JP2010077215A - バイオディーゼル燃料と直接接触する成形部材 - Google Patents
バイオディーゼル燃料と直接接触する成形部材 Download PDFInfo
- Publication number
- JP2010077215A JP2010077215A JP2008244727A JP2008244727A JP2010077215A JP 2010077215 A JP2010077215 A JP 2010077215A JP 2008244727 A JP2008244727 A JP 2008244727A JP 2008244727 A JP2008244727 A JP 2008244727A JP 2010077215 A JP2010077215 A JP 2010077215A
- Authority
- JP
- Japan
- Prior art keywords
- diamine
- temperature
- polyamide resin
- acid
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Fuel-Injection Apparatus (AREA)
- Polyamides (AREA)
Abstract
【課題】耐バイオディーゼル燃料性に優れ、低吸水性、耐加水分解性にも優れ、かつ成形可能温度幅が広く、成形加工性に優れたバイオディーゼル燃料と直接接触するポリアミド樹脂成形部材を提供すること。
【解決手段】ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下、「C9ジアミン混合物」という。)及び1,6−ヘキサンジアミン(以下、「C6ジアミン」という。)からなり、C9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるポリアミド樹脂を含むことを特徴とするバイオディーゼル燃料と直接接触する成形部材。
【選択図】なし
【解決手段】ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下、「C9ジアミン混合物」という。)及び1,6−ヘキサンジアミン(以下、「C6ジアミン」という。)からなり、C9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるポリアミド樹脂を含むことを特徴とするバイオディーゼル燃料と直接接触する成形部材。
【選択図】なし
Description
本発明は、バイオディーゼル燃料と直接接触する成形部材に関する。詳しくは、耐バイオディーゼル燃料に優れ、低吸水性、耐加水分解性にも優れ、かつ成形可能温度幅が広く、成形加工性に優れた新規なポリアミド樹脂を用いたバイオディーゼル燃料と直接接触する成形部材に関するものである。
ナイロン6、ナイロン66などに代表される結晶性ポリアミドは、その優れた特性と溶融成形の容易さから、衣料用、産業資材用繊維、あるいは汎用のエンジニアリングプラスチックとして広く用いられているが、特にナイロン11、ナイロン12は、耐油性、燃料透過防止性の燃料部品用途に必要な基本性能を有しているので、ガソリン燃料や軽油燃料の燃料タンク、燃料パイプ、燃料搬送ユニット、燃料ポンプモジュール、バルブなどの燃料と接触する成形部材に使用されている。
近年、二酸化炭素排出量削減に対する期待から、ディーゼルエンジンが見直され、特に、バイオベースの燃料を用いるディーゼルエンジンが積極的に検討されている。ディーゼルエンジン用バイオベースの燃料は、「バイオディーゼル燃料」と呼ばれており、菜種油、ひまわり油、大豆油、コーン油などの植物油や、それらの廃食用油を原油としてメタノールでエステル化してグリセリンを分離除去した脂肪酸メチルエステルからなる燃料である。バイオディーゼル燃料は、一般的には、この脂肪酸メチルエステルを軽油にある割合で混ぜて用いられる。
しかし、ポリアミド樹脂のバイオディーゼル燃料に対する抵抗性は、ガソリンや軽油の従来の燃料に対する抵抗性と異なるものと考えられており、従来の燃料抵抗性を有するものが必ずしもバイオディーゼル燃料に対する抵抗性に優れるとは限らないので、ポリアミド樹脂のバイオディーゼル燃料に対する抵抗性に優れたポリアミド樹脂で製造した燃料部品に需要がある。
また、燃料部品に用いられるポリアミド樹脂においても、耐燃料性のほかに、吸水による物性変化、酸、高温のアルコール中での劣化などの問題点もあり、より寸法安定性に優れたポリアミドへの要求がある。
ジカルボン酸成分として蓚酸を用いるポリアミド樹脂はポリオキサミド樹脂と呼ばれ、同じアミノ基濃度の他のポリアミド樹脂と比較して融点が高いこと、吸水率が低いことが知られ(特許文献1)、吸水による物性変化が問題となっていた従来のポリアミドが使用困難な分野での活用が期待される。
これまでに、ジアミン成分として種々の脂肪族直鎖ジアミンを用いたポリオキサミド樹脂が提案されている。しかしながら、例えば、ジアミン成分として1,6−ヘキサンジアミンを用いたポリオキサミド樹脂は融点(約320℃)が熱分解温度(窒素中の1%重量減少温度;約310℃)より高いため(非特許文献1)、溶融重合、溶融成形が困難であり実用に耐えうるものではなかった。
ジアミン成分が1,9−ノナンジアミンであるポリオキサミド樹脂(以後、PA92と略称する)については、L. Francoらが蓚酸源として蓚酸ジエチルを用いた場合の製造法とその結晶構造を開示している(非特許文献2)。ここで得られるPA92は固有粘度が0.97dL/g、融点が246℃のポリマーであるが、強靭な成形体が成形出来ない程度の低分子量体しか得られていない。また、ジカルボン酸エステルとして蓚酸ジブチルを用いた場合について、固有粘度が0.99dL/g、融点が248℃のPA92を製造したことが示されている(特許文献2)。この場合も強靭な成形体が成形出来ない程度の低分子量体しか得られていないという問題点がある。
本発明者らは、ジカルボン酸成分として蓚酸を用い、ジアミン成分として1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンを特定の比率で用いたポリアミド樹脂が低吸水性でありながら、溶融成形温度幅が広く、しかも諸特性に優れるポリアミド樹脂(PA92C)であることを開示した(特許文献3)。
しかしながら、このポリアミド樹脂は、ジカルボン酸成分として蓚酸を用い、ジアミン成分として1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミンの3種のジアミンを特定の比率で用いたポリオキサミド樹脂ではない。
S. W. Shalaby., J. Polym. Sci., 11, 1(1973) L. Franco et al., Macromolecules., 31, 3912(1988) 特開2006−57033号公報
特表平5−506466号公報
WO2008/072754
しかしながら、このポリアミド樹脂は、ジカルボン酸成分として蓚酸を用い、ジアミン成分として1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミンの3種のジアミンを特定の比率で用いたポリオキサミド樹脂ではない。
S. W. Shalaby., J. Polym. Sci., 11, 1(1973) L. Franco et al., Macromolecules., 31, 3912(1988)
本発明が解決しようとする課題は、耐バイオディーゼル燃料性に優れ、十分な高分子量化が達成され、融点と熱分解温度の差から見積もられる成形可能温度幅がより広く、溶融成形性により優れ、さらに、脂肪族直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、耐薬品性、柔軟性、耐加水分解性などに優れたバイオディーゼル燃料と直接接触するポリアミド樹脂成形部材を提供することにある。
