JP2004508064A - コドン最適化hiv1−gag、pol、nefおよび修飾体を発現する増強された第1世代アデノウイルスワクチン - Google Patents
コドン最適化hiv1−gag、pol、nefおよび修飾体を発現する増強された第1世代アデノウイルスワクチン Download PDFInfo
- Publication number
- JP2004508064A JP2004508064A JP2002526335A JP2002526335A JP2004508064A JP 2004508064 A JP2004508064 A JP 2004508064A JP 2002526335 A JP2002526335 A JP 2002526335A JP 2002526335 A JP2002526335 A JP 2002526335A JP 2004508064 A JP2004508064 A JP 2004508064A
- Authority
- JP
- Japan
- Prior art keywords
- adenovirus
- vector
- hiv
- nef
- seq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images














































































































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/525—Virus
- A61K2039/5256—Virus expressing foreign proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10341—Use of virus, viral particle or viral elements as a vector
- C12N2710/10343—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10351—Methods of production or purification of viral material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16111—Human Immunodeficiency Virus, HIV concerning HIV env
- C12N2740/16134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16311—Human Immunodeficiency Virus, HIV concerning HIV regulatory proteins
- C12N2740/16334—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/42—Vector systems having a special element relevant for transcription being an intron or intervening sequence for splicing and/or stability of RNA
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Virology (AREA)
- General Health & Medical Sciences (AREA)
- Zoology (AREA)
- Medicinal Chemistry (AREA)
- Immunology (AREA)
- Wood Science & Technology (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Biomedical Technology (AREA)
- Biophysics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Microbiology (AREA)
- Veterinary Medicine (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Oncology (AREA)
- Gastroenterology & Hepatology (AREA)
- Communicable Diseases (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
増強された安定性および増殖特性ならびにより大きな細胞性免疫を示す第1世代アデノウイルスベクターおよび関連した組換えアデノウイルスに基づくHIVワクチンを本明細書に記載する。これらのアデノウイルスベクターは、細胞培養により、HIV−1 gag、HIV−1 polおよび/またはHIV−1 nefポリヌクレオチド医薬品およびそれらの生物学的に関連した修飾体を含有する種々のアデノウイルスに基づくHIV−1ワクチンを作製および製造するために使用される。これらのアデノウイルスワクチンは、生きた脊椎動物組織、好ましくは哺乳動物宿主(例えばヒト、または商業的もしくは家畜獣医学的に重要な非ヒト哺乳類)内に直接導入されると、HIV1−Gag、Polおよび/またはNefタンパク質あるいはそれらの生物学的な修飾体を発現して、HIV−1を特異的に認識する細胞性免疫応答を誘導する。例示されている本発明のポリヌクレオチドは、HIV−1 Gag、コード化コドン最適化HIV−1 Pol、最適化HIV−1 Polの誘導体(HIV−1 Polのプロテアーゼ、逆転写酵素、RNアーゼHおよびインテグラーゼ活性が不活性化された構築物を含む)、HIV−1 Nefおよび最適化HIV−1 Nefの誘導体(宿主CD4のダウンレギュレーションおよびミリスチル化のようなNefの野生型特性に影響を及ぼすnef突然変異体を含む)をコードする合成DNA分子である。本発明のアデノウイルスワクチンは、単独または組合せ様式法で投与されると、これまでに未感染の個体に対して予防的利点をもたらし、および/または感染個体内のウイルス負荷レベルを減少させてHIV−1感染の無症候期を延長することにより治療効果をもたらすであろう。
Description
【0001】
(発明の分野)
本発明は、他の複製欠損型ベクターと比較して増強した増殖特性およびより大きな細胞性免疫を示すことが見出された組換え複製欠損型第1世代アデノウイルスワクチンに関する。本発明はまた、追加的な5’アデノウイルス配列の取り込みにより、本明細書に記載の組換え複製欠損型アデノウイルスの大規模生産効率を増加させる本明細書に記載の関連第1世代アデノウイルスベクターに関する。本発明のもう1つの態様は、ヒトサイトメガロウイルス(hCMV)プロモーターのイントロンA部分がアデノウイルスベクター構築物における不安定性の領域を構成するという驚くべき知見にある。アデノウイルス発現構築物からこの領域を除去すると、著しく改善されたベクター安定性が得られる。したがって、イントロンA欠失CMVプロモーターの制御下でトランスジーンを発現する改良されたベクターは、本発明のもう1つの態様を構成する。これらのアデノウイルスベクターは、ヒト免疫不全ウイルス(HIV)に対する組換えアデノウイルスワクチンの製造に有用である。特に、本明細書に記載の第1世代アデノウイルスベクターは、HIV−1 Gag、HIV−1 Polおよび/またはHIV−1 Nefポリヌクレオチド医薬品ならびにそれらの生物学的に活性な修飾体を含有する、アデノウイルスに基づくHIV−1ワクチンを構築し生成するために使用される。本明細書に記載の組換え複製欠損型アデノウイルスワクチンの宿主投与は、HIV−Gag、HIV−1−Polおよび/またはNefタンパク質またはそれらの免疫学的に関連した修飾体の発現をもたらして、HIV−1を特異的に認識する細胞性免疫応答を誘導する。例示されている本発明のポリヌクレオチドは、コドン最適化HIV−1 Gag、HIV−1 Pol、最適化HIV−1 Polの誘導体(HIV−1 Polのプロテアーゼ、逆転写酵素、RNアーゼHおよびインテグラーゼ活性が不活性化された構築物を含む)、HIV−1 Nef、および最適化HIV−1 Nefの誘導体(宿主CD4のダウンレギュレーションおよびミリスチル化のようなNefの野生型特性に影響を及ぼすnef突然変異体を含む)をコードする合成DNA分子である。本発明のHIVアデノウイルスワクチンは、単独で或いは組合せ様式および/または初回抗原刺激/追加抗原刺激法で投与されると、これまでに未感染の個体に予防上の利点をもたらし、および/または、感染個体内のウイルス負荷レベルを減少させてHIV−1感染の無症候期を延長させることにより治療効果をもたらすであろう。
【0002】
(発明の背景)
ヒト免疫不全ウイルス−1(HIV−1)は後天性ヒト免疫不全症候群(エイズ)および関連障害の病原体である。HIV−1はレトロウイルス科のRNAウイルスであり、すべてのレトロウイルスの5’−LTR−gag−pol−env−LTR3’体制を示す。プロウイルスとして公知の組込み形態のHIV−1は約9.8Kb長である。該ウイルスゲノムの各末端は、長末端反復配列(LTR)として公知のフランキング配列を含有する。該HIV遺伝子は少なくとも9個のタンパク質をコードし、それらは主要構造タンパク質(Gag、PolおよびEnv)、調節タンパク質(TatおよびRev)および補助タンパク質(Vpu、Vpr、VifおよびNef)の3つのクラスに分けられる。
【0003】
gag遺伝子は55キロダルトン(kDa)の前駆体タンパク質(p55)をコードしており、これは、スプライスされていないウイルスmRNAから発現され、HIVプロテアーゼ(pol遺伝子の産物)によるタンパク質分解によりプロセシングされる。成熟p55タンパク質産物はp17(マトリックス)、p24(カプシド)、p9(ヌクレオカプシド)およびp6である。
【0004】
pol遺伝子は、ウイルスの複製に必要なタンパク質、すなわち、逆転写酵素、プロテアーゼ、インテグラーゼおよびRNアーゼHをコードしている。これらのウイルスタンパク質は、リボソームフレームシフトにより産生される160kDaの前駆体タンパク質であるGag−Pol融合タンパク質として発現される。該ウイルスコード化プロテアーゼは、タンパク質分解により該Gag−Pol融合体からPolポリペプチドを切り離し、さらに該Polポリペプチドを成熟タンパク質にまで切断し、該成熟タンパク質はプロテアーゼ(Pro、P10)、逆転写酵素(RT、P50)、インテグラーゼ(IN、p31)およびRNアーゼH(RNアーゼ、p15)活性をもたらす。
【0005】
nef遺伝子は、CD4発現のダウンレギュレーション、T細胞活性化の阻害およびHIV感染性の促進のような幾つかの活性を有することが示されている初期補助(アクセサリー)HIVタンパク質(Nef)をコードする。
【0006】
env遺伝子は、ウイルスエンベロープ糖タンパク質をコードしており、該糖タンパク質は、160キロダルトン(kDa)の前駆体(gp160)として翻訳され、ついで細胞プロテアーゼにより切断されて、外部120kDaエンベロープ糖タンパク質(gp120)および膜貫通41kDaエンベロープ糖タンパク質(gp41)を与える。Gp120およびgp41は会合したままであり、ウイルス粒子上およびHIV感染細胞表面上に提示される。
【0007】
tat遺伝子は、HIV−1の複製に必須の転写トランスアクチベーターであるRNA結合タンパク質であるTatタンパク質の長い形態および短い形態をコードしている。
【0008】
rev遺伝子は、RNA結合タンパク質である13kDaのRevタンパク質をコードしている。Revタンパク質は、Rev応答配列(RRE)と称されるウイルスRNAの領域に結合する。Revタンパク質は、核から細胞質への、スプライスされていないウイルスRNAの輸送を促進する。Revタンパク質は、HIV後期遺伝子発現に要求され、そしてHIVの複製に要求される。
【0009】
Gp120は、他の補助受容体(コレセプター)分子に加えて、ヘルパーTリンパ球、マクロファージおよび他の標的細胞の表面上に存在するCD4/ケモカイン受容体に結合する。X4(マクロファージ指向性)ウイルスはCD4/CXCR4複合体に対する指向性を示し、R5(T細胞系指向性)ウイルスはCD4/CCR5受容体複合体と相互作用する。gp120がCD4に結合した後、gp41が、ウイルスの侵入をもたらす融合事象を媒介する。該ウイルスは該標的細胞と融合しそれに侵入し、ついでRNA依存性DNAポリメラーゼにより、その一本鎖RNAゲノムから二本鎖DNAへの逆転写が生じる。プロウイルスとして公知の該ウイルスDNAは細胞核に侵入し、そこで、該ウイルスDNAは、該核内での新たなウイルスRNAの産生、初期および後期HIVウイルスタンパク質の発現、ならびにそれに続く新たなウイルス粒子の産生および細胞放出を指令する。該一次感染は、該ウイルスの非常に高い産生および組織分布ならびにそれに続くウイルスの定常状態レベル(ただし、これはこの相における連続的なウイルス産生および代謝回転によるものである)を与え、最終的には、臨床的エイズ発症を招く更なるウイルス負荷の放出をもたらすことが、宿主内のウイルス負荷の検出能における最近の進歩から示されている。生産的感染細胞は数日間の半減期を有し、一方、慢性または潜伏感染細胞は3週間の半減期を有し、それに続く非生産的感染細胞は、長い半減期(100日以上)を有するが、疾患経過の全体にわたり認められる日々のウイルス負荷に有意には寄与しない。
【0010】
免疫防御に決定的に重要なCD4ヘルパーTリンパ球の破壊が、HIV感染の特徴である進行性免疫機能不全の主要原因である。CD4 T細胞の喪失は、ほとんどの侵入体と戦う身体能力を著しく損なうが、それは、ウイルス、真菌、寄生生物および或る細菌(マイコバクテリアを含む)に対する防御に対して特に深刻な影響を及ぼす。
【0011】
最近、HIV−1感染個体に対する有効な治療法が利用可能となった。しかし、これらの薬物は、世界の多数の地域においては該疾患に対して有意な影響を及ぼさず、それらは、ヒト集団内の感染の広がりの阻止において最低限の影響しか及ぼさないであろう。多数の他の感染症の場合と同様、HIV−1感染の広がりに対する有意な疫学的効果は、有効なワクチンの開発および導入の後でしか見出されないであろう。現在までにワクチン開発が成功していないのには、多数の要因が関与している。前記のとおり、慢性感染者においては、抗HIV−1体液性および細胞性免疫応答の存在ならびにウイルス感染細胞の破壊にもかかわらず、絶え間ないウイルス産生が生じている。他の感染症の場合と同様に、疾患の結末は、該免疫応答の速度論および規模と、該病原体複製速度および該免疫応答による影響の受け易さとの平衡がどのようになるかによって決まる。急性感染における既存免疫は、確立された感染における生成免疫応答より有効かもしれない。第2の要因は、該ウイルスの際立った遺伝的可変性である。細胞培養内ではHIV−1感染性を中和しうる抗HIV−1抗体が存在するが、これらの抗体は、一般には、それらの活性において、ウイルス分離体に特異的である。伝統的な方法を用いてHIV−1の血清学的分類を定義することは不可能であることが判明している。むしろ、該ウイルスは血清学的「連続体(continuum)」を特徴づけるようであり、そのため、個々の中和抗体応答は、せいぜい、少数のウイルス変異体に対して有効であるに過ぎない。この後者の見解を考慮すると、抗HIV−1細胞性免疫応答を惹起しうる免疫原および関連送達技術を同定することが有用であろう。CTL応答を得るためには、抗原が細胞内で合成されるか又は細胞内に導入され、ついでプロテアソーム複合体により、小さなペプチドにプロセシングされ、主要組織適合遺伝子複合体(MHC)クラスIタンパク質との最終的会合のために小胞体/ゴルジ複合体分泌経路に輸送されなければならないことが公知である。CD8+ Tリンパ球は、T細胞受容体(TCR)およびCD8細胞表面タンパク質を介して、MHCクラスIと会合した抗原を認識する。活性化エフェクターまたは記憶細胞へのナイーブCD8+ T細胞の活性化は、一般には、前記のとおりの抗原のTCR関与および共刺激タンパク質の関与の両方を要する。CTL応答の最適な誘導は、通常、TCRおよびCD4の関与を介してMHCクラスII分子と会合した抗原を認識するCD4+ Tリンパ球からのサイトカインの形態での「援助」を要する。
【0012】
欧州特許出願第0 638 316号(1995年2月15日付け公開)および第0 586 076号(1994年3月9日付け公開)(共にAmerican Home Products Corporationに譲渡されている)は、envまたはgagを含むHIV遺伝子を保持する複製型アデノウイルスベクターを記載している。チンパンジーおよびイヌでの種々の治療計画が用いられ、それらのいくつかはブースターアデノウイルスまたはタンパク質+ミョウバンでの治療を含むものであった。
【0013】
E1領域内に欠失を保持する複製欠損型アデノウイルスベクターが公知であり、最近のアデノウイルスベクターは、これらのベクター内に公知パッケージング反復配列を組込んでいる。例えば、とりわけ、塩基対459〜3328のE1配列において欠失したアデノウイルスベクターを開示しているEP 0707071、および、とりわけ、塩基対459〜3510が欠失したアデノウイルスベクターを開示している米国特許第6,033,908号を参照されたい。アデノウイルスのパッケージング効率は、組込まれる個々のA(パッケージング)反復配列の数に左右されると教示されている。例えば、GrableおよびHearing,1990 J.Virol.64(5):2047−2056;GrableおよびHearing,1992 J.Virol.66(2):723−731を参照されたい。
【0014】
Larderら(1987,Nature 327:716−717)およびLarderら(1989,Proc.Natl.Acad.Sci.86:4803−4807)は、HIV−1 RTの部位特異的突然変異誘発を開示しており、また、そのような変化が、RTの公知インヒビターとの相互作用に関連したインビトロ活性および感染性に及ぼす影響を開示している。
【0015】
Daviesら(1991,Science 252:,88−95)は、HIV−1 PolのRNアーゼHドメインの結晶構造を開示している。
【0016】
Schatzら(1989,FEBS Lett.257−311−314)は、完全なHIV−1 RT/RNアーゼH DNA断片内の突然変異Glu478GlnおよびHis539Pheが、RT活性に影響を及ぼすことなく欠損RNアーゼ活性を与えることを開示している。
【0017】
Mizrahiら(1990,Nucl.Acids.Res.18:pp.5359−5353)は、同様に欠損RNアーゼ活性を与えるpol遺伝子のRNアーゼ領域内の追加的突然変異Asp443AsnおよびAsp498Asnを開示している。該著者は、Asp498Asn突然変異体の特徴づけが、この突然変異体タンパク質の不安定性のために困難であったことを記載している。
【0018】
Leavittら(1993,J.Biol.Chem.268:2113−2119)は、HIV−1インテグラーゼ(IN)活性に対して異なる効果を示すAsp64Val突然変異を含む幾つかの突然変異を開示している。
【0019】
Wiskerchenら(1995,J.Virol.69:376−386)は、HIV−1 INおよびウイルス複製機能に影響を及ぼすアスパラギン酸残基の突然変異を含む一重および二重突然変異体を開示している。
【0020】
HIV感染に対して強力な細胞性免疫応答を産生する予防および/または治療に基づくHIVワクチンを得ることが、エイズとの闘いにおいて非常に重要であろう。本発明は、宿主に投与されるとHIV−1遺伝子gag、polおよびnefのコドン最適化および修飾形態を発現するアデノウイルスワクチンのクラスを開示することにより、この要求を検討し、それを満足させるものである。これらの複製欠損型組換えアデノウイルスワクチンは、単独で、あるいは本発明の成分および/または別個のウイルスHIV DNAワクチン、非ウイルス性HIV DNAワクチン、HIVサブユニットワクチン、HIV全不活化ワクチンおよび/または生弱毒化HIVワクチンでの初回抗原刺激ワクチン接種法および/または組合せ様式の一部として、ヒトのような宿主に投与することができる。
【0021】
(発明の概要)
本発明は、HIV−1 Gag、HIV−1 PolおよびHIV−1 Nefの免疫学的に関連した修飾体を含む種々の形態のHIV−1 Gag、HIV−1 Polおよび/またはHIV−1 Nefをコードする増強された複製欠損型組換えアデノウイルスワクチンベクターおよび関連組換え複製欠損型アデノウイルスワクチンに関する。本発明のアデノウイルスワクチンはHIV抗原を発現し、宿主投与に際して、改善された細胞性免疫応答をもたらす。潜在的なワクチン被接種体には、霊長類、特にヒトおよび非ヒト霊長類が含まれ、また、商業的または家畜獣医学的に重要な任意の非ヒト哺乳動物が含まれるが、これらに限定されるものではない。本発明の改良された組換えアデノウイルスに基づくワクチンの効果は、これまでに未感染の個体に対する、より低い伝染率(すなわち、予防的用途)、および/または、感染個体内のウイルス負荷のレベルの減少によるHIV−1感染の無症候期の延長(すなわち、治療的用途)でなければならない。特に、本発明は、種々の形態のコドン最適化HIV−1 Gag(p55形態のコドン最適化完全長(FL)GagおよびtPA−Gag融合タンパク質を含むが、何らこれらに限定されるものではない)、HIV−1 Pol、HIV−1 Nefおよび免疫学的に関連のある選択された修飾体をコードする、アデノウイルスに基づくワクチンに関する。これらのアデノウイルスワクチンの投与、細胞内運搬および発現は、宿主CTLおよびTh応答を惹起する。好ましい複製欠損型組換えアデノウイルスワクチンベクターには、(1)コドン最適化形態の野生型HIV−1 Gagをコードする、(2)コドン最適化形態のHIV−1 Polをコードする、(3)コドン最適化形態のHIV−1 Pol融合タンパク質をコードする、(4)コドン最適化形態の修飾HIV−1 Polタンパク質および融合タンパク質、例えば宿主細胞内でRT、RNアーゼおよびIN活性をもたらす触媒領域内の残基を含むpol修飾体(これらに限定されるものではない)をコードする、(5)コドン最適化形態の野生型HIV−1 Nefをコードする、(6)コドン最適化形態のHIV−1 Nef融合タンパク質をコードする、および/または(7)コドン最適化形態のHIV−1 Nef誘導体、例えばアミノ末端リーダー配列の導入、アミノ末端ミリスチル化部位の除去および/またはジロイシンモチーフ突然変異の導入を含むnef修飾体(これらに限定されるものではない)をコードする合成DNA分子が含まれるが、これらに限定されるものではない。アデノウイルスに基づくベクターワクチンから発現される本明細書に開示するNefに基づく融合体および修飾タンパク質は、宿主MHC I複合体に適切に提示されて宿主CTLおよびTh応答を惹起する能力を保有する一方、改変された輸送(トラフィッキング)および/または宿主細胞機能を有しうる。HIV−1 Gag、Polおよび/またはNef融合タンパク質の具体例には、該ウイルス抗原コード領域のNH2末端部分でのリーダーまたはシグナルペプチドの融合体が含まれるが、これらに限定されるものではない。そのようなリーダーペプチドには、tPAリーダーペプチドが含まれるが、これに限定されるものではない。
【0022】
本発明のHIV−1 Gag、HIV−1 Polおよび/またはHIV−1 Nefに基づくワクチンの構築において使用するアデノウイルスベクターは、該組換えウイルスの大規模な製造および精製にわたる該組換えアデノウイルスゲノムの遺伝的安定性の増強をもたらす任意の複製欠損型アデノウイルスベクターを含みうる。すなわち、本発明のHIV−1 Gag、PolまたはNefに基づくアデノウイルスワクチンは、細胞培養における多数の継代にわたり遺伝的に安定であり従って大規模な製造および精製操作中に維持されることが示される精製された組換え複製欠損型アデノウイルスである。そのような組換えアデノウイルスベクターおよび回収されたアデノウイルスワクチンは、有効な単価または多価HIVワクチンに要求される大規模用量の充填およびそれに続く世界的な流通に有用である。本発明は、この基本要件を満足し、野生型アデノウイルスゲノムの塩基対約1から塩基対約342から(より好ましくは400から)塩基対約458までの間の野生型アデノウイルス・シス作用性パッケージング領域を含みE1において少なくとも部分的に欠失した複製欠損型アデノウイルスベクターおよびそれに由来するベクターとして記載される。本発明の好ましい実施形態は、野生型アデノウイルスの塩基対1〜450を含む。他の好ましい実施形態においては、該複製欠損型アデノウイルスベクターは、それに加えて、塩基対3511〜3523を含むE1欠失領域の3’側の領域を有する。塩基対342〜450(より詳しくは400〜450)は、ウイルス抗原、特にHIV抗原を保持する既に開示されているベクターの5’領域の伸長を構成する(例えば、1999年7月6日および1999年8月13日付け出願のそれぞれ米国仮特許出願第60/142,631号および第60/148,981号に基づき優先権を主張している2001年1月11日付け公開のPCT国際出願PCT/US00/18332(WO 01/02067)を参照されたい;これらの文書を参照により本明細書に組み入れることとする)。本出願人は、該5’領域を該開示ワクチン内のE1遺伝子内に更に伸長させると該ウイルスのパッケージングの最適化に重要であることが見い出された要素が組込まれること、を見出した。
【0023】
野生型アデノウイルスゲノムの塩基対約1からと塩基対約342(より好ましくは400)から塩基対約458との間までを含まない従来のベクターと比較して、前記領域を含むベクターは、より強力なウイルス効果である約5〜10倍大きな増幅速度を伴う増強された増殖特性を示して、より低いウイルス使用量で同等の免疫生成を可能にし、この領域(塩基対1〜450)を含まない複製欠損型ベクターより大きな細胞性免疫応答を可能にする。より一層重要なことは、それに由来するアデノウイルス構築物、特に、イントロンAを欠くhCMVプロモーターの制御下で発現カセットを含むものは、大規模生産において遺伝的に非常に安定である。本出願人は、驚くべきことに、hCMVプロモーターのイントロンA部分が、アデノウイルスベクター中で使用された場合に不安定性の領域を構成することを見出しており、前記の安定性はこれに基づくものである。したがって、本出願人は、遺伝子治療における使用に特に適した増強されたアデノウイルスベクター、および好ましいことに大規模増殖に有用な、ヌクレオチドに基づくワクチンベクターを特定した。
【0024】
本発明の好ましい実施形態は、(a)タンパク質または生物学的に活性な及び/又はその免疫学的に関連した部分をコードする核酸と、(b)a)部の核酸に作動的に結合した異種プロモーターと、(c)転写ターミネーターと、を含む遺伝子発現カセットの形態で該遺伝子が挿入されている、前記のとおりの複製欠損型アデノウイルスベクターである。
【0025】
好ましい実施形態においては、塩基対1〜450内に含有されているもの以外または塩基対1〜450および3511〜3523内に含有されているもの以外のE1遺伝子が該アデノウイルスベクターから欠失しており、該欠失E1遺伝子は該遺伝子発現カセットで置換されている。他の好ましい実施形態においては、該複製欠損アデノウイルスゲノムは機能的E3遺伝子を有さず、あるいは該E3遺伝子が欠失している。最も好ましくは、該E3領域は該アデノウイルスゲノム内に存在する。さらに好ましい実施形態においては、該遺伝子発現カセットは、E1に逆平行(該ベクターバックボーンに対して3’から5’への方向で転写される)な配向、より好ましくは、E1に平行(該ベクターバックボーンに対して5’から3’への方向で転写される)な配向で存在する。
【0026】
さらなる実施形態は、アデノウイルス部分とプラスミド部分とを含むシャトルプラスミドベクターに関する。ここで、該アデノウイルス部分は、a)野生型アデノウイルスゲノムの塩基対約1からと塩基対約342(より好ましくは400)から塩基対約458までの間との野生型アデノウイルス・シス作用性パッケージング領域(好ましくは1〜450)および好ましくはそれに加えて野生型アデノウイルス配列の塩基対3511〜3523を含みE1において少なくとも部分的に欠失した複製欠損型アデノウイルスゲノム、ならびにb)(a)タンパク質またはその生物学的に活性な及び/又は免疫学的に関連した部分をコードする核酸と、(b)a)部の核酸に作動的に結合した異種プロモーターと、(c)転写終結および/またはポリアデニル化部位と、を含む遺伝子発現カセットを含む。
【0027】
本発明の他の態様は、該アデノウイルスベクターおよび/または該シャトルプラスミドベクターを含む宿主細胞;該ベクターを含むワクチン組成物;および(a)アデノウイルスE1タンパク質を発現する宿主細胞内に該アデノウイルスベクターを導入し、(b)得られたアデノウイルスベクターを回収することを含む該ベクターの製造方法を含む。
【0028】
この目的において、本発明は特に、本明細書に記載のHIV−1抗原を含む(これらに限定されるものではない)付随トランスジーを伴う又は伴わないMRKAd5ベクターバックボーンのいずれかに関連した回収されたウイルスを含む(これらに限定されるものではない)、293細胞またはPER.C6(登録商標)(これらに限定されるものではない)のような宿主細胞に由来する回収された組換え複製欠損型ウイルスに関する。HIV−1ワクチンは、本明細書に開示するHIV−1抗原の任意の1以上を発現する任意の回収された組換えアデノウイルス物質により表される。ついで、この回収された物質は、宿主投与の前に精製され製剤化され保存されうる。
【0029】
本発明のもう1つの態様は、タンパク質に対する細胞性免疫応答を個体において生成させる方法であって、
a)塩基対約1からと塩基対約342(より好ましくは400)から塩基対約458まで(好ましくは1〜450)の間との野生型アデノウイルス・シス作用性アデノウイルスパッケージング領域および好ましくはそれに加えて野生型アデノウイルス配列の塩基対3511〜3523を含む、E1において少なくとも部分的に欠失した組換え複製欠損型アデノウイルベクター、ならびに
b)(i)タンパク質またはその生物学的に活性な及び/又は免疫学的に関連した部分をコードする核酸と、(ii)a)部の核酸に作動的に結合した異種プロモーターと、(iii)転写ターミネーターおよび/またはポリアデニル化部位とを含む遺伝子発現カセットを含むアデノウイルスワクチンベクターを該個体に投与することを含んでなる方法である。
【0030】
本明細書に記載のアデノウイルスおよび/またはDNAプラスミドワクチンの有効な性質を考慮して、本発明は、有効な免疫予防を得るための、またはHIV−1ウイルスに対する曝露後のHIV−1感染の確立を妨げるため、または急性HIV−1感染を軽減して、有益な長期的結果を伴う、より低いウイルス負荷の確立をもたらすためのHIV感染後の治療用ワクチンとしての、これらのアデノウイルスおよび/またはDNAプラスミドワクチンの1以上の投与に関するすべての方法に関する。本明細書に記載されているとおり、そのような治療計画は、単価または多価組成物、種々の組合せ形態の適用、および/または初回/追加抗原刺激法(それにより、生きた脊椎動物組織内への接種後の抗原発現および付随的な細胞性および/または体液性免疫応答が最適化される)を含みうる。したがって、本発明は、哺乳類組織内に導入されるとgag、polおよび/またはnefに基づくベクターの細胞内発現を誘発する、本明細書に開示する種々のパラメーターおよび当技術分野で公知の追加的パラメーターの範囲内の本明細書に開示するアデノウイルスおよび/またはDNAプラスミドワクチンの使用方法を提供する。
【0031】
この目的において、本発明は、1つには、ワクチン被接種体、好ましくはヒトのワクチン被接種者において細胞性免疫応答を生成させる方法に関する。この場合、該個体には、アデノウイルスワクチンベクターの2以上の投与が行われ、それは、プラスミドワクチンの投与を伴う計画において投与されうる。該プラスミドワクチン(本明細書中では「DNAプラスミドワクチン」または「ワクチンプラスミド」とも称される)は、タンパク質またはその免疫学的に関連した部分をコードする核酸、該核酸配列に作動的に結合した異種プロモーター、および転写ターミネーターまたはポリアデニル化シグナル(例えば、それぞれbGHまたはSPA)を含む。それらの投与を隔てる所定の最低限度の長さの時間を設けることが可能である。該個体に、第1用量のプラスミドワクチン、ついで第2用量のプラスミドワクチンを投与することができる。あるいは、該個体に、第1用量のアデノウイルスワクチン、ついで第2用量のアデノウイルスワクチンを投与することができる。他の実施形態においては、まず、該プラスミドワクチンを投与し、ついで幾らかの時間の後、該アデノウイルスワクチンを投与する。逆に、まず、該アデノウイルスワクチンを投与し、ついで幾らかの時間の後、プラスミドワクチンを投与することができる。これらの実施形態においては、個体に、ウイルスベクターまたはプラスミド形態として複数用量の同じアデノウイルス血清型を投与することが可能であり、代替的態様では、該ウイルスは、異なる血清型のものでありうる。該代替的態様においては、まず、関心のあるウイルス抗原を、アデノウイルスに基づくワクチン以外のウイルスワクチンを介して運搬し、ついで、開示されているアデノウイルスワクチンで行うことができる。代替的ウイルスワクチンには、ポックスウイルスおよびベネズエラウマ脳炎ウイルスが含まれるが、これらに限定されるものではない。
【0032】
本発明はまた、本明細書に記載のGag、PolおよびNef成分を含む多価アデノウイルスワクチン組成物に関する。例えば、実施例29および表25を参照されたい。そのような組成物は、ヒトMHCの及び循環ウイルスの遺伝的多様性を仮定した場合には特に、宿主への投与の後、増強した細胞性免疫応答をもたらすであろう。具体例には、二価(すなわち、gagおよびnef、gagおよびpol、またはpolおよびnef成分)または三価ワクチン(すなわち、gag、polおよびnef成分)組成物を与えるMRKAd5ベクターに基づく多価ワクチン組成物が含まれるが、これらに限定されるものではない。そのような多価ワクチンは1回量用に充填されたり、各個に充填された成分の複数の接種よりなることが可能であり、また、前段落に記載のウイルス性または非ウイルス性ベクターワクチンでの初回/追加抗原刺激法の一部でありうる。この目的のためには、好ましい組成物は、複数の異なるHIV抗原クラスと組合せて使用されるMRKAd5アデノウイルスである。各HIV抗原クラスは配列操作に付され、それにより多数の潜在的なワクチンの組合せを与え、そのような組合せは本発明の範囲内である。そのような組合せ様式のワクチン製剤および投与の利用は、単一様式の方法での接種より一層強力な細胞性免疫応答を惹起する可能性を増大させる。
【0033】
本明細書に開示する「組合せ様式」の概念は、複数のオープンリーディングフレームを含む前アデノウイルスプラスミドの生成のために適当なシャトルプラスミドに複数のHIV−1ウイルス抗原が連結されうる代替的投与様式をも含む。例えば、三価ベクターは、E3(−)またはE3(+)バックグラウンド、好ましくはE3欠失バックボーンにおけるgag−pol−nef融合体、あるいは、おそらくは、「2+1」二価ワクチン、例えば、同じMRKAd5バックボーン内のgag−pol融合体(すなわち、コドン最適化p55gagおよび不活性化最適化pol;実施例29および表25)(各オープンリーディングフレームは別個のプロモーターおよび転写終結配列に作動的に結合している)を含みうる。あるいは、それらの2つのオープンリーディングフレームは単一のプロモーターに作動的に結合しており、該オープンリーディングフレームはIRES(internal ribosome entry sequence)に作動的に連結していることが可能である。したがって、単一の又は恐らくは第2の回収された組換え複製欠損型アデノウイルスとして運搬される多価ワクチンは、本発明の一部と意図される。
【0034】
したがって、本発明のアデノウイルスワクチンおよびプラスミドDNAワクチンは単独で投与することが可能であり、あるいは初回および追加抗原刺激投与法の一部でありうる。混合様式の初回および追加接種法は、既存の抗ベクター免疫応答が存在する場合には特に、増強された免疫応答をもたらすであろう。本発明のこの1つの態様は、該プラスミドワクチンを少なくとも1回投与することにより該プラスミドワクチンで対象を初回抗原刺激し、所定の長さの時間を経過させ、ついで該アデノウイルスワクチンを投与することにより追加抗原刺激する方法である。通常、複数の初回抗原刺激、典型的には1〜4回の初回抗原刺激を行うが、より多数の初回抗原刺激を行うことが可能である。初回抗原刺激と追加抗原刺激との間の時間の長さは、典型的には約4ヶ月〜1年と様々となりうるが、他の時間枠を用いることも可能である。アカゲザルでの実験においては、該動物をプラスミドワクチンで4回初回抗原刺激し、ついで4ヶ月後に該アデノウイルスワクチンで追加抗原刺激した。それらの細胞性免疫応答は、アデノウイルスワクチンのみが投与された動物の場合より顕著に高かった。初回抗原刺激法の利用は、既存の抗アデノウイルス免疫応答を対象者が有する場合には特に好ましいかもしれない。
【0035】
増強された複製欠損型組換えアデノウイルスワクチンベクターバックボーンを提供することが、本発明の目的の1つである。これらの組換えアデノウイルスバックボーンは1以上のトランスジーンを受容することが可能であり、それは、増殖、増幅および回収のために細胞培養を介して継代されうる。
【0036】
種々のトランスジーンをコードする増強された複製欠損型組換えアデノウイルスワクチンベクターを提供することが、もう1つの目的である。
【0037】
細胞培養における連続継代後のウイルス安定性の増加と共に増殖および増幅速度の増加を示す回収された組換え複製欠損型アデノウイルスを提供することも、本発明の目的の1つである。そのような組換えアデノウイルスは、遺伝子治療における及びヌクレオチドに基づくワクチンベクターにおける使用に特に適しており、有利なことに、大規模増殖に有用である。
【0038】
この目的において、(1)種々の形態のHIV−1 Gag、HIV−1 Polおよび/またはHIV−1 Nef(HIV−1 Gag、HIV−1 PolおよびHIV−1 Nefの免疫学的に関連した修飾体を含む)をコードする本明細書に記載の増強された複製欠損型組換えアデノウイルスワクチンベクター、ならびに(2)細胞培養による1回または複数回の継代(293細胞またはPER.C6(登録商標)細胞による継代を含むが、これらに限定されるものではない)を介する(1)のアデノウイルスベクターの継代により産生された回収され精製された組換え複製欠損型アデノウイルスを提供することが、本発明の目的の1つである。
【0039】
細胞培養による1回または複数回の継代により回収された組換えアデノウイルスを提供することも、本発明の目的の1つである。組換えアデノウイルスワクチンベクターの場合と同様に、この組換えウイルスを、後続の宿主投与のために回収し製剤化する。
【0040】
また、本発明の目的の1つは、(a)タンパク質またはその生物学的に活性な及び/又は免疫学的に関連した部分をコードする核酸と、(b)a)部の核酸に作動的に結合した異種プロモーターと、(c)転写ターミネーターと、を含む遺伝子発現カセットの形態で少なくとも1つの遺伝子が挿入された複製欠損型アデノウイルスベクターを提供することである。
【0041】
また、本発明の目的の1つは、該アデノウイルスベクターおよび/または該シャトルプラスミドベクターを含む宿主細胞;該ベクターを含むワクチン組成物;および(a)アデノウイルスE1タンパク質を発現する宿主細胞内に該アデノウイルスベクターを導入し、(b)得られたアデノウイルスベクターを回収することを含む該ベクターの製造方法提供することである。本発明のもう1つの目的は、タンパク質に対する細胞性免疫応答を個体において生成させる方法であって、a)塩基対約1からと塩基対約342(より好ましくは400)から約450まで(好ましくは1〜450)の間との野生型アデノウイルス・シス作用性アデノウイルスパッケージング領域および好ましくは野生型アデノウイルス配列の3511〜3523を含みE1において少なくとも部分的に欠失した複製欠損型アデノウイルベクター、ならびにb)(i)タンパク質またはその生物学的に活性な及び/又は免疫学的に関連した部分をコードする核酸と、(ii)a)部の核酸に作動的に結合した異種プロモーターと、(iii)転写ターミネーターおよび/またはポリアデニル化部位と、を含む遺伝子発現カセットを含むアデノウイルスワクチンベクターを該個体に投与することを含んでなる方法を提供することである。
【0042】
ワクチン投与法、すなわち、未感染個体に対する有効的な免疫予防またはHIV感染患者に対する治療的処置をもたらす本明細書に記載の1以上のアデノウイルスおよび/またはDNAプラスミドワクチンの投与のための種々の代替手段を提供することが、本発明の目的の1つである。そのような方法は、多価HIV−1ワクチン組成物、種々の組合せ様式の方法および種々の初回/追加抗原刺激代替手段を含むが、これらに限定されるものではない。ワクチン組成物および/または計画された投与に関するこれらの投与方法は、単一様式の方法での接種より一層強力な細胞性免疫応答を惹起する可能性を増大させるであろう。
【0043】
本明細書および特許請求の範囲の全体にわたり、以下の定義および略語を用いる。
【0044】
「HAART」は、高度に活性な抗レトロウイルス療法を意味する。
【0045】
「第1世代」ベクターは、複製欠損型であるとして特徴づけられる。それは、典型的には、欠失した又は不活性化されたE1遺伝子領域を有し、好ましくは、欠失した又は不活性化されたE3遺伝子領域をも有する。
【0046】
「AEX」は陰イオン交換クロマトグラフィーを意味する。
【0047】
「QPA」はQuick PCR−based Potency Assayを意味する。
【0048】
「bp」は塩基対を意味する。
【0049】
「s」または「str」は、該トランスジーンが「E1平行」または「直線的」配向で位置することを示す。
【0050】
「PBMC」は末梢血単球細胞を意味する。
【0051】
「FL」は完全長を意味する。
【0052】
「FLgag」は、図2に示すとおりの完全長最適化gag遺伝子を意味する。
【0053】
「Ad5−Flgag」は、CMVプロモーターの制御下で完全長最適化gag遺伝子を含む発現カセットを保持するアデノウイルス血清型5複製欠損型ウイルスを意味する。
【0054】
「プロモーター」は、RNAポリメラーゼが結合するDNA鎖上の認識部位を意味する。該プロモーターは、RNAポリメラーゼとの開始複合体を形成して、転写活性を開始させ駆動する。該複合体は、「エンハンサー」のような配列を活性化することにより、あるいは「サイレンサー」のような配列を抑制することにより修飾されうる。
【0055】
「リーダー」は、構造遺伝子の5’末端の、該遺伝子と共に転写されるDNA配列を意味する。これは、通常、プロ配列としばしば称されるN末端ペプチド伸長を有するタンパク質を与える。
【0056】
「イントロン」は、遺伝子産物中のアミノ酸をコードしていない遺伝子の中間に見出されるDNAの区分を意味する。該イントロンの前駆体RNAは切り出され、したがって、タンパク質に翻訳されないmRNAには転写されない。
【0057】
「免疫学的に関連」または「生物学的に活性」は、(1)ウイルスタンパク質に関しては、該タンパク質が、投与されると、該ウイルスの増殖および/または広がりを遅延させるのに及び/又は個体内に存在するウイルス負荷を減少させるのに十分な個体内における測定可能な免疫応答を惹起する能力を有すること、または(2)ヌクレオチド配列に関しては、該配列が、前記の能力を有するタンパク質をコードしうることを意味する。
【0058】
「カセット」なる語は、転写および翻訳制御配列を伴う、発現される核酸配列を意味する。該カセットを交換することにより、ベクターは異なる配列を発現しうる。
【0059】
「bGHpA」は、ウシ成長ホルモン転写ターミネーター/ポリアデニル化配列を意味する。
【0060】
「tPAgag」は、DNAに基づくワクチンベクターの場合かアデノウイルスに基づくワクチンベクターの場合かに無関係に、図30A〜Bに例示されている組織プラスミノーゲンアクチベーターリーダー配列のリーダー配列と最適化HIV gag遺伝子との融合体を意味する。
【0061】
「IA」または「inact」は、用いられている場合には、遺伝子の不活性化形態(例えば、IApol)を意味する。
【0062】
「MCS」は「マルチクローニング部位」である。
【0063】
一般に、アデノウイルス構築物、遺伝子構築物は、それらに含有される遺伝子を参照することにより命名される。
【0064】
例えば、「Ad5 HIV−1 gag」は、元のHIV−1 gagアデノウイルスベクターとも称され、hCMVイントロンAプロモーター、完全長形態のヒトコドン最適化HIV−1 gag遺伝子およびウシ成長ホルモンポリアデニル化シグナルから構成されるトランスジーンカセットを含有するベクターである。該トランスジーンは、E1およびE3欠失アデノベクター内にE1逆平行配向で挿入された。
【0065】
「MRK Ad5 HIV−1 gag」は、「MRKAd5gag」または「Ad5gag2」とも称され、塩基対1〜450および3511〜3523を含みヒトコドン最適化HIV−1遺伝子をイントロンA無しのCMVプロモーターの制御下でE1平行配向で有しE1が欠失した本明細書に教示するアデノウイルスベクターである。該構築物はまた、ウシ成長ホルモンポリアデニル化シグナルを含む。
【0066】
「pV1JnsHIVgag」は、「HIVFLgagPR9901」とも称され、CMV最初期(IE)プロモーターおよびイントロンA、完全長コドン最適化HIV gag遺伝子、ウシ成長ホルモン由来ポリアデニル化および転写終結配列ならびに最小pUCバックボーンを含むプラスミドである。
【0067】
「pV1JnsCMV(無イントロン)−FLgag−bGHpA」は、完全長HIV gag遺伝子を含みCMVのイントロンA部分が欠失したpV1JnsHIVgag由来のプラスミドである。このプラスミドは、「pV1JnsHIVgag−bGHpA」、「pV1Jns−hCMV−FL−gag−bGHpA」および「pV1JnsCMV(無イントロン)+FLgag+bGHpA」と称される。
【0068】
「pV1JnsCMV(無イントロン)−Flgag−SPA」は、bGHpA終結配列がSPA終結配列で置換されている以外はpV1JnsCMV(無イントロン)−FLgag−bGHpAと同じ組成のプラスミドである。このプラスミドは、「pV1Jns−HIVgag−SPA」および「pV1Jns−hCMV−FLgag−SPA」とも称される。
【0069】
「pdelE1sp1A」は、発現カセットを有さない(すなわち、プロモーターもポリAも有さない)普遍的シャトルベクターである。該ベクターは、bp1〜bp341およびbp3524〜bp5798の野生型アデノウイルス血清型5(Ad5)配列を含み、341bpで終結するAd5配列と3524bpで開始するAd5配列との間にマルチクローニング部位を有する。このプラスミドは、元のAd5シャトルベクターとも称される。「MRKpdelE1sp1A」または「MRKpdelE1(Pac/pIX/pack−450)」または「MRKpdelE1(Pac/pIX/pack450)Cla1」は、bp1〜bp450およびbp3511〜bp5798の野生型アデノウイルス血清型5(Ad5)配列を含み発現カセットを有さない(すなわち、プロモーターもポリAも有さない)普遍的シャトルベクターである。該ベクターは、450bpで終結するAd5配列と3511bpで開始するAd5配列との間にマルチクローニング部位を有する。このシャトルベクターを使用して、該CMVプロモーターおよび該bGHpA断片を直線的(「str」またはE1平行)配向または逆(oppまたはE1逆平行)配向の両方で挿入することができる。
【0070】
「MRKpdeE1(Pac/pIX/pack450)+CMVmin+BGHpA(str.)」は、該CMVプロモーター(イントロンA無し)および該bGHpA断片を含有する修飾されたベクターである更にもう1つのシャトルベクターである。唯一のBglII部位における選択された遺伝子の挿入が、MRKpAd5(E1−/E3+)Cla1前プラスミド内への挿入の場合に、該トランスジーンの転写の方向がAd5 E1に平行となることを保証するよう、該hCMVプロモーター(イントロンA無し)および該ウシ成長ホルモンポリアデニル化シグナルを含有する発現単位が該シャトルベクター内に挿入されている。このシャトルベクターは、図22および23に示すとおり、それぞれのIApolおよびG2A、LLAA nef遺伝子を直接挿入するするために使用された。
【0071】
「MRKpdelE1−CMV(無イントロン)−FLgag−bGHpA」は、イントロンAを伴わないヒトCMV、完全長ヒトコドン最適化HIV gag遺伝子およびウシ成長ホルモンポリアデニル化シグナルを含有する発現カセットと共に塩基対1〜450および3511〜5798のAd5配列を含むシャトルである。このプラスミドは、「MRKpdeE1シャトル+hCMV−FL−gag−BGHpA」とも称される。
【0072】
「MRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpA」は、E1領域を含むヌクレオチド以外のすべてのAd5配列(451〜3510)、イントロン無しのヒトCMVプロモーター、完全長ヒトコドン最適化HIV gag遺伝子およびウシ成長ホルモンポリアデニル化シグナルを含むアデノウイルスベクターである。このベクターは、「MRKpAdHVE3+hCMV−FL−gag−BGHpA」、「MRKpAd5HIV−1gag」、「MRKpAd5gag」、「pMRKAd5gag」または「pAd5gag2」とも称される。
【0073】
「pV1Jns−HIV−pol inact(opt)」または「pV1Jns−HIV IA pol(opt)」は、V1JnsのBglII部位内にクローニングされた不活性化Pol遺伝子(配列番号3中に含有されている)である。本明細書に記載のとおり、HIV−1 polの種々の誘導体を、V1JnsまたはV1Jns−tPAのようなプラスミド発現ベクター内にクローニングして、直接的にDNAワクチン候補として又は適当なアデノウイルスベクター内へのサブクローニングのための起源として使用することができる。
【0074】
「MRKpdel+hCMVmin+FL−pol+bGHpA(s)」は、適切な配向でIA pol遺伝子を含有する前記の「MRKpdelE1(Pac/pIX/pack450)+CMVmin+BGHpA(str.)」シャトルである。このシャトルベクターは、MRKpAd(E1−/E3+)Cla1との細菌組換えにおいて使用される。
【0075】
本発明で「pMRKAd5pol」とも称される「MRKpAd+hCMVmin+FL−pol+bGHpA(S)E3+」は、CMV−pol inact(opt)−pGHpA構築物を含む前アデノウイルスプラスミドである。この前アデノウイルスプラスミドの構築を図22に示す。
【0076】
「pV1Jns/nef(G2A,LLAA)」または「V1Jns/opt nef(G2A,LLAA)」は、アミノ末端ミリスチル化部位における修飾(Gly−2からAla−2)およびAla−174−Ala−175へのLeu−174−Leu−175ジロイシンモチーフの置換をオープンリーディングフレームがコードするコドン最適化HIV−1 Nefを含む(配列番号13;これは、ヌクレオチド12〜14の開始メチオニン残基およびヌクレオチド660〜662の「TAA」終結コドンを含む)。この断片をV1Jnsおよび/またはV1Jns−tPAのBglII部位内にサブクローニングする(図16A〜B)。HIV−1 polに関して前記したとおり、HIV−1 nef構築物は、V1JnsまたはV1Jns−tPAのようなプラスミド発現ベクター内にクローニングして、直接的にDNAワクチン候補として又は適当なアデノウイルスベクター内へのサブクローニングのための起源として使用することができる。
【0077】
本発明で「pMRKAd5nef」とも称される「MRKpdelE1hCMVminFL−nefBGHpA(s)」は、CMV−nef(G2A,LLAA)コドン最適化配列を含む前アデノウイルスプラスミドである。この前アデノウイルスプラスミドの構築を図23に示す。
【0078】
(図面の簡単な記載)
図1は、元のHIV−1 gagアデノベクター(Ad5HIV−1gag)を示す。このベクターは、1999年7月6日付け出願の米国仮特許出願第60/142,631号および1999年8月13日付けの米国特許出願第60/148,981号に基づいて優先権を主張している2000年7月3日付け出願のPCT国際出願番号PCT/US00/18332(WO01/02607)に開示されており、これらの3つの出願をすべて、参照により本明細書に組み入れることとする。
【0079】
図2は、該最適化ヒトHIV−1 gagオープンリーディングフレームの核酸配列(配列番号29)を示す。
【0080】
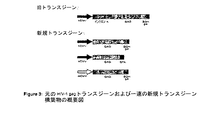
図3は、元のgagトランスジーンと比較して新規トランスジーン構築物を図示する。
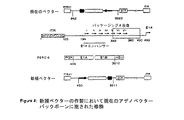
【0081】
図4は、本発明の新規ベクターの作製において元のアデノベクターバックボーンに施された修飾を示す。
【0082】
図5は、該パッケージングシグナル領域に対して施された付加(実験#1)およびウイルス増殖に対するE3遺伝子(実験2)の効果を判定するために行ったウイルス混合実験を示す。棒線は、該E1欠失に対して施された修飾の領域を示す。
【0083】
図6は、実施例6および7に記載するウイルス混合実験後のウイルスDNA分析のオートラジオグラフを示す。
【0084】
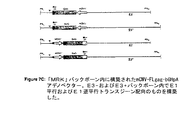
図7A、7Bおよび7Cは以下のとおりである。図7Aは、MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたhCMV−Flgag−bGHpAアデノベクターを示す。E1平行およびE1逆平行のトランスジーン配向が示されている。図7Bは、MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたhCMV−Flgag−SPAアデノベクターを示す。この場合もまた、E1平行およびE1逆平行の両方のトランスジーン配向が示されている。図7Cは、MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたmCMV−Flgag−bGHpAアデノベクターを示す。この場合もまた、E1平行およびE1逆平行の両方のトランスジーン配向が示されている。
【0085】
図8Aは、トランスジーン配向の効果を試験するよう意図された実験を示す。図8Bは、ポリアデニル化シグナルの効果を試験するよう意図された実験を示す。
【0086】
図9は、BstE11消化後のP5における試験された4つのアデノウイルスベクターからのウイルスDNA(実施例12)を示す。
【0087】
図10は、MRKpAdHVE3、MRKAd5HIV−1gagおよびMRKAd5HIV−1gagE3−の継代11および12のウイルスDNA分析を示す。
【0088】
図11は、ウイルス競合研究を開始するために使用した継代6のMRKpAdHVE3およびMRKAd5HIV−1gagのウイルスDNA分析(HindIII消化)を示す。最後の2つのレーンは、該競合研究の二重継代の継代11分析である(各ウイルスは280ウイルス粒子のMOIである)。
【0089】
図12は、特定された時点で回収が行われた血清含有培地中のMRKAd5HIV−1gagに関する高継代数上のHindIII消化によるウイルスDNA分析を示す。第1レーンは1kb DNAサイズマーカーを示す。その他のレーンは、前プラスミド対照(Pac1およびHindIIIで消化されたもの)、P16、P19およびP21におけるMRKAd5HIV−1gagを示す。
【0090】
図13は、種々の用量のいくつかのAdgag構築物でbalb/cマウス(n=10)をi.m.免疫した3週間後の血清抗p24レベルを示す:(A)MRK Ad5 HIV−1 gag(継代5)、(B)MRKAd5hCMV−FLgag−bGHpA(E3−)、(C)MRKAd5 hCMV−FLgag−SPA(E3+)、(D)MRKAd5 mCMV−FLgag−bGHpA(E3+)、(E)Ad5HIV−1 gagの研究ロット(293細胞由来)、および(F)Ad5HIV−1 gagの臨床ロット(Ad5gagFN0001)。示されているのは、各コホートの幾何平均力価(GMT)および標準誤差線である。
【0091】
図14は、pMRKAd5HIV−1 gagベクターの制限地図を示す。
【0092】
図15A〜Xは、pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【0093】
図16A〜Bは、DNAワクチン発現ベクターV1Jns(A)およびV1Jns−tPA(B)の概要図を示す。これらは、本明細書に記載の種々のDNA/ウイルスベクター組合せ様式法におけるHIV−1 gag、polおよびnef構築物に使用される。
【0094】
図17A〜Cは、IA−Polのヌクレオチド(配列番号3)およびアミノ酸配列(配列番号4)を示す。下線付きのコドンおよびアミノ酸は、表1に列挙された突然変異を示す。
【0095】
図18は、tPA−pol inact(opt)の融合結合部にわたるコドン最適化ヌクレオチドおよびアミノ酸配列(それぞれ配列番号7および8中に含有される)を示す。下線付き部分はIA−PolのNH2末端領域を示す。
【0096】
図19A〜Bは、野生型nef(jrfl)とコドン最適化nefとのヌクレオチド配列比較を示す。該jrfl分離体からの野生型nef遺伝子は、216アミノ酸のポリペプチドをコードしうる648ヌクレオチドよりなる。WT,野生型配列(配列番号19);opt,コドン最適化配列(配列番号1中に含有されている)。Nefアミノ酸配列は1文字記号で示されている(配列番号2)。
【0097】
図20A〜Cは、nef発現ベクターV1Jns/nef(図20A)、V1Jns/nef(G2A,LLAA)(図20B)、V1Jns/tpanef(図20C)およびV1Jns/tpanef(LLAA)(同様に図20C)のプラスミドバックボーンとnefコード配列との間の結合部のヌクレオチド配列を示す。コドン最適化nefまたはコドン最適化nef突然変異遺伝子の5’および3’フランキング配列が太字/イタリック体で示されており、nefおよびnef突然変異体コード配列が無修飾文字で示されている。また(下線付きで)、それぞれのnef発現ベクターの構築に関わる制限エンドヌクレアーゼ部位が示されている。V1Jns/tpanefおよびV1Jns/tpanef(LLAA)は該結合部位に同一配列を有する。
【0098】
図21は、nefおよびnef誘導体の概要図を示す。Nef誘導体に含まれるアミノ酸残基が示されている。グリシン2ならびにロイシン174および175は、それぞれミリスチル化およびジロイシンモチーフに関わる部位である。該tpanef融合遺伝子の両形態に関して、推定リーダーペプチド切断部位は「*」で示されており、該突然変異の構築中に導入された外因性セリン残基は下線で示されている。
【0099】
図22は、前アデノウイルスプラスミド構築物MRKAd5Polの構築の概要図を示す。
【0100】
図23は、前アデノウイルスプラスミド構築物MRKAd5Nefの構築の概要図を示す。
【0101】
図24は、クレードB HIV感染個体におけるクレードBとクレードCとの抗gag T細胞応答の比較を示す。
【0102】
図25は、クレードB HIV感染個体におけるクレードBとクレードCとの抗nef T細胞応答の比較を示す。
【0103】
図26A〜AOは、不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【0104】
図27A〜AMは、不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【0105】
図28は、E3(+)またはE3(−)バックボーン中に種々のプロモーター断片(hCMVまたはmCMV)および終結シグナル(bGHまたはSPA)を含むMRKAd5ベクターの安定性を示す。
【0106】
図29AおよびBは、24、36、48および60hpiの時点における凍結融解回収細胞結合ウイルスの陰イオン交換HPLCウイルス粒子濃度(図29A)、ならびにMRKAd5gag、MRKAd5polおよびMRKA5nefに関する経時的QPA上清力価(図29B)を示す。
【0107】
図30は、本明細書に開示するDNAおよび/またはアデノウイルスワクチンにおいて使用する代表的なtPA−gag融合体のオープンリーディングフレームをオープンリーディングフレーム含むヌクレオチド配列(配列番号36)およびアミノ酸配列(配列番号37)を示す。
【0108】
図31は、第10週(DNA初回抗原刺激後)および第30週(Ad追加抗原刺激後)に集めたPBMCの細胞内γIFN染色を示す。該細胞は、該gagペプチドプールの存在下または不存在下で一晩にわたり刺激した。ついで、蛍光標識抗CD3、抗CD8、抗CD4および抗γIFNモノクローナル抗体を使用して、それらを染色した。各プロットは、表面CD8およびγIFN産生についての陽性染色に関して分離された全CD3+T細胞を示す。各プロットの右上および右下の四分域中の数字は、それぞれCD8+γIFN+およびCD4+γIFN+であったCD3+細胞の割合(%)である。
【0109】
図32は、単一様式アデノウイルス免疫とDNA+アジュバント初回/アデノウイルス追加免疫との比較を示す。
【0110】
図33A〜Bは、実施例29のgag−IApol融合体のオープンリーディングフレームのヌクレオチド配列(配列番号38)を示す。
【0111】
図34A〜Bは、該gag−IApol融合フレームのタンパク質配列(配列番号39)を示す。
【0112】
(発明の詳細な記載)
遺伝子治療における又はヌクレオチドに基づくワクチンベクターにおける使用に適した新規複製欠損型または「第1世代」アデノウイルスベクターを記載する。このベクターは、E1において少なくとも部分的に欠失しており、野生型アデノウイルス配列の塩基対約1からと塩基対約342(より好ましくは400)〜塩基対約458までの間との野生型アデノウイルス・シス作用性パッケージング領域(好ましくは1〜450)および好ましくは塩基対3511〜3523を含む。この記載のベクターは、約5〜10倍大きな増幅速度を伴う増強された増殖特性を有し、より強力であり、より低いウイルス用量で同等の免疫生成を可能にすることが判明している。該ベクターは更に、この領域(塩基対342〜450)を含まない複製欠損型ベクターより大きな細胞性免疫応答を示す回収された組換えアデノウイルスを与える。さらに、これらのベクターに由来するアデノウイルス構築物、特に、イントロンAを欠くhCMVプロモーターの制御下でトランスジーンを含むものは、遺伝的に非常に安定である。ここに記載のウイルスを連続的に継代し、分析した。実施例12を参照されたい。分析した各ウイルスは、適切な遺伝的構造でそれを維持した。また、大規模生産での実施と同様の増殖条件下で分析を行った。この場合もまた、該ベクターは、増強した遺伝的安定性を有することが判明した。実施例12を参照されたい。継代21の後、該ウイルスDNAは再構成の証拠を示さず、1つの製造ロットから次の製造ロットに高度に再生可能であった。すべての関連試験の結果は、図28に関連したデータで本明細書中に示されるとおり、該アデノウイルスベクターが組換え複製欠損型アデノウイルスの大規模生産に非常に良く適合することを示している。
【0113】
この記載の好ましいアデノウイルスベクターは、E3において欠失しており塩基対1〜450を含むベクターである。このベクターは、約7,500塩基対までの外来DNAインサート(または外因性遺伝物質)を収容しうる。もう1つの好ましいベクターは、塩基対1〜450を含むE3を保有するベクターである。この記載の好ましいベクターは、塩基対1〜450および3511〜3523を含むE3+ベクターである。このベクターは、塩基対451〜3510に及ぶ領域が欠失している場合には、約4,850塩基対までの外来DNAインサート(または外因性遺伝物質)を収容しうる。前記ベクターのクローニング容量は、ゲノムサイズの上限として105%の野生型Ad5配列を使用して測定されている。
【0114】
野生型アデノウイルス血清型5は、本明細書の全体にわたり記載されている特定の塩基対番号の基準として用いられる。野生型アデノウイルス血清型5配列は公知であり、当技術分野において記載されている。参照により本明細書に組み入れるChroboczekら,1992 J.Virology 186:280を参照されたい。したがって、本発明の特定の実施形態は、アデノウイルス血清型5配列に基づくベクターである。当業者は、他のアデノウイルス血清型(例えば、血清型2、4、6、12、16、17、24、31、33および42)における前記領域、アデノウイルス血清型5に関して与えられた前記塩基対位置に対応する塩基対により定められる領域を容易に同定することが可能である。したがって、本発明は、野生型アデノウイルス血清型5(Ad5)核酸配列の1〜450(特に、342〜450)に対応する塩基対および好ましくは3511〜3523を含みE1において部分的に欠失したすべてのアデノウイルスベクターを含む。本発明の特に好ましい実施形態は、亜群Cに分類されるAd5に類似したアデノウイルス(例えば、Ad2)に由来するものである。
【0115】
本発明のベクターは、E1において少なくとも部分的に欠失している。好ましくは、該E1領域は、完全に欠失しているか又は不活性化されている。最も好ましくは、E1が欠失した領域は塩基対451〜3510内に存在する。開示しているベクターの伸長5’および3’領域が、使用する細胞系、すなわち、塩基対459〜3510でトランスフェクトされたPER.C6(登録商標)細胞系内に存在するE1A/E1B遺伝子のいずれの部分とも重複することなく従来の構築物のE1欠失のサイズを有効に減少させると考えられることは注目すべきことである。アデノウイルス配列の重複が避けられるのは、組換えの可能性によるものである。したがって、疑いなく、当業者は、異なるアデノウイルスDNAセグメントでトランスフェクトされた異なる細胞系を使用する場合には本発明を修飾しうると理解することが可能である。例示目的で挙げると、塩基対450〜3510のアデノウイルス配列で細胞系をトランスフェクトする場合には、塩基対1から449までの5’領域が、より適切である。このことは、該E1欠失の3’側のセグメントを考える場合にも当てはまる。
【0116】
本発明の好ましい実施形態は、無傷E3領域(すなわち、機能的E3をコードしうるE3遺伝子)を有する。別の実施形態は、部分的に欠失したE3、不活性化されたE3領域、またはE3が完全に欠失した配列を有する。本出願人は、本発明において、該E3遺伝子を含むウイルスが、E3遺伝子を含まないウイルスより迅速に増幅しうることを見出した。図6を参照されたい。この図においては、試験したE3+ウイルスに対応する特徴的CsClバンド(5,665bp)が、E3−ウイルスに対応する3,010bpの特徴的バンドより大量に存在した。これらの結果は、E3遺伝子を含むアデノベクターおよびE3遺伝子を含まないアデノベクターの両方を等しいMOI比(1:1)で混合することを含むウイルス競合研究の後で得られた。ついで増殖研究で、このE3+アデノベクターの増幅能の増加が確認された。表4Aを参照されたい。この表においては、E3−構築物は115および40〜50の増殖率を示しているのに対して、E3+ウイルスは470、420および320の増幅率を示している。
【0117】
前記のとおり、本発明のベクターは、E3+ベクターに関しては約4,850塩基対までの外因性遺伝物質を、そしてE3−ベクターに関しては約7,500塩基対を収容しうる。好ましくは、該インサートは、該アデノウイルスベクターを、野生型ゲノムサイズ(例えば、Ad5の場合、35,935塩基対)に可能な限り近づける。アデノウイルスは、それがその野生型ゲノムサイズに近い場合に最も良く増幅することが良く知られている。
【0118】
図7A、7Bおよび図8Aに例示されているとおり、該遺伝物質はE1平行またはE1逆平行配向で挿入することができる。本発明の特に好ましい実施形態は、E1平行配向で該インサートを有する。本出願人は、異なる配向でトランスジーンを含有するプラスミドとの競合実験により(図8A)、該外来DNAインサートをE1平行配向で有するベクター構築物が、E1逆平行配向トランスジーンより良く増幅し、競合において実際にそれを凌ぐことを見出した。継代3において、そしてもちろん継代6においても、該混合物のウイルスDNA分析は、E1平行配向で該トランスジーンを保持するウイルスの、E1逆平行形態より大きな比率を示した。継代10までは、観察される唯一のウイルス種は、試験した両方のトランスジーンに関してE1平行配向で該トランスジーンを有するアデノベクターであった。
【0119】
本発明のアデノウイルスベクターは、所望のタンパク質(その一例は、HIVタンパク質、特にHIV完全長gagタンパク質である)の発現の達成に特に良く適合する。関心のあるタンパク質をコードする外因性遺伝物質は発現カセットの形態で存在しうる。遺伝子発現カセットは、好ましくは、(a)関心のあるタンパク質をコードする核酸、(b)該タンパク質をコードする核酸に作動的に結合した異種プロモーター、および(c)転写ターミネーターを含む。
【0120】
該転写プロモーターは、好ましくは、真核性RNAポリメラーゼにより認識される。好ましい実施形態においては、該プロモーターは「強力」または「効率的」なプロモーターである。強力なプロモーターの一例は、好ましくはイントロン配列を伴わない、最初期ヒトサイトメガロウイルスプロモーター(参照により本明細書に組み入れるChapmanら,1991 Nucl.Acids Res 19:3979−3986)である。本アデノウイルスベクターにおける使用に最も好ましいのは、イントロンAのようなイントロン配列を伴わないヒトCMVプロモーターである。本出願人は、イントロンA(ヒトサイトメガロウイルスプロモーター(hCMV)の一部である)がアデノウイルスベクターの不安定性の領域を構成することを見出した。イントロンAを伴わないCMVは、HIV gag発現の駆動に際して、比較しうるインビトロ発現能をもたらすことが判明しており(実施例1〜3)、さらには、試験したプラスミドDNAの両方の投与量(20μgおよび200μg)で、それらの抗体およびT細胞応答に関して、Balb/cマウスにおいてインビボでイントロンA含有構築物と同等に挙動した。当業者は、多数の他の公知プロモーターのいずれか、例えば強力な免疫グロブリンまたは他の真核性遺伝子プロモーター、例えばEF1αプロモーター、マウスCMVプロモーター、ラウス肉腫ウイルス(RSV)プロモーター、SV40初期/後期プロモーターおよびβアクチンプロモーターを使用することが可能であると理解するであろう。
【0121】
好ましい実施形態においては、該プロモーターはまた、Tetオペレーター配列のような調節可能な配列を含みうる。これは、例えば、望まない結果を遺伝子産物がもたらしており抑制が求められる場合に非常に有用であろう。
【0122】
該遺伝子配列カセット内に存在する好ましい転写終結配列としては、ウシ成長ホルモンターミネーター/ポリアデニル化シグナル(bGHpA)および以下のとおりに定められる50ヌクレオチド長の短い合成ポリAシグナル(SPA)がが挙げられる:AATAAAAGATCTTTATTTTCATTAGATCTGTGTGTTGGTTTTTTGTGTG(配列番号26)。
【0123】
該CMVプロモーター(イントロンA領域を欠くもの)とBGHターミネーターとの組合せが特に好ましいが、FGアデノウイルスの場合における他のプロモーター/ターミネーターの組合せも用いることができる。
【0124】
他の実施形態は、リーダーまたはシグナルペプチドを該トランスジーン内に組込むものである。好ましいリーダーは、組織特異的プラスミノーゲンアクチベータータンパク質tPA由来のものである。具体例には、本明細書の全体にわたり開示されている種々のtPA−gag、tPA−polおよびtPA−nefアデノウイルスに基づくワクチンが含まれるが、これらに限定されるものではない。
【0125】
本明細書に記載の改善されたアデノウイルスベクターを考慮すると、本発明の必須部分は、予防的または治療的状況において哺乳類宿主、好ましくはヒト宿主に投与されうる、前記アデノウイルスバックボーンを含む、アデノウイルスに基づくHIVワクチンである。本発明のHIVワクチンは、単独で投与されるか他のウイルスまたは非ウイルスに基づくDNAワクチンとの組合せ法で投与されるかにかかわらず、HIVに対して強力かつ広域な細胞性免疫応答を惹起するはずであり、これは、持続的ウイルス感染の可能性を減少させ及び/又はHIV感染を受ける臨床的に有意な低下したウイルス負荷の確立を招くか、あるいはHAART療法と組合せて、既に確立されたHIV感染の影響を軽減するであろう(抗ウイルス免疫療法(ARI))。本明細書に記載の組換えアデノウイルスベクターにおいては任意のHIV抗原(例えば、gag、pol、nef、gp160、gp41、gp120、tat、revなど)を使用することが可能であるが、好ましい実施形態には、コドン最適化p55 gag抗原(本発明ではMRKAd5gag)、polおよびnefが含まれる。HIV−1の種々のクレードに基づく配列が本発明での使用に適しており、それらのうちで最も好ましいのはクレードBおよびクレードCである。特に好ましい実施形態は、コンセンサス・クレードB配列に基づく配列(特に、コドン最適化配列)である。一連のMRKAd5polおよびMRKAd5nefのアデノウイルスワクチンの好ましい形態は、本明細書に記載のpolまたはnefの修飾形態をコードする。HIVエンベロープ遺伝子およびその修飾体を保持するMRKAd5HIV−1ベクターの好ましい実施形態は、それぞれ1997年8月28日(WO 97/31115)および1997年12月24日付けで公開されたPCT国際出願PCT/US97/02294およびPCT/US97/10517(それらの両方の文書を参照により本明細書に組み入れることとする)のHIVコドン最適化env配列を含む。
【0126】
本発明の最も好ましい態様は、HIV gagの発現を達成するための前記のアデノウイルスベクターの開示されている使用である。多数のHIV株の多数の遺伝子の配列がGENBANKにおいて公に入手可能であり、主として、HIVの野外分離体は、これらの株を入手可能にするようQuality Biological(Gaithersburg,MD)に委託しているNational Institute of Allergy and Infectious Diseases(NIAID)から入手可能である。また、株は、World Health Organization(WHO),Geneva Switzerlandからも入手可能である。該gag遺伝子はHIV−1株由来であることが好ましい(CAM−1;Myersら編 “Human Retroviruses and AIDS:1995,IIA3−IIA19(これを参照により本明細書に組み入れることとする))。この遺伝子は、クレードB(北米/欧州)配列のコンセンサスアミノ酸配列に酷似している。したがって、特異的HIV gagまたはその免疫学的に関連した部分をコードする適当なヌクレオチド配列を選択することは当業者の認識範囲内である。実施例25に示すとおり、クレードBまたはクレードCに基づくp55 gag抗原は地球的規模で潜在的に有用であろう。本明細書に記載のとおり、本発明のDNAまたはMRKAdに基づくアデノウイルスベクター内への挿入のために選択されるトランスジーンは、p55 gagのコドン最適化形態である。そのようなMRKAd5gagアデノウイルスベクターは実施例11に記載されており、少なくとも本発明においてはMRKAd5HIV−1gagと称される。もちろん、追加的な形態も意図され、それらには、プロモーター(例えば、hCMVの代わりにmCMV)および/またはpA終結シグナル(bGHの代わりにSPA)の置換のような修飾、ならびにAd5E3遺伝子の欠失を伴う又は伴わないMRKAd5バックボーンの作製が含まれるが、これらに限定されるものではない。
【0127】
本発明はまた、マウスおよび非ヒト霊長類での研究における投与後に細胞性免疫応答を生成することが本発明において示された一連のMRKAd5polに基づくアデノウイルスワクチンに関する。一連のMRKAd5polのいくつかは本明細書中に例示されている。1つのそのようなアデノウイルスベクターはMRKAd5hCMV−inact opt pol(E3+)と称され、これは、MRKAd5バックボーン、hCMVプロモーター(イントロンAを有さない)、不活性化polトランスジーンを含み、該アデノウイルスバックボーン中にAd5 E3遺伝子を含有する。第2の例示されている前アデノウイルスプラスミドおよび付随ウイルスはMRKAd5hCMV−inact opt pol(E3−)と称され、これは、該E3が欠失している以外は前記アデノウイルスベクターと同一である。どちらの構築物もHIV−1 Polのコドン最適化不活性化形態を含有し、ここで、少なくとも全コード領域は本発明では配列番号3として開示されており、該発現タンパク質は配列番号4として示される(不活性化polの標的化欠失を示す図17A〜Cおよび表1も参照されたい)。本明細書に記載のHIV Polのこの及び他の好ましいコドン最適化形態は、実質的には、2000年12月21日付け出願の米国特許出願第09/745,221号および同様に2000年12月21日付け出願のPCT国際出願PCT/US00/34724(それらの両方の文書を参照により本明細書に組み入れることとする)に記載のとおりである。前記文書に開示されているとおり、これらのコドン最適化HIV−1 Polに基づくDNAワクチンのオープンリーディングフレームは、コドン最適化HIV−1 Pol(例えば、配列番号2)、アミノ末端局在化リーダー配列に融合したコドン最適化HIV−1 Pol(例えば、配列番号6)および特に好ましくは、例えば実施例19におけるMRKAd5−Pol構築物により具体的に示された、野生型Polに関連した有意なPR、RT、RNアーゼまたはIN活性を欠く生物学的に不活性化されたpol(「inact opt Pol」;例えば配列番号4)により例示される。また、IA Polタンパク質のアミノ末端領域にリーダーペプチドを含有する配列番号4に関連した構築物が意図される。機能的リーダーペプチドをコードするヌクレオチド配列を伴って又は伴わないでそれぞれのHIV−1 Polコード領域に作動的に結合した調節領域を含有する適当なDNAプラスミドベクター内に、特定の構築物を連結する。この目的において、本明細書に開示する種々のHIV−1 Pol構築物は、野生型Pol(配列番号2に記載のとおりWT opt PolをコードするDNA分子を含む)、tPA−opt WTPol(配列番号6に記載のとおりtPA PolをコードするDNA分子を含む)、inact opt Pol(配列番号4に記載のとおりIA PolをコードするDNA分子を含む)およびtPA−inact opt Pol(配列番号8に記載のとおりtPA−inact opt PolをコードするDNA分子を含む)を含む(これらに限定されるものではない)本発明の増強された第1世代Adベクター(例えば、一連のMRKAd5polアデノウイルスワクチンベクター)にクローニングするためのオープンリーディングフレームに関する。polに基づく形態の増強された第1世代アデノウイルスワクチンは、霊長類、特にヒトを含む宿主に投与されると、CTLおよびTh細胞性免疫応答を惹起する。前記のとおり、本発明の細胞性免疫誘導性ワクチンの効果は、これまでに未感染の個体に対する、より低い伝達率、および/またはHIV−1感染の無症候期を延長するような感染個体内のウイルス負荷のレベルの減少にあるはずである。
【0128】
本発明は更に、HIV gagおよびpol抗原と同様に、マウスおよび非ヒト霊長類での研究において投与された後に細胞性免疫応答を生成する一連のMRKAd5nefに基づくアデノウイルスワクチンに関する。その一連のMRKAd5nefは、本発明では、HIV nefの修飾形態と組合せて、改善されたMRKアデノウイルスバックボーンを使用することにより例示される。これらの例示されているMRKAd5nefベクターは以下のとおりである:(1)改善されたMRKAd5バックボーン、ヒトCMVプロモーター、無傷Ad5 E3遺伝子および修飾されたnef遺伝子を含むMRKAd5hCMV−nef(G2A,LLAA)(E3+);(2)ヒトCMVプロモーターがマウスCMVプロモーターで置換されている以外は前記(1)と同じであるMRKAd5mCMV−nef(G2A,LLAA)(E3+);および(3)該nefトランスジーンがtpanef(LLAA)である以外は(2)と同じであるMRKAd5mCMV−tpanef(LLAA)(E3+)。HIV−1 NefおよびHIV−1 Nef修飾体のコドン最適化形態は、実質的には、2000年12月15日付け出願の米国特許出願第09/738,782号および同様に2000年12月15日付け出願のPCT国際出願PCT/US00/34162(両方の文書を参照により本明細書に組み入れることとする)に記載のとおりである。コドン最適化NefおよびNef修飾体の特定の実施形態は、HIV−1 jfrl分離体からのHIV−1 NefをコードするDNA分子に関する。この場合、該コドンは、ヒトのような哺乳類系内での発現に関して最適化されている。このタンパク質をコードするDNA分子は本発明では配列番号9として開示されており、発現されるオープンリーディングフレームは本発明では配列番号10として開示されている。本発明のアデノウイルスベクターにおける使用のためのNefに基づくコード領域のもう1つの実施形態は、該HIV−1 NefポリペプチドのNH2末端に融合したヒトプラスミノーゲンアクチベーター(tpa)リーダーペプチドを含有するタンパク質をコードするコドン最適化DNA分子を含む。このタンパク質をコードするDNA分子は本発明では配列番号11として開示されており、発現されるオープンリーディングフレームは本発明では配列番号12として開示されている。もう1つの修飾されたNef最適化コード領域は、最適化HIV−1 NefをコードするDNA分子に関する。この場合、該オープンリーディングフレームは、アミノ末端ミリスチル化部位における修飾(Gly−2からAla−2)、およびLeu−174−Leu−175ジロイシンモチーフからAla−174−Ala−175への置換をコードする(本発明ではopt nef(G2A,LLAA)として記載されている)。このタンパク質をコードするDNA分子は本発明では配列番号13として開示されており、発現されるオープンリーディングフレームは本発明では配列番号14として開示されている。MRKAd5nefベクターである(1)MRKAd5hCMV−nef(G2A,LLAA)(E3+)および(2)MRKAd5mCMV−nef(G2A,LLAA)(E3+)はこのトランスジーンを含有する。更なる実施形態は、最適化HIV−1 NefをコードするDNA分子に関する。この場合、アミノ末端ミリスチル化部位およびジロイシンモチーフが欠失しており、tPAリーダーペプチドを含む。このDNA分子opt tpanef(LLAA)は、HIV−1 Nef(jfrl)のアミノ酸残基6−216に融合したtPAリーダー配列を含有するNefタンパク質をコードするオープンリーディングフレームを含み、ここで、Leu−174およびLeu−175はAla−174およびAla−175で置換されており、これは本発明ではopt tpanef(LLAA)と称され、本発明では配列番号15として開示されており、発現されるオープンリーディングフレームは本発明では配列番号16として開示されている。MRKAd5nefベクター「MRKAd5mCMV−tpanef(LLAA)(E3+)」はこのトランスジーンを含有する。
【0129】
本明細書に記載の改善されたMRKA5gagアデノウイルスワクチンベクターに加えて、MRKAd5polおよびMRKAd5nefアデノウイルスベクターの作製は、増強したHIVワクチン能力をもたらす。すなわち、このトリオのアデノウイルスワクチンベクターの作製はすべて、宿主投与後に有効な細胞性免疫応答を生成することが示されており、これらのワクチン候補を、単独で投与する可能性だけでなく、好ましくは、二価(すなわち、gagおよびnef、gagおよびpol、またはpolおよびnef成分)または三価ワクチン(すなわち、gag、polおよびnef成分)の一部として投与する可能性をも与える。したがって、本発明の好ましい態様はワクチン製剤、ならびに投与およびそれに付随する3つの異なる一連のMRKAd5に基づくアデノウイルスベクターワクチンの製剤化に関連した宿主細胞性免疫応答の生成の関連方法である。もちろん、異なるHIV抗原に基づくこの一連のMRKAd5ワクチンは、二価または三価ワクチンの製剤化の機会、あるいは恐らくは、合理的な時間枠内の1以上の一価または二価製剤の別々の製剤の投与の機会の拡大を促進する。また、特異的抗原由来の複数であるが異なる成分を含む組合せ様式法を開始することも、本発明の範囲内である。決して限定的ではない具体例としては、1つのワクチンベクターが野生型Pol(配列番号2)を発現しもう1つのMRKAd5polベクターが不活性化Pol(配列番号6)を発現する別々のMRKAd5polベクターが挙げられるであろう。もう1つの具体例としては、1つのワクチンベクターがNefのtPA/LLAA形態(配列番号16)を発現しもう1つのMRKAd5nefベクターがNefのG2A,LLAA修飾形態(配列番号14)を発現する別々のMRKAd5nefベクターが挙げられるであろう。したがって、本発明のMRKAd5アデノウイルスベクターは、複数の異なるHIV抗原クラスと組合せて使用することができる。各HIV抗原クラスを配列操作に付して、多数の潜在的なワクチンの組合せを得る。そのような組合せは本発明の範囲内である。そのような組合せ様式ワクチン製剤および投与の利用は、単一様式法での接種と比較して、より一層強力な細胞性免疫応答を惹起する可能性を増加させる。
【0130】
本発明はまた、初回/追加ワクチン接種計画における一連のMRKAd5gag、polおよびnefアデノウイルスワクチンの一重、二重または三重様式の投与法の適用に関する。この初回/追加計画は、本明細書に開示されている一連のMRKAd5gag、polおよびnefアデノウイルスワクチンの任意の合理的な組合せを含みうる。さらに、初回/追加法はまた、他のウイルスおよび/または非ウイルスDNAワクチンを含みうる。アデノウイルスワクチンベクター法に加えられる好ましいものは、プラスミドDNAワクチン、特に、本明細書に開示されているコドン最適化gag、polおよびnef構築物の少なくとも1つを含有するDNAプラスミドワクチンを含むが、これに限定されるものではない。
【0131】
したがって、本発明の1つの態様は、gagをコードするプラスミドDNAと組合された初回/追加抗原刺激法における最適化gag遺伝子を含有するアデノウイルスベクターの投与である。このプラスミドを、アデノウイルスベクターの構築において使用するアデノウイルス含有シャトルプラスミドから区別するために、このプラスミドを「ワクチンプラスミド」または「DNAプラスミドワクチン」と称することにする。この投与法において使用する好ましいワクチンプラスミドは、1998年2月3日付け出願の係属中の米国特許出願第09/017,981号および1998年8月13日付け公開のWO98/34640(それらの両方を参照により本明細書に組み入れることとする)に開示されている。簡単に説明すると、好ましいワクチンプラスミドはV1Jns−FLgagと称され、これは、本発明のアデノウイルスベクターと同じコドン最適化gag遺伝子を発現する(完全長p55 gagの例示最適化コドン形態のヌクレオチド配列に関しては、図2を参照されたい)。V1Jnsと称されるワクチンプラスミドバックボーンは、CMV最初期(IE)プロモーターおよびイントロンA、遺伝子発現調節要素としてのウシ成長ホルモン由来ポリアデニル化および転写終結配列、ならびに最小pUCバックボーンを含有する(Montgomeryら,1993,DNA Cell Biol.12:777−783を参照されたい)。該pUC配列は、大腸菌(E.coli)内での高レベルのプラスミド産生を可能にし、アンピシリン耐性遺伝子の代わりにネオマイシン耐性遺伝子を有していてカナマイシンの存在下での選択増殖をもたらす。あるいは、イントロンAが欠失したCMVプロモーターを有するワクチンプラスミドを使用することが可能である。当業者は、これらの特定の構築物が代替的ワクチンプラスミドベクターで容易に置換されうると認識するであろう。本発明においては特に、そのような代替的プラスミドDNAワクチンベクターの使用が予想される。
【0132】
本発明のもう1つの態様は、HIV pol抗原、好ましくは、コドン最適化形態のpolをコードするワクチンプラスミドを含む、および同様に好ましくは、配列番号2、4、6および8に示すPol抗原の群から選ばれるPol抗原をコードするヌクレオチド配列を含んでなるワクチンプラスミドを含む初回/追加抗原刺激法である。種々の生物学的に活性な形態のHIV−1 Polをコードする種々の潜在的DNAプラスミドワクチンにおいては、関心のあるHIV−1 Pol遺伝子の投与、細胞内運搬および発現は宿主CTLおよびTh応答を惹起する。本発明の好ましい合成DNA分子は、コドン最適化野生型Pol(Pro活性を有さない)および種々のコドン最適化不活性化HIV−1 Polタンパク質をコードする。本明細書に開示されているHIV−1 polオープンリーディングフレームは、医薬用途、特に組換えアデノウイルスワクチン、特に、本明細書に記載の増強された第1世代組換えアデノウイルスワクチンにより運搬されるヒトへの投与に、特に好ましい。本発明のこの態様のいくつかの実施形態、すなわち、完全長polをコードするか又は本明細書に記載の修飾体もしくは融合体をコードするかにかかわらず、哺乳類、特にヒトにおける発現に関してコドン使用頻度が最適化されたHIV−1 polオープンリーディングフレームを含むDNA分子は、後記に詳しく説明されている。この場合もまた、関心のあるそれぞれのHIV−1 Pol遺伝子の発現を促進し、投与後に、宿主CTLおよびTh応答を惹起するよう、これらのDNA配列は、本明細書に記載の例示されている組換えアデノウイルスベクターのような組換えアデノウイルスベクター内に適切に配置されている。この場合もまた、これらの好ましい(決して限定的ではない)pol遺伝子は本明細書に開示されているとおりであり、実質的には、2000年12月21日付け出願の米国特許出願第09/745,221号および同様に2000年12月21日付け出願のPCT国際出願PCT/US00/34724(両方の文書を参照により本明細書に組み入れることとする)に記載されているとおりである。
【0133】
組合せ様式および/または初回/追加抗原刺激法において有用な第3の一連のワクチンプラスミドは、HIV nef抗原またはその生物学的および/または免疫学的に関連した修飾体をコードするワクチンプラスミドである。他の箇所に記載されているとおり、好ましいワクチンプラスミドは、nefのコドン最適化形態を含有し、また、好ましくは、配列番号10、12、14および16に示すNef抗原の群から選ばれるNef抗原をコードするヌクレオチド配列を含む。これらの好ましいnefコード領域は本明細書に開示されており、2000年12月15日付け出願の米国特許出願第09/738,782号および同様に2000年12月15日付け出願のPCT国際出願PCT/US00/34162(両方の文書を参照により本明細書に組み入れることとする)に記載されている。
【0134】
したがって、本発明のアデノウイルスワクチンおよびプラスミドDNAワクチンは単独で投与することが可能であり、あるいは初回抗原刺激および追加抗原刺激投与法の一部でありうる。混合様式の初回および追加接種計画は、既存の抗ベクター免疫応答が存在する場合には特に、増強された免疫応答をもたらす。本発明のこの1つの態様は、該プラスミドワクチンを少なくとも1回投与することにより該プラスミドワクチンで対象を初回抗原刺激し、所定の長さの時間を経過させ、ついで該アデノウイルスワクチンを投与することにより追加抗原刺激する方法である。通常、複数の初回抗原刺激、典型的には1〜4回の初回抗原刺激を行うが、より多数の初回抗原刺激を行うことが可能である。初回抗原刺激と追加抗原刺激との間の時間の長さは、典型的には約4ヶ月〜1年と様々となりうるが、他の時間枠を用いることも可能である。アカゲザルでの実験においては、該動物をプラスミドワクチンで4回初回抗原刺激し、ついで4ヶ月後に該アデノウイルスワクチンで追加抗原刺激した。それらの細胞性免疫応答は、アデノウイルスワクチンのみが投与された動物の場合より顕著に高かった。初回抗原刺激法の利用は、既存の抗アデノウイルス免疫応答を対象者が有する場合には特に好ましいかもしれない。
【0135】
それに加えて及びその代わりに、本明細書に開示されているMRKAd5アデノウイルスワクチンのような複数のHIV−1ウイルス抗原を適当なシャトルプラスミドに連結させて、複数のオープンリーディングフレームを含む前アデノウイルスプラスミドを生成させることができる。例えば、三価ベクターは、E3(−)またはE3(+)バックグラウンド、好ましくはE3欠失バックボーンにおけるgag−pol−nef融合体、あるいは、おそらくは、「2+1」二価ワクチン、例えば、同じMRKAd5バックボーン内のgag−pol融合体(すなわち、コドン最適化p55 gagおよび不活性化最適化pol;実施例29および表25)(各オープンリーディングフレームは別個のプロモーターおよび転写終結配列に作動的に結合している)を含みうる。あるいは、それらの2つのオープンリーディングフレームが単一のプロモーターに作動的に結合しており、該オープンリーディングフレームがIRES(internal ribosome entry sequence)に作動的に連結していることが可能である(参照により本明細書に組み入れる国際公開番号WO95/24485に開示されているとおりである)。図9は、複数のプロモーターおよび終結配列の使用が、同様の増殖特性をもたらすことを示しており、図28は、これらのMRKAd5gagに基づくベクターがまた、少なくとも継代21にわたり安定であることを示している。IRESに基づく技術を用いない場合には、ベクター安定性が最良に維持されるよう、それぞれのオープンリーディングフレームを支持するために、異なるプロモーターを使用することが好ましい。決して限定的ではない例として挙げると、潜在的な多トランスジーンワクチンは、E3欠失バックボーン中のhCMV−gagpol−bGHpA+mCMV−nef−SPAまたはhCMV−gagpol−bGHpA+mCMV−nef−SPA(E3+)のような3トランスジーンベクターを含みうる。本発明の潜在的な「2+1」二価ワクチンは、すべてのトランスジーンがE1平行配向であるE3+バックボーン中のhCMV−pol−bGHpA(ベクター#2)と組合されたE3+バックボーン中のhCMV−gag−bGHpA+mCMV−nef−SPA(ベクター#1)でありうる。前記のgag−pol融合体以外の融合構築物も、種々の二価ワクチン法での使用に適しており、お互いに融合した任意の2つのHIV抗原(例えば、nef−polおよびgag−nef)から構成されうる。これらのアデノウイルス組成物は、前記のとおり、好ましくは、投与後に生成する免疫応答を多様化するために追加的なHIV抗原を含むアデノウイルス組成物と共に運搬される。したがって、単一の又は可能な第2のアデノウイルスベクターとして運搬される多価ワクチンが本発明の一部と意図されることは明らかである。この場合もまた、この投与様式は、アデノウイルスに基づく有効なHIV−1ワクチンが組合せ様式法により投与されうるもう1つの例である。しかし、開示されているアデノウイルスベクター用のインサートの決定に関しては、アデノウイルスベクターの有効パッケージング限界を十分に考慮する必要があることに注目することは重要である。アデノウイルスは、該野生型Ad5配列の約105%のクローニング容量上限を示すことが示されている。
【0136】
発現用に選択した遺伝子にかかわらず、該配列をヒト細胞環境中の発現に関して「最適化」することが好ましい。4つの可能なヌクレオチド塩基の「トリプレット」コドンは、異なる64個の形態で存在しうる。これらの形態が僅か20個の異なるアミノ酸(ならびに転写の開始および終結)のメッセージを与えるに過ぎないことは、いくつかのアミノ酸が2以上のコドンによりコードされうることを意味する。実際に、いくつかのアミノ酸は6つもの「重複」した代替的コドンを有し、一方、他のいくつかは、単一の必要なコドンを有する。完全には理解されていない理由により、代替的コドンは種々の細胞型の内因性DNA内に決して一様には存在せず、或る細胞型においては或るコドンに関する可変的天然階層または「優先性」が存在するようである。例えば、アミノ酸ロイシンは、CTA、CTC、CTG、CTT、TTAおよびTTG(これらはそれぞれ、mRNコドンCUA、CUC、CUG、CUU、UUAおよびUUGに対応する)を含む6つのDNAコドンのいずれかにより特定される。大腸菌(E.coli)の内因性DNAは、最も一般的には、ロイシン特定コドンCTGを含有し、一方、酵母および粘菌のDNAは、最も一般的には、ロイシン特定コドンTTAを含有することが、微生物についてのゲノムコドン出現頻度の詳細な分析から示されている。この階層を考慮すると、大腸菌(E.coli)宿主によるロイシンに富むポリペプチドの高レベルの発現が得られる可能性は、コドン使用頻度に或る程度は左右されると、一般に考えられている。例えば、TTAコドンに富む遺伝子は、十中八九は大腸菌(E.coli)内では発現されにくく、一方、CTGに富む遺伝子は、おそらく該ポリペプチドを高度に発現するであろう。同様に、酵母細胞が、ロイシンに富むポリペプチドの発現のための意図される形質転換宿主細胞である場合には、挿入されるDNA内で使用するための好ましいコドンはTTAであろう。
【0137】
組換えDNA技術へのコドン優先性現象の関与は明白であり、該現象は、成功裏に形質転換された宿主生物において外因性遺伝子の高い発現レベルがこれまで多くの場合に達成されていないことを説明するのに役立ちうる。すなわち、該挿入遺伝子内には、それほど「好まし」くないコドンが反復的に存在している可能性があり、発現用の宿主細胞装置が、それほど効率的には機能していない可能性があるのである。この現象は、意図される宿主細胞の好ましいコドンを含むように設計された合成遺伝子が、組換えDNA技術の実施のための好ましい形態の外来遺伝物質を与えることを示唆している。したがって、本発明の1つの態様は、アデノウイルスベクター、またはワクチンプラスミドとアデノウイルスベクターとの何らかの組合せであり、この場合、それらの両方は特に、ヒト細胞環境中での発現に関してコドンが最適化された遺伝子を含む。本明細書に記載のとおり、本発明での使用のための好ましい遺伝子はコドン最適化HIV遺伝子であり、特に、HIV gag、polまたはnefである。
【0138】
本発明のアデノウイルスベクターは、Hittら,1997“Human Adenovirus Vectors for Gene Transfer into Mammalian Cells”Advances in Pharmacology 40:137−206(これを参照により本明細書に組み入れることとする)に概説されているような公知技術を用いて構築することができる。
【0139】
本発明のアデノウイルスベクターを構築する際には、プラスミドまたはシャトルベクター内にそれを挿入することがしばしば簡便である。これらの技術は公知であり、Hittら,前掲に記載されている。本発明は特に、該アデノウイルスと、シャトルプラスミド内に挿入された場合のアデノウイルスとの両方を含む。
【0140】
好ましいシャトルベクターは、アデノウイルス部分およびプラスミド部分を含有する。該アデノウイルス部分は、実質的には、アデノウイルス配列(非機能的または欠失したE1およびE3領域を伴う)と簡便な制限部位に隣接した遺伝子発現カセットとを含有する前記のアデノウイルスベクターと同じである。該シャトルベクターのプラスミド部分は、しばしば、原核性プロモーターの転写制御下で抗生物質耐性マーカーを含有していて、該抗生物質の発現は真核細胞内では生じない。アンピシリン耐性遺伝子、ネオマイシン耐性遺伝子および他の医薬上許容される抗生物質耐性マーカーを使用することができる。原核生物における発酵による該ポリヌクレオチドの高レベルの産生を補助するためには、該シャトルベクターが原核性複製起点を含有し高コピー数であることが有利である。多数の商業的に入手可能な原核性クローニングベクターがこれらの利点をもたらす。非必須DNA配列を除去することが望ましい。また、該ベクターは真核細胞内で複製不能であることが望ましい。これは、受容者のゲノム内へのポリヌクレオチドワクチン配列の組込みの危険性を最小限に抑える。該ポリヌクレオチドの発現を特定の組織型に限定することが望ましい場合には、組織特異的プロモーターまたはエンハンサーを使用することができる。
【0141】
本発明の1つの実施形態においては、該前プラスミド、例えば、pMRKAd5pol、pMRKAd5nefおよびpMRKAd5gagは、MRKHVE3(およびE3−形態用にはMRKHVO)バックボーンおよび適当なシャトルベクターを使用する相同組換えにより作製することが可能であり、pMRKAd5polに関しては図22に、そしてpMRKAd5nefに関しては図23に示されているとおりである。線状形態のプラスミドは、PER.C6(登録商標)細胞への侵入後に複製されることが可能であり、ウイルスが産生される。感染細胞および培地は、ウイルス複製が完了した後に回収した。
【0142】
ウイルスベクターは、公知細胞系293およびPER.C6(登録商標)を含む種々のE1相補性細胞系内で増殖しうる。これらの細胞系は共に、アデノウイルスE1遺伝子産物を発現する。PER.C6(登録商標)は、WO 97/00326(1997年1月3日付け公開)、および発行されている米国特許第6,033,908号(それらの両方を参照により本明細書に組み入れることとする)に記載されている。それは、複製欠損型(FG)アデノウイルスの産生を相補するE1遺伝子セグメントで形質導入された初代ヒト網膜芽細胞系であるが、相同組換えによる複製許容型アデノウイルスの生成を妨げるように設計される。特に関心が持たれる細胞は、PER.C6(登録商標)のように、459bp〜3510bp(これを含む)のAD5E1AおよびE1B遺伝子をコードするトランスジーンで安定に形質転換されている。293細胞は、参照により本明細書に組み入れるGrahamら,1997 J.Gen.Virol 36:59−72に記載されている。前記のとおり、使用する相補性細胞系内に存在するアデノウイルス配列を考慮しなければならない。組換えの可能性を最小限に抑えたい場合には、該配列は、該ベクター内に存在する配列と重複しないことが重要である。
【0143】
前記に従い作製したベクターは、免疫応答の誘導において、より有効であることが判明しており、したがって非常に有望なワクチン候補に相当する。より詳しくは、強力な異種プロモーターで調節されるコドン最適化HIV gag遺伝子を保持する前記の第1世代アデノウイルスベクターはヒト抗HIVワクチンとして使用することが可能であり、免疫応答を誘導しうることが判明している。
【0144】
DNA構築物を製造し精製するための分子生物学の標準的な技術は本発明のDNA免疫原の製造を可能にする。
【0145】
本発明のアデノウイルスベクターを含むワクチン組成物は、バッファー、正常食塩水またはリン酸緩衝食塩水、スクロース、他の塩およびポリソルベートのような生理的に許容される成分を含有しうる。1つの好ましい製剤は、2.5〜10mM TRISバッファー、好ましくは約5mM TRISバッファー;25〜100mM NaCl、好ましくは約75mM NaCl;2.5〜10% スクロース、好ましくは約5% スクロース;0.01〜2mM MgCl2;および0.001%〜0.01% ポリソルベート80(植物由来)を含有する。pHは、約7.0〜9.0、好ましくは約8.0であるべきである。当業者は、該製剤を製造するために他の通常のワクチン賦形剤も使用しうると理解するであろう。好ましい製剤は、5mM TRIS、75mM NaCl、5% スクロース、1mM MgCl2、0.005% ポリソルベート80をpH8.0で含有する。これは、ガラス表面へのウイルスの吸着の可能性を最小限に抑えAd5の安定性にとって最適に近いpHおよび二価陽イオン組成を有する。それは、筋肉注射の際に組織刺激を引き起こさない。それは、好ましくは、使用まで凍結される。
【0146】
ワクチン受容者に導入されるワクチン組成物中のアデノウイルス粒子の量は、使用する転写および翻訳プロモーターの強度ならびに発現遺伝子産物の免疫原性に左右されるであろう。一般には、免疫学的または予防的に有効な用量である1×107〜1×1012粒子、好ましくは約1×1010〜1×1011粒子を筋肉組織内に直接投与する。皮下注射、皮内導入、皮膚を介した圧入(impression)および他の投与様式、例えば腹腔内、静脈内または吸入運搬も意図される。また、追加ワクチン接種が行われると意図される。HIVアデノウイルスベクターでのワクチン接種の後、後続のHIVアデノウイルスベクターおよび/またはプラスミドでの追加抗原刺激が望ましいかもしれない。本発明のワクチン組成物の非経口的導入と同時またはその後のインターロイキン12タンパク質の非経口投与、例えば静脈内、筋肉内、皮下または他の手段の投与も有利である。
【0147】
本発明のポリヌクレオチドのアデノウイルスベクターおよび/またはワクチンプラスミドは、受容者の免疫系に影響を及ぼすいずれのタンパク質、アジュバントまたは他の物質とも会合していないことが可能である。この場合、該ベクターは、生理的に許容される溶液、例えば無菌食塩水または無菌緩衝食塩水(これらに限定されるものではない)中に存在することが望ましい。あるいは、該ベクターは、免疫応答を増強する当技術分野で公知のアジュバント(すなわち、「生物学的に有効」なアジュバント)、例えばタンパク質または他の担体と会合していることが可能である。本発明のワクチンプラスミドは、例えば、アジュバントの存在下または不存在下、食塩水(例えば、PBS)中で運搬することができる。好ましいアジュバントはAlumまたはCRL 1005 Block Copolymerである。DNAの細胞取り込みを補助する物質、例えばカルシウムイオン(これに限定されるものではない)を有利に使用することができる。これらの物質は、本発明では一般には、トランスフェクション促進試薬および医薬上許容される担体と称される。ポリヌクレオチドでコーティングされたマイクロプロジェクタイル(microprojectile)をコーティングするための技術は当技術分野で公知であり、本発明においても有用である。
【0148】
本発明はまた、第1アデノウイルスベクターを投与し次いで追加抗原刺激投与を行う初回抗原刺激および追加抗原刺激法を含む。該追加抗原刺激投与は、選択された時間間隔で反復することができる。あるいは、好ましい接種計画は、第1アデノウイルス血清型で初回抗原刺激し、ついで第2アデノウイルス血清型で追加抗原刺激することを含む。より好ましくは、該接種計画は、第1アデノウイルス血清型で初回抗原刺激し、ついで第2アデノウイルス血清型で追加抗原刺激することを含み、ここで、該第1および第2アデノウイルス血清型はアデノウイルスの別々の亜群内に分類される。前記の初回/追加抗原刺激計画は、選択したアデノウイルスベクターに対して既存免疫が確認されている場合に特に好ましい。このタイプの計画においては、個体または個体集団を、既存免疫が確認されている血清型以外の血清型のアデノウイルスで初回抗原刺激する。これは、該第1アデノウイルスが、該第2アデノウイルスに対する既存免疫を避けつつ該トランスジーンの十分な発現を達成するのを可能にし、さらに、該追加抗原刺激アデノウイルスを介した該トランスジーンの後続運搬が、より有効となるのを可能にする。アデノウイルス血清型5は、そのような計画が望ましいと考えられるウイルスの一例である。したがって、本発明では、非C群アデノウイルス(例えば、A群アデノウイルスであるAd12、D群アデノウイルスであるAd24、またはB群アデノウイルスであるAd35)で初回抗原刺激して抗Ad5免疫を回避し、ついでC群アデノウイルスであるAd5で追加抗原刺激することを選択しうるであろう。もう1つの好ましい実施形態は、異なるアデノウイルス(非ヒトアデノウイルスを含む)ワクチンの投与およびそれに続く、開示されているアデノウイルスワクチンの投与を含む。別法においては、まず、関心のあるウイルス抗原を、アデノウイルスに基づくワクチン以外のウイルスワクチンを介して運搬し、ついで、開示されているアデノウイルスワクチンで行う。別のウイルスワクチンは、ポックスウイルスおよびベネズエラウマ脳炎ウイルスを含むが、これらに限定されるものではない。
【0149】
膨大なヒトおよび動物データは、HIV感染の抑制(または排除)における細胞性免疫応答、特にCTLの重要性を支持している。ヒトにおいては、初感染後に非常に高いレベルのCTLが生じ、それはウイルス血症の抑制と相関している。HIVに繰返し曝露されたにもかかわらず未感染のままである幾つかの小さな個体群が記載されている。これらのコホートの幾つかにおいて、CTLが注目されている。HIV感染のSIVモデルにおいては、初感染後に同様にCTLが生じ、抗CD8モノクローナル抗体の添加はこの感染抑制を妨げ疾患の進行を招くことが示されている。本発明では、CTLを誘導するためにアデノウイルスワクチンを単独で又はプラスミドワクチンと組合せて使用する。
【0150】
本発明を更に例示するために、以下に非限定的な実施例を記載する。
【0151】
実施例1
hCMVプロモーターのイントロンA部分の除去
hCMVプロモーターを増幅するための出発物質として、GMP等級のpVIJnsHIVgagを使用した。pVIJnsHIVgagは、CMV最初期(IE)プロモーターおよびイントロンA、完全長コドン最適化HIV gag遺伝子、ウシ成長ホルモン由来ポリアデニル化および転写終結配列、ならびに最小pUCバックボーンを含むプラスミドである。該プラスミドバックボーンの説明としては、Montgomeryら,前掲を参照されたい。該増幅は、hCMVプロモーターに隣接するよう適切に配置されるプライマーで行った。5’プライマーはhCMVプロモーターのMsc1部位の上流に配置され、3’プライマー(BglII認識配列を含有するよう設計されている)はhCMVプロモーターの3’側に配置された。全hCMVプロモーター(−イントロンA)を含む得られたPCR産物(高忠実度Taqポリメラーゼを使用)を、TOPO PCR平滑ベクター内にクローニングし、ついでMsc1およびBglIIでの二重消化により取り出した。ついでこの断片を、Msc1およびBglII消化後に元のプロモーター、イントロンAおよびgag遺伝子が除去された元のGMP等級のpV1JnsHIVgagプラスミド内に再びクローニングした。この連結反応は、元のpV1JnsHIVgagベクターバックボーン内のhCMVプロモーター(−イントロンA)+bGHpA発現カセットの構築をもたらした。このベクターはpVIJnsCMV(無イントロン)と称される。
【0152】
BglII消化を用いてpV1JnsHIVgagからFLgag遺伝子を切り出し、その1,526bpの遺伝子をゲル精製し、pVIJnsCMV(無イントロン)内にBglII部位においてクローニングした。正しい配向でFlgag遺伝子を保持するクローンを同定するために、Sma1制限酵素を使用してコロニーをスクリーニングした。pV1JnsCMV(無イントロン)−FLgag−bGHpAと称されるこのプラスミドを完全に配列決定して、配列の完全性を確認した。
【0153】
また、2つの追加的なトランスジーンを構築した。プラスミドpV1JnsCMV(無イントロン)−FLgag−SPAは、50ヌクレオチド長の短い合成ポリAシグナル(SPA)でウシ成長ホルモンポリアデニル化シグナルが置換されていること以外はpV1JnsCMV(無イントロン)−FLgag−bGHpAと同一である。該SPAの配列は以下に示すとおりであり、ここでは、必須成分(それぞれ、ポリ(A)部位、(GH)nおよび(T)n)が下線で示されている:AATAAAAGATCTTTATTTTCATTAGATCTGTGTGTTGGTTTTTTGTGTG(配列番号18)。
【0154】
プラスミドpV1Jns−mGMV−FLgag−bGHpAは、hCMVプロモーターが除去されてマウスCMV(mCMV)プロモーターで置換されていること以外はpV1JnsCMV(無イントロン)−FLgag−bGHpAと同一である。
【0155】
図3は、元のトランスジーンと比較して新規トランスジーン構築物を図示する。
【0156】
実施例2
修飾gagトランスジーンに関するgag発現アッセイ
一過性組織培養トランスフェクション実験から得た培養上清についてGag Elisaを行った。この場合、共にイントロンAを欠く2つの新規hCMV含有プラスミド構築物pV1JnsCMV(無イントロン)−FLgag−bGHpAおよびpV1JnsCMV(無イントロン)−FLgag−SPAを、前記のとおりhCMVプロモーターの一部としてイントロンAを有するpV1JnsHIVgagと比較した。以下の表2は、GMP等級の元のプラスミドと比較して該新規gagプラスミドのインビトロgag発現データを示している。表2に示す結果は、それらの新規hCMV gagプラスミド構築物の両方が、hCMVプロモーターのイントロンA部分を含有する元のプラスミド構築物と比較しうる発現能を有することを示している。
【0157】
【表1】
 【0158】
【0158】
実施例3
修飾gagトランスジーンに関するげっ歯類(Balb/c)研究
前記の2つの新規プラスミド構築物pV1JnsCMV(無イントロン)−FLgag−bGHpAおよびpV1JnsCMV(無イントロン)−FLgag−SPAに関して、CMVプロモーターのイントロンA部分を有する前記構築物pV1JnsHIVgagとそれらを比較するために、げっ歯類での研究を行った。gag抗体およびElispot応答(1999年7月6日付け出願の米国仮特許出願第60/142,631号および1999年8月13日付け出願の米国特許出願第60/148,981号に基づく優先権を主張している2000年7月3日付け出願のPCT国際出願番号PCT/US00/18332(WO 01/02607)に記載されている;これらの3つの出願を参照により本明細書に組み入れることとする)を測定した。以下の表3に示す結果は、該新規プラスミド構築物が、試験したプラスミドDNAの両方の用量(20μgおよび200μg)で、その抗体およびT細胞応答に関して、Balc/cマウスにおいて元の構築物と同等に挙動したことを示している。
【0159】
実施例4
【0160】
【表2】
【0161】
修飾シャトルベクター「MRKpdelE1シャトル」の構築
元のAd5シャトルベクター(pdelElsp1A;Ad5のヌクレオチド341〜3524の複数のクローニング領域と共に塩基対1〜341および3524〜5798のAd5配列を含むベクター)に対する修飾は、以下のとおりに連続的なクローニング工程において行う以下の3つの操作を含んでいた:
(1)左ITR領域を、該ベクターバックボーンと該アデノウイルス左ITR配列との間の結合部にPac1部位を含むよう伸長させた。これは、細菌相同組換え系を用いる、より容易な操作を可能にする。
(2)パッケージング領域を、342bp〜450bpの野生型(WT)アデノウイルスの配列を含むよう伸長させた。
(3)pIXの下流の領域を13ヌクレオチド(すなわち、ヌクレオチド3511〜3523に)伸長させた。
これらの修飾(図4)は、形質転換PER.C6(登録商標)細胞系内に存在するE1A/E1B遺伝子のいずれの部分とも重複することなくE1欠失のサイズを有効に減少させた。すべての操作は、AdシャトルベクターpdelElsp1Aを修飾することにより行った。
【0162】
該修飾を該シャトルベクターに施したら、該変化は、大腸菌(E.coli)BJ5183化学的コンピテント細胞を使用する細菌相同組換えにより元のAd5アデノベクターバックボーン(pAdHVOおよびpAdHVE3)内に組み込まれた。
【0163】
実施例5
修飾アデノベクターバックボーン(E3+およびE3−)の構築
元のアデノベクターpAdHVO(E1およびE3領域を含むヌクレオチド以外のすべてのAd5配列を含む)およびpADHVE3(E1領域を含むヌクレオチド以外のすべてのAd5配列を含む)をそれぞれ、それらがE1領域への修飾を含有するように再構築した。これは、新たに修飾されたシャトルベクター(MRKpdelE1シャトル)をPac1およびBstZ1101で消化し、該アデノウイルス配列に対応する2,734bpの断片を単離することにより達成された。この断片を、Cla1線状化pAdHVO(E3−アデノベクター)またはCla1線状化pAdHVE3(E3+アデノベクター)からのDNAと共に大腸菌(E.coli)BJ5183コンピテント細胞内に同時形質転換した。各形質転換からの少なくとも2つのコロニーを選択し、濁り状態に達するまで6〜8時間、Terrific(商標)ブロス内で成長させた。DNAを各細胞ペレットから抽出し、ついで大腸菌(E.coli)XL1コンピテント細胞内に形質転換した。各形質転換からただ1つのコロニーを選択し、プラスミドDNA精製のために成長させた。正しいクローンを確認するために、該プラスミドを制限消化により分析した。該修飾アデノベクターをMRKpAdHVO(E3−プラスミド)およびMRKpAdHVE3(E3+プラスミド)と命名した。古い形態のアデノベクターおよびこれらの新規アデノベクターからのウイルス(それぞれMRKHVOおよびMRKHVE3)を、以下の一連のウイルス競合実験に対応させるために、PER.C6(登録商標)細胞系内で産生させた。また、元のシャトルベクターのマルチクローニング部位はClaI、BamHI、XhoI、EcoRV、HindIII、SalIおよびBglII部位を含有していた。このMCSを、NotI、ClaI、EcoRVおよびAscI部位を含有する新規MCSで置換した。この新規MCSは、該パッケージング領域およびpIX遺伝子に施された修飾と共に、MRKpAdHVOおよびMRKpAdHVE3前プラスミドに導入されている。
【0164】
実施例6
パッケージングシグナル伸長の効果の分析
E1欠失領域に施された修飾の効果を調べるために、元のバックボーン(pAdHVE3)および新規バックボーン(MRKpAdHVE3)から得られたウイルスを等しいMOI比(1:1および5:5)で混合し、数ラウンドにわたり継代した。図5、実験#1を参照されたい。該実験におけるウイルスの両方は、E3遺伝子を無傷で含有しており、トランスジーンを含有していなかった。それらの2つのウイルスの間の唯一の相違はE1欠失の領域内に存在した。P1(継代1)における該ウイルスの同時感染の後、該混合物を更に4継代にわたり増殖させ、その時点で、該細胞を回収し、該ウイルスを抽出し、CsClバンディングにより精製した。該ウイルスDNAを抽出し、HindIIIで消化し、ついで該消化産物を放射能標識した。また、対照のために、それぞれの前プラスミド(pAdHVE3(「OLD E3+」)、MRKpAdHVE3(「NEW E3+」))をHindIII(および該ベクターバックボーンを取り出すためにPac1)で消化し、ついで[33P]dATPで標識した。該放射能標識消化産物をゲル電気泳動に付し、該ゲルをWhatman紙上で乾燥させた後、オートラジオグラフィーフィルムにさらした。図6は、パッケージングシグナル領域に施された付加を有する新規アデノウイルスが、元のアデノウイルスと比べて増殖上の利点を有することを、明らかに示している。行った実験(試験したいずれの比においても)においては、新たに修飾されたウイルスに関する消化バンドのみが存在した。3,206のサイズの特徴的バンド(該新規ウイルス由来のもの)が明らかに存在した。しかし、元のウイルスから予想される2,737bpのサイズの特徴的バンドの証拠は認められなかった。
【0165】
実施例7
E3遺伝子の効果の分析
第2組の該ウイルス競合研究は、MRKpAdHVOおよびMRKpAdHVE3から得られた等しいMOI比(1:1)の新規修飾ウイルスを混合することを含むものであった(図5、実験#2)。この組においては、両方のウイルスは、E1欠失に施された新たな修飾を有していた。第1ウイルス(MRKpAdHVO由来)はE3遺伝子を含有しない。第2ウイルス(MRKpAdHVE3由来)はE3遺伝子を含有しない。該ウイルスはいずれも、トランスジーンを含有しない。該ウイルスの同時感染の後、該混合物を更に4継代にわたり増殖させ、その時点で、該細胞を回収し、該全ウイルスを抽出し、CsClバンディングにより精製した。該ウイルスDNAを抽出し、HindIIIで消化し、ついで該消化産物を放射能標識した。また、対照のために、それぞれの前プラスミドMRKpAdHVO(「NEW E3−」)、MRKpAdHVE3(「NEW E3+」))をHindIII(および該ベクターバックボーンを取り出すためにPac1)で消化し、ついで[33P]dATPで標識した。該放射能標識消化産物をゲル電気泳動に付し、該ゲルをWhatman紙上で乾燥させた後、オートラジオグラフィーフィルムにさらした。図6は、該E3+ウイルスおよびE3−ウイルス混合実験のウイルスDNA分析の結果を示す。該E3+ウイルスに対応する特徴的バンド(5,665bp)は、該E3−ウイルスに対応する3,010bpの特徴的バンドより大量に存在した。このことは、該E3遺伝子を含有するウイルスが、E3遺伝子を含有しないウイルスより迅速に増幅しうることを示している。この増幅率の増加は増殖研究により確認されている。後記の表4を参照されたい。
【0166】
実施例8
修飾gagトランスジーンを含有する新規シャトルベクター「MRKpdel E1−CMV(無イントロン)−FLgag−bGHpA」の構築
修飾プラスミドpV1JnsCMV(無イントロン)−FLgag−bGHpAをMsc1で一晩消化し、ついでSfi1で50℃で2時間消化した。ついで該DNAをマングマメヌクレアーゼで30℃で30分間処理した。該DNA混合物を、Qiaex IIキットを使用して脱塩し、ついで37℃で30分間にわたりクレノウ処理して、該トランスジーン断片の末端を完全に平滑化した。ついでその2,559bpのトランスジーン断片をゲル精製した。該修飾シャトルベクター(MRKpdelE1シャトル)を、EcoRVでの消化により線状化し、仔ウシ腸アルカリホスファターゼで処理し、ついで、得られた6,479bpの断片をゲル精製した。ついでその2つの精製断片を互いに連結し、数十個のクローンをスクリーニングして、該シャトルベクター内の該トランスジーンの挿入に関して確認した。特徴的制限消化を行って、該トランスジーンをE1平行およびE1逆平行配向で保持するクローンを同定した。この方法に従って、該MRKpdelE1シャトルベクター内のその他のgagトランスジーンにおけるクローニングを行った。
【0167】
実施例9
MRK FGアデノベクターの構築
HIV−1 gagトランスジーンをE1平行配向で含有するシャトルベクターであるMRKpdelE1−CMV(無イントロン)−FLgag−bGHpAをPac1で消化した。該反応混合物をBsfZ171で消化した。その5,291bpの断片をゲル抽出により精製した。MRKpAdHVE3プラスミドをCla1で37℃で一晩消化し、ゲル精製した。約100ngの5,290bpシャトル+トランスジーン断片および〜100ngの線状化MRKpAdHVE3 DNAを大腸菌(E.coli)BJ5183化学コンピテント細胞内に同時形質転換した。いくつかのクローンを選択し、濁り状態に達するまで6〜8時間、2mlのTerrific(商標)ブロス内で増殖させた。該細胞ペレットからの全DNAを、Qiagenアルカリ細胞溶解およびフェノールクロロホルム法を用いて精製した。該DNAをイソプロパノールで沈殿させ、20μl dH2Oに再懸濁させた。このDNAの2μl アリコートを大腸菌(E.coli)XL−1コンピテント細胞内に形質転換した。それぞれの別々の形質転換からの単コロニーを選択し、3mlのLB+100μg/ml アンピシリン中で一晩成長させた。Qiagenカラムを使用して、該DNAを単離した。該gag遺伝子および該プラスミドバックボーン中で切断する制限酵素BstEIIでの消化により、陽性クローンを同定した。該前プラスミドクローンはMRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpAと称され、37,498bpのサイズを有する。この方法に従って、E1平行およびE1逆平行形態のその他のgagトランスジーン構築物のそれぞれのE3−およびE3+形態を作製した。図7A、7Bおよび7Cは、構築したアデノベクターの種々の組合せを示す。
【0168】
実施例10
プラスミド競合研究
一連のプラスミド競合研究を行った。簡単に説明すると、2つの競合プラスミドのそれぞれの等量を混合することにより、新規構築物の種々の組合せのスクリーニングを行った。図8Aに示す実験においては、同じトランスジーンを異なる配向で含有するプラスミドを互いに混合して、それらの2つのプラスミドの「競合」を行った。その目的は、トランスジーン配向の効果を観察することにあった。図8Bに示す実験においては、種々のポリアデニル化シグナルを(同じ配向で)含有するプラスミドを互いに等量で混合した。その目的は、ポリAシグナルの効果を評価することにあった。該初期トランスフェクションの後、該ウイルスを10ラウンドにわたり継代し、該ウイルスDNAを放射性制限分析により分析した。
【0169】
該プラスミド混合実験(図8A)からのウイルス種の分析は、E1平行配向で挿入されたトランスジーンを有するアデノベクターが、E1逆平行配向で挿入されたトランスジーンを有するアデノウイルスより良く増殖し、競合においてそれを凌ぎうることを示した。継代3において、そしてもちろん継代6においても、該混合物のウイルスDNA分析は、E1平行配向で該トランスジーンを保持するウイルスの、E1逆平行形態より大きな比率を示した。継代10までは、観察される唯一のウイルス種は、試験した両方のトランスジーン(hCMV(無イントロン)−FLgag−bGHpAおよびhCMV(無イントロン)−FLgag−SPA)に関してE1平行配向で該トランスジーンを有するアデノベクターであった。
【0170】
継代3および6におけるプラスミド混合物実験#2(図8B)からのウイルス種の分析は、試験したポリアデニル化シグナル(bGHpAおよびSPA)はウイルスの増殖に影響を及ぼさないことを示した。継代10でさえ、該混合物中の2つのウイルス種は尚も等量で存在した。
【0171】
実施例11
増強されたアデノウイルス構築物「MRK Ad5 HIV−1gag」のウイルス産生
該競合研究から得た結果から、本発明者らは以下のとおりに結論づけた:(1)該パッケージングシグナル伸長は有益である;(2)E3の存在はウイルス増殖を増強する;(3)E1平行配向が推奨される;および(4)ポリAシグナルは該アデノウイルスの増殖に影響を及ぼさない。
【0172】
MRK Ad5 HIV−1 gagは最も望ましい結果を示した。この構築物は、新規E3+アデノベクターバックボーンMRKpAdHVE3内にE1平行配向で挿入されたhCMV(無イントロン)−FLgag−bGHpAトランスジーンを含有する。本発明者らは、このアデノベクターをMRK Ad5 HIV−1 gagと命名した。この構築物は以下のとおりに調製した。
【0173】
前プラスミドMRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpAをPac1で消化して該ベクターバックボーンを遊離させ、〜60%コンフルエントのPER.C6(登録商標)細胞を含有する6cmディッシュ中で3.3μgをリン酸カルシウム法(Amersham Pharmacia Biotech)によりトランスフェクトした。CPEが得られたら(7〜10日)、該培養を3回凍結/融解し、該細胞片をペレット化した。1mlのこの細胞ライセートを使用して、80〜90% コンフルエントのPER.C6(登録商標)細胞を含有する6cmディッシュ内に感染させた。CPEが得られたら、該培養を3回凍結/融解し、該細胞片をペレット化した。ついで該細胞ライセートを使用して、80〜90% コンフルエントのPER.C6(登録商標)細胞を含有する15cmディッシュに感染させた。この感染手法を継続し、6継代に拡張した。ついで該ウイルスをCsCl法により該細胞ペレットから抽出した。2回のバンディングを行った(3勾配CsClおよびそれに続く連続的CsCl勾配)。該第2バンディングの後、該ウイルスをA105バッファー中で透析した。プロナーゼ処理およびそれに続くフェノールクロロホルム処理を用いて、ウイルスDNAを抽出した。ついで該ウイルスDNAをHindIIIで消化し、[33P]dATPで放射能標識した。該消化産物を分離するためのゲル電気泳動の後、該ゲルをWatman紙上で乾燥させ、ついでオートラジオグラフィーに付した。該消化産物を、(標識前にPac1/HindIIIで消化された)前プラスミドからの消化産物と比較した。予想されたサイズが観察され、このことは、該ウイルスが成功裏のうちにレスキューされたことを示している。この方法を用いて、調製した種々のアデノベクタープラスミド構築物のそれぞれからウイルスをレスキューした。
【0174】
実施例12
安定性分析
種々のアデノベクター構築物(例えば、MRK Ad5 HIV−1 gag)が遺伝的安定性を示すか否かを判定するために、該ウイルスをそれぞれ連続継代した。該ウイルスDNAを継代3、6および10で分析した。各ウイルスは、その正しい遺伝的構造を維持していた。また、大規模生産で行うのと同様の増殖条件下で該MRK Ad5 HIV−1 gagの安定性を分析した。この分析のために、MRK Ad5 HIV−1 gagおよび3つの他のアデノウイルスベクターのトランスフェクションを繰返し、該ウイルスをP3で精製した。その3つの他のアデノベクターは以下のとおりであった:(1)E3−アデノベクターバックボーン中にbGHpAターミネーターと共にhCMV(無イントロン)−Flgagを含むもの、(2)E3+アデノベクターバックボーン中にSPA終結シグナルと共にhCMV(無イントロン)−Flgagを含むもの、およびE3+アデノベクターバックボーン中にbGHpAターミネーターと共にmCMV−Flgagを含むもの。該ベクターのすべては、E1平行配向で挿入されたトランスジーンを有する。ウイルスDNAを放射性制限分析により分析して、それが適当であることを確認してから、無血清培地内での連続継代のための発酵細胞培養に移した。P5において、それらの4つのウイルスのそれぞれを精製し、該ウイルスDNAを制限消化および放射能標識法による分析用に抽出した。後記のとおり、このウイルスは後に、一連の研究(COS細胞内のインビトロgag発現、げっ歯類での研究およびアカゲザルでの研究)において使用されている。P5からのウイルスを図9に示す。
【0175】
MRKHVE3(MRKpAdHVE3前プラスミドから得られた無トランスジーン)およびMRKAd5HIV−1gag(MRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpA前プラスミド)ウイルスに関して、無血清条件下での継代を継続した。図10は、MRKHVE3、MRKAd5HIV−1gagE3−については継代11での、MRKAd5HIV−1gagについては継代11および12での、放射性制限消化によるウイルスDNA分析を示す。DNAマーカーレーンである第1レーンに加えて、次の3つのレーンは、前プラスミド対照(元のウイルスに基づく対照)、すなわち、それぞれMRKpAdHVE3(「pMRKHVE3」)、MRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpAおよびpMRKAd5gag(E3−)からのウイルスである。図10に示すとおり、該ウイルスDNAサンプルのそれぞれは、示される外来バンドを伴わない予想バンドを示している。このことは、オートラジオグラフィーにより検出されうる主要変異体アデノウイルス種が存在しないことを示している。
【0176】
図11は、MRKHVE3とMRKAd5HIV−1gagとの間のウイルス競合研究の結果を示す。これらのウイルスを等しいMOI(それぞれウイルス粒子140個;合計280 vp)で継代6において互いに混合し、P11まで継代し続けた。該DNAマーカーレーンである第1レーンに加えて、次の2つのレーンは、MRKpAdHVE3およびMRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpAから得られた前プラスミド対照である。次の2つのレーンは、継代6における出発ウイルス物質からのウイルスDNAである。最後の2つのレーンは、二重に行った競合研究である。図11のデータは、培養内でのgagトランスジーンの効果を示している。MRKAd5gagウイルスの増殖を「無トランスジーン」MRKHVE3の増殖と比較した。これらの2つのウイルスを、継代6において、同じMOI(すなわち、それぞれ140vp)で感染させ、ついで継代11まで継代し、該ウイルスプールを放射性制限分析により分析した。該データは、競合において一方のウイルスが他方のウイルスを凌ぐことがなかったことを示している。したがって、該gagトランスジーンは、該アデノウイルスに対する明白な毒性の徴候を示さなかった。
【0177】
HindIII消化による分析は、各ウイルス種がほぼ等量で存在することを示している。前記のとおり、いずれの外来バンドの徴候も存在しないらしい。図12は、血清含有条件下で増殖させたMRKAd5HIV−1gagに関して、より高い継代数を示している。この場合もまた、該ゲノム完全性が維持されており、最高継代レベル(P21)においてさえも再構成の証拠は認められない。
【0178】
図9に示す4つのベクターのそれぞれを、増幅能に関して分析した。以下の表4は、P4におけるウイルス増幅率の評価において用いたQPA分析を示す。元のHIV−1 gag構築物に関する増幅率の測定は、P12の臨床ロットに基づく。増幅率は、元のウイルスに関しては、継代数の増加と共に増加することが示されている。この観察の理由は、無傷アデノベクターと比較して増殖率の増加を示す変異体の出現によるものである。元のAd gagベクターの連続継代により、変異体のレベルは増加し、したがって増幅率も増加する。
【0179】
該MRK Ad5 HIV−1 gagウイルスは、プロセス条件下(すなわち、無血清培地)で連続的に継代されている。継代11および12から抽出されたウイルスDNAは再構成の証拠を示さない。
【0180】
【表3】
【0181】
実施例13
増強されたAd5構築物の分析的評価
ウイルス増幅に対する該トランスジーンおよびE3遺伝子の効果を調べるために、増強されたアデノウイルスベクターMRK Ad5 HIV−1 gagを、その無トランスジーン形態(MRKpAdHVE3)およびそのE3形態(MRK Ad5 HIV−1 gag E3−)と共に、無血清条件下でいくつかの継代にわたり調べた。表5Aは、MRK Ad5 HIV−1 gagに関して継代P3〜P8にわたり測定した増幅率を示している。あるMOI範囲内では、該ウイルス産生量は該ウイルス投入量に正比例することが確認された。したがって、感染時の細胞当たりのウイルス粒子の数が増えれば増えるほど、産生されるウイルス量は増える。一方、ウイルス増幅率は該ウイルス投入量に反比例する。該ウイルス投入量が少なければ少ないほど、該増幅率は大きくなる。
【0182】
表5Bは、新規E3+ベクターバックボーンMRKpAdHVE3の増幅率を示している。それは、該gagトランスジーン含有形態より有意に低い増幅率を有していた。これは、MRK Ad5 HIV−1 gagが該トランスジーンを含有しているため、より大きなサイズを有することに帰されうる。このように該トランスジーンを含むと、該アデノウイルスのサイズは、野生型Ad5ウイルスのサイズに、より近づく。アデノウイルスは、それがその野生型ゲノムサイズに近い場合に最も良く増幅することが、よく知られている。野生型Ad5は35,935bpである。MRKpAdHVE3は32,905bp長である。増強されたアデノベクターMRK Ad5 HIV−1 gagは35,453bpである(ベクター地図に関しては図14を参照されたい;また、図15A〜Xは、該ベクターバックボーンの追加的な2,021bpを含む完全な前アデノウイルスベクター配列を示す)。
【0183】
表5Cは、新規E3−gag含有ウイルスMRK Ad5 HIV−1 gag E3−の増幅率を示す。この場合もまた、このウイルスは、増強されたアデノウイルスベクターより低い増殖率を示す。これは、野生型Ad5と比較した場合のこのウイルスのサイズの減少(E3遺伝子の欠失による)に帰されうる。該MRK Ad5 HIV−1 gag E3−ウイルスは32,810bp長である。これは、35,935bp長の野生型Ad5および35,453bp長のMRK Ad5 HIV−1 gagに匹敵する。
【0184】
【表4】
【0185】
実施例14
該新規構築物のgag発現分析
MRK Ad5 HIV−1 gagおよび元のHIV−gagベクター(研究および臨床ロット)のインビトロgag分析は、比較しうるgag発現を示している。該臨床ロットは、少しだけ減少したgag発現レベルを示している。最も注目すべき相違は、mCMVベクターでのものである。このベクターは、試験したその他のベクター(これらはすべてhCMVプロモーターを含有する)と比較して約3倍低い発現レベルを示している。bGHpAを伴うmCMV−FLgagのアッセイは、種々の増殖および精製ロットを使用して3回行ったが、それは一貫して、より弱いgag発現を示した。
【0186】
実施例15
Balc/cマウスにおけるMRK Ad5 HIV−1 gagおよび他のgag含有アデノベクターの評価
10匹のbalb/cマウスのコホートを、漸増量のMRK Ad5 HIV−1 gagならびに元のAd5HIV−gagの研究および臨床ロットで筋肉内にワクチン接種した。血清サンプルを、第1投与の3週間後に集め、抗p24サンドイッチELISAにより分析した。
【0187】
MRK Ad5 HIV−1 gag(107および109vp(ウイルス粒子)の用量)を投与したウスにおける抗p24力価は、初期のアカゲザルのデータの多くが得られたAd5HIV−1 gagの研究ロットのものと比較しうるものであった(図13)。また、E3が欠失している場合(MRKAd5hCMVgagbGHpA(E3−))またはbGHpAターミネーターの代わりにSPAを使用した場合(MRKAd5 hCMV−gag−SPA(E3+))または該MRKAd5バックボーンにおいてhCMVの代わりにマウスCMVプロモーターを使用した場合(MRKAd5 mCMV−gag−bGHpA(E3+))には、これらの力価は比較しうるものであった。
【0188】
表7に示す結果は、好ましいベクターMRK Ad5 HIV−1 gagに加えて、それらの3つの他のベクターも、マウスにおいて強力な抗gag抗体応答を誘導しうることを示している。非常に興味深いことに、bGHpAおよびE3+をE1平行配向で含有するmCMV−Flgag構築物は、試験したその他のベクターと比較してCOS細胞インビトロ感染において最低のgag発現を示したが(表6)、それは、このインビボBalb/c研究において最大の抗gag抗体応答を引き起こした。また、表7は、該研究ロットおよび該臨床ロットの両方における抗gag抗体産生の用量反応関係を示している。予想どおり、該臨床ロットは、該研究ロットに使用したのと同じ用量と比較して、各用量レベルで抗gag抗体誘導の減少を示している。
【0189】
【表5】
【0190】
【表6】
【0191】
実施例16
アカゲザルにおける元のAd−gag構築物と新規MRK Ad5 HIV−1 gagとに対する体液性および細胞性応答の比較
3匹のアカゲザルのコホートを、MRK Ad5 HIV−1 gagまたは臨床用Ad5gagバルクで2つの用量(1011vpおよび109vp)で筋肉内にワクチン接種した。免疫は、第0週、4週および25週に行った。血清およびPBMCサンプルを、選択された時点で集めた。該血清サンプルを抗p24Ab力価に関して(競合に基づくアッセイを使用)、そしてgag20マーペプチドプールでの一晩の刺激後に該PBMCを抗原特異的IFNγ分泌に関して(ELISpotアッセイによる)アッセイした。
【0192】
表8に示す結果は、抗gag抗体の生成に関する比較しうる応答を示している。表9に要約した末梢血中のgag特異的T細胞の度数は、新規構築物MRK Ad5 HIV−1 gagを1回投与した後に生じた強力な細胞性免疫応答を示している。該応答はまた、同じベクターの第2投与により増強されうる。該ベクターはまた、細胞傷害性活性を引き起こすCD8+T細胞応答(PBMCのCD4+枯渇後の残りのスポット計数により示される)を誘導しうる。
【0193】
【表7】
【0194】
【表8】
【0195】
本明細書に記載のアデノベクター、特にMRK Ad5 HIV−1 gagは、血清中およびより重要なことには無血清培地条件中でのそれらの増強された増殖特性に関して非常に有望なHIV−gagアデノベクターを代表する。現在のHIV−1 gagアデノベクター構築物と比較して、MRK Ad5 HIV−1 gagは、5〜10倍増加した増幅率を示す。本発明者らは、それが継代21において遺伝的に安定であることを示している。この構築物は、比較的低い用量である10∧9vpにおいてさえもインビボで有意な細胞性免疫応答を産生しうる。該MRKAd5gag構築物の効力は、このアカゲザル研究において示されているとおり、元のHIV−1 gagベクターより良くはないとしてもそれと比較しうるものである。
【0196】
実施例17
コドン最適化HIV−1 POLおよびコドン最適化HIV−1 POL修飾
本明細書に開示する種々の合成pol遺伝子のオープンリーディングフレームは、逆転写酵素(またはポリメラーゼおよびRNアーゼH活性よりなるRT)およびインテグラーゼ(IN)のコード配列を含む。該タンパク質配列は、IIIBのクローン単離物であるHxb2rの配列に基づく。この配列はコンセンサス・クレードB配列に最も近く、848個の残基中で16個の不一致残基を有するに過ぎないことが示されている(Korberら,1998,Human retroviruses and AIDS,Los Alamon National Laboratory,Los Alamons,New Mexico)。当業者は、本明細書を精査した後、利用可能な任意のHIV−1またはHIV−2株が、本明細書に開示するHIV pol DNAワクチン構築物の潜在的鋳型を与えると理解するであろう。突然変異不活性化にもかかわらず、いずれかの残留プロテアーゼ活性からの安全性を保証するために、該プロテアーゼ遺伝子が本発明のDNAワクチン構築物から除去されることが、更に注目される。インビボ哺乳類発現を最高にするために、野生型(wt−pol)および不活性化pol(IA−pol)の両方の遺伝子配列の設計においては、該配列内の各アミノ酸残基に関するヒトにとって好ましい(「ヒト化」)コドンの使用が含まれる(Lathe,1985,J.Mol.Biol.183:1−12)。配列番号1、3、5および7におけるコドン使用頻度を調べることにより理解されうるとおり、哺乳類における最適化のためには以下のコドンの使用が好ましい:Met(ATG)、Gly(GGC)、Lys(AAG)、Trp(TGG)、Ser(TCC)、Arg(AGG)、Val(GTG)、Pro(CCC)、Thr(ACC)、Glu(GAG);Leu(CTG)、His(CAC)、Ile(ATC)、Asn(AAC)、Cys(TGC)、Ala(GCC)、Gln(CAG)、Phe(TTC)およびTyr(TAC)。哺乳類(ヒト)におけるコドンの最適化に関して更に考察するためには、WO 97/31115(PCT/US97/02294)(本明細書の他の箇所にも示されているとおり、これを参照により本明細書に組み入れることとする)を参照されたい。当業者は別の形態のコドン最適化を用いたり、あるいは本発明の範囲内のHIV polワクチン構築物の作製の際にはこの工程を省略しうると予想される。したがって、本発明はまた、本明細書に開示するHIV Polタンパク質の種々の野生型および修飾形態をコードするDNA分子および関連組換えアデノウイルスHIVワクチンの非コドン最適化形態に関する。しかし、これらの構築物のコドン最適化が、本発明の好ましい実施形態である。
【0197】
本発明のこの態様の特に好ましい実施形態は、プロテアーゼ(PR)活性をコードするDNA配列が欠失しており、RT(逆転写酵素およびRNアーゼH活性)およびINインテグラーゼ活性をコードするコドン最適化「野生型」配列が残された、wt−pol DNA構築物(本発明においては、「wt−pol」または「wt−pol(コドン最適化)」と称される)をコードするコドン最適化ヌクレオチド配列を含む。このタンパク質をコードするDNA分子は本明細書中には配列番号1として開示されており、該オープンリーディングフレームは、ヌクレオチド10−12の開始Met残基からヌクレオチド2560−2562の終結コドンにわたり含有される。配列番号1は以下のとおりである。
【0198】
【化1】
【0199】
配列番号1として開示される野生型pol構築物のオープンリーディングフレームは、以下のとおり本明細書中で配列番号2として開示される850アミノ酸を含有する。
【0200】
【化2】
【0201】
本発明は特に、該プロテアーゼ活性をコードする野生型配列部分の欠失に加えて、該発現タンパク質のHIV−1 pol(RT−RH−IN)活性に有害な活性部位残基突然変異の組合せが導入されたコドン最適化HIV−1 DNA pol構築物を含むアデノウイルスベクターワクチンに関する。したがって、本発明は、好ましくは、アデノウイルスHIV−1 DNA polに基づくワクチンであって、該構築物が、RT、RNアーゼおよび/またはIN活性を少なくとも部分的に、好ましくは実質的に無効にする突然変異を含有し、いずれかのPR活性をコードするDNA配列を欠くことを特徴とする前記ワクチンに関する。アデノウイルスベクターワクチンの一部および一団であるHIV−1 pol突然変異体の1つの型は、HIV−1 PolのRT、RNアーゼHおよび/またはIN機能に関する少なくとも実質的に減少した酵素活性を与えるよう該発現タンパク質のRT、RNアーゼおよび/またはIN領域内の活性部位を有効に改変する点突然変異を引き起こす少なくとも1つのヌクレオチド置換を含む突然変異DNA分子を含みうるが、これに限定されるものではない。本発明のこの態様の好ましい実施形態においては、HIV−1 DNA pol構築物は、RT、RNアーゼHおよびIN活性を有効に無効にするPolコード領域内に突然変異を含有する。特に好ましいHIV−1 DNA pol構築物は、各活性を少なくとも実質的に無効にするようPolのRT、RNアーゼHおよびINドメインの活性部位を改変する少なくとも1つの点突然変異を含有するDNA分子である。そのようなHIV−1 Pol突然変異体は、十中八九、それぞれRT、RNアーゼHおよびIN活性をもたらす各触媒ドメイン内またはその周囲に少なくとも1つの点突然変異を含むであろう。この目的において、特に好ましいHIV−1 DNA pol構築物が本発明において例示され、それは、PR、RT、RNアーゼまたはIN活性を有さない不活性化Polタンパク質(IA Pol:配列番号4、図17A〜C)を与える9個のコドン置換突然変異を含有し、その場合、3個のそのような点突然変異は該RT、RNアーゼおよびIN触媒ドメインのそれぞれの中に存在する。したがって、特に好ましい例は、以下の表1に示す全9個の突然変異を含有する、IA−polをコードするDNA分子を適当な様態で含むアデノウイルスワクチンである。置換のための追加的な好ましいアミノ酸残基は、PolのRNアーゼドメイン内に位置するAsp551である。本明細書に開示する突然変異の任意の組合せが適している可能性があり、したがって、本発明のIA−Polに基づくワクチンとして利用されうる。付加および欠失突然変異が意図され本発明の範囲内にあるが、好ましい突然変異は、野生型アミノ酸から別のアミノ酸残基への置換をもたらす点突然変異である。
【0202】
【表9】
【0203】
HIV−1 Polの活性部位内またはその周囲のエピトープを改変する可能性が少なくなるよう、本発明のIApol突然変異体ワクチン内に点突然変異を組込むのが好ましい。
【0204】
この目的において、配列番号3は、表1に示す9個の突然変異に加えてコドン最適化polをコードするヌクレオチド配列を開示しており、これは、以下のとおり開示され、本発明において「IApol」と称される。
【0205】
【化3】
【0206】
本発明のIA−polに基づくアデノウイルスワクチンを製造するために、該酵素サブユニットからの9個の活性部位残基の全てをアラニン側鎖で置換することにより、酵素機能の不活性化を行った。表1に示すとおり、該ポリメラーゼの触媒性3残基、すなわちAsp112、Asp187およびAsp188を含む全ての残基を、アラニン(Ala)残基で置換した(Larderら,Nature 1987,327:716−717;Larderら,1989,Proc.Natl.Acad.Sci.1989,86:4803−4807)。RNアーゼH活性を無効にするために、3つの追加的な突然変異をAsp445、Glu480およびAsp500に導入し(このIA Pol構築物においてはAsp551は不変のままとした)、ここで、各残基を、それぞれAla残基で置換した(Daviesら,1991,Science 252:,88−95;Schatzら,1989,FEBS Lett.257:311−314;Mizrahiら,1990,Nucl.Acids.Res.18:pp.5359−5353)。HIV polインテグラーゼ機能は、Asp626、Asp678およびGlu714における3つの突然変異により無効となった。この場合も、これらの残基のそれぞれをAla残基で置換した(Wiskerchenら1995,J.Virol.69:376−386;Leavittら,1993,J.Biol.Chem.268:2113−2119)。配列番号4のアミノ酸残基Pro3は該RT遺伝子の開始部位を示す。IA−Polの完全なアミノ酸配列は、以下のとおりに本発明においては配列番号4および図17A〜Cとして開示される。
【0207】
【化4】
【0208】
前記のとおり、前記で開示した突然変異の任意の組合せが適している可能性があり、したがって、単独または組合せ様式法および/または初回−追加法で投与される場合に本発明のIA−polに基づくワクチンとして使用されうる。例えば、それぞれの逆転写酵素、RNアーゼHおよびインテグラーゼコード領域内の3個の残基中の2個だけしか突然変異させないで、尚もこれらの酵素活性を無効にすることが可能でありうる。しかしながら、前記の及び配列番号3として開示されているIA−pol構築物ならびに該発現タンパク質(配列番号4)が好ましい。それらの3つの触媒ドメインのそれぞれにおいて少なくとも1つの突然変異が存在することも好ましい。
【0209】
本発明のこの形態のもう1つの態様は、真核性輸送(トラフィッキング)シグナルペプチド、例えばtPA(組織プラスミノーゲンアクチベーター)由来のもの又はリーダーペプチド(例えば、免疫グロブリンリーダーペプチドのような高度に発現される哺乳類タンパク質内で見出されるもの)を含むコドン最適化HIV−1 Polに基づくワクチン構築物である。任意の機能的リーダーペプチドを、有効性に関して試験することが可能である。しかしながら、本明細書中に示すHIV−1 Nef構築物の場合と同様に本発明の好ましい実施形態は、polコード領域またはその一部がリーダーペプチド、好ましくはヒトtPA由来のリーダーペプチドに作動的に結合した本明細書に開示するHIV−1 Pol突然変異体アデノウイルスワクチン構築物を提供することにある。すなわち、IA−Pol(配列番号4)のようなコドン最適化HIV−1 Pol突然変異体は、該タンパク質のアミノ末端部分にリーダーペプチドをも含むことが可能であり、これは、該宿主細胞内の該発現タンパク質の細胞輸送および従ってその免疫原性に影響を及ぼしうる。図16A〜Bに示すとおり、本発明を実施するために使用しうるDNAベクターを、関心のあるリーダーシグナルペプチドを含有するよう公知組換えDNA法により修飾して、関心のある修飾HIV−1タンパク質の下流クローニングが、修飾HIV−1 tPA/Polタンパク質をコードするヌクレオチド配列を与えるようにすることが可能である。別法においては、前記のとおり、リーダーペプチドをコードするヌクレオチド配列の挿入を、関心のあるPolタンパク質のオープンリーディングフレームを収容するDNAベクター内に挿入することができる。該クローニング方法には無関係に、該最終産物は、関心のある修飾HIV−1 Polタンパク質(リーダーペプチドを含有するHIV−1 Polタンパク質を含むが、これに限定されるものではない)をコードするヌクレオチド配列と共に、有効な遺伝子発現のためのベクター成分を含むポリヌクレオチドワクチンである。本発明で用いるヒトtPAリーダーのアミノ酸配列は以下のとおりである:MDAMKRGLCCVLLLCGAVFVSPSEISS(配列番号17)。したがって、本発明のもう1つの態様は、真核性輸送シグナルペプチド(例えば、tPA由来のもの)を含む、HIV−1 Polに基づくワクチン構築物を製造することにある。この目的において、本発明は、コドン最適化wt−pol DNA構築物をコードするDNA分子であって、該プロテアーゼ(PR)活性が欠失しており、ヒトtPAリーダー配列が該コード領域の5’末端に融合していることを特徴とするDNA分子に関する。このタンパク質をコードするDNA分子は本明細書中には配列番号5として開示されており、該オープンリーディングフレームは本明細書中には配列番号6として開示されている。
【0210】
この目的において、本発明は、コドン最適化wt−pol DNA構築物をコードするDNA分子であって、該プロテアーゼ(PR)活性が欠失しており、ヒトtPAリーダー配列が該コード領域の5’末端に融合していることを特徴とするDNA分子(本発明においては、「tPA−wt−pol」と称される)に関する。このタンパク質をコードするDNA分子は本明細書中には配列番号5として開示されており、該オープンリーディングフレームは、ヌクレオチド8−10の開始Met残基からヌクレオチド2633−2635の終結コドンにわたり含まれる。配列番号5は以下のとおりである。
【0211】
【化5】
【0212】
配列番号5として開示される野生型pol構築物のオープンリーディングフレームは、以下のとおり本明細書中で配列番号6として開示される875アミノ酸を含有する。
【0213】
【化6】
【0214】
本発明はまた、宿主細胞内の発現タンパク質の細胞輸送および従ってその免疫原性に影響を及ぼしうるヒトtPAリーダーのようなリーダーペプチドをアミノ末端部分に含む、IA−Pol(配列番号4)のような組換えアデノウイルスベクター内に含有されるコドン最適化HIV−1 Pol突然変異体に関する。前記段落に開示している任意のそのようなアデノウイルスに基づくHIV−1 DNA pol突然変異体は、ヒトtPAリーダー配列を含む(これに限定されるものではない)リーダーペプチドのようなリーダーペプチドの下流における融合に適している。したがって、任意のそのようなリーダーペプチドに基づくHIV−1 pol突然変異体構築物は、HIV−1 PolのRT、RNアーゼHおよび/またはIN機能の1以上に関する少なくとも実質的に減少した酵素活性を与えるよう該発現タンパク質のRT、RNアーゼおよび/またはIN領域の触媒活性を有効に改変する突然変異体DNA分子を含みうるが、これに限定されるものではない。本発明のこの形態の好ましい実施形態においては、リーダーペプチド/HIV−1 DNA pol構築物は、RT、RNアーゼHおよびIN活性を有効に無効にする該Polコード領域内に突然変異を含有する。特に好ましいHIV−1 DNA pol構築物は、各活性を少なくとも実質的に無効にするよう(好ましくは、完全に無効にするよう)PolのRT、RNアーゼHおよびINドメイン内の活性部位および触媒活性を改変する少なくとも1つの点突然変異を含有するDNA分子である。そのようなHIV−1 Pol突然変異体は、十中八九、それぞれRT、RNアーゼHおよびIN活性をもたらす各触媒ドメイン内またはその周囲に少なくとも1つの点突然変異を含むであろう。本発明のこの形態の特に好ましい実施形態は、表1に示す9個の突然変異を含むIA−Polタンパク質に融合したヒトtPAリーダーに関する。該DNA分子は本明細書中には配列番号7として開示されており、該発現tPA−IA Polタンパク質は、図18に示すとおりの融合結合部を含む。該発現タンパク質の完全なアミノ酸配列を配列番号8に示す。この目的において、配列番号7は、表1に示す9個の突然変異を含むIA Polタンパク質に融合したヒトtPAリーダーをコードするヌクレオチド配列(本発明においては、「tPA−opt−IApol」と称される)を開示する。該オープンリーディングフレームは、開始Met(ヌクレオチド8−10)で始まり、ヌクレオチド2633−2635の「TAA」コドンで終結する。tPA−IAPolをコードするヌクレオチド配列はまた、以下のとおりに開示される。
【0215】
【化7】
【0216】
配列番号7として開示されるtPA−IA−pol構築物のオープンリーディングフレームは、以下のとおりにtPA−IA−Polおよび配列番号8として本明細書中に開示される875アミノ酸を含有する。
【0217】
【化8】
【0218】
実施例18
コドン最適化HIV−1 NEFおよびコドン最適化HIV−1 NEF修飾体
HIV−1 NefおよびHIV−1 Nef修飾体のコドン最適化形態は、実質的には、2000年12月15日付け出願の米国特許出願第09/738,782号および同様に2000年12月15日付け出願のPCT国際出願PCT/US00/34162(両方の文書を参照により本明細書に組み入れることとする)に記載のとおりである。前記文書に開示されているとおり、コドン最適化NefおよびNef修飾体の特定の実施形態は、HIV−1 jfrl分離体からのHIV−1 NefをコードするDNA分子に関する。この場合、該コドンは、ヒトのような哺乳類系内での発現に関して最適化されている。このタンパク質をコードするDNA分子は本発明では配列番号9として開示されており、発現されるオープンリーディングフレームは本発明では配列番号10として開示されている。本発明のアデノウイルスベクターにおける使用のためのNefに基づくコード領域のもう1つの実施形態は、該HIV−1 NefポリペプチドのNH2末端に融合したヒトプラスミノーゲンアクチベーター(tpa)リーダーペプチドを含有するタンパク質をコードするコドン最適化DNA分子を含む。このタンパク質をコードするDNA分子は本発明では配列番号11として開示されており、発現されるオープンリーディングフレームは本発明では配列番号12として開示されている。もう1つの修飾されたNef最適化コード領域は、最適化HIV−1 NefをコードするDNA分子に関する。この場合、該オープンリーディングフレームは、アミノ末端ミリスチル化部位における修飾(Gly−2からAla−2)、およびLeu−174−Leu−175ジロイシンモチーフからAla−174−Ala−175への置換をコードする(本発明ではopt nef(G2A,LLAA)として記載されている)。このタンパク質をコードするDNA分子は本発明では配列番号13として開示されており、発現されるオープンリーディングフレームは本発明では配列番号14として開示されている。更なる実施形態は、最適化HIV−1 NefをコードするDNA分子に関する。この場合、アミノ末端ミリスチル化部位およびジロイシンモチーフが欠失しており、tPAリーダーペプチドを含む。このDNA分子opt tpanef(LLAA)は、HIV−1 Nef(jfrl)のアミノ酸残基6−216に融合したtPAリーダー配列を含有するNefタンパク質をコードするオープンリーディングフレームを含み、ここで、Leu−174およびLeu−175はAla−174およびAla−175で置換されており、これは本発明ではopt tpanef(LLAA)と称され、本発明では配列番号15として開示されており、発現されるオープンリーディングフレームは本発明では配列番号16として開示されている。
【0219】
前記文書(米国特許出願第09/738,782号およびPCT国際出願PCT/US00/34162)に開示されており本明細書中で繰返し記載されているとおり、それぞれのオープンリーディングフレームを含むnefに基づく以下のヌクレオチドおよびアミノ酸配列は以下のとおりである。
【0220】
1.コドン最適化形態のHIV−1 jrfl nef遺伝子のヌクレオチド配列は、以下に示すとおり本発明では配列番号9として開示されている。
【0221】
【化9】
【0222】
好ましいコドンの使用は以下のとおりである:Met(ATG)、Gly(GGC)、Lys(AAG)、Trp(TGG)、Ser(TCC)、Arg(AGG)、Val(GTG)、Pro(CCC)、Thr(ACC)、Glu(GAG);Leu(CTG)、His(CAC)、Ile(ATC)、Asn(AAC)、Cys(TGC)、Ala(GCC)、Gln(CAG)、Phe(TTC)およびTyr(TAC)。哺乳類(ヒト)におけるコドンの最適化に関して更に考察するためには、WO 97/31115(PCT/US97/02294)(これを参照により本明細書に組み入れることとする)を参照されたい。また、HIV−Nefのオープンリーディングフレームを含む野生型対コドン最適化ヌクレオチドの比較に関しては図19〜Bを参照されたい。
【0223】
前記の配列番号9のオープンリーディングフレームは、ヌクレオチド12−14の開始メチオニン残基およびヌクレオチド660−662の「TAA」終結コドンを含む。配列番号9のオープンリーディングフレームは、コドン最適化DNAワクチンベクターの使用により発現される216アミノ酸のHIV−1 Nefタンパク質を与える。その216アミノ酸のHIV−1 Nef(jfrl)タンパク質は、本発明では配列番号10として開示されており、以下のとおりである。
【0224】
【化10】
【0225】
HIV−1 Nefは、Gly−2のミリスチル化を介して宿主細胞膜の内部表面と会合する216アミノ酸のサイトゾルタンパク質である(Franchiniら,1986,Virology 155:593−599)。すべての考えられうるNef機能が解明されているわけではないが、細胞内膜へのNefの適切な輸送は、HIV−1の生活環の初期相を促進するために宿主の細胞内環境を改変することにより及び後代ウイルス粒子の感染性を増加させることにより、ウイルス複製を促進することが明らかになっている。コドンが最適化されたタンパク質修飾ポリペプチドに関する本発明の1つの態様においては、本発明のアデノウイルスベクターのnefコード領域を修飾して、異種リーダーペプチドをコードするヌクレオチド配列を含有させて、発現タンパク質のアミノ末端領域が該リーダーペプチドを含有するようにする。真核細胞を典型づける機能の多様性は、その膜境界の構造的相違に依存する。これらの構造を生成し維持するためには、タンパク質が、小胞体内のその合成部位から該細胞の全体にわたる所定の目的地へ輸送される必要がある。このためには、主要輸送経路へのアクセスポイントに位置する経路選択をもたらす分子装置により認識されるソーティングシグナルを該輸送タンパク質が示すことが必要である。ほとんどのタンパク質についてのソーティング決定は、それらがそれらの生合成経路を通過するにつれて1回だけ行われる必要がある。なぜなら、それらの最終目的地、すなわち、それらがそれらの機能を果たす細胞位置は、それらの永久的な存在位置となるからである。細胞内完全性の維持は、1つには、タンパク質の選択的ソーティングおよびその正しい目的地への正確な輸送に依存する。タンパク質中には、「住所標識」として機能しうる一定の配列モチーフが存在する。多数のソーティングシグナルは、膜タンパク質の細胞質ドメインと会合していることが判明している。CTL応答の有効な誘導は、しばしば、抗原の持続的な高レベルの内因性発現を必要とした。ミリスチル化を介した膜会合は、ほとんどのNef機能のための必須要件であるため、本明細書に記載のとおりのグリシンからアラニンへの変化、ジロイシンモチーフの変化および/またはtpaリーダー配列での置換によりミリスチル化を欠く突然変異体は機能的に欠損しており、したがって、HIV−1ワクチン成分として使用するための、野生型Nefと比較して改善された安全性プロフィールを有するであろう。
【0226】
本発明のこの態様のもう1つの実施形態においては、ヒト組織特異的プラスミノーゲンアクチベーター(tPA)リーダーを含むよう、該DNAベクターまたは該HIV−1 nefヌクレオチド配列を修飾する。図16A〜Bに示すとおり、DNAベクターを、関心のあるリーダーシグナルペプチドを含有するよう公知組換えDNA法により修飾して、関心のある修飾HIV−1タンパク質の下流クローニングが、修飾HIV−1 tPA/Nefタンパク質をコードするヌクレオチド配列を与えるようにすることが可能である。別法においては、前記のとおり、リーダーペプチドをコードするヌクレオチド配列の挿入を、関心のあるNefタンパク質のオープンリーディングフレームを収容するDNAベクター内に挿入することができる。該クローニング方法には無関係に、該最終産物は、リーダーペプチドを含有するHIV−1 Nefタンパク質を含む(これに限定されるものではない)関心のある修飾HIV−1 Nefタンパク質をコードするヌクレオチド配列と共に、有効な遺伝子発現のためのベクター成分を含むポリヌクレオチドワクチンである。本発明で用いるヒトtPAリーダーのアミノ酸配列は以下のとおりである:MDAMKRGLCCVLLLCGAVFVSPSEISS(配列番号17)。
【0227】
該タンパク質のカルボキシ領域内のジロイシンモチーフと結びついたGly−2のミリスチル化は、エンドサイトーシスを介したCD4のNef誘導性ダウンレギュレーションに必須であることが示されている(Aikenら,1994,Cell 76:853−864)。また、Nef発現は、エンドサイトーシスを介したMHCIのダウンレギュレーションを促進することが示されている(Schwartzら,1996,Natue Medicine 2(3):338−342)。本発明は、1つには、輸送および/または機能特性において改変した修飾Nefタンパク質をコードするDNAワクチンに関する。本発明のアデノウイルスベクターHIVワクチン内に導入される修飾は、アミノ末端リーダーペプチドを含む修飾Nefタンパク質の発現をもたらすnefオープンリーディングフレームに対する付加、欠失または置換、アミノ末端ミリスチル化部位の修飾または欠失、およびNefタンパク質内のジロイシンモチーフの修飾または欠失のうち感染宿主細胞内の機能を改変するものを含むが、これらに限定されるものではない。したがって、本発明のDNA分子および組換えアデノウイルスHIVワクチンの中心的主題は、(1)nefに基づくコドン最適化アデノウイルスHIVワクチンの宿主投与および細胞内運搬、(2)CTL応答およびTh応答の両方を惹起する点で免疫原性である修飾Nefタンパク質の発現、ならびに(3)感染宿主内でのHIV−1の複製および負荷(load)を促進することが示されているNefの公知初期ウイルス機能の抑制または少なくとも改変にある。したがって、別のアミノ酸残基を発現するようアミノ末端Gly−2ミリスチル化残基が欠失または修飾された修飾Nefタンパク質を発現するDNAワクチンを与えるよう、該nefコード領域を改変することができる。また、別のアミノ酸残基を発現するようジロイシンモチーフが欠失または修飾された修飾Nefタンパク質を発現するDNAワクチンを与えるよう、該nefコード領域を改変することができる。さらに、本発明のアデノウイルスベクターHIVワクチンはまた、野生型または修飾Nefタンパク質(本明細書に記載のとおりのもの)、例えば、Gly2の欠失または置換、Leu174およびLeu175の欠失または置換および/またはリーダー配列の含有を含む修飾Nefタンパク質(これに限定されるものではない)を発現する、コドン使用頻度には無関係な単離されたDNA分子に関する。
【0228】
したがって、特定のNefに基づく構築物は更に、非限定的な例示として以下のものを含む。例えば、本発明は、HIV−1の修飾形態をコードするアデノウイルスベクターワクチンに関する。HIV−1 Nef(jfrl)のアミノ酸残基6−216に融合したtPAリーダー配列を含むNefタンパク質をコードするオープンリーディングフレームは、opt tpanefと称される。opt tpanefのオープンリーディングフレームを含むヌクレオチド配列は、本発明では配列番号11として開示されており、以下のとおりである。
【0229】
【化11】
【0230】
配列番号11のオープンリーディングフレームは、ヌクレオチド2−4の開始メチオニン残基、およびヌクレオチド713−715の「TAA」終結コドンを含む。配列番号3のオープンリーディングフレームは、アミノ酸残基174および175のジロイシンモチーフを含む(これに限定されるものではない)HIV−1 Nefのアミノ酸6−216に融合したtPAリーダー配列を含む237アミノ酸のHIV−1 Nefタンパク質を与える。この237アミノ酸のtPA/Nef(jfrl)融合タンパク質は、本発明では配列番号12として開示されており、以下に示されるとおりである。
【0231】
【化12】
【0232】
したがって、この例示されているNefタンパク質Opt tPA−Nefは、ヒトのような哺乳類系内での発現に関してコドンが最適化されたHIV−1 jfrl分離体からのHIV−1 NefをコードするGly−2A DNA分子のミリスチル化部位の欠失およびtPAリーダー配列の両方を含有する。
【0233】
本発明のもう1つの特定の実施形態においては、組換えアデノウイルスHIVワクチンのオープンリーディングフレームがアミノ末端ミリスチル化部位における修飾(Gly−2からAla−2)およびLeu−174−Leu−175ジロイシンモチーフからAla−174−Ala−175への置換をコードしている最適化HIV−1 NefをコードするDNA分子を開示する。このオープンリーディングフレームは本発明ではopt nef(G2A,LLAA)として記載され、配列番号13として開示され、それは、ヌクレオチド12−14の開始メチオニン残基およびヌクレオチド660−662の「TAA」終結コドンを含む。前記の修飾を有するHIV−1 jfrl nef遺伝子のこのコドン最適化形態のヌクレオチド配列は、本発明では以下のとおりの配列番号13として開示されている。
【0234】
【化13】
【0235】
配列番号13のオープンリーディングフレームは、本発明では以下のとおりの配列番号14として開示されるNef(G2A,LLAA)をコードしている。
【0236】
【化14】
【0237】
本発明の更なる実施形態は、アミノ末端ミリスチル化部位およびジロイシンモチーフが欠失しておりtPAリーダーペプチドを含む最適化HIV−1 Nefをコードするもう1つのDNA分子に関する。このDNA分子opt tpanef(LLAA)は、HIV−1 Nef(jfrl)のアミノ酸残基6−216に融合したtPAリーダー配列を含有しLeu−174およびLeu−175がAla−174およびAla−175(このtPAに基づく融合タンパク質ではAla−195およびAla−196)で置換されたNefタンパク質をコードするオープンリーディングフレームを含む。opt tpanef(LLAA)のオープンリーディングフレームを含むヌクレオチド配列は、本発明では以下のとおりの配列番号15として開示されている。
【0238】
【化15】
【0239】
本発明では配列番号16として開示されるtPA−Nef(LLAA)をコードする配列番号7のオープンリーディングフレームは、以下のとおりである。
【0240】
【化16】
【0241】
本発明のアデノウイルスベクターは、野生型または修飾Nefタンパク質(本明細書に記載のとおり)、例えば、Gly2の欠失または置換、Leu174およびLeu175の欠失または置換、および/またはリーダー配列の付加を含む修飾Nefタンパク質(これに限定されるものではない)を発現する、コドン使用頻度とは無関係のDNA配列を含みうる。したがって、部分的または完全にコドンが最適化されたDNAワクチン発現ベクター構築物が好ましい。なぜなら、そのような構築物は宿主発現の増加をもたらすはずだからである。しかし、本明細書に開示する構築物の「非コドン最適化」形態、特に、宿主投与後に実質的な細胞性免疫応答を促進することが示されているHIV Nefの修飾形態を使用することも、本発明の範囲内である。
【0242】
図20A〜Cは、nef発現ベクターV1Jns/nef(図20A)、V1Jns/nef(G2A,LLAA)(図20B)、V1Jns/tpanef(図20C)およびV1Jns/tpanef(LLAA)(同様に図20C)のプラスミドバックボーンとnefコード配列との間の結合部のヌクレオチド配列を示す。コドン最適化nefまたはコドン最適化nef突然変異遺伝子の5’および3’フランキング配列が太字/イタリック体で示されており、nefおよびnef突然変異体コード配列が無修飾文字で示されている。また(下線付きで)、それぞれのnef発現ベクターの構築に関わる制限エンドヌクレアーゼ部位が示されている。V1Jns/tpanefおよびV1Jns/tpanef(LLAA)は該結合部位に同一配列を有する。
【0243】
図21は、nefおよびnef誘導体の概要図を示す。Nef誘導体に含まれるアミノ酸残基が示されている。グリシン2ならびにロイシン174および175は、それぞれミリスチル化およびジロイシンモチーフに関わる部位である。
【0244】
実施例19
MRKAd5polの構築およびウイルスレスキュー
ベクター:シャトルプラスミドおよび前アデノウイルスプラスミドの構築
MRKAd5polと称される前アデノウイルスプラスミドを含む該ベクターの構築における鍵工程を、図22に示す。簡単に説明すると、完全長不活性化HIV−1 pol遺伝子用のアデノウイルスシャトルベクターは以下のとおりである。ベクターMRKpdeE1(Pac/pIX/pack450)+CMVmin+BGHpA(str.)は、MEKAd5gagアデノウイルス前プラスミドの構築に使用するシャトルベクターの誘導体である。該ベクターは、hCMVプロモーター(無イントロンA)とウシ成長ホルモンポリアデニル化シグナルとを有する発現カセットを含有する。唯一のBglII部位における選択された遺伝子の挿入が、MRKpAd5(E1−/E3+)Cla1(またはMRKpAdHVE3)前プラスミド内への挿入の場合に、該トランスジーンの転写の方向がAd5 E1に平行となることを保証するよう、該発現単位は該シャトルベクター内に挿入されている。該ベクターは、元のシャトルベクターと同様に、PacI部位、パッケージングシグナル領域への伸長、およびpIX遺伝子への伸長を含有する。合成完全長コドン最適化HIV−1 pol遺伝子をプラスミドpV1Jns−HIV−pol−inact(opt)から直接単離した。BglIIでのこのプラスミドの消化は、無傷のpol遺伝子(配列番号3に記載のコドン最適化IA pol配列を含む)を遊離する。該pol断片をゲル精製し、MRKpdelE1(Pac/pIX/pack450)+CMVmin+BGHpA(str.)シャトルベクター内にBglII部位で連結した。制限酵素DraIII/Not1を使用することにより、該遺伝子の配向が正しいか否かに関して該クローンを調べた。陽性クローンを単離し、MRKpdel+hCMVmin+FL−pol+bGHpA(s)と命名した。このプラスミドの遺伝的構造を、PCR、制限酵素およびDNA配列決定により確認した。該前アデノウイルスプラスミドは以下のとおりに構築した。シャトルプラスミドMRKpdel+hCMVmin+FL−pol+bGHpA(S)を制限酵素Pac1およびBst1107I(またはそのアイソシゾマーであるBstZ107I)で消化し、ついで線状化(Cla1消化)アデノウイルスバックボーンプラスミドMRKpAd(E1−/E3+)Cla1と共に大腸菌(E.coli)株BJ5183内に同時形質転換した。元々はMRKpAd+hCMVmin+FL−pol+bGHpA(S)E3+と称されていた得られた前プラスミドを、新たに「pMRKAd5pol」と改称した。得られたpMRKAd5polの遺伝的構造を、PCR、制限酵素およびDNA配列分析により確認した。予備的産生のために、該ベクターをコンピテント大腸菌(E.coli)XL−1 Blue内に形質転換した。回収されたプラスミドを、制限酵素消化およびDNA配列分析により、ならびに一過性トランスフェクション細胞培養内の該polトランスジーンの発現により確認した。このpMRKAd5HIV−1polアデノウイルスベクターの完全なヌクレオチド配列を図26A〜AOに示す。
【0245】
研究等級組換えアデノウイルスの作製
前アデノウイルスプラスミドpMRKAd5polを、PER.C6(登録商標)接着単層細胞培養内で感染ビリオンとしてレスキューした。感染性ウイルスをレスキューするために、12μgのpMRKAd5polを制限酵素PacI(New England Biolabs)で消化し、リン酸カルシウム共沈技術(Cell Phect Transfection Kit,Amersham Pharmacia Biotech Inc.)を用いてPER.C6(登録商標)の6cmディッシュ当たり3.3μgをトランスフェクトした。PacI消化はプラスミド配列からウイルスゲノムを遊離して、PER.C6(登録商標)細胞内への侵入後にウイルス複製を生じさせる。トランスフェクションの6〜10日後、完全なウイルス細胞変性効果(CPE)が観察された後で、感染細胞および培地を回収した。感染細胞および培地を−60℃以下で保存した。このpol含有組換えアデノウイルスは本発明では「MRKAd5pol」と称される。この組換えアデノウイルスは、配列番号6に示すとおりの不活性化HIV−1 Polタンパク質を発現する。
【0246】
実施例20
MRKAd5Nefの構築およびウイルスレスキュー
ベクター:シャトルプラスミドおよび前アデノウイルスプラスミドの構築
MRKAd5nefと称される前アデノウイルスプラスミドを含む該ベクターの構築における鍵工程を、図23に示す。簡単に説明すると、前記実施例19に示したとおり、ベクターMRKpdeE1(Pac/pIX/pack450)+CMVmin+BGHpA(str.)は、MEKAd5gagアデノウイルス前プラスミドの構築に使用するシャトルベクターである。それは、PacI部位、パッケージングシグナル領域への伸長、およびpIX遺伝子への伸長を含有するよう修飾されている。それは、hCMVプロモーター(無イントロンA)とウシ成長ホルモンポリアデニル化シグナルとを有する発現カセットを含有する。唯一のBglII部位における選択された遺伝子の挿入が、MRKpAd5(E1−/E3+)Cla1前プラスミド内への挿入の場合に、該トランスジーンの転写の方向がAd5 E1に平行となることを保証するよう、該発現単位は該シャトルベクター内に挿入されている。合成完全長コドン最適化HIV−1 nef遺伝子をプラスミドpV1Jns/nef(G2A,LLAA)から直接単離した。BglIIでのこのプラスミドの消化は、配列番号13に開示するヌクレオチド配列を含む無傷のpol遺伝子を遊離する。該nef断片をゲル精製し、MRKpdelE1+CMVmin+BGHpA(str.)シャトルベクター内にBglII部位で連結した。制限酵素Sca1を使用することにより、該遺伝子の配向が正しいか否かに関して該クローンを調べた。陽性クローンを単離し、MRKpdelE1hCMVminFL−nefBGHpA(s)と命名した。このプラスミドの遺伝的構造を、PCR、制限酵素およびDNA配列決定により確認した。該前アデノウイルスプラスミドは以下のとおりに構築した。シャトルプラスミドMRKpdelE1hCMVminFL−nefBGHpA(s)を制限酵素Pac1およびBst1107I(またはそのアイソシゾマーであるBstZ107I)で消化し、ついで線状化(Cla1消化)アデノウイルスバックボーンプラスミドMRKpAd(E1−/E3+)Cla1と共に大腸菌(E.coli)株BJ5183内に同時形質転換した。元々はMRKpdelE1hCMVminFL−nefBGHpA(s)と称されていた得られた前プラスミドを、新たに「pMRKAd5nef」と改称した。得られたpMRKAd5nefの遺伝的構造を、PCR、制限酵素およびDNA配列分析により確認した。予備的産生のために、該ベクターをコンピテント大腸菌(E.coli)XL−1 Blue内に形質転換した。回収されたプラスミドを、制限酵素消化およびDNA配列分析により、ならびに一過性トランスフェクション細胞培養内の該nefトランスジーンの発現により確認した。このpMRKAd5HIV−1nefアデノウイルスベクターの完全なヌクレオチド配列を図27A〜AMに示す。
【0247】
研究等級組換えアデノウイルスの作製
前アデノウイルスプラスミドpMRKAd5nefを、PER.C6(登録商標)接着単層細胞培養内で感染ビリオンとしてレスキューした。感染性ウイルスをレスキューするために、12μgのpMRKAd5nefを制限酵素PacI(New England Biolabs)で消化し、リン酸カルシウム共沈技術(Cell Phect Transfection Kit,Amersham Pharmacia Biotech Inc.)を用いてPER.C6(登録商標)の6cmディッシュ当たり3.3μgをトランスフェクトした。Pac1消化はプラスミド配列からウイルスゲノムを遊離して、PER.C6(登録商標)細胞内への侵入後にウイルス複製を生じさせる。トランスフェクションの6〜10日後、完全なウイルス細胞変性効果(CPE)が観察された後で、感染細胞および培地を回収した。感染細胞および培地を−60℃以下で保存した。ここで、このnef含有組換えアデノウイルスは、「MRKAd5nef」と称される。
【0248】
実施例21
不活性化PolおよびNef/G2A,LLAA用のマウスCMVプロモーター含有シャトルベクターの構築
プライマーセット:mCMV(NotI)フォワード5’−ATA AGA ATG CGG CCG CCA TAT ACT GAG TCA TTA GG−3’(配列番号20);mCMV(Bgl II)リバース:5’−AAG GAA GAT CTA CCG ACG CTG GTC GCG CCT C−3’(配列番号21)を使用して、プラスミドpMH4(Frank Graham,McMaster Universityから供給された)からマウスCMV(mCMV)を増幅した。該下線付きヌクレオチドは、各プライマーのそれぞれNotIおよびBglII部位を表す。該トランスジーンをE1平行配向で含有するmCMVシャトルベクターの構築のために、このPCRアンプリコンを使用した。NotIおよびBglIIでの消化により、元のシャトルベクター(hCMV−gag−bGHpAトランスジーンをE1平行配向で含有する)からhCMVプロモーターを除去した。mCMVプロモーター(NotI/BglII消化PCR産物)を該シャトルベクター内に一定方向で挿入した。ついで該シャトルベクターをBglIIで消化し、gagレポーター遺伝子(BglII断片)を該シャトルベクター内に再挿入した。いくつかのクローンを、該レポーター遺伝子の方向の正しさに関してスクリーニングした。E1逆平行配向のmCMV−gagの構築のためには、プライマーセット:mCMV(AscI)フォワード5’−ATA AGA ATG GCG CGC CAT ATA CTG AGT CAT TAG G(配列番号22);mCMV(BglII)リバース:5’AAG GAA GAT CTA CCG ACG CTG GTC GCG CCT C(配列番号23)を使用して、プラスミドpMH4からmCMVプロモーターを増幅した。該下線付きヌクレオチドは、各プライマーのそれぞれAscIおよびBglII部位を表す。hCMV−gagトランスジーンをE1逆平行配向で含有するシャトルベクターをAsc1およびBgl11で消化して、該トランスジーンのhCMV−gag部分を除去した。mCMVプロモーター(Asc1/Bgl11消化PCR産物)を該シャトルベクター内に一定方向で挿入した。ついで該ベクターをBgl11で消化し、該gagレポーター遺伝子(Bgl11断片)を再挿入した。いくつかのクローンを、該レポーター遺伝子の配向の正しさに関してスクリーニングした。それぞれの完全長IAおよび完全長nef/G2A,LLAA遺伝子に関しては、該mCMV−bGHpAシャトルベクター内の唯一のBglII部位を使用して、クローニングを行った。該polおよびnef遺伝子を、BglII消化により、それらのそれぞれのpV1Jnsプラスミドから切り出した。
【0249】
実施例22
mCMV完全長不活性化Polおよび完全長nef/G2A.LLAAアデノベクターの構築
実施例21のこれらのトランスジーンのそれぞれを該修飾シャトルベクター内にE1平行およびE1逆平行の両方の配向で挿入した。各シャトルベクターのPac1およびBstZ110I消化を行い、フランキングAd5配列を含有する各特異的トランスジーン断片を単離し、ClaIで消化されたMRKpAd5(E3+)またはMRKpAd5(E3−)アデノベクタープラスミドと共に、BJ5183大腸菌(E.coli)細胞内で細菌相同組換えにより同時形質転換した。ついで、種々のトランスジーンをE3−およびE3+の両方の形態(ならびにE1平行およびE1逆平行配向)で含有する組換え前プラスミドアデノベクターを、XL−1 Blue大腸菌(E.coli)細胞内への形質転換の後で大規模で調製し、制限分析および配列決定により分析した。
【0250】
実施例23
hCMV−tpa−nef(LLAA)アデノベクターの構築
プライマーセット:Tpanef(BamHI)F5’−ATT GGA T CC ATG GAT GCA ATG AAG AGA GGG(配列番号24);Tpanef(BamHI)R5’−ATA GGA TCC TTA GCA GTC CTT GTA GTA CTC G(配列番号25)を使用して、GMP等級pV1Jns−tpanef(LLAA)ベクターからtpa−nef遺伝子を増幅した。得られたPCR産物をBamHIで消化し、ゲル精製し、MRKAd5CMV−bGHpAシャトルベクター(BglIIで消化され仔ウシ腸ホスファターゼで処理されたもの)のBglII部位内にクローニングした。hCMVプロモーターに対して正しい配向でtpanef(LLAA)遺伝子(完全なコード領域に関しては配列番号15を参照されたい)を含有するクローンを、ScaI消化後に選択した。得られたMRKAd5tpanefシャトルベクターをPacIおよびBstZ1101で消化し、細菌相同組換え技術によりE3+MRKAd5アデノベクター内にクローニングした。
【0251】
実施例24
MRKAd5polおよびMRKAd5nefワクチンの免疫原性
材料および方法−げっ歯類への免疫
N=10のBALB/cマウスの群の筋肉内(i.m.)に以下のベクターで免疫した:(1)10∧7vpおよび10∧9vpのうちのいずれかのMRKAd5hCMV−IApol(E3+)、ならびに(2)10∧7vpおよび10∧9vpのうちのいずれかのMRKAd5hCMV−IApol(E3−)。投与の7週間後、10匹/コホートのうちの5匹のマウスを、それらが最初に投与されたのと同じベクターおよび用量で追加抗原刺激した。該第2投与の3週間後、それらのすべての動物から血清および脾臓をそれぞれRT ELISAおよびIFNg ELIspot分析のために集めた。げっ歯類への全ての免疫のために、該Ad5ベクターを5mM Tris、5% スクロース、75mM NaCl、1mM MgCl2、0.005% ポリソルベート80(pH8.0)中で希釈した。28−1/2G針付きの0.3mL インスリンシリンジ(Becton−Dickinson,Franklin Lakes,NJ)を使用して、全用量を50μM アリコート中で両方の方形筋に注射した。
【0252】
N=10のC57/BL6マウスの群の筋肉内(i.m.)に以下のベクターで免疫した:(1)10∧7vpおよび10∧9vpのうちのいずれかのMRKAd5hCMV−nef(G2A,LLAA)(E3+)、(2)10∧7vpおよび10∧9vpのうちのいずれかのMRKAd5mCMV−nef(G2A,LLAA)(E3+)、ならびに(3)10∧7vpおよび10∧9vpのうちのいずれかのMRKAd5mCMV−tpanef(LLAA)(E3+)。投与の7週間後、10匹/コホートのうちの5匹のマウスを、それらが最初に投与されたのと同じベクターおよび用量で追加抗原刺激した。該第2投与の3週間後、それらのすべての動物から血清および脾臓をそれぞれRT ELISAおよびIFNg ELIspot分析のために集めた。
【0253】
非ヒト霊長類への免疫
3匹のアカゲザル(2〜3kg)のコホートを以下のAdベクターでワクチン接種した:(1)10∧9vpおよび10∧11vp用量のうちのいずれかのMRKAd5hCMV−IApol(E3+)、ならびに(2)10∧9vpおよび10∧11vpのうちのいずれかのMRKAd5hCMV−IApol(E3−)、(3)10∧9vpおよび10∧11vpのうちのいずれかのMRKAd5hCMV−nef(G2A,LLAA)(E3+)、ならびに(4)10∧9vpおよび10∧11vpのうちのいずれかのMRKAd5mCMV−nef(G2A,LLAA)(E3+)。該ワクチンを化学的拘束サル(10mg/kg ケタミン)に、2つの0.5mLアリコートの該Adベクター(5mM Tris、5% スクロース、75mM NaCl、1mM MgCl2、0.005% ポリソルベート80(pH8.0)中)の両三角筋への針注射により投与した。該動物は、4週間隔で2回免疫した(T=0、4週)。
【0254】
マウス抗RTおよび抗nefELISA
標準的な二次抗体に基づくELISAにより、抗RT力価を得た。PBS中の100μLの1μg/mL HIV−1 RTタンパク質(Advanced Biotechnologies,Columbia,MD)と共に一晩インキュベートすることにより、Maxisorpプレート(NUNC Rochester,NY)をコートした。該プレートを、Titertek MAP装置(Hunstville,AL)を使用してPBS/0.05% Tween 20で洗浄し、200μL/ウェルのブロッキング溶液(PBS/0.05% tween/1% BSA)と共に2時間インキュベートした。100倍の初期血清希釈を行い、ついで4倍の系列希釈を行った。系列希釈サンプルの100μL アリコートをウェルごとに加え、室温で2時間インキュベートした。該プレートを洗浄し、100μLの1/1000希釈HRPウサギ抗マウスIgG(ZYMED,San Francisco,CA)を、1時間インキュベートしながら加えた。該プレートを十分に洗浄し、100μL 1,2−フェニレンジアミン二塩酸塩/過酸化水素(DAKO,Norway)溶液に15分間浸した。ウェル当たり100μLの0.5M H2SO4を加えることにより、該反応をクエンチした。S20スタッカと共にTitertek Multiskan MCC/340を使用して、OD492の読み取り値を記録した。終点力価は、0.1 OD492(該バックグラウンド値の2.5倍)以上の吸光度値を与える最高血清希釈度と定義した。
【0255】
非ヒト霊長類およびマウスELIspotアッセイ:
酵素結合イムノスポット(ELISpot)アッセイを用いて、アカゲザルPBMCまたはマウス脾臓からの抗原特異的INFγ分泌細胞(Miyahiraら,1995,J.Immunol.Methods 1995,181,45−54)を計数した。マウス脾臓を5匹/コホートのマウスからプールし、8mLの完全RPMI培地(RPMI1640,10% FBS,2mM L−グルタミン,100U/mL ペニシリン,100u/mL ストレプトマイシン,10mM Hepes,50uM β−ME)中、5×106/mLで単細胞懸濁液を調製した。標準的なフィコール勾配分離(Coliganら,1998,Current Protocols in Immunology.John Wiley & Sons,Inc.)により、アカゲザルPBMCを8〜15mLのヘパリン化血から調製した。Multiscreen不透明プレート(Millipore,France)をPBS中の100μL/ウェルの5μg/mL 精製ラット抗マウスIFN−γ IgG1、クローンR4−6A2(Pharmingen,San Diego,CA)または15μg/mL マウス抗ヒトINF−γ IgG2a(Cat.No.1598−00,R&D Systems,Minneapolis,MN)で、それぞれマウスまたはサルでのアッセイのために4℃で一晩にわたりコートした。該プレートを、PBS/ペニシリン/ストレプトマイシンで洗浄し、200μL/ウェルの完全RPMI培地で37℃で少なくとも2時間にわたりブロッキングした。
【0256】
各ウェルに、50μLの細胞サンプル(4〜5×105細胞/ウェル)および50μLの該抗原溶液を加えた。該対照ウェルには、50μLのDMSO含有培地を加え、特異的応答のために、選択されたペプチドまたはペプチドプール(ペプチドの最終濃度ごとに4μg/mL)を加えた。該pol構築物で免疫したBALB/cマウスに関しては、CD4+−エピトープ含有20量体ペプチド(aa21−40,aa411−430,aa641−660,aa731−750,aa771−790)のプールまたはCD8+−エピトープ含有ペプチド(aa201−220,aa311−330,aa781−800)を使用して刺激を行った。該nef構築物で免疫したC57/BL6マウスに関しては、nef配列に由来するaa51−70(CD8+T細胞エピトープ)またはaa81−100(CD4+)ペプチドを特異的刺激のために加えた。サルにおいては、全pol配列を含み10アミノ酸で重複する20−aaペプチドの2つのプール(LおよびR)を使用して、polに対する応答を評価した。該nef構築物でワクチン接種したサルにおいては、全HIV−1 nef配列を含み10aaで重複する20量体ペプチドを含有する単一のプールを使用した。各サンプル/抗原混合物は、マウスサンプルについては三重ウェル中で、アカゲザルPBMCについては二重ウェル中で実施された。プレートを37℃、5% CO2、90% 湿度で20〜24時間インキュベートした。該プレートをPBS/0.05% Tween 20で洗浄し、100μL/ウェルの1.25μg/mL ビオチン共役ラット抗マウスIFN−γmAb、クローンXMG1.2(Pharmingen)または0.1μg/mL ビオチン化抗ヒトIFNγヤギポリクローナル抗体(R&D Systems)と共に4℃で一晩インキュベートした。該プレートを洗浄し、PBS/0.005% Tween/5% FBS中、100μL/ウェルの1/2500希釈度のストレプトアビジン−アルカリホスファターゼ共役体(Pharmingen)と共に37℃で30分間インキュベートした。洗浄後、100μl/ウェルの1工程(1−step)NBT/BCIP(Pierce Chemicals)と共に6〜10分間インキュベートすることにより、スポットを現像した。該プレートを水で洗浄し、風乾した。解剖顕微鏡を使用して各ウェル内のスポットの数を測定し、該データを106個の細胞の入力に対して正規化した。
【0257】
非ヒト霊長類抗RT ELISA
該サルにおけるpol特異的抗体を競合RT EIAアッセイで測定し、この場合、プレートウェル上のコート抗体への結合からRT抗原を遮断する能力によりサンプルの活性を測定した。簡単に説明すると、Mxisorpプレートを飽和量のpol陽性ヒト血清(#97111234)でコートした。250μLの各サンプルを15μLの266ng/mL RT組換えタンパク質(RCM 563,1% BSA,0.1% トゥイーン,0.1% NaN3中)および20μLの細胞溶解バッファー(Coulter p24抗原アッセイキット)と共に室温で15分間インキュベートする。最大RT結合を定める陰性対照および標準の系列希釈サンプルを使用して、同様の混合物を調製する。200μL/ウェルの各サンプルおよび標準を該洗浄プレートに加え、該プレートを室温で16〜24時間インキュベートした。結合RTを、Coulter p24アッセイキットに記載の方法に従い定量し、選択された標準により任意に定められる1mL当たりのミリ・メルク(milliMerck)単位で表す。
【0258】
結果−げっ歯類での研究
BALB/cマウス(n=マウス5匹/コホート)を種々の用量のMRKAd5dhCMV−IApol(E3+)およびMRKAd5hCMV−IApol(E3−)で1回または2回免疫した。該第2投与の3週間後、代替抗原としてRTを用いるELISAアッセイにより、抗pol IgGレベルを測定した。polエピトープ含有ペプチドのプールに対するINFγELISpotアッセイにより、細胞性応答を定量した。これらのアッセイの結果を表10に要約する。該結果は、該ワクチン接種マウスが、僅か10∧7vpのアデノベクター用量で、検出可能な抗RT IgGを示している。該体液性応答は高度に用量依存的であり、第2免疫で増強可能である。1または2用量のpolベクターは、高度数の抗原特異的CD4+およびCD8+T細胞を惹起し、該応答は、低度に用量依存的であるが、第2免疫で増強可能である。
【0259】
【表10】
【0260】
C57/BL6マウスを、種々の用量のMRKAd5hCMV−nef(G2A,LLAA)(E3+)、MRKAd5mCMV−nef(G2A,LLAA)(E3+)で10∧7vpにて、および(3)MRKAd5mCMV−tpanef(LLAA)(E3+)で10∧7vpおよび10∧9vpのうちのいずれかで免疫した。同様のプロトコールを用いて該免疫応答を分析し、結果を表11に示す。このモデル系においては該構築物のいずれにおいても抗nef IgG応答を検出することができなかったが、ELIspotアッセイによれば、nefに対して生成した細胞性免疫の強力な指標が存在する。
【0261】
【表11】
【0262】
サルでの研究
3匹のアカゲザルのコホートを2用量のMRKAd5hCMV−IApol(E3+)およびMRKAd5hcMV−IApol(E3−)で免疫した。全pol配列を含む2つのペプチドプール(LおよびR)のうちの一方を使用して、抗原特異的T細胞の数(百万個のPBMC当たり)を計数した。結果を表12に示す。いずれかの構築物を10∧9vpの低用量で使用した場合にも、ワクチン被接種体において中等度〜強力なT細胞応答が検出された。該動物における抗RT抗体力価の縦断的分析は、体液性応答を惹起するよう該polトランスジーン産物が効率的に発現されることを示唆している(表13)。E3−構築物が投与された動物においてはE3+ウイルスの場合と比較して、一般に、より高い免疫応答が観察されるようであった。
【0263】
【表12】
【0264】
【表13】
【0265】
アカゲザルを2用量のMRKAd5nef構築物で筋肉内に免疫した場合には、12匹中8匹のワクチン被投与体において、百万当たり100〜1100の範囲の強力なT細胞応答が観察された(表14)。これまでに得られたデータに基づけば、mCMVおよびhCMVにより駆動されるnef構築物の効力は比較しうるものである。
【0266】
【表14】
【0267】
実施例25
HIV感染対象におけるクレードBとクレードCとのT細胞応答の比較
米国においてHIV−1に感染した24人の患者から集めたPBMCサンプルを、10アミノ酸により重複する20量体ペプチドのペプチドプールを使用するELISPOTアッセイにおいて試験した。4個の異なるペプチドプールを交差クレード認識に関して試験した。それらは、クレードBに基づく分離体(gag H−b;nef−b)またはクレードCに基づく分離体(gag H−c,nef−c)から誘導された。表15のデータは、恐らくクレードB HIV−1に感染したこれらの患者からのT細胞が、ELISPOTアッセイにおいてクレードC gagおよびnef抗原を認識しうることを示している。相関分析は更に、クレードC gagペプチドプールに対するこれらのT細胞応答がクレードB対応物の約60%であり、クレードC nefに対するT細胞応答がクレードB対応物の約85%であることを示した(図25)。これらの結果は、クレードB HIV−1に感染した患者において生成した細胞性免疫応答がクレードC HIV−1由来のgagおよびnef抗原を認識しうることを示唆している。これらのデータは、クレードB gagおよび/またはnef抗原を発現するDNAまたはMRKAdに基づくワクチンのようなHIVワクチンが、地球規模での予防的および/または治療的利点をもたらす能力を潜在的に有することを示している。
【0268】
【表15】
【0269】
実施例26
ローラーボトルにおけるのMRKAd5polおよびMRKAd5nefベクターの特徴づけおよび生産
nefおよびpolアデノベクターの増殖
nefおよびpol CsCl精製MRKAd5種(シード)を使用してローラーボトルに感染させて、更なる実験の種(シード)として使用するP4ウイルスを製造した。P4 MRKAd5 polおよびnefベクターを使用してMOI 280vp/細胞でローラーボトルに感染させた。ただし、hCMV−tpa−nef[E3+]は例外で、これは、P4で得た種の力価が低かったため125のMOIで感染させた。
【0270】
【表16】
【0271】
ローラーボトル継代
polおよびnef構築物の継代を継代7まで継続した。全継代において得られた細胞結合(凍結/融解細胞溶解)および全ブロス(トリトン細胞溶解)力価は非常に一貫したものであった。一般には、MRKAd5polはMRKA5gagの約70%生産性であり、MRKA5nefはMRKAd5gagの約25%生産性であった。両構築物のP7ウイルスのサンプルを制限消化分析によるV&CBにより分析し、それらはいずれの再構成をも示さなかった。
【0272】
【表17】
【0273】
【表18】
【0274】
MRKAd5nefおよびMRKAd5polウイルス生産速度論
MRKAd5polおよびMRKAd5nefベクターのウイルス生産速度論がMRKAd5gagのものに類似しているか否かを判定するために、ローラーボトル内で経時的実験を行った。ローラーボトル培養内のPER.C6(登録商標)細胞に、P5 MRKAd5pol、P5 MRKAd5nefおよびP7 MRKAd5gagを280vp/細胞のMOIで感染させた。各アデノベクターに関して、感染の24、36、48および60時間後、2つの感染ボトルからサンプルを採取した。また、I相(Phase I)法条件下で回収する場合には、48hpiまで、2つのボトルをサンプル未採取のまま残した。24、36、48および60hpiの時点における凍結融解回収細胞結合ウイルスの陰イオン交換HPLCウイルス粒子濃度を図29A〜Bに示す。QPA力価は、類似した傾向を示している(データは示していない)。
【0275】
hCMVおよびmCMV−FL−nefの比較
MRKAd5nef構築物(hCMV−FL−nef)で得られた力価は、MRKAd5gagまたはMRKAd5polで得られたものより低かったため、ウイルス生産性比較実験はmCMV−FL−nefで行った。2つのアデノベクター(hCMV−およびmCMV−FL−nef)のそれぞれに関して、2つのローラーボトルに継代5の清澄化ライセートを280vp/細胞のMOIで感染させた。それらの4つのローラーボトルの肉眼的および顕微鏡的観察は、回収の時点で同じであった。得られた清澄化ライセートの分析は、表19に示すとおり、mCMV−FL−nefに感染したボトル内の、より高いウイルス粒子濃度を示した。mCMVプロモーターにより駆動されるnefベクターでの、より高い生産性は、PER.C6(登録商標)細胞における、より低いnef発現レベルによるものと認められる。nef発現レベルを測定するための実験がV&CBで進行中である。
【0276】
【表19】
【0277】
実施例27
バイオリアクターにおけるMRKAd5nefウイルスの特徴づけおよび大規模生産
材料および方法
本実施例の実験は、以下の条件下で2回実施した:36.5℃、DO 30%、pH7.30、150rpm攪拌速度、散布無し、Life Technologies(Gibco Invitrogen)293 SFM II(6mM L−グルタミン含有)、pH制御用の塩基としての0.5M NaOH。第1の実施(B20010115)においては、2つの10L 攪拌容器バイオリアクターにPER.C6(登録商標)細胞を0.2×106細胞/mlの濃度で接種した。細胞を、約1×106細胞/mlの細胞濃度に達するまで増殖させた。該細胞に未クローン化MRKAd5nef(G2A,LLAA)を280ウイルス粒子(vp)/細胞で感染させた。第2バッチ(B20010202)には、該細胞にクローン化MRAd5nefを感染させたこと以外は該第1実施と同じ方法を用いた。両方の実施中、該バイオリアクターを感染の48時間後に回収した。サンプルを採取し、全ブロス(トリトン細胞溶解を伴う)、上清および細胞ペレット(3×凍結/融解)から、AEXおよびQPAアッセイでウイルス濃度を測定した。該方法の全体にわたり、BioProfile250で代謝産物を測定した。
【0278】
【表20】
【0279】
【表21】
【0280】
結果−表22および表23は、MRKAd5nefのバイオリアクター増殖によるスケールアップ生産の能力を示す。
【0281】
【表22】
【0282】
【表23】
【0283】
本発明の範囲は、本明細書に記載の特定の実施形態により限定されるものではない。実際のところ、前記の説明から、本明細書に記載されているものに加えて本発明の種々の修飾が当業者に明らかとなるであろう。そのような修飾も、特許請求の範囲の範囲内に含まれると意図される。
【0284】
実施例28
DNA初回抗原刺激動物のMRKAdHIV−1gag追加抗原刺激
3〜5匹のアカゲザルの群を、第0、4および8週に、(a)5mgのV1Jns−Flgag(pVIJnsCMV(無イントロン)−FL−gag−bGHpA)、(b)45mgの非イオン性ブロック共重合体CRL1005で製剤化された5mgのV1Jns−Flgag、または(c)7.5mgのCRL1005および0.6mM 塩化ベンザルコニウムで製剤化された5mgのV1Jns−Flgagで免疫した。すべての動物に、1回量の10e7 ウイルス粒子(vp)の該MRKAd5HIV−1gagを第26週に投与した。注:単一様式での投与の場合には(用量は、アデノウイルスへの前曝露を模擬するよう選択される)、10e7は余りにも低すぎて、有効に初回抗原刺激または追加抗原刺激することができない;図32を参照されたい。
【0285】
血液サンプルを全動物から幾つかの時点で集め、末梢血単核細胞(PBMC)を、標準的なフィコール法を用いて調製した。該PBMCを計数し、ELISpotアッセイを用いてγインターフェロン分泌に関して分析した(表24)。各サルに関して、全HIV−1 gag配列を含む50個の20アミノ酸長ペプチドのプールの不存在下(培地)または存在下(「gag H」と称される)、該PBMCを一晩インキュベートした。
【0286】
結果は、これらのサルにおけるT細胞免疫応答の増強においてMRKAd5HIV−1gagが非常に有効であったことを示している。該ウイルス追加抗原刺激の28週間後または2週間後、百万個のPBMC当たりのgag特異的T細胞数は、該追加抗原刺激の24週間前または2週間前に観察されたレベルと比較して2〜48倍増加した。
【0287】
また、該MRKAd5gag追加抗原刺激の前(第10週)および後(第30週)に、PBMCを細胞内γインターフェロン染色により分析した。選択した動物に関する結果を図31に示す。結果は、(a)DNA/アジュバント製剤での免疫が、平衡的、CD4+偏向的またはCD8+偏向的でありうるT細胞応答を惹起したこと、および(b)MRKAd5gag構築物での追加刺激が、すべての場合において、強くCD8+偏向した応答を引き起こしたことをを示している。これらの結果は、MRKAd5HIV−1gag構築物での追加刺激が抗原特異的CD8+T細胞のレベルを改善しうることを示唆している。
【0288】
【表24】
【0289】
実施例29
MRKAd5gagpol融合構築物の構築
コドン最適化HIV−1 gag遺伝子のオープンリーディングフレームを、段階的PCRにより、IA pol遺伝子(RT、RNアーゼおよびインテグラーゼドメインよりなる)のオープンリーディングフレームに直接的に融合させた。該遺伝子(配列番号38)は該プロテアーゼ遺伝子およびフレームシフト配列を含まないため、それは、p55、RT、RNアーゼHおよびインテグラーゼが組合されたサイズ(1350アミノ酸;配列番号39)の単一ポリペプチドをコードしている。
【0290】
該gag遺伝子内のBstEII部位から最後の非終結コドンまで伸長する断片を、該IApolの開始コドンから唯一のBamHI部位まで伸長する断片にPCRにより連結した。この断片をBstEIIおよびBamHIで消化した。gag−IApol融合体の構築は、PstI−BstEII gag消化断片、BstEII/BamHI消化PCR産物および長いPstI/BamHI/V1R−FLpolバックボーン断片を含む3断片連結により達成された。
【0291】
該MRKAd5−gagpolアデノウイルスベクターは、gag−IApol融合遺伝子の全ORFを含有するV1R−gagpolのBglII断片を使用して構築した。
【0292】
実施例30
非ヒト霊長類における免疫原性研究
3匹のアカゲザルのコホートを、10e8または10e10ウイルス粒子(vp)の以下のMRKAd5 HIV−1ワクチンの1つで免疫した:(1)MRKAd5gag;(2)MRKAd5pol;(3)MRKAd5nef;(4)等量のMRKAd5gag、MRKAd5polおよびMRKAd5nefを含有する混合物、または(5)等量のMRKAd5gagpolおよびMRKAd5nefの混合物。
【0293】
該HIV−1抗原のそれぞれに対するT細胞応答を、各抗原の全タンパク質配列を含む20−aaのペプチドを使用するIFNγELISpotアッセイによりアッセイした。結果(表25)は、該ペプチドプールのそれぞれに応答する百万個の末梢血単核細胞(PBMC)当たりのスポット形成細胞(sfc)の数として表されている。
【0294】
結果は以下の観察を示している:(1)該単一遺伝子構築物(MRKAd5gag,MRKAd5polまたはMRKAd5nef)のそれぞれは、サルにおいて、高レベルの抗原特異的T細胞を惹起しうる;(2)該単一遺伝子MRKAd5構築物は、gag、polおよびnefに対して非常に広範なT細胞応答を惹起しうる多カクテル製剤として混合されうる;(3)gagとIA polとの融合タンパク質を発現するMRKAd5ベクターは、gagおよびpolの両方に対して強力なT細胞応答を誘導しうる。
【0295】
【表25】
【図面の簡単な説明】
【図1】
元のHIV−1 gagアデノベクター(Ad5HIV−1gag)を示す。
【図2】
該最適化ヒトHIV−1 gagオープンリーディングフレームの核酸配列(配列番号29)を示す。
【図3】
元のgagトランスジーンと比較して新規トランスジーン構築物を図示する。
【図4】
本発明の新規ベクターの作製において元のアデノベクターバックボーンに施された修飾を示す。
【図5】
該パッケージングシグナル領域に対して施された付加(実験#1)およびウイルス増殖に対するE3遺伝子(実験2)の効果を判定するために行ったウイルス混合実験を示す。
【図6】
実施例6および7に記載するウイルス混合実験後のウイルスDNA分析のオートラジオグラフを示す。
【図7A】
MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたhCMV−Flgag−bGHpAアデノベクターを示す。
【図7B】
MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたhCMV−Flgag−SPAアデノベクターを示す。
【図7C】
MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたmCMV−Flgag−bGHpAアデノベクターを示す。
【図8A】
トランスジーン配向の効果を試験するよう意図された実験を示す。
【図8B】
ポリアデニル化シグナルの効果を試験するよう意図された実験を示す。
【図9】
BstE11消化後のP5における試験された4つのアデノウイルスベクターからのウイルスDNA(実施例12)を示す。
【図10】
MRKpAdHVE3、MRKAd5HIV−1gagおよびMRKAd5HIV−1gagE3−の継代11および12のウイルスDNA分析を示す。
【図11】
ウイルス競合研究を開始するために使用した継代6のMRKpAdHVE3およびMRKAd5HIV−1gagのウイルスDNA分析(HindIII消化)を示す。
【図12】
特定された時点で回収が行われた血清含有培地中のMRKAd5HIV−1gagに関する高継代数上のHindIII消化によるウイルスDNA分析を示す。
【図13】
種々の用量のいくつかのAdgag構築物でbalb/cマウス(n=10)をi.m.免疫した3週間後の血清抗p24レベルを示す:(A)MRK Ad5 HIV−1 gag(継代5)、(B)MRKAd5hCMV−FLgag−bGHpA(E3−)、(C)MRKAd5 hCMV−FLgag−SPA(E3+)、(D)MRKAd5 mCMV−FLgag−bGHpA(E3+)、(E)Ad5HIV−1 gagの研究ロット(293細胞由来)、および(F)Ad5HIV−1 gagの臨床ロット(Ad5gagFN0001)。示されているのは、各コホートの幾何平均力価(GMT)および標準誤差線である。
【図14】
pMRKAd5HIV−1 gagベクターの制限地図を示す。
【図15A】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15B】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15C】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15D】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15E】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15F】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15G】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15H】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15I】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15J】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15K】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15L】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15M】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15N】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15O】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15P】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15Q】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15R】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15S】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15T】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15U】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15V】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15W】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15X】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図16】
DNAワクチン発現ベクターV1Jns(A)およびV1Jns−tPA(B)の概要図を示す。
【図17A】
IA−Polのヌクレオチド(配列番号3)およびアミノ酸配列(配列番号4)を示す。
【図17B】
IA−Polのヌクレオチド(配列番号3)およびアミノ酸配列(配列番号4)を示す。
【図17C】
IA−Polのヌクレオチド(配列番号3)およびアミノ酸配列(配列番号4)を示す。
【図18】
tPA−pol inact(opt)の融合結合部にわたるコドン最適化ヌクレオチドおよびアミノ酸配列(それぞれ配列番号7および8中に含有される)を示す。
【図19A】
野生型nef(jrfl)とコドン最適化nefとのヌクレオチド配列比較を示す。
【図19B】
野生型nef(jrfl)とコドン最適化nefとのヌクレオチド配列比較を示す。
【図20】
nef発現ベクターV1Jns/nef(図20A)、V1Jns/nef(G2A,LLAA)(図20B)、V1Jns/tpanef(図20C)およびV1Jns/tpanef(LLAA)(同様に図20C)のプラスミドバックボーンとnefコード配列との間の結合部のヌクレオチド配列を示す。
【図21】
nefおよびnef誘導体の概要図を示す。
【図22】
前アデノウイルスプラスミド構築物MRKAd5Polの構築の概要図を示す。
【図23】
前アデノウイルスプラスミド構築物MRKAd5Nefの構築の概要図を示す。
【図24】
クレードB HIV感染個体におけるクレードBとクレードCとの抗gag T細胞応答の比較を示す。
【図25】
クレードB HIV感染個体におけるクレードBとクレードCとの抗nef T細胞応答の比較を示す。
【図26A】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26B】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26C】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26D】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26E】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26F】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26G】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26H】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26I】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26J】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26K】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26L】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26M】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26N】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26O】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26P】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26Q】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26R】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26S】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26T】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26U】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26V】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26W】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26X】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26Y】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26Z】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AA】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AB】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AC】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AD】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AE】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AF】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AG】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AH】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AI】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AJ】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AK】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AL】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AM】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AN】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AO】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図27A】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27B】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27C】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27D】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27E】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27F】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27G】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27H】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27I】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27J】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27K】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27L】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27M】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27N】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27O】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27P】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27Q】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27R】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27S】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27T】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27U】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27V】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27W】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27X】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27Y】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27Z】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AA】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AB】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AC】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AD】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AE】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AF】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AG】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AH】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AI】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AJ】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AK】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AL】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AM】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図28】
E3(+)またはE3(−)バックボーン中に種々のプロモーター断片(hCMVまたはmCMV)および終結シグナル(bGHまたはSPA)を含むMRKAd5ベクターの安定性を示す。
【図29A】
24、36、48および60hpiの時点における凍結融解回収細胞結合ウイルスの陰イオン交換HPLCウイルス粒子濃度を示す。
【図29B】
MRKAd5gag、MRKAd5polおよびMRKA5nefに関する経時的QPA上清力価を示す。
【図30A】
本明細書に開示するDNAおよび/またはアデノウイルスワクチンにおいて使用する代表的なtPA−gag融合体のオープンリーディングフレームをオープンリーディングフレーム含むヌクレオチド配列(配列番号36)およびアミノ酸配列(配列番号37)を示す。
【図30B】
本明細書に開示するDNAおよび/またはアデノウイルスワクチンにおいて使用する代表的なtPA−gag融合体のオープンリーディングフレームをオープンリーディングフレーム含むヌクレオチド配列(配列番号36)およびアミノ酸配列(配列番号37)を示す。
【図31】
第10週(DNA初回抗原刺激後)および第30週(Ad追加抗原刺激後)に集めたPBMCの細胞内γIFN染色を示す。
【図32】
単一様式アデノウイルス免疫とDNA+アジュバント初回/アデノウイルス追加免疫との比較を示す。
【図33A】
実施例29のgag−IApol融合体のオープンリーディングフレームのヌクレオチド配列(配列番号38)を示す。
【図33B】
実施例29のgag−IApol融合体のオープンリーディングフレームのヌクレオチド配列(配列番号38)を示す。
【図34A】
該gag−IApol融合フレームのタンパク質配列(配列番号39)を示す。
【図34B】
該gag−IApol融合フレームのタンパク質配列(配列番号39)を示す。
(発明の分野)
本発明は、他の複製欠損型ベクターと比較して増強した増殖特性およびより大きな細胞性免疫を示すことが見出された組換え複製欠損型第1世代アデノウイルスワクチンに関する。本発明はまた、追加的な5’アデノウイルス配列の取り込みにより、本明細書に記載の組換え複製欠損型アデノウイルスの大規模生産効率を増加させる本明細書に記載の関連第1世代アデノウイルスベクターに関する。本発明のもう1つの態様は、ヒトサイトメガロウイルス(hCMV)プロモーターのイントロンA部分がアデノウイルスベクター構築物における不安定性の領域を構成するという驚くべき知見にある。アデノウイルス発現構築物からこの領域を除去すると、著しく改善されたベクター安定性が得られる。したがって、イントロンA欠失CMVプロモーターの制御下でトランスジーンを発現する改良されたベクターは、本発明のもう1つの態様を構成する。これらのアデノウイルスベクターは、ヒト免疫不全ウイルス(HIV)に対する組換えアデノウイルスワクチンの製造に有用である。特に、本明細書に記載の第1世代アデノウイルスベクターは、HIV−1 Gag、HIV−1 Polおよび/またはHIV−1 Nefポリヌクレオチド医薬品ならびにそれらの生物学的に活性な修飾体を含有する、アデノウイルスに基づくHIV−1ワクチンを構築し生成するために使用される。本明細書に記載の組換え複製欠損型アデノウイルスワクチンの宿主投与は、HIV−Gag、HIV−1−Polおよび/またはNefタンパク質またはそれらの免疫学的に関連した修飾体の発現をもたらして、HIV−1を特異的に認識する細胞性免疫応答を誘導する。例示されている本発明のポリヌクレオチドは、コドン最適化HIV−1 Gag、HIV−1 Pol、最適化HIV−1 Polの誘導体(HIV−1 Polのプロテアーゼ、逆転写酵素、RNアーゼHおよびインテグラーゼ活性が不活性化された構築物を含む)、HIV−1 Nef、および最適化HIV−1 Nefの誘導体(宿主CD4のダウンレギュレーションおよびミリスチル化のようなNefの野生型特性に影響を及ぼすnef突然変異体を含む)をコードする合成DNA分子である。本発明のHIVアデノウイルスワクチンは、単独で或いは組合せ様式および/または初回抗原刺激/追加抗原刺激法で投与されると、これまでに未感染の個体に予防上の利点をもたらし、および/または、感染個体内のウイルス負荷レベルを減少させてHIV−1感染の無症候期を延長させることにより治療効果をもたらすであろう。
【0002】
(発明の背景)
ヒト免疫不全ウイルス−1(HIV−1)は後天性ヒト免疫不全症候群(エイズ)および関連障害の病原体である。HIV−1はレトロウイルス科のRNAウイルスであり、すべてのレトロウイルスの5’−LTR−gag−pol−env−LTR3’体制を示す。プロウイルスとして公知の組込み形態のHIV−1は約9.8Kb長である。該ウイルスゲノムの各末端は、長末端反復配列(LTR)として公知のフランキング配列を含有する。該HIV遺伝子は少なくとも9個のタンパク質をコードし、それらは主要構造タンパク質(Gag、PolおよびEnv)、調節タンパク質(TatおよびRev)および補助タンパク質(Vpu、Vpr、VifおよびNef)の3つのクラスに分けられる。
【0003】
gag遺伝子は55キロダルトン(kDa)の前駆体タンパク質(p55)をコードしており、これは、スプライスされていないウイルスmRNAから発現され、HIVプロテアーゼ(pol遺伝子の産物)によるタンパク質分解によりプロセシングされる。成熟p55タンパク質産物はp17(マトリックス)、p24(カプシド)、p9(ヌクレオカプシド)およびp6である。
【0004】
pol遺伝子は、ウイルスの複製に必要なタンパク質、すなわち、逆転写酵素、プロテアーゼ、インテグラーゼおよびRNアーゼHをコードしている。これらのウイルスタンパク質は、リボソームフレームシフトにより産生される160kDaの前駆体タンパク質であるGag−Pol融合タンパク質として発現される。該ウイルスコード化プロテアーゼは、タンパク質分解により該Gag−Pol融合体からPolポリペプチドを切り離し、さらに該Polポリペプチドを成熟タンパク質にまで切断し、該成熟タンパク質はプロテアーゼ(Pro、P10)、逆転写酵素(RT、P50)、インテグラーゼ(IN、p31)およびRNアーゼH(RNアーゼ、p15)活性をもたらす。
【0005】
nef遺伝子は、CD4発現のダウンレギュレーション、T細胞活性化の阻害およびHIV感染性の促進のような幾つかの活性を有することが示されている初期補助(アクセサリー)HIVタンパク質(Nef)をコードする。
【0006】
env遺伝子は、ウイルスエンベロープ糖タンパク質をコードしており、該糖タンパク質は、160キロダルトン(kDa)の前駆体(gp160)として翻訳され、ついで細胞プロテアーゼにより切断されて、外部120kDaエンベロープ糖タンパク質(gp120)および膜貫通41kDaエンベロープ糖タンパク質(gp41)を与える。Gp120およびgp41は会合したままであり、ウイルス粒子上およびHIV感染細胞表面上に提示される。
【0007】
tat遺伝子は、HIV−1の複製に必須の転写トランスアクチベーターであるRNA結合タンパク質であるTatタンパク質の長い形態および短い形態をコードしている。
【0008】
rev遺伝子は、RNA結合タンパク質である13kDaのRevタンパク質をコードしている。Revタンパク質は、Rev応答配列(RRE)と称されるウイルスRNAの領域に結合する。Revタンパク質は、核から細胞質への、スプライスされていないウイルスRNAの輸送を促進する。Revタンパク質は、HIV後期遺伝子発現に要求され、そしてHIVの複製に要求される。
【0009】
Gp120は、他の補助受容体(コレセプター)分子に加えて、ヘルパーTリンパ球、マクロファージおよび他の標的細胞の表面上に存在するCD4/ケモカイン受容体に結合する。X4(マクロファージ指向性)ウイルスはCD4/CXCR4複合体に対する指向性を示し、R5(T細胞系指向性)ウイルスはCD4/CCR5受容体複合体と相互作用する。gp120がCD4に結合した後、gp41が、ウイルスの侵入をもたらす融合事象を媒介する。該ウイルスは該標的細胞と融合しそれに侵入し、ついでRNA依存性DNAポリメラーゼにより、その一本鎖RNAゲノムから二本鎖DNAへの逆転写が生じる。プロウイルスとして公知の該ウイルスDNAは細胞核に侵入し、そこで、該ウイルスDNAは、該核内での新たなウイルスRNAの産生、初期および後期HIVウイルスタンパク質の発現、ならびにそれに続く新たなウイルス粒子の産生および細胞放出を指令する。該一次感染は、該ウイルスの非常に高い産生および組織分布ならびにそれに続くウイルスの定常状態レベル(ただし、これはこの相における連続的なウイルス産生および代謝回転によるものである)を与え、最終的には、臨床的エイズ発症を招く更なるウイルス負荷の放出をもたらすことが、宿主内のウイルス負荷の検出能における最近の進歩から示されている。生産的感染細胞は数日間の半減期を有し、一方、慢性または潜伏感染細胞は3週間の半減期を有し、それに続く非生産的感染細胞は、長い半減期(100日以上)を有するが、疾患経過の全体にわたり認められる日々のウイルス負荷に有意には寄与しない。
【0010】
免疫防御に決定的に重要なCD4ヘルパーTリンパ球の破壊が、HIV感染の特徴である進行性免疫機能不全の主要原因である。CD4 T細胞の喪失は、ほとんどの侵入体と戦う身体能力を著しく損なうが、それは、ウイルス、真菌、寄生生物および或る細菌(マイコバクテリアを含む)に対する防御に対して特に深刻な影響を及ぼす。
【0011】
最近、HIV−1感染個体に対する有効な治療法が利用可能となった。しかし、これらの薬物は、世界の多数の地域においては該疾患に対して有意な影響を及ぼさず、それらは、ヒト集団内の感染の広がりの阻止において最低限の影響しか及ぼさないであろう。多数の他の感染症の場合と同様、HIV−1感染の広がりに対する有意な疫学的効果は、有効なワクチンの開発および導入の後でしか見出されないであろう。現在までにワクチン開発が成功していないのには、多数の要因が関与している。前記のとおり、慢性感染者においては、抗HIV−1体液性および細胞性免疫応答の存在ならびにウイルス感染細胞の破壊にもかかわらず、絶え間ないウイルス産生が生じている。他の感染症の場合と同様に、疾患の結末は、該免疫応答の速度論および規模と、該病原体複製速度および該免疫応答による影響の受け易さとの平衡がどのようになるかによって決まる。急性感染における既存免疫は、確立された感染における生成免疫応答より有効かもしれない。第2の要因は、該ウイルスの際立った遺伝的可変性である。細胞培養内ではHIV−1感染性を中和しうる抗HIV−1抗体が存在するが、これらの抗体は、一般には、それらの活性において、ウイルス分離体に特異的である。伝統的な方法を用いてHIV−1の血清学的分類を定義することは不可能であることが判明している。むしろ、該ウイルスは血清学的「連続体(continuum)」を特徴づけるようであり、そのため、個々の中和抗体応答は、せいぜい、少数のウイルス変異体に対して有効であるに過ぎない。この後者の見解を考慮すると、抗HIV−1細胞性免疫応答を惹起しうる免疫原および関連送達技術を同定することが有用であろう。CTL応答を得るためには、抗原が細胞内で合成されるか又は細胞内に導入され、ついでプロテアソーム複合体により、小さなペプチドにプロセシングされ、主要組織適合遺伝子複合体(MHC)クラスIタンパク質との最終的会合のために小胞体/ゴルジ複合体分泌経路に輸送されなければならないことが公知である。CD8+ Tリンパ球は、T細胞受容体(TCR)およびCD8細胞表面タンパク質を介して、MHCクラスIと会合した抗原を認識する。活性化エフェクターまたは記憶細胞へのナイーブCD8+ T細胞の活性化は、一般には、前記のとおりの抗原のTCR関与および共刺激タンパク質の関与の両方を要する。CTL応答の最適な誘導は、通常、TCRおよびCD4の関与を介してMHCクラスII分子と会合した抗原を認識するCD4+ Tリンパ球からのサイトカインの形態での「援助」を要する。
【0012】
欧州特許出願第0 638 316号(1995年2月15日付け公開)および第0 586 076号(1994年3月9日付け公開)(共にAmerican Home Products Corporationに譲渡されている)は、envまたはgagを含むHIV遺伝子を保持する複製型アデノウイルスベクターを記載している。チンパンジーおよびイヌでの種々の治療計画が用いられ、それらのいくつかはブースターアデノウイルスまたはタンパク質+ミョウバンでの治療を含むものであった。
【0013】
E1領域内に欠失を保持する複製欠損型アデノウイルスベクターが公知であり、最近のアデノウイルスベクターは、これらのベクター内に公知パッケージング反復配列を組込んでいる。例えば、とりわけ、塩基対459〜3328のE1配列において欠失したアデノウイルスベクターを開示しているEP 0707071、および、とりわけ、塩基対459〜3510が欠失したアデノウイルスベクターを開示している米国特許第6,033,908号を参照されたい。アデノウイルスのパッケージング効率は、組込まれる個々のA(パッケージング)反復配列の数に左右されると教示されている。例えば、GrableおよびHearing,1990 J.Virol.64(5):2047−2056;GrableおよびHearing,1992 J.Virol.66(2):723−731を参照されたい。
【0014】
Larderら(1987,Nature 327:716−717)およびLarderら(1989,Proc.Natl.Acad.Sci.86:4803−4807)は、HIV−1 RTの部位特異的突然変異誘発を開示しており、また、そのような変化が、RTの公知インヒビターとの相互作用に関連したインビトロ活性および感染性に及ぼす影響を開示している。
【0015】
Daviesら(1991,Science 252:,88−95)は、HIV−1 PolのRNアーゼHドメインの結晶構造を開示している。
【0016】
Schatzら(1989,FEBS Lett.257−311−314)は、完全なHIV−1 RT/RNアーゼH DNA断片内の突然変異Glu478GlnおよびHis539Pheが、RT活性に影響を及ぼすことなく欠損RNアーゼ活性を与えることを開示している。
【0017】
Mizrahiら(1990,Nucl.Acids.Res.18:pp.5359−5353)は、同様に欠損RNアーゼ活性を与えるpol遺伝子のRNアーゼ領域内の追加的突然変異Asp443AsnおよびAsp498Asnを開示している。該著者は、Asp498Asn突然変異体の特徴づけが、この突然変異体タンパク質の不安定性のために困難であったことを記載している。
【0018】
Leavittら(1993,J.Biol.Chem.268:2113−2119)は、HIV−1インテグラーゼ(IN)活性に対して異なる効果を示すAsp64Val突然変異を含む幾つかの突然変異を開示している。
【0019】
Wiskerchenら(1995,J.Virol.69:376−386)は、HIV−1 INおよびウイルス複製機能に影響を及ぼすアスパラギン酸残基の突然変異を含む一重および二重突然変異体を開示している。
【0020】
HIV感染に対して強力な細胞性免疫応答を産生する予防および/または治療に基づくHIVワクチンを得ることが、エイズとの闘いにおいて非常に重要であろう。本発明は、宿主に投与されるとHIV−1遺伝子gag、polおよびnefのコドン最適化および修飾形態を発現するアデノウイルスワクチンのクラスを開示することにより、この要求を検討し、それを満足させるものである。これらの複製欠損型組換えアデノウイルスワクチンは、単独で、あるいは本発明の成分および/または別個のウイルスHIV DNAワクチン、非ウイルス性HIV DNAワクチン、HIVサブユニットワクチン、HIV全不活化ワクチンおよび/または生弱毒化HIVワクチンでの初回抗原刺激ワクチン接種法および/または組合せ様式の一部として、ヒトのような宿主に投与することができる。
【0021】
(発明の概要)
本発明は、HIV−1 Gag、HIV−1 PolおよびHIV−1 Nefの免疫学的に関連した修飾体を含む種々の形態のHIV−1 Gag、HIV−1 Polおよび/またはHIV−1 Nefをコードする増強された複製欠損型組換えアデノウイルスワクチンベクターおよび関連組換え複製欠損型アデノウイルスワクチンに関する。本発明のアデノウイルスワクチンはHIV抗原を発現し、宿主投与に際して、改善された細胞性免疫応答をもたらす。潜在的なワクチン被接種体には、霊長類、特にヒトおよび非ヒト霊長類が含まれ、また、商業的または家畜獣医学的に重要な任意の非ヒト哺乳動物が含まれるが、これらに限定されるものではない。本発明の改良された組換えアデノウイルスに基づくワクチンの効果は、これまでに未感染の個体に対する、より低い伝染率(すなわち、予防的用途)、および/または、感染個体内のウイルス負荷のレベルの減少によるHIV−1感染の無症候期の延長(すなわち、治療的用途)でなければならない。特に、本発明は、種々の形態のコドン最適化HIV−1 Gag(p55形態のコドン最適化完全長(FL)GagおよびtPA−Gag融合タンパク質を含むが、何らこれらに限定されるものではない)、HIV−1 Pol、HIV−1 Nefおよび免疫学的に関連のある選択された修飾体をコードする、アデノウイルスに基づくワクチンに関する。これらのアデノウイルスワクチンの投与、細胞内運搬および発現は、宿主CTLおよびTh応答を惹起する。好ましい複製欠損型組換えアデノウイルスワクチンベクターには、(1)コドン最適化形態の野生型HIV−1 Gagをコードする、(2)コドン最適化形態のHIV−1 Polをコードする、(3)コドン最適化形態のHIV−1 Pol融合タンパク質をコードする、(4)コドン最適化形態の修飾HIV−1 Polタンパク質および融合タンパク質、例えば宿主細胞内でRT、RNアーゼおよびIN活性をもたらす触媒領域内の残基を含むpol修飾体(これらに限定されるものではない)をコードする、(5)コドン最適化形態の野生型HIV−1 Nefをコードする、(6)コドン最適化形態のHIV−1 Nef融合タンパク質をコードする、および/または(7)コドン最適化形態のHIV−1 Nef誘導体、例えばアミノ末端リーダー配列の導入、アミノ末端ミリスチル化部位の除去および/またはジロイシンモチーフ突然変異の導入を含むnef修飾体(これらに限定されるものではない)をコードする合成DNA分子が含まれるが、これらに限定されるものではない。アデノウイルスに基づくベクターワクチンから発現される本明細書に開示するNefに基づく融合体および修飾タンパク質は、宿主MHC I複合体に適切に提示されて宿主CTLおよびTh応答を惹起する能力を保有する一方、改変された輸送(トラフィッキング)および/または宿主細胞機能を有しうる。HIV−1 Gag、Polおよび/またはNef融合タンパク質の具体例には、該ウイルス抗原コード領域のNH2末端部分でのリーダーまたはシグナルペプチドの融合体が含まれるが、これらに限定されるものではない。そのようなリーダーペプチドには、tPAリーダーペプチドが含まれるが、これに限定されるものではない。
【0022】
本発明のHIV−1 Gag、HIV−1 Polおよび/またはHIV−1 Nefに基づくワクチンの構築において使用するアデノウイルスベクターは、該組換えウイルスの大規模な製造および精製にわたる該組換えアデノウイルスゲノムの遺伝的安定性の増強をもたらす任意の複製欠損型アデノウイルスベクターを含みうる。すなわち、本発明のHIV−1 Gag、PolまたはNefに基づくアデノウイルスワクチンは、細胞培養における多数の継代にわたり遺伝的に安定であり従って大規模な製造および精製操作中に維持されることが示される精製された組換え複製欠損型アデノウイルスである。そのような組換えアデノウイルスベクターおよび回収されたアデノウイルスワクチンは、有効な単価または多価HIVワクチンに要求される大規模用量の充填およびそれに続く世界的な流通に有用である。本発明は、この基本要件を満足し、野生型アデノウイルスゲノムの塩基対約1から塩基対約342から(より好ましくは400から)塩基対約458までの間の野生型アデノウイルス・シス作用性パッケージング領域を含みE1において少なくとも部分的に欠失した複製欠損型アデノウイルスベクターおよびそれに由来するベクターとして記載される。本発明の好ましい実施形態は、野生型アデノウイルスの塩基対1〜450を含む。他の好ましい実施形態においては、該複製欠損型アデノウイルスベクターは、それに加えて、塩基対3511〜3523を含むE1欠失領域の3’側の領域を有する。塩基対342〜450(より詳しくは400〜450)は、ウイルス抗原、特にHIV抗原を保持する既に開示されているベクターの5’領域の伸長を構成する(例えば、1999年7月6日および1999年8月13日付け出願のそれぞれ米国仮特許出願第60/142,631号および第60/148,981号に基づき優先権を主張している2001年1月11日付け公開のPCT国際出願PCT/US00/18332(WO 01/02067)を参照されたい;これらの文書を参照により本明細書に組み入れることとする)。本出願人は、該5’領域を該開示ワクチン内のE1遺伝子内に更に伸長させると該ウイルスのパッケージングの最適化に重要であることが見い出された要素が組込まれること、を見出した。
【0023】
野生型アデノウイルスゲノムの塩基対約1からと塩基対約342(より好ましくは400)から塩基対約458との間までを含まない従来のベクターと比較して、前記領域を含むベクターは、より強力なウイルス効果である約5〜10倍大きな増幅速度を伴う増強された増殖特性を示して、より低いウイルス使用量で同等の免疫生成を可能にし、この領域(塩基対1〜450)を含まない複製欠損型ベクターより大きな細胞性免疫応答を可能にする。より一層重要なことは、それに由来するアデノウイルス構築物、特に、イントロンAを欠くhCMVプロモーターの制御下で発現カセットを含むものは、大規模生産において遺伝的に非常に安定である。本出願人は、驚くべきことに、hCMVプロモーターのイントロンA部分が、アデノウイルスベクター中で使用された場合に不安定性の領域を構成することを見出しており、前記の安定性はこれに基づくものである。したがって、本出願人は、遺伝子治療における使用に特に適した増強されたアデノウイルスベクター、および好ましいことに大規模増殖に有用な、ヌクレオチドに基づくワクチンベクターを特定した。
【0024】
本発明の好ましい実施形態は、(a)タンパク質または生物学的に活性な及び/又はその免疫学的に関連した部分をコードする核酸と、(b)a)部の核酸に作動的に結合した異種プロモーターと、(c)転写ターミネーターと、を含む遺伝子発現カセットの形態で該遺伝子が挿入されている、前記のとおりの複製欠損型アデノウイルスベクターである。
【0025】
好ましい実施形態においては、塩基対1〜450内に含有されているもの以外または塩基対1〜450および3511〜3523内に含有されているもの以外のE1遺伝子が該アデノウイルスベクターから欠失しており、該欠失E1遺伝子は該遺伝子発現カセットで置換されている。他の好ましい実施形態においては、該複製欠損アデノウイルスゲノムは機能的E3遺伝子を有さず、あるいは該E3遺伝子が欠失している。最も好ましくは、該E3領域は該アデノウイルスゲノム内に存在する。さらに好ましい実施形態においては、該遺伝子発現カセットは、E1に逆平行(該ベクターバックボーンに対して3’から5’への方向で転写される)な配向、より好ましくは、E1に平行(該ベクターバックボーンに対して5’から3’への方向で転写される)な配向で存在する。
【0026】
さらなる実施形態は、アデノウイルス部分とプラスミド部分とを含むシャトルプラスミドベクターに関する。ここで、該アデノウイルス部分は、a)野生型アデノウイルスゲノムの塩基対約1からと塩基対約342(より好ましくは400)から塩基対約458までの間との野生型アデノウイルス・シス作用性パッケージング領域(好ましくは1〜450)および好ましくはそれに加えて野生型アデノウイルス配列の塩基対3511〜3523を含みE1において少なくとも部分的に欠失した複製欠損型アデノウイルスゲノム、ならびにb)(a)タンパク質またはその生物学的に活性な及び/又は免疫学的に関連した部分をコードする核酸と、(b)a)部の核酸に作動的に結合した異種プロモーターと、(c)転写終結および/またはポリアデニル化部位と、を含む遺伝子発現カセットを含む。
【0027】
本発明の他の態様は、該アデノウイルスベクターおよび/または該シャトルプラスミドベクターを含む宿主細胞;該ベクターを含むワクチン組成物;および(a)アデノウイルスE1タンパク質を発現する宿主細胞内に該アデノウイルスベクターを導入し、(b)得られたアデノウイルスベクターを回収することを含む該ベクターの製造方法を含む。
【0028】
この目的において、本発明は特に、本明細書に記載のHIV−1抗原を含む(これらに限定されるものではない)付随トランスジーを伴う又は伴わないMRKAd5ベクターバックボーンのいずれかに関連した回収されたウイルスを含む(これらに限定されるものではない)、293細胞またはPER.C6(登録商標)(これらに限定されるものではない)のような宿主細胞に由来する回収された組換え複製欠損型ウイルスに関する。HIV−1ワクチンは、本明細書に開示するHIV−1抗原の任意の1以上を発現する任意の回収された組換えアデノウイルス物質により表される。ついで、この回収された物質は、宿主投与の前に精製され製剤化され保存されうる。
【0029】
本発明のもう1つの態様は、タンパク質に対する細胞性免疫応答を個体において生成させる方法であって、
a)塩基対約1からと塩基対約342(より好ましくは400)から塩基対約458まで(好ましくは1〜450)の間との野生型アデノウイルス・シス作用性アデノウイルスパッケージング領域および好ましくはそれに加えて野生型アデノウイルス配列の塩基対3511〜3523を含む、E1において少なくとも部分的に欠失した組換え複製欠損型アデノウイルベクター、ならびに
b)(i)タンパク質またはその生物学的に活性な及び/又は免疫学的に関連した部分をコードする核酸と、(ii)a)部の核酸に作動的に結合した異種プロモーターと、(iii)転写ターミネーターおよび/またはポリアデニル化部位とを含む遺伝子発現カセットを含むアデノウイルスワクチンベクターを該個体に投与することを含んでなる方法である。
【0030】
本明細書に記載のアデノウイルスおよび/またはDNAプラスミドワクチンの有効な性質を考慮して、本発明は、有効な免疫予防を得るための、またはHIV−1ウイルスに対する曝露後のHIV−1感染の確立を妨げるため、または急性HIV−1感染を軽減して、有益な長期的結果を伴う、より低いウイルス負荷の確立をもたらすためのHIV感染後の治療用ワクチンとしての、これらのアデノウイルスおよび/またはDNAプラスミドワクチンの1以上の投与に関するすべての方法に関する。本明細書に記載されているとおり、そのような治療計画は、単価または多価組成物、種々の組合せ形態の適用、および/または初回/追加抗原刺激法(それにより、生きた脊椎動物組織内への接種後の抗原発現および付随的な細胞性および/または体液性免疫応答が最適化される)を含みうる。したがって、本発明は、哺乳類組織内に導入されるとgag、polおよび/またはnefに基づくベクターの細胞内発現を誘発する、本明細書に開示する種々のパラメーターおよび当技術分野で公知の追加的パラメーターの範囲内の本明細書に開示するアデノウイルスおよび/またはDNAプラスミドワクチンの使用方法を提供する。
【0031】
この目的において、本発明は、1つには、ワクチン被接種体、好ましくはヒトのワクチン被接種者において細胞性免疫応答を生成させる方法に関する。この場合、該個体には、アデノウイルスワクチンベクターの2以上の投与が行われ、それは、プラスミドワクチンの投与を伴う計画において投与されうる。該プラスミドワクチン(本明細書中では「DNAプラスミドワクチン」または「ワクチンプラスミド」とも称される)は、タンパク質またはその免疫学的に関連した部分をコードする核酸、該核酸配列に作動的に結合した異種プロモーター、および転写ターミネーターまたはポリアデニル化シグナル(例えば、それぞれbGHまたはSPA)を含む。それらの投与を隔てる所定の最低限度の長さの時間を設けることが可能である。該個体に、第1用量のプラスミドワクチン、ついで第2用量のプラスミドワクチンを投与することができる。あるいは、該個体に、第1用量のアデノウイルスワクチン、ついで第2用量のアデノウイルスワクチンを投与することができる。他の実施形態においては、まず、該プラスミドワクチンを投与し、ついで幾らかの時間の後、該アデノウイルスワクチンを投与する。逆に、まず、該アデノウイルスワクチンを投与し、ついで幾らかの時間の後、プラスミドワクチンを投与することができる。これらの実施形態においては、個体に、ウイルスベクターまたはプラスミド形態として複数用量の同じアデノウイルス血清型を投与することが可能であり、代替的態様では、該ウイルスは、異なる血清型のものでありうる。該代替的態様においては、まず、関心のあるウイルス抗原を、アデノウイルスに基づくワクチン以外のウイルスワクチンを介して運搬し、ついで、開示されているアデノウイルスワクチンで行うことができる。代替的ウイルスワクチンには、ポックスウイルスおよびベネズエラウマ脳炎ウイルスが含まれるが、これらに限定されるものではない。
【0032】
本発明はまた、本明細書に記載のGag、PolおよびNef成分を含む多価アデノウイルスワクチン組成物に関する。例えば、実施例29および表25を参照されたい。そのような組成物は、ヒトMHCの及び循環ウイルスの遺伝的多様性を仮定した場合には特に、宿主への投与の後、増強した細胞性免疫応答をもたらすであろう。具体例には、二価(すなわち、gagおよびnef、gagおよびpol、またはpolおよびnef成分)または三価ワクチン(すなわち、gag、polおよびnef成分)組成物を与えるMRKAd5ベクターに基づく多価ワクチン組成物が含まれるが、これらに限定されるものではない。そのような多価ワクチンは1回量用に充填されたり、各個に充填された成分の複数の接種よりなることが可能であり、また、前段落に記載のウイルス性または非ウイルス性ベクターワクチンでの初回/追加抗原刺激法の一部でありうる。この目的のためには、好ましい組成物は、複数の異なるHIV抗原クラスと組合せて使用されるMRKAd5アデノウイルスである。各HIV抗原クラスは配列操作に付され、それにより多数の潜在的なワクチンの組合せを与え、そのような組合せは本発明の範囲内である。そのような組合せ様式のワクチン製剤および投与の利用は、単一様式の方法での接種より一層強力な細胞性免疫応答を惹起する可能性を増大させる。
【0033】
本明細書に開示する「組合せ様式」の概念は、複数のオープンリーディングフレームを含む前アデノウイルスプラスミドの生成のために適当なシャトルプラスミドに複数のHIV−1ウイルス抗原が連結されうる代替的投与様式をも含む。例えば、三価ベクターは、E3(−)またはE3(+)バックグラウンド、好ましくはE3欠失バックボーンにおけるgag−pol−nef融合体、あるいは、おそらくは、「2+1」二価ワクチン、例えば、同じMRKAd5バックボーン内のgag−pol融合体(すなわち、コドン最適化p55gagおよび不活性化最適化pol;実施例29および表25)(各オープンリーディングフレームは別個のプロモーターおよび転写終結配列に作動的に結合している)を含みうる。あるいは、それらの2つのオープンリーディングフレームは単一のプロモーターに作動的に結合しており、該オープンリーディングフレームはIRES(internal ribosome entry sequence)に作動的に連結していることが可能である。したがって、単一の又は恐らくは第2の回収された組換え複製欠損型アデノウイルスとして運搬される多価ワクチンは、本発明の一部と意図される。
【0034】
したがって、本発明のアデノウイルスワクチンおよびプラスミドDNAワクチンは単独で投与することが可能であり、あるいは初回および追加抗原刺激投与法の一部でありうる。混合様式の初回および追加接種法は、既存の抗ベクター免疫応答が存在する場合には特に、増強された免疫応答をもたらすであろう。本発明のこの1つの態様は、該プラスミドワクチンを少なくとも1回投与することにより該プラスミドワクチンで対象を初回抗原刺激し、所定の長さの時間を経過させ、ついで該アデノウイルスワクチンを投与することにより追加抗原刺激する方法である。通常、複数の初回抗原刺激、典型的には1〜4回の初回抗原刺激を行うが、より多数の初回抗原刺激を行うことが可能である。初回抗原刺激と追加抗原刺激との間の時間の長さは、典型的には約4ヶ月〜1年と様々となりうるが、他の時間枠を用いることも可能である。アカゲザルでの実験においては、該動物をプラスミドワクチンで4回初回抗原刺激し、ついで4ヶ月後に該アデノウイルスワクチンで追加抗原刺激した。それらの細胞性免疫応答は、アデノウイルスワクチンのみが投与された動物の場合より顕著に高かった。初回抗原刺激法の利用は、既存の抗アデノウイルス免疫応答を対象者が有する場合には特に好ましいかもしれない。
【0035】
増強された複製欠損型組換えアデノウイルスワクチンベクターバックボーンを提供することが、本発明の目的の1つである。これらの組換えアデノウイルスバックボーンは1以上のトランスジーンを受容することが可能であり、それは、増殖、増幅および回収のために細胞培養を介して継代されうる。
【0036】
種々のトランスジーンをコードする増強された複製欠損型組換えアデノウイルスワクチンベクターを提供することが、もう1つの目的である。
【0037】
細胞培養における連続継代後のウイルス安定性の増加と共に増殖および増幅速度の増加を示す回収された組換え複製欠損型アデノウイルスを提供することも、本発明の目的の1つである。そのような組換えアデノウイルスは、遺伝子治療における及びヌクレオチドに基づくワクチンベクターにおける使用に特に適しており、有利なことに、大規模増殖に有用である。
【0038】
この目的において、(1)種々の形態のHIV−1 Gag、HIV−1 Polおよび/またはHIV−1 Nef(HIV−1 Gag、HIV−1 PolおよびHIV−1 Nefの免疫学的に関連した修飾体を含む)をコードする本明細書に記載の増強された複製欠損型組換えアデノウイルスワクチンベクター、ならびに(2)細胞培養による1回または複数回の継代(293細胞またはPER.C6(登録商標)細胞による継代を含むが、これらに限定されるものではない)を介する(1)のアデノウイルスベクターの継代により産生された回収され精製された組換え複製欠損型アデノウイルスを提供することが、本発明の目的の1つである。
【0039】
細胞培養による1回または複数回の継代により回収された組換えアデノウイルスを提供することも、本発明の目的の1つである。組換えアデノウイルスワクチンベクターの場合と同様に、この組換えウイルスを、後続の宿主投与のために回収し製剤化する。
【0040】
また、本発明の目的の1つは、(a)タンパク質またはその生物学的に活性な及び/又は免疫学的に関連した部分をコードする核酸と、(b)a)部の核酸に作動的に結合した異種プロモーターと、(c)転写ターミネーターと、を含む遺伝子発現カセットの形態で少なくとも1つの遺伝子が挿入された複製欠損型アデノウイルスベクターを提供することである。
【0041】
また、本発明の目的の1つは、該アデノウイルスベクターおよび/または該シャトルプラスミドベクターを含む宿主細胞;該ベクターを含むワクチン組成物;および(a)アデノウイルスE1タンパク質を発現する宿主細胞内に該アデノウイルスベクターを導入し、(b)得られたアデノウイルスベクターを回収することを含む該ベクターの製造方法提供することである。本発明のもう1つの目的は、タンパク質に対する細胞性免疫応答を個体において生成させる方法であって、a)塩基対約1からと塩基対約342(より好ましくは400)から約450まで(好ましくは1〜450)の間との野生型アデノウイルス・シス作用性アデノウイルスパッケージング領域および好ましくは野生型アデノウイルス配列の3511〜3523を含みE1において少なくとも部分的に欠失した複製欠損型アデノウイルベクター、ならびにb)(i)タンパク質またはその生物学的に活性な及び/又は免疫学的に関連した部分をコードする核酸と、(ii)a)部の核酸に作動的に結合した異種プロモーターと、(iii)転写ターミネーターおよび/またはポリアデニル化部位と、を含む遺伝子発現カセットを含むアデノウイルスワクチンベクターを該個体に投与することを含んでなる方法を提供することである。
【0042】
ワクチン投与法、すなわち、未感染個体に対する有効的な免疫予防またはHIV感染患者に対する治療的処置をもたらす本明細書に記載の1以上のアデノウイルスおよび/またはDNAプラスミドワクチンの投与のための種々の代替手段を提供することが、本発明の目的の1つである。そのような方法は、多価HIV−1ワクチン組成物、種々の組合せ様式の方法および種々の初回/追加抗原刺激代替手段を含むが、これらに限定されるものではない。ワクチン組成物および/または計画された投与に関するこれらの投与方法は、単一様式の方法での接種より一層強力な細胞性免疫応答を惹起する可能性を増大させるであろう。
【0043】
本明細書および特許請求の範囲の全体にわたり、以下の定義および略語を用いる。
【0044】
「HAART」は、高度に活性な抗レトロウイルス療法を意味する。
【0045】
「第1世代」ベクターは、複製欠損型であるとして特徴づけられる。それは、典型的には、欠失した又は不活性化されたE1遺伝子領域を有し、好ましくは、欠失した又は不活性化されたE3遺伝子領域をも有する。
【0046】
「AEX」は陰イオン交換クロマトグラフィーを意味する。
【0047】
「QPA」はQuick PCR−based Potency Assayを意味する。
【0048】
「bp」は塩基対を意味する。
【0049】
「s」または「str」は、該トランスジーンが「E1平行」または「直線的」配向で位置することを示す。
【0050】
「PBMC」は末梢血単球細胞を意味する。
【0051】
「FL」は完全長を意味する。
【0052】
「FLgag」は、図2に示すとおりの完全長最適化gag遺伝子を意味する。
【0053】
「Ad5−Flgag」は、CMVプロモーターの制御下で完全長最適化gag遺伝子を含む発現カセットを保持するアデノウイルス血清型5複製欠損型ウイルスを意味する。
【0054】
「プロモーター」は、RNAポリメラーゼが結合するDNA鎖上の認識部位を意味する。該プロモーターは、RNAポリメラーゼとの開始複合体を形成して、転写活性を開始させ駆動する。該複合体は、「エンハンサー」のような配列を活性化することにより、あるいは「サイレンサー」のような配列を抑制することにより修飾されうる。
【0055】
「リーダー」は、構造遺伝子の5’末端の、該遺伝子と共に転写されるDNA配列を意味する。これは、通常、プロ配列としばしば称されるN末端ペプチド伸長を有するタンパク質を与える。
【0056】
「イントロン」は、遺伝子産物中のアミノ酸をコードしていない遺伝子の中間に見出されるDNAの区分を意味する。該イントロンの前駆体RNAは切り出され、したがって、タンパク質に翻訳されないmRNAには転写されない。
【0057】
「免疫学的に関連」または「生物学的に活性」は、(1)ウイルスタンパク質に関しては、該タンパク質が、投与されると、該ウイルスの増殖および/または広がりを遅延させるのに及び/又は個体内に存在するウイルス負荷を減少させるのに十分な個体内における測定可能な免疫応答を惹起する能力を有すること、または(2)ヌクレオチド配列に関しては、該配列が、前記の能力を有するタンパク質をコードしうることを意味する。
【0058】
「カセット」なる語は、転写および翻訳制御配列を伴う、発現される核酸配列を意味する。該カセットを交換することにより、ベクターは異なる配列を発現しうる。
【0059】
「bGHpA」は、ウシ成長ホルモン転写ターミネーター/ポリアデニル化配列を意味する。
【0060】
「tPAgag」は、DNAに基づくワクチンベクターの場合かアデノウイルスに基づくワクチンベクターの場合かに無関係に、図30A〜Bに例示されている組織プラスミノーゲンアクチベーターリーダー配列のリーダー配列と最適化HIV gag遺伝子との融合体を意味する。
【0061】
「IA」または「inact」は、用いられている場合には、遺伝子の不活性化形態(例えば、IApol)を意味する。
【0062】
「MCS」は「マルチクローニング部位」である。
【0063】
一般に、アデノウイルス構築物、遺伝子構築物は、それらに含有される遺伝子を参照することにより命名される。
【0064】
例えば、「Ad5 HIV−1 gag」は、元のHIV−1 gagアデノウイルスベクターとも称され、hCMVイントロンAプロモーター、完全長形態のヒトコドン最適化HIV−1 gag遺伝子およびウシ成長ホルモンポリアデニル化シグナルから構成されるトランスジーンカセットを含有するベクターである。該トランスジーンは、E1およびE3欠失アデノベクター内にE1逆平行配向で挿入された。
【0065】
「MRK Ad5 HIV−1 gag」は、「MRKAd5gag」または「Ad5gag2」とも称され、塩基対1〜450および3511〜3523を含みヒトコドン最適化HIV−1遺伝子をイントロンA無しのCMVプロモーターの制御下でE1平行配向で有しE1が欠失した本明細書に教示するアデノウイルスベクターである。該構築物はまた、ウシ成長ホルモンポリアデニル化シグナルを含む。
【0066】
「pV1JnsHIVgag」は、「HIVFLgagPR9901」とも称され、CMV最初期(IE)プロモーターおよびイントロンA、完全長コドン最適化HIV gag遺伝子、ウシ成長ホルモン由来ポリアデニル化および転写終結配列ならびに最小pUCバックボーンを含むプラスミドである。
【0067】
「pV1JnsCMV(無イントロン)−FLgag−bGHpA」は、完全長HIV gag遺伝子を含みCMVのイントロンA部分が欠失したpV1JnsHIVgag由来のプラスミドである。このプラスミドは、「pV1JnsHIVgag−bGHpA」、「pV1Jns−hCMV−FL−gag−bGHpA」および「pV1JnsCMV(無イントロン)+FLgag+bGHpA」と称される。
【0068】
「pV1JnsCMV(無イントロン)−Flgag−SPA」は、bGHpA終結配列がSPA終結配列で置換されている以外はpV1JnsCMV(無イントロン)−FLgag−bGHpAと同じ組成のプラスミドである。このプラスミドは、「pV1Jns−HIVgag−SPA」および「pV1Jns−hCMV−FLgag−SPA」とも称される。
【0069】
「pdelE1sp1A」は、発現カセットを有さない(すなわち、プロモーターもポリAも有さない)普遍的シャトルベクターである。該ベクターは、bp1〜bp341およびbp3524〜bp5798の野生型アデノウイルス血清型5(Ad5)配列を含み、341bpで終結するAd5配列と3524bpで開始するAd5配列との間にマルチクローニング部位を有する。このプラスミドは、元のAd5シャトルベクターとも称される。「MRKpdelE1sp1A」または「MRKpdelE1(Pac/pIX/pack−450)」または「MRKpdelE1(Pac/pIX/pack450)Cla1」は、bp1〜bp450およびbp3511〜bp5798の野生型アデノウイルス血清型5(Ad5)配列を含み発現カセットを有さない(すなわち、プロモーターもポリAも有さない)普遍的シャトルベクターである。該ベクターは、450bpで終結するAd5配列と3511bpで開始するAd5配列との間にマルチクローニング部位を有する。このシャトルベクターを使用して、該CMVプロモーターおよび該bGHpA断片を直線的(「str」またはE1平行)配向または逆(oppまたはE1逆平行)配向の両方で挿入することができる。
【0070】
「MRKpdeE1(Pac/pIX/pack450)+CMVmin+BGHpA(str.)」は、該CMVプロモーター(イントロンA無し)および該bGHpA断片を含有する修飾されたベクターである更にもう1つのシャトルベクターである。唯一のBglII部位における選択された遺伝子の挿入が、MRKpAd5(E1−/E3+)Cla1前プラスミド内への挿入の場合に、該トランスジーンの転写の方向がAd5 E1に平行となることを保証するよう、該hCMVプロモーター(イントロンA無し)および該ウシ成長ホルモンポリアデニル化シグナルを含有する発現単位が該シャトルベクター内に挿入されている。このシャトルベクターは、図22および23に示すとおり、それぞれのIApolおよびG2A、LLAA nef遺伝子を直接挿入するするために使用された。
【0071】
「MRKpdelE1−CMV(無イントロン)−FLgag−bGHpA」は、イントロンAを伴わないヒトCMV、完全長ヒトコドン最適化HIV gag遺伝子およびウシ成長ホルモンポリアデニル化シグナルを含有する発現カセットと共に塩基対1〜450および3511〜5798のAd5配列を含むシャトルである。このプラスミドは、「MRKpdeE1シャトル+hCMV−FL−gag−BGHpA」とも称される。
【0072】
「MRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpA」は、E1領域を含むヌクレオチド以外のすべてのAd5配列(451〜3510)、イントロン無しのヒトCMVプロモーター、完全長ヒトコドン最適化HIV gag遺伝子およびウシ成長ホルモンポリアデニル化シグナルを含むアデノウイルスベクターである。このベクターは、「MRKpAdHVE3+hCMV−FL−gag−BGHpA」、「MRKpAd5HIV−1gag」、「MRKpAd5gag」、「pMRKAd5gag」または「pAd5gag2」とも称される。
【0073】
「pV1Jns−HIV−pol inact(opt)」または「pV1Jns−HIV IA pol(opt)」は、V1JnsのBglII部位内にクローニングされた不活性化Pol遺伝子(配列番号3中に含有されている)である。本明細書に記載のとおり、HIV−1 polの種々の誘導体を、V1JnsまたはV1Jns−tPAのようなプラスミド発現ベクター内にクローニングして、直接的にDNAワクチン候補として又は適当なアデノウイルスベクター内へのサブクローニングのための起源として使用することができる。
【0074】
「MRKpdel+hCMVmin+FL−pol+bGHpA(s)」は、適切な配向でIA pol遺伝子を含有する前記の「MRKpdelE1(Pac/pIX/pack450)+CMVmin+BGHpA(str.)」シャトルである。このシャトルベクターは、MRKpAd(E1−/E3+)Cla1との細菌組換えにおいて使用される。
【0075】
本発明で「pMRKAd5pol」とも称される「MRKpAd+hCMVmin+FL−pol+bGHpA(S)E3+」は、CMV−pol inact(opt)−pGHpA構築物を含む前アデノウイルスプラスミドである。この前アデノウイルスプラスミドの構築を図22に示す。
【0076】
「pV1Jns/nef(G2A,LLAA)」または「V1Jns/opt nef(G2A,LLAA)」は、アミノ末端ミリスチル化部位における修飾(Gly−2からAla−2)およびAla−174−Ala−175へのLeu−174−Leu−175ジロイシンモチーフの置換をオープンリーディングフレームがコードするコドン最適化HIV−1 Nefを含む(配列番号13;これは、ヌクレオチド12〜14の開始メチオニン残基およびヌクレオチド660〜662の「TAA」終結コドンを含む)。この断片をV1Jnsおよび/またはV1Jns−tPAのBglII部位内にサブクローニングする(図16A〜B)。HIV−1 polに関して前記したとおり、HIV−1 nef構築物は、V1JnsまたはV1Jns−tPAのようなプラスミド発現ベクター内にクローニングして、直接的にDNAワクチン候補として又は適当なアデノウイルスベクター内へのサブクローニングのための起源として使用することができる。
【0077】
本発明で「pMRKAd5nef」とも称される「MRKpdelE1hCMVminFL−nefBGHpA(s)」は、CMV−nef(G2A,LLAA)コドン最適化配列を含む前アデノウイルスプラスミドである。この前アデノウイルスプラスミドの構築を図23に示す。
【0078】
(図面の簡単な記載)
図1は、元のHIV−1 gagアデノベクター(Ad5HIV−1gag)を示す。このベクターは、1999年7月6日付け出願の米国仮特許出願第60/142,631号および1999年8月13日付けの米国特許出願第60/148,981号に基づいて優先権を主張している2000年7月3日付け出願のPCT国際出願番号PCT/US00/18332(WO01/02607)に開示されており、これらの3つの出願をすべて、参照により本明細書に組み入れることとする。
【0079】
図2は、該最適化ヒトHIV−1 gagオープンリーディングフレームの核酸配列(配列番号29)を示す。
【0080】
図3は、元のgagトランスジーンと比較して新規トランスジーン構築物を図示する。
【0081】
図4は、本発明の新規ベクターの作製において元のアデノベクターバックボーンに施された修飾を示す。
【0082】
図5は、該パッケージングシグナル領域に対して施された付加(実験#1)およびウイルス増殖に対するE3遺伝子(実験2)の効果を判定するために行ったウイルス混合実験を示す。棒線は、該E1欠失に対して施された修飾の領域を示す。
【0083】
図6は、実施例6および7に記載するウイルス混合実験後のウイルスDNA分析のオートラジオグラフを示す。
【0084】
図7A、7Bおよび7Cは以下のとおりである。図7Aは、MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたhCMV−Flgag−bGHpAアデノベクターを示す。E1平行およびE1逆平行のトランスジーン配向が示されている。図7Bは、MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたhCMV−Flgag−SPAアデノベクターを示す。この場合もまた、E1平行およびE1逆平行の両方のトランスジーン配向が示されている。図7Cは、MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたmCMV−Flgag−bGHpAアデノベクターを示す。この場合もまた、E1平行およびE1逆平行の両方のトランスジーン配向が示されている。
【0085】
図8Aは、トランスジーン配向の効果を試験するよう意図された実験を示す。図8Bは、ポリアデニル化シグナルの効果を試験するよう意図された実験を示す。
【0086】
図9は、BstE11消化後のP5における試験された4つのアデノウイルスベクターからのウイルスDNA(実施例12)を示す。
【0087】
図10は、MRKpAdHVE3、MRKAd5HIV−1gagおよびMRKAd5HIV−1gagE3−の継代11および12のウイルスDNA分析を示す。
【0088】
図11は、ウイルス競合研究を開始するために使用した継代6のMRKpAdHVE3およびMRKAd5HIV−1gagのウイルスDNA分析(HindIII消化)を示す。最後の2つのレーンは、該競合研究の二重継代の継代11分析である(各ウイルスは280ウイルス粒子のMOIである)。
【0089】
図12は、特定された時点で回収が行われた血清含有培地中のMRKAd5HIV−1gagに関する高継代数上のHindIII消化によるウイルスDNA分析を示す。第1レーンは1kb DNAサイズマーカーを示す。その他のレーンは、前プラスミド対照(Pac1およびHindIIIで消化されたもの)、P16、P19およびP21におけるMRKAd5HIV−1gagを示す。
【0090】
図13は、種々の用量のいくつかのAdgag構築物でbalb/cマウス(n=10)をi.m.免疫した3週間後の血清抗p24レベルを示す:(A)MRK Ad5 HIV−1 gag(継代5)、(B)MRKAd5hCMV−FLgag−bGHpA(E3−)、(C)MRKAd5 hCMV−FLgag−SPA(E3+)、(D)MRKAd5 mCMV−FLgag−bGHpA(E3+)、(E)Ad5HIV−1 gagの研究ロット(293細胞由来)、および(F)Ad5HIV−1 gagの臨床ロット(Ad5gagFN0001)。示されているのは、各コホートの幾何平均力価(GMT)および標準誤差線である。
【0091】
図14は、pMRKAd5HIV−1 gagベクターの制限地図を示す。
【0092】
図15A〜Xは、pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【0093】
図16A〜Bは、DNAワクチン発現ベクターV1Jns(A)およびV1Jns−tPA(B)の概要図を示す。これらは、本明細書に記載の種々のDNA/ウイルスベクター組合せ様式法におけるHIV−1 gag、polおよびnef構築物に使用される。
【0094】
図17A〜Cは、IA−Polのヌクレオチド(配列番号3)およびアミノ酸配列(配列番号4)を示す。下線付きのコドンおよびアミノ酸は、表1に列挙された突然変異を示す。
【0095】
図18は、tPA−pol inact(opt)の融合結合部にわたるコドン最適化ヌクレオチドおよびアミノ酸配列(それぞれ配列番号7および8中に含有される)を示す。下線付き部分はIA−PolのNH2末端領域を示す。
【0096】
図19A〜Bは、野生型nef(jrfl)とコドン最適化nefとのヌクレオチド配列比較を示す。該jrfl分離体からの野生型nef遺伝子は、216アミノ酸のポリペプチドをコードしうる648ヌクレオチドよりなる。WT,野生型配列(配列番号19);opt,コドン最適化配列(配列番号1中に含有されている)。Nefアミノ酸配列は1文字記号で示されている(配列番号2)。
【0097】
図20A〜Cは、nef発現ベクターV1Jns/nef(図20A)、V1Jns/nef(G2A,LLAA)(図20B)、V1Jns/tpanef(図20C)およびV1Jns/tpanef(LLAA)(同様に図20C)のプラスミドバックボーンとnefコード配列との間の結合部のヌクレオチド配列を示す。コドン最適化nefまたはコドン最適化nef突然変異遺伝子の5’および3’フランキング配列が太字/イタリック体で示されており、nefおよびnef突然変異体コード配列が無修飾文字で示されている。また(下線付きで)、それぞれのnef発現ベクターの構築に関わる制限エンドヌクレアーゼ部位が示されている。V1Jns/tpanefおよびV1Jns/tpanef(LLAA)は該結合部位に同一配列を有する。
【0098】
図21は、nefおよびnef誘導体の概要図を示す。Nef誘導体に含まれるアミノ酸残基が示されている。グリシン2ならびにロイシン174および175は、それぞれミリスチル化およびジロイシンモチーフに関わる部位である。該tpanef融合遺伝子の両形態に関して、推定リーダーペプチド切断部位は「*」で示されており、該突然変異の構築中に導入された外因性セリン残基は下線で示されている。
【0099】
図22は、前アデノウイルスプラスミド構築物MRKAd5Polの構築の概要図を示す。
【0100】
図23は、前アデノウイルスプラスミド構築物MRKAd5Nefの構築の概要図を示す。
【0101】
図24は、クレードB HIV感染個体におけるクレードBとクレードCとの抗gag T細胞応答の比較を示す。
【0102】
図25は、クレードB HIV感染個体におけるクレードBとクレードCとの抗nef T細胞応答の比較を示す。
【0103】
図26A〜AOは、不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【0104】
図27A〜AMは、不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【0105】
図28は、E3(+)またはE3(−)バックボーン中に種々のプロモーター断片(hCMVまたはmCMV)および終結シグナル(bGHまたはSPA)を含むMRKAd5ベクターの安定性を示す。
【0106】
図29AおよびBは、24、36、48および60hpiの時点における凍結融解回収細胞結合ウイルスの陰イオン交換HPLCウイルス粒子濃度(図29A)、ならびにMRKAd5gag、MRKAd5polおよびMRKA5nefに関する経時的QPA上清力価(図29B)を示す。
【0107】
図30は、本明細書に開示するDNAおよび/またはアデノウイルスワクチンにおいて使用する代表的なtPA−gag融合体のオープンリーディングフレームをオープンリーディングフレーム含むヌクレオチド配列(配列番号36)およびアミノ酸配列(配列番号37)を示す。
【0108】
図31は、第10週(DNA初回抗原刺激後)および第30週(Ad追加抗原刺激後)に集めたPBMCの細胞内γIFN染色を示す。該細胞は、該gagペプチドプールの存在下または不存在下で一晩にわたり刺激した。ついで、蛍光標識抗CD3、抗CD8、抗CD4および抗γIFNモノクローナル抗体を使用して、それらを染色した。各プロットは、表面CD8およびγIFN産生についての陽性染色に関して分離された全CD3+T細胞を示す。各プロットの右上および右下の四分域中の数字は、それぞれCD8+γIFN+およびCD4+γIFN+であったCD3+細胞の割合(%)である。
【0109】
図32は、単一様式アデノウイルス免疫とDNA+アジュバント初回/アデノウイルス追加免疫との比較を示す。
【0110】
図33A〜Bは、実施例29のgag−IApol融合体のオープンリーディングフレームのヌクレオチド配列(配列番号38)を示す。
【0111】
図34A〜Bは、該gag−IApol融合フレームのタンパク質配列(配列番号39)を示す。
【0112】
(発明の詳細な記載)
遺伝子治療における又はヌクレオチドに基づくワクチンベクターにおける使用に適した新規複製欠損型または「第1世代」アデノウイルスベクターを記載する。このベクターは、E1において少なくとも部分的に欠失しており、野生型アデノウイルス配列の塩基対約1からと塩基対約342(より好ましくは400)〜塩基対約458までの間との野生型アデノウイルス・シス作用性パッケージング領域(好ましくは1〜450)および好ましくは塩基対3511〜3523を含む。この記載のベクターは、約5〜10倍大きな増幅速度を伴う増強された増殖特性を有し、より強力であり、より低いウイルス用量で同等の免疫生成を可能にすることが判明している。該ベクターは更に、この領域(塩基対342〜450)を含まない複製欠損型ベクターより大きな細胞性免疫応答を示す回収された組換えアデノウイルスを与える。さらに、これらのベクターに由来するアデノウイルス構築物、特に、イントロンAを欠くhCMVプロモーターの制御下でトランスジーンを含むものは、遺伝的に非常に安定である。ここに記載のウイルスを連続的に継代し、分析した。実施例12を参照されたい。分析した各ウイルスは、適切な遺伝的構造でそれを維持した。また、大規模生産での実施と同様の増殖条件下で分析を行った。この場合もまた、該ベクターは、増強した遺伝的安定性を有することが判明した。実施例12を参照されたい。継代21の後、該ウイルスDNAは再構成の証拠を示さず、1つの製造ロットから次の製造ロットに高度に再生可能であった。すべての関連試験の結果は、図28に関連したデータで本明細書中に示されるとおり、該アデノウイルスベクターが組換え複製欠損型アデノウイルスの大規模生産に非常に良く適合することを示している。
【0113】
この記載の好ましいアデノウイルスベクターは、E3において欠失しており塩基対1〜450を含むベクターである。このベクターは、約7,500塩基対までの外来DNAインサート(または外因性遺伝物質)を収容しうる。もう1つの好ましいベクターは、塩基対1〜450を含むE3を保有するベクターである。この記載の好ましいベクターは、塩基対1〜450および3511〜3523を含むE3+ベクターである。このベクターは、塩基対451〜3510に及ぶ領域が欠失している場合には、約4,850塩基対までの外来DNAインサート(または外因性遺伝物質)を収容しうる。前記ベクターのクローニング容量は、ゲノムサイズの上限として105%の野生型Ad5配列を使用して測定されている。
【0114】
野生型アデノウイルス血清型5は、本明細書の全体にわたり記載されている特定の塩基対番号の基準として用いられる。野生型アデノウイルス血清型5配列は公知であり、当技術分野において記載されている。参照により本明細書に組み入れるChroboczekら,1992 J.Virology 186:280を参照されたい。したがって、本発明の特定の実施形態は、アデノウイルス血清型5配列に基づくベクターである。当業者は、他のアデノウイルス血清型(例えば、血清型2、4、6、12、16、17、24、31、33および42)における前記領域、アデノウイルス血清型5に関して与えられた前記塩基対位置に対応する塩基対により定められる領域を容易に同定することが可能である。したがって、本発明は、野生型アデノウイルス血清型5(Ad5)核酸配列の1〜450(特に、342〜450)に対応する塩基対および好ましくは3511〜3523を含みE1において部分的に欠失したすべてのアデノウイルスベクターを含む。本発明の特に好ましい実施形態は、亜群Cに分類されるAd5に類似したアデノウイルス(例えば、Ad2)に由来するものである。
【0115】
本発明のベクターは、E1において少なくとも部分的に欠失している。好ましくは、該E1領域は、完全に欠失しているか又は不活性化されている。最も好ましくは、E1が欠失した領域は塩基対451〜3510内に存在する。開示しているベクターの伸長5’および3’領域が、使用する細胞系、すなわち、塩基対459〜3510でトランスフェクトされたPER.C6(登録商標)細胞系内に存在するE1A/E1B遺伝子のいずれの部分とも重複することなく従来の構築物のE1欠失のサイズを有効に減少させると考えられることは注目すべきことである。アデノウイルス配列の重複が避けられるのは、組換えの可能性によるものである。したがって、疑いなく、当業者は、異なるアデノウイルスDNAセグメントでトランスフェクトされた異なる細胞系を使用する場合には本発明を修飾しうると理解することが可能である。例示目的で挙げると、塩基対450〜3510のアデノウイルス配列で細胞系をトランスフェクトする場合には、塩基対1から449までの5’領域が、より適切である。このことは、該E1欠失の3’側のセグメントを考える場合にも当てはまる。
【0116】
本発明の好ましい実施形態は、無傷E3領域(すなわち、機能的E3をコードしうるE3遺伝子)を有する。別の実施形態は、部分的に欠失したE3、不活性化されたE3領域、またはE3が完全に欠失した配列を有する。本出願人は、本発明において、該E3遺伝子を含むウイルスが、E3遺伝子を含まないウイルスより迅速に増幅しうることを見出した。図6を参照されたい。この図においては、試験したE3+ウイルスに対応する特徴的CsClバンド(5,665bp)が、E3−ウイルスに対応する3,010bpの特徴的バンドより大量に存在した。これらの結果は、E3遺伝子を含むアデノベクターおよびE3遺伝子を含まないアデノベクターの両方を等しいMOI比(1:1)で混合することを含むウイルス競合研究の後で得られた。ついで増殖研究で、このE3+アデノベクターの増幅能の増加が確認された。表4Aを参照されたい。この表においては、E3−構築物は115および40〜50の増殖率を示しているのに対して、E3+ウイルスは470、420および320の増幅率を示している。
【0117】
前記のとおり、本発明のベクターは、E3+ベクターに関しては約4,850塩基対までの外因性遺伝物質を、そしてE3−ベクターに関しては約7,500塩基対を収容しうる。好ましくは、該インサートは、該アデノウイルスベクターを、野生型ゲノムサイズ(例えば、Ad5の場合、35,935塩基対)に可能な限り近づける。アデノウイルスは、それがその野生型ゲノムサイズに近い場合に最も良く増幅することが良く知られている。
【0118】
図7A、7Bおよび図8Aに例示されているとおり、該遺伝物質はE1平行またはE1逆平行配向で挿入することができる。本発明の特に好ましい実施形態は、E1平行配向で該インサートを有する。本出願人は、異なる配向でトランスジーンを含有するプラスミドとの競合実験により(図8A)、該外来DNAインサートをE1平行配向で有するベクター構築物が、E1逆平行配向トランスジーンより良く増幅し、競合において実際にそれを凌ぐことを見出した。継代3において、そしてもちろん継代6においても、該混合物のウイルスDNA分析は、E1平行配向で該トランスジーンを保持するウイルスの、E1逆平行形態より大きな比率を示した。継代10までは、観察される唯一のウイルス種は、試験した両方のトランスジーンに関してE1平行配向で該トランスジーンを有するアデノベクターであった。
【0119】
本発明のアデノウイルスベクターは、所望のタンパク質(その一例は、HIVタンパク質、特にHIV完全長gagタンパク質である)の発現の達成に特に良く適合する。関心のあるタンパク質をコードする外因性遺伝物質は発現カセットの形態で存在しうる。遺伝子発現カセットは、好ましくは、(a)関心のあるタンパク質をコードする核酸、(b)該タンパク質をコードする核酸に作動的に結合した異種プロモーター、および(c)転写ターミネーターを含む。
【0120】
該転写プロモーターは、好ましくは、真核性RNAポリメラーゼにより認識される。好ましい実施形態においては、該プロモーターは「強力」または「効率的」なプロモーターである。強力なプロモーターの一例は、好ましくはイントロン配列を伴わない、最初期ヒトサイトメガロウイルスプロモーター(参照により本明細書に組み入れるChapmanら,1991 Nucl.Acids Res 19:3979−3986)である。本アデノウイルスベクターにおける使用に最も好ましいのは、イントロンAのようなイントロン配列を伴わないヒトCMVプロモーターである。本出願人は、イントロンA(ヒトサイトメガロウイルスプロモーター(hCMV)の一部である)がアデノウイルスベクターの不安定性の領域を構成することを見出した。イントロンAを伴わないCMVは、HIV gag発現の駆動に際して、比較しうるインビトロ発現能をもたらすことが判明しており(実施例1〜3)、さらには、試験したプラスミドDNAの両方の投与量(20μgおよび200μg)で、それらの抗体およびT細胞応答に関して、Balb/cマウスにおいてインビボでイントロンA含有構築物と同等に挙動した。当業者は、多数の他の公知プロモーターのいずれか、例えば強力な免疫グロブリンまたは他の真核性遺伝子プロモーター、例えばEF1αプロモーター、マウスCMVプロモーター、ラウス肉腫ウイルス(RSV)プロモーター、SV40初期/後期プロモーターおよびβアクチンプロモーターを使用することが可能であると理解するであろう。
【0121】
好ましい実施形態においては、該プロモーターはまた、Tetオペレーター配列のような調節可能な配列を含みうる。これは、例えば、望まない結果を遺伝子産物がもたらしており抑制が求められる場合に非常に有用であろう。
【0122】
該遺伝子配列カセット内に存在する好ましい転写終結配列としては、ウシ成長ホルモンターミネーター/ポリアデニル化シグナル(bGHpA)および以下のとおりに定められる50ヌクレオチド長の短い合成ポリAシグナル(SPA)がが挙げられる:AATAAAAGATCTTTATTTTCATTAGATCTGTGTGTTGGTTTTTTGTGTG(配列番号26)。
【0123】
該CMVプロモーター(イントロンA領域を欠くもの)とBGHターミネーターとの組合せが特に好ましいが、FGアデノウイルスの場合における他のプロモーター/ターミネーターの組合せも用いることができる。
【0124】
他の実施形態は、リーダーまたはシグナルペプチドを該トランスジーン内に組込むものである。好ましいリーダーは、組織特異的プラスミノーゲンアクチベータータンパク質tPA由来のものである。具体例には、本明細書の全体にわたり開示されている種々のtPA−gag、tPA−polおよびtPA−nefアデノウイルスに基づくワクチンが含まれるが、これらに限定されるものではない。
【0125】
本明細書に記載の改善されたアデノウイルスベクターを考慮すると、本発明の必須部分は、予防的または治療的状況において哺乳類宿主、好ましくはヒト宿主に投与されうる、前記アデノウイルスバックボーンを含む、アデノウイルスに基づくHIVワクチンである。本発明のHIVワクチンは、単独で投与されるか他のウイルスまたは非ウイルスに基づくDNAワクチンとの組合せ法で投与されるかにかかわらず、HIVに対して強力かつ広域な細胞性免疫応答を惹起するはずであり、これは、持続的ウイルス感染の可能性を減少させ及び/又はHIV感染を受ける臨床的に有意な低下したウイルス負荷の確立を招くか、あるいはHAART療法と組合せて、既に確立されたHIV感染の影響を軽減するであろう(抗ウイルス免疫療法(ARI))。本明細書に記載の組換えアデノウイルスベクターにおいては任意のHIV抗原(例えば、gag、pol、nef、gp160、gp41、gp120、tat、revなど)を使用することが可能であるが、好ましい実施形態には、コドン最適化p55 gag抗原(本発明ではMRKAd5gag)、polおよびnefが含まれる。HIV−1の種々のクレードに基づく配列が本発明での使用に適しており、それらのうちで最も好ましいのはクレードBおよびクレードCである。特に好ましい実施形態は、コンセンサス・クレードB配列に基づく配列(特に、コドン最適化配列)である。一連のMRKAd5polおよびMRKAd5nefのアデノウイルスワクチンの好ましい形態は、本明細書に記載のpolまたはnefの修飾形態をコードする。HIVエンベロープ遺伝子およびその修飾体を保持するMRKAd5HIV−1ベクターの好ましい実施形態は、それぞれ1997年8月28日(WO 97/31115)および1997年12月24日付けで公開されたPCT国際出願PCT/US97/02294およびPCT/US97/10517(それらの両方の文書を参照により本明細書に組み入れることとする)のHIVコドン最適化env配列を含む。
【0126】
本発明の最も好ましい態様は、HIV gagの発現を達成するための前記のアデノウイルスベクターの開示されている使用である。多数のHIV株の多数の遺伝子の配列がGENBANKにおいて公に入手可能であり、主として、HIVの野外分離体は、これらの株を入手可能にするようQuality Biological(Gaithersburg,MD)に委託しているNational Institute of Allergy and Infectious Diseases(NIAID)から入手可能である。また、株は、World Health Organization(WHO),Geneva Switzerlandからも入手可能である。該gag遺伝子はHIV−1株由来であることが好ましい(CAM−1;Myersら編 “Human Retroviruses and AIDS:1995,IIA3−IIA19(これを参照により本明細書に組み入れることとする))。この遺伝子は、クレードB(北米/欧州)配列のコンセンサスアミノ酸配列に酷似している。したがって、特異的HIV gagまたはその免疫学的に関連した部分をコードする適当なヌクレオチド配列を選択することは当業者の認識範囲内である。実施例25に示すとおり、クレードBまたはクレードCに基づくp55 gag抗原は地球的規模で潜在的に有用であろう。本明細書に記載のとおり、本発明のDNAまたはMRKAdに基づくアデノウイルスベクター内への挿入のために選択されるトランスジーンは、p55 gagのコドン最適化形態である。そのようなMRKAd5gagアデノウイルスベクターは実施例11に記載されており、少なくとも本発明においてはMRKAd5HIV−1gagと称される。もちろん、追加的な形態も意図され、それらには、プロモーター(例えば、hCMVの代わりにmCMV)および/またはpA終結シグナル(bGHの代わりにSPA)の置換のような修飾、ならびにAd5E3遺伝子の欠失を伴う又は伴わないMRKAd5バックボーンの作製が含まれるが、これらに限定されるものではない。
【0127】
本発明はまた、マウスおよび非ヒト霊長類での研究における投与後に細胞性免疫応答を生成することが本発明において示された一連のMRKAd5polに基づくアデノウイルスワクチンに関する。一連のMRKAd5polのいくつかは本明細書中に例示されている。1つのそのようなアデノウイルスベクターはMRKAd5hCMV−inact opt pol(E3+)と称され、これは、MRKAd5バックボーン、hCMVプロモーター(イントロンAを有さない)、不活性化polトランスジーンを含み、該アデノウイルスバックボーン中にAd5 E3遺伝子を含有する。第2の例示されている前アデノウイルスプラスミドおよび付随ウイルスはMRKAd5hCMV−inact opt pol(E3−)と称され、これは、該E3が欠失している以外は前記アデノウイルスベクターと同一である。どちらの構築物もHIV−1 Polのコドン最適化不活性化形態を含有し、ここで、少なくとも全コード領域は本発明では配列番号3として開示されており、該発現タンパク質は配列番号4として示される(不活性化polの標的化欠失を示す図17A〜Cおよび表1も参照されたい)。本明細書に記載のHIV Polのこの及び他の好ましいコドン最適化形態は、実質的には、2000年12月21日付け出願の米国特許出願第09/745,221号および同様に2000年12月21日付け出願のPCT国際出願PCT/US00/34724(それらの両方の文書を参照により本明細書に組み入れることとする)に記載のとおりである。前記文書に開示されているとおり、これらのコドン最適化HIV−1 Polに基づくDNAワクチンのオープンリーディングフレームは、コドン最適化HIV−1 Pol(例えば、配列番号2)、アミノ末端局在化リーダー配列に融合したコドン最適化HIV−1 Pol(例えば、配列番号6)および特に好ましくは、例えば実施例19におけるMRKAd5−Pol構築物により具体的に示された、野生型Polに関連した有意なPR、RT、RNアーゼまたはIN活性を欠く生物学的に不活性化されたpol(「inact opt Pol」;例えば配列番号4)により例示される。また、IA Polタンパク質のアミノ末端領域にリーダーペプチドを含有する配列番号4に関連した構築物が意図される。機能的リーダーペプチドをコードするヌクレオチド配列を伴って又は伴わないでそれぞれのHIV−1 Polコード領域に作動的に結合した調節領域を含有する適当なDNAプラスミドベクター内に、特定の構築物を連結する。この目的において、本明細書に開示する種々のHIV−1 Pol構築物は、野生型Pol(配列番号2に記載のとおりWT opt PolをコードするDNA分子を含む)、tPA−opt WTPol(配列番号6に記載のとおりtPA PolをコードするDNA分子を含む)、inact opt Pol(配列番号4に記載のとおりIA PolをコードするDNA分子を含む)およびtPA−inact opt Pol(配列番号8に記載のとおりtPA−inact opt PolをコードするDNA分子を含む)を含む(これらに限定されるものではない)本発明の増強された第1世代Adベクター(例えば、一連のMRKAd5polアデノウイルスワクチンベクター)にクローニングするためのオープンリーディングフレームに関する。polに基づく形態の増強された第1世代アデノウイルスワクチンは、霊長類、特にヒトを含む宿主に投与されると、CTLおよびTh細胞性免疫応答を惹起する。前記のとおり、本発明の細胞性免疫誘導性ワクチンの効果は、これまでに未感染の個体に対する、より低い伝達率、および/またはHIV−1感染の無症候期を延長するような感染個体内のウイルス負荷のレベルの減少にあるはずである。
【0128】
本発明は更に、HIV gagおよびpol抗原と同様に、マウスおよび非ヒト霊長類での研究において投与された後に細胞性免疫応答を生成する一連のMRKAd5nefに基づくアデノウイルスワクチンに関する。その一連のMRKAd5nefは、本発明では、HIV nefの修飾形態と組合せて、改善されたMRKアデノウイルスバックボーンを使用することにより例示される。これらの例示されているMRKAd5nefベクターは以下のとおりである:(1)改善されたMRKAd5バックボーン、ヒトCMVプロモーター、無傷Ad5 E3遺伝子および修飾されたnef遺伝子を含むMRKAd5hCMV−nef(G2A,LLAA)(E3+);(2)ヒトCMVプロモーターがマウスCMVプロモーターで置換されている以外は前記(1)と同じであるMRKAd5mCMV−nef(G2A,LLAA)(E3+);および(3)該nefトランスジーンがtpanef(LLAA)である以外は(2)と同じであるMRKAd5mCMV−tpanef(LLAA)(E3+)。HIV−1 NefおよびHIV−1 Nef修飾体のコドン最適化形態は、実質的には、2000年12月15日付け出願の米国特許出願第09/738,782号および同様に2000年12月15日付け出願のPCT国際出願PCT/US00/34162(両方の文書を参照により本明細書に組み入れることとする)に記載のとおりである。コドン最適化NefおよびNef修飾体の特定の実施形態は、HIV−1 jfrl分離体からのHIV−1 NefをコードするDNA分子に関する。この場合、該コドンは、ヒトのような哺乳類系内での発現に関して最適化されている。このタンパク質をコードするDNA分子は本発明では配列番号9として開示されており、発現されるオープンリーディングフレームは本発明では配列番号10として開示されている。本発明のアデノウイルスベクターにおける使用のためのNefに基づくコード領域のもう1つの実施形態は、該HIV−1 NefポリペプチドのNH2末端に融合したヒトプラスミノーゲンアクチベーター(tpa)リーダーペプチドを含有するタンパク質をコードするコドン最適化DNA分子を含む。このタンパク質をコードするDNA分子は本発明では配列番号11として開示されており、発現されるオープンリーディングフレームは本発明では配列番号12として開示されている。もう1つの修飾されたNef最適化コード領域は、最適化HIV−1 NefをコードするDNA分子に関する。この場合、該オープンリーディングフレームは、アミノ末端ミリスチル化部位における修飾(Gly−2からAla−2)、およびLeu−174−Leu−175ジロイシンモチーフからAla−174−Ala−175への置換をコードする(本発明ではopt nef(G2A,LLAA)として記載されている)。このタンパク質をコードするDNA分子は本発明では配列番号13として開示されており、発現されるオープンリーディングフレームは本発明では配列番号14として開示されている。MRKAd5nefベクターである(1)MRKAd5hCMV−nef(G2A,LLAA)(E3+)および(2)MRKAd5mCMV−nef(G2A,LLAA)(E3+)はこのトランスジーンを含有する。更なる実施形態は、最適化HIV−1 NefをコードするDNA分子に関する。この場合、アミノ末端ミリスチル化部位およびジロイシンモチーフが欠失しており、tPAリーダーペプチドを含む。このDNA分子opt tpanef(LLAA)は、HIV−1 Nef(jfrl)のアミノ酸残基6−216に融合したtPAリーダー配列を含有するNefタンパク質をコードするオープンリーディングフレームを含み、ここで、Leu−174およびLeu−175はAla−174およびAla−175で置換されており、これは本発明ではopt tpanef(LLAA)と称され、本発明では配列番号15として開示されており、発現されるオープンリーディングフレームは本発明では配列番号16として開示されている。MRKAd5nefベクター「MRKAd5mCMV−tpanef(LLAA)(E3+)」はこのトランスジーンを含有する。
【0129】
本明細書に記載の改善されたMRKA5gagアデノウイルスワクチンベクターに加えて、MRKAd5polおよびMRKAd5nefアデノウイルスベクターの作製は、増強したHIVワクチン能力をもたらす。すなわち、このトリオのアデノウイルスワクチンベクターの作製はすべて、宿主投与後に有効な細胞性免疫応答を生成することが示されており、これらのワクチン候補を、単独で投与する可能性だけでなく、好ましくは、二価(すなわち、gagおよびnef、gagおよびpol、またはpolおよびnef成分)または三価ワクチン(すなわち、gag、polおよびnef成分)の一部として投与する可能性をも与える。したがって、本発明の好ましい態様はワクチン製剤、ならびに投与およびそれに付随する3つの異なる一連のMRKAd5に基づくアデノウイルスベクターワクチンの製剤化に関連した宿主細胞性免疫応答の生成の関連方法である。もちろん、異なるHIV抗原に基づくこの一連のMRKAd5ワクチンは、二価または三価ワクチンの製剤化の機会、あるいは恐らくは、合理的な時間枠内の1以上の一価または二価製剤の別々の製剤の投与の機会の拡大を促進する。また、特異的抗原由来の複数であるが異なる成分を含む組合せ様式法を開始することも、本発明の範囲内である。決して限定的ではない具体例としては、1つのワクチンベクターが野生型Pol(配列番号2)を発現しもう1つのMRKAd5polベクターが不活性化Pol(配列番号6)を発現する別々のMRKAd5polベクターが挙げられるであろう。もう1つの具体例としては、1つのワクチンベクターがNefのtPA/LLAA形態(配列番号16)を発現しもう1つのMRKAd5nefベクターがNefのG2A,LLAA修飾形態(配列番号14)を発現する別々のMRKAd5nefベクターが挙げられるであろう。したがって、本発明のMRKAd5アデノウイルスベクターは、複数の異なるHIV抗原クラスと組合せて使用することができる。各HIV抗原クラスを配列操作に付して、多数の潜在的なワクチンの組合せを得る。そのような組合せは本発明の範囲内である。そのような組合せ様式ワクチン製剤および投与の利用は、単一様式法での接種と比較して、より一層強力な細胞性免疫応答を惹起する可能性を増加させる。
【0130】
本発明はまた、初回/追加ワクチン接種計画における一連のMRKAd5gag、polおよびnefアデノウイルスワクチンの一重、二重または三重様式の投与法の適用に関する。この初回/追加計画は、本明細書に開示されている一連のMRKAd5gag、polおよびnefアデノウイルスワクチンの任意の合理的な組合せを含みうる。さらに、初回/追加法はまた、他のウイルスおよび/または非ウイルスDNAワクチンを含みうる。アデノウイルスワクチンベクター法に加えられる好ましいものは、プラスミドDNAワクチン、特に、本明細書に開示されているコドン最適化gag、polおよびnef構築物の少なくとも1つを含有するDNAプラスミドワクチンを含むが、これに限定されるものではない。
【0131】
したがって、本発明の1つの態様は、gagをコードするプラスミドDNAと組合された初回/追加抗原刺激法における最適化gag遺伝子を含有するアデノウイルスベクターの投与である。このプラスミドを、アデノウイルスベクターの構築において使用するアデノウイルス含有シャトルプラスミドから区別するために、このプラスミドを「ワクチンプラスミド」または「DNAプラスミドワクチン」と称することにする。この投与法において使用する好ましいワクチンプラスミドは、1998年2月3日付け出願の係属中の米国特許出願第09/017,981号および1998年8月13日付け公開のWO98/34640(それらの両方を参照により本明細書に組み入れることとする)に開示されている。簡単に説明すると、好ましいワクチンプラスミドはV1Jns−FLgagと称され、これは、本発明のアデノウイルスベクターと同じコドン最適化gag遺伝子を発現する(完全長p55 gagの例示最適化コドン形態のヌクレオチド配列に関しては、図2を参照されたい)。V1Jnsと称されるワクチンプラスミドバックボーンは、CMV最初期(IE)プロモーターおよびイントロンA、遺伝子発現調節要素としてのウシ成長ホルモン由来ポリアデニル化および転写終結配列、ならびに最小pUCバックボーンを含有する(Montgomeryら,1993,DNA Cell Biol.12:777−783を参照されたい)。該pUC配列は、大腸菌(E.coli)内での高レベルのプラスミド産生を可能にし、アンピシリン耐性遺伝子の代わりにネオマイシン耐性遺伝子を有していてカナマイシンの存在下での選択増殖をもたらす。あるいは、イントロンAが欠失したCMVプロモーターを有するワクチンプラスミドを使用することが可能である。当業者は、これらの特定の構築物が代替的ワクチンプラスミドベクターで容易に置換されうると認識するであろう。本発明においては特に、そのような代替的プラスミドDNAワクチンベクターの使用が予想される。
【0132】
本発明のもう1つの態様は、HIV pol抗原、好ましくは、コドン最適化形態のpolをコードするワクチンプラスミドを含む、および同様に好ましくは、配列番号2、4、6および8に示すPol抗原の群から選ばれるPol抗原をコードするヌクレオチド配列を含んでなるワクチンプラスミドを含む初回/追加抗原刺激法である。種々の生物学的に活性な形態のHIV−1 Polをコードする種々の潜在的DNAプラスミドワクチンにおいては、関心のあるHIV−1 Pol遺伝子の投与、細胞内運搬および発現は宿主CTLおよびTh応答を惹起する。本発明の好ましい合成DNA分子は、コドン最適化野生型Pol(Pro活性を有さない)および種々のコドン最適化不活性化HIV−1 Polタンパク質をコードする。本明細書に開示されているHIV−1 polオープンリーディングフレームは、医薬用途、特に組換えアデノウイルスワクチン、特に、本明細書に記載の増強された第1世代組換えアデノウイルスワクチンにより運搬されるヒトへの投与に、特に好ましい。本発明のこの態様のいくつかの実施形態、すなわち、完全長polをコードするか又は本明細書に記載の修飾体もしくは融合体をコードするかにかかわらず、哺乳類、特にヒトにおける発現に関してコドン使用頻度が最適化されたHIV−1 polオープンリーディングフレームを含むDNA分子は、後記に詳しく説明されている。この場合もまた、関心のあるそれぞれのHIV−1 Pol遺伝子の発現を促進し、投与後に、宿主CTLおよびTh応答を惹起するよう、これらのDNA配列は、本明細書に記載の例示されている組換えアデノウイルスベクターのような組換えアデノウイルスベクター内に適切に配置されている。この場合もまた、これらの好ましい(決して限定的ではない)pol遺伝子は本明細書に開示されているとおりであり、実質的には、2000年12月21日付け出願の米国特許出願第09/745,221号および同様に2000年12月21日付け出願のPCT国際出願PCT/US00/34724(両方の文書を参照により本明細書に組み入れることとする)に記載されているとおりである。
【0133】
組合せ様式および/または初回/追加抗原刺激法において有用な第3の一連のワクチンプラスミドは、HIV nef抗原またはその生物学的および/または免疫学的に関連した修飾体をコードするワクチンプラスミドである。他の箇所に記載されているとおり、好ましいワクチンプラスミドは、nefのコドン最適化形態を含有し、また、好ましくは、配列番号10、12、14および16に示すNef抗原の群から選ばれるNef抗原をコードするヌクレオチド配列を含む。これらの好ましいnefコード領域は本明細書に開示されており、2000年12月15日付け出願の米国特許出願第09/738,782号および同様に2000年12月15日付け出願のPCT国際出願PCT/US00/34162(両方の文書を参照により本明細書に組み入れることとする)に記載されている。
【0134】
したがって、本発明のアデノウイルスワクチンおよびプラスミドDNAワクチンは単独で投与することが可能であり、あるいは初回抗原刺激および追加抗原刺激投与法の一部でありうる。混合様式の初回および追加接種計画は、既存の抗ベクター免疫応答が存在する場合には特に、増強された免疫応答をもたらす。本発明のこの1つの態様は、該プラスミドワクチンを少なくとも1回投与することにより該プラスミドワクチンで対象を初回抗原刺激し、所定の長さの時間を経過させ、ついで該アデノウイルスワクチンを投与することにより追加抗原刺激する方法である。通常、複数の初回抗原刺激、典型的には1〜4回の初回抗原刺激を行うが、より多数の初回抗原刺激を行うことが可能である。初回抗原刺激と追加抗原刺激との間の時間の長さは、典型的には約4ヶ月〜1年と様々となりうるが、他の時間枠を用いることも可能である。アカゲザルでの実験においては、該動物をプラスミドワクチンで4回初回抗原刺激し、ついで4ヶ月後に該アデノウイルスワクチンで追加抗原刺激した。それらの細胞性免疫応答は、アデノウイルスワクチンのみが投与された動物の場合より顕著に高かった。初回抗原刺激法の利用は、既存の抗アデノウイルス免疫応答を対象者が有する場合には特に好ましいかもしれない。
【0135】
それに加えて及びその代わりに、本明細書に開示されているMRKAd5アデノウイルスワクチンのような複数のHIV−1ウイルス抗原を適当なシャトルプラスミドに連結させて、複数のオープンリーディングフレームを含む前アデノウイルスプラスミドを生成させることができる。例えば、三価ベクターは、E3(−)またはE3(+)バックグラウンド、好ましくはE3欠失バックボーンにおけるgag−pol−nef融合体、あるいは、おそらくは、「2+1」二価ワクチン、例えば、同じMRKAd5バックボーン内のgag−pol融合体(すなわち、コドン最適化p55 gagおよび不活性化最適化pol;実施例29および表25)(各オープンリーディングフレームは別個のプロモーターおよび転写終結配列に作動的に結合している)を含みうる。あるいは、それらの2つのオープンリーディングフレームが単一のプロモーターに作動的に結合しており、該オープンリーディングフレームがIRES(internal ribosome entry sequence)に作動的に連結していることが可能である(参照により本明細書に組み入れる国際公開番号WO95/24485に開示されているとおりである)。図9は、複数のプロモーターおよび終結配列の使用が、同様の増殖特性をもたらすことを示しており、図28は、これらのMRKAd5gagに基づくベクターがまた、少なくとも継代21にわたり安定であることを示している。IRESに基づく技術を用いない場合には、ベクター安定性が最良に維持されるよう、それぞれのオープンリーディングフレームを支持するために、異なるプロモーターを使用することが好ましい。決して限定的ではない例として挙げると、潜在的な多トランスジーンワクチンは、E3欠失バックボーン中のhCMV−gagpol−bGHpA+mCMV−nef−SPAまたはhCMV−gagpol−bGHpA+mCMV−nef−SPA(E3+)のような3トランスジーンベクターを含みうる。本発明の潜在的な「2+1」二価ワクチンは、すべてのトランスジーンがE1平行配向であるE3+バックボーン中のhCMV−pol−bGHpA(ベクター#2)と組合されたE3+バックボーン中のhCMV−gag−bGHpA+mCMV−nef−SPA(ベクター#1)でありうる。前記のgag−pol融合体以外の融合構築物も、種々の二価ワクチン法での使用に適しており、お互いに融合した任意の2つのHIV抗原(例えば、nef−polおよびgag−nef)から構成されうる。これらのアデノウイルス組成物は、前記のとおり、好ましくは、投与後に生成する免疫応答を多様化するために追加的なHIV抗原を含むアデノウイルス組成物と共に運搬される。したがって、単一の又は可能な第2のアデノウイルスベクターとして運搬される多価ワクチンが本発明の一部と意図されることは明らかである。この場合もまた、この投与様式は、アデノウイルスに基づく有効なHIV−1ワクチンが組合せ様式法により投与されうるもう1つの例である。しかし、開示されているアデノウイルスベクター用のインサートの決定に関しては、アデノウイルスベクターの有効パッケージング限界を十分に考慮する必要があることに注目することは重要である。アデノウイルスは、該野生型Ad5配列の約105%のクローニング容量上限を示すことが示されている。
【0136】
発現用に選択した遺伝子にかかわらず、該配列をヒト細胞環境中の発現に関して「最適化」することが好ましい。4つの可能なヌクレオチド塩基の「トリプレット」コドンは、異なる64個の形態で存在しうる。これらの形態が僅か20個の異なるアミノ酸(ならびに転写の開始および終結)のメッセージを与えるに過ぎないことは、いくつかのアミノ酸が2以上のコドンによりコードされうることを意味する。実際に、いくつかのアミノ酸は6つもの「重複」した代替的コドンを有し、一方、他のいくつかは、単一の必要なコドンを有する。完全には理解されていない理由により、代替的コドンは種々の細胞型の内因性DNA内に決して一様には存在せず、或る細胞型においては或るコドンに関する可変的天然階層または「優先性」が存在するようである。例えば、アミノ酸ロイシンは、CTA、CTC、CTG、CTT、TTAおよびTTG(これらはそれぞれ、mRNコドンCUA、CUC、CUG、CUU、UUAおよびUUGに対応する)を含む6つのDNAコドンのいずれかにより特定される。大腸菌(E.coli)の内因性DNAは、最も一般的には、ロイシン特定コドンCTGを含有し、一方、酵母および粘菌のDNAは、最も一般的には、ロイシン特定コドンTTAを含有することが、微生物についてのゲノムコドン出現頻度の詳細な分析から示されている。この階層を考慮すると、大腸菌(E.coli)宿主によるロイシンに富むポリペプチドの高レベルの発現が得られる可能性は、コドン使用頻度に或る程度は左右されると、一般に考えられている。例えば、TTAコドンに富む遺伝子は、十中八九は大腸菌(E.coli)内では発現されにくく、一方、CTGに富む遺伝子は、おそらく該ポリペプチドを高度に発現するであろう。同様に、酵母細胞が、ロイシンに富むポリペプチドの発現のための意図される形質転換宿主細胞である場合には、挿入されるDNA内で使用するための好ましいコドンはTTAであろう。
【0137】
組換えDNA技術へのコドン優先性現象の関与は明白であり、該現象は、成功裏に形質転換された宿主生物において外因性遺伝子の高い発現レベルがこれまで多くの場合に達成されていないことを説明するのに役立ちうる。すなわち、該挿入遺伝子内には、それほど「好まし」くないコドンが反復的に存在している可能性があり、発現用の宿主細胞装置が、それほど効率的には機能していない可能性があるのである。この現象は、意図される宿主細胞の好ましいコドンを含むように設計された合成遺伝子が、組換えDNA技術の実施のための好ましい形態の外来遺伝物質を与えることを示唆している。したがって、本発明の1つの態様は、アデノウイルスベクター、またはワクチンプラスミドとアデノウイルスベクターとの何らかの組合せであり、この場合、それらの両方は特に、ヒト細胞環境中での発現に関してコドンが最適化された遺伝子を含む。本明細書に記載のとおり、本発明での使用のための好ましい遺伝子はコドン最適化HIV遺伝子であり、特に、HIV gag、polまたはnefである。
【0138】
本発明のアデノウイルスベクターは、Hittら,1997“Human Adenovirus Vectors for Gene Transfer into Mammalian Cells”Advances in Pharmacology 40:137−206(これを参照により本明細書に組み入れることとする)に概説されているような公知技術を用いて構築することができる。
【0139】
本発明のアデノウイルスベクターを構築する際には、プラスミドまたはシャトルベクター内にそれを挿入することがしばしば簡便である。これらの技術は公知であり、Hittら,前掲に記載されている。本発明は特に、該アデノウイルスと、シャトルプラスミド内に挿入された場合のアデノウイルスとの両方を含む。
【0140】
好ましいシャトルベクターは、アデノウイルス部分およびプラスミド部分を含有する。該アデノウイルス部分は、実質的には、アデノウイルス配列(非機能的または欠失したE1およびE3領域を伴う)と簡便な制限部位に隣接した遺伝子発現カセットとを含有する前記のアデノウイルスベクターと同じである。該シャトルベクターのプラスミド部分は、しばしば、原核性プロモーターの転写制御下で抗生物質耐性マーカーを含有していて、該抗生物質の発現は真核細胞内では生じない。アンピシリン耐性遺伝子、ネオマイシン耐性遺伝子および他の医薬上許容される抗生物質耐性マーカーを使用することができる。原核生物における発酵による該ポリヌクレオチドの高レベルの産生を補助するためには、該シャトルベクターが原核性複製起点を含有し高コピー数であることが有利である。多数の商業的に入手可能な原核性クローニングベクターがこれらの利点をもたらす。非必須DNA配列を除去することが望ましい。また、該ベクターは真核細胞内で複製不能であることが望ましい。これは、受容者のゲノム内へのポリヌクレオチドワクチン配列の組込みの危険性を最小限に抑える。該ポリヌクレオチドの発現を特定の組織型に限定することが望ましい場合には、組織特異的プロモーターまたはエンハンサーを使用することができる。
【0141】
本発明の1つの実施形態においては、該前プラスミド、例えば、pMRKAd5pol、pMRKAd5nefおよびpMRKAd5gagは、MRKHVE3(およびE3−形態用にはMRKHVO)バックボーンおよび適当なシャトルベクターを使用する相同組換えにより作製することが可能であり、pMRKAd5polに関しては図22に、そしてpMRKAd5nefに関しては図23に示されているとおりである。線状形態のプラスミドは、PER.C6(登録商標)細胞への侵入後に複製されることが可能であり、ウイルスが産生される。感染細胞および培地は、ウイルス複製が完了した後に回収した。
【0142】
ウイルスベクターは、公知細胞系293およびPER.C6(登録商標)を含む種々のE1相補性細胞系内で増殖しうる。これらの細胞系は共に、アデノウイルスE1遺伝子産物を発現する。PER.C6(登録商標)は、WO 97/00326(1997年1月3日付け公開)、および発行されている米国特許第6,033,908号(それらの両方を参照により本明細書に組み入れることとする)に記載されている。それは、複製欠損型(FG)アデノウイルスの産生を相補するE1遺伝子セグメントで形質導入された初代ヒト網膜芽細胞系であるが、相同組換えによる複製許容型アデノウイルスの生成を妨げるように設計される。特に関心が持たれる細胞は、PER.C6(登録商標)のように、459bp〜3510bp(これを含む)のAD5E1AおよびE1B遺伝子をコードするトランスジーンで安定に形質転換されている。293細胞は、参照により本明細書に組み入れるGrahamら,1997 J.Gen.Virol 36:59−72に記載されている。前記のとおり、使用する相補性細胞系内に存在するアデノウイルス配列を考慮しなければならない。組換えの可能性を最小限に抑えたい場合には、該配列は、該ベクター内に存在する配列と重複しないことが重要である。
【0143】
前記に従い作製したベクターは、免疫応答の誘導において、より有効であることが判明しており、したがって非常に有望なワクチン候補に相当する。より詳しくは、強力な異種プロモーターで調節されるコドン最適化HIV gag遺伝子を保持する前記の第1世代アデノウイルスベクターはヒト抗HIVワクチンとして使用することが可能であり、免疫応答を誘導しうることが判明している。
【0144】
DNA構築物を製造し精製するための分子生物学の標準的な技術は本発明のDNA免疫原の製造を可能にする。
【0145】
本発明のアデノウイルスベクターを含むワクチン組成物は、バッファー、正常食塩水またはリン酸緩衝食塩水、スクロース、他の塩およびポリソルベートのような生理的に許容される成分を含有しうる。1つの好ましい製剤は、2.5〜10mM TRISバッファー、好ましくは約5mM TRISバッファー;25〜100mM NaCl、好ましくは約75mM NaCl;2.5〜10% スクロース、好ましくは約5% スクロース;0.01〜2mM MgCl2;および0.001%〜0.01% ポリソルベート80(植物由来)を含有する。pHは、約7.0〜9.0、好ましくは約8.0であるべきである。当業者は、該製剤を製造するために他の通常のワクチン賦形剤も使用しうると理解するであろう。好ましい製剤は、5mM TRIS、75mM NaCl、5% スクロース、1mM MgCl2、0.005% ポリソルベート80をpH8.0で含有する。これは、ガラス表面へのウイルスの吸着の可能性を最小限に抑えAd5の安定性にとって最適に近いpHおよび二価陽イオン組成を有する。それは、筋肉注射の際に組織刺激を引き起こさない。それは、好ましくは、使用まで凍結される。
【0146】
ワクチン受容者に導入されるワクチン組成物中のアデノウイルス粒子の量は、使用する転写および翻訳プロモーターの強度ならびに発現遺伝子産物の免疫原性に左右されるであろう。一般には、免疫学的または予防的に有効な用量である1×107〜1×1012粒子、好ましくは約1×1010〜1×1011粒子を筋肉組織内に直接投与する。皮下注射、皮内導入、皮膚を介した圧入(impression)および他の投与様式、例えば腹腔内、静脈内または吸入運搬も意図される。また、追加ワクチン接種が行われると意図される。HIVアデノウイルスベクターでのワクチン接種の後、後続のHIVアデノウイルスベクターおよび/またはプラスミドでの追加抗原刺激が望ましいかもしれない。本発明のワクチン組成物の非経口的導入と同時またはその後のインターロイキン12タンパク質の非経口投与、例えば静脈内、筋肉内、皮下または他の手段の投与も有利である。
【0147】
本発明のポリヌクレオチドのアデノウイルスベクターおよび/またはワクチンプラスミドは、受容者の免疫系に影響を及ぼすいずれのタンパク質、アジュバントまたは他の物質とも会合していないことが可能である。この場合、該ベクターは、生理的に許容される溶液、例えば無菌食塩水または無菌緩衝食塩水(これらに限定されるものではない)中に存在することが望ましい。あるいは、該ベクターは、免疫応答を増強する当技術分野で公知のアジュバント(すなわち、「生物学的に有効」なアジュバント)、例えばタンパク質または他の担体と会合していることが可能である。本発明のワクチンプラスミドは、例えば、アジュバントの存在下または不存在下、食塩水(例えば、PBS)中で運搬することができる。好ましいアジュバントはAlumまたはCRL 1005 Block Copolymerである。DNAの細胞取り込みを補助する物質、例えばカルシウムイオン(これに限定されるものではない)を有利に使用することができる。これらの物質は、本発明では一般には、トランスフェクション促進試薬および医薬上許容される担体と称される。ポリヌクレオチドでコーティングされたマイクロプロジェクタイル(microprojectile)をコーティングするための技術は当技術分野で公知であり、本発明においても有用である。
【0148】
本発明はまた、第1アデノウイルスベクターを投与し次いで追加抗原刺激投与を行う初回抗原刺激および追加抗原刺激法を含む。該追加抗原刺激投与は、選択された時間間隔で反復することができる。あるいは、好ましい接種計画は、第1アデノウイルス血清型で初回抗原刺激し、ついで第2アデノウイルス血清型で追加抗原刺激することを含む。より好ましくは、該接種計画は、第1アデノウイルス血清型で初回抗原刺激し、ついで第2アデノウイルス血清型で追加抗原刺激することを含み、ここで、該第1および第2アデノウイルス血清型はアデノウイルスの別々の亜群内に分類される。前記の初回/追加抗原刺激計画は、選択したアデノウイルスベクターに対して既存免疫が確認されている場合に特に好ましい。このタイプの計画においては、個体または個体集団を、既存免疫が確認されている血清型以外の血清型のアデノウイルスで初回抗原刺激する。これは、該第1アデノウイルスが、該第2アデノウイルスに対する既存免疫を避けつつ該トランスジーンの十分な発現を達成するのを可能にし、さらに、該追加抗原刺激アデノウイルスを介した該トランスジーンの後続運搬が、より有効となるのを可能にする。アデノウイルス血清型5は、そのような計画が望ましいと考えられるウイルスの一例である。したがって、本発明では、非C群アデノウイルス(例えば、A群アデノウイルスであるAd12、D群アデノウイルスであるAd24、またはB群アデノウイルスであるAd35)で初回抗原刺激して抗Ad5免疫を回避し、ついでC群アデノウイルスであるAd5で追加抗原刺激することを選択しうるであろう。もう1つの好ましい実施形態は、異なるアデノウイルス(非ヒトアデノウイルスを含む)ワクチンの投与およびそれに続く、開示されているアデノウイルスワクチンの投与を含む。別法においては、まず、関心のあるウイルス抗原を、アデノウイルスに基づくワクチン以外のウイルスワクチンを介して運搬し、ついで、開示されているアデノウイルスワクチンで行う。別のウイルスワクチンは、ポックスウイルスおよびベネズエラウマ脳炎ウイルスを含むが、これらに限定されるものではない。
【0149】
膨大なヒトおよび動物データは、HIV感染の抑制(または排除)における細胞性免疫応答、特にCTLの重要性を支持している。ヒトにおいては、初感染後に非常に高いレベルのCTLが生じ、それはウイルス血症の抑制と相関している。HIVに繰返し曝露されたにもかかわらず未感染のままである幾つかの小さな個体群が記載されている。これらのコホートの幾つかにおいて、CTLが注目されている。HIV感染のSIVモデルにおいては、初感染後に同様にCTLが生じ、抗CD8モノクローナル抗体の添加はこの感染抑制を妨げ疾患の進行を招くことが示されている。本発明では、CTLを誘導するためにアデノウイルスワクチンを単独で又はプラスミドワクチンと組合せて使用する。
【0150】
本発明を更に例示するために、以下に非限定的な実施例を記載する。
【0151】
実施例1
hCMVプロモーターのイントロンA部分の除去
hCMVプロモーターを増幅するための出発物質として、GMP等級のpVIJnsHIVgagを使用した。pVIJnsHIVgagは、CMV最初期(IE)プロモーターおよびイントロンA、完全長コドン最適化HIV gag遺伝子、ウシ成長ホルモン由来ポリアデニル化および転写終結配列、ならびに最小pUCバックボーンを含むプラスミドである。該プラスミドバックボーンの説明としては、Montgomeryら,前掲を参照されたい。該増幅は、hCMVプロモーターに隣接するよう適切に配置されるプライマーで行った。5’プライマーはhCMVプロモーターのMsc1部位の上流に配置され、3’プライマー(BglII認識配列を含有するよう設計されている)はhCMVプロモーターの3’側に配置された。全hCMVプロモーター(−イントロンA)を含む得られたPCR産物(高忠実度Taqポリメラーゼを使用)を、TOPO PCR平滑ベクター内にクローニングし、ついでMsc1およびBglIIでの二重消化により取り出した。ついでこの断片を、Msc1およびBglII消化後に元のプロモーター、イントロンAおよびgag遺伝子が除去された元のGMP等級のpV1JnsHIVgagプラスミド内に再びクローニングした。この連結反応は、元のpV1JnsHIVgagベクターバックボーン内のhCMVプロモーター(−イントロンA)+bGHpA発現カセットの構築をもたらした。このベクターはpVIJnsCMV(無イントロン)と称される。
【0152】
BglII消化を用いてpV1JnsHIVgagからFLgag遺伝子を切り出し、その1,526bpの遺伝子をゲル精製し、pVIJnsCMV(無イントロン)内にBglII部位においてクローニングした。正しい配向でFlgag遺伝子を保持するクローンを同定するために、Sma1制限酵素を使用してコロニーをスクリーニングした。pV1JnsCMV(無イントロン)−FLgag−bGHpAと称されるこのプラスミドを完全に配列決定して、配列の完全性を確認した。
【0153】
また、2つの追加的なトランスジーンを構築した。プラスミドpV1JnsCMV(無イントロン)−FLgag−SPAは、50ヌクレオチド長の短い合成ポリAシグナル(SPA)でウシ成長ホルモンポリアデニル化シグナルが置換されていること以外はpV1JnsCMV(無イントロン)−FLgag−bGHpAと同一である。該SPAの配列は以下に示すとおりであり、ここでは、必須成分(それぞれ、ポリ(A)部位、(GH)nおよび(T)n)が下線で示されている:AATAAAAGATCTTTATTTTCATTAGATCTGTGTGTTGGTTTTTTGTGTG(配列番号18)。
【0154】
プラスミドpV1Jns−mGMV−FLgag−bGHpAは、hCMVプロモーターが除去されてマウスCMV(mCMV)プロモーターで置換されていること以外はpV1JnsCMV(無イントロン)−FLgag−bGHpAと同一である。
【0155】
図3は、元のトランスジーンと比較して新規トランスジーン構築物を図示する。
【0156】
実施例2
修飾gagトランスジーンに関するgag発現アッセイ
一過性組織培養トランスフェクション実験から得た培養上清についてGag Elisaを行った。この場合、共にイントロンAを欠く2つの新規hCMV含有プラスミド構築物pV1JnsCMV(無イントロン)−FLgag−bGHpAおよびpV1JnsCMV(無イントロン)−FLgag−SPAを、前記のとおりhCMVプロモーターの一部としてイントロンAを有するpV1JnsHIVgagと比較した。以下の表2は、GMP等級の元のプラスミドと比較して該新規gagプラスミドのインビトロgag発現データを示している。表2に示す結果は、それらの新規hCMV gagプラスミド構築物の両方が、hCMVプロモーターのイントロンA部分を含有する元のプラスミド構築物と比較しうる発現能を有することを示している。
【0157】
【表1】
実施例3
修飾gagトランスジーンに関するげっ歯類(Balb/c)研究
前記の2つの新規プラスミド構築物pV1JnsCMV(無イントロン)−FLgag−bGHpAおよびpV1JnsCMV(無イントロン)−FLgag−SPAに関して、CMVプロモーターのイントロンA部分を有する前記構築物pV1JnsHIVgagとそれらを比較するために、げっ歯類での研究を行った。gag抗体およびElispot応答(1999年7月6日付け出願の米国仮特許出願第60/142,631号および1999年8月13日付け出願の米国特許出願第60/148,981号に基づく優先権を主張している2000年7月3日付け出願のPCT国際出願番号PCT/US00/18332(WO 01/02607)に記載されている;これらの3つの出願を参照により本明細書に組み入れることとする)を測定した。以下の表3に示す結果は、該新規プラスミド構築物が、試験したプラスミドDNAの両方の用量(20μgおよび200μg)で、その抗体およびT細胞応答に関して、Balc/cマウスにおいて元の構築物と同等に挙動したことを示している。
【0159】
実施例4
【0160】
【表2】
修飾シャトルベクター「MRKpdelE1シャトル」の構築
元のAd5シャトルベクター(pdelElsp1A;Ad5のヌクレオチド341〜3524の複数のクローニング領域と共に塩基対1〜341および3524〜5798のAd5配列を含むベクター)に対する修飾は、以下のとおりに連続的なクローニング工程において行う以下の3つの操作を含んでいた:
(1)左ITR領域を、該ベクターバックボーンと該アデノウイルス左ITR配列との間の結合部にPac1部位を含むよう伸長させた。これは、細菌相同組換え系を用いる、より容易な操作を可能にする。
(2)パッケージング領域を、342bp〜450bpの野生型(WT)アデノウイルスの配列を含むよう伸長させた。
(3)pIXの下流の領域を13ヌクレオチド(すなわち、ヌクレオチド3511〜3523に)伸長させた。
これらの修飾(図4)は、形質転換PER.C6(登録商標)細胞系内に存在するE1A/E1B遺伝子のいずれの部分とも重複することなくE1欠失のサイズを有効に減少させた。すべての操作は、AdシャトルベクターpdelElsp1Aを修飾することにより行った。
【0162】
該修飾を該シャトルベクターに施したら、該変化は、大腸菌(E.coli)BJ5183化学的コンピテント細胞を使用する細菌相同組換えにより元のAd5アデノベクターバックボーン(pAdHVOおよびpAdHVE3)内に組み込まれた。
【0163】
実施例5
修飾アデノベクターバックボーン(E3+およびE3−)の構築
元のアデノベクターpAdHVO(E1およびE3領域を含むヌクレオチド以外のすべてのAd5配列を含む)およびpADHVE3(E1領域を含むヌクレオチド以外のすべてのAd5配列を含む)をそれぞれ、それらがE1領域への修飾を含有するように再構築した。これは、新たに修飾されたシャトルベクター(MRKpdelE1シャトル)をPac1およびBstZ1101で消化し、該アデノウイルス配列に対応する2,734bpの断片を単離することにより達成された。この断片を、Cla1線状化pAdHVO(E3−アデノベクター)またはCla1線状化pAdHVE3(E3+アデノベクター)からのDNAと共に大腸菌(E.coli)BJ5183コンピテント細胞内に同時形質転換した。各形質転換からの少なくとも2つのコロニーを選択し、濁り状態に達するまで6〜8時間、Terrific(商標)ブロス内で成長させた。DNAを各細胞ペレットから抽出し、ついで大腸菌(E.coli)XL1コンピテント細胞内に形質転換した。各形質転換からただ1つのコロニーを選択し、プラスミドDNA精製のために成長させた。正しいクローンを確認するために、該プラスミドを制限消化により分析した。該修飾アデノベクターをMRKpAdHVO(E3−プラスミド)およびMRKpAdHVE3(E3+プラスミド)と命名した。古い形態のアデノベクターおよびこれらの新規アデノベクターからのウイルス(それぞれMRKHVOおよびMRKHVE3)を、以下の一連のウイルス競合実験に対応させるために、PER.C6(登録商標)細胞系内で産生させた。また、元のシャトルベクターのマルチクローニング部位はClaI、BamHI、XhoI、EcoRV、HindIII、SalIおよびBglII部位を含有していた。このMCSを、NotI、ClaI、EcoRVおよびAscI部位を含有する新規MCSで置換した。この新規MCSは、該パッケージング領域およびpIX遺伝子に施された修飾と共に、MRKpAdHVOおよびMRKpAdHVE3前プラスミドに導入されている。
【0164】
実施例6
パッケージングシグナル伸長の効果の分析
E1欠失領域に施された修飾の効果を調べるために、元のバックボーン(pAdHVE3)および新規バックボーン(MRKpAdHVE3)から得られたウイルスを等しいMOI比(1:1および5:5)で混合し、数ラウンドにわたり継代した。図5、実験#1を参照されたい。該実験におけるウイルスの両方は、E3遺伝子を無傷で含有しており、トランスジーンを含有していなかった。それらの2つのウイルスの間の唯一の相違はE1欠失の領域内に存在した。P1(継代1)における該ウイルスの同時感染の後、該混合物を更に4継代にわたり増殖させ、その時点で、該細胞を回収し、該ウイルスを抽出し、CsClバンディングにより精製した。該ウイルスDNAを抽出し、HindIIIで消化し、ついで該消化産物を放射能標識した。また、対照のために、それぞれの前プラスミド(pAdHVE3(「OLD E3+」)、MRKpAdHVE3(「NEW E3+」))をHindIII(および該ベクターバックボーンを取り出すためにPac1)で消化し、ついで[33P]dATPで標識した。該放射能標識消化産物をゲル電気泳動に付し、該ゲルをWhatman紙上で乾燥させた後、オートラジオグラフィーフィルムにさらした。図6は、パッケージングシグナル領域に施された付加を有する新規アデノウイルスが、元のアデノウイルスと比べて増殖上の利点を有することを、明らかに示している。行った実験(試験したいずれの比においても)においては、新たに修飾されたウイルスに関する消化バンドのみが存在した。3,206のサイズの特徴的バンド(該新規ウイルス由来のもの)が明らかに存在した。しかし、元のウイルスから予想される2,737bpのサイズの特徴的バンドの証拠は認められなかった。
【0165】
実施例7
E3遺伝子の効果の分析
第2組の該ウイルス競合研究は、MRKpAdHVOおよびMRKpAdHVE3から得られた等しいMOI比(1:1)の新規修飾ウイルスを混合することを含むものであった(図5、実験#2)。この組においては、両方のウイルスは、E1欠失に施された新たな修飾を有していた。第1ウイルス(MRKpAdHVO由来)はE3遺伝子を含有しない。第2ウイルス(MRKpAdHVE3由来)はE3遺伝子を含有しない。該ウイルスはいずれも、トランスジーンを含有しない。該ウイルスの同時感染の後、該混合物を更に4継代にわたり増殖させ、その時点で、該細胞を回収し、該全ウイルスを抽出し、CsClバンディングにより精製した。該ウイルスDNAを抽出し、HindIIIで消化し、ついで該消化産物を放射能標識した。また、対照のために、それぞれの前プラスミドMRKpAdHVO(「NEW E3−」)、MRKpAdHVE3(「NEW E3+」))をHindIII(および該ベクターバックボーンを取り出すためにPac1)で消化し、ついで[33P]dATPで標識した。該放射能標識消化産物をゲル電気泳動に付し、該ゲルをWhatman紙上で乾燥させた後、オートラジオグラフィーフィルムにさらした。図6は、該E3+ウイルスおよびE3−ウイルス混合実験のウイルスDNA分析の結果を示す。該E3+ウイルスに対応する特徴的バンド(5,665bp)は、該E3−ウイルスに対応する3,010bpの特徴的バンドより大量に存在した。このことは、該E3遺伝子を含有するウイルスが、E3遺伝子を含有しないウイルスより迅速に増幅しうることを示している。この増幅率の増加は増殖研究により確認されている。後記の表4を参照されたい。
【0166】
実施例8
修飾gagトランスジーンを含有する新規シャトルベクター「MRKpdel E1−CMV(無イントロン)−FLgag−bGHpA」の構築
修飾プラスミドpV1JnsCMV(無イントロン)−FLgag−bGHpAをMsc1で一晩消化し、ついでSfi1で50℃で2時間消化した。ついで該DNAをマングマメヌクレアーゼで30℃で30分間処理した。該DNA混合物を、Qiaex IIキットを使用して脱塩し、ついで37℃で30分間にわたりクレノウ処理して、該トランスジーン断片の末端を完全に平滑化した。ついでその2,559bpのトランスジーン断片をゲル精製した。該修飾シャトルベクター(MRKpdelE1シャトル)を、EcoRVでの消化により線状化し、仔ウシ腸アルカリホスファターゼで処理し、ついで、得られた6,479bpの断片をゲル精製した。ついでその2つの精製断片を互いに連結し、数十個のクローンをスクリーニングして、該シャトルベクター内の該トランスジーンの挿入に関して確認した。特徴的制限消化を行って、該トランスジーンをE1平行およびE1逆平行配向で保持するクローンを同定した。この方法に従って、該MRKpdelE1シャトルベクター内のその他のgagトランスジーンにおけるクローニングを行った。
【0167】
実施例9
MRK FGアデノベクターの構築
HIV−1 gagトランスジーンをE1平行配向で含有するシャトルベクターであるMRKpdelE1−CMV(無イントロン)−FLgag−bGHpAをPac1で消化した。該反応混合物をBsfZ171で消化した。その5,291bpの断片をゲル抽出により精製した。MRKpAdHVE3プラスミドをCla1で37℃で一晩消化し、ゲル精製した。約100ngの5,290bpシャトル+トランスジーン断片および〜100ngの線状化MRKpAdHVE3 DNAを大腸菌(E.coli)BJ5183化学コンピテント細胞内に同時形質転換した。いくつかのクローンを選択し、濁り状態に達するまで6〜8時間、2mlのTerrific(商標)ブロス内で増殖させた。該細胞ペレットからの全DNAを、Qiagenアルカリ細胞溶解およびフェノールクロロホルム法を用いて精製した。該DNAをイソプロパノールで沈殿させ、20μl dH2Oに再懸濁させた。このDNAの2μl アリコートを大腸菌(E.coli)XL−1コンピテント細胞内に形質転換した。それぞれの別々の形質転換からの単コロニーを選択し、3mlのLB+100μg/ml アンピシリン中で一晩成長させた。Qiagenカラムを使用して、該DNAを単離した。該gag遺伝子および該プラスミドバックボーン中で切断する制限酵素BstEIIでの消化により、陽性クローンを同定した。該前プラスミドクローンはMRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpAと称され、37,498bpのサイズを有する。この方法に従って、E1平行およびE1逆平行形態のその他のgagトランスジーン構築物のそれぞれのE3−およびE3+形態を作製した。図7A、7Bおよび7Cは、構築したアデノベクターの種々の組合せを示す。
【0168】
実施例10
プラスミド競合研究
一連のプラスミド競合研究を行った。簡単に説明すると、2つの競合プラスミドのそれぞれの等量を混合することにより、新規構築物の種々の組合せのスクリーニングを行った。図8Aに示す実験においては、同じトランスジーンを異なる配向で含有するプラスミドを互いに混合して、それらの2つのプラスミドの「競合」を行った。その目的は、トランスジーン配向の効果を観察することにあった。図8Bに示す実験においては、種々のポリアデニル化シグナルを(同じ配向で)含有するプラスミドを互いに等量で混合した。その目的は、ポリAシグナルの効果を評価することにあった。該初期トランスフェクションの後、該ウイルスを10ラウンドにわたり継代し、該ウイルスDNAを放射性制限分析により分析した。
【0169】
該プラスミド混合実験(図8A)からのウイルス種の分析は、E1平行配向で挿入されたトランスジーンを有するアデノベクターが、E1逆平行配向で挿入されたトランスジーンを有するアデノウイルスより良く増殖し、競合においてそれを凌ぎうることを示した。継代3において、そしてもちろん継代6においても、該混合物のウイルスDNA分析は、E1平行配向で該トランスジーンを保持するウイルスの、E1逆平行形態より大きな比率を示した。継代10までは、観察される唯一のウイルス種は、試験した両方のトランスジーン(hCMV(無イントロン)−FLgag−bGHpAおよびhCMV(無イントロン)−FLgag−SPA)に関してE1平行配向で該トランスジーンを有するアデノベクターであった。
【0170】
継代3および6におけるプラスミド混合物実験#2(図8B)からのウイルス種の分析は、試験したポリアデニル化シグナル(bGHpAおよびSPA)はウイルスの増殖に影響を及ぼさないことを示した。継代10でさえ、該混合物中の2つのウイルス種は尚も等量で存在した。
【0171】
実施例11
増強されたアデノウイルス構築物「MRK Ad5 HIV−1gag」のウイルス産生
該競合研究から得た結果から、本発明者らは以下のとおりに結論づけた:(1)該パッケージングシグナル伸長は有益である;(2)E3の存在はウイルス増殖を増強する;(3)E1平行配向が推奨される;および(4)ポリAシグナルは該アデノウイルスの増殖に影響を及ぼさない。
【0172】
MRK Ad5 HIV−1 gagは最も望ましい結果を示した。この構築物は、新規E3+アデノベクターバックボーンMRKpAdHVE3内にE1平行配向で挿入されたhCMV(無イントロン)−FLgag−bGHpAトランスジーンを含有する。本発明者らは、このアデノベクターをMRK Ad5 HIV−1 gagと命名した。この構築物は以下のとおりに調製した。
【0173】
前プラスミドMRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpAをPac1で消化して該ベクターバックボーンを遊離させ、〜60%コンフルエントのPER.C6(登録商標)細胞を含有する6cmディッシュ中で3.3μgをリン酸カルシウム法(Amersham Pharmacia Biotech)によりトランスフェクトした。CPEが得られたら(7〜10日)、該培養を3回凍結/融解し、該細胞片をペレット化した。1mlのこの細胞ライセートを使用して、80〜90% コンフルエントのPER.C6(登録商標)細胞を含有する6cmディッシュ内に感染させた。CPEが得られたら、該培養を3回凍結/融解し、該細胞片をペレット化した。ついで該細胞ライセートを使用して、80〜90% コンフルエントのPER.C6(登録商標)細胞を含有する15cmディッシュに感染させた。この感染手法を継続し、6継代に拡張した。ついで該ウイルスをCsCl法により該細胞ペレットから抽出した。2回のバンディングを行った(3勾配CsClおよびそれに続く連続的CsCl勾配)。該第2バンディングの後、該ウイルスをA105バッファー中で透析した。プロナーゼ処理およびそれに続くフェノールクロロホルム処理を用いて、ウイルスDNAを抽出した。ついで該ウイルスDNAをHindIIIで消化し、[33P]dATPで放射能標識した。該消化産物を分離するためのゲル電気泳動の後、該ゲルをWatman紙上で乾燥させ、ついでオートラジオグラフィーに付した。該消化産物を、(標識前にPac1/HindIIIで消化された)前プラスミドからの消化産物と比較した。予想されたサイズが観察され、このことは、該ウイルスが成功裏のうちにレスキューされたことを示している。この方法を用いて、調製した種々のアデノベクタープラスミド構築物のそれぞれからウイルスをレスキューした。
【0174】
実施例12
安定性分析
種々のアデノベクター構築物(例えば、MRK Ad5 HIV−1 gag)が遺伝的安定性を示すか否かを判定するために、該ウイルスをそれぞれ連続継代した。該ウイルスDNAを継代3、6および10で分析した。各ウイルスは、その正しい遺伝的構造を維持していた。また、大規模生産で行うのと同様の増殖条件下で該MRK Ad5 HIV−1 gagの安定性を分析した。この分析のために、MRK Ad5 HIV−1 gagおよび3つの他のアデノウイルスベクターのトランスフェクションを繰返し、該ウイルスをP3で精製した。その3つの他のアデノベクターは以下のとおりであった:(1)E3−アデノベクターバックボーン中にbGHpAターミネーターと共にhCMV(無イントロン)−Flgagを含むもの、(2)E3+アデノベクターバックボーン中にSPA終結シグナルと共にhCMV(無イントロン)−Flgagを含むもの、およびE3+アデノベクターバックボーン中にbGHpAターミネーターと共にmCMV−Flgagを含むもの。該ベクターのすべては、E1平行配向で挿入されたトランスジーンを有する。ウイルスDNAを放射性制限分析により分析して、それが適当であることを確認してから、無血清培地内での連続継代のための発酵細胞培養に移した。P5において、それらの4つのウイルスのそれぞれを精製し、該ウイルスDNAを制限消化および放射能標識法による分析用に抽出した。後記のとおり、このウイルスは後に、一連の研究(COS細胞内のインビトロgag発現、げっ歯類での研究およびアカゲザルでの研究)において使用されている。P5からのウイルスを図9に示す。
【0175】
MRKHVE3(MRKpAdHVE3前プラスミドから得られた無トランスジーン)およびMRKAd5HIV−1gag(MRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpA前プラスミド)ウイルスに関して、無血清条件下での継代を継続した。図10は、MRKHVE3、MRKAd5HIV−1gagE3−については継代11での、MRKAd5HIV−1gagについては継代11および12での、放射性制限消化によるウイルスDNA分析を示す。DNAマーカーレーンである第1レーンに加えて、次の3つのレーンは、前プラスミド対照(元のウイルスに基づく対照)、すなわち、それぞれMRKpAdHVE3(「pMRKHVE3」)、MRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpAおよびpMRKAd5gag(E3−)からのウイルスである。図10に示すとおり、該ウイルスDNAサンプルのそれぞれは、示される外来バンドを伴わない予想バンドを示している。このことは、オートラジオグラフィーにより検出されうる主要変異体アデノウイルス種が存在しないことを示している。
【0176】
図11は、MRKHVE3とMRKAd5HIV−1gagとの間のウイルス競合研究の結果を示す。これらのウイルスを等しいMOI(それぞれウイルス粒子140個;合計280 vp)で継代6において互いに混合し、P11まで継代し続けた。該DNAマーカーレーンである第1レーンに加えて、次の2つのレーンは、MRKpAdHVE3およびMRKpAdHVE3+CMV(無イントロン)−FLgag−bGHpAから得られた前プラスミド対照である。次の2つのレーンは、継代6における出発ウイルス物質からのウイルスDNAである。最後の2つのレーンは、二重に行った競合研究である。図11のデータは、培養内でのgagトランスジーンの効果を示している。MRKAd5gagウイルスの増殖を「無トランスジーン」MRKHVE3の増殖と比較した。これらの2つのウイルスを、継代6において、同じMOI(すなわち、それぞれ140vp)で感染させ、ついで継代11まで継代し、該ウイルスプールを放射性制限分析により分析した。該データは、競合において一方のウイルスが他方のウイルスを凌ぐことがなかったことを示している。したがって、該gagトランスジーンは、該アデノウイルスに対する明白な毒性の徴候を示さなかった。
【0177】
HindIII消化による分析は、各ウイルス種がほぼ等量で存在することを示している。前記のとおり、いずれの外来バンドの徴候も存在しないらしい。図12は、血清含有条件下で増殖させたMRKAd5HIV−1gagに関して、より高い継代数を示している。この場合もまた、該ゲノム完全性が維持されており、最高継代レベル(P21)においてさえも再構成の証拠は認められない。
【0178】
図9に示す4つのベクターのそれぞれを、増幅能に関して分析した。以下の表4は、P4におけるウイルス増幅率の評価において用いたQPA分析を示す。元のHIV−1 gag構築物に関する増幅率の測定は、P12の臨床ロットに基づく。増幅率は、元のウイルスに関しては、継代数の増加と共に増加することが示されている。この観察の理由は、無傷アデノベクターと比較して増殖率の増加を示す変異体の出現によるものである。元のAd gagベクターの連続継代により、変異体のレベルは増加し、したがって増幅率も増加する。
【0179】
該MRK Ad5 HIV−1 gagウイルスは、プロセス条件下(すなわち、無血清培地)で連続的に継代されている。継代11および12から抽出されたウイルスDNAは再構成の証拠を示さない。
【0180】
【表3】
実施例13
増強されたAd5構築物の分析的評価
ウイルス増幅に対する該トランスジーンおよびE3遺伝子の効果を調べるために、増強されたアデノウイルスベクターMRK Ad5 HIV−1 gagを、その無トランスジーン形態(MRKpAdHVE3)およびそのE3形態(MRK Ad5 HIV−1 gag E3−)と共に、無血清条件下でいくつかの継代にわたり調べた。表5Aは、MRK Ad5 HIV−1 gagに関して継代P3〜P8にわたり測定した増幅率を示している。あるMOI範囲内では、該ウイルス産生量は該ウイルス投入量に正比例することが確認された。したがって、感染時の細胞当たりのウイルス粒子の数が増えれば増えるほど、産生されるウイルス量は増える。一方、ウイルス増幅率は該ウイルス投入量に反比例する。該ウイルス投入量が少なければ少ないほど、該増幅率は大きくなる。
【0182】
表5Bは、新規E3+ベクターバックボーンMRKpAdHVE3の増幅率を示している。それは、該gagトランスジーン含有形態より有意に低い増幅率を有していた。これは、MRK Ad5 HIV−1 gagが該トランスジーンを含有しているため、より大きなサイズを有することに帰されうる。このように該トランスジーンを含むと、該アデノウイルスのサイズは、野生型Ad5ウイルスのサイズに、より近づく。アデノウイルスは、それがその野生型ゲノムサイズに近い場合に最も良く増幅することが、よく知られている。野生型Ad5は35,935bpである。MRKpAdHVE3は32,905bp長である。増強されたアデノベクターMRK Ad5 HIV−1 gagは35,453bpである(ベクター地図に関しては図14を参照されたい;また、図15A〜Xは、該ベクターバックボーンの追加的な2,021bpを含む完全な前アデノウイルスベクター配列を示す)。
【0183】
表5Cは、新規E3−gag含有ウイルスMRK Ad5 HIV−1 gag E3−の増幅率を示す。この場合もまた、このウイルスは、増強されたアデノウイルスベクターより低い増殖率を示す。これは、野生型Ad5と比較した場合のこのウイルスのサイズの減少(E3遺伝子の欠失による)に帰されうる。該MRK Ad5 HIV−1 gag E3−ウイルスは32,810bp長である。これは、35,935bp長の野生型Ad5および35,453bp長のMRK Ad5 HIV−1 gagに匹敵する。
【0184】
【表4】
実施例14
該新規構築物のgag発現分析
MRK Ad5 HIV−1 gagおよび元のHIV−gagベクター(研究および臨床ロット)のインビトロgag分析は、比較しうるgag発現を示している。該臨床ロットは、少しだけ減少したgag発現レベルを示している。最も注目すべき相違は、mCMVベクターでのものである。このベクターは、試験したその他のベクター(これらはすべてhCMVプロモーターを含有する)と比較して約3倍低い発現レベルを示している。bGHpAを伴うmCMV−FLgagのアッセイは、種々の増殖および精製ロットを使用して3回行ったが、それは一貫して、より弱いgag発現を示した。
【0186】
実施例15
Balc/cマウスにおけるMRK Ad5 HIV−1 gagおよび他のgag含有アデノベクターの評価
10匹のbalb/cマウスのコホートを、漸増量のMRK Ad5 HIV−1 gagならびに元のAd5HIV−gagの研究および臨床ロットで筋肉内にワクチン接種した。血清サンプルを、第1投与の3週間後に集め、抗p24サンドイッチELISAにより分析した。
【0187】
MRK Ad5 HIV−1 gag(107および109vp(ウイルス粒子)の用量)を投与したウスにおける抗p24力価は、初期のアカゲザルのデータの多くが得られたAd5HIV−1 gagの研究ロットのものと比較しうるものであった(図13)。また、E3が欠失している場合(MRKAd5hCMVgagbGHpA(E3−))またはbGHpAターミネーターの代わりにSPAを使用した場合(MRKAd5 hCMV−gag−SPA(E3+))または該MRKAd5バックボーンにおいてhCMVの代わりにマウスCMVプロモーターを使用した場合(MRKAd5 mCMV−gag−bGHpA(E3+))には、これらの力価は比較しうるものであった。
【0188】
表7に示す結果は、好ましいベクターMRK Ad5 HIV−1 gagに加えて、それらの3つの他のベクターも、マウスにおいて強力な抗gag抗体応答を誘導しうることを示している。非常に興味深いことに、bGHpAおよびE3+をE1平行配向で含有するmCMV−Flgag構築物は、試験したその他のベクターと比較してCOS細胞インビトロ感染において最低のgag発現を示したが(表6)、それは、このインビボBalb/c研究において最大の抗gag抗体応答を引き起こした。また、表7は、該研究ロットおよび該臨床ロットの両方における抗gag抗体産生の用量反応関係を示している。予想どおり、該臨床ロットは、該研究ロットに使用したのと同じ用量と比較して、各用量レベルで抗gag抗体誘導の減少を示している。
【0189】
【表5】
【表6】
実施例16
アカゲザルにおける元のAd−gag構築物と新規MRK Ad5 HIV−1 gagとに対する体液性および細胞性応答の比較
3匹のアカゲザルのコホートを、MRK Ad5 HIV−1 gagまたは臨床用Ad5gagバルクで2つの用量(1011vpおよび109vp)で筋肉内にワクチン接種した。免疫は、第0週、4週および25週に行った。血清およびPBMCサンプルを、選択された時点で集めた。該血清サンプルを抗p24Ab力価に関して(競合に基づくアッセイを使用)、そしてgag20マーペプチドプールでの一晩の刺激後に該PBMCを抗原特異的IFNγ分泌に関して(ELISpotアッセイによる)アッセイした。
【0192】
表8に示す結果は、抗gag抗体の生成に関する比較しうる応答を示している。表9に要約した末梢血中のgag特異的T細胞の度数は、新規構築物MRK Ad5 HIV−1 gagを1回投与した後に生じた強力な細胞性免疫応答を示している。該応答はまた、同じベクターの第2投与により増強されうる。該ベクターはまた、細胞傷害性活性を引き起こすCD8+T細胞応答(PBMCのCD4+枯渇後の残りのスポット計数により示される)を誘導しうる。
【0193】
【表7】
【表8】
本明細書に記載のアデノベクター、特にMRK Ad5 HIV−1 gagは、血清中およびより重要なことには無血清培地条件中でのそれらの増強された増殖特性に関して非常に有望なHIV−gagアデノベクターを代表する。現在のHIV−1 gagアデノベクター構築物と比較して、MRK Ad5 HIV−1 gagは、5〜10倍増加した増幅率を示す。本発明者らは、それが継代21において遺伝的に安定であることを示している。この構築物は、比較的低い用量である10∧9vpにおいてさえもインビボで有意な細胞性免疫応答を産生しうる。該MRKAd5gag構築物の効力は、このアカゲザル研究において示されているとおり、元のHIV−1 gagベクターより良くはないとしてもそれと比較しうるものである。
【0196】
実施例17
コドン最適化HIV−1 POLおよびコドン最適化HIV−1 POL修飾
本明細書に開示する種々の合成pol遺伝子のオープンリーディングフレームは、逆転写酵素(またはポリメラーゼおよびRNアーゼH活性よりなるRT)およびインテグラーゼ(IN)のコード配列を含む。該タンパク質配列は、IIIBのクローン単離物であるHxb2rの配列に基づく。この配列はコンセンサス・クレードB配列に最も近く、848個の残基中で16個の不一致残基を有するに過ぎないことが示されている(Korberら,1998,Human retroviruses and AIDS,Los Alamon National Laboratory,Los Alamons,New Mexico)。当業者は、本明細書を精査した後、利用可能な任意のHIV−1またはHIV−2株が、本明細書に開示するHIV pol DNAワクチン構築物の潜在的鋳型を与えると理解するであろう。突然変異不活性化にもかかわらず、いずれかの残留プロテアーゼ活性からの安全性を保証するために、該プロテアーゼ遺伝子が本発明のDNAワクチン構築物から除去されることが、更に注目される。インビボ哺乳類発現を最高にするために、野生型(wt−pol)および不活性化pol(IA−pol)の両方の遺伝子配列の設計においては、該配列内の各アミノ酸残基に関するヒトにとって好ましい(「ヒト化」)コドンの使用が含まれる(Lathe,1985,J.Mol.Biol.183:1−12)。配列番号1、3、5および7におけるコドン使用頻度を調べることにより理解されうるとおり、哺乳類における最適化のためには以下のコドンの使用が好ましい:Met(ATG)、Gly(GGC)、Lys(AAG)、Trp(TGG)、Ser(TCC)、Arg(AGG)、Val(GTG)、Pro(CCC)、Thr(ACC)、Glu(GAG);Leu(CTG)、His(CAC)、Ile(ATC)、Asn(AAC)、Cys(TGC)、Ala(GCC)、Gln(CAG)、Phe(TTC)およびTyr(TAC)。哺乳類(ヒト)におけるコドンの最適化に関して更に考察するためには、WO 97/31115(PCT/US97/02294)(本明細書の他の箇所にも示されているとおり、これを参照により本明細書に組み入れることとする)を参照されたい。当業者は別の形態のコドン最適化を用いたり、あるいは本発明の範囲内のHIV polワクチン構築物の作製の際にはこの工程を省略しうると予想される。したがって、本発明はまた、本明細書に開示するHIV Polタンパク質の種々の野生型および修飾形態をコードするDNA分子および関連組換えアデノウイルスHIVワクチンの非コドン最適化形態に関する。しかし、これらの構築物のコドン最適化が、本発明の好ましい実施形態である。
【0197】
本発明のこの態様の特に好ましい実施形態は、プロテアーゼ(PR)活性をコードするDNA配列が欠失しており、RT(逆転写酵素およびRNアーゼH活性)およびINインテグラーゼ活性をコードするコドン最適化「野生型」配列が残された、wt−pol DNA構築物(本発明においては、「wt−pol」または「wt−pol(コドン最適化)」と称される)をコードするコドン最適化ヌクレオチド配列を含む。このタンパク質をコードするDNA分子は本明細書中には配列番号1として開示されており、該オープンリーディングフレームは、ヌクレオチド10−12の開始Met残基からヌクレオチド2560−2562の終結コドンにわたり含有される。配列番号1は以下のとおりである。
【0198】
【化1】
配列番号1として開示される野生型pol構築物のオープンリーディングフレームは、以下のとおり本明細書中で配列番号2として開示される850アミノ酸を含有する。
【0200】
【化2】
本発明は特に、該プロテアーゼ活性をコードする野生型配列部分の欠失に加えて、該発現タンパク質のHIV−1 pol(RT−RH−IN)活性に有害な活性部位残基突然変異の組合せが導入されたコドン最適化HIV−1 DNA pol構築物を含むアデノウイルスベクターワクチンに関する。したがって、本発明は、好ましくは、アデノウイルスHIV−1 DNA polに基づくワクチンであって、該構築物が、RT、RNアーゼおよび/またはIN活性を少なくとも部分的に、好ましくは実質的に無効にする突然変異を含有し、いずれかのPR活性をコードするDNA配列を欠くことを特徴とする前記ワクチンに関する。アデノウイルスベクターワクチンの一部および一団であるHIV−1 pol突然変異体の1つの型は、HIV−1 PolのRT、RNアーゼHおよび/またはIN機能に関する少なくとも実質的に減少した酵素活性を与えるよう該発現タンパク質のRT、RNアーゼおよび/またはIN領域内の活性部位を有効に改変する点突然変異を引き起こす少なくとも1つのヌクレオチド置換を含む突然変異DNA分子を含みうるが、これに限定されるものではない。本発明のこの態様の好ましい実施形態においては、HIV−1 DNA pol構築物は、RT、RNアーゼHおよびIN活性を有効に無効にするPolコード領域内に突然変異を含有する。特に好ましいHIV−1 DNA pol構築物は、各活性を少なくとも実質的に無効にするようPolのRT、RNアーゼHおよびINドメインの活性部位を改変する少なくとも1つの点突然変異を含有するDNA分子である。そのようなHIV−1 Pol突然変異体は、十中八九、それぞれRT、RNアーゼHおよびIN活性をもたらす各触媒ドメイン内またはその周囲に少なくとも1つの点突然変異を含むであろう。この目的において、特に好ましいHIV−1 DNA pol構築物が本発明において例示され、それは、PR、RT、RNアーゼまたはIN活性を有さない不活性化Polタンパク質(IA Pol:配列番号4、図17A〜C)を与える9個のコドン置換突然変異を含有し、その場合、3個のそのような点突然変異は該RT、RNアーゼおよびIN触媒ドメインのそれぞれの中に存在する。したがって、特に好ましい例は、以下の表1に示す全9個の突然変異を含有する、IA−polをコードするDNA分子を適当な様態で含むアデノウイルスワクチンである。置換のための追加的な好ましいアミノ酸残基は、PolのRNアーゼドメイン内に位置するAsp551である。本明細書に開示する突然変異の任意の組合せが適している可能性があり、したがって、本発明のIA−Polに基づくワクチンとして利用されうる。付加および欠失突然変異が意図され本発明の範囲内にあるが、好ましい突然変異は、野生型アミノ酸から別のアミノ酸残基への置換をもたらす点突然変異である。
【0202】
【表9】
HIV−1 Polの活性部位内またはその周囲のエピトープを改変する可能性が少なくなるよう、本発明のIApol突然変異体ワクチン内に点突然変異を組込むのが好ましい。
【0204】
この目的において、配列番号3は、表1に示す9個の突然変異に加えてコドン最適化polをコードするヌクレオチド配列を開示しており、これは、以下のとおり開示され、本発明において「IApol」と称される。
【0205】
【化3】
本発明のIA−polに基づくアデノウイルスワクチンを製造するために、該酵素サブユニットからの9個の活性部位残基の全てをアラニン側鎖で置換することにより、酵素機能の不活性化を行った。表1に示すとおり、該ポリメラーゼの触媒性3残基、すなわちAsp112、Asp187およびAsp188を含む全ての残基を、アラニン(Ala)残基で置換した(Larderら,Nature 1987,327:716−717;Larderら,1989,Proc.Natl.Acad.Sci.1989,86:4803−4807)。RNアーゼH活性を無効にするために、3つの追加的な突然変異をAsp445、Glu480およびAsp500に導入し(このIA Pol構築物においてはAsp551は不変のままとした)、ここで、各残基を、それぞれAla残基で置換した(Daviesら,1991,Science 252:,88−95;Schatzら,1989,FEBS Lett.257:311−314;Mizrahiら,1990,Nucl.Acids.Res.18:pp.5359−5353)。HIV polインテグラーゼ機能は、Asp626、Asp678およびGlu714における3つの突然変異により無効となった。この場合も、これらの残基のそれぞれをAla残基で置換した(Wiskerchenら1995,J.Virol.69:376−386;Leavittら,1993,J.Biol.Chem.268:2113−2119)。配列番号4のアミノ酸残基Pro3は該RT遺伝子の開始部位を示す。IA−Polの完全なアミノ酸配列は、以下のとおりに本発明においては配列番号4および図17A〜Cとして開示される。
【0207】
【化4】
前記のとおり、前記で開示した突然変異の任意の組合せが適している可能性があり、したがって、単独または組合せ様式法および/または初回−追加法で投与される場合に本発明のIA−polに基づくワクチンとして使用されうる。例えば、それぞれの逆転写酵素、RNアーゼHおよびインテグラーゼコード領域内の3個の残基中の2個だけしか突然変異させないで、尚もこれらの酵素活性を無効にすることが可能でありうる。しかしながら、前記の及び配列番号3として開示されているIA−pol構築物ならびに該発現タンパク質(配列番号4)が好ましい。それらの3つの触媒ドメインのそれぞれにおいて少なくとも1つの突然変異が存在することも好ましい。
【0209】
本発明のこの形態のもう1つの態様は、真核性輸送(トラフィッキング)シグナルペプチド、例えばtPA(組織プラスミノーゲンアクチベーター)由来のもの又はリーダーペプチド(例えば、免疫グロブリンリーダーペプチドのような高度に発現される哺乳類タンパク質内で見出されるもの)を含むコドン最適化HIV−1 Polに基づくワクチン構築物である。任意の機能的リーダーペプチドを、有効性に関して試験することが可能である。しかしながら、本明細書中に示すHIV−1 Nef構築物の場合と同様に本発明の好ましい実施形態は、polコード領域またはその一部がリーダーペプチド、好ましくはヒトtPA由来のリーダーペプチドに作動的に結合した本明細書に開示するHIV−1 Pol突然変異体アデノウイルスワクチン構築物を提供することにある。すなわち、IA−Pol(配列番号4)のようなコドン最適化HIV−1 Pol突然変異体は、該タンパク質のアミノ末端部分にリーダーペプチドをも含むことが可能であり、これは、該宿主細胞内の該発現タンパク質の細胞輸送および従ってその免疫原性に影響を及ぼしうる。図16A〜Bに示すとおり、本発明を実施するために使用しうるDNAベクターを、関心のあるリーダーシグナルペプチドを含有するよう公知組換えDNA法により修飾して、関心のある修飾HIV−1タンパク質の下流クローニングが、修飾HIV−1 tPA/Polタンパク質をコードするヌクレオチド配列を与えるようにすることが可能である。別法においては、前記のとおり、リーダーペプチドをコードするヌクレオチド配列の挿入を、関心のあるPolタンパク質のオープンリーディングフレームを収容するDNAベクター内に挿入することができる。該クローニング方法には無関係に、該最終産物は、関心のある修飾HIV−1 Polタンパク質(リーダーペプチドを含有するHIV−1 Polタンパク質を含むが、これに限定されるものではない)をコードするヌクレオチド配列と共に、有効な遺伝子発現のためのベクター成分を含むポリヌクレオチドワクチンである。本発明で用いるヒトtPAリーダーのアミノ酸配列は以下のとおりである:MDAMKRGLCCVLLLCGAVFVSPSEISS(配列番号17)。したがって、本発明のもう1つの態様は、真核性輸送シグナルペプチド(例えば、tPA由来のもの)を含む、HIV−1 Polに基づくワクチン構築物を製造することにある。この目的において、本発明は、コドン最適化wt−pol DNA構築物をコードするDNA分子であって、該プロテアーゼ(PR)活性が欠失しており、ヒトtPAリーダー配列が該コード領域の5’末端に融合していることを特徴とするDNA分子に関する。このタンパク質をコードするDNA分子は本明細書中には配列番号5として開示されており、該オープンリーディングフレームは本明細書中には配列番号6として開示されている。
【0210】
この目的において、本発明は、コドン最適化wt−pol DNA構築物をコードするDNA分子であって、該プロテアーゼ(PR)活性が欠失しており、ヒトtPAリーダー配列が該コード領域の5’末端に融合していることを特徴とするDNA分子(本発明においては、「tPA−wt−pol」と称される)に関する。このタンパク質をコードするDNA分子は本明細書中には配列番号5として開示されており、該オープンリーディングフレームは、ヌクレオチド8−10の開始Met残基からヌクレオチド2633−2635の終結コドンにわたり含まれる。配列番号5は以下のとおりである。
【0211】
【化5】
配列番号5として開示される野生型pol構築物のオープンリーディングフレームは、以下のとおり本明細書中で配列番号6として開示される875アミノ酸を含有する。
【0213】
【化6】
本発明はまた、宿主細胞内の発現タンパク質の細胞輸送および従ってその免疫原性に影響を及ぼしうるヒトtPAリーダーのようなリーダーペプチドをアミノ末端部分に含む、IA−Pol(配列番号4)のような組換えアデノウイルスベクター内に含有されるコドン最適化HIV−1 Pol突然変異体に関する。前記段落に開示している任意のそのようなアデノウイルスに基づくHIV−1 DNA pol突然変異体は、ヒトtPAリーダー配列を含む(これに限定されるものではない)リーダーペプチドのようなリーダーペプチドの下流における融合に適している。したがって、任意のそのようなリーダーペプチドに基づくHIV−1 pol突然変異体構築物は、HIV−1 PolのRT、RNアーゼHおよび/またはIN機能の1以上に関する少なくとも実質的に減少した酵素活性を与えるよう該発現タンパク質のRT、RNアーゼおよび/またはIN領域の触媒活性を有効に改変する突然変異体DNA分子を含みうるが、これに限定されるものではない。本発明のこの形態の好ましい実施形態においては、リーダーペプチド/HIV−1 DNA pol構築物は、RT、RNアーゼHおよびIN活性を有効に無効にする該Polコード領域内に突然変異を含有する。特に好ましいHIV−1 DNA pol構築物は、各活性を少なくとも実質的に無効にするよう(好ましくは、完全に無効にするよう)PolのRT、RNアーゼHおよびINドメイン内の活性部位および触媒活性を改変する少なくとも1つの点突然変異を含有するDNA分子である。そのようなHIV−1 Pol突然変異体は、十中八九、それぞれRT、RNアーゼHおよびIN活性をもたらす各触媒ドメイン内またはその周囲に少なくとも1つの点突然変異を含むであろう。本発明のこの形態の特に好ましい実施形態は、表1に示す9個の突然変異を含むIA−Polタンパク質に融合したヒトtPAリーダーに関する。該DNA分子は本明細書中には配列番号7として開示されており、該発現tPA−IA Polタンパク質は、図18に示すとおりの融合結合部を含む。該発現タンパク質の完全なアミノ酸配列を配列番号8に示す。この目的において、配列番号7は、表1に示す9個の突然変異を含むIA Polタンパク質に融合したヒトtPAリーダーをコードするヌクレオチド配列(本発明においては、「tPA−opt−IApol」と称される)を開示する。該オープンリーディングフレームは、開始Met(ヌクレオチド8−10)で始まり、ヌクレオチド2633−2635の「TAA」コドンで終結する。tPA−IAPolをコードするヌクレオチド配列はまた、以下のとおりに開示される。
【0215】
【化7】
配列番号7として開示されるtPA−IA−pol構築物のオープンリーディングフレームは、以下のとおりにtPA−IA−Polおよび配列番号8として本明細書中に開示される875アミノ酸を含有する。
【0217】
【化8】
実施例18
コドン最適化HIV−1 NEFおよびコドン最適化HIV−1 NEF修飾体
HIV−1 NefおよびHIV−1 Nef修飾体のコドン最適化形態は、実質的には、2000年12月15日付け出願の米国特許出願第09/738,782号および同様に2000年12月15日付け出願のPCT国際出願PCT/US00/34162(両方の文書を参照により本明細書に組み入れることとする)に記載のとおりである。前記文書に開示されているとおり、コドン最適化NefおよびNef修飾体の特定の実施形態は、HIV−1 jfrl分離体からのHIV−1 NefをコードするDNA分子に関する。この場合、該コドンは、ヒトのような哺乳類系内での発現に関して最適化されている。このタンパク質をコードするDNA分子は本発明では配列番号9として開示されており、発現されるオープンリーディングフレームは本発明では配列番号10として開示されている。本発明のアデノウイルスベクターにおける使用のためのNefに基づくコード領域のもう1つの実施形態は、該HIV−1 NefポリペプチドのNH2末端に融合したヒトプラスミノーゲンアクチベーター(tpa)リーダーペプチドを含有するタンパク質をコードするコドン最適化DNA分子を含む。このタンパク質をコードするDNA分子は本発明では配列番号11として開示されており、発現されるオープンリーディングフレームは本発明では配列番号12として開示されている。もう1つの修飾されたNef最適化コード領域は、最適化HIV−1 NefをコードするDNA分子に関する。この場合、該オープンリーディングフレームは、アミノ末端ミリスチル化部位における修飾(Gly−2からAla−2)、およびLeu−174−Leu−175ジロイシンモチーフからAla−174−Ala−175への置換をコードする(本発明ではopt nef(G2A,LLAA)として記載されている)。このタンパク質をコードするDNA分子は本発明では配列番号13として開示されており、発現されるオープンリーディングフレームは本発明では配列番号14として開示されている。更なる実施形態は、最適化HIV−1 NefをコードするDNA分子に関する。この場合、アミノ末端ミリスチル化部位およびジロイシンモチーフが欠失しており、tPAリーダーペプチドを含む。このDNA分子opt tpanef(LLAA)は、HIV−1 Nef(jfrl)のアミノ酸残基6−216に融合したtPAリーダー配列を含有するNefタンパク質をコードするオープンリーディングフレームを含み、ここで、Leu−174およびLeu−175はAla−174およびAla−175で置換されており、これは本発明ではopt tpanef(LLAA)と称され、本発明では配列番号15として開示されており、発現されるオープンリーディングフレームは本発明では配列番号16として開示されている。
【0219】
前記文書(米国特許出願第09/738,782号およびPCT国際出願PCT/US00/34162)に開示されており本明細書中で繰返し記載されているとおり、それぞれのオープンリーディングフレームを含むnefに基づく以下のヌクレオチドおよびアミノ酸配列は以下のとおりである。
【0220】
1.コドン最適化形態のHIV−1 jrfl nef遺伝子のヌクレオチド配列は、以下に示すとおり本発明では配列番号9として開示されている。
【0221】
【化9】
好ましいコドンの使用は以下のとおりである:Met(ATG)、Gly(GGC)、Lys(AAG)、Trp(TGG)、Ser(TCC)、Arg(AGG)、Val(GTG)、Pro(CCC)、Thr(ACC)、Glu(GAG);Leu(CTG)、His(CAC)、Ile(ATC)、Asn(AAC)、Cys(TGC)、Ala(GCC)、Gln(CAG)、Phe(TTC)およびTyr(TAC)。哺乳類(ヒト)におけるコドンの最適化に関して更に考察するためには、WO 97/31115(PCT/US97/02294)(これを参照により本明細書に組み入れることとする)を参照されたい。また、HIV−Nefのオープンリーディングフレームを含む野生型対コドン最適化ヌクレオチドの比較に関しては図19〜Bを参照されたい。
【0223】
前記の配列番号9のオープンリーディングフレームは、ヌクレオチド12−14の開始メチオニン残基およびヌクレオチド660−662の「TAA」終結コドンを含む。配列番号9のオープンリーディングフレームは、コドン最適化DNAワクチンベクターの使用により発現される216アミノ酸のHIV−1 Nefタンパク質を与える。その216アミノ酸のHIV−1 Nef(jfrl)タンパク質は、本発明では配列番号10として開示されており、以下のとおりである。
【0224】
【化10】
HIV−1 Nefは、Gly−2のミリスチル化を介して宿主細胞膜の内部表面と会合する216アミノ酸のサイトゾルタンパク質である(Franchiniら,1986,Virology 155:593−599)。すべての考えられうるNef機能が解明されているわけではないが、細胞内膜へのNefの適切な輸送は、HIV−1の生活環の初期相を促進するために宿主の細胞内環境を改変することにより及び後代ウイルス粒子の感染性を増加させることにより、ウイルス複製を促進することが明らかになっている。コドンが最適化されたタンパク質修飾ポリペプチドに関する本発明の1つの態様においては、本発明のアデノウイルスベクターのnefコード領域を修飾して、異種リーダーペプチドをコードするヌクレオチド配列を含有させて、発現タンパク質のアミノ末端領域が該リーダーペプチドを含有するようにする。真核細胞を典型づける機能の多様性は、その膜境界の構造的相違に依存する。これらの構造を生成し維持するためには、タンパク質が、小胞体内のその合成部位から該細胞の全体にわたる所定の目的地へ輸送される必要がある。このためには、主要輸送経路へのアクセスポイントに位置する経路選択をもたらす分子装置により認識されるソーティングシグナルを該輸送タンパク質が示すことが必要である。ほとんどのタンパク質についてのソーティング決定は、それらがそれらの生合成経路を通過するにつれて1回だけ行われる必要がある。なぜなら、それらの最終目的地、すなわち、それらがそれらの機能を果たす細胞位置は、それらの永久的な存在位置となるからである。細胞内完全性の維持は、1つには、タンパク質の選択的ソーティングおよびその正しい目的地への正確な輸送に依存する。タンパク質中には、「住所標識」として機能しうる一定の配列モチーフが存在する。多数のソーティングシグナルは、膜タンパク質の細胞質ドメインと会合していることが判明している。CTL応答の有効な誘導は、しばしば、抗原の持続的な高レベルの内因性発現を必要とした。ミリスチル化を介した膜会合は、ほとんどのNef機能のための必須要件であるため、本明細書に記載のとおりのグリシンからアラニンへの変化、ジロイシンモチーフの変化および/またはtpaリーダー配列での置換によりミリスチル化を欠く突然変異体は機能的に欠損しており、したがって、HIV−1ワクチン成分として使用するための、野生型Nefと比較して改善された安全性プロフィールを有するであろう。
【0226】
本発明のこの態様のもう1つの実施形態においては、ヒト組織特異的プラスミノーゲンアクチベーター(tPA)リーダーを含むよう、該DNAベクターまたは該HIV−1 nefヌクレオチド配列を修飾する。図16A〜Bに示すとおり、DNAベクターを、関心のあるリーダーシグナルペプチドを含有するよう公知組換えDNA法により修飾して、関心のある修飾HIV−1タンパク質の下流クローニングが、修飾HIV−1 tPA/Nefタンパク質をコードするヌクレオチド配列を与えるようにすることが可能である。別法においては、前記のとおり、リーダーペプチドをコードするヌクレオチド配列の挿入を、関心のあるNefタンパク質のオープンリーディングフレームを収容するDNAベクター内に挿入することができる。該クローニング方法には無関係に、該最終産物は、リーダーペプチドを含有するHIV−1 Nefタンパク質を含む(これに限定されるものではない)関心のある修飾HIV−1 Nefタンパク質をコードするヌクレオチド配列と共に、有効な遺伝子発現のためのベクター成分を含むポリヌクレオチドワクチンである。本発明で用いるヒトtPAリーダーのアミノ酸配列は以下のとおりである:MDAMKRGLCCVLLLCGAVFVSPSEISS(配列番号17)。
【0227】
該タンパク質のカルボキシ領域内のジロイシンモチーフと結びついたGly−2のミリスチル化は、エンドサイトーシスを介したCD4のNef誘導性ダウンレギュレーションに必須であることが示されている(Aikenら,1994,Cell 76:853−864)。また、Nef発現は、エンドサイトーシスを介したMHCIのダウンレギュレーションを促進することが示されている(Schwartzら,1996,Natue Medicine 2(3):338−342)。本発明は、1つには、輸送および/または機能特性において改変した修飾Nefタンパク質をコードするDNAワクチンに関する。本発明のアデノウイルスベクターHIVワクチン内に導入される修飾は、アミノ末端リーダーペプチドを含む修飾Nefタンパク質の発現をもたらすnefオープンリーディングフレームに対する付加、欠失または置換、アミノ末端ミリスチル化部位の修飾または欠失、およびNefタンパク質内のジロイシンモチーフの修飾または欠失のうち感染宿主細胞内の機能を改変するものを含むが、これらに限定されるものではない。したがって、本発明のDNA分子および組換えアデノウイルスHIVワクチンの中心的主題は、(1)nefに基づくコドン最適化アデノウイルスHIVワクチンの宿主投与および細胞内運搬、(2)CTL応答およびTh応答の両方を惹起する点で免疫原性である修飾Nefタンパク質の発現、ならびに(3)感染宿主内でのHIV−1の複製および負荷(load)を促進することが示されているNefの公知初期ウイルス機能の抑制または少なくとも改変にある。したがって、別のアミノ酸残基を発現するようアミノ末端Gly−2ミリスチル化残基が欠失または修飾された修飾Nefタンパク質を発現するDNAワクチンを与えるよう、該nefコード領域を改変することができる。また、別のアミノ酸残基を発現するようジロイシンモチーフが欠失または修飾された修飾Nefタンパク質を発現するDNAワクチンを与えるよう、該nefコード領域を改変することができる。さらに、本発明のアデノウイルスベクターHIVワクチンはまた、野生型または修飾Nefタンパク質(本明細書に記載のとおりのもの)、例えば、Gly2の欠失または置換、Leu174およびLeu175の欠失または置換および/またはリーダー配列の含有を含む修飾Nefタンパク質(これに限定されるものではない)を発現する、コドン使用頻度には無関係な単離されたDNA分子に関する。
【0228】
したがって、特定のNefに基づく構築物は更に、非限定的な例示として以下のものを含む。例えば、本発明は、HIV−1の修飾形態をコードするアデノウイルスベクターワクチンに関する。HIV−1 Nef(jfrl)のアミノ酸残基6−216に融合したtPAリーダー配列を含むNefタンパク質をコードするオープンリーディングフレームは、opt tpanefと称される。opt tpanefのオープンリーディングフレームを含むヌクレオチド配列は、本発明では配列番号11として開示されており、以下のとおりである。
【0229】
【化11】
配列番号11のオープンリーディングフレームは、ヌクレオチド2−4の開始メチオニン残基、およびヌクレオチド713−715の「TAA」終結コドンを含む。配列番号3のオープンリーディングフレームは、アミノ酸残基174および175のジロイシンモチーフを含む(これに限定されるものではない)HIV−1 Nefのアミノ酸6−216に融合したtPAリーダー配列を含む237アミノ酸のHIV−1 Nefタンパク質を与える。この237アミノ酸のtPA/Nef(jfrl)融合タンパク質は、本発明では配列番号12として開示されており、以下に示されるとおりである。
【0231】
【化12】
したがって、この例示されているNefタンパク質Opt tPA−Nefは、ヒトのような哺乳類系内での発現に関してコドンが最適化されたHIV−1 jfrl分離体からのHIV−1 NefをコードするGly−2A DNA分子のミリスチル化部位の欠失およびtPAリーダー配列の両方を含有する。
【0233】
本発明のもう1つの特定の実施形態においては、組換えアデノウイルスHIVワクチンのオープンリーディングフレームがアミノ末端ミリスチル化部位における修飾(Gly−2からAla−2)およびLeu−174−Leu−175ジロイシンモチーフからAla−174−Ala−175への置換をコードしている最適化HIV−1 NefをコードするDNA分子を開示する。このオープンリーディングフレームは本発明ではopt nef(G2A,LLAA)として記載され、配列番号13として開示され、それは、ヌクレオチド12−14の開始メチオニン残基およびヌクレオチド660−662の「TAA」終結コドンを含む。前記の修飾を有するHIV−1 jfrl nef遺伝子のこのコドン最適化形態のヌクレオチド配列は、本発明では以下のとおりの配列番号13として開示されている。
【0234】
【化13】
配列番号13のオープンリーディングフレームは、本発明では以下のとおりの配列番号14として開示されるNef(G2A,LLAA)をコードしている。
【0236】
【化14】
本発明の更なる実施形態は、アミノ末端ミリスチル化部位およびジロイシンモチーフが欠失しておりtPAリーダーペプチドを含む最適化HIV−1 Nefをコードするもう1つのDNA分子に関する。このDNA分子opt tpanef(LLAA)は、HIV−1 Nef(jfrl)のアミノ酸残基6−216に融合したtPAリーダー配列を含有しLeu−174およびLeu−175がAla−174およびAla−175(このtPAに基づく融合タンパク質ではAla−195およびAla−196)で置換されたNefタンパク質をコードするオープンリーディングフレームを含む。opt tpanef(LLAA)のオープンリーディングフレームを含むヌクレオチド配列は、本発明では以下のとおりの配列番号15として開示されている。
【0238】
【化15】
本発明では配列番号16として開示されるtPA−Nef(LLAA)をコードする配列番号7のオープンリーディングフレームは、以下のとおりである。
【0240】
【化16】
本発明のアデノウイルスベクターは、野生型または修飾Nefタンパク質(本明細書に記載のとおり)、例えば、Gly2の欠失または置換、Leu174およびLeu175の欠失または置換、および/またはリーダー配列の付加を含む修飾Nefタンパク質(これに限定されるものではない)を発現する、コドン使用頻度とは無関係のDNA配列を含みうる。したがって、部分的または完全にコドンが最適化されたDNAワクチン発現ベクター構築物が好ましい。なぜなら、そのような構築物は宿主発現の増加をもたらすはずだからである。しかし、本明細書に開示する構築物の「非コドン最適化」形態、特に、宿主投与後に実質的な細胞性免疫応答を促進することが示されているHIV Nefの修飾形態を使用することも、本発明の範囲内である。
【0242】
図20A〜Cは、nef発現ベクターV1Jns/nef(図20A)、V1Jns/nef(G2A,LLAA)(図20B)、V1Jns/tpanef(図20C)およびV1Jns/tpanef(LLAA)(同様に図20C)のプラスミドバックボーンとnefコード配列との間の結合部のヌクレオチド配列を示す。コドン最適化nefまたはコドン最適化nef突然変異遺伝子の5’および3’フランキング配列が太字/イタリック体で示されており、nefおよびnef突然変異体コード配列が無修飾文字で示されている。また(下線付きで)、それぞれのnef発現ベクターの構築に関わる制限エンドヌクレアーゼ部位が示されている。V1Jns/tpanefおよびV1Jns/tpanef(LLAA)は該結合部位に同一配列を有する。
【0243】
図21は、nefおよびnef誘導体の概要図を示す。Nef誘導体に含まれるアミノ酸残基が示されている。グリシン2ならびにロイシン174および175は、それぞれミリスチル化およびジロイシンモチーフに関わる部位である。
【0244】
実施例19
MRKAd5polの構築およびウイルスレスキュー
ベクター:シャトルプラスミドおよび前アデノウイルスプラスミドの構築
MRKAd5polと称される前アデノウイルスプラスミドを含む該ベクターの構築における鍵工程を、図22に示す。簡単に説明すると、完全長不活性化HIV−1 pol遺伝子用のアデノウイルスシャトルベクターは以下のとおりである。ベクターMRKpdeE1(Pac/pIX/pack450)+CMVmin+BGHpA(str.)は、MEKAd5gagアデノウイルス前プラスミドの構築に使用するシャトルベクターの誘導体である。該ベクターは、hCMVプロモーター(無イントロンA)とウシ成長ホルモンポリアデニル化シグナルとを有する発現カセットを含有する。唯一のBglII部位における選択された遺伝子の挿入が、MRKpAd5(E1−/E3+)Cla1(またはMRKpAdHVE3)前プラスミド内への挿入の場合に、該トランスジーンの転写の方向がAd5 E1に平行となることを保証するよう、該発現単位は該シャトルベクター内に挿入されている。該ベクターは、元のシャトルベクターと同様に、PacI部位、パッケージングシグナル領域への伸長、およびpIX遺伝子への伸長を含有する。合成完全長コドン最適化HIV−1 pol遺伝子をプラスミドpV1Jns−HIV−pol−inact(opt)から直接単離した。BglIIでのこのプラスミドの消化は、無傷のpol遺伝子(配列番号3に記載のコドン最適化IA pol配列を含む)を遊離する。該pol断片をゲル精製し、MRKpdelE1(Pac/pIX/pack450)+CMVmin+BGHpA(str.)シャトルベクター内にBglII部位で連結した。制限酵素DraIII/Not1を使用することにより、該遺伝子の配向が正しいか否かに関して該クローンを調べた。陽性クローンを単離し、MRKpdel+hCMVmin+FL−pol+bGHpA(s)と命名した。このプラスミドの遺伝的構造を、PCR、制限酵素およびDNA配列決定により確認した。該前アデノウイルスプラスミドは以下のとおりに構築した。シャトルプラスミドMRKpdel+hCMVmin+FL−pol+bGHpA(S)を制限酵素Pac1およびBst1107I(またはそのアイソシゾマーであるBstZ107I)で消化し、ついで線状化(Cla1消化)アデノウイルスバックボーンプラスミドMRKpAd(E1−/E3+)Cla1と共に大腸菌(E.coli)株BJ5183内に同時形質転換した。元々はMRKpAd+hCMVmin+FL−pol+bGHpA(S)E3+と称されていた得られた前プラスミドを、新たに「pMRKAd5pol」と改称した。得られたpMRKAd5polの遺伝的構造を、PCR、制限酵素およびDNA配列分析により確認した。予備的産生のために、該ベクターをコンピテント大腸菌(E.coli)XL−1 Blue内に形質転換した。回収されたプラスミドを、制限酵素消化およびDNA配列分析により、ならびに一過性トランスフェクション細胞培養内の該polトランスジーンの発現により確認した。このpMRKAd5HIV−1polアデノウイルスベクターの完全なヌクレオチド配列を図26A〜AOに示す。
【0245】
研究等級組換えアデノウイルスの作製
前アデノウイルスプラスミドpMRKAd5polを、PER.C6(登録商標)接着単層細胞培養内で感染ビリオンとしてレスキューした。感染性ウイルスをレスキューするために、12μgのpMRKAd5polを制限酵素PacI(New England Biolabs)で消化し、リン酸カルシウム共沈技術(Cell Phect Transfection Kit,Amersham Pharmacia Biotech Inc.)を用いてPER.C6(登録商標)の6cmディッシュ当たり3.3μgをトランスフェクトした。PacI消化はプラスミド配列からウイルスゲノムを遊離して、PER.C6(登録商標)細胞内への侵入後にウイルス複製を生じさせる。トランスフェクションの6〜10日後、完全なウイルス細胞変性効果(CPE)が観察された後で、感染細胞および培地を回収した。感染細胞および培地を−60℃以下で保存した。このpol含有組換えアデノウイルスは本発明では「MRKAd5pol」と称される。この組換えアデノウイルスは、配列番号6に示すとおりの不活性化HIV−1 Polタンパク質を発現する。
【0246】
実施例20
MRKAd5Nefの構築およびウイルスレスキュー
ベクター:シャトルプラスミドおよび前アデノウイルスプラスミドの構築
MRKAd5nefと称される前アデノウイルスプラスミドを含む該ベクターの構築における鍵工程を、図23に示す。簡単に説明すると、前記実施例19に示したとおり、ベクターMRKpdeE1(Pac/pIX/pack450)+CMVmin+BGHpA(str.)は、MEKAd5gagアデノウイルス前プラスミドの構築に使用するシャトルベクターである。それは、PacI部位、パッケージングシグナル領域への伸長、およびpIX遺伝子への伸長を含有するよう修飾されている。それは、hCMVプロモーター(無イントロンA)とウシ成長ホルモンポリアデニル化シグナルとを有する発現カセットを含有する。唯一のBglII部位における選択された遺伝子の挿入が、MRKpAd5(E1−/E3+)Cla1前プラスミド内への挿入の場合に、該トランスジーンの転写の方向がAd5 E1に平行となることを保証するよう、該発現単位は該シャトルベクター内に挿入されている。合成完全長コドン最適化HIV−1 nef遺伝子をプラスミドpV1Jns/nef(G2A,LLAA)から直接単離した。BglIIでのこのプラスミドの消化は、配列番号13に開示するヌクレオチド配列を含む無傷のpol遺伝子を遊離する。該nef断片をゲル精製し、MRKpdelE1+CMVmin+BGHpA(str.)シャトルベクター内にBglII部位で連結した。制限酵素Sca1を使用することにより、該遺伝子の配向が正しいか否かに関して該クローンを調べた。陽性クローンを単離し、MRKpdelE1hCMVminFL−nefBGHpA(s)と命名した。このプラスミドの遺伝的構造を、PCR、制限酵素およびDNA配列決定により確認した。該前アデノウイルスプラスミドは以下のとおりに構築した。シャトルプラスミドMRKpdelE1hCMVminFL−nefBGHpA(s)を制限酵素Pac1およびBst1107I(またはそのアイソシゾマーであるBstZ107I)で消化し、ついで線状化(Cla1消化)アデノウイルスバックボーンプラスミドMRKpAd(E1−/E3+)Cla1と共に大腸菌(E.coli)株BJ5183内に同時形質転換した。元々はMRKpdelE1hCMVminFL−nefBGHpA(s)と称されていた得られた前プラスミドを、新たに「pMRKAd5nef」と改称した。得られたpMRKAd5nefの遺伝的構造を、PCR、制限酵素およびDNA配列分析により確認した。予備的産生のために、該ベクターをコンピテント大腸菌(E.coli)XL−1 Blue内に形質転換した。回収されたプラスミドを、制限酵素消化およびDNA配列分析により、ならびに一過性トランスフェクション細胞培養内の該nefトランスジーンの発現により確認した。このpMRKAd5HIV−1nefアデノウイルスベクターの完全なヌクレオチド配列を図27A〜AMに示す。
【0247】
研究等級組換えアデノウイルスの作製
前アデノウイルスプラスミドpMRKAd5nefを、PER.C6(登録商標)接着単層細胞培養内で感染ビリオンとしてレスキューした。感染性ウイルスをレスキューするために、12μgのpMRKAd5nefを制限酵素PacI(New England Biolabs)で消化し、リン酸カルシウム共沈技術(Cell Phect Transfection Kit,Amersham Pharmacia Biotech Inc.)を用いてPER.C6(登録商標)の6cmディッシュ当たり3.3μgをトランスフェクトした。Pac1消化はプラスミド配列からウイルスゲノムを遊離して、PER.C6(登録商標)細胞内への侵入後にウイルス複製を生じさせる。トランスフェクションの6〜10日後、完全なウイルス細胞変性効果(CPE)が観察された後で、感染細胞および培地を回収した。感染細胞および培地を−60℃以下で保存した。ここで、このnef含有組換えアデノウイルスは、「MRKAd5nef」と称される。
【0248】
実施例21
不活性化PolおよびNef/G2A,LLAA用のマウスCMVプロモーター含有シャトルベクターの構築
プライマーセット:mCMV(NotI)フォワード5’−ATA AGA ATG CGG CCG CCA TAT ACT GAG TCA TTA GG−3’(配列番号20);mCMV(Bgl II)リバース:5’−AAG GAA GAT CTA CCG ACG CTG GTC GCG CCT C−3’(配列番号21)を使用して、プラスミドpMH4(Frank Graham,McMaster Universityから供給された)からマウスCMV(mCMV)を増幅した。該下線付きヌクレオチドは、各プライマーのそれぞれNotIおよびBglII部位を表す。該トランスジーンをE1平行配向で含有するmCMVシャトルベクターの構築のために、このPCRアンプリコンを使用した。NotIおよびBglIIでの消化により、元のシャトルベクター(hCMV−gag−bGHpAトランスジーンをE1平行配向で含有する)からhCMVプロモーターを除去した。mCMVプロモーター(NotI/BglII消化PCR産物)を該シャトルベクター内に一定方向で挿入した。ついで該シャトルベクターをBglIIで消化し、gagレポーター遺伝子(BglII断片)を該シャトルベクター内に再挿入した。いくつかのクローンを、該レポーター遺伝子の方向の正しさに関してスクリーニングした。E1逆平行配向のmCMV−gagの構築のためには、プライマーセット:mCMV(AscI)フォワード5’−ATA AGA ATG GCG CGC CAT ATA CTG AGT CAT TAG G(配列番号22);mCMV(BglII)リバース:5’AAG GAA GAT CTA CCG ACG CTG GTC GCG CCT C(配列番号23)を使用して、プラスミドpMH4からmCMVプロモーターを増幅した。該下線付きヌクレオチドは、各プライマーのそれぞれAscIおよびBglII部位を表す。hCMV−gagトランスジーンをE1逆平行配向で含有するシャトルベクターをAsc1およびBgl11で消化して、該トランスジーンのhCMV−gag部分を除去した。mCMVプロモーター(Asc1/Bgl11消化PCR産物)を該シャトルベクター内に一定方向で挿入した。ついで該ベクターをBgl11で消化し、該gagレポーター遺伝子(Bgl11断片)を再挿入した。いくつかのクローンを、該レポーター遺伝子の配向の正しさに関してスクリーニングした。それぞれの完全長IAおよび完全長nef/G2A,LLAA遺伝子に関しては、該mCMV−bGHpAシャトルベクター内の唯一のBglII部位を使用して、クローニングを行った。該polおよびnef遺伝子を、BglII消化により、それらのそれぞれのpV1Jnsプラスミドから切り出した。
【0249】
実施例22
mCMV完全長不活性化Polおよび完全長nef/G2A.LLAAアデノベクターの構築
実施例21のこれらのトランスジーンのそれぞれを該修飾シャトルベクター内にE1平行およびE1逆平行の両方の配向で挿入した。各シャトルベクターのPac1およびBstZ110I消化を行い、フランキングAd5配列を含有する各特異的トランスジーン断片を単離し、ClaIで消化されたMRKpAd5(E3+)またはMRKpAd5(E3−)アデノベクタープラスミドと共に、BJ5183大腸菌(E.coli)細胞内で細菌相同組換えにより同時形質転換した。ついで、種々のトランスジーンをE3−およびE3+の両方の形態(ならびにE1平行およびE1逆平行配向)で含有する組換え前プラスミドアデノベクターを、XL−1 Blue大腸菌(E.coli)細胞内への形質転換の後で大規模で調製し、制限分析および配列決定により分析した。
【0250】
実施例23
hCMV−tpa−nef(LLAA)アデノベクターの構築
プライマーセット:Tpanef(BamHI)F5’−ATT GGA T CC ATG GAT GCA ATG AAG AGA GGG(配列番号24);Tpanef(BamHI)R5’−ATA GGA TCC TTA GCA GTC CTT GTA GTA CTC G(配列番号25)を使用して、GMP等級pV1Jns−tpanef(LLAA)ベクターからtpa−nef遺伝子を増幅した。得られたPCR産物をBamHIで消化し、ゲル精製し、MRKAd5CMV−bGHpAシャトルベクター(BglIIで消化され仔ウシ腸ホスファターゼで処理されたもの)のBglII部位内にクローニングした。hCMVプロモーターに対して正しい配向でtpanef(LLAA)遺伝子(完全なコード領域に関しては配列番号15を参照されたい)を含有するクローンを、ScaI消化後に選択した。得られたMRKAd5tpanefシャトルベクターをPacIおよびBstZ1101で消化し、細菌相同組換え技術によりE3+MRKAd5アデノベクター内にクローニングした。
【0251】
実施例24
MRKAd5polおよびMRKAd5nefワクチンの免疫原性
材料および方法−げっ歯類への免疫
N=10のBALB/cマウスの群の筋肉内(i.m.)に以下のベクターで免疫した:(1)10∧7vpおよび10∧9vpのうちのいずれかのMRKAd5hCMV−IApol(E3+)、ならびに(2)10∧7vpおよび10∧9vpのうちのいずれかのMRKAd5hCMV−IApol(E3−)。投与の7週間後、10匹/コホートのうちの5匹のマウスを、それらが最初に投与されたのと同じベクターおよび用量で追加抗原刺激した。該第2投与の3週間後、それらのすべての動物から血清および脾臓をそれぞれRT ELISAおよびIFNg ELIspot分析のために集めた。げっ歯類への全ての免疫のために、該Ad5ベクターを5mM Tris、5% スクロース、75mM NaCl、1mM MgCl2、0.005% ポリソルベート80(pH8.0)中で希釈した。28−1/2G針付きの0.3mL インスリンシリンジ(Becton−Dickinson,Franklin Lakes,NJ)を使用して、全用量を50μM アリコート中で両方の方形筋に注射した。
【0252】
N=10のC57/BL6マウスの群の筋肉内(i.m.)に以下のベクターで免疫した:(1)10∧7vpおよび10∧9vpのうちのいずれかのMRKAd5hCMV−nef(G2A,LLAA)(E3+)、(2)10∧7vpおよび10∧9vpのうちのいずれかのMRKAd5mCMV−nef(G2A,LLAA)(E3+)、ならびに(3)10∧7vpおよび10∧9vpのうちのいずれかのMRKAd5mCMV−tpanef(LLAA)(E3+)。投与の7週間後、10匹/コホートのうちの5匹のマウスを、それらが最初に投与されたのと同じベクターおよび用量で追加抗原刺激した。該第2投与の3週間後、それらのすべての動物から血清および脾臓をそれぞれRT ELISAおよびIFNg ELIspot分析のために集めた。
【0253】
非ヒト霊長類への免疫
3匹のアカゲザル(2〜3kg)のコホートを以下のAdベクターでワクチン接種した:(1)10∧9vpおよび10∧11vp用量のうちのいずれかのMRKAd5hCMV−IApol(E3+)、ならびに(2)10∧9vpおよび10∧11vpのうちのいずれかのMRKAd5hCMV−IApol(E3−)、(3)10∧9vpおよび10∧11vpのうちのいずれかのMRKAd5hCMV−nef(G2A,LLAA)(E3+)、ならびに(4)10∧9vpおよび10∧11vpのうちのいずれかのMRKAd5mCMV−nef(G2A,LLAA)(E3+)。該ワクチンを化学的拘束サル(10mg/kg ケタミン)に、2つの0.5mLアリコートの該Adベクター(5mM Tris、5% スクロース、75mM NaCl、1mM MgCl2、0.005% ポリソルベート80(pH8.0)中)の両三角筋への針注射により投与した。該動物は、4週間隔で2回免疫した(T=0、4週)。
【0254】
マウス抗RTおよび抗nefELISA
標準的な二次抗体に基づくELISAにより、抗RT力価を得た。PBS中の100μLの1μg/mL HIV−1 RTタンパク質(Advanced Biotechnologies,Columbia,MD)と共に一晩インキュベートすることにより、Maxisorpプレート(NUNC Rochester,NY)をコートした。該プレートを、Titertek MAP装置(Hunstville,AL)を使用してPBS/0.05% Tween 20で洗浄し、200μL/ウェルのブロッキング溶液(PBS/0.05% tween/1% BSA)と共に2時間インキュベートした。100倍の初期血清希釈を行い、ついで4倍の系列希釈を行った。系列希釈サンプルの100μL アリコートをウェルごとに加え、室温で2時間インキュベートした。該プレートを洗浄し、100μLの1/1000希釈HRPウサギ抗マウスIgG(ZYMED,San Francisco,CA)を、1時間インキュベートしながら加えた。該プレートを十分に洗浄し、100μL 1,2−フェニレンジアミン二塩酸塩/過酸化水素(DAKO,Norway)溶液に15分間浸した。ウェル当たり100μLの0.5M H2SO4を加えることにより、該反応をクエンチした。S20スタッカと共にTitertek Multiskan MCC/340を使用して、OD492の読み取り値を記録した。終点力価は、0.1 OD492(該バックグラウンド値の2.5倍)以上の吸光度値を与える最高血清希釈度と定義した。
【0255】
非ヒト霊長類およびマウスELIspotアッセイ:
酵素結合イムノスポット(ELISpot)アッセイを用いて、アカゲザルPBMCまたはマウス脾臓からの抗原特異的INFγ分泌細胞(Miyahiraら,1995,J.Immunol.Methods 1995,181,45−54)を計数した。マウス脾臓を5匹/コホートのマウスからプールし、8mLの完全RPMI培地(RPMI1640,10% FBS,2mM L−グルタミン,100U/mL ペニシリン,100u/mL ストレプトマイシン,10mM Hepes,50uM β−ME)中、5×106/mLで単細胞懸濁液を調製した。標準的なフィコール勾配分離(Coliganら,1998,Current Protocols in Immunology.John Wiley & Sons,Inc.)により、アカゲザルPBMCを8〜15mLのヘパリン化血から調製した。Multiscreen不透明プレート(Millipore,France)をPBS中の100μL/ウェルの5μg/mL 精製ラット抗マウスIFN−γ IgG1、クローンR4−6A2(Pharmingen,San Diego,CA)または15μg/mL マウス抗ヒトINF−γ IgG2a(Cat.No.1598−00,R&D Systems,Minneapolis,MN)で、それぞれマウスまたはサルでのアッセイのために4℃で一晩にわたりコートした。該プレートを、PBS/ペニシリン/ストレプトマイシンで洗浄し、200μL/ウェルの完全RPMI培地で37℃で少なくとも2時間にわたりブロッキングした。
【0256】
各ウェルに、50μLの細胞サンプル(4〜5×105細胞/ウェル)および50μLの該抗原溶液を加えた。該対照ウェルには、50μLのDMSO含有培地を加え、特異的応答のために、選択されたペプチドまたはペプチドプール(ペプチドの最終濃度ごとに4μg/mL)を加えた。該pol構築物で免疫したBALB/cマウスに関しては、CD4+−エピトープ含有20量体ペプチド(aa21−40,aa411−430,aa641−660,aa731−750,aa771−790)のプールまたはCD8+−エピトープ含有ペプチド(aa201−220,aa311−330,aa781−800)を使用して刺激を行った。該nef構築物で免疫したC57/BL6マウスに関しては、nef配列に由来するaa51−70(CD8+T細胞エピトープ)またはaa81−100(CD4+)ペプチドを特異的刺激のために加えた。サルにおいては、全pol配列を含み10アミノ酸で重複する20−aaペプチドの2つのプール(LおよびR)を使用して、polに対する応答を評価した。該nef構築物でワクチン接種したサルにおいては、全HIV−1 nef配列を含み10aaで重複する20量体ペプチドを含有する単一のプールを使用した。各サンプル/抗原混合物は、マウスサンプルについては三重ウェル中で、アカゲザルPBMCについては二重ウェル中で実施された。プレートを37℃、5% CO2、90% 湿度で20〜24時間インキュベートした。該プレートをPBS/0.05% Tween 20で洗浄し、100μL/ウェルの1.25μg/mL ビオチン共役ラット抗マウスIFN−γmAb、クローンXMG1.2(Pharmingen)または0.1μg/mL ビオチン化抗ヒトIFNγヤギポリクローナル抗体(R&D Systems)と共に4℃で一晩インキュベートした。該プレートを洗浄し、PBS/0.005% Tween/5% FBS中、100μL/ウェルの1/2500希釈度のストレプトアビジン−アルカリホスファターゼ共役体(Pharmingen)と共に37℃で30分間インキュベートした。洗浄後、100μl/ウェルの1工程(1−step)NBT/BCIP(Pierce Chemicals)と共に6〜10分間インキュベートすることにより、スポットを現像した。該プレートを水で洗浄し、風乾した。解剖顕微鏡を使用して各ウェル内のスポットの数を測定し、該データを106個の細胞の入力に対して正規化した。
【0257】
非ヒト霊長類抗RT ELISA
該サルにおけるpol特異的抗体を競合RT EIAアッセイで測定し、この場合、プレートウェル上のコート抗体への結合からRT抗原を遮断する能力によりサンプルの活性を測定した。簡単に説明すると、Mxisorpプレートを飽和量のpol陽性ヒト血清(#97111234)でコートした。250μLの各サンプルを15μLの266ng/mL RT組換えタンパク質(RCM 563,1% BSA,0.1% トゥイーン,0.1% NaN3中)および20μLの細胞溶解バッファー(Coulter p24抗原アッセイキット)と共に室温で15分間インキュベートする。最大RT結合を定める陰性対照および標準の系列希釈サンプルを使用して、同様の混合物を調製する。200μL/ウェルの各サンプルおよび標準を該洗浄プレートに加え、該プレートを室温で16〜24時間インキュベートした。結合RTを、Coulter p24アッセイキットに記載の方法に従い定量し、選択された標準により任意に定められる1mL当たりのミリ・メルク(milliMerck)単位で表す。
【0258】
結果−げっ歯類での研究
BALB/cマウス(n=マウス5匹/コホート)を種々の用量のMRKAd5dhCMV−IApol(E3+)およびMRKAd5hCMV−IApol(E3−)で1回または2回免疫した。該第2投与の3週間後、代替抗原としてRTを用いるELISAアッセイにより、抗pol IgGレベルを測定した。polエピトープ含有ペプチドのプールに対するINFγELISpotアッセイにより、細胞性応答を定量した。これらのアッセイの結果を表10に要約する。該結果は、該ワクチン接種マウスが、僅か10∧7vpのアデノベクター用量で、検出可能な抗RT IgGを示している。該体液性応答は高度に用量依存的であり、第2免疫で増強可能である。1または2用量のpolベクターは、高度数の抗原特異的CD4+およびCD8+T細胞を惹起し、該応答は、低度に用量依存的であるが、第2免疫で増強可能である。
【0259】
【表10】
C57/BL6マウスを、種々の用量のMRKAd5hCMV−nef(G2A,LLAA)(E3+)、MRKAd5mCMV−nef(G2A,LLAA)(E3+)で10∧7vpにて、および(3)MRKAd5mCMV−tpanef(LLAA)(E3+)で10∧7vpおよび10∧9vpのうちのいずれかで免疫した。同様のプロトコールを用いて該免疫応答を分析し、結果を表11に示す。このモデル系においては該構築物のいずれにおいても抗nef IgG応答を検出することができなかったが、ELIspotアッセイによれば、nefに対して生成した細胞性免疫の強力な指標が存在する。
【0261】
【表11】
サルでの研究
3匹のアカゲザルのコホートを2用量のMRKAd5hCMV−IApol(E3+)およびMRKAd5hcMV−IApol(E3−)で免疫した。全pol配列を含む2つのペプチドプール(LおよびR)のうちの一方を使用して、抗原特異的T細胞の数(百万個のPBMC当たり)を計数した。結果を表12に示す。いずれかの構築物を10∧9vpの低用量で使用した場合にも、ワクチン被接種体において中等度〜強力なT細胞応答が検出された。該動物における抗RT抗体力価の縦断的分析は、体液性応答を惹起するよう該polトランスジーン産物が効率的に発現されることを示唆している(表13)。E3−構築物が投与された動物においてはE3+ウイルスの場合と比較して、一般に、より高い免疫応答が観察されるようであった。
【0263】
【表12】
【表13】
アカゲザルを2用量のMRKAd5nef構築物で筋肉内に免疫した場合には、12匹中8匹のワクチン被投与体において、百万当たり100〜1100の範囲の強力なT細胞応答が観察された(表14)。これまでに得られたデータに基づけば、mCMVおよびhCMVにより駆動されるnef構築物の効力は比較しうるものである。
【0266】
【表14】
実施例25
HIV感染対象におけるクレードBとクレードCとのT細胞応答の比較
米国においてHIV−1に感染した24人の患者から集めたPBMCサンプルを、10アミノ酸により重複する20量体ペプチドのペプチドプールを使用するELISPOTアッセイにおいて試験した。4個の異なるペプチドプールを交差クレード認識に関して試験した。それらは、クレードBに基づく分離体(gag H−b;nef−b)またはクレードCに基づく分離体(gag H−c,nef−c)から誘導された。表15のデータは、恐らくクレードB HIV−1に感染したこれらの患者からのT細胞が、ELISPOTアッセイにおいてクレードC gagおよびnef抗原を認識しうることを示している。相関分析は更に、クレードC gagペプチドプールに対するこれらのT細胞応答がクレードB対応物の約60%であり、クレードC nefに対するT細胞応答がクレードB対応物の約85%であることを示した(図25)。これらの結果は、クレードB HIV−1に感染した患者において生成した細胞性免疫応答がクレードC HIV−1由来のgagおよびnef抗原を認識しうることを示唆している。これらのデータは、クレードB gagおよび/またはnef抗原を発現するDNAまたはMRKAdに基づくワクチンのようなHIVワクチンが、地球規模での予防的および/または治療的利点をもたらす能力を潜在的に有することを示している。
【0268】
【表15】
実施例26
ローラーボトルにおけるのMRKAd5polおよびMRKAd5nefベクターの特徴づけおよび生産
nefおよびpolアデノベクターの増殖
nefおよびpol CsCl精製MRKAd5種(シード)を使用してローラーボトルに感染させて、更なる実験の種(シード)として使用するP4ウイルスを製造した。P4 MRKAd5 polおよびnefベクターを使用してMOI 280vp/細胞でローラーボトルに感染させた。ただし、hCMV−tpa−nef[E3+]は例外で、これは、P4で得た種の力価が低かったため125のMOIで感染させた。
【0270】
【表16】
ローラーボトル継代
polおよびnef構築物の継代を継代7まで継続した。全継代において得られた細胞結合(凍結/融解細胞溶解)および全ブロス(トリトン細胞溶解)力価は非常に一貫したものであった。一般には、MRKAd5polはMRKA5gagの約70%生産性であり、MRKA5nefはMRKAd5gagの約25%生産性であった。両構築物のP7ウイルスのサンプルを制限消化分析によるV&CBにより分析し、それらはいずれの再構成をも示さなかった。
【0272】
【表17】
【表18】
MRKAd5nefおよびMRKAd5polウイルス生産速度論
MRKAd5polおよびMRKAd5nefベクターのウイルス生産速度論がMRKAd5gagのものに類似しているか否かを判定するために、ローラーボトル内で経時的実験を行った。ローラーボトル培養内のPER.C6(登録商標)細胞に、P5 MRKAd5pol、P5 MRKAd5nefおよびP7 MRKAd5gagを280vp/細胞のMOIで感染させた。各アデノベクターに関して、感染の24、36、48および60時間後、2つの感染ボトルからサンプルを採取した。また、I相(Phase I)法条件下で回収する場合には、48hpiまで、2つのボトルをサンプル未採取のまま残した。24、36、48および60hpiの時点における凍結融解回収細胞結合ウイルスの陰イオン交換HPLCウイルス粒子濃度を図29A〜Bに示す。QPA力価は、類似した傾向を示している(データは示していない)。
【0275】
hCMVおよびmCMV−FL−nefの比較
MRKAd5nef構築物(hCMV−FL−nef)で得られた力価は、MRKAd5gagまたはMRKAd5polで得られたものより低かったため、ウイルス生産性比較実験はmCMV−FL−nefで行った。2つのアデノベクター(hCMV−およびmCMV−FL−nef)のそれぞれに関して、2つのローラーボトルに継代5の清澄化ライセートを280vp/細胞のMOIで感染させた。それらの4つのローラーボトルの肉眼的および顕微鏡的観察は、回収の時点で同じであった。得られた清澄化ライセートの分析は、表19に示すとおり、mCMV−FL−nefに感染したボトル内の、より高いウイルス粒子濃度を示した。mCMVプロモーターにより駆動されるnefベクターでの、より高い生産性は、PER.C6(登録商標)細胞における、より低いnef発現レベルによるものと認められる。nef発現レベルを測定するための実験がV&CBで進行中である。
【0276】
【表19】
実施例27
バイオリアクターにおけるMRKAd5nefウイルスの特徴づけおよび大規模生産
材料および方法
本実施例の実験は、以下の条件下で2回実施した:36.5℃、DO 30%、pH7.30、150rpm攪拌速度、散布無し、Life Technologies(Gibco Invitrogen)293 SFM II(6mM L−グルタミン含有)、pH制御用の塩基としての0.5M NaOH。第1の実施(B20010115)においては、2つの10L 攪拌容器バイオリアクターにPER.C6(登録商標)細胞を0.2×106細胞/mlの濃度で接種した。細胞を、約1×106細胞/mlの細胞濃度に達するまで増殖させた。該細胞に未クローン化MRKAd5nef(G2A,LLAA)を280ウイルス粒子(vp)/細胞で感染させた。第2バッチ(B20010202)には、該細胞にクローン化MRAd5nefを感染させたこと以外は該第1実施と同じ方法を用いた。両方の実施中、該バイオリアクターを感染の48時間後に回収した。サンプルを採取し、全ブロス(トリトン細胞溶解を伴う)、上清および細胞ペレット(3×凍結/融解)から、AEXおよびQPAアッセイでウイルス濃度を測定した。該方法の全体にわたり、BioProfile250で代謝産物を測定した。
【0278】
【表20】
【表21】
結果−表22および表23は、MRKAd5nefのバイオリアクター増殖によるスケールアップ生産の能力を示す。
【0281】
【表22】
【表23】
本発明の範囲は、本明細書に記載の特定の実施形態により限定されるものではない。実際のところ、前記の説明から、本明細書に記載されているものに加えて本発明の種々の修飾が当業者に明らかとなるであろう。そのような修飾も、特許請求の範囲の範囲内に含まれると意図される。
【0284】
実施例28
DNA初回抗原刺激動物のMRKAdHIV−1gag追加抗原刺激
3〜5匹のアカゲザルの群を、第0、4および8週に、(a)5mgのV1Jns−Flgag(pVIJnsCMV(無イントロン)−FL−gag−bGHpA)、(b)45mgの非イオン性ブロック共重合体CRL1005で製剤化された5mgのV1Jns−Flgag、または(c)7.5mgのCRL1005および0.6mM 塩化ベンザルコニウムで製剤化された5mgのV1Jns−Flgagで免疫した。すべての動物に、1回量の10e7 ウイルス粒子(vp)の該MRKAd5HIV−1gagを第26週に投与した。注:単一様式での投与の場合には(用量は、アデノウイルスへの前曝露を模擬するよう選択される)、10e7は余りにも低すぎて、有効に初回抗原刺激または追加抗原刺激することができない;図32を参照されたい。
【0285】
血液サンプルを全動物から幾つかの時点で集め、末梢血単核細胞(PBMC)を、標準的なフィコール法を用いて調製した。該PBMCを計数し、ELISpotアッセイを用いてγインターフェロン分泌に関して分析した(表24)。各サルに関して、全HIV−1 gag配列を含む50個の20アミノ酸長ペプチドのプールの不存在下(培地)または存在下(「gag H」と称される)、該PBMCを一晩インキュベートした。
【0286】
結果は、これらのサルにおけるT細胞免疫応答の増強においてMRKAd5HIV−1gagが非常に有効であったことを示している。該ウイルス追加抗原刺激の28週間後または2週間後、百万個のPBMC当たりのgag特異的T細胞数は、該追加抗原刺激の24週間前または2週間前に観察されたレベルと比較して2〜48倍増加した。
【0287】
また、該MRKAd5gag追加抗原刺激の前(第10週)および後(第30週)に、PBMCを細胞内γインターフェロン染色により分析した。選択した動物に関する結果を図31に示す。結果は、(a)DNA/アジュバント製剤での免疫が、平衡的、CD4+偏向的またはCD8+偏向的でありうるT細胞応答を惹起したこと、および(b)MRKAd5gag構築物での追加刺激が、すべての場合において、強くCD8+偏向した応答を引き起こしたことをを示している。これらの結果は、MRKAd5HIV−1gag構築物での追加刺激が抗原特異的CD8+T細胞のレベルを改善しうることを示唆している。
【0288】
【表24】
実施例29
MRKAd5gagpol融合構築物の構築
コドン最適化HIV−1 gag遺伝子のオープンリーディングフレームを、段階的PCRにより、IA pol遺伝子(RT、RNアーゼおよびインテグラーゼドメインよりなる)のオープンリーディングフレームに直接的に融合させた。該遺伝子(配列番号38)は該プロテアーゼ遺伝子およびフレームシフト配列を含まないため、それは、p55、RT、RNアーゼHおよびインテグラーゼが組合されたサイズ(1350アミノ酸;配列番号39)の単一ポリペプチドをコードしている。
【0290】
該gag遺伝子内のBstEII部位から最後の非終結コドンまで伸長する断片を、該IApolの開始コドンから唯一のBamHI部位まで伸長する断片にPCRにより連結した。この断片をBstEIIおよびBamHIで消化した。gag−IApol融合体の構築は、PstI−BstEII gag消化断片、BstEII/BamHI消化PCR産物および長いPstI/BamHI/V1R−FLpolバックボーン断片を含む3断片連結により達成された。
【0291】
該MRKAd5−gagpolアデノウイルスベクターは、gag−IApol融合遺伝子の全ORFを含有するV1R−gagpolのBglII断片を使用して構築した。
【0292】
実施例30
非ヒト霊長類における免疫原性研究
3匹のアカゲザルのコホートを、10e8または10e10ウイルス粒子(vp)の以下のMRKAd5 HIV−1ワクチンの1つで免疫した:(1)MRKAd5gag;(2)MRKAd5pol;(3)MRKAd5nef;(4)等量のMRKAd5gag、MRKAd5polおよびMRKAd5nefを含有する混合物、または(5)等量のMRKAd5gagpolおよびMRKAd5nefの混合物。
【0293】
該HIV−1抗原のそれぞれに対するT細胞応答を、各抗原の全タンパク質配列を含む20−aaのペプチドを使用するIFNγELISpotアッセイによりアッセイした。結果(表25)は、該ペプチドプールのそれぞれに応答する百万個の末梢血単核細胞(PBMC)当たりのスポット形成細胞(sfc)の数として表されている。
【0294】
結果は以下の観察を示している:(1)該単一遺伝子構築物(MRKAd5gag,MRKAd5polまたはMRKAd5nef)のそれぞれは、サルにおいて、高レベルの抗原特異的T細胞を惹起しうる;(2)該単一遺伝子MRKAd5構築物は、gag、polおよびnefに対して非常に広範なT細胞応答を惹起しうる多カクテル製剤として混合されうる;(3)gagとIA polとの融合タンパク質を発現するMRKAd5ベクターは、gagおよびpolの両方に対して強力なT細胞応答を誘導しうる。
【0295】
【表25】
【図面の簡単な説明】
【図1】
元のHIV−1 gagアデノベクター(Ad5HIV−1gag)を示す。
【図2】
該最適化ヒトHIV−1 gagオープンリーディングフレームの核酸配列(配列番号29)を示す。
【図3】
元のgagトランスジーンと比較して新規トランスジーン構築物を図示する。
【図4】
本発明の新規ベクターの作製において元のアデノベクターバックボーンに施された修飾を示す。
【図5】
該パッケージングシグナル領域に対して施された付加(実験#1)およびウイルス増殖に対するE3遺伝子(実験2)の効果を判定するために行ったウイルス混合実験を示す。
【図6】
実施例6および7に記載するウイルス混合実験後のウイルスDNA分析のオートラジオグラフを示す。
【図7A】
MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたhCMV−Flgag−bGHpAアデノベクターを示す。
【図7B】
MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたhCMV−Flgag−SPAアデノベクターを示す。
【図7C】
MRKpAdHVE3およびMRKpAdHVOアデノベクターバックボーン内に構築されたmCMV−Flgag−bGHpAアデノベクターを示す。
【図8A】
トランスジーン配向の効果を試験するよう意図された実験を示す。
【図8B】
ポリアデニル化シグナルの効果を試験するよう意図された実験を示す。
【図9】
BstE11消化後のP5における試験された4つのアデノウイルスベクターからのウイルスDNA(実施例12)を示す。
【図10】
MRKpAdHVE3、MRKAd5HIV−1gagおよびMRKAd5HIV−1gagE3−の継代11および12のウイルスDNA分析を示す。
【図11】
ウイルス競合研究を開始するために使用した継代6のMRKpAdHVE3およびMRKAd5HIV−1gagのウイルスDNA分析(HindIII消化)を示す。
【図12】
特定された時点で回収が行われた血清含有培地中のMRKAd5HIV−1gagに関する高継代数上のHindIII消化によるウイルスDNA分析を示す。
【図13】
種々の用量のいくつかのAdgag構築物でbalb/cマウス(n=10)をi.m.免疫した3週間後の血清抗p24レベルを示す:(A)MRK Ad5 HIV−1 gag(継代5)、(B)MRKAd5hCMV−FLgag−bGHpA(E3−)、(C)MRKAd5 hCMV−FLgag−SPA(E3+)、(D)MRKAd5 mCMV−FLgag−bGHpA(E3+)、(E)Ad5HIV−1 gagの研究ロット(293細胞由来)、および(F)Ad5HIV−1 gagの臨床ロット(Ad5gagFN0001)。示されているのは、各コホートの幾何平均力価(GMT)および標準誤差線である。
【図14】
pMRKAd5HIV−1 gagベクターの制限地図を示す。
【図15A】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15B】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15C】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15D】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15E】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15F】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15G】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15H】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15I】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15J】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15K】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15L】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15M】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15N】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15O】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15P】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15Q】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15R】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15S】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15T】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15U】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15V】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15W】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図15X】
pMRKAd5HIV−1gagベクター(配列番号27[コード化]および配列番号28[非コード化])のヌクレオチド配列を示す。
【図16】
DNAワクチン発現ベクターV1Jns(A)およびV1Jns−tPA(B)の概要図を示す。
【図17A】
IA−Polのヌクレオチド(配列番号3)およびアミノ酸配列(配列番号4)を示す。
【図17B】
IA−Polのヌクレオチド(配列番号3)およびアミノ酸配列(配列番号4)を示す。
【図17C】
IA−Polのヌクレオチド(配列番号3)およびアミノ酸配列(配列番号4)を示す。
【図18】
tPA−pol inact(opt)の融合結合部にわたるコドン最適化ヌクレオチドおよびアミノ酸配列(それぞれ配列番号7および8中に含有される)を示す。
【図19A】
野生型nef(jrfl)とコドン最適化nefとのヌクレオチド配列比較を示す。
【図19B】
野生型nef(jrfl)とコドン最適化nefとのヌクレオチド配列比較を示す。
【図20】
nef発現ベクターV1Jns/nef(図20A)、V1Jns/nef(G2A,LLAA)(図20B)、V1Jns/tpanef(図20C)およびV1Jns/tpanef(LLAA)(同様に図20C)のプラスミドバックボーンとnefコード配列との間の結合部のヌクレオチド配列を示す。
【図21】
nefおよびnef誘導体の概要図を示す。
【図22】
前アデノウイルスプラスミド構築物MRKAd5Polの構築の概要図を示す。
【図23】
前アデノウイルスプラスミド構築物MRKAd5Nefの構築の概要図を示す。
【図24】
クレードB HIV感染個体におけるクレードBとクレードCとの抗gag T細胞応答の比較を示す。
【図25】
クレードB HIV感染個体におけるクレードBとクレードCとの抗nef T細胞応答の比較を示す。
【図26A】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26B】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26C】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26D】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26E】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26F】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26G】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26H】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26I】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26J】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26K】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26L】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26M】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26N】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26O】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26P】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26Q】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26R】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26S】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26T】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26U】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26V】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26W】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26X】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26Y】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26Z】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AA】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AB】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AC】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AD】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AE】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AF】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AG】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AH】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AI】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AJ】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AK】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AL】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AM】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AN】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図26AO】
不活性化pol遺伝子(配列番号3)のコード領域を含むpMRKAd5HIV−1polアデノウイルスベクターのヌクレオチド配列(配列番号32[コード化]および配列番号33[非コード化])を示す。
【図27A】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27B】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27C】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27D】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27E】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27F】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27G】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27H】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27I】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27J】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27K】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27L】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27M】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27N】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27O】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27P】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27Q】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27R】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27S】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27T】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27U】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27V】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27W】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27X】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27Y】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27Z】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AA】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AB】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AC】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AD】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AE】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AF】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AG】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AH】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AI】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AJ】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AK】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AL】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図27AM】
不活性化pol遺伝子(配列番号13)のコード領域を含むpMRKAd5HIV−1 nefアデノウイルスベクターのヌクレオチド配列(配列番号34[コード化]および配列番号35[非コード化])を示す。
【図28】
E3(+)またはE3(−)バックボーン中に種々のプロモーター断片(hCMVまたはmCMV)および終結シグナル(bGHまたはSPA)を含むMRKAd5ベクターの安定性を示す。
【図29A】
24、36、48および60hpiの時点における凍結融解回収細胞結合ウイルスの陰イオン交換HPLCウイルス粒子濃度を示す。
【図29B】
MRKAd5gag、MRKAd5polおよびMRKA5nefに関する経時的QPA上清力価を示す。
【図30A】
本明細書に開示するDNAおよび/またはアデノウイルスワクチンにおいて使用する代表的なtPA−gag融合体のオープンリーディングフレームをオープンリーディングフレーム含むヌクレオチド配列(配列番号36)およびアミノ酸配列(配列番号37)を示す。
【図30B】
本明細書に開示するDNAおよび/またはアデノウイルスワクチンにおいて使用する代表的なtPA−gag融合体のオープンリーディングフレームをオープンリーディングフレーム含むヌクレオチド配列(配列番号36)およびアミノ酸配列(配列番号37)を示す。
【図31】
第10週(DNA初回抗原刺激後)および第30週(Ad追加抗原刺激後)に集めたPBMCの細胞内γIFN染色を示す。
【図32】
単一様式アデノウイルス免疫とDNA+アジュバント初回/アデノウイルス追加免疫との比較を示す。
【図33A】
実施例29のgag−IApol融合体のオープンリーディングフレームのヌクレオチド配列(配列番号38)を示す。
【図33B】
実施例29のgag−IApol融合体のオープンリーディングフレームのヌクレオチド配列(配列番号38)を示す。
【図34A】
該gag−IApol融合フレームのタンパク質配列(配列番号39)を示す。
【図34B】
該gag−IApol融合フレームのタンパク質配列(配列番号39)を示す。
Claims (89)
- E1において少なくとも部分的に欠失していてE1活性を欠き、
a)野生型アデノウイルスゲノムの塩基対約1からと塩基対約400から塩基対約458までとの間に対応するアデノウイルス・シス作用性パッケージング領域、および
b)HIVタンパク質またはその免疫学的に関連する修飾体をコードする遺伝子、
を含んでなる、組換えアデノウイルスワクチンベクター。 - 野生型アデノウイルスゲノムの塩基対約1から塩基対約450までに対応するパッケージング領域を含む、請求項1記載のベクター。
- 野生型アデノウイルスゲノムの塩基対約3511から約3524までからと塩基対約5798までとの間に対応するヌクレオチドを更に含む、請求項1記載のベクター。
- 野生型アデノウイルスゲノムの1〜450からと3511〜5798までとの間に対応する塩基対を含む、請求項3記載のベクター。
- 塩基対451〜3510が欠失している、請求項4記載のベクター。
- E3において少なくとも部分的に欠失している、請求項1記載のベクター。
- 該E3欠失領域が塩基対28,133〜30,818である、請求項6記載のベクター。
- 該HIVタンパク質またはその修飾体をコードする遺伝子が、ヒトにおける発現に関して最適化されたコドンを含む、請求項1記載のベクター。
- 該ベクターが、
a)タンパク質をコードする核酸と、
b)該タンパク質をコードする核酸に作動的に結合した異種プロモーターと、
c)転写終結配列と
を含む遺伝子発現カセットを含む、請求項1記載のベクター。 - 該遺伝子発現カセットが該E1領域内に挿入されている、請求項9記載のベクター。
- 該遺伝子発現カセットがE1に平行な配向で存在する、請求項9記載のアデノウイルスベクター。
- 該遺伝子発現カセットがE1に逆平行な配向で存在する、請求項9記載のアデノウイルスベクター。
- 該プロモーターが、イントロン配列を欠くサイトメガロウイルスプロモーターである、請求項9記載のアデノウイルスベクター。
- 該プロモーターが最初期ヒトサイトメガロウイルスプロモーターである、請求項13記載のアデノウイルスベクター。
- 該プロモーターがマウスサイトメガロウイルスプロモーターである、請求項9記載のアデノウイルスベクター。
- 該転写終結配列がウシ成長ホルモンポリアデニル化および転写終結配列である、請求項9記載のアデノウイルスベクター。
- 該転写終結配列が合成ポリアデニル化シグナル(SPA)である、請求項9記載のアデノウイルスベクター。
- 請求項1記載のアデノウイルスベクターを含んでなる細胞。
- アデノウイルスE1タンパク質を相補的レベルで発現する細胞系内への請求項1記載のアデノウイルスベクターのトランスフェクションの後に回収され精製された組換え複製欠損型アデノウイルス粒子。
- 請求項19記載の精製されたアデノウイルス粒子を含んでなるHIVワクチン組成物。
- 生理的に許容される担体を含む、請求項20記載のHIVワクチン組成物。
- アデノウイルスE1タンパク質を発現する宿主細胞内に該アデノウイルスベクターを導入し、得られた組換え複製欠損型アデノウイルスを回収することを含んでなる、請求項1記載のアデノウイルスベクターのアデノウイルスゲノムを含有する組換え複製欠損型アデノウイルス粒子の製造方法。
- 該細胞がPER.C6(登録商標)細胞である、請求項22記載の製造方法。
- 請求項21記載のワクチンを該個体に投与することを含んでなる個体においてHIVに対する細胞性免疫応答を生成させる方法。
- DNAプラスミドワクチンを該個体に投与することを更に含み、この場合、それと共に、所望により、該免疫応答を増強しうる生物学的に有効なアジュバント、タンパク質または他の物質を投与しうる、請求項24記載の方法。
- アデノウイルスワクチンの投与の前に該DNAプラスミドワクチンを該個体に投与する、請求項25記載の方法。
- 異なる血清型のアデノウイルスワクチンが該アデノウイルスワクチンに先行する、請求項24記載の方法。
- 該アデノウイルスワクチンベクターを該個体に投与および再投与することを含む、請求項24記載の方法。
- 該HIVタンパク質がHIV gagまたはその免疫学的に関連した修飾体である、請求項1記載のアデノウイルスベクター。
- 該遺伝子発現カセットが、HIV gagタンパク質またはその免疫学的に関連した修飾体をコードするオープンリーディングフレームを含む、請求項9記載のアデノウイルスベクター。
- E1において少なくとも部分的に欠失していてE1活性を欠き、
a)野生型アデノウイルスゲノムの塩基対約451から塩基対約3510までに対応する塩基対が欠失している、野生型アデノウイルスゲノムの塩基対約1から塩基対約450までおよび約3511から約5798までに対応するアデノウイルス・シス作用性パッケージング領域、および
b)i)配列番号29と、
ii)i)に作動的に結合した異種プロモーターと、
iii)転写終結配列と、を含む遺伝子発現カセット、
を含んでなる組換えアデノウイルスワクチンベクター。 - 該遺伝子発現カセットがE1に平行な配向で存在する、請求項31記載のアデノウイルスベクター。
- 該遺伝子発現カセットがE1に逆平行な配向で存在する、請求項31記載のアデノウイルスベクター。
- 該プロモーターが、イントロン配列を欠くサイトメガロウイルスプロモーターである、請求項31記載のアデノウイルスベクター。
- 該転写終結配列がウシ成長ホルモンポリアデニル化および転写終結配列である、請求項31記載のアデノウイルスベクター。
- E3において少なくとも部分的に欠失した、請求項31記載のアデノウイルスベクター。
- 請求項30記載のアデノウイルスベクターを含んでなる細胞。
- アデノウイルスE1タンパク質を相補的レベルで発現する細胞系内への請求項30記載のアデノウイルスベクターのトランスフェクションの後に回収され精製された組換え複製欠損型アデノウイルス粒子。
- 請求項38記載の精製されたアデノウイルス粒子を含んでなるHIVワクチン組成物。
- 生理的に許容される担体を含む、請求項39記載のHIVワクチン組成物。
- アデノウイルスE1タンパク質を発現する宿主細胞内に該アデノウイルスベクターを導入し、得られた組換え複製欠損型アデノウイルスを回収することを含んでなる、請求項30記載のアデノウイルスベクターのアデノウイルスゲノムを含有する組換え複製欠損型アデノウイルス粒子の製造方法。
- 該細胞がPER.C6(登録商標)細胞である、請求項41記載の製造方法。
- 請求項21記載のワクチンを該個体に投与することを含んでなる、個体においてHIVに対する細胞性免疫応答を生成させる方法。
- DNAプラスミドワクチンを該個体に投与することを更に含み、この場合、それと共に、所望により、該免疫応答を増強しうる生物学的に有効なアジュバント、タンパク質または他の物質を投与しうる、請求項43記載の方法。
- アデノウイルスワクチンの投与の前に該DNAプラスミドワクチンを該個体に投与する、請求項44記載の方法。
- 異なる血清型のアデノウイルスワクチンが該アデノウイルスワクチンに先行する、請求項43記載の方法。
- 該アデノウイルスワクチンベクターを該個体に投与および再投与することを含む、請求項43記載の方法。
- 該HIVタンパク質がHIV polまたはその免疫学的に関連した修飾体である、請求項1記載のアデノウイルスベクター。
- 該遺伝子発現カセットが、HIV polタンパク質またはその免疫学的に関連した修飾体をコードするオープンリーディングフレームを含む、請求項9記載のアデノウイルスベクター。
- E1において少なくとも部分的に欠失していてE1活性を欠き、
a)野生型アデノウイルスゲノムの塩基対約451から塩基対約3510までに対応する塩基対が欠失している、野生型アデノウイルスゲノムの塩基対約1から塩基対約450までおよび約3511から約5798までに対応するアデノウイルス・シス作用性パッケージング領域、および
b)i)配列番号1、配列番号5および配列番号7よりなる群から選ばれるヌクレオチド配列と、
ii)i)に作動的に結合した異種プロモーターと、
iii)転写終結配列と、を含む遺伝子発現カセット、
を含んでなる組換えアデノウイルスワクチンベクター。 - 該遺伝子発現カセットがE1に平行な配向で存在する、請求項50記載のアデノウイルスベクター。
- 該遺伝子発現カセットがE1に逆平行な配向で存在する、請求項50記載のアデノウイルスベクター。
- 該プロモーターが、イントロン配列を欠くサイトメガロウイルスプロモーターである、請求項50記載のアデノウイルスベクター。
- 該転写終結配列がウシ成長ホルモンポリアデニル化および転写終結配列である、請求項50記載のアデノウイルスベクター。
- E3において少なくとも部分的に欠失した、請求項50記載のアデノウイルスベクター。
- 請求項49記載のアデノウイルスベクターを含んでなる細胞。
- アデノウイルスE1タンパク質を相補的レベルで発現する細胞系内への請求項49記載のアデノウイルスベクターのトランスフェクションの後に回収され精製された組換え複製欠損型アデノウイルス粒子。
- 請求項57記載の精製されたアデノウイルス粒子を含んでなるHIVワクチン組成物。
- 生理的に許容される担体を含む、請求項58記載のHIVワクチン組成物。
- アデノウイルスE1タンパク質を発現する宿主細胞内に該アデノウイルスベクターを導入し、得られた組換え複製欠損型アデノウイルスを回収することを含んでなる、請求項49記載のアデノウイルスベクターのアデノウイルスゲノムを含有する組換え複製欠損型アデノウイルス粒子の製造方法。
- 該細胞がPER.C6(登録商標)細胞である、請求項60記載の製造方法。
- 請求項59記載のワクチンを該個体に投与することを含んでなる、個体においてHIVに対する細胞性免疫応答を生成させる方法。
- DNAプラスミドワクチンを該個体に投与することを更に含み、この場合、それと共に、所望により、該免疫応答を増強しうる生物学的に有効なアジュバント、タンパク質または他の物質を投与しうる、請求項62記載の方法。
- アデノウイルスワクチンの投与の前に該DNAプラスミドワクチンを該個体に投与する、請求項63記載の方法。
- 異なる血清型のアデノウイルスワクチンが該アデノウイルスワクチンに先行する、請求項62記載の方法。
- 該アデノウイルスワクチンベクターを該個体に投与および再投与することを含む、請求項62記載の方法。
- 該HIVタンパク質がHIV nefまたはその免疫学的に関連した修飾体である、請求項1記載のアデノウイルスベクター。
- 該遺伝子発現カセットが、HIV nefタンパク質またはその免疫学的に関連した修飾体をコードするオープンリーディングフレームを含む、請求項9記載のアデノウイルスベクター。
- E1において少なくとも部分的に欠失していてE1活性を欠き、
a)野生型アデノウイルスゲノムの塩基対約451から塩基対約3510までに対応する塩基対が欠失している、野生型アデノウイルスゲノムの塩基対約1から塩基対約450までおよび約3511から約5798までに対応するアデノウイルス・シス作用性パッケージング領域、および
b)i)配列番号9、配列番号11、配列番号13および配列番号15よりなる群から選ばれるヌクレオチド配列と、
ii)i)に作動的に結合した異種プロモーターと、
iii)転写終結配列と、を含む遺伝子発現カセット、
を含んでなる組換えアデノウイルスワクチンベクター。 - 該遺伝子発現カセットがE1に平行な配向で存在する、請求項69記載のアデノウイルスベクター。
- 該遺伝子発現カセットがE1に逆平行な配向で存在する、請求項69記載のアデノウイルスベクター。
- 該プロモーターが、イントロン配列を欠くサイトメガロウイルスプロモーターである、請求項69記載のアデノウイルスベクター。
- 該転写終結配列がウシ成長ホルモンポリアデニル化および転写終結配列である、請求項69記載のアデノウイルスベクター。
- E3において少なくとも部分的に欠失した、請求項69記載のアデノウイルスベクター。
- 請求項68記載のアデノウイルスベクターを含んでなる細胞。
- アデノウイルスE1タンパク質を相補的レベルで発現する細胞系内への請求項68記載のアデノウイルスベクターのトランスフェクションの後に回収され精製された組換え複製欠損型アデノウイルス粒子。
- 請求項76記載の精製されたアデノウイルス粒子を含んでなるHIVワクチン組成物。
- 生理的に許容される担体を含む、請求項77記載のHIVワクチン組成物。
- アデノウイルスE1タンパク質を発現する宿主細胞内に該アデノウイルスベクターを導入し、得られた組換え複製欠損型アデノウイルスを回収することを含んでなる、請求項68記載のアデノウイルスベクターのアデノウイルスゲノムを含有する組換え複製欠損型アデノウイルス粒子の製造方法。
- 該細胞がPER.C6(登録商標)細胞である、請求項79記載の製造方法。
- 請求項78記載のワクチンを該個体に投与することを含んでなる、個体においてHIVに対する細胞性免疫応答を生成させる方法。
- DNAプラスミドワクチンを該個体に投与することを更に含み、この場合、それと共に、所望により、該免疫応答を増強しうる生物学的に有効なアジュバント、タンパク質または他の物質を投与しうる、請求項81記載の方法。
- アデノウイルスワクチンの投与の前に該DNAプラスミドワクチンを該個体に投与する、請求項82記載の方法。
- 異なる血清型のアデノウイルスワクチンが該アデノウイルスワクチンに先行する、請求項81記載の方法。
- 該アデノウイルスワクチンベクターを該個体に投与および再投与することを含む、請求項81記載の方法。
- 組換え複製欠損型アデノウイルス粒子を含んでなり、該アデノウイルス粒子は、アデノウイルスE1タンパク質を発現する細胞系から回収され精製され、該粒子は、請求項9記載のアデノウイルスベクターでの該細胞のトランスフェクションの後に回収され、該ベクターが、
a)3つの個々のベクターから、独立して発現されるgag、polおよびnef、
b)1つのベクターから、独立して発現されるgag、polおよびnef(これらのコード核酸配列は別個のプロモーターおよび転写終結配列に作動的に結合している)、
c)2つのベクター(一方はpol−nef融合体を発現し、もう一方はgagを発現する)を介して発現されるgag、polおよびnef、
d)2つのベクター(一方はgag−pol融合体を発現し、もう一方はnefを発現する)を介して発現されるgag、polおよびnef、
e)2つのベクター(一方はnef−gag融合体を発現し、もう一方はpolを発現する)を介して発現されるgag、polおよびnef、
f)gag−pol−nef融合体を発現する1つのベクターを介して発現されるgag、polおよびnef、
g)2つの個々のベクターから、独立して発現されるgagおよびpol、
h)1つのベクターから、独立して発現されるgagおよびpol(これらのコード核酸配列は別個のプロモーターおよび転写終結配列に作動的に結合している)、
i)2つの個々のベクターから、独立して発現されるpolおよびnef、
j)1つのベクターから、独立して発現されるpolおよびnef(これらのコード核酸配列は別個のプロモーターおよび転写終結配列に作動的に結合している)、
k)2つの個々のベクターから、独立して発現されるnefおよびgag、
l)1つのベクターから、独立して発現されるnefおよびgag(これらのコード核酸配列は別個のプロモーターおよび転写終結配列に作動的に結合している)、
m)gag−pol融合体を発現する1つのベクターを介して発現されるgagおよびpol、
n)pol−nef融合体を発現する1つのベクターを介して発現されるpolおよびnef、ならびに
o)nef−gag融合体を発現する1つのベクターを介して発現されるnefおよびgag
よりなる群から選ばれるHIVタンパク質をコードするヌクレオチド配列を含む遺伝子発現カセットを含む、多価アデノウイルスワクチン組成物。 - 該gag−pol融合体が配列番号39よりなる、請求項86記載の多価アデノウイルスワクチン組成物。
- 該融合配列が、別個のプロモーターおよび転写終結配列に作動的に結合したコード核酸配列を有する、請求項86記載の多価アデノウイルスワクチン組成物。
- 該融合配列が、単一のプロモーターに作動的に結合したコード核酸配列、およびIRES(internal ribosome entry sequence)に作動的に結合したコード核酸配列を有する、請求項86記載の多価アデノウイルスワクチン組成物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US23318000P | 2000-09-15 | 2000-09-15 | |
| PCT/US2001/028861 WO2002022080A2 (en) | 2000-09-15 | 2001-09-14 | Enhanced first generation adenovirus vaccines expressing codon optimized hiv1-gag, pol, nef and modifications |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004508064A true JP2004508064A (ja) | 2004-03-18 |
| JP2004508064A5 JP2004508064A5 (ja) | 2005-08-04 |
Family
ID=22876220
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002526335A Withdrawn JP2004508064A (ja) | 2000-09-15 | 2001-09-14 | コドン最適化hiv1−gag、pol、nefおよび修飾体を発現する増強された第1世代アデノウイルスワクチン |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1320621A4 (ja) |
| JP (1) | JP2004508064A (ja) |
| AU (2) | AU2001294562B2 (ja) |
| CA (1) | CA2422882A1 (ja) |
| WO (1) | WO2002022080A2 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007532656A (ja) * | 2004-04-12 | 2007-11-15 | アメリカ合衆国 | アデノウイルスベクターを用いて免疫応答を誘導するための方法 |
| JP2008539746A (ja) * | 2005-05-12 | 2008-11-20 | グラクソ グループ リミテッド | ワクチン組成物 |
| JP2018035177A (ja) * | 2008-11-18 | 2018-03-08 | ベス イスラエル デアコネス メディカル センター インコーポレイテッド | 細胞性免疫原性が向上した抗ウイルスワクチン |
Families Citing this family (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1242441A4 (en) * | 1999-12-17 | 2004-04-14 | Merck & Co Inc | POLYNUCLEOTIDE VACCINES EXPRESSING CODON-OPTIMIZED NEF HIV-1 PROTEIN AND MODIFIED HIV-1 NEF PROTEIN |
| JP4475561B2 (ja) | 2001-10-11 | 2010-06-09 | メルク・シャープ・エンド・ドーム・コーポレイション | C型肝炎ウイルスワクチン |
| CN1606625A (zh) | 2001-10-31 | 2005-04-13 | 南非医学研究会 | Hiv-1亚型分离株调节/附加基因及其修饰物和衍生物 |
| EP1485124A4 (en) * | 2002-03-13 | 2006-03-22 | Merck & Co Inc | METHOD FOR INDUCING AN ENHANCED IMMUNE RESPONSE AGAINST HIV |
| WO2003085138A1 (en) | 2002-03-29 | 2003-10-16 | Merck & Co., Inc. | Methods of virus production |
| ATE360683T1 (de) | 2002-05-14 | 2007-05-15 | Merck & Co Inc | Verfahren zur reinigung von adenovirus |
| CN1490056A (zh) * | 2002-10-18 | 2004-04-21 | ��¡���ɵ°��̲��о����� | 针对hiv-1的免疫方法和组合物 |
| AU2003288273A1 (en) | 2002-10-23 | 2004-05-13 | Crucell Holland B.V. | New settings for recombinant adenoviral-based vaccines |
| US20080153083A1 (en) | 2003-10-23 | 2008-06-26 | Crucell Holland B.V. | Settings for recombinant adenoviral-based vaccines |
| CA2507915C (en) | 2002-12-17 | 2013-07-02 | Crucell Holland B.V. | Recombinant viral-based malaria vaccines |
| JP2006525787A (ja) | 2003-01-03 | 2006-11-16 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | アカゲザルHER2/neu、これをコードするヌクレオチド及びその使用 |
| JP4772674B2 (ja) | 2003-07-21 | 2011-09-14 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | ヒト上皮成長因子2/neu抗原をコードする合成遺伝子およびその用途 |
| GB2406336A (en) * | 2003-09-24 | 2005-03-30 | Oxxon Pharmaccines Ltd | HIV Pharmaccines |
| ES2387850T3 (es) | 2004-02-11 | 2012-10-02 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.R.L. | Proteína de fusión al antígeno carcinoembrionario y sus usos |
| CA2555412C (en) | 2004-02-23 | 2013-06-25 | Crucell Holland B.V. | Virus purification methods |
| GB0417494D0 (en) | 2004-08-05 | 2004-09-08 | Glaxosmithkline Biolog Sa | Vaccine |
| US7741099B2 (en) | 2004-10-13 | 2010-06-22 | Beth Israel Deaconess Medical Center Inc. | Adenoviral vectors and uses thereof |
| WO2006108707A1 (en) | 2005-04-11 | 2006-10-19 | Crucell Holland B.V. | Virus purification using ultrafiltration |
| AU2012201827B2 (en) * | 2005-05-12 | 2014-09-04 | Glaxo Group Limited | Vaccine composition |
| JP5361386B2 (ja) | 2005-10-07 | 2013-12-04 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・エルレ・エルレ | マトリックスメタロプロテイナーゼ11ワクチン |
| US9717788B2 (en) | 2007-03-02 | 2017-08-01 | Glaxosmithkline Biologicals Sa | Method of inducing an immune response against HIV employing HIV immunogens, adenoviral vectors encoding said immunogens, and adjuvant |
| AR065523A1 (es) * | 2007-03-02 | 2009-06-10 | Glaxosmithkline Biolog Sa | Procedimiento para fomentar una respuesta inmunitaria frente a un patogeno y composiciones |
| EP2047861B1 (en) * | 2007-10-12 | 2019-07-31 | Institut Pasteur | Lentiviral gene transfer vectors suitable for iterative administration and their medicinal applications |
| CN108114276A (zh) | 2007-08-03 | 2018-06-05 | 巴斯德研究院 | 慢病毒基因转移载体及其医学应用 |
| EP2185195A2 (en) | 2007-08-16 | 2010-05-19 | Tripep Ab | Immunogen platform |
| CA2832022C (en) | 2011-04-06 | 2019-01-08 | Biovaxim Limited | Pharmaceutical compositions for preventing and/or treating an hiv disease in humans |
| PL2825640T3 (pl) | 2012-03-12 | 2016-10-31 | Partie rekombinowanych adenowirusów o zmienionych końcach | |
| US8932607B2 (en) | 2012-03-12 | 2015-01-13 | Crucell Holland B.V. | Batches of recombinant adenovirus with altered terminal ends |
| AU2013344512B2 (en) | 2012-11-16 | 2018-06-28 | Beth Israel Deaconess Medical Center, Inc. | Recombinant adenoviruses and use thereof |
| AU2016232050B2 (en) | 2015-03-18 | 2022-04-07 | Janssen Vaccines & Prevention B.V. | Assays for recombinant expression systems |
| PL3283634T3 (pl) | 2015-04-14 | 2019-10-31 | Janssen Vaccines & Prevention Bv | Rekombinowany adenowirus eksprymujący dwa transgeny z dwukierunkowym promotorem |
| BR112018075969A2 (pt) | 2016-06-20 | 2019-04-02 | Janssen Vaccines & Prevention B.V. | promotor bidirecional potente e equilibrado |
| AU2018359492B2 (en) | 2017-10-31 | 2023-12-14 | Janssen Vaccines & Prevention B.V. | Adenovirus and uses thereof |
| SG11202003398SA (en) | 2017-10-31 | 2020-05-28 | Janssen Vaccines & Prevention Bv | Adenovirus vectors and uses thereof |
| BR112020007695A2 (pt) | 2017-10-31 | 2020-10-20 | Janssen Vaccines & Prevention B.V. | adenovírus e seus usos |
| CA3078771A1 (en) | 2017-10-31 | 2019-05-09 | Janssen Vaccines & Prevention B.V. | Adenovirus and uses thereof |
| EP3723771A4 (en) | 2017-12-11 | 2022-04-06 | Beth Israel Deaconess Medical Center, Inc. | RECOMBINANT ADENOVIRUS AND THEIR USES |
| CN111057716B (zh) * | 2019-07-27 | 2022-04-15 | 中国人民解放军军事科学院军事医学研究院 | 用于包装重组人4型腺病毒的单质粒载体系统及其应用 |
| AU2020358474A1 (en) | 2019-10-03 | 2022-04-07 | Batavia Biosciences B.V. | Adenovirus vectors and uses thereof |
| CN112522276B (zh) * | 2020-12-15 | 2022-07-15 | 武汉纽福斯生物科技有限公司 | 一种emc1核苷酸序列及其应用 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5643579A (en) * | 1984-11-01 | 1997-07-01 | American Home Products Corporation | Oral vaccines |
| FR2705686B1 (fr) * | 1993-05-28 | 1995-08-18 | Transgene Sa | Nouveaux adénovirus défectifs et lignées de complémentation correspondantes. |
| AU3551195A (en) * | 1994-09-23 | 1996-04-09 | Somatix Therapy Corporation | Chimeric adenovirus for gene delivery |
| US5872005A (en) * | 1994-11-03 | 1999-02-16 | Cell Genesys Inc. | Packaging cell lines for adeno-associated viral vectors |
| WO1996039178A1 (en) * | 1995-06-05 | 1996-12-12 | The Wistar Institute Of Anatomy And Biology | A replication-defective adenovirus human type 5 recombinant as a vaccine carrier |
| US6019978A (en) * | 1995-06-05 | 2000-02-01 | The Wistar Institute Of Anatomy And Biology | Replication-defective adenovirus human type 5 recombinant as a vaccine carrier |
| EP1003895A2 (de) * | 1997-07-10 | 2000-05-31 | Hepavec AG fur Gentherapie | Klonierungsvektoren für die herstellung von adenoviralen minimalviren |
| GB9803351D0 (en) * | 1998-02-17 | 1998-04-15 | Oxford Biomedica Ltd | Anti-viral vectors |
| US6670188B1 (en) * | 1998-04-24 | 2003-12-30 | Crucell Holland B.V. | Packaging systems for human recombinant adenovirus to be used in gene therapy |
| WO2000015819A1 (en) * | 1998-09-11 | 2000-03-23 | The Children's Medical Center Corporation | Packaging cell lines for hiv-derived retroviral vector particles |
| EP1980617A1 (en) * | 1998-12-31 | 2008-10-15 | Novartis Vaccines and Diagnostics, Inc. | Improved expression of HIV polypeptides and production of virus-like particles |
| GB9906177D0 (en) * | 1999-03-17 | 1999-05-12 | Oxford Biomedica Ltd | Anti-viral vectors |
| GB9912965D0 (en) * | 1999-06-03 | 1999-08-04 | Oxford Biomedica Ltd | In vivo selection method |
| CA2395429A1 (en) * | 1999-12-22 | 2001-06-28 | Merck & Co., Inc. | Polynucleotide vaccines expressing codon optimized hiv-1 pol and modified hiv-1 pol |
| EP1156112B1 (en) * | 2000-05-18 | 2006-03-01 | Geneart GmbH | Synthetic gagpol genes and their uses |
| EP1311686A2 (en) * | 2000-08-14 | 2003-05-21 | THE GOVERNMENT OF THE UNITED STATES OF AMERICA as represented by THE SECRETARY, DEPARTMENT OF HEALTH AND HUMAN SERVICES | Modifications of hiv env, gag, and pol enhance immunogenicity for genetic immunization |
-
2001
- 2001-09-14 WO PCT/US2001/028861 patent/WO2002022080A2/en active IP Right Grant
- 2001-09-14 AU AU2001294562A patent/AU2001294562B2/en not_active Ceased
- 2001-09-14 EP EP01975214A patent/EP1320621A4/en not_active Withdrawn
- 2001-09-14 JP JP2002526335A patent/JP2004508064A/ja not_active Withdrawn
- 2001-09-14 AU AU9456201A patent/AU9456201A/xx active Pending
- 2001-09-14 CA CA002422882A patent/CA2422882A1/en not_active Abandoned
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007532656A (ja) * | 2004-04-12 | 2007-11-15 | アメリカ合衆国 | アデノウイルスベクターを用いて免疫応答を誘導するための方法 |
| JP2008539746A (ja) * | 2005-05-12 | 2008-11-20 | グラクソ グループ リミテッド | ワクチン組成物 |
| JP2013046613A (ja) * | 2005-05-12 | 2013-03-07 | Glaxo Group Ltd | ワクチン組成物 |
| JP2018035177A (ja) * | 2008-11-18 | 2018-03-08 | ベス イスラエル デアコネス メディカル センター インコーポレイテッド | 細胞性免疫原性が向上した抗ウイルスワクチン |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1320621A2 (en) | 2003-06-25 |
| WO2002022080A2 (en) | 2002-03-21 |
| AU2001294562B8 (en) | 2002-03-26 |
| AU2001294562B2 (en) | 2007-05-24 |
| EP1320621A4 (en) | 2005-11-23 |
| WO2002022080A8 (en) | 2003-01-16 |
| WO2002022080A3 (en) | 2002-05-02 |
| WO2002022080A9 (en) | 2003-03-06 |
| CA2422882A1 (en) | 2002-03-21 |
| AU9456201A (en) | 2002-03-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2004508064A (ja) | コドン最適化hiv1−gag、pol、nefおよび修飾体を発現する増強された第1世代アデノウイルスワクチン | |
| US20050070017A1 (en) | Enhanced first generation adenovirus vaccines expressing codon optimized HIV1-Gag, Pol, Nef and modifications | |
| JP2003530307A (ja) | gag遺伝子保有アデノウイルスHIVワクチン | |
| AU2001294562A1 (en) | Enhanced First Generation Adenovirus Vaccines Expressing Codon Optimized HIV1-Gag, Pol, Nef and Modifications | |
| US20070054395A1 (en) | Enhanced first generation adenovirus vaccines expressing codon optimized HIV1-Gag, Pol, Nef and modifications | |
| US20080063656A1 (en) | Adenoviral Vector Compositions | |
| CA2597404A1 (en) | Adenovirus serotype 26 vectors, nucleic acid and viruses produced thereby | |
| AU2003262790A1 (en) | Adenovirus serotype 24 vectors, nucleic acids and virus produced thereby | |
| AU2003298554A1 (en) | Adenovirus serotype 34 vectors, nucleic acids and virus produced thereby | |
| US20060165664A1 (en) | Method of inducing an enhanced immune response against hiv | |
| US20070077257A1 (en) | Enhanced first generation adenovirus vaccines expressing condon optimized HIV1-Gag, Pol, Nef and modifications | |
| US20050106123A1 (en) | Method of inducing an enhanced immune response against hiv | |
| ZA200603029B (en) | Combination approaches for generating immune responses |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A300 | Withdrawal of application because of no request for examination |
Free format text: JAPANESE INTERMEDIATE CODE: A300 Effective date: 20081202 |