JP2008543749A - 結晶形アジスロマイシンl−リンゴ酸一水和物及びこれを含む医薬組成物 - Google Patents
結晶形アジスロマイシンl−リンゴ酸一水和物及びこれを含む医薬組成物 Download PDFInfo
- Publication number
- JP2008543749A JP2008543749A JP2008515621A JP2008515621A JP2008543749A JP 2008543749 A JP2008543749 A JP 2008543749A JP 2008515621 A JP2008515621 A JP 2008515621A JP 2008515621 A JP2008515621 A JP 2008515621A JP 2008543749 A JP2008543749 A JP 2008543749A
- Authority
- JP
- Japan
- Prior art keywords
- azithromycin
- malic acid
- pharmaceutical composition
- formula
- acid monohydrate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 0 CC[C@]([C@](C)[C@](*)[C@@](C)C(C*)C[C@](*)C[C@](*)([C@@]([C@@](C)[C@]([C@]1CIC(C[C@]2C)O[C@@](C)[C@]2O)O)O[C@@]([C@]2O)O[C@](C)C[C@@]2N(*)*)*(C)=C)OC1=O Chemical compound CC[C@]([C@](C)[C@](*)[C@@](C)C(C*)C[C@](*)C[C@](*)([C@@]([C@@](C)[C@]([C@]1CIC(C[C@]2C)O[C@@](C)[C@]2O)O)O[C@@]([C@]2O)O[C@](C)C[C@@]2N(*)*)*(C)=C)OC1=O 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Dermatology (AREA)
- Pulmonology (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
熱安定性及び溶解度に優れておりかつ吸湿性がないので、多様な細菌感染性疾患の治療に有効な化合物の提供することを目的とし、結晶形アジスロマイシンL−リンゴ酸一水和物、その製造方法及びこれを含む医薬組成物により解決される。
Description
本発明は、1分子のアジスロマイシン、2分子のL−リンゴ酸及び1分子の水から構成された結晶形アジスロマイシンL−リンゴ酸一水和物、その製造方法及びこれを含む医薬組成物に関する。
式(II)に表されるアジスロマイシン、9−デオキソ−9a−アザ−9a−メチル−9a−ホモエリスロマイシンA(USAN)は米国特許第4,517,359号及び米国特許第4,474,768号に既に開示されており、気管支炎、性的接触による感染及び皮膚感染症などの治療に有効なアザライド型準合成マクロライド系抗生物質である(参照:H.A. Kirst 及び G.D. Sides、 Antimicrob. Agents Chemother. 1989、 33、 1419-1422)。

前記特許に開示されたアジスロマイシンは吸湿性が高く、不安定な結晶形無水物または一水和物であって、これらは薬剤として不向きである。
このような問題点を解決するために、ヨーロッパ特許第0 298 605号は非吸湿性の結晶形アジスロマイシン二水和物を開示している。ヨーロッパ特許第0 984 020号及び国際公開特許第WO2002/085898号はアジスロマイシンの無毒性アルコールとの溶媒和物を開示している。
このような問題点を解決するために、ヨーロッパ特許第0 298 605号は非吸湿性の結晶形アジスロマイシン二水和物を開示している。ヨーロッパ特許第0 984 020号及び国際公開特許第WO2002/085898号はアジスロマイシンの無毒性アルコールとの溶媒和物を開示している。
しかし、アジスロマイシン二水和物は37℃で1.1mg/mlの低い水溶解度を有するので、カプセルまたは錠剤形態のような高用量医薬組成物の形態で投与される場合には薬物溶出速度及び生体内吸収率が不十分であり、従って、注射投与の場合などにはアジスロマイシン二水和物の生体内における薬物吸収率を向上させるために可溶化剤をともに用いることが報告されている。
アジスロマイシンは2つの第3級アミンを有しており、これらを酸付加塩の形態に転換させるようになったその溶解度を高めることができる。例えば、米国特許第4,474,768号は多様な有機酸または無機酸を用いたアジスロマイシンの酸付加塩を開示している。また、塩酸、ヨウ化水素酸、酢酸、L−アスパラギン酸及びラクトビオン酸の付加塩などのような多様なジスロマイシン酸付加塩が報告されている(参照:S. Djokicら、 J. Chem. Research (S)、 1988、 152-153、 または J. Chem. Research (M)、 1988、 1239-1261).また、中国公開特許第1,123,279号、第1,157,824号、第1,205,338号及び第1,334,541号にはグルタミン酸、アスパラギン酸、乳酸、クエン酸、酢酸、グルクロン酸、N−アセチルシステイン、メチル硫酸、アスコルビン酸、及び硫酸などのアジスロマイシン塩が開示されている。
しかし、前述の酸付加塩は、凍結乾燥、噴霧乾燥または減圧蒸留による塩形成段階で用いられる溶媒を取り除いて得られる無定形塩である。ヨーロッパ特許第0 677 530号は沈澱法により製造された無定形のアジスロマイシン二塩酸塩を提供する。この無定形塩はある程度の残留水または有機溶媒を含んでいるだけではなく、吸湿性が非常に大きく、不安定である。従って、このような塩は医薬組成物に用いるには望ましくない
しかし、前述の酸付加塩は、凍結乾燥、噴霧乾燥または減圧蒸留による塩形成段階で用いられる溶媒を取り除いて得られる無定形塩である。ヨーロッパ特許第0 677 530号は沈澱法により製造された無定形のアジスロマイシン二塩酸塩を提供する。この無定形塩はある程度の残留水または有機溶媒を含んでいるだけではなく、吸湿性が非常に大きく、不安定である。従って、このような塩は医薬組成物に用いるには望ましくない
国際公開特許WO 2004/106355号はクエン酸を用いて製造したアジスロマイシンの結晶形塩、すなわちクエン酸水素アジスロマイシンを提供する。しかし、この塩の含水量は湿度が変ると、それにつれて変化するという問題がある。
従って、本発明者らはアジスロマイシンの改善した酸付加塩を開発するために研究を重ねた結果、公知のアジスロマイシン二水和物に比べて優れた安定性、非吸湿性及び溶解度を有するアジスロマイシンの結晶形塩を発見し、本発明を完成した。
米国特許第4,517,359号
米国特許第4,474,768号
ヨーロッパ特許第0 298 605号
ヨーロッパ特許第0 984 020号
WO 2002/085898号
中国公開特許第1,123,279号
中国公開特許第1,157,824号
中国公開特許第1,205,338号
中国公開特許第1,334,541号
ヨーロッパ特許第0677530号
WO2004/106355号
H.A. Kirst及びG.D. Sides、 Antimicrob. Agents Chemother. 1989、 33、 1419-1422
S. Djokicなど、 J. Chem. Research (S)、 1988、 152-153
J. Chem. Research (M)、 1988、 1239-1261
従って、本発明者らはアジスロマイシンの改善した酸付加塩を開発するために研究を重ねた結果、公知のアジスロマイシン二水和物に比べて優れた安定性、非吸湿性及び溶解度を有するアジスロマイシンの結晶形塩を発見し、本発明を完成した。
本発明の目的は溶解度、安定性及び非吸湿性に優れたアジスロマイシンの酸付加塩及びその製造方法を提供することである。
本発明は、式(I)の結晶形アジスロマイシンL−リンゴ酸一水和物及びその製造方法を提供する。
また、本発明は式(I)の結晶形アジスロマイシンL−リンゴ酸一水和物を活性成分として含む細菌感染性疾患治療用医薬組成物を提供する。
本発明による結晶形アジスロマイシンL−リンゴ酸一水和物は公知のアジスロマイシン二水和物に比べて優れた溶解度、安定性及び非吸湿性を有するので、多様な細菌感染性疾患の治療に有用である。
本発明による式(I)のアジスロマイシンL−リンゴ酸一水和物は、a)水性有機溶媒内で式(II)のアジスロマイシンを式(III)のリンゴ酸と反応させるか、またはb)水性有機溶媒から式(IV)のアジスロマイシンL−リンゴ酸無水物を再結晶化させて製造することができる。
具体的に、本発明による式(I)の化合物は、水性有機溶媒に式(II)のアジスロマイシンを懸濁させ、この懸濁液に式(III)のL−リンゴ酸を添加した後、室温ないし用いられた溶媒の沸点範囲の温度に加熱して得られた澄明な溶液を0℃ないし室温に冷却した後、沈澱した結晶を濾過、乾燥する段階を含む方法によって製造することができる。
本発明で用いられた式(II)のアジスロマイシンは、無水物、一水和物、二水和物または溶媒和物の形態であり得る。
本発明で用いられた式(II)のアジスロマイシンは、無水物、一水和物、二水和物または溶媒和物の形態であり得る。
本発明で用いられる式(III)のリンゴ酸は、L−リンゴ酸、ラセミ体のDL−リンゴ酸またはこれらの混合物であり得、L−リンゴ酸を用いることが好ましい。
本発明の前記方法によれば、ラセミ体であるDL−リンゴ酸を用いる場合にも、立体化学的な理由でL−リンゴ酸のみがキラルアジスロマイシンと選択的に反応して式(I)のアジスロマイシンL−リンゴ酸一水和物が生成される。アジスロマイシンとD−リンゴ酸またはDL−リンゴ酸とアジスロマイシンL−リンゴ酸の塩を他の方法、例えば非水性有機溶媒を用いて得られてその塩は無水物形態である。
本発明の前記方法によれば、ラセミ体であるDL−リンゴ酸を用いる場合にも、立体化学的な理由でL−リンゴ酸のみがキラルアジスロマイシンと選択的に反応して式(I)のアジスロマイシンL−リンゴ酸一水和物が生成される。アジスロマイシンとD−リンゴ酸またはDL−リンゴ酸とアジスロマイシンL−リンゴ酸の塩を他の方法、例えば非水性有機溶媒を用いて得られてその塩は無水物形態である。
従って、式(I)のアジスロマイシンL−リンゴ酸一水和物には、L−リンゴ酸の不斉炭素がS−構造を有するL−(−)−リンゴ酸の塩を意味する。
本発明において、L−リンゴ酸はアジスロマイシン1モル当量に対して2モル〜2.5モル当量で用いることが望ましい。
本発明において、L−リンゴ酸はアジスロマイシン1モル当量に対して2モル〜2.5モル当量で用いることが望ましい。
本発明で用いられた水性有機溶媒としては、2〜10体積%の含水量を有するアセトン、メチルエチルケトン、メチルイソブチルケトン、エタノール、1−プロパノール、2−プロパノール、1−ブタノール、テトラヒドロフラン、1,4−ジオキサン、酢酸メチル、及び酢酸エチル、望ましくはアセトン及び2−プロパノールが挙げられる。
本発明において、前記水性有機溶媒は式(II)のアジスロマイシン1gに対して3〜20ml、望ましくは4〜10mlの量で用いることができる。
本発明において、前記水性有機溶媒は式(II)のアジスロマイシン1gに対して3〜20ml、望ましくは4〜10mlの量で用いることができる。
また、アジスロマイシンL−リンゴ酸一水和物はアジスロマイシンL−リンゴ酸無水物を前記水性有機溶媒に溶解してから再結晶させることで製造することができる。
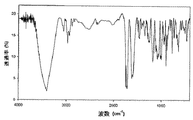
このように製造された、本発明による式(I)のアジスロマイシンL−リンゴ酸一水和物は、図1及び2に示したように、1分子のアジスロマイシン、2分子のL−リンゴ酸及び1分子の水からなる結晶構造を形成する。具体的に、本発明による化合物はX線粉末回折(XRPD)スペクトル(図1)において、I/Io値(Iは各回折角におけるピークの強度、Ioは最も大きいピークの強度である)が10%以上であるピークが9.6、10.6、11.2、12.0、12.4、14.3、14.6、15.0、16.6、17.5、18.1、18.6、19.3、19.7、20.2、20.5、21.4、22.6、23.6、24.0、24.6、27.1、27.7、34.4の回折角(2θ±0.2)で現われ、赤外(IR)吸収スペクトル(図2)における特徴的な吸収ピークは波数(cm−1)として3411、3059、2971、1742、1716、1619、1594、1493、1457、1345、1286、1177、1112、1080、1056、1013、1001、900、773、637などの値を有する。また、
本発明による結晶形アジスロマイシンL−リンゴ酸一水和物は173〜176℃の融点を示し、これは熱に対して安定であることを示す。
本発明によるアジスロマイシンL−リンゴ酸一水和物の結晶構造は、前記一水和物形態を
本発明によるアジスロマイシンL−リンゴ酸一水和物の結晶構造は、前記一水和物形態を
100℃以上の温度で数時間減圧乾燥(1.0mmHg)下で乾燥及び脱水させるか、非水性有機溶媒でアジスロマイシンをL−リンゴ酸で反応させることで得られる無水物形態の結晶構造(図3のXRPDスペクトル及び図4のIR吸収スペクトル参照)と異なる。前記無水物形態は約180〜184℃の融点を示す。
本発明による結晶形アジスロマイシンL−リンゴ酸一水和物は、40℃の温度及び75%の相対湿度の条件下で24時間実施した吸湿性試験から得た下記表1の結果から明らかなように、減圧濃縮、凍結乾燥、噴霧乾燥などの溶媒蒸発法によって得られる通常の無定形塩や沈澱によって形成される他の種類の結晶塩とは異なり、非吸湿性を示す。
表1
表1
また、本発明による式(I)の結晶形アジスロマイシンL−リンゴ酸一水和物は今まで当該分野において薬学成分として用いられている唯一の公知のアジスロマイシン二水和物と比べて水溶解度が非常に高く、このような溶解度の向上はアジスロマイシンの薬物動態プロファイルを著しく改善させて多様な細菌感染性疾患治療のための改良された医薬組成物を製剤に有用である。
従って、本発明は式(I)の結晶形アジスロマイシンL−リンゴ酸一水和物を活性成分として含む細菌感染性疾患治療用医薬組成物を提供する。
前記細菌感染性疾患としては、肺炎連鎖球菌(Streptococcus pneumoniae)、ヘモフィルス・ヘモフィルス・インフルエンザ菌(Haemophilus influenzae)、マイコプラズマ・ニューモニエ(Mycoplasma pneumoniae)またはクラミジア・ニューモニエ(Chlamydia pneumoniae)による市中感染型の肺炎(community acquired pneumonia);化膿連鎖球菌(Streptococcus pyogenes)による咽頭炎(pharyngitis)及びへんとう炎(tonsillitis);ヘモフィルス・インフルエンザ菌(Haemophilus influenzae)、モラクセラ・カタラーリス(Moraxella catarrhalis)または肺炎連鎖球菌(Streptococcus pneumoniae)による慢性閉塞性肺疾患(chronic obstructive pulmonary disease)及び急性中耳炎(acute otitis);黄色ブドウ球菌(Staphylococcus aureus)、化膿連鎖球菌(Streptococcus pyogenes)または無乳性連鎖球菌(Streptococcus agalactiae)による単純性皮膚感染症(uncomplicated skin infections);淋菌(Neisseria gonorrhoeae)またはクラミジア・トラコマチス(Chlamydia trachomatis)による泌尿生殖器系感染症(genitourinary tract infections);及びトリ型結核菌(Mycobacterium avium)による播種性(disseminated)非定型抗酸菌症(Mycobacterium Avium Complex; MAC)が挙げられる。
本発明によるアジスロマイシンL−リンゴ酸一水和物を活性成分として含む医薬組成物は経口用、注射用及び眼科用などの多様な形態で投与可能である。
経口投与のために、本発明の医薬組成物は錠剤、カプセル剤、懸濁剤及び粉末剤などの形態に製剤化されることができ、これらは単回投与用または分割投与用であり得る。このような経口投与用組成物は、結合剤、充填剤、緩衝剤、潤滑剤、崩壊剤、甘味剤、着臭剤(odorant)、界面活性剤またはコート剤などのような担体、希釈剤及び賦形剤を含むことができる。
経口投与のために、本発明の医薬組成物は錠剤、カプセル剤、懸濁剤及び粉末剤などの形態に製剤化されることができ、これらは単回投与用または分割投与用であり得る。このような経口投与用組成物は、結合剤、充填剤、緩衝剤、潤滑剤、崩壊剤、甘味剤、着臭剤(odorant)、界面活性剤またはコート剤などのような担体、希釈剤及び賦形剤を含むことができる。
前記崩壊剤としては、例えば、澱粉、ゼラチン化澱粉、澱粉グリコール酸ナトリウム、カルボキシメチルセルロースナトリウム、クロスカルメロースナトリウム、微結晶性セルロース、アルギン酸塩、樹脂、界面活性剤、発泡性組成物、水性ケイ酸アルミニウム及び架橋ポリビニルピロリドンが挙げられる。前記結合剤としては、例えばアカシア;メチルセルロース、カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース及びヒドロキシエチルセルロースのようなセルロース誘導体;ゼラチン、グルコース、デキストロース、キシリトール、ポリメタクリレート、ポリビニルピロリドン、ソルビトール、澱粉、ゼラチン化澱粉、キサンタン樹脂、アルギン酸塩、ケイ酸マグネシウム−アルミニウム、ポリエチレングリコール及びベントナイトが挙げられる。
前記充填剤としては、例えば乳糖、無水乳糖、乳糖一水和物、スクロース、デキストロース、マンニトール、ソルビトール、澱粉、微結晶性セルロースのようなセルロース類、無水または二水和物形態のリン酸カルシウム、炭酸カルシウム及び硫酸カルシウムが挙げられる。前記潤滑剤としては、例えばステアリン酸マグネシウム、滑石、ポリエチレン・グリコール、酸化エチレンの重合体、ラウリル硫酸ナトリウムまたはマグネシウム、オレイン酸ナトリウム、フマル酸ステアリルナトリウム、DL−ロイシン及びコロイド性二酸化ケイ素が挙げられる。前記着臭剤としては、例えば油、花、果物及びこれらの混合物の抽出物、及び人工または天然芳香油を含む。
前記コート剤としては、飲み込みを容易にし、放出性を調節でき、外形を改善しかつ不快な味を遮断するのに適当なものであって、例えばヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース及びアクリル酸−メチルアクリル酸の共重合体が挙げられる。前記甘味剤としては、アスパルテーム、サッカリン、サッカリンナトリウム、サイクラミン酸ナトリウム、キシリトール、マンニトール、ソルビトール、ラクトース(乳糖)及びスクロースが挙げられる。前記緩衝剤としては、例えばクエン酸、クエン酸ナトリウム、炭酸水素ナトリウム、第二リン酸ナトリウム、酸化マグネシウム、炭酸カルシウム及び水酸化マグネシウムが挙げられる。前記界面活性剤の例としては、ラウリル硫酸ナトリウム及びポリソルベートが挙げられる。
経口投与用医薬組成物は、50mg〜700mg範囲のアジスロマイシンを含む分割投与用、または700mg〜3,500mg範囲のアジスロマイシンを含む単回投与用に製剤化され、組成物100重量部に対し20〜80重量部のアジスロマイシンL−リンゴ酸一水和物を含むものが望ましい。例えば、組成物の全量500mg(100%)に対しアジスロマイシン250mg(50%)を含んだ医薬組成物は式(I)のアジスロマイシンL−リンゴ酸一水和物345.53mg(69.1%)と適切な担体、希釈剤または賦形剤などの添加剤とを154.47mg(30.9%)に含むことができる。
無菌注射投与のために、本発明の医薬組成物は無菌条件下で本発明の結晶形アジスロマイシンL−リンゴ酸一水和物及び薬学的に許容可能な担体を直接バイアルに充填するか、滅菌水にアジスロマイシンL−リンゴ酸一水和物を溶解させてから凍結乾燥して得た無定形粉末をバイアルに充填させることで製造することができ、前記組成物は投与直前に滅菌水を加えて溶解させる。注射投与のための医薬組成物は式(I)の結晶形アジスロマイシンL−リンゴ酸一水和物を50mg/ml〜250mg/mlの量で含むことが望ましい。
眼投与のために、本発明の医薬組成物は、等張食塩水、リン酸またはホウ酸塩緩衝液にアジスロマイシンL−リンゴ酸一水和物を0.05%〜1.0%の濃度で溶解させた水溶液として製造することができ、この時、亜硫酸ナトリウムまたは亜硫酸水素ナトリウムなどのような抗酸化剤を添加してもよい。
以下、本発明を下記実施例によりさらに詳しく説明する。但し、これらの実施例は、本発明を例示するためのものに過ぎず、本発明の範囲を限定することはない。
以下、本発明を下記実施例によりさらに詳しく説明する。但し、これらの実施例は、本発明を例示するためのものに過ぎず、本発明の範囲を限定することはない。
実施例1
L−リンゴ酸からのアジスロマイシンL−リンゴ酸一水和物の製造
アジスロマイシン二水和物100.0g(127mmol)を95%2−プロパノール1,000mlに溶解させてからL−リンゴ酸34.1g(254mmol、99.7%eeの光学純度)を加えて室温で一晩中攪拌した後、0℃〜5℃で2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、45℃で乾燥して白色結晶の標題化合物118.3g(収率90%)を得た。
融点:173〜175℃
比旋光度、[α]D 20:−32.8゜(c=1、メタノール)
含水量(カールフィッシャー水分計):1.80%(一水和物の理論値の1.74%)
塩形成後のリンゴ酸の光学純度(HPLC):99.9%eeのL−リンゴ酸
アジスロマイシン相対含量(HPLC):74.6%(1分子の理論値の72.35%)
L−リンゴ酸相対含量(0.1N KOH滴定):25.8%(2分子の理論値の25.91%)
IR(KBr、cm−1):3411、3059、2971、1742、1716、1619、1594、1493、1457、1345、1286、1177、1112、1080、1056、1013、1001、900、773、637
L−リンゴ酸からのアジスロマイシンL−リンゴ酸一水和物の製造
アジスロマイシン二水和物100.0g(127mmol)を95%2−プロパノール1,000mlに溶解させてからL−リンゴ酸34.1g(254mmol、99.7%eeの光学純度)を加えて室温で一晩中攪拌した後、0℃〜5℃で2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、45℃で乾燥して白色結晶の標題化合物118.3g(収率90%)を得た。
融点:173〜175℃
比旋光度、[α]D 20:−32.8゜(c=1、メタノール)
含水量(カールフィッシャー水分計):1.80%(一水和物の理論値の1.74%)
塩形成後のリンゴ酸の光学純度(HPLC):99.9%eeのL−リンゴ酸
アジスロマイシン相対含量(HPLC):74.6%(1分子の理論値の72.35%)
L−リンゴ酸相対含量(0.1N KOH滴定):25.8%(2分子の理論値の25.91%)
IR(KBr、cm−1):3411、3059、2971、1742、1716、1619、1594、1493、1457、1345、1286、1177、1112、1080、1056、1013、1001、900、773、637
得られた結晶形アジスロマイシンL−リンゴ酸一水和物の結晶状態は、X線粉末回折分光器で測定し(図1参照)、X線粉末回折スペクトルに示した特徴的な回折角でのピーク(peak)のうち、相対的なピークの相対強度(I/Io)が10%以上であるものの回折角(2θ)及び結晶面同士の距離(d)を表2に示した。
表2
表2
実施例2〜実施例6
アジスロマイシンL−リンゴ酸一水和物の製造
アジスロマイシン、L−リンゴ酸及び溶媒を下記表3に示したように変化させることを除いては、実施例1と同様な工程を行って標題化合物を得た。
表3
アジスロマイシンL−リンゴ酸一水和物の製造
アジスロマイシン、L−リンゴ酸及び溶媒を下記表3に示したように変化させることを除いては、実施例1と同様な工程を行って標題化合物を得た。
表3
得られた化合物の融点、XRPD及びIR吸収スペクトルの結果は実施例1で測定したものと同一である。
実施例7
アジスロマイシンL−リンゴ酸無水物からのアジスロマイシンL−リンゴ酸一水和物の製造
アジスロマイシンL−リンゴ酸無水物50.0g(含水量0.4%)を95%2−プロパノール400mlに加え、加温して溶解させてから室温に冷却し一晩中攪拌してから0℃〜5℃で2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、45℃で乾燥して白色結晶の標題化合物43.1g(収率:85%)を得た。
融点:173〜175℃
含水量(カールフィッシャー水分計):1.83%(一水和物の理論値の1.74%)
得られた化合物のXRPD及びIR吸収スペクトルの結果は実施例1で測定した結果と同一である。
アジスロマイシンL−リンゴ酸無水物からのアジスロマイシンL−リンゴ酸一水和物の製造
アジスロマイシンL−リンゴ酸無水物50.0g(含水量0.4%)を95%2−プロパノール400mlに加え、加温して溶解させてから室温に冷却し一晩中攪拌してから0℃〜5℃で2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、45℃で乾燥して白色結晶の標題化合物43.1g(収率:85%)を得た。
融点:173〜175℃
含水量(カールフィッシャー水分計):1.83%(一水和物の理論値の1.74%)
得られた化合物のXRPD及びIR吸収スペクトルの結果は実施例1で測定した結果と同一である。
実施例8
DL−リンゴ酸からのアジスロマイシンL−リンゴ酸一水和物の製造
アジスロマイシン二水和物100.0g(127mmol)を95%2−プロパノール1,000mlに溶解させてからDL−リンゴ酸34.1g(254mmol、L−リンゴ酸の光学純度:1.7%ee)を加えて室温で一晩中攪拌した後、0℃〜5℃で2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、45℃で乾燥して白色結晶粉末61.8g(収率:47%)を得た。
融点:170〜174℃
比旋光度、[α]D 20:−33.7゜(c=1、メタノール)
含水量(カールフィッシャー水分計):1.85%
塩形成後のリンゴ酸の光学純度(HPLC):80.0%eeのL−リンゴ酸
DL−リンゴ酸からのアジスロマイシンL−リンゴ酸一水和物の製造
アジスロマイシン二水和物100.0g(127mmol)を95%2−プロパノール1,000mlに溶解させてからDL−リンゴ酸34.1g(254mmol、L−リンゴ酸の光学純度:1.7%ee)を加えて室温で一晩中攪拌した後、0℃〜5℃で2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、45℃で乾燥して白色結晶粉末61.8g(収率:47%)を得た。
融点:170〜174℃
比旋光度、[α]D 20:−33.7゜(c=1、メタノール)
含水量(カールフィッシャー水分計):1.85%
塩形成後のリンゴ酸の光学純度(HPLC):80.0%eeのL−リンゴ酸
前記で得られた結晶性粉末56.0gを95%2−プロパノールから再結晶して45.2g(収率80%)の標題化合物を得た。
融点:172〜175℃
比旋光度、[α]D 20:−33.0゜(c=1、メタノール)
含水量(カールフィッシャー水分計):1.81%
リンゴ酸の光学純度(HPLC):98.9%eeのL−リンゴ酸
得られた化合物のXRPD及びIR吸収スペクトルの結果は実施例1で測定した結果と同一である。
融点:172〜175℃
比旋光度、[α]D 20:−33.0゜(c=1、メタノール)
含水量(カールフィッシャー水分計):1.81%
リンゴ酸の光学純度(HPLC):98.9%eeのL−リンゴ酸
得られた化合物のXRPD及びIR吸収スペクトルの結果は実施例1で測定した結果と同一である。
参照例1
アジスロマイシンL−リンゴ酸無水物の製造
方法A)
前記実施例1〜8のうちいずれか一つで得られたアジスロマイシンL−リンゴ酸一水和物10.0gを100℃で10時間減圧下(1mmHg)で乾燥して白色粉末の標題化合物を得た。
アジスロマイシンL−リンゴ酸無水物の製造
方法A)
前記実施例1〜8のうちいずれか一つで得られたアジスロマイシンL−リンゴ酸一水和物10.0gを100℃で10時間減圧下(1mmHg)で乾燥して白色粉末の標題化合物を得た。
方法B)
アジスロマイシン無水物37.5g(50mmol、含水量0.2%)を無水2−プロパノール400mlに溶解させてからL−リンゴ酸13.4g(100mmol)を加えて室温で一晩中攪拌した後、0℃〜5℃で2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、45℃で乾燥して白色結晶の標題化合物47.3g(収率:93%)を得た。
融点:182〜184℃
比旋光度、[α]D 20:−32.8゜(c=1、メタノール)
含水量(カールフィッシャー水分計):0.4%以下(乾燥直後)
IR(KBr、cm−1):3415、3057、2980、2932、2884、1736、1607、1462、1386、1326、1177、1084、1060、1000、939、895、726、637
アジスロマイシン無水物37.5g(50mmol、含水量0.2%)を無水2−プロパノール400mlに溶解させてからL−リンゴ酸13.4g(100mmol)を加えて室温で一晩中攪拌した後、0℃〜5℃で2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、45℃で乾燥して白色結晶の標題化合物47.3g(収率:93%)を得た。
融点:182〜184℃
比旋光度、[α]D 20:−32.8゜(c=1、メタノール)
含水量(カールフィッシャー水分計):0.4%以下(乾燥直後)
IR(KBr、cm−1):3415、3057、2980、2932、2884、1736、1607、1462、1386、1326、1177、1084、1060、1000、939、895、726、637
前記で得られたアジスロマイシンL−リンゴ酸化合物をX線粉末回折分析し(図3参照)、その結果前記化合物はI/Io値が10%以上の主要ピークが6.0、10.0、11.0、11.4、12.5、13.9、15.5、16.2、17.3、18.0、19.2、20.0、20.5、20.8、21.2、22.6、24.5、25.7の回折角(2θ±0.2)で現われる結晶構造を有することが確認され、含水量の測定結果から無水物形態であることが確認された。
このように得られたアジスロマイシンL−リンゴ酸無水物を40℃の温度及び75%の相対湿度の条件下で10時間露出させた結果、約2.0%まで含水量が増加した。すなわち、本発明によるアジスロマイシンL−リンゴ酸無水物は高湿の条件下で長時間露出される場合、水和物の形態に徐々に転換されることが確認された。
参照例2
アジスロマイシンD−リンゴ酸無水物の製造
アジスロマイシン二水和物10.0g(12.7mmol)を無水2−プロパノール100mlに溶解させてからD−リンゴ酸3.41g(25.4mmol、98.2%eeの光学純度)を加えて室温で一晩中攪拌した後、0℃〜5℃で2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、45℃で乾燥して白色結晶の標題化合物10.4g(収率:79%)を得た。
融点:160〜163℃
比旋光度、[α]D 25:−39.5゜(c=1、メタノール)
塩形成後のリンゴ酸の光学純度(HPLC):98.9%eeのD−リンゴ酸
含水量(カールフィッシャー水分計):0.4%以下(乾燥直後)
IR(KBr、cm−1):3427、2974、2937、2882、1735、1598、1466、1385、1179、1171、1080、1060、1013、1002、899、726
アジスロマイシンD−リンゴ酸無水物の製造
アジスロマイシン二水和物10.0g(12.7mmol)を無水2−プロパノール100mlに溶解させてからD−リンゴ酸3.41g(25.4mmol、98.2%eeの光学純度)を加えて室温で一晩中攪拌した後、0℃〜5℃で2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、45℃で乾燥して白色結晶の標題化合物10.4g(収率:79%)を得た。
融点:160〜163℃
比旋光度、[α]D 25:−39.5゜(c=1、メタノール)
塩形成後のリンゴ酸の光学純度(HPLC):98.9%eeのD−リンゴ酸
含水量(カールフィッシャー水分計):0.4%以下(乾燥直後)
IR(KBr、cm−1):3427、2974、2937、2882、1735、1598、1466、1385、1179、1171、1080、1060、1013、1002、899、726
前記で得られたアジスロマイシンD−リンゴ酸化合物をX線粉末回折分析し、その結果、前記化合物はI/Io値が10%以上の主要ピークが5.7、9.9、10.9、11.3、12.3、15.9、17.1、17.8、18.2、19.9、20.6及び22.2の回折角(2θ±0.2)で現われる結晶構造を有することが確認され、含水量の測定結果は無水物形態であることが確認された。しかし、40℃の温度及び75%の相対湿度の条件下で10時間露出の時、8%以上に含水量が増加した。
前記で得られたアジスロマイシンD−リンゴ酸無水物は実施例1〜8で用いられた水性溶媒条件下で水和物形態に転換されなかった。
前記で得られたアジスロマイシンD−リンゴ酸無水物は実施例1〜8で用いられた水性溶媒条件下で水和物形態に転換されなかった。
参照例3
アジスロマイシンDL−リンゴ酸無水物の製造
アジスロマイシン二水和物10.0g(12.7mmol)を無水2−プロパノール100mlに溶解させてからDL−リンゴ酸3.41g(25.4mmol、L−リンゴ酸の光学純度:1.7%ee超過)を入れて室温で一晩中攪拌した後、0℃〜5℃から2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、40℃のオーブンで乾燥して白色結晶粉末状の標題化合物10.3g(収率:78%)を得た。
融点:169〜172℃
比旋光度、[α]D 25:−35.5゜(c=1、メタノール)
リンゴ酸の光学純度(HPLC):3.4%eeのL−リンゴ酸
含水量(カールフィッシャー水分計):0.5%以下(乾燥直後)
IR(KBr、cm−1):3410、2973、2937、2882、1736、1603、1458、1385、1170、1076、1060、1016、1008、895、641
アジスロマイシンDL−リンゴ酸無水物の製造
アジスロマイシン二水和物10.0g(12.7mmol)を無水2−プロパノール100mlに溶解させてからDL−リンゴ酸3.41g(25.4mmol、L−リンゴ酸の光学純度:1.7%ee超過)を入れて室温で一晩中攪拌した後、0℃〜5℃から2時間攪拌した。得られた沈殿物を濾過し、冷たい2−プロパノールで洗浄した後、40℃のオーブンで乾燥して白色結晶粉末状の標題化合物10.3g(収率:78%)を得た。
融点:169〜172℃
比旋光度、[α]D 25:−35.5゜(c=1、メタノール)
リンゴ酸の光学純度(HPLC):3.4%eeのL−リンゴ酸
含水量(カールフィッシャー水分計):0.5%以下(乾燥直後)
IR(KBr、cm−1):3410、2973、2937、2882、1736、1603、1458、1385、1170、1076、1060、1016、1008、895、641
前記で得られたアジスロマイシンD−リンゴ酸化合物をX線粉末回折分析し、その結果、前記化合物はI/Io値が10%以上の主要ピークが5.9、9.9、10.9、11.3、12.4、16.0、17.2、17.9、19.9、20.6、22.5、24.4の回折角(2θ±0.2)で現われる結晶構造を有することが確認され、含水量の測定結果は無水物形態であることが確認された。しかし、含水量が40℃の温度及び75%の相対湿度の条件下で10時間露出される場合、6%以上に増加した。
一方、実施例8で見られるように、水性溶媒条件下ではアジスロマイシンDL−リンゴ酸無水物の代りに非吸湿性のアジスロマイシンL−リンゴ酸一水和物が決定化された。
一方、実施例8で見られるように、水性溶媒条件下ではアジスロマイシンDL−リンゴ酸無水物の代りに非吸湿性のアジスロマイシンL−リンゴ酸一水和物が決定化された。
実験例1
水溶解度の試験
本発明のアジスロマイシンL−リンゴ酸一水和物およびアジスロマイシン二水和物のそれぞれを脱イオン水およびリン酸緩衝液(pH7)のそれぞれに飽和状態に到逹するように溶解させた後、それぞれの溶液を米国薬局方に記載された分析方法によってHPLCで分析してアジスロマイシンを基準として溶解された量を測定した。その結果を下記表4に示した。
表4
水溶解度の試験
本発明のアジスロマイシンL−リンゴ酸一水和物およびアジスロマイシン二水和物のそれぞれを脱イオン水およびリン酸緩衝液(pH7)のそれぞれに飽和状態に到逹するように溶解させた後、それぞれの溶液を米国薬局方に記載された分析方法によってHPLCで分析してアジスロマイシンを基準として溶解された量を測定した。その結果を下記表4に示した。
表4
前記表4から、本発明によるアジスロマイシンL−リンゴ酸一水和物の溶解度が公知のアジスロマイシン二水和物に比べて著しく向上し、これは本発明のアジスロマイシン塩が生体内への適用にさらに望ましいことを意味する。
実験例2
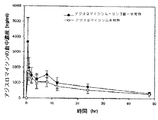
非吸湿性の試験
本発明のアジスロマイシンL−リンゴ酸一水和物を25℃または40℃の温度及び40%〜90%の相対湿度の条件下で15日以上連続的に露出させた後、0、3、7及び15日目にカールフィッシャー水分計器で含水量を測定して、その結果を下記表5及び図5に示した。
表5
非吸湿性の試験
本発明のアジスロマイシンL−リンゴ酸一水和物を25℃または40℃の温度及び40%〜90%の相対湿度の条件下で15日以上連続的に露出させた後、0、3、7及び15日目にカールフィッシャー水分計器で含水量を測定して、その結果を下記表5及び図5に示した。
表5
前記表5から、本発明の結晶形アジスロマイシンL−リンゴ酸一水和物は高湿条件下でも水分を吸収しない非吸湿性塩であることを確認することができ、また低温条件下でも本来の含水量を保持する安定した水和物であることを確認した。
実験例3
熱安定性の試験
本発明によるアジスロマイシンL−リンゴ酸一水和物に対して高温での経時による安定性を公知のアジスロマイシン二水和物と比べて測定した。
具体的に、本発明のアジスロマイシンL−リンゴ酸一水和物及びアジスロマイシン二水和物のそれぞれを60℃の温度、75%の相対湿度の苛酷条件下で密封状態で保管しながら、それぞれ7日、14日、21日及び28日が経った後の試料に対して米国薬局方に記載された分析方法によってHPLCで分析して活性アジスロマイシンの残量を測定した。その結果を表6及び図6に示した。
表6
熱安定性の試験
本発明によるアジスロマイシンL−リンゴ酸一水和物に対して高温での経時による安定性を公知のアジスロマイシン二水和物と比べて測定した。
具体的に、本発明のアジスロマイシンL−リンゴ酸一水和物及びアジスロマイシン二水和物のそれぞれを60℃の温度、75%の相対湿度の苛酷条件下で密封状態で保管しながら、それぞれ7日、14日、21日及び28日が経った後の試料に対して米国薬局方に記載された分析方法によってHPLCで分析して活性アジスロマイシンの残量を測定した。その結果を表6及び図6に示した。
表6
前記表6から、アジスロマイシン二水和物は28日間分解が活発に行われたが、本発明のアジスロマイシンL−リンゴ酸一水和物は非常に安定した。
実験例4
アジスロマイシンの時間別血中濃度の測定(薬物動態解析)
本発明による結晶形アジスロマイシンL−リンゴ酸一水和物の向上した溶解度による生体内における薬物動態作用をビーグル犬(beagle dog)を用いて試し、その結果をアジスロマイシン二水和物と比べた。
アジスロマイシンの時間別血中濃度の測定(薬物動態解析)
本発明による結晶形アジスロマイシンL−リンゴ酸一水和物の向上した溶解度による生体内における薬物動態作用をビーグル犬(beagle dog)を用いて試し、その結果をアジスロマイシン二水和物と比べた。
具体的に、12匹のビーグル犬(中国北京産、平均体重9.5±0.5kg)を6匹ずつ二つの群に分けた後、本発明のアジスロマイシンL−リンゴ酸一水和物(試験群:T群)とアジスロマイシン二水和物(対照群:C群)を20mg/kgずつゼラチンカプセルに収めて16時間絶食させたそれぞれの個体に単回経口投与した。投与の後、時間別に血液を採取した後、血漿を分離した。血漿から抽出されたアジスロマイシン試料をLC/MS/MS方法によって分析して、アジスロマイシンの含有量を測定した後、薬物動態パラメーターを算出した。その結果は表7に示した通りである。
表7
表7
前記表7に示したように、本発明のアジスロマイシンL−リンゴ酸一水和物はアジスロマイシン二水和物に比べて改善した薬物動態パラメーター値を示した。例えば、本発明のアジスロマイシンL−リンゴ酸一水和物のCmax値はアジスロマイシン二水和物に比べて約2倍増加した。このように、本発明のアジスロマイシンL−リンゴ酸一水和物は初期に高い血中濃度を示すので、耐性病源菌による感染症を治療するのに有効である。
本発明のアジスロマイシンL−リンゴ酸一水和物は、単独または薬学的に許容される賦形剤とともに通常の製剤方法によって軟質または硬質カプセル剤、錠剤、懸濁剤、粉末剤及び液剤などのような組成物に製剤化されることができる。
次に組成物の製剤化の製造例を例示するが、本発明による組成物がこれらに限定するのではない。
本発明のアジスロマイシンL−リンゴ酸一水和物は、単独または薬学的に許容される賦形剤とともに通常の製剤方法によって軟質または硬質カプセル剤、錠剤、懸濁剤、粉末剤及び液剤などのような組成物に製剤化されることができる。
次に組成物の製剤化の製造例を例示するが、本発明による組成物がこれらに限定するのではない。
製造例1
アジスロマイシンカプセル
下記成分を用いてゼラチンカプセルを製造した。
アジスロマイシンカプセル
下記成分を用いてゼラチンカプセルを製造した。
製造例2
アジスロマイシン錠剤
下記成分を用いて錠剤を製造した。
アジスロマイシン錠剤
下記成分を用いて錠剤を製造した。
製造例3
アジスロマイシンの懸濁による経口投与用粉末
下記成分を用いて経口投与用粉末を製造した。
アジスロマイシンの懸濁による経口投与用粉末
下記成分を用いて経口投与用粉末を製造した。
前述したように、本発明によるアジスロマイシンL−リンゴ酸一水和物は公知のアジスロマイシン二水和物と比べて水溶解度が非常に高いだけでなく、熱安定性及び非吸湿性に優れる。また、本発明の塩は動物実験で薬物動態作用が公知の塩に比べて優れた。従って、本発明のアジスロマイシンL−リンゴ酸一水和物は多様な感染性疾患の治療に有用である。
以上、本発明を具体的な実施例と関連付けて記述したが、添付された特許請求の範囲の技術的思想を遺脱しない範囲内で当該分野における熟練者により多様に変形及び修正し得ることは勿論である。
Claims (15)
- 式(I)の結晶形アジスロマイシンL−リンゴ酸一水和物:
- X線粉末回折スペクトルにおいてI/Io値が10%以上である主要ピークが9.6、10.6、11.2、12.0、12.4、14.3、14.6、15.0、16.6、17.5、18.1、18.6、19.3、19.7、20.2、20.5、21.4、22.6、23.6、24.0、24.6、27.1、27.7、34.4の回折角(2θ±0.2)で現われることを特徴とする請求項1に記載の結晶形アジスロマイシンL−リンゴ酸一水和物。
- a)水性有機溶媒中で式(II)のアジスロマイシンと式(III)のリンゴ酸を反応させるか、b)式(IV)のアジスロマイシンL−リンゴ酸無水物を水性有機溶媒に溶解させた後、再結晶化させることを特徴とする請求項1に記載の結晶形アジスロマイシンL−リンゴ酸一水和物の製造方法:
- 前記式(III)のリンゴ酸がL−リンゴ酸、ラセミ体のDL−リンゴ酸またはこれらの混合物であることを特徴とする請求項3に記載の方法。
- 前記水性有機溶媒がアセトン、メチルエチルケトン、メチルイソブチルケトン、エタノール、1−プロパノール、2−プロパノール、1−ブタノール、テトラヒドロフラン、1,4−ジオキサン、酢酸メチル、酢酸エチル及びこれらの混合物で構成された群から選ばれることを特徴とする請求項3に記載の方法。
- 前記水性有機溶媒が2〜10体積%の含水量を有することを特徴とする請求項3に記載の方法。
- 前記反応段階a)において、前記水性有機溶媒が式(II)のアジスロマイシン1gに対して3〜20mlの量で用いられることを特徴とする請求項3に記載の方法。
- 前記反応段階a)において、式(III)のL−リンゴ酸が式(II)のアジスロマイシン1モルに対して2〜2.5モルの量で用いられることを特徴とする請求項3に記載の方法。
- 請求項1の結晶形アジスロマイシンL−リンゴ酸一水和物を活性成分として含む細菌感染性疾患治療用医薬組成物。
- 経口用、注射用及び眼科用製剤形態で投与されることを特徴とする請求項9に記載の医薬組成物。
- 前記経口用製剤が錠剤、カプセル剤、懸濁剤または粉末形態であることを特徴とする請求項10に記載の医薬組成物。
- 前記経口用製剤が結合剤、充填制、緩衝剤、潤滑剤、崩壊剤、甘味剤、着臭剤(odorant)、界面活性剤、コート剤及びこれらの混合物で構成された群から選ばれる担体、希釈剤及び賦形剤を含むことを特徴とする請求項10に記載の医薬組成物。
- 前記アジスロマイシンL−リンゴ酸一水和物が前記組成物100重量部に対して20〜80重量部の量で含まれることを特徴とする請求項12に記載の医薬組成物。
- 前記錠剤またはカプセル剤がアジスロマイシンを50〜3,500mgの含量で含むことを特徴とする請求項11に記載の医薬組成物。
- 前記細菌感染性疾患が肺炎、咽頭炎、へんとう炎、慢性閉塞性肺疾患、急性中耳炎、単純性皮膚感染症、泌尿生殖器系感染症及び播種性非定型抗酸菌症で構成された群から選ばれることを特徴とする請求項9に記載の医薬組成物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020050048923A KR100666091B1 (ko) | 2005-06-08 | 2005-06-08 | 아지스로마이신 l-말산염 일수화물 및 이를 포함하는 약학조성물 |
| PCT/KR2006/002157 WO2006132486A1 (en) | 2005-06-08 | 2006-06-05 | Crystalline azithromycin l-malate monohydrate and pharmaceutical composition containing same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2008543749A true JP2008543749A (ja) | 2008-12-04 |
Family
ID=37498652
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008515621A Withdrawn JP2008543749A (ja) | 2005-06-08 | 2006-06-05 | 結晶形アジスロマイシンl−リンゴ酸一水和物及びこれを含む医薬組成物 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20090318375A1 (ja) |
| EP (1) | EP1910392B1 (ja) |
| JP (1) | JP2008543749A (ja) |
| KR (1) | KR100666091B1 (ja) |
| CN (1) | CN101193904A (ja) |
| AT (1) | ATE449102T1 (ja) |
| AU (1) | AU2006255914B2 (ja) |
| CA (1) | CA2610449A1 (ja) |
| DE (1) | DE602006010565D1 (ja) |
| ES (1) | ES2333610T3 (ja) |
| IL (1) | IL187639A0 (ja) |
| RU (1) | RU2348644C1 (ja) |
| WO (1) | WO2006132486A1 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8106111B2 (en) | 2009-05-15 | 2012-01-31 | Eastman Chemical Company | Antimicrobial effect of cycloaliphatic diol antimicrobial agents in coating compositions |
| RU2512683C2 (ru) | 2012-06-08 | 2014-04-10 | Общество с ограниченной ответственностью "ВИК-здоровье животных" | Антибактериальная инъекционная фармацевтическая композиция |
| CN115057814A (zh) * | 2021-12-07 | 2022-09-16 | 山东新时代药业有限公司 | 一种米力农苹果酸盐晶体 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4474768A (en) * | 1982-07-19 | 1984-10-02 | Pfizer Inc. | N-Methyl 11-aza-10-deoxo-10-dihydro-erytromycin A, intermediates therefor |
| PT1246831E (pt) * | 2000-01-04 | 2008-03-28 | Teva Pharma | Método de preparação de di-hidrato de azitromicina |
| ES2162764B1 (es) * | 2000-05-17 | 2003-04-01 | Ercros Ind Sa | Forma polimorfica del dihidrato de azitromicina, y su procedimiento deobtencion. |
| US6949519B2 (en) * | 2000-11-27 | 2005-09-27 | Sandoz Ag | Macrolide solvates |
| KR100491183B1 (ko) * | 2001-03-21 | 2005-05-25 | 한미약품 주식회사 | 아지트로마이신의 제조방법 및 이 방법에 사용되는9-데옥소-9에이-아자-9에이-호모에리트로마이신 에이의결정성 수화물 |
| KR100431431B1 (ko) * | 2001-04-25 | 2004-05-14 | 한미약품 주식회사 | 아지트로마이신 수화물의 1,2-프로필렌글리콜 내포화합물,그의 제조방법 및 그의 약학적 조성물 |
| US6855813B2 (en) * | 2002-07-19 | 2005-02-15 | Alembic Limited | Process for the preparation of azithromycin monohydrate |
| ES2220229B1 (es) | 2003-05-29 | 2005-10-16 | Quimica Sintetica, S.A. | Sales de adicion de azitromicina y acido citrico y procedimiento para su obtencion. |
| GB2395482A (en) * | 2003-07-03 | 2004-05-26 | Jubilant Organosys Ltd | Process for preparing non-hygroscopic azithromycin dihydrate |
-
2003
- 2003-06-06 US US11/915,929 patent/US20090318375A1/en not_active Abandoned
-
2005
- 2005-06-08 KR KR1020050048923A patent/KR100666091B1/ko not_active IP Right Cessation
-
2006
- 2006-06-05 DE DE602006010565T patent/DE602006010565D1/de active Active
- 2006-06-05 JP JP2008515621A patent/JP2008543749A/ja not_active Withdrawn
- 2006-06-05 EP EP06747495A patent/EP1910392B1/en not_active Not-in-force
- 2006-06-05 CN CNA2006800201470A patent/CN101193904A/zh active Pending
- 2006-06-05 AT AT06747495T patent/ATE449102T1/de not_active IP Right Cessation
- 2006-06-05 ES ES06747495T patent/ES2333610T3/es active Active
- 2006-06-05 CA CA002610449A patent/CA2610449A1/en not_active Abandoned
- 2006-06-05 WO PCT/KR2006/002157 patent/WO2006132486A1/en active Application Filing
- 2006-06-05 RU RU2007148467/04A patent/RU2348644C1/ru not_active IP Right Cessation
- 2006-06-05 AU AU2006255914A patent/AU2006255914B2/en not_active Expired - Fee Related
-
2007
- 2007-11-26 IL IL187639A patent/IL187639A0/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CA2610449A1 (en) | 2006-12-14 |
| CN101193904A (zh) | 2008-06-04 |
| DE602006010565D1 (de) | 2009-12-31 |
| IL187639A0 (en) | 2008-03-20 |
| WO2006132486A1 (en) | 2006-12-14 |
| ATE449102T1 (de) | 2009-12-15 |
| US20090318375A1 (en) | 2009-12-24 |
| KR100666091B1 (ko) | 2007-01-10 |
| EP1910392B1 (en) | 2009-11-18 |
| AU2006255914B2 (en) | 2009-09-03 |
| AU2006255914A1 (en) | 2006-12-14 |
| EP1910392A1 (en) | 2008-04-16 |
| RU2348644C1 (ru) | 2009-03-10 |
| ES2333610T3 (es) | 2010-02-24 |
| EP1910392A4 (en) | 2008-10-29 |
| KR20060128066A (ko) | 2006-12-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20220144825A1 (en) | Solid state forms of ripretinib | |
| KR101626506B1 (ko) | 에리트로마이신염의 수화물, 그의 제조방법 및 용도 | |
| US20160046603A1 (en) | Crystalline Forms of D-Glucitol, 1-Deoxy-1-(Methylamino)-, 1-(6-Amino-3,5-Difluoropyridine-2-Yl)-8-Chloro-6-Fluoro-1,4-Dihydro-7-(3-Hydroxyazetidin-1-Yl)-4-Oxo-3-Quinolinecarboxylate | |
| JP2011527292A6 (ja) | エリスロマイシン塩の水和物、その製造方法及び使用 | |
| JP2008543749A (ja) | 結晶形アジスロマイシンl−リンゴ酸一水和物及びこれを含む医薬組成物 | |
| US7569549B2 (en) | Isostructural pseudopolymorphs of 9-deoxo-9a-aza-9a-methyl-9a-homoerythromycin A | |
| JP2005516984A (ja) | 結晶質及び非晶質ムピロシンカルシウムを生産する方法 | |
| US20040092460A1 (en) | Novel amorphous 9-deoxo-9a-aza-9a-methyl-9a-homoerythromycin A, process for preparing the same, and uses thereof | |
| US20220411371A1 (en) | Solid state forms of lucerastat and process for preparation thereof | |
| US20060154953A1 (en) | Amorphous tacrolimus and preparation thereof | |
| KR101423630B1 (ko) | 비칼루타미드와 니코틴아미드의 공결정 | |
| WO2011027988A2 (en) | Novel polymorphic form of prasugrel-hydrogen sulfate | |
| KR20230087557A (ko) | 로레시비빈트의 고상형 | |
| EP3870298A1 (en) | New crystalline polymorphs of rigosertib sodium | |
| US20060270685A1 (en) | Anhydrous ziprasidone mesylate and a process for its preparation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20090917 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20100929 |