JP2010143864A - 光応答型アゾベンゼン化合物 - Google Patents
光応答型アゾベンゼン化合物 Download PDFInfo
- Publication number
- JP2010143864A JP2010143864A JP2008323606A JP2008323606A JP2010143864A JP 2010143864 A JP2010143864 A JP 2010143864A JP 2008323606 A JP2008323606 A JP 2008323606A JP 2008323606 A JP2008323606 A JP 2008323606A JP 2010143864 A JP2010143864 A JP 2010143864A
- Authority
- JP
- Japan
- Prior art keywords
- group
- formula
- site
- spacer
- nucleic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 Cc(cc1)ccc1C#Cc1c(*)ccc(C#Cc(cc2)ccc2N=Nc2ccc(*)cc2)c1 Chemical compound Cc(cc1)ccc1C#Cc1c(*)ccc(C#Cc(cc2)ccc2N=Nc2ccc(*)cc2)c1 0.000 description 5
- JXLYWLFMXFHTGS-WUKNDPDISA-N Bc(cc1)ccc1/N=N/c(cc1)ccc1SCC(O)=O Chemical compound Bc(cc1)ccc1/N=N/c(cc1)ccc1SCC(O)=O JXLYWLFMXFHTGS-WUKNDPDISA-N 0.000 description 1
- AHCCZWRNFZAWIF-UHFFFAOYSA-N Nc(cc1)ccc1SCCO Chemical compound Nc(cc1)ccc1SCCO AHCCZWRNFZAWIF-UHFFFAOYSA-N 0.000 description 1
Images









Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
【解決手段】
式(1):

で表されるアゾベンゼン化合物である。
【選択図】図1
Description
"Allosteric control of an ionotropic glutamate receptor with an optical switch", M. Volgraf et al., Nature Chemical Biology, (2006), Vol.2, No.1, 47-51. "Mechanisms of photoswitch conjugation and light activation of an ionotropic glutamate receptor", P. Gorostiza et al., PNAS, (2007), Vol.104, No.26, 10865-10870. "Azobenzene-tethered T7 promoter for efficient photoregulation of transcription", M Liu, et al., J. Am. Chem. Soc., (2006), 128, 1009-1015. "Two-photon absorption in azoaromatic compounds", L. D. Boni et al., Chem. Phys. Lett., (2002), 361, 209-213. "Degenerate Two-photon absorption spectra in azoaromatic compounds", L. D. Boni et al., Chem. Phys. Chem., (2005), 6, 1121-1125.
で表される基であり、
Aは、低級アルキレン基、低級アルケニレン基、低級アルキニレン基、又は直接結合であり、
Xは、−O−R2a、−N−(R2b)2又は−S(O)k−R2cであり、
R2aは、低級アルキル基;ペプチド核酸部位を有する基;蛍光部位又は消光部位を有するスペーサーを有する基;アミノ基、カルボキシル基、低級アルコキシカルボニル基、ヒドロキシル基、ペプチド核酸部位を有する基、又は蛍光部位もしくは消光部位を有するスペーサーを有する基が置換されていてもよい低級アルキル基であり、
R2bは、同じであっても異なっていてもよく、水素原子;低級アルキル基;ペプチド核酸部位を有する基;蛍光部位又は消光部位を有するスペーサーを有する基;アミノ基、カルボキシル基、低級アルコキシカルボニル基、ヒドロキシル基、ペプチド核酸部位を有する基、又は蛍光部位もしくは消光部位を有するスペーサーを有する基が置換されていてもよい低級アルキル基であり、
R2cは、低級アルキル基;ペプチド核酸部位を有する基;蛍光部位又は消光部位を有するスペーサーを有する基;アミノ基、カルボキシル基、低級アルコキシカルボニル基、ヒドロキシル基、ペプチド核酸部位を有する基又は蛍光部位もしくは消光部位を有するスペーサーを有する基、が置換されていてもよい低級アルキル基であって、
kは0〜3の整数である)
で表されるアゾベンゼン化合物。
と、亜硝酸塩及び酸を添加して、ジアゾニウム塩を形成させ、さらに、前記ジアゾニウム塩と、式(a2):
を反応させることを特徴とする項1に記載のアゾベンゼン化合物の製造方法。
と、式(b2):
を酸性条件下で反応させることを特徴とする項1に記載のアゾベンゼン化合物の製造方法。
と、トリメチルシリルアセチレンを反応させ、脱シリル化することによって、式(c2):
を反応させることを特徴とする項1に記載のアゾベンゼン化合物の製造方法。
で表される基であることが好ましく、より具体的には、
であることが好ましい。
R2a、R2b及びR2cにおける低級アルキル基、低級アルコキシ基、低級アルコキシカルボニル基は、前記のものが挙げられる。
式(1-2):
式(1-3):
式(1-4):
式(1-5):
が挙げられる。
と、亜硝酸塩及び酸を添加して、ジアゾニウム塩を形成させ、さらに、前記ジアゾニウム塩と、式(a2):
を反応させる方法である反応式(a):
によって製造され、また、式(b1):
と、式(b2):
を酸性条件下で反応させる反応式(b):
によって製造される。
式(c1):
と、トリメチルシリルアセチレンとを反応させた後、脱シリル化することによって、式(c2):
を得、さらに、式(c3):
を反応させることによって製造する方法が挙げられる。
1H NMR (400 MHz, CDCl3): 7.87 (d, 2H,J=9.0 Hz), 7.48 (tm, 2H), 7.38 (tm, 1H, J=7.3 Hz), 6.79 (d, 2H, J=9.1 Hz), 3.51 (t, 2H, J=6.6 Hz), 3.10 (s, 3H), 2.98 (t, 2H, J=6.6 Hz), 1.40(brs, 2H);
13C NMR (100 MHz, CDCl3): 153.1, 151.6, 143.7, 129.4, 128.9, 125.0, 122.2, 111.4, 55.5, 39.6, 39.0
1H NMR (400 MHz, CDCl3): 7.87 (d, 2H, J=9.3Hz), 7.72 (d, 2H, J=8.8 Hz), 7.59 (d, 1H, J=8.8 Hz), 6.79 (d, 2H, J=9.3 Hz), 3.53 (t, 2H, J=6.8Hz), 3.11 (s, 3H), 2.99 (t, 2H, J=6.8 Hz), 1.40(br, 2H);
13C NMR (100 MHz, CDCl3): 1152.1, 143.7, 132.2, 125.4, 123.9, 123.5, 111.6, 70.7, 55.7, 39.8, 39.2
1H NMR (400 MHz, CDCl3): 7.87 (d, 2H, J=9.1 Hz), 7.84 (d, 2H, J=8.4 Hz), 7.80 (d, 1H, J=7.9 Hz), 7.16 (dd, 1H, J=7.9 Hz, 1.3 Hz), 7.14 (br,1H), 3.94 (s, 3H), 3.89 (s, 3H), 3.51 (t, 2H, J=6.6 Hz), 3.09 (s, 3H), 2.98 (t, 2H, J=6.6 Hz), 1.37(br, 2H);
13C NMR (100 MHz, CDCl3):166.1, 158.9, 152.8, 151.9, 143.7, 132.5, 131.8, 128.4, 125.3, 123.4, 123.12, 122.3, 119.6, 114.8, 111.4, 91.9, 90.1, 56.1, 55.5, 39.6, 39.0
1H NMR (400 MHz, CDCl3): 7.98 (d, 2H, J=8.1 Hz), 7.88 (d, 2H, J=9.0 Hz), 7.83 (d, 2H, J=8.3 Hz), 7.75 (br, 1H), 7.62 (d, 2H, J=8.3 Hz), 7.57 (d, 2H, J=8.1 Hz), 7.52 (tm, 2H, J=7.5 Hz), 7.36 (tm, 1H, J=7.5 Hz), 6.8 (d, 2H, J=9.0 Hz), 3.55 (t, 2H, J=6.4 Hz), 3.11 (s, 3H), 3.01 (t, 2H, J=6.4 Hz), 2.89 (br, 2H), 1.61 (s, 9H);
13C NMR(100 MHz, CDCl3): 165.1, 152.6, 151.8, 143.7, 134.7, 132.4, 131.7, 131.5, 131.4, 131.3, 129.3, 128.6, 127.1, 125.3, 123.6, 123.6, 123.1, 122.3, 111.4, 91.0, 90.3, 90.0, 89.4, 81.3, 55.3, 39.5, 39.0, 28.1
合成例1〜5で合成したアミノアゾベンゼン(AZO1〜5)10nmolをジクロロメタン1mlにそれぞれ添加し、濃度10μmol/lのAZO1〜5溶液をそれぞれ調製した。調製した各アミノアゾベンゼン水溶液を可視・紫外分光光度計により、300〜700nmの波長領域で25℃にて測定した。
合成例4で合成したAZO4を10nmol、ジクロロメタン1ml、プロトンのトラップ試薬としてトリエチルアミンを触媒量添加し、濃度10μmol/lのAZO4溶液のサンプルを調製した。調製したAZO4溶液に450nmの可視光をキセノンランプ光源(朝日分光(株)製のMAX-302)を用いて、光照射時間として、0,20, 60, 100, 140, 260, 500 秒間照射し、照射後のUVスペクトルを測定した。UVスペクトルを図1に示す。
試験例7(AZO4の赤外線二光子(800nm)照射後のUVスペクトル測定)
試験例6で調製した濃度10μmol/lのAZO4溶液のサンプルにTi-spphire レーザー (Ti:S レーザー)により、160 fs のパルスレーザーから800 nm の光を照射し、集光レンズから焦点までの距離を15 cm に設定し、集光レンズより12 cm の位置で照射することによって、800nmの赤外線二光子を光照射時間として、0, 200, 440 秒間照射し、照射後のUVスペクトルを測定した。UVスペクトルを図2に示す。
(AZO搭載FRETペプチドの分子設計)
FRETペプチド分子の設計には、光エネルギーのDonorであるFAM(5'-fluorescein:490nmを吸収して520nmの蛍光を発光する)とAcceptorである(QXLTM520 :520nmの蛍光を吸収して消光する)の距離を、アゾベンゼンがトランス体のときにはFAMの蛍光強度が約50%となる約50Åに、アゾベンゼンがシスの時にはFAMの蛍光が約100%消光される30Åになるように設計した。
を表す。
Fmoc-XAL-PEG-PS 樹脂 (2 μmol: PE Biosystems)(ここで、Ddeとは、4,4 - dimethyl - 2,6 - dioxocyclohex - 1 - ylidine]ethylを示し、Fmocとは、9-fluorenylmethylを示し、XALとは、Xanthenylamideを示し、PEGとは、ポリエチレングリコールを示す。) をDMF中に10分ほど放置して湿らせ, Fmoc-保護基をDeblock solution (20% piperidine in DMF : 500 μL)中で5 分間、振トウ反応させることで脱保護し、DMF (500μL)にて5回洗浄した。その後、樹脂をBase solution (2,6-lutidine (0.3 M)及び Diisopropyl ethylamine(0.2 M)/DMF) (500μL) 中で5分インキュベートすることでN末端のアミンの求核性を高めた。一方で、Dde-Lys(Fmoc)-OH (5.3 mg, 10 μmol)にアクティベーター(0.02M HATU/ DMF: 500μL)を加え、5分間インキュベートすることでカルボン酸の反応性を高め、これをBase solution溶液中の樹脂に加えて15分間反応させた。反応後、樹脂をDMF (500μL)にて5回洗浄した。
Dde-Lys (Fmoc)-XAL-PEG-PS 樹脂に対して上記と同様の操作で、リシンのε-アミノ基にあるFmoc-基を脱保護し、先と同様にBase solutionにてアミンの求核性を高めた。これにQXLTM520 520 acid SE, (6.5 mg, 10 μmol, SE:スクシンイミドエステル) を加え、15分インキュベートした。反応後、樹脂をDMF (500μL)にて5回洗浄した。
Dde-Lys (QXL TM520)-XAL-PEG-PS樹脂のDde-基を2%のヒドラジン(hydrazine)と10% のアリルアルコール(allylalcohol)のDMF溶液中(500 μL)で10分間 インキュベートすることで脱保護し、その後 DMF (500 μL)で5回洗浄した。続いて上記と同様の操作でリシンのN-末端アミノ基をbase solution で活性化し, Fmoc-6-aminocaproic acid (3.5 mg, 10 μmol)のカルボン酸をアクティベーター添加により活性化させた。反応後、樹脂をDMF (500μL)にて5回洗浄した。
Fmoc-CAP-Lys (QXL TM520)-XAL-PEG-PS 樹脂に対して上記と同様にFmoc-基の脱保護、Base solutionによるアミノ基の活性化、アクティベーターによるFmoc-AZO4-OH (5.3 mg, 7.5 μmol) のカルボン酸を活性化させたものを調製し、それぞれを反応させた。反応後、樹脂をDMF (500μL)にて5回洗浄した。
Fmoc-AZO4-CAP-Lys (QXL TM520)-XAL-PEG-PS 樹脂に対して、上記と同様の操作を用いてFmoc-6-aminocaproic acid (3.5 mg, 10μmol) を伸長した。反応後、樹脂をDMF (500μL)にて5回洗浄した。
Fmoc-CAP-AZO4-CAP-Lys (QXLTM520)-XAL-PEG-PS 樹脂に対して、上記と同様の操作を用いてFmoc-Lys (Boc)-OH (5.3 mg, 10 μmol) を伸長した。反応後、樹脂をDMF (500μL)にて5回洗浄した。
Fmoc-Lys (Boc)-CAP-AZO4-CAP-Lys (QXL TM520)-XAL-PEG-PS 樹脂に対して、上記と同様の操作でFmoc-基を脱保護した。
Fmoc-Lys (Boc)-CAP-AZO4-CAP-Lys (QXL TM520)-XAL-PEG-PS 樹脂に対して、上記と同様の操作で、FAM (5.3 mg, 10 μmol)を伸長した。
