ES2930458T3 - Compuestos de aluminio para su uso en agentes terapéuticos y vacunas - Google Patents
Compuestos de aluminio para su uso en agentes terapéuticos y vacunas Download PDFInfo
- Publication number
- ES2930458T3 ES2930458T3 ES17185526T ES17185526T ES2930458T3 ES 2930458 T3 ES2930458 T3 ES 2930458T3 ES 17185526 T ES17185526 T ES 17185526T ES 17185526 T ES17185526 T ES 17185526T ES 2930458 T3 ES2930458 T3 ES 2930458T3
- Authority
- ES
- Spain
- Prior art keywords
- aqueous composition
- aluminum
- protein
- composition
- content
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/104—Pseudomonadales, e.g. Pseudomonas
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/525—Virus
- A61K2039/5252—Virus inactivated (killed)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55505—Inorganic adjuvants
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/24011—Flaviviridae
- C12N2770/24111—Flavivirus, e.g. yellow fever virus, dengue, JEV
- C12N2770/24134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Inorganic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Virology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicinal Preparation (AREA)
Abstract
La invención se refiere a medios y métodos para preparar una composición acuosa que comprende aluminio y una proteína. La invención se refiere además a composiciones acuosas que comprenden una proteína y una sal de aluminio, comprendiendo dicha composición menos de 350 ppb de metal pesado basado en el peso de la composición acuosa. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Compuestos de aluminio para su uso en agentes terapéuticos y vacunas
La invención se refiere a los campos de productos farmacéuticos y vacunas. Más en particular, la invención se refiere al campo de los compuestos y composiciones que se coadministran con el medicamento y/o antígeno.
Los compuestos de aluminio (también denominados en el presente documento “ aluminio” ), incluidos el fosfato de aluminio (AlPO4), el hidróxido de aluminio (Al(OH)3) y otras vacunas precipitadas con aluminio, son actualmente los adyuvantes más utilizados con vacunas humanas y veterinarias. Los adyuvantes con frecuencia se denominan “ alumbre” en la literatura.
Los adyuvantes de aluminio se han utilizado en la vacunación práctica durante más de medio siglo. Inducen una inmunidad protectora temprana, de alto título y duradera. A lo largo de los años, se han administrado miles de millones de dosis de vacunas con adyuvantes de aluminio. Su seguridad y eficacia los han convertido en los adyuvantes más populares en vacunas hasta la fecha. En general, los adyuvantes de aluminio se consideran seguros cuando se utilizan de acuerdo con los calendarios de vacunación vigentes.
En las vacunaciones humanas, antiguamente se han utilizado adyuvantes de aluminio en las vacunas contra el tétanos, la difteria, la tos ferina y la poliomielitis como parte de los programas convencionales de vacunación infantil. También se han introducido adyuvantes de aluminio en las vacunas contra el virus de la hepatitis A y la hepatitis B y contra el virus de la encefalitis japonesa (también denominado en el presente documento “ JEV” ). Otras vacunas adsorbidas en aluminio contra, por ejemplo, el ántrax, están disponibles para grupos de riesgo especiales. En medicina veterinaria, los adyuvantes de aluminio se han utilizado en un gran número de formulaciones de vacunas contra enfermedades víricas y bacterianas, y en intentos de fabricar vacunas antiparasitarias.
Los adyuvantes normalmente sirven para poner en contacto el antígeno, la sustancia que estimula la respuesta inmunitaria protectora específica, con el sistema inmunitario e influir en el tipo de inmunidad producida, así como en la calidad de la respuesta inmunitaria (magnitud o duración); los adyuvantes también pueden disminuir la toxicidad de determinados antígenos; y proporcionar solubilidad a algunos componentes de las vacunas. Los estudios han demostrado que muchas vacunas que contienen aluminio provocan respuestas de anticuerpos mayores y más prolongadas que las vacunas comparables sin el adyuvante. El beneficio de los adyuvantes por lo general se ha observado durante la serie de inmunización inicial en lugar de con dosis de refuerzo.
Existen tres tipos generales de adyuvantes que contienen aluminio:
Hidróxido de aluminio, Fosfato de aluminio y Sulfato de aluminio y potasio (denominados colectivamente con frecuencia “Alumbre” )
La eficacia de cada sal como adyuvante depende de las características de la vacuna específica y de cómo el fabricante prepara la vacuna. Para funcionar como adyuvante, el antígeno normalmente se adsorbe al aluminio; es decir, se agrupa con la sal de aluminio para mantener el antígeno en sitio de inyección.
No todas las vacunas contienen sales de aluminio. En ocasiones puede no haber sido necesario un adyuvante o se seleccionó otro adyuvante. Son ejemplos de vacunas comerciales que no contienen sales de aluminio son la vacuna inactivada contra el virus de la polio (IPV), la vacuna contra el sarampión, las paperas y la rubéola (MMR), la vacuna contra la varicela, la vacuna contra el meningococo conjugado (MCV4) y las vacunas contra la gripe. Que las vacunas comerciales no contengan sales de aluminio normalmente no significa que una sal de aluminio no funcione. Simplemente significa que, por alguna razón, se seleccionó otro adyuvante.
Son ejemplos de vacunas para niños autorizadas en EE. UU. que contienen adyuvantes de aluminio: DTP (vacuna contra la difteria, el tétanos y la tos ferina); DTaP (vacuna contra la difteria, el tétanos y la tos ferina acelular); algunas, pero no todas, las vacunas conjugadas contra el Hib (Haemophilus influenzae de tipo b); vacuna antineumocócica conjugada; vacunas contra la hepatitis B; vacunas contra la hepatitis A; vacuna contra el virus del papiloma humano; vacuna contra el ántrax; y vacuna contra la rabia.
El aluminio es un elemento muy abundante en nuestro entorno. Está en muchos alimentos que comemos, muchos productos de higiene personal que aplicamos en nuestra piel (desodorantes, por ejemplo) y muchos medicamentos que ingerimos. Diversas agencias gubernamentales establecen directrices para la exposición a sustancias potencialmente tóxicas. Estas directrices se denominan “ niveles mínimos de riesgo” : la cantidad máxima a la que uno puede estar expuesto a lo largo del tiempo, por lo general a diario, sin daño esperado.
La Agencia de los EE. UU. para el Registro de Sustancias Tóxicas y Enfermedades (ATSDR) estimó estos niveles para bebés teniendo en cuenta la cantidad de aluminio (por ejemplo, en forma de sal) que un niño comería y recibiría mediante inyección de vacunas. La carga corporal de aluminio de ambas fuentes está por debajo del nivel de riesgo mínimo, excepto de forma transitoria después de las vacunaciones; puesto que el 50-70 % del aluminio inyectado se excreta en 24 horas, se cree que esto no tiene ningún efecto negativo.
Los adyuvantes de hidróxido de aluminio y fosfato de aluminio se preparan generalmente exponiendo soluciones acuosas de iones de aluminio a condiciones por lo general ligeramente alcalinas en un entorno químico bien definido y controlado. Se pueden usar diversas sales de aluminio solubles para la producción de hidróxido de aluminio. Los aniones presentes en el momento de la precipitación pueden coprecipitar (para consultar una revisión, véase Lindblad, EB (2004) Immunol. and Cell Biol. Vol. 82: 497-505).
La sal de aluminio también se utiliza en la fabricación y composición de medicamentos. Por ejemplo, el factor VIII se purifica a partir de crioprecipitado de plasma. El precipitado se solubiliza, se absorbe en hidróxido de aluminio y después se trata para inactivar virus con envuelta lipídica. Después de varios otras etapas de procesamiento, el concentrado se utiliza para tratar pacientes con hemofilia A (Burnouf T, (1991) Vox Sang. Vol 60: págs. 8-15).
Estey et al. (2009. Journal of Pharmaceutical Sciences, 98(9): 2994-3012) describe la evaluación de la degradación química de una vacuna de proteína recombinante trivalente contra la neurotoxina botulínica mediante cartografiado de péptido LysC y espectrometría de masas MALDI-TOF. Los aspectos y realizaciones de la presente invención se exponen en las reivindicaciones.
En la presente invención se ha demostrado que la estabilidad de un producto biológico en una composición que también comprende una sal de aluminio no siempre es la misma. La presente invención, por ejemplo, muestra que la estabilidad de un componente proteínico (por ejemplo, como tal o dentro de un complejo tal como, por ejemplo, un virus u otro patógeno) en el contexto de una composición acuosa que también comprende sal de aluminio depende del contenido de metales pesados. Para estimar a priori si la proteína será estable en esta composición, la presente invención establece que es necesario determinar el contenido residual de metales pesados en la composición (de lo contrario, la composición acuosa que comprende una proteína corre el riesgo de degradarse con el tiempo, en particular, la invención prevé que este riesgo sea considerable cuando el contenido de metales pesados residuales sea superior a 350 ppb (es decir, aproximadamente 350 ng por ml) en dicha composición acuosa). Además, la invención también reveló que este contenido residual de metales pesados no puede retirarse fácilmente del compuesto de aluminio. Para este fin, la descripción proporciona un método para preparar una composición acuosa que comprende aluminio y una proteína, comprendiendo dicho método
- combinar una sal de aluminio, dicha proteína y agua para producir dicha composición acuosa y
- determinar el nivel de un metal pesado en la composición acuosa y/o la sal de aluminio. Las composiciones que comprenden menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa, se pueden almacenar en una fase líquida a una temperatura de entre 0 y 30 grados centígrados, durante al menos 1 mes, tal como, por ejemplo, 20 meses a 2-8 °C. El componente proteínico de dicha composición es estable durante al menos 1 mes en dicha fase líquida. Las composiciones que comprenden más de 350 ppb de metal pesado basándose en el peso de la composición acuosa no se pueden almacenar durante un período prolongado en condiciones tales como que el componente proteínico en dicha composición cambie en al menos un aspecto durante el período de tiempo indicado. Por lo tanto, un mililitro o un gramo de composición acuosa contiene preferiblemente no más de 350 nanogramos de metal pesado. La composición acuosa comprende preferiblemente entre 0,1 mg/ml y 2,5 mg/ml de aluminio. La dosis promedio de aluminio por administración es preferiblemente de no más de 1,25 miligramos (mg). En una realización particularmente preferida, la dosis de aluminio por administración es de no más de 0,25 mg de aluminio. Una dosis normalmente comprende entre 0,5 y 1 ml de la composición acuosa.
En un aspecto adicional de la presente invención, se ha demostrado que la estabilidad de un producto biológico en una composición que comprende una sal de aluminio y un compuesto reactivo no siempre es la misma. La presente invención, por ejemplo, muestra que la estabilidad de un componente proteínico (por ejemplo, como tal o dentro de un complejo tal como, por ejemplo, un virus u otro patógeno) en el contexto de una composición acuosa que también comprende sal de aluminio y un compuesto reactivo tal como, por ejemplo, un sulfito, depende críticamente del contenido de metales pesados. Para estimar a priori si el componente proteínico (tal como, por ejemplo, el componente proteínico dentro de un complejo, tal como, por ejemplo, una partícula de virus; en el presente documento también denominada simplemente proteína) será estable en esta composición, es necesario determinar el contenido de metales pesados en la composición. Para este fin, la descripción proporciona un método para preparar una composición acuosa que comprende aluminio y una proteína, comprendiendo dicho método
- combinar una sal de aluminio, dicha proteína y agua para producir dicha composición acuosa y
- determinar el nivel de un metal pesado en la composición acuosa y/o la sal de aluminio. Las composiciones que comprenden menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa, se pueden almacenar en una fase líquida a una temperatura de entre 0 y 30 grados centígrados, durante al menos 1 mes, tal como, por ejemplo, 20 meses a 2-8 °C. El componente de proteína en dicha composición es estable durante al menos 1 mes en dicha fase líquida, tal como, por ejemplo, 20 meses a 2-8 °C. Las composiciones que comprenden más de 350 ppb de metal pesado basándose en el peso de la composición acuosa no se pueden almacenar durante un período prolongado en condiciones tales como que el componente proteínico en dicha composición cambie en al menos un aspecto durante el período de tiempo indicado. Por lo tanto, un mililitro o un gramo de composición acuosa contiene preferiblemente no más de 350 nanogramos de metal pesado. La composición acuosa comprende preferiblemente
entre 0,1 mg/ml (miligramo por mililitro) y 2,5 mg/ml de aluminio. La dosis promedio de aluminio por administración es preferiblemente de no más de 1,25 miligramos (mg). En una realización particularmente preferida, la dosis de aluminio por administración es de no más de 0,25 mg de aluminio. Una dosis normalmente comprende entre 0,5 y 1 ml de la composición acuosa. Una composición acuosa que comprende una proteína, una sal de aluminio y, opcionalmente, un compuesto reactivo, comprendiendo dicha composición menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa, también se denomina en el presente documento “ una composición acuosa que comprende una proteína según la descripción” o “ una composición que comprende una proteína según la descripción” .
El contenido de aluminio de la composición acuosa por lo general es de aproximadamente 0,1 mg/ml a 2,5 mg/ml, comprendiendo la vacuna promedio aproximadamente de 0,5 a 1,5 mg/ml del adyuvante de aluminio. Para este fin, la descripción proporciona una composición acuosa, una composición farmacéutica acuosa o de vacuna que comprende aluminio, un compuesto reactivo y una proteína, que comprende entre 5 mcgramo/ml y 50 mg/ml de aluminio y que comprende no más de 700 ppm de un metal pesado en comparación con el contenido de aluminio (gramos/gramos). En este contexto, 700 ppm de un metal pesado equivalen a 700 mcgramo de metal pesado por gramo de aluminio.
En una realización preferida, la composición acuosa, la composición farmacéutica acuosa o de vacuna que comprende aluminio, un compuesto reactivo y una proteína, comprende entre 5 mcgramo/ml y 50 mg/ml de aluminio y comprende no más de 450 ppm de un metal pesado en comparación con el contenido de aluminio (gramos/gramos). En este contexto, 450 ppm de un metal pesado equivalen a 450 mcgramo de metal pesado por gramo de aluminio. En una realización preferida, la composición acuosa, la composición farmacéutica acuosa o de vacuna que comprende aluminio, un compuesto reactivo y una proteína, comprende entre 5 mcgramo/ml y 50 mg/ml de aluminio y comprende no más de 700 ppm de Fe en comparación con el contenido de aluminio (gramos/gramos). Preferiblemente, el contenido de Fe es inferior a 420 ppm de Fe en comparación con el contenido de aluminio. Preferiblemente, el contenido de Fe es inferior a 350 ppm, preferiblemente inferior a 100 y más preferiblemente inferior a 50 ppm en comparación con el contenido de aluminio (1 ppm = 1 mcgramo/gramo de Al). En una realización particularmente preferida, el contenido de Fe de la composición acuosa, el producto farmacéutico acuoso o vacuna es inferior a 420 ppm en comparación con el contenido de aluminio. En una realización preferida, la composición acuosa, el producto farmacéutico acuoso o vacuna comprende más de 10 ppm de Fe en comparación con el contenido de aluminio.
En una realización preferida, la composición acuosa, la composición farmacéutica acuosa o de vacuna que comprende aluminio, un compuesto reactivo y una proteína, comprende entre 5 mcgramo/ml y 50 mg/ml de aluminio y comprende no más de 35 ppm de Ni en comparación con el contenido de aluminio (gramos/gramos). Preferiblemente, el contenido de Ni es inferior a 18 ppm de Ni en comparación con el contenido de aluminio. Preferiblemente, el contenido de Ni es inferior a 9 ppm, preferiblemente inferior a 3 y más preferiblemente inferior a 1 ppm en comparación con el contenido de aluminio (1 ppm = 1 mcgramo/gramo de Al). En una realización particularmente preferida, el contenido de Ni de la composición acuosa, el producto farmacéutico acuoso o vacuna es inferior a 18 ppm en comparación con el contenido de aluminio. En una realización preferida, la composición acuosa, una composición farmacéutica acuosa o de vacuna comprende al menos 200 ppb de Ni en comparación con el contenido de aluminio.
En una realización preferida, la composición acuosa, la composición farmacéutica acuosa o de vacuna que comprende aluminio, un compuesto reactivo y una proteína, comprende entre 5 mcgramo/ml y 50 mg/ml de aluminio y comprende no más de 5 ppm de Cu en comparación con el contenido de aluminio (gramos/gramos). Preferiblemente, el contenido de Cu es inferior a 2,5 ppm de Cu en comparación con el contenido de aluminio. Preferiblemente, el contenido de Cu es inferior a 1 ppm, preferiblemente inferior a 0,5 y más preferiblemente inferior a 0,25 ppm en comparación con el contenido de aluminio (1 ppm = 1 mcgramo/gramo de Al). En una realización particularmente preferida, el contenido de Cu de la composición acuosa, el producto farmacéutico acuoso o vacuna es inferior a 2,5 ppm en comparación con el contenido de aluminio. En una realización preferida, la composición acuosa, la composición farmacéutica acuosa o de vacuna comprende al menos 50 ppb de Cu en comparación con el contenido de aluminio.
En una realización particularmente preferida, el contenido de Fe de la composición acuosa, el producto farmacéutico acuoso o vacuna es inferior a 420 ppm en comparación con el contenido de aluminio, el contenido de Ni de la composición acuosa, el producto farmacéutico acuoso o vacuna es inferior a 18 ppm en comparación con al contenido de aluminio y al contenido de Cu de la composición acuosa, el producto farmacéutico acuoso o vacuna es inferior a 2,5 ppm en comparación con el contenido de aluminio. En una realización preferida, la composición acuosa, el producto farmacéutico acuoso o vacuna comprende más de 10 ppm de Fe en comparación con el contenido de aluminio, al menos 200 ppb de Ni en comparación con el contenido de aluminio y al menos 50 ppb de Cu en comparación con el contenido de aluminio.
En un método para preparar una composición acuosa, una composición farmacéutica acuosa o de vacuna que comprende aluminio, un compuesto reactivo y una proteína según la descripción, o un método para preparar o seleccionar una sal de aluminio según la invención, o un método para preparar una precipitado de sal de aluminio de calidad clínica según la descripción, se prefiere que el contenido de metales pesados con respecto al contenido de aluminio sea como se ha indicado en el presente documento anteriormente. Por lo tanto, la descripción proporciona un método para preparar una composición acuosa que comprende aluminio, un compuesto reactivo y una proteína, comprendiendo dicho método
- preparar o seleccionar una sal de aluminio que sea capaz de proporcionar una composición acuosa que tenga menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa y
- combinar dicha sal de aluminio, dicho compuesto reactivo, dicha proteína y agua para producir dicha composición acuosa,
por lo que la composición acuosa comprende entre 5 mcgramo/ml y 50 mg/ml de aluminio, preferiblemente entre 0,1 mg/ml y 2,5 mg/ml; y no más de 700 ppm de metal pesado y preferiblemente no más de 450 ppm de metal pesado en comparación con el contenido de aluminio; en donde dicha composición acuosa comprende no más de 420 ppm de Fe, preferiblemente no más de 100 ppm, en comparación con el contenido de aluminio; no más de 18 ppm, preferiblemente no más de 3 ppm, de Ni en comparación con el contenido de aluminio; y/o no más de 2,5 ppm de Cu, preferiblemente no más de 0,5 ppm, en comparación con el contenido de aluminio. En un ejemplo, dicha composición acuosa comprende no más de 700 ppm de metal pesado y preferiblemente no más de 450 ppm de metal pesado en comparación con el contenido de aluminio; no más de 420 ppm de Fe, preferiblemente no más de 100 ppm, en comparación con el contenido de aluminio; no más de 18 ppm, preferiblemente no más de 3 ppm de Ni en comparación con el contenido de aluminio; y no más de 2,5 ppm de Cu, preferiblemente no más de 0,5 ppm, en comparación con el contenido de aluminio.
Se ha observado que el componente de aluminio es una fuente importante para el metal pesado en la composición acuosa. Por lo tanto, una forma de controlar la cantidad de metal pesado en la composición acuosa es controlar la cantidad de metal pesado en la fuente de aluminio utilizada para generar la composición acuosa. Por lo tanto, la descripción proporciona además un método para preparar una composición acuosa que comprende aluminio y una proteína, comprendiendo dicho método
- preparar o seleccionar una solución de sal de aluminio (tal como, por ejemplo, 10 mg/ml de líquido de hidróxido de aluminio (tal como, por ejemplo, alhydrogel® 2 % de Brenntag Biosector, número de catálogo 843261) que en la formulación proteínica final comprenda no más de 350 ppb de metal pesado basándose en el peso de la composición acuosa (por ejemplo, para el alhydrogel® 2 % y una cantidad final de 0,25 mgramos de hidróxido de aluminio en la composición acuosa de 0,5 ml (= dosis), la solución de sal de aluminio seleccionada o preparada no debe contener más de 7 microgramos de metales pesados por mililitro de la solución de alhydrogel® 2 %, aproximadamente 7 ppm de contenido de metales pesados), y
- combinar dicha solución de sal de aluminio, dicha proteína, agua y, opcionalmente, un compuesto reactivo para producir dicha composición acuosa (con no más de 350 ppb de metal pesado en la composición acuosa).
Por ejemplo, la solución de sal de aluminio, por ejemplo, el líquido de hidróxido de aluminio (utilizado como un componente que ha de mezclarse para dar como resultado la composición acuosa final) no debe tener un contenido de metales pesados superior a 7 ppm (proporcionado como un ejemplo en el presente documento donde los 10 mg/ml de líquido de hidróxido de aluminio se diluirán a 0,5 mg/ml para dar como resultado un contenido de metales pesados de aproximadamente 350 ppb con respecto a la composición acuosa, suponiendo que 1 ppm = aproximadamente 1 mg/ml) sobre la base del peso del líquido de hidróxido de aluminio. El límite de contenido aceptable de metales pesados también puede expresarse con respecto al peso de la sal de aluminio tal como el hidróxido de aluminio en solución (denominado también “ compuesto de aluminio de partida” ). El contenido aceptable de metales pesados en este ejemplo entonces no puede superar los 7 microgramos de metales pesados por cada gramo de solución de hidróxido de aluminio (es decir, aproximadamente 7 ppm), es decir, la solución de alhydrogel® 2 %. Como se ha indicado anteriormente en el presente documento, la composición acuosa que comprende la proteína preferiblemente (tal como, por ejemplo, si se utiliza como vacuna) comprende entre 0,1 y 2,5 mg/ml del compuesto de aluminio. Sin embargo, la concentración de metales pesados en la composición acuosa, según la composición, no debe superar 350 ppb, es decir, aproximadamente 350 ng por ml de la composición final y, por lo tanto, la selección o preparación del compuesto de aluminio de partida debe realizarse en consecuencia. Con el fin de ilustrar adicionalmente la selección de una solución de compuesto de aluminio de partida adecuada (por ejemplo, en forma de una solución concentrada (véase anteriormente alhydrogel® 2 % = 10 mg/ml), se muestra que una solución de compuesto de aluminio de 10 mg/ml de hidróxido de aluminio que tiene aproximadamente 7 ppm de impurezas de metales pesados corresponde a una concentración de metales pesados en la composición proteínica cuando la concentración de aluminio es de aproximadamente 0,1 mg/ml de aproximadamente 70 nanogramos/ml o 70 ppb (muy por debajo de las 350 ppb proporcionadas como límite del contenido de metales pesados como se enseña en la invención). Una concentración de 2,5 mg/ml del hidróxido de aluminio en la composición acuosa final partiendo de una solución de aluminio de 10 mg/ml de hidróxido de aluminio que tenga aproximadamente 7 ppm de impurezas de metales pesados dará como resultado un contenido de metales pesados en la composición proteínica acuosa que corresponde a una concentración de metales pesado de aproximadamente 1,75 microgramos/ml o aproximadamente 1.750 ppm (muy por encima de las 350 ppb proporcionadas como límite de contenido de metales pesados como enseña la invención). Por lo tanto, la solución de aluminio de 10 mg/ml de hidróxido de aluminio en este caso (el contenido final de hidróxido de aluminio es de 2,5 mg/ml) no debería tener más de 1,4 ppm de impurezas de metales pesados.
Un método de preparación de una composición acuosa que comprende una proteína comprende preferiblemente además envasar alícuotas de dicha composición acuosa que tiene menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa en recipientes de almacenamiento herméticos separados. La proteína en los recipientes de almacenamiento herméticos es estable y se puede almacenar durante al menos tres meses, tal como,
por ejemplo, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 meses, preferiblemente 20 o 24 meses, más preferiblemente 20 meses a una temperatura de entre 2 a 8 °C grados centígrados.
Sin quedar ligados a la teoría, la degradación de antígenos en composiciones acuosas, tal como la composición inmunogénica, que comprende iones de metales pesados presentes en una sal de aluminio, tal como hidróxido de aluminio, podría explicarse con una vía de degradación subyacente asumiendo radicales libres tales como, por ejemplo, radicales libres de sulfito. La formación de radicales libres puede ser catalizada por iones de metales pesados presentes en, por ejemplo, el hidróxido de aluminio y este efecto (en el caso de una determinada cantidad de metal pesado como se indica según la invención) podría ser el mecanismo de causa raíz subyacente para problemas de estabilidad como se identifica como parte de la contribución inventiva. La parte experimental de la presente solicitud muestra con gran detalle la evidencia de esta causa raíz para la vacuna contra la encefalitis japonesa (también conocida como “JEV” ) y presenta una demostración similar para una composición de polipéptido con adyuvante de aluminio simple que comprende un compuesto reactivo tal como el sulfito. Por lo tanto, es evidente que también se puede producir una reacción similar en otra composición acuosa que comprenda aluminio (con un contenido elevado de metales pesados, por ejemplo, superior a 350 ppb basándose en el peso de la composición), proteína y posiblemente un componente reactivo tal como el sulfito y/o otras partículas formadoras de radicales. La oxidación catalizada por metales pesados es una vía de degradación que da como resultado la modificación covalente de las proteínas. Las propiedades fisicoquímicas modificadas de la proteína o antígeno oxidados/modificados pueden dar como resultado la pérdida de la actividad biológica (Li et al., 1995; Mayo et al., 2003; Stadtmann, 1990).
Los siguientes esquemas de reacción (que posiblemente ocurran en el producto de JEV como se describe en la parte experimental) son indicativos de las composiciones de la descripción que comprenden además de la proteína y el metal pesado también sulfito y/u otro compuesto reactivo tal como, por ejemplo, sulfito y/u formaldehído.
El metabisulfito de sodio (Na2S2 Os) en solución se disuelve en bisulfito (S2 O52-), en una solución alcalina el equilibrio del bisulfito es hacia el sulfito (SO32-) y en una solución ácida hacia H2SO3/SO2. Después de la disolución de Na2S2O5 en agua, se hidroliza en NaHSO3 de la siguiente manera
Na2S2O5 o 2 NaHSO3
A pH neutro se puede asumir el siguiente equilibrio:
HSO3- o H+ SO32- pKa=7,2
Esto significa que a pH 7 el equilibrio se desplaza hacia HSO3- y en condiciones más básicas (por ejemplo, pH 8) hacia SO32-.
El formaldehído forma un aducto de bisulfito durante la neutralización según la siguiente ecuación:
CH2O HSO3- ^ CH2(OH)(SO3)-
Después de que se agota el bisulfato, la reacción transcurre hasta que se alcanza el equilibrio de la siguiente manera:
CH2O SO32- H2O ^ CH2(OH)(SO3)- OH-
El formaldehído y el sulfito reaccionan entre sí, sin embargo, se ha encontrado que el formaldehído y el sulfito todavía están presentes en equilibrio en la vacuna contra el JEV y se pueden detectar en el siguiente intervalo (n=49):

Según la literatura (Ranguelova et al., 2010), los iones de metales de transición catalizan la auto-oxidación de (bi)sulfito a través de la formación de radical anión trióxido de azufre (•SO 3"):
Mn+ SO32" ^ M(n-1)+ •SO^
donde M puede ser cobre (Cu2+), hierro (Fe3+), oxivanadio (VO2+), manganeso (Mn2+), níquel (Ni2+) o anión cromato (CrO42-) (Alipazaga et al. 2004; Berglund et al. 1993; Brandt y Elding 1998; Lima et al. 2002; Shi 1994).
Se demostró que dichos radicales sulfito son altamente reactivos y pueden oxidar diversas sustancias, tales como ascorbato, Hidroquinona e Histidina (Huie et al., 1985). Neta y Huie, 1985, publicaron una revisión de la química de los radicales libres del sulfito. Los autores también muestran que la formación de radicales también puede ser catalizada por fotoionización de sulfito de la siguiente manera:
SO32- hv ^ *SO3- e-
La formación de radicales catalizada por la luz también podría explicar las diferencias observadas en la potencia y los resultados de ELISA de las jeringas desnudas sin etiquetar (utilizadas para los ensayos de liberación y con fines de referencia) y las muestras del lote de vacuna final totalmente envasadas para el producto de JEV. Las muestras totalmente envasadas están completamente protegidas de la luz, mientras que las jeringas sin etiquetar pueden estar expuestas a la luz durante el almacenamiento y la manipulación.
Una reacción importante del radical sulfito en los sistemas de autooxidación es con el oxígeno molecular para formar un radical peroxilo que es mucho más reactivo:
•SO3- O2 ^ *SO5-
La solubilidad de O2 en agua a 0 °C y 20 °C es de 0,4 mM y 0,25 mM, respectivamente. Asumiendo que la solubilidad de O2 en una composición de la descripción está en un intervalo similar, hay presente una cantidad considerable de oxígeno para formar el radical peroxilo. Este radical es un oxidante mucho más fuerte en comparación con ^SO3- y puede oxidar determinados sustratos que no están unidos por ^SO3- en absoluto y que, de hecho, pueden formar radicales que oxidan iones sulfito. En dichos casos, cuando el potencial redox del sustrato es intermedio entre los de •SO3- y •SOs-, es probable que se desarrolle una cadena de reacción en presencia de O2 siguiendo el patrón general:
•SO3- O2 ^ ^SO5-
•SO5- X ^ SO52- •X*
•X+ SO32- ^ X ^SO3-
La intermediación de un sustrato X puede potenciar el proceso en cadena de oxidación de sulfitos por oxígeno. La reducción de un electrón de ^SO5- rinde HSO5-(peroximonosulfato), un oxidante muy fuerte que es capaz de oxidar muchos compuestos orgánicos (Lambeth et al., 1973; Ito y Kawanashi, 1991). El peroximonosulfato es también un radical anión sulfato precursor ^SO4-.
•SO5- HSO3- ^ ^SO4-. HSO4-
El radical ^SO4- es un oxidante muy fuerte, casi tan fuerte como el radical hidroxilo (•OH), y es muy probable que oxide otras biomoléculas por oxidación de un electrón.
En una realización preferida, la composición que comprende una proteína según la descripción es una composición terapéutica o una composición inmunogénica, tal como una vacuna. Las composiciones terapéuticas se administran a individuos, tales como un ser humano o un animal. En particular para dichas composiciones, es importante que la proteína dentro de la composición aún tenga su efecto terapéutico en el momento en que se administra a dicho individuo. La degradación de la proteína o los cambios en la estructura de la proteína pueden dar como resultado que la proteína pierda su actividad terapéutica. De forma similar, la degradación o los cambios estructurales de la composición inmunogénica también conducirán a una reducción de la eficacia de la composición para inducir y/o potenciar una respuesta inmunitaria en un individuo. Una composición inmunogénica se administra preferiblemente a un individuo para contrarrestar o prevenir una infección vírica o bacteriana. La proteína contenida dentro de la composición inmunogénica acuosa puede ser una única proteína o una proteína multimérica o parte de un complejo que comprende dicha proteína (por ejemplo, tal como parte de un virus o una célula, por ejemplo, una célula bacteriana).
En una realización preferida, dicha proteína o complejo proteínico comprende una bacteria o un virus vivo atenuado, inactivado o mutado o una proteína vírica o bacteriana inmunogénica o una parte inmunogénica de dicha proteína (por ejemplo, un péptido inmunogénico), una bacteria o un virus dividido, o células bacterianas completas. Si dicha composición inmunogénica se administra para proporcionar protección frente a una infección vírica o bacteriana, la degradación de la proteína puede dar como resultado la pérdida de la capacidad protectora de la composición inmunogénica. Como se utiliza en el presente documento, un virus o una bacteria “vivos atenuados” es un virus o una bacteria que son menos patógenos que el virus o la bacteria de tipo silvestre, pero que ha mantenido propiedades inmunogénicas. Como se utiliza en el presente documento, un virus o una bacteria “ inactivados” se refieren a un virus o una bacteria que se han inactivado para que ya no sea infecciosos, mientras que las propiedades inmunogénicas se han mantenido al menos en parte. Un virus o una bacteria inactivados pueden estar en forma de virus o células bacterianas completos inactivados. Sin embargo, la inactivación puede dar como resultado la alteración de virus o células bacterianas. Por lo tanto, el virus o la bacteria inactivados también pueden estar en forma alterada. Como se utiliza en el presente documento, un virus o una bacteria “ dividido” se refieren a un virus o una bacteria que se alteran utilizando, por ejemplo, un detergente.
La composición acuosa de la descripción puede comprender un compuesto reactivo, tal como formaldehído, que generalmente está presente para inactivar un virus o una bacteria. También puede haber presente un compuesto reactivo adicional o alternativo para inactivar cualquier formaldehído residual en la solución acuosa, tal como el compuesto reactivo sulfito. Como se ha detallado anteriormente, se piensa que los metales pesados catalizan la oxidación del (bi)sulfito y la formación de radicales sulfito presentes en la composición acuosa, lo que a su vez puede inducir la degradación de la proteína presente en la composición. Por lo tanto, es probable que las composiciones que comprenden virus o bacterias inactivados o partes inmunogénicas de los mismos comprendan uno o más compuestos reactivos. Por lo tanto, la ausencia de metales pesados o la presencia por debajo de los niveles identificados por la presente invención, es de particular relevancia cuando la composición acuosa de la descripción comprende un virus o una bacteria inactivados o una parte inmunogénica de los mismos. En una realización particularmente preferida, una composición inmunogénica según la descripción, por lo tanto, comprende un virus inactivado. En una realización preferida, la composición acuosa, preferiblemente la composición inmunogénica, según la descripción comprende virus o bacterias inactivados. Dicho virus o bacteria es inactivado, por ejemplo, por un compuesto reactivo tal como formaldehído como se describe por la presente descripción.
En otra realización preferida, la composición acuosa, composición inmunogénica o vacuna según la descripción comprende un toxoide.
Como se utiliza en el presente documento, un “toxoide” se refiere a una toxina bacteriana inactivada, tal como una exotoxina, como resultado de la cual se reduce o suprime la toxicidad mientras se mantienen, al menos en parte, las propiedades inmunogénicas. Dicha toxina es inactivada, por ejemplo, por un compuesto reactivo tal como formaldehído como se describe por la presente descripción. Los ejemplos de toxoides presentes en una composición inmunogénica según la descripción incluyen, pero sin limitación, toxinas de difteria, tétanos y botulismo.
La expresión “ proteína vírica o bacteriana inmunogénica” se refiere a una proteína vírica o bacteriana que es capaz de desencadenar una respuesta inmunitaria. La expresión “ parte inmunogénica” , como se utiliza en el presente documento, se refiere a una parte de una proteína vírica o bacteriana que es capaz de desencadenar una respuesta inmunitaria. Preferiblemente, la respuesta inmunitaria desencadenada reconoce tanto dicha parte de la proteína como la proteína entera. Por lo tanto, una composición acuosa que comprende una proteína según la descripción es una composición inmunogénica. Dicha composición es preferiblemente una composición terapéutica y/o una composición profiláctica tal como una vacuna. También se proporciona una vacuna que es una composición acuosa que comprende una proteína según la descripción
Una “ composición inmunogénica” se define en el presente documento como una composición que es capaz de desencadenar una respuesta inmunitaria cuando se administra a un individuo. La respuesta inmunitaria desencadenada puede ser humoral, celular o una combinación de las mismas e incluye, pero sin limitación, la producción de anticuerpos, linfocitos B tales como linfocitos B activados y linfocitos T tales como linfocitos T activados. Una respuesta inmunitaria tal como se utiliza en el presente documento se dirige preferiblemente de forma específica a uno o más inmunógenos dentro de una composición que comprende una proteína según la descripción. Una composición inmunogénica de la presente descripción puede administrarse a un individuo mediante cualquier técnica conocida en la técnica incluyendo, pero sin limitación, inyección intramuscular (IM), intradérmica (ID), subcutánea (SC), intracraneal (IC), intraperitoneal (IP) o intravenosa (IV), administración transdérmica, oral, intranasal o rectal, y combinaciones de las mismas, se prefieren la inyección intramuscular (IM), intradérmica (ID), subcutánea (SC), intracraneal (IC), intraperitoneal (IP) o intravenosa (IV). En una realización preferida, se utiliza una composición inmunogénica que comprende una proteína según la descripción para desencadenar una respuesta inmunitaria que puede ser útil en un entorno crónico (tal como el tratamiento del cáncer) o profiláctico (tal como una vacuna típica). Se prefiere que la composición inmunogénica se use como vacuna, es decir, uso profiláctico. En esta realización, el aluminio está normalmente presente como adyuvante.
Un “ adyuvante” , como se utiliza en el presente documento, se refiere a un agente farmacológico o inmunológico que modifica el efecto de otros agentes, tal como un agente inmunológico que aumenta la respuesta antigénica. Los adyuvantes normalmente sirven para poner en contacto el antígeno -la sustancia que estimula la respuesta inmunitaria protectora específica- con el sistema inmunitario e influir en el tipo de inmunidad producida, así como en la calidad de la respuesta inmunitaria (magnitud o duración); disminuir la toxicidad de determinados antígenos; y proporcionar solubilidad a algunos componentes de las vacunas
Un “ individuo” se define en el presente documento como un ser humano o un animal. Los individuos incluyen, pero sin limitación, pollos, patos, gansos, pavos, cisnes, emúes, gallinas de Guinea y faisanes, seres humanos, cerdos, hurones, focas, conejos, gatos, perros y caballos. En una realización preferida de la descripción, un individuo es un mamífero, preferiblemente un ser humano.
El “ micro” en microgramo o microlitro u otra unidad en ocasiones se denomina mc, el símbolo p o la letra u. Un valor de 3 mcgramo es, por lo tanto, 3 microgramos, 3 pl son 3 microlitros y 3 ugramo son 3 microgramos.
Con frecuencia se prepara un adyuvante de aluminio mediante la exposición controlada de una solución acuosa de iones de aluminio a condiciones alcalinas (para consultar una revisión, véase Lindblad, EB (2004) Immunol. and Cell Biol. Vol.
82: 497-505). En la presente invención se ha encontrado para el producto de JEV (véase la parte experimental) que una
gran cantidad del metal pesado en esta solución acuosa de iones de aluminio termina en el precipitado de sal de aluminio para el adyuvante de aluminio. Se ha encontrado además que la cantidad de metal pesado en el precipitado de aluminio afecta a la estabilidad de la vacuna durante el almacenamiento de la vacuna. La cantidad de metal pesado que está presente en la sal de aluminio se puede controlar determinando la cantidad de metal pesado en la sal pero además, y preferiblemente, controlando la cantidad de metal pesado en la solución acuosa de iones de aluminio. Por lo tanto, la descripción proporciona además un método para preparar un precipitado de sal de aluminio de grado clínico para su incorporación en un medicamento y/o vacuna, comprendiendo dicho método preparar una solución acuosa de iones de aluminio y precipitar dichos iones de aluminio en dicha solución, y determinar el nivel de un metal pesado en la solución y/o el precipitado de sal de aluminio, preferiblemente en donde se determina que dicha solución y/o el precipitado de sal de aluminio comprenden una cantidad que da como resultado menos de 350 ppb de metal pesado en la composición final, por ejemplo, cuando se resuspenden en la composición final. En una realización preferida, la descripción proporciona un método para preparar un precipitado de sal de aluminio de grado clínico para su incorporación en un medicamento y/o vacuna, comprendiendo dicho método preparar una solución acuosa de iones de aluminio y precipitar dichos iones de aluminio en dicha solución, y determinar el nivel de un metal pesado en la solución y/o el precipitado de sal de aluminio, en donde se selecciona el precipitado que es capaz de proporcionar una composición acuosa que comprende entre 5 mcgramo/ml y 50 mg/ml de aluminio, preferiblemente entre 0,1 mg/ml y 2,5 mg/ml, y que comprende no más de 700 ppm de metal pesado y preferiblemente no más de 450 ppm de metal pesado en comparación con el contenido de aluminio, en donde dicha composición comprende no más de 420 ppm de Fe, preferiblemente no más de 100 ppm, en comparación con el contenido de aluminio, no más de 18 ppm, preferiblemente no más de 3 ppm, de Ni en comparación con el contenido de aluminio, y/o no más de 2,5 ppm de Cu, preferiblemente no más de 0,5 ppm, en comparación con el contenido de aluminio. En una realización preferida, dicho precipitado comprende no más de 700 ppm de metal pesado y preferiblemente no más de 450 ppm de metal pesado en comparación con el contenido de aluminio, no más de 420 ppm de Fe, preferiblemente no más de 100 ppm, en comparación con el contenido de aluminio, no más de 18 ppm, preferiblemente no más de 3 ppm, de Ni en comparación con el contenido de aluminio, y no más de 2,5 ppm de Cu, preferiblemente no más de 0,5 ppm, en comparación con el contenido de aluminio.
También se proporciona una composición farmacéutica que comprende una proteína según la descripción, opcionalmente que comprende además un portador y/o diluyente farmacéuticamente aceptable. “ Un diluyente farmacéuticamente aceptable” , como se utiliza en el presente documento, se define como cualquier solución, sustancia o combinación de las mismas que no tiene actividad biológica o no deseada, lo que significa que se puede administrar a un individuo junto con otros componentes de una composición inmunológica sin provocar una reacción adversa sustancial. Los ejemplos de portadores adecuados comprenden, por ejemplo, hemocianina de lapa californiana (KLH), albúmina sérica (por ejemplo, BSA o RSA) y ovoalbúmina. En una realización preferida, dicho portador adecuado comprende una solución, como, por ejemplo, solución salina.
En un aspecto, se utiliza un método según la invención para prolongar la vida de almacenamiento o la vida útil de una composición acuosa que comprende una proteína según la descripción. Como se utiliza en el presente documento, la expresión “vida útil” se define como el período de tiempo que una composición que comprende una proteína según la descripción puede almacenarse sin volverse inadecuada para su uso, por ejemplo, debido a la degradación de la proteína (por ejemplo, está dentro de la especificación de potencia de la composición, por ejemplo, vacuna, según lo exija la agencia reguladora que aprobó o aprobará la vacuna). Durante el almacenamiento de composiciones acuosas como se describe en el presente documento, puede ocurrir la degradación de la proteína, en particular cuando se supera un determinado nivel (como se describe en el presente documento) de metales pesados. La degradación generalmente aumenta con el tiempo cuando se almacenan dichas composiciones acuosas. Ahora que se ha encontrado que la degradación de la proteína se reduce en una composición acuosa que comprende además de dicha proteína una sal de aluminio, si dicha composición comprende menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa, se ha vuelto posible contrarrestar la degradación de proteína en composiciones acuosas. Al contrarrestar la degradación de la proteína con un método según la invención, se aumenta la estabilidad de dicha proteína dentro de dicha composición y se prolonga la vida de almacenamiento de las composiciones acuosas que comprenden dicha proteína. Una composición acuosa según la presente descripción proporciona la ventaja de que es estable y no experimenta degradación de proteínas durante un período prolongado. Dicha composición acuosa que comprende una proteína según la descripción es estable durante al menos un mes a temperatura elevada tal como, por ejemplo, 20 o 37 °C, preferiblemente durante al menos tres meses a temperatura elevada, tal como, por ejemplo, 20 o 37 °C.
Una composición acuosa que comprende una proteína según la descripción se almacena preferiblemente a una temperatura de entre 0 °C y 20 °C para contribuir a una mayor vida útil, más preferiblemente entre 2 °C y 15 °C, más preferiblemente entre 2 °C y 10 °C, mucho más preferiblemente entre 2 °C y 8 °C. La vida útil a una temperatura entre 2 °C y 8 °C es estable preferiblemente durante al menos tres meses, tal como, por ejemplo, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 meses, preferiblemente 20 o 24 meses, más preferiblemente 20 meses a una temperatura de entre 2 a 8 °C grados centígrados. Por lo tanto, se proporciona una composición acuosa que comprende una proteína según la descripción que tiene una vida útil de aproximadamente 12 a 24 meses. La descripción proporciona además una solución acuosa según la descripción que se ha almacenado durante al menos un mes, preferiblemente durante al menos dos meses, más preferiblemente durante al menos tres meses, más preferiblemente durante al menos seis meses. Una composición acuosa comprende preferiblemente una proteína y una sal de aluminio, comprendiendo dicha composición menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa, entre 5 mcgramo/ml y 50 mg/ml de aluminio,
preferiblemente entre 0,1 mg/ml y 2,5 mg/ml, y no más de 700 ppm de metal pesado y preferiblemente no más de 450 ppm de metal pesado en comparación con el contenido de aluminio, en donde dicha composición comprende no más de 420 ppm de Fe, preferiblemente no más de 100 ppm, en comparación con el contenido de aluminio, no más de 18 ppm, preferiblemente no más de 3 ppm, de Ni en comparación con el contenido de aluminio, y/o no más de 2,5 ppm de Cu, preferiblemente no más de 0,5 ppm, en comparación con el contenido de aluminio. En una realización preferida, dicha composición comprende no más de 700 ppm más de 100 ppm, en comparación con el contenido de aluminio, no más de 18 ppm, preferiblemente no más de 3 ppm, de Ni en comparación con el contenido de aluminio, y no más de 2,5 ppm de Cu, preferiblemente no más de 0,5 ppm, en comparación con el contenido de aluminio.
