JP2017507899A - バイオベースのアルキルおよびフラン系ジオールエーテル、アセタート、エーテル−アセタート、ならびにカーボナートの直接合成 - Google Patents
バイオベースのアルキルおよびフラン系ジオールエーテル、アセタート、エーテル−アセタート、ならびにカーボナートの直接合成 Download PDFInfo
- Publication number
- JP2017507899A JP2017507899A JP2016538518A JP2016538518A JP2017507899A JP 2017507899 A JP2017507899 A JP 2017507899A JP 2016538518 A JP2016538518 A JP 2016538518A JP 2016538518 A JP2016538518 A JP 2016538518A JP 2017507899 A JP2017507899 A JP 2017507899A
- Authority
- JP
- Japan
- Prior art keywords
- carbonate
- diol compound
- ether
- alkyl
- deprotonating agent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- -1 furan diol ethers Chemical class 0.000 title claims abstract description 90
- 125000000217 alkyl group Chemical group 0.000 title claims description 11
- 150000004649 carbonic acid derivatives Chemical class 0.000 title abstract description 4
- 230000015572 biosynthetic process Effects 0.000 title description 18
- 238000003786 synthesis reaction Methods 0.000 title description 16
- CJMRDWKLOVHYSM-UHFFFAOYSA-N 8-epi-furandiol Natural products CC1(O)CC2=COC=C2C(O)C2CC(C)(C)CC21 CJMRDWKLOVHYSM-UHFFFAOYSA-N 0.000 title description 4
- 150000001242 acetic acid derivatives Chemical class 0.000 title description 4
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims abstract description 139
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims abstract description 51
- 238000000034 method Methods 0.000 claims abstract description 44
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 20
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 127
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 32
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 19
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 16
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 9
- 239000007848 Bronsted acid Substances 0.000 claims description 7
- 125000005910 alkyl carbonate group Chemical group 0.000 claims description 6
- OWBTYPJTUOEWEK-UHFFFAOYSA-N butane-2,3-diol Chemical compound CC(O)C(C)O OWBTYPJTUOEWEK-UHFFFAOYSA-N 0.000 claims description 6
- ZFTFAPZRGNKQPU-UHFFFAOYSA-N dicarbonic acid Chemical compound OC(=O)OC(O)=O ZFTFAPZRGNKQPU-UHFFFAOYSA-N 0.000 claims description 6
- 230000008569 process Effects 0.000 claims description 6
- 239000003341 Bronsted base Substances 0.000 claims description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 claims description 4
- 125000003118 aryl group Chemical group 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 4
- 150000007524 organic acids Chemical class 0.000 claims description 4
- 150000001412 amines Chemical class 0.000 claims description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 claims description 2
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims 2
- TVYABKWHFSGGMF-UHFFFAOYSA-N [2-(hydroxymethyl)oxolan-3-yl]methanol Chemical compound OCC1CCOC1CO TVYABKWHFSGGMF-UHFFFAOYSA-N 0.000 claims 2
- 229910052806 inorganic carbonate Inorganic materials 0.000 claims 1
- 229910000029 sodium carbonate Inorganic materials 0.000 claims 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 abstract description 40
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 abstract description 34
- 239000003054 catalyst Substances 0.000 abstract description 18
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 abstract description 17
- 239000002253 acid Substances 0.000 abstract description 7
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 36
- 238000006243 chemical reaction Methods 0.000 description 31
- 239000000047 product Substances 0.000 description 26
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 18
- 238000006266 etherification reaction Methods 0.000 description 18
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical class COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 17
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- 239000000203 mixture Substances 0.000 description 13
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 238000002474 experimental method Methods 0.000 description 11
- 238000009835 boiling Methods 0.000 description 10
- 239000003153 chemical reaction reagent Substances 0.000 description 10
- 150000002009 diols Chemical class 0.000 description 10
- PPPFYBPQAPISCT-UHFFFAOYSA-N 2-hydroxypropyl acetate Chemical compound CC(O)COC(C)=O PPPFYBPQAPISCT-UHFFFAOYSA-N 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- 239000002585 base Substances 0.000 description 8
- 150000002170 ethers Chemical class 0.000 description 8
- 125000002947 alkylene group Chemical group 0.000 description 7
- 239000008241 heterogeneous mixture Substances 0.000 description 7
- 239000004810 polytetrafluoroethylene Substances 0.000 description 7
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 7
- 239000003921 oil Substances 0.000 description 6
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- 239000012230 colorless oil Substances 0.000 description 5
- 238000010586 diagram Methods 0.000 description 5
- 238000000921 elemental analysis Methods 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical group [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 4
- 238000005804 alkylation reaction Methods 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 238000003818 flash chromatography Methods 0.000 description 4
- 150000002334 glycols Chemical class 0.000 description 4
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical group CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 4
- ZLMJMSJWJFRBEC-OUBTZVSYSA-N potassium-40 Chemical compound [40K] ZLMJMSJWJFRBEC-OUBTZVSYSA-N 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- CJMRDWKLOVHYSM-HTUGSXCWSA-N (5s,5as,8ar,9s)-5,7,7-trimethyl-4,5a,6,8,8a,9-hexahydroazuleno[5,6-c]furan-5,9-diol Chemical compound C[C@]1(O)CC2=COC=C2[C@@H](O)[C@@H]2CC(C)(C)C[C@@H]21 CJMRDWKLOVHYSM-HTUGSXCWSA-N 0.000 description 3
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 150000005215 alkyl ethers Chemical class 0.000 description 3
- 230000029936 alkylation Effects 0.000 description 3
- 150000001450 anions Chemical class 0.000 description 3
- 239000012300 argon atmosphere Substances 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- PYGSKMBEVAICCR-UHFFFAOYSA-N hexa-1,5-diene Chemical group C=CCCC=C PYGSKMBEVAICCR-UHFFFAOYSA-N 0.000 description 3
- KKQAVHGECIBFRQ-UHFFFAOYSA-N methyl propyl carbonate Chemical compound CCCOC(=O)OC KKQAVHGECIBFRQ-UHFFFAOYSA-N 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 239000000376 reactant Substances 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 238000010189 synthetic method Methods 0.000 description 3
- FRBJEDLSFZZCFY-OCAPTIKFSA-N (2S,5R)-2,5-bis(methoxymethyl)oxolane Chemical compound COC[C@@H]1O[C@@H](CC1)COC FRBJEDLSFZZCFY-OCAPTIKFSA-N 0.000 description 2
- QWUWMCYKGHVNAV-UHFFFAOYSA-N 1,2-dihydrostilbene Chemical group C=1C=CC=CC=1CCC1=CC=CC=C1 QWUWMCYKGHVNAV-UHFFFAOYSA-N 0.000 description 2
- DZEPFNDOOWFEKN-UHFFFAOYSA-N 2,5-bis(methoxymethyl)furan Chemical compound COCC1=CC=C(COC)O1 DZEPFNDOOWFEKN-UHFFFAOYSA-N 0.000 description 2
- HXDLWJWIAHWIKI-UHFFFAOYSA-N 2-hydroxyethyl acetate Chemical compound CC(=O)OCCO HXDLWJWIAHWIKI-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- JOFDQONITRMYMX-UHFFFAOYSA-N [5-(methoxymethyl)furan-2-yl]methanol Chemical compound COCC1=CC=C(CO)O1 JOFDQONITRMYMX-UHFFFAOYSA-N 0.000 description 2
- 239000000370 acceptor Substances 0.000 description 2
- 230000021736 acetylation Effects 0.000 description 2
- 238000006640 acetylation reaction Methods 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- 150000001346 alkyl aryl ethers Chemical class 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- 125000006267 biphenyl group Chemical group 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 150000001983 dialkylethers Chemical class 0.000 description 2
- ROORDVPLFPIABK-UHFFFAOYSA-N diphenyl carbonate Chemical compound C=1C=CC=CC=1OC(=O)OC1=CC=CC=C1 ROORDVPLFPIABK-UHFFFAOYSA-N 0.000 description 2
- VUPKGFBOKBGHFZ-UHFFFAOYSA-N dipropyl carbonate Chemical compound CCCOC(=O)OCCC VUPKGFBOKBGHFZ-UHFFFAOYSA-N 0.000 description 2
- SNRUBQQJIBEYMU-UHFFFAOYSA-N dodecane Chemical group CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 2
- 238000006735 epoxidation reaction Methods 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N hexan-1-ol Chemical compound CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- REOJLIXKJWXUGB-UHFFFAOYSA-N mofebutazone Chemical group O=C1C(CCCC)C(=O)NN1C1=CC=CC=C1 REOJLIXKJWXUGB-UHFFFAOYSA-N 0.000 description 2
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 2
- 150000002924 oxiranes Chemical class 0.000 description 2
- 239000003973 paint Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- YTMCUIACOKRXQA-UHFFFAOYSA-N (2-aminoacetyl) 2-aminoacetate Chemical class NCC(=O)OC(=O)CN YTMCUIACOKRXQA-UHFFFAOYSA-N 0.000 description 1
- KNPKBOGRBZGQDS-UHFFFAOYSA-N 6-benzhydryl-1,3,5-trioxepane-2,4-dione Chemical compound C1(=O)OCC(C(C2=CC=CC=C2)C2=CC=CC=C2)OC(O1)=O KNPKBOGRBZGQDS-UHFFFAOYSA-N 0.000 description 1
- 239000002028 Biomass Substances 0.000 description 1
- RGTKJHVGIPUVAL-RNFRBKRXSA-N COC[C@@H]1O[C@@H](CO)CC1 Chemical compound COC[C@@H]1O[C@@H](CO)CC1 RGTKJHVGIPUVAL-RNFRBKRXSA-N 0.000 description 1
- RGTKJHVGIPUVAL-BQBZGAKWSA-N COC[C@H]1O[C@H](CO)CC1 Chemical compound COC[C@H]1O[C@H](CO)CC1 RGTKJHVGIPUVAL-BQBZGAKWSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- UXWLTDLXJUWSTK-NKWVEPMBSA-N OCC[C@H]1O[C@@H](CO)CC1 Chemical compound OCC[C@H]1O[C@@H](CO)CC1 UXWLTDLXJUWSTK-NKWVEPMBSA-N 0.000 description 1
- YCZZQSFWHFBKMU-WDSKDSINSA-N OC[C@H]1O[C@H](CO)CC1 Chemical compound OC[C@H]1O[C@H](CO)CC1 YCZZQSFWHFBKMU-WDSKDSINSA-N 0.000 description 1
- DSLRVRBSNLHVBH-UHFFFAOYSA-N OCc1ccc(CO)[o]1 Chemical compound OCc1ccc(CO)[o]1 DSLRVRBSNLHVBH-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- 229910021536 Zeolite Inorganic materials 0.000 description 1
- WPRAHVNHWSRKKO-RYUDHWBXSA-N [(2S,5S)-5-(propoxycarbonyloxymethyl)oxolan-2-yl]methyl propyl carbonate Chemical compound C(OCCC)(OC[C@H]1O[C@@H](CC1)COC(OCCC)=O)=O WPRAHVNHWSRKKO-RYUDHWBXSA-N 0.000 description 1
- OMZSQHQZAILHNR-UHFFFAOYSA-N [5-(hydroxymethyl)furan-2-yl]methyl propyl carbonate Chemical compound C(OCC=1OC(=CC1)CO)(OCCC)=O OMZSQHQZAILHNR-UHFFFAOYSA-N 0.000 description 1
- YCZZQSFWHFBKMU-UHFFFAOYSA-N [5-(hydroxymethyl)oxolan-2-yl]methanol Chemical compound OCC1CCC(CO)O1 YCZZQSFWHFBKMU-UHFFFAOYSA-N 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 238000010306 acid treatment Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 230000002528 anti-freeze Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 238000006254 arylation reaction Methods 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229920001429 chelating resin Polymers 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 238000005237 degreasing agent Methods 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 230000005595 deprotonation Effects 0.000 description 1
- 238000010537 deprotonation reaction Methods 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000011143 downstream manufacturing Methods 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 238000007210 heterogeneous catalysis Methods 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- QWPPOHNGKGFGJK-UHFFFAOYSA-N hypochlorous acid Chemical compound ClO QWPPOHNGKGFGJK-UHFFFAOYSA-N 0.000 description 1
- 238000005286 illumination Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 239000004434 industrial solvent Substances 0.000 description 1
- 239000000976 ink Substances 0.000 description 1
- 239000011229 interlayer Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 150000005677 organic carbonates Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- 230000000475 sunscreen effect Effects 0.000 description 1
- 239000000516 sunscreening agent Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 1
- 238000005809 transesterification reaction Methods 0.000 description 1
- 238000007039 two-step reaction Methods 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/08—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides with the hydroxy or O-metal group of organic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/16—Preparation of ethers by reaction of esters of mineral or organic acids with hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/03—Ethers having all ether-oxygen atoms bound to acyclic carbon atoms
- C07C43/14—Unsaturated ethers
- C07C43/15—Unsaturated ethers containing only non-aromatic carbon-to-carbon double bonds
- C07C43/16—Vinyl ethers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C68/00—Preparation of esters of carbonic or haloformic acids
- C07C68/06—Preparation of esters of carbonic or haloformic acids from organic carbonates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/02—Esters of acyclic saturated monocarboxylic acids having the carboxyl group bound to an acyclic carbon atom or to hydrogen
- C07C69/12—Acetic acid esters
- C07C69/16—Acetic acid esters of dihydroxylic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/96—Esters of carbonic or haloformic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/10—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/12—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/38—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/40—Radicals substituted by oxygen atoms
- C07D307/42—Singly bound oxygen atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Furan Compounds (AREA)
- Heterocyclic Compounds That Contain Two Or More Ring Oxygen Atoms (AREA)
Abstract
2つの経路のうちのどちらか一方を含む、アルキレングリコール、HMFまたはその還元誘導体生成物(すなわち、FDM、bHMTHF)からグリコールモノエーテルまたはモノアセタート、またはカーボナートを調製する方法を提供する。特に、1つの経路によれば、アルキレングリコール、HMFまたはFDM、bHMTHFとジアルキルカーボナートを、脱プロトン化剤の存在下で外部触媒を実質的に存在させずに、反応させて、エーテルを生成し、引き続いてエーテルと酸塩基を反応させる。もう1つの経路によれば、アルキレングリコールとアセタート供与体を、酸、塩基の存在下で反応させて、アルキレンモノアセタートを生じ、カーボナートを用いて脱プロトン化剤の存在下でエーテル化する。
Description
優先権の利益
本出願は、2013年12月20日出願の米国特許仮出願第61/918,795号、2014年12月5日出願の国際出願PCT/US2014/68809号、および2014年12月18日出願の米国特許仮出願第62/093683号の優先権の利益を主張し、それぞれの内容は参照により本明細書に組み込まれる。
本出願は、2013年12月20日出願の米国特許仮出願第61/918,795号、2014年12月5日出願の国際出願PCT/US2014/68809号、および2014年12月18日出願の米国特許仮出願第62/093683号の優先権の利益を主張し、それぞれの内容は参照により本明細書に組み込まれる。
本発明は、生物学的に誘導されたグリコールを有用な生成物に転換する方法に関する。詳細には、本発明は、種々の化合物をアルキレングリコールまたはフラン系ジオールから合成する、簡単で環境に優しい方法に関する。
グリコールエーテルは、同一分子にエーテルとアルコールの両方の官能基を有しているので、最も用途の広い有機溶媒クラスの1つである。これらの分子はアルコールとエーテルの最良の溶解力という特徴を併せもち、これによって広範囲の有機化学薬品および油における良好な混和性および溶解力、ならびに水に対する溶解性がある。グリコールエーテルはまた、より高い沸点も有する。このような理由で、グリコールエーテルは、(i)表面コーティング産業において樹脂用活性溶媒として、(ii)ブレーキ液産業において溶媒として、(iii)石油産業において様々な石油系燃料中の防氷液として、(iv)自動車産業において不凍剤として、(v)家庭用品に使用するための特殊製品で特にみられる。
典型的には、グリコールエーテルは、エチレンから生成されるかそれともプロピレンから生成されるかに応じてそれぞれ「e−シリーズ」または「p−シリーズ」グリコールエーテルとして分類される。典型的には、e−シリーズのグリコールエーテルは医薬品、日焼け止め剤、化粧品、インク、染料および水性塗料でみられ、p−シリーズのグリコールエーテルは、脱脂剤、清浄剤、エアロゾル塗料および接着剤で使用される。E−シリーズのグリコールエーテルは分子量がより大きく、さらなる化学反応を受ける中間体として使用することができる。P−シリーズのグリコールエーテルは一般に高性能工業用溶媒である。
グリコールエーテルの調製は通常、アルキレンオキシドの生成を伴うものである。例えば、エチレンオキシド(EO)またはプロピレンオキシド(PO)をそれぞれe−シリーズおよびp−シリーズ中でアルコールと反応させることができる。グリコールエーテル分子は、1つまたは複数のEOまたはPO分子を含むことができる。使用される典型的なアルコールとしては、メタノール、エタノール、プロパノール、ブタノール、ペンタノールおよびヘキサノールが挙げられる。この反応は、反応のモル比、ならびに反応で使用される温度および圧力に応じて異なる鎖長のグリコールエーテルを生成することができる。より穏やかな条件および、アルコールに対してより低いアルキレンオキシドのモル比では、モノアルキレングリシルエーテルが生成され、より多いアルキレンオキシドならびにより高い温度および圧力を使用すると、ジアルキレングリコールエーテルおよびトリアルキレングリコールエーテルが生成される。生成物は蒸留により精製される。次いで、グリコールエーテルをさらに酢酸と反応(エステル化)させて、対応する酢酸エステル生成物を生成することができる。したがって、考え得る複数の組合せによる生成物のファミリーがすべて存在する(概括的には、例えばHenry Chinn et al., "Marketing Research Report: Glycol Ethers," CHEMICAL ECONOMICS HANDBOOK, 663.5000A-633.5005Q (Nov. 2010), SRI Consultingを参照のこと)。
あるいは、アルキレンオキシドは、アルキレンと次亜塩素酸の水和と、その後に続く塩基触媒エポキシ化によって、またはアルキレンとt−ブチルヒドロペルオキシドの直接エポキシ化によって合成することができる。
別の方法では、アルコールとオレフィンオキシドを酸性または塩基性触媒の存在下で反応させることによって、グリコールエーテルを生成することができる。例えば、米国特許第6,124,506号には、層状構造が損なわれておらず、層間アニオンを有し、それらの少なくとも一部が金属アニオンまたは(ポリ)オキソメタラートアニオンである層状複水酸化物(LDH)粘土を含む触媒上で、オレフィンオキシドとアルコールを反応させることを伴う、グリコールエーテル合成の別法が記載されている。同様に、米国特許第8,748,635号B2には、固相ゼオライト触媒を使用したアンヒドロ糖アルコールのアルキル化によるアンヒドロ糖エーテルの調製方法が記載されている。
アルキレングリコールを多種多様な方法で生じることができる。例えば、一経路において、グルコースを水素化および水素化分解にかけて、プロピレングリコール(PG)またはエチレングリコール(EG)を生じる。別の経路において、グルコースを発酵させて、エタノールおよびCO2を生成する。次いで、銀触媒を用いて、エタノールをエチレンオキシドに転換し、次いで、CO2と反応させて、環式エチレンカーボナートを形成し、アルコールと反応すると対応するジアルキルカーボナートを生じる。エポキシドを作製する脱水/還元工程において、追加の反応工程が必要とされる。これらの方法はすべて、所望の生成物を生成する複雑さとコストの両方を増大させる複数の工程を必要とする。
商業的製造業者は、より簡単な一工程エーテル化方法を求めている。しかし、現在利用可能な合成方法では、バイオベース資源に由来するアルキレングリコール(例えば、エチレングリコール(EG)およびプロピレングリコール(PG))からエーテルを直接作製できない。いくつかの先行するまたは介在する工程をまず行わなければならない。現時点では、選択的にバイオベースアルキレングリコールを、酸化することなく直接にそれぞれのモノエーテルにすることができる方法は知られていない。したがって、出発物質としてアルキレングリコールだけでなく環式(フラン系)ジオールをも直接エーテル化する経路をもたらす新規方法は、歓迎される進歩となるはずである。
本開示は、モノエーテルをジオール化合物から調製する方法であって、第1の経路または第2の経路を含む方法に関する。第1の経路において、ジオール化合物とR1有機酸を、ブレンステッド酸の存在下で、ジオール化合物のR1モノエステルを形成するのに十分な温度で十分な時間接触させ、次いでジオール化合物のR1モノエステルと式R2(CO3)R2のR2アルキルジエステルを、脱プロトン化剤の存在下で、モノエステルエーテルを形成するのに十分な温度で十分な時間接触させる。第2の経路において、ジオール化合物と式R2(CO3)R2のR2アルキルジエステルを、脱プロトン化剤の存在下で、ジオール化合物のモノエステルを形成するのに十分な温度で十分な時間接触させ、次いでジオール化合物のモノエステルとR1有機酸を、ブレンステッド酸の存在下で、モノエステルエーテルを形成するのに十分な温度で十分な時間接触させる。R1およびR2は同じかまたは異なるアルキル、シクロアルキルまたは芳香族部分である。
本方法の追加の特徴および利点を以下の詳細な説明で開示する。前述の概要と以下の詳細な説明および実施例は共に本発明を代表するものにすぎず、特許請求されている本発明を理解するための概観を提供するよう意図されているが理解されよう。
セクションI.− 説明
A.
本合成方法は、エーテルおよび/またはアセタートを、再生可能なバイオベース材料からの出発物質を脱水または還元する必要がなく、アルキルまたはフラン系ジオールから直接調製する簡単で、清浄で、かつ洗練された方法を提供する。通常のエーテル合成のしばしば複雑で厳しい条件とは対照的に、本方法は、アルキルグリコールとジアルキルカーボナート試薬溶液を、脱プロトン化剤の存在下でかつ他の外部触媒を実質的に存在させずに、反応させるものである。本明細書では「実質的に存在しない」という用語は、外部触媒が概ねもしくは完全に存在せず、または触媒効率未満の僅少量もしくは微量で存在している状態を指す。言い換えれば、外部触媒は存在していない、または反応におけるジアルキルカーボナート試薬の量に対して5%、3%、もしくは1%(重量/重量)未満のレベルでしか存在していない。
A.
本合成方法は、エーテルおよび/またはアセタートを、再生可能なバイオベース材料からの出発物質を脱水または還元する必要がなく、アルキルまたはフラン系ジオールから直接調製する簡単で、清浄で、かつ洗練された方法を提供する。通常のエーテル合成のしばしば複雑で厳しい条件とは対照的に、本方法は、アルキルグリコールとジアルキルカーボナート試薬溶液を、脱プロトン化剤の存在下でかつ他の外部触媒を実質的に存在させずに、反応させるものである。本明細書では「実質的に存在しない」という用語は、外部触媒が概ねもしくは完全に存在せず、または触媒効率未満の僅少量もしくは微量で存在している状態を指す。言い換えれば、外部触媒は存在していない、または反応におけるジアルキルカーボナート試薬の量に対して5%、3%、もしくは1%(重量/重量)未満のレベルでしか存在していない。
方法は、再生可能なアルキレン、アルキルまたはフラン系ジオールから、酸化して、オキシドを形成する必要なく、または、脱水、還元して、エポキシドを形成する必要なく、モノエーテル、モノエステル、およびアルコキシ−エステルを作製することに使用ができる。ジオールの例は、エチレングリコール(EG)、プロピレングリコール(PG)、および2,3−ブタンジオール(BDO)などのグリコールである。あるいは、反応物質材料はエチレングリコールモノアセタート、プロピレングリコールモノアセタート、またはそれらの混合物であってもよい。フラン系ジオール反応物質は、HMF−フラン−2,−5−ジメタノール(FDM)および/または2,5−ビスヒドロキシメチルテトラヒドロフラン(bHMTHF)の還元類似体とすることができる。あるいは、HMFが本反応条件下で反応物質であるとき、HMF自体をエーテル化またはアセチル化してもよい。
本方法によれば、通常、グリコールモノエーテルは塩基が媒介する方法によって合成される。実施形態によれば、モノアセタート、またはエーテル−アセタートもしくはグリコール、モノカーボナートもしくはジカーボナートは、アルキルカーボナートをアルキル化剤として用いて、かつ/または酸が触媒するフィッシャー(Fischer)アセチル化を使用する簡単な直接方式で、アルキレングリコール前駆体から直接調製される。別の実施形態において、方法によって、エーテル、アセタート、凝集体エーテル−アセタート、モノカーボナートおよびジカーボナートをフラン系ジオールから選択的に調製することも可能になる。いくつかの実施形態によれば、モノエーテルは、反応によって生じる、好ましくかつ主な生成物である。
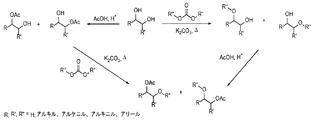
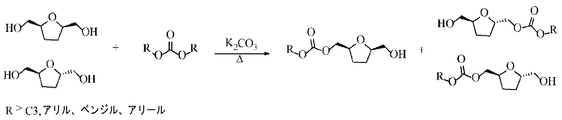
図1は、グリコールモノエーテルまたはモノ酢酸エステルを調製する、本発明による2つの代替経路の概略図を表す。両経路によって、エーテルまたはアセタート生成物を生じることが可能になる。第1の経路において、アルキレングリコールとジカーボナート試薬の溶液を、脱プロトン化剤の存在下で外部触媒を実質的に存在させずに、反応させて、エーテルを生成し、引き続いて酸、塩基または酵素触媒を用いてエーテルをアセチル化する。第2の経路において、アルキレングリコールとアセタート供与体を酸、塩基または酵素触媒の存在下で反応させて、アルキレンモノアセタートを生じ、次いで脱プロトン化剤または塩基の存在下でカーボナートを用いてエーテル化する。その後の工程において、第1の経路のエーテル生成物または第2の経路のアセタート生成物とC3鎖以上、アリル、フェニル、またはベンジルを含むカーボナートを反応させて、モノカーボナートもしくはジカーボナートまたは両方を生成する。
したがって、第1の経路に従って、アルキレングリコールを出発物とすると、第1の工程においてエーテルが生じる。あるいは、他方の第2の経路の第1の工程において、アセタートを作製する。特に、第1の経路によれば、アルキレングリコールとジアルキルカーボナート試薬を、脱プロトン化剤の存在下で外部触媒を実質的に存在させずに、接触させて、エーテルを生成する。引き続いて、エーテル生成物を、図示したように酸(例えば、酢酸)を用いて、あるいは塩基(例えば、任意のアルコキシド塩基−メトキシド)または酵素触媒を用いてアセチル化する。第2の経路によれば、アルキレングリコールとアセタート供与体(例えば、遊離酸、無水物、エーテル)を鉱酸(あるいは、塩基または酵素触媒)の存在下で反応させて、アルキレンモノアセタートを生じ、次いで脱プロトン化剤または塩基の存在下でカーボナートを用いてエーテル化する。次工程において、中間体のエーテルまたはアセタート生成物をそれぞれアセチル化またはエーテル化して、最終生成物を得る。
ジアルキルカーボナート試薬は、炭素原子1〜20個のR基を有することができる。R基がメチル、エチル、プロピル基であるとき、エーテルは通常、反応の生成物である。R基がC4〜C20基であるとき、モノアルキルカーボナートが生じる。R部分が大きくなるまたは嵩高くなるほど、モノアルキルカーボナートの形成が促進される傾向がある。エーテル化剤がアリル、フェニル、もしくはベンジル部分であるまたはC4以上の鎖を有するR基を含むとき、生成物はモノアルキルもしくはジアルキルカーボナートまたは両方の混合物である傾向がある。
他の実施形態において、本方法では、カーボナートが炭素約3個以下のアルキルR基を有する場合、アルキルまたはフラン系ジオールまたはグリコールアセタートとアルキルカーボナートを弱塩基(例えば、pKa=8〜11)の存在下で直接反応させて、対応するモノエーテルまたはジエーテル化合物を生じる。
反応は、脱プロトン化剤、またはブレンステッド塩基などのプロトン受容体によって援助される。様々なプロトン受容体としては、例えば以下の炭酸カルシウム、カリウムまたはナトリウム、アミン、アンモニアなどのうちの少なくとも1つを挙げることができる。鉱物炭酸塩は特に、反応器媒体に対して低溶解性を示し、これによって、下流処理において最終生成物からカーボナートを分離することが容易になる。
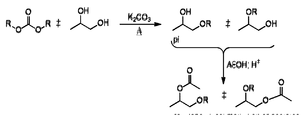
経路を反転することができる。すなわち、グリコールを、上記のようにしてまずモノアセチル化し、次いでエーテル化することができる。エーテル化は、外部触媒を用いることなく、ブレンステッド塩基を使用するだけで、アルキル化を容易にできることによって行われる。ブレンステッド塩基はpKaが少なくとも4であり、ポリオールの−OH脱プロトン化を援助する。
反応で使用されるジアルキルカーボナート試薬の量は、アルキレングリコール1分子当たり少なくとも1〜約3化学量論当量の量とすることができる。モノエーテルの調製では、ジアルキルカーボナート試薬の量は、アルキルジオールのヒドロキシル(OH)基1個当たり約2化学量論当量で存在する。
いくつかの実施形態において、カーボナート試薬はモノプロピル、モノブチル、モノペンチル、モノヘキシル、モノベンジル、モノフェニル、モノアリル、ジプロピル、ジブチル、ジペンチル、ジヘキシル、ジベンジル、ジフェニル、ジアリル官能基のうちの1つとすることができる。得られるエーテルまたはカーボナート生成物はそれぞれ、モノアルキルエーテルまたはジアルキルエーテル、あるいはモノアルキル、モノアリル、モノアリールカーボナート、またはジアルキル、ジアリル、もしくはジアリールカーボナートとすることができる。
別の態様において、本開示は、前述の方法に従って合成されたエーテル、アセタートおよびアルキルカーボナートに関する。一般に、アルキレングリコール化合物のモノエーテルは、以下のエチレングリコール(EG)、プロピレングリコール(PG)、または2,3−ブタンジオール(BDO)のモノエーテルのうちの少なくとも1つである。アルキレングリコール化合物のモノアセタートは、以下のエチレングリコール、プロピレングリコールモノアセタート、または2,3−ブタンジオール(BDO)のうちの少なくとも1つである。
一般に、アルキレングリコール化合物のアセタートは、以下のエチレングリコール(EG)、プロピレングリコール(PG)、2,3−ブタンジオール(BDO)、エチレングリコールモノエーテル、またはプロピレングリコールモノエーテル、それぞれ2,3−ブタンジオールのうちの少なくとも1つである。
一般に、モノアルキルまたはジアルキルカーボナート生成物は、以下のアルキル、アリルまたはアリール基:モノブチル、モノペンチル、モノヘキシル、モノベンジル、モノフェニル、モノアリル、ジブチル、ジペンチル、ジヘキシル、ジベンジル、ジフェニル、ジアリル、またはC3〜C20の炭素原子のモノアルキルもしくはジアルキル基のうちの少なくとも1つを含むことができる。
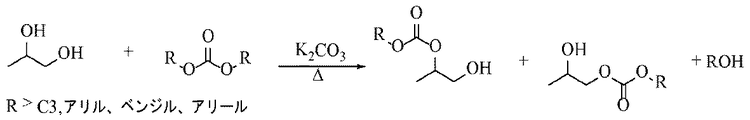
B.
本方法の実施形態による合成を図2に示す。この実施形態に示すように、プロピレングリコールとジカーボナートを加熱下に炭酸カリウムなどの求核試薬の存在下で反応させて、プロピレングリコールアルキルエーテルを生じる。
本方法の実施形態による合成を図2に示す。この実施形態に示すように、プロピレングリコールとジカーボナートを加熱下に炭酸カリウムなどの求核試薬の存在下で反応させて、プロピレングリコールアルキルエーテルを生じる。
これらのエーテルは、アセチル−アルコールおよび酸で処理することによってプロピレングリコールアルキルエーテルアセタートを作製するようにさらに処理することができる。同様に、図3および4に描かれたアルキル化反応はそれぞれフラン系ジオール、FDMおよびbHMTHFを使用した代替の実施形態を示す。図3では、FDMはジアルキルカーボナートと反応して、FDMアルキルエーテルを生成し、引き続いてFDMアルキルエーテル−アセタートに転換される。図4では、2つのbHMTHF異性体をエタノールおよび酸触媒と反応させることによって3つのbHMTHFアルキルエーテル異性体に転換し、引き続いて酢酸を用いて酸処理した後3つのbHMTHFアルキルエーテルアセタート異性体に転換する。
本方法の利点は、エーテルをアルキレングリコール、特に生物学的に誘導されたアルキレングリコールから直接調製する簡単で、清浄で、かつ洗練された方法を提供できることである。本明細書では「生物学的に誘導された」または「バイオベースの」という用語は、いわゆる化石系または石油系炭化水素とは対照的に、植物、セルロース質もしくは農業バイオマスまたはそれらの誘導体などの再生可能な生物資源から生成された炭化水素分子を指す。清浄な方法は、下流の分離および精製プロセスを簡単にするのに役立つことができる。
実施形態によれば、エーテル化がジアルキルカーボナート中ニートで実施される場合、ジアルキルエーテル類似体は唯一認められる生成物である。エーテル化が、約1当量のジアルキルカーボナート(すなわち、化学量論量のアルキル化剤)を用いて実施される場合、比較的低収率(例えば、≦10%)ではあるもののモノエーテル生成物しか生じない。プロピレングリコールまたはエチレングリコールの大部分は未反応のままである。しかし、条件の最適化によって、目標収率を改善することができる。例えば、標的モノアルキルエーテルの収率改善は、約2または3当量のジアルキルカーボナートを使用して、かつ温度の低下または反応時間の長期化など他の反応パラメータを改変して達成することができる。
方法は、比較的穏和な温度および周囲圧力下で行われる制御反応に従って、グリコールのエーテル化のための環境に優しい手法を提供する。反応は一般に約70℃〜150℃の間の温度で行われる。典型的には、反応は約70℃または80℃〜約130℃または140℃の範囲の温度で行われる。さらに典型的には、反応温度は約80℃または90℃〜約110℃または120℃の範囲である。大半の反応において、温度は約125℃未満である。これらの穏和な反応条件は、副生化合物または他の潜在的異性体および不純物の形成を制御し、最小限に抑制するのに役立つ。
本エーテル化反応が約130℃〜約150℃などのより高い温度で約24または40時間という長期の反応時間操作される場合、かなりの収量のエーテル生成物を比較的高い選択性および純度レベルで生成することができる。
C.
1.アルキレングリコールエーテル化
種々のよくみられるグリコールエーテルを作製するように、本明細書に記載された一般合成方法を変更することができる。例えば、表1に、よくみられる工業的に有用な一部のグリコールエーテルをそれらの略語および化学名と共に列挙する。
1.アルキレングリコールエーテル化
種々のよくみられるグリコールエーテルを作製するように、本明細書に記載された一般合成方法を変更することができる。例えば、表1に、よくみられる工業的に有用な一部のグリコールエーテルをそれらの略語および化学名と共に列挙する。

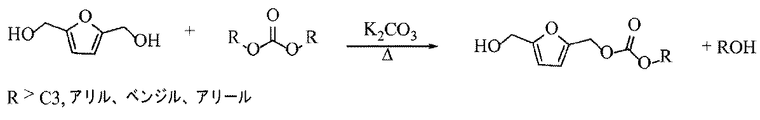
2.フラン系エーテル化
本反応は、フラン系化合物を用いて使用することもできる。図3は、FDMとジアルキルカーボナートを反応させて、FDMモノアルキルエーテルを形成する実施形態による合成反応の概略図を示す。引き続いて、エーテルをアセチル化して、対応するFDMアルキルエーテルアセタートを生じる。図4には、bHMTHF(THF−ジオール)を用いた同様の2工程反応が描かれ、bHMTHFが対応するTHFアルキルエーテルに転換され、次いでTHFアルキルエーテルアセタートにアセチル化される。
本反応は、フラン系化合物を用いて使用することもできる。図3は、FDMとジアルキルカーボナートを反応させて、FDMモノアルキルエーテルを形成する実施形態による合成反応の概略図を示す。引き続いて、エーテルをアセチル化して、対応するFDMアルキルエーテルアセタートを生じる。図4には、bHMTHF(THF−ジオール)を用いた同様の2工程反応が描かれ、bHMTHFが対応するTHFアルキルエーテルに転換され、次いでTHFアルキルエーテルアセタートにアセチル化される。
一般に、フラン系ジオールは以下のFDM、それぞれbHMTHFジアステレオマー;FDMモノアセタート、それぞれbHMTHFモノアセタートジアステレオマーのうちの少なくとも1つである。エーテル生成物は、以下のアルキル基:モノアルキル、モノエチル、モノアリルのうちの少なくとも1つを有する。
3.アルキルカーボナート形成
別の態様において、本反応は、特にエステル交換、アルキル化、またはアリール化で多様な有用性を有する反応性プラットフォームの一群である有機カーボナートを作製するように変更可能である。
別の態様において、本反応は、特にエステル交換、アルキル化、またはアリール化で多様な有用性を有する反応性プラットフォームの一群である有機カーボナートを作製するように変更可能である。
図5〜7は、ジアルキルカーボナート試薬のR基がC3以上、アリル、ベンジル、またはアリールであるときの、異なる実施形態によるカーボナートを調製する3つの別々な一般的反応を表す。図5では、プロピレングリコールを対応するジアルキルカーボナート(diaklokycarbonate)に転換する。図6では、FDMをフランカーボナートに転換し、図7では、bHMTHFを異性体THFカーボナートに転換する。
セクションII.
以下の実施例は、エーテルのプロピレングリコールおよびエチレングリコールからの合成をさらに説明するものとして、また本開示の他の態様として記載されている。パラメータおよび条件の変化(例えば、温度、時間および試薬濃度の変化、特定の出発種および触媒ならびにそれらの量)は、本発明の完全な実施に影響を及ぼし、それを拡大することがある。
以下の実施例は、エーテルのプロピレングリコールおよびエチレングリコールからの合成をさらに説明するものとして、また本開示の他の態様として記載されている。パラメータおよび条件の変化(例えば、温度、時間および試薬濃度の変化、特定の出発種および触媒ならびにそれらの量)は、本発明の完全な実施に影響を及ぼし、それを拡大することがある。
A.グリコールモノアセタート
以下の実施例は、プロピレングリコールモノアセタートを合成する反応の例示となる。グリコールアセタートは、溶媒、添加剤の前駆体、結合剤、可塑剤、潤滑剤および界面活性剤などの用途において有用である材料を構成する。
以下の実施例は、プロピレングリコールモノアセタートを合成する反応の例示となる。グリコールアセタートは、溶媒、添加剤の前駆体、結合剤、可塑剤、潤滑剤および界面活性剤などの用途において有用である材料を構成する。
実施例1
プロピレングリコールモノアセタートの合成
ディーンスターク装置を装備した500mLの丸底フラスコに、プロピレングリコール100g、酢酸75g、および高温不均一系触媒作用で使用するマクロポーラスポリマー触媒(商業的にDow Chemical, Inc.社のAmberlyst(商標)70として知られている)5gを加えた。反応混合物を120℃に加熱し、水を反応混合物から除去した。残渣は主にプロピレングリコールモノアセタートを含有するものであった。
プロピレングリコールモノアセタートの合成
ディーンスターク装置を装備した500mLの丸底フラスコに、プロピレングリコール100g、酢酸75g、および高温不均一系触媒作用で使用するマクロポーラスポリマー触媒(商業的にDow Chemical, Inc.社のAmberlyst(商標)70として知られている)5gを加えた。反応混合物を120℃に加熱し、水を反応混合物から除去した。残渣は主にプロピレングリコールモノアセタートを含有するものであった。
実施例2
プロピレングリコールモノアセタートの合成
ディーンスターク装置を装備した500mLの丸底フラスコに、プロピレングリコール100g、酢酸エチル115g、およびナトリウムメトキシド0.5gを加えた。反応混合物を90℃に加熱し、エタノールを反応混合物から除去した。残渣は主にプロピレングリコールモノアセタートを含有するものであった。
プロピレングリコールモノアセタートの合成
ディーンスターク装置を装備した500mLの丸底フラスコに、プロピレングリコール100g、酢酸エチル115g、およびナトリウムメトキシド0.5gを加えた。反応混合物を90℃に加熱し、エタノールを反応混合物から除去した。残渣は主にプロピレングリコールモノアセタートを含有するものであった。
実施例3
プロピレングリコールモノアセタートの合成
1Lのオートクレーブエンジニア反応器に、プロピレングリコール200g、酢酸150mLおよび濃H2SO42滴を加えた。反応器本体を組立て、反応器を130℃まで3時間加熱した。反応器を冷却した。生成物は、大部分がプロピレングリコールモノアセタートからなるものであった。
プロピレングリコールモノアセタートの合成
1Lのオートクレーブエンジニア反応器に、プロピレングリコール200g、酢酸150mLおよび濃H2SO42滴を加えた。反応器本体を組立て、反応器を130℃まで3時間加熱した。反応器を冷却した。生成物は、大部分がプロピレングリコールモノアセタートからなるものであった。
B.線状アルキレングリコールモノエーテル
実施例1:メタノール中におけるPGメチルエーテル化(1:1 PG/DMC)
実験:100mLの沸騰フラスコに、プロピレングリコール5g(PG、65.7mmol)、炭酸ジメチル5.53mL(65.7mmol)、炭酸カリウム18.2g、およびメタノール40mLを加えた。混合物にフリードリヒ冷却器を装備し、終夜還流した(約80℃)。この後、不均一な混合物を室温に冷却し、過剰の炭酸カリウムを濾過し、濾液を貯蔵した。濾液の試料を定量分析にかけた。その結果は、PGの約10%が対応するモノメチルエーテルAおよびBに均等に転換したことを示した。PGジメチルエーテルも他の生成物もみられなかった。
実施例2:メタノール中におけるPGメチルエーテル化(1:2 PG/DMC)
実験:実施例1に記載された反応と同様にして、プロピレングリコールと炭酸ジメチルの比を1:2とした別の反応で、より多くの量のメチルエーテルを生成した。100mLの沸騰フラスコに、プロピレングリコール5g(PG、65.7mmol)、炭酸ジメチル11.06mL(131.4mmol)、炭酸カリウム18.2g、およびメタノール40mLを加えた。混合物にフリードリヒ冷却器を装備し、終夜還流した(約80℃)。この後、不均一な混合物を室温に冷却し、過剰の炭酸カリウムを濾過し、濾液を貯蔵した。濾液の試料をGC/MSにより定量分析した。その結果は、約40%のPGが対応するモノメチルエーテル(18%Aおよび18%B)に均等に転換し、ジメチルエーテルへの転換率は約4%であったことを示した。
実験:実施例1に記載された反応と同様にして、プロピレングリコールと炭酸ジメチルの比を1:2とした別の反応で、より多くの量のメチルエーテルを生成した。100mLの沸騰フラスコに、プロピレングリコール5g(PG、65.7mmol)、炭酸ジメチル11.06mL(131.4mmol)、炭酸カリウム18.2g、およびメタノール40mLを加えた。混合物にフリードリヒ冷却器を装備し、終夜還流した(約80℃)。この後、不均一な混合物を室温に冷却し、過剰の炭酸カリウムを濾過し、濾液を貯蔵した。濾液の試料をGC/MSにより定量分析した。その結果は、約40%のPGが対応するモノメチルエーテル(18%Aおよび18%B)に均等に転換し、ジメチルエーテルへの転換率は約4%であったことを示した。
実施例3:PGメチルエーテル化、ニート
実験:PTFE被覆した電磁撹拌子を装備した100mLの一口沸騰フラスコに、プロピレングリコール1g(PG、13.1mmol)、炭酸カリウム7.27g(52.6mmol)、および炭酸ジメチル50mLを加えた。水で冷却したフリードリヒ冷却器を沸騰フラスコに装着し、次いで混合物を終夜90℃に加熱した。この後、一定分量を取り、濾過し、GC/MS分析した。それによって、PGはすべてジメチルエーテル類似体に変換され、モノメチルエーテル生成物の形跡はないことが明らかになった。
実施例4:メタノール中におけるEGメチルエーテル化
実験:100mLの沸騰フラスコに、エチレングリコール1g(EG、16.1mmol)、炭酸ジメチル1.35mL(16.1mmol)、炭酸カリウム11.13g(52.6mmol)、およびメタノール40mLを加えた。混合物にフリードリヒ冷却器を装備し、終夜還流した(約80℃)。この後、不均一な混合物を室温に冷却し、過剰の炭酸カリウムを濾過し、濾液を貯蔵した。濾液の試料をGC/MSにより定量分析した。その結果は、約15%のEGが対応するモノメチルエーテルAおよびBに均等に転換したことを示唆した。EGジメチルエーテルも他の生成物も認められなかった。
実施例5:EGメチルエーテル化、ニート
実験:PTFE被覆した電磁撹拌子を装備した100mLの一口沸騰フラスコに、エチレングリコール1g(EG、16.1mmol)、炭酸カリウム11.13g(52.6mmol)、および炭酸ジメチル50mLを加えた。水で冷却したフリードリヒ冷却器を沸騰フラスコに装着し、次いで混合物を終夜90℃に加熱した。この後、一定分量を取り、濾過し、GC/MS分析した。それによって、EGはすべてジメチルエーテル類似体に変換され、モノメチルエーテル生成物を示すものはないことが明らかになった。
実施例6:メタノール中におけるEGメチルエーテル化(1:1 EG/DMC)
実験:100mLの沸騰フラスコに、エチレングリコール1g(EG、16.1mmol)、炭酸ジメチル1.35mL(16.1mmol)、炭酸カリウム11.13g(52.6mmol)、およびメタノール40mLを加えた。混合物にフリードリヒ冷却器を装備し、終夜還流した(約80℃)。この後、不均一な混合物を室温に冷却し、過剰の炭酸カリウムを濾過し、濾液を貯蔵した。濾液の試料をGC/MSにより定量分析した。その結果は、約15%のEGが対応するモノメチルエーテルAおよびBに均等に転換したことを示唆した。EGジメチルエーテルも他の生成物も認められなかった。
C.線状アルキレングリコールカーボナート
C.線状アルキレングリコールカーボナート
実施例7:ジフェニルプロパン−1,2−ジイルジカーボナート、PGジフェニルカーボナートCの合成
実験:楕円形のPTFE電磁撹拌子を装備した25mLの丸底フラスコに、プロピレングリコールA 1g(13.1mmol)、ジフェニルカーボナートB 5.65g(25.2mmol)、および炭酸カリウム3.65g(25.2mmol)を加えた。撹拌しながら、アルゴン雰囲気下で、不均一な混合物を100℃に終夜加熱した。この後、混合物を塩化メチレン20mLで希釈し、濾過して、過剰の固形物を除去し、TLC(酢酸エチル中2%メタノール、UV−Visおよび過マンガン酸カリウム照明)で分析した。それによって、プロピレングリコールはすべて消費され、さらに1スポットしか示されなかったことが示唆された。母液の一定分量を取り、CDC13で希釈し、NMRにより分析した。1H NMR(CDCl3,400MHz)δ(ppm)7.29〜7.27(m,4H)、7.17〜7.15(m,4H)、7.13〜7.11、4.70〜4.69(m,1H)、4.10〜4.08(m,1H)、4.01〜3.99(m,1H)、1.47(s,3H);13C NMR(CDCl3,125MHz)δ(ppm)158.54、157.51、153.38、151.15、129.78、126.16、121.32、116.14、114.65、74.05、73.02、16.55。
D.フラン系ジオール(FDMおよびbHMTHF)エーテル
D.フラン系ジオール(FDMおよびbHMTHF)エーテル
実施例1.(5−(メトキシメチル)フラン−2−イル)メタノールB、2,5−ビス(メトキシメチル)フランCの合成
実験:PTFE被覆した電磁撹拌子を装備した10mLの一口沸騰フラスコに、A 100mg(FDM、0.780mmol)、炭酸カリウム539mg(3.902mmol)、および炭酸ジメチル5mL(413mmol)を加えた。還流冷却器をフラスコに備え付け、撹拌しながら、不均一な混合物を、90℃に8時間加熱した。この後、残存している炭酸カリウムを濾過により除去し、濾液を減圧濃縮した。得られた淡黄色油を最少量の塩化メチレンに溶解し、予め作製済みのシリカゲルカラムに加え、酢酸エチルを用いたフラッシュクロマトグラフィーにより、2組の画分が得られた。A)半透明油としてCを含む画分、Rf=0.72、濃縮後重量26mg。この材料の元素分析によって、以下の結果が明らかになった。C8H12O3の期待値、C 61.52;H 7.74。測定値、C 61.43;H 7.85。B)ワックス状ベージュ色固体としてBを表す画分、Rf=0.54、濃縮後の重量21mg。この物質の元素分析によって、以下の結果が開示された。C7H10O3の期待値、C 59.15;H 7.09。測定値、C 59.28;H 7.07。
実施例2: ((2S,5R)−5−(メトキシメチル)テトラヒドロフラン−2−イル)メタノール、((2S,5S)−5−(メトキシメチル)テトラヒドロフラン−2−イル)メタノール、((2R,5R)−5−(メトキシメチル)テトラヒドロフラン−2−イル)メタノールB;(2R,5S)−2,5−ビス(メトキシメチル)テトラヒドロフラン、(2S,5S)−2,5−ビス(メトキシ−メチル)テトラヒドロフランCの合成
実験:PTFE被覆した電磁撹拌子を装備した25mLの一口丸底フラスコに、A 250mg(シス/トランス 9:1、1.89mmol)、炭酸カリウム1.05g(7.57mmol)、および炭酸ジメチル15mLを加えた。還流冷却器をフラスコに備え付け、撹拌しながら、不均一な混合物を、90℃に12時間加熱した。この後、淡黄色残渣を減圧濃縮し、緩い透明油を得た。次いで、この油を最少量の塩化メチレンに溶解し、予め作製済みのシリカゲルカラムに加え、酢酸エチル溶出液を用いたフラッシュクロマトグラフィーにより、2組の画分が得られた。A)Cを構成する画分(Rf=0.67、濃縮後緩い無色油68mg)、以下の元素分析結果を開示した:C8H16O3の期待値、C 59.98;H 10.07。測定値、C 59.87;H 10.01。B)Bを含む画分(Rf=0.46、濃縮後緩い無色油94mg)、以下の元素分析結果を明らかにした:C7H14O3の期待値、C 57.51;H 9.65。測定値、C 57.70;H 9.53。
E.フラン系ジオールカーボナート
E.フラン系ジオールカーボナート
実施例1.(5−(ヒドロキシメチル)フラン−2−イル)メチルプロピルカーボナートB、フラン−2,5−ジイルビス(メチレン)ジプロピルビス(カーボナート)C
実験:PTFE被覆した電磁撹拌子を装備した5mLの一口丸底フラスコに、A 100mg(0.780mmol)、ジプロピルカーボナート1.21mL(DPC、7.80mmol)、およびDIEA 543μL(3.12mmol)を加えた。アルゴン入口に取り付けられたゴムセプタムで口を塞ぎ、混合物をアルゴン雰囲気下で激しく撹拌しながら120℃に終夜加熱した。この後、過剰のDPCおよびDIEAを高真空下で除去し、混合物を塩化メチレン1mLに溶解し、予め作製済みのシリカゲルカラムに加え、ヘキサン/酢酸エチル溶出液をグラジエントで用いたフラッシュクロマトグラフィーにより、C(Rf1=0.72)に特有の画分が重量22mg、濃縮後粘着性の半透明半固体として得られた。1H NMR分析(400MHz、CDC13)によって、以下のシグナルδ(ppm)が明らかになった。6.23(d,J=8.2Hz,1H)、6.15(d,J=8.2Hz,1H)、5.21(s,2H)、5.10(t,J=6.8Hz,1H)、4.24(d,J=6.2Hz,2H)、4.10(t,J=7.4Hz,2H)、1.59(m,2H)、1.10(t,J=7.0Hz,3H)。
さらに、B(Rf=0.54)に特有の溶出分画を単離し、濃縮後緩い無色油28mgを得た。混合物の1H NMR分析によって、以下のシグナルδ(ppm)が明らかになった。6.25(s,2H)、5.20(s,2H)、4.22(d,J=6.2Hz,2H)、1.61(m,2H)、1.03(t,J=6.8Hz,3H)。
実施例2:((2R,5S)−5−(ヒドロキシメチル)テトラヒドロフラン−2−イル)メチルプロピルカーボナート、((2S,5S)−5−(ヒドロキシメチル)テトラヒドロフラン−2−イル)メチルプロピルカーボナート、((2R,5R)−5−(ヒドロキシメチル)テトラヒドロフラン−2−イル)メチルプロピルカーボナートB;ジプロピル(((2R,5S)−テトラヒドロフラン−2,5−ジイル)ビス(メチレン))ビス(カーボナート)、ジプロピル(((2S,5S)-テトラヒドロフラン−2,5−ジイル)ビス(メチレン))ビス(カーボナート)Cの合成
実験:PTFE被覆した電磁撹拌子を装備した5mLの一口丸底フラスコに、A 100mg(0.751mmol)、ジプロピルカーボナート1.17mL(DPC、7.51mmol)、およびDIEA 522μL(3.00mmol)を加えた。アルゴン入口に取り付けられたゴムセプタムで口を塞ぎ、混合物をアルゴン雰囲気下で激しく撹拌しながら120℃に終夜加熱した。この後、過剰のDPCおよびDIEAを高真空下で除去し、粘着性の黄色油を最少量の塩化メチレンに溶解し、予め作製済みのシリカゲルカラムに加えた。酢酸エチルを溶出液として用いたフラッシュクロマトグラフィーにより、2組の画分が得られた。A)緩い無色油、Rf=0.70、濃縮後重量18mg、元素分析により分析した:14H24O7の期待値、C 55.25;H 7.95。測定値、C 55.12;H 7.84。B)緩い無色油、Rf=0.52、濃縮後重量26mg:C10H18O5の期待値、C 55.03;H 8.31。測定値、C 55.16;H 8.24。
本発明を概括的に、また例により詳細に説明してきた。当業者は、本発明が具体的に開示されている実施形態に必ずしも限定されないこと、本発明の範囲内で使用することができる現在公知のまたは開発されるであろう他の等価成分を含めて、以下の特許請求の範囲またはそれらの等価物によって画定される本発明の範囲から逸脱することなく修正および変形を行うことができることを了解する。したがって、変更が特段に本発明の範囲を逸脱しない限り、変更は本明細書に含まれるものと解釈されるべきである。
Claims (15)
- 第1の経路または第2の経路を含む、モノエーテルをジオール化合物から調製する方法であって、
前記第1の経路において、ジオール化合物とR1有機酸を、ブレンステッド酸の存在下で、ジオール化合物のR1モノエステルを形成するのに十分な温度で十分な時間接触させ、次いでジオール化合物のR1モノエステルと式R2(CO3)R2のR2アルキルジエステルを、脱プロトン化剤の存在下で、モノエステルエーテルを形成するのに十分な温度で十分な時間接触させ、または
前記第2の経路において、ジオール化合物と式R2(CO3)R2のR2アルキルジエステルを、脱プロトン化剤の存在下で、ジオール化合物のモノエステルを形成するのに十分な温度で十分な時間接触させ、次いでジオール化合物のモノエステルとR1有機酸を、ブレンステッド酸の存在下で、モノエステルエーテルを形成するのに十分な温度で十分な時間接触させ、
R1およびR2が同じかまたは異なるアルキル、シクロアルキルまたは芳香族部分である、方法。 - ジオール化合物が、エチレングリコール(EG)、プロピレングリコール(PG)および2,3−ブタンジオール(BDO)からなる群より選択される少なくとも1種である、請求項1に記載の方法。
- ジオール化合物が、フランジメタノール(FDM)およびテトラヒドロフランジメタノール(THFジオール)からなる群より選択される少なくとも1種である、請求項1に記載の方法。
- 前記R1有機酸が酢酸であり、かつ前記モノエステルがエーテルアセタート化合物である、請求項1に記載の方法。
- R1およびR2が、同じであるか異なるC2〜C8アルキル部分である、請求項1に記載の方法。
- 前記モノカーボナートおよび/またはジカーボナートがC3〜C8のR基を有する、請求項10に記載の方法。
- 前記脱プロトン化剤がブレンステッド塩基である、請求項1に記載の方法。
- 前記脱プロトン化剤が、炭酸カリウム、炭酸ナトリウム、炭酸カルシウム、およびアミンからなる群より選択される、請求項1に記載の方法。
- ブレンステッド酸および脱プロトン化剤と接触させるための前記温度が、約70℃〜150℃の間の温度である、請求項1に記載の方法。
- ブレンステッド酸および脱プロトン化剤と接触させるための前記温度が、約80℃〜130℃の間の温度である、請求項1に記載の方法。
- ブレンステッド酸および脱プロトン化剤と接触させるための前記温度が、約90℃〜約120℃の間の温度である、請求項1に記載の方法。
- 前記脱プロトン化剤が、ジオール化合物1個当たり少なくとも1〜約3化学量論当量の量で存在している無機カーボナートである、請求項1に記載の方法。
- ジオール化合物と式R2(CO3)R2のアルキルジエステルを、脱プロトン化剤の存在下で、ジオール化合物のR2アルキルカーボナートを形成するのに十分な温度で十分な時間接触させる工程を含む、ジオール化合物のアルキルカーボナートを作製する方法。
- ジオール化合物が、エチレングリコール(EG)、プロピレングリコール(PG)および2,3−ブタンジオール(BDO)からなる群より選択される少なくとも1種である、請求項13に記載の方法。
- ジオール化合物が、フランジメタノール(FDM)およびテトラヒドロフランジメタノール(THFジオール)からなる群より選択される少なくとも1種である、請求項13に記載の方法。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361918795P | 2013-12-20 | 2013-12-20 | |
| US61/918,795 | 2013-12-20 | ||
| PCT/US2014/068809 WO2015094716A1 (en) | 2013-12-20 | 2014-12-05 | Synthesis of isohexide ethers and carbonates |
| USPCT/US2014/068809 | 2014-12-05 | ||
| US201462093683P | 2014-12-18 | 2014-12-18 | |
| US62/093,683 | 2014-12-18 | ||
| PCT/US2014/071512 WO2015095710A1 (en) | 2013-12-20 | 2014-12-19 | Direct synthesis of bio-based alkyl & furanic diol ethers, acetates, ether-acetates, and carbonates |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017507899A true JP2017507899A (ja) | 2017-03-23 |
| JP2017507899A5 JP2017507899A5 (ja) | 2018-01-18 |
Family
ID=53403742
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016538518A Withdrawn JP2017507899A (ja) | 2013-12-20 | 2014-12-19 | バイオベースのアルキルおよびフラン系ジオールエーテル、アセタート、エーテル−アセタート、ならびにカーボナートの直接合成 |
Country Status (9)
| Country | Link |
|---|---|
| EP (1) | EP3083548A4 (ja) |
| JP (1) | JP2017507899A (ja) |
| KR (1) | KR20160099646A (ja) |
| CN (1) | CN105849080A (ja) |
| AU (1) | AU2014369062A1 (ja) |
| BR (1) | BR112016014267A2 (ja) |
| CA (1) | CA2934512A1 (ja) |
| MX (1) | MX2016008061A (ja) |
| WO (1) | WO2015095710A1 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2019001840A (es) | 2016-08-16 | 2019-05-09 | Ppg Ind Ohio Inc | Composicion polimerizable para articulos opticos. |
| US10865172B1 (en) * | 2019-09-04 | 2020-12-15 | Eastman Chemical Company | Aromatic enol ethers |
| US10858304B1 (en) * | 2019-09-04 | 2020-12-08 | Eastman Chemical Company | Aromatic enol ethers |
| CN111349492B (zh) * | 2020-02-28 | 2021-05-25 | 浙江糖能科技有限公司 | 2,5-四氢呋喃二甲醇脂肪酸二酯在柴油添加剂的应用 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2963489A (en) * | 1953-07-30 | 1960-12-06 | American Cyanamid Co | 3, 4-furandimethanol and derivatives |
| US4012424A (en) * | 1975-08-07 | 1977-03-15 | Chem Systems Inc. | Cracking of mixtures containing hydroxyesters |
| US4170596A (en) * | 1976-10-05 | 1979-10-09 | Kowa Company, Ltd. | Novel monoesters of cis-cyclopentenediol, process for preparation thereof, and process for preparation of lactones from the monoesters |
| US4192949A (en) * | 1977-06-28 | 1980-03-11 | Basf Aktiengesellschaft | Preparation of aralkyl phenyl ethers and alkyl phenyl ethers |
| JPH07100702B2 (ja) * | 1987-07-22 | 1995-11-01 | 三井石油化学工業株式会社 | 環状カ−ボネ−トの製造方法 |
| JPH09227435A (ja) * | 1996-02-26 | 1997-09-02 | Mitsui Toatsu Chem Inc | (ポリ)エチレングリコールジエーテルの製造法 |
| KR100593529B1 (ko) * | 2004-03-31 | 2006-06-28 | 주식회사 엘지생활건강 | 에스테르계 표백활성화제 화합물의 제조방법 |
| FR2950892B1 (fr) * | 2009-10-01 | 2011-11-18 | Roquette Freres | Procede de preparation de di(alkylcarbonate) de dianhydrohexitol |
| WO2012003501A2 (en) * | 2010-07-02 | 2012-01-05 | Reviva Pharmaceuticals, Inc. | Compositions, synthesis, and methods of using cycloalkylmethylamine derivatives |
-
2014
- 2014-12-19 KR KR1020167018951A patent/KR20160099646A/ko not_active Application Discontinuation
- 2014-12-19 JP JP2016538518A patent/JP2017507899A/ja not_active Withdrawn
- 2014-12-19 CN CN201480071558.7A patent/CN105849080A/zh active Pending
- 2014-12-19 EP EP14871997.4A patent/EP3083548A4/en not_active Withdrawn
- 2014-12-19 AU AU2014369062A patent/AU2014369062A1/en not_active Abandoned
- 2014-12-19 MX MX2016008061A patent/MX2016008061A/es unknown
- 2014-12-19 CA CA2934512A patent/CA2934512A1/en not_active Abandoned
- 2014-12-19 BR BR112016014267A patent/BR112016014267A2/pt not_active IP Right Cessation
- 2014-12-19 WO PCT/US2014/071512 patent/WO2015095710A1/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| MX2016008061A (es) | 2017-02-27 |
| CA2934512A1 (en) | 2015-06-25 |
| CN105849080A (zh) | 2016-08-10 |
| AU2014369062A1 (en) | 2016-06-30 |
| WO2015095710A1 (en) | 2015-06-25 |
| EP3083548A4 (en) | 2017-08-30 |
| BR112016014267A2 (pt) | 2017-08-08 |
| KR20160099646A (ko) | 2016-08-22 |
| EP3083548A1 (en) | 2016-10-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Teng et al. | A review on the performance of glycerol carbonate production via catalytic transesterification: Effects of influencing parameters | |
| EA017997B1 (ru) | 5-замещенные 2-(алкоксиметил)фураны | |
| EA017536B1 (ru) | Простые гидроксиметилфурфуралевые эфиры сахаров или hmf и смешанных спиртов | |
| US9890131B2 (en) | Direct synthesis of bio-based alkyl and furanic diol ethers, acetates, ether-acetates, and carbonates | |
| US20170044123A1 (en) | Synthesis of r-glucosides, sugar alcohols, reduced sugar alcohols, and furan derivatives of reduced sugar alcohols | |
| JP2017507899A (ja) | バイオベースのアルキルおよびフラン系ジオールエーテル、アセタート、エーテル−アセタート、ならびにカーボナートの直接合成 | |
| Lluna‐Galán et al. | Catalytic Reductive Alcohol Etherifications with Carbonyl‐Based Compounds or CO2 and Related Transformations for the Synthesis of Ether Derivatives | |
| Roze et al. | Catalytic etherification of glycerol with alcohols | |
| US10519124B2 (en) | Synthesis of R-glucosides, sugar alcohols, reduced sugar alcohols, and furan derivatives of reduced sugar alcohols | |
| EP3221301A1 (en) | Acid-catalyzed acylation of 5-(hydroxylmethyl)-furfural reduction products | |
| Sathicq et al. | Alkyl carbonate derivatives of furanics: A family of bio-based stable compounds | |
| WO2014008301A1 (en) | Biorefining compounds and organocatalytic upgrading methods | |
| JP2017525659A (ja) | モノアンヒドロ−ヘキシトールのモノアルキルエーテルの組成物、その製造方法、ならびにその使用 | |
| WO2017091412A1 (en) | Oligomers of fdca and glycols from a one-pot esterification-transesterification process using water-tolerant metal triflate catalyst | |
| Giomi et al. | A convenient method for producing mono-and dichlorohydrins from glycerol | |
| US20170121258A1 (en) | Synthesis of r-glucosides, sugar alcohols, reduced sugar alcohols, and furan derivatives of reduced sugar alcohols | |
| Kawale et al. | Applications of Glycerol as Green Solvent | |
| Hattori et al. | An efficient method for the refinement of 1, 3-methyleneglycerol via bridged acetal exchange and the synthesis of a symmetrically branched glycerol trimer | |
| WO2017030685A1 (en) | One-pot synthesis of anhydropentitol mono- and diethers | |
| JP2022504061A (ja) | アルカンジオールの製造方法 | |
| Jia et al. | Novel Synthesis of 4-chloro-3-hydroxy-2-pyrone by using Glyceraldehyde Acetonide | |
| WO2017065980A1 (en) | Preparation of a sugar-derived ester, glycol and polymers therefrom | |
| Kishore | Applications of Glycerol as Green Solvent | |
| US20110173877A1 (en) | Process for preparing a hydrocarbon or mixture of hydrocarbons | |
| Marsden | New Methods for the Deoxygenation of 1, 2-diols and Epoxides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171128 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20171128 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20180719 |