本発明者らは、上記の課題を解決するために鋭意検討を重ねた結果、蓚酸源として蓚酸ジエステルを用い、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下において「C9ジアミン混合物」ともいう。)及び1,6−ヘキサンジアミン(以下において「C6ジアミン」ともいう。)からなり、かつC9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1である混合物を用いたポリアミド樹脂(以下において「PA92/62T」ともいう)が、耐バイオディーゼル燃料に優れ、かつ直鎖ポリオキサミド樹脂に見られる低吸水性を損なうことなく、耐加水分解性に優れ、しかも、高分子量で、融点と熱分解温度の差が大きく溶融成形性に優れたてポリアミド樹脂であること、したがって、このポリアミド樹脂を用いれば目的のバイオディーゼル燃料と直接接触するポリアミド樹脂成形部材が提供されることを見出し、本発明を完成した。
本発明によれば、耐バイオディーゼル燃料性に優れ、低吸水性、耐加水分解性にも優れ、かつ成形可能温度幅が広く、成形加工性に優れたバイオディーゼル燃料と直接接触するポリアミド樹脂成形部材が提供される。
(1)ポリアミド樹脂の構成成分
本発明に用いられるポリアミドPA92/62Tは、ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下、「C9ジアミン混合物」という。)及び1,6−ヘキサンジアミン(以下、「C6ジアミン」という。)からなり、C9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるジアミン混合物であるポリアミド樹脂である。
本発明に用いられるポリアミドPA92/62Tは、ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下、「C9ジアミン混合物」という。)及び1,6−ヘキサンジアミン(以下、「C6ジアミン」という。)からなり、C9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるジアミン混合物であるポリアミド樹脂である。
このポリアミドの製造に用いられる蓚酸源としては、蓚酸ジエステルが用いられ、これらはアミノ基との反応性を有するものであれば特に制限はなく、蓚酸ジメチル、蓚酸ジエチル、蓚酸ジn−(またはi−)プロピル、蓚酸ジn−(またはi−、またはt−)ブチル等の脂肪族1価アルコールの蓚酸ジエステル、蓚酸ジシクロヘキシル等の脂環式アルコールの蓚酸ジエステル、蓚酸ジフェニル等の芳香族アルコールの蓚酸ジエステル等が挙げられる。
上記の蓚酸ジエステルの中でも炭素原子数が3を超える脂肪族1価アルコールの蓚酸ジエステル、脂環式アルコールの蓚酸ジエステル、芳香族アルコールの蓚酸ジエステルが好ましく、その中でも蓚酸ジブチル及び蓚酸ジフェニルが特に好ましい。
本発明のポリアミド樹脂に用いるC9ジアミン混合物における1,9−ノナンジアミン成分と2−メチル−1,8−オクタンジアミン成分のモル比は、一般的には1:99〜99:1であり、好ましくは5:95〜95:5、より好ましくは5:95〜40:60又は60:40〜95:5、特に5:95〜30:70又は70:30〜90:10である。1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミンを上記の特定量共重合することにより、成形可能温度幅が広く、溶融成形性に優れ、かつ低吸水性、耐薬品性、耐加水分解性、透明性などにも優れたポリアミドが得られる。
本発明のポリアミド樹脂においては、ジアミン成分として、上記C9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)に1,6−ヘキサンジアミンを混合したものを用いる。C9ジアミン混合物と1,6−ヘキサンジアミンのモル比は、1:99〜99:1である。C9ジアミン混合物に対して1,6−ヘキサンジアミンをモル比で1/99以上混合することにより、ジカルボン酸成分として蓚酸、ジアミン成分としてC9ジアミン混合物からなるポリアミド樹脂(PA92C)の上記の優れた効果を実質的に保持しながら(特に溶融成形性、低吸水性を損なうことなく)、ポリアミド樹脂の融点が上昇し特に力学的物性を向上させることができる。C9ジアミン混合物と1,6−ヘキサンジアミンのモル比は、好ましくは5.1:94.9〜99:1、より好ましくは10:90〜99:1、さらに好ましくは20:80〜99:1である。特に30:70〜98:2、さらに30:70〜90:10(さらに30:70〜70:30)であることが好ましい。本ポリアミド樹脂においては、C9ジアミン混合物に対して1,6−ヘキサンジアミンをモル比で1/99以上共重合することによって融点への変化は明瞭に現れ、樹脂の融点は上昇するが、1,6−ヘキサンジアミンがモル比で99/1以内であれば溶融成形性は許容できるものが得られる。また、1,6−ヘキサンジアミンがモル比で80/20以内であればポリアミド樹脂の融点は300℃以下となり、重合及び成形加工(溶融成形性)がより容易であり、70/30以内であれば融点が280℃以下になって、溶融成形性がより容易となるのでより好ましい。
(2)ポリアミド樹脂の製造
本発明で用いるポリアミド樹脂PA92/62Tは、ポリアミドを製造する方法として知られている任意の方法を用いて製造することができる。本発明者らの研究によれば、ジアミン及び蓚酸ジエステルをバッチ式又は連続式で重縮合反応させることにより得ることができる。具体的には、以下の操作で示されるような、(i)前重縮合工程、(ii)後重縮合工程の順で行うのが好ましい。
本発明で用いるポリアミド樹脂PA92/62Tは、ポリアミドを製造する方法として知られている任意の方法を用いて製造することができる。本発明者らの研究によれば、ジアミン及び蓚酸ジエステルをバッチ式又は連続式で重縮合反応させることにより得ることができる。具体的には、以下の操作で示されるような、(i)前重縮合工程、(ii)後重縮合工程の順で行うのが好ましい。
(i)前重縮合工程:まず反応器内を窒素置換した後、ジアミン(ジアミン成分)及び蓚酸ジエステル(蓚酸源)を混合する。混合する場合にジアミン及び蓚酸ジエステルが共に可溶な溶媒を用いても良い。ジアミン成分及び蓚酸源が共に可溶な溶媒としては、特に制限されないが、トルエン、キシレン、トリクロロベンゼン、フェノール、トリフルオロエタノールなどを用いることができ、特にトルエンを好ましく用いることができる。例えば、ジアミンを溶解したトルエン溶液を50℃に加熱した後、これに対して蓚酸ジエステルを加える。このとき、蓚酸ジエステルと上記ジアミンの仕込み比は、蓚酸ジエステル/上記ジアミンで、0.8〜1.5(モル比)、好ましくは0.91〜1.1(モル比)、更に好ましくは0.99〜1.01(モル比)である。
このように仕込んだ反応器内を攪拌及び/又は窒素バブリングしながら、常圧下で昇温する。反応温度は、最終到達温度が80〜150℃、好ましくは100〜140℃の範囲になるように制御するのが好ましい。最終到達温度での反応時間は3時間〜6時間である。
(ii)後重縮合工程:更に高分子量化を図るために、前重縮合工程で生成した重合物を常圧下において反応器内で徐々に昇温する。昇温過程において前重縮合工程の最終到達温度、すなわち80〜150℃から、最終的に220℃以上300℃以下、好ましくは230℃以上280℃以下、更に好ましくは240℃以上270℃以下の温度範囲にまで到達させる。昇温時間を含めて1〜8時間、好ましくは2〜6時間保持して反応を行うことが好ましい。さらに後重合工程において、必要に応じて減圧下での重合を行うこともできる。減圧重合を行う場合の好ましい最終到達圧力は0.1MPa未満〜13.3Paである。
本発明に用いるポリアミド樹脂の製造方法の具体的例を説明する。
まず原料の蓚酸ジエステルを容器内に仕込む。容器は、後に行う重縮合反応の温度および圧力に耐え得るものであれば、特に制限されない。その後、容器を原料のジアミンと混合する温度まで昇温させ、次いでジアミンを注入し重縮合反応を開始させる。原料を混合する温度は、原料の蓚酸ジエステルおよびジアミンの融点以上、沸点未満の温度であり、かつ蓚酸ジエステルとジアミンの重縮合反応によって生じるポリオキサミドが熱分解しない温度であれば特に制限されない。例えば、1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン、1,6−ヘキサンジアミンの混合物からなり、かつC9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)と1,6−ヘキサンジアミンのモル比が1:99〜99:1であるジアミンと蓚酸ジブチルを原料とするポリオキサミド樹脂の場合、上記混合温度は15℃から300℃が好ましい。また、C9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)と1,6−ヘキサンジアミンのモル比は、5:95〜90:10の場合、常温で液状か又は50℃程度に加温するだけで液化するので取り扱いやすいためより好ましい。混合温度が縮合反応によって生成するアルコールの沸点以上の場合、アルコールを留去、凝縮する装置を備えた容器を用いるのが望ましい。また、縮合反応によって生成するアルコール存在下で加圧重合する場合には、耐圧容器を用いる。蓚酸ジエステルとジアミンの仕込み比は、蓚酸ジエステル/上記ジアミンで、0.8〜1.2(モル比)、好ましくは0.91〜1.09(モル比)、更に好ましくは0.98〜1.02(モル比)である。
まず原料の蓚酸ジエステルを容器内に仕込む。容器は、後に行う重縮合反応の温度および圧力に耐え得るものであれば、特に制限されない。その後、容器を原料のジアミンと混合する温度まで昇温させ、次いでジアミンを注入し重縮合反応を開始させる。原料を混合する温度は、原料の蓚酸ジエステルおよびジアミンの融点以上、沸点未満の温度であり、かつ蓚酸ジエステルとジアミンの重縮合反応によって生じるポリオキサミドが熱分解しない温度であれば特に制限されない。例えば、1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン、1,6−ヘキサンジアミンの混合物からなり、かつC9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)と1,6−ヘキサンジアミンのモル比が1:99〜99:1であるジアミンと蓚酸ジブチルを原料とするポリオキサミド樹脂の場合、上記混合温度は15℃から300℃が好ましい。また、C9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)と1,6−ヘキサンジアミンのモル比は、5:95〜90:10の場合、常温で液状か又は50℃程度に加温するだけで液化するので取り扱いやすいためより好ましい。混合温度が縮合反応によって生成するアルコールの沸点以上の場合、アルコールを留去、凝縮する装置を備えた容器を用いるのが望ましい。また、縮合反応によって生成するアルコール存在下で加圧重合する場合には、耐圧容器を用いる。蓚酸ジエステルとジアミンの仕込み比は、蓚酸ジエステル/上記ジアミンで、0.8〜1.2(モル比)、好ましくは0.91〜1.09(モル比)、更に好ましくは0.98〜1.02(モル比)である。
次に、容器内をポリオキサミド樹脂の融点以上かつ熱分解しない温度以下に昇温する。例えば、1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンからなり、かつC9ジアミン混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物)と1,6−ヘキサンジアミンのモル比が50:50であり、さらに1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンのモル比が50:50であるジアミンと蓚酸ジブチルを原料とするポリオキサミド樹脂の場合、融点は261℃であることから270℃から300℃に昇温するのが好ましい(圧力は、2MPa〜4MPa)。生成したアルコールを留去しながら、必要に応じて常圧窒素気流下もしくは減圧下において継続して重縮合反応を行う。耐圧容器内で原料を混合し、縮合反応によって生成するアルコール存在下で加圧重合する場合は、まず生成したアルコールを留去しながら放圧する。その後、必要に応じて常圧窒素気流下もしくは減圧下において継続して重縮合反応を行う。減圧重合を行う場合の好ましい最終到達圧力は760〜0.1Torrである。温度は、270〜300℃が好ましい。また、アルコールは水冷コンデンサで冷却して液化し、回収する。
(3)ポリアミド樹脂の性状及び物性
本発明に用いるポリアミド樹脂PA92/62Tの分子量に特別の制限はないが、96%濃硫酸を溶媒とし、ポリアミド樹脂濃度が1.0g/dlの96%濃硫酸溶液を用い、25℃で測定した相対粘度ηrが1.8〜6.0の範囲内である。好ましくは2.0〜5.5であり、2.5〜4.5が特に好ましい。ηrが1.8より低いと成形物が脆くなり物性が低下する。一方、ηrが6.0より高いと溶融粘度が高くなり、成形加工性が悪くなる。
本発明に用いるポリアミド樹脂PA92/62Tの分子量に特別の制限はないが、96%濃硫酸を溶媒とし、ポリアミド樹脂濃度が1.0g/dlの96%濃硫酸溶液を用い、25℃で測定した相対粘度ηrが1.8〜6.0の範囲内である。好ましくは2.0〜5.5であり、2.5〜4.5が特に好ましい。ηrが1.8より低いと成形物が脆くなり物性が低下する。一方、ηrが6.0より高いと溶融粘度が高くなり、成形加工性が悪くなる。
本発明に用いるポリアミド樹脂は、カルボン酸成分として蓚酸を用い、ジアミン成分として1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンを共重合することで、蓚酸と1,9−ノナンジアミンからなるポリアミドと比べて、上記相対粘度を増加させること、すなわち分子量を増加させることが可能である。また、本発明に用いるポリアミド樹脂PA92/62Tは、カルボン酸成分として蓚酸を用い、ジアミン成分として1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンにさらに1,6−ヘキサメチレンジアミンを共重合することで、蓚酸と1,9−ノナンジアミン及び2−メチル−1,8−オクタンジアミンからなるポリアミドと比べて、樹脂の融点を上昇させることが可能である。また、実質的な熱分解の指標である1%重量減少温度(以下、Tdと略す)と融点(以下、Tmと略す)の差(Td−Tm)で表される成形可能温度範囲が、蓚酸と1,9−ノナンジアミンからなるポリアミドと比べて拡大し、好ましくは50℃以上、より好ましくは60℃以上であることができ、さらには90℃以上も可能である。本発明のポリアミド樹脂は、Tdが好ましくは280℃以上、より好ましくは300℃以上、さらに好ましくは320℃以上であり、高い耐熱性を有することを特徴とする。
さらに、本発明は、ポリアミド樹脂PA92/62Tがバイオディーゼル燃料性に優れることを確認し、完成されたものである。バイオディーゼル燃料は、菜種油、ひまわり油、大豆油、コーン油などの植物油や、それらの廃食用油を原油としてメタノールでエステル化してグリセリンを分離除去した脂肪酸メチルエステルからなる燃料である。
(4)本発明のポリアミド樹脂に配合できる成分
本発明に用いられるポリアミド樹脂PA92/62Tには、本発明の効果を損なわない範囲で他のジカルボン酸成分を混合する事が出来る。蓚酸以外の他のジカルボン酸成分としては、マロン酸、ジメチルマロン酸、コハク酸、グルタル酸、アジピン酸、2−メチルアジピン酸、トリメチルアジピン酸、ピメリン酸、2,2−ジメチルグルタル酸、3,3−ジエチルコハク酸、アゼライン酸、セバシン酸、スベリン酸などの脂肪族ジカルボン酸、また、1,3−シクロペンタンジカルボン酸、1,4−シクロヘキサンジカルボン酸などの脂環式ジカルボン酸、さらにテレフタル酸、イソフタル酸、2,6−ナフタレンジカルボン酸、2,7−ナフタレンジカルボン酸、1,4−ナフタレンジカルボン酸、1,4−フェニレンジオキシジ酢酸、1,3−フェニレンジオキシジ酢酸、ジ安息香酸、4,4’−オキシジ安息香酸、ジフェニルメタン−4,4’−ジカルボン酸、ジフェニルスルホン−4,4’−ジカルボン酸、4,4’−ビフェニルジカルボン酸などの芳香族ジカルボン酸などを単独で、あるいはこれらの任意の混合物を重縮合反応時に添加することもできる。さらに、トリメリット酸、トリメシン酸、ピロメリット酸などの多価カルボン酸を溶融成形が可能な範囲内で用いることもできる。蓚酸以外の他のジカルボン酸成分の配合量は全ジカルボン酸成分基準に5モル%以下である。
本発明に用いられるポリアミド樹脂PA92/62Tには、本発明の効果を損なわない範囲で他のジカルボン酸成分を混合する事が出来る。蓚酸以外の他のジカルボン酸成分としては、マロン酸、ジメチルマロン酸、コハク酸、グルタル酸、アジピン酸、2−メチルアジピン酸、トリメチルアジピン酸、ピメリン酸、2,2−ジメチルグルタル酸、3,3−ジエチルコハク酸、アゼライン酸、セバシン酸、スベリン酸などの脂肪族ジカルボン酸、また、1,3−シクロペンタンジカルボン酸、1,4−シクロヘキサンジカルボン酸などの脂環式ジカルボン酸、さらにテレフタル酸、イソフタル酸、2,6−ナフタレンジカルボン酸、2,7−ナフタレンジカルボン酸、1,4−ナフタレンジカルボン酸、1,4−フェニレンジオキシジ酢酸、1,3−フェニレンジオキシジ酢酸、ジ安息香酸、4,4’−オキシジ安息香酸、ジフェニルメタン−4,4’−ジカルボン酸、ジフェニルスルホン−4,4’−ジカルボン酸、4,4’−ビフェニルジカルボン酸などの芳香族ジカルボン酸などを単独で、あるいはこれらの任意の混合物を重縮合反応時に添加することもできる。さらに、トリメリット酸、トリメシン酸、ピロメリット酸などの多価カルボン酸を溶融成形が可能な範囲内で用いることもできる。蓚酸以外の他のジカルボン酸成分の配合量は全ジカルボン酸成分基準に5モル%以下である。
また、本発明に用いられるポリアミド樹脂PA92/62Tには本発明の効果を損なわない範囲で、他のジアミン成分を混合する事が出来る。1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミン以外の他のジアミン成分としては、エチレンジアミン、プロピレンジアミン、1,4−ブタンジアミン、1,8−オクタンジアミン、1,10−デカンジアミン、1,12−ドデカンジアミン、3−メチル−1,5−ペンタンジアミン、2,2,4−トリメチル−1,6−ヘキサンジアミン、2,4,4−トリメチル−1,6−ヘキサンジアミン、5−メチル−1,9−ノナンジアミンなどの脂肪族ジアミン、さらにシクロヘキサンジアミン、メチルシクロヘキサンジアミン、イソホロンジアミンなどの脂環式ジアミン、さらにp−フェニレンジアミン、m−フェニレンジアミン、p−キシレンジアミン、m−キシレンジアミン、4,4’−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルエーテルなどの芳香族ジアミンなどを単独で、あるいはこれらの任意の混合物を重縮合反応時に添加することもできる。1,9−ノナンジアミン、2−メチル−1,8−オクタンジアミン及び1,6−ヘキサンジアミン以外の他のジアミン成分の配合量は全ジアミン成分基準に5モル%以下である。
本発明の耐バイオディーゼル燃料性に優れる成形部品には、本発明の効果を損なわない範囲で、上記ポリアミド樹脂の一部を、他のポリオキサミドや、芳香族ポリアミド、脂肪族ポリアミド、脂環式ポリアミドなどポリアミド類で置換することが可能である。更に、ポリアミド以外の熱可塑性ポリマー、エラストマーでも置換することができる。しかし、ポリアミド樹脂PA92/62T以外の置換する樹脂は50質量%以下であることが好ましい。
特に、本発明の耐バイオディーゼル燃料性に優れる成形部品には、バイオディーゼル燃料透過性を減少させる目的で層状ケイ酸塩を添加することができる。
層状ケイ酸塩とは、ケイ酸マグネシウム又はケイ酸アルミニウムの層で構成される層状フィロケイ酸塩等を挙げることができ、1辺の長さが0.002〜1μmで、厚さが6〜20Åの平板が層を形成したものである。
層状フィロケイ酸塩の具体例としては、たとえば、モンモリロナイト、サボナイト、バイデライト、ノントロナイト、ヘクトライト、スティブンサイト等のスメクタイト系粘土鉱物やバーミキュライト、ハロサイトなどを挙げることができる。これらは天然物でも合成物でもよい。
層状ケイ酸塩は、組成物中に分散した際に、平均20Å以上の層間距離を保ち、均一に分散している。ここで層間距離とは層状ケイ酸塩の平板の重心間の距離をいい、均一に分散するとは、層状ケイ酸塩の平板が、平均的に5層以下で重なった多層物が平行に、もしくはランダムに、もしくは平行とランダムが混在した状態で、50質量%以上が、好ましくは70質量%以上が局所的な塊を形成することなく分散した状態をいう。
層状ケイ酸塩の配合量は、ポリアミド樹脂100質量部に対して、0.05質量部〜10質量部が好ましく、0.05質量部〜5質量部がより好ましい。0.05質量部未満では燃料透過抑制効果が十分でなく、10質量部を超えると、成形が困難になったり、衝撃強度や柔軟性が低下する恐れがある。
本発明の耐バイオディーゼル燃料性に優れる成形部品には、さらに可塑剤を添加することが好ましい。可塑剤としては、たとえば、ベンゼンスルホン酸ブチルアミド、p−ヒドロキシ安息鉱酸と炭素数6〜21の直鎖又は分岐鎖アルコールとのエステル(たとえば、2−エチルヘキシルp−ヒドロキシベンゾエート)等を挙げることができる。
可塑剤の配合量は、ポリアミド樹脂100質量部に対して、0〜30質量部が好ましい。30質量部を超えるときは、ブリードアウトの可能性がある。
可塑剤の配合量は、ポリアミド樹脂100質量部に対して、0〜30質量部が好ましい。30質量部を超えるときは、ブリードアウトの可能性がある。
さらに、本発明の耐バイオディーゼル燃料性に優れる成形部品には、必要に応じて、ガラス繊維などの補強材、充填材、銅化合物などの安定剤、着色剤、紫外線吸収剤、光安定化剤、酸化防止剤、帯電防止剤、難燃剤、結晶化促進剤、導電材、潤滑剤などを重縮合反応時、またはその後に添加することもできる。
(5)成形部品の成形加工
本発明の耐バイオディーゼル燃料性に優れる成形部品の成形方法としては、ポリアミド樹脂あるいはそれに各種の配合成分や添加剤を配合したポリアミド樹脂組成物を射出、押出、中空、プレス、ロール、発泡、真空・圧空、延伸などポリアミドに適用できる公知の成形加工法はすべて可能であり、これらの成形法によってフィルム、シート、成形品、繊維などに加工することができる。
本発明の耐バイオディーゼル燃料性に優れる成形部品の成形方法としては、ポリアミド樹脂あるいはそれに各種の配合成分や添加剤を配合したポリアミド樹脂組成物を射出、押出、中空、プレス、ロール、発泡、真空・圧空、延伸などポリアミドに適用できる公知の成形加工法はすべて可能であり、これらの成形法によってフィルム、シート、成形品、繊維などに加工することができる。
(6)耐バイオディーゼル燃料性に優れる成形部品の用途
本発明によって得られる耐バイオディーゼル燃料性に優れる成形部品は、従来のポリアミド樹脂製燃料部品が用いられてきた各種成形部品のいずれにも用いることが可能である。バイオディーゼル燃料と接触する成形部材としては、たとえば、燃料タンク、燃料チューブ、燃料パイプ、燃料搬送ユニット、燃料ポンプモジュール、バルブなどがある。
本発明によって得られる耐バイオディーゼル燃料性に優れる成形部品は、従来のポリアミド樹脂製燃料部品が用いられてきた各種成形部品のいずれにも用いることが可能である。バイオディーゼル燃料と接触する成形部材としては、たとえば、燃料タンク、燃料チューブ、燃料パイプ、燃料搬送ユニット、燃料ポンプモジュール、バルブなどがある。
[物性測定、成形、評価方法]
以下、実施例を挙げて本発明を具体的に説明するが、本発明はこれらにより何ら制限されるものではない。なお、実施例中の測定は以下の方法により行った。
以下、実施例を挙げて本発明を具体的に説明するが、本発明はこれらにより何ら制限されるものではない。なお、実施例中の測定は以下の方法により行った。
(1)相対粘度(ηr)
ηrはポリアミドの96%硫酸溶液(濃度:1.0g/dl)を使用してオストワルド型粘度計を用いて25℃で測定した。
ηrはポリアミドの96%硫酸溶液(濃度:1.0g/dl)を使用してオストワルド型粘度計を用いて25℃で測定した。
(2)融点(Tm)及び結晶化温度(Tc)
Tm及びTcは、PerkinELmer社製PYRIS Diamond DSC用いて窒素雰囲気下で測定した。30℃から300℃まで10℃/分の速度で昇温し(昇温ファーストランと呼ぶ)、300℃で3分保持したのち、−100℃まで10℃/分の速度で降温し(降温ファーストランと呼ぶ)、次に300℃まで10℃/分の速度で昇温した(昇温セカンドランと呼ぶ)。得られたDSCチャートから降温ファーストランの発熱ピーク温度をTc、昇温セカンドランの吸熱ピーク温度をTmとした。
Tm及びTcは、PerkinELmer社製PYRIS Diamond DSC用いて窒素雰囲気下で測定した。30℃から300℃まで10℃/分の速度で昇温し(昇温ファーストランと呼ぶ)、300℃で3分保持したのち、−100℃まで10℃/分の速度で降温し(降温ファーストランと呼ぶ)、次に300℃まで10℃/分の速度で昇温した(昇温セカンドランと呼ぶ)。得られたDSCチャートから降温ファーストランの発熱ピーク温度をTc、昇温セカンドランの吸熱ピーク温度をTmとした。
(3)1%重量減少温度(Td)
Tdは島津製作所社製THERMOGRAVIMETRIC ANALYZER TGA−50を用い、熱重量分析(TGA)により測定した。20ml/分の窒素気流下室温から500℃まで10℃/分の昇温速度で昇温し、Tdを測定した。
Tdは島津製作所社製THERMOGRAVIMETRIC ANALYZER TGA−50を用い、熱重量分析(TGA)により測定した。20ml/分の窒素気流下室温から500℃まで10℃/分の昇温速度で昇温し、Tdを測定した。
(4)溶融粘度
溶融粘度はティー・エイ・インスツルメント・ジャパン社製溶融粘弾性測定装置ARESに25mmのコーン・プレートを装着して、窒素中、290℃、せん断速度0.1s-1の条件で測定した。
溶融粘度はティー・エイ・インスツルメント・ジャパン社製溶融粘弾性測定装置ARESに25mmのコーン・プレートを装着して、窒素中、290℃、せん断速度0.1s-1の条件で測定した。
(5)フィルム成形
東邦マシナリー社製真空プレス機TMB−10を用いてフィルム成形を行った。500〜700Paの減圧雰囲気下290℃(ナイロン6を用いた場合は260℃、ナイロン66を用いた場合は290℃、ナイロン12を用いた場合は230℃)で5分間加熱溶融させた後、5MPaで1分間プレスを行いフィルム成形した。次に減圧雰囲気を常圧まで戻したのち室温5MPaで1分間冷却結晶化させてフィルムを得た。
東邦マシナリー社製真空プレス機TMB−10を用いてフィルム成形を行った。500〜700Paの減圧雰囲気下290℃(ナイロン6を用いた場合は260℃、ナイロン66を用いた場合は290℃、ナイロン12を用いた場合は230℃)で5分間加熱溶融させた後、5MPaで1分間プレスを行いフィルム成形した。次に減圧雰囲気を常圧まで戻したのち室温5MPaで1分間冷却結晶化させてフィルムを得た。
(6)吸水率(飽和吸水率、平衡吸水率)
ポリアミド樹脂を(5)の条件で成形したフィルム(寸法:20mm×10mm、厚さ0.25mm;重量約0.05g)を23℃のイオン交換水に浸漬し、所定時間ごとにフィルムを取り出し、フィルムの重量を測定した。フィルム重量の増加率が0.2%の範囲で3回続いた場合にポリアミド樹脂フィルムへの水分の吸収が飽和に達したと判断して、水に浸漬する前のフィルムの重量(Xg)と飽和に達した時のフィルムの重量(Yg)から式(1)により飽和吸水率(%)を算出した。
飽和吸水率(%)=100(Y−X)/X (1)
なお、上記フィルムの成形直後の重量(Xg)と上記フィルムを成形後に湿度65%、温度23℃で平衡に達したときの重量(Yg)から式(1)により算出した吸水率(平衡吸水率)をウェットでの吸水率として表中に記載した。
ポリアミド樹脂を(5)の条件で成形したフィルム(寸法:20mm×10mm、厚さ0.25mm;重量約0.05g)を23℃のイオン交換水に浸漬し、所定時間ごとにフィルムを取り出し、フィルムの重量を測定した。フィルム重量の増加率が0.2%の範囲で3回続いた場合にポリアミド樹脂フィルムへの水分の吸収が飽和に達したと判断して、水に浸漬する前のフィルムの重量(Xg)と飽和に達した時のフィルムの重量(Yg)から式(1)により飽和吸水率(%)を算出した。
飽和吸水率(%)=100(Y−X)/X (1)
なお、上記フィルムの成形直後の重量(Xg)と上記フィルムを成形後に湿度65%、温度23℃で平衡に達したときの重量(Yg)から式(1)により算出した吸水率(平衡吸水率)をウェットでの吸水率として表中に記載した。
(7)耐薬品性
本発明によって得られるポリアミドの熱プレスフィルムを以下に列挙する薬品中に7日間浸漬した後に、フィルムの重量残存率(%)及び外観の変化を観測した。濃塩酸、64%硫酸、氷酢酸のそれぞれの溶液においては23℃において浸漬した試料について試験を行った。
本発明によって得られるポリアミドの熱プレスフィルムを以下に列挙する薬品中に7日間浸漬した後に、フィルムの重量残存率(%)及び外観の変化を観測した。濃塩酸、64%硫酸、氷酢酸のそれぞれの溶液においては23℃において浸漬した試料について試験を行った。
(8)耐加水分解性
本発明によって得られるポリアミドの熱プレスフィルムをオートクレーブに入れ、水(pH=7)、0.5mol/l硫酸(pH=1)、1mol/l水酸化ナトリウム水溶液(pH=14)中でそれぞれ121℃、60分間処理した後の重量残存率(%)、及び外観変化を調べた。
本発明によって得られるポリアミドの熱プレスフィルムをオートクレーブに入れ、水(pH=7)、0.5mol/l硫酸(pH=1)、1mol/l水酸化ナトリウム水溶液(pH=14)中でそれぞれ121℃、60分間処理した後の重量残存率(%)、及び外観変化を調べた。
(9)機械的物性
以下に示す〔1〕〜〔4〕の測定は、下記の試験片を樹脂温度290℃(ナイロン6を用いた場合は260℃、ナイロン66を用いた場合は290℃、ナイロン12を用いた場合は230℃)、金型温度80℃の射出成形により成形し、これを用いて行った。成形後直ちに調湿せずに23℃で評価したものをドライ、成形後に湿度65%、温度23℃で調湿した後に23℃で評価したものをウェットとして表中に記載した。
〔1〕 引張試験(引張降伏点強度):ASTM D638に記載のTypeIの試験片を用いてASTM D638に準拠して測定した。
〔2〕 曲げ試験(曲げ弾性率):試験片寸法127mm×12.7mm×3.2mmの試験片を用いてASTM D790に準拠し、23℃で測定した。
〔3〕 アイゾット衝撃強度:試験片寸法127mm×12.7mm×3.2mmの試験片を用いてASTM D256に準拠し、23℃で測定した。
〔4〕 荷重たわみ温度:試験片寸法127mm×12.7mm×3.2mmの試験片を用いてASTM D648に準拠し、荷重1.82MPaで測定した。
以下に示す〔1〕〜〔4〕の測定は、下記の試験片を樹脂温度290℃(ナイロン6を用いた場合は260℃、ナイロン66を用いた場合は290℃、ナイロン12を用いた場合は230℃)、金型温度80℃の射出成形により成形し、これを用いて行った。成形後直ちに調湿せずに23℃で評価したものをドライ、成形後に湿度65%、温度23℃で調湿した後に23℃で評価したものをウェットとして表中に記載した。
〔1〕 引張試験(引張降伏点強度):ASTM D638に記載のTypeIの試験片を用いてASTM D638に準拠して測定した。
〔2〕 曲げ試験(曲げ弾性率):試験片寸法127mm×12.7mm×3.2mmの試験片を用いてASTM D790に準拠し、23℃で測定した。
〔3〕 アイゾット衝撃強度:試験片寸法127mm×12.7mm×3.2mmの試験片を用いてASTM D256に準拠し、23℃で測定した。
〔4〕 荷重たわみ温度:試験片寸法127mm×12.7mm×3.2mmの試験片を用いてASTM D648に準拠し、荷重1.82MPaで測定した。
(10)耐バイオディーゼル燃料性
有限会社AKM製造のバイオディーゼル燃料を用いて、上記(9)の試験片を140℃で100時間浸漬させた。用いたバイオディーゼル燃料の脂肪酸メチルエステル総量は90.2wt%、密度(15℃)は0.8864g/mL、酸価は0.09mgKOH/gであった。100時間後に取り出した試験片の表面のクラック発生状態を目視で観察した。また、燃料浸漬試験前および燃料浸漬100時間後の試験片の引張強度および引張伸度の保持率を評価した。引張強度(伸度)保持率(%)=[燃料浸漬100時間後の引張強度(伸度)]/[燃料浸漬前の引張強度(伸度)]×100で計算した。
有限会社AKM製造のバイオディーゼル燃料を用いて、上記(9)の試験片を140℃で100時間浸漬させた。用いたバイオディーゼル燃料の脂肪酸メチルエステル総量は90.2wt%、密度(15℃)は0.8864g/mL、酸価は0.09mgKOH/gであった。100時間後に取り出した試験片の表面のクラック発生状態を目視で観察した。また、燃料浸漬試験前および燃料浸漬100時間後の試験片の引張強度および引張伸度の保持率を評価した。引張強度(伸度)保持率(%)=[燃料浸漬100時間後の引張強度(伸度)]/[燃料浸漬前の引張強度(伸度)]×100で計算した。
[製造例1:PA92/62T−1]
攪拌機、温度計、トルクメーター、圧力計、ダイアフラムポンプを直結した原料投入口、窒素ガス導入口、放圧口、圧力調節装置及びポリマー抜出し口を備えた内容積が約150リットルの圧力容器に蓚酸ジブチル28.230kg(139.56モル)を仕込み、圧力容器の内部を純度が99.9999%の窒素ガスで0.5MPaに加圧した後、次に常圧まで窒素ガスを放出する操作を5回繰り返し、窒素置換を行った後、封圧下、攪拌しながら系内を昇温した。約30分間かけて蓚酸ジブチルの温度を100℃にした後、1,9−ノナンジアミン1.241kg(7.84モル)と2−メチル−1,8−オクタンジアミン19.639kg(124.04モル)と1,6−ヘキサンジアミン0.893kg(7.68モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が5.62:88.88:5.50)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を235℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.75MPaに調節した。重縮合物の温度が235℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を260℃にし、260℃において4.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.13であった。
攪拌機、温度計、トルクメーター、圧力計、ダイアフラムポンプを直結した原料投入口、窒素ガス導入口、放圧口、圧力調節装置及びポリマー抜出し口を備えた内容積が約150リットルの圧力容器に蓚酸ジブチル28.230kg(139.56モル)を仕込み、圧力容器の内部を純度が99.9999%の窒素ガスで0.5MPaに加圧した後、次に常圧まで窒素ガスを放出する操作を5回繰り返し、窒素置換を行った後、封圧下、攪拌しながら系内を昇温した。約30分間かけて蓚酸ジブチルの温度を100℃にした後、1,9−ノナンジアミン1.241kg(7.84モル)と2−メチル−1,8−オクタンジアミン19.639kg(124.04モル)と1,6−ヘキサンジアミン0.893kg(7.68モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が5.62:88.88:5.50)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を235℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.75MPaに調節した。重縮合物の温度が235℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を260℃にし、260℃において4.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.13であった。
[製造例2:PA92/62T−2]
蓚酸ジブチル28.462kg(140.71モル)を仕込み、1,9−ノナンジアミン16.448kg(103.88モル)と2−メチル−1,8−オクタンジアミン2.903kg(18.34モル)と1,6−ヘキサンジアミン2.150kg(18.50モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が73.83:13.03:13.14)を仕込んだほかは、製造例1と同様に反応を行ってポリアミドを得た。得られたポリアミドは白色の強靭なポリマーで、ηr=2.97であった。
蓚酸ジブチル28.462kg(140.71モル)を仕込み、1,9−ノナンジアミン16.448kg(103.88モル)と2−メチル−1,8−オクタンジアミン2.903kg(18.34モル)と1,6−ヘキサンジアミン2.150kg(18.50モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が73.83:13.03:13.14)を仕込んだほかは、製造例1と同様に反応を行ってポリアミドを得た。得られたポリアミドは白色の強靭なポリマーで、ηr=2.97であった。
[製造例3:PA92/62T−3]
蓚酸ジブチル30.238kg(149.49モル)を仕込み、1,9−ノナンジアミン4.486kg(28.33モル)と2−メチル−1,8−オクタンジアミン4.486kg(28.33モル)と1,6−ヘキサンジアミン10.79kg(92.85モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が18.95:18.95:62.10)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1.5時間かけて温度を270℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を1.00MPaに調節した。重縮合物の温度が270℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を285℃にし、285℃において1.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=2.88であった。
蓚酸ジブチル30.238kg(149.49モル)を仕込み、1,9−ノナンジアミン4.486kg(28.33モル)と2−メチル−1,8−オクタンジアミン4.486kg(28.33モル)と1,6−ヘキサンジアミン10.79kg(92.85モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が18.95:18.95:62.10)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1.5時間かけて温度を270℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を1.00MPaに調節した。重縮合物の温度が270℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を285℃にし、285℃において1.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=2.88であった。
[製造例4:PA92/62T−4]
蓚酸ジブチル29.864kg(147.64モル)を仕込み、1,9−ノナンジアミン5.598kg(35.36モル)と2−メチル−1,8−オクタンジアミン5.598kg(35.36モル)と1,6−ヘキサンジアミン8.941kg(76.92モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が23.95:23.95:52.10)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を250℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を1.00MPaに調節した。重縮合物の温度が250℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を270℃にし、270℃において2時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=2.83であった。
蓚酸ジブチル29.864kg(147.64モル)を仕込み、1,9−ノナンジアミン5.598kg(35.36モル)と2−メチル−1,8−オクタンジアミン5.598kg(35.36モル)と1,6−ヘキサンジアミン8.941kg(76.92モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が23.95:23.95:52.10)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を250℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を1.00MPaに調節した。重縮合物の温度が250℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を270℃にし、270℃において2時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=2.83であった。
[製造例5:PA92/62T−5]
蓚酸ジブチル29.107kg(143.89モル)を仕込み、1,9−ノナンジアミン5.641kg(35.63モル)と2−メチル−1,8−オクタンジアミン10.028kg(63.34モル)と1,6−ヘキサンジアミン5.223kg(44.93モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が24.76:44.02:31.22)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を250℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.75MPaに調節した。重縮合物の温度が240℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を265℃にし、265℃において3時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.11であった。
蓚酸ジブチル29.107kg(143.89モル)を仕込み、1,9−ノナンジアミン5.641kg(35.63モル)と2−メチル−1,8−オクタンジアミン10.028kg(63.34モル)と1,6−ヘキサンジアミン5.223kg(44.93モル)の混合物(1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンと1,6−ヘキサンジアミンのモル比が24.76:44.02:31.22)をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を250℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.75MPaに調節した。重縮合物の温度が240℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を265℃にし、265℃において3時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.11であった。
[参考製造例1:PA92C]
蓚酸ジブチル28.40kg(140.4モル)を仕込み、1,9−ノナンジアミン11.11kg(70.2モル)と2−メチル−1,8−オクタンジアミン11.11kg(70.2モル)の混合物をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を235℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.5MPaに調節した。重縮合物の温度が235℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を260℃にし、260℃において4.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.35であった。
蓚酸ジブチル28.40kg(140.4モル)を仕込み、1,9−ノナンジアミン11.11kg(70.2モル)と2−メチル−1,8−オクタンジアミン11.11kg(70.2モル)の混合物をダイアフラムフポンプにより流速1.49リットル/分で約17分間かけて反応容器内に供給すると同時に昇温した。供給直後の圧力容器内の内圧は、重縮合反応により生成したブタノールによって0.35MPaまで上昇し、重縮合物の温度は約170℃まで上昇した。その後、1時間かけて温度を235℃まで昇温した。その間、生成したブタノールを放圧口より抜き出しながら、内圧を0.5MPaに調節した。重縮合物の温度が235℃に達した直後から放圧口よりブタノールを約20分間かけて抜き出し、内圧を常圧にした。常圧にしたところから、1.5リットル/分で窒素ガスを流しながら昇温を開始し、約1時間かけて重縮合物の温度を260℃にし、260℃において4.5時間反応させた。その後、攪拌を止めて系内を窒素で1MPaに加圧して約10分間静置した後、内圧0.5MPaまで放圧し、重縮合物を圧力容器下部抜出口より紐状に抜き出した。紐状の重合物は直ちに水冷し、水冷した紐状の樹脂はペレタイザーによってペレット化した。得られたポリアミドは白色の強靭なポリマーであり、ηr=3.35であった。
[比較製造例1:PA92の製造]
ジアミン原料として1,9−ノナンジアミン22.25kg(140.4モル)だけを用いて、製造例1と同様に反応を行ってポリアミドを得た。得られた重合物は黄白色のポリマーであり、ηr=2.78であった。
ジアミン原料として1,9−ノナンジアミン22.25kg(140.4モル)だけを用いて、製造例1と同様に反応を行ってポリアミドを得た。得られた重合物は黄白色のポリマーであり、ηr=2.78であった。
製造例1〜5、参考製造例1及び比較製造例1で製造したポリアミドPA92/62T−1〜PA92/62T−5、PA92C、PA92、並びにナイロン6(宇部興産製、UBEナイロン1015B:PA6)、ナイロン66(宇部興産製、UBEナイロン2020B:PA66)及びナイロン12(宇部興産製、UBESTA3020U:PA12)について、相対粘度、融点、結晶化温度、1%重量減少温度、溶融粘度、飽和吸水率、耐薬品性、耐加水分解性、ドライ及びウェットにおける機械的特性を測定した。結果を表1に示す。
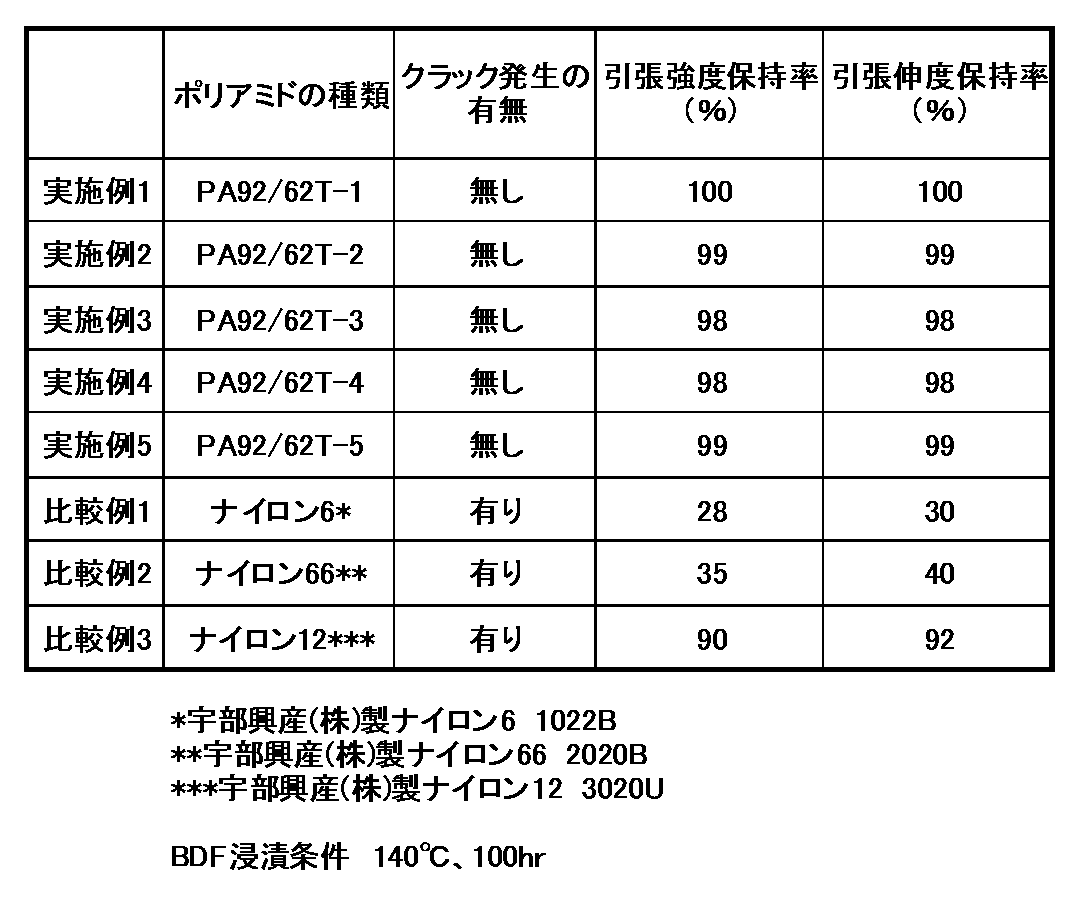
実施例1〜5のポリアミド樹脂PA92/62T−1〜PA92/62T−5、並びに比較例1〜3のナイロン6(宇部興産製、UBEナイロン1015B)、ナイロン66(宇部興産製、UBEナイロン2020B)及びナイロン12(宇部興産製、UBESTA3020U)について、さらに耐バイオディーゼル燃料性を評価した。結果を表2に示す。
本発明による成形部品は、耐バイオディーゼル燃料性に優れ、かつ低吸水性、成形加工性等に優れているので、バイオディーゼル燃料と直接に接触する部品に大いに利用されることが期待される。
Claims (4)
- ジカルボン酸成分が蓚酸からなり、ジアミン成分が1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンの混合物(以下、「C9ジアミン混合物」という。)及び1,6−ヘキサンジアミン(以下、「C6ジアミン」という。)からなり、C9ジアミン混合物とC6ジアミンのモル比が1:99〜99:1であるポリアミド樹脂を含むことを特徴とするバイオディーゼル燃料と直接接触する成形部材。
- 前記ポリアミド樹脂は、96%硫酸を溶媒とし、濃度が1.0g/dlのポリアミド樹脂溶液を用いて25℃で測定した相対粘度(ηr)が1.8〜6.0である請求項1記載のバイオディーゼル燃料と直接接触する成形部材。
- 前記ポリアミド樹脂は、窒素雰囲気下、10℃/分の昇温速度で測定した熱重量分析における1%重量減少温度と窒素雰囲気下、10℃/分の昇温速度で測定した示差走査熱量法により測定した融点との温度差が50℃以上である請求項1または2に記載のバイオディーゼル燃料と直接接触する成形部材。
- 前記ポリアミド樹脂は、1,9−ノナンジアミンと2−メチル−1,8−オクタンジアミンのモル比が5:95〜95:5であるジアミン成分とからなる請求項1〜3のいずれか1項に記載のバイオディーゼル燃料と直接接触する成形部材。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008244727A JP2010077215A (ja) | 2008-09-24 | 2008-09-24 | バイオディーゼル燃料と直接接触する成形部材 |
| PCT/JP2009/060979 WO2009151145A1 (ja) | 2008-06-10 | 2009-06-10 | 新規なポリアミド樹脂組成物及びポリアミド樹脂含有製品 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008244727A JP2010077215A (ja) | 2008-09-24 | 2008-09-24 | バイオディーゼル燃料と直接接触する成形部材 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2010077215A true JP2010077215A (ja) | 2010-04-08 |
Family
ID=42208032
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008244727A Pending JP2010077215A (ja) | 2008-06-10 | 2008-09-24 | バイオディーゼル燃料と直接接触する成形部材 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2010077215A (ja) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05506466A (ja) * | 1990-02-20 | 1993-09-22 | エクソン・ケミカル・パテンツ・インク | 酸素バリヤー |
| JPH07228689A (ja) * | 1994-02-16 | 1995-08-29 | Kuraray Co Ltd | ポリアミド樹脂 |
| JP2000191771A (ja) * | 1998-12-25 | 2000-07-11 | Kuraray Co Ltd | ポリアミドおよびその組成物 |
| JP2004083817A (ja) * | 2002-08-29 | 2004-03-18 | Kuraray Co Ltd | ポリアミド |
| JP2006057033A (ja) * | 2004-08-23 | 2006-03-02 | Ube Ind Ltd | 低吸水性部材 |
-
2008
- 2008-09-24 JP JP2008244727A patent/JP2010077215A/ja active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05506466A (ja) * | 1990-02-20 | 1993-09-22 | エクソン・ケミカル・パテンツ・インク | 酸素バリヤー |
| JPH07228689A (ja) * | 1994-02-16 | 1995-08-29 | Kuraray Co Ltd | ポリアミド樹脂 |
| JP2000191771A (ja) * | 1998-12-25 | 2000-07-11 | Kuraray Co Ltd | ポリアミドおよびその組成物 |
| JP2004083817A (ja) * | 2002-08-29 | 2004-03-18 | Kuraray Co Ltd | ポリアミド |
| JP2006057033A (ja) * | 2004-08-23 | 2006-03-02 | Ube Ind Ltd | 低吸水性部材 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5056763B2 (ja) | ポリアミド樹脂 | |
| JP5796573B2 (ja) | ポリアミド樹脂 | |
| JP2009235225A (ja) | ポリアミド樹脂 | |
| JP5146124B2 (ja) | 燃料チューブ | |
| JPWO2012036303A1 (ja) | 耐衝撃性に優れるポリオキサミド樹脂及び耐衝撃性部品 | |
| JP2009298870A (ja) | 自動車エンジンルーム内部品 | |
| JP5572921B2 (ja) | バイオディーゼル燃料と直接接触する成形部材 | |
| JP5347930B2 (ja) | 電子写真用部材 | |
| JP2009298853A (ja) | ポリアミド樹脂組成物 | |
| JP2009299206A (ja) | フィラメント | |
| JP5458846B2 (ja) | 新規なポリアミド樹脂を含む電子写真用部材 | |
| JP2009298857A (ja) | ポリアミド樹脂組成物及び該ポリアミド樹脂組成物から形成された成形物 | |
| JP2009235223A (ja) | 自動車部材用ポリアミド樹脂 | |
| JP5621220B2 (ja) | 導電性ポリアミド樹脂組成物及びケーブルハウジング | |
| JP5584963B2 (ja) | ポリアミド樹脂組成物 | |
| JP2010077215A (ja) | バイオディーゼル燃料と直接接触する成形部材 | |
| JP5584964B2 (ja) | ポリアミド樹脂、層状珪酸塩を含む複合材料及び中空成形部品 | |
| JP5446795B2 (ja) | Icトレイ用ポリアミド樹脂組成物及びicトレイ | |
| JP2009298867A (ja) | ポリアミドフィルム | |
| JP2010018794A (ja) | 導電性ポリアミド樹脂組成物 | |
| JP2009298865A (ja) | 燃料タンク部品 | |
| JP2009298854A (ja) | ポリアミド樹脂及び層状珪酸塩を含む複合材料 | |
| JP2011116886A (ja) | 産業用チューブ | |
| JP2009298856A (ja) | 耐熱剤含有樹脂組成物及び該耐熱剤含有樹脂組成物から形成された成形物 | |
| JP5577574B2 (ja) | 新規なポリアミド樹脂を含む射出成形材料及びそれらから作製した射出成形体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110830 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130828 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20140129 |