RU2151765C1 - Способ получения лактама - Google Patents
Способ получения лактама Download PDFInfo
- Publication number
- RU2151765C1 RU2151765C1 RU97114807/04A RU97114807A RU2151765C1 RU 2151765 C1 RU2151765 C1 RU 2151765C1 RU 97114807/04 A RU97114807/04 A RU 97114807/04A RU 97114807 A RU97114807 A RU 97114807A RU 2151765 C1 RU2151765 C1 RU 2151765C1
- Authority
- RU
- Russia
- Prior art keywords
- equal
- alumina
- specific surface
- catalyst
- pore volume
- Prior art date
Links
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D201/00—Preparation, separation, purification or stabilisation of unsubstituted lactams
- C07D201/02—Preparation of lactams
- C07D201/08—Preparation of lactams from carboxylic acids or derivatives thereof, e.g. hydroxy carboxylic acids, lactones or nitriles
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/584—Recycling of catalysts
Abstract
Изобретение относится к получению алифатических лактамов, в частности Е-капролактама, используемых в производстве полиамидов. Лактам получают гидролизом водой в паровой фазе при 200 - 450°С алифатического аминонитрила, включающего алкил C3-C12. В качестве катализатора используют оксид алюминия с удельной поверхностью 10 - 280 м2/г и с объемом пор, имеющих диаметр пор более 500
Description
Настоящее изобретение касается получения лактама путем циклизирующего гидролиза соответствующего аминонитрила.
Алифатические лактамы, например, ε - капролактам, представляет собой основные соединения для получения полиамидов (полиамид 6 из капролактама).
Один из известных способов получения лактамов заключается в проведении циклизирующего гидролиза соответствующих аминонитрилов, в частности, алифатических, не разветвленных аминонитрилов путем пропускания через паровую фазу с присутствием воды на твердом катализаторе.
В патенте US 2 357 484 описан способ получения лактама в паровой фазе, при котором смесь из воды и аминонитрила пропускают через катализатор обезвоживания, которым является, например, активированная окись алюминия, гель двуокиси кремния или борофосфорная кислота.
Патент US 4 628 085 предлагает способ получения лактама в паровой фазе, при котором приводят в контакт алифатический или ароматический аминонитрил и воду с катализатором на основе двуокиси кремния в виде сферических частиц с удельной поверхностью BET более 250 м2г при среднем диаметре пор менее 20 нм и, как правило, в присутствии водорода, и аммиака.
Катализаторы, используемые в известных из уровня техники способах, позволяют в некоторых случаях достигать достаточно высоких результатов по селективности лактама, но в то же время может происходить также быстрая потеря их активности, что вызывает очень большую трудность при промышленном применении указанных способов.
Кроме того, способ по патенту US 4 628 085 предусматривает использование очень сложной реакционной смеси с проведением в конце реакции операций по разделению и рециклированию, существенно усложняющих данный способ.
Настоящее изобретение предлагает новые катализаторы из оксида алюминия, которые при обеспечении достаточно высокой селективности реакции превращения аминонитрилов в лактамы характеризуются большим сроком службы и, следовательно, нуждаются в менее частой регенерации.
Более конкретно, изобретение относится к способу получения лактама путем взаимодействия в паровой фазе алифатического аминонитрила общей формулы (1)
N≡C-R-NH2, (1)
в которой R означает алкиленовый радикал с 3-12 атомами углерода, с водой в присутствии твердого катализатора, заключающемуся в том, что катализатором является оксид алюминия с удельной поверхностью, измеряемой по методу BET, превышающей или равной 10 м2/г.
N≡C-R-NH2, (1)
в которой R означает алкиленовый радикал с 3-12 атомами углерода, с водой в присутствии твердого катализатора, заключающемуся в том, что катализатором является оксид алюминия с удельной поверхностью, измеряемой по методу BET, превышающей или равной 10 м2/г.
Предпочтительно, чтобы используемый в способе согласно изобретению оксид алюминия имел удельную поверхность, равную или менее 500 м2/г.
Среди аминонитрилов формулы (1) наиболее подходящими являются аминонитрилы, позволяющие получать лактамы, используемые в качестве исходного сырья для получения полиамидов 4, 5, 6 и 10, т.е. аминонитрилы, в формуле которых символ означает линейный алкиленовый радикал с 3, 4, 5 и 9 атомами углерода.
Предпочтительным соединением формулы (1) является 6-аминокапронитрил (или ε-капронитрил), позволяющий получать капролактам, полимеризация которого обеспечивает получение полиамида 6.
Оксиды алюминия, пригодные для использования в данном способе, это в первую очередь те, которые имеют удельную поверхность более или равную 10 м2/г и менее или равную 280 м2/г с объемом пор диаметром более 500 Ангстрем, превышающим или равным 10 мл/100 г.
Удельная поверхность по BET - это поверхность, которая определяется адсорбцией азота согласно стандарту ASTM D 3663-78, установленному на основании метода BRUNAUER-EMMET-TELLER, описанного в периодическом издании "The Journal of the American Society" (журнал Американского общества), 60, 309 (1938г.).
Объем пор диаметром свыше 500  представляет собой суммарный объем, образованный всеми порами диаметром более 500
представляет собой суммарный объем, образованный всеми порами диаметром более 500  Этот объем замеряется методом пенетрации ртути, при котором используется закон Кельвина.
Этот объем замеряется методом пенетрации ртути, при котором используется закон Кельвина.
Предпочтительно, чтобы оксиды алюминия указанной первой группы имели объем пор диаметром свыше 500  превышающий или равный 20 мл/100 г, еще более предпочтительно - более или равный 30 мл/100 г.
превышающий или равный 20 мл/100 г, еще более предпочтительно - более или равный 30 мл/100 г.
Предпочтительно также, чтобы оксиды алюминия указанной первой группы имели удельную поверхность более или равную 50 м2/г.
Оксидами алюминия, также пригодными для применения в настоящем способе, являются оксиды, у которых удельная поверхность превышает или равна 50 м2/г и ниже или равна 280 м2/г, а объем пор диаметром более 70  превышает или равен 30 мл/100 г.
превышает или равен 30 мл/100 г.
Предпочтительно, чтобы оксиды алюминия этой второй группы имели объем пор диаметром более 70  превышающий или равный 45 мл/100 г.
превышающий или равный 45 мл/100 г.
Предпочтительно также, чтобы оксиды алюминия указанной второй группы имели удельную поверхность более или равную 80 м2/г.
Оксидами алюминия, пригодными для применения в данном способе, являются также оксиды, у которых удельная поверхность превышает или равна 280 м2/г, а общий объем пор превышает или равен 15 мл/100 г.
Предпочтительно, чтобы оксиды алюминия такой третьей группы имели общий объем пор более или равный 22 мл/100 г, а еще более предпочтительно, чтобы он превышал или был равен 30 мл/100 г.
Кроме того, оксиды алюминия характеризуются и своей кислотностью.
Упомянутая кислотность может измеряться путем теста на изомеризацию 1-бутена в 2-бутен.
Такой тест основывается на реакции изомеризации 1-бутена до образования смеси цис-2-бутена с транс-2-бутеном при температуре Т (в данном случае Т= 400oC).
Реакция изомеризации характеризуется термодинамическим равновесием. Можно выделить две константы:
- константа теоретического равновесия КT(Т), рассчитываемая по формуле

где [бутен] эк. обозначает концентрацию каждого изомера в равновесном состоянии при температуре Т;
- константа фактического равновесия К(Т), рассчитываемая по результату замеров

где [бутен] означает концентрацию каждого изомера на выходе из реактора при температуре Т.
- константа теоретического равновесия КT(Т), рассчитываемая по формуле
где [бутен] эк. обозначает концентрацию каждого изомера в равновесном состоянии при температуре Т;
- константа фактического равновесия К(Т), рассчитываемая по результату замеров
где [бутен] означает концентрацию каждого изомера на выходе из реактора при температуре Т.
Изомеризующая активность (A) оксидов алюминия определяется активностью по отношению к равновесному состоянию
 .
.
На практике тестирование проводят в реакторе с паровой фазой, работающем в импульсном режиме, при этом в него вводят 500 мг измельченного оксида алюминия (размер частиц от 400 до 500 мкм). Оксид алюминия выдерживают в течение 2 часов при температуре 250oC в потоке гелия при расходе 2,5 л/ч. Затем оксид алюминия нагревают до температуры 400oC и над окисью алюминия впрыскивают в поток гелия 1 мл 1-бутена. Анализ газов на выходе производили с помощью газофазовой хроматографии, позволяющей замерять количества уловленных 1-бутена и 2-бутена цис и транс.
Полученная изомеризующая активность (A) корректируется по изомеризующей активности катализатора, полученной при тех же условиях в пустом реакторе. Скорректированная изомеризующая активность AC представляет собой кислотность упомянутых оксидов алюминия.
Когда содержание щелочного или щелочно-земельного металла, содержащегося в оксиде алюминия, составляет менее 60 ммол на 100 г оксида алюминия, то, чем выше значение AC, тем выше кислотность оксида алюминия.
Как правило, оксид алюминия получают дегидратацией гиббсита, байерита, нордстрандита или их смесей. Можно, например, в этой связи указать на энциклопедию KIR-OTHMER, т.2, стр. 291-297.
Оксиды алюминия, применяемые в данном способе, могут быть получены путем контактирования гидратированного оксида алюминия в тонко измельченном виде с потоком горячего газа при температуре от 400 до 1000oC, при продолжительности указанного контакта от доли секунды до 10 секунд с последующим разделением частично дегидратированного оксида алюминия и горячих газов. Здесь можно сослаться в особенности на способ, который описан в американском патенте US 2 915 365.
Можно также применить автоклавную обработку агломерата полученных выше оксидов алюминия в водной среде, при необходимости в присутствии кислоты, при температуре свыше 100oC, предпочтительно при температуре от 150 до 250oC, в течение времени 1-20 часов, затем его сушку и прокаливание.
Температура прокаливания регулируется с таким расчетом, чтобы можно было получить удельные поверхности и объемы пор, соответствующие диапазонам указанных выше величин.
В связи с их основными принципами получения оксиды алюминия, применяемые в данном способе, чаще всего содержат натрий, содержание которого выражается обычно в массе Na2O к массе оксида алюминия.
Катализатор может применяться в разных видах, например, в виде порошка, шариков, дробленого материала, продуктов экструзии и таблетирования, формование может производиться при необходимости с помощью связующего вещества.
Могут применяться предпочтительно шарики из оксида алюминия, получаемые капельной коагуляцией. Такой тип шариков можно получать, например, по способу, описанному в патентах EP-A-0 015 801 и EP-A-0 097 539. Контроль за пористостью может осуществляться, в частности, по способу, описанному в патенте EP-A-0 097 539, посредством капельной коагуляции водной суспензии или водной дисперсии оксида алюминия или раствора основной соли алюминия, находящейся в виде эмульсии, состоящей из органической фазы, водной фазы, поверхностно-активного вещества или эмульгатора. Указанной органической фазой может быть, в частности, углеводород.
Может применяться и дробленый оксид алюминия. Дробленый материал может быть получен путем дробления любого материала на основе оксида алюминия, например, шариков, получаемых любым методом (капельной коагуляцией, во вращающемся барабане) или продуктов экструзии. Контроль за пористостью дробленых материалов достигается выбором материала на основе оксида алюминия.
Могут применяться и продукты экструзии из оксида алюминия. Последние могут быть получены растиранием с последующей экструзией материала на основе оксида алюминия, причем указанный материал может быть получен в результате быстрой дегидратации гидраргиллита или осаждения геля оксида алюминия. Контроль за пористостью таких экструдатов может производиться путем выбора используемого оксида алюминия и условиями получения указанного оксида алюминия или режимом растирания оксида алюминия перед экструзией так, что оксид алюминия может быть смешан в процессе растирания с порообразующими агентами. В качестве примера экструдаты могут быть приготовлены по способу, раскрытому в патенте US 3 856 708.
В некоторых случаях может оказаться целесообразным, чтобы по меньшей мере часть свободного объема реактора была заполнена инертным твердым веществом, например, кварцем, с тем, чтобы повысить парообразование и диспергирование реактивов.
Для протекания реакции циклизирующего гидролиза требуется присутствие воды. Молярное соотношение между вводимыми водой и аминонитрилом составляет обычно 0,5-50, предпочтительно 1-20. Соотношение с большим значением не является критичным для изобретения, однако более высокие соотношения почти не представляют интереса с точки зрения экономичности.
Аминонитрил и вода могут вводиться в виде смеси в парообразном состоянии или вводиться в реактор раздельно. Можно производить предварительное испарение реактивов, которые затем подают в смесительную камеру.
Можно использовать без всякого ущерба любой инертный газ в качестве газа-носителя, например, азот, гелий или аргон.
Температура осуществления способа согласно изобретению должна быть достаточной для поддержания реактивов в парообразном состоянии. Она составляет, как правило, от 200 до 450oC, предпочтительно от 250 до 400oC.
Время контактирования аминонитрила с катализатором не является критическим. Оно может колебаться в зависимости от применяемой аппаратуры. Контактное время составляет преимущественно от 0,5 до 200 секунд, предпочтительно от 1 до 100 секунд.
Давление не является критическим параметром способа. Так, например, процесс можно вести при давлениях от 10-3 бар до 200 бар. Предпочтительно, чтобы способ осуществлялся при давлении от 0,1 до 20 бар.
Возможно использование инертного в условиях реакции растворителя, например, алкана, циклоалкана, ароматического углеводорода или одного из их галогенированных производных и обеспечить таким образом жидкую фазу в реакционном потоке.
Приводимые ниже примеры служат иллюстрацией к изобретению.
ПРИМЕРЫ 1-4
В цилиндрический реактор емкостью 20 мл из пирексового стекла, установленный вертикально и снабженный средствами нагрева, отверстиями для подачи и выхода газовых потоков и системой подачи реактивов, последовательно загружали 10 мл кварца, 1 мл катализатора в виде порошка с гранулометрией 0,8-1,25 мкм (природа катализатора приведена ниже в таблице 1) и снова 10 мл кварца.
В цилиндрический реактор емкостью 20 мл из пирексового стекла, установленный вертикально и снабженный средствами нагрева, отверстиями для подачи и выхода газовых потоков и системой подачи реактивов, последовательно загружали 10 мл кварца, 1 мл катализатора в виде порошка с гранулометрией 0,8-1,25 мкм (природа катализатора приведена ниже в таблице 1) и снова 10 мл кварца.
Загруженный таким образом реактор нагревали до 400oС в потоке воздуха (при расходе 1,5 л/ч) в течение 2 часов. Затем реактор охлаждали до 320oC (выбранная температура реакции) и помещали под поток азота (при расходе 1 л/ч).
Затем с помощью насоса подавали смесь, состоящую из 6-аминокапронитрила (ACN) и воды (весовое соотношение: 50/50, т.е. молярное соотношение вода/ACN : 6,2). Скорость подачи смеси составляла 1,2 мл/ч.
На выходе из реактора пары конденсировали в стеклянной ловушке при температуре окружающей среды в течение 2 часов.
Конечная реакционная смесь анализировалась при хроматографии в паровой фазе.
Определяли степень преобразования (ТТ) аминокапронитрила, выход (RT) капролактама по отношению к преобразованному аминокапронитрилу и активность катализатора в течение 2 часов протекания реакции, измеряемой в граммах образованного капролактама на миллиметр каталитического слоя в час.
Оксиды алюминия, используемые в качестве катализаторов, характеризовались следующими показателями:
- оксид алюминия 7: - кислотность AC(400oC)=62%; удельная поверхность (SS)=81 м2/г; 0,0714% Na2O; объем пор диаметром более 500 27 мл/100 г.
27 мл/100 г.
- оксид алюминия 7: - кислотность AC(400oC)=62%; удельная поверхность (SS)=81 м2/г; 0,0714% Na2O; объем пор диаметром более 500
- оксид алюминия 6: - кислотность AC(400oC) = 65%; удельная поверхность (SS) = 244 м2/г; 0,0730% Na2O; объем пор диаметром более 500  12 мл/100 г.
12 мл/100 г.
- оксид алюминия 16: - кислотность AC(400oC) = 65%; удельная поверхность (SS): 314 м2/г; 0,3640% Na2O; общий объем пор 40 мл/100 г.
- оксид алюминия 10: - кислотность AC(400oC) = 99%; удельная поверхность (SS): 217 м2/г; 0,0030% Na2O; объем пор диаметром более 70  45 мл/100 г.
45 мл/100 г.
В таблице 1 приведены полученные результаты.
ПРИМЕРЫ 5-7
Повторяют опыт согласно примерам 1-3, исследуя активность различных катализаторов, используемых в течение 32 часов.
Повторяют опыт согласно примерам 1-3, исследуя активность различных катализаторов, используемых в течение 32 часов.
В таблице 2 представлены значения активности каждого катализатора при возрастающем времени реакции.
Можно видеть, что используемые оксиды алюминия не теряли своей каталитической активности на протяжении по меньшей мере 32 часов.
ПРИМЕРЫ 8-21 И СРАВНИТЕЛЬНЫЙ ПРИМЕР 1
В цилиндрический реактор емкостью 20 мл из периксового стекла, установленный вертикально и снабженный средствами нагрева, отверстиями для подачи и выхода газовых потоков и системой подачи реактивов, последовательно загружали 2 мл кварца, 5 мл катализатора с гранулометрией 1-5 мм и снова 5 мл кварца.
В цилиндрический реактор емкостью 20 мл из периксового стекла, установленный вертикально и снабженный средствами нагрева, отверстиями для подачи и выхода газовых потоков и системой подачи реактивов, последовательно загружали 2 мл кварца, 5 мл катализатора с гранулометрией 1-5 мм и снова 5 мл кварца.
Загруженный таким образом реактор нагревали до 350oC в потоке азота (при расходе 5,2 л/ч) в течение 2 часов. Затем реактор охлаждали до 250oC (выбранная температура реакции) и помещали в поток азота (при расходе 5,2 л/ч).
Затем с помощью насоса подавали смесь, состоящую из 6-аминокапронитрила (ACN) и воды (молярное соотношение вода/ACN: 2,9). Скорость подачи жидкой смеси составляла 14 г/ч.
На выходе из реактора пары конденсировали в стеклянной ловушке при температуре окружающей среды в течение 2 часов.
Конечная реакционная смесь анализировалась методом хроматографии в паровой фазе.
Определяли степень образования (TT) аминокапронитрила, выход (RT) капролактама по отношению к преобразованному аминокапронитрилу и активность катализатора в течение 2 часов протекания реакции, измеряемой в граммах образовавшегося капролактама на грамм катализатора в час (активность a) и в граммах образовавшегося капролактама на миллилитр каталитического слоя в час (активность b).
Степень преобразования 6-аминокапронитрила составляет от 25 до 40% в разных опытах, выход RTкапролактама составил более 90% в примерах 8-20 и 15% в сранительном опыте 1.
Характеристики оксидов алюминия, использованных в качестве катализаторов (удельная поверхность SS, общий объем пор VPT, объем пор диаметром более 500  = V500A
= V500A  объем пор диаметром более 70
объем пор диаметром более 70  = V70
= V70  , и значения активности а и b разных видов оксидов алюминия указаны в таблице 3.
, и значения активности а и b разных видов оксидов алюминия указаны в таблице 3.
ПРИМЕРЫ 22 - 28
В реактор, описанный в предыдущих примерах, последовательно загружали 3 мл кварца, 2 мл катализатора с гранулометрией от 1 до 5 мм и снова 5 мл кварца.
В реактор, описанный в предыдущих примерах, последовательно загружали 3 мл кварца, 2 мл катализатора с гранулометрией от 1 до 5 мм и снова 5 мл кварца.
Загруженный таким образом реактор нагревали до 350oC в потоке азота (при расходе 5,2 л/ч) в течение 2 часов. Затем реактор выдерживали при 350oC (выбранная температура реакции) и помещали в поток азота (при расходе 5,2 л/ч).
Затем с помощью насоса подавали смесь, состоящую из 6-аминокапронитрила (ACN) и воды (молярное соотношение вода/ ACN: 1,1). Скорость подачи смеси составляла 11 г/ч.
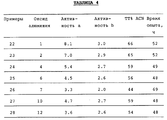
На выходе из реактора пары конденсировали в стеклянной ловушке при температуре окружающей среды в течение времени, приведенном в нижеследующей таблице 4.
Конечную реакционную смесь анализировали путем хроматографии в паровой фазе.
Определяли степень преобразования (TT) аминокапронитрила, выход (RT) капролактама по отношению к преобразованному аминокапронитрилу и активность катализатора в течение времени протекания реакции, измеряемой в граммах образовавшегося капролактама на грамм катализатора в час (активность a) и в граммах образовавшегося капролактама на миллилитр каталитического слоя в час (активность b).
Степень преобразования 6-аминокапронитрила представлена в таблице 4, выход RTкапролактама в примерах 22-28 составил более 90%.
Claims (8)
1. Способ получения лактама путем взаимодействия в паровой фазе алифатического аминонитрила общей формулы:
N ≡ C-R-NH2
в которой R означает алкиленовый радикал с 3 - 12 атомами углерода, с водой в присутствии твердого катализатора, отличающийся тем, что катализатором является оксид алюминия с удельной поверхностью, измеряемой методом ВЕТ, выше или равной 10 м2/г, при этом оксид выбирают из: а) оксидов алюминия с удельной поверхностью менее или равной 280 м2/г и с объемом пор, имеющих диаметр более 500 выше или равным 10 мл/100 г; б) оксидов алюминия с удельной поверхностью более или равной 50 м2/г и менее или равной 280 м2/г, а также с объемом пор, имеющих диаметр более 70
выше или равным 10 мл/100 г; б) оксидов алюминия с удельной поверхностью более или равной 50 м2/г и менее или равной 280 м2/г, а также с объемом пор, имеющих диаметр более 70 выше или равным 30 мл/100 г.
выше или равным 30 мл/100 г.
N ≡ C-R-NH2
в которой R означает алкиленовый радикал с 3 - 12 атомами углерода, с водой в присутствии твердого катализатора, отличающийся тем, что катализатором является оксид алюминия с удельной поверхностью, измеряемой методом ВЕТ, выше или равной 10 м2/г, при этом оксид выбирают из: а) оксидов алюминия с удельной поверхностью менее или равной 280 м2/г и с объемом пор, имеющих диаметр более 500
2. Способ по п.1, отличающийся тем, что оксид алюминия выбирают из оксидов алюминия а) с объемом пор, имеющих диаметр более 500 выше или равным 20 мл/100 г, предпочтительно более или равным 30 мл/100 г.
выше или равным 20 мл/100 г, предпочтительно более или равным 30 мл/100 г.
3. Способ по п.1 или 2, отличающийся тем, что оксид алюминия выбирают из оксидов алюминия а) с удельной поверхностью, выше или равной 50 м2/г.
4. Способ по п.1, отличающийся тем, что оксид алюминия выбирают из оксидов алюминия б) имеющих объем пор диаметров свыше 70 выше или равным 45 мл/100 г.
выше или равным 45 мл/100 г.
5. Способ по пп.1 - 4, отличающийся тем, что оксид алюминия выбирают из оксидов алюминия б) с удельной поверхностью, выше или равной 80 м2/г.
6. Способ по пп.1 - 5, отличающийся тем, что аминонитрилом формулы (I) является 6-аминокапронитрил.
7. Способ по пп.1 - 6, отличающийся тем, что молярное соотношение между вводимыми водой и аминонитрилом составляет от 0,5 до 50, предпочтительно от 1 до 20.
8. Способ по пп.1 - 7, отличающийся тем, что температура, при которой осуществляют способ, составляет от 200 до 450oC, предпочтительно от 250 до 400oC.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9501183A FR2729949A1 (fr) | 1995-01-27 | 1995-01-27 | Procede de preparation de lactame |
| FR9501183 | 1995-01-27 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU97114807A RU97114807A (ru) | 1999-07-10 |
| RU2151765C1 true RU2151765C1 (ru) | 2000-06-27 |
Family
ID=9475751
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU97114807/04A RU2151765C1 (ru) | 1995-01-27 | 1996-01-22 | Способ получения лактама |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US6262259B1 (ru) |
| EP (1) | EP0805801B1 (ru) |
| JP (1) | JP3053114B2 (ru) |
| KR (1) | KR100369885B1 (ru) |
| CN (1) | CN1083832C (ru) |
| AR (1) | AR000777A1 (ru) |
| BR (1) | BR9606939A (ru) |
| CA (1) | CA2211015C (ru) |
| DE (1) | DE69615660T2 (ru) |
| FR (1) | FR2729949A1 (ru) |
| MY (1) | MY132442A (ru) |
| RU (1) | RU2151765C1 (ru) |
| TW (1) | TW396154B (ru) |
| WO (1) | WO1996022974A1 (ru) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2755132B1 (fr) * | 1996-10-24 | 1998-11-27 | Rhone Poulenc Fibres | Procede de traitement de lactames |
| DE19718706A1 (de) * | 1997-05-02 | 1998-11-05 | Basf Ag | Verfahren zur Herstellung cyclischer Lactame |
| DE19738463C2 (de) * | 1997-09-03 | 1999-09-23 | Basf Ag | Verfahren zur Herstellung von Caprolactam |
| DE19753301A1 (de) * | 1997-12-01 | 1999-06-02 | Basf Ag | Verfahren zur Herstellung von Lactamen |
| DE19811880A1 (de) * | 1998-03-18 | 1999-09-23 | Basf Ag | Verfahren zur Herstellung von Lactamen |
| FR2780401B1 (fr) * | 1998-06-25 | 2001-02-09 | Rhone Poulenc Fibres | Procede de vaporisation d'aminonitrile |
| FR2781480B1 (fr) * | 1998-07-22 | 2001-06-01 | Rhone Poulenc Fibres | Procede d'hydrolyse cyclisante d'un compose aminonitrile en lactame |
| FR2781393B1 (fr) * | 1998-07-22 | 2000-08-25 | Rhone Poulenc Fibres | Procede de regeneration d'un catalyseur d'hydrolyse cyclisante d'un aminonitrile en lactame et utilisation du catalyseur regenere pour la fabrication de lactames |
| FR2781796B1 (fr) * | 1998-07-28 | 2000-09-22 | Rhone Poulenc Fibres | Procede de deshydratation de lactame |
| DE19842905A1 (de) * | 1998-09-18 | 2000-03-23 | Basf Ag | Verfahren zur gleichzeitigen Herstellung eines cyclischen Lactams und eines cyclischen Amins |
| FR2786180B1 (fr) | 1998-11-19 | 2001-11-23 | Rhone Poulenc Fibres | Procede de traitement de lactames et procede de purification d'un lactame |
| DE10021201A1 (de) * | 2000-05-03 | 2001-11-08 | Basf Ag | Verfahren zur Herstellung cyclischer Lactame |
| DE10021193A1 (de) * | 2000-05-03 | 2001-11-08 | Basf Ag | Verfahren zur Herstellung cyclischer Lactame |
| US6686465B2 (en) | 2000-05-03 | 2004-02-03 | Basf Aktiengesellschaft | Preparation of cyclic lactams |
| DE10021192A1 (de) | 2000-05-03 | 2001-11-08 | Basf Ag | Verfahren zur Herstellung von Caprolactam |
| US6716977B1 (en) * | 2003-06-17 | 2004-04-06 | E. I. Du Pont De Nemours And Company | Method for making caprolactam from impure ACN wherein ammonia and water are removed from crude caprolactam in a simple separation step and then THA is removed from the resulting caprolactam melt |
| FR2915997B1 (fr) | 2007-05-07 | 2009-07-03 | Rhodia Recherches & Tech | Traitement anti-graffitis. |
| FR2944791B1 (fr) | 2009-04-27 | 2012-02-10 | Rhodia Operations | Procede de preparation de lactames. |
| WO2017132938A1 (en) * | 2016-02-04 | 2017-08-10 | Rhodia Operations | Macroporous catalyst for the preparation of aliphatic amines |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2357484A (en) * | 1941-09-12 | 1944-09-05 | Du Pont | Process for producing compounds containing an n-substituted amide group |
| FR1166597A (fr) * | 1956-02-18 | 1958-11-13 | Degussa | Procédé pour préparer des lactames |
| AT201578B (de) * | 1956-02-18 | 1959-01-10 | Degussa | Verfahren zur Herstellung von Lactamen |
| JPS4821958B1 (ru) * | 1969-01-28 | 1973-07-02 | ||
| FR2449474A1 (fr) * | 1979-02-26 | 1980-09-19 | Rhone Poulenc Ind | Billes d'alumine a double porosite, leur procede de preparation et leurs applications comme supports de catalyseurs |
| FR2527197B1 (fr) * | 1982-05-19 | 1985-06-21 | Rhone Poulenc Spec Chim | Procede de fabrication de billes d'alumine mises en forme par coagulation en gouttes |
| EP0150295A3 (en) * | 1983-12-19 | 1988-03-30 | Allied Corporation | Selective production of n-substituted amides by use of cu(o)/metallic oxides catalyst compositions |
| DE3403574A1 (de) * | 1984-02-02 | 1985-08-08 | Basf Ag, 6700 Ludwigshafen | Verfahren zur gewinnung von caprolactam aus (epsilon)-aminocapronsaeure |
| US4628085A (en) * | 1985-09-03 | 1986-12-09 | Allied Corporation | Use of silica catalyst for selective production of lactams |
| US4625023A (en) | 1985-09-03 | 1986-11-25 | Allied Corporation | Selective conversion of aliphatic and aromatic aminonitriles and/or dinitriles into lactams |
| DE4319134A1 (de) * | 1993-06-09 | 1994-12-15 | Basf Ag | Verfahren zur Herstellung von Caprolactam |
| DE4339648A1 (de) * | 1993-11-20 | 1995-05-24 | Basf Ag | Verfahren zur Herstellung von Caprolactam |
| FR2714379B1 (fr) * | 1993-12-23 | 1996-02-02 | Rhone Poulenc Chimie | Procédé de préparation de lactame. |
-
1995
- 1995-01-27 FR FR9501183A patent/FR2729949A1/fr active Granted
-
1996
- 1996-01-22 US US08/875,451 patent/US6262259B1/en not_active Expired - Lifetime
- 1996-01-22 CA CA002211015A patent/CA2211015C/fr not_active Expired - Fee Related
- 1996-01-22 WO PCT/FR1996/000102 patent/WO1996022974A1/fr active IP Right Grant
- 1996-01-22 DE DE69615660T patent/DE69615660T2/de not_active Expired - Lifetime
- 1996-01-22 KR KR1019970705106A patent/KR100369885B1/ko not_active IP Right Cessation
- 1996-01-22 EP EP96901830A patent/EP0805801B1/fr not_active Expired - Lifetime
- 1996-01-22 CN CN96191604A patent/CN1083832C/zh not_active Expired - Lifetime
- 1996-01-22 JP JP8522668A patent/JP3053114B2/ja not_active Expired - Fee Related
- 1996-01-22 BR BR9606939A patent/BR9606939A/pt not_active Application Discontinuation
- 1996-01-22 RU RU97114807/04A patent/RU2151765C1/ru not_active IP Right Cessation
- 1996-01-23 AR ARP960101111A patent/AR000777A1/es not_active Application Discontinuation
- 1996-01-25 MY MYPI96000284A patent/MY132442A/en unknown
- 1996-02-03 TW TW085101343A patent/TW396154B/zh not_active IP Right Cessation
Non-Patent Citations (1)
| Title |
|---|
| Производство капролактама. /Под ред.Овчинникова В.И. и Ручинского В.Р. - М.: Химия, 1977, с.218 - 244. * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0805801A1 (fr) | 1997-11-12 |
| FR2729949A1 (fr) | 1996-08-02 |
| AR000777A1 (es) | 1997-08-06 |
| KR19980701710A (ko) | 1998-06-25 |
| MY132442A (en) | 2007-10-31 |
| CN1083832C (zh) | 2002-05-01 |
| WO1996022974A1 (fr) | 1996-08-01 |
| TW396154B (en) | 2000-07-01 |
| EP0805801B1 (fr) | 2001-10-04 |
| CA2211015A1 (fr) | 1996-08-01 |
| FR2729949B1 (ru) | 1997-02-28 |
| CN1169146A (zh) | 1997-12-31 |
| CA2211015C (fr) | 2003-04-22 |
| BR9606939A (pt) | 1997-12-23 |
| DE69615660T2 (de) | 2002-04-18 |
| KR100369885B1 (ko) | 2003-05-17 |
| US6262259B1 (en) | 2001-07-17 |
| JPH10506123A (ja) | 1998-06-16 |
| JP3053114B2 (ja) | 2000-06-19 |
| DE69615660D1 (de) | 2001-11-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2151765C1 (ru) | Способ получения лактама | |
| CA1142907A (en) | Hydrogenation catalyst and process | |
| US3647916A (en) | Isoparaffin-olefin alkylation with crystalline zeolite catalysts at low isoparaffin to olefin ratios | |
| RU2119912C1 (ru) | Способ получения циклических лактамов | |
| US4628085A (en) | Use of silica catalyst for selective production of lactams | |
| JPH05103995A (ja) | オレフインの不均化触媒およびその触媒を用いたオレフインの不均化方法 | |
| KR100450103B1 (ko) | 락탐의제조방법 | |
| Kob et al. | Catalysis of the formation of-caprolactam from cyclohexanone oxime by tungsten oxide solid acids | |
| EP0463673B1 (en) | Process for oligomerizing light olefins | |
| US6479658B1 (en) | Method for cyclizing hydrolysis of an aminonitrile compound into lactam | |
| Curtin et al. | The relationship between acidity of aluminas and their selectivity in the conversion of cyclohexanone oxime into caprolactam | |
| RU2205690C2 (ru) | Способ регенерации катализатора циклизирующего гидролиза аминонитрила в лактам и использование регенерированного катализатора для производства лактамов | |
| EP0320570B1 (en) | Process for preparing internal olefins | |
| US3631030A (en) | Production of epsilon-caprolactam | |
| US4085130A (en) | Ammonialytic cleavage of lactams to ω-aminonitriles | |
| MXPA01000749A (en) | Method for cyclizing hydrolysis of an aminonitrile compound into lactam | |
| RU2177827C1 (ru) | Катализатор для дегидрирования парафиновых углеводородов | |
| CN116237049A (zh) | 包括水热处理步骤的用于制备低聚催化剂的方法 | |
| CA1226006A (en) | Process for the production of ethane and/or ethylene from methane and device for its completion | |
| Wilkes | Ethylene addition to lower n-olefins with potassium-graphite catalysts | |
| CZ2001256A3 (cs) | Způsob cyklizační hydrolýzy aminonitrilové sloučeniny na laktam | |
| JPH0684318B2 (ja) | 内部オレフィンの製造法 | |
| JPH0674220B2 (ja) | アルケニルノルボルネンの異性化方法 | |
| JPH0674221B2 (ja) | アルケニルノルボルネンの異性化法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20140123 |