Como se utiliza en el presente documento, “ una composición de proteína estable” significa que, en comparación con una composición de partida, no más del 50 %, preferiblemente no más del 40 %, incluso más preferiblemente no más del 30 %, incluso más preferiblemente no más del 20 %, incluso más preferiblemente no más del 10, incluso más preferiblemente no más del 5 % de la proteína en dicha composición se degrada. “ Degradado” en este contexto se refiere a cualquier modificación detectable de la proteína en comparación con la proteína en la composición de partida. Por ejemplo, una disminución en la detección de la proteína con un anticuerpo monoclonal tal como, por ejemplo, un anticuerpo que reconoce un epítopo neutralizante, es adecuada y puede medirse con cualquier método conocido en la técnica, tal como ELISA (véase el Ejemplo 4). El nivel detectado se puede comparar, por ejemplo, con el nivel detectado con un policlonal específico para la misma proteína tal como, por ejemplo, que representa la cantidad de proteína total (cambiada o sin cambios). También se proporciona un método de prolongación de la vida útil de una composición acuosa que comprende una proteína y una sal de aluminio, comprendiendo dicho método seleccionar y/o preparar una sal de aluminio dando como resultado una composición acuosa con un contenido de metales pesados de menos de 350 ppb basándose en la base del peso de la composición acuosa y combinando dicha sal de aluminio, dicha proteína y agua para producir dicha composición acuosa.
Además se proporciona un método de mejora de la reproducibilidad de la vida útil de preparaciones de composiciones acuosas que comprenden una proteína y una sal de aluminio, comprendiendo dicho método obtener al menos dos preparaciones de sal de aluminio diferentes, determinando la cantidad de al menos un metal pesado en dichas preparaciones de sal de aluminio, seleccionando entre dichas preparaciones de sal de aluminio, preparaciones de sal de aluminio que comprenden menos de 350 ppb de dicho al menos un metal pesado; y combinar la sal de aluminio de dichas preparaciones seleccionadas con dicha proteína y agua para producir dichas composiciones acuosas. Preferiblemente un metal pesado; y combinar la sal de aluminio de dichas preparaciones seleccionadas con dicha proteína y agua para producir dichas composiciones acuosas. Preferiblemente se selecciona una composición que comprende entre 5 mcgramo/ml y 50 mg/ml de aluminio, preferiblemente entre 0,1 mg/ml y 2,5 mg/ml, y que comprende no más de 700 ppm de metal pesado y preferiblemente no más de 450 ppm de metal pesado en comparación con el contenido de aluminio, en donde dicha composición comprende no más de 420 ppm de Fe, preferiblemente no más de 100 ppm, en comparación con el contenido de aluminio; no más de 18 ppm, preferiblemente no más de 3 ppm, de Ni en comparación con el contenido de aluminio; y/o no más de 2,5 ppm de Cu, preferiblemente no más de 0,5 ppm, en comparación con el contenido de aluminio. Más preferiblemente, dicha composición comprende no más de 700 ppm de metal pesado y preferiblemente no más de 450 ppm de metal pesado en comparación con el contenido de aluminio, en donde dicha composición comprende no más de 420 ppm de Fe, preferiblemente no más de 100 ppm, en comparación con el contenido de aluminio; no más de 18 ppm, preferiblemente no más de 3 ppm, de Ni en comparación con el contenido de aluminio; y no más de 2,5 ppm de Cu, preferiblemente no más de 0,5 ppm, en comparación con el contenido de aluminio.
En otro aspecto, la descripción proporciona un método para analizar la estabilidad al almacenamiento de una composición que comprende aluminio y un compuesto terapéutico o profiláctico, comprendiendo dicho método combinar en una composición una cantidad predeterminada de producto terapéutico o vacuna y una cantidad predeterminada de un compuesto de una sal de aluminio, comprendiendo dicho método además almacenar dicha composición durante al menos 2 semanas, preferiblemente al menos 4 semanas y preferiblemente al menos un mes, a una temperatura de más de 20 °C, preferiblemente a una temperatura de aproximadamente 22 °C y determinar la estabilidad y/o la cantidad de proteína, preferiblemente compuesto terapéutico o profiláctico, en dicha composición. Como se demuestra en los Ejemplos 1 y 2, una temperatura de 22 °C, que es una temperatura más alta en comparación con las condiciones normales de almacenamiento o aproximadamente 2-8 °C, da como resultado una degradación acelerada de la proteína en una composición acuosa que comprende proteína, una sal de aluminio y más de 350 ppb de dicho metal pesado basándose en el peso con respecto a dicha composición. Por lo tanto, una temperatura de aproximadamente 22 °C y una duración de almacenamiento de al menos 2 semanas, preferiblemente al menos 4 semanas, son adecuadas para determinar la estabilidad al almacenamiento de las composiciones acuosas que comprenden una proteína según la descripción. La estabilidad al almacenamiento se puede determinar mediante cualquier método conocido en la técnica. La estabilidad al almacenamiento de una composición acuosa que comprende una proteína según la descripción se analiza preferiblemente determinando la estabilidad al almacenamiento de dicha proteína, preferiblemente determinando un epítopo sensible al almacenamiento en dicha proteína. Por ejemplo, como se describe en los Ejemplos 1 y 2, la estabilidad de una proteína, preferiblemente un antígeno, se determina determinando la relación de epítopo intacto sensible al almacenamiento, tal como el contenido de epítopo antigénico intacto (por ejemplo, epítopo de un epítopo neutralizante) y el contenido de proteína total. “ Epítopo sensible al almacenamiento intacto” o “ epítopo antigénico intacto” , como se utilizan en el presente documento, significa que no se ha producido degradación dentro de dicho epítopo. El contenido de epítopo antigénico intacto se mide, por ejemplo, determinando la proteína unida a un anticuerpo monoclonal dirigido específicamente contra dicho epítopo en, por ejemplo, un ELISA. El contenido de
proteína total se mide, por ejemplo, determinando la proteína unida al anticuerpo policlonal que se dirige contra diversos epítopos dentro de la proteína, por ejemplo, mediante ELISA. El contenido de epítopo específico relativo puede expresarse entonces como la relación del contenido de antígeno total determinado mediante la unión a anticuerpo monoclonal dividido por el contenido de antígeno total determinado por la unión al anticuerpo policlonal. Una relación alta indica un contenido alto de epítopos antigénicos y una relación baja indica un contenido bajo de epítopos antigénicos. Una relación baja medida para una composición acuosa después de un almacenamiento de al menos 20 °C, preferiblemente 22 °C, durante al menos 2 semanas, preferiblemente 4 semanas, en comparación con la relación medida para dicha composición acuosa antes del almacenamiento, indica que han tenido lugar cambios estructurales dentro del epítopo antigénico. Los cambios estructurales dentro de dicho epítopo antigénico indican una estabilidad al almacenamiento reducida de la composición acuosa.
El contenido de metales pesados de una composición acuosa preparada según la descripción es inferior a 350 ppb basándose en el peso de la composición acuosa. Generalmente, la composición de composición acuosa con menos metales pesados, más preferiblemente menos de 300 ppb, más preferiblemente menos de 275, más preferiblemente menos de 250 ppb y más preferiblemente menos de 235 ppb basándose en el peso de la composición acuosa.
Como se utiliza en el presente documento, la expresión “ metal pesado” se refiere a la cantidad total de elementos que presentan propiedades metálicas e incluye los metales de transición, metaloides, lantánidos y actínidos. Los metales de transición son elementos cuyo átomo tiene una subcapa d incompleta, o que pueden dar lugar a cationes con una subcapa d incompleta, e incluyen cinc, molibdeno, cadmio, escandio, titanio, tecnecio, paladio, vanadio, cromo, manganeso, hierro, cobalto, rodio, hafnio, cobre, níquel, itrio, niobio, circonio, rugenio, plata, tantalio, renio, wolframio, osmio, meitnerio, platino, iridio, mercurio, bohrio, seaborgio, hassio. Los metaloides son boro (B), silicio (Si), germanio (Ge), arsénico (As), antimonio (Sb), telurio (Te), polonio (Po). Los lantánidos son los quince elementos químicos metálicos con números atómicos del 57 al 71, es decir, lantano, cerio, praseodimio, neodimio, prometio, samario, europio, gadolinio, terbio, disprosio, holmio, erbio, tulio, iterbio y lutecio. Los actínidos son los quince elementos químicos metálicos con números atómicos del 89 al 103, actinio, torio, protactinio, uranio, neptunio, plutonio, americio, curio, berkelio, californio, einstenio, fermio, mendelevio, nobelio y laurencio. El metal pesado es preferiblemente un metal del bloque D de la tabla periódica. El metal pesado es preferiblemente un metal del grupo 3-12 de la tabla periódica que incluye los lantánidos y los actínidos. En una realización particularmente preferida, el metal pesado es un metal del bloque D, o del grupo 3-12 de la tabla periódica y sin incluir los lantánidos y los actínidos. Preferiblemente, dicho metal pesado es un elemento seleccionado entre los metales de transición. En otra realización preferida el metal pesado se selecciona de Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, Cr, Pb, Rb y Mo. En otra realización preferida, dicho metal pesado es un metal que tiene una masa molar de entre 21 y 83, preferiblemente dicho metal pesado es un metal del bloque D de la tabla periódica con una masa molecular de entre 21 y 83, más masa de entre 21 y 83, preferiblemente dicho metal pesado es un metal del bloque D de la tabla periódica con una masa molecular de entre 21 y 83, más preferiblemente de Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, Cr y Mo. Preferiblemente, dicho metal pesado es un metal del bloque D de la tabla periódica con una masa molecular de entre 21-30 y 39-48, más preferiblemente de Cu, Ni, Co, Ru, Cd, Ag, Fe, V, Cr y Mo. En un aspecto aún más preferido, el metal pesado se selecciona de los metales pesados Cu, Ni y Fe.
Como se demuestra en los Ejemplos, se identificó que el Lote 4230 de Hidróxido de aluminio (Alumbre) contribuye significativamente a la degradación del antígeno en la vacuna contra el JEV FVL09L37. En el Ejemplo 3 se muestra que este lote de Alumbre comprende al menos los siguientes metales: Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V. Se observaron niveles mayores de iones de Fe, Ni y Cu en el lote de Alumbre 4230 en comparación con otros lotes investigados. El lote 4230 fue el único en el que se detectaron iones de Cu residuales. Por lo tanto, preferiblemente dicho metal pesado se selecciona del grupo que consiste en Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, más preferiblemente en Fe, Ni y Cu.
La cantidad de metal pesado en la composición acuosa es preferiblemente inferior a 350 ppb basándose en el peso de la composición acuosa. Preferiblemente, la cantidad de metal pesado en la composición acuosa es inferior a 250 ppb, preferiblemente inferior a 225, más preferiblemente inferior a 200, más preferiblemente inferior a 150, más preferiblemente inferior a 100, más preferiblemente inferior a 50, más preferiblemente inferior a 25 ppb basándose en el peso de la composición acuosa. La cantidad de metal pesado en una composición acuosa de la descripción se ha definido anteriormente en el presente documento. Esta cantidad es normalmente para el total de metales pesados determinados, o para los metales pesados Fe, Cr y Ni, o una combinación de los mismos que constituyen los principales metales pesados en peso en la composición acuosa de la descripción. Para metales pesados específicos pueden preferirse diferentes cantidades máximas. Por ejemplo, se prefiere que la cantidad de Fe en la composición acuosa de la descripción sea inferior a 350 ppb basándose en el peso de la composición acuosa. En una realización preferida, la cantidad de Fe es inferior a 250 ppb, preferiblemente inferior a 210 ppb de Fe basándose en el peso de la composición acuosa.
Existe una fuerte evidencia de que muchos de los efectos proinflamatorios de los adyuvantes de aluminio están mediados a través de la formación de especies reactivas de oxígeno (ROS). El aluminio puede, en condiciones fisiológicas, promover la reducción de Fe(III) a Fe(II) y la oxidación de este último. Por lo tanto, la combinación de Fe y Al en el adyuvante potenciará la formación y actividades de ROS (Exley, C (2010). Trends in Immunol. Vol. 31: págs. 103-109). En la presente invención se ha encontrado que puede haber presente Fe en una composición acuosa de la descripción sin afectar significativamente a la estabilidad al almacenamiento de la composición. En esta realización de la descripción, se prefiere que la composición acuosa de la descripción comprenda entre 5 ppb y 250 ppb de Fe basándose en el peso
de la composición acuosa. En estas cantidades, la formación de ROS durante el almacenamiento debido a la presencia de dicha cantidad de Fe (si hay ROS) no afecta significativamente a la estabilidad al almacenamiento de la composición acuosa como se define en otra parte del presente documento. Sin embargo, las cantidades son suficientes para permitir efectos proinflamatorios después de la administración de la vacuna in vivo.
En la presente invención se ha encontrado que particularmente la presencia del metal pesado Cu afecta seriamente a la estabilidad al almacenamiento de la composición de la descripción. La cantidad de Cu en una composición que contiene proteína o método para producir la composición de proteína de la descripción es preferiblemente inferior a 25 ppb. Preferiblemente, menos de 5 ppb, más preferiblemente menos de 1 ppb, más preferiblemente menos de 0,2 ppb basándose en el peso de la composición acuosa. En una realización particularmente preferida, una composición acuosa de la descripción comprende, por lo tanto, menos de 3 ppb de Cu basándose en el peso de la composición acuosa. Preferiblemente, menos de 2,5 ppb. La cantidad de Cu en una composición o método de la descripción es, en una realización particularmente preferida, inferior a 1,25 ppb basándose en el peso de la composición acuosa. En una realización particularmente preferida, dicha composición acuosa comprende Cu a un nivel que está por debajo del límite de detección del método para la detección de cobre como se describe en los Ejemplos.
En la presente invención se ha encontrado que particularmente el metal pesado Ni afecta a la estabilidad al almacenamiento de la composición de la descripción. La cantidad de Ni en una composición o método de la descripción es preferiblemente inferior a 200 ppb. Preferiblemente menos de 40 ppb, preferiblemente menos de 9 ppb y preferiblemente menos de 2 ppb basándose en el peso de la composición acuosa. En una realización particularmente preferida, una composición acuosa de la descripción comprende, por lo tanto, menos de 40 ppb de Ni basándose en el peso de la composición acuosa. Preferiblemente, menos de 30 ppb, más preferiblemente menos de 20 ppb y más preferiblemente menos de 15 ppb de Ni basándose en el peso de la composición acuosa. En una realización particularmente preferida, dicha composición acuosa comprende Ni a un nivel que está por debajo del límite de detección del método para la detección de níquel como se describe en los Ejemplos.
El metal pesado puede estar presente en forma electrónica neutra o puede estar ionizado. Normalmente y preferiblemente, el metal pesado está presente en forma iónica en una composición acuosa de la descripción.
El contenido de metal de una composición se puede determinar de diversas maneras. En un aspecto, un método según la descripción comprende determinar el nivel de un metal pesado en una composición acuosa y/o la sal de aluminio presente en dicha composición acuosa. Se conocen en la técnica métodos para medir el nivel de uno o más metales pesados en una solución acuosa. Los ejemplos de dichos métodos incluyen la espectrometría de masas, tal como la plasma acoplado inductivamente-espectrometría de masas (ICP-MS), la espectrometría de absorción atómica de llama (F-AAs ) y/o la espectrometría de absorción atómica en horno de grafito (GF-AAS).
En el Ejemplo 3 se describe un ejemplo de un ensayo que puede utilizarse para determinar el contenido de metales pesados. El ensayo implica tratar una muestra de una solución acuosa que contiene una sal de aluminio con HNO3 concentrado en calor hasta obtener una solución transparente. Después, la solución transparente se puede diluir y analizar adicionalmente, por ejemplo, mediante ICP-Ms , F-AAS y/o GF-AAS, para determinar la presencia y el contenido de iones metálicos, incluyendo Pb, Cd, Cr, Co, Fe, Cu, Ni, Ag, W y Al.
Los Ejemplos 1 y 2 demuestran que el antígeno de JEV muestra una mayor estabilidad a pH 7,5-8 en comparación con el pH 7. En los Ejemplos, la estabilidad del antígeno se expresa como la relación de ELISA monoclonal/policlonal. Se demostró que el anticuerpo monoclonal utilizado (clon 52-2-5) reconoce un epítopo neutralizante en la vacuna contra la encefalitis japonesa (JEV). El contenido de epítopo específico relativo puede expresarse como la relación del contenido de antígeno total determinado mediante “ ELISA monoclonal” dividido por el contenido de antígeno total determinado mdiante “ ELISA policlonal” . Sin quedar ligados a la teoría, el efecto de una mayor estabilidad del antígeno a pH 7,5-8 puede explicarse basándose en la química de reacción compleja asumida subyacente de los sulfitos. El pH podría influir en las condiciones de reacción relacionadas con el equilibrio de la reacción de sulfito/formaldehído y la carga superficial de determinadas cadenas laterales de proteínas/aminoácidos accesibles a la modificación. El pH puede afectar la oxidación por influencia directa sobre los potenciales redox de los restos de aminoácidos y los agentes oxidantes, por ejemplo, los radicales libres. Por lo tanto, en una realización, un método según la descripción comprende tamponar dicha composición acuosa a un pH de entre 7,5 y 8,5.
Se están utilizando diversas sales de aluminio en composiciones para la administración de un individuo. El adyuvante de aluminio normalmente contiene un óxido o sulfato de aluminio o una combinación de los mismos. En una realización preferida, la sal de aluminio comprende óxido de aluminio (AhO3), hidróxido de aluminio (Al(OH)3) o fosfato de aluminio (AlPO4).
En una realización preferida, la composición acuosa de la descripción comprende además un compuesto reactivo. Normalmente, aunque no necesariamente, el compuesto reactivo está presente como resultado de una manipulación de la composición acuosa, por ejemplo, para tratar o inactivar un agente infeccioso, si lo hay en la composición. El compuesto reactivo también puede estar presente por otra razón. El sulfito, por ejemplo, en ocasiones está presente para inactivar cualquier formaldehído residual en la solución acuosa. El formaldehído es normalmente un producto químico que se utiliza con frecuencia para inactivar cualquier agente infeccioso.
El compuesto reactivo es preferiblemente un compuesto activo redox, un compuesto formador de radicales y/o un compuesto estabilizante. En una realización preferida, dicha composición acuosa de la descripción comprende formaldehído, etanol, cloroformo, tricloroetileno, acetona, triton-X-100, desoxicolato, dietilpirocarbonato, sulfito, Na2S2O5, beta-propriolactona, polisorbato tal como Tween 20®, Tween 80®, O2 , fenol, copolímeros de tipo pluronic o una combinación de los mismos.
El sulfito está preferiblemente presente en una cantidad de entre 0,1 mM y 5 mM, o preferiblemente entre 0,5-2 mM. El formol está preferiblemente presente en una cantidad de entre 0,1 mM y 5 mM, más preferiblemente entre 0,5 mM y 2 mM. El oxígeno está preferiblemente presente en una cantidad equivalente a la solubilidad del O2 a la temperatura medida, el O2 está preferiblemente presente en una cantidad de entre 10 y 250 uM cuando se mide a 20 grados centígrados. Cuando se mide a una temperatura de 0 grados centígrados, el O2 está preferiblemente presente en una cantidad de entre 10 y 400 uM. Un compuesto estabilizante está preferiblemente presente en una cantidad de entre 10 y 400 uM. De manera similar, un compuesto activo redox está presente en una cantidad de entre 0,1 mM y 5 mM, o preferiblemente entre 0,5-2 mM. Un compuesto formador de radicales está preferiblemente presente en una cantidad de entre 0,1 mM y 5 mM, o preferiblemente entre 0,5-2 mM. En este contexto y en aras de la claridad, es importante señalar que el compuesto activo redox, el compuesto formador de radicales y/o el compuesto estabilizante se consumen en la producción de un radical, mientras que el metal pesado indicado anteriormente en el presente documento, es un catalizador en la producción de un radical y no se consume, como tal. Por lo tanto, el compuesto activo redox, el compuesto formador de radicales y/o el compuesto estabilizante no es un metal pesado.
La cantidad total de compuesto activo redox, el compuesto formador de radicales y/o el compuesto estabilizante, aunque pequeña en cantidades absolutas, aún puede ser significativa con respecto al antígeno o la proteína en la composición acuosa de la descripción. El antígeno/proteína está preferiblemente presente en una cantidad de entre 0,1 nmol y 1 umol, más preferiblemente entre 1 nmol y 100 nmol.
La concentración de proteína, preferiblemente una proteína terapéutica o de vacuna, en una composición acuosa que comprende una proteína según la descripción es preferiblemente de entre 1 ng/ml y 10 mg/ml, preferiblemente entre 10 ng/ml y 1 mg/ml, más preferiblemente entre 100 ng/ml y 100 mcg/ml, tal como entre 1 mcg/ml y 100 mcg/ml. La concentración es preferiblemente de al menos 1 ng/ml para garantizar que la proteína terapéutica o vacuna esté en una concentración suficiente para ejercer su efecto terapéutico cuando se administre a un individuo. Sin embargo, la concentración no debería superar preferiblemente 10 mg/ml para prevenir o reducir la aparición de posibles efectos secundarios asociados a la administración de dicha proteína a un individuo. En particular, la concentración de proteína vírica en una composición acuosa según la descripción que comprende JEV está preferiblemente entre 0,01 pg/ml y 1 mg/ml, más preferiblemente entre 0,1 mcg/ml y 100 mcg/ml. En una realización de ejemplo de la descripción, una composición acuosa según la descripción comprende aproximadamente 10 mcg/ml de JEV. La dosis de una única administración de una composición acuosa que comprende una proteína, preferiblemente una proteína terapéutica o de vacuna, según la descripción, es preferiblemente de entre 0,1 ml y 10 ml, preferiblemente entre 0,5 ml y 5 ml, tal como 0,5 ml, 1 ml, 1,5 ml, 2 ml, 2,5 ml, porque dicha dosis permite una administración conveniente a un individuo, tal como un ser humano.
