RU2540860C2 - Нитроимидазооксазиновые и нитроимидазооксазольные аналоги и их применение - Google Patents
Нитроимидазооксазиновые и нитроимидазооксазольные аналоги и их применение Download PDFInfo
- Publication number
- RU2540860C2 RU2540860C2 RU2012107180/04A RU2012107180A RU2540860C2 RU 2540860 C2 RU2540860 C2 RU 2540860C2 RU 2012107180/04 A RU2012107180/04 A RU 2012107180/04A RU 2012107180 A RU2012107180 A RU 2012107180A RU 2540860 C2 RU2540860 C2 RU 2540860C2
- Authority
- RU
- Russia
- Prior art keywords
- methyl
- nitro
- oxazine
- imidazo
- dihydro
- Prior art date
Links
- UOHVERHTRSLFEH-UHFFFAOYSA-N CC1(COCc(cc2)ccc2OC(F)(F)F)Oc2nc([N+]([O-])=O)c[n]2CC1 Chemical compound CC1(COCc(cc2)ccc2OC(F)(F)F)Oc2nc([N+]([O-])=O)c[n]2CC1 UOHVERHTRSLFEH-UHFFFAOYSA-N 0.000 description 1
- ALHDHSLKVWVIHN-UHFFFAOYSA-N CC1(COCc2cccc(-c(cc3)ccc3OC(F)(F)F)c2)Oc2nc([N+]([O-])=O)c[n]2CC1 Chemical compound CC1(COCc2cccc(-c(cc3)ccc3OC(F)(F)F)c2)Oc2nc([N+]([O-])=O)c[n]2CC1 ALHDHSLKVWVIHN-UHFFFAOYSA-N 0.000 description 1
- JOWOAMMSABIQDN-UHFFFAOYSA-N CC1(COc(cc2)ccc2-c(cc2)ccc2C#N)Oc2nc([N+]([O-])=O)c[n]2C1 Chemical compound CC1(COc(cc2)ccc2-c(cc2)ccc2C#N)Oc2nc([N+]([O-])=O)c[n]2C1 JOWOAMMSABIQDN-UHFFFAOYSA-N 0.000 description 1
- ZCQDJADLEOACNA-UHFFFAOYSA-N CC1(COc(nc2)cnc2-c(cc2)ccc2F)Oc2nc([N+]([O-])=O)c[n]2C1 Chemical compound CC1(COc(nc2)cnc2-c(cc2)ccc2F)Oc2nc([N+]([O-])=O)c[n]2C1 ZCQDJADLEOACNA-UHFFFAOYSA-N 0.000 description 1
- XDFSBIUYBLENFF-UHFFFAOYSA-N [O-][N+](c1c[n](CC(COCc2cccc(-c(cc3)ccc3OC(F)(F)F)c2)CO2)c2n1)=O Chemical compound [O-][N+](c1c[n](CC(COCc2cccc(-c(cc3)ccc3OC(F)(F)F)c2)CO2)c2n1)=O XDFSBIUYBLENFF-UHFFFAOYSA-N 0.000 description 1
- QUMAYTSFYCTSLG-UHFFFAOYSA-N [O-][N+](c1c[n](CC(COc(cc2)ccc2OC(F)(F)F)O2)c2n1)=O Chemical compound [O-][N+](c1c[n](CC(COc(cc2)ccc2OC(F)(F)F)O2)c2n1)=O QUMAYTSFYCTSLG-UHFFFAOYSA-N 0.000 description 1
- YRZTVAYXNMPEHK-UHFFFAOYSA-N [O-][N+](c1c[n](CCC(COc(cc2)ccc2-c(cc2)ccc2OC(F)(F)F)O2)c2n1)=O Chemical compound [O-][N+](c1c[n](CCC(COc(cc2)ccc2-c(cc2)ccc2OC(F)(F)F)O2)c2n1)=O YRZTVAYXNMPEHK-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
- A01N43/90—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having two or more relevant hetero rings, condensed among themselves or with a common carbocyclic ring system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Epidemiology (AREA)
- Pest Control & Pesticides (AREA)
- Plant Pathology (AREA)
- Engineering & Computer Science (AREA)
- Dentistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Environmental Sciences (AREA)
- Pulmonology (AREA)
- Agronomy & Crop Science (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Изобретение относится к области органической химии, а именно к производным нитроимидазооксазина общей формулы I, где n равно 1, V и W независимо представляют собой Н или СН3, и один из Х или Y представляет собой Н, и другой представляет собой одну из формул IIa или IIb, где формула IIa включает единственное кольцо, отмеченное в положении 3 и положении 4 и содержащее R1 в качестве заместителя, и формула IIb включает первое кольцо, отмеченное в положении 3 и положении 4 и содержащее в качестве заместителей как R2, так и концевое кольцо, отмеченное в положении 4 и содержащее R1 в качестве заместителя, где единственное кольцо формулы IIa и первое кольцо и концевое кольцо формулы IIb включают С, СН или N в каждом положении в кольце, где единственное кольцо формулы IIa и первое кольцо и концевое кольцо формулы IIb независимо содержат не более двух атомов азота; Z в формулах IIa и IIb представляет собой СН2 или прямую связь, R1 независимо представляет собой любой один или два из Н, F, С1, CF3, OCF3 или OCH2Ph, и R2 представляет собой Н. Также изобретение относится к фармацевтической композиции на основе соединения формулы I, способу предупреждения и лечения микробной инфекции, основанному на использовании соединения формулы I, конкретным производным нитроимидазооксазина. Технический результат: получены новые соединения, обладающие полезной биологической активностью. 4 н. и 3 з.п. ф-лы, 21 ил., 3 табл.


Description
Предшествующий уровень техники
[0001] По настоящей заявке испрашивается приоритет на основании временной патентной заявки США, серийный номер 61/230422, озаглавленной "Нитроимидазооксазиновые и нитроимидазооксазольные аналоги и их применения", поданной 31 июля 2009 года, полное содержание которой включено в настоящее описание посредством ссылки.
[0002] Настоящее изобретение относится к нитроимидазооксазиновым и нитроимидазооксазольным аналогам, к их получению и к их применению в качестве лекарственных средств, эффективных против Mycobacterium tuberculosis, и в качестве антипротозойных средств, либо отдельно, либо в комбинации с другими лечениями.
[0003] Туберкулез остается основной инфекцией, являющейся причиной смертности, распространенной по всему миру (согласно оценкам, смертность составила 1,3 миллиона в 2008 году), при этом в последнее время эта инфекция активизировалась, что относят за счет повышенной восприимчивости ВИЧ пациентов, а также за счет роста числа случаев возникновения штаммов с множественной лекарственной резистентностью и возникновения штаммов с сильной лекарственной резистентностью. Существующая в настоящее время лекарственная терапия для лечения туберкулеза является длительной и комплексной и включает сочетания нескольких лекарственных средств (обычно исониазида, рифампина, пиразинамида и этамбутола), вводимых ежедневно на протяжении более 6 месяцев. Кроме того, эти лекарственные средства относительно неэффективны против персистентной формы заболевания, которая, как предполагают, возникает в значительной части случаев (Ferrara et al., 2006). Лекарственные средства второй очереди, используемые в длительных комбинированных терапиях для лечения полилекарственно-резистентных заболеваний (обычно более 2 лет), в большинстве случаев обладают пониженной активностью или более высокой токсичностью по сравнению с существующими первоочередными средствами. Часто лечение является неполным, что приводит к большому числу рецидивов и повышенной лекарственной резистентности, что подчеркивает настоятельную необходимость в новых, более эффективных лекарственных средствах.
[0004] Болезнь Шагаса поражает около 9 миллионов людей, преимущественно в Южной Америке, и приводит к около 14000 случаям смертности ежегодно. Причиной, вызывающей эту болезнь, является протозойный паразит Trypanosoma cruzi, который передается человеку кровососущими насекомыми. Два существующих в настоящее время лекарственных средства для лечения, нифуртимокс и бензнидазол, демонстрируют эффективность, которая ограничивается острой фазой заболевания и только некоторыми патогенными штаммами. Эти лекарственные средства также имеют серьезные побочные эффекты, и это, вместе с необходимым длительным и дорогим лечением, приводит к тому, что пациенты неадекватно поддаются лечению, а также к развитию лекарственной резистентности (Cavalli et al., 2009).
[0005] Лейшманиозы поражают почти 12 миллионов людей примерно в 90 странах и приводят к около 51000 случаям смертности ежегодно. Они особенно распространены на Индийском субконтиненте и в восточной Африке, где паразит Leishmania donovani является причиной этого заболевания. Этот паразит передается человеку через укус самок москитов, и он является ответственным за наиболее тяжелую форму, висцеральный лейшманиоз (кала-азар), который вызывает хроническое заболевание печени и селезенки и является гибельным, если его не лечить при помощи химиотерапии. Первоочередное лечение включает препараты на основе сурьмы меглумин антимонат (глюкантим) и стибоглюконата натрия (пентостам), открытые более чем 50 лет назад, которые имеют тяжелые нежелательные побочные эффекты. Их введение в низких дозах в течение длительного периода времени привело к увеличивающемуся числу случаев лекарственной резистентности, такой, что их больше нельзя использовать в Индии (Cavalli et al., 2009). Средства второй очереди имеют такие же проблемы токсичности, что говорит о действительной необходимости в более безопасных, более эффективных лечениях.
[0006] Поэтому крайне желательно обеспечить новые нитроимидазооксазиновые и нитроимидазооксазольные аналоги с неожиданно высокой активностью как против аэробных (реплицирующих), так и гипоксических (латентных или персистентных) культур Mycobacterium tuberculosis для применения в качестве противотуберкулезных лекарственных средств, и/или с неожиданно высокой активностью против Trypanosoma cruzi или Leishmania donovani для применения в качестве антипротозойных средств, и для лечения других микробных инфекций.
Краткое описание изобретения
[0007] Настоящее изобретение относится к нитроимидазооксазиновым и нитроимидазооксазольным аналогам, к способам их получения и к применению таких соединений в качестве средств для лечения Mycobacterium tuberculosis, для применения в качестве противотуберкулезных лекарственных средств, для применения в качестве анти-протозойных средств с неожиданно высокой активностью против Trypanosoma cruzi или Leishmania donovani и для лечения других микробных инфекций.
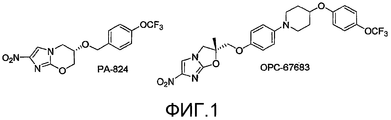
[0008] Тот факт, что нитроимидазооксазин PA-824 был недавно введен в стадию клинических испытаний, является очень важным, поскольку это соединение демонстрирует хорошую in vitro и in vivo активность против Mycobacterium tuberculosis, как в активной, так и в персистентной формах (Tyagi et al., 2005). Родственный 2-нитроимидазо[2,1-b]оксазол, OPC-67683, также находится в стадии клинических испытаний (Sasaki et al., 2006). Структуры этих соединений представлены на фиг.1. Не желая быть связанными теорией, предполагают, что механизм действия PA-824 включает высвобождение оксида азота (Singh et al., 2008), после восстановительной стадии, в способе, зависимом от бактериальной глюкоза-6-фосфат-дегидрогеназы (FGD1) и ее кофактора F420 (Stover et al., 2000). Исследования с использованием микроматрицы на мутантных штаммах дикого типа, как для FGD1, так и F420, показали, что 151-аминокислотный (17,37 кДа) белок с неизвестной функцией, Rv3547, по-видимому, является критическим для этой активации (Manjunatha et al., 2006). Осуществленные недавно механистические исследования восстановительной химии PA-824 подтверждают такую точку зрения (Anderson et al., 2008). Нитроимидазооксазиновые аналоги и нитроимидазооксазольные аналоги и их применение при туберкулезе были описаны ранее (патенты США 5668127 и 6087358; Jiricek et al., WO 2007075872A2; Li et al., 2008; Kim et al., 2009; Nagarajan et al., 1989; Ashtekar et al., 1993; Sasaki et al., 2006; Matsumoto et al., 2006; Tsubochi et al., WO 2005042542A1 и WO 2004033463 A1; JP 2005330266A; EP 1555267A1).
[0009] В первом аспекте настоящее изобретение относится к соединению, имеющему общую структурную формулу I:

где n равно 0 или 1,
V и W независимо представляют собой H или CH3, и
один из X или Y представляет собой H, и другой представляет собой одну из формул IIa или IIb, где формулы IIa и IIb имеют общие структуры:

где формула IIb включает первое кольцо, отмеченное в положении 3 и положении 4 и содержащее в качестве заместителей как R2, так и концевое кольцо, отмеченное в положении 4 и содержащее R1 в качестве заместителя,
Z в формулах IIa и IIb представляет собой CH2 или прямую связь, и
R1 и R2 каждый представляет собой любой один или два из H, F, Cl, I, CN, CF3, OCF3, OCH3, OCH2Ph, аза (-CH= заменена группой -N=) или диаза (-CH=CH- заменена группой -N=N-, -CH=CH-CH= заменена группой -N=CH-N=, или -CH=CH-CH=CH- заменена группой -N=CH-CH=N-) в любом из доступных положений в кольце;
при условии, что, когда n равно 0, V, W и X, все представляют собой H, и Y представляет собой формулу IIa, где Z представляет собой либо CH2, либо прямую связь, тогда R1 не является H;
и при условии, что, когда n равно 0, V и X оба представляют собой H, W представляет собой CH3, и Y представляет собой формулу IIa, где Z представляет собой прямую связь, тогда R1 не является H, 4-Cl, 4-I, 4-CF3, 4-OCH3 или 4-OCF3;
и при условии, что, когда n равно 0, V и X, оба представляют собой H, W представляет собой CH3, и Y представляет собой формулу IIb, где Z представляет собой прямую связь, концевое кольцо расположено в положении 4 на первом кольце, и R2 представляет собой H, тогда R1 не является H или 4-аза.
[0010] Более предпочтительный подкласс соединений включает соединения, имеющие общую структурную формулу I, как определено выше, где:
n равно 0 или 1,
V и W независимо представляют собой H или CH3, и
один из X или Y представляет собой H, и другой представляет собой одну из формул IIa или IIb, где формулы IIa и IIb имеют общие структуры:
где формула IIb включает первое кольцо, отмеченное в положении 3 и положении 4 и содержащее в качестве заместителей как R2, так и концевое кольцо, отмеченное в положении 4 и содержащее R1 в качестве заместителя,
Z в формулах IIa и IIb представляет собой CH2 или прямую связь,
R1 представляет собой 4-F, 4-CN, 4-I, 4-CF3, 3-OCF3, 4-OCF3, 4-OCH2Ph или 3-аза-4-OMe, и
R2 представляет собой H, аза (-CH= заменена группой -N=) или диаза (-CH=CH- заменена группой -N=N-, -CH=CH-CH= заменена группой -N=CH-N=, или -CH=CH-CH=CH- заменена группой -N=CH-CH=N-) в любом из доступных положений в кольце;
при условии, что, когда n равно 0, V, W и X все представляют собой H, и Y представляет собой формулу IIa, где Z представляет собой либо CH2, либо прямую связь, тогда R1 не является H;
и при условии, что, когда n равно 0, V и X оба представляют собой H, W представляет собой CH3, и Y представляет собой формулу IIa, где Z представляет собой прямую связь, тогда R1 не является H, 4-Cl, 4-I, 4-CF3, 4-OCH3 или 4-OCF3;
и при условии, что, когда n равно 0, V и X оба представляют собой H, W представляет собой CH3, и Y представляет собой формулу IIb, где Z представляет собой прямую связь, концевое кольцо расположено в положении 4 на первом кольце, и R2 представляет собой H, тогда R1 не является H или 4-аза.
[0011] Эти соединения, а также их смеси, изомеры, физиологически функциональные солевые производные и пролекарства являются полезными для предупреждения или лечения Mycobacterium tuberculosis, для применения в качестве противотуберкулезных лекарственных средств, для применения в качестве антипротозойных средств с неожиданно высокой активностью против Trypanosoma cruzi или Leishmania donovani и для лечения других микробных инфекций.
Краткое описание чертежей
[0012] На фиг.1 представлены структуры соединений PA-824 и OPC-67683;
[0013] на фиг.2 представлены общие структуры репрезентативных соединений, указанных в таблице 1;
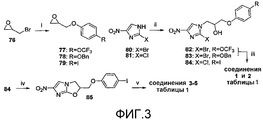
[0014] на фиг.3 представлена общая схема синтеза для получения репрезентативных соединений;
[0015] на фиг.4 представлена общая схема синтеза для получения репрезентативных соединений;
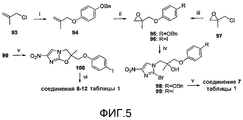
[0016] на фиг.5 представлена общая схема синтеза для получения репрезентативных соединений;
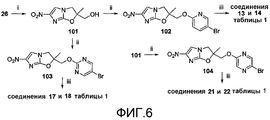
[0017] на фиг.6 представлена общая схема синтеза для получения репрезентативных соединений;
[0018] на фиг.7 представлена общая схема синтеза для получения репрезентативных соединений;
[0019] на фиг.8 представлена общая схема синтеза для получения репрезентативных соединений;
[0020] на фиг.9 представлена общая схема синтеза для получения репрезентативных соединений;
[0021] на фиг.10 представлена общая схема синтеза для получения репрезентативных соединений;
[0022] на фиг.11 представлена общая схема синтеза для получения репрезентативных соединений;
[0023] на фиг.12 представлена общая схема синтеза для получения репрезентативных соединений;
[0024] на фиг.13 представлена общая схема синтеза для получения репрезентативных соединений;
[0025] на фиг.14 представлена общая схема синтеза для получения репрезентативных соединений;
[0026] на фиг.15 представлена общая схема синтеза для получения репрезентативных соединений;
[0027] на фиг.16 представлена общая схема синтеза для получения репрезентативных соединений;
[0028] на фиг.17 представлена общая схема синтеза для получения репрезентативных соединений;
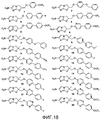
[0029] на фиг.18 представлена структура репрезентативных соединений 1-20, указанных в таблице 1 и примерах 1-3;
[0030] на фиг.19 представлены структуры репрезентативных соединений 21-38, указанных в таблице 1 и примерах 1-3;
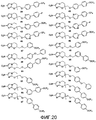
[0031] на фиг.20 представлены структуры репрезентативных соединений 39-58, указанных в таблице 1 и примерах 1-3; и
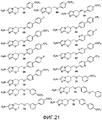
[0032] на фиг.21 представлены структуры репрезентативных соединений 59-75, указанных в таблице 1 и примерах 1-3.
Подробное описание предпочтительных вариантов осуществления изобретения
[0033] Настоящее изобретение относится к нитроимидазооксазиновым и нитроимидазооксазольным аналогам, к способам их получения и к применению соединений для предупреждения или лечения Mycobacterium tuberculosis, для применения в качестве противотуберкулезных лекарственных средств, для применения в качестве антипротозойных средств с неожиданно высокой активностью против Trypanosoma cruzi или Leishmania donovani и для лечения других микробных инфекций.
[0034] В первом аспекте настоящее изобретение относится к соединению, имеющему общую структурную формулу I:
где n равно 0 или 1,
V и W независимо представляют собой H или CH3, и
один из X или Y представляет собой H, и другой представляет собой одну из формул IIa или IIb, где формулы IIa и IIb имеют общие структуры:
где формула IIb включает первое кольцо, отмеченное в положении 3 и положении 4 и содержащее в качестве заместителей как R2, так и концевое кольцо, отмеченное в положении 4 и содержащее R1 в качестве заместителя,
Z в формуле IIa и IIb представляет собой CH2 или прямую связь, и
R1 и R2 каждый представляет собой любой один или два из H, F, Cl, I, CN, CF3, OCF3, OCH3, OCH2Ph, аза (-CH= заменена группой -N=) или диаза (-CH=CH- заменена группой -N=N-, -CH=CH-CH= заменена группой N=CH-N=, или -CH=CH-CH=CH- заменена группой -N=CH-CH=N-) в любом из доступных положений в кольце;
при условии, что, когда n равно 0, V, W и X все представляют собой H, и Y представляет собой формулу IIa, где Z представляет собой либо CH2, либо прямую связь, тогда R1 не является H;
и при условии, что, когда n равно 0, V и X оба представляют собой H, W представляет собой CH3, и Y представляет собой формулу IIa, где Z представляет собой прямую связь, тогда R1 не является H, 4-Cl, 4-I, 4-CF3, 4-OCH3 или 4-OCF3;
и при условии, что, когда n равно 0, V и X оба представляют собой H, W представляет собой CH3, и Y представляет собой формулу IIb, где Z представляет собой прямую связь, концевое кольцо расположено в положении 4 на первом кольце, и R2 представляет собой H, тогда R1 не является H или 4-аза.
[0035] Более предпочтительный подкласс соединений включает соединения, имеющие общую структурную формулу I, как определено выше, где:
n равно 0 или 1,
V и W независимо представляют собой H или CH3, и
один из X или Y представляет собой H, и другой представляет собой одну из формул IIa или IIb, где формулы IIa и IIb имеют общие структуры:
где формула IIb включает первое кольцо, отмеченное в положении 3 и положении 4 и содержащее в качестве заместителей как R2, так и концевое кольцо, отмеченное в положении 4 и содержащее R1 в качестве заместителя,
Z в формулах IIa и IIb представляет собой CH2 или прямую связь,
R1 представляет собой 4-F, 4-CN, 4-I, 4-CF3, 3-OCF3, 4-OCF3, 4-OCH2Ph или 3-аза-4-OMe, и
R2 представляет собой H, аза (-CH= заменена группой -N=) или диаза (-CH=CH- заменена группой -N=N-, -CH=CH-CH= заменена группой -N=CH-N=, или -CH=CH-CH=CH- заменена группой -N=CH-CH=N-) в любом из доступных положений в кольце;
при условии, что, когда n равно 0, V, W и X все представляют собой H, и Y представляет собой формулу IIa, где Z представляет собой либо CH2 или прямую связь, тогда R1 не является H;
и при условии, что, когда n равно 0, V и X оба представляют собой H, W представляет собой CH3, и Y представляет собой формулу IIa, где Z представляет собой прямую связь, тогда R1 не является H, 4-Cl, 4-I, 4-CF3, 4-OCH3 или 4-OCF3;
и при условии, что, когда n равно 0, V и X оба представляют собой H, W представляет собой CH3, и Y представляет собой формулу IIb, где Z представляет собой прямую связь, концевое кольцо расположено в положении 4 на первом кольце, и R2 представляет собой H, тогда R1 не является H или 4-аза.
[0036] Наиболее предпочтительными из соединений формулы I являются следующие:
A. 6-нитро-2-{[4-(трифторметокси)фенокси]метил}-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 1 таблицы 1 и фиг.18);
B. 2-{[4-(бензилокси)фенокси]метил}-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 2 таблицы 1 и фиг.18);
C. 2-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 3 таблицы 1 и фиг.18);
D. 6-нитро-2-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 4 таблицы 1 и фиг.18);
E. 6-нитро-2-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 5 таблицы 1 и фиг.18);
F. 6-нитро-2-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 6 таблицы 1 и фиг.18);
G. 2-{[4-(бензилокси)фенокси]метил}-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 7 таблицы 1 и фиг.18);
H. 2-{[4-(6-метокси-3-пиридинил)фенокси]метил}-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 8 таблицы 1 и фиг.18);
I. 4'-[(2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол-2-ил)метокси][1,1'-бифенил]-4-карбонитрил (соединение 9 таблицы 1 и фиг.18);
J. 2-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 10 таблицы 1 и фиг.18);
K. 2-метил-6-нитро-2-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 11 таблицы 1 и фиг.18);
L. 2-метил-6-нитро-2-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 12 таблицы 1 и фиг.18);
M. 2-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 13 таблицы 1 и фиг.18);
N. 2-метил-6-нитро-2-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 14 таблицы 1 и фиг.18);
O. 2-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 15 таблицы 1 и фиг.18);
P. 2-метил-6-нитро-2-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 16 таблицы 1 и фиг.18);
Q. 2-({[5-(4-фторфенил)-2-пиримидинил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 17 таблицы 1 и фиг.18);
R. 2-метил-6-нитро-2-[({5-[4-(трифторметокси)фенил]-2-пиримидинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 18 таблицы 1 и фиг.18);
S. 2-({[2-(4-фторфенил)-5-пиримидинил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 19 таблицы 1 и фиг.18);
T. 2-метил-6-нитро-2-[({2-[4-(трифторметокси)фенил]-5-пиримидинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 20 таблицы 1 и фиг.18);
U. 2-({[5-(4-фторфенил)-2-пиразинил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 21 таблицы 1 и фиг.19);
V. 2-метил-6-нитро-2-[({5-[4-(трифторметокси)фенил]-2-пиразинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 22 таблицы 1 и фиг.19);
W. 6-нитро-2-({[4-(трифторметокси)бензил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 23 таблицы 1 и фиг.19);
X. 2-({[4-(бензилокси)бензил]окси}метил)-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 24 таблицы 1 и фиг.19);
Y. 2-метил-6-нитро-2-({[4-(трифторметокси)бензил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 25 таблицы 1 и фиг.19);
Z. 2-({[4-(бензилокси)бензил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (соединение 26 таблицы 1 и фиг.19);
AA. 2-нитро-7-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 27 таблицы 1 и фиг.19);
BB. 7-{[4-(бензилокси)фенокси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 28 таблицы 1 и фиг.19);
CC. 7-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 29 таблицы 1 и фиг.19);
DD. 2-нитро-7-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 30 таблицы 1 и фиг.19);
EE. 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 31 таблицы 1 и фиг.19);
FF. 7-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 32 таблицы 1 и фиг.19);
GG. 2-нитро-7-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 33 таблицы 1 и фиг.19);
ΗΗ. 7-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 34 таблицы 1 и фиг.19);
II. 2-нитро-7-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 35 таблицы 1 и фиг.19);
JJ. 7-метил-2-нитро-7-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 36 таблицы 1 и фиг.19);
KK. 7-{[4-(бензилокси)фенокси]метил}-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 37 таблицы 1 и фиг.19);
LL. 7-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 38 таблицы 1 и фиг.19);
MM. 7-метил-2-нитро-7-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 39 таблицы 1 и фиг.20);
NN. 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 40 таблицы 1 и фиг.20);
OO. 7-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 41 таблицы 1 и фиг.20);
PP. 7-метил-2-нитро-7-[({5-[4-(трифторметил)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 42 таблицы 1 и фиг.20);
QQ. 7-метил-2-нитро-7-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 43 таблицы 1 и фиг.20);
RR. 7-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 44 таблицы 1 и фиг.20);
SS. 7-метил-2-нитро-7-[({6-[4-(трифторметил)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 45 таблицы 1 и фиг.20);
TT. 7-метил-2-нитро-7-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 46 таблицы 1 и фиг.20);
UU. 2-нитро-7-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 47 таблицы 1 и фиг.20);
VV. 2-нитро-7-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 48 таблицы 1 и фиг.20);
WW. 7-({[4-(бензилокси)бензил]окси}метил)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 49 таблицы 1 и фиг.20);
XX. 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-3-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 50 таблицы 1 и фиг.20);
YY. 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 51 таблицы 1 и фиг.20);
ZZ. 7-метил-2-нитро-7-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 52 таблицы 1 и фиг.20);
AAA. 7-метил-2-нитро-7-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 53 таблицы 1 и фиг.20);
BBB. 7-({[4-(бензилокси)бензил]окси}метил)-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 54 таблицы 1 и фиг.20);
CCC. 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-3-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 55 таблицы 1 и фиг.20);
DDD. 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 56 таблицы 1 и фиг.20);
EEE. (7R)-7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 57 таблицы 1 и фиг.20);
FFF. (7S)-7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 58 таблицы 1 и фиг.20);
GGG. 2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 59 таблицы 1 и фиг.21);
HHH. (6R)-2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 60 таблицы 1 и фиг.21);
III. (6S)-2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 61 таблицы 1 и фиг.21);
JJJ. 6-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 62 таблицы 1 и фиг.21);
KKK. 2-нитро-6-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 63 таблицы 1 и фиг.21);
LLL. 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 64 таблицы 1 и фиг.21);
MMM. 6-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 65 таблицы 1 и фиг.21);
NNN. 2-нитро-6-[({5-[4-(трифторметил)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 66 таблицы 1 и фиг.21);
OOO. 2-нитро-6-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 67 таблицы 1 и фиг.21);
PPP. 6-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 68 таблицы 1 и фиг.21);
QQQ. 2-нитро-6-[({6-[4-(трифторметил)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 69 таблицы 1 и фиг.21);
RRR. 2-нитро-6-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 70 таблицы 1 и фиг.21);
SSS. 2-нитро-6-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 71 таблицы 1 и фиг.21);
TTT. 2-нитро-6-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 72 таблицы 1 и фиг.21);
UUU. 6-({[4-(бензилокси)бензил]окси}метил)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 73 таблицы 1 и фиг.21);
VVV. 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-3-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 74 таблицы 1 и фиг.21) и
WWW. 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 75 таблицы 1 и фиг.21).
[0037] Соединения формулы I могут существовать в различных геометрических и энантиомерных формах, и как чистые формы, так и смеси этих отдельных изомеров включены в объем настоящего изобретения, а также их любые физиологически функциональные или фармакологически приемлемые солевые производные или пролекарства. Получение таких альтернативных форм не должно составлять трудностей для специалистов в данной области.
[0038] Настоящее изобретение также относится к способам предупреждения или лечения туберкулезной, протозойных и других микробных инфекций, таких как Mycobacterium tuberculosis, Trypanosoma cruzi и Leishmania donovani, включающим стадию введения соединения формулы I.
[0039] В другом аспекте настоящего изобретения обеспечивается фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы I, определенного выше, и фармацевтически приемлемый эксципиент, адъювант, носитель, буфер или стабилизатор. Под "терапевтически эффективным количеством" следует понимать количество соединения формулы I, которое является достаточным для проявления антибактериального или антимикробного эффектов. Действительное количество, частота и продолжительность введения зависят от природы и тяжести заболевания, подлежащего лечению. Лечение прописывают врачи общей практики и другие лечащие врачи. Фармацевтически приемлемые эксципиент, адъювант, носитель, буфер или стабилизатор должны быть нетоксичными и не должны негативно влиять на эффективность активного ингредиента. Конкретная природа носителя или другого вещества зависит от пути введения, который может представлять собой пероральный путь введения или при помощи инъекции, такой как внутрикожная, подкожная, внутривенная инъекция, или с использованием ингалятора сухого порошка.
[0040] Фармацевтические композиции для перорального введения могут быть в форме таблетки, капсулы, порошка или жидкости. Таблетка может включать твердый носитель или адъювант. Жидкие фармацевтические композиции, как правило, содержат жидкий носитель, такой как вода, нефтяные, животные или растительные масла, минеральное масло или синтетическое масло. Могут быть включены физиологический солевой раствор, раствор декстрозы или другого сахарида или гликоли, такие как этиленгликоль, пропиленгликоль или полиэтиленгликоль. Капсула может содержать твердый носитель, такой как желатин. Для внутривенной, внутрикожной или подкожной инъекции активный ингредиент должен быть в форме парентерально приемлемого водного раствора, который является апирогенным и имеет подходящее значение pH, изотоничность и стабильность. Специалисты, обладающие соответствующей квалификацией, смогут получить подходящие растворы с использованием, например, изотонических носителей, таких как раствор хлорида натрия для инъекций, раствор Рингера для инъекций, содержащий лактат раствор Рингера для инъекций. Консерванты, стабилизаторы, буферы, антиоксиданты и/или другие добавки могут быть включены, по мере необходимости.
[0041] Фармацевтическая композиция, кроме того, может содержать одно или несколько дополнительных терапевтических средств против инфекции. Такие терапевтические средства против инфекции могут представлять собой любое подходящее, коммерчески доступное терапевтическое средство или доступное из других источников, которое известно как эффективное для предупреждения или лечения микробных инфекций, таких как Mycobacterium tuberculosis, Trypanosoma cruzi и/или Leishmania donovani.
[0042] В другом аспекте обеспечивается применение для получения лекарственного средства, включающего терапевтически эффективное количество соединения формулы I, определенного выше, для введения субъекту. Также обеспечивается способ получения соединения формулы I.
[0043] Термин "фармакологически приемлемая соль", используемый в настоящем описании, следует рассматривать как означающий любую соль кислоты или основания, образованную из хлористоводородной, серной, фосфорной, уксусной, лимонной, щавелевой, малоновой, салициловой, яблочной, фумаровой, янтарной, аскорбиновой, малеиновой, метансульфоновой, изоэтоновой кислот и подобных, и карбоната калия, гидроксида натрия или калия, аммиака, триэтиламина, триэтаноламина и подобных.
[0044] Термин "пролекарство" означает фармакологическое вещество, которое вводят в неактивной или по существу менее активной форме. После введения пролекарство метаболизируется in vivo в активный метаболит.
[0045] Термин "терапевтически эффективное количество" означает нетоксичное количество лекарственного средства, но которое является достаточным для обеспечения желаемого терапевтического эффекта. Количество, которое является "эффективным", для разных субъектов будет различным, в зависимости от возраста и общего состояния субъекта, конкретной вводимой концентрации и композиции, и т.п. Таким образом, не всегда можно точно определить, какое количество является эффективным. Однако подходящее эффективное количество в каждом конкретном случае сможет определить специалист в данной области, обладающий средней квалификацией, с использованием рутинного экспериментирования. Кроме того, эффективное количество представляет собой концентрацию, находящуюся в пределах, достаточных, чтобы можно было легко использовать композицию для доставки количества лекарственного средства, находящегося в пределах терапевтически эффективного количества.
[0046] Термин "аза" означает группу -CH=, которая заменена -N=, в соединении. Термин "диаза" означает группу -CH=CH-, которая заменена -N=N-, группу -CH=CH-CH=, которая заменена -N=CH-N=, или группу -CH=CH-CH=CH-, которая заменена -N=CH-CH=N-, в соединении.
[0047] Другие аспекты настоящего изобретения будут очевидны из следующего описания, представленного исключительно при помощи примеров, со ссылкой на сопровождающие схемы синтеза.
ПРИМЕР 1. Общие схемы синтеза
[0048] Соединения могут быть получены общими способами, схематически показанными на схемах 1-15, которые представлены на фиг.3-17, или с использованием любого другого подходящего способа. В представленном ниже описании схем 1-15 делаются ссылки на репрезентативные соединения, представленные в таблице 1 ниже и на фиг.2 и 18-21.
|
Таблица 1
Репрезентативные соединения |
||||||
| No | Фиг.2 Структура | R | Формула | Т.пл. (°C) | Анализы | |
| 1 | A | 4-OCF3 (X=H) | C13H10F3N3O5 | 170-172 | C,H,N | |
| 2 | A | 4-OCH2Ph (X=H) | C19H17N3O5 | 208-210 | C,H,N | |
| 3 | A | 4-[(4-F)фенил] (X=H) | C18H14FN3O4 | 224-226 | ||
| 4 | A | 4-[(4-CF3)фенил] (X-H) | C19H14F3N3O4 | 210-211 | ||
| 5 | A | 4-[(4-OCF3)фенил] (X=H) | C19H14F3N3O5 | 200-201 | C,H,N | |
| 6 | A | 2-аза, 4-[(4-OCF3)фенил] (X=H) | Cl8H13F3N4O5 | 127-130 | ||
| 7 | A | 4-OCH2Ph (X=Me) | C20H19N3O5 | 162-165 | C,H,N | |
| 8 | A | 4-[(6-OMe)3-пиридил)] (X=Me) | C19H18N4O5 | 217-219 | C,H,N | |
| 9 | A | 4-[(4-CN)фенил] (X=Me) | C20H16N4O4 | 180-181 | C,H,N | |
| 10 | A | 4-[(4-F)фенил] (X=Me) | C19H16FN3O4 | 180-181 | C,H,N | |
| 11 | A | 4-[(4-CF3)фенил] (X=Me) | C20H16F3N3O4 | 219-220 | C,H,N | |
| 12 | A | 4-[(4-OCF3)фенил] (X=Me) | C20H16F3N3O5 | 209-211 | C,H,N | |
| 13 | A | 2-аза, 4-[(4-F)фенил] (X=Me) | C18H15FN4O4 | 162-164 | ||
| 14 | A | 2-аза, 4-[(4-OCF3)фенил] (X=Me) | C19H15F3N4O5 | 172-174 | C,H,N | |
| 15 | A | 3-аза, 4-[(4-F)фенил] (X=Me) | C18H15FN4O4 | 180-181 | ||
| 16 | A | 3-аза, 4-[(4-OCF3)фенил] (X=Me) | C19H15F3N4O5 | 209-211 | C,H,N | |
| 17 | A | 2,6-диаза, 4-[(4-F)фенил] (X=Me) | C17H14FN5O4 | 196 разл. | C,H,N | |
| 18 | A | 2,6-диаза, 4-[(4-OCF3)фенил] (X=Me) | C18H14F3N5O5 | 227 разл. | C,H,N | |
| 19 | A | 3,5-диаза, 4-[(4-F)фенил] (X=Me) | C17H14FN5O4 | 201-203 | ||
| 20 | A | 3,5-диаза, 4-[(4-OCF3)фенил] (X=Me) | C18H14F3N5O5 | 223-225 | C,H,N | |
| 21 | A | 2,5-диаза, 4-[(4-F)фенил] (X=Me) | C17H14FN5O4 | 200-201 | C,H,N | |
| 22 | A | 2,5-диаза, 4-[(4-OCF3)фенил)] (X=Me) | C18H14F3N5O5 | 222-224 | C,H,N | |
| 23 | B | 4-OCF3 (X=H) | C14H12F3N3O5 | 134-135 | C,H,N | |
| 24 | B | 4-OCH2Ph (X=H) | C20H19N3O5 | 123-124 | C,H,N | |
| 25 | B | 4-OCF3 (X=Me) | C15H14F3N3O5 | 110-111 | C,H,N | |
| 26 | B | 4-OCH2Ph (X=Me) | C21H21N3O5 | 130-131 | C,H,N | |
| 27 | C | 4-OCF3 (X=H) | C14H12F3N3O5 | 138-140 | C,H,N | |
| 28 | C | 4-OCH2Ph (X=H) | C20H19N3O5• 0,25H2O |
222-224 | C,H,N | |
| 29 | C | 4-[(4-F)фенил] (X=H) | C19H16FN3O4 | 217-219 | C,H,N | |
| 30 | C | 4-[(4-CF3)фенил] (X=H) | C20H16F3N3O4 | 242-245 | C,H,N | |
| 31 | C | 4-[(4-OCF3)фенил] (X=H) | C20H16F3N3O5 | 197-199 | C,H,N | |
| 32 | C | 2-аза, 4-[(4-F)фенил] (X=H) | C18H15FN4O4 | 180-181 | ||
| 33 | C | 2-аза, 4-[(4-OCF3)фенил] (X=H) | C19H15F3N4O5 | 161-163 | C,H,N | |
| 34 | C | 3-аза, 4-[(4-F)фенил] (X=H) | C18H15FN4O4 | 204-206 | ||
| 35 | C | 3-аза, 4-[(4-OCF3)фенил] (X=H) | C19H15F3N4O5 | 161-163 | C,H,N | |
| 36 | C | 4-OCF3 (X=Me) | C15H14F3N3O5 | 134-136 | C,H,N | |
| 37 | C | 4-OCH2Ph (X=Me) | C21H21N3O5 | 174-176 | C,H,N | |
| 38 | C | 4-[(4-F)фенил] (X=Me) | C20H18FN3O4 | 160-162 | C,H,N | |
| 39 | C | 4-[(4-CF3)фенил] (X=Me) | C21H18F3N3O4 | 196-198 | C,H,N | |
| 40 | C | 4-[(4-OCF3)фенил] (X=Me) | C21H18F3N3O5 | 186-188 | C,H,N | |
| 41 | C | 2-аза, 4-[(4-F)фенил] (X=Me) | C19H17FN4O4 | 145-147 | C,H,N | |
| 42 | C | 2-аза, 4-[(4-CF3)фенил] (X=Me) | C20H17F3N4O4 | 212-214 | C,H,N | |
| 43 | C | 2-аза, 4-[(4-OCF3)фенил] (X=Me) | C20H17F3N4O5 | 195-198 | C,H,N | |
| 44 | C | 3-аза, 4-[(4-F)фенил] (X=Me) | C19H17FN4O4 | 203-204 | C,H,N | |
| 45 | C | 3-аза, 4-[(4-CF3)фенил] (X=Me) | C20H17F3N4O4 | 215-217 | C,H,N | |
| 46 | C | 3-аза, 4-[(4-OCF3)фенил] (X=Me) | C20H17F3N4O5 | 202-203 | C,H,N | |
| 47 | D | 3-OCF3 (X=H) | C15H14F3N3O5 | 100-112 | C,H,N | |
| 48 | D | 4-OCF3 (X=H) | C15H14F3N3O5 | 158-160 | C,H,N | |
| 49 | D | 4-OCH2Ph (X=H) | C21H21N3O5 | 151-153 | C,H,N | |
| 50 | D | 3-[(4-OCF3)фенил] (X=H) | C21H18F3N3O5 | 117-119 | C,H,N | |
| 51 | D | 4-[(4-OCF3)фенил] (X=H) | C21H18F3N3O5 | 159-161 | C,H,N | |
| 52 | D | 3-OCF3 (X=Me) | C16H16F3N3O5 | 108-110 | C,H,N | |
| 53 | D | 4-OCF3 (X-Me) | C16H16F3N3O5 | 100-101 | C,H,N | |
| 54 | D | 4-OCH2Ph (X=Me) | C22H23N3O5 | 109-111 | C,H,N | |
| 55 | D | 3-[(4-OCF3)фенил] (X=Me) | C22H20F3N3O5 | 80-82 | C,H,N | |
| 56 | D | 4-[(4-OCF3)фенил] (X=Me) (рац) | C22H20F3N3O5 | 150-152 | C,H,N | |
| 57 | D | 4-[(4-OCF3)фенил] (X=Me) (7-R) | C22H20F3N3O5 | 165-167 | C,H,N | |
| 58 | D | 4-[(4-OCF3)фенил] (X=Me) (7-S) | C22H20F3N3O5 | 162-164 | C,H,N | |
| 59 | E | 4-OCF3 (рац) | C14H12F3N3O5 | 141-143 | C,H,N | |
| 60 | E | 4-OCF3 (6-R) | C14H12F3N3O5 | 138-139 | C,H,N | |
| 61 | E | 4-OCF3 (6-S) | C14H12F3N3O5 | 139-140 | C,H,N | |
| 62 | E | 4-[(4-F)фенил] | C19H16FN3O4 | 201-203 | C,H,N | |
| 63 | E | 4-[(4-CF3)фенил] | C20H16F3N3O4 | 218-221 | C,H,N | |
| 64 | E | 4-[(4-OCF3)фенил] | C20H16F3N3O5 | 192-194 | C,H,N | |
| 65 | E | 2-аза, 4-[(4-F)фенил] | C18H15FN4O4 | 160-161 | C,H,N | |
| 66 | E | 2-аза, 4-[(4-CF3)фенил] | C19H15F3N4O4 | 180-182 | C,H,N | |
| 67 | E | 2-аза, 4-[(4-OCF3)фенил] | C19H15F3N4O5 | 182-183 | C,H,N | |
| 68 | E | 3-аза, 4-[(4-F)фенил] | C18H15FN4O4 | 214-216 | C,H,N | |
| 69 | E | 3-аза, 4-[(4-CF3)фенил] | C19H15F3N4O4 | 233-235 | C,H,N | |
| 70 | E | 3-аза, 4-[(4-OCF3)фенил] | C19H15F3N4O5 | 180-181 | C,H,N | |
| 71 | F | 3-OCF3 | C15H14F3N3O5 | 60-61 | C,H,N | |
| 72 | F | 4-OCF3 | C15H14F3N3O5 | 92-93 | C,H,N | |
| 73 | F | 4-OCH2Ph | C21H21N3O5 | 150-151 | C,H,N | |
| 74 | F | 3-[(4-OCF3)фенил] | C21H18F3N3O5 | 78-80 | C,H,N | |
| 75 | F | 4-[(4-OCF3)фенил] | C21H18F3N3O5 | 135-138 | C,H,N | |
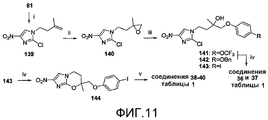
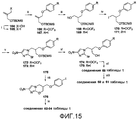
[0049] На схеме 1, представленной на фиг.3, использовали следующие реагенты и условия: (i) RPhOH, K2CO3, ацетон, кипячение с обратным холодильником, 36-52 час; (ii) соединения 77, 78 или 79, DIPEA, 105°C, 6,5-12 час; (iii) NaH, ДМФА, 0°C, 45 мин; (iv) NaH, ДМФА, 0°C, 80 мин, затем 17°C, 60 мин; (v) ArB(OH)2, 2M Na2CO3, толуол, EtOH, ДМФА, Pd(dppf)Cl2 в атмосфере N2, 88-90°C, 50-90 мин. Катализируемые основанием взаимодействия 2-бром-4(5)-нитроимидазола (80) или 2-хлор-4(5)-нитроимидазола (81) с эпоксидами 77-79 [получены путем алкилирования соответствующих 4-замещенных фенолов при использовании 2-(бромметил)оксирана (76)] давали спирты 82-84, которые подвергали реакциям замыкания кольца при использовании NaH, с получением соединений 1 и 2 таблицы 1 и йодида 85 соответственно. Реакции сочетания по методу Сузуки соединения 85 с арилбороновыми кислотами затем давали соединения 3-5 таблицы 1.
[0050] На схеме 2, представленной на фиг.4, использовали следующие реагенты и условия: (i) 70°C, 16 час; (ii) NaH, ДМФА, от -20 до -10°C, 50 мин; (iii) 1% HCl в 95% EtOH, 20°C, 6 час, затем 4°C, 2,5 дня; (iv) 4-OCF3PhB(OH)2, 2M Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 85-88°C, 3 час; (v) 90, NaH, ДМФА, 0-20°C, 2,5 часов. Взаимодействие 2,4-динитроимидазола (86) с эпоксидом 87 давало спирт 88, который подвергали реакции замыкания кольца при использовании NaH, с последующим катализируемым кислотой десилилированием, с получением спирта 90. Осуществляемое при помощи NaH алкилирование соединения 90 фторпиридином 92 [получен путем реакции сочетания Сузуки 5-бром-2-фторпиридина (91) с 4-(трифторметокси)фенилбороновой кислотой] затем давало соединение 6 таблицы 1.
[0051] На схеме 3, представленной на фиг.5, использовали следующие реагенты и условия: (i) A-BnOPhOH, K2CO3, ацетон, кипячение с обратным холодильником, 24 час; (ii) m-CPBA, Na2HPO4, CH2Cl2, 0-20°C, 3,5 час; (iii) 4-IPhOH, K2CO3, NaI, ДМФА, 70-73°C, 32 час; (iv) соединение 80, DIPEA, 107°C, 14-15 час; (v) NaH, ДМФА, 0°C, 50-75 мин; (vi) ArB(OH)2, 2M Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 90°C, 45 мин. Катализируемые основанием взаимодействия 2-бром-4(5)-нитроимидазола (80) с эпоксидами 95 и 96 [получены путем металлилирования 4-(бензилокси)фенола при помощи хлорида 93, с последующим эпоксидированием, или путем алкилирования 4-иодфенола при помощи 2-(хлорметил)-2-метилоксирана (97)] давали спирты 98 и 99, которые подвергали реакциям замыкания кольца при использовании NaH, с получением соединения 7 таблицы 1 и йодида 100 соответственно. Реакции сочетания Сузуки соединения 100 с арилбороновыми кислотами затем давали соединения 8-12 таблицы 1.
[0052] На схеме 4, представленной на фиг.6, использовали следующие реагенты и условия: (i) TFA, анизол, CH2Cl2, 20°C, 4 час; (ii) соединение 91 или 5-Br,2-Сlпиримидин или 2,5-диBrпиразин, NaH, ДМФА, 0-20°C, 2-3 час; (iii) ArB(OH)2, 2M Na2CO3, толуол, EtOH, (ДМФА), Pd(dppf)Cl2 в атмосфере N2, 89-90°C, 1,8-3 часов. Осуществляемые при помощи NaH реакции алкилирования спирта 101 [получен из соединения 26 путем катализируемого кислотой отщепления 4-(бензилокси)бензилэфирной боковой цепи] 5-бром-2-фторпиридином (91), 5-бром-2-хлорпиримидином и 2,5-дибромпиразином давали бромиды 102-104, которые подвергали реакции сочетания Сузуки с арилбороновыми кислотами, с получением соединений 13, 14, 17, 18, 21 и 22 таблицы 1.
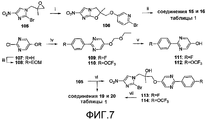
[0053] На схеме 5, представленной на фиг.7, использовали следующие реагенты и условия: (i) 6-Br-3-пиридинол, NaH, ДМФА, 0-20°C, 10 мин, затем 50°C, 4 час; (ii) ArB(OH)2, 2M Na2CO3, толуол, EtOH, ДМФА, Pd(dppf)Cl2 в атмосфере N2, 90°C, 3 час; (iii) EtOCH2 CH2Cl, K2CO3, ДМФА, 20°C, 16 час; (iv) ArB(OH)2, 2M Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 86°C, 2-2,5 час; (v) 1,25M HCl в MeOH, 20°C, 0-12 час, затем 53°C, 2-4 час; (vi) 111 или 112, NaH, ДМФА, 0-20°C, 10-30 мин, затем 50-60°C, 3 час; (vii) NaH, ДМФА, 0°C, 35-80 мин. Катализируемое основанием взаимодействие эпоксида 105 (получен в 2 стадии из соединения 80 через эпоксидирование соответствующего алкена, как описано Ding et al., в WO 2008008480A2) с 6-бром-3-пиридинолом давало бромид 106, который подвергали реакции сочетания Сузуки с арилбороновыми кислотами, с получением соединений 15 и 16 таблицы 1. Подобные реакции соединения 105 с арилпиримидинолами 111 и 112 [получены из 2-хлор-5-пиримидинола (107) через последовательное осуществление этоксиметильной защиты гидроксильной группы, реакции сочетания Сузуки с арилбороновыми кислотами, с последующим катализируемым кислотой удалением защитной группы] давали смеси спиртовых предшественников (113 или 114) и конечных соединений 19 или 20 таблицы 1. Осуществляемая при помощи NaH реакция замыкания кольца этих смесей (или, предпочтительно, очищенных спиртов 113 или 114) затем давала соединения 19 и 20 таблицы 1.
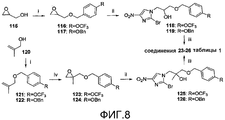
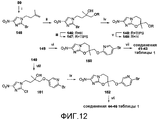
[0054] На схеме 6, представленной на фиг.8, использовали следующие реагенты и условия: (i) 4-OCF3BnBr или 4-BnOBnCl, NaH, ДМФА, 0-20°C, 7-21 час; (ii) соединение 80, DIPEA, 107-108°C, 13-16 час; (iii) NaH, ДМФА, 0°C, 65-80 мин; (iv) m-CPBA, Na2HPO4, CH2Cl2, 0-20°C, 2,5-3,5 часов. Осуществляемые при помощи NaH реакции алкилирования глицидола (115) при помощи замещенных бензилгалогенидов давали эпоксиды 116 и 117, которые последовательно подвергали катализируемому основанием взаимодействию с 2-бром-4(5)-нитроимидазолом (80) с последующим осуществляемым при помощи NaH замыканием кольца промежуточных спиртов (118 и 119), с получением соединений 23 и 24 таблицы 1. Подобные реакции эпоксидов 123 и 124 [получены из 2-метил-2-пропен-1-ола (120) через алкилирование при помощи замещенных бензилгалогенидов, с последующим эпоксидированием] с соединением 80 давали спирты 125 и 126, которые также подвергали реакции замыкания кольца при использовании NaH, с получением соединений 25 и 26 таблицы 1.
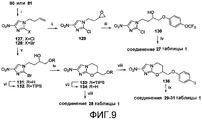
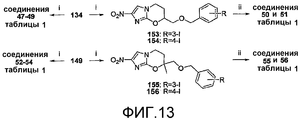
[0055] На схеме 7, представленной на фиг.9, использовали следующие реагенты и условия: (i) Br(CH2)2CH=CH2, K2CO3, ДМФА, 66-73°C, 4,5-12 час; (ii) m-CPBA, Na2HPO4, CH2Cl2, 0-20°C, 50 час; (iii) 4-OCF3PhOH, K2CO3, MEK, 81°C, 12 час; (iv) NaH, ДМФА, 0-20°C, 2-2,5 час; (v) OsO4, NMO, CH2Cl2, 20°C, 4 час; (vi) TIPSCl, имидазол, ДМФА, 20°C, 18 час; (vii) 1% HCl в 95% EtOH, 20°C, 35 час; (viii) 4-BnOPhOH или 4-IPhOH, DEAD, PPh3, ТГФ, 0-20°C, 32-51 час; (ix) ArB(OH)2, 2M Na2CO3, толуол, EtOH, ДМФА, Pd(dppf)Cl2 в атмосфере N2, 90°C, 90 мин. Катализируемое основанием взаимодействие эпоксида 129 [получен из 2-хлор-4(5)-нитроимидазола (81) через алкилирование при помощи 4-бром-1-бутена, с последующим эпоксидированием] с 4-трифторметоксифенолом давало спирт 130, который подвергали реакции замыкания кольца при использовании NaH, с получением соединения 27 таблицы 1. Подобная реакция замыкания кольца монозащищенного диола 132 [получен из 2-бром-4(5)-нитроимидазола (80) через алкилирование при помощи 4-бром-1-бутена, с последующим дигидроксилированием и TIPS защитой первичного спирта] и катализируемое кислотой десилилирование давали спирт 134. Реакции Мицунобу соединения 134 с подходящими фенолами давали соединение 28 таблицы 1 и иодид 135. Реакции сочетания Сузуки соединения 135 с арилбороновыми кислотами затем давали соединения 29-31 таблицы 1.
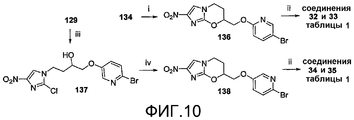
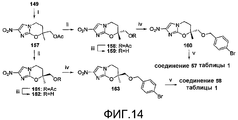
[0056] На схеме 8, представленной на фиг.10, использовали следующие реагенты и условия: (i) соединение 91, NaH, ДМФА, 0-20°C, 2,5 час; (ii) ArB(OH)2, 2M Na2CO3, толуол, EtOH, ДМФА, Pd(dppf)Cl2 в атмосфере N2, 90°C, 2,5 час; (iii) 6-Br-3-пиридинол, K2CO3, MEK, 82-85°C, 28 час; (iv) NaH, ДМФА, 0-20°C, 2,5 часов. Осуществляемое при помощи NaH алкилирование спирта 134 при использовании 5-бром-2-фторпиридина (91) давало бромид 136, который подвергали реакции сочетания Сузуки с арилбороновыми кислотами, с получением соединений 32 и 33 таблицы 1. Альтернативно, катализируемое основанием взаимодействие эпоксида 129 с 6-бром-3-пиридинолом, с последующим осуществляемым при помощи NaH замыканием кольца полученного спирта 137 давали бромид 138, который также подвергали реакции сочетания Сузуки с арилбороновыми кислотами, с получением соединений 34 и 35 таблицы 1.
[0057] На схеме 9, представленной на фиг.11, использовали следующие реагенты и условия: (i) I(CH2)2C(CH3)=CH2, K2CO3, ДМФА, 61°C, 20 час; (ii) m-CPBA, Na2HPO4, CH2Cl2, 0-20°C, 4 час; (iii) RPhOH, K2CO3, MEK, 82-83°C, 8-10 час; (iv) NaH, ДМФА, 0-20°C, 2-2,5 час; (v) ArB(OH)2, 2M Na2CO3, толуол, EtOH, ДМФА, Pd(dppf)Cl2 в атмосфере N2, 90°C, 100-105 мин. Катализируемые основанием взаимодействия эпоксида 140 [получен из 2-хлор-4(5)-нитроимидазола (81) через алкилирование при помощи 4-иод-2-метил-1-бутена (получен путем йодирования 3-метил-3-бутен-1-ола, как описано Helmboldt et al., 2006), с последующим эпоксидированием] с подходящими фенолами давали спирты 141-143, которые подвергали реакциям замыкания кольца при использовании NaH, с получением соединений 36 и 37 таблицы 1 и йодида 144 соответственно. Реакции сочетания Сузуки соединения 144 с арилбороновыми кислотами затем давали соединения 38-40 таблицы 1.
[0058] На схеме 10, представленной на фиг.12, использовали следующие реагенты и условия: (i) I(CH2)2C(CH3)=CH2, K2CO3, ДМФА, 60°C, 11 час; (ii) OsO4, NMO, CH2Cl2, 20°C, 4 час; (iii) TIPSCl, имидазол, ДМФА, 20°C, 6 дней; (iv) NaH, ДМФА, 0-20°C, 2,5 час, затем 46°C, 3,2 час; (v) 1% HCl в 95% EtOH, 44°C, 3 дня; (vi) соединение 91, NaH, ДМФА, 0-20°C, 2,5 час; (vii) ArB(OH)2, 2M Na2CO3, толуол, EtOH, ДМФА, Pd(dppf)Cl2 в атмосфере N2, 90°C, 120-135 мин; (viii) 6-Br-3-пиридинол, K2CO3, MEK, 84°C, 18,5 час; (ix) NaH, ДМФА, 0-20°C, 2,5 часов. Осуществляемое при помощи NaH замыкание кольца монозащищенного диола 147 [получен из 2-бром-4(5)-нитроимидазола (80) через алкилирование при помощи 4-иод-2-метил-1-бутена (получен путем йодирования 3-метил-3-бутен-1-ола, как описано Helmboldt et al., 2006), с последующим дигидроксилированием и TIPS-защитой первичного спирта] и катализируемое кислотой десилилирование давали спирт 149. Осуществляемое при помощи NaH алкилирование соединения 149 при помощи 5-бром-2-фторпиридина (91) давало бромид 150, который подвергали реакции сочетания Сузуки с арилбороновыми кислотами, с получением соединений 41-43 таблицы 1. Альтернативно, катализируемое основанием взаимодействие эпоксида 140 с 6-бром-3-пиридинолом, с последующим осуществляемым при помощи NaH замыканием кольца полученного спирта 151 давали бромид 152, который также подвергали реакции сочетания Сузуки с арилбороновыми кислотами, с получением соединений 44-46 таблицы 1.
[0059] На схеме 11, представленной на фиг.13, использовали следующие реагенты и условия: (i) RBnBr или 4-BnOBnCl, NaH, ДМФА, 0-20°C, 2,5-7 час; (ii) 4-OCF3PhB(OH)2, 2M Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 90°C, 20-25 мин. Осуществляемые при помощи NaH реакции алкилирования спиртов 134 и 149 при помощи замещенных бензилгалогенидов давали соединения 47-49 и 52-54 таблицы 1 и иодиды 153-156. Реакции сочетания Сузуки соединений 153-156 с 4-трифторметоксифенилбороновой кислотой затем давали соединения 50, 51, 55 и 56 таблицы 1.
[0060] На схеме 12, представленной на фиг.14, использовали следующие реагенты и условия: (i) Ac2O, пиридин, 20°C, 38 час; (ii) препаративная хиральная ВЭЖХ (ChiralPak IA, 40% EtOH/гексан); (iii) K2CO3, водный раствор MeOH, 20°C, 4 час; (iv) 4-BrBnBr, NaH, ДМФА, 0-20°C, 3 час; (v) 4-OCF3PhB(OH)2, 2M Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 88°C, 75 мин. Препаративная хиральная ВЭЖХ рацемического ацетата соединения 157 [получен из спирта 149 путем ацетилирования] давала энантиомеры соединений 158 и 161, которые гидролизовали до энантиомерных спиртов 159 и 162. Осуществляемые при помощи NaH реакции алкилирования этих спиртов при помощи 4-бромбензилбромида давали бромиды 160 и 163, которые подвергали реакции Сузуки для сочетания с 4-трифторметоксифенилбороновой кислотой, с получением соединений 57 и 58 таблицы 1.
[0061] На схеме 13, представленной на фиг.15, использовали следующие реагенты и условия: (i) I2, PPh3, имидазол, CH2Cl2, 0-8°C, 5 час; (ii) RPhOH, K2CO3, ацетон, 50°C, 6-11 час; (iii) I25NaBH4, ТГФ, 0°C, 3-4 час, затем 20°C, 13 час, затем 30% H2O2, 3н. NaOH, 0-20°C, 3 час; (iv) I2, PPh3, имидазол, CH2Cl2, 20°C, 12-15 час; (v) 80, K2CO3, ДМФА, 84-88°C, 33-37 час; (vi) 1% HCl в 95% EtOH, 20°C, 7-12 час; (vii) NaH, ДМФА, 0-20°C, 4-5 час; (viii) препаративная хиральная ВЭЖХ (ChiralPak IA, 27% EtOH/гексан); (ix) ArB(OH)2, 2M Na2CO3, толуол, EtOH, ДМФА, Pd(dppf)Cl2 в атмосфере N2, 90°C, 90 мин. Гидроборирование алкенов 166 и 167 [получены путем алкилирования соответствующих фенолов при помощи йодида 165, полученного путем йодирования 2-({[трет-бутил(диметил)силил]окси}метил)-2-пропен-1-ола (164) (Chen et al., US 2007213341 A1, путем моносилилирования 2-метилен-1,3-пропандиола)] давало спирты 168 и 169, которые преобразовывали в иодиды 170 и 171. Алкилирование 2-бром-4(5)-нитроимидазола (80) этими йодидами, катализируемое кислотой десилилирование и осуществляемое при помощи NaH замыкание кольца полученных спиртов 174 и 175 затем давали соединение 59 таблицы 1 и йодид 176 соответственно. Реакции сочетания Сузуки соединения 176 с арилбороновыми кислотами также давали соединения 62-64 таблицы 1.
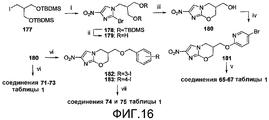
[0062] На схеме 14, представленной на фиг.16, использовали следующие реагенты и условия: (i) соединение 80, K2CO3, ДМФА, 82°C, 24 час; (ii) 1% HCl в 95% EtOH, 20°C, 4 час, затем 4°C, 12 час; (iii) NaH, ДМФА, 0-20°C, 3,5 час; (iv) соединение 91, NaH, ДМФА, 0-20°C, 3 час; (v) ArB(OH)2, 2M Na2CO3, толуол, EtOH, ДМФА, Pd(dppf)Cl2 в атмосфере N2, 90°C, 2-2,5 час; (vi) RBnBr или 4-BnOBnI, NaH, ДМФА, 0-20°C, 0,5-3 час; (vii) 4-OCF3PhB(OH)2, 2M Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 90°C, 20 мин. Алкилирование 2-бром-4(5)-нитроимидазола (80) известным иодидом 177 (см. Curran et al., 1998, на стадии 4, исходя из 2-метилен-1,3-пропандиола) и катализируемое кислотой десилилирование продукта (178) давали диол 179, который подвергали реакции замыкания кольца при использовании NaH, с получением спирта 180. Осуществляемое при помощи NaH алкилирование соединения 180 при помощи 5-бром-2-фторпиридина (91) давало бромид 181, который подвергали реакции сочетания Сузуки с арилбороновыми кислотами, с получением соединений 65-67 таблицы 1. Подобное алкилирование спирта 180 при помощи подходящих, замещенных бензилгалогенидов давало соединения 71-73 таблицы 1, а также иодбензиловые эфиры 182 и 183. Реакции сочетания Сузуки соединений 182 и 183 с 4-(трифторметокси)фенилбороновой кислотой затем давали соединения 74 и 75 таблицы 1.
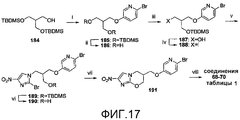
[0063] На схеме 15, представленной на фиг.17, использовали следующие реагенты и условия: (i) 6-Br-3-пиридинол, DEAD, PPh3, ТГФ, 0°C, 1 час, затем 20°C, 41 час; (ii) 1% HCl в 95% EtOH, 20°C, 13 час; (iii) NaH, ТГФ, 20°C, 1 час, затем TBDMSCl, 20°C, 100 мин; (iv) I2, PPh3, имидазол, CH2Cl2, 20°C, 18 час; (v) соединение 80, K2CO3, ДМФА, 87°C, 42 час; (vi) TBAF, ТГФ, 20°C, 4 час; (vii) NaH, ДМФА, 0-20°C, 200 мин; (viii) ArB(OH)2, 2M Na2CO3, толуол, EtOH, ДМФА, Pd(dppf)Cl2 в атмосфере N2, 90°C, 140 мин. Реакция Мицунобу 6-бром-3-пиридинола с известным спиртом 184 (см. Kim et al., 2001, через силилирование и гидроборирование 2-метилен-1,3-пропандиола) и катализируемое кислотой десилилирование продукта (185) давали диол 186. Моносилилирование соединения 186 давало спирт 187, который преобразовывали в иодид 188. Алкилирование 2-бром-4(5)-нитроимидазола (80) при помощи соединения 188, десилилирование и осуществляемое при помощи NaH замыкание кольца полученного спирта 190 давали бромид 191. Реакции сочетания Сузуки соединения 191 с арилбороновыми кислотами затем давали соединения 68-70 таблицы 1.
ПРИМЕР 2. Способы получения
[0064] A. Синтез 6-нитро-2-{[4-(трифторметокси)фенокси]метил}-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 1 таблицы 1)
при использовании способа схемы 1
[0065] Смесь 4-трифторметоксифенола (0,152 мл, 1,17 ммоль), K2CO3 (260 мг, 1,17 ммоль) и 2-(бромметил)оксирана (76) (0,30 мл, 3,51 ммоль) в безводном ацетоне (3 мл) перемешивали в герметично закрытой пробирке при 59°C в течение 36 часов. Полученную смесь фильтровали, промывали CH2Cl2, затем фильтрат упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-15% CH2Cl2/пентан сначала давало головные фракции, и последующее элюирование смесью 20-25% CH2Cl2/пентан давало 2-{[4-(трифторметокси)фенокси]метил}оксиран (77) (аналогично полученному Cao et al., WO 2008112483A2 при использовании эпихлоргидрина) (260 мг, 95%) в виде масла;
1H ЯМР (CDCl3) δ 7,14 (ушир.дд, J=9,0, 0,6 Гц, 2H), 6,91 (дт, J=9,1, 3,0 Гц, 2H), 4,23 (дд, J=11,1, 3,1 Гц, 1H), 3,94 (дд, J=11,1, 5,7 Гц, 1H), 3,34 (м, 1H), 2,91 (дд, J=4,8, 4,2 Гц, 1H), 2,75 (дд, J=4,9, 2,6 Гц, 1H).
[0066] Смесь эпоксида 77 (200 мг, 0,854 ммоль), 2-бром-4(5)-нитроимидазола (80) (180 мг, 0,938 ммоль) и диизопропилэтиламина (0,75 мл, 4,31 ммоль) перемешивали в герметично закрытой пробирке при 105°C в течение 6,5 часов и затем охлаждали. Продукт растворяли в CH2Cl2 (15 мл), промывали водным раствором NaHCO3 (15 мл) и водный слой затем экстрагировали CH2Cl2 (4×15 мл). Объединенные органические экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 0-1% EtOAc/CH2Cl2 давало 1-(2-бром-4-нитро-1Н-имидазол-1-ил)-3-[4-(трифторметокси)фенокси]-2-пропанол (82) (255 мг, 70%) в виде белого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 139-141°C;
1H ЯМР [(CD3)2SO] δ 8,52 (с, 1H), 7,30 (ушир.дд, J=9,1, 0,7 Гц, 2H), 7,05 (дт, J=9,2, 3,1 Гц, 2H), 5,66 (ушир.с, 1H), 4,28 (дд, J=13,3, 3,3 Гц, 1H), 4,21 (м, 1H), 4,13 (дд, J=13,3, 8,0 Гц, 1H), 4,01 (д, J=5,0 Гц, 2H); элементный анализ: (C13H11BrF3N3O5) C, H, N.
[0067] Раствор спирта 82 (242 мг, 0,568 ммоль) в безводном ДМФА (5 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (36 мг, 0,90 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при 0°C в течение 45 минут реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (15 мл), добавляли к насыщенному раствору соли (40 мл) и экстрагировали CH2Cl2 (6×50 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле, элюируя CH2Cl2, с получением соединения 1 (171 мг, 87%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 170-172°C;
1H ЯМР [(CD3)2SO] δ 8,16 (с, 1H), 7,31 (ушир.дд, J=9,1, 0,8 Гц, 2H), 7,05 (дт, J=9,2, 3,1 Гц, 2H), 5,74 (м, 1H), 4,50 (дд, J=10,8, 8,9 Гц, 1H), 4,46 (дд, J=11,5, 2,8 Гц, 1H), 4,39 (дд, J=11,5, 5,2 Гц, 1H), 4,22 (дд, J=10,8, 6,5 Гц, 1H); элементный анализ: (C13H10F3N3O5) C, H, N.
[0068] B. Синтез 2-{[4-(бензилокси)фенокси]метил}-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 2 таблицы 1) при использовании способа схемы 1

[0069] Алкилирование 4-(бензилокси)фенола при помощи 2-(бромметил)оксирана (76), как в примере 2A, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-25% CH2Cl2/петролейный эфир (головные фракции) и затем смесью 25% CH2Cl2/петролейный эфир давали 2-{[4-(бензилокси)фенокси]метил}оксиран (78) (описано Kopka et al., 2003, при использовании эпихлоргидрина) (79%) в виде белого твердого вещества: т.пл. (CH2Cl2/пентан) 61-62°C;
1H ЯМР (CDCl3) δ 7,44-7,28 (м, 5H), 6,90 (дт, J=9,3, 2,8 Гц, 2H), 6,85 (дт, J=9,3, 2,8 Гц, 2H), 4,16 (дд, J=11,1, 3,3 Гц, 1H), 3,92 (дд, J=11,1, 5,6 Гц, 1H), 3,32 (м, 1H), 2,89 (дд, J=4,8, 4,3 Гц, 1H), 2,73 (дд, J=5,0, 2,7 Гц, 1H).
[0070] Взаимодействие эпоксида 78 с 2-бром-4(5)-нитроимидазолом (80), как в примере 2A, в течение 12 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 1-2% EtOAc/CH2Cl2 давали 1-[4-(бензилокси)фенокси]-3-(2-бром-4-нитро-1Н-имидазол-1-ил)-2-пропанол (83) (74%) в виде бледно-желтого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 160-162°C;
1H ЯМР [(CD3)2SO] δ 8,50 (с, 1H), 7,46-7,28 (м, 5H),6,94 (дт, J=9,2, 2,9 Гц, 2H), 6,88 (дт, J=9,2, 2,9 Гц, 2H), 5,60 (ушир.д, J=4,6 Гц, 1H), 5,04 (с, 2H), 4,27 (дд, J=13,0, 2,7 Гц, 1H), 4,16 (м, 1H), 4,11 (дд, J=13,1, 8,2 Гц, 1H), 3,93 (дд, J=10,0, 4,8 Гц, 1H), 3,89 (дд, J=10,1, 5,3 Гц, 1H); элементный анализ: (C19H18BrN3O5) C, H, N.
[0071] Замыкание кольца спирта 83 при помощи NaH, как в примере 2A, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 1-2% MeOH/CH2Cl2 давали соединение 2 (94%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 208-210°C;
1H ЯМР [(CD3)2SO] δ 8,15 (с, 1H), 7,45-7,28 (м, 5H), 6,95 (дт, J=9,2, 3,0 Гц, 2H), 6,88 (дт, J=9,2, 3,0 Гц, 2H), 5,70 (м, 1H), 5,05 (с, 2H), 4,48 (дд, J=10,7, 8,9 Гц, 1H), 4,35 (дд, J=11,6, 2,8 Гц, 1H), 4,28 (дд, J=11,6, 5,1 Гц, 1H), 4,20 (дд, J=10,8, 6,5 Гц, 1H); элементный анализ: (Cl9H17N3O5) C, H, N.
[0072] C. Синтез 2-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 3 таблицы 1) при использовании способа схемы 1
[0073] Алкилирование 4-иодфенола при использовании 2-(бромметил)оксирана (76), как в примере 2A, в течение 52 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-10% CH2Cl2/петролейный эфир (головные фракции) и затем смесью 20-25% CH2Cl2/петролейный эфир давали 2-[(4-иодфенокси)метил]оксиран (79) (описано Apparu et al., 2000 при использовании глицидилтозилата) (89%) в виде белого твердого вещества: т.пл. (CH2Cl2/петролейный эфир) 67-68°C;
1H ЯМР (CDCl3) δ 7,56 (дт, J=9,0, 2,7 Гц, 2H), 6,70 (дт, J=9,0, 2,7 Гц, 2H), 4,20 (дд, J=11,1, 3,1 Гц, 1H), 3,92 (дд, J=11,1, 5,7 Гц, 1H), 3,33 (м, 1H), 2,90 (дд, J=4,8, 4,3 Гц, 1H), 2,74 (дд, J=4,9, 2,6 Гц, 1H).
[0074] Взаимодействие эпоксида 79 с 2-хлор-4(5)-нитроимидазолом (81) и диизопропилэтиламином, как в примере 2A (но с экстракцией водной промывки 6 раз смесью 10% MeOH/CH2Cl2), с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% MeOH/CH2Cl2 (головные фракции) и затем смесью 0,5-1% MeOH/CH2Cl2 давали 1-(2-хлор-4-нитро-1H-имидазол-1-ил)-3-(4-иодфенокси)-2-пропанол (84) (83%) в виде желтого твердого вещества: т.пл. (MeOH/H2O) 174-176°C;
1H ЯМР [(CD3)2SO] δ 8,49 (с, 1H), 7,60 (дт, J=8,9, 2,7 Гц, 2H), 6,81 (дт, J=9,0, 2,7 Гц, 2H), 5,66 (ушир.с, 1H), 4,28 (дд, J=12,8, 2,6 Гц, 1H), 4,19 (м, 1H), 4,14 (дд, J=12,9, 8,0 Гц, 1H), 3,97 (д, J=4,6 Гц, 2H); HRESIMS вычислено для C12H11ClIN3NaO4 m/z [M+Na]+ 447,9346, 445,9375, найдено 447,9322, 445,9366.
[0075] Замыкание кольца спирта 84 при помощи NaH, как в примере 2A, при 0°C в течение 80 минут и затем при 17°C в течение 60 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-40% EtOAc/петролейный эфир (головные фракции) и затем смесью 40% EtOAc/петролейный эфир и 0-0,5% MeOH/CH2Cl2 давали 2-[(4-иодфенокси)метил]-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (85) (77%) в виде бледно-желтого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 198-199°C;
1H ЯМР [(CD3)2SO] δ 8,15 (с, 1H), 7,61 (дт, J=8,9, 2,6 Гц, 2H), 6,80 (дт, J=9,0, 2,6 Гц, 2H), 5,72 (м, 1H), 4,49 (дд, J=10,7, 9,0 Гц, 1H), 4,41 (дд, J=11,6, 2,7 Гц, 1H), 4,35 (дд, J=11,6, 5,2 Гц, 1H), 4,20 (дд, J=10,8, 6,5 Гц, 1H); элементный анализ: (C12H10IN3O4) C, H, N.
[0076] Перемешиваемую смесь йодида 85 (250 мг, 0,646 ммоль), 4-фторфенилбороновой кислоты (163 мг, 1,16 ммоль) и Pd(dppf)Cl2 (95 мг, 0,13 ммоль) в ДМФА (5,6 мл), толуоле (4,4 мл) и EtOH (2,5 мл) дегазировали в течение 11 минут (вакуумный насос) и затем вводили N2. Добавляли 2М водный раствор Na2CO3 (1,3 мл, 2,6 ммоль) через шприц и перемешиваемую смесь снова дегазировали в течение 11 минут, затем вводили N2. Полученную смесь перемешивали при 88°C в течение 70 минут, затем охлаждали, разбавляли водным раствором NaHCO3 (50 мл) и экстрагировали CH2Cl2 (6×50 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле, элюируя смесью 0-0,5% MeOH/CH2Cl2 (головные фракции) и затем смесью 0,5% MeOH/CH2Cl2, с получением соединения 3 (191 мг, 83%) в виде бледно-коричневого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 224-226°C;
1H ЯМР [(CD3)2SO] δ 8,18 (с, 1H), 7,65 (ддт, J=8,9, 5,4, 2,7 Гц, 2H), 7,59 (дт, J=8,8, 2,6 Гц, 2H), 7,25 (тт, J=8,9, 2,7 Гц, 2H), 7,03 (дт, J=8,8, 2,6 Гц, 2H), 5,76 (м, 1H), 4,51 (дд, J=10,8, 9,0 Гц, 1H), 4,47 (дд, J=11,6, 2,8 Гц, 1H), 4,41 (дд, J=11,6, 5,3 Гц, 1H), 4,23 (дд, J=10,8, 6,5 Гц, 1H); APCI МС m/z 356 [M+H].
[0077] D. Синтез 6-нитро-2-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 4 таблицы 1) при использовании способа схемы 1

[0078] Реакция сочетания Сузуки йодида 85 и 4-(трифторметил)фенилбороновой кислоты, как в примере 2C, в течение 90 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% MeOH/CH2Cl2 (головные фракции) и затем смесью 0,5% MeOH/CH2Cl2 давали соединение 4 (77%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 210-211°C;
1H ЯМР [(CD3)2SO] δ 8,19 (с, 1H), 7,86 (ушир.д, J=8,2 Гц, 2H), 7,77 (ушир.д, J=8,4 Гц, 2H), 7,71 (дт, J=8,8, 2,5 Гц, 2H), 7,09 (дт, J=8,9, 2,6 Гц, 2H), 5,77 (м, 1H), 4,52 (дд, J=10,7, 9,0 Гц, 1H), 4,50 (дд, J=11,6, 2,8 Гц, 1H), 4,43 (дд, J=11,6, 5,3 Гц, 1H), 4,24 (дд, J=10,8, 6,5 Гц, 1H); APCI МС m/z 406 [M+H]+.
[0079] E. Синтез 6-нитро-2-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 5 таблицы 1) при использовании способа схемы 1

[0080] Реакция сочетания Сузуки йодида 85 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2C, в течение 50 минут, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 давали соединение 5 (83%) в виде бледно-розового твердого вещества: т.пл. (CH2Cl2/гексан) 200-201°C;
1H ЯМР [(CD3)2SO] δ 8,18 (с, 1H), 7,74 (ушир.д, J=8,8 Гц, 2H), 7,64 (ушир.д, J=8,8 Гц, 2H), 7,41 (ушир.д, J=8,1 Гц, 2H), 7,06 (ушир.д, J=8,8 Гц, 2H), 5,76 (м, 1H), 4,52 (дд, J=10,5, 9,0 Гц, 1H), 4,49 (дд, J=11,6, 2,6 Гц, 1H), 4,42 (дд, J=11,6, 5,2 Гц, 1H), 4,23 (дд, J=10,7, 6,5 Гц, 1H); элементный анализ: (C19H14F3N3O5) C, H, N.
[0081] F. Синтез 6-нитро-2-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 6 таблицы 1) при использовании способа схемы 2

[0082] Смесь 2,4-динитроимидазола (86) (2,02 г, 12,8 ммоль) и трет-бутил(диметил)силил-2-оксиранилметилового эфира (87) (3,84 г, 20,4 ммоль) перемешивали при 70°C в течение 16 часов. Полученную охлажденную смесь разбавляли EtOAc (300 мл), промывали водным раствором NaHCO3 (3×50 мл), водой (2×50 мл) и насыщенным раствором соли (50 мл) и затем растворитель удаляли. Хроматография остатка на силикагеле при элюировании смесью 10-20% EtOAc/петролейный эфир давала 1-{[трет-бутил(диметил)силил]окси}-3-(2,4-динитро-1H-имидазол-1-ил)-2-пропанол (88) (описано Otera et al., US 2006063929A1, исходя из 2,4-динитроимидазола и глицидола) (2,63 г, 60%) в виде желтого масла;
1H ЯМР (CDCl3) δ 8,01 (с, 1H), 4,78 (дд, J=13,9, 2,9 Гц, 1H), 4,46 (дд, J=14,0, 8,3 Гц, 1H), 4,08 (м, 1H), 3,76 (дд, J=10,4, 4,6 Гц, 1H), 3,67 (дд, J=10,5, 5,0 Гц, 1H), 2,60 (ушир.с, 1H), 0,92 (с, 9H), 0,11 (с, 6H); APCI МС m/z 300 [M+H-HNO2]+.
[0083] Раствор спирта 88 (2,04 г, 5,89 ммоль) в безводном ДМФА (20 мл) в атмосфере N2 при -20°C обрабатывали 60% NaH (0,34 г, 8,50 ммоль). После перемешивания при температуре от -20 до -10°C в течение 50 минут реакционную смесь гасили EtOAc и водой (150 мл) и экстрагировали EtOAc (500 мл). Экстракт промывали водой (2×100 мл) и насыщенным раствором соли (100 мл), снова экстрагировали EtOAc (100 мл) и затем растворитель удаляли. Хроматография остатка на силикагеле при элюировании смесью 40-67% EtOAc/петролейный эфир давала 2-({[трет-бутил(диметил)силил]окси}метил)-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (89) (1,13 г, 64%) в виде бледно-желтого твердого вещества: т.пл. (EtOAc/петролейный эфир) 142-144°C;
1H ЯМР (CDCl3) δ 7,52 (с, 1H), 5,33 (м, 1H), 4,29 (д, J=7,2 Гц, 2H), 4,05 (дд, J=11,9, 3,5 Гц, 1H), 3,86 (дд, J=11,9, 2,8 Гц, 1H), 0,81 (с, 9H), 0,08, 0,03 (2с, 2× 3H); элементный анализ: (C12H21N3O4Si) C, H, N.
[0084] Суспензию силилового эфира 89 (503 мг, 1,68 ммоль) в 1% растворе HCl в 95% EtOH (условия десилилирования описаны Cunico et al., 1980) (27 мл) перемешивали при комнатной температуре в течение 6 часов и затем хранили при 4°C в течение 2,5 дней. Полученный раствор нейтрализовали добавлением по каплям 7M NH3 в MeOH (2 мл) при перемешивании, затем концентрировали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-2% MeOH/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 2-5% MeOH/CH2Cl2 давало (6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол-2-ил)метанол (90) (описано Sehgal et al., 1981, через взаимодействие 2,4-динитроимидазола и глицидола) (299 мг, 97%) в виде белого твердого вещества (после растирания с CH2Cl2): т.пл. (CH2Cl2) 166-169°C;
1H ЯМР [(CD3)2SO] δ 8,10 (с, 1H), 5,40 (м, 1H), 5,27 (т, J=5,6 Гц, 1H), 4,36 (дд, J=10,5, 8,8 Гц, 1H), 4,11 (дд, J=10,5, 6,4 Гц, 1H), 3,80 (ддд, J=12,8, 5,4, 3,0 Гц, 1H), 3,65 (дд, J=12,8, 5,8, 3,9 Гц, 1H); элементный анализ: (C6H7N3O4) C, H, N.
[0085] Перемешиваемую смесь 4-(трифторметокси)фенилбороновой кислоты (1,55 г, 7,53 ммоль) и Pd(dppf)Cl2 (367 мг, 0,502 ммоль) в толуоле (50 мл) и EtOH (25 мл) дегазировали в течение 15 минут (вакуумный насос) и затем вводили N2. Добавляли 2М водный раствор Na2CO3 (12,5 мл, 25,0 ммоль) через шприц, перемешиваемую смесь снова дегазировали в течение 15 минут и затем вводили N2, с последующим добавлением 5-бром-2-фторпиридина (91) (0,53 мл, 5,15 ммоль). Полученную смесь перемешивали при 85-88°C в течение 3 часов, затем охлаждали, разбавляли водным раствором NaHCO3 (100 мл) и экстрагировали CH2Cl2 (4×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-10% CH2Cl2/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 10-20% CH2Cl2/петролейный эфир давало 2-фтор-5-[4-(трифторметокси)фенил]пиридин (92) (1,32 г, 100%) в виде белого твердого вещества: т.пл. (CH2Cl2/петролейный эфир) 58-60°C;
1H ЯМР (CDCl3) δ 8,40 (д, J=2,5 Гц, 1H), 7,94 (ддд, J=8,4, 7,6, 2,6 Гц, 1H), 7,55 (дт, J=8,8, 2,5 Гц, 2H), 7,33 (ушир.д, J=8,0 Гц, 2H), 7,02 (дд, J=8,5, 3,0 Гц, 1H); HRESIMS вычислено для C12H8F4NO m/z (MH+) 258,0537, найдено 258,0531.
[0086] Смесь спирта 90 (300 мг, 1,62 ммоль) и фторпиридина 92 (1,255 г, 4,88 ммоль) в безводном ДМФА (6 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (96 мг, 2,40 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 2,5 часов реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (10 мл), добавляли к насыщенному раствору соли (40 мл) и экстрагировали CH2Cl2 (6×50 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-20% CH2Cl2/петролейный эфир сначала давало головные фракции (включая восстановленное соединение 92) и последующее элюирование смесью CH2Cl2 давало соединение 6 (5,5 мг, 0,8%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 127-130°C;
1H ЯМР (CDCl3) δ 8,32 (дд, J=2,5, 0,7 Гц, 1H), 7,80 (дд, J=8,5, 2,5 Гц, 1H), 7,58 (с, 1H), 7,53 (дт, J=8,8, 2,5 Гц, 2H), 7,31 (ушир.дд, J=8,7, 0,8 Гц, 2H), 6,83 (дд, J=8,6, 0,7 Гц, 1H), 5,69 (м, 1H), 4,80 (дд, J=12,4, 4,0 Гц, 1H), 4,75 (дд, J=12,4, 4,1 Гц, 1H), 4,45 (дд, J=10,2, 8,7 Гц, 1H), 4,35 (дд, J=10,2, 6,5 Гц, 1H); APCI МС m/z 423 [M+H]+.
[0087] G. Синтез 2-{[4-(бензилокси)фенокси]метил}-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 7 таблицы 1) при использовании способа схемы 3

[0088] Смесь 4-(бензилокси)фенола (2,01 г, 10,1 ммоль), K2CO3 (1,60 г, 11,6 ммоль) и 3-хлор-2-метилпропена (93) (2,00 мл, 20,4 ммоль) в безводном ацетоне (2,5 мл) перемешивали в герметично закрытой пробирке при 58°C в течение 24 часов. Полученную смесь фильтровали, промывали CH2Cl2, затем фильтрат упаривали досуха и остаток хроматографировали на силикагеле. Элюирование петролейным эфиром сначала давало головные фракции, и последующее элюирование смесью 25% CH2Cl2/петролейный эфир давало 1-(бензилокси)-4-[(2-метил-2-пропенил)окси]бензол (94) (Karrer, F. DE 2312518) (1,74 г, 68%) в виде белого твердого вещества: т.пл. (CH2Cl2/гексан) 62-64°C;
1H ЯМР (CDCl3) δ 7,45-7,29 (м, 5H), 6,90 (дт, J=9,3, 2,8 Гц, 2H), 6,85 (дт, J=9,3, 2,8 Гц, 2H), 5,08 (м, 1H), 5,01 (с, 2H), 4,79 (м, 1H), 4,38 (с, 2H), 1,82 (с, 3H).
[0089] 3-Хлорпербензойную кислоту (1,43 г 50% раствора, 4,14 ммоль) добавляли к охлажденной льдом смеси соединения 94 (500 мг, 1,97 ммоль) и порошкообразного Na2HPO4 (974 мг, 6,86 ммоль) в CH2Cl2 (20 мл), и полученную смесь перемешивали при комнатной температуре в течение 3,5 часов. Добавляли охлажденный водный раствор Na2SO3 (50 мл 10% раствора) и смесь экстрагировали CH2Cl2 (3×50 мл). Экстракты последовательно промывали охлажденным водным раствором Na2SO3 (50 мл 10% раствора), водным раствором NaHCO3 (50 мл) и насыщенным раствором соли (50 мл). Объединенные экстракты затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 25% CH2Cl2/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 25-33% CH2Cl2/петролейный эфир давало 2-{[4-(бензилокси)фенокси]метил}-2-метилоксиран (95) (460 мг, 87%) в виде белого твердого вещества: т.пл. (CH2Cl2/пентан) 105-107°C;
1H ЯМР (CDCl3) δ 7,44-7,28 (м, 5H), 6,90 (дт, J=9,3, 2,9 Гц, 2H), 6,85 (дт, J=9,3, 2,9 Гц, 2H), 5,01 (с, 2H), 3,97 (д, J=10,5 Гц, 1H), 3,90 (д, J=10,5 Гц, 1H), 2,85 (д, J=4,8 Гц, 1H), 2,71 (д, J=4,8 Гц, 1H), 1,47 (с, 3H); элементный анализ: (C17H18O3) C, H, N.
[0090] Взаимодействие эпоксида 95 с 2-бром-4(5)-нитроимидазолом (80), как в примере 2A, при 107°C в течение 14 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0-1% EtOAc/CH2Cl2 давали 1-[4-(бензилокси)фенокси]-3-(2-бром-4-нитро-1Н-имидазол-1-ил)-2-метил-2-пропанол (98) (86%) в виде бледно-желтого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 148-150°C;
1H ЯМР [(CD3)2SO] δ 8,31 (с, 1H), 7,45-7,28 (м, 5H), 6,94 (дт, J=9,2, 3,0 Гц, 2H), 6,87 (дт, J=9,2, 3,0 Гц, 2H), 5,41 (с, 1H), 5,04 (с, 2H), 4,22 (д, J=14,3 Гц, 1H), 4,15 (д, J=14,3 Гц, 1H), 3,76 (д, J=9,5 Гц, 1H), 3,72 (д, J=9,4 Гц, 1H), 1,19 (с, 3H); элементный анализ: (C20H20BrN3O5) C, H, N.
[0091] Замыкание кольца спирта 98 при помощи NaH, как в примере 2A, в течение 50 минут, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0-1% EtOAc/CH2Cl2 давали соединение 7 (92%) в виде бледно-желтого твердого вещества: т.пл. (CH2Cl2/гексан) 162-165°C;
1H ЯМР (CDCl3) δ 7,54 (с, 1H), 7,43-7,28 (м, 5H), 6,89 (дт, J=9,1, 3,0 Гц, 2H), 6,78 (дт, J=9,1, 3,1 Гц, 2H), 5,01 (с, 2H), 4,48 (д, J=10,2 Гц, 1H), 4,17 (д, J=10,1 Гц, 1H), 4,03 (д, J=10,2 Гц, 1H), 4,00 (д, J=10,2 Гц, 1H), 1,76 (с, 3H); элементный анализ: (C20H19O5) C, H, N.
[0092] Н. Синтез 2-{[4-(6-метокси-3-пиридинил)фенокси]метил}-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 8 таблицы 1) при использовании способа схемы 3

[0093] Смесь 4-иодфенола (2,00 г, 9,09 ммоль), порошкообразного K2CO3 (2,54 г, 18,4 ммоль), NaI (364 мг, 2,43 ммоль) и 2-(хлорметил)-2-метилоксирана (97) (0,90 мл, 9,31 ммоль) в безводном ДМФА (5 мл) перемешивали в герметично закрытой пробирке при 70°C в течение 15 часов. Снова добавляли 2-(хлорметил)-2-метилоксиран (97) (0,18 мл, 1,86 ммоль) и смесь затем перемешивали при 73°C в течение 17 часов. Охлажденную смесь добавляли к смеси лед/водный раствор NaHCO3 (100 мл) и экстрагировали Et2O (5×100 мл). Экстракты промывали водой (100 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-15% CH2Cl2/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 15-20% CH2Cl2/петролейный эфир давало 2-[(4-иодфенокси)метил]-2-метилоксиран (96) (1,81 г, 69%) в виде белого твердого вещества: т.пл. (CH2Cl2/пентан) 40-41°C;
1H ЯМР (CDCl3) δ 7,55 (дт, J=9,0, 2,7 Гц, 2H), 6,70 (дт, J=9,0, 2,7 Гц, 2H), 4,01 (д, J=10,5 Гц, 1H), 3,90 (д, J=10,5 Гц, 1H), 2,85 (д, J=4,7 Гц, 1H), 2,72 (д, J=4,7 Гц, 1H), 1,47 (с, 3H); элементный анализ: (C10H11IO2) C, H, N.
[0094] Взаимодействие эпоксида 96 с 2-бром-4(5)-нитроимидазолом (80), как в примере 2A, при 107°C в течение 15 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0-1% EtOAc/CH2Cl2 давали 1-(2-бром-4-нитро-1H-имидазол-1-ил)-3-(4-иодфенокси)-2-метил-2-пропанол (99) (85%) в виде пены (после растирания в смеси Et2O/пентан);
1H ЯМР (CDCl3) δ 8,04 (с, 1H), 7,59 (дт, J=9,0, 2,7 Гц, 2H), 6,66 (дт, J=9,0, 2,7 Гц, 2H), 4,27 (д, J=14,5 Гц, 1H), 4,16 (д, J=14,5 Гц, 1H), 3,86 (д, J=9,2 Гц, 1H), 3,82 (д, J=9,2 Гц, 1H), 2,44 (с, 1H), 1,35 (с, 3H); элементный анализ: (C13H13BrIN3O4.0,1Et2O) C, H, N.
[0095] Замыкание кольца спирта 99 при помощи NaH, как в примере 2A, в течение 75 минут, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 давали 2-[(4-иодфенокси)метил]-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (100) (92%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 181-183°C;
1H ЯМР (CDCl3) δ 7,57 (дт, J=9,0, 2,7 Гц, 2H), 7,54 (с, 1H), 6,63 (дт, J=9,0, 2,7 Гц, 2H), 4,46 (д, J=10,2 Гц, 1H), 4,20 (д, J=10,1 Гц, 1H), 4,05 (д, J=9,9 Гц, 1H), 4,03 (д, J=10,1 Гц, 1H), 1,78 (с, 3H); элементный анализ: (C13H12IN3O4) C, H, N.
[0096] Перемешиваемую смесь йодида 100 (40,1 мг, 0,100 ммоль), 6-метокси-3-пиридинилбороновой кислоты (23,8 мг, 0,156 ммоль) и Pd(dppf)Cl2 (7,3 мг, 9,98 мкмоль) в толуоле (1,7 мл) и EtOH (0,6 мл) дегазировали в течение 4 минут (вакуумный насос) и затем вводили N2. Добавляли 2М водный раствор Na2CO3 (0,30 мл, 0,60 ммоль) через шприц, перемешиваемую смесь снова дегазировали в течение 4 минут и затем вводили N2. Полученную смесь перемешивали при 90°C в течение 45 минут, затем охлаждали, разбавляли водным раствором NaHCO3 (50 мл) и экстрагировали CH2Cl2 (4×50 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-3% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 4% EtOAc/CH2Cl2 давало соединение 8 (32 мг, 84%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/пентан) 217-219°C;
1H ЯМР (CDCl3) δ 8,32 (ушир.д, J=2,2Гц, 1H), 7,72 (дд, J=8,6, 2,6 Гц, 1H), 7,56 (с, 1H), 7,44 (дт, J=8,8, 2,5 Гц, 2H), 6,92 (дт, J=8,8, 2,5 Гц, 2H), 6,79 (д, J=8,5 Гц, 1H), 4,51 (д, J=10,2 Гц, 1H), 4,27 (д, J=10,1 Гц, 1H), 4,13 (д, J=10,1 Гц, 1H), 4,05 (д, J=10,2 Гц, 1H), 3,97 (с, 3H), 1,80 (с, 3H); элементный анализ: (C19H18N4O5) C, H, N.
[0097] I. Синтез 4'-[(2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол-2-ил)метокси][1,1'-бифенил]-4-карбонитрила (соединение 9 таблицы 1) при использовании способа схемы 3

[0098] Реакция сочетания Сузуки йодида 100 и 4-цианофенилбороновой кислоты, как в примере 2H, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% EtOAc/CH2Cl2 (головные фракции) и затем смесью 0,5-1% EtOAc/CH2Cl2 давали соединение 9 (45%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 180-181°C;
1H ЯМР (CDCl3) δ 7,70 (дт, J=8,6, 1,8 Гц, 2H), 7,62 (дт, J=8,6, 1,8 Гц, 2H), 7,56 (с, 1H), 7,53 (дт, J=8,9, 2,6 Гц, 2H), 6,95 (дт, J=8,9, 2,6 Гц, 2H), 4,51 (д, J=10,2 Гц, 1H), 4,30 (д, J=10,2 Гц, 1H), 4,15 (д, J=10,2 Гц, 1H), 4,06 (д, J=10,2 Гц, 1H), 1,81 (с, 3H); элементный анализ: (C20H16N4O4) C, H, N.
[0099] J. Синтез 2-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 10 таблицы 1) при использовании способа схемы 3

[00100] Реакция сочетания Сузуки йодида 100 и 4-фторфенилбороновой кислоты, как в примере 2H, с последующей хроматографией продукта на силикагеле при элюировании смесью CH2Cl2 (головные фракции) и затем смесью 0-1% EtOAc/CH2Cl2 давали соединение 10 (84%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/пентан) 180-181°C;
1H ЯМР (CDCl3) δ 7,56 (с, 1H), 7,50-7,43 (м, 4 H), 7,10 (тт, J=8,7, 2,6 Гц, 2H), 6,91 (дт, J=8,8, 2,6 Гц, 2H), 4,51 (д, J=10,2 Гц, 1H), 4,27 (д, J=10,1 Гц, 1H), 4,13 (д, J=10,1 Гц, 1H), 4,05 (д, J=10,2 Гц, 1H), 1,80 (с, 3 H); элементный анализ: (C19Hl6FN3O4) C, H, N.
[0100] K. Синтез 2-метил-6-нитро-2-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 11 таблицы 1) при использовании способа схемы 3

[0101] Реакция сочетания Сузуки йодида 100 и 4-(трифторметил)фенилбороновой кислоты, как в примере 2H, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% EtOAc/CH2Cl2 (головные фракции) и затем смесью 0,5% EtOAc/CH2Cl2 давали соединение 11 (88%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 219-220°C;
1H ЯМР (CDCl3) δ 7,67 (d, J=8,5 Гц, 2H), 7,63 (д, J=8,5 Гц, 2H), 7,56 (с, 1 H), 7,53 (дт, J=8,8, 2,5 Гц, 2H), 6,95 (дт, J=8,8, 2,5 Гц, 2H), 4,51 (д, J=10,2 Гц, 1H), 4,29 (д, J=10,1 Гц, 1H), 4,14 (д, J=10,1 Гц, 1H), 4,06 (д, J=10,2 Гц, 1 H), 1,81 (с, 3H); элементный анализ: (C20H16F3N3O4) C, H, N.
[0102] L. Синтез 2-метил-6-нитро-2-({[4'-трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 12 таблицы 1) при использовании способа схемы 3

[0103] Реакция сочетания Сузуки йодида 100 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2H, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 давали соединение 12 (90%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/пентан) 209-211°C;
1H ЯМР (CDCl3) δ 7,56 (с, 1H), 7,53 (дт, J=8,8, 2,5 Гц, 2H), 7,48 (дт, J=8,8, 2,5 Гц, 2H), 7,26 (м, 2H), 6,92 (дт, J=8,8, 2,5 Гц, 2H), 4,51 (д, J=10,2 Гц, 1H), 4,28 (д, J=10,1 Гц, 1H), 4,13 (д, J=10,1 Гц, 1H), 4,05 (д, J=10,2 Гц, 1H), 1,81 (с, 3H); элементный анализ: (C20H16F3N3O5) C, H, N.
[0104] M. Синтез 2-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 13 таблицы 1) при использовании способа схемы 4

[0105] Трифторуксусную кислоту (25,4 мл, 0,342 моль) добавляли по каплям к перемешиваемой смеси 2-({[4-(бензилокси)бензил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (26) (см. пример 2Z) (2,53 г, 6,40 ммоль) и анизола (7,0 мл, 64 ммоль) в CH2Cl2 (100 мл) (охлаждение на водяной бане). После перемешивания при комнатной температуре в течение 4 часов растворители удаляли продувкой в токе N2. Маслянистый остаток обрабатывали избыточным количеством твердого NaHCO3, затем разбавляли 15% MeOH/CH2Cl2 (100 мл) и смесь перемешивали при комнатной температуре в течение 30 минут, затем фильтровали. Фильтрат упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% MeOH/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1-2% MeOH/CH2Cl2 давало (2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол-2-ил)метанол (101) (описано Tsubouchi et al., WO 2004033463A1, через 3 стадии, исходя из 2-хлор-4(5)-нитроимидазола (81) и 2-[(метоксиметокси)метил]-2-метилоксирана) (1,215 г, 95%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 174-176°C;
1H ЯМР [(CD3)2SO] δ 8,09 (с, 1H), 5,41 (т, J=5,7 Гц, 1H), 4,24 (д, J=10,6 Гц, 1H), 4,03 (д, J=10,7 Гц, 1H), 3,64 (дд, J=12,2, 5,6 Гц, 1H), 3,54 (дд, J=12,2, 5,9 Гц, 1H), 1,51 (с, 3H); элементный анализ: (C7H9N3O4) C, H, N.
[0106] 5-Бром-2-фторпиридин (91) (0,25 мл, 2,43 ммоль) добавляли к раствору спирта 101 (200 мг, 1,01 ммоль) в безводном ДМФА (4,5 мл) в атмосфере N2 при 0°C. Полученную смесь обрабатывали 60% NaH (64 мг, 1,60 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. Снова добавляли 5-бром-2-фторпиридин (91) (0,25 мл, 2,43 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов, затем охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (15 мл), добавляли к насыщенному раствору соли (40 мл) и экстрагировали CH2Cl2 (8×40 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 0-1,5% EtOAc/CH2Cl2 давало 2-{[(5-бром-2-пиридинил)окси]метил}-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (102) (130 мг, 36%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 151-153°C;
1H ЯМР (CDCl3) δ 8,17 (дд, J=2,5, 0,5 Гц, 1H), 7,68 (дд, J=8,8, 2,5 Гц, 1H), 7,52 (с, 1H), 6,60 (дд, J=8,7, 0,6 Гц, 1H), 4,58 (д, J=12,0 Гц, 1H), 4,50 (д, J=12,0 Гц, 1H), 4,41 (д, J=10,2 Гц, 1H), 4,01 (д, J=10,2 Гц, 1H), 1,76 (с, 3H); элементный анализ: (C12H11BrN4O4) C, H, N.
[0107] Перемешиваемую смесь бромида 102 (77,2 мг, 0,217 ммоль), 4-фторфенилбороновой кислоты (58 мг, 0,415 ммоль) и Pd(dppf)Cl2 (43,5 мг, 59,4 мкмоль) в ДМФА (2,3 мл), толуоле (1,6 мл) и EtOH (1,1 мл) дегазировали в течение 9 минут (вакуумный насос) и затем вводили N2. Добавляли 2М водный раствор Na2CO3 (0,55 мл, 1,1 ммоль) через шприц, перемешиваемую смесь снова дегазировали в течение 9 минут и затем вводили N2. Полученную смесь перемешивали при 90°C в течение 3 часов, затем охлаждали, разбавляли водным раствором NaHCO3 (50 мл) и экстрагировали CH2Cl2 (6×50 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1-2% EtOAc/CH2Cl2 давало соединение 13 (60 мг, 74%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 162-164°C;
1H ЯМР (CDCl3) δ 8,28 (дд, J=2,5, 0,6 Гц, 1H), 7,76 (дд, J=8,5, 2,5 Гц, 1H), 7,55 (с, 1H), 7,46 (ддт, J=8,9, 5,2, 2,6 Гц, 2H), 7,14 (тт, J=8,7, 2,6 Гц, 2H), 6,75 (дд, J=8,5, 0,7 Гц, 1H), 4,67 (д, J=11,9 Гц, 1H), 4,58 (д, J=11,9 Гц, 1H), 4,47 (д, J=10,2 Гц, 1H), 4,04 (д, J=10,2 Гц, 1H), 1,79 (с, 3H); APCI МС m/z 371 [M+H]+.
[0108] N. Синтез 2-метил-6-нитро-2-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 14 таблицы 1) при использовании способа схемы 4

[0109] Реакция сочетания Сузуки бромида 102 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2M, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2, давали соединение 14 (80%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 172-174°C;
1H ЯМР (CDCl3) δ 8,31 (д, J=2,1 Гц, 1H), 7,78 (дд, J=8,6, 2,5Гц, 1H), 7,55 (с, 1H), 7,52 (ушир.д, J=8,8 Гц, 2H), 7,30 (ушир.д, J=8,2 Гц, 2H), 6,76 (д, J=8,7 Гц, 1H), 4,68 (д, J=11,9 Гц, 1H), 4,58 (д, J=11,9 Гц, 1H), 4,47 (д, J=10,2 Гц, 1H), 4,04 (д, J=10,2 Гц, 1H), 1,80 (с, 3H); элементный анализ: (C19H15N4O5) C, H, N.
[0110] O. Синтез 2-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 15 таблицы 1) при использовании способа схемы 5

[0111] Смесь 2-бром-1-[(2-метил-2-оксиранил)метил]-4-нитро-1Н-имидазола (105) (получен на стадии 2 из соединения 80 через эпоксидирование соответствующего алкена, как описано у Ding et al., WO 2008008480A2) (1,011 г, 3,86 ммоль) и 6-бром-3-пиридинола (615 мг, 3,53 ммоль) в безводном ДМФА (12 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (180 мг, 4,50 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 10 минут и затем при 50°C в течение 4 часов реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (20 мл), добавляли к насыщенному раствору соли (100 мл) и экстрагировали CH2Cl2 (6×100 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-50% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 50-75% EtOAc/петролейный эфир и EtOAc давало неочищенное твердое вещество, которое затем хроматографировали на силикагеле. Элюирование CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 0,3-0,5% MeOH/CH2Cl2 давало 2-{[(6-бром-3-пиридинил)окси]метил}-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (106) (описано Ding et al., WO 2009120789A1, исходя из соединения 105 через аналогичную процедуру) (564 мг, 45%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 148-150°C;
1H ЯМР [(CD3)2SO] δ 8,14 (с, 1H), 8,10 (дд, J=3,2, 0,3 Гц, 1H), 7,56 (дд, J=8,7, 0,4 Гц, 1H), 7,39 (дд, J=8,8, 3,2 Гц, 1H), 4,42 (д, J=11,1 Гц, 1H), 4,39 (д, J=11,1 Гц, 1H), 4,38 (д, J=11,0 Гц, 1H), 4,19 (д, J=11,0 Гц, 1H), 1,68 (с, 3H); элементный анализ: (C12H11BrNO4) C, H, N.
[0112] Реакция сочетания Сузуки бромида 106 и 4-фторфенилбороновой кислоты, как в примере 2M, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% MeOH/CH2Cl2 (головные фракции) и затем смесью 0,5% MeOH/CH2Cl2 давали соединение 15 (74%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 180-181°C;
1H ЯМР [(CD3)2SO] δ 8,32 (д, J=2,8 Гц, 1H), 8,18 (с, 1H), 8,05 (ддт, J=8,9, 5,6, 2,6 Гц, 2H), 7,91 (д, J=8,8 Гц, 1H), 7,48 (дд, J=8,8, 3,0 Гц, 1H), 7,27 (тт, J=8,9, 2,6 Гц, 2H), 4,47 (д, J=11,0 Гц, 1H), 4,43 (д, J=11,1 Гц, 1H), 4,41 (д, J=11,0 Гц, 1H), 4,22 (д, J=11,0 Гц, 1H), 1,71 (с, 3H); APCI МС m/z 371 [M+H]+.
[0113] P. Синтез 2-метил-6-нитро-2-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 16 таблицы 1) при использовании способа схемы 5

[0114] Реакция сочетания Сузуки бромида 106 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2M, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,33% MeOH/CH2Cl2 (головные фракции) и затем смесью 0,33% MeOH/CH2Cl2 давали соединение 16 (67%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 209-211°C;
1H ЯМР [(CD3)2SO] δ 8,35 (д, J=2,9 Гц, 1H), 8,18 (с, 1H), 8,13 (ушир.д, J=8,9 Гц, 2H), 7,97 (д, J=8,8 Гц, 1H), 7,51 (дд, J=8,8, 3,0 Гц, 1H), 7,44 (ушир.д, J=8,2 Гц, 2H), 4,48 (д, J=11,1 Гц, 1H), 4,44 (д, J=11,2 Гц, 1H), 4,42 (д, J=11,1 Гц, 1H), 4,22 (д, J=11,0 Гц, 1H), 1,71 (с, 3H); элементный анализ: (C19H15F3N4O5) C, H, N.
[0115] Q. Синтез 7-({[5-(4-фторфенил)-2-пиримидинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 17 таблицы 1) при использовании способа схемы 4

[0116] Смесь спирта 101 (см. пример 2M) (100 мг, 0,502 ммоль) и 5-бром-2-хлорпиримидина (156 мг, 0,806 ммоль) в безводном ДМФА (2,5 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (32 мг, 0,80 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 140 минут реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (10 мл), добавляли к насыщенному раствору соли (40 мл) и экстрагировали CH2Cl2 (6×50 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-0,25% MeOH/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 0,25-0,5% MeOH/CH2Cl2 давало 2-{[(5-бром-2-пиримидинил)окси]метил}-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (103) (описано Ding et al., WO 2009120789 A1, исходя из соединения 101, через аналогичную процедуру) (163 мг, 91%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 224-226°C;
1H ЯМР [(CD3)2SO] δ 8,77 (с, 2H), 8,14 (с, 1H), 4,61 (с, 2H), 4,41 (д, J=11,0 Гц, 1H), 4,20 (д, J=11,1 Гц, 1H), 1,70 (с, 3H); элементный анализ: (C11H10BrN5O4) C, H, N.
[0117] Реакция сочетания Сузуки бромида 103 и 4-фторфенилбороновой кислоты, как в примере 2M, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0,5% MeOH/CH2Cl2 давали соединение 17 (22%) в виде бледно-желтого твердого вещества: т.пл. (CH2Cl2/пентан) 196°С разл.;
1H ЯМР [(CD3)2SO] δ 8,92 (с, 2H), 8,18 (с, 1H), 7,79 (ушир.дд, J=8,8, 5,4 Гц, 2H), 7,34 (ушир.т, J=8,9 Гц, 2H), 4,69 (д, J=12,0 Гц, 1H), 4,65 (д, J=12,0 Гц, 1H), 4,44 (д, J=11,0 Гц, 1H), 4,22 (д, J=11,0 Гц, 1H), 1,72 (с, 3H); элементный анализ: (C17H14FN5O4) C, H, N.
[0118] R. Синтез 2-метил-6-нитро-2-[({5-[4-(трифторметокси)фенил]-2-пиримидинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 18 таблицы 1) при использовании способа схемы 4

[0119] Реакция сочетания Сузуки бромида 103 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2M, в течение 2 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0,25% MeOH/CH2Cl2 давали соединение 18 (80%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 227°C разл.;
1H ЯМР [(CD3)2SO] δ 8,96 (с, 2H), 8,18 (с, 1H), 7,87 (ушир.д, J=8,7 Гц, 2H), 7,50 (ушир.д, J=8,2 Гц, 2H), 4,70 (д, J=12,0 Гц, 1H), 4,67 (д, J=12,0 Гц, 1H), 4,44 (д, J=11,0 Гц, 1H), 4,22 (д, J=11,0 Гц, 1H), 1,72 (с, 3H); элементный анализ: (C18H14F3N5O5) C, H, N.
[0120] S. Синтез 2-({[2-(4-фторфенил)-5-пиримидинил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 19 таблицы 1) при использовании способа схемы 5

[0121] Перемешиваемую смесь 2-хлор-5-пиримидинола (107) (1,00 г, 7,66 ммоль) и хлорметилэтилового эфира (1,75 мл, 18,9 ммоль) в безводном ДМФА (2,5 мл) обрабатывали K2CO3 (2,15 г, 15,6 ммоль). После перемешивания при комнатной температуре в течение 16 часов смесь добавляли к смеси лед/водный раствор NaHCO3 (100 мл) и экстрагировали смесью 50% Et2O/петролейный эфир (5×100 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% Et2O/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 1-10% Et2O/петролейный эфир давало 2-хлор-5-(этоксиметокси)пиримидин (108) (1,27 г, 88%) в виде масла;
1H ЯМР (CDCl3) δ 8,43 (с, 2H), 5,27 (с, 2H), 3,74 (кв, J=7,1 Гц, 2H), 1,23 (т, J=7,1 Гц, 3H); HRESIMS вычислено для C7Hl0ClN2O2 m/z [M+H]+ 191,0396, 189,0425, найдено 191,0397, 189,0426.
[0122] Перемешиваемую смесь 4-фторфенилбороновой кислоты (282 мг, 2,02 ммоль) и Pd(dppf)Cl2 (199 мг, 0,272 ммоль) в толуоле (14 мл) и EtOH (7 мл) дегазировали в течение 10 минут (вакуумный насос) и затем вводили N2. Добавляли 2М водный раствор Na2CO3 (3,3 мл, 6,6 ммоль) через шприц, перемешиваемую смесь снова дегазировали в течение 10 минут и затем вводили N2, с последующим добавлением хлорпиримидина 108 (260 мг, 1,38 ммоль). Полученную смесь перемешивали при 86°C в течение 2,5 часов, затем охлаждали, разбавляли водным раствором NaHCO3 (50 мл) и экстрагировали CH2Cl2 (5×50 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-2% Et2O/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 2% Et2O/петролейный эфир давало 5-(этоксиметокси)-2-(4-фторфенил)пиримидин (109) (312 мг, 91%) в виде белого твердого вещества: т.пл. (петролейный эфир) 42-44°C;
1H ЯМР (CDCl3) δ 8,58 (с, 2H), 8,36 (ддт, J=9,0, 5,6, 2,5 Гц, 2H), 7,14 (тт, J=8,8, 2,5 Гц, 2H), 5,30 (с, 2H), 3,77 (кв, J=7,1 Гц, 2H), 1,25 (т, J=7,1 Гц, 3H); HRESIMS вычислено для C13H13FN2O2 m/z [M+H]+ 249,1034, найдено 249,1039.
[0123] Простой эфир 109 (301 мг, 1,21 ммоль) обрабатывали 1,25M раствором HCl в MeOH (10 мл) и смесь перемешивали при 53°C в течение 4 часов. Полученный охлажденный раствор разбавляли ледяной водой (100 мл) и экстрагировали CH2Cl2 (5×80 мл). Объединенные экстракты упаривали досуха, и остаток растирали в пентане, с получением 2-(4-фторфенил)-5-пиримидинола (111) (225 мг, 98%) в виде белого твердого вещества: т.пл. (пентан) 200-202°C;
1H ЯМР [(CD3)2SO] δ 10,55 (очень ушир.с, 1H), 8,42 (с, 2H), 8,29 (ддт, J=9,1, 5,7, 2,6 Гц, 2H), 7,28 (тт, J=9,0, 2,6 Гц, 2H); HRESIMS вычислено для C10H8FN2O m/z [M+H]+ 191,0615, найдено 191,0616.
[0124] Смесь 2-бром-1-[(2-метил-2-оксиранил)метил]-4-нитро-1Н-имидазола (105) (получен в 2 стадии из соединения 80, через эпоксидирование соответствующего алкена, как описано Ding et al., WO 2008008480A2) (279,5 мг, 1,07 ммоль) и пиримидинола 111 (201 мг, 1,06 ммоль) в безводном ДМФА (3 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (54 мг, 1,35 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 30 минут и затем при 60°C в течение 3 часов реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (5 мл), добавляли к насыщенному раствору соли (50 мл) и экстрагировали CH2Cl2 (8×50 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-33% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 50% EtOAc/петролейный эфир давало 1-(2-бром-4-нитро-1H-имидазол-1-ил)-3-{[2-(4-фторфенил)-5-пиримидинил]окси}-2-метил-2-пропанол (113) (76 мг, 16%) в виде масла;
1H ЯМР (CDCl3) δ 8,46 (с, 2H), 8,36 (ддт, J=9,0, 5,5, 2,5 Гц, 2H), 8,09 (с, 1H), 7,15 (тт, J=8,7, 2,5 Гц, 2H), 4,32 (д, J=14,6 Гц, 1H), 4,21 (д, J=14,5 Гц, 1H), 4,03 (д, J=9,2 Гц, 1H), 3,99 (д, J=9,1 Гц, 1H), 2,59 (с, 1H), 1,43 (с, 3H); HRESIMS вычислено для C17H16BrFN5O4 m/z [M+H]+ 454,0345, 452,0364, найдено 454,0342, 452,0358.
[0125] Дальнейшее элюирование описанной выше колонки EtOAc давало неочищенное твердое вещество, которое затем очищали хроматографией на силикагеле. Элюирование смесью 0-2% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 2-5% EtOAc/CH2Cl2 давало соединение 19 (135 мг, 34%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 201-203°C;
1H ЯМР 8,63 (с, 2H), 8,33 (ддт, J=9,0, 5,7, 2,6 Гц, 2H), 8,18 (с, 1H), 7,32 (тт, J=8,9, 2,6 Гц, 2H), 4,57 (д, J=11,1 Гц, 1H), 4,53 (д, J=11,1 Гц, 1H), 4,42 (д, J=11,0 Гц, 1H), 4,22 (д, J=11,0 Гц, 1H), 1,71 (с, 3H); APCI МС m/z 372 [M+H]+.
[0126] Замыкание кольца спирта 113 при помощи NaH (1,8 экв.), как в примере 2A, в течение 35 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-2% EtOAc/CH2Cl2 (головные фракции) и затем смесью 2-5% EtOAc/CH2Cl2 давали дополнительное количество соединения 19 (67%).
[0127] T. Синтез 2-метил-6-нитро-2-[({2-[4-(трифторметокси)фенил]-5-пиримидинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 20 таблицы 1) при использовании способа схемы 5

[0128] Реакция сочетания Сузуки хлорпиримидина 108 (см. пример 2S) и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2S выше, в течение 2 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-2% Et2O/петролейный эфир (головные фракции) и затем смесью 2% Et2O/петролейный эфир давали 5-(этоксиметокси)-2-[4-(трифторметокси)фенил]пиримидин (110) (91%) в виде белого твердого вещества: т.пл. (петролейный эфир) 41-43°C;
1H ЯМР (CDCl3) δ 8,60 (с, 2H), 8,41 (дт, J=9,0, 2,4 Гц, 2H), 7,30 (ушир.дд, J=9,0,0,9 Гц, 2H), 5,31 (с, 2H), 3,77 (кв, J=7,1 Гц, 2H), 1,25 (т, J=7,1 Гц, 3H); HRESIMS вычислено для C14H14F3N2O3 m/z [M+H]+ 315,0951, найдено 315,0944.
[0129] Простой эфир 110 (379 мг, 1,21 ммоль) обрабатывали 1,25M раствором HCl в MeOH (11 мл) и смесь перемешивали при комнатной температуре в течение 12 часов и затем при 53°C в течение 2 часов. Полученный охлажденный раствор разбавляли водой (50 мл) и экстрагировали CH2Cl2 (5×50 мл). Объединенные экстракты упаривали досуха, и остаток растирали в пентане, с получением 2-[4-(трифторметокси)фенил]-5-пиримидинола (112) (305 мг, 99%) в виде белого твердого вещества: т.пл. (пентан) 156-157°C;
1H ЯМР (CDCl3) δ 8,45 (с, 2H), 8,38 (дт, J=8,9, 2,4 Гц, 2H), 7,29 (ушир.дд, J=8,9, 0,7 Гц, 2H), 5,60 (ушир.с, 1H); HRESIMS вычислено для C11H8N2O2 m/z [M+H]+ 257,0532, найдено 257,0526.
[0130] Смесь 2-бром-1-[(2-метил-2-оксиранил)метил]-4-нитро-1Н-имидазола (105) (получен в 2 стадии из соединения 80, через эпоксидирование соответствующего алкена, как описано Ding et al., WO 2008008480A2) (165 мг, 0,630 ммоль) и пиримидинола 112 (160 мг, 0,625 ммоль) в безводном ДМФА (2 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (33,5 мг, 0,838 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 10 минут и затем при 50°C в течение 3 часов реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (5 мл), добавляли к насыщенному раствору соли (50 мл) и экстрагировали CH2Cl2 (6×50 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-33% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование EtOAc давало неочищенную смесь соединения 20 и спирта 114 с незамкнутым кольцом (95 мг). Раствор полученной смеси в безводном ДМФА (2 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (6,3 мг, 0,158 ммоль), затем дегазировали, снова герметично закрывали в атмосфере N2 и перемешивали при 0°C в течение 80 минут. Реакционную смесь гасили и подвергали обработке, как описано выше, затем продукт очищали хроматографией на силикагеле, элюируя смесью 0-2% EtOAc/CH2Cl2 (головные фракции) и затем смесью 3-5% EtOAc/CH2Cl2, с получением соединения 20 (62 мг, 23%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 223-225°C;
1H ЯМР [(CD3)2SO] δ 8,66 (с, 2H), 8,40 (дт, J=8,9, 2,4 Гц, 2H), 8,18 (с, 1H), 7,49 (ушир.д, J=8,2 Гц, 2H), 4,59 (д, J=11,1 Гц, 1H), 4,55 (д, J=11,1 Гц, 1H), 4,43 (д, J=11,1 Гц, 1H), 4,23 (д, J=11,0 Гц, 1H), 1,72 (с, 3H); элементный анализ: (C18H14F3N5O5) C, H, N.
[0131] U. Синтез 2-({[5-(4-фторфенил)-2-пиразинил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 21 таблицы 1) при использовании способа схемы 4

[0132] Раствор спирта 101 (см. пример 2M) (350 мг, 1,76 ммоль) в безводном ДМФА (7 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (104 мг, 2,60 ммоль) и 2,5-дибромпиразина (837 мг, 3,52 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 3 часов реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (20 мл), добавляли к насыщенному раствору соли (80 мл) и экстрагировали CH2Cl2 (6×100 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 0-2% EtOAc/CH2Cl2 давало 2-{[(5-бром-2-пиразинил)окси]метил}-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазол (104) (428 мг, 68%) в виде белого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 198-200°C;
1H ЯМР [(CD3)2SO] δ 8,44 (д, J=1,3 Гц, 1H), 8,16 (д, J=1,3 Гц, 1H), 8,14 (с, 1H), 4,62 (с, 2H), 4,40 (д, J=11,0 Гц, 1H), 4,19 (д, J=11,0 Гц, 1H), 1,70 (с,3H); элементный анализ: (C11H10BrN5O4) C, H, N.
[0133] Перемешиваемую смесь бромида 104 (140,2 мг, 0,394 ммоль), 4-фторфенилбороновой кислоты (104 мг, 0,743 ммоль) и Pd(dppf)Cl2 (29,8 мг, 40,7 мкмоль) в толуоле (6 мл) и EtOH (2,4 мл) дегазировали в течение 8 минут (вакуумный насос) и затем вводили N2. Добавляли 2М водный раствор Na2CO3 (1,0 мл, 2,0 ммоль) через шприц и перемешиваемую смесь снова дегазировали в течение 8 минут и затем вводили N2. Полученную смесь перемешивали при 89°C в течение 110 минут и затем охлаждали, разбавляли водным раствором NaHCO3 (50 мл) и экстрагировали CH2Cl2 (5×50 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 2-3% EtOAc/CH2Cl2 давало соединение 21 (112 мг, 77%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 200-201°C;
1H ЯМР [(CD3)2SO] δ 8,80 (д, J=1,2 Гц, 1H), 8,33 (д, J=1,3 Гц, 1H), 8,17 (с, 1H), 8,08 (ушир.дд, J=8,8, 5,5 Гц, 2H), 7,33 (ушир.т, J=8,9 Гц, 2H), 4,70 (д, J=12,5 Гц, 1H), 4,66 (д, J=12,5 Гц, 1H), 4,43 (д, J=11,1 Гц, 1H), 4,22 (д, J=11,0 Гц, 1H), 1,72 (с, 3H); элементный анализ: (C17H14FN5O4) C, H, N.
[0134] V. Синтез 2-метил-6-нитро-2-[({5-[4-(трифторметокси)фенил]-2-пиразинил}окси)метил]-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 22 таблицы 1) при использовании способа схемы 4

[0135] Реакция сочетания Сузуки бромида 104 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2U, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 1-2,5% EtOAc/CH2Cl2 давали соединение 22 (81%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 222-224°C;
1H ЯМР [(CD3)2SO] δ 8,85 (д, J=1,3 Гц, 1H), 8,36 (д, J=1,4 Гц, 1H), 8,17 (с, 1H), 8,16 (ушир.д, J=9,1 Гц, 2H), 7,49 (ушир.д, J=8,2 Гц, 2H), 4,69 (с, 2H), 4,44 (д, J=11,0 Гц, 1H), 4,22 (д, J=11,0 Гц, 1H), 1,73 (с,3H); элементный анализ: (C18H14F3N5O5) C, H, N.
[0136] W. Синтез 6-нитро-2-({[4-(трифторметокси)бензил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 23 таблицы 1) при использовании способа схемы 6

[0137] Смесь глицидола (115) (303 мг, 4,09 ммоль) и 4-(трифторметокси)бензилбромида (0,810 мл, 5,06 ммоль) в безводном ДМФА (6 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (246 мг, 6,15 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 7 часов смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (20 мл), добавляли к воде (100 мл) и экстрагировали EtOAc (4×100 мл). Экстракты промывали насыщенным раствором соли (100 мл), упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-5% Et2O/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 5-10% Et2O/петролейный эфир давало 2-({[4-(трифторметокси)бензил]окси}метил)оксиран (116) (625 мг, 62%) в виде масла;
1H ЯМР (CDCl3) δ 7,38 (дт, J=8,7, 2,3 Гц, 2H), 7,20 (ушир.дд, J=8,7, 0,7 Гц, 2H), 4,62 (д, J=12,0 Гц, 1H), 4,56 (д, J=12,0 Гц, 1H), 3,82 (дд, J=11,5, 2,8 Гц, 1H), 3,43 (дд, J=11,5, 6,0 Гц, 1H), 3,21 (м, 1H), 2,82 (дд, J=4,9, 4,2 Гц, 1H), 2,63 (дд, J=5,0, 2,7 Гц, 1H); HRESIMS вычислено для C11H11F3NaO3 m/z [M+Na]+ 271,0552, найдено 271,0557.
[0138] Взаимодействие эпоксида 116 с 2-бром-4(5)-нитроимидазолом (80), как в примере 2A, при 107°C в течение 13 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1-2% EtOAc/CH2Cl2 давали 1-(2-бром-4-нитро-1H-имидазол-1-ил)-3-{[4-(трифторметокси)бензил]окси}-2-пропанол (118) (61%) в виде белого твердого вещества: т.пл. (CΗ2Cl2/пентан) 80-81°C;
1H ЯМР (CDCl3) δ 7,95 (с, 1H), 7,35 (дт, J=8,7, 2,3 Гц, 2H), 7,23 (ушир.д, J=8,7 Гц, 2H), 4,57 (с, 2H), 4,20 (дд, J=13,6, 2,9 Гц, 1H), 4,14 (м, 1H), 4,07 (дд, J=13,4, 7,1 Гц, 1H), 3,59 (дд, J=9,6, 4,2 Гц, 1H), 3,46 (дд, J=9,6, 5,3 Гц, 1H), 2,61 (д, J=5,0 Гц, 1H); HRESIMS вычислено для C14H14BrF3N3O5 m/z [M+H]+ 442,0044, 440,0063, найдено 442,0044, 440,0061; элементный анализ: (C14H13BrF3N3O5) H, N. C: вычислено, 38,20; найдено, 38,61.
[0139] Замыкание кольца спирта 118 при помощи NaH, как в примере 2A, в течение 65 минут, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 давали соединение 23 (90%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 134-135°C;
1H ЯМР (CDCl3) δ 7,53 (с, 1H), 7,29 (дт, J=8,7, 2,1 Гц, 2H), 7,20 (ушир.д, J=8,0 Гц, 2H), 5,42 (м, 1H), 4,32 (дд, J=10,0, 8,6 Гц, 1H), 4,26 (дд, J=10,0, 6,5 Гц, 1H), 3,89 (дд, J=11,3, 3,9 Гц, 1H), 3,78 (дд, J=11,3, 3,5 Гц, 1H); элементный анализ: (C14H12F3N3O5) C, H, N.
[0140] X. Синтез 2-({[4-(бензилокси)бензил]окси}метил)-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 24 таблицы 1) при использовании способа схемы 6

[0141] Алкилирование глицидола (115) при помощи 4-(бензилокси)бензилхлорида, как в примере 2W, в течение 10 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-7,5% Et2O/петролейный эфир (головные фракции) и затем смесью 7,5-10% Et2O/петролейный эфир давали неочищенное масло, которое затем хроматографировали на силикагеле. Элюирование смесью 0-50% CH2Cl2/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 50-66% CH2Cl2/петролейный эфир давало 2-({[4-(бензилокси)бензил]окси}метил)оксиран (117) (32%) (описано Cousse et al., EP 187096A1, исходя из эпихлоргидрина и 4-(бензилокси)бензилового спирта) в виде масла;
1H ЯМР (CDCl3) δ 7,45-7,29 (м, 5H), 7,27 (дт, J=8,8, 2,3 Гц, 2H), 6,95 (дт, J=8,7, 2,5 Гц, 2H), 4,54 (д, J=11,6 Гц, 1H), 4,48 (д, J=11,6 Гц, 1H), 3,72 (дд, J=11,5, 3,2 Гц, 1H), 3,42 (дд, J=11,4, 5,8 Гц, 1H), 3,17 (м, 1H), 2,79 (дд, J=5,0, 4,2 Гц, 1H), 2,60 (дд, J=5,1, 2,7 Гц, 1H); HRESIMS вычислено для C17H18NaO3 m/z [M+Na]+ 293,1148, найдено 293,1143.
[0142] Взаимодействие эпоксида 117 с 2-бром-4(5)-нитроимидазолом (80), как в примере 2A, при 108°C в течение 14 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 2-4% EtOAc/CH2Cl2 давали 1-{[4-(бензилокси)бензил]окси}-3-(2-бром-4-нитро-1Н-имидазол-1-ил)-2-пропанол (119) (73%) в виде белого твердого вещества: т.пл. (CH2Cl2/гексан) 122-123°C;
1H ЯМР (CDCl3) δ 7,88 (с, 1H), 7,46-7,30 (м, 5H), 7,24 (дт, J=8,6, 2,4 Гц, 2H), 6,98 (дт, J=8,7, 2,4 Гц, 2H), 5,08 (с, 2H), 4,52 (д, J=11,5 Гц, 1H), 4,48 (д, J=11,5 Гц, 1H), 4,18-4,01 (м, 3H), 3,55 (дд, J=9,7, 4,0 Гц, 1H), 3,39 (дд, J=9,6, 5,1 Гц, 1H), 2,48 (д, J=5,3 Гц, 1H); элементный анализ: (C20H20BrN3O5) C, H, N.
[0143] Замыкание кольца спирта 119 при помощи NaH, как в примере 2A, в течение 80 минут, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 1-2% EtOAc/CH2Cl2 давали соединение 24 (88%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 123-124°C;
1H ЯМР (CDCl3) δ 7,50 (с, 1H), 7,45-7,29 (м, 5H), 7,19 (дт, J=8,7, 2,4 Гц, 2H), 6,95 (дт, J=8,6, 2,4 Гц, 2H), 5,37 (м, 1H), 4,54 (д, J=11,7 Гц, 1H), 4,50 (д, J=11,7 Гц, 1H), 4,27 (дд, J=10,0, 8,5 Гц, 1H), 4,22 (дд, J=10,0, 6,5 Гц, 1H), 3,82 (дд, J=11,2, 4,2 Гц, 1H), 3,73 (дд, J=11,2, 3,6 Гц, 1H); элементный анализ: (C20H19N3O5) C, H, N.
[0144] Y. Синтез 2-метил-6-нитро-2-({[4-(трифторметокси)бензил]окси}метил)-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 25 таблицы 1) при использовании способа схемы 6

[0145] Раствор 2-метил-2-пропен-1-ола (120) (2,34 мл, 27,8 ммоль) в безводном ДМФА (10 мл, затем 2×2 мл для промывки) добавляли к суспензии 60% NaH (1,32 г, 33,1 ммоль) в безводном ДМФА (10 мл) в атмосфере N2 при 0°C, и полученную смесь перемешивали при 0°C в течение 30 минут. Добавляли 4-(трифторметокси)бензилбромид (5,1 мл, 31,9 ммоль) и смесь перемешивали при комнатной температуре в течение 21 часа. Полученную смесь добавляли к смеси лед/водный раствор NaHCO3 (200 мл) и экстрагировали 25% EtOAc/петролейный эфир (2×200 мл) и 50% EtOAc/петролейный эфир (3×200 мл). Экстракты промывали водой (200 мл), летучие растворители удаляли и остаточное масло хроматографировали на силикагеле. Элюирование петролейным эфиром сначала давало головные фракции, затем последующее элюирование смесью 0-15% CH2Cl2/петролейный эфир давало 1-{[(2-метил-2-пропенил)окси]метил}-4-(трифторметокси)бензол (121) (6,57 г, 96%) в виде масла, которое использовали непосредственно на следующей стадии;
1H ЯМР (CDCl3) δ 7,37 (дт, J=8,7, 2,3 Гц, 2H), 7,19 (ушир.д, J=8,0 Гц, 2H), 5,00 (м, 1H), 4,94 (м, 1H), 4,48 (с, 2H), 3,94 (с, 2H), 1,77 (с, 3H).
[0146] Эпоксидирование алкена 121 при использовании 3-хлорпербензойной кислоты, как в примере 2G, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-15% CH2Cl2/петролейный эфир (головные фракции) и затем смесью 15-75% CH2Cl2/петролейный эфир и CH2Cl2 давали 2-метил-2-({[4-(трифторметокси)бензил]окси}метил)оксиран (123) (93%) в виде масла;
1H ЯМР (CDCl3) δ 7,37 (дт, J=8,7, 2,4 Гц, 2H), 7,19 (ушир.д, J=7,9 Гц, 2H), 4,59 (д, J=12,1 Гц, 1H), 4,54 (д, J=12,1 Гц, 1H), 3,61 (д, J=11,1 Гц, 1H), 3,44 (д, J=11,1 Гц, 1H), 2,75 (д, J=4,9 Гц, 1H), 2,64 (д, J=4,9 Гц, 1H), 1,40 (с, 3H); HRESIMS (NH3) вычислено для C12H17F3O3N m/z [M+H+NH3]+ 280,1161, найдено 280,1144.
[0147] Взаимодействие эпоксида 123 с 2-бром-4(5)-нитроимидазолом (80), как в примере 2A, при 108°C в течение 15 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 давали 1-(2-бром-4-нитро-1H-имидазол-1-ил)-2-метил-3-{[4-(трифторметокси)бензил]окси}-2-пропанол (125) (94%) в виде бледно-желтого масло;
1H ЯМР (CDCl3) δ 8,00 (с, 1H), 7,33 (дт, J=8,6, 2,3 Гц, 2H), 7,22 (ушир.д, J=8,0 Гц, 2H), 4,56 (с, 2H), 4,15 (д, J=14,8 Гц, 1H), 4,04 (д, J=14,5 Гц, 1H), 3,39 (с, 2H), 2,51 (с, 1H), 1,22 (с, 3H); HRESIMS вычислено для C15H16BrF3N3O5 m/z [M+H]+ 456,0200, 454,0220, найдено 456,0197, 454,0221.
[0148] Замыкание кольца спирта 125 при помощи NaH, как в примере 2A, в течение 80 минут, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0-1% EtOAc/CH2Cl2 давали соединение 25 (87%) в виде бледно-желтого твердого вещества: т.пл. (CH2Cl2/гексан) 110-111°C;
1H ЯМР (CDCl3) δ 7,50 (с, 1H), 7,26 (ушир.д, J=8,4 Гц, 2H), 7,19 (ушир.д, J=8,3 Гц, 2H), 4,59 (д, J=12,3 Гц, 1H), 4,56 (д, J=12,3 Гц, 1H), 4,36 (д, J=10,0 Гц, 1H), 3,91 (д, J=10,0 Гц, 1H), 3,72 (д, J=10,7 Гц, 1H), 3,59 (д, J=10,6 Гц, 1H), 1,65 (с, 3H); элементный анализ: (C15H14F3N3O5) C, H, N.
[0149] Z. Синтез 2-({[4-(бензилокси)бензил]окси}метил)-2-метил-6-нитро-2,3-дигидроимидазо[2,1-b][1,3]оксазола (соединение 26 таблицы 1) при использовании способа схемы 6

[0150] Раствор 2-метил-2-пропен-1-ола (120) (1,17 мл, 13,9 ммоль) в безводном ДМФА (5 мл, затем 2×1 мл для промывки) добавляли к суспензии 60% NaH (674 мг, 16,9 ммоль) в безводном ДМФА (5 мл) в атмосфере N2 при 0°C и смесь перемешивали при 0°C в течение 30 минут. Добавляли раствор 4-(бензилокси)бензилхлорида (3,87 г, 16,6 ммоль) в безводном ДМФА (6 мл, затем 2×2 мл для промывки) и смесь перемешивали при комнатной температуре в течение 16 часов. Полученную смесь добавляли к смеси лед/водный раствор NaHCO3 (100 мл) и экстрагировали EtOAc (4×100 мл). Экстракты промывали водой (100 мл), EtOAc удаляли и остаточное масло хроматографировали на силикагеле. Элюирование петролейным эфиром сначала давало головные фракции, затем последующее элюирование смесью 0-25% CH2Cl2/петролейный эфир давало 1-(бензилокси)-4-{[(2-метил-2-пропенил)окси]метил}бензол (122) (описано Wennerberg et al., 1999, через алкилирование 4-(бензилокси)бензилового спирта) (3,48 г, 93%) в виде масла, которое использовали непосредственно на следующей стадии;
1H ЯМР (CDCl3) δ 7,45-7,28 (м, 5H), 7,27 (дт, J=8,5, 2,4 Гц, 2H), 6,95 (дт, J=8,7, 2,4 Гц, 2H), 5,07 (с, 2H), 4,99 (м, 1H), 4,91 (м, 1H), 4,42 (с, 2H), 3,91 (с, 2H), 1,76 (с, 3H).
[0151] Эпоксидирование алкена 122 при использовании 3-хлорпербензойной кислоты, как в примере 2G, в течение 2,5 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 50% CH2Cl2/петролейный эфир (головные фракции) и затем смесью 50-80% CH2Cl2/петролейный эфир и CH2Cl2 давали 2-({[4-(бензилокси)бензил]окси}метил)-2-метилоксиран (124) (95%) в виде масла;
1H ЯМР (CDCl3) δ 7,45-7,29 (м, 5H), 7,26 (дт, J=8,7, 2,4 Гц, 2H), 6,95 (дт, J=8,7, 2,5 Гц, 2H), 5,07 (с, 2H), 4,52 (д, J=11,6 Гц, 1H), 4,47 (д, J=11,6 Гц, 1H), 3,54 (д, J=11,0 Гц, 1H), 3,42 (д, J=11,0 Гц, 1H), 2,73 (д, J=4,9 Гц, 1H), 2,62 (д, J=4,9 Гц, 1H), 1,39 (с, 3H); HRESIMS вычислено для C18H20O3 m/z (М+) 284,1412, найдено 284,1416.
[0152] Взаимодействие эпоксида 124 с 2-бром-4(5)-нитроимидазолом (80), как в примере 2A, при 108°C в течение 16 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0-2% EtOAc/CH2Cl2 давали 1-{[4-(бензилокси)бензил]окси}-3-(2-бром-4-нитро-1H-имидазол-1-ил)-2-метил-2-пропанол (126) (100%) в виде светло-желтого масла;
1H ЯМР (CDCl3) δ 7,94 (с, 1H), 7,46-7,30 (м, 5H), 7,22 (дт, J=8,6, 2,4 Гц, 2H), 6,98 (дт, J=8,7, 2,5 Гц, 2H), 5,08 (с, 2H), 4,50 (д, J=11,5 Гц, 1H), 4,47 (д, J=11,5 Гц, 1H), 4,11 (д, J=14,4 Гц, 1H), 4,00 (д, J=14,4 Гц, 1H), 3,34 (с, 2H), 2,55 (с, 1H), 1,17 (с, 3H); HRESIMS вычислено для C21H23BrN3O5 m/z [M+H]+ 478,0796, 476,0816, найдено 478,0792, 476,0809.
[0153] Замыкание кольца спирта 126 при помощи NaH (1,5 экв.), как в примере 2A, в течение 80 минут, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 давали соединение 26 (97%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 130-131°C;
1H ЯМР (CDCl3) δ 7,48 (с, 1H), 7,45-7,29 (м, 5H), 7,16 (дт, J=8,7, 2,4 Гц, 2H), 6,94 (дт, J=8,7, 2,4 Гц, 2H), 5,06 (с, 2H), 4,52 (д, J=11,7 Гц, 1H), 4,47 (д, J=11,7 Гц, 1H), 4,32 (д, J=10,0 Гц, 1H), 3,86 (д, J=10,0 Гц, 1H), 3,67 (д, J=10,6 Гц, 1H), 3,53 (д, J=10,6 Гц, 1H), 1,62 (с, 3H); элементный анализ: (C21H21N3O5) C, H, N.
[0154] AA. Синтез 2-нитро-7-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 27 таблицы 1) при использовании способа схемы 7

[0155] 4-Бром-1-бутен (2,65 мл, 26,1 ммоль) добавляли к смеси 2-хлор-4(5)-нитроимидазола (81) (2,50 г, 17,0 ммоль) и K2CO3 (7,88 г, 57,0 ммоль) в безводном ДМФА (12 мл) в атмосфере N2 и смесь перемешивали при 66°C в течение 12 часов. Полученную охлажденную смесь добавляли к смеси лед/водный раствор NaHCO3 (140 мл) и экстрагировали смесью 50% EtOAc/петролейный эфир (5×100 мл). Экстракты промывали водой (100 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-10% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 10-20% EtOAc/петролейный эфир давало 1-(3-бутенил)-2-хлор-4-нитро-1Н-имидазол (127) (2,82 г, 82%) в виде кремового твердого вещества: т.пл. (Et2O/пентан) 56-58°C;
1H ЯМР (CDCl3) δ 7,72 (с, 1H), 5,74 (ддт, J=17,1, 10,2, 6,9 Гц, 1H), 5,18 (дкв, J=10,3, 1,0 Гц, 1H), 5,12 (дкв, J=17,1, 1,3 Гц, 1H), 4,09 (т, J=6,9 Гц, 2H), 2,58 (квт, J=6,9, 1,1 Гц, 2H); HRESIMS вычислено для C7H9ClN3O2 m/z [M+H]+ 204,0349, 202,0378, найдено 204,0350, 202,0377.
[0156] Эпоксидирование алкена 127 при использовании 3-хлорпербензойной кислоты, как в примере 2G, в течение 50 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-10% EtOAc/петролейный эфир (головные фракции) и затем смесью 20-30% EtOAc/петролейный эфир сначала давали восстановленный алкен 127 (0,49 г, 17%). Элюирование смесью 0-5% Et2O/CH2Cl2 давало неочищенный продукт, который затем хроматографировали на силикагеле, элюируя CH2Cl2 (головные фракции) и затем смесью 0-5% Et2O/CH2Cl2, с получением 2-хлор-4-нитро-1-[2-(2-оксиранил)этил]-1H-имидазола (129) (73%) в виде бледно-желтого твердого вещества: т.пл. (CΗ2Cl2/гексан) 51-52°C;
1H ЯМР (CDCl3) δ 7,81 (с, 1H), 4,28-4,16 (м, 2H), 2,98-2,92 (м, 1H), 2,85 (дд, J=4,7, 4,0 Гц, 1H), 2,53 (дд, J=4,8, 2,6 Гц, 1H), 2,35-2,25 (м, 1H), 1,87-1,77 (м, 1H); HRESIMS вычислено для C7H9ClN3O3 m/z [M+H]+ 220,0298, 218,0327, найдено 220,0297, 218,0322.
[0157] 4-Трифторметоксифенол (0,375 мл, 2,89 ммоль) добавляли к смеси эпоксида 129 (250 мг, 1,15 ммоль) и порошкообразного K2CO3 (558 мг, 4,04 ммоль) в безводном 2-бутаноне (3 мл) в атмосфере N2 и смесь перемешивали при 81°C в течение 12 часов. Полученную охлажденную смесь разбавляли водой (50 мл) и экстрагировали CH2Cl2 (4×50 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-25% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 25-33% EtOAc/петролейный эфир давало 4-(2-хлор-4-нитро-1Н-имидазол-1-ил)-1-[4-(трифторметокси)фенокси]-2-бутанол (130) (306 мг, 67%) в виде бледно-желтого масла;
1H ЯМР (CDCl3) δ 7,85 (с, 1H), 7,16 (ушир.дд, J=9,1, 0,8 Гц, 2H), 6,88 (дт, J=9,2, 3,0 Гц, 2H), 4,37-4,24 (м, 2H), 4,02-3,93 (м, 2H), 3,86 (дд, J=10,0, 7,8 Гц, 1H), 2,47 (дд, J=4,2, 1,1 Гц, 1H), 2,13-1,98 (м, 2H); HRESIMS вычислено для C14H14ClF3N3O3 m/z [M+H]+ 398,0540, 396,0569, найдено 398,0538, 396,0567.
[0158] Дальнейшее элюирование указанной выше колонки смесью 66% EtOAc/петролейный эфир давало неочищенное твердое вещество (72 мг), которое затем хроматографировали на силикагеле. Элюирование CH2Cl2 давало головные фракции, и последующее элюирование смесью 0-3% EtOAc/CH2Cl2 давало соединение 27 (61 мг, 15%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 138-140°C;
1H ЯМР (CDCl3) δ 7,45 (с, 1H), 7,17 (ушир.дд, J=9,1, 0,7 Гц, 2H), 6,91 (дт, J=9,2, 3,0 Гц, 2H), 4,75 (м, 1H), 4,31 (дд, J=10,2, 4,3 Гц, 1H), 4,26-4,09 (м, 3H), 2,52-2,32 (м, 2H); элементный анализ: (C14H12F3N3O5) C, H, N.
[0159] Перемешиваемый раствор спирта 130 (305 мг, 0,771 ммоль) в безводном ДМФА (5 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (49 мг, 1,23 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 2,5 часов реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (10 мл), добавляли к насыщенному раствору соли (40 мл) и экстрагировали CH2Cl2 (6×50 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 25-40% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 50-66% EtOAc/петролейный эфир давало дополнительное количество соединения 27 (217 мг, 78%) в виде бледно-желтого твердого вещества (см. данные выше).
[0160] BB. Синтез 7-{[4-(бензилокси)фенокси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 28 таблицы 1) при использовании способа схемы 7

[0161] Смесь 2-бром-4(5)-нитроимидазола (80) (2,50 г, 13,0 ммоль), 4-бром-1-бутена (2,00 мл, 19,7 ммоль) и K2CO3 (5,39 г, 39,0 ммоль) в безводном ДМФА (25 мл) в атмосфере N2 перемешивали при 73°C в течение 4,5 часов. Полученную охлажденную смесь добавляли к смеси лед/водный раствор NaHCO3 (200 мл) и экстрагировали EtOAc (4×200 мл). Экстракты промывали водой (200 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-10% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 20% EtOAc/петролейный эфир давало 2-бром-1-(3-бутенил)-4-нитро-1H-имидазол (128) (2,96 г, 92%) в виде бледно-желтого воскообразного твердого вещества: т.пл. 28-30°C;
1H ЯМР (CDCl3) δ 7,77 (с, 1H), 5,75 (ддт, J=17,1, 10,2, 6,9 Гц, 1H), 5,18 (дкв, J=10,2, 1,1 Гц, 1H), 5,12 (дкв, J=17,1, 1,4 Гц, 1H), 4,09 (т, J=7,0 Гц, 2H), 2,59 (квт, J=6,9, 1,2 Гц, 2H); HRFABMS вычислено для C7H9BrN3O2 m/z [M+H]+ 247,9858, 245,9878, найдено 247,9860, 245,9882.
[0162] Тетраоксид осмия (3,75 мл 4% водного раствора, 0,614 ммоль) добавляли к раствору алкена 128 (3,00 г, 12,2 ммоль) и 4-метилморфолин N-оксида (2,16 г, 18,4 ммоль) в CH2Cl2 (75 мл) и затем смесь перемешивали при комнатной температуре в течение 4 часов. Полученный осадок выделяли фильтрованием, промывали CH2Cl2 и воды и затем сушили, с получением 4-(2-бром-4-нитро-1Н-имидазол-1-ил)-1,2-бутандиола (131) (2,39 г, 70%) в виде кремового твердого вещества: т.пл. (ТГФ/Et2O/пентан) 99-101°C;
1H ЯМР [(CD3)2SO] δ 8,55 (с, 1H), 4,77 (д, J=5,0 Гц, 1H), 4,58 (т, J=5,6 Гц, 1H), 4,14 (м, 2H), 3,42 (м, 1H), 3,34 (дт, J=10,7, 5,4 Гц, 1H), 3,24 (дт, J=10,7, 5,9 Гц, 1H), 1,98 (дтд, J=13,7, 7,9, 3,2 Гц, 1H), 1,69 (дддд, J=13,6, 9,1, 7,4, 6,0 Гц, 1H); элементный анализ: (C7H10BrN3O4) C, H, N.
[0163] Полученный выше фильтрат добавляли к смеси лед/водный раствор Na2SO3 (100 мл) и экстрагировали EtOAc (3×100 мл). Водный слой насыщали солью и затем экстрагировали EtOAc (7×100 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 50-67% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 67% EtOAc/петролейный эфир и EtOAc давало дополнительное количество диола 131 (728 мг, 21%).
[0164] Триизопропилсилилхлорид (2,50 мл, 11,7 ммоль) добавляли к раствору диола 131 (3,11 г, 11,1 ммоль) и имидазола (1,66 г, 24,4 ммоль) в безводном ДМФА (30 мл) в атмосфере N2 и затем смесь перемешивали при комнатной температуре в течение 18 часов. Полученную смесь добавляли к смеси лед-вода (200 мл) и экстрагировали EtOAc (4×200 мл). Экстракты промывали водой (200 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-20% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 20-33% EtOAc/петролейный эфир давало 4-(2-бром-4-нитро-1H-имидазол-1-ил)-1-[(триизопропилсилил)окси]-2-бутанол (132) (4,60 г, 95%) в виде белого твердого вещества: т.пл. (CΗ2Cl2/пентан) 90-91°C;
1H ЯМР (CDCl3) δ 7,89 (с, 1H), 4,24 (дд, J=7,7, 6,2 Гц, 2H), 3,74 (дд, J=9,6, 3,5 Гц, 1H), 3,62 (м, 1H), 3,53 (дд, J=9,6, 6,8 Гц, 1H), 2,59 (д, J=3,8 Гц, 1H), 1,95-1,82 (м, 2H), 1,17-1,03 (м, 21H); элементный анализ: (C16H30BrN3O4Si) C, H, N.
[0165] Перемешиваемый раствор спирта 132 (2,45 г, 5,61 ммоль) в безводном ДМФА (25 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (388 мг, 9,70 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 2 часов реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (20 мл), разбавляли ледяной водой (150 мл) и экстрагировали EtOAc (8×80 мл). Экстракты промывали насыщенным раствором соли (100 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-25% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 25% EtOAc/петролейный эфир давало 2-нитро-7-{[(триизопропилсилил)окси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (133) (1,77 г, 89%) в виде бледно-желтого твердого вещества: т.пл. (CH2Cl2/пентан) 121-123°C;
1H ЯМР (CDCl3) δ 7,42 (с, 1H), 4,45 (м, 1H), 4,17 (ддд, J=12,3, 5,8, 3,7 Гц, 1H), 4,06 (ддд, J=12,3, 10,3, 5,4 Гц, 1H), 4,03 (дд, J=10,7, 4,1 Гц, 1H), 3,95 (дд, J=10,7, 5,8 Гц, 1H), 2,37 (дддд, J=14,5, 5,5, 3,6, 2,8 Гц, 1H), 2,27 (дтд, J=14,5, 10,1, 5,8 Гц, 1H), 1,17-1,03 (м, 21H); элементный анализ: (C16H29N3O4Si) C, H, N.
[0166] Суспензию силилового эфира 133 (1,627 г, 4,58 ммоль) в 1% растворе HCl в 95% EtOH (условия десилилирования описаны Cunico et al., 1980) (58 мл) перемешивали при комнатной температуре в течение 35 часов. Полученный раствор охлаждали (CO2/ацетон), нейтрализовали добавлением по каплям 7M NH3 в MeOH (7 мл) при перемешивании, затем концентрировали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-2% MeOH/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 2% MeOH/CH2Cl2 давало (2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин-7-ил)метанол (134) (877 мг, 96%) в виде бледно-желтого твердого вещества: т.пл. (ТГФ/MeOH/CH2Cl2/гексан) 179-181°C;
1H ЯМР [(CD3)2SO] δ 8,04 (с, 1H), 5,12 (т, J=5,8 Гц, 1H), 4,48 (дтд, J=10,2, 4,7, 2,5 Гц, 1H), 4,13 (ддд, J=12,5, 5,8, 3,0 Гц, 1H), 4,04 (ддд, J=12,4, 11,0, 5,1 Гц, 1H), 3,64 (м, 2H), 2,18 (дтд, J=14,4, 5,0, 2,8 Гц, 1H), 2,03 (дтд, J=14,4, 10,6, 5,7 Гц, 1H); элементный анализ: (C7H9N3O4) C, H, N.
[0167] Диэтилазодикарбоксилат (0,070 мл, 0,45 ммоль) добавляли по каплям к суспензии спирта 134 (52,4 мг, 0,263 ммоль), трифенилфосфина (104 мг, 0,397 ммоль) и 4-(бензилокси)фенола (79,5 мг, 0,397 ммоль) в безводном ТГФ (1,0 мл) и полученную смесь перемешивали при комнатной температуре в течение 51 часа. Растворитель удаляли и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 4% EtOAc/CH2Cl2 давало неочищенное твердое вещество, которое затем хроматографировали на силикагеле. Элюирование смесью 0-33% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 4% MeOH/CH2Cl2 давало соединение 28 (36 мг, 36%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 222-224°C;
1H ЯМР [(CD3)2SO] δ 8,07 (с, 1H), 7,46-7,28 (м, 5H), 6,99-6,89 (м, 4H), 4,86 (м, 1H), 4,27-4,14 (м, 3H), 4,09 (ддд, J=12,5, 10,9, 5,2 Гц, 1H), 2,35-2,25 (м, 1H), 2,25-2,12 (м, 1H); элементный анализ: (C20H19N3O5.0,25H2O) C, H, N.
[0168] CC. Синтез 7-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 29 таблицы 1) при использовании способа схемы 7

[0169] Диэтилазодикарбоксилат (0,070 мл, 0,45 ммоль) добавляли по каплям к суспензии оксазинового спирта 134 (см. пример 2BB выше) (251 мг, 1,26 ммоль), трифенилфосфина (448 мг, 1,71 ммоль) и 4-иодфенола (377 мг, 1,71 ммоль) в безводном ТГФ (3,0 мл) при 0°C в атмосфере N2 и полученную смесь перемешивали при комнатной температуре в течение 32 часов. Растворитель удаляли и остаток хроматографировали на силикагеле. Элюирование CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 0-2% EtOAc/CH2Cl2 давало неочищенное твердое вещество, которое затем хроматографировали на силикагеле. Элюирование смесью 0-50% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 10% MeOH/CH2Cl2 давало 7-[(4-иодфенокси)метил]-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (135) (433 мг, 86%) в виде кремового твердого вещества: т.пл. (MeOΗ/CΗ2Cl2/гексан) 224-227°C;
1H ЯМР [(CD3)2SO] δ 8,08 (с, 1H), 7,62 (дт, J=9,0, 2,7 Гц, 2H), 6,86 (дт, J=9,0, 2,7 Гц, 2H), 4,89 (м, 1H), 4,31 (дд, J=11,1, 3,4 Гц, 1H), 4,25 (дд, J=11,1, 5,8 Гц, 1H), 4,18 (ддд, J=12,6, 5,8, 3,0 Гц, 1H), 4,09 (ддд, J=12,5, 10,8, 5,2 Гц, 1H), 2,35-2,26 (м, 1H), 2,25-2,12 (м, 1H); элементный анализ: (C13H12IN3O4) C, H, N.
[0170] Перемешиваемую смесь йодида 135 (50,1 мг, 0,125 ммоль), 4-фторфенилбороновой кислоты (31,5 мг, 0,225 ммоль) и Pd(dppf)Cl2 (14,1 мг, 0,019 ммоль) в толуоле (1 мл), EtOH (0,6 мл) и ДМФА (1,5 мл) дегазировали в течение 5 минут (вакуумный насос) и затем вводили N2. Добавляли 2М водный раствор Na2CO3 (0,40 мл, 0,80 ммоль) через шприц, перемешиваемую смесь снова дегазировали в течение 5 минут и затем вводили N2. Полученную смесь перемешивали при 90°C в течение 90 минут, затем охлаждали, разбавляли водным раствором NaHCO3 (50 мл) и экстрагировали CH2Cl2 (4×50 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1-2% EtOAc/CH2Cl2 давало соединение 29 (42 мг, 91%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/пентан) 217-219°C;
1H ЯМР [(CD3)2SO] δ 8,09 (с, 1H), 7,66 (ддт, J=8,9, 5,4, 2,7 Гц, 2H), 7,60 (дт, J=8,8, 2,6 Гц, 2H), 7,25 (тт, J=8,9, 2,7 Гц, 2H), 7,09 (дт, J=8,8, 2,6 Гц, 2H), 4,93 (м, 1H), 4,37 (дд, J=11,1, 3,4 Гц, 1H), 4,32 (дд, J=11,1, 5,7 Гц, 1H), 4,20 (ддд, J=12,6, 5,8, 3,0 Гц, 1H), 4,11 (ддд, J=12,5, 10,8, 5,2 Гц, 1H), 2,38-2,30 (м, 1H), 2,29-2,16 (м, 1H); элементный анализ: (C19H16FN3O4) C, H, N.
[0171] DD. Синтез 2-нитро-7-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 30 таблицы 1) при использовании способа схемы 7

[0172] Реакция сочетания Сузуки йодида 135 и 4-(трифторметил)фенилбороновой кислоты, как в примере 2CC выше, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1-2% EtOAc/CH2Cl2 давали соединение 30 (88%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/пентан) 242-245°C;
1H ЯМР [(CD3)2SO] δ 8,09 (с, 1 H), 7,86 (ушир.д, J=8,2 Гц, 2H), 7,77 (ушир.д, J=8,3 Гц, 2H), 7,72 (дтт, J=8,9, 2,5 Гц, 2H), 7,14 (дт, J=8,8, 2,5 Гц, 2 H), 4,94 (м, 1H), 4,40 (дд, J=11,1, 3,4 Гц, 1H), 4,34 (дд, J=11,1, 5,8 Гц, 1 H), 4,21 (ддд, J=12,5, 5,8, 3,0 Гц, 1H), 4,12 (ддд, J=12,5, 10,9, 5,2 Гц, 1H), 2,39-2,30 (м, 1H), 2,29-2,17 (м, 1H); элементный анализ: (C20H16F3N3O4) C, H, N.
[0173] EE. Синтез 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 31 таблицы 1) при использовании способа схемы 7

[0174] Реакция сочетания Сузуки йодида 135 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2CC выше, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1-2% EtOAc/CH2Cl2 давали соединение 31 (89%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 197-199°C;
1H ЯМР [(CD3)2SO] δ 8,12 (с, 1H), 7,75 (дт, J=8,8, 2,5 Гц, 2H), 7,65 (дт, J=8,9, 2,5 Гц, 2H), 7,42 (ушир.д, J=8,0 Гц, 2H), 7,11 (дт, J=8,9, 2,5 Гц, 2H), 4,94 (м, 1H), 4,38 (дд, J=11,1, 3,3 Гц, 1H), 4,32 (дд, J=11,1, 5,8 Гц, 1H), 4,20 (ддд, J=12,5, 5,7, 2,8 Гц, 1H), 4,11 (ддд, J=12,4, 11,0, 5,1 Гц, 1H), 2,38-2,29 (м, 1H), 2,28-2,15 (м, 1H); элементный анализ: (C20H16F3N3O5) C, H, N.
[0175] FF. Синтез 7-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 32 таблицы 1) при использовании способа схемы 8

[0176] 5-Бром-2-фторпиридин (91) (0,52 мл, 5,05 ммоль) добавляли к раствору оксазинового спирта 134 (см. пример 2BB) (500 мг, 2,51 ммоль) в безводном ДМФА (10 мл) в атмосфере N2 при 0°C. Полученную смесь обрабатывали 60% NaH (151 мг, 3,78 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. Снова добавляли 5-бром-2-фторпиридин (91) (0,52 мл, 5,05 ммоль), смесь перемешивали при комнатной температуре в течение 2,5 часов, затем охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (30 мл), добавляли к насыщенному раствору соли (100 мл) и экстрагировали CH2Cl2 (8×100 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 2-4% EtOAc/CΗ2Cl2 давало 7-{[(5-бром-2-пиридинил)окси]метил}-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (136) (778 мг, 87%) в виде белого твердого вещества: т.пл. (MeOΗ/CΗ2Cl2/гексан) 182-184°C;
1H ЯМР [(CD3)2SO] δ 8,30 (дд, J=2,6, 0,5 Гц, 1H), 8,07 (с, 1H), 7,95 (дд, J=8,8, 2,6 Гц, 1H),6,91 (дд, J=8,8, 0,6 Гц, 1H), 4,90 (м, 1H), 4,58 (дд, J=12,0, 3,3 Гц, 1H), 4,52 (дд, J=12,0, 6,0 Гц, 1H), 4,17 (ддд, J=12,6, 5,8, 2,8 Гц, 1H), 4,09 (ддд, J=12,5, 11,0, 5,2 Гц, 1H), 2,34-2,26 (м, 1H), 2,23-2,11 (м, 1H); элементный анализ: (C12H11BrN4O4) C, H, N.
[0177] Реакция сочетания Сузуки бромида 136 и 4-фторфенилбороновой кислоты (2,0 экв.), как в примере 2M, в течение 2,5 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-3% EtOAc/CΗ2Cl2 (головные фракции) и затем смесью 3% EtOAc/CH2Cl2 давали соединение 32 (91%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 180-181°C;
1H ЯМР [(CD3)2SO] δ 8,47 (дд, J=2,5, 0,5 Гц, 1H), 8,09 (с, 1H), 8,05 (дд, J=8,6, 2,6 Гц, 1H), 7,72 (ддт, J=8,9, 5,4, 2,7 Гц, 2H), 7,30 (тт, J=8,9, 2,7 Гц, 2H), 6,98 (дд, J=8,6, 0,6 Гц, 1H), 4,94 (м, 1H), 4,64 (дд, J=12,0, 3,4 Гц, 1H), 4,58 (дд, J=12,0, 6,1 Гц, 1H), 4,19 (ддд, J=12,6, 5,8, 2,7 Гц, 1H), 4,10 (ддд, J=12,4, 11,1, 5,1 Гц, 1H), 2,37-2,28 (м, 1H), 2,26-2,13 (м, 1H); APCI МС m/z 371 [M+H]+.
[0178] GG. Синтез 2-нитро-7-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 33 таблицы 1) при использовании способа схемы 8

[0179] Реакция сочетания Сузуки бромида 136 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2M, в течение 2,5 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0-2,5% EtOAc/CH2Cl2 давали соединение 33 (90%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 161-163°C;
1H ЯМР (CDCl3) δ 8,32 (д, J=2,0 Гц, 1H), 7,79 (дд, J=8,5, 2,5 Гц, 1H), 7,53 (ушир.д, J=8,7 Гц, 2H), 7,45 (с, 1H), 7,30 (ушир.д, J=8,1 Гц, 2H), 6,86 (д, J=8,6 Гц, 1H), 4,84 (м, 1H), 4,72 (дд, J=11,7, 5,1 Гц, 1H), 4,66 (дд, J=11,7, 4,9 Гц, 1H), 4,21 (ддд, J=12,4, 5,8, 3,4 Гц, 1H), 4,13 (ддд, J=12,4, 10,4, 5,5 Гц, 1H), 2,48-2,30 (м, 2H); элементный анализ: (C19H15F3N4O5) C, H, N.
[0180] HH. Синтез 7-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 34 таблицы 1) при использовании способа схемы 8

[0181] Смесь эпоксида 129 (см. пример 2AA) (1,004 г, 4,61 ммоль), 6-бром-3-пиридинола (4,015 г, 23,1 ммоль) и порошкообразного K2CO3 (3,319 г, 24,0 ммоль) в безводном 2-бутаноне (10 мл) в атмосфере N2 перемешивали при 82-85°C в течение 28 часов. Полученную охлажденную смесь разбавляли водой (100 мл) и экстрагировали смесью 25% EtOAc/CH2Cl2 (3×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-40% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 40% EtOAc/петролейный эфир давало 1-[(6-бром-3-пиридинил)окси]-4-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-бутанол (137) (667 мг, 37%) в виде кремового твердого вещества: т.пл. (MeOΗ/CΗ2Cl2/пентан) 112-114°C;
1H ЯМР [(CD3)2SO] δ 8,56 (с, 1H), 8,12 (д, J=3,1 Гц, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,39 (дд, J=8,8, 3,2 Гц, 1H), 5,28 (ушир.д, J=4,5 Гц, 1H), 4,24 (дд, J=14,0, 5,8 Гц, 1H), 4,18 (дд, J=14,2, 7,3 Гц, 1H), 3,99 (дд, J=10,0, 4,9 Гц, 1H), 3,96 (дд, J=10,0, 5,5 Гц, 1H), 3,82 (м, 1H), 2,06 (дтд, J=13,9, 7,7, 3,4 Гц, 1H), 1,90 (ддт, J=13,7, 9,2, 6,7 Гц, 1H); HRESIMS вычислено для C12H13BrClN4O4 m/z [M+H]+ 394,9754, 392,9782, 390,9803, найдено 394,9753, 392,9777, 390,9797.
[0182] Дальнейшее элюирование указанной выше колонки EtOAc давало неочищенное вещество с замкнутым кольцом, которое затем хроматографировали на силикагеле. Элюирование смесью 0-0,5% MeOH/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 0,5% MeOH/CH2Cl2 давало 7-{[(6-бром-3-пиридинил)окси]метил}-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (138) (51 мг, 3%) в виде кремового твердого вещества: т.пл. (MeOΗ/CΗ2Cl2/гексан) 200-202°C;
1H ЯМР [(CD3)2SO] δ 8,19 (д, J=3,2 Гц, 1H), 8,08 (с, 1H), 7,58 (д, J=8,7 Гц, 1H), 7,47 (дд, J=8,8, 3,2 Гц, 1H), 4,92 (м, 1H), 4,43 (дд, J=11,2, 3,3 Гц, 1H), 4,37 (дд, J=11,2, 5,8 Гц, 1H), 4,19 (ддд, J=12,5, 5,8, 3,0 Гц, 1H), 4,10 (ддд, J=12,5, 10,9, 5,2 Гц, 1H), 2,36-2,27 (м, 1H), 2,26-2,13 (м, 1H); элементный анализ: (C12H11BrN4O4) C, H, N.
[0183] Замыкание кольца спирта 137 при помощи NaH (1,6 экв.), как в примере 2AA, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% MeOH/CH2Cl2 (головные фракции) и затем смесью 0,5-0,75% MeOH/CH2Cl2 давали дополнительное количество соединения 138 (87%) в виде бледно-желтого твердого вещества (см. выше).
[0184] Реакция сочетания Сузуки бромида 138 и 4-фторфенилбороновой кислоты, как в примере 2M, в течение 2,5 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% MeOH/CH2Cl2 (головные фракции) и затем смесью 0,5% MeOH/CH2Cl2 давали соединение 34 (87%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 204-206°C;
1H ЯМР [(CD3)2SO] δ 8,43 (ушир.д, J=2,7 Гц, 1H) 8,11 (с, 1H), 8,07 (ддт, J=8,9, 5,6, 2,7 Гц, 2H), 7,94 (ушир.д, J=8,7 Гц, 1H), 7,56 (дд, J=8,8 3,0 Гц, 1H), 7,28 (тт, J=8,9, 2,6 Гц, 2H), 4,96 (м, 1H), 4,47 (дд, J=11,2, 3,2 Гц, 1H), 4,41 (дд, J=11,2, 5,8 Гц, 1H), 4,20 (ддд, J=12,5, 5,7, 2,9 Гц, 1H), 4,11 (ддд, J=12,4, 11,0, 5,1 Гц, 1H), 2,38-2,30 (м, 1H), 2,29-2,16 (м, 1H); APCI МС m/z 371 [M+H]+.
[0185] II. Синтез 2-нитро-7-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 35 таблицы 1) при использовании способа схемы 8

[0186] Реакция сочетания Сузуки бромида 138 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2M, в течение 2,5 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,33% MeOH/CH2Cl2 (головные фракции) и затем смесью 0,33% MeOH/CH2Cl2 давали соединение 35 (87%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 161-163°C;
1H ЯМР [(CD3)2SO] δ 8,46 (д, J=2,9 Гц, 1H), 8,15 (ушир.д, J=8,8 Гц, 2H), 8,11 (с, 1H), 7,99 (д, J=8,8 Гц, 1H), 7,59 (дд, J=8,8, 3,0 Гц, 1H), 7,45 (ушир.д, J=8,2 Гц, 2H), 4,96 (м, 1H), 4,49 (дд, J=11,2, 3,2 Гц, 1H), 4,43 (дд, J=11,2, 5,8 Гц, 1H), 4,20 (ддд, J=12,5, 5,6, 2,7 Гц, 1H), 4,12 (ддд, J=12,4, 11,0, 5,2 Гц, 1H), 2,39-2,30 (м, 1H), 2,29-2,16 (м, 1H); элементный анализ: (C19H15F3N4O5) C, H, N.
[0187] JJ. Синтез 7-метил-2-нитро-7-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 36 таблицы 1) при использовании способа схемы 9

[0188] Раствор 4-иод-2-метил-1-бутена (получен путем йодирования 3-метил-3-бутен-1-ола, как описано Helmboldt et al., 2006) (2,01 г, 10,3 ммоль) в безводном ДМФА (3 мл, затем 3×1 мл для промывки) добавляли к перемешиваемой смеси 2-хлор-4(5)-нитроимидазола (81) (1,00 г, 6,80 ммоль) и порошкообразного K2CO3 (2,83 г, 20,5 ммоль) в безводном ДМФА (6,5 мл) в атмосфере N2 и смесь перемешивали при 61°C в течение 20 часов. Полученную охлажденную смесь добавляли к смеси лед/водный раствор NaHCO3 (100 мл) и экстрагировали EtOAc (4×100 мл). Экстракты промывали разбавленным раствором соли (100 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-10% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 10-15% EtOAc/петролейный эфир давало 2-хлор-1-(3-метил-3-бутенил)-4-нитро-1H-имидазол (139) (1,15 г, 78%) в виде белого твердого вещества: т.пл. (CΗ2Cl2/пентан) 68-69°C;
1H ЯМР (CDCl3) δ 7,71 (с, 1H), 4,90 (м, 1H), 4,69 (м, 1H), 4,13 (т, J=7,1 Гц, 2H), 2,52 (ушир.т, J=7,1 Гц, 2H), 1,80 (с, 3H); HRFABMS вычислено для C8H11ClN3O2 m/z [M+H]+ 218,0510, 216,0540, найдено 218,0512, 216,0544.
[0189] Эпоксидирование алкена 139 при использовании 3-хлорпербензойной кислоты, как в примере 2G, в течение 4 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0-5% EtOAc/CH2Cl2 давали 2-хлор-1-[2-(2-метил-2-оксиранил)этил]-4-нитро-1Н-имидазол (140) (88%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 82-85°C;
1H ЯМР (CDCl3) δ 7,78 (с, 1H), 4,12 (т, J=7,6 Гц, 1H), 2,66 (д, J=4,4 Гц, 1H), 2,62 (д, J=4,4 Гц, 1H), 2,19 (дт, J=14,3, 7,7 Гц, 1H), 2,04 (дт, J=14,3, 7,4 Гц, 1H), 1,39 (с, 3H); HRFABMS вычислено для C8H11ClN3O3 m/z [M+H]+ 234,0459, 232,0489, найдено 234,0466, 232,0488.
[0190] Взаимодействие эпоксида 140 с 4-трифторметоксифенолом, как в примере 2AA, при 82°C в течение 10 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0-2% EtOAc/CH2Cl2 давали 4-(2-хлор-4-нитро-1Н-имидазол-1-ил)-2-метил-1-[4-(трифторметокси)фенокси]-2-бутанол (141) (77%) в виде бледно-желтого масла;
1H ЯМР (CDCl3) δ 7,81 (с, 1H), 7,17 (ушир.дд, J=9,1, 0,7 Гц, 2H), 6,90 (дт, J=9,2, 3,1 Гц, 2H), 4,29 (ддд, J=14,1, 9,5, 6,3 Гц, 1H), 4,24 (ддд, J=14,1, 9,6, 6,5 Гц, 1H), 3,85 (д, J=9,0 Гц, 1H), 3,82 (д, J=9,0 Гц, 1H), 2,23 (ддд, J=13,8, 9,3, 6,5 Гц, 1H), 2,21 (с, 1H), 1,40 (с, 3H); HRESIMS вычислено для C15H16ClF3N3O5 m/z [M+H]+ 412,0697, 410,0725, найдено 412,0700, 410,0722.
[0191] Замыкание кольца спирта 141 при помощи NaH, как в примере 2AA, в течение 2 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 25-33% EtOAc/петролейный эфир (головные фракции) и затем смесью 50% EtOAc/петролейный эфир давали соединение 36 (61%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 134-136°C;
1H ЯМР (CDCl3) δ 7,45 (с, 1H), 7,16 (ушир.дд, J=9,1, 0,8 Гц, 2H), 6,87 (дт, J=9,2, 3,0 Гц, 2H), 4,21-4,02 (м, 4H), 2,51 (ддд, J=14,5, 7,4, 6,0 Гц, 1H), 2,25 (дт, J=14,5, 6,2 Гц, 1H), 1,60 (с, 3H); элементный анализ: (C15H14F3N3O5) C, H, N.
[0192] KK. Синтез 7-{[4-(бензилокси)фенокси]метил}-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 37 таблицы 1) при использовании способа схемы 9

[0193] Взаимодействие эпоксида 140 (см. пример 2U) с 4-(бензилокси)фенолом, как в примере 2AA, при 82°C в течение 10 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 2% EtOAc/CH2Cl2 давали 1-[4-(бензилокси)фенокси]-4-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-метил-2-бутанол (142) (79%) в виде масла;
1H ЯМР (CDCl3) δ 7,79 (с, 1H), 7,44-7,29 (м, 5H), 6,92 (дт, J=9,2, 3,0 Гц, 2H), 6,83 (дт, J=9,2, 3,0 Гц, 2H), 4,28 (ддд, J=14,0, 9,7, 6,1 Гц, 1H), 4,23 (ддд, J=14,0, 9,7, 6,3 Гц, 1H), 3,81 (д, J=9,1 Гц, 1H), 3,77 (д, J=9,0 Гц, 1H), 2,29 (с, 1H), 2,22 (ддд, J=13,8, 9,6, 6,2 Гц, 1H), 2,02 (ддд, J=13,6, 9,7, 6,5 Гц, 1H), 1,38 (с, 3H); HRESIMS вычислено для C21H23ClN3O5 m/z [M+H]+ 434,1293, 432,1321, найдено 434,1298, 432,1319.
[0194] Замыкание кольца спирта 142 при помощи NaH (1,4 экв.), как в примере 2AA, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-33% EtOAc/петролейный эфир (головные фракции) и затем EtOAc давали неочищенный продукт, который затем хроматографировали на силикагеле. Элюирование смесью 0-2,5% EtOAc/CH2Cl2 сначала давало головные фракции, затем последующее элюирование смесью 2,5% EtOAc/CH2Cl2 давало соединение 37 (53%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 174-176°C;
1H ЯМР [(CD3)2SO] δ 8,07 (с, 1H), 7,45-7,28 (м, 5H), 6,94 (дт, J=9,3, 2,9 Гц, 2H), 6,89 (дт, J=9,3, 2,9 Гц, 2H), 5,04 (с, 2H), 4,21-4,06 (м, 2H), 4,10 (с, 2H), 2,37 (ддд, J=14,5, 7,9, 6,2 Гц, 1H), 2,17 (дт, J=14,4, 5,8 Гц, 1H), 1,48 (с, 3H); элементный анализ: (C21H21N3O5) C, H, N.
[0195] LL. Синтез 7-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 38 таблицы 1) при использовании способа схемы 9

[0196] Взаимодействие эпоксида 140 (см. пример 2JJ) с 4-иодфенолом, как в примере 2AA, при 83°C в течение 8 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-2% EtOAc/CH2Cl2 (головные фракции) и затем смесью 5% EtOAc/CH2Cl2 давали 4-(2-хлор-4-нитро-1Н-имидазол-1-ил)-1-(4-иодфенокси)-2-метил-2-бутанол (143) (81%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 91-93°C;
1H ЯМР (CDCl3) δ 7,80 (с, 1H), 7,59 (дт, J=8,9, 2,6 Гц, 1H), 6,69 (дт, J=8,9, 2,6 Гц, 1H), 4,28 (ддд, J=14,1, 9,6, 6,3 Гц, 1H), 4,23 (ддд, J=14,1, 9,4, 6,5 Гц, 1H), 3,82 (д, J=9,0 Гц, 1H), 3,79 (д, J=9,0 Гц, 1H), 2,22 (ддд, J=13,8, 9,2, 6,5 Гц, 1H), 2,20 (с, 1H), 2,02 (ддд, J=13,8, 9,6, 6,6 Гц, 1H), 1,39 (с, 3H); HRESIMS вычислено для C14H16ClIN3O4 m/z [M+H]+ 453,9840, 451,9869, найдено 453,9832, 451,9857.
[0197] Замыкание кольца спирта 143 при помощи NaH (1,5 экв.), как в примере 2AA, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-33% EtOAc/петролейный эфир (головные фракции) и затем смесью 0-5% EtOAc/CH2Cl2 давали 7-[(4-иодфенокси)метил]-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (144) (74%) в виде бледно-желтого твердого вещества: т.пл. (CH2Cl2/гексан) 170-172°C;
1H ЯМР (CDCl3) δ 7,57 (дт, J=9,0, 2,7 Гц, 2H), 7,44 (с, 1H 6,64 (дт, J=9,0, 2,7 Гц, 2H), 4,19-4,05 (м, 2H), 4,07 (д, J=9,6 Гц, 1H), 4,02 (д, J=9,6 Гц, 1H), 2,49 (ддд, J=14,5, 7,4, 6,0 Гц, 1H), 2,24 (ддд, J=14,5, 6,4, 5,9 Гц, 1H), 1,58 (с, 3H); элементный анализ: (C14H14IN3O4) C, H, N.
[0198] Реакция сочетания Сузуки йодида 144 и 4-фторфенилбороновой кислоты, как в примере 2CC, в течение 100 минут с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1-2% EtOAc/CH2Cl2 давали соединение 38 (90%) в виде бледно-желто-оранжевого твердого вещества: т.пл. (CH2Cl2/пентан) 160-162°C;
1H ЯМР (CDCl3) δ 7,51-7,44 (м, 5H), 7,10 (тт, J=8,7, 2,6 Гц, 2H), 6,92 (дт, J=8,8, 2,6 Гц, 2H), 4,23-4,06 (м, 4H), 2,53 (ддд, J=14,4, 7,2, 6,0 Гц, 1H), 2,28 (ддд, J=14,5, 6,8, 5,9 Гц, 1H), 1,62 (с, 3H); элементный анализ: (C20H18FN3O4) C, H, N.
[0199] MM. Синтез 7-метил-2-нитро-7-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 39 таблицы 1) при использовании способа схемы 9

[0200] Реакция сочетания Сузуки йодида 144 (см. пример 2LL) и 4-(трифторметил)фенилбороновой кислоты, как в примере 2CC, в течение 100 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1% EtOAc/CH2Cl2 давали соединение 39 (87%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 196-198°C;
1H ЯМР (CDCl3) δ 7,67 (ушир.д, J=8,5 Гц, 2H), 7,63 (ушир.д, J=8,4 Гц, 2H), 7,54 (дт, J=8,8, 2,6 Гц, 2H), 7,46 (с, 1H), 6,96 (дт, J=8,8, 2,6 Гц, 2H), 4,23-4,08 (м, 4H), 2,54 (ддд, J=14,5, 7,3, 6,0 Гц, 1H), 2,28 (ддд, J=14,5, 6,6, 5,9 Гц, 1H), 1,62 (с, 3H); элементный анализ: (C21H18F3N3O4) C, H, N.
[0201] NN. Синтез 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 40 таблицы 1) при использовании способа схемы 9

[0202] Реакция сочетания Сузуки йодида 144 (см. пример 2LL) и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2CC, в течение 105 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1% EtOAc/CH2Cl2 давали соединение 40 (89%) в виде бледно-желто-розового твердого вещества: т.пл. (CH2Cl2/пентан) 186-188°C;
1H ЯМР (CDCl3) δ 7,53 (дт, J=8,8, 2,5 Гц, 2H), 7,49 (дд, J=8,8, 2,6 Гц, 2H), 7,46 (с, 1H), 7,26 (ушир.дд, J=8,7, 0,8 Гц, 2H), 6,94 (дт, J=8,8, 2,6 Гц, 2H), 4,23-4,07 (м, 4H), 2,53 (ддд, J=14,5, 7,2, 6,0 Гц, 1H), 2,28 (ддд, J=14,5, 6,7, 5,9 Гц, 1H), 1,62 (с, 3H); элементный анализ: (C21H18F3N3O5) C, H, N.
[0203] OO. Синтез 7-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 41 таблицы 1) при использовании способа схемы 10

[0204] Раствор 4-иод-2-метил-1-бутена (получен путем йодирования 3-метил-3-бутен-1-ола, как описано Helmboldt et al., 2006) (2,68 г, 13,7 ммоль) в безводном ДМФА (5 мл, затем 2×2 мл+1 мл для промывки) добавляли к перемешиваемой смеси 2-бром-4(5)-нитроимидазола (80) (2,00 г, 10,4 ммоль) и порошкообразного K2CO3 (4,35 г, 31,5 ммоль) в безводном ДМФА (10 мл) в атмосфере N2, и полученную смесь перемешивали при 60°C в течение 11 часов. Полученную охлажденную смесь добавляли к смеси лед/водный раствор NaHCO3 (120 мл) и экстрагировали EtOAc (3×100 мл). Экстракты промывали разбавленным раствором соли (100 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-10% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 20-25% EtOAc/петролейный эфир давало 2-бром-1-(3-метил-3-бутенил)-4-нитро-1Н-имидазол (145) (2,296 г, 85%) в виде белого твердого вещества: т.пл. (CΗ2Cl2/пентан) 90-92°C;
1H ЯМР (CDCl3) δ 7,76 (с, 1H), 4,90 (м, 1H), 4,70 (м, 1H), 4,12 (т, J=7,2 Гц, 2H), 2,52 (ушир.т, J=7,1 Гц, 2H), 1,81 (с, 3H); элементный анализ: (C8H10BrN3O2) C, H, N.
[0205] Тетраоксид осмия (2,55 мл 4% водного раствора, 0,417 ммоль) добавляли к раствору алкена 145 (2,15 г, 8,27 ммоль) и 4-метилморфолин N-оксида (1,49 г, 12,7 ммоль) в CH2Cl2 (55 мл) и затем смесь перемешивали при комнатной температуре в течение 4 часов. Смесь охлаждали (-20°C), медленно разбавляли петролейным эфиром (70 мл), снова охлаждали (-20°C) и полученный осадок выделяли фильтрованием, промывали петролейным эфиром и водой, сушили, с получением 4-(2-бром-4-нитро-1H-имидазол-1-ил)-2-метил-1,2-бутандиола (146) (1,53 г, 63%) в виде бледно-серовато-коричневого твердого вещества: т.пл. (MeOH/CH2Cl2/пентан) 121-123°C;
1H ЯМР [(CD3)2SO] δ 8,58 (с, 1H), 4,69 (ушир.т, J=5,3 Гц, 1H), 4,41 (ушир.с, 1H), 4,13 (т, J=8,1 Гц, 2H), 3,24 (дд, J=10,6, 5,6 Гц, 1H), 3,18 (дд, J=10,7, 5,6 Гц, 1H), 1,89 (дт, J=13,3, 8,1 Гц, 1H), 1,82 (дт, J=13,3, 8,1 Гц, 1H), 1,09 (с, 3H); элементный анализ: (C8H12BrN3O4) C, H, N.
[0206] Полученную на предыдущей стадии водную часть насыщали солью и экстрагировали EtOAc (6×100 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 50-67% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 67-80% EtOAc/петролейный эфир давало дополнительное количество соединения 146 (882 мг, 36%).
[0207] Триизопропилсилилхлорид (2,00 мл, 9,35 ммоль) добавляли к раствору диола 146 (2,507 г, 8,52 ммоль) и имидазола (1,278 г, 18,8 ммоль) в безводном ДМФА (25 мл) в атмосфере N2 и затем смесь перемешивали при комнатной температуре в течение 3 дней. Снова добавляли триизопропилсилилхлорид (0,50 мл, 2,34 ммоль) и смесь перемешивали при комнатной температуре в течение 3 дней. Полученную смесь добавляли в ледяную воду (130 мл) и экстрагировали EtOAc (4×100 мл). Экстракты промывали водой (100 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-10% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 33% EtOAc/петролейный эфир давало 4-(2-бром-4-нитро-1H-имидазол-1-ил)-2-метил-1-[(триизопропилсилил)окси]-2-бутанол (147) (3,658 г, 95%) в виде белого твердого вещества: т.пл. (CΗ2Cl2/пентан) 73-75°C;
1H ЯМР (CDCl3) δ 7,85 (с, 1H), 4,26 (ддд, J=14,1, 10,3, 5,8 Гц, 1H), 4,19 (ддд, J=14,1, 10,3, 6,0 Гц, 1H), 3,56 (с, 2H), 2,52 (с, 1H), 2,11 (ддд, J=13,6, 10,3, 5,8 Гц, 1H), 1,87 (ддд, J=13,6, 10,3, 6,0 Гц, 1H), 1,25 (с, 3H), 1,21-1,04 (м, 21H); элементный анализ: (C17H32BrN3O4Si) C, H, N.
[0208] Перемешиваемый раствор спирта 147 (3,60 г, 8,00 ммоль) в безводном ДМФА (35 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (550 мг, 13,8 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 2,5 часов и затем при 46°C в течение 190 минут реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (40 мл), разбавляли ледяной водой (140 мл) и экстрагировали EtOAc (5×80 мл). Объединенные экстракты промывали насыщенным раствором соли (80 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-15% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 15-25% EtOAc/петролейный эфир давало 7-метил-2-нитро-7-{[(триизопропилсилил)окси]метил}-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (148) (2,599 г, 88%) в виде бледно-желтого твердого вещества: т.пл. (CH2Cl2/пентан) 112-114°C;
1H ЯМР (CDCl3) δ 7,41 (с, 1H), 4,14 (ддд, J=12,4, 6,9, 5,8 Гц, 1H), 4,03 (ддд, J=12,4, 7,3, 5,8 Гц, 1H), 3,84 (д, J=10,2 Гц, 1H), 3,77 (д, J=10,2 Гц, 1H), 2,37 (ддд, J=14,4, 7,2, 5,8 Гц, 1H), 2,11 (ддд, J=14,4, 6,9, 5,9 Гц, 1H), 1,45 (с, 3H), 1,16-0,97 (м, 21H); элементный анализ: (C17H31N3O4Si) C, H, N.
[0209] Суспензию силилового эфира 148 (2,518 г, 6,81 ммоль) в 1% растворе HCl в 95% EtOH (условия десилилирования описаны Cunico et al., 1980) (90 мл) перемешивали при 44°C в течение 3 дней. Полученный раствор охлаждали (CO2/ацетон), нейтрализовали добавлением по каплям 7M NH3 в MeOH (8 мл) и NaHCO3 (0,10 г, 1,19 ммоль) при перемешивании, затем концентрировали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% MeOH/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1,5% MeOH/CH2Cl2 давало (7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин-7-ил)метанол (149) (1,285 г, 88%) в виде бледно-желтого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 199-201°C;
1H ЯМР [(CD3)2SO] δ 8,03 (с, 1H), 5,22 (т, J=5,7 Гц, 1H), 4,13 (дт, J=12,9, 6,0 Гц, 1H), 4,05 (ддд, J=13,0, 8,1, 5,6 Гц, 1H), 3,54 (дд, J=11,6, 5,5 Гц, 1H), 3,48 (дд, J=11,6, 5,8 Гц, 1H), 2,21 (ддд, J=14,4, 8,1, 5,9 Гц, 1H), 2,00 (дт, J=14,4, 5,8 Гц, 1H), 1,32 (с, 3H); элементный анализ: (C8H11N3O4) C, H, N.
[0210] Раствор спирта 149 (200 мг, 0,938 ммоль) в безводном ДМФА (4 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (53,8 мг, 1,35 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. Добавляли 5-бром-2-фторпиридин (91) (0,245 мл, 2,38 ммоль), смесь перемешивали при комнатной температуре в течение 2,5 часов и затем охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (10 мл), добавляли к насыщенному раствору соли (40 мл) и экстрагировали CH2Cl2 (10×50 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1-3% EtOAc/CH2Cl2 давало 7-{[(5-бром-2-пиридинил)окси]метил}-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (150) (269 мг, 78%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 172-174°C;
1H ЯМР (CDCl3) δ 8,17 (ушир.д, J=2,2 Гц, 1H), 7,67 (дд, J=8,8, 2,5 Гц, 1H), 6,64 (дд, J=8,7, 0,4 Гц, 1H), 4,49 (д, J=11,5 Гц, 1H), 4,42 (д, J=11,4 Гц, 1H), 4,17 (дт, J=12,7, 6,1 Гц, 1H), 4,09 (ддд, J=12,6, 7,7, 5,8 Гц, 1H), 2,45 (ддд, J=14,5, 7,6, 5,9 Гц, 1H), 2,18 (дт, J=14,6, 6,1 Гц, 1H), 1,57 (с, 3H); элементный анализ: (C13H13BrN4O4) C, H, N.
[0211] Реакция сочетания Сузуки бромида 150 и 4-фторфенилбороновой кислоты, как в примере 2M, в течение 135 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-3% EtOAc/CH2Cl2 (головные фракции) и затем смесью 3-5% EtOAc/CH2Cl2 давали соединение 41 (92%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 145-147°C;
1H ЯМР (CDCl3) δ 8,28 (д, J=2,5 Гц, 1H), 7,76 (дд, J=8,6, 2,5 Гц, 1H), 7,46 (ддт, J=8,8, 5,1, 2,6 Гц, 2H), 7,45 (с, 1H), 7,14 (тт, J=8,6, 2,6 Гц, 2H), 6,88 (д, J=8,6 Гц, 1H), 4,58 (д, J=11,4 Гц, 1H), 4,50 (д, J=11,4 Гц, 1H), 4,20 (ддд, J=12,6, 6,5, 6,1 Гц, 1H), 4,10 (ддд, J=12,6, 7,3, 5,8 Гц, 1H), 2,49 (ддд, J=14,4, 7,3, 6,0 Гц, 1H), 2,21 (ддд, J=14,4, 6,6, 6,0 Гц, 1H), 1,61 (с, 3H); элементный анализ: (C19H17FN4O4) C, H, N.
[0212] PP. Синтез 7-метил-2-нитро-7-[({5-[4-(трифторметил)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 42 таблицы 1) при использовании способа схемы 10

[0213] Реакция сочетания Сузуки бромида 150 и 4-(трифторметил)фенилбороновой кислоты, как в примере 2M, в течение 2 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-2% EtOAc/CH2Cl2 (головные фракции) и затем смесью 3-5% EtOAc/CH2Cl2 давали соединение 42 (91%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 212-214°C;
1H ЯМР (CDCl3) δ 8,35 (дд, J=2,5, 0,4 Гц, 1H), 7,82 (дд, J=8,6, 2,5 Гц, 1H), 7,71 (ушир.д, J=8,2 Гц, 2H), 7,62 (ушир.д, J=8,1 Гц, 2H), 7,46 (с, 1H), 6,82 (дд, J=8,7, 0,4 Гц, 1H), 4,60 (д, J=11,4 Гц, 1H), 4,52 (д, J=11,4 Гц, 1H), 4,21 (ддд, J=12,6, 6,5, 6,0 Гц, 1H), 4,11 (ддд, J=12,7, 7,4, 5,8 Гц, 1H), 2,50 (ддд, J=14,6, 7,4, 5,9 Гц, 1H), 2,22 (ддд, J=14,5, 6,5, 6,0 Гц, 1H), 1,61 (с, 3H); элементный анализ: (C20H17F3N4O4) C, H, N.
[0214] QQ. Синтез 7-метил-2-нитро-7-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 43 таблицы 1) при использовании способа схемы 10

[0215] Реакция сочетания Сузуки бромида 150 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2M, в течение 2 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-2% EtOAc/CH2Cl2 (головные фракции) и затем смесью 2-3,5% EtOAc/CH2Cl2 давали соединение 43 (92%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 195-198°C;
1H ЯМР (CDCl3) δ 8,31 (дд, J=2,5, 0,7 Гц, 1H), 7,78 (дд, J=8,5, 2,6 Гц, 1H), 7,52 (дт, J=8,8, 2,5 Гц, 2H), 7,45 (с, 1H), 7,30 (ушир.дд, J=8,7, 0,8 Гц, 2H), 6,79 (дд, J=8,6, 0,7 Гц, 1H), 4,59 (д, J=11,4 Гц, 1H), 4,50 (д, J=11,4 Гц, 1H), 4,20 (ддд, J=12,6, 6,7, 5,9 Гц, 1H), 4,10 (ддд, J=12,6, 7,4, 5,8 Гц, 1H), 2,49 (ддд, J=14,5, 7,4, 5,9 Гц, 1H), 2,21 (ддд, J=14,5, 6,6, 5,9 Гц, 1H), 1,61 (с, 3H); элементный анализ: (C20H17F3N4O5) C, H, N.
[0216] RR. Синтез 7-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 44 таблицы 1) при использовании способа схемы 10

[0217] Взаимодействие эпоксида 140 (см. пример 2JJ) с 6-бром-3-пиридинолом, как в примере 2AA, при 84°C в течение 18,5 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 25-40% EtOAc/петролейный эфир (головные фракции) и затем смесью 40-50% EtOAc/петролейный эфир давали 1-[(6-бром-3-пиридинил)окси]-4-(2-хлор-4-нитро-1H-имидазол-1-ил)-2-метил-2-бутанол (151) (70%) в виде бледно-желто-коричневой пены;
1H ЯМР (CDCl3) δ 8,09 (дд, J=3,0, 0,3 Гц, 1H), 7,80 (с, 1H), 7,41 (дд, J=8,7, 0,4 Гц, 1H), 7,13 (дд, J=8,7, 3,2 Гц, 1H), 4,29 (ддд, J=14,2, 9,4, 6,4 Гц, 1H), 4,25 (ддд, J=14,1, 9,4, 6,7 Гц, 1H), 3,89 (д, J=8,9 Гц, 1H), 3,86 (д, J=9,0 Гц, 1H), 2,22 (ддд, J=13,9, 9,3, 6,5 Гц, 1H), 2,18 (с, 1H), 2,04 (ддд, J=13,8, 9,4, 6,7 Гц, 1H), 1,42 (с, 3H); HRESIMS вычислено для C13H15BrClN4O4) m/z [M+H]+ 408,9910, 406,9939, 404,9960, найдено 408,9920, 406,9945, 404,9966.
[0218] Замыкание кольца спирта 151 при помощи NaH (1,5 экв.), как в примере 2AA, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-50% EtOAc/петролейный эфир (головные фракции) и затем смесью 0-2% MeOH/CH2Cl2 давали 7-{[(6-бром-3-пиридинил)окси]метил}-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (152) (66%) в виде светло-желтого твердого вещества: т.пл. (CH2Cl2/гексан) 170-171°C;
1H ЯМР (CDCl3) δ 8,06 (дд, J=3,1, 0,3 Гц, 1H), 7,46 (с, 1H), 7,40 (дд, J=8,7, 0,3 Гц, 1H), 7,11 (дд, J=8,7, 3,2 Гц, 1H), 4,21-4,07 (м, 4H), 2,52 (ддд, J=14,5, 8,1, 6,3 Гц, 1H), 2,24 (дт, J=14,5, 5,7 Гц, 1H), 1,60 (с, 3H); элементный анализ: (C13H13BrN4O4) C, H, N.
[0219] Реакция сочетания Сузуки бромида 152 и 4-фторфенилбороновой кислоты, как в примере 2M, в течение 2 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-3% EtOAc/CH2Cl2 (головные фракции) и затем смесью 3-7% EtOAc/CH2Cl2 давали соединение 44 (88%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 203-204°C;
1H ЯМР [(CD3)2SO] δ 8,39 (д, J=2,8 Гц, 1H), 8,10 (с, 1H), 8,06 (ддт, J=9,0, 5,6, 2,6 Гц, 2H), 7,92 (д, J=8,8 Гц, 1H), 7,53 (дд, J=8,8, 3,0 Гц, 1H), 7,27 (тт, J=8,9, 2,6 Гц, 2H), 4,33 (с, 2H), 4,25-4,11 (м, 2H), 2,42 (ддд, J=14,5, 8,2, 6,2 Гц, 1H), 2,21 (дт, J=14,4, 5,7 Гц, 1H), 1,52 (с, 3H); элементный анализ: (C19H17FN4O4) C, H, N.
[0220] SS. Синтез 7-метил-2-нитро-7-[({6-[4-(трифторметил)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 45 таблицы 1) при использовании способа схемы 10

[0221] Реакция сочетания Сузуки бромида 152 (см. пример 2RR) и 4-(трифторметил)фенилбороновой кислоты, как в примере 2M, в течение 130 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-3% EtOAc/CH2Cl2 (головные фракции) и затем смесью 4-7% EtOAc/CH2Cl2 давали соединение 45 (65%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 215-217°C;
1H ЯМР [(CD3)2SO] δ 8,46 (д, J=2,9 Гц, 1H), 8,25 (ушир.д, J=8,1 Гц, 2H), 8,10 (с, 1H), 8,06 (д, J=8,8 Гц, 1H), 7,81 (ушир.д, J=8,3 Гц, 2H), 7,59 (дд, J=8,8, 3,0 Гц, 1H), 4,36 (с, 2H), 4,26-4,11 (м, 2H), 2,42 (ддд, J=14,5, 8,1, 6,0 Гц, 1H), 2,21 (дт, J=14,4, 5,7 Гц, 1H), 1,53 (с, 3H); элементный анализ: (C20H17F3N4O4) C, H, N.
[0222] TT. Синтез 7-метил-2-нитро-7-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 46 таблицы 1) при использовании способа схемы 10

[0223] Реакция сочетания Сузуки бромида 152 (см. пример 2RR) и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2M, в течение 130 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-4% EtOAc/CH2Cl2 (головные фракции) и затем смесью 5-7% EtOAc/CH2Cl2 давали соединение 46 (84%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 202-203°C;
1H ЯМР [(CD3)2SO] δ 8,42 (д, J=2,8 Гц, 1H), 8,14 (дт, J=8,9, 2,6 Гц, 2H), 8,10 (с, 1H), 7,97 (д, J=8,8 Гц, 1H), 7,56 (дд, J=8,8, 3,0 Гц, 1H), 7,44 (ушир.дд, J=8,8, 0,8 Гц, 2H), 4,34 (с, 2H), 4,25-4,11 (м, 2H), 2,42 (ддд, J=14,5, 8,2, 6,1 Гц, 1H), 2,21 (дт, J=14,4, 5,7 Гц, 1H), 1,52 (с, 3H); элементный анализ: (C20H17F3N4O5) C, H, N.
[0224] UU. Синтез 2-нитро-7-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 47 таблицы 1) при использовании способа схемы 11

[0225] Смесь оксазинового спирта 134 (см. пример 2BB выше) (31,8 мг, 0,160 ммоль) и 3-(трифторметокси)бензилбромида (0,040 мл, 0,247 ммоль) в безводном ДМФА (3 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (9,5 мг, 0,238 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 2,5 часов смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (10 мл), добавляли к насыщенному раствору соли (40 мл) и экстрагировали CH2Cl2 (4×50 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1-2% EtOAc/CH2Cl2 давало соединение 47 (44 мг, 74%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 110-112°C;
1H ЯМР (CDCl3) δ 7,41 (с, 1H), 7,41-7,35 (м, 1H), 7,23 (ушир.д, J=7,8 Гц, 1H), 7,19-7,13 (м, 2H), 4,62 (с, 2H), 4,58 (м, 1H), 4,15 (ддд, J=12,4, 5,8, 3,7 Гц, 1H), 4,06 (ддд, J=12,3, 10,1, 5,6 Гц, 1H), 3,84 (дд, J=10,6, 4,3 Гц, 1H), 3,78 (дд, J=10,6, 5,1 Гц, 1H), 2,40-2,21 (м, 2H); элементный анализ: (C15H14F3N3O5) C, H, N.
[0226] VV. Синтез 2-нитро-7-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 48 таблицы 1) при использовании способа схемы 11

[0227] Алкилирование оксазинового спирта 134 (см. пример 2BB выше) при использовании 4-(трифторметокси)бензилбромида (1,9 экв.) и NaH (1,7 экв.), как в примере 2UU выше, в течение 165 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% MeOH/CH2Cl2 (головные фракции) и затем смесью 0,5% MeOH/CH2Cl2 давали соединение 48 (69%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 158-160°C;
1H ЯМР (CDCl3) δ 7,41 (с, 1H), 7,34 (дт, J=8,8, 2,3 Гц, 2H), 7,20 (ушир.д, J=7,9 Гц, 2H), 4,61 (с, 2H), 4,61-4,54 (м, 1H), 4,14 (ддд, J=12,4, 5,7, 3,7 Гц, 1H), 4,06 (ддд, J=12,3, 10,0, 5,8 Гц, 1H), 3,82 (дд, J=10,7, 4,4 Гц, 1H), 3,78 (дд, J=10,7, 4,9 Гц, 1H), 2,38-2,21 (м, 1H); элементный анализ: (C15H14F3N3O5) C, H, N.
[0228] WW. Синтез 7-({[4-(бензилокси)бензил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 49 таблицы 1) при использовании способа схемы 11

[0229] Алкилирование оксазинового спирта 134 (см. пример 2BB выше) при использовании 4-(бензилокси)бензилхлорида (3,0 экв.) и NaH (1,5 экв.), как в примере 2UU выше, в течение 3 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1% EtOAc/CH2Cl2 давали соединение 49 (20 мг, 25%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 151-153°C;
1H ЯМР (CDCl3) δ 7,45-7,29 (м, 6H), 7,22 (дт, J=8,7, 2,4 Гц, 2H), 6,95 (дт, J=8,7, 2,4 Гц, 2H), 5,07 (с, 2H), 4,54 (м, 1H), 4,52 (с, 2H), 4,11 (ддд, J=12,3, 5,8, 3,9 Гц, 1H), 4,02 (ддд, J=12,3, 10,0, 5,5 Гц, 1H), 3,78 (дд, J=10,5, 4,3 Гц, 1H), 3,71 (дд, J=10,5, 5,5 Гц, 1H), 2,33 (дддд, J=14,5, 5,4, 3,8, 3,0 Гц, 1H), 2,23 (дтд, J=14,6, 9,8, 5,9 Гц, 1H); элементный анализ: (C21H21N3O5) C, H, N.
[0230] XX. Синтез 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-3-ил]метокси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 50 таблицы 1) при использовании способа схемы 11

[0231] Алкилирование оксазинового спирта 134 (см. пример 2BB выше) при использовании 3-иодбензилбромида (1,36 экв.) и NaH (1,5 экв.), как в примере 2UU выше, в течение 3 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1,5-2% EtOAc/CH2Cl2, давало 7-{[(3-иодбензил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (153) (65%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 131-133°C;
1H ЯМР (CDCl3) δ 7,65 (ушир.с, 1H), 7,64 (ушир.д, J=7,5 Гц, 1H), 7,41 (с, 1H), 7,26 (м, 1H), 7,09 (тд, J=7,4, 1,0 Гц, 1H), 4,57 (м, 1H), 4,54 (с, 2H), 4,15 (ддд, J=12,3, 5,8, 3,8 Гц, 1H), 4,06 (ддд, J=12,3, 10,0, 5,5 Гц, 1H), 3,82 (дд, J=10,6, 4,3 Гц, 1H), 3,76 (дд, J=10,6, 5,1 Гц, 1H), 2,39-2,21 (м, 2H); элементный анализ: (C14H14IN3O4) C, H, N.
[0232] Перемешиваемую смесь йодида 153 (30,2 мг, 0,0727 ммоль), 4-(трифторметокси)фенилбороновой кислоты (20,8 мг, 0,101 ммоль) и Pd(dppf)Cl2 (2,3 мг, 3,14 мкмоль) в толуоле (1,7 мл) дегазировали в течение 4 минут (вакуумный насос) и затем вводили N2. Добавляли EtOH (0,6 мл) и 2М водный раствор Na2CO3 (0,30 мл, 0,60 ммоль) через шприц, полученную смесь перемешивали при 90°C в течение 20 минут, затем охлаждали, разбавляли водным раствором NaHCO3 (50 мл) и экстрагировали CH2Cl2 (4×50 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1-1,5% EtOAc/CH2Cl2 давало соединение 50 (30 мг, 92%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 117-119°C;
1H ЯМР (CDCl3) δ 7,60 (дт, J=8,7, 2,4 Гц, 2H), 7,52-7,47 (м, 2H), 7,44 (т, J=7,8 Гц, 1H), 7,40 (с, 1H), 7,32-7,26 (м, 3H), 4,67 (с, 2H), 4,59 (м, 1H), 4,14 (ддд, J=12,3, 5,7, 3,8 Гц, 1H), 4,05 (ддд, J=12,3, 10,0, 5,6 Гц, 1H), 3,86 (дд, J=10,7,4,3 Гц, 1H), 3,80 (дд, J=10,7, 5,0 Гц, 1H), 2,40-2,22 (м, 2H); элементный анализ: (C2lH18F3N305) C, H, N.
[0233] YY. Синтез 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 51 таблицы 1) при использовании способа схемы 11

[0234] Алкилирование оксазинового спирта 134 (см. пример 2BB выше) при использовании 4-иодбензилбромида (1,35 экв.) и NaH (1,5 экв.), как в примере 2UU выше, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 1-1,5% EtOAc/CH2Cl2 давали 7-{[(4-иодбензил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (154) (61%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 169-171°C;
1H ЯМР (CDCl3) δ 7,68 (дт, J=8,3, 2,0 Гц, 2H), 7,41 (с, 1H), 7,05 (ушир.д, J=8,3 Гц, 2H), 4,56 (м, 1H), 4,54 (с, 2H), 4,14 (ддд, J=12,3, 5,7, 3,8 Гц, 1H), 4,05 (ддд, J=12,3, 10,0, 5,6 Гц, 1H), 3,80 (дд, J=10,6, 4,3 Гц, 1H), 3,75 (дд, J=10,6, 5,0 Гц, 1H), 2,37-2,20 (м, 2H); HRFABMS вычислено для C14H15IN3O4 m/z [M+H]+ 416,0107, найдено 416,0105.
[0235] Реакция сочетания Сузуки йодида 154 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2XX выше, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1-1,5% EtOAc/CH2Cl2 давали соединение 51 (85%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 159-161°C;
1H ЯМР (CDCl3) δ 7,59 (дт, J=8,8, 2,5 Гц, 2H), 7,54 (ушир.д, J=8,2 Гц, 2H), 7,41 (с, 1H), 7,39 (ушир.д, J=8,3 Гц, 2H), 7,29 (ушир.д, J=8,0 Гц, 2H), 4,65 (с, 2H), 4,59 (м, 1H), 4,15 (ддд, J=12,3, 5,8, 3,8 Гц, 1H), 4,06 (ддд, J=12,3, 10,0, 5,6 Гц, 1H), 3,85 (дд, J=10,6, 4,3 Гц, 1H), 3,80 (дд, J=10,6, 5,1 Гц, 1H), 2,41-2,23 (м, 2H); элементный анализ: (C21H18F3N3O5) C, H, N.
[0236] ZZ. Синтез 7-метил-2-нитро-7-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 52 таблицы 1) при использовании способа схемы 11

[0237] Алкилирование оксазинового спирта 149 (см. пример 200) при использовании 3-(трифторметокси)бензилбромида (1,6 экв.) и NaH (2,0 экв.), как в примере 2UU выше, в течение 3 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1-1,5% EtOAc/CH2Cl2 давали соединение 52 (83%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 108-110°C;
1H ЯМР (CDCl3) δ 7,38 (с, 1H), 7,36 (т, J=8,0 Гц, 1H), 7,19-7,10 (м, 3H), 4,58 (с, 2H), 4,10 (ддд, J=12,5, 6,9, 5,9 Гц, 1H), 4,02 (ддд, J=12,5, 7,1, 5,9 Гц, 1H), 3,65 (д, J=10,2 Гц, 1H), 3,61 (д, J=10,2 Гц, 1H), 2,38 (ддд, J=14,4, 7,1, 6,0 Гц, 1H), 2,12 (ддд, J=14,5, 6,9, 6,0 Гц, 1H), 1,48 (с, 3H); элементный анализ: (C16H16F3N3O5) C, H, N.
[0238] AAA. Синтез 7-метил-2-нитро-7-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 53 таблицы 1) при использовании способа схемы 11

[0239] Алкилирование оксазинового спирта 149 (см. пример 200) при использовании 4-(трифторметокси)бензилбромида (1,6 экв.) и NaH (1,8 экв.), как в примере 2UU выше, в течение 3 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 1,5% EtOAc/CH2Cl2 давали соединение 53 (83%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 100-101°C;
1H ЯМР (CDCl3) δ 7,39 (с, 1H), 7,28 (ушир.д, J=8,7 Гц, 2H), 7,18 (ушир.д, J=8,0 Гц, 2H), 4,56 (с, 2H), 4,09 (ддд, J=12,5, 6,8, 5,9 Гц, 1H), 4,02 (ддд, J=12,5, 7,3, 5,9 Гц, 1H), 3,64 (д, J=10,2 Гц, 1H), 3,60 (д, J=10,2 Гц, 1H), 2,38 (ддд, J=14,5, 7,2, 5,9 Гц, 1H), 2,11 (ддд, J=14,5, 6,7, 6,0 Гц, 1H), 1,47 (с, 3H); элементный анализ: (C16H16N3O5) C, H, N.
[0240] BBB. Синтез 7-({[4-(бензилокси)бензил]окси}метил)-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]аксазина (соединение 54 таблицы 1) при использовании способа схемы 11

[0241] Алкилирование оксазинового спирта 149 (см. пример 200) при использовании 4-(бензилокси)бензилхлорида (2,8 экв.) и NaH (1,6 экв.), как в примере 2UU выше, в течение 7 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1% EtOAc/CH2Cl2 давали соединение 54 (41%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 109-111°C;
1H ЯМР (CDCl3) δ 7,45-7,29 (м, 6H), 7,16 (дт, J=8,6, 2,3 Гц, 2H), 6,93 (дт, J=8,6, 2,4 Гц, 2H), 5,06 (с, 2H), 4,48 (д, J=11,5 Гц, 1H), 4,45 (д, J=11,6 Гц, 1H), 4,03 (ддд, J=12,5, 7,6, 5,8 Гц, 1H), 3,95 (дт, J=12,5, 6,2 Гц, 1H), 3,58 (д, J=10,1 Гц, 1H), 3,54 (д, J=10,1 Гц, 1H), 2,34 (дт, J=14,5, 6,2 Гц, 1H), 2,08 (ддд, J=14,4, 7,6, 6,0 Гц, 1H), 1,45 (с, 3H); элементный анализ: (C22H23N3O5) C, H, N.
[0242] CCC. Синтез 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-3-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 55 таблицы 1) при использовании способа схемы 11

[0243] Алкилирование оксазинового спирта 149 (см. пример 200) при использовании 3-иодбензилбромида (1,6 экв.) и NaH (1,8 экв.), как в примере 2UU выше, в течение 3,5 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 0-2% EtOAc/CH2Cl2 давали 7-{[(3-иодбензил)окси]метил}-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (155) (69%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 122-125°C (разл.);
1H ЯМР (CDCl3) δ 7,63 (ушир.д, J=7,9 Гц, 1H), 7,58 (м, 1H), 7,40 (с, 1H), 7,19 (ушир.д, J=7,7 Гц, 1H), 7,06 (т, J=7,7 Гц, 1H), 4,49 (с, 2H), 4,11 (ддд, J=12,4, 7,3, 5,8 Гц, 1H), 4,01 (ддд, J=12,5, 6,7, 6,0 Гц, 1H), 3,63 (д, J=10,2 Гц, 1H), 3,60 (д, J=10,2 Гц, 1H), 2,37 (ддд, J=14,4, 6,7, 6,0 Гц, 1H), 2,12 (ддд, J=14,4, 7,3, 6,0 Гц, 1H), 1,47 (с, 3H); элементный анализ: (C15H16IN3O4) C, H, N.
[0244] Реакция сочетания Сузуки йодида 155 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2H, в течение 25 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1% EtOAc/CH2Cl2 давали соединение 55 (94%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 80-82°C;
1H ЯМР (CDCl3) δ 7,58 (дт, J=8,8, 2,5 Гц, 2H), 7,49 (ушир.дт, J=6A, 1,5 Гц, 1H), 7,43 (ушир.с, 1H), 7,41 (т, J=7,6 Гц, 1H), 7,37 (с, 1H), 7,30 (ушир.дд, J=8,7, 0,8 Гц, 2H), 7,25 (м, 1H), 4,62 (с, 2H), 4,11 (ддд, J=12,4, 7,1, 5,8 Гц, 1H), 4,00 (ддд, J=12,5, 6,9, 5,9 Гц, 1H), 3,68 (д, J=10,2 Гц, 1H), 3,63 (д, J=10,2 Гц, 1H), 2,39 (ддд, J=14,4, 6,9, 6,0 Гц, 1H), 2,12 (ддд, J=14,4, 7,1, 6,0 Гц, 1H), 1,48 (с, 3H); элементный анализ: (C22H20F3N3O5) C, H, N.
[0245] DDD. Синтез 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 56 таблицы 1) при использовании способа схемы 11

[0246] Алкилирование оксазинового спирта 149 (см. пример 200) при использовании 4-иодбензилбромида (1,7 экв.) и NaH (1,9 экв.), как в примере 2UU выше, в течение 3 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 1% EtOAc/CH2Cl2 давали 7-{[(4-иодбензил)окси]метил}-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (156) (54%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 130-132°C;
1H ЯМР (CDCl3) δ 7,67 (дт, J=8,3, 2,0 Гц, 2H), 7,39 (с, 1H), 6,99 (ушир.д, J=8,3 Гц, 2H), 4,49 (с, 2H), 4,09 (ддд, J=12,5, 7,0, 5,9 Гц, 1H), 4,01 (ддд, J=12,5, 7,1, 5,9 Гц, 1H), 3,62 (д, J=10,2 Гц, 1H), 3,58 (д, J=10,2 Гц, 1H), 2,37 (ддд, J=14,4, 7,0, 6,0 Гц, 1H), 2,10 (ддд, J=14,4, 6,9, 6,0 Гц, 1H), 1,46 (с, 3H); элементный анализ: (C15H16IN3O4) C, H, N.
[0247] Реакция сочетания Сузуки йодида 156 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2H, в течение 25 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% EtOAc/CH2Cl2 (головные фракции) и затем смесью 0,5-1% EtOAc/CH2Cl2 давали соединение 56 (92%) в виде кремового твердого вещества: т.пл. (CΗ2Cl2/пентан) 150-152°C;
1H ЯМР (CDCl3) δ 7,58 (дт, J=8,8, 2,5 Гц, 2H), 7,52 (дт, J=8,3, 1,9 Гц, 2H), 7,38 (с, 1H), 7,32 (ушир.д, J=8,3 Гц, 2H), 7,28 (ушир.дд, J=8,8, 0,8 Гц, 2H), 4,61 (д, J=12,0 Гц, 1H), 4,58 (д, J=12,0 Гц, 1H), 4,11 (ддд, J=12,4, 7,3, 5,8 Гц, 1H), 4,01 (ддд, J=12,5, 6,7, 6,0 Гц, 1H), 3,67 (д, J=10,2 Гц, 1H), 3,63 (д, J=10,2 Гц, 1H), 2,40 (ддд, J=14,5, 6,7, 6,0 Гц, 1H), 2,13 (ддд, J=14,5, 7,3, 6,0 Гц, 1H), 1,48 (с, 3H); элементный анализ:(C22H20F3N3O5) C, H, N.
[0248] EEE. Синтез (7R)-7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 57 таблицы 1) при использовании способа схемы 12

[0249] Ac2O (3,6 мл, 38,1 ммоль) добавляли к перемешиваемой суспензии спирта 149 (см. пример 200) (807 мг, 3,79 ммоль) в безводном пиридине (7,0 мл). После перемешивания при комнатной температуре в течение 38 часов смесь разбавляли CH2Cl2, добавляли в ледяную воду (150 мл) и экстрагировали CH2Cl2 (5×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1-6% EtOAc/CH2Cl2 давало 7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин-7-ил]метилацетат (157) (962 мг, 100%) в виде кремового твердого вещества: т.пл. (CΗ2Cl2/гексан) 145-147°C;
1H ЯМР (CDCl3) δ 7,44 (с, 1H), 4,27 (д, J=11,9 Гц, 1H), 4,20 (д, J=11,9 Гц, 1H), 4,14 (дт, J=12,7, 5,9 Гц, 1H), 4,08 (ддд, J=12,7, 8,3, 5,6 Гц, 1H), 2,32 (ддд, J=14,5, 8,3, 6,1 Гц, 1H), 2,10 (дт, J=14,5, 5,7 Гц, 1H), 2,09 (с, 3H), 1,50 (с, 3H); HRFABMS вычислено для C10H14N3O5 w/z [M+H]+ 256,0934, найдено 256,0941.
[0250] Рацемический ацетат 157 (990 мг) разделяли на чистые энантиомеры при помощи препаративной хиральной ВЭЖХ, используя колонку ChiralPak IA и изократную систему растворителя 40% EtOH в гексане, при скорости потока 6 мл/минута, с получением, во-первых, [(7S)-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин-7-ил]метилацетата (161) (427 мг, 43%) в виде кремового твердого вещества, которое использовали непосредственно на следующей стадии;
1H ЯМР (CDCl3) δ 7,44 (с, 1H), 4,27 (д, J=11,9 Гц, 1H), 4,20 (д, J=11,9 Гц, 1H), 4,14 (дт, J=12,7, 5,9 Гц, 1H), 4,08 (ддд, J=12,7, 8,3, 5,6 Гц, 1H), 2,32 (ддд, J=14,5, 8,3, 6,1 Гц, 1H), 2,10 (дт, J=14,5, 5,7 Гц, 1H), 2,09 (с, 3H), 1,50 (с, 3H); [α]D 26 -6,0° (c 1,00, CHCl3).
[0251] Описанная выше препаративная хиральная ВЭЖХ рацемического ацетата 157, во-вторых, давала [(7R)-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин-7-ил]метилацетат (158) (428 мг, 43%) в виде кремового твердого вещества, которое использовали непосредственно на следующей стадии;
1H ЯМР (CDCl3) δ 7,44 (с, 1H), 4,27 (д, J=11,9 Гц, 1H), 4,20 (д, J=11,8 Гц, 1H), 4,14 (дт, J=12,7, 5,9 Гц, 1H), 4,08 (ддд, J=12,7, 8,3, 5,6 Гц, 1H), 2,32 (ддд, J=14,5, 8,3, 6,1 Гц, 1H), 2,10 (дт, J=14,5, 5,7 Гц, 1H), 2,09 (с, 3H), 1,50 (с, 3H); [α]D 26 6,0° (c 1,00, CHCl3).
[0252] Воду (4 мл) добавляли по каплям к перемешиваемой смеси (R)-ацетата 158 (427 мг, 1,67 ммоль) и K2CO3 (256 мг, 1,85 ммоль) в MeOH (36 мл). После перемешивания при комнатной температуре в течение 4 часов смесь охлаждали на льду и обрабатывали 0,1M HCl (37 мл, 3,70 ммоль). Растворители удаляли при пониженном давлении и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% MeOH/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1-2,5% MeOH/CH2Cl2 давало [(7R)-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин-7-ил]метанол (159) (343 мг, 96%) в виде бледно-желтого твердого вещества, которое использовали непосредственно на следующей стадии;
1H ЯМР [(CD3)2SO] δ 8,03 (с, 1H), 5,23 (ушир.т, J=5,4 Гц, 1H), 4,13 (дт, J=13,0, 6,0 Гц, 1H), 4,05 (ддд, J=12,9, 8,1, 5,6 Гц, 1H), 3,54 (дд, J=11,6, 4,9 Гц, 1H), 3,48 (дд, J=11,6, 5,2 Гц, 1H), 2,21 (ддд, J=14,4, 8,1, 5,9 Гц, 1H), 2,00 (дт, J=14,4, 5,8 Гц, 1H), 1,32 (с, 3H); [α]D 27 -16,0° (c 1,00, ДМФА).
[0253] Алкилирование (R)-спирта 159 с использованием 4-бромбензилбромида (1,3 экв.) и NaH (1,5 экв.), как в примере 2UU выше, в течение 3 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 1% EtOAc/CH2Cl2 давали (7R)-7-{[(4-бромбензил)окси]метил}-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (160) (349 мг, 57%) в виде белого твердого вещества: т.пл. (CH2Cl2/гексан) 157-159°C;
1H ЯМР (CDCl3) δ 7,46 (дт, J=8,3, 2,0 Гц, 2H), 7,39 (с, 1H), 7,12 (ушир.д, J=8,3 Гц, 2H), 4,50 (с, 2H), 4,09 (ддд, J=12,5, 6,9, 6,0 Гц, 1H), 4,01 (ддд, J=12,5, 7,0, 6,0 Гц, 1H), 3,62 (д, J=10,2 Гц, 1H), 3,58 (д, J=10,2 Гц, 1H), 2,37 (ддд, J=14,4, 7,0, 6,0 Гц, 1H), 2,10 (ддд, J=14,4, 6,9, 6,1 Гц, 1H), 1,46 (с, 3H); [α]D 27 30,0° (c 1,00, CHCl3); HRFABMS вычислено для C15H17BrN3O4 m/z [M+H]+ 384,0382, 382,0402, найдено 384,0385, 382,0398.
[0254] Перемешиваемую смесь бромида 160 (347,5 мг, 0,909 ммоль), 4-(трифторметокси)фенилбороновой кислоты (283 мг, 1,37 ммоль) и Pd(dppf)Cl2 (101 мг, 0,138 ммоль) в толуоле (16 мл) и EtOH (6 мл) дегазировали в течение 10 минут (вакуумный насос) и затем вводили N2. Добавляли 2М водный раствор Na2CO3 (3,0 мл, 6,0 ммоль) через шприц, перемешиваемую смесь снова дегазировали в течение 10 минут и затем вводили N2. Полученную смесь перемешивали при 88°C в течение 75 минут, затем охлаждали, разбавляли водным раствором NaHCO3 (100 мл) и экстрагировали CH2Cl2 (6×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-0,5% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 0,5-1,5% EtOAc/CH2Cl2 давало соединение 57 (381 мг, 90%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 165-167°C;
1H ЯМР (CDCl3) δ 7,58 (дт, J=8,7, 2,4 Гц, 2H), 7,52 (ушир.д, J=8,2 Гц, 2H), 7,38 (с, 1H), 7,32 (ушир.д, J=8,1 Гц, 2H), 7,28 (ушир.д, J=8,1 Гц, 2H), 4,61 (д, J=12,1 Гц, 1H), 4,58 (д, J=12,1 Гц, 1H), 4,11 (ддд, J=12,4, 7,2, 5,8 Гц, 1H), 4,01 (ддд, J=12,6, 6,5, 6,1 Гц, 1H), 3,67 (д, J=10,2 Гц, 1H), 3,63 (д, J=10,2 Гц, 1H), 2,40 (ддд, J=14,4, 6,6, 6,1 Гц, 1H), 2,13 (ддд, J=14,5, 7,3, 6,0 Гц, 1H), 1,48 (с, 3H); [α]D 27 37,0° (c 1,00, CHCl3); элементный анализ: (C22H20F3N3O5) C, H, N.
[0255] FFF. Синтез (7S)-7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 58 таблицы 1) при использовании способа схемы 12

[0256] Гидролиз (S)-ацетата 161 (426 мг, 1,67 ммоль) при использовании K2CO3 в смеси MeOH/вода, как в примере 2EEE выше, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% MeOH/CH2Cl2 (головная фракция) и затем смесью 1-2,5% MeOH/CH2Cl2 давали [(7S)-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин-7-ил]метанол (162) (343 мг, 96%) в виде бледно-желтого твердого вещества, которое использовали непосредственно на следующей стадии;
1H ЯМР [(CD3)2SO] δ 8,03 (с, 1H), 5,22 (ушир.т, J=5,7 Гц, 1H), 4,13 (дт, J=13,0, 6,0 Гц, 1H), 4,05 (ддд, J=12,9, 8,1, 5,6 Гц, 1H), 3,54 (дд, J=11,6, 5,4 Гц, 1H), 3,48 (дд, J=11,6, 5,7 Гц, 1H), 2,21 (ддд, J=14,4, 8,1, 5,9 Гц, 1H), 2,00 (дт, J=14,4, 5,8 Гц, 1H), 1,32 (с, 3H); [α]D 27 18,0° (c 1,00, ДМФА).
[0257] Алкилирование (S)-спирта 162 при использовании 4-бромбензилбромида (1,35 экв.) и NaH (1,55 экв.), как в примере 2UU выше, в течение 3 часов, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 (головные фракции) и затем смесью 1% EtOAc/CH2Cl2 давали (7S)-7-{[(4-бромбензил)окси]метил}-7-метил-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (163) (373 мг, 61%) в виде белого твердого вещества: т.пл. (CΗ2Cl2/гексан) 159-161°C;
1H ЯМР (CDCl3) δ 7,46 (дт, J=8,4, 2,1 Гц, 2H), 7,39 (с, 1H), 7,12 (дт, J=8,4, 2,1 Гц, 2H), 4,50 (с, 2H), 4,09 (ддд, J=12,5, 7,0, 5,8 Гц, 1H), 4,01 (ддд, J=12,5, 7,1, 5,9 Гц, 1H), 3,62 (д, J=10,2 Гц, 1H), 3,58 (д, J=10,2 Гц, 1H), 2,37 (ддд, J=14,4, 7,1,5,9 Гц, 1H), 2,10 (ддд, J=14,5, 7,0, 5,9 Гц, 1H), 1,46 (с, 3H); [α]D 27 -32,0° (c 1,00, CHCl3); HRFABMS вычислено для C15H17BrN3O4 m/z [M+H]+ 384,0382, 382,0402, найдено 384,0374, 382,0393.
[0258] Реакция сочетания Сузуки бромида 163 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2EEE, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,5% EtOAc/CH2Cl2 (головные фракции) и затем смесью 0,5-1% EtOAc/CH2Cl2 давали соединение 58 (415 мг, 92%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 162-164°C;
1H ЯМР (CDCl3) δ 7,58 (дт, J=8,8, 2,5 Гц, 2H), 7,52 (ушир.д, J=8,3 Гц, 2H), 7,38 (с, 1H), 7,32 (ушир.д, J=8,3 Гц, 2H), 7,28 (ушир.дд, J=8,8, 0,8 Гц, 2H), 4,61 (д, J=12,1 Гц, 1H), 4,58 (д, J=12,1 Гц, 1H), 4,11 (ддд, J=12,4, 7,3, 5,8 Гц, 1H), 4,01 (ддд, J=12,5, 6,6, 6,1 Гц, 1H), 3,67 (д, J=10,2 Гц, 1H), 3,63 (д, J=10,2 Гц, 1H), 2,40 (ддд, J=14,4, 6,7, 6,0 Гц, 1H), 2,13 (ддд, J=14,4, 7,3, 6,0 Гц, 1H), 1,48 (с, 3H), [α]D 27 -36,0° (c 1,00, CHCl3); элементный анализ: (C22H20F3N3O5) C, H, N.
[0259] GGG. Синтез 2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 59 таблицы 1) при использовании способа схемы 13

[0260] Раствор йода (1,49 г, 5,85 ммоль) в безводном CH2Cl2 (3×10 мл, затем 4×1 мл для промывки) добавляли по каплям к перемешиваемой смеси имидазола (0,441 г, 6,48 ммоль) и трифенилфосфина (1,50 г, 5,71 ммоль) в безводном CH2Cl2 (3 мл) при 0°C в атмосфере N2. После перемешивания при 0°C в течение 30 минут добавляли раствор 2-({[трет-бутил(диметил)силил]окси}метил)-2-пропен-1-ола (164) (описано Chen et at., US 2007213341 A1, путем моносилилирования 2-метилен-1,3-пропандиола) (1,00 г, 4,94 ммоль) в безводном CH2Cl2 (4 мл, затем 4×1 мл для промывки) и смесь перемешивали при 0-8°C в течение 5 часов. Полученную смесь осторожно концентрировали при пониженном давлении и остаточное масло хроматографировали на силикагеле. Элюирование пентаном сначала давало головные фракции, и последующее элюирование смесью 10% CH2Cl2/пентан давало трет-бутил(диметил)силил-2-(иодметил)-2-пропениловый эфир (165) (1,46 г, 95%) в виде летучего розового масла, которое использовали непосредственно на следующей стадии;
1H ЯМР (CDCl3) δ 5,31 (ушир.с, 1H), 5,19 (д, J=1,3 Гц, 1H), 4,31 (с, 2H), 3,95 (с, 2H), 0,92 (с, 9H), 0,10 (с, 6H).
[0261] Смесь йодида 165 (4,63 г, 14,8 ммоль), 4-(трифторметокси)фенола (3,10 мл, 23,9 ммоль) и порошкообразного K2CO3 (3,56 г, 25,8 ммоль) в ацетоне (10 мл) перемешивали при 50°C в течение 11 часов. Полученную охлажденную смесь разбавляли ледяной водой (100 мл) и экстрагировали CH2Cl2 (4×100 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-3% CH2Cl2/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 5-10% CH2Cl2/петролейный эфир давало трет-бутил(диметил)[(2-{[4-(трифторметокси)фенокси]метил}-2-пропенил)окси]силан (166) (3,12 г, 58%) в виде бесцветного масло;
1H ЯМР (CDCl3) δ 7,12 (ушир.д, J=9,0 Гц, 2H), 6,90 (дт, J=9,1, 2,9 Гц, 2H), 5,27 (д, J=0,7 Гц, 1H), 5,20 (д, J=1,1 Гц, 1H), 4,54 (с, 2H), 4,24 (с, 2H), 0,91 (с, 9H), 0,07 (с, 6H); HRFABMS вычислено для C17H26F3O3Si m/z [M+H]+ 363,1603, найдено 363,1604.
[0262] Раствор йода (825 мг, 3,25 ммоль) в безводном ТГФ (5 мл, затем 2×3 мл для промывки) добавляли по каплям (в течение 70 минут) к перемешиваемой смеси алкена 166 (5,21 г, 14,4 ммоль) и порошкообразного NaBH4 (257 мг, 6,79 ммоль) в безводном ТГФ (18 мл) при 0°C в атмосфере N2. После перемешивания при 0°C в течение 3 часов и затем при комнатной температуре в течение 13 часов смесь снова охлаждали до 0°C, обрабатывали 30% раствором H2O2 (6,8 мл) и 3н. раствором NaOH (6,8 мл) и затем перемешивали при комнатной температуре в течение 3 часов. Затем добавляли воду (160 мл) и смесь экстрагировали EtOAc (4×160 мл). Экстракты промывали насыщенным раствором соли (80 мл), упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-3,5% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 4-8% EtOAc/петролейный эфир давало 3-{[трет-бутил(диметил)силил]окси}-2-{[4-трифторметокси)фенокси]метил}-1-пропанол (168) (3,32 г, 61%) в виде бесцветного масла;
1H ЯМР (CDCl3) δ 7,13 (ушир.д, J=9,0 Гц, 2H), 6,89 (дт, J=9,1, 3,0 Гц, 2H), 4,08 (дд, J=9,3, 6,6 Гц, 1H), 4,04 (дд, J=9,3, 6,0 Гц, 1H), 3,94-3,81 (м, 4H), 2,36 (дд, J=6,1, 5,2 Гц, 1H), 2,19 (м, 1H), 0,89 (с, 9H), 0,07, 0,05 (2с, 6H); HRFABMS вычислено для C17H28F3O4Si m/z [M+H]+ 381,1709, найдено 381,1707.
[0263] Раствор йода (2,89 г, 11,4 ммоль) в безводном CH2Cl2 (6×10 мл, затем 5 мл+2 мл для промывки) добавляли по каплям (в течение 100 минут) к перемешиваемой смеси спирта 168 (3,28 г, 8,62 ммоль), имидазола (1,50 г, 22,0 ммоль) и трифенилфосфина (2,83 г, 10,8 ммоль) в безводном CH2Cl2 (20 мл) в атмосфере N2. После перемешивания при комнатной температуре в течение 15 часов полученную смесь концентрировали при пониженном давлении и остаток хроматографировали на силикагеле. Элюирование петролейным эфиром сначала давало головные фракции, и последующее элюирование смесью 5-20% CH2Cl2/петролейный эфир давало трет-бутил(3-иод-2-{[4-(трифторметокси)фенокси]метил}пропокси)диметилсилан (170) (3,90 г, 92%) в виде бледно-коричневого масла;
1H ЯМР (CDCl3) δ 7,13 (ушир.дд, J=9,1, 0,7 Гц, 2H), 6,89 (дт, J=9,1, 3,0 Гц, 2H), 4,01 (дд, J=9,3, 5,7 Гц, 1H), 3,94 (дд, J=9,3, 6,2 Гц, 1H), 3,75 (дд, J=10,1, 5,6 Гц, 1H), 3,70 (дд, J=10,1, 5,6 Гц, 1H), 3,39 (дд, J=10,1, 5,9 Гц, 1H), 3,37 (дд, J=10,1, 6,0 Гц, 1H), 2,10 (септ., J=5,8 Гц, 1H), 0,89 (с, 9H), 0,07, 0,06 (2с, 6H); HRCIMS вычислено для C17H27F3IO3Si m/z [M+H]+ 491,0726, найдено 491,0721.
[0264] Смесь йодида 170 (3,89 г, 7,93 ммоль), 2-бром-4(5)-нитроимидазола (80) (1,68 г, 8,77 ммоль) и порошкообразного K2CO3 (1,90 г, 13,7 ммоль) в безводном ДМФА (20 мл) перемешивали при 84-88°C в течение 37 часов. Полученную охлажденную смесь разбавляли ледяной водой (100 мл) и экстрагировали EtOAc (5×100 мл). Экстракты промывали насыщенным раствором соли (100 мл), снова экстрагировали EtOAc (50 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-7% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 7-15% EtOAc/петролейный эфир давало 2-бром-1-(3-{[трет-бутил(диметил)силил]окси}-2-{[4-(трифторметокси)фенокси]метил}пропил)-4-нитро-1H-имидазол (172) (3,57 г, 81%) в виде бледно-желтого масла;
1H ЯМР (CDCl3) δ 7,82 (с, 1H), 7,16 (ушир.дд, J=9,1, 0,7 Гц, 2H), 6,85 (дт, J=9,1, 3,0 Гц, 2H), 4,26 (д, J=7,1 Гц, 2H), 3,96 (д, J=5,6 Гц, 2H), 3,77 (дд, J=10,6, 5,1 Гц, 1H), 3,67 (дд, J=10,6, 4,7 Гц, 1H), 2,51 (м, 1H), 0,92 (с, 9H), 0,08, 0,07 (с, 6H); HRFABMS вычислено для C20H28BrF3N3O5Si m/z [M+H]+ 556,0913, 554,0934, найдено 556,0921, 554,0938.
[0265] Силиловый эфир 172 (3,42 г, 6,17 ммоль) обрабатывали раствором 1% HCl в 95% EtOH (условия десилилирования описаны Cunico et al., 1980) (31 мл) и смесь перемешивали при комнатной температуре в течение 12 часов. Полученный раствор охлаждали (CO2/ацетон), нейтрализовали добавлением по каплям 7M NH3 в MeOH (6,6 мл) при перемешивании, затем концентрировали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-30% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 40-50% EtOAc/петролейный эфир давало 3-(2-бром-4-нитро-1Н-имидазол-1-ил)-2-{[4-(трифторметокси)фенокси]метил}-1-пропанол (174) (2,48 г, 91%) в виде белого твердого вещества: т.пл. (CH2Cl2/пентан) 97-99°C;
1H ЯМР (CDCl3) δ 7,88 (с, 1H), 7,17 (ушир.д, J=8,5 Гц, 2H), 6,88 (дт, J=9,1, 3,0 Гц, 2H), 4,34 (дд, J=14,4, 7,4 Гц, 1H), 4,3l (дд, J=14,4, 7,1 Гц, 1H), 4,06 (дд, J=9,6, 5,7 Гц, 1H), 4,03 (дд, J=9,6, 4,8 Гц, 1H), 3,88 (дт, J=10,8, 4,4 Гц, 1H), 3,76 (дт, J=10,8, 4,9 Гц, 1H), 2,54 (м, 1H), 1,72 (т, J=4,4 Гц, 1H); HRFABMS вычислено для C14H14BrF3N3O5 m/z [M+H]+ 442,0049, 440,0069, найдено 442,0053, 440,0063.
[0266] Перемешиваемый раствор спирта 174 (2,48 г, 5,64 ммоль) в безводном ДМФА (50 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (345 мг, 8,63 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 4 часов реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (30 мл), добавляли к насыщенному раствору соли (200 мл) и экстрагировали CH2Cl2 (8×100 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1,5-2% EtOAc/CH2Cl2 давало соединение 59 (1,407 г, 69%) в виде бледно-желтого твердого вещества: т.пл. (CH2Cl2/гексан) 141-143°C;
1H ЯМР (CDCl3) δ 7,45 (с, 1H), 7,17 (ушир.дд, J=9,1, 0,7 Гц, 2H), 6,88 (дт, J=9,2, 3,0 Гц, 2H), 4,62 (ддд, J=11,5, 3,2, 0,8 Гц, 1H), 4,50 (дд, J=11,6, 7,3 Гц, 1H), 4,27 (ддд, J=12,5, 5,6, 0,7 Гц, 1H), 4,17 (дд, J=12,4, 7,1 Гц, 1H), 4,13 (дд, J=9,6, 5,7 Гц, 1H), 4,07 (дд, J=9,6, 6,7 Гц, 1H), 2,88 (м, 1H); элементный анализ: (C14H12F3N3O5) C, H, N.
[0267] HHH. Синтез (6R)-2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 60 таблицы 1) при использовании способа схемы 13

[0268] Рацемический эфир 59 (1,18 г) разделяли на чистые энантиомеры препаративной хиральной ВЭЖХ, используя колонку ChiralPak IA и изократную систему растворителя 27% EtOH в гексане, с получением, во-первых, соединения 60 (510 мг, 43%) в виде белого твердого вещества: т.пл. (CH2Cl2/гексан) 138-139°C;
1H ЯМР (CDCl3) δ 7,45 (с, 1H), 7,17 (ушир.дд, J=9,0, 0,6 Гц, 2H), 6,88 (дт, J=9,2, 3,0 Гц, 2H), 4,62 (ддд, J=11,5, 3,2, 0,7 Гц, 1H), 4,50 (дд, J=11,5, 7,3 Гц, 1H), 4,27 (ушир.дд, J=12,4, 5,6 Гц, 1H), 4,17 (дд, J=12,4, 7,0 Гц, 1H), 4,13 (дд, J=9,6, 5,7 Гц, 1H), 4,07 (дд, J=9,6, 6,7 Гц, 1H), 2,88 (м, 1H); [α]26 14° {c, 1,00, CHCl3); элементный анализ: (C14H12F3N3O5) C, H, N.
[0269] III. Синтез (6S)-2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 61 таблицы 1) при использовании способа схемы 13

[0270] Препаративная хиральная ВЭЖХ эфира 59 (см. пример 2HHH выше) вторично давала соединение 61 (509 мг, 43%) в виде белого твердого вещества: т.пл. (CH2Cl2/гексан) 139-140°C;
1H ЯМР (CDCl3) δ 7,45 (с, 1H), 7,17 (ушир.дд, J=9,1, 0,6 Гц, 2H), 6,88 (дт, J=9,1, 3,0 Гц, 2H), 4,62 (ддд, J=11,5, 3,2, 0,6 Гц, 1H), 4,50 (дд, J=11,5, 7,3 Гц, 1H), 4,27 (ушир.дд, J=12,4, 5,2 Гц, 1H), 4,17 (дд, J=12,5, 7,1 Гц, 1H), 4,13 (дд, J=9,6, 5,7 Гц, 1H), 4,07 (дд, J=9,6, 6,7 Гц, 1H), 2,88 (м, 1H); [α]26 -14° (c, 1,00, CHCl3); элементный анализ: (C14H12F3N3O5) C, H, N.
[0271] JJJ. Синтез 6-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 62 таблицы 1) при использовании способа схемы 13

[0272] Алкилирование 4-иодфенола при использовании йодида 165 (см. пример 2GGG) и K2CO3, как в примере 2GGG выше, в течение 6 часов, с последующей хроматографией продукта на силикагеле при элюировании петролейным эфиром (головные фракции) и затем смесью 5% CH2Cl2/петролейный эфир давали трет-бутил({2-[(4-иодфенокси)метил]-2-пропенил}окси)диметилсилан (167) (94%) в виде масла;
1H ЯМР (CDCl3) δ 7,54 (дт, J=8,9, 2,7 Гц, 2H), 6,70 (дт, J=8,9, 2,7 Гц, 2H), 5,25 (д, J=1,0 Гц, 1H), 5,19 (д, J=1,2 Гц, 1H), 4,51 (с, 2H), 4,23 (с, 2H), 0,91 (с, 9H), 0,07 (2с, 6H); HRFABMS вычислено для C16H26IO2Si m/z [M+H]+ 405,0747, найдено 405,0739.
[0273] Раствор йода (282 мг, 1,11 ммоль) в безводном ТГФ (1,5 мл, затем 2×0,75 мл для промывки) добавляли по каплям (в течение 40 минут) к перемешиваемой смеси алкена 167 (1,71 г, 4,23 ммоль) и порошкообразного NaBH4 (90 мг, 2,38 ммоль) в безводном ТГФ (5,5 мл) при 0°C в атмосфере N2. После перемешивания при 0°C в течение 4 часов и затем при комнатной температуре в течение 13 часов смесь снова охлаждали до 0°C, обрабатывали 30% раствором H2O2 (2,4 мл) и 3н. раствором NaOH (2,4 мл) и затем перемешивали при комнатной температуре в течение 3 часов. Затем добавляли воду (50 мл) и смесь экстрагировали EtOAc (4×50 мл). Экстракты промывали насыщенным раствором соли (50 мл), упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-2% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 4-5% EtOAc/петролейный эфир давало 3-{[трет-бутил(диметил)силил]окси}-2-[(4-иодфенокси)метил]-1-пропанол (169) (1,26 г, 71%) в виде бледно-желтого масла;
1H ЯМР (CDCl3) δ 7,55 (дт, J=9,0, 2,7 Гц, 2H), 6,69 (дт, J=9,0, 2,7 Гц, 2H), 4,06 (дд, J=9,3, 6,7 Гц, 1H), 4,01 (дд, J=9,3, 5,9 Гц, 1H), 3,93-3,80 (м, 4H), 2,36 (дд, J=6,3, 5,1 Гц, 1H), 2,17 (септ., J=5,4 Гц, 1H), 0,89 (с, 9H), 0,06, 0,05 (2с, 6H); HRFABMS вычислено для C16H28IO3Si m/z [M+H]+ 423,0853, найдено 423,0849.
[0274] Йодирование спирта 169 при использовании I2, PPh3 и имидазола, как в примере 2GGG выше, в течение 12 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-5% CH2Cl2/петролейный эфир (головные фракции) и затем смесью 5-10% CH2Cl2/петролейный эфир давали трет-бутил{3-иод-2-[(4-иодфенокси)метил]пропокси}диметилсилан (171) (94%) в виде бесцветного масла;
1H ЯМР (CDCl3) δ 7,55 (дт, J=9,0, 2,7 Гц, 2H), 6,68 (дт, J=9,0, 2,7 Гц, 2H), 3,98 (дд, J=9,4, 5,7 Гц, 1H), 3,92 (дд, J=9,4, 6,2 Гц, 1H), 3,74 (дд, J=10,1, 5,6 Гц, 1H), 3,69 (дд, J=10,1, 5,6 Гц, 1H), 3,38 (ддJ=10,0, 5,9 Гц, 1H), 3,35 (дд, J=10,0, 6,1 Гц, 1H), 2,09 (септ., J=5,8 Гц, 1H), 0,89 (с, 9H), 0,06 (2с, 6H); HRFABMS вычислено для C16H27I2O2Si m/z [M+H]+ 532,9870, найдено 532,9864.
[0275] Алкилирование 2-бром-4(5)-нитроимидазола (80) с использованием йодида 171 и K2CO3, как в примере 2GGG выше, в течение 33 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-7% EtOAc/петролейный эфир (головные фракции) и затем смесью 8-15% EtOAc/петролейный эфир давали 2-бром-1-{3-{[трет-бутил(диметил)силил]окси}-2-[(4-иодфенокси)метил]пропил}-4-нитро-1H-имидазол (173) (80%) в виде белого твердого вещества: т.пл. (CH2Cl2/пентан) 81-83°C;
1H ЯМР (CDCl3) δ 7,81 (с, 1H), 7,57 (дт, J=9,0, 2,7 Гц, 2H), 6,64 (дт, J=9,0, 2,6 Гц, 2H), 4,24 (д, J=7,1 Гц, 1H), 3,93 (д, J=5,6 Гц, 1H), 3,76 (дд, J=10,6, 5,1 Гц, 1H), 3,66 (дд, J=10,6, 4,7 Гц, 1H), 2,50 (м, 1H), 0,91 (с, 9H), 0,07 (2с, 6H); HRFABMS вычислено для C19H28BrIN3O4Si m/z [M+H]+ 598,0057, 596,0077, найдено 598,0070, 596,0082.
[0276] Гидролиз силилового эфира 173 при использовании 1% HCl в 95% EtOH, как в примере 2GGG выше, в течение 7 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-30% EtOAc/петролейный эфир (головные фракции) и затем смесью 40-50% EtOAc/петролейный эфир давали 3-(2-бром-4-нитро-1Н-имидазол-1-ил)-2-[(4-иодфенокси)метил]-1-пропанол (175) (86%) в виде белого твердого вещества: т.пл. (CH2Cl2/пентан) 109-111°C;
1H ЯМР (CDCl3) δ 7,87 (с, 1H), 7,58 (дт, J=9,0, 2,7 Гц, 2H), 6,66 (дт, J=9,0, 2,7 Гц, 2H), 4,33 (дд, J=14,4, 7,3 Гц, 1H), 4,29 (дд, J=14,4, 7,1 Гц, 1H), 4,03 (дд, J=9,6, 5,7 Гц, 1H), 4,00 (дд, J=9,6, 7,8 Гц, 1H), 3,86 (ддд, J=10,9, 4,6, 4,3 Гц, 1H), 3,75 (дт, J=10,8, 4,9 Гц, 1H), 2,52 (м, 1H), 1,72 (т, J=4,4 Гц, 1H); HRFABMS вычислено для C13H14BrIN3O4 m/z [M+H]+ 483,9192, 481,9212, найдено 483,9200, 481,9211.
[0277] Замыкание кольца спирта 175 при помощи NaH, как в примере 2GGG, в течение 5 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1,5-2% EtOAc/CH2Cl2 давали 6-[(4-иодфенокси)метил]-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (176) (78%) в виде бледно-желтого твердого вещества: т.пл. (CH2Cl2/пентан, растирание) 239-240°C;
1H ЯМР [(CD3)2SO] δ 8,09 (с, 1H), 7,60 (дт, J=9,0, 2,7 Гц, 2H), 6,82 (дт, J=9,0, 2,7 Гц, 2H), 4,59 (дд, J=10,9, 2,9 Гц, 1H), 4,44 (дд, J=11,0, 7,2 Гц, 1H), 4,28 (дд, J=12,5, 5,4 Гц, 1H), 4,09 (дд, J=10,0, 6,7 Гц, 1H), 4,06 (дд, J=10,0, 6,7 Гц, 1H), 4,03 (дд, J=12,5, 7,0 Гц, 1H), 2,82 (м, 1H); элементный анализ: (C13H12IN3O4) C, H, N.
[0278] Реакция сочетания Сузуки йодида 176 и 4-фторфенилбороновой кислоты, как в примере 2CC выше, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1-2% EtOAc/CH2Cl2 давали соединение 62 (92%) в виде бледно-розового твердого вещества: т.пл. (CH2Cl2/пентан) 201-203°C;
1H ЯМР [(CD3)2SO] δ 8,11 (с, 1H), 7,65 (дт, J=8,9, 2,7 Гц, 2H), 7,64 (дт, J=8,8, 2,7 Гц, 2H), 7,59 (дт, J=8,8, 2,5 Гц, 2H), 7,25 (тт, 8,9, 2,7 Гц, 2H), 7,05 (дт, J=8,8, 2,6 Гц, 2H), 4,63 (дд, J=10,9, 2,9 Гц, 1H), 4,48 (дд, J=11,0, 7,3 Гц, 1H), 4,31 (дд, J=12,5, 5,4 Гц, 1H), 4,16 (дд, J=10,0, 6,7 Гц, 1H), 4,12 (дд, J=10,0, 6,7 Гц, 1H), 4,07 (дд, J=12,6, 7,0 Гц, 1H), 2,86 (м, 1H); элементный анализ: (C19H16FN3O4) C, H, N.
[0279] KKK. Синтез 2-нитро-6-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 63 таблицы 1) при использовании способа схемы 13

[0280] Реакция сочетания Сузуки йодида 176 (см. пример 2JJJ выше) и 4-(трифторметил)фенилбороновой кислоты, как в примере 2CC выше, с последующей хроматографией продукта на силикагеле при элюировании 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1-2% EtOAc/CH2Cl2 давали соединение 63 (90%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 218-221°C;
1H ЯМР [(CD3)2SO] δ 8,11 (с, 1H), 7,85 (ушир.д, J=8,2 Гц, 2H), 7,77 (ушир.д, J=8,3 Гц, 2H), 7,71 (дт, J=8,8, 2,5 Гц, 2H), 7,10 (дт, J=8,8, 2,5 Гц, 2H), 4,63 (дд, J=10,9, 2,9 Гц, 1H), 4,48 (дд, J=11,0, 7,2 Гц, 1H), 4,32 (дд, J=12,5, 5,5 Гц, 1H), 4,18 (дд, J=10,0, 6,7 Гц, 1H), 4,15 (дд, J=10,0, 6,7 Гц, 1H), 4,08 (дд, J=12,6, 7,0 Гц, 1H), 2,87 (м, 1H); элементный анализ: (C20H16F3N3O4) C, H, N.
[0281] LLL. Синтез 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 64 таблицы 1) при использовании способа схемы 13

[0282] Реакция сочетания Сузуки йодида 176 (см. пример 2JJJ выше) и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2CC выше, с последующей хроматографией продукта на силикагеле при элюировании CH2Cl2 давали соединение 64 (93%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 192-194°C;
1H ЯМР (CDCl3) δ 7,54 (дт, J=8,7, 2,4 Гц, 2H), 7,50 (дт, J=8,7, 2,5 Гц, 2H), 7,46 (с, 1H), 7,26 (м, 2H), 6,96 (дт, J=8,7, 2,4 Гц, 2H), 4,63 (дд, J=11,5, 3,1 Гц, 1H), 4,52 (дд, J=11,5, 7,4 Гц, 1H), 4,28 (дд, J=12,4, 5,6 Гц, 1H), 4,20 (м, 1H), 4,18 (дд, J=10,0, 5,7 Гц, 1H), 4,12 (дд, J=9,7, 6,7 Гц, 1H), 2,91 (м, 1H); элементный анализ: (C20Hl6F3N3O5) C, H, N.
[0283] MMM. Синтез 6-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 65 таблицы 1) при использовании способа схемы 14

[0284] Смесь 2-бром-4(5)-нитроимидазола (80) (3,373 г, 17,6 ммоль), 6-(иодметил)-2,2,3,3,9,9,10,10-октаметил-4,8-диокса-3,9-дисилаундекана (177) (описано Curran et al., 1998 в 4 стадии, исходя из 2-метилен-1,3-пропандиола) (6,79 г, 15,3 ммоль) и порошкообразного K2CO3 (5,10 г, 36,9 ммоль) в безводном ДМФА (40 мл) в атмосфере N2 перемешивали при 82°C в течение 24 часов. Полученную охлажденную смесь добавляли в ледяную воду (200 мл) и экстрагировали EtOAc (3×200 мл). Экстракты промывали водой (200 мл), снова экстрагировали EtOAc (200 мл) и затем снова промывали насыщенным раствором соли (150 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-2% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 3-5% EtOAc/петролейный эфир давало 2-бром-1-[3-{[трет-бутил(диметил)силил]окси}-2-({[трет-бутил(диметил)силил]окси}метил)пропил]-4-нитро-1Н-имидазол (178) (7,35 г, 95%) в виде белого твердого вещества: т.пл. (пентан) 51-53°C;
1H ЯМР (CDCl3) δ 7,83 (с, 1H), 4,12 (д, J=7,2 Гц, 2H), 3,61 (дд, J=10,4, 5,5 Гц, 2H), 3,56 (дд, J=10,4, 5,0 Гц, 2H), 2,15 (м, 1H), 0,91 (с, 18H), 0,07 (2с, 2× 6H); элементный анализ: (C19H38BrN3O4Si2) C, H, N.
[0285] Суспензию силилового эфира 178 (7,35 г, 14,5 ммоль) в 1% растворе HCl в 95% EtOH (условия десилилирования описаны Cunico et al., 1980) (150 мл) перемешивали при комнатной температуре в течение 4 часов и затем выдерживали при 4°C в течение 12 часов. Полученный раствор охлаждали (CO2/ацетон), нейтрализовали добавлением по каплям 7M NH3 в MeOH (9,8 мл) при перемешивании, затем концентрировали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 33-75% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 75% EtOAc/петролейный эфир и EtOAc давало 2-[(2-бром-4-нитро-1H-имидазол-1-ил)метил]-1,3-пропандиол (179) (3,42, 85%) в виде белого твердого вещества: т.пл. (MeOΗ/CΗ2Cl2/гексан) 110-112°C;
1H ЯМР [(CD3)2SO] δ 8,50 (с, 1H), 4,65 (т, J=5,0 Гц, 2H), 4,07 (д, J=7,3 Гц, 2H), 3,41 (м, 4H), 2,06 (м, 1H); элементный анализ: (C7H10BrN3O4) C, H, N.
[0286] Перемешиваемый раствор диола 179 (3,44 г, 12,3 ммоль) в безводном ДМФА (30 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (1,72 г, 43,0 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 3,5 часов реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NH4Cl (20 мл) и водным раствором NaHCO3 (20 мл), добавляли к насыщенному раствору соли (150 мл) и экстрагировали CH2Cl2 (3×150 мл), 10% MeOH/CH2Cl2 (6×150 мл) и EtOAc (15×150 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-2% MeOH/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 2-3% MeOH/CH2Cl2 давало неочищенный продукт (1,88 г), который затем хроматографировали на силикагеле. Элюирование смесью 50-90% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 90% EtOAc/петролейный эфир и EtOAc давало (2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин-6-ил)метанол (180) (1,649 г, 67%) в виде бледно-желтого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 130-131°C;
1H ЯМР [(CD3)2SO] δ 8,06 (с, 1H), 4,96 (т, J=5,1 Гц, 1H), 4,49 (ддд, J=10,9, 3,3, 0,9 Гц, 1H), 4,30 (дд, J=10,9, 7,9 Гц, 1H), 4,15 (ддд, J=12,5, 5,4, 0,8 Гц, 1H), 3,90 (дд, J=12,5, 7,7 Гц, 1H), 3,47 (м, 2H), 2,40 (м, 1H); элементный анализ: (C7H9N3O4) C, H, N.
[0287] Алкилирование оксазинового спирта 180 при использовании 5-бром-2-фторпиридина (91) (2,0 экв.) и NaH (1,74 экв.), как в примере 200, в течение 3 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-0,25% MeOH/CH2Cl2 (головные фракции) и затем смесью 0,25-0,5% MeOH/CH2Cl2 давали 6-{[(5-бром-2-пиридинил)окси]метил}-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (181) (67%) в виде белого твердого вещества: т.пл. (MeOΗ/CΗ2Cl2/гексан) 233-235°C;
1H ЯМР [(CD3)2SO] δ 8,27 (дд, J=2,5, 0,4 Гц, 1H), 8,07 (с, 1H), 7,93 (дд, J=8,8, 2,6 Гц, 1H),6,85 (дд, J=8,9, 0,5 Гц, 1H), 4,60 (дд, J=11,0, 2,7 Гц, 1H), 4,44 (дд, J=11,1, 7,4 Гц, 1H), 4,36 (дд, J=11,0, 6,7 Гц, 1H), 4,33 (дд, J=11,0, 6,7 Гц, 1H), 4,27 (дд, J=12,5, 5,4 Гц, 1H), 4,04 (дд, J=12,6, 7,1 Гц, 1H), 2,85 (м, 1H); элементный анализ: (C12H11BrN4O4) C, H, N.
[0288] Реакция сочетания Сузуки бромида 181 и 4-фторфенилбороновой кислоты (2,0 экв.), как в примере 2M, в течение 2,5 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-5% EtOAc/CH2Cl2 (головные фракции) и затем смесью 5-6% EtOAc/CH2Cl2 давали соединение 65 (93%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 160-161°C;
1H ЯМР (CDCl3) δ 8,29 (дд, J=2,5, 0,6 Гц, 1H), 7,78 (дд, J=8,8, 2,5 Гц, 1H), 7,46 (ддт, J=8,9, 5,2, 2,6 Гц, 2H), 7,44 (с, 1H), 7,14 (тт, J=6,5, 2,6 Гц, 2H), 6,83 (дд, J=8,6, 0,7 Гц, 1H), 4,64 (ддд, J=11,4, 3,3, 1,0 Гц, 1H), 4,54 (дд, J=11,3, 6,2 Гц, 1H), 4,49 (дд, J=11,3, 6,5 Гц, 1H), 4,45 (дд, J=11,5, 7,9 Гц, 1H), 4,26 (ддд, J=12,4, 5,6, 0,9 Гц, 1H), 4,10 (дд, J=12,4, 7,7 Гц, 1H), 2,94 (м, 1H); элементный анализ: (C18H15FN4O4) C, H, N.
[0289] NNN. Синтез 2-нитро-6-[({5-[4-(трифторметил)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 66 таблицы 1) при использовании способа схемы 14

[0290] Реакция сочетания Сузуки бромида 181 (см. пример 2MMM) и 4-(трифторметил)фенилбороновой кислоты, как в примере 2M, в течение 130 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-4% EtOAc/CH2Cl2 (головные фракции) и затем смесью 5-6% EtOAc/CH2Cl2 давали соединение 66 (94%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 180-182°C;
1H ЯМР (CDCl3) δ 8,38 (дд, J=2,5, 0,5 Гц, 1H), 7,87 (дд, J=8,6, 2,6 Гц, 1H), 7,73 (ушир.д, J=8,2 Гц, 2H), 7,64 (ушир.д, J=8,1 Гц, 2H), 7,48 (с, 1H), 6,89 (дд, J=8,6, 0,6 Гц, 1H), 4,66 (ддд, J=11,4, 3,3, 0,9 Гц, 1H), 4,57 (дд, J=11,3, 6,3 Гц, 1H), 4,52 (дд, J=11,3, 6,4 Гц, 1H), 4,49 (дд, J=11,5, 7,9 Гц, 1H), 4,29 (ддд, J=12,4, 5,6, 0,8 Гц, 1H), 4,13 (дд, J=12,4, 7,6 Гц, 1H), 2,98 (м, 1H); элементный анализ: (C19H15F3N4O4) C, H, N.
[0291] OOO. Синтез 2-нитро-6-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 67 таблицы 1) при использовании способа схемы 14

[0292] Реакция сочетания Сузуки бромида 181 (см. пример 2MMM) и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2M, в течение 2 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-4% EtOAc/CH2Cl2 (головные фракции) и затем смесью 4-5% EtOAc/CH2Cl2 давали соединение 67 (93%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 182-183°C;
1H ЯМР (CDCl3) δ 8,31 (дд, J=2,5, 0,7 Гц, 1H), 7,80 (дд, J=8,6, 2,6 Гц, 1H), 7,52 (дт, J=8,8, 2,6 Гц, 2H), 7,44 (с, 1H), 7,30 (ушир.дд, J=8,7, 0,8 Гц, 2H), 6,84 (дд, J=8,6, 0,6 Гц, 1H), 4,64 (ддд, J=11,5, 3,3, 0,9 Гц, 1H), 4,54 (дд, J=11,3, 6,2 Гц, 1H), 4,50 (дд, J=11,2, 6,4 Гц, 1H), 4,46 (дд, J=11,4, 7,9 Гц, 1H), 4,26 (ддд, J=12,4, 5,6, 0,8 Гц, 1H), 4,10 (дд, J=12,4, 7,6 Гц, 1H), 2,95 (м, 1H); элементный анализ: (C19H15F3N4O5) C, H, N.
[0293] PPP. Синтез 6-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 68 таблицы 1) при использовании способа схемы 15

[0294] Диэтилазодикарбоксилат (3,445 мл, 22,2 ммоль) добавляли по каплям к перемешиваемой смеси 3-{[трет-бутил(диметил)силил]окси}-2-({[трет-бутил(диметил)силил]окси}метил)-1-пропанола (184) (описано Kim et al., 2001, через силилирование и гидроборирование 2-метилен-1,3-пропандиола) (5,706 г, 17,1 ммоль), 6-бром-3-пиридинола (3,571 г, 20,5 ммоль) и трифенилфосфина (5,386 г, 20,5 ммоль) в безводном ТГФ (55 мл) при 0°C в атмосфере N2. После перемешивания при 0°C в течение 1 часа и затем при комнатной температуре в течение 41 часов смесь концентрировали при пониженном давлении и остаток хроматографировали на силикагеле. Элюирование смесью 0-5% Et2O/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 5% Et2O/петролейный эфир давало 2-бром-5-[3-{[трет-бутил(диметил)силил]окси}-2-({[трет-бутил(диметил)силил]окси}метил)пропокси]пиридин (185) (8,09 г, 97%) в виде бесцветного масла;
1H ЯМР (CDCl3) δ 8,06 (д, J=3,0 Гц, 1H), 7,35 (дд, J=8,7, 0,3 Гц, 1H), 7,11 (дд, J=8,7, 3,2 Гц, 1H), 4,03 (д, J=5,9 Гц, 2H), 3,73 (дд, J=10,1, 5,7 Гц, 1H), 3,69 (дд, J=10,0, 6,0 Гц, 1H), 2,16 (септ., J=5,8 Гц, 1H), 0,88 (с, 18H), 0,03 (2с, 12H); HRESIMS вычислено для C21H41BrNO3Si2 m/z [M+H]+ 492,1783, 490,1803, найдено 492,1786, 490,1804.
[0295] Силиловый эфир 185 (11,06 г, 22,5 ммоль) обрабатывали 1% раствором HCl в 95% EtOH (условия десилилирования описаны Cunico et al., 1980) (200 мл) и смесь перемешивали при комнатной температуре в течение 13 часов. Полученный раствор охлаждали (CO2/ацетон), нейтрализовали добавлением по каплям 7M NH3 в MeOH (10 мл) при перемешивании, затем концентрировали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-3% MeOH/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 5% MeOH/CH2Cl2 давало 2-{[(6-бром-3-пиридинил)окси]метил}-1,3-пропандиол (186) (5,56 г, 94%) в виде белого твердого вещества: т.пл. (CH2Cl2) 90-91°C;
1H ЯМР (CDCl3) δ 8,07 (д, J=3,1 Гц, 1H), 7,36 (д, J=8,7 Гц, 1H), 7,12 (дд, J=8,7, 3,1 Гц, 1H), 4,15(д, J=6,1 Гц, 1H), 3,95 (дт, J=10,8, 4,9 Гц, 1H), 3,92 (дт, J=10,8, 5,3 Гц, 1H), 2,24 (м, 1H), 1,99 (т, J=5,1 Гц, 2H); элементный анализ: (C9H12BrNO3) C, H, N.
[0296] Суспензию диола 186 (5,25 г, 20,0 ммоль) в безводном ТГФ (66 мл) в атмосфере N2 перемешивали при комнатной температуре до полного растворения твердого вещества (~10 минут), затем обрабатывали 60% NaH (0,829 г, 20,7 ммоль), быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 60 минут (до получения белого осадка) добавляли трет-бутилдиметилсилилхлорид (3,21 г, 21,3 ммоль) и смесь перемешивали при комнатной температуре в течение 100 минут. Полученную смесь концентрировали при пониженном давлении и остаток хроматографировали на силикагеле. Элюирование смесью 0-33% Et2O/петролейный эфир сначала давало головные фракции, и последующее элюирование смесью 33-50% Et2O/петролейный эфир давало 3-[(6-бром-3-пиридинил)окси]-2-({[трет-бутил(диметил)силил]окси}метил)-1-пропанол (187) (5,97 г, 79%) в виде бесцветного масла;
1H ЯМР (CDCl3)8 8,07 (д, J=3,1 Гц, 1H), 7,36 (д, J=8,7 Гц, 1H), 7,12 (дд, J=8,7, 3,1 Гц, 1H), 4,12 (дд, J=9,2, 6,6 Гц, 1H), 4,09 (дд, J=9,2, 5,9 Гц, 1H), 3,94-3,80 (м, 4H), 2,27 (дд, J=6,3, 4,8 Гц, 1H), 2,18 (септ., J=5,4 Гц, 1H), 0,89 (с, 9H), 0,06, 0,05 (2с, 6H); HRESIMS вычислено для C15H27BrNO3Si m/z [M+H]+ 378,0918, 376,0938, найдено 378,0912, 376,0931,
[0297] Йодирование спирта 187 использованием I2, PPh3 и имидазола, как в примере 2GGG выше, в течение 18 часов, с последующей хроматографией продукта на силикагеле при элюировании петролейным эфиром и пентаном (головные фракции) и затем смесью 5-25% Et2O/пентан давали 2-бром-5-[3-{[трет-бутил(диметил)силил]окси}-2-(иодметил)пропокси]пиридин (188) (97%) в виде бесцветного масла;
1H ЯМР (CDCl3) δ 8,07 (д, J=3,0 Гц, 1H), 7,37 (дд, J=8,7, 0,3 Гц, 1H), 7,11 (дд, J=8,7, 3,2 Гц, 1H), 4,06 (дд, J=9,2, 5,7 Гц, 1H), 3,99 (дд, J=9,2, 6,1 Гц, 1H), 3,74 (дд, J=10,1, 5,6 Гц, 1H), 3,70 (дд, J=10,1, 5,5 Гц, 1H), 3,36 (д, J=6,0 Гц, 2H), 2,12 (септ., J=5,8 Гц, 1H), 0,89 (с, 9H), 0,06 (2с, 6H); HRESIMS вычислено для C15H26BrINO2Si m/z [M+H]+ 487,9935, 485,9955, найдено 487,9931, 485,9952.
[0298] Алкилирование 2-бром-4(5)-нитроимидазола (80) при использовании йодида 188 и K2CO3, как в примере 2GGG выше, в течение 42 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-20% EtOAc/петролейный эфир (головные фракции) и затем смесью 20-33% EtOAc/петролейный эфир давали 2-бром-5-[3-(2-бром-4-нитро-1H-имидазол-1-ил)-2-({[трет-бутил(диметил)силил]окси}метил)пропокси]пиридин (189) (73%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 132-134°C;
1H ЯМР (CDCl3) δ 8,05 (д, J=3,0 Гц, 1H), 7,82 (с, 1H), 7,40 (дд, J=8,7, 0,4 Гц, 1H), 7,06 (дд, J=8,7, 3,2 Гц, 1H), 4,25 (д, J=7,2 Гц, 2H), 4,01 (д, J=5,7 Гц, 2H), 3,77 (дд, J=10,7, 4,9 Гц, 1H), 3,66 (дд, J=10,6, 4,6 Гц, 1H), 2,53 (м, 1H), 0,91 (с, 9H), 0,08, 0,07 (2с, 6H); элементный анализ: (C18H26Br2N4O4Si) C, H, N.
[0299] Тетра-н-бутиламмонийфторид (13,0 мл 1M раствора в ТГФ, 13,0 ммоль) добавляли по каплям к перемешиваемому раствору силилового эфира 189 (6,78 г, 12,3 ммоль) в безводном ТГФ (140 мл) и смесь перемешивали при комнатной температуре в течение 4 часов. Полученный раствор концентрировали при пониженном давлении, затем разбавляли ледяной водой (120 мл) и экстрагировали EtOAc (5×120 мл). Экстракты промывали насыщенным раствором соли (100 мл), затем упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-80% Et2O/петролейный эфир, петролейным эфиром и смесью 0-1% MeOH/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 2-3% MeOH/CH2Cl2 давало 3-(2-бром-4-нитро-1Н-имидазол-1-ил)-2-{[(6-бром-3-пиридинил)окси]метил}-1-пропанол (190) (5,36 г, 100%) в виде бледно-желтой пены;
1H ЯМР (CDCl3) δ 8,07 (д, J=3,0 Гц, 1H), 7,89 (с, 1H), 7,40 (д, J=8,7 Гц, 1H), 7,09 (дд, J=8,7, 3,2 Гц, 1H), 4,32 (д, J=7,2 Гц, 2H), 4,09 (д, J=5,5 Гц, 2H), 3,87 (дд, J=10,7, 4,7 Гц, 1H), 3,75 (дд, J=10,8, 4,8 Гц, 1H), 2,57 (м, 1H); HRESIMS вычислено для C12H13Br2N4O4 m/z [M+H]+ 438,9258, 436,9278, 434,9298, найдено 438,9262, 436,9279, 434,9299.
[0300] Замыкание кольца спирта 190 при помощи NaH (1,35 экв.), как в примере 2GGG, в течение 200 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% MeOH/CH2Cl2 (головные фракции) и затем смесью 1-3% MeOH/CH2Cl2 давали 6-{[(6-бром-3-пиридинил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (191) (71%) в виде бледно-желтого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 197-199°C;
1H ЯМР [(CD3)2SO] δ 8,15 (ушир.д, J=3,0 Гц, 1H), 8,10 (с, 1H), 7,56 (дд, J=8,7, 0,3 Гц, 1H), 7,42 (дд, J=8,8, 3,2 Гц, 1H), 4,60 (дд, J=11,0, 2,7 Гц, 1H), 4,45 (дд, J=11,0, 7,0 Гц, 1H), 4,29 (дд, J=12,5, 5,5 Гц, 1H), 4,20 (дд, J=10,0, 6,8 Гц, 1H), 4,17 (дд, J=10,0, 6,7 Гц, 1H), 4,05 (дд, J=12,6, 6,8 Гц, 1H), 2,85 (м, 1H); элементный анализ: (C12H11BrN4O4) C, H, N.
[0301] Реакция сочетания Сузуки бромида 191 и 4-фторфенилбороновой кислоты, как в примере 2M, в течение 140 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-4% EtOAc/CH2Cl2 (головные фракции) и затем смесью 5-6% EtOAc/CH2Cl2 давали соединение 68 (85%) в виде бледно-желто-коричневого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 214-216°C;
1H ЯМР [(CD3)2SO] δ 8,38 (д, J=2,8 Гц, 1H), 8,11 (с, 1H), 8,06 (ддт, J=8,9, 5,6, 2,6 Гц, 2H), 7,91 (д, J=8,8 Гц, 1H), 7,51 (дд, J=8,8, 3,0 Гц, 1H), 7,27 (дт, J=8,9, 2,6 Гц, 2H), 4,63 (дд, J=11,0, 2,9 Гц, 1H), 4,49 (дд, J=11,1, 7,1 Гц, 1H), 4,32 (дд, J=12,5, 5,5 Гц, 1H), 4,25 (дд, J=10,0, 6,8 Гц, 1H), 4,22 (дд, J=10,0, 6,7 Гц, 1H), 4,08 (дд, J=12,6, 6,9 Гц, 1H), 2,89 (м, 1H); элементный анализ: (C18H15FN4O4)C, H, N.
[0302] QQQ. Синтез 2-нитро-6-[({6-[4-(трифторметил)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 69 таблицы 1) при использовании способа схемы 15

[0303] Реакция сочетания Сузуки бромида 191 (см. пример 2PPP) и 4-(трифторметил)фенилбороновой кислоты, как в примере 2M, в течение 140 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-4% EtOAc/CH2Cl2 (головные фракции) и затем смесью 4-5% EtOAc/CH2Cl2 давали соединение 69 (41 мг, 69%) в виде бледно-желтого твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 233-235°C;
1H ЯМР [(CD3)2SO] δ 8,45 (д, J=2,7 Гц, 1H), 8,24 (ушир.д, J=8,1 Гц, 2H), 8,12 (с, 1H), 8,05 (д, J=8,9 Гц, 1H), 7,81 (ушир.д, J=8,3 Гц, 2H), 7,57 (дд, J=8,8, 3,0 Гц, 1H), 4,64 (дд, J=11,0, 2,9 Гц, 1H), 4,50 (дд, J=11,1, 7,1 Гц, 1H), 4,32 (дд, J=12,5, 5,4 Гц, 1H), 4,28 (дд, J=10,0, 6,7 Гц, 1H), 4,25 (дд, J=10,1, 6,7 Гц, 1H), 4,09 (дд,, J=12,6, 6,8 Гц, 1H), 2,90 (м, 1H); элементный анализ: (C19H15F3N4O4) C, H, N.
[0304] RRR. Синтез 2-нитро-6-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 70 таблицы 1) при использовании способа схемы 15

[0305] Реакция сочетания Сузуки бромида 191 (см. пример 2PPP) и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2M, в течение 140 минут, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-4% EtOAc/CH2Cl2 (головные фракции) и затем смесью 4-5% EtOAc/CH2Cl2 давали соединение 70 (55 мг, 89%) в виде кремового твердого вещества: т.пл. (MeOH/CH2Cl2/гексан) 180-181°C;
1H ЯМР [(CD3)2SO] δ 8,41 (д, J=2,7 Гц, 1H), 8,14 (дт, J=8,9, 2,5 Гц, 2H), 8,11 (с, 1H), 7,96 (д, J=8,7 Гц, 1H), 7,54 (дд, J=8,8, 3,0 Гц, 1H), 7,44 (ушир.дд, J=8,8, 0,8 Гц, 2H), 4,64 (дд, J=10,9, 2,9 Гц, 1H), 4,49 (дд, J=11,0, 7,1 Гц, 1H), 4,32 (дд, J=12,5, 5,4 Гц, 1H), 4,26 (дд, J=10,1, 6,7 Гц, 1H), 4,23 (дд, J=10,1, 6,7 Гц, 1H), 4,09 (дд, J=12,6, 6,8 Гц, 1H), 2,89 (м, 1H); элементный анализ: (C19H15F3N4O5) C, H, N.
[0306] SSS. Синтез 2-нитро-6-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 71 таблицы 1) при использовании способа схемы 14

[0307] Алкилирование оксазинового спирта 180 (см. пример 2MMM) при использовании 3-(трифторметокси)бензилбромида и NaH (1,6 экв.), как в примере 2UU выше, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1,5% EtOAc/CH2Cl2 давали соединение 71 (56%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 60-61°C;
1H ЯМР (CDCl3) δ 7,40 (с, 1H), 7,39 (т, J=7,8 Гц, 1H), 7,21 (ушир.д, J=7,8 Гц, 1H), 7,20-7,14 (м, 2H), 4,54 (с, 2H), 4,51 (ддд, J=11,5, 3,4, 0,9 Гц, 1H), 4,36 (дд, J=11,4, 7,8 Гц, 1H), 4,15 (ддд, J=12,3, 5,6, 0,8 Гц, 1H), 4,03 (дд, J=12,3, 7,5 Гц, 1H), 3,62 (дд, J=9,6, 5,8 Гц, 1H), 3,57 (дд, J=9,6, 6,6 Гц, 1H), 2,68 (м, 1H); элементный анализ: (C15H14F3N3O5) C, H, N.
[0308] TTT. Синтез 2-нитро-6-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 72 таблицы 1) при использовании способа схемы 14

[0309] Алкилирование оксазинового спирта 180 (см. пример 2MMM) при использовании 4-(трифторметокси)бензилбромида (1,9 экв.) и NaH (1,6 экв.), как в примере 2UU выше, в течение 3 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 2% EtOAc/CH2Cl2 давали соединение 72 (59%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 92-93°C;
1H ЯМР (CDCl3) δ 7,40 (с, 1H), 7,32 (дт, J=8,7, 2,3 Гц, 2H), 7,21 (ушир.д, J=8,0 Гц, 2H), 4,52 (с, 2H), 4,51 (ддд, J=11,3, 3,3, 0,9 Гц, 1H), 4,36 (дд, J=11,4, 7,8 Гц, 1H), 4,15 (ддд, J=12,3, 5,6, 0,8 Гц, 1H), 4,02 (дд, J=12,3, 7,5 Гц, 1H), 3,62 (дд, J=9,6, 5,9 Гц, 1H), 3,56 (дд, J=9,6, 6,5 Гц, 1H), 2,67 (м, 1H); элементный анализ: (C15H14F3N3O5) C, H, N.
[0310] UUU. Синтез 6-({[4-(бензилокси)бензил]окси}метил)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 73 таблицы 1) при использовании способа схемы 14

[0311] Раствор 4-(бензилокси)бензилйодида (описан в литературе Cativiela et al., 1995, через йодирование 4-(бензилокси)бензилового спирта) (98 мг, 0,302 ммоль) в безводном ДМФА (0,3 мл, затем 2×0,4 мл для промывки) добавляли к раствору оксазинового спирта 180 (см. пример 2MMM) (30,7 мг, 0,154 ммоль) в безводном ДМФА (1 мл) в атмосфере N2 при 0°C. Смесь обрабатывали 60% NaH (8,8 мг, 0,22 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 35 минут смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (10 мл), добавляли к насыщенному раствору соли (40 мл) и экстрагировали CH2Cl2 (3×50 мл) и EtOAc (3×50 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-2% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 2-3% EtOAc/CH2Cl2 давало неочищенный продукт (20 мг), который затем хроматографировали на силикагеле. Элюирование смесью 25-40% EtOAc/петролейный эфир сначала давало головные фракции, и последующее элюирование EtOAc давало соединение 73 (15 мг, 25%) в виде белого твердого вещества: т.пл. (CH2Cl2/гексан) 150-151°C;
1H ЯМР (CDCl3) δ 7,46-7,29 (м, 6H), 7,20 (дт, J=8,6, 2,4 Гц, 2H), 6,96 (дт, J=8,7, 2,4 Гц, 2H), 5,07 (с, 2H), 4,48 (ддд, J=11,4, 3,3, 0,8 Гц, 1H), 4,46 (д, J=11,6 Гц, 1H), 4,43 (д, J=11,6 Гц, 1H), 4,32 (дд, J=11,4, 7,9 Гц, 1H), 4,09 (ушир.дд, J=12,3, 5,5 Гц, 1H), 3,99 (дд, J=12,3, 7,6 Гц, 1H), 3,56 (дд, J=9,6, 5,7 Гц, 1H), 3,50 (дд, J=9,6, 6,7 Гц, 1H), 2,62 (м, 1H); элементный анализ: (C21H21N3O5) C, H, N.
[0312] VVV. Синтез 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-3-ил]метокси}метил)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (соединение 74 таблицы 1) при использовании способа схемы 14

[0313] Смесь оксазинового спирта 180 (см. пример 2MMM) (200,3 мг, 1,01 ммоль) и 3-йодбензилбромида (406 мг, 1,37 ммоль) в безводном ДМФА (7,5 мл) в атмосфере N2 при 0°C обрабатывали 60% NaH (57 мг, 1,43 ммоль), затем быстро дегазировали и снова герметично закрывали в атмосфере N2. После перемешивания при комнатной температуре в течение 140 минут смесь охлаждали (CO2/ацетон), гасили смесью лед/водный раствор NaHCO3 (10 мл) и разбавляли водой (40 мл) для осаждения неочищенного твердого вещества, которое отделяли фильтрованием и промывали водой и петролейным эфиром (0,49 г). Фильтрат экстрагировали EtOAc (3×80 мл) и затем экстракты промывали насыщенным раствором соли (50 мл). Объединенные экстракты упаривали досуха, остаток объединяли с полученным ранее твердым веществом и хроматографировали на силикагеле. Элюирование смесью 0-1% EtOAc/CH2Cl2 сначала давало головные фракции, и последующее элюирование смесью 1-2% EtOAc/CH2Cl2 давало 6-{[(3-йодбензил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (182) (184 мг, 44%) в виде кремового твердого вещества: т.пл. (CH2Cl2/гексан) 127-130°C;
1H ЯМР (CDCl3) δ 7,69-7,62 (м, 2H), 7,40 (с, 1H), 7,24 (м, 1H), 7,10 (ушир.т, J=8,0 Гц, 1H), 4,51 (дд, J=11,4, 3,1 Гц, 1H), 4,48 (д, J=12,3 Гц, 1H), 4,44 (д, J=12,2 Гц, 1H), 4,35 (дд, J=11,4, 7,7 Гц, 1H), 4,14 (дд, J=12,3, 5,5 Гц, 1H), 4,02 (дд, J=12,3, 7,4 Гц, 1H), 3,60 (дд, J=9,6, 5,8 Гц, 1H), 3,54 (дд, J=9,6, 6,7 Гц, 1H), 2,66 (м, 1H); элементный анализ: (C14H14IN3O4) C, H, N.
[0314] Реакция сочетания Сузуки йодида 182 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2XX выше, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1-1,5% EtOAc/CH2Cl2 давали соединение 74 (92%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 78-80°C;
1H ЯМР (CDCl3) δ 7,58 (дт, J=8,8, 2,5 Гц, 2H), 7,51 (дт, J=7,8, 1,5 Гц, 1H), 7,48-7,42 (м, 2H), 7,37 (с, 1H), 7,33-7,26 (м, 3H), 4,61 (д, J=11,9 Гц, 1H), 4,57 (д, J=12,0 Гц, 1H), 4,51 (ддд, J=11,4, 3,2, 0,7 Гц, 1H), 4,37 (дд, J=11,4, 7,6 Гц, 1H), 4,13 (дд, J=12,4, 5,5 Гц, 1H), 4,03 (дд, J=12,3, 7,4 Гц, 1H), 3,64 (дд, J=9,6, 5,8 Гц, 1H), 3,58 (дд, J=9,6, 6,7 Гц, 1H), 2,67 (м, 1H); элементный анализ: (C21H18F3N3O5) C, H, N.
[0315] WWW. Синтез 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (соединение 75 таблицы 1) при использовании способа схемы 14

[0316] Алкилирование оксазинового спирта 180 (см. пример 2MMM) при использовании 4-йодбензилбромида и NaH (1,4 экв.), как в примере 2UU выше, в течение 3 часов, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 3% EtOAc/CH2Cl2 давали 6-{[(4-йодбензил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (183) (42%) в виде белого твердого вещества: т.пл. (CH2Cl2/гексан) 161-163°C;
1H ЯМР (CDCl3) δ 7,70 (дт, J=8,3, 2,0 Гц, 2H), 7,40 (с, 1H), 7,03 (ушир.д, J=8,3 Гц, 2H), 4,50 (ддд, J=11,4, 3,3, 0,8 Гц, 1H), 4,46 (с, 2H), 4,34 (дд, J=11,4, 7,8 Гц, 1H), 4,13 (ддд, J=12,3, 5,6, 0,8 Гц, 1H), 4,00 (дд, J=12,3, 7,6 Гц, 1H), 3,59 (дд, J=9,6, 5,8 Гц, 1H), 3,53 (дд, J=9,6, 6,5 Гц, 1H), 2,65 (м, 1H); HRFABMS вычислено для C14H15IN3O4 m/z [M+H]+ 416,0107, найдено 416,0108.
[0317] Реакция сочетания Сузуки йодида 183 и 4-(трифторметокси)фенилбороновой кислоты, как в примере 2XX выше, с последующей хроматографией продукта на силикагеле при элюировании смесью 0-1% EtOAc/CH2Cl2 (головные фракции) и затем смесью 1-1,5% EtOAc/CH2Cl2 давали соединение 75 (95%) в виде кремового твердого вещества: т.пл. (CH2Cl2/пентан) 135-138°C;
1H ЯМР (CDCl3) δ 7,59 (дт, J=8,8, 2,5 Гц, 2H), 7,55 (дт, J=8,3, 1,9 Гц, 2H), 7,40 (с, 1H), 7,37 (ушир.д, J=8,3 Гц, 2H), 7,29 (ушир.дд, J=8,7, 0,8 Гц, 2H), 4,57 (с, 2H), 4,52 (ддд, J=11,4, 3,3, 0,8 Гц, 1H), 4,37 (дд, J=11,4, 7,8 Гц, 1H), 4,15 (ддд, J=12,3, 5,6, 0,7 Гц, 1H), 4,04 (дд, J=12,3, 7,5 Гц, 1H), 3,64 (дд, J=9,6, 5,8 Гц, 1H), 3,58 (дд, J=9,6, 6,6 Гц, 1H), 2,68 (м, 1H); элементный анализ: (C21H18F3N3O5) C, H, N.
ПРИМЕР 3. Биологическая активность и стабильность
[0318] Биологическую активность соединений по настоящему изобретению оценивали следующим образом. Результаты представлены ниже в таблице 2.
[0319] (a) Минимальные ингибирующие концентрации (MIC)
Соединения оценивали на их активность против реплицирующего Mycobacterium tuberculosis в 8-дневном микропланшетном анализе с использованием реагента Alamar blue (добавляли в день 7) для определения роста (MABA) (Collins et al., 1997; Falzari et al., 2005). Наименьшую концентрацию соединения, обеспечивающую ингибирование >90%, рассматривали как MIC. Для скрининга на активность соединений против бактерий в нереплицирующем состоянии, которое моделирует клиническую персистентность, использовали 11-дневный высокопроизводительный люминесцентный анализ с низкой регенерацией кислорода (LORA), где бактерии M. tuberculosis, содержащие плазмиду с ацетамидазным промотором, управляющим бактериальным геном люциферазы, сначала адаптировали к условиям с низким содержанием кислорода при помощи продленной культуры (Cho et al., 2007).
[0320] (b) Анализ цитотоксичности с использованием клеток млекопитающих
Это оценивали против VERO клеток (CCL-81, American Type Culture Collection) при воздействии в течение 72 часов в анализе с использованием тетразолиевого красителя (Falzari et al., 2005).
[0321] (c) Антипротозойный скрининг
Соединения оценивали на их активность как против амастигот Trypanosoma cruzi, так и против амастигот Leishmania donovani (свободные или инкапсулированные в макрофагах), в соответствии со следующими протоколами:
[0322] (i) Анализ Trypanosoma cruzi
L-6 клетки (2×103) в среде (100 мкл RPMI 1640, дополненной 2 мМ 1-глутамина плюс 10% термоактивированная фетальная телячья сыворотка) высевали в 96-луночные микротитровальные планшеты (Costar™) и инкубировали при 37°C (5% CO2) в течение 1 дня. Добавляли суспензию (50 мкл) трипомастигот Trypanosoma cruzi (5×103 штамма Tulahuen C2C4, содержащего ген β-галактозидазы) и клетки инкубировали при 37°C (5% CO2) еще в течение 48 часов для установления инфекции. Среду удаляли и заменяли свежей средой, и инфицированные клетки затем инкубировали при 37°C (5% CO2) в течение 96 часов либо в среде как таковой, либо в присутствии серийных (3-кратные) разведений испытуемых соединений (изначально получали в виде 10 мг/мл исходных растворов в ДМСО и разводили в среде). Бензнидазол использовали в качестве стандарта в каждом анализе. После инкубации добавляли хлорфеноловый красный гликозид (100 мМ) в 0,1% Nonidet P40/PBS (50 мкл) и (через 6 часов) измеряли спектральную поглощательную способность при 540 нм и использовали для расчета значений IC50.
[0323] (ii) Анализ аксенических Leishmania donovani
Выращенные в аксенических условиях амастиготы L. donovani (MHOM-ET-67/L82) из здоровой культуры в log фазе высевали при плотности 1×106/мл среды (SM, pH 5,4 плюс 10% термоактивированная фетальная телячья сыворотка) в 96-луночные микротитровальные планшеты (Costar™) и инкубировали при 37°C (5% CO2) в течение 70 часов либо в среде как таковой, либо в присутствии серийных (3-кратные) разведений испытуемых соединений (изначально получали в виде 10 мг/мл исходных растворов в ДМСО и разводили в среде). Милтефосин использовали в качестве стандарта в каждом анализе. После инкубации в каждую лунку добавляли ресазуриновый флуоресцентный краситель и инкубацию продолжали еще в течение 2 часов. Значения IC50 определяли на основании измерений флуоресценции.
[0324] (iii) Анализ макрофагов, инфицированных Leishmania donovani
Свежехарвестированные мышиные макрофаги в среде (RPMI 1640 плюс 10% термоактивированная фетальная телячья сыворотка) инкубировали при 37°C (5% CO2) в течение 24 часов и затем инфицировали (1:3 макрофагов к амастиготам) с аксенической культурой амастигот L. donovani (MHOM-ET-67/L82) в среде (SM, pH 5,4 плюс 10% термоактивированная фетальная телячья сыворотка). Инфицированные макрофаги высевали при плотности 1,2×106/мл (путем разведения в RPMI+10% FCS) в 16-луночные предметные стекла (Lab-tek™) и инкубировали при 37°C (5% CO2) в течение 24 часов. Среду удаляли и заменяли свежей средой (RPMI 1640+10% FCS), и это повторяли после смешивания. Инфицированные макрофаги затем инкубировали при 37°C (5% CO2) в течение 96 часов либо в среде как таковой, либо в присутствии серийных (3-кратные) разведений испытуемых соединений (изначально получали в виде 10 мг/мл исходных растворов в ДМСО и разводили в среде). Милтефосин использовали в качестве стандарта в каждом анализе. После удаления среды и лунок предметные стекла фиксировали (5 минут в 100% MeOH) и окрашивали (10% Giemsa, 10 минут). Отношение инфицированных макрофагов к неинфицированным определяли путем исследования под микроскопом и затем рассчитывали значения IC50 при помощи анализа линейной регрессии.
|
Таблица 2
In vitro биологическая активность выбранных соединений таблицы 1 |
||||||
| No | MIC (мкМ) | IC 50 (нМ) | IC 50 (нг/мл) | |||
| MABA (аэробные) | LORA (аэробные) | VERO | T. cruzi | L. donovani | ||
| аксен | макро | |||||
| 7 | 0,53 | 24 | ND | 3,2 | 0,016 | 0,40 |
| 8 | 0,04 | 14 | >128 | 4,1 | 0,017 | 0,38 |
| 9 | 0,04 | 3,7 | >128 | 1,2 | 0,016 | 0,20 |
| 10 | 0,045 | 11 | >128 | 2,0 | 0,040 | 0,23 |
| 11 | 0,08 | >128 | >128 | 18 | 0,028 | 0,47 |
| 12 | 0,087 | 64 | >128 | 9,4 | 0,016 | 0,74 |
| 25 | 0,25 | 34 | >128 | 0,24 | 0,029 | 0,22 |
| 26 | 0,30 | 50 | >128 | 0,34 | 0,013 | 0,36 |
| 47 | 3,8 | 15 | ND | 1,1 | 0,041 | 0,084 |
| 49 | 0,46 | 3,0 | >128 | 0,55 | 0,048 | 0,065 |
| 50 | 0,24 | 5,1 | >128 | 6,3 | 0,041 | 0,15 |
| 51 | 0,055 | 1,5 | >128 | 15 | 0,046 | 0,17 |
| 54 | 0,20 | 1,4 | >128 | 0,74 | 0,047 | 0,095 |
| 71 | 0,33 | 15 | >128 | 0,56 | 0,20 | 0,65 |
| 72 | 2,4 | 7,9 | 46 | 0,62 | 0,089 | 0,55 |
| 73 | 3,1 | 35 | 113 | 2,8 | 0,16 | 0,53 |
| 74 | 0,22 | 2,9 | >128 | 0,94 | 0,16 | 0,54 |
| 75 | 0,30 | >128 | >128 | 0,47 | 0,20 | 0,67 |
[0325] In vitro микросомальную стабильность и in vivo биологическую активность выбранных соединений по настоящему изобретению также оценивали следующим образом, и результаты представлены в таблице 3.
[0326] (а) Стабильность соединений в человеческих и мышиных микросомах
Испытуемые соединения (1 мкМ) инкубировали при 37°С с объединенными препаратами микросом печени человека или CD-1 мышей (конечная концентрация белка 0,5 мг/мл) и NADPH регенерирующей системой (MgCl2, 3,3 мМ; G6P, 3,3 мМ; G6PD, 0,4 Ед./мл; NADP+, 1,3 мМ) в фосфатном буфере (75 мМ, рН 7,4), с использованием конечного объема 200 мкл. Соединения растворяли в ДМСО таким образом, чтобы конечная концентрация ДМСО составляла 0,5%. Реакции останавливали в точке времени 0 и 60 минут путем добавления MeCN (100 мкл), содержащего 0,2 мкМ метопропола, в качестве внутреннего стандарта. Образцы разводили 10× и центрифугировали, затем проводили анализ ЖХ-МС/МС с использованием ионизации электрораспылением и SRM мониторинг с использованием градиентного метода ЖХ. ЖХ площади пиков интегрировали и выражали как отношение площадей пиков аналит/IS (PAR), и среднее значение для каждой точки времени рассчитывали из данных, полученных в двух повторах. Остаточное процентное содержание рассчитывали следующим образом:
% остаточн.=100×(Среднее PART60/Среднее PART0)
[0327] (b) In vivo анализ острой ТВ инфекции у мышей
BALB/c мышей инфицировали через аэрозоль с суспензией ~2×106 колониеобразующих единиц (КОЕ) М.tuberculosis Erdman/мл (Falzari et al., 2005). Каждое соединение вводили перорально группе из 7 или 8 мышей при 100 мг/кг ежедневно, 5 дней в неделю в течение 3 недель, начиная со дня 11 после инфицирования. Соединения вводили в виде суспензии в 0,5% СМС/0,08% Tween 80 в воде. Мышей умерщвляли в день 31 и определяли количество КОЕ в легких и сравнивали с КОЕ у мышей, которым вводили только носитель, в данной точке времени. РА-824 использовали в качестве положительного контроля в каждом эксперименте и результаты представляли в виде отношения среднее уменьшение КОЕ у мышей, обработанных соединением/среднее уменьшение КОЕ у мышей, обработанных РА-824. В данном анализе РА-824 вызывал до 2,5-3 log уменьшения КОЕ.
Таблица 3
Микросомальная стабильность и in vivo биологическая активность соединений, выбранных из таблицы 1
| No |
Микросомы
(% остаточн.) |
In vivo эффективность против РА-824 | |
| Человек | Мышь | ||
| PA-824 | 82 | 94 | 1,00 |
| 12 | 82 | 81 | 112 |
| 25 | 68 | 30 | 0,025 |
| 72 | 71 | 41 | ND |
ССЫЛОЧНЫЕ ДОКУМЕНТЫ
Содержание каждого из документов, перечисленных ниже, включено в настоящее описание посредством ссылки.
Патентные документы США
Патент США 5668127
Патент США 6087358
US 2006063929A1
Международные патентные документы
DE 2312518
EP 1555267
JP 2005/330266
WO 2004/033463
WO 2005/042542
WO 2007/075872
WO 2008/112483
WO 2009/120789
Непатентные публикации
Anderson et al., Org. Biomol. Chem. 6, 1973-1980 (2008).
Apparu et al., Tetrahedron: Asymmetry 11, 2885-2898 (2000).
Ashtekar et al., Antimicrob. Agents Chemother. 37, 183-186 (1993).
Cavalli et al., J. Med. Chem. 52, 7339-7359 (2009).
Cativiela et al., J. Org. Chem. 60, 3074-3083 (1995).
Cho et al., Antimicrob. Agents Chemother. 51, 1380-1385 (2007).
Collins et al, Antimicrob. Agents Chemother. 41, 1004-1009 (1997).
Cunico et al., J Org. Chem. 45, 4797-4798 (1980).
Curran et al., J Am. Chem. Soc. 120, 342-351 (1998).
Falzari et al., Antimicrob. Agents Chemother. 49, 1447-1454 (2005).
Ferrara et al., Lancet 367, 1328-1334 (2006).
Helmboldt et al., Org. Lett. 8, 1573-1576 (2006).
Kim et al., J. Med Chem. 52, 1317-1328 и 1329-1344 (2009).
Kim et al., J Med. Chem. 44, 3092-3108 (2001).
Kopka et al., Bioorg. Med. Chem. 11, 3513-3527 (2003).
Li et al., Bioorg. Med. Chem. Lett. 18, 2256-2262 (2008).
Manjunatha et al., Proc. Natl. Acad. ScL USA 103, 431-436 (2006).
Matsumoto et al., PLoS Medicine 3, 2131-2143 (2006).
Nagarajan et al., Eur. J. Med. Chem. 24, 631-633 (1989).
Sasaki et al., J Med. Chem. 49, 7854-7860 (2006).
Sehgal et al., J. Med. Chem. 24, 601-604 (1981).
Singh et al., Science 322, 1392-1395 (2008).
Stover et al., Nature 405, 962-966 (2000).
Tyagi et al., Antimicrob. Agents Chemother. 49, 2289-2293 (2005).
Wennerberg et al., J Org. Chem. 64, 54-59 (1999).
Claims (7)
1. Соединение, имеющее общую структурную формулу I:
где n равно 1,
V и W независимо представляют собой Н или СН3, и
один из Х или Y представляет собой Н, и другой представляет собой одну из формул IIa или IIb, где формулы IIa и IIb имеют общие структуры:
где формула IIa включает единственное кольцо, отмеченное в положении 3 и положении 4 и содержащее R1 в качестве заместителя, и формула IIb включает первое кольцо, отмеченное в положении 3 и положении 4 и содержащее в качестве заместителей как R2, так и концевое кольцо, отмеченное в положении 4 и содержащее R1 в качестве заместителя,
где единственное кольцо формулы IIa и первое кольцо и концевое кольцо формулы IIb включают С, СН или N в каждом положении в кольце, где единственное кольцо формулы IIa и первое кольцо и концевое кольцо формулы IIb независимо содержат не более двух атомов азота;
Z в формулах IIa и IIb представляет собой СН2 или прямую связь,
R1 независимо представляют собой любой один или два из Н, F, С1, CF3, OCF3 или OCH2Ph, и
R2 представляет собой Н.
где n равно 1,
V и W независимо представляют собой Н или СН3, и
один из Х или Y представляет собой Н, и другой представляет собой одну из формул IIa или IIb, где формулы IIa и IIb имеют общие структуры:
где формула IIa включает единственное кольцо, отмеченное в положении 3 и положении 4 и содержащее R1 в качестве заместителя, и формула IIb включает первое кольцо, отмеченное в положении 3 и положении 4 и содержащее в качестве заместителей как R2, так и концевое кольцо, отмеченное в положении 4 и содержащее R1 в качестве заместителя,
где единственное кольцо формулы IIa и первое кольцо и концевое кольцо формулы IIb включают С, СН или N в каждом положении в кольце, где единственное кольцо формулы IIa и первое кольцо и концевое кольцо формулы IIb независимо содержат не более двух атомов азота;
Z в формулах IIa и IIb представляет собой СН2 или прямую связь,
R1 независимо представляют собой любой один или два из Н, F, С1, CF3, OCF3 или OCH2Ph, и
R2 представляет собой Н.
2. Соединение по п.1, где:
n равно 1,
V и W независимо представляют собой Н или СН3, и
один из Х или Y представляет собой Н, и другой представляет собой одну из формул IIa или IIb, где формулы IIa и IIb имеют общие структуры:

где формула IIa включает единственное кольцо, отмеченное в положении 3 и положении 4 и содержащее R1 в качестве заместителя, и формула IIb включает первое кольцо, отмеченное в положении 3 и положении 4 и содержащее в качестве заместителей как R2, так и концевое кольцо, отмеченное в положении 4 и содержащее R1 в качестве заместителя,
где первое кольцо формулы IIb включает С, СН или N в каждом положении в кольце, где первое кольцо формулы IIb включает не более двух атомов азота, и единственное кольцо формулы IIa и концевое кольцо формулы IIb оба включают С или СН в каждом положении в кольце,
Z в формулах IIa и IIb представляет собой CH2 или прямую связь,
R1 представляет собой 4-F, 4-CF3, 3-OCF3, 4-OCF3 или 4-OCH2Ph, и
R2 представляет собой Н.
n равно 1,
V и W независимо представляют собой Н или СН3, и
один из Х или Y представляет собой Н, и другой представляет собой одну из формул IIa или IIb, где формулы IIa и IIb имеют общие структуры:
где формула IIa включает единственное кольцо, отмеченное в положении 3 и положении 4 и содержащее R1 в качестве заместителя, и формула IIb включает первое кольцо, отмеченное в положении 3 и положении 4 и содержащее в качестве заместителей как R2, так и концевое кольцо, отмеченное в положении 4 и содержащее R1 в качестве заместителя,
где первое кольцо формулы IIb включает С, СН или N в каждом положении в кольце, где первое кольцо формулы IIb включает не более двух атомов азота, и единственное кольцо формулы IIa и концевое кольцо формулы IIb оба включают С или СН в каждом положении в кольце,
Z в формулах IIa и IIb представляет собой CH2 или прямую связь,
R1 представляет собой 4-F, 4-CF3, 3-OCF3, 4-OCF3 или 4-OCH2Ph, и
R2 представляет собой Н.
3. Фармацевтическая композиция, предназначенная для предупреждения и лечения микробной инфекции, содержащая терапевтически эффективное количество соединения по п.1.
4. Фармацевтическая композиция по п.3, дополнительно содержащая фармацевтически приемлемый эксципиент, адъювант, носитель, буфер, стабилизатор или их смесь.
5. Способ предупреждения и лечения микробной инфекции, включающий введение фармацевтической композиции по п.3.
6. Способ по п.5, где микробная инфекция вызвана Mycobacterium tuberculosis, Trypanosoma cruzi или Leishmania donovani.
7. Соединение, выбранное из группы, включающей:
А. 2-нитро-7-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
B. 7-{[4-(бензилокси)фенокси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
С. 7-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
D. 2-нитро-7-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Е. 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
F. 7-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
G. 2-нитро-7-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Н. 7-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
I. 2-нитро-7-[({б-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
J. 7-метил-2-нитро-7-{[4-(трифторметокси)фенокси]метил}-6,7 дигидро-5Н-имидазо[2,1-b] [1,3]оксазин;
К. 7-{[4-(бензилокси)фенокси]метил}-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
L. 7-{[(4'-фтор[1,1-бифенил]-4-ил)окси]метил}-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
М. 7-метил-2-нитро-7-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
N. 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
О. 7-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-7-метил-2-нитро-6, 7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Р. 7-метил-2-нитро-7-[({5-[4-(трифторметил)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Q. 7-метил-2-нитро-7-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
R. 7-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
S. 7-метил-2-нитро-7-[({6-[4-(трифторметил)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Т. 7-метил-2-нитро-7-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
U. 2-нитро-7-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
V. 2-нитро-7-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
W. 7-({[4-(бензилокси)бензил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
X. 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-3-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Y. 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Z. 7-метил-2-нитро-7-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b] [1,3]оксазин;
АА. 7-метил-2-нитро-7-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b] [1,3]оксазин;
ВВ. 7-({[4-(бензилокси)бензил]окси}метил)-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
СС. 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]3-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
DD. 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
ЕЕ. (7R)-7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
FF. (7S)-7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
GG. 2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
HH. (6R)-2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
II. (6S)-2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
JJ. 6-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
КК. 2-нитро-6-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
LL. 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
MM. 6-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
NN. 2-нитро-6-[({5-[4-(трифторметил)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
OO. 2-нитро-6-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
PP. 6-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
QQ. 2-нитро-6-[({6-[4-(трифторметил)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
RR. 2-нитро-6-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
SS. 2-нитро-6-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
ТТ. 2-нитро-6-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
UU. 6-({[4-(бензилокси)бензил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
VV. 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-3-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин и
WW. 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин; и
их смеси, оптические или геометрические изомеры, фармакологически приемлемые солевые производные.
А. 2-нитро-7-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
B. 7-{[4-(бензилокси)фенокси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
С. 7-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
D. 2-нитро-7-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Е. 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
F. 7-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
G. 2-нитро-7-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Н. 7-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
I. 2-нитро-7-[({б-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
J. 7-метил-2-нитро-7-{[4-(трифторметокси)фенокси]метил}-6,7 дигидро-5Н-имидазо[2,1-b] [1,3]оксазин;
К. 7-{[4-(бензилокси)фенокси]метил}-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
L. 7-{[(4'-фтор[1,1-бифенил]-4-ил)окси]метил}-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
М. 7-метил-2-нитро-7-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
N. 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
О. 7-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-7-метил-2-нитро-6, 7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Р. 7-метил-2-нитро-7-[({5-[4-(трифторметил)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Q. 7-метил-2-нитро-7-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
R. 7-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
S. 7-метил-2-нитро-7-[({6-[4-(трифторметил)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Т. 7-метил-2-нитро-7-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
U. 2-нитро-7-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
V. 2-нитро-7-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
W. 7-({[4-(бензилокси)бензил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
X. 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-3-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Y. 2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
Z. 7-метил-2-нитро-7-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b] [1,3]оксазин;
АА. 7-метил-2-нитро-7-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b] [1,3]оксазин;
ВВ. 7-({[4-(бензилокси)бензил]окси}метил)-7-метил-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
СС. 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]3-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
DD. 7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
ЕЕ. (7R)-7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
FF. (7S)-7-метил-2-нитро-7-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
GG. 2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
HH. (6R)-2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
II. (6S)-2-нитро-6-{[4-(трифторметокси)фенокси]метил}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
JJ. 6-{[(4'-фтор[1,1'-бифенил]-4-ил)окси]метил}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
КК. 2-нитро-6-({[4'-(трифторметил)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
LL. 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
MM. 6-({[5-(4-фторфенил)-2-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
NN. 2-нитро-6-[({5-[4-(трифторметил)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
OO. 2-нитро-6-[({5-[4-(трифторметокси)фенил]-2-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
PP. 6-({[6-(4-фторфенил)-3-пиридинил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
QQ. 2-нитро-6-[({6-[4-(трифторметил)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
RR. 2-нитро-6-[({6-[4-(трифторметокси)фенил]-3-пиридинил}окси)метил]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
SS. 2-нитро-6-({[3-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
ТТ. 2-нитро-6-({[4-(трифторметокси)бензил]окси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
UU. 6-({[4-(бензилокси)бензил]окси}метил)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин;
VV. 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-3-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин и
WW. 2-нитро-6-({[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}метил)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин; и
их смеси, оптические или геометрические изомеры, фармакологически приемлемые солевые производные.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US23042209P | 2009-07-31 | 2009-07-31 | |
| US61/230,422 | 2009-07-31 | ||
| PCT/US2010/043908 WO2011014776A1 (en) | 2009-07-31 | 2010-07-30 | Nitroimidazooxazine and nitroimidazooxazole analogues and their uses |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2012107180A RU2012107180A (ru) | 2013-09-10 |
| RU2540860C2 true RU2540860C2 (ru) | 2015-02-10 |
Family
ID=42829983
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2012107180/04A RU2540860C2 (ru) | 2009-07-31 | 2010-07-30 | Нитроимидазооксазиновые и нитроимидазооксазольные аналоги и их применение |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US8293734B2 (ru) |
| EP (1) | EP2459570B1 (ru) |
| JP (1) | JP5685254B2 (ru) |
| KR (1) | KR101738868B1 (ru) |
| CN (1) | CN102741259B (ru) |
| AU (1) | AU2010278779B2 (ru) |
| BR (1) | BR112012002214B1 (ru) |
| CA (1) | CA2769263C (ru) |
| ES (1) | ES2530266T3 (ru) |
| NZ (1) | NZ598165A (ru) |
| RU (1) | RU2540860C2 (ru) |
| WO (1) | WO2011014776A1 (ru) |
| ZA (1) | ZA201201259B (ru) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2744784C1 (ru) * | 2017-03-28 | 2021-03-15 | Инститьют Оф Матириа Медика, Чайниз Акэдеми Оф Медикал Сайенсиз | Бензоксазин-оксазолидиноновое соединение, замещенное азотсодержащим гетероциклом, способ его получения и применение |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201012209D0 (en) * | 2010-05-31 | 2010-09-08 | Ge Healthcare Ltd | In vivo imaging agent |
| US9051333B2 (en) | 2011-04-15 | 2015-06-09 | Otsuka Pharmaceutical Co., Ltd. | 6,7-dihydroimidazo [2,1-b] [1,3]oxazine bactericides |
| JP2015006994A (ja) * | 2011-10-28 | 2015-01-15 | 大正製薬株式会社 | ジヒドロイミダゾオキサゾール誘導体 |
| KR101918469B1 (ko) * | 2012-06-04 | 2018-11-15 | 한국화학연구원 | 바이사이클릭 니트로이미다졸 유도체 또는 이들의 광학이성질체, 이의 약학적으로 허용가능한 염, 이의 제조방법 및 이를 유효성분으로 함유하는 약제학적 조성물 |
| US10301294B2 (en) * | 2013-03-13 | 2019-05-28 | The Broad Institute Inc. | Compounds for the treatment of tuberculosis |
| CN104059082B (zh) * | 2013-03-21 | 2016-08-03 | 苏州迈泰生物技术有限公司 | 硝基咪唑杂环类化合物及其在制备治疗结核病药物中的应用 |
| KR101564425B1 (ko) | 2013-11-26 | 2015-10-30 | 한국화학연구원 | 신규한 바이사이클릭 니트로이미다졸 카바메이트 화합물, 이를 제조하는 방법 및 이를 유효성분으로 함유하는 결핵 질환의 예방 또는 치료용 약제학적 조성물 |
| AR101704A1 (es) | 2014-08-28 | 2017-01-04 | Otsuka Pharma Co Ltd | Compuestos heterocíclicos fusionados |
| KR101682356B1 (ko) | 2014-11-04 | 2016-12-06 | 한국화학연구원 | 이미다조 옥사진 유도체, 이의 약학적으로 허용 가능한 염 또는 이의 광학이성질체 및 이를 유효성분으로 함유하는 약제학적 조성물 |
| US10227362B2 (en) | 2015-01-29 | 2019-03-12 | Medshine Discovery Inc. | Anti-pulmonary tuberculosis nitroimidazole derivative |
| GB201519054D0 (en) * | 2015-10-28 | 2015-12-09 | Univ Dundee | Composition |
| CN108884044B (zh) | 2016-02-26 | 2023-01-31 | 大塚制药株式会社 | 哌啶衍生物 |
| EP3962903A1 (en) * | 2019-05-01 | 2022-03-09 | Boehringer Ingelheim International GmbH | (r)-(2-methyloxiran-2-yl)methyl 4-bromobenzenesulfonate |
| CN114249747A (zh) * | 2021-12-27 | 2022-03-29 | 苏州虞美景盛新药开发有限公司 | 一种用于治疗广泛耐药性结核病的硝基咪唑吡喃类药物的新合成工艺 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SU1271861A1 (ru) * | 1985-05-07 | 1986-11-23 | Иркутский институт органической химии СО АН СССР | Способ получени 2,2-диметил-3-цианометилиденбензимидазо @ 2,1- @ -1,3-оксазолидина |
| EP1555267A1 (en) * | 2002-10-11 | 2005-07-20 | Otsuka Pharmaceutical Co., Ltd. | 2,3-DIHYDRO-6-NITROIMIDAZO 2,1-b OXAZOLES |
| EP1678185B1 (en) * | 2003-10-31 | 2008-10-08 | Otsuka Pharmaceutical Co., Ltd. | 2,3-dihydro-6-nitroimidazo [2,1-b] oxazole compounds for the treatment of tuberculosis |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL41744A0 (en) | 1972-03-16 | 1973-05-31 | Ciba Geigy Ag | New aryl ether derivatives,their production and their use as pesticides |
| CH568715A5 (ru) | 1972-03-16 | 1975-11-14 | Ciba Geigy Ag | |
| FR2575158B1 (fr) | 1984-12-20 | 1987-10-02 | Pf Medicament | Arylalcoyloxymethyl-2 morpholines, leur preparation et leur application en tant que medicaments utiles dans le traitement des troubles du systeme nerveux central |
| US5668127A (en) * | 1995-06-26 | 1997-09-16 | Pathogenesis Corporation | Nitroimidazole antibacterial compounds and methods of use thereof |
| US6087358A (en) | 1995-06-26 | 2000-07-11 | Pathogenesis Corporation | Nitro-[2,1-b]imidazopyran compounds and antibacterial uses thereof |
| JP4787529B2 (ja) | 2004-04-09 | 2011-10-05 | 大塚製薬株式会社 | 医薬組成物 |
| JP4789966B2 (ja) | 2004-04-09 | 2011-10-12 | 大塚製薬株式会社 | 医薬組成物 |
| US7115736B2 (en) | 2004-09-17 | 2006-10-03 | Daiso Co., Ltd. | Process for preparing imidazopyran derivatives |
| CN101268084A (zh) * | 2005-07-28 | 2008-09-17 | 沃泰克斯药物股份有限公司 | 天冬氨酸特异性半胱氨酸蛋白酶抑制剂前体药物 |
| JP2009521464A (ja) | 2005-12-23 | 2009-06-04 | ノバルティス アクチエンゲゼルシャフト | ニトロイミダゾール化合物 |
| US20070213341A1 (en) | 2006-03-13 | 2007-09-13 | Li Chen | Spiroindolinone derivatives |
| US7678791B2 (en) | 2006-07-12 | 2010-03-16 | Cumbre Ip Ventures, L.P. | Nitroheteroaryl-containing rifamycin derivatives |
| MX2009009447A (es) | 2007-03-09 | 2009-09-14 | Sanofi Aventis | Dihidro y tetrahidro oxazolopirimidinonas sustituidas, preparacion y uso de las mismas. |
| CN101965353B (zh) | 2008-03-26 | 2014-12-17 | 丹诺医药(苏州)有限公司 | 共价连接到取代苯基噁唑烷酮的双环硝基咪唑类 |
-
2010
- 2010-07-30 RU RU2012107180/04A patent/RU2540860C2/ru active
- 2010-07-30 US US12/847,459 patent/US8293734B2/en active Active
- 2010-07-30 AU AU2010278779A patent/AU2010278779B2/en active Active
- 2010-07-30 JP JP2012523088A patent/JP5685254B2/ja active Active
- 2010-07-30 BR BR112012002214-8A patent/BR112012002214B1/pt active IP Right Grant
- 2010-07-30 KR KR1020127005331A patent/KR101738868B1/ko active IP Right Grant
- 2010-07-30 CN CN201080041655.3A patent/CN102741259B/zh active Active
- 2010-07-30 CA CA2769263A patent/CA2769263C/en active Active
- 2010-07-30 NZ NZ598165A patent/NZ598165A/xx unknown
- 2010-07-30 WO PCT/US2010/043908 patent/WO2011014776A1/en active Application Filing
- 2010-07-30 ES ES10740807.2T patent/ES2530266T3/es active Active
- 2010-07-30 EP EP10740807.2A patent/EP2459570B1/en active Active
-
2012
- 2012-02-20 ZA ZA2012/01259A patent/ZA201201259B/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SU1271861A1 (ru) * | 1985-05-07 | 1986-11-23 | Иркутский институт органической химии СО АН СССР | Способ получени 2,2-диметил-3-цианометилиденбензимидазо @ 2,1- @ -1,3-оксазолидина |
| EP1555267A1 (en) * | 2002-10-11 | 2005-07-20 | Otsuka Pharmaceutical Co., Ltd. | 2,3-DIHYDRO-6-NITROIMIDAZO 2,1-b OXAZOLES |
| EP1678185B1 (en) * | 2003-10-31 | 2008-10-08 | Otsuka Pharmaceutical Co., Ltd. | 2,3-dihydro-6-nitroimidazo [2,1-b] oxazole compounds for the treatment of tuberculosis |
Non-Patent Citations (1)
| Title |
|---|
| Hirofumi Saaki, et al: ";Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles";, J.Med.Chem., 2006, 49, pp.7854-7860 . * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2744784C1 (ru) * | 2017-03-28 | 2021-03-15 | Инститьют Оф Матириа Медика, Чайниз Акэдеми Оф Медикал Сайенсиз | Бензоксазин-оксазолидиноновое соединение, замещенное азотсодержащим гетероциклом, способ его получения и применение |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5685254B2 (ja) | 2015-03-18 |
| US20110028466A1 (en) | 2011-02-03 |
| EP2459570B1 (en) | 2014-12-24 |
| ES2530266T3 (es) | 2015-02-27 |
| CN102741259A (zh) | 2012-10-17 |
| CA2769263C (en) | 2017-03-07 |
| WO2011014776A1 (en) | 2011-02-03 |
| CN102741259B (zh) | 2016-04-13 |
| AU2010278779A1 (en) | 2012-03-08 |
| US8293734B2 (en) | 2012-10-23 |
| CA2769263A1 (en) | 2011-02-03 |
| ZA201201259B (en) | 2013-08-28 |
| BR112012002214B1 (pt) | 2022-01-25 |
| NZ598165A (en) | 2013-05-31 |
| BR112012002214A2 (pt) | 2020-08-18 |
| KR20120084714A (ko) | 2012-07-30 |
| RU2012107180A (ru) | 2013-09-10 |
| EP2459570A1 (en) | 2012-06-06 |
| KR101738868B1 (ko) | 2017-05-23 |
| AU2010278779B2 (en) | 2014-08-14 |
| JP2013500997A (ja) | 2013-01-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2540860C2 (ru) | Нитроимидазооксазиновые и нитроимидазооксазольные аналоги и их применение | |
| AU2014221775B2 (en) | TETRAHYDROIMIDAZO[1,5-d][1,4]OXAZEPINE DERIVATIVE | |
| EP3116858B1 (en) | Azaspiro derivatives as trpm8 antagonists | |
| JP5820921B2 (ja) | 1,2−二置換複素環式化合物 | |
| EP3544979B1 (en) | Oxadiazolones as transient receptor potential channel inhibitors | |
| EP2895490B1 (en) | Glucopyranosyl derivatives and their uses in medicine | |
| DE60019158T2 (de) | Neue oxabispidin-verbindungen zur behandlung von herzarrhythmien | |
| WO2016173425A1 (en) | A glucopyranosyl derivative and preparation method and uses thereof | |
| RU2542988C2 (ru) | Нитроимидазооксазины и их применения при противотуберкулезной терапии | |
| EP3768260A1 (en) | Oxadiazole transient receptor potential channel inhibitors | |
| WO2007075872A2 (en) | Nitroimidazole compounds | |
| CN111094267B (zh) | 作为RORγ抑制剂的磺酰基取代的双环化合物 | |
| US20230159521A1 (en) | 1h-pyrazolo[4,3-g]isoquinoline and 1h-pyrazolo[4,3-g]quinoline derivatives as alpha-1-antitrypsin modulators for treating alpha-1-antitrypsin deficiency (aatd) | |
| CN118632850A (zh) | Apol1的螺环抑制剂和其使用方法 | |
| WO2020125627A1 (zh) | 苯基吡咯烷类化合物及其用途 | |
| JP6231107B2 (ja) | マクロライド誘導体、その製造およびその治療的使用 | |
| US11161837B2 (en) | Benzofuran derivatives for the treatment of hepatitis C | |
| US9198913B2 (en) | Nitroimidazooxazines and their uses in anti-tubercular therapy |