JP4471663B2 - ペプチドデホルミラーゼ阻害剤 - Google Patents
ペプチドデホルミラーゼ阻害剤 Download PDFInfo
- Publication number
- JP4471663B2 JP4471663B2 JP2003575966A JP2003575966A JP4471663B2 JP 4471663 B2 JP4471663 B2 JP 4471663B2 JP 2003575966 A JP2003575966 A JP 2003575966A JP 2003575966 A JP2003575966 A JP 2003575966A JP 4471663 B2 JP4471663 B2 JP 4471663B2
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- ylmethyl
- group
- butyl
- dioxo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/72—Two oxygen atoms, e.g. hydantoin
- C07D233/76—Two oxygen atoms, e.g. hydantoin with substituted hydrocarbon radicals attached to the third ring carbon atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/72—Two oxygen atoms, e.g. hydantoin
- C07D233/76—Two oxygen atoms, e.g. hydantoin with substituted hydrocarbon radicals attached to the third ring carbon atom
- C07D233/78—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Communicable Diseases (AREA)
- Animal Behavior & Ethology (AREA)
- Oncology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Description
本発明は、新規な抗菌性N−ホルミル−N−ヒドロキシルアミン化合物、および該化合物を含有する医薬組成物のペプチドデホルミラーゼ阻害剤としての使用に関する。
細菌性イニシエーター、メチオニルtRNAは、メチオニルtRNAホルミルトランスフェラーゼ(FMT)によって修飾されてホルミル−メチオニルtRNAを生成する。次いで、ホルミルメチオニン(f−Met)が、新たに合成されるポリペプチドのN末端に組み込まれる。次いで、ポリペプチドデホルミラーゼ(PDFまたはDef)が第一翻訳産物を脱ホルミル化してN−メチオニルポリペプチドを生じる。ほとんどの細胞内タンパク質はさらに、メチオニンアミノペプチダーゼ(MAP)によって処理されて成熟ペプチドおよび遊離メチオニンを生産し、それが再循環する。PDFおよびMAPはどちらも、細菌増殖に必須であり、PDFはMAP活性に必要とされる。この一連の反応をメチオニンサイクルという(図1)。
PDFはイン・ビトロでの細菌増殖に必要であることが明らかにされ(Mazel, D. et al, EMBO J. 13 (4), 914-923, 1994)、真核生物タンパク質合成に関与しないと考えられ(Rajagopalan et al, J. Am. Chem. Soc. 119, 12418-12419, 1997)、また、原核生物において普遍的に保存されている(Kozak, M. Microbiol. Rev. 47, 1-45, 1983)ので、該酵素は魅力的な抗菌標的であると認められる。したがって、PDF阻害剤は潜在的に幅広いスペクトルの抗菌剤として働くことができる。
本発明は、下記の式(I)で示される新規な抗菌化合物およびPDF阻害剤としてのその使用を含む。
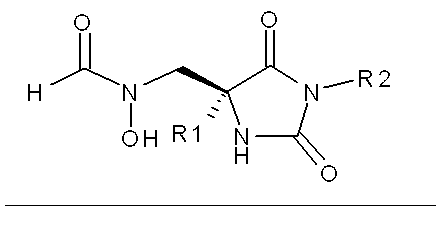
本発明の方法において有用な化合物は下記式(I):
R1は、C1−C9アルキル、C1−2アルキルArおよびArからなる群から選択され;
R2は、水素、C1−C9アルキル、C1−4アルキルAr’、NR4、NC(O)R4、C2−4アルキルNR3R4、C1−3アルキルC(O)NR3R4、C1−3アルキルC(O)Ar’、C2−3アルキルNHC(O)NR3R4、C2−3アルキルNHC(O)Ar’およびC1−2アルキルSO2R4からなる群から選択され;
R3は、C1−C9アルキル、C1−2アルキルArおよびArからなる群から選択され;
R4はR3、Ar’であるか、またはR4はR3およびそれらが結合している窒素原子と一緒になって、C1−3アルキル、アリール、C1−3アルコキシ(1ないし3個のFで置換されていてもよい)、アリールオキシ、カルボキシ、オキソ、ヒドロキシ、アミノ、ニトロおよびシアノからなる群から選択される1、2または3個の置換基で置換されていてもよく、またはアリール、ヘテロアリールもしくは第2の複素環に縮合していてもよい複素環を形成していてもよく;
Arは、フェニル、フリルおよびチエニルからなる群から選択され、その各々がC1−C3アルキル、CN、F、Cl、BrおよびIからなる群から選択される1、2または3個の置換基で置換されていてもよく;
Ar’は、フェニル、ナフチル、フリル、ピリジル、チエニル、チアゾリル、イソチアゾリル、ピラゾリル、トリアゾリル、テトラゾリル、イミダゾリル、イミダゾリジニル、ベンゾフラニル、インドリル、チアゾリジニル、イソキサゾリル、オキサジアゾリル、チアジアゾリル、ピロリルおよびピリミジルからなる群から選択され、その各々がC1−C6アルキル、C1−C6アルコキシ、(CH2)0−5CO2R1、C(O)N(R1)2、CN、(CH2)0−5OH、NO2、F、Cl、Br、I、CF3、N(R1)2およびNHC(O)R1からなる群から選択される1、2または3個の置換基で置換されていてもよく;
Aは、C(O)NHOHまたはN(CHO)OHの群から選択され;
YはC(O)であるとき、XはNHであり、またはYがC(O)またはCH2であるとき、XはCH2である]
で示される化合物から選択される。
本明細書で使用される場合、「アリール」なる語は、2個までの共役または縮合した環系を含有する少なくとも1つの共役型パイ電子系を有する環を有する置換されていてもよい芳香族基を示す。「アリール」は、炭素環式アリール、複素環式アリールおよびビアリール基を包含し、その各々は置換されていてもよい。
N−[(S)−1−ベンジル−4−ペンチル−2,5−ジオキソイミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−[(S)−1,4−ジベンジル−2,5−ジオキソイミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−[(S)−1−ベンジル−4−ブチル−2,5−ジオキソイミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−[(S)−2,5−ジオキソ−4−ペンチル−1−フェニルイミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−[(S)−4−ブチル−1−(3,4−ジクロロベンジル)−2,5−ジオキソイミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−[(S)−4−ブチル−2,5−ジオキソ−1−(2−オキソ−2−フェニルエチル)−イミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−[(S)−1−ビフェニル4−イルメチル−4−ブチル−2,5−ジオキソイミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−[(S)−1−ベンジル−4−シクロヘキシルメチル−2,5−ジオキソイミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−{(S)−4−ブチル−1−[2−(5−クロロ−3−メチル1−ベンゾ[b]チオフェン−2−イル)−2−オキソエチル]−2,5−ジオキソイミダゾリジン−4−イルメチル}−N−ヒドロキシホルムアミド;
2−{(S)−4−ブチル−4−[(ホルミルヒドロキシアミノ)メチル]−2,5−ジオキソイミダゾリジン−1−イル}−N−(3,5−ジクロロフェニル)アセトアミド;
2−{(S)−4−ブチル−4−[(ホルミルヒドロキシアミノ)メチル]−2,5−ジオキソイミダゾリジン−1−イルメチル}安息香酸メチルエステル;
N−[(S)−4−ブチル−1−(2−モルホリン−4−イル−エチル)−2,5−ジオキソイミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−[(S)−1−(2−ベンゾフラン−2−イル−2−オキソエチル)−4−ブチル−2,5−ジオキソイミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;および
2−{(S)−4−ブチル−4−[(ホルミルヒドロキシアミノ)−メチル]−2,5−ジオキソイミダゾリジン−1−イルメチル}安息香酸
からなる群から選択される。
1a. (2R,4S)−4−ブチル−2−tert−ブチル−3−ホルミルオキサゾリジン−4−カルボン酸メチルエステル
(2R,4S)−2−tert−ブチル−3−ホルミルオキサゾリジン−4−カルボン酸メチルエステル(4.6g,21.4mmol)[D. Seebach, J. D. Aebi, M. Gander-Coquoz and R. Naef, Helv. Chim. Acta., 70, 1194 (1987)参照]の乾燥THF(120mL)中溶液に、N2下で、1−ヨードブタン(12.2mL,106.8mmol)およびHMPA(12mL)を加えた。該混合物を−78℃に冷却し、ナトリウムビス(トリメチルシリル)アミドのTHF(1M,32mL,32mmol)中溶液を15分かけて滴下した。2時間後、反応混合物を0℃に加温し、飽和水性NH4Cl(200mL)でクエンチした。該クエンチした反応混合物をエーテル(400mL)で希釈し、水(3x200mL)およびブライン(200mL)で洗浄し、次いで乾燥させ(Na2SO4)、ろ過した。ろ液の濃縮および残渣のフラッシュクロマトグラフィー(20%酢酸エチル/ヘキサン)により、標題化合物を薄茶色固体として得た(4.0g,69%)。
MS(ES) m/e272[M+H]+
実施例1aの化合物(4.0g,14.7mmol)の40mL水性濃HCl/ジオキサン(1:1)中溶液を2時間熱還流した。室温に冷却後、反応混合物を真空下で濃縮し、水(15mL)中に再溶解した。該溶液に、水酸化カリウム(1g,17.8mmol)およびシアン酸カリウム(2.39g,29.4mmol)を加え、混合物を115℃に1時間加熱した。反応物を室温に冷却し、水性濃HCl(5mL)でゆっくりと処理し、次いで2時間還流した。溶媒を真空下で除去し、得られた固体をCH2Cl2/H2O(2:1)(3x20mL)で抽出した。合わせた有機層を濃縮して白色固体を得(4.8g)、それをGilson自動化HPLCによって精製して標題化合物を白色固体として得た(0.85g,31%)。MS(ES) m/e187[M+H]+
実施例1bの化合物(0.13g,0.70mmol)のDMF(3mL)中溶液に、K2CO3(0.1g,0.74mmol)および臭化ベンジル(0.087mL,0.74mmol)を加えた。反応混合物を室温で一晩攪拌した。固体をろ過し、有機溶液をGilson自動化HPLCによって精製して標題化合物を白色固体として得た(0.14g,73%)。MS(ES) m/e277[M+H]+
実施例1cの化合物(0.14g,0.51mmol)の10mLアセトニトリル/ジクロロメタン(1:1)中攪拌溶液に、0℃にて、デス−マーチン ペルヨージナン(0.32g,0.77mmol)を加えた。反応混合物を0℃で1時間攪拌し、次いで、室温に加温した。室温で一晩攪拌後、有機溶媒を真空下で除去した。白色固体をピリジン(10mL)中に溶解し、O−ベンジルヒドロキシルアミン塩酸塩(0.123g,0.77mmol)で処理した。室温で1時間後、反応混合物を濃縮乾固した。残渣をジクロロメタン(25mL)中に溶解し、飽和水性NaHCO3(20mL)、水(20mL)で洗浄し、乾燥させ(Na2SO4)、濃縮した。固体をGilson自動化HPLCによって精製して標題化合物を茶色がかった固体として得た(0.09g,47%)。
MS(ES) m/e380[M+H]+
攪拌しながら、シアノ水素化ホウ素ナトリウム(45mg,0.72mmol)を、実施例1dの化合物(0.09g,0.24mmol)の酢酸(5mL)中溶液にゆっくりと加えた。室温で2時間攪拌後、反応混合物を真空下で濃縮した。残渣を飽和水性NaHCO3(15mL)中に溶解し、ジクロロメタン(3x10mL)で抽出した。合わせた有機抽出物を乾燥させ(Na2SO4)、ろ過し、濃縮して、粗(S)−3−ベンジル−5−(ベンジルオキシアミノ−メチル)−5−ブチル−イミダゾリジン−2,4−ジオンを得た。MS(ES) m/e382[M+H]+
上記の粗中間体をジクロロメタン(5mL)中に溶解した。該溶液に、トリエチルアミン(0.035mL)を加え、次いで、新たに調製した混合無水物(0.019mLギ酸および0.045mL無水酢酸を50℃で1時間加熱し、室温に冷却することによって調製した)を加えた。該反応混合物を室温で1時間攪拌し、次いで、濃縮乾固した。残渣をGilson自動化HPLCによって精製して標題化合物を白色固体として得た(0.06g,62%)。MS(ES) m/e410[M+H]+
実施例1eの化合物(0.06g,0.15mmol)をメタノール(5mL)中に溶解し、活性炭素上のパラジウム(0.02g)の存在下、バルーン水素圧下で4時間攪拌した。反応混合物をろ過および濃縮し、残渣をGilson自動化HPLCによって精製して標題化合物を白色固体として得た(0.04g,84%)。
MS(ES) m/e320[M+H]+
N−[(S)−1−ベンジル−4−シクロヘキシルメチル−2,5−ジオキソ−イミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド,MS(ES)m/e360[M+H]+
2−{(S)−4−ブチル−4−[(ホルミルヒドロキシアミノ)メチル]−2,5−ジオキ−ソイミダゾリジン−1−イル}−N−(3,5−ジクロロフェニル)アセトアミド,MS(ES)m/e431[M+H]+
2−{(S)−4−ブチル−4−[(ホルミルヒドロキシアミノ)メチル]−2,5−ジオキソ−イミダゾリジン−1−イルメチル}安息香酸メチルエステル,MS(ES)m/e378[M+H]+
N−[(S)−4−ブチル−1−(2−モルホリン−4−イル−エチル)−2,5−ジオキソ−イミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド,MS(ES)m/e343[M+H]+
2−{(S)−4−ブチル−4−[(ホルミルヒドロキシアミノ)メチル]−2,5−ジオキソ−イミダゾリジン−1−イルメチル}安息香酸,MS(ES)m/e364[M+H]+
2a. (S)−5−ペンチル−5−ヒドロキシメチル−3−フェニル−イミダゾリジン−2,4−ジオン
ヨードブタンに代えてヨウ化ペンチルを用いることを除き、実施例1aの手法にしたがい、次いで、シアン酸カリウムの代わりにイソシアン酸フェニルを用いることを除き、実施例1bの手法にしたがって、標題化合物を白色固体として調製した(75%)。
MS(ES)m/e277[M+H]+
実施例1cの化合物の代わりに実施例2aの化合物を用いることを除き、実施例1d−fの手法にしたがって、標題化合物を白色固体として調製した。
MS(ES)m/e320[M+H]+
上記の中間体に代えて適当な中間体を用いること以外は、同様の方法により、下記の化合物を調製した。
N−[(S)−1−ベンジル−4−ペンチル−2,5−ジオキソ−イミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド,MS(ES)m/e334[M+H]+
N−[(S)−1,4−ジベンジル−2,5−ジオキソ−イミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド,MS(ES)m/e354[M+H]+
3a. (S)−5−ブチル−5−ヒドロキシメチル−3−(2−オキソ−2−フェニル−エチル)−イミダゾリジン−2,4−ジオン
臭化ベンジルの代わりに2−ブロモ−1−フェニルエタノンを用いることを除き、実施例1cの手法にしたがって、標題化合物を白色固体として調製した(93%)。
MS(ES)m/e305[M+H]+
実施例3aの化合物(0.15g,0.49mmol)およびtert−ブチルN−(tert−ブトキシカルボキシカルボニルオキシ)カルバメート(0.18g,0.74mmol)をN2下0℃にて、THF(3mL)中に溶解した。該溶液にN2下0℃にて、予め混合したトリブチルホスフィン(0.19mL,0.74mmol)およびジ−t−ブチルアゾジカルボキシレート(0.176g,0.74mmol)のTHF(2mL)中溶液を加えた。反応混合物を0℃で1時間維持し、室温に加温し、一晩攪拌した。反応物を飽和水性NaHCO3(15mL)でクエンチし、ジクロロメタン(3x10mL)で抽出した。合わせた有機層を乾燥させ(Na2SO4)、ろ過し、濃縮した。残渣をGilson自動化HPLCによって精製して標題化合物を白色固体として得た(0.11g,42%)。MS(ES)m/e520[M+H]+
実施例3bの化合物(0.11g,0.21mmol)を15%TFA/1,2−ジクロロエタン(5mL)中に溶解した。室温で1時間攪拌後、反応混合物を真空下で濃縮した。残渣をジクロロメタン(5mL)中に溶解し、トリエチルアミン(0.3mL)、次いで、新たに調製した混合無水物(0.031mLギ酸および0.069mL無水酢酸、50℃、1時間)で処理した。室温で1時間攪拌後、有機溶媒を真空下で除去し、メタノール(10mL)を加え、次いで、飽和水性Na2CO3(3mL)を勢いよく攪拌しながら加えた。3時間後、反応混合物を濃縮乾固し、固体を30%メタノール/ジクロロメタン(3x5mL)で抽出した。合わせた抽出物をろ過および濃縮し、得られた残渣をGilson自動化HPLCによって精製して標題化合物を白色固体として得た(0.03g,41%)。MS(ES)m/e348[M+H]+
N−[(S)−4−ブチル−1−(3,4−ジクロロベンジル)−2,5−ジオキソ−イミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド,MS(ES)m/e388[M+H]+
N−[(S)−1−ビフェニル−4−イルメチル−4−ブチル−2,5−ジオキソ−イミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド,MS(ES)m/e396[M+H]+
N−{(S)−4−ブチル−1−[2−(5−クロロ−3−メチル−1−ベンゾ[b]チオフェン−2−イル)−2−オキソ−エチル]−2,5−ジオキソ−イミダゾリジン−4−イルメチル}−N−ヒドロキシホルムアミド,MS(ES)m/e452[M+H]+
N−[(S)−1−(2−ベンゾフラン−2−イル−2−オキソ−エチル)−4−ブチル−2,5−ジオキソイミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド,MS(ES)m/e388[M+H]+
本発明の化合物は、細菌感染の治療に有用である。式(I)の化合物またはその医薬上許容される塩を使用するために、通常、それは標準的な製薬基準にしたがって医薬組成物として処方される。
式(I)の化合物およびその医薬上許容される塩は、抗生物質のための標準的な方法において、例えば、経口的に、非経口的に、舌下から、皮膚から、経皮的に、直腸から、吸入によって、またはバッカル投与によって投与されうる。
吸入用の典型的な組成物は、ジクロロジフルオロメタンまたはトリクロロフルオロメタンなどの通常のプロペラントを用いて、乾燥粉末としてまたはエーロゾルの形態において投与されうる溶液、懸濁液またはエマルジョンの形態である。
典型的な座剤処方は、この方法で投与された場合に活性な式(I)の化合物またはその医薬上許容される塩と結合剤および/または滑沢剤、例えば、重合グリコール、ゼラチン、ココア脂または他の低融点植物性ワックスもしくは油脂またはそれらの合成類似物を含んでなる。
好ましくは、該組成物は、患者が単回投与量を服用すればよいように、単位投与形態、例えば、錠剤、カプセルまたは計量されるエーロゾル投与量の形態である。
経口投与用の各投与単位は、式(I)の化合物または遊離の酸として計算されるその医薬上許容される塩として、適当には0.1mg〜500mg/kg、好ましくは1mg〜100mg/kg、非経口投与用の各投与単位は、式(I)の化合物または遊離の酸として計算されるその医薬上許容される塩として、適当には0.1mg〜100mg/kgを含有する。鼻腔内投与用の各投与単位は、一人につき、適当には、1〜400mg、好ましくは、10〜200mgを含有する。局所処方は、適当には0.01〜5.0%の式(I)の化合物を含有する。
鼻腔内投与および経口吸入についての一日の投与方針は、適当には、一人につき、約10〜約500mgである。活性成分は、所望の活性を示すのに十分なように、1日に1〜6回投与されてもよい。
本発明の化合物を本発明にしたがって投与する場合、許容されない毒物学的効果は予想されない。
生物学的アッセイ:
S.aureusまたはE.coli PDF活性を25℃にて、Lazennec & Meinnel, (1997) "Formate dehydrogenase-coupled spectrophotometric assay of peptide deformylase" Anal. Biochem. 244, pp.180-182によって開発された連続的酵素結合アッセイに少しの変更を加えたものを用いて測定する。反応混合物は、50μLの50mMリン酸カリウムバッファー(pH7.6)、15mM NAD、0.25Uホルメートデヒドロゲナーゼに含有される。基質ペプチド、f−Met−Ala−Serは、KM濃度で含まれる。反応は、10nM Def1酵素の添加によって引き起こされ、吸光度を340nmで20分間モニターする。
全細胞抗菌活性は、the National Committee for Clinical Laboratory Standards(NCCLS)が推奨する手法、ドキュメントM7−A4、"Methods for Dilution Susceptibility Tests for Bacteria that Grow Aerobically"(出典明示により本明細書の一部とされる)を用いて、ブイヨン微量希釈によって決定した。化合物は、0.06〜64mcg/mlの範囲の連続2倍希釈において試験された。12株のパネルをアッセイで評価した。該パネルは、下記の研究室株: Staphylococcus aureus Oxford、Staphylococcus aureus WCUH29、Enterococcus faecalis I、Enterococcus faecalis 7、Haemophilus influenzae Q1、Haemophilus influenzae NEMC1、Moraxella catarrhalis 1502、Streptococcus pneumoniae 1629、Streptococcus pneumoniae N1387、Streptococcus pneumoniae N1387、E. coli 7623 (AcrABEFD+)および E. coli 120 (AcrAB-)から構成された。最少阻害濃度(MIC)は、目で見える増殖を阻害した化合物の最低濃度として決定した。MIC終点の決定を助けるために、ミラーリーダー(mirror reader)を用いた。
上述の記載は、その好ましい具体例を包含する本発明を完全に開示するものである。本明細書中に特別に開示される具体例の修飾および改良は、特許請求の範囲の範囲内にある。さらに工夫することなく、当業者は、上述の記載を用いて、本発明をその完全な範囲まで利用することができると確信する。したがって、本明細書の実施例は、単なる例示として解釈されるべきであり、いかなる方法においても、本発明の範囲を制限するものではない。排他的な所有権または権利が主張される具体的な本発明は特許請求の範囲において定義される。
Claims (2)
- 式(I):
[式中、
R1は、C1−C9アルキル、C1−2アルキルArおよびArからなる群から選択され;
R2は、水素、C1−C9アルキル、C1−4アルキルAr’、NHR4、NHC(O)R4、C2−4アルキルNR3R4、C1−3アルキルC(O)NR3R4、C1−3アルキルC(O)Ar’、C2−3アルキルNHC(O)NR3R4、C2−3アルキルNHC(O)Ar’およびC1−2アルキルSO2R4からなる群から選択され;
R3は、C1−C9アルキル、C1−2アルキルArおよびArからなる群から選択され;
R4はR3、Ar’であるか、またはR4はR3およびそれらが結合している窒素原子と一緒になって、C1−3アルキル、アリール、C1−3アルコキシ(1ないし3個のFで置換されていてもよい)、アリールオキシ、カルボキシ、オキソ、ヒドロキシ、アミノ、ニトロおよびシアノからなる群から独立して選択される1、2または3個の置換基で置換されていてもよく、またはアリール、ヘテロアリールもしくは第2の複素環に縮合していてもよい複素環を形成していてもよく;
Arは、フェニル、フリルおよびチエニルからなる群から選択され、その各々がC1−C3アルキル、CN、F、Cl、BrおよびIからなる群から独立して選択される1、2または3個の置換基で置換されていてもよく;
Ar’は、 ナフチル、フリル、ピリジル、チエニル、チアゾリル、イソチアゾリル、ピラゾリル、トリアゾリル、テトラゾリル、イミダゾリル、イミダゾリジニル、ベンゾフラニル、インドリル、チアゾリジニル、イソキサゾリル、オキサジアゾリル、チアジアゾリル、ピロリルおよびピリミジルからなる群から選択され、その各々がC1−C6アルキル、C1−C6アルコキシ、(CH2)0−5CO2R1、C(O)N(R1)2、CN、(CH2)0−5OH、NO2、F、Cl、Br、I、CF3、N(R1)2およびNHC(O)R1からなる群から独立して選択される1、2または3個の置換基で置換されていてもよい]
で示される化合物またはその医薬上許容される塩。 -
N−[(S)−2,5−ジオキソ−4−ペンチル−1−フェニル−イミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−[(S)−1−ビフェニル−4−イルメチル−4−ブチル−2,5−ジオキソ−イミダゾリジン−4−イルメチル]−N−ヒドロキシホルムアミド;
N−{(S)−4−ブチル−1−[2−(5−クロロ−3−メチル−1−ベンゾ[b]チオフェン−2−イル)−2−オキソ−エチル]−2,5−ジオキソ−イミダゾリジン−4−イルメチル}−N−ヒドロキシホルムアミド;および
2−{(S)−4−ブチル−4−[(ホルミル−ヒドロキシ−アミノ)メチル]−2,5−ジオキソ−イミダゾリジン−1−イル}−N−(3,5−ジクロロフェニル)アセトアミド
またはその医薬上許容される塩からなる群から選択される 化合物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US36442302P | 2002-03-13 | 2002-03-13 | |
| PCT/US2003/007509 WO2003077913A1 (en) | 2002-03-13 | 2003-03-13 | Peptide deformylase inhibitors |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2005525383A JP2005525383A (ja) | 2005-08-25 |
| JP2005525383A5 JP2005525383A5 (ja) | 2006-04-27 |
| JP4471663B2 true JP4471663B2 (ja) | 2010-06-02 |
Family
ID=28041913
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003575966A Expired - Fee Related JP4471663B2 (ja) | 2002-03-13 | 2003-03-12 | ペプチドデホルミラーゼ阻害剤 |
Country Status (20)
| Country | Link |
|---|---|
| US (2) | US20050124677A1 (ja) |
| EP (1) | EP1482930B1 (ja) |
| JP (1) | JP4471663B2 (ja) |
| KR (1) | KR100980145B1 (ja) |
| CN (1) | CN100336809C (ja) |
| AT (1) | ATE514683T1 (ja) |
| AU (1) | AU2003216554A1 (ja) |
| BR (1) | BR0308321A (ja) |
| CA (1) | CA2478331A1 (ja) |
| ES (1) | ES2366396T3 (ja) |
| IL (1) | IL163899A0 (ja) |
| IS (1) | IS7441A (ja) |
| MX (1) | MXPA04008799A (ja) |
| NO (1) | NO328933B1 (ja) |
| NZ (1) | NZ534839A (ja) |
| PL (1) | PL371016A1 (ja) |
| RU (1) | RU2287525C2 (ja) |
| TW (1) | TWI294778B (ja) |
| WO (1) | WO2003077913A1 (ja) |
| ZA (1) | ZA200406973B (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7208595B2 (en) * | 2003-07-25 | 2007-04-24 | The Ohio State University Research Foundation | Peptide deformylase inhibitors as novel antibiotics |
| BRPI0611694A2 (pt) * | 2005-06-07 | 2010-09-28 | Novartis Ag | inibidores peptìdicos da deformilase 4 (pdf) |
| EP2195656A4 (en) * | 2007-08-21 | 2011-10-19 | Senomyx Inc | HUMAN T2R BITTERITY RECEPTORS AND USES THEREOF |
| DE102009045969B4 (de) * | 2009-10-23 | 2019-01-31 | Technische Universität Bergakademie Freiberg | Verfahren und Mittel zur Spaltung von Estern, Amiden und Thioestern der Ameisensäure |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3420193A1 (de) * | 1984-05-30 | 1985-12-05 | Boehringer Ingelheim KG, 6507 Ingelheim | Neue substituierte pyrrolidinone, verfahren zu ihrer herstellung und arzneimittel |
| TW448172B (en) * | 1996-03-08 | 2001-08-01 | Pharmacia & Upjohn Co Llc | Novel hydroxamic acid derivatives useful for the treatment of diseases related to connective tissue degradation |
| JP3382127B2 (ja) * | 1996-07-04 | 2003-03-04 | エフ.ホフマン−ラ ロシュ アーゲー | キラルなコハク酸誘導体の製造方法 |
| EP1165546A2 (en) * | 1999-04-02 | 2002-01-02 | Du Pont Pharmaceuticals Company | Novel lactam inhibitors of matrix metalloproteinases, tnf-alpha, and aggrecanase |
| WO2002028829A2 (en) | 2000-09-25 | 2002-04-11 | Questcor Pharmaceuticals, Inc. | Peptide deformylase inhibitors |
| CA2434205A1 (en) * | 2001-01-11 | 2002-07-18 | Dupont Pharmaceuticals Company | 1,1-disubstituted cyclic inhibitors of matrix metalloprotease and tnf-.alpha. |
-
2003
- 2003-03-11 TW TW092105141A patent/TWI294778B/zh not_active IP Right Cessation
- 2003-03-12 EP EP03744644A patent/EP1482930B1/en not_active Expired - Lifetime
- 2003-03-12 ES ES03744644T patent/ES2366396T3/es not_active Expired - Lifetime
- 2003-03-12 JP JP2003575966A patent/JP4471663B2/ja not_active Expired - Fee Related
- 2003-03-12 CA CA002478331A patent/CA2478331A1/en not_active Abandoned
- 2003-03-12 US US10/507,510 patent/US20050124677A1/en not_active Abandoned
- 2003-03-12 CN CNB038106507A patent/CN100336809C/zh not_active Expired - Fee Related
- 2003-03-12 AU AU2003216554A patent/AU2003216554A1/en not_active Abandoned
- 2003-03-12 AT AT03744644T patent/ATE514683T1/de not_active IP Right Cessation
- 2003-03-12 RU RU2004130454/04A patent/RU2287525C2/ru not_active IP Right Cessation
- 2003-03-12 PL PL03371016A patent/PL371016A1/xx not_active Application Discontinuation
- 2003-03-12 NZ NZ534839A patent/NZ534839A/en not_active IP Right Cessation
- 2003-03-13 BR BR0308321-7A patent/BR0308321A/pt not_active IP Right Cessation
- 2003-03-13 IL IL16389903A patent/IL163899A0/xx unknown
- 2003-03-13 MX MXPA04008799A patent/MXPA04008799A/es active IP Right Grant
- 2003-03-13 WO PCT/US2003/007509 patent/WO2003077913A1/en not_active Ceased
- 2003-03-13 KR KR1020047014184A patent/KR100980145B1/ko not_active Expired - Fee Related
-
2004
- 2004-09-01 ZA ZA200406973A patent/ZA200406973B/en unknown
- 2004-09-08 IS IS7441A patent/IS7441A/is unknown
- 2004-09-30 NO NO20044186A patent/NO328933B1/no not_active IP Right Cessation
-
2007
- 2007-02-16 US US11/675,788 patent/US7745637B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| EP1482930B1 (en) | 2011-06-29 |
| ES2366396T3 (es) | 2011-10-19 |
| ZA200406973B (en) | 2005-06-23 |
| WO2003077913A1 (en) | 2003-09-25 |
| RU2287525C2 (ru) | 2006-11-20 |
| TW200405810A (en) | 2004-04-16 |
| KR20040091120A (ko) | 2004-10-27 |
| CN100336809C (zh) | 2007-09-12 |
| CA2478331A1 (en) | 2003-09-25 |
| US7745637B2 (en) | 2010-06-29 |
| MXPA04008799A (es) | 2004-11-26 |
| IL163899A0 (en) | 2005-12-18 |
| RU2004130454A (ru) | 2005-04-10 |
| NZ534839A (en) | 2006-09-29 |
| EP1482930A1 (en) | 2004-12-08 |
| BR0308321A (pt) | 2004-12-28 |
| US20070155810A1 (en) | 2007-07-05 |
| AU2003216554A1 (en) | 2003-09-29 |
| ATE514683T1 (de) | 2011-07-15 |
| CN1652775A (zh) | 2005-08-10 |
| JP2005525383A (ja) | 2005-08-25 |
| US20050124677A1 (en) | 2005-06-09 |
| NO20044186L (no) | 2004-09-30 |
| IS7441A (is) | 2004-09-08 |
| KR100980145B1 (ko) | 2010-09-03 |
| EP1482930A4 (en) | 2006-06-28 |
| PL371016A1 (en) | 2005-06-13 |
| NO328933B1 (no) | 2010-06-21 |
| TWI294778B (en) | 2008-03-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7745637B2 (en) | Peptide deformylase inhibitors | |
| US6806369B2 (en) | Peptide deformylase inhibitors | |
| US20040267015A1 (en) | Peptide deformylase inhibitors | |
| US6797730B2 (en) | Peptide deformylase inhibitors | |
| US6897233B2 (en) | Peptide deformylase inhibitors | |
| JP2005532358A (ja) | ペプチドデホルミラーゼ阻害剤 | |
| US7456211B2 (en) | Peptide deformylase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060309 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060309 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090901 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20091201 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20091208 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20091224 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100202 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100302 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130312 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130312 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140312 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |