ES2941348T3 - Composición de proenzima - Google Patents
Composición de proenzima Download PDFInfo
- Publication number
- ES2941348T3 ES2941348T3 ES16863236T ES16863236T ES2941348T3 ES 2941348 T3 ES2941348 T3 ES 2941348T3 ES 16863236 T ES16863236 T ES 16863236T ES 16863236 T ES16863236 T ES 16863236T ES 2941348 T3 ES2941348 T3 ES 2941348T3
- Authority
- ES
- Spain
- Prior art keywords
- trypsinogen
- chymotrypsinogen
- cancer
- amount
- study
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21004—Trypsin (3.4.21.4)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
- A61K38/48—Hydrolases (3) acting on peptide bonds (3.4)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
- A61K38/48—Hydrolases (3) acting on peptide bonds (3.4)
- A61K38/482—Serine endopeptidases (3.4.21)
- A61K38/4826—Trypsin (3.4.21.4) Chymotrypsin (3.4.21.1)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21001—Chymotrypsin (3.4.21.1)
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Enzymes And Modification Thereof (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Compounds Of Unknown Constitution (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Medicines Containing Plant Substances (AREA)
Abstract
La presente invención se refiere a composiciones, métodos, usos y kits para el tratamiento del cáncer. En particular, la invención se refiere a composiciones y métodos para tratar el cáncer en un sujeto que comprende administrar quimotripsinógeno en ciertas cantidades, por ejemplo, superiores a aproximadamente 0,1 mg/kg, y tripsinógeno en una cantidad, por ejemplo, superior a aproximadamente 0,02 mg/kg, tratando así el cáncer. La invención también se refiere a composiciones y métodos para tratar el cáncer en un sujeto que comprende quimotripsinógeno y tripsinógeno en los que la proporción en peso de quimotripsinógeno:tripsinógeno es superior a 8:1. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Composición de proenzima
Campo de la invención
La presente invención se refiere a composiciones para tratar cáncer.
Antecedentes de la invención
El uso de proteasas en el tratamiento de cáncer se ha sugerido desde hace algún tiempo. Inicialmente, se contemplaron extractos de enzimas pancreáticas recientes como posible terapia contra el cáncer y se llevaron a cabo algunos experimentos satisfactorios con modelo de sarcoma de ratón de Jersen. Tras inyectar al ratón la enzima proteasa tripsina, se observó una regresión de tumores. Los resultados obtenidos produjeron gran interés y se usaron extractos de enzimas en bruto preparados a partir de páncreas de oveja para tratar a pacientes humanos con cáncer para reducir la progresión tumoral y prolongar el tiempo de supervivencia.
El uso de proenzimas (forma de precursor inactivo de enzimas) se ha usado para intentar superar problemas encontrados con la administración oral de enzimas con resultados mixtos. Se ha mostrado que una mezcla de proenzimas que incluye tripsinógeno, que es la forma de proenzima del inhibidor de serina proteasa tripsina, es útil en el tratamiento de carcinomas y se cree que se activa selectivamente en la superficie de células tumorales. Se cree que el mecanismo de acción de la tripsina se produce mediante proteolisis de las células tumorales. Se ha mostrado que una composición que incluye quimotripsinógeno y tripsinógeno en un intervalo de razón en peso de entre 4:1 y 8:1 es eficaz en ensayos para el cáncer, incluyendo cáncer pancreático y cáncer de colon (documento WO 2011/047434).
Novak et al. (Anticancer Research vol. 25, 2005, págs. 1157-1177) describen una mezcla de tripsinógeno y quimotripsinógeno a una razón en peso de 1:1, en combinación con amilasa.
Sin embargo, existe una necesidad de proporcionar tratamientos contra el cáncer nuevos o mejorados.
La referencia a cualquier técnica anterior en la memoria descriptiva no es un reconocimiento o sugerencia de que esta técnica anterior forme parte del conocimiento general común en ninguna jurisdicción o de que esta técnica anterior pueda esperarse razonablemente que se entienda, se considere como relevante y/o se combine con otros documentos de la técnica anterior por un experto en la técnica.
Sumario de la invención
La presente invención proporciona una composición farmacéutica para su uso en el tratamiento de cáncer, comprendiendo la composición quimotripsinógeno, tripsinógeno y un diluyente, excipiente o portador farmacéuticamente aceptable, en la que el quimotripsinógeno se administra en una cantidad igual a, o mayor de, 3,5 mg/kg, y de no más de 41 mg/kg y el tripsinógeno se administra en una cantidad igual a, o mayor de, 0,6 mg/kg y de no más de 7 mg/kg.
En cualquier aspecto de la invención, la composición para su uso en el tratamiento de cáncer en un ser humano comprende o consiste en quimotripsinógeno y tripsinógeno, en la que la cantidad de quimotripsinógeno que va a administrarse es igual a, o mayor de, 3,5 mg/kg, 10 mg/kg, 20 mg/kg o 40 mg/kg, y de no más de 41 mg/kg.
En cualquier aspecto de la invención, la composición para su uso en el tratamiento de cáncer en un ser humano comprende o consiste en quimotripsinógeno y tripsinógeno, en la que la cantidad de tripsinógeno que va a administrarse es igual a, o mayor de, 0,6 mg/kg, 1,5 mg/kg, 3 mg/kg o 6 mg/kg y de no más de 7 mg/kg.
En una realización, los únicos principios activos antitumorales presentes en la composición son quimotripsinógeno y tripsinógeno.
La invención proporciona una composición farmacéutica para su uso en el tratamiento de cáncer pancreático en un sujeto, comprendiendo la composición quimotripsinógeno, tripsinógeno y un diluyente, excipiente o portador farmacéuticamente aceptable, en la que la composición está adaptada para administrar quimotripsinógeno en una cantidad igual a, o mayor de, 5 mg/kg, preferiblemente mayor de 13 mg/kg, y de no más de 41 mg/kg, y tripsinógeno en una cantidad igual a, o mayor de, 1,5 mg/kg, preferiblemente mayor de 2 mg/kg, y de no más de 7 mg/kg.
La invención proporciona una composición farmacéutica para su uso en el tratamiento de cáncer de ovarios en un sujeto, comprendiendo la composición quimotripsinógeno, tripsinógeno y un diluyente, excipiente o portador farmacéuticamente aceptable, en la que la composición está adaptada para administrar quimotripsinógeno en una cantidad igual a, o mayor de, 4 mg/kg, y de no más de 41 mg/kg, y tripsinógeno en una cantidad igual a, o mayor de, 0,7 mg/kg, y de no más de 7 mg/kg.
El presente documento describe además un método de tratamiento de cáncer en un sujeto que comprende administrar quimotripsinógeno en una única dosis o en múltiples dosis.
Preferiblemente, el cáncer es cáncer pancreático o cáncer de ovarios. Incluso más preferiblemente, el cáncer pancreático es un adenocarcinoma.
El presente documento describes un método de tratamiento de cáncer en un ser humano que comprende administrar quimotripsinógeno en una cantidad de 40 mg/kg y tripsinógeno en una cantidad de 7 mg/kg, tratando de ese modo el cáncer. Preferiblemente, el cáncer es cáncer pancreático. Incluso más preferiblemente, el cáncer pancreático es un adenocarcinoma.
El presente documento describes un método de tratamiento de cáncer en un ser humano que comprende administrar quimotripsinógeno en una cantidad de 4 mg/kg o 13 mg/kg y tripsinógeno en una cantidad de 0,7 mg/kg o 2 mg/kg, tratando de ese modo el cáncer. Preferiblemente, el cáncer es cáncer de ovarios.
En cualquier aspecto de la invención, la administración de la cantidad de tripsinógeno y quimotripsinógeno no da como resultado ningún acontecimiento adverso clínicamente observable en el sujeto 1 semana después de la administración, 1 día después de la administración o preferiblemente 1 hora después de la administración. El acontecimiento adverso clínicamente observable puede ser uno cualquiera o más de pérdida de peso, enrojecimiento en el sitio de la inyección y cambios de comportamiento, o cualquier otro acontecimiento descrito en el presente documento, particularmente en los ejemplos.
Pueden administrarse quimotripsinógeno y tripsinógeno en una razón en peso en el intervalo de o de aproximadamente 4: 1 a o a aproximadamente 8: 1, a o a aproximadamente 5: 1 a o a aproximadamente 7: 1, o a aproximadamente 6: 1.
El presente documento también describe un kit para tratar cáncer que comprende al menos una unidad de dosificación, en el que la unidad de dosificación comprende quimotripsinógeno, tripsinógeno y un diluyente, excipiente o portador farmacéuticamente aceptable, en el que la unidad de dosificación está adaptada para administrar quimotripsinógeno y tripsinógeno igual a, o mayor que, cualquier cantidad o valor en mg/kg descrito en el presente documento, incluyendo en las tablas 1, 4, 8 y 10, particularmente en el ejemplo 5.
Opcionalmente, el kit también incluye instrucciones escritas que indican al usuario que administre una unidad de dosificación de quimotripsinógeno en una cantidad tal como se describe en el presente documento, particularmente en el ejemplo 5.
Las composiciones farmacéuticas para su uso según la invención son útiles para tratar cánceres y carcinomas metastásicos incluyendo cáncer pancreático, cáncer de esófago, cáncer de colon, cáncer de intestino, cáncer de próstata, cáncer de ovarios, cáncer de cerebro, cáncer de estómago, cáncer de mama, cáncer de hígado, melanoma maligno o cáncer de pulmón. Preferiblemente, el cáncer es cáncer pancreático, cáncer de colon o cáncer de ovarios. Más preferiblemente, el cáncer es cáncer pancreático.
En cualquier aspecto de un uso de la invención, el uso puede comprender además la etapa de identificar a un sujeto que tiene, o en riesgo de desarrollar, cáncer. Preferiblemente, el cáncer es uno cualquiera descrito en el presente documento.
En cualquier aspecto de la invención, la composición no contiene, o el uso no administra, amilasa.
En cualquier aspecto, realización o forma de la invención descrita en el presente documento, la cantidad de quimotripsinógeno administrada puede ser mayor de, o igual a, 3,5 mg/kg, 5 mg/kg, 15 mg/kg, 20 mg/kg o 40 mg/kg. En cualquier aspecto, realización o forma de la invención descrita en el presente documento, la cantidad de tripsinógeno administrada puede ser mayor de, o igual a, 0,6 mg/kg, 0,8 mg/kg, 2 mg/kg, 2,5 mg/kg, 3 mg/kg o 5 mg/kg.
En cualquier composición de la invención anterior, la composición puede estar adaptada para administrar los mg, o mg/kg, relevantes de quimotripsinógeno y tripsinógeno al sujeto.
La invención también proporciona que cualquier aspecto de la invención anterior que incluye valores tiene "aproximadamente" el valor mencionado anteriormente. Por ejemplo, un aspecto adicional de la invención es cualquiera de los aspectos anteriores en los que la referencia a 500 mg/kg es aproximadamente 500 mg/kg. Esto se contempla para todos los aspectos o realizaciones de la invención y para todos los valores.
La invención proporciona una composición para su uso en el tratamiento de cáncer en un sujeto que comprende quimotripsinógeno y tripsinógeno en la que la razón en peso de quimotripsinógeno:tripsinógeno es mayor de 8:1 (es decir, 8 o más: 1). Preferiblemente, la razón en peso es mayor de 10:1. Preferiblemente, la razón en peso es de 10:1. La razón en peso puede ser de 8:1, 10:1 o entre 8: 1 y 10:1. En cualquiera de las realizaciones, la composición preferiblemente no contiene amilasa, es decir, comprende la administración de una composición libre de amilasa.
Preferiblemente, el cáncer es uno cualquiera o más de cáncer pancreático, cáncer de esófago, cáncer de colon, cáncer de intestino, cáncer de próstata, cáncer de ovarios, cáncer de estómago, cáncer de mama, cáncer de hígado, melanoma maligno, fibrosarcoma o cáncer de pulmón. Preferiblemente, el cáncer es de ovarios, melanoma, de cerebro, de próstata, colorrectal, de hígado o de pulmón. Preferiblemente, el cáncer es cáncer pancreático, cáncer de colon o cáncer de ovarios. Más preferiblemente, el cáncer es cáncer pancreático.
En cualquier aspecto de la invención, se administran quimotripsinógeno y tripsinógeno por vía intravenosa, por vía subcutánea o por vía intramuscular.
En cualquier aspecto de la invención, pueden administrarse quimotripsinógeno y tripsinógeno de manera simultánea o secuencial.
Tal como se usa en el presente documento, excepto cuando el contexto requiera lo contrario, no se pretende que el término "comprender" y variaciones del término, tales como "que comprende", "comprende" y "comprendido", excluyan aditivos, componentes, números enteros o etapas adicionales.
Aspectos adicionales de la presente invención y realizaciones adicionales de los aspectos descritos en los párrafos anteriores resultarán evidentes a partir de la siguiente descripción, facilitada a modo de ejemplo y con referencia a los dibujos adjuntos.
Breve descripción de los dibujos
Figura 1: Peso corporal medio ± EEM (g) para cada grupo durante el estudio n.° 1. Se administraron tratamientos como una única inyección i.v., una vez al día durante siete días consecutivos. El tratamiento se inició para cada grupo de una manera escalonada, comenzando con la dosis más baja y después aumentando la dosis en cada día posterior. Para todos los cálculos y la presentación, el primer día de tratamiento para cada grupo se designa como el día del estudio 0.
Figura 2: Peso corporal medio ± EEM (g) para cada grupo durante el estudio n.° 2. Se administraron tratamientos como una única inyección i.v., una vez al día durante siete días consecutivos. El tratamiento se inició para cada grupo de una manera escalonada, comenzando con la dosis más baja en el primer día y después aumentando la dosis hasta tripsinógeno/quimotripsinógeno A a 86,8/500 mg/kg en el segundo día. El tratamiento con tripsinógeno/quimotripsinógeno A a 43,4/250 mg/kg comenzó en el tercer día. Para todos los cálculos y la presentación, el primer día de tratamiento para cada grupo se designa como el día del estudio 0.
Figura 3: Resultados de peso de tumor medio ± EEM (mg) de ratones de control con PBS (grupo 1, n=8) y ratones que recibieron T:C, 27,5 mg/kg:165 mg/kg (grupo 2, n=9) o T:C, 83,3 mg/kg:500 mg/kg (grupo 3, n=10) en el estudio descrito en el ejemplo 3 realizado para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación frente a células de cáncer pancreático de ratón Pan02, inoculadas de manera ortotópica en ratones C57BL/6 hembra. Se administraron los tratamientos como una única inyección i.v., una vez al día en los días del estudio de 0 a 25. Se sacrificaron los animales en el día del estudio 26.
Figura 4: Imágenes de tumores escindidos a partir de ratones en el grupo 1 (PBS, 10 ml/kg, i.v. diaria) el día 26 en el estudio descrito en el ejemplo 3 realizado para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación frente a células de cáncer pancreático de ratón Pan02, inoculadas de manera ortotópica en ratones C57BL/6 hembra.
Figura 5: Imágenes de tumores escindidos a partir de ratones en el grupo 2 (T:C, 27,5 mg/kg: 165 mg/kg) el día 26 en el estudio descrito en el ejemplo 3 realizado para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación frente a células de cáncer pancreático de ratón Pan02, inoculadas de manera ortotópica en ratones C57BL/6 hembra.
Figura 6: Imágenes de tumores escindidos a partir de ratones en el grupo 3 (T:C, 83,3 mg/kg:500 mg/kg) el día 26 en el estudio descrito en el ejemplo 3 realizado para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación frente a células de cáncer pancreático de ratón Pan02, inoculadas de manera ortotópica en ratones C57BL/6 hembra.
Figura 7: Peso de tumor medio ± EEM (g) en el grupo 2: control de vehículo (PBS). Grupo 3: tripsinógeno/quimotripsinógeno A, 83,3/500 mg/kg. Grupo 4: tripsinógeno/quimotripsinógeno A, 27,5/165 mg/kg. Grupo 5: tripsinógeno/quimotripsinógeno A, 9,1/54 mg/kg, en el estudio descrito en el ejemplo 4 realizado para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación, frente a células de cáncer de ovarios humano A2780 inoculadas de manera ortotópica en ratones atímicos desnudos-Foxn1nu hembra. Se administraron los tratamientos como una única inyección i.v., una vez al día (días del estudio de 0 a 20). Se sacrificaron estos animales en el día del estudio 21.
Figura 8: Imágenes de tumores escindidos en la terminación del estudio para el grupo 2: control de vehículo (PBS) en el estudio descrito en el ejemplo 4 realizado para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación, frente a células de cáncer de ovarios humano A2780 inoculadas de manera ortotópica en ratones atímicos desnudos-Foxn1nu hembra.
Figura 9: Imágenes de tumores escindidos en la terminación del estudio para el grupo 3: tripsinógeno/quimotripsinógeno A, 83,3/500 mg/kg, en el estudio descrito en el ejemplo 4 realizado para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación, frente a células de cáncer de ovarios humano A2780 inoculadas de manera ortotópica en ratones atímicos desnudos-Foxn1nu hembra.
Figura 10: Imágenes de tumores escindidos en la terminación del estudio para el grupo 4: tripsinógeno/quimotripsinógeno A, 27,5/165 mg/kg, en el estudio descrito en el ejemplo 4 realizado para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación, frente a células de cáncer de ovarios humano A2780 inoculadas de manera ortotópica en ratones atímicos desnudos-Foxn1nu hembra.
Figura 11: Imágenes de tumores escindidos en la terminación del estudio para el grupo 5: tripsinógeno/quimotripsinógeno A, 9,1/54 mg/kg, en el estudio descrito en el ejemplo 4 realizado para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación, frente a células de cáncer de ovarios humano A2780 inoculadas de manera ortotópica en ratones atímicos desnudos-Foxn1nu hembra.
Figura 12: El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de ovarios humano A2780. Se realizaron ensayos de combinación de tripsinógeno y quimotripsinógeno a razones de 1:1, 1:2, 1:4, 1:6, 1:8 y 1:10 basándose en la CI50 anteriormente determinada de tripsinógeno (3,174 mg/ml). Se calcularon valores de CDI tal como se describe.
Figura 13: El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de melanoma humano C8161.9. Se realizaron ensayos de combinación de tripsinógeno y quimotripsinógeno a razones de 1:1, 1:2, 1:4, 1:6, 1:8 y 1:10 basándose en la CI50 anteriormente determinada de tripsinógeno (3,917 mg/ml). Se calcularon valores de CDI tal como se describe.
Figura 14: El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de cerebro humano DAOY. Se realizaron ensayos de combinación de tripsinógeno y quimotripsinógeno a razones de 1:1, 1:2, 1:4, 1:6, 1:8 y 1:10 basándose en la CI50 anteriormente determinada de tripsinógeno (2,654 mg/ml). Se calcularon valores de CDI tal como se describe.
Figura 15: El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de próstata humano DU145. Se realizaron ensayos de combinación de tripsinógeno y quimotripsinógeno a razones de 1:1, 1:2, 1:4, 1:6, 1:8 y 1:10 basándose en la CI50 anteriormente determinada de tripsinógeno (3,843 mg/ml). Se calcularon valores de CDI tal como se describe.
Figura 16: El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor colorrectal humano h Ct 116. Se realizaron ensayos de combinación de tripsinógeno y quimotripsinógeno a razones de 1:1, 1:2, 1:4, 1:6, 1:8 y 1:10 basándose en la CI50 anteriormente determinada de tripsinógeno (16,43 mg/ml). Se calcularon valores de CDI tal como se describe.
Figura 17: El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de hígado humano Hep3B2.1-7. Se realizaron ensayos de combinación de tripsinógeno y quimotripsinógeno a razones de 1:1, 1:2, 1:4, 1:6, 1:8 y 1:10 basándose en la CI50 anteriormente determinada de tripsinógeno (2,483 mg/ml). Se calcularon valores de CDI tal como se describe.
Figura 18: El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor colorrectal humano HT-29. Se realizaron ensayos de combinación de tripsinógeno y quimotripsinógeno a razones de 1:1, 1:2, 1:4, 1:6, 1:8 y 1:10 basándose en la CI50 anteriormente determinada de tripsinógeno (15,12 mg/ml). Se calcularon valores de CDI tal como se describe.
Figura 19: El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de hígado humano HuH-7. Se realizaron ensayos de combinación de tripsinógeno y quimotripsinógeno a razones de 1:1, 1:2, 1:4, 1:6, 1:8 y 1:10 basándose en la CI50 anteriormente determinada de tripsinógeno (3,934 mg/ml). Se calcularon valores de CDI tal como se describe.
Figura 20: El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de pulmón humano NCI-H460. Se realizaron ensayos de combinación de tripsinógeno y
quimotripsinógeno a razones de 1:1, 1:2, 1:4, 1:6, 1:8 y 1:10 basándose en la CI50 anteriormente determinada de tripsinógeno (4,028 mg/ml). Se calcularon valores de CDI tal como se describe.
Descripción detallada de las realizaciones
Ahora se hará referencia en detalle a determinadas realizaciones de la invención. Aunque la invención se describirá junto con las realizaciones, se entenderá que no se pretende limitar la invención a esas realizaciones. Por el contrario, se pretende que la invención cubra todas las alternativas, modificaciones y equivalentes, que puedan incluirse dentro del alcance de la presente invención tal como se define por las reivindicaciones.
Con fines de interpretar esta memoria descriptiva, los términos usados en singular también incluirán el plural y viceversa.
La presente invención se basa en el sorprendente hallazgo de que pueden administrarse quimotripsinógeno y tripsinógeno a cantidades significativamente superiores a las usadas anteriormente. Esto puede realizarse en una única administración o a lo largo de un periodo de un único día. Sorprendentemente, estas cantidades significativamente superiores de quimotripsinógeno y tripsinógeno, incluso cantidades cien veces superiores, pueden administrarse sin ningún acontecimiento adverso clínico significativo. Además, se muestra que estas cantidades superiores reducen el peso de varios tipos de tumor in vivo. La presente invención proporciona un beneficio importante en el tratamiento de cáncer ya que puede administrarse de manera segura una dosis eficaz de quimotripsinógeno y tripsinógeno más grande que la anteriormente usada, proporcionando de ese modo un mayor intervalo terapéutico.
El quimotripsinógeno (que puede abreviarse como "C" en el presente documento) es una forma de proenzima de la enzima quimotripsina, que escinde preferiblemente proteínas en los siguientes aminoácidos: tirosina, triptófano, fenilalanina y leucina. La quimotripsina puede denominarse, o incluye, quimotripsina A, quimotripsina B (incluyendo formas B1 y B2), quimotripsina C, a-quimaroft, avazima, quimar, quimotest, enzeon, quimar, quimotrasa, a-quimar, a-quimotripsina A, aquimotripsina. La quimotripsina C puede formarse a partir de quimotripsinógeno C de cerdo o a partir de la subunidad II de ganado de procarboxipeptidasa A, y preferiblemente escinde proteínas en los siguientes aminoácidos: tirosina, triptófano, fenilalanina, leucina, metionina, glutamina y asparagina. El quimotripsinógeno incluye quimotripsinógeno B1 y quimotripsinógeno B2.
El tripsinógeno (que puede abreviarse como "T" en el presente documento) es una forma de proenzima de tripsina, que escinde preferiblemente proteínas en arginina y lisina. La tripsina puede denominarse, o incluir, a-tripsina, p-tripsina, cocoonasa, parenzima, parenzimol, triptar, tripura, pseudotripsina, triptasa, tripcellim, hidrolasa de receptor de espermatozoides p-tripsina puede formarse a partir de tripsinógeno mediante escisión de un enlace peptídico. Escisiones de enlaces peptídicos adicionales producen la forma a y otras iso-formas. Pueden aislarse múltiples tripsinas catiónicas y aniónicas a partir del páncreas de muchos vertebrados y a partir de especies inferiores incluyendo cangrejo de patas rojas, insectos (cocoonasa) y microorganismos (Streptomyces griseus). En procesos normales durante la digestión, el tripsinógeno inactivo se activa mediante enteropeptidasa presente en la mucosa intestinal para formar la enzima tripsina, que al ser una serina proteasa actúa entonces para escindir los enlaces peptídicos en el lado carboxilo de aminoácidos/proteínas básicos.
El tripsinógeno y el quimotripsinógeno usados en cualquier aspecto de la invención pueden estar aislados, purificados, sustancialmente purificados, ser recombinantes o sintéticos.
Las proenzimas tripsinógeno y quimotripsinógeno pueden ser precursores de las enzimas seleccionadas de las clases de quimotripsina 3.4.21.1 o 3.4.21.2 o tripsina de la clase 3.4.21.4, o seleccionadas de cualquier otra fuente adecuada (clases agrupadas según la clasificación del Comité de nomenclatura de la Unión internacional de bioquímica y biología molecular). Estas enzimas están comercialmente disponibles y pueden ser de origen humano, bovino o porcino.
Tal como se usa en el presente documento, "cualquier otra cantidad tal como se describe en el presente documento" o "cualquier valor en mg/kg descrito en el presente documento" incluye cualquier cantidad o valor en mg/kg descrito en cualquiera de los ejemplos incluyendo en las tablas 1, 4, 8 y 10.
La presente invención incluye conversiones a ser humano para todas las cantidades en mg/kg mencionadas en el presente documento basándose en pesos corporales de ser humano de 50, 60, 70, 80, 90, 100 o más kg y área de superficie corporal de 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8 o más m2 Específicamente, las cantidades por kg para administración a seres humanos, y métodos para el cálculo, se mencionan en el ejemplo 5.
Tal como se menciona, la forma de proenzima proporciona esencialmente una forma inactivada de la enzima que se activa in situ (por ejemplo, activación in vivo o in vitro). Por ejemplo, la activación de la proenzima (conversión de proenzima en enzima activa) puede producirse al entrar en contacto con la superficie de la célula tumoral. Se cree que las proenzimas tripsinógeno y quimotripsinógeno se activan selectivamente para dar las enzimas tripsina y quimotripsina al entrar en contacto con células tumorales y no al entrar en contacto con células sanas. El uso de proenzimas reduce problemas asociados con proporcionar, in situ, una enzima activa, tales como reacciones no deseadas o inactivación de la enzima antes de alcanzar la diana prevista de una célula tumoral.
Con respecto a las células tumorales, las enzimas proteasa pueden actuar para descomponer la pared celular de células malignas escindiendo los enlaces amida presentes en cadenas peptídicas de las paredes celulares (proteolisis). También se entiende que los inhibidores de proteasa, que están presentes en células no malignas e inhiben o reducen el efecto de las enzimas en la descomposición de las paredes celulares, están ausentes en células tumorales malignas. Además de proporcionar actividad proteolítica, las proenzimas de proteasa pueden regular por incremento la expresión de p-catenina y E-cadherina en células tumorales. La adhesión de célula a célula se facilita mediante la formación de complejo o unión entre p-catenina y E-cadherina en la superficie celular y, por tanto, la expresión aumentada de p-catenina y E-cadherina conduce a una adhesión potenciada de célula a célula y, de ese modo, reduce la metástasis de células tumorales. Las proenzimas de proteasa también pueden proporcionar otra actividad celular tal como aumento del inmunorreconocimiento o diferenciación.
Una reacción, experiencia o acontecimiento adverso significativo es cualquier manifestación médica desfavorable que, a cualquier dosis: da como resultado la muerte, es potencialmente mortal (pone al sujeto en riesgo inmediato de muerte), requiere hospitalización del sujeto o prolongación de una hospitalización existente, da como resultado discapacidad persistente o significativa, incapacidad o es una anomalía congénita/defecto de nacimiento.
Un acontecimiento adverso clínicamente observable es cualquier manifestación médica desfavorable en un sujeto o sujeto de investigación clínica al que se le administra un producto farmacéutico. Un acontecimiento adverso clínicamente observable puede incluir uno cualquiera de los acontecimientos u observaciones descritos en el presente documento, particularmente en los ejemplos. Normalmente, el periodo de observación es de aproximadamente 1 semana después de la administración, aproximadamente 1 día después de la administración o de manera preferible aproximadamente 1 hora después de la administración. El periodo de observación puede ser el tiempo entre administración de dosis.
"Tratar" o "tratamiento" se refiere tanto al tratamiento terapéutico como a medidas profilácticas o preventivas, en los que el objetivo es prevenir, mejorar, reducir o ralentizar (disminuir) el cáncer o la diseminación (metástasis) del mismo.
En cualquier método de la presente divulgación, puede observarse uno o más de los siguientes efectos: reducción de la reaparición de tumores malignos, reducción de la metástasis de tumores malignos, reducción del número o tamaño de tumores, diferenciación de células tumorales, expresión de p-catenina y E-cadherina en tumores malignos para facilitar la adhesión de célula a célula y la reducción de la metástasis, reducción de la capacidad de células tumorales para prevenir el inmunorreconocimiento.
"Prevenir", "prevención", "preventivo" o "profiláctico" se refiere a evitar que se produzca, o a dificultar, defender frente a, o proteger frente a la aparición de un estado, enfermedad, trastorno o fenotipo, incluyendo una anomalía o síntoma. Un sujeto que necesita prevención puede ser propenso a desarrollar el estado.
El término "mejorar" o "mejora" se refiere a una disminución, reducción o eliminación de un estado, enfermedad, trastorno o fenotipo, incluyendo una anomalía o síntoma. Un sujeto que necesita tratamiento puede tener ya el estado, o puede ser propenso a tener el estado o puede ser uno en el que debe prevenirse el estado.
El "sujeto" incluye un mamífero. El mamífero puede ser un ser humano o puede ser un animal doméstico, de zoológico o de compañía. Aunque se contempla particularmente que los métodos de la invención son adecuados para el tratamiento médico de seres humanos, también son aplicables al tratamiento veterinario, incluyendo el tratamiento de animales de compañía tales como perros y gatos, y animales domésticos tales como caballos, ganado y ovejas, o animales de zoológico tales como félidos, cánidos, bóvidos y ungulados. Un sujeto puede estar afectado por cáncer u otro trastorno o puede no estar afectado por cáncer u otro trastorno (es decir, libre de enfermedad detectable).
El peso corporal típico de un sujeto humano puede ser mayor de, o igual a, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105 o 110 kg.
El término "cantidad terapéuticamente eficaz" se refiere a una cantidad de composición, o agente o compuesto en la composición, que puede tratar, prevenir o mejorar el cáncer o la diseminación (metástasis) del mismo. Una cantidad terapéuticamente eficaz puede determinarse empíricamente y de una manera rutinaria con respecto al tratamiento de cáncer, y dará como resultado un aumento de la esperanza de vida.
Tal como se usa en el presente documento, "adaptada para administrar" se refiere a una composición que tiene la capacidad de administrar tripsinógeno y quimotripsinógeno a la cantidad o concentración especificada en una única dosis o múltiples dosis.
Tal como se describe en el presente documento, los métodos incluyen tratamiento de neoplasias y estados, cánceres, tumores, estados malignos y metastásicos relacionados. Los tejidos y órganos asociados con tumores sólidos y metástasis que pueden tratarse con un método o una composición farmacéutica de la invención incluyen, pero no se limitan a, tracto biliar, vejiga, sangre, cerebro, mama, cuello uterino, colon, endometrio, esófago, cabeza, cuello, riñón, laringe, hígado, pulmón, médula, melanina, ovario, páncreas, próstata, recto, renal, retina, piel, estómago, testículos, tiroides, tracto urinario y útero. El cáncer puede ser un carcinoma y desarrollarse en un epitelio o a partir de células epiteliales. El cáncer puede ser un sarcoma y desarrollarse en tejido conjuntivo. El cáncer de cerebro puede ser glioblastoma.
Las composiciones farmacéuticas para su uso de la invención son útiles para tratar cánceres y carcinomas metastásicos de los siguientes tipos: cáncer pancreático, cáncer de esófago, cáncer de colon, cáncer de intestino, cáncer de próstata, cáncer de ovarios, cáncer de estómago, cáncer de mama, melanoma maligno o cáncer de pulmón. Preferiblemente, el cáncer es cáncer pancreático, cáncer de colon o cáncer de ovarios. Más preferiblemente, el cáncer es cáncer pancreático. El carcinoma metastásico puede tener un potencial metastásico bajo, moderado o alto.
Las composiciones farmacéuticas pueden proporcionar un enfoque de múltiples efectos para tratar cáncer, por ejemplo aumentando en células tumorales la apoptosis, la adhesión de célula a célula, la diferenciación y la inmunogenicidad (selección como diana y eliminación por parte del sistema inmunitario). Por tanto, resulta beneficioso llevar a cabo el tratamiento en ausencia de cualquier otro tratamiento que pueda suprimir o dañar al sistema inmunitario.
Las composiciones para su uso de la invención pueden complementarse con otros enfoques terapéuticos anticancerígenos convencionales dirigidos al tratamiento o a la prevención de trastornos proliferativos (por ejemplo, tumor). Por ejemplo, tales métodos pueden usarse en la prevención profiláctica del cáncer, prevención de recidiva de cáncer y metástasis tras la cirugía, y como adyuvante de otra terapia contra el cáncer convencional.
Las composiciones farmacéuticas para su uso de la invención pueden formularse, por ejemplo, empleando vehículos o diluyentes sólidos o líquidos convencionales, así como aditivos farmacéuticos de un tipo apropiado para el modo de administración deseada (por ejemplo, excipientes, aglutinantes, conservantes, estabilizantes, aromatizantes, etc.) según técnicas tales como las bien conocidas en la técnica de la formulación farmacéutica.
Las composiciones farmacéuticas para su uso de la invención pueden administrarse mediante cualquier medio adecuado, por ejemplo, por vía oral, tal como en forma de comprimidos, cápsulas, gránulos o polvos; por vía sublingual; por vía bucal; por vía parenteral, tal como mediante técnicas de inyección o infusión subcutánea, intravenosa, intramuscular o intracisternal (por ejemplo, como disoluciones o suspensiones acuosas o no acuosas inyectables estériles); por vía nasal tal como mediante pulverización de inhalación; por vía tópica, tal como en forma de una crema o pomada; o por vía rectal tal como en forma de supositorios; en formulaciones de unidad de dosificación que contienen vehículos o diluyentes no tóxicos farmacéuticamente aceptables. Por ejemplo, pueden administrarse en una forma adecuada para liberación inmediata o liberación sostenida, por ejemplo, mediante el uso de dispositivos tales como implantes subcutáneos, esferoides encapsulados o bombas osmóticas.
Además de primates, tales como seres humanos, puede tratarse a una variedad de otros mamíferos según los métodos del décimo aspecto. Por ejemplo, puede tratarse a mamíferos incluyendo, pero sin limitarse a, vacas, ovejas, cabras, caballos, perros, gatos, cobayas, ratas u otras especies bovinas, ovinas, equinas, caninas, felinas, de roedores o murinas.
El término "farmacéuticamente aceptable" tal como se usa en el presente documento significa que el portador, diluyente o excipiente no es perjudicial para el receptor del mismo.
Debe entenderse que los términos "administración de" y/o "administrar" significan proporcionar a un individuo que necesita tratamiento.
Un individuo que necesita tratamiento puede ser uno con diagnóstico de, o en riesgo de desarrollar, uno cualquiera de los cánceres descritos en el presente documento.
Las composiciones farmacéuticas para su uso de la invención, y preparaciones o formulaciones de las mismas, pueden prepararse mezclando entre sí los componentes de la composición, en concreto quimotripsinógeno y tripsinógeno. El mezclado puede realizarse de manera secuencial o simultánea.
Las composiciones farmacéuticas para su uso de la invención pueden presentarse convenientemente en forma de unidad de dosificación y pueden prepararse mediante cualquiera de los métodos bien conocidos en la técnica de la farmacia. Todos los métodos incluyen la etapa de poner los agentes activos y la proenzima de proteasa en asociación con el portador que constituye uno o más componentes auxiliares. En general, las composiciones farmacéuticas se preparan poniendo de manera uniforme e íntima los agentes activos y las proenzimas de proteasa en asociación con un portador líquido o un portador sólido finamente dividido o ambos, y después, si es necesario, conformando el producto para dar la formulación deseada. Los agentes activos y las proenzimas de proteasa se proporcionan en una forma de unidad de dosificación en una cantidad suficiente para producir el efecto deseado sobre el proceso o estado de enfermedades tras una administración única o repetida.
Las composiciones farmacéuticas para su uso de la invención pueden estar en una forma adecuada para uso oral, por ejemplo, como comprimidos, trociscos, pastillas para chupar, suspensiones acuosas o aceitosas, polvos o gránulos dispersables, emulsiones, cápsulas duras o blandas o jarabes o elixires. Las composiciones destinadas para su uso oral pueden prepararse según cualquier método conocido en la técnica para la fabricación de composiciones farmacéuticas y tales composiciones pueden contener uno o más agentes seleccionados del grupo que consiste en agentes edulcorantes, agentes aromatizantes, agentes colorantes y agentes conservantes con el fin de proporcionar preparaciones farmacéuticamente agradables y sabrosas. Los comprimidos contienen la proenzima de proteasa y agente activo del
primer y segundo aspectos en mezcla con excipientes no tóxicos farmacéuticamente aceptables que son adecuados para la fabricación de comprimidos. Estos excipientes pueden ser, por ejemplo, diluyentes inertes, tales como carbonato de calcio, carbonato de sodio, lactosa, fosfato de calcio o fosfato de sodio; agentes de granulación y disgregación, por ejemplo, almidón de maíz o ácido algínico; agentes aglutinantes, por ejemplo almidón, gelatina o goma arábiga, y agentes lubricantes, por ejemplo estearato de magnesio, ácido esteárico o talco. Los comprimidos pueden no estar recubiertos o pueden recubrirse mediante técnicas conocidas para retardar la disgregación y absorción en el tracto gastrointestinal y de ese modo proporcionar una acción sostenida a lo largo de un periodo más prolongado. Por ejemplo, puede emplearse un material de retardo en el tiempo tal como monoestearato de glicerilo o diestearato de glicerilo. También pueden recubrirse para formar comprimidos terapéuticos osmóticos para liberación controlada.
Las formulaciones para uso oral también pueden presentarse como cápsulas de gelatina dura en las que la proenzima de proteasa y el agente activo del primer y segundo aspectos se mezclan con un diluyente sólido inerte, por ejemplo, carbonato de calcio, fosfato de calcio o caolín, o como cápsulas de gelatina blanda en las que la proenzima de proteasa y el agente activo del primer y segundo aspectos se mezclan con agua o un medio aceitoso, por ejemplo aceite de cacahuete, parafina líquida o aceite de oliva.
Las suspensiones acuosas contienen el agente activo y proenzima de proteasa en mezcla con excipientes adecuados para la fabricación de suspensiones acuosas. Tales excipientes son agentes de suspensión, por ejemplo carboximetilcelulosa de sodio, metilcelulosa, hidroxipropilmetilcelulosa, alginato de sodio, polivinilpirrolidona, n-metilpirrolidona, goma tragacanto y goma arábiga; los agentes dispersantes o humectantes pueden ser un fosfátido que se produce de manera natural, por ejemplo lecitina, o productos de condensación de un óxido de alquileno con ácidos grasos, por ejemplo estearato de polioxietileno, o productos de condensación de óxido de etileno con alcoholes alifáticos de cadena larga, por ejemplo heptadecaetilenoxicetanol, o productos de condensación de óxido de etileno con ésteres parciales derivados de ácidos grasos y un hexitol tales como monooleato de polioxietilensorbitol, o productos de condensación de óxido de etileno con ésteres parciales derivados de ácidos grasos y anhídridos de hexitol, por ejemplo monooleato de polietilensorbitano. Las suspensiones acuosas también pueden contener uno o más conservante, por ejemplo p-hidroxibenzoato de etilo, o n-propilo, uno o más agentes colorantes, uno o más agentes aromatizantes y uno o más agentes edulcorantes, tales como sacarosa o sacarina.
Las suspensiones aceitosas pueden formularse suspendiendo el agente activo y la proenzima de proteasa en un aceite vegetal, por ejemplo aceite de maní, aceite de oliva, aceite de sésamo o aceite de coco, o en un aceite mineral tal como parafina líquida. Las suspensiones aceitosas pueden contener un agente espesante, por ejemplo cera de abeja, parafina dura o alcohol cetílico. Pueden añadirse agentes edulcorantes, tales como los expuestos anteriormente, y agentes aromatizantes para proporcionar una preparación oral sabrosa. Estas pueden conservarse mediante la adición de un antioxidante tal como ácido ascórbico.
Los polvos y gránulos dispersables adecuados para la preparación de una suspensión acuosa mediante la adición de agua proporcionan la proenzima de proteasa y el agente activo del primer y segundo aspectos en mezcla con un agente dispersante o humectante, agente de suspensión y uno o más conservantes. Los agentes dispersantes o humectantes y agentes de suspensión adecuados se muestran a modo de ejemplo mediante los ya mencionados anteriormente. También pueden estar presentes excipientes adicionales, por ejemplo agentes edulcorantes, aromatizantes y colorantes.
Las composiciones farmacéuticas para su uso de la invención también pueden estar en forma de emulsiones de aceite en agua. La fase aceitosa puede ser un aceite vegetal, por ejemplo aceite de oliva o aceite de maní, o un aceite mineral, por ejemplo parafina líquida o mezclas de los mismos. Los agentes emulsionantes adecuados pueden ser gomas que se producen de manera natural, por ejemplo goma arábiga o goma tragacanto, fosfátidos que se producen de manera natural, por ejemplo semilla de soja, lecitina y ésteres o ésteres parciales derivados de ácidos grasos y anhídridos de hexitol, por ejemplo monooleato de sorbitano, y productos de condensación de dichos ésteres parciales con óxido de etileno, por ejemplo monooleato de polioxietilensorbitano. Las emulsiones también pueden contener agentes edulcorantes y aromatizantes.
Los jarabes y elixires pueden formularse con agentes edulcorantes, por ejemplo glicerol, propilenglicol, sorbitol o sacarosa. También pueden contener un demulcente, un conservante y agentes aromatizantes y colorantes.
Las composiciones farmacéuticas para su uso de la invención pueden estar en forma de una suspensión acuosa u oleaginosa inyectable estéril. Esta suspensión puede formularse según la técnica conocida usando los agentes dispersantes o humectantes y agentes de suspensión adecuados que se mencionaron anteriormente. Las composiciones farmacéuticas del primer y segundo aspectos también pueden ser una disolución o suspensión inyectable estéril en un diluyente o disolvente no tóxico parenteralmente aceptable, por ejemplo como disolución en 1,3-butanodiol. Entre los vehículos y disolventes aceptables que pueden emplearse se encuentran agua, disolución de Ringer y disolución isotónica de cloruro de sodio. Además, convencionalmente se emplean aceites fijos estériles como disolvente o medio de suspensión. Con este fin, puede emplearse cualquier aceite fijo insípido incluyendo mono o diglicéridos sintéticos. Además, ácidos grasos tales como ácido oleico encuentran uso en la preparación de productos inyectables.
En una realización particular, las composiciones farmacéuticas para su uso de la invención se formulan como supositorios para administración rectal del fármaco. Estas formulaciones pueden prepararse mezclando la proenzima de proteasa y el
agente activo del primer y segundo aspectos con un excipiente no irritante adecuado que es sólido a temperaturas habituales pero líquido a la temperatura rectal y, por tanto, se fundirá en el recto para liberar el fármaco. Tales materiales incluyen manteca de cacao y polietilenglicoles. Puede usarse administración rectal para eliminar el efecto de primer paso enterohepático en el tracto gastrointestinal relacionado con la administración oral de enzimas.
Las composiciones farmacéuticas para su uso de la invención, también pueden formularse en liposomas. Tal como se conoce en la técnica, los liposomas se derivan generalmente a partir de fosfolípidos u otras sustancias lipídicas. Los liposomas se están formados por cristales líquidos hidratados mono o multilamelares que se dispersan en un medio acuoso. Puede usarse cualquier lípido no tóxico fisiológicamente aceptable y metabolizable que puede formar liposomas. La formulación de liposoma puede contener estabilizadores, conservantes, excipientes y similares. Los lípidos preferidos son los fosfolípidos y fosfatidilcolinas, tanto naturales como sintéticas. En la técnica se conocen métodos para formar liposomas.
Las composiciones farmacéuticas para su uso de la invención pueden incluirse en un recipiente, envase o dispensador junto con instrucciones de administración. Las proenzimas de proteasa y los agentes activos, y opcionalmente agente activo adicional, de la composición farmacéutica pueden proporcionarse como componentes independientes en el recipiente, envase o dispensador, para tomarse por separado o juntos al mismo tiempo o en un momento diferente en un uso o método de la invención descrito en el presente documento.
Ejemplos
Ejemplo 1
Se completó un estudio de dosificación para determinar la dosis máxima tolerada y viable en la que no se observaba ningún acontecimiento adverso clínico, o en la que cualquier acontecimiento adverso clínico se resolvía antes de la dosificación posterior. A continuación se expone el protocolo del estudio de dosificación.
Se disolvieron tripsinógeno y quimotripsinógeno A en solución salina tamponada con fosfato (PBS) en cada día de tratamiento para proporcionar disoluciones madre de 1 mg/ml. Se prepararon disoluciones de dosificación de las concentraciones requeridas de tripsinógeno/quimotripsinógeno A diluyendo las disoluciones madre juntas en PBS para lograr la concentración final deseada de cada uno.
Para el estudio n.° 1, se prepararon disoluciones de dosificación de tripsinógeno/quimotripsinógeno A a 0,0254/0,1521, 0,0381/0,2282, 0,0572/0,3423 y 0,0858/0,5135 mg/ml.
Para el estudio, se administraron los tratamientos a animales en cada grupo una vez al día durante siete días consecutivos en un volumen de dosificación de 10 ml/kg. El volumen de disolución de dosificación administrado a cada animal se calculó y se ajustó basándose en el peso corporal individual medido inmediatamente antes de la dosificación.
Para el estudio n.° 1, se aleatorizaron ratones mediante el peso corporal en cuatro grupos de cuatro ratones. El régimen de dosificación usado para el estudio se resume en la tabla 1. Se administraron tripsinógeno y quimotripsinógeno A en combinación como una única inyección intravenosa en la vena de la cola (i.v.) a dosis de 10 ml/Kg. La concentración final de tripsinógeno y quimotripsinógeno para cada grupo fue de 0,254/1,521, 0,381/2,282, 0,572/3,423 y 0,858/5,135 mg/kg (grupos 1, 2, 3 y 4, respectivamente).
El tratamiento para cada grupo en el estudio n.° 1 se inició de una manera escalonada, comenzando con la dosis más baja de tripsinógeno/quimotripsinógeno A y después aumentando la dosis en cada día del estudio posterior. Para todos los cálculos y la presentación, el primer día de tratamiento para cada grupo se designa como el día del estudio 0.
En el primer día de cada estudio, se trató a un ratón en el grupo 1 con tripsinógeno/quimotripsinógeno A a la dosis más baja (0,254/1,521 mg/kg para el estudio n.° 1). Dado que no hubo ningún efecto adverso intenso prolongado dentro del plazo de dos horas desde el tratamiento, se trataron los tres ratones restantes en cada uno de estos grupos de manera similar.
Dado que no hubo ningún efecto adverso intenso prolongado dentro del plazo de 24 horas desde el tratamiento inicial en los ratones del grupo 1, se trató a un ratón en el grupo 2 con tripsinógeno/quimotripsinógeno A a 0,381/2,282 mg/kg para el estudio n.° 1. Dado que no hubo ningún signo visible de toxicidad dentro del plazo de dos horas desde este tratamiento, los ratones restantes en cada uno de estos grupos recibieron el mismo tratamiento.
Se continuó el tratamiento de esta manera para las dos dosis restantes de tripsinógeno/quimotripsinógeno A para el estudio n.° 1 (0,572/3,423 y 0,858/5,135 mg/kg) para los animales en los grupos 3 y 4, respectivamente.
Todos los animales en cada grupo en cada estudio recibieron siete tratamientos diarios consecutivos.
Mortalidad
Se realizaron comprobaciones de mortalidad una vez al día por la mañana durante el estudio.
Observaciones clínicas
Se comprobaron los animales para detectar signos clínicos (tales como mala salud y cambios de comportamiento) dos veces al día durante el estudio.
Pesos corporales
Se registraron los pesos corporales para todos los animales el primer día de tratamiento, después diariamente durante el periodo de tratamiento y durante siete días tras el tratamiento final.
Observaciones clínicas:

Criterios de evaluación para observaciones clínicas:
Marcha: Claudicante, paresia, parálisis = anómala
Temperatura corporal: Caliente o frío = anómala
Tipo de respiración: Rápida; superficial o fatigosa = anómala
Diarrea: Heces sueltas en el suelo de la jaula, charcos de heces en el suelo de la jaula o derrame al manipularlo = anómala
Estado corporal (ref: Ullman-Cullere & Foltz. Lab. Animal Science 1999; 49:319): BC1 = el animal está demacrado; BC2 = el animal presenta un estado inferior al adecuado; BC3 = el animal presenta un buen estado; BC4 = el animal presenta un estado superior al adecuado; BC5 = el animal está obeso.
Observaciones clínicas y acontecimientos adversos para el estudio n.° 1
No se observó ningún signo clínico adverso en ningún animal durante el estudio.
Cambios del peso corporal
Hubo un aumento de peso corporal medio a lo largo del transcurso del estudio (días del estudio de 0 a 13) para cada grupo (el 2,59%, el 7,48%, el 7,97% y el 2,03% del peso inicial para los animales tratados con tripsinógeno y quimotripsinógeno A a 0,254/1,521, 0,381/2,282, 0,572/3,423 y 0,858/5,135 mg/kg; grupos 1, 2, 3 y 4, respectivamente) (tabla 2 y figura 1). Ningún animal perdió peso corporal en más del 15% del peso inicial (tabla 3).
Ejemplo 2
Sorprendentemente, las cantidades de tripsinógeno y quimotripsinógeno A en el estudio n.° 1 se toleraron bien tal como se muestra a partir de los resultados en el ejemplo 2 anterior. Esto era inesperado dado que estas cantidades eran significativamente superiores a cantidades anteriormente usadas. A la vista de estos resultados, se realizó un estudio adicional, el estudio n.° 2, usando cantidades de tripsinógeno y quimotripsinógeno A, en combinación, incluso superiores a las cantidades anteriormente usadas.
Para el estudio n.° 2, se prepararon disoluciones madre de tripsinógeno y quimotripsinógeno A a 5 y 6 mg/ml, respectivamente. Se diluyeron adicionalmente las disoluciones madre en Pb S para preparar las disoluciones de dosificación de tripsinógeno/quimotripsinógeno A para el grupo 1 de 0,256/1,5 mg/ml. Para los grupos 2 y 3, se prepararon disoluciones madre de tripsinógeno y quimotripsinógeno A a 30 y 100 mg/ml, respectivamente.
Se diluyeron adicionalmente las disoluciones madre en PBS para preparar las disoluciones de dosificación de tripsinógeno/quimotripsinógeno A para el grupo 2 de 8,68/50 mg/ml y para el grupo 3 de 4,34/25 mg/ml. Se observó que, a una concentración de 50 mg/ml, el quimotripsinógeno A estaba casi al límite de viscosidad para la inyección en la vena de la cola.
Para el estudio n.° 2, se aleatorizaron 12 ratones mediante el peso corporal en tres grupos de cuatro ratones. El régimen de dosificación usado para el estudio se resume en la tabla 4. Se administraron tripsinógeno y quimotripsinógeno A en combinación como una única inyección intravenosa en la vena de la cola (i.v.) a dosis de 10 ml/Kg. La concentración final de tripsinógeno y quimotripsinógeno para cada grupo fue de 2,6/15, 86,8/500 y 43,4/250 mg/kg (grupos 1, 2 y 3, respectivamente)
El tratamiento para cada grupo en el estudio n.° 2 se inició de una manera escalonada, comenzando con la dosis más baja de tripsinógeno/quimotripsinógeno A y después aumentando la dosis en cada día del estudio posterior. Para todos los cálculos y la presentación, el primer día de tratamiento para cada grupo se designa como el día del estudio 0.
En el primer día de cada estudio, se trató a un ratón en el grupo 1 con tripsinógeno/quimotripsinógeno A a la dosis más baja de 2,6/15 mg/kg. Dado que no hubo ningún efecto adverso intenso prolongado dentro del plazo de dos horas desde el tratamiento, se trataron los tres ratones restantes en este grupo de manera similar.
Dado que no hubo ningún efecto adverso intenso prolongado dentro del plazo de 24 horas desde el tratamiento inicial en los ratones del grupo 1, se trató a un ratón en el grupo 2 con tripsinógeno/quimotripsinógeno A a 86,8/500 mg/kg para el estudio n.° 2. Dado que no hubo ningún signo visible de toxicidad dentro del plazo de dos horas desde este tratamiento, los ratones restantes en cada uno de estos grupos recibieron el mismo tratamiento.
Se continuó el tratamiento de esta manera para la dosis restante de tripsinógeno/quimotripsinógeno A para el estudio n.° 2 (43,4/250 mg/kg) para los animales en el grupo 3.
Observaciones clínicas y acontecimientos adversos para el estudio n.° 2
No se observó ningún signo clínico adverso en los animales tratados con tripsinógeno y quimotripsinógeno A a 2,6/15, y 43,4/250 mg/kg; grupos 1 y 3, respectivamente) (tabla 5).
El enrojecimiento de la piel en la cola y formaciones de coágulos minoritarias temporales en la vena de la cola en el sitio de la inyección fueron los únicos signos adversos observados, que se produjeron inmediatamente tras la dosificación en el grupo de dosis más alta (tripsinógeno/quimotripsinógeno A a 86,8/500 mg/kg) y se resolvieron antes del momento de la siguiente administración de dosis (tabla 5). Los signos clínicos no interfirieron con una administración de dosis apropiada.
Cambios del peso corporal
Hubo un aumento de peso corporal medio a lo largo del transcurso del estudio (días del estudio de 0 a 13) para cada grupo (el 6,98%, el 4,30% y el 6,61% del peso inicial para los animales tratados con tripsinógeno y quimotripsinógeno A a 2,6/15, 86,8/500 y 43,4/250 mg/kg; grupos 1, 2 y 3, respectivamente) (tabla 6 y figura 2). Ningún animal perdió peso corporal en más del 15% del peso inicial (tabla 7).
Tabla 1: Régimen de dosificación para el estudio n.° 1

Tabla 2: Mediciones de peso corporal medio ± EEM (g) para cada grupo de tratamiento al inicio y la terminación del estudio n.° 1

Tabla 3: Peso corporal inicial y aparición del peso corporal más bajo (g) para cada animal durante el transcurso del estudio n.° 1

Grupo Peso inicial Peso más Días tras el Delta de

, , , Se administraron tratamientos como una única inyección i.v., una vez al día durante siete días
consecutivos.
El tratamiento se inició para cada grupo de una manera escalonada, comenzando con la dosis más
baja y después aumentando la dosis en cada día posterior. Para todos los cálculos y la
presentación, el primer día de tratamiento para cada grupo se designa como el día del estudio 0. 
Tabla 4: R im n ifi i n r l i n.° 2

Tabla 5: Observaciones clínicas y acontecimientos adversos para el estudio n.° 2


Tabla 6 : Mediciones de peso corporal medio ± EEM (g) para cada grupo de tratamiento al inicio y la terminación del estudio n.° 2

Tabla 7: Peso corporal inicial y aparición del peso corporal más bajo (g) para cada animal durante el transcurso del estudio n.° 2
Gr

Se administraron tratamientos como una única inyección i.v., una vez al día duran consecutivos.
El tratamiento se inició para cada grupo de una manera escalonada, comenzando c
baja en el primer día y después aumentando la dosis hasta tripsinógeno/quimotrip
86,8/500 mg/kg en el segundo día. El tratamiento con tripsinógeno/quimotripsinógen
mg/kg comenzó en el tercer día. Para todos los cálculos y la presentación, el pri
tratamiento para cada grupo se designa como el día del estudio 0. 
Ejemplo 3
El estudio se realizó para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación frente a células de cáncer pancreático de ratón Pan02, inoculadas de manera ortotópica en ratones C57BL/6 hembra.
Línea celular
Las células de tumor pancreático de ratón Pan02 se obtuvieron del Instituto nacional del cáncer (Frederick, MD, EE.UU.).
Cultivo de células tumorales
Se cultivaron células de tumor pancreático de ratón Pan02 (disolución madre de trabajo VP-Stock 1235) en medio de cultivo celular RPMI 1640 complementado con FBS al 10%, GlutaMAX™ al 1% y penicilina-estreptomicina al 1%, y se hicieron crecer a 37°C en una incubadora de cultivo celular humidificada a la que se le suministró el 5% de CO2. Se recogieron las células (pase 7) mediante tripsinización, se lavaron dos veces en HBSS y se contaron (usando exclusión por azul de tripano). Se ajustó la densidad celular final con HBSS:Matrigel™ (1:1, v/v) hasta 5 * 107 células Pan02/ml. Se sabe que el uso de Matrigel™ en la suspensión de inoculación soporta la vascularización temprana lo cual puede aumentar la tasa de implantación de tumores y, por tanto, tiene el potencial para reducir la variabilidad del tamaño de tumor.
Inoculación de células tumorales
Cuarenta ratones hembra C57BL/6 se sometieron a inoculación mientras estaban con anestesia inyectada por vía intraperitoneal (ketamina (14 mg/ml)/xilazina (0,9 mg/ml)). Antes de la inoculación, se limpió con hisopo la piel en el sitio de la incisión con disolución de yoduro de povidona tópica y luego con alcohol. Se realizó una incisión para exponer el páncreas. Se introdujo una aguja directamente en la cola del páncreas en la que se descargaron 20 |jl de suspensión celular, que consistía en 1 * 106 células Pan02.
A los ratones se les administró una dosis en bolo de 200 j l de Buprenex (buprenorfina HCl, 0,01 mg/ml) por vía subcutánea para alivio del dolor en el momento de la cirugía y al día siguiente.
Determinación de la tasa de implantación de tumor
Se sacrificaron diez animales seleccionados aleatoriamente (grupo 0) siete días después de la inoculación para evaluar el tamaño y la tasa de implantación de tumores en el páncreas. Se escindió el páncreas y se examinó para detectar la presencia de un tumor.
Se confirmó la presencia de un tumor en el páncreas de los 10 de estos animales.
Aleatorización
Dos días tras la confirmación de una tasa de implantación de tumor satisfactoria, se aleatorizaron 30 de los animales restantes, basándose en el peso corporal, en tres grupos de 10 (nueve días tras la inoculación; día del estudio 0).
Formulación de compuesto
Se disolvieron tripsinógeno y quimotripsinógeno A en solución salina tamponada con fosfato (PBS) en cada día de tratamiento para proporcionar disoluciones madre de 1 mg/ml. Se prepararon las disoluciones de dosificación de tripsinógeno/quimotripsinógeno A a 2,75/16,5 y 8,33/50 mg/ml (razón de 1:6) diluyendo en primer lugar la disolución de tripsinógeno en PBS y después añadiendo quimotripsinógeno A.
Administración de compuesto
El régimen de dosificación usado en este estudio se resume en la tabla 8.
Los animales asignados para la evaluación de la tasa de implantación (grupo 0) permanecieron sin tratar.
El control de vehículo (PBS; grupos 1) y tripsinógeno/quimotripsinógeno A (en combinación en una única inyección) a dosis de 27,5/165 (grupo 2) y 83,3/500 mg/kg (grupos 3) se administraron una vez al día i.v. en un volumen de dosificación de 10 ml/kg. El volumen de disolución de dosificación administrado a cada animal se calculó y se ajustó basándose en el peso corporal individual medido inmediatamente antes de la dosificación.
Los tratamientos comenzaron en el día del estudio 0 (nueve días tras la inoculación). Se administraron tratamientos a los animales en los grupos 1,2 y 3 durante 26 días consecutivos.
Procedimiento de terminación
Todos los animales de evaluación de tasa de implantación y aquellos en los grupos 1, 2 y 3 se sacrificaron mediante inhalación de dióxido de carbono mediante procedimientos convencionales aprobados.
Recogida de muestras
T ras la terminación, se escindió el páncreas de todos los animales con el tumor intacto. Se aisló el tumor a partir del tejido de páncreas, se pesó y después se fotografió (figuras 4 a 6). Se almacenó una porción de 25 mg de tumor a partir de cada una de las muestras en los grupos 4 y 5 a -80°C y se envió a Adaptive Biotechnologies Corp. para su análisis adicional.
Cálculos
El cambio en porcentaje medio del peso corporal (% de cambio de PC) entre el día 0 y cualquier día dado (día X) se calculó usando la ecuación:
% de cambio de PC = media(PCDíadei estudio X) / media(PCDíadei estudio o) x 100% - 100%
donde PCdíb dei estudio o = valor inicial en el día 0 y PCüíadei estudio x = valor actual en el día X
El cambio en porcentaje del peso de tumor medio para grupos tratados con respecto al grupo de control se calculó usando la siguiente ecuación:
Cambio en porcentaje = ((Mediacontroi - MedíaTratamiento) / Mediacontroi) x 100
Cálculos estadísticos
Todos los cálculos estadísticos se realizaron usando Prism 6 para Mac OS X (GraphPad Software Inc, La Jolla, CA, EE.UU.).
Se sometió a prueba la normalidad de todos los conjuntos de datos usando la prueba de normalidad Kolmogorov-Smirnov (KS).
Se usó una prueba de la t para datos emparejados para determinar si el peso corporal cambió significativamente dentro de un grupo de tratamiento entre el día del estudio 0 y el día de terminación de cada grupo.
Se realizó la comparación del peso de tumor en la terminación del estudio entre los grupos 1, 2 y 3 usando análisis de la varianza de un factor (ANOVA). Se determinaron diferencias significativas entre los grupos usando la prueba de comparaciones múltiples de Holm-Sidak.
Se consideró que un valor de p de < 0,05 era significativo.
Resultados y observaciones
Cambios del peso corporal
Hubo una pequeña pérdida de peso corporal medio (no significativa) (el 0,54% del peso inicial) en los animales tratados con control de vehículo durante 26 días (PBS; grupo 1).
Todos los demás grupos mostraron aumento de peso corporal medio; el 0,82% y el 2,72% del peso inicial para los grupos tratados con tripsinógeno/quimotripsinógeno A a 27,5/165 y 83,3/500 mg/kg durante 26 días (grupos 2 y 3, respectivamente).
Eficacia de compuestos
Hubo una reducción significativa (p<0,05) del peso de tumor medio en animales tratados durante 26 días con tripsinógeno/quimotripsinógeno A a 83,3/500 mg/kg (30,2 mg; inhibición del 85,9%; grupo 3) en comparación con el control de vehículo (PBS; 214,8 mg; grupo 1), pero no entre tripsinógeno/quimotripsinógeno A a 27,5/165 mg/kg (196,5 mg; inhibición del 8,5%; grupo 2) y el control de vehículo (tabla 9 y figura 3).
Conclusión
Se evaluó la eficacia antitumoral del tratamiento diario con tripsinógeno y quimotripsinógeno A, administrados en combinación como una única inyección intravenosa a dosis de 27,5/165 o 83,3/500 mg/kg, frente a células de cáncer pancreático de ratón Pan02, inoculadas de manera ortotópica en ratones C57BL/6 hembra.
Las mediciones del peso de tumor en la terminación mostraron una eficacia antitumoral significativa (p<0,05) en animales tratados con tripsinógeno/quimotripsinógeno A a dosis alta en comparación con animales tratados con control después de 26 días de tratamiento en este estudio.
Tabla 8: Ré imen de dosificación

Tabla 9: P m r m i ± EEM n l rmin i n m r r

Se administraron los tratamientos como una única inyección i.v., una vez al día (días del estudio de 0 a 25 para los grupos 1,2 y 3). Los grupos 1, 2 y 3 se terminaron en el día del estudio 26. No se recogieron los tumores de los animales que se encontraron muertos (uno en cada uno de los grupos 1 y 2). El valor para el único animal en el grupo 1 que se sacrificó en el día del estudio 6 no se incluyó en los análisis.
a p<0,05 en comparación con el grupo 1 (p<0,05; prueba de comparaciones múltiples de Holm-Sidak).
b p<0,05 en comparación con el grupo 2 (p<0,05; prueba de comparaciones múltiples de Holm-Sidak).
El peso de tumor medio ± EEM para cada grupo se representa en la figura 3.
* El % de inhibición de tumor se calculó para los grupos 2 y 3 usando el grupo 1 como control.
N/A: no aplicable
Ejemplo 4
Se realizó el estudio para evaluar la eficacia antitumoral de tripsinógeno y quimotripsinógeno A, administrados en combinación, frente a células de cáncer de ovarios humano A2780 inoculadas de manera ortotópica en ratones atímicos desnudos-Foxn1nu hembra.
Línea celular
Las células de carcinoma de ovarios humano A2780 se obtuvieron del Instituto nacional del cáncer (Frederick, MD, EE.UU.).
Cultivo de células tumorales
Se cultivaron células de cáncer de ovarios humano A2780 (disolución madre de trabajo VP-Stock 1277) en DMEM complementado con FBS al 10%, GlutaMAX™ al 1% y penicilina-estreptomicina al 1%, y se hicieron crecer a 37°C en una incubadora de cultivo celular humidificada a la que se le suministró el 5% de CO2. Se recogieron las células (pase 8) mediante tripsinización, se lavaron dos veces en HBSS y se contaron (usando exclusión por azul de tripano). Se ajustó la densidad celular final con HBSS:Matrigel™ (1:1, v/v) hasta 2,0 * 108 células/ml.
Se sabe que el uso de Matrigel™ en la suspensión de inoculación soporta la vascularización temprana lo cual puede aumentar la tasa de implantación de tumores y, por tanto, tiene el potencial para reducir la variabilidad del tamaño de tumor.
Inoculación de células tumorales
Sesenta ratones atímicos desnudos-Foxn1nu se sometieron a inoculación mientras estaban con anestesia inyectada por vía intraperitoneal (ketamina (14 mg/ml)/xilazina (0,9 mg/ml)). Antes de la inoculación, se limpió con hisopo la piel en el sitio de la incisión con alcohol. Se realizó una incisión para exponer el ovario. Se introdujo una aguja directamente en el ovario en el que se descargaron 5 |jl de suspensión celular que consistía en 1 * 106 células A2780.
A los ratones se les administró una dosis en bolo de 200 j l de Buprenex (buprenorfina HCl, 0,01 mg/ml) por vía subcutánea para alivio del dolor en el momento de la cirugía y al día siguiente.
Aleatorización
Se aleatorizaron los animales, basándose en el peso corporal, en cinco grupos de 12 y un grupo de 24, 7 días tras la inoculación (día del estudio 0).
Formulación de compuesto
Se disolvieron tripsinógeno y quimotripsinógeno A en solución salina tamponada con fosfato (PBS) en cada día de tratamiento para proporcionar disoluciones madre de 30 y 100 mg/ml, respectivamente. Se preparó la disolución de dosificación de tripsinógeno/quimotripsinógeno A a 8,33/50, 2,75/16,5 y 0,91/5,4 mg/ml (razón de 1:6) diluyendo en primer lugar la disolución de tripsinógeno en PBS y después añadiendo quimotripsinógeno A.
Administración de compuesto
El régimen de dosificación usado en este estudio se resume en la tabla 10.
Los animales en el grupo 1 permanecieron sin tratar para la evaluación de la tasa de implantación de tumor.
Se administraron control de vehículo (PBS; grupo 2) y tripsinógeno y quimotripsinógeno A a dosis de 83,3/500, 27,5/165 y 9,1/54 mg/kg (grupos 3, 4 y 5, respectivamente; en combinación en una única inyección) una vez al día mediante inyección intravenosa (i.v.) en la vena de la cola en un volumen de dosificación de 10 ml/kg (12 animales por grupo). El volumen de disolución de dosificación administrado a cada animal se calculó y se ajustó basándose en el peso corporal individual medido inmediatamente antes de la dosificación. Los tratamientos comenzaron en el día del estudio 0 y se administraron durante 21 días consecutivos.
Determinación de la tasa de implantación de tumor
Los animales sin tratar en el grupo 1 se sacrificaron siete días tras el inicio del tratamiento (14 días tras la inoculación) para evaluar el tamaño y la tasa de implantación de tumores en el ovario.
Se confirmó la presencia de un tumor en el ovario de los 12 animales, después se aisló el tumor a partir del ovario escindido y se pesó para todos los animales. Se presentan imágenes fotográficas de tumores e las figuras 8 a 11.
Procedimiento de terminación
Se sacrificaron todos los animales mediante inhalación de dióxido de carbono según procedimientos convencionales aprobados (grupos 1 a 5).
Recogida de muestras
Tras la terminación, se escindió el ovario de todos los animales con el tumor intacto. Se aisló el tumor a partir del tejido de ovario y se pesó. También se fotografiaron tumores de animales que recibieron 21 días de tratamiento (grupos 2 a 5).
Cálculos
El cambio en porcentaje medio del peso corporal (% de cambio de PC) entre el día 0 y cualquier día dado (día X) se calculó usando la ecuación:
% de cambio de PC = media(PCDíadei estudio X) / media(PCDíadei estudio o) x 100% -100%
donde PCdíb dei estudio o = valor inicial en el día 0 y PCdíb dei estudio x = valor actual en el día X
La inhibición en porcentaje del crecimiento de tumor para grupos tratados con respecto al grupo de control se calculó usando la siguiente ecuación:
Inhibición en porcentaje = ((Media control — Mediairatamíento) / Mediacontroi) x 100
Cálculos estadísticos
Todos los cálculos estadísticos se realizaron usando Prism 6 para Mac OS X (GraphPad Software Inc, La Jolla, CA, EE.UU.).
Se sometió a prueba la normalidad de todos los conjuntos de datos usando la prueba omnibus de normalidad de D'Agostino y Pearson.
Se usó una prueba de la t para datos emparejados para determinar si el peso corporal cambió significativamente dentro de grupos que recibieron 21 días de tratamiento (grupos 2 a 5) entre el día del estudio 0 y la terminación del estudio. Cuando los datos no presentaron una distribución normal, se determinó la significación usando la prueba de rangos con signos para muestras emparejadas de Wilcoxon.
Se realizó la comparación del peso de tumor en la terminación del estudio entre grupos que recibieron 21 días de tratamiento (grupos 2 a 5) usando análisis de la varianza de un factor (ANOVA).
Se consideró que un valor de p de < 0,05 era significativo.
Resultados y observaciones
Cambios del peso corporal
Hubo un aumento de peso corporal medio significativo (p<0,05) entre los días del estudio 0 y 7 en los animales sin tratar (el 4,33% del peso inicial; grupo 1).
Hubo un aumento de peso corporal medio entre los días del estudio 0 y 21 en los grupos tratados durante 21 días con control de vehículo (PBS; el 6,56% del peso inicial; grupo 2) y todas las dosis de tripsinógeno/quimotripsinógeno A (83,3/500, 27,5/165 y 9,1/54 mg/kg; el 8,60%, el 6,00% y el 3,89% del peso inicial; grupos 3, 4 y 5, respectivamente). El aumento de peso corporal fue significativo (p<0,05) en todos los grupos excepto por el tratamiento a dosis baja (grupo 5).
Eficacia de compuestos
Hubo una reducción significativa (p<0,05) del peso de tumor medio en animales tratados durante 21 días con tripsinógeno/quimotripsinógeno A a dosis media y baja (27,5/165 y 9,1/54 mg/kg; 957,3 y 1074,2 mg; el 53,6% y el 47,9%; grupos 4 y 5, respectivamente) en comparación con control de vehículo (PBS; 2062,2 mg; grupo 2), pero no con tripsinógeno/quimotripsinógeno A a dosis alta (83,3/500 mg/kg; 1762,2 mg; el 14,5%; grupo 3) (tabla 11 y figura 7). El peso de tumores para animales sin tratar (grupo 1) sacrificados en el día 7 para la evaluación de la tasa de implantación no se incluyó en el análisis.
Conclusión
Se evaluó la eficacia antitumoral del tratamiento diario con tripsinógeno y quimotripsinógeno A, administrados en combinación como una única inyección intravenosa a dosis de 83,3/500, 27,5/165 o 9,1/54 mg/kg, frente a células de cáncer de ovarios humano A2780, inoculadas de manera ortotópica en ratones atímicos desnudos-Foxn1nu hembra. Las mediciones del peso de tumor en la terminación mostraron una eficacia antitumoral significativa (p<0,05) en animales tratados con tripsinógeno/quimotripsinógeno A a dosis media y baja en comparación con animales tratados con control de vehículo después de 21 días de tratamiento en este estudio. No se observó eficacia antitumoral significativa tras el tratamiento a dosis alta.
Tabla 10: Régimen de dosificación

Tabla 11: P m r m i ± EEM n l rmin i n m r r

Los animales sin tratar en el grupo 1 se sacrificaron en el día del estudio 7 (14 días tras la inoculación) para la evaluación de la tasa de implantación de tumor. El peso de tumores para animales en el grupo 1 no se incluyó en el análisis estadístico. Se administraron los tratamientos a los animales en los grupos 2 a 5 como una única inyección i.v., una vez al día (días del estudio de 0 a 20). Se sacrificaron estos animales en el día del estudio 21.
1. a: p<0,05 en comparación con el grupo 2 (prueba de comparaciones múltiples de Holm-Sidak).
El peso de tumor medio ± EEM para cada grupo se representa en la figura 7.
N/A: no aplicable
Ejemplo 5
La conversión a dosis para seres humanos puede realizarse mediante cualquiera de los métodos relevantes descritos en U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER), Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Rockville, MD 2005. Por ejemplo, la dosis equivalente en seres humanos a partir de una dosis que se ha administrado a un ratón puede determinarse de la siguiente manera:
Dosis equivalente en seres humanos (HED en mg/kg) = dosis en animales (mg/kg) * K de animales K de seres humanos, donde K es un factor de corrección que refleja la relación entre el peso corporal y el área de superficie corporal.
Para un adulto típico (peso corporal de 60 kg, área de superficie corporal de 1,6 m2), K es de 37 y la K de ratón es de 3. Por ejemplo, una cantidad de 500 mg/kg descrita en el presente documento puede convertirse en aproximadamente 41 mg/kg para una dosis equivalente en seres humanos, y otras conversiones a modo de ejemplo usando esta fórmula incluyen:
Quimotri psinógeno:
Ratón (mg/kg): 1,5, 2, 3,5, 5, 15, 45, 135, 250 y 500.
Ser humano (mg/kg): 0,12, 0,16, 0,28, 0,4, 1,2, 3,6, 11, 20 y 41
Tri psinógeno:
Ratón (mg/kg): 0,25, 0,4, 0,6, 0,8, 2,5, 8, 20, 40 y 80.
Ser humano (mg/kg): 0,02, 0,03, 0,05, 0,065, 0,2, 0,65, 1,6, 3,2 y 6,5.
Quimotri psinógeno:
Ratón (mg/kg): 15, 54, 165, 250, 500.
Ser humano (mg/kg): 1,2, 4,4, 13, 20 y 41.
Tripsinógeno:
Ratón (mg/kg): 2,6, 9, 27,5, 43,4, 83,3, 86,8.
Ser humano (mg/kg): 0,2, 0,7, 2,2, 3,5, 6,75 y 7.
La presente invención incluye conversiones a ser humano de todas las cantidades en mg/kg mencionadas en el presente documento, incluyendo en las tablas 1, 4, 8 y 10 y en cualquier otra parte en este documento, basándose en pesos corporales de ser humano de 50, 60, 70, 80, 90, 100 o más kg y área de superficie corporal de 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8 o más m2
Las dosis equivalentes en seres humanos descritas en el presente documento se derivan usando los métodos descritos en "Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers" del CDER para la conversión de dosis basándose en el área de superficie corporal. Se apreciará que también puede usarse cualquier variación en los métodos descritos en el documento del CDER para determinar la dosis en seres humanos apropiada a partir de las dosificaciones administradas a ratones, tal como se describe en el presente documento (por ejemplo, en algunas circunstancias puede resultar apropiado ajustar la dosis basándose en el peso corporal en lugar de en el área de superficie corporal). Además, el experto también estará familiarizado con métodos para determinar la dosis apropiada para un ser humano joven (es decir, ser humano no adulto), así como métodos para determinar la dosis apropiada en un organismo no humano, al que se le pueden aplicar los métodos de la presente invención. El experto también estará familiarizado con métodos para ajustar la dosis apropiada dependiendo del método de administración previsto (por ejemplo, intravenosa, intramuscular, subcutánea, tópica, oral u otro método de administración).
Ejemplo 6
Los siguientes experimentos se realizaron para examinar razones de quimotripsinógeno con respecto a tripsinógeno mayores de 8:1.
Materiales y métodos
Materiales


Cultivo celular
La siguiente tabla (tabla 12) muestra las condiciones de crecimiento y las densidades de siembra celular iniciales en células por pocillo usadas en todos los ensayos de determinación de CI50 y de combinación. Se cultivaron las células a 37°C en una incubadora de cultivo celular humidificada a la que se le suministró el 95% de aire/el 5% de CO2.


Todas de las siguientes líneas celulares se obtuvieron de la Colección americana de cultivos tipo (ATCC) (Rockville, MD, EE.UU.): 786-0, ACHN, BT-474, DAOY, DU 145, G-361, HCT 116, HCT-15, Hep3B2.1-7, HL-60, HT-1080, HT-29, MCF-7, MDA-MB-231, MES-SA, NCI-H460, NCI-H82, PC-3, Raji, SK-OV-3, U-87 MG.
La línea celular A2780 se obtuvo del Instituto nacional del cáncer (NCI) (Bethesda, MD, EE.UU.).
La línea celular C8161.9 se obtuvo del laboratorio del Dr. Gavin Robertson (College of Medicine, Pennsylvania State University, Hershey, PA, EE.UU.)).
La línea celular HuH-7 se obtuvo del banco de células de la Colección japonesa de biorrecursos de investigación (JCRB) (Osaka, Japón).
La línea celular SNB-19 se obtuvo de Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) (colección alemana de microorganismos y cultivos celulares) (Braunschweig, Alemania).
Todas las líneas celulares se usaron en los ensayos hasta el pase 10.
Artículos de prueba
Artículo de prueba 1

Artículo de prueba 2

Formulación de artículo de prueba
Se disolvieron tripsinógeno y quimotripsinógeno directamente en el medio de cultivo celular apropiado y se añadieron inmediatamente a las células.
Ensayos de crecimiento celular
Para ensayos de combinación, se añadieron artículos de prueba a células 24 horas tras la siembra. Se sometieron a prueba concentraciones de artículo de prueba por triplicado para cada línea celular. Setenta y dos horas tras la adición de los artículos de prueba, se llevó a cabo el ensayo de CellTiter-Blue® en todas las placas.
La concentración de tripsinógeno usada en los ensayos de combinación se basó en una CI50 calculada a partir de experimentos con un único artículo de prueba. La concentración de tripsinógeno para cada línea celular individual determinó la concentración de quimotripsinógeno a las siguientes razones: 1:1, 1:2, 1:4, 1:6, 1:8 y 1:10 (tripsinógeno:quimotripsinógeno). Los controles consistieron en medio de crecimiento solo y células sin tratar más medio de crecimiento (control sin tratar).
Los controles de ensayo usados fueron el control de medio de crecimiento como vehículo y medio de crecimiento solo (fondo) en contraposición a control de solución salina tamponada con fosfato como vehículo y Triton-x 100 como control positivo. No se realizó la resta de fondo para la determinación de valores de CI50.
Tras la incubación de células en medios que contenían artículo de prueba, se añadieron 10 | j l de CellTiter-Blue® a cada pocillo, después se incubaron con células durante hasta 6 horas. Se midió la fluorescencia usando un fluorómetro Spectramax Gemini XPS (560 nm de excitación, 590 nm de emisión). Se registraron todos los datos y se introdujeron en hojas de cálculo de Microsoft® Excel para su interpretación.
Cálculos
Los datos recopilados a partir de ensayos de CellTiter-Blue® se representaron gráficamente como curvas de dosisrespuesta para la determinación de CI50. Se representaron gráficamente unidades de fluorescencia relativa (UFR) frente a concentraciones de compuesto. En estas representaciones gráficas, el eje de las X (concentración de compuesto) se representó en una escala logarítmica. Se calculó la concentración CI50 como la mitad de la máxima (50%) concentración inhibitoria (CI) para cada compuesto mediante un algoritmo de ajuste de la curva de pendiente variable usando GraphPad Prism versión 6.0e para Mac OSX (GraphPad Software, San Diego California, EE.UU.).
Para estudios de combinación, se calculó el coeficiente de interacción de fármacos (CDI) según la siguiente ecuación:

donde TC es la inhibición del crecimiento de la combinación de tripsinógeno y quimotripsinógeno, T es la inhibición del crecimiento del agente individual tripsinógeno y C es la inhibición del crecimiento del agente individual quimotripsinógeno. Los valores de CDI por debajo de 1 indican sinergia de fármacos, mientras que valores por encima de 1 indican una interacción antagonista de tripsinógeno y quimotripsinógeno.
Inhibición del crecimiento celular mediante tripsinógeno y quimotripsinógeno en cáncer humano
El efecto de tripsinógeno (T) y quimotripsinógeno (C) solos o en combinación sobre el crecimiento de células de tumor de ovarios humano A2780 se muestra en la figura 12. El mayor nivel de inhibición del crecimiento de células de tumor de
ovarios A2780 se observó para razones mayores de 1:8 (T:C), por ejemplo 1:10 (T:C), véase la tabla 13 (justo a continuación .

El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de melanoma C8161.9 se muestra en la figura 13. El mayor nivel de inhibición del crecimiento de células de melanoma C8161.9 se observó ara razones ma ores de 1:8 T:C or eem lo 1:10 T:C véase la tabla 14 usto a continuación .

C8161.9 Repeticiones (UFR) 
Promedio EEM Inhibición del crecimiento CDI 1 2 3
Control (solo células) 7676,335 7617,46 7233,554 7509,12 138,83 Solo medio 701,358 680,206 678,760 686,77 7,30 
El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de cerebro DAOY se muestra en la figura 14. El mayor nivel de inhibición del crecimiento de células de tumor de cerebro DAOY se o rv r r z n m r 1: T: r m l 1:1 T: v l l 1 nin i n.

El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de próstata DU145 se muestra en la figura 15. El mayor nivel de inhibición del crecimiento de células de tumor de próstata DU145 se observó ara razones ma ores de 1:8 T:C or eem lo 1:10 T:C véase la tabla 16 usto a continuación.
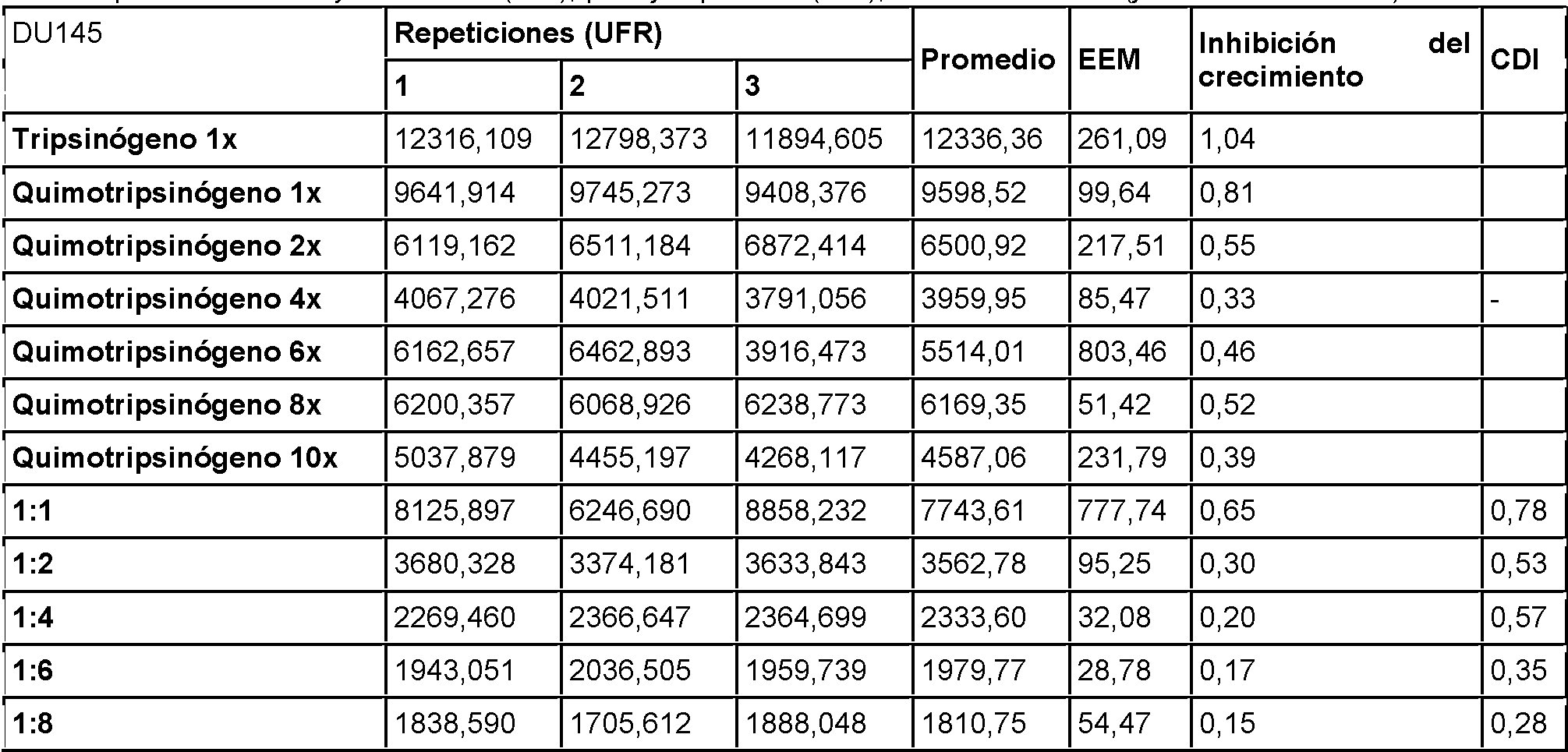

El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor colorrectal HCT 116 se muestra en la figura 16. El mayor nivel de inhibición del crecimiento de células de tumor colorrectal HCT 116 rv r r z n m r 1: T: r m l 1:1 T: v l l 17 nin i n.

El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de hígado Hep3B2.1-7 se muestra en la figura 17. El mayor nivel de inhibición del crecimiento de células de tumor de hígado He 3B2.1-7 se observó ara razones ma ores de 1:8 T:C or eem lo 1:10 T:C véase la tabla 18 usto a continuación.


El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor colorrectal HT-29 se muestra en la figura 18. El mayor nivel de inhibición del crecimiento de células de tumor colorrectal HT-29 se rv r r z n m r 1: T: r m l 1:1 T: v l l 1 nin i n.

El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de hígado HuH-7 se muestra en la figura 19. El mayor nivel de inhibición del crecimiento de células de tumor de hígado HuH-7 se rv r r z n m r 1: T: r m l 1:1 T: v l l 2 nin i n.


El efecto de tripsinógeno y quimotripsinógeno solos o en combinación sobre el crecimiento de células de tumor de pulmón NCI-H460 se muestra en la figura 20. El mayor nivel de inhibición del crecimiento de células de tumor de pulmón NCI-H460 se observó ara razones ma ores de 1:8 T:C or eem lo 1:10 T:C véase la tabla 21 usto a continuación.

Claims (17)
1. Composición farmacéutica para su uso en el tratamiento de cáncer, comprendiendo la composición quimotripsinógeno, tripsinógeno y un diluyente, excipiente o portador farmacéuticamente aceptable, en la que el quimotripsinógeno se administra en una cantidad igual a, o mayor de, 3,5 mg/kg, y de no más de 41 mg/kg y el tripsinógeno se administra en una cantidad igual a, o mayor de, 0,6 mg/kg y de no más de 7 mg/kg.
2. Composición farmacéutica para el uso según la reivindicación 1, en la que el cáncer es uno cualquiera de cáncer pancreático, cáncer de esófago, cáncer de colon, cáncer de intestino, cáncer de próstata, cáncer de ovarios, cáncer de estómago, cáncer de mama, cáncer de hígado, melanoma maligno, fibrosarcoma o cáncer de pulmón, preferiblemente en la que el cáncer es cáncer pancreático o cáncer de ovarios.
3. Composición farmacéutica para el uso según la reivindicación 1 o 2, en la que la cantidad de quimotripsinógeno es mayor de 10 mg/kg, 20 mg/kg o 40 mg/kg;
y/o
en la que la cantidad de tripsinógeno es mayor de 1,5 mg/kg, 3 mg/kg o 6 mg/kg.
4. Composición farmacéutica para el uso según la reivindicación 3, en la que la cantidad de quimotripsinógeno es igual a, o mayor de, 13 mg/kg, y la cantidad de tripsinógeno es igual a, o una cantidad mayor de, 2,2 mg/kg.
5. Composición farmacéutica para el uso según la reivindicación 3, en la que la cantidad de quimotripsinógeno es de 41 mg/kg y la cantidad tripsinógeno es de 6,75 mg/kg, por ejemplo, en la que el quimotripsinógeno está en una cantidad de aproximadamente 41 mg/kg y el tripsinógeno está en una cantidad de aproximadamente 7 mg/kg.
6. Composición farmacéutica para su uso en el tratamiento de cáncer pancreático, comprendiendo la composición quimotripsinógeno, tripsinógeno y un diluyente, excipiente o portador farmacéuticamente aceptable, en la que la composición está adaptada para administrar quimotripsinógeno en una cantidad igual a, o mayor de, 5 mg/kg, y de no más de 41 mg/kg y tripsinógeno en una cantidad igual a, o mayor de, 1,5 mg/kg y de no más de 7 mg/kg.
7. Composición farmacéutica para el uso según la reivindicación 6, en la que el quimotripsinógeno está en una cantidad mayor de 13 mg/kg, y tripsinógeno en una cantidad mayor de 2 mg/kg.
8. Composición farmacéutica para el uso según la reivindicación 6 o 7, en la que el quimotripsinógeno se administra en una cantidad de aproximadamente 41 mg/kg y el tripsinógeno se administra en una cantidad de aproximadamente 7 mg/kg.
9. Composición farmacéutica para su uso en el tratamiento de cáncer de ovarios, comprendiendo la composición quimotripsinógeno, tripsinógeno y un diluyente, excipiente o portador farmacéuticamente aceptable, en la que la composición está adaptada para administrar quimotripsinógeno en una cantidad igual a, o mayor de, 4 mg/kg y de no más de 41 mg/kg y tripsinógeno en una cantidad igual a, o mayor de, 0,7 mg/kg, y de no más de 7 mg/kg.
10. Composición farmacéutica para el uso según la reivindicación 9, en la que el quimotripsinógeno está en una cantidad de no más de 13 mg/kg y el tripsinógeno está en una cantidad de no más de 2,5 mg/kg, por ejemplo, en la que el quimotripsinógeno está en una cantidad de 13 mg/kg y tripsinógeno está en una cantidad de 2,2 mg/kg.
11. Composición farmacéutica para el uso según una cualquiera de las reivindicaciones 1 a 10, en la que el quimotripsinógeno y el tripsinógeno se administran en una razón en peso en el intervalo de entre aproximadamente 4: 1 y 8: 1.
12. Composición farmacéutica para el uso según una cualquiera de las reivindicaciones 1 a 10, en la que el quimotripsinógeno y el tripsinógeno se administran en una razón en peso de entre aproximadamente 5: 1 y 7: 1.
13. Composición farmacéutica para el uso según una cualquiera de las reivindicaciones 1 a 10, en la que el quimotripsinógeno y el tripsinógeno se administran en una razón en peso de aproximadamente 6: 1.
14. Composición farmacéutica para el uso según una cualquiera de las reivindicaciones 1 a 10, en la que el quimotripsinógeno y el tripsinógeno se administran en una razón en peso mayor de 8: 1.
15. Composición farmacéutica para el uso según la reivindicación 14, en la que la razón en peso es mayor de 10:1.
16. Composición farmacéutica para el uso según la reivindicación 14, en la que la razón en peso es de 10: 1.
17. Composición farmacéutica para el uso según una cualquiera de las reivindicaciones 1 a 16, en la que la composición no contiene amilasa.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2015904678A AU2015904678A0 (en) | 2015-11-12 | Proenzyme composition | |
| US201662321377P | 2016-04-12 | 2016-04-12 | |
| PCT/AU2016/051082 WO2017079802A1 (en) | 2015-11-12 | 2016-11-11 | Proenzyme composition |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2941348T3 true ES2941348T3 (es) | 2023-05-22 |
Family
ID=58694564
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16863236T Active ES2941348T3 (es) | 2015-11-12 | 2016-11-11 | Composición de proenzima |
Country Status (13)
| Country | Link |
|---|---|
| EP (1) | EP3373956B1 (es) |
| JP (1) | JP7479119B2 (es) |
| CN (1) | CN108367054A (es) |
| AU (1) | AU2016353539B2 (es) |
| CA (1) | CA3003835A1 (es) |
| DK (1) | DK3373956T3 (es) |
| ES (1) | ES2941348T3 (es) |
| IL (1) | IL259259B2 (es) |
| MY (1) | MY196737A (es) |
| NZ (1) | NZ742020A (es) |
| SG (1) | SG11201803531XA (es) |
| WO (1) | WO2017079802A1 (es) |
| ZA (1) | ZA201803855B (es) |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4514388A (en) * | 1983-03-22 | 1985-04-30 | Psaledakis Nicholas G | Proteolytic enzymes in the zymogen form to treat sarcoma cells |
| CZ283972B6 (cs) * | 1995-05-17 | 1998-07-15 | František Mudr. Trnka | Farmaceutický prostředek s modulačním účinkem na zhoubné nádory a jeho použití |
| RU2149021C1 (ru) * | 1998-05-07 | 2000-05-20 | Научный центр акушерства, гинекологии и перинатологии РАМН | Способ фармакологического лечения аутоиммунного мужского бесплодия |
| ES2692377T3 (es) * | 2009-10-22 | 2018-12-03 | Propanc Pty Ltd | Composición farmacéutica para tratar el cáncer que comprende tripsinógeno y quimotripsinógeno |
| CZ307195B6 (cs) * | 2013-11-18 | 2018-03-14 | František Trnka | Farmaceutická kompozice obsahující směs proenzymů a enzymů |
| JP7004665B2 (ja) * | 2016-04-12 | 2022-02-04 | プロパンク・ピーティーワイ・リミテッド | 癌治療用の前酵素の組成物 |
-
2016
- 2016-11-11 ES ES16863236T patent/ES2941348T3/es active Active
- 2016-11-11 WO PCT/AU2016/051082 patent/WO2017079802A1/en active Application Filing
- 2016-11-11 DK DK16863236.2T patent/DK3373956T3/da active
- 2016-11-11 SG SG11201803531XA patent/SG11201803531XA/en unknown
- 2016-11-11 EP EP16863236.2A patent/EP3373956B1/en active Active
- 2016-11-11 CA CA3003835A patent/CA3003835A1/en active Pending
- 2016-11-11 CN CN201680070907.2A patent/CN108367054A/zh active Pending
- 2016-11-11 JP JP2018524804A patent/JP7479119B2/ja active Active
- 2016-11-11 AU AU2016353539A patent/AU2016353539B2/en active Active
- 2016-11-11 MY MYPI2018000728A patent/MY196737A/en unknown
- 2016-11-11 NZ NZ742020A patent/NZ742020A/en unknown
-
2018
- 2018-05-09 IL IL259259A patent/IL259259B2/en unknown
- 2018-06-08 ZA ZA2018/03855A patent/ZA201803855B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| EP3373956A1 (en) | 2018-09-19 |
| CA3003835A1 (en) | 2017-05-18 |
| DK3373956T3 (da) | 2023-04-03 |
| JP7479119B2 (ja) | 2024-05-08 |
| IL259259B2 (en) | 2023-07-01 |
| EP3373956A4 (en) | 2019-05-08 |
| MY196737A (en) | 2023-05-03 |
| SG11201803531XA (en) | 2018-05-30 |
| IL259259A (en) | 2018-07-31 |
| WO2017079802A1 (en) | 2017-05-18 |
| IL259259B1 (en) | 2023-03-01 |
| ZA201803855B (en) | 2024-04-24 |
| JP2018533612A (ja) | 2018-11-15 |
| NZ742020A (en) | 2022-10-28 |
| EP3373956B1 (en) | 2023-01-04 |
| AU2016353539A1 (en) | 2018-05-17 |
| CN108367054A (zh) | 2018-08-03 |
| AU2016353539B2 (en) | 2020-04-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9295682B2 (en) | Adjuvant immunotherapy for the preventive, curative or palliative treatment of chronic systemic diseases such as cancer, of clinical manifestations associated with diseases like cachexia and correction of adverse effects of drugs such as immunosuppression, neutropenia and lymphopenia, comprising the association or combination of a biological response modifier specially selected and other substances with antineoplastic action and/or other treatments | |
| ES2373963T3 (es) | Tratamiento de neoplasmas con neurotoxina. | |
| US20190388369A1 (en) | Use of N-acetylcysteine Amide in the Treatment Of Disease and Injury | |
| ES2692377T3 (es) | Composición farmacéutica para tratar el cáncer que comprende tripsinógeno y quimotripsinógeno | |
| US20100178273A1 (en) | Methods and compositions for treating mucositis | |
| JP2003530320A (ja) | 甲状腺疾患の治療法 | |
| JP2012082231A (ja) | 子宮障害を処置するためのクロストリジウム神経毒 | |
| EP3442564B1 (en) | Composition of proenzymes for cancer treatment | |
| JP3954644B2 (ja) | 医療に使用する化合物 | |
| ES2434943T3 (es) | Uso de antagonista de bombesina/péptido liberador de gastrina para el tratamiento de septicemia, choque séptico, lesión pulmonar aguda o artritis reumatoide | |
| ES2941348T3 (es) | Composición de proenzima | |
| ES2965358T3 (es) | Tratamiento del cáncer | |
| JP2004002429A (ja) | 組成物およびその治療用途 | |
| KR20210068455A (ko) | 종양 치료에서 오존화 오일을 포함하는 제제 | |
| US20190262435A1 (en) | Proenzyme composition | |
| KR20230084242A (ko) | 피부과적 병태의 치료 | |
| DE69732784T2 (de) | Inhibierung von invasiver remodulierung | |
| US20220249425A1 (en) | Methods for treating breast cancer | |
| KR20240025512A (ko) | 건선의 치료 방법 | |
| 김경수 | Radiation-induced esophagitis in vivo and in vitro reveals that epidermal growth factor (EGF) is a potential candidate for therapeutic intervention strategy | |
| WO2023150720A2 (en) | Medicinal uses of oligopeptides in combination with an antiandrogen | |
| EA046351B1 (ru) | Состав, содержащий озонированное масло, для лечения опухоли |