JP2011500780A - 新規結晶形 - Google Patents
新規結晶形 Download PDFInfo
- Publication number
- JP2011500780A JP2011500780A JP2010530564A JP2010530564A JP2011500780A JP 2011500780 A JP2011500780 A JP 2011500780A JP 2010530564 A JP2010530564 A JP 2010530564A JP 2010530564 A JP2010530564 A JP 2010530564A JP 2011500780 A JP2011500780 A JP 2011500780A
- Authority
- JP
- Japan
- Prior art keywords
- bosentan
- crystalline
- crystal form
- solid
- until
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000013078 crystal Substances 0.000 title claims description 66
- GJPICJJJRGTNOD-UHFFFAOYSA-N bosentan Chemical compound COC1=CC=CC=C1OC(C(=NC(=N1)C=2N=CC=CN=2)OCCO)=C1NS(=O)(=O)C1=CC=C(C(C)(C)C)C=C1 GJPICJJJRGTNOD-UHFFFAOYSA-N 0.000 claims abstract description 178
- 229960003065 bosentan Drugs 0.000 claims abstract description 178
- 238000004519 manufacturing process Methods 0.000 claims abstract description 22
- 201000010099 disease Diseases 0.000 claims abstract description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 21
- 208000024172 Cardiovascular disease Diseases 0.000 claims abstract description 20
- 102000010180 Endothelin receptor Human genes 0.000 claims abstract description 20
- 108050001739 Endothelin receptor Proteins 0.000 claims abstract description 20
- 230000001404 mediated effect Effects 0.000 claims abstract description 20
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 19
- 206010020772 Hypertension Diseases 0.000 claims abstract description 12
- 206010002383 Angina Pectoris Diseases 0.000 claims abstract description 11
- 206010047163 Vasospasm Diseases 0.000 claims abstract description 11
- 208000028867 ischemia Diseases 0.000 claims abstract description 11
- 238000000034 method Methods 0.000 claims description 70
- 239000007787 solid Substances 0.000 claims description 60
- 239000000203 mixture Substances 0.000 claims description 59
- 239000002904 solvent Substances 0.000 claims description 38
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 26
- 239000000725 suspension Substances 0.000 claims description 24
- 238000001914 filtration Methods 0.000 claims description 22
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 claims description 20
- 238000001816 cooling Methods 0.000 claims description 19
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 19
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 18
- 239000003814 drug Substances 0.000 claims description 17
- 239000002244 precipitate Substances 0.000 claims description 17
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 14
- 238000010438 heat treatment Methods 0.000 claims description 11
- 239000003960 organic solvent Substances 0.000 claims description 11
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 claims description 10
- 238000002441 X-ray diffraction Methods 0.000 claims description 10
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 claims description 10
- 238000001035 drying Methods 0.000 claims description 10
- PGMYKACGEOXYJE-UHFFFAOYSA-N pentyl acetate Chemical compound CCCCCOC(C)=O PGMYKACGEOXYJE-UHFFFAOYSA-N 0.000 claims description 10
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 10
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 claims description 9
- XYIBRDXRRQCHLP-UHFFFAOYSA-N ethyl acetoacetate Chemical compound CCOC(=O)CC(C)=O XYIBRDXRRQCHLP-UHFFFAOYSA-N 0.000 claims description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 9
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 claims description 8
- 239000000706 filtrate Substances 0.000 claims description 8
- 230000001376 precipitating effect Effects 0.000 claims description 8
- 238000003756 stirring Methods 0.000 claims description 8
- 238000002156 mixing Methods 0.000 claims description 6
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 5
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 claims description 5
- 230000002265 prevention Effects 0.000 claims description 4
- 241000124008 Mammalia Species 0.000 claims description 3
- 238000005303 weighing Methods 0.000 claims 1
- 238000002560 therapeutic procedure Methods 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 38
- 239000002552 dosage form Substances 0.000 description 14
- 235000019441 ethanol Nutrition 0.000 description 12
- 239000000047 product Substances 0.000 description 12
- 239000007788 liquid Substances 0.000 description 11
- 239000004480 active ingredient Substances 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 239000008187 granular material Substances 0.000 description 8
- 239000003826 tablet Substances 0.000 description 8
- 239000000126 substance Substances 0.000 description 7
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 238000000113 differential scanning calorimetry Methods 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 229920002472 Starch Polymers 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 238000007907 direct compression Methods 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 239000008107 starch Substances 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- 108010010803 Gelatin Proteins 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 4
- 229920002125 Sokalan® Polymers 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000001913 cellulose Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229940014259 gelatin Drugs 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 229920000609 methyl cellulose Polymers 0.000 description 4
- 239000001923 methylcellulose Substances 0.000 description 4
- 235000010981 methylcellulose Nutrition 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 239000007909 solid dosage form Substances 0.000 description 4
- 239000008247 solid mixture Substances 0.000 description 4
- 229940032147 starch Drugs 0.000 description 4
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 3
- -1 2-hydroxyethoxy Chemical group 0.000 description 3
- 241000220479 Acacia Species 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 229920002907 Guar gum Polymers 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 3
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 3
- 229920000881 Modified starch Polymers 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 239000008186 active pharmaceutical agent Substances 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 239000000783 alginic acid Substances 0.000 description 3
- 229960001126 alginic acid Drugs 0.000 description 3
- 150000004781 alginic acids Chemical class 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- 238000007906 compression Methods 0.000 description 3
- 230000006835 compression Effects 0.000 description 3
- 235000010417 guar gum Nutrition 0.000 description 3
- 239000000665 guar gum Substances 0.000 description 3
- 229960002154 guar gum Drugs 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 229960002900 methylcellulose Drugs 0.000 description 3
- 239000008108 microcrystalline cellulose Substances 0.000 description 3
- 229940016286 microcrystalline cellulose Drugs 0.000 description 3
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 3
- 229920003124 powdered cellulose Polymers 0.000 description 3
- 235000019814 powdered cellulose Nutrition 0.000 description 3
- 235000010413 sodium alginate Nutrition 0.000 description 3
- 239000000661 sodium alginate Substances 0.000 description 3
- 229940005550 sodium alginate Drugs 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- XPCTZQVDEJYUGT-UHFFFAOYSA-N 3-hydroxy-2-methyl-4-pyrone Chemical compound CC=1OC=CC(=O)C=1O XPCTZQVDEJYUGT-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical compound OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 229920001353 Dextrin Polymers 0.000 description 2
- 239000004375 Dextrin Substances 0.000 description 2
- 239000004097 EU approved flavor enhancer Substances 0.000 description 2
- 239000001856 Ethyl cellulose Substances 0.000 description 2
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 2
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 2
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 239000005913 Maltodextrin Substances 0.000 description 2
- 229920002774 Maltodextrin Polymers 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 2
- XAAHAAMILDNBPS-UHFFFAOYSA-L calcium hydrogenphosphate dihydrate Chemical compound O.O.[Ca+2].OP([O-])([O-])=O XAAHAAMILDNBPS-UHFFFAOYSA-L 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 229960001631 carbomer Drugs 0.000 description 2
- 229940084030 carboxymethylcellulose calcium Drugs 0.000 description 2
- 229940063834 carboxymethylcellulose sodium Drugs 0.000 description 2
- 229940082500 cetostearyl alcohol Drugs 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000011109 contamination Methods 0.000 description 2
- 235000019425 dextrin Nutrition 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 235000019325 ethyl cellulose Nutrition 0.000 description 2
- 229920001249 ethyl cellulose Polymers 0.000 description 2
- CBOQJANXLMLOSS-UHFFFAOYSA-N ethyl vanillin Chemical group CCOC1=CC(C=O)=CC=C1O CBOQJANXLMLOSS-UHFFFAOYSA-N 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 235000019264 food flavour enhancer Nutrition 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- UBHWBODXJBSFLH-UHFFFAOYSA-N hexadecan-1-ol;octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO.CCCCCCCCCCCCCCCCCCO UBHWBODXJBSFLH-UHFFFAOYSA-N 0.000 description 2
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 2
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 2
- 229940071826 hydroxyethyl cellulose Drugs 0.000 description 2
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 2
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 229940035034 maltodextrin Drugs 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 229920000191 poly(N-vinyl pyrrolidone) Polymers 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000193 polymethacrylate Polymers 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229940069328 povidone Drugs 0.000 description 2
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 2
- 239000004299 sodium benzoate Substances 0.000 description 2
- 235000010234 sodium benzoate Nutrition 0.000 description 2
- 229920003109 sodium starch glycolate Polymers 0.000 description 2
- 239000008109 sodium starch glycolate Substances 0.000 description 2
- 229940079832 sodium starch glycolate Drugs 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- OULAJFUGPPVRBK-UHFFFAOYSA-N tetratriacontyl alcohol Natural products CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCO OULAJFUGPPVRBK-UHFFFAOYSA-N 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- 238000005550 wet granulation Methods 0.000 description 2
- NOOLISFMXDJSKH-UTLUCORTSA-N (+)-Neomenthol Chemical compound CC(C)[C@@H]1CC[C@@H](C)C[C@@H]1O NOOLISFMXDJSKH-UTLUCORTSA-N 0.000 description 1
- AEQDJSLRWYMAQI-UHFFFAOYSA-N 2,3,9,10-tetramethoxy-6,8,13,13a-tetrahydro-5H-isoquinolino[2,1-b]isoquinoline Chemical compound C1CN2CC(C(=C(OC)C=C3)OC)=C3CC2C2=C1C=C(OC)C(OC)=C2 AEQDJSLRWYMAQI-UHFFFAOYSA-N 0.000 description 1
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 239000004255 Butylated hydroxyanisole Substances 0.000 description 1
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- 108010076119 Caseins Proteins 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- NOOLISFMXDJSKH-UHFFFAOYSA-N DL-menthol Natural products CC(C)C1CCC(C)CC1O NOOLISFMXDJSKH-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 102000002322 Egg Proteins Human genes 0.000 description 1
- 108010000912 Egg Proteins Proteins 0.000 description 1
- 229940118365 Endothelin receptor antagonist Drugs 0.000 description 1
- YIKYNHJUKRTCJL-UHFFFAOYSA-N Ethyl maltol Chemical compound CCC=1OC=CC(=O)C=1O YIKYNHJUKRTCJL-UHFFFAOYSA-N 0.000 description 1
- 229920003134 Eudragit® polymer Polymers 0.000 description 1
- 229940124602 FDA-approved drug Drugs 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- HYMLWHLQFGRFIY-UHFFFAOYSA-N Maltol Natural products CC1OC=CC(=O)C1=O HYMLWHLQFGRFIY-UHFFFAOYSA-N 0.000 description 1
- 229920003091 Methocel™ Polymers 0.000 description 1
- 241000238367 Mya arenaria Species 0.000 description 1
- 229920003072 Plasdone™ povidone Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- HDSBZMRLPLPFLQ-UHFFFAOYSA-N Propylene glycol alginate Chemical compound OC1C(O)C(OC)OC(C(O)=O)C1OC1C(O)C(O)C(C)C(C(=O)OCC(C)O)O1 HDSBZMRLPLPFLQ-UHFFFAOYSA-N 0.000 description 1
- WINXNKPZLFISPD-UHFFFAOYSA-M Saccharin sodium Chemical compound [Na+].C1=CC=C2C(=O)[N-]S(=O)(=O)C2=C1 WINXNKPZLFISPD-UHFFFAOYSA-M 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 241000256011 Sphingidae Species 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000012296 anti-solvent Substances 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- 229940092782 bentonite Drugs 0.000 description 1
- 235000012216 bentonite Nutrition 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 1
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 1
- 229940043253 butylated hydroxyanisole Drugs 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 229940078456 calcium stearate Drugs 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 229920003123 carboxymethyl cellulose sodium Polymers 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 229960004106 citric acid Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 239000011549 crystallization solution Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- RBLGLDWTCZMLRW-UHFFFAOYSA-K dicalcium phosphate dihydrate Substances O.O.[Ca+2].[Ca+2].[O-]P([O-])([O-])=O RBLGLDWTCZMLRW-UHFFFAOYSA-K 0.000 description 1
- GXGAKHNRMVGRPK-UHFFFAOYSA-N dimagnesium;dioxido-bis[[oxido(oxo)silyl]oxy]silane Chemical compound [Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O GXGAKHNRMVGRPK-UHFFFAOYSA-N 0.000 description 1
- 229960001484 edetic acid Drugs 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 235000013345 egg yolk Nutrition 0.000 description 1
- 210000002969 egg yolk Anatomy 0.000 description 1
- 239000002308 endothelin receptor antagonist Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- MVPICKVDHDWCJQ-UHFFFAOYSA-N ethyl 3-pyrrolidin-1-ylpropanoate Chemical compound CCOC(=O)CCN1CCCC1 MVPICKVDHDWCJQ-UHFFFAOYSA-N 0.000 description 1
- 229960004667 ethyl cellulose Drugs 0.000 description 1
- 229940093503 ethyl maltol Drugs 0.000 description 1
- 229940073505 ethyl vanillin Drugs 0.000 description 1
- 238000013265 extended release Methods 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 229960002737 fructose Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229960002598 fumaric acid Drugs 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- FETSQPAGYOVAQU-UHFFFAOYSA-N glyceryl palmitostearate Chemical compound OCC(O)CO.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O FETSQPAGYOVAQU-UHFFFAOYSA-N 0.000 description 1
- 229940046813 glyceryl palmitostearate Drugs 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 229940071676 hydroxypropylcellulose Drugs 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229960004903 invert sugar Drugs 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 235000014380 magnesium carbonate Nutrition 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- 235000012245 magnesium oxide Nutrition 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940057948 magnesium stearate Drugs 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 229940043353 maltol Drugs 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229940041616 menthol Drugs 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000003605 opacifier Substances 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 229960000292 pectin Drugs 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229960000540 polacrilin potassium Drugs 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229940068984 polyvinyl alcohol Drugs 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- WVWZXTJUCNEUAE-UHFFFAOYSA-M potassium;1,2-bis(ethenyl)benzene;2-methylprop-2-enoate Chemical compound [K+].CC(=C)C([O-])=O.C=CC1=CC=CC=C1C=C WVWZXTJUCNEUAE-UHFFFAOYSA-M 0.000 description 1
- 230000003334 potential effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 235000010409 propane-1,2-diol alginate Nutrition 0.000 description 1
- 239000000770 propane-1,2-diol alginate Substances 0.000 description 1
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 1
- 229940032159 propylene carbonate Drugs 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 239000000700 radioactive tracer Substances 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229960003885 sodium benzoate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000002639 sodium chloride Nutrition 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 239000000176 sodium gluconate Substances 0.000 description 1
- 235000012207 sodium gluconate Nutrition 0.000 description 1
- 229940005574 sodium gluconate Drugs 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 229940080313 sodium starch Drugs 0.000 description 1
- 229940045902 sodium stearyl fumarate Drugs 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229960004793 sucrose Drugs 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960001367 tartaric acid Drugs 0.000 description 1
- 238000002411 thermogravimetry Methods 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
Images










Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/69—Benzenesulfonamido-pyrimidines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description

(a)1つ又は複数の有機溶媒にボセンタンを溶解する工程;
(b)工程(a)において得られる溶液から結晶固体を沈殿させる工程;及び
(c)工程(b)から得られる結晶固体を単離する工程
を含む、ボセンタンの結晶形5の製造方法を提供する。
(a)透明溶液が得られるまでエチルアセトアセテート中でボセンタンを加熱する工程;
(b)沈殿が形成するまで工程(a)で得られた溶液を冷却する工程;及び
(c)工程(b)で得られた懸濁物を濾過して、得られた固体を真空条件下20〜40℃、好ましくは25℃で重量が一定になるまで乾燥させる工程
を含む、ボセンタンの結晶形5の製造方法を提供する。
(a)1つ又は複数の有機溶媒及び任意に水にボセンタンを溶解する工程;
(b)工程(a)で得られた溶液から結晶固体を沈殿させる工程;及び
(c)工程(b)から得られた結晶個体を単離する工程
を含む、ボセンタンの結晶形6の製造方法が提供される。
(a)1つ又は複数のC1−C6アルコールを含む溶媒系にボセンタンを溶解又は懸濁する工程;
(b)工程(a)で得られた溶液又は懸濁物に、透明溶液が得られるまで水を添加する工程;
(c)工程(b)で得られた溶液を約20〜40℃まで冷却する工程;
(d)工程(c)で得られた溶液に沈殿が形成するまでシクロヘキサンを添加する工程;及び
(e)得られた沈殿固体を濾過して、約20〜30℃で重量が一定になるまで真空条件下で乾燥させる工程
を含む、ボセンタンの結晶形6の製造方法を提供する。
(a)1つ又は複数の有機溶媒にボセンタンを溶解する工程;
(b)工程(a)で得られた溶液から結晶固体を沈殿させる工程;及び
(c)工程(b)から得られた結晶個体を単離する工程
を含む、ボセンタンの結晶形7の製造方法が提供する。
(a)シクロヘキサノンとC1−C6アルコールとの混合物にボセンタンを溶解する工程;
(b)攪拌しながら約30分以内に約30℃まで工程(a)で得られた溶液を冷却する工程;
(c)工程(b)で得られた溶液にn−へキサンを添加して、沈殿を形成させ、約2〜5時間に亘って懸濁物を攪拌する工程;及び
(d)工程(c)で得られた懸濁物を濾過して、得られた固体を真空条件下で約20〜40℃、好ましくは25℃で、重量が一定になるまで乾燥させる工程
を含む、ボセンタンの結晶形7の製造方法を提供する。
(a)シクロヘキサノンとC1−C6アルコールとの混合物にボセンタンを溶解する工程;
(b)攪拌しながら約20分以内に約30℃まで、工程(a)で得られた溶液を冷却する工程;
(c)工程(b)で得られた溶液にn−へキサンを添加し、沈殿を形成させ、約15〜25時間の間に亘って懸濁物を攪拌する工程;及び
(d)工程(c)で得られた懸濁物を濾過して、真空条件下で約20〜40℃、好ましくは約25℃で、重量が一定になるまで乾燥させる工程
を含む、ボセンタンの結晶形7の製造方法を提供する。
(a)1つ又は複数の有機溶媒中でボセンタンを混合する工程;
(b)工程(a)で得られた混合物を濾過する工程;及び
(c)工程(b)で得られた濾過物から結晶個体を単離する工程
を含む、ボセンタンの結晶形8の製造方法を提供する。
(a)1つ又は複数の有機溶媒中でボセンタンを混合する工程;
(b)工程(a)で得られた混合物を濾過する工程;
(c)工程(b)で得られた濾過物から結晶固体を沈殿させる工程;及び
(d)工程(c)で得られた結晶個体を単離する工程
を含む。
(a)約90〜110℃、好ましくは約100℃まで、エチルアセテート、オクタノール、及びn−ペンチルアセテートの混合物中でボセンタンを加熱する工程;
(b)真空条件下において工程(a)で得られた溶液を濾過する工程;
(c)沈殿が形成するまで工程(b)で得られた濾過物を冷却する工程;
(d)工程(c)で得られた沈殿物を濾過して、前記沈殿を真空条件下で約20〜40℃、好ましくは25℃で、重量が一定になるまで乾燥させる工程
を含む、ボセンタンの結晶形8の製造方法を提供する。
ボセンタン結晶形5の製造方法
ボセンタン及びエチルアセトアセテート(5容量部(vol))を、透明溶液が得られるまで50℃に加熱した。次いで、その溶液を約30℃まで約50分以内に冷却し、その温度で約17時間に亘って攪拌した。得られた懸濁物を濾過して、所望の結晶固体を得、前記固体を真空条件下、25℃で約4時間に亘って又は重量が一定になるまで乾燥させた。XRPD及びDSC分析データによって、得られた生成物がボセンタン結晶形5であることを確認した。
モル収率=0.18g
化学純度>99%(HPLCで測定)
多形体純度=高(DSCによって測定)
ボセンタン結晶形6の製造方法
ボセンタン、ブタン−2−オール(2.6vol)、及びアミルアルコール(2.6vol)の混合物を約80℃に加熱して、溶媒中のボセンタンの懸濁物を得た。水(1vol)を当該懸濁物に添加して、ボセンタンを溶解し、透明溶液を得た。次いで該溶液を30℃まで約45分以内に冷却した。シクロヘキサン(10vol)を冷却した当該溶液に添加して、温度を約30℃に約30分間に亘って維持しながら攪拌した。次いで、得られた懸濁物を濾過して、所望の結晶固体を得、前記固体を真空条件下、25℃で約2時間に亘って又は重量が一定になるまで乾燥させた。XRPD及びDSC分析によって、得られた生成物がボセンタン結晶形6であることを確認した。
モル収率=0.81g
化学純度>99%(HPLCで測定)
多形体純度=高(DSCで測定)
ボセンタン結晶形7の製造方法
ボセンタン、シクロヘキサノン(2vol)、及びn−ブタノール(2vol)の混合物を約50℃に加熱して、透明溶液を得た。その溶液を約30℃まで約30分以内に冷却して、その温度で30分間に亘って攪拌した。次いで、n−ヘキサン(15vol)を冷却した溶液に添加して、固体沈殿物が生じた。その懸濁物を更に約2.5時間に亘って攪拌した。次いで、その懸濁物を濾過して、所望の結晶固体を得、前記固体を真空条件下、25℃で約23時間に亘って又は重量が一定になるまで乾燥させた。XRPD及びDSC分析データによって、得られた生成物がボセンタン結晶形7であることを確認した。
モル収率=0.83g
化学純度>99%(HPLCで測定)
多形体純度=高(DSCによって測定)
ボセンタン結晶形7の製造方法
ボセンタン、シクロヘキサノン(2vol)、及びエタノール(2vol)の混合物を約80まで加熱して、透明溶液を得た。当該溶液を約30℃まで約30分内に冷却して、その温度で30分間に亘って攪拌した。次いで、n−へキサン(15vol)を冷却した溶液に添加して、固体沈殿が生じた。その懸濁物を更に約21時間に亘って攪拌した。次いで、その懸濁物を濾過して所望の結晶固体物を得、前記固体を真空条件下、25℃で約13時間に亘って又は重量が一定になるまで乾燥させた。XRPD及びDSC分析データによって、得られた生成物がボセンタン結晶形7であることを確認した。
モル収率=0.54g
化学純度>99%(HPLCで測定)
多形体純度=高(DSCによって測定)
ボセンタン結晶形8の製造方法
ボセンタン、エチルアセトアセテート(0.8vol)、オクタノール(2vol)、及びn−ペンチルアセテート(1.2vol)を約100℃まで加熱した。得られた懸濁物を当該温度で真空条件下において濾過した。次いで、その濾過物を約26℃まで約15分以内に冷却して、固体沈殿の形成が生じ、その温度で約4時間に亘って攪拌した。次いで、その懸濁物を濾過して所望の結晶固体を得、前記固体を真空条件下、25℃で約13時間に亘って又は重量が一定になるまで乾燥させた。XRPD及びDSC分析データによって、得られた生成物がボセンタン結晶形8であることを確認した。
モル収率=0.42g
化学純度>99%(HPLCで測定)
多形体純度=高(DSCによって測定)
Claims (78)
- 4.02、6.12、8.38、9.39、10.04、15.26、17.72、17.98、18.81、19.28、20.31、21.05、27.57、31.91、及び45.65±0.2°θの2θ値のピークから選択される2以上のピークを含むX線回折パターンによって特徴付けられる、ボセンタンの結晶形5。
- 実質的に図1に示すXRPDトレースを有する、ボセンタンの結晶形5。
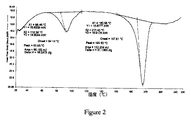
- 約93℃±2℃及び約196℃±2℃の吸熱ピークを含むDSCトレースによって特徴付けられる、ボセンタンの結晶形5。
- 実質的に図2に示すDSCトレースを有する、ボセンタンの結晶形5。
- 実質的に図3に示すTGAトレースを有する、ボセンタンの結晶形5。
- (a)1つ又は複数の有機溶媒にボセンタンを溶解する工程;
(b)工程(a)で得られた溶液から結晶固体を沈殿させる工程;及び
(c)工程(b)で得られた結晶固体を単離する工程
を含む、請求項1から5のいずれか一項に記載のボセンタンの結晶形5の製造方法。 - 工程(a)における前記溶媒がエチルアセトアセテートを含む、請求項6に記載の方法。
- 前記溶媒が、工程(a)において、透明な溶液が得られるまで加熱される、請求項6又は7に記載の方法。
- 前記溶液を、工程(b)において、冷却することによって結晶固体を沈殿させる、請求項6から8のいずれか一項に記載の方法。
- 前記沈殿した固体が、工程(c)において、濾過によって単離される、請求項6から9のいずれか一項に記載の方法。
- 工程(c)で単離した固体を、重量が一定になるまで真空条件下で乾燥させる、請求項6から10のいずれか一項に記載の方法。
- (a)透明溶液が得られるまでエチルアセトアセテート中でボセンタンを加熱する工程;
(b)沈殿が形成するまで工程(a)で得られた溶液を冷却する工程;及び
(c)工程(b)で得られた懸濁物を濾過して、得られた固体を真空条件下、約20〜40℃で重量が一定になるまで乾燥させる工程
を含む、請求項1から5のいずれか一項に記載のボセンタンの結晶形5の製造方法。 - 3.87、7.51、8.84、11.14、18.74、及び23.30±0.2°θの2θ値のピークから選択される2以上のピークを含むX線回折パターンによって特徴付けられる、ボセンタンの結晶形6。
- 実質的に図4に示すXRPDトレースを有する、ボセンタンの結晶形6。
- 約78℃±2℃及び約134℃±2℃の吸熱ピークを含むDSCトレースによって特徴付けられる、ボセンタンの結晶形6。
- 実質的に図5に示すDSCトレースを有する、ボセンタンの結晶形6。
- 実質的に図6に示すTGAトレースを有する、ボセンタンの結晶形6。
- (a)1つ又は複数の有機溶媒及び任意に水にボセンタンを溶解する工程;
(b)工程(a)で得られた溶液から結晶固体を沈殿させる工程;及び
(c)工程(b)で得られた結晶個体を単離する工程
を含む、請求項13から17のいずれか一項に記載のボセンタンの結晶形6の製造方法。 - 工程(a)における前記溶媒、前記溶媒の各々、又は前記溶媒の1つ又は複数がC1−C6アルコールである、請求項18に記載の方法。
- ボセンタンが、工程(a)において、ブタン−2−オール及びアミルアルコール中で加熱し、水を添加して溶解される、請求項18又は19に記載の方法。
- 前記固体を、工程(b)において、工程(a)で得られた溶液を冷却し、シクロヘキサンを添加して沈殿させる、請求項18から20のいずれか一項に記載の方法。
- 工程(b)で沈殿した固体を濾過によって単離する、請求項18から21のいずれか一項に記載の方法。
- 工程(c)で単離した固体を、真空条件下で重量が一定になるまで乾燥させる、請求項18から22のいずれか一項に記載の方法。
- (a)1又は複数のC1−C6アルコールを含む溶媒系にボセンタンを溶解又は懸濁する工程;
(b)工程(a)で得られた溶液又は懸濁物に、透明溶液が得られるまで水を添加する工程;
(c)工程(b)で得られた溶液を約20〜40℃まで冷却する工程;
(d)工程(c)で得られた溶液に沈殿が形成するまでシクロヘキサンを添加する工程;及び
(e)得られた沈殿固体を濾過して、真空条件下、約20〜30℃で重量が一定になるまで乾燥させる工程
を含む、請求項13から17のいずれか一項に記載のボセンタンの結晶形6の製造方法。 - 工程(a)の溶媒系が、ブタン−2−オール及びアミルアルコールを含む、請求項24に記載の方法。
- 前記溶媒が工程(a)で約80℃まで加熱される、請求項24又は25に記載の方法。
- 3.64、4.23、4.95、7.04、7.68、8.23、9.05、9.66、10.48、13.95、15.20、16.17、17.37、18.06、20.03、22.13、及び23.62±0.2°θの2θ値のピークから選択される2以上のピークを含むX線回折パターンによって特徴付けられる、ボセンタンの結晶形7。
- 実質的に図7に示すXRPDトレースを有する、ボセンタンの結晶形7。
- 約75℃±2℃及び約130℃±2℃の吸熱ピークを含むDSCトレースによって特徴付けられる、ボセンタンの結晶形7。
- 実質的に図8に示すDSCトレースを有する、ボセンタンの結晶形7。
- 実質的に図9に示すTGAトレースを有する、ボセンタンの結晶形7。
- (a)1つ又は複数の有機溶媒にボセンタンを溶解する工程;
(b)工程(a)で得られた溶液から結晶固体を沈殿させる工程;及び
(c)工程(b)で得られた結晶個体を単離する工程
を含む、請求項27から31のいずれか一項に記載のボセンタンの結晶形7の製造方法。 - ボセンタンが、工程(a)において、透明な溶液が得られるまで前記溶媒又は前記溶媒の各々に溶解される、請求項32に記載の方法。
- ボセンタンが、工程(a)において、前記溶媒又は前記溶媒の各々を加熱することによって溶解される、請求項32又は33に記載の方法。
- 工程(a)の前記溶媒、前記溶媒の各々、又は前記溶媒の1つ若しくは複数がC1−C6アルコールである、請求項32から34のいずれか一項に記載の方法。
- 工程(a)の前記溶媒がシクロヘキサノンとn−ブタノールとの混合物である、請求項32から35のいずれか一項に記載の方法。
- 工程(a)の前記溶媒がシクロヘキサノンとエタノールとの混合物である、請求項32から35のいずれか一項に記載の方法。
- 前記結晶固体を、工程(a)で得られた溶液にn−へキサンを添加することによって沈殿させる、請求項32から37のいずれか一項に記載の方法。
- 前記固体が、工程(c)において、濾過によって単離される、請求項32から38のいずれか一項に記載の方法。
- 工程(c)で単離した固体を、真空条件下で重量が一定になるまで乾燥させる、請求項32から39のいずれか一項に記載の方法。
- (a)シクロヘキサノンとC1−C6アルコールとの混合物にボセンタンを溶解する工程;
(b)攪拌しながら約30分以内に約30℃まで工程(a)で得られた溶液を冷却する工程;
(c)工程(b)で得られた溶液にn−へキサンを添加して、沈殿を形成させ、約2〜5時間に亘って懸濁物を攪拌する工程;及び
(d)工程(c)で得られた懸濁物を濾過して、得られた固体を、真空条件下、約20〜40℃で重量が一定になるまで乾燥させる工程
を含む、請求項27から31のいずれか一項に記載のボセンタンの結晶形7の製造方法。 - 工程(a)におけるC1−C6アルコールがn−ブタノールである、請求項41に記載の方法。
- ボセンタンが、工程(a)において、透明溶液が得られるまで混合物を加熱することによって溶解される、請求項41又は42に記載の方法。
- (a)シクロヘキサノンとC1−C6アルコールとの混合物にボセンタンを溶解する工程;
(b)攪拌しながら約30分以内に約30℃まで、工程(a)で得られた溶液を冷却する工程;
(c)工程(b)で得られた溶液にn−へキサンを添加し、沈殿を形成させ、約15〜25時間の間に亘って懸濁物を攪拌する工程;及び
(d)工程(c)で得られた懸濁物を濾過して、得られた固体を、真空条件下、約20〜40℃で重量が一定になるまで乾燥させる工程
を含む、請求項27から31のいずれか一項に記載のボセンタンの結晶形7の製造方法。 - 工程(a)のC1−C6アルコールがエタノールである、請求項44に記載の方法。
- ボセンタンが、工程(a)において、透明溶液が得られるまで混合物を加熱することによって溶解される、請求項44又は45に記載の方法。
- 9.47、13.41、14.52、15.46、15.73、16.35、16.88、17.99、18.87、19.25、20.53、21.82、23.02、23.83、24.61、24.86、25.13、及び26.03±0.2°θの2θ値のピークから選択される2以上のピークを含むX線回折パターンによって特徴付けられる、ボセンタンの結晶形8。
- 実質的に図10に示すXRPDトレースを有する、ボセンタンの結晶形8。
- 約58℃±2℃及び約110℃±2℃の吸熱ピークを含むDSCトレースによって特徴付けられる、ボセンタンの結晶形8。
- 実質的に図11に示すDSCトレースを有する、ボセンタンの結晶形8。
- 実質的に図12に示すTGAトレースを有する、ボセンタンの結晶形8。
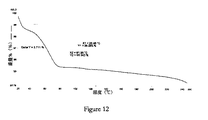
- (a)1つ又は複数の有機溶媒中でボセンタンを混合する工程;
(b)工程(a)で得られた混合物を濾過する工程;
(c)工程(b)で得られた濾過物から結晶固体を沈殿させる工程;及び
(d)工程(c)で得られた結晶個体を単離する工程
を含む、請求項47から51のいずれか一項に記載のボセンタンの結晶形8の製造方法。 - ボセンタンが、工程(a)において、エチルアセトアセテート、オクタノール、及びn−ペンチルアセテートの混合物中で加熱される、請求項52に記載の方法。
- 工程(b)において、前記混合物を依然として加熱しながら真空条件下で濾過する、請求項52又は53に記載の方法。
- 前記結晶固体が、工程(c)で濾過物を冷却することによって得られる、請求項52から54のいずれか一項に記載の方法。
- 前記結晶固体を、工程(d)で濾過し、真空条件下で重量が一定になるまで乾燥させる、請求項52から55のいずれか一項に記載の方法。
- (a)エチルアセトアセテート、オクタノール、及びn−ペンチルアセテートの混合物にボセンタンを溶解する工程;
(b)真空条件下において工程(a)で得られた溶液を濾過する工程;
(c)沈殿が形成するまで工程(b)で得られた濾過物を冷却する工程;及び
(d)工程(c)で得られた沈殿物を濾過して、前記沈殿物を真空条件下、約20〜40℃で重量が一定になるまで乾燥させる工程
を含む、請求項47から51のいずれか一項に記載のボセンタンの結晶形8の製造方法。 - ボセンタンが、工程(a)において、約90〜110℃まで混合物を加熱することによって溶解される、請求項57に記載の方法。
- 10%未満の他の形状のボセンタンを含む、請求項1から5、13から17、27から31、又は47から51のいずれか一項に記載のボセンタンの結晶形。
- 5%未満の他の形状のボセンタンを含む、請求項59に記載のボセンタンの結晶形。
- 1%未満の他の形状のボセンタンを含む、請求項60に記載のボセンタンの結晶形。
- 0.1%未満の他の形状のボセンタンを含む、請求項61に記載のボセンタンの結晶形。
- 医薬において使用するための、請求項1から5、13から17、27から31、47から51、又は59から62のいずれか一項に記載のボセンタンの結晶形。
- エンドセリン受容体媒介疾患を治療又は予防するための、請求項63に記載のボセンタンの結晶形。
- 前記エンドセリン受容体媒介疾患が心臓血管疾患である、請求項64に記載のボセンタンの結晶形。
- 前記心臓血管疾患が高血圧、肺高血圧、虚血、血管痙攣、又は狭心症である、請求項65に記載のボセンタンの結晶形。
- 前記心臓血管疾患が肺動脈高血圧である、請求項66に記載のボセンタンの結晶形。
- 請求項1から5、13から17、27から31、47から51、又は59から67のいずれか一項に記載のボセンタンの結晶形の1つ又は複数、及び医薬品として許容される賦形剤の1つ又は複数を含む、医薬組成物。
- エンドセリン受容体媒介疾患の治療又は予防のための、請求項1から5、13から17、27から31、47から51、若しくは59から67のいずれか一項に記載のボセンタンの結晶形の1つ又は複数の使用、又は請求項68に記載の組成物の使用。
- 心臓血管疾患の治療又は予防のための、請求項69に記載の使用。
- 前記心臓血管疾患が高血圧、肺高血圧、虚血、血管痙攣、又は狭心症である、請求項70に記載の使用。
- 前記心臓血管疾患が肺動脈高血圧である、請求項71に記載の使用。
- 請求項1から5、13から17、27から31、47から51、若しくは59から67のいずれか一項に記載のボセンタンの結晶形の1つ若しくは複数、又は請求項68に記載の組成物の治療上又は予防上有効量を、必要な患者に投与する工程を含む、エンドセリン受容体媒介疾患を治療又は予防する方法。
- 前記エンドセリン受容体媒介疾患が心臓血管疾患である、請求項73に記載の方法。
- 前記心臓血管疾患が高血圧、肺高血圧、虚血、血管痙攣、又は狭心症である、請求項74に記載の方法。
- 前記心臓血管疾患が肺動脈高血圧である、請求項75に記載の方法。
- 前記患者が哺乳動物である、請求項73から76のいずれか一項に記載の方法。
- 前記哺乳動物がヒトである、請求項77に記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN2108MU2007 | 2007-10-24 | ||
| PCT/GB2008/050986 WO2009053748A2 (en) | 2007-10-24 | 2008-10-24 | Novel crystalline forms |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2011500780A true JP2011500780A (ja) | 2011-01-06 |
| JP2011500780A5 JP2011500780A5 (ja) | 2011-12-08 |
Family
ID=40262729
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010530564A Pending JP2011500780A (ja) | 2007-10-24 | 2008-10-24 | 新規結晶形 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US8530488B2 (ja) |
| EP (1) | EP2222649A2 (ja) |
| JP (1) | JP2011500780A (ja) |
| CN (1) | CN101939303B (ja) |
| AU (1) | AU2008315757A1 (ja) |
| CA (1) | CA2703230A1 (ja) |
| NZ (2) | NZ585438A (ja) |
| WO (1) | WO2009053748A2 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011508767A (ja) * | 2008-01-01 | 2011-03-17 | シプラ・リミテッド | ボセンタン、その多形形態及びその塩の合成方法 |
| JP2020143080A (ja) * | 2012-04-04 | 2020-09-10 | アルカヘスト インコーポレイテッド | Ccr3アンタゴニストを含む医薬製剤 |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2686457A1 (en) * | 2007-05-08 | 2008-11-13 | Generics [Uk] Limited | Novel polymorphic forms |
| AU2008272685B2 (en) * | 2007-06-29 | 2013-04-18 | Generics [Uk] Limited | Process for introduction of hydroxyethoxy side chain in bosentan |
| EP2222649A2 (en) * | 2007-10-24 | 2010-09-01 | Generics (UK) Limited | Novel crystalline forms |
| EP2240469A2 (en) | 2008-01-24 | 2010-10-20 | Actavis Group PTC EHF | Substantially pure and a stable crystalline form of bosentan |
| JP5683276B2 (ja) | 2008-02-08 | 2015-03-11 | ジェネリクス・[ユーケー]・リミテッド | ボセンタンの製造方法 |
| WO2010032261A1 (en) | 2008-08-12 | 2010-03-25 | Cadila Healthcare Limited | Process for preparation of bosentan |
| US8975402B2 (en) | 2008-11-03 | 2015-03-10 | Generics [Uk] Limited | HPLC method for the analysis of bosetan and related substances and use of these substances as reference standards and markers |
| WO2012002547A1 (ja) * | 2010-07-02 | 2012-01-05 | 富士化学工業株式会社 | ボセンタン固体分散体 |
| PL402305A1 (pl) * | 2012-12-30 | 2014-07-07 | Instytut Farmaceutyczny | Sposób wytwarzania bozentanu w postaci monohydratu o czystości farmaceutycznej |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6185370A (ja) * | 1984-09-24 | 1986-04-30 | ルセル‐ユクラフ | 4h‐1,2,4‐トリアゾール誘導体の新製造法、得られる新トリアゾール、それらの薬剤としての使用及びそれらを含む製薬組成物 |
| JPH05222003A (ja) * | 1991-06-13 | 1993-08-31 | F Hoffmann La Roche Ag | スルホンアミド類および薬物としてのそれらの使用 |
| US6136971A (en) * | 1998-07-17 | 2000-10-24 | Roche Colorado Corporation | Preparation of sulfonamides |
| JP2003520857A (ja) * | 2000-01-25 | 2003-07-08 | エフ.ホフマン−ラ ロシュ アーゲー | スルホンアミドの製造 |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5276004A (en) * | 1987-03-20 | 1994-01-04 | Dai Nippon Insatsu Kabushiki Kaisha | Process for heat transfer recording |
| US5739333A (en) * | 1995-05-16 | 1998-04-14 | Tanabe Seiyaku Co., Ltd. | Sulfonamide derivative and process for preparing the same |
| PT1535613E (pt) | 1999-11-17 | 2010-10-04 | Teva Pharma | Processo de preparação de uma forma polimórfica de atorvastatina de cálcio |
| CN1411373A (zh) | 1999-12-16 | 2003-04-16 | 特瓦制药工业有限公司 | 制备来氟米特的新方法和新晶形的来氟米特 |
| US6479692B1 (en) * | 2001-05-02 | 2002-11-12 | Nobex Corporation | Methods of synthesizing acylanilides including bicalutamide and derivatives thereof |
| WO2004076443A1 (en) | 2003-02-25 | 2004-09-10 | Hetero Drugs Limited | Amorphous form of losartan potassium |
| EP1603920A1 (en) | 2003-03-12 | 2005-12-14 | Cadila Healthcare Ltd. | Polymorph and amorphous form of (s)-(+)-clopidogrel bisulfate |
| US7772399B2 (en) | 2003-04-02 | 2010-08-10 | Hetero Drugs Limited | Process for amorphous form of donepezil hydrochloride |
| US20080188663A1 (en) * | 2007-01-29 | 2008-08-07 | Ashok Kumar | Process for the preparation of crystalline clopidogrel hydrogen sulphate Form I |
| JP2010523584A (ja) * | 2007-04-02 | 2010-07-15 | オースペックス・ファーマシューティカルズ・インコーポレイテッド | 置換ピリミジン |
| CA2686457A1 (en) | 2007-05-08 | 2008-11-13 | Generics [Uk] Limited | Novel polymorphic forms |
| AU2008272685B2 (en) | 2007-06-29 | 2013-04-18 | Generics [Uk] Limited | Process for introduction of hydroxyethoxy side chain in bosentan |
| WO2009047637A1 (en) | 2007-10-11 | 2009-04-16 | Actavis Group Ptc Ehf | Novel polymorphs of bosentan |
| EP2222649A2 (en) * | 2007-10-24 | 2010-09-01 | Generics (UK) Limited | Novel crystalline forms |
| ATE530531T1 (de) * | 2007-12-18 | 2011-11-15 | Dipharma Francis Srl | Verfahren zur herstellung von bosentan |
| WO2009095933A2 (en) | 2008-01-10 | 2009-08-06 | Msn Laboratories Limited | Improved and novel process for the preparation of bosentan |
| EP2240469A2 (en) | 2008-01-24 | 2010-10-20 | Actavis Group PTC EHF | Substantially pure and a stable crystalline form of bosentan |
| JP5683276B2 (ja) | 2008-02-08 | 2015-03-11 | ジェネリクス・[ユーケー]・リミテッド | ボセンタンの製造方法 |
| EP2268634A2 (en) | 2008-03-13 | 2011-01-05 | Actavis Group PTC EHF | Processes for the preparation of bosentan and related compounds using novel intermediates |
| WO2009141167A1 (en) * | 2008-05-23 | 2009-11-26 | Synthon B.V. | Bosentan salts |
| US8975402B2 (en) | 2008-11-03 | 2015-03-10 | Generics [Uk] Limited | HPLC method for the analysis of bosetan and related substances and use of these substances as reference standards and markers |
| IT1393136B1 (it) * | 2009-03-11 | 2012-04-11 | Sifa Vitor S R L | Procedimento per la preparazione del bosentan |
-
2008
- 2008-10-24 EP EP08806793A patent/EP2222649A2/en not_active Withdrawn
- 2008-10-24 NZ NZ585438A patent/NZ585438A/xx not_active IP Right Cessation
- 2008-10-24 WO PCT/GB2008/050986 patent/WO2009053748A2/en active Application Filing
- 2008-10-24 CA CA2703230A patent/CA2703230A1/en not_active Abandoned
- 2008-10-24 CN CN200880122464.2A patent/CN101939303B/zh not_active Expired - Fee Related
- 2008-10-24 NZ NZ600010A patent/NZ600010A/xx not_active IP Right Cessation
- 2008-10-24 US US12/739,303 patent/US8530488B2/en not_active Expired - Fee Related
- 2008-10-24 JP JP2010530564A patent/JP2011500780A/ja active Pending
- 2008-10-24 AU AU2008315757A patent/AU2008315757A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6185370A (ja) * | 1984-09-24 | 1986-04-30 | ルセル‐ユクラフ | 4h‐1,2,4‐トリアゾール誘導体の新製造法、得られる新トリアゾール、それらの薬剤としての使用及びそれらを含む製薬組成物 |
| JPH05222003A (ja) * | 1991-06-13 | 1993-08-31 | F Hoffmann La Roche Ag | スルホンアミド類および薬物としてのそれらの使用 |
| US6136971A (en) * | 1998-07-17 | 2000-10-24 | Roche Colorado Corporation | Preparation of sulfonamides |
| JP2003520857A (ja) * | 2000-01-25 | 2003-07-08 | エフ.ホフマン−ラ ロシュ アーゲー | スルホンアミドの製造 |
Non-Patent Citations (5)
| Title |
|---|
| BIOORGANIC & MEDICINAL CHEMISTRY, vol. 9, JPN6013032141, 2001, pages 2955 - 2968, ISSN: 0002572428 * |
| CHIMIA, vol. 50, JPN6013032143, 1996, pages 519 - 524, ISSN: 0002572429 * |
| CHINESE JOURNAL OF MEDICINAL CHEMISTRY, vol. 15, no. 4, JPN6013032145, 2005, pages 230 - 233, ISSN: 0002572430 * |
| ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 6, no. 2, JPN6013032138, 2002, pages 120 - 124, ISSN: 0002572427 * |
| PHARM TECH JAPAN, vol. 18, no. 10, JPN7014002019, 2002, pages 81 - 96, ISSN: 0002848776 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011508767A (ja) * | 2008-01-01 | 2011-03-17 | シプラ・リミテッド | ボセンタン、その多形形態及びその塩の合成方法 |
| JP2020143080A (ja) * | 2012-04-04 | 2020-09-10 | アルカヘスト インコーポレイテッド | Ccr3アンタゴニストを含む医薬製剤 |
| US11612596B2 (en) | 2012-04-04 | 2023-03-28 | Alkahest, Inc. | Pharmaceutical formulations comprising CCR3 antagonists |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2008315757A1 (en) | 2009-04-30 |
| EP2222649A2 (en) | 2010-09-01 |
| CN101939303A (zh) | 2011-01-05 |
| NZ600010A (en) | 2013-11-29 |
| US20100331352A1 (en) | 2010-12-30 |
| US8530488B2 (en) | 2013-09-10 |
| WO2009053748A2 (en) | 2009-04-30 |
| WO2009053748A3 (en) | 2009-06-18 |
| NZ585438A (en) | 2012-09-28 |
| CN101939303B (zh) | 2014-06-11 |
| CA2703230A1 (en) | 2009-04-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2011500780A (ja) | 新規結晶形 | |
| AU2008247169B2 (en) | Polymorphic forms of bosentan | |
| KR20080055990A (ko) | 시나칼셋 HCl의 결정형 및 이의 제조 방법 | |
| JP2007526251A (ja) | エゼチミベ多形体 | |
| US20100249162A1 (en) | Process for the introduction of hydroxyethoxy side chain in bosentan | |
| US20080161607A1 (en) | Processes for preparation of polymorphic form II of sertraline hydrochloride | |
| JP2012507496A (ja) | レナリドマイドの結晶形およびその調製方法 | |
| KR20040077872A (ko) | 카베디롤의 결정질 고체 및 그 제조 방법 | |
| JP2008539278A (ja) | 結晶性ロスバスタチンカルシウム | |
| WO2015139386A1 (zh) | 坎格列净一水合物及其晶型、它们的制备方法和用途 | |
| US20150038721A1 (en) | Solid forms of dabigatran etexilate mesylate and processes for their preparation | |
| US20060293377A1 (en) | Amorphous and polymorphic forms of telmisartan sodium | |
| JP2013528206A (ja) | 結晶形態のサリドマイド及びその調製方法 | |
| JP2004526706A (ja) | オクスカルバゼピンの新しい結晶形態及びそれらの調製方法 | |
| JP2004526714A (ja) | ラモトリジンの新しい結晶形およびそれらの調製方法 | |
| EP4426678A1 (en) | Compounds for the treatment of kinase-dependent disorders | |
| US20060052350A1 (en) | Crystalline forms of 1,24(S)-dihydroxy vitamin D2 | |
| KR20070088507A (ko) | 카베딜올의 결정질 형태 및 이것의 제조 방법 | |
| KR101423630B1 (ko) | 비칼루타미드와 니코틴아미드의 공결정 | |
| EP2610239A1 (en) | Preparation Of Rasagiline Hemitartrate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100929 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20111021 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20111021 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130702 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130930 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20131007 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131225 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20131225 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140707 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20150126 |