JP5442202B2 - プロパンをプロピレンに接触的に脱水素する方法 - Google Patents
プロパンをプロピレンに接触的に脱水素する方法 Download PDFInfo
- Publication number
- JP5442202B2 JP5442202B2 JP2007540581A JP2007540581A JP5442202B2 JP 5442202 B2 JP5442202 B2 JP 5442202B2 JP 2007540581 A JP2007540581 A JP 2007540581A JP 2007540581 A JP2007540581 A JP 2007540581A JP 5442202 B2 JP5442202 B2 JP 5442202B2
- Authority
- JP
- Japan
- Prior art keywords
- hydrogen
- oxygen
- propane
- gas mixture
- reactor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C5/00—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms
- C07C5/32—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by dehydrogenation with formation of free hydrogen
- C07C5/327—Formation of non-aromatic carbon-to-carbon double bonds only
- C07C5/333—Catalytic processes
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Description
C3H8 ⇔ C3H6+ H2 (1)
一般に気相において540℃〜820℃の温度で実施される化学反応(1)は強い吸熱平衡反応であり、その転化率は熱力学的に制限されておりそしてその都度の分圧及び温度に左右される。この脱水素反応は炭化水素の低い分圧及び高い温度によって促進される。副反応で分解生成物が生じ、これが炭素堆積物として触媒上に堆積しそして触媒を失活させてしまうので、工業的に運転する場合には触媒を周期的に再生しなければならない。
− プロパン、水蒸気及び水素を含有する工業的に酸素不含の少なくとも400℃、好ましくは少なくとも500℃の温度の第一のガス混合物を、少なくとも一つの触媒床を持ち、かつ、通例の脱水素条件を示す反応装置に導入し;
− アンモニアも含有してもよい、プロパン及び酸素を含有し、かつ、酸素含有量に比較してプロパン含有量が過剰である別のガス混合物を同じ反応装置に案内し、そこで該ガス混合物をプロピレン、水蒸気及び水素を形成しながら第一のガス混合物と反応させ、
− そしてプロピレン、プロパン、水蒸気及び水素を含有する生じたガス混合物を反応装置から引き出す。

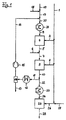
2 第一の部分流
3 ループガス
4 第一の酸化脱水素化反応器
5 反応ガス
6 水−水蒸気混合物
7 ガス混合物
8 第二の部分流
9 第二の酸化脱水素化反応器
10 プロピレンリッチで水素リッチのガス
11 循環ガス
12 循環ガス冷却器
13 水性凝縮液
14 凝縮液分離器
15 ループガス圧縮器
16 ループガス
17 混合物
18 水蒸気
19 プロパン
20 ループガス
21 加熱器
22 プロピレンリッチのガス
23 ガス冷却器
24 冷却されたガス
25 水素燃料反応器
26 酸素
27 残留部分流
28 生成物ガス
Claims (10)
- プロパンからプロピレンを製造する方法において、
− プロパン、水蒸気及び水素を含有する工業的に酸素不含の少なくとも400℃の温度の第一のガス混合物を、少なくとも一つの触媒床を持ち、かつ、通例の脱水素条件を有する反応装置に導入し;
− プロパン及び酸素を含有し、アンモニアも含有していてもよく、かつ、プロパン含有量が酸素含有量に比較して過剰である別のガス混合物を、同じ反応装置に案内し、そこで該ガス混合物をプロピレン、水蒸気及び水素を形成しながら第一のガス混合物と反応させ、
− そして次いでプロピレン、プロパン、水蒸気及び水素を含有する生じたガス混合物を該反応装置から引き出すことを特徴とする、上記方法。 - 第一のガス混合物に、それを反応装置に導入する前に過熱した水蒸気を供給する、請求項1に記載の方法。
- プロピレン、プロパン、水蒸気及び水素を含有する生じたガス混合物の少なくとも一部を加えて、プロパン、水蒸気及び水素を含有しそして少なくとも400℃の温度で反応装置に供給される第一のガス混合物とする、請求項1または2に記載の方法。
- プロパン、水蒸気及び水素を含有する工業的に酸素不含の少なくとも500℃の温度の第一のガス混合物を、少なくとも一つの触媒床を持ち、かつ、通例の脱水素条件を示す反応装置に導入する、請求項1〜3のいずれか一つに記載の方法。
- 反応装置が連続して連結された2つの触媒床で形成されており、触媒床のそれぞれにプロパン及び酸素を含有する別のガス混合物が供給されそしてその際に供給される酸素の量が供給される水素に比較して酸素と水素とが反応して水蒸気を形成する反応に関して化学量論的に不足している、請求項1〜4のいずれか一つに記載の方法。
- 反応装置が連続して連結された3つの触媒床で形成されており、最初の2つの触媒床のそれぞれにプロパン及び酸素を含有する別のガス混合物が供給されそしてその際に供給される酸素の量が最初の2つの触媒床に供給される水素に比較して、酸素と水素とが反応して水蒸気を形成する反応に関して化学量論的に不足しており、そして第三の触媒床に、プロパン及び酸素を含有する別のガス混合物であってもよいしまたはこれを含有していてもよい酸素含有ガスを供給しそしてその際に供給される酸素の量が第三の触媒床に供給される水素に比較して、酸素と水素とが反応して水蒸気を形成する反応に関して化学量論量である、請求項1〜4のいずれか一つに記載の方法。
- 連続して連結された第一の触媒床と第二の触媒床との間に水または水蒸気または水−水蒸気混合物を供給する、請求項5または6に記載の方法。
- 第三の触媒床において水素の完全酸化を200〜500℃の温度で実施する、請求項6に記載の方法。
- 第三の触媒床において水素の完全酸化を300〜400℃の温度で実施する、請求項8に記載の方法。
- アクリルニトリルまたはプロピレンオキサイドまたはアクロレインまたはアクリル酸の製法を出所とする、プロパン及び酸素含有ガス混合物を請求項1〜9のいずれか一つに記載の方法の原料として使用する方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102004054657A DE102004054657A1 (de) | 2004-11-11 | 2004-11-11 | Verfahren zur katalytischen Dehydrierung von Propan zu Propylen |
| DE102004054657.6 | 2004-11-11 | ||
| PCT/EP2005/012070 WO2006050957A1 (de) | 2004-11-11 | 2005-11-10 | Verfahren zur katalytischen dehydrierung von propan zu propylen |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2008519792A JP2008519792A (ja) | 2008-06-12 |
| JP2008519792A5 JP2008519792A5 (ja) | 2008-12-25 |
| JP5442202B2 true JP5442202B2 (ja) | 2014-03-12 |
Family
ID=35788038
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007540581A Expired - Fee Related JP5442202B2 (ja) | 2004-11-11 | 2005-11-10 | プロパンをプロピレンに接触的に脱水素する方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US7678956B2 (ja) |
| EP (1) | EP1809587A1 (ja) |
| JP (1) | JP5442202B2 (ja) |
| DE (1) | DE102004054657A1 (ja) |
| EA (1) | EA010463B1 (ja) |
| EG (1) | EG25078A (ja) |
| NO (1) | NO20072980L (ja) |
| UA (1) | UA92902C2 (ja) |
| WO (1) | WO2006050957A1 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7582268B1 (en) * | 2006-07-12 | 2009-09-01 | Uop Llc | Reactor system with interstage product removal |
| DE102006035718A1 (de) | 2006-07-28 | 2008-01-31 | Basf Ag | Verfahren zum Langzeitbetrieb einer kontinuierlich betriebenen heterogen katalysierten partiellen Dehydrierung eines zu dehydrierenden Kohlenwasserstoffs |
| WO2008095860A2 (de) * | 2007-02-06 | 2008-08-14 | Basf Se | Verfahren zur bereitstellung eines sauerstoff enthaltenden gasstromes für die endotherme umsetzung eines ausgangsstromes, enthaltend einen oder mehrere kohlenwasserstoffe |
| DE102008010422A1 (de) * | 2008-02-21 | 2009-09-03 | Uhde Gmbh | Fixiervorrichtung für Katalysatorpartikel |
| MX2011006487A (es) * | 2008-12-18 | 2011-09-30 | Uhde Gmbh | Variacion de la impregnacion de estaño de un catalizador para la deshidrogenacion de alcanos. |
| DE102009056539A1 (de) | 2009-12-03 | 2011-06-09 | Uhde Gmbh | Variation der Zinnimprägnierung eines Katalysators zur Alkandehydrierung |
| DE102008062782A1 (de) | 2008-12-18 | 2010-07-01 | Uhde Gmbh | Variation der Zinnimprägnierung eines Katalysators zur Alkandehydrierung |
| DE102009012452A1 (de) * | 2009-03-12 | 2010-09-16 | Uhde Gmbh | Verfahren zur Verminderung von Olefinverlusten bei der Entfernung von Kohlendioxid aus einem Olefinstrom aus Dehydrierungsreaktionen |
| CA2833822C (en) * | 2013-11-21 | 2020-08-04 | Nova Chemicals Corporation | Inherently safe odh operation |
| EP4192802A1 (en) | 2020-08-06 | 2023-06-14 | ExxonMobil Chemical Patents Inc. | Processes for upgrading alkanes and alkyl aromatic hydrocarbons |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0117146B1 (en) * | 1983-02-22 | 1986-12-30 | The Halcon Sd Group, Inc. | Conversion of propane to acrylic acid |
| US4609502A (en) * | 1985-02-14 | 1986-09-02 | The Halcon Sd Group, Inc. | Process for preparing unsaturated nitriles from alkanes |
| US4599471A (en) | 1985-09-16 | 1986-07-08 | Uop Inc. | Method for oxygen addition to oxidative reheat zone of hydrocarbon dehydrogenation process |
| US4990632A (en) * | 1988-03-23 | 1991-02-05 | The Boc Group, Inc. | Process for the production of oxides |
| US5233118A (en) * | 1988-12-05 | 1993-08-03 | Uop | Steam dehydrogenation process |
| US5235121A (en) | 1991-08-02 | 1993-08-10 | Phillips Petroleum Company | Method for reforming hydrocarbons |
| US5527979A (en) | 1993-08-27 | 1996-06-18 | Mobil Oil Corporation | Process for the catalytic dehydrogenation of alkanes to alkenes with simultaneous combustion of hydrogen |
| NO300117B1 (no) | 1994-12-22 | 1997-04-14 | Norske Stats Oljeselskap | Reaktor for dehydrogenering av hydrokarboner med selektiv oksidasjon av hydrogen |
| DE19858747A1 (de) | 1998-12-18 | 2000-06-21 | Linde Ag | Verfahren und Katalysatorstation zur Dehydrierung von Alkanen |
| DE10240129B4 (de) * | 2002-08-30 | 2004-11-11 | Basf Ag | Integriertes Verfahren zur Synthese von Propylenoxid |
| AU2003278286A1 (en) | 2002-09-10 | 2004-04-30 | Arkema | Method for producing acrylic acid from propane, in the presence of molecular oxygen |
| DE10251135B4 (de) | 2002-10-31 | 2006-07-27 | Uhde Gmbh | Verfahren zur katalytischen Dehydrierung von leichten Paraffinen zu Olefinen |
-
2004
- 2004-11-11 DE DE102004054657A patent/DE102004054657A1/de not_active Ceased
-
2005
- 2005-11-10 WO PCT/EP2005/012070 patent/WO2006050957A1/de active Application Filing
- 2005-11-10 UA UAA200706356A patent/UA92902C2/ru unknown
- 2005-11-10 EP EP05810736A patent/EP1809587A1/de not_active Withdrawn
- 2005-11-10 US US11/667,518 patent/US7678956B2/en not_active Expired - Fee Related
- 2005-11-10 JP JP2007540581A patent/JP5442202B2/ja not_active Expired - Fee Related
- 2005-11-10 EA EA200701037A patent/EA010463B1/ru not_active IP Right Cessation
-
2007
- 2007-05-13 EG EGNA2007000471 patent/EG25078A/xx active
- 2007-06-11 NO NO20072980A patent/NO20072980L/no not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| UA92902C2 (ru) | 2010-12-27 |
| JP2008519792A (ja) | 2008-06-12 |
| EA200701037A1 (ru) | 2007-10-26 |
| US20080300440A1 (en) | 2008-12-04 |
| EP1809587A1 (de) | 2007-07-25 |
| US7678956B2 (en) | 2010-03-16 |
| EG25078A (en) | 2011-07-31 |
| WO2006050957A1 (de) | 2006-05-18 |
| EA010463B1 (ru) | 2008-08-29 |
| NO20072980L (no) | 2007-08-07 |
| DE102004054657A1 (de) | 2006-05-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5442202B2 (ja) | プロパンをプロピレンに接触的に脱水素する方法 | |
| KR101263087B1 (ko) | Co2를 온화한 산화제로 사용하는 에틸벤젠의 산화성 탈수소화에 의한 스티렌 모노머 공정 | |
| US7740829B2 (en) | Synthesis gas production and use | |
| US4910228A (en) | Methanol | |
| EP0001329B1 (en) | Process and plant for producing ammonia | |
| CN107428650B (zh) | 用于生产甲醛的方法 | |
| WO1999003807A1 (en) | Process for the preparation of methanol and hydrogen | |
| WO2000009441A2 (en) | Steam reforming | |
| JPS5953245B2 (ja) | メタンフユウガスノ セイゾウホウホウ | |
| JP3221938B2 (ja) | エタンの酢酸への流動層酸化法 | |
| KR101581054B1 (ko) | 알칸 탈수소화용 촉매의 재생방법 | |
| US7816576B2 (en) | Method for catalytically dehydrating hydrocarbons | |
| CA3137256A1 (en) | Process for synthesising methanol | |
| CN106999844B (zh) | 从含烃气体混合物中除去氧气 | |
| US6667409B2 (en) | Process and apparatus for integrating an alkene derivative process with an ethylene process | |
| WO2020176650A1 (en) | Integrated indirect heat transfer process for the production of syngas and olefins by catalytic partial oxidation and catalytic selective dehydrogenation | |
| US20220135506A1 (en) | Methanol production process | |
| WO2023041396A1 (en) | A method for producing syngas using catalytic reverse water gas shift | |
| KR20240055754A (ko) | 메탄의 산화적 커플링을 수행하기 위한 방법 및 시스템 | |
| WO2022238671A1 (en) | Process for synthesising methanol | |
| CN113710613A (zh) | 具有提高的能效的甲醇生产方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20081107 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20081107 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20100525 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20110803 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110906 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20111202 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20111209 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120305 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120612 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20121011 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20121012 |
|
| A911 | Transfer of reconsideration by examiner before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20121105 |
|
| A912 | Removal of reconsideration by examiner before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A912 Effective date: 20121122 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20131218 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |