JP7340680B2 - Irak4及びbtkマルチターゲット阻害剤としてのオキサゾール化合物 - Google Patents
Irak4及びbtkマルチターゲット阻害剤としてのオキサゾール化合物 Download PDFInfo
- Publication number
- JP7340680B2 JP7340680B2 JP2022500996A JP2022500996A JP7340680B2 JP 7340680 B2 JP7340680 B2 JP 7340680B2 JP 2022500996 A JP2022500996 A JP 2022500996A JP 2022500996 A JP2022500996 A JP 2022500996A JP 7340680 B2 JP7340680 B2 JP 7340680B2
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- compound
- pharmaceutically acceptable
- acceptable salt
- optionally substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Description
CN201910619602.8、出願日は2019年7月10日であり;
CN201911240843.8、出願日は2019年12月6日であり;
CN202010470469.7、出願日は2020年5月28日である。
本発明はIRAK4及びBTKのマルチターゲット阻害剤、並びにIRAK4及びBTKに関連する疾患を治療する医薬の製造におけるその使用に関する。具体的に、式(II)で表される化合物、その異性体又はその薬学的に許容される塩に関する。
インターロイキン-1受容体関連キナーゼ4(IRAK4)は、セリン/スレオニン特異的プロテインキナーゼであり、チロシンキナーゼ(TLK)ファミリーのメンバーに属し、インターロイキン-1、18、33受容体及びToll様受容体が関与する自然免疫応答の重要なノードである。細胞外シグナル伝達分子がインターロイキン受容体又はToll様受容体に結合した後、動員してMyD88:IRAK4:IRAK1/2ポリタンパク質複合体を形成し、IRAK1/2のリン酸化を引き起こし、一連の下流シグナル伝達を仲介し、それによってp38、JNK及びNF-κBシグナル伝達経路を活性化させ、最終的に炎症性サイトカインの発現を引き起こす。臨床病理学的研究は、IRAK4突然変異を持つ個人は慢性肺疾患、炎症性腸疾患に対して保護効果を持っていることを示した。IRAK4の欠陥自体は致命的ではなく、個人は成人期まで生き残ることができ、感染のリスクは年齢とともに減少する。従って、IRAK4は重要な治療標的となり、炎症性疾患、免疫疾患、腫瘍性疾患等の様々な疾患の治療に広く使用できる。以下の図のように、BAY-1830839とBAY-1834845はバイエル社が開発した小分子IRAK4阻害剤であり、現在、免疫疾患と腫瘍性疾患の臨床研究は既に行われていた。

0%を占めるMyD88L265P点突然変異をメーインとし、IRAK4阻害剤は、異常に活性化されたMyD88シグナル伝達経路を効果的にブロックでき、NF-κB経路の異常な活性化をさらにブロックする。ただし、MyD88L265P突然変異を有するABC-DLBCL患者は、異常なMyD88シグナル伝達経路を有するため、BCR阻害剤に対する反応が悪く、バイエル、ニンバス及びアストラゼネカの多量の研究データは、ABC-DLBCL異種移植動物モデルにおいてIRAK4阻害剤とBTK阻害剤の併用はイブルチニブの生体内有効性を大幅に向上できることを示している。BCR経路とMyD88経路の異常を同時に効果的に抑制できれば、ABC-DLBCLを治療するより効果的な経路であり、RAK4とびBTKの二重標的阻害剤の開発によりNF-κB経路の遮断において2倍の効果が得られ、薬理学的メカニズムの観点から、これは非常に効率的かつ効果的な戦略であり、ABC-DLBCL患者に潜在的に効果的な新しい治療法を提供する。
本発明は式(II)で表される化合物、その異性体又はその薬学的に許容される塩を提供し、

R1はH、F、Cl、Br、I、OH、NH2、CN、C1-6アルキル、シクロプロピル、及び-C(=O)-NH2であり、前記C1-6アルキル、シクロプロピル、及び-C(=O)-NH2は1、2又は3個のRaで任意に置換され;
R2はチエニル、フェニル、ピリジル、シクロプロピル、シクロヘキシル、及び

から選択され、前記チエニル、フェニル、ピリジル、シクロプロピル、シクロヘキシル、及び

は1、2、3、4又は5個のRbで任意に置換され;
R3はC1-6アルキルから選択され、前記C1-6アルキルは1、2又は3個のRcで任意に置換され;
Raはそれぞれ独立してF、OH、NH2及びCNから選択され;
Rbはそれぞれ独立してH、D、F、Cl、Br、I、OH、NH2、CN、C1-3アルキル、COOH、-C(=O)-C1-3アルキル、-C(=O)-O-C1-3アルキル、及び-C(=O)-NH2から選択され、前記OH、NH2、C1-3アルキル、-C(=O)-C1-3アルキル、-C(=O)-O-C1-3アルキル及び-C(=O)-NH2は任意に1、2又は3個のRで任意に置換され;
Rcはそれぞれ独立してF、OH、NH2、CN、CH3、COOH及び-SO2CH3から選択され;
Rはそれぞれ独立してF、OH、NH2及びCH3から選択される。

R1はH、F、Cl、Br、I、OH、NH2、CN、C1-6アルキル及びシクロプロピルであり、前記C1-6アルキル及びシクロプロピルは1、2又は3個のRaで任意に置換され;
R2はチエニル、フェニル、ピリジル、シクロプロピル、シクロヘキシル、及び

から選択され、前記チエニル、フェニル、ピリジル、シクロプロピル、シクロヘキシル、及び

は1、2、3、4又は5個のRbで任意に置換され;
R3はC1-6アルキルから選択され、前記C1-6アルキルは1、2又は3個のRcで任意に置換され;
Raはそれぞれ独立してF、OH、NH2及びCNから選択され;
Rbはそれぞれ独立してH、D、F、Cl、Br、I、OH、NH2、CN、C1-3アルキル、-C(=O)-C1-3アルキル及び-C(=O)-O-C1-3アルキルから選択され、前記OH、NH2、C1-3アルキル、-C(=O)-C1-3アルキル及び-C(=O)-O-C1-3アルキルは1、2又は3個のRで任意に置換され;
Rcはそれぞれ独立してF、OH、NH2、CN、CH3、COOH及び-SO2CH3から選択され;
Rはそれぞれ独立してF、OH、NH2及びCH3から選択される。
本発明の幾つかの実施の態様において、前記R1はH、F、Cl、Br、I、OH、NH2、CN、C1-3アルキル及びシクロプロピルから選択され、前記C1-3アルキル及びシクロプロピルは1、2又は3個のRaで任意に置換され、他の変数は本発明で定義された通りである。

及び-C(=O)-NH2から選択され、他の変数は本発明で定義された通りである。

から選択され、他の変数は本発明で定義された通りである。

から選択され、前記OH、NH2、CH3、CH2CH3、CH2CH2CH3、CH(CH3)2、

は1、2又は3個のRで任意に置換され、他の変数は本発明で定義された通りである。

から選択され、前記OH、NH2、CH3、CH2CH3、CH2CH2CH3、CH(CH3)2、

は1、2又は3個のRで任意に置換され、他の変数は本発明で定義された通りである。

から選択され、他の変数は本発明で定義された通りである。

から選択され、前記チエニル、フェニル、ピリジル、シクロプロピル、シクロヘキシル、

は1、2、3、4又は5個のRbで任意に置換され、他の変数は本発明で定義された通りである。

から選択され、他の変数は本発明で定義された通りである。

から選択され、他の変数は本発明で定義された通りである。

から選択され、他の変数は本発明で定義された通りである。
された通りである。

から選択され、他の変数は本発明で定義された通りである。

R1はH、F、Cl、Br、I、OH、NH2、CN、C1-6アルキル及びシクロプロピルであり、前記C1-6アルキル及びシクロプロピルは1、2又は3個のRaで任意に置換され;
R2はフェニル、ピリジル、シクロプロピル、シクロヘキシル及び

から選択され、前記フェニル、ピリジル、シクロプロピル、シクロヘキシル、及び

は1、2又は3個のRbで任意に置換され;;
R3はC1-6アルキルから選択され、前記C1-6アルキルは1、2又は3個のRcで任意に置換され;
Raはそれぞれ独立してF、OH、NH2及びCNから選択され;
Rbはそれぞれ独立してH、F、OH、NH2、CN、CH3、-C(=O)-C1-3アルキル及び-C(=O)-O-C1-3アルキルから選択され、前記CH3、-C(=O)-C1-3アルキル及び-C(=O)-O-C1-3アルキルは1、2又は3個のRで任意に置換され;
Rcはそれぞれ独立してF、OH、NH2、CN、CH3及びC(=O)から選択され;
Rはそれぞれ独立してF、OH及びNH2から選択される。

から選択され、他の変数は本発明で定義された通りである。

から選択され、前記CH3、

はRで任意に置換され、他の変数は本発明で定義された通りである。

から選択され、他の変数は本発明で定義された通りである。

から選択され、前記フェニル、ピリジル、シクロプロピル、シクロヘキシル、

は1、2又は3個のRbで任意に置換され、他の変数は本発明で定義された通りである。

から選択され、他の変数は本発明で定義された通りである。

から選択され、他の変数は本発明で定義された通りである。

から選択され、他の変数は本発明で定義された通りである。

R1はH、F、Cl、Br、I、OH、NH2、CN及びC1-6アルキルであり、前記C1-6アルキルは1、2又は3個のRaで任意に置換され;
R2はC3-8シクロアルキル及び3~8員ヘテロシクロアルキルから選択され、前記C3~8シクロアルキル、3~8員ヘテロシクロアルキルは1、2又は3個のRbで任意に置換され;
L1はC1-6アルキルから選択され、前記C1-6アルキルは1、2又は3個のRcで任意に置換され;
Raはそれぞれ独立してF、Cl、Br、I、OH、NH2及びCNから選択され;
Rbはそれぞれ独立してH、F、Cl、Br、I、OH、NH2、CN、CH3、-C(=O)-C1-3アルキル及び-C(=O)-C1-3アルコキシから選択され;
Rcはそれぞれ独立してF、Cl、Br、I、OH、NH2、CN及びCH3から選択され;
前記3~8員ヘテロシクロアルキルは1、2又は3個の独立して-NH-、N及びOから選択されるヘテロ原子又はヘテロ原子団を含む。
本発明の幾つかの実施の態様において、前記RbはH、F、Cl、Br、I、OH、NH2、CN、CH3、

から選択され、他の変数は本発明で定義された通りである。

から選択され、他の変数は本発明で定義された通りである。

であり、他の変数は本発明で定義された通りである。

又、本発明の幾つかの実施の態様は前記変数の任意の組み合わせからなるものである。


本発明は又、IRAK4及びBTKに関連する疾患を治療する医薬の製造における、前記化合物、その異性体又はその薬学的に許容される塩、或いは前記医薬組成物の使用を提供する。
本発明の化合物は、IRAK4、BTKに対して一般的に良好な阻害活性を示す。本発明の化合物はTHP-1細胞において、一般的に良好な細胞TNF-α生成を阻害する活性を示し、OCI-LY10、OCI-LY3及びTMD-8細胞において、良好な細胞増殖を阻害する活性を示し、ヒトB細胞リンパ腫OCI-LY10細胞皮下異種移植腫瘍モデルにおいて良好な生体内有効性を示した。
別途に説明しない限り、本明細書で用いられる以下の用語及び連語は以下の意味を有する。一つの特定の用語又は連語は、特別に定義されていない限り、不確定又は不明瞭ではなく、普通の定義として理解されるべきである。本明細書で商品名が出た場合、相応の商品又はその活性成分を指す。
酢酸、プロピオン酸、イソ酪酸、マレイン酸、マロン酸、安息香酸、コハク酸、スベリン酸、フマル酸、乳酸、マンデル酸、フタル酸、ベンゼンスルホン酸、p-トルエンスルホン酸、クエン酸、酒石酸やメタンスルホン酸等の類似の酸を含む。本発明の一部の特定の化合物は、塩基性及び酸性の官能基を含有するため、任意の塩基付加塩又は酸付加塩に転換することができる。
別途に説明しない限り、用語「エナンチオマー」又は「光学異性体」とは互いに鏡像の関係にある立体異性体である。
別途に説明しない限り、用語「ジアステレオマー」とは分子が二つ又は複数のキラル中心を有し、かつ分子同士は非鏡像の関係にある立体異性体である。
別途に説明しない限り、楔形実線結合

及び楔形点線結合

で一つの立体中心の絶対配置を、棒状実線結合

及び棒状点線結合

で立体中心の相対配置を、波線

で楔形実線結合

又は楔形点線結合

を、或いは波線

で棒状実線結合

及び棒状点線結合

を表す。

で連結している場合、当該化合物の(Z)形異性体、(E)形異性体、又は2つの異性体の混合物を意味する。例えば、下記の式(A)は、当該化合物が式(A-1)又は式(A-2)の単一の異性体の形で存在するか、又は式(A-1)と式(A-2)の2つの異性体の形で存在することを意味し;下記の式(B)は、当該化合物が式(B-1)又は式(B-2)の単一の異性体の形で存在するか、又は式(B-1)と式(B-2)の2つの異性体の形で存在することを意味する。下記の式(C)は、当該化合物が式(C-1)又は式(C-2)の単一の異性体の形で存在するか、又は式(C-1)と式(C-2)の2つの異性体の形で存在することを意味する。

な組み合わせでのみに安定した化合物になる場合のみ許容される。
一つの置換基がない場合、当該置換基が存在しないことを表し、例えばA-XにおけるXがない場合、当該構造が実際にAとなることを表す。挙げられた置換基に対してその中のどの原子が置換された基に連結されたかを明示しない場合、その置換基はいずれかの原子を通じて結合され、例えば、ピリジルを置換基とする場合、ピリジル上のいずれかの炭素原子を通じて置換された基に連結されることができる。
挙げられた連結基に対してその連結方向を明示しない場合、その連結方向は任意で、例えば、

における連結基Lが-M-W-である場合、-M-W-は左から右への読む順と同様の方向で環Aと環Bを連結して

を構成してもよく、左から右への読む順と反対の方向で環Aと環Bを連結して

を構成してもよい。前記連結基、置換基及び/又はその変形体の組み合わせは、このような組み合わせで安定した化合物になる場合のみ許容される。

直線破線結合

又は波線

で表すことができる。例えば、-OCH3の直線実線結合は、該基の酸素原子を介して他の基に結合されていることを意味する。

中の直線の破線結合は、該基内の窒素原子の両端が他の基に結合されていることを意味する。

中の波線は、当該フェニル基の部位1と2の炭素原子を介して他の基に結合されていることを意味する。

は、当該ピペリジニル基の任意の結合可能な部位が1つの化学結合によって他の基に結合できることを意味し、少なくとも

の四つの結合形態を含み、H原子が-N-に描かれていても、

には

この結合形態の基が含まれるが、1つの化学結合が接続されると、その部位のHは1つ減少して対応する一価ピペリジン基になる。
て分子の残りの部分に結合した1~3個の炭素原子を含むアルキルを指す。前記C1-3アルコキシはC1-2、C2-3、C2及びC3アルコキシ等を含む。C1-3アルコキシの例には、メトキシ、エトキシ、プロポキシ(n-プロポキシ及びイソプロポキシを含む)等が含まれるが、これらに限定されない。
別途に定義しない限り、「C3-8シクロアルキル」という用語は、3~8個の炭素原子で構成される飽和環状炭化水素基を表し、それは単環、二環系を含み、ここで、二環はスピロ環、縮合環及び架橋環を含む。前記C3-8シクロアルキルには、C3-6、C3-5、C4-8、C4-6、C4-5、C5-8又はC5-6シクロアルキル等が含まれ;それは一価、二価又は多価であり得る。C3-8シクロアルキルの実例には、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、ノルボルニルアルキル、[2.2.2]ジシクロオクチル等が含まれるが、これらに限定されない。
シベンジル(PMB)、9-フルオレニルメチル(Fm)及びジフェニルメチル(DPM)のようなアリールメチル、トリメチルシリル(TMS)及びt-ブチルジメチルシリル(TBS)のようなシリル等を含むが、これらに限定されない。


エチルスクシニルクロリド(50.0g)をアセトニトリル(500.0mL)に添加し、均一に攪拌し、トリメチルシリルジアゾメタン(2M、227.8mL)を反応系に滴下し、25℃で0.5時間攪拌した。その後、0℃で臭化水素酸酢酸溶液(93.1g、33%を含有)を反応系に滴下し、25℃で0.5時間攪拌した。反応溶液を減圧濃縮してアセトニトリルを除去し、残留物を酢酸エチル(500.0mL)に注ぎ、飽和炭酸水素ナトリウム水溶液で洗浄した(100mL×3)。有機相を分離し、適量の無水硫酸ナトリウムを添加して乾燥させた。濾過して乾燥剤を除去し、濾液を乾燥するまで減圧濃縮して粗生成物を得た。粗生成物をカラムクロマトグラフィー(溶離液:石油エーテル~石油エーテル:酢酸エチル=10:1)で精製した後、化合物A1を得た。


4-ブロモ-2-メチルピリジン(8.5g)、オキサゾール-4-カルボン酸エチル(7.0g)及びN,N-ジメチルホルムアミド(70.0mL)の混合溶液に酢酸セシウム(2.2g)炭酸セシウム(32.3g)及びトリ(o-トリル)ホスフィン(6.0g)を添加した。混合物を窒素ガスで3回置換した後、100℃で16時間攪拌した。反応溶液を室温まで冷却させた後、珪藻土パッドで濾過した。濾液を減圧濃縮して粗生成物を得た。粗生成物をカラムクロマトグラフィー(溶離液:石油エーテル:酢酸エチル=10:1~0:1)で精製した後、化合物A2-1を得た。
化合物A2-1(6.5g)をメタノール(35.0mL)及び水(35.0mL)に溶解させ、均一に攪拌し、反応系に水酸化ナトリウム(2.2g)を添加し、15℃で2時間攪拌した。減圧濃縮してメタノールを除去し、水層をメチルtert-ブチルエーテルで抽出した(10.0mL×1)。水層を分離し、1Mの塩酸でpH=3に調節した。
水層を減圧濃縮した後、残留物にトルエン(10.0mL)を添加し、均一に攪拌した。濾過し、濾液を減圧濃縮して、化合物A2を得た。LCMS (ESI) m/z = 205.2[M+H]+.1H NMR (400MHz, MeOH-d4) δ =
8.87-8.86 (m, 2H), 8.53 (s, 1H), 8.45 (d, J=6.0 Hz, 1H), 2.89 (s, 3H)。






4-クロロ-5-ニトロ-ピリジン-2-アミン(25.0g)をテトラヒドロフラン(200.0mL)に溶解させた後、ピペリジン(61.3g)を添加した。10℃で12時間攪拌した後、反応溶液を乾燥するまで減圧濃縮し、残留物に酢酸エチル(100.0mL)を添加し、スラリー化させた。濾過し、濾液を収集した。濾液を乾燥するまで減圧濃縮して粗生成物を得、粗生成物をカラムクロマトグラフィー(石油エーテル:酢酸エ
チル=5:1~0:1)で精製して化合物WX001-1を得た。
化合物WX001-1(5.0g)及び中間体A1(5.0g)の混合物を窒素ガスで3回置換した後、在100℃で12時間攪拌した。反応溶液を室温まで冷却させた後、反応溶液を水(200.0mL)に注ぎ、ジクロロメタン(200.0mL×3)を添加して抽出した。有機層を合わせ、適量の無水硫酸ナトリウムで乾燥させた。濾過して乾燥剤を除去し、濾液を乾燥するまで減圧濃縮して粗生成物を得た。粗生成物をカラムクロマトグラフィー(溶離液:ジクロロメタン:メタノール=100:0~10:1)で精製して化合物WX001-2を得た。
ラネーニッケル(3.0g)をWX001-2(3.0g)のEtOH(50.0mL)溶液に添加し、H2(50Psi)、30℃で続いて1時間攪拌した。濾過して触媒を除去し、濾液を乾燥するまで減圧濃縮して化合物WX001-3を得た。
化合物WX001-3(3.0g)、A2(2.9g)、O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロホスフェート(6.5g)及びN,N-ジイソプロピルエチルアミン(3.7g)をジクロロメタン(50.0mL)に添加し、20℃で12時間攪拌した。反応終了後、反応溶液に飽和炭酸水素ナトリウム水溶液(50.0mL)を注ぎ、均一に攪拌した。有機層を分離し、適量の無水硫酸ナトリウムを添加して乾燥させた。濾過して乾燥剤を除去し、濾液を乾燥するまで減圧濃縮して粗生成物を得た。粗生成物をカラムクロマトグラフィー(純粋な石油エーテル、石油エーテル:酢酸エチル=1:1、酢酸エチル:メタノール=10:1)で精製して化合物WX001-4を得た。
化合物WX001-4(3.3g)を無水テトラヒドロフラン(70.0mL)に溶解させ、反応溶液を10℃に冷却させた。メチルマグネシウムブロミド(3M、15.4mL)のエーテル溶液を反応系に滴下し、15℃で20分間攪拌した。反応溶液を飽和塩化アンモニウム水溶液(30.0mL)に注ぎ、酢酸エチルで抽出した(20.0mL×3)。有機層を合わせ、適量の無水硫酸ナトリウムを添加して乾燥させた。濾過して乾燥剤を除去し、濾液を乾燥するまで減圧濃縮して粗生成物を得た。粗生成物をカラムクロマトグラフィー(純粋な石油エーテル、石油エーテル:酢酸エチル=1:1、酢酸エチル:メタノール=10:1)で精製した後、有機精製(カラム:Welch Xtimate C18 250*50mm*10μm;移動相:A:10mMのNH4HCO3を含む水溶液、B:アセトニトリル;勾配:B%:30%~55%、10分)して化合物WX001を得た。LCMS(ESI)m/z=489.3[M+H]+.1H NMR(400MHz, DMSO-d6) δ = 9.25 (s, 1H), 8.70 (s,
1H), 8.63 (d, J=4.8 Hz, 1H), 7.93 (s, 1H), 7.83(d, J=4.8 Hz, 1H), 7.50 (s, 1H),
7.31 (s, 1H), 7.14 (s, 1H), 4.14-4.12(m, 1H), 2.99-2.97(m, 4H), 2.81-2.77 (m, 2H), 2.64 (s, 3H), 1.99-1.96 (m, 4H), 1.91-1.89 (m, 2H), 1.87 (br s, 2H), 1.27 (s, 6H)。
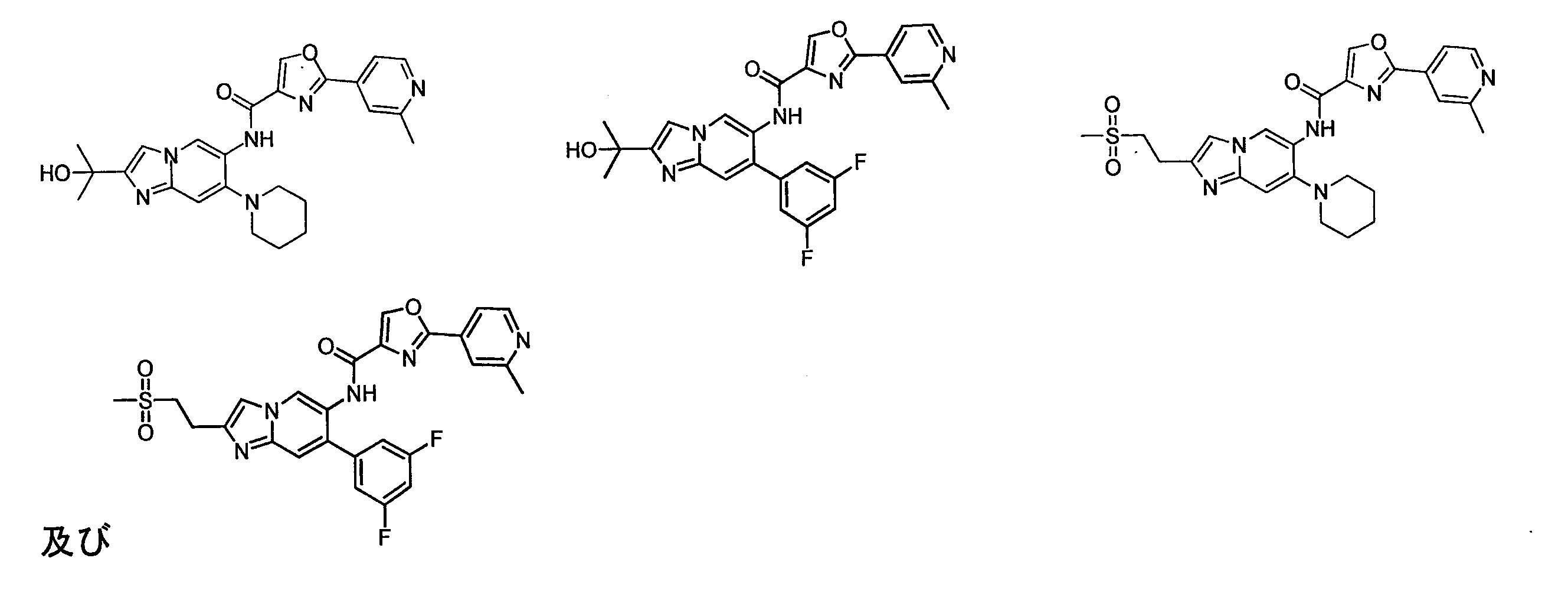
成ステップはBocの脱離、加水分解、メチルグリニャール試薬とエステルを使用して第三級アルコールを生成させるか、又は鈴木カップリング等の一般的な操作を使用することができ、最後に下記表3の各実施例を合成した。
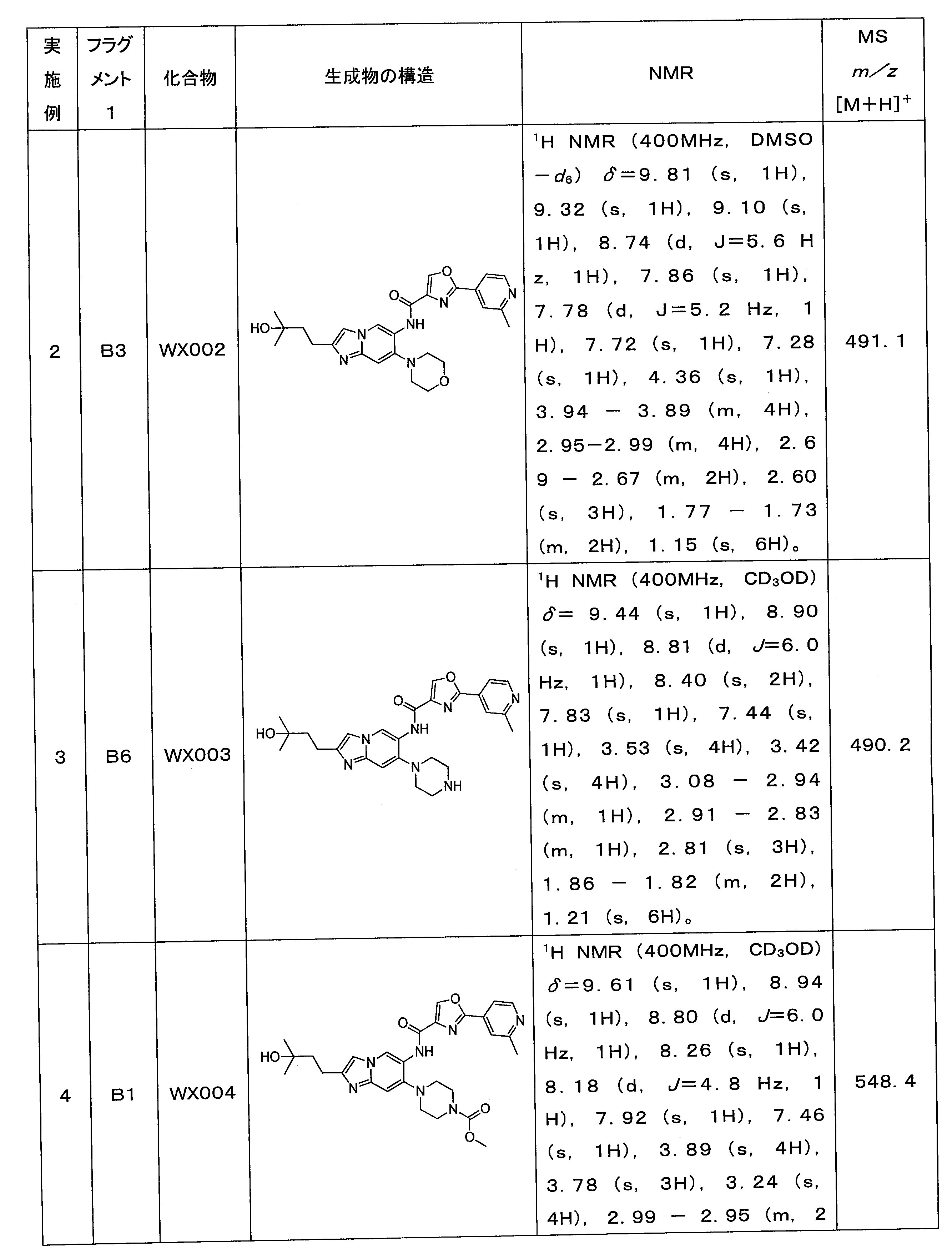







ステップ1:化合物16の合成
化合物WX016-1(15.0mg)を水酸化ナトリウム(2.3mg)の水(1.0mL)溶液に溶解させた後、メタノール(1.0mL)を添加し、25℃で2時間反応させた。反応溶液を1.0Mの塩酸を使用してpH=6~7に調節した後、酢酸エチル(30.0mL×4)を使用して抽出し、有機層を合わせた後、無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮した。有機精製(カラム:Welch Xtimate C18
150*25mm*5μm移動相:[(10.0mM)のNH4HCO3を含む水溶液)-アセトニトリル];勾配:B%:15%~50%、10.5分)して化合物WX016を得た。LCMS (ESI) m/z: 503.3 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ= 9.74 (s, 1H), 9.35 (s, 1H), 9.06 (s, 1H), 8.67 (d, J=5.2
Hz, 1H), 7.83 (s, 1H),7.76 (d, J=5.2 Hz, 1H), 7.70-7.68 (m, 1H), 7.29 (s, 1H), 2.92-2.89 (m, 4H), 2.87-2.83 (m, 4H), 2.65-2.55 (m, 4H), 1.68 (s, 4H), 1.07 (s, 6H)。
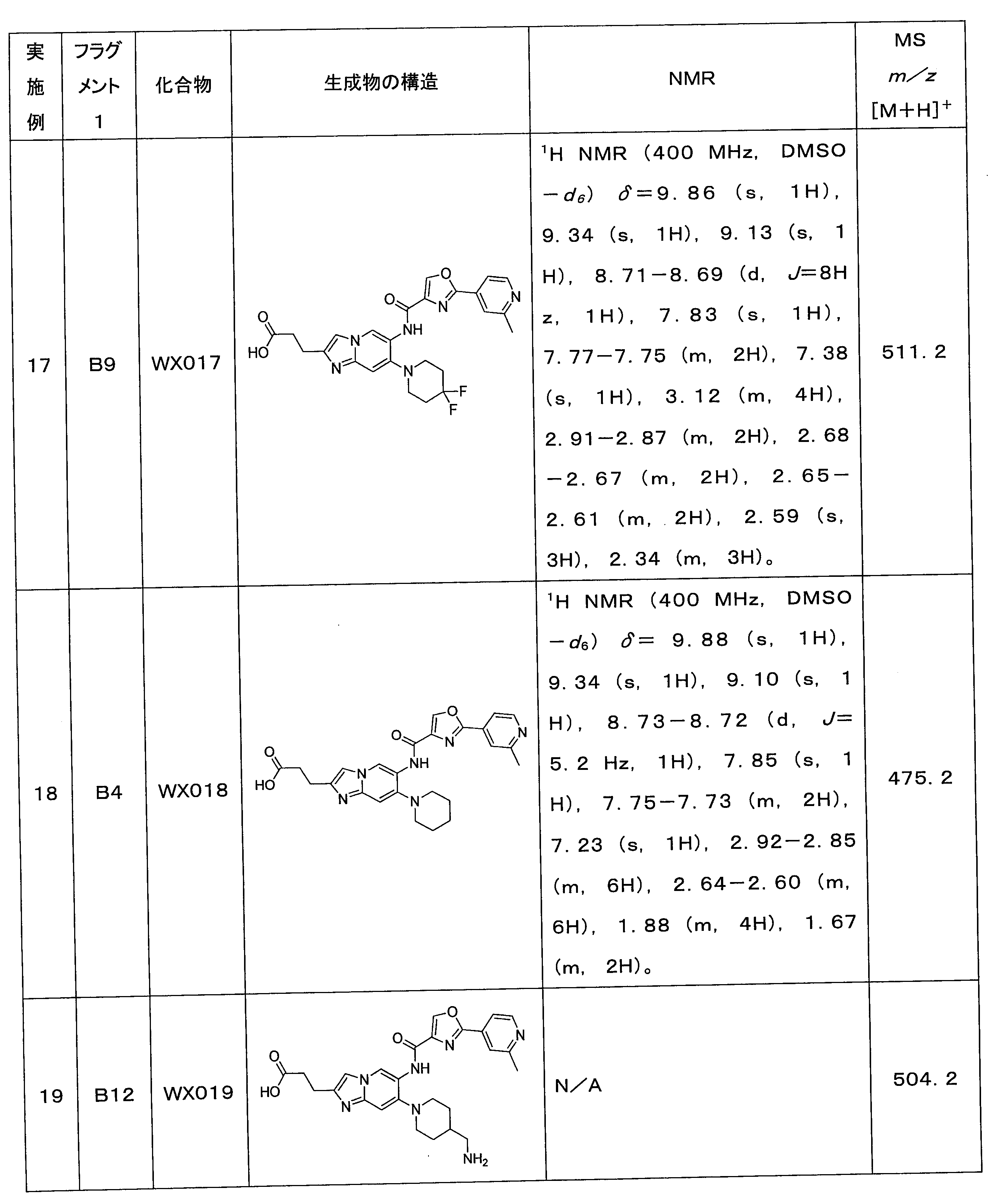



のステップ1で、ピペリジンを使用して塩素原子を置換しなかったことである。
ステップ1:化合物WX021の合成
反応フラスコにX020-1(3.3mg)、B10(3.33mg)及びトルエン(1.0mL)、エタノール(0.5mL)、水(0.3mL)を添加した後、炭酸水素ナトリウム(5.7mg)及びテトラ(トリフェニルホスフィン)パラジウム(5.3mg)を添加した。窒素ガスで3回置換した後、80℃で12時間攪拌した。濾過し、濾液を収集し、減圧濃縮して乾燥させた。粗生成物をシリカゲルプレートで精製(溶離液:ジクロロメタン:メタノール=10:1)して化合物WX021を得た。LCMS (ESI) m/z: 482.3[M+H]+.1H NMR (400 MHz, DMSO-d6) δ=9.70 (s, 1H), 8.99 (s, 1H), 8.93 (s, 1 H), 8.68-8.67 (d, J=4Hz, 1H), 7.80
(s, 1H), 7.72 (s, 1H), 7.65-7.64 (d, J=4Hz, 1H), 7.57-7.45 (m, 6H), 4.34 (s, 1H), 2.78-2.74 (m, 2H), 2.59 (s, 3H), 1.82-1.78 (m, 2H), 1.17 (s, 6H)。




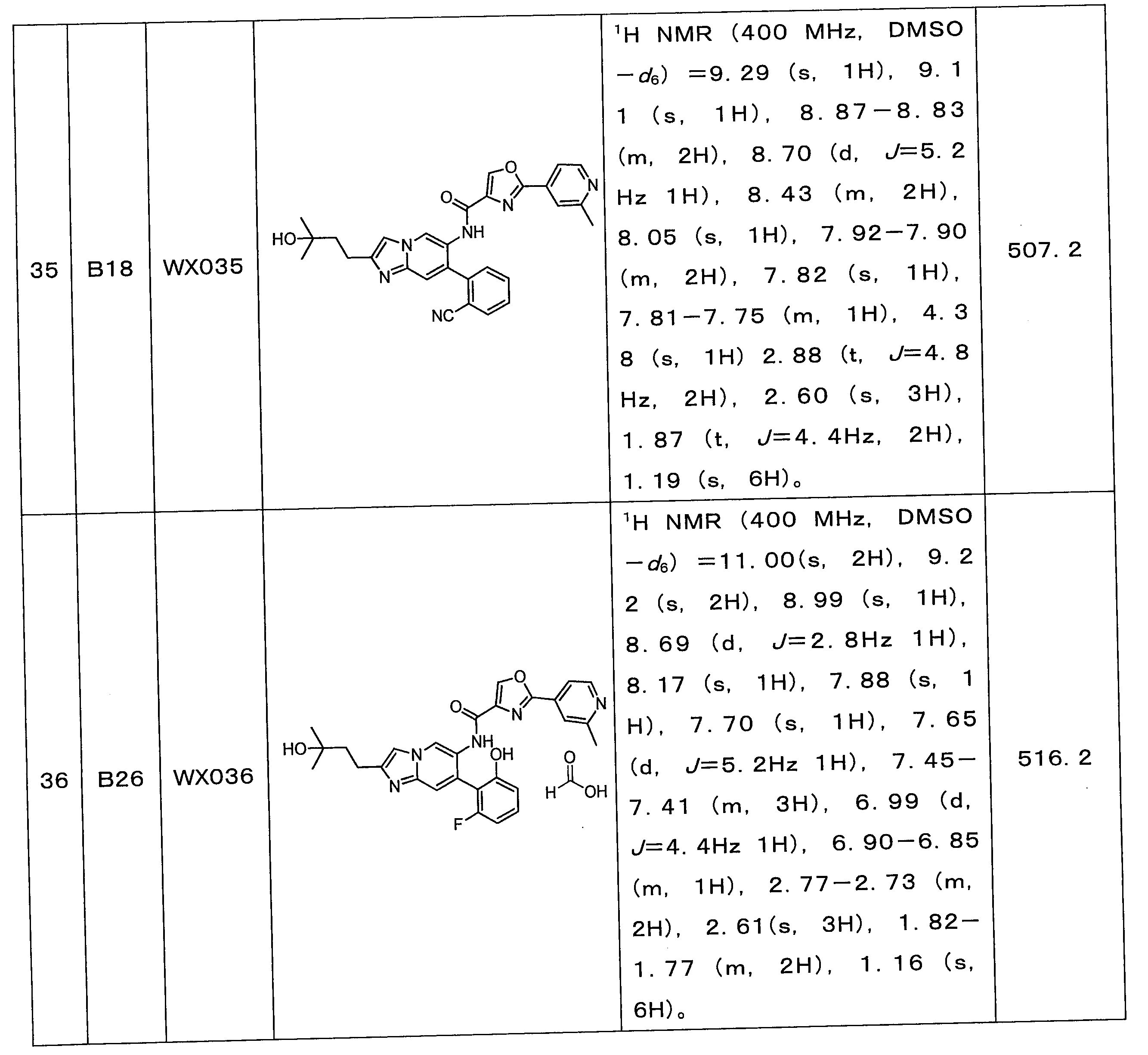

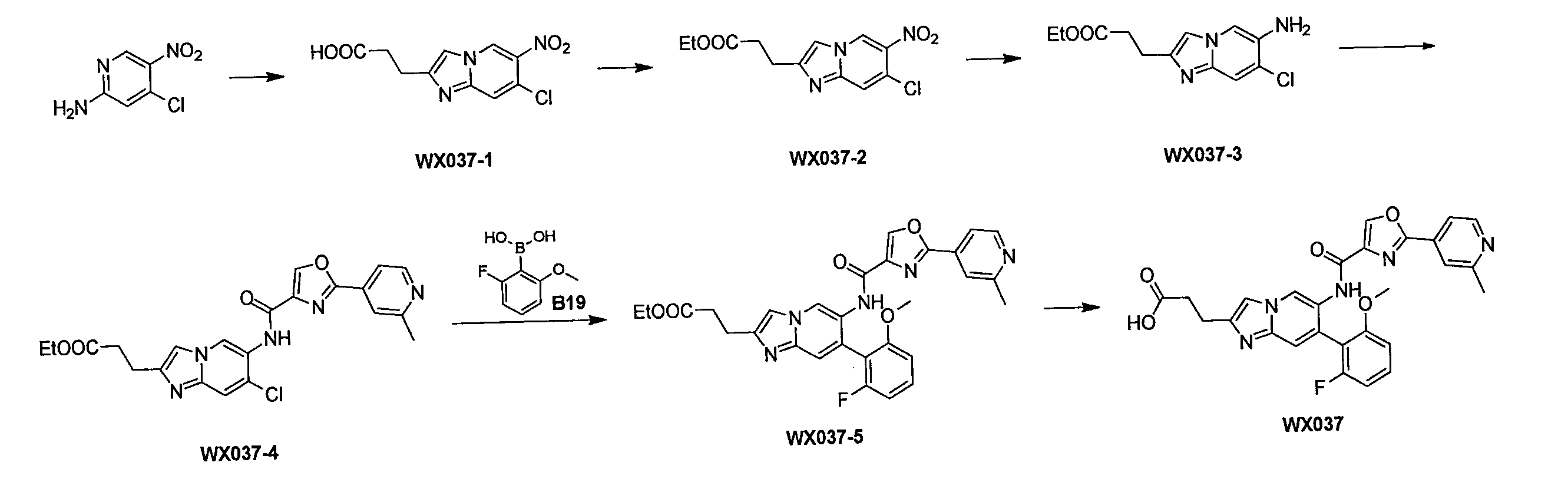
一つの反応フラスコに2-アミノ-4-クロロ-5-ニトロピリジン(10.0g、57.62mmol)及びブロモジオキソ吉草酸エチル(21.5g、97.95mmol)を添加し、混合物を窒素ガスで3回置換し、反応溶液を続いて110℃で16時間攪拌した。反応溶液にエタノール(80mL)を添加して続いて2時間攪拌した後、濾過し、固体を収集し、減圧濃縮して乾燥させ、化合物WX037-1を得た。
一つの反応フラスコにWX037-1(10.0g、37.1mmol)及び無水エタノール(100.0mL)を添加した後、濃硫酸(3.7g、37.1mmol、2.0mL、純度:98%)を添加し、反応溶液を80℃で続いて12時間攪拌した。反応溶液を減圧濃縮して乾燥させた。酢酸エチル(300.0mL)を添加して溶解させた後、飽和炭酸ナトリウム水溶液でpHを8に調節し、液を分離し、有機層を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して乾燥させた。粗生成物をカラムクロマトグラフィーで精製(ジクロロメタン:メタノール=100:0~10:1)して、WX037-2を得た。
一つの反応フラスコにWX037-2(7.5g、25.2mmol)及び酢酸イソプロピル(140.0mL)を添加した後、塩化スズ二水和物(34.1g、151.2mmol)を添加した。混合物を50℃で12時間攪拌した。反応溶液に酢酸エチル(200.0mL)を添加した後、アンモニア水をpH=9になるまで滴下し、その後、無水硫酸ナトリウムを添加して砂の状態になるまで攪拌し、濾過し、濾液を収集し、減圧濃縮して乾燥させた。粗生成物をカラムクロマトグラフィーで精製(溶離液:ジクロロメタン:メタノール=100:0~10:1)してWX037-3を得た。
一つの反応フラスコにWX037-3(3g、11.21mmol、1eq)、A2(3.0g、14.6mmol)、N,N-ジイソプロピルエチルアミン(5.8g、44.8mmol、7.8mL)、トリ-n-プロピル環状リン酸無水物の50%の酢酸エチル溶液(21.9g、33.6mmol、20.0mL、純度:50%)及びTHF(50.0mL)を添加した。混合物を50℃で12時間攪拌した。酢酸エチル(100.0mL)を添加し、飽和炭酸ナトリウム水溶液でpHを8に調節した後、有機層を収集し、無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮して乾燥させた。粗生成物をカラムクロマトグラフィーで精製(ジクロロメタン:メタノール=100:0~10:1)してWX037-4を得た。
一つの反応フラスコにWX037-4(1.0g、2.2mmol)、B19(486.8mg、2.9mmol)、リン酸カリウム(1.4g、6.6mmol)、[メタンスルホン酸(2-ジシクロヘキシルホスフィン)-3,6-ジメトキシ-2,4,6-トリイソプロピル-1,1-ビフェニル)(2-アミノ-1,1-ビフェニル-2-イル)パラジウム(II)(299.6mg、330.5μmol)、テトラヒドロフラン(10.0mL)、水(3.0mL)を添加し、窒素ガスで3回置換した後、80℃で12時間攪拌した。濾過し、濾液を収集し、減圧濃縮して乾燥させた。粗生成物をカラムクロマトグラフィー(ジクロロメタン:メタノール=100:0~10:1)で精製してWX037-5を得た。
一つの反応フラスコにWX037-5(0.05g、92.9μmol)、水酸化ナトリウム(2M、919.9μL)、メタノール(5.0mL)を添加し、窒素ガスで3回置換した後、25℃で2時間攪拌した。メタノールを減圧濃縮して乾燥させた後、2Nの塩酸でpHを7に調節し、その後、減圧濃縮して乾燥させた。粗生成物をカラムクロマトグラフィーで精製(カラム:Phenomenex Gemini NX-C18(75*30mm*3μm);移動相:[(10.0mM)を含むNH4HCO3の水溶液)-アセトニトリル];勾配B%:15%~40%、8分間)してWX037を得た。
1H NMR (400 MHz, DMSO-d6) δ=11.5 (s, 1H), 9.21 (s, 1H), 9.02 (s, 1H), 8.98 (s, 1H), 8.70-8.69 (d, J=4.0Hz, 1H), 7.90 (s,
1H), 7.65-7.58 (m, 3H), 7.49 (s, 1H), 7.17-7.15 (d, J=8.0Hz, 1H), 7.09-7.04 (t,
1H), 3.80 (s, 3H), 2.97-2.93 (t, 2H), 2.69-2.65 (t, 2H), 2.61 (s, 3H)。
LCMS (ESI) m/z: 516.1[M+H]+。



WX001-1(5.4g、24.3mmol)をブロモピルビン酸エチル(47.4g、243.0mmol、30.4mL)が入っているボトルに添加し、当該反応溶液を90℃で12時間攪拌した。熱いうちに反応溶液を酢酸エチル(150.0mL)に注ぎ、15℃で15分間攪拌し、吸引濾過し、ケーキを酢酸エチル(20.0mL×3)で洗い流し、ケーキを減圧濃縮して乾燥させ、WX040-1を得た。
ラネーニッケル(942.0mg)をアルゴン保護下の水素化ボトルに添加し、その後、エタノール(30.0mL)で濡らせ、WX040-1(1.0g、3.1mmol)を反応系に添加し、当該反応を25℃、水素ガス50Psi下で2時間攪拌した。反応溶液を珪藻土で吸引濾過し、濾液を減圧濃縮して乾燥させ、WX040-2を得た。
WX040-2(200mg、693.6μmol)、A2(170.0mg、832.34μmol)、O-(7-アザベンゾトリアゾール-1-イル)-N,N,N,N-テトラメチルウロニウムヘキサフルオロホスフェート(395.6mg、1.0mmol)、N,N-ジイソプロピルエチルアミン(268.9mg、2.1mmol、362.
4μL)を無水ジクロロメタン(15.0mL)が入っているボトルに添加し、当該反応溶液を25℃で2時間攪拌した。反応溶液を飽和塩化アンモニウム溶液(20.0mL)に注ぎ、液を分離し、有機層を乾燥させ、濾過し、減圧濃縮した。粗生成物をカラムクロマトグラフィーで分離(石油エーテルから石油エーテル:酢酸エチル=1:1から酢酸エチル)し、洗浄してWX040-3を得た。
メチルマグネシウムクロリド(3.0mol/L、4.2mL)を無水テトラヒドロフラン(15.0mL)が入っているボトルに添加し、窒素ガスの保護、20℃下でWX040-3(100.0mg、210.7μmol)を無水テトラヒドロフラン(9.0mL)に溶解させて前記反応溶液に滴下し、当該反溶溶液を20℃で0.5時間攪拌した。反応溶液を飽和塩化アンモニウム溶液(20.0mL)に添加してクエンチングさせ、酢酸エチル(10.0mL×4)で抽出し、有機層を乾燥させ、濾過し、減圧濃縮した。粗生成物をプレートで分離(酢酸エチル:メタノール=10:1)・精製してWX040を得た。
1H NMR 400 MHz, CD3OD-d4) δ=9.41 (s, 1H), 8.75 (s, 1H), 8.66 (d, J=5.2Hz 1H), 7.99 (s, 1H), 7.90 (d, J=5.6Hz 1H), 7.66 (s, 1H), 7.20 (s, 1H), 3.15-2.99(m, 4H), 2.67 (s, 3H), 3.11-1.98 (m, 4H), 1.85-1.64 (m, 2H), 1.62 (s, 6H)。
LCMS (ESI) m/z: 461.3 [M+H]+。



ステップ1:化合物WX042-2の合成
化合物WX042-1(0.8g、1.64mmol)をテトラヒドロフラン(10.0mL)に溶解させ、-10℃に冷却させ、リチウム四水素アルミニウム(155.4mg)をバッチで反応系に添加し、当該反応溶液を-10℃で1時間攪拌した。反応溶液を塩化アンモニウム水溶液(50.0mL)に注ぎ、その後、酢酸エチル(50.0mL×4)で抽出し、有機層を合わせ、飽和食塩水(100.0mL)で有機層を洗浄し、有機層を無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮した。粗生成物をカラムクロマトグラフィー:ジクロロメタン:メタノール=100:0~100:0.25で精製してWX042-2を得た。
化合物WX042-2(200.0mg)をクロロホルム(10.0mL)に溶解させた後、トリエチルアミン(136.0mg)を添加し、10℃に冷却させた後、10分間攪拌し、その後、ゆっくりとメタンスルホニルクロリド(77.0mg)のクロロホルム(1.0mL)溶液を滴下した。当該反応溶液をゆっくりと25℃に昇温させ、続いて2
0分間攪拌した。反応溶液を減圧濃縮した後WX042-3を得た。
化合物WX042-3(0.2g)、メタンスルフィン酸ナトリウム(70.1mg、686.3μmol)をN,N-ジメチルホルムアミド(10.0mL)に溶解させた後、ヨウ化カリウム(189.8mg)を添加した。当該反応溶液をマイクロ波装置80℃(0bar)で1時間反応させた。反応溶液に10.0mLの酢酸エチルを添加して希釈させた後、半飽和食塩水(50.0mL)に注ぎ、液を分離し、水層を酢酸エチル(50.0mL×4)で抽出した後、有機層を合わせ、無水硫酸ナトリウムで乾燥させ、濾過し、減圧濃縮した。粗生成物を有機精製(カラム:Welch Xtimate BEH C18 100*30mm*10μm;移動相:A:10mMのNH4HCO3を含む水溶液、B:アセトニトリル;勾配:B%:30%~50%、6分間)した後、凍結乾燥させてWX042を得た。
1H NMR (400 MHz, DMSO-d6) δ= 9.87(s, 1H), 9.35 (s, 1H), 9.09(s, 1H), 8.71 (s, 1H), 7.83 (s, 2H), 7.73(s, 1H), 7.23(s, 1H), 3.48 - 3.46 (m, 2H), 3.44(s, 2H), 3.09 - 2.99 (m, 3H), 2.91 - 2.90 (m, 4H), 2.60 - 2.58 (m, 3H), 1.87 - 1.86 (m, 4H),
1.67(s, 2H)。
LCMS (ESI) m/z: 509.1 [M+H]+。

33P同位体標識キナーゼ活性試験(Reaction Biology Corp)
を使用してIC50値を検出し、ヒトIRAK4に対する試験化合物の阻害能力を評価した。


試験例2:体外BTKキナーゼ活性評価:
33P同位体標識キナーゼ活性試験(Reaction Biology Corp)を使用してIC50値を検出し、ヒトBTKに対する試験化合物の阻害能力を評価した。
technology; nanoliter range)を使用してキナーゼ反応混合物に添加した。室温で20分間インキュベーションした後、33P-ATPを添加して反応を開始させた。室温で2時間反応させた後、反応液点をP81イオン交換濾紙を使用して、濾過-結合方法で放射能を検出した。キナーゼ活性データは、試験化合物を含有するキナーゼ活性と空白群(DMSOのみを含む)のキナーゼ活性を比較することで表され、Prism4ソフトウェア(GraphPad)を使用したカーブフィッティングによってIC50値を得、実験結果は表10に示される通りであった。


試験例3:体外THP-1細胞学の活性評価
THP-1細胞学TNFa ELISA実験
1.実験材料:
THP-1ヒト急性単球性白血病細胞系はATCC(Cat#TIB-202)から購入し、37℃、5%のCO2のインキュベーターで培養した。培地の成分はRPMI1640(Gibco、Cat#22400-105)であり、添加した成分は10%のFBS(Gibco、Cat#10091148);1%のPenStrep(Gibco、Cat#15140);0.05mMの2-Mercaptoethanol(Sigma、Cat#M6250)である。
2.実験方法:
TNF-αElisaキットを使用して、細胞培養上清サンプルの中のTNF-αの含有量を検出した。TNF-αは、150ng/mLのLPS(Sigma、Cat#L6529)でTHP-1細胞を刺激して産生した。
3.データ分析:
OD450-OD570信号値を抑制率%に変換した。
「HPE」はLPS刺激のない細胞の対照ウェルのOD450-OD570信号値を表し、「ZPE」はLPS刺激のある細胞の対照ウェルのOD450-OD570信号値を表した。ExcelアドインのXLFitを使用して化合物のIC50値を計算した。
結果の要約は表11に示された通りである。

試験例4:体外OCI-LY10及びTMD-8細胞学の活性評価
1.実験材料
OCI-LY10ヒトB細胞リンパ腫細胞であり、37℃、5%のCO2のインキュベーターで培養した。培地の成分はIMDM(GIBCO、Cat#12440053)であり、添加した成分は20%のFBS(Hyclone、Cat#SH30084.03);1%のPenStrep(Thermo、Cat#SV30010)であった。
TMD8ヒトB細胞リンパ腫細胞であり、37℃、5%のCO2のインキュベーターで培養した。培地の成分はRPMI1640(GIBCO、Cat#22400-089)であり、添加した成分は10%のFBS(Hyclone、Cat#SH30084.03);1%のPenStrep(Thermo、Cat#SV30010)であった。
2.実験方法
腫瘍細胞株OCI-LY10及びTMD8を使用して、化合物の体外腫瘍細胞の増殖を阻害する効果を検出した。腫瘍細胞株を37℃、5%のCO2インキュベーターで指定の培養条件下で培養し、定期的に継代培養し、対数増殖期の細胞を取り、カウントして、96ウェルプレートに広げた(各ウェルの細胞を適切な濃度に調整し、各ウェルに合計90個の細胞懸濁液を添加した)。37℃、5%のCO2インキュベーターで一晩培養した後、異なる濃度の薬物(10μLの薬物溶液を添加)を添加して3日間処理した後、各ウェルに50μLのCellTiter-Gloワーキングソリューションを添加し、光を避けるために細胞プレートをアルミホイルで包んだ。オービタルシェーカーで培養プレートを2分間振とうして細胞溶解を誘導し、室温で10分間放置して発光信号を安定させ、2104EnVisionプレートリーダーで発光信号を検出した。
3.データ分析
下記公式を使用して、試験化合物の阻害率を計算(Inhibition rate、IR)した:
IR(%)=(1-(RLU化合物-RLU空白対照群)/(RLU溶媒対照群-RLU空白対照群))*100%。
4.実験結果
結果は表12に示めされた通りである。

注:「/」は未検出を表す。
1.実験細胞株情報と細胞培養
当該実験で使用された腫瘍細胞株は、Nanjing Kebai Biotechnology Co.,Ltd.から提供されたものであり、具体的な情報は下記表13に示される通りである。

2.実験方法
3.データ処理及び分析
各薬物濃度下で測定された発光値結果を、空白対照群の発光値で正規化され、当該値とDMSO群の比率を細胞阻害率(%)にした。GraphPadソフトウェアを使用して、薬物濃度の対数(log薬物濃度)を阻害率に対してプロッし、ソフトウェアは、非線形回帰(Nonlinear Regression)のlog(inhibitor)対正規化応答アルゴリズムを自動的に適合させて、IC50値と95%信頼限界の値を計算した。
4.実験結果
実験結果は表14に示された通りである。

試験例6:リポポリコラーゲン(LPS)によって誘発されたSDラットにおけるTNF-α分泌の生体内薬力学的研究
1.モデリングと薬物投与
SDラットそれぞれに溶媒、陽性薬物デキサメタゾン(DEX、0.5mg/kg)及び試験化合物を経口投与し、投与0.5時間後にLPS(1mg/kg)を腹腔内注射した。LPS注射の2時間後、動物をCO2で安楽死させ、心臓の血液を採取し、EDTA-K2を含む抗凝固チューブに入れ、抗凝固血液の一部を遠心分離し、血漿を-80℃で凍結保存した。
2.TNF-αの検出
血漿を-80℃で冷蔵庫から取り出し、室温で解凍し、血漿中のTNF-αの濃度をELISAキットの説明書に従って検出した。
3.統計学処理
実験データは平均±標準誤差(平均±SEM)で表され、TNF-αのレベルは一元配置分散(One-way ANOVA)で分析し、p<0.05は有意差があると見なされた。リポポリコラーゲン(LPS)によって誘発されたSDラットにおけるTNF-α分泌の生体内薬力学的研究の結果は図1に示された通りである。
4.実験結果
図1の結果は、SDラットにWX001を経口投与した後、化合物はリポポリコラーゲン(LPS)によって誘発されたTNF-α分泌に対して有意な阻害効果を示し、20mpkの用量での有効性は、0.5mpkの用量でのデキサメタゾン(DEX)有効性と同じであることを示した。
1.実験目的
本実験の目的は、CB17SCIDマウスモデルにおけるヒトB細胞リンパ腫OCI-LY10細胞皮下異種移植腫瘍に対するWX001試験薬の有効性を研究することである。
2.実験材料
OCI-LY10ヒトB細胞リンパ腫細胞であり、37℃、5%のCO2のインキュベーターで培養した。培地の成分はIMDM(GIBCO、Cat#12440053)であり、添加した成分は20%のFBS(Hyclone、Cat#SH30084.03);1%のPenStrep(Thermo、Cat#SV30010)である。
3.実験方法
OCI-LY10腫瘍細胞を培養継代し、0.2mL(1×107個)のOCI-LY10細胞を各ヌードマウスの右背部に皮下接種し(マトリゲル、体積比1:1)、平均腫瘍体積が167mm3に達したときに群分け投与を開始した。毎日動物の健康と死亡をモニタリングし、腫瘍の成長の観察と行動活動、食物と水の摂取量、体重の変化(週に2回の体重の測定)、腫瘍の大きさ(週に2回腫瘍の体積を測定)、身体的兆候又はその他の異常などの動物の日常の行動に対する薬物治療の効果を定期的に検査した。
4.データ分析
実験的指標は、腫瘍の成長が抑制、遅延、又は治癒されたか否かを調査することである。腫瘍体積(TV)の測定、化合物の抗腫瘍効果TGI(%)又は相対腫瘍増殖率T/C(%)の計算などが含まれた。
TGI(%)=(1-(特定の治療群の投与終了時の平均腫瘍体積-当該治療群の投与開始時の平均腫瘍体積))/(溶媒対照群の治療終了時の平均腫瘍体積-溶媒対照群の治療開始時の平均腫瘍体積))×100%。
5.実験結果
5.1.死亡率、罹患率、及び体重の変化
実験動物の体重は、薬物毒性を間接的に測定する参照指標とした。18日間の投与(PG-D1-D18)後、実験群のすべてのマウスは正常であり、良好な薬剤耐性を示した。
5.2.腫瘍増殖曲線
図4は、WX001化合物の投与後のヒトB細胞リンパ腫OCI-LY10細胞皮下異種移植腫瘍モデル担癌マウスの腫瘍増殖曲線を示す。データポイントは群内の平均腫瘍体積を表し、誤差線は標準誤差(SEM)を表す。
6.実験結果及び考察
本実験では、ヒトB細胞リンパ腫OCI-LY10細胞皮下異種移植腫瘍モデルにおけるWX001化合物の生体内有効性を評価した。異なる時点での各郡の腫瘍体積は図4に示された通りである。
Claims (15)
- 式(II)で表される化合物又はその薬学的に許容される塩。

R1はH、F、Cl、Br、I、OH、NH2、CN、C1-6アルキル、シクロプロピル、及び-C(=O)-NH2であり、前記C1-6アルキル、シクロプロピル、及び-C(=O)-NH2は1、2又は3個のRaで任意に置換され;
R2はチエニル、フェニル、ピリジル、シクロプロピル、シクロヘキシル、及び


T1はCH2、NH及びOから選択され;
R3はC1-6アルキルから選択され、前記C1-6アルキルは1、2又は3個のRcで任意に置換され;
Raはそれぞれ独立してF、OH、NH2及びCNから選択され;
Rbはそれぞれ独立してH、D、F、Cl、Br、I、OH、NH2、CN、C1-3アルキル、COOH、-C(=O)-C1-3アルキル、-C(=O)-O-C1-3アルキル、及び-C(=O)-NH2から選択され、前記OH、NH2、C1-3アルキル、COOH、-C(=O)-C1-3アルキル、-C(=O)-O-C1-3アルキル及び-C(=O)-NH2は1、2又は3個のRで任意に置換され;
Rcはそれぞれ独立してF、OH、NH2、CN、CH3、COOH及び-SO2CH3から選択され;
Rはそれぞれ独立してF、OH、NH2及びCH3から選択される。) - R1はH、F、Cl、Br、I、OH、NH2、CN、C1-3アルキル、シクロプロピル及び-C(=O)-NH2から選択され、前記C1-3アルキル、シクロプロピル、及び-C(=O)-NH2は1、2又は3個のRaで任意に置換される、請求項1に記載の化合物又はその薬学的に許容される塩。
- R1はCN、CH3、CF3、

- Rbはそれぞれ独立してH、D、F、Cl、Br、I、OH、NH2、CN、CH3、CH2CH3、CH2CH2CH3、CH(CH3)2、COOH、


- Rbはそれぞれ独立してH、D、F、Cl、OH、OCH3、CN、CH3、CH2OH、CH2NH2、COOH、

- R2はチエニル、フェニル、ピリジル、シクロプロピル、シクロヘキシル、


- R2は

- R2は

- R3はC2-5アルキルから選択され、前記C2-5アルキルは1、2又は3個のRcで任意に置換される、請求項1~3のいずれか1項に記載の化合物又はその薬学的に許容される塩。
- R3はCH2CH3、CH2CH2CH3、CH(CH3)2、CH2CH2CH2CH3、CH2CH(CH3)2及びCH2CH2CH(CH3)2から選択され、前記CH2CH3、CH2CH2CH3、CH(CH3)2、CH2CH2CH2CH3、CH2CH(CH3)2及びCH2CH2CH(CH3)2は1、2又は3個のRcで任意に置換される、請求項9に記載の化合物又はその薬学的に許容される塩。
- R3は

- 下記から選択される、請求項1~11のいずれか1項に記載の化合物又はその薬学的に許容される塩。

- 下記式で表される化合物又はその薬学的に許容される塩。



- 活性成分として治療有効量の請求項1~13のいずれか1項に記載の化合物又はその薬学的に許容される塩、及び薬学的に許容される担体を含む、医薬組成物。
- IRAK4及びBTKに関連する疾患を治療する医薬の製造における、請求項1~13のいずれか1項に記載の化合物又はその薬学的に許容される塩、或いは請求項14に記載の医薬組成物の使用。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201910619602.8 | 2019-07-10 | ||
| CN201910619602 | 2019-07-10 | ||
| CN201911240843 | 2019-12-06 | ||
| CN201911240843.8 | 2019-12-06 | ||
| CN202010470469.7 | 2020-05-28 | ||
| CN202010470469 | 2020-05-28 | ||
| PCT/CN2020/101369 WO2021004533A1 (zh) | 2019-07-10 | 2020-07-10 | 作为irak4和btk多靶点抑制剂的噁唑类化合物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2022540464A JP2022540464A (ja) | 2022-09-15 |
| JP7340680B2 true JP7340680B2 (ja) | 2023-09-07 |
Family
ID=74114367
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022500996A Active JP7340680B2 (ja) | 2019-07-10 | 2020-07-10 | Irak4及びbtkマルチターゲット阻害剤としてのオキサゾール化合物 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20220267322A1 (ja) |
| EP (1) | EP3998264A4 (ja) |
| JP (1) | JP7340680B2 (ja) |
| CN (1) | CN114072401B (ja) |
| WO (1) | WO2021004533A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20230391776A1 (en) * | 2020-12-25 | 2023-12-07 | Medshine Discovery Inc. | Amide oxazole compound |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005016928A1 (ja) | 2003-08-15 | 2005-02-24 | Banyu Pharmaceutical Co., Ltd. | イミダゾピリジン誘導体 |
| JP2017502088A (ja) | 2014-01-13 | 2017-01-19 | アウリジーン ディスカバリー テクノロジーズ リミテッド | Irak4阻害剤としての二環式ヘテロシクリル誘導体 |
| JP2017505337A (ja) | 2014-01-10 | 2017-02-16 | アウリジーン ディスカバリー テクノロジーズ リミテッド | Irak4阻害剤としてのインダゾール化合物 |
| JP2018524372A (ja) | 2015-07-15 | 2018-08-30 | アウリジーン ディスカバリー テクノロジーズ リミテッド | Irak−4阻害剤としてのインダゾール及びアザインダゾール化合物 |
| WO2018178947A2 (en) | 2017-03-31 | 2018-10-04 | Aurigene Discovery Technologies Limited | Compounds and compositions for treating hematological disorders |
| CN110835338A (zh) | 2018-08-17 | 2020-02-25 | 浙江海正药业股份有限公司 | 咪唑并吡啶类衍生物及其制备方法和其在医药上的用途 |
-
2020
- 2020-07-10 JP JP2022500996A patent/JP7340680B2/ja active Active
- 2020-07-10 US US17/625,599 patent/US20220267322A1/en active Pending
- 2020-07-10 WO PCT/CN2020/101369 patent/WO2021004533A1/zh unknown
- 2020-07-10 CN CN202080047753.1A patent/CN114072401B/zh active Active
- 2020-07-10 EP EP20836225.1A patent/EP3998264A4/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005016928A1 (ja) | 2003-08-15 | 2005-02-24 | Banyu Pharmaceutical Co., Ltd. | イミダゾピリジン誘導体 |
| JP2017505337A (ja) | 2014-01-10 | 2017-02-16 | アウリジーン ディスカバリー テクノロジーズ リミテッド | Irak4阻害剤としてのインダゾール化合物 |
| JP2017502088A (ja) | 2014-01-13 | 2017-01-19 | アウリジーン ディスカバリー テクノロジーズ リミテッド | Irak4阻害剤としての二環式ヘテロシクリル誘導体 |
| JP2018524372A (ja) | 2015-07-15 | 2018-08-30 | アウリジーン ディスカバリー テクノロジーズ リミテッド | Irak−4阻害剤としてのインダゾール及びアザインダゾール化合物 |
| WO2018178947A2 (en) | 2017-03-31 | 2018-10-04 | Aurigene Discovery Technologies Limited | Compounds and compositions for treating hematological disorders |
| CN110835338A (zh) | 2018-08-17 | 2020-02-25 | 浙江海正药业股份有限公司 | 咪唑并吡啶类衍生物及其制备方法和其在医药上的用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2022540464A (ja) | 2022-09-15 |
| WO2021004533A1 (zh) | 2021-01-14 |
| CN114072401A (zh) | 2022-02-18 |
| EP3998264A4 (en) | 2023-07-12 |
| EP3998264A1 (en) | 2022-05-18 |
| US20220267322A1 (en) | 2022-08-25 |
| CN114072401B (zh) | 2023-11-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN105189456B (zh) | Kras g12c的共价抑制剂 | |
| JP2022550215A (ja) | ジアザインドール誘導体及びそのChk1阻害剤としての使用 | |
| CN116348466A (zh) | 吡嗪硫联苯基类化合物及其应用 | |
| KR102345208B1 (ko) | 퀴놀리노-피롤리딘-2-온 유도체 및 이의 응용 | |
| CN114761411B (zh) | 作为erk抑制剂的螺环类化合物及其应用 | |
| JP2021514997A (ja) | ピラゾロピリミジン誘導体及びその使用 | |
| JP7474319B2 (ja) | ヘテロ環式アミド化合物及びその製造方法並びに使用 | |
| CN113527299B (zh) | 一类含氮稠环类化合物、制备方法和用途 | |
| WO2020259626A1 (zh) | 作为irak4抑制剂的咪唑并吡啶类化合物 | |
| WO2021129817A1 (zh) | 具有果糖激酶(khk)抑制作用的嘧啶类化合物 | |
| TW202317097A (zh) | 戊二醯亞胺類化合物與其應用 | |
| WO2022063297A1 (zh) | 喹唑啉衍生物及其制备方法和用途 | |
| CN115667258A (zh) | 氟代吡咯并吡啶类化合物及其应用 | |
| JP7545622B2 (ja) | 5,6-ジドヒロチエノ[3,4-h]キナゾリン系化合物 | |
| JP7340680B2 (ja) | Irak4及びbtkマルチターゲット阻害剤としてのオキサゾール化合物 | |
| JP2022536188A (ja) | Fgfrとvegfr二重阻害剤としての縮合環系化合物 | |
| JP7292588B2 (ja) | Ccr2/ccr5アンタゴニストとしてのヘテロシクロアルキル系化合物 | |
| CN113874379B (zh) | 作为Cdc7抑制剂的四并环类化合物 | |
| WO2023143147A1 (zh) | 一种哒嗪并吡啶酮类化合物、其药物组合物及应用 | |
| JP2004269547A (ja) | キノキサリンジオン類の製造方法 | |
| TWI847445B (zh) | 一種噠嗪類化合物、其藥物組合物及應用 | |
| US20240279232A1 (en) | Use of substituted 5-(4-methyl-6-phenyl-4h-benzo[f]imidazo[1,5-a][1,4] diazepin-3-yl)-1,2,4-oxadiazoles in the treatment of inflammatory conditions | |
| WO2023237012A1 (zh) | 双环取代的芳香羧酸类氘代化合物 | |
| CN117263950A (zh) | 一种哒嗪类化合物、其药物组合物及应用 | |
| JP2024510647A (ja) | オキサジアゾール置換スピロ環系化合物とその使用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20220303 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20230216 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20230314 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230531 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230801 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230828 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7340680 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |