JP4098697B2 - Exhaust gas treatment method - Google Patents
Exhaust gas treatment method Download PDFInfo
- Publication number
- JP4098697B2 JP4098697B2 JP2003362597A JP2003362597A JP4098697B2 JP 4098697 B2 JP4098697 B2 JP 4098697B2 JP 2003362597 A JP2003362597 A JP 2003362597A JP 2003362597 A JP2003362597 A JP 2003362597A JP 4098697 B2 JP4098697 B2 JP 4098697B2
- Authority
- JP
- Japan
- Prior art keywords
- mercury
- catalyst
- exhaust gas
- oxide
- titanium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 238000000034 method Methods 0.000 title claims description 54
- 229910052753 mercury Inorganic materials 0.000 claims description 139
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 claims description 137
- 239000003054 catalyst Substances 0.000 claims description 130
- 239000007789 gas Substances 0.000 claims description 92
- MWUXSHHQAYIFBG-UHFFFAOYSA-N nitrogen oxide Inorganic materials O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 claims description 89
- 238000005658 halogenation reaction Methods 0.000 claims description 40
- 230000026030 halogenation Effects 0.000 claims description 36
- 229910052720 vanadium Inorganic materials 0.000 claims description 36
- LEONUFNNVUYDNQ-UHFFFAOYSA-N vanadium atom Chemical compound [V] LEONUFNNVUYDNQ-UHFFFAOYSA-N 0.000 claims description 35
- -1 mercury halide Chemical class 0.000 claims description 30
- 229910006295 Si—Mo Inorganic materials 0.000 claims description 14
- 150000002366 halogen compounds Chemical class 0.000 claims description 13
- 229910052751 metal Inorganic materials 0.000 claims description 11
- 239000002184 metal Substances 0.000 claims description 11
- 239000011206 ternary composite Substances 0.000 claims description 6
- 239000002131 composite material Substances 0.000 description 53
- 239000010936 titanium Substances 0.000 description 48
- 229910052719 titanium Inorganic materials 0.000 description 39
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 38
- 229910052750 molybdenum Inorganic materials 0.000 description 33
- 239000000243 solution Substances 0.000 description 29
- 229910052721 tungsten Inorganic materials 0.000 description 26
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 25
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 23
- 229910004339 Ti-Si Inorganic materials 0.000 description 22
- 229910010978 Ti—Si Inorganic materials 0.000 description 22
- 239000000843 powder Substances 0.000 description 22
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 22
- 239000000203 mixture Substances 0.000 description 21
- 239000007864 aqueous solution Substances 0.000 description 20
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 19
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 17
- 229910010413 TiO 2 Inorganic materials 0.000 description 17
- XHCLAFWTIXFWPH-UHFFFAOYSA-N [O-2].[O-2].[O-2].[O-2].[O-2].[V+5].[V+5] Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[V+5].[V+5] XHCLAFWTIXFWPH-UHFFFAOYSA-N 0.000 description 17
- 229940008718 metallic mercury Drugs 0.000 description 17
- 229910001935 vanadium oxide Inorganic materials 0.000 description 17
- 238000002441 X-ray diffraction Methods 0.000 description 16
- 238000004519 manufacturing process Methods 0.000 description 16
- 239000011733 molybdenum Substances 0.000 description 16
- 229910052782 aluminium Inorganic materials 0.000 description 15
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 15
- 239000010937 tungsten Substances 0.000 description 15
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 14
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 14
- 238000010521 absorption reaction Methods 0.000 description 12
- 229910052710 silicon Inorganic materials 0.000 description 12
- 239000000126 substance Substances 0.000 description 12
- 239000011148 porous material Substances 0.000 description 11
- 239000002002 slurry Substances 0.000 description 11
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 10
- 229910044991 metal oxide Inorganic materials 0.000 description 10
- 150000004706 metal oxides Chemical class 0.000 description 10
- 238000002156 mixing Methods 0.000 description 10
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 9
- 239000005708 Sodium hypochlorite Substances 0.000 description 9
- QGLKJKCYBOYXKC-UHFFFAOYSA-N nonaoxidotritungsten Chemical compound O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1 QGLKJKCYBOYXKC-UHFFFAOYSA-N 0.000 description 9
- 239000010703 silicon Substances 0.000 description 9
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- 229910001930 tungsten oxide Inorganic materials 0.000 description 9
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- 150000003609 titanium compounds Chemical class 0.000 description 8
- 230000002140 halogenating effect Effects 0.000 description 7
- 150000002484 inorganic compounds Chemical class 0.000 description 7
- 238000000465 moulding Methods 0.000 description 7
- 150000002894 organic compounds Chemical class 0.000 description 7
- 229910052726 zirconium Inorganic materials 0.000 description 7
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- 235000011114 ammonium hydroxide Nutrition 0.000 description 6
- 239000011218 binary composite Substances 0.000 description 6
- XAYGUHUYDMLJJV-UHFFFAOYSA-Z decaazanium;dioxido(dioxo)tungsten;hydron;trioxotungsten Chemical compound [H+].[H+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O XAYGUHUYDMLJJV-UHFFFAOYSA-Z 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 229910000476 molybdenum oxide Inorganic materials 0.000 description 6
- PQQKPALAQIIWST-UHFFFAOYSA-N oxomolybdenum Chemical compound [Mo]=O PQQKPALAQIIWST-UHFFFAOYSA-N 0.000 description 6
- 229910052814 silicon oxide Inorganic materials 0.000 description 6
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 5
- 150000003863 ammonium salts Chemical class 0.000 description 5
- UNTBPXHCXVWYOI-UHFFFAOYSA-O azanium;oxido(dioxo)vanadium Chemical compound [NH4+].[O-][V](=O)=O UNTBPXHCXVWYOI-UHFFFAOYSA-O 0.000 description 5
- 239000011230 binding agent Substances 0.000 description 5
- 238000006243 chemical reaction Methods 0.000 description 5
- 150000004820 halides Chemical class 0.000 description 5
- 238000004898 kneading Methods 0.000 description 5
- 235000006408 oxalic acid Nutrition 0.000 description 5
- 238000002459 porosimetry Methods 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 239000008107 starch Substances 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- 229910004298 SiO 2 Inorganic materials 0.000 description 4
- 229910002796 Si–Al Inorganic materials 0.000 description 4
- 229910006774 Si—W Inorganic materials 0.000 description 4
- 229910011214 Ti—Mo Inorganic materials 0.000 description 4
- RCTYPNKXASFOBE-UHFFFAOYSA-M chloromercury Chemical compound [Hg]Cl RCTYPNKXASFOBE-UHFFFAOYSA-M 0.000 description 4
- 238000010304 firing Methods 0.000 description 4
- 150000004679 hydroxides Chemical class 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- VLAPMBHFAWRUQP-UHFFFAOYSA-L molybdic acid Chemical compound O[Mo](O)(=O)=O VLAPMBHFAWRUQP-UHFFFAOYSA-L 0.000 description 4
- 238000001179 sorption measurement Methods 0.000 description 4
- 229910000349 titanium oxysulfate Inorganic materials 0.000 description 4
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 4
- 229910052723 transition metal Inorganic materials 0.000 description 4
- 150000003624 transition metals Chemical class 0.000 description 4
- 150000003755 zirconium compounds Chemical class 0.000 description 4
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 3
- 229910008341 Si-Zr Inorganic materials 0.000 description 3
- 229910006682 Si—Zr Inorganic materials 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 239000008119 colloidal silica Substances 0.000 description 3
- 239000012456 homogeneous solution Substances 0.000 description 3
- 238000005070 sampling Methods 0.000 description 3
- VSZWPYCFIRKVQL-UHFFFAOYSA-N selanylidenegallium;selenium Chemical compound [Se].[Se]=[Ga].[Se]=[Ga] VSZWPYCFIRKVQL-UHFFFAOYSA-N 0.000 description 3
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 3
- 150000003377 silicon compounds Chemical class 0.000 description 3
- CMPGARWFYBADJI-UHFFFAOYSA-L tungstic acid Chemical compound O[W](O)(=O)=O CMPGARWFYBADJI-UHFFFAOYSA-L 0.000 description 3
- BNGXYYYYKUGPPF-UHFFFAOYSA-M (3-methylphenyl)methyl-triphenylphosphanium;chloride Chemical compound [Cl-].CC1=CC=CC(C[P+](C=2C=CC=CC=2)(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 BNGXYYYYKUGPPF-UHFFFAOYSA-M 0.000 description 2
- VXEGSRKPIUDPQT-UHFFFAOYSA-N 4-[4-(4-methoxyphenyl)piperazin-1-yl]aniline Chemical compound C1=CC(OC)=CC=C1N1CCN(C=2C=CC(N)=CC=2)CC1 VXEGSRKPIUDPQT-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- BOTDANWDWHJENH-UHFFFAOYSA-N Tetraethyl orthosilicate Chemical compound CCO[Si](OCC)(OCC)OCC BOTDANWDWHJENH-UHFFFAOYSA-N 0.000 description 2
- 229910004349 Ti-Al Inorganic materials 0.000 description 2
- 229910004692 Ti—Al Inorganic materials 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 2
- 229910007729 Zr W Inorganic materials 0.000 description 2
- HDYRYUINDGQKMC-UHFFFAOYSA-M acetyloxyaluminum;dihydrate Chemical compound O.O.CC(=O)O[Al] HDYRYUINDGQKMC-UHFFFAOYSA-M 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- DIZPMCHEQGEION-UHFFFAOYSA-H aluminium sulfate (anhydrous) Chemical compound [Al+3].[Al+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O DIZPMCHEQGEION-UHFFFAOYSA-H 0.000 description 2
- 229940009827 aluminum acetate Drugs 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 238000000975 co-precipitation Methods 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 238000000151 deposition Methods 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000010419 fine particle Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 229910021331 inorganic silicon compound Inorganic materials 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- 150000003891 oxalate salts Chemical class 0.000 description 2
- BBJSDUUHGVDNKL-UHFFFAOYSA-J oxalate;titanium(4+) Chemical compound [Ti+4].[O-]C(=O)C([O-])=O.[O-]C(=O)C([O-])=O BBJSDUUHGVDNKL-UHFFFAOYSA-J 0.000 description 2
- DCKVFVYPWDKYDN-UHFFFAOYSA-L oxygen(2-);titanium(4+);sulfate Chemical compound [O-2].[Ti+4].[O-]S([O-])(=O)=O DCKVFVYPWDKYDN-UHFFFAOYSA-L 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 239000002574 poison Substances 0.000 description 2
- 231100000614 poison Toxicity 0.000 description 2
- 235000019353 potassium silicate Nutrition 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 239000005049 silicon tetrachloride Substances 0.000 description 2
- NTHWMYGWWRZVTN-UHFFFAOYSA-N sodium silicate Chemical compound [Na+].[Na+].[O-][Si]([O-])=O NTHWMYGWWRZVTN-UHFFFAOYSA-N 0.000 description 2
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 2
- 229910000348 titanium sulfate Inorganic materials 0.000 description 2
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 2
- PMTRSEDNJGMXLN-UHFFFAOYSA-N titanium zirconium Chemical compound [Ti].[Zr] PMTRSEDNJGMXLN-UHFFFAOYSA-N 0.000 description 2
- DUNKXUFBGCUVQW-UHFFFAOYSA-J zirconium tetrachloride Chemical compound Cl[Zr](Cl)(Cl)Cl DUNKXUFBGCUVQW-UHFFFAOYSA-J 0.000 description 2
- ZXAUZSQITFJWPS-UHFFFAOYSA-J zirconium(4+);disulfate Chemical compound [Zr+4].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O ZXAUZSQITFJWPS-UHFFFAOYSA-J 0.000 description 2
- YLZOPXRUQYQQID-UHFFFAOYSA-N 3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]propan-1-one Chemical compound N1N=NC=2CN(CCC=21)CCC(=O)N1CCN(CC1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F YLZOPXRUQYQQID-UHFFFAOYSA-N 0.000 description 1
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- NIPNSKYNPDTRPC-UHFFFAOYSA-N N-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 NIPNSKYNPDTRPC-UHFFFAOYSA-N 0.000 description 1
- AFCARXCZXQIEQB-UHFFFAOYSA-N N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CCNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 AFCARXCZXQIEQB-UHFFFAOYSA-N 0.000 description 1
- 229910021536 Zeolite Inorganic materials 0.000 description 1
- GEIAQOFPUVMAGM-UHFFFAOYSA-N ZrO Inorganic materials [Zr]=O GEIAQOFPUVMAGM-UHFFFAOYSA-N 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 238000007664 blowing Methods 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000000498 cooling water Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000006477 desulfuration reaction Methods 0.000 description 1
- 230000023556 desulfurization Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000001125 extrusion Methods 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 150000002730 mercury Chemical class 0.000 description 1
- NGYIMTKLQULBOO-UHFFFAOYSA-L mercury dibromide Chemical compound Br[Hg]Br NGYIMTKLQULBOO-UHFFFAOYSA-L 0.000 description 1
- LWJROJCJINYWOX-UHFFFAOYSA-L mercury dichloride Chemical compound Cl[Hg]Cl LWJROJCJINYWOX-UHFFFAOYSA-L 0.000 description 1
- 229910000474 mercury oxide Inorganic materials 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000005078 molybdenum compound Substances 0.000 description 1
- 150000002752 molybdenum compounds Chemical class 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- CBXWGGFGZDVPNV-UHFFFAOYSA-N so4-so4 Chemical compound OS(O)(=O)=O.OS(O)(=O)=O CBXWGGFGZDVPNV-UHFFFAOYSA-N 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000008400 supply water Substances 0.000 description 1
- 238000004056 waste incineration Methods 0.000 description 1
- 239000002351 wastewater Substances 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
Images







Description
本発明は、窒素酸化物とともに水銀をも含む排ガスを効率よく処理することができる、排ガス処理方法に関する。 The present invention relates to an exhaust gas treatment method capable of efficiently treating exhaust gas containing mercury as well as nitrogen oxides.
従来、排ガス中の窒素酸化物(NOx)を処理する方法として、アンモニアまたは尿素などの還元剤を用いて排ガス中の窒素酸化物を脱硝触媒上で接触還元し、無害な窒素と水とに分解する選択的触媒還元法(いわゆるSCR法)が知られており、これを採用した排ガス処理システムが実用化されている。
しかし、排ガス中には、窒素酸化物(NOx)とともに、有害物質である水銀が、金属水銀(Hg0)もしくは塩化水銀(HgCl2)のようなハロゲン化水銀として含まれていることがある。このように窒素酸化物とともに水銀(金属水銀やハロゲン化水銀)をも含む排ガスを前述したSCR法で処理すると、経時的に脱硝触媒に水銀が蓄積してしまい、蓄積した水銀が触媒毒として作用して脱硝触媒の劣化を促進することとなり、高い排ガス処理効果を長期にわたり持続することができない、といった問題を生じる。
Conventionally, as a method of treating nitrogen oxides (NOx) in exhaust gas, nitrogen oxides in exhaust gas are contact-reduced on a denitration catalyst using a reducing agent such as ammonia or urea, and decomposed into harmless nitrogen and water. A selective catalytic reduction method (so-called SCR method) is known, and an exhaust gas treatment system employing this method has been put into practical use.
However, in some cases, mercury, which is a harmful substance, is contained in the exhaust gas as mercury halides such as metallic mercury (Hg 0 ) or mercury chloride (HgCl 2 ) together with nitrogen oxides (NOx). When exhaust gas containing mercury (metal mercury or mercury halide) together with nitrogen oxides is treated by the SCR method as described above, mercury accumulates over time in the denitration catalyst, and the accumulated mercury acts as catalyst poison. As a result, deterioration of the denitration catalyst is promoted, resulting in a problem that a high exhaust gas treatment effect cannot be maintained for a long time.
また、水銀は、ハロゲン化水銀として存在する場合には、水に容易に吸収されるために捕捉・除去することは比較的容易であるが、金属水銀として存在する場合には、水にほとんど吸収されないため除去が困難である。このため、窒素酸化物とともに水銀をも含む排ガスを従来の前記SCR法による排ガス処理システムで処理した場合、金属水銀が蒸気として大気中に排出される恐れもあった。
これまで、排ガス中から水銀(金属水銀を含む)を除去する技術としては、例えば、活性炭吸着法や次亜塩素酸ソーダ吸収法が知られている(例えば、特許文献1、2など参照)。詳しくは、活性炭吸着法としては、例えば、排ガス中に活性炭粉末を吹き込んでバグフィルターで回収する方法が実用化されており、一方、次亜塩素酸ソーダ吸収法としては、例えば、排ガス処理システムにおける冷却塔の冷却水、脱硫吸収塔の吸収液、湿式電気集じん機の供給水や循環水等に、次亜塩素酸ソーダを直接添加する方法が実用化されている。
Mercury is easily absorbed and removed by mercury when it is present as mercury halide, but it is almost absorbed by water when it is present as metallic mercury. This is difficult to remove. For this reason, when the exhaust gas containing both nitrogen oxides and mercury is treated by the conventional exhaust gas treatment system based on the SCR method, the metal mercury may be discharged into the atmosphere as vapor.
Conventionally, as a technique for removing mercury (including metallic mercury) from exhaust gas, for example, an activated carbon adsorption method and a sodium hypochlorite absorption method are known (see, for example, Patent Documents 1 and 2). Specifically, as the activated carbon adsorption method, for example, a method of blowing activated carbon powder into exhaust gas and collecting it with a bag filter has been put to practical use. On the other hand, as the sodium hypochlorite absorption method, for example, in an exhaust gas treatment system A method of directly adding sodium hypochlorite to cooling water in a cooling tower, absorption liquid in a desulfurization absorption tower, supply water or circulating water in a wet electric dust collector has been put into practical use.
しかしながら、前記活性炭吸着法は、水銀を吸着させた活性炭を再生して繰り返し使用することができないので、活性炭に多大なコストがかかるものであり、しかも使用済み活性炭の処分も問題となる。一方、前記次亜塩素酸ソーダ吸収法は、排ガス処理システムの主要機器に次亜塩素酸ソーダを加えるものであるので、装置内の腐食が懸念されると同時に、次亜塩素酸ソーダに多大なコストがかかるものであり、しかも該次亜塩素酸ソーダによる2次公害の恐れがあり、生じた排水の処分も問題となる。そのため、活性炭吸着法および次亜塩素酸ソーダ吸収法はいずれも、ゴミ焼却排ガス等の少量の排ガス処理においてのみ実用化されているものであり、発電所排ガス等の大容量ガスの処理には適用し難いものであった。 However, since the activated carbon adsorption method cannot regenerate and repeatedly use activated carbon on which mercury is adsorbed, the activated carbon is very expensive, and disposal of the used activated carbon is also a problem. On the other hand, the sodium hypochlorite absorption method adds sodium hypochlorite to the main equipment of the exhaust gas treatment system. The cost is high, and there is a risk of secondary pollution due to the sodium hypochlorite, and disposal of the generated waste water is also a problem. Therefore, both the activated carbon adsorption method and the sodium hypochlorite absorption method have been put into practical use only for the treatment of a small amount of exhaust gas such as waste incineration exhaust gas, and are applicable to the treatment of large-capacity gas such as power plant exhaust gas. It was difficult.
したがって、水銀(金属水銀を含む)除去のために活性炭や次亜塩素酸ソーダを用いることなく大容量の排ガス処理にも適用することができ、窒素酸化物とともに金属水銀をも効率よく処理することができる新たな方法が求められている。
そこで、本発明が解決しようとする課題は、窒素酸化物と金属水銀とを含む排ガスを長期にわたり効率よく処理することができ、大容量の排ガス処理にも適用可能な新たな排ガス処理方法を提供することにある。 Therefore, the problem to be solved by the present invention is to provide a new exhaust gas treatment method that can efficiently treat exhaust gas containing nitrogen oxides and metal mercury over a long period of time and can be applied to large-capacity exhaust gas treatment. There is to do.
本発明者は上記課題を解決するべく、鋭意検討を行った結果、下記(1)式
Hg0+2HCl+1/2O2⇔HgCl2+H2O (1)
に示す平衡反応において、HCl存在下、触媒を用いることにより平衡が右へ移動することに着目し、まず、脱硝触媒を用いて窒素酸化物を処理したのち、ハロゲン化合物の存在下で水銀ハロゲン化触媒を用いて前記(1)式の平衡反応を右へ移動させて金属水銀を捕捉・除去が容易なハロゲン化水銀に変換すれば、窒素酸化物と水銀とをともに処理できることを見出した。さらに、窒素酸化物とともに金属水銀をも含む排ガスに対してまず窒素酸化物の処理を行なう場合に懸念される水銀の蓄積による脱硝触媒の性能低下を回避するには、特定の全細孔容積をもつ脱硝触媒を用いればよいこと、窒素酸化物の処理と金属水銀のハロゲン化とを効率よく行なうためには、各触媒の触媒温度を特定範囲に設定すればよいこと、を見出した。そして、これらの知見に基づき本発明を完成した。
As a result of intensive studies to solve the above problems, the inventor of the present invention has the following formula (1): Hg 0 + 2HCl + 1 / 2O 2 ⇔HgCl 2 + H 2 O (1)
In the equilibrium reaction shown in Fig. 1, paying attention to the fact that the equilibrium shifts to the right by using a catalyst in the presence of HCl, first, nitrogen oxides were treated with a denitration catalyst, then mercury halogenated in the presence of a halogen compound. It has been found that both nitrogen oxide and mercury can be treated by moving the equilibrium reaction of the formula (1) to the right using a catalyst and converting the metal mercury into mercury halide that can be easily captured and removed. Furthermore, in order to avoid degradation of the performance of the denitration catalyst due to mercury accumulation, which is a concern when first treating nitrogen oxides with exhaust gas that also contains metallic mercury together with nitrogen oxides, a specific total pore volume should be set. The present inventors have found that a denitration catalyst having a low temperature should be used, and that the catalyst temperature of each catalyst may be set within a specific range in order to efficiently perform the treatment of nitrogen oxides and the halogenation of metallic mercury. And based on these knowledge, this invention was completed.
すなわち、本発明にかかる排ガス処理方法は、窒素酸化物と金属水銀とを含む排ガスを処理する排ガス処理方法であって、脱硝触媒を用いて窒素酸化物を処理したのち、ハロゲン化合物の存在下で水銀ハロゲン化触媒を用いて金属水銀をハロゲン化水銀に変換するに当り、前記水銀ハロゲン化触媒としてTi−Si−Moなる三元系複合酸化物と活性種としてのバナジウムを含有するものを用いるとともに、前記脱硝触媒の触媒温度は300℃よりも高い温度、前記水銀ハロゲン化触媒の触媒温度は300℃以下とすることを特徴とする。 That is, the exhaust gas treatment method according to the present invention is an exhaust gas treatment method for treating exhaust gas containing nitrogen oxides and metallic mercury, and after treating nitrogen oxides using a denitration catalyst, in the presence of a halogen compound. per to convert metallic mercury to halogenated mercury with mercury halogenating catalyst, those containing vanadium as T i-Si-Mo becomes ternary composite oxide and the active species by said mercury halide catalyst In addition, the catalyst temperature of the denitration catalyst is higher than 300 ° C., and the catalyst temperature of the mercury halogenated catalyst is 300 ° C. or less.
本発明の新たな排ガス処理方法によれば、窒素酸化物と金属水銀とを含む排ガスを長期にわたり効率よく処理することができる。しかも、本発明の排ガス処理方法は、水銀除去のために活性炭や次亜塩素酸ソーダを用いる必要がないので、大容量の排ガス処理にも適用することができる。 According to the new exhaust gas treatment method of the present invention, exhaust gas containing nitrogen oxides and metallic mercury can be efficiently treated over a long period of time. Moreover, since the exhaust gas treatment method of the present invention does not require the use of activated carbon or sodium hypochlorite for mercury removal, it can also be applied to large-volume exhaust gas treatment.
以下、本発明にかかる排ガス処理方法について詳しく説明するが、本発明の範囲はこれらの説明に拘束されることはなく、以下の例示以外についても、本発明の趣旨を損なわない範囲で適宜変更実施し得る。
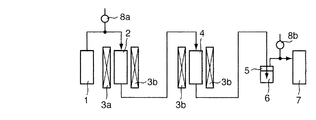
本発明の排ガス処理方法は、窒素酸化物と金属水銀とを含有する排ガスに対して、まず、脱硝触媒を用いて窒素酸化物の処理(以下「脱硝処理」と称することもある。)を施し、その後、ハロゲン化合物の存在下で水銀ハロゲン化触媒を用いて金属水銀をハロゲン化水銀に変換する処理(以下「水銀ハロゲン化処理」と称することもある。)を施すものである。以下、各処理について、詳しく説明する。
Hereinafter, the exhaust gas treatment method according to the present invention will be described in detail. However, the scope of the present invention is not limited to these descriptions, and modifications other than the following examples are made as appropriate without departing from the spirit of the present invention. Can do.
In the exhaust gas treatment method of the present invention, first, exhaust gas containing nitrogen oxides and metallic mercury is first treated with nitrogen oxides (hereinafter also referred to as “denitration treatment”) using a denitration catalyst. Thereafter, a treatment for converting metallic mercury to mercury halide using a mercury halogenation catalyst in the presence of a halogen compound (hereinafter sometimes referred to as “mercury halogenation treatment”) is performed. Hereinafter, each process will be described in detail.
脱硝処理においては、脱硝触媒を用いて排ガス中の窒素酸化物を処理する。具体的には、前記脱硝処理は、脱硝触媒をアンモニアや尿素などの還元剤の存在下、排ガスと接触させ、排ガス中の窒素酸化物を還元して無害化すればよい。なお、前記脱硝処理は、脱硝のため通常の排ガスシステムに設けられている脱硝装置で行なえばよい。
前記脱硝触媒としては、特に制限はなく、従来公知の脱硝触媒を用いることができるが、水銀に対する耐性が高いという点では、バナジウム(V)および/またはチタン(Ti)を含有する触媒が好ましい。以下、バナジウム(V)および/またはチタン(Ti)を含有する触媒の特に好ましい態様について詳しく説明する。
In the denitration treatment, nitrogen oxides in the exhaust gas are treated using a denitration catalyst. Specifically, in the denitration treatment, a denitration catalyst may be brought into contact with exhaust gas in the presence of a reducing agent such as ammonia or urea, and nitrogen oxides in the exhaust gas may be reduced and rendered harmless. The denitration process may be performed by a denitration apparatus provided in a normal exhaust gas system for denitration.
There is no restriction | limiting in particular as said denitration catalyst, Although a conventionally well-known denitration catalyst can be used, the catalyst containing vanadium (V) and / or titanium (Ti) is preferable at the point that the tolerance with respect to mercury is high. Hereinafter, particularly preferred embodiments of the catalyst containing vanadium (V) and / or titanium (Ti) will be described in detail.
前記脱硝触媒は、バナジウム(V)および/またはチタン(Ti)のほかに、ケイ素(Si)、ジルコニウム(Zr)、アルミニウム(Al)、タングステン(W)およびモリブデン(Mo)からなる群より選ばれる1種以上の遷移金属をも含有することが好ましい。これら遷移金属は、独立した金属酸化物として含有されていてもよいが、チタンとの複合酸化物を形成して含有されていることがより好ましい。すなわち、前記脱硝触媒は、ケイ素、ジルコニウム、アルミニウム、タングステンおよびモリブデンからなる群より選ばれる1種または2種とチタンとの二元系または三元系複合酸化物を含有するものであることがより好ましいのである。具体的には、例えば、Ti−Si、Ti−Zr、Ti−Al、Ti−W、Ti−Mo等の二元系複合酸化物;Ti−Si−Mo、Ti−Si−W、Ti−Si−Zr、Ti−Si−Al、Ti−Zr−Al、Ti−Zr−Mo、Ti−Zr−W、Ti−Al−Mo、Ti−W−Mo等の三元系複合酸化物;等が、安定な構造を維持でき水銀に対する高い耐性を発揮しうる点で好ましい。これらの中でも、Ti−Siの二元系複合酸化物や、Ti−Si−Mo、Ti−Si−W、Ti−Si−Zr、Ti−Si−Alのような三元系複合酸化物など、ケイ素(Si)を必須とする遷移金属とチタン(Ti)との二元系または三元系複合酸化物が、脱硝活性がより高いことから、より好ましい。なお、ここで、複合酸化物とは、X線回折パターンにおいてTiO2以外の物質に帰属される明らかな固有のピークを示さず、TiO2についてはアナターゼ型酸化チタンに帰属される固有のピークを示さないか、もしくは示してもアナターゼ型酸化チタンの回折ピークよりもブロードな回折ピークを示すものを言う。 The denitration catalyst is selected from the group consisting of silicon (Si), zirconium (Zr), aluminum (Al), tungsten (W) and molybdenum (Mo) in addition to vanadium (V) and / or titanium (Ti). It is also preferable to contain one or more transition metals. These transition metals may be contained as independent metal oxides, but are more preferably contained by forming a composite oxide with titanium. That is, the denitration catalyst more preferably contains a binary or ternary composite oxide of one or two selected from the group consisting of silicon, zirconium, aluminum, tungsten and molybdenum and titanium. Is preferred. Specifically, for example, binary complex oxides such as Ti-Si, Ti-Zr, Ti-Al, Ti-W, Ti-Mo; Ti-Si-Mo, Ti-Si-W, Ti-Si -Ternary complex oxides such as -Zr, Ti-Si-Al, Ti-Zr-Al, Ti-Zr-Mo, Ti-Zr-W, Ti-Al-Mo, Ti-W-Mo; This is preferable in that a stable structure can be maintained and high resistance to mercury can be exhibited. Among these, Ti-Si binary complex oxides, ternary complex oxides such as Ti-Si-Mo, Ti-Si-W, Ti-Si-Zr, Ti-Si-Al, etc. A binary or ternary composite oxide of a transition metal essentially containing silicon (Si) and titanium (Ti) is more preferable because of its higher denitration activity. Here, the composite oxide exhibited no apparent specific peak attributed to substance other than TiO 2 in the X-ray diffraction pattern, a unique peak for TiO 2 is attributable to anatase titanium oxide It means not showing or showing a broader diffraction peak than that of anatase-type titanium oxide.
前記脱硝触媒がチタン系複合酸化物を含有するものである場合、該チタン系複合酸化物における各元素の組成は、特に制限されるものではないが、例えば、チタン系複合酸化物に占めるチタン(Ti)以外の元素(Si、Zr、Al、W、Moなど)の含有量が、各元素の酸化物換算重量比で、それぞれ、0.5〜30重量%であることが好ましく、1〜30重量%であることがより好ましく、1〜25重量%であることがさらに好ましい。
前記脱硝触媒がチタン系複合酸化物を含有するものである場合、該チタン系複合酸化物の調製方法としては、特に制限はなく、例えば、沈殿法(共沈法)、沈着法、混練法などの従来公知の方法を採用することができる。例えば、Ti−Siであれば、コロイド状シリカなどのケイ素化合物をアンモニア水溶液に分散させて水溶液(A)を得、この水溶液(A)に攪拌下でチタン化合物の水溶液を徐々に滴下し、得られたスラリーを濾過、洗浄し、さらに乾燥した後、300〜600℃の高温で焼成することにより、得ることができる。また、例えば、Ti−Si−Mo、Ti−Si−Zr、Ti−Si−Al、Ti−Si−Wであれば、前記水溶液(A)に、さらにMo、Zr、Al、Wなどの塩の水溶液を加え、得られた水溶液に攪拌下でチタン化合物の水溶液を徐々に滴下し、得られたスラリーを濾過、洗浄し、さらに乾燥した後、300〜600℃の高温で焼成することにより、得ることができる。
When the denitration catalyst contains a titanium composite oxide, the composition of each element in the titanium composite oxide is not particularly limited. For example, titanium ( The content of elements (Si, Zr, Al, W, Mo, etc.) other than (Ti) is preferably 0.5 to 30% by weight in terms of oxide-converted weight ratio of each element. It is more preferable that it is weight%, and it is still more preferable that it is 1-25 weight%.
When the denitration catalyst contains a titanium complex oxide, the method for preparing the titanium complex oxide is not particularly limited, and examples thereof include a precipitation method (coprecipitation method), a deposition method, and a kneading method. The conventionally known method can be employed. For example, in the case of Ti—Si, a silicon compound such as colloidal silica is dispersed in an aqueous ammonia solution to obtain an aqueous solution (A), and an aqueous solution of a titanium compound is gradually added dropwise to the aqueous solution (A) with stirring. The obtained slurry can be filtered, washed, dried, and then fired at a high temperature of 300 to 600 ° C. to obtain the slurry. Further, for example, in the case of Ti—Si—Mo, Ti—Si—Zr, Ti—Si—Al, Ti—Si—W, a salt of Mo, Zr, Al, W or the like is further added to the aqueous solution (A). An aqueous solution is added, and an aqueous solution of a titanium compound is gradually added dropwise to the obtained aqueous solution with stirring. The obtained slurry is filtered, washed, dried, and then fired at a high temperature of 300 to 600 ° C. be able to.
前記脱硝触媒がチタン系複合酸化物を含有するものである場合、前記チタン系複合酸化物を調製する際に用いる各元素の供給源は、特に制限されない。例えば、チタン(Ti)源としては、無機および有機のいずれの化合物も使用可能であり、例えば、四塩化チタン、硫酸チタンなどの無機チタン化合物または蓚酸チタン、テトライソプロピルチタネートなどの有機チタン化合物を用いることができる。ケイ素(Si)源としては、コロイド状シリカ、水ガラス、微粒子ケイ素、四塩化ケイ素、シリカゲルなどの無機ケイ素化合物およびテトラエチルシリケートなどの有機ケイ素化合物から適宜選択して使用することができる。ジルコニウム(Zr)源としては、例えば、塩化ジルコニウム、硫酸ジルコニウムなどの無機系ジルコニウム化合物や、シュウ酸ジウコニウムなどの有機系ジルコニウム化合物から適宜選択して使用することができる。アルミニウム(Al)源としては、例えば、硝酸アルミニウム、硫酸アルミニウムなどの無機系アルミニウム化合物や、酢酸アルミニウムなどの有機系アルミニウム化合物から適宜選択して使用することができる。タングステン(W)源としては、例えば、酸化タングステン、パラタングステン酸アンモニウム、メタタングステン酸アンモニウム、タングステン酸などから適宜選択して使用することができる。モリブデン(Mo)源としては、無機および有機のいずれの化合物でもよく、例えば、モリブデンを含む酸化物、水酸化物、アンモニウム塩、ハロゲン化物などから適宜用いることができ、具体的には、パラモリブデン酸アンモニウム、モリブデン酸等が挙げられる。 When the denitration catalyst contains a titanium-based composite oxide, the supply source of each element used when preparing the titanium-based composite oxide is not particularly limited. For example, as the titanium (Ti) source, both inorganic and organic compounds can be used. For example, inorganic titanium compounds such as titanium tetrachloride and titanium sulfate or organic titanium compounds such as titanium oxalate and tetraisopropyl titanate are used. be able to. The silicon (Si) source can be appropriately selected from inorganic silicon compounds such as colloidal silica, water glass, fine particle silicon, silicon tetrachloride, and silica gel, and organic silicon compounds such as tetraethyl silicate. As the zirconium (Zr) source, for example, an inorganic zirconium compound such as zirconium chloride or zirconium sulfate, or an organic zirconium compound such as diuconium oxalate can be appropriately selected and used. As the aluminum (Al) source, for example, an inorganic aluminum compound such as aluminum nitrate or aluminum sulfate or an organic aluminum compound such as aluminum acetate can be appropriately selected and used. As the tungsten (W) source, for example, tungsten oxide, ammonium paratungstate, ammonium metatungstate, tungstic acid and the like can be appropriately selected and used. As the molybdenum (Mo) source, any of inorganic and organic compounds may be used. For example, oxides, hydroxides, ammonium salts, halides and the like containing molybdenum can be used as appropriate. Specifically, para-molybdenum can be used. Examples thereof include ammonium acid and molybdic acid.
さらに、前記脱硝触媒は、前記チタン系複合酸化物に、活性種としてバナジウム(V)を添加した触媒であることが好ましい。また、特に、前記チタン系複合酸化物が前記Ti−Si二元系複合酸化物である場合、活性種としてバナジウム(V)とともに、タングステン(W)、モリブデン(Mo)のうちの少なくとも1種(以下「W・Mo」と略す)を添加した触媒であることが、脱硝活性がより高い点で好ましい。
前記脱硝触媒が前記チタン系複合酸化物に前記活性種(V、W、Mo)を添加したものである場合(前記チタン系複合酸化物にVを添加したものである場合、もしくは前記Ti−Si二元系複合酸化物にVとW・Moとを添加したものである場合)、これらVもしくはVとW・Moの含有量は、特に限定されないが、例えば、脱硝触媒の全重量に対し、各元素(V、W、Mo)の酸化物換算重量比で0.1〜25重量%であることが好ましく、1〜20重量%であることがより好ましい。
Furthermore, the denitration catalyst is preferably a catalyst obtained by adding vanadium (V) as an active species to the titanium-based composite oxide. In particular, when the titanium-based composite oxide is the Ti-Si binary composite oxide, at least one of tungsten (W) and molybdenum (Mo) as active species together with vanadium (V) ( A catalyst to which “W · Mo” is added) is preferable from the viewpoint of higher denitration activity.
When the denitration catalyst is obtained by adding the active species (V, W, Mo) to the titanium-based composite oxide (when V is added to the titanium-based composite oxide, or when the Ti-Si is used) When V and W · Mo are added to the binary composite oxide), the content of these V or V and W · Mo is not particularly limited. For example, with respect to the total weight of the denitration catalyst, The oxide-converted weight ratio of each element (V, W, Mo) is preferably 0.1 to 25% by weight, and more preferably 1 to 20% by weight.
前記脱硝触媒が前記チタン系複合酸化物に前記活性種(V、W、Mo)を添加したものである場合、前記チタン系複合酸化物にVもしくはVとW・Moを添加する方法としては、特に限定されず、例えば、前記チタン系複合酸化物の粉末に、添加しようとする活性種源(バナジウム源、タングステン源、モリブデン源のうちの1種以上)を含む水溶液を、一般にこの種の成形を行う際に用いられる有機または無機の成形助剤とともに加え、混合、混錬しつつ加熱して水分を蒸発させ、押出し可能なペースト状とし、これを押出し成形機でハニカム状等に成形した後、乾燥し空気中にて高温(好ましくは200〜600℃)で焼成する方法等が挙げられる。また、別の方法として、前記チタン系複合酸化物の粉末を予め球状、円柱状のペレット、格子状のハニカムなどの形に成形し、焼成した後、添加しようとする活性種源(バナジウム源、タングステン源、モリブデン源のうちの1種以上)を含む水溶液を含浸させる方法も採用することができる。また、前記チタン系複合酸化物の粉末を、添加しようとする活性種の酸化物(バナジウム酸化物、タングステン酸化物、モリブデン酸化物のうちの1種以上)の粉体と直接混練する方法で調製することもできる。 When the denitration catalyst is obtained by adding the active species (V, W, Mo) to the titanium composite oxide, as a method of adding V or V and W · Mo to the titanium composite oxide, For example, an aqueous solution containing an active species source (one or more of a vanadium source, a tungsten source, and a molybdenum source) to be added to the titanium composite oxide powder is generally formed of this type. It is added together with organic or inorganic forming aids used in the process of mixing, mixing and kneading and heating to evaporate the water, forming an extrudable paste, and forming this into a honeycomb or the like with an extruder The method of drying and baking at high temperature (preferably 200-600 degreeC) in the air etc. are mentioned. As another method, the titanium-based composite oxide powder is previously formed into a spherical, cylindrical pellet, lattice-shaped honeycomb, and the like, fired, and then an active species source (vanadium source, A method of impregnating an aqueous solution containing one or more of a tungsten source and a molybdenum source can also be employed. Further, the titanium composite oxide powder is prepared by directly kneading with a powder of the active species oxide (one or more of vanadium oxide, tungsten oxide, molybdenum oxide) to be added. You can also
前記脱硝触媒が前記チタン系複合酸化物に前記活性種(V、W、Mo)を添加したものである場合、前記V、W、Moの各供給源は、特に制限されない。例えば、バナジウム源(V)としては、バナジウム酸化物のほか、焼成によってバナジウム酸化物を生成するものであれば、無機および有機のいずれの化合物も用いることができ、具体的には、バナジウムを含む水酸化物、アンモニウム塩、蓚酸塩、ハロゲン化物、硫酸塩などを用いることができる。タングステン(W)源としては、例えば、酸化タングステン、パラタングステン酸アンモニウム、メタタングステン酸アンモニウム、タングステン酸などから適宜選択して用いることができる。モリブデン源(Mo)としては、焼成によりモリブデン酸化物を生成するものであれば、無機および有機のいずれの化合物でもよく、例えば、モリブデンを含む酸化物、水酸化物、アンモニウム塩、ハロゲン化物などから適宜用いることができ、具体的には、パラモリブデン酸アンモニウム、モリブデン酸等が挙げられる。 When the denitration catalyst is obtained by adding the active species (V, W, Mo) to the titanium-based composite oxide, the supply sources of the V, W, and Mo are not particularly limited. For example, as the vanadium source (V), in addition to the vanadium oxide, any inorganic and organic compounds can be used as long as they generate vanadium oxide by firing, and specifically include vanadium. Hydroxides, ammonium salts, oxalates, halides, sulfates, and the like can be used. As the tungsten (W) source, for example, tungsten oxide, ammonium paratungstate, ammonium metatungstate, tungstic acid and the like can be appropriately selected and used. The molybdenum source (Mo) may be any inorganic or organic compound as long as it generates molybdenum oxide by firing. For example, from molybdenum-containing oxide, hydroxide, ammonium salt, halide, etc. It can be used as appropriate, and specific examples include ammonium paramolybdate, molybdic acid and the like.
前記脱硝触媒の形状は、特に限定されるものではなく、ハニカム状、板状、網状、円柱状、円筒状など所望の形状に成形して使用することができる。
前記脱硝触媒は、水銀圧入法により測定される全細孔容積が0.20〜0.80cm3/gであることが重要である。前記脱硝触媒の全細孔容積が前記範囲であることにより、水銀(金属水銀やハロゲン化水銀)をも含む排ガスを脱硝処理に供しても、脱硝触媒に触媒毒となる水銀が蓄積することを抑制することができ、長期にわたって良好な脱硝性能を維持することができるのである。前記脱硝触媒の水銀圧入法により測定される全細孔容積は、好ましくは0.25〜0.75cm3/g、より好ましくは0.30〜0.60cm3/gであるのがよい。
The shape of the denitration catalyst is not particularly limited, and can be used after being formed into a desired shape such as a honeycomb shape, a plate shape, a net shape, a columnar shape, or a cylindrical shape.
It is important that the denitration catalyst has a total pore volume of 0.20 to 0.80 cm 3 / g as measured by mercury porosimetry. When the total pore volume of the denitration catalyst is within the above range, even if exhaust gas containing mercury (metal mercury or mercury halide) is subjected to denitration treatment, mercury that becomes a catalyst poison accumulates in the denitration catalyst. It can be suppressed and good denitration performance can be maintained over a long period of time. The total pore volume measured by the mercury intrusion method of the denitration catalyst is preferably 0.25 to 0.75 cm 3 / g, more preferably 0.30 to 0.60 cm 3 / g.
前記脱硝触媒のBET比表面積は、特に制限されないが、20〜300m2/gであることが好ましく、より好ましくは30〜250m2/gである。
前記脱硝処理における処理温度、すなわち前記脱硝触媒の触媒温度は、300℃よりも高い温度、すなわち300℃を超える温度とすることが重要である。これにより、高い脱硝効率を得ることができるのである。前記脱硝触媒の触媒温度は、好ましくは310〜550℃、より好ましくは310〜500℃、さらに好ましくは330〜500℃とするのがよい。
前記脱硝処理における排ガスの空間速度は、特に制限されないが、100〜100000Hr−1が好ましく、より好ましくは200〜50000Hr−1である。
The BET specific surface area of the denitration catalyst is not particularly limited, but is preferably 20 to 300 m 2 / g, more preferably 30 to 250 m 2 / g.
It is important that the treatment temperature in the denitration treatment, that is, the catalyst temperature of the denitration catalyst is higher than 300 ° C., that is, a temperature exceeding 300 ° C. Thereby, high denitration efficiency can be obtained. The catalyst temperature of the denitration catalyst is preferably 310 to 550 ° C, more preferably 310 to 500 ° C, and still more preferably 330 to 500 ° C.
The space velocity of the exhaust gas in the denitration treatment is not particularly limited but is preferably 100~100000Hr -1, more preferably 200~50000Hr -1.
前記水銀ハロゲン化処理は、詳しくは、排ガス中の金属水銀をハロゲン化合物の存在下でハロゲン化水銀に変化させる反応(以下「水銀ハロゲン化反応」と称することもある。)を行なう処理である。前記ハロゲン化合物としては、HCl、HBr等が挙げられ、前記水銀ハロゲン化反応において、金属水銀は、ハロゲン化合物がHClである場合には塩化水銀に、ハロゲン化合物がHBrである場合は臭化水銀に、変換されることになる。例えば、塩化水銀への変換反応は、具体的には、前記(1)式に示される平衡反応を右へ移動させるものである。なお、水銀ハロゲン化処理は、水銀ハロゲン化触媒を備えた水銀ハロゲン化装置に排ガスを通すことによって行なう。 More specifically, the mercury halogenation treatment is a treatment for carrying out a reaction (hereinafter sometimes referred to as “mercury halogenation reaction”) in which metallic mercury in exhaust gas is changed to mercury halide in the presence of a halogen compound. Examples of the halogen compound include HCl and HBr. In the mercury halogenation reaction, metallic mercury is converted to mercury chloride when the halogen compound is HCl, and to mercury bromide when the halogen compound is HBr. Will be converted. For example, in the conversion reaction to mercury chloride, specifically, the equilibrium reaction represented by the formula (1) is moved to the right. The mercury halogenation treatment is performed by passing exhaust gas through a mercury halogenation apparatus equipped with a mercury halogenation catalyst.
前記水銀ハロゲン化反応において、前記ハロゲン化合物の量は、化学量論量以上、すなわち排ガス中の金属水銀1モルに対して2モル以上とすることが好ましい。なお、通常、排ガス中にはガス状ハロゲン化合物が存在していることが多く、処理に供する排ガスが前記範囲の量のガス状ハロゲン化合物を含む場合には、水銀ハロゲン化処理は、水銀ハロゲン化触媒を備えた水銀ハロゲン化装置に排ガスを通すことによって行なうことができる。一方、処理に供する排ガスがガス状ハロゲン化合物を含まない場合もしくは含む場合であってもその量が前記範囲に満たない場合には、水銀ハロゲン化処理は、水銀ハロゲン化触媒を備えた水銀ハロゲン化装置にハロゲン化合物をガス状もしくは液状で供給した状態で排ガスを通すことによって行なうようにすればよい。 In the mercury halogenation reaction, the amount of the halogen compound is preferably a stoichiometric amount or more, that is, 2 mol or more with respect to 1 mol of metallic mercury in the exhaust gas. Normally, gaseous halogen compounds are often present in the exhaust gas, and when the exhaust gas to be treated contains gaseous halogen compounds in the above-mentioned range, mercury halogenation treatment is This can be done by passing the exhaust gas through a mercury halogenator equipped with a catalyst. On the other hand, even if the exhaust gas to be treated does not contain or contain a gaseous halogen compound, if the amount is less than the above range, the mercury halogenation treatment is a mercury halogenation equipped with a mercury halogenation catalyst. What is necessary is just to make it carry out by letting exhaust gas pass in the state which supplied the halogen compound in gaseous or liquid state to the apparatus.
前記水銀ハロゲン化触媒としては、TiO2、SiO2、ZrO2、Al2O3、WO3、MoO3、チタン系複合酸化物、ゼオライトからなる群より選ばれる1種以上(以下「金属酸化物類A」と称する)に、Pt、Ru、Rh、Pd、Ir、V、W、Mo、Ni、Co、Fe、Cr、Cu、Mnのうちの少なくとも1種からなる活性種を添加した触媒が好ましく挙げられる。
前記水銀ハロゲン化触媒は、前記金属酸化物類Aとして、チタン系複合酸化物を用いた触媒であることが、安定な構造を維持でき、水銀に対する高い耐性を発揮しうる点で好ましい。前記チタン系複合酸化物としては、ケイ素、ジルコニウム、アルミニウム、タングステンおよびモリブデンからなる群より選ばれる1種または2種とチタンとの二元系または三元系複合酸化物が好ましく、具体的には、例えば、Ti−Si、Ti−Zr、Ti−Al、Ti−W、Ti−Mo等の二元系複合酸化物;Ti−Si−Mo、Ti−Si−W、Ti−Si−Zr、Ti−Si−Al、Ti−Zr−Al、Ti−Zr−Mo、Ti−Zr−W、Ti−Al−Mo、Ti−W−Mo等の三元系複合酸化物;等が挙げられる。これらの中でも、Ti−Moの二元系複合酸化物や、Ti−Si−Mo、Ti−Zr−Mo、、Ti−Al−Mo、Ti−W−Moのような三元系複合酸化物など、モリブデン(Mo)を必須とする遷移金属とチタン(Ti)との二元系または三元系複合酸化物が、水銀ハロゲン化活性がより高いことから、より好ましい。なお、ここで、複合酸化物とは、X線回折パターンにおいてTiO2以外の物質に帰属される明らかな固有のピークを示さず、TiO2についてはアナターゼ型酸化チタンに帰属される固有のピークを示さないか、もしくは示してもアナターゼ型酸化チタンの回折ピークよりもブロードな回折ピークを示すものを言う。
The mercury halogenation catalyst may be one or more selected from the group consisting of TiO 2 , SiO 2 , ZrO 2 , Al 2 O 3 , WO 3 , MoO 3 , titanium-based composite oxide, and zeolite (hereinafter “metal oxide”). A catalyst having an active species composed of at least one of Pt, Ru, Rh, Pd, Ir, V, W, Mo, Ni, Co, Fe, Cr, Cu, and Mn. Preferably mentioned.
The mercury halogenation catalyst is preferably a catalyst using a titanium-based composite oxide as the metal oxides A in terms of maintaining a stable structure and exhibiting high resistance to mercury. The titanium-based composite oxide is preferably a binary or ternary composite oxide of titanium and one or two selected from the group consisting of silicon, zirconium, aluminum, tungsten, and molybdenum. Specifically, For example, binary complex oxides such as Ti-Si, Ti-Zr, Ti-Al, Ti-W, Ti-Mo; Ti-Si-Mo, Ti-Si-W, Ti-Si-Zr, Ti Ternary complex oxides such as -Si-Al, Ti-Zr-Al, Ti-Zr-Mo, Ti-Zr-W, Ti-Al-Mo, Ti-W-Mo; and the like. Among these, Ti-Mo binary complex oxides, ternary complex oxides such as Ti-Si-Mo, Ti-Zr-Mo, Ti-Al-Mo, Ti-W-Mo, etc. A binary or ternary composite oxide of a transition metal essentially containing molybdenum (Mo) and titanium (Ti) is more preferable because of its higher mercury halogenation activity. Here, the composite oxide exhibited no apparent specific peak attributed to substance other than TiO 2 in the X-ray diffraction pattern, a unique peak for TiO 2 is attributable to anatase titanium oxide It means not showing or showing a broader diffraction peak than that of anatase-type titanium oxide.
前記水銀ハロゲン化触媒における前記金属酸化物類Aがチタン系複合酸化物である場合、該チタン系複合酸化物における各元素の組成は、特に制限されるものではないが、例えば、チタン系複合酸化物に占めるチタン(Ti)以外の元素(Si、Zr、Al、W、Moなど)の含有量が、各元素の酸化物換算重量比で、それぞれ、0.5〜30重量%であることが好ましく、1〜30重量%であることがより好ましく、1〜25重量%であることがさらに好ましい。
前記水銀ハロゲン化触媒における前記金属酸化物類Aがチタン系複合酸化物である場合、前記チタン系複合酸化物の調製方法としては、特に制限はなく、例えば、沈殿法(共沈法)、沈着法、混練法などの従来公知の方法を採用することができる。例えば、Ti−Moであれば、パラモリブデン酸アンモニウムやモリブデン酸等のモリブデン化合物をアンモニア水溶液に分散させて水溶液(B)を得、この水溶液(B)に攪拌下でチタン化合物の水溶液を徐々に滴下し、得られたスラリーを濾過、洗浄し、さらに乾燥した後、300〜600℃の高温で焼成することにより、得ることができる。また、例えば、Ti−Si−Mo、Ti−Zr−Mo、Ti−Al−Mo、Ti−W−Moであれば、前記水溶液(B)に、さらにSi、Zr、Al、Wなどの塩の水溶液を加え、得られた水溶液に攪拌下でチタン化合物の水溶液を徐々に滴下し、得られたスラリーを濾過、洗浄し、さらに乾燥した後、300〜600℃の高温で焼成することにより、得ることができる。
When the metal oxide A in the mercury halogenated catalyst is a titanium composite oxide, the composition of each element in the titanium composite oxide is not particularly limited. The content of elements (Si, Zr, Al, W, Mo, etc.) other than titanium (Ti) in the material is 0.5 to 30% by weight in terms of the oxide equivalent weight ratio of each element. Preferably, it is 1-30 weight%, More preferably, it is 1-25 weight%.
When the metal oxide A in the mercury halogenation catalyst is a titanium composite oxide, the method for preparing the titanium composite oxide is not particularly limited. For example, precipitation (coprecipitation), deposition Conventionally known methods such as a method and a kneading method can be employed. For example, in the case of Ti—Mo, a molybdenum compound such as ammonium paramolybdate or molybdic acid is dispersed in an aqueous ammonia solution to obtain an aqueous solution (B). The aqueous solution of the titanium compound is gradually added to the aqueous solution (B) with stirring. The slurry is added dropwise, and the resulting slurry is filtered, washed, dried, and then fired at a high temperature of 300 to 600 ° C. to obtain the slurry. Further, for example, in the case of Ti—Si—Mo, Ti—Zr—Mo, Ti—Al—Mo, and Ti—W—Mo, the aqueous solution (B) is further added with a salt such as Si, Zr, Al, or W. An aqueous solution is added, and an aqueous solution of a titanium compound is gradually added dropwise to the obtained aqueous solution with stirring. The obtained slurry is filtered, washed, dried, and then fired at a high temperature of 300 to 600 ° C. be able to.
前記水銀ハロゲン化触媒における前記金属酸化物類Aがチタン系複合酸化物である場合、前記チタン系複合酸化物を調製する際に用いる各元素の供給源は、特に制限されない。例えば、チタン(Ti)源としては、無機および有機のいずれの化合物も使用可能であり、例えば、四塩化チタン、硫酸チタンなどの無機チタン化合物または蓚酸チタン、テトライソプロピルチタネートなどの有機チタン化合物を用いることができる。ケイ素(Si)源としては、コロイド状シリカ、水ガラス、微粒子ケイ素、四塩化ケイ素、シリカゲルなどの無機ケイ素化合物およびテトラエチルシリケートなどの有機ケイ素化合物から適宜選択して使用することができる。ジルコニウム(Zr)源としては、例えば、塩化ジルコニウム、硫酸ジルコニウムなどの無機系ジルコニウム化合物や、シュウ酸ジウコニウムなどの有機系ジルコニウム化合物から適宜選択して使用することができる。アルミニウム(Al)源としては、例えば、硝酸アルミニウム、硫酸アルミニウムなどの無機系アルミニウム化合物や、酢酸アルミニウムなどの有機系アルミニウム化合物から適宜選択して使用することができる。タングステン(W)源としては、例えば、酸化タングステン、パラタングステン酸アンモニウム、メタタングステン酸アンモニウム、タングステン酸などから適宜選択して使用することができる。モリブデン(Mo)源としては、無機および有機のいずれの化合物でもよく、例えば、モリブデンを含む酸化物、水酸化物、アンモニウム塩、ハロゲン化物などから適宜用いることができ、具体的には、パラモリブデン酸アンモニウム、モリブデン酸等が挙げられる。 When the metal oxides A in the mercury halogenated catalyst are titanium-based composite oxides, the supply source of each element used when preparing the titanium-based composite oxide is not particularly limited. For example, as the titanium (Ti) source, both inorganic and organic compounds can be used. For example, inorganic titanium compounds such as titanium tetrachloride and titanium sulfate or organic titanium compounds such as titanium oxalate and tetraisopropyl titanate are used. be able to. The silicon (Si) source can be appropriately selected from inorganic silicon compounds such as colloidal silica, water glass, fine particle silicon, silicon tetrachloride, and silica gel, and organic silicon compounds such as tetraethyl silicate. As the zirconium (Zr) source, for example, an inorganic zirconium compound such as zirconium chloride or zirconium sulfate, or an organic zirconium compound such as diuconium oxalate can be appropriately selected and used. As the aluminum (Al) source, for example, an inorganic aluminum compound such as aluminum nitrate or aluminum sulfate or an organic aluminum compound such as aluminum acetate can be appropriately selected and used. As the tungsten (W) source, for example, tungsten oxide, ammonium paratungstate, ammonium metatungstate, tungstic acid and the like can be appropriately selected and used. As the molybdenum (Mo) source, any of inorganic and organic compounds may be used. For example, oxides, hydroxides, ammonium salts, halides and the like containing molybdenum can be used as appropriate. Specifically, para-molybdenum can be used. Examples thereof include ammonium acid and molybdic acid.
また、前記水銀ハロゲン化触媒は、前記活性種の中でも、バナジウム(V)を添加した触媒であることが、水銀に対する耐性が高い点で、特に好ましい。
前記水銀ハロゲン化触媒における前記バナジウム(V)の含有量は、特に限定されないが、例えば、水銀ハロゲン化触媒の全重量に対し、酸化物換算重量比で、0.1〜25重量%であることが好ましく、1〜20重量%であることがより好ましい。
前記水銀ハロゲン化触媒が前記金属酸化物類Aにバナジウム(V)を添加したものである場合、バナジウム(V)を添加する方法としては、特に限定されず、例えば、前記金属酸化物類Aの粉末に、バナジウム源を含む水溶液を、一般にこの種の成形を行う際に用いられる有機または無機の成形助剤とともに加え、混合、混錬しつつ加熱して水分を蒸発させ、押出し可能なペースト状とし、これを押出し成形機でハニカム状等に成形した後、乾燥し空気中にて高温(好ましくは200〜600℃)で焼成する方法等が挙げられる。また、別の方法として、前記金属酸化物類Aの粉末を予め球状、円柱状のペレット、格子状のハニカムなどの形に成形し、焼成した後、バナジウム源を含む水溶液を含浸させる方法も採用することができる。また、前記金属酸化物類Aの粉末を、バナジウム酸化物の粉体と直接混練する方法で調製することもできる。
In addition, the mercury halogenation catalyst is particularly preferably a catalyst to which vanadium (V) is added among the active species, from the viewpoint of high resistance to mercury.
The content of the vanadium (V) in the mercury halogenation catalyst is not particularly limited, but is, for example, 0.1 to 25% by weight in terms of oxide relative to the total weight of the mercury halogenation catalyst. Is preferable, and it is more preferable that it is 1 to 20 weight%.
When the mercury halogenation catalyst is obtained by adding vanadium (V) to the metal oxides A, the method for adding vanadium (V) is not particularly limited. An aqueous solution containing a vanadium source is added to the powder, together with organic or inorganic molding aids generally used for this type of molding, and mixed and kneaded to heat and evaporate the moisture, allowing the paste to be extruded. And a method of forming this into a honeycomb or the like with an extrusion molding machine, drying and firing in air at a high temperature (preferably 200 to 600 ° C.). As another method, the powder of the metal oxides A is previously formed into a spherical or cylindrical pellet, a lattice-shaped honeycomb, and fired, and then impregnated with an aqueous solution containing a vanadium source. can do. Alternatively, the metal oxides A powder can be prepared by directly kneading with the vanadium oxide powder.
前記水銀ハロゲン化触媒が前記金属酸化物類Aにバナジウム(V)を添加したものである場合、バナジウム源としては、特に制限されないが、例えば、バナジウム酸化物のほか、焼成によってバナジウム酸化物を生成するものであれば、無機および有機のいずれの化合物も用いることができ、具体的には、バナジウムを含む水酸化物、アンモニウム塩、蓚酸塩、ハロゲン化物、硫酸塩などを用いることができる。
前記水銀ハロゲン化触媒の形状は、特に限定されるものではなく、ハニカム状、板状、網状、円柱状、円筒状など所望の形状に成形して使用することができる。
前記水銀ハロゲン化触媒の全細孔容積は、特に制限されないが、水銀圧入法により測定される全細孔容積が、0.20〜0.80cm3/gであることが好ましく、より好ましくは0.25〜0.80cm3/g、さらに好ましくは0.25〜0.70cm3/gであるのがよい。水銀ハロゲン化触媒の水銀圧入法により測定される全細孔容積が0.20cm3/g未満であると、触媒内部へのガスの拡散が不充分になり、金属水銀をハロゲン化水銀に変換する反応が効率よく進行させることができず、その結果、水銀の除去効率が低下することになる。一方、水銀ハロゲン化触媒の全細孔容積が0.80cm3/gを超えると、触媒の機械的強度が低下して僅かな衝撃で形状がくずれやすく、触媒としての使用に耐えないこととなる。
When the mercury halogenation catalyst is obtained by adding vanadium (V) to the metal oxides A, the vanadium source is not particularly limited. For example, in addition to vanadium oxide, vanadium oxide is generated by firing. Any inorganic and organic compounds can be used as long as they are used. Specifically, hydroxides containing ammonium, ammonium salts, oxalates, halides, sulfates, and the like can be used.
The shape of the mercury halogenation catalyst is not particularly limited, and can be used after being formed into a desired shape such as a honeycomb shape, a plate shape, a net shape, a columnar shape, or a cylindrical shape.
The total pore volume of the mercury halogenated catalyst is not particularly limited, but the total pore volume measured by mercury porosimetry is preferably 0.20 to 0.80 cm 3 / g, more preferably 0. .25 to 0.80 cm 3 / g, more preferably 0.25 to 0.70 cm 3 / g. If the total pore volume of the mercury halogenated catalyst measured by the mercury intrusion method is less than 0.20 cm 3 / g, gas diffusion into the catalyst becomes insufficient, and metallic mercury is converted to mercury halide. The reaction cannot proceed efficiently, and as a result, the mercury removal efficiency decreases. On the other hand, when the total pore volume of the mercury halogenated catalyst exceeds 0.80 cm 3 / g, the mechanical strength of the catalyst is lowered, and the shape is easily deformed by a slight impact, so that it cannot be used as a catalyst. .
前記水銀ハロゲン化触媒のBET比表面積は、特に制限されないが、20〜400m2/gであることが好ましく、より好ましくは30〜350m2/gである。
前記水銀ハロゲン化処理における処理温度、すなわち前記水銀ハロゲン化触媒の触媒温度は、300℃以下とすることが重要である。これにより、水銀ハロゲン化反応を効率よく進行させることができるのである。前記水銀ハロゲン化触媒の触媒温度は、好ましくは60〜300℃とすることが好ましく、より好ましくは100〜300℃、さらに好ましくは100〜280℃とするのがよい。
前記水銀ハロゲン化処理における排ガスの空間速度は、特に制限されないが、100〜90000Hr−1が好ましく、より好ましくは200〜50000Hr−1である。100Hr−1未満では、処理装置が大きくなりすぎるため非効率となり、一方、90000Hr−1を超えると、ハロゲン化水銀への変換反応の効率が低下する。
BET specific surface area of the mercury halogenating catalyst is not particularly limited, is preferably from 20 to 400 m 2 / g, more preferably 30~350m 2 / g.
It is important that the treatment temperature in the mercury halogenation treatment, that is, the catalyst temperature of the mercury halogenation catalyst is 300 ° C. or less. Thereby, mercury halogenation reaction can be advanced efficiently. The catalyst temperature of the mercury halogenation catalyst is preferably 60 to 300 ° C, more preferably 100 to 300 ° C, still more preferably 100 to 280 ° C.
The space velocity of the exhaust gas in the mercury halogenation treatment is not particularly limited, but is preferably 100 to 90000 Hr −1 , more preferably 200 to 50000 Hr −1 . If it is less than 100 Hr −1 , the processing apparatus becomes too large to be inefficient. On the other hand, if it exceeds 90000 Hr −1 , the efficiency of the conversion reaction to mercury halide decreases.
本発明の排ガス処理方法においては、前記水銀ハロゲン化処理で生じたハロゲン化水銀を吸収液で捕捉することによって除去するようにすることが好ましい。ハロゲン化水銀は、吸収液中に溶解させることが容易であり、吸収液に溶解させることで非ガス中から除去することができる。前記吸収液としては、例えば、水、アルカリ水溶液(具体的には、炭酸カルシウム水溶液、水酸化ナトリウム水溶液、炭酸ナトリウム水溶液など)等を用いればよい。
本発明の排ガス処理方法においては、吸収液に吸収された水銀(ハロゲン化水銀)を回収し、回収した水銀を資源として再利用することが好ましい。具体的には、例えば、ハロゲン化水銀を吸収させたのちの吸収液を加熱して水銀蒸気を発生させ、これを急冷することにより、水銀を回収することができる。
In the exhaust gas treatment method of the present invention, it is preferable to remove the mercury halide generated by the mercury halogenation treatment by capturing it with an absorbing solution. Mercury halide can be easily dissolved in the absorbing solution, and can be removed from the non-gas by dissolving in the absorbing solution. As the absorbing solution, for example, water, an aqueous alkali solution (specifically, an aqueous calcium carbonate solution, an aqueous sodium hydroxide solution, an aqueous sodium carbonate solution, etc.) may be used.
In the exhaust gas treatment method of the present invention, it is preferable to recover mercury (mercury halide) absorbed in the absorbing solution and reuse the recovered mercury as a resource. Specifically, for example, mercury can be recovered by heating the absorbing solution after absorbing mercury halide to generate mercury vapor and quenching it.
本発明の排ガス処理方法において、処理に供する排ガスは、少なくとも窒素酸化物および金属水銀を含むものであるが、勿論、例えばハロゲン化水銀など、窒素酸化物および金属水銀以外の成分(化合物)を含むものであってもよい。
本発明の排ガス処理方法においては、処理に供する排ガス中の水銀(金属水銀およびハロゲン化水銀)濃度は100mg/m3N以下であることが好ましく、50mg/m3N以下であることがより好ましく、40mg/m3N以下であることがさらに好ましい。なお、一般に、排ガス中に存在する除去対象物の濃度が低すぎると、除去効果が充分に認められないことがあるが、本発明の排ガス処理方法においては、10μg/m3N以下のような極めて低い水銀(金属水銀およびハロゲン化水銀)濃度であっても、充分に除去効果を発揮することができる。
In the exhaust gas treatment method of the present invention, the exhaust gas to be used for treatment contains at least nitrogen oxides and metal mercury. Of course, for example, it contains components (compounds) other than nitrogen oxides and metal mercury such as mercury halides. There may be.
In the exhaust gas treatment method of the present invention, the concentration of mercury (metal mercury and mercury halide) in the exhaust gas to be treated is preferably 100 mg / m 3 N or less, more preferably 50 mg / m 3 N or less. 40 mg / m 3 N or less is more preferable. In general, if the concentration of the removal target present in the exhaust gas is too low, the removal effect may not be sufficiently observed. However, in the exhaust gas treatment method of the present invention, the concentration is 10 μg / m 3 N or less. Even at very low mercury (metal mercury and mercury halide) concentrations, a sufficient removal effect can be exhibited.
以下に、実施例により、本発明をさらに具体的に説明するが、本発明はこれらにより何ら限定されるものではない。
〔製造例1−1−脱硝触媒(1)の製造〕
まず、Ti−Si複合酸化物を次のように調製した。シリカゾル(「スノーテックス−30」日産化学社製、SiO2換算30wt%含有)10kg、工業用アンモニア水(25wt%NH3含有)104kg、および水73リットルを混合し、均一溶液を調製した。この溶液に、硫酸チタニルの硫酸溶液(テイカ社製:TiO2として70g/リットル、H2SO4として287g/リットル含有)243リットルを撹拌しながら徐々に滴下した。得られたスラリーを20時間静置したのち、濾過し、水で充分洗浄した後、続いて150℃で1時間乾燥した。さらに、空気雰囲気下、550℃で5時間焼成し、Ti−Si複合酸化物粉体を得た。該Ti−Si複合酸化物粉体の組成は、酸化物換算重量比で、チタン酸化物:ケイ素酸化物=85:15であった。
Hereinafter, the present invention will be described more specifically by way of examples. However, the present invention is not limited to these examples.
[Production Example 1-1 Production of DeNOx Catalyst (1)]
First, a Ti—Si composite oxide was prepared as follows. Silica sol ( "Snowtex -30" manufactured by Nissan Chemical Industries, Ltd., SiO 2 in terms of 30 wt% containing) 10 kg, industrial aqueous ammonia (25 wt% NH 3 containing) 104 kg, and water were mixed 73 liters to prepare a homogeneous solution. To this solution, 243 liters of a sulfuric acid solution of titanyl sulfate (manufactured by Teika Corporation: 70 g / liter as TiO 2 and 287 g / liter as H 2 SO 4 ) was gradually added dropwise with stirring. The obtained slurry was allowed to stand for 20 hours, filtered, sufficiently washed with water, and then dried at 150 ° C. for 1 hour. Further, it was fired at 550 ° C. for 5 hours in an air atmosphere to obtain a Ti—Si composite oxide powder. The composition of the Ti—Si composite oxide powder was titanium oxide: silicon oxide = 85: 15 in terms of weight ratio in terms of oxide.
次いで、上記Ti−Si複合酸化物にバナジウムとタングステンを次のようにして添加した。8リットルの水に、メタバナジン酸アンモニウム1.29kg、パラタングステン酸アンモニウム1.12kg、シュウ酸1.67kg、およびモノエタノールアミン0.85kgを混合して溶解させ、均一なバナジウムおよびタングステン含有溶液を調製した。上記で得られたTi−Si複合酸化物粉体18kgをニーダーに投入後、有機バインダー(デンプン1.5kg)を含む成形助材とともに上記バナジウムおよびタングステン含有溶液全量を加え、よく攪拌した。さらに適量の水を加えつつブレンダーでよく混合した後、連続ニーダーで充分混練りし、ハニカム状に押し出し成形した。得られた成形物を60℃で乾燥後、450℃で5時間焼成して、脱硝触媒(1)を得た。 Next, vanadium and tungsten were added to the Ti—Si composite oxide as follows. Mix and dissolve 1.29 kg of ammonium metavanadate, 1.12 kg of ammonium paratungstate, 1.67 kg of oxalic acid, and 0.85 kg of monoethanolamine in 8 liters of water to prepare a uniform vanadium and tungsten-containing solution. did. After putting 18 kg of the Ti-Si composite oxide powder obtained above into a kneader, the above vanadium and tungsten-containing solution was added together with a molding aid containing an organic binder (starch 1.5 kg) and stirred well. Furthermore, after adding a proper amount of water and mixing well with a blender, the mixture was sufficiently kneaded with a continuous kneader and extruded into a honeycomb shape. The obtained molded product was dried at 60 ° C. and then calcined at 450 ° C. for 5 hours to obtain a denitration catalyst (1).
得られた脱硝触媒(1)の組成は、酸化物換算重量比で、Ti−Si複合酸化物:バナジウム酸化物:タングステン酸化物=90:5:5(酸化物換算重量比で、チタン酸化物:ケイ素酸化物:バナジウム酸化物:タングステン酸化物=76.5:13.5:5:5)であり、水銀圧入法により測定した全細孔容積は0.45cm3/gであった。
なお、脱硝触媒(1)のX線回折パターンを図1に示す。図1において、TiO2以外の物質に帰属される明らかな固有のピークは認められず、かつ、アナターゼ型酸化チタンに帰属されるブロードなピークが認められることから、脱硝触媒(1)は複合酸化物であることが確認できた。参考として、TiO2のX線回折パターンを図7に示す(なお、以下の製造例でも該図7を参考とする)。
The composition of the obtained denitration catalyst (1) is a weight ratio in terms of oxide, Ti—Si composite oxide: vanadium oxide: tungsten oxide = 90: 5: 5 (weight ratio in terms of oxide, titanium oxide) : Silicon oxide: vanadium oxide: tungsten oxide = 76.5: 13.5: 5: 5), and the total pore volume measured by mercury porosimetry was 0.45 cm 3 / g.
The X-ray diffraction pattern of the denitration catalyst (1) is shown in FIG. In FIG. 1, no obvious intrinsic peak attributed to substances other than TiO 2 is observed, and a broad peak attributed to anatase-type titanium oxide is observed, so that the denitration catalyst (1) is complex oxidized. It was confirmed to be a thing. For reference, an X-ray diffraction pattern of TiO 2 is shown in FIG. 7 (note that FIG. 7 is also referred to in the following production examples).
〔製造例1−2−脱硝触媒(2)の製造〕
製造例1−1と同様にしてTi−Si複合酸化物を得たのち、パラタングステン酸アンモニウム1.12kgの代わりにパラモリブデン酸アンモニウム1.23kgを用いたこと以外は製造例1−1と同様にして、Ti−Si複合酸化物にバナジウムとモリブデンを添加した脱硝触媒(2)を得た。
得られた脱硝触媒(2)の組成は、酸化物換算重量比で、Ti−Si複合酸化物:バナジウム酸化物:モリブデン酸化物=90:5:5(酸化物換算重量比で、チタン酸化物:ケイ素酸化物:バナジウム酸化物:モリブデン酸化物=76.5:13.5:5:5)であり、水銀圧入法により測定した全細孔容積は0.44cm3/gであった。
[Production Example 1-2 Production of Denitration Catalyst (2)]
Similar to Production Example 1-1, after obtaining a Ti—Si composite oxide in the same manner as in Production Example 1-1, except that 1.23 kg of ammonium paramolybdate was used instead of 1.12 kg of ammonium paratungstate. Thus, a denitration catalyst (2) in which vanadium and molybdenum were added to the Ti—Si composite oxide was obtained.
The composition of the obtained denitration catalyst (2) is an oxide equivalent weight ratio, Ti—Si composite oxide: vanadium oxide: molybdenum oxide = 90: 5: 5 (oxide equivalent weight ratio, titanium oxide : Silicon oxide: vanadium oxide: molybdenum oxide = 76.5: 13.5: 5: 5), and the total pore volume measured by mercury porosimetry was 0.44 cm 3 / g.
なお、脱硝触媒(2)のX線回折パターンを図2に示す。図2において、TiO2以外の物質に帰属される明らかな固有のピークは認められず、かつ、アナターゼ型酸化チタンに帰属されるブロードなピークが認められることから、脱硝触媒(2)は複合酸化物であることが確認できた。
〔製造例1−3−脱硝触媒(3)の製造〕
市販のチタン酸化物粉体にバナジウムとタングステンを次のようにして添加した。8リットルの水に、メタバナジン酸アンモニウム1.29kg、パラタングステン酸アンモニウム1.12kg、シュウ酸1.67kg、およびモノエタノールアミン0.85kgを混合して溶解させ、均一なバナジウムおよびタングステン含有溶液を調製した。市販のチタン酸化物粉体(「DT−51」ミレニアム社製)18kgをニーダーに投入後、有機バインダー(デンプン1.5kg)を含む成形助材とともに上記バナジウムおよびタングステン含有溶液全量を加え、よく攪拌した。さらに適量の水を加えつつブレンダーでよく混合した後、連続ニーダーで充分混練りし、ハニカム状に押し出し成形した。得られた成形物を60℃で乾燥後、450℃で5時間焼成して、脱硝触媒(3)を得た。
An X-ray diffraction pattern of the denitration catalyst (2) is shown in FIG. In FIG. 2, no obvious intrinsic peak attributed to substances other than TiO 2 is observed, and a broad peak attributed to anatase-type titanium oxide is observed, so that the denitration catalyst (2) is complex oxidized. It was confirmed to be a thing.
[Production Example 1-3 Production of Denitration Catalyst (3)]
Vanadium and tungsten were added to commercially available titanium oxide powder as follows. Mix and dissolve 1.29 kg of ammonium metavanadate, 1.12 kg of ammonium paratungstate, 1.67 kg of oxalic acid, and 0.85 kg of monoethanolamine in 8 liters of water to prepare a uniform vanadium and tungsten-containing solution. did. After putting 18 kg of commercially available titanium oxide powder (“DT-51” manufactured by Millennium) into a kneader, add the above vanadium and tungsten-containing solution together with a molding aid containing an organic binder (starch 1.5 kg) and stir well. did. Furthermore, after adding a proper amount of water and mixing well with a blender, the mixture was sufficiently kneaded with a continuous kneader and extruded into a honeycomb shape. The obtained molded product was dried at 60 ° C. and then calcined at 450 ° C. for 5 hours to obtain a denitration catalyst (3).
得られた脱硝触媒(3)の組成は、酸化物換算重量比で、チタン酸化物:バナジウム酸化物:タングステン酸化物=90:5:5)であり、水銀圧入法により測定した全細孔容積は0.13cm3/gであった。
なお、脱硝触媒(3)のX線回折パターンを図3に示す。図3において、TiO2以外の物質に帰属される明らかな固有のピークは認められず、かつ、アナターゼ型酸化チタンに帰属されるシャープなピークが認められることから、脱硝触媒(3)はチタン単一酸化物を主とするものであることが確認できた。
〔製造例2−1−水銀ハロゲン化触媒(1)の製造〕
まず、Ti−W複合酸化物を次のように調製した。工業用アンモニア水(25wt%NH3含有)190kgおよび水140リットルの混合溶液に、メタタングステン酸アンモニウム水溶液(日本無機化学工業(株)製:WO3として50wt%含有)20kgを加えてよく攪拌し、均一溶液を調製した。この溶液に、硫酸チタニルの硫酸溶液(テイカ社製:TiO2として70g/リットル、H2SO4として287g/リットル含有)571リットルを攪拌しながら徐々に滴下し、沈殿を生成させた後、適量のアンモニア水を加えてpHを4に調整した。この共沈スラリーを約40時間静置したのち、濾過し、水で充分洗浄した後、150℃で1時間乾燥させた。さらに、空気雰囲気下、500℃で5時間焼成し、Ti−W複合酸化物粉体を得た。該Ti−W複合酸化物粉体の組成は、酸化物換算重量比で、チタン酸化物:タングステン酸化物=80:20であった。
The composition of the obtained denitration catalyst (3) is a weight ratio of oxide, titanium oxide: vanadium oxide: tungsten oxide = 90: 5: 5), and the total pore volume measured by mercury porosimetry. Was 0.13 cm 3 / g.
The X-ray diffraction pattern of the denitration catalyst (3) is shown in FIG. 3, not observed apparent specific peak attributed to substance other than TiO 2, and, since a sharp peak assigned to anatase titanium oxide is observed, the denitration catalyst (3) is Titanium single It was confirmed that it was mainly composed of monoxide .
[Production Example 2-1 Production of Mercury Halogenated Catalyst (1)]
First, a Ti—W composite oxide was prepared as follows. 20 kg of ammonium metatungstate aqueous solution (manufactured by Nippon Inorganic Chemical Industry Co., Ltd .: containing 50 wt% as WO 3 ) is added to a mixed solution of 190 kg of industrial ammonia water (containing 25 wt% NH 3 ) and 140 liters of water and stirred well. A homogeneous solution was prepared. To this solution, 571 liters of a sulfuric acid solution of titanyl sulfate (manufactured by Teika: 70 g / liter as TiO 2 and 287 g / liter as H 2 SO 4 ) was gradually added dropwise with stirring to form a precipitate, and then an appropriate amount Was added to adjust the pH to 4. The coprecipitated slurry was allowed to stand for about 40 hours, filtered, thoroughly washed with water, and dried at 150 ° C. for 1 hour. Further, it was fired at 500 ° C. for 5 hours in an air atmosphere to obtain a Ti—W composite oxide powder. The composition of the Ti—W composite oxide powder was titanium oxide: tungsten oxide = 80: 20 in terms of weight ratio in terms of oxide.
次いで、上記Ti−W複合酸化物にバナジウムを次のようにして添加した。8リットルの水に、メタバナジン酸アンモニウム1.29kg、シュウ酸1.67kg、およびモノエタノールアミン0.4kgを混合して溶解させ、均一なバナジウム含有溶液を調製した。上記で得られたTi−W複合酸化物粉体19kgをニーダーに投入後、有機バインダー(デンプン1.5kg)を含む成形助材とともに上記バナジウム含有溶液全量を加え、よく攪拌した。さらに適量の水を加えつつブレンダーでよく混合した後、連続ニーダーで充分混練りし、ハニカム状に押し出し成形した。得られた成形物を60℃で乾燥後、450℃で5時間焼成して、水銀ハロゲン化触媒(1)を得た。 Next, vanadium was added to the Ti—W composite oxide as follows. A uniform vanadium-containing solution was prepared by mixing and dissolving 1.29 kg of ammonium metavanadate, 1.67 kg of oxalic acid, and 0.4 kg of monoethanolamine in 8 liters of water. After adding 19 kg of the Ti-W composite oxide powder obtained above to a kneader, the above vanadium-containing solution was added together with a molding aid containing an organic binder (starch 1.5 kg) and stirred well. Furthermore, after adding a proper amount of water and mixing well with a blender, the mixture was sufficiently kneaded with a continuous kneader and extruded into a honeycomb shape. The obtained molded product was dried at 60 ° C. and then calcined at 450 ° C. for 5 hours to obtain a mercury halogenated catalyst (1).
得られた水銀ハロゲン化触媒(1)の組成は、酸化物換算重量比で、Ti−W複合酸化物:バナジウム酸化物=95:5(酸化物換算重量比で、チタン酸化物:タングステン酸化物:バナジウム酸化物=76:19:5)であった。
なお、水銀ハロゲン化触媒(1)のX線回折パターンを図4に示す。図4において、TiO2以外の物質に帰属される明らかな固有のピークは認められず、かつ、アナターゼ型酸化チタンに帰属されるブロードなピークが認められることから、水銀ハロゲン化触媒(1)は複合酸化物であることが確認できた。
〔製造例2−2−水銀ハロゲン化触媒(2)の製造〕
まず、Ti−Si−Mo複合酸化物を次のように調製した。シリカゾル(「スノーテックス−30」日産化学社製、SiO2換算30wt%含有)3.3kg、工業用アンモニア水(25wt%NH3含有)103kg、および水58リットルの混合溶液に、モリブデン酸3.4kgを加えてよく攪拌し、均一溶液を調製した。この溶液に、硫酸チタニルの硫酸溶液(テイカ社製:TiO2として70g/リットル、H2SO4として287g/リットル含有)228リットルを攪拌しながら徐々に滴下し、沈殿を生成させた後、適量のアンモニア水を加えてpHを4に調整した。この共沈スラリーを約40時間静置したのち、濾過し、水で充分洗浄した後、100℃で1時間乾燥させた。さらに、空気雰囲気下、500℃で5時間焼成し、Ti−Si−Mo複合酸化物粉体を得た。該Ti−Si−Mo複合酸化物粉体の組成は、酸化物換算重量比で、チタン酸化物:ケイ素酸化物:モリブデン酸化物=80:5:15であった。
The composition of the obtained mercury halogenation catalyst (1) is an oxide equivalent weight ratio, Ti—W composite oxide: vanadium oxide = 95: 5 (oxide equivalent weight ratio, titanium oxide: tungsten oxide). : Vanadium oxide = 76: 19: 5).
The X-ray diffraction pattern of the mercury halogenated catalyst (1) is shown in FIG. In FIG. 4, no obvious intrinsic peak attributed to substances other than TiO 2 is observed, and a broad peak attributed to anatase-type titanium oxide is observed, so that the mercury halogenated catalyst (1) is It was confirmed to be a complex oxide.
[Production Example 2-2 Production of Mercury Halogenated Catalyst (2)]
First, a Ti—Si—Mo composite oxide was prepared as follows. In a mixed solution of 3.3 kg of silica sol (“Snowtex-30” manufactured by Nissan Chemical Co., containing 30 wt% in terms of SiO 2 ), 103 kg of industrial ammonia water (containing 25 wt% NH 3 ), and 58 liters of water, 3. 4 kg was added and stirred well to prepare a homogeneous solution. To this solution, 228 liters of a titanyl sulfate sulfuric acid solution (manufactured by Teika Corporation: 70 g / liter as TiO 2 and 287 g / liter as H 2 SO 4 ) was gradually added dropwise with stirring to form a precipitate, and then an appropriate amount Was added to adjust the pH to 4. The coprecipitated slurry was allowed to stand for about 40 hours, filtered, thoroughly washed with water, and dried at 100 ° C. for 1 hour. Further, it was fired at 500 ° C. for 5 hours in an air atmosphere to obtain a Ti—Si—Mo composite oxide powder. The composition of the Ti—Si—Mo composite oxide powder was titanium oxide: silicon oxide: molybdenum oxide = 80: 5: 15 in terms of weight ratio in terms of oxide.
次いで、上記Ti−Si−Mo複合酸化物にバナジウムを次のようにして添加した。8リットルの水に、メタバナジン酸アンモニウム1.29kg、シュウ酸1.67kg、およびモノエタノールアミン0.4kgを混合して溶解させ、均一なバナジウム含有溶液を調製した。上記で得られたTi−Si−Mo複合酸化物粉体19kgをニーダーに投入後、有機バインダー(デンプン1.5kg)を含む成形助材とともに上記バナジウム含有溶液全量を加え、よく攪拌した。さらに適量の水を加えつつブレンダーでよく混合した後、連続ニーダーで充分混練りし、ハニカム状に押し出し成形した。得られた成形物を60℃で乾燥後、450℃で5時間焼成して、水銀ハロゲン化触媒(2)を得た。 Next, vanadium was added to the Ti—Si—Mo composite oxide as follows. A uniform vanadium-containing solution was prepared by mixing and dissolving 1.29 kg of ammonium metavanadate, 1.67 kg of oxalic acid, and 0.4 kg of monoethanolamine in 8 liters of water. After adding 19 kg of the Ti—Si—Mo composite oxide powder obtained above to a kneader, the above vanadium-containing solution was added together with a molding aid containing an organic binder (starch 1.5 kg) and stirred well. Furthermore, after adding a proper amount of water and mixing well with a blender, the mixture was sufficiently kneaded with a continuous kneader and extruded into a honeycomb shape. The obtained molded product was dried at 60 ° C. and then calcined at 450 ° C. for 5 hours to obtain a mercury halogenated catalyst (2).
得られた水銀ハロゲン化触媒(2)の組成は、酸化物換算重量比で、Ti−Si−Mo複合酸化物:バナジウム酸化物=95:5(酸化物換算重量比で、チタン酸化物:ケイ素酸化物:モリブデン酸化物:バナジウム酸化物=76:4.8:14.2:5)であった。
なお、水銀ハロゲン化触媒(2)のX線回折パターンを図5に示す。図5において、TiO2以外の物質に帰属される明らかな固有のピークは認められず、かつ、アナターゼ型酸化チタンに帰属されるブロードなピークが認められることから、水銀ハロゲン化触媒(2)は複合酸化物であることが確認できた。
The composition of the obtained mercury halogenation catalyst (2) is an oxide equivalent weight ratio, Ti—Si—Mo composite oxide: vanadium oxide = 95: 5 (oxide oxide weight ratio, titanium oxide: silicon). Oxide: molybdenum oxide: vanadium oxide = 76: 4.8: 14.2: 5).
The X-ray diffraction pattern of the mercury halogenated catalyst (2) is shown in FIG. In FIG. 5, no obvious intrinsic peak attributed to substances other than TiO 2 is observed, and a broad peak attributed to anatase-type titanium oxide is observed, so that the mercury halogenated catalyst (2) is It was confirmed to be a complex oxide.
〔製造例2−3−水銀ハロゲン化触媒(3)の製造〕
まず、Ti−Si複合酸化物を次のように調製した。シリカゾル(「スノーテックス−30」日産化学社製、SiO2換算30wt%含有)6.7kg、工業用アンモニア水(25wt%NH3含有)110kg、および水70リットルを混合し、均一溶液を調製した。この溶液に、硫酸チタニルの硫酸溶液(テイカ社製:TiO2として70g/リットル、H2SO4として287g/リットル含有)257リットルを撹拌しながら徐々に滴下した。得られたスラリーを20時間静置したのち、濾過し、水で充分洗浄した後、続いて100℃で1時間乾燥した。さらに、空気雰囲気下、500℃で5時間焼成し、Ti−Si複合酸化物粉体を得た。該Ti−Si複合酸化物粉体の組成は、酸化物換算重量比で、チタン酸化物:ケイ素酸化物=90:10であった。
[Production Example 2-3 Production of Mercury Halogenation Catalyst (3)]
First, a Ti—Si composite oxide was prepared as follows. 6.7 kg of silica sol (“Snowtex-30” manufactured by Nissan Chemical Industries, containing 30 wt% in terms of SiO 2 ), 110 kg of industrial aqueous ammonia (containing 25 wt% NH 3 ), and 70 liters of water were mixed to prepare a uniform solution. . To this solution, 257 liters of a sulfuric acid solution of titanyl sulfate (manufactured by Teika: 70 g / liter as TiO 2 and 287 g / liter as H 2 SO 4 ) was gradually added dropwise with stirring. The obtained slurry was allowed to stand for 20 hours, filtered, sufficiently washed with water, and then dried at 100 ° C. for 1 hour. Further, it was fired at 500 ° C. for 5 hours in an air atmosphere to obtain a Ti—Si composite oxide powder. The composition of the Ti—Si composite oxide powder was titanium oxide: silicon oxide = 90: 10 in terms of weight ratio in terms of oxide.
次いで、上記Ti−Si複合酸化物にバナジウムを次のようにして添加した。8リットルの水に、メタバナジン酸アンモニウム1.29kg、シュウ酸1.67kg、およびモノエタノールアミン0.4kgを混合して溶解させ、均一なバナジウム含有溶液を調製した。上記で得られたTi−Si複合酸化物粉体19kgをニーダーに投入後、有機バインダー(デンプン1.5kg)を含む成形助材とともに上記バナジウム含有溶液全量を加え、よく攪拌した。さらに適量の水を加えつつブレンダーでよく混合した後、連続ニーダーで充分混練りし、ハニカム状に押し出し成形した。得られた成形物を60℃で乾燥後、500℃で5時間焼成して、水銀ハロゲン化触媒(3)を得た。 Next, vanadium was added to the Ti—Si composite oxide as follows. A uniform vanadium-containing solution was prepared by mixing and dissolving 1.29 kg of ammonium metavanadate, 1.67 kg of oxalic acid, and 0.4 kg of monoethanolamine in 8 liters of water. After adding 19 kg of the Ti-Si composite oxide powder obtained above to a kneader, the above vanadium-containing solution was added together with a molding aid containing an organic binder (starch 1.5 kg) and stirred well. Furthermore, after adding a proper amount of water and mixing well with a blender, the mixture was sufficiently kneaded with a continuous kneader and extruded into a honeycomb shape. The obtained molded product was dried at 60 ° C. and then calcined at 500 ° C. for 5 hours to obtain a mercury halogenated catalyst (3).
得られた水銀ハロゲン化触媒(3)の組成は、酸化物換算重量比で、Ti−Si複合酸化物:バナジウム酸化物=95:5(酸化物換算重量比で、チタン酸化物:ケイ素酸化物:バナジウム酸化物=85.5:9.5:5)であった。
なお、水銀ハロゲン化触媒(3)のX線回折パターンを図6に示す。図6において、TiO2以外の物質に帰属される明らかな固有のピークは認められず、かつ、アナターゼ型酸化チタンに帰属されるブロードなピークが認められることから、水銀ハロゲン化触媒(3)は複合酸化物であることが確認できた。
〔実施例1、参考例1〕
図8に示す排ガス処理システムにて、表1に示す水銀ハロゲン化触媒および脱硝触媒をそれぞれ表1に示す触媒温度で用い、下記ガス組成の模擬排ガスを処理した。なお、処理に際しては、水銀ハロゲン化装置における空間速度は3000Hr−1とし、脱硝装置における空間速度は19000Hr−1とした。
The composition of the obtained mercury halogenation catalyst (3) is an oxide equivalent weight ratio, Ti-Si composite oxide: vanadium oxide = 95: 5 (oxide oxide weight ratio, titanium oxide: silicon oxide). : Vanadium oxide = 85.5: 9.5: 5).
The X-ray diffraction pattern of the mercury halogenated catalyst (3) is shown in FIG. In FIG. 6, no obvious intrinsic peak attributed to substances other than TiO 2 is observed, and a broad peak attributed to anatase-type titanium oxide is observed, so that the mercury halogenated catalyst (3) is It was confirmed to be a complex oxide.
[Example 1 and Reference Example 1 ]
In the exhaust gas treatment system shown in FIG. 8, a simulated exhaust gas having the following gas composition was treated using the mercury halogenation catalyst and the denitration catalyst shown in Table 1 at the catalyst temperatures shown in Table 1, respectively. In the treatment, the space velocity in the mercury halogenating apparatus was 3000 Hr −1, and the space velocity in the denitration apparatus was 19000 Hr −1 .
[ガス組成]
NOx:100ppm
NH3:100ppm
HCl:5ppm
Hg:30μg/m3N(内、金属水銀(Hg0)は18μg/m3N)
O2:9%
H2O:10%
詳しくは、まず、排ガス容器1に入れた模擬排ガスを、脱硝装置2に導き、該装置にて窒素酸化物を処理した。このとき、脱硝装置2中の脱硝触媒の温度は、温度制御装置3aによって制御した。次いで、模擬排ガスを脱硝装置2から水銀ハロゲン化触媒を備えた水銀ハロゲン化装置4に導き、該装置にて金属水銀をハロゲン化水銀に変換させた。このとき、水銀ハロゲン化装置4中の水銀ハロゲン化触媒の温度は、温度制御装置3bによって制御した。次いで、水銀ハロゲン化装置4を通った模擬排ガスを、吸収瓶5中の吸収液(3%炭酸カルシウム水溶液)6の中に導き、模擬排ガス中のハロゲン化水銀を吸収液に吸収させた。その後、模擬排ガスを吸収瓶5から回収瓶7に導き、回収した。
[Gas composition]
NOx: 100ppm
NH 3 : 100 ppm
HCl: 5ppm
Hg: 30 μg / m 3 N (including 18 μg / m 3 N for metallic mercury (Hg 0 ))
O 2 : 9%
H 2 O: 10%
Specifically, first, the simulated exhaust gas put in the exhaust gas container 1 was guided to the
そして、処理を開始して10時間後および500時間後に、排ガス容器1と脱硝装置2の間に設けたガスサンプリング口8aから採取した処理前のガスaと、吸収瓶5と回収瓶7の間に設けたガスサンプリング口8bから採取した処理前のガスbとについて、それぞれに含まれるNOx濃度およびHg濃度(Hg0およびHgCl2)を測定し、下記式に従って脱硝率および水銀除去率を求めた。結果を表1に示す。
<脱硝率>
脱硝率(%)={(ガスaのNOx濃度)−(ガスbのNOx濃度)}÷(ガスaのNOx濃度)×100
<水銀除去率>
水銀除去率(%)={(ガスaのHg濃度)−(ガスbのHg濃度)}÷(ガスaのHg濃度)×100
〔比較例1〜3〕
実施例1、参考例1と同様の排ガス処理システムにて、表1に示す水銀ハロゲン化触媒および脱硝触媒をそれぞれ表1に示す触媒温度で用い、実施例1、参考例1と同様の模擬排ガスを処理した。なお、処理に際し、水銀ハロゲン化装置における空間速度および脱硝装置における空間速度は実施例1、参考例1と同様とした。そして、実施例1、参考例1と同様に脱硝率および水銀除去率を求めた。結果を表1に示す。
Then, 10 hours and 500 hours after the start of the treatment, the gas a before being collected from the
<Denitration rate>
Denitration rate (%) = {(NOx concentration of gas a) − (NOx concentration of gas b)} ÷ (NOx concentration of gas a) × 100
<Mercury removal rate>
Mercury removal rate (%) = {(Hg concentration of gas a) − (Hg concentration of gas b)} ÷ (Hg concentration of gas a) × 100
[Comparative Examples 1-3]
Example 1 In the same exhaust gas treatment system as in Reference Example 1 , the mercury halide catalyst and the denitration catalyst shown in Table 1 were used at the catalyst temperatures shown in Table 1, respectively , and the simulated exhaust gas as in Example 1 and Reference Example 1 was used. Processed. In the treatment, the space velocity in the mercury halogenating apparatus and the space velocity in the denitration apparatus were the same as those in Example 1 and Reference Example 1 . Then, the denitration rate and the mercury removal rate were determined in the same manner as in Example 1 and Reference Example 1 . The results are shown in Table 1.

本発明にかかる排ガス処理方法は、例えば、ボイラ、焼却炉、ガスタービン、ディーゼルエンジンおよび各種工業プロセスから排出される各種排ガスの処理に適用することができる。 The exhaust gas treatment method according to the present invention can be applied to, for example, treatment of various exhaust gases discharged from boilers, incinerators, gas turbines, diesel engines, and various industrial processes.
1 排ガス容器
2 脱硝装置
3 温度制御装置
4 水銀ハロゲン化装置
5 吸収瓶
6 吸収液
7 回収瓶
8 ガスサンプリング口
1
Claims (2)
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003362597A JP4098697B2 (en) | 2003-10-22 | 2003-10-22 | Exhaust gas treatment method |
| TW093130901A TWI306410B (en) | 2003-10-22 | 2004-10-12 | Method for treating exhaust gas |
| US10/963,969 US7264784B2 (en) | 2003-10-22 | 2004-10-12 | Method for treating exhaust gas |
| EP04024961A EP1525913A3 (en) | 2003-10-22 | 2004-10-20 | Method for treating exhaust gas |
| KR1020040083827A KR100782387B1 (en) | 2003-10-22 | 2004-10-20 | Method for treating exhaust gas |
| CNB2004100870152A CN100375646C (en) | 2003-10-22 | 2004-10-22 | Method for treating exhaust gas |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003362597A JP4098697B2 (en) | 2003-10-22 | 2003-10-22 | Exhaust gas treatment method |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2005125212A JP2005125212A (en) | 2005-05-19 |
| JP4098697B2 true JP4098697B2 (en) | 2008-06-11 |
Family
ID=34642176
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003362597A Expired - Fee Related JP4098697B2 (en) | 2003-10-22 | 2003-10-22 | Exhaust gas treatment method |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4098697B2 (en) |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08229412A (en) * | 1995-11-30 | 1996-09-10 | Nippon Shokubai Co Ltd | Catalyst and method for removing nitrogen oxide |
| JPH10323570A (en) * | 1997-05-26 | 1998-12-08 | Babcock Hitachi Kk | Catalyst for denitration of flue gas and its production |
| JP3023102B1 (en) * | 1999-01-11 | 2000-03-21 | 川崎重工業株式会社 | Method and apparatus for removing mercury from exhaust gas |
| JP4831801B2 (en) * | 2001-08-09 | 2011-12-07 | 三菱重工業株式会社 | Method and apparatus for removing mercury from exhaust gas |
| JP4175465B2 (en) * | 2003-02-07 | 2008-11-05 | 三菱重工業株式会社 | Method and system for removing mercury from exhaust gas |
| JP2004337781A (en) * | 2003-05-16 | 2004-12-02 | Mitsubishi Heavy Ind Ltd | Exhaust gas treating method, exhaust gas treating system, and catalyst oxidation apparatus |
-
2003
- 2003-10-22 JP JP2003362597A patent/JP4098697B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2005125212A (en) | 2005-05-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100782387B1 (en) | Method for treating exhaust gas | |
| JP4113090B2 (en) | Exhaust gas treatment method | |
| JPWO2006132097A1 (en) | Titanium oxide, exhaust gas treatment catalyst, and exhaust gas purification method | |
| JP4098703B2 (en) | Nitrogen oxide removing catalyst and nitrogen oxide removing method | |
| KR100456748B1 (en) | Catalyst for purification of exhaust gases, production process therefor, and process for purification of exhaust gases | |
| JP4822740B2 (en) | Exhaust gas treatment catalyst and exhaust gas treatment method | |
| US6858562B1 (en) | Catalyst for decomposing organic harmful substances and method for decomposing organic halides by use thereof | |
| JP4098698B2 (en) | Exhaust gas treatment method | |
| JP2001286734A (en) | Method for decomposing chlorinated organic compound and method for treating combustion exhaust gas | |
| JP4098697B2 (en) | Exhaust gas treatment method | |
| KR100382050B1 (en) | Catalyst for Removing Dioxin and Nitrogen Oxides in Flue Gas and Method for Treating Combustion Exhaust Gases Using the Same | |
| JP2001252562A (en) | Low temperature denitration catalyst and low temperature denitration method | |
| JP2001038206A (en) | Catalyst for treating exhaust gas and method and apparatus for treating exhaust gas | |
| JP3860734B2 (en) | Exhaust gas treatment catalyst and exhaust gas treatment method | |
| JP2001286733A (en) | Method for decomposing chlorinated organic compound and method for treating combustion exhaust gas | |
| JP3739659B2 (en) | Exhaust gas treatment catalyst, exhaust gas treatment method, and exhaust gas treatment catalyst manufacturing method | |
| JP4499511B2 (en) | Method for treating exhaust gas containing nitrogen oxides | |
| JP6906420B2 (en) | Manufacturing method of inorganic composite oxide and exhaust gas treatment method using this | |
| JP6016572B2 (en) | Exhaust gas treatment catalyst and exhaust gas treatment method | |
| JP4499512B2 (en) | Method for treating exhaust gas containing odor components | |
| JP4283092B2 (en) | Exhaust gas treatment catalyst and exhaust gas treatment method | |
| JP2017177051A (en) | Exhaust gas treatment catalyst and exhaust gas treatment method | |
| JP2001113170A (en) | Method for manufacturing for denitration catalyst | |
| JP2004081995A (en) | Denitration catalyst and denitration method using the same | |
| JP4002437B2 (en) | Exhaust gas treatment catalyst and exhaust gas treatment method |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050215 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20070926 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20071009 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20071207 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080115 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080212 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20080311 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20080313 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 4098697 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110321 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120321 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120321 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130321 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140321 Year of fee payment: 6 |
|
| LAPS | Cancellation because of no payment of annual fees |