JP2017123794A - アルカリホスファターゼ及びその製造方法 - Google Patents
アルカリホスファターゼ及びその製造方法 Download PDFInfo
- Publication number
- JP2017123794A JP2017123794A JP2016003867A JP2016003867A JP2017123794A JP 2017123794 A JP2017123794 A JP 2017123794A JP 2016003867 A JP2016003867 A JP 2016003867A JP 2016003867 A JP2016003867 A JP 2016003867A JP 2017123794 A JP2017123794 A JP 2017123794A
- Authority
- JP
- Japan
- Prior art keywords
- alkaline phosphatase
- seq
- dna
- polynucleotide
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images



Landscapes
- Enzymes And Modification Thereof (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
Description
すなわち本発明は、以下の態様を包含する。
(1) 配列番号4から8のいずれかに記載のアミノ酸配列を含むアルカリホスファターゼ(以下、本発明のアルカリホスファターゼと称する)。
(2) 本発明のアルカリホスファターゼをコードするポリヌクレオチド(以下、本発明のポリヌクレオチドと称する)。
(3)本発明のポリヌクレオチドを含有する発現ベクター(以下、本発明の発現ベクターと称する。)。
(4)本発明の発現ベクターを有する形質転換細胞((以下、本発明の細胞と称する。)。
(5)本発明の細胞が、大腸菌、もしくはチャイニーズハムスター卵巣細胞である(4)に記載の細胞。
(6)本発明の細胞を培養し、得られた培養液からアルカリホスファターゼを回収する、アルカリホスファターゼの生産方法(以下、本発明のアルカリホスファターゼ生産方法と称する。)。
(1)K323N (2)S385G/N
具体的には、配列番号4から8のいずれかに記載のアミノ酸配列を含むアルカリホスファターゼを例示でき、当該ポリペプチドのN末端側および/またはC末端側にオリゴペプチドの付加された誘導体も包含している。bIAPを含め哺乳動物由来のアルカリホスファターゼの多くは膜結合性のため、アルカリホスファターゼのC末端側20から30アミノ酸残基にGPIアンカー領域を有しているが、分泌発現させるためにGPIアンカー領域を除去してもよい。
本検討に用いたプラスミド(発現ベクター)を、それぞれ以下に示す方法で作製した。
(A−1)N末端側に仔牛小腸由来のアルカリホスファターゼ(CIP)のシグナルペプチド(配列番号18)を、C末端側にヒスチジン6個からなるHisタグを、配列番号1に記載のアミノ酸配列からなるbIAPIIに付加したポリペプチドをコードする配列番号19に記載のポリヌクレオチドを含むプラスミドである、pCIP−1をOPERON Biotechnologies社で合成し、これを鋳型とし、配列番号20に記載の配列からなるオリゴヌクレオチド(Primer−#1)と配列番号21に記載の配列からなるオリゴヌクレオチド(Primer−#2)をプライマーセットとして、Taq DNA polymerase(ライフテクノロジー社製)を用いてPCRを行なった。
(A−2)得られた1.5kbpのポリヌクレオチド(DNA−1)を、CMVプロモーターを用いた発現ベクターであるpOptiVEC−TOPO(ライフテクノロジーズ社製)のクローニングサイトに挿入した。
(A−3)(A−2)でDNA−1を挿入したプラスミドの塩基配列を調べた。結果、配列番号1に記載のアミノ酸配列からなるポリペプチドをコードするポリヌクレオチド(配列番号22)が挿入されたプラスミド(5.9kbp)であることを確認し、当該プラスミドをpCIP1000と名付けた。
pCIP−1を制限酵素EcoRIとMluIで二重消化して得た1.5kbpのポリヌクレオチド(DNA−2)と、pCIP1000を制限酵素EcoRIとMluIで二重消化して得た4.4kbpのポリヌクレオチド(DNA−3)をDNA Ligation Kit Mighty Mix(タカラバイオ社製)を用いてライゲーションすることで得たプラスミド(5.9kbp)をpCIP1000Hと名付けた。
(C−1)pTrc99Aを制限酵素HindIIIで消化後、DNA BluntingKit(タカラバイオ社製)で平滑化し、それを制限酵素NcoIで消化して4.1kbpのポリヌクレオチド(DNA−4)を得た。
(C−2)配列番号23に記載の配列からなるオリゴヌクレオチド(Primer−#3)、配列番号24に記載の配列からなるオリゴヌクレオチド(Primer−#4)、配列番号25に記載の配列からなるオリゴヌクレオチド(Primer−#5)、配列番号26に記載の配列からなるオリゴヌクレオチド(Primer−#6)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、配列番号27に記載のアミノ酸配列からなるSalmonella typhimurium由来mglBのシグナルペプチドをコードする0.085kbpのオリゴヌクレオチド(DNA−5)を得た。
(C−4)DNA−5とDNA−6を鋳型セットにして、配列番号23に記載の配列からなるオリゴヌクレオチド(Primer−#3)と配列番号30に記載の配列からなるオリゴヌクレオチド(Primer−#9)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、配列番号27に記載のアミノ酸配列からなるmglBシグナルペプチド、Mycタグおよび配列番号1に記載のアミノ酸配列からなるbIAPIIのN末端6アミノ酸をコードする0.138kbpのオリゴヌクレオチド(DNA−7)を得た。
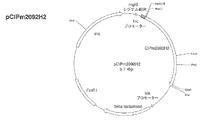
(C−5)DNA−4と、NA−7を制限酵素BspHIとBbvCIで二重消化して得たオリゴヌクレオチド(DNA−8)と、pCIP1000Hを制限酵素BbvCIとHpaIで二重消化して得た1.5kbpのポリヌクレオチド(DNA−9)をライゲーションすることで得たプラスミド(5.7kbp)をpCIPm2092H(図1)と名付けた。
(D−1)pCIPm2092Hを鋳型とし、配列番号31に記載の配列からなるオリゴヌクレオチド(Primer−#10)と配列番号32に記載の配列からなるオリゴヌクレオチド(Primer−#11)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、1.5kbpのポリヌクレオチド(DNA−10)を得た。
(D−2)DNA−10を鋳型とし、配列番号31に記載の配列からなるオリゴヌクレオチド(Primer−#10)と配列番号33に記載の配列からなるオリゴヌクレオチド(Primer−#12)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、1.5kbpのポリヌクレオチド(DNA−11)を得た。
(D−3)DNA−11を制限酵素BbvCIとAgeIで二重消化して得た1.5kbpのポリヌクレオチド(DNA−12)とpCIPm2092Hを制限酵素BbvCIとAgeIで二重消化して得た4.2kbpのポリヌクレオチド(DNA−13)をライゲーションすることで得たプラスミド(5.7kbp)をpCIPm2092H2と名付けた。pCIPm2092H2は、配列番号27に記載のアミノ酸配列からなるmglBシグナルペプチド、Mycタグおよび配列番号1に記載のアミノ酸配列からなるbIAPII、および配列番号10に記載のアミノ酸配列からなるヒスタグをコードするポリヌクレオチドを含むプラスミドである(図1)。
(E−1)配列番号34に記載の配列からなるオリゴヌクレオチド(Primer−#13)、配列番号35に記載の配列からなるオリゴヌクレオチド(Primer−#14)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、0.045kbpのオリゴヌクレオチド(DNA−14)を得た。
(E−2)pCIPm2092H2を鋳型とし、配列番号36に記載の配列からなるオリゴヌクレオチド(Primer−#15)と配列番号37に記載の配列からなるオリゴヌクレオチド(Primer−#16)、配列番号32に記載の配列からなるオリゴヌクレオチド(Primer−#11)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、0.535kbpのポリヌクレオチド(DNA−15)を得た。
(E−4)DNA−16を制限酵素XmaIとSacIIで二重消化して得た0.5kbpのポリヌクレオチド(DNA−17)とpCIPm2092H2を制限酵素XmaIとSacIIで二重消化して得た5.2kbpのポリヌクレオチド(DNA−18)をライゲーションすることで得たプラスミド(5.7kbp)をpCIPm2093H2と名付けた。
pCIPm2093H2は、配列番号27に記載のアミノ酸配列からなるmglBシグナルペプチド、Mycタグ、配列番号2に記載のアミノ酸配列からなるbIAPII変異体(配列番号1に記載のアミノ酸配列からなるbIAPIIの322位がアスパラギン酸に置換された変異体)および配列番号10に記載のアミノ酸配列からなるヒスタグをコードするポリヌクレオチドを含むプラスミドである。
(F−1)プラスミドpECEdhfr(Yasukawaら、J.Biochem.、108,673−676(1990))を鋳型とし、配列番号38に記載の配列からなるオリゴヌクレオチド(Primer−17)および配列番号39に記載の配列からなるオリゴヌクレオチド(Primer−18)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、0.37kbpの増幅産物(ポリヌクレオチド)(DNA−18)を得た。
(F−2)DNA−18を鋳型とし、配列番号38に記載の配列からなるオリゴヌクレオチド(Primer−17)および配列番号40に記載の配列からなるオリゴヌクレオチド(Primer−19)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、0.39kbpの増幅産物(ポリヌクレオチド)(DNA−19)を得た。
(F−3)プラスミドpECEdhfrを鋳型とし、配列番号41に記載の配列からなるオリゴヌクレオチド(Primer−20)および配列番号42に記載の配列からなるオリゴヌクレオチド(Primer−21)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、0.69kbpの増幅産物(ポリヌクレオチド)(DNA−20)を得た。
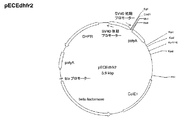
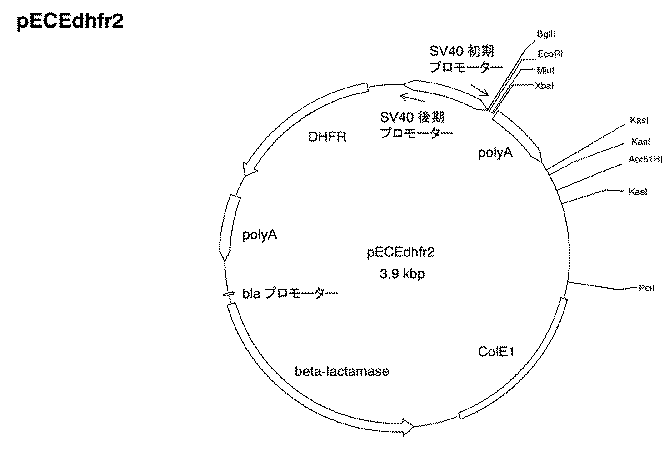
(F−5)DNA−19を制限酵素MfeIとXbaIで二重消化し得られたポリヌクレオチド(DNA−22)と、DNA−21を制限酵素XbaIとPciIで二重消化して得られたポリヌクレオチド(DNA−23)と、プラスミドpECEdhfrを制限酵素EcoRIとPciIで二重消化し得られた2.8kbpのポリヌクレオチド(DNA−24)を、ライゲーションすることで、配列番号44に記載の塩基配列を含んだプラスミドpECEdhfr2を作製した。なおプラスミドpECEdhfr2は、プラスミドpECEdhfrのSV40後期(late)プロモーターとdhfr遺伝子の間にある制限酵素EcoRIの認識部位をなくし、かつ、SV40初期(early)プロモーターからSV40ポリAまでの領域を置換したプラスミドである(図2)。
(G−1)pECEdhfr2を制限酵素KasIで消化後、DNA Ligation Kit Mighty Mix(タカラバイオ社製)を用いてライゲーションすることで、配列番号37に記載の塩基配列を含んだプラスミドpECEdhfr3を作製した。
(H−1)pTrc99Aを鋳型とし、配列番号45に記載の配列からなるオリゴヌクレオチド(Primer−23)および配列番号46に記載の配列からなるオリゴヌクレオチド(Primer−24)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、0.063kbpの増幅産物(ポリヌクレオチド)(DNA−25)を得た。
(H−2)配列番号47に記載の配列からなるオリゴヌクレオチドを鋳型とし、配列番号48に記載の配列からなるオリゴヌクレオチド(Primer−25)および配列番号49に記載の配列からなるオリゴヌクレオチド(Primer−26)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、マウスMMP−9由来のシグナルペプチド(配列番号50)をコードする0.075kbpの増幅産物(ポリヌクレオチド)(DNA−26)を得た。
(H−3)pTrc99Aを鋳型とし、DNA−25とDNA−26をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、0.15kbpの増幅産物(ポリヌクレオチド)(DNA−27)を得た。
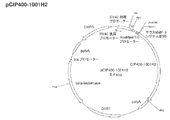
スミドである(図3)。
(I−1)pCIP400−1001H2を鋳型とし、配列番号51に記載の配列からなるオリゴヌクレオチド(Primer−27)および配列番号52に記載の配列からなるオリゴヌクレオチド(Primer−28)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、0.3kbpの増幅産物(ポリヌクレオチド)(DNA−31)を得た。
(I−2)DNA−31と配列番号53に記載の配列からなるオリゴヌクレオチド(Primer−29)をプライマーセットとして、PrimeSTAR HS DNA polymerase(タカラバイオ社製)を用いてPCRを行ない、0.33kbpの増幅産物(ポリヌクレオチド)(DNA−32)を得た。
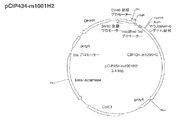
(I−3)DNA−32を制限酵素SfiIとAor51HIで二重消化して得られた0.3kbpのポリヌクレオチド(DNA−33)と、pCIP400−1001H2を制限酵素SfiIとAor51HIで二重消化して得られた5.1kbpのポリヌクレオチド(DNA−34)をライゲーションすることで得たプラスミド(5.4kbp)をpCIP434−m1001H2と名付けた。pCIP434−m1001H2は、配列番号50に記載のアミノ酸配列からなるマウスMMP−9由来のシグナルペプチド、配列番号9に記載のアミノ酸配列からなるMycタグ、配列番号1に記載のアミノ酸配列からなるbIAPII、および配列番号10に記載のアミノ酸配列からなるヒスタグをコードするポリヌクレオチドを含むプラスミドである(図4)。
エラープローンPCR法による変異導入
pCIPm2093H2を鋳型(50ng/μl)として、配列番号54に記載の配列からなるオリゴヌクレオチド(Primer−30)(0.4μM)、配列番号55に記載の配列からなるオリゴヌクレオチド(Primer−31)(0.4μM)、0.08mM MnCl2、5mM MgCl2、0.2mM dATP、0.2mM dGTP、1mM dCTP、1mM dTTP、5unit Go Taq DNA Polymerase(プロメガ株式会社)を混合し、95℃で2分加熱後、95℃(30秒)、60℃(30秒)、72℃(88秒)のPCR反応を30サイクル繰り返した。1.4kbpの増幅産物をアガロース電気泳動により回収後、制限酵素BbvCIとSacIIで二重消化した。
実施例2のエラープローンPCR法による変異導入で調製した制限酵素消化物とpCIPm2093H2を制限酵素BbvCIとSacIIで二重消化して得た4.3kbpのポリヌクレオチド(DNA−35)をライゲーションして得たプラスミドを大腸菌DH5α(ニッポンジーン社製)にトランスフェクションし、2mM MgCl2、40μg/ml BCIP(5−bromo−4−chloro−3−indolyl phosphate disodium salt)、50μg/ml Carbenicillin Naを含むLB寒天培地にて28℃で培養した。青く呈色したコロニーを、1mM MgCl2、25μg/ml Carbenicillin Naを含む1/2濃度のLB培地にて28℃で培養し、菌濁度(OD600)と培養液中のアルカリホスファターゼ活性を測定し、菌濁度あたりのアルカリホスファターゼ活性の高い菌を回収した。
実施例3で回収した大腸菌を5mM MgCl2、50μg/ml Carbenicillin Naを含む4xYT培地(BactoTrypton 3.2%、Yeast Extract 2%、NaCl 0.5%)にて28℃で培養後、遠心分離(3,500rpm、10min)して菌を回収した。
抗c−Myc抗体(Roche社製)を0.4μg/mlに希釈し100μlずつ96ウェルイムノプレート(NUNC社製 MAXISORP 430341)に添加し、抗体固定化プレートを作製した。それに実施例4で調製したペリプラズム画分を添加し、一時間インキュベートした。プレートを洗浄後、pNPP溶液を添加し室温下で波長405nmの吸光度をモニタリングし吸光度増加速度(OD405/min)を求めた。抗c−Myc抗体固定化プレートに実施例4で調製したペリプラズム画分を添加し、一時間インキュベートした後、プレートを洗浄し、HRP標識抗ヒスタグ抗体(Bethyl社製)を添加し、一時間インキュベートした。プレートを洗浄後、TMB溶液を添加し室温下で波長620nmの吸光度をモニタリングし吸光度増加速度(OD620/min)を求めた。pNPPを基質にした吸光度増加速度(OD405/min)の値を、TMBを基質にした吸光度増加速度(OD620/min)の値で割った値(OD405/OD620)を比活性として表し、対照サンプルと比較した。
実施例5の比活性測定の結果選択されたbIAPII変異体を生産する大腸菌よりプラスミドを抽出し、配列解析を行った。その結果、配列番号2に記載のアミノ酸配列の430位のグルタミン酸がアラニンに置換されていた。このプラスミドをpCIPm2103H2と名付けた。
実施例6で得られたpCIPm2103H2を鋳型として用い、実施例2に記載の変異導入、実施例3に記載の大腸菌での高活性変異体のスクリーニング、実施例4に記載の大腸菌ペリプラズム画分の調製、実施例5に記載の比活性測定、実施例6に記載の配列解析を実施した結果、配列番号3に記載のアミノ酸配列の323位がアスパラギン(N)、385位のセリンがグリシン(G)、アスパラギン(N)に置換された多重変異体が得られた。これらのプラスミドをそれぞれpCIPm2157H2、pCIPm2155H2、pCIPm2156H2と名付けた。
実施例7で得られたbIAPII多重変異体のアミノ酸置換を掛け合わせた多重変異体を作製した。pCIPm2155H2、pCIPm2156H2を制限酵素PstIとMluIで二重消化して得た0.4kbpのポリヌクレオチド(DNA−36、DNA−37)とpCIPm2157H2を制限酵素PstIとMluIで二重消化して得た5.3kbpのポリヌクレオチド(DNA−38)をライゲーションすることで得たプラスミド(5.7kbp)をpCIPm2158H2、pCIPm2159H2と名付けた。
実施例3から8の結果、得られたbIAPII多重変異体のプラスミドを制限酵素BbvCIとMluIで二重消化し、1.4kbpのポリヌクレオチド(DNA−39)を回収した。実施例1(H)で作製したpCIP400−1001H2を制限酵素BbvCIとMluIで二重消化し、3.9kbpのポリヌクレオチド(DNA−40)を回収した。実施例1(I)で作製したpCIP434−m1001H2を制限酵素BbvCIとMluIで二重消化し、3.9kbpのポリヌクレオチド(DNA−41)を回収した。DNA−39とDNA−40、DNA−39とDNA−41をライゲーションすることで、以下に示すプラスミドを作製した。
(1)実施例9で作製したプラスミドを制限酵素PvuIで消化することで直鎖化後、FreeStyle MAX試薬(ライフテクノロジーズ社製)を使用したリポフェクション法によりチャイニーズハムスター卵巣細胞(CHO細胞)であるDG44細胞に導入した。
(2)プラスミドを導入したDG44細胞を、8mMのグルタミンおよび0.18%のPluronic F−68(ライフテクノロジーズ社製)を含んだCD CHO培地(培地1)で、温度37℃、CO2濃度8%、回転振とう速度135rpmで3日間培養した。
(3)培養液を低速(900rpm)で遠心分離することで培養細胞を回収後、8mM グルタミンおよび0.18% Pluronic F−68を含んだCD OptiCHO培地(培地2)に懸濁して(培地交換1回目)、7日間培養を継続した。
(4)(3)と同様にして、培養細胞を回収後、培地2に懸濁して(培地交換2回目)、7日間培養を継続した。
(5)(3)と同様にして、培養細胞を回収後、培地2に懸濁して(培地交換3回目)、7日間培養を継続した。
(6)(3)と同様にして、培養細胞を回収後、培地2に懸濁して(培地交換4回目)、7日間培養を継続した。
(7)(6)の7日目の培養液を遠心分離(12,000rpm、10min)して培養上清を回収した。
抗ヒスタグ抗体(和光純薬社製)を0.5μg/mlに希釈し100μlずつ96ウェルイムノプレート(NUNC社製 MAXISORP 430341)に添加し、抗体固定化プレートを作製した。それに実施例10で調製した、N末端に配列番号9に記載のアミノ酸配列からなるMycタグ、C末端に配列番号10に記載のアミノ酸配列からなるヒスタグが付加された実施例9表5に記載のbIAPII変異体を含んだ培養上清を添加し、一時間インキュベートした。プレートを洗浄後、pNPP溶液を添加し室温下で波長405nmの吸光度をモニタリングし吸光度増加速度(OD405/min)を求めた。抗ヒスタグ抗体固定化プレートに実施例10で調製した、N末端に配列番号9に記載のアミノ酸配列からなるMycタグ、C末端に配列番号10に記載のアミノ酸配列からなるヒスタグが付加された実施例9表5に記載のbIAPII変異体を含んだ培養上清を添加し、一時間インキュベートした後、プレートを洗浄し、HRP標識抗c−Myc抗体(和光純薬社製)を添加し、一時間インキュベートした。プレートを洗浄後、TMB溶液を添加し室温下で波長620nmの吸光度をモニタリングし吸光度増加速度(OD620/min)を求めた。pNPPを基質にした吸光度増加速度(OD405/min)の値を、TMBを基質にした吸光度増加速度(OD620/min)の値で割った値(OD405/OD620)を比活性として表し、対照サンプルと比較した。
比活性測定の結果を図5に示す。配列番号4から8に記載のbIAPII変異体は、配列番号1に記載のbIAPIIと同等の比活性を示した。
実施例10で調製した、bIAPII変異体を含んだ培養上清をアルカリホスファターゼ活性が同等になるように実施例10に記載の培地2で希釈後、7.5μlずつPCR8連チューブ(eppendorf社製)に分注した。それらをGeneAmp PCR system9700(Applied Biosystems社製)で55℃から70℃で10分間加温後、氷冷した。それらにpNPP溶液を150μL添加し、室温で一定時間保温後、それらのうちの100μLを0.5Mの水酸化ナトリウム100μLと混合することで反応を停止し、波長405nmにおける吸光度を測定した。未加温のサンプルの活性を100%としたときの各温度での加温サンプル中の残存活性を示す。
Claims (6)
- 配列番号4から8のいずれかに記載のアミノ酸配列を含むアルカリホスファターゼ。
- 請求項1に記載のアルカリホスファターゼをコードするポリヌクレオチド。
- 請求項2に記載のポリヌクレオチドを含有する発現ベクター。
- 請求項3に記載の発現ベクターを有する形質転換細胞。
- 大腸菌またはチャイニーズハムスター卵巣細胞である請求項4に記載の形質転換細胞。
- 請求項4または5に記載の形質転換細胞を培養し、得られた培養液からアルカリホスファターゼを回収する、アルカリホスファターゼの生産方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016003867A JP6732368B2 (ja) | 2016-01-12 | 2016-01-12 | アルカリホスファターゼ及びその製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016003867A JP6732368B2 (ja) | 2016-01-12 | 2016-01-12 | アルカリホスファターゼ及びその製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017123794A true JP2017123794A (ja) | 2017-07-20 |
| JP6732368B2 JP6732368B2 (ja) | 2020-07-29 |
Family
ID=59363349
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016003867A Active JP6732368B2 (ja) | 2016-01-12 | 2016-01-12 | アルカリホスファターゼ及びその製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6732368B2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2022196538A1 (ja) * | 2021-03-17 | 2022-09-22 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002253269A (ja) * | 2000-07-25 | 2002-09-10 | F Hoffmann La Roche Ag | 酵母におけるアルカリホスファターゼの発現 |
| JP2008005734A (ja) * | 2006-06-28 | 2008-01-17 | Kikkoman Corp | アルカリホスファターゼ |
| JP2010524496A (ja) * | 2007-04-27 | 2010-07-22 | アーエム−ファルマ ベー.フェー. | 修飾ホスファターゼ |
| WO2014007229A1 (ja) * | 2012-07-06 | 2014-01-09 | 東洋紡株式会社 | 改変型アルカリホスファターゼ |
| JP2015078161A (ja) * | 2013-10-18 | 2015-04-23 | 国立大学法人九州大学 | タンパク質検出用融合タンパク質およびタンパク質の検出方法 |
-
2016
- 2016-01-12 JP JP2016003867A patent/JP6732368B2/ja active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002253269A (ja) * | 2000-07-25 | 2002-09-10 | F Hoffmann La Roche Ag | 酵母におけるアルカリホスファターゼの発現 |
| JP2008005734A (ja) * | 2006-06-28 | 2008-01-17 | Kikkoman Corp | アルカリホスファターゼ |
| JP2010524496A (ja) * | 2007-04-27 | 2010-07-22 | アーエム−ファルマ ベー.フェー. | 修飾ホスファターゼ |
| WO2014007229A1 (ja) * | 2012-07-06 | 2014-01-09 | 東洋紡株式会社 | 改変型アルカリホスファターゼ |
| JP2015078161A (ja) * | 2013-10-18 | 2015-04-23 | 国立大学法人九州大学 | タンパク質検出用融合タンパク質およびタンパク質の検出方法 |
Non-Patent Citations (2)
| Title |
|---|
| JOURNAL OF BIOLOGICAL CHEMISTRY, 1998, VOL.273, NO.36, PP.23353-23360, JPN6020002315, ISSN: 0004200233 * |
| JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, 2012, VOL.134, PP.229-246, JPN6020002313, ISSN: 0004200232 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2022196538A1 (ja) * | 2021-03-17 | 2022-09-22 | ||
| WO2022196538A1 (ja) * | 2021-03-17 | 2022-09-22 | 味の素株式会社 | 改変アルカリホスファターゼ |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6732368B2 (ja) | 2020-07-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8097701B2 (en) | Aldehyde tags, uses thereof in site-specific protein modification | |
| US8729232B2 (en) | Aldehyde tags, uses thereof in site-specific protein modification | |
| US20170226495A1 (en) | Sortase molecules and uses thereof | |
| US10227595B2 (en) | Universal protein overexpression tag comprising ramp function, and application thereof | |
| JP2013509867A5 (ja) | ||
| JP2001505783A (ja) | 分泌ヒトタンパク質 | |
| WO2022214065A1 (zh) | Rna修饰嵌合蛋白及其应用 | |
| JP6732368B2 (ja) | アルカリホスファターゼ及びその製造方法 | |
| Terada et al. | Escherichia coli cell-free protein synthesis and isotope labeling of mammalian proteins | |
| JP2017192381A (ja) | 変異型アルカリホスファターゼをコードする遺伝子 | |
| US20150291683A1 (en) | Modified apol1 polypeptides | |
| JP5865002B2 (ja) | 組換えプラスミドベクターおよびそれを用いたタンパク質の製造方法 | |
| Chen et al. | High-efficiency secretory expression of human neutrophil gelatinase-associated lipocalin from mammalian cell lines with human serum albumin signal peptide | |
| JP4441170B2 (ja) | S12リボソームタンパク質に変異を有する大腸菌細胞抽出液及びそれを用いる無細胞系によるタンパク質の製造方法 | |
| JP6131074B2 (ja) | 分泌シグナル配列 | |
| CN109880840B (zh) | 一种重组蛋白大肠杆菌体内生物素化标记系统 | |
| EP1836223B1 (en) | Method for identifying a nucleic acid encoding a hemopexin-like structure which specifically binds a predetermined target molecule. | |
| CN120322561A (zh) | 新型转座酶系统 | |
| Terada et al. | Cell-free protein production for structural biology | |
| US20040209323A1 (en) | Protein expression by codon harmonization and translational attenuation | |
| RU2625010C1 (ru) | Экспрессионный плазмидный вектор для экспрессии активной формы TNFR1-Fc и способ получения рекомбинантного белка | |
| JP2022515427A (ja) | タンパク質生成に対するキメラシグナルペプチド | |
| WO2025163019A1 (en) | Tags for enhanced expression of recombinant proteins | |
| WO2025104126A1 (en) | Tags for enhanced expression of recombinant proteins | |
| CN104011212B (zh) | 利用大肠杆菌yiat蛋白质的靶蛋白的表面表达方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20160115 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20181217 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200204 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200226 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20200707 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20200707 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6732368 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |