JP2005298631A - シクロデキストリン誘導体およびその製造方法、ロタキサンおよびその製造方法、並びに屈折率変換材料および光−熱変換蓄積材料 - Google Patents
シクロデキストリン誘導体およびその製造方法、ロタキサンおよびその製造方法、並びに屈折率変換材料および光−熱変換蓄積材料 Download PDFInfo
- Publication number
- JP2005298631A JP2005298631A JP2004115330A JP2004115330A JP2005298631A JP 2005298631 A JP2005298631 A JP 2005298631A JP 2004115330 A JP2004115330 A JP 2004115330A JP 2004115330 A JP2004115330 A JP 2004115330A JP 2005298631 A JP2005298631 A JP 2005298631A
- Authority
- JP
- Japan
- Prior art keywords
- cyclodextrin
- cyclodextrin derivative
- rotaxane
- refractive index
- norbornadiene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- CGXBVDDCNQEPCA-UHFFFAOYSA-N COC(C(C1C=CC2C1)=C2c1ccccc1)=O Chemical compound COC(C(C1C=CC2C1)=C2c1ccccc1)=O CGXBVDDCNQEPCA-UHFFFAOYSA-N 0.000 description 1
Images
Landscapes
- Optical Modulation, Optical Deflection, Nonlinear Optics, Optical Demodulation, Optical Logic Elements (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
【解決手段】 本発明のシクロデキストリン誘導体は、一般式(1)で表されるシクロデキストリン誘導体であって、シクロデキストリンにおける全水酸基に対するエーテル化率が60%以上である。
【化1】
〔但し、R1 〜R3 は、水素原子または式(a)の基、mは6〜9の整数である。〕
【化2】
【選択図】 なし
Description
また、NBD構造を有する化合物は、異性化したQC構造を有する化合物と異なる屈折率を有する、すなわち光の照射によって屈折率が変化する特性を有することから、例えば光記憶素子や光スイッチシステムに用いられる屈折率変換材料への応用が期待されている(非特許文献3参照)。
しかしながら、従来のNBD構造が導入されたポリマーは、光照射による屈折率の変化量が0.01程度またはそれ以下であり、屈折率の変化量が大きいものではない。
本発明の第2の目的は、上記のシクロデキストリン誘導体を環状分子として有するロタキサンおよびその製造方法を提供することにある。
本発明の第3の目的は、屈折率の変化量が大きく、しかも、容易に成膜することができる屈折率変換材料を提供することにある。
本発明の第4の目的は、光照射による蓄熱量が大きく、しかも、容易に成膜することができる熱エネルギー変換蓄積材料を提供することにある。
また、本発明のロタキサンにおいては、下記一般式(2)で表される化合物であることが好ましい。
また、本発明の屈折率変換材料は、上記のシクロデキストリン誘導体を環状分子として有するロタキサンよりなることを特徴とする。
また、本発明の光−熱エネルギー変換蓄積材料は、上記のシクロデキストリン誘導体を環状分子として有するロタキサンよりなることを特徴とする。
本発明に係るシクロデキストリン誘導体の製造方法によれば、上記のシクロデキストリンを有利に製造することができる。
本発明に係るロタキサンは、環状分子として、上記のシクロデキストリン誘導体を有するため、光照射によって屈折率が変化し、かつ、屈折率の変化量が極めて大きく、また、光照射による蓄熱量が大きいものであり、しかも、容易に成膜することができるものであり、従って、屈折率変換材料または光−熱エネルギー変換蓄積材料として有用である。
本発明に係るロタキサンの製造方法によれば、上記のロタキサンを有利に製造することができる。
本発明の屈折率変換材料は、上記のシクロデキストリン誘導体またはロタキサンよりなるため、屈折率の変化量が極めて大きく、しかも、容易に成膜することができるものである。
本発明の光−熱エネルギー変換蓄積材料は、上記のシクロデキストリン誘導体またはロタキサンよりなるため、光照射による蓄熱量が大きく、しかも、容易に成膜することができるものであである。
〔シクロデキストリン誘導体〕
本発明に係るシクロデキストリン誘導体は、上記一般式(1)で表される化合物であって、シクロデキストリンにおける全水酸基に対するエーテル化率が60%以上、好ましくは70%以上であるシクロデキストリン誘導体(以下、「特定のシクロデキストリン誘導体」という。)である。
一般式(1)において、R1 、R2 およびR3 は、互いに独立して水素原子または上記式(a)で表される基であり、R1 、R2 およびR3 の合計の60%以上が上記式(a)で表される基とされる。また、ピラノース環の数を示すmは6〜9の整数である。
また、シクロデキストリンにおける全水酸基に対するエーテル化率は、 1H−NMRスペクトルにおいて、シクロデキストリンにおけるアセタールに基づく4.84ppmのメチンプロトンのシグナルを基準とし、NBDのオレフィン部位に基づく6.84〜7.03ppmのメチンプロトンのシグナルの積分強度比から求めることができる。
先ず、適宜の溶媒中において、3−フェニル−2,5−ノルボルナジエン−2−カルボン酸と、1−ブロモ−2−クロロエタンとを反応させることにより、3−フェニル−2,5−ノルボルナジエン−2−カルボキシクロロエチル(以下、「特定のノルボルナジエン誘導体」という。)を合成する。
この特定のノルボルナジエン誘導体を得るための反応工程(以下、「反応工程(a−1)」という。)において、溶媒としては、ジメチルスルホキシド、ジメチルフォルムアミド、ジオキサンなどを用いることができる。
3−フェニル−2,5−ノルボルナジエン−2−カルボン酸と1−ブロモ−2−クロロエタンとの使用割合は、3−フェニル−2,5−ノルボルナジエン−2−カルボン酸1モルに対して1−ブロモ−2−クロロエタンが1.0〜2.0モルであることが好ましい。 また、反応工程(a−1)においては、例えば1,8−ジアザビシクロ[5.4.0]ウンデセン−7等のアルカリ剤を添加することが好ましく、その使用割合は、3−フェニル−2,5−ノルボルナジエン−2−カルボン酸1モルに対して1.0〜2.0モルである。
また、反応工程(a−1)における反応条件としては、例えば反応温度が60〜80℃、反応時間が24〜48時間である。
触媒としては、テトラブチルアンモニウムブロミド、テトラフェニルフォスフォニウムブロミドなどを用いることができる。また、触媒の使用割合は、シクロデキストリンにおける全水酸基1モルに対して0.03〜0.15モルである。
シクロデキストリンとしては、具体的には、α−シクロデキストリン、β−シクロデキストリン、γ−シクロデキストリン、δ−シクロデキストリンが用いられる。これらの化合物は、単独でまたは2種以上を組み合わせて用いることができる。
シクロデキストリンと特定のノルボルナジエン誘導体との使用割合は、シクロデキストリンにおける全水酸基1モルに対して特定のノルボルナジエン誘導体が0.7モル以上とされ、好ましくは0.7〜2モルとされる。
また、反応工程(a−2)においては、例えば水素化ナトリウム等のアルカリ剤を添加することが好ましく、その使用割合は、特定のノルボルナジエン1モルに対して1.0〜1.5モルである。
また、反応工程(a−2)における反応条件としては、例えば反応温度が40〜80℃、反応時間が2〜48時間である。
具体的には、特定のシクロデキストリン誘導体を適宜の溶媒に溶解し、得られた溶液を適宜の支持体上に塗布して乾燥処理することにより、成膜することができる。
特定のシクロデキストリン誘導体を溶解するための溶媒としては、ジメチルホルムアミド、テトラヒドロフラン、N−メチルピロリドン、ジメチルホルムアルデヒド、クロロホルム、塩化メチレンなどを用いることができる。
本発明のロタキサンは、環状分子として特定のシクロデキストリン誘導体を有してなるロタキサン(以下、「特定のロタキサン」という。)である。
この特定のロタキサンにおいて、軸分子としては、種々の鎖状高分子を用いることができるが、両末端にジニトロフェニル基が結合された鎖状高分子が好ましい。
また、特定のロタキサン1分子中に存在する特定のシクロデキストリン誘導体の数は、10〜40であることが好ましい。
また、特定のロタキサンは、ゲルパーミエーションクロマトグラフ法によって測定される数平均分子量が15000〜20000であることが好ましい。
特定のロタキサンとして好ましい具体例としては、上記一般式(2)で表されるロタキサンを挙げることができる。
上記一般式(2)において、R1 、R2 およびR3 は、互いに独立して水素原子または上記式(a)で表される基であり、R1 、R2 およびR3 の合計の60%以上が上記式(a)で表される基とされる。Xは−CH2 CH2 O−で表される繰り返し単位からなる高分子鎖である。ピラノース環の数を示すmは6〜9の整数である。特定のシクロデキストリンの数を示すkは、10〜40である。
先ず、環状分子として上記一般式(3)で表されるシクロデキストリンを有するロタキサンよりなる中間体を合成する。
この中間体を合成する方法は、軸分子を形成する鎖状高分子の種類によって異なるが、上記一般式(2)で表されるロタキサンにおける軸分子を有する中間体を合成する場合を例に挙げると、以下の通りである。
両末端に3−アミノプロピル基を有するポリエチレングリコールを用意し、このポリエチレングリコールとシクロデキストリンとを適宜の溶媒に溶解し、得られた溶液に対して例えば超音波によって攪拌処理することにより、プソイドロタキサンを調製する。
ここで、プソイドロタキサンの調製に用いられる溶媒としては、蒸留水を挙げることができる。
また、シクロデキストリンとしては、具体的には、α−シクロデキストリン、β−シクロデキストリン、γ−シクロデキストリン、δ−シクロデキストリンが用いられる。これらの化合物は、単独でまたは2種以上を組み合わせて用いることができる。
また、ポリエチレングリコールとシクロデキストリンとの使用割合は、製造すべき特定のロタキサン1分子中に存在する特定のシクロデキストリン誘導体の数に応じて選択されるが、例えばポリエチレングリコール1モル(数平均分子量換算)に対し、シクロデキストリン10〜40モルである。
また、プソイドロタキサンの調製は、15〜40℃の温度で行うことが好ましい。
次いで、得られたプソイドロタキサンと2,4−ジニトロフルオロベンゼンとを適宜の溶媒中で反応させることにより、両末端にジニトロフェニル基が結合された軸分子と、シクロデキストリンよりなる環状分子とを有するロタキサンよりなる中間体が得られる。
ここで、プソイドロタキサンと2,4−ジニトロフルオロベンゼンとの反応に用いられる溶媒としては、ジメチルホルムアミド、ジメチルアセトアミドなどを挙げることができる。
また、プソイドロタキサンと2,4−ジニトロフルオロベンゼンとの使用割合は、例えばプソイドロタキサン1モル(数平均分子量換算)に対し、2,4−ジニトロフルオロベンゼン90〜100モルである。
また、プソイドロタキサンと2,4−ジニトロフルオロベンゼンとの反応条件は、例えば反応温度が25〜40℃、反応時間が2〜24時間である。
この特定のロタキサンを得るための反応工程において、溶媒としては、N−メチル−2−ピロリドン、ジメチルフォルムアミド、ジオキサンなどを用いることができる。
触媒としては、テトラブチルアンモニウムブロミド、テトラフェニルフォスフォニウムブロミドなどを用いることができる。また、触媒の使用割合は、中間体中に存在するシクロデキストリンにおける全水酸基1モルに対して0.03〜0.15モルである。
中間体と特定のノルボルナジエン誘導体との使用割合は、中間体中に存在するシクロデキストリンにおける全水酸基1モルに対して特定のノルボルナジエン誘導体が0.7モル以上とされ、好ましくは0.7〜2モルとされる。
また、この反応工程においては、例えば水素化ナトリウム等のアルカリ剤を添加することが好ましく、その使用割合は、特定のノルボルナジエン1モルに対して1.0〜2.0モルである。
また、この反応工程における反応条件としては、例えば反応温度が40〜80℃、反応時間が4〜48時間である。
(1)テトラブチル−n−アンモニウムブロミド(以下、「TBAB」という。)としては、市販品を、脱水酢酸エチルを用いて2回再結晶したものを使用した。
(2)3−フェニル−2,5−ノルボルナジエン−2−カルボン酸(以下、「PNC」という。)としては、市販品を、酢酸エチル/n−ヘキサン混合溶媒を用いて1回再結晶したものを使用した。
(3)α−シクロデキストリン(以下、「α−CD」という。)としては、市販品を、蒸留水を用いて2回再結晶したものを使用した。
(4)β−シクロデキストリン(以下、「β−CD」という。)としては、市販品を、蒸留水を用いて2回再結晶したものを使用した。
(5)γ−シクロデキストリン(以下、「γ−CD」という。)としては、市販品を、蒸留水を用いて2回再結晶したものを使用した。
(6)1−ブロモ−2−クロロエタン(以下、「BCE」という。)としては、市販品を、塩化カルシウム(乾燥剤)を用いて蒸留精製したものを使用した。
(7)N−メチル−2−ピロリドン(以下、「NMP」という。)としては、市販品を、水素化カルシウム(乾燥剤)を用いて蒸留精製したものを使用した。
(8)ジメチルスルホキシド(以下、「DMSO」という。)としては、市販品を、水素化カルシウム(乾燥剤)を用いて蒸留精製したものを使用した。
(9)ジメチルホルムアミド(以下、「DMF」という。)としては、市販品を、水素化カルシウム(乾燥剤)を用いて蒸留精製したものを使用した。
(10)1,8−ジアザビシクロ[5.4.0]ウンデセン−7(以下、「DBU」という。)としては、市販品を、水素化カルシウム(乾燥剤)を用いて蒸留精製したものを使用した。
(11)両末端に3−アミノプロピル基を有するポリエチレングリコール(以下,「PEG」という。)としては、数平均分子量が1500の市販品をそのまま使用した。
(12)2,4−ジニトロフルオロベンゼン、シクロヘキサノンおよび水素化ナトリウムとしては、市販品をそのまま使用した。
(1)赤外分光光度計:日本分光(株)製「FT/IR−420」
(2)紫外分光光度計:(株)島津製作所製「UV−2500PC」
(3) 1H核磁気共鳴装置:日本電子(株)製「JNM−α−500」(500MHz)および「JNM−α−600」(600MHz)
(4)リサイクル分取高速液体クロマトグラフィー(以下、「分取HPLC」という:日本分析工業(株)製「HPLC−908型」(カラム:JAIgel1HA−FおよびJAIgel1HA−A,展開溶媒:クロロホルム)
(5)エリプソメーター:ガードナー社製「L115Bエリプソメーター」
PNC2.16g(10mmol)およびDBU1.72g(12mmol)の混合物に、DMSO10mLを添加して溶解し、その後、BCE2.92g(20mmol)を滴下し、さらに室温で4時間の条件で反応させた。
反応が終了した後、反応溶液を酢酸エチルによって希釈し、この希釈溶液に対して蒸留水による洗浄を5回行い、さらに酢酸エチル相に乾燥剤として無水硫酸マグネシウムを添加して乾燥処理を行った。次いで、乾燥剤をろ別した後、酢酸エチル相を減圧留去し、その後、酢酸エチルおよびn−ヘキサンの混合物(混合比1:10)を展開溶媒とするシリカゲルカラムクロマトグラフィーによって、単離精製することにより、黄色の粘性液体1.94gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(b)で表される3−フェニル−2,5−ノルボルナジエン−2−ガルボキシクロロエチル(特定のノルボルナジエン誘導体)であると同定された。収率は71%であった。
○IR(film,cm-1):
1698(νC=C ester ),
1612(νC=C NBD),
1594,1491(νC=C aromatic),
1180,1021(νC−O−C ester ),
760(νC−Cl)
○ 1H NMR(500MHz,CDCl3 ,TMS)δ(ppm):
1.98(d,J=6.0,1.0H,Ha ),
2.17(d,J=6.0,1.0H,Ha ' ),
3.78(t,J=5.5,2.0H,Hj ),
4.00(s,1.0H,Hb ),
4.29(s,1.0H,Hc ),
6.98〜7.04(m,2.0H,Hd ,He ),
7.34〜7.56(m,5.0H,Hg ,Hf ,Hh )
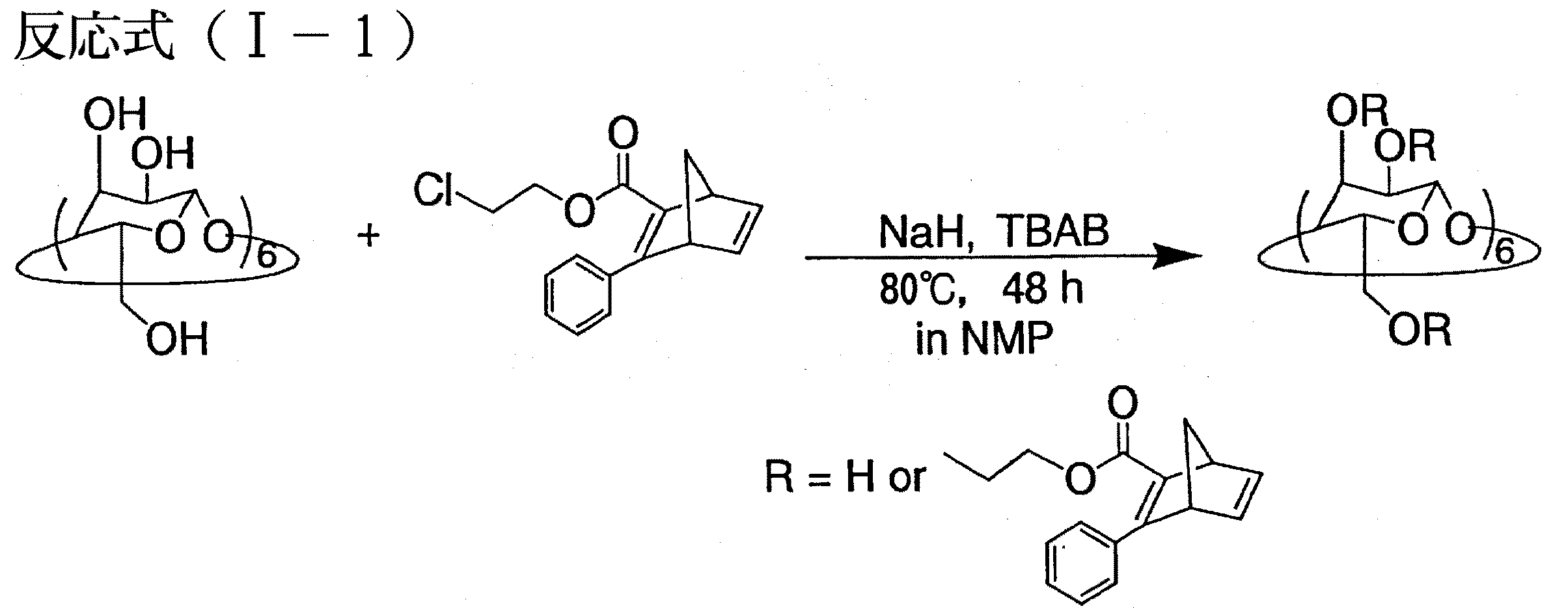
α−CD0.05g(0.05mmol)、TBAB0.03g(5mol%)および水素化ナトリウム0.04g(1.8mmol)の混合物に、NMP4mLを添加し、室温で1時間攪拌した。その後、特定のノルボルナジエン誘導体0.49g(1.8mmol)を添加し、さらに80℃で48時間の条件で反応させた。
反応が終了した後、反応溶液をクロロホルムで希釈し、0.5N塩酸水溶液で1回、蒸留水で5回洗浄し、有機相を乾燥剤として無水硫酸マグネシウムを用いて乾燥処理した。乾燥剤をろ別した後、分取HPLCによって単離精製し、60℃で24時間減圧乾燥することにより、茶褐色の粉末固体0.15gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(I)で表される化合物であって、α−CDにおける全水酸基に対するエーテル化率が89%であるものと同定された。収率は45%であった。この生成物を「シクロデキストリン誘導体(I)」とする。また、シクロデキストリン誘導体(I)の合成工程を下記反応式(I−1)に示す。
○IR(film,cm-1):
3447(νOH),
1704(νC=C ester ),
1615(νC=C NBD),
1491,1444(νC=C aromatic),
1149,1034(νC−O−C)
○ 1H NMR(500MHz,DMSO−d6 )δ(ppm):
1.94〜2.14(m,2.00H,H9 ),
3.15〜4.75(m,6.99H,H1 ,H2 ,H3 ,H4 ,H5 ,H6 ,H7 ,H8 ,H10,H11),
5.40〜6.05(m,0.12H,OH in CD),
6.94〜7.42(m,7.00H,H12,H13,H14,H15,H16)
β−CD0.06g(0.05mmol)、TBAB0.04g(5mol%)および水素化ナトリウム0.05g(2.1mmol)の混合物に、NMP4mLを添加し、室温で1時間攪拌した。その後、特定のノルボルナジエン誘導体0.59g(2.1mmol)を添加し、さらに80℃で48時間の条件で反応させた。
反応が終了した後、反応溶液をクロロホルムで希釈し、0.5N塩酸水溶液で1回、蒸留水で5回洗浄し、有機相を乾燥剤として無水硫酸マグネシウムを用いて乾燥処理した。乾燥剤をろ別した後、分取HPLCによって単離精製し、60℃で24時間減圧乾燥することにより、茶褐色の粉末固体0.15gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(II)で表される化合物であって、β−CDにおける全水酸基に対するエーテル化率が87%であるものと同定された。収率は44%であった。この生成物を「シクロデキストリン誘導体(II)」とする。また、シクロデキストリン誘導体(II)の合成工程を下記反応式(II−1)に示す。
○IR(film,cm-1):
3423(νOH),
1699(νC=C ester ),
1615(νC=C NBD),
1491,1444(νC=C aromatic),
1149,1033(νC−O−C)
○ 1H NMR(500MHz,DMSO−d6 )δ(ppm):
1.92〜2.13(m,2.00H,H9 ),
3.15〜4.82(m,8.85H,H1 ,H2 ,H3 ,H4 ,H5 ,H6 ,H7 ,H8 ,H10,H11),
5.58〜5.88(m,0.14H,OH in CD),
6.96〜7.49(m,7.00H,H12,H13,H14,H15,H16)
γ−CD0.07g(0.05mmol)、TBAB0.04g(5mol%)および水素化ナトリウム0.06g(2.4mmol)の混合物に、NMP4mLを添加し、室温で1時間攪拌した。その後、特定のノルボルナジエン誘導体0.66g(2.4mmol)を添加し、さらに80℃で48時間の条件で反応させた。
反応が終了した後、反応溶液をクロロホルムで希釈し、0.5N塩酸水溶液で1回、蒸留水で5回洗浄し、有機相を乾燥剤として無水硫酸マグネシウムを用いて乾燥処理した。乾燥剤をろ別した後、分取HPLCによって単離精製し、60℃で24時間減圧乾燥することにより、茶褐色の粉末固体0.11gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(III)で表される化合物であって、γ−CDにおける全水酸基に対するエーテル化率が84%であるものと同定された。収率は35%であった。この生成物を「シクロデキストリン誘導体(III)」とする。また、シクロデキストリン誘導体(III)の合成工程を下記反応式(III −1)に示す。
○IR(film,cm-1):
3414(νOH),
1701(νC=C ester ),
1615(νC=C NBD),
1491,1444(νC=C aromatic),
1149,1033(νC−O−C)
○ 1H NMR(500MHz,DMSO−d6 )δ(ppm):
1.96〜2.10(m,2.00H,H9 ),
3.15〜4.75(m,9.26H,H1 ,H2 ,H3 ,H4 ,H5 ,H6 ,H7 ,H8 ,H10,H11,),
5.58〜5.88(m,0.18H,OH in CD),
6.94〜7.42(m,7.00H,H12,H13,H14,H15,H16)
β−CD0.23g(0.2mmol)、TBAB0.06g(5mol%)および水素化ナトリウム0.09g(3.0mmol)の混合物に、NMP6mLを添加し、室温で1時間攪拌した。その後、特定のノルボルナジエン誘導体0.83g(3.0mmol)を添加し、さらに80℃で48時間の条件で反応させた。
反応が終了した後、反応溶液を蒸留水に滴下し、沈殿物をろ別した後、良溶媒としてテトラヒドロフランを用い、貧溶媒としてジエチルエーテルを用いて2回再沈精製を行い、60℃で24時間減圧乾燥することにより、茶褐色の粉末固体0.30gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(IV)で表される化合物であって、β−CDにおける全水酸基に対するエーテル化率が62%であるものと同定された。収率は35%であった。この生成物を「シクロデキストリン誘導体(IV)」とする。
○IR(film,cm-1):
3377(νOH),
1695(νC=C ester ),
1606(νC=C NBD),
1594,1491(νC=C aromatic),
1153,1032(νC−O−C ester )
○ 1H NMR(500MHz,DMSO−d6 )δ(ppm):
1.84〜2.23(m,2.0H,CH2 in NBD),
3.29〜4.75(m,10.9H,Ha ,Hb ,Hc ,Hd ,Hf ,Hg ,Hh ,CH in NBD,H2 O),
4.80〜4.84(m,0.66H,He ),
5.58〜5.88(m,0.81H,OH in CD),
6.84〜7.03(m,2.0H,CH in NBD),
7.34〜7.47(m,5.0H,aromatic)
α−CD0.29g(0.3mmol)、TBAB0.04g(5mol%)および水素化ナトリウム0.07g(2.7mmol)の混合物に、NMP6mLを添加し、室温で1時間攪拌した。その後、特定のノルボルナジエン誘導体0.74g(2.7mmol)を添加し、さらに50℃で48時間の条件で反応させた。
反応が終了した後、反応溶液を蒸留水に滴下し、沈殿物をろ別した後、良溶媒としてテトラヒドロフランを用い、貧溶媒としてジエチルエーテルを用いて2回再沈精製を行い、60℃で24時間減圧乾燥することにより、茶褐色の粉末固体0.14gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(V)で表される化合物であって、α−CDにおける全水酸基に対するエーテル化率が35%であるものと同定された。収率は20%であった。この生成物を「シクロデキストリン誘導体(V)」とする。
○IR(film,cm-1):
3393(νOH),
1696(νC=C ester ),
1615(νC=C NBD),
1594,1491(νC=C aromatic),
1331,1034(νC−O−C ester ),
1151,1034(νC−O−C ether )
○ 1H NMR(500MHz,DMSO−d6 )δ(ppm):
1.84〜2.23(m,2.0H,CH2 in NBD),
3.29〜4.75(m,19.3H,Ha ,Hb ,Hc ,Hd ,Hf ,Hg ,Hh ,CH in NBD,H2 O),
4.80〜4.84(m,1.0H,He ),
5.58〜5.88(m,1.95H,OH in CD),
6.84〜7.03(m,2.0H,CH in NBD),
7.34〜7.47(m,5.0H,aromatic)
β−CD0.34g(0.3mmol)、TBAB0.04g(5mol%)および水素化ナトリウム0.08g(3.1mmol)の混合物に、NMP6mLを添加し、室温で1時間攪拌した。その後、特定のノルボルナジエン誘導体0.85g(3.1mmol)を添加し、さらに50℃で48時間の条件で反応させた。
反応が終了した後、反応溶液を蒸留水に滴下し、沈殿物をろ別した後、良溶媒としてテトラヒドロフランを用い、貧溶媒としてジエチルエーテルを用いて2回再沈精製を行い、60℃で24時間減圧乾燥することにより、茶褐色の粉末固体0.24gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(VI)で表される化合物であって、β−CDにおける全水酸基に対するエーテル化率が33%であるものと同定された。収率は27%であった。この生成物を「シクロデキストリン誘導体(VI)」とする。
○IR(film,cm-1):
3394(νOH),
1698(νC=C ester ),
1615(νC=C NBD),
1593,1491(νC=C aromatic),
1333,1033(νC−O−C ester ),
1151,1078(νC−O−C ether )
○ 1H NMR(500MHz,DMSO−d6 )δ(ppm):
1.84〜2.23(m,2.0H,CH2 in NBD),
3.29〜4.75(m,19.3H,Ha ,Hb ,Hc ,Hd ,Hf ,Hg ,Hh ,CH in NBD,H2 O),
4.80〜4.84(m,1.0H,He ),
5.58〜5.88(m,1.95H,OH in CD),
6.84〜7.03(m,2.0H,CH in NBD),
7.34〜7.47(m,5.0H,aromatic)
γ−CD0.39g(0.3mmol)、TBAB0.06g(5mol%)および水素化ナトリウム0.09g(3.6mmol)の混合物に、NMP6mLを添加し、室温で1時間攪拌した。その後、特定のノルボルナジエン誘導体0.99g(3.6mmol)を添加し、さらに50℃で48時間の条件で反応させた。
反応が終了した後、反応溶液を蒸留水に滴下し、沈殿物をろ別した後、良溶媒としてテトラヒドロフランを用い、貧溶媒としてジエチルエーテルを用いて2回再沈精製を行い、60℃で24時間減圧乾燥することにより、茶褐色の粉末固体0.14gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(VII)で表される化合物であって、γ−CDにおける全水酸基に対するエーテル化率が36%であるものと同定された。収率は15%であった。この生成物を「シクロデキストリン誘導体(VII)」とする。
○IR(film,cm-1):
3394(νOH),
1711(νC=C ester ),
1615(νC=C NBD),
1596,1492(νC=C aromatic),
1331,1030(νC−O−C ester ),
1151,1080(νC−O−C ether )
○ 1H NMR(500MHz,DMSO−d6 )δ(ppm):
1.84〜2.23(m,2.0H,CH2 in NBD),
3.29〜4.75(m,10.9H,Ha ,Hb ,Hc ,Hd ,Hf ,Hg ,Hh ,CH in NBD,H2 O),
4.80〜4.84(m,1.0H,He ),
5.58〜5.88(m,1.75H,OH in CD),
6.84〜7.03(m,2.0H,CH in NBD),
7.34〜7.47(m,5.0H,aromatic)
(1)屈折率変化:
実施例1〜4に係るシクロデキストリン誘導体(I)〜シクロデキストリン誘導体(IV)および参考例1〜3に係るシクロデキストリン誘導体(V)〜シクロデキストリン誘導体(VII)の各々をシクロヘキサノンに溶解し、得られた溶液を、スピンコーターによってシリコンウエハーに塗布して乾燥処理することにより、厚みが約1.0mmの薄膜を形成した。得られた薄膜に対し、500Wキセノンランプを用いて30分間光照射し、エリプソメーターを用い、波長632.8nmのレーザー光により、光照射前後における屈折率を測定し、光照射前後における屈折率の変化量を求めた。
以上、結果を表2に示す。
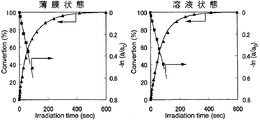
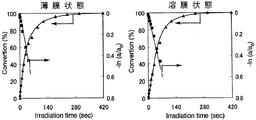
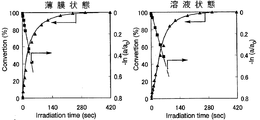
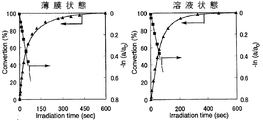
実施例1〜3に係るシクロデキストリン誘導体(I)〜シクロデキストリン誘導体(III)および参考例1〜3に係るシクロデキストリン誘導体(V)〜シクロデキストリン誘導体(VII)の各々をテトラヒドロフランに溶解し、得られた溶液の各々を、石英セルの内壁面に塗布し、室温で2時間減圧乾燥処理することにより、薄膜を形成した。石英セル内に形成された薄膜に対して、500Wキセノンランプ「UXL−500D−O」(ウシオ電機(株)製)および熱線カットフィルター「HA50」(HOYA(株)製)を用い、1.20mW/cm2 (313nm)の条件で、光照射時間を変えながら光照射処理を行うと共に、紫外分光光度計により、当該薄膜における紫外線の吸光度の変化を測定した。結果を、図1(シクロデキストリン誘導体(I)、図2(シクロデキストリン誘導体(II))、図3(シクロデキストリン誘導体(III))、図4(シクロデキストリン誘導体(V))、図5(シクロデキストリン誘導体(VI))および図6(シクロデキストリン誘導体(VII))に示す。
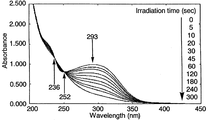
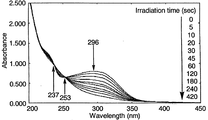
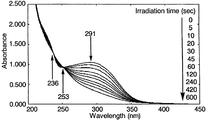
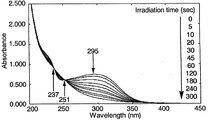
図2の結果から、シクロデキストリン誘導体(II)よりなる薄膜においては、NBD構造に基づく最大吸収波長296nmの紫外線の吸収が、光照射時間の経過に伴って減少することが確認され、また、波長237nmおよび波長253nmに等吸収点が確認されたことにより、NBD構造からこれに対応するQC構造への光異性化反応は、副反応が生じることなしに進行することが理解される。また、光異性化反応は、光照射時間が約7分間で完了することが確認された。
図3の結果から、シクロデキストリン誘導体(III)よりなる薄膜においては、NBD構造に基づく最大吸収波長291nmの紫外線の吸収が、光照射時間の経過に伴って減少することが確認され、また、波長236nmおよび波長253nmに等吸収点が確認されたことにより、NBD構造からこれに対応するQC構造への光異性化反応は、副反応が生じることなしに進行することが理解される。また、光異性化反応は、光照射時間が約10分間で完了することが確認された。
図4の結果から、シクロデキストリン誘導体(V)よりなる薄膜においては、NBD構造に基づく最大吸収波長295nmの紫外線の吸収が、光照射時間の経過に伴って減少することが確認され、また、波長237nmおよび251nmに等吸収点が確認されたことにより、NBD構造からこれに対応するQC構造への光異性化反応は、副反応が生じることなしに進行することが理解される。また、光異性化反応は、光照射時間が約5分間で完了することが確認された。
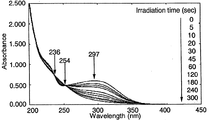
図5の結果から、シクロデキストリン誘導体(VI)よりなる薄膜においては、NBD構造に基づく最大吸収波長297nmの紫外線の吸収が、光照射時間の経過に伴って減少することが確認され、また、波長236nmおよび254nmに等吸収点が確認されたことにより、NBD構造からこれに対応するQC構造への光異性化反応は、副反応が生じることなしに進行することが理解される。また、光異性化反応は、光照射時間が約5分間で完了することが確認された。
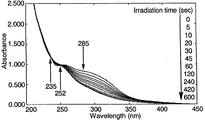
図6の結果から、シクロデキストリン誘導体(VI)よりなる薄膜においては、NBD構造に基づく最大吸収波長285nmの紫外線の吸収が、光照射時間の経過に伴って減少することが確認され、また、波長235nmおよび252nmに等吸収点が確認されたことにより、NBD構造からこれに対応するQC構造への光異性化反応は、副反応が生じることなしに進行することが理解される。また、光異性化反応は、光照射時間が約10分間で完了することが確認された。
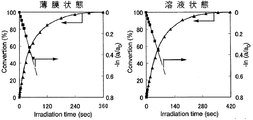
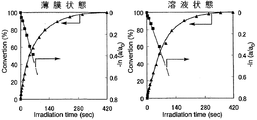
また、シクロデキストリン誘導体(I)〜シクロデキストリン誘導体(III)およびシクロデキストリン誘導体(V)〜シクロデキストリン誘導体(VII)の各々を、NBD残基の濃度が1×10-4mol/Lとなるようテトラヒドロフランに溶解した。得られた溶液の各々を石英セルに入れ、この溶液に対して、500Wキセノンランプおよび熱線カットフィルター「HA50」(HOYA(株)製)を用い、1.20mW/cm2 (313nm)の条件で、光照射時間を変えながら光照射処理を行うと共に、紫外線分光光度により、当該溶液における紫外線の吸光度の変化を測定し、これらのシクロデキストリンの各々の溶液状態における異性化反応率を一次速度式にプロットした。ここで、異性化反応率は、最大吸収波長における吸光度の変化から求めた。
以上の結果を、図7(シクロデキストリン誘導体(I))、図8(シクロデキストリン誘導体(II))、図9(シクロデキストリン誘導体(III))、図10(シクロデキストリン誘導体(V))、図11(シクロデキストリン誘導体(VI))および図12(シクロデキストリン誘導体(VII))に示す。
図7〜図12の結果から、シクロデキストリン誘導体(I)〜シクロデキストリン誘導体(III)およびシクロデキストリン誘導体(V)〜シクロデキストリン誘導体(VII)の各々における光異性化反応は、薄膜状態および溶液状態のいずれにおいても一次で進行していることが理解される。
α−CD9.72g(10mmol)を蒸留水70mLに溶解し、その後、PEG1.45g(0.91mmol)を添加し、超音波を用いて30分間攪拌し、その後、室温で24時間静置した。次いで、析出した固体をろ過によって回収し、蒸留水で洗浄した後、24時間減圧乾燥することにより、白色粉末固体6.7gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(X−1)で表される、PEGよりなる軸分子の周りに、α−CDよりなる環状分子が存在するプソイドロタキサンであると同定された。収率は45%であった。また、 1H−NMR分析の結果から、プソイドロタキサン1分子内に存在するα−CDの数を測定したところ、平均で11個であった。
○IR(film,cm-1):
3397(νOH),
1645(νN−H),
1149,1077,1024(νC−O−C),
861(νC−N)
○ 1H NMR(600MHz,DMSO−d6 )δ(ppm):
1.35〜1.64(m,4.00H,H11),
3.28〜3.30(m,67.2H,H4 ),
3.48〜3.51(m,197.1H,H10),
4.02〜4.45(m,39.4H,H5 ,H6 ,H12),
4.48〜4.51(m,65.7H,H3 ),
4.79〜4.80(m,65.6H,H2 ),
5.44〜5.52(m,128.3H,H1 ,H7 ,H8 ,H9 )
反応が終了した後、反応溶液にエーテルを加え、析出した固体をろ過により回収し、その後、良溶媒としてDMSOを用い、貧溶媒として蒸留水を用いて1回再沈精製を行い、減圧乾燥することにより、黄色の粉末固体0.32gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(X−2)で表される、両末端が2,4−ジニトロフェニル基が結合したPEGよりなる軸分子と、α−CDよりなる環状分子とよりなるロタキサンであると同定された。収率は16%であった。このロタキサンを「中間体A」とする。
○IR(film,cm-1):
3440(νOH),
1525,1389,1335(νN−O),
1491(νC=C aromatic),
1149,1077,1024(νC−O−C),
861(νC−N)
○ 1H NMR(600MHz,DMSO−d6 )δ(ppm):
2.80〜3.91(m,567.7H,H3 ,H4 ,H5 ,H6 ,H10,H11,H12),
4.19〜4.55(m,59.1H,H2 ),
4.56〜4.87(m,58.8H,H1 ),
5.39〜5.73(m,110.0H,H7 ,H8 ,H9 ),
7.74〜8.82(m,6.00H,H13,H14,H15)
以上において、中間体Aと特定のノルボルナジエン誘導体との使用割合は、中間体A中に存在するα−シクロデキストリンにおける全水酸基1モルに対して特定のノルボルナジエン誘導体1.8モルとなる割合である。
反応が終了した後、反応溶液を蒸留水に滴下し、沈殿物をろ別した後、分取HPLCによって単離精製し、60℃で24時間減圧乾燥することにより、粉末固体0.10gを得た。
IR分析および 1H−NMR分析の結果から、得られた生成物は、下記式(X)で表されるロタキサンであって、α−CDにおける全水酸基に対するエーテル化率が77%であるものと同定された。収率は5%であった。また、得られたロタキサンの分子量を、DMFを溶媒とするゲルパーミエーションクロマトグラフ法によって測定したところ、数平均分子量Mnが1.8×104 、重量平均分子量Mwに対する数平均分子量Mnの比Mn/Mwが1.38であった。また、このロタキサンの合成工程を下記反応式(X−3)に示す。
○IR(film,cm-1):
3440(νOH),
1704(νC=O ster),
1525,1389,1335(νN−O),
1594,1491(νC=C aromatic),
1331,1034(νC−O−C ster),
1235,1072(νC−O−C ether),
861(νC−N)
○ 1H NMR(600MHz,DMSO−d6 )δ(ppm):
1.75〜2.07(m,2.00H,H18),
2.80〜3.91(m,9.09H,H2 ,H3 ,H4 ,H5 ,H6 ,H10,H11,H12),
4.88〜4.89(m,0.30H,H1 ),
5.39〜5.73(m,0.29H,H7 ,H8 ,H9 ),
6.96〜7.05(m,2.00H,H21,H22),
7.74〜8.82(m,5.14H,H13,H14,H15 aromatic in NBD)
Claims (10)
- 下記一般式(1)で表されるシクロデキストリン誘導体であって、シクロデキストリンにおける全水酸基に対するエーテル化率が60%以上であることを特徴とするシクロデキストリン誘導体。
- シクロデキストリンと、3−フェニル−2,5−ノルボルナジエン−2−カルボキシシクロクロロエチルとを、それぞれの使用割合がシクロデキストリンにおける全水酸基1モルに対して3−フェニル−2,5−ノルボルナジエン−2−カルボキシシクロクロロエチルが0.7モル以上となる条件で反応させることにより、請求項1に記載のシクロデキストリン誘導体を得ることを特徴とするシクロデキストリン誘導体の製造方法。
- 環状分子として、請求項1に記載のシクロデキストリン誘導体を有することを特徴とするロタキサン。
- 軸分子として、両末端にジニトロフェニル基を有する高分子を有することを特徴とする請求項3に記載のロタキサン。
- 下記一般式(2)で表される化合物であることを特徴とする請求項4に記載のロタキサン。
- 環状分子としてシクロデキストリンを有するロタキサンよりなる中間体を合成し、この中間体と、3−フェニル−2,5−ノルボルナジエン−2−カルボキシシクロクロロエチルとを、それぞれの使用割合が中間体のシクロデキストリンにおける全水酸基1モルに対して3−フェニル−2,5−ノルボルナジエン−2−カルボキシシクロクロロエチルが0.7モル以上となる条件で反応させることにより、請求項3乃至請求項5のいずれかに記載のロタキサンを得ることを特徴とするロタキサンの製造方法。
- 請求項1に記載のシクロデキストリン誘導体よりなることを特徴とする屈折率変換材料。
- 請求項3乃至請求項5のいずれかに記載のロタキサンよりなることを特徴とする屈折率変換材料。
- 請求項1に記載のシクロデキストリン誘導体よりなることを特徴とする光−熱エネルギー変換蓄積材料。
- 請求項3乃至請求項5のいずれかに記載のロタキサンよりなることを特徴とする光−熱エネルギー変換蓄積材料。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004115330A JP4593157B2 (ja) | 2004-04-09 | 2004-04-09 | 屈折率変換材料 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004115330A JP4593157B2 (ja) | 2004-04-09 | 2004-04-09 | 屈折率変換材料 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2005298631A true JP2005298631A (ja) | 2005-10-27 |
| JP4593157B2 JP4593157B2 (ja) | 2010-12-08 |
Family
ID=35330569
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004115330A Expired - Fee Related JP4593157B2 (ja) | 2004-04-09 | 2004-04-09 | 屈折率変換材料 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4593157B2 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007026578A1 (ja) * | 2005-08-31 | 2007-03-08 | Nissan Motor Co., Ltd. | 疎水性修飾ポリロタキサン及び架橋ポリロタキサン |
| JP2007254570A (ja) * | 2006-03-23 | 2007-10-04 | Lintec Corp | 擬ポリロタキサンおよびポリロタキサンの製造方法 |
| JP2009270120A (ja) * | 2005-08-31 | 2009-11-19 | Nissan Motor Co Ltd | 疎水性修飾ポリロタキサン含有溶液 |
| CN116217573A (zh) * | 2023-02-24 | 2023-06-06 | 南开大学 | 一种垂直单分子膜场效应控制开关及其制备方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003306470A (ja) * | 2002-04-17 | 2003-10-28 | Univ Kanagawa | カリックスアレーン誘導体およびその製造方法、シクロデキストリン誘導体およびその製造方法並びに屈折率変換材料および光−熱エネルギー変換蓄積材料 |
-
2004
- 2004-04-09 JP JP2004115330A patent/JP4593157B2/ja not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003306470A (ja) * | 2002-04-17 | 2003-10-28 | Univ Kanagawa | カリックスアレーン誘導体およびその製造方法、シクロデキストリン誘導体およびその製造方法並びに屈折率変換材料および光−熱エネルギー変換蓄積材料 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007026578A1 (ja) * | 2005-08-31 | 2007-03-08 | Nissan Motor Co., Ltd. | 疎水性修飾ポリロタキサン及び架橋ポリロタキサン |
| JP2007091938A (ja) * | 2005-08-31 | 2007-04-12 | Nissan Motor Co Ltd | 疎水性修飾ポリロタキサン及び架橋ポリロタキサン |
| JP2009270120A (ja) * | 2005-08-31 | 2009-11-19 | Nissan Motor Co Ltd | 疎水性修飾ポリロタキサン含有溶液 |
| US7943718B2 (en) | 2005-08-31 | 2011-05-17 | Nissan Motor Co., Ltd. | Hydrophobic modified polyrotaxane and crosslinked polyrotaxane |
| JP2007254570A (ja) * | 2006-03-23 | 2007-10-04 | Lintec Corp | 擬ポリロタキサンおよびポリロタキサンの製造方法 |
| CN116217573A (zh) * | 2023-02-24 | 2023-06-06 | 南开大学 | 一种垂直单分子膜场效应控制开关及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP4593157B2 (ja) | 2010-12-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Nishikubo et al. | Successful synthesis of polymers containing pendant norbornadiene moieties and norbornadiene photochemical valence isomerization | |
| JP3643583B2 (ja) | フォトクロミック蛍光重合体およびその製造方法 | |
| Aguirresarobe et al. | UV-light responsive waterborne polyurethane based on coumarin: synthesis and kinetics of reversible chain extension | |
| JP4593157B2 (ja) | 屈折率変換材料 | |
| CN113402536A (zh) | 一种卟啉桥联双bodipy衍生物及其制备方法 | |
| Griesser et al. | Refractive index modulation in polymers bearing photoreactive phenyl and naphthyl ester units using different UV wavelengths | |
| Balaji et al. | Studies on photocrosslinkable polymers having bromo-substituted pendant cinnamoyl group | |
| JP3989281B2 (ja) | カリックスアレーン誘導体およびその製造方法並びに屈折率変換材料および光−熱エネルギー変換蓄積材料 | |
| Sidharthan et al. | Synthesis and characterization of photo-crosslinkable liquid crystalline copolyesters containing arylidene-keto and chalcone moieties | |
| Kawashima et al. | Synthesis and photochemical reaction of polystyrenes with pendant donor–acceptor type norbornadienes containing carbamoyl chromophores | |
| Kudo et al. | New large refractive-index change materials: synthesis and photochemical valence isomerization of the calixarene derivatives containing norbornadiene moieties | |
| JP5481017B2 (ja) | ラダーポリマー誘導体の製造方法 | |
| CN113024540B (zh) | 一种D-π-A结构非线性化合物的制备方法及应用 | |
| Zhang et al. | Synthesis and characterization of dendrons and dendrimers skeleton-constructed with azobenzene moiety | |
| JP4669720B2 (ja) | カリックスレゾルシンアレーン誘導体およびその製造方法 | |
| Perala et al. | Orthogonally clickable hyperbranched polymers: effect of reactant size and polarity on core-functionalization of peripherally jacketed HBPs | |
| JP2008222854A (ja) | ノボラック誘導体およびその製造方法並びに屈折率変換材料 | |
| JP4634834B2 (ja) | シルセスキオキサン重合体並びに屈折率変換材料および光−熱エネルギー変換蓄積材料 | |
| JP2004262822A (ja) | カリックスレゾルシンアレーン誘導体およびその製造方法並びに屈折率変換材料および光−熱エネルギー変換蓄積材料 | |
| Kudo et al. | Refractive-index changes of thin films of photo-reactive α-, β-, and γ-cyclodextrin derivatives upon photo-irradiation | |
| JP2007238665A (ja) | シクロデキストリン誘導体およびその製造方法ならびに光硬化性組成物 | |
| JP2010084123A (ja) | 不飽和脂環式ポリカルボナート及びその製造方法 | |
| Wang et al. | Synthesis and Properties of a Photoresponsive Azobenzene-Containing Hyperbranched Polymer. | |
| US9834723B2 (en) | Pentaarylbiimidazole compound and production method for said compound | |
| JP4166201B2 (ja) | 車輪状マルチポルフィリンデンドリマー化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20061023 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100622 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100818 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100907 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100915 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130924 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4593157 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |