C1.O=COO[K].[KH] HLEXQDSKYXTIML-UHFFFAOYSA-M 0.000 description 1
- KOZZSYONHRBSKR-UHFFFAOYSA-N BrC1=CN=CN=C1.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(F)C=CC=C2)C=C1.OB(O)C1=C(F)C=CC=C1 Chemical compound BrC1=CN=CN=C1.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(F)C=CC=C2)C=C1.OB(O)C1=C(F)C=CC=C1 KOZZSYONHRBSKR-UHFFFAOYSA-N 0.000 description 1
- CWYHXHUXYBGLEX-UHFFFAOYSA-N BrC1=NC=CC=N1.C.COC1=NC=C(C2=NC=CC=N2)C=C1.COC1=NC=C(OBO)C=C1.O=C1C=CC(C2=NC=CC=N2)=CN1 Chemical compound BrC1=NC=CC=N1.C.COC1=NC=C(C2=NC=CC=N2)C=C1.COC1=NC=C(OBO)C=C1.O=C1C=CC(C2=NC=CC=N2)=CN1 CWYHXHUXYBGLEX-UHFFFAOYSA-N 0.000 description 1
- RPMZKPJZXKKKFL-UHFFFAOYSA-N BrC1=NC=CN=C1.CCO.COC1=NC=C(B(O)O)C=C1.COC1=NC=C(C2=NC=CC=N2)C=C1.O=C1C=CC(C2=NC=CC=N2)=CN1 Chemical compound BrC1=NC=CN=C1.CCO.COC1=NC=C(B(O)O)C=C1.COC1=NC=C(C2=NC=CC=N2)C=C1.O=C1C=CC(C2=NC=CC=N2)=CN1 RPMZKPJZXKKKFL-UHFFFAOYSA-N 0.000 description 1
- NESKOYUEWYYNDP-UHFFFAOYSA-N C.C.C.CC(C)(C)OC(=O)OC(=O)OC(C)(C)C.CC1=CC=CN=C1N.CCO.CCOC(=O)C1(O)CC2=C(N=CC=C2)N1C.CCOC(=O)C1=CC2=C(N1C)[N+]([O-])=CC=C2.CCOC(=O)C1=CC2=C(N=C(OC(C)=O)C=C2)N1.CCOC(=O)C1=CC2=C(N=CC=C2)N1.CCOC(=O)C1=CC2=C(N=CC=C2)N1C.CCOC(=O)C1=CC2=C(NC(=O)C=C2)N1.CNC1=NC=CC=C1C Chemical compound C.C.C.CC(C)(C)OC(=O)OC(=O)OC(C)(C)C.CC1=CC=CN=C1N.CCO.CCOC(=O)C1(O)CC2=C(N=CC=C2)N1C.CCOC(=O)C1=CC2=C(N1C)[N+]([O-])=CC=C2.CCOC(=O)C1=CC2=C(N=C(OC(C)=O)C=C2)N1.CCOC(=O)C1=CC2=C(N=CC=C2)N1.CCOC(=O)C1=CC2=C(N=CC=C2)N1C.CCOC(=O)C1=CC2=C(NC(=O)C=C2)N1.CNC1=NC=CC=C1C NESKOYUEWYYNDP-UHFFFAOYSA-N 0.000 description 1
- IZHPUOSSTGOQIT-UHFFFAOYSA-N C.C.C.CC1=CC=CN=C1N.CCO.CCOC(=O)C1(O)CC2=C(N=CC=C2)N1C.CCOC(=O)C1=CC2=C(N1)[N+]([O-])=CC=C2.CCOC(=O)C1=CC2=C(N=C(OC(C)=O)C=C2)N1.CCOC(=O)C1=CC2=C(N=CC=C2)N1.CCOC(=O)C1=CC2=C(NC(=O)C=C2)N1.CNC1=NC=CC=C1C Chemical compound C.C.C.CC1=CC=CN=C1N.CCO.CCOC(=O)C1(O)CC2=C(N=CC=C2)N1C.CCOC(=O)C1=CC2=C(N1)[N+]([O-])=CC=C2.CCOC(=O)C1=CC2=C(N=C(OC(C)=O)C=C2)N1.CCOC(=O)C1=CC2=C(N=CC=C2)N1.CCOC(=O)C1=CC2=C(NC(=O)C=C2)N1.CNC1=NC=CC=C1C IZHPUOSSTGOQIT-UHFFFAOYSA-N 0.000 description 1
- VSBAXGHTRLWXOI-UHFFFAOYSA-N C.C.C=C1C=CC(C2=CC=C(CO)C=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=CC=CC(CO)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CC(F)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CC=C2F)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(N[SH](=O)=O)=CN1C1=CC=C(OCC)C=C1C.CC(=O)NC1=CC(N2C=C(C3=C(C)NN=C3C)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=NC=CN3)C=CC2=O)=CC=C1 Chemical compound C.C.C=C1C=CC(C2=CC=C(CO)C=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=CC=CC(CO)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CC(F)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CC=C2F)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(N[SH](=O)=O)=CN1C1=CC=C(OCC)C=C1C.CC(=O)NC1=CC(N2C=C(C3=C(C)NN=C3C)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=NC=CN3)C=CC2=O)=CC=C1 VSBAXGHTRLWXOI-UHFFFAOYSA-N 0.000 description 1
- POEAWQULSYATQK-UHFFFAOYSA-N C.C.C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CN(C)N=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CN(C)N=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=NC=CS2)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=NC=C3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CN=CN=C3)C=CC2=O)=CC=C1 Chemical compound C.C.C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CN(C)N=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CN(C)N=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=NC=CS2)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=NC=C3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CN=CN=C3)C=CC2=O)=CC=C1 POEAWQULSYATQK-UHFFFAOYSA-N 0.000 description 1
- WXIOJUPAHQGUCG-UHFFFAOYSA-N C.C.CC1=CC(=O)N(C2=CC=CC=C2)C=C1Br.CC1=CC(=O)NC=C1Br.COC1=NC=C(Br)C(C)=C1 Chemical compound C.C.CC1=CC(=O)N(C2=CC=CC=C2)C=C1Br.CC1=CC(=O)NC=C1Br.COC1=NC=C(Br)C(C)=C1 WXIOJUPAHQGUCG-UHFFFAOYSA-N 0.000 description 1
- WXRGZAATHLVMTF-UHFFFAOYSA-M C.C.CCO.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC(Cl)=CC=C2)C=C1.O=C1C=CC(C2=CC(Cl)=CC=C2)=CN1.O=C1C=CC(C2=CC(Cl)=CC=C2)=CN1[Ar].O=COO[K].OB(O)C1=CC=CC(Cl)=C1.[KH] Chemical compound C.C.CCO.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC(Cl)=CC=C2)C=C1.O=C1C=CC(C2=CC(Cl)=CC=C2)=CN1.O=C1C=CC(C2=CC(Cl)=CC=C2)=CN1[Ar].O=COO[K].OB(O)C1=CC=CC(Cl)=C1.[KH] WXRGZAATHLVMTF-UHFFFAOYSA-M 0.000 description 1
- SBLGDAIQZHVATR-UHFFFAOYSA-N C.C.COC1=CC=C(C2=CN([Ar])C(=O)C=C2)C=C1.COC1=CC=C(C2=CN=C(OC)C=C2)C=C1.COC1=CC=C(C2=CNC(=O)C=C2)C=C1.COC1=NC=C(Br)C=C1.OBO.[Ar] Chemical compound C.C.COC1=CC=C(C2=CN([Ar])C(=O)C=C2)C=C1.COC1=CC=C(C2=CN=C(OC)C=C2)C=C1.COC1=CC=C(C2=CNC(=O)C=C2)C=C1.COC1=NC=C(Br)C=C1.OBO.[Ar] SBLGDAIQZHVATR-UHFFFAOYSA-N 0.000 description 1
- UULBVLGVRCUOCK-UHFFFAOYSA-N C.C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(Cl)C=CC=C2)C=C1.O=C1C=CC(C2=C(Cl)C=CC=C2)=CN1.O=C1C=CC(C2=C(Cl)C=CC=C2)=CN1[Ar].OB(O)C1=C(Cl)C=CC=C1 Chemical compound C.C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(Cl)C=CC=C2)C=C1.O=C1C=CC(C2=C(Cl)C=CC=C2)=CN1.O=C1C=CC(C2=C(Cl)C=CC=C2)=CN1[Ar].OB(O)C1=C(Cl)C=CC=C1 UULBVLGVRCUOCK-UHFFFAOYSA-N 0.000 description 1
- RUGWNNRZIYGUQO-UHFFFAOYSA-N C.C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=C(Cl)C=C2)C=C1.O=C1C=CC(C2=CC=C(Cl)C=C2)=CN1.O=C1C=CC(C2=CC=C(Cl)C=C2)=CN1[Ar].OBO.[Ar] Chemical compound C.C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=C(Cl)C=C2)C=C1.O=C1C=CC(C2=CC=C(Cl)C=C2)=CN1.O=C1C=CC(C2=CC=C(Cl)C=C2)=CN1[Ar].OBO.[Ar] RUGWNNRZIYGUQO-UHFFFAOYSA-N 0.000 description 1
- MDNXVFBBGQBNIX-UHFFFAOYSA-N C.C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=C(N[SH](=O)=O)C=C2)C=C1.COC1=NC=C(C2=CC=CC=C2)C=C1.O=C1C=CC(C2=CC=C(N[SH](=O)=O)C=C2)=CN1.OB(O)C1=CC=CC=C1 Chemical compound C.C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=C(N[SH](=O)=O)C=C2)C=C1.COC1=NC=C(C2=CC=CC=C2)C=C1.O=C1C=CC(C2=CC=C(N[SH](=O)=O)C=C2)=CN1.OB(O)C1=CC=CC=C1 MDNXVFBBGQBNIX-UHFFFAOYSA-N 0.000 description 1
- BMQTZAMLXPKVHS-UHFFFAOYSA-L C.C.COC1=NC=C(F)C=C1.COC1=NC=C(N)C=C1.COC1=NC=C([N+]#N)C=C1.FB(F)(F)F.FB(F)F.O=C1C=CC(F)=CN1.O=NO[Na].[F-] Chemical compound C.C.COC1=NC=C(F)C=C1.COC1=NC=C(N)C=C1.COC1=NC=C([N+]#N)C=C1.FB(F)(F)F.FB(F)F.O=C1C=CC(F)=CN1.O=NO[Na].[F-] BMQTZAMLXPKVHS-UHFFFAOYSA-L 0.000 description 1
- ACXUNAYZTIRNNW-UHFFFAOYSA-N C.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(F)C=CC=C2)C=C1.O=C1=NC=C(C2=C(F)C=CC=C2)C=C1.OB(O)C1=CC=CC=C1F.[Pd] Chemical compound C.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.C1=CC=C(P(C2=CC=CC=C2)C2=CC=CC=C2)C=C1.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(F)C=CC=C2)C=C1.O=C1=NC=C(C2=C(F)C=CC=C2)C=C1.OB(O)C1=CC=CC=C1F.[Pd] ACXUNAYZTIRNNW-UHFFFAOYSA-N 0.000 description 1
- VKWIGCMGHHWSBW-UHFFFAOYSA-N C.C=C1C=CC(C2=C(O)C=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(OCC)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(S(=O)(=O)N(C)C)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C(CN(C)C)C=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C3OCOC3=C2)=CN1C1=CC=CC=C1 Chemical compound C.C=C1C=CC(C2=C(O)C=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(OCC)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(S(=O)(=O)N(C)C)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C(CN(C)C)C=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C3OCOC3=C2)=CN1C1=CC=CC=C1 VKWIGCMGHHWSBW-UHFFFAOYSA-N 0.000 description 1
- LXEFUCBZNBVNBW-UHFFFAOYSA-N C.C=C1C=CC(C2=CC=CS2)=CN1C1=CC=CC=C1.CC(=O)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.CC(=O)C1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1.CC(=O)NC1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.CC(F)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.CN(C)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.COCC1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(C(F)(F)F)C=C1.O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(C(F)F)C=C1 Chemical compound C.C=C1C=CC(C2=CC=CS2)=CN1C1=CC=CC=C1.CC(=O)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.CC(=O)C1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1.CC(=O)NC1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.CC(F)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.CN(C)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.COCC1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(C(F)(F)F)C=C1.O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(C(F)F)C=C1 LXEFUCBZNBVNBW-UHFFFAOYSA-N 0.000 description 1
- AIZGLZHBHQHEEV-UHFFFAOYSA-N C.C=C1C=CC2=C(NC(C(=O)N(C)C)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)N(C)C3=CC=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.CNC(=O)C1=CC2=C(N1)N(C1=CC(O)=CC=C1)C(=O)C=C2 Chemical compound C.C=C1C=CC2=C(NC(C(=O)N(C)C)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)N(C)C3=CC=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.CNC(=O)C1=CC2=C(N1)N(C1=CC(O)=CC=C1)C(=O)C=C2 AIZGLZHBHQHEEV-UHFFFAOYSA-N 0.000 description 1
- UWSAHARYXVQNES-FHJHGPAASA-N C.C=COC.CCO/C=C/C(=O)C(F)(F)F.O=C(OC(=O)C(F)(F)F)C(F)(F)F Chemical compound C.C=COC.CCO/C=C/C(=O)C(F)(F)F.O=C(OC(=O)C(F)(F)F)C(F)(F)F UWSAHARYXVQNES-FHJHGPAASA-N 0.000 description 1
- PAXYTSWZPVNAKY-UHFFFAOYSA-N C.CC1=NOC(C)=C1B1OC(C)(C)C(C)(C)O1.CC1=NOC(C)=C1C1=CNC(=O)C=C1.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(C)ON=C2C)C=C1 Chemical compound C.CC1=NOC(C)=C1B1OC(C)(C)C(C)(C)O1.CC1=NOC(C)=C1C1=CNC(=O)C=C1.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(C)ON=C2C)C=C1 PAXYTSWZPVNAKY-UHFFFAOYSA-N 0.000 description 1
- BOPCMZXTMJHPGF-UHFFFAOYSA-M C.CCO.CN(C)S(=O)(=O)C1=CC=C(Br)C=C1.CN(C)S(=O)(=O)C1=CC=C(C2=CNC(=O)C=C2)C=C1.COC1=NC=C(C2=CC=C(S(=O)(=O)N(C)C)C=C2)C=C1.COC1=NC=C(OBO)C=C1.O=COO[K].[KH] Chemical compound C.CCO.CN(C)S(=O)(=O)C1=CC=C(Br)C=C1.CN(C)S(=O)(=O)C1=CC=C(C2=CNC(=O)C=C2)C=C1.COC1=NC=C(C2=CC=C(S(=O)(=O)N(C)C)C=C2)C=C1.COC1=NC=C(OBO)C=C1.O=COO[K].[KH] BOPCMZXTMJHPGF-UHFFFAOYSA-M 0.000 description 1
- BOEAGSLCEAVLPY-UHFFFAOYSA-M C.CCO.CN(C)S(=O)(=O)C1=CC=CC(Br)=C1.CN(C)S(=O)(=O)C1=CC=CC(C2=CNC(=O)C=C2)=C1.COC1=NC=C(C2=CC(S(=O)(=O)N(C)C)=CC=C2)C=C1.COC1=NC=C(OBO)C=C1.O=COO[K].[KH] Chemical compound C.CCO.CN(C)S(=O)(=O)C1=CC=CC(Br)=C1.CN(C)S(=O)(=O)C1=CC=CC(C2=CNC(=O)C=C2)=C1.COC1=NC=C(C2=CC(S(=O)(=O)N(C)C)=CC=C2)C=C1.COC1=NC=C(OBO)C=C1.O=COO[K].[KH] BOEAGSLCEAVLPY-UHFFFAOYSA-M 0.000 description 1
- RBNRGSNETVYJPM-NXZCPFRHSA-N C.CCO/C=C/C(=O)C(F)(F)F.N#CCCl.O=C1C=C(C(F)(F)F)C=CN1 Chemical compound C.CCO/C=C/C(=O)C(F)(F)F.N#CCCl.O=C1C=C(C(F)(F)F)C=CN1 RBNRGSNETVYJPM-NXZCPFRHSA-N 0.000 description 1
- BFPOQPUBJDBXML-UHFFFAOYSA-M C.CCS[Na].OB(O)C1=CC=CC=C1.[C-]#[N+]C1=CN(C2=CC=CC=C2)C(=O)C=C1.[C-]#[N+]C1=CN=C(OC)C=C1.[C-]#[N+]C1=CNC(=O)C=C1 Chemical compound C.CCS[Na].OB(O)C1=CC=CC=C1.[C-]#[N+]C1=CN(C2=CC=CC=C2)C(=O)C=C1.[C-]#[N+]C1=CN=C(OC)C=C1.[C-]#[N+]C1=CNC(=O)C=C1 BFPOQPUBJDBXML-UHFFFAOYSA-M 0.000 description 1
- DARULQUICPDVLF-UHFFFAOYSA-N C.CN1C=C(B2OC(C)(C)C(C)(C)O2)C=N1.CN1C=C(C2=CNC(=O)C=C2)C=N1.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CN(C)N=C2)C=C1 Chemical compound C.CN1C=C(B2OC(C)(C)C(C)(C)O2)C=N1.CN1C=C(C2=CNC(=O)C=C2)C=N1.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CN(C)N=C2)C=C1 DARULQUICPDVLF-UHFFFAOYSA-N 0.000 description 1
- BEXHWPAKFSBXFJ-UHFFFAOYSA-N C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=C(F)C=C2)C=C1.O=C1C=CC(C2=CC=C(F)C=C2)=CN1.OB(O)C1=CC=C(F)C=C1 Chemical compound C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=C(F)C=C2)C=C1.O=C1C=CC(C2=CC=C(F)C=C2)=CN1.OB(O)C1=CC=C(F)C=C1 BEXHWPAKFSBXFJ-UHFFFAOYSA-N 0.000 description 1
- JNQKZRGPXHKPET-UHFFFAOYSA-M C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=NC=C2)C=C1.O=C1C=CC(C2=CC=NC=C2)=CN1.O=COO[K].OB(O)C1=CC=NC=C1.[KH] Chemical compound C.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=NC=C2)C=C1.O=C1C=CC(C2=CC=NC=C2)=CN1.O=COO[K].OB(O)C1=CC=NC=C1.[KH] JNQKZRGPXHKPET-UHFFFAOYSA-M 0.000 description 1
- DRCQKDINRRJZOX-QHHAFSJGSA-N C/C=C/C1=CC(=O)N(C2=CC=CC=C2)C=C1 Chemical compound C/C=C/C1=CC(=O)N(C2=CC=CC=C2)C=C1 DRCQKDINRRJZOX-QHHAFSJGSA-N 0.000 description 1
- FBOAZEYJAOTYNQ-FARCUNLSSA-N C/C=C/C1=CC=CC(=O)N1C1=CC=CC=C1 Chemical compound C/C=C/C1=CC=CC(=O)N1C1=CC=CC=C1 FBOAZEYJAOTYNQ-FARCUNLSSA-N 0.000 description 1
- JAEJPWBHSIAMJS-FARCUNLSSA-N C/C=C/C1=CC=CN(C2=CC=CC=C2)C1=O Chemical compound C/C=C/C1=CC=CN(C2=CC=CC=C2)C1=O JAEJPWBHSIAMJS-FARCUNLSSA-N 0.000 description 1
- MGNZGRCXSMBZBN-NSCUHMNNSA-N C/C=C/C1=CN(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C1 Chemical compound C/C=C/C1=CN(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C1 MGNZGRCXSMBZBN-NSCUHMNNSA-N 0.000 description 1
- CMSABKKJVJLRIN-NSCUHMNNSA-N C/C=C/C1=CN(C2=CC=C(C(F)F)C=C2)C(=O)C=C1 Chemical compound C/C=C/C1=CN(C2=CC=C(C(F)F)C=C2)C(=O)C=C1 CMSABKKJVJLRIN-NSCUHMNNSA-N 0.000 description 1
- GCYSYZUQBVLILM-OXFMIOTJSA-N C/C=C/C1=CN(C2=CC=C(C(F)F)C=C2)C(=O)C=C1.C/C=C/C1=CN(C2=CC=C(C=O)C=C2)C(=O)C=C1.C/C=C/C1=CNC(=O)C=C1.O=CC1=CC=C(B(O)O)C=C1 Chemical compound C/C=C/C1=CN(C2=CC=C(C(F)F)C=C2)C(=O)C=C1.C/C=C/C1=CN(C2=CC=C(C=O)C=C2)C(=O)C=C1.C/C=C/C1=CNC(=O)C=C1.O=CC1=CC=C(B(O)O)C=C1 GCYSYZUQBVLILM-OXFMIOTJSA-N 0.000 description 1
- VQSRMWBKHIHLAW-NSCUHMNNSA-N C/C=C/C1=CN(C2=CC=C(Cl)C=C2)C(=O)C=C1 Chemical compound C/C=C/C1=CN(C2=CC=C(Cl)C=C2)C(=O)C=C1 VQSRMWBKHIHLAW-NSCUHMNNSA-N 0.000 description 1
- BARAGOJTXCUNDV-SNAWJCMRSA-N C/C=C/C1=CN(C2=CC=C(OC(C)C)C=C2)C(=O)C=C1 Chemical compound C/C=C/C1=CN(C2=CC=C(OC(C)C)C=C2)C(=O)C=C1 BARAGOJTXCUNDV-SNAWJCMRSA-N 0.000 description 1
- AASGRNAFGHVVDU-NSCUHMNNSA-N C/C=C/C1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 Chemical compound C/C=C/C1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 AASGRNAFGHVVDU-NSCUHMNNSA-N 0.000 description 1
- UUDMRWVBGKZRIX-ONEGZZNKSA-N C/C=C/C1=CN(C2=CC=C(OC)C=C2)C(=O)C=C1 Chemical compound C/C=C/C1=CN(C2=CC=C(OC)C=C2)C(=O)C=C1 UUDMRWVBGKZRIX-ONEGZZNKSA-N 0.000 description 1
- GWLQCFFIAXAMPJ-GQCTYLIASA-N C/C=C/C1=CN(C2=CC=CC(N(C)C)=C2)C(=O)C=C1 Chemical compound C/C=C/C1=CN(C2=CC=CC(N(C)C)=C2)C(=O)C=C1 GWLQCFFIAXAMPJ-GQCTYLIASA-N 0.000 description 1
- RKKXEDYVDMXCRD-QHHAFSJGSA-N C/C=C/C1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound C/C=C/C1=CN(C2=CC=CC=C2)C(=O)C=C1 RKKXEDYVDMXCRD-QHHAFSJGSA-N 0.000 description 1
- LQGBANWYUFEKSE-UHFFFAOYSA-N C=C1C=C(C)C(C2=CN(C)N=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound C=C1C=C(C)C(C2=CN(C)N=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 LQGBANWYUFEKSE-UHFFFAOYSA-N 0.000 description 1
- ZPJQFXJLFXASTN-UHFFFAOYSA-N C=C1C=C(C)C(C2=CN(C)N=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=C(C)C(C2=CN(C)N=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=C(OCC)C=C1C.CC(=O)NC1=CC(N2C=C(C3=CC=C(CO)C=C3)C=CC2=O)=CC=C1 Chemical compound C=C1C=C(C)C(C2=CN(C)N=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=C(C)C(C2=CN(C)N=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=C(OCC)C=C1C.CC(=O)NC1=CC(N2C=C(C3=CC=C(CO)C=C3)C=CC2=O)=CC=C1 ZPJQFXJLFXASTN-UHFFFAOYSA-N 0.000 description 1
- IVMZPBSOMWXESS-UHFFFAOYSA-N C=C1C=C(C)C(C2=CN(C)N=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CNN(C)=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=CNN=C2)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1.CCCC.CCCC Chemical compound C=C1C=C(C)C(C2=CN(C)N=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CNN(C)=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=CNN=C2)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1.CCCC.CCCC IVMZPBSOMWXESS-UHFFFAOYSA-N 0.000 description 1
- FPUYTTASQBUJDR-UHFFFAOYSA-N C=C1C=C(C)C(C2=CN(C)N=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=C(CO)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CN(C)N=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=NC=CC=N2)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=CC(F)=C3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CN(C)N=C3)C(C)=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=NC=CC=N3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=NC=CO3)C=CC2=O)=CC=C1 Chemical compound C=C1C=C(C)C(C2=CN(C)N=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=C(CO)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CN(C)N=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=NC=CC=N2)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=CC(F)=C3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CN(C)N=C3)C(C)=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=NC=CC=N3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=NC=CO3)C=CC2=O)=CC=C1 FPUYTTASQBUJDR-UHFFFAOYSA-N 0.000 description 1
- COFHAUGZWXCAQO-UHFFFAOYSA-N C=C1C=CC(C(C)=O)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=CO2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CO2)=CN1C1=CC=CC=C1.C=C1C=CC(C2CC2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(S(=O)(=O)N(C)C)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(S(=O)(=O)N(C)C)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3CC3)C=CC2=O)=CC=C1.CC(C)(F)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.CC(F)(F)C1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 Chemical compound C=C1C=CC(C(C)=O)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=CO2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CO2)=CN1C1=CC=CC=C1.C=C1C=CC(C2CC2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(S(=O)(=O)N(C)C)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(S(=O)(=O)N(C)C)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3CC3)C=CC2=O)=CC=C1.CC(C)(F)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1.CC(F)(F)C1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 COFHAUGZWXCAQO-UHFFFAOYSA-N 0.000 description 1
- FWDDLNZXJBSZCC-UHFFFAOYSA-N C=C1C=CC(C2=C(C)C=CS2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=C(C)C=CS2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC(C)=CS2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=C(C)C=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=C(C)C=CC=C1.C=C1C=CC(C2=CC=C(NC=O)C=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C3C(=C2)C=CN3C)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C3C(=C2)C=CN3C)=CN1C1=CC=CC=C1 Chemical compound C=C1C=CC(C2=C(C)C=CS2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=C(C)C=CS2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC(C)=CS2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=C(C)C=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=C(C)C=CC=C1.C=C1C=CC(C2=CC=C(NC=O)C=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C3C(=C2)C=CN3C)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C3C(=C2)C=CN3C)=CN1C1=CC=CC=C1 FWDDLNZXJBSZCC-UHFFFAOYSA-N 0.000 description 1
- ADZPBUDHANFKHM-UHFFFAOYSA-N C=C1C=CC(C2=C(C)N(C)N=C2C)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(S(C)(=O)=O)C=C1.C=C1C=CC(C2=NC=CC=N2)=CN1C1=CC=C(OC(F)(F)F)C=C1.CC(=O)NC1=CC(N2C=C(C3=C(C)N(C)N=C3C)C=CC2=O)=CC=C1.CC1=CC(N2C=C(C3=CNN=C3)C=CC2=O)=CC=C1.CS(=O)(=O)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 Chemical compound C=C1C=CC(C2=C(C)N(C)N=C2C)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(S(C)(=O)=O)C=C1.C=C1C=CC(C2=NC=CC=N2)=CN1C1=CC=C(OC(F)(F)F)C=C1.CC(=O)NC1=CC(N2C=C(C3=C(C)N(C)N=C3C)C=CC2=O)=CC=C1.CC1=CC(N2C=C(C3=CNN=C3)C=CC2=O)=CC=C1.CS(=O)(=O)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 ADZPBUDHANFKHM-UHFFFAOYSA-N 0.000 description 1
- JWCKHRZKKSERDI-UHFFFAOYSA-N C=C1C=CC(C2=C(C)NN=C2C)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=CC(CO)=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=CC=CC=C2OC)=CN1C1=CC=C(OC(F)(F)F)C=C1.CC1=CC(N2C=C(C3=CC=C(Cl)C=C3)C=CC2=O)=CC=C1.CC1=CC(N2C=C(C3=CC=CC(CO)=C3)C=CC2=O)=CC=C1 Chemical compound C=C1C=CC(C2=C(C)NN=C2C)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=CC(CO)=C2)=CN1C1=CC=C(OCC)C=C1C.C=C1C=CC(C2=CC=CC=C2OC)=CN1C1=CC=C(OC(F)(F)F)C=C1.CC1=CC(N2C=C(C3=CC=C(Cl)C=C3)C=CC2=O)=CC=C1.CC1=CC(N2C=C(C3=CC=CC(CO)=C3)C=CC2=O)=CC=C1 JWCKHRZKKSERDI-UHFFFAOYSA-N 0.000 description 1
- BLPUDEJLIQKJOZ-UHFFFAOYSA-N C=C1C=CC(C2=C(C)ON=C2C)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=C(C)ON=C2C)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=NC=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=NC=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CN(C)N=C2)=CN1C1=CC=C(S(N)(=O)=O)C=C1.C=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=C(S(N)(=O)=O)C=C1.C=C1C=CC(N2CCOCC2)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=C(C)ON=C3C)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CN(C)N=C3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=NC=CS3)C=CC2=O)=CC=C1 Chemical compound C=C1C=CC(C2=C(C)ON=C2C)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=C(C)ON=C2C)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CC=NC=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=NC=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CN(C)N=C2)=CN1C1=CC=C(S(N)(=O)=O)C=C1.C=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=C(S(N)(=O)=O)C=C1.C=C1C=CC(N2CCOCC2)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=C(C)ON=C3C)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CN(C)N=C3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=NC=CS3)C=CC2=O)=CC=C1 BLPUDEJLIQKJOZ-UHFFFAOYSA-N 0.000 description 1
- MWODIXIEQNLFQI-UHFFFAOYSA-N C=C1C=CC(C2=C(NC(C)=O)C=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(C)=CS2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(Cl)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(NC(C)=O)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(NC(C)=O)C=C1.C=C1C=CC(C2=CC=C3OCOC3=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=NN2C)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CN=C(CO)C=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=COC=C2)=CN1C1=CC(C)=CC=C1 Chemical compound C=C1C=CC(C2=C(NC(C)=O)C=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(C)=CS2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(Cl)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(NC(C)=O)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(NC(C)=O)C=C1.C=C1C=CC(C2=CC=C3OCOC3=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=NN2C)=CN1C1=CC=CC=C1.C=C1C=CC(C2=CN=C(CO)C=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=COC=C2)=CN1C1=CC(C)=CC=C1 MWODIXIEQNLFQI-UHFFFAOYSA-N 0.000 description 1
- RQXUAHBDHYMOBL-UHFFFAOYSA-N C=C1C=CC(C2=CC(C(N)=O)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(O)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C(CO)N=C2)=CN1C1=CC(C)=CC=C1 Chemical compound C=C1C=CC(C2=CC(C(N)=O)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC(O)=CC=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=C(CO)N=C2)=CN1C1=CC(C)=CC=C1 RQXUAHBDHYMOBL-UHFFFAOYSA-N 0.000 description 1
- GPVUALUBXOOFSL-UHFFFAOYSA-N C=C1C=CC(C2=CC=C(C[SH](=O)=O)C=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=CC(Cl)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.CC1=CC(N2C=C(C3=CC(S(C)(=O)=O)=CC=C3)C=CC2=O)=CC=C1.CC1=CC(N2C=C(C3=CC=CC=C3Cl)C=CC2=O)=CC=C1.CC1=CC(N2C=C(C3=CSC=C3)C=CC2=O)=CC=C1.COC1=CC=CC=C1C1=CN(C2=CC=CC(C)=C2)C(=O)C=C1.O=C1C=CC(C2=CSC=C2)=CN1C1=CC=CC=C1 Chemical compound C=C1C=CC(C2=CC=C(C[SH](=O)=O)C=C2)=CN1C1=CC(C)=CC=C1.C=C1C=CC(C2=CC=CC(Cl)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.CC1=CC(N2C=C(C3=CC(S(C)(=O)=O)=CC=C3)C=CC2=O)=CC=C1.CC1=CC(N2C=C(C3=CC=CC=C3Cl)C=CC2=O)=CC=C1.CC1=CC(N2C=C(C3=CSC=C3)C=CC2=O)=CC=C1.COC1=CC=CC=C1C1=CN(C2=CC=CC(C)=C2)C(=O)C=C1.O=C1C=CC(C2=CSC=C2)=CN1C1=CC=CC=C1 GPVUALUBXOOFSL-UHFFFAOYSA-N 0.000 description 1
- CPYSJJRONFKVAT-UHFFFAOYSA-N C=C1C=CC(C2=CC=C(Cl)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=C(Cl)C=C2)=CN1C1=CC=C(OCC)C=C1C.CC1=CC(N2N=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1.[C-]#[N+]C1=CC(C2=CN(C3=CC=CC(C)=C3)C(=O)C=C2)=CC=C1.[C-]#[N+]C1=CC=C(C2=CN(C3=CC=CC(C)=C3)C(=O)C=C2)C=C1 Chemical compound C=C1C=CC(C2=CC=C(Cl)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=C(Cl)C=C2)=CN1C1=CC=C(OCC)C=C1C.CC1=CC(N2N=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1.[C-]#[N+]C1=CC(C2=CN(C3=CC=CC(C)=C3)C(=O)C=C2)=CC=C1.[C-]#[N+]C1=CC=C(C2=CN(C3=CC=CC(C)=C3)C(=O)C=C2)C=C1 CPYSJJRONFKVAT-UHFFFAOYSA-N 0.000 description 1
- GBWFPMMQCYZRQP-UHFFFAOYSA-N C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(Cl)C=C1.C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(F)C=C1.C=C1C=CC(C2=CNN=C2)=CN1C1=CC=CC=C1.CC1=CC(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1.CCCC.CCCC Chemical compound C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(Cl)C=C1.C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(F)C=C1.C=C1C=CC(C2=CNN=C2)=CN1C1=CC=CC=C1.CC1=CC(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1.CCCC.CCCC GBWFPMMQCYZRQP-UHFFFAOYSA-N 0.000 description 1
- NPLAIGQZSIEOIE-UHFFFAOYSA-N C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(S(N)(=O)=O)C=C1.C=C1C=CC(C2=CC=C(N[SH](=O)=O)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2CC2)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=CO3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(S(=O)(=O)N(C)C)C=CC2=O)=CC=C1.NS(=O)(=O)C1=CC=C(N2C=C(C3CC3)C=CC2=O)C=C1 Chemical compound C=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(S(N)(=O)=O)C=C1.C=C1C=CC(C2=CC=C(N[SH](=O)=O)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2CC2)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=CO3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(S(=O)(=O)N(C)C)C=CC2=O)=CC=C1.NS(=O)(=O)C1=CC=C(N2C=C(C3CC3)C=CC2=O)C=C1 NPLAIGQZSIEOIE-UHFFFAOYSA-N 0.000 description 1
- WTXMRBPQQCWFPO-UHFFFAOYSA-N C=C1C=CC(C2=CC=CC=C2F)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=C(S(N)(=O)=O)C=C1.C=C1C=CC(C2=CNN=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=NC=CO2)=CN1C1=CC=CC=C1.C=C1C=CC(N[SH](=O)=O)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(N[SH](=O)=O)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=CC=C3F)C=CC2=O)=CC=C1 Chemical compound C=C1C=CC(C2=CC=CC=C2F)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=C(S(N)(=O)=O)C=C1.C=C1C=CC(C2=CNN=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=NC=CO2)=CN1C1=CC=CC=C1.C=C1C=CC(N[SH](=O)=O)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(N[SH](=O)=O)=CN1C1=CC=CC=C1.CC(=O)NC1=CC(N2C=C(C3=CC=CC=C3F)C=CC2=O)=CC=C1 WTXMRBPQQCWFPO-UHFFFAOYSA-N 0.000 description 1
- POWHAHCSYIFTEP-UHFFFAOYSA-N C=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=NC=CN2)=CN1C1=CC=CC=C1.C=C1C=CC(N2CCOCC2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound C=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=CC=C1.C=C1C=CC(C2=NC=CN2)=CN1C1=CC=CC=C1.C=C1C=CC(N2CCOCC2)=CN1C1=CC=C(OC(F)(F)F)C=C1 POWHAHCSYIFTEP-UHFFFAOYSA-N 0.000 description 1
- VKXQPRUODWQZRR-UHFFFAOYSA-N C=C1C=CC(C2=NC=CS2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(N2CCCC2)=CN1C1=CC=CC=C1 Chemical compound C=C1C=CC(C2=NC=CS2)=CN1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC(N2CCCC2)=CN1C1=CC=CC=C1 VKXQPRUODWQZRR-UHFFFAOYSA-N 0.000 description 1
- QLLUAIZAEQGKGS-UHFFFAOYSA-N C=C1C=CC2=C(N(C)C(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(C)(=O)=O)=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(C)(=O)=O)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=NC4=C(C=CC=C4)N3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NCCN3CCCC3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.CC1=CC(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(Cl)=CC=C2)=C3)=CC=C1 Chemical compound C=C1C=CC2=C(N(C)C(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(C)(=O)=O)=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(C)(=O)=O)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=NC4=C(C=CC=C4)N3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NCCN3CCCC3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.CC1=CC(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(Cl)=CC=C2)=C3)=CC=C1 QLLUAIZAEQGKGS-UHFFFAOYSA-N 0.000 description 1
- LGTXKNANBVGOTJ-UHFFFAOYSA-N C=C1C=CC2=C(N(C)C(C(=O)O)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCCC3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCOCC3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(OC4=CC=CC=C4)C=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(OC4=CC=CC=C4)C=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC(OC)=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)O)=C2)N1C1=CC=C(Cl)C=C1 Chemical compound C=C1C=CC2=C(N(C)C(C(=O)O)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCCC3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCOCC3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(OC4=CC=CC=C4)C=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(OC4=CC=CC=C4)C=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC(OC)=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)O)=C2)N1C1=CC=C(Cl)C=C1 LGTXKNANBVGOTJ-UHFFFAOYSA-N 0.000 description 1
- NBNNHDAOQSBYGI-UHFFFAOYSA-N C=C1C=CC2=C(NC(Br)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)N(C)C(C)C)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)N(C)C(C)C)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)N(C)C(C)C)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(C)=O)=C2)N1C1=CC=C(O)C=C1.C=C1C=CC2=C(NC(C(C)=O)=C2)N1C1=CC=CC=C1 Chemical compound C=C1C=CC2=C(NC(Br)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)N(C)C(C)C)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)N(C)C(C)C)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)N(C)C(C)C)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(C)=O)=C2)N1C1=CC=C(O)C=C1.C=C1C=CC2=C(NC(C(C)=O)=C2)N1C1=CC=CC=C1 NBNNHDAOQSBYGI-UHFFFAOYSA-N 0.000 description 1
- BPRKMRRRJIGMRV-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)N3CCCC3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCN(C)CC3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(Cl)=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(=O)(=O)OC)=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(OC4=CC=CC=C4)C=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)O)=C2)N1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)N3CCCC3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCN(C)CC3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(Cl)=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(=O)(=O)OC)=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(OC4=CC=CC=C4)C=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)O)=C2)N1C1=CC=C(OC(F)(F)F)C=C1 BPRKMRRRJIGMRV-UHFFFAOYSA-N 0.000 description 1
- PHSYKIRYGZALHS-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)N3CCCC3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(OC4=CC=CC=C4)C=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)O)=C2)N1C1=CC=C(F)C=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)N3CCCC3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(OC4=CC=CC=C4)C=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)O)=C2)N1C1=CC=C(F)C=C1 PHSYKIRYGZALHS-UHFFFAOYSA-N 0.000 description 1
- WSQNNCNADXZGEE-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)N3CCCC3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCCC3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCN(C)CC3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCOCC3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(OC(C)C)C=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)N3CCCC3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCCC3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCN(C)CC3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCOCC3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(OC(C)C)C=C1 WSQNNCNADXZGEE-UHFFFAOYSA-N 0.000 description 1
- CKLGWFZCDBTMFA-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)N3CCN(C)CC3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(Cl)=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(Cl)=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(C(F)(F)F)C=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)N3CCN(C)CC3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(Cl)=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(Cl)=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(C(F)(F)F)C=C1 CKLGWFZCDBTMFA-UHFFFAOYSA-N 0.000 description 1
- ZNXWVYAHIWZJOY-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)N3CCN(C)CC3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCOCC3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(S(N)(=O)=O)C=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)O)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)O)=C2)N1C1=CC=C(OC(C)C)C=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)N3CCN(C)CC3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCOCC3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(S(N)(=O)=O)C=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)O)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)O)=C2)N1C1=CC=C(OC(C)C)C=C1 ZNXWVYAHIWZJOY-UHFFFAOYSA-N 0.000 description 1
- SGLINQATLZSKLH-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)N3CCN(C)CC3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCOCC3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)N3CCN(C)CC3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)N3CCOCC3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1 SGLINQATLZSKLH-UHFFFAOYSA-N 0.000 description 1
- ZWVYWXASDLUZBR-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)N3CCNCC3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(OC4=CC=CC=C4)C=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=NC4=C(C=CC=C4)N3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCCN3CCCC3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(N)=O)=C2)N1C1=CC=C(OC(C)C)C=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)N3CCNCC3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC=C(OC4=CC=CC=C4)C=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=NC4=C(C=CC=C4)N3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCCN3CCCC3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(N)=O)=C2)N1C1=CC=C(OC(C)C)C=C1 ZWVYWXASDLUZBR-UHFFFAOYSA-N 0.000 description 1
- ZFEXAWIWKVXIKJ-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)N3CCNCC3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(Cl)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(=O)(=O)N(C)C)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(C)(=O)=O)=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=NC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCCN3CCCC3)=C2)N1C1=CC=C(OC(C)C)C=C1.CC1=CC(N2C(=O)C=CC3=C2NC(C(=O)N2CCCC2)=C3)=CC=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)N3CCNCC3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(Cl)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(=O)(=O)N(C)C)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(C)(=O)=O)=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=NC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCCN3CCCC3)=C2)N1C1=CC=C(OC(C)C)C=C1.CC1=CC(N2C(=O)C=CC3=C2NC(C(=O)N2CCCC2)=C3)=CC=C1 ZFEXAWIWKVXIKJ-UHFFFAOYSA-N 0.000 description 1
- FSWZIPBTRPTYBC-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)N3CCOCC3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC(C)C3=CC=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(OC(C)C)C=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)N3CCOCC3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC(C)C3=CC=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3CCCO3)=C2)N1C1=CC=C(OC(C)C)C=C1 FSWZIPBTRPTYBC-UHFFFAOYSA-N 0.000 description 1
- HHNQSTOZORUKAY-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)NC(C)C3=CC=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(F)=C(OC)C=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(N)(=O)=O)=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=NC4=C(C=CC=C4)N3)=C2)N1C1=CC=C(OC(C)C)C=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)NC(C)C3=CC=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(F)=C(OC)C=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(N)(=O)=O)=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=NC=CS3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=NC4=C(C=CC=C4)N3)=C2)N1C1=CC=C(OC(C)C)C=C1 HHNQSTOZORUKAY-UHFFFAOYSA-N 0.000 description 1
- YENXECFBUYELRU-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)NC(C)C3=CC=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC4=CC=CC=C4)=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC(OC)=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC(OC)=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=NC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.CC1=CC(N2C(=O)C=CC3=C2NC(C(=O)O)=C3)=CC=C1.CC1=CC(N2C(=O)C=CC3=C2NC(C(=O)O)=C3)=CC=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)NC(C)C3=CC=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC4=CC=CC=C4)=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC(OC)=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC(OC)=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=CC=C3)=C2)N1C1=CC=C(Cl)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=NC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.CC1=CC(N2C(=O)C=CC3=C2NC(C(=O)O)=C3)=CC=C1.CC1=CC(N2C(=O)C=CC3=C2NC(C(=O)O)=C3)=CC=C1 YENXECFBUYELRU-UHFFFAOYSA-N 0.000 description 1
- OYNBRFOOYPVCKC-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)NC3=CC(Cl)=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC4=CC=CC=C4)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC4=CC=CC=C4)=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(N)(=O)=O)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=NC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)NC3=CC(Cl)=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC4=CC=CC=C4)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC4=CC=CC=C4)=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(N)(=O)=O)=CC=C3)=C2)N1C1=CC=C(OC(C)C)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=CC=NC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1 OYNBRFOOYPVCKC-UHFFFAOYSA-N 0.000 description 1
- DRIVYPHQOVJRGI-UHFFFAOYSA-N C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC4=CC=CC=C4)=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC4=CC=CC=C4)=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(C)(=O)=O)=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=NC4=C(C=CC=C4)N3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=NC4=C(C=CC=C4)N3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.CC1=CC(N2C(=O)C=CC3=C2NC(C(=O)NCC2=CC=CC=C2)=C3)=CC=C1 Chemical compound C=C1C=CC2=C(NC(C(=O)NC3=CC(OC)=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC4=CC=CC=C4)=CC=C3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(OC4=CC=CC=C4)=CC=C3)=C2)N1C1=CC=C(F)C=C1.C=C1C=CC2=C(NC(C(=O)NC3=CC(S(C)(=O)=O)=CC=C3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=NC4=C(C=CC=C4)N3)=C2)N1C1=CC=C(C(F)(F)F)C=C1.C=C1C=CC2=C(NC(C(=O)NCC3=NC4=C(C=CC=C4)N3)=C2)N1C1=CC=C(OC(F)(F)F)C=C1.CC1=CC(N2C(=O)C=CC3=C2NC(C(=O)NCC2=CC=CC=C2)=C3)=CC=C1 DRIVYPHQOVJRGI-UHFFFAOYSA-N 0.000 description 1
- SSSFFBMSJSKRRS-UHFFFAOYSA-N C=CC1=CC(N2C=C(C)C=CC2=O)=CC=C1 Chemical compound C=CC1=CC(N2C=C(C)C=CC2=O)=CC=C1 SSSFFBMSJSKRRS-UHFFFAOYSA-N 0.000 description 1
- TWUUSPYWLFDWNV-UHFFFAOYSA-N C=CC1=CC=C(N2C=C(C)C=CC2=O)C=C1 Chemical compound C=CC1=CC=C(N2C=C(C)C=CC2=O)C=C1 TWUUSPYWLFDWNV-UHFFFAOYSA-N 0.000 description 1
- PQPVNUNCCZMKTF-UHFFFAOYSA-N C=CC1=CC=CC=C1N1C=C(C)C=CC1=O Chemical compound C=CC1=CC=CC=C1N1C=C(C)C=CC1=O PQPVNUNCCZMKTF-UHFFFAOYSA-N 0.000 description 1
- DRNDEDMVBHXAID-UHFFFAOYSA-N CB(O)N1C=C(B(O)O)C=N1.CC(=O)NC1=CC=CC(N2C=C(I)C=CC2=O)=C1.CCC(=O)C1=CC(N2C=C(C3=CCN=C3)C=CC2=O)=CC=C1 Chemical compound CB(O)N1C=C(B(O)O)C=N1.CC(=O)NC1=CC=CC(N2C=C(I)C=CC2=O)=C1.CCC(=O)C1=CC(N2C=C(C3=CCN=C3)C=CC2=O)=CC=C1 DRNDEDMVBHXAID-UHFFFAOYSA-N 0.000 description 1
- UGROLZKXYQCJDK-UHFFFAOYSA-N CC(=O)C1=CC(N2C=C(C)C=CC2=O)=CC=C1 Chemical compound CC(=O)C1=CC(N2C=C(C)C=CC2=O)=CC=C1 UGROLZKXYQCJDK-UHFFFAOYSA-N 0.000 description 1
- CIFXDWRLVACKQO-UHFFFAOYSA-N CC(=O)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 CIFXDWRLVACKQO-UHFFFAOYSA-N 0.000 description 1
- UGIYSSIKCHWQIU-UHFFFAOYSA-N CC(=O)C1=CC2=C(N1)N(C1=CC=C(O)C=C1)C(=O)C=C2 Chemical compound CC(=O)C1=CC2=C(N1)N(C1=CC=C(O)C=C1)C(=O)C=C2 UGIYSSIKCHWQIU-UHFFFAOYSA-N 0.000 description 1
- NNYKNRFUTHDPSJ-UHFFFAOYSA-N CC(=O)C1=CC2=C(N1)N(C1=CC=CC=C1)C(=O)C=C2 Chemical compound CC(=O)C1=CC2=C(N1)N(C1=CC=CC=C1)C(=O)C=C2 NNYKNRFUTHDPSJ-UHFFFAOYSA-N 0.000 description 1
- ARTREYSZGAZYDB-UHFFFAOYSA-N CC(=O)C1=CC=C(N2C=C(C)C=CC2=O)C=C1 Chemical compound CC(=O)C1=CC=C(N2C=C(C)C=CC2=O)C=C1 ARTREYSZGAZYDB-UHFFFAOYSA-N 0.000 description 1
- CODQMKINZKBDQG-UHFFFAOYSA-N CC(=O)C1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 Chemical compound CC(=O)C1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 CODQMKINZKBDQG-UHFFFAOYSA-N 0.000 description 1
- TYQLMCHHXGRACW-UHFFFAOYSA-N CC(=O)NC1=C(C2=CN(C3=CC(C)=CC=C3)C(=O)C=C2)C=CC=C1 Chemical compound CC(=O)NC1=C(C2=CN(C3=CC(C)=CC=C3)C(=O)C=C2)C=CC=C1 TYQLMCHHXGRACW-UHFFFAOYSA-N 0.000 description 1
- QFIDRRXUIBGJAT-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C)C=C(Cl)C2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C)C=C(Cl)C2=O)=CC=C1 QFIDRRXUIBGJAT-UHFFFAOYSA-N 0.000 description 1
- PULZVVDGXSGYOD-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C)C=CC2=O)=CC=C1 PULZVVDGXSGYOD-UHFFFAOYSA-N 0.000 description 1
- YGOZGSKNKAINOW-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=C(C)N(C)N=C3C)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=C(C)N(C)N=C3C)C=CC2=O)=CC=C1 YGOZGSKNKAINOW-UHFFFAOYSA-N 0.000 description 1
- KCKWYESHHIXBJN-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=C(C)NN=C3C)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=C(C)NN=C3C)C=CC2=O)=CC=C1 KCKWYESHHIXBJN-UHFFFAOYSA-N 0.000 description 1
- HSNNRURMNULPPO-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=C(C)ON=C3C)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=C(C)ON=C3C)C=CC2=O)=CC=C1 HSNNRURMNULPPO-UHFFFAOYSA-N 0.000 description 1
- HBTPVWYBNSTXBR-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC=C(CO)C=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC=C(CO)C=C3)C=CC2=O)=CC=C1 HBTPVWYBNSTXBR-UHFFFAOYSA-N 0.000 description 1
- MDWVOQYYVYGWAN-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC=C(Cl)C=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC=C(Cl)C=C3)C=CC2=O)=CC=C1 MDWVOQYYVYGWAN-UHFFFAOYSA-N 0.000 description 1
- AIMFDVYSOCNGIO-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1 AIMFDVYSOCNGIO-UHFFFAOYSA-N 0.000 description 1
- AZXSXIWJYZVJFT-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC=CC(CO)=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC=CC(CO)=C3)C=CC2=O)=CC=C1 AZXSXIWJYZVJFT-UHFFFAOYSA-N 0.000 description 1
- ZXPDHGOAAWIAJY-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC=CC(F)=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC=CC(F)=C3)C=CC2=O)=CC=C1 ZXPDHGOAAWIAJY-UHFFFAOYSA-N 0.000 description 1
- MOYWZZGAWMXJFC-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC=CC=C3Cl)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC=CC=C3Cl)C=CC2=O)=CC=C1 MOYWZZGAWMXJFC-UHFFFAOYSA-N 0.000 description 1
- ZUDQHGZIEKCDMC-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC=CC=C3F)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC=CC=C3F)C=CC2=O)=CC=C1 ZUDQHGZIEKCDMC-UHFFFAOYSA-N 0.000 description 1
- RDGCYVMCBIDUNF-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC=CO3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC=CO3)C=CC2=O)=CC=C1 RDGCYVMCBIDUNF-UHFFFAOYSA-N 0.000 description 1
- XOEDOOXAGZNHPO-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CC=NC=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CC=NC=C3)C=CC2=O)=CC=C1 XOEDOOXAGZNHPO-UHFFFAOYSA-N 0.000 description 1
- YKLHXOVFBWXOJD-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CN(C)N=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CN(C)N=C3)C=CC2=O)=CC=C1 YKLHXOVFBWXOJD-UHFFFAOYSA-N 0.000 description 1
- FDHOBUOZJTXRRY-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CN=CN=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CN=CN=C3)C=CC2=O)=CC=C1 FDHOBUOZJTXRRY-UHFFFAOYSA-N 0.000 description 1
- ZAJLZDXSKYJWSF-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CNN=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CNN=C3)C=CC2=O)=CC=C1 ZAJLZDXSKYJWSF-UHFFFAOYSA-N 0.000 description 1
- RACWTLFQUIRJOX-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=CSC=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=CSC=C3)C=CC2=O)=CC=C1 RACWTLFQUIRJOX-UHFFFAOYSA-N 0.000 description 1
- AROMFMQGYQSUFI-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=NC=CN3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=NC=CN3)C=CC2=O)=CC=C1 AROMFMQGYQSUFI-UHFFFAOYSA-N 0.000 description 1
- FRAGMIGEFVBFQJ-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=NC=CO3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=NC=CO3)C=CC2=O)=CC=C1 FRAGMIGEFVBFQJ-UHFFFAOYSA-N 0.000 description 1
- YEDOAQIDBGBIKR-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=NC=CO3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(I)C=CC2=O)=CC=C1.CCCC[Sn](CCCC)(CCCC)C1=CC=CC1.N#N Chemical compound CC(=O)NC1=CC(N2C=C(C3=NC=CO3)C=CC2=O)=CC=C1.CC(=O)NC1=CC(N2C=C(I)C=CC2=O)=CC=C1.CCCC[Sn](CCCC)(CCCC)C1=CC=CC1.N#N YEDOAQIDBGBIKR-UHFFFAOYSA-N 0.000 description 1
- UNCAHSXLPUUWOJ-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3=NC=CS3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3=NC=CS3)C=CC2=O)=CC=C1 UNCAHSXLPUUWOJ-UHFFFAOYSA-N 0.000 description 1
- FQYODSXLSGBXLH-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(C3CC3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(C3CC3)C=CC2=O)=CC=C1 FQYODSXLSGBXLH-UHFFFAOYSA-N 0.000 description 1
- VZYQPHSIZGNJQS-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(F)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(F)C=CC2=O)=CC=C1 VZYQPHSIZGNJQS-UHFFFAOYSA-N 0.000 description 1
- XTSQHYDDNZZHOE-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=C(S(=O)(=O)N(C)C)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=C(S(=O)(=O)N(C)C)C=CC2=O)=CC=C1 XTSQHYDDNZZHOE-UHFFFAOYSA-N 0.000 description 1
- MYDYWCNGFBAQRT-UHFFFAOYSA-N CC(=O)NC1=CC(N2C=CC=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2C=CC=CC2=O)=CC=C1 MYDYWCNGFBAQRT-UHFFFAOYSA-N 0.000 description 1
- UGOYEJWDGZPMPY-UHFFFAOYSA-N CC(=O)NC1=CC(N2N=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1 Chemical compound CC(=O)NC1=CC(N2N=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1 UGOYEJWDGZPMPY-UHFFFAOYSA-N 0.000 description 1
- REOCUGAUIPOPIS-UHFFFAOYSA-N CC(=O)NC1=CC=C(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)C=C1 Chemical compound CC(=O)NC1=CC=C(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)C=C1 REOCUGAUIPOPIS-UHFFFAOYSA-N 0.000 description 1
- JWERIBINNGPEGP-UHFFFAOYSA-N CC(=O)NC1=CC=CC(C2=CN(C3=CC(C)=CC=C3)C(=O)C=C2)=C1 Chemical compound CC(=O)NC1=CC=CC(C2=CN(C3=CC(C)=CC=C3)C(=O)C=C2)=C1 JWERIBINNGPEGP-UHFFFAOYSA-N 0.000 description 1
- MUPDFCOPQVQVOV-UHFFFAOYSA-N CC(=O)NC1=CC=CC(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=C1 Chemical compound CC(=O)NC1=CC=CC(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=C1 MUPDFCOPQVQVOV-UHFFFAOYSA-N 0.000 description 1
- QSIUOSLPTGPCGG-UHFFFAOYSA-N CC(=O)NC1=CC=CC=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound CC(=O)NC1=CC=CC=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 QSIUOSLPTGPCGG-UHFFFAOYSA-N 0.000 description 1
- VYGBKWLBXRICNH-UHFFFAOYSA-N CC(C)N(C)C(=O)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound CC(C)N(C)C(=O)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 VYGBKWLBXRICNH-UHFFFAOYSA-N 0.000 description 1
- RJZSIAIHAJLRMW-UHFFFAOYSA-N CC(C)N(C)C(=O)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 Chemical compound CC(C)N(C)C(=O)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 RJZSIAIHAJLRMW-UHFFFAOYSA-N 0.000 description 1
- GUPBURXNWKGEIU-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N(C)C(C)C)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N(C)C(C)C)=C3)C=C1 GUPBURXNWKGEIU-UHFFFAOYSA-N 0.000 description 1
- ANPKTWOIAVLVTK-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N(C)C)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N(C)C)=C3)C=C1 ANPKTWOIAVLVTK-UHFFFAOYSA-N 0.000 description 1
- PZOJXIDDOPHBFN-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N2CCCC2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N2CCCC2)=C3)C=C1 PZOJXIDDOPHBFN-UHFFFAOYSA-N 0.000 description 1
- AIFVTBXAHKCZKO-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N2CCN(C)CC2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N2CCN(C)CC2)=C3)C=C1 AIFVTBXAHKCZKO-UHFFFAOYSA-N 0.000 description 1
- NOQJUETXTWKRKX-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N2CCNCC2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N2CCNCC2)=C3)C=C1 NOQJUETXTWKRKX-UHFFFAOYSA-N 0.000 description 1
- JYQVJWXXMBXCTO-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N2CCOCC2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N2CCOCC2)=C3)C=C1 JYQVJWXXMBXCTO-UHFFFAOYSA-N 0.000 description 1
- UPHODXKWQUHYMD-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC(C)C2=CC=CC=C2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC(C)C2=CC=CC=C2)=C3)C=C1 UPHODXKWQUHYMD-UHFFFAOYSA-N 0.000 description 1
- DSKKGAHDWVTFPP-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(Cl)=CC=C2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(Cl)=CC=C2)=C3)C=C1 DSKKGAHDWVTFPP-UHFFFAOYSA-N 0.000 description 1
- DPQVCKKKTKNMCF-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(OC4=CC=CC=C4)=CC=C2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(OC4=CC=CC=C4)=CC=C2)=C3)C=C1 DPQVCKKKTKNMCF-UHFFFAOYSA-N 0.000 description 1
- CDUNGLSMNJTRBC-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(S(=O)(=O)N(C)C)=CC=C2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(S(=O)(=O)N(C)C)=CC=C2)=C3)C=C1 CDUNGLSMNJTRBC-UHFFFAOYSA-N 0.000 description 1
- MOFDTXHYFJSMPW-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(S(C)(=O)=O)=CC=C2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(S(C)(=O)=O)=CC=C2)=C3)C=C1 MOFDTXHYFJSMPW-UHFFFAOYSA-N 0.000 description 1
- XCZCVZJECJXROL-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(S(N)(=O)=O)=CC=C2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC(S(N)(=O)=O)=CC=C2)=C3)C=C1 XCZCVZJECJXROL-UHFFFAOYSA-N 0.000 description 1
- DVAUPAVOXULYBX-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC=C(OC4=CC=CC=C4)C=C2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=CC=C(OC4=CC=CC=C4)C=C2)=C3)C=C1 DVAUPAVOXULYBX-UHFFFAOYSA-N 0.000 description 1
- DKGUCCZKKNXWSH-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=NC=CS2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NC2=NC=CS2)=C3)C=C1 DKGUCCZKKNXWSH-UHFFFAOYSA-N 0.000 description 1
- AQBJJHMHEWWKDC-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NCC2=CC=CC=C2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NCC2=CC=CC=C2)=C3)C=C1 AQBJJHMHEWWKDC-UHFFFAOYSA-N 0.000 description 1
- BBTKFUXAZGNQOS-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NCC2=CC=NC=C2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NCC2=CC=NC=C2)=C3)C=C1 BBTKFUXAZGNQOS-UHFFFAOYSA-N 0.000 description 1
- ICFCATDMBVRLOT-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NCC2=NC4=C(C=CC=C4)N2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NCC2=NC4=C(C=CC=C4)N2)=C3)C=C1 ICFCATDMBVRLOT-UHFFFAOYSA-N 0.000 description 1
- KQTGYWPNLFNHSY-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NCC2CCCO2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NCC2CCCO2)=C3)C=C1 KQTGYWPNLFNHSY-UHFFFAOYSA-N 0.000 description 1
- WKNCJOAFCPPJNL-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NCCN2CCCC2)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)NCCN2CCCC2)=C3)C=C1 WKNCJOAFCPPJNL-UHFFFAOYSA-N 0.000 description 1
- FPAZPAMCWKBDNJ-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)O)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)O)=C3)C=C1 FPAZPAMCWKBDNJ-UHFFFAOYSA-N 0.000 description 1
- VGGRBJQDMAURMO-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(N)=O)=C3)C=C1 Chemical compound CC(C)OC1=CC=C(N2C(=O)C=CC3=C2NC(C(N)=O)=C3)C=C1 VGGRBJQDMAURMO-UHFFFAOYSA-N 0.000 description 1
- PZYGSZABUBBCIT-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 Chemical compound CC(C)OC1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 PZYGSZABUBBCIT-UHFFFAOYSA-N 0.000 description 1
- PEMHOGQVMHYHST-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C=CC(C(F)(F)F)=CC2=O)C=C1 Chemical compound CC(C)OC1=CC=C(N2C=CC(C(F)(F)F)=CC2=O)C=C1 PEMHOGQVMHYHST-UHFFFAOYSA-N 0.000 description 1
- YSELILQPDBCQQA-UHFFFAOYSA-N CC(C)OC1=CC=C(N2C=CC(C(F)F)=CC2=O)C=C1 Chemical compound CC(C)OC1=CC=C(N2C=CC(C(F)F)=CC2=O)C=C1 YSELILQPDBCQQA-UHFFFAOYSA-N 0.000 description 1
- YVLNCCRTGYJBHK-UHFFFAOYSA-N CC(C)OC1=CC=CC=C1N1C=C(C2=CC=CC=C2)C=CC1=O Chemical compound CC(C)OC1=CC=CC=C1N1C=C(C2=CC=CC=C2)C=CC1=O YVLNCCRTGYJBHK-UHFFFAOYSA-N 0.000 description 1
- WGCQHNTWZKRBDI-UHFFFAOYSA-N CC(C)OC1=CC=CC=C1N1C=CC(C(F)(F)F)=CC1=O Chemical compound CC(C)OC1=CC=CC=C1N1C=CC(C(F)(F)F)=CC1=O WGCQHNTWZKRBDI-UHFFFAOYSA-N 0.000 description 1
- SIUPHGWNVGDWTJ-UHFFFAOYSA-N CC(C1=CN(C2=CC=CC=C2)C(=O)C=C1)C(F)(F)F Chemical compound CC(C1=CN(C2=CC=CC=C2)C(=O)C=C1)C(F)(F)F SIUPHGWNVGDWTJ-UHFFFAOYSA-N 0.000 description 1
- SLVQZLWZGOUXEE-UHFFFAOYSA-N CC(F)(F)C1=CC(N2C=C(C(F)F)C=CC2=O)=CC=C1 Chemical compound CC(F)(F)C1=CC(N2C=C(C(F)F)C=CC2=O)=CC=C1 SLVQZLWZGOUXEE-UHFFFAOYSA-N 0.000 description 1
- DLIWQFQDOIERKW-UHFFFAOYSA-N CC(F)(F)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 Chemical compound CC(F)(F)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 DLIWQFQDOIERKW-UHFFFAOYSA-N 0.000 description 1
- PKXKSJJMGZYDPZ-UHFFFAOYSA-N CC(F)(F)C1=CC(N2C=CC(C(F)F)=CC2=O)=CC=C1 Chemical compound CC(F)(F)C1=CC(N2C=CC(C(F)F)=CC2=O)=CC=C1 PKXKSJJMGZYDPZ-UHFFFAOYSA-N 0.000 description 1
- GAEJBRCLLKAMJB-UHFFFAOYSA-N CC(F)(F)C1=CC=C(N2C=C(C(F)F)C=CC2=O)C=C1 Chemical compound CC(F)(F)C1=CC=C(N2C=C(C(F)F)C=CC2=O)C=C1 GAEJBRCLLKAMJB-UHFFFAOYSA-N 0.000 description 1
- AFIOLZLQJIQUII-UHFFFAOYSA-N CC(F)(F)C1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 Chemical compound CC(F)(F)C1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 AFIOLZLQJIQUII-UHFFFAOYSA-N 0.000 description 1
- TUBIWCZGHXCNMB-UHFFFAOYSA-N CC(F)(F)C1=CC=C(N2C=CC(C(F)F)=CC2=O)C=C1 Chemical compound CC(F)(F)C1=CC=C(N2C=CC(C(F)F)=CC2=O)C=C1 TUBIWCZGHXCNMB-UHFFFAOYSA-N 0.000 description 1
- NOGHHMAGKZMJMW-UHFFFAOYSA-N CC(F)C1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound CC(F)C1=CN(C2=CC=CC=C2)C(=O)C=C1 NOGHHMAGKZMJMW-UHFFFAOYSA-N 0.000 description 1
- RRELCSXABTWIFG-UHFFFAOYSA-N CC(NC(=O)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2)C1=CC=CC=C1 Chemical compound CC(NC(=O)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2)C1=CC=CC=C1 RRELCSXABTWIFG-UHFFFAOYSA-N 0.000 description 1
- BIAXMYPXFSWQQZ-UHFFFAOYSA-N CC(Nc1cccc(N(C=C(C=C2)c3ncccn3)C2=O)c1)=O Chemical compound CC(Nc1cccc(N(C=C(C=C2)c3ncccn3)C2=O)c1)=O BIAXMYPXFSWQQZ-UHFFFAOYSA-N 0.000 description 1
- RMDQXZDOYICYKC-UHFFFAOYSA-N CC.CC.O=C1C=CC(I)=CN1C1=CC=CC=C1.OB(O)C1=CC=CC=C1.[H]N1C=C(I)C=CC1=O Chemical compound CC.CC.O=C1C=CC(I)=CN1C1=CC=CC=C1.OB(O)C1=CC=CC=C1.[H]N1C=C(I)C=CC1=O RMDQXZDOYICYKC-UHFFFAOYSA-N 0.000 description 1
- YZMZWQNUDQKSSP-UHFFFAOYSA-N CC1=C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)SC=C1 Chemical compound CC1=C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)SC=C1 YZMZWQNUDQKSSP-UHFFFAOYSA-N 0.000 description 1
- ZTKKXSRFAOKONH-UHFFFAOYSA-N CC1=C(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)C=CC=C1 Chemical compound CC1=C(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)C=CC=C1 ZTKKXSRFAOKONH-UHFFFAOYSA-N 0.000 description 1
- NMXRDMGYEKHNCV-UHFFFAOYSA-N CC1=CC(=O)N(C2=CC=C(OC(F)(F)F)C=C2)C=C1C1=CN(C)N=C1 Chemical compound CC1=CC(=O)N(C2=CC=C(OC(F)(F)F)C=C2)C=C1C1=CN(C)N=C1 NMXRDMGYEKHNCV-UHFFFAOYSA-N 0.000 description 1
- LPZWUECCZRSOAQ-UHFFFAOYSA-N CC1=CC(=O)N(C2=CC=CC=C2)C=C1 Chemical compound CC1=CC(=O)N(C2=CC=CC=C2)C=C1 LPZWUECCZRSOAQ-UHFFFAOYSA-N 0.000 description 1
- XRNGRGNNQILHAD-UHFFFAOYSA-N CC1=CC(=O)N(C2=CC=CC=C2)C=C1Br Chemical compound CC1=CC(=O)N(C2=CC=CC=C2)C=C1Br XRNGRGNNQILHAD-UHFFFAOYSA-N 0.000 description 1
- UEFMEEILRIFPHH-UHFFFAOYSA-N CC1=CC(=O)N(C2=CC=CC=C2)C=C1C1=CN(C)C=N1 Chemical compound CC1=CC(=O)N(C2=CC=CC=C2)C=C1C1=CN(C)C=N1 UEFMEEILRIFPHH-UHFFFAOYSA-N 0.000 description 1
- WIMYHGCMIPWGOS-UHFFFAOYSA-M CC1=CC(B(O)O)=CC=C1.CCO.COC1=CC=CC(C2=CN=C(OC)C=C2)=C1.COC1=CC=CC(C2=CNC(=O)C=C2)=C1.COC1=NC=C(Br)C=C1.O=COO[K].[KH] Chemical compound CC1=CC(B(O)O)=CC=C1.CCO.COC1=CC=CC(C2=CN=C(OC)C=C2)=C1.COC1=CC=CC(C2=CNC(=O)C=C2)=C1.COC1=NC=C(Br)C=C1.O=COO[K].[KH] WIMYHGCMIPWGOS-UHFFFAOYSA-M 0.000 description 1
- NDGCMBKTLPWHDG-UHFFFAOYSA-N CC1=CC(N2C=C(C)C=CC2=O)=CC=C1 Chemical compound CC1=CC(N2C=C(C)C=CC2=O)=CC=C1 NDGCMBKTLPWHDG-UHFFFAOYSA-N 0.000 description 1
- VEAHWIKRUNSCMX-UHFFFAOYSA-N CC1=CC=C(C2=CN(C3=CC(C)=CC=C3)C(=O)C=C2)C=C1 Chemical compound CC1=CC=C(C2=CN(C3=CC(C)=CC=C3)C(=O)C=C2)C=C1 VEAHWIKRUNSCMX-UHFFFAOYSA-N 0.000 description 1
- QFWOAKZLGUFOLH-UHFFFAOYSA-N CC1=CC=C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)C=C1 Chemical compound CC1=CC=C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)C=C1 QFWOAKZLGUFOLH-UHFFFAOYSA-N 0.000 description 1
- ZVEJTXODUXFIAO-UHFFFAOYSA-N CC1=CC=C(N2C=C(C)C=CC2=O)C=C1 Chemical compound CC1=CC=C(N2C=C(C)C=CC2=O)C=C1 ZVEJTXODUXFIAO-UHFFFAOYSA-N 0.000 description 1
- YBOFHXJDTXZWNJ-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=C(C)C=CS3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=C(C)C=CS3)C=CC2=O)=C1 YBOFHXJDTXZWNJ-UHFFFAOYSA-N 0.000 description 1
- MRWYGDPKBCNZSI-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC(C(N)=O)=CC=C3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC(C(N)=O)=CC=C3)C=CC2=O)=C1 MRWYGDPKBCNZSI-UHFFFAOYSA-N 0.000 description 1
- XIAUXEVBIOIMAN-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC(C)=CS3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC(C)=CS3)C=CC2=O)=C1 XIAUXEVBIOIMAN-UHFFFAOYSA-N 0.000 description 1
- NMNPIAWVGKMNNB-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC(Cl)=CC=C3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC(Cl)=CC=C3)C=CC2=O)=C1 NMNPIAWVGKMNNB-UHFFFAOYSA-N 0.000 description 1
- CXBPZVWWFLLBCZ-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC(O)=CC=C3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC(O)=CC=C3)C=CC2=O)=C1 CXBPZVWWFLLBCZ-UHFFFAOYSA-N 0.000 description 1
- IOKFEGRBTQVWRX-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC(S(=O)(=O)N(C)C)=CC=C3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC(S(=O)(=O)N(C)C)=CC=C3)C=CC2=O)=C1 IOKFEGRBTQVWRX-UHFFFAOYSA-N 0.000 description 1
- JTRFWLDUUZBDTA-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC=C(CN(C)C)C=C3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC=C(CN(C)C)C=C3)C=CC2=O)=C1 JTRFWLDUUZBDTA-UHFFFAOYSA-N 0.000 description 1
- BCHMPLLQKDYLLW-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC=C(CO)N=C3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC=C(CO)N=C3)C=CC2=O)=C1 BCHMPLLQKDYLLW-UHFFFAOYSA-N 0.000 description 1
- BXHROHRTJQYZMU-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC=C(C[SH](=O)=O)C=C3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC=C(C[SH](=O)=O)C=C3)C=CC2=O)=C1 BXHROHRTJQYZMU-UHFFFAOYSA-N 0.000 description 1
- JFGDJFHGANOKPI-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC=C(NC=O)C=C3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC=C(NC=O)C=C3)C=CC2=O)=C1 JFGDJFHGANOKPI-UHFFFAOYSA-N 0.000 description 1
- YJKWOKAAMVTNEA-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC=C4C(=C3)C=CN4C)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC=C4C(=C3)C=CN4C)C=CC2=O)=C1 YJKWOKAAMVTNEA-UHFFFAOYSA-N 0.000 description 1
- XAGREDMOFKFCHG-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=CC=C4OCOC4=C3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=CC=C4OCOC4=C3)C=CC2=O)=C1 XAGREDMOFKFCHG-UHFFFAOYSA-N 0.000 description 1
- JNDUAENKJCMBDQ-UHFFFAOYSA-N CC1=CC=CC(N2C=C(C3=COC=C3)C=CC2=O)=C1 Chemical compound CC1=CC=CC(N2C=C(C3=COC=C3)C=CC2=O)=C1 JNDUAENKJCMBDQ-UHFFFAOYSA-N 0.000 description 1
- ORDALLFBTLCGJF-UHFFFAOYSA-N CC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(F)C=C2)C(=O)C=C3)=C1 Chemical compound CC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(F)C=C2)C(=O)C=C3)=C1 ORDALLFBTLCGJF-UHFFFAOYSA-N 0.000 description 1
- IMTXBILLEULLTF-UHFFFAOYSA-N CC1=CN(C2=CC(OC(C)C)=CC(F)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC(OC(C)C)=CC(F)=C2)C(=O)C=C1 IMTXBILLEULLTF-UHFFFAOYSA-N 0.000 description 1
- YGNLAKNPHZNQGX-UHFFFAOYSA-N CC1=CN(C2=CC=C(C#N)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C#N)C=C2)C(=O)C=C1 YGNLAKNPHZNQGX-UHFFFAOYSA-N 0.000 description 1
- FENQDWMLJSTNRP-UHFFFAOYSA-N CC1=CN(C2=CC=C(C(=O)C(F)(F)F)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C(=O)C(F)(F)F)C=C2)C(=O)C=C1 FENQDWMLJSTNRP-UHFFFAOYSA-N 0.000 description 1
- CUVCOZANPVODBG-UHFFFAOYSA-N CC1=CN(C2=CC=C(C(C)(C)C)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C(C)(C)C)C=C2)C(=O)C=C1 CUVCOZANPVODBG-UHFFFAOYSA-N 0.000 description 1
- UYWOQCPXDMYRKT-UHFFFAOYSA-N CC1=CN(C2=CC=C(C(C)(F)F)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C(C)(F)F)C=C2)C(=O)C=C1 UYWOQCPXDMYRKT-UHFFFAOYSA-N 0.000 description 1
- BWDUXQVQOMNYAZ-UHFFFAOYSA-N CC1=CN(C2=CC=C(C(F)(F)C(F)(F)F)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C(F)(F)C(F)(F)F)C=C2)C(=O)C=C1 BWDUXQVQOMNYAZ-UHFFFAOYSA-N 0.000 description 1
- ABZLYMASRYCZDM-UHFFFAOYSA-N CC1=CN(C2=CC=C(C(F)C(F)(F)F)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C(F)C(F)(F)F)C=C2)C(=O)C=C1 ABZLYMASRYCZDM-UHFFFAOYSA-N 0.000 description 1
- RYEWVWYUVFHOIG-UHFFFAOYSA-N CC1=CN(C2=CC=C(C(F)F)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C(F)F)C=C2)C(=O)C=C1 RYEWVWYUVFHOIG-UHFFFAOYSA-N 0.000 description 1
- SNYUXDDWRYXRCD-UHFFFAOYSA-N CC1=CN(C2=CC=C(C(O)(C(F)(F)F)C(F)(F)F)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C(O)(C(F)(F)F)C(F)(F)F)C=C2)C(=O)C=C1 SNYUXDDWRYXRCD-UHFFFAOYSA-N 0.000 description 1
- GEIRYRNIYKSXKR-UHFFFAOYSA-N CC1=CN(C2=CC=C(C(O)C(F)(F)F)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C(O)C(F)(F)F)C=C2)C(=O)C=C1 GEIRYRNIYKSXKR-UHFFFAOYSA-N 0.000 description 1
- RCFFXPCMJKQOFV-UHFFFAOYSA-N CC1=CN(C2=CC=C(C3=CC=CC=C3)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C3=CC=CC=C3)C=C2)C(=O)C=C1 RCFFXPCMJKQOFV-UHFFFAOYSA-N 0.000 description 1
- QXXFBSGFQDBPTF-UHFFFAOYSA-N CC1=CN(C2=CC=C(C3CCCCC3)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C3CCCCC3)C=C2)C(=O)C=C1 QXXFBSGFQDBPTF-UHFFFAOYSA-N 0.000 description 1
- AZKOOLALAJMZRW-UHFFFAOYSA-N CC1=CN(C2=CC=C(C=O)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(C=O)C=C2)C(=O)C=C1 AZKOOLALAJMZRW-UHFFFAOYSA-N 0.000 description 1
- WHHRHQAYJGLIBF-UHFFFAOYSA-N CC1=CN(C2=CC=C(CC(C)C)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(CC(C)C)C=C2)C(=O)C=C1 WHHRHQAYJGLIBF-UHFFFAOYSA-N 0.000 description 1
- KZESGAYSFUZRJO-UHFFFAOYSA-N CC1=CN(C2=CC=C(Cl)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(Cl)C=C2)C(=O)C=C1 KZESGAYSFUZRJO-UHFFFAOYSA-N 0.000 description 1
- OQMDNPAGUMKWKE-UHFFFAOYSA-N CC1=CN(C2=CC=C(Cl)C=C2)C(=O)C=C1.F Chemical compound CC1=CN(C2=CC=C(Cl)C=C2)C(=O)C=C1.F OQMDNPAGUMKWKE-UHFFFAOYSA-N 0.000 description 1
- QZOUMXAKIOWPLG-UHFFFAOYSA-N CC1=CN(C2=CC=C(N(C)C)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(N(C)C)C=C2)C(=O)C=C1 QZOUMXAKIOWPLG-UHFFFAOYSA-N 0.000 description 1
- LIZDKTCDGFZOER-UHFFFAOYSA-N CC1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C(Cl)=C1 Chemical compound CC1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C(Cl)=C1 LIZDKTCDGFZOER-UHFFFAOYSA-N 0.000 description 1
- XOUDHZGYKRJJKF-UHFFFAOYSA-N CC1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 XOUDHZGYKRJJKF-UHFFFAOYSA-N 0.000 description 1
- PBMZKKPUHCIAGD-UHFFFAOYSA-N CC1=CN(C2=CC=C(OC3=CC=CC=C3)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(OC3=CC=CC=C3)C=C2)C(=O)C=C1 PBMZKKPUHCIAGD-UHFFFAOYSA-N 0.000 description 1
- AIZOFIUTSSIOBR-UHFFFAOYSA-N CC1=CN(C2=CC=C(OCC3=CC=CC=C3)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(OCC3=CC=CC=C3)C=C2)C(=O)C=C1 AIZOFIUTSSIOBR-UHFFFAOYSA-N 0.000 description 1
- DUVBCRQQZMKDBA-UHFFFAOYSA-N CC1=CN(C2=CC=C(S(C)(=O)=O)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(S(C)(=O)=O)C=C2)C(=O)C=C1 DUVBCRQQZMKDBA-UHFFFAOYSA-N 0.000 description 1
- KJGBVWJOUIZZOO-UHFFFAOYSA-N CC1=CN(C2=CC=C(SC(C)C)C=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C(SC(C)C)C=C2)C(=O)C=C1 KJGBVWJOUIZZOO-UHFFFAOYSA-N 0.000 description 1
- HXBBAUQRRCICNX-UHFFFAOYSA-N CC1=CN(C2=CC=C3OCCOC3=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C3OCCOC3=C2)C(=O)C=C1 HXBBAUQRRCICNX-UHFFFAOYSA-N 0.000 description 1
- TXFRJCKCUTXTLK-UHFFFAOYSA-N CC1=CN(C2=CC=C3OCOC3=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=C3OCOC3=C2)C(=O)C=C1 TXFRJCKCUTXTLK-UHFFFAOYSA-N 0.000 description 1
- XUQZEWRNLTZRBK-UHFFFAOYSA-N CC1=CN(C2=CC=CC(C(C)(F)F)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(C(C)(F)F)=C2)C(=O)C=C1 XUQZEWRNLTZRBK-UHFFFAOYSA-N 0.000 description 1
- IOHVTSRZEOUPCB-UHFFFAOYSA-N CC1=CN(C2=CC=CC(C(F)F)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(C(F)F)=C2)C(=O)C=C1 IOHVTSRZEOUPCB-UHFFFAOYSA-N 0.000 description 1
- WYDNWLVKQFQESQ-UHFFFAOYSA-N CC1=CN(C2=CC=CC(C3=CC=CC=C3)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(C3=CC=CC=C3)=C2)C(=O)C=C1 WYDNWLVKQFQESQ-UHFFFAOYSA-N 0.000 description 1
- MACUBSHVKMSFML-UHFFFAOYSA-N CC1=CN(C2=CC=CC(Cl)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(Cl)=C2)C(=O)C=C1 MACUBSHVKMSFML-UHFFFAOYSA-N 0.000 description 1
- JDZYVVUJIQYGRX-UHFFFAOYSA-N CC1=CN(C2=CC=CC(F)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(F)=C2)C(=O)C=C1 JDZYVVUJIQYGRX-UHFFFAOYSA-N 0.000 description 1
- SKKBKUVQAFHQMF-UHFFFAOYSA-N CC1=CN(C2=CC=CC(N(C)C)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(N(C)C)=C2)C(=O)C=C1 SKKBKUVQAFHQMF-UHFFFAOYSA-N 0.000 description 1
- LLYSZFOHZAXQRD-UHFFFAOYSA-N CC1=CN(C2=CC=CC(O)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(O)=C2)C(=O)C=C1 LLYSZFOHZAXQRD-UHFFFAOYSA-N 0.000 description 1
- LRQRYLKHIJSYGH-UHFFFAOYSA-N CC1=CN(C2=CC=CC(OC(C)C)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(OC(C)C)=C2)C(=O)C=C1 LRQRYLKHIJSYGH-UHFFFAOYSA-N 0.000 description 1
- USRHWUQNSFPIDJ-UHFFFAOYSA-N CC1=CN(C2=CC=CC(OCC3=CC=CC=C3)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(OCC3=CC=CC=C3)=C2)C(=O)C=C1 USRHWUQNSFPIDJ-UHFFFAOYSA-N 0.000 description 1
- QRVXODFUZMIRQD-UHFFFAOYSA-N CC1=CN(C2=CC=CC(S(C)(=O)=O)=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC(S(C)(=O)=O)=C2)C(=O)C=C1 QRVXODFUZMIRQD-UHFFFAOYSA-N 0.000 description 1
- RXTODUWTKFLLKQ-UHFFFAOYSA-N CC1=CN(C2=CC=CC=C2)C(=O)C(Cl)=C1 Chemical compound CC1=CN(C2=CC=CC=C2)C(=O)C(Cl)=C1 RXTODUWTKFLLKQ-UHFFFAOYSA-N 0.000 description 1
- PRWIETUEFNQJSQ-UHFFFAOYSA-N CC1=CN(C2=CC=CC=C2)C(C)NC1=O Chemical compound CC1=CN(C2=CC=CC=C2)C(C)NC1=O PRWIETUEFNQJSQ-UHFFFAOYSA-N 0.000 description 1
- RRHMWKBGPCGEOI-UHFFFAOYSA-N CC1=CN(C2=CC=CC=C2C(F)(F)F)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC=C2C(F)(F)F)C(=O)C=C1 RRHMWKBGPCGEOI-UHFFFAOYSA-N 0.000 description 1
- VYXSRCVSKXRDAN-UHFFFAOYSA-N CC1=CN(C2=CC=CC=C2C)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC=C2C)C(=O)C=C1 VYXSRCVSKXRDAN-UHFFFAOYSA-N 0.000 description 1
- SSWPWTTWPDRNCD-UHFFFAOYSA-N CC1=CN(C2=CC=CC=C2C2=CC=CC=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC=C2C2=CC=CC=C2)C(=O)C=C1 SSWPWTTWPDRNCD-UHFFFAOYSA-N 0.000 description 1
- YFHFQXYJYNXDRA-UHFFFAOYSA-N CC1=CN(C2=CC=CC=C2Cl)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC=C2Cl)C(=O)C=C1 YFHFQXYJYNXDRA-UHFFFAOYSA-N 0.000 description 1
- KOPPMZQGQZIVND-UHFFFAOYSA-N CC1=CN(C2=CC=CC=C2O)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC=C2O)C(=O)C=C1 KOPPMZQGQZIVND-UHFFFAOYSA-N 0.000 description 1
- BHMTUDNKCGXNJM-UHFFFAOYSA-N CC1=CN(C2=CC=CC=C2OC(C)C)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC=C2OC(C)C)C(=O)C=C1 BHMTUDNKCGXNJM-UHFFFAOYSA-N 0.000 description 1
- QZRSJYUFGMGCBQ-UHFFFAOYSA-N CC1=CN(C2=CC=CC=C2OC2=CC=CC=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC=C2OC2=CC=CC=C2)C(=O)C=C1 QZRSJYUFGMGCBQ-UHFFFAOYSA-N 0.000 description 1
- GYYBTWWHWZSWSA-UHFFFAOYSA-N CC1=CN(C2=CC=CC=C2OCC2=CC=CC=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC=C2OCC2=CC=CC=C2)C(=O)C=C1 GYYBTWWHWZSWSA-UHFFFAOYSA-N 0.000 description 1
- BJJXCNNYFLIUIW-UHFFFAOYSA-N CC1=CN(C2=CC=CC=N2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=CC=N2)C(=O)C=C1 BJJXCNNYFLIUIW-UHFFFAOYSA-N 0.000 description 1
- LXAVUJBSQRLHMU-UHFFFAOYSA-N CC1=CN(C2=CC=NC=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CC=NC=C2)C(=O)C=C1 LXAVUJBSQRLHMU-UHFFFAOYSA-N 0.000 description 1
- XUEAITVYLOKCBX-UHFFFAOYSA-N CC1=CN(C2=CN=CN=C2)C(=O)C(Cl)=C1 Chemical compound CC1=CN(C2=CN=CN=C2)C(=O)C(Cl)=C1 XUEAITVYLOKCBX-UHFFFAOYSA-N 0.000 description 1
- QURLOEDJTRZTSU-UHFFFAOYSA-N CC1=CN(C2=CN=CN=C2)C(=O)C=C1 Chemical compound CC1=CN(C2=CN=CN=C2)C(=O)C=C1 QURLOEDJTRZTSU-UHFFFAOYSA-N 0.000 description 1
- UUVBODZPAWVKFO-UHFFFAOYSA-N CC1=CSC(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=C1 Chemical compound CC1=CSC(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=C1 UUVBODZPAWVKFO-UHFFFAOYSA-N 0.000 description 1
- ZYZHCFWCDHNLOH-UHFFFAOYSA-N CC1=NN(C)C(C)=C1B1OC(C)(C)C(C)(C)O1.CC1=NN(C)C(C)=C1C1=CNC(=O)C=C1.CCO.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(C)N(C)N=C2C)C=C1 Chemical compound CC1=NN(C)C(C)=C1B1OC(C)(C)C(C)(C)O1.CC1=NN(C)C(C)=C1C1=CNC(=O)C=C1.CCO.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(C)N(C)N=C2C)C=C1 ZYZHCFWCDHNLOH-UHFFFAOYSA-N 0.000 description 1
- GPODOGHKEDMKLO-UHFFFAOYSA-N CC1=NN(C)C(C)=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound CC1=NN(C)C(C)=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 GPODOGHKEDMKLO-UHFFFAOYSA-N 0.000 description 1
- COYLJFXQSYYVPJ-UHFFFAOYSA-N CC1=NN(C)C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=C1 Chemical compound CC1=NN(C)C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=C1 COYLJFXQSYYVPJ-UHFFFAOYSA-N 0.000 description 1
- NZXHSLUUYJFOKD-UHFFFAOYSA-N CC1=NNC(C)=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound CC1=NNC(C)=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 NZXHSLUUYJFOKD-UHFFFAOYSA-N 0.000 description 1
- CQYTXKPQRFLMPQ-UHFFFAOYSA-N CC1=NOC(C)=C1C1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 Chemical compound CC1=NOC(C)=C1C1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 CQYTXKPQRFLMPQ-UHFFFAOYSA-N 0.000 description 1
- NZTPDPNRKHQCSA-UHFFFAOYSA-N CC1=NOC(C)=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound CC1=NOC(C)=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 NZTPDPNRKHQCSA-UHFFFAOYSA-N 0.000 description 1
- KBUDITDELRMMKB-UHFFFAOYSA-N CC1=NOC(C)=C1C1=CN(C2=CC=NC=C2)C(=O)C=C1 Chemical compound CC1=NOC(C)=C1C1=CN(C2=CC=NC=C2)C(=O)C=C1 KBUDITDELRMMKB-UHFFFAOYSA-N 0.000 description 1
- VTDIFRFOVVWMTI-UHFFFAOYSA-N CC1=NOC(C)=C1C1=CN(C2=CN=CN=C2)C(=O)C=C1 Chemical compound CC1=NOC(C)=C1C1=CN(C2=CN=CN=C2)C(=O)C=C1 VTDIFRFOVVWMTI-UHFFFAOYSA-N 0.000 description 1
- BVKHQAWKMRHWMW-NERMPLDQSA-N CC1=[Y]C(C2=C(C3=CC=C(F)C=C3)N=CN2C2CCNCC2)=CC=N1.COC1=C2C(=NC(N)=C1)CC(C1=CC=C(F)C=C1)=C2C1=CC=NC=C1.C[C@H](NC1=CC(C2=C(C3=CC(C(F)(F)F)=CC=C3)N=C(C3CCNCC3)N2C)=CC=N1)C1=CC=CC=C1.C[S@H](O)C1=CC=C(C2=CC(C3=CC=NC=C3)=C(C3=CC=C(F)C=C3)C2)C=C1.FC1=CC=C(C2=C(C3=CC=NC=C3)N3CCSC3=N2)C=C1 Chemical compound CC1=[Y]C(C2=C(C3=CC=C(F)C=C3)N=CN2C2CCNCC2)=CC=N1.COC1=C2C(=NC(N)=C1)CC(C1=CC=C(F)C=C1)=C2C1=CC=NC=C1.C[C@H](NC1=CC(C2=C(C3=CC(C(F)(F)F)=CC=C3)N=C(C3CCNCC3)N2C)=CC=N1)C1=CC=CC=C1.C[S@H](O)C1=CC=C(C2=CC(C3=CC=NC=C3)=C(C3=CC=C(F)C=C3)C2)C=C1.FC1=CC=C(C2=C(C3=CC=NC=C3)N3CCSC3=N2)C=C1 BVKHQAWKMRHWMW-NERMPLDQSA-N 0.000 description 1
- JDXBTKBNEMYUPB-UHFFFAOYSA-N CCC1=CC(N2C=C(C)C=CC2=O)=CC=C1 Chemical compound CCC1=CC(N2C=C(C)C=CC2=O)=CC=C1 JDXBTKBNEMYUPB-UHFFFAOYSA-N 0.000 description 1
- CVEZCRFYNAOOJE-UHFFFAOYSA-N CCC1=CC=C(N2C=C(C(F)F)C=CC2=O)C=C1 Chemical compound CCC1=CC=C(N2C=C(C(F)F)C=CC2=O)C=C1 CVEZCRFYNAOOJE-UHFFFAOYSA-N 0.000 description 1
- PSADCXAAVHFKCC-UHFFFAOYSA-N CCC1=CC=C(N2C=C(CF)C=CC2=O)C=C1 Chemical compound CCC1=CC=C(N2C=C(CF)C=CC2=O)C=C1 PSADCXAAVHFKCC-UHFFFAOYSA-N 0.000 description 1
- OIEBZGFHQSUGFT-UHFFFAOYSA-N CCC1=CC=CC=C1N1C=C(C)C=CC1=O Chemical compound CCC1=CC=CC=C1N1C=C(C)C=CC1=O OIEBZGFHQSUGFT-UHFFFAOYSA-N 0.000 description 1
- VANOHWLVAFCHBO-UHFFFAOYSA-N CCC1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound CCC1=CN(C2=CC=CC=C2)C(=O)C=C1 VANOHWLVAFCHBO-UHFFFAOYSA-N 0.000 description 1
- XAALVNFFCNNDGX-UHFFFAOYSA-N CCCC[Sn](CCCC)(CCCC)C1=CN(C)N=C1C(F)(F)F.CN1C=C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)C(C(F)(F)F)=N1.N#N.O=C1C=CC(I)=CN1C1=CC=CC=C1 Chemical compound CCCC[Sn](CCCC)(CCCC)C1=CN(C)N=C1C(F)(F)F.CN1C=C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)C(C(F)(F)F)=N1.N#N.O=C1C=CC(I)=CN1C1=CC=CC=C1 XAALVNFFCNNDGX-UHFFFAOYSA-N 0.000 description 1
- MPJABXAGVNDNSQ-UHFFFAOYSA-N CCCC[Sn](CCCC)(CCCC)C1=NC=CO1.N#N.O=C1C=CC(C2=NC=CO2)=CN1C1=CC=CC=C1.O=C1C=CC(I)=CN1C1=CC=CC=C1 Chemical compound CCCC[Sn](CCCC)(CCCC)C1=NC=CO1.N#N.O=C1C=CC(C2=NC=CO2)=CN1C1=CC=CC=C1.O=C1C=CC(I)=CN1C1=CC=CC=C1 MPJABXAGVNDNSQ-UHFFFAOYSA-N 0.000 description 1
- VNYIOMXQMHLAQL-UHFFFAOYSA-N CCCC[Sn](CCCC)(CCCC)C1=NC=CS1.N#N.O=C1C=CC(C2=NC=CS2)=CN1C1=CC=C(OC(F)(F)F)C=C1.O=C1C=CC(I)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound CCCC[Sn](CCCC)(CCCC)C1=NC=CS1.N#N.O=C1C=CC(C2=NC=CS2)=CN1C1=CC=C(OC(F)(F)F)C=C1.O=C1C=CC(I)=CN1C1=CC=C(OC(F)(F)F)C=C1 VNYIOMXQMHLAQL-UHFFFAOYSA-N 0.000 description 1
- KJIZQEVSUUHUML-UHFFFAOYSA-N CCCC[Sn](CCCC)(CCCC)C1=NC=CS1.N#N.O=C1C=CC(C2=NC=CS2)=CN1C1=CC=NC=C1.O=C1C=CC(I)=CN1C1=CC=NC=C1 Chemical compound CCCC[Sn](CCCC)(CCCC)C1=NC=CS1.N#N.O=C1C=CC(C2=NC=CS2)=CN1C1=CC=NC=C1.O=C1C=CC(I)=CN1C1=CC=NC=C1 KJIZQEVSUUHUML-UHFFFAOYSA-N 0.000 description 1
- LGCOYJXBRPIYNS-UHFFFAOYSA-N CCOC(c([nH]1)cc(C=C2)c1N(c1ccccc1)C2=O)=O Chemical compound CCOC(c([nH]1)cc(C=C2)c1N(c1ccccc1)C2=O)=O LGCOYJXBRPIYNS-UHFFFAOYSA-N 0.000 description 1
- FSKFEYOTQPJVMD-UHFFFAOYSA-N CCOC1=CC(N2C=C(C)C=CC2=O)=CC=C1 Chemical compound CCOC1=CC(N2C=C(C)C=CC2=O)=CC=C1 FSKFEYOTQPJVMD-UHFFFAOYSA-N 0.000 description 1
- QRDXZUJBYMZYLB-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C)C=CC2=O)C(C)=C1 Chemical compound CCOC1=CC=C(N2C=C(C)C=CC2=O)C(C)=C1 QRDXZUJBYMZYLB-UHFFFAOYSA-N 0.000 description 1
- IKTZNGQRWWEBPS-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C)C=CC2=O)C=C1Cl Chemical compound CCOC1=CC=C(N2C=C(C)C=CC2=O)C=C1Cl IKTZNGQRWWEBPS-UHFFFAOYSA-N 0.000 description 1
- GLKXIBWLCWDUSY-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C)C=CC2=O)C=C1F Chemical compound CCOC1=CC=C(N2C=C(C)C=CC2=O)C=C1F GLKXIBWLCWDUSY-UHFFFAOYSA-N 0.000 description 1
- LIUFVXDXMVQKPK-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C3=CC=C(CO)C=C3)C=CC2=O)C(C)=C1 Chemical compound CCOC1=CC=C(N2C=C(C3=CC=C(CO)C=C3)C=CC2=O)C(C)=C1 LIUFVXDXMVQKPK-UHFFFAOYSA-N 0.000 description 1
- RTBNPJDUCJISCZ-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C3=CC=C(Cl)C=C3)C=CC2=O)C(C)=C1 Chemical compound CCOC1=CC=C(N2C=C(C3=CC=C(Cl)C=C3)C=CC2=O)C(C)=C1 RTBNPJDUCJISCZ-UHFFFAOYSA-N 0.000 description 1
- NTTFXUNFDNCRAR-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)C=C1 Chemical compound CCOC1=CC=C(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)C=C1 NTTFXUNFDNCRAR-UHFFFAOYSA-N 0.000 description 1
- UIBWDARHYCOSJK-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C3=CC=CC(CO)=C3)C=CC2=O)C(C)=C1 Chemical compound CCOC1=CC=C(N2C=C(C3=CC=CC(CO)=C3)C=CC2=O)C(C)=C1 UIBWDARHYCOSJK-UHFFFAOYSA-N 0.000 description 1
- LUCJRMYXZPTHMA-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C3=CC=CC=C3F)C=CC2=O)C(C)=C1 Chemical compound CCOC1=CC=C(N2C=C(C3=CC=CC=C3F)C=CC2=O)C(C)=C1 LUCJRMYXZPTHMA-UHFFFAOYSA-N 0.000 description 1
- QVBFZBPATBANQK-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C3=CC=CC=C3OC)C=CC2=O)C(C)=C1 Chemical compound CCOC1=CC=C(N2C=C(C3=CC=CC=C3OC)C=CC2=O)C(C)=C1 QVBFZBPATBANQK-UHFFFAOYSA-N 0.000 description 1
- ZCJWFMKDNMYCKK-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C3=CN(C)N=C3)C(C)=CC2=O)C(C)=C1 Chemical compound CCOC1=CC=C(N2C=C(C3=CN(C)N=C3)C(C)=CC2=O)C(C)=C1 ZCJWFMKDNMYCKK-UHFFFAOYSA-N 0.000 description 1
- DWMXNLFXOAYXFK-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C3=CN(C)N=C3)C=CC2=O)C(C)=C1.CN1C=C(B2OC(C)(C)C(C)(C)O2)C=N1.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CN(C)N=C2)C=C1 Chemical compound CCOC1=CC=C(N2C=C(C3=CN(C)N=C3)C=CC2=O)C(C)=C1.CN1C=C(B2OC(C)(C)C(C)(C)O2)C=N1.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CN(C)N=C2)C=C1 DWMXNLFXOAYXFK-UHFFFAOYSA-N 0.000 description 1
- LNIWDWWQNXXANB-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(C3=CN=CN=C3)C=CC2=O)C(C)=C1 Chemical compound CCOC1=CC=C(N2C=C(C3=CN=CN=C3)C=CC2=O)C(C)=C1 LNIWDWWQNXXANB-UHFFFAOYSA-N 0.000 description 1
- LVRKYFLRCIEOOG-UHFFFAOYSA-N CCOC1=CC=C(N2C=C(N[SH](=O)=O)C=CC2=O)C(C)=C1 Chemical compound CCOC1=CC=C(N2C=C(N[SH](=O)=O)C=CC2=O)C(C)=C1 LVRKYFLRCIEOOG-UHFFFAOYSA-N 0.000 description 1
- YPIXKLYHUOHDKG-UHFFFAOYSA-N CCOC1=CC=CC(C2=CN(C3=CC(C)=CC=C3)C(=O)C=C2)=C1 Chemical compound CCOC1=CC=CC(C2=CN(C3=CC(C)=CC=C3)C(=O)C=C2)=C1 YPIXKLYHUOHDKG-UHFFFAOYSA-N 0.000 description 1
- DEWMFGALALGPKP-UHFFFAOYSA-N CCOc(cc1C)ccc1N(C=C(C=C1)c(cccc2)c2C(C)=O)C1=O Chemical compound CCOc(cc1C)ccc1N(C=C(C=C1)c(cccc2)c2C(C)=O)C1=O DEWMFGALALGPKP-UHFFFAOYSA-N 0.000 description 1
- XXFJESILQAIWCR-OGKIIUBNSA-M CCS[Na].COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=CO2)C=C1.O=C1C=CC(C2=CC=CO2)=CN1.OB(O)C1=CC=CO1.[2H]CF Chemical compound CCS[Na].COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=CO2)C=C1.O=C1C=CC(C2=CC=CO2)=CN1.OB(O)C1=CC=CO1.[2H]CF XXFJESILQAIWCR-OGKIIUBNSA-M 0.000 description 1
- HWTAAHAHWRYYJW-UHFFFAOYSA-M CCS[Na].COC1=NC=C(Br)C=C1.COC1=NC=C(C2CC2)C=C1.O=C1C=CC(C2CC2)=CN1.OB(O)C1CC1 Chemical compound CCS[Na].COC1=NC=C(Br)C=C1.COC1=NC=C(C2CC2)C=C1.O=C1C=CC(C2CC2)=CN1.OB(O)C1CC1 HWTAAHAHWRYYJW-UHFFFAOYSA-M 0.000 description 1
- XERHDMJFTRHZFI-UHFFFAOYSA-N CFC1=CN(C2=CC=C(OC(C)C)C=C2)C(=O)C=C1 Chemical compound CFC1=CN(C2=CC=C(OC(C)C)C=C2)C(=O)C=C1 XERHDMJFTRHZFI-UHFFFAOYSA-N 0.000 description 1
- CNQPROBYLBERPL-UHFFFAOYSA-N CN(C(=O)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2)C1=CC=CC=C1 Chemical compound CN(C(=O)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2)C1=CC=CC=C1 CNQPROBYLBERPL-UHFFFAOYSA-N 0.000 description 1
- AXZQBOQQEZDTTR-UHFFFAOYSA-N CN(C)C1=CC(N2C=C(C(F)F)C=CC2=O)=CC=C1 Chemical compound CN(C)C1=CC(N2C=C(C(F)F)C=CC2=O)=CC=C1 AXZQBOQQEZDTTR-UHFFFAOYSA-N 0.000 description 1
- DULHTPJBEJGSCF-UHFFFAOYSA-N CN(C)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 Chemical compound CN(C)C1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 DULHTPJBEJGSCF-UHFFFAOYSA-N 0.000 description 1
- KZJZPWSZYKJDLV-UHFFFAOYSA-N CN(C)C1=CC(N2C=CC(C(F)(F)F)=CC2=O)=CC=C1 Chemical compound CN(C)C1=CC(N2C=CC(C(F)(F)F)=CC2=O)=CC=C1 KZJZPWSZYKJDLV-UHFFFAOYSA-N 0.000 description 1
- SQUHKGXCMZQJCM-UHFFFAOYSA-N CN(C)C1=CC(N2C=CC(C(F)F)=CC2=O)=CC=C1.O=C1C=CC=CN1C1=CC=CC=C1 Chemical compound CN(C)C1=CC(N2C=CC(C(F)F)=CC2=O)=CC=C1.O=C1C=CC=CN1C1=CC=CC=C1 SQUHKGXCMZQJCM-UHFFFAOYSA-N 0.000 description 1
- JOLLVPGCQNEITL-UHFFFAOYSA-N CN(C)CC1=CC=C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)C=C1 Chemical compound CN(C)CC1=CC=C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)C=C1 JOLLVPGCQNEITL-UHFFFAOYSA-N 0.000 description 1
- IKPGSWGWHFGNAE-UHFFFAOYSA-N CN(C)S(=O)(=O)C1=CC=CC(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=C1 Chemical compound CN(C)S(=O)(=O)C1=CC=CC(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=C1 IKPGSWGWHFGNAE-UHFFFAOYSA-N 0.000 description 1
- OALIEXKAZCTRLU-UHFFFAOYSA-N CN(C)S(=O)(=O)C1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 Chemical compound CN(C)S(=O)(=O)C1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 OALIEXKAZCTRLU-UHFFFAOYSA-N 0.000 description 1
- FOLQHPRJZAQWLT-UHFFFAOYSA-N CN(C)S(=O)(=O)C1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound CN(C)S(=O)(=O)C1=CN(C2=CC=CC=C2)C(=O)C=C1 FOLQHPRJZAQWLT-UHFFFAOYSA-N 0.000 description 1
- NZSXYCVENLWBIY-UHFFFAOYSA-N CN1C(C(=O)NC2=NC=CS2)=CC2=C1N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound CN1C(C(=O)NC2=NC=CS2)=CC2=C1N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 NZSXYCVENLWBIY-UHFFFAOYSA-N 0.000 description 1
- QNNAFELXJYFQPW-UHFFFAOYSA-N CN1C(C(=O)O)=CC2=C1N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound CN1C(C(=O)O)=CC2=C1N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 QNNAFELXJYFQPW-UHFFFAOYSA-N 0.000 description 1
- ZQPGZVXJCHADAW-UHFFFAOYSA-N CN1C=C(C2=CN(C3=CC=C(OC(F)(F)F)C=C3)C(=O)C=C2)C=N1 Chemical compound CN1C=C(C2=CN(C3=CC=C(OC(F)(F)F)C=C3)C(=O)C=C2)C=N1 ZQPGZVXJCHADAW-UHFFFAOYSA-N 0.000 description 1
- SNCPPHYJUZWQAD-UHFFFAOYSA-N CN1C=C(C2=CN(C3=CC=C(S(N)(=O)=O)C=C3)C(=O)C=C2)C=N1 Chemical compound CN1C=C(C2=CN(C3=CC=C(S(N)(=O)=O)C=C3)C(=O)C=C2)C=N1 SNCPPHYJUZWQAD-UHFFFAOYSA-N 0.000 description 1
- DMMANHCURJEECJ-UHFFFAOYSA-N CN1C=C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)C=N1 Chemical compound CN1C=C(C2=CN(C3=CC=CC=C3)C(=O)C=C2)C=N1 DMMANHCURJEECJ-UHFFFAOYSA-N 0.000 description 1
- SBKLMLRENOCDNI-UHFFFAOYSA-N CN1C=C(C2=CN(C3=CC=NC=C3)C(=O)C=C2)C=N1 Chemical compound CN1C=C(C2=CN(C3=CC=NC=C3)C(=O)C=C2)C=N1 SBKLMLRENOCDNI-UHFFFAOYSA-N 0.000 description 1
- WMYLPEGUAHTZJN-UHFFFAOYSA-N CN1C=C(C2=CN(C3=CN=CN=C3)C(=O)C=C2)C=N1 Chemical compound CN1C=C(C2=CN(C3=CN=CN=C3)C(=O)C=C2)C=N1 WMYLPEGUAHTZJN-UHFFFAOYSA-N 0.000 description 1
- BWHMNSOQZWBGNY-UHFFFAOYSA-N CN1C=CC2=CC(C3=CN(C4=CC=CC=C4)C(=O)C=C3)=CC=C21 Chemical compound CN1C=CC2=CC(C3=CN(C4=CC=CC=C4)C(=O)C=C3)=CC=C21 BWHMNSOQZWBGNY-UHFFFAOYSA-N 0.000 description 1
- RGIRWQAPNXZBLX-UHFFFAOYSA-N CN1CCN(C(=O)C2=CC3=C(N2)N(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C3)CC1 Chemical compound CN1CCN(C(=O)C2=CC3=C(N2)N(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C3)CC1 RGIRWQAPNXZBLX-UHFFFAOYSA-N 0.000 description 1
- WMIRJJMVIKWJFW-UHFFFAOYSA-N CN1CCN(C(=O)C2=CC3=C(N2)N(C2=CC=C(F)C=C2)C(=O)C=C3)CC1 Chemical compound CN1CCN(C(=O)C2=CC3=C(N2)N(C2=CC=C(F)C=C2)C(=O)C=C3)CC1 WMIRJJMVIKWJFW-UHFFFAOYSA-N 0.000 description 1
- PGQWQKYZXWWITA-UHFFFAOYSA-N CN1CCN(C(=O)C2=CC3=C(N2)N(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C3)CC1.COC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N2CCN(C)CC2)=C3)C=C1 Chemical compound CN1CCN(C(=O)C2=CC3=C(N2)N(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C3)CC1.COC1=CC=C(N2C(=O)C=CC3=C2NC(C(=O)N2CCN(C)CC2)=C3)C=C1 PGQWQKYZXWWITA-UHFFFAOYSA-N 0.000 description 1
- WJBZOBIZRQFVAZ-UHFFFAOYSA-N CN1N=CC=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound CN1N=CC=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 WJBZOBIZRQFVAZ-UHFFFAOYSA-N 0.000 description 1
- LUAZFUMMEFJGBV-UHFFFAOYSA-N CNC(=O)C1=CC2=C(N1)C(C1=CC(O)=CC=C1)C(=O)C=C2 Chemical compound CNC(=O)C1=CC2=C(N1)C(C1=CC(O)=CC=C1)C(=O)C=C2 LUAZFUMMEFJGBV-UHFFFAOYSA-N 0.000 description 1
- VKPJRMQOHHDTQO-UHFFFAOYSA-N CNC(=O)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound CNC(=O)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 VKPJRMQOHHDTQO-UHFFFAOYSA-N 0.000 description 1
- RFXNCAQVASYSGA-UHFFFAOYSA-N CNC(=O)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 Chemical compound CNC(=O)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 RFXNCAQVASYSGA-UHFFFAOYSA-N 0.000 description 1
- WODSEQAAIKYIAC-UHFFFAOYSA-N CNC(=O)C1=CC2=C(N1)N(C1=CC=C(OC(C)C)C=C1)C(=O)C=C2 Chemical compound CNC(=O)C1=CC2=C(N1)N(C1=CC=C(OC(C)C)C=C1)C(=O)C=C2 WODSEQAAIKYIAC-UHFFFAOYSA-N 0.000 description 1
- SCODMJOQVFKHAM-UHFFFAOYSA-N CNC(=O)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound CNC(=O)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 SCODMJOQVFKHAM-UHFFFAOYSA-N 0.000 description 1
- XTFRMQCHRVEWHR-UHFFFAOYSA-N COC1=C(C2=CN([Ar])C(=O)C=C2)C=CC=C1.COC1=C(C2=CNC(=O)C=C2)C=CC=C1.COC1=CC=CC=C1B(O)O.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(OC)C=CC=C2)C=C1 Chemical compound COC1=C(C2=CN([Ar])C(=O)C=C2)C=CC=C1.COC1=C(C2=CNC(=O)C=C2)C=CC=C1.COC1=CC=CC=C1B(O)O.COC1=NC=C(Br)C=C1.COC1=NC=C(C2=C(OC)C=CC=C2)C=C1 XTFRMQCHRVEWHR-UHFFFAOYSA-N 0.000 description 1
- MEEHKRPRDZXAIY-UHFFFAOYSA-N COC1=C(F)C=C(NC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(C)C)C=C2)C(=O)C=C3)C=C1 Chemical compound COC1=C(F)C=C(NC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(C)C)C=C2)C(=O)C=C3)C=C1 MEEHKRPRDZXAIY-UHFFFAOYSA-N 0.000 description 1
- ACXUKSXBXYXYPY-UHFFFAOYSA-N COC1=CC(CNC(=O)C2=CC3=C(N2)N(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C3)=CC=C1 Chemical compound COC1=CC(CNC(=O)C2=CC3=C(N2)N(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C3)=CC=C1 ACXUKSXBXYXYPY-UHFFFAOYSA-N 0.000 description 1
- CIXMLZFGWKVRQS-UHFFFAOYSA-N COC1=CC(CNC(=O)C2=CC3=C(N2)N(C2=CC=C(Cl)C=C2)C(=O)C=C3)=CC=C1 Chemical compound COC1=CC(CNC(=O)C2=CC3=C(N2)N(C2=CC=C(Cl)C=C2)C(=O)C=C3)=CC=C1 CIXMLZFGWKVRQS-UHFFFAOYSA-N 0.000 description 1
- TUBIPDMNYGCHBH-UHFFFAOYSA-N COC1=CC(CNC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C3)=CC=C1 Chemical compound COC1=CC(CNC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C3)=CC=C1 TUBIPDMNYGCHBH-UHFFFAOYSA-N 0.000 description 1
- LNDXEMIYLOJHHE-UHFFFAOYSA-N COC1=CC(N2C(=O)C=CC3=C2NC(C(=O)O)=C3)=CC=C1 Chemical compound COC1=CC(N2C(=O)C=CC3=C2NC(C(=O)O)=C3)=CC=C1 LNDXEMIYLOJHHE-UHFFFAOYSA-N 0.000 description 1
- YMKPPOITVHTKFO-UHFFFAOYSA-N COC1=CC(N2C=C(C)C=CC2=O)=CC(CO)=C1OC Chemical compound COC1=CC(N2C=C(C)C=CC2=O)=CC(CO)=C1OC YMKPPOITVHTKFO-UHFFFAOYSA-N 0.000 description 1
- NUUFOILDYXVXLL-UHFFFAOYSA-N COC1=CC(N2C=C(C)C=CC2=O)=CC=C1 Chemical compound COC1=CC(N2C=C(C)C=CC2=O)=CC=C1 NUUFOILDYXVXLL-UHFFFAOYSA-N 0.000 description 1
- YSIHFAJWAGCJMJ-UHFFFAOYSA-N COC1=CC(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1 Chemical compound COC1=CC(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)=CC=C1 YSIHFAJWAGCJMJ-UHFFFAOYSA-N 0.000 description 1
- CFLAUZSFDIRHKF-UHFFFAOYSA-N COC1=CC=C(N2C=C(C)C=CC2=O)C(C)=C1 Chemical compound COC1=CC=C(N2C=C(C)C=CC2=O)C(C)=C1 CFLAUZSFDIRHKF-UHFFFAOYSA-N 0.000 description 1
- XMDITNSOBKTMIO-UHFFFAOYSA-N COC1=CC=C(N2C=C(C)C=CC2=O)C=C1 Chemical compound COC1=CC=C(N2C=C(C)C=CC2=O)C=C1 XMDITNSOBKTMIO-UHFFFAOYSA-N 0.000 description 1
- DBMFSBOHDGSKGH-UHFFFAOYSA-N COC1=CC=C(N2C=C(C)C=CC2=O)C=C1C Chemical compound COC1=CC=C(N2C=C(C)C=CC2=O)C=C1C DBMFSBOHDGSKGH-UHFFFAOYSA-N 0.000 description 1
- QWSPDEZQOQLIJX-UHFFFAOYSA-N COC1=CC=C(N2C=C(C)C=CC2=O)C=C1C=O Chemical compound COC1=CC=C(N2C=C(C)C=CC2=O)C=C1C=O QWSPDEZQOQLIJX-UHFFFAOYSA-N 0.000 description 1
- SNDXNXFCVRIYBT-UHFFFAOYSA-N COC1=CC=C(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)C=C1 Chemical compound COC1=CC=C(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)C=C1 SNDXNXFCVRIYBT-UHFFFAOYSA-N 0.000 description 1
- BFYWEBLYBVPPNY-UHFFFAOYSA-N COC1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 Chemical compound COC1=CC=C(N2C=C(C3=CC=CC=C3)C=CC2=O)C=C1 BFYWEBLYBVPPNY-UHFFFAOYSA-N 0.000 description 1
- NZDFKACTNSNACF-UHFFFAOYSA-N COC1=CC=C(N2C=CC(C(F)(F)F)=CC2=O)C=C1 Chemical compound COC1=CC=C(N2C=CC(C(F)(F)F)=CC2=O)C=C1 NZDFKACTNSNACF-UHFFFAOYSA-N 0.000 description 1
- KGCCXYPIANVDHW-UHFFFAOYSA-N COC1=CC=C(N2C=CC(C(F)F)=CC2=O)C=C1 Chemical compound COC1=CC=C(N2C=CC(C(F)F)=CC2=O)C=C1 KGCCXYPIANVDHW-UHFFFAOYSA-N 0.000 description 1
- QOZZMYRDBQWNLV-UHFFFAOYSA-N COC1=CC=CC(CNC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(C)C)C=C2)C(=O)C=C3)=C1 Chemical compound COC1=CC=CC(CNC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(C)C)C=C2)C(=O)C=C3)=C1 QOZZMYRDBQWNLV-UHFFFAOYSA-N 0.000 description 1
- QNYHDMHKPLWNRJ-UHFFFAOYSA-N COC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C3)=C1 Chemical compound COC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C3)=C1 QNYHDMHKPLWNRJ-UHFFFAOYSA-N 0.000 description 1
- AUSOARXRTJZUAJ-UHFFFAOYSA-N COC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(C)C)C=C2)C(=O)C=C3)=C1 Chemical compound COC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(C)C)C=C2)C(=O)C=C3)=C1 AUSOARXRTJZUAJ-UHFFFAOYSA-N 0.000 description 1
- YMMMSOVDXNIXCF-UHFFFAOYSA-N COC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C3)=C1 Chemical compound COC1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C3)=C1 YMMMSOVDXNIXCF-UHFFFAOYSA-N 0.000 description 1
- RVQVWQLDZYWSPA-UHFFFAOYSA-N COC1=CC=CC=C1C1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 Chemical compound COC1=CC=CC=C1C1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 RVQVWQLDZYWSPA-UHFFFAOYSA-N 0.000 description 1
- NFYUWPRPEQYRPV-UHFFFAOYSA-N COC1=CC=CC=C1C1=CN(C2=CC=CC(NC(C)=O)=C2)C(=O)C=C1 Chemical compound COC1=CC=CC=C1C1=CN(C2=CC=CC(NC(C)=O)=C2)C(=O)C=C1 NFYUWPRPEQYRPV-UHFFFAOYSA-N 0.000 description 1
- WAPWYJYNFDUDGF-UHFFFAOYSA-N COC1=CC=CC=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound COC1=CC=CC=C1C1=CN(C2=CC=CC=C2)C(=O)C=C1 WAPWYJYNFDUDGF-UHFFFAOYSA-N 0.000 description 1
- HJZZWZBSSKITRG-UHFFFAOYSA-N COC1=CC=CC=C1C1=CN(C2=CC=NC=C2)C(=O)C=C1 Chemical compound COC1=CC=CC=C1C1=CN(C2=CC=NC=C2)C(=O)C=C1 HJZZWZBSSKITRG-UHFFFAOYSA-N 0.000 description 1
- MJWRQZILSOKOTB-UHFFFAOYSA-N COC1=CC=CC=C1N1C=C(C)C=CC1=O Chemical compound COC1=CC=CC=C1N1C=C(C)C=CC1=O MJWRQZILSOKOTB-UHFFFAOYSA-N 0.000 description 1
- FGPGLHCBGSVHLI-UHFFFAOYSA-N COC1=CC=CC=C1N1C=C(C2=CC=CC=C2)C=CC1=O Chemical compound COC1=CC=CC=C1N1C=C(C2=CC=CC=C2)C=CC1=O FGPGLHCBGSVHLI-UHFFFAOYSA-N 0.000 description 1
- BDWJCFRIHFDQET-UHFFFAOYSA-N COC1=CC=CC=C1N1C=CC(C(F)(F)F)=CC1=O Chemical compound COC1=CC=CC=C1N1C=CC(C(F)(F)F)=CC1=O BDWJCFRIHFDQET-UHFFFAOYSA-N 0.000 description 1
- CQHGIZNCUIJKFX-UHFFFAOYSA-N COC1=CC=CN(C2=CC=CC=C2)C1=O Chemical compound COC1=CC=CN(C2=CC=CC=C2)C1=O CQHGIZNCUIJKFX-UHFFFAOYSA-N 0.000 description 1
- RHTUYRNDPZWIOU-UHFFFAOYSA-N COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC(F)=CC=C2)C=C1.O=C1C=CC(C2=CC(F)=CC=C2)=CN1.O=C1C=CC(C2=CC(F)=CC=C2)=CN1[Ar].OBO.[Ar] Chemical compound COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC(F)=CC=C2)C=C1.O=C1C=CC(C2=CC(F)=CC=C2)=CN1.O=C1C=CC(C2=CC(F)=CC=C2)=CN1[Ar].OBO.[Ar] RHTUYRNDPZWIOU-UHFFFAOYSA-N 0.000 description 1
- GLAPECXQSJIFQP-UHFFFAOYSA-N COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=C(F)C=C2)C=C1.O=C1C=CC(C2=CC=C(F)C=C2)=CN1.OB(O)C1=CC=C(F)C=C1 Chemical compound COC1=NC=C(Br)C=C1.COC1=NC=C(C2=CC=C(F)C=C2)C=C1.O=C1C=CC(C2=CC=C(F)C=C2)=CN1.OB(O)C1=CC=C(F)C=C1 GLAPECXQSJIFQP-UHFFFAOYSA-N 0.000 description 1
- FJNARCCMMVFFMS-UHFFFAOYSA-N COC1=NC=C(N)C=C1.COC1=NC=C(S(=O)(=O)Cl)C=C1.COC1=NC=C(S(N)(=O)=O)C=C1.N Chemical compound COC1=NC=C(N)C=C1.COC1=NC=C(S(=O)(=O)Cl)C=C1.COC1=NC=C(S(N)(=O)=O)C=C1.N FJNARCCMMVFFMS-UHFFFAOYSA-N 0.000 description 1
- HMSJWHKZNQBTEP-UHFFFAOYSA-N COCC1=CC(N2C=C(C)C=CC2=O)=CC=C1 Chemical compound COCC1=CC(N2C=C(C)C=CC2=O)=CC=C1 HMSJWHKZNQBTEP-UHFFFAOYSA-N 0.000 description 1
- JOBDKGRSANDMPH-UHFFFAOYSA-N COCC1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 Chemical compound COCC1=CC(N2C=C(C3=CC=CC=C3)C=CC2=O)=CC=C1 JOBDKGRSANDMPH-UHFFFAOYSA-N 0.000 description 1
- BUQQUZLNAJQAJC-UHFFFAOYSA-N COCC1=CC(N2C=CC(C(F)(F)F)=CC2=O)=CC=C1 Chemical compound COCC1=CC(N2C=CC(C(F)(F)F)=CC2=O)=CC=C1 BUQQUZLNAJQAJC-UHFFFAOYSA-N 0.000 description 1
- PANQGOFNMRYSPY-UHFFFAOYSA-N COCC1=CC=C(N2C=C(C)C=CC2=O)C=C1 Chemical compound COCC1=CC=C(N2C=C(C)C=CC2=O)C=C1 PANQGOFNMRYSPY-UHFFFAOYSA-N 0.000 description 1
- IPWFEWRONIOGFV-UHFFFAOYSA-N COc(cc1)ncc1-c1ccncc1 Chemical compound COc(cc1)ncc1-c1ccncc1 IPWFEWRONIOGFV-UHFFFAOYSA-N 0.000 description 1
- OMLIVJOTRYWHNY-UHFFFAOYSA-N COc(cc1)ncc1S(Cl)(=O)=O Chemical compound COc(cc1)ncc1S(Cl)(=O)=O OMLIVJOTRYWHNY-UHFFFAOYSA-N 0.000 description 1
- RDPYEAKRRIRVQI-UHFFFAOYSA-N COc(cc1)ncc1S(N)(=O)=O Chemical compound COc(cc1)ncc1S(N)(=O)=O RDPYEAKRRIRVQI-UHFFFAOYSA-N 0.000 description 1
- GAUAFWJVOYEYFH-UHFFFAOYSA-N COc(nc1)ccc1C(C=C1)=CN(c2ccccc2)C1=O Chemical compound COc(nc1)ccc1C(C=C1)=CN(c2ccccc2)C1=O GAUAFWJVOYEYFH-UHFFFAOYSA-N 0.000 description 1
- UUVDJIWRSIJEBS-UHFFFAOYSA-N COc(nc1)ccc1N Chemical compound COc(nc1)ccc1N UUVDJIWRSIJEBS-UHFFFAOYSA-N 0.000 description 1
- WNVQCAKDASVGKR-UHFFFAOYSA-N CS(=O)(=O)C1=CC(N2C=C(C(F)F)C=CC2=O)=CC=C1 Chemical compound CS(=O)(=O)C1=CC(N2C=C(C(F)F)C=CC2=O)=CC=C1 WNVQCAKDASVGKR-UHFFFAOYSA-N 0.000 description 1
- PZHJCILCZPKCOJ-UHFFFAOYSA-N CS(=O)(=O)C1=CC(N2C=CC(C(F)F)=CC2=O)=CC=C1 Chemical compound CS(=O)(=O)C1=CC(N2C=CC(C(F)F)=CC2=O)=CC=C1 PZHJCILCZPKCOJ-UHFFFAOYSA-N 0.000 description 1
- BOLIKSCLAQLPLO-UHFFFAOYSA-N CS(=O)(=O)C1=CC=C(N2C=C(C(F)F)C=CC2=O)C=C1 Chemical compound CS(=O)(=O)C1=CC=C(N2C=C(C(F)F)C=CC2=O)C=C1 BOLIKSCLAQLPLO-UHFFFAOYSA-N 0.000 description 1
- CXEJHZFNGDNORB-UHFFFAOYSA-N CS(=O)(=O)C1=CC=C(N2C=CC(C(F)F)=CC2=O)C=C1 Chemical compound CS(=O)(=O)C1=CC=C(N2C=CC(C(F)F)=CC2=O)C=C1 CXEJHZFNGDNORB-UHFFFAOYSA-N 0.000 description 1
- MAVLXCAJDPYVPK-UHFFFAOYSA-N CS(=O)(=O)C1=CC=CC(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=C1 Chemical compound CS(=O)(=O)C1=CC=CC(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=C1 MAVLXCAJDPYVPK-UHFFFAOYSA-N 0.000 description 1
- RNOFKGJCCVUCDZ-UHFFFAOYSA-N CS(=O)(=O)C1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C3)=C1 Chemical compound CS(=O)(=O)C1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(C(F)(F)F)C=C2)C(=O)C=C3)=C1 RNOFKGJCCVUCDZ-UHFFFAOYSA-N 0.000 description 1
- SNDCVYKALDZJBJ-UHFFFAOYSA-N CS(=O)(=O)C1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(Cl)C=C2)C(=O)C=C3)=C1 Chemical compound CS(=O)(=O)C1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(Cl)C=C2)C(=O)C=C3)=C1 SNDCVYKALDZJBJ-UHFFFAOYSA-N 0.000 description 1
- RMOODPVSQIELPP-UHFFFAOYSA-N CS(=O)(=O)C1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C3)=C1 Chemical compound CS(=O)(=O)C1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C3)=C1 RMOODPVSQIELPP-UHFFFAOYSA-N 0.000 description 1
- VOGUIBDHPLEEAK-UHFFFAOYSA-N CSC1=CC(N2C=C(C)C=CC2=O)=CC=C1 Chemical compound CSC1=CC(N2C=C(C)C=CC2=O)=CC=C1 VOGUIBDHPLEEAK-UHFFFAOYSA-N 0.000 description 1
- OEDGZSCTEQRGCZ-UHFFFAOYSA-N CSC1=CC=C(N2C=C(C(F)F)C=CC2=O)C=C1 Chemical compound CSC1=CC=C(N2C=C(C(F)F)C=CC2=O)C=C1 OEDGZSCTEQRGCZ-UHFFFAOYSA-N 0.000 description 1
- WQDUUOGJLFTGHW-UHFFFAOYSA-N CSC1=CC=C(N2C=C(CF)C=CC2=O)C=C1 Chemical compound CSC1=CC=C(N2C=C(CF)C=CC2=O)C=C1 WQDUUOGJLFTGHW-UHFFFAOYSA-N 0.000 description 1
- WQHNCVPSVBQFGT-UHFFFAOYSA-N CSC1=CC=CC=C1N1C=C(C)C=CC1=O Chemical compound CSC1=CC=CC=C1N1C=C(C)C=CC1=O WQHNCVPSVBQFGT-UHFFFAOYSA-N 0.000 description 1
- UMELMYFEIWUROC-UHFFFAOYSA-N N#CC1=CC=CN(C2=CC=CC=C2)C1=O Chemical compound N#CC1=CC=CN(C2=CC=CC=C2)C1=O UMELMYFEIWUROC-UHFFFAOYSA-N 0.000 description 1
- NLLAEARKYSJHFN-UHFFFAOYSA-N NC1=NC(=O)N(C2=CC=CC=C2)C=N1 Chemical compound NC1=NC(=O)N(C2=CC=CC=C2)C=N1 NLLAEARKYSJHFN-UHFFFAOYSA-N 0.000 description 1
- RSMNBJUUUDBQCA-UHFFFAOYSA-N NOOSC1=CC=C(C2=CN(C3=CN=CN=C3)C(=O)C=C2)C=C1 Chemical compound NOOSC1=CC=C(C2=CN(C3=CN=CN=C3)C(=O)C=C2)C=C1 RSMNBJUUUDBQCA-UHFFFAOYSA-N 0.000 description 1
- ZPIGWSFGYYHKAG-UHFFFAOYSA-N NOOSC1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 Chemical compound NOOSC1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 ZPIGWSFGYYHKAG-UHFFFAOYSA-N 0.000 description 1
- JQNKAHQBKGEXAG-UHFFFAOYSA-N NS(=O)(=O)C1=CC=C(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)C=C1 Chemical compound NS(=O)(=O)C1=CC=C(N2C=C(C3=CC=C(F)C=C3)C=CC2=O)C=C1 JQNKAHQBKGEXAG-UHFFFAOYSA-N 0.000 description 1
- QHQIEUNUJOGHFW-UHFFFAOYSA-N NS(=O)(=O)C1=CC=C(N2C=C(C3=CC=CN=C3)C=CC2=O)C=C1 Chemical compound NS(=O)(=O)C1=CC=C(N2C=C(C3=CC=CN=C3)C=CC2=O)C=C1 QHQIEUNUJOGHFW-UHFFFAOYSA-N 0.000 description 1
- LQKOKHUMHMQIHC-UHFFFAOYSA-N NS(=O)(=O)C1=CC=C(N2C=C(C3=CN=CN=C3)C=CC2=O)C=C1 Chemical compound NS(=O)(=O)C1=CC=C(N2C=C(C3=CN=CN=C3)C=CC2=O)C=C1 LQKOKHUMHMQIHC-UHFFFAOYSA-N 0.000 description 1
- RHHDROMMBULTBB-UHFFFAOYSA-N NS(=O)(=O)C1=CC=C(N2C=C(C3CC3)C=CC2=O)C=C1 Chemical compound NS(=O)(=O)C1=CC=C(N2C=C(C3CC3)C=CC2=O)C=C1 RHHDROMMBULTBB-UHFFFAOYSA-N 0.000 description 1
- SKCKNYSGAASEOV-UHFFFAOYSA-N NS(=O)(=O)C1=CC=C(N2C=CC=CC2=O)C=C1 Chemical compound NS(=O)(=O)C1=CC=C(N2C=CC=CC2=O)C=C1 SKCKNYSGAASEOV-UHFFFAOYSA-N 0.000 description 1
- FTALVKOYETXMEY-UHFFFAOYSA-N NS(=O)(=O)C1=CC=C(NC(=O)C2=CC3=C(N2)N(C2=CC=C(F)C=C2)C(=O)C=C3)C=C1 Chemical compound NS(=O)(=O)C1=CC=C(NC(=O)C2=CC3=C(N2)N(C2=CC=C(F)C=C2)C(=O)C=C3)C=C1 FTALVKOYETXMEY-UHFFFAOYSA-N 0.000 description 1
- IQXLIOADOAHXIQ-UHFFFAOYSA-N NS(=O)(=O)C1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(Cl)C=C2)C(=O)C=C3)=C1 Chemical compound NS(=O)(=O)C1=CC=CC(NC(=O)C2=CC3=C(N2)N(C2=CC=C(Cl)C=C2)C(=O)C=C3)=C1 IQXLIOADOAHXIQ-UHFFFAOYSA-N 0.000 description 1
- RTFIQGPKNRMENK-UHFFFAOYSA-N O=C(C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2)N1CCCC1 Chemical compound O=C(C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2)N1CCCC1 RTFIQGPKNRMENK-UHFFFAOYSA-N 0.000 description 1
- CBAPJAYEYSAWTP-UHFFFAOYSA-N O=C(C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2)N1CCCC1 Chemical compound O=C(C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2)N1CCCC1 CBAPJAYEYSAWTP-UHFFFAOYSA-N 0.000 description 1
- GFFAEVPWYWXNRS-UHFFFAOYSA-N O=C(C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2)N1CCNCC1 Chemical compound O=C(C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2)N1CCNCC1 GFFAEVPWYWXNRS-UHFFFAOYSA-N 0.000 description 1
- SHLVLGHWBSMUEP-UHFFFAOYSA-N O=C(C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2)N1CCOCC1 Chemical compound O=C(C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2)N1CCOCC1 SHLVLGHWBSMUEP-UHFFFAOYSA-N 0.000 description 1
- GJABTZXQFSWUBA-UHFFFAOYSA-N O=C(C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2)N1CCCC1 Chemical compound O=C(C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2)N1CCCC1 GJABTZXQFSWUBA-UHFFFAOYSA-N 0.000 description 1
- YSNLXVTXCBPXAY-UHFFFAOYSA-N O=C(C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2)N1CCOCC1 Chemical compound O=C(C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2)N1CCOCC1 YSNLXVTXCBPXAY-UHFFFAOYSA-N 0.000 description 1
- JOSGRYWVCRWXLV-UHFFFAOYSA-N O=C(C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2)N1CCCC1 Chemical compound O=C(C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2)N1CCCC1 JOSGRYWVCRWXLV-UHFFFAOYSA-N 0.000 description 1
- UZZDVBNJKLMTOK-UHFFFAOYSA-N O=C(C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2)N1CCOCC1 Chemical compound O=C(C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2)N1CCOCC1 UZZDVBNJKLMTOK-UHFFFAOYSA-N 0.000 description 1
- SGCUGGBKTIYHNT-UHFFFAOYSA-N O=C(C1=CC2=C(N1)N(C1=CC=CC(O)=C1)C(=O)C=C2)N1CCCC1 Chemical compound O=C(C1=CC2=C(N1)N(C1=CC=CC(O)=C1)C(=O)C=C2)N1CCCC1 SGCUGGBKTIYHNT-UHFFFAOYSA-N 0.000 description 1
- MHPQKDWIECPKES-UHFFFAOYSA-N O=C(C=C1)NC=C1c1cccnc1 Chemical compound O=C(C=C1)NC=C1c1cccnc1 MHPQKDWIECPKES-UHFFFAOYSA-N 0.000 description 1
- JLEJXZDPNYUTIR-UHFFFAOYSA-N O=C(C=C1)NC=C1c1ccncc1 Chemical compound O=C(C=C1)NC=C1c1ccncc1 JLEJXZDPNYUTIR-UHFFFAOYSA-N 0.000 description 1
- LMPQVKCDSYKYOX-UHFFFAOYSA-N O=C(C=C1)NC=C1c1cncnc1 Chemical compound O=C(C=C1)NC=C1c1cncnc1 LMPQVKCDSYKYOX-UHFFFAOYSA-N 0.000 description 1
- UYHFGXNZZNLOFX-UHFFFAOYSA-N O=C(C=CC(C(F)(F)F)=C1)N1c1ccncc1 Chemical compound O=C(C=CC(C(F)(F)F)=C1)N1c1ccncc1 UYHFGXNZZNLOFX-UHFFFAOYSA-N 0.000 description 1
- MNGAKJAKXWYVMV-UHFFFAOYSA-N O=C(NC1=CC(Cl)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC(Cl)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 MNGAKJAKXWYVMV-UHFFFAOYSA-N 0.000 description 1
- VIRFQKYJLSQTAX-UHFFFAOYSA-N O=C(NC1=CC(Cl)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC(Cl)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 VIRFQKYJLSQTAX-UHFFFAOYSA-N 0.000 description 1
- UYLNPCPKCJDWJB-UHFFFAOYSA-N O=C(NC1=CC(Cl)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC(Cl)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 UYLNPCPKCJDWJB-UHFFFAOYSA-N 0.000 description 1
- IZIIILPWTTWBTM-UHFFFAOYSA-N O=C(NC1=CC(Cl)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC(Cl)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 IZIIILPWTTWBTM-UHFFFAOYSA-N 0.000 description 1
- RIJDIDBAFJVSLS-UHFFFAOYSA-N O=C(NC1=CC(Cl)=CC=C1)C1=CC2=C(N1)N(C1=CC=CC(O)=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC(Cl)=CC=C1)C1=CC2=C(N1)N(C1=CC=CC(O)=C1)C(=O)C=C2 RIJDIDBAFJVSLS-UHFFFAOYSA-N 0.000 description 1
- SWRKKKQRMJFYTI-UHFFFAOYSA-N O=C(NC1=CC(OC2=CC=CC=C2)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC(OC2=CC=CC=C2)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 SWRKKKQRMJFYTI-UHFFFAOYSA-N 0.000 description 1
- YGIQPBMVNAOBKH-UHFFFAOYSA-N O=C(NC1=CC(OC2=CC=CC=C2)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC(OC2=CC=CC=C2)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 YGIQPBMVNAOBKH-UHFFFAOYSA-N 0.000 description 1
- SKDYIMRDUJSJIU-UHFFFAOYSA-N O=C(NC1=CC(OC2=CC=CC=C2)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC(OC2=CC=CC=C2)=CC=C1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 SKDYIMRDUJSJIU-UHFFFAOYSA-N 0.000 description 1
- GXGWQUKEDQUSDY-UHFFFAOYSA-N O=C(NC1=CC=C(OC2=CC=CC=C2)C=C1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC=C(OC2=CC=CC=C2)C=C1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 GXGWQUKEDQUSDY-UHFFFAOYSA-N 0.000 description 1
- PEEXYCGJPLMMHU-UHFFFAOYSA-N O=C(NC1=CC=C(OC2=CC=CC=C2)C=C1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC=C(OC2=CC=CC=C2)C=C1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 PEEXYCGJPLMMHU-UHFFFAOYSA-N 0.000 description 1
- LBYFMZFYQHPMLC-UHFFFAOYSA-N O=C(NC1=CC=C(OC2=CC=CC=C2)C=C1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC=C(OC2=CC=CC=C2)C=C1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 LBYFMZFYQHPMLC-UHFFFAOYSA-N 0.000 description 1
- MRAWWSNWOCBELR-UHFFFAOYSA-N O=C(NC1=CC=C(OC2=CC=CC=C2)C=C1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=CC=C(OC2=CC=CC=C2)C=C1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 MRAWWSNWOCBELR-UHFFFAOYSA-N 0.000 description 1
- TXJXLUUXDUEWQD-UHFFFAOYSA-N O=C(NC1=NC=CS1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=NC=CS1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 TXJXLUUXDUEWQD-UHFFFAOYSA-N 0.000 description 1
- JMPGTTVCLFYJEV-UHFFFAOYSA-N O=C(NC1=NC=CS1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=NC=CS1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 JMPGTTVCLFYJEV-UHFFFAOYSA-N 0.000 description 1
- JATJMYWISKNEND-UHFFFAOYSA-N O=C(NC1=NC=CS1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=NC=CS1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 JATJMYWISKNEND-UHFFFAOYSA-N 0.000 description 1
- QQLBSUIKAJDWEL-UHFFFAOYSA-N O=C(NC1=NC=CS1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NC1=NC=CS1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 QQLBSUIKAJDWEL-UHFFFAOYSA-N 0.000 description 1
- DAABLZZPFZUUCL-UHFFFAOYSA-N O=C(NCC1=CC=CC=C1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1=CC=CC=C1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 DAABLZZPFZUUCL-UHFFFAOYSA-N 0.000 description 1
- CWBVNMHMVRDIHB-UHFFFAOYSA-N O=C(NCC1=CC=CC=C1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1=CC=CC=C1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 CWBVNMHMVRDIHB-UHFFFAOYSA-N 0.000 description 1
- JMVNYMOJDQCOFA-UHFFFAOYSA-N O=C(NCC1=CC=CC=C1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1=CC=CC=C1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 JMVNYMOJDQCOFA-UHFFFAOYSA-N 0.000 description 1
- NKAIUCNPVJRNPP-UHFFFAOYSA-N O=C(NCC1=CC=CC=C1)C1=CC2=C(N1)N(C1=CC=CC(O)=C1)C(=O)C=C2 Chemical compound O=C(NCC1=CC=CC=C1)C1=CC2=C(N1)N(C1=CC=CC(O)=C1)C(=O)C=C2 NKAIUCNPVJRNPP-UHFFFAOYSA-N 0.000 description 1
- KQOWKZKEEDRAIG-UHFFFAOYSA-N O=C(NCC1=CC=NC=C1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1=CC=NC=C1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 KQOWKZKEEDRAIG-UHFFFAOYSA-N 0.000 description 1
- QBIYGHDEISYYEK-UHFFFAOYSA-N O=C(NCC1=CC=NC=C1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1=CC=NC=C1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 QBIYGHDEISYYEK-UHFFFAOYSA-N 0.000 description 1
- LITCYSROJJFYFW-UHFFFAOYSA-N O=C(NCC1=NC2=C(C=CC=C2)N1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1=NC2=C(C=CC=C2)N1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 LITCYSROJJFYFW-UHFFFAOYSA-N 0.000 description 1
- XTYUZJKTRADJQE-UHFFFAOYSA-N O=C(NCC1=NC2=C(C=CC=C2)N1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1=NC2=C(C=CC=C2)N1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 XTYUZJKTRADJQE-UHFFFAOYSA-N 0.000 description 1
- WJJHADMPMKXWGB-UHFFFAOYSA-N O=C(NCC1=NC2=C(C=CC=C2)N1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1=NC2=C(C=CC=C2)N1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 WJJHADMPMKXWGB-UHFFFAOYSA-N 0.000 description 1
- UJMDHFQWQBLYOI-UHFFFAOYSA-N O=C(NCC1=NC2=C(C=CC=C2)N1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1=NC2=C(C=CC=C2)N1)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 UJMDHFQWQBLYOI-UHFFFAOYSA-N 0.000 description 1
- OXPSZRXHFQWRJF-UHFFFAOYSA-N O=C(NCC1CCCO1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1CCCO1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 OXPSZRXHFQWRJF-UHFFFAOYSA-N 0.000 description 1
- KBYXURCMKHGGEU-UHFFFAOYSA-N O=C(NCC1CCCO1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1CCCO1)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 KBYXURCMKHGGEU-UHFFFAOYSA-N 0.000 description 1
- JFZQAMUNIFCMFI-UHFFFAOYSA-N O=C(NCC1CCCO1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 Chemical compound O=C(NCC1CCCO1)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 JFZQAMUNIFCMFI-UHFFFAOYSA-N 0.000 description 1
- QDEYGWAHFVFOAN-UHFFFAOYSA-N O=C(NCCN1CCCC1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(NCCN1CCCC1)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 QDEYGWAHFVFOAN-UHFFFAOYSA-N 0.000 description 1
- HZQMXAGXZCIVOA-UHFFFAOYSA-N O=C(O)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(O)C1=CC2=C(N1)N(C1=CC=C(C(F)(F)F)C=C1)C(=O)C=C2 HZQMXAGXZCIVOA-UHFFFAOYSA-N 0.000 description 1
- CSIMFTKAUXVEIR-UHFFFAOYSA-N O=C(O)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2.O=C(O)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 Chemical compound O=C(O)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2.O=C(O)C1=CC2=C(N1)N(C1=CC=C(Cl)C=C1)C(=O)C=C2 CSIMFTKAUXVEIR-UHFFFAOYSA-N 0.000 description 1
- CFYXQIRGGLZKNO-UHFFFAOYSA-N O=C(O)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 Chemical compound O=C(O)C1=CC2=C(N1)N(C1=CC=C(F)C=C1)C(=O)C=C2 CFYXQIRGGLZKNO-UHFFFAOYSA-N 0.000 description 1
- YDTQXDPXQBXLCD-UHFFFAOYSA-N O=C(O)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 Chemical compound O=C(O)C1=CC2=C(N1)N(C1=CC=C(OC(F)(F)F)C=C1)C(=O)C=C2 YDTQXDPXQBXLCD-UHFFFAOYSA-N 0.000 description 1
- XTSXMISXZZOSFW-UHFFFAOYSA-N O=C(O)C1=CC2=C(N1)N(C1=CC=CC(O)=C1)C(=O)C=C2 Chemical compound O=C(O)C1=CC2=C(N1)N(C1=CC=CC(O)=C1)C(=O)C=C2 XTSXMISXZZOSFW-UHFFFAOYSA-N 0.000 description 1
- VGZLDHXMYYHAKO-BUHFOSPRSA-N O=C1C(/C=C/C2=CC=CC=C2)=CC=CN1C1=CC=CC=C1 Chemical compound O=C1C(/C=C/C2=CC=CC=C2)=CC=CN1C1=CC=CC=C1 VGZLDHXMYYHAKO-BUHFOSPRSA-N 0.000 description 1
- BIIRXKVOOYFIIL-UHFFFAOYSA-N O=C1C(C(F)(F)F)=CC=CN1C1=CC=CC=C1 Chemical compound O=C1C(C(F)(F)F)=CC=CN1C1=CC=CC=C1 BIIRXKVOOYFIIL-UHFFFAOYSA-N 0.000 description 1
- KPHXJZCISPLFPS-UHFFFAOYSA-N O=C1C(C2=CC=CS2)=CC=CN1C1=CC=CC=C1 Chemical compound O=C1C(C2=CC=CS2)=CC=CN1C1=CC=CC=C1 KPHXJZCISPLFPS-UHFFFAOYSA-N 0.000 description 1
- KVDOBODYYDVBGT-VAWYXSNFSA-N O=C1C=C(/C=C/C2=CC=CC=C2)C=CN1C1=CC=CC=C1 Chemical compound O=C1C=C(/C=C/C2=CC=CC=C2)C=CN1C1=CC=CC=C1 KVDOBODYYDVBGT-VAWYXSNFSA-N 0.000 description 1
- KIPNNSSACIMIPP-UHFFFAOYSA-N O=C1C=C(C(F)(F)F)C=CN1C1=CC=C(C(F)(F)F)C=C1 Chemical compound O=C1C=C(C(F)(F)F)C=CN1C1=CC=C(C(F)(F)F)C=C1 KIPNNSSACIMIPP-UHFFFAOYSA-N 0.000 description 1
- RRSBVTWPEZGNBT-UHFFFAOYSA-N O=C1C=C(C(F)(F)F)C=CN1C1=CC=C(Cl)C=C1 Chemical compound O=C1C=C(C(F)(F)F)C=CN1C1=CC=C(Cl)C=C1 RRSBVTWPEZGNBT-UHFFFAOYSA-N 0.000 description 1
- NYRBUAOMZBUBDV-UHFFFAOYSA-N O=C1C=C(C(F)(F)F)C=CN1C1=CC=CC=C1 Chemical compound O=C1C=C(C(F)(F)F)C=CN1C1=CC=CC=C1 NYRBUAOMZBUBDV-UHFFFAOYSA-N 0.000 description 1
- FGFPXIGRWVVXHS-UHFFFAOYSA-N O=C1C=C(C(F)F)C=CN1C1=CC=C(C(F)(F)F)C=C1 Chemical compound O=C1C=C(C(F)F)C=CN1C1=CC=C(C(F)(F)F)C=C1 FGFPXIGRWVVXHS-UHFFFAOYSA-N 0.000 description 1
- VJTYORUWFQGGAK-UHFFFAOYSA-N O=C1C=C(C(F)F)C=CN1C1=CC=C(C(F)F)C=C1 Chemical compound O=C1C=C(C(F)F)C=CN1C1=CC=C(C(F)F)C=C1 VJTYORUWFQGGAK-UHFFFAOYSA-N 0.000 description 1
- IGWVMIBGPQTSCR-UHFFFAOYSA-N O=C1C=C(C(F)F)C=CN1C1=CC=C(Cl)C=C1 Chemical compound O=C1C=C(C(F)F)C=CN1C1=CC=C(Cl)C=C1 IGWVMIBGPQTSCR-UHFFFAOYSA-N 0.000 description 1
- KKNBHNNXUPKQNI-UHFFFAOYSA-N O=C1C=C(C(F)F)C=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=C(C(F)F)C=CN1C1=CC=C(OC(F)(F)F)C=C1 KKNBHNNXUPKQNI-UHFFFAOYSA-N 0.000 description 1
- ITLMXDOXDZLXEH-UHFFFAOYSA-N O=C1C=C(C(F)F)C=CN1C1=CC=CC(C(F)F)=C1 Chemical compound O=C1C=C(C(F)F)C=CN1C1=CC=CC(C(F)F)=C1 ITLMXDOXDZLXEH-UHFFFAOYSA-N 0.000 description 1
- AFQPZPPTEAYKAE-UHFFFAOYSA-N O=C1C=C(C2=CC=CC=C2)C=CN1C1=CC=CC=C1 Chemical compound O=C1C=C(C2=CC=CC=C2)C=CN1C1=CC=CC=C1 AFQPZPPTEAYKAE-UHFFFAOYSA-N 0.000 description 1
- CUIIAJPRPOOEDJ-UHFFFAOYSA-N O=C1C=C(C2=CC=CS2)C=CN1C1=CC=CC=C1 Chemical compound O=C1C=C(C2=CC=CS2)C=CN1C1=CC=CC=C1 CUIIAJPRPOOEDJ-UHFFFAOYSA-N 0.000 description 1
- RQNYJZFHBQTKOF-UHFFFAOYSA-N O=C1C=C(O)C=CN1C1=CC=CC=C1.O=C1C=C(OCC2=CC=CC=C2)C=CN1.O=C1C=C(OCC2=CC=CC=C2)C=CN1C1=CC=CC=C1 Chemical compound O=C1C=C(O)C=CN1C1=CC=CC=C1.O=C1C=C(OCC2=CC=CC=C2)C=CN1.O=C1C=C(OCC2=CC=CC=C2)C=CN1C1=CC=CC=C1 RQNYJZFHBQTKOF-UHFFFAOYSA-N 0.000 description 1
- SYGBOMJQPVJCIX-UHFFFAOYSA-N O=C1C=C(OCC2=CC=CC=C2)C=CN1C1=CC=CC=C1 Chemical compound O=C1C=C(OCC2=CC=CC=C2)C=CN1C1=CC=CC=C1 SYGBOMJQPVJCIX-UHFFFAOYSA-N 0.000 description 1
- NKBVATUCJGOYHO-VAWYXSNFSA-N O=C1C=CC(/C=C/C2=CC=CC=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(/C=C/C2=CC=CC=C2)=CN1C1=CC=CC=C1 NKBVATUCJGOYHO-VAWYXSNFSA-N 0.000 description 1
- ZBRAYWIGAVNERT-UHFFFAOYSA-N O=C1C=CC(Br)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(Br)=CN1C1=CC=NC=C1 ZBRAYWIGAVNERT-UHFFFAOYSA-N 0.000 description 1
- KLKVAWRLNZWMSF-UHFFFAOYSA-N O=C1C=CC(C(F)(F)C(F)(F)F)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C(F)(F)C(F)(F)F)=CN1C1=CC=CC=C1 KLKVAWRLNZWMSF-UHFFFAOYSA-N 0.000 description 1
- KVSNMHHUKSILND-UHFFFAOYSA-N O=C1C=CC(C(F)C(F)(F)F)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C(F)C(F)(F)F)=CN1C1=CC=CC=C1 KVSNMHHUKSILND-UHFFFAOYSA-N 0.000 description 1
- QKXSRXXQSRQYKD-UHFFFAOYSA-N O=C1C=CC(C(F)F)=CN1C1=CC=C(C(F)F)C=C1 Chemical compound O=C1C=CC(C(F)F)=CN1C1=CC=C(C(F)F)C=C1 QKXSRXXQSRQYKD-UHFFFAOYSA-N 0.000 description 1
- BHAHRXCXMRRRMA-UHFFFAOYSA-N O=C1C=CC(C(F)F)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C(F)F)=CN1C1=CC=C(OC(F)(F)F)C=C1 BHAHRXCXMRRRMA-UHFFFAOYSA-N 0.000 description 1
- KMBYCTGWWBZMRA-UHFFFAOYSA-N O=C1C=CC(C(F)F)=CN1C1=CC=CC(C(F)(F)F)=C1 Chemical compound O=C1C=CC(C(F)F)=CN1C1=CC=CC(C(F)(F)F)=C1 KMBYCTGWWBZMRA-UHFFFAOYSA-N 0.000 description 1
- WISZRLILWIRQTE-UHFFFAOYSA-N O=C1C=CC(C(F)F)=CN1C1=CC=CC(C(F)F)=C1 Chemical compound O=C1C=CC(C(F)F)=CN1C1=CC=CC(C(F)F)=C1 WISZRLILWIRQTE-UHFFFAOYSA-N 0.000 description 1
- YMDGHGFBVXJSFS-UHFFFAOYSA-N O=C1C=CC(C(F)F)=CN1C1=CC=CC=N1 Chemical compound O=C1C=CC(C(F)F)=CN1C1=CC=CC=N1 YMDGHGFBVXJSFS-UHFFFAOYSA-N 0.000 description 1
- JWRCICKKLSFWGJ-UHFFFAOYSA-N O=C1C=CC(C(F)F)=CN1C1=CC=CN=C1 Chemical compound O=C1C=CC(C(F)F)=CN1C1=CC=CN=C1 JWRCICKKLSFWGJ-UHFFFAOYSA-N 0.000 description 1
- ZAPWLNRWNFOIIH-UHFFFAOYSA-N O=C1C=CC(C(F)F)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C(F)F)=CN1C1=CC=NC=C1 ZAPWLNRWNFOIIH-UHFFFAOYSA-N 0.000 description 1
- MIUBYTKRNODPRE-UHFFFAOYSA-N O=C1C=CC(C(O)(C(F)(F)F)C(F)(F)F)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C(O)(C(F)(F)F)C(F)(F)F)=CN1C1=CC=CC=C1 MIUBYTKRNODPRE-UHFFFAOYSA-N 0.000 description 1
- GNTPWVXYTYDYPJ-UHFFFAOYSA-N O=C1C=CC(C(O)C(F)(F)F)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C(O)C(F)(F)F)=CN1C1=CC=CC=C1 GNTPWVXYTYDYPJ-UHFFFAOYSA-N 0.000 description 1
- XEUIWMRMROBWMY-UHFFFAOYSA-N O=C1C=CC(C2=CC(O)=CC=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC(O)=CC=C2)=CN1C1=CC=CC=C1 XEUIWMRMROBWMY-UHFFFAOYSA-N 0.000 description 1
- TWXLHRSJLXNBPR-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(CO)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=C(CO)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 TWXLHRSJLXNBPR-UHFFFAOYSA-N 0.000 description 1
- GBRRLNCDXYJDGI-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(CO)C=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=C(CO)C=C2)=CN1C1=CC=CC=C1 GBRRLNCDXYJDGI-UHFFFAOYSA-N 0.000 description 1
- MQWZLCTVPYWMNU-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(CO)C=C2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=CC=C(CO)C=C2)=CN1C1=CC=NC=C1 MQWZLCTVPYWMNU-UHFFFAOYSA-N 0.000 description 1
- BYZKGKOBVDJDEY-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(C[SH](=O)=O)C=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=C(C[SH](=O)=O)C=C2)=CN1C1=CC=CC=C1 BYZKGKOBVDJDEY-UHFFFAOYSA-N 0.000 description 1
- HRBKTCPRCOAKAU-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(Cl)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=C(Cl)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 HRBKTCPRCOAKAU-UHFFFAOYSA-N 0.000 description 1
- MHMOOKJQWSMVGB-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(Cl)C=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=C(Cl)C=C2)=CN1C1=CC=CC=C1 MHMOOKJQWSMVGB-UHFFFAOYSA-N 0.000 description 1
- MTGALYWLAHGKGW-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(Cl)C=C2)=CN1C1=CN=CN=C1 Chemical compound O=C1C=CC(C2=CC=C(Cl)C=C2)=CN1C1=CN=CN=C1 MTGALYWLAHGKGW-UHFFFAOYSA-N 0.000 description 1
- OQGBKUHHFBHTOZ-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=C(CO)C=CC=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=C(CO)C=CC=C1 OQGBKUHHFBHTOZ-UHFFFAOYSA-N 0.000 description 1
- PJEQHRRFRIPIQQ-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(Cl)C=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(Cl)C=C1 PJEQHRRFRIPIQQ-UHFFFAOYSA-N 0.000 description 1
- LAYPIRXZCZTIEW-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(F)C=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(F)C=C1 LAYPIRXZCZTIEW-UHFFFAOYSA-N 0.000 description 1
- WDCDDDQVPKQIAR-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 WDCDDDQVPKQIAR-UHFFFAOYSA-N 0.000 description 1
- VJHSJQOTIVMMGV-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=CC(Cl)=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=CC(Cl)=C1 VJHSJQOTIVMMGV-UHFFFAOYSA-N 0.000 description 1
- YCXUMJOXOMKFSN-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=CC(F)=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=CC(F)=C1 YCXUMJOXOMKFSN-UHFFFAOYSA-N 0.000 description 1
- RKVKSKMKIUXKIH-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=CC=C1 RKVKSKMKIUXKIH-UHFFFAOYSA-N 0.000 description 1
- ATOBIXZTKIUOSR-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CC=NC=C1 ATOBIXZTKIUOSR-UHFFFAOYSA-N 0.000 description 1
- FPQWALPYYGTBPW-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CN=CN=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2)=CN1C1=CN=CN=C1 FPQWALPYYGTBPW-UHFFFAOYSA-N 0.000 description 1
- NPRIKPBPECXMFE-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2)=NN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2)=NN1C1=CC=CC=C1 NPRIKPBPECXMFE-UHFFFAOYSA-N 0.000 description 1
- YTVCONJNWSVONS-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(F)C=C2F)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=C(F)C=C2F)=CN1C1=CC=CC=C1 YTVCONJNWSVONS-UHFFFAOYSA-N 0.000 description 1
- CHGUHEAXXPIHEZ-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(N[SH](=O)=O)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=C(N[SH](=O)=O)C=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 CHGUHEAXXPIHEZ-UHFFFAOYSA-N 0.000 description 1
- SAOHCGFWOZPPMP-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(N[SH](=O)=O)C=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=C(N[SH](=O)=O)C=C2)=CN1C1=CC=CC=C1 SAOHCGFWOZPPMP-UHFFFAOYSA-N 0.000 description 1
- HHLJTTBOJWEOLM-UHFFFAOYSA-N O=C1C=CC(C2=CC=C(O)C=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=C(O)C=C2)=CN1C1=CC=CC=C1 HHLJTTBOJWEOLM-UHFFFAOYSA-N 0.000 description 1
- DCGMDTGBPQCWKU-UHFFFAOYSA-N O=C1C=CC(C2=CC=C3OCOC3=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=C3OCOC3=C2)=CN1C1=CC=CC=C1 DCGMDTGBPQCWKU-UHFFFAOYSA-N 0.000 description 1
- QQUXMXMJMGVPHW-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC(CO)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=CC(CO)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 QQUXMXMJMGVPHW-UHFFFAOYSA-N 0.000 description 1
- YCCQTBDAUFVXDZ-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC(CO)=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=CC(CO)=C2)=CN1C1=CC=CC=C1 YCCQTBDAUFVXDZ-UHFFFAOYSA-N 0.000 description 1
- BAGOXQOHZRSKQI-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC(CO)=C2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=CC=CC(CO)=C2)=CN1C1=CC=NC=C1 BAGOXQOHZRSKQI-UHFFFAOYSA-N 0.000 description 1
- OVGKYIORCMBURR-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC(Cl)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=CC(Cl)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 OVGKYIORCMBURR-UHFFFAOYSA-N 0.000 description 1
- AEFKLWDJYPIINJ-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC(Cl)=C2)=CN1C1=CN=CN=C1 Chemical compound O=C1C=CC(C2=CC=CC(Cl)=C2)=CN1C1=CN=CN=C1 AEFKLWDJYPIINJ-UHFFFAOYSA-N 0.000 description 1
- SAKUULGBQOVBLU-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC(F)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=CC(F)=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 SAKUULGBQOVBLU-UHFFFAOYSA-N 0.000 description 1
- CRGNJLKBLAYHPX-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC(F)=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=CC(F)=C2)=CN1C1=CC=CC=C1 CRGNJLKBLAYHPX-UHFFFAOYSA-N 0.000 description 1
- LKRPTEPMDOSSFI-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(C(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(C(F)(F)F)C=C1 LKRPTEPMDOSSFI-UHFFFAOYSA-N 0.000 description 1
- OMDMBXMGLGTERJ-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(C(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(C(F)F)C=C1 OMDMBXMGLGTERJ-UHFFFAOYSA-N 0.000 description 1
- LSZOXCDSYFXHEC-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(Cl)C=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(Cl)C=C1 LSZOXCDSYFXHEC-UHFFFAOYSA-N 0.000 description 1
- KEQHZSMDTLOACU-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 KEQHZSMDTLOACU-UHFFFAOYSA-N 0.000 description 1
- LSUOSRFDLSUSOV-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=CC(C(F)F)=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2)=CN1C1=CC=CC(C(F)F)=C1 LSUOSRFDLSUSOV-UHFFFAOYSA-N 0.000 description 1
- RPWKUWNZPJKTKG-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2Cl)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2Cl)=CN1C1=CC=CC=C1 RPWKUWNZPJKTKG-UHFFFAOYSA-N 0.000 description 1
- BESATBAIJCTWIS-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2Cl)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2Cl)=CN1C1=CC=NC=C1 BESATBAIJCTWIS-UHFFFAOYSA-N 0.000 description 1
- FINTYSUOFGJPIU-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2F)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2F)=CN1C1=CC=C(OC(F)(F)F)C=C1 FINTYSUOFGJPIU-UHFFFAOYSA-N 0.000 description 1
- SDQTXLBSFPUHFG-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2F)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2F)=CN1C1=CC=CC=C1 SDQTXLBSFPUHFG-UHFFFAOYSA-N 0.000 description 1
- PJDHRUGMGRLLGS-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2F)=CN1C1=CN=CN=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2F)=CN1C1=CN=CN=C1 PJDHRUGMGRLLGS-UHFFFAOYSA-N 0.000 description 1
- SBLFYNTUMQMFQY-UHFFFAOYSA-N O=C1C=CC(C2=CC=CC=C2O)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=CC=C2O)=CN1C1=CC=CC=C1 SBLFYNTUMQMFQY-UHFFFAOYSA-N 0.000 description 1
- CPXUPBCAUHEBAU-UHFFFAOYSA-N O=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 CPXUPBCAUHEBAU-UHFFFAOYSA-N 0.000 description 1
- HEOSIUZNFCYUDL-UHFFFAOYSA-N O=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=CC=C1 HEOSIUZNFCYUDL-UHFFFAOYSA-N 0.000 description 1
- KQNFXCOFRHADDD-UHFFFAOYSA-N O=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=CC=CN=C2)=CN1C1=CC=NC=C1 KQNFXCOFRHADDD-UHFFFAOYSA-N 0.000 description 1
- YPYGKBXSVDQVAX-UHFFFAOYSA-N O=C1C=CC(C2=CC=CO2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=CO2)=CN1C1=CC=C(OC(F)(F)F)C=C1 YPYGKBXSVDQVAX-UHFFFAOYSA-N 0.000 description 1
- HOBXXCSYRXVYJG-UHFFFAOYSA-N O=C1C=CC(C2=CC=CO2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=CO2)=CN1C1=CC=CC=C1 HOBXXCSYRXVYJG-UHFFFAOYSA-N 0.000 description 1
- OCODDGUVONTFSX-UHFFFAOYSA-N O=C1C=CC(C2=CC=CO2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=CC=CO2)=CN1C1=CC=NC=C1 OCODDGUVONTFSX-UHFFFAOYSA-N 0.000 description 1
- DVTIIIZUDWEALN-UHFFFAOYSA-N O=C1C=CC(C2=CC=CO2)=CN1C1=CN=CN=C1 Chemical compound O=C1C=CC(C2=CC=CO2)=CN1C1=CN=CN=C1 DVTIIIZUDWEALN-UHFFFAOYSA-N 0.000 description 1
- FMJZZESOMXWLLC-UHFFFAOYSA-N O=C1C=CC(C2=CC=CS2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=CS2)=CN1C1=CC=CC=C1 FMJZZESOMXWLLC-UHFFFAOYSA-N 0.000 description 1
- YKGUKAMWOVTELC-UHFFFAOYSA-N O=C1C=CC(C2=CC=NC=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CC=NC=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 YKGUKAMWOVTELC-UHFFFAOYSA-N 0.000 description 1
- HEEPKQRHQVTGGC-UHFFFAOYSA-N O=C1C=CC(C2=CC=NC=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CC=NC=C2)=CN1C1=CC=CC=C1 HEEPKQRHQVTGGC-UHFFFAOYSA-N 0.000 description 1
- SRSUBBZAMGGTMX-UHFFFAOYSA-N O=C1C=CC(C2=CN=C(CO)C=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CN=C(CO)C=C2)=CN1C1=CC=CC=C1 SRSUBBZAMGGTMX-UHFFFAOYSA-N 0.000 description 1
- HUINIQQKJKKCKW-UHFFFAOYSA-N O=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=C(OC(F)(F)F)C=C1 HUINIQQKJKKCKW-UHFFFAOYSA-N 0.000 description 1
- ASVCOOPQZZSIEC-UHFFFAOYSA-N O=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=CC=C1 ASVCOOPQZZSIEC-UHFFFAOYSA-N 0.000 description 1
- LIBNCLXBZCOVLT-UHFFFAOYSA-N O=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=CN=C1 Chemical compound O=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=CN=C1 LIBNCLXBZCOVLT-UHFFFAOYSA-N 0.000 description 1
- OLZFDCSPUWGZNY-UHFFFAOYSA-N O=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=CN=CN=C2)=CN1C1=CC=NC=C1 OLZFDCSPUWGZNY-UHFFFAOYSA-N 0.000 description 1
- QXDPRKYSNCJLQC-UHFFFAOYSA-N O=C1C=CC(C2=CNN=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CNN=C2)=CN1C1=CC=CC=C1 QXDPRKYSNCJLQC-UHFFFAOYSA-N 0.000 description 1
- PTHMUXYSAGDJBG-UHFFFAOYSA-N O=C1C=CC(C2=CNN=C2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=CNN=C2)=CN1C1=CC=NC=C1 PTHMUXYSAGDJBG-UHFFFAOYSA-N 0.000 description 1
- IUPNYEWHAHLZIL-UHFFFAOYSA-N O=C1C=CC(C2=CSC=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=CSC=C2)=CN1C1=CC=CC=C1 IUPNYEWHAHLZIL-UHFFFAOYSA-N 0.000 description 1
- CQBRXNOKJINDRW-UHFFFAOYSA-N O=C1C=CC(C2=NC=CC=N2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2=NC=CC=N2)=CN1C1=CC=C(OC(F)(F)F)C=C1 CQBRXNOKJINDRW-UHFFFAOYSA-N 0.000 description 1
- PWNMBPLVSCOSEN-UHFFFAOYSA-N O=C1C=CC(C2=NC=CC=N2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=NC=CC=N2)=CN1C1=CC=CC=C1 PWNMBPLVSCOSEN-UHFFFAOYSA-N 0.000 description 1
- XDWGURYXKJXNDK-UHFFFAOYSA-N O=C1C=CC(C2=NC=CC=N2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=NC=CC=N2)=CN1C1=CC=NC=C1 XDWGURYXKJXNDK-UHFFFAOYSA-N 0.000 description 1
- WCDVAEOIBGNFOT-UHFFFAOYSA-N O=C1C=CC(C2=NC=CN2)=CN1.O=C1C=CC(C2=NC=CN2)=CN1C1=CC=CC=C1.O=C1C=CC(C2=NC=CN2C2=CC=CC=C2)=CN1C1=CC=CC=C1.OB(O)C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=NC=CN2)=CN1.O=C1C=CC(C2=NC=CN2)=CN1C1=CC=CC=C1.O=C1C=CC(C2=NC=CN2C2=CC=CC=C2)=CN1C1=CC=CC=C1.OB(O)C1=CC=CC=C1 WCDVAEOIBGNFOT-UHFFFAOYSA-N 0.000 description 1
- MEMHDLJOVNXUOV-UHFFFAOYSA-N O=C1C=CC(C2=NC=CN2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=NC=CN2)=CN1C1=CC=CC=C1 MEMHDLJOVNXUOV-UHFFFAOYSA-N 0.000 description 1
- MONCKFMYOVSSNY-UHFFFAOYSA-N O=C1C=CC(C2=NC=CN2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=NC=CN2)=CN1C1=CC=NC=C1 MONCKFMYOVSSNY-UHFFFAOYSA-N 0.000 description 1
- OILNOTQSBYJZGC-UHFFFAOYSA-N O=C1C=CC(C2=NC=CN2C2=CC=CC=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=NC=CN2C2=CC=CC=C2)=CN1C1=CC=CC=C1 OILNOTQSBYJZGC-UHFFFAOYSA-N 0.000 description 1
- QVRZYJHFCNPLGN-UHFFFAOYSA-N O=C1C=CC(C2=NC=CO2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=NC=CO2)=CN1C1=CC=CC=C1 QVRZYJHFCNPLGN-UHFFFAOYSA-N 0.000 description 1
- HXUCVVBZWISANQ-UHFFFAOYSA-N O=C1C=CC(C2=NC=CS2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2=NC=CS2)=CN1C1=CC=CC=C1 HXUCVVBZWISANQ-UHFFFAOYSA-N 0.000 description 1
- VPRQCYJNDFGRLG-UHFFFAOYSA-N O=C1C=CC(C2=NC=CS2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2=NC=CS2)=CN1C1=CC=NC=C1 VPRQCYJNDFGRLG-UHFFFAOYSA-N 0.000 description 1
- OJOUHWLLHGEMIX-UHFFFAOYSA-N O=C1C=CC(C2CC2)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(C2CC2)=CN1C1=CC=C(OC(F)(F)F)C=C1 OJOUHWLLHGEMIX-UHFFFAOYSA-N 0.000 description 1
- SAKAMMUEPPRFEZ-UHFFFAOYSA-N O=C1C=CC(C2CC2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(C2CC2)=CN1C1=CC=CC=C1 SAKAMMUEPPRFEZ-UHFFFAOYSA-N 0.000 description 1
- YRYUZMZHAJMOBU-UHFFFAOYSA-N O=C1C=CC(C2CC2)=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC(C2CC2)=CN1C1=CC=NC=C1 YRYUZMZHAJMOBU-UHFFFAOYSA-N 0.000 description 1
- VVXABNKRUYTERV-UHFFFAOYSA-N O=C1C=CC(C2CC2)=CN1C1=CN=CN=C1 Chemical compound O=C1C=CC(C2CC2)=CN1C1=CN=CN=C1 VVXABNKRUYTERV-UHFFFAOYSA-N 0.000 description 1
- ONKGHRHKQUPDKH-UHFFFAOYSA-N O=C1C=CC(CF)=CN1C1=CC=C(C(F)(F)F)C=C1 Chemical compound O=C1C=CC(CF)=CN1C1=CC=C(C(F)(F)F)C=C1 ONKGHRHKQUPDKH-UHFFFAOYSA-N 0.000 description 1
- SYGOSVIVLUTJIQ-UHFFFAOYSA-N O=C1C=CC(CF)=CN1C1=CC=CC(C(F)(F)F)=C1 Chemical compound O=C1C=CC(CF)=CN1C1=CC=CC(C(F)(F)F)=C1 SYGOSVIVLUTJIQ-UHFFFAOYSA-N 0.000 description 1
- NTKLDYMOWZPYDY-UHFFFAOYSA-N O=C1C=CC(Cl)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(Cl)=CN1C1=CC=CC=C1 NTKLDYMOWZPYDY-UHFFFAOYSA-N 0.000 description 1
- OKVPJRHFHXNHEY-UHFFFAOYSA-N O=C1C=CC(F)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(F)=CN1C1=CC=C(OC(F)(F)F)C=C1 OKVPJRHFHXNHEY-UHFFFAOYSA-N 0.000 description 1
- NCJOKGIABAWOLW-UHFFFAOYSA-N O=C1C=CC(F)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(F)=CN1C1=CC=CC=C1 NCJOKGIABAWOLW-UHFFFAOYSA-N 0.000 description 1
- WMYVTHFYFXRGDR-UHFFFAOYSA-N O=C1C=CC(I)=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC(I)=CN1C1=CC=C(OC(F)(F)F)C=C1 WMYVTHFYFXRGDR-UHFFFAOYSA-N 0.000 description 1
- KDJVNBNMWQHEDC-UHFFFAOYSA-N O=C1C=CC(I)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(I)=CN1C1=CC=CC=C1 KDJVNBNMWQHEDC-UHFFFAOYSA-N 0.000 description 1
- ZVXAFCPBVBMWQY-UHFFFAOYSA-N O=C1C=CC(N2CCCC2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(N2CCCC2)=CN1C1=CC=CC=C1 ZVXAFCPBVBMWQY-UHFFFAOYSA-N 0.000 description 1
- COWRFWRIYKSXNK-UHFFFAOYSA-N O=C1C=CC(N2CCOCC2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(N2CCOCC2)=CN1C1=CC=CC=C1 COWRFWRIYKSXNK-UHFFFAOYSA-N 0.000 description 1
- HFOXYISHORSMRJ-UHFFFAOYSA-N O=C1C=CC(N[SH](=O)=O)=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC(N[SH](=O)=O)=CN1C1=CC=CC=C1 HFOXYISHORSMRJ-UHFFFAOYSA-N 0.000 description 1
- WZISIOSNQAMPDG-CCEZHUSRSA-N O=C1C=CC=C(/C=C/C2=CC=CC=C2)N1C1=CC=CC=C1 Chemical compound O=C1C=CC=C(/C=C/C2=CC=CC=C2)N1C1=CC=CC=C1 WZISIOSNQAMPDG-CCEZHUSRSA-N 0.000 description 1
- SJSXYXSGOIPBBA-UHFFFAOYSA-N O=C1C=CC=C(C2=CC=CC=C2)N1C1=CC=CC=C1 Chemical compound O=C1C=CC=C(C2=CC=CC=C2)N1C1=CC=CC=C1 SJSXYXSGOIPBBA-UHFFFAOYSA-N 0.000 description 1
- OIWKRXKPOMVROT-UHFFFAOYSA-N O=C1C=CC=C(C2=CC=CS2)N1C1=CC=CC=C1 Chemical compound O=C1C=CC=C(C2=CC=CS2)N1C1=CC=CC=C1 OIWKRXKPOMVROT-UHFFFAOYSA-N 0.000 description 1
- YTFMBMOTHNMJHR-UHFFFAOYSA-N O=C1C=CC=CN1C1=CC=C(OC(F)(F)F)C=C1 Chemical compound O=C1C=CC=CN1C1=CC=C(OC(F)(F)F)C=C1 YTFMBMOTHNMJHR-UHFFFAOYSA-N 0.000 description 1
- HQWNTNFZAGSJAX-UHFFFAOYSA-N O=C1C=CC=CN1C1=CC=CC=C1 Chemical compound O=C1C=CC=CN1C1=CC=CC=C1 HQWNTNFZAGSJAX-UHFFFAOYSA-N 0.000 description 1
- GAYVJHFPDKXUGX-UHFFFAOYSA-N O=C1C=CC=CN1C1=CC=NC=C1 Chemical compound O=C1C=CC=CN1C1=CC=NC=C1 GAYVJHFPDKXUGX-UHFFFAOYSA-N 0.000 description 1
- KELXKKNILLDRLS-UHFFFAOYSA-N O=C1C=CN(C2=CC=CC=C2)C(=O)N1 Chemical compound O=C1C=CN(C2=CC=CC=C2)C(=O)N1 KELXKKNILLDRLS-UHFFFAOYSA-N 0.000 description 1
- BPVMTLUBESQBCS-UHFFFAOYSA-N O=C1C=NC(C2=CC=C(F)C=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=NC(C2=CC=C(F)C=C2)=CN1C1=CC=CC=C1 BPVMTLUBESQBCS-UHFFFAOYSA-N 0.000 description 1
- SJEYGSKVPBOQQR-UHFFFAOYSA-N O=C1C=NC(C2=CC=CC=C2)=CN1C1=CC=CC=C1 Chemical compound O=C1C=NC(C2=CC=CC=C2)=CN1C1=CC=CC=C1 SJEYGSKVPBOQQR-UHFFFAOYSA-N 0.000 description 1
- AGBICDQAIFBBMD-UHFFFAOYSA-N O=C1N=CC(Br)=CN1C1=CC=CC=C1 Chemical compound O=C1N=CC(Br)=CN1C1=CC=CC=C1 AGBICDQAIFBBMD-UHFFFAOYSA-N 0.000 description 1
- FADDGUDTESIUNX-UHFFFAOYSA-N O=C1N=CC=CN1C1=CC=CC=C1 Chemical compound O=C1N=CC=CN1C1=CC=CC=C1 FADDGUDTESIUNX-UHFFFAOYSA-N 0.000 description 1
- OVNNLTJZRQSHFU-UHFFFAOYSA-N O=C1N=CN2NC(SC3=C(F)C=C(F)C=C3)C=CC2=C1C1=C(Cl)C=CC=C1Cl Chemical compound O=C1N=CN2NC(SC3=C(F)C=C(F)C=C3)C=CC2=C1C1=C(Cl)C=CC=C1Cl OVNNLTJZRQSHFU-UHFFFAOYSA-N 0.000 description 1
- JFLJYEIXHVHRRO-UHFFFAOYSA-N O=C1NCC=CN1C1=CC=CC=C1 Chemical compound O=C1NCC=CN1C1=CC=CC=C1 JFLJYEIXHVHRRO-UHFFFAOYSA-N 0.000 description 1
- UMIOVNLOMRBFII-UHFFFAOYSA-N O=C1NCCCN1C1=CC=CC=C1 Chemical compound O=C1NCCCN1C1=CC=CC=C1 UMIOVNLOMRBFII-UHFFFAOYSA-N 0.000 description 1
- ABMYEXAYWZJVOV-UHFFFAOYSA-N OB(c1cccnc1)O Chemical compound OB(c1cccnc1)O ABMYEXAYWZJVOV-UHFFFAOYSA-N 0.000 description 1
- QLULGIRFKAWHOJ-UHFFFAOYSA-N OB(c1ccncc1)O Chemical compound OB(c1ccncc1)O QLULGIRFKAWHOJ-UHFFFAOYSA-N 0.000 description 1
- OECGHRZCVMITJN-UHFFFAOYSA-N [C-]#[N+]C1=CC(C2=CN(C3=CC=CC(NC(C)=O)=C3)C(=O)C=C2)=CC=C1 Chemical compound [C-]#[N+]C1=CC(C2=CN(C3=CC=CC(NC(C)=O)=C3)C(=O)C=C2)=CC=C1 OECGHRZCVMITJN-UHFFFAOYSA-N 0.000 description 1
- WEAGHDUYOAXCOA-UHFFFAOYSA-N [C-]#[N+]C1=CC(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=CC=C1 Chemical compound [C-]#[N+]C1=CC(C2=CN(C3=CC=CC=C3)C(=O)C=C2)=CC=C1 WEAGHDUYOAXCOA-UHFFFAOYSA-N 0.000 description 1
- XRXIKIBYZKKPQY-UHFFFAOYSA-N [C-]#[N+]C1=CC=C(C2=CN(C3=CC=CC(NC(C)=O)=C3)C(=O)C=C2)C=C1 Chemical compound [C-]#[N+]C1=CC=C(C2=CN(C3=CC=CC(NC(C)=O)=C3)C(=O)C=C2)C=C1 XRXIKIBYZKKPQY-UHFFFAOYSA-N 0.000 description 1
- XPUSYIBFJDFDCY-UHFFFAOYSA-N [C-]#[N+]C1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 Chemical compound [C-]#[N+]C1=CN(C2=CC=C(OC(F)(F)F)C=C2)C(=O)C=C1 XPUSYIBFJDFDCY-UHFFFAOYSA-N 0.000 description 1
- CKRFGQGULQKDBG-UHFFFAOYSA-N [C-]#[N+]C1=CN(C2=CC=CC(NC(C)=O)=C2)C(=O)C=C1 Chemical compound [C-]#[N+]C1=CN(C2=CC=CC(NC(C)=O)=C2)C(=O)C=C1 CKRFGQGULQKDBG-UHFFFAOYSA-N 0.000 description 1
- LXYOKPKJWBWFDP-UHFFFAOYSA-N [C-]#[N+]C1=CN(C2=CC=CC=C2)C(=O)C=C1 Chemical compound [C-]#[N+]C1=CN(C2=CC=CC=C2)C(=O)C=C1 LXYOKPKJWBWFDP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/08—Antiseborrheics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/08—Antibacterial agents for leprosy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
- A61P5/16—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4 for decreasing, blocking or antagonising the activity of the thyroid hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/12—Antidiuretics, e.g. drugs for diabetes insipidus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/06—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom containing only hydrogen and carbon atoms in addition to the ring nitrogen atom
- C07D213/22—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom containing only hydrogen and carbon atoms in addition to the ring nitrogen atom containing two or more pyridine rings directly linked together, e.g. bipyridyl
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/64—One oxygen atom attached in position 2 or 6
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/69—Two or more oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/70—Sulfur atoms
- C07D213/71—Sulfur atoms to which a second hetero atom is attached
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/84—Nitriles
- C07D213/85—Nitriles in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/14—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/34—One oxygen atom
- C07D239/36—One oxygen atom as doubly bound oxygen atom or as unsubstituted hydroxy radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/52—Two oxygen atoms
- C07D239/54—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/18—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D251/00—Heterocyclic compounds containing 1,3,5-triazine rings
- C07D251/02—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings
- C07D251/10—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- This invention relates to compounds and methods useful in treating various inflammatory and fibrotic conditions, including those associated with enhanced activity of kinase p38.
- cytokines participate in this response, including IL-1, IL-6, IL-8 and TNF ⁇ . It appears that the activity of these cytokines in the regulation of inflammation may be associated with the activation of an enzyme on the cell signaling pathway, a member of the MAP kinase family generally known as p38 and also known as SAPK, CSBP and RK.
- inhibitors of p38 such as NPC 31169, SB239063, SB203580, FR-167653, and pirfenidone have been tested in vitro and/or in vivo and found to be effective for modulating inflammatory responses.
- M is N or CR 1 ;
- A is N or CR 2 ;
- L is N or CR 3 ;
- B is N or CR 4 ;
- E is N or CX 4 ;
- G is N or CX 3 ;
- J is N or CX 2 ;
- K is N or CX 1 ;
- R 1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl, alkenyl, cyano, sulfonamido, halo, aryl, alkenylenearyl, and heteroaryl;
- R 2 is selected from the group consisting of hydrogen, alkyl, haloalkyl, halo, cyano, aryl, alkenyl, alkenylenearyl, heteroaryl, haloalkylcarbonyl, cycloalkyl, hydroxylalkyl, sulfonamido, and
- the compounds of formula (I) have a structure of formula (II) or (III):
- X 6 and X 7 are independently selected from the group consisting of hydrogen, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, alkylenylaryl, alkylenylheteroaryl, alkylenylheterocycloalkyl, alkylenylcycloalkyl, or X 6 and X 7 together form an optionally substituted 5 or 6 membered heterocyclic ring.
- the compound of formula (I) is a compound selected from the group recited in Table 1, below.
- a compound disclosed herein preferably exhibits an IC 50 in the range of about 0.1 ⁇ M to about 1000 ⁇ M, and preferably about 1 ⁇ M to about 800 ⁇ M, about 1 ⁇ M to about 500 ⁇ M, about 1 ⁇ M to about 300 ⁇ M, about 1 ⁇ M to about 200 ⁇ M, or about 1 ⁇ M to about 100 ⁇ M for inhibition of p38 MAPK.
- composition including the compound of formula (I) and a pharmaceutically acceptable excipient.
- SAPK stress activated protein kinase
- MAPK mitogen-activated protein kinase
- the contacting is conducted at a SAPK-modulating concentration that is less than an IC 30 for inhibition of the p38 MAPK by the compound.
- Contemplated p38 MAPKs include, but are not limited to, p38 ⁇ , p38 ⁇ , p38 ⁇ , and p38 ⁇ .
- the concentration of the compound disclosed herein is effective to alter TNF ⁇ release in whole blood by at least 15%.
- a SAPK system in a subject in need thereof, comprising administering to the subject a therapeutically effective amount of a compound as disclosed herein, wherein the compound exhibits an IC 50 in the range of about 0.1 ⁇ M to about 1000 ⁇ M for inhibition of p38 MAPK; and the therapeutically effective amount produces a blood or serum concentration of the compound that is less than an IC 30 for inhibition of p38 mitogen-activated protein kinase (MAPK).
- the subject suffers from an inflammatory condition.
- the subject preferably is a mammal, more preferably human.
- the compound can be administered to the subject on a schedule selected from the group consisting of three times a day, twice a day, once a day, once every two days, three times a week, twice a week, and once a week.
- MAPKs extracellular signal-regulated kinase
- ERK extracellular signal-regulated kinase
- p38 extracellular signal-regulated kinase
- SAPK/JNK SAPK/JNK
- MAPKs are activated by their specific MAPK kinases (MAPKKs): ERK by MEK1 and MEK2, p38 by MKK3 and MKK6, and SAPK/JNK by SEK1 (also known as MKK4) and MKK7 (SEK2).
- MAPKKs MAPK kinases
- SEK1 also known as MKK4
- SEK7 SEK7
- the MAPK network involves at least twelve cloned highly conserved, proline-directed serine-threonine kinases which, when activated by cell stresses (e.g., oxidative stress, DNA damage, heat or osmotic shock, ultraviolet irradiation, ischemia-reperfusion), exogenous agents (e.g., anisomycin, Na arsenite, lipopolysaccharide, LPS) or pro-inflammatory cytokines, TNF- ⁇ and IL-1 ⁇ , can phosphorylate and activate other kinases or nuclear proteins such as transcription factors in either the cytoplasm or the nucleus.
- cell stresses e.g., oxidative stress, DNA damage, heat or osmotic shock, ultraviolet irradiation, ischemia-reperfusion
- exogenous agents e.g., anisomycin, Na arsenite, lipopolysaccharide, LPS
- pro-inflammatory cytokines TNF- ⁇ and IL-1 ⁇
- p38 MAP kinase pathway is directly or indirectly activated by cell surface receptors, such as receptor tyrosine kinases, chemokines or G protein-coupled receptors, which have been activated by a specific ligand, e.g., cytokines, chemokines or lipopolysaccharide (LPS) binding to a cognate receptor.
- a specific ligand e.g., cytokines, chemokines or lipopolysaccharide (LPS) binding to a cognate receptor.
- p38 MAP kinase is activated by phosphorylation on specific threonine and tyrosine residues.
- p38 MAP kinase can phosphorylate other intracellular proteins, including protein kinases, and can be translocated to the cell nucleus, where it phosphorylates and activates transcription factors leading to the expression of pro-inflammatory cytokines and other proteins that contribute to the inflammatory response, cell adhesion, and proteolytic degradation.
- pro-inflammatory cytokines and other proteins that contribute to the inflammatory response, cell adhesion, and proteolytic degradation.
- cytokines and other proteins that contribute to the inflammatory response, cell adhesion, and proteolytic degradation.
- myeloid lineage such as macrophages and monocytes
- both IL-1 ⁇ and TNF ⁇ are transcribed in response to p38 activation.
- Subsequent translation and secretion of these and other cytokines initiates a local or systemic inflammatory response in adjacent tissue and through infiltration of leukocytes.
- cytokine gene products In alveolar macrophages, inhibition of p38 kinases with p38 inhibitor, SB203580, reduces cytokine gene products. It is believed that inflammatory cytokines (TNF- ⁇ , IFN- ⁇ , IL-4, IL-5) and chemokines (IL-8, RANTES, eotaxin) are capable of regulating or supporting chronic airway inflammation. The production and action of many of the potential mediators of airway inflammation appear to be dependent upon the stress-activated MAP kinase system (SAPK) or p38 kinase cascade (Underwood et al. Prog Respir Res 31:342-345 (2001)).
- SAPK stress-activated MAP kinase system
- p38 kinase cascade Underwood et al. Prog Respir Res 31:342-345 (2001)
- Protein kinase substrates of p38 ⁇ or p38 ⁇ include MAP kinase-activated protein kinase 2 (MAPKAPK2 or M2), MAP kinase interaction protein kinase (MNK1), p38 regulated/activated kinase (PRAK), mitogen- and stress-activated kinase (MSK: RSK-B or RLPK).
- MAPKAPK2 or M2 MAP kinase-activated protein kinase 2
- MNK1 MAP kinase interaction protein kinase
- PRAK p38 regulated/activated kinase
- MSK mitogen- and stress-activated kinase
- halo used herein refers to fluoro, chloro, bromo, or iodo.
- alkenylene is defined identical as “alkylene,” except the group contains at least one carbon-carbon double bond.
- aryl refers to a monocyclic or polycyclic aromatic group, preferably a monocyclic or bicyclic aromatic group, e.g., phenyl or naphthyl. Unless otherwise indicated, an aryl group can be unsubstituted or substituted with one or more, and in particular one to four groups independently selected from, for example, halo, alkyl, alkenyl, OCF 3 , NO 2 , CN, NC, OH, alkoxy, haloalkoxy, amino, CO 2 H, CO 2 alkyl, aryl, and heteroaryl.
- aryl groups include, but are not limited to, phenyl, naphthyl, tetrahydronaphthyl, chlorophenyl, methylphenyl, methoxyphenyl, trifluoromethylphenyl, nitrophenyl, 2,4-methoxychlorophenyl, and the like.
- heteroaryl refers to a monocyclic or bicyclic ring system containing one or two aromatic rings and containing at least one nitrogen, oxygen, or sulfur atom in an aromatic ring. Unless otherwise indicated, a heteroaryl group can be unsubstituted or substituted with one or more, and in particular one to four, substituents selected from, for example, halo, alkyl, alkenyl, OCF 3 , NO 2 , CN, NC, OH, alkoxy, haloalkoxy, amino, CO 2 H, CO 2 alkyl, aryl, and heteroaryl.
- heteroaryl groups include, but are not limited to, thienyl, furyl, pyridyl, oxazolyl, quinolyl, thiophenyl, isoquinolyl, indolyl, triazinyl, triazolyl, isothiazolyl, isoxazolyl, imidazolyl, benzothiazolyl, pyrazinyl, pyrimidinyl, thiazolyl, and thiadiazolyl.
- haloalkyl used herein refers to one or more halo groups appended to an alkyl group.
- nitroalkyl used herein refers to one or more nitro groups appended to an alkyl group.
- hydroxyalkyl refers to one or more hydroxy groups appended to an alkyl group.
- alkoxy refers to straight or branched chain alkyl group covalently bonded to the parent molecule through an —O— linkage.
- alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, isopropoxy, butoxy, n-butoxy, sec-butoxy, t-butoxy and the like.
- amido refers to —NHC(O)alkyl or —NHC(O)H.
- a non-limiting example of an amido group is —NHC(O)CH 3 .
- carboxy or “carboxyl” used herein refers to —COOH or its deprotonated form —COO ⁇ .
- alkylcarbonyl refers to —(CO)-alkyl.
- alkylcarbonyl groups include, but are not limited to, methylcarbonyl group, ethylcarbonyl group, propylcarbonyl group, and the like.
- sulfonyl refers to —SO 2 alkyl.
- a sulfonyl group is methylsulfonyl (e.g., —SO 2 CH 3 ).
- uronide refers to a monosaccharide having a carboxyl group on the carbon that is not part of the ring.
- the uronide name retains the root of the monosaccharide, but the -ose sugar suffix is changed to -uronide.
- the structure of glucuronide corresponds to glucose.
- a radical indicates species with a single, unpaired electron such that the species containing the radical can be covalently bonded to another species.
- a radical is not necessarily a free radical. Rather, a radical indicates a specific portion of a larger molecule.
- the term “radical” can be used interchangeably with the term “group.”
- Asymmetric carbon atoms can be present. All such isomers, including diastereomers and enantiomers, as well as the mixtures thereof, are intended to be included in the scope of the disclosure herein. In certain cases, compounds can exist in tautomeric forms. All tautomeric forms are intended to be included in the scope of the disclosure herein. Likewise, when compounds contain an alkenyl or alkenylene group, there exists the possibility of cis- and trans-isomeric forms of the compounds. Both cis- and trans-isomers, as well as the mixtures of cis- and trans-isomers, are contemplated.
- M is N or CR 1 ;
- A is N or CR 2 ;
- L is N or CR 3 ;
- B is N or CR 4 ;
- E is N or CX 4 ;
- G is N or CX 3 ;
- J is N or CX 2 ;
- K is N or CX 1 ;
- R 1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl, alkenyl, cyano, sulfonamido, halo, aryl, alkenylenearyl, and heteroaryl;
- R 2 is selected from the group consisting of hydrogen, alkyl, haloalkyl, halo, cyano, aryl, alkenyl, alkenylenearyl, heteroaryl, haloalkylcarbonyl, cycloalkyl, hydroxyalkyl, sulfonamido, and
- only A is N. In various embodiments, only E and J are each N. In some embodiments, only B is N. In various embodiments, only G is N. In some embodiments, only K is N. In various embodiments, only E is N. In some embodiments, only J is N. In some embodiments, only L is N. In various embodiments, only M is N.
- R 1 is selected from the group consisting of hydrogen, 4-pyridyl, cyclopropanyl, 4-fluorophenyl, 2-furanyl, cyano, H 2 NSO 2 , (CH 3 ) 2 NSO 2 , 4-sulfonamido-phenyl, fluoro, 4-(3,5-dimethyl)-isoxazolyl, 4-pyrazolyl, 4-(1-methyl)-pyrazolyl, 5-pyrimidinyl, 1-piperazinyl, 1-morpholinyl, 1-pyrrolidinyl, 2-imidazolyl, and thiazolyl.
- the compound of formula (I) is a compound of formula (II):
- R 1 , R 2 , R 3 , or R 4 is not selected from the group consisting of hydrogen, alkyl, alkenyl, haloalkyl, hydroxyalkyl, alkoxy, phenyl, substituted phenyl, halo, hydroxy, and alkoxyalkyl.
- R 1 and R 2 together form an optionally substituted 5-membered nitrogen-containing heterocyclic ring.
- the compound of formula (I) is a compound of formula (III) or formula (IV):
- X 8 is hydrogen or alkyl
- X 6 and X 7 are independently selected from the group consisting of hydrogen, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, alkylenylaryl, alkylenylheteroaryl, alkylenylheterocycloalkyl, alkylenylcycloalkyl, or X 6 and X 7 together form an optionally substituted 5 or 6 membered heterocyclic ring.
- X 7 is hydrogen.
- X 8 is methyl.
- At least one of X 1 , X 2 , X 3 , X 4 , or X 5 is alkyl, for example, haloalkyl. In various embodiments, at least one of X 1 , X 2 , X 3 , X 4 , or X 5 is alkenyl. In some embodiments, at least one of X 1 , X 2 , X 3 , X 4 , or X 5 is amino. In various embodiments, at least one of X 1 , X 2 , X 3 , X 4 , or X 5 is thioalkyl.
- At least one of X 1 , X 2 , X 3 , X 4 , or X 5 is aryloxy. In various embodiments, at least one of X 1 , X 2 , X 3 , X 4 , or X 5 is arylalkoxy. In some embodiments, at least one of X 1 , X 2 , X 3 , X 4 , or X 5 is alkoxyalkyl. In various embodiments, at least one of X 1 , X 2 , X 3 , X 4 , or X 5 is alkylcarbonyl.
- the compound can also be synthesized using a scheme as depicted below, where a triflate intermediate is used in the Suzuki coupling.
- step 1 the procedure is the same as for general procedure A.
- step 2 a solution of the intermediate benzyl protected phenol (3.5 g, 10.8 mmol) in methanol (200 ml) is added to a Pd/C (300 mg) catalyst under N 2 atmosphere, and then stirred for 2 h under H 2 atmosphere (1 atm, 25° C.). The catalyst is filtered off through a celite pad, and the filtrate is concentrated in vacuo to give the free phenolic hydroxyl.
- step 3 a solution of the resulting phenol intermediate (2.2 g, 11.8 mmol) in dichloromethane (DCM, 120 mL) is added to triethylamine (1.7 g, 16.8 mmol) at ⁇ 78° C., followed by the addition of trifluoromethanesulfonic anhydride (4.76 g, 16.9 mmol). The resulting mixture is stirred at ⁇ 78° C. for 15 min and quenched with ammonium chloride solution (10 mL). After warming to room temperature, water (30 mL) and DCM (50 mL) are added and separated. The target product is obtained by washing the crude mixture with methanol.
- DCM dichloromethane
- step 4 a solution of trifluoromethanesulfonic acid intermediate (0.79 mmol) and tetrakis(triphenylphosphine)palladium (0.011 g, 0.0095 mmol) in dimethoxyethane (DME, 1 mL) is stirred at room temperature for 15 min followed by the addition of the solution arylboronic acid (0.21 mmol) in DME (1 mL) and 2M sodium carbonate (1 mL). The resulting mixture is refluxed for 14 hr and cooled down to room temperature. Water and ethyl acetate are added. After separation, the aqueous layer is extracted with ethyl acetate. The combined ethyl acetate solution is dried over sodium sulfate and filtered. The filtrate is concentrated in vacuo to dryness. Target products are isolated by prep-TLC.
- the compound of formula (I) has at least one fluorine atom as a substituent.
- Introduction of the fluorine can be accomplished, as outlined in Scheme 6.
- the compounds of formula I are prepared using a Chan-Lam reaction, as depicted in Scheme 8, above.
- the Chan-Lam synthetic procedure is as follows (Method HIA H1A—where reagent A is a solid): to a solution of A (0.5 mmol) in 6 mL of DCM and 2 mL of DMF, copper (II) acetate (1.0 mmol, 2 eq), boronic acid B (1.2 eq), pyridine (2 eq) and finely ground, activated 4 ⁇ molecular sieves (600 mg) are added.
- the reagent A is a hydrobromide salt
- TEA (2 mL) is added.
- the mixture is stirred at room temperature in the open air for 12 hours up to about 4 days. Additional boronic acid B can be added to the reaction mixture. Then, concentrated NH 4 OH is added. The solvents are evaporated under vacuum, and the crude product absorbed on a silica pad and purified by chromatographic column. In some specific cases, the product is further purified on reverse-phase preparative HPLC.
- Method H1B starts with reagent A as a solution in DMF.
- This procedure is as follows: to a solution of A (2.5 mL of DMF solution, 0.74 mmol) in 5 mL of DMF, copper (II) acetate (1.48 mmol, 2 eq), boronic acid B (1.2 eq), pyridine (2 eq) and finely ground, activated 4 ⁇ molecular sieves (600 mg) are added. The mixture is stirred at room temperature in the open air for 12 hours up to about 4 days. Additional boronic acid B can be further added. Then, concentrated NH 4 OH is added. The solvents are evaporated under vacuum, and the crude product is absorbed on silica pad and purified by chromatographic column. In some specific cases, the product is further purified on reverse-phase preparative HPLC.
- the Chan-Lam reaction proceeds as follows (Method H2): to a 0.3 M solution of A in DMF, copper (II) acetate (2 eq), boronic acid B (1.2 eq) and pyridine (2 eq) are added. The mixture is heated 1 h at 100° C. under microwave irradiation, then concentrated NH 4 OH is added. The reaction mixture then is diluted with EA and filtered through a celite pad. Solvents are evaporated, and the crude mixture absorbed on silica pad and purified by chromatographic column. In some specific cases, the product is further purified on reverse-phase preparative HPLC.
- reaction was quenched by slowly adding 700 mL of water and extracted with EA, washed with brine and dried over Na 2 SO 4 to obtain 100 g of 3 (1-tert-butyl 2-(ethoxycarbonyl)-2-hydroxy-2,3-dihydro-1H-pyrrolo[2,3-b]pyridine-1,2-dicarboxylate) as a crude orange oil.
- 5-bromo-pyridin-2-one (1 eq.) is dissolved in DCM (5 mL/mmol of aryl halide) and N,N-dimethylformammide (0.7 mL/mmol of aryl halide).
- the appropriate boronic acid (1.2 eq.), copper(II) acetate (2.0 eq.), pyridine (2.0 eq.) and 4 ⁇ molecular sieves are added to the solution and the reaction is stirred at room temperature in an open vessel for 3 days. The reaction is monitored by UPLC-MS. At the end of the reaction a concentrated solution of NH 4 OH is added.
- Method L2 is as follows: ⁇ BINAP (0.2 eq.) is suspended in dry toluene (7.5 mL/mmol of aryl halide) and Pd 2 dba 3 (0.1 eq) is added. The mixture is stirred for 15 minutes, then the appropriate bromopyridone (1 eq.) is added, followed by the appropriate amine (5 eq.) and NaOtBu (1.4 eq.). The reaction is heated at 80° C. for 15 h, and then cooled at room temperature. Solvents are evaporated and the crude product is purified by flash chromatography (SiO 2 ; Pet. Ether/EtOAc 3:1 up to pure EtOAc) then by reverse-phase preparative HPLC.
- methods for modulating a SAPK system, in vitro or in vivo.
- the methods include contacting a SAPK-modulating concentration of a compound with a p38 MAPK (e.g., by contacting the compound with a cell or tissue containing the p38 MAPK), wherein the compound has a relatively low potency for inhibition of the p38 MAPK, corresponding to a relatively high inhibitory concentration for inhibition of the p38 MAPK by the compound.
- the inhibitory concentration (IC) is a concentration that results in a reduction in the activity of p38 MAPK by a specified percentage (e.g., 50%, 40%, 30%, 20%, 10%) on a dose-response curve.
- IC 50 , IC 40 , IC 30 , IC 20 and IC 10 are determined as concentrations that result in reductions in the activity of p38 MAPK by 50%, 40%, 30%, 20% and 10%, respectively on a dose-response curve.
- modulation of the SAPK system may involve contacting a compound (e.g., a compound of Formula I) with a p38 MAPK at a concentration that is less than an IC 40 , preferably less than IC 30 , more preferably less than IC 20 , even preferably less than IC 10 for inhibition of the p38 MAPK by the compound as determined on a concentration-response curve.
- a compound e.g., a compound of Formula I
- a p38 MAPK at a concentration that is less than an IC 40 , preferably less than IC 30 , more preferably less than IC 20 , even preferably less than IC 10 for inhibition of the p38 MAPK by the compound as determined on a concentration-response curve.
- Contacting a cell refers to a condition in which a compound or other composition of matter is in direct contact with a cell or tissue, or is close enough to induce a desired biological effect in a cell or tissue.
- contacting a cell or tissue containing p38 MAPK with a compound may be conducted in any manner that permits an interaction between p38 MAPK and the compound, resulting in the desired biological effect in a cell.
- Contacting a cell or tissue may be accomplished, for example, by intermixing or administering a compound (such as a compound of Formula I; and/or a salt, ester, prodrug and/or intermediate thereof, and/or a pharmaceutical composition comprising one or more of the foregoing).
- contacting a cell or tissue may be accomplished by introducing a compound in a manner such that the compound will be targeted, directly or indirectly, to a cell or tissue containing p38 MAPK.
- Contacting a cell or tissue may be accomplished under conditions such that a compound binds to the p38 MAPK. Such conditions may include proximity of the compound and p38-containing cell or tissue, pH, temperature, or any condition that affects the binding of a compound to p38 MAPK.
- the effective concentration is a concentration that results in a reduction in the activity of a p38 MAPK by a specified percentage (e.g., 50%, 40%, 30%, 20%, 10%) as measured by a specific physiological response which depends on the reduction of the activity of the p38 MAPK.
- physiological response may be, for example, reduction in blood or other bodily fluid concentration of TNF ⁇ .
- EC 50 , EC 40 , EC 30 , EC 20 and EC 10 are determined as concentrations that result in reductions in the activity of a p38 MAPK as measured by reduction in TNF ⁇ concentration by 50%, 40%, 30%, 20% and 10%, respectively on a dose-response curve.
- the EC 50 of the SAPK system-modulating compound is preferably in the range of about 100 ⁇ M to about 1000 ⁇ M, more preferably about 200 ⁇ M to about 800 ⁇ M for inhibition of the p38 MAPK.
- modulation of the SAPK system may involve contacting a compound (e.g., a compound of Formula I) with a p38 MAPK at a concentration that is less than an EC 40 , preferably less than EC 30 , more preferably less than EC 20 , even preferably less than EC 10 for inhibition of the p38 MAPK by the compound as determined on a dose-response curve in vivo.
- the compound can be provided in the form of a pharmaceutical composition, together with a pharmaceutically acceptable carrier.
- the methods further comprise contacting a p38 MAPK with the plurality of compounds, and determining whether the compounds inhibit the activity of cytokines.
- the p38 MAPK is preferably selected from the group consisting of p38 ⁇ , p38 ⁇ , p38 ⁇ , and p38 ⁇ .
- the contacting step takes place in vitro.
- the contacting step comprises contacting a cell comprising p38 MAPK with the compound.
- methods are provided for inhibiting the activity of a p38 MAPK in a cell, in vitro or in vivo.
- such methods include contacting a cell containing a p38 MAPK with an effective p38-inhibiting amount of a compound (e.g., a compound of Formula I), under conditions such that p38 activity in the cell is inhibited. Examples of such methods are provided in the EXAMPLES section below.
- the method of identifying a pharmaceutically active compound may further include determining a mammalian toxicity of the selected compound. Such methods are generally known to those skilled in the art.
- the method of identifying a pharmaceutically active compound may also include administering the selected compound to a test subject, either in conjunction with the determination of mammalian toxicity or for other reasons.
- the test subject test subject has or is at risk for having an inflammatory condition.
- the test subject is a mammal, and can be a human.
- p38-associated condition means a disease or other deleterious condition in which the p38 MAP kinase signaling pathway is implicated, whether directly or indirectly.
- Examples of p38-associated conditions include conditions caused by IL-1 ⁇ , TNF ⁇ , IL-6 or IL-8 dysregulation or overexpression resulting from sustained, prolonged, enhanced or elevated levels of p38 activity.
- a “fibrotic condition,” “fibroproliferative condition,” “fibrotic disease,” “fibroproliferative disease,” “fibrotic disorder,” and “fibroproliferative disorder” are used interchangeably to refer to a condition, disease or disorder that is characterized by dysregulated proliferation or activity of fibroblasts and/or pathologic or excessive accumulation of collagenous tissue. Typically, any such disease, disorder or condition is amenable to treatment by administration of a compound having anti-fibrotic activity.
- Fibrotic disorders include, but are not limited to, pulmonary fibrosis, including idiopathic pulmonary fibrosis (IPF) and pulmonary fibrosis from a known etiology, liver fibrosis, and renal fibrosis.
- Other exemplary fibrotic conditions include musculoskeletal fibrosis, cardiac fibrosis, post-surgical adhesions, scleroderma, glaucoma, and skin lesions such as keloids.
- Such conditions include, without limitation, inflammatory diseases, autoimmune diseases, fibrotic diseases, destructive bone disorders, proliferative disorders, infectious diseases, neurodegenerative diseases, allergies, reperfusion/ischemia in stroke, heart attacks, angiogenic disorders, organ hypoxia, vascular hyperplasia, cardiac hypertrophy, thrombin-induced platelet aggregation, and conditions associated with the cyclooxygenase and lipoxygenase signaling pathways, such as prostaglandin endoperoxide synthase.
- a cytokine-associated condition can include any condition associated with or mediated by IL-1 (particularly IL-1 ⁇ ), TNF ⁇ , IL-6 or IL-8, or any other cytokine which can be regulated by p38.
- the cytokine associated condition is a condition associated with TNF ⁇ .
- the methods described herein may also be used to treat autoimmune diseases and diseases associated with acute and chronic inflammation.
- diseases include, but are not limited to: chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), rheumatoid arthritis; rheumatoid spondylitis; osteoarthritis; gout, other arthritic conditions; sepsis; septic shock; endotoxic shock; gram-negative sepsis; toxic shock syndrome; myofacial myofascial pain syndrome (MPS); Shigellosis; asthma; adult respiratory distress syndrome; inflammatory bowel disease; Crohn's disease; psoriasis; eczema; ulcerative colitis; glomerular nephritis; scleroderma; chronic thyroiditis; Grave's disease; Ormond's disease; autoimmune gastritis; myasthenia gravis; autoimmune hemolytic anemia; autoimmune neutropenia;
- NSCLC non-small cell lung cancer
- pain disorders including neuromuscular pain, headache, cancer pain, dental pain, and arthritis pain
- angiogenic disorders including solid tumor angiogenesis, ocular neovascularization, and infantile hemangioma
- conditions associated with the cyclooxygenase and lipoxygenase signaling pathways including conditions associated with prostaglandin endoperoxide synthase-2 (including edema, fever, analgesia, and pain)
- organ hypoxia thrombin-induced platelet aggregation.
- the methods described herein may be useful for the treatment of protozoal diseases in animals, including mammals.
- a subject may include one or more cells or tissues, or organisms.
- a preferred subject is a mammal.
- a mammal can include any mammal.
- preferred mammals include cattle, pigs, sheep, goats, horses, camels, buffalo, cats, dogs, rats, mice, and humans.
- a highly preferred subject mammal is a human.
- the compound(s) can be administered to the subject via any drug delivery route.
- Specific exemplary administration routes include oral, ocular, rectal, buccal, topical, nasal, ophthalmic, subcutaneous, intramuscular, intravenous (bolus and infusion), intracerebral, transdermal, and pulmonary.
- therapeutically effective amount and “prophylactically effective amount,” as used herein, refer to an amount of a compound sufficient to treat, ameliorate, or prevent the identified disease or condition, or to exhibit a detectable therapeutic, prophylactic, or inhibitory effect. The effect can be detected by, for example, the assays disclosed in the following examples.
- the precise effective amount for a subject will depend upon the subject's body weight, size, and health; the nature and extent of the condition; and the therapeutic or combination of therapeutics selected for administration.
- Therapeutically and prophylactically effective amounts for a given situation can be determined by routine experimentation that is within the skill and judgment of the clinician.
- the effective amount of the compound of the embodiments produces a blood or serum or another bodily fluid concentration that is less than an IC 30 , IC 20 or IC 10 for inhibition of p38 MAP kinase.
- the effective amount of the compound of the embodiments produces a blood or serum or another bodily fluid concentration that is effective to alter TNF ⁇ secretion from whole blood by 10%, 15%, 20%, 30%, 40% or 50%.
- the therapeutically or prophylactically effective amount can be estimated initially either in cell culture assays, e.g., of neoplastic cells, or in animal models, usually rats, mice, rabbits, dogs, or pigs.
- the animal model may also be used to determine the appropriate concentration range and route of administration. Such information can then be used to determine useful doses and routes for administration in humans.
- Therapeutic/prophylactic efficacy and toxicity may be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., ED 50 (the dose therapeutically effective in 50% of the population) and LD 50 (the dose lethal to 50% of the population).
- the dose ratio between therapeutic and toxic effects is the therapeutic index, and it can be expressed as the ratio, ED 50 /LD 50 .
- Pharmaceutical compositions that exhibit large therapeutic indices are preferred. However, pharmaceutical compositions that exhibit narrow therapeutic indices are also within the scope of the invention.
- the data obtained from cell culture assays and animal studies may be used in formulating a range of dosage for human use.
- the dosage contained in such compositions is preferably within a range of circulating concentrations that include an ED 50 with little or no toxicity. The dosage may vary within this range depending upon the dosage form employed, sensitivity of the patient, and the route of administration.
- the maximum plasma concentrations (Cmax) can range from about 0.1 ⁇ M to about 200 ⁇ M.
- Cmax can be about 0.5 ⁇ M to about 175 ⁇ M, about 65 ⁇ M to about 115 ⁇ M, or about 75 ⁇ M to about 105 ⁇ M, or about 85 ⁇ M to about 95 ⁇ M, or about 85 ⁇ M to about 90 ⁇ M depending upon the route of administration.
- Cmax can be about 1 ⁇ M to about 50 ⁇ M, about 1 ⁇ M to about 25 ⁇ M, about 1 ⁇ M to about 20 ⁇ M, about 1 ⁇ M to about 15 ⁇ M, about 1 ⁇ M to about 10 ⁇ M, about 1 ⁇ M to about 5 ⁇ M.
- Specific Cmax values can be about 1 ⁇ M, about 2 ⁇ M, about 3 ⁇ M, about 4 ⁇ M, about 5 ⁇ M, about 6 ⁇ M, about 7 ⁇ M, about 8 ⁇ M, about 9 ⁇ M, about 10 ⁇ M, about 11 ⁇ M, about 12 ⁇ M, about 13 ⁇ M, about 14 ⁇ M, about 15 ⁇ M, about 16 ⁇ M, about 17 ⁇ M, about 18 ⁇ M, about 19 ⁇ M, about 20 ⁇ M, about 21 ⁇ M, about 22 ⁇ M, about 23 ⁇ M, about 24 ⁇ M, or about 25 ⁇ M.
- the dose will be in the range of about 100 mg/day to about 10 g/day, or about 200 mg to about 5 g/day, or about 400 mg to about 3 g/day, or about 500 mg to about 2 g/day, in single, divided, or continuous doses for a patient weighing between about 40 to about 100 kg (which dose may be adjusted for patients above or below this weight range, particularly children under 40 kg).
- the dose will be in the range of about 1 mg/kg to about 100 mg/kg of body weight per day.
- the exact dosage will be determined by the practitioner, in light of factors related to the subject that requires treatment. Dosage and administration are adjusted to provide sufficient levels of the active agent(s) or to maintain the desired effect. Factors which may be taken into account include the severity of the disease state, general health of the subject, age, weight, and gender of the subject, diet, time and frequency of administration, drug combination(s), reaction sensitivities, and tolerance/response to therapy. Long-acting pharmaceutical compositions may be administered every 3 to 4 days, every week, or once every two weeks depending on half-life and clearance rate of the particular formulation.
- treatment as described herein includes preventing a disease, ameliorating symptoms, slowing disease progression, reversing damage, or curing a disease.
- treating an inflammatory condition results in a decrease in the mortality rate of a population of treated subjects in comparison to a population of subjects receiving carrier alone.
- treating an inflammatory condition results in a decrease in the mortality rate of a population of treated subjects in comparison to an untreated population.
- treating an inflammatory condition results a decrease in the mortality rate of a population of treated subjects in comparison to a population receiving monotherapy with a drug that is not a compound of the embodiments, or a pharmaceutically acceptable salt, metabolite, analog or derivative thereof.
- the mortality rate is decreased by more than about 2%; more preferably, by more than about 5%; more preferably, by more than about 10%; and most preferably, by more than about 25%.
- a decrease in the mortality rate of a population of treated subjects may be measured by any reproducible means.
- a decrease in the mortality rate of a population may be measured, for example, by calculating for a population the average number of disease-related deaths per unit time following initiation of treatment with an active compound.
- a decrease in the mortality rate of a population may also be measured, for example, by calculating for a population the average number of disease related deaths per unit time following completion of a first round of treatment with an active compound.
- treating an inflammatory condition results in a decrease in growth rate of a tumor.
- tumor growth rate is reduced by at least about 5% relative to/number prior to treatment; more preferably, tumor growth rate is reduced by at least about 10%; more preferably, reduced by at least about 20%; more preferably, reduced by at least about 30%; more preferably, reduced by at least about 40%; more preferably, reduced by at least about 50%; even more preferably, reduced by at least 60%; and most preferably, reduced by at least about 75%.
- Tumor growth rate may be measured by any reproducible means of measurement. In a preferred aspect, tumor growth rate is measured according to a change in tumor diameter per unit time.
- treating an inflammatory condition results in a reduction in the rate of cellular proliferation.
- the rate of cellular proliferation is reduced by at least about 5%; more preferably, by at least about 10%; more preferably, by at least about 20%; more preferably, by at least about 30%; more preferably, by at least about 40%; more preferably, by at least about 50%; even more preferably, by at least about 60%; and most preferably, by at least about 75%.
- the rate of cellular proliferation may be measured by any reproducible means of measurement.
- the rate of cellular proliferation is measured, for example, by measuring the number of dividing cells in a tissue sample per unit time.
- treating an inflammatory condition results in a reduction in the proportion of proliferating cells.
- the proportion of proliferating cells is reduced by at least about 5%; more preferably, by at least about 10%; more preferably, by at least about 20%; more preferably, by at least about 30%; more preferably, by at least about 40%; more preferably, by at least about 50%; even more preferably, by at least about 60%; and most preferably, by at least about 75%.
- the proportion of proliferating cells may be measured by any reproducible means of measurement.
- the proportion of proliferating cells is measured, for example, by quantifying the number of dividing cells relative to the number of nondividing cells in a tissue sample.
- the proportion of proliferating cells is equivalent to the mitotic index.
- treating an inflammatory condition results in a decrease in size of an area or zone of cellular proliferation.
- size of an area or zone of cellular proliferation is reduced by at least 5% relative to its size prior to treatment; more preferably, reduced by at least about 10%; more preferably, reduced by at least about 20%; more preferably, reduced by at least about 30%; more preferably, reduced by at least about 40%; more preferably, reduced by at least about 50%; even more preferably, reduced by at least about 60%; and most preferably, reduced by at least about 75%.
- Size of an area or zone of cellular proliferation may be measured by any reproducible means of measurement.
- size of an area or zone of cellular proliferation may be measured as a diameter or width of an area or zone of cellular proliferation.
- the methods described herein may include identifying a subject in need of treatment.
- the methods include identifying a mammal in need of treatment.
- the methods include identifying a human in need of treatment. Identifying a subject in need of treatment may be accomplished by any means that indicates a subject who may benefit from treatment. For example, identifying a subject in need of treatment may occur by clinical diagnosis, laboratory testing, or any other means known to one of skill in the art, including any combination of means for identification.
- a tissue biopsy sample may be taken from a subject suffering from an inflammatory condition, e.g., a p38-associated or cytokine-associated condition.
- the biopsy sample can be tested to determine the level of p38 activity (or cytokine levels) present in the sample; the sample can then be contacted with a selected compound of the invention, and the p38 activity (or cytokine levels) measured to determine whether the compound has a desired effect (e.g., inhibition of p38 or cytokine activity with an IC 50 in the range of about about 0.1 ⁇ M to about 1000 ⁇ M, and preferably about 1 ⁇ M to about 800 ⁇ M, about 1 ⁇ M to about 500 ⁇ M, about 1 ⁇ M to about 300 ⁇ M, about 1 ⁇ M to about 200 ⁇ M, or about 1 ⁇ M to about 100 ⁇ M for inhibition of p38 MAPK).
- a desired effect e.g., inhibition of p38 or cytokine activity with an IC 50 in the
- renal fibrosis is considered to be the common final pathway by which kidney disease progresses to end-stage renal failure.
- the compounds and methods described herein are useful in the treatment of renal fibrosis.
- autoimmune diseases and diseases associated with chronic inflammation, as well as acute responses have been linked to activation of p38 MAP kinase and overexpression or dysregulation of inflammatory cytokines.
- diseases include, but are not limited to: rheumatoid arthritis; rheumatoid spondylitis; osteoarthritis; gout, other arthritic conditions; sepsis; septic shock; endotoxic shock; gram-negative sepsis; toxic shock syndrome; asthma; adult respiratory distress syndrome; chronic obstructive pulmonary disease; chronic pulmonary inflammation; inflammatory bowel disease; Crohn's disease; psoriasis; eczema; ulcerative colitis; pancreatic fibrosis; hepatic fibrosis; acute and chronic renal disease; irritable bowel syndrome; pyresis; restenosis; cerebral malaria; stroke and ischemic injury; neural trauma; Alzheimer's disease; Huntington's disease; Parkinson's disease; acute and chronic pain; allergic
- Cardiovascular disease Inflammation and leukocyte activation/infiltration play a major role in the initiation and progression of cardiovascular diseases including atherosclerosis and heart failure.
- Acute p38 mitogen-activated protein kinase (MAPK) pathway inhibition attenuates tissue damage and leukocyte accumulation in myocardial ischemia/reperfusion injury.
- the compounds and methods described herein are useful for treating cardiovascular disease.
- Inflammation in the central nervous system occurs in diseases such as multiple sclerosis and leads to axon dysfunction and destruction.
- NO nitric oxide
- p38 MAP kinase is activated by NO exposure and inhibition of p38 signalling was shown to lead to neuronal and axonal survival effects.
- OCM and IGF-1 reduced p38 activation in NO-exposed cortical neurons and improved axon survival in cultures exposed to NO, a process dependent on mitogen-activated protein kinase/extracellular signal-related kinase signalling.
- the compounds and methods described herein are useful for treating multiple sclerosis.
- Nonspecific inflammation is associated with primary graft nonfunction (PNF).
- PNF primary graft nonfunction
- Inflammatory islet damage is mediated at least partially by pro-inflammatory cytokines, such as interleukin-1 ⁇ (IL-1 ⁇ ) and tumor necrosis factor- ⁇ (TNF- ⁇ ) produced by resident islet macrophages.
- IL-1 ⁇ interleukin-1 ⁇
- TNF- ⁇ tumor necrosis factor- ⁇
- the p38 pathway is known to be involved in cytokine production in the cells of the monocyte-macrophage lineage. Inhibition of the p38 pathway by a chemical p38 inhibitor, SB203580, suppresses IL-1 ⁇ and TNF- ⁇ production in human islets exposed to lipopolysaccharide (LPS) and/or inflammatory cytokines.
- LPS lipopolysaccharide
- IL-1 ⁇ is predominantly produced by resident macrophages, ductal cells and islet vascular endothelial cells were found to be another cellular source of IL-1 ⁇ in isolated human islets.
- SB203580 also inhibited the expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) in the treated islets.
- iNOS inducible nitric oxide synthase
- COX-2 cyclooxygenase-2
- human islets treated with SB203580 for 1 h prior to transplantation showed significantly improved graft function.
- the compounds and methods described herein are useful for improving graft survival in clinical islet transplantation.
- Cisplatin is an important chemotherapeutic agent but can cause acute renal injury. Part of this acute renal injury is mediated through tumor necrosis factor- ⁇ (TNF- ⁇ ).
- TNF- ⁇ tumor necrosis factor- ⁇
- Cisplatin activates p38 MAPK and induces apoptosis in cancer cells.
- p38 MAPK activation leads to increased production of TNF- ⁇ in ischemic injury and in macrophages.
- cisplatin caused a dose dependent activation of p38 MAPK in proximal tubule cells. Inhibition of p38 MAPK activation led to inhibition of TNF- ⁇ production.
- mice treated with a single dose of cisplatin developed severe renal dysfunction, which was accompanied by an increase in kidney p38 MAPK activity and an increase in infiltrating leukocytes.
- animals treated with the p38 MAPK inhibitor SKF86002 along with cisplatin showed less renal dysfunction, less severe histologic damage and fewer leukocytes compared with cisplatin+vehicle treated animals.
- the compounds and methods described herein are useful for preventing acute renal injury.
- bradykinin BK
- IL-8 interleukin-8
- MAPK mitogen-activated protein kinase
- compositions useful in the methods of the invention are provided. More particularly, the pharmaceutical compositions described herein may be useful, inter alia, for treating or preventing inflammatory conditions, e.g., conditions associated with p38 activity or cytokine activity or any combination thereof.
- a pharmaceutical composition is any composition that may be administered in vitro or in vivo or both to a subject in order to treat or ameliorate a condition.
- a pharmaceutical composition may be administered in vivo.
- a subject may include one or more cells or tissues, or organisms.
- a preferred subject is a mammal.
- a mammal includes any mammal, such as by way of non-limiting example, cattle, pigs, sheep, goats, horses, camels, buffalo, cats, dogs, rats, mice, and humans.
- a highly preferred subject mammal is a human.
- the pharmaceutical compositions may be formulated with pharmaceutically acceptable excipients such as carriers, solvents, stabilizers, adjuvants, diluents, etc., depending upon the particular mode of administration and dosage form.
- the pharmaceutical compositions should generally be formulated to achieve a physiologically compatible pH, and may range from a pH of about 3 to a pH of about 11, preferably about pH 3 to about pH 7, depending on the formulation and route of administration. In alternative embodiments, it may be preferred that the pH is adjusted to a range from about pH 5.0 to about pH 8. More particularly, the pharmaceutical compositions may comprise a therapeutically or prophylactically effective amount of at least one compound as described herein, together with one or more pharmaceutically acceptable excipients.
- the pharmaceutical compositions may comprise a combination of the compounds described herein, or may include a second active ingredient useful in the treatment or prevention of bacterial infection (e.g., anti-bacterial or anti-microbial agents).
- Formulations are most typically solids, liquid solutions, emulsions or suspensions, while inhalable formulations for pulmonary administration are generally liquids or powders, with powder formulations being generally preferred.
- a preferred pharmaceutical composition may also be formulated as a lyophilized solid that is reconstituted with a physiologically compatible solvent prior to administration.
- Alternative pharmaceutical compositions may be formulated as syrups, creams, ointments, tablets, and the like.
- pharmaceutically acceptable excipient refers to an excipient for administration of a pharmaceutical agent, such as the compounds described herein.
- the term refers to any pharmaceutical excipient that may be administered without undue toxicity.
- compositions are determined in part by the particular composition being administered, as well as by the particular method used to administer the composition. Accordingly, there exists a wide variety of suitable formulations of pharmaceutical compositions (see, e.g., Remington's Pharmaceutical Sciences).
- Suitable excipients may be carrier molecules that include large, slowly metabolized macromolecules such as proteins, polysaccharides, polylactic acids, polyglycolic acids, polymeric amino acids, amino acid copolymers, and inactive virus particles.
- Other exemplary excipients include antioxidants (e.g., ascorbic acid), chelating agents (e.g., EDTA), carbohydrates (e.g., dextrin, hydroxyalkylcellulose, and/or hydroxyalkylmethylcellulose), stearic acid, liquids (e.g., oils, water, saline, glycerol and/or ethanol) wetting or emulsifying agents, pH buffering substances, and the like.
- Liposomes are also included within the definition of pharmaceutically acceptable excipients.
- compositions described herein may be formulated in any form suitable for an intended method of administration.
- tablets, troches, lozenges, aqueous or oil suspensions, non-aqueous solutions, dispersible powders or granules (including micronized particles or nanoparticles), emulsions, hard or soft capsules, syrups or elixirs may be prepared.
- Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions, and such compositions may contain one or more agents including sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide a palatable preparation.
- compositions particularly suitable for use in conjunction with tablets include, for example, inert diluents, such as celluloses, calcium or sodium carbonate, lactose, calcium or sodium phosphate; disintegrating agents, such as cross-linked povidone, maize starch, or alginic acid; binding agents, such as povidone, starch, gelatin or acacia; and lubricating agents, such as magnesium stearate, stearic acid or talc.
- inert diluents such as celluloses, calcium or sodium carbonate, lactose, calcium or sodium phosphate
- disintegrating agents such as cross-linked povidone, maize starch, or alginic acid
- binding agents such as povidone, starch, gelatin or acacia
- lubricating agents such as magnesium stearate, stearic acid or talc.
- Tablets may be uncoated or may be coated by known techniques including microencapsulation to delay disintegration and adsorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate alone or with a wax may be employed.
- Formulations for oral use may be also presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example celluloses, lactose, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with non-aqueous or oil medium, such as glycerin, propylene glycol, polyethylene glycol, peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example celluloses, lactose, calcium phosphate or kaolin
- non-aqueous or oil medium such as glycerin, propylene glycol, polyethylene glycol, peanut oil, liquid paraffin or olive oil.
- compositions may be formulated as suspensions comprising a compound of the embodiments in admixture with at least one pharmaceutically acceptable excipient suitable for the manufacture of a suspension.
- compositions may be formulated as dispersible powders and granules suitable for preparation of a suspension by the addition of suitable excipients.
- Excipients suitable for use in connection with suspensions include suspending agents (e.g., sodium carboxymethylcellulose, methylcellulose, hydroxypropyl methylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth, gum acacia); dispersing or wetting agents (e.g., a naturally occurring phosphatide (e.g., lecithin), a condensation product of an alkylene oxide with a fatty acid (e.g., polyoxyethylene stearate), a condensation product of ethylene oxide with a long chain aliphatic alcohol (e.g., heptadecaethyleneoxycethanol), a condensation product of ethylene oxide with a partial ester derived from a fatty acid and a hexitol anhydride (e.g., polyoxyethylene sorbitan monooleate)); and thickening agents (e.g., carbomer, beeswax, hard paraffin or cetyl alcohol).
- suspending agents
- the suspensions may also contain one or more preservatives (e.g., acetic acid, methyl or n-propyl p-hydroxy-benzoate); one or more coloring agents; one or more flavoring agents; and one or more sweetening agents such as sucrose or saccharin.
- preservatives e.g., acetic acid, methyl or n-propyl p-hydroxy-benzoate
- coloring agents e.g., acetic acid, methyl or n-propyl p-hydroxy-benzoate
- flavoring agents e.g., methyl or n-propyl p-hydroxy-benzoate
- sweetening agents such as sucrose or saccharin.
- the pharmaceutical compositions may also be in the form of oil-in water emulsions.
- the oily phase may be a vegetable oil, such as olive oil or arachis oil, a mineral oil, such as liquid paraffin, or a mixture of these.
- Suitable emulsifying agents include naturally-occurring gums, such as gum acacia and gum tragacanth; naturally occurring phosphatides, such as soybean lecithin, esters or partial esters derived from fatty acids; hexitol anhydrides, such as sorbitan monooleate; and condensation products of these partial esters with ethylene oxide, such as polyoxyethylene sorbitan monooleate.
- the emulsion may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, such as glycerol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, a flavoring or a coloring agent.
- sweetening agents such as glycerol, sorbitol or sucrose.
- Such formulations may also contain a demulcent, a preservative, a flavoring or a coloring agent.
- compositions may be in the form of a sterile injectable preparation, such as a sterile injectable aqueous emulsion or oleaginous suspension.
- a sterile injectable preparation such as a sterile injectable aqueous emulsion or oleaginous suspension.
- This emulsion or suspension may be formulated by a person of ordinary skill in the art using those suitable dispersing or wetting agents and suspending agents, including those mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, such as a solution in 1,2-propane-diol.
- the sterile injectable preparation may also be prepared as a lyophilized powder.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution, and isotonic sodium chloride solution.
- sterile fixed oils may be employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids e.g., oleic acid
- a pharmaceutically acceptable salt of a compound described herein may be dissolved in an aqueous solution of an organic or inorganic acid, such as 0.3 M solution of succinic acid, or more preferably, citric acid. If a soluble salt form is not available, the compound may be dissolved in a suitable co-solvent or combination of co-solvents. Examples of suitable co-solvents include alcohol, propylene glycol, polyethylene glycol 300, polysorbate 80, glycerin and the like in concentrations ranging from about 0 to about 60% of the total volume. In one embodiment, the active compound is dissolved in DMSO and diluted with water.
- the pharmaceutical composition may also be in the form of a solution of a salt form of the active ingredient in an appropriate aqueous vehicle, such as water or isotonic saline or dextrose solution.
- an appropriate aqueous vehicle such as water or isotonic saline or dextrose solution.
- compounds which have been modified by substitutions or additions of chemical or biochemical moieties which make them more suitable for delivery e.g., increase solubility, bioactivity, palatability, decrease adverse reactions, etc.
- esterification glycosylation, PEGylation, etc.
- the compounds described herein may be formulated for oral administration in a lipid-based formulation suitable for low solubility compounds.
- Lipid-based formulations can generally enhance the oral bioavailability of such compounds.
- a preferred pharmaceutical composition comprises a therapeutically or prophylactically effective amount of a compound described herein, together with at least one pharmaceutically acceptable excipient selected from the group consisting of medium chain fatty acids and propylene glycol esters thereof (e.g., propylene glycol esters of edible fatty acids, such as caprylic and capric fatty acids) and pharmaceutically acceptable surfactants, such as polyoxyl 40 hydrogenated castor oil.
- a pharmaceutically acceptable excipient selected from the group consisting of medium chain fatty acids and propylene glycol esters thereof (e.g., propylene glycol esters of edible fatty acids, such as caprylic and capric fatty acids) and pharmaceutically acceptable surfactants, such as polyoxyl 40 hydrogenated castor oil.
- cyclodextrins may be added as aqueous solubility enhancers.
- Preferred cyclodextrins include hydroxypropyl, hydroxyethyl, glucosyl, maltosyl and maltotriosyl derivatives of ⁇ -, ⁇ -, and ⁇ -cyclodextrin.
- a particularly preferred cyclodextrin solubility enhancer is hydroxypropyl-o-cyclodextrin (BPBC), which may be added to any of the above-described compositions to further improve the aqueous solubility characteristics of the compounds of the embodiments.
- BPBC hydroxypropyl-o-cyclodextrin
- the composition comprises about 0.1% to about 20% hydroxypropyl-o-cyclodextrin, more preferably about 1% to about 15% hydroxypropyl-o-cyclodextrin, and even more preferably from about 2.5% to about 10% hydroxypropyl-o-cyclodextrin.
- solubility enhancer employed will depend on the amount of the compound of the invention in the composition.
- a pharmaceutical composition contains a total amount of the active ingredient(s) sufficient to achieve an intended therapeutic effect. More specifically, in some embodiments, the pharmaceutical composition contains a therapeutically effective amount (e.g., an amount of an SAPK-modulating compound that is effective in the prevention or treatment of the symptoms of an inflammatory disease or condition, wherein the compound exhibits an IC 50 in the range of about 0.1 ⁇ M to about 1000 ⁇ M, and preferably about 1 ⁇ M to about 800 ⁇ M, about 1 ⁇ M to about 500 ⁇ M, about 1 ⁇ M to about 300 ⁇ M, about 1 ⁇ M to about 200 ⁇ M, or about 1 ⁇ M to about 100 ⁇ M for inhibition of p38 MAPK).
- a therapeutically effective amount e.g., an amount of an SAPK-modulating compound that is effective in the prevention or treatment of the symptoms of an inflammatory disease or condition, wherein the compound exhibits an IC 50 in the range of about 0.1 ⁇ M to about 1000 ⁇ M, and preferably about 1 ⁇ M to
- compositions are formulated so that a dose of between 0.01 to 100 mg/kg body weight/day of an SAPK-modulating compound is administered to a patient receiving the compositions.
- Trimethylchlorosilane (26.5 mL, 150 mmol) was added to the solution of zinc powder (10 g, 150 mmol) in anhydrous THF (150 ml) under N 2 .
- a solution of chloroacetonitrile (6.35 mL, 100 mmol) and trifluoroacetylvinyl ether (8.4 g, 50 mmol) in anhydrous THF (75 mL) was added dropwise slowly to keep the temperature at 40° C.
- the mixture was refluxed for 2 h.
- concentrated HCl 25 mL was added.
- the mixture was refluxed for 1 h, then cooled to room temperature.
- Compound 103 was prepared from compound 102. A mixture of compound 102 (2 g, 10 mmol) and trimethyl-trifluoromethyl-silane (7 ml, 2M in THF, 15 mmol) in THF (40 mL) was cooled to 0° C. in an ice bath and then treated with tetrabutylammonium fluoride (0.5 ml, 1 M in THF, 0.5 mmol) under nitrogen atmosphere at 0° C. for 30 min. The mixture was warmed to room temperature and stirred 24 h. Then, 1 M HCl (50 mL) was added and the mixture was stirred overnight. The aqueous layer was extracted with EA (70 mL ⁇ 2) and the organic layer was concentrated. The desired product was separated by column chromatography to give pure compound 103 (1.5 g, 45% yields) as a white solid.
- Compound 104 was prepared from compound 103. Potassium bromate (16.6 g, 0.1 mol) was added over 0.5 h to a vigorously stirred mixture of 2-iodobenzoic acid (20 g, 0.08 mmol) and 180 mL 0.73 M H 2 SO 4 (0.13 mol) in a 55° C. bath. The mixture was stirred for 4 h at 68° C., and the Br 2 formed was removed by reduced pressure in the reaction process. The reaction was cooled to room temperature with an ice bath. Filtration and washing of the solid with ice water and iced ethanol gave the desired compound IBX (16 g, 70% yield).
- Compound 105 was prepared from compound 104.
- Compound 104 (80 mg, 0.24 mmol) in dry DCM (1.5 mL) was added at the temperature of ⁇ 78° C. under N 2 atmosphere to a solution of DAST (50 mg, 0.31 mmol) in DCM (0.5 mL). The mixture was stirred at ⁇ 78° C. for 2 h, and then warmed to room temperature overnight.
- the reaction mixture was diluted with DCM (20 mL), and poured into saturated NaHCO 3 (30 mL). The organic phase was separated and dried over Na 2 SO 4 and concentrated in vacuo.
- Compound 105 was isolated by thin-layer chromatography (42 mg, 50% yields) as a white solid.
- Compound 107 was prepared from compound 104.
- a mixture of Compound 104 (80 mg, 0.24 mmol) and trimethyl-trifluoromethyl-silane (0.07 mL pure, 0.49 mmol) in THF (2.5 mL) cooled to 0° C. in an ice bath is treated with tetrabutylammonium fluoride (0.5 mL, 0.024 M in THF, 0.012 mmol) under nitrogen atmosphere at 0° C. for 30 min.
- the mixture was raised to room temperature and stirred 24 h.
- 1 M HCl (20 mL) was added and the mixture was stirred overnight.
- Compound 109 was prepared from compound 108. Sodium borohydride (200 mg 5.26 mmol) was added slowly to a solution of Compound 108 (90 mg, 0.526 mmol) in acetic acid (32 mL) and the mixture was stirred for 30 min at room temperature. The reaction mixture was neutralized cautiously with aqueous sodium hydroxide, on an ice-water bath, and then extracted with dichloromethane and dried over anhydrous magnesium sulfate.
- a for compound 115 was prepared in the following manner. A solution of 5-cyano-2-methoxy pyridine (1 eq), sodium ethylsulfide (EtSNa) (2 eq) in DMF (5 ml/eq) was heated to 60° C. for 4 h. To the reaction mixture was added HCl-Et 2 O until the mixture reached a pH of about 6, under nitrogen flush, in order to remove volatiles (Et2O Et 2 O and EtSH). The mixture was centrifuged and filtered, which removed the sodium chloride. The DMF solution was used as prepared in General procedure H2 to provide compound 115 in 21% yield.
- 1 H NMR 300 MHz, DMSO-d6) ppm 8.63 (d, 1H), 7.77 (dd, 1H), 7.64 (m, 2H), 7.55 (m, 2H), 6.61 (d, 1H)
- the 5-Furan-2-yl-1H-pyridin-2-one product was obtained by reaction of 2.66 g (14 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO 2 ; Pet. Ether/AcOEt 9:1) 1.83 g (75% yield) of pure product were obtained as white solid. The obtained product was de-methylated using EtSNa. The obtained A in a DMF solution (10 mmol/30 ml) was used for the Chan Lam reaction, following General Procedure H2 to provide compound 121 in 19% yield.
- the 2-Methoxy-5-phenyl-pyridine was obtained by reaction of 1.9 g (10 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO 2 ; Pet. Ether/EA 9:1) 1.8 g (97% yield) of pure product were obtained as white solid.
- the 2-Methoxy-5-phenyl-pyridine (1 g, 5.4 mmol) was added to HSO 3 Cl (2 ml) at 0° C. The dark solution was stirred at room temperature for 4 h and then poured onto ice. Concentrated ammonia was added, while maintaining the temperature ⁇ 10° C. The intermediate was extracted with EA (1.2 g, 84% yield) and used for the next step without further purification.
- Ether/EA 9:1) 1.8 g (92% yield) of a 1:1 mixture of intermediate and the dimeric 2-methoxy-pyridine were obtained and used for the next step.
- the mixture (1.1 g) was dissolved in HBr 48% (10 ml) and EtOH (3 ml) and the solution was heated at reflux for 3 h. After evaporation of volatiles, the crude was purified by column chromatography (SiO 2 ; Pet. Ether/EA 9:1) leading to the desired pyridone A (350 mg). Following general procued procedure H1A, compound 138 was prepared in 35% yield.
- 6-Methoxy-pyridin-3-ylamine (2.5 g 2 mmol) was dissolved in 48% HBF 4 (10 ml) and cooled at 0° C.
- NaNO 2 (2.4 g, 3.4 mmol) was added portionwise maintaining the temperature ⁇ 5° C.
- the dark solution was stirred at low temperature for 1 h.
- the solid was collected by filtration and washed with water and then dried under vacuum.
- the desired diazonium salt was obtained (3.18 g, 72%) as white crystalline solid.
- the diazonium salt (2 g, 8.9 mmol) and celite (4 g) were finely mixed in a mortar, transferred to a reaction vessel, then gradually heated to 150° C., whereupon a rapid evolution of fumes occurred.
- the 2-methoxy-pyridine-5-boronic acid (1.9 g, 12 mmol), the 5-bromo-pyrimidine (1.2 eq) and K 2 CO 3 (3 eq) were dissolved in a 10:1 mixture of DME/H 2 O (4 mL/mmol). The solution was degassed by bubbling nitrogen for 15 min and then Pd(PPh 3 ) 4 (0.05 eq) was added. The reaction mixture was heated at 90° C. for 8 h and then cooled at room temperature, diluted with EtOAc and filtered on a celite plug. The filtrate was washed with brine. The separated organic phase was dried over Na 2 SO4 and concentrated under reduced pressure. The obtained residue was purified by column chromatography.
- 6-methoxynicotinaldehyde (1.0 g, 7.2 mmol) was dissolved in HBr 48% (10 mL) and EtOH (3 mL) and the solution was heated at reflux for 2 h. After evaporation of volatiles, 1.6 g of the desired pyridone intermediate was obtained. The intermediate was used in the next step without further purification.
- the iodopyridone intermediate above was obtained by reaction of 600 mg (2.7 mmol) of 5-iodo-2-pyridone with phenylboronic acid. After purification (SiO 2 ; Hexane/Acetate/MeOH 1/1/0 to 0/10/1). 600 mg (75% yield) of pure intermediate were obtained as a pale yellow solid.
- 1 H NMR 300 MHz, DMSO-d6) ppm 7.98 (s, 2H), 7.91 (dd, 1H), 7.83 (dd, 1 H), 7.36-7.62 (m, 5H), 6.54 (dd, 1H)
- 6-Methoxy-pyridine-3-sulfonyl chloride (5.0 g, 0.025 mol) was dissolved in DCM and cooled at 0° C. Gasseous Gaseous NH 3 was bubbled in the solution for 10 min. The resulting pale brown suspension was filtered and the solid was triturated with water. The resulting white solid was filtered and dried under vacuum to afford 3.2 g (70.6% yield) of pure 6-Methoxy-pyridine-3-sulfonamide.
- 6-Methoxy-pyridine-3-sulfonamide (0.752 g, 4.0 mmol) was dissolved in EtOH (6 mL). An excess of 48% HBr aqueous solution (12 mL) was added and the reaction was heated at 90° C.
- 5-(2-fluorophenyl)-2-methoxypyridine was obtained by reaction of 3 g (16 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO 2 ; Pet. Ether/EtOAc 1/1 to 0/1), 750 mg (31% yield) of pure product was obtained as a white solid.
- 5-(2-fluorophenyl)-2-methoxypyridine 750 mg was dissolved in HBr 48% (10 mL) and EtOH (3 mL) and the solution was heated at reflux for 3 h. After evaporation of volatiles, 700 mg (quantitative yield) of the desired pyridone were obtained as a white solid.
- the iodopyridone (1 eq), the appropriate boronic acid (1.2 eq) and K 2 CO 3 (3 eq) were dissolved in a 10:1 mixture of DME/H 2 O (4 mL/mmol).
- the solution was degassed by bubbling N 2 for 15 min and then Pd(PPh 3 ) 4 (0.05 eq) was added.
- the reaction mixture was heated at 90° C. for 18 h, after which time, BOC protecting group was completely cleaved.
- Mixture was cooled at room temperature, diluted with EtOAc and filtered on a celite plug. The filtrate was washed with brine. The separated organic phase was dried over Na 2 SO 4 and concentrated under reduced pressure.
- Compound 341 was prepared from compound 338 as follows. Compound 338 (50 mg, 0.19 mmol) and manganese dioxide (165 mg, 1.9 mmol) were stirred overnight at room temperature in DCM (5 ml). The reaction was detected by TLC. Upon completion, the crude mixture was filtered through a pad of celite and the filtrate was concentrated. The desired compound was isolated by washing the crude with PE to give pure intermediate product (36 mg, 70% yield) as a white solid.
- Reagent 3 (0.3-0.5 mmol, 1 eq.) was dissolved in acetonitrile (3 mL/1 mmol reagent 3), DAST (2 eq.) was added slowly at room temperature. The resulting solution was stirred at 80° C. in a capped plastic tube overnight. After cooling to room temperature, it was diluted with DCM, washed with aqueous solution of saturated sodium bicarbonate, water and brine, dried over Na 2 SO 4 , concentrated to give a residue, which was purified by pre-TLC (using EA/PE as solvent) to give target compound. Following this procedure, compound 359 was prepared in 13.9% yield as a white solid.
- the product was obtained by reaction of 963 mg (6.3 mmol) of 2-methoxy-pyridine-5-boronic acid. After purification (SiO 2 ; Hexanes:EtOAc 1:1) 747 mg (65% yield) of pure product were obtained as white solid.
- 2-(6-methoxypyridin-3-yl)pyrimidine (747 mg) was dissolved in HBr 48% (10 ml) and EtOH (5 ml) and the solution was heated at reflux overnight. After evaporation of volatiles the desired pyridone was obtained as white solid (1.016 g, quantitative yield).
- N-(3-(5-iodo-2-oxopyridin-1(2H)-yl)phenyl)acetamide (0.050 g, 0.14 mmol) was dissolved in dry and degassed toluene (3 mL). The catalyst was then added (0.008 g, 0.007 mmol) and the mixture was stirred for 10 minutes. 2-(tributylstannyl)oxazole (0.050 g, 0.14 mmol) was added and the reaction was heated at 90° C. for 18 h under nitrogen atmosphere. Conc. NH 4 OH was added. The solvent was removed at reduced pressure and the crude was purified by preparative HPLC.
- the intermediate was obtained by reaction of 420 mg (3 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO 2 ; Hexanes:EtOAc 95:5) 390 mg (65% yield) of pure product was obtained as white solid.
- the intermediate (390 mg) was dissolved in HBr 48% (5 ml) and EtOH (5 ml) and the solution was heated at reflux for 24 h. After evaporation of volatiles the desired pyridone was obtained as white solid (359 mg, quantitative yield).
- 6-methoxynicotinaldehyde (1.0 g, 7.2 mmol) was dissolved in HBr 48% (10 mL) and EtOH (3 mL) and the solution was heated at reflux for 2 h. After evaporation of volatiles 1.6 g of the desired pyridone was obtained. The product was used in the next step without further purification.
- 6-Methoxy-pyridine-3-sulfonyl chloride (5.0 g, 0.025 mol) was dissolved in DCM and cooled at 0° C. Gasseous Gaseous NH 3 was bubbled in the solution for 10 min. The resulting pale brown suspension was filtered and the solid was triturated with water. The resulting white solid was filtered and dried under vacuum to afford 3.2 g (70.6% yield) of pure 6-Methoxy-pyridine-3-sulfonamide.
- 6-Methoxy-pyridine-3-sulfonamide (0.752 g, 4.0 mmol) was dissolved in EtOH (6 mL). An excess of 48% HBr aqueous solution (12 mL) was added and the reaction was heated at 90° C. for 20 h. The solvent was removed under reduced pressure and the residual hydrobromic acid was further dried under reduced pressure, at 40° C. Quantitative yield.
- the intermediate was obtained by reaction of 3 g (16 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO 2 ; Hexanes:EtOAc 1:1 to 100% EtOAc) 3.99 g (95% yield) of pure product was obtained as white solid.
- the intermediate (3.99 g) was dissolved in HBr 48% (12 ml) and EtOH (6 ml) and the solution was heated at reflux for 24 h. After evaporation of volatiles the desired pyridone was obtained as white solid (3.72 g, quantitative yield).
- 5-bromo-2-methoxy-4-methylpyridine (1.0 g, 4.95 mmol) was dissolved in HBr 48% (10 mL) and EtOH (10 mL) and the solution was heated at 90° C. for 24 h. After evaporation of volatiles, 930 mg (quantitative yield) of the desired pyridone were obtained as a white solid.
- 5-bromo-4-methyl-1-phenylpyridin-2(1H)-one was obtained by reaction of 450 mg (2.39 mmol) of 5-bromo-4-methylpyridin-2(1H)-one with phenylboronic acid.
- the Transcreener KinasePlus assay determines p38 activity by measuring ATP consumption in the presence of a relevant peptide substrate. This assay is commonly used in the characterization of kinases (Lowrey and Kleman-Leyer, Expert Opin Ther Targets 10(1):179-90 (2006)).
- the Transcreener KinasePlus assay measures the p38 catalyzed conversion of ATP to ADP using a florescence polarization-based approach. The p38 reaction is performed as usual and stopped by addition of Stop-Detect reagents.
- p38 gamma was obtained from Millipore, Inc (Billerica, Ma.).
- p38 MAP Kinases are recombinant human full-length proteins with an amino-terminal GST fusion, expressed in and purified from E. coli. Proteins were aliquoted and stored at ⁇ 80° C. Assays for p38 activity were performed in the presence of an EGF receptor peptide (sequence KRELVEPLTPSGEAPNQALLR—SEQ ID NO: 1) that was obtained from Midwest Biotech (Fishers, Ind.). EGFR peptide was aliquotted and stored at ⁇ 20° C.
- p38 MAP kinase assays were performed using p38 assay buffer containing 20 mM HEPES, pH 7.5, 10 mM MgCl 2 , 2 mM DTT, 0.01% Triton X-100, 10% glycerol, and 0.0002% bovine serum albumin (BSA). This buffer was supplemented with 10 ⁇ ATP, 25 ⁇ M EGFR peptide and 1 nM p38- ⁇ . Compounds were weighed and dissolved to a known final concentration in DMSO.
- the assay and compound dilutions were conducted on a Janus liquid handling platform (Perkin Elmer, Waltham, Mass.) at room temperature (about 25° C.).
- Compounds in DMSO were placed in column 1 of a Costar V-bottom 96 well plate and diluted serially across the plate (3.3 ⁇ dilutions). Columns 11 and 12 contain DMSO only (no inhibitor). Each compound dilution was 30-fold higher than the desired final concentration.
- a daughter plate was created by placing 180 ⁇ L of p38 assay buffer in each well of a second Costar V-bottom 96 well plate and 20 ⁇ L of the diluted compound stocks in DMSO were transferred and mixed.
- the assay was conducted in a black Proxipate Proxiplate F-Plus 384 well plate (Perkin Elmer, Waltham, Mass.). All subsequent transfers were conducted using a 96 well head such that the final assay was quad mapped with 4 replicates of each reaction. 5 ⁇ L of the compound mixture was transferred from the daughter plate to the assay plate. 5 ⁇ L of a mixture containing enzyme and EGFR peptide at 3-fold the desired final concentration in p38 assay buffer was then added to the appropriate wells. The reactions in the final two columns of the 384 well plate received a mixture of EGFR peptide in p38 assay buffer in the absence of enzyme. These wells served as a control for complete inhibition of enzyme.
- the plates were read for fluorescence polarization (FP) on a PerkinElmer EnVision using 3 filters (Cy5 Ex 620/40; Cy5 Em FP P-pol 688 nm; Cy5 Em FP S-pol 688 nm), and a mirror (Cy5 FP D658/fp688). Each read was integrated for 100 flashes.
- mP output from the EnVision was transferred to a plot of mP versus compound concentration.
- XLfit (IDBS, Guildford, England) was used to apply a 4-parameter logistic fit to the data and determine the median inhibitory concentration (IC 50 ).
- Preferred compounds exhibit IC 50 values of between about 0.05 ⁇ M and about 10 ⁇ M, preferably about 0.1 ⁇ M to about 5 ⁇ M.
- TNF ⁇ levels were measured using ELISA kits (R&D systems PDTA00C). A SpectraMAX M5 was used as the plate reader. The calculated amount of TNF ⁇ released was expressed as a percentage of the vehicle+LPS control.
- Test compounds were prepared at 2.5 times the final concentration. Final DMSO concentration was no more than 0.5% (v/v). Cells were preincubated with compound for 60 minutes at 37° C., 5% CO 2 prior to stimulation with lipopolysaccharide (LPS) (Sigma L-2880, 1 mg/ml stock in PBS). The final LPS concentration in each well was 200 ng/ml for TNF ⁇ release. Unstimulated control cell suspensions received DMSO/RPMI Media vehicle only. Cell mixtures were incubated for 4 hours for TNF ⁇ release. 80 ⁇ l of supernatants were taken and transferred to a fresh plate and stored at ⁇ 70° C. until further analysis. TNF ⁇ levels were measured using ELISA kits (R&D systems PDTA00C). A SpectraMAX M5 was used as the plate reader. Analysis was performed by non-linear regression to generate a dose response curve. The calculated IC 50 value was the concentration of the test compound that caused a 50% decrease in TNF ⁇ levels.
- LPS lip
- Compounds inhibit the release of TNF ⁇ in this in vitro assay.
- Preferred compounds exhibit IC 50 values for TNF ⁇ between about 1 ⁇ M and about 1000 ⁇ M, preferably about 1 ⁇ M to about 800 ⁇ M.
- THP-1 cells were treated with compounds as described for TNF ⁇ tests. 4 hours after LPS addition, 80 ⁇ l of media is removed for ELISA. 48 hrs after LPS addition of media and cells were mixed with 100 ⁇ l of ATPlite reagent. The mixture was shaken for 2 minutes then read for luminescence. A SpectraMAX M5 is used as the plate reader.
- the calculated cytotoxicity is expressed as a percentage of the LPS/DMSO control compound.
- Compounds which had a low score in ATPlite compared to the LPS/DMSO control were classified as cytotoxic rather than TNF ⁇ inhibitors. Where appropriate compounds were tested at 5-10 fold lower concentrations to determine whether the compound had activity at lower, non cytotoxic concentrations.
- PBMC peripheral blood mononuclear cells
- LPS lipopolysaccharide
- TNF ⁇ inhibition in this model is due to inhibition of p38 MAP kinase by the compound.
- Preferred compounds inhibit the release of TNF ⁇ in this in vivo assay.
- Preferred compounds exhibit an ED 50 value of less than 500 mg/kg, preferably less than 400 mg/kg, preferably less than 200 mg/kg, preferably less than 100 mg/kg, more preferably, less than 50 mg/kg, more preferably, less than 40 mg/kg, more preferably, less than 30 mg/kg, more preferably, less than 20 mg/kg, more preferably, less than 10 mg/kg.
- the methods of determining the IC 50 of the inhibition of p38 by a compound include any methods known in the art that allow the quantitative detection of any of the downstream substrates of p38 MAPK as described above. Therefore, these methods additionally include but limited to detection of expression of genes known to be regulated by p38 either individually, or by gene arrays.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Physical Education & Sports Medicine (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Virology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Pulmonology (AREA)
- Immunology (AREA)
- Pain & Pain Management (AREA)
- Tropical Medicine & Parasitology (AREA)
- Dermatology (AREA)
- Cardiology (AREA)
- Molecular Biology (AREA)
- Urology & Nephrology (AREA)
- Heart & Thoracic Surgery (AREA)
- Endocrinology (AREA)
- Hospice & Palliative Care (AREA)
- Psychology (AREA)
- Emergency Medicine (AREA)
- Ophthalmology & Optometry (AREA)
Abstract
Description

wherein M is N or CR1; A is N or CR2; L is N or CR3; B is N or CR4; E is N or CX4; G is N or CX3; J is N or CX2; K is N or CX1; a dashed line is a single or double bond, except when B is CR4, then each dashed line is a double bond;
R1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl, alkenyl, cyano, sulfonamido, halo, aryl, alkenylenearyl, and heteroaryl;
R2 is selected from the group consisting of hydrogen, alkyl, haloalkyl, halo, cyano, aryl, alkenyl, alkenylenearyl, heteroaryl, haloalkylcarbonyl, cycloalkyl, hydroxylalkyl, sulfonamido, and cycloheteroalkyl or R2 and R1 together form an optionally substituted 5-membered nitrogen-containing heterocyclic ring;
R3 is selected from the group consisting of hydrogen, aryl, alkenylenearyl, heteroaryl, alkyl, alkenyl, haloalkyl, amino, and hydroxy;
R4 is selected from the group consisting of hydrogen, alkyl, haloalkyl, cyano, alkoxy, aryl, alkenyl, alkenylenearyl, and heteroaryl; and
X1, X2, X3, X4, and X5 are independently selected from the group consisting of hydrogen, alkyl, alkenyl, halo, hydroxy, amino, aryl, cycloalkyl, thioalkyl, alkoxy, haloalkyl, haloalkoxy, alkoxyalkyl, cyano, aldehydo, alkylcarbonyl, amido, haloalkylcarbonyl, sulfonyl, and sulfonamide, or X2 and X3 together form a 5- or 6-membered ring comprising —O(CH2)nO—, wherein n is 1 or 2,
with the proviso that when all of A, B, E, G, J, K, L, and M are not N, then (a) at least one of X1, X2, X3, X4, and X5 is not selected from the group consisting of hydrogen, halo, alkoxy, and hydroxy or (b) at least one of R1, R2, R3, or R4 is not selected from the group consisting of hydrogen, alkyl, alkenyl, haloalkyl, hydroxyalkyl, alkoxy, phenyl, substituted phenyl, halo, hydroxy, and alkoxyalkyl,
or a pharmaceutically acceptable salt, ester, or solvate thereof.

wherein X6 and X7 are independently selected from the group consisting of hydrogen, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, alkylenylaryl, alkylenylheteroaryl, alkylenylheterocycloalkyl, alkylenylcycloalkyl, or X6 and X7 together form an optionally substituted 5 or 6 membered heterocyclic ring. In a specific class of embodiments, the compound of formula (I) is a compound selected from the group recited in Table 1, below.



wherein M is N or CR1; A is N or CR2; L is N or CR3; B is N or CR4; E is N or CX4; G is N or CX3; J is N or CX2; K is N or CX1; a dashed line is a single or double bond, except when B is CR4, then each dashed line is a double bond;
R1 is selected from the group consisting of hydrogen, alkyl, cycloalkyl, alkenyl, cyano, sulfonamido, halo, aryl, alkenylenearyl, and heteroaryl;
R2 is selected from the group consisting of hydrogen, alkyl, haloalkyl, halo, cyano, aryl, alkenyl, alkenylenearyl, heteroaryl, haloalkylcarbonyl, cycloalkyl, hydroxyalkyl, sulfonamido, and cycloheteroalkyl or R2 and R1 together form an optionally substituted 5-membered nitrogen-containing heterocyclic ring;
R3 is selected from the group consisting of hydrogen, aryl, alkenylenearyl, heteroaryl, alkyl, alkenyl, haloalkyl, amino, and hydroxy;
R4 is selected from the group consisting of hydrogen, alkyl, haloalkyl, cyano, alkoxy, aryl, alkenyl, alkenylenearyl, and heteroaryl; and
X1, X2, X3, X4, and X5 are independently selected from the group consisting of hydrogen, alkyl, alkenyl, halo, hydroxy, amino, aryl, cycloalkyl, thioalkyl, alkoxy, haloalkyl, haloalkoxy, alkoxyalkyl, cyano, aldehydo, alkylcarbonyl, amido, haloalkylcarbonyl, sulfonyl, and sulfonamide, or X2 and X3 together form a 5- or 6-membered ring comprising —O(CH2)nO—, wherein n is 1 or 2, with the proviso that when all of A, B, E, G, J, K, L, and M are not N, then either (a) at least one of X1, X2, X3, X4, and X5 is not selected from the group consisting of hydrogen, halo, alkoxy, and hydroxy or (b) at least one of R1, R2, R3, or R4 is not selected from the group consisting of hydrogen, alkyl, alkenyl, haloalkyl, hydroxyalkyl, alkoxy, phenyl, substituted phenyl, halo, hydroxy, and alkoxyalkyl.

wherein at least one of R1, R2, R3, or R4 is not selected from the group consisting of hydrogen, alkyl, alkenyl, haloalkyl, hydroxyalkyl, alkoxy, phenyl, substituted phenyl, halo, hydroxy, and alkoxyalkyl.

wherein X8 is hydrogen or alkyl; X6 and X7 are independently selected from the group consisting of hydrogen, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, alkylenylaryl, alkylenylheteroaryl, alkylenylheterocycloalkyl, alkylenylcycloalkyl, or X6 and X7 together form an optionally substituted 5 or 6 membered heterocyclic ring. In some embodiments, X7 is hydrogen. In various embodiments, X8 is methyl.
| TABLE 1 | ||
| Cmpd | ||
| No. | Structure | |
| 1 |  | |
| 2 |  | |
| 3 |  | |
| 4 |  | |
| 5 |  | |
| 6 |  | |
| 7 |  | |
| 8 |  | |
| 9 |  | |
| 10 |  | |
| 11 |  | |
| 12 |  | |
| 13 |  | |
| 14 |  | |
| 15 |  | |
| 16 |  | |
| 17 |  | |
| 18 |  | |
| 19 |  | |
| 20 |  | |
| 21 |  | |
| 22 |  | |
| 23 |  | |
| 24 |  | |
| 25 |  | |
| 26 |  | |
| 27 |  | |
| 28 |  | |
| 29 |  | |
| 30 |  | |
| 31 |  | |
| 32 |  | |
| 33 |  | |
| 34 |  | |
| 35 |  | |
| 36 |  | |
| 37 |  | |
| 38 |  | |
| 39 |  | |
| 40 |  | |
| 41 |  | |
| 42 |  | |
| 43 |  | |
| 44 |  | |
| 45 |  | |
| 46 |  | |
| 47 |  | |
| 48 |  | |
| 49 |  | |
| 50 |  | |
| 51 |  | |
| 52 |  | |
| 53 |  | |
| 54 |  | |
| 55 |  | |
| 56 |  | |
| 57 |  | |
| 58 |  | |
| 59 |  | |
| 60 |  | |
| 61 |  | |
| 62 |  | |
| 63 |  | |
| 64 |  | |
| 65 |  | |
| 66 |  | |
| 67 |  | |
| 68 |  | |
| 69 |  | |
| 70 |  | |
| 71 |  | |
| 72 |  | |
| 73 |  | |
| 74 |  | |
| 75 |  | |
| 76 |  | |
| 77 |  | |
| 78 |  | |
| 79 |  | |
| 80 |  | |
| 81 |  | |
| 82 | 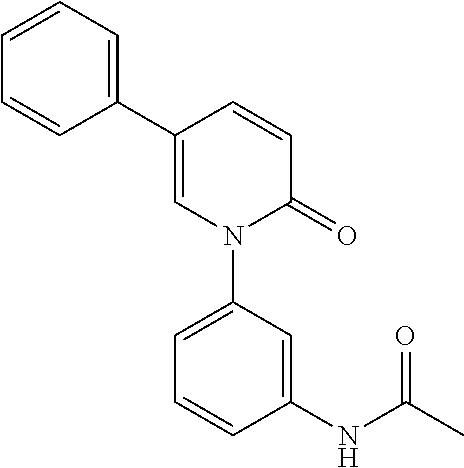 | |
| 83 |  | |
| 84 |  | |
| 85 |  | |
| 86 |  | |
| 87 |  | |
| 88 |  | |
| 89 |  | |
| 90 |  | |
| 91 |  | |
| 92 |  | |
| 93 |  | |
| 94 |  | |
| 95 |  | |
| 96 |  | |
| 97 |  | |
| 98 |  | |
| 99 |  | |
| 100 |  | |
| 101 |  | |
| 102 |  | |
| 103 |  | |
| 104 |  | |
| 105 |  | |
| 106 |  | |
| 107 |  | |
| 108 |  | |
| 109 |  | |
| 110 |  | |
| 111 |  | |
| 112 |  | |
| 113 |  | |
| 114 |  | |
| 115 |  | |
| 116 |  | |
| 117 |  | |
| 118 |  | |
| 119 |  | |
| 120 |  | |
| 121 |  | |
| 122 |  | |
| 123 |  | |
| 124 |  | |
| 125 |  | |
| 126 |  | |
| 127 |  | |
| 128 |  | |
| 129 |  | |
| 130 |  | |
| 131 |  | |
| 132 |  | |
| 133 |  | |
| 134 |  | |
| 135 |  | |
| 136 |  | |
| 137 |  | |
| 138 |  | |
| 139 |  | |
| 140 |  | |
| 141 |  | |
| 142 |  | |
| 143 |  | |
| 144 |  | |
| 145 |  | |
| 146 |  | |
| 147 |  | |
| 148 |  | |
| 149 |  | |
| 150 |  | |
| 151 |  | |
| 152 |  | |
| 153 |  | |
| 154 |  | |
| 155 |  | |
| 156 |  | |
| 157 |  | |
| 158 |  | |
| 159 |  | |
| 160 |  | |
| 161 |  | |
| 162 |  | |
| 163 |  | |
| 164 |  | |
| 165 |  | |
| 166 |  | |
| 167 |  | |
| 168 |  | |
| 169 |  | |
| 170 |  | |
| 171 |  | |
| 172 |  | |
| 173 |  | |
| 174 |  | |
| 175 |  | |
| 176 |  | |
| 177 |  | |
| 178 |  | |
| 179 |  | |
| 180 |  | |
| 181 |  | |
| 182 |  | |
| 183 |  | |
| 184 |  | |
| 185 |  | |
| 186 |  | |
| 187 |  | |
| 188 |  | |
| 189 |  | |
| 190 |  | |
| 191 |  | |
| 192 |  | |
| 193 |  | |
| 194 |  | |
| 195 |  | |
| 196 |  | |
| 197 |  | |
| 198 |  | |
| 199 |  | |
| 200 |  | |
| 201 |  | |
| 202 |  | |
| 203 |  | |
| 204 |  | |
| 205 |  | |
| 206 |  | |
| 207 |  | |
| 208 |  | |
| 209 |  | |
| 210 |  | |
| 211 |  | |
| 212 |  | |
| 213 |  | |
| 214 |  | |
| 215 |  | |
| 216 |  | |
| 217 |  | |
| 218 |  | |
| 219 |  | |
| 220 |  | |
| 221 |  | |
| 222 |  | |
| 223 |  | |
| 224 |  | |
| 225 |  | |
| 226 |  | |
| 227 |  | |
| 228 |  | |
| 229 |  | |
| 230 |  | |
| 231 |  | |
| 232 |  | |
| 233 |  | |
| 234 |  | |
| 235 |  | |
| 236 |  | |
| 237 |  | |
| 238 |  | |
| 239 |  | |
| 240 |  | |
| 241 |  | |
| 242 |  | |
| 243 |  | |
| 244 |  | |
| 245 |  | |
| 246 |  | |
| 247 |  | |
| 248 |  | |
| 249 |  | |
| 250 |  | |
| 251 |  | |
| 252 |  | |
| 253 |  | |
| 254 |  | |
| 255 |  | |
| 256 |  | |
| 257 |  | |
| 258 |  | |
| 259 |  | |
| 260 |  | |
| 261 |  | |
| 262 |  | |
| 263 |  | |
| 264 |  | |
| 265 |  | |
| 266 |  | |
| 267 |  | |
| 268 |  | |
| 269 |  | |
| 270 |  | |
| 271 |  | |
| 272 |  | |
| 273 |  | |
| 274 |  | |
| 275 |  | |
| 276 |  | |
| 277 |  | |
| 278 |  | |
| 279 |  | |
| 280 |  | |
| 281 |  | |
| 282 |  | |
| 283 |  | |
| 284 |  | |
| 285 |  | |
| 286 |  | |
| 287 |  | |
| 288 |  | |
| 289 |  | |
| 290 |  | |
| 291 |  | |
| 292 |  | |
| 293 |  | |
| 294 |  | |
| 295 |  | |
| 296 |  | |
| 297 |  | |
| 298 |  | |
| 299 |  | |
| 300 |  | |
| 301 |  | |
| 302 |  | |
| 303 |  | |
| 304 |  | |
| 305 |  | |
| 306 |  | |
| 307 |  | |
| 308 |  | |
| 309 |  | |
| 310 |  | |
| 311 |  | |
| 312 |  | |
| 313 |  | |
| 314 |  | |
| 315 |  | |
| 316 |  | |
| 317 |  | |
| 318 |  | |
| 319 |  | |
| 320 |  | |
| 321 |  | |
| 322 |  | |
| 323 |  | |
| 324 | 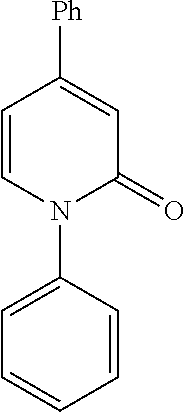 | |
| 325 |  | |
| 326 |  | |
| 327 |  | |
| 328 |  | |
| 329 |  | |
| 330 |  | |
| 331 |  | |
| 332 |  | |
| 333 |  | |
| 334 |  | |
| 335 |  | |
| 336 |  | |
| 337 |  | |
| 338 |  | |
| 339 |  | |
| 340 |  | |
| 341 |  | |
| 342 |  | |
| 343 |  | |
| 344 |  | |
| 345 |  | |
| 346 |  | |
| 347 |  | |
| 348 |  | |
| 349 |  | |
| 350 |  | |
| 351 |  | |
| 352 |  | |
| 353 |  | |
| 354 |  | |
| 355 |  | |
| 356 |  | |
| 357 |  | |
| 358 |  | |
| 359 |  | |
| 360 |  | |
| 361 |  | |
| 362 |  | |
| 363 |  | |
| 364 |  | |
| 365 |  | |
| 366 |  | |
| 367 |  | |
| 368 |  | |
| 369 |  | |
| 370 |  | |
| 371 |  | |
| 372 |  | |
| 373 |  | |
| 374 |  | |
| 375 | Intentionally blank | |
| 376 |  | |
| 377 |  | |
| 378 |  | |
| 379 |  | |
| 380 |  | |
| 381 |  | |
| 382 |  | |
| 383 |  | |
| 384 |  | |
| 385 |  | |
| 386 |  | |
| 387 |  | |
| 388 |  | |
| 389 |  | |
| 390 |  | |
| 391 |  | |
| 392 |  | |
| 393 |  | |
| 394 |  | |
| 395 |  | |
| 396 |  | |
| 397 |  | |
| 398 |  | |
| 399 |  | |
| 400 |  | |
| 401 |  | |
| 402 |  | |
| 403 |  | |
| 404 |  | |
| 405 |  | |
| 406 |  | |
| 407 |  | |
| 408 |  | |
| 409 |  | |
| 410 |  | |
| 411 |  | |
| 412 |  | |
| 413 |  | |
| 414 |  | |
| 415 |  | |
| 416 |  | |
| 417 |  | |
| 418 |  | |
| 419 |  | |
| 420 |  | |
| 421 |  | |
| 422 |  | |
| 423 |  | |
| 424 |  | |
| 425 |  | |
| 426 |  | |
| 427 |  | |
| 428 |  | |
| 429 |  | |
| 430 |  | |
| 431 |  | |
| 432 |  | |
| 433 |  | |
| 434 |  | |
| 435 | 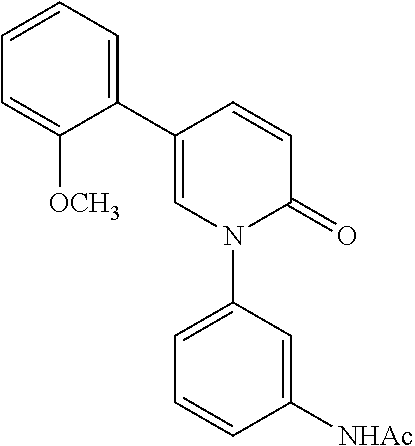 | |
| 436 |  | |
| 437 |  | |
| 438 |  | |
| 439 |  | |
| 440 |  | |
| 441 |  | |
| 442 |  | |
| 443 |  | |
| 444 |  | |
| 445 |  | |
| 446 |  | |
| 447 |  | |
| 448 |  | |
| 449 |  | |
| 450 |  | |
| 451 |  | |
| 452 |  | |
| 453 |  | |
| 454 |  | |
| 455 |  | |
| 456 |  | |
| 457 |  | |
| 458 |  | |
| 459 |  | |
| 460 |  | |
| 461 |  | |
| 462 |  | |
| 463 |  | |
| 464 |  | |
| 465 |  | |
| 466 |  | |
| 467 |  | |
| 468 |  | |
| 469 |  | |
| 470 |  | |
| 471 |  | |
| 472 |  | |
| 473 |  | |
| 474 |  | |
| 475 |  | |
| 476 |  | |
| 477 |  | |
| 478 |  | |
| 479 |  | |
| 480 |  | |
| 481 |  | |
| 482 |  | |
| 483 |  | |
| 484 |  | |
| 485 |  | |
| 486 |  | |
| 487 |  | |
Synthetic Processes














A solution of 1 (134 mg, 1 mmol), EtSNa (168 mg, 2 mmol) in DMF (5 ml) was heated to 60° C. for 4 h. To the reaction mixture was added HCl (aq.) until pH was about 6. The mixture was evaporated in vacuo to give 2. (110 mg, 92%). Pyridine (140 mg, 1.8 mmol) was slowly added to a mixture of 2 (110 mg, 0.9 mmol), phenylboronic acid (220 mg, 1.8 mmol) and Cu(OAc)2 (18 mg) in DCM (5 mL). After the suspension was stirred overnight at room temperature, it was monitored by TLC. When no starting material was detected, the mixture was washed with saturated NaHCO3. The organic layer was dried over sodium sulfate, evaporated in vacuo to afford the crude product, which was purified by preparative TLC to give compound 46 (50 mg, 25% yield) as a white solid. MS-ESI: m/z=197.3 [M+1]+

Following general procedure E, compound 57 was synthesized (yield of first step 51%; yield of second step 17%). MS-ESI: m/z=274.3 [M+1]+

A mixture of 2, 6-dibromopyridine (1) (4 g, 17 mmol), potassium t-butoxide (20 g, 0.27 mol), and redistilled t-butyl alcohol (100 mL) was refluxed overnight. After cooling, the solvent was removed in vacuo, ice/water was carefully added, and the aqueous layer was extracted with chloroform (100 mL×2), which removed the unreacted staring material. The aqueous layer was acidified with 3 N HCl, extracted with chloroform (100 mL×2), washed with brine, dried over anhydrous Na2SO4 and concentrated affording pure 6-bromo-2-pyridone (2.5 g, 85% yields) as a white solid. The preparation of 3 followed the general procedure A, in a 73% yield. 3 was then subjected to the conditions of general procedure A to prepare compound 59 in 35% yield as a yellowish oil. MS-ESI: m/z=274.3 [M+1]+

To a solution of ethyl vinyl ether (1, 40 mL) in 100 ml of dichloromethane, pyridine (36 mL) was added. Then a solution of trifluoroacetic anhydride (87.6 g) in 50 mL of dichloromethane was added at 0° C. After stirring at room temperature for 30 min, the solution was poured into 40 mL of H2O. The layers were separated and the aqueous layer was extracted again with 40 mL of dichloromethane. The organic layers were combined, washed with H2O and dried over MgSO4. Removal of solvent the gave crude 3, which was used directly in the next step.

Trimethylchlorosilane (26.5 mL, 150 mmol) was added to the solution of zinc powder (10 g, 150 mmol) in anhydrous THF (150 ml) under N2. After stirring for 0.5 h, a solution of chloroacetonitrile (6.35 mL, 100 mmol) and trifluoroacetylvinyl ether (8.4 g, 50 mmol) in anhydrous THF (75 mL) was added dropwise slowly to keep the temperature at 40° C. The mixture was refluxed for 2 h. After cooling to room temperature, concentrated HCl (25 mL) was added. The mixture was refluxed for 1 h, then cooled to room temperature. The reaction mixture was then poured into ice water. The product was extracted with EA, and washed with brine. The organic layer was dried over anhydrous Na2SO4, filtered and evaporated to dryness to give the residue. The residue was purified by column chromatography to afford 8.3 g of 5. Following general procedure A, compound 61 was prepared in 52% yield as a white solid. MS-ESI: m/z=284.0 [M+1]+

Following general procedure F, compound 72 was prepared in 89% for the first step and 53% for the second step, where in the second step, 4 eq of DAST was used. MS-ESI: m/z=272.0 [M+1]+

3, above, was prepared using general procedure A. 4 was prepared in the following manner. To a solution of 3 (1 eq) in tetrahydrofuran-methanol (10:1) was added sodium borohydride (5 eq) at 0° C. The mixture was stirred at room temperature for 30 min. Water was added and then mixture was extracted with EA. The organics were washed with brine, dried over Na2SO4 and concentrated in vacuo. 4 was isolated by prep-TLC. 5 was prepared according to general procedure F. Compound 91 was prepared under these reaction conditions to provide 86% yield of first step, 70% yield of second step, and 30% yield of third step. MS-ESI: m/z=272.2 [M+1]+

To a solution of 5-bromo-2-methoxy-pyridine (2.4 g, 8.94 mmol), (E)-prop-1-enylboronic acid (1 g, 11.6 mmol), K3PO4 (6.6 g, 31.3 mmol) and tricyclohexylphosphine (250 mg, 0.894 mmol) in toluene (40 mL) and water (2 mL) under a nitrogen atmosphere was added palladium acetate (100 mg, 0.447 mmol). The mixture was heated to 100° C. for 3 h and then cooled to room temperature. Water (100 mL) was added and the mixture extracted with EA (2×150 mL), the combined organics were washed with brine (100 mL), dried over Na2SO4 and concentrated in vacuo. The product was purified by column chromatography to give 1.3 g of compound 3 (68.4%, yield). Compound 3 (1.3 g, 8.72 mmol) was added to a stirred solution of hydrobromic acid (9.7 mL) in absolute ethanol (234 mL) under nitrogen and the mixture was heated under reflux for 5 hours. The cooled solution was evaporated in vacuo, and the residue partitioned between 10% sodium carbonate solution and DCM. The organic phase was dried with Na2SO4 and evaporated in vacuo to give 1.04 g of compound 4 as a white solid. (89% yield). Compound 5 was prepared using general procedure A. Compound 95 was prepared to give 85% yield of an oil. MS-ESI: m/z=255.3 [M+1]+

Using the procedure as outlined for compound 95 and general procedure F, compound 97 was prepared in 82% yield (first step) and 59% yield (second step). MS-ESI: m/z=262.2 [M+1]+

Meta-chloroperbenzoic acid (mCPBA, 5 eq.) was added to the solution of 1 in DCM at −78° C. The reaction was stirred at 0° C. for 20 minutes, then filtered. The reaction filtrate was purified by prep-TLC to give 2. Following this general procedure, compound 18 was subjected to these conditions to provide compound 98 in 22% yield as a white solid. MS-ESI: m/z=263.9 [M+1]+

2 is prepared using general procedure A in 84% yield. A mixture of 2 (1 g, 5 mmol) and trimethyl-trifluoromethylsilane (3.5 mL, 2M in THF, 7 mmol) in THF (20 mL) was cooled to 0° C. in an ice bath and treated with tetrabutylammonium fluoride (0.25 mL, 1 M in THF, 0.25 mmol) under nitrogen atmosphere at 0° C. for 30 min. The mixture was warmed to room temperature and stirred 24 h. Then, 1 M HCl (50 mL) was added, and the mixture was stirred overnight. The aqueous layer was extracted with EA (50 mL×2) and the organic layer was concentrated. The desired product was separated by column chromatography to give pure intermediate (0.94 g, 70% yield) as yellow solid. MS-ESI: m/z=270.2 [M+1]+ The intermediate (50 mg, 0.19 mmol) and manganese dioxide (165 mg, 1.9 mmol) were stirred overnight at room temperature in DCM (5 mL). The progress of the reaction was detected by TLC. Upon completion, the crude mixture was filtered through a pad of celite and the filtrate was concentrated. Compound 101 was isolated by washing the crude with petroleum ether to give pure product (36 mg, 70% yields) as a white solid. MS-ESI: m/z=268.2 [M+1]+

The 5-bromo-2-methoxy-pyridine (750 mg, 4 mmol), cyclopropyl boronic acid (1.08 g, 12.5 mmol) KF (760 mg, 13 mmol) and Pd(PPh3)4 were dissolved in toluene (12 ml) and the reaction mixture was heated at 150° C. by microwave for 1.5 h. The crude was purified by column chromatography to give the intermediate (1.33 g 74% yield) as colorless oil. To a magnetically stirred solution of 5-cyclopropyl-2-methoxypyridine (1.33 g, 8.9 mmol), in 30 mL of DMF, EtSNa (1.502 g, 17.8 mmol) was added. The mixture was heated at 90° C. for 24 h. The reaction was cooled at room temperature and HCl/Et2O was added until pH 6. EtSH formed. The remaining HCl/Et2O and EtSH was evaporated by bubbling N2 at 40° C. The solution of A (concentration 40 mg/ml) was use as such for the next step. Following general procedure H1A, compound 116 was prepared in 25% yield. 1H NMR (300 MHz, DMSO-d6) ppm 8.65-8.77 (m, 2H), 7.51-7.57 (m, 2H), 7.48 (d, 1H), 7.30 (dd, 1H), 6.46 (d, 1H), 1.69-1.85 (m, 1H), 0.76-0.87 (m, 2H), 0.57-0.66 (m, 2H)

The 5-Furan-2-yl-1H-pyridin-2-one product was obtained by reaction of 2.66 g (14 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Pet. Ether/AcOEt 9:1) 1.83 g (75% yield) of pure product were obtained as white solid. The obtained product was de-methylated using EtSNa. The obtained A in a DMF solution (10 mmol/30 ml) was used for the Chan Lam reaction, following General Procedure H2 to provide compound 121 in 19% yield. 1H NMR (300 MHz, DMSO-d6) ppm 7.85-7.94 (m, 2H), 7.66 (dd, 1H), 7.42-7.59 (m, 5H), 6.80 (dd, 1H), 6.60 (dd, 1H), 6.55 (dd, 1H)

Following standard Suzuki coupling, the 2-Methoxy-5-phenyl-pyridine was obtained by reaction of 1.9 g (10 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Pet. Ether/EA 9:1) 1.8 g (97% yield) of pure product were obtained as white solid. The 2-Methoxy-5-phenyl-pyridine (1 g, 5.4 mmol) was added to HSO3Cl (2 ml) at 0° C. The dark solution was stirred at room temperature for 4 h and then poured onto ice. Concentrated ammonia was added, while maintaining the temperature<10° C. The intermediate was extracted with EA (1.2 g, 84% yield) and used for the next step without further purification. To a magnetically stirred solution of the intermediate (4-(6-Methoxy-pyridin-3-yl)benzenesulfonamide, 1.2 g, 4.5 mmol), in 3 mL of EtOH, 15 mL of HBr were added. The mixture was heated at 80° C. for 20 h. The reaction was cooled at room temperature and poured into KHCO3 saturated solution. EA was added and the mixture was transferred into a separator funnel. The aqueous layer was separated and extracted with additional portion of EA. The combined organics were washed once with water. The organic layer was dried with sodium sulfate, filtered and evaporated under vacuum, affording 300 mg of A. The aqueous layer was acidified and the solvent was evaporated under vacuum. Purification by flash column chromatography (EA) afforded 750 mg of A (89% of yield). A was used for the Chan Lam reaction, following General Procedure H1A to provide compound 130 in 77% yield. 1H NMR (300 MHz, DMSO-d6) ppm 8.12 (d, 1H), 8.00 (dd, 1H), 7.83 (m, 4H), 7.42-7.64 (m, 5H), 7.34 (s, 2H), 6.64 (d, 1H)

Following standard Suzuki coupling, an intermediate was obtained by reaction of 2.82 g (15 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Pet. Ether/EA 9:1) 2.8 g (92% yield) of pure intermediate were obtained as white solid. The intermediate (900 mg) was dissolved in HBr 48% (10 ml) and EtOH (3 ml) and the solution was heated at reflux for 3 h. After evaporation of volatiles the desired A pyridone was obtained as white solid (780 mg, 93% yield). A was used in the Chan Lam reaction, following General Procedure H1A to provide compound 134 in 33% yield. 1H NMR (300 MHz, DMSO-d6) ppm 8.02 (d, 1H), 7.87-7.99 (m, 3H), 7.73 (m, 2H), 7.69 (m, 2H), 7.50 (s, 2H), 7.25 (m, 2H), 6.62 (d, 1H)

1.53 g (10 mmol) of 2-methoxy-pyridine-5-boronic acid and 2.46 g (15 mmol) of 2-Bromo-thiazole and K2CO3 (3 eq) were dissolved in a 10:1 mixture of DME/H2O (4 ml/mmol). The solution was degassed by bubbling N2 for 15 min and then Pd(PPh3)4 (0.05 eq) was added. The reaction mixture was heated at 90° C. for 8 h an and then cooled at room temperature, diluted with EA and filtered on a celite plug. The filtrate was washed with brine. The separated organic phase was dried over Na2SO4 and concentrated under reduced pressure. After purification (SiO2; Pet. Ether/EA 9:1) 1.8 g (92% yield) of a 1:1 mixture of intermediate and the dimeric 2-methoxy-pyridine were obtained and used for the next step. The mixture (1.1 g) was dissolved in HBr 48% (10 ml) and EtOH (3 ml) and the solution was heated at reflux for 3 h. After evaporation of volatiles, the crude was purified by column chromatography (SiO2; Pet. Ether/EA 9:1) leading to the desired pyridone A (350 mg). Following general procued procedure H1A, compound 138 was prepared in 35% yield. 1H NMR (300 MHz, DMSO-d6) ppm 8.23 (d, 1H), 8.07 (dd, 1H), 7.84 (d, 1H), 7.70 (d, 1H), 7.41-7.63 (m, 5H), 6.64 (d, 1 H)

Following standard procedure for Suzuki coupling, the intermediate was obtained by reaction of 3 g (16 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexane/EA 30/1 to EA) 2.2 g (51% yield) of the intermediate were obtained as white solid. To a magnetically stirred solution of 2-Methoxy-5-(1-methyl-1H-pyrazol-4-yl)-pyridine (1.2 g, 6.3 mmol), in 3 mL of EtOH, 15 mL of HBr were added. The mixture was heated at 80° C. for 20 h. The reaction was cooled at room temperature. The solvent was evaporated under vacuum. Purification by flash column chromatography (EA) afforded 1.1 g of A (quantitative yield). Following general procedure H1A, compound 139 was prepared in 63% yield. 1H NMR (300 MHz, DMSO-d6) ppm 8.04 (d, 1H), 7.94 (dd, 1H), 7.79 (d, 1H), 7.79 (dd, 1H), 7.62 (m, 2 H), 7.54 (m, 2H), 6.56 (d, 1H), 3.82 (s, 3H)

6-Methoxy-pyridin-3-ylamine (2.5 g 2 mmol) was dissolved in 48% HBF4 (10 ml) and cooled at 0° C. NaNO2 (2.4 g, 3.4 mmol) was added portionwise maintaining the temperature<5° C. The dark solution was stirred at low temperature for 1 h. The solid was collected by filtration and washed with water and then dried under vacuum. The desired diazonium salt was obtained (3.18 g, 72%) as white crystalline solid. The diazonium salt (2 g, 8.9 mmol) and celite (4 g) were finely mixed in a mortar, transferred to a reaction vessel, then gradually heated to 150° C., whereupon a rapid evolution of fumes occurred. The resulting solid was washed several times with abundant diethyl ether. The organic solution was washed with Et2O/HCl then evaporated to provide the desired intermediate as its hydrochloric salt (1.2 g) as pale yellow viscous oil. The obtained fluoromethoxy pyridine (1.2 g) was dissolved in HBr 48% (10 ml) and EtOH (3 ml) and the solution was heated at reflux for 6 h. After evaporation of volatiles the desired pyridone A was obtained as amorphous solid in quantitative yield, and used in General procedure H1A to provide compound 142 in 28% yield. 1H NMR (300 MHz, DMSO-d6) ppm 7.85-7.97 (m, 1H), 7.59-7.73 (m, 1H), 7.34-7.57 (m, 5H), 6.45-6.57 (m, 1H)

The 2-methoxy-pyridine-5-boronic acid (1.9 g, 12 mmol), the 5-bromo-pyrimidine (1.2 eq) and K2CO3 (3 eq) were dissolved in a 10:1 mixture of DME/H2O (4 ml/mmol). The solution was degassed by bubbling N2 for 15 min and then Pd(PPh3)4 (0.05 eq) was added. The reaction mixture was heated at 90° C. for 8 h an and then cooled at room temperature, diluted with EA and filtered on a celite plug. The filtrate was washed with brine. The separated organic phase was dried over Na2SO4 and concentrated under reduced pressure. The obtained residue was purified by column chromatography. (SiO2; Hexane/EA 30/1 to EA) 1.29 g (56% yield) of intermediate were obtained as white solid. A solution of 5-(6-Methoxy-pyridin-3-yl)-pyrimidine (1.29 g, 6.9 mmol) in EtOH (4 ml) and HBr 48% (10 ml) was stirred at 90° C. for 7 h. The solvent was evaporated and the crude A (as hydrobromide salt) was utilized in the next step without any purification. Following general procedure H1A, compound 147 was prepared in 22% yield. 1H NMR (300 MHz, DMSO-d6) ppm 9.12 (s, 2H), 9.11 (s, 1H), 8.32 (d, 1H), 8.06 (dd, 1H), 7.69 (m, 2H), 7.56 (m, 2H), 6.68 (d, 1H)

Following standard procedure for Suzuki coupling, the intermediate product was obtained by reaction of 2.5 g (13.3 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexane/EA 30/1 to EA) 2.1 g (87% yield) of the intermediate product were obtained as white solid. A solution of 6-Methoxy-[3,4′]bipyridinyl (2.1 g, 11.3 mmol) in EtOH (6 ml) and HBr 48% (12 ml) was stirred at 90° C. for 6 h. The solvent was evaporated and crude A (as hydrobromide salt) was utilized in the next step without any purification. Following general procedure H1A, compound 151 was prepared in 12% yield. 1H NMR (300 MHz, DMSO-d6) ppm 10.15 (s, 1H), 8.57 (br. s., 2H), 8.25 (d, 1H), 8.06 (dd, 1H), 7.75 (dd, 1 H), 7.66-7.73 (m, 2H), 7.62 (ddd, 1H), 7.46 (dd, 1H), 7.15 (ddd, 1H), 6.64 (d, 1H), 2.07 (s, 3H)

The 2-methoxy-pyridine-5-boronic acid (1.9 g, 12 mmol), the 5-bromo-pyrimidine (1.2 eq) and K2CO3 (3 eq) were dissolved in a 10:1 mixture of DME/H2O (4 mL/mmol). The solution was degassed by bubbling nitrogen for 15 min and then Pd(PPh3)4 (0.05 eq) was added. The reaction mixture was heated at 90° C. for 8 h and then cooled at room temperature, diluted with EtOAc and filtered on a celite plug. The filtrate was washed with brine. The separated organic phase was dried over Na2SO4 and concentrated under reduced pressure. The obtained residue was purified by column chromatography. (SiO2; Hexanes/EtOAc 30/1 to EtOAc) 1.29 g (56% yield) of pure product were obtained as white solid. A solution of 5-(6-Methoxy-pyridin-3-yl)-pyrimidine (1.29 g, 6.9 mmol) in EtOH (4 ml) and HBr 48% (10 ml) was stirred at 90° C. for 7 h. The solvent was evaporated and the crude compound (as hydrobromide salt) was utilized in the next step without any purification. A was used in the Chan Lam reaction, following General Procedure H1A to provide compound 157 in 11% yield. 1H NMR (300 MHz, DMSO-d6) ppm 9.10-9.17 (m, 3H), 8.73-8.81 (m, 2H), 8.31 (dd, 1H), 8.07 (dd, 1H), 7.63-7.70 (m, 2H), 6.70 (dd, 1H)

Following standard procedure for Suzuki coupling, the intermediate was obtained by reaction of 2.82 g (15 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexane/EA 8/2) 1.8 g (59% yield) of pure intermediate were obtained as white solid. To a magnetically stirred solution of 2-Methoxy-5-(1-methyl-1H-pyrazol-4-yl)-pyridine (1 g, 4.9 mmol), in 3 mL of EtOH, 10 mL of HBr were added. The mixture was heated at 90° C. for 4 h. The reaction was cooled at room temperature. The solvent was evaporated under vacuum, afforded 1.34 g of A (quantitative yield). Following general procedure H1A, compound 159 was prepared in 11% yield. 1H NMR (300 MHz, DMSO-d6) ppm 10.13 (s, 1H), 7.74 (dd, 1 H), 7.69 (dd, 1H), 7.52-7.62 (m, 2H), 7.44 (dd, 1H), 7.11 (ddd, 1H), 6.58 (dd, 1H), 2.39 (s, 3H), 2.22 (s, 3H), 2.06 (s, 3H)

Following standard procedure for Suzuki coupling, the intermediate was obtained by reaction of 2 g (10.64 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; hexane/EA 20/1 to EA) 2.1 g (87% yield) of pure intermediate were obtained as white solid. A solution of 6-methoxy-3,3′-bipyridine (1.7 g, 11.3 mmol) in EtOH (6 ml) and HBr 48% (12 ml) was stirred at 80° C. for 20 h. The solvent was evaporated and crude A (as hydrobromide salt) was utilized in the next step without any purification (quantitative yield). Following general procedure H1A, with the addition of triethylamine, compound 169 was prepared in 14% yield. 1H NMR (300 MHz, DMSO-d6) ppm 8.89 (br. s., 1H), 8.51 (d, 1H), 8.18 (dd, 1H), 8.07 (ddd, 1H), 8.01 (dd, 1H), 7.64-7.74 (m, 2H), 7.49-7.60 (m, 2H), 7.44 (dd, 1H), 6.65 (dd, 1H)

6-methoxynicotinaldehyde (1.0 g, 7.2 mmol) was dissolved in HBr 48% (10 mL) and EtOH (3 mL) and the solution was heated at reflux for 2 h. After evaporation of volatiles, 1.6 g of the desired pyridone intermediate was obtained. The intermediate was used in the next step without further purification. To a solution of 6-oxo-1,6-dihydropyridine-3-carbaldehyde (640 mg, 5.2 mmol) in DCM (6 mL) and DMF (2 mL), Cu(OAc)2 (1.8 g, 10.4 mmol), phenyl boronic acid (1.2 g, 10.4 mmol), pyridine (0.8 g, 10.4 mmol) and finely grounded, activated 4 Å molecular sieves (1 g) were added. The mixture was stirred at room temperature for 24 h. A concentrated solution of NH4OH was added. The solvents were evaporated under vacuum, and the resulting crude was purified by chromatographic column (SiO2; Pet. Ether/EtOAc 10/1 to 0/1). 300 mg (48% yield) of the second intermediate were obtained as a white solid. To a solution of the second intermediate (6-oxo-1-phenyl-1,6-dihydropyridine-3-carbaldehyde, 300 mg, 2.5 mmol) in of MeOH (20 mL), glyoxal (0.89 g, 10.4 mmol) was added at 0° C. Gaseous NH3 was bubbled into the mixture at 0° C. for 1 h. The reaction was warmed at room temperature and stirred for 24 h. The solvent was evaporated under vacuum and the resulting crude was purified by flash chromatography (SiO2, Pet. Ether/EtOAc 10/1 to 0/1) and by reverse-phase preparative HPLC. 80 mg (14% yield) of compound 173 were obtained. 1H NMR (300 MHz, DMSO-d6) ppm 14.16 (br. S., 1H), 8.49 (d, 1H), 8.03 (dd, 1H), 7.66 (s, 2H, 7.44-7.63 (m, 5H), 6.75 (d, 1H)

5-iodo-1-(pyridin-4-yl)pyridin-2(1H)-one (0.120 g, 0.4 mmol) was dissolved in dry and degassed toluene (10 mL), previously degassed. Pd(PPh3)4 (0.023 g, 0.02 mmol) was then added and the mixture was stirred for 10 minutes. 2-(tributylstannyl)thiazole (0.15 g, 0.4 mmol) was added and the reaction was heated at 90° C. for 4 h under nitrogen atmosphere. A large excess of a KF/H2O solution was added and the mixture was stirred for 1 h. The aqueous phase was extracted with EtOAc. The solvent was removed under reduced pressure and the crude was purified by flash chromatography (SiO2; EtOAc/MeOH 95:5) and then through titration in CH3CN. 38.7 mg (38% yield) of compound 174 were obtained as a white solid. 1H NMR (300 MHz, DMSO-d6) ppm 8.65-8.86 (m, 2H), 8.31 (dd, 1H), 8.10 (dd, 1H), 7.86 (d, 1H), 7.72 (d, 1H), 7.58-7.68 (m, 2H), 6.60-6.74 (m, 1 H)

The iodopyridone intermediate above was obtained by reaction of 600 mg (2.7 mmol) of 5-iodo-2-pyridone with phenylboronic acid. After purification (SiO2; Hexane/Acetate/MeOH 1/1/0 to 0/10/1). 600 mg (75% yield) of pure intermediate were obtained as a pale yellow solid. The Suzuki coupling, as outlined for compound 189, below, provided compound 183 in 38% yield. 1H NMR (300 MHz, DMSO-d6) ppm 7.98 (s, 2H), 7.91 (dd, 1H), 7.83 (dd, 1 H), 7.36-7.62 (m, 5H), 6.54 (dd, 1H)

A mixture of 2-methoxy-5-aminopyridine (10 g, 0.08 mol) in AcOH (125 mL), and concentrated HCl (150 mL) was cooled at 0° C. in an ice/water bath. A solution of NaNO2 (4.0 g, 0.058 mol) in water (15 mL) was added dropwise at 0° C. The resulting mixture was stirred for 45 minutes at 0° C. In a separate round bottom flask, 150 mL of concentrated HCl was added dropwise to a sodium bisulphite solution. The gaseous SO2 thus formed was purged for 2-3 h into a third round bottom flask containing AcOH cooled at −20° C. CuCl2 (18 g) was added, and the reaction was stirred for 20 minutes at −20° C. The mixture was added dropwise to the 2-methoxy-5-aminopyridine/AcOH/concentrated HCl mixture maintained at 0° C. The reaction was allowed to warm up to room temperature and stirred overnight. The mixture was quenched with water and the solid thus formed was filtered, re-dissolved in DCM and filtered through celite. The clear solution was dried over Na2SO4 and concentrated under vacuum to afford 10.2 g (61% yield) of pure 6-methoxy-pyridine-3-sulfonyl chloride. 6-Methoxy-pyridine-3-sulfonyl chloride (5.0 g, 0.025 mol) was dissolved in DCM and cooled at 0° C. Gasseous Gaseous NH3 was bubbled in the solution for 10 min. The resulting pale brown suspension was filtered and the solid was triturated with water. The resulting white solid was filtered and dried under vacuum to afford 3.2 g (70.6% yield) of pure 6-Methoxy-pyridine-3-sulfonamide. 6-Methoxy-pyridine-3-sulfonamide (0.752 g, 4.0 mmol) was dissolved in EtOH (6 mL). An excess of 48% HBr aqueous solution (12 mL) was added and the reaction was heated at 90° C. for 20 h. The solvent was removed under reduced pressure and the residual hydrobromic acid was further dried under reduced pressure, at 40° C., to provide the intermediate sulfonamide in quantitative yield. The sulfonamide was used in General Procedure H1A to provide compound 184 in 10% yield. 1H NMR (300 MHz, DMSO-d6) ppm 8.05 (dd, 1H), 7.79 (dd, 1H), 7.62 (m, 2H), 7.55 (m, 2H), 7.36 (s, 2H), 6.67 (dd, 1H)

Following the general procedure I, 5-(2-fluorophenyl)-2-methoxypyridine was obtained by reaction of 3 g (16 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Pet. Ether/EtOAc 1/1 to 0/1), 750 mg (31% yield) of pure product was obtained as a white solid. 5-(2-fluorophenyl)-2-methoxypyridine (750 mg) was dissolved in HBr 48% (10 mL) and EtOH (3 mL) and the solution was heated at reflux for 3 h. After evaporation of volatiles, 700 mg (quantitative yield) of the desired pyridone were obtained as a white solid. Following general procedure H1A, compound 185 was prepared in 58% yield. 1H NMR (300 MHz, DMSO-d6) ppm 10.13 (s, 1H), 7.69-7.90 (m, 3H), 7.51-7.65 (m, 2H), 7.20-7.50 (m, 4 H), 7.14 (dd, 1H), 6.60 (d, 1H), 2.06 (s, 3H)

5-(1H-imidazol-2-yl)pyridin-2(1H)-one (0.097 g, 0.6 mmol) was dissolved in DCM (3 mL) and N,N-dimethylformammide (3 mL). Phenylboronic acid (0.087 g, 0.72 mmol), copper(II) acetate (0.21 g, 1.2 mmol), pyridine (0.095 g, 1.2 mmol) and 4 Å molecular sieves were added and the reaction was stirred at room temperature in an open vessel for nine days. The reaction was monitored by UPLC-MS. At the end of the reaction, a concentrated solution of NH4OH was added. Solvents were removed at reduced pressure, and the crude was purified by flash chromatography (SiO2; EtOAc/MeOH 1:0 to 95:5). Two main products were recovered: 24 mg of compound 173 (10% yield) and 7 mg of compound 188 (2% yield). 1H NMR (300 MHz, DMSO-d6) ppm 7.81-7.94 (m, 1H), 7.75 (d, 1H), 7.71 (br. s., 1H), 7.40-7.66 (m, 8H), 7.33 (dd, 1H), 7.22-7.30 (m, 2H), 6.50 (d, 1H)

To a solution of 5-iodo-pyridin-2-one (1 eq) in DCM (5 mL/mmol of aryl halide) and DMF (0.7 mL/mmol of aryl halide), Cu(OAc)2 (2 eq), the appropriate boronic acid (1.2 eq), pyridine (2 eq) and finely grounded, activated 4 Å molecular sieves were added. The mixture was stirred at room temperature in an open vessel for a variable time (from 12 hours to 7 days). Fresh Boronic acid was further added in sluggish reactions. A concentrated solution of NH4OH was added. The solvents were evaporated under vacuum and the resulting crude was absorbed on silica pad and purified by flash chromatographic column (SiO2; Pet. Ether/EtOAc mixture).
800 mg (4.2 mmol) of 5-iodo-2-pyridone with 4-pyridine-boronic acid. After purification (SiO2; Pet. Ether/EtOAc/MeOH 1/1/0 to 0/10/1). 387 mg (31% yield) of pure product were obtained as a pale yellow solid. MS-ESI+: m/z=299 [MH+]

For the Suzuki coupling, the iodopyridone (1 eq), the appropriate boronic acid (1.2 eq) and K2CO3 (3 eq) were dissolved in a 10:1 mixture of DME/H2O (4 mL/mmol). The solution was degassed by bubbling N2 for 15 min and then Pd(PPh3)4 (0.05 eq) was added. The reaction mixture was heated at 90° C. for 18 h, after which time, BOC protecting group was completely cleaved. Mixture was cooled at room temperature, diluted with EtOAc and filtered on a celite plug. The filtrate was washed with brine. The separated organic phase was dried over Na2SO4 and concentrated under reduced pressure. The obtained residue was purified by column chromatography (SiO2; Pet. Ether/EtOAc mixture).
Compound 189 was obtained in 42% yield. 1H NMR (300 MHz, DMSO-d6) ppm 8.71-8.92 (m, 2H), 8.00 (s, 2H), 7.98 (dd, 1H), 7.88 (dd, 1H), 7.66-7.78 (m, 2H), 6.60 (dd, 1H), 5.74 (br. s., 1H)

5-iodo-1-phenylpyridin-2(1H)-one (0.088 g, 0.3 mmol) was dissolved in dry and degassed toluene (7.5 mL/mmol). The catalyst was then added (0.017 g, 0.015 mmol) and the mixture was stirred for 10 minutes. 2-(tributylstannyl)oxazole (0.107 g, 0.3 mmol) was added and the reaction was heated at 90° C. for 18 h under nitrogen atmosphere. Conc. NH4OH was added. The solvent was removed at reduced pressure and the crude was purified by flash chromatography (SiO2; Pet. Ether/EtOAc 1:1) and then through titration in di-isopropylether. The residual product present in the mother liquor was recovered after purification with preparative. 36 mg (30% yield) of compound 193 were obtained as a pale yellow solid. 1H NMR (300 MHz, DMSO-d6) ppm 8.19 (dd, 1H), 8.14 (d, 1H), 8.02 (dd, 1H), 7.43-7.61 (m, 5H), 7.32 (d, 1H), 6.66 (dd, 1H)

The product was obtained by reaction of 500 mg (2.25 mmol) of 5-iodo-2-pyridone with 4-trifluoromethoxy-phenyl-boronic acid. After flash chromatography (SiO2; Pet. Ether/EtOAc 2:1) 300 mg (35% yield) of the intermediate were obtained as a white solid. MS-ESI+: m/z=380.9 [MH+] The iodopyridone was then used in a Stille coupling.

5-iodo-1-(4-(trifluoromethoxy)phenyl)pyridin-2(1H)-one (0.19 g, 0.5 mmol) was dissolved in dry and degassed toluene (10 mL). Pd(PPh3)4 (0.029 g, 0.025 mmol) was then added and the mixture was stirred for 10 minutes. 2-(tributylstannyl) thiazole (0.187 g, 0.5 mmol) was added and the reaction was heated at 90° C. for 4 h under nitrogen atmosphere. A large excess of a KF/H2O solution was added and the mixture was stirred for 1 h. The aqueous phase was extracted with EtOAc. The solvent was removed under reduced pressure and the crude was purified by flash chromatography (SiO2; Pet. Ether/EtOAc 7:3 to Hex/EtOAc 1:1) and then through titration in a Pet. Ether/EtOAc mixture. 62.5 mg (37% yield) of compound 203 were obtained as a white solid. 1H NMR (300 MHz, DMSO-d6) ppm 8.30 (dd, 1H), 8.09 (dd, 1H), 7.85 (d, 1H), 7.60-7.75 (m, 3H), 7.55 (m, 2H), 6.66 (dd, 1H)


To a mixture of 209-1 (45.0 g, 381.4 mmol), para-toluene-sulfonyl chloride (80.1 g, 421.6 mmol) and a catalytic amount of tetrabutyl ammonium bromide (TBABr) in toluene (540 ml) was added aq. NaOH (288.0 g in 900 ml water, 7.2 mol). The biphasic solution was stirred at ambient temperature for 4 h, and then extracted twice with toluene. The organic phase was dried over anhydrous Na2SO4 and concentrated. The crude product was triturated in ethyl acetate/petroleum ether (V:V=1:20) and filtrated to afford the compound 209-2 (90 g, 87% yield). MS-ESI: m/z=273.1 [M+1]+
A solution of 209-2 (50.0 g, 183.8 mmol) in dry THF cooled to −78° C. and n-BuLi (81 ml, 2.5 M in hexane) was added over 20 minutes. The resulted solution was maintained at −78° C. for 1 h, and then a solution of BrCl2CCCl2Br (71.0 g, 220.5 mmol) in dry THF was added. The mixture was stirred −78° C. for 30 min and allowed to warm slowly to room temperature. The solvent was removed under vacuum and the residue was partitioned between EtOAc and water. The organic layer was dried over anhydrous Na2SO4 and concentrated. The crude product was purified by column chromatography (5% ethyl acetate in petroleum ether to 20% ethyl acetate in petroleum ether as the eluent) to afford 209-3 (37 g, 58% yield). MS-ESI: m/z=351.0 [M+1]+
A mixture of 209-3 (30 g, 0.085 mol), methanol (85OmL850 mL) and aqueous potassium hydroxide (5 mol/L, 100 mL) was heated under reflux overnight. The majority of the solvent was removed under vacuum, and the residue was partitioned between EtOAc and water. The organic layer was dried over anhydrous Na2SO4 and concentrated to give 209-4 (24 g, 80% yield) which was used without further purification. 1H 1H NMR (400 MHz, DMSO): 6.550 (s, 1H); 7.039 (dd, J=5.2 Hz, J=3.2 Hz, 1H); 7.857 (dd, J=1.6 Hz, J=3.2 Hz, 1H); 8.155 (q, J=1.6 Hz, 1H); 12.418 (br, 1H)
mCPBA (14.0 g, 81.4 mmol) was added into a solution of 209-4 (8.0 g, 40.8 mmol) in THF (140 ml) at 0° C., and then the reaction was warmed up the room temperature for 1 h and quenched with saturated Na2S2O3. The solution was concentrated after filtering. The crude was purified by column chromatography (0-10% methanol in ethyl acetate as the eluent) to afford 209-5 (6.3 g, 73% yield). 1H NMR (400 MHz, DMSO): 6.698 (s, 1H); 7.055 (t, J=6.4 Hz, 1H); 7.660 (d, J=6.0 Hz, 1H); 8.099 (d, J=6.0 Hz, 1H)
A mixture of 209-5 (3.5 g, 16.5 mmol) and acetic acid anhydride was heated at its reflux temperature for 1.5 h. The solution was then evaporated. The residue was mixed with methanol and Et3N at room temperature for 2 h. The solution was concentrated and the residue was partitioned between EtOAc and water. The organic layer was dried over anhydrous Na2SO4 and concentrated. The crude product was purified by column chromatography (10% ethyl acetate/petroleum) to afford 209-6 (1.4 g, 40% yield). 1H NMR (300 MHz, DMSO): 6.337 (d, J=8.1 Hz, 1H); 6.359 (s, 1H); 7.662 (d, J=8.1 Hz, 1H) MS-ESI: m/z=214.1 [M+1]+
A solution of 209-6 (150 mg, 0.71 mmol) and triethylamine (470 mg, 4.2 mmol) in THF (5 mL) was stirred for 15 min before the addition of (Boc)2O (0.907 g, 4.2 mmol). The solution was stirred at room temperature overnight. Most of the solvent was removed under vacuum to get a residue, then it was partitioned between water (50 mL) and DCM (100 mL), the organic layer was separated and the aqueous layer was extracted with DCM (50 mL×2). The combined organic layer was washed with water (100 mL) and brine (100 mL), dried over Na2SO4 and concentrated to give a residue, which was purified by Prep-TLC (25% ethyl acetate in petroleum ether as the eluent) to give 209-7 (250 mg, yield 85%). 1H NMR (400 MHz, CDCl3): 1.558 (s, 9H); 1.684 (s, 9H); 6.673(s, 1H); 6.970 (d, J=8.4 Hz, 1H); 7.824 (d, J=8.4 Hz, 1H); MS-ESI: m/z=436.9 [M+23]+
A mixture of 209-7 (550 mg, 1.33 mmol), K2CO3 (200 mg, 1.45 mmol) and methanol (8 mL) was stirred at rt for 1 h. Methanol was removed before the addition of water (50 mL), then it was extracted with DCM (50 mL×3). Combined DCM was washed with water and brine, dried over Na2SO4 and concentrated to give a residue, which was isolated by prep-TLC (50% ethyl acetate in petroleum ether as the eluent) to give 209-8 (130 mg, 31.2%) as a white solid.
A solution of 209-9 (50 mg, 0.112 mmol) in DCM/TFA (V:V=1:1) was stirred at room temperature for 3 h. All the solvents were removed by evaporation to give a residue. It was isolated by Prep-TLC (25% ethyl acetate in petroleum ether as the eluent) to give compound 209 (30 mg, 68.5%) as a white solid. 1H NMR (400 MHz, CDCl3): 1.350 (d, J=6.0 Hz, 6H); 4.509 (m, J=6.0 Hz, 1H); 6.440 (d, J=2.0 Hz, 1H); 6.674 (d, J=8.4 Hz, 1H); 6.899 (d, J=8.8 Hz, 2H); 7.053 (d, J=8.8 Hz, 2H); 7.774 (d, J=8.4 Hz, 1H); 8.683 (br, 1H); MS-ESI: m/z=349.2 [M+1]+

To a solution of 4 (5 g crude) in dry DMF (50 mL) anhydrous potassium carbonate (10.9 g, 79 mmol) and methyl iodide (2.5 mL, 0.039 mol) were added. The reaction was stirred at room temperature overnight. The mixture was filtered and the residue was washed with methanol. Mother liquors were concentrated and the obtained crude product was purified by column chromatography (SiO2, hexanes:EtOAc 7:3) to obtain 1.5 g of a yellow solid (9:1 mixture of 1-Methyl-1H-pyrrolo[2,3-b]pyridine-2-carboxylic acid methyl ester and 1-Methyl-1H-pyrrolo[2,3-b]pyridine-2-carboxylic acid ethyl ester. This intermediate was then used in the subsequent reactions of general procedure K to obtain a methyl version of intermediate 8 for use in general procedure J. Following this modified procedure, compound 223 was obtained in 94% yield. 1H NMR (300 MHz, DMSO-d6) ppm 12.98 (br. s., 1H) 8.18 (d, 1H) 7.44 (m, 2H) 7.36 (m, 2H) 7.20 (s, 1H) 6.88 (d, 1H) 3.82 (s, 3H)

Following general procedure A, the intermediate compound was prepared in 78% yield as a white solid. MS-ESI: m/z=278.1 [M+1]+ To a solution of the intermediate (3.5 g, 10.8 mmol) in methanol (200 ml) was added Pd/C (300 mg) catalyst under N2 atmosphere, and then stirred for 2 h under H2 atmosphere (1 atm, 25° C.). The catalyst was filtered off through the celite pad, and the filtrate was concentrated in vacuo to give compound 318 (2.2 g, 93% yields) as a white solid. MS-ESI: m/z=188.2 [M+1]+

To a solution Br-substitution-1-Phenyl-1H-pyridin-2-one (1 eq), the appropriate boromic acid (1.2 eq), potassium phosphate (3.5 eq) and tricyclohexylphosphine (0.1 eq) in toluene/water (2:/1, V:V) under a nitrogen atmosphere was added palladium acetate (0.05 eq). The mixture was heated to 100° C. for 2-3 h, and then cooled to room temperature, water was added and the mixture was extracted with EtOAc, the combined organics were washed with brine and water, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by pre-TLC to afford the desired compound 322 in 70% yield as a pink solid. MS-ESI: m/z=212.2 [M+1]+

A mixture of 2,6-dibromopyridine (4 g, 17 mmol), potassium t-butoxide (20 g, 0.27 mol), and redistilled t-butyl alcohol (100 ml) was refluxed overnight. After cooling, the solvent was removed in vacuo, ice/water was carefully added, and the aqueous layer was extracted with chloroform (100 ml×2), which removed the unreacted staring material. The aqueous layer was acidified with 3 N HCl, extracted with chloroform (100 ml×2), washed with brine, dried over anhydrous Na2SO4 and concentrated affording pure 6-bromo-2-pyridone (2.5 g, 85% yield) as a white solid. Intermediate 3 was prepared following general procedure A in 73% yield. Intermediate 3 was then reacted with the appropriate boronic acid, Pd (OAc)2, PCy3, K3PO4 at 100° C. to afford compound 327 in 40% yield as an oil. MS-ESI: m/z=248.3 [M+1]+

A mixture of compound 1 (200 mg, 1.3 mmol) in AcOH (4 ml) was added HBr (aq. 40%, 1 ml), then heated to reflux for 2 h. The compound 2 was obtained by evaporated in vacuo (160 mg, 90%). To a mixture of compound 2 (160 mg, 1.2 mmol), phenylboronic acid (293 mg, 2.4 mmol) and Cu(OAc)2 (36 mg, 0.2 mmol) in DCM, pyridine (190 mg, 2.4 mmol) was added slowly. After the suspension was stirred overnight at room temperature, it was checked by TLC and the starting material was completely vanished, and then washed with saturated NaHCO3. The DCM layer was dried over sodium sulfate, and evaporated to obtain the crude product. The crude product was purified by preparative TLC to afford the compound 3 (110 mg, 43%). A mixture of compound 3 (110 mg,0.5 mmol) in DAST (2.5 ml) was heated to 80° C. for 4 h. The reaction mixture was extracted by DCM and saturated NaHCO3, and the crude product was purified by preparative TLC to give compound 335 (40 mg, 34% yield) as yellow solid. MS-ESI: m/z=236.3 [M+1]+
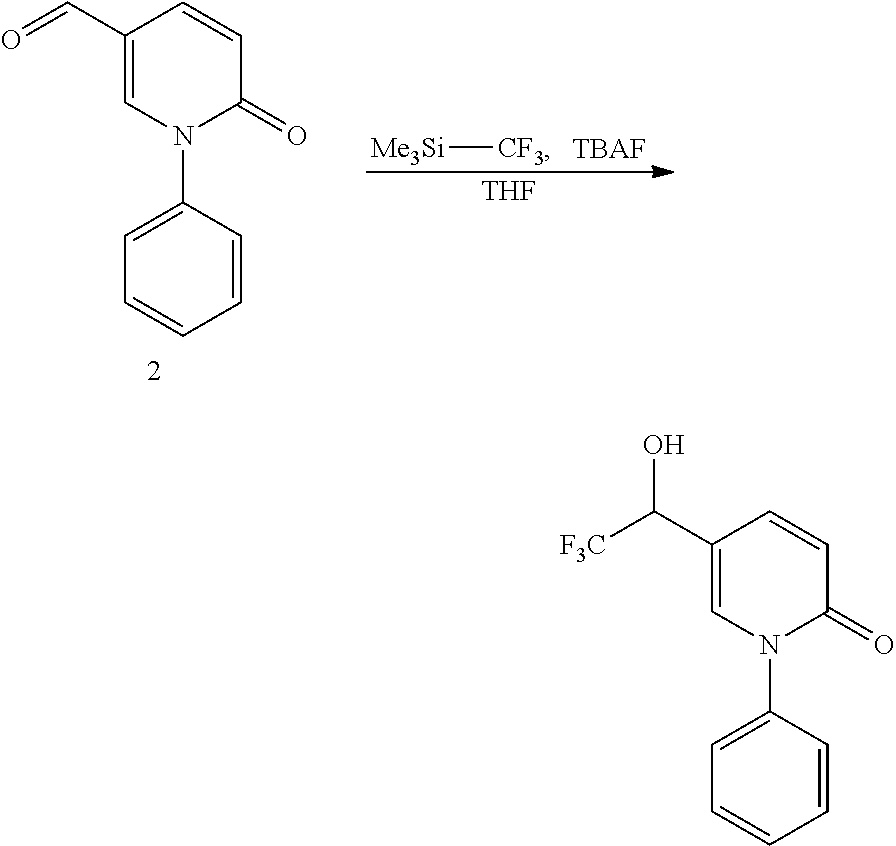
A mixture of compound 2 (1 g, 5 mmol) and trimethyl-trifluoromethyl-silane (3.5 ml, 2M in THF, 7 mmol) in THF (20 ml) cooled to 0° C. in an ice bath was treated with tetrabutylammonium fluoride (0.25 ml, 1 m in THF, 0.25 mmol) under nitrogen atmosphere at 0° C. for 30 min. The mixture was raised to room temperature and stirred 24 h. Then 1 M HCl (50 ml) was added and the mixture was stirred overnight. The aqueous layer was extracted with EtOAc (50 ml×2) and the organics was concentrated. The desired product was separated by columnar chromatography to give compound 338 (0.94 g, 70% yields) as yellow solid. MS-ESI: m/z=270.2 [M+1]+

Intermediate 3 was prepared thus. To a solution of compound 1 (3.0 g, 16 mmol), compound 2 (2.5 g, 21 mmol), K3PO4 (12.5 g, 57 mmol) in toluene/water (60 ml/3 ml) under a nitro nitrogen atmosphere was added Pd(PPh3)4 (2.0 g, 1.6 mmol). The mixture was heated to reflux for 3 h and then cooled to room temperature. Water was added and the mixture extracted with EtOAc, the combined organics were washed with brine, dried over Na2SO4 and concentrated in vacuo. The product was isolated by column chromatography afforded the compound 3. (2.1 g, 69%). Intermediate 3 (2.0 g, 11 mmol) in HBr (aq. 40%)/EtOH (20 ml/4 ml) was heated to reflux for 2 h, the reaction was monitored by TLC, when completed, the mixture was cooled to r.t. The reaction mixture was neutralized by NaHCO3, then extracted with EtOAc several times. The combined organics was washed with brine, dried over Na2SO4 and concentrated in vacuo to afford the compound 4 (1.7 g, 91%) Following general procedure A, compound 343 was prepared from intermediate 4 in 50% as an oil. MS-ESI: m/z=306.0 [M+1]+

A mixture of reagent 1 (0.5-1 mmol, 1 eq.), boronic acids 2 (2 eq.), copper(II) acetate (0.4-0.6 eq.), pyridine (2 eq.) and molecular sieves 4 A in dichloromethane (5 ml/1 mmol reagent 1) was stirred for overnight at the room temperature opened to the air. The reactions were monitored by TLC, and when found to be completed washed with saturated sodium bicarbonate with EDTA and dried over sodium sulfate. Compounds 3 were isolated by pre-TLC (using EA/PE as solvent). Reagent 3 (0.3-0.5 mmol, 1 eq.) was dissolved in acetonitrile (3 mL/1 mmol reagent 3), DAST (2 eq.) was added slowly at room temperature. The resulting solution was stirred at 80° C. in a capped plastic tube overnight. After cooling to room temperature, it was diluted with DCM, washed with aqueous solution of saturated sodium bicarbonate, water and brine, dried over Na2SO4, concentrated to give a residue, which was purified by pre-TLC (using EA/PE as solvent) to give target compound. Following this procedure, compound 359 was prepared in 13.9% yield as a white solid. 1H NMR (400 MHz, CDCl3): 2.98 (s, 6H); 3.323˜6.341 (t, J=5.6 Hz, 1H); 6.460˜6.599 (d, J=55.6 Hz, 1H); 6.639˜6.657 (d, J=7.2 Hz, 2H); 6.765˜6.790 (d, J=10.0 Hz, 2H); 7.314˜7.353 (t, J=8.0 Hz, 1H); 7.446˜7.464 (d, J=7.2 Hz, 1H) MS-ESI: m/z=265.1 [M+1]+

5-bromo-2-methoxypyridine (0.66 g, 3.49 mmol) and 1,3,5-trimethyl-1 h-pyrazole-4-boronic acid pinacol ester (0.99 g, 4.19 mmol) were dissolved in a degassed DME/H2O mixture (14 mL, 10:1 ratio). Solid Na2CO3 (1.1 g, 10.47 mmol) was added, followed by Pd(PPh3)4 (0.2 g, 0.17 mmol). The reaction mixture was heated at 80° C. for 18 h. Water was added until complete dissolution of the residual carbonate and the solution was stirred for additional 6 h at the same temperature. The organic layer was separated and evaporated under reduced pressure and the resulting crude mixture was purified by flash chromatography (SiO2; DCM/MeOH 20:1). 440 mg (66% yield) of pure product were obtained as a pale yellow solid. MS-ESI: m/z=218.3 [M+1]+5-(1,3,5-trimethyl-1H-pyrazol-4-yl)pyridin-2(1H)-one (0.44 g, 2.3 mmol) was dissolved in EtOH (3 mL). An excess of 48% HBr aqueous solution (10 mL) was added and the reaction was heated at 90° C. for 24 h. The solvent was removed under reduced pressure and the crude was purified by flash chromatography (SiO2; AcOEt to AcOEt/MeOH 3.5:1). 400 mg (92% yield) of pure intermediate product were obtained as an off-white foam.

2-bromo pyrimidine (0.55 g, 3.49 mmol) and 2-methoxy-5-pyridineboronic acid (0.53 g, 3.49 mmol) were dissolved in a degassed mixture of DME/H2O (11 mL, 10:1 ratio). Solid K2CO3 (1.4 g, 10.47 mmol) was added, followed by Pd(PPh3)4 (0.2 g, 0.17 mmol). The reaction mixture was heated at 90° C. for 18 h. The organic layer was separated and evaporated under vacuum. The resulting crude was purified by flash chromatography (SiO2; Pet. Ether/AcOEt 1:1). 420 mg (65% yield) of pure product were obtained as a pale yellow solid. MS-ESI: m/z=188 [M+1]+ 2-(6-methoxypyridin-3-yl)pyrimidine (0.78 g, 4 mmol) was dissolved in EtOH (5 mL). An excess of 48% HBr aqueous solution (10 mL) was added and the reaction was heated at 90° C. for 24 h. The solvent was removed under reduced pressure and the residual hydrobromic acid was stripped at reduced pressure, at 40° C. The resulting off white solid was used in the next step without further purification. MS-ESI: m/z=174 [M+1]+

The 5-iodo-1-arylpyridin-2(1H)-one (1 eq), the boronic acid (1.2 eq) and K2CO3 (3 eq) were dissolved in a 10:1 mixture of DME/H2O (4 ml/mmol). The solution was degassed by bubbling N2 for 15 min and then Pd(PPh3)4 (0.05 eq) was added. The reaction mixture was heated at 90° C. for 18 h, after which time, BOC protecting group was completely cleaved. Mixture was cooled at room temperature, diluted with AcOEt and filtered on a celite plug. The filtrate was washed with brine. The separated organic phase was dried over Na2SO4 and concentrated under reduced pressure. The obtained residue was purified by column chromatography (EtOAc:Hexanes 3:7 to 1:1) to afford compound 379 as a pale yellow solid (11% yield). 1H NMR (300 MHz, DMSO-d6) ppm 10.12 (s, 1H), 7.98 (s, 2H), 7.80-7.87 (m, 1H), 7.69 (t, 1H), 7.56-7.64 (m, 1H), 7.10 (ddd, 1H), 6.54 (dd, 1H), 2.06 (s, 3H)

Following the standard procedure for Suzuki coupling the intermediate was obtained by reaction of 3 g (16 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 9:1) 1.4 g (31% yield) of pure product were obtained as white solid. 2-methoxy-5-(4-methoxyphenyl)pyridine (1.4 g, 4.96 mmol) was dissolved in HBr 48% (12 ml) and EtOH (6 ml) and the solution was heated at reflux for 24 h. After evaporation of volatiles the desired pyridone was obtained as white solid (0.99 g, quantitative yield).

The product was obtained by reaction of 963 mg (6.3 mmol) of 2-methoxy-pyridine-5-boronic acid. After purification (SiO2; Hexanes:EtOAc 1:1) 747 mg (65% yield) of pure product were obtained as white solid. 2-(6-methoxypyridin-3-yl)pyrimidine (747 mg) was dissolved in HBr 48% (10 ml) and EtOH (5 ml) and the solution was heated at reflux overnight. After evaporation of volatiles the desired pyridone was obtained as white solid (1.016 g, quantitative yield).

5-iodo-1-phenyl-1H-pyridin-2-one (0.34 g, 1.13 mmol) was dissolved in dry and degassed toluene 5 mL). The catalyst was then added (0.065 g, 0.057 mmol) and the mixture was stirred for 10 minutes. 1-methyl-4-(tributylstannyl)-3-(trifluoromethyl)-1H-pyrazole (0.49 g, 1.13 mmol) was added and the reaction was heated at 90° C. for 18 h under nitrogen atmosphere. Conc. NH4OH was added. The solvent was removed at reduced pressure and the crude was purified by elution through basic alumina (Hexanes:EtOAc 1:1). 37 mg (10% yield) of compound 383 were obtained as a pale yellow solid. 1H NMR (300 MHz, DMSO-d6) d ppm 7.99 (d, 1H), 7.75 (dd, 1H), 7.38-7.62 (m, 5H), 6.91 (s, 1H), 6.62 (dd, 1H), 3.94 (s, 3H)

N-(3-(5-iodo-2-oxopyridin-1(2H)-yl)phenyl)acetamide (0.050 g, 0.14 mmol) was dissolved in dry and degassed toluene (3 mL). The catalyst was then added (0.008 g, 0.007 mmol) and the mixture was stirred for 10 minutes. 2-(tributylstannyl)oxazole (0.050 g, 0.14 mmol) was added and the reaction was heated at 90° C. for 18 h under nitrogen atmosphere. Conc. NH4OH was added. The solvent was removed at reduced pressure and the crude was purified by preparative HPLC. 16 mg (38.7% yield) of compound 384 were obtained as a pale yellow solid. 1H NMR (300 MHz, DMSO-d6) ppm 10.15 (br. s., 1H), 8.16-8.21 (m, 1H), 8.14 (d, 1H), 8.02 (dd, 1H), 7.76 (t, 1H), 7.61 (ddd, 1H), 7.46 (dd, 1H), 7.32 (d, 1H), 7.16 (ddd, 1H), 6.65 (dd, 1H), 2.07 (s, 3H)

Following standard Suzuki coupling, the product was obtained by reaction of 2.82 g (15 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 9:1) 2.8 g (92% yield) of pure product were obtained as white solid. The intermediate (900 mg) was dissolved in HBr 48% (10 ml) and EtOH (3 ml) and the solution was heated at reflux for 3 h. After evaporation of volatiles the desired pyridone was obtained as white solid (780 mg, 93% yield).

Following standard procedure for Suzuki coupling, the intermediate was obtained by reaction of 3 g (16 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 20:1 to 100% EtOAc) 2.2 g (51% yield) of pure product were obtained as white solid. To a magnetically stirred solution of 2-Methoxy-5-(1-methyl-1H-pyrazol-4-yl)-pyridine (1.2 g, 6.3 mmol), in 3 mL of EtOH, 15 mL of HBr were added. The mixture was heated at 80° C. for 20 h. The reaction was cooled at room temperature. The solvent was evaporated under vacuum. Purification by flash column chromatography (SiO2; 100% AcOEt) afforded 1.1 g of the intermediate compound (quantitative yield).

Following the standard procedure for Suzuki coupling, the intermediate was obtained by reaction of 3.2 g (16 mmol) of 5-bromo-2-methoxy-4-methylpyridine. After purification (SiO2; Hexanes:EtOAc 20:1 to 100% EtOAc) 2 g (62% yield) of pure product was obtained as white solid. A solution of 2-methoxy-4-methyl-5-(1-methyl-1H-pyrazol-4-yl)pyridine (2 g, 9.9 mmol) in EtOH (6 ml) and HBr 48% (12 ml) was stirred at 90° C. for 24 h. The solvent was evaporated and the crude compound (as hydrobromide salt) was utilized in the next step without any purification. Quantitative yield.

Following standard procedure for Suzuki coupling, the intermediate was obtained by reaction of 420 mg (3 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 95:5) 390 mg (65% yield) of pure product was obtained as white solid. The intermediate (390 mg) was dissolved in HBr 48% (5 ml) and EtOH (5 ml) and the solution was heated at reflux for 24 h. After evaporation of volatiles the desired pyridone was obtained as white solid (359 mg, quantitative yield).

Following the standard procedure for Suzuki coupling, the product was obtained by reaction of 2.7 g (14.4 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 1:1 to 100% EtOAc) 1.29 g mg (48% yield) of pure product was obtained as white solid. A solution of 5-(6-Methoxy-pyridin-3-yl)-pyrimidine (1.29 g, 6.9 mmol) in EtOH (4 ml) and HBr 48% (10 ml) was stirred at 90° C. for 7 h. The solvent was evaporated and the crude compound (as hydrobromide salt) was utilized in the next step without any purification.

Following the standard procedure for Suzuki coupling, the intermediate was obtained by reaction of 3 g (16 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 1:1 to 100% EtOAc) 750 mg (31% yield) of pure product was obtained as white solid. 5-(2-fluorophenyl)-2-methoxypyridine (750 mg) was dissolved in HBr 48% (10 ml) and EtOH (3 ml) and the solution was heated at reflux for 3 h. After evaporation of volatiles the desired pyridone was obtained as white solid (700 mg, quantitative yield).

6-methoxynicotinaldehyde (1.0 g, 7.2 mmol) was dissolved in HBr 48% (10 mL) and EtOH (3 mL) and the solution was heated at reflux for 2 h. After evaporation of volatiles 1.6 g of the desired pyridone was obtained. The product was used in the next step without further purification. To a solution of 6-oxo-1,6-dihydropyridine-3-carbaldehyde (300 mg, 2.4 mmol) in DMF (10 mL), Cu(OAc)2 (0.88 g, 4.8 mmol), 3-acetamidophenyl boronic acid (0.5 g, 2.8 mmol), pyridine (0.42 mL, 2.8 mmol) and finely grounded, activated 4 A molecular sieves (1 g) were added. The mixture was stirred at room temperature for 24 h. A concentrated solution of NH4OH was added. The solvents were evaporated under vacuum, and the resulting crude was purified by chromatographic column (SiO2; Hexanes:EtOAc 9:1 to 100% EtOAc). 370 mg (38.5% yield) of pure product were obtained as a white solid. To a solution of N-(3-(5-formyl-2-oxopyridin-1(2H)-yl)phenyl)acetamide (370 mg, 0.94 mmol) in MeOH (20 mL), glioxal (0.4 mL, 3.4 mmol) was added at 0° C. Gaseous NH3 was bubbled into the mixture at 0° C. for 1 h. The reaction was warmed at room temperature and stirred for 24 h. The solvent was evaporated under vacuum and the resulting crude was purified by flash chromatography (SiO2, Hexanes:EtOAc 9:1 to 100% EtOAc). 100 mg (24.6% yield) of compound 401 were obtained. 1H NMR (300 MHz, DMSO-d6) ppm 10.20 (s, 1H), 8.49 (d, 1H), 8.03 (dd, 1H), 7.83-7.91 (m, 1H), 7.67 (s, 2H), 7.54-7.62 (m, 1H), 7.50 (dd, 1H), 7.15 (ddd, 1H), 6.75 (d, 1H), 2.07 (s, 3H)

A mixture of 2-methoxy-5-aminopyridine (10 g, 0.08 mol) in AcOH (125 mL), and concentrated HCl (150 mL) was cooled at 0° C. in an ice/water bath. A solution of NaNO2 (4.0 g, 0.058 mol) in water (15 mL) was added dropwise at 0° C. The resulting mixture was stirred for 45 minutes at 0° C. In the meantime, in a separate round bottom flask, 150 mL of concentrated HCl was added dropwise to a sodium bisulphite solution. The gaseous SO2 thus formed was purged for 2-3 h into a third round bottom flask containing AcOH cooled at −20° C. CuCl2 (18 g) was added, and the reaction was stirred for 20 minutes at −20° C. The mixture was added dropwise to the 2-methoxy-5-aminopyridine/AcOH/concentrated HCl mixture maintained at 0° C. The reaction was allowed to warm up to room temperature and stirred overnight. The mixture was quenched with water and the solid thus formed was filtered, re-dissolved in DCM and filtered through celite. The clear solution was dried over Na2SO4 and concentrated under vacuum to afford 10.2 g (61% yield) of pure 6-methoxy-pyridine-3-sulfonyl chloride. 6-Methoxy-pyridine-3-sulfonyl chloride (5.0 g, 0.025 mol) was dissolved in DCM and cooled at 0° C. Gasseous Gaseous NH3 was bubbled in the solution for 10 min. The resulting pale brown suspension was filtered and the solid was triturated with water. The resulting white solid was filtered and dried under vacuum to afford 3.2 g (70.6% yield) of pure 6-Methoxy-pyridine-3-sulfonamide. 6-Methoxy-pyridine-3-sulfonamide (0.752 g, 4.0 mmol) was dissolved in EtOH (6 mL). An excess of 48% HBr aqueous solution (12 mL) was added and the reaction was heated at 90° C. for 20 h. The solvent was removed under reduced pressure and the residual hydrobromic acid was further dried under reduced pressure, at 40° C. Quantitative yield.

Following the standard procedure for Suzuki coupling, the product was obtained by reaction of 1.02 g (5.4 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 20:1 to 100% EtOAc) 1.12 g (96% yield) of pure product were obtained as white solid. A solution of 2-Methoxy-5-(4-methoxy-phenyl)-pyridine (1.12 g, 5.2 mmol) in EtOH (5 ml) and HBr 48% (10 ml) was stirred at 80° C. for 48 h. The solvent was evaporated and the crude compound (as hydrobromide salt) was utilized in the next step without any purification (quantitative yield).

Following standard procedure for Suzuki coupling the intermediate was obtained by reaction of 3 g (16 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 9:1) 3.1 g (70% yield) of pure product were obtained as white solid. The intermediate (3.1 g) was dissolved in HBr 48% (10 ml) and EtOH (5 ml) and the solution was heated at reflux for 24 h. After evaporation of volatiles the desired pyridone was obtained as white solid (2.9 g, quantitative yield).

To a suspension of A (2.9 mmol) in a MeOH:Water (10 mL:1 mL) mixture, a solution of NaOH in water (2.9 mmol in 2 mL of Water) was added at −30° C. To the stirred reaction, a solution of 2-aminoacetamide (2.9 mmol) in MeOH (2 mL) was added. The mixture was stirred at the same temperature for 1 h, then warmed at room temperature and stirred for additional 3 h. AcOH was added until pH 5 and the volatile portion was evaporated under vacuum. The remaining mixture was portioned between Water (10 mL) and Ethyl Acetate (10 mL). The organic layer was dried over Na2SO4, filtered and evaporated under vacuum.

Following the standard procedure for Suzuki coupling, the product was obtained by reaction of 1.02 g (5.4 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 20:1 to 100% EtOAc)) 1.06 g (89% yield) of pure product were obtained as white solid. A solution of 2-Methoxy-5-(4-methoxy-phenyl)-pyridine (1.12 g, 5.2 mmol) in EtOH (5 ml) and HBr 48% (10 ml) was stirred at 80° C. overnight. The solvent was evaporated and the crude compound (as hydrobromide salt) was utilized in the next step without any purification (quantitative yield).


In a round bottom flask, glyoxylic acid acid B (22 mmol) and 4-F acetophenone A (8 mmol) were mixed together and the reaction was heated at 115° C. overnight, then allowed to cool down at room temperature. Water (5 mL) and concentrated NH4OH (1 mL), were poured into the reaction vessel and the mixture was extracted with DCM (3×5 mL). To the aqueous basic solution hydrazine (8 mmol) was added and the reaction was stirred at 100° C. for 2 h. The precipitate thus formed was collected by filtration and washed with plenty of water. The desired compound was recovered as a light yellow solid (45% yield). 1H NMR (300 MHz, DMSO-d6) ppm 13.15 (br. s., 1H) 8.01 (d, 1H) 7.91 (m, 2H) 7.31 (m, 2H) 6.97 (d, 1H)

Following standard procedure for Suzuki coupling, the intermediate was obtained by reaction of 3 g (16 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 1:1 to 100% EtOAc) 3.99 g (95% yield) of pure product was obtained as white solid. The intermediate (3.99 g) was dissolved in HBr 48% (12 ml) and EtOH (6 ml) and the solution was heated at reflux for 24 h. After evaporation of volatiles the desired pyridone was obtained as white solid (3.72 g, quantitative yield).

Following standard procedure for Suzuki coupling, the product was obtained by reaction of 1.02 g (5.4 mmol) of 5-bromo-2-methoxy-pyridine. After purification (SiO2; Hexanes:EtOAc 20:1 to 100% EtOAc) 1.06 g (80% yield) of pure product were obtained as white solid. A solution of 2-Methoxy-5-(4-methoxy-phenyl)-pyridine (949 mg, 4.3 mmol) in EtOH (10 ml) and HBr 48% (10 ml) was stirred at 80° C. overnight. The solvent was evaporated and the crude compound (as hydrobromide salt) was utilized in the next step without any purification (quantitative yield).

5-bromo-2-methoxy-4-methylpyridine (1.0 g, 4.95 mmol) was dissolved in HBr 48% (10 mL) and EtOH (10 mL) and the solution was heated at 90° C. for 24 h. After evaporation of volatiles, 930 mg (quantitative yield) of the desired pyridone were obtained as a white solid. 5-bromo-4-methyl-1-phenylpyridin-2(1H)-one was obtained by reaction of 450 mg (2.39 mmol) of 5-bromo-4-methylpyridin-2(1H)-one with phenylboronic acid. After purification (SiO2; Hexanes: EtOAc 9:1 to 1:1) 250 mg (39.7% yield) of compound 438 were obtained as a white solid. 1H NMR (300 MHz, DMSO-d6) ppm 7.93 (s, 1H), 7.32-7.58 (m, 5H), 6.47-6.57 (m, 1H), 2.24 (d, 3H)

Following standard procedure for Suzuki coupling, the product was obtained by reaction of 264 mg (1.0 mmol) of 3-bromo-N,N-dimethylbenzenesulfonamide. After purification (SiO2; Hexanes:EtOAc 1:1) 293 mg (quantitative yield) of pure product were obtained as white solid. A solution of 3-(6-methoxypyridin-3-yl)-N,N-dimethylbenzenesulfonamide (292 mg, 1.0 mmol) in EtOH (4 ml) and HBr 48% (4 ml) was stirred at 80° C. overnight. The solvent was evaporated and the crude compound (as hydrobromide salt) was purified by flash chromatography (DCM:MeOH 9:1). 278 mg were obtained as a pale yellow solid (quantitative yield).

Following the standard procedure for Suzuki coupling, the product was obtained by reaction of 264 mg (1.0 mmol) of 4-bromo-N,N-dimethylbenzenesulfonamide. After purification (SiO2; Hexanes:EtOAc 1:1) 292 mg (quantitative yield) of pure product were obtained as white solid. A solution of 4-(6-methoxypyridin-3-yl)-N,N-dimethylbenzenesulfonamide (292 mg, 1.0 mmol) in EtOH (4 ml) and HBr 48% (4 ml) was stirred at 80° C. overnight. The solvent was evaporated and the crude compound (as hydrobromide salt) was purified by flash chromatography (DCM:MeOH 9:1). 278 mg were obtained as a pale yellow solid (quantitative yield).
1000*(S−G*P)/(S+G*P)
was used to convert the 2 emission readouts into mP; S—S-pol filter signal, P—P-pol filter signal, and G=gain.
| TABLE 2 | |||
| Example | Bin | ||
| 1 | A | ||
| 2 | C | ||
| 3 | C | ||
| 4 | C | ||
| 5 | C | ||
| 6 | A | ||
| 7 | C | ||
| 8 | B | ||
| 9 | C | ||
| 10 | C | ||
| 11 | C | ||
| 12 | C | ||
| 13 | C | ||
| 14 | C | ||
| 15 | C | ||
| 16 | C | ||
| 17 | C | ||
| 18 | B | ||
| 19 | A | ||
| 20 | C | ||
| 21 | C | ||
| 22 | A | ||
| 23 | C | ||
| 24 | C | ||
| 25 | C | ||
| 26 | C | ||
| 27 | C | ||
| 28 | C | ||
| 29 | A | ||
| 30 | C | ||
| 31 | C | ||
| 32 | C | ||
| 33 | C | ||
| 34 | C | ||
| 35 | C | ||
| 36 | C | ||
| 37 | C | ||
| 38 | C | ||
| 39 | C | ||
| 40 | C | ||
| 41 | A | ||
| 42 | C | ||
| 43 | C | ||
| 44 | C | ||
| 45 | C | ||
| 46 | C | ||
| 47 | A | ||
| 48 | C | ||
| 49 | C | ||
| 50 | B | ||
| 51 | C | ||
| 52 | C | ||
| 53 | C | ||
| 54 | C | ||
| 56 | C | ||
| 58 | C | ||
| 59 | C | ||
| 60 | A | ||
| 61 | C | ||
| 62 | C | ||
| 63 | C | ||
| 64 | C | ||
| 65 | C | ||
| 66 | C | ||
| 67 | C | ||
| 68 | C | ||
| 69 | C | ||
| 70 | C | ||
| 72 | A | ||
| 73 | A | ||
| 74 | A | ||
| 75 | C | ||
| 77 | C | ||
| 78 | C | ||
| 79 | C | ||
| 80 | A | ||
| 81 | C | ||
| 82 | A | ||
| 83 | C | ||
| 84 | A | ||
| 85 | C | ||
| 86 | C | ||
| 87 | C | ||
| 88 | C | ||
| 89 | A | ||
| 90 | B | ||
| 91 | C | ||
| 92 | C | ||
| 93 | A | ||
| 94 | A | ||
| 96 | A | ||
| 97 | A | ||
| 98 | C | ||
| 99 | C | ||
| 100 | C | ||
| 101 | C | ||
| 102 | A | ||
| 103 | C | ||
| 104 | C | ||
| 105 | A | ||
| 106 | A | ||
| 107 | C | ||
| 108 | C | ||
| 109 | A | ||
| 110 | C | ||
| 111 | C | ||
| 112 | C | ||
| 113 | C | ||
| 114 | B | ||
| 115 | C | ||
| 116 | C | ||
| 117 | A | ||
| 118 | A | ||
| 119 | C | ||
| 120 | B | ||
| 121 | A | ||
| 122 | C | ||
| 123 | C | ||
| 124 | C | ||
| 125 | C | ||
| 126 | C | ||
| 127 | B | ||
| 128 | A | ||
| 129 | B | ||
| 130 | A | ||
| 131 | C | ||
| 132 | A | ||
| 133 | C | ||
| 134 | C | ||
| 135 | A | ||
| 136 | A | ||
| 137 | A | ||
| 138 | A | ||
| 139 | A | ||
| 140 | B | ||
| 141 | A | ||
| 142 | C | ||
| 143 | C | ||
| 144 | C | ||
| 145 | A | ||
| 147 | A | ||
| 148 | C | ||
| 149 | C | ||
| 150 | C | ||
| 151 | C | ||
| 152 | A | ||
| 153 | A | ||
| 154 | C | ||
| 155 | A | ||
| 156 | C | ||
| 158 | C | ||
| 159 | A | ||
| 160 | C | ||
| 161 | C | ||
| 162 | C | ||
| 163 | B | ||
| 164 | C | ||
| 166 | C | ||
| 167 | B | ||
| 168 | B | ||
| 169 | A | ||
| 171 | C | ||
| 172 | A | ||
| 173 | C | ||
| 174 | C | ||
| 177 | A | ||
| 178 | A | ||
| 179 | C | ||
| 182 | C | ||
| 183 | A | ||
| 184 | C | ||
| 185 | C | ||
| 186 | C | ||
| 187 | C | ||
| 188 | C | ||
| 189 | B | ||
| 190 | C | ||
| 192 | C | ||
| 193 | B | ||
| 195 | C | ||
| 197 | C | ||
| 200 | A | ||
| 201 | C | ||
| 202 | C | ||
| 203 | A | ||
| 209 | C | ||
| 210 | C | ||
| 211 | A | ||
| 212 | C | ||
| 213 | C | ||
| 214 | A | ||
| 215 | C | ||
| 216 | B | ||
| 217 | A | ||
| 218 | C | ||
| 219 | C | ||
| 220 | C | ||
| 221 | C | ||
| 222 | C | ||
| 223 | C | ||
| 224 | C | ||
| 225 | B | ||
| 226 | C | ||
| 227 | C | ||
| 228 | A | ||
| 229 | C | ||
| 230 | C | ||
| 231 | C | ||
| 232 | B | ||
| 233 | C | ||
| 234 | C | ||
| 235 | A | ||
| 236 | A | ||
| 237 | C | ||
| 238 | B | ||
| 239 | C | ||
| 240 | B | ||
| 241 | A | ||
| 242 | A | ||
| 243 | C | ||
| 244 | C | ||
| 245 | A | ||
| 246 | C | ||
| 247 | C | ||
| 248 | C | ||
| 249 | A | ||
| 250 | B | ||
| 251 | C | ||
| 252 | A | ||
| 253 | C | ||
| 254 | A | ||
| 255 | C | ||
| 256 | C | ||
| 257 | A | ||
| 258 | A | ||
| 259 | C | ||
| 260 | B | ||
| 261 | C | ||
| 262 | B | ||
| 263 | C | ||
| 264 | C | ||
| 265 | C | ||
| 266 | C | ||
| 267 | A | ||
| 268 | A | ||
| 269 | C | ||
| 270 | C | ||
| 271 | C | ||
| 272 | A | ||
| 273 | A | ||
| 274 | A | ||
| 275 | C | ||
| 276 | C | ||
| 277 | A | ||
| 278 | A | ||
| 279 | A | ||
| 280 | C | ||
| 281 | C | ||
| 282 | B | ||
| 283 | A | ||
| 284 | C | ||
| 285 | A | ||
| 286 | C | ||
| 287 | B | ||
| 288 | A | ||
| 289 | C | ||
| 290 | A | ||
| 291 | A | ||
| 292 | C | ||
| 293 | C | ||
| 294 | A | ||
| 295 | A | ||
| 296 | C | ||
| 297 | C | ||
Claims (19)










































Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/534,450 USRE47142E1 (en) | 2008-06-03 | 2014-11-06 | Compounds and methods for treating inflammatory and fibrotic disorders |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US5843608P | 2008-06-03 | 2008-06-03 | |
| US7444608P | 2008-06-20 | 2008-06-20 | |
| US12/477,715 US8304413B2 (en) | 2008-06-03 | 2009-06-03 | Compounds and methods for treating inflammatory and fibrotic disorders |
| US14/534,450 USRE47142E1 (en) | 2008-06-03 | 2014-11-06 | Compounds and methods for treating inflammatory and fibrotic disorders |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/477,715 Reissue US8304413B2 (en) | 2008-06-03 | 2009-06-03 | Compounds and methods for treating inflammatory and fibrotic disorders |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| USRE47142E1 true USRE47142E1 (en) | 2018-11-27 |
Family
ID=41398503
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/477,715 Ceased US8304413B2 (en) | 2008-06-03 | 2009-06-03 | Compounds and methods for treating inflammatory and fibrotic disorders |
| US13/652,247 Expired - Fee Related US8969347B2 (en) | 2008-06-03 | 2012-10-15 | Compounds and methods for treating inflammatory and fibrotic disorders |
| US14/263,822 Expired - Fee Related US9290450B2 (en) | 2008-06-03 | 2014-04-28 | Compounds and methods for treating inflammatory and fibrotic disorders |
| US14/534,450 Expired - Fee Related USRE47142E1 (en) | 2008-06-03 | 2014-11-06 | Compounds and methods for treating inflammatory and fibrotic disorders |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/477,715 Ceased US8304413B2 (en) | 2008-06-03 | 2009-06-03 | Compounds and methods for treating inflammatory and fibrotic disorders |
| US13/652,247 Expired - Fee Related US8969347B2 (en) | 2008-06-03 | 2012-10-15 | Compounds and methods for treating inflammatory and fibrotic disorders |
| US14/263,822 Expired - Fee Related US9290450B2 (en) | 2008-06-03 | 2014-04-28 | Compounds and methods for treating inflammatory and fibrotic disorders |
Country Status (8)
| Country | Link |
|---|---|
| US (4) | US8304413B2 (en) |
| EP (1) | EP2296653B1 (en) |
| JP (2) | JP5627574B2 (en) |
| CN (1) | CN102099036B (en) |
| CA (2) | CA3034994A1 (en) |
| ES (1) | ES2567283T3 (en) |
| MX (1) | MX2010012848A (en) |
| WO (1) | WO2009149188A1 (en) |
Families Citing this family (65)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2600460T3 (en) | 2005-05-10 | 2017-02-09 | Intermune, Inc. | Pyridone-2-one derivatives as modulators of the stress-activated protein kinase system |
| CN102099036B (en) | 2008-06-03 | 2015-05-27 | 英特芒尼公司 | Compounds and methods for treating inflammatory and fibrotic disorders |
| US7635707B1 (en) | 2008-11-10 | 2009-12-22 | Intermune, Inc. | Pirfenidone treatment for patients with atypical liver function |
| AP2011005824A0 (en) * | 2009-01-26 | 2011-08-31 | Intermune Inc | Methods for treating acute myocardial infarctions and associated disorders. |
| SG176053A1 (en) * | 2009-05-15 | 2011-12-29 | Intermune Inc | Methods of treating hiv patients with anti-fibrotics |
| WO2010135972A1 (en) * | 2009-05-25 | 2010-12-02 | 中南大学 | Preparation methods and uses of 1-(substituted aryl)-5-trifluoromethyl-2-(1h)-pyridone compounds and their salts |
| KR101478133B1 (en) | 2009-05-25 | 2014-12-31 | 센트럴 사우스 유니버시티 | Preparation of 1-(substituted benzyl)-5-trifluoromethyl-2(1H)pyridone compounds and salts thereof and their applications |
| US8957254B2 (en) | 2009-07-06 | 2015-02-17 | Solvay Sa | Process for chemical synthesis from an alkenone made from a halogenated precursor |
| US7816383B1 (en) | 2009-12-04 | 2010-10-19 | Intermune, Inc. | Methods of administering pirfenidone therapy |
| US20120245188A1 (en) * | 2010-11-15 | 2012-09-27 | Translational Genomics Research Institute | Methods of treating memory loss and enhancing memory performance |
| US20130253012A1 (en) | 2010-12-10 | 2013-09-26 | Basf Se | Pyrazole Compounds for Controlling Invertebrate Pests |
| WO2012106382A1 (en) | 2011-01-31 | 2012-08-09 | Genoa Pharmaceuticals, Inc. | Aerosol pirfenidone and pyridone analog compounds and uses thereof |
| US10105356B2 (en) | 2011-01-31 | 2018-10-23 | Avalyn Pharma Inc. | Aerosol pirfenidone and pyridone analog compounds and uses thereof |
| FI20115234A0 (en) | 2011-03-08 | 2011-03-08 | Biotie Therapies Corp | New pyridazinone and pyridone compounds |
| BR112014030284B1 (en) * | 2012-07-18 | 2021-11-30 | Sunshine Lake Pharma Co., Ltd | COMPOUND, PHARMACEUTICAL COMPOSITION, AND, USE OF A COMPOUND OR PHARMACEUTICAL COMPOSITION |
| CA2819967C (en) | 2012-08-31 | 2016-03-22 | Intermune, Inc. | Use of pirfenidone concomitantly with ciprofloxacin |
| AR092742A1 (en) * | 2012-10-02 | 2015-04-29 | Intermune Inc | ANTIFIBROTIC PYRIDINONES |
| US20140094456A1 (en) * | 2012-10-02 | 2014-04-03 | Intermune, Inc. | Anti-fibrotic pyridinones |
| US20160045625A1 (en) | 2013-04-04 | 2016-02-18 | Eisai R&D Management Co., Ltd. | [11c] and [18f] labeled 1,3-diphenyl-5-(pyrimidin-2-yl)-pyridin-2(1h)-one derivatives and their use for pet imaging of the ampa receptor |
| US9902712B2 (en) | 2013-12-19 | 2018-02-27 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous heterocyclic derivatives and their application in drugs |
| AU2015204558B2 (en) | 2014-01-10 | 2020-04-30 | Avalyn Pharma Inc. | Aerosol pirfenidone and pyridone analog compounds and uses thereof |
| MX382781B (en) * | 2014-04-02 | 2025-03-13 | Intermune Inc | ANTI-FIBROTIC PYRIDINONES. |
| EA036342B1 (en) | 2014-06-13 | 2020-10-29 | Инвентива | Use of pan-ppar agonist, in particular 5-chloro-1-[(6-benzothiazolyl)sulfonyl]-1h-indole-2-butanoic acid, in the treatment of fibrotic conditions |
| EP3166610B1 (en) * | 2014-07-09 | 2022-11-02 | Eip Pharma, LLC | Methods for treating neurologic disorders |
| CR20170077A (en) | 2014-08-04 | 2017-06-26 | Nuevolution As | OPTIONALLY CONDENSED HEREROCICLYL PYRIMIDINE DERIVATIVES USEFUL FOR THE TREATMENT OF INFLAMMATORY, METABOLIC, ONCOLOGICAL AND AUTOINMUNITY DISEASES |
| PT3263133T (en) * | 2015-02-27 | 2020-05-21 | Univ Kyushu Nat Univ Corp | Pyridinone compound and use thereof |
| CN105130884B (en) * | 2015-07-30 | 2017-09-01 | 四川大学 | 5-methyl-2 (1H) pyridone derivatives and their preparation methods and uses |
| CN105085383B (en) * | 2015-08-19 | 2017-09-01 | 四川大学 | 5-methyl-2 (1H) pyridone derivatives and their preparation methods and uses |
| WO2017066758A1 (en) * | 2015-10-15 | 2017-04-20 | The Schepens Eye Research Institute, Inc. | P38 map kinase inhibitors for wound healing |
| WO2017075013A1 (en) * | 2015-10-26 | 2017-05-04 | Eip Pharma, Llc | Methods and compositions for recovery from stroke |
| CN105669562A (en) * | 2016-04-05 | 2016-06-15 | 叶芳 | Fluoro-substited pyrimidine compound and preparation method thereof |
| RU2738844C2 (en) * | 2016-04-14 | 2020-12-17 | Гуанчжоу Джойо Фарматек Ко., Лтд | Containing a substitute, which is butane, in the heterocyclic ring a pyridone derivative for treating fibrosis and inflammatory diseases |
| CN105884680B (en) * | 2016-06-16 | 2019-06-25 | 杨若一 | Pirfenidone derivative and preparation method thereof and purposes |
| CN107556234A (en) * | 2016-06-30 | 2018-01-09 | 陕西合成药业股份有限公司 | A kind of new pyridine ketone compound and preparation method thereof and application medically |
| BR112019000897A2 (en) * | 2016-07-27 | 2019-04-30 | Sunshine Lake Pharma Co., Ltd. | acid addition salt, pharmaceutical composition, use of the acid addition salt or pharmaceutical composition, and method for preventing, treating or attenuating a tissue or organ fibrosis disorder. |
| CN116023327A (en) * | 2016-08-08 | 2023-04-28 | 北京康蒂尼药业股份有限公司 | A kind of preparation method of oxynidone |
| CN107698499A (en) * | 2016-08-08 | 2018-02-16 | 罗楹 | A kind of preparation method of oxynidone |
| GB201614934D0 (en) * | 2016-09-02 | 2016-10-19 | Glaxosmithkline Intellectual Property (No 2) Ltd | Chemical compounds |
| US12365651B2 (en) | 2017-07-31 | 2025-07-22 | Washington University | Use of pirfenidone and derivatives for modulation of B lymphocyte activity and organ protection from acute tissue damage |
| US11466008B2 (en) | 2017-09-18 | 2022-10-11 | Eip Pharma, Llc | Co-crystals of neflamapimod (VX-745) |
| WO2019072236A1 (en) * | 2017-10-13 | 2019-04-18 | 石家庄智康弘仁新药开发有限公司 | Crystal form and salt form of pyridone compound and preparation method therefor |
| JP7253498B2 (en) | 2017-12-22 | 2023-04-06 | 住友化学株式会社 | Heterocyclic compound and harmful arthropod control agent containing the same |
| EP3632855A1 (en) | 2018-10-02 | 2020-04-08 | Solvay Sa | A method for providing aqueous compositions with reduced content of organic fluorinated compounds |
| MX2021008661A (en) | 2019-01-18 | 2021-08-19 | Astrazeneca Ab | PCSK9 INHIBITORS AND METHODS OF USE THEREOF. |
| BR112021013807A2 (en) | 2019-01-18 | 2021-11-30 | Astrazeneca Ab | pcsk9 inhibitors and their methods of use |
| JP7428693B2 (en) | 2019-02-20 | 2024-02-06 | 住友化学株式会社 | Ether compound and harmful arthropod control composition containing the same |
| WO2021108999A1 (en) * | 2019-12-03 | 2021-06-10 | 辽宁凯莱英医药化学有限公司 | Method for continuous synthesis of 4-ethoxy-1,1,1-trifluoro-3-buten-2-one |
| UY38994A (en) | 2019-12-20 | 2021-07-30 | Nuevolution As | ACTIVE COMPOUNDS AGAINST NUCLEAR RECEPTORS |
| MX2022007265A (en) | 2019-12-20 | 2022-09-09 | Nuevolution As | Compounds active towards nuclear receptors. |
| CN111303034A (en) * | 2020-03-18 | 2020-06-19 | 徐州圣元化工有限公司 | Preparation method of 3- (difluoromethyl) -1-methyl-1H-pyrazole-4-carboxylic acid |
| AU2021245397A1 (en) | 2020-03-31 | 2022-10-20 | Nuevolution A/S | Compounds active towards nuclear receptors |
| WO2021198956A1 (en) | 2020-03-31 | 2021-10-07 | Nuevolution A/S | Compounds active towards nuclear receptors |
| WO2022208370A1 (en) | 2021-03-31 | 2022-10-06 | Pi Industries Ltd. | Fused heterocyclic compounds and their use as pest control agents |
| CN113173921B (en) * | 2021-04-01 | 2022-09-27 | 常州大学 | A kind of 6-thienyl pyridone compound and its synthetic method |
| CN115364097B (en) * | 2021-05-20 | 2023-07-25 | 广州嘉越医药科技有限公司 | Application of a pyridone derivative containing a heteroatom cyclobutane substituent |
| CA3225945A1 (en) * | 2021-07-16 | 2023-01-19 | Tuan Trang | Sulfamoyl benzene derivatives and uses thereof |
| TW202315625A (en) * | 2021-08-13 | 2023-04-16 | 大陸商深圳信立泰藥業股份有限公司 | Terpyridine diketone compound or salt thereof, preparation method therefor and application thereof |
| CN114716365B (en) * | 2022-01-04 | 2024-03-01 | 大连理工大学 | Application of N-substituted phenyl-2-pyridone compound or pharmaceutically acceptable salt thereof in treatment of pulmonary fibrosis |
| EP4257133A1 (en) * | 2022-04-05 | 2023-10-11 | Institut Pasteur | Oxo-azaheterocyclic derivatives for use in the treatment of malaria |
| EP4556472A1 (en) * | 2022-07-14 | 2025-05-21 | Guangzhou Joyo Pharmatech Co., Ltd | Use of pyridone derivative |
| CN115286568B (en) * | 2022-08-24 | 2023-12-12 | 常州沃腾化工科技有限公司 | Preparation method of 2-hydroxy-4-trifluoromethyl pyridine |
| CN115385852A (en) * | 2022-09-05 | 2022-11-25 | 湖南阿斯迪康药业有限公司 | A kind of efficient synthetic method of 2-amino-4-trifluoromethylpyridine |
| WO2024235145A1 (en) * | 2023-05-12 | 2024-11-21 | 大连理工大学 | Preparation and use of n-substituted phenyl-2-pyridone and endoperoxide |
| WO2025107982A1 (en) * | 2023-11-23 | 2025-05-30 | 广东中科药物研究有限公司 | Pirfenidone derivative, preparation method therefor, and use thereof |
| WO2025209440A1 (en) * | 2024-04-02 | 2025-10-09 | 广州嘉越医药科技有限公司 | Pharmaceutical composition of pyridone derivative and use thereof |
Citations (482)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CH312531A (en) | 1952-06-20 | 1955-12-31 | Ciba Geigy | Process for making a pyridazone. |
| CH312530A (en) | 1952-06-20 | 1955-12-31 | Ciba Geigy | Process for making a pyridazone. |
| GB788393A (en) | 1953-04-30 | 1958-01-02 | Ciba Ltd | Process for the manufacture of pyridazone compounds |
| CH333366A (en) | 1953-09-04 | 1958-10-15 | Ciba Geigy | Process for the production of new pyridazones |
| DE1070639B (en) | 1959-12-10 | Farbenfabriken Bayer Aktiengesellschaft, Levenkusen-Bayerwerk | Process for the preparation of Peptiderj | |
| US3014034A (en) | 1959-01-22 | 1961-12-19 | Ciba Pharm Prod Inc | 1, 3-diaryl, 5-amino-pyridazinones |
| GB889317A (en) | 1959-01-22 | 1962-02-14 | Ciba Ltd | New phenyl-diazines and a process for their manufacture |
| DE1936231A1 (en) | 1968-08-12 | 1970-10-29 | Eastman Kodak Co | Photosensitive silver halide photographic emulsion |
| FR2046068A5 (en) | 1969-05-05 | 1971-03-05 | Eastman Kodak Co | Stabilised photographic emulsions |
| DE2112026A1 (en) | 1970-03-12 | 1971-09-23 | Rhone Poulenc Sa | 3,4-dihydroisoquinoline derivatives, their preparation and the medicinal compositions containing them |
| DE2143744A1 (en) | 1971-09-01 | 1973-03-08 | Hoechst Ag | 3,4-DIHYDRO-2H-ISOCHINOLIN-1-ONE AND THE METHOD FOR MANUFACTURING IT |
| US3839346A (en) | 1972-12-18 | 1974-10-01 | Affiliated Med Res | N-substituted pyridone and general method for preparing pyridones |
| GB1388001A (en) | 1971-08-13 | 1975-03-19 | Thiemann Chem Pharm Fab | 2-pyridones and preparations comprising them |
| US3974281A (en) | 1972-12-18 | 1976-08-10 | Affiliated Medical Research, Inc. | 5-Methyl-1-phenyl-2-(1H)-pyridone compositions and methods of use |
| JPS51128438A (en) | 1975-04-26 | 1976-11-09 | Yamanouchi Pharmaceut Co Ltd | An antibacterial drug against fish diseases |
| GB1458049A (en) | 1972-12-18 | 1976-12-08 | Affiliated Med Res | Process for preparing aryl-substituted pyridones |
| AT333774B (en) | 1974-09-24 | 1976-12-10 | Chemie Linz Ag | PROCESS FOR THE PREPARATION OF 3-PHENYLPYRIDAZONES |
| DE2557342A1 (en) | 1975-12-19 | 1977-06-30 | Hoechst Ag | BASIC SUBSTITUTED INDOLDER DERIVATIVES AND THE PROCESS FOR THEIR PRODUCTION |
| US4042699A (en) | 1972-12-18 | 1977-08-16 | Affiliated Medical Research, Inc. | Method for reducing serum glucose levels |
| US4052509A (en) | 1972-12-18 | 1977-10-04 | Affiliated Medical Research, Inc. | Method for reducing serum uric acid levels |
| DE2707268A1 (en) | 1977-02-19 | 1978-08-31 | Hoechst Ag | Indole-3-carboxaldehyde oxime derivs. - with hypouricaemic, analgesic, antiinflammatory, hypoglycaemic, cardiovascular and diuretic activity |
| DE2830700A1 (en) | 1977-07-15 | 1979-02-01 | Rohm & Haas | NEW 2-PYRIDON |
| US4256640A (en) | 1978-05-23 | 1981-03-17 | Shionogi & Co., Ltd. | Tetrahydrothiopyrano[2,3-b]indole derivatives |
| US4258052A (en) | 1976-08-17 | 1981-03-24 | Yu Ruey J | Treatment of psoriasis with nicotinamide analogues |
| DD149666A1 (en) | 1979-06-27 | 1981-07-22 | Karl Gewald | PROCESS FOR THE PREPARATION OF 6-AMINO AND 6-HYDROXY-1-ARYL-5-CYAN-2 (1H) -PYRIDINONE AND THIONES |
| JPS5721388A (en) | 1980-07-11 | 1982-02-04 | Nippon Nohyaku Co Ltd | Condensed pyrazole derivative |
| US4325863A (en) | 1979-02-05 | 1982-04-20 | Sandoz Ltd. | Benzofuranone or indolinone compounds useful as stabilizers for organic materials |
| JPS5777671A (en) | 1980-10-31 | 1982-05-15 | Yasumitsu Tamura | Preparation of hydroxyindoles ( or 1,4-dihydroisoquinolones) and their intermediate |
| US4397854A (en) | 1981-05-14 | 1983-08-09 | Warner-Lambert Company | Substituted 6-phenyl-3(2H)-pyridazinones useful as cardiotonic agents |
| US4404203A (en) | 1981-05-14 | 1983-09-13 | Warner-Lambert Company | Substituted 6-phenyl-3(2H)-pyridazinones useful as cardiotonic agents |
| EP0104860A1 (en) | 1982-09-20 | 1984-04-04 | Pfizer Inc. | 1-Phenyl-2(1H,3H)-indolone psycho-therapeutic agents |
| US4473696A (en) | 1982-10-07 | 1984-09-25 | Ici Americas Inc. | Synthesis of 2-substituted-5-methyl-pyridines |
| US4576942A (en) | 1984-07-12 | 1986-03-18 | Usv Pharmaceutical Corp. | Anti-allergic and anti-inflammatory bi- and tri- cyclo-1,4-thiazine derivatives, composition, and method of use therefor |
| US4645839A (en) | 1982-12-17 | 1987-02-24 | Ici Americas Inc. | Sulphur dehydrogenation process to yield 5-methyl-2-pyridone |
| US4650804A (en) | 1984-03-30 | 1987-03-17 | Fujisawa Pharmaceutical Co., Ltd. | Quinolizinone compounds and pharmaceutical composition comprising the same, useful as anti-ulcerative and anti-allergic agents |
| EP0241006A2 (en) | 1986-04-10 | 1987-10-14 | E.I. Du Pont De Nemours And Company | 3,3-Disubstituted indolines |
| EP0259048A2 (en) | 1986-09-03 | 1988-03-09 | Imperial Chemical Industries Plc | Chemical compounds |
| JPS63290821A (en) | 1987-05-25 | 1988-11-28 | Otsuka Pharmaceut Co Ltd | Antiarrhythmic |
| US4820309A (en) | 1987-03-05 | 1989-04-11 | Sandoz Ltd. | 1:2-chromium complex dyes for tanned leather |
| EP0311010A2 (en) | 1987-10-06 | 1989-04-12 | The Du Pont Merck Pharmaceutical Company | Alpha,Alpha-disubstituted aromatics and heteroaromatics as cognition enhancers |
| EP0319957A2 (en) | 1987-12-07 | 1989-06-14 | Gluetech Aps | A method for modifying the surface of a polymer |
| US4898654A (en) | 1988-05-27 | 1990-02-06 | Otsuka Kagaku Kabushiki Kaisha | Method of producing optically active β-lactam derivatives |
| EP0381374A1 (en) | 1989-01-27 | 1990-08-08 | Les Laboratoires Beecham S.A. | Phosphodiesterase inhibitors |
| EP0393936A1 (en) | 1989-04-18 | 1990-10-24 | Pfizer Inc. | 3-substituted-2-oxindole derivatives as antiinflammatory agents |
| EP0409435A1 (en) | 1989-07-07 | 1991-01-23 | Pfizer Inc. | Heteroaryl piperazine antipsychotic agents |
| JPH0343744A (en) | 1989-07-12 | 1991-02-25 | Hitachi Chem Co Ltd | Electrophotographic sensitive body |
| US5019365A (en) | 1988-11-29 | 1991-05-28 | The Dow Chemical Company | Quaternary polyamines as sulfite oxidation inhibitors |
| WO1991014674A2 (en) | 1990-03-27 | 1991-10-03 | Smithkline Beecham Corporation | 5-lipoxygenase inhibitors |
| US5077142A (en) | 1989-04-20 | 1991-12-31 | Ricoh Company, Ltd. | Electroluminescent devices |
| US5080710A (en) | 1988-03-05 | 1992-01-14 | Basf Aktiengesellschaft | Novel n-aryltetrahydrophthalimide compounds |
| EP0478195A1 (en) | 1990-09-21 | 1992-04-01 | Rohm And Haas Company | Dihydropyridazinones, pyridazinones and related compounds as fungicides |
| JPH04223457A (en) | 1990-12-26 | 1992-08-13 | Konica Corp | Silver halide photographic sensitive material |
| WO1992013451A1 (en) | 1991-02-11 | 1992-08-20 | Schering Agrochemicals Limited | Imidazole pesticides |
| WO1992020642A1 (en) | 1991-05-10 | 1992-11-26 | Rhone-Poulenc Rorer International (Holdings) Inc. | Bis mono-and bicyclic aryl and heteroaryl compounds which inhibit egf and/or pdgf receptor tyrosine kinase |
| WO1992020816A1 (en) | 1991-05-15 | 1992-11-26 | Yale University | Determination of prodrugs metabolizable by the liver and therapeutic use thereof |
| US5167941A (en) | 1988-11-29 | 1992-12-01 | The Dow Chemical Company | Quaternary polyamines as sulfite oxidation inhibitors in amine scrubbing of SO2 |
| EP0531578A1 (en) | 1991-09-10 | 1993-03-17 | Agfa-Gevaert N.V. | Thermally transferable fluorescent compounds |
| EP0548680A1 (en) | 1991-12-26 | 1993-06-30 | Mitsubishi Chemical Corporation | Beta-oxo-beta-benzenepropanethioamide derivatives |
| US5241065A (en) | 1992-02-25 | 1993-08-31 | Schering Corporation | 2,3,4,5-tetrahydro-1h-3-benzazepines having anti-psychotic activity |
| WO1993021185A1 (en) | 1992-04-16 | 1993-10-28 | The Du Pont Merck Pharmaceutical Company | Substituted nitrogen containing spiro compounds for use in treating cognitive deficits |
| WO1993023404A1 (en) | 1992-05-19 | 1993-11-25 | Immunopharmaceutics, Inc. | Compounds that modulate endothelin activity |
| EP0577325A1 (en) | 1992-07-02 | 1994-01-05 | Sawai Pharmaceutical Co., Ltd. | Carbostyril derivatives and antiallergic agent |
| EP0579059A1 (en) | 1992-07-02 | 1994-01-19 | Tanabe Seiyaku Co., Ltd. | Pyridazinone derivatives and processes for preparing the same |
| US5310562A (en) | 1989-11-22 | 1994-05-10 | Margolin Solomon B | Composition and method for reparation and prevention of fibrotic lesions |
| EP0602515A1 (en) | 1992-12-16 | 1994-06-22 | BASF Aktiengesellschaft | Process for the preparation of 5-membered heterocycles |
| WO1994017059A1 (en) | 1993-01-29 | 1994-08-04 | Nippon Soda Co., Ltd. | Heterocyclic derivative |
| JPH06256187A (en) | 1993-03-02 | 1994-09-13 | Fujisawa Pharmaceut Co Ltd | Antitussive expectorant containing qunolizinone compound or its salt |
| US5356904A (en) | 1992-10-07 | 1994-10-18 | Merck & Co., Inc. | Carbostyril oxytocin receptor antagonists |
| WO1994026249A1 (en) | 1993-05-07 | 1994-11-24 | Margolin Solomon B | Compositions and methods for reparation and prevention of fibrotic lesions |
| EP0626377A1 (en) | 1993-05-26 | 1994-11-30 | Shionogi & Co., Ltd. | Method for producing benzylidene derivatives |
| DE4423934A1 (en) | 1993-09-02 | 1995-03-09 | Basf Ag | 3(2H)-Pyridazinone derivatives, their preparation and their use |
| US5401738A (en) | 1992-06-13 | 1995-03-28 | Merck Patent Gesellschaft Mit Beschrankter | Benzimidazole compounds |
| EP0648760A2 (en) | 1993-10-15 | 1995-04-19 | Takeda Chemical Industries, Ltd. | Triazine derivative, production and use thereof |
| JPH07128793A (en) | 1993-11-05 | 1995-05-19 | Konica Corp | Fine solid particle dispersion of dye and silver halide photographic sensitive material containing the same |
| WO1995016712A1 (en) | 1993-12-15 | 1995-06-22 | Smithkline Beecham Corporation | Compounds and methods |
| WO1995018128A1 (en) | 1993-12-29 | 1995-07-06 | Fujisawa Pharmaceutical Co., Ltd. | Pyrazolopyridine adenosine antagonists |
| JPH07233072A (en) | 1993-12-28 | 1995-09-05 | Tanabe Seiyaku Co Ltd | Pharmaceutical composition |
| JPH07295165A (en) | 1994-04-27 | 1995-11-10 | Konica Corp | Method for processing silver halide photographic sensitive material |
| JPH07295166A (en) | 1994-04-28 | 1995-11-10 | Konica Corp | Method for processing silver halide photographic sensitive material |
| US5466697A (en) | 1994-07-13 | 1995-11-14 | Syntex (U.S.A.) Inc. | 8-phenyl-1,6-naphthyridin-5-ones |
| US5518729A (en) | 1989-11-22 | 1996-05-21 | Margolin; Solomon B. | Compositions and methods for reparation and prevention of fibrotic lesions |
| JPH08134371A (en) | 1994-11-02 | 1996-05-28 | Konica Corp | Dispersion of solid fine particle, silver halide photosensitive material and image forming method |
| WO1996018770A2 (en) | 1994-12-16 | 1996-06-20 | Lignozym Gmbh | Multicomponent system for modifying, decomposing or bleaching lignin, lignin-containing materials or similar substances and method of using this system |
| WO1996027374A1 (en) | 1995-03-03 | 1996-09-12 | Margolin Solomon B | Treatment of cytokine growth factor caused disorders |
| EP0733629A1 (en) | 1995-03-13 | 1996-09-25 | Ishihara Sangyo Kaisha, Ltd. | Pyridonesulfonylurea compounds, process for their production and herbicides containing them |
| EP0738716A2 (en) | 1995-04-21 | 1996-10-23 | Rohm And Haas Company | Dihydropyridazinones and pyridazinones and their use as fungicides and insecticides |
| WO1996033994A1 (en) | 1995-04-28 | 1996-10-31 | Nippon Soda Co., Ltd. | Amino-substituted derivatives, process for the preparation thereof, and herbicide |
| WO1997005137A1 (en) | 1995-07-31 | 1997-02-13 | Novo Nordisk A/S | Heterocyclic compounds, their preparation and use |
| WO1997005109A1 (en) | 1995-07-31 | 1997-02-13 | Novo Nordisk A/S | Heterocyclic compounds, their preparation and use |
| EP0760208A2 (en) | 1995-08-25 | 1997-03-05 | Rohm And Haas Company | Compositions having synergistic fungitoxic effects |
| WO1997010712A1 (en) | 1995-09-19 | 1997-03-27 | Margolin Solomon B | Inhibition of tumor necrosis factor alpha |
| WO1997029107A1 (en) | 1996-02-09 | 1997-08-14 | Applied Research Systems Ars Holding N.V. | BENZO[C]QUINOLIZINE DERIVATIVES, THEIR PREPARATION AND USE AS 5α-REDUCTASES INHIBITORS |
| JPH09244235A (en) | 1996-03-14 | 1997-09-19 | Toshiba Corp | Resist for alkaline development |
| JPH09249567A (en) | 1996-01-12 | 1997-09-22 | Otsuka Pharmaceut Co Ltd | Medicinal composition |
| WO1997036863A1 (en) | 1996-03-29 | 1997-10-09 | Merck Frosst Canada Inc. | Bisarylcyclobutene derivates as cyclooxygenase inhibitors |
| WO1997041830A1 (en) | 1996-05-09 | 1997-11-13 | Margolin Solomon B | Reparation and prevention of fibrotic lesions |
| JPH09319023A (en) | 1996-05-27 | 1997-12-12 | Fuji Photo Film Co Ltd | Thermosensitive recording material |
| US5719155A (en) | 1993-11-10 | 1998-02-17 | Japan Tobacco Inc. | Chroman derivative and pharmaceutical use thereof |
| US5731106A (en) | 1996-01-25 | 1998-03-24 | Fujitsu Limited | Electrolytic solution for lithium secondary battery and lithium secondary battery using the same |
| WO1998013361A1 (en) | 1996-09-26 | 1998-04-02 | Novartis Ag | Herbicidal composition |
| EP0835865A1 (en) | 1996-10-11 | 1998-04-15 | Rohm And Haas Company | Dihydropyridazinones and pyridazinones and their use as fungicides and insecticides |
| DE19754348A1 (en) | 1996-12-11 | 1998-06-18 | Ciba Geigy Ag | New 2-(hetero)aryl-2(3H)-pyridazinone derivatives |
| WO1998029119A1 (en) | 1996-12-30 | 1998-07-09 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| EP0856255A2 (en) | 1997-01-30 | 1998-08-05 | Rohm And Haas Company | Pyridazinones as marine antifouling agents |
| US5808015A (en) | 1995-09-25 | 1998-09-15 | Bayer Aktiengesellschaft | Pyridone-methide azo dyestuffs with improved dyeing and printing properties for hydrophobic synethic fiber materials |
| US5817700A (en) | 1996-03-29 | 1998-10-06 | Merck Frosst Canada, Inc. | Bisaryl cyclobutenes derivatives as cyclooxygenase inhibitors |
| WO1998051772A1 (en) | 1997-05-12 | 1998-11-19 | Call, Krimhild | Enzymatic bleaching system containing new compounds for intensifying enzymatic action |
| DE19821263A1 (en) | 1997-05-12 | 1998-11-19 | Call Krimhild | Enzymatic bleach system containing mediator to enhance performance in bleaching textile fabric |
| WO1998052948A1 (en) | 1997-05-19 | 1998-11-26 | The Regents Of The University Of California | Compounds for inhibition of ceramide-mediated signal transduction |
| DE19726241A1 (en) | 1997-06-20 | 1998-12-24 | Call Krimhild | Enhanced multi-component enzymatic system for the treatment of waste water, for the production of wood composites, for deinking waste paper, color stripping of waste paper, for use as an oxidation system in organic synthesis and for use in coal liquefaction |
| US5859041A (en) | 1996-06-10 | 1999-01-12 | Merck & Co., Inc. | Substituted imidazoles having cytokine inhibitory activity |
| DE19729061A1 (en) | 1997-07-08 | 1999-01-14 | Agfa Gevaert Ag | Colour photographic material sensitised without increased fogging |
| WO1999002501A1 (en) | 1997-07-09 | 1999-01-21 | Astra Pharmaceuticals Ltd. | Novel compounds |
| WO1999005123A1 (en) | 1997-07-24 | 1999-02-04 | Astrazeneca Uk Limited | Novel pyrimidin derivatives |
| WO1999005125A1 (en) | 1997-07-24 | 1999-02-04 | Bayer Aktiengesellschaft | Substituted n-aryl-n-thioxocarbonyl sulfonamides as herbicides |
| WO1999005913A1 (en) | 1997-08-01 | 1999-02-11 | Applied Research Systems Ars Holding N.V. | Use of benzo[c]quinolizine derivatives as plant growth regulators |
| JPH1149755A (en) | 1997-07-30 | 1999-02-23 | Nippon Kayaku Co Ltd | New nitrogen containing heterocyclic derivative and acaricide composition using the same as active component |
| WO1999010332A1 (en) | 1997-08-22 | 1999-03-04 | Abbott Laboratories | Prostaglandin endoperoxide h synthase biosynthesis inhibitors |
| WO1999010331A1 (en) | 1997-08-22 | 1999-03-04 | Abbott Laboratories | Arylpyridazinones as prostaglandin endoperoxide h synthase biosynthesis inhibitors |
| WO1999012903A1 (en) | 1997-09-09 | 1999-03-18 | Du Pont Pharmaceuticals Company | Benzimidazolinones, benzoxazolinones, benzopiperazinones, indanones, and derivatives thereof as inhibitors of factor xa |
| WO1999021837A1 (en) | 1997-10-27 | 1999-05-06 | Isk Americas Incorporated | Substituted benzene compounds, process for their preparation, and herbicidal and defoliant compositions containing them |
| WO1999026944A1 (en) | 1997-11-21 | 1999-06-03 | Astrazeneka Uk Limited | New compounds which are p2-purinoceptor 7-transmembrane (tm) g-protein coupled receptor antagonists |
| WO1999028313A1 (en) | 1996-12-05 | 1999-06-10 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| WO1999032448A1 (en) | 1997-12-19 | 1999-07-01 | Amgen Inc. | Substituted pyridine and pyridazine compounds and their pharmaceutical use |
| JPH11180952A (en) | 1997-12-19 | 1999-07-06 | Maruho Co Ltd | 2-oxindole derivative |
| WO1999038857A2 (en) | 1998-01-30 | 1999-08-05 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Method for producing 5-alkoxy (or 5-aroxy)-2,3-dihydrofuran-2-ones |
| FR2774986A1 (en) | 1998-02-16 | 1999-08-20 | Rhodia Chimie Sa | Masked isocyanates made by reacting an isocyanate starting material with a masking agent having a pyronoid structure and a reactive hydrogen atom |
| WO1999047140A1 (en) | 1998-03-17 | 1999-09-23 | Margolin Solomon B | Topical antiseptic compositions and methods |
| US5962478A (en) | 1995-09-19 | 1999-10-05 | Margolin; Solomon B. | Inhibition of tumor necrosis factor α |
| WO1999050263A1 (en) | 1998-03-31 | 1999-10-07 | Warner-Lambert Company | Quinolones as serine protease inhibitors |
| WO1999052878A1 (en) | 1998-04-09 | 1999-10-21 | Bayer Aktiengesellschaft | Substituted phenyl pyridazinones |
| WO1999055676A1 (en) | 1998-04-27 | 1999-11-04 | Centre National De La Recherche Scientifique | 3-(amino- or aminoalkyl)pyridinone derivatives and their use for the treatment of hiv related diseases |
| WO1999062900A1 (en) | 1998-06-03 | 1999-12-09 | Sanofi-Synthelabo | Oxindole derivatives used as neurokinin receptor antagonists |
| WO2000016775A1 (en) | 1998-09-18 | 2000-03-30 | Mepha Ag | Topical formulation of alkyl-, phenyl-pyridone |
| WO2000025789A1 (en) | 1998-10-29 | 2000-05-11 | Merck & Co., Inc. | A method of treating endometriosis |
| US6090822A (en) | 1995-03-03 | 2000-07-18 | Margolin; Solomon B. | Treatment of cytokine growth factor caused disorders |
| WO2000044381A1 (en) | 1999-01-28 | 2000-08-03 | Margolin Solomon B | Treatment of lymphomas, leukemias, and leiomyomas |
| US6117973A (en) | 1997-02-24 | 2000-09-12 | Georgia Tech Research Corp. | PNA monomers with electron donor or acceptor |
| DE19918725A1 (en) | 1999-04-24 | 2000-10-26 | Bayer Ag | New heterocyclic-substituted N-cyano-sulfonanilide compounds, useful as total or selective pre- or post-emergence herbicides, showing desiccant, defoliant, germination inhibiting and especially weed-killing activities |
| WO2000068188A1 (en) | 1999-05-07 | 2000-11-16 | Texas Biotechnology Corporation | Propanoic acid derivatives that inhibit the binding of integrins to their receptors |
| US6174901B1 (en) | 1998-12-18 | 2001-01-16 | Amgen Inc. | Substituted pyridine and pyridazine compounds and methods of use |
| WO2001012600A1 (en) | 1999-08-12 | 2001-02-22 | Cor Therapeutics, Inc. | INHIBITORS OF FACTOR Xa |
| FR2797629A1 (en) | 1999-08-19 | 2001-02-23 | Rhodia Chimie Sa | Isocyanate used in polyisocyanate production has a masked isocyanate function(s) susceptible to action by an isocyanate of a derivative of pyrone structure and with active hydrogen |
| US6265350B1 (en) | 1998-06-16 | 2001-07-24 | Hoechst Schering Agrevo Gmbh | 1,3-oxazoline and 1,3-thiazoline derivatives, their preparation, and their use as pesticides |
| WO2001057019A1 (en) | 2000-02-01 | 2001-08-09 | Cor Therapeutics, Inc. | INDALONE AND BENZIMIDAZOLONE INHIBITORS OF FACTOR Xa |
| WO2001057021A2 (en) | 2000-02-01 | 2001-08-09 | Cor Therapeutics, Inc. | 2-[1H]-QUINOLONE AND 2-[1H]-QUINOXALONE INHIBITORS OF FACTOR Xa |
| WO2001056992A2 (en) | 2000-02-07 | 2001-08-09 | Novartis Ag | Dibenzo (b,f) azepine intermediates |
| WO2001057037A1 (en) | 2000-02-04 | 2001-08-09 | Cor Therapeutics, Inc. | Platelet adp receptor inhibitors |
| WO2001058448A1 (en) | 2000-02-09 | 2001-08-16 | Shionogi & Co., Ltd. | Apoptosis inhibitor |
| WO2001062253A1 (en) | 2000-02-21 | 2001-08-30 | Cymar, Inc. | Compositions and methods for treatment of epilepsy |
| WO2001070746A1 (en) | 2000-03-23 | 2001-09-27 | Takeda Chemical Industries, Ltd. | Furoisoquinoline derivatives, process for producing the same and use thereof |
| WO2001072708A2 (en) | 2000-03-24 | 2001-10-04 | Cor Therapeutics, Inc. | OXINDOLE INHIBITORS OF FACTOR Xa |
| US6307047B1 (en) | 1997-08-22 | 2001-10-23 | Abbott Laboratories | Prostaglandin endoperoxide H synthase biosynthesis inhibitors |
| DE10024938A1 (en) | 2000-05-19 | 2001-11-22 | Bayer Ag | New substituted iminoazine derivatives useful as herbicides, especially for weed control in crops |
| WO2001096308A1 (en) | 2000-06-12 | 2001-12-20 | Eisai Co., Ltd. | 1,2-dihydropyridine compounds, process for preparation of the same and use thereof |
| WO2002006244A1 (en) | 2000-07-18 | 2002-01-24 | Bayer Aktiengesellschaft | Heterocyclic substituted herbicidal sulphonic acid anilides |
| EP1186318A2 (en) | 2000-09-06 | 2002-03-13 | Pfizer Products Inc. | Combination, for treating depression and anxiety, containing a 5HT1d receptor antagonist and aCNS penetrant NK-1 receptor antagonist |
| WO2002022587A1 (en) | 2000-09-18 | 2002-03-21 | Eisai Co., Ltd. | Pyridazinones and triazinones and medicinal use thereof |
| WO2002024650A2 (en) | 2000-09-19 | 2002-03-28 | Centre National De La Recherche Scientifique (Cnrs) | Pyridinone and pyridinethione derivatives having hiv inhibiting properties |
| CN1349982A (en) | 2000-10-24 | 2002-05-22 | 大连化学工业股份有限公司 | Process for the preparation of lactams |
| WO2002040448A1 (en) | 2000-11-20 | 2002-05-23 | Bristol-Myers Squibb Company | Pyridone derivatives as ap2 inhibitors |
| EP1213288A1 (en) | 2000-11-06 | 2002-06-12 | Texas Biotechnology Corporation | Propanoic acid derivatives that inhibit the binding of integrins to their receptors |
| US20020077486A1 (en) | 2000-02-04 | 2002-06-20 | Scarborough Robert M. | Platelet ADP receptor inhibitors |
| WO2002060446A1 (en) | 2001-01-29 | 2002-08-08 | Shionogi & Co., Ltd. | Medicinal preparation containing 5-methyl-1-phenyl-2-(1h)-pyridone as active ingredient |
| WO2002067675A2 (en) | 2000-10-27 | 2002-09-06 | Dow Agrosciences Llc | Substituted 4,5-dihydro-1,2,4-triazin-6-ones, 1,2,4-triazin-6-ones, and their use as fungicides and insecticides |
| WO2002085858A1 (en) | 2001-04-20 | 2002-10-31 | Asahi Glass Company, Limited | Process for producing purified piperidine derivative |
| WO2002090334A1 (en) | 2001-05-08 | 2002-11-14 | Kudos Pharmaceuticals Limited | Isoquinolinone derivatives as parp inhibitors |
| WO2002098853A2 (en) | 2001-06-01 | 2002-12-12 | The Regents Of The University Of California | Inhibitors of cell proliferation, angiogenesis, fertility, and muscle contraction |
| CN1386737A (en) | 2002-06-11 | 2002-12-25 | 中南大学湘雅医学院 | Antifibrosis pyridinone medicine and its prepaing process |
| JP2002371078A (en) | 2001-06-12 | 2002-12-26 | Sankyo Co Ltd | Quinoline derivative and quinolone derivative |
| JP2003012645A (en) | 2001-06-29 | 2003-01-15 | Japan Science & Technology Corp | Method for producing nitrogen-containing 6-membered ring |
| US6509354B1 (en) | 1998-04-27 | 2003-01-21 | Kumiai Chemical Industry Co., Ltd. | 3-arylphenyl sulfide derivative and insecticide and miticide |
| US6521656B1 (en) | 1998-06-01 | 2003-02-18 | Fujisawa Pharmaceutical Co., Ltd. | Remedies for male sterility |
| WO2003014087A1 (en) | 2001-08-06 | 2003-02-20 | Asahi Glass Company, Limited | Process for preparation of 5-methyl-1-phenyl-2(1h) -pyridinone |
| US20030065175A1 (en) | 2000-12-26 | 2003-04-03 | Dr. Reddy's Research Foundation | Novel heterocyclic compounds having antibacterial activity: process for their preparation and pharmaceutical compositions containing them |
| US6551963B1 (en) | 1995-06-06 | 2003-04-22 | Bayer Aktiengesellschaft | Phenylpyridazinones |
| WO2003033502A1 (en) | 2001-10-16 | 2003-04-24 | Celltech R & D Limited | Bicyclic oxopyridine and oxopyrimidine derivatives |
| WO2003035650A1 (en) | 2001-09-25 | 2003-05-01 | Takeda Chemical Industries, Ltd. | Entry inhibitor |
| CN1417209A (en) | 2001-11-08 | 2003-05-14 | 中国科学院上海药物研究所 | A class of tetrahydroquinolinone-piperidine compounds and their preparation and use |
| WO2003045912A1 (en) | 2001-11-29 | 2003-06-05 | Warner-Lambert Company Llc | Inhibitors of factor xa and other serine proteases involved in the coagulation cascade |
| WO2003047577A2 (en) | 2001-12-06 | 2003-06-12 | Eisai Co Ltd | Pharmaceutical compositions comprising dihydropyridinone compounds and an immunoregulatory or an antiinflammatory agent and their uses |
| WO2003047347A1 (en) | 2001-12-07 | 2003-06-12 | Syngenta Participations Ag | Microbiocidal n-phenyl-n-[4-(4-pyridyl)-2-pyrimidin-2-yl]-amine derivatives |
| US6586447B1 (en) | 1999-04-01 | 2003-07-01 | Pfizer Inc | 3,3-disubstituted-oxindole derivatives useful as anticancer agents |
| WO2003059891A1 (en) | 2002-01-18 | 2003-07-24 | Pharmacia Corporation | Substituted pyridazinones as inhibitors of p38 |
| WO2003059871A1 (en) | 2002-01-09 | 2003-07-24 | Ajinomoto Co.,Inc. | N-alkylsulfonyl-substituted amide derivatives |
| US6602826B1 (en) | 1997-03-05 | 2003-08-05 | Bayer Aktiengesellschaft | Heterocyclically substituted aromatic amino compounds with a herbicidal effect |
| WO2003068230A1 (en) | 2002-02-14 | 2003-08-21 | Pharmacia Corporation | Substituted pyridinones as modulators of p38 map kinase |
| JP2003238611A (en) | 2002-02-18 | 2003-08-27 | Japan Polyolefins Co Ltd | Catalyst component for olefin polymerization, catalyst for olefin polymerization, and method for producing polyolefin |
| US20030162130A1 (en) | 2001-07-05 | 2003-08-28 | Yasubumi Murota | Process for photopolymerization of photosensitive lithographic printing plate |
| WO2003076405A1 (en) | 2002-03-14 | 2003-09-18 | Bayer Healthcare Ag | Monocyclic aroylpyridinones as antiinflammatory agents |
| JP2003261535A (en) | 2002-03-08 | 2003-09-19 | Mitsubishi Chemicals Corp | Method for producing 2-hydroxy-5-methylpyridine |
| WO2003082265A2 (en) | 2002-04-02 | 2003-10-09 | Fujisawa Pharmaceutical Co | Pharmaceutical composition for treating or preventing virus infectious diseases |
| US20030194748A1 (en) | 2000-05-29 | 2003-10-16 | Tohru Nagasaki | Method for labeling with tritium |
| WO2003093273A1 (en) | 2002-05-02 | 2003-11-13 | Merck Sharp & Dohme Limited | Imidazo-triazine derivatives as ligands for gaba receptors |
| US20030220368A1 (en) | 2000-01-20 | 2003-11-27 | Fumihiro Ozaki | Novel piperidine compouds and drugs containing the same |
| WO2003097062A1 (en) | 2002-05-13 | 2003-11-27 | Merck & Co., Inc. | Phenyl substituted imidazopyridines and phenyl substituted benzimidazoles |
| WO2003106452A2 (en) | 2002-06-12 | 2003-12-24 | Millennium Pharmaceuticals, Inc. | Antagonists of melanin concentrating hormone receptor |
| WO2004000355A1 (en) | 2002-06-19 | 2003-12-31 | Pfizer Products Inc. | Combination treatment for depression and anxiety by nk1 and nk3 antagonists |
| WO2004000846A1 (en) | 2002-06-20 | 2003-12-31 | Celltech R & D Limited | Arylamine substututed bicyclic heteroaromatic compounds as p38 kinase inhibitors |
| US20040006082A1 (en) | 2000-08-11 | 2004-01-08 | Hitoshi Harada | 2-Aminopyridine compounds and use thereof as drugs |
| WO2004005286A2 (en) | 2002-07-03 | 2004-01-15 | K.U.Leuven Research & Development | Viral inhibitors |
| WO2004006906A2 (en) | 2002-07-15 | 2004-01-22 | Combinatorx, Incorporated | Methods for the treatment of neoplasms |
| US20040014986A1 (en) | 1999-12-27 | 2004-01-22 | Wolfram Hendel | Method for producing oxindoles |
| WO2004009560A1 (en) | 2002-07-22 | 2004-01-29 | Orchid Chemicals & Pharmaceuticals Ltd | Novel bio-active molecules |
| WO2004014859A2 (en) | 2002-08-10 | 2004-02-19 | Tanabe Seiyaku Co., Ltd. | Process for preparing quinolin antibiotic intermediates |
| WO2004014892A1 (en) | 2002-08-13 | 2004-02-19 | Warner-Lambert Company Llc | Monocyclic derivatives as matrix metalloproteinase inhibitors |
| WO2004019863A2 (en) | 2002-08-28 | 2004-03-11 | Intermune, Inc. | Combination therapy for treatment of fibrotic disorders |
| EP1400243A1 (en) | 2002-09-19 | 2004-03-24 | Tanabe Seiyaku Co., Ltd. | Calcium-activated K channel activator |
| WO2004024152A1 (en) | 2002-09-13 | 2004-03-25 | Mepha Ag | Stable cream preparations of phenyl-pyridone compounds for topical application |
| WO2004024078A2 (en) | 2002-09-11 | 2004-03-25 | Merck & Co., Inc. | Dihydroxypyridopyrazine-1,6-dione compounds useful as hiv integrase inhibitors |
| US20040063955A1 (en) | 1999-05-07 | 2004-04-01 | Biediger Ronald J. | Carboxylic acid derivatives that inhibit the binding of integrins to their receptors |
| WO2004031188A1 (en) | 2002-10-01 | 2004-04-15 | Celltech R & D Limited | Bicyclic heteroaromatic compounds as kinase inhibitors |
| WO2004031145A2 (en) | 2002-10-02 | 2004-04-15 | Bristol-Myers Squibb Company | Lactam-containing diaminoalkyl, beta-aminoacids, alpha-aminoacids and derivatives thereof as factor xa inhibitors |
| US20040082619A1 (en) | 2000-12-28 | 2004-04-29 | Yukio Tada | Pyridone derivatives having affinity for cannabinoid 2-type receptor |
| WO2004037159A2 (en) | 2002-10-23 | 2004-05-06 | Obetherapy Biotechnology | Compounds, compositions and methods for modulating fat metabolism |
| US20040097560A1 (en) | 2002-11-09 | 2004-05-20 | The Procter & Gamble Company | N-alkyl-4-methyleneamino-3-hydroxy-2-pyridones |
| US20040102494A1 (en) | 2000-12-26 | 2004-05-27 | Dr. Reddy's Laboratories Limited | Novel heterocyclic compounds having antibacterial activity: process for their preparation and pharmaceutical compositions containing them |
| WO2004048314A1 (en) | 2002-11-26 | 2004-06-10 | Novartis Ag | Substituted amino phenylacetic acids, derivatives thereof, their preparation and their use as cyclooxygenase 2 (cox-2) inhibitors |
| WO2004058256A1 (en) | 2002-12-26 | 2004-07-15 | Kdl, Inc. | Pharmaceutical liquid composition containing pyridone derivative |
| WO2004060306A2 (en) | 2002-12-31 | 2004-07-22 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US20040142950A1 (en) | 2003-01-17 | 2004-07-22 | Bunker Amy Mae | Amide and ester matrix metalloproteinase inhibitors |
| US20040157738A1 (en) | 2003-02-12 | 2004-08-12 | Ishihara Sangyo Kaisha, Ltd. | Novel oxygen containing fused cyclic derivatives and herbicidal, desiccant and defoliate compositions containing them |
| WO2004072033A2 (en) | 2003-02-12 | 2004-08-26 | Biogen Idec Ma Inc. | Pyrazoles and methods of making and using the same |
| WO2004074282A1 (en) | 2003-02-24 | 2004-09-02 | Pharmacia & Upjohn Company Llc | Antibacterial indolone oxazolidinones, intermediates for their preparation and pharmaceutical compositions containing them |
| WO2004073628A2 (en) | 2003-02-14 | 2004-09-02 | Smithkline Beecham Corporation | Novel compounds |
| WO2004078174A1 (en) | 2003-02-28 | 2004-09-16 | Morgan Lee R | Resonance modulator for diagnosis and therapy |
| JP2004269469A (en) | 2003-03-12 | 2004-09-30 | Yamanouchi Pharmaceut Co Ltd | Pyrimidine derivative or salt thereof |
| JP2004315594A (en) | 2003-04-14 | 2004-11-11 | Konica Minolta Holdings Inc | Polymerization initiator, polymer composition, method for free radical generation, lithographic printing plate material and method for manufacturing lithographic printing plate |
| US20040235886A1 (en) | 2003-01-31 | 2004-11-25 | Charifson Paul S. | Gyrase inhibitors and uses thereof |
| WO2004103296A2 (en) | 2003-05-16 | 2004-12-02 | Intermune, Inc. | Methods of treating idiopathic pulmonary fibrosis |
| WO2004105684A2 (en) | 2003-05-16 | 2004-12-09 | Intermune, Inc. | Combination therapy for proliferative disorders |
| US20040259865A1 (en) | 2001-10-22 | 2004-12-23 | Hitoshi Harada | Pyrimidone compounds and pharmaceutical compositions containing the same |
| US20040259864A1 (en) | 2001-02-23 | 2004-12-23 | Herve Geneste | Substituted pyrimidinone derivatives as ligands of integrin receptors |
| WO2004110245A2 (en) | 2003-05-16 | 2004-12-23 | Intermune, Inc. | Combination therapy for cancer treatment |
| JP2004359641A (en) | 2003-06-06 | 2004-12-24 | Ono Pharmaceut Co Ltd | CCR5 activator |
| WO2004113347A1 (en) | 2003-06-20 | 2004-12-29 | Celltech R & D Limited | Thienopyridone derivatives as kinase inhibitors |
| US20050004149A1 (en) | 2001-10-22 | 2005-01-06 | Hitoshi Harada | Pyrimidine compound and medicinal composition thereof |
| WO2005000227A2 (en) | 2003-06-06 | 2005-01-06 | Intermune, Inc. | Methods of treating tnf-mediated disorders |
| WO2005000818A1 (en) | 2003-06-27 | 2005-01-06 | Warner-Lambert Company Llc | 5-substituted-4-`(substituted phenyl)!amino!-2-pyridone deviatives for use as mek inhibitors |
| WO2005002672A2 (en) | 2003-07-01 | 2005-01-13 | President And Fellows Of Harvard College | Sirt1 modulators for manipulating cells/organism lifespan/stress response |
| JP2005013152A (en) | 2003-06-27 | 2005-01-20 | Asahi Kasei Corp | Cell differentiation inhibitor and cell culture method, culture solution and cultured cells using the same |
| WO2005007632A1 (en) | 2003-07-18 | 2005-01-27 | Pharmacia Corporation | Substituted pyridazinones as inhibitors of p38 |
| WO2005012288A1 (en) | 2003-08-01 | 2005-02-10 | Genelabs Technologies, Inc | Bicyclic imidazol derivatives against flaviviridae |
| WO2005013917A2 (en) | 2003-02-28 | 2005-02-17 | Intermune, Inc. | Combination therapy for treating alphavirus infection and liver fibrosis |
| US20050038247A1 (en) | 2003-01-31 | 2005-02-17 | Charifson Paul S. | Gyrase inhibitors and uses thereof |
| WO2005018557A2 (en) | 2003-08-13 | 2005-03-03 | Pharmacia Corporation | Substituted pyridinones |
| US20050054631A1 (en) | 2003-09-04 | 2005-03-10 | Aventis Pharmaceuticals Inc. | Substituted indoles as inhibitors of poly (ADP-ribose) polymerase (PARP) |
| WO2005026123A1 (en) | 2003-09-18 | 2005-03-24 | Astrazeneca Ab | 2-pyridone derivatives as neutrophil elastase inhibitors and their use |
| US20050065144A1 (en) | 2003-09-08 | 2005-03-24 | Syrrx, Inc. | Dipeptidyl peptidase inhibitors |
| WO2005026124A1 (en) | 2003-09-18 | 2005-03-24 | Astrazeneca Ab | 2-pyridone derivatives as netrophil elastase inhibitors and their use |
| DE10345648A1 (en) | 2003-10-01 | 2005-04-21 | Studiengesellschaft Kohle Mbh | Production of new or known tetrahydropyranyl-pyridone compounds, for use as phosphatase inhibitors, e.g. for treating cancer, from pyridinone and pyranone compounds via several new intermediates |
| WO2005039598A1 (en) | 2003-10-24 | 2005-05-06 | Intermune, Inc. | Method of treating alcoholic liver disease |
| WO2005040758A2 (en) | 2003-10-24 | 2005-05-06 | Intermune, Inc. | Use of pirfenidone in therapeutic regimens |
| US20050101590A1 (en) | 2002-02-19 | 2005-05-12 | Kiyoshi Yasui | Antipruritics |
| JP2005145882A (en) | 2003-11-14 | 2005-06-09 | Ichimaru Pharcos Co Ltd | Cosmetic composition |
| US20050130976A1 (en) | 2003-11-19 | 2005-06-16 | Eli Wallace | Bicyclic inhibitors of MEK and methods of use thereof |
| WO2005053707A1 (en) | 2003-12-05 | 2005-06-16 | Ono Pharmaceutical Co., Ltd. | Blood flow promoters for cauda equina tissues |
| EP1544194A1 (en) | 2002-07-24 | 2005-06-22 | Kyorin Pharmaceutical Co., Ltd. | 4-(substituted aryl)-5-hydroxyisoquinolinone derivative |
| US20050153941A1 (en) | 2003-06-27 | 2005-07-14 | Tomoyuki Miyabayashi | Cell differntiation inhibiting agent, cell culture method using the same, culture medium, and cultured cell line |
| WO2005073222A1 (en) | 2004-01-29 | 2005-08-11 | Pfizer Japan, Inc. | 1-isopropyl-2-oxo-1,2-dihydropyridine-3-carboxamide derivatives having 5-ht4 receptor agonistic activity |
| WO2005075438A1 (en) | 2004-02-09 | 2005-08-18 | Tanabe Seiyaku Co., Ltd. | Alpha-4 integrin mediated cell adhesion inhibitors for the treatment or prevention of inflammatory diseases |
| WO2005085200A1 (en) | 2004-03-05 | 2005-09-15 | Banyu Pharmaceutical Co., Ltd. | Pyridone derivative |
| JP2005255675A (en) | 2004-02-09 | 2005-09-22 | Tanabe Seiyaku Co Ltd | Pharmaceutical composition |
| WO2005090294A2 (en) | 2004-02-20 | 2005-09-29 | Japan Science And Technology Agency | PROCESS OF MAKING α-AMINOOXYKETONE/α-AMINOOXYALDEHYDE AND α-HYDROXYKETONE/α-HYDROXYALDEHYDE COMPOUNDS AND A PROCESS OF MAKING REACTION PRODUCTS FROM CYCLIC α,ß-UNSATURATED KETONE SUBSTRATES AND NITROSO SUBSTRATES |
| CN1676518A (en) | 2005-03-17 | 2005-10-05 | 南开大学 | 4-Substituted phenylpyridazines and their herbicidal activity |
| WO2005096784A2 (en) | 2004-04-08 | 2005-10-20 | Targegen, Inc. | Benzotriazine inhibitors of kinases |
| WO2005097750A1 (en) | 2004-03-30 | 2005-10-20 | Aventis Pharmaceuticals Inc. | Substituted pyridones as inhibitors of poly(adp-ribose) polymerase (parp) |
| WO2005099688A2 (en) | 2004-04-07 | 2005-10-27 | Takeda Pharmaceutical Company Limited | Cyclic compounds |
| WO2005105790A1 (en) | 2004-04-28 | 2005-11-10 | Tanabe Seiyaku Co., Ltd. | 4- 2- (cycloalkylamino) pyrimidin-4-yl ! - (phenyl) - imidazolin-2- one derivatives as p38 map- kinase inhibitors for the treatment of inflammatory diseases |
| WO2005105743A1 (en) | 2004-04-28 | 2005-11-10 | Ono Pharmaceutical Co., Ltd. | Nitrogen-containing heterocyclic compounds and medicinal use thereof |
| US20050256136A1 (en) | 2003-01-31 | 2005-11-17 | Charifson Paul S | Gyrase inhibitors and uses thereof |
| US20050256123A1 (en) | 2003-11-19 | 2005-11-17 | Marlow Allison L | Heterocyclic inhibitors of MEK and methods of use thereof |
| US20050288286A1 (en) | 2003-12-24 | 2005-12-29 | Flynn Daniel L | Anti-inflammatory medicaments |
| WO2005123687A1 (en) | 2004-06-16 | 2005-12-29 | Sanofi-Aventis Deutschland Gmbh | Substituted tetrahydro-2h-isoquinolin-1-one derivatives, method for the production thereof, and use of the same as medicaments |
| WO2006004107A1 (en) | 2004-07-06 | 2006-01-12 | Eisai R & D Management Co., Ltd. | Crystal of 1,2-dihydropyridine compound and method for producing same |
| US20060025337A1 (en) | 2003-07-01 | 2006-02-02 | President And Fellows Of Harvard College | Sirtuin related therapeutics and diagnostics for neurodegenerative diseases |
| US20060025424A1 (en) | 2003-01-31 | 2006-02-02 | Paul Charifson | Gyrase inhibitors and uses thereof |
| WO2006011024A2 (en) | 2004-07-19 | 2006-02-02 | Glenmark Pharmaceuticals Ltd. | New tricyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| WO2006017443A2 (en) | 2004-08-02 | 2006-02-16 | Osi Pharmaceuticals, Inc. | Aryl-amino substituted pyrrolopyrimidine multi-kinase inhibiting compounds |
| WO2006020145A2 (en) | 2004-07-19 | 2006-02-23 | The Johns Hopkins University | Flt3 inhibitors for immune suppression |
| WO2006026305A1 (en) | 2004-08-31 | 2006-03-09 | Biogen Idec Ma Inc | Pyrimidinylpyrazoles as tgf-beta inhibitors |
| WO2006030032A1 (en) | 2004-09-17 | 2006-03-23 | Janssen Pharmaceutica N.V. | Novel pyridinone derivatives and their use as positive allosteric modulators of mglur2-receptors |
| US20060069260A1 (en) | 2004-09-28 | 2006-03-30 | Huiping Zhang | Preparation of N-aryl pyridones |
| WO2006032631A1 (en) | 2004-09-22 | 2006-03-30 | Janssen Pharmaceutica N.V. | Inhibitors of the interaction between mdm2 and p53 |
| WO2006038734A1 (en) | 2004-10-08 | 2006-04-13 | Astellas Pharma Inc. | Pyridazinone derivatives cytokines inhibitors |
| WO2006044405A1 (en) | 2004-10-12 | 2006-04-27 | Decode Genetics, Inc. | Aryl sulfonamide peri-substituted bicyclics for occlusive artery disease |
| WO2006046778A1 (en) | 2004-10-28 | 2006-05-04 | Shionogi & Co., Ltd. | 3-carbamoyl-2-pyridone derivative |
| US20060100193A1 (en) | 2004-06-18 | 2006-05-11 | Millennium Pharmaceuticals, Inc. | Factor Xa inhibitors |
| US20060111355A1 (en) | 2004-11-23 | 2006-05-25 | Wyeth | Gonadotropin releasing hormone receptor antagonists |
| WO2006055918A2 (en) | 2004-11-19 | 2006-05-26 | Pharmacopeia Drug Discovery, Inc. | One-dimensional qsar models |
| WO2006056427A1 (en) | 2004-11-24 | 2006-06-01 | Laboratoires Serono S.A. | Novel 4-arylamino pyridone derivatives as mek inhibitors for the treatment of hyperproliferative disorders |
| WO2006060122A2 (en) | 2004-11-30 | 2006-06-08 | Artesian Therapeutics, Inc. | Cardiotonic compounds with inhibitory activity against beta-adrenergic receptors and phosphodiesterase |
| JP2006142666A (en) | 2004-11-19 | 2006-06-08 | Mitsubishi Chemicals Corp | Dye for optical recording media |
| WO2006066079A2 (en) | 2004-12-17 | 2006-06-22 | Anadys Pharmaceuticals, Inc. | Pyridazinone compounds |
| WO2006072039A1 (en) | 2004-12-29 | 2006-07-06 | E. I. Du Pont De Nemours And Company | Nepetalactams and n-substituted derivatives thereof |
| WO2006072037A1 (en) | 2004-12-29 | 2006-07-06 | E. I. Du Pont De Nemours And Company | Dihydronepetalactams and n-substituted derivatives thereof |
| US20060160862A1 (en) | 2004-11-24 | 2006-07-20 | Jean-Damien Charrier | Caspase inhibitors and uses thereof |
| WO2006076681A2 (en) | 2005-01-13 | 2006-07-20 | Sirtris Pharmaceuticals, Inc. | Novel compositions for preventing and treating neurodegenerative and blood coagulation disorders |
| WO2006079021A2 (en) | 2005-01-20 | 2006-07-27 | Sirtris Pharmaceuticals, Inc. | Use of sirtuin-activating compounds for treating flushing and drug induced weight gain |
| CN1817862A (en) | 2006-03-15 | 2006-08-16 | 浙江省医学科学院 | Production of pyriphenanthrenone as anti-fibrosis medicine |
| US20060189616A1 (en) | 2005-02-18 | 2006-08-24 | Wyeth | 7-Substituted imidazo[4,5-c]pyridine antagonists of gonadotropin releasing hormone receptor |
| US20060189617A1 (en) | 2005-02-18 | 2006-08-24 | Wyeth | Imidazo[4,5-b]pyridine antagonists of gonadotropin releasing hormone receptor |
| US20060211577A1 (en) | 2003-04-08 | 2006-09-21 | Basf Aktiengesellschaft | Benzenesulphonamide derivatives as herbicides or desiccant/defoliant compounds |
| WO2006107859A2 (en) | 2005-04-04 | 2006-10-12 | Eisai Co., Ltd. | Dihydropyridine compounds for neurodegenerative diseases and dementia |
| WO2006108354A1 (en) | 2005-04-13 | 2006-10-19 | Xiangya Hospital Of Central South University | 1-(substituted phenyl)-5- methyl- 2 - (1h) pyridone in the manufacture of medicaments for treating fibrosis in organs or tissues |
| WO2006116713A1 (en) | 2005-04-27 | 2006-11-02 | Amgen Inc. | Substituted amide derivatives as protein kinase inhibitors |
| US20060247269A1 (en) | 2003-06-20 | 2006-11-02 | Brookings Daniel C | Thienopyridone derivatives as kinase inhibitors |
| US20060258861A1 (en) | 2005-04-28 | 2006-11-16 | Boehringer Ingelheim International Gmbh | Compounds for the treatment of inflammatory diseases |
| WO2006122154A2 (en) | 2005-05-10 | 2006-11-16 | Intermune, Inc. | Pyridone derivatives for modulating stress-activated protein kinase system |
| WO2006129076A1 (en) | 2005-06-01 | 2006-12-07 | Sterix Limited | Use of a steroid sulphatase inhibitor for inhibiting the synthesis of androstenedione and/or testosterone |
| WO2006131186A1 (en) | 2005-06-10 | 2006-12-14 | Merck Patent Gmbh | Oxindoles as kinase inhibitors |
| WO2006133147A2 (en) | 2005-06-08 | 2006-12-14 | Novartis Ag | Organic compounds |
| US20060287319A1 (en) | 2005-06-15 | 2006-12-21 | Shibo Jiang | Anti-viral compositions comprising heterocyclic substituted phenyl furans and related compounds |
| WO2006138418A2 (en) | 2005-06-14 | 2006-12-28 | President And Fellows Of Harvard College | Improvement of cognitive performance with sirtuin activators |
| WO2007008548A2 (en) | 2005-07-07 | 2007-01-18 | Sirtris Pharmaceuticals, Inc. | Methods and related compositions for treating or preventing obesity, insulin resistance disorders, and mitochondrial-associated disorders |
| WO2007006591A2 (en) | 2005-07-13 | 2007-01-18 | Bayer Cropscience Sa | Dihalogenation of n,o-disubstituted hydroxipyridones and uses thereof |
| US20070037822A1 (en) | 2005-06-07 | 2007-02-15 | Pharmacopeia Drug Discovery, Inc. | Azinone and diazinone V3 inhibitors for depression and stress disorders |
| US20070049624A1 (en) | 2003-11-14 | 2007-03-01 | Xianghui Yi | Derivatives of pyridone and the use of them |
| WO2007024021A1 (en) | 2005-08-26 | 2007-03-01 | Fuji Film Corporation | Impregnating oil composition for sintered bearing, bearing apparatus and sliding member |
| WO2007026950A1 (en) | 2005-09-01 | 2007-03-08 | Astellas Pharma Inc. | Pyridazinone derivatives used for the treatment of pain |
| CN1927923A (en) | 2006-09-06 | 2007-03-14 | 北京理工大学 | Ion liquid, non-aqueous liquid and polymer electrolyte material based on the same |
| JP2007063268A (en) | 2005-08-05 | 2007-03-15 | Tanabe Seiyaku Co Ltd | Pharmaceutical composition |
| WO2007037543A1 (en) | 2005-09-29 | 2007-04-05 | Banyu Pharmaceutical Co., Ltd. | Biarylamide derivative |
| WO2007044796A2 (en) | 2005-10-11 | 2007-04-19 | Nps Pharmaceuticals, Inc. | Pyridazinone compounds as calcilytics |
| WO2007048065A2 (en) | 2005-10-21 | 2007-04-26 | Exelixis, Inc. | Pyrimidinones as casein kinase ii (ck2) modulators |
| WO2007053685A2 (en) | 2005-11-01 | 2007-05-10 | Intermune, Inc. | Methods of treating atrial fibrillation with p38 inhibitor compounds |
| US20070105890A1 (en) | 2003-11-26 | 2007-05-10 | Dainippon Sumitomo Pharma Co., Ltd | Novel condensed imidazole derivative |
| CN1962642A (en) | 2006-11-21 | 2007-05-16 | 南开大学 | Trifluoromethylphenylpyridazine derivative and its preparation method |
| US20070112038A1 (en) | 2003-11-19 | 2007-05-17 | Marlow Allison L | Heterocyclic inhibitors of MEK and methods of use thereof |
| WO2007058392A1 (en) | 2005-11-21 | 2007-05-24 | Japan Tobacco Inc. | Heterocyclic compound and medicinal application thereof |
| WO2007057329A1 (en) | 2005-11-18 | 2007-05-24 | F. Hoffmann-La Roche Ag | Azaindole-2-carboxamide derivatives |
| WO2007062167A2 (en) | 2005-11-23 | 2007-05-31 | Intermune, Inc. | Method of modulating stress-activated protein kinase system |
| WO2007064797A2 (en) | 2005-11-30 | 2007-06-07 | Vertex Pharmaceuticals Incorporated | Inhibitors of c-met and uses thereof |
| JP2007145819A (en) | 2005-10-28 | 2007-06-14 | Tanabe Seiyaku Co Ltd | Pharmaceutical composition |
| US20070149513A1 (en) | 2005-12-23 | 2007-06-28 | Ning Chen | Vanilloid receptor ligands and their use in treatments |
| US20070185092A1 (en) | 2004-06-18 | 2007-08-09 | Millennium Pharmaceuticals, Inc. | FACTOR Xa INHIBITORS |
| WO2007088996A1 (en) | 2006-01-31 | 2007-08-09 | Mitsubishi Tanabe Pharma Corporation | Trisubstituted amine compounds as inhibitors of -cholesteryl ester transfer protein cetp |
| US20070191336A1 (en) | 2003-12-24 | 2007-08-16 | Flynn Daniel L | Anti-inflammatory medicaments |
| WO2007100990A2 (en) | 2006-02-24 | 2007-09-07 | Abbott Laboratories | Octahydro-pyrrolo[3,4-b] pyrrole derivatives and their use as histamine-3 receptor ligands |
| WO2007104034A2 (en) | 2006-03-08 | 2007-09-13 | Takeda San Diego, Inc. | Glucokinase activators |
| US20070219182A1 (en) | 2003-12-11 | 2007-09-20 | Wilfried Lubisch | Keto Lactam Compounds and Use Thereof |
| WO2007107545A1 (en) | 2006-03-22 | 2007-09-27 | Janssen Pharmaceutica N.V. | Cyclic-alkylaminederivatives as inhibitors of the interaction between mdm2 and p53 |
| WO2007108968A2 (en) | 2006-03-13 | 2007-09-27 | Merck & Co., Inc. | Ophthalmic compositions for treating ocular hypertension |
| WO2007117778A2 (en) | 2006-02-24 | 2007-10-18 | Kalypsys, Inc. | Quinolones useful as inducible nitric oxide synthase inhibitors |
| WO2007117482A2 (en) | 2006-04-05 | 2007-10-18 | Vitae Pharmaceuticals, Inc. | Renin inhibitors |
| WO2007117559A2 (en) | 2006-04-05 | 2007-10-18 | Vitae Pharmaceuticals, Inc. | Renin inhibitors |
| WO2007120842A2 (en) | 2006-04-13 | 2007-10-25 | Cornell Research Foundation, Inc. | Methods and compositions for targeting c-rel |
| WO2007127475A2 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Pyridazines for demyelinating diseases and neuropathic pain |
| WO2007127474A2 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Compositions and treatments using pyridazine compounds and cholinesterase inhibitors |
| US20070259924A1 (en) | 2006-05-05 | 2007-11-08 | Millennium Pharmaceuticals, Inc. | Factor xa inhibitors |
| US20070265308A1 (en) | 2004-11-10 | 2007-11-15 | Ono Pharmaceutical Co., Ltd. | Nitrogenous Heterocyclic Compound and Pharmaceutical Use Thereof |
| WO2007129040A1 (en) | 2006-05-04 | 2007-11-15 | Chroma Therapeutics Ltd. | p38 MAP KINASE INHIBITORS |
| WO2007139150A1 (en) | 2006-05-30 | 2007-12-06 | The University Of Tokushima | ANTI-INFLUENZA VIRAL AGENT COMPRISING TNF-α INHIBITOR |
| WO2007146712A2 (en) | 2006-06-09 | 2007-12-21 | Kemia, Inc. | Therapy using cytokine inhibitors |
| WO2007147297A1 (en) | 2006-06-15 | 2007-12-27 | Shanghai Genomics, Inc. | The use of derviate of pyridone for preventing and treating radioactive injury of lungs |
| WO2007148093A1 (en) | 2006-06-22 | 2007-12-27 | Prolysis Ltd. | Antibacterial compositions |
| WO2008002490A2 (en) | 2006-06-23 | 2008-01-03 | Radius Health, Inc. | Treatment of vasomotor symptoms with selective estrogen receptor modulators |
| US20080020010A1 (en) | 2006-07-19 | 2008-01-24 | The University Of Georgia Research Foundation, Inc. | Pyridinone diketo acids: inhibitors of HIV replication |
| WO2008013838A2 (en) | 2006-07-25 | 2008-01-31 | Cephalon, Inc. | Pyridizinone derivatives |
| WO2008016239A1 (en) | 2006-08-02 | 2008-02-07 | Lg Life Sciences Ltd. | Caspase inhibitors based on pyridazinone scaffold |
| JP2008039883A (en) | 2006-08-02 | 2008-02-21 | Konica Minolta Opto Inc | Optical film, polarizing plate using the same and liquid crystal display |
| JP2008076948A (en) | 2006-09-25 | 2008-04-03 | Konica Minolta Medical & Graphic Inc | Photosensitive lithographic printing plate material |
| US20080114033A1 (en) | 2006-11-08 | 2008-05-15 | Bristol-Myers Squibb Company | Pyridinone compounds |
| KR20080045538A (en) | 2006-11-20 | 2008-05-23 | 에스케이케미칼주식회사 | Pharmaceutical composition for the treatment of inflammation and immune disease, including pyridine compound |
| WO2008065428A2 (en) | 2006-12-01 | 2008-06-05 | Sterix Limited | Steroid sulphatase inhibitors for treating hormone dependent cancer |
| WO2008068265A1 (en) | 2006-12-05 | 2008-06-12 | Janssen Pharmaceutica N.V. | Novel substituted diaza spiro pyridinone derivatives for use in mch-1 mediated diseases |
| US20080139557A1 (en) | 2006-09-11 | 2008-06-12 | Blomgren Peter A | Certain substituted amides, method of making, and method of use thereof |
| WO2008072784A1 (en) | 2006-12-14 | 2008-06-19 | Astellas Pharma Inc. | Polycyclic acid compounds useful as crth2 antagonists and antiallergic agents |
| WO2008076562A1 (en) | 2006-12-14 | 2008-06-26 | Eli Lilly And Company | 5- [4- (azetidin-3-yl0xy) -phenyl] -2-phenyl-5h-thiaz0l0 [5,4-c] pyridin-4-0ne derivatives and their use as mch receptor antagonists |
| WO2008079787A2 (en) | 2006-12-20 | 2008-07-03 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2008079277A1 (en) | 2006-12-22 | 2008-07-03 | Millennium Pharmaceuticals, Inc. | Certain pyrazoline derivatives with kinase inhibitory activity |
| WO2008080056A2 (en) | 2006-12-21 | 2008-07-03 | Sloan-Kettering Institute For Cancer Research | Pyridazinones and furan-containing compounds |
| WO2008077550A1 (en) | 2006-12-27 | 2008-07-03 | Sanofi-Aventis | Substituted isoquinoline and isoquinolinone derivatives as inhibitors of rho-kinase |
| WO2008091555A2 (en) | 2007-01-22 | 2008-07-31 | Gtx, Inc. | Nuclear receptor binding agents |
| CN101235030A (en) | 2007-01-30 | 2008-08-06 | 中南大学 | 1-substituted-5-trifluoromethyl-2-(1H)pyridone compound, preparation method and use thereof |
| WO2008099000A2 (en) | 2007-02-16 | 2008-08-21 | Boehringer Ingelheim International Gmbh | Substituted arylsulphonylglycines, the preparation thereof and the use thereof as pharmaceutical compositions |
| WO2008103277A2 (en) | 2007-02-16 | 2008-08-28 | Amgen Inc. | Nitrogen-containing heterocyclyl ketones and their use as c-met inhibitors |
| WO2008106202A1 (en) | 2007-02-27 | 2008-09-04 | Housey Gerard M | Theramutein modulators |
| WO2008107480A1 (en) | 2007-03-07 | 2008-09-12 | Ortho-Mcneil-Janssen Pharmaceuticals, Inc | 1,4-disubstituted 3-cyano-pyridone derivatives and their use as positive mglur2 -receptor modulators |
| WO2008112715A2 (en) | 2007-03-12 | 2008-09-18 | Vm Discovery Inc. | Novel agents of calcium ion channel modulators |
| WO2008110793A1 (en) | 2007-03-12 | 2008-09-18 | Biolipox Ab | Piperidinones useful in the treatment of inflammation |
| JP2008214225A (en) | 2007-03-01 | 2008-09-18 | Tosoh Corp | Process for producing 5,5-disubstituted-3-pyrrolin-2-one derivatives |
| US20080234332A1 (en) | 2007-03-20 | 2008-09-25 | Xiong Cai | Raf kinase inhibitors containing a zinc binding moiety |
| US20080249090A1 (en) | 2006-07-20 | 2008-10-09 | Amgen Inc. | Substituted pyridone compounds and methods of use |
| WO2008121877A2 (en) | 2007-04-02 | 2008-10-09 | Institute For Oneworld Health | Cftr inhibitor compounds and uses thereof |
| WO2008121407A1 (en) | 2007-03-30 | 2008-10-09 | The Regents Of The University Of California | In vivo imaging of sulfotransferases |
| US20080255083A1 (en) | 2006-08-25 | 2008-10-16 | Dirk Stenkamp | New pyridone derivates with mch antagonistic activity and medicaments comprising these compounds |
| WO2008124323A1 (en) | 2007-04-03 | 2008-10-16 | Array Biopharma Inc. | Imidazo[1,2-a]pyridine compounds as receptor tyrosine kinase inhibitors |
| WO2008124582A1 (en) | 2007-04-05 | 2008-10-16 | Smithkline Beecham Corporation | Renin inhibitors |
| WO2008124575A1 (en) | 2007-04-05 | 2008-10-16 | Smithkline Beecham Corporation | Renin inhibitors |
| US20080269287A1 (en) | 2004-12-01 | 2008-10-30 | Norikazu Ohtake | Substituted Pyridone Derivative |
| WO2008132155A2 (en) | 2007-04-26 | 2008-11-06 | Janssen Pharmaceutica Nv | Inhibitors of the binding between hdm2 and the proteasome |
| US20080280930A1 (en) | 2004-12-23 | 2008-11-13 | Hui Yao | Pyrimidinone Compounds and Preparation and Use Thereof |
| WO2008140066A2 (en) | 2007-05-03 | 2008-11-20 | Astellas Pharma Inc. | Pyridone derivatives as p38a mapk inhibitors |
| WO2008141119A2 (en) | 2007-05-09 | 2008-11-20 | Vertex Pharmaceuticals Incorporated | Modulators of cftr |
| WO2008144720A2 (en) | 2007-05-21 | 2008-11-27 | University Of Washington | Inhibitors of igf-1r signaling for the treatment of respiratory disorders |
| WO2008147170A1 (en) | 2007-05-29 | 2008-12-04 | Cell Therapy Technology, S.A. De C.V. | New process of synthesis for obtaining 5-methyl-1-phenyl-2 (ih) -pyridone, composition and use of the same |
| WO2008147169A2 (en) | 2007-05-29 | 2008-12-04 | Cell Therapy Technology, S.A. De C.V. | Microemulsion containing pirfenidone |
| US20080306053A1 (en) | 2007-06-08 | 2008-12-11 | Senomyx, Inc. | Modulation of chemosensory receptors and ligands associated therewith |
| US20080306093A1 (en) | 2007-06-08 | 2008-12-11 | Senomyx, Inc. | Modulation of chemosensory receptors and ligands associated therewith |
| US20080312284A1 (en) | 2005-12-21 | 2008-12-18 | Eisai R&D Management Co., Ltd. | Amorphous Form of 1,2-Dihydropyridine Compound |
| WO2008157786A1 (en) | 2007-06-20 | 2008-12-24 | Auspex Pharmaceutical, Inc. | Substituted n-aryl pyridinones as fibrotic inhibitors |
| WO2008155069A2 (en) | 2007-06-20 | 2008-12-24 | Bayer Schering Pharma Aktiengesellschaft | Substituted oxazolidinones and use thereof |
| WO2009012275A1 (en) | 2007-07-17 | 2009-01-22 | Bristol-Myers Squibb Company | Pyridone gpr119 g protein-coupled receptor agonists |
| WO2009011410A1 (en) | 2007-07-13 | 2009-01-22 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists and aldose reductase inhibitors for neuropathic pain |
| WO2009011411A1 (en) | 2007-07-13 | 2009-01-22 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists and zonisamide for neuropathic pain |
| WO2009011412A2 (en) | 2007-07-13 | 2009-01-22 | Eisai R & D Management Co., Ltd. | Combination of ampa receptor antagonists and acetylcholinesterase inhibitors for the treatment of neuropathic pain |
| US20090030017A1 (en) | 2005-04-08 | 2009-01-29 | Eisai R & D Management Co., Ltd | Therapeutic agent for dyskinesia |
| WO2009016118A1 (en) | 2007-07-27 | 2009-02-05 | Boehringer Ingelheim International Gmbh | Substituted arylsulfonylaminomethylphosphonic acid derivatives, their preparation and their use in the treatment of type i and ii diabetes mellitus |
| WO2009026816A1 (en) | 2007-08-23 | 2009-03-05 | Central South University | The application of 1-aryl-2(1h)-pyridone compounds in preparation of medincines for treating itching of the skin |
| WO2009029625A1 (en) | 2007-08-27 | 2009-03-05 | Kalypsys, Inc. | 4- [heterocyclyl-methyl] -8-fluoro-quinolin-2-ones useful as nitric oxide synthase inhibitors |
| WO2009035598A1 (en) | 2007-09-10 | 2009-03-19 | Concert Pharmaceuticals, Inc. | Deuterated pirfenidone |
| WO2009033703A1 (en) | 2007-09-14 | 2009-03-19 | Ortho-Mcneil-Janssen Pharmaceuticals, Inc. | 1,3-disubstituted-4-phenyl-1 h-pyridin-2-ones |
| WO2009039773A1 (en) | 2007-09-19 | 2009-04-02 | Central South University | New medicine use of 1-substituted aryl -2(1h)-pyridone |
| US20090088574A1 (en) | 2005-12-21 | 2009-04-02 | Eisai R&D Management Co., Ltd. | Crystal of 1,2-dihydropyridine compound (type iv) |
| WO2009054543A1 (en) | 2007-10-26 | 2009-04-30 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists and zonisamide for parkinson's disease and movement disorders |
| WO2009054544A1 (en) | 2007-10-26 | 2009-04-30 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists for parkinson's disease and movement disorders |
| WO2009057827A1 (en) | 2007-10-31 | 2009-05-07 | Nissan Chemical Industries, Ltd. | Pyridazinone derivatives and use thereof as p2x7 receptor inhibitors |
| WO2009060835A1 (en) | 2007-11-05 | 2009-05-14 | Kyoto University | Novel ubiquilin-binding small molecule |
| WO2009065922A2 (en) | 2007-11-22 | 2009-05-28 | Boehringer Ingelheim International Gmbh | Novel compounds |
| WO2009073620A2 (en) | 2007-11-30 | 2009-06-11 | Newlink Genetics | Ido inhibitors |
| WO2009076529A1 (en) | 2007-12-11 | 2009-06-18 | Research Development Foundation | Small molecules for neuronal differentiation of embryonic stem cells |
| WO2009074810A1 (en) | 2007-12-13 | 2009-06-18 | Prolysis Ltd | Antibacterial compositions |
| WO2009082039A1 (en) | 2007-12-26 | 2009-07-02 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists for epilepsy, mental disorders or deficits in sensory organ |
| US20090170861A1 (en) | 2007-03-15 | 2009-07-02 | Schering Corporation And Albany Molecular Research, Inc. | Pyridazinone Derivatives Useful as Glucan Synthase Inhibitors |
| WO2009082038A2 (en) | 2007-12-26 | 2009-07-02 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists and zonisamide for epilepsy |
| WO2009094427A1 (en) | 2008-01-23 | 2009-07-30 | Bristol-Myers Squibb Company | 4-pyridinone compounds and their use for cancer |
| WO2009097309A1 (en) | 2008-01-30 | 2009-08-06 | Cephalon, Inc. | Substituted spirocyclic piperidine derivatives as histamine-3 (h3) receptor ligands |
| WO2009108499A1 (en) | 2008-02-25 | 2009-09-03 | Merck & Co., Inc. | Tetrahydro-1h-pyrrolo fused pyridones |
| WO2009111785A2 (en) | 2008-03-07 | 2009-09-11 | Solanan, Inc. | Treatment of sepsis with 5-ethyl-1-phenyl-2(1h)-pyridone and novel methods for synthesis |
| US20090247566A1 (en) | 2008-03-25 | 2009-10-01 | Alexander Kornienko | Pyrano [3,2-C] Pyridones and Related Heterocyclic Compounds as Pharmaceutical Agents for Treating Disorders Responsive to Apoptosis, Antiproliferation or Vascular Disruption, and the Use Thereof |
| US20090246198A1 (en) | 2008-03-31 | 2009-10-01 | Takeda Pharmaceutical Company Limited | Mapk/erk kinase inhibitors and methods of use thereof |
| WO2009124119A2 (en) | 2008-04-01 | 2009-10-08 | The Trustes of Columbia University in the City of New York | Phosphodiesterase inhibitors and uses thereof |
| WO2009124553A2 (en) | 2008-04-09 | 2009-10-15 | Neurokey A/S | Use of hypothermia inducing drugs |
| WO2009135299A1 (en) | 2008-05-05 | 2009-11-12 | Merck Frosst Canada Ltd. | 3, 4 - substituted piperidine derivatives as renin inhibitors |
| WO2009142732A2 (en) | 2008-05-20 | 2009-11-26 | Cephalon, Inc. | Substituted pyridazinone derivatives as histamine-3 (h3) receptor ligands |
| WO2009149188A1 (en) | 2008-06-03 | 2009-12-10 | Intermune, Inc. | Compounds and methods for treating inflammatory and fibrotic disorders |
| WO2009158393A1 (en) | 2008-06-25 | 2009-12-30 | Envivo Pharmaceuticals, Inc. | 1, 2 disubstituted heterocyclic compounds |
| WO2009156484A2 (en) | 2008-06-27 | 2009-12-30 | Novartis Ag | Organic compounds |
| WO2010006191A1 (en) | 2008-07-11 | 2010-01-14 | Irm Llc | 4-phenoxymethylpiperidines as modulators of gpr119 activity |
| WO2010009183A1 (en) | 2008-07-16 | 2010-01-21 | Bristol-Myers Squibb Company | Pyridone and pyridazone analogues as gpr119 modulators |
| WO2010021693A2 (en) | 2008-08-18 | 2010-02-25 | Yale University | Mif modulators |
| WO2010025087A1 (en) | 2008-08-25 | 2010-03-04 | Smithkline Beecham Corporation | Prolyl hydroxylase inhibitors |
| WO2010027567A2 (en) | 2008-07-23 | 2010-03-11 | Schering Corporation | Tricyclic spirocycle derivatives and methods of use thereof |
| US20100063104A1 (en) | 2005-10-03 | 2010-03-11 | Ono Pharmaceutical Co., Ltd., | Nitrogen-containing heterocyclic compound and pharmaceutical application thereof |
| WO2010029299A1 (en) | 2008-09-12 | 2010-03-18 | Biolipox Ab | Pyrimidinone derivaties for use as medicaments |
| WO2010044885A2 (en) | 2008-10-17 | 2010-04-22 | Whitehead Institute For Biomedical Research | Soluble mtor complexes and modulators thereof |
| WO2010048716A1 (en) | 2008-10-29 | 2010-05-06 | Pacific Therapeutics Ltd. | Composition and method for treating fibrosis |
| US20100120862A1 (en) | 2008-10-24 | 2010-05-13 | Laykea Tafesse | Monocyclic compounds and their use as trpv1 ligands |
| WO2010065755A1 (en) | 2008-12-04 | 2010-06-10 | Concert Pharmaceuticals, Inc. | Deuterated pyridinones |
| WO2010066028A1 (en) | 2008-12-10 | 2010-06-17 | Merck Frosst Canada Ltd. | 3,4 - substituted piperidine derivatives as renin inhibitors |
| WO2010068253A1 (en) | 2008-12-11 | 2010-06-17 | Shionogi & Co., Ltd. | Synthesis of carbamoylpyridone hiv integrase inhibitors and intermediates |
| WO2010075561A1 (en) | 2008-12-23 | 2010-07-01 | President And Fellows Of Harvard College | Small molecule inhibitors of necroptosis |
| WO2010077680A2 (en) | 2008-12-08 | 2010-07-08 | Vm Discovery Inc. | Compositions of protein receptor tyrosine kinase inhibitors |
| WO2010080795A2 (en) | 2009-01-07 | 2010-07-15 | Henkel Corporation | Hydrogen peroxide complexes and their use in the cure system of anaerobic adhesives |
| WO2010085805A1 (en) | 2009-01-26 | 2010-07-29 | Intermune, Inc. | Methods for treating acute myocardial infarctions and associated disorders |
| US20100197651A1 (en) | 2009-02-05 | 2010-08-05 | Takahiko Taniguchi | Pyridazinone compounds |
| WO2010088177A1 (en) | 2009-02-02 | 2010-08-05 | Merck Sharp & Dohme Corp. | Inhibitors of akt activity |
| US20100222592A1 (en) | 2006-02-02 | 2010-09-02 | Kumiai Chemical Industry Co., Ltd. | Pyridone Derivative and Herbicide |
| WO2010104830A1 (en) | 2009-03-09 | 2010-09-16 | Bristol-Myers Squibb Company | Pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| WO2010104818A1 (en) | 2009-03-09 | 2010-09-16 | Bristol-Myers Squibb Company | Aza pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| US20100233710A1 (en) | 2009-03-16 | 2010-09-16 | Mcdougall Mark | Nucleic acid binding dyes and uses therefor |
| WO2010108652A1 (en) | 2009-03-27 | 2010-09-30 | Ardea Biosciences Inc. | Dihydropyridin sulfonamides and dihydropyridin sulfamides as mek inhibitors |
| US20100267767A1 (en) | 2007-01-22 | 2010-10-21 | Ramesh Narayanan | Nuclear receptor binding agents |
| US20100266717A1 (en) | 2009-04-20 | 2010-10-21 | Marrone Bio Innovations, Inc. | Chemical and biological agents for the control of molluscs |
| WO2010126104A1 (en) | 2009-04-28 | 2010-11-04 | 日産化学工業株式会社 | 4-substituted pyridazinone compound and p2x7 receptor inhibitor |
| WO2010129467A1 (en) | 2009-05-04 | 2010-11-11 | Plexxikon, Inc. | Compounds and methods for inhibition of renin, and indications therefor |
| WO2010135470A1 (en) | 2009-05-19 | 2010-11-25 | Intermune, Inc. | Pifenidone derivatives for treating bronchial asthma |
| US20100305326A1 (en) | 2009-06-02 | 2010-12-02 | Marquette University | Chemical Fragment Screening and Assembly Utilizing Common Chemistry for NMR Probe Introduction and Fragment Linkage |
| WO2010141545A1 (en) | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| WO2010141540A1 (en) | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| WO2010141539A1 (en) | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| WO2010141538A1 (en) | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| US20110009407A1 (en) | 2009-04-27 | 2011-01-13 | Elan Pharmaceuticals, Inc. | Pyridinone antagonists of alpha-4 integrins |
| US20110067612A1 (en) | 2007-08-10 | 2011-03-24 | Kumiai Chemical Industry Co., Ltd. | Pesticidal composition and method for controlling pest |
| US20110224265A1 (en) | 2007-08-14 | 2011-09-15 | Cell Therapy and Technology S.A. DE C.V. | Gel containing pirfenidone |
| US20120014917A1 (en) | 2009-05-15 | 2012-01-19 | University Of Minnesota | Methods of treating hiv patients with anti-fibrotics |
| US20120016133A1 (en) | 2009-06-03 | 2012-01-19 | Intermune, Inc. | Method for Synthesizing Pirfenidone |
| US20120015984A1 (en) | 2005-09-22 | 2012-01-19 | Intermune, Inc. | Granulate Formulation of Pirfenidone and Pharmaceutically Acceptable Excipients |
| US20120077850A1 (en) | 2008-11-10 | 2012-03-29 | Intermune, Inc. | Pirfenidone treatment for patients with atypical liver function |
| US8377932B2 (en) | 2009-05-25 | 2013-02-19 | Central South University | Preparation of 1-(substituted benzyl)-5-trifluoromethyl-2(1H)pyridone compounds and salts thereof and their applications |
| US20150266899A1 (en) | 2012-10-02 | 2015-09-24 | Intermune, Inc. | Anti-fibrotic pyridinones |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1070639C2 (en) | 1958-09-30 | 1964-04-16 | ||
| DE1936231U (en) | 1965-12-06 | 1966-04-07 | Anton Poettgens | CONCRETE RUETTEL BOTTLE. |
| JPS422264Y1 (en) | 1966-08-10 | 1967-02-10 |
-
2009
- 2009-06-03 CN CN200980128312.8A patent/CN102099036B/en not_active Expired - Fee Related
- 2009-06-03 EP EP09759347.9A patent/EP2296653B1/en not_active Not-in-force
- 2009-06-03 CA CA3034994A patent/CA3034994A1/en not_active Abandoned
- 2009-06-03 ES ES09759347.9T patent/ES2567283T3/en active Active
- 2009-06-03 CA CA2726588A patent/CA2726588C/en not_active Expired - Fee Related
- 2009-06-03 WO PCT/US2009/046136 patent/WO2009149188A1/en not_active Ceased
- 2009-06-03 US US12/477,715 patent/US8304413B2/en not_active Ceased
- 2009-06-03 MX MX2010012848A patent/MX2010012848A/en active IP Right Grant
- 2009-06-03 JP JP2011512616A patent/JP5627574B2/en not_active Expired - Fee Related
-
2012
- 2012-10-15 US US13/652,247 patent/US8969347B2/en not_active Expired - Fee Related
-
2014
- 2014-04-28 US US14/263,822 patent/US9290450B2/en not_active Expired - Fee Related
- 2014-08-07 JP JP2014161019A patent/JP2014210809A/en active Pending
- 2014-11-06 US US14/534,450 patent/USRE47142E1/en not_active Expired - Fee Related
Patent Citations (561)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1070639B (en) | 1959-12-10 | Farbenfabriken Bayer Aktiengesellschaft, Levenkusen-Bayerwerk | Process for the preparation of Peptiderj | |
| CH312531A (en) | 1952-06-20 | 1955-12-31 | Ciba Geigy | Process for making a pyridazone. |
| CH312530A (en) | 1952-06-20 | 1955-12-31 | Ciba Geigy | Process for making a pyridazone. |
| GB788393A (en) | 1953-04-30 | 1958-01-02 | Ciba Ltd | Process for the manufacture of pyridazone compounds |
| CH333366A (en) | 1953-09-04 | 1958-10-15 | Ciba Geigy | Process for the production of new pyridazones |
| US3014034A (en) | 1959-01-22 | 1961-12-19 | Ciba Pharm Prod Inc | 1, 3-diaryl, 5-amino-pyridazinones |
| GB889317A (en) | 1959-01-22 | 1962-02-14 | Ciba Ltd | New phenyl-diazines and a process for their manufacture |
| DE1936231A1 (en) | 1968-08-12 | 1970-10-29 | Eastman Kodak Co | Photosensitive silver halide photographic emulsion |
| US3622340A (en) | 1968-08-12 | 1971-11-23 | Eastman Kodak Co | 4-thiouracil compounds as fog inhibitors |
| FR2046068A5 (en) | 1969-05-05 | 1971-03-05 | Eastman Kodak Co | Stabilised photographic emulsions |
| DE2112026A1 (en) | 1970-03-12 | 1971-09-23 | Rhone Poulenc Sa | 3,4-dihydroisoquinoline derivatives, their preparation and the medicinal compositions containing them |
| US3759924A (en) | 1970-03-12 | 1973-09-18 | Rhone Poulenc Sa | 1-amino-3(or4)-phenyl-3,4-dehydroisoquinolines |
| GB1388001A (en) | 1971-08-13 | 1975-03-19 | Thiemann Chem Pharm Fab | 2-pyridones and preparations comprising them |
| DE2143744A1 (en) | 1971-09-01 | 1973-03-08 | Hoechst Ag | 3,4-DIHYDRO-2H-ISOCHINOLIN-1-ONE AND THE METHOD FOR MANUFACTURING IT |
| AU4610372A (en) | 1971-09-01 | 1974-03-07 | Farewehke Hoechst Aktiengesellschaft | 3, 4-dihydro-2h-isoquinoline-1-ones and process for preparing them |
| US3839346A (en) | 1972-12-18 | 1974-10-01 | Affiliated Med Res | N-substituted pyridone and general method for preparing pyridones |
| US3974281A (en) | 1972-12-18 | 1976-08-10 | Affiliated Medical Research, Inc. | 5-Methyl-1-phenyl-2-(1H)-pyridone compositions and methods of use |
| GB1458049A (en) | 1972-12-18 | 1976-12-08 | Affiliated Med Res | Process for preparing aryl-substituted pyridones |
| GB1458048A (en) | 1972-12-18 | 1976-12-08 | Affiliated Med Res | Pharmaceutical composition |
| US4042699A (en) | 1972-12-18 | 1977-08-16 | Affiliated Medical Research, Inc. | Method for reducing serum glucose levels |
| US4052509A (en) | 1972-12-18 | 1977-10-04 | Affiliated Medical Research, Inc. | Method for reducing serum uric acid levels |
| AT333774B (en) | 1974-09-24 | 1976-12-10 | Chemie Linz Ag | PROCESS FOR THE PREPARATION OF 3-PHENYLPYRIDAZONES |
| JPS51128438A (en) | 1975-04-26 | 1976-11-09 | Yamanouchi Pharmaceut Co Ltd | An antibacterial drug against fish diseases |
| DE2557342A1 (en) | 1975-12-19 | 1977-06-30 | Hoechst Ag | BASIC SUBSTITUTED INDOLDER DERIVATIVES AND THE PROCESS FOR THEIR PRODUCTION |
| US4258052A (en) | 1976-08-17 | 1981-03-24 | Yu Ruey J | Treatment of psoriasis with nicotinamide analogues |
| DE2707268A1 (en) | 1977-02-19 | 1978-08-31 | Hoechst Ag | Indole-3-carboxaldehyde oxime derivs. - with hypouricaemic, analgesic, antiinflammatory, hypoglycaemic, cardiovascular and diuretic activity |
| DE2830700A1 (en) | 1977-07-15 | 1979-02-01 | Rohm & Haas | NEW 2-PYRIDON |
| CA1085857A (en) | 1977-07-15 | 1980-09-16 | Glenn R. Carlson | 1-aryl-5-carboxy-2 pyridones and derivatives thereof |
| GB1596887A (en) | 1977-07-15 | 1981-09-03 | Rohm & Haas | 2-pyridone derivatives and their use as plant growth regulators |
| US4256640A (en) | 1978-05-23 | 1981-03-17 | Shionogi & Co., Ltd. | Tetrahydrothiopyrano[2,3-b]indole derivatives |
| US4325863A (en) | 1979-02-05 | 1982-04-20 | Sandoz Ltd. | Benzofuranone or indolinone compounds useful as stabilizers for organic materials |
| DD149666A1 (en) | 1979-06-27 | 1981-07-22 | Karl Gewald | PROCESS FOR THE PREPARATION OF 6-AMINO AND 6-HYDROXY-1-ARYL-5-CYAN-2 (1H) -PYRIDINONE AND THIONES |
| JPS5721388A (en) | 1980-07-11 | 1982-02-04 | Nippon Nohyaku Co Ltd | Condensed pyrazole derivative |
| JPS5777671A (en) | 1980-10-31 | 1982-05-15 | Yasumitsu Tamura | Preparation of hydroxyindoles ( or 1,4-dihydroisoquinolones) and their intermediate |
| US4397854A (en) | 1981-05-14 | 1983-08-09 | Warner-Lambert Company | Substituted 6-phenyl-3(2H)-pyridazinones useful as cardiotonic agents |
| US4404203A (en) | 1981-05-14 | 1983-09-13 | Warner-Lambert Company | Substituted 6-phenyl-3(2H)-pyridazinones useful as cardiotonic agents |
| EP0104860A1 (en) | 1982-09-20 | 1984-04-04 | Pfizer Inc. | 1-Phenyl-2(1H,3H)-indolone psycho-therapeutic agents |
| US4473696A (en) | 1982-10-07 | 1984-09-25 | Ici Americas Inc. | Synthesis of 2-substituted-5-methyl-pyridines |
| US4645839A (en) | 1982-12-17 | 1987-02-24 | Ici Americas Inc. | Sulphur dehydrogenation process to yield 5-methyl-2-pyridone |
| US4650804A (en) | 1984-03-30 | 1987-03-17 | Fujisawa Pharmaceutical Co., Ltd. | Quinolizinone compounds and pharmaceutical composition comprising the same, useful as anti-ulcerative and anti-allergic agents |
| US4698349A (en) | 1984-03-30 | 1987-10-06 | Fujisawa Pharmaceutical Co., Ltd. | Quinolizinone compounds, and pharmaceutical composition comprising the same, useful as anti-ulcerative and anti-allergic agents |
| US4576942A (en) | 1984-07-12 | 1986-03-18 | Usv Pharmaceutical Corp. | Anti-allergic and anti-inflammatory bi- and tri- cyclo-1,4-thiazine derivatives, composition, and method of use therefor |
| EP0241006A2 (en) | 1986-04-10 | 1987-10-14 | E.I. Du Pont De Nemours And Company | 3,3-Disubstituted indolines |
| EP0259048A2 (en) | 1986-09-03 | 1988-03-09 | Imperial Chemical Industries Plc | Chemical compounds |
| US4820309A (en) | 1987-03-05 | 1989-04-11 | Sandoz Ltd. | 1:2-chromium complex dyes for tanned leather |
| JPS63290821A (en) | 1987-05-25 | 1988-11-28 | Otsuka Pharmaceut Co Ltd | Antiarrhythmic |
| EP0311010A2 (en) | 1987-10-06 | 1989-04-12 | The Du Pont Merck Pharmaceutical Company | Alpha,Alpha-disubstituted aromatics and heteroaromatics as cognition enhancers |
| EP0319957A2 (en) | 1987-12-07 | 1989-06-14 | Gluetech Aps | A method for modifying the surface of a polymer |
| US5080710A (en) | 1988-03-05 | 1992-01-14 | Basf Aktiengesellschaft | Novel n-aryltetrahydrophthalimide compounds |
| US4898654A (en) | 1988-05-27 | 1990-02-06 | Otsuka Kagaku Kabushiki Kaisha | Method of producing optically active β-lactam derivatives |
| US5167941A (en) | 1988-11-29 | 1992-12-01 | The Dow Chemical Company | Quaternary polyamines as sulfite oxidation inhibitors in amine scrubbing of SO2 |
| US5019365A (en) | 1988-11-29 | 1991-05-28 | The Dow Chemical Company | Quaternary polyamines as sulfite oxidation inhibitors |
| EP0381374A1 (en) | 1989-01-27 | 1990-08-08 | Les Laboratoires Beecham S.A. | Phosphodiesterase inhibitors |
| EP0393936A1 (en) | 1989-04-18 | 1990-10-24 | Pfizer Inc. | 3-substituted-2-oxindole derivatives as antiinflammatory agents |
| US5077142A (en) | 1989-04-20 | 1991-12-31 | Ricoh Company, Ltd. | Electroluminescent devices |
| EP0409435A1 (en) | 1989-07-07 | 1991-01-23 | Pfizer Inc. | Heteroaryl piperazine antipsychotic agents |
| JPH0343744A (en) | 1989-07-12 | 1991-02-25 | Hitachi Chem Co Ltd | Electrophotographic sensitive body |
| US5310562A (en) | 1989-11-22 | 1994-05-10 | Margolin Solomon B | Composition and method for reparation and prevention of fibrotic lesions |
| US5518729A (en) | 1989-11-22 | 1996-05-21 | Margolin; Solomon B. | Compositions and methods for reparation and prevention of fibrotic lesions |
| US5716632A (en) | 1989-11-22 | 1998-02-10 | Margolin; Solomon B. | Compositions and methods for reparation and prevention of fibrotic lesions |
| WO1991014674A2 (en) | 1990-03-27 | 1991-10-03 | Smithkline Beecham Corporation | 5-lipoxygenase inhibitors |
| EP0478195A1 (en) | 1990-09-21 | 1992-04-01 | Rohm And Haas Company | Dihydropyridazinones, pyridazinones and related compounds as fungicides |
| US5552409A (en) | 1990-09-21 | 1996-09-03 | Rohm And Haas Company | Dihydropyridazinones, pyridazinones and related compounds as fungicides |
| JPH04223457A (en) | 1990-12-26 | 1992-08-13 | Konica Corp | Silver halide photographic sensitive material |
| WO1992013451A1 (en) | 1991-02-11 | 1992-08-20 | Schering Agrochemicals Limited | Imidazole pesticides |
| WO1992020642A1 (en) | 1991-05-10 | 1992-11-26 | Rhone-Poulenc Rorer International (Holdings) Inc. | Bis mono-and bicyclic aryl and heteroaryl compounds which inhibit egf and/or pdgf receptor tyrosine kinase |
| WO1992020816A1 (en) | 1991-05-15 | 1992-11-26 | Yale University | Determination of prodrugs metabolizable by the liver and therapeutic use thereof |
| EP0531578A1 (en) | 1991-09-10 | 1993-03-17 | Agfa-Gevaert N.V. | Thermally transferable fluorescent compounds |
| EP0548680A1 (en) | 1991-12-26 | 1993-06-30 | Mitsubishi Chemical Corporation | Beta-oxo-beta-benzenepropanethioamide derivatives |
| US5241065A (en) | 1992-02-25 | 1993-08-31 | Schering Corporation | 2,3,4,5-tetrahydro-1h-3-benzazepines having anti-psychotic activity |
| WO1993021185A1 (en) | 1992-04-16 | 1993-10-28 | The Du Pont Merck Pharmaceutical Company | Substituted nitrogen containing spiro compounds for use in treating cognitive deficits |
| WO1993023404A1 (en) | 1992-05-19 | 1993-11-25 | Immunopharmaceutics, Inc. | Compounds that modulate endothelin activity |
| US5543521A (en) | 1992-05-19 | 1996-08-06 | Immunopharmaceutics, Inc. | Compounds that modulate endothelin activity |
| US5401738A (en) | 1992-06-13 | 1995-03-28 | Merck Patent Gesellschaft Mit Beschrankter | Benzimidazole compounds |
| US5457099A (en) | 1992-07-02 | 1995-10-10 | Sawai Pharmaceutical Co., Ltd. | Carbostyril derivatives and antiallergic agent |
| EP0579059A1 (en) | 1992-07-02 | 1994-01-19 | Tanabe Seiyaku Co., Ltd. | Pyridazinone derivatives and processes for preparing the same |
| EP0577325A1 (en) | 1992-07-02 | 1994-01-05 | Sawai Pharmaceutical Co., Ltd. | Carbostyril derivatives and antiallergic agent |
| US5356904A (en) | 1992-10-07 | 1994-10-18 | Merck & Co., Inc. | Carbostyril oxytocin receptor antagonists |
| EP0602515A1 (en) | 1992-12-16 | 1994-06-22 | BASF Aktiengesellschaft | Process for the preparation of 5-membered heterocycles |
| WO1994017059A1 (en) | 1993-01-29 | 1994-08-04 | Nippon Soda Co., Ltd. | Heterocyclic derivative |
| JPH06256187A (en) | 1993-03-02 | 1994-09-13 | Fujisawa Pharmaceut Co Ltd | Antitussive expectorant containing qunolizinone compound or its salt |
| JPH08510251A (en) | 1993-05-07 | 1996-10-29 | ビー マーゴリン、ソロモン | Compositions and methods for repair and prevention of fibrotic lesions |
| WO1994026249A1 (en) | 1993-05-07 | 1994-11-24 | Margolin Solomon B | Compositions and methods for reparation and prevention of fibrotic lesions |
| EP0626377A1 (en) | 1993-05-26 | 1994-11-30 | Shionogi & Co., Ltd. | Method for producing benzylidene derivatives |
| DE4423934A1 (en) | 1993-09-02 | 1995-03-09 | Basf Ag | 3(2H)-Pyridazinone derivatives, their preparation and their use |
| EP0648760A2 (en) | 1993-10-15 | 1995-04-19 | Takeda Chemical Industries, Ltd. | Triazine derivative, production and use thereof |
| JPH07128793A (en) | 1993-11-05 | 1995-05-19 | Konica Corp | Fine solid particle dispersion of dye and silver halide photographic sensitive material containing the same |
| US5719155A (en) | 1993-11-10 | 1998-02-17 | Japan Tobacco Inc. | Chroman derivative and pharmaceutical use thereof |
| WO1995016712A1 (en) | 1993-12-15 | 1995-06-22 | Smithkline Beecham Corporation | Compounds and methods |
| JPH07233072A (en) | 1993-12-28 | 1995-09-05 | Tanabe Seiyaku Co Ltd | Pharmaceutical composition |
| WO1995018128A1 (en) | 1993-12-29 | 1995-07-06 | Fujisawa Pharmaceutical Co., Ltd. | Pyrazolopyridine adenosine antagonists |
| JPH07295165A (en) | 1994-04-27 | 1995-11-10 | Konica Corp | Method for processing silver halide photographic sensitive material |
| JPH07295166A (en) | 1994-04-28 | 1995-11-10 | Konica Corp | Method for processing silver halide photographic sensitive material |
| US5466697A (en) | 1994-07-13 | 1995-11-14 | Syntex (U.S.A.) Inc. | 8-phenyl-1,6-naphthyridin-5-ones |
| JPH08134371A (en) | 1994-11-02 | 1996-05-28 | Konica Corp | Dispersion of solid fine particle, silver halide photosensitive material and image forming method |
| WO1996018770A2 (en) | 1994-12-16 | 1996-06-20 | Lignozym Gmbh | Multicomponent system for modifying, decomposing or bleaching lignin, lignin-containing materials or similar substances and method of using this system |
| WO1996027374A1 (en) | 1995-03-03 | 1996-09-12 | Margolin Solomon B | Treatment of cytokine growth factor caused disorders |
| US6114353A (en) | 1995-03-03 | 2000-09-05 | Margolin; Solomon B. | Compositions and method for treatment of lymphomas, leukemias, and leiomyomas |
| US6090822A (en) | 1995-03-03 | 2000-07-18 | Margolin; Solomon B. | Treatment of cytokine growth factor caused disorders |
| JPH11501911A (en) | 1995-03-03 | 1999-02-16 | ビー マーゴリン、ソロモン | Treatment of diseases caused by cytokine growth factors |
| EP0733629A1 (en) | 1995-03-13 | 1996-09-25 | Ishihara Sangyo Kaisha, Ltd. | Pyridonesulfonylurea compounds, process for their production and herbicides containing them |
| EP0738716A2 (en) | 1995-04-21 | 1996-10-23 | Rohm And Haas Company | Dihydropyridazinones and pyridazinones and their use as fungicides and insecticides |
| WO1996033994A1 (en) | 1995-04-28 | 1996-10-31 | Nippon Soda Co., Ltd. | Amino-substituted derivatives, process for the preparation thereof, and herbicide |
| US6551963B1 (en) | 1995-06-06 | 2003-04-22 | Bayer Aktiengesellschaft | Phenylpyridazinones |
| WO1997005109A1 (en) | 1995-07-31 | 1997-02-13 | Novo Nordisk A/S | Heterocyclic compounds, their preparation and use |
| WO1997005137A1 (en) | 1995-07-31 | 1997-02-13 | Novo Nordisk A/S | Heterocyclic compounds, their preparation and use |
| EP0760208A2 (en) | 1995-08-25 | 1997-03-05 | Rohm And Haas Company | Compositions having synergistic fungitoxic effects |
| US5741793A (en) | 1995-08-25 | 1998-04-21 | Rohm And Haas Company | Compositions having synergistic fungitoxic effects |
| US5962478A (en) | 1995-09-19 | 1999-10-05 | Margolin; Solomon B. | Inhibition of tumor necrosis factor α |
| JPH11512699A (en) | 1995-09-19 | 1999-11-02 | ビー マーゴリン、ソロモン | Inhibition of tumor necrosis factor alpha |
| WO1997010712A1 (en) | 1995-09-19 | 1997-03-27 | Margolin Solomon B | Inhibition of tumor necrosis factor alpha |
| US6300349B1 (en) | 1995-09-19 | 2001-10-09 | Solomon B. Margolin | Inhibition of tumor necrosis factor alpha |
| US5808015A (en) | 1995-09-25 | 1998-09-15 | Bayer Aktiengesellschaft | Pyridone-methide azo dyestuffs with improved dyeing and printing properties for hydrophobic synethic fiber materials |
| JPH09249567A (en) | 1996-01-12 | 1997-09-22 | Otsuka Pharmaceut Co Ltd | Medicinal composition |
| US5731106A (en) | 1996-01-25 | 1998-03-24 | Fujitsu Limited | Electrolytic solution for lithium secondary battery and lithium secondary battery using the same |
| WO1997029107A1 (en) | 1996-02-09 | 1997-08-14 | Applied Research Systems Ars Holding N.V. | BENZO[C]QUINOLIZINE DERIVATIVES, THEIR PREPARATION AND USE AS 5α-REDUCTASES INHIBITORS |
| JPH09244235A (en) | 1996-03-14 | 1997-09-19 | Toshiba Corp | Resist for alkaline development |
| US5817700A (en) | 1996-03-29 | 1998-10-06 | Merck Frosst Canada, Inc. | Bisaryl cyclobutenes derivatives as cyclooxygenase inhibitors |
| WO1997036863A1 (en) | 1996-03-29 | 1997-10-09 | Merck Frosst Canada Inc. | Bisarylcyclobutene derivates as cyclooxygenase inhibitors |
| KR20000010676A (en) | 1996-05-09 | 2000-02-25 | 솔로몬 비. 마르골린 | Reparation and prevention of fibrotic lesions |
| WO1997041830A1 (en) | 1996-05-09 | 1997-11-13 | Margolin Solomon B | Reparation and prevention of fibrotic lesions |
| JPH09319023A (en) | 1996-05-27 | 1997-12-12 | Fuji Photo Film Co Ltd | Thermosensitive recording material |
| US5859041A (en) | 1996-06-10 | 1999-01-12 | Merck & Co., Inc. | Substituted imidazoles having cytokine inhibitory activity |
| WO1998013361A1 (en) | 1996-09-26 | 1998-04-02 | Novartis Ag | Herbicidal composition |
| EP0835865A1 (en) | 1996-10-11 | 1998-04-15 | Rohm And Haas Company | Dihydropyridazinones and pyridazinones and their use as fungicides and insecticides |
| WO1999028313A1 (en) | 1996-12-05 | 1999-06-10 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| DE19754348A1 (en) | 1996-12-11 | 1998-06-18 | Ciba Geigy Ag | New 2-(hetero)aryl-2(3H)-pyridazinone derivatives |
| WO1998029119A1 (en) | 1996-12-30 | 1998-07-09 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| EP0856255A2 (en) | 1997-01-30 | 1998-08-05 | Rohm And Haas Company | Pyridazinones as marine antifouling agents |
| US6225052B1 (en) | 1997-02-24 | 2001-05-01 | Roche Diagnostics Gmbh | Method for determining a nucleic acid |
| US6117973A (en) | 1997-02-24 | 2000-09-12 | Georgia Tech Research Corp. | PNA monomers with electron donor or acceptor |
| US6602826B1 (en) | 1997-03-05 | 2003-08-05 | Bayer Aktiengesellschaft | Heterocyclically substituted aromatic amino compounds with a herbicidal effect |
| DE19821263A1 (en) | 1997-05-12 | 1998-11-19 | Call Krimhild | Enzymatic bleach system containing mediator to enhance performance in bleaching textile fabric |
| WO1998051772A1 (en) | 1997-05-12 | 1998-11-19 | Call, Krimhild | Enzymatic bleaching system containing new compounds for intensifying enzymatic action |
| WO1998052948A1 (en) | 1997-05-19 | 1998-11-26 | The Regents Of The University Of California | Compounds for inhibition of ceramide-mediated signal transduction |
| DE19726241A1 (en) | 1997-06-20 | 1998-12-24 | Call Krimhild | Enhanced multi-component enzymatic system for the treatment of waste water, for the production of wood composites, for deinking waste paper, color stripping of waste paper, for use as an oxidation system in organic synthesis and for use in coal liquefaction |
| DE19729061A1 (en) | 1997-07-08 | 1999-01-14 | Agfa Gevaert Ag | Colour photographic material sensitised without increased fogging |
| WO1999002501A1 (en) | 1997-07-09 | 1999-01-21 | Astra Pharmaceuticals Ltd. | Novel compounds |
| WO1999005123A1 (en) | 1997-07-24 | 1999-02-04 | Astrazeneca Uk Limited | Novel pyrimidin derivatives |
| WO1999005125A1 (en) | 1997-07-24 | 1999-02-04 | Bayer Aktiengesellschaft | Substituted n-aryl-n-thioxocarbonyl sulfonamides as herbicides |
| JPH1149755A (en) | 1997-07-30 | 1999-02-23 | Nippon Kayaku Co Ltd | New nitrogen containing heterocyclic derivative and acaricide composition using the same as active component |
| WO1999005913A1 (en) | 1997-08-01 | 1999-02-11 | Applied Research Systems Ars Holding N.V. | Use of benzo[c]quinolizine derivatives as plant growth regulators |
| WO1999010331A1 (en) | 1997-08-22 | 1999-03-04 | Abbott Laboratories | Arylpyridazinones as prostaglandin endoperoxide h synthase biosynthesis inhibitors |
| US6307047B1 (en) | 1997-08-22 | 2001-10-23 | Abbott Laboratories | Prostaglandin endoperoxide H synthase biosynthesis inhibitors |
| WO1999010332A1 (en) | 1997-08-22 | 1999-03-04 | Abbott Laboratories | Prostaglandin endoperoxide h synthase biosynthesis inhibitors |
| WO1999012903A1 (en) | 1997-09-09 | 1999-03-18 | Du Pont Pharmaceuticals Company | Benzimidazolinones, benzoxazolinones, benzopiperazinones, indanones, and derivatives thereof as inhibitors of factor xa |
| WO1999021837A1 (en) | 1997-10-27 | 1999-05-06 | Isk Americas Incorporated | Substituted benzene compounds, process for their preparation, and herbicidal and defoliant compositions containing them |
| WO1999026944A1 (en) | 1997-11-21 | 1999-06-03 | Astrazeneka Uk Limited | New compounds which are p2-purinoceptor 7-transmembrane (tm) g-protein coupled receptor antagonists |
| JPH11180952A (en) | 1997-12-19 | 1999-07-06 | Maruho Co Ltd | 2-oxindole derivative |
| WO1999032448A1 (en) | 1997-12-19 | 1999-07-01 | Amgen Inc. | Substituted pyridine and pyridazine compounds and their pharmaceutical use |
| WO1999038857A2 (en) | 1998-01-30 | 1999-08-05 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Method for producing 5-alkoxy (or 5-aroxy)-2,3-dihydrofuran-2-ones |
| FR2774986A1 (en) | 1998-02-16 | 1999-08-20 | Rhodia Chimie Sa | Masked isocyanates made by reacting an isocyanate starting material with a masking agent having a pyronoid structure and a reactive hydrogen atom |
| WO1999047140A1 (en) | 1998-03-17 | 1999-09-23 | Margolin Solomon B | Topical antiseptic compositions and methods |
| WO1999050263A1 (en) | 1998-03-31 | 1999-10-07 | Warner-Lambert Company | Quinolones as serine protease inhibitors |
| WO1999052878A1 (en) | 1998-04-09 | 1999-10-21 | Bayer Aktiengesellschaft | Substituted phenyl pyridazinones |
| WO1999055676A1 (en) | 1998-04-27 | 1999-11-04 | Centre National De La Recherche Scientifique | 3-(amino- or aminoalkyl)pyridinone derivatives and their use for the treatment of hiv related diseases |
| US6509354B1 (en) | 1998-04-27 | 2003-01-21 | Kumiai Chemical Industry Co., Ltd. | 3-arylphenyl sulfide derivative and insecticide and miticide |
| US6521656B1 (en) | 1998-06-01 | 2003-02-18 | Fujisawa Pharmaceutical Co., Ltd. | Remedies for male sterility |
| WO1999062900A1 (en) | 1998-06-03 | 1999-12-09 | Sanofi-Synthelabo | Oxindole derivatives used as neurokinin receptor antagonists |
| US6265350B1 (en) | 1998-06-16 | 2001-07-24 | Hoechst Schering Agrevo Gmbh | 1,3-oxazoline and 1,3-thiazoline derivatives, their preparation, and their use as pesticides |
| WO2000016775A1 (en) | 1998-09-18 | 2000-03-30 | Mepha Ag | Topical formulation of alkyl-, phenyl-pyridone |
| WO2000025789A1 (en) | 1998-10-29 | 2000-05-11 | Merck & Co., Inc. | A method of treating endometriosis |
| US6174901B1 (en) | 1998-12-18 | 2001-01-16 | Amgen Inc. | Substituted pyridine and pyridazine compounds and methods of use |
| WO2000044381A1 (en) | 1999-01-28 | 2000-08-03 | Margolin Solomon B | Treatment of lymphomas, leukemias, and leiomyomas |
| US6586447B1 (en) | 1999-04-01 | 2003-07-01 | Pfizer Inc | 3,3-disubstituted-oxindole derivatives useful as anticancer agents |
| DE19918725A1 (en) | 1999-04-24 | 2000-10-26 | Bayer Ag | New heterocyclic-substituted N-cyano-sulfonanilide compounds, useful as total or selective pre- or post-emergence herbicides, showing desiccant, defoliant, germination inhibiting and especially weed-killing activities |
| US20040063955A1 (en) | 1999-05-07 | 2004-04-01 | Biediger Ronald J. | Carboxylic acid derivatives that inhibit the binding of integrins to their receptors |
| WO2000068188A1 (en) | 1999-05-07 | 2000-11-16 | Texas Biotechnology Corporation | Propanoic acid derivatives that inhibit the binding of integrins to their receptors |
| US20030199692A1 (en) | 1999-05-07 | 2003-10-23 | Biediger Ronald J. | Propanoic acid derivatives that inhibit the binding of integrins to their receptors |
| WO2001012600A1 (en) | 1999-08-12 | 2001-02-22 | Cor Therapeutics, Inc. | INHIBITORS OF FACTOR Xa |
| FR2797629A1 (en) | 1999-08-19 | 2001-02-23 | Rhodia Chimie Sa | Isocyanate used in polyisocyanate production has a masked isocyanate function(s) susceptible to action by an isocyanate of a derivative of pyrone structure and with active hydrogen |
| US20040014986A1 (en) | 1999-12-27 | 2004-01-22 | Wolfram Hendel | Method for producing oxindoles |
| US20030220368A1 (en) | 2000-01-20 | 2003-11-27 | Fumihiro Ozaki | Novel piperidine compouds and drugs containing the same |
| WO2001057021A2 (en) | 2000-02-01 | 2001-08-09 | Cor Therapeutics, Inc. | 2-[1H]-QUINOLONE AND 2-[1H]-QUINOXALONE INHIBITORS OF FACTOR Xa |
| WO2001057019A1 (en) | 2000-02-01 | 2001-08-09 | Cor Therapeutics, Inc. | INDALONE AND BENZIMIDAZOLONE INHIBITORS OF FACTOR Xa |
| US20020077486A1 (en) | 2000-02-04 | 2002-06-20 | Scarborough Robert M. | Platelet ADP receptor inhibitors |
| WO2001057037A1 (en) | 2000-02-04 | 2001-08-09 | Cor Therapeutics, Inc. | Platelet adp receptor inhibitors |
| WO2001056992A2 (en) | 2000-02-07 | 2001-08-09 | Novartis Ag | Dibenzo (b,f) azepine intermediates |
| WO2001058448A1 (en) | 2000-02-09 | 2001-08-16 | Shionogi & Co., Ltd. | Apoptosis inhibitor |
| WO2001062253A1 (en) | 2000-02-21 | 2001-08-30 | Cymar, Inc. | Compositions and methods for treatment of epilepsy |
| WO2001070746A1 (en) | 2000-03-23 | 2001-09-27 | Takeda Chemical Industries, Ltd. | Furoisoquinoline derivatives, process for producing the same and use thereof |
| WO2001072708A2 (en) | 2000-03-24 | 2001-10-04 | Cor Therapeutics, Inc. | OXINDOLE INHIBITORS OF FACTOR Xa |
| DE10024938A1 (en) | 2000-05-19 | 2001-11-22 | Bayer Ag | New substituted iminoazine derivatives useful as herbicides, especially for weed control in crops |
| US20030194748A1 (en) | 2000-05-29 | 2003-10-16 | Tohru Nagasaki | Method for labeling with tritium |
| US20040023973A1 (en) | 2000-06-12 | 2004-02-05 | Satoshi Nagato | 1,2-Dihydropyridine compounds, process for preparation of the same and use thereof |
| US20050245581A1 (en) | 2000-06-12 | 2005-11-03 | Satoshi Nagato | 1, 2-Dihydropyridine compounds, manufacturing method thereof and use thereof |
| US6949571B2 (en) * | 2000-06-12 | 2005-09-27 | Eisai Co., Ltd. | 1,2-dihydropyridine compounds, process for preparation of the same and use thereof |
| US7939549B2 (en) | 2000-06-12 | 2011-05-10 | Eisai R&D Management Co., Ltd. | 1,2-dihydropyridine compounds, manufacturing method thereof and use thereof |
| WO2001096308A1 (en) | 2000-06-12 | 2001-12-20 | Eisai Co., Ltd. | 1,2-dihydropyridine compounds, process for preparation of the same and use thereof |
| WO2002006244A1 (en) | 2000-07-18 | 2002-01-24 | Bayer Aktiengesellschaft | Heterocyclic substituted herbicidal sulphonic acid anilides |
| US20040006082A1 (en) | 2000-08-11 | 2004-01-08 | Hitoshi Harada | 2-Aminopyridine compounds and use thereof as drugs |
| US6750232B2 (en) | 2000-08-11 | 2004-06-15 | Eisai Co., Ltd. | 2-aminopyridine compounds and use thereof as drugs |
| EP1186318A2 (en) | 2000-09-06 | 2002-03-13 | Pfizer Products Inc. | Combination, for treating depression and anxiety, containing a 5HT1d receptor antagonist and aCNS penetrant NK-1 receptor antagonist |
| WO2002022587A1 (en) | 2000-09-18 | 2002-03-21 | Eisai Co., Ltd. | Pyridazinones and triazinones and medicinal use thereof |
| WO2002024650A2 (en) | 2000-09-19 | 2002-03-28 | Centre National De La Recherche Scientifique (Cnrs) | Pyridinone and pyridinethione derivatives having hiv inhibiting properties |
| CN1349982A (en) | 2000-10-24 | 2002-05-22 | 大连化学工业股份有限公司 | Process for the preparation of lactams |
| WO2002067675A2 (en) | 2000-10-27 | 2002-09-06 | Dow Agrosciences Llc | Substituted 4,5-dihydro-1,2,4-triazin-6-ones, 1,2,4-triazin-6-ones, and their use as fungicides and insecticides |
| EP1213288A1 (en) | 2000-11-06 | 2002-06-12 | Texas Biotechnology Corporation | Propanoic acid derivatives that inhibit the binding of integrins to their receptors |
| WO2002040448A1 (en) | 2000-11-20 | 2002-05-23 | Bristol-Myers Squibb Company | Pyridone derivatives as ap2 inhibitors |
| US20040102494A1 (en) | 2000-12-26 | 2004-05-27 | Dr. Reddy's Laboratories Limited | Novel heterocyclic compounds having antibacterial activity: process for their preparation and pharmaceutical compositions containing them |
| US20030065175A1 (en) | 2000-12-26 | 2003-04-03 | Dr. Reddy's Research Foundation | Novel heterocyclic compounds having antibacterial activity: process for their preparation and pharmaceutical compositions containing them |
| US20040082619A1 (en) | 2000-12-28 | 2004-04-29 | Yukio Tada | Pyridone derivatives having affinity for cannabinoid 2-type receptor |
| WO2002060446A1 (en) | 2001-01-29 | 2002-08-08 | Shionogi & Co., Ltd. | Medicinal preparation containing 5-methyl-1-phenyl-2-(1h)-pyridone as active ingredient |
| US20040048902A1 (en) | 2001-01-29 | 2004-03-11 | Gakuji Kiyonaka | Medicinal preparation containing 5-methyl-1-phenyl-2-(1h)-pyridone as active ingredient |
| US20040259864A1 (en) | 2001-02-23 | 2004-12-23 | Herve Geneste | Substituted pyrimidinone derivatives as ligands of integrin receptors |
| WO2002085858A1 (en) | 2001-04-20 | 2002-10-31 | Asahi Glass Company, Limited | Process for producing purified piperidine derivative |
| WO2002090334A1 (en) | 2001-05-08 | 2002-11-14 | Kudos Pharmaceuticals Limited | Isoquinolinone derivatives as parp inhibitors |
| WO2002098853A2 (en) | 2001-06-01 | 2002-12-12 | The Regents Of The University Of California | Inhibitors of cell proliferation, angiogenesis, fertility, and muscle contraction |
| JP2002371078A (en) | 2001-06-12 | 2002-12-26 | Sankyo Co Ltd | Quinoline derivative and quinolone derivative |
| JP2003012645A (en) | 2001-06-29 | 2003-01-15 | Japan Science & Technology Corp | Method for producing nitrogen-containing 6-membered ring |
| US20030162130A1 (en) | 2001-07-05 | 2003-08-28 | Yasubumi Murota | Process for photopolymerization of photosensitive lithographic printing plate |
| WO2003014087A1 (en) | 2001-08-06 | 2003-02-20 | Asahi Glass Company, Limited | Process for preparation of 5-methyl-1-phenyl-2(1h) -pyridinone |
| WO2003035650A1 (en) | 2001-09-25 | 2003-05-01 | Takeda Chemical Industries, Ltd. | Entry inhibitor |
| WO2003033502A1 (en) | 2001-10-16 | 2003-04-24 | Celltech R & D Limited | Bicyclic oxopyridine and oxopyrimidine derivatives |
| US20050004149A1 (en) | 2001-10-22 | 2005-01-06 | Hitoshi Harada | Pyrimidine compound and medicinal composition thereof |
| US20040259865A1 (en) | 2001-10-22 | 2004-12-23 | Hitoshi Harada | Pyrimidone compounds and pharmaceutical compositions containing the same |
| CN1417209A (en) | 2001-11-08 | 2003-05-14 | 中国科学院上海药物研究所 | A class of tetrahydroquinolinone-piperidine compounds and their preparation and use |
| WO2003045912A1 (en) | 2001-11-29 | 2003-06-05 | Warner-Lambert Company Llc | Inhibitors of factor xa and other serine proteases involved in the coagulation cascade |
| WO2003047577A2 (en) | 2001-12-06 | 2003-06-12 | Eisai Co Ltd | Pharmaceutical compositions comprising dihydropyridinone compounds and an immunoregulatory or an antiinflammatory agent and their uses |
| WO2003047347A1 (en) | 2001-12-07 | 2003-06-12 | Syngenta Participations Ag | Microbiocidal n-phenyl-n-[4-(4-pyridyl)-2-pyrimidin-2-yl]-amine derivatives |
| WO2003059871A1 (en) | 2002-01-09 | 2003-07-24 | Ajinomoto Co.,Inc. | N-alkylsulfonyl-substituted amide derivatives |
| WO2003059891A1 (en) | 2002-01-18 | 2003-07-24 | Pharmacia Corporation | Substituted pyridazinones as inhibitors of p38 |
| US7067540B2 (en) | 2002-02-14 | 2006-06-27 | Pharmacia Corporation | Substituted pyridinones |
| WO2003068230A1 (en) | 2002-02-14 | 2003-08-21 | Pharmacia Corporation | Substituted pyridinones as modulators of p38 map kinase |
| US20040058964A1 (en) | 2002-02-14 | 2004-03-25 | Balekudru Devadas | Substituted pyridinones |
| JP2003238611A (en) | 2002-02-18 | 2003-08-27 | Japan Polyolefins Co Ltd | Catalyst component for olefin polymerization, catalyst for olefin polymerization, and method for producing polyolefin |
| US20050101590A1 (en) | 2002-02-19 | 2005-05-12 | Kiyoshi Yasui | Antipruritics |
| JP2003261535A (en) | 2002-03-08 | 2003-09-19 | Mitsubishi Chemicals Corp | Method for producing 2-hydroxy-5-methylpyridine |
| WO2003076405A1 (en) | 2002-03-14 | 2003-09-18 | Bayer Healthcare Ag | Monocyclic aroylpyridinones as antiinflammatory agents |
| JP2005526068A (en) | 2002-03-14 | 2005-09-02 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | Monocyclic aroylpyridinones as anti-inflammatory agents |
| WO2003082265A2 (en) | 2002-04-02 | 2003-10-09 | Fujisawa Pharmaceutical Co | Pharmaceutical composition for treating or preventing virus infectious diseases |
| WO2003093273A1 (en) | 2002-05-02 | 2003-11-13 | Merck Sharp & Dohme Limited | Imidazo-triazine derivatives as ligands for gaba receptors |
| WO2003097062A1 (en) | 2002-05-13 | 2003-11-27 | Merck & Co., Inc. | Phenyl substituted imidazopyridines and phenyl substituted benzimidazoles |
| CN1386737A (en) | 2002-06-11 | 2002-12-25 | 中南大学湘雅医学院 | Antifibrosis pyridinone medicine and its prepaing process |
| WO2003106452A2 (en) | 2002-06-12 | 2003-12-24 | Millennium Pharmaceuticals, Inc. | Antagonists of melanin concentrating hormone receptor |
| WO2004000355A1 (en) | 2002-06-19 | 2003-12-31 | Pfizer Products Inc. | Combination treatment for depression and anxiety by nk1 and nk3 antagonists |
| WO2004000846A1 (en) | 2002-06-20 | 2003-12-31 | Celltech R & D Limited | Arylamine substututed bicyclic heteroaromatic compounds as p38 kinase inhibitors |
| WO2004005286A2 (en) | 2002-07-03 | 2004-01-15 | K.U.Leuven Research & Development | Viral inhibitors |
| WO2004006906A2 (en) | 2002-07-15 | 2004-01-22 | Combinatorx, Incorporated | Methods for the treatment of neoplasms |
| WO2004009560A1 (en) | 2002-07-22 | 2004-01-29 | Orchid Chemicals & Pharmaceuticals Ltd | Novel bio-active molecules |
| EP1544194A1 (en) | 2002-07-24 | 2005-06-22 | Kyorin Pharmaceutical Co., Ltd. | 4-(substituted aryl)-5-hydroxyisoquinolinone derivative |
| WO2004014859A2 (en) | 2002-08-10 | 2004-02-19 | Tanabe Seiyaku Co., Ltd. | Process for preparing quinolin antibiotic intermediates |
| WO2004014892A1 (en) | 2002-08-13 | 2004-02-19 | Warner-Lambert Company Llc | Monocyclic derivatives as matrix metalloproteinase inhibitors |
| WO2004019863A2 (en) | 2002-08-28 | 2004-03-11 | Intermune, Inc. | Combination therapy for treatment of fibrotic disorders |
| WO2004024078A2 (en) | 2002-09-11 | 2004-03-25 | Merck & Co., Inc. | Dihydroxypyridopyrazine-1,6-dione compounds useful as hiv integrase inhibitors |
| WO2004024152A1 (en) | 2002-09-13 | 2004-03-25 | Mepha Ag | Stable cream preparations of phenyl-pyridone compounds for topical application |
| EP1400243A1 (en) | 2002-09-19 | 2004-03-24 | Tanabe Seiyaku Co., Ltd. | Calcium-activated K channel activator |
| WO2004031188A1 (en) | 2002-10-01 | 2004-04-15 | Celltech R & D Limited | Bicyclic heteroaromatic compounds as kinase inhibitors |
| WO2004031145A2 (en) | 2002-10-02 | 2004-04-15 | Bristol-Myers Squibb Company | Lactam-containing diaminoalkyl, beta-aminoacids, alpha-aminoacids and derivatives thereof as factor xa inhibitors |
| WO2004037159A2 (en) | 2002-10-23 | 2004-05-06 | Obetherapy Biotechnology | Compounds, compositions and methods for modulating fat metabolism |
| US20040097560A1 (en) | 2002-11-09 | 2004-05-20 | The Procter & Gamble Company | N-alkyl-4-methyleneamino-3-hydroxy-2-pyridones |
| WO2004048314A1 (en) | 2002-11-26 | 2004-06-10 | Novartis Ag | Substituted amino phenylacetic acids, derivatives thereof, their preparation and their use as cyclooxygenase 2 (cox-2) inhibitors |
| WO2004058256A1 (en) | 2002-12-26 | 2004-07-15 | Kdl, Inc. | Pharmaceutical liquid composition containing pyridone derivative |
| US20040180906A1 (en) | 2002-12-31 | 2004-09-16 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20070037808A1 (en) | 2002-12-31 | 2007-02-15 | Flynn Daniel L | Anti-inflammatory medicaments |
| WO2004060306A2 (en) | 2002-12-31 | 2004-07-22 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US20040142950A1 (en) | 2003-01-17 | 2004-07-22 | Bunker Amy Mae | Amide and ester matrix metalloproteinase inhibitors |
| US20040235886A1 (en) | 2003-01-31 | 2004-11-25 | Charifson Paul S. | Gyrase inhibitors and uses thereof |
| US20060025424A1 (en) | 2003-01-31 | 2006-02-02 | Paul Charifson | Gyrase inhibitors and uses thereof |
| US20050256136A1 (en) | 2003-01-31 | 2005-11-17 | Charifson Paul S | Gyrase inhibitors and uses thereof |
| US20050038247A1 (en) | 2003-01-31 | 2005-02-17 | Charifson Paul S. | Gyrase inhibitors and uses thereof |
| WO2004072033A2 (en) | 2003-02-12 | 2004-08-26 | Biogen Idec Ma Inc. | Pyrazoles and methods of making and using the same |
| US20040157738A1 (en) | 2003-02-12 | 2004-08-12 | Ishihara Sangyo Kaisha, Ltd. | Novel oxygen containing fused cyclic derivatives and herbicidal, desiccant and defoliate compositions containing them |
| JP2006517976A (en) | 2003-02-14 | 2006-08-03 | スミスクライン・ビーチャム・コーポレイション | New compounds |
| WO2004073628A2 (en) | 2003-02-14 | 2004-09-02 | Smithkline Beecham Corporation | Novel compounds |
| WO2004074282A1 (en) | 2003-02-24 | 2004-09-02 | Pharmacia & Upjohn Company Llc | Antibacterial indolone oxazolidinones, intermediates for their preparation and pharmaceutical compositions containing them |
| WO2005013917A2 (en) | 2003-02-28 | 2005-02-17 | Intermune, Inc. | Combination therapy for treating alphavirus infection and liver fibrosis |
| WO2004078174A1 (en) | 2003-02-28 | 2004-09-16 | Morgan Lee R | Resonance modulator for diagnosis and therapy |
| JP2004269469A (en) | 2003-03-12 | 2004-09-30 | Yamanouchi Pharmaceut Co Ltd | Pyrimidine derivative or salt thereof |
| US20060211577A1 (en) | 2003-04-08 | 2006-09-21 | Basf Aktiengesellschaft | Benzenesulphonamide derivatives as herbicides or desiccant/defoliant compounds |
| JP2004315594A (en) | 2003-04-14 | 2004-11-11 | Konica Minolta Holdings Inc | Polymerization initiator, polymer composition, method for free radical generation, lithographic printing plate material and method for manufacturing lithographic printing plate |
| WO2004103296A2 (en) | 2003-05-16 | 2004-12-02 | Intermune, Inc. | Methods of treating idiopathic pulmonary fibrosis |
| WO2004105684A2 (en) | 2003-05-16 | 2004-12-09 | Intermune, Inc. | Combination therapy for proliferative disorders |
| WO2004110245A2 (en) | 2003-05-16 | 2004-12-23 | Intermune, Inc. | Combination therapy for cancer treatment |
| US20070092488A1 (en) | 2003-05-16 | 2007-04-26 | Intermune Inc. | Methods of treating idiopathic pulmonary fibrosis |
| WO2005000227A2 (en) | 2003-06-06 | 2005-01-06 | Intermune, Inc. | Methods of treating tnf-mediated disorders |
| JP2004359641A (en) | 2003-06-06 | 2004-12-24 | Ono Pharmaceut Co Ltd | CCR5 activator |
| US20060247269A1 (en) | 2003-06-20 | 2006-11-02 | Brookings Daniel C | Thienopyridone derivatives as kinase inhibitors |
| WO2004113347A1 (en) | 2003-06-20 | 2004-12-29 | Celltech R & D Limited | Thienopyridone derivatives as kinase inhibitors |
| US20050153941A1 (en) | 2003-06-27 | 2005-07-14 | Tomoyuki Miyabayashi | Cell differntiation inhibiting agent, cell culture method using the same, culture medium, and cultured cell line |
| WO2005000818A1 (en) | 2003-06-27 | 2005-01-06 | Warner-Lambert Company Llc | 5-substituted-4-`(substituted phenyl)!amino!-2-pyridone deviatives for use as mek inhibitors |
| JP2005013152A (en) | 2003-06-27 | 2005-01-20 | Asahi Kasei Corp | Cell differentiation inhibitor and cell culture method, culture solution and cultured cells using the same |
| WO2005002672A2 (en) | 2003-07-01 | 2005-01-13 | President And Fellows Of Harvard College | Sirt1 modulators for manipulating cells/organism lifespan/stress response |
| US20060025337A1 (en) | 2003-07-01 | 2006-02-02 | President And Fellows Of Harvard College | Sirtuin related therapeutics and diagnostics for neurodegenerative diseases |
| US20060084135A1 (en) | 2003-07-01 | 2006-04-20 | Howitz Konrad T | Compositions for manipulating the lifespan and stress response of cells and organisms |
| US20050020594A1 (en) | 2003-07-18 | 2005-01-27 | Pharmacia Corporation | Substituted pyridazinones |
| WO2005007632A1 (en) | 2003-07-18 | 2005-01-27 | Pharmacia Corporation | Substituted pyridazinones as inhibitors of p38 |
| WO2005012288A1 (en) | 2003-08-01 | 2005-02-10 | Genelabs Technologies, Inc | Bicyclic imidazol derivatives against flaviviridae |
| US20050176775A1 (en) | 2003-08-13 | 2005-08-11 | Balekudru Devadas | Substituted pyridinones |
| WO2005018557A2 (en) | 2003-08-13 | 2005-03-03 | Pharmacia Corporation | Substituted pyridinones |
| US20050054631A1 (en) | 2003-09-04 | 2005-03-10 | Aventis Pharmaceuticals Inc. | Substituted indoles as inhibitors of poly (ADP-ribose) polymerase (PARP) |
| US20050065144A1 (en) | 2003-09-08 | 2005-03-24 | Syrrx, Inc. | Dipeptidyl peptidase inhibitors |
| WO2005026124A1 (en) | 2003-09-18 | 2005-03-24 | Astrazeneca Ab | 2-pyridone derivatives as netrophil elastase inhibitors and their use |
| WO2005026123A1 (en) | 2003-09-18 | 2005-03-24 | Astrazeneca Ab | 2-pyridone derivatives as neutrophil elastase inhibitors and their use |
| DE10345648A1 (en) | 2003-10-01 | 2005-04-21 | Studiengesellschaft Kohle Mbh | Production of new or known tetrahydropyranyl-pyridone compounds, for use as phosphatase inhibitors, e.g. for treating cancer, from pyridinone and pyranone compounds via several new intermediates |
| WO2005040758A2 (en) | 2003-10-24 | 2005-05-06 | Intermune, Inc. | Use of pirfenidone in therapeutic regimens |
| US20070117841A1 (en) | 2003-10-24 | 2007-05-24 | Ozes Osman N | Use of pirfenidone in therapeutic regimens |
| WO2005039598A1 (en) | 2003-10-24 | 2005-05-06 | Intermune, Inc. | Method of treating alcoholic liver disease |
| JP2005145882A (en) | 2003-11-14 | 2005-06-09 | Ichimaru Pharcos Co Ltd | Cosmetic composition |
| US20070049624A1 (en) | 2003-11-14 | 2007-03-01 | Xianghui Yi | Derivatives of pyridone and the use of them |
| US7825133B2 (en) | 2003-11-14 | 2010-11-02 | Shanghai Genomics, Inc. | Derivatives of pyridone and the use of them |
| US20050130976A1 (en) | 2003-11-19 | 2005-06-16 | Eli Wallace | Bicyclic inhibitors of MEK and methods of use thereof |
| US20050250782A1 (en) | 2003-11-19 | 2005-11-10 | Marlow Allison L | Heterocyclic inhibitors of MEK and methods of use thereof |
| US20050153942A1 (en) | 2003-11-19 | 2005-07-14 | Eli Wallace | Heterocyclic inhibitors of MEK and methods of use thereof |
| US20050256123A1 (en) | 2003-11-19 | 2005-11-17 | Marlow Allison L | Heterocyclic inhibitors of MEK and methods of use thereof |
| US20070112038A1 (en) | 2003-11-19 | 2007-05-17 | Marlow Allison L | Heterocyclic inhibitors of MEK and methods of use thereof |
| US20050130943A1 (en) | 2003-11-19 | 2005-06-16 | Eli Wallace | Bicyclic inhibitors of MEK and methods of use thereof |
| US20070105890A1 (en) | 2003-11-26 | 2007-05-10 | Dainippon Sumitomo Pharma Co., Ltd | Novel condensed imidazole derivative |
| WO2005053707A1 (en) | 2003-12-05 | 2005-06-16 | Ono Pharmaceutical Co., Ltd. | Blood flow promoters for cauda equina tissues |
| US20070219182A1 (en) | 2003-12-11 | 2007-09-20 | Wilfried Lubisch | Keto Lactam Compounds and Use Thereof |
| US20050288286A1 (en) | 2003-12-24 | 2005-12-29 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20070191336A1 (en) | 2003-12-24 | 2007-08-16 | Flynn Daniel L | Anti-inflammatory medicaments |
| WO2005073222A1 (en) | 2004-01-29 | 2005-08-11 | Pfizer Japan, Inc. | 1-isopropyl-2-oxo-1,2-dihydropyridine-3-carboxamide derivatives having 5-ht4 receptor agonistic activity |
| WO2005075438A1 (en) | 2004-02-09 | 2005-08-18 | Tanabe Seiyaku Co., Ltd. | Alpha-4 integrin mediated cell adhesion inhibitors for the treatment or prevention of inflammatory diseases |
| JP2005255675A (en) | 2004-02-09 | 2005-09-22 | Tanabe Seiyaku Co Ltd | Pharmaceutical composition |
| US20070037973A1 (en) | 2004-02-20 | 2007-02-15 | Norie Momiyama | Process of making alpha-aminooxyketone/alpha-aminooxyaldehyde and alpha-hydroxyketone/alpha-hydroxyaldehyde compounds and a process making reaction products from cyclic alpha,beta-unsaturated ketone substrates and nitroso substrates |
| WO2005090294A2 (en) | 2004-02-20 | 2005-09-29 | Japan Science And Technology Agency | PROCESS OF MAKING α-AMINOOXYKETONE/α-AMINOOXYALDEHYDE AND α-HYDROXYKETONE/α-HYDROXYALDEHYDE COMPOUNDS AND A PROCESS OF MAKING REACTION PRODUCTS FROM CYCLIC α,ß-UNSATURATED KETONE SUBSTRATES AND NITROSO SUBSTRATES |
| WO2005085200A1 (en) | 2004-03-05 | 2005-09-15 | Banyu Pharmaceutical Co., Ltd. | Pyridone derivative |
| WO2005097750A1 (en) | 2004-03-30 | 2005-10-20 | Aventis Pharmaceuticals Inc. | Substituted pyridones as inhibitors of poly(adp-ribose) polymerase (parp) |
| US20070179165A1 (en) | 2004-04-07 | 2007-08-02 | Gyorkos Albert C | Cyclic compounds |
| WO2005099688A2 (en) | 2004-04-07 | 2005-10-27 | Takeda Pharmaceutical Company Limited | Cyclic compounds |
| WO2005096784A2 (en) | 2004-04-08 | 2005-10-20 | Targegen, Inc. | Benzotriazine inhibitors of kinases |
| WO2005105743A1 (en) | 2004-04-28 | 2005-11-10 | Ono Pharmaceutical Co., Ltd. | Nitrogen-containing heterocyclic compounds and medicinal use thereof |
| US20080081825A1 (en) | 2004-04-28 | 2008-04-03 | Hisao Nakai | Nitrogen-Containing Heterocyclic Compounds and Medicinal Use Thereof |
| WO2005105790A1 (en) | 2004-04-28 | 2005-11-10 | Tanabe Seiyaku Co., Ltd. | 4- 2- (cycloalkylamino) pyrimidin-4-yl ! - (phenyl) - imidazolin-2- one derivatives as p38 map- kinase inhibitors for the treatment of inflammatory diseases |
| WO2005123687A1 (en) | 2004-06-16 | 2005-12-29 | Sanofi-Aventis Deutschland Gmbh | Substituted tetrahydro-2h-isoquinolin-1-one derivatives, method for the production thereof, and use of the same as medicaments |
| US20110021462A1 (en) | 2004-06-16 | 2011-01-27 | Sanofi-Aventis Deutschland Gmbh | Substituted tetrahydro-2h-isoquinolin-1-one derivatives, and methods for the production and use thereof |
| US20060100193A1 (en) | 2004-06-18 | 2006-05-11 | Millennium Pharmaceuticals, Inc. | Factor Xa inhibitors |
| US20070185092A1 (en) | 2004-06-18 | 2007-08-09 | Millennium Pharmaceuticals, Inc. | FACTOR Xa INHIBITORS |
| WO2006004107A1 (en) | 2004-07-06 | 2006-01-12 | Eisai R & D Management Co., Ltd. | Crystal of 1,2-dihydropyridine compound and method for producing same |
| US20070142640A1 (en) | 2004-07-06 | 2007-06-21 | Eisai R&D Management Co., Ltd. | Method for producing 1, 2-dihydropyridine-2-one compound |
| WO2006011024A2 (en) | 2004-07-19 | 2006-02-02 | Glenmark Pharmaceuticals Ltd. | New tricyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation and pharmaceutical compositions containing them |
| WO2006020145A2 (en) | 2004-07-19 | 2006-02-23 | The Johns Hopkins University | Flt3 inhibitors for immune suppression |
| WO2006017443A2 (en) | 2004-08-02 | 2006-02-16 | Osi Pharmaceuticals, Inc. | Aryl-amino substituted pyrrolopyrimidine multi-kinase inhibiting compounds |
| WO2006026305A1 (en) | 2004-08-31 | 2006-03-09 | Biogen Idec Ma Inc | Pyrimidinylpyrazoles as tgf-beta inhibitors |
| WO2006030032A1 (en) | 2004-09-17 | 2006-03-23 | Janssen Pharmaceutica N.V. | Novel pyridinone derivatives and their use as positive allosteric modulators of mglur2-receptors |
| WO2006032631A1 (en) | 2004-09-22 | 2006-03-30 | Janssen Pharmaceutica N.V. | Inhibitors of the interaction between mdm2 and p53 |
| US20060069260A1 (en) | 2004-09-28 | 2006-03-30 | Huiping Zhang | Preparation of N-aryl pyridones |
| WO2006038734A1 (en) | 2004-10-08 | 2006-04-13 | Astellas Pharma Inc. | Pyridazinone derivatives cytokines inhibitors |
| WO2006044405A1 (en) | 2004-10-12 | 2006-04-27 | Decode Genetics, Inc. | Aryl sulfonamide peri-substituted bicyclics for occlusive artery disease |
| WO2006046778A1 (en) | 2004-10-28 | 2006-05-04 | Shionogi & Co., Ltd. | 3-carbamoyl-2-pyridone derivative |
| US20080103139A1 (en) | 2004-10-28 | 2008-05-01 | Natsuki Ishizuka | 3-Carbamoyl-2-Pyridone Derivative |
| US20070265308A1 (en) | 2004-11-10 | 2007-11-15 | Ono Pharmaceutical Co., Ltd. | Nitrogenous Heterocyclic Compound and Pharmaceutical Use Thereof |
| JP2006142666A (en) | 2004-11-19 | 2006-06-08 | Mitsubishi Chemicals Corp | Dye for optical recording media |
| WO2006055918A2 (en) | 2004-11-19 | 2006-05-26 | Pharmacopeia Drug Discovery, Inc. | One-dimensional qsar models |
| US20060111355A1 (en) | 2004-11-23 | 2006-05-25 | Wyeth | Gonadotropin releasing hormone receptor antagonists |
| WO2006056427A1 (en) | 2004-11-24 | 2006-06-01 | Laboratoires Serono S.A. | Novel 4-arylamino pyridone derivatives as mek inhibitors for the treatment of hyperproliferative disorders |
| US20060160862A1 (en) | 2004-11-24 | 2006-07-20 | Jean-Damien Charrier | Caspase inhibitors and uses thereof |
| WO2006060122A2 (en) | 2004-11-30 | 2006-06-08 | Artesian Therapeutics, Inc. | Cardiotonic compounds with inhibitory activity against beta-adrenergic receptors and phosphodiesterase |
| US20080269287A1 (en) | 2004-12-01 | 2008-10-30 | Norikazu Ohtake | Substituted Pyridone Derivative |
| WO2006066079A2 (en) | 2004-12-17 | 2006-06-22 | Anadys Pharmaceuticals, Inc. | Pyridazinone compounds |
| US20080280930A1 (en) | 2004-12-23 | 2008-11-13 | Hui Yao | Pyrimidinone Compounds and Preparation and Use Thereof |
| WO2006072039A1 (en) | 2004-12-29 | 2006-07-06 | E. I. Du Pont De Nemours And Company | Nepetalactams and n-substituted derivatives thereof |
| WO2006072037A1 (en) | 2004-12-29 | 2006-07-06 | E. I. Du Pont De Nemours And Company | Dihydronepetalactams and n-substituted derivatives thereof |
| WO2006076681A2 (en) | 2005-01-13 | 2006-07-20 | Sirtris Pharmaceuticals, Inc. | Novel compositions for preventing and treating neurodegenerative and blood coagulation disorders |
| WO2006079021A2 (en) | 2005-01-20 | 2006-07-27 | Sirtris Pharmaceuticals, Inc. | Use of sirtuin-activating compounds for treating flushing and drug induced weight gain |
| US20060189617A1 (en) | 2005-02-18 | 2006-08-24 | Wyeth | Imidazo[4,5-b]pyridine antagonists of gonadotropin releasing hormone receptor |
| US20060189616A1 (en) | 2005-02-18 | 2006-08-24 | Wyeth | 7-Substituted imidazo[4,5-c]pyridine antagonists of gonadotropin releasing hormone receptor |
| CN1676518A (en) | 2005-03-17 | 2005-10-05 | 南开大学 | 4-Substituted phenylpyridazines and their herbicidal activity |
| WO2006107859A2 (en) | 2005-04-04 | 2006-10-12 | Eisai Co., Ltd. | Dihydropyridine compounds for neurodegenerative diseases and dementia |
| WO2006107860A2 (en) | 2005-04-04 | 2006-10-12 | Eisai Co., Ltd. | Dihydropyridine compounds and compositions for headaches |
| US20090030017A1 (en) | 2005-04-08 | 2009-01-29 | Eisai R & D Management Co., Ltd | Therapeutic agent for dyskinesia |
| WO2006108354A1 (en) | 2005-04-13 | 2006-10-19 | Xiangya Hospital Of Central South University | 1-(substituted phenyl)-5- methyl- 2 - (1h) pyridone in the manufacture of medicaments for treating fibrosis in organs or tissues |
| CA2603763A1 (en) | 2005-04-13 | 2006-10-19 | Xiangya Hospital Of Central South University | 5-methyl-1-(substituted phenyl)-2-(1h)-pyridone in the manufacture of medicaments for treating fibrosis in organ or tissues |
| WO2006116713A1 (en) | 2005-04-27 | 2006-11-02 | Amgen Inc. | Substituted amide derivatives as protein kinase inhibitors |
| US20060258861A1 (en) | 2005-04-28 | 2006-11-16 | Boehringer Ingelheim International Gmbh | Compounds for the treatment of inflammatory diseases |
| US9527816B2 (en) | 2005-05-10 | 2016-12-27 | Intermune, Inc. | Method of modulating stress-activated protein kinase system |
| US8741936B2 (en) | 2005-05-10 | 2014-06-03 | Intermune, Inc. | Method of modulating stress-activated protein kinase system |
| US20140228310A1 (en) | 2005-05-10 | 2014-08-14 | Intermune, Inc. | Method of modulating stress-activated protein kinase system |
| US7728013B2 (en) | 2005-05-10 | 2010-06-01 | Intermune, Inc. | Method of modulating stress-activated protein kinase system |
| WO2006122154A2 (en) | 2005-05-10 | 2006-11-16 | Intermune, Inc. | Pyridone derivatives for modulating stress-activated protein kinase system |
| US20120258924A1 (en) | 2005-05-10 | 2012-10-11 | Intermune, Inc. | Method of modulating stress-activated protein kinase system |
| US20100240704A1 (en) | 2005-05-10 | 2010-09-23 | Blatt Lawrence M | Method of modulating stress-activated protein kinase system |
| US20060270612A1 (en) | 2005-05-10 | 2006-11-30 | Blatt Lawrence M | Method of modulating stress-activated protein kinase system |
| WO2006129076A1 (en) | 2005-06-01 | 2006-12-07 | Sterix Limited | Use of a steroid sulphatase inhibitor for inhibiting the synthesis of androstenedione and/or testosterone |
| US20070037822A1 (en) | 2005-06-07 | 2007-02-15 | Pharmacopeia Drug Discovery, Inc. | Azinone and diazinone V3 inhibitors for depression and stress disorders |
| WO2006133147A2 (en) | 2005-06-08 | 2006-12-14 | Novartis Ag | Organic compounds |
| WO2006131186A1 (en) | 2005-06-10 | 2006-12-14 | Merck Patent Gmbh | Oxindoles as kinase inhibitors |
| WO2006138418A2 (en) | 2005-06-14 | 2006-12-28 | President And Fellows Of Harvard College | Improvement of cognitive performance with sirtuin activators |
| US20060287319A1 (en) | 2005-06-15 | 2006-12-21 | Shibo Jiang | Anti-viral compositions comprising heterocyclic substituted phenyl furans and related compounds |
| WO2007008548A2 (en) | 2005-07-07 | 2007-01-18 | Sirtris Pharmaceuticals, Inc. | Methods and related compositions for treating or preventing obesity, insulin resistance disorders, and mitochondrial-associated disorders |
| WO2007006591A2 (en) | 2005-07-13 | 2007-01-18 | Bayer Cropscience Sa | Dihalogenation of n,o-disubstituted hydroxipyridones and uses thereof |
| JP2007063268A (en) | 2005-08-05 | 2007-03-15 | Tanabe Seiyaku Co Ltd | Pharmaceutical composition |
| WO2007024021A1 (en) | 2005-08-26 | 2007-03-01 | Fuji Film Corporation | Impregnating oil composition for sintered bearing, bearing apparatus and sliding member |
| WO2007026950A1 (en) | 2005-09-01 | 2007-03-08 | Astellas Pharma Inc. | Pyridazinone derivatives used for the treatment of pain |
| US20120015984A1 (en) | 2005-09-22 | 2012-01-19 | Intermune, Inc. | Granulate Formulation of Pirfenidone and Pharmaceutically Acceptable Excipients |
| WO2007037543A1 (en) | 2005-09-29 | 2007-04-05 | Banyu Pharmaceutical Co., Ltd. | Biarylamide derivative |
| US20100063104A1 (en) | 2005-10-03 | 2010-03-11 | Ono Pharmaceutical Co., Ltd., | Nitrogen-containing heterocyclic compound and pharmaceutical application thereof |
| WO2007044796A2 (en) | 2005-10-11 | 2007-04-19 | Nps Pharmaceuticals, Inc. | Pyridazinone compounds as calcilytics |
| WO2007048065A2 (en) | 2005-10-21 | 2007-04-26 | Exelixis, Inc. | Pyrimidinones as casein kinase ii (ck2) modulators |
| JP2007145819A (en) | 2005-10-28 | 2007-06-14 | Tanabe Seiyaku Co Ltd | Pharmaceutical composition |
| WO2007053685A2 (en) | 2005-11-01 | 2007-05-10 | Intermune, Inc. | Methods of treating atrial fibrillation with p38 inhibitor compounds |
| US20120046321A1 (en) | 2005-11-01 | 2012-02-23 | Intermune, Inc. | Methods of treating atrial fibrillation with p38 inhibitor compounds |
| WO2007057329A1 (en) | 2005-11-18 | 2007-05-24 | F. Hoffmann-La Roche Ag | Azaindole-2-carboxamide derivatives |
| WO2007058392A1 (en) | 2005-11-21 | 2007-05-24 | Japan Tobacco Inc. | Heterocyclic compound and medicinal application thereof |
| WO2007062167A2 (en) | 2005-11-23 | 2007-05-31 | Intermune, Inc. | Method of modulating stress-activated protein kinase system |
| US20110034495A1 (en) | 2005-11-23 | 2011-02-10 | Intermune, Inc. | Method of Modulating Stress-Activated Protein Kinase System |
| WO2007064797A2 (en) | 2005-11-30 | 2007-06-07 | Vertex Pharmaceuticals Incorporated | Inhibitors of c-met and uses thereof |
| US20090088574A1 (en) | 2005-12-21 | 2009-04-02 | Eisai R&D Management Co., Ltd. | Crystal of 1,2-dihydropyridine compound (type iv) |
| US20080312284A1 (en) | 2005-12-21 | 2008-12-18 | Eisai R&D Management Co., Ltd. | Amorphous Form of 1,2-Dihydropyridine Compound |
| US20070149513A1 (en) | 2005-12-23 | 2007-06-28 | Ning Chen | Vanilloid receptor ligands and their use in treatments |
| WO2007088996A1 (en) | 2006-01-31 | 2007-08-09 | Mitsubishi Tanabe Pharma Corporation | Trisubstituted amine compounds as inhibitors of -cholesteryl ester transfer protein cetp |
| US20090029994A1 (en) | 2006-01-31 | 2009-01-29 | Mitsubishi Tanabe Pharma Corporation | Trisubstituted amine compound |
| US20100222592A1 (en) | 2006-02-02 | 2010-09-02 | Kumiai Chemical Industry Co., Ltd. | Pyridone Derivative and Herbicide |
| WO2007100990A2 (en) | 2006-02-24 | 2007-09-07 | Abbott Laboratories | Octahydro-pyrrolo[3,4-b] pyrrole derivatives and their use as histamine-3 receptor ligands |
| WO2007117778A2 (en) | 2006-02-24 | 2007-10-18 | Kalypsys, Inc. | Quinolones useful as inducible nitric oxide synthase inhibitors |
| WO2007104034A2 (en) | 2006-03-08 | 2007-09-13 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2007108968A2 (en) | 2006-03-13 | 2007-09-27 | Merck & Co., Inc. | Ophthalmic compositions for treating ocular hypertension |
| CN1817862A (en) | 2006-03-15 | 2006-08-16 | 浙江省医学科学院 | Production of pyriphenanthrenone as anti-fibrosis medicine |
| WO2007107545A1 (en) | 2006-03-22 | 2007-09-27 | Janssen Pharmaceutica N.V. | Cyclic-alkylaminederivatives as inhibitors of the interaction between mdm2 and p53 |
| WO2007117482A2 (en) | 2006-04-05 | 2007-10-18 | Vitae Pharmaceuticals, Inc. | Renin inhibitors |
| WO2007117559A2 (en) | 2006-04-05 | 2007-10-18 | Vitae Pharmaceuticals, Inc. | Renin inhibitors |
| WO2007120842A2 (en) | 2006-04-13 | 2007-10-25 | Cornell Research Foundation, Inc. | Methods and compositions for targeting c-rel |
| WO2007127474A2 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Compositions and treatments using pyridazine compounds and cholinesterase inhibitors |
| WO2007127475A2 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Pyridazines for demyelinating diseases and neuropathic pain |
| WO2007129040A1 (en) | 2006-05-04 | 2007-11-15 | Chroma Therapeutics Ltd. | p38 MAP KINASE INHIBITORS |
| US20070259924A1 (en) | 2006-05-05 | 2007-11-08 | Millennium Pharmaceuticals, Inc. | Factor xa inhibitors |
| WO2007139150A1 (en) | 2006-05-30 | 2007-12-06 | The University Of Tokushima | ANTI-INFLUENZA VIRAL AGENT COMPRISING TNF-α INHIBITOR |
| WO2007146712A2 (en) | 2006-06-09 | 2007-12-21 | Kemia, Inc. | Therapy using cytokine inhibitors |
| US20080161361A1 (en) | 2006-06-15 | 2008-07-03 | Jun Wu | Use of pyridone derivatives in the prevention or treatment of tissue or organ toxicity induced by cytotoxic agents and radiation |
| WO2007147297A1 (en) | 2006-06-15 | 2007-12-27 | Shanghai Genomics, Inc. | The use of derviate of pyridone for preventing and treating radioactive injury of lungs |
| WO2007148093A1 (en) | 2006-06-22 | 2007-12-27 | Prolysis Ltd. | Antibacterial compositions |
| WO2008002490A2 (en) | 2006-06-23 | 2008-01-03 | Radius Health, Inc. | Treatment of vasomotor symptoms with selective estrogen receptor modulators |
| US20080020010A1 (en) | 2006-07-19 | 2008-01-24 | The University Of Georgia Research Foundation, Inc. | Pyridinone diketo acids: inhibitors of HIV replication |
| US20080249090A1 (en) | 2006-07-20 | 2008-10-09 | Amgen Inc. | Substituted pyridone compounds and methods of use |
| WO2008013838A2 (en) | 2006-07-25 | 2008-01-31 | Cephalon, Inc. | Pyridizinone derivatives |
| JP2008039883A (en) | 2006-08-02 | 2008-02-21 | Konica Minolta Opto Inc | Optical film, polarizing plate using the same and liquid crystal display |
| WO2008016239A1 (en) | 2006-08-02 | 2008-02-07 | Lg Life Sciences Ltd. | Caspase inhibitors based on pyridazinone scaffold |
| US20080255083A1 (en) | 2006-08-25 | 2008-10-16 | Dirk Stenkamp | New pyridone derivates with mch antagonistic activity and medicaments comprising these compounds |
| CN1927923A (en) | 2006-09-06 | 2007-03-14 | 北京理工大学 | Ion liquid, non-aqueous liquid and polymer electrolyte material based on the same |
| US20080139557A1 (en) | 2006-09-11 | 2008-06-12 | Blomgren Peter A | Certain substituted amides, method of making, and method of use thereof |
| JP2008076948A (en) | 2006-09-25 | 2008-04-03 | Konica Minolta Medical & Graphic Inc | Photosensitive lithographic printing plate material |
| US20080114033A1 (en) | 2006-11-08 | 2008-05-15 | Bristol-Myers Squibb Company | Pyridinone compounds |
| KR20080045538A (en) | 2006-11-20 | 2008-05-23 | 에스케이케미칼주식회사 | Pharmaceutical composition for the treatment of inflammation and immune disease, including pyridine compound |
| CN1962642A (en) | 2006-11-21 | 2007-05-16 | 南开大学 | Trifluoromethylphenylpyridazine derivative and its preparation method |
| WO2008065428A2 (en) | 2006-12-01 | 2008-06-05 | Sterix Limited | Steroid sulphatase inhibitors for treating hormone dependent cancer |
| WO2008068265A1 (en) | 2006-12-05 | 2008-06-12 | Janssen Pharmaceutica N.V. | Novel substituted diaza spiro pyridinone derivatives for use in mch-1 mediated diseases |
| WO2008072784A1 (en) | 2006-12-14 | 2008-06-19 | Astellas Pharma Inc. | Polycyclic acid compounds useful as crth2 antagonists and antiallergic agents |
| WO2008076562A1 (en) | 2006-12-14 | 2008-06-26 | Eli Lilly And Company | 5- [4- (azetidin-3-yl0xy) -phenyl] -2-phenyl-5h-thiaz0l0 [5,4-c] pyridin-4-0ne derivatives and their use as mch receptor antagonists |
| WO2008079787A2 (en) | 2006-12-20 | 2008-07-03 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2008080056A2 (en) | 2006-12-21 | 2008-07-03 | Sloan-Kettering Institute For Cancer Research | Pyridazinones and furan-containing compounds |
| WO2008079277A1 (en) | 2006-12-22 | 2008-07-03 | Millennium Pharmaceuticals, Inc. | Certain pyrazoline derivatives with kinase inhibitory activity |
| WO2008077550A1 (en) | 2006-12-27 | 2008-07-03 | Sanofi-Aventis | Substituted isoquinoline and isoquinolinone derivatives as inhibitors of rho-kinase |
| WO2008086188A2 (en) | 2007-01-05 | 2008-07-17 | Millennium Pharmaceuticals, Inc. | Thiophene carboxamides as factor xa inhibitors |
| US20090030036A1 (en) | 2007-01-22 | 2009-01-29 | Dalton James T | Nuclear receptor binding agents |
| US20100267767A1 (en) | 2007-01-22 | 2010-10-21 | Ramesh Narayanan | Nuclear receptor binding agents |
| WO2008091555A2 (en) | 2007-01-22 | 2008-07-31 | Gtx, Inc. | Nuclear receptor binding agents |
| CN101235030A (en) | 2007-01-30 | 2008-08-06 | 中南大学 | 1-substituted-5-trifluoromethyl-2-(1H)pyridone compound, preparation method and use thereof |
| WO2008103277A2 (en) | 2007-02-16 | 2008-08-28 | Amgen Inc. | Nitrogen-containing heterocyclyl ketones and their use as c-met inhibitors |
| WO2008099000A2 (en) | 2007-02-16 | 2008-08-21 | Boehringer Ingelheim International Gmbh | Substituted arylsulphonylglycines, the preparation thereof and the use thereof as pharmaceutical compositions |
| US20080280917A1 (en) | 2007-02-16 | 2008-11-13 | Amgen Inc. | Nitrogen-Containing heterocyclyl ketones and methods of use |
| WO2008106202A1 (en) | 2007-02-27 | 2008-09-04 | Housey Gerard M | Theramutein modulators |
| JP2008214225A (en) | 2007-03-01 | 2008-09-18 | Tosoh Corp | Process for producing 5,5-disubstituted-3-pyrrolin-2-one derivatives |
| WO2008107480A1 (en) | 2007-03-07 | 2008-09-12 | Ortho-Mcneil-Janssen Pharmaceuticals, Inc | 1,4-disubstituted 3-cyano-pyridone derivatives and their use as positive mglur2 -receptor modulators |
| WO2008112715A2 (en) | 2007-03-12 | 2008-09-18 | Vm Discovery Inc. | Novel agents of calcium ion channel modulators |
| WO2008110793A1 (en) | 2007-03-12 | 2008-09-18 | Biolipox Ab | Piperidinones useful in the treatment of inflammation |
| US20090170861A1 (en) | 2007-03-15 | 2009-07-02 | Schering Corporation And Albany Molecular Research, Inc. | Pyridazinone Derivatives Useful as Glucan Synthase Inhibitors |
| US20080234332A1 (en) | 2007-03-20 | 2008-09-25 | Xiong Cai | Raf kinase inhibitors containing a zinc binding moiety |
| WO2008121407A1 (en) | 2007-03-30 | 2008-10-09 | The Regents Of The University Of California | In vivo imaging of sulfotransferases |
| WO2008121877A2 (en) | 2007-04-02 | 2008-10-09 | Institute For Oneworld Health | Cftr inhibitor compounds and uses thereof |
| WO2008124323A1 (en) | 2007-04-03 | 2008-10-16 | Array Biopharma Inc. | Imidazo[1,2-a]pyridine compounds as receptor tyrosine kinase inhibitors |
| WO2008124575A1 (en) | 2007-04-05 | 2008-10-16 | Smithkline Beecham Corporation | Renin inhibitors |
| WO2008124582A1 (en) | 2007-04-05 | 2008-10-16 | Smithkline Beecham Corporation | Renin inhibitors |
| WO2008132155A2 (en) | 2007-04-26 | 2008-11-06 | Janssen Pharmaceutica Nv | Inhibitors of the binding between hdm2 and the proteasome |
| WO2008140066A2 (en) | 2007-05-03 | 2008-11-20 | Astellas Pharma Inc. | Pyridone derivatives as p38a mapk inhibitors |
| WO2008141119A2 (en) | 2007-05-09 | 2008-11-20 | Vertex Pharmaceuticals Incorporated | Modulators of cftr |
| WO2008144720A2 (en) | 2007-05-21 | 2008-11-27 | University Of Washington | Inhibitors of igf-1r signaling for the treatment of respiratory disorders |
| WO2008147169A2 (en) | 2007-05-29 | 2008-12-04 | Cell Therapy Technology, S.A. De C.V. | Microemulsion containing pirfenidone |
| WO2008147170A1 (en) | 2007-05-29 | 2008-12-04 | Cell Therapy Technology, S.A. De C.V. | New process of synthesis for obtaining 5-methyl-1-phenyl-2 (ih) -pyridone, composition and use of the same |
| US20080306093A1 (en) | 2007-06-08 | 2008-12-11 | Senomyx, Inc. | Modulation of chemosensory receptors and ligands associated therewith |
| US20080306053A1 (en) | 2007-06-08 | 2008-12-11 | Senomyx, Inc. | Modulation of chemosensory receptors and ligands associated therewith |
| WO2008157786A1 (en) | 2007-06-20 | 2008-12-24 | Auspex Pharmaceutical, Inc. | Substituted n-aryl pyridinones as fibrotic inhibitors |
| US20080319026A1 (en) | 2007-06-20 | 2008-12-25 | Auspex Pharmaceuticals, Inc. | Substituted n-aryl pyridinones |
| WO2008155069A2 (en) | 2007-06-20 | 2008-12-24 | Bayer Schering Pharma Aktiengesellschaft | Substituted oxazolidinones and use thereof |
| US20100298293A1 (en) | 2007-06-20 | 2010-11-25 | Bayer Schering Pharma Atiengesellschaft | Substituted oxazolidinones and their use |
| WO2009011410A1 (en) | 2007-07-13 | 2009-01-22 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists and aldose reductase inhibitors for neuropathic pain |
| WO2009011411A1 (en) | 2007-07-13 | 2009-01-22 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists and zonisamide for neuropathic pain |
| WO2009011412A2 (en) | 2007-07-13 | 2009-01-22 | Eisai R & D Management Co., Ltd. | Combination of ampa receptor antagonists and acetylcholinesterase inhibitors for the treatment of neuropathic pain |
| US20090023702A1 (en) | 2007-07-17 | 2009-01-22 | Bristol-Myers Squibb Company | Pyridone gpr119 g protein-coupled receptor agonists |
| WO2009012275A1 (en) | 2007-07-17 | 2009-01-22 | Bristol-Myers Squibb Company | Pyridone gpr119 g protein-coupled receptor agonists |
| US20090042919A1 (en) | 2007-07-17 | 2009-02-12 | Bristol-Myers Squibb Company | Method for modulating gpr119 g protein-coupled receptor and selected compounds |
| WO2009016118A1 (en) | 2007-07-27 | 2009-02-05 | Boehringer Ingelheim International Gmbh | Substituted arylsulfonylaminomethylphosphonic acid derivatives, their preparation and their use in the treatment of type i and ii diabetes mellitus |
| US20110067612A1 (en) | 2007-08-10 | 2011-03-24 | Kumiai Chemical Industry Co., Ltd. | Pesticidal composition and method for controlling pest |
| US20110224265A1 (en) | 2007-08-14 | 2011-09-15 | Cell Therapy and Technology S.A. DE C.V. | Gel containing pirfenidone |
| WO2009026816A1 (en) | 2007-08-23 | 2009-03-05 | Central South University | The application of 1-aryl-2(1h)-pyridone compounds in preparation of medincines for treating itching of the skin |
| WO2009029625A1 (en) | 2007-08-27 | 2009-03-05 | Kalypsys, Inc. | 4- [heterocyclyl-methyl] -8-fluoro-quinolin-2-ones useful as nitric oxide synthase inhibitors |
| US20090131485A1 (en) | 2007-09-10 | 2009-05-21 | Concert Pharmaceuticals, Inc. | Deuterated pirfenidone |
| WO2009035598A1 (en) | 2007-09-10 | 2009-03-19 | Concert Pharmaceuticals, Inc. | Deuterated pirfenidone |
| WO2009033703A1 (en) | 2007-09-14 | 2009-03-19 | Ortho-Mcneil-Janssen Pharmaceuticals, Inc. | 1,3-disubstituted-4-phenyl-1 h-pyridin-2-ones |
| WO2009039773A1 (en) | 2007-09-19 | 2009-04-02 | Central South University | New medicine use of 1-substituted aryl -2(1h)-pyridone |
| WO2009054543A1 (en) | 2007-10-26 | 2009-04-30 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists and zonisamide for parkinson's disease and movement disorders |
| WO2009054544A1 (en) | 2007-10-26 | 2009-04-30 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists for parkinson's disease and movement disorders |
| WO2009057827A1 (en) | 2007-10-31 | 2009-05-07 | Nissan Chemical Industries, Ltd. | Pyridazinone derivatives and use thereof as p2x7 receptor inhibitors |
| WO2009060835A1 (en) | 2007-11-05 | 2009-05-14 | Kyoto University | Novel ubiquilin-binding small molecule |
| WO2009065922A2 (en) | 2007-11-22 | 2009-05-28 | Boehringer Ingelheim International Gmbh | Novel compounds |
| US20120149698A1 (en) | 2007-11-22 | 2012-06-14 | Boehringer Ingelheim International Gmbh | Nouvel compounds |
| WO2009073620A2 (en) | 2007-11-30 | 2009-06-11 | Newlink Genetics | Ido inhibitors |
| WO2009076529A1 (en) | 2007-12-11 | 2009-06-18 | Research Development Foundation | Small molecules for neuronal differentiation of embryonic stem cells |
| WO2009074810A1 (en) | 2007-12-13 | 2009-06-18 | Prolysis Ltd | Antibacterial compositions |
| WO2009082039A1 (en) | 2007-12-26 | 2009-07-02 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists for epilepsy, mental disorders or deficits in sensory organ |
| WO2009082038A2 (en) | 2007-12-26 | 2009-07-02 | Eisai R & D Management Co., Ltd. | Ampa receptor antagonists and zonisamide for epilepsy |
| WO2009094427A1 (en) | 2008-01-23 | 2009-07-30 | Bristol-Myers Squibb Company | 4-pyridinone compounds and their use for cancer |
| WO2009097309A1 (en) | 2008-01-30 | 2009-08-06 | Cephalon, Inc. | Substituted spirocyclic piperidine derivatives as histamine-3 (h3) receptor ligands |
| WO2009108499A1 (en) | 2008-02-25 | 2009-09-03 | Merck & Co., Inc. | Tetrahydro-1h-pyrrolo fused pyridones |
| WO2009111785A2 (en) | 2008-03-07 | 2009-09-11 | Solanan, Inc. | Treatment of sepsis with 5-ethyl-1-phenyl-2(1h)-pyridone and novel methods for synthesis |
| US20090247566A1 (en) | 2008-03-25 | 2009-10-01 | Alexander Kornienko | Pyrano [3,2-C] Pyridones and Related Heterocyclic Compounds as Pharmaceutical Agents for Treating Disorders Responsive to Apoptosis, Antiproliferation or Vascular Disruption, and the Use Thereof |
| US20090246198A1 (en) | 2008-03-31 | 2009-10-01 | Takeda Pharmaceutical Company Limited | Mapk/erk kinase inhibitors and methods of use thereof |
| WO2009124119A2 (en) | 2008-04-01 | 2009-10-08 | The Trustes of Columbia University in the City of New York | Phosphodiesterase inhibitors and uses thereof |
| WO2009124553A2 (en) | 2008-04-09 | 2009-10-15 | Neurokey A/S | Use of hypothermia inducing drugs |
| WO2009135299A1 (en) | 2008-05-05 | 2009-11-12 | Merck Frosst Canada Ltd. | 3, 4 - substituted piperidine derivatives as renin inhibitors |
| WO2009142732A2 (en) | 2008-05-20 | 2009-11-26 | Cephalon, Inc. | Substituted pyridazinone derivatives as histamine-3 (h3) receptor ligands |
| US8969347B2 (en) | 2008-06-03 | 2015-03-03 | Intermune, Inc. | Compounds and methods for treating inflammatory and fibrotic disorders |
| WO2009149188A1 (en) | 2008-06-03 | 2009-12-10 | Intermune, Inc. | Compounds and methods for treating inflammatory and fibrotic disorders |
| US20090318455A1 (en) | 2008-06-03 | 2009-12-24 | Intermune, Inc. | Compounds and methods for treating inflammatory and fibrotic disorders |
| US9290450B2 (en) | 2008-06-03 | 2016-03-22 | Intermune, Inc. | Compounds and methods for treating inflammatory and fibrotic disorders |
| US8304413B2 (en) | 2008-06-03 | 2012-11-06 | Intermune, Inc. | Compounds and methods for treating inflammatory and fibrotic disorders |
| US20140235637A1 (en) | 2008-06-03 | 2014-08-21 | Intermune, Inc. | Compounds and methods for treating inflammatory and fibrotic disorders |
| US20130102597A1 (en) | 2008-06-03 | 2013-04-25 | Intermune, Inc. | Compounds and methods for treating inflammatory and fibrotic disorders |
| US20100137317A1 (en) | 2008-06-25 | 2010-06-03 | Envivo Pharmaceuticals, Inc. | 1,2-Disubstituted Heterocyclic Compounds |
| WO2009158393A1 (en) | 2008-06-25 | 2009-12-30 | Envivo Pharmaceuticals, Inc. | 1, 2 disubstituted heterocyclic compounds |
| WO2009158473A1 (en) | 2008-06-25 | 2009-12-30 | Envivo Pharmaceuticals, Inc. | 5- and 6-membered heterocyclic compounds |
| WO2009156484A2 (en) | 2008-06-27 | 2009-12-30 | Novartis Ag | Organic compounds |
| WO2010006191A1 (en) | 2008-07-11 | 2010-01-14 | Irm Llc | 4-phenoxymethylpiperidines as modulators of gpr119 activity |
| WO2010009183A1 (en) | 2008-07-16 | 2010-01-21 | Bristol-Myers Squibb Company | Pyridone and pyridazone analogues as gpr119 modulators |
| WO2010027567A2 (en) | 2008-07-23 | 2010-03-11 | Schering Corporation | Tricyclic spirocycle derivatives and methods of use thereof |
| WO2010021693A2 (en) | 2008-08-18 | 2010-02-25 | Yale University | Mif modulators |
| WO2010025087A1 (en) | 2008-08-25 | 2010-03-04 | Smithkline Beecham Corporation | Prolyl hydroxylase inhibitors |
| WO2010029299A1 (en) | 2008-09-12 | 2010-03-18 | Biolipox Ab | Pyrimidinone derivaties for use as medicaments |
| WO2010044885A2 (en) | 2008-10-17 | 2010-04-22 | Whitehead Institute For Biomedical Research | Soluble mtor complexes and modulators thereof |
| US20100120862A1 (en) | 2008-10-24 | 2010-05-13 | Laykea Tafesse | Monocyclic compounds and their use as trpv1 ligands |
| WO2010048716A1 (en) | 2008-10-29 | 2010-05-06 | Pacific Therapeutics Ltd. | Composition and method for treating fibrosis |
| US20120077850A1 (en) | 2008-11-10 | 2012-03-29 | Intermune, Inc. | Pirfenidone treatment for patients with atypical liver function |
| WO2010065755A1 (en) | 2008-12-04 | 2010-06-10 | Concert Pharmaceuticals, Inc. | Deuterated pyridinones |
| WO2010077680A2 (en) | 2008-12-08 | 2010-07-08 | Vm Discovery Inc. | Compositions of protein receptor tyrosine kinase inhibitors |
| WO2010066028A1 (en) | 2008-12-10 | 2010-06-17 | Merck Frosst Canada Ltd. | 3,4 - substituted piperidine derivatives as renin inhibitors |
| WO2010068253A1 (en) | 2008-12-11 | 2010-06-17 | Shionogi & Co., Ltd. | Synthesis of carbamoylpyridone hiv integrase inhibitors and intermediates |
| WO2010075561A1 (en) | 2008-12-23 | 2010-07-01 | President And Fellows Of Harvard College | Small molecule inhibitors of necroptosis |
| WO2010080795A2 (en) | 2009-01-07 | 2010-07-15 | Henkel Corporation | Hydrogen peroxide complexes and their use in the cure system of anaerobic adhesives |
| US20100190731A1 (en) | 2009-01-26 | 2010-07-29 | Jeff Olgin | Methods for treating acute myocardial infarctions and associated disorders |
| WO2010085805A1 (en) | 2009-01-26 | 2010-07-29 | Intermune, Inc. | Methods for treating acute myocardial infarctions and associated disorders |
| WO2010088177A1 (en) | 2009-02-02 | 2010-08-05 | Merck Sharp & Dohme Corp. | Inhibitors of akt activity |
| US20100197651A1 (en) | 2009-02-05 | 2010-08-05 | Takahiko Taniguchi | Pyridazinone compounds |
| WO2010104830A1 (en) | 2009-03-09 | 2010-09-16 | Bristol-Myers Squibb Company | Pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| WO2010104818A1 (en) | 2009-03-09 | 2010-09-16 | Bristol-Myers Squibb Company | Aza pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| US20100233710A1 (en) | 2009-03-16 | 2010-09-16 | Mcdougall Mark | Nucleic acid binding dyes and uses therefor |
| WO2010108652A1 (en) | 2009-03-27 | 2010-09-30 | Ardea Biosciences Inc. | Dihydropyridin sulfonamides and dihydropyridin sulfamides as mek inhibitors |
| US20100266717A1 (en) | 2009-04-20 | 2010-10-21 | Marrone Bio Innovations, Inc. | Chemical and biological agents for the control of molluscs |
| US20110009407A1 (en) | 2009-04-27 | 2011-01-13 | Elan Pharmaceuticals, Inc. | Pyridinone antagonists of alpha-4 integrins |
| WO2010126104A1 (en) | 2009-04-28 | 2010-11-04 | 日産化学工業株式会社 | 4-substituted pyridazinone compound and p2x7 receptor inhibitor |
| WO2010129467A1 (en) | 2009-05-04 | 2010-11-11 | Plexxikon, Inc. | Compounds and methods for inhibition of renin, and indications therefor |
| US20120014917A1 (en) | 2009-05-15 | 2012-01-19 | University Of Minnesota | Methods of treating hiv patients with anti-fibrotics |
| WO2010135470A1 (en) | 2009-05-19 | 2010-11-25 | Intermune, Inc. | Pifenidone derivatives for treating bronchial asthma |
| US8377932B2 (en) | 2009-05-25 | 2013-02-19 | Central South University | Preparation of 1-(substituted benzyl)-5-trifluoromethyl-2(1H)pyridone compounds and salts thereof and their applications |
| US20100305326A1 (en) | 2009-06-02 | 2010-12-02 | Marquette University | Chemical Fragment Screening and Assembly Utilizing Common Chemistry for NMR Probe Introduction and Fragment Linkage |
| US20120016133A1 (en) | 2009-06-03 | 2012-01-19 | Intermune, Inc. | Method for Synthesizing Pirfenidone |
| WO2010141545A1 (en) | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| WO2010141540A1 (en) | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| WO2010141539A1 (en) | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| WO2010141538A1 (en) | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| US20150266899A1 (en) | 2012-10-02 | 2015-09-24 | Intermune, Inc. | Anti-fibrotic pyridinones |
| US20160263090A1 (en) | 2012-10-02 | 2016-09-15 | Intermune, Inc. | Anti-fibrotic pyridinones |
Non-Patent Citations (187)
| Title |
|---|
| Altman et al., Orthogonal Pd-and Cu-based catalyst systems for C- and N-arylation of oxindoles, J Am Chem Soc. (Jul. 2008) 130(29): 9613-9620. |
| Ammar et al., Cyanoacetanilides Intermediates in Heterocyclic Syntheses. Part 1: A Facile Synthesis of Polysubstituted and Condensed Pyridones. J Chinese Chem Society (2004) 51: 975-981. |
| Ammar et al., Novel Pirfenidone Analogues: Synthesis of Pyridin-2-ones for the Treatment of Pulmonary Fibrosis, Arch Pharm Chem Life Sci., (Apr. 2006) 339(8): 429-426. |
| Anonymous, Verfahren zur Hestellung von 6-Arylyridazinon-3 Verbindungen, (Mar. 1999), Research Disclosure No. 311123, The Industry Standard Disclosure Publication Service, Questel Ireland Ltd., pp. 1-5. |
| Azuma et al., A placebo control and double blind phase II clinical study of pirfenidone in patients with idiopathic pulmanory fibrosis in Japan, Am J Respir Crit Care Med., (2002) 165: A729. |
| Badger et al., Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J Pharmacol Exp Ther., (Dec. 1996) 279(3): 1453-1461. |
| Barluenga et al., Easy Preparation of 2-Methyl-1,3-Dimorpholino-1,3-Butadiene and an Overview of Its Synthetic Applications. Tetrahed Lttrs. (1995) 36(36): 6551-6554. |
| Beccalli et al., Pd-catalyzed intramolecular cyclization of pyrrolo-2carboxamindes: regiodivergent routes to pyrrolo-pyrazines and pyrrolo-pyridines. Tetrahedron (2005) 61: 1077-1082. |
| Boehm et al., New Inhibitors of p38 Kinase. Expert Opin Ther Pat (2000), 10(1): 25-37. |
| Border et al., Transforming growth factor beta in tissue fibrosis, N Engl J Med., (1994) 331(19): 1286-1292. |
| Brinkman et al., Engagement of Tumor Necrosis Factor (TNF) Receptor 1 Leads to ATF-2- and p38 Mitogen-activated Protein Kinase-dependent TNF-alpha Gene Expression, J Biol Chem., (Oct. 1999) 274(43): 20882-30886. |
| Buysens et al., Intramolecular Diels-Alder Reactions of 2(1H)-Pyrazinones: Synthesis of New Furo/Pyrano-pyridinones and -pyridines. Tetrahedron (1995) 51(45): 12463-12478. |
| Cambpell et al., A Novel Mechanism for TNF-alpha Regulation by p38 MAPK: Involvement of NF-kB with Implications for Therapy in Rheumatoid Arthritis, J Immunol., (2004) 173(11): 6928-6937. |
| Canadian Office Action dated Jul. 13, 2015 for Application No. 2726588, filed Jun. 3, 2009. |
| CAS Database Accession No. 2000:723577, "Electroluminescent devices"; Daigo Aoki ; Dai Nippon Printing Co., Ltd., Japan; XP-002756228; 1 pages. |
| CAS Database Accession No. 2004:117844, "4-Aryl-5-hydroxyisoquinolinones, their preparation, and poly(ADP-ribose) polymerase inhibitors containing them", Asano et al.; Jpn. Kokai Tokkyo Koho, Japan; XP-002756230; 15 pages. |
| CAS Database Accession No. 2004:252487, "Preparation of 4-(substituted aryl)-5-hydroxyisoquinolinone derivatives aas poly(ADP-ribose) polymerase inhibitors" Shiga et al., Kyorin Pharmaceutical Co., Ltd. Japan; XP-002756231; 30 pages. |
| CAS Registry No. 1011358-02-3, STN Entry Date: Apr. 1, 2008, 1-cycloproyl-N-[2-(diethylamino)ethyl]-1,2,5,6,7,8-hexahydro-4-hydroxy-2-oxo-3quinolinecarboxamide, 1 page. |
| CAS Registry No. 131521-89-6, STN Entry Date: Jan. 18, 1991, 2,7-Naphthyridine-4-carboxamide, 1,2,5,6,7,8-hexohydro-1,6,8-trioxo-N,2,7-triphenyl, 2 pages. |
| CAS Registry No. 586387-14-6, STN Entry Date: Sep. 16, 2003, 1-Benzyl-4-(benzylthio)-5-methylpyridin-2(1H)-one, 1 page. |
| Chemical Abstracts Services; CAS Registry No. 102718-41-2; 3,4-Pyridinedicarboxylic acid, 1,6--dihydro-1-(2-hydroxyphenyl)-6-oxo-, 3,4-dimethyl ester; Entered: Jun. 14, 1986; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1073610-66-8; 2(1H)-Pyridinone, 1-(3-chlorophenyl)-5- methyl; Entered: Nov. 21, 2008; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1076199-03-5; 2(1H)-Pyridinone, 1-[4-(phenylmethoxy) phenyl]; Entered Nov. 26, 2008; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1099106-04-3; 2(1H)-Pyridinone, 1-[3-(difluoromethoxy) phenyl]-5-methyl; Entered: Feb. 1, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1099106-08-7; 2(1H)-Pyridinone, 5-methyl-1-[3-(trifluoromethoxy) phenyl]; Entered: Feb. 1, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1099106-10-1; 2(1H)-Pyridinone, 5-ethyl-1-[trifluoromethoxy)pheyl]; Entered: Feb. 1, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-10-4; 4-Pyridinecarbonitrile, 1-[4-chloro-2-fluoro-5-[(1-methyl-2-propyn-1-yl) oxy] phenyl]-1,2-dihydro-2-oxo; Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-78-4; Acetic adid, 2-[2-chloro-4-fluoro-5-[5-methyl-2-oxo-4-(trifluoromethyl)-1(2H)-pyridinyl]phenoxy', ethyl ester; Entered Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-80-8; 2(1H)-Pyridinone, 1-[4-chloro-2-fluoro-5-[C1-methyl-2-propyn-1-yl) oxy] phenyl]-5-methyl-4-(trifluoromethyl); Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-81-9; 2(1H)-Pyridinone, 1-[4-chloro-2-fluoro--5-(2-propen-1-yloxy)phenyl]-5-methyl-4-(trifluoromethyl); Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-83-1; 2(1H)-Pyridinone, 5-chloro-1-[4-chloro-2-fluoro-5-(1-methylethoxy) phenyl]-4-(trifluoromethyl); Entered Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-84-2; 2(1H)-Pyridinone, 5-chloro-1-[4-chloro-2-fluoro-5-[1-methyl-2-propyn-1yl) oxy] phenyl]-4-(trifluoromethyl); Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-85-3; 2(1H)-Pyridinone, 5-chloro-1-[4-chloro-2-fluoro-5-(2-propen-1-yloxy) phenyl]-4-(trifluoromethyl); Entered Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-86-4; Propanoic acid, 2-[2-chloro-5p[5-chloro-2-oxo-4-(trifluoromethyl)-1 (2H)-pyridinyl]-4-fluorophenoxy], ethyl ester; Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-90-0; 2(1H)-Pyridinone, 1-[4-chloro-2-fluoro-5-(1-methylethoxy) phenyl]-4-(trifluoromethyl); Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-91-1; 2(1H)-Pyridinone, 1-[4-chloro-2-fluoro-5-(2-propen-1-yloxy) phenyl]-4-(trifluoromethyl); Entered: Feb. 10, 2006; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139505-92-2; 2(1H)-Pyridinone, 1-(4-chloro-2-fluoro-5-[(1-methyl-2-propyn-1-yl) oxy] phenyl]-4-(trifluoromethyl); Entered Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139506-08-3; Acetic Acid, 2-[2-chloro-4-fluoro-5-[2-oxo-4-(trifluoromethyl)-1 (2H)-pyridinyl] phenoxyl], methyl ester; Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139506-09-4; Acetic acid, 2-{2-chloro-4-5-[2-oxo-4-(trifluoromethyl)-1 (2H)-phyridinyl] phenoxy], ethyl ester; Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139506-10-7; Acetic acid, 2-[2-chloro-4-fluoro-5-[2-oxo-4-(trifluoromethyl)-1 (2H)-pyridinyl] phenoxy], propyl ester; Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139506-11-8; Acetic acid, 2-[2-chloro-4-fluoro-5-[2-oxo-4-(trifluoromethyl)-1 (2H)-pyridinyl' phenoxyl', 1-methylethyl ester; Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139506-12-9; Propanoic acid, 2-[2-chloro-4-fluoro-5-[2-oxo-4-(trifluoromethyl)-1 (2H)-pyridinyl] phenoxy], methyl ester; Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139506-13-0; Propanoic acid, 2-[2-chloro-4-fluoro-5-[2-oxo-4-(trifluoromethyl)-1 (2H)-pyridinyl] phenoxyl], ethyl ester; Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139506-14-1; 2(1H)-Pyridinone, 1-(4-chloro-2-fluoro-5-(methoxymethoxy) phenyl]-4-(trifluoromethyl); Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139506-15-2; 2(1H)-Pyridinone, 1-(4-chloro-2-fluoro-5-hydroxyphenyl)-4-(trifluoromethyl); Entered Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139506-16-3; 2(1H)-Pyridinone, 1-(4-chloro-2-fluoro-5-phenoxyphenyl)-4-(trifluoromethyl); Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1139506-17-4; 2(1H)-Pyridinone, 1-[4-chloro-2-fluoro-5-(2-fluoropheoxy)pheyl]-4-(trifluoromethyl); Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 115551-60-5; 2(1H)-Pyridinone, 1-(2,3,4,5,6-pentafluorophenyl)-4-(trifluoromethyl); Entered: Jul. 30, 1988; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 115551-94-5; 2(1H)-Pyridinone, 1-(4-chloro-2-fluoro-5-hydroxyphenyl)-4-(trifluoromethyl); Entered Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 1239505-79-5; Propanoic acid, 2-[2-chloro-4-fluoro-5-[5-methyl-2-oxo-4-(trifluoromethyl)-1 (2H) -pyridinyl ]phenoxy], ethyl ester; Entered: Apr. 27, 2009; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 130879-34-4; 3,4-Pyridinedicarboxylic acid, 5-chloro-1, 6-dihydro-1-(4-methoxyphenyl)-6-0×0, 3,4-dimethyl ester; Entered: Dec. 7, 1990; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 13179-26-5; 2(1H)-Pyridinone, 1-(4-chlorophenyl); Entered: Nov. 16, 1984; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 145705-06-2; 2(1H)-Pyridinone, 1-(2-fluoro-4-hydroxyphenyl)-4-(trifluoromethyl); Entered: Feb. 4, 1993; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 145705-07-3; 2(1H)-Pyridinone, 1-(2-fluoro-4-hydroxyphenyl)-5-methyl-4-(trifluoromethyl); Entered: Feb. 4, 1993; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 145705-09-5; 2(1H)-Pyridinone, 5-chloro-1-(2-fluoro-4-hydroxyphenyl)-4-(trifluoromethyl phenyl]; Entered: Feb. 4, 1993; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 145705-10-8; 2(1H)-Pyridinone, 5-bromo-1-(2-fluoro-4-hydroxyphenyl)-4-(trifluoromethyl); Entered: Feb. 4, 1993; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 145705-11-9; 2(1H)-Pyridinone, 5-ETHYL-1-(2-fluoro-4hydroxyphenyl)-4-(trifluoromethyl); Entered: Feb. 4, 1993; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 222978-30-5; 2(1H)-Pyridinone, 1-(2-methoxyphenyl); Entered: May 14, 1999; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 222978-31-6; 2(1H)-Pyridinone, 1-(2-hydroxyphenyl); Entered: May 14, 1999; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 3512-19-4; 2(1H)-Pyridinone, 1-(3-chloropheyl); Entered: Nov. 16, 1984; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 3512-20-7; 2(1H)-Pyridinone, 1-(3-bromophenyl); Entered: Nov. 16, 1984; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 3534-60-9; 2(1H)-Pyridinone, 1-(3-hydroxyphenyl); Entered: Nov. 16, 1984; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 53427-77-3; 2(1H)-Pyridinone, 1-(4-methoxyphenyl)-5-methyl; Entered: Nov. 16, 1984; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 53427-80-8; 2(1H)-Pyridinone, 1-(4-chlorophenyl)-5-methyl; Entered: Nov. 16, 1984; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 60532-42-5; 2(1H)-Pyridinone, 1-(4-fluoropheyl); Entered: Nov. 16, 1984; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-34-8; 2(1H)-Pyridinone, 1-(3-methoxyphenyl); Entered: Aug. 11, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-35-9; 2(1H)-Pyridinone, 1-(3-methoxyphenyl)-5-methyl; Entered: Aug. 11, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-36-0; 2(1H)-Pyridinone, 5-chloro-1-(4-methoxyphenyl); Entered: Aug. 11, 2004; American Chemical Society; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-37-1; 3-Pyridinecarboxylic acid, 1,6-dihydro-1-(3-methoxyphenyl)-6-oxo, ethyl ester; Entered: Aug. 11, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-38-2; 2(1H)-Pyridinone, 5-chloro-1-(3-methoxyphenyl); Entered: Aug. 11, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-40-6; 2(1H)-Pyridinone, 1-(4-methoxyphenyl); Entered: Aug. 11, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-41-7; 2(1H)-Pyridinone, 5-methoxy-1-(4-methoxyphenyl); Entered: Aug. 11, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-42-8; 3-Pyridinecarbosylic acid, 1,6-dihydro-1-(4-methoxyphenyl)-64-oxo-, ethyl ester; Entered: Aug. 11, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-43-9; 2(1h)-Pyridinone, 5-chloro-1-(4-methoxyphenyl); Entered: Aug. 11, 2004; American Chemical Society; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-50-8; 2(1H)-Pyridinone, 1-(4-chlorophenyl)-5-methoxy; Entered: Aug. 11, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-51-9; 3-Pyridinecarboxylic acid, 1-(4-chlorophenyl)-1,6-dihydro-6-oxo-, ethyl ester; Entered: Aug. 11, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 725256-52-0; 2(1H)-Pyridinone, 5-chloro-1-(4-chlorophenyl); Entered: Aug. 11, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 766556-75-6; 2(1H)-Pyridinone, 1-(4-iodophenyl); Entered: Oct. 21, 2004; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 77095-38-6; 3-Pyridinecarboxylic acid, 5-bromo-1-(5-bromophenyl)-1,6-dihydro-4-methyl-6-oxo; Entered: Nov. 16, 1984; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 848353-85-5; 2(1H)-Pyridinone, 1-(3-fluorophenyl)-5-methyl; Entered: Apr. 12, 2005; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 851518-71-3; 2(1H)-Pyridinone, 1-(4-hydoxyphenyl)-5-methyl; Entered: Jun. 2, 2005; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 859538-51-5; 2(1H)-Pyridinone, 1-(4-hydroxyphenyl); Entered Aug. 11, 2005; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 873969-21-2; 2(1H)-Pyridinone, 1-(2,5-dimethoxyphenyl); Entered: Feb. 10, 2006; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 873969-22-3; 2(1H)-Pyridinone, 1-(2,3-dimethoxyphenyl); Entered: Feb. 10, 2006; 1 page. |
| Chemical Abstracts Services; CAS Registry No. 912570-13-9; 2(1H)-Pyridinone, 1-(3-bromophenyl)-5-methyl; Entered: Nov. 7, 2006; 1 page. |
| CHEMICAL ABSTRACTS, 12 February 2004, Columbus, Ohio, US; ASANO JUN, SHIGA FUTOSHI, TAKANO YASUO, ISHIYAMA JUNICHI: "4-Aryl-5- hydroxyisoquinolinones, their preparation, and poly(ADP-ribose) polymerase inhibitors containing them" XP002756230 |
| CHEMICAL ABSTRACTS, 13 October 2000, Columbus, Ohio, US; AOKI DAIGO: "Electroluminescent devices" XP002756228 |
| CHEMICAL ABSTRACTS, 25 March 2004, Columbus, Ohio, US; SHIGA FUTOSHI, KANDA TAKAHIRO, TAKANO YASUO, ISHIYAMA JUNICHI: "Preparation of 4-(substituted aryl)-5-hydroxyisoquinolinone derivatives as poly(ADP-ribose) polymerase inhibitors" XP002756231 |
| Chinese Office Action dated Aug. 7, 2014 for Application No. 200980128312.8, filed Jun. 3, 2009. |
| Chinese Office Action dated Feb. 5, 2013 for Application No. 200980128312.8, filed Jun. 3, 2009. |
| Chinese Office Action dated Jun. 6, 2012 for Application No. 200980128312.8, filed Jun. 3, 2009. |
| Clive et al., "Studies related to furopyridinone antibiotics. Synthesis of 2-epi-CJ-16,170", Tetrahedron (2002) 58: 10243-10250. |
| Dong et al., MAP kinases in the immune response, Annu Rev Immunol., (2002) 20: 55-72. |
| Eiden et al., 6-Styryl-4-methoxy-2-pyridon-Derivate. Archiv der Pharmazie & Berichte der Deutschen Pharmazeutischen Gesellschaft (1971) 304(10): 723-729. |
| Elbashir et al., Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells, Nature (2001) 411(6836): 494-498. |
| English et al., Pharmacological inhibitors of MAPK pathways, Trends Pharmacol Sci., (2002) 23(1): 40-45. |
| Erian et al., Beta-Enaminonitriles in heterocyclic synthesis; Pyridines, pyrimidines and pyrazoles as antibacterial agents, Scientia Pharmaceutica (Dec. 1999) 67(4): 253-261. |
| European Extended Search Report dated Feb. 6, 2012 for Application No. 09759347.9, filed Jun. 3, 2009. |
| European Office Action dated Dec. 9, 2013 for Application No. 09759347.9, filed Jun. 3, 2009. |
| European Office Action dated Oct. 10, 2012 for Application No. 09759347.9, filed Jun. 3, 2009. |
| European Office Action dated Sep. 18, 2014 for Application No. 09759347.9, filed Jun. 3, 2009. |
| European Patent Application No. 09759347.9 Search Report dated Feb. 6, 2012. |
| Faid-Allah, H. "Synthesis of pyridones and dihydropyridines from pyrylium salts and nitriles from aldimines" Croatica Chemica Acta, 1987, 60(4), 717-733. * |
| Fitzgerald et al., Structural basis for p38alpha MAP kinase quinazolinone and pyridol-pyrimidine inhibitor specificity, Nat Struct Biol (2003) 10(9): 764-769. |
| Fuchs et al., Stability of the ATF2 Transcription Factor is Regulated by Phosphorylation and Dephosphorylation, J Biol Chem., (Apr. 2000), 275(17): 12560-12564. |
| Furukawa et al., p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts, Hepatology (2003) 38(4): 879-889. |
| Gahl et al., Effect of pirfenidone on the pulmonary fibrosis of Hermansky-Pudlak syndrome, Mol Genet Metab., (Jul. 2002) 76(3): 234-242. |
| Giri et al., Pharmacokinetics and Metabolism of a Novel Antifibrotic Drug Pirfenidone, in Mice Following Intravenous Administration, Biopharm Drug Disp. (2002) 23: 203-211. |
| Greene et al, Protective Groups in Organic Synthesis. John Wiley & Sons, 3rd Edition, 1999, Table of Contents Only. |
| Griswold et al., Differentiation in vivo of Classical Non-Steroidal Antiinflammatory Drugs from Cytokine Suppressive Antiinflammatory Drugs and other Pharmacological Classes Using Mouse Tumour Necrosis Factor Alpha Production, Drugs Exp Clin Res. (1993) 19(6): 243-248. |
| Higuchi et al., Pro-drugs as a Novel Drug Delivery System, (1975) vol. 14, A.C.S. Symposium Series, Table of Content Only. |
| Hoogenband van den, et al., An efficient synthesis of 4-bromo-N-substituted oxindoles by an intramolecular copper-catalyzed amidation reaction, Tetra Lttrs. (May 2007) 48(26): 4461-4465. |
| Hoogenband van den, et al., An efficient synthesis of 4-bromo-N-substituted oxindoles by an intramolecular copper-catalyzed amidation reation, Tetra Lttrs. (May 2007) 48(26): 4461-4465. |
| International Search Report dated Oct. 2, 2009 for PCT/US2009/046136, filed Jun. 3, 2009. |
| IPRP and WO dated Dec. 6, 2010 for PCT/US2009/046136, filed Jun. 3, 2009. |
| Japanese Office Action dated Dec. 4, 2013 for Application No. 2011-512616, filed Jun. 3, 2009. |
| Japanese Office Action dated May 7, 2014 for Application No. 2011-512616, filed Jun. 3, 2009. |
| Jiang et al., Characterization of the Structure and Function of a New Mitogen-activated Protein Kinase (p38-beta). J Biol Chem. (Jul. 1996) 271(30): 17920-17926. |
| Joe et al., Animal Models of Rheumatoid Arthritis and Related Inflammation, Curr Rheumatol Rep. (1999) 1: 139-148. |
| Kaminska et al., TGF beta signalling and its role in tumour pathogenesis, Acta Biochim Pol., (Jun. 2005) 52(2): 329-337. |
| Kane et al., "Reactions of Diazenediyl Compounds with Pyridones: A Novel [4+2] Cycloaddition," J Heter Chem. (1976) 13(3): 673-674. |
| Kappe et al., "Syntheses of Heterocycles, CLXII: The Synthesis of N-Malonylheterocycles", Monatsheft Chemie (1972); 103(2)586-598. |
| Katritzky et al., J Hetero Chem., Synthesis and Some Transformations 4-Benzotriazolyl-3,4-dihydropyrid-2-ones (1996) 33(6): 2031-2036. |
| Kisteneva, "Azomethine Dyes from Oxindole Derivatives. I", The Journal of General Chemistry of the U.S.S.R. (1956) 26(3): 1327-1332. |
| Kisteneva, "Azomethine Dyes from Oxindole Derivatives. II", The Journal of General Chemistry of the U.S.S.R. (1956) 26(7): 2251-2255. |
| Ko et al., "A New and Facile Synthesis of 2-Pyridones", Bull Korean Chem Soc. (2001) 22(2): 234-236. |
| Korb et al., Differential tissue expression and activation of p38 MAPK alpha, beta, gamma, and delta isoforms in rheumatoid arthritis, Arthritis & Rheumatism (Sep. 2006) 54(9): 2745-2756. |
| Kumar et al., Novel Homologues of CSBP/P38 MAP Kinase: Activation, Substrate, Specificity and Sensitivity to Inhibition by Pyridinyl Imidazoles, Biochem Biophys Res Comm., (1997) 235: 533-538. |
| Lam et al., Copper-catalyzed general C—N and C—O bond cross-coupling with arylboronic acid, Tetrahedron Lett. (2001) 42: 3415-3418. |
| Lam et al., Copper-promoted C—N bond cross-coupling with phenylstannane, Tetrahedron Lettrs (2002) 43: 3091-3094. |
| Laufer et al., An in-vitro screening assay for the detection of inhibitors of proinflammatory cytokine synthesis: a useful tool for the development of new antiarthritic and disease modifying drugs, Osteoarth Cartilage (2002) 10: 961-967. |
| Laufer et al., From Imidazoles to Pyrimidines: New Inhibitors of Cytokine Release, J Med Chem., (2002) 45: 2733-2740. |
| Laurent, Biochemical pathways leading to collagen deposition in pulmonary fibrosis, Ciba Found Symp. (1985) 114: 222-233. |
| Lee et al., A protein kinase involved in the regulation of inflammatory cytokine biosynthesis, Nature (Dec. 1994), 372(22): 740-746. |
| Lee et al., Bicyclic imidazoles as a novel class of cytokine biosynthesis inhibitors, Ann NY Acad Sci., (1993) 696: 149-170. |
| Lee et al., Inhibition of Monocyte IL-1 Production by the Anti-inflammatory Compound, SK&F 86002, Int J Immunopharmac., (1988) 10(7): 835-843. |
| Lee et al., Inhibition of p38 MAP Kinase as a Therapeutic Strategy, Immunopharmacol. (2000) 47: 185-201. |
| Lee et al., MAP Kinase p38 Inhibitors: Clinical Results and an Intimate Look at Their Interactions with p38alpha Protein, Curr Med Chem. (2005) 12: 2979-2994. |
| Lee et al., p38 Mitogen-activated Protein Kinase Inhibitors—Mechanisms and Therapeutic Potentials, Pharmacol. Ther., (1999) 82(2-3): 389-397. |
| Lee et al., Pirfenidone: A Novel Pharmacological Agent That Inhibits Leiomyoma Cell Proliferation and Collagen Production, J Clin Endocrinol Metab. (1998) 83(1): 219-223. |
| Li et al., The Primary Structure of p38gamma: A New Member of p38 Group of MAP Kinases, Biochem Biophys Res Comm., (1996) 228: 334-340. |
| Liverton et al., Design and Synthesis of Potent, Selective, and Orally Bioavailable Tetrasubstituted Imidazole Inhibitors of p38 Mitogen-Activated Protein Kinase, J Med Chem. (1999)42(12): 2180-2190. |
| Lowery et al., Transcreener: screening enzymes involved in covalent regulation, Expert Opin Ther Targets (Feb. 2006) 10(1): 179-190. |
| Ma et al., "Synthesis and biological evaluation of the pirfenidone derivatives as antifibrotic agents", Bioorg Med Chem Lttrs (2013)—Available Online Nov. 25, 2013 at http://dx.doi.org/10.1016/j.bmcl.2013.11.038: 14 pages. |
| Majumdar et al., "Palladium-Mediated Direct Synthesis of N-Substituted 4-Methyl- and 4-Ethylisoquinolone Derivatives", Synthesis, (Aug. 2008) Issue 18, 2991-2995. |
| Matsuoka, et al., A p38 MAPK inhibitor, FR-167653, ameliorates murine bleomycin-induced pulmonary fibrosis, Am J Physiol Lung Cell Mol Physiol., (2002) 283: L103-L112. |
| Mayer et al., p38 MAP kinase inhibitors: A future therapy for inflammatory diseases, Drug Discovery Today: Therapeutic Strategies/Immunological disorders and autoimmunity, (2006) 3(1): 50-54. |
| McIntyre et al., Pyridazine Based Inhibitors of p38 MAPK, Bioorg Med Chem Lttr., (2002) 12: 689-692. |
| Mederski et al., N-Aryl Heterocycles via Coupling Reactions with Arylboronic Acids, Tetrahedron (1999) 55(44): 12757-12770. |
| Mexican Office Action dated Aug. 27, 2012 for Application No. MX/a/2010/012848, filed Jun. 3, 2009. |
| Mexican Office Action dated Feb. 22, 2013 for Application No. MX/a/2010/012848, filed Jun. 3, 2009. |
| Muddu et al., Resolving fibrosis in the diseased liver: translating the scientific promise to the clinic, Int J Biochem Cell Biol., (2007) 39(4): 695-714 [Online Oct. 7, 2006]. |
| Nagai et al., Open-label Compassionate Use One Year-treatment with Pirfenidone to Patients with Chronic Pulmonary Fibrosis, Intern Med., (2002) 41(12): 1118-1123. |
| Newton et al., New aspects of p38 mitogen activated protein kinase (MAPK) biology in lung inflammation, Drug Discovery Today: Disease Mechanisms, (2006) 3: 53-61. |
| Noble et al., Idiopathic pulmonary fibrosis: new insights into pathogenesis, Clin Chest Med. (2004) 25(4): 749-758. |
| Ono et al., The p38 Signal Transduction Pathway Activation and Function, Cell Signal., (2000) 12: 1-13. |
| Ozes et al., 697 Preclinical activity of pirfenidone (5-methyl-1pheny1-2(IH)-pyridone) in cell-based models of nonalcoholic steatohepatitis, Hepatology, (2003) 38: 495-. |
| Pargellis et al., Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site, Nat Struct Biol. (2002) 9(4): 268-272. |
| Patani et al., Bioisosterism: A Rational Approach in Drug Design. Chem. Rev. (1996), 96(8): 3147-3176. |
| Pednekar et al., Synthesis of Substituted Pyridazinones and Some Novel Fused Heterocycles from Pyran-2-One and Pyridin-2-One Systems, Indian J Heter Chem. (1998) 8(2): 89-94. |
| Przheval'skii et al., Mono(m-substituted) Chloroacetyldiarylamines in the Stollé´ Reaction, Chem Heterocycl Compd. (1982) 18(7): 716-719. |
| Raghu et al., Treatment of Idiopathic Pulmonary Fibrosis with a New Antifibrotic Agent, Pirenidone—Results of a Prospective, Open-label Phase II Study, Am J Respir Crit Care Med, (1999) 159: 1061-1069. |
| Raingeaud et al., Pro-inflammatory Cytokines and Environmental Stress Cause p38 Mitogen-activated Protein Kinase Activation by Dual Phosphorylation on Tyrosine and Threonine, J Biol Chem., (1995) 270(13): 7420-7426. |
| Results from Chemical Abstract Services Search of May 9, 2012 in REGISTRY/CAPLUS 1907-date, WPINDEX DCR 1999-date, USPATFULL 1971-date, USPATOLD 1790-1975, and REAXYSFILE 1771-2009. |
| Results from Chemical Abstract Services Search of Sep. 4, 2013 in REGISTRY/CAPLUS 1907-date, CASREACT, 1840-date, and WPINDEX DCR 1999-date. Answers 1-130 are to patent documents and answers 131-214 are to non-patent literature. One exemplary structure was displaced for Answers 1-130; pp. 291. |
| Results from Chemical Abstract Services Search of Sep. 4, 2013 showing 3161 structures that were registered from other sources such as chemical libraries and do not have any literature associated with them. Each compound is identified by CA Index name, molecular formula, and structure; pp. 1051. |
| Richards et al., Biochemical and cellular mechanisms of pulmonary fibrosis, Toxicol Pathol. (1991) 19: 526-539. |
| Roche et al. (Eds.), Bioveversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press (1987) Table of Contents Only. |
| Salituro et al., Inhibitors of p38 MAP Kinase: Therapeutic Intervention in Cytokine-Mediated Diseases, Curr Med Chem., (1999) 6: 807-823. |
| Sarges et al., A Novel Class of "GABAergic" Agents: 1-Aryl-3-(aminoalkylidene)oxindoles, (1989) 32(2): 437-444. |
| Shi-Wen et al., "Endothelin-1 Induces Expression of Matrix-associated Genes in Lung Fibroblasts through MEK/ERK". J Biol Chem. (Mar. 23, 2004) 279(22): 23098-23103. |
| Simonsen et al., Ethyl 6-Methyl-2-pyrone-3, 5-dicarboxylate and its derivatives. J Chem Soc Trans. (1908) 93: 1022-1032. |
| Stambe et al., The Role of p38alpha Mitogen-Activated Protein Kinase Activation in Renal Fibrosis, J Am Soc Nephrol., (2004) 15: 370-379. |
| Stein et al., p38-2, a Novel Mitogen-activated Protein Kinase with Distinct Properties, J Biol Chem., (1997) 272(31): 19509-19517. |
| Stolle, Über N-substituierte Oxindole und Isatine, Journal für Praktische Chemie, (1930), 128: 1-43. |
| Sugahara et al., A facile copper-catalyzed Ullmann condensation: N-arylation of heterocyclic compounds containing an -NHCO-moiety, Chem Pharm Bull., (1997) 45(4): 719-721. |
| Ting et al., Substituted 1,3-Dihydro-2H-pyrrolo[2,3-b]pyridin-2-ones as Potential Antiinflammatory Agents, J Med Chem. (1990) 33(10): 2697-2706. |
| Trifilieff et al., CGH2466, a combined adenosine receptor antagonist, p38 mitogen-activated protein kinase and phosphodiesterase type 4 inhibitor with potent in vitro and in vivo anti-inflammatory activities, Br J Pharmacol., (Jan. 2005) 144: 1002-1010. |
| Underwood et al., Inhibition of p38 MAP Kinase, Prog Respir Res., (2001) 3′1: 342-345. |
| Underwood et al., SB 239063, A p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung, Am J Physiol Lung Cell Mol Physiol., (2000) 279: L895-L902. |
| Vippagunta et al., Crystalline solids, Advanced Drug Delivery Reviews (2001) 48: 3-26. |
| Wang et al., "An improved Ullmann-Ukita-Buchwald-Li conditions for Cul-catalyzed coupling reaction of 2-pyridones with aryl halides". Tetrahedron (2005) 61(11): 2931-2939. |
| Wang et al., Molecular Cloning and Characterization of a Novel p38 Mitogen-activated Protein Kinase, J Biol Chem., (1997) 272(38): 23668-23674. |
| Wang et al., Requirement of Mitogen-activated Protein Kinase Kinase 3 (MKK3) for Activation of p38α and p38δ MAPK Isoforms of TGF-β1 in Murine Mesangial Cells (2002) 277: 47257-47262. |
| Yan et al., Pirfenidone effect and mechanism in the treatment of fibrotic diseases. West China Medical Journal (2004) 19(1):169-170 [with English machine translation]. |
| Zhang et al., "Convenient Synthesis of 2,7-Naphthyridine Lophocladines A and B and their Analogues", J Comb Chem (2007) 9: 916-919. |
| Zohdi et al., Heterocyclic Synthesis with Isothiocyanate and sulfur: A novel route for the synthesis of Pyridino[2,3-d]Pyridazine and Thiazolo[4,5-b]Isoquinoline derivatives. Phosphorus, Sulfur, and Silicon (1995) 101(1-4): 179-187. |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2296653A1 (en) | 2011-03-23 |
| CA2726588C (en) | 2019-04-16 |
| JP5627574B2 (en) | 2014-11-19 |
| US8969347B2 (en) | 2015-03-03 |
| EP2296653A4 (en) | 2012-03-07 |
| US20130102597A1 (en) | 2013-04-25 |
| CN102099036A (en) | 2011-06-15 |
| US9290450B2 (en) | 2016-03-22 |
| CA2726588A1 (en) | 2009-12-10 |
| WO2009149188A1 (en) | 2009-12-10 |
| US20090318455A1 (en) | 2009-12-24 |
| EP2296653B1 (en) | 2016-01-27 |
| US8304413B2 (en) | 2012-11-06 |
| HK1155374A1 (en) | 2012-05-18 |
| JP2011522056A (en) | 2011-07-28 |
| US20140235637A1 (en) | 2014-08-21 |
| CA3034994A1 (en) | 2009-12-10 |
| MX2010012848A (en) | 2011-03-01 |
| CN102099036B (en) | 2015-05-27 |
| JP2014210809A (en) | 2014-11-13 |
| ES2567283T3 (en) | 2016-04-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| USRE47142E1 (en) | Compounds and methods for treating inflammatory and fibrotic disorders | |
| US10010536B2 (en) | Method of modulating stress-activated protein kinase system | |
| KR20040062557A (en) | Aminobenzamide derivatives as glycogen synthase kinase 3β inhibitors | |
| JP2001520655A (en) | 5,7-Disubstituted 4-aminopyrido [2,3-D] pyrimidine compounds and their use as adenosine kinase inhibitors | |
| CN101237869A (en) | Pyridone derivatives for modulating stress-activated protein kinase system | |
| HK1155374B (en) | Compounds and methods for treating inflammatory and fibrotic disorders | |
| AU2012216834A1 (en) | Pyridone Derivatives for Modulating Stress-Activated Protein Kinase System | |
| HK1185285B (en) | Pyridine-2-one-derivatives as modulators of stress-activated protein kinase system | |
| HK1185285A (en) | Pyridine-2-one-derivatives as modulators of stress-activated protein kinase system |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: INTERMUNE, INC., CALIFORNIA Free format text: CERTIFICATE OF CHANGE OF COMPANY'S ADDRESS;ASSIGNOR:INTERMUNE, INC.;REEL/FRAME:046638/0466 Effective date: 20180711 |
|
| AS | Assignment |
Owner name: INTERMUNE, INC., CALIFORNIA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:KOSSEN, KARL;SEIWERT, SCOTT D.;SEREBRYANY, VLADIMIR;AND OTHERS;SIGNING DATES FROM 20090708 TO 20090722;REEL/FRAME:046647/0860 |
|
| CC | Certificate of correction | ||
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 8TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1552); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 8 |
|
| FEPP | Fee payment procedure |
Free format text: MAINTENANCE FEE REMINDER MAILED (ORIGINAL EVENT CODE: REM.); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| LAPS | Lapse for failure to pay maintenance fees |
Free format text: PATENT EXPIRED FOR FAILURE TO PAY MAINTENANCE FEES (ORIGINAL EVENT CODE: EXP.); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |