JP5102002B2 - アセナピン合成中間体の製造方法 - Google Patents
アセナピン合成中間体の製造方法 Download PDFInfo
- Publication number
- JP5102002B2 JP5102002B2 JP2007313406A JP2007313406A JP5102002B2 JP 5102002 B2 JP5102002 B2 JP 5102002B2 JP 2007313406 A JP2007313406 A JP 2007313406A JP 2007313406 A JP2007313406 A JP 2007313406A JP 5102002 B2 JP5102002 B2 JP 5102002B2
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound
- mol
- compound represented
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- IUJAAIZKRJJZGQ-UHFFFAOYSA-N OC(Cc(cccc1)c1Cl)=O Chemical compound OC(Cc(cccc1)c1Cl)=O IUJAAIZKRJJZGQ-UHFFFAOYSA-N 0.000 description 1
- ALMJMOPIBISVHZ-UHFFFAOYSA-N OC(Cc(cccc1)c1Oc(cc1)ccc1Cl)=O Chemical compound OC(Cc(cccc1)c1Oc(cc1)ccc1Cl)=O ALMJMOPIBISVHZ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/02—Preparation of carboxylic acid amides from carboxylic acids or from esters, anhydrides, or halides thereof by reaction with ammonia or amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/367—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by introduction of functional groups containing oxygen only in singly bound form
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Description
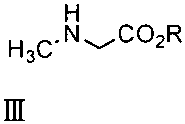
(1) 式[I]:
(2) 式[I]:
(3) 縮合反応を式[II]:
(4) 式[II]:
(5) 式[I]:
(6) 式[II]:
(7) 式[II]:
(8) Rがメチル基またはエチル基である前記(2)〜(6)のいずれかに記載の製造方法、
に関する。
化合物[II]は化合物[I]と4−クロロフェノールとを反応させることにより製造することができる。本反応は、触媒存在下で行なうのが好ましい。
4−クロロフェノールの使用量は、化合物[I]1モルに対し、通常1モル〜2モル程度、好ましくは1モル〜1.5モル程度である。
化合物[II]と化合物[III]とを縮合させて化合物[IV]を製造する方法は、化合物[II]またはそのカルボキシル基における反応性誘導体と化合物[III]とを反応させることにより実施することができる。
なお、化合物[III]において、記号Rで示される炭素数1〜6のアルキル基としては、直鎖状でも分岐状でもよく、とりわけメチル基、エチル基が好ましい。
(1−a) 2−(2−(4−クロロフェノキシ)フェニル)酢酸[II]の合成
2−クロロフェニル酢酸[I](1.0g,5.9mmol)、4−クロロフェノール(0.78g,6.5mmol)、炭酸セシウム(3.8g,11.7mmol),臭化銅(I)(42mg,0.29mmol)およびジエチレングリコールジメチルエーテル(5ml)を混合し、145℃で8時間加熱した。冷却後、反応マスに水(15ml)を流入した後、濃塩酸(約2g)を加え酸性とし、トルエン(20ml×2)にて抽出を行った。トルエン層を水(20ml)、飽和食塩水(20ml)で洗浄後、無水硫酸マグネシウムで脱水し、その後硫酸マグネシウムを濾過により除去し、[II]のトルエン溶液を得た(標品を用いた定量収率59%)。一部スペクトルデータ用に濃縮後シリカゲルカラムクロマトグラフィーにより精製をおこない化合物[II]を得た。
炭酸セシウムの代わりに炭酸カリウム(1.6g,11.7mmol)を用いて上記と同条件で行った場合の収率は62%、また、臭化銅(I)の代わりに塩化銅(I)(29mg,0.29mmol)を用いて上記と同条件で反応を行った場合の収率は59%であった。
2−クロロフェニル酢酸[I](10.0g,58.6mmol)、4−クロロフェノール(7.8g,64.5mmol)、炭酸カリウム(16.2g,117.2mmol)、N,N−ジメチルグリシン(0.41g,2.9mmol),臭化銅(I)(0.42g,2.9mmol)およびジエチレングリコールジメチルエーテル(30ml)を混合し、140℃で4時間加熱した。冷却後、反応マスに水(50ml)を流入した後、濃塩酸(18g)を加え酸性とし、トルエン(30ml×2)にて抽出を行った。トルエン層を水(30ml×2)、飽和食塩水(30ml)で洗浄後、無水硫酸マグネシウムで脱水し、その後硫酸マグネシウムを濾過により除去し、化合物[II]のトルエン溶液を得た。この溶液をそのまま次の工程に使用した。一部スペクトルデータ用に濃縮後シリカゲルカラムクロマトグラフィーにより精製を行い、化合物[II]を得た。
上記実施例(1−b)で得た化合物[II]のトルエン溶液にN,N−ジメチルホルムアミド(0.5ml)を加えた後、50℃〜65℃に温度を保ちながら、塩化チオニル(8.4g,70.3mmol)を約30分かけて滴下した。滴下終了後さらに2時間、50℃〜65℃で反応を行った。冷却後、トルエン(約25ml)を減圧留去し化合物[II]の酸クロライドのトルエン溶液を得た。トルエン(10ml)、水(30ml)および炭酸水素ナトリウム(14.8g,175.8mmol)の混合溶液にN−メチルグリシンエチルエステル塩酸塩(10.4g,67.4mmol)を水(15ml)に溶解させた液を加えた後、上記で得た化合物[II]の酸クロライドのトルエン溶液を0〜10℃に保ちながら滴下した。
滴下終了後、0〜10℃で4時間攪拌した後、水(30ml)およびトルエン(30ml)を流入し抽出を行った。トルエン層を飽和食塩水(30ml)で洗浄後、無水硫酸マグネシウムで脱水した。硫酸マグネシウムを濾過により除去後、減圧濃縮によりトルエンを留去(約100ml)し、化合物[IV]のトルエン溶液を得た。この溶液をそのまま次の工程に使用した。一部スペクトルデータ用に濃縮後シリカゲルカラムクロマトグラフィーにより精製をおこない化合物[IV]を得た。
7.41〜7.12(5H,m)、6.91〜6.85(3H,m)、4.16(2H,q,2J=7Hz)、4.08(2H,s)、3.76(2H,s)、3.06(3H,s)、1.25(3H,t,J=7Hz)
2−(2−(4−クロロフェノキシ)フェニル)酢酸[II]の合成
2−クロロフェニル酢酸メチルエステル(1.85g、10mmol)と4−クロロフェノール(1.29g、10mmol)とジグライム(30ml)と炭酸カリウム(2.76g、20mmol)を混合させた後、反応容器で窒素置換し、臭化銅(I)(72mg、0.5mmol)を加え、120℃で24時間加熱した。高速液体クロマトグラフィーにて分析したところ、化合物[II]のメチルエステルの生成は認められなかった。2−クロロフェニル酢酸メチルエステルが91%残存し、化合物[II]が4%生成していた。
2−(2−(4−クロロフェノキシ)フェニル)酢酸[II]の合成
4−クロロフェニル酢酸(3.4g、20mmol)、4−クロロフェノール(3.1g、24mmol)、炭酸カリウム(5.5g、40mmol)、臭化銅(I)(57mg、0.4mmol)およびジグライム(17ml)を混合し、130℃で6時間加熱した。冷却後、反応マスを高速液体クロマトグラフィーで分析したところ、原料以外の生成物のピークは認められなかった。
(1) 3−(2−(4−クロロフェノキシ)フェニル)−4−ヒドロキシ−1−メチル−1H−ピロール−2(5H)−オン[V]の合成
上記実施例(2)で得られた化合物[IV]のトルエン溶液に20%ナトリウムメチラートメタノール溶液(13.4g,70.3mmol)を加え、50〜65℃で1.5時間攪拌した。その後、水(50ml)を加えた後、濃塩酸(8g)によりpH1以下とした。析出した結晶を濾過し、水(50ml×2)で洗浄を行った後、減圧乾燥し淡黄色結晶の化合物[V]を3.9g得た。
上記(1)で得た化合物[V](10.0g,31.7mmol)と105%リン酸(44g)を混合し、145〜155℃で14時間加熱した。原料が残留していたため、五酸化二リン(10.7g)を加え、145〜155℃で4時間加熱した。まだ原料が残留していたため、さらに五酸化二リン(10.7g)を加え、155〜165℃で3時間加熱した。反応マスに水(3ml)とアセトン(57ml)の混合液を60〜80℃で滴下し、さらに水(50ml)を滴下した。滴下終了後、さらに、水(20ml)を流入し5℃まで冷却後、濾過を行った。得られた結晶を水(20ml×5)で洗浄後、減圧乾燥し淡緑色固体の生成物[V]を8.4g得た。収率88%。
1H−NMRデータ(ppm in DMSO−d6):3.09(3H,d,J=2Hz),4.61(2H,d,J=4Hz),7.30〜7.60(6H,m),8.04(1H,t−like,J=ca.3Hz).
上記(2)で得られた5−クロロ−2,3−ジヒドロ−2−メチル−1H−ジベンゾ[2,3:6,7]オキセピノ[4,5−c]ピロール−1−オン[VI]を国際公開2006/106136号パンフレット記載の方法により、化合物[VII]および化合物[VIII]を経て、アセナピン(トランス−5−クロロ−2,3,3a,12b−テトラヒドロ−2−メチル−1H−ジベンゾ[2,3:6,7]オキセピノ[4,5−c]ピロール)を得る。
上記(3)で得られたトランス−5−クロロ−2,3,3a,12b−テトラヒドロ−2−メチル−1H−ジベンゾ[2,3:6,7]オキセピノ[4,5−c]ピロールを99.5%エタノールに溶解させ、マレイン酸のエタノール溶液を室温で滴下する。析出する固体をろ過、乾燥して、トランス−5−クロロ−2,3,3a,12b−テトラヒドロ−2−メチル−1H−ジベンゾ[2,3:6,7]オキセピノ[4,5−c]ピロール マレエートを得る。mp.141℃
Claims (8)
- 式[I]:
- 式[I]:
- 縮合反応を式[II]:
- 式[II]:
- 式[I]:
- 式[II]:
- 式[II]:
- Rがメチル基またはエチル基である請求項2〜6のいずれかに記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007313406A JP5102002B2 (ja) | 2006-12-22 | 2007-12-04 | アセナピン合成中間体の製造方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006346735 | 2006-12-22 | ||
| JP2006346735 | 2006-12-22 | ||
| JP2007313406A JP5102002B2 (ja) | 2006-12-22 | 2007-12-04 | アセナピン合成中間体の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2008174547A JP2008174547A (ja) | 2008-07-31 |
| JP5102002B2 true JP5102002B2 (ja) | 2012-12-19 |
Family
ID=39562275
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007313406A Expired - Fee Related JP5102002B2 (ja) | 2006-12-22 | 2007-12-04 | アセナピン合成中間体の製造方法 |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP5102002B2 (ja) |
| CN (1) | CN101563312A (ja) |
| WO (1) | WO2008078482A1 (ja) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101851242B (zh) * | 2010-05-25 | 2013-07-24 | 上海皓元生物医药科技有限公司 | 阿塞那平中间体的制备方法 |
| WO2012038975A2 (en) * | 2010-09-22 | 2012-03-29 | Msn Laboratories Limited | Process for the preparation of (3ars,12brs)-5-chloro-2-methyl-2,3,3a12b-tetrahydro-1hdibenzo[2,3:6,7] oxepino [4,5-c]pyrrole maleate and it's pharmaceutical composition thereof |
| CN102746209A (zh) * | 2012-06-20 | 2012-10-24 | 盛世泰科生物医药技术(苏州)有限公司 | 一种3-(2-(4-氯苯氧基)苯基)-1-甲基-2,4-二酮的合成方法 |
| CN103760258A (zh) * | 2014-01-07 | 2014-04-30 | 万特制药(海南)有限公司 | 一种用液相色谱法分离测定马来酸阿塞那平有关物质的方法 |
| US11337932B2 (en) | 2016-12-20 | 2022-05-24 | Lts Lohmann Therapie-Systeme Ag | Transdermal therapeutic system containing asenapine and polysiloxane or polyisobutylene |
| CN110087640A (zh) | 2016-12-20 | 2019-08-02 | 罗曼治疗系统股份公司 | 包含阿塞那平的透皮治疗系统 |
| JP2020525545A (ja) | 2017-06-26 | 2020-08-27 | エルテーエス ローマン テラピー−ジステーメ アーゲー | アセナピンおよびシリコーンアクリルハイブリッドポリマーを含有する経皮治療システム |
| US12329862B2 (en) | 2018-06-20 | 2025-06-17 | Lts Lohmann Therapie-Systeme Ag | Transdermal therapeutic system containing asenapine |
| CN112704672A (zh) | 2018-06-20 | 2021-04-27 | 罗曼治疗系统股份公司 | 含有阿塞那平的透皮治疗系统 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5775947A (en) * | 1980-10-28 | 1982-05-12 | Ihara Chem Ind Co Ltd | Production of 2-(substituted phenoxy)phenylacetic acid |
| JPS5775948A (en) * | 1980-10-28 | 1982-05-12 | Ihara Chem Ind Co Ltd | Production of 2-(substituted phenoxy)phenylacetic acid |
| WO2006106136A1 (en) * | 2005-04-07 | 2006-10-12 | N.V. Organon | Intermediate compounds for the preparation of trans-5-chloro-2-methyl-2,3,3a,12b-tetrahydro-1h-dibenz[2,3:6,7]oxepino[4,5-c]pyrrole |
-
2007
- 2007-11-15 WO PCT/JP2007/072601 patent/WO2008078482A1/ja not_active Ceased
- 2007-11-15 CN CNA2007800467159A patent/CN101563312A/zh active Pending
- 2007-12-04 JP JP2007313406A patent/JP5102002B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| CN101563312A (zh) | 2009-10-21 |
| JP2008174547A (ja) | 2008-07-31 |
| WO2008078482A1 (ja) | 2008-07-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5102002B2 (ja) | アセナピン合成中間体の製造方法 | |
| JP4528123B2 (ja) | ナプロキセンのニトロオキシ誘導体の製造法 | |
| WO2007046554A1 (ja) | ジベンゾオキセピノピロール化合物の製造方法、その中間体及び該中間体の製造方法 | |
| WO2010122794A1 (ja) | ピラジンカルボン酸誘導体の製造方法及びその中間体 | |
| CN101605773B (zh) | 二苯并氧杂*化合物的制造方法 | |
| JP2008056615A (ja) | ビニルエチニルアリールカルボン酸類、その製造方法及びそれを用いた熱架橋性化合物の製造方法 | |
| JP2009242244A (ja) | ピラジン誘導体類の製造方法及びその中間体類 | |
| WO2006090783A1 (ja) | 4-フルオロイソキノリン-5-スルホニルハライド又はその塩の製造方法 | |
| JP5027477B2 (ja) | ジベンゾオキセピノピロール化合物およびその中間体の製造方法ならびに新規中間体 | |
| JP2009286717A (ja) | ビスベンゾオキサジノン化合物の製造方法 | |
| EP4081503A1 (en) | A process for the synthesis of melphalan | |
| US20050283021A1 (en) | Production method of O-substituted tyrosine compound | |
| CN101160288B (zh) | 烟酸衍生物或其盐的制造方法 | |
| JP4061333B2 (ja) | 2−(ピラゾール−1−イル)ピリジン誘導体 | |
| WO1999041214A1 (en) | Halogenating agent and process for halogenating hydroxyl group | |
| CN116082155A (zh) | 一种利用对称环氧化合物制备唑啉草酯中间体的方法 | |
| JP2006045138A (ja) | 1−アミノシクロプロパンカルボン酸の精製方法及び製造方法 | |
| JP4379970B2 (ja) | オレフィン化合物の製造法 | |
| JP6004307B2 (ja) | 3−(2−(1−ベンゾチオフェン−5−イル)エトキシ)プロピオン酸の塩またはその水和物およびその製造法 | |
| EP1698611A1 (en) | Process for producing phenylacetic acid derivative | |
| JP3961049B2 (ja) | 3−アミノ−4−(1−ヒドロキシアルキル)ピラゾリン化合物、その製造方法およびそれを使用した製造方法 | |
| KR100525358B1 (ko) | 카르복실 벤조트리아졸 알킬에스테르의 제조방법 | |
| CN120485299A (zh) | 一种中间体的制备方法 | |
| JP3819473B2 (ja) | 4,4−ビスハロメチル−3−オキソアルカンカルボン酸誘導体とそれを用いる3−シクロプロピル−3−オキソプロピオン酸誘導体の製造方法 | |
| JP3332171B2 (ja) | チエノ〔3,2−b〕ピリジン誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100624 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120724 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120925 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120927 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151005 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |