RU2569491C2 - Кристаллическая форма бензил-бензольного ингибитора sglt - Google Patents
Кристаллическая форма бензил-бензольного ингибитора sglt Download PDFInfo
- Publication number
- RU2569491C2 RU2569491C2 RU2013101580/04A RU2013101580A RU2569491C2 RU 2569491 C2 RU2569491 C2 RU 2569491C2 RU 2013101580/04 A RU2013101580/04 A RU 2013101580/04A RU 2013101580 A RU2013101580 A RU 2013101580A RU 2569491 C2 RU2569491 C2 RU 2569491C2
- Authority
- RU
- Russia
- Prior art keywords
- crystalline form
- mixture
- benzyl
- peaks
- compound
- Prior art date
Links
Images







Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/351—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom not condensed with another ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/10—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
- C07H1/06—Separation; Purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/04—Carbocyclic radicals
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Molecular Biology (AREA)
- Obesity (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Cardiology (AREA)
- Endocrinology (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Emergency Medicine (AREA)
- Vascular Medicine (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Child & Adolescent Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pyrane Compounds (AREA)
Abstract
Изобретение относится к новой кристаллической форме ингибитора натрий-зависимого котранспортера глюкозы SGLT2 формулы

характеризущейся порошковой рентгеновской дифрактограммой (XRPD) с использованием излучения CuKα1, включающей пики при 11,2, 12,9, 15,5, 17,8, 19,1, 20,0 и 20,7 градусах 2θ (±0,1 градуса 2θ), спектром комбинационного рассеяния, включающим пики при 353, 688, 825, 1178, 1205, 1212, 1608, 2945, 3010 и 3063 см-1, а также эндотермой, полученной методом дифференциальной сканирующей калориметрии (DSC), при 136°С, ее получению перекристаллизацией из этанола или метанола добавлением воды и фармацевтическим композициям на ее основе для лечения заболеваний, патогенез которых требует ингибирования натрий-зависимого котранспортера глюкозы 2 (SGLT2). Предложена новая кристаллическая форма биологически активного вещества с низкой гигроскопичностью. 6 н. и 27 з.п. ф-лы, 8 ил., 5 табл., 12 пр.
Description
Перекрестные ссылки на родственные заявки
Для настоящего изобретения испрашивается приоритет по заявке PCT/CN2010/073865, поданной 12 июня, 2010, включенной в настоящий документ во всей своей полноте для всех возможных целей.
Уровень техники
Натрий-зависимые («активные») котранспортеры глюкозы (SGLT), включая SGLT1 (обнаруживается преимущественно в щеточной каемке кишечника) и SGLT2 (локализован в проксимальных почечных канальцах), являются предметом тщательных исследований. В частности, было обнаружено, что SGLT2 ответственен за большую часть обратного захвата глюкозы почками. В настоящее время считается, что ингибирование почечного SGLT представляет собой плодотворный подход для лечения гипергликемии путем увеличения количества выводимой с мочой глюкозы (Arakawa К, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999). Потенциал этого терапевтического подхода дополнительно подтверждается недавними данными о мутациях в гене SGLT2, обнаруживаемых в случаях наследственной почечной глюкозурии, неопасного, по всей видимости, синдрома, для которого характерно выведение глюкозы с мочой при нормальных уровнях глюкозы в сыворотке и отсутствии системных нарушений функции почек или других заболеваний (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Таким образом, соединения, ингибирующие SGLT, в особенности SGLT2, являются перспективными кандидатами для применения в качестве антидиабетических средств (см. обзор Washbum WN, Expert Opin Ther Patents 19:1485-99, 2009). Кроме того, поскольку раковые клетки характеризуются усиленным поглощением глюкозы по сравнению с нормальными клетками, ингибирование SGLT предлагалось в качестве способа лечения рака путем лишения раковых клеток питательных веществ. Например, исследования показывают, что SGLT2 играет определенную роль в захвате глюкозы в метастатических очагах при раке легкого (Ishikawa N, et al., Jpn J Cancer Res 92:874-9, 2001). Таким образом, ингибиторы SGLT2 также могут быть полезны в качестве противораковых средств.
Помимо, собственно, фармацевтической активности, успешная разработка лекарственного препарата дополнительно подразумевает рассмотрение параметров, связанных с физической природой активного вещества. Некоторые из этих параметров представляют собой стабильность активного вещества в различных условиях окружающей среды, стабильность активного вещества в ходе производства фармацевтической композиции и стабильность активного вещества в конечных композициях лекарственного препарата. Для обеспечения необходимой стабильности фармацевтически активное вещество, применяемое в лекарственном препарате, должно быть как можно более чистым, что приводит к его стабильности при долговременном хранении в различных условиях окружающей среды.
Другой фактор, который необходимо учитывать, представляет собой однородность распределения активного вещества в композиции, в особенности, если активное вещество предназначено для введения в малых дозах. Для обеспечения однородного распределения размер частиц активного вещества должен быть уменьшен до подходящего уровня, например путем измельчения. Однако следует избегать разрушения фармацевтически активного вещества, которое может происходить как побочный эффект измельчения (или микронизации). В результате, в связи с жесткими условиями, налагаемыми этим процессом, активное вещество должно быть стабильным на протяжении всего процесса измельчения. Более того, если активное вещество не стабильно в ходе процесса измельчения, то, скорее всего, способ получения гомогенной фармацевтической композиции с заданным количеством активного вещества окажется слабо воспроизводимым.
Еще одно соображение, связанное с процессом измельчения для получения желаемой фармацевтической композиции, заключается в затратах энергии на этот процесс и нагрузках на поверхностях кристаллов. Этот фактор может в некоторых обстоятельствах приводить к полиморфным превращениям, переходу в аморфное состояние или к изменению в кристаллической решетке. Поскольку фармацевтическая композиция фармацевтического качества всегда должна иметь активное вещество с одинаковой кристаллической морфологией, стабильностью и свойствами, к с этой точки зрения к кристаллическому активному веществу также предъявляют строгие требования.
Другое соображение в отношении фармацевтически активного вещества касается его стабильности в композиции, что в свою очередь приводит к более длительному сроку годности конкретного лекарственного препарата. В данном случае под сроком годности понимается продолжительность временного периода, в течение которого лекарственный препарат может быть введен без риска того, что активное вещество деградировало. Высокая стабильность лекарственного препарата в упомянутых выше фармацевтических композициях при различных условиях хранения, таким образом, представляет собой дополнительное преимущество, как для пациента, так и для производителя.
Кроме того, наличие четко определенной кристаллической формы позволяет проводить очистку вещества лекарственного соединения путем перекристаллизации.
Наряду с указанными выше требованиями необходимо в целом учитывать, что любые изменения в твердом состоянии фармацевтической композиции, способные улучшить ее физическую и химическую стабильность, дают значительное преимущество по сравнению с менее стабильными формами того же лекарственного препарата.
Соединение по настоящему изобретению было получено в соответствии со способами, раскрытыми в публикации США №2009/0118201, дата подачи 22 августа 2008, в заявке США №12/545,400 и в PCT/US 2009/054585, в настоящее время WO 2010/022313, обе из которых поданы 21 августа 2009. Цель настоящего изобретения заключается в получении стабильной кристаллической формы соединения, которая отвечает важным требованиям, предъявляемым к фармацевтически активным веществам, как указано выше.
Раскрытие изобретения
Настоящее изобретение относится к кристаллическим формам (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола, обладающего способностью ингибировать натрий-зависимый котранспортер глюкозы 2 (SGLT2). Изобретение также относится к фармацевтическим композициям, способам получения кристаллической формы соединения и способам применения соединений, отдельно или в комбинации с другими терапевтическими агентами, для лечения заболеваний и состояний, на которые влияет ингибирование SGLT2.
Краткое описание чертежей
На фиг.1 представлены спектры кристаллического (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола, полученные методом рентгеновской порошковой дифрактометрии (XRPD).
На фиг.2 представлена таблица данных XRPD для спектров XRPD, представленных на фиг.1.
На фиг.3 представлены спектры комбинационного рассеяния для кристаллического (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола.
На фиг.4 представлен перечень пиков интенсивности для спектров комбинационного рассеяния, представленных на фиг.3.
На фиг.5 представлены результаты термического гравиметрического анализа (TGA) для кристаллического (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола.
На фиг.6 представлены спектры дифференциальной сканирующей калориметрии (DSC) для кристаллического (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола.
На фиг.7 представлена таблица данных по элементарным ячейкам для кристаллического (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола.
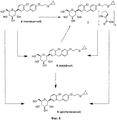
На фиг.8 представлена схема получения кристаллического (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола.
Осуществление изобретения
I. Определения
В настоящем изобретении термины «лечить» и «лечение» означают задержку наступления, замедление или обращение прогрессирования, или облегчение, или предупреждение заболевания или состояния, к которому этот термин применяется, или одного или нескольких симптомов такого заболевания или состояния.
В настоящем изобретении термин «введение» означает введение субъекту перорально, в виде суппозиториев, местно, внутривенно, внутрибрюшинно, внутримышечно, в очаг поражения, интраназально или подкожно лекарственного средства или имплантацию субъекту устройства с замедленным высвобождением лекарственного средства, например, осмотического мини-насоса. Введение осуществляют любым способом, включая парентеральный и трансмукозальный (например пероральный, назальный, вагинальный, ректальный или трансдермальный). Парентеральное введение включает, например, внутривенное, внутримышечное, интраарериолярное, внутрикожное, подкожное, внутрибрюшинное, внутрижелудочковое и внутричерепное введение. Другие способы доставки включают применение липосомальных композиций, внутривенную инфузию, трансдермальные пластыри и т.п., но не ограничиваются этим.
В настоящем изобретении термин «пролекарство» означает соединение-предшественник, которое после его введения высвобождает биологически активное соединение in vivo в результате некоего химического или физиологического процесса (например, пролекарство превращается в биологически активное соединение при достижении физиологического pH или под действием ферментов). Пролекарство само по себе может как обладать, так и не обладать желаемой биологической активностью.
В настоящем изобретении термин «соединение» означает молекулу, полученную любым способом, включая синтез in vitro или образование in situ или in vivo, но не ограничиваясь только этим.
Предполагается, что термины «контролируемое высвобождение», «замедленное высвобождение», «пролонгированное высвобождение» и «высвобождение в определенный момент времени» взаимозаменяемо относятся к любой содержащей лекарственное средство композиции, в которой высвобождение лекарственного средства происходит не безотлагательно, т.е. при применении композиции с «контролируемым высвобождением», а пероральное введение не приводит к незамедлительному высвобождению лекарственного средства в абсорбционный пул. Эти термины применяют взаимозаменяемо с термином «немгновенное высвобождение» как он определен в Remington: The Science и Practice of Pharmacy, 21st Ed., Gennaro, Ed., Lippencott Williams & Wilkins (2003). Как указано в этом документе, немедленное и немгновенное высвобождение можно разделить кинетически с помощью следующего уравнения:

Термин «абсорбционный пул» означает раствор лекарственного средства, введенный в определенное место абсорбирования, a kr, ka и ke представляют собой константы скорости первого порядка для, соответственно, (1) высвобождения лекарственного средства из композиции, (2) абсорбции и (3) выведения. Для лекарственных форм с мгновенным высвобождением константа скорости высвобождения лекарственного средства kr значительно превышает константу скорости абсорбции ka. Для композиций с контролируемым высвобождением верно обратное, т.е. kr<<kg, так что высвобождение лекарственного средства из лекарственной формы представляет собой лимитирующую стадию в процессе доставки лекарственного средства к целевой области.
Термины «замедленное высвобождение» и «пролонгированное высвобождение» применяют в общепринятом смысле этих слов для отсылки к композиции лекарственного средства, обеспечивающей постепенное высвобождение лекарственного средства в течение длительного периода времени, например 12-ти часов или более, и преимущественно, хотя и не обязательно, приводящей к практически постоянному уровню лекарственного средства в крови в течение длительного периода времени.
В настоящем изобретении термин «отсроченное высвобождение» означает фармацевтический препарат, который проходит через желудок в неизмененном виде и растворяется в тонком кишечнике.
В настоящем изобретении термин «фармацевтически приемлемое вспомогательное вещество» означает вещество, способствующее введению активного агента и абсорбированию его субъектом. Фармацевтические вспомогательные вещества, применяемые в настоящем изобретении, включают связующие вещества, наполнители, разрыхлители, лубриканты, покрытия, подсластители, ароматизаторы и красители, но не ограничиваются только ими. Специалисту в данной области техники известны и другие фармацевтические вспомогательные вещества, которые могут быть использованы в настоящем изобретении.
В настоящем изобретении термин «субъект» означает животных, таких как млекопитающие, включая приматов (например, людей), коров, овец, коз, лошадей, собак, кошек, кроликов, крыс, мышей и тому подобных, но не ограничиваясь только ими. В определенных вариантах осуществления субъект представляет собой человека.
В настоящем изобретении термины «терапевтически эффективное количество или доза» или «терапевтически достаточное количество или доза» или «эффективное или достаточное количество или доза» означает дозу, которая позволяет достичь тех терапевтических эффектов, ради которых она вводится. Точная доза будет зависеть от цели лечения, и будет установлена специалистом в данной области техники с помощью известных методик (см., например, Lieberman, Pharmaceutical Dosage Forms (vols.1-3, 1992); Lloyd, The Art, Science and Technology of Pharmaceutical Compounding (1999); Pickar, Dosage Calculations (1999); и Remington: The Science and Practice of Pharmacy, 20th Edition, 2003, Gennaro, Ed., Lippincott, Williams & Wilkins).
II. Кристаллические формы
Настоящее изобретение относится к кристаллической форме соединения, обладающего ингибирующим эффектом на натрий-зависимый котранспортер глюкозы SGLT, предпочтительно SGLT2. Таким образом, кристаллическое соединение по настоящему изобретению подходит для предупреждения и лечения заболеваний и состояний, в особенности метаболических нарушений, включая сахарный диабет 1-го и 2-го типов, гипергликемию, осложнения, вызываемые диабетом (такие как ретинопатия, нефропатия, например прогрессирующее заболевание почек, нейропатия, язвы, микро- и макроангиопатии и синдром диабетической стопы), резистентность к инсулину, метаболический синдром (синдром X), гиперинсулинемия, повышенное артериальное давление, гиперурикемия, ожирение, отечность, дислипидемия, хроническая сердечная недостаточность, атеросклероз и связанные с ними заболевания, но не ограничиваясь только указанным.
Настоящее изобретение также относится к фармацевтическим композициям и пролекарствам кристаллической формы по настоящему изобретению.
Настоящее изобретение, кроме того, относится к способам синтеза для получения кристаллического соединения по настоящему изобретению.
Настоящее изобретение также относится к способам применения кристаллической формы соединения по настоящему изобретению, отдельно или в комбинации с другими терапевтическими агентами, для лечения заболевания и состояния, на которые может воздействовать ингибирование SGLT.
Настоящее изобретение также относится к способам применения соединения по настоящему изобретению для получения лекарственного препарата для лечения заболеваний и состояний, на которые может воздействовать ингибирование SGLT.
В одном из аспектов настоящее изобретение относится к кристаллической форме (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола. Его химическая структура приведена ниже:

Кристаллическое соединение по настоящему изобретению может быть охарактеризовано порошковой рентгеновской дифрактограммой (XRPD), спектрами комбинационного рассеяния, эндотермой, полученной методом дифференциальной сканирующей калориметрии (DSC), термогравиметрическим анализом (TGA), демонстрирующим температуру разложения, и элементарной ячейкой кристаллической структуры.
В некоторых вариантах осуществления настоящее изобретение относится к кристаллической форме соединения, XRPD-спектр которой по существу соответствуют показанному на фиг.1, и XRPD пики которого по существу соответствуют пикам, приведенным на фиг.2. Кристаллическое соединение по настоящему изобретению может характеризоваться любой комбинацией пиков, по существу соответствующих пикам на фиг.2. Кроме того, величина каждого пика, приведенного на фиг.2, может быть определена с погрешностью, лежащей в диапазоне ±0,2 градуса 2θ, предпочтительно ±0,1 градуса 2θ.
В других вариантах осуществления кристаллическая форма соединения характеризуется порошковой рентгеновской дифрактограммой, включающей один или несколько пиков при 5,4, 11,2, 11,3, 11,9, 12,9, 15,5, 16,3, 17,8, 19,1, 20,0, 20.6, 20,7, 21,2, 22,8, 23,0, 23,4, 23,6, 23,9, 24,7, 25,4, 25,8, 27,8 и 28,2 градусах 2θ (±0,1 градуса 2θ), при этом указанный XRPD-спектр получают с использованием излучения CuKα1. В другом воплощении кристаллическая форма соединения характеризуется XRPD-спектром, включающим два или более, три или более, четыре или более, или пять или более пиков при 5,4, 11,2, 11,3, 11,9, 12,9, 15,5, 16,3, 17,8, 19,1, 20,0, 20,6, 20,7, 21,2, 22,8, 23,0, 23,4, 23,6, 23,9, 24,7, 25,4, 25,8, 27,8 и 28,2 градусах 2θ (±0,1 градуса 2θ). В некоторых других вариантах осуществления кристаллическая форма соединения характеризуется XRPD-спектром, включающим пики при 12,9, 19,1 и 20,7 градусах 2θ (±0,1 градуса 2θ). В еще одних вариантах осуществления кристаллическая форма соединения характеризуется XRPD-спектром, включающим пики при 11,2, 12,9, 15,5, 17,8, 19,1, 20,0 и 20,7 градусах 2θ (±0,1 градуса 2θ). Еще в одних вариантах осуществления кристаллическая форма соединения характеризуется XRPD-спектром, включающим пики при 5,4, 11,2, 11,9, 12,9, 15,5, 16,3, 17,8 и 19,1 градусах 2θ (±0,1 градуса 2θ). Еще в одних вариантах осуществления кристаллическая форма соединения характеризуется XRPD-спектром, включающим пики при 5,4, 11,2, 11,9 и 12,9 градусах 2θ (±0,1 градуса 2θ). В другом воплощении кристаллическая форма соединения характеризуется XRPD-спектром, включающим пики при 11,2 и 12,9 градусах 2θ (±0,1 градуса 2θ). В других вариантах осуществления кристаллическая форма соединения характеризуется пиками XRPD, которые по существу соответствуют пикам фиг.2.
Кристаллическое соединение по настоящему изобретению также характеризуется спектрами комбинационного рассеяния, которые по существу соответствуют спектрам, представленным на фиг.3, а пики которых по существу соответствуют пикам, представленным на фиг.4. В некоторых вариантах осуществления кристаллическая форма соединения характеризуется спектрами комбинационного рассеяния, включающим один или несколько пиков при примерно 353, 688, 825,1178, 1205, 1212, 1608, 2945, 3010 и 3063 см-1. В другом воплощении кристаллическая форма соединения характеризуется спектрами комбинационного рассеяния, включающими два или более, три или более, четыре или более или пять или более пиков. В других вариантах осуществления кристаллическая форма соединения характеризуется спектрами комбинационного рассеяния, включающими пики при примерно 353, 688 и 825 см-1. В некоторых других вариантах осуществления кристаллическая форма соединения характеризуется пиками на спектрах комбинационного рассеяния, которые по существу соответствуют пикам на фиг.4.
Кристаллическое соединение по настоящему изобретению также характеризуется эндотермой, определенной методом дифференциальной сканирующей калориметрии (DSC). В некоторых вариантах осуществления кристаллическая форма соединения характеризуется эндотермой DSC при примерно 136°С.
Кристаллическое соединение по настоящему изобретению также характеризуется данными об элементарной ячейке, которые по существу соответствуют данным, представлены на фиг.7. Для определения кристаллического соединения по настоящему изобретению также может быть применен термогравиметрический анализ (TGA). Например, типичный TGA по существу соответствует приведенному на фиг.5 и демонстрирует термическую устойчивость кристаллического соединения при температуре выше 200°С.
В некоторых вариантах осуществления кристаллическое соединение характеризуется по меньшей мере одним из следующих: по меньшей мере одним пиком XRPD, как описано выше, по меньшей мере одним пиком в спектре комбинационного рассеяния, как описано выше, эндотермой DSC, такой как описана выше, данными TGA относительно термической устойчивости, как описано выше, и данными об элементарной ячейке, как они описаны выше и приведены на фиг.7. В других вариантах осуществления кристаллическое соединение характеризуется по меньшей мере двумя характеристиками из следующих: по меньшей мере одним пиком XRPD, как описано выше, по меньшей мере одним пиком в спектре комбинационного рассеяния, как описано выше, эндотермой DSC, такой как описанная выше, данными TGA относительно термической устойчивости, как описано выше, и данными об элементарной ячейке, такими как описанные выше и приведенные на фиг.7. Например, кристаллическое соединение может быть охарактеризовано по меньшей мере одним пиком XRPD и по меньшей мере одним пиком в спектре комбинационного рассеяния, или по меньшей мере одним пиком XRPD и эндотермой DSC, или по меньшей мере одним пиком в спектре комбинационного рассеяния и эндотермой DSC, или по меньшей мере одним пиком XRPD и данными об элементарной ячейке, или по меньшей мере одним пиком в спектре комбинационного рассеяния и данными об элементарной ячейке и т.д.
В некоторых вариантах осуществления кристаллическое соединение по настоящему изобретению характеризуется порошковой рентгеновской дифрактограммой (XRPD), включающей один или несколько пиков при 5,4, 11,2, 11,3, 11,9, 12,9, 15,5, 16,3, 17,8, 19,1, 20,0, 20,6, 20,7, 21,2, 22,8, 23,0, 23,4, 23,6, 23,9, 24,7, 25,4, 25,8, 27,8 и 28,2 градусах 2θ (±0,1 градуса 2θ), при этом указанную XRPD получают, используя излучение CuKα1, и спектром комбинационного рассеяния, включающим один или несколько пиков при примерно 353, 688, 825, 1178, 1205, 1212, 1608, 2945, 3010 и 3063 см-1. В других вариантах осуществления кристаллическое соединение по настоящему изобретению характеризуется порошковой рентгеновской дифрактограммой (XRPD), включающей один или несколько пиков при 11,2, 12,9, 15,5, 17,8, 19,1, 20,0, 20,6, 20,7, 21,2 и 22,8 и 28,2 градусах 2θ (±0,1 градуса 2θ), при этом указанную XRPD получают, используя излучение CuKα1, и спектром комбинационного рассеяния, включающим один или несколько пиков при примерно 353, 688 и 825 см-1. В некоторых других вариантах осуществления кристаллическое соединение по настоящему изобретению характеризуется порошковой рентгеновской дифрактограммой (XRPD), включающей один или несколько пиков при 11,2 и 12,9 градусах 2θ (±0,1 градуса 2θ), при этом указанную XRPD получают, используя излучение CuKα1, и спектром комбинационного рассеяния, включающим один или несколько пиков при примерно 353, 688 и 825 см-1.
В других вариантах осуществления настоящее изобретение относится к соединению (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол в кристаллической форме.
Настоящее изобретение также включает меченные изотопами формы (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-иклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола, в которых один или несколько атомов замещены одним или несколькими атомами, обладающими определенными атомными массами или массовыми числами. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фтора, серы и хлора (такие как 2H, 3H, 13С, 14С, 15N, 18O, 17O, 18F, 35S и 36Cl), но не ограничиваются только ими. Меченные изотопами соединения и их пролекарства, а также меченные изотопами фармацевтически приемлемые соли и их пролекарства, входят в объем настоящего изобретения. Меченные изотопами соединения по настоящему изобретению применяются в анализах для изучения тканевого распределения соединений и их пролекарств и метаболитов; предпочтительные изотопы для таких анализов включают 3H и 14C. Кроме того, в некоторых случаях замещение более тяжелыми изотопами, такими как дейтерий (2H) может придать повышенную метаболическую стабильность, которая привносит терапевтические преимущества, такие как увеличение периода полувыведения in vivo или снижение требуемой дозы. Меченные изотопами соединения по настоящему изобретению и их пролекарства как правило могут быть получены в соответствии со способами, описанными в настоящем документе, путем замены немеченого изотопами реагента реагентом, меченным изотопами.
II.1. Способы получения кристаллической формы
Кристаллическая форма по настоящему изобретению может быть получена с помощью различных способов, как показано на фиг.8. Например, кристаллическое соединение 8 может быть получено непосредственно из комплекса L-пролина 7. В качестве альтернативного варианта L-пролин может быть удален из комплекса L-пролина 7, что позволит получить аморфное соединение 8, которое затем кристаллизуют до кристаллического соединения 8. Кристаллическое соединение 8 также может быть получено непосредственно из неочищенного соединения 6, путем исходного выделения и последующей кристаллизации аморфного соединения 8 для получения кристаллического соединения 8, или непосредственно из неочищенного соединения 6.
Специалистам в данной области техники известны и другие способы получения кристаллического соединения 8. Кроме того, каждый процесс кристаллизации может быть повторен для удаления примесей. В некоторых вариантах осуществления для получения кристаллического соединения по настоящему изобретению может быть применен более чем один из различных способов кристаллизации.
В некоторых вариантах осуществления кристаллическое соединение 8 может быть получено из продукта совместной кристаллизации бис-L-пролина и (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2Я-пиран-3,4,5-триола, как описано в примерах. Вкратце, исходные вещества для совместной кристаллизации помещают в подходящий растворитель (например в метанол или этанол) и получают раствор, к которому для кристаллизации желаемого соединения добавляют осаждающий растворитель (например воду).
Соответственно, настоящее изобретение, кроме того, относится еще и способу получения кристаллической формы (2S,3R,4R,5S,6R)-2-(4-xnop-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола, включающему (а) объединение при перемешивании комплекса (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола и бис(L-пролина) и подходящего растворителя с получением раствора; (b) добавление к раствору осаждающего растворителя для получения смеси; и (c) выделение кристаллической формы из смеси стадии (b).
В некоторых вариантах осуществления настоящее изобретение относится к способу получения кристаллической формы (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола, включающему (а) объединение при перемешивании (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола и подходящего растворителя с получением раствора; (b) добавление к раствору осаждающего растворителя для получения смеси; и (с) выделение кристаллической формы из смеси стадии (b).
В других вариантах осуществления настоящее изобретение относится к способу получения кристаллической формы (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-иклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола, включающему (а) объединение при перемешивании аморфного (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола и подходящего растворителя с получением раствора; (b) добавление к раствору осаждающего растворителя для получения смеси; и (с) выделение кристаллической формы из смеси стадии (b).
На стадии (а) перечисленных выше способов растворитель может представлять собой любой растворитель, подходящий для образования раствора, который легко смешивается с применяемым на стадии (b) осаждающим растворителем. Обычно растворитель на стадии (а) представляет собой полярный растворитель, который в некоторых вариантах осуществления представляет собой протонный растворитель. Подходящие растворители включают C1-C4-спирты, этиленгликоль и полиэтиленгликоль, такой как PEG400, алканоаты, такие как этилацетат, изопропилацетат, пропилацетат и бутилацетат, ацетонитрил, алканоны, такие как ацетон, бутанон, метилэтилкетон и метилпропилкетон, и смеси из двух или нескольких таких растворителей. Более предпочтительные растворители выбирают из группы, состоящей из метанола, этанола, изопропанола, этилацетата, ацетона, и смеси из двух или нескольких таких растворителей. Метанол и этанол представляют собой еще более предпочтительные растворители. В одном из конкретных воплощений растворитель, применяемый на стадии (а), представляет собой метанол.
Стадия (а) перечисленных выше способов может быть осуществлена при температуре, как правило, от примерно 0°С до температуры кипения растворителя (например 65°С для метанола). Предпочтительный диапазон температур находится примерно между 35°С и 100°С, еще более предпочтительный - от примерно 45°С до 80°С. Как только раствор получен, добавляют осаждающий растворитель. Осаждающий растворитель представляет собой растворитель, в котором продукт значительно хуже растворим, чем в исходном растворителе. Подходящие осаждающие растворители включают воду, простые эфиры, циклические простые эфиры, алканы, циклоалканы, фенилы и их смеси, в частности С4-С6-алифатические простые эфиры, С6-С8-алканы, С6-C8-циклоалканы, такие фенилы, как бензол, толуол и ксилол, и их смеси. Примеры осаждающих растворителей включают диизопропиловый эфир, трет-бутилметиловый эфир (ТВМЕ), циклогексан, метилциклогексан, гексан, гептан, октан и их смеси. В одном из конкретных воплощений осаждающий растворитель представляет собой воду.
Точное соотношение растворителей и исходного вещества не столь важно для изобретения, однако оптимизированные отношения могут дать более высокие выходы и более однородный кристаллизованный продукт. Отношение растворителей в описанных выше способах может представлять собой любое подходящее отношение от примерно 1:1 до примерно 1:9, включая примерно 1:2, 1:3, 1:4, 1:5, 1:6, 1:7 и примерно 1:8. Отношения растворителей находятся в диапазоне предпочтительно от примерно 1:1 до примерно 1:9, более предпочтительно от примерно 1:2 до примерно 1:7, еще более предпочтительно от примерно 1:2 до примерно 1:5. В одной из групп воплощений, если в качестве растворителя применяют метанол, а осаждающий растворитель представляет собой воду, то отношение метанола к воде в смеси на стадии (b) равно от примерно 1:1 до примерно 1:9 (по объему), более предпочтительно примерно 1:5 (по объему).
Отношение комплекса к растворителю, такому как смесь метанола и воды, может представлять собой любое подходящее отношение, способствующее кристаллизации. Например, отношение комплекса к растворителю может быть равно от примерно 1:5 (масса/объем или масс./об.) до примерно 1:50 (масс./об.), включая примерно 1:6, 1:7, 1:8, 1:9, 1:10, 1:11, 1:12, 1:13, 1:14, 1:15, 1:20, 1:25, 1:30, 1:35, 1:40 и примерно 1:45 (масс./об.). Отношение комплекса к растворителю предпочтительно равно от примерно 1:10 до примерно 1:25 (масс./об.), более предпочтительно от примерно 1:10 до примерно 1:15 (масс./об.). В другой группе воплощений отношение комплекса к растворителю и осаждающему растворителю в смеси на стадии (b) равно от примерно 1:10 до примерно 1:25 (масс./об.). В других вариантах осуществления отношение комплекса к метанолу и воде в смеси на стадии (b) равно от примерно 1:10 до примерно 1:25 (масс./об.). В некоторых других вариантах осуществления отношение комплекса к метанолу и воде в смеси на стадии (b) равно от примерно 1:2:7 (масс./об./об.) до примерно 1:3:10 (масс./об./об.), предпочтительно примерно 1:2:10 (масс./об./об.).
Смесь для кристаллизации кристаллического соединения 8 также может содержать множество других компонентов, таких как кислоты, основания и соли. Кислоты, применяемые в настоящем изобретении, включают уксусную кислоту, муравьиную кислоту, соляную кислоту, серную кислоту и другие слабые и сильные кислоты но не ограничиваются только ими. Основания, применяемые в настоящем изобретении, включают аммиак, гидроксид натрия и другие, но не ограничиваются только ими. Соли, применяемые в настоящем изобретении, включают хлорид натрия, хлорид калия, карбонат калия и другие, но не ограничиваются только ими. В некоторых вариантах осуществления смесь стадии (b) в перечисленных выше способах включает гидроксид натрия. В других вариантах осуществления смесь стадии (b) в перечисленных выше способах включает хлорид натрия.
После добавления осаждающего растворителя смесь, как правило, хранят при комнатной температуре или охлаждают в течение достаточного периода времени, чтобы дать возможность процессу формирования кристаллов продукта пройти полностью. Температура смеси на стадии (b) предпочтительно примерно равна температуре на стадии (а) или ниже ее. Во время хранения температуру раствора, содержащего продукт, предпочтительно снижают до температуры в диапазоне от -10°С до 25°С или даже ниже, еще более предпочтительно - в диапазоне от -5°С до 15°С. Стадия (b) может быть проведена с перемешиванием или без него. Как отмечено выше, условия проведения стадии (b) могут повлиять на размер, форму и качество полученных кристаллов.
Кристаллизацию можно индуцировать с помощью способов, известных в данной области техники, например, механическим способом, царапая или потирая контактирующую поверхность реакционного сосуда, например, стеклянной палочкой. В насыщенный или пересыщенный раствор могут необязательно быть внесены затравочные кристаллы. Смесь для кристаллизации кристаллического соединения 8 также может содержать затравочные кристаллы кристаллического соединения 8. В некоторых вариантах осуществления раствор или смесь в перечисленных выше способах включают затравочный кристалл кристаллического соединения по настоящему изобретению.
Выделение желаемой кристаллической формы может быть завершено удалением из кристаллов растворителя и осаждающего растворителя. Как правило, это осуществляют с помощью известных способов, например, таких как фильтрация, фильтрация с отсасыванием, декантация или центрифугирование. Кроме того, выделить кристаллическую форму можно удалением избытка растворителя(растворителей) из кристаллической формы с помощью способов, известных специалисту в данной области техники, например, таких как применение пониженного давления, и/или нагреванием до температуры выше 20°С, предпочтительно в температурном диапазоне ниже 80°С, еще более предпочтительно ниже 50°С.
В других вариантах осуществления настоящее изобретение относится к способу получения кристаллической формы (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола, включающему (а) объединение (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола и подходящего растворителя при перемешивании с получением раствора; и (b) выделение кристаллической формы из раствора. В других вариантах осуществления этот способ также включает добавление к раствору осаждающего растворителя. На стадии (а) перечисленных выше способов растворитель может представлять собой любой растворитель, подходящий для образования раствора. Подходящие растворители включают алканоаты, такие как этилацетат, изопропилацетат, пропилацетат и бутилацетат, простые эфиры, такие как этиловый эфир, метил-трет-бутиловый эфир, и смеси из двух или более таких растворителей. Более предпочтительные растворители выбирают из группы, состоящей из этилацетата, этилового эфира, метил-трет-бутилового эфира и смеси двух или более из этих растворителей. Еще более предпочтительные растворители представляют собой этилацетат и метил-трет-бутиловый эфир. (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол может иметь любую подходящую форму, включая аморфную, кристаллическую или их комбинацию. Кроме того, (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол может иметь любую подходящую степень чистоты, например быть очищенным или неочищенным.
В некоторых вариантах осуществления (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол представляет собой аморфный (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол. Аморфный (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол, аморфное соединение 8, может быть получен с помощью различных способов, известных в данной области техники. Например, аморфное соединение 8 может быть выделено из неочищенной смеси 6 с помощью известных способов выделения. В альтернативном варианте аморфное соединение 8 может быть получено из комплекса 7 путем удаления L-пролина способами, известными в данной области техники. В некоторых вариантах осуществления аморфный (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол получают из (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола бис(L-пролина) путем объединения при перемешивании комплекса (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол бис(L-пролин) и смеси подходящих растворителей с получением раствора, и выделения из раствора аморфного (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола. Подходящие растворители и смеси растворителей описаны выше.
III. Фармацевтические композиции
Настоящее изобретение, кроме того, относится к фармацевтической композиции, включающей эффективное количество кристаллической формы (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола в фармацевтически приемлемом вспомогательном веществе.
Кристаллическая форма по настоящему изобретению может быть включена в различные композиции для терапевтического введения. В частности, кристаллическая форма по настоящему изобретению может быть введена в состав фармацевтических композиций путем получения композиции с подходящим фармацевтически приемлемыми вспомогательными веществами или разбавителями, и может быть введена в состав препаратов в твердой, полужидкой, жидкой или газообразной форме, таких как таблетки, капсулы, пилюли, порошки, гранулы, драже, гели, суспензии, мази, растворы, суппозитории, растворы для инъекций, средства для ингаляции и аэрозоли. В связи с этим, кристаллические формы по настоящему изобретению могут быть введены различными путями, включая пероральный, буккальный, парентеральный, внутривенный, внутридермальный (например подкожное, внутримышечное введение), трансдермальный и т.д. Кроме того, кристаллическая форма может быть введена преимущественно локально, а не системно, например в виде депо-композиции или композиции с замедленным высвобождением.
Подходящие композиции для применения в настоящем изобретении можно найти в руководстве-справочнике Remington: The Science and Practice of Pharmacy, 21st Ed., Gennaro, Ed., Lippencott Williams & Wilkins (2003), включенном в настоящий документ посредством ссылки. Фармацевтические композиции, описанные в этом документе, могут быть получены с помощью способов, известных специалистам в данной области техники, т.е. с помощью традиционных способов смешивания, растворения, гранулирования, изготовления драже, растирания в порошок, эмульгирования, инкапсулирования, захватывания или лиофилизации. Описанные далее способы и вспомогательные вещества приведены только для иллюстрации и никоим образом не для ограничения.
В одном из предпочтительных воплощений кристаллическую форму по настоящему изобретению готовят в виде композиции с пролонгированным высвобождением, контролируемым высвобождением, замедленным высвобождением, задержанным высвобождением или отсроченным высвобождением, например в полупроницаемых матрицах из твердых гидрофобных полимеров, содержащих терапевтический агент. Для пролонгированного высвобождения были созданы различные типы материалов, и они хорошо известны специалистам в данной области техники. Современные композиции с замедленным высвобождением используют покрытые пленкой таблетки, системы, состоящие из множества частиц, или системы из гранул, матриксные технологии с применением гидрофильных или гидрофобных материалов и таблетки на основе воска с вспомогательными веществами, формирующими поры (см., например, Huang, et al. Drug Dev. Ind. Pharm. 29:79 (2003); Peamchob, et al. Drug Dev. Ind. Pharm. 29:925 (2003); Maggi, et al. Eur. J. Pharm. Biopharm. 55:99 (2003); Khanvilkar, et al., Drug Dev. Ind. Pharm. 228:601 (2002); и Schmidt, et al., Int. J. Pharm. 216:9 (2001)). Системы доставки с пролонгированным высвобождением могут, в зависимости от их конструкции, высвобождать соединения на протяжении часов или дней, например в течение 4, 6, 8, 10, 12, 16, 20, 24 или более часов. Обычно композиции с замедленным высвобождением могут быть получены с помощью природных или синтетических полимеров, например полимерных винилпирролидонов, таких как поливинилпирролидон (PVP); карбоксивиниловых гидрофильных полимеров; гидрофобных и/или гидрофильных гидроколлоидов, таких как метилцеллюлоза, этилцеллюлоза, гидроксипропилцеллюлоза и гидроксипропилметилцеллюлоза; и карбоксиполиметилен.
Композиции с пролонгированным или замедленным высвобождением также могут быть получены с помощью природных ингредиентов, таких как минералы, включая диоксид титана, диоксид кремния, оксид цинка и глина (см. патент США №6,638,521, включенный в настоящий документ посредством ссылки). Типичные композиции с пролонгированным высвобождением, которые могут быть применены для доставки соединения по настоящему изобретению, включают композиции, описанные в патентах США №№6,635,680; 6,624,200; 6,613,361; 6,613,358, 6,596,308; 6,589,563; 6,562,375; 6,548,084; 6,541,020; 6,537,579; 6,528,080 и 6,524,621, каждый из которых включен в настоящий документ посредством ссылки. Композиции с контролируемым высвобождением, представляющие особый интерес, включают композиции, описанные в патентах США №№6,607,751; 6,599,529; 6,569,463; 6,565,883; 6,482,440; 6,403,597; 6,319,919; 6,150,354; 6,080,736; 5,672,356; 5,472,704; 5,445,829; 5,312,817 и 5,296,483, каждый из которых включен в настоящий документ посредством ссылки. Специалисты в данной области техники легко могут определить другие композиции с замедленным высвобождением, которые могут быть применены аналогичным образом.
Для перорального введения кристаллическая форма по настоящему изобретению может быть легко введена в лекарственную форму путем объединения с фармацевтически приемлемым вспомогательными веществами, хорошо известными в данной области техники. Такие вспомогательные вещества позволяют ввести соединение в состав таблеток, пилюль, драже, капсул, эмульсий, гидрофобных и гидрофильных суспензий, жидкостей, гелей, сиропов, взвесей, суспензий и тому подобного, для перорального приема пациентом, который получает лечение. Фармацевтические препараты для перорального применения могут быть получены с помощью смешивания соединения с твердым вспомогательным веществом, необязательно измельчения полученной смеси, и переработки смеси гранул после добавления подходящих вспомогательных веществ, при необходимости, для получения таблеток или ядер драже. Подходящие вспомогательные вещества представляют собой, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; целлюлозные препараты, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, натриевая соль карбоксиметилцеллюлозы и/или поливинилпирролидон (PVP). При необходимости, могут быть добавлены вещества для улучшения распадаемости таблеток, такие как поперечно-сшитый поливинилпирролидон, агар или альгиновая кислота, или ее соль, такая как альгинат натрия.
Фармацевтические препараты, которые могут применяться перорально, включают твердые капсулы, выполненные из желатина, а также мягкие, запечатанные капсулы, выполненные из желатина и пластификатора, такого как глицерин или сорбит. Твердые капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими веществами, такими как крахмалы, и/или со смазывающими веществами, такими как тальк или стеарат магния, и, необязательно, со стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы. Дозировки всех композиций для перорального введение должны быть подходящими для такого введения.
Ядра драже обеспечиваются подходящими оболочками. Для этих целей могут быть использованы концентрированные растворы сахара, которые необязательно могут содержать гуммиарабик, тальк, поливинилпирролидон, гель карбомера, полиэтиленгликоль и/или диоксид титана, растворы лака и подходящие органические растворители или смеси растворителей. Для идентификации или характеризации различных комбинаций доз активного соединения к оболочкам таблеток или драже могут быть добавлены красители или пигменты.
Кристаллические формы, описанные в настоящем документе, также могут быть введены в состав лекарственных форм для парентерального введения с помощью инъекции, например, болюсной инъекции или непрерывной инфузии. Для инъекции соединение может быть введено в состав препарата путем растворения, суспендирования или эмульгирования в водном или неводном растворителе, таком как растительные или другие похожие масла, синтетические глицериды алифатических кислот, сложные эфиры высших алифатических кислот или пропиленгликоля, при необходимости, с традиционными добавками, такими как солюбилизаторы, изотонические средства, суспендирующие средства, эмульгирующие средства, стабилизаторами и консерванты. В некоторых вариантах осуществления кристаллические формы по изобретению могут входить в состав водных растворов, предпочтительно в физиологически совместимых буферах, таких как раствор Хэнкса, раствор Рингера или физиологический солевой буфер. Композиции для инъекций могут быть представлены в виде стандартной лекарственной формы, например в ампулах или в многодозовых контейнерах, с добавленным консервантом. Композиции могут иметь такие формы, как суспензии, растворы или эмульсии в масляных или водных основах, и могут содержать вспомогательные средства, такие как средства для суспендирования, стабилизирующие и/или диспергирующие средства.
Фармацевтические композиции для парентерального введения включают водные растворы активных соединений в водорастворимой форме. Кроме того, суспензии активных соединений могут быть получены в виде подходящих масляных суспензий для инъекций. Подходящие гидрофобные растворители или основы включают жирные масла, такие как кунжутное масло, или синтетические эфиры жирных кислот, такие как этилолеат или триглицериды, или липосомы. Водные суспензии для инъекций могут содержать вещества, которые увеличивают вязкость суспензии, такие как карбоксиметилцеллюлоза натрия, сорбит или декстран. Суспензия также может необязательно содержать подходящие стабилизаторы или средства, увеличивающие растворимость соединений, что позволяет получать высококонцентрированные растворы. В альтернативном варианте активный ингредиент может находиться в форме порошка, предназначенного для объединения, перед употреблением, с подходящим носителем, например, со стерильной апирогенной водой.
Системное введение также может быть осуществлено трансмукозальным или трансдермальным способом. Для трансмукозального или трансдермального введения в композиции применяют вещества, подходящие для проникновения через барьер, который нужно сделать проницаемым в данном случае. Для местного введения агенты вводят в состав мазей, кремов, бальзамов, порошков и гелей. В одном варианте осуществления средство для трансдермальной доставки может представлять собой DMSO. Трансдермальные системы доставки могут включать, например, пластыри. Для трансмукозального введения в композиции применяют вещества, подходящие для проникновения через барьер, который нужно сделать проницаемым в данном случае. Такие проникающие вещества, как правило, известны в данной области техники. Типичные композиции для трансдермальной доставки, которые могут найти применение в настоящем изобретении, включают композиции, описанные в патентах США №№6,589,549; 6,544,548; 6,517,864; 6,512,010; 6,465,006; 6,379,696; 6,312,717 и 6,310,177, каждый из которых включен в настоящий документ посредством ссылки.
Для буккального введения композиции могут принимать форму таблеток или пастилок, изготавливаемых традиционным образом.
Помимо описанных выше композиций кристаллическая форма по настоящему изобретению также может быть введена в состав лекарственных препаратов-депо. Такие композиции длительного действия могут быть введены путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. Таким образом, например, соединения могут быть смешаны с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или с ионообменными смолами, или в виде умеренно растворимых производных, например в виде умеренно растворимой соли.
Фармацевтические композиции также могут включать подходящие носители или вспомогательные вещества в твердом виде или в виде гелевой фазы. Примеры таких носителей или вспомогательных веществ включают кальция карбонат, кальция фосфат, различные сахара, крахмалы, производные целлюлозы, желатины и полимеры, такие как полиэтиленгликоли, но не ограничиваются только ими.
Фармацевтические композиции, подходящие для применения в настоящем изобретении, включают композиции, содержащие активные ингредиенты в терапевтически эффективном количестве. Настоящее изобретение также рассматривает фармацевтические композиции, включающие кристаллические формы по изобретению в смеси с эффективным количеством других терапевтических агентов в качестве партнеров по комбинации, в особенности тех, которые применяют для лечения заболевания и состояния, на которые можно оказывать влияния посредством ингибирования SGLT, такие как антидиабетические средства, средства, снижающие/модулирующие уровень липидов, средства для лечения осложнений, вызываемых диабетом, средства против ожирения, антигипертензивные средства, противогиперурикемические средства и средства для лечения хронической сердечной недостаточности, атеросклероза или связанных с ними заболеваний. Эффективное количество соединения и/или партнера по комбинации, конечно, будут зависеть от субъекта, получающего лечение, тяжести состояния и способа введения. Определение эффективного количества находится в пределах компетенции специалистов в данной области техники, в особенности, в свете детального раскрытия, предоставленного в настоящем документе. Как правило, действенное или эффективное количество соединения определяют путем первоначального введения низкой дозы или небольшого количества и последующего постепенного увеличения вводимой дозы или доз до тех пор, пока не получают желаемый терапевтический эффект у получающего лечение субъекта, с минимальными или отсутствующими токсическими побочными эффектами. Применяемые для определения подходящей дозы и режима дозирования для введения по настоящему изобретению способы описаны, например, в руководствах «Goodman и Oilman's The Pharmacological Basis of Therapeutics», 11th Ed., Brunton, Lazo и Parker, Eds., McGraw-Hill (2006), и Remington: The Science и Practice of Pharmacy, 21st Ed., Gennaro, Ed., Lippencott Williams & Wilkins (2003), оба из которых включены в настоящий документ посредством ссылки.
IV. Способы применения
Настоящее изобретение, кроме того, относится к способам применения кристаллических форм (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола для предупреждения и лечения заболевания. В одном варианте осуществления настоящее изобретение относится к способу лечения заболевания или состояния, на которое влияет ингибирование SGLT2, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества композиции, включающей кристаллическую форму соединения по настоящему изобретению. Заболевания, на которые влияет ингибирование SGLT2, включают (но не ограничиваются только ими) сахарный диабет 1-го и 2-го типа, гипергликемию, осложнения, вызываемые диабетом (такие как ретинопатия, нефропатия, нейропатия, язвы, микро- и макроангиопатии, подагра и синдром диабетической стопы), резистентность к инсулину, метаболический синдром (синдром X), гиперинсулинемию, повышенное артериальное давление, гиперурикемию, ожирение, отечность, дислипидемию, хроническую сердечную недостаточность, атеросклероз, рак и связанные с ними заболевания, которые включают введение эффективного количества (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола нуждающемуся в этом субъекту. В другом воплощении изобретение относится к способу применения кристаллического соединения для получения лекарственного препарата для лечения сахарного диабета 1-го и 2-го типа, гипергликемии, осложнений, вызываемых диабетом, резистентности к инсулину, метаболического синдрома, гиперинсулинемии, повышенного артериального давления, гиперурикемии, ожирения, отечности, дислипидемии, хронической сердечной недостаточности, атеросклероза, рака и связанных с ними заболеваний. В других вариантах осуществления изобретение относится к способу лечения сахарного диабета 1-го типа, сахарного диабета 2-го типа, гипергликемии, осложнений, вызываемых диабетом, резистентности к инсулину, метаболического синдрома, гиперинсулинемии, повышенного артериального давления, гиперурикемии, ожирения, отечности, дислипидемии, хронической сердечной недостаточности, атеросклероза и рака.
В других вариантах осуществления настоящее изобретение относится к способу лечения диабета, включающему введение нуждающемуся в этом субъекту терапевтически эффективного количества композиции, включающей кристаллическую форму соединения по настоящему изобретению. Диабет может представлять собой любую подходящую форму диабета, включая сахарный диабет 1-го типа, сахарный диабет 2-го типа и осложнения, вызываемые диабетом, но не ограничиваясь только этим. В некоторых вариантах осуществления диабет представляет собой сахарный диабет 1-го типа. В некоторых других вариантах осуществления диабет представляет собой сахарный диабет 2-го типа.
Настоящее изобретение также включает применение кристаллических форм (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола в комбинации с другими терапевтическими агентами, в особенности с теми, которые применяют для лечения вышеупомянутых заболеваний и состояний, таких как антидиабетические средства, средства, снижающие/модулирующие уровень липидов, средства для лечения осложнений, вызываемых диабетом, средства против ожирения, антигипертензивные средства, средства против гиперурикемии и средства для лечения хронической сердечной недостаточности, атеросклероза или связанных с ними нарушений. Специалистам в данной области техники понятно, что другие терапевтические средства, которые будут обсуждаться ниже, могут иметь несколько терапевтических применений и перечисление средства в одной из конкретных категорий никоим образом не должно рассматриваться ограничение его полезности в комбинированной терапии с соединением по настоящему изобретению.
Примеры антидиабетических средств, подходящих для применения в комбинации с кристаллической формой (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола по настоящему изобретению, включают инсулин и соединения, имитирующие действие инсулина, сульфонилмочевины (такие как ацетогексамид, карбутамид, хлорпропамид, глибенкламид, глиборнурид, гликлазид, глимепирид, глипизид, гликвидон, глизоксепид, глибурид, гликлопирамид, толазамид, толцикламид, толбутамид и т.п.), усилители секреции инсулина (такие как JTT-608, глибузол и т.п.), бигуаниды (такие как метформин, буформин, фенформин и т.п.), комбинации сульфонилмочевина/бигуанид (такие как глибурид/метформин и т.п.), меглитиниды (такие как репаглинид, натеглинид, митиглинид и т.п.), тиазолидиндионы (такие как розиглитазон, пиоглитазон, изаглитазон, нетоглитазон, ривоглитазон, балаглитазон, дарглитазон, CLX-0921 и т.п.), комбинации тиазолидиндион/бигуанид (такие как пиоглитазон/метформин и т.п.), оксадиазолидиндионы (такие как YM440 и т.п.), агонисты рецептора, активируемого пролифератором пероксисом (РРАР)-гамма (такие как фарглитазар, метаглидазен, МВХ-2044, GI 262570, GW1929, GW7845 и т.п.), двойные агонисты PPAR-альфа/гамма (такие как мураглитазар, навеглитазар, тезаглитазар, пелиглитазар, JTT-501, GW-409544, GW-501516 и т.п.), пан-агонисты PPAR-альфа/гамма/дельта (такие как PLX204, GlaxoSmithKline 625019, GlaxoSmithKline 677954 и т.п.), агонисты рецептора (RXR) ретиноидов-х (такие как ALRT-268, AGN-4204, MX-6054, AGN-194204, LG-100754, бексаротен и т.п.), ингибиторы альфа-глюкозидазы (такие как акарбоза, миглитол и т.п.), стимуляторы инсулинового рецептора тирозинкиназы (такие как TER-17411, L-783281, KRX-613 и т.п.), ингибиторы трипептидилпептидазы II (такие как UCL-1397 и т.п.), ингибиторы дипептидилпептидазы IV (такие как ситаглиптин, вилдаглиптин, денаглиптин, саксаглиптин, алоглиптин, дутоглиптин, NVP-DPP728, Р93/01, Р32/98, FE 99901, TS-021, TSL-225, GRC8200, соединения, описанные в патентах США №№6,869,947; 6,727,261; 6,710,040; 6,432,969; 6,172,081; 6,011,155 и т.п.), активаторы глюкокиназы (такие как ARRY-403, пираглиатин (R04389620), R00281675, МК-0941, ТТР355, GKA50, GKA60, GKM-001, PSN010, PSN-GK1, соединения, описанные в работе Sarabu, R., et al., Expert Opinion on Therapeutic Patents, Vol.21, No.1, 2011, pp.13-33, и т.п.), ингибиторы протеинтирозинфосфатазы-lB (такие как KR61639, IDD-3, РТР-3848 РТР-112, ОС-86839, PNU-177496, соединения, описанные в работе Vats, R.K., et al., Current Science, Vol.88, No.2, 25 January 2005, pp.241-249, и т.п.), ингибиторы гликогенфосфорилазы (такие как NN-4201, СР-368296 и т.п.), ингибиторы глюкозо-6-фосфатазы, ингибиторы фруктозо-1,6-бисфосфатазы (такие как CS-917, MB05032 и т.п.), ингибиторы пируватдегидрогеназы (такие как AZD-7545 и т.п.), производные имидазолина (такие как BL11282 и т.п.), ингибиторы глюконеогенеза в печени (такие как FR-225659 и т.п.), D-хироинозитол, ингибиторы киназы-3 гликогенсинтетазы (такие как соединения, описанные в работе Vats, R.K., et al., Current Science, Vol.88, No.2,25 January 2005, pp.241-249, и т.п.), ингибиторы дегидрогеназы 11-бета-гидроксистероидов типа 1 (такие как карбеноксолон, INCB 13739 и т.п.), антагонисты глюкагонового рецептора (такие как BAY-27-9955, NN-2501, NNC-92-1687 и т.п.), глюкагон-подобный пептид-1 (GLP-1), агонисты рецептора GLP-1 (такие как эксенатид, лираглутид, CJC-1131, AVE-0100, AZM-134, LY-315902, GlaxoSmithKline 716155 и т.п.), амилин, аналоги и агонисты амилина (такие как прамлинтид и т.п.), ингибиторы связывающего жирную кислоту белка (аР2) (такие как соединения, описанные в патентах США №№6,984,645; 6,919,323; 6,670,380; 6,649,622; 6,548,529 и т.п.), агонисты бета-3 адренергического рецептора (такие как солабегрон, CL-316243, L-771047, FR-149175 и т.п.), и другие усилители чувствительности к инсулину (такие как регликсан, ONO-5816, MBX-102, CRE-1625, FK-614, CLX-0901, CRE-1633, NN-2344, ВМ-13125, ВМ-501050, HQL-975, CLX-0900, МВХ-668, МВХ-675, S-15261, GW-544, AZ-242, LY-510929, AR-H049020, GW-501516 и т.п.).
Примеры средств для лечения осложнений, вызываемых диабетом, подходящих для применения в комбинации с кристаллическим соединением по настоящему изобретению, включают ингибиторы альдозоредуктазы (такие как эпалрестат, имирестат, толрестат, миналрестат, поналрестат, зополрестат, фидарестат, аскорбил гамоленат, ADN-138, BAL-ARI8, ZD-5522, ADN-311, GP-1447, IDD-598, ризарестат, зенарестат, метосорбинил, AL-1567, М-16209, TAT, AD-5467, AS-3201, NZ-314, SG-210, JTT-811, линдолрестат, сорбинил и т.п.), ингибиторы образования конечных продуктов гликирования (AGE) (такие как пиридоксамин, ОРВ-9195, ALT-946, ALT-711, пимагедин и т.п.), соединения, разщепляющие AGE (такие как ALT-711 и т.п.), сулодескид, 5-гидрокси-1-метилгидантоин, инсулин-подобный фактор роста-1, фактор роста тромбоцитов, аналоги фактора роста тромбоцитов, эпидермальный фактор роста, фактор роста нервов, уридин, ингибиторы протеинкиназы C (такие как рубоксистаурин, мидостаурин и т.п.), антагонисты натриевого канала (такие как мексилетин, окскарбазепин и т.п.), ингибиторы ядерного фактора каппа-В (NF-каппа В) (такие как декслипотам и т.п.), ингибиторы пероксидазы липидов (такие как тирилазада мезилат и т.п.), ингибиторы N-ацетилированной-альфа-связанной кислотной дипептидазы (такие как GPI-5232, GPI-5693 и т.п.), и производные карнитина (такие как карнитин, левацекамин, левокарнитин, ST-261 и т.п.).
Примеры средств против гиперурикемии, подходящих для применения в комбинации с кристаллическим соединением по настоящему изобретению, включают ингибиторы синтеза мочевой кислоты (такие как аллопуринол, оксипуринол и т.п.), средства, способствующие выведению мочевой кислоты (такие как пробенецид, сульфинпиразон, бензбромарон и т.п.) и средства для снижения pH мочи (такие как гидрокарбонат натрия, цитрат калия, цитрат натрия и т.п.).
Примеры средств, снижающих/модулирующих уровень липидов, подходящих для применения в комбинации с кристаллическим соединением по настоящему изобретению, включают ингибиторы гидроксиметилглутарил-коэнзим A редуктазы (такие как ацитемат, аторвастатин, бервастатин, карвастатин, церивастатин, колестолон, крилвастатин, далвастатин, флувастатин, гленвастатир, ловастатин, мевастатин, нисвастатин, питавастатин правастатин, ритонавир, розувастатин, саквинавир, симвастатин, визастатин, SC-45355, SQ-33600, СР-83101, ВВ-476, L-669262, S-2468, DMP-565, U-20685, BMS-180431, BMY-21950, соединения, описанные в патентах США №№5,753,675; 5,691,322; 5,506,219; 4,686,237; 4,647,576; 4,613,610; 4,499,289 и т.п.), производные фиброевой кислоты (такие как гемфиброзил, фенофибрат, безафибрат, беклобрат, бинифибрат, ципрофибрат, клинофибрат, клофибрат, этофибрат, никофибрат, пирифибрат, ронифибрат, симфибрат, теофибрат, AHL-157 и т.п.), агонисты PPAR-альфа (такие как GlaxoSmithKline 590735 и т.п.), агонисты PPAR-дельта (такие как GlaxoSmithKline 501516 и т.п.), ингибиторы ацил-коэнзим А:холестерин ацилтрансферазы (такие как авасимиб, эфлюсимиб, элдацимиб, лецимибид, NTE-122, MCC-147, PD-132301-2, C1-1011, DUP-129, U-73482, U-76807, TS-962, RP-70676, Р-06139, СР-113818, RP-73163, FR-129169, FY-038, ЕАВ-309, KY-455, LS-3115, FR-145237, T-2591, J-104127, R-755, FCE-27677, FCE-28654, YIC-C8-434, CI-976, RP-64477, F-1394, CS-505, CL-283546, YM-17E, 447С88, YM-750, Е-5324, KW-3033, HL-004 и т.п.), пробукол, агонисты рецептора тиреоидного гормона (такие как лиотиронин, левотироксин, КВ-2611, GC-1 и т.п.), ингибиторы абсорбции холестерина (такие как эзетимиб, SCH48461 и т.п.), ингибиторы ассоциированной с липопротеином фосфолипазы А2 (такие как рилапладиб, дарапладиб и т.п.), ингибиторы микросомального белка-переносчика триглицеридов (такие как СР-346086, BMS-201038, соединения, описанные в патентах США №№5,595,872; 5,739,135; 5,712,279; 5,760,246; 5,827,875; 5,885,983; 5,962,440; 6,197,798; 6,617,325; 6,821,967; 6,878,707 и т.п.), активаторы рецептора липопротеинов низкой плотности (такие как LY295427, MD-700 и т.п.), ингибиторы липоксигеназы (такие как соединения, описанные в WO 97/12615, WO 97/12613, WO 96/38144 и т.п.), ингибиторы карнитин-пальмитоил-трансферазы (такие как этомоксир и т.п.), ингибиторы сквален-синтазы (такие как YM-53601, TAK-475, SDZ-268-198, BMS-188494, A-87049, RPR-101821, ZD-9720, RPR-107393, ER-27856, соединения, описанные в патентах США №№5,712,396; 4,924,024; 4,871,721 и т.п.), производные никотиновой кислоты (такие как аципимокс, никотиновая кислота, никотинамид, никомол, ницеритрол, никорандил и т.п.), соединения, способствующие выведению желчных кислот (такие как колестипол, холестирамин, колестилан, колесевелам, GT-102-279 и т.п.), ингибиторы котранспортера натрия/жирной кислоты (такие как 264W94, S-8921, SD-5613 и т.п.), и ингибиторы транспортного белка эфиров холестерина (такие как торцеграпиб, JTT-705, PNU-107368E, SC-795, СР-529414 и т.п.).
Примеры средств против ожирения, подходящих для применения в комбинации с кристаллическим соединением по настоящему изобретению, включают ингибиторы обратного захвата серотонина-норэпинефрина (такие как сибутрамин, милнаципран, миртазапин, венлафаксин, дулоксетин, десвенлафаксин и т.п.), ингибиторы обратного захвата норэпинефрина-допамина (такие как радафаксин, бупропион, аминептин и т.п.), ингибиторы обратного захвата серотонина-норэпинефрина-допамина (такие как тезофенсин и т.п.), селективные ингибиторы обратного захвата серотонина (такие как циталопрам, эсциталопрам, флуоксетин, флувоксамин, пароксетин, сертралин и т.п.), селективные ингибиторы обратного захвата норэпинефрина (такие как ребоксетин, атомоксетин и т.п.), стимуляторы высвобождения норэпинефрина (такие как ролипрам, YM-992 и т.п.), средства, снижающие аппетит (такие как амфетамин, метамфетамин, декстроамфетамин, фентермин, бензфетамин, фендиметразин, фенметразин, диэтилпропион, мазиндол, фенфлурамин, дексфенфлурамин, фенилпропаноламин и т.п.), агонисты допаминовых рецепторов (такие как ER-230, допрексин, бромкриптина мезилат и т.п.), антагонисты Нз-гистаминового рецептора (такие как импентамин, тиоперамид, ципроксифан, клобенпропит, GT-2331, GT-2394, А-331440, и т.п.), агонисты рецептора 5-НТ2 с (такие как 1-(м-хлорфенил)пиперазин (м-СРР), миртазапин, APD-356 (лоркасерин), SCA-136 (вабикасерин), ORG-12962, ORG-37684, ORG-36262, ORG-8484, Ro-60-175, Ro-60-0332, VER-3323, VER-5593, VER-5384, VER-8775, LY-448100, WAY-161503, WAY-470, WAY-163909, MK-212, BVT.933, YM-348, IL-639, IK-264, ATH-88651, ATHX-105 и т.п. (см., например, работу Nilsson BM, J. Med. Chem. 2006, 49:4023-4034)), агонисты бета-3 адренергического рецептора (такие как L-796568, CGP 12177, BRL-28410, SR-58611A, ICI-198157, ZD-2079, BMS-194449, BRL-37344, CP-331679, CP-331648, CP-114271, L-750355, BMS-187413, SR-59062A, BMS-210285, LY-377604, SWR-0342SA, AZ-40140, SB-226552, D-7114, BRL-35135, FR-149175, BRL-26830A, CL-316243, AJ-9677, GW-427353, N-5984, GW-2696 и т.п.), агонисты холецистокининовых рецепторов (такие как SR-146131, SSR-125180, BP-3,200, A-71623, A-71378, FPL-15849, GI-248573, GW-7178, GI-181771, GW-7854, GW-5823, и т.п.), комбинации антидепрессантов/ингибиторов ацетилхолинэстеразы (такие как венлафаксин/ривастигмин, сертралин/галантамин и т.п.), ингибиторы липазы (такие как орлистат, ATL-962 и т.п.), противоэпилептические средства (такие как топирамат, зонисамид и т.п.), лептин, аналоги лептина и агонисты рецептора лептина (такие как LY-355101 и т.п.), антагонисты рецептора нейропептида Y (NPY) и модуляторы рецептора нейропептида Y (такие как SR-120819-A, PD-160170, NGD-95-1, BIBP-3226, 1229-U-91, CGP-71683, BIBO-3304, СР-671906-01, J-115814 и т.п.), цилиарный нейротрофический фактор (такие как «Axokine» и т.п.), агонисты бета-рецептора тиреоидного гормона (такие как КВ-141, GC-1, GC-24, GB98/284425 и т.п.), антагонисты каннабиоидного рецептора СВ1 (такие как римонабант, SR147778, SLV 319 и т.п. (см., например, работу Antel J et al., J. Med. Chem. 2006, 49:4008-4016)), антагонисты рецептора меланиноконцентрирующего гормона (такие как GlaxoSmithKline 803430X, GlaxoSmithKline 856464, SNAP-7941, T-226296 и т.п. (см., например, Handlon AL и Zhou Н, J. Med. Chem. 2006, 49:4017-4022)), агонисты рецептора меланокортина-4 (включая РТ-15, Ro27-3225, THIQ, NBI 55886, NBI 56297, NBI 56453, NBI 58702, NBI 58704, MB243 и т.п. (см., например, работу Nargund RP et al., J. Med. Chem. 2006, 49:4035-4043)), антагонисты селективного мускаринового рецептора Mi (такие как телензепин, пирензепин и т.п.), антагонисты опиоидного рецептора (такие как налтрексон, метилналтрексон, налмефен, налоксон, алвимопан, норбиналторфимин, налорфин и т.п.) и их комбинации.
Примеры антигипертензивных средств и средств для лечения хронической сердечной недостаточности, атеросклероза или связанных с ними заболеваний, подходящих для применения в комбинации с кристаллическим соединением по настоящему изобретению, включают бимокломол, ингибиторы ангиотензин-конвертирующего фермента (такие как каптоприл, эналаприл, фозиноприл, лизиноприл, периндоприл, хинаприл, рамиприл и т.п.), ингибиторы нейтральной эндопептидазы (такие как тиорфан, омапатрилат, MDL-100240, фасидотрил, сампатрилат, GW-660511, миксанприл, SA-7060, E-4030, SLV-306, экадотрил и т.п.), антагонисты рецептора ангиотензина II (такие как кандесартана цилексетил, эпросартан, ирбесартан, лозартан, олмесартана медоксомил, телмисартан, вальсартан, тасосартан, энолтасосартан и т.п.), ингибиторы эндотелин-конвертирующего фермента (такие как CGS 35066, CGS 26303, CGS-31447, SM-19712 и т.п.), антагонисты эндотелинового рецептора (такие как траклир, ситаксентан амбрисентан, L-749805, TBC-3214, BMS-182874, BQ-610, TA-0201, SB-215355, PD-180988, BMS-193884, дарусентан, ТВС-3711, босентан, тезосентан, J-104132, YM-598, S-0139, SB-234551, RPR-118031A, ATZ-1993, RO-61-1790, ABT-546, энласентан BMS-207940 и т.п.), диуретические средства (такие как гидрохлортиазид, бендрофлуметиазид, трихлорметиазид, индапамид, метолазон, фуросемид, буметанид, торасемид, хлорталидон, метолазон, гидрофлуметиазид, трипамид, мефрузид, бензилгидрохлортиазид, пенфлутизид, метиклотиазид, азосемид, этакриновая кислота, торасемид, пиретанид, метикран, калия канреноат, спиронолактон, триамтерен, аминофиллин, циклетанин, LLU-альфа, PNU-80873A, изосорбид, D-маннит, D-сорбит, фруктоза, глицерин, ацетазоламид, метазоламид, FR-179544, ОРС-31260, ликсиваптан, кониваптан и т.п.), антагонисты кальциевых каналов (такие как амлодипин, бепридил, дилтиазем, фелодипин, исрадипин, никардипин, нимодипин, верапамил, S-верапамил, аранидипин, эфонидипин, барнидипин, бенидипин, манидипин, цилнидипин, низолдипин, нитрендипин, нифедипин, нилвадипин, фелодипин, пранидипин, лерканидипин, исрадипин, элгодипин, азелнидипин, лацидипин, ватанидипин, лемилдипин, дилтиазем, клентиазем, фасудил, бепридил, галлопамил и т.п.), сосудорасширяющие антигипертензивные средства (такие как индапамид, тодралазин, гидралазин, кадралазин, будралазин и т.п.), бетаблокаторы (такие как ацебутолол, бисопролол, эсмолол, пропранолол, атенолол, лабеталол, карведиол, метопролол и т.п.), симпатические блокирующие средства (такие как амосулалол, теразозин, буназозин, празозин, доксазозин, пропранолол, атенолол, метопролол, карведилол, нипрадилол, целипролол, небиволол, бетаксолол, пиндолол, тертатолол, бевантолол, тимолол, картеолол, небиволол, бопиндоло, нипрадилол, пенбутолол, ацебутолол, тилисолол, надолол, урапидил, индорамин и т.п.), агонисты альфа-2-адренорецептора (такие как клонидин, метилдопа, CHF-1035, гуанабенз ацетат, гуанфацин, моксонидин, лофексидин, талипексол и т.п.), антигипертензивные средства центрального действия (такие как резерпин и т.п.), ингибиторы агрегации тромбоцитов (такие как варфарин, дикумарол, фенпрокумон, аценокумарол, анизиндион, фениндион, ксимелагатран и т.п.), и антитромбоцитарные средства (такие как аспирин, клопидогрель, тиклопидин, дипиридамол, цилостазол, этиликозапентат, сарпогрелат, дилазеп, трапидил, берапрост и т.п.).
Кроме того, в другом аспекте изобретение относится к фармацевтической композиции, включающей эффективное количество кристаллической формы (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола и по меньшей мере одно из группы терапевтических средств, перечисленных выше в качестве партнеров по комбинации, в фармацевтически приемлемом вспомогательном веществе.
Кристаллическое соединение по настоящему изобретению также полезно для лечения нарушений обмена глюкозы. В некоторых вариантах осуществления настоящее изобретение относится к способу снижения уровня глюкозы в крови нуждающегося в этом субъекта, включающему введение субъекту эффективного количества композиции, содержащей кристаллическую форму соединения по настоящему изобретению. В других вариантах осуществления настоящее изобретение относится к способу снижения уровня гликозилированного гемоглобина (HbA1c) в плазме крови нуждающегося в этом субъекта, включающему введение субъекту эффективного количества композиции, содержащей кристаллическую форму соединения по настоящему изобретению. В еще одних вариантах осуществления настоящее изобретение относится к способу повышения экскреции глюкозы с мочой у нуждающегося в этом субъекта, включающему введение субъекту эффективного количества композиции, содержащей кристаллическую форму соединения по настоящему изобретению.
Лечение по настоящему изобретению может быть проведено профилактически для предупреждения или задержки наступления или прогрессирования заболевания или состояния (такого как гипергликемия) или терапевтически для достижения требуемого эффекта (такого как требуемый уровень глюкозы в сыворотке крови) в течение длительного периода времени.
Кристаллическая форма (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола может быть введена субъекту, например, пациенту-человеку, пациенту-домашнему животному, такому как кошка или собака, отдельно или совместно с партнером по комбинации, в форме их фармацевтически приемлемых солей или пролекарств, или в форме фармацевтических композиций, в которых соединения и/или партнеры по комбинации смешивают с подходящими носителями или вспомогательным веществом (вспомогательными веществами) в терапевтически эффективном количестве. Таким образом, кристаллическая форма (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола и дополнительный активный агент, который с ней объединен, могут присутствовать в единственной композиции, например в капсуле или в таблетке, или в двух отдельных композициях, которые могут быть одинаковыми или различными, например в форме набора, включающего определенное число доз каждого средства.
Подходящая дозировка соединения буде меняться в зависимости от выбранного способа введения и состава композиции наряду с другими факторами, такими как реакция пациента. Дозировка может быть увеличена или снижена через некоторое время, в соответствии с требованиями конкретного пациента. Исходно, пациенту могут давать низкую дозу, которую затем увеличивают до эффективной дозы, которую способен перенести пациент. Обычно полезная дозировка для взрослых равна от 1 до 2000 мг, предпочтительно от 1 до 200 мг, при введении пероральным путем, и от 0,1 до 100 мг, предпочтительно от 1 до 30 мг, при введении внутривенным путем, в каждом случае введение осуществляют от 1 до 4 раз в день. Если соединение по изобретению вводят в комбинации с другим терапевтическим средством, полезная дозировка партнера по комбинации может быть равна от 20% до 100% дозы, рекомендуемой обычно.
Величина дозы и частота приема может быть подобрана индивидуально для достижения уровня активных соединений в плазме крови, достаточного для поддержания терапевтического эффекта. Предпочтительно терапевтически эффективного уровня в сыворотке крови достигают путем введения однократной суточной дозы, однако режим получения многократных эффективных суточных доз также включается в настоящее изобретение. В случаях местного введения или избирательного поглощения, эффективная локальная концентрация лекарственного средства может быть не связана с концентрацией в плазме крови. Специалист в данной области техники способен оптимизировать терапевтически эффективные локальные дозы без излишнего экспериментирования.
Все публикации и патентные заявки, приведенные в настоящем описании, включены в него посредством ссылки, как если бы было специально и индивидуально указано, что каждая отдельная публикация или патентная заявка включена посредством ссылки. Все противоречия между ссылками, процитированными в настоящем документе, и данным описанием должны разрешаться в пользу последнего. Аналогично, любой конфликт между признанным в данной области техники определением слова или выражения и определением слова или выражения, примененным в данном изобретении, должен быть разрешен в пользу последнего. Несмотря на то, что вышеизложенное изобретение достаточно подробно описано с помощью иллюстраций и примеров для ясности понимания, специалисты в данной области техники в свете идей настоящего изобретения могут легко внести в него некоторые изменения и модификации без отклонения от сущности или объема прилагаемой формулы изобретения. Изобретение далее описано более подробно с помощью конкретных примеров.
Примеры
Приведенные далее примеры предлагаются в иллюстративных целях и не предназначены для ограничения изобретения каким-либо образом. Специалисты в данной области техники легко определят некоторые некритичные параметры, которые могут быть изменены или модифицированы с получением по существу тех же результатов.
Названия соединений, указанные в приведенных далее примерах, получены из показанных структур с помощью алгоритма Struct=Name компании «CambridgeSoft», реализованного в программе «ChemDraw Ultra», версия 10.0. Если не указано иное, структуры соединений, синтезированных по представленным ниже примерам, были подтверждены приведенными далее методами:
(1) Газовую хроматографию-масс-спектрометрию с ионизацией электрораспылением (MS ESI) проводили с помощью масс-спектрометра «Agilent 5973N», оборудованного газовым хроматографом «Agilent 6890» с колонкой HP-5 MS (0,25 мкм покрытие; 30 м×0,25 мм). Ионный источник поддерживали при 230°С и сканирование спектров проводили от 25-500 а.е.м. со скоростью 3,09 сек на скан.
(2) Жидкостную хроматографию высокого давления/масс-спектрометрию (LC-MS) проводили с помощью HPLC «Finnigan Surveyor», оборудованного насосом для четырехкомпонентных смесей, детектором с перестраиваемой длиной волны, установленным на 254 нм, колонкой ХВ-С18 (4,6×50 мм, 5 мкм), и масс-спектрометрической ионной ловушки «Finnigan LCQ» с ионизацией электрораспылением. Сканирование спектров проводили с 80-2000 а.е.м., применяя переменное ионное время в соответствии с числом ионов в источнике. В качестве элюентов применяли смеси B: ацетонитрил и D:вода. Применяли градиент элюции от 10% до 90% B в течение 8 мин со скоростью потока, равной 1,0 мл/мин с конечным удерживанием при 90% B, равным 7 мин. Общее время хроматографии равно 15 мин.
(3) Стандартную одномерную ЯМР-спектроскопию проводят на спектрометрах 400 MHz или 300 MHz Varian Mercury-Plus. Образцы растворяют в дейтерированных растворителях, полученных от компании «Qingdao Tenglong Weibo Technology Co., Ltd.», и переносят в 5 мм Ю ампулы для ЯМР-спектроскопии. Спектры получают при 293 К. Химические сдвиги регистрируют в шкале ррт и соотносят с подходящими сигналами растворителя, такими как 2,49 ppm для DMSO-d6, 1,93 ppm для CD3CN, 3,30 ppm для CD3OD, 5,32 ppm для CD2Cl2 и 7,26 ppm для CDCl3 для 1Н спектров.
Если в тексте применяются аббревиатуры и сокращения, то они имеют приведенные далее значения: ACN, ацетонитрил; Ac2O, уксусный ангидрид; AcOEt, этилацетат; АсОН, уксусная кислота; AlBr3, бромид алюминия; AlCl3, хлорид алюминия; BBr3, трибромид бора; BF3·Et2O, эфират трифтористого бора; n-BuLi, н-бутиллитий; s-BuLi, втор-бутиллитий; t-BuLi, трет-бутиллитий; t-BuOK, трет-бутоксид калия; CaCl2, хлорид кальция; рассч., рассчитанный; CD3OD, метанол-d4; CDCl3, хлорформ-d; CF3SO3H, трифторметансульфоновая кислота; CH2Cl2, метилен хлорид; CH2I2, метилен иодид; CH3CN, ацетонитрил; (COCl)2, оксалилхлорид; DAST, трифторид (диэтиламино)серы; DCM, дихлорметан; DIAD, диизопропил азодикарбоксилат; DMAP, 4-диметиламинопиридин; DMEM, модифицированная по способу Дульбекко среда Игла; DMF, N,N-диметилформамид; DMP, периодинан Десса-Мартина; DMSO, диметилсульфоксид; ЕА, этилацетат; eq, эквиваленты; ESI, ионизация электрораспылением; Et, этил; Et3SiH, триэтилсилан; EtOAc, этилацетат; EtOH, этанол; FBS, эмбриональная бычья сыворотка; h, час; Н2, газообразный водород; H2SO4, серная кислота; Hepes, 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота; 1H ЯМР, протонный ядерный магнитный резонанс; HPLC, высокоэффективная жидкостная хроматография; IPA, изопропиловый спирт (2-пропанол); IPC, контроль в технологическом процессе; K2CO3, карбонат калия; K2CrO7, дихромат калия; KOH, гидроксид калия; LC-ESI-MS, жидкостная хроматография-ионизация электрораспылением-масс-спектрометрия; LC-MS, жидкостная хроматография - масс-спектроскопия; Me, метил; MeOH, метанол; MeSO3H, метансульфоновая кислота; Mg, магний; MgCl2, хлорид магния; мин, минута; MS, масс-спектроскопия; MsOH, метансульфоновая кислота; NaH, гидрид натрия; NaHCO3, бикарбонат натрия; NaOAc, ацетат натрия; NaOH, гидроксид натрия; Na2SO4, сульфат натрия; NH4Cl, хлорид аммония; Pd/C, палладиевый катализатор на углеродном носителе; PE, петролейный эфир; Ph, фенил; POCl3, оксихлорид фосфора; PPh3, трифенилфосфин; Rf, коэффициент удерживания; rt, комнатная температура; SOCl2, тионилхлорид; TBAI, иодид тетрабутиламмония; TFA, трифторуксусная кислота; THF, тетрагидрофуран; TLC, тонкослойная хроматография; TMS, триметилсилил; Tris, трисгидроксиметиламинометан (или 2-амино-2-(гидроксиметил)пропан-1,3-диол).
Пример 1. Получение комплекса (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол, бис(L-пролин)

Пример 1А. Получение 2-циклопропоксиэтанола (1)

К суспензии Mg-порошка (86,7 г, 3,6 моль) и йода (кат.) в безводном THF (0,7 л) медленно добавляли 1,2-дибромэтан (460 г, 2,4 моль) в безводном THF (2 л), здесь медленно означает с такой скоростью, чтобы поддерживать внутреннюю температуру между 40 и 55°С. После окончания добавления по каплям добавляли раствор 2-(2-бромэтил)-1,3-диоксолана (100 г, 0,56 моль) в безводном THF (750 мл). Реакционную смесь поддерживали при 40-55°С в течение 16-ти часов и реакцию останавливали добавлением водного раствора хлорида аммония. Смесь экстрагировали метиленхлоридом. Органический слой высушивали над сульфатом натрия и концентрировали с получением указанного в подзаголовке продукта (27 г) в виде желтого масла, которое применяли непосредственно без последующей очистки.
Пример 1В. Получение 2-циклопропоксиэтил-4-метилбензолсульфоната (2)

К перемешиваемому раствору гидроксида натрия (32 г, 0,8 моль) в воде (180 мл) и THF (180 мл) добавляли соединение по примеру 1А (27 г, 0,26 моль) при температуре от -5 до 0°С. После этого добавляли по каплям раствор и-толуолсульфонилхлорида (52 г, 0,27 моль) в THF (360 мл). Реакционную смесь поддерживали при температуре от -5 до 0°С в течение 16 часов. Реакционную смесь затем хранили при комнатной температуре в течение 30 мин. Органический слой отделяли и водный слой экстрагировали этилацетатом (2×1,0 л). Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4 и концентрировали с получением неочищенного продукта в виде желтого масла (53,3 г). Его применяли непосредственно без последующей очистки.
Пример 1C. Получение 4-(5-бром-2-хлорбензил)фенола (3)

К перемешиваемому раствору 4-бром-1-хлор-2-(4-этоксибензил)бензола (747 г, 2,31 моль) в дихлорметане медленно при -78°С добавляли трибромид бора (1,15 кг, 4,62 моль). Реакционную смесь оставляли для того, чтобы ее температура достигла комнатной температуры. После завершения реакции, протекание которой контролировали с помощью TLC, реакцию останавливали водой. Смесь экстрагировали дихлорметаном. Органический слой промывали водным раствором насыщенного натрия бикарбоната, водой, солевым раствором, высушивали над Na2SO4 и концентрировали. Осадок перекристаллизовывали в петролейном эфире с получением указанного в подзаголовке соединение в виде белого твердого вещества (460 г, выход 68%). 1H ЯМР (CDCl3, 400 MHz): δ 7,23~7,29 (m, 3Н), 7,08 (d, J=8,8 Hz, 2H), 6,79 (d, J=8,8 Hz, 2H), 5,01 (s, 1H), 4,00 (s, 2H).
Пример 1D. Получение 4-бром-1-хлор-2-(4-(2-циклопропоксиэтокси)бензил)бензола (4)

Смесь по примеру 1C (56,7 г, 210 ммоль) и Cs2CO3 (135 г, 420 ммоль) в DMF (350 мл) перемешивали при комнатной температуре в течение 0,5 часов. Добавляли соединение по примеру 1В (53,3 г, 210 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Ее разводили водой (3 л) и экстрагировали EtOAc. Органический слой промывали водой, солевым раствором, высушивали над Na2SO4 и концентрировали. Осадок очищали с помощью колоночной флэш-хроматографии на силикагеле, элюировали смесью петролейный эфир:этилацетат (10:1) с получением указанного в подзаголовке соединение в виде жидкости (51 г, выход 64%). 1Н ЯМР (CDCl3, 400 MHz): δ 7,22~7,29 (m, 3Н), 7,08 (d, J=8,8 Hz, 2H), 6,88 (d, J=8,8 Hz, 2H), 4,10 (t, J=4,8 Hz, 2H), 3,86 (t, J=4,8 Hz, 2H), 3,38-3,32 (m, 1H), 0,62-0,66 (m, 2H), 0,49-0,52 (m, 2H).
Пример 1Е. Получение (2S,3R,4S,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триола (5)

К перемешиваемому раствору по примеру 1D (213 г) в смеси безводный THF/толуол (1:2 (объем/объем), 1,7 л) под аргоном по каплям добавляли н-BuLi (2,5 М гексан, 245,9 мл) при -60±5°С. Смесь перемешивали в течение 30 мин перед внесением в перемешиваемый раствор 2,3,4,6-тетра-O-триметилсилил-β-D-глюколактона (310,5 г) в толуоле (1,6 л) при -60±5°С. Реакционную смесь непрерывно перемешивали при -60±5°С в течение 1-го часа перед тем, как реакцию останавливали водным раствором насыщенного аммония хлорида (1,5 л). Затем смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1-го часа. Органический слой отделяли, и водный слой экстрагировали этилацетатом (3×500 мл). Объединенные органические слои промывали солевым раствором (1 л), высушивали над Na2SO4 и концентрировали. Осадок растворяли в метаноле (450 мл) и при 0°С добавляли метансульфоновую кислоту (9,2 мл). Раствор оставляли нагреваться до комнатной температуры и перемешивали в течение 20-ти часов. Реакцию останавливали водным раствором бикарбоната натрия (50 г) в воде (500 мл) и добавляли дополнительную воду (900 мл). Смесь экстрагировали этилацетатом (3×1,0 л). Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, концентрировали и использовали на следующей стадии непосредственно без последующей очистки.
Пример 1F. Получение комплекса (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол,бис(L-пролин)(7)

К перемешиваемому раствору соединения по примеру 1Е в CH2Cl2/CH3CN (650 мл:650 мл) при -5°С добавляли триэтилсилан (28,2 мл, 563 ммоль), а затем добавляли BF3·Et2O (52,3 мл, 418,9 ммоль). Реакционную смесь перемешивали в течение 16-ти часов, в течение которых ее температура постепенно повышалась до комнатной температуры. Реакцию останавливали доведением рН до 8,0 водным раствором насыщенного бикарбоната натрия. Органические летучие соединения удаляли при пониженном давлении. Осадок разделяли между этилацетатом (2,25 л) и водой (2,25 л). Органический слой отделяли, промывали солевым раствором, высушивали над Na2SO4 и концентрировали с получением неочищенного продукта 6 (230 г, чистота 82,3%). Этот продукт и L-пролин (113,7 г) в EtOH/H2O (15:1 объем/объем, 2,09 л) перемешивали при 80°С в течение 1-го часа, пока раствор не становился прозрачным. Гексан (3,0 л) добавляли по каплям в вышеуказанный горячий раствор в течение 50-ти мин, а температуру поддерживали при примерно 60°С. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Твердое вещество фильтровали и промывали смесью EtOH/H2O (15:1 (объем/объем), 2×300 мл), гексаном (2×900 мл), и высушивали при 45°С при пониженном давлении в течение 10-ти часа и получали указанное в подзаголовке чистое соединение 7 в виде белого твердого вещества (209 г). Чистота (HPLC) 99,2% (UV). 1H ЯМР (CD3OD, 400 MHz): δ 7,25~7,34 (m, 3H), 7,11 (d, J=8,8 Hz, 2H), 6,84 (d, J=8,8 Hz, 2H), 4,03-4,11 (m, 5H), 3,96-4,00 (m, 2H), 3,83-3,90 (m, 3H), 3,68-3,72 (m, 1H), 3,36-3,46 (m, 6H), 3,21-3,30 (m, 3H), 2,26-2,34 (m, 2H), 2,08-2,17 (m, 2H),1,94-2,02 (m, 4H), 0,56-0,57 (m, 2H), 0,52-0,53 (m, 2H).
Пример 2. Прямое получение кристаллического соединения 8 из комплекса 7 Этот пример иллюстрирует получение кристаллической формы (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола.

В 5,0 л 4-горлую колбу, оборудованную механической мешалкой, добавляли исходные кристаллы для совместной кристаллизации (150,0 г) и метанол (300 мл). Смесь перемешивали при комнатной температуре с помощью механического перемешивания (якорная мешалка, 2-лопастная 9 см) до тех пор, пока не сформировался мутный раствор/суспензия, к которому по каплям добавляли дистиллированную воду (1500 мл) со скоростью -12,5 мл/мин. Поскольку в результате экзотермического эффекта от добавления воды к метанолу происходило нагревание смеси, то смесь просветлялась после добавления примерно от 1/5 до 1/3 объема воды. После завершения добавления реакционную смесь непрерывно перемешивали при 80 rpm в течение дополнительных 5-ти часов. Реакционную смесь фильтровали через фильтровальную бумагу со средней скоростью фильтрования, осадок с фильтра промывали дистиллированной водой (450 мл и затем 300 мл) и высушивали при пониженном давлении, применяя масляный насос (~6 мм Hg) при 45°С в течение 48-ми часов и получали целевой продукт в виде белого кристаллического твердого вещества (94,2 г, 93,9% выход, чистота (HPLC): 99,3%).
Пример 3. Прямое получение кристаллического соединения 8 из комплекса 7
Этот пример иллюстрирует альтернативные условия получения кристаллического (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
Процедура А:
В 250 мл 4-горлую колбу помещали исходный комплекс (10,0 г) и метанол (33,5 мл). После нагревания с обратным холодильником в течение 20 мин при механическом перемешивании сформировался прозрачный раствор. К нему медленно по каплям в течение 20-ти мин добавляли воду (67,0 мл). Реакционную смесь медленно охлаждали до комнатной температуры (25°С) на масляной бане и перемешивали еще в течение 3-х часов при комнатной температуре. Реакционную смесь фильтровали через фильтровальную бумагу, и отфильтрованный осадок промывали водой (2×20 мл), высушивали при пониженном давлении при 65°С в течение 8-ми часов и получали белое кристаллическое твердое вещество. Выход: 6,0 г (89,6%)
Процедура B:
В 250 мл 4-горлую колбу помещают исходный комплекс (10,0 г) и метанол (33,5 мл). После перемешивания в течение 20-ти мин с помощью механического мешалки, твердые вещества полностью не растворялись. В колбу медленно по каплям в течение 20-ти мин добавляли воду (67,0 мл). Сначала все оставшиеся твердые вещества растворялись, а затем начинали формироваться новые кристаллы. Реакционную смесь перемешивали еще в течение 3-х часов при комнатной температуре. Реакционную смесь фильтровали через фильтровальную бумагу, и отфильтрованный осадок промывали водой (2×20 мл), высушивали при пониженном давлении при 65°С в течение 8-ми часов и получали белое кристаллическое твердое вещество. Выход: 6,0 г (89,6%).
Процедуры A и B суммированы в приведенной ниже таблице 1 с другими условиями получения кристаллического соединения 8 непосредственно из комплекса 7.
Таблица 1. Сводная таблица условий кристаллизации
| Комплекс (г) | Метанол (мл) | Вода (мл) | Темп. (°С) | Выход (%) |
| 4,0 | 20,0 | 80,0 | 70 | 87,4 |
| 10,0 | 33,5 | 67,0 | 25 | 89,6 |
| 10,0 | 33,5 | 100,0 | 25 | 91,1 |
| 10,0 | 33,5 | 67,0 | 70 | 89,6 |
Пример 4. Прямое получение кристаллического соединения 8 из комплекса 7
Этот пример иллюстрирует получение кристаллической формы (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5 -триола.
Соединение 7 (14,0 кг) растворяли в метаноле (36,2 кг) и деионизированной (DI) воде (11,2 кг) и затем фильтровали. Добавляли дополнительную DI воду (41,3 кг) и затем при 35±5°С добавляли затравочные кристаллы для кристаллизации соединения 8 из раствора. Для завершения осаждения добавляли дополнительную DI воду (41,3 кг). Полученную суспензию фильтровали, и продукт, полученный в виде твердого вещества, промывали на фильтре DI водой, переносили на лотки и высушивали при пониженном давлении при ~65°С и получали 8,75 кг соединения 8.
Пример 5. Непрямое получение кристаллического соединения 8 из комплекса 7

К 200-литровому реакционному сосуду, облицованному с внутренней стороны эмалью и оборудованному двухуровневой лопастной мешалкой и стеклянным холодильником, последовательно добавляли комплекс 7 (7,33 кг), этилацетат (67,5 кг) и чистую воду (74,0 кг). Смесь кипятили с обратным холодильником и перемешивали с обратным холодильником в течение 30-ти мин. Реакционную смесь охлаждали до приблизительно 50°С и отделяли органический слой, а водный слой экстрагировали этилацетатом (34,0 кг). Объединенные органические слои промывали чистой водой (3×74,0 кг) (IPC-тест показал, что IPC-показатель для остатка L-пролина достигался после трех промывок водой). Смесь концентрировали при 40°С при пониженном давлении (~15 мм Hg) в течение 3-х часов до тех пор, пока уровень жидкости не падал ниже лопасти мешалки нижнего уровня. Смесь (18 кг) извлекали и переносили в 20-литровый роторный испаритель. Смесь концентрировали при пониженном давлении (40°С, ~5 мм Hg) до минимального объема. Оставшиеся следовые количества этилацетата удаляли азеотропно при 40°С при пониженном давлении с метанолом (10 кг). Осадок высушивали при пониженном давлении, которое создавали при помощи масляного насоса (~6 мм Hg) при 40°С в течение 10-ти часа и получали соединение 8 в виде белого аморфного твердого вещества (4,67 кг, чистота (HPLC): 99,2%), которое применяли на следующей стадии без последующей очистки.
Перекристаллизацию завершали с помощью приведенных далее стадий. К 100-литровому реакционному сосуду, облицованному с внутренней стороны стеклом и оборудованному двухуровневой лопастной мешалкой и стеклянным холодильником, добавляли описанное выше аморфное соединение 8 (4,67 кг) и метанол (18,0 кг). Смесь кипятили с обратным холодильником при 70°С в течение 30-ти мин до тех пор, пока не получали прозрачный раствор, к которому добавляли чистую воду (45,0 кг) в течение 2-х часов. После завершения добавления (температура реакционной среды равна 41°С), реакционную смесь охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение 15-ти часов. Реакционную смесь фильтровали, осадок промывали чистой водой (2×15 кг) и высушивали при пониженном давлении при 55~60°С в течение 12-ти часов и получали целевой продукт в виде грязно-белого кристаллического твердого вещества (3,93 кг, выход: 84% в двух стадиях; чистота (HPLC): 99,7%).
Пример 6. Прямое получение кристаллического соединения 8 из аморфного соединения 8

В 5-литровую 4-горлую колбу загружали соединение 8 (аморфное), 116 г, и метанол (580 мл). Реакционную смесь нагревали до 60°С с помощью механического перемешивания, и раствор становился прозрачным. К реакционному раствору добавляли по каплям воду (2320 мл) со скоростью 40 мл/мин при 50°С. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь фильтровали, и отфильтрованный осадок промывали водой (2×200 мл), высушивали при пониженном давлении при 55°С в течение 12-ти часов, с получением белого кристаллического соединения 8. Выход равен 112,8 г (97,2%).
Пример 7. Прямое получение кристаллического соединения 8 из неочищенного соединения 6 (с затравочным кристаллом)

Этот пример иллюстрирует получение кристаллической формы (2S, 3R,4R,5S, 6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола.
В 250-милилитровую 4-горлую колбу загружали соединение 6 (12,0 г, чистота согласно HPLC: 88,3%) и метанол (48 мл). После нагревания с обратным холодильником в течение 30 мин при перемешивании с помощью магнитной мешалки (120 RPM), к указанному выше раствору добавляли по каплям воду (72 мл) в течение 20-ти минут. После нагревания с обратным холодильником в течение дополнительных 30-ти минут, смесь медленно охлаждали до 40-45°С и добавляли затравочный кристалл (10 мг). После перемешивания еще в течение 2-х часов при 35-40°С, смесь охлаждали медленно до 20-25°С и перемешивали в течение дополнительных 16-ти часов. Смесь фильтровали, и отфильтрованный осадок промывали водой (2×24 мл), высушивали при пониженном давлении при 60-65°С в течение 12-ти часов, и получали грязно-белое кристаллическое соединение 8. Выход равен 10,6 г (88,3%). Чистота согласно HPLC: 91,8%.
Пример 8. Прямое получение кристаллического соединения 8 из неочищенного соединения 6 (без затравочного кристалла)

Этот пример иллюстрирует получение кристаллической формы (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола.
В 100-милилитровую 3-горлую колбу загружали соединение 6 (5,0 г, чистота согласно HPLC: 90,7%) и метанол (20 мл). После нагревания в течение 30 мин при перемешивании с помощью магнитной мешалки (120 RPM), к указанному выше раствору добавляли по каплям воду (30 мл) в течение 20-ти мин. После нагревания с обратным холодильником в течение дополнительных 30-ти мин, смесь медленно охлаждали до 20-25°С в течение 3-х часов. После перемешивания в течение дополнительных 60-ти часов при 20-25°С смесь фильтровали, и отфильтрованный осадок промывали водой (2×10 мл), высушивали при пониженном давлении при 60-65°С в течение 12-ти часов, и получали грязно-белое кристаллическое соединение 8. Выход равен 4,3 г (86%). Чистота согласно HPLC: 92,6%.
Пример 9. Получение кристаллического соединения 8 с помощью единственного растворителя

Этот пример иллюстрирует получение кристаллической формы (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола.
В 40-миллилитровый стеклянный флакон загружали (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (300 мг, чистота согласно HPLC: 99,6%) и этанол (10 мл). После взбалтывания в течение 15-ти минут при 20-25°С, твердое вещество полностью растворялось. Раствор поддерживали в спокойном состоянии, но позволяли растворителю медленно испаряться. Через 2 недели, оставалось только примерно 2 мл этанола и формировалось множество игольчатых кристаллов. Смесь фильтровали, высушивали при пониженном давлении при 60-65°С в течение 12-ти часов, с получением белого кристаллического соединения 8. Выход равен 246 мг (82%). Чистота согласно HPLC: 99,7%.
Пример 10. Перекристаллизация соединения 8
Примерно 100 мг кристаллического соединения 8 растворяли в минимальном количестве растворителя при примерно 60°С. Раствор фильтровали и разделяли на две части, при этом одну часть охлаждали на ледяной бане и перемешивали (быстрый способ), и другую оставляли охлаждаться естественно под воздействием окружающей атмосферы и температуры (медленный способ). Твердые вещества собирали на фильтре, высушивали и анализировали с помощью XRPD. В приведенной ниже таблице суммированы растворители и результаты перекристаллизации. Все сформировавшиеся кристаллы были идентичны XRPD исходного вещества.
| Таблица 2 | ||
| Сводная таблица способов кристаллизации соединения 8 из горячих насыщенных растворов | ||
| Растворители | Способы1 | Результаты2 |
| ACN | Быстрый Медленный | Кристаллы, без изменения Кристаллы, без изменения |
| 95% EtOH | Быстрый Медленный | Кристаллы, без изменения Кристаллы, без изменения |
| EtOAc | Быстрый Медленный | Кристаллы, без изменения Кристаллы, без изменения |
| IPA | Быстрый Медленный | Кристаллы, без изменения Кристаллы, без изменения |
| Бутанол | Быстрый Медленный | Кристаллы, без изменения Кристаллы, без изменения |
| Бутанон | Быстрый Медленный | Кристаллы, без изменения Кристаллы, без изменения |
| 1,4-диоксан-гептан (1:1) | Быстрый Медленный | Кристаллы, без изменения Нет кристаллов |
| IPA-EtOAc (1:1) | Быстрый Медленный | Кристаллы, без изменения Кристаллы, без изменения |
| 1 Быстрый = охлаждение на ледяной бане; медленный = охлаждение под воздействием окружающей атмосферы и температуры. | ||
| 2 Без изменения = XRPD продукта идентична XRPD исходного вещества. | ||
Пример 11. Низкая гигроскопичность кристаллического соединения 8
Тенденцию к гигроскопичности кристаллического соединения 8 в форме порошка тестировали при относительной влажности 75% и 92,5%, при 25°С в течение вплоть до 10-ти дней. Реагенты включали: (1) воду: для местного пользования, Milli,18,2MΩ; (2) NaCl: степень чистоты AR (аналитическая); (3) KNO3: степень чистоты AR; (4) насыщенный раствор NaCl с дополнительным твердым NaCl для контроля условий, соответствующих 25°С/75% RH; и (5) насыщенный раствор KNO3 с дополнительным твердым KNO3 для контроля условий, соответствующих 25°C/92,5%RH. Использованное оборудование включало: (1) эксикаторы, 240 мм ID; и (2) стаканчики для взвешивания с крышками: 50 мм ID×30 мм высотой.
Насыщенные солевые растворы и твердые вещества помещали в индивидуальные эксикаторы, и уравновешивали при 25°С по меньшей мере в течение ночи для достижения желаемых показаний относительной влажности. В каждый эксикатор помещали четыре стаканчика для взвешивания, и уравновешивали в течение ночи. Пустые стаканчики для взвешивания взвешивали, и записывали массы тары (W1). Кристаллическое соединение 8 (0,5 г) добавляли в три стаканчика для взвешивания в каждый эксикатор для формирования тонкого слоя толщиной 1-2 мм. Массу образца в каждом стаканчике (W2) записывают. Один пустой стаканчик для взвешивания применяют для холостой калибровки. В каждом эксикаторе стаканчики оставляют с открытыми крышками. Крышки закрывали, и каждый стаканчик аккуратно взвешивали на день 1, 5 и 10 (W3). Стаканчики возвращали в каждый эксикатор с открытыми крышками сразу же после взвешивания.
Приведенную далее формулу применяли для расчета увеличения массы:

где WB=увеличение массы пустого стаканчика для взвешивания.
| Таблица 3 | |||||||
| Результаты анализа кристаллического соединения 8 при 25°С/75% RH | |||||||
| Масса | Пустой стаканчик | Масса образца | Внешний вид | Стаканчик + Образец | Масса образца | Увеличение массы | Внешний вид |
| Исходная | 1 день | ||||||
| 1 | 30,36142 | 0,50434 | Белый порошок | 30,86727 | 0,50585 | 0,32914% | Белый порошок |
| 2 | 32,96588 | 0,50631 | Белый порошок | 33,47323 | 0,50735 | 0,23503% | Белый порошок |
| 3 | 31,27798 | 0,50066 | Белый порошок | 31,77934 | 0,50136 | 0,16978% | Белый порошок |
| Холост. | 31,92783 | 31,92768 | -0,00015 | ||||
| Среднее | 0,24465% | ||||||
| 5 дней | |||||||
| 1 | 30,86648 | 0,50506 | 0,10509% | Белый порошок | |||
| 2 | 33,47339 | 0,50751 | 0,19948% | Белый порошок | |||
| 3 | 31,77962 | 0,50164 | 0,15779 | Белый порошок | |||
| Холост. | 31,92802 | 0,00019 | |||||
| Среднее | 0,15412 | ||||||
| 10 дней | |||||||
| 1 | 30,86738 | 0,50596 | 0,35095% | Белый порошок | |||
| 2 | 33,47321 | 0,50733 | 0,23108% | Белый порошок | |||
| 3 | 31,77947 | 0,50149 | 0,19574% | Белый порошок | |||
| Холост. | 31,92768 | -0,00015 | |||||
| Среднее | 0,25926% | ||||||
| Таблица 4 | |||||||
| Результаты анализа кристаллического соединения 8 при 5°C/92,5% RH | |||||||
| Масса | Пустой стаканчик | Масса образца | Внешний вид | Стаканчик + Образец | Масса образца | Увеличение массы | Внешний вид |
| Исходная | 1 день | ||||||
| 1 | 34,11948 | 0,50356 | Белый порошок | 34,62330 | 0,50382 | 0,05362% | Белый порошок |
| 2 | 30,13094 | 0,50215 | Белый порошок | 30,63360 | 0,50266 | 0,10355% | Белый порошок |
| 3 | 33,01277 | 0,50546 | Белый порошок | 33,51923 | 0,50646 | 0,19982% | Белый порошок |
| Холост. | 40,35822 | 40,35821 | -0,00001 | ||||
| Среднее | 0,11900% | ||||||
| 5 дней | |||||||
| 1 | 34,62358 | 0,50410 | 0,04170% | Белый порошок | |||
| 2 | 30,63446 | 0,50352 | 0,20711% | Белый порошок | |||
| 3 | 33,51977 | 0,50700 | 0,23939% | Белый порошок | |||
| Холост. | 40,35855 | 0,00033 | |||||
| Среднее | 0,16273 | ||||||
| 10 дней | |||||||
| 1 | 34,62472 | 0,50524 | 0,26015% | Белый порошок | |||
| 2 | 30,63428 | 0,50334 | 0,16330% | Белый порошок | |||
| 3 | 33,51994 | 0,50717 | 0,26511% | Белый порошок | |||
| Холост. | 40,35859 | 0,00037 | |||||
| Среднее | 0,22952% | ||||||
Увеличение массы кристаллического соединения 8 в порошкообразных образцах при относительной влажности 75% и 92,5% при 25°С в течение 10 дней было ниже 0,26%. Таким образом, кристаллическое соединение 8 в порошкообразной форме в условиях эксперимента демонстрирует низкую гигроскопичность.
Пример 12. Получение капсул, содержащих кристаллическое соединение 8
Для получения капсул, содержащих кристаллическое соединение 8, соединение и силицифицированную микрокристаллическую целлюлозу (Prosolv HD90) смешивают и затем просеивают через сетчатый фильтр #30 в полиэтиленовый мешок. Часть смеси кристаллического соединения 8/Prosolv HD90 отбирали. Стеарат магния просеивали через сетчатый фильтр #30 в эту порцию, и смесь перемешивали. 3-хкомпонентную порцию возвращали к большей части смеси соединение 8/Prosolv HD90, и смесь дополнительно перемешивали. Окончательную смесь высыпали в полиэтиленовый мешок. Пустые оболочки капсул (размер 2) взвешивали для определения средней массы капсулы. Конечную смесь в полиэтиленовом мешке засыпали в инкапсулятор «MG2 Planeta» и в каждую капсулу помещали приблизительно по 100 мг конечной смеси. Примерно каждые 5-10 минут отбирали заполненные капсулы для определения приемлемой массы заполнения. Этого делали путем отбора 10 заполненных капсул, взвешивания каждой заполненной капсулы и сравнения результатов с теоретической средней массой капсулы плюс целевая нагрузка в 100 мг смеси. Другие 10 капсул контролировали визуально на наличие трещин, сколов, вмятин, щелей, непредвиденных пятен и включений. Если необходимо, то способ инкапсуляция и загрузки смеси может быть подобран так, чтобы поддерживать надлежащую целевую массу, помещаемую в капсулы. Приемлемые капсулы помещают в полиэтиленовый мешок с двойной выстилкой для дополнительной обработки. Заполненные капсулы сортируют по массе с помощью электронного устройства, отбрасывая недостаточно и чрезмерно заполненные капсулы, и затем капсулы, заполненные приемлемой массой, направляют на дополнительную обработку. Проверенные капсулы с приемлемой массой полируют и помещают в двойные полиэтиленовые мешки.
| Таблица 5 | |||
| Компоненты 20 мг капсул с кристаллическим соединением 8 | |||
| Компонент | Количество на единицу (мг/капсула) | Функция | Стандарт качества |
| Активный ингредиент | |||
| Кристаллическое соединение 8 | 20,00 | Местный | |
| Силицифицированная | Скользящее вещество и гидрофильный матрикс | USP/NF; Ph. Eur, JP. | |
| микрокристаллическая целлюлоза (Prosolv® HD90) | 79,40 | ||
| Стеарат магния (HyQual®, растительной степени чистоты) |
Смазывающее вещество | USP/NF; Ph. Eur., JP | |
| 0,60 | |||
| Размер 2, Желатин, Белое, непрозрачное тело и крышка Coni-Snap®(0999) | |||
| 1 | Оболочка капсулы | Местный | |
Хотя вышеуказанное изобретение было детально описано с помощью иллюстраций и примеров для ясности понимания, специалисту в данной области техники должно быть понятно, что возможны определенные изменения и модификации, осуществляемые в рамках прилагаемой формулы изобретения. Кроме того, каждый документ, упомянутый в настоящем описании, посредством ссылки включен во всей своей полноте в той же степени, как если бы каждый из них был индивидуально включен посредством ссылки. Если возникает противоречие между настоящим описанием и документом, упоминаемым в настоящем описании, предпочтение следует отдавать сведениям, изложенным в настоящем описании.
Claims (33)
1. Кристаллическая форма соединения, имеющего формулу:

характеризуемая порошковой рентгеновской дифрактограммой (XRPD), включающей пики при 11,2, 12,9, 15,5, 17,8, 19,1, 20,0 и 20,7 градусах 2θ (±0,1 градуса 2θ), причем указанную XRPD получают с использованием излучения CuKα1.
характеризуемая порошковой рентгеновской дифрактограммой (XRPD), включающей пики при 11,2, 12,9, 15,5, 17,8, 19,1, 20,0 и 20,7 градусах 2θ (±0,1 градуса 2θ), причем указанную XRPD получают с использованием излучения CuKα1.
2. Кристаллическая форма по п. 1, характеризуемая XRPD, дополнительно включающей один или несколько пиков при 5,4, 11,3, 11,9, 16,3, 20,6, 21,2, 22,8, 23,0, 23,4, 23,6, 23,9, 24,7, 25,4, 25,8, 27,8 и 28,2 градусах 2θ (±0,1 градуса 2θ), причем указанную XRPD получают с использованием излучения CuKα1.
3. Кристаллическая форма по п. 2, характеризуемая XRPD, включающей пики при 11,2, 12,9, 15,5, 17,8, 19,1, 20,0, 20,6, 20,7, 21,2 и 22,8 градусах 2θ (±0,1 градуса 2θ), причем указанную XRPD получают с использованием излучения CuKα1.
4. Кристаллическая форма по п. 2, характеризуемая XRPD, включающей пики при 5,4, 11,2, 11,9, 12,9, 15,5, 16,3, 17,8, 19,1, 20,0 и 20,7 градусах 2θ (±0,1 градуса 2θ), причем указанную XRPD получают с использованием излучения CuKα1.
5. Кристаллическая форма по п. 1, характеризуемая пиками XRPD, которые по существу соответствуют пикам на фиг. 2.
6. Кристаллическая форма по п. 1, характеризуемая спектрами комбинационного рассеяния, включающими один или несколько пиков при примерно 353, 688, 825, 1178, 1205, 1212, 1608, 2945, 3010 и 3063 см-1.
7. Кристаллическая форма по п. 1, характеризуемая спектрами комбинационного рассеяния, включающими пики при примерно 353, 688 и 825 см-1.
8. Кристаллическая форма по п. 1, характеризуемая пиками в спектре комбинационного рассеяния, которые по существу соответствуют пикам на фиг. 4.
9. Кристаллическая форма по п. 1, характеризуемая
порошковой рентгеновской дифрактограммой (XRPD), дополнительно включающей один или несколько пиков при 5,4, 11,3, 11,9, 16,3, 20,6, 21,2, 22,8, 23,0, 23,4, 23,6, 23,9, 24,7, 25,4, 25,8, 27,8 и 28,2 градусах 2θ (±0,1 градуса 2θ), причем указанную XRPD получают с использованием излучения CuKα1; и
спектрами комбинационного рассеяния, включающими один или несколько пиков при примерно 353, 688, 825, 1178, 1205, 1212, 1608, 2945, 3010 и 3063 см-1.
порошковой рентгеновской дифрактограммой (XRPD), дополнительно включающей один или несколько пиков при 5,4, 11,3, 11,9, 16,3, 20,6, 21,2, 22,8, 23,0, 23,4, 23,6, 23,9, 24,7, 25,4, 25,8, 27,8 и 28,2 градусах 2θ (±0,1 градуса 2θ), причем указанную XRPD получают с использованием излучения CuKα1; и
спектрами комбинационного рассеяния, включающими один или несколько пиков при примерно 353, 688, 825, 1178, 1205, 1212, 1608, 2945, 3010 и 3063 см-1.
10. Кристаллическая форма по п. 9, характеризуемая
порошковой рентгеновской дифрактограммой (XRPD), дополнительно включающей один или несколько пиков при 20,6, 21,2 и 22,8 и 28,2 градусах 2θ (±0,1 градуса 2θ), причем указанную XRPD получают с использованием излучения CuKα1; и
спектрами комбинационного рассеяния, включающими один или несколько пиков при примерно 353, 688 и 825 см-1.
порошковой рентгеновской дифрактограммой (XRPD), дополнительно включающей один или несколько пиков при 20,6, 21,2 и 22,8 и 28,2 градусах 2θ (±0,1 градуса 2θ), причем указанную XRPD получают с использованием излучения CuKα1; и
спектрами комбинационного рассеяния, включающими один или несколько пиков при примерно 353, 688 и 825 см-1.
11. Кристаллическая форма по п. 1, характеризуемая эндотермой, полученной методом дифференциальной сканирующей калориметрии (DSC), при примерно 136°С.
12. Кристаллическая форма по п. 1, характеризуемая параметрами элементарной ячейки, которые по существу соответствуют данным на фиг. 7.
13. Фармацевтическая композиция, способная ингибировать натрий-зависимый котранспортер глюкозы 2 (SGLT2), включающая фармацевтически приемлемое вспомогательное вещество и кристаллическую форму соединения (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола по п. 1.
14. Способ получения кристаллической формы соединения по п. 1, включающий стадии, на которых:
(a) объединяют при перемешивании комплекс (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола и бис(L-пролина) и подходящий растворитель, выбранный из группы, состоящей из метанола и этанола, для получения раствора;
(b) к указанному раствору добавляют воду для получения смеси; и
(c) указанную кристаллическую форму выделяют из указанной смеси.
(a) объединяют при перемешивании комплекс (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола и бис(L-пролина) и подходящий растворитель, выбранный из группы, состоящей из метанола и этанола, для получения раствора;
(b) к указанному раствору добавляют воду для получения смеси; и
(c) указанную кристаллическую форму выделяют из указанной смеси.
15. Способ по п. 14, в котором растворитель на стадии (а) представляет собой метанол.
16. Способ по п. 15, в котором отношение метанола к воде в смеси на стадии (b) равно от примерно 1:1 до примерно 1:9 по объему.
17. Способ по п. 15, в котором отношение метанола к воде в смеси на стадии (b) равно примерно 1:5 по объему.
18. Способ по п. 14, в котором отношение комплекса к растворителю и воде в смеси на стадии (b) равно от примерно 1:10 до примерно 1:25 (масс./об.).
19. Способ по п. 14, в котором смесь на стадии (b) дополнительно включает основание.
20. Способ по п. 14, в котором смесь на стадии (b) дополнительно включает соль.
21. Способ по п. 14, в котором смесь на стадии (b) дополнительно включает затравочный кристалл соединения по п. 1.
22. Способ получения кристаллической формы соединения по п. 1, включающий стадии, на которых:
(a) объединяют при перемешивании (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол и подходящий растворитель, выбранный из группы, состоящей из метанола и этанола, для получения раствора; и
(b) из указанного раствора выделяют указанную кристаллическую форму.
(a) объединяют при перемешивании (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол и подходящий растворитель, выбранный из группы, состоящей из метанола и этанола, для получения раствора; и
(b) из указанного раствора выделяют указанную кристаллическую форму.
23. Способ по п. 22, включающий стадии, на которых:
(a) объединяют аморфный (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол и подходящий растворитель, выбранный из группы, состоящей из метанола и этанола, при перемешивании для получения раствора;
(b) к указанному раствору добавляют воду для получения смеси; и
(c) из указанной смеси выделяют указанную кристаллическую форму.
(a) объединяют аморфный (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол и подходящий растворитель, выбранный из группы, состоящей из метанола и этанола, при перемешивании для получения раствора;
(b) к указанному раствору добавляют воду для получения смеси; и
(c) из указанной смеси выделяют указанную кристаллическую форму.
24. Способ по п. 22, дополнительно включающий стадию добавления воды к указанному раствору.
25. Способ по п. 24, в котором растворитель на стадии (а) представляет собой метанол.
26. Способ по п. 25, в котором отношение метанола к воде в смеси на стадии (b) равно от примерно 1:1 до примерно 1:9 по объему.
27. Способ по п. 25, в котором отношение метанола к воде в смеси на стадии (b) равно примерно 1:5 по объему.
28. Способ по п. 24, в котором отношение комплекса к растворителю и воде в смеси на стадии (b) равно от примерно 1:1 до примерно 1:9 по объему.
29. Способ по п. 24, в котором раствор дополнительно включает затравочный кристалл соединения по п. 1.
30. Способ по п. 24, в котором (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол представляет собой аморфный (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол.
31. Способ по п. 30, в котором аморфный (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол получают из (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола бис(L-пролина) путем:
(а2) объединения при перемешивании комплекса (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола и бис(L-пролина), этилацетата и воды для получения раствора; и
(а3) выделения аморфного (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола из указанного раствора стадии (а2).
(а2) объединения при перемешивании комплекса (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола и бис(L-пролина), этилацетата и воды для получения раствора; и
(а3) выделения аморфного (2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-(2-циклопропоксиэтокси)бензил)фенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола из указанного раствора стадии (а2).
32. Композиция для использования в лечении заболевания или состояния, на которое влияет ингибирование SGLT2, содержащая кристаллическую форму соединения по п. 1, где указанное заболевание или состояние выбрано из группы, состоящей из сахарного диабета 1-го типа, сахарного диабета 2-го типа, гипергликемии, осложнений, вызываемых диабетом, резистентности к инсулину, метаболического синдрома, гиперинсулинемии, повышенного артериального давления, гиперурикемии, ожирения, отечности, дислипидемии, хронической сердечной недостаточности, атеросклероза и рака.
33. Композиция для использования в лечении у субъекта заболевания или состояния, на которое влияет ингибирование SGLT2, содержащая кристаллическую форму соединения по п. 1.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNPCT/CN2010/073865 | 2010-06-12 | ||
| PCT/CN2010/073865 WO2011153712A1 (en) | 2010-06-12 | 2010-06-12 | Crystalline form of benzylbenzene sglt2 inhibitor |
| PCT/CN2011/075554 WO2011153953A1 (en) | 2010-06-12 | 2011-06-10 | Crystalline form of benzylbenzene sglt2 inhibitor |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2013101580A RU2013101580A (ru) | 2014-07-20 |
| RU2569491C2 true RU2569491C2 (ru) | 2015-11-27 |
Family
ID=45097470
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2013101580/04A RU2569491C2 (ru) | 2010-06-12 | 2011-06-10 | Кристаллическая форма бензил-бензольного ингибитора sglt |
Country Status (31)
| Country | Link |
|---|---|
| US (4) | US8987323B2 (ru) |
| EP (1) | EP2580225B1 (ru) |
| JP (1) | JP5842191B2 (ru) |
| KR (1) | KR101831675B1 (ru) |
| CN (1) | CN102933592B (ru) |
| AR (1) | AR121997A2 (ru) |
| AU (1) | AU2011264220B2 (ru) |
| BR (1) | BR112012031616A2 (ru) |
| CA (1) | CA2800379C (ru) |
| CO (1) | CO6640246A2 (ru) |
| CY (1) | CY1120690T1 (ru) |
| DK (1) | DK2580225T3 (ru) |
| ES (1) | ES2683123T3 (ru) |
| HR (1) | HRP20181695T1 (ru) |
| HU (1) | HUE040355T2 (ru) |
| IL (1) | IL223205B (ru) |
| LT (1) | LT2580225T (ru) |
| MA (1) | MA34371B1 (ru) |
| MX (1) | MX342665B (ru) |
| MY (1) | MY184925A (ru) |
| NZ (1) | NZ605570A (ru) |
| PL (1) | PL2580225T3 (ru) |
| PT (1) | PT2580225T (ru) |
| RS (1) | RS57653B1 (ru) |
| RU (1) | RU2569491C2 (ru) |
| SG (1) | SG186206A1 (ru) |
| SI (1) | SI2580225T1 (ru) |
| TW (1) | TWI589295B (ru) |
| UA (1) | UA108887C2 (ru) |
| WO (2) | WO2011153712A1 (ru) |
| ZA (1) | ZA201300004B (ru) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ591818A (en) * | 2008-08-22 | 2013-01-25 | Theracos Inc | Processes for the preparation of sglt2 inhibitors |
| WO2011153712A1 (en) | 2010-06-12 | 2011-12-15 | Theracos, Inc. | Crystalline form of benzylbenzene sglt2 inhibitor |
| US9145434B2 (en) * | 2012-07-26 | 2015-09-29 | Boehringer Ingelheim International Gmbh | Crystalline complex of 1-cyano-2-(4-cyclopropyl-benzyl)-4-(ss-d-glucopyranos-1-yl)-benzene, methods for its preparation and the use thereof for preparing medicaments |
| PL2981269T3 (pl) * | 2013-04-04 | 2024-02-05 | Boehringer Ingelheim Vetmedica Gmbh | Leczenie zaburzeń metabolicznych u zwierząt koniowatych |
| CN105611920B (zh) * | 2013-10-12 | 2021-07-16 | 泰拉科斯萨普有限责任公司 | 羟基-二苯甲烷衍生物的制备 |
| HUE053906T2 (hu) | 2013-12-17 | 2021-07-28 | Boehringer Ingelheim Vetmedica Gmbh | SGLT2-inhibitorok macskafélék közé tartozó állatok anyagcsere-rendellenességeinek kezelésére |
| US10603300B2 (en) | 2014-01-23 | 2020-03-31 | Boehringer Ingelheim Vetmedica Gmbh | Treatment of metabolic disorders in canine animals |
| WO2015145815A1 (ja) * | 2014-03-27 | 2015-10-01 | 三菱電機株式会社 | 半導体装置、半導体装置の製造方法 |
| PT3125882T (pt) | 2014-04-01 | 2020-08-17 | Boehringer Ingelheim Vetmedica Gmbh | Tratamento de distúrbios metabólicos em animais equinos |
| KR102539788B1 (ko) * | 2014-09-25 | 2023-06-07 | 베링거잉겔하임베트메디카게엠베하 | 말과 동물의 대사 장애를 예방하기 위한 sglt2 억제제와 도파민 작용제의 병용 치료 |
| CN107995862B8 (zh) | 2015-08-27 | 2021-12-03 | 勃林格殷格翰维特梅迪卡有限公司 | 包含sglt-2抑制剂之液态药物组合物 |
| CN108239055B (zh) * | 2016-12-23 | 2023-07-18 | 杭州领业医药科技有限公司 | 一种thr1442 l-天冬氨酸共晶、其制备方法及药物组合物 |
| US11031285B2 (en) | 2017-10-06 | 2021-06-08 | Invensas Bonding Technologies, Inc. | Diffusion barrier collar for interconnects |
| US20190343853A1 (en) * | 2018-04-25 | 2019-11-14 | Theracos Sub, Llc | Methods of treating hypertension |
| EP4106744A1 (en) | 2020-02-17 | 2022-12-28 | Boehringer Ingelheim Vetmedica GmbH | Use of sglt-2 inhibitors for the prevention and/or treatment of cardiac diseases in felines |
| WO2022073151A1 (en) * | 2020-10-05 | 2022-04-14 | Theracos Sub, Llc | Pharmaceutical formulations |
| GB202213849D0 (en) * | 2022-09-22 | 2022-11-09 | Macfarlan Smith Ltd | Crystalline forms of bexagliflozin, processes for the preparation and use thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000026537A1 (en) * | 1998-11-05 | 2000-05-11 | Frantz Medical Development Ltd. | Detecting obstructions in enteral/parenteral feeding tubes and automatic removal of clogs therefrom |
| RU2262507C2 (ru) * | 1999-10-12 | 2005-10-20 | Бристол-Маерс Сквибб Компани | С-арилглюкозидные ингибиторы sglt2 |
| WO2008144346A2 (en) * | 2007-05-18 | 2008-11-27 | Bristol-Myers Squibb Company | Crystal structures of sglt2 inhibitors and processes for their preparation |
| WO2010009243A1 (en) * | 2008-07-15 | 2010-01-21 | Theracos, Inc. | Deuterated benzylbenzene derivatives and methods of use |
| CA2734295A1 (en) * | 2008-08-22 | 2010-02-25 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
Family Cites Families (137)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA254853A (en) | 1925-10-20 | Wyatt Prentice Frank | Radio train stop and telephone despatching system | |
| US4499289A (en) | 1982-12-03 | 1985-02-12 | G. D. Searle & Co. | Octahydronapthalenes |
| US4933438A (en) | 1984-02-29 | 1990-06-12 | University Of Florida | Brain-specific analogues of centrally acting amines |
| US4613610A (en) | 1984-06-22 | 1986-09-23 | Sandoz Pharmaceuticals Corp. | Cholesterol biosynthesis inhibiting pyrazole analogs of mevalonolactone and its derivatives |
| US4686237A (en) | 1984-07-24 | 1987-08-11 | Sandoz Pharmaceuticals Corp. | Erythro-(E)-7-[3'-C1-3 alkyl-1'-(3",5"-dimethylphenyl)naphth-2'-yl]-3,5-dihydroxyhept-6-enoic acids and derivatives thereof |
| US4647576A (en) | 1984-09-24 | 1987-03-03 | Warner-Lambert Company | Trans-6-[2-(substitutedpyrrol-1-yl)alkyl]-pyran-2-one inhibitors of cholesterol synthesis |
| US6150354A (en) | 1987-01-15 | 2000-11-21 | Bonnie Davis | Compounds for the treatment of Alzheimer's disease |
| US4871721A (en) | 1988-01-11 | 1989-10-03 | E. R. Squibb & Sons, Inc. | Phosphorus-containing squalene synthetase inhibitors |
| US4924024A (en) | 1988-01-11 | 1990-05-08 | E. R. Squibb & Sons, Inc. | Phosphorus-containing squalene synthetase inhibitors, new intermediates and method |
| US5506219A (en) | 1988-08-29 | 1996-04-09 | E. R. Squibb & Sons, Inc. | Pyridine anchors for HMG-CoA reductase inhibitors |
| US5753675A (en) | 1989-03-03 | 1998-05-19 | Novartis Pharmaceuticals Corporation | Quinoline analogs of mevalonolactone and derivatives thereof |
| US5133974A (en) | 1989-05-05 | 1992-07-28 | Kv Pharmaceutical Company | Extended release pharmaceutical formulations |
| CN1020944C (zh) | 1990-01-30 | 1993-05-26 | 阿图尔-费希尔股份公司费希尔厂 | 紧固件 |
| ATE183084T1 (de) | 1991-05-14 | 1999-08-15 | Ernir Snorrason | Behandlung des ermüdungssyndroms mit cholinesteraseinhibitoren |
| IT1250421B (it) | 1991-05-30 | 1995-04-07 | Recordati Chem Pharm | Composizione farmaceutica a rilascio controllato con proprieta' bio-adesive. |
| US5595872A (en) | 1992-03-06 | 1997-01-21 | Bristol-Myers Squibb Company | Nucleic acids encoding microsomal trigyceride transfer protein |
| US5712396A (en) | 1992-10-28 | 1998-01-27 | Magnin; David R. | α-phosphonosulfonate squalene synthetase inhibitors |
| EP0671953A4 (en) | 1992-11-13 | 1996-01-10 | Univ Ohio State Res Found | ANALOGS OF N- (4-HYDROXYPHENYL) RETINAMIDE-O-GLUCURONIDE ARYLAMIDE. |
| US6537579B1 (en) | 1993-02-22 | 2003-03-25 | American Bioscience, Inc. | Compositions and methods for administration of pharmacologically active compounds |
| US5739135A (en) | 1993-09-03 | 1998-04-14 | Bristol-Myers Squibb Company | Inhibitors of microsomal triglyceride transfer protein and method |
| FR2710265B1 (fr) | 1993-09-22 | 1995-10-20 | Adir | Composition pharmaceutique bioadhésive pour la libération contrôlée de principes actifs. |
| US5620997A (en) | 1995-05-31 | 1997-04-15 | Warner-Lambert Company | Isothiazolones |
| US6548084B2 (en) | 1995-07-20 | 2003-04-15 | Smithkline Beecham Plc | Controlled release compositions |
| US6245347B1 (en) | 1995-07-28 | 2001-06-12 | Zars, Inc. | Methods and apparatus for improved administration of pharmaceutically active compounds |
| AU6966696A (en) | 1995-10-05 | 1997-04-28 | Warner-Lambert Company | Method for treating and preventing inflammation and atherosclerosis |
| DE19541260A1 (de) | 1995-11-06 | 1997-05-07 | Lohmann Therapie Syst Lts | Therapeutische Zubereitung zur transdermalen Applikation von Wirkstoffen durch die Haut |
| US5962440A (en) | 1996-05-09 | 1999-10-05 | Bristol-Myers Squibb Company | Cyclic phosphonate ester inhibitors of microsomal triglyceride transfer protein and method |
| US5885983A (en) | 1996-05-10 | 1999-03-23 | Bristol-Myers Squibb Company | Inhibitors of microsomal triglyceride transfer protein and method |
| US5827875A (en) | 1996-05-10 | 1998-10-27 | Bristol-Myers Squibb Company | Inhibitors of microsomal triglyceride transfer protein and method |
| US6512010B1 (en) | 1996-07-15 | 2003-01-28 | Alza Corporation | Formulations for the administration of fluoxetine |
| US6011155A (en) | 1996-11-07 | 2000-01-04 | Novartis Ag | N-(substituted glycyl)-2-cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| US5760246A (en) | 1996-12-17 | 1998-06-02 | Biller; Scott A. | Conformationally restricted aromatic inhibitors of microsomal triglyceride transfer protein and method |
| AU6024998A (en) | 1997-01-15 | 1998-08-07 | Glycomed Incorporated | Aryl c-glycoside compounds and sulfated esters thereof |
| CA2301883A1 (en) | 1997-09-11 | 1999-03-18 | Nycomed Danmark A/S | Modified release multiple-units compositions of non-steroid anti-inflammatory drug substances (nsaids) |
| US6624200B2 (en) | 1998-08-25 | 2003-09-23 | Columbia Laboratories, Inc. | Bioadhesive progressive hydration tablets |
| US6607751B1 (en) | 1997-10-10 | 2003-08-19 | Intellipharamaceutics Corp. | Controlled release delivery device for pharmaceutical agents incorporating microbial polysaccharide gum |
| US6403597B1 (en) | 1997-10-28 | 2002-06-11 | Vivus, Inc. | Administration of phosphodiesterase inhibitors for the treatment of premature ejaculation |
| US6248864B1 (en) | 1997-12-31 | 2001-06-19 | Adherex Technologies, Inc. | Compounds and methods and modulating tissue permeability |
| US6613358B2 (en) | 1998-03-18 | 2003-09-02 | Theodore W. Randolph | Sustained-release composition including amorphous polymer |
| US6312717B1 (en) | 1998-07-07 | 2001-11-06 | Bristol-Myers Squibb Company | Method for treatment of anxiety and depression |
| US6197798B1 (en) | 1998-07-21 | 2001-03-06 | Novartis Ag | Amino-benzocycloalkane derivatives |
| US20020009478A1 (en) | 1998-08-24 | 2002-01-24 | Douglas Joseph Dobrozsi | Oral liquid mucoadhesive compositions |
| SE9802864D0 (sv) | 1998-08-27 | 1998-08-27 | Pharmacia & Upjohn Ab | Transdermally administered tolterodine as antimuscarinic agent for the treatment of overactive bladder |
| US6069238A (en) | 1998-09-30 | 2000-05-30 | Eli Lilly And Company | Spirocyclic C-glycosides |
| ATE286725T1 (de) | 1998-10-01 | 2005-01-15 | Novartis Pharma Gmbh | Neue oral anzuwendende arzneizubereitungen für rivastigmine mit kontrollierter wirkstoffabgabe |
| US6248363B1 (en) | 1999-11-23 | 2001-06-19 | Lipocine, Inc. | Solid carriers for improved delivery of active ingredients in pharmaceutical compositions |
| US6548529B1 (en) | 1999-04-05 | 2003-04-15 | Bristol-Myers Squibb Company | Heterocyclic containing biphenyl aP2 inhibitors and method |
| US7407978B2 (en) | 1999-04-06 | 2008-08-05 | Theracos, Inc. | Heterocyclic analogs of diphenylethylene compounds |
| FR2792527B1 (fr) | 1999-04-22 | 2004-08-13 | Ethypharm Lab Prod Ethiques | Microgranules de ketoprofene, procede de preparation et compositions pharmaceutiques |
| US6172081B1 (en) | 1999-06-24 | 2001-01-09 | Novartis Ag | Tetrahydroisoquinoline 3-carboxamide derivatives |
| US6541020B1 (en) | 1999-07-09 | 2003-04-01 | Trimeris, Inc. | Methods and compositions for administration of therapeutic reagents |
| DE19933926A1 (de) | 1999-07-20 | 2001-01-25 | Boehringer Ingelheim Pharma | Biphenylderivate, ihre Herstellung und ihre Verwendung als Arzneimittel |
| US6562375B1 (en) | 1999-08-04 | 2003-05-13 | Yamanouchi Pharmaceuticals, Co., Ltd. | Stable pharmaceutical composition for oral use |
| JP2003508422A (ja) | 1999-09-02 | 2003-03-04 | ノストラム・ファーマスーティカルズ・インコーポレイテッド | 放出制御ペレット製剤 |
| US6544548B1 (en) | 1999-09-13 | 2003-04-08 | Keraplast Technologies, Ltd. | Keratin-based powders and hydrogel for pharmaceutical applications |
| US6515117B2 (en) | 1999-10-12 | 2003-02-04 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| US6080736A (en) | 1999-10-27 | 2000-06-27 | Janus Pharmaceuticals, Inc. | Methods and compositions for treating and preventing anxiety and anxiety disorders using optically pure (R) tofisopam |
| US6461631B1 (en) | 1999-11-16 | 2002-10-08 | Atrix Laboratories, Inc. | Biodegradable polymer composition |
| DE19963234A1 (de) | 1999-12-27 | 2002-01-24 | Boehringer Ingelheim Pharma | Substituierte Piperazinderivate, ihre Herstellung und ihre Verwendung als Arzneimittel |
| US6482439B2 (en) | 1999-12-29 | 2002-11-19 | Nanodelivery, Inc. | Drug delivery system exhibiting permeability control |
| ES2240420T3 (es) | 2000-01-18 | 2005-10-16 | Novartis Ag | Carboxamidas utiles como inhibidores de la proteina de transferencia de trigliceridos microsomicos y de la secrecion de apolipoproteina b. |
| JP2004501066A (ja) | 2000-01-28 | 2004-01-15 | ブリストル−マイヤーズ スクイブ カンパニー | 脂肪酸結合タンパク質のテトラヒドロピリミドンインヒビターおよび方法 |
| US6555519B2 (en) | 2000-03-30 | 2003-04-29 | Bristol-Myers Squibb Company | O-glucosylated benzamide SGLT2 inhibitors and method |
| US6683056B2 (en) | 2000-03-30 | 2004-01-27 | Bristol-Myers Squibb Company | O-aryl glucoside SGLT2 inhibitors and method |
| US6589549B2 (en) | 2000-04-27 | 2003-07-08 | Macromed, Incorporated | Bioactive agent delivering system comprised of microparticles within a biodegradable to improve release profiles |
| US6524621B2 (en) | 2000-05-01 | 2003-02-25 | Aeropharm Technology Inc. | Core formulation |
| US6432969B1 (en) | 2000-06-13 | 2002-08-13 | Novartis Ag | N-(substituted glycyl)-2 cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| US6482440B2 (en) | 2000-09-21 | 2002-11-19 | Phase 2 Discovery, Inc. | Long acting antidepressant microparticles |
| EP1340749A4 (en) | 2000-11-17 | 2007-09-05 | Takeda Pharmaceutical | isoxazole |
| HUP0500543A2 (hu) | 2000-11-20 | 2005-09-28 | Bristol-Myers Squibb Company | Piridonszármazékok mint aP2 inhibitorok és a vegyületeket tartalmazó gyógyszerkészítmények |
| TWI255817B (en) | 2001-02-14 | 2006-06-01 | Kissei Pharmaceutical | Glucopyranosyloxybenzylbenzene derivatives and medicinal use thereof |
| US6524615B2 (en) | 2001-02-21 | 2003-02-25 | Kos Pharmaceuticals, Incorporated | Controlled release pharmaceutical composition |
| US6936590B2 (en) | 2001-03-13 | 2005-08-30 | Bristol Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| ES2258141T3 (es) | 2001-04-11 | 2006-08-16 | Bristol-Myers Squibb Company | Complejos de aminoacidos de glucosidos c-arilo para el tratamiento de la diabetes y procedimiento. |
| US6869947B2 (en) | 2001-07-03 | 2005-03-22 | Novo Nordisk A/S | Heterocyclic compounds that are inhibitors of the enzyme DPP-IV |
| EP1414461A4 (en) | 2001-07-13 | 2005-10-26 | Bristol Myers Squibb Co | PYRAZINONE INHIBITORS OF FATTY ACID BINDING PROTEIN AND METHOD |
| WO2003020737A1 (en) | 2001-09-05 | 2003-03-13 | Bristol-Myers Squibb Company | O-pyrazole glucoside sglt2 inhibitors and method of use |
| JP2003080635A (ja) * | 2001-09-12 | 2003-03-19 | Teijin Meton Kk | 合成樹脂複合構造体、それからなる浴室構成品及びその製造方法 |
| EP1443919A4 (en) | 2001-11-16 | 2006-03-22 | Bristol Myers Squibb Co | DOUBLE INHIBITORS OF THE FATTY ACID BINDING PROTEIN OF THE ADIPOCYTES AND THE FATTY ACID BINDING PROTEIN OF KERATINOCYTES |
| US6727261B2 (en) | 2001-12-27 | 2004-04-27 | Hoffman-La Roche Inc. | Pyrido[2,1-A]Isoquinoline derivatives |
| PL210710B1 (pl) | 2002-03-22 | 2012-02-29 | Kissei Pharmaceutical | Krystaliczne postaci 2-(4-metoksybenzylo) fenylo-6-O-etoksykarbonylo-ß-D-glukopiranozydu, ich zastosowanie oraz zawierająca je kompozycja farmaceutyczna |
| US6710040B1 (en) | 2002-06-04 | 2004-03-23 | Pfizer Inc. | Fluorinated cyclic amides as dipeptidyl peptidase IV inhibitors |
| DE60332743D1 (de) | 2002-08-08 | 2010-07-08 | Kissei Pharmaceutical | Pyrazolderivat, dieses enthaltende medizinische zusammensetzung, medizinische verwendung davon, und zwischenprodukt für dessen herstellung |
| EP1528066A4 (en) | 2002-08-09 | 2007-05-02 | Taisho Pharmaceutical Co Ltd | ARYL-t-THIO-B-GLUCOPYRANOSIDE DERIVATIVES AND DIABETES CONTAINING THEM |
| US6700238B1 (en) * | 2002-08-13 | 2004-03-02 | Wei Tong | Generator gas shield and related method |
| DE10258007B4 (de) | 2002-12-12 | 2006-02-09 | Sanofi-Aventis Deutschland Gmbh | Aromatische Fluorglycosidderivate, diese Verbindungen enthaltende Arzneimittel und Verfahren zur Herstellung dieser Arzneimittel |
| DE10258008B4 (de) | 2002-12-12 | 2006-02-02 | Sanofi-Aventis Deutschland Gmbh | Heterocyclische Fluorglycosidderivate, diese Verbindungen enthaltende Arzneimittel und Verfahren zur Herstellung dieser Arzneimittel |
| US7375213B2 (en) | 2003-01-03 | 2008-05-20 | Bristol-Myers Squibb Company | Methods of producing C-aryl glucoside SGLT2 inhibitors |
| WO2004080990A1 (ja) | 2003-03-14 | 2004-09-23 | Astellas Pharma Inc. | C-グリコシド誘導体又はその塩 |
| JP2004300102A (ja) | 2003-03-31 | 2004-10-28 | Kissei Pharmaceut Co Ltd | 縮合複素環誘導体、それを含有する医薬組成物およびその医薬用途 |
| EP1679966A4 (en) | 2003-08-01 | 2009-05-27 | Janssen Pharmaceutica Nv | BENZIMIDAZOLE-, BENZTRIAZOLE- AND BENZIMIDAZOLONE-SUBSTITUTED O-GLUCOSIDES |
| AR048376A1 (es) | 2003-08-01 | 2006-04-26 | Janssen Pharmaceutica Nv | C- glicosidos heterociclos fusionados sustituidos |
| DK2896397T4 (da) | 2003-08-01 | 2020-11-16 | Mitsubishi Tanabe Pharma Corp | Nye forbindelser med hæmmende aktivitet mod natriumafhængig glukosetransportør |
| US7375090B2 (en) | 2003-08-26 | 2008-05-20 | Boehringer Ingelheim International Gmbh | Glucopyranosyloxy-pyrazoles, pharmaceutical compositions containing these compounds, the use thereof and processed for the preparation thereof |
| WO2005021566A2 (de) | 2003-08-26 | 2005-03-10 | Boehringer Ingelheim International Gmbh | Glucopyranosyloxy-pyrazole, diese verbindungen enthaltende arzneimittel, deren verwendung und verfahren zu ihrer herstellung |
| DE10361133A1 (de) | 2003-12-22 | 2005-07-21 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Glucopyranosyloxy-substituierte Aromaten, diese Verbindungen enthaltende Arzneimittel, deren Verwendung und Verfahren zu ihrer Herstellung |
| US7371732B2 (en) | 2003-12-22 | 2008-05-13 | Boehringer Ingelheim International Gmbh | Glucopyranosyloxy-substituted aromatic compounds, medicaments containing such compounds, their use and process for their manufacture |
| US8058245B2 (en) | 2004-03-04 | 2011-11-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| CA2557801C (en) | 2004-03-16 | 2013-06-25 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzol derivatives, drugs containing said compounds, the use thereof and method for the production thereof |
| US20070185197A1 (en) | 2004-03-31 | 2007-08-09 | Hideki Fujikura | Phenol derivative, medicinal composition containing the same, and medicinal use thereof |
| US7393836B2 (en) | 2004-07-06 | 2008-07-01 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| DE102004034690A1 (de) | 2004-07-17 | 2006-02-02 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Methyliden-D-xylopyranosyl-und Oxo-D-xylopyranosyl-substituierte Phenyle, diese Verbindungen enthaltende Arzneimittel, deren Verwendung und Verfahren zu ihrer Herstellung |
| TW200606129A (en) | 2004-07-26 | 2006-02-16 | Chugai Pharmaceutical Co Ltd | Novel cyclohexane derivative, its prodrug, its salt and diabetic therapeutic agent containing the same |
| EP1773800A1 (de) | 2004-07-27 | 2007-04-18 | Boehringer Ingelheim International GmbH | D-glucopyranosyl-phenyl-substituierte cyclen, diese verbindungen enthaltende arzneimittel, deren verwendung und verfahren zu ihrer herstellung |
| WO2006018150A1 (de) | 2004-08-11 | 2006-02-23 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituierte cyclen, diese verbindungen enthaltende arzneimittel, deren verwendung und verfahren zu ihrer herstellung |
| AR051446A1 (es) | 2004-09-23 | 2007-01-17 | Bristol Myers Squibb Co | Glucosidos de c-arilo como inhibidores selectivos de transportadores de glucosa (sglt2) |
| DE102004048388A1 (de) | 2004-10-01 | 2006-04-06 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | D-Pyranosyl-substituierte Phenyle, diese Verbindungen enthaltende Arzneimittel, deren Verwendung und Verfahren zu ihrer Herstellung |
| US7687469B2 (en) | 2004-12-16 | 2010-03-30 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| TW200637839A (en) | 2005-01-07 | 2006-11-01 | Taisho Pharmaceutical Co Ltd | 1-thio-d-glucitol derivatives |
| TW200637869A (en) | 2005-01-28 | 2006-11-01 | Chugai Pharmaceutical Co Ltd | The spiroketal derivatives and the use as therapeutical agent for diabetes of the same |
| ATE445608T1 (de) * | 2005-02-23 | 2009-10-15 | Boehringer Ingelheim Int | Glucopyranosylsubstituierte ((hetero)arylethynyl- benzyl)-benzenderivative und deren verwendung als inhibitoren des natriumabhängigen glucose- cotransporters typ 2 (sglt2) |
| MY145694A (en) | 2005-04-11 | 2012-03-30 | Xenon Pharmaceuticals Inc | Spiroheterocyclic compounds and their uses as therapeutic agents |
| ATE453656T1 (de) | 2005-04-15 | 2010-01-15 | Boehringer Ingelheim Int | Glucopyranosyl-substituierte (heteroaryloxy- benzyl)-benzen-derivate als sglt-inhibitoren |
| UA91546C2 (ru) | 2005-05-03 | 2010-08-10 | Бьорінгер Інгельхайм Інтернаціональ Гмбх | КРИСТАЛЛИЧЕСКАЯ ФОРМА 1-ХЛОР-4-(b-D-ГЛЮКОПИРАНОЗ-1- ИЛ)-2-[4-((S)- ТЕТРАГИДРОФУРАН-3-ИЛОКСИ)-БЕНЗИЛ]-БЕНЗОЛА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ ПРИ ПРИГОТАВЛЕНИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ |
| US7723309B2 (en) | 2005-05-03 | 2010-05-25 | Boehringer Ingelheim International Gmbh | Crystalline forms of 1-chloro-4-(β-D-glucopyranos-1-yl)-2-[4-((R)-tetrahydrofuran-3-yloxy)-benzyl]-benzene, a method for its preparation and the use thereof for preparing medicaments |
| US7772191B2 (en) | 2005-05-10 | 2010-08-10 | Boehringer Ingelheim International Gmbh | Processes for preparing of glucopyranosyl-substituted benzyl-benzene derivatives and intermediates therein |
| WO2007000445A1 (en) | 2005-06-29 | 2007-01-04 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| JP2007007628A (ja) * | 2005-07-04 | 2007-01-18 | Hokkaido Univ | セメントコンクリートの中性化促進方法 |
| TW200726755A (en) | 2005-07-07 | 2007-07-16 | Astellas Pharma Inc | A crystalline choline salt of an azulene derivative |
| JP5128474B2 (ja) | 2005-07-27 | 2013-01-23 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | グルコピラノシル−置換((ヘテロ)シクロアルキルエチニル−ベンジル)−ベンゼン誘導体、該化合物を含有する薬剤、それらの使用及びそれらの製造方法 |
| GB0516156D0 (en) | 2005-08-05 | 2005-09-14 | Eisai London Res Lab Ltd | JNK inhibitors |
| DE602006017566D1 (de) * | 2005-08-30 | 2010-11-25 | Boehringer Ingelheim Pharma | Glucopyranosyl-substituierte benzyl-derivate, medikamente mit solchen verbindungen, ihre verwendung und herstellungsverfahren dafür |
| EP1926720B1 (en) * | 2005-09-08 | 2010-12-15 | Boehringer Ingelheim International GmbH | CRYSTALLINE FORMS OF 1-CHLORO-4-(ß-D-GLUCOPYRANOS-1-YL)-2-(4-ETHYNYL-BENZYL)-BENZENE, METHODS FOR ITS PREPARATION AND THE USE THEREOF FOR PREPARING MEDICAMENTS |
| TWI370818B (en) | 2006-04-05 | 2012-08-21 | Astellas Pharma Inc | Cocrystal of c-glycoside derivative and l-proline |
| AU2007252432B2 (en) | 2006-05-19 | 2011-11-17 | Taisho Pharmaceutical Co., Ltd. | C-phenyl glycitol compound |
| US7803778B2 (en) | 2006-05-23 | 2010-09-28 | Theracos, Inc. | Glucose transport inhibitors and methods of use |
| US7919598B2 (en) | 2006-06-28 | 2011-04-05 | Bristol-Myers Squibb Company | Crystal structures of SGLT2 inhibitors and processes for preparing same |
| US20080027014A1 (en) | 2006-07-28 | 2008-01-31 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| CA2667550A1 (en) * | 2006-10-27 | 2008-05-02 | Boehringer Ingelheim International Gmbh | Crystalline form of 4-(.beta.-d-glucopyranos-1-yl)-1-methyl-2-[4-((s)-tetrahydrofuran-3-yloxy)-benzyl]-benzene, a method for its preparation and the use thereof for preparing medicaments |
| UY30730A1 (es) | 2006-12-04 | 2008-07-03 | Mitsubishi Tanabe Pharma Corp | Forma cristalina del hemihidrato de 1-(b (beta)-d-glucopiranosil) -4-metil-3-[5-(4-fluorofenil) -2-tienilmetil]benceno |
| AU2008232419B2 (en) | 2007-04-02 | 2013-06-20 | Theracos, Inc. | Benzylic glycoside derivatives and methods of use |
| EP2183263B1 (en) * | 2007-07-26 | 2011-10-26 | Lexicon Pharmaceuticals, Inc. | Methods and compounds useful for the preparation of sodium glucose co-transporter 2 inhibitors |
| JP4809931B2 (ja) * | 2007-08-23 | 2011-11-09 | セラコス・インコーポレイテッド | ベンジルベンゼン誘導体およびその使用方法 |
| ES2647504T3 (es) | 2007-09-10 | 2017-12-21 | Janssen Pharmaceutica N.V. | Proceso para la preparación de compuestos útiles como inhibidores de SGLT |
| AR070701A1 (es) * | 2008-02-13 | 2010-04-28 | Sanofi Aventis | Derivados de fluoroglicosidos aromaticos, medicamentos que comprenden estos compuestos y uso de los mismos para producir un medicamento |
| TW201023858A (en) * | 2008-09-18 | 2010-07-01 | Ortho Mcneil Janssen Pharm | Synergistic combinations of a macrocyclic inhibitor of HCV and a nucleoside |
| WO2011153712A1 (en) | 2010-06-12 | 2011-12-15 | Theracos, Inc. | Crystalline form of benzylbenzene sglt2 inhibitor |
-
2010
- 2010-06-12 WO PCT/CN2010/073865 patent/WO2011153712A1/en active Application Filing
-
2011
- 2011-06-10 PT PT11791943T patent/PT2580225T/pt unknown
- 2011-06-10 NZ NZ605570A patent/NZ605570A/en unknown
- 2011-06-10 CN CN201180028665.8A patent/CN102933592B/zh active Active
- 2011-06-10 SI SI201131551T patent/SI2580225T1/sl unknown
- 2011-06-10 RS RS20181044A patent/RS57653B1/sr unknown
- 2011-06-10 SG SG2012089710A patent/SG186206A1/en unknown
- 2011-06-10 CA CA2800379A patent/CA2800379C/en active Active
- 2011-06-10 DK DK11791943.1T patent/DK2580225T3/en active
- 2011-06-10 MY MYPI2012005263A patent/MY184925A/en unknown
- 2011-06-10 EP EP11791943.1A patent/EP2580225B1/en active Active
- 2011-06-10 ES ES11791943.1T patent/ES2683123T3/es active Active
- 2011-06-10 KR KR1020137000844A patent/KR101831675B1/ko active IP Right Grant
- 2011-06-10 WO PCT/CN2011/075554 patent/WO2011153953A1/en active Application Filing
- 2011-06-10 AU AU2011264220A patent/AU2011264220B2/en active Active
- 2011-06-10 MX MX2012014423A patent/MX342665B/es active IP Right Grant
- 2011-06-10 BR BR112012031616A patent/BR112012031616A2/pt not_active Application Discontinuation
- 2011-06-10 JP JP2013513540A patent/JP5842191B2/ja active Active
- 2011-06-10 LT LTEP11791943.1T patent/LT2580225T/lt unknown
- 2011-06-10 PL PL11791943T patent/PL2580225T3/pl unknown
- 2011-06-10 RU RU2013101580/04A patent/RU2569491C2/ru active
- 2011-06-10 HU HUE11791943A patent/HUE040355T2/hu unknown
- 2011-06-10 MA MA35549A patent/MA34371B1/fr unknown
- 2011-06-13 TW TW100120591A patent/TWI589295B/zh active
- 2011-06-13 US US13/158,724 patent/US8987323B2/en active Active
- 2011-10-06 UA UAA201300352A patent/UA108887C2/ru unknown
-
2012
- 2012-11-22 IL IL223205A patent/IL223205B/en active IP Right Grant
- 2012-11-23 CO CO12212781A patent/CO6640246A2/es not_active Application Discontinuation
-
2013
- 2013-01-02 ZA ZA2013/00004A patent/ZA201300004B/en unknown
-
2015
- 2015-02-20 US US14/627,693 patent/US9834573B2/en active Active
-
2017
- 2017-11-06 US US15/804,074 patent/US10533032B2/en active Active
-
2018
- 2018-09-20 CY CY181100972T patent/CY1120690T1/el unknown
- 2018-10-17 HR HRP20181695TT patent/HRP20181695T1/hr unknown
-
2019
- 2019-11-25 US US16/694,642 patent/US10981942B2/en active Active
-
2021
- 2021-05-03 AR ARP210101202A patent/AR121997A2/es unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000026537A1 (en) * | 1998-11-05 | 2000-05-11 | Frantz Medical Development Ltd. | Detecting obstructions in enteral/parenteral feeding tubes and automatic removal of clogs therefrom |
| RU2262507C2 (ru) * | 1999-10-12 | 2005-10-20 | Бристол-Маерс Сквибб Компани | С-арилглюкозидные ингибиторы sglt2 |
| WO2008144346A2 (en) * | 2007-05-18 | 2008-11-27 | Bristol-Myers Squibb Company | Crystal structures of sglt2 inhibitors and processes for their preparation |
| WO2010009243A1 (en) * | 2008-07-15 | 2010-01-21 | Theracos, Inc. | Deuterated benzylbenzene derivatives and methods of use |
| CA2734295A1 (en) * | 2008-08-22 | 2010-02-25 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10981942B2 (en) | Crystalline form of benzylbenzene SGLT2 inhibitor | |
| US7838499B2 (en) | Benzylbenzene derivatives and methods of use | |
| TWI482779B (zh) | Sglt2抑制劑之結晶型 | |
| CA2707909C (en) | Benzylphenyl cyclohexane derivatives and methods of use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PC41 | Official registration of the transfer of exclusive right |
Effective date: 20160314 |