La composición acuosa según la descripción puede comprender además una molécula de ácido nucleico. El ácido nucleico puede administrarse con fines terapéuticos. En ese caso, la composición acuosa según la descripción comprende preferiblemente una molécula de ácido nucleico, tal como un plásmido que comprende la secuencia de ácido nucleico que codifica un antígeno o antígenos o virus o bacteria o parte inmunogénica de los mismos contra los que se busca una respuesta inmunitaria. La incorporación de dicha molécula de ácido nucleico se basa en la producción in situ del antígeno, virus, bacteria o parte inmunogénica diana. En otra realización preferida, la composición acuosa según la descripción comprende un ácido nucleico en forma de un ARN antisentido, ARNi o mimético del mismo. En otra realización más, la composición acuosa según la descripción comprende el ácido nucleico en forma de un agente infeccioso como tal, un virus o un virus modificado, como es el caso en muchos enfoques de terapia génica. La concentración de ácido nucleico en la composición acuosa, composición inmunogénica o vacuna según la descripción está preferiblemente en el intervalo de aproximadamente 1 ng/ml a aproximadamente 10 mg/ml, preferiblemente de aproximadamente 0,1 mcg/ml a aproximadamente 1 mg/ml, más preferiblemente aproximadamente 1 mcg/ml a aproximadamente 100 mcg/ml. La dosificación adecuada dependerá del sujeto al que se le administre la composición y del tamaño de las secuencias de ácido nucleico presentes en la composición.
Una composición acuosa según la descripción puede comprender además un polisacárido y/o un oligosacárido, preferiblemente la cápsula de polisacárido y/u oligosacárido de bacterias encapsuladas contra las que se busca una respuesta inmunitaria. Los ejemplos de polisacáridos y oligosacáridos que pueden estar presentes en una composición acuosa según la descripción incluyen polisacáridos neumocócicos, polisacáridos meningocócicos, polisacárido de Haemophilus influenzae de tipo b, polisacáridos estreptocócicos del grupo B, polisacárido de Salmonella typhi Vi, polisacáridos u oligosacáridos derivados del estreptococo del grupo A, estafilococos, Neisseria meningitidis, Klebsiella pneumoniae, enterococos, E. coli, Pseudomonas aeruginosa y Bacillus Anthracis. Dicho polisacárido y/u oligosacárido pueden estar presentes en la composición acuosa según la descripción como tales o, como alternativa, el polisacárido y/u oligosacárido pueden conjugarse con una proteína. La concentración de polisacárido y/u oligosacárido en la composición acuosa, composición inmunogénica o vacuna según la descripción está preferiblemente en el intervalo de aproximadamente 10 ng/ml a aproximadamente 500 mcg/ml, más preferiblemente de aproximadamente 0,1 mcg/ml
a aproximadamente 500 mcg/ ml, más preferiblemente de aproximadamente 1 mcg/ml a aproximadamente 50 mcg/ml. La dosificación adecuada dependerá del sujeto al que se le administre la composición.
Se utiliza preferiblemente un método según la invención para aumentar la estabilidad de una composición inmunogénica, preferiblemente una vacuna, que comprende un adyuvante a base de sal de aluminio. Son ejemplos de dichas vacunas las dirigidas contra la infección por Bacillus Anthracis (que provoca ántrax), Corynebacterium diphtheriae (que provoca difteria), Clostridium tetani (que provoca tétanos), pseudomonas tales como Pseudomonas aeruginosa, estafilococos tales como estafilococo aureus o Staphylococcus epidermidis, bacterias Haemophilus influenzae de tipo B (Hib), virus de la poliomielitis, virus de la hepatitis A, virus de la hepatitis B, virus del papiloma humano, virus de la gripe, virus de la encefalitis japonesa, rotavirus, bacterias Rickettsia (causantes del tifus), virus de la fiebre amarilla, virus de la varicela zóster, meningococo o combinaciones de las mismas, tales como DTP (difteria, tétanos, poliomielitis). Por lo tanto, una composición acuosa que comprende una proteína según la descripción comprende preferiblemente una proteína que es una proteína vírica o bacteriana, preferiblemente una proteína de Bacillus Anthracis, Corynebacterium diphtheriae, Clostridium tetani, bacterias Haemophilus influenzae de tipo B (Hib), virus de la poliomielitis, virus de la hepatitis A, virus de la hepatitis B, virus del papiloma humano, virus de la gripe, virus de la encefalitis japonesa, rotavirus, bacterias Rickettsia, virus de la fiebre amarilla, virus de la varicela zóster y/o meningococo. En una realización preferida, dicha proteína contenida en una composición que comprende una proteína según la descripción es una proteína vírica de un virus de la familia Flaviviridae, preferiblemente de un virus de la encefalitis japonesa (JEV). Como se demuestra en los Ejemplos, la estabilidad de las composiciones acuosas que comprenden una proteína de JEV, una sal de aluminio y que comprenden menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa y, en particular, en donde la cantidad de Cu es inferior a 3 ppb basándose en el peso de la composición acuosa, aumenta en comparación con la composición acuosa que comprende más de 350 ppb de metal pesado y más de 3 ppb de Cu. Por lo tanto, un método según la invención es particularmente adecuado para aumentar la estabilidad de una composición acuosa que comprende una proteína de j Ev . En las tablas 25 y 26 se proporciona una lista de ejemplos de vacunas a base de aluminio, tanto para su uso humano como para su uso veterinario. También se describe un método particularmente adecuado para aumentar la estabilidad de una vacuna enumerada en la tabla 25 y/o la tabla 26. Por lo tanto, una vacuna enumerada en la tabla 25 y/o la tabla 26 se prepara preferiblemente utilizando un método de la invención.
En otra realización o aspecto preferido de la descripción, dicha proteína contenida en una composición que comprende una proteína según la descripción es una proteína bacteriana de una bacteria de la familia Pseudomonas, preferiblemente de Pseudomonas aeruginosa. Como se demuestra en los Ejemplos, la estabilidad de las composiciones acuosas que comprenden proteína de fusión de Pseudomonas aeruginosa (SEQ ID NO: 1) y una sal de aluminio se reduce cuando hay presente más de 350 ppb de metal pesado basándose en el peso de la composición acuosa.
Una composición acuosa que comprende una proteína según la descripción es particularmente adecuada para su uso como vacuna o composición inmunogénica. Por ejemplo, dichas composiciones son particularmente útiles para inmunizar a un individuo para tratar o prevenir una infección vírica o bacteriana. En una realización, la invención por lo tanto proporciona un método para el tratamiento de un individuo que comprende la obtención de una composición acuosa inmunogénica que comprende una proteína y una sal de aluminio, teniendo dicha sal de aluminio menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa, preferiblemente menos de 3 ppb de Cu, y administrar la composición acuosa inmunogénica a un individuo que lo necesite. También se proporciona un método para el tratamiento profiláctico de un individuo que comprende la obtención de una composición acuosa inmunogénica que comprende una proteína y una sal de aluminio, teniendo dicha sal de aluminio menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa, preferiblemente menos de 3 ppb de Cu, y administrar la composición acuosa inmunogénica a un individuo que lo necesite. Además se proporciona un método para inducir y/o potenciar una respuesta inmunitaria hacia un antígeno en un individuo, comprendiendo dicho método obtener una composición acuosa que comprende una proteína que comprende dicho antígeno y una sal de aluminio, teniendo dicha sal de aluminio inferior a 350 ppb de metal pesado basándose en el peso de la composición acuosa, preferiblemente inferior a 3 ppb de Cu, y administrar la composición acuosa a un individuo que lo necesite. En otro aspecto, la invención proporciona un método para inmunizar un individuo que comprende administrar a dicho individuo al menos dos composiciones inmunogénicas con un intervalo de al menos dos semanas entre cada administración, y en donde cada una de dichas al menos dos composiciones inmunogénicas comprende el mismo antígeno, y en donde al menos una de dichas composiciones inmunogénicas comprende además una sal de aluminio que tiene menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa, preferiblemente menos de 3 ppb de Cu, y administrar la composición acuosa inmunogénica a un individuo que lo necesite. Las composiciones de ácido nucleico en ocasiones también se coadministran con aluminio. Por lo tanto, para la presente descripción es posible reemplazar “ proteína” en una composición acuosa de la descripción con ácido nucleico. Por lo tanto, en una realización, la descripción proporciona un método para preparar una composición acuosa que comprende aluminio y un ácido nucleico, comprendiendo dicho método
- combinar una sal de aluminio, dicho ácido nucleico y agua para producir dicha composición acuosa y
- determinar el nivel de un metal pesado en la composición acuosa y/o la sal de aluminio.
La descripción también proporciona un método para preparar una composición acuosa que comprende aluminio y un ácido nucleico, comprendiendo dicho método
- preparar o seleccionar una sal de aluminio que tenga menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa final, preferiblemente menos de 3 ppb de Cu, y administrar la composición acuosa inmunogénica a un individuo que lo necesite y
- combinar dicha sal de aluminio, dicho ácido nucleico y agua para producir dicha composición acuosa.
En una realización preferida, dichos métodos comprenden además tamponar dicha composición acuosa a un pH de entre 7,5 y 8,5. En una realización particularmente preferida, dichos métodos comprenden además envasar alícuotas de dicha composición acuosa que tiene menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa en recipientes de almacenamiento herméticos separados. El ácido nucleico puede administrarse con fines terapéuticos. Por ejemplo, en la forma de un ARN antisentido, ARNi o mimético del mismo. El ácido nucleico también se puede administrar en forma de un agente infeccioso, normalmente un virus o un virus modificado, como es el caso en muchos enfoques de terapia génica. En ese caso, el ácido nucleico está encerrado en una partícula que comprende proteína. Por lo tanto, la descripción proporciona además una composición acuosa que comprende un ácido nucleico y una sal de aluminio, comprendiendo dicha composición menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa. La concentración de ácido nucleico en la composición acuosa, composición inmunogénica o vacuna según la descripción está preferiblemente en el intervalo de aproximadamente 1 ng/ml a aproximadamente 10 mg/ml, preferiblemente de aproximadamente 0,1 mcg/ml a aproximadamente 1 mg/ml, más preferiblemente aproximadamente 1 mcg/ml a aproximadamente 100 mcg/ml. La dosificación apropiada dependerá del sujeto al que se le administre la composición y del tamaño de las secuencias de ácido nucleico presentes en la composición.
Las composiciones que comprenden conjugados de polisacárido, oligosacárido o polisacárido-polipéptido en ocasiones también se administran junto con aluminio. Por lo tanto, para la presente descripción es posible reemplazar “ proteína” en una composición acuosa de la descripción con polisacárido, oligosacárido o conjugado polisacárido-polipéptido o una combinación de los mismos. Dicho polisacárido u oligosacárido es preferiblemente el polisacárido u oligosacárido de la cápsula de bacterias encapsuladas contra las que se busca una respuesta inmunitaria. Los ejemplos de polisacáridos y oligosacáridos que pueden estar presentes en una composición acuosa según la descripción incluyen polisacáridos neumocócicos, polisacáridos meningocócicos, polisacárido de Haemophilus influenzae de tipo b, polisacáridos estreptocócicos del grupo B, polisacárido de Salmonella typhi Vi, polisacáridos u oligosacáridos derivados del estreptococo del grupo A, estafilococos, Neisseria meningitidis, Klebsiella pneumoniae, enterococos, E. coli, Pseudomonas aeruginosa y Bacillus Anthracis. Dicho polisacárido y/u oligosacárido pueden estar presentes en la composición acuosa según la descripción como tales o, como alternativa, el polisacárido y/u oligosacárido pueden conjugarse con una proteína. Por lo tanto, en una realización, la descripción proporciona un método para preparar una composición acuosa que comprende aluminio y un polisacárido u oligosacárido, comprendiendo dicho método
- combinar una sal de aluminio, dicho polisacárido u oligosacárido y agua para producir dicha composición acuosa y
- determinar el nivel de un metal pesado en la composición acuosa y/o la sal de aluminio.
La descripción también proporciona un método para preparar una composición acuosa que comprende aluminio y un polisacárido u oligosacárido, comprendiendo dicho método
- preparar o seleccionar una sal de aluminio que tenga menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa final, preferiblemente menos de 3 ppb de Cu, y administrar la composición acuosa inmunogénica a un individuo que lo necesite y
- combinar dicha sal de aluminio, dicho polisacárido u oligosacárido y agua para producir dicha composición acuosa.
La concentración de polisacárido y/u oligosacárido en la composición acuosa, composición inmunogénica o vacuna según la descripción está preferiblemente en el intervalo de aproximadamente 10 ng/ml a aproximadamente 500 pg/ml, más preferiblemente de aproximadamente 0,1 mcg/ml a aproximadamente 500 mcg/ ml, más preferiblemente de aproximadamente 1 mcg/ml a aproximadamente 50 mcg/ml. La dosificación apropiada dependerá del sujeto al que se le administre la composición.
El conjugado polisacárido-polipéptido según la presente descripción comprende al menos un polisacárido y al menos un polipéptido. El polisacárido según la descripción es preferiblemente un polisacárido capsular bacteriano. Los polisacáridos capsulares se pueden preparar mediante técnicas convencionales conocidas por los expertos en la materia.
En una realización, el polisacárido es un polisacárido capsular de S. pneumoniae. En otra realización, el polisacárido capsular de S. pneumoniae se selecciona del grupo que consiste en los serotipos 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 1OA, HA, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F y 33F, que representan los 23 serotipos neumocócicos que causan la gran mayoría de las enfermedades neumocócicas en todos los grupos de edad, de los más de 90 serotipos conocidos hasta el momento.
El término “ polisacárido” , como se utiliza en el presente documento, se refiere a polisacáridos y/u oligosacáridos. Los polisacáridos se aíslan de bacterias y se pueden despolimerizar a un intervalo de tamaño preferido mediante métodos conocidos (véanse, por ejemplo, los documentos EP 497524 y EP 497525). Los oligosacáridos tienen un bajo número de unidades repetidas (normalmente de 5 a 30 unidades repetidas) y normalmente son polisacáridos hidrolizados.
Los polisacáridos capsulares de Streptococcus pneumoniae comprenden unidades repetidas de oligosacáridos que pueden contener hasta 8 restos de azúcar. Para consultar una revisión de las unidades de oligosacáridos para los serotipos clave de Streptococcus pneumoniae, véase Jones et al., An. Acad. Bras. Cienc, 2005, 77(2):293-324. En una realización, un antígeno de sacárido capsular puede ser un polisacárido de longitud completa, sin embargo, en otras puede ser una unidad de oligosacárido, o una cadena de sacárido de longitud más corta que la nativa de unidades de oligosacárido repetidas. Los polisacáridos de longitud completa se pueden “ dimensionar” o “ despolimerizar” , es decir, su tamaño puede reducirse mediante diversos métodos conocidos en la técnica (como se ha descrito anteriormente). El término “ despolimerización” incluye la despolimerización parcial.
Después, la despolimerización de los polisacáridos puede ir seguida de una etapa de activación antes de la conjugación con un polipéptido portador. Por “ activación” se entiende el tratamiento químico del polisacárido para proporcionar grupos químicos capaces de reaccionar con el polipéptido portador. En la técnica se conocen métodos apropiados.
Por “ polipéptido” o “ proteína” se entiende cualquier cadena de aminoácidos que tenga al menos 10 y preferiblemente al menos 100 aminoácidos en uniones peptídicas entre sí, independientemente de la modificación postraduccional. Los portadores polipeptídicos adecuados incluyen toxina diftérica, toxoide diftérico, CRM 197, toxoide tetánico, toxoide pertussis, lT de E. coli, ST de E. coli, exotoxina A, complejo de membrana externa c (OMPC), porina, proteína de unión a transferrina, neumólisis, proteína de superficie neumocócica A (PspA), proteína adhesina neumocócica (PsaA), ovoalbúmina, hemocianina de lapa californiana (KLH), albúmina sérica bovina (BSA) o derivado proteínico purificado de tuberculina (PPD). Los polipéptidos portadores son preferiblemente polipéptidos que no son tóxicos ni reactogénicos y que pueden obtenerse en cantidad y pureza suficientes. En una realización particularmente preferida, el polipéptido portador comprende un toxoide tetánico. En otra realización preferida, el polipéptido transportador comprende un derivado de cualquiera de los polipéptidos transportadores mencionados anteriormente, por ejemplo, una subunidad o una versión mutada de LT de E. coli, tal como LT o la subunidad A de LT (LTA) que tiene una sustitución de aminoácido en la posición del aa 192 (por ejemplo, LTG 192, LTT 192, LTS 192, LTA 192), Lt K 63 LTR 72 u otros mutantes como se describe, por ejemplo, en los documentos WO 98/42375, WO 02/64162, US 4.761.372, US 5.308.835. Los polipéptidos también pueden contener alargamientos en el extremo carboxi o amino del polipéptido que facilitan la interacción con el compuesto o compuestos policatiónicos o el compuesto o compuestos inmunoestimuladores.
Además, los polipéptidos también se pueden derivatizar para incluir moléculas que potencien la presentación de antígenos y el direccionamiento de antígenos a células presentadoras de antígenos.
El polipéptido puede activarse antes de la conjugación.
La naturaleza y el tamaño del sacárido, la naturaleza y el tamaño de la proteína o el polipéptido, la relación del sacárido y la proteína/polipéptido, así como otros factores y condiciones para la preparación de un conjugado según la presente descripción pueden ser determinados por un experto, como se describe, por ejemplo, en Robbins et al., JAMA, 1996, 276(14): 1181-5. Por ejemplo, para un polisacárido de neumococo y un toxoide tetánico, una relación preferida es de aproximadamente 2:1.
Por “ conjugado” se entiende un compuesto en el que el polisacárido está unido covalentemente a un polipéptido portador. Existen muchas reacciones de conjugación conocidas en la técnica anterior que se han empleado para unir covalentemente polisacáridos a polipéptidos con el fin de producir un conjugado polisacárido-polipéptido. Tres de los métodos más comúnmente empleados incluyen: 1) aminación reductora, en donde el grupo aldehído o cetona en un componente de la reacción reacciona con el grupo amino o hidrazida en el otro componente, y el doble enlace C-N formado se reduce posteriormente a un enlace sencillo C-N mediante un agente reductor; 2) conjugación de cianilación, en donde el polisacárido se activa por bromuro de cianógeno (CNBr) o por tetrafluoroborato de l-ciano-4-dimetilamoniopiridinio (CDAP) para introducir un grupo cianato en el grupo hidroxilo, que forma un enlace covalente con el grupo amino o hidrazida tras la adición del componente proteínico; y 3) una reacción de carbodiimida, en donde la carbodiimida activa el grupo carboxilo en un componente de la reacción de conjugación, y el grupo carbonilo activado reacciona con el grupo amino o hidrazida en el otro componente. Estas reacciones también se emplean frecuentemente para activar los componentes del conjugado antes de la reacción de conjugación. El polisacárido se puede conjugar con el polipéptido directamente o a través de un enlazador. La unión a través de un grupo enlazador se puede realizar utilizando cualquier procedimiento conocido, por ejemplo, los procedimientos descritos en los documentos US 4.882.317 y US 4.695.624. Los enlazadores adecuados incluyen carbonilo, ácido adípico, B-propionamido (documento WO 00/10599), nitrofenil-etilamina (Gever et al., Med. Microbiol. Immunol, 1979, 165:171-288), haluros de haloacilo (documento US 4.057.685), uniones glucosídicas (documentos US 4.673.574; US 4.761.283; US 4.808.700), ácido 6-aminocaproico (documento US 4.459.286), ADH (documento US 4.965.338) y restos C4 a Ci2 (documento US 4.663.160).
Después de la conjugación del polisacárido con el polipéptido portador, el conjugado polisacárido-polipéptido puede purificarse (enriquecerse con respecto a la cantidad de conjugado polisacárido-polipéptido) mediante una diversidad de técnicas conocidas en la técnica. Un objetivo de la etapa de purificación es retirar el polisacárido y/o polipéptido no unido del conjugado polisacárido-polipéptido. Los métodos de purificación incluyen, por ejemplo, ultrafi ltración en presencia de sulfato de amonio, cromatografía de exclusión por tamaño, centrifugación en gradiente de densidad y cromatografía de interacción hidrófoba.
Los adyuvantes de aluminio, tales como los adyuvantes de hidróxido de aluminio y fosfato de aluminio, se preparan generalmente exponiendo soluciones acuosas de iones de aluminio a condiciones por lo general ligeramente alcalinas en un entorno químico bien definido y controlado. Se pueden usar diversas sales de aluminio solubles para la producción de hidróxido de aluminio. Los aniones presentes en el momento de la precipitación pueden coprecipitar con el hidróxido o sulfato de aluminio. Después de la precipitación de la sal de amonio por desplazamiento de pH utilizando, por ejemplo, NaOH, no es posible retirar ningún metal pesado presente en el adyuvante de aluminio. Incluso un lavado exhaustivo no da como resultado una reducción suficiente del contenido de metales pesados. Por lo tanto, es importante controlar el metal pesado antes de la precipitación de la sal de aluminio, es decir, mediante la selección de materias primas apropiadas, el control de las condiciones del proceso, por ejemplo, como el alumbre de amoníaco y el sulfato de aluminio u otras fuentes de aluminio están en solución, no debe haber metal añadido, por ejemplo, otras sales tales como CuSO3. Como alternativa o adicionalmente, cualquier metal pesado presente en la solución se retira antes de la precipitación del adyuvante de aluminio. Esto se puede conseguir, por ejemplo, a través de cristalización o intercambio iónico (cationes), que son métodos bien conocidos en la técnica. El intercambio iónico se refiere a un proceso a través del cual los iones (metálicos) en solución se transfieren a una matriz sólida que, a su vez, libera iones de un tipo diferente pero de la misma polaridad en la solución. Por lo tanto, los iones en solución se reemplazan por diferentes iones presentes originariamente en la matriz sólida. La cristalización de metales se refiere a la precipitación de cristales metálicos insolubles de iones metálicos en solución. Por lo tanto, en una realización preferida, la descripción proporciona un método para preparar una composición acuosa que comprende aluminio, un compuesto reactivo y una proteína, comprendiendo dicho método
- preparar una sal de aluminio que sea capaz de proporcionar una composición acuosa que tenga menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa y
- combinar dicha sal de aluminio, dicho compuesto reactivo, dicha proteína y agua para producir dicha composición acuosa,
por lo que dicha sal de aluminio se prepara preparando una solución acuosa de iones de aluminio, retirando los metales pesados de dicha solución acuosa tal como mediante cristalización o intercambio iónico, preferiblemente intercambio catiónico, y precipitando dichos iones de aluminio de dicha solución, preferiblemente utilizando una base.
Además se proporciona un método para preparar un precipitado de sal de aluminio de grado clínico para su incorporación en un medicamento y/o vacuna, comprendiendo dicho método preparar una solución acuosa de iones de aluminio, retirar los metales pesados de dicha solución acuosa tal como mediante cristalización o intercambio iónico, preferiblemente intercambio catiónico, y precipitar dichos iones de aluminio de dicha solución, y determinar el nivel de un metal pesado en la solución y/o el precipitado de sal de aluminio, en donde se selecciona el precipitado que es capaz de proporcionar una composición acuosa que comprende menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa.
La preparación de hidróxido de aluminio a partir de materias primas se describe, entre otros, en los documentos CN101734698 y WO98/14401.
La descripción proporciona además un método para preparar una composición farmacéutica acuosa o de vacuna que comprende aluminio, un compuesto reactivo y una proteína, comprendiendo dicho método
- seleccionar una sal de aluminio que sea capaz de proporcionar una composición acuosa que tenga menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa y
- combinar dicha sal de aluminio, dicho compuesto reactivo, dicha proteína y agua para producir dicha composición acuosa (i) que tiene menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa y (ii) que comprende entre 5 pg/ml y 50 mg/ml de aluminio;
en donde el compuesto reactivo se selecciona del grupo que consiste en un compuesto activo redox, un compuesto formador de radicales, un compuesto estabilizante y una combinación de cualquiera de los mismos.
Preferiblemente, el método comprende además tamponar dicha composición acuosa a un pH de entre 6,5 y 8,5.
Preferiblemente, el método comprende además envasar alícuotas de dicha composición acuosa que tiene menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa en recipientes de almacenamiento herméticos separados.
La descripción proporciona además un método para seleccionar un precipitado de sal de aluminio de grado clínico para su incorporación en un medicamento y/o vacuna, comprendiendo dicho método preparar una solución acuosa de iones de aluminio y precipitar dichos iones de aluminio de dicha solución, y determinar el nivel de un metal pesado en la solución y/o el precipitado de sal de aluminio, en donde se selecciona el precipitado que es capaz de proporcionar una composición acuosa que comprende (i) menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa y (ii) entre 5 pg/ml y 50 mg/ml de aluminio. También se proporciona una composición farmacéutica acuosa o de vacuna que comprende una proteína, un compuesto reactivo y una sal de aluminio, comprendiendo dicha composición (i) menos de 350 ppb de metal pesado basándose en el peso de la composición acuosa y (ii) entre 5 pg/ ml y 50 mg/ml de aluminio, en donde el compuesto reactivo se selecciona del grupo que consiste en un compuesto activo redox, un compuesto formador de radicales, un compuesto estabilizante y una combinación de cualquiera de los mismos. En una realización preferida, dicho metal pesado se selecciona de Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, Cr, Pb, Rb y Mo. El metal pesado se selecciona preferiblemente de Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V. En una realización particularmente preferida, el metal pesado se selecciona de Cu o Ni. El metal pesado está preferiblemente presente en forma iónica. La sal de aluminio es preferiblemente hidróxido de aluminio (Al(OH)3) o fosfato de aluminio (AlPO4). La sal de aluminio es preferiblemente hidróxido de aluminio (Al(OH)3). El compuesto reactivo se selecciona preferiblemente del grupo que consiste en formaldehído, etanol, cloroformo, tricloroetileno, acetona, 4-(1,1,3,3-tetrametilbutil)fenil-polietilenglicol, desoxicolato, dietilpirocarbonato, sulfito, Na2S2Os, beta-propriolactona, polisorbato tal como monolaurato de polietilenglicol sorbitano, monooleato de polietilenglicol sorbitano, O2 , fenol, copolímeros de tipo pluronic y una combinación de cualquiera de los mismos. La composición farmacéutica acuosa de vacuna comprende preferiblemente entre 50 pg/ml y 5 mg/ml de aluminio. La composición farmacéutica acuosa o de vacuna comprende preferiblemente entre 5 ppb y 250 ppb de Fe basándose en el peso de la composición acuosa. La composición farmacéutica acuosa o de vacuna comprende preferiblemente menos de 3 ppb de Cu basándose en el peso de la composición acuosa. La composición farmacéutica acuosa o de vacuna comprende preferiblemente menos de 40 ppb de Ni basándose en el peso de la composición acuosa. La proteína en dicha composición farmacéutica o de vacuna comprende preferiblemente menos de 40 ppb de Ni basándose en el peso de la composición acuosa. La proteína en dicha composición farmacéutica acuosa o de vacuna es preferiblemente un agente terapéutico y/o una vacuna. La proteína es preferiblemente una proteína vírica o bacteriana. La proteína vírica es preferiblemente una proteína del virus de la encefalitis japonesa o una proteína de la bacteria Pseudomonas aeruginosa. La proteína es preferiblemente una proteína dentro de una partícula de virus inactivada con formaldehído. La composición farmacéutica acuosa o de vacuna comprende preferiblemente además sulfito. La descripción proporciona además una vacuna que comprende una composición de vacuna acuosa según la descripción.
Donde en el presente documento se indica un intervalo entre los valores X e Y, el intervalo incluye los valores X e Y.
La descripción proporciona además un método para preparar una composición acuosa que comprende aluminio, un compuesto reactivo y una proteína, comprendiendo dicho método
- preparar o seleccionar una sal de aluminio que sea capaz de proporcionar una composición acuosa que tenga menos de 450 ppm de metal pesado basándose en el peso del aluminio (gramos/gramos) y
- combinar dicha sal de aluminio, dicho compuesto reactivo, dicha proteína y agua para producir dicha composición acuosa. Preferiblemente, el contenido de Fe de la composición acuosa es inferior a 700 ppm basándose en el peso del aluminio en la composición; el contenido de Ni es inferior a 18 ppm basándose en el peso del aluminio en la composición; o el contenido de Cu es inferior a 2,5 ppm basándose en el peso del aluminio en la composición acuosa, o una combinación de los mismos. Preferiblemente, el método comprende además tamponar dicha composición acuosa a un pH de entre 6,5 y 8,5. Preferiblemente, el método comprende además envasar alícuotas de dicha composición acuosa que tiene menos de 450 ppm de metal pesado basándose en el peso del aluminio en la composición en recipientes de almacenamiento herméticos separados.
La descripción proporciona además un método para preparar un precipitado de sal de aluminio de grado clínico para su incorporación en un medicamento y/o vacuna, comprendiendo dicho método preparar una solución acuosa de iones de aluminio y precipitar dichos iones de aluminio de dicha solución, y determinar el nivel de un metal pesado en la solución y/o el precipitado de sal de aluminio, en donde se selecciona el precipitado que es capaz de proporcionar una composición acuosa que comprende menos de 450 ppm de metal pesado basándose en el peso de los iones de aluminio (gramos/gramos) en la solución.
Además se proporciona una composición acuosa que comprende una proteína y una sal de aluminio, comprendiendo dicha composición menos de 450 ppm de metal pesado basándose en el peso del aluminio en la composición. La composición acuosa preferiblemente se ha almacenado a temperaturas superiores a 20 °C durante al menos 1 mes. El metal pesado se selecciona preferiblemente de Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, Cr y Mo. Preferiblemente, el metal pesado se selecciona de Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V. En una realización particularmente preferida, el metal pesado se selecciona de Cu o Ni. Preferiblemente, el metal pesado es Cu.
La invención se explica además en los siguientes ejemplos.
Referencias
Alipazaga MV, Moreno RGM, Coichev N. 2004. Synergistic effect of Ni(II) and Co(II) ions on the sulphite induced autoxidation of Cu(II)/tetraglycine complex. Dalton Trans 13:2036-2040.
Arunee Wittayanukulluk, Dongping Jiang, Fred E. Regnier, Stanley L. Hem, “ Effect of microenvironment pH of aluminum hydroxide adjuvant on the chemical stability of adsorbed antigen” , Vaccine 22 (2004) 1172-1176 Brandt C, Elding LI. 1998. Role of chromium and vanadium in the atmospheric oxidation of sulfur (IV). Atmos Environ 32(4):797-800.
Exley, C (2010). Trends in Immunol. Vol. 31: págs. 103-109.
Ito, Kimiko y Kawanashi, Shosuke. Site-specific fragmentation and modification of Albumin by sulphite in presence of metal ions or peroxidase/H2O2: Role of Sulphate radical. Biochem and Biophys Res Comm., 1991, 176, 1306-1312 Huie R.E., Neta P. One-electron redox reaction in aqueous solutions of sulphite with hydroquinone and other hydroxyphenols. J. Phys. Chem., 1985, 89 (18), 3918-3921
Kalina Ranguelova, Marcelo G. Bonini y Ronald P. Mason: (Bi)sulphite Oxidation by Copper, Zinc-Superoxide Dismutase: Sulphite- Derived, Radical-Initiated Protein Radical Formation. Environmental Health Perspectives 2010, 118 (7), 970-975 Lambeth D.O., Palmer G. The kinetics and mechanism of reduction of electron transfer proteins and other compounds of biological interest by dithionite. J. Biochem. Chem. 1973, 248, 6095-6103
Li S, Schoneich C, Borchardt RT. Chemical instability of protein pharmaceuticals: Mechanisms of oxidation and strategies for stabilization. Biotechnol Bioeng. 5 de diciembre de 1995;48(5):490-500
Lindblad, EB (2004) Immunol. y Cell Biol. Vol. 82: 497-505.
Lima S, Bonifacio RL, Azzellini GC, Coichev N. 2002. Ruthenium(II) tris(bipyridyl) ion as a luminescent probe for oxygen uptake on the catalyzed oxidation of HSO3-. Talanta 56:547-556.
Mayo JC, Tan DX, Sainz RM, Natarajan M, Lopez-Burillo S, Reiter RJ. Protection against oxidative protein damage induced by metal-catalyzed reaction or alkylperoxyl radicals: comparative effects of melatonin and other antioxidants. Biochim Biophys Acta. 17 de marzo de 2003;1620(1-3):139-50.
Neta P., Huie RE: Free Radical Chemistry of Sulphite. Environmental Health Perspectives 1985, 64, 209-217 Shi X. 1994. Generation of •SOa- and OH radicals in SO32- reactions with inorganic environmental pollutants and its implications to SO32- toxicity. J Inorg Biochem 56(3):155-165.
Stadtman ER. Metal ion-catalyzed oxidation of proteins: biochemical mechanism and biological consequences. Free Radic Biol Med. 1990;9(4):315-25.
Breve descripción de los dibujos
Figura 1: Perfiles de elución por RP-HPLC del extracto de tapón de clorobutilo (diluido 1:4) y JEV09L37 SN.
Figura 2: Perfiles de elución por HPLC SEC de PS (2 mg/ml) antes y después de la escisión con tripsina.
Figura 3: Perfiles de elución por HPLC SEC de PS tratado con tripsina y PS degradado como está presente en NIV11A74. Téngase en cuenta que los perfiles de elución se normalizaron a una altura de pico similar para permitir una mejor comparación.
Figura 4: Evaluación de DOE de la relación de ELISA monoclonal/policlonal mediante análisis de gráficos de Pareto y gráficos de efectos principales (4 semanas a 22 °C).
Figura 5: Evaluación de DOE de la relación de ELISA monoclonal/policlonal mediante análisis de gráficos de Pareto y gráficos de efectos principales ( 8 semanas a 22 °C).
Figura 6 : Gráfico de contorno de la respuesta estimada
Figura 7: Gráfico residual de la respuesta estimada
Figura 8: Relación de ELISA (monoclonal/policlonal) para formulaciones de JEV a pH 7 en presencia de Ni, Cu y Cr. Las muestras se almacenaron a 22 °C durante 5 semanas.
Figura 9: Sumario de resultados obtenidos después de 7 semanas a 22 °C. Se muestran los datos en bruto de la relación en función del pH y el tipo de ion metálico y los resultados combinados para cada parámetro.
Figura 10: Relación media de formulaciones de DP (medicamento, del inglés drug product) preparadas con diferentes lotes de Alumbre. Las muestras se almacenaron durante 6 semanas a 22 °C. Las barras de error representan el intervalo de confianza del 95 % calculado basándose en la desviación estándar agrupada. Ejemplos de izquierda a derecha: Alumbre 3877, Alumbre 4074, Alumbre 4230 no GI, Alumbre 4230 GI, Alumbre 4470, Alumbre 4563, Alumbre 4621, Mezcla de Alumbre 4074_4230.
Figura 11: Distribución de tamaño de partícula de muestras de Alhydrogel®.
Figura 12: Curvas de titulación de Alhydrogel® en PBS. ■ AIOH no irradiado (RQCS0890), A A AIOH GI (RQCS1200), ♦ AIOH GI (RQCS1342), AIOH GI (RQCS0448)
Figura 13: Visión de conjunto de los lotes de Alhydrogel® sometidos a ensayo. Se muestra la concentración total de iones metálicos contaminantes en ng/ml y la proporción de los principales iones metálicos Fe, Cr y Ni.
Figura 14: Secuencia de aminoácidos de Ala-(His)6-OprF190-342-OprI21-83 (SEQ ID NO: 1) - en el presente documento también denominada “ proteína A” .
Ejemplos
Ejemplo 1
Se identificó previamente que el Lote 4230 de Hidróxido de aluminio (Alumbre) contribuye significativamente a la degradación del antígeno en FVL09L37. En este lote de Alumbre en particular se observó un contenido de iones metálicos residuales mucho mayor en comparación con otros lotes de Alumbre utilizados para la formulación del antígeno de JEV inactivado. Este Ejemplo demuestra estudios adicionales realizados para identificar adicionalmente el mecanismo de causa raíz subyacente y la influencia de los iones metálicos en la vía de degradación de JEV. Se realizó un diseño de experimentos (DOE, del inglés design of experiments) para determinar la influencia de los parámetros individuales en la estabilidad del antígeno.
Los parámetros sometidos a ensayo en un DOE factorial completo de 25 fueron
• Lote 4230 de Hidróxido de aluminio frente a Lote 4074 de Hidróxido de aluminio
• Presencia de exceso de fragmentos de sulfato de protamina
• Presencia de lixiviables del tapón de caucho de clorobutilo
• Intervalo de pH 7 a 8
• Contenido de formaldehído residual
El lote 4230 de Alumbre contiene niveles mucho mayores de impurezas de iones metálicos residuales en comparación con otros lotes de Alumbre utilizados para la formulación de JEV. Se seleccionó un “ diseño de experimento” (DOE) para investigar adicionalmente el posible mecanismo de causa raíz y la interacción de los parámetros que finalmente podrían conducir a la degradación del producto. En los diseños factoriales, se investigan múltiples factores simultáneamente durante el ensayo. Como en los diseños de un factor, se pueden considerar factores cualitativos y/o cuantitativos. El objetivo de estos diseños es identificar los factores que tienen un efecto significativo sobre la respuesta, así como investigar el efecto de las interacciones (dependiendo del diseño del experimento utilizado). Las predicciones también se pueden realizar cuando hay presentes factores cuantitativos, pero se debe tener cuidado puesto que determinados diseños son muy limitados en la elección del modelo predictivo. Para consultar información sobre el DOE en general, véase (Siebertz, Karl; van Bebber, David, Hochkirchen, Thomas: Statistische Versuchsplanung: Design of Experiments (DoE). Editor: Springer Berlín Heidelberg; 1.a edición (2010), ISBN-10: 3642054927).
1.1 Diseño del estudio DOE
1.1.1 Definición de Parámetros y niveles para el Diseño DOE
Se tomaron en consideración los siguientes parámetros y niveles para diseñar un experimento DOE apropiado:
• Contenido de iones metálicos residuales de Alumbre: Se seleccionaron los lotes 4230 y 4074 de hidróxido de aluminio como representativos de los dos extremos de calidad con respecto al contenido de iones metálicos residuales de hidróxido de aluminio. El punto central fue una mezcla de 50/50 % de ambos lotes de Alumbre. El análisis inicial de las impurezas de iones metálicos restantes en la solución madre de hidróxido de aluminio al 2 % mediante ICP-MS mostró diferencias significativas en el contenido de iones de Cr, Fe, Ni y Cu entre estos dos lotes (véase la Tabla 1).
• Fragmentos de sulfato de protamina: Los fragmentos de sulfato de protamina (PS) están presentes en cantidades bajas (<5 pg/ml) en el lote de vacuna final. Se sometió a ensayo si los fragmentos de PS podrían contribuir a la modificación de la superficie del virus (por ejemplo, interacción/unión covalente con las proteínas de la superficie del virus) en combinación con Alumbre y otros factores utilizados en este estudio. Por lo tanto, se preparó una solución madre de fragmentos de PS mediante digestión con Tripsina seguida de inactivación por calor y ultrafiltración utilizando una membrana de 5 kDa para la inactivación de la proteasa y la retirada de la enzima. Esta solución madre se utilizó para hacer adiciones de fragmentos de PS adicionales en las formulaciones respectivas al nivel alto de 50 pg/ml. En las muestras de nivel bajo no se hicieron adiciones de fragmentos de PS adicionales y el nivel real en las formulaciones fue < 5 pg/ml según el análisis por HPLC.
• pH: El nivel inferior y superior de pH en las formulaciones fue de 7 y 8 con el punto central a pH 7,5.
• Lixiviables/Extraíbles del émbolo de la jeringa: Émbolos de jeringa (hechos de clorobutilo PH701/50 negro) que se utilizan actualmente en el sistema de cierre de recipientes. Se sometió a ensayo si los lixiviados del caucho de clorobutilo en la formulación podrían contribuir a la modificación del antígeno. Por lo tanto, se preparó una solución madre de lixiviables y se utilizó para experimentos de adiciones. Se estimó que el nivel alto de lixiviables adicionados en la formulación era en promedio 1,4 veces mayor en comparación con el Lote de Vacuna Final (FVL) comercial. Debido a las condiciones de extracción severas, se detectaron picos adicionales que no estaban presentes en las muestras de FVL. Por lo tanto, las formulaciones adicionadas representan el “ peor de los casos” con respecto a los lixiviables y extraíbles. Las formulaciones en el nivel bajo no contenían lixiviables del caucho de clorobutilo.
• Formaldehído residual: Para formulaciones de nivel bajo, no se añadió formaldehído adicional a las muestras de formulación. El nivel más bajo era el formaldehído residual que todavía estaba presente en la muestra de NIV diluida después de la inactivación/neutralización y la dilución doble estaba en el intervalo de aprox. 37 ppm (recalculado a partir del certificado analítico GMP de liberación de DS comercial). Para el nivel alto, se adicionaron 40 ppm adicionales de formaldehído a la formulación correspondiente (contenido final total aprox. 77 ppm). Se sometió a ensayo si el formaldehído residual en combinación con un nivel más alto de iones metálicos presentes en Alumbre 4230 y otros posibles factores podrían reaccionar adicionalmente con el virus, lo que provocaría un hiperentrecruzamiento de las proteínas de la superficie y la pérdida de epítopos pertinentes.
Determinación de otras impurezas residuales relacionadas con el proceso:
El formaldehído, el sulfito y la sacarosa residuales en las formulaciones finales se estimaron basándose en los certificados GMP para el principio activo comercial JEV11A74. Los resultados se volvieron a calcular mediante la dilución doble real de NIV a DS utilizada en los experimentos DOE.
Sulfito Residual: La concentración de sulfito residual fue constante en todas las formulaciones con aprox. 93 ppm.
Sacarosa residual: La concentración de sacarosa residual fue constante en todas las formulaciones con aprox. 1 % v/p.
1.1.2 Diseño DOE
Estos 5 factores se combinaron en un plan de 25 DOE dando como resultado un número total de 34 experimentos que incluyen 2 puntos centrales con el siguiente diseño base. La planificación y evaluación del DOE se realizó con un software apropiado (Statgraphic Plus 3.0)
Diseño base: Factorial 25
Número de factores experimentales: 5
Número de bloques: 1
Número de respuestas: 1
Número de puntos centrales por bloque: 2
Número de ejecuciones: 34
Grados de libertad de error: 18
Aleatorizado: Sí
Factores continuos1* Bajo Alto Unidades
pH 7,0 8,0
Sí
Alumbre2) 0,0 100,0 % de Alumbre 4230
Sí
Fragmentos de PS adicionados3 0 50 pg/ml
No
Lixiviables adicionados4) 0 1,4 contenido relativo en comparación con FVL No Formaldehído adicionado5) 0 40 Ppm
No
Respuestas Unidades
ELISA (monoclonal, policlonal) de antígeno desorbido UA/ml
1) Continuo significa que un punto central (valor medio de nivel alto y bajo) está presente en el diseño del estudio. Sin punto central significa que solo los niveles alto y bajo están presentes en el diseño del estudio.
2) Nivel bajo (0 %) significa que la formulación se preparó con el Lote 4074 de Alumbre. Nivel alto (100 %) significa que la formulación se preparó con el lote 4230 de Alumbre. Para las formulaciones de punto central se utilizó una mezcla igual (50/50 %) de ambos lotes de Alumbre.
3) Puesto que el PS está presente en el NIV utilizado para la preparación de muestras de medicamentos, la concentración real de PS en las formulaciones sin adiciones fue de <5 pg/ml y ~50-55 pg/ml para las formulaciones adicionadas con PS.
4) No se asumieron lixiviables en formulaciones sin adiciones puesto que las muestras se prepararon/almacenaron en tubos Eppendorf de baja unión. El contenido total de lixiviables del émbolo de la jeringa de caucho de clorobutilo en las muestras adicionadas fue aprox. 1,4 veces mayor en comparación con FVL.
5) La concentración real de formaldehído en muestras de DP sin adiciones fue de aprox. 37 ppm, la concentración total de formaldehído en las muestras adicionadas fue de aproximadamente 77 ppm.
2 Definiciones y abreviaturas
AcCN Acetonitrilo
DOE Diseño de experimentos
DS Principio activo
FVL Lote de vacuna final
HPLC Cromatografía de líquidos de alto rendimiento
ICP-MS Plasma acoplado inductivamente-Espectrometría de masas
PS Sulfato de protamina
RP Fase Inversa
SEC Cromatografía de exclusión por tamaño
SN Sobrenadante
TFA Ácido trifluoroacético
s/ sin
3 Materiales y Métodos
3.1 Estudios DO
3.1.1 Materiales
• Tapones de émbolo de jeringa: PH701/50/C negro Sil67002-1051 (obtenido en West, N.° de pedido 2116) • Frasco de vidrio de 100 ml (Schott) Tapón de rosca recubierto con teflón
• Papel de aluminio
• Agua HQ
• Baño María eléctrico (IKA, HBR 4 digital)
• Tubos Eppendorf LoBind de 2 ml (Eppendorf, N.° de Cat. 0030 108.132)
• Speed Vac (Cristo, RVC-2-25)
• Viales de HPLC, vidrio transparente, 900 pl, Chromacol (VWR, N.° de Cat.: 548-1124)
• Viales de HPLC, PP, 900 pl, (Agilent, N.° de artículo 5182-0567)
• Tapones para viales de HPLC, precortados (VWR, N.° de Cat.: 548-1260)
• Tubos Falcon de 15 ml (Greiner, N.° de Cat. 188724)
• Alumbre lote 4470 (RQCS 1342); Lote 4230 de Alumbre (RQCS 1200)
• 10xPBS (Gibco, número de pedido 14200-091)
• Parafilm
• columna Waters Atlantis T3; diámetro de partícula de 3 pm; diámetro/longitud de la columna 2,1 x 100 mm (N.° de pedido 186003718; Lote 0107372331)
• Acetonitrilo (Merck, N.° de Cat. 1.13358.2500)
• TFA (Sigma, N.° de pedido 302031
• Sistema HPLC Dionex 3000
• Estante de disolvente SR-3000
• Pump UltiMate-3000, bomba analítica de gradiente de baja presión
• Automuestreador WPS-3000 TSL, automuestreador analítico - temperatura controlada
• Compartimento de columna TCC-3200, temperatura controlada
• PDA-Detector PDA-3000
• Solución de formaldehído al 37 % (Merck, N.° de Cat. 1.040031000)
• Sulfato de protamina (Intercell Biomedical Ltd, Lote N.° 086056)
• Dispositivo de ultrafilatrión (Amicon® Ultra 3 kDa) (Millipore, N.° de Cat. UFC900324)
• Incubadora Infors HT Incubator Multitron Standard (InforsAG)
• Tripsina (Sigma, N.° de pedido: T0303)
3.1.2 Procedimiento para la preparación de extraíbles de émbolo de jeringa
Se obtuvieron émbolos de jeringa (hechos de clorobutilo PH701/50 negro) que se utilizan actualmente en el sistema de cierre de recipientes de West (Alemania). Por lo tanto, se preparó una solución madre de lixiviables mediante tratamiento térmico del émbolo de jeringa en agua (90 °C/2 h) seguido de concentración en un speed-vac. El contenido relativo de lixiviables en esta solución madre se estimó mediante RP-HPLC utilizando una columna C18 (columna Atlantis T3) y se comparó con el sobrenadante de FVL JEV09L37.
