ES2941981T3 - Derivados de aminopirimidina/pirazina como inhibidores de CTPS1 - Google Patents
Derivados de aminopirimidina/pirazina como inhibidores de CTPS1 Download PDFInfo
- Publication number
- ES2941981T3 ES2941981T3 ES19789997T ES19789997T ES2941981T3 ES 2941981 T3 ES2941981 T3 ES 2941981T3 ES 19789997 T ES19789997 T ES 19789997T ES 19789997 T ES19789997 T ES 19789997T ES 2941981 T3 ES2941981 T3 ES 2941981T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- pharmaceutically acceptable
- cell
- lymphoma
- solvate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Pyridine Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Compuestos de fórmula (I), y aspectos relacionados. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Derivados de aminopirimidina/pirazina como inhibidores de CTPS1
Campo de la invención
La invención se refiere a un compuesto que es la 4-(2-(ciclopropanosulfonamido)pirimidin-4-il)-W-(5-(6-etoxipirazin-2-il)piridin-2-il)tetrahidro-2H-piran-4-carboxamida o una de sus sales y/o solvatos, y aspectos relacionados que incluyen compuestos intermedios, composiciones que comprenden dicho compuesto y al uso de dicho compuesto como un inhibidor de citidina trifosfato sintasa 1, particularmente en el tratamiento o profilaxis de trastornos asociados con la proliferación celular.
Antecedentes de la invención
Los nucleótidos son una unidad estructural clave para los procesos metabólicos celulares, tales como la síntesis de ácido desoxirribonucleico (ADN) y ácido ribonucleico (ARN). Hay dos clases de nucleótidos, que contienen bases de purina o pirimidina, las cuales son ambas importantes para los procesos metabólicos. Basándose en esto, se han desarrollado muchas terapias para dirigirse a diferentes aspectos de la síntesis de nucleótidos, inhibiendo algunas la generación de nucleótidos de purina y algunas de nucleótidos de pirimidina o ambas.
El nucleótido de pirimidina 5'-trifosfato de citidina (CTP) es un precursor necesario no solo para el anabolismo del ADN y el ARN, sino también para los fosfolípidos y la sialización de proteínas. El CTP se origina a partir de dos fuentes: una ruta de recuperación y una ruta de síntesis nueva que depende de dos enzimas, las CTP sintasas (o sintetasas) 1 y 2 (CTPS1 y CTPS2) (Evans y Guy 2004; Higgins, et al. 2007; Ostrador, et al. 1998).
La CTPS1 y CTPS2 catalizan la conversión de trifosfato de uridina (UTP) y glutamina en trifosfato de citidina (CTP) y L-glutamato:

Ambas enzimas tienen dos dominios, un dominio de sintetasa N-terminal y un dominio de glutaminasa C-terminal (Kursula, et al. 2006). El dominio de sintetasa transfiere un fosfato del trifosfato de adenosina (ATP) a la posición 4 de UTP para crear un compuesto intermedio activado, 4-fosfo-UTP. El dominio de glutaminasa genera amoniaco a partir de glutamina, a través de un tioéster covalente intermedio con un sitio activo conservado de cisteína, generando glutamato. Este amonio se transfiere del dominio glutaminasa al dominio sintetasa a través de un túnel o puede derivarse de amonio externo. Este amonio después es utilizado por el dominio de sintetasa para generar CTP a partir del 4-fosfo-UTP (Lieberman, 1956).
Aunque la CTPS existe como dos isoenzimas en los seres humanos y otros organismos eucariotas, CTPS1 y CTPS2, las diferencias funcionales entre las dos isoenzimas aún no se han dilucidado por completo (van Kuilenburg, et al.
2000).
El sistema inmunitario proporciona protección contra las infecciones y, por lo tanto, ha evolucionado para responder rápidamente a la amplia variedad de patógenos a los que el individuo puede estar expuesto. Esta respuesta puede tomar muchas formas, pero la expansión y diferenciación de las poblaciones inmunes es un elemento crítico y, por lo tanto, está estrechamente relacionado con la rápida proliferación celular. Dentro de esto, la actividad de la CTP sintasa parece desempeñar un papel importante en la síntesis de ADN y la rápida expansión de los linfocitos después de la activación (Fairbanks, et al. 1995; van den Berg, et al. 1995).
La importante validación clínica de que la CTPS1 es la enzima crítica en la proliferación de linfocitos humanos llegó con la identificación de una mutación homocigota de pérdida de función (rs145092287) en esta enzima que causa una inmunodeficiencia distinta y potencialmente mortal, caracterizada por una capacidad deteriorada de que las células T y B activadas proliferen en respuesta a la activación mediada por el receptor de antígeno. Se mostró que las células deficientes en CTPS1 activadas tenían niveles reducidos de CTP. La proliferación normal de células T se restableció en células deficientes en CTPS1 mediante la expresión de CTPS1 de tipo natural o mediante la adición de citidina. Se encontró que la expresión de CTPS1 era baja en los linfocitos en reposo, pero era regulada por aumento rápidamente después de la activación de estas células. La expresión de CTPS1 en otros tejidos generalmente era baja. La CTPS2 parece expresarse de manera ubicua en una variedad de células y tejidos, pero en niveles bajos, y el fallo de CTPS2
que aún está intacta en los pacientes, para compensar la CTPS1 mutada, respalda que la CTPS1 sea la enzima crítica para las poblaciones inmunitarias afectadas en los pacientes (Martin, et al. 2014).
En general, estos hallazgos sugieren que la CTPS1 es una enzima crítica necesaria para satisfacer las demandas de suministro de CTP requeridas por varias poblaciones de células inmunitarias importantes.
Normalmente, la respuesta inmunitaria está estrictamente regulada para garantizar la protección contra la infección, mientras se controla cualquier respuesta dirigida a los tejidos del hospedante. En determinadas situaciones, el control de este proceso no es eficaz, dando lugar a una patología inmunomediada. Se cree que una amplia variedad de enfermedades humanas se deben a dichas respuestas inapropiadas mediadas por diferentes elementos del sistema inmunitario.
Dada la función que se cree que desempeñan las poblaciones de células, tales como los linfocitos T y B, en una amplia variedad de enfermedades autoinmunitarias y de otro tipo, la CTPS1 representa un objetivo para una nueva clase de agentes inmunosupresores. Por lo tanto, la inhibición de CTPS1 proporciona un enfoque novedoso para la inhibición de los linfocitos activados y otras poblaciones de células inmunitarias seleccionadas, tales como las células citolíticas naturales, las células T invariantes asociadas a la mucosa (MAIT) y las células T citolíticas naturales invariantes, destacadas por el fenotipo de los pacientes humanos con mutaciones (Martín, et al. 2014).
El cáncer puede afectar a múltiples tipos de células y tejidos, pero la causa subyacente es un fallo en el control de la división celular. Este proceso es muy complejo y requiere una cuidadosa coordinación de múltiples rutas, muchas de las cuales aún no se han caracterizado por completo. La división celular requiere la replicación efectiva del ADN de la célula y otros constituyentes. Interferir con la capacidad de replicación de una célula dirigiéndose a la síntesis de ácido nucleico ha sido un enfoque central en la terapia del cáncer durante muchos años. Ejemplos de terapias que actúan de esta manera son 6-tioguanina, 6-mecaptopurina, 5-fluorouracilo, citarabina, gemcitabina y pemetrexed.
Como se indicó anteriormente, las rutas implicadas en proporcionar las unidades estructurales básicas clave para la replicación de ácidos nucleicos son las rutas de síntesis de purina y pirimidina, y se ha observado que la biosíntesis de pirimidina está regulada por aumento en tumores y células neoplásicas.
La actividad de CTPS está regulada por aumento en una variedad de tipos de tumores de origen hematológico y no hematológico, aunque se observa heterogeneidad entre los pacientes. También se han establecido vínculos entre los niveles elevados de enzimas y la resistencia a los agentes quimioterapéuticos.
Actualmente, el papel preciso que pueden desempeñar la CTPS1 y CTPS2 en el cáncer no está completamente claro. Se han desarrollado varios inhibidores de CTPS no selectivos para indicaciones oncológicas hasta ensayos clínicos de fase I/II, pero se suspendieron debido a problemas de toxicidad y eficacia.
La mayoría de los inhibidores desarrollados son profármacos análogos de nucleósidos (3-desazauridina, CPEC, carbodina), que son convertidos en el metabolito trifosforilado activo por las quinasas implicadas en la biosíntesis de pirimidina: uridina/citidina quinasa, nucleósido monofosfato-quinasa (NMP-quinasa) y nucleósido difosfatoquinasa (NDP-quinasa). Los inhibidores restantes (acivicina, DON) son análogos reactivos de la glutamina, que inhiben de forma irreversible el dominio glutaminasa de CTPS. También se describe que la gemcitibina tiene cierta actividad inhibitoria contra la CTPS (McClusky et al., 2016).
Por lo tanto, la CTPS parece ser un objetivo importante en el campo del cáncer. La naturaleza de todos los compuestos anteriores es tal que es probable que los efectos sobre otras rutas contribuyan a la eficacia que muestran en la inhibición de tumores.
Por lo tanto, los inhibidores selectivos de CTPS ofrecen un enfoque alternativo atractivo para el tratamiento de tumores. Los compuestos con diferentes potencias contra CTPS1 y CTPS2 pueden ofrecer oportunidades importantes para dirigirse a diferentes tumores dependiendo de su dependencia relativa de estas enzimas.
También se ha sugerido que CTPS1 juega un papel en la proliferación de células del músculo liso vascular después de una lesión vascular o cirugía (Tang, et al. 2013).
Por lo que se sabe hasta la fecha, no se han desarrollado inhibidores selectivos de CTPS1. Recientemente, se ha identificado el péptido inhibidor selectivo de CTPS1 CTpep-3. Sin embargo, los efectos inhibidores de CTpep-3 se observaron en ensayos sin células, pero no en el contexto celular. Sin embargo, esto no fue inesperado, ya que es poco probable que el péptido entre en la célula y, por lo tanto, no se puede desarrollar fácilmente como agente terapéutico (Sakamoto, et al. 2017).
En resumen, la información y los datos disponibles sugieren claramente que los inhibidores de CTPS1 reducirán la proliferación de una serie de poblaciones de células cancerosas e inmunitarias, con el potencial de tener un efecto sobre otros tipos de células seleccionadas, tales como las células del músculo liso vascular. Por lo tanto, se puede esperar que los inhibidores de CTPS1 tengan utilidad para el tratamiento o la profilaxis en una amplia variedad de indicaciones en donde la patología es impulsada por estas poblaciones.
Los inhibidores de CTPS1 representan un enfoque novedoso para inhibir componentes seleccionados del sistema inmunitario en varios tejidos y las patologías o afecciones patológicas relacionadas tales como, en términos generales, rechazo de células y tejidos trasplantados, enfermedades o trastornos relacionados con injerto, alergias y enfermedades autoinmunitarias. Además, los inhibidores de CTPS1 ofrecen potencial terapéutico en una variedad de indicaciones de cáncer y para mejorar la recuperación de una lesión o cirugía vascular y reducir la morbilidad y la mortalidad asociadas con la neoíntima y la reestenosis.
Las solicitudes de patentes internacionales WO2019/106156, WO2019/106146, WO2019/179652 y WO2019/180244 describen inhibidores de CTPS1.
Existe la necesidad de más inhibidores de CTPS1 que puedan demostrar propiedades beneficiosas tales como: - alta potencia;
- inhibición selectiva de CTPS1 frente a CTPS2;
- buena permeabilidad celular;
- fracción libre alta;
- parámetros farmacodinámicos y farmacocinéticos deseables;
- metabolitos distintos;
- lipofilia de baja a moderada.
Sumario de la invención
La invención proporciona un compuesto que es la 4-(2-(ciclopropanosulfonamido)pirimidin-4-il)-W-(5-(6-etoxipirazin-2-il)piridin-2-il)tetrahidro-2H-piran-4-carboxamida:

en el presente documento denominada "el compuesto de la invención".
El compuesto de la invención se puede proporcionar en forma de una de sus sales y/o solvatos. Adecuadamente, el compuesto de la invención se puede proporcionar en forma de una de sus sales y/o solvatos farmacéuticamente aceptables. En particular, el compuesto de la invención se puede proporcionar en forma de una sal y/o solvato farmacéuticamente aceptable, tal como una sal farmacéuticamente aceptable.
También se proporciona el compuesto de la invención, o una de sus sales y/o solvatos farmacéuticamente aceptables, para usar como medicamento, en particular para usar en la inhibición de CTPS1 en un sujeto o la profilaxis o tratamiento de enfermedades o trastornos asociados, tales como aquellos en los que sería beneficiosa una reducción en la proliferación de células T y/o células B.
Adecuadamente, la enfermedad o trastorno se selecciona de: enfermedades inflamatorias de la piel tales como psoriasis o liquen plano; GVHD aguda y/o crónica tal como GVHD aguda resistente a esteroides; síndrome linfoproliferativo agudo (ALPS); lupus eritematoso sistémico, nefritis lúpica o lupus cutáneo; y trasplante. Además, la enfermedad o trastorno puede seleccionarse de miastenia grave, esclerosis múltiple y esclerodermia/esclerosis sistémica.
También se proporciona el compuesto de la invención, o una de sus sales y/o solvatos farmacéuticamente aceptables, para usar en el tratamiento del cáncer.
También se proporciona el compuesto de la invención, o una de sus sales y/o solvatos farmacéuticamente aceptables, para usar para mejorar la recuperación de una lesión o cirugía vascular y reducir la morbilidad y mortalidad asociadas con la neoíntima y la reestenosis en un sujeto.
También se proporcionan composiciones farmacéuticas que contienen el compuesto de la invención, o una de sus sales y/o solvatos farmacéuticamente aceptables, y un vehículo o excipiente farmacéuticamente aceptable.
Descripción detallada de la invención
La invención proporciona un compuesto que es:
4-(2-(ciclopropanosulfonamido)pirimidin-4-il)-W-(5-(6-etoxipirazin-2-il)piridin-2-il)tetrahidro-2H-piran-4-carboxamida:

o una de sus sales y/o solvatos.
El compuesto de la invención se puede proporcionar en forma de una de sus sales y/o solvatos farmacéuticamente aceptables. En particular, el compuesto de la invención se puede proporcionar en forma de una sal y/o solvato farmacéuticamente aceptable, tal como una sal farmacéuticamente aceptable.
Se apreciará que para usar en medicina, las sales del compuesto de la invención deben ser farmacéuticamente aceptables. Las sales no farmacéuticamente aceptables del compuesto de la invención pueden ser útiles en otros contextos tales como durante la preparación del compuesto de la invención. Las sales farmacéuticamente aceptables adecuadas serán evidentes para los expertos en la técnica. Las sales farmacéuticamente aceptables incluyen las descritas por Berge et al. (1977). Dichas sales farmacéuticamente aceptables incluyen sales de adición de ácidos y bases. Se pueden formar sales adicionales de ácido farmacéuticamente aceptables con ácidos inorgánicos, p. ej., ácido clorhídrico, bromhídrico, sulfúrico, nítrico o fosfórico y ácidos orgánicos, p. ej., ácido succínico, maleico, acético, fumárico, cítrico, tartárico, benzoico, p-toluenosulfónico, metanosulfónico o naftalenosulfónico. Otras sales, p. ej., oxalatos o formiatos, pueden usarse por ejemplo en el aislamiento del compuesto de la invención y están incluidas dentro del alcance de esta invención.
El compuesto de la invención puede formar sales de adición de ácido o base con uno o más equivalentes del ácido o la base. La presente invención incluye dentro de su alcance todas las formas estequiométricas y no estequiométricas posibles.
El compuesto de la invención se puede preparar en forma cristalina o no cristalina y, si es cristalina, se puede solvatar opcionalmente, p. ej. como el hidrato. Esta invención incluye dentro de su alcance solvatos estequiométricos (p. ej., hidratos) así como compuestos que contienen cantidades variables de disolvente (p. ej., agua).
Debe entenderse que la presente invención abarca todos los isómeros geométricos, isómeros tautómeros y formas ópticas, y mezclas de los mismos (p. ej., mezclas racémicas) de fórmula (I). Las diferentes formas isoméricas pueden separarse o resolverse unas de otras por métodos convencionales, o cualquier isómero dado puede obtenerse por métodos sintéticos convencionales o por síntesis estereoespecífica o asimétrica.
La presente descripción incluye todas las formas isotópicas del compuesto de la invención proporcionadas en el presente documento, ya sea en una forma (i) en donde todos los átomos de un número atómico dado tienen un número de masa (o mezcla de números de masa) que predomina en la naturaleza (mencionado en el presente documento como la "forma isotópica natural") o (ii) en donde uno o más átomos son reemplazados por átomos que tienen el mismo número atómico, pero un número de masa diferente del número de masa de los átomos que predominan en la naturaleza (referido en el presente documento como una "forma isotópica variante no natural"). Se entiende que un átomo puede existir naturalmente como una mezcla de números de masa. La expresión "forma isotópica variante no natural" también incluye realizaciones en las que la proporción de un átomo de número atómico dado que tiene un número de masa que se encuentra de forma menos común en la naturaleza (denominado en el presente documento como "isótopo no común") se ha incrementado en relación con el que es de origen natural, p. ej., al nivel de >20%, >50%, >75%, >90%, >95% o >99% en número de átomos de ese número atómico (la última realización se denomina "forma variante enriquecida isotópicamente"). La expresión "forma isotópica variante no natural" también incluye realizaciones en las que la proporción de un isótopo no común se ha reducido en relación con la que se produce de forma natural. Las formas isotópicas pueden incluir formas radiactivas (es decir, incorporan radioisótopos) y formas no radiactivas. Las formas radiactivas serán típicamente formas variantes enriquecidas isotópicamente.
Por lo tanto, una forma isotópica variante no natural del compuesto de la invención puede contener uno o más isótopos artificiales o no comunes, tales como el deuterio (2H o D), carbono-11 (11C), carbono-13 (13C), carbono-14 (14C), nitrógeno-13 (13N), nitrógeno-15 (15N), oxígeno-15 (15O), oxígeno-17 (17O), oxígeno-18 (18O), fósforo-32 (32P), azufre-35 (35S), cloro-36 (36Cl), cloro-37 (37Cl), flúor-18 (18F) yodo-123 (123I), yodo-125 (125I), en uno o más átomos o puede contener una proporción aumentada de dichos isótopos en comparación con la proporción que predomina en la naturaleza en uno o más átomos.
Las formas isotópicas variantes no naturales que comprenden radioisótopos pueden usarse, por ejemplo, para estudios de distribución de fármacos y/o sustratos en tejidos. Los isótopos radiactivos tritio, es decir, 3H, y carbono-14, es decir, 14C, son particularmente útiles para este propósito en vista de su facilidad de incorporación y medios de detección disponibles. Formas isotópicas variantes no naturales que incorporan deuterio, es decir, 2H o D pueden ofrecer ciertas ventajas terapéuticas que resultan de una mayor estabilidad metabólica, por ejemplo, mayor semivida in vivo o requisitos de dosificación reducidos y, por lo tanto, pueden ser preferibles en algunas circunstancias. Además, se pueden preparar formas isotópicas variantes no naturales que incorporan isótopos emisores de positrones, tales como 11C, 18F, 15O y 13N, y serían útiles en los estudios de topografía por emisión de positrones (PET) para examinar la ocupación del receptor por el sustrato.
En una realización, el compuesto de la invención se proporciona en forma isotópica natural.
En una realización, el compuesto de la invención se proporciona en una forma isotópica variante no natural. En una realización específica, la forma isotópica variante no natural es una forma en la que el deuterio (es decir, 2H o D) se incorpora donde se especifica hidrógeno en la estructura química en uno o más átomos del compuesto de la invención. En una realización, los átomos del compuesto de la invención están en una forma isotópica que no es radiactiva. En una realización, uno o más átomos del compuesto de la invención están en una forma isotópica que es radiactiva. Los isótopos adecuadamente radiactivos son isótopos estables. Adecuadamente, la forma isotópica variante no natural es una forma farmacéuticamente aceptable.
En una realización, el compuesto de la invención se proporciona de modo que existe un solo átomo del compuesto en una forma isotópica variante no natural. En otra realización, el compuesto de la invención se proporciona de modo que existen dos o más átomos en una forma isotópica variante no natural.
Las formas variantes isotópicas no naturales pueden prepararse generalmente mediante técnicas convencionales conocidas por los expertos en la técnica o mediante procedimientos descritos en el presente documento, p. ej., procedimientos análogos a los descritos en los Ejemplos adjuntos para preparar formas isotópicas naturales. Por lo tanto, las formas variantes isotópicas no naturales podrían prepararse usando reactivos variantes isotópicamente apropiados (o marcados) en lugar de los reactivos normales empleados en los Ejemplos. Dado que el compuesto de la invención está destinado a su uso en composiciones farmacéuticas, se entenderá fácilmente que se proporciona preferiblemente en forma sustancialmente pura, por ejemplo, con una pureza de al menos 60%, más adecuadamente de al menos 75% y preferiblemente de al menos 85%, especialmente al menos 98% puro (los % son peso por peso). Pueden usarse preparaciones impuras del compuesto para preparar las formas más puras usadas en las composiciones farmacéuticas.
En general, el compuesto de la invención se puede preparar según las técnicas de síntesis orgánica conocidas por los expertos en este campo, así como por los métodos representativos expuestos en los Ejemplos y modificaciones de los mismos.
Compuestos intermedios de la invención
La presente invención también se refiere a compuestos de fórmula (II):

en donde R es H o alquilo C1-6 (p. ej., metilo y etilo).
Están incluidas como un aspecto de la invención sales tales como sales farmacéuticamente aceptables de uno cualquiera de los compuestos de fórmula (II).
Métodos Terapéuticos
El compuesto de la presente invención tiene utilidad como inhibidor de CTPS1.
Por lo tanto, la invención también proporciona un compuesto de la invención, o una de sus sales y/o solvatos farmacéuticamente aceptables (p. ej., sal), para su uso como medicamento, en particular en el tratamiento o profilaxis de una enfermedad o trastorno en donde es beneficioso un inhibidor de CTPS1, por ejemplo, aquellas enfermedades y trastornos mencionados en el presente documento a continuación.
Más adecuadamente, la enfermedad o trastorno en donde es beneficioso un inhibidor de CTPS1 es una enfermedad o trastorno en donde sería beneficiosa una reducción de la proliferación de células T y/o células B.
La invención también proporciona el compuesto de la invención, o una de sus sales y/o solvatos farmacéuticamente aceptables (p. ej., sal), para su uso en la inhibición de CTPS1 en un sujeto.
La invención también proporciona el compuesto de la invención, o una de sus sales y/o solvatos farmacéuticamente aceptables (p. ej., sal), para su uso en la reducción de la proliferación de células T y/o células B en un sujeto.
Más adecuadamente, la enfermedad o trastorno en donde es beneficioso un inhibidor de CTPS1 es una enfermedad o trastorno en donde sería beneficiosa una reducción de la proliferación de células T y/o células B.
El término “tratamiento” o “tratar” como se usa en el presente documento incluye el control, mitigación, reducción o modulación del estado de la enfermedad o sus síntomas.
El término "profilaxis" o "prevenir" se usa en el presente documento para significar la prevención de síntomas de una enfermedad o trastorno en un sujeto o la prevención de la recurrencia de síntomas de una enfermedad o trastorno en un sujeto afectado y no se limita a la prevención completa de una dolencia.
Adecuadamente, la enfermedad o trastorno se selecciona de rechazo de células y tejidos trasplantados, enfermedades o trastornos relacionados con injertos, alergias y enfermedades autoinmunitarias.
En una realización, la enfermedad o trastorno es el rechazo de células y tejidos trasplantados. El sujeto puede haber sido sometido a trasplante de un injerto seleccionado del grupo que consiste en corazón, riñón, pulmón, hígado, páncreas, islotes pancreáticos, tejido cerebral, estómago, intestino grueso, intestino delgado, córnea, piel, tráquea, hueso, médula ósea (o cualquier otra fuente de células precursoras y células madre hematopoyéticas, incluidas las células hematopoyéticas movilizadas desde la médula ósea hacia la sangre periférica o células sanguíneas del cordón umbilical), músculo o vejiga. El compuesto de la invención puede ser útil para prevenir o suprimir una respuesta inmunitaria asociada con el rechazo de un trasplante de tejido, célula, injerto u órgano de un donante en un sujeto.
En una realización adicional, la enfermedad o trastorno es una enfermedad o trastorno relacionado con injerto. Las enfermedades o trastornos relacionados con injerto incluyen la enfermedad de injerto contra huésped (GVHD, por sus siglas en inglés Graft Versus Host Disease), tal como la GVHD asociada con el trasplante de médula ósea, y los trastornos inmunitarios resultantes de o asociados con el rechazo del trasplante de órganos, tejidos o células (p. ej., aloinjertos o xenoinjertos de tejidos o células), incluyendo, p. ej., injertos de piel, músculo, neuronas, islotes, órganos, células parenquimatosas del hígado, etc., y la enfermedad de huésped contra injerto (HVGD, por sus siglas en inglés Host Versus Graft Disease). El compuesto de la invención puede ser útil para prevenir o suprimir el rechazo agudo de dicho trasplante en el receptor y/o para la terapia de mantenimiento a largo plazo para prevenir el rechazo de dicho trasplante en el receptor (p. ej., inhibiendo el rechazo del trasplante de células de los islotes productoras de insulina de un donante en el sujeto receptor que sufre diabetes). Por lo tanto, el compuesto de la invención tiene utilidad en la prevención de la enfermedad de huésped contra injerto (HVGD) y la enfermedad de injerto contra huésped (GVHD).
El compuesto de la invención se puede administrar al sujeto antes, después del trasplante y/o durante el trasplante. En algunas realizaciones, el compuesto de la invención se puede administrar al sujeto periódicamente antes y/o después del trasplante.
En otra realización, la enfermedad o trastorno es una alergia.
En realizaciones adicionales, la enfermedad o trastorno inmunorrelacionado es una enfermedad autoinmunitaria. Como se usa en el presente documento, una "enfermedad autoinmunitaria" es una enfermedad o trastorno dirigido a los propios tejidos de un sujeto. Los ejemplos de enfermedades autoinmunitarias incluyen, pero no se limitan a la enfermedad de Addison, enfermedad de Still del adulto, alopecia areata, enfermedad de Alzheimer, vasculitis asociada a los anticuerpos anti-citoplasma de neutrófilos (ANCA), espondilitis anquilosante, síndrome antifosfolípido (síndrome de Hughes), anemia aplásica, artritis, asma, aterosclerosis, placa aterosclerótica, dermatitis atópica, anemia hemolítica autoinmunitaria, hepatitis autoinmunitaria, hipofisitis autoinmunitaria (hipofisitis linfocítica), enfermedad del oído interno autoinmunitaria, síndrome linfoproliferativo autoinmunitario, miocarditis autoinmunitaria, neutropenia autoinmunitaria, ooforitis autoinmunitaria, orquitis autoinmunitaria, enfermedades autoinflamatorias que requieren un tratamiento inmunosupresor, azoospermia, enfermedad de Bechet, enfermedad de Berger, penfigoide ampolloso, miocardiopatía, enfermedad cardiovascular, enfermedad celíaca incluida la enfermedad celíaca refractaria (tipo I y tipo II), síndrome de disfunción inmunitaria de fatiga crónica (CFIDS), polineuritis idiopática crónica, polineuropatía desmielinizante inflamatoria crónica (CIPD), polineuropatía crónica recidivante (síndrome de Guillain-Barre), síndrome de Churg-Strauss (CSS), penfigoide cicatricial, enfermedad por crioaglutininas (CAD), enfermedad pulmonar obstructiva crónica (COPD), síndrome CREST, síndromes de crioglobulina, lupus cutáneo, dermatitis herpetiforme, dermatomiositis, eccema, epidermólisis ampollosa adquirida, crioglobulinemia mixta esencial, síndrome de Evan, exoftalmos, fibromialgia, síndrome de Goodpasture, enfermedad de Grave, linfohistiocitosis hemofagocítica (HLH) (incluida la linfohistiocitosis hemofagocítica tipo 1), histiocitosis/trastornos histiocíticos, tiroiditis de Hashimoto, fibrosis pulmonar idiopática, púrpura trombocitopénica idiopática (ITP), nefropatía por IgA, enfermedades o trastornos inmunoproliferativos, enfermedad inflamatoria intestinal (IBD), enfermedad pulmonar intersticial, artritis juvenil, artritis idiopática juvenil (AIJ), enfermedad de Kawasaki, síndrome miasténico de Lambert-Eaton, liquen plano, esclerodermia localizada, nefritis lúpica, enfermedad de Méniére, anemia hemolítica microangiopática, poliangitis microscópica, síndrome de Miller Fischer/encefalomielorradiculopatía diseminada aguda, enfermedad mixta del tejido conjuntivo, esclerosis múltiple (EM), reumatismo muscular, encefalomielitis miálgica (ME), miastenia grave, inflamación ocular, pénfigo foliáceo, pénfigo vulgar, anemia perniciosa, poliarteritis nodosa, policondritis, síndromes poliglandulares
(síndrome de Whitaker), polimialgia reumática, polimiositis, agammaglobulinemia primaria, cirrosis biliar primaria/colangiopatía autoinmunitaria, glomerulonefritis primaria, colangitis esclerosante primaria, psoriasis, artritis psoriásica, anemia pura de glóbulos rojos, fenómeno de Raynaud, síndrome de Reiter/artritis reactiva, policondritis recidivante, reestenosis, fiebre reumática, enfermedad reumática, artritis reumatoide, sarcoidosis, síndrome de Schmidt, esclerodermia/esclerosis sistémica, síndrome de Sjorgen, síndrome de la persona rígida, síndrome de Sweet (dermatosis neutrofílica febril), lupus eritematoso sistémico (SLE), esclerodermia sistémica, arteritis de Takayasu, arteritis temporal/arteritis de células gigantes, tiroiditis, diabetes tipo 1, diabetes tipo 2, uveítis, vasculitis, vitíligo, granulomatosis de Wegener y enfermedad linfoproliferativa ligada al cromosoma X.
De particular interés son las enfermedades y trastornos que son estimulados principalmente por la activación y proliferación de células T, que incluyen:
- enfermedades y trastornos que no están conectados con la alorreactividad, que incluyen:
■ alopecia areata, dermatitis atópica, eccema, psoriasis, liquen plano, artritis psoriásica, vitíligo;
■ uveítis;
■ espondilitis anquilosante, síndrome de Reiter/artritis reactiva;
■ anemia aplásica, síndrome/trastornos linfoproliferativos autoinmunitarios, linfohistiocitosis hemofagocítica;
■ diabetes tipo 1; y
■ enfermedad celíaca refractaria;
- rechazo agudo de tejidos injertados y órganos trasplantados; enfermedad aguda de injerto contra huésped (GVHD) después del trasplante de células de médula ósea o cualquier otra fuente de células alogénicas, incluidas células precursoras y/o células madre hematopoyéticas.
También son de interés las enfermedades y trastornos que son estimulados por la activación y proliferación de células T y B, con una participación importante de las células B, que incluyen:
- enfermedades y trastornos para los cuales la implicación de autoanticuerpos patógenos está bien caracterizada, que incluyen:
■ alergia;
■ penfigoide cicatricial, penfigoide ampolloso, epidermólisis ampollosa adquirida, pénfigo foliáceo, pénfigo vulgar, dermatitis herpetiforme;
■ vasculitis asociada a ANCA y poliangitis microscópica, vasculitis, granulomatosis de Wegener; síndrome de Churg-Strauss (CSS), poliarteritis nodosa, síndromes de crioglobulina y criglobulinemia mixta esencial; ■ lupus eritematoso sistémico (SLE), síndrome antifosfolípido (síndrome de Hughes), lupus cutáneo, nefritis lúpica, enfermedad mixta del tejido conjuntivo;
■ tiroiditis, tiroiditis de Hashimoto, enfermedad de Grave, exoftalmos;
■ - anemia hemolítica autoinmunitaria, neutropenia autoinmunitaria, ITP, anemia perniciosa, anemia pura de glóbulos rojos, anemia hemolítica microangiopática;
■ glomerulonefritis primaria, enfermedad de Berger, síndrome de Goodpasture, nefropatía por IgA; y
■ polineuritis idiopática crónica, polineuropatía desmielinizante inflamatoria crónica (CIPD), polineuropatía crónica recidivante (síndrome de Guillain-Barré), síndrome de Miller Fischer, síndrome de la persona rígida, síndrome miasténico de Lambert-Eaton, miastenia grave.
- enfermedades y trastornos en los que la implicación de las células B está menos claramente caracterizada (aunque a veces ilustrada por la eficacia de los anticuerpos monoclonales anti-CD20 o las infusiones de inmunoglobulina intravenosa) y pueden no corresponder o estar limitados a la producción de anticuerpos patógenos (sin embargo, a veces se describen anticuerpos no patógenos o incluso a menudo están presentes y se usan como biomarcadores de diagnóstico), que incluyen:
■ enfermedad de Addison, ooforitis autoinmunitaria y azoospermia, síndromes poliglandulares (síndrome de Whitaker), síndrome de Schmidt;
■ miocarditis autoinmunitaria, cardiomiopatía, enfermedad de Kawasaki;
■ artritis reumatoide, síndrome de Sjogren, enfermedad mixta del tejido conjuntivo, polimiositis y dermatomiositis; policondritis;
■ glomerulonefritis primaria;
■ esclerosis múltiple;
■ hepatitis autoinmunitaria, cirrosis biliar primaria/colangiopatía autoinmunitaria,
■ rechazo hiperagudo de órganos trasplantados;
■ rechazo crónico de injertos o trasplantes;
■ reacción/enfermedad crónica de injerto contra huésped después del trasplante de células de médula ósea o células precursoras hematopoyéticas.
Además, son de interés las enfermedades y trastornos en los que el mecanismo es compartido entre la activación/proliferación de células T y la activación/proliferación de células inmunitarias innatas y otras subpoblaciones celulares inflamatorias (incluidas células mieloides, tales como macrófagos o granulocitos) y células residentes (tales como fibroblastos y células endoteliales), incluyendo:
■ COPD, fibrosis pulmonar idiopática, enfermedad pulmonar intersticial, sarcoidosis;
■ enfermedad de Still del adulto, artritis idiopática juvenil, esclerosis sistémica, síndrome CREST donde las células B y los anticuerpos patógenos también pueden desempeñar un papel; el síndrome de Sweet; arteritis de Takayasu, arteritis temporal/arteritis de células gigantes;
■ colangitis ulcerosa, enfermedad inflamatoria intestinal (IBD), incluida la enfermedad de Crohn y la colitis ulcerosa, colangitis esclerosante primaria.
También son de interés las enfermedades y trastornos en los que el mecanismo sigue estando poco caracterizado pero implica la activación y proliferación de células T, que incluyen:
■ enfermedad de Alzheimer, síndrome cardiovascular, diabetes tipo 2, reestenosis, síndrome de disfunción inmunitaria de fatiga crónica (CFIDS).
- trastornos linfoproliferativos autoinmunitarios, que incluyen:
■ síndrome linfoproliferativo autoinmunitario y enfermedad linfoproliferativa ligada al X.
Adecuadamente, la enfermedad o trastorno se selecciona de: enfermedades inflamatorias de la piel tales como psoriasis o liquen plano; GVHD aguda y/o crónica tal como GVHD aguda resistente a esteroides; síndrome linfoproliferativo agudo; lupus eritematoso sistémico, nefritis lúpica o lupus cutáneo; o trasplante. Además, la enfermedad o trastorno puede seleccionarse de miastenia grave, esclerosis múltiple y esclerodermia/esclerosis sistémica.
El compuesto de la invención puede usarse en el tratamiento del cáncer.
Por lo tanto, en una realización se proporciona el compuesto de la invención, o una de sus sales y/o solvatos farmacéuticamente aceptables, para usar en el tratamiento del cáncer.
Convenientemente, el cáncer es un cáncer hematológico, tal como la leucemia mieloide aguda, linfoma de células T angioinmunoblástico, leucemia linfoblástica aguda de células B, síndrome de Sweet, linfoma no Hodgkin de células T (incluido el linfoma de células T/citolíticas naturales, linfoma/leucemia de células T del adulto, linfoma de células T tipo enteropatía, linfoma hepatoesplénico de células T y linfoma de células T cutáneo), leucemia linfoblástica aguda de células T, linfoma no Hodgkin de células B (incluido el linfoma de Burkitt, linfoma difuso de células B grandes, linfoma folicular, linfoma de células del manto, linfoma de la zona marginal), leucemia de células pilosas, linfoma de Hodgkin, linfoma linfoblástico, linfoma linfoplasmocitario, linfoma del tejido linfoide asociado a mucosas, mieloma múltiple, síndrome mielodisplásico, mieloma de células plasmáticas, linfoma mediastinal primario de células B grandes, trastornos mieloproliferativos crónicos (tales como leucemia mieloide crónica, mielofibrosis primaria, trombocitemia esencial, policitemia vera) y leucemia linfocítica crónica.
Alternativamente, el cáncer es un cáncer no hematológico, tal como el seleccionado del grupo que consiste en cáncer de vejiga, mama, melanoma, neuroblastoma, mesotelioma pleural maligno y sarcoma.
Además, el compuesto de la invención se puede usar para mejorar la recuperación de una lesión vascular o cirugía y reducir la morbilidad y la mortalidad asociadas con la neoíntima y la reestenosis en un sujeto. Por ejemplo, el compuesto de la invención se puede usar para prevenir, reducir o inhibir la formación de neoíntima. Un dispositivo médico puede tratarse antes de la inserción o implantación con una cantidad eficaz de una composición que comprende el compuesto de la invención con el fin de prevenir, reducir o inhibir la formación de neoíntima después de
la inserción o implantación del dispositivo o injerto en el sujeto. El dispositivo puede ser un dispositivo que se inserta en el sujeto de forma transitoria o un dispositivo que se implanta de forma permanente. En algunas realizaciones, el dispositivo es un dispositivo quirúrgico. Los ejemplos de dispositivos médicos incluyen, pero no se limitan a agujas, cánulas, catéteres, derivaciones, globos e implantes, tales como endoprótesis y válvulas.
Convenientemente, el sujeto es un mamífero, en particular, el sujeto es un ser humano.
Composiciones farmacéuticas
Para usar en terapia, el compuesto de la invención normalmente se administra como una composición farmacéutica. La invención también proporciona una composición farmacéutica que comprende el compuesto de la invención, o una de sus sales y/o solvatos farmacéuticamente aceptables (p. ej., sal), y un vehículo o excipiente farmacéuticamente aceptable.
En una realización, se proporciona una composición farmacéutica que comprende el compuesto de la invención, o una de sus sales y/o solvatos farmacéuticamente aceptables (p. ej., sal), para usar en el tratamiento o profilaxis de una enfermedad o trastorno como se describe en el presente documento.
El compuesto de la invención o sus sales y/o solvatos farmacéuticamente aceptables pueden administrarse por cualquier método conveniente, p. ej., por administración oral, parenteral, bucal, sublingual, nasal, rectal o transdérmica, y las composiciones farmacéuticas adaptadas en consecuencia.
El compuesto de la invención o sus sales y/o solvatos farmacéuticamente aceptables pueden administrarse por vía tópica, por ejemplo en el ojo, intestino o piel. Por lo tanto, en una realización se proporciona una composición farmacéutica que comprende el compuesto de la invención opcionalmente en combinación con uno o más diluyentes o vehículos aceptables por vía tópica.
Una composición farmacéutica de la invención puede administrarse por vía tópica en la piel. Las composiciones adecuadas para la administración transdérmica incluyen ungüentos, geles y parches. Dicha composición farmacéutica también puede estar adecuadamente en forma de crema, loción, espuma, polvo, pasta o tintura.
La composición farmacéutica puede incluir adecuadamente análogos de vitamina D3 (p. ej., calcipotriol y maxacalcitol), esteroides (p. ej., propionato de fluticasona, valerato de betametasona y propionato de clobetasol), retinoides (p. ej., tazaroteno), alquitrán de hulla y ditranol. Los medicamentos tópicos a menudo se usan en combinación entre sí (p. ej., una vitamina D3 y un esteroide) o con otros agentes tales como ácido salicílico.
Una composición farmacéutica de la invención puede administrarse por vía tópica en el ojo. Dicha composición farmacéutica puede estar adecuadamente en forma de colirios o una pomada.
Una composición farmacéutica de la invención puede administrarse por vía tópica en el intestino. Dicha composición farmacéutica puede administrarse adecuadamente por vía oral, tal como en forma de un comprimido o una cápsula, o por vía rectal, tal como en forma de un supositorio.
Convenientemente, las formulaciones de liberación retardada están en forma de una cápsula.
El compuesto de la invención o sus sales y/o solvatos farmacéuticamente aceptables que son activos cuando se administran por vía oral se pueden formular como líquidos o sólidos, p. ej., como jarabes, suspensiones, emulsiones, comprimidos, cápsulas o pastillas.
Una formulación líquida consistirá generalmente en una suspensión o solución del ingrediente activo (tal como el compuesto de la invención o una de sus sales y/o solvatos farmacéuticamente aceptables (p. ej., sal)) en un vehículo(s) líquido(s) adecuado(s), p. ej. un disolvente acuoso tal como agua, etanol o glicerina, o un disolvente no acuoso, tal como polietilenglicol o un aceite. La formulación también puede contener un agente de suspensión, conservante, aromatizante y/o agente colorante.
Se puede preparar una composición en forma de un comprimido usando cualquier vehículo(s) farmacéutico(s) adecuado(s) usado(s) habitualmente para preparar formulaciones sólidas, tales como estearato de magnesio, almidón, lactosa, sacarosa y celulosa.
Se puede preparar una composición en forma de una cápsula usando procedimientos de encapsulación rutinarios, p. ej. los gránulos que contienen el ingrediente activo (tal como el compuesto de la invención o una de sus sales y/o solvatos farmacéuticamente aceptables (p. ej., sal)) pueden prepararse usando vehículos convencionales y luego cargarlos en una cápsula de gelatina dura; alternativamente, se puede preparar una dispersión o suspensión utilizando cualquier vehículo(s) farmacéutico(s) adecuado(s), p. ej. gomas acuosas, celulosas, silicatos o aceites y luego llenar con la dispersión o suspensión una cápsula de gelatina blanda.
Las composiciones parenterales típicas consisten en una solución o suspensión del ingrediente activo (tal como el compuesto de la invención o una de sus sales y/o solvatos farmacéuticamente aceptables (p. ej., sal)) en un vehículo acuoso estéril o aceite aceptable por vía parenteral, p. ej. polietilenglicol, polivinilpirrolidona, lecitina, aceite de
cacahuete o aceite de sésamo. Alternativamente, la solución se puede liofilizar y luego reconstituir con un disolvente adecuado justo antes de la administración.
Las composiciones para administración nasal pueden formularse convenientemente como aerosoles, gotas, geles y polvos. Las formulaciones en aerosol típicamente comprenden una solución o suspensión fina del ingrediente activo en un disolvente acuoso o no acuoso farmacéuticamente aceptable y generalmente se presentan en cantidades de dosis unitaria o multidosis en forma estéril en un recipiente sellado que puede tener la forma de un cartucho o recarga para usar con un dispositivo atomizador. Alternativamente, el recipiente sellado puede ser un dispositivo dispensador desechable tal como un inhalador nasal de dosis única o un dispensador de aerosol equipado con una válvula dosificadora. Cuando la forma farmacéutica comprende un dispensador de aerosol, contendrá un propulsor que puede ser un gas comprimido, p. ej. aire, o un propulsor orgánico tal como un fluoro-cloro-hidrocarburo o hidrofluorocarburo. Las formas farmacéuticas en aerosol también pueden tener la forma de atomizadores de bomba.
Las composiciones adecuadas para administración bucal o sublingual incluyen comprimidos, pastillas para chupar y pastillas donde el ingrediente activo se formula con un vehículo tal como azúcar y goma arábiga, tragacanto o gelatina y glicerina.
Las composiciones para administración rectal están convenientemente en forma de supositorios que contienen una base de supositorio convencional tal como manteca de cacao.
Convenientemente, la composición está en forma de dosis unitaria tal como una comprimido, cápsula o ampolla.
La composición puede contener, por ejemplo, de 0,1% a 100% en peso, por ejemplo de 10 a 60% en peso, de material activo, dependiendo del método de administración. La composición puede contener de 0% a 99% en peso, por ejemplo de 40% a 90% en peso, de vehículo, dependiendo del método de administración. La composición puede contener de 0,05 mg a 2000 mg, por ejemplo de 1,0 mg a 500 mg, de material activo, dependiendo del método de administración. La composición puede contener de 50 mg a 1000 mg, por ejemplo de 100 mg a 400 mg de vehículo, dependiendo del método de administración. La dosis del compuesto utilizada en el tratamiento o profilaxis de los trastornos antes mencionados variará de forma habitual con la gravedad de los trastornos, el peso del que lo padece y otros factores similares. Sin embargo, como guía general, las dosis unitarias adecuadas pueden ser de 0,05 mg a 1000 mg, más adecuadamente de 1,0 mg a 500 mg, y dichas dosis unitarias pueden administrarse más de una vez al día, por ejemplo, dos o tres al día. Tal terapia puede prolongarse durante una serie de semanas o meses.
La invención proporciona, en otro aspecto, una combinación que comprende el compuesto de la invención o una sal 0 solvato farmacéuticamente aceptable junto con un ingrediente o ingredientes activos farmacéuticamente aceptables adicionales.
La invención proporciona el compuesto de la invención, para usar en combinación con otro ingrediente o ingredientes activos farmacéuticamente aceptables.
Cuando el compuesto de la invención se usa en combinación con otros agentes terapéuticos, el compuesto de la invención puede administrarse por separado, secuencial o simultáneamente por cualquier vía conveniente.
Las combinaciones óptimas pueden depender de la enfermedad o trastorno. Las posibles combinaciones incluyen aquellas con uno o más agentes activos seleccionados de la lista que consiste en: ácido 5-aminosalicílico, o un profármaco del mismo (tal como sulfasalazina, olsalazina o bisalazida); corticosteroides (p. ej., prednisolona, metilprednisolona o budesonida); inmunosupresores (p. ej., ciclosporina, tacrolimus, sirolimus, metotrexato, azatioprina micofenolato mofetilo, leflunomida, ciclofosfamida, 6-mercaptopurina o globulinas antilinfocitos (o timocitos)); anticuerpos anti-TNF-alfa (p. ej., infliximab, adalimumab, certolizumab pegol o golimumab); anticuerpos anti-IL12/IL23 (p. ej., ustekinumab); anticuerpos anti-IL6 o anti-IL6R, anticuerpos anti-IL17 o inhibidores de IL12/IL23 de molécula pequeña (p. ej., apilimod); anticuerpos anti-alfa-4-beta-7 (p. ej., vedolizumab); bloqueadores de MAdCAM-1 (p. ej., PF-00547659); anticuerpos contra la molécula de adhesión celular alfa-4-integrina (p. ej., natalizumab); anticuerpos contra la subunidad alfa del receptor de IL2 (p. ej., daclizumab o basiliximab); inhibidores de JAK, incluidos inhibidores de JAK1 y JAK3 (p. ej., tofacitinib, baricitinib, R348); inhibidores de Syk y profármacos de los mismos (p. ej., fostamatinib y R-406); inhibidores de fosfodiesterasa-4 (p. ej., tetomilast); HMPL-004; probióticos; dersalazina; semapimod/CPSI-2364; e inhibidores de proteína quinasa C (p. ej., AEB-071).
Para el cáncer, el ingrediente activo farmacéuticamente aceptable adicional puede seleccionarse de agentes antimitóticos tales como vinblastina, paclitaxel y docetaxel; agentes alquilantes, por ejemplo, cisplatino, carboplatino, dacarbazina y ciclofosfamida; antimetabolitos, por ejemplo, 5-fluorouracilo, arabinósido de citosina e hidroxiurea; agentes intercalantes, por ejemplo, adriamicina y bleomicina; inhibidores de topoisomerasa, por ejemplo, etopósido, topotecán e irinotecán; inhibidores de timidilato sintasa, por ejemplo, raltitrexed; inhibidores de la quinasa PI3, por ejemplo, idelalisib; inhibidores de mTor, por ejemplo, everolimus y temsirolimus; inhibidores de proteosomas, por ejemplo, bortezomib; inhibidores de histona desacetilasa, por ejemplo, panobinostat o vorinostat; y bloqueadores de la vía hedgehog tales como vismodegib.
El ingrediente activo farmacéuticamente aceptable adicional puede seleccionarse de inhibidores de tirosina quinasa tales como, por ejemplo, axitinib, dasatinib, erlotinib, imatinib, nilotinib, pazopanib y sunitinib.
Los anticuerpos contra el cáncer pueden incluirse en una terapia combinada y pueden seleccionarse del grupo que consiste en olaratumab, daratumumab, necitumumab, dinutuximab, traztuzumab emtansina, pertuzumab, obinutuzumab, brentuximab, ofatumumab, panitumumab, catumaxomab, bevacizumab, cetuximab, tositumomab, traztuzumab, gentuzumab ozogamicina y rituximab.
El compuesto de la invención o las composiciones farmacéuticas del mismo también se pueden usar en combinación con radioterapia.
Algunas de las combinaciones a las que se ha hecho referencia anteriormente pueden presentarse convenientemente para usar en forma de una formulación farmacéutica y, por lo tanto, las formulaciones farmacéuticas que comprenden una combinación como se define anteriormente junto con un vehículo o excipiente farmacéuticamente aceptable comprenden un aspecto adicional de la invención. Los componentes individuales de dichas combinaciones pueden administrarse secuencial o simultáneamente en formulaciones farmacéuticas separadas o combinadas. Los componentes individuales de las combinaciones también pueden administrarse por separado, a través de la misma vía o de vías diferentes.
Cuando el compuesto de la invención se usa en combinación con un segundo agente terapéutico activo contra el mismo estado patológico, la dosis de cada compuesto puede diferir de la que se usa cuando el compuesto se usa solo. Los expertos en la técnica apreciarán fácilmente las dosis apropiadas.
Dispositivos médicos
En una realización, el compuesto de la invención o las composiciones farmacéuticas que comprenden dicho compuesto pueden formularse para permitir la incorporación en el dispositivo médico, proporcionando así la aplicación del compuesto o composición directamente al sitio para prevenir o tratar las afecciones descritas en el presente documento.
En una realización, el compuesto de la invención o la composición farmacéutica del mismo se formula incluyéndolo dentro de un recubrimiento sobre el dispositivo médico. Hay varios recubrimientos que se pueden utilizar tales como, por ejemplo, recubrimientos poliméricos que pueden liberar el compuesto durante un período de tiempo prescrito. El compuesto, o una composición farmacéutica del mismo, puede incorporarse directamente dentro del dispositivo médico. En algunas realizaciones, el compuesto se aplica como recubrimiento sobre o dentro del dispositivo en un vehículo de administración tal como una micropartícula o un liposoma que facilita su liberación y administración. En algunas realizaciones, el compuesto o composición farmacéutica es miscible en el recubrimiento.
En algunas realizaciones, el dispositivo médico es un implante vascular tal como una endoprótesis. Las endoprótesis se utilizan en medicina para prevenir o eliminar las restricciones vasculares. Los implantes pueden insertarse en un vaso restringido de modo que el vaso restringido se ensancha. El crecimiento excesivo de las células adyacentes después de la implantación vascular da como resultado una restricción del vaso, particularmente en los extremos de los implantes, lo que da como resultado una eficacia reducida de los implantes. Si se inserta un implante vascular en una arteria humana para eliminar, por ejemplo, una estenosis arterioesclerótica, se puede producir hiperplasia de la íntima en el plazo de un año en los extremos del implante vascular y da como resultado una estenosis renovada (“reestenosis”).
Por consiguiente, en algunas realizaciones, las endoprótesis se recubren o cargan con una composición que incluye el compuesto de la invención o su composición farmacéutica y, opcionalmente, una señal de direccionamiento, un vehículo de administración o una combinación de los mismos. Muchas endoprótesis están disponibles comercialmente o son conocidas en la técnica.
En algunas realizaciones, la endoprótesis es una endoprótesis liberadora de fármacos. En la técnica se conocen varias endoprótesis liberadoras de fármacos que administran simultáneamente una sustancia terapéutica en el sitio de tratamiento a la vez que proporcionan un soporte radial artificial al tejido de la pared. Los dispositivos endoluminales que incluyen endoprótesis a veces están recubiertos en sus superficies exteriores con una sustancia tal como un agente de liberación de fármacos, factor de crecimiento o similar. También se han desarrollado endoprótesis que tienen una estructura tubular hueca con orificios o puertos cortados a través de la pared lateral para permitir la elución del fármaco desde una luz central. Aunque la naturaleza hueca de la endoprótesis permite que la luz central se cargue con una solución de fármaco que se administra a través de los puertos u orificios en la pared lateral de la endoprótesis, es posible que la estructura tubular hueca no tenga la resistencia mecánica adecuada para proporcionar un andamiaje adecuado en el vaso.
En algunas realizaciones, los dispositivos también están recubiertos o impregnados con el compuesto de la invención, o una composición farmacéutica del mismo y uno o más agentes terapéuticos adicionales, incluyendo, pero no limitados a agentes antiplaquetarios, agentes anticoagulantes, agentes antiinflamatorios, agentes antimicrobianos, agentes antimetabólicos, agentes anti-neoíntima adicionales, agentes antiproliferativos adicionales, inmunomoduladores, agentes antiproliferativos, agentes que afectan a la migración y la producción de matriz extracelular, agentes que afectan a la deposición de plaquetas o la formación de trombos, y agentes que promueven la cicatrización vascular y la reendotelización, tales como como los descritos en Sousa et al. (2003) y Salu et al. (2004) y otros.
Los ejemplos de agentes antitrombina incluyen, pero no se limitan a, heparina (incluida la heparina de bajo peso molecular), R-hirudina, hirulog, argatroban, efegatran, péptido anticoagulante Tick y Ppack.
Los ejemplos de agentes antiproliferativos incluyen, pero no se limitan a, paclitaxel (taxol), QP-2 vincristina, metotrexato, angiopeptina, mitomicina, BCP 678, c-myc antisentido, ABT 578, actinomicina-D, RestenASE, 1 clordesoxiadenosina, ribozima PCNA y celecoxib.
Los ejemplos de agentes anti-reestenosis incluyen, pero no se limitan a, inmunomoduladores tales como sirolimus (rapamicina), tacrolimus, biorest, mizoribina, ciclosporina, interferón-Y Ib, leflunomida, tranilast, corticosteroide, ácido micofenólico y bifosfonato.
Los ejemplos de agentes antimigratorios y moduladores de la matriz extracelular incluyen, pero no se limitan a, halofuginona, inhibidores de propilhidroxilasa, inhibidores de proteinasa C, inhibidores de MMP, batimastat, probucol.
Los ejemplos de agentes antiplaquetarios incluyen, pero no se limitan a heparina.
Ejemplos de agentes de cicatrización de heridas y promotores de endotelización incluyen el factor de crecimiento epitelial vascular ("VEGF"), 17-estradiol, inhibidores de Tkasa, BCP 671, estatinas, donadores de óxido nítrico ("NO") y células progenitoras endoteliales ("EPC")-anticuerpos.
Además de las aplicaciones coronarias, se pueden incorporar fármacos y agentes activos a la endoprótesis o al recubrimiento de la endoprótesis para otras indicaciones. Por ejemplo, en aplicaciones urológicas, se pueden incorporar agentes antibióticos en la endoprótesis o en el recubrimiento de la endoprótesis para la prevención de infecciones. En aplicaciones gastroenterológicas y urológicas, los agentes activos pueden incorporarse a la endoprótesis o al recubrimiento de la endoprótesis para el tratamiento local de carcinoma. También puede ser ventajoso incorporar en o sobre la endoprótesis un agente de contraste, marcadores radiopacos u otros aditivos para permitir que se formen imágenes de la endoprótesis in vivo para el seguimiento, posicionamiento y otros fines. Dichos aditivos podrían añadirse a la composición absorbible utilizada para fabricar la endoprótesis o el recubrimiento de la endoprótesis, o ser absorbidos en, fundirse en o ser pulverizados sobre la superficie de una parte o la totalidad de la endoprótesis. Los aditivos preferidos para este propósito incluyen plata, yodo y compuestos marcados con yodo, sulfato de bario, óxido de gadolinio, derivados de bismuto, dióxido de circonio, cadmio, tungsteno, oro-tántalo, bismuto, platino, iridio y rodio. Estos aditivos pueden ser, pero no se limitan partículas de tamaño micrométrico o nanométrico o nanopartículas. La radioopacidad puede determinarse por fluoroscopia o por análisis de rayos X.
El compuesto de la invención y uno o más agentes adicionales, o composición farmacéutica del mismo, pueden incorporarse a la endoprótesis, ya sea cargando el compuesto y uno o más agentes adicionales, o su composición farmacéutica en el material absorbible antes del procesamiento, y/ o recubrir la superficie de la endoprótesis con el(los) agente(s). La velocidad de liberación del agente puede controlarse mediante una serie de métodos que incluyen la variación de lo siguiente: la proporción del material absorbible al compuesto y uno o más agentes adicionales, o la composición farmacéutica, el peso molecular del material absorbible, la composición del compuesto y uno o más agentes adicionales, o la composición farmacéutica, la composición del polímero absorbible, el espesor del recubrimiento, el número de capas de recubrimiento y sus espesores relativos, y/o el compuesto y uno o más agentes adicionales, o la concentración de la composición farmacéutica. También se pueden aplicar capas superiores de polímeros y otros materiales, incluidos los polímeros absorbibles, a los recubrimientos de agente activo para controlar la velocidad de liberación. Por ejemplo, se puede aplicar P4HB como una capa superior sobre una endoprótesis metálica recubierta con P4HB que incluye un agente activo para retardar la liberación del agente activo.
La invención se ilustra adicionalmente mediante los siguientes ejemplos no limitantes.
Ejemplos
Las abreviaturas utilizadas en el presente documento se definen a continuación. Cualquier abreviatura no definida pretende transmitir su significado generalmente aceptado.
Abreviaturas
Ac acetilo (C(O)CH3)
ac. acuoso
Ar Anillo aromático
BEH híbrido con puente de etileno
Bz bencilo (CH2-fenilo)
Boc grupo protector ferc-butiloxicarbonilo
CSH híbrido de superficie cargada
d doblete
DCM diclorometano
dioxano 1,4-dioxano
DMF N,N-dimetilformamida
DMSO dimetilsulfóxido
(ES+) ionización por electropulverización, modo positivo
(ES-) ionización por electropulverización, modo negativo
ESI ionización por electropulverización
Et etilo
g gramos
Hal halógeno
HATU hexafluorofosfato del 3-óxido de 1 -[bis(dimetilamino)metilen]-1 H-1,2,3-triazolo[4,5-£>]piridinio HPLC cromatografía líquida de alta resolución
h hora(s)
CI50 concentración inhibidora de 50%
iPr isopropilo
LCMS cromatografía líquida-espectrometría de masas
LHMDS hexametildisilazida de litio
(M+H)+ ion molecular protonado
(M-H)- ion molecular no protonado
M concentración molar
ml mililitro
mm milímetro
mmol milimoles
Me metilo
MHz megahercios
min minuto(s)
MSD detector selectivo de masas
m/z relación masa-carga
N2 nitrógeno gaseoso
nm nanómetro
RMN (espectroscopía) resonancia magnética nuclear
P4HB poli-4-hidroxibutirato
PDA matriz de fotodiodos
PD 174 triflato de alil(2-di-fer-butilfosfino-2',4',6'-triisopropil-1,1'-bifenil)paladio(II) o [tBuXPhosPd(alil)]OTf
PdCl2(dppf) [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II)
Pd(PPh3)4 tetrakis(trifenilfosfina)paladio(0)
PMB 4-metoxibencilo
HPLC prep cromatografía líquida de alta resolución preparativa
Ph fenilo
pos/neg positivo/negativo
q cuartete
RF/EM Espectrometría de masas RapidFire
TA temperatura ambiente
Rt tiempo de retención
RP fase inversa
s singlete
SNAr sustitución nucleofílica aromática
sat. saturado
SCX intercambio catiónico soportado sobre sólido (resina)
Selectfluor Bis(tetrafluoroborato) de W-clorometil-W-fluorotrietilendiamonio
t triplete
tBu íerc-butilo
T3P 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosforinano-2,4,6-trióxido
TFA Ácido trifluoroacético
[í-BuXPhos Pd(alilo)]OTf triflato de alil(2-di-ferc-butilfosfino-2',4',6'-triisopropil-1, 1 '-bifenil)paladio(II)
THF tetrahidrofurano
TMSOK trimetilsilanolato de potasio
UPLC cromatografía líquida de ultra rendimiento
UV ultravioleta
v/v volumen/volumen
VWD detector de onda variable
p peso
um micrómetro
ul microlitro
°C grados Celsius
Procedimientos generales
Todos los materiales de partida y los disolventes se obtuvieron de fuentes comerciales o se prepararon según la bibliografía. A menos que se indique lo contrario, todas las reacciones se agitaron. Las soluciones orgánicas se secaron de forma rutinaria sobre sulfato de magnesio anhidro. Las hidrogenaciones se realizaron en un reactor de flujo Thales H-cube en las condiciones indicadas.
La cromatografía en columna se realizó en cartuchos de sílice preempaquetada (n.° de malla 230-400, 40-63 um) usando la cantidad indicada. El SCX se adquirió de Supelco y se trató con ácido clorhídrico 1 M antes de su uso. A menos que se indique lo contrario, la mezcla de reacción para purificar se diluyó primero con MeOH y se acidificó con unas pocas gotas de AcOH. Esta solución se cargó directamente en el SCX y se lavó con MeOH. Luego, el material deseado se eluyó lavando con NH30,7 M en MeOH.
Cromatografía líquida de alta resolución de fase inversa preparativa
HPLC preparativa
Preparación ácida
Columna Waters X-Select CSH C18, 5 um (19 x 50 mm), caudal 28 ml.min-1 eluyendo con una gradiente de H2O-MeCN que contiene ácido fórmico al 0,1% v/v durante 6,5 min usando detección UV a 254 nm.
Preparación básica
Columna Waters X-Bridge Prep C18, 5 um (19 x 50 mm), caudal 28 ml.min-1 eluyendo con un gradiente de NH4HCO3 10 mM-MeCN durante 6,5 min usando detección UV a 254 nm.
Métodos analíticos
Condiciones de HPLC de fase inversa para los métodos analíticos de LCMS
HPLC ácida: LCMS ácida 4 minutos (5-95%)
La LCMS analítica se llevó a cabo utilizando una columna Waters X-Select CSH C18, 2,5 um, 4,6 x 30 mm, eluyendo con un gradiente de ácido fórmico al 0,1% en MeCN en ácido fórmico al 0,1% en agua. El gradiente de 5-95% de ácido fórmico al 0,1% en MeCN ocurre entre 0,00-3,00 minutos a 2,5 ml/min con un lavado de 3,01-3,5 minutos a 4,5 ml/min. El reequilibrio de una columna a MeCN al 5% es de 3,60-4,00 minutos a 2,5 ml/min. Los espectros UV de los picos eluidos se midieron utilizando un Agilent 1260 Infinity VWD a 254 nm. Los espectros de masas se midieron utilizando un MSD Agilent 6120 funcionando con conmutación positiva/negativa.
HPLC básica: LCMS básica 4 minutos (5-95%)
La LCMS analítica se llevó a cabo utilizando una columna Waters X-Select BEH C18, 2,5 um, 4,6 x 30 mm, eluyendo con un gradiente de MeCN en bicarbonato de amonio 10 mM acuoso. El gradiente de 5-95% de MeCN ocurre entre 0,00-3,00 minutos a 2,5 ml/min con un lavado de 3,01 -3,5 minutos a 4,5 ml/min. El reequilibrio de una columna a MeCN al 5% es de 3,60-4,00 minutos a 2,5 ml/min. Los espectros UV de los picos eluidos se midieron usando un Agilent 1260 Infinity VWD a 254 nm. Los espectros de masas se midieron utilizando un MSD Agilent 6120 funcionando con conmutación positiva/negativa.
Condiciones de HPLC de fase inversa para los métodos analíticos de UPLC
UPLC ácida: UPLC ácida 3 minutos
La UPLC/MS analítica se llevó a cabo utilizando una columna Waters Acquity CSH C18, 1,7 um, 2,1 x 30 mm, eluyendo con un gradiente de ácido fórmico al 0,1% en MeCN en ácido fórmico al 0,1% en agua. El gradiente está estructurado con un punto de partida de 5% de MeCN mantenido 0,0-0,11 minutos. El gradiente de 5-95% ocurre entre 0,11-2,15 minutos con un lavado de 2,15-2,56 minutos. El reequilibrio de una columna a MeCN al 5% es de 2,56-2,83 minutos. Los espectros UV de los picos eluidos se midieron usando un Acquity PDA y los espectros de masas se registraron usando un detector Acquity QDa con conmutación de ESI pos/neg.
UPLC ácida 2: UPLC ácida 1 minuto
La UPLC/MS analítica se llevó a cabo utilizando una columna Waters Acquity CSH C18, 1,7 um, 2,1 x 30 mm, eluyendo con un gradiente de ácido fórmico al 0,1% en MeCN en ácido fórmico al 0,1% en agua. El gradiente está estructurado con un punto de partida de 5% de MeCN mantenido 0,0-0,08 minutos. El gradiente de 5-95% ocurre entre 0,08 y 0,70 minutos con un lavado de 0,7 a 0,8 minutos. El reequilibrio de una columna al 5% de MeCN es de 0,8-0,9 minutos. Los espectros UV de los picos eluidos se midieron usando un Acquity PDA y los espectros de masas se registraron usando un detector Acquity QDa con conmutación de ESI pos/neg.
UPLC básica: UPLC básica 3 minutos
La UPLC/MS analítica se llevó a cabo utilizando una columna Waters Acquity BEH C18, 1,7 um, 2,1 x 30 mm, eluyendo con un gradiente de MeCN en bicarbonato de amonio 10 mM acuoso. El gradiente está estructurado con un punto de partida de 5% de MeCN mantenido 0,0-0,11 minutos. El gradiente de 5-95% ocurre entre 0,11-2,15 minutos con un lavado de 2,15-2,56 minutos. El reequilibrio de una columna a MeCN al 5% es de 2,56-2,83 minutos. Los espectros UV de los picos eluidos se midieron usando un Acquity PDA y los espectros de masas se registraron usando un detector Acquity QDa con conmutación de ESI pos/neg.
UPLC básica 2: UPLC básica 1 minuto
La UPLC/MS analítica se llevó a cabo utilizando una columna Waters Acquity BEH C18, 1,7 um, 2,1 x 30 mm, eluyendo con un gradiente de MeCN en bicarbonato de amonio 10 mM acuoso. El gradiente está estructurado con un punto de partida de 5% de MeCN mantenido 0,0-0,08 minutos. El gradiente de 5-95% ocurre entre 0,08-0,70 minutos con un
lavado de 0,7-0,8 minutos. El reequilibrio de una columna a MeCN al 5% es de 0,8-0,9 minutos. Los espectros UV de los picos eluidos se midieron usando un Acquity PDA y los espectros de masas se registraron usando un detector Acquity QDa con conmutación de ESI pos/neg.
La temperatura de la columna es de 40°C en todas las ejecuciones. El volumen de inyección es de 3 ul y el caudal es de 0,77 ml/min. Barrido de PDA de 210 a 400 nm en todas las ejecuciones.
Condiciones de HPLC de fase normal para los métodos analíticos quirales
Método quiral IC3: HPLC quiral (Diacel Chiralpak IC, 5 um, 4,6 x 250 mm, 1,0 ml/min, 25-70% de EtOH (TFA al 0,2%) en isohexano (TFA al 0,2%)
Método quiral IC5: HPLC quiral (Diacel Chiralpak IC, 5 um, 4,6 x 250 mm, 1,0 ml/min, EtOH al 20% (TFA al 0,2%) en isohexano (TFA al 0,2%).
Condiciones de HPLC de fase inversa para los métodos analíticos quirales
Método quiral IC6: HPLC quiral (Diacel Chiralpak IC, 5 um, 4,6 x 250 mm, 1,0 ml/min, MeCN al 50% (ácido fórmico al 0,1%) en agua (ácido fórmico al 0,1%).
Espectroscopía de RMN 1H
Los espectros de RMN 1H se adquirieron en un espectrómetro Bruker Avance III a 400 MHz o un espectrómetro Bruker Avance III HD a 500 MHz utilizando disolvente no deuterado residual como referencia y, a menos que se especifique lo contrario, se realizaron en DMSO-d6.
Preparación de compuestos intermedios
Los intermedios sintéticos conocidos se consiguieron de fuentes comerciales o se obtuvieron usando procedimientos publicados en la bibliografía. Se prepararon compuestos intermedios adicionales por los procedimientos sintéticos representativos descritos en el presente documento.
2-(2-Cloropirimidin-4-il)malonato de 1-(tere-butilo) y 3-metilo INTC1

Se añadió en porciones NaH (al 60% en peso en aceite mineral, 5,10 g, 128 mmol) a una solución agitada enfriada con hielo de malonato de tere-butilo y metilo (20,5 ml, 121 mmol) en THF (160 ml). La reacción se agitó a 0°C durante 20 min y luego a TA durante 60 min hasta que cesó el desprendimiento de hidrógeno. Luego se añadió 2,4-dicloropirimidina (10 g, 67,1 mmol) y la mezcla resultante se agitó a 70°C durante 3 h. La reacción se dejó enfriar, se repartió entre NH4Cl (ac. sat., 500 ml) y EtOAc (500 ml), las dos fases se separaron y la capa orgánica se pasó a través de un separador de fases. El producto bruto se purificó por cromatografía en gel de sílice (columna de 220 g, EtOAc/isohexano 0-30%) para proporcionar el 2-(2-cloropirimidin-4-il)malonato de 1 -tere-butilo y 3-metilo (13,1 g, 44,3 mmol, 66% de rendimiento) como un aceite transparente de color amarillo pálido; Rt 2,09 min (HPLC ácida); m/z 230 (M+H-tBu)+ (ES+) y 287 (M+H)+ (ES+); RMN 1H (400 MHz, DMSO-d6) 58,83 (d, J = 5,1 Hz, 1 H), 7,65 (d, J = 5,1 Hz, 1H), 5,21 (s, 1 H), 3,73 (s, 3H), 1,42 (s, 9H).
2-(2-Cloropirimidin-4-il)acetato de metilo INTC4

Se añadió gota a gota TFA (55,3 ml, 717 mmol) a una solución agitada enfriada con hielo de 2-(2-cloropirimidin-4-il)malonato de 1 -tere-butilo y 3-metilo INTC1 (12,1 g, 42,2 mmol) en DCM (50 ml). La reacción se agitó a 25°C durante 1 h y luego se concentró al vacío. El residuo se disolvió en EtOAc (200 ml) y se hizo básico con NaHCO3 (200 ml), la capa orgánica se aisló y se pasó por un separador de fases, el disolvente se eliminó al vacío. El producto bruto se purificó por cromatografía en gel de sílice (cartucho de 220 g, EtOAc/isohexano 0-50%) para proporcionar el 2-(2-cloropirimidin-4-il)acetato de metilo (7,12 g, 37,8 mmol, 90% de rendimiento) como un aceite amarillo pálido. Rt 1,16 min (HPLC ácida); m/z 187 (M+H)+ (ES+); RMN 1 H (500 MHz, DMSO-d6) 58,76 (d, J = 5,0 Hz, 1 H), 7,60 (d, J = 5,0 Hz, 1H), 3,96 (s, 2H), 3,66 (s, 3H).
4-(2-Cloropirimidin-4-il)tetrahidro-2H-piran-4-carboxilato de metilo INTC52

A una solución de 2-(2-cloropirimidin-4-il)acetato de metilo INTC4 (2,0 g, 10,7 mmol) en DMF (10 ml, 10,7 mmol) a 0°C se añadió NaOH (0,986 g, 24,6 mmol). La mezcla de reacción se agitó a 0°C durante 20 min y luego se añadió 1-bromo-2-(2-bromoetoxi)etano (1,8 ml, 12,9 mmol). La reacción se agitó a TA durante 23 h. La mezcla de reacción se acidificó usando HCl 1 M (ac., 53,6 ml, 53,6 mmol) antes de extraer con DCM (70 ml). Las fases se separaron utilizando un cartucho separador de fases y la fase acuosa se extrajo con DCM adicional (2 x 50 ml). Los compuestos orgánicos combinados se concentraron al vacío. El producto bruto se purificó por cromatografía en gel de sílice (columna de 80 g, EtOAc/isohexano 0-50%) para producir el 4-(2-cloropirimidin-4-il)tetrahidro-2H-piran-4-carboxilato de metilo (1,83 g, 5,57 mmol, 52% de rendimiento) como un aceite amarillo. Rt 1,56 min (HPLC, ácida); m/z 257 (35Cl M+H)+ (ES+); RMN 1H (500 MHz, DMSO-d6) 58,80 (d, J = 5,3 Hz, 1H), 7,69 (d, J = 5,3 Hz, 1 H), 3,72-3,67 (m, 2H), 3,66 (s, 3H), 3,55-3,50 (m, 2H), 2,33 - 2,22 (m, 2H), 2,16 - 2,06 (m, 2H).
Método C: Formación de sulfonamidas a partir de haluros aromáticos

Se disolvieron la 2-cloropirimidina intermedia (1 equiv.), sulfonamida (1,2 equiv.) y base (2 equiv.) en dioxano (40 volúmenes). La mezcla se desgasificó (N2, 5 min) y luego se añadió catalizador (5% en moles). La mezcla resultante se calentó en atmósfera de nitrógeno a 90°C durante 2 h. La mezcla se filtró, se lavó con EtOAc o DCM y el filtrado resultante se concentró. El producto bruto se purificó por cromatografía en fase normal o trituración utilizando un disolvente adecuado.
Tabla 1: El siguiente compuesto intermedio se preparó de acuerdo con el Método C.

Derivado de tetrahidropirano a través de tioéter
4-(2-(Metiltio)pirimidin-4-il)tetrahidro-2H-piran-4-carboxilato de metilo INTC178

A una solución de 4-cloro-2-(metiltio)pirimidina (0,55 g, 3,42 mmol) y tetrahidro-2H-piran-4-carboxilato de metilo (494 mg, 3,42 mmol) en THF (5 ml) a 30°C se añadió gota a gota LHMDS (1 M en THF) (4,11 ml, 4,11 mmol). La mezcla de reacción se agitó a 30°C durante 5 min, luego se vertió en agua (100 ml) y se extrajo con EtOAc (2 x 200 ml). El extracto orgánico se lavó con salmuera (1 x 100 ml), se secó (MgSO4), se filtró y el disolvente se separó al vacío para proporcionar el 4-(2-(metiltio)pirimidin-4-il)tetrahidro-2H-piran-4-carboxilato de metilo (915 mg, 3,24 mmol, 95% de
rendimiento) como un aceite amarillo pálido. Rt 1,74 min (HPLC ácida); m/z 269 (M+H)+ (ES+); RMN 1H (500 MHz, DMSO-d6) 58,62 (d, J = 5,3 Hz, 1 H), 7,27 (d, J = 5,3 Hz, 1H), 3,76-3,70 (m, 2H), 3,67 (s, 3H) , 3,54-3,46 (m, 2H), 2,49 (s, 3H), 2,27-2,20 (m, 2H), 2,14-2,04 (m, 2H).
4-(2-(Metilsulfonil)pirimidin-4-il)tetrahidro-2H-piran-4-carboxilato de metilo INTC179

Se añadió en porciones mCPBA (1,60 g, 7,13 mmol) a una solución en agitación de 4-(2-(metiltio)pirimidin-4-il)tetrahidro-2H-piran-4-carboxilato de metilo INTC178 (915 mg, 3,24 mmol) en DCM (50 ml) y la mezcla de reacción resultante se agitó a TA durante 3 h. La mezcla de reacción se vertió en solución sat. de NaHCO3 (ac, 200 ml) y se extrajo con DCM (3 x 100 ml). El extracto orgánico se lavó secuencialmente con solución sat. de NaHCO3 (ac, 100 ml) y salmuera (100 ml), se secó (MgSO4), se filtró y se separó el disolvente al vacío para dar el 4-(2-(metilsulfonil)pirimidin-4-il)tetrahidro-2H-piran-4-carboxilato de metilo (1,10 g, 3,30 mmol, rendimiento cuantitativo) como goma espesa. Rt 1,20 min (HPLC ácida); m/z 301 (M+H)+ (ES+); RMN 1H (500 MHz, DMSO-d6) 59,09 (d, J = 5,3 Hz, 1H), 7,95 (d, J = 5,3 Hz, 1 H), 3,77-3,70 (m, 2H), 3,68 (s, 3H) , 3,60-3,49 (m, 2H), 3,42 (s, 3H), 2,34-2,24 (m, 2H), 2,23 -2,13 (m, 2H).
4-(2-(Ciclopropanosulfonamido)pirimidin-4-il)tetrahidro-2H-piran-4-carboxilato de metilo INTC53

A una solución de 4-(2-(metilsulfonil)pirimidin-4-il)tetrahidro-2H-piran-4-carboxilato de metilo INTC179 (1,0 g, 3,33 mmol) y ciclopropanosulfonamida (0,52 g, 4,33 mmol) en NMP (100 ml) se añadió carbonato de cesio (3,25 g, 9,99 mmol) y se calentó a 90°C durante 1 h. La mezcla de reacción se enfrió a TA y se diluyó con agua (100 ml) y la mezcla se lavó con MTBE (2 x 100 ml) y la fase acuosa se acidificó lentamente a pH 3 usando HCl diluido (20 ml). El precipitado resultante se filtró para proporcionar el 4-(2-(ciclopropanosulfonamido)pirimidin-4-il)tetrahidro-2H-piran-4-carboxilato de metilo (755 mg, 2,21 mmol, 66% de rendimiento) como un sólido incoloro. Rt 0,88 (UPLC, ácida), m/z 342 (M+H)+ (ES+); RMN 1H (500 MHz, DMSO-d6) 511,30 (s, 1H), 8,60 (d, J = 5,3 Hz, 1 H), 7,20 (d, J = 5,3 Hz, 1 H), 3,79-3,72 (m, 2H), 3,67 (s, 3H), 3,52-3,44 (m, 2H), 3,25 - 3,14 (m, 1H), 2,30 - 2,17 (m, 2H), 2,12-2,04 (m, 2H), 1,14 - 1,01 (m, 4H).
Preparación de amina intermedia
Método F: Acoplamiento de Suzuki de haluros heteroaromáticos con boronatos de anilina
Se añadió catalizador de paladio (5% en moles) a una solución desgasificada (N2, 5 min) de (hetero)aril-X (1 eq; X = B(OH)2, B (pin)2), etoxipirazina-Z (1 eq; Z = Br, Cl) y base (3 eq, 6,85 mmol) en disolvente (3 volúmenes). Luego, la solución se desgasificó adicionalmente (N2, 5 min) y luego se calentó a 90°C durante 2 h y luego se dejó enfriar a TA. En general, el compuesto deseado se purificó por cromatografía en columna.
Tabla 2: El siguiente compuesto intermedio se preparó de acuerdo con el método F.

Preparación del Ejemplo P115
Método 2: acoplamiento de amida mediado por AlMe3 a partir de éster
A una solución enfriada con hielo de anilina (2 eq) en tolueno (40 volúmenes) se añadió AlMe3 (2,0 M en heptano, 2 eq). La mezcla se agitó a esta temperatura durante 5 min y luego a TA durante 10 min. A esta solución se añadió éster (1 eq) en una porción y la mezcla resultante se calentó y agitó a 80°C durante 2 h. La mezcla de reacción se enfrió en un baño de hielo y se inactivó cuidadosamente con MeOH (10 volúmenes). Después de agitar durante 20 min, la mezcla se diluyó en una mezcla de DCM/MeOH (10 volúmenes), se filtró a través de celite y el filtrado se concentró. El producto bruto se purificó por cromatografía de fase inversa o normal.
El siguiente compuesto se preparó de acuerdo con el Método 2:
Tabla 3: Métodos de preparación y datos de caracterización del Ejemplo P115

Los nombres químicos, fórmulas estructurales y datos de caracterización de los ejemplos de referencia se describen en la Tabla 4.
Tabla 4: Ejemplos de referencia



Ejemplos biológicos
Ejemplo biológico 1 - Inhibición de la enzima CTPS1 humana
Las actividades inhibidoras de enzimas de los compuestos inventados contra el objetivo de interés se determinaron utilizando el ensayo ADP-Glo™ Max (Promega, Reino Unido). Los ensayos para la CTPS1 humana se realizaron en 1 x tampón de ensayo que contenía Tris 50 mM, MgCl2 10 mM, Tween-20 al 0,01%, pH a 8,0 consecuentemente. Finalmente, inmediatamente antes del uso, se añadió L-cisteína al 1 x tampón de ensayo hasta una concentración final de 2 mM. Todos los reactivos son de Sigma-Aldrich a menos que se especifique lo contrario. La CTPS1 marcada con FLAG-His8 C-terminal activa de longitud completa humana (UniProtKB - P17812, CTPS[1-591]-GGDYKDDDDKGGHHHHHHHH) se obtuvo de Proteros biostructures GmbH.
Procedimiento de ensayo
Se prepararon 3x proteína CTPS1 humana en 1x tampón de ensayo hasta la concentración de proteína de trabajo final requerida para la reacción. Se mezcló un volumen de 2 ul por pocillo de 3x proteína CTPS1 humana con 2 ul por pocillo de 3x compuesto de ensayo (compuesto preparado en 1x tampón de ensayo hasta una 3x concentración de compuesto final apropiada correspondiente a la curva de respuesta a la concentración diseñada para los compuestos de ensayo) durante 10 minutos a 25°C. A continuación, se inició la reacción enzimática mediante la adición de un volumen de 2 ul por pocillo de una mezcla de sustrato premezclada (ATP UltraPuro del kit ADP-Glo™ Max (0,31 mM), GTP (0,034 mM), UTP (0,48 mM) y L-glutamina (0,186 mM)) y la mezcla se incubó durante un período de tiempo adecuado dentro de la fase lineal determinada de la reacción a 25°C en condiciones de placa sellada con agitación constante a 500 revoluciones por minuto (rpm). Se añadió el reactivo ADP-Glo™ Max durante 60 minutos (6 gl por pocillo) y posteriormente se añadió el reactivo de desarrollo ADP-Glo™ Max durante 60 minutos (12 ul por pocillo) antes de la detección de la señal en un lector de microplacas (EnVision® Multilabel Reader, Perkin Elmer). Después de cada adición de reactivo en el transcurso del ensayo, las placas de ensayo se centrifugaron por pulsos durante 30 segundos a 500 rpm.
En todos los casos, la enzima convierte el ATP en ADP y el reactivo ADP-Glo™ Max posteriormente agota cualquier ATP endógeno restante en el sistema de reacción. El reactivo de detección ADP-Glo™ Max convierte el ADP que se ha producido enzimáticamente de nuevo en ATP y, utilizando ATP como sustrato junto con luciferina para la enzima luciferasa, se genera luz que produce una luminiscencia detectable. La señal luminiscente medida es directamente proporcional a la cantidad de ADP producida por la reacción enzimática y una reducción de esta señal tras el tratamiento del compuesto demuestra la inhibición de la enzima. El porcentaje de inhibición producido por cada concentración de compuesto se calculó utilizando la ecuación que se muestra a continuación:
% de Inhibición = i - (Median - Media**,) x 100
(MediaMín - MediaMáx)
A continuación, se representó gráficamente el porcentaje de inhibición frente a la concentración del compuesto, y la concentración inhibidora de 50% (CI50) se determinó a partir de la curva de concentración-respuesta resultante.
Los datos para el compuesto de la invención y ciertos ejemplos de referencia ensayados se presentan a continuación.
Tabla 5: Datos de inhibición de la enzima CTPS1 humana agrupados por intervalo de potencia (++ indica CI50 en el intervalo >0,1 a 1 micromolar, ++ indica CI50 á0,1 micromolar)

Se encontró que todos los compuestos de referencia y el compuesto de la invención que se habían ensayado demostraban la inhibición de la enzima CTPS1 en este ensayo. En consecuencia, se puede esperar que estos compuestos tengan utilidad en la inhibición de CTPS1. También se espera que los compuestos de referencia y el compuesto de la invención tengan utilidad como herramientas de investigación, por ejemplo, para uso en ensayos de CTPS.
Ejemplo biológico 2 - Ensayos de selectividad enzimática basados en RapidFire/MS.
Evaluación de la selectividad de CTPS1 frente a CTPS2 humanas mediante análisis de RapidFire/MS.
Las actividades inhibidoras de la enzima frente a cada isoforma diana de interés pueden determinarse para los compuestos de la invención utilizando un formato de ensayo de espectrometría de masas de alto rendimiento-RapidFire optimizado (RF/MS). Los ensayos de RF/MS para CTPS1 y CTPS2 humanas se pueden realizar en un tampón de ensayo que consiste en HEPES 50 mM (Merck), MgCl220 mM, KCI 5 mM, DTT 1 mM, Tween-20 al 0,01%, pH a 8,0 consecuentemente. La CTPS1 marcada con FLAG-His C-terminal activa de longitud completa humana (UniProtKB - P17812, CTPS[1-591]-GGDYKDDDDKGGHHHHHHHH) se puede obtener de Proteros biostructures GmbH. La CTPS2 marcada con FLAG-His-Avi C-terminal activa de longitud completa humana (UniProtKB - Q9NRF8, CTPS2 [1-586]-DYKDDDDKHHHHHHGLNDIFEAQKIEWHE) se puede obtener de Harker Bio.
Procedimiento de ensayo
La proteína CTPS humana (1 o 2) se puede preparar en 1x tampón de ensayo hasta la concentración de proteína de trabajo final requerida para la reacción. Se puede mezclar un volumen de 2 ul por pocillo de 2x proteína CTPS (1 o 2) con 40 nl de compuesto usando administración acústica (ECHO) e incubar durante 10 minutos a 25°C. Cada reacción enzimática de isoforma puede iniciarse posteriormente mediante la adición de 2 ul por pocillo de una 2x mezcla de sustrato en tampón de ensayo. Para hCTPS1: ATP (0,3 mM), UTP (0,2 mM), GTP (0,07 mM) y L-glutamina (0,1 mM). Para hCTPS2: At P (0,1 mM), UTP (0,04 mM), GTP (0,03 mM) y L-glutamina (0,1 mM). Cada mezcla se puede incubar durante una cantidad de tiempo apropiada por isoforma dentro de la fase lineal determinada de la reacción a 25°C. Se puede añadir un volumen de 60 ul de solución de parada (ácido fórmico al 1% con 13C9-15N3-CTP 0,5 uM en H2O) y sellar inmediatamente con calor la placa y centrifugar durante 10 minutos a 4000 rpm. Después de la centrifugación, las placas se pueden cargar en el sistema de extracción en fase sólida de microfluidos Agilent RapidFire acoplado a un espectrómetro de masas (RF/MS) de triple cuadrupolo API4000 para su análisis.
En todos los casos, la enzima convierte UTP en CTP. Los métodos de MS de seguimiento de reacciones múltiples (MRM) altamente específicos y sensibles pueden optimizarse para la detección del producto de la reacción enzimática, CTP y el patrón de producto marcado con isótopos estables 13C9-15N3-CTP. La lectura para el análisis de datos puede calcularse como la relación entre el área del pico del producto CTP y el patrón interno 13C9-15N3-CTP. Para el informe de datos, se puede utilizar la siguiente ecuación:

(R = relación/lectura, P = área de señal del producto, IS = área de la señal del patrón interno)
Para cada placa de cribado, se utilizaron las medias de los valores de control negativo (DMSO) y positivo para el cálculo de la ventana de ensayo respectiva (S/B) y los valores Z'. La mediana de los respectivos valores de control se utilizó para calcular el porcentaje de inhibición de acuerdo con la siguiente ecuación:

(I = Inhibición, Rneg = mediana de los valores de lectura del control negativo, Rpos = mediana de los valores de lectura del control positivo, Rmuestra = valor de lectura de la muestra)
A continuación, se representó gráficamente el porcentaje de inhibición frente a la concentración del compuesto, y la concentración inhibidora de 50% (CI50) se determinó a partir de la curva de concentración-respuesta resultante.
El número de veces de selectividad entre CTPS1 y CTPS2 se calculó posteriormente de acuerdo con la siguiente ecuación:
N.° veces selectividad = CTPS2 CL
CTPS1 Cl50
Ciertos compuestos de referencia y el compuesto de la invención se ensayaron en el ensayo anterior. Los datos para todos los compuestos ensayados se presentan a continuación.
Tabla 6: Datos de selectividad divididos en grupos de 2-30 veces (+) o >60 veces (+++)


Se encontró que todos los compuestos de referencia y el compuesto de la invención analizados en el ensayo descrito en el Ejemplo biológico 2 tenían una selectividad de al menos 2 veces por CTPS1 frente a CTPS2, y muchos compuestos tenían una selectividad por CTPS1 de más de 60 veces. En particular, puede esperarse que estos compuestos tengan utilidad en el tratamiento de enfermedades en las que es beneficioso un compuesto selectivo para CTPS1.
A lo largo de la memoria descriptiva y las reivindicaciones que siguen, a menos que el contexto requiera lo contrario, la palabra “comprender”, y variaciones tales como “comprende” y “que comprende”, se entenderá que implica la inclusión de un número entero, etapa, grupo de números enteros o grupo de etapas expuestos pero sin exclusión de cualquier otro número entero, etapa, grupo de números enteros o grupo de etapas.
La solicitud de la que forman parte esta descripción y reivindicaciones puede utilizarse como base para la prioridad con respecto a cualquier solicitud posterior. Las reivindicaciones de dicha solicitud posterior pueden estar dirigidas a cualquier característica o combinación de características descritas en el presente documento. Pueden tomar la forma de reivindicaciones de producto, composición, procedimiento o uso y pueden incluir, a modo de ejemplo y sin limitación, las reivindicaciones que siguen.
Referencias
Evans, D. R. & Guy, H. I. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J. Biol. Chem.
279, 33035-33038 (2004).
Fairbanks, L. D. et al. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans. Disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. J. Biol. Chem.
270, 29682-29689 (1995).
Higgins, M. J. et al. Regulation of human cytidine triphosphate synthetase 1 by glycogen synthase kinase 3. J. Biol. Chem. 282, 29493-29503 (2007).
Kursula, P. et al. Structure of the synthetase domain of human CTP synthetase, a target for anticancer therapy. Acta Crystallogr Sect F Struct Biol Cryst Commun. 62 (Pt7): 613-617 (2006).
Lieberman I. Enzymatic amination of uridine triphosphate to cytidine triphosphate. The J. Biol. Chem. 222 (2): 765 75 (1956).
Martin E. et al. CTP synthase 1 deficiency in humans reveals its central role in lymphocytes proliferation. Nature. Jun 12; 510(7504):288-92 (2014). Erratum in: Nature. Jul 17; 511(7509):370 (2014).
McCluskey GD et al., Exploring the Potent Inhibition of CTP Synthase by Gemcitabine-5'-Triphosphate. Chembiochem. 17, 2240-2249 (2016).
Ostrander, D. B. et al. Effect of CTP synthetase regulation by CTP on phospholipid synthesis in Saccharomyces cerevisiae. J. Biol. Chem. 273, 18992-19001 (1998).
Sakamoto K. et al. Identification of cytidine-5-triphosphate synthase1 -selective inhibitory peptide from random peptide library displayed on T7 phage. Peptides. 2017; 94:56-63 (2017).
Salu et al. Drug-eluting stents: a new treatment in the prevention of restenosis Part I: experimental studies. Acta Cardiol, 59, 51-61 (2004).
Sousa J. E. et al. Drug-Eluting Stents. Circulation, 107 (2003) 2274 (Part I), 2283 (Part II).
Tang R. et al. CTP synthase 1, a smooth muscle-sensitive therapeutic target for effective vascular repair. Arterioscler Thromb Vasc Biol. 33(10), 1-19, (2013).
van den Berg, A. A. et al. Cytidine triphosphate (CTP) synthetase activity during cell cycle progression in normal and malignant T-lymphocytic cells. Eur. J. Cancer 31, 108-112 (1995).
van Kuilenburg, A.B.P. et al. Identification of a cDNA encoding an isoform of human CTP synthetase. Biochimica et Biophysica Acta 1492548-552 (2000).
Claims (17)
1. Un compuesto que es la 4-(2-(ciclopropanosulfonamido)pirimidin-4-il)-W-(5-(6-etoxipirazin-2-il)piridin-2-il)tetrahidro-2H-piran-4-carboxamida:

o una de sus sales y/o solvatos.
2. El compuesto 4-(2-(ciclopropanosulfonamido)pirimidin-4-il)-W-(5-(6-etoxipirazin-2-il)piridin-2-il)tetrahidro-2H-piran-4-carboxamida:

o una de sus sales y/o solvatos farmacéuticamente aceptables según la reivindicación 1.
3. La sal y el solvato farmacéuticamente aceptables de la 4-(2-(ciclopropanosulfonamido)pirimidin-4-il)-W-(5-(6-etoxipirazin-2-il)piridin-2-il)tetrahidro-2H-piran-4- carboxamida:
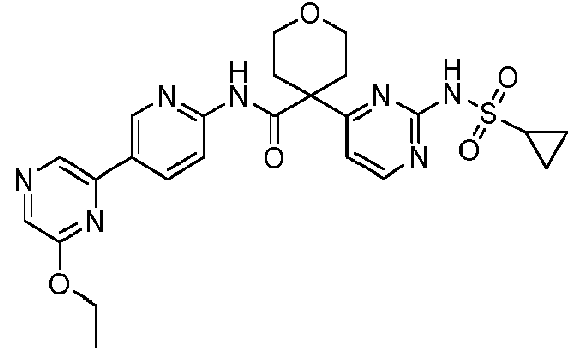
según la reivindicación 2.
4. El solvato farmacéuticamente aceptable de la 4-(2-(ciclopropanosulfonamido)pirimidin-4-il)-W-(5-(6-etoxipirazin-2-il)piridin-2-il)tetrahidro-2H-piran-4-carboxamida:

según la reivindicación 2.
5. La sal farmacéuticamente aceptable de la 4-(2-(ciclopropanosulfonamido)pirimidin-4-il)-W-(5-(6-etoxipirazin-2-il)piridin-2-il)tetrahidro-2H-piran-4-carboxamida:

según la reivindicación 2.
6. El compuesto según la reivindicación 1 que es la 4-(2-(ciclopropanosulfonamido)pirimidin-4-il)-W-(5-(6-etoxipirazin-2-il)piridin-2-il)tetrahidro-2H-piran-4-carboxamida:

7. El compuesto, sal y/o solvato farmacéuticamente aceptable del mismo según una cualquiera de las reivindicaciones 2 a 6, para usar como medicamento.
8. El compuesto, sal y/o solvato farmacéuticamente aceptable del mismo según la reivindicación 7, para usar en la reducción de la proliferación de células T y/o células B en un sujeto.
9. El compuesto, sal y/o solvato farmacéuticamente aceptable del mismo según la reivindicación 7, para usar en el tratamiento o profilaxis de: enfermedades inflamatorias de la piel tales como psoriasis o liquen plano; GVHD aguda y/o crónica tal como GVHD aguda resistente a esteroides; síndrome linfoproliferativo agudo (ALPS); lupus eritematoso sistémico, nefritis lúpica o lupus cutáneo; o trasplante; o para usar en el tratamiento o profilaxis de miastenia grave, esclerosis múltiple o esclerodermia/esclerosis sistémica; o para usar para mejorar la recuperación de una lesión vascular o cirugía y reducir la morbilidad y mortalidad asociadas con la neoíntima y la reestenosis en un sujeto.
10. El compuesto, sal y/o solvato farmacéuticamente aceptable del mismo según la reivindicación 7, para usar en el tratamiento del cáncer.
11. El compuesto, sal y/o solvato farmacéuticamente aceptable del mismo según la reivindicación 10, en donde el cáncer es un cáncer hematológico.
12. El compuesto, sal y/o su solvato farmacéuticamente aceptable del mismo según la reivindicación 11, en donde el cáncer hematológico se selecciona del grupo que consiste en leucemia mieloide aguda, linfoma de células T angioinmunoblásticas, leucemia linfoblástica aguda de células B, síndrome de Sweet, linfoma no Hodgkins de células T (incluido linfoma de células T/citolíticas naturales, leucemia/linfoma de células T del adulto, linfoma de células T tipo enteropatía, linfoma hepatoesplénico de células T y linfoma cutáneo de células T), leucemia linfoblástica aguda de células T, linfoma no Hodgkins de células B (incluido el linfoma de Burkitt, linfoma difuso de células B grandes, linfoma folicular, linfoma de células del manto, linfoma de la zona marginal), leucemia de células pilosas, linfoma de Hodgkin, linfoma linfoblástico, linfoma linfoplasmocitario, linfoma de tejido linfoide asociado a mucosas, mieloma múltiple, síndrome mielodisplásico, mieloma de células plasmáticas, linfoma mediastinal primario de células B grandes, trastornos mieloproliferativos crónicos (tales como leucemia mieloide crónica, mielofibrosis primaria, trombocitemia esencial, policitemia vera) y leucemia linfocítica crónica.
13. El compuesto, sal y/o solvato farmacéuticamente aceptable del mismo según la reivindicación 10, en donde el cáncer es un cáncer no hematológico.
14. El compuesto, sal y/o solvato farmacéuticamente aceptable del mismo según la reivindicación 13, en donde el cáncer no hematológico se selecciona del grupo que consiste en cáncer de vejiga, mama, melanoma, neuroblastoma, mesotelioma pleural maligno y sarcoma.
15. El compuesto, sal y/o solvato farmacéuticamente aceptable del mismo según una cualquiera de las reivindicaciones 7 a 14, para usar en un sujeto humano.
16. Una composición farmacéutica que comprende el compuesto, sal y/o solvato farmacéuticamente aceptable del mismo según una cualquiera de las reivindicaciones 2 a 6.
17. Un compuesto de fórmula (II) que es:

en donde R es H o alquilo Ci-6, o sales del mismo.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18202136 | 2018-10-23 | ||
| PCT/EP2018/086617 WO2019179652A1 (en) | 2018-03-23 | 2018-12-21 | Aminopyrimidine derivatives as ctps1 inhibitors |
| PCT/EP2019/057320 WO2019180244A1 (en) | 2018-03-23 | 2019-03-22 | Aminopyrimidine derivatives as ctps1 inhibitors |
| PCT/EP2019/078848 WO2020083975A1 (en) | 2018-10-23 | 2019-10-23 | Aminopyrimidine/pyrazine derivatives as ctps1 inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2941981T3 true ES2941981T3 (es) | 2023-05-29 |
Family
ID=63965420
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19789997T Active ES2941981T3 (es) | 2018-10-23 | 2019-10-23 | Derivados de aminopirimidina/pirazina como inhibidores de CTPS1 |
Country Status (27)
| Country | Link |
|---|---|
| US (3) | US11987573B2 (es) |
| EP (1) | EP3870574B1 (es) |
| JP (1) | JP7308263B2 (es) |
| CN (1) | CN112839938B (es) |
| AU (1) | AU2019367191B2 (es) |
| BR (1) | BR112021005986A2 (es) |
| CA (1) | CA3115568C (es) |
| CR (1) | CR20210265A (es) |
| DK (1) | DK3870574T3 (es) |
| EA (1) | EA202191104A1 (es) |
| ES (1) | ES2941981T3 (es) |
| FI (1) | FI3870574T3 (es) |
| HR (1) | HRP20230328T1 (es) |
| HU (1) | HUE061652T2 (es) |
| IL (1) | IL282386B2 (es) |
| LT (1) | LT3870574T (es) |
| MX (1) | MX2021004659A (es) |
| MY (1) | MY203303A (es) |
| NZ (1) | NZ774355A (es) |
| PL (1) | PL3870574T3 (es) |
| PT (1) | PT3870574T (es) |
| RS (1) | RS64081B1 (es) |
| SG (1) | SG11202103823PA (es) |
| SI (1) | SI3870574T1 (es) |
| SM (1) | SMT202300090T1 (es) |
| WO (1) | WO2020083975A1 (es) |
| ZA (1) | ZA202101788B (es) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11505547B2 (en) | 2017-11-30 | 2022-11-22 | Step Pharma S.A.S. | Compounds |
| US11655246B2 (en) | 2017-11-30 | 2023-05-23 | Step Pharma S.A.S. | Aminothiazole compounds as inhibitors of CTPS1 |
| US20230086703A1 (en) | 2019-06-04 | 2023-03-23 | Step Pharma S.A.S. | N-(4-(5-chloropyridin-3-yl)phenyl)-2-(2-(cyclopropanesulfonamido)pyrimidin-4-yl) butanamide derivatives and related compounds as human CTPS1 inhibitors for the treatment of proliferative diseases |
| WO2020245664A1 (en) | 2019-06-04 | 2020-12-10 | Step Pharma S.A.S. | N-(5-(6-ethoxypyrazin-2-yl)pyridin-2-yl)-4-(2-(methylsulfonamido)pyrimidin-4-yl) tetrahydro-2h-pyran-4-carboxamide derivatives and related compounds as human ctps1 inhibitors for the treatment of proliferative diseases |
| US20230002352A1 (en) * | 2019-09-20 | 2023-01-05 | Step Pharma S.A.S. | Sulfonamide derivatives as ctps1 inhibitors |
| EP4041715A1 (en) | 2019-09-20 | 2022-08-17 | Step Pharma S.A.S. | Sulfonamide inhibitors as ctps 1 inhibitors |
| JP2025508896A (ja) | 2022-03-01 | 2025-04-10 | ステップ ファーマ エス.エー.エス | 癌のためのctps1阻害剤とbcl2阻害剤の組合せ |
| WO2023166080A1 (en) | 2022-03-01 | 2023-09-07 | Step Pharma S.A.S. | Combination treatments comprising a ctps1 inhibitor and a wee1 inhibitor |
| WO2023166078A1 (en) | 2022-03-01 | 2023-09-07 | Step Pharma S.A.S. | Combination treatments comprising a ctps1 inhibitor and a chek1 inhibitor |
| WO2023166077A1 (en) | 2022-03-01 | 2023-09-07 | Step Pharma S.A.S. | Combination of a ctps1 inhibitor and a atr inhibitor in cancer therapy |
| KR20250118853A (ko) | 2022-12-21 | 2025-08-06 | 스텝 파마 에스.에이.에스. | Ctps2 결핍성 암의 치료에 사용하기 위한 ctps1 억제제 |
| WO2024133721A1 (en) | 2022-12-21 | 2024-06-27 | Step Pharma S.A.S. | Combinations of ctps1 inhibitor with iap inhibitor for use in the treatment of cancer |
| CN118359588A (zh) * | 2023-01-19 | 2024-07-19 | 杭州英创医药科技有限公司 | 作为ctps1抑制剂的化合物 |
| WO2026027753A1 (en) * | 2024-08-02 | 2026-02-05 | Step Pharma S.A.S. | Ctps1 inhibitors for the treatment of myeloproliferative neoplasms |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1575803A (en) | 1976-03-09 | 1980-10-01 | Fujisawa Pharmaceutical Co | 3,7 disubstituted 3 cephem 4 carboxylic acid compounds andprocesses for the preparation thereof |
| GB1555007A (en) | 1976-05-25 | 1979-11-07 | Fujisawa Pharmaceutical Co | Intermediates in the preparation of substituted cepham carboxylic acid derivatives and the preparation thereof |
| KR100564902B1 (ko) | 2000-08-21 | 2006-03-30 | 주식회사 태평양 | 신규 티오우레아 유도체 및 이를 함유하는 약제학적 조성물 |
| JP2004509874A (ja) | 2000-09-25 | 2004-04-02 | アクテリオン ファマシューティカルズ リミテッド | 新規なアリールアルカン−スルフォンアミド類 |
| US20030158218A1 (en) | 2001-12-21 | 2003-08-21 | Nantermet Philippe G. | Thrombin inhibitors |
| ES2325836T3 (es) | 2004-07-26 | 2009-09-21 | Merck Serono Sa | Derivados de la n.hidroxiamida y uso de los mismos. |
| EP1659113A1 (en) * | 2004-11-08 | 2006-05-24 | Evotec AG | Inhibitors of 11beta-hydroxy steroid dehydrogenase type 1 (11beta-HSD1) |
| AR063706A1 (es) | 2006-09-11 | 2009-02-11 | Cgi Pharmaceuticals Inc | Determinadas amidas sustituidas, el uso de las mismas para el tratamiento de enfermedades mediadas por la inhibicion de la actividad de btk y composiciones farmaceuticas que las comprenden. |
| WO2009075874A1 (en) * | 2007-12-13 | 2009-06-18 | Amgen Inc. | Gamma secretase modulators |
| KR101097312B1 (ko) | 2009-08-10 | 2011-12-23 | 삼성모바일디스플레이주식회사 | 유기 발광 소자 |
| MX2015007291A (es) | 2012-12-13 | 2015-09-10 | Hoffmann La Roche | Derivados de tiazol como inhibidores de la tirosina cinasa de bruton. |
| JP2016028795A (ja) | 2013-03-28 | 2016-03-03 | 国立研究開発法人科学技術振興機構 | 触媒金属ナノ粒子含有複合体及びその利用 |
| CN105473136A (zh) * | 2013-04-18 | 2016-04-06 | 国家健康与医学研究院 | 用于在有此需要的受试者中抑制淋巴细胞增殖的方法和药物组合物 |
| WO2014186435A2 (en) * | 2013-05-14 | 2014-11-20 | University Of Georgia Research Foundation, Inc. | Compositions and methods for reducing neointima formation |
| GB201322333D0 (en) | 2013-12-17 | 2014-01-29 | Agency Science Tech & Res | WNT pathway modulators |
| CN104262071A (zh) | 2014-10-16 | 2015-01-07 | 中国科学院山西煤炭化学研究所 | 一种联苯类化合物的光催化合成方法 |
| US11655246B2 (en) | 2017-11-30 | 2023-05-23 | Step Pharma S.A.S. | Aminothiazole compounds as inhibitors of CTPS1 |
| US11505547B2 (en) | 2017-11-30 | 2022-11-22 | Step Pharma S.A.S. | Compounds |
| WO2019180244A1 (en) | 2018-03-23 | 2019-09-26 | Step Pharma S.A.S. | Aminopyrimidine derivatives as ctps1 inhibitors |
| IL277455B2 (en) | 2018-03-23 | 2024-05-01 | Step Pharma S A S | Aminopyrimidine derivatives as ctps1 inhibitors |
| US20230192673A1 (en) * | 2018-06-04 | 2023-06-22 | Step Pharma S.A.S. | Compounds |
| EP4232425A4 (en) | 2020-10-23 | 2024-07-24 | Nimbus Clotho, Inc. | Ctps1 inhibitors and uses thereof |
-
2019
- 2019-10-23 PL PL19789997.4T patent/PL3870574T3/pl unknown
- 2019-10-23 MY MYPI2021002215A patent/MY203303A/en unknown
- 2019-10-23 JP JP2021522085A patent/JP7308263B2/ja active Active
- 2019-10-23 EA EA202191104A patent/EA202191104A1/ru unknown
- 2019-10-23 LT LTEPPCT/EP2019/078848T patent/LT3870574T/lt unknown
- 2019-10-23 DK DK19789997.4T patent/DK3870574T3/da active
- 2019-10-23 ES ES19789997T patent/ES2941981T3/es active Active
- 2019-10-23 PT PT197899974T patent/PT3870574T/pt unknown
- 2019-10-23 HU HUE19789997A patent/HUE061652T2/hu unknown
- 2019-10-23 WO PCT/EP2019/078848 patent/WO2020083975A1/en not_active Ceased
- 2019-10-23 SG SG11202103823PA patent/SG11202103823PA/en unknown
- 2019-10-23 SM SM20230090T patent/SMT202300090T1/it unknown
- 2019-10-23 FI FIEP19789997.4T patent/FI3870574T3/fi active
- 2019-10-23 CA CA3115568A patent/CA3115568C/en active Active
- 2019-10-23 EP EP19789997.4A patent/EP3870574B1/en active Active
- 2019-10-23 AU AU2019367191A patent/AU2019367191B2/en active Active
- 2019-10-23 MX MX2021004659A patent/MX2021004659A/es unknown
- 2019-10-23 IL IL282386A patent/IL282386B2/en unknown
- 2019-10-23 CR CR20210265A patent/CR20210265A/es unknown
- 2019-10-23 BR BR112021005986-5A patent/BR112021005986A2/pt active Search and Examination
- 2019-10-23 US US17/287,137 patent/US11987573B2/en active Active
- 2019-10-23 NZ NZ774355A patent/NZ774355A/en unknown
- 2019-10-23 HR HRP20230328TT patent/HRP20230328T1/hr unknown
- 2019-10-23 SI SI201930504T patent/SI3870574T1/sl unknown
- 2019-10-23 CN CN201980063914.3A patent/CN112839938B/zh active Active
- 2019-10-23 RS RS20230239A patent/RS64081B1/sr unknown
-
2021
- 2021-03-17 ZA ZA2021/01788A patent/ZA202101788B/en unknown
-
2024
- 2024-04-16 US US18/636,731 patent/US12371419B2/en active Active
-
2025
- 2025-05-23 US US19/217,739 patent/US20250282756A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2941981T3 (es) | Derivados de aminopirimidina/pirazina como inhibidores de CTPS1 | |
| ES3044436T3 (en) | N-(5-(6-ethoxypyrazin-2-yl)pyridin-2-yl)-4-(2-(ethylsulfonamido)pyrimidin-4-yl)tetrahydro-2h-pyran-4-carboxamide as a human ctps1 inhibitor for the treatment of proliferative diseases | |
| US12319680B2 (en) | Compounds | |
| ES2974445T3 (es) | Derivados de aminopirimidina como inhibidores de CTPS1 | |
| WO2019106156A1 (en) | Compounds | |
| EP4041715A1 (en) | Sulfonamide inhibitors as ctps 1 inhibitors | |
| EP4041723A2 (en) | Sulfonamide derivatives as ctps1 inhibitors | |
| HK40059463B (en) | Aminopyrimidine/pyrazine derivatives as ctps1 inhibitors | |
| HK40059463A (en) | Aminopyrimidine/pyrazine derivatives as ctps1 inhibitors | |
| EA050418B1 (ru) | Производные аминопиридина/пиразина в качестве ингибиторов ctps1 |