本発明のエネルギーデバイスは、集電体層と、前記集電体層上に直接又は下地層を介して設けられた負極活物質薄膜とを含む。そして、前記負極活物質薄膜がシリコンを主成分として含む2以上のシリコン薄膜を含む多層構造を有する。
真空成膜法などによってシリコン薄膜を形成すると、シリコンは一般に厚さ方向を中心軸方向とする略逆円錐台状の柱状粒子を形成する。電池容量を増加させるためにシリコン薄膜の厚みを増大させると、前記略逆円錐台状粒子の高さが増加し、その結果、その径が粗大化する。シリコン薄膜をリチウムイオン2次電池の負極活物質として使用して充放電を行うと、シリコン薄膜内でリチウムイオンの吸蔵/放出が繰り返され、シリコン粒子が膨張/収縮を繰り返す。シリコン薄膜内に径が粗大化したシリコン粒子が存在すると、シリコン粒子の膨張/収縮がシリコン薄膜と負極集電体層との界面での剥離、シリコン粒子の歪みや破壊を生じさせ、サイクル特性を低下させる。
本発明では、負極活物質薄膜が2以上のシリコン薄膜の多層構造を有しているので、負極活物質薄膜の厚みを増大させても、シリコン薄膜の層数を増大させることにより、1層あたりのシリコン薄膜の厚みの増大を防止できる。従って、シリコン薄膜内でシリコン粒子径が粗大化しない。よって、負極活物質薄膜を厚くしてもサイクル特性の劣化を防止することが出来る。その結果、大容量とサイクル特性とが両立したエネルギーデバイスを実現することができる。
本発明において、「シリコンを主成分として含む」とは、シリコンの含有量が50at%以上であることを意味する。電池容量を向上させるためには、シリコンの含有量は、望ましくは70at%以上、更に望ましくは80at%以上、最も望ましくは90at%以上である。シリコン含有量が高いほど電池容量を向上できるからである。
前記シリコン薄膜は厚さ方向を長手方向とする柱状粒子を有し、厚さ方向に隣り合う前記シリコン薄膜間で前記柱状粒子が不連続であることが好ましい。これにより、径が粗大化したシリコン粒子の発生を抑えることができる。従って、充放電の繰返しに伴うリチウムイオンの吸蔵/放出によってシリコン粒子が膨張/収縮しても、シリコン薄膜と負極集電体層との界面での剥離、シリコン粒子の歪みや破壊を抑制できる。その結果、サイクル特性の劣化を防止できる。
隣り合う前記シリコン薄膜間に界面層が存在することが好ましい。これにより、界面層を挟む柱状粒子を不連続にすることができるので、径が粗大化したシリコン粒子の発生を抑えることができる。
前記界面層内にシリコンと気体元素との化合物が存在することが好ましい。これにより、界面層を挟む柱状粒子をより確実に不連続にすることができる。
前記化合物が窒化物又は酸化物であることが好ましい。このような化合物は、シリコン薄膜の形成中に容易に形成することができ、しかも、不連続なシリコンの柱状粒子を確実に形成できる。
あるいは、隣り合う前記シリコン薄膜間にシリコンが50at%未満である中間層が存在していても良い。これにより、中間層を挟む柱状粒子を不連続にすることができるので、径が粗大化したシリコン粒子の発生を抑えることができる。
前記中間層がリチウムを含むことが好ましい。これにより、中間層がリチウムイオンを補完することができるので、電池容量を増大させることができる。
前記中間層が不連続膜又は島状であることが好ましい。これによりイオンの移動が容易になるので、サイクル特性を向上できる。
前記中間層がシリコンより低融点の元素を主成分として含むことが好ましい。これにより、不連続膜又は島状の中間層の形成が容易になる。
前記シリコン薄膜に含まれるシリコンの一部が酸化物であることが好ましい場合がある。ここでいうシリコンの酸化物とは、厚さ方向に隣り合うシリコン薄膜間に存在する上記界面層に含まれるシリコンの酸化物を含まない。厚さ方向において、上下の界面層を除いた中間領域にシリコンの酸化物が含まれていることを意味する。シリコン薄膜中のシリコンの含有量が多く、電池容量が大きい場合には、充放電時のシリコン薄膜の膨張/収縮の程度が大きくなり、サイクル特性が低下する場合がある。シリコン薄膜がシリコンの酸化物を含むと、シリコンの酸化物は充放電時の膨張/収縮が少ないから、充放電時のシリコン薄膜の膨張/収縮を抑えることができ、サイクル特性を向上させることができる。
次に、本発明のエネルギーデバイスの製造方法は、負極活物質薄膜と正極活物質薄膜とが電子伝導を呈さない層を介して対向したエネルギーデバイスの製造方法であって、集電体層上に直接又は下地層を介して前記負極活物質薄膜を真空成膜法により形成する工程と、前記正極活物質薄膜を形成する工程とを含む。そして、前記負極活物質薄膜の形成は、経時的に分割された2以上のシリコン堆積工程を含む。
シリコン堆積工程を経時的に複数回に分割して単一の負極活物質薄膜を形成することにより、この単一の負極活物質薄膜中に厚さ方向に不連続なシリコン柱状粒子を容易に形成できる。従って、負極活物質薄膜の厚みを増大させても、シリコン堆積工程の数を増大させることにより、略逆円錐台状のシリコン粒子の厚さ方向の寸法の増大を防止できる。従って、負極活物質薄膜内でシリコン粒子径が粗大化しない。よって、負極活物質層を厚くしてもサイクル特性の劣化を防止することが出来る。その結果、大容量とサイクル特性とが両立したエネルギーデバイスを実現することができる。
ここで、「真空成膜法」とは、蒸着法、スパッタ法、CVD法、イオンプレーティング法、レーザーアブレーション法などの各種真空薄膜製造プロセスを含む。薄膜の種類に応じて最適な成膜法を選択することができる。真空成膜法により薄い負極活物質薄膜を効率よく製造できる。その結果、小型薄型のエネルギーデバイスが得られる。
2以上の前記シリコン堆積工程の間に、形成されたシリコンを主成として含むシリコン薄膜の表面に表面改質処理を行うことが好ましい。表面改質処理により、負極活物質層中にシリコン柱状粒子を不連続にする界面層を容易に形成できる。従って、径が粗大化したシリコン粒子の発生を抑えることができる。
前記表面改質処理が前記シリコン薄膜の表面へのガス導入であることが好ましい。このような表面改質処理は、真空成膜法において容易に行うことができる。
前記ガスが窒素又は酸素を主成分として含むことが好ましい。これにより、簡単な方法で表面改質処理の前後で形成されるシリコン粒子を容易に不連続にすることができる。
前記ガスをイオン化又はプラズマ化させることが好ましい。これにより、表面改質処理の前後で形成されるシリコン粒子をより確実に不連続にすることができる。
あるいは、2以上の前記シリコン堆積工程の間に、シリコンが50at%未満である中間層を形成しても良い。これにより、中間層の形成の前後で形成されるシリコン薄膜中の柱状粒子を不連続にすることができるので、径が粗大化したシリコン粒子の発生を抑えることができる。
前記シリコン堆積工程に引き続き、前記中間層を真空雰囲気中で真空成膜法により形成することが好ましい。真空雰囲気を破壊することなくシリコン堆積工程と中間層形成工程とを行うことにより、層間の密着性が向上し剥離を防止できるので、電池容量の低下を防止できる。また、いずれの工程をも真空成膜法で行うことにより、負極活物質薄膜を効率よく形成できる。
前記中間層を真空雰囲気中で真空成膜法により形成し、引き続いて前記シリコン堆積工程を行うことが好ましい。真空雰囲気を破壊することなく中間層形成工程とシリコン堆積工程とを行うことにより、層間の密着性が向上し剥離を防止できるので、電池容量の低下を防止できる。また、いずれの工程をも真空成膜法で行うことにより、負極活物質薄膜を効率よく形成できる。
前記中間層がシリコンより低融点の元素を主成分として含むことが好ましい。これにより、成膜中又は成膜後に加熱することにより、不連続膜又は島状の中間層を容易に形成できる。このような中間層を形成することにより、イオンの移動が容易になるので、サイクル特性を向上できる。
前記中間層を形成するための材料の融点をTm(℃)としたとき、前記中間層の形成時における被成膜面温度をTm/3(℃)以上に維持することが好ましい。これにより、不連続膜又は島状の中間層を効率よく形成できる。
あるいは、前記中間層を形成した後、前記中間層をその融点以上に加熱しても良い。これによっても、不連続膜又は島状の中間層を容易に形成できる。
前記シリコン堆積工程を不活性ガス又は窒素雰囲気中で行うことが好ましい。これにより、被堆積面と平行な方向に隣り合うシリコン柱状粒子が合併して成長して、シリコン粒子径が粗大化するのを防止できる。その結果、充放電時にシリコン粒子の膨張/収縮の程度が激しくなり、サイクル特性が低下するのを抑制できる。雰囲気ガスとしては、実用性及び上記の効果の顕著性の観点から、アルゴンが最も好ましい。
以下、図面を参照しながら本発明の実施の形態について説明する。
(参考実施形態1)
参考実施形態1にかかるエネルギーデバイスを説明する。
本参考実施形態1のエネルギーデバイスは、両面に正極活物質が形成された正極集電体と、電子伝導を呈さない層としてのセパレータと、両面に負極活物質が形成された負極集電体とを、正極集電体と負極集電体との間にセパレータが介在するようにして巻回した円筒状巻回物を電池缶に収め、この電池缶を電解液で満たしてなる。
正極集電体としては、Al、Cu、Ni、ステンレススチールの厚さ10〜80μmの箔、網などを用いることが出来る。あるいは、表面に金属薄膜が形成されたポリエチレンテレフタレート、ポリエチレンナフタレートなどの高分子基板を用いることも出来る。
正極活物質はリチウムイオンの出入が出来ることが必要であり、Co、Ni、Mo、Ti、Mn、Vなどの遷移金属を含むリチウム含有遷移金属酸化物や、これにアセチレンブラックなどの導電性補助剤とニトリルゴム、ブチルゴム、ポリテトラフルオロエチレン、ポリフッ化ビニリデンなどの結着剤とを混合した混合ペーストを用いることも出来る。
負極集電体としては、Cu、Ni、ステンレススチールの厚さ10〜80μmの箔、網などを用いることが出来る。あるいは、表面に金属薄膜が形成されたポリエチレンテレフタレート、ポリエチレンナフタレートなどの高分子基板を用いることも出来る。
セパレーターは機械的強度とイオン透過性とに優れることが好ましく、ポリエチレン、ポリプロピレン、ポリフッ化ビニリデンなどを用いることが出来る。セパレーターの孔径は例えば0.01〜10μmであり、その厚さは例えば5〜200μmである。
電解液としては、エチレンカーボネート、プロピレンカーボネート、メチルエチルカーボネート、6フッ化メチルアセテート、又はテトロヒドロフラン等の溶媒に、LiPF6、LiBF4、LiClO4などの電解質塩を溶解させた溶液を用いることが出来る。
電池缶としては、ステンレススチール、鉄、アルミニウム、ニッケルメッキスチールなどの金属材料を用いることができるが、電池用途に応じてプラスチック材料を用いることもできる。
負極活物質は、シリコンを主成分とするシリコン薄膜である。シリコン薄膜はアモルファスまたは微結晶であることが好ましく、スパッタリング法、蒸着法、CVD法をはじめとする真空成膜法で形成することができる。
[参考例1〜3,比較例1]
参考実施形態1に対応する参考例を説明する。
まず、正極の作製方法を述べる。Li2CO3とCoCO3とを所定のモル比で混合し、大気中において900℃で加熱することによって合成してLiCoO2を得た。これを100メッシュ以下に分級して正極活物質を得た。この正極活物質100g、導電剤として炭素粉末10g、結着剤としてポリ4フッ化エチレンディスパージョン8g、及び純水を混ぜ合わせてペースト状にした。この正極活物質含有ペーストを、正極集電体としての厚さ15μmの帯状のアルミニウム箔の両面に塗布し、乾燥して正極を得た。
負極集電体として厚さ30μmの帯状の銅箔を用い、その両面に負極活物質としてシリコン薄膜をスパッタリング法により形成した。詳細は後述する。
セパレ−タとして、厚さ25μmで、正極集電体及び負極集電体よりも広幅の帯状の多孔性ポリエチレンを用いた。
正極集電体にこれと同材質の正極リードをスポット溶接にて取り付けた。また、負極集電体にこれと同材質の負極リードをスポット溶接にて取り付けた。
上記によって得た正極と負極との間にセパレータが介在するようにこれらを重ね合わせて渦巻き状に巻回した。この円筒状巻回物の上下面に、ポリプロピレン製の絶縁板をそれぞれ配して有底の円筒状電池缶内に収納し、電池缶の開口近傍に段部を形成した後、非水電解液として、LiPF6を濃度1×103モル/m3で溶解したエチレンカーボネートとジエチルカーボネートの等比体積混合溶液を電池缶に注入し、封口板で開口を密閉してリチウムイオン二次電池を得た。
負極活物質としてのシリコン薄膜の形成方法を図1を用いて説明する。
図1に示す真空成膜装置10は、隔壁1aにより上下に仕切られた真空槽1を備える。隔壁1aより上側の部屋(搬送室)1bには、捲き出しロール11,円筒状のキャンロール13,捲き取りロール14,搬送ロール12a,12bが配置される。隔壁1aより下側の部屋(薄膜形成室)1cには、第1スパッタ成膜源51、ガス導入ノズル55、第2スパッタ成膜源52が配置されている。隔壁1aの中央部にはマスク4が設けられ、マスク4の開口を介してキャンロール13の下面が薄膜形成室1c側に露出している。真空槽1内は、真空ポンプ16により所定の真空度に維持される。
捲き出しロール11から捲き出された帯状の負極集電体5は、搬送ロール12a、キャンロール13、搬送ロール12bによって順に搬送され、捲き取りロール14に捲き取られる。この過程で、第1スパッタ成膜源51及び第2スパッタ成膜源52から生成された原子、分子、又はクラスタなどの粒子(以下、「スパッタ粒子」という)が隔壁1aのマスク4を通過して、キャンロール13上を走行している負極集電体5の表面上に付着して薄膜6を形成する。負極集電体5に対向して、その搬送方向の上流側から下流側に向かって、第1スパッタ成膜源51、ガス導入ノズル55、第2スパッタ成膜源52が配置されている。ガス導入ノズル55はキャンロール13の近傍にて、負極集電体5の幅方向(図1の紙面に垂直方向)の全幅にわたって伸びている。従って、ガス導入ノズル55は第2のマスクとして機能して、キャンロール13の外表面のうち、ガス導入ノズル55が対向する領域へのスパッタ粒子の堆積を防止する。従って、負極集電体5の表面には、最初に第1スパッタ成膜源51からのスパッタ粒子が主として堆積し、わずかな時間を挟んで、第2スパッタ成膜源52からのスパッタ粒子が主として堆積する。
参考例1では、このような装置を用いて、第1スパッタ成膜源51及び第2スパッタ成膜源52のそれぞれにおいてアルゴンイオンによりシリコンをスパッタして、負極集電体5としての銅箔上に、厚さ8μmのシリコン薄膜を形成した。シリコン薄膜の堆積速度を概ね2nm/sに設定した。第1スパッタ成膜源51及び第2スパッタ成膜源52のスパッタ源としていずれも直流マグネトロンスパッタを使用した。成膜中にガス導入ノズル55からのガス導入は行わなかった。
参考例2では、成膜中にガス導入ノズル55からN2ガスを0.05Pa・m3/sで導入する以外は参考例1と同様にしてシリコン薄膜を形成した。
参考例3では、成膜中にガス導入ノズル55からO2ガスを0.05Pa・m3/sで導入する以外は参考例1と同様にしてシリコン薄膜を形成した。
比較例1では、第2スパッタ成膜源52及びガス導入ノズル55を取り除き、第1スパッタ成膜源51のみを用い、堆積速度が参考例1と同じになるように負極集電体5の走行速度を調節する以外は参考例1と同様にして厚さ8μmのシリコン薄膜を形成した。
図2(A)〜(D)は、順に、比較例1,参考例1,2,3のシリコン薄膜(負極活物質薄膜)の厚さ方向の断面を撮影したSEM写真を模式的に示した図である。各図において、紙面下側が負極集電体(銅箔)5側、上側はシリコン薄膜の表面である。
比較例1(図2(A))では、シリコン粒子が負極集電体との界面から徐々に粒子径が増大しながら略逆円錐台状の柱状粒子として成長し、シリコン薄膜の表面近傍で粒子径が最も大きい。
これに対して、参考例1〜3(順に図2(B)〜(D))では、シリコン粒子が同様に略逆円錐台状の柱状粒子として成長しているが、厚さ方向の中央部分でシリコン粒子の成長が不連続となっている。粒子成長の不連続性はガス導入を行った参考例2,3(図2(C),(D))においてより顕著である。シリコン粒子が不連続である部分の厚さ方向の位置はいずれのシリコン粒子についてもほぼ一致している。参考例1〜3では、シリコン薄膜は、この不連続部分である界面層50によって分割され、上下方向に2層構造を有している。界面層50よりも下側(銅箔側)の層は主として第1スパッタ成膜源51により形成されたシリコン薄膜であり、界面層50よりも上側の層は主として第2スパッタ成膜源52により形成されたシリコン薄膜であると考えられる。界面層50よりも上側の層のシリコン粒子は、界面層50から新たに略逆円錐台状に成長を開始するために、シリコン薄膜の表面近傍での粒子径は比較例1と比して小さい。なお、参考例1〜3,比較例1のそれぞれのシリコン薄膜は、X線回折測定(CuKα、40kV)でいずれも顕著なシリコン結晶に相当するピークが検出されず、アモルファス又は微結晶であることが推定出来た。
図3は参考例1のシリコン薄膜(負極活物質薄膜)のオージェデプスプロファイルを示す図である。オージェデプスプロファイルは、フィリップス社製のSAM670を用いて測定した。電子銃の加速電圧を10kV、照射電流10nAとし、エッチング用のイオンガンの加速電圧3kV、スパッタレート0.4nm/sにて測定した。図の横軸の「膜表面からの深さ」は、サンプルと同一のSi膜とCu膜をスパッタエッチングして形成された段差を段差計で測定して得たスパッタレートを用いて、サンプルのスパッタエッチング時間を厚さ方向のエッチング深さに換算して得た。図3から分かるように、成膜の途中にガスを導入していない参考例1では、界面層に相当する位置にわずかなアルゴンのピークが見られる程度で顕著な組成の不連続性は確認できなかった。図2(B)において粒子成長の不連続性が認められるのは、オージェデプスプロファイルでは検出不能又は微量に検出できる程度のわずかなガス付着などによってもシリコン粒子の成長が不連続になる場合があることを示唆している。
図4は参考例2のシリコン薄膜(負極活物質薄膜)のオージェデプスプロファイルを示す図である。図4から分かるように、第1スパッタ成膜源51による成膜と第2スパッタ成膜源52による成膜との間にN2ガス雰囲気に曝される参考例2では、界面層に相当する位置に窒素分布のピークが検出されている。即ち、界面層にシリコンの窒化物が存在している。参考例2は、シリコン薄膜の形成途中でシリコンの窒化物が形成されると、シリコン粒子の成長が不連続になる可能性が高くなることを示唆している。
図示を省略するが、第1スパッタ成膜源51による成膜と第2スパッタ成膜源52による成膜との間にO2ガス雰囲気に曝される参考例3のシリコン薄膜(負極活物質薄膜)においても、図4と同様に、界面層に相当する位置に酸素分布のピークが検出された。即ち、界面層にシリコンの酸化物が存在していた。参考例3より、シリコン薄膜の形成途中でシリコンの酸化物が形成されることによっても、シリコン粒子の成長が不連続になる可能性が高くなることが分かった。
参考例1〜3及び比較例1で形成したリチウムイオン二次電池に対し、試験温度20℃、充放電電流3mA/cm2、充放電電圧範囲4.2V〜2.5Vで充放電サイクル試験を行った。初回放電容量に対する、50サイクル後、及び200サイクル後の放電容量の割合を電池容量維持率(サイクル特性)として求めた。結果を表1に示す。
表1から分かるように、厚さ方向のほぼ中央の界面層でシリコン粒子の成長が不連続である参考例1〜3は、界面層が存在せず厚さ方向の全範囲にわたってシリコン粒子の成長が連続している比較例1に比べて、50サイクル後及び200サイクル後の電池容量維持率が大きい。また、第1スパッタ成膜源51による成膜と第2スパッタ成膜源52による成膜との間にガス導入される結果、シリコン粒子の成長の不連続性がより顕著に認められる参考例2,3は、ガス導入されない参考例1に比べて、良好な電池容量維持率を有している。
シリコン粒子の不連続性が電池容量維持率(サイクル特性)を向上させるのは以下のような理由によると考えられる。上述のように、真空成膜法により形成されたシリコン薄膜内において、シリコンは厚さ方向を中心軸方向とする略逆円錐台形状の柱状粒子を形成している。このシリコン薄膜をリチウムイオン2次電池の負極活物質として使用すると、充放電によりリチウムイオンの吸蔵/放出が繰り返され、シリコン粒子が膨張/収縮を繰り返す。これがシリコン薄膜と負極集電体層との界面での剥離、シリコン粒子の歪みや破壊を生じさせ、電池容量維持率(サイクル特性)を低下させる。電池容量を増大させようとしてシリコン薄膜の厚みを厚くするとサイクル特性が低下するのは、厚みの増大に応じてシリコン粒子が成長し、その最大径も増大するので、シリコン粒子間の相互干渉が増大するためであると考えられる。参考例1〜3のように、シリコン薄膜中にシリコン粒子の成長を不連続にする界面層を形成すると、シリコン粒子は界面層から新たに成長を開始する。従って、シリコン粒子の最大径の増大が抑制され、シリコン粒子間の相互干渉が抑制される。このために、良好な電池容量維持率(サイクル特性)が得られるのである。
このように、シリコンの柱状粒子形状を不連続にする界面層をシリコン薄膜中に形成することにより、シリコンを主成分とする2以上のシリコン薄膜を含む多層シリコン薄膜を得ることができる。このような多層シリコン薄膜をリチウムイオン2次電池の負極活物質層として使用した場合には、負極活物質層の全厚みを増加させてもサイクル特性が劣化しない。従って、電池容量の増大とサイクル特性とが両立したエネルギーデバイスを提供できる。
ガス導入ノズル55より導入するガスをイオン化やプラズマ化することで導入ガスと薄膜粒子との反応性が増し、ガス導入の効果を高めることが出来る。導入ガスのイオン化はガス導入ノズル55にイオン源やプラズマ源の機能を追加付与することで実現できる。イオン源としては、熱陰極型、ホローカソード陰極型など様々な方式のものを用いることができ、その方式に特に制限はない。酸素ガスの導入時には例えば熱陰極が酸化により劣化しないように不活性ガスとの混合ガスを用いるなどの工夫をすることが好ましい。プラズマ生成のための印加電圧は直流、交流、高周波のいずれであっても良い。導入ガスのイオン化やプラズマ化を行った場合にも断面SEM形状、オージェデプスプロファイル、電池容量維持率(サイクル特性)で参考例2,3と同様の効果が得られるが、200サイクル後の容量維持率は参考例2,3に比べて2%程度向上した。これは導入したガスをイオン化やプラズマ化することで導入ガスと薄膜粒子との反応性が増し、シリコン粒子の成長を不連続にするというガス導入の効果が高められたためと思われる。
[参考例4,5,比較例2]
参考実施形態1に対応する参考例を説明する。
まず、正極の作製方法を述べる。Li2CO3とCoCO3とを所定のモル比で混合し、大気中において900℃で加熱することによって合成してLiCoO2を得た。これを100メッシュ以下に分級して正極活物質を得た。この正極活物質100g、導電剤として炭素粉末12g、結着剤としてポリ4フッ化エチレンディスパージョン10g、及び純水を混ぜ合わせてペースト状にした。この正極活物質含有ペーストを、正極集電体としての厚さ25μmの帯状のアルミニウム箔の両面に塗布し、乾燥して正極を得た。
負極集電体として厚さ20μmの帯状の銅箔を用い、その両面に負極活物質としてシリコン薄膜を真空蒸着法により形成した。詳細は後述する。
セパレ−タとして、厚さが35μmで、正極集電体及び負極集電体よりも広幅の帯状の多孔性ポリエチレンを用いた。
正極集電体にこれと同材質の正極リードをスポット溶接にて取り付けた。また、負極集電体にこれと同材質の負極リードをスポット溶接にて取り付けた。
上記によって得た正極と負極との間にセパレータが介在するようにこれらを重ね合わせて渦巻き状に巻回した。この円筒状巻回物の上下面に、ポリプロピレン製の絶縁板をそれぞれ配して有底の円筒状電池缶内に収納し、電池缶の開口近傍に段部を形成した後、非水電解液として、LiPF6を濃度1×103モル/m3で溶解したエチレンカーボネートとジエチルカーボネートの等比体積混合溶液を電池缶に注入し、封口板で開口を密閉してリチウムイオン二次電池を得た。
負極活物質としてのシリコン薄膜の形成方法を図5を用いて説明する。
図5の装置では、薄膜形成室1cに、第1スパッタ成膜源51、ガス導入ノズル55、及び第2スパッタ成膜源52に代えて、第1蒸着源61、中間層蒸着源65、及び第2蒸着源62が配置されている点で図1の装置と異なる。図5において図1と同一の構成要素には同一の符号を付してそれらについての説明を省略する。
捲き出しロール11から捲き出された帯状の負極集電体5は、搬送ロール12a、キャンロール13、搬送ロール12bによって順に搬送され、捲き取りロール14に捲き取られる。この過程で、第1蒸着源61、中間層蒸着源65、及び第2蒸着源62から生成された原子、分子、又はクラスタなどの粒子(以下、「蒸発粒子」という)が隔壁1aのマスク4を通過して、キャンロール13上を走行している負極集電体5の表面上に付着して薄膜6を形成する。負極集電体5に対向して、その搬送方向の上流側から下流側に向かって、第1蒸着源61、中間層蒸着源65、及び第2蒸着源62が配置されている。中間層蒸着源65はキャンロール13の近傍にて、負極集電体5の幅方向(図5の紙面に垂直方向)の全幅にわたって伸びている。従って、中間層蒸着源65は第2のマスクとして機能して、キャンロール13の外表面のうち、中間層蒸着源65が対向する領域への第1蒸着源61及び第2蒸着源62からの蒸発粒子の堆積を防止する。従って、負極集電体5の表面には、最初に第1蒸着源61からの蒸発粒子が主として堆積し、次いで中間層蒸着源65からの蒸発粒子が主として堆積し、最後に第2蒸着源62からの蒸発粒子が主として堆積する。
参考例4,5では、このような装置を用いて、第1蒸着源61及び第2蒸着源62からそれぞれシリコンを電子ビーム蒸着して、負極集電体5としての銅箔上に、厚さ6μmのシリコン薄膜を形成した。シリコン薄膜の堆積速度を概ね0.1μm/sに設定した。第1蒸着源61及び第2蒸着源62として、270度偏向型電子ビーム蒸着源を使用した。
更に、参考例4では、中間層蒸着源65からアルミニウムを蒸着した。アルミニウムの堆積量は、アルミニウムのみを蒸着したときに厚み0.1μmの薄膜が形成されるのと同等とした。
また、参考例5では、中間層蒸着源65からリチウムを蒸着した。リチウムの堆積量は、リチウムのみを蒸着したときに厚み0.1μmの薄膜が形成されるのと同等とした。
比較例2では、第2蒸着源62及び中間層蒸着源65を取り除き、第1蒸着源61のみを用い、堆積速度が参考例4,5と同じになるように負極集電体5の走行速度を調節する以外は参考例4,5と同様にして厚さ6μmのシリコン薄膜を形成した。
図6(A)〜(C)は、順に、比較例2,参考例4,5のシリコン薄膜(負極活物質薄膜)の厚さ方向の断面を撮影したSEM写真を模式的に示した図である。各図において、紙面下側が負極集電体(銅箔)5側、上側はシリコン薄膜の表面である。
比較例2(図6(A))では、シリコン粒子が負極集電体との界面から徐々に粒子径が増大しながら略逆円錐台状の柱状粒子として成長し、負極活物質の表面近傍で粒子径が最も大きい。
これに対して、参考例4,5(順に図6(B),(C))では、シリコン粒子が同様に略逆円錐台状の柱状粒子として成長しているが、厚さ方向の中央部分に存在するわずかな厚みを有する層(中間層)60の両側でシリコン粒子の成長が不連続となっている。シリコン薄膜は、この中間層60によって分割され、上下方向に2層構造を有している。中間層60よりも下側(銅箔側)の層は主として第1蒸着源61により形成されたシリコン薄膜であり、中間層60は中間層蒸着源65によって形成されたアルミニウム薄膜(参考例4)又はリチウム薄膜(参考例5)であり、中間層60よりも上側の層は主として第2蒸着源62により形成されたシリコン薄膜であると考えられる。中間層60よりも上側の層のシリコン粒子は、中間層60から新たに略逆円錐台状に成長を開始するために、シリコン薄膜の表面近傍での粒子径は比較例2と比して小さい。なお、参考例4,5,比較例2のそれぞれのシリコン薄膜は、X線回折測定(CuKα、40kV)でいずれも顕著なシリコン結晶に相当するピークが検出されず、アモルファス又は微結晶であることが推定出来た。
図7は参考例4のシリコン薄膜(負極活物質薄膜)のオージェデプスプロファイルを示す図である。図7から分かるように、シリコン薄膜の成膜の途中にアルミニウムの成膜を行う参考例4では、中間層に相当する位置にアルミニウムの顕著なピークが検出されている。参考例4は、シリコン薄膜の形成途中でアルミニウム薄膜が形成されると、シリコン粒子の成長が不連続になる可能性が高くなることを示唆している。
図示を省略するが、シリコン薄膜(負極活物質薄膜)の成膜の途中にリチウムの成膜を行う参考例5においても、図7と同様に、中間層に相当する位置にリチウムのピークが検出された。参考例5より、シリコン薄膜の形成途中でリチウム薄膜が形成されることによっても、シリコン粒子の成長が不連続になる可能性が高くなることが分かった。
参考例4のオージェデプスプロファイルと参考例5のオージェデプスプロファイルとを比較すると、参考例5のリチウムの方が参考例4のアルミニウムよりもピーク分布がなだらかであった。これはリチウム原子がアルミニウム原子に比べて小形軽量であるために成膜中や成膜後において分散移動する傾向が強いためであると思われる。
参考例4,5及び比較例2で形成したリチウムイオン二次電池に対し、試験温度20℃、充放電電流7.5mA/cm2、充放電電圧範囲4.2V〜2.5Vで充放電サイクル試験を行った。初回放電容量に対する、50サイクル後、及び200サイクル後の容量の割合を電池容量維持率(サイクル特性)として求めた。結果を表2に示す。
表2から分かるように、厚さ方向のほぼ中央の中間層でシリコン粒子の成長が不連続である参考例4,5は、中間層が存在せず厚さ方向の全範囲にわたってシリコン粒子の成長が連続している比較例2に比べて、50サイクル後及び200サイクル後の電池容量維持率が大きい。
また、初回放電容量を比較すると、参考例4と比較例2とはほぼ同等であったが、参考例5は参考例4に比べて5%程度高容量であった。これは中間層を構成するリチウムの一部が、最初の充電後に何らかの理由によりシリコン薄膜内で自由な動きが出来なくなった一部のリチウムイオンを補完する役割を果たしたためではないかと推察される。
このように、シリコンの柱状粒子形状を不連続にする中間層をシリコン薄膜中に形成することにより、シリコンを主成分とする2以上のシリコン薄膜を含む多層シリコン薄膜を得ることができる。このような多層シリコン薄膜をリチウムイオン2次電池の負極活物質層として使用した場合には、負極活物質層の全厚みを増加させてもサイクル特性が劣化しない。従って、電池容量の増大とサイクル特性とが両立したエネルギーデバイスを提供できる。
(実施の形態1)
本発明の実施の形態1にかかるエネルギーデバイスを説明する。
本発明の実施の形態1にかかるエネルギーデバイスの概略構成の一例を図8に示す。本実施の形態のエネルギーデバイスは、基板22上に、電池要素20が積層されてなる。電池要素20は、正極集電体27、正極活物質26、電子伝導を呈さない層としての固体電解質25、負極活物質24、負極集電体23がこの順に形成されている。図8では、基板22は、電池要素20の正極集電体27側に配されているが、負極集電体23側に配されていてもよい。
基板22としては、例えばポリイミド(PI)、ポリアミド(PA)、ポリエチレンナフタレート(PEN)、ポリエチレンテレフタレート(PET)やその他の高分子フィルム、又はステンレス金属箔、又はニッケル、銅、アルミニウムやその他の金属元素を含む金属箔などの可撓性を有する材料を用いることが出来る。更に、各種形状のシリコン、ガラス、セラミック、プラスチックなどを用いることも出来、本発明では基板の材質や形状に特に限定はない。エネルギーデバイスに求められる特性に応じて適宜選択すればよい。
正極集電体27としては、例えばニッケル、銅、アルミニウム、白金、白金−パラジウム、金、銀、チタン、ITO(インジウム−スズ酸化物)で代表される金属を用いることが出来る。エネルギーデバイスの最終形態によっては、正極側に基板22を配置し、且つ基板22として導電性材料を用いる場合には、正極集電体27を省略し、基板22を正極集電体27としても機能させることができる。
正極活物質26としては、例えばコバルト酸リチウム、ニッケル酸リチウムなどを用いることが出来る。但し、本発明の正極活物質26の材料は上記に限定されず、その他の材料を用いることも出来る。
固体電解質25としては、イオン伝導性があり、電子伝導性が無視できるほど小さい材料を用いることが出来る。特にエネルギーデバイスがリチウムイオン2次電池である場合には、リチウムイオンが可動イオンであるため、Li3PO4や、Li3PO4に窒素を混ぜて(あるいはLi3PO4の元素の一部を窒素で置換して)得られる材料(LiPON:代表的な組成はLi2.9PO3.3N0.36)などからなる固体電解質はリチウムイオン伝導性に優れるので好ましい。同様に、Li2S−SiS2、Li2S−P2S5、Li2S−B2S3などの硫化物からなる固体電解質も有効である。更にこれらの固体電解質にLiIなどのハロゲン化リチウムや、Li3PO4等のリチウム酸素酸塩をドープした固体電解質も有効である。本発明の固体電解質25の材料は上記に限定されず、その他の材料を固体電解質25として用いることも出来る。電解質として固体電解質を用いることにより、従来の液型電解質で必須の液漏れ対策が不要となり、エネルギーデバイスの小型化、薄型化が容易になる。
負極活物質24としては、シリコンを主成分として含む2以上のシリコン薄膜を含む多層構造のシリコン薄膜を用いることが出来る。
負極集電体23としては、正極集電体27と同様に、例えばニッケル、銅、アルミニウム、白金、白金−パラジウム、金、銀、ITO(インジウム−スズ酸化物)で代表される金属を用いることが出来る。エネルギーデバイスの最終形態によっては、負極側に基板22を配置し、且つ基板22として導電性材料を用いる場合には、負極集電体23を省略し、基板22を負極集電体23としても機能させることができる。
エネルギーデバイスの製品形態としては特に制限されず、種々のものが考えられる。例えば、可撓性の長尺の基板22上に図8に示した電池要素20を積層したものを図9に示すようにして平板状に巻回しても良い。このとき、巻回体30の内周に平板状の内芯31を配置しても良い。
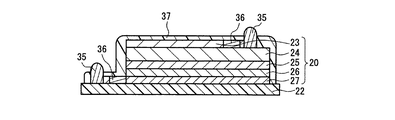
図10は図9に示した平板状エネルギーデバイスの斜視図である。図10において、32は巻回体30の両端に設けられる一対の外部電極である。外部電極32の材料としては、ニッケル、亜鉛、スズ、はんだ合金、導電性樹脂などの各種導電材料を用いることが出来る。その形成方法としては、溶射、メッキ、塗布などを用いることが出来る。一方の外部電極32には負極集電体23が電気的に接続され、他方の外部電極32には正極集電体27が電気的に接合される。このとき、一対の外部電極32が相互に絶縁されるように、負極集電体23及び正極集電体27の幅方向(巻回中心方向)の形成領域をパターニングしておく必要がある。
図11は、エネルギーデバイスの別の製品形態を示す断面図である。図11において、35は一対の外部電極であり、一方の外部電極35には負極集電体23が電気的に接続され、他方の外部電極35には正極集電体27が電気的に接合される。その材料及び形成方法は図10に示した外部電極32と同様である。
36は、負極集電体23の外部電極35との接続部近傍、及び正極集電体27の外部電極35との接続部近傍に設けられたヒューズ部である。ヒューズ部36は、過電流が流れたときに溶断して過電流を遮断して発火などに至るのを事前に防止する安全装置として機能する。本実施の形態では、ヒューズ部36は、負極集電体23及び正極集電体27に設けられているが、いずれか一方のみであっても良い。
図12(A)はヒューズ部36の一例を示した平面図、図12(B)は、図12(A)における12B−12B線での矢視断面図である。集電体23,27に、電流が流れる部分の幅が狭くなるようなパターンを付与してヒューズ部36を形成している。過電流が流れると、ヒューズ部36がジュール熱により発熱し、溶断して、過電流を遮断する。従って、発火などの重大な事態に至るのを未然に防止することができる。なお、ヒューズ部36の構成は図12(A)及び図12(B)に限定されない。例えば、負極集電体23及び正極集電体27の厚みを部分的に薄くすることによりヒューズ部36を構成しても良い。あるいは、負極集電体23及び正極集電体27内に、電気抵抗の温度係数の大きな異種材料を特定パターンで形成することによりヒューズ部を形成しても良い。異種材料の電気抵抗の温度係数が、負極集電体23及び正極集電体27の電気抵抗の温度係数よりも大きいので、過電流時にヒューズ部36がわずかに温度上昇すると異種材料の抵抗値が急激に増大する。よって、ヒューズ部36内の異種材料以外の負極集電体23及び正極集電体27の材料内に電流が集中して流れる結果、ヒューズ部36がジュール熱により発熱し、溶断して、過電流を遮断する。
図11において、37は、機械的保護、耐湿性向上、層間剥離の防止などを目的として設けられる保護層である。保護層37の材料は特に限定されないが、例えばシランカップリング剤等の表面処理剤、光あるいは熱硬化性樹脂、金属、金属酸化物、金属窒化物などを用いることができる。形成方法としては、塗布、ディップ(浸漬)、スプレーなどの湿式プロセスや、蒸着、スパッタなどのドライプロセスを採ることができる。また、保護層37は異種又は同種の材料からなる多層の複合膜であっても良い。保護層37は、外部電極35を除くエネルギーデバイスの外表面に形成することができる。基板22の材料によっては、図11に示すように基板22の表面には保護層37を形成しなくてもよい。
さらに、電池要素20を必要な数だけ繰り返して積層してエネルギーデバイスを構成してもよい。
[実施例1,参考例6,比較例3]
実施の形態1に対応する実施例を説明する。
基板22として厚さ25μmのポリイミドフィルムを用い、この上に、正極集電体27として厚さ0.5μmのニッケル、正極活物質26として厚さ8μmのコバルト酸リチウムをそれぞれ真空蒸着法により積層し、更に、厚さ2μmのリン酸リチウム系の固体電解質25を真空蒸着法により積層した。
その後、固体電解質25の表面に負極活物質24として5μmのシリコン薄膜を形成した。詳細は後述する。
次いで、負極活物質24の表面に、負極集電体23として厚さ0.5μmのニッケルを形成して、図8に示す積層構造を有する帯状の積層物を得た。
これを平板状に巻回し、正極集電体27及び負極集電体23にそれぞれ電気的に接続されるように一対の外部電極33を形成して、図10に示すような平板状のリチウムイオン二次電池を得た。
負極活物質24としてのシリコン薄膜の形成方法を図13を用いて説明する。
図13の装置では、薄膜形成室1cに、第1スパッタ成膜源51、ガス導入ノズル55、及び第2スパッタ成膜源52に代えて、蒸着源71及び中間層蒸着源75が配置されている点で図1の装置と異なる。図13において図1と同一の構成要素には同一の符号を付してそれらについての説明を省略する。
正極集電体27、正極活物質26、固体電解質25が形成された帯状の上記基板22は、捲き出しロール11から捲き出され、搬送ロール12a、キャンロール13、搬送ロール12bによって順に搬送され、捲き取りロール14に捲き取られる。この過程で、蒸着源71及び中間層蒸着源75から生成された原子、分子、又はクラスタなどの粒子(以下、「蒸発粒子」という)が隔壁1aのマスク4を通過して、キャンロール13上を走行している基板22の固体電解質25の表面上に付着して薄膜6を形成する。基板22に対向して、蒸着源71及び中間層蒸着源75が配置されている。蒸着源71から放出されマスク4を通過する蒸発粒子流中の、基板22の走行方向のほぼ中央の位置に、中間層蒸着源75が配置されている。中間層蒸着源75はキャンロール13の近傍にて、基板22の幅方向(図13の紙面に垂直方向)の全幅にわたって伸びている。従って、中間層蒸着源75は第2のマスクとして機能して、キャンロール13の外表面のうち、中間層蒸着源75が対向する領域への蒸着源71からの蒸発粒子の堆積を防止する。従って、基板22の固体電解質25の表面には、最初に蒸着源71からの蒸発粒子が主として堆積し、次いで中間層蒸着源75からの蒸発粒子が主として堆積し、再度蒸着源71からの蒸発粒子が主として堆積する。
実施例1、参考例6では、このような装置を用いて、蒸着源71からシリコンを、中間層蒸着源75から銅―アルミニウム系材料(融点Tm=548℃)を、それぞれ真空蒸着法により蒸発させて、合計厚さ5μmの多層構造の負極活物質24を形成した。
更に、参考例6では、負極活物質24を形成時のキャンロール13の外周面温度を、図示しない蓄熱媒体をキャンロール13の内部で循環させることにより、20℃に維持した。
また、実施例1では、負極活物質24を形成時のキャンロール13の外周面温度を280℃に維持した以外は参考例6と同様にした。
比較例3では、中間層蒸着源75を取り除き、蒸着源71のみを用い、負極活物質24を形成時のキャンロール13の外周面温度を20℃に維持して、厚さ5μmの単層構造の負極活物質24を形成した以外は参考例6と同様にした。
図14(A),(B)は、順に、参考例6,実施例1の負極活物質(シリコン薄膜)24の厚さ方向の断面を撮影したSEM写真を模式的に示した図である。各図において、紙面下側が固体電解質25側、上側は負極活物質24の表面である。
参考例6,実施例1(順に図14(A),(B))では、シリコン粒子が固体電解質25との界面から徐々に粒子径が増大しながら略逆円錐台状の柱状粒子として成長しているが、厚さ方向の中央部分に存在するわずかな厚みを有する層(中間層)70の両側でシリコン粒子の成長が不連続となっている。負極活物質24は、この中間層70によって分割され、上下方向に2層構造を有している。中間層70は中間層蒸着源75によって形成された銅―アルミニウム系薄膜である。中間層70よりも上側の層のシリコン粒子は、中間層70から新たに略逆円錐台状に成長を開始するために、負極活物質(シリコン薄膜)24の表面近傍での粒子径の拡大が抑えられている。
更に、キャンロール13の外周面温度を20℃にして形成された負極活物質24(参考例6、図14(A))では中間層70が連続膜であるが、キャンロール13の外周面温度を280℃にして形成された負極活物質24(実施例1、図14(B))では中間層70が連続膜でなく網目状もしくは島状に分断されていることが分かった。これは銅―アルミニウム系材料の融点がシリコンの融点よりも低いために、高温下で薄膜を形成すると網目状もしくは島状に分断されやすいためであると思われる。参考例6と実施例1との比較で認められる中間層70の分断は、中間層70が銅―アルミニウム系材料である場合に限られず、他の低融点の材料を用いた場合にも同様に確認された。
また、参考例6と同じ条件で負極活物質24を形成した後に、280℃に加熱し、冷却して厚さ方向の断面のSEM写真を撮影したところ、実施例1(図14(B))の中間層70と同様に網目状もしくは島状に分断されていた。即ち、低融点材料からなる連続する中間膜を形成した後、加熱することによっても網目状もしくは島状に分断された中間層が得られることを確認した。
なお、図示を省略するが、中間層蒸着源75を使用しない比較例3では、比較例2(図6(A))と同様に、シリコン粒子が固体電解質25との界面から徐々に粒子径が増大しながら略逆円錐台状の柱状粒子として成長し、負極活物質24の表面近傍で粒子径が最も大きかった。
参考例6,実施例1及び比較例3で得たリチウムイオン二次電池に対し、試験温度20℃、充放電電流3mA/cm2、充放電電圧範囲4.2V〜2.5Vで充放電サイクル試験を行った。初回放電容量に対する、50サイクル後、及び200サイクル後の容量の割合を電池容量維持率(サイクル特性)として求めた。結果を表3に示す。
表3から分かるように、厚さ方向のほぼ中央の中間層でシリコン粒子の成長が不連続である参考例6,実施例1は、中間層が存在せず厚さ方向の全範囲にわたってシリコン粒子の成長が連続している比較例3に比べて、50サイクル後及び200サイクル後の電池容量維持率が大きい。
表3では、中間層が連続膜である参考例6と分断膜である実施例1とでは、サイクル特性に顕著な相違が見られない。
充放電レートを5Cと高速にして充放電を行った場合の50サイクル後の容量維持率は、参考例6が58%であったのに対して実施例1は65%と実施例1で優れた結果が得られた。これは、中間層が網目状もしくは島状に分断されることにより、負極活物質24内でのイオンの移動が容易になるためではないかと思われる。
なお、本実施例では、1つの蒸着源71を用いて2層構造の負極活物質24を形成したが、図5に示すように2つの蒸着源を用いて2層構造の負極活物質24を形成しても良い。
上記の参考実施形態1,実施の形態1では、負極活物質薄膜(シリコン薄膜)は、集電体層の表面に直接設けられていたが、本発明はこれに限定されず、集電体層の表面に設けられた下地層を介して設けられていてもよい。下地層としては、例えば、集電体層と負極活物質薄膜(シリコン薄膜)との間の接着力強化や防錆処理などを目的とする層であってもよく、具体的には、例えばシリコン−銅薄膜やクロメート処理層などを用いることが出来る。あるいは、後述する表面処理により形成された層であってもよい。
また、上記の参考実施形態1,実施の形態1では、負極活物質薄膜(シリコン薄膜)は、スパッタ法又は蒸着法により形成する例を示したが、本発明はこれに限定されず、CVD法をはじめとする他の真空成膜法を用いてもよく、その場合であっても同様の効果が得られる。
また、上記の実施例で示した負極活物質薄膜は、2層のシリコン薄膜からなる2層構造であったが、本発明はこれに限定されず、3層以上の多層構造であってもよい。多層構造にすることによって本発明の効果であるサイクル特性の向上が得られる。シリコン薄膜の1層あたりの厚さは12μm以下、更には6μm以下、特に2μm以下であることが好ましい。シリコン薄膜の1層あたりの厚さが12μmを超えると、そのシリコン薄膜内で柱状粒子の粒子径が大きくなり、サイクル特性の低下が顕著となる。従って、1層あたりの厚さが12μmを超える場合には、負極活物質薄膜を多層化することで、多層に分割しない場合に対してはサイクル特性が向上するものの、長期の実用に供するには課題が残る。負極活物質薄膜の層数は形成しようとする負極活物質薄膜の厚みに応じて設定することができる。
上記の実施例において得られた典型的なシリコン粒子の直径は、リチウム吸蔵による膨張前の状態で2〜15μm程度であり、通常、この各シリコン粒子は1〜6μmの複数のシリコン微粒子が合体してなる。但し、本発明におけるシリコン粒子径はこれに限定されない。
また、参考例1〜5において負極集電体として用いた銅箔には表面処理が施されていてもよい。銅箔に施すことができる表面処理としては、亜鉛メッキ、スズ、銅、ニッケル、若しくはコバルトと亜鉛との合金メッキ、ベンゾトリアゾールなどのアゾール誘導体を用いた被覆層の形成、クロム酸若しくは二クロム酸塩を含む溶液などによるクロム含有被膜の形成、またはこれらの組み合わせを用いることが出来る。あるいは、銅箔に代えて、他の基材の表面に銅被覆を施したものを用いることも出来る。この銅被覆の表面に上記の表面処理を施してもよい。
上記の実施の形態及び実施例の説明では言及しなかったが、シリコン薄膜の成膜を不活性ガス又は窒素雰囲気で行うことが望ましい。雰囲気ガスは、被成膜面(上記の実施例ではマスク4の開口)に向けて導入してもよく、あるいは、真空槽(上記の実施例では薄膜形成室1c)内全体に行き渡るように導入してもよいが、被成膜面に向けて導入する方が効率的で好ましい。
シリコン薄膜をこのような雰囲気ガス中で成膜することにより、被成膜面と平行な方向に隣り合うシリコン柱状粒子が合併して成長して、シリコン粒子径が粗大化するのを防止できる。その結果、充放電時にシリコン粒子の膨張/収縮の程度が激しくなり、サイクル特性が低下するのを抑制することができる。本発明者らの実験によれば、詳細な実験結果を示すグラフを省略するが、上記のガス雰囲気でシリコン薄膜を成膜することにより、エネルギーデバイスの電池容量維持率を80%にまで低下させる充放電サイクル数が例えば15〜50%増加した。
ガスの好ましい導入量はシリコン薄膜の成膜条件、特に薄膜堆積速度R(nm/s)に応じて設定される。例えば、被成膜面に向けてガスを導入する場合には、成膜幅100mmあたりのガス導入量Q(m3/s)は、1×10-10×R〜1×10-6×R、特に1×10-9×R〜1×10-7×Rであることが好ましい。ガス導入量が少なすぎると上記の効果が得られない。逆にガス導入量が多すぎるとシリコン薄膜の堆積速度が低下する。
使用するガスとしては、実用性及び上記の効果の顕著性の観点から、アルゴンが最も好ましい。
また、シリコン薄膜に含まれるシリコンの一部が酸化物であることが好ましい場合がある。シリコン薄膜中のシリコンの含有量が多く、電池容量が大きい場合には、充放電時のシリコン薄膜の膨張/収縮の程度が大きくなり、サイクル特性が低下する場合がある。シリコン薄膜がシリコンの酸化物を含むと、シリコンの酸化物は充放電時の膨張/収縮が少ないから、充放電時のシリコン薄膜の膨張/収縮を抑えることができ、サイクル特性を向上させることができる。例えば、シリコン薄膜に含まれるシリコンの20〜50%が酸化物になるように成膜することが好ましい。本発明者らの実験によれば、詳細な実験結果を示すグラフを省略するが、シリコン薄膜がシリコンの酸化物を含むことにより、シリコン薄膜にもよるが、エネルギーデバイスの電池容量維持率を80%にまで低下させる充放電サイクル数が例えば10〜140%増加した。
シリコンの一部を酸化物にするには、例えば真空雰囲気下でシリコン薄膜の成膜中に、被成膜面の近傍に酸素系のガスを導入し、シリコン原子と反応させることによって可能である。反応性を高めるために、オゾンを用いたり、プラズマや基板電位などによってエネルギー付与を行うことは有効である。
ガスの好ましい導入量はシリコン薄膜の成膜条件、特に薄膜堆積速度R(nm/s)に応じて設定される。例えば、被成膜面に向けてガスを導入する場合には、成膜幅100mmあたりのガス導入量P(m3/s)は、1×10-11×R〜1×10-5×R、特に1×10-10×R〜1×10-6×R、更には1×10-9×R〜1×10-7×Rであることが好ましい。但し、設備形態等によりガス導入量Pはこの限りではない。ガス導入量が少なすぎると上記の効果が得られない。逆にガス導入量が多すぎるとシリコン薄膜全体が酸化物となってしまい電池容量が低下する。