Método de extracción
Se lavó con agua caliente un frasco de vidrio Schott de 100 ml con una tapa de rosca recubierta con teflón y un trozo de papel de aluminio y se aclaró minuciosamente con agua HQ. Se cargaron 30 tapones en el frasco y se añadieron 30 ml de agua HQ. El frasco se cerró con el papel de aluminio ajustado entre el frasco y la rosca y se selló
adicionalmente con Parafilm. El frasco se calentó en el baño de agua a 90 °C durante 2 horas y se dejó enfriar a temperatura ambiente. El extracto se transfirió a 14 tubos Eppendorf de baja unión (se recuperó un total de 28 ml de extracto). Se concentraron doce viales (un total de 24 ml) en un Speed Vac durante aproximadamente 44 horas y se agruparon en un tubo falcon para obtener 6 ml de extracto de tapón concentrado 4x. De la misma manera se preparó una muestra de control que contenía 30 ml de agua HQ sin tapón para evaluar cualquier posible contaminación.
Método de RP-HPLC C18
Los lixiviables se separaron mediante una columna RP-HPLC C18 (Atlantis T3) operada a 40 °C y 0,25 ml/min. El disolvente A era TFA al 0,1 % en H2O, el disolvente B era TFA al 0,1 % en AcCN. La separación se realizó mediante un gradiente lineal que variaba entre el 0 y el 95 % de B en 30 min. La detección se realizó a 214 nm, 254 nm y 280 nm. Se estimó que la concentración relativa total del extracto de tapón concentrado era 80 veces mayor en comparación con los picos detectados en el sobrenadante del Lote de Vacuna Final (FVL SN; obtenido por retirada de partículas de Alumbre por centrifugación a 5000 g/5 min) detectadas a 254 nm. Por lo tanto, se asignó un contenido relativo total de 80 U/ml (Unidades arbitrarias U) para la solución madre, mientras que la concentración relativa total de lixiviables en FVL SN se fijó en 1 U/ml. Para los estudios DOE, la solución madre se diluyó 16 veces en las formulaciones respectivas, produciendo aprox. 5 U/ml de extraíbles totales.
3.1.3 Preparación de fragmentos de sulfato de protamina
Se preparó una solución madre de fragmentos de PS digiriendo una solución de PS (2 mg/ml en PBS) con Tripsina (200 ng/ml durante 60 min a 37 °C). Posteriormente, la enzima se inactivó mediante calor (90 °C durante 10 min), seguido de ultrafiltración utilizando una membrana de 3 kDa (filtro centrífugo Amicon® Ultra). Debido al corte de la membrana, la Tripsina permaneció en la porción retenida, mientras que los fragmentos de PS estaban presentes en la fracción permeada. La inactivación completa de la enzima se evaluó adicionando 500 pg/ml de PS de longitud completa en una alícuota del fragmento de PS obtenido seguido de incubación a 37 °C durante 18 h. No se observó degradación de PS de longitud completa, lo que indica una inactivación/retirada completa de la Tripsina. La degradación se controló mediante PS-HPLC SEC.
3.1.4 Plan de DOE
Las muestras se prepararon según el esquema de pipeteo que se muestra en la Tabla 2. Se utilizó el lote de NIV JEV11A74 obtenido de una serie de producción comercial como muestra inicial. Se diluyó NIV 2 veces a DS utilizando tampón de PBS seguido de ajuste de pH. Se extrajeron alícuotas de 5 ml y se adyuvaron con el correspondiente lote 4230, 4074 de Alumbre o una mezcla al 50/50 % de ambos. La cantidad final de solución madre de Alumbre (Al2O3 al 2 %) añadida fue de 500 pg/ml de aluminio (Al2O3 al 0,1 %). Cada formulación (5 ml) se dividió en dos partes (2 x 2,5 ml) utilizando tubos Eppendorf Lo-bind. Una alícuota se almacenó a 2-8 °C, otra alícuota se almacenó a 22 ± 1 °C (Incubadora Infors HT) con agitación suave (20 rpm).
3.2 ELISA de JEV inactivado (basado en anticuerpos policlonales)
La desorción del antígeno del Alumbre y el análisis por ELISA se realizaron utilizando anticuerpos anti-JEV de oveja policlonales para recubrir las placas de ELISA de 96 pocillos como se describe en el Ejemplo 4.
3.3 ELISA de JEV inactivado (basado en anticuerpos monoclonales)
Se desarrolló un ELISA de JEV basado en anticuerpos monoclonales (mAb). El ensayo se basa principalmente en el formato de ensayo “ ELISA de JEV policlonal” , solo se utiliza un anticuerpo monoclonal anti-JEV (clon 52-2-5) para el recubrimiento. Se demostró que el mab 52-2-5 empleado era específico para JEV y reconocía un epítopo neutralizante. El clon 52-2-5 de Mab se obtuvo mediante la inmunización subcutánea de ratones BALB/c con el lote de vacuna comercialmente disponible JEV08J14B. Se fusionaron células de bazo de los ratones con células de mieloma. A partir de las células de hibridoma resultantes, se seleccionaron y subclonaron clones individuales. Los clones se cribaron negativamente frente a albúmina sérica bovina, sulfato de protamina y un extracto de la estirpe celular de producción de la vacuna JE (células Vero). Se realizó un cribado positivo contra el Virus Inactivado Neutralizado (NIV) del lote de vacuna JEV08M20. Para el cribado, se recubrieron placas de microtitulación con el antígeno relevante y se hicieron reaccionar con sobrenadante de cultivos de los clones seleccionados. Para la detección se utilizó un anticuerpo policlonal de cabra anti-ratón conjugado con fosfatasa alcalina. Se demostró que el clon 52-2-5 de Mab reconoce un epítopo neutralizante en el dominio III de la proteína de la envuelta (E) de JEV que contiene Ser331 y Asp332 (Lin C.-W. y Wu W.-C. J Virol. 2003;77(4):2600-6). La unión del mab al epítopo neutralizante indicado se determina, por ejemplo, como se describe en Lin y Wu (2003) mediante mutagénesis dirigida al sitio del dominio III en la posición 331 (por ejemplo: S^-R), y/o mediante mutaciones de alanina en o cerca de la posición 331 del dominio III, por ejemplo, de los restos Ser 331 y Asp332, seguidas de inmunotransferencias para determinar la unión del mab a las proteínas mutadas. Los resultados de unión negativos indican que el epítopo del mab es el epítopo neutralizante identificado por Lin y Wu (2003). La característica neutralizante del epítopo da lugar a la suposición de que el epítopo puede ser importante para que el antígeno desencadene una respuesta inmunitaria protectora.
Las muestras de JEV se analizaron mediante ensayos ELISA, tanto policlonal como monoclonal. El contenido de epítopo específico relativo puede expresarse como la relación del contenido de antígeno total determinado mediante “ ELISA monoclonal” (clon 52-2-5) dividido por el contenido de antígeno total determinado mdiante “ ELISA policlonal” . Cualquier diferencia en la relación puede indicar diferencias en el contenido de epítopos específicos 52-2-5. Los resultados cercanos a 1 corresponderían a un contenido alto de epítopos, y los resultados cercanos a 0 corresponderían a un contenido de epítopos relativo bajo. Una relación baja indica la presencia de cambios estructurales del epítopo neutralizante.
En el curso del desarrollo de este “ ELISA de mAb” , se detectaron diferencias entre lotes de vacunas, lo que podría correlacionarse con los resultados de potencia de estos lotes.
3.4 SEC-HPLC de sulfato de protamina
El PS (longitud completa) y sus fragmentos se analizaron mediante HPLC de exclusión por tamaño (SEC-HPLC) utilizando un Superdex Peptide 10/300 GL, 10 * 300 mm, 13 pm (GE Healthcare) utilizando ácido trifluoroacético (TFA) al 0,1 % (v/v) en acetinitrilo (CAN) al 30 % como fase móvil a un caudal de 0,6 ml/min. Se prepararon muestras que contenían PS por duplicado, es decir, se diluyeron con fase móvil antes de la inyección.
4 Resultados
4.1 Análisis de lixiviables de tapón utilizados para adicionar Experimentos
En la Figura 1 se muestran perfiles de elución por RP-HPLC de la solución madre concentrada obtenida después de la extracción de tapones con calor en comparación con FVL SN. Patrón de pico similar al observado para ambas muestras. Debido a las condiciones de extracción severas, se detectaron picos adicionales en el concentrado que no estaban presentes en las muestras de FVL o presentes solo en un contenido relativo mucho más bajo. Por lo tanto, las formulaciones adicionadas representan “ el peor de los casos” con respecto a los lixiviables y extraíbles. El contenido relativo total de picos individuales en el extracto concentrado y la formulación adicionada en comparación con FVL SN se resume en la Tabla 3. La cantidad total de lixiviables en la solución madre se calculó como la suma de todos los picos detectados y se expresó en unidades arbitrarias como 67 U/ml. Puesto que la solución madre se diluyó 16 veces en la formulación respectiva, el contenido total resultante de lixiviables se estimó en 4,2 U/ml. Esto corresponde a un aumento promedio de 1,4 veces en comparación con el sobrenadante de FVL JEV09L37 (3,0 U/ml).
4.2 Análisis de fragmentos de sulfato de protamina
Los fragmentos de PS obtenidos después de la escisión de PS de longitud completa por Tripsina se muestran en la Figura 2. Se obtuvieron perfiles de pico similares de PS tratado con Tripsina y PS ya degradado presente en NIV11A74 mediante HPLC (véase la Figura 3).
4.3 Evaluación del DOE
Las formulaciones preparadas para este DOE se analizaron después de 4 y 8 semanas de incubación en condiciones aceleradas (22 °C). Se consideró que cualquier reacción de degradación se aceleraría cuando se almacenara a una temperatura más alta en comparación con las condiciones normales de almacenamiento (2-8 °C). Sin embargo, las muestras aún se almacenan a 2-8 °C y se analizarán en un momento posterior (~4-6 meses). El primer análisis de las muestras almacenadas a 22 °C durante 4 y 8 semanas se muestra en la Tabla 4.
4.3.1 Evaluación de DOE después de 4 semanas a 22 °C
La evaluación estadística de los resultados de la matriz de DOE obtenidos después de 4 semanas a 22 °C mostró que el contenido de epítopo específico 52-2-5 (expresado como la relación de antígeno desorbido analizado mediante ELISA monoclonal/policlonal) se vio influenciado estadísticamente de manera significativa (nivel de confianza del 95 %, véase la Tabla 5) por los siguientes factores:
• menor contenido de epítopo específico 52-2-5 en presencia de lote 4230 de Alumbre
• menor contenido de epítopo específico 52-2-5 a un pH menor de 7
• Mayor contenido de epítopo específico 52-2-5 a mayor concentración de formaldehído
La presencia de una mayor concentración de fragmentos de PS y lixiviables de caucho de clorobutilo no mostró ninguna influencia en el contenido de epítopos específicos. No se detectaron interacciones de segundo orden o mayor entre parámetros individuales.
La tabla ANOVA divide la variabilidad en “ Relación de 4 semanas” en partes separadas para cada uno de los efectos. Después somete a ensayo la importancia estadística de cada efecto comparando el cuadrado de la media con una estimación del error experimental. En este caso, 3 efectos (Alumbre, pH, formaldehído) tienen valores P inferiores a 0,05,
lo que indica que son significativamente diferentes de cero al nivel de confianza del 95,0 %. El estadístico R Cuadrado indica que el modelo ajustado explica el 74,85 % de la variabilidad en la Relación de 4 semanas. El estadístico R cuadrado ajustado, que es más adecuado para comparar modelos con diferente número de variables independientes, es del 51,27 %. El error estándar de la estimación muestra que la desviación estándar de los valores residuales es de 0,063. El error absoluto medio (EAM) de 0,0353 es el valor promedio de los valores residuales. El estadístico de Durbin-Watson (DW) somete a ensayo los valores residuales para determinar si existe alguna correlación significativa basándose en el orden en que ocurren en su archivo de datos. Puesto que el valor de DW es superior a 1,4, probablemente no exista una autocorrelación grave en los valores residuales. Los efectos también se muestran mediante gráficos de Pareto estandarizados y gráficos de efectos principales, como se muestra en la Figura 4.
4.3.2 Evaluación de DOE después de 8 semanas a 22 °C
La evaluación estadística de los resultados de la matriz de DOE obtenidos después de 8 semanas a 22 °C fue similar a los resultados obtenidos después de 4 semanas. La evaluación muestra que el contenido de epítopo específico 52-2-5 (expresado como la relación de antígeno desorbido analizado mediante ELISA monoclonal/policlonal) se vio influenciado estadísticamente de manera significativa (nivel de confianza del 95 %, véase la Tabla 6) por los siguientes factores:
• menor contenido de epítopo específico 52-2-5 en presencia de lote 4230 de Alumbre
• menor contenido de epítopo específico 52-2-5 a un pH menor de 7
La presencia de una mayor concentración de fragmentos de PS, lixiviables de caucho de clorobutilo y formaldehído no mostró ninguna influencia en el contenido de epítopos específicos. Téngase en cuenta que el valor P para el formaldehído (0,08) está bastante cerca de ser significativo. No se detectaron interacciones de segundo orden o mayor entre parámetros individuales.
La tabla ANOVA divide la variabilidad en “ Relación de 8 semanas” en partes separadas para cada uno de los efectos. Después somete a ensayo la importancia estadística de cada efecto comparando el cuadrado de la media con una estimación del error experimental. En este caso, 2 efectos (Alumbre y pH) tienen valores P inferiores a 0,05, lo que indica que son significativamente diferentes de cero al nivel de confianza del 95,0 %. El estadístico R Cuadrado indica que el modelo ajustado explica el 75,9 % de la variabilidad en la “ Relación de 8 semanas” . El estadístico R cuadrado ajustado, que es más adecuado para comparar modelos con diferente número de variables independientes, es del 53,3 %. El error estándar de la estimación muestra que la desviación estándar de los valores residuales es de 0,095. El error absoluto medio (EAM) de 0,057 es el valor promedio de los valores residuales. Los efectos también se muestran mediante gráficos de Pareto estandarizados y gráficos de efectos principales, como se muestra en la Figura 5.
También se realizó un análisis de regresión a los datos ajustados y los coeficientes de regresión calculados se muestran en la Tabla 7. A continuación se muestra la ecuación de regresión que se ha ajustado a los datos, incluyendo el pH, el Alumbre y el Formaldehído. La ecuación del modelo ajustado es
“ Relación 8 semanas” = 0,0228125 0,113125*pH - 0,00185625*Alumbre+0,0315625*Formaldehído
donde los valores de las variables se especifican en sus unidades originales, excepto los factores categóricos que toman los valores -1 para el nivel bajo y 1 para el nivel alto. El contorno de la respuesta estimada y la gráfica de valores residuales se muestran en la Figura 6 y la Figura 7. La relación aumenta cuando el contenido relativo de Alumbre Lote 4230 disminuye y el pH aumenta.
La Tabla 8 contiene información sobre los valores de “ Relación de 8 semanas” generados utilizando el modelo ajustado. La tabla incluye:
(1) el valor observado de “ Relación de 8 semanas”
(2) el valor predicho de “ Relación de 8 semanas” utilizando el modelo ajustado
(3) Límites de confianza del 95,0 % para la respuesta media
Como se muestra, los resultados experimentales están bien predichos por el modelo de regresión.
5. Sumario
De los parámetros sometidos a ensayo, se demostró que el lote 4230 de Alumbre contribuye significativamente a la degradación del antígeno, según se analizó mediante ELISA monoclonal/policlonal en condiciones aceleradas (22 °C, puntos temporales de ensayo de 4 y 8 semanas). Los resultados del DOE obtenidos después de 4 y 8 semanas a 22 °C muestran que Alumbre 4230 es el factor más significativo con respecto a la degradación del antígeno, según lo detectado por la relación de ELISA monoclonal/policlonal. Las formulaciones preparadas con Alumbre 4074 (mucha mayor pureza con respecto a los iones metálicos residuales) muestran en general un contenido de epítopo específico mucho mayor.
El formaldehído y el pH también contribuyeron a la estabilidad del antígeno, pero en menor medida. El efecto de una mayor estabilidad del antígeno en muestras formuladas con Alumbre 4230 a un nivel de formaldehído mayor quedó bien demostrado (por ejemplo, muestras n.° 19 y 29). Sin embargo, la influencia del formaldehído fue menos pronunciada después de un período prolongado de almacenamiento (8 semanas a 22 °C).
Se observó una mejor estabilidad del antígeno a pH 8 en comparación con el pH 7. El sulfato de protamina y los lixiviables del tapón de caucho de clorobutilo no contribuyeron a la degradación del antígeno.
Ejemplo 2
En estudios previos (véase el Ejemplo 1), el lote 4230 de Hidróxido de Aluminio se identificó como un factor importante que contribuye a la degradación del antígeno observada en FVL09L37. En este lote particular de hidróxido de aluminio (Alumbre), se observó un contenido de iones metálicos residuales mucho mayor en comparación con otros lotes de Alumbre utilizados para la formulación del antígeno de JEV inactivado. Este Ejemplo resume estudios adicionales realizados para evaluar la influencia de los iones metálicos en la estabilidad del j Ev inactivado. Se realizaron estudios de adiciones con el antígeno presente en una solución de virus neutralizado inactivado (NIV) o en una suspensión de medicamento (DP) después de la formulación del antígeno con hidróxido de aluminio.
1 Descripción del estudio
Previamente se demostró que el lote 4230 de Alumbre contiene niveles mucho mayores de impurezas de iones metálicos residuales en comparación con otros lotes de Alumbre utilizados para la formulación de JEV (véase también el Ejemplo 3). Se realizaron estudios adicionales para evaluar la influencia de los iones metálicos en la estabilidad y en una posible modificación de la superficie de JEV. El antígeno inactivado estaba presente en la solución de virus inactivado neutralizado (NIV) o en la suspensión de medicamento (DP) después de la dilución adicional de NIV y la formulación con hidróxido de aluminio. En otro conjunto de experimentos, se utilizaron y formularon diferentes lotes de Alumbre que cubrían un amplio intervalo de contenido de iones metálicos residuales con un único lote de NIV definido. Todas estas formulaciones aún contenían formaldehído residual y bisulfito en concentraciones representativas en comparación con el producto comercial. La solución madre de sales metálicas se disolvió en agua y se añadió a las muestras a la concentración final deseada. 2 Definiciones y abreviaturas
AcCN Acetonitrilo
ANOVA Análisis de varianza
DOE Diseño de experimentos
DP Medicamento
DS Principio activo
FBV Vacuna final a granel
FVL Lote de vacuna final
GI Irradiado con rayos gamma
HPLC Cromatografía de líquidos de alto rendimiento
LSD Diferencia mínima significativa de Fisher
mAb Anticuerpo monoclonal
NIV Virus inactivado neutralizado
PS Sulfato de protamina
RP Fase Inversa
SEC Cromatografía de exclusión por tamaño
SN Sobrenadante
TFA Ácido trifluoroacético
s/ sin
3 Materiales y Métodos
3.1 Materiales
Tetrahidrato de cloruro de hierro (II) (Sigma, N.° de pedido 44939)
Hexahidrato de cloruro de hierro (III) (Sigma, N.° de pedido 31232)
Hexahidrato de sulfato de níquel (II) (Sigma, N.° de pedido N4882)
Hexahidrato de cloruro de cobalto (II) (Sigma, N.° de pedido 31277)
Deshidrato de cloruro de cobre (II) (Sigma, N.° de pedido 807483)
Heptahidrato de sulfato de cinc (Sigma, N.° de pedido 24750)
Hexahidrato de crom (III) (AlfaAesar, N.° de pedido 42114)
Deshidrato de sal disódica del ácido etilendiaminotetraacético (EDTA) (Sigma, E5134)
Agua bidest. (Fresenius Kabi, N.° de Art. 0712221/01 A)
10xPBS (Gibco, número de pedido 14200-091)
Solución de formaldehído al 37 % (Merck, N.° de Cat. 1.040031000)
Sulfato de protamina (Intercell Biomedical Ltd, Lote N.° 086056)
Tubos Eppendorf LoBind de 2 ml (Eppendorf, N.° de Cat. 0030 108.132)
Tubos Falcon de 15 ml (Greiner, N.° de Cat. 188724)
Incubadora Infors HT Incubator Multitron Standard (InforsAG)
Filtro de 0,2 pm Mini Kleenpak 25 mm (Pall)
El NIV11A74 y la vacuna final a granel (FBV, formulada con lote 4539 de Alumbre) JEV 11D87 de ciclos e producción comercial se obtuvieron en Intercell Biomedical (Livingston, Reino Unido) y se almacenaron a 2-8 °C asta su posterior procesamiento
Se prepararon soluciones madre de sales metálicas en agua (concentración final 1 mM) utilizadas para los experimentos de adiciones y se almacenaron a 2-8 °C hasta su uso.
Las muestras de hidróxido de aluminio (Al2O3 al 2 %, Brenntag Biosector) se obtuvieron de Intercell Biomedical o se adquirieron directamente en Brenntag. Las muestras de Alumbre se almacenaron a 2-8 °C. En este estudio se utilizaron los siguientes lotes de Alumbre: 4470, 4563, 4621, 3877, 4230 (no irradiados con rayos gamma e irradiados con rayos gamma);
3.2 Preparación de soluciones madre de metales
3.2.1 Solución madre de hierro (II)
Se preparó una solución madre de hierro (II) 20 mM disolviendo 397 mg de tetrahidrato de cloruro de hierro (II) en 100 ml de agua bidestilada.
3.2.2 Solución madre de hierro (III)
Se preparó una solución madre de hierro (III) 20 mM disolviendo 540 mg de hexahidrato de cloruro de hierro (III) en 100 ml de agua bidestilada.
3.2.3 Solución madre de níquel (II)
Se preparó una solución madre de níquel (II) 20 mM disolviendo 525 mg de hexahidrato de sulfato de níquel (II) en 100 ml de agua bidestilada.
3.2.4 Solución madre de cobalto (II)
Se preparó una solución madre de cobalto (II) 20 mM disolviendo 476 mg de hexahidrato de cloruro de cobalto (II) en 100 ml de agua bidestilada.
3.2.5 Solución madre de cobre (II)
Se preparó una solución madre de cobre (II) 20 mM disolviendo 341 mg de dihidrato de cloruro de cobre (II) en 100 ml de agua bidestilada.
3.2.6 Solución madre de cinc
Se preparó una solución madre de cinc 20 mM disolviendo 575 mg de heptahidrato de sulfato de cinc en 100 ml de agua bidestilada.
3.2.7 Solución madre de Crom (III)
Se preparó solución madre de Crom (III) 20 mM disolviendo 533 mg de hexahidrato de cloruro de Crom (III) en 100 ml de agua bidestilada.
3.3 Preparación de soluciones de trabajo
Se prepararon soluciones de trabajo de iones metálicos (concentración final de 1 mM si no se indica lo contrario) mediante dilución de soluciones madre de iones metálicos con agua bidestilada y filtración estéril a través de un filtro de jeringa de 0,2 pm.
3.4 Preparación de la Formulación
Todas las formulaciones se prepararon en condiciones estériles. El NIV y la FBV obtenidos de series de producción comercial se ajustaron al pH deseado y se adicionaron con alícuotas de solución madre de metal. Todas las muestras se almacenaron en tubos de plástico si no se indica lo contrario. En todas las formulaciones que utilizaron Alumbre, el contenido final de Al fue de 500 pg/ml, lo que corresponde a Al2O3 al 0,1 %. Cabe señalar que los iones metálicos, especialmente el hierro (II), el hierro (III) y, en cierta medida, el Cr (III), formaron un precipitado con los iones de fosfato presentes en el tampón, lo que provocó una coprecipitación parcial del virus inactivado, representada por la baja recuperación determinada por HPLC de exclusión por tamaño (SEC-HPLC).
3.4.1 Experimento 20110913 (NIV): Formulación de NIV a diferentes concentraciones de iones metálicos de Ni (II), Cu (II), Cr (III) con o sin presencia de fragmentos de PS
El NIV 11A74 se ajustó a pH 7 y pH 8 seguido de adiciones de iones metálicos (Ni (II), Cu (II), Cr (III)) a una concentración final de 100/500/1000 ng/ml. Todas las formulaciones se almacenaron en tubos Eppendorf de baja unión a 22 °C. También se prepararon alícuotas de todas las formulaciones en presencia de fragmentos de sulfato de protamina (50 pg/ml). Esto se hizo para evaluar cualquier efecto de los fragmentos de PS sobre la estabilidad de JEV en presencia de metales. La preparación de fragmentos de Sulfato de Protamina (PS) se describe en el Ejemplo 1. Las muestras se prepararon el mismo día (véase la Tabla 9) y se analizaron tres semanas después. Todas las muestras se analizaron mediante SEC-HPLC, pero solo las muestras a pH 8 (n.° 21-40) se analizaron mediante ELISA.
3.4.2 Experimento 20110913 (DP): Formulación de DP a diferentes concentraciones de iones metálicos de Ni (II), Cu (II), Cr (III)
En este estudio se utilizó FBV 11D87 (formulada con Lote 4539 de Alumbre). La FBV se ajustó a pH 7 y pH 8 y se adicionó con Ni (II)/Cu (II)/Cr (III) a 100, 500 y 1000 ng/ml para evaluar cualquier efecto dependiente de la concentración de iones metálicos/pH. La Tabla 10 muestra el diseño experimental de este experimento. Todas las formulaciones se almacenaron en tubos Falcon a 2-8 °C y 22 °C. Las muestras almacenadas a 22 °C se analizaron mediante SEC-HPLC y ELISA después de 5 semanas.
3.4.3 Experimento 20110812-DP adicionado con metal
Se obtuvo vacuna final a granel 11D87 (formulada con lote 4539 de Alumbre) de un ciclo de producción comercial y se utilizó en este estudio. El formol residual en el DS se analizó como 28,1 ppm, los sulfitos residuales fueron 92,2 ppm. Se puede considerar que el contenido real en el DP está en el mismo intervalo. Se ajustó FBV JEV11D87 a pH
7,0/7,4/7,8 y se adicionó con 500 ng/ml (concentración final) de Fe (II), Fe (III), Ni (II), Co(II), Cu (II), Zn (II). También se preparó una formulación de mezcla de iones metálicos que contenía todos los iones metálicos individuales juntos en solución. Posteriormente se prepararon formulaciones con Cr (III) y el Cr (III) no se incluyó en la mezcla de iones metálicos. Las formulaciones de control solo se ajustaron al pH deseado, pero no se adicionaron con metales. Todas las formulaciones (n.° 1-24) se prepararon el mismo día y se almacenaron en tubos Falcon a 2-8 °C y 22 °C.
Se prepararon muestras adicionadas con Cr (III) adicionales (n.° 25-27) tomando alícuotas de las muestras de control almacenadas a 2-8 °C y adicionadas con Cr (III) hasta una concentración final de 500 ng/ml. Las formulaciones se almacenaron únicamente en condiciones aceleradas (22 °C). La Tabla 11 muestra el montaje experimental de este experimento. Todas las muestras almacenadas a 22 °C se analizaron mediante ELISA (monoclonal y policlonal) después de 4 y 7 semanas.
3.4.4 Experimento 20110819: Formulación de DP utilizando diversos lotes de Alumbre
Los estudios de adiciones como se ha descrito anteriormente pueden proporcionar una primera evidencia de la posible inestabilidad del antígeno formulado en presencia de determinados metales, pero pueden no ser completamente representativos de las condiciones reales en las que los metales presentes en el Hidróxido de aluminio se incorporan en la estructura tridimensional del gel dando como resultado diferentes concentraciones locales y orientación/accesibilidad. Para superar estas limitaciones se comenzó un estudio inicial para simular las condiciones reales. Se formuló un único lote de NIV (11A74) obtenido de un ciclo de producción comercial con diversos lotes de Alumbre producidos por Brenntag que cubren una amplia gama de metales residuales. Se mezclaron 4,75 ml de NIV con 0,25 ml de Alumbre (2 %) en tubos Falcon. La concentración final de hidróxido de aluminio fue de 500 pg/ml (=Al2O3 al 0,1 %). Las muestras de vacunas formuladas se almacenaron a 2-8 °C y en condiciones aceleradas a 22 °C. Todos estos lotes de Alumbre contenían iones metálicos residuales en diferentes concentraciones. El lote 4230 de Alumbre tiene el nivel más alto de Fe, Cu, Ni y V (véase el Ejemplo 3). Téngase en cuenta que ICP-MS no puede especificar las valencias de los iones metálicos. También se preparó una muestra mixta de Alumbre que contenía cantidades iguales de 4230 y 4074 para obtener un nivel “ intermedio” para Ni (II) y Cu (II). Las muestras se analizaron después de 6 semanas de almacenamiento a 22 °C. La cantidad residual de formaldehído y sulfito estimada mediante el recálculo de los resultados de análisis de DS disponibles corregidos por el factor de dilución de NIV a DS fue de 76 ppm de formaldehído y 192 ppm de sulfito, respectivamente.
3.1 Desorción de antígeno de hidróxido a partir de aluminio para análisis por SEC-MALLS
Se desorbieron partículas víricas a partir de Hidróxido de aluminio. Se centrifugaron ~625 pl de DP (8 °C, 5 min, 3300 xg) y el sobrenadante se descartó si no se indica lo contrario o se analizó mediante JEV-SEC-MALLS para detectar la concentración de antígeno no unido. Se desorbieron partículas víricas suspendiendo las partículas de Hidróxido de aluminio con 62,5 pl de tampón de fosfato de potasio 0,8 M (pH 8) que contenía BSA (50 pg/ml). Se añadió BSA al tampón de desorción para el análisis por SEC-MALLS para minimizar las pérdidas provocadas por la adsorción inespecífica del antígeno. Después de agitar (500 rpm) las partículas de Hidróxido de aluminio durante 10 minutos a temperatura ambiente, las partículas se retiraron por centrifugación y el sobrenadante se recogió en un tubo Eppendorf LoBind y el procedimiento de desorción se repitió en la muestra restante. El antígeno desorbido agrupado (muestra concentrada ~5 veces; volumen final 125 pl; volumen de partida ~625 pl) después se analizó adicionalmente mediante SEC-MALLS.
3.2 Método de HPLC SEC-MALLS
El antígeno desorbido se analizó mediante SEC-MALLS. En resumen, después de la desorción del antígeno a partir de Hidróxido de aluminio, se cargaron 100 pl del material desorbido agrupado (concentrado ~5 veces) en una columna de SEC Superose 610/300 GL. Como fase móvil se utilizó 1* PBS NaCl 250 mM. Las señales ultravioleta (UV) de 214 nm y MALLS de partículas víricas se registraron y analizaron utilizando los paquetes de software Chromeleon y ASTRA.
3.3 ELISA de JEV inactivado (basado en anticuerpos policlonales)
La desorción del antígeno del Alumbre y el análisis por ELISA se realizaron utilizando anticuerpos anti-JEV de oveja policlonales para recubrir las placas de ELISA de 96 pocillos como se describe en el Ejemplo 4.
3.4 ELISA de JEV inactivado (basado en anticuerpos monoclonales)
Durante el curso de este ensayo de investigación, se desarrolló un ELISA de JEV basado en anticuerpos monoclonales (mAb). El ensayo se basa principalmente en el formato de ensayo “ ELISA de JEV policlonal” , solo se utiliza un anticuerpo monoclonal anti-JEV (clon 52-2-5) para el recubrimiento y el anticuerpo policlonal actual para la detección. Se demostró que el mab 52-2-5 empleado era específico para JEV y reconocía un epítopo neutralizante. El clon 52-2 5 de Mab se obtuvo mediante la inmunización subcutánea de ratones BALB/c con el lote de vacuna comercialmente disponible JEV08J14B. Se fusionaron células de bazo de los ratones con células de mieloma. A partir de las células de hibridoma resultantes, se seleccionaron y subclonaron clones individuales. Los clones se cribaron negativamente frente a albúmina sérica bovina, sulfato de protamina y un extracto de la estirpe celular de producción de la vacuna JE (células Vero). Se realizó un cribado positivo contra el Virus Inactivado Neutralizado (NIV) del lote de vacuna JEV08M20. Para el cribado, se recubrieron placas de microtitulación con el antígeno relevante y se hicieron reaccionar
con sobrenadante de cultivos de los clones seleccionados. Para la detección se utilizó un anticuerpo policlonal de cabra anti-ratón conjugado con fosfatasa alcalina. Se demostró que el clon 52-2-5 de Mab reconoce un epítopo neutralizante en el dominio III de la proteína de la envuelta (E) de JEV que contiene Ser331 y Asp332 (Lin C.-W. y Wu W.-C. J Virol. 2003;77(4):2600-6). La unión del mab al epítopo neutralizante indicado se determina, por ejemplo, como se describe en Lin y Wu (2003) mediante mutagénesis dirigida al sitio del dominio III en la posición 331 (por ejemplo: S^-R), y/o mediante mutaciones de alanina en o cerca de la posición 331 del dominio III, por ejemplo, de los restos Ser 331 y Asp332, seguidas de inmunotransferencias para determinar la unión del mab a las proteínas mutadas. Los resultados de unión negativos indican que el epítopo del mab es el epítopo neutralizante identificado por Lin y Wu (2003). La característica neutralizante del epítopo da lugar a la suposición de que el epítopo puede ser importante para que el antígeno desencadene una respuesta inmunitaria protectora. Las muestras de JEV se analizaron mediante ensayos ELISA, tanto policlonal como monoclonal. El contenido de epítopo específico relativo puede expresarse como la relación del contenido de antígeno total determinado mediante “ ELISA monoclonal” (clon 52-2-5) dividido por el contenido de antígeno total determinado mdiante “ ELISA policlonal” . Cualquier diferencia en la relación puede indicar diferencias en el contenido de epítopos específicos 52-2-5. Los resultados cercanos a 1 corresponderían a un contenido alto de epítopos, y los resultados cercanos a 0 corresponderían a un contenido de epítopos relativo bajo. Una relación baja indica la presencia de cambios estructurales del epítopo neutralizante.
En el curso del desarrollo de este “ ELISA de mAb” , se detectaron diferencias entre lotes de vacunas, lo que podría correlacionarse con los resultados de potencia de estos lotes.
3.5 Evaluación estadística
La evaluación estadística se realizó con Statgraphic Plus 3.0.
4 Resultados
4.1 Experimento 20110913 (NIV): Formulación de NIV a diferentes concentraciones de iones metálicos de Ni (II), Cu (II), Cr (III) con o sin presencia de fragmentos de PS
Los resultados de SEC-HPLC de formulaciones de NIV (pH 7 y pH 8) que contenían iones metálicos [Ni (II), Cu (II), Cr (III)] con y sin fragmentos de PS se resumen en la Tabla 12. Los resultados de SEC-HPLC muestran que la recuperación de antígenos de la mayoría de las muestras fue >80 %. Algunas muestras (n.° 7, n.° 36, n.° 38) mostraron recuperaciones ligeramente reducidas en el intervalo del 70-80 %. Cabe señalar que el contenido real de virus es bastante bajo y la precisión de los resultados de HPLC se puede estimar en aproximadamente ± 20 %. Puesto que para las muestras n.° 36 y n.° 38 las recuperaciones para las siguientes formulaciones (n.° 37, n.° 39) en el siguiente nivel de contenido de iones metálicos individuales fueron nuevamente más altas, estas diferencias pueden deberse a la variabilidad del ensayo y no se consideraron significativas. Basándose en los resultados obtenidos no fue posible aclarar la influencia de los iones metálicos con respecto a la recuperación de JEV inactivado soluble. Sin embargo, SEC-HPLC solo proporciona información sobre el contenido de virus soluble, pero no proporciona información sobre cualquier posible modificación de la superficie. Solo las formulaciones preparadas a pH 8 también se analizaron mediante ELISA (análisis por duplicado). Se calculó la relación de ELISA monoclonal/policlonal y se puede utilizar con fines de comparación de resultados. El análisis de muestras mediante ELISA (véase la Tabla 13) no muestra ninguna influencia significativa de los metales sometidos a ensayo en la degradación de JEV inactivado a pH 8 después de tres semanas a 22 °C. Puede haber una tendencia a la disminución de la relación en presencia de Cu (II), pero en general parece que un tiempo de incubación de tres semanas a 22 °C no parece ser suficiente para detectar una degradación significativa. Como también se muestra en el experimento DOE (Ejemplo 1), el JEV inactivado parece tener una mayor estabilidad a pH 8 cuando se almacena en condiciones aceleradas a 22 °C y esto también contribuiría a que no se observaran efectos significativos. En este experimento también se demostró que los fragmentos de PS no tienen ninguna influencia sobre la estabilidad de JEV. Esto también está muy de acuerdo con los resultados del DOE. Las muestras de NIV 1-20 formuladas a pH 7 mostraron una reducción significativa en el contenido de epítopos monoclonales en presencia de Cu (II). A la concentración más alta analizada (1000 ng/ml), la relación fue cercana a cero, lo que indica cambios estructurales significativos del antígeno.
4.2 Experimento 20110913 (DP): Formulación de DP con diferentes concentraciones de iones metálicos de Ni (II), Cu (II), Cr (III)
El análisis del antígeno JEV desorbido se resume en la Tabla 14 (SEC-HPLC) y la Tabla 15 (ELISA). Las recuperaciones de antígeno para todas las muestras determinadas mediante SEC-HPLC fueron >80 % después de 5 semanas a 22 °C, lo que indica que no hay una influencia significativa de los iones metálicos sometidos a ensayo en la recuperación por desorción. Como se muestra en la Figura 8, existe una tendencia a la disminución de la relación según lo analizado mediante ELISA en presencia de Cu (II) y Cr (III) a pH 7. Las formulaciones a pH 8 parecen ser más estables.
4.3 Experimento 20110812 (DP): DP adicionado con iones metálicos
En este experimento se utilizó FBV11D87 como material de partida. El valor de pH de la formulación se ajustó en un intervalo más estrecho (pH 7,0, 7,4, 7,8) y se utilizaron iones metálicos adicionales para adicionar, cada uno a 500 ng/ml (concentración final). La mezcla de iones metálicos contenía todos los iones metálicos individuales con la excepción de
Cr (III) en formulaciones individuales (cada metal a 500 ng/ml). Los resultados de ELISA obtenidos después de 4 y 7 semanas a 22 °C se resumen en la Tabla 16. Los resultados también se muestran como gráficos en la Figura 9.
Se realizó una evaluación estadística de la estabilidad de las muestras almacenadas a 22 °C durante 7 semanas. El ANOVA (análisis de varianza) mostró efectos significativos de parámetros (pH y tipo de metal) sobre la estabilidad del antígeno, que se expresa como la relación de ELISA monoclonal/policlonal (véase la Tabla 17). La tabla ANOVA descompone la variabilidad de Relación en contribuciones debidas a diversos factores. Puesto que se han elegido sumas de cuadrados de Tipo III, la contribución de cada factor se mide retirando los efectos de todos los demás factores. Los valores P someten a ensayo la significación estadística de cada uno de los factores. Puesto que los valores P para el pH y el tipo de ion metálico son inferiores a 0,05, estos factores tienen un efecto estadísticamente significativo sobre la Relación al nivel de confianza del 95,0 %.
En la Tabla 18 se aplica un procedimiento de comparación múltiple para determinar la significancia de las diferencias observadas con respecto a las medias. Se mostró un efecto significativo sobre la relación para Cu (II) y la mezcla de metales en comparación con las formulaciones de control sin adiciones. La mitad inferior de los resultados muestra la diferencia estimada entre cada par de medias. Se ha colocado un asterisco junto a 7 pares, lo que indica que estos pares muestran diferencias estadísticamente significativas al nivel de confianza del 95,0 %. En la parte superior de la página, se identifican 3 grupos homogéneos utilizando columnas de X. Dentro de cada columna, los niveles que contienen X forman un grupo de medias dentro de las cuales no existen diferencias estadísticamente significativas. El método que se utiliza actualmente para discriminar entre las medias es el procedimiento de diferencia mínima significativa (LSD) de Fisher. Con este método, existe un riesgo del 5,0 % de llamar a cada par de medias significativamente diferente cuando la diferencia real es igual a 0.
Se mostró un efecto significativo sobre la relación para Cu (II) y la mezcla de iones metálicos. La influencia de otros iones metálicos puede volverse significativa en períodos de almacenamiento más prolongados. La mezcla de iones metálicos contenía la concentración total más alta y podría representar el peor de los casos. Sin embargo, se concluyó que varios iones metálicos presentes en el Alumbre podrían contribuir a la degradación, cada uno en diferente medida. Estos resultados respaldan adicionalmente la causa raíz propuesta de la degradación del antígeno catalizada por iones metálicos. Debe tenerse en cuenta que el experimento de adiciones podría no simular totalmente las condiciones reales de las impurezas de iones metálicos residuales presentes en Alumbre 4230. Los iones metálicos se incorporan en la estructura de Alumbre y la concentración y orientación locales pueden ser diferentes de los iones metálicos utilizados en los experimentos de adiciones. También se sabe que los iones metálicos (por ejemplo, Fe) tienen una baja solubilidad en presencia de iones fosfato (PO43-). Por lo tanto, se desconoce la concentración real de iones metálicos solubles y la contribución de los metales presentes como complejo metal-fosfato en la degradación de JEV.
4.4 Experimento 20110819: Preparación de muestras de DP con diferentes lotes de Alumbre
Los estudios de adiciones como se ha descrito anteriormente pueden proporcionar una primera evidencia de la posible inestabilidad del antígeno formulado en presencia de algunos iones metálicos, pero pueden no ser completamente representativos de las condiciones reales en las que se espera que estos iones metálicos presentes en el Hidróxido de aluminio se incorporen en la estructura tridimensional del gel de Hidróxido de aluminio dando como resultado diferentes concentraciones locales y orientación/accesibilidad. Para superar estas limitaciones se comenzó un estudio inicial para simular las condiciones reales. Se formuló un único NIV (11A74) con diversos lotes de Alumbre obtenidos de Brenntag que cubren una amplia gama de metales residuales. Las muestras de vacunas formuladas se almacenaron a 2-8 °C y en condiciones aceleradas a 22 °C. Todos estos lotes de Alumbre contenían iones metálicos residuales en diferentes niveles. El lote 4230 tuvo el nivel más alto de Fe, Cu, Ni y V (véase la Tabla 19). Téngase en cuenta que el contenido real de iones metálicos presentes en el producto formulado es solo 1/20 de la concentración en la solución madre de Alumbre (2 %). Téngase en cuenta que ICP-MS no puede especificar las valencias de los iones metálicos. El análisis del antígeno desorbido mediante ELISA de muestras almacenadas a 22 °C durante 6 semanas se muestra en la Tabla 20.
La desviación estándar agrupada se calculó a partir de todas las muestras (agrupada ~ 0,075) como medida de la incertidumbre experimental. Los valores medios para la relación y los intervalos de confianza del 95 % (calculados basándose en agrupada) se representaron frente a las formulaciones individuales (véase la Figura 10). Las muestras formuladas con Alumbre 4230 mostraron una tendencia a una relación más baja en comparación con las otras muestras. Sin embargo, las diferencias no fueron tan grandes como para mostrar diferencias estadísticamente significativas entre las diversas formulaciones.
5 Sumario
Se demostró que los iones metálicos contribuyen a la degradación del JEV inactivado en condiciones de almacenamiento acelerado (22°). En los estudios de adición, se utilizó una concentración más alta de iones metálicos residuales (intervalo 100-1000 ng/ml) que la presente en FVL formulada con lote 4230 de Alumbre (por ejemplo, Fe~310 ng/ml; Cr~64 ng/ml; Ni~52 ng/ml). El contenido de Cu en FVL solo se puede estimar en 3 ng/ml basándose en los datos de ICP-MS de una solución madre de Alumbre al 2 %, puesto que el LDD es de 25 ng/ml. Se eligieron concentraciones de metal y temperaturas de almacenamiento más altas para aumentar la velocidad de cualquier posible reacción de degradación. De hecho, para FVL JEV09L37, la pérdida de potencia ocurrió después
de 11 meses de almacenamiento a 2-8 °C. También se demostró que los metales pueden formar complejos insolubles con iones de fosfato, lo que dificulta la estimación de los niveles reales de metales presentes.