合成例6で合成したFAM-AZO4-QXL TM520(FAM-Lys (NH2)-CAP-AZO4-CAP-Lys (QXL TM520))を10nmol、トリエチルアミン(ペプチドに対して過剰)、ジクロロメタン1mlに添加し、濃度10μmol/lのFAM-AZO4-QXL TM520溶液のサンプルを調製した。調製したFAM-AZO4-QXL TM520溶液にキセノンランプ光源から450nmの可視光を60秒間し、AZO4部位におけるアゾ部位の幾何構造をトランス体からシス体へと異性化させた。そののち、このサンプルを蛍光分析装置に入れ、490nmの励起光によりFAM由来の蛍光発光(520nm)を測定した。図3に光照射前後における蛍光スペクトル変化を示す。
K’K’K’-CTTCTTCT-AZO4-TCTTC-K’K’K’:
K’K’K’−CTTCTTCT−は、
式中、−Cは、シトシン:
−Tは、チミン:
“AZO4”搭載型ペプチド核酸は、Fmoc-固相合成法により調製した。合成に使用した、樹脂、カップリング試薬、脱保護試薬、洗浄操作、樹脂からの切り出し、HPLCによる精製は定法に従った。PNAの分子量は、MALDI-TOFMSにより解析し、計算分子量が5375.0であるのに対して、検出された分子量は5371.0であった。
試験例9(PNA−AZOの可視光(450nm)照射後のUVスペクトル測定)
合成例8で合成したPNA−AZOを10mMのリン酸ナトリウム緩衝液(pH 6.9)に溶解して、濃度300nMのPNA−AZO溶液のサンプルを調製した。調製したPNA−AZO溶液におけるアゾ骨格の幾何構造を完全にトランス体にするために、95℃で5分加温し、その後サンプルを室温にまで戻した後、キセノンランプ光源により450nmの可視光を10分間、20分間及び30分間それぞれ照射し、光照射後のUVスペクトルを測定した。UVスペクトルを図4に示す。なお、図4における曲線は上から順に0分間、10分間、20分間、30分間光照射した後のUVスペクトルを示す。
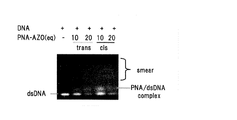
PNA(5’-CTTCTTCT-AZO4-TCTTC-3’)の標的配列を含む、DNA(a)鎖:3’- CATCCTCGAAGAAGATGGTGCGCTCCTGGA-5’、およびそれに対して核酸塩基相補的なDNA(b)鎖:5’- TCCAGGAGCGCACCATCTTCTTCGAGGATG-3’(Sigma-Genosis製)を10mM リン酸ナトリウム溶液(pH6.9)に溶解し、95℃に5分間加熱後、0.5℃/minの割合で冷却(アニーリング)することで二重鎖DNA(a,b)を形成させた。そして、PNAと二重鎖DNA(a,b)の終濃度がそれぞれ750nM, 250nMになるように混合して、2時間、25℃でインキュベートした。
蛍光顕微鏡観察用のグラスチャンバーにイヌ腎臓(MDCK)細胞を播種し、接着するまで約6時間インキュベートした。次に、1.6μg のGFP発現プラスミドDNA(clontech社製のpAcGFP-C1)を含む10mM リン酸ナトリウム溶液(pH6.9)に溶解させた試料を調製し、ここに予め可視光450nmを照射したPNA-AZO(アゾ骨格がシス型)と非照射のPNA-AZO(アゾ骨格がトランス型)を核酸塩基の数にして100倍等量になるように加えた。このPNA-AZO/GFP発現プラスミドDNAの会合体に対して、Fugene 6(ロシュ・ダイアグノスティック社製)を加え、30分インキュベートし、MDCK細胞に対してトランスフェクションした。蛍光顕微鏡により8時間後のGFP蛋白発現を観測した。図6にPNA-AZOのトランス体及びシス体を作用させたプラスミドDNAの蛍光顕微鏡写真を示す。
合成例9(2−(4−((4−ブロモフェニル)ジアゼニル)フェニルアミノ)エタノール(2-(4-((4-bromophenyl)diazenyl)phenylamino)ethanol)(以下、N-type,-OHともいう)の合成)
1H-NMR(400 MHz, CDCl3): 7.82(d, 2H, J=8.9), 7.71(d, 2H, J=8.6), 7.59(d, 2H, J=8.6), 6.69(d, 2H, J=8.9), 3.89(brs, 2H), 3.41(t, 2H, J=5.2)
合成例10−1
合成例10−2
1H-NMR(400MHz, CDCl3):7.91(d, 2H, J=9.0), 7.76(d, 2H,J=8.7), 7.63(d, 2H, J=8.7), 7.04(d, 2H, J=9.0), 4.18(t, 2H, J=4.2), 4.02(brs, 2H)
1H-NMR(400 MHz, CDCl3): 7.86(d, 2H, J=8.3), 7.78(d, 2H, J=8.5), 7.64(d, 2H, J=8.5), 7.36(d, 2H, J=8.3), 3.05(t, 2H, J=7.7), 2.74(t, 2H, J=7.7)
1H-NMR(400 MHz, CDCl3): 7.90 (d, 2H, J=7.0), 7.86 (d, 2H, J=8.3), 7.50(d, 2H, J=7.7), 7.35 (d, 2H, J=8.3), 3.71 (t, 2H, J=6.3), 2.80 (t, 2H, J=7.0), 1.94 (m, 2H)
1H-NMR(400 MHz, CDCl3): 7.87(d ,2H, J=8.6), 7.78(d, 2H, J=8.7), 7.64(d, 2H, J=8.7), 7.49 (d, 2H, J=8.6), 3.79 ( s, 2H)
1H-NMR(400 MHz, CDCl3): 7.85 (d, 2H, J=8.3), 7.78 (d, 2H,J=8.7), 7.64 (d, 2H, J=8.6), 7.46 (d, 2H, J=8.3), 3.84 (t, 2H,J=11), 3.24 (t, 2H, J=12)
合成例9、10、12及び14で合成したS-type,-OH、C-type,-OH、O-type,-OH及びN-type,-OHを水に添加して濃度20μmol/lの各水溶液を調製し、紫外−可視吸収スペクトルをそれぞれ測定した。紫外−可視吸収スペクトルを図8に示し、表2に測定結果を示す。
合成例11及び13で合成したS-type,-COOH及びC-type,-COOHを水にそれぞれ添加し、濃度20μmol/lの各水溶液を調製し、紫外−可視吸収スペクトルを測定した。紫外−可視吸収スペクトルを図9に示し、表3に測定結果を示す。
合成例9及び13で合成したS-type,-COOH及びC-type,-COOHの量子収率を以下の方法により算出した。
モル吸光係数の算出:サンプルをジメチルスルホキシドに溶解し濃度、40μmol/l, 30μmol/l, 20μmol/l, 15μmol/l, 10μmol/lの各濃度で調整し、95 ℃で30 分加熱した。その後サンプルを石英セルにいれ、UV-Vis分光分析装置でそれぞれの吸光度を測定した。その結果から380 nmの吸収を縦軸に、濃度を横軸にとり、その直線の傾きからモル吸光係数を算出した。
量子収率の算出:パワーメータ(OPHIR Nova, フォトダオードヘッド)でキセノンランプ光源の光量を測定し、その結果から、サンプルを透過した光量を算出した。サンプルをジメチルスルホキシドに溶かし、20μmol/lに調整したサンプルを95 ℃で30分加熱し、その後20 ℃に冷やした。サンプル1.5 ml を石英セルに入れ、UV-Vis分光分析装置でそれぞれの吸光度を測定した。このときの波長 380 nmにおける吸光度から前記で求めたモル吸光係数からサンプルのモル濃度を算出した。サンプルを氷浴中ですみやかに室温に戻し、石英セルに1.5mlはかりとり、キセノンランプ光源から380nmの波長光を一定時間(10,20,30,40秒)照射し、吸光度変化を測定した。測定したデータから横軸に時間(秒)、縦軸に異性化したモル濃度をプロットしたグラフの傾きから異性化速度を算出した。
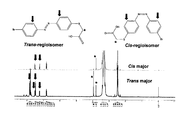
合成例13で合成したS-type,-COOHの450nmの光照射前後によるシス−トランス異性化挙動をNMRにより測定した。図10にNMRチャートを示す。
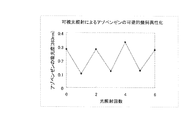
合成例13で合成したS-type,-OHをジメチルスルホキシドに溶解し、20μmol/lに濃度を調整し、UV測定用の石英セルに1.5ml入れ、キセノンランプ光源(朝日分光(株)製:ハイパワーキセノン光源Max-302, 300W)を用いて、波長380nmのバンドパスフィルター(半値幅10nm±2nm)を透過した光のみを5分程度照射した。トランス体のS-type,-OHのスペクトルが減少したことを確認したのち、波長470nmのバンドパスフィルター(半値幅10nm±2nm)を透過した光を5分間照射した。同様の操作を3回ずつ繰り返し、アゾ骨格の異性化が可逆的に起こることを確認した。図11に光照射におけるS-type,-OH溶液の各吸光度を示す。
合成例9、10、12及び14で合成したS-type,-OH、C-type,-OH、O-type,-OH及びN-type,-OHをそれぞれジメチルスルホキシドに溶解し、20μmol/lの濃度に調整した。上記のキセノンランプ光源を使用し、トランス体由来の最大吸収波長に相当する波長光を各トランス体のスペクトルが減少するまで照射し続けた。その後、各サンプルを恒温セルフォルダの装着したUV-Vis分光分析装置に静置し、40℃で加温した状態で各アゾベンゼンのトランス体由来の最大吸収波長における吸光度の経時変化を調べた。経時変化に対するアゾ骨格のトランス体の存在比を表すグラフを図12に示す。
Claims (6)
- 式(1):
で表される基であり、
Aは、低級アルキレン基、低級アルケニレン基、低級アルキニレン基、又は直接結合であり、
Xは、−O−R2a、−N−(R2b)2又は−S(O)k−R2cであり、
R2aは、低級アルキル基;ペプチド核酸部位を有する基;蛍光部位又は消光部位を有するスペーサーを有する基;アミノ基、カルボキシル基、低級アルコキシカルボニル基、ヒドロキシル基、ペプチド核酸部位を有する基、又は蛍光部位もしくは消光部位を有するスペーサーを有する基が置換されていてもよい低級アルキル基であり、
R2bは、同じであっても異なっていてもよく、水素原子;低級アルキル基;ペプチド核酸部位を有する基;蛍光部位又は消光部位を有するスペーサーを有する基;アミノ基、カルボキシル基、低級アルコキシカルボニル基、ヒドロキシル基、ペプチド核酸部位を有する基、又は蛍光部位もしくは消光部位を有するスペーサーを有する基が置換されていてもよい低級アルキル基であり、
R2cは、低級アルキル基;ペプチド核酸部位を有する基;蛍光部位又は消光部位を有するスペーサーを有する基;アミノ基、カルボキシル基、低級アルコキシカルボニル基、ヒドロキシル基、ペプチド核酸部位を有する基又は蛍光部位もしくは消光部位を有するスペーサーを有する基、が置換されていてもよい低級アルキル基であって、
kは0〜3の整数である)
で表されるアゾベンゼン化合物。 - 式(a1):
と、亜硝酸塩及び酸を添加して、ジアゾニウム塩を形成させ、さらに、前記ジアゾニウム塩と、式(a2):
を反応させることを特徴とする請求項1に記載のアゾベンゼン化合物の製造方法。 - 式(b1):
と、式(b2):
を酸性条件下で反応させることを特徴とする請求項1に記載のアゾベンゼン化合物の製造方法。 - 式(c1):
と、トリメチルシリルアセチレンを反応させた後、脱シリル化することによって、式(c2):
を反応させることを特徴とする請求項1に記載のアゾベンゼン化合物の製造方法。 - 光スイッチング材料として用いられる請求項1に記載のアゾベンゼン化合物。
- mRNA転写反応促進剤又はmRNA転写反応抑制剤として用いられる請求項1に記載のアゾベンゼン化合物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008323606A JP2010143864A (ja) | 2008-12-19 | 2008-12-19 | 光応答型アゾベンゼン化合物 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008323606A JP2010143864A (ja) | 2008-12-19 | 2008-12-19 | 光応答型アゾベンゼン化合物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2010143864A true JP2010143864A (ja) | 2010-07-01 |
Family
ID=42564668
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008323606A Pending JP2010143864A (ja) | 2008-12-19 | 2008-12-19 | 光応答型アゾベンゼン化合物 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2010143864A (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012075376A (ja) * | 2010-09-30 | 2012-04-19 | Osaka Univ | アゾベンゼン架橋型ペプチド核酸を用いたインフルエンザウイルスを測定する方法 |
| JPWO2012029434A1 (ja) * | 2010-08-31 | 2013-10-28 | 国立大学法人名古屋大学 | オリゴヌクレオチドおよびその利用 |
| JP2015040198A (ja) * | 2013-08-23 | 2015-03-02 | 公立大学法人首都大学東京 | 異性化反応制御方法、及び異性体製造方法 |
| WO2017047807A1 (ja) * | 2015-09-17 | 2017-03-23 | 国立大学法人大阪大学 | トラン化合物 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0256456A (ja) * | 1988-05-13 | 1990-02-26 | Mitsubishi Kasei Corp | アゾ系化合物及び該化合物を含有する液晶組成物 |
| JPH10287635A (ja) * | 1997-04-10 | 1998-10-27 | Dainippon Ink & Chem Inc | 重合性光学活性光異性化液晶性化合物 |
| WO2001021637A1 (fr) * | 1999-09-20 | 2001-03-29 | Makoto Komiyama | Oligonucleotide sensible a la lumiere |
| JP2004196662A (ja) * | 2002-11-19 | 2004-07-15 | Credia Japan:Kk | 新規な機能性ペプチド核酸およびその製法 |
| EP1462484A1 (en) * | 2003-03-05 | 2004-09-29 | Universita Degli Studi di Firenze | Colouring agent containing a mono- or disaccharide |
| JP2004277416A (ja) * | 2003-02-27 | 2004-10-07 | Masahiro Irie | ジアリールエテン系化合物、フォトクロミック材料および光機能素子 |
| JP2007326846A (ja) * | 2006-05-11 | 2007-12-20 | Institute Of Physical & Chemical Research | アゾベンゼン誘導体、蛍光性粒子およびその製造方法 |
| WO2007145213A1 (ja) * | 2006-06-12 | 2007-12-21 | Osaka University | Dna二本鎖形成制御 |
-
2008
- 2008-12-19 JP JP2008323606A patent/JP2010143864A/ja active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0256456A (ja) * | 1988-05-13 | 1990-02-26 | Mitsubishi Kasei Corp | アゾ系化合物及び該化合物を含有する液晶組成物 |
| JPH10287635A (ja) * | 1997-04-10 | 1998-10-27 | Dainippon Ink & Chem Inc | 重合性光学活性光異性化液晶性化合物 |
| WO2001021637A1 (fr) * | 1999-09-20 | 2001-03-29 | Makoto Komiyama | Oligonucleotide sensible a la lumiere |
| JP2004196662A (ja) * | 2002-11-19 | 2004-07-15 | Credia Japan:Kk | 新規な機能性ペプチド核酸およびその製法 |
| JP2004277416A (ja) * | 2003-02-27 | 2004-10-07 | Masahiro Irie | ジアリールエテン系化合物、フォトクロミック材料および光機能素子 |
| EP1462484A1 (en) * | 2003-03-05 | 2004-09-29 | Universita Degli Studi di Firenze | Colouring agent containing a mono- or disaccharide |
| JP2007326846A (ja) * | 2006-05-11 | 2007-12-20 | Institute Of Physical & Chemical Research | アゾベンゼン誘導体、蛍光性粒子およびその製造方法 |
| WO2007145213A1 (ja) * | 2006-06-12 | 2007-12-21 | Osaka University | Dna二本鎖形成制御 |
Non-Patent Citations (4)
| Title |
|---|
| JPN6013045970; SURESH,S. et al.: 'Polyacrylates with new azo dye pendants for nonlinear optics' MCLC S&T, Section B: Nonlinear Optics Vol.23, 2000, p.133-148 * |
| JPN6013045971; KALASHNIKOV,V.V. and SAMUKOV,V.V.: 'Synthesis of C-terminal peptide fragments of the heavy chain of the hemagglutinin of influenza virus' Khimiya Prirondnykh Soedinenii No.5, 1988, p.732-738 * |
| JPN6013045972; 澤田慎二郎、外2名: '可視光応答型アゾベンゼンの合成とその機能評価' 日本化学会第88春季年会-講演予稿集 I , 20080312, p.582 * |
| JPN6013045973; 澤田慎二郎、外2名: '可視光応答型アゾベンゼンを利用した生体機能制御分子の開発' 2007年 日本化学会西日本大会 講演要旨集 , 2007, p.172 * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2012029434A1 (ja) * | 2010-08-31 | 2013-10-28 | 国立大学法人名古屋大学 | オリゴヌクレオチドおよびその利用 |
| JP2012075376A (ja) * | 2010-09-30 | 2012-04-19 | Osaka Univ | アゾベンゼン架橋型ペプチド核酸を用いたインフルエンザウイルスを測定する方法 |
| JP2015040198A (ja) * | 2013-08-23 | 2015-03-02 | 公立大学法人首都大学東京 | 異性化反応制御方法、及び異性体製造方法 |
| WO2017047807A1 (ja) * | 2015-09-17 | 2017-03-23 | 国立大学法人大阪大学 | トラン化合物 |
| CN108026018A (zh) * | 2015-09-17 | 2018-05-11 | 国立大学法人大阪大学 | 二苯乙炔化合物 |
| JPWO2017047807A1 (ja) * | 2015-09-17 | 2018-08-09 | 国立大学法人大阪大学 | トラン化合物 |
| US10577396B2 (en) | 2015-09-17 | 2020-03-03 | Osaka University | Tolan compound |
| CN108026018B (zh) * | 2015-09-17 | 2021-01-15 | 国立大学法人大阪大学 | 二苯乙炔化合物 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Volkova et al. | Spectroscopic study of squaraines as protein-sensitive fluorescent dyes | |
| US8753570B2 (en) | Structure, synthesis, and applications for oligo phenylene ethynylenes | |
| CA2748313A1 (fr) | Composes dipyrromethenes-borosubstitues fluorescents et leur utilisation pour le diagnostic | |
| Talukder et al. | Tryptophan-based fluorophores for studying protein conformational changes | |
| KR101717953B1 (ko) | 신규 아조화합물, 이의 이용 및 이의 제조방법 | |
| JP2010143864A (ja) | 光応答型アゾベンゼン化合物 | |
| JP2016050296A (ja) | 蛍光色素結合セレンテラジン | |
| US20070184559A1 (en) | Method and compounds for the fluorescent labelling of biomolecules and polymer particles | |
| Gebhard et al. | Hybridization-Sensitive Fluorescent Probes for DNA and RNA by a Modular “Click” Approach | |
| Buschbeck et al. | Orthogonally protected diaminoterephthalate scaffolds: installation of two functional units at the chromophore | |
| JP4397659B2 (ja) | 蛍光性分子のモノマー発光とエキシマー発光のスイッチングを利用した分子ビーコンを用いるdna検出法 | |
| Ghosh et al. | Design, Synthesis, and DNA Binding Properties of Photoisomerizable Azobenzene− Distamycin Conjugates: An Experimental and Computational Study | |
| FR2934595A1 (fr) | Reactifs de marquage ayant un noyau pyridine portant une fonction diazomethyle, procedes de synthese de tels reactifs et procedes de detection de molecules biologiques | |
| KR101590527B1 (ko) | 새로운 인도시아닌 유도체 화합물, 상기 화합물을 포함하는 조성물, 및 싸이올기 함유 화합물 검출용 센서 | |
| Ikeda et al. | Hybridization-sensitive fluorescence control in the near-infrared wavelength range | |
| EP2397464B1 (en) | Synthesis of novel azo-dyes and their use in oligonucleotide synthesis | |
| Moustafa et al. | An azo-based PNA monomer: synthesis and spectroscopic study | |
| WO2008075718A1 (ja) | 蛍光発生分子 | |
| Aparin et al. | 1-Phenylethynylpyrene (PEPy) as a novel blue-emitting dye for qPCR assay | |
| JP5881509B2 (ja) | アゾベンゼン化合物 | |
| WO2019012963A1 (ja) | シアニン化合物及びそれを用いた蛍光色素 | |
| CN104479396B (zh) | 氨基酸类双光子荧光染料 | |
| Wang et al. | Design and synthesis of efficient fluorescent dyes for incorporation into DNA backbone and biomolecule detection | |
| KR101125058B1 (ko) | 물질 표지용 화합물 및 그 제조방법 | |
| McGrory et al. | Fluorescent α-amino acids via Heck–Matsuda reactions of phenylalanine-derived arenediazonium salts |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20110325 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20110325 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20111219 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20130827 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130917 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20140204 |