En los experimentos de adición, los resultados de ELISA mostraron cambios estructurales estadísticamente significativos en la superficie del virus en tan solo 4 semanas a 22 °C en presencia de iones metálicos. Se demostró que las relaciones de ELISA para formulaciones que contenían Cu (II) y mezcla de iones metálicos (que contenían Fe (II), Fe (III), Co (II), Cu (II) y Zn (II)) fueron estadísticamente significativamente más bajas en comparación con la formulación de control sin adiciones. También se encontró Cu (II) en el lote 4230 de Alumbre (solución madre al 2 %) a 64 ng/ml, lo que corresponde a ~3 ng/ml en FVL. En todos los demás lotes de Alumbre (2 %), el contenido de Cu (II) fue < 25 ng/ml (por debajo del límite de detección). Para experimentos de formulación del antígeno utilizando diferentes lotes de Alumbre, se requiere un tiempo de almacenamiento más prolongado (>6 semanas a 22 °C) en condiciones aceleradas. Existe una tendencia de que las formulaciones preparadas con Alumbre 4230 mostraron relaciones de ELISA más bajas en comparación con otros lotes. La tasa de degradación más lenta en comparación con la formulación adicionada podría estar contribuyendo a un menor contenido de iones metálicos en los lotes comerciales de Alumbre. También se observó que el antígeno muestra una mayor estabilidad a pH 7,5-8 en comparación con pH 7 y que los fragmentos de PS no contribuyen a ninguna reacción de degradación. Estos resultados concuerdan bien con los resultados del DOE descritos en el Ejemplo 1.
Ejemplo 3
Como parte de la investigación fuera de especificación relativa a FVL JEV09L37, se determinó que el lote de Hidróxido de aluminio utilizado (lote 4230) era la causa raíz más probable de la pérdida de potencia observada. El hidróxido de aluminio (denominado Alumbre durante el proceso de fabricación de JE-PIV) se adquiere en Brenntag Biosector en forma de una suspensión esterilizada en autoclave denominada “Adyuvante de gel de Hidróxido de aluminio Alhydrogel®” . Cada lote se esteriliza por radiación antes de su uso en el proceso de producción de JEV. Se analizó el aspecto, el contenido de iones metálicos y las propiedades físicas de varios lotes diferentes de Alhydrogel®.
1. Introducción
1.1 Hidróxido de aluminio
Alhydrogel® de Brenntag Biosector tiene un contenido de aluminio específico de 10 mg/ml, lo que se traduce en Al2O3 al 2 % y Al(OH)3 al 3 %, respectivamente. Otras especificaciones son nitrógeno (máx. 0,005 %), sulfato libre (máx. 0,05 %), sulfato total (máx. 0,1 %) y pH (6,5 ± 0,5). Tiene una vida útil de 26 meses cuando se almacena a temperatura ambiente.
1.2 Generación de Hidróxido de Aluminio
Alhydrogel® 2 % (denominado Hidróxido de aluminio) es fabricado por Brenntag (n.° de CAS 21645-51-2).
1.3 Uso de lotes de hidróxido de aluminio en la fabricación de JEV
Para la producción de lotes comerciales de vacunas contra el JEV, hasta ahora se han utilizado un total de 5 lotes diferentes de Brenntag Alhydrogel® 2 %.
2 Definiciones y abreviaturas
Solución de hidróxido de aluminio Alhydrogel 2 % (también denominada Alumbre)
DS/DP Principio activo/Medicamento
ESG Grupo de científicos ambientales
F-AAS Espectrometría de absorción atómica de llama
FVL Lote de Vacuna Final
GF-AAS Espectrometría de absorción atómica en horno de grafito
ICP-MS Plasma Acoplado Inductivamente-Espectrometría de Masas
JEV Virus de la encefalitis japonesa
JE-PIV Virus inactivado purificado de la encefalitis japonesa
LDC Límite de cuantificación
P&TD Parche y desarrollo técnico
PSD Distribución de tamaño de partícula
PZC Punto de carga cero
QCI Inmunología de control de calidad
3 Materiales y Métodos
3.1 Lotes de Alhydrogel®
Lotes de Alhydrogel® 2 %: 3877, 4074, 4187, 4230, 4414, 4470, 4539, 4563, 4587, 4621 (no todos los lotes de Alumbre enumerados se usaron en la formulación de JE-PIV) lotes de Alhydrogel® 2 % lavados 7 veces: 4577, 4580, 4596 (procedente de Brenntag, no típico del Alumbre al 2 % recibido para la formulación)
3.2 Mediciones de PSD de Alhydrogel®
La distribución de tamaño de partícula (PSD) de hidróxido de aluminio se analizó en un sistema Malvern Mastersizer 2000 |jP con una celda de muestra de 20 ml. La sustancia a granel Alhydrogel® 2 % se diluyó 1:20 en agua y se añadió 1 ml a la celda de muestra. Por lo tanto, la dilución final de la muestra en la celda de muestra fue de 400 veces (hidróxido de aluminio al 0,005 %).
3.3 Mediciones del potencial zeta de Alhydrogel®
El potencial zeta y el punto de carga cero (PZC) se midieron en un sistema Malvern Zetasizer ZS equipado con un autotitulador MPT-2. La sustancia a granel Alhydrogel® 2 % se diluyó 1:20 en PBS y se equilibró durante la noche a temperatura ambiente. Para registrar la curva de titulación de la carga, se ajustó el pH utilizando soluciones de HCl 100 mM y NaOH 100 mM. El PZC se determinó por extrapolación de la carga cero en el gráfico de titulación (intersección de la curva de titulación y el eje x). El punto de carga cero corresponde al valor de pH donde la superficie de la muestra no tiene carga neta.
3.4 Análisis de iones metálicos en hidróxido de aluminio
Los iones metálicos seleccionados se analizaron mediante plasma acoplado inductivamente-espectrometría de masas (ICP-MS), espectrometría de absorción atómica de llama (F-AAS) y espectrometría de absorción atómica en horno de grafito (GF-AAS) en el Medical Laboratory Bremen (Alemania). En resumen, las muestras que contenían Hidróxido de aluminio se trataron con HNO3 conc. con calor hasta obtener una solución transparente. Después, la solución transparente se diluye y analiza adicionalmente. Se determinó la presencia y el contenido de los siguientes iones metálicos: Pb, Cd, Cr, Co, Fe, Cu, Ni, Ag, W y Al. Dependiendo de la dilución de la muestra, el límite de cuantificación (LDC) fue de 5 a 25 ng/ml.
Además, ESG (Reino Unido) realizó un barrido semicuantitativo de 70 elementos utilizando una combinación de ICP-MS (Agilent 7500ce) e ICP-AES (Perkin Elmer Optima 4300DV), que se calibraron utilizando patrones certificados. El barrido de elementos es un método de cribado y no tan sensible como el análisis de trazas de metales para metales seleccionados como lo realiza Medical Laboratory Bremen. Sin embargo, dicho cribado proporciona una buena visión de conjunto sobre la presencia y los niveles de determinados metales.
4 Resultados
4.1 Determinación de la distribución de tamaño de partícula de Alhydrogel®
La Tabla 21 resume los datos de PSD de dos sublotes de cada uno de los lotes 4230 y 4740 de Alhydrogel®. Los resultados de la distribución se muestran en la Figura 11. La partícula media fue de ~2-4 jm con poblaciones de partículas más pequeñas (<1 jm ) y más grandes (>20 jm ) presentes en las cuatro muestras. Las cuatro muestras de Alhydrogel® sometidas a ensayo no mostraron diferencias significativas en la distribución de tamaño de partícula medio.
4.2 Mediciones de potencial zeta
Se analizaron dos sublotes de cada uno de los lotes 4230 y 4740 de Alhydrogel® (solución madre al 2 % diluida 20 veces en PBS y equilibrada durante la noche a TA antes del análisis) para determinar el punto de carga cero. La Tabla 22 resume los resultados de PZC para las cuatro muestras que muestran PZC muy similar en tampón de PBS. Las curvas de titulación se muestran en la Figura 12. No se pudo observar ninguna diferencia en las curvas de titulación y PZC entre las cuatro muestras analizadas.
4.3 Determinación del contenido de iones metálicos residuales en lotes de Alhydrogel®
Los límites actuales para Fe en soluciones de hidróxido de aluminio al 2 % según la Ph. Eur. son 15 ppm (= 15 pg/ml) y un máximo total de 20 ppm (= 20 pg/ml) para otros metales pesados (tales como Pb). Sin embargo, una concentración de 15 ppm de Fe correspondería a Fe 0,27 mM en solución. Teniendo en cuenta que incluso pequeñas cantidades de iones metálicos residuales pueden catalizar una diversidad de reacciones de degradación de proteínas (por ejemplo, oxidación y activación de proteasas) y que los metales permanecen estables en solución, las diferencias en el contenido de iones metálicos entre lotes de hidróxido de aluminio podrían provocar diferencias en la estabilidad del antígeno en el tiempo.
Las concentraciones de varios iones metálicos en lotes de hidróxido de aluminio disponibles en el mercado se analizaron utilizando ICP-MS. Los resultados de estos análisis se resumen en la Tabla 23. Se utilizaron los lotes 4074, 4230, 4470, 4414 y 4539 en la producción de lotes de JEV comerciales. Como una solución madre de Alhydrogel® 2 % equivale a una concentración de Al de 10 mg/ml, la cuantificación del contenido de Al en las diferentes muestras puede utilizarse como referencia para los resultados obtenidos para los otros iones metálicos. De hecho, se pudo medir un contenido promedio de Aluminio de 10,3 mg/ml, lo que demuestra la precisión y la reproducibilidad del método.
Cuando se compararon los diferentes lotes de Alhydrogel® se observaron grandes variaciones en la cantidad de iones metálicos contaminantes. Los iones metálicos contaminantes más notables son Fe, Cr y Ni, que se detectaron en todos los lotes. Además, el lote 4230 contenía cantidades detectables de Cu que estaban por debajo del LDC en todos los demás lotes.
Sin embargo, debe tenerse en cuenta que ninguno de estos metales se detectó en cantidades cercanas a las especificaciones de Alhydrogel® mencionadas anteriormente. Por ejemplo, la concentración más alta de hierro encontrada en el lote 4230 fue de 5,6 pg/ml o aproximadamente el 40 % de la concentración permitida.
Un Alhydrogel® “ mejorado” se lava 7 veces con agua durante la etapa de purificación en lugar de solo 4 veces para el Alhydrogel® convencional. Para someter a ensayo si estas etapas de lavado adicionales darían como resultado una contaminación de iones metálicos reducida, se analizaron tres lotes diferentes (4580, 4596 y 4577). Los resultados se incluyen en la Tabla 23. No se pudo observar ninguna diferencia en los iones metálicos en comparación con el Alhydrogel® de grado convencional, lo que sugiere que o bien los iones metálicos están fuertemente unidos a la superficie de las partículas de Hidróxido de aluminio o en realidad coprecipitan durante el proceso de producción.
La Figura 13 muestra una comparación de los diferentes lotes de Alhydrogel® analizados. El contenido total de iones metálicos contaminantes para todos los elementos contaminantes sometidos a ensayo se muestra con las proporciones absolutas de los tres metales principales Fe, Cr y Ni representados en diferentes colores. Como puede observarse, el lote 4074 tiene muy pocos iones metálicos contaminantes en comparación con la mayoría de los otros lotes analizados. Solo el lote 3877 mostró una contaminación de lote similar, mientras que el lote 4230 muestra, con mucho, la contaminación más alta de todos los lotes analizados durante esta investigación.
Durante los ensayos de investigación, se observó una gran variación en el contenido de iones metálicos entre diferentes lotes de Alhydrogel® (véase la Figura 13). Para someter a ensayo si estos iones metálicos contaminantes están ubicados en la fracción de Hidróxido de aluminio o en el sobrenadante, el Lote 4230 se separó en una fracción sobrenadante y otra de sedimento (véase la Tabla 24). Como puede observarse, se pudo detectar menos del 2 % de los iones metálicos en el sobrenadante, lo que indica que todos los iones metálicos contaminantes están unidos a la superficie de la partícula de Hidróxido de aluminio o dentro de las estructuras de la partícula. Por lo tanto, se puede estimar que la concentración local de iones metálicos es al menos 50-100 veces mayor, puesto que la fracción de volumen sólido (volumen de gránulos de Alumbre después de la centrifugación) de Al2O3 al 0,1 % (que corresponde a 0,5 mg/ml de Al) utilizada en la formulación de vacuna contra el JEV es aprox. 10-20 pl por 1000 pl de FVL.
5 Sumario
Alhydrogel® se utiliza en una concentración final del 0,1 % como adyuvante en la formulación actual de la vacuna contra el JEV. Durante la investigación de un resultado de potencia fuera de especificación (OOS, del inglés out-ofspecification) para la producción de FVL JEV09L37, se inició una evaluación del proceso de producción de Alhydrogel®. Se analizó un total de 13 lotes diferentes de Alhydrogel® para detectar la presencia de iones metálicos contaminantes que podrían reducir la estabilidad proteínica.
Se observaron grandes variaciones en la concentración de varios iones metálicos para diferentes lotes de Alhydrogel®. Cuando se analizaron las materias primas, se demostró que estas contaminaciones estaban presentes en la misma concentración que se encuentran dentro del Alhydrogel.
Se observaron niveles mayores de iones Fe, Ni y Cu en el lote 4230 de Alhydrogel® en comparación con los otros lotes investigados. El lote 4230 fue el único en el que se detectaron iones de Cu residuales. Este lote 4230 se utilizó para la formulación de FVL JEV09L37.
Cuando se analizó el sobrenadante y la fracción insoluble de un lote de Alhydrogel®, estos iones metálicos contaminantes solo se pudieron encontrar en el precipitado, lo que indica que estos iones están unidos a la superficie de la partícula de Hidróxido de aluminio o, en realidad, son parte de la partícula.
Aunque macroscópicamente y en composición diferente de otros lotes de Alhydrogel® utilizados para la producción de JEV, el lote 4230 cumplió con todos los requisitos detallados por la Ph. Eur. Además, la caracterización física (distribución de tamaño de partícula y punto de carga cero) no mostró diferencias entre el lote 4230 y otros lotes de Alhydrogel® que no mostraban estas contaminaciones altas de iones metálicos.
Ejemplo 4
1.1. Materiales, Equipo y Métodos
1.2. Equipo
Balanza analítica (legibilidad de 0,1 mg; por ejemplo, Mettler Toledo XP205DR/M)
Balanza de precisión (legibilidad de 0,1 g; por ejemplo, Mettler Toledo, Modelo N° XS6002S Gama Delta) Unidades de filtro de 0,22 pm (por ejemplo, Stericup N.° de Cat. SCGPV01RE) o sistema de filtro de 0,2 pm (por ejemplo, Steriflip Millipore de 50 ml)
Congelador (-20 °C) y Ultracongelador (-80 °C)
Frigorífico (+2 a 8 °C)
Agitador magnético (por ejemplo, KIKA Labortechnik RCT basic) y barras de agitación magnéticas
Lavadora de microplacas: por ejemplo, BioTek ELx405
Lector de microplacas: por ejemplo, BioTek Synergy 2 y software Gen5 Secure
Incubadora de microplacas (37 °C)
Cinta de sellado de microtitulación (por ejemplo, Thermo Electron 9503130)
Pipetas y puntas multicanal (por ejemplo, Eppendorf Research Pro 50-1200 pl,
Eppendorf Research, 10-100 pl)
Medidor de pH (por ejemplo, Serie WTW ino Lab, Terminal 740 y pH/Cond. 740)
Pipetas y puntas (por ejemplo, Eppendorf Research, 0,5-10 pl, 2-20 pl, 20-200 pl, 100-1000 pl, 500-5000 pl) Micropipeta (por ejemplo, IBS Biosciences Pipetboy)
Tubos de PP de 15 ml (por ejemplo, Sarstedt 62.515.006) o tubos de PP de 50 ml (por ejemplo, Greiner 227261) Depósito de reactivos de 50 ml (por ejemplo, Corning Incorporated 4870)
Pipetas serológicas (por ejemplo, Falcon, 2 ml, 5 ml, 10 ml, 25 ml, 50 ml)
Microtubos Titertube: a granel (BioRad 223-9391)
Mezclador de vórtice (por ejemplo, mezclador VWR Analog Vortex Mixer, modelo N.° 945304)
Tubos Eppendorf LoBind de 1,5 ml o 2,0 ml (N.° de Cat. 0030 108.116, N.° de Cat 0030 108.132, respectivamente) Microplaca de 96 pocillos (F96 Cert. Maxisorp Nunc-Immunoplates)
Para el análisis de muestras de DP además:
Centrífuga de sobremesa (por ejemplo, Beckman Coulter, Microfuge 16 Centrifuge, N.° de Cat. A46473) Agitador orbital (por ejemplo, Eppendorfer Thermomixer compact)
Tubos de PP de 50 ml (por ejemplo, Greiner 227261)
1.3. Reactivos
PBS 10x (por ejemplo, Gibco, N.° de Cat. 14200-083)
Tween 20 (por ejemplo, Sigma N.° de Cat. P7949)
Ácido sulfúrico 2 M (solución volumétrica, por ejemplo, Fisher, N.° de Cat. J/8410/17) Agua desionizada, por ejemplo, (Milli-Q, 18,2 O)
Carbonato de sodio - cápsulas de bicarbonato (por ejemplo, Sigma, N.° de Cat. C3041) Ácido clorhídrico (HCl) 1 mol/l (por ejemplo, Merck, N.° de Cat. 1.09057.1000)
Hidróxido de sodio (NaOH) 1 mol/l (por ejemplo, Merck, N.° de Cat. 1.09132.1000) Glicerol (por ejemplo, Sigma) Para el análisis de muestras de DP además:
Trihidrato de fosfato de hidrógeno y potasio (por ejemplo, Sigma, N.° de Cat. P5504) Dihidrogenofosfato de potasio (por ejemplo, VWR, AnalaR Normapur, N.° de Cat. 26936.260)
Albúmina, suero bovino (BSA), grado ELISA (por ejemplo, Sigma, N.° de Cat. A3059) Sustrato TMB (por ejemplo, BioFX, TMBW-1000-01)
Conjugado de HRP de burro anti IgG de conejo (Jackson Immuno Research, N.° de Cat. 711-035-152) Reconstitución:
El contenido de 1 vial (0,4 mg) se reconstituye en 0,5 ml de agua desionizada y se mezcla bien hasta la disolución total. Añadir 0.5 ml de Glicerol y mezclar más hasta homogeneidad. Las alícuotas se almacenan a -20 °C hasta su uso.
Patrón de referencia de JEV inactivado (Intercell Biomedical Ltd.)
Anti-JEV de oveja purificado (Intercell Biomedical Ltd.)
Anti-JEV de conejo purificado (Intercell Biomedical Ltd.)
1.4. Soluciones
a) Tampón de carbonato 0,05 M a pH 9,6 (utilizado para el recubrimiento de placas de ELISA) Para 100 ml de tampón, disolver una cápsula de tampón bicarbonato/carbonato en 100 ml de agua desionizada. Comprobar el pH y ajustar a 9,6 ± 0,1 con HCl o NaOH si es necesario. Usar únicamente el día de la preparación. Mantener el tampón de recubrimiento de ELISA a TA durante el día de uso y después desechar.
b) Tampón de lavado de ELISA y parte del bloque / diluyente de muestra (PBS-T)
Preparar aproximadamente 1 litro por cada placa utilizada. Diluir solución madre de PBS 10x 1+9 en agua desionizada, mezclar bien y verificar el pH (7,4 /- 0,1), ajustar con HCl 1 M o NaOH 1 M según sea necesario. Añadir Tween20 al 0,05 % (v/v), mezclar bien.
por ejemplo, tampón de lavado de ELISA (PBS-T) [1 l]:
100 ml PBS 10x
900 ml agua desionizada
Mezclar bien, controlar/ajustar el pH (7,4 /- 0,1).
0,5 ml de Tween20
Mezclar bien.
Usar únicamente el día de la preparación; mantener el tampón de lavado de ELISA a TA durante el día de uso y después desechar.
c) Solución de bloqueo: BSA al 5 % en PBS-T
Preparar aproximadamente 25 ml para cada placa. Medir la cantidad requerida de PBS-T en un frasco de vidrio limpio utilizando una pipeta serológica. Añadir una barra de agitación magnética limpia. Pesar la cantidad requerida de BSA, añadir a la superficie del PBS-T y mezclar suavemente con un agitador magnético hasta que todo el BSA se haya disuelto. Filtrar la solución con un filtro de 0,2 pm (ya sea un sistema de filtro Steriflip o un filtro de jeringa). por ejemplo, solución de bloqueo [100 ml]
5 g BSA
100 ml PBS-T
Usar únicamente el día de la preparación; mantener la solución de bloqueo a temperatura ambiente durante el día de uso y después desechar.
d) Diluyente de la muestra: BSA al 1 % en PBS-T
Preparar como anteriormente, pero con 1 g de BSA por 100 ml de PBS-T, se requieren aproximadamente 25 ml por placa. por ejemplo, diluyente de muestra [100 ml]
1 g BSA
100 ml PBS-T
Usar únicamente el día de la preparación; mantener el diluyente de muestra a temperatura ambiente durante el día de uso y después desechar.
Para el análisis de muestras de DP además:
e) PBS 1x
Preparar 1 parte de PBS 10x con 9 partes de agua desionizada
por ejemplo, PBS 1x [100 ml]
10 ml de PBS 10x
90 ml de agua desionizada
Usar únicamente el día de la preparación; mantener PBS 1x a TA durante el día de uso, después desechar.
f) Tampón de ELISA 20x
Pesar una cantidad adecuada de BSA en un recipiente adecuado para preparar una solución 20x. Añadir el volumen apropiado de PBS 1x. Añadir Tween20 a una concentración final del 0,05 % Mezclar en el agitador magnético hasta que la BSA se disuelva totalmente. Filtrar la solución a través de un filtro de 0,2 pm (utilizando un sistema de filtro Steriflip o un filtro de jeringa) en un recipiente estéril (y dividir en alícuotas según sea necesario).
por ejemplo, tampón de ELISA 20x [25 ml]
5 g de BSA
25 ml de PBS 1x
12,5 pl de Tween20
La solución se puede almacenar a 2-8 °C durante 1 semana.
g) El tampón de ELISA 2x se prepara mediante dilución del Tampón de ELISA 20x con PBS 1x (1 parte de Tampón de ELISA 20x y 9 partes de Pb S 1x).
por ejemplo, Tampón de ELISA 2x [20 ml]
Tampón de ELISA 20x de 2 ml
18 ml de PBS 1x
Usar únicamente el día de la preparación; mantener el tampón de ELISA 2x a TA durante el día de uso y después desechar. h) Tampón de desorción
o Solución madre de fosfato de potasio: Preparar una solución madre 3x de fosfato de potasio (2,4 M) disolviendo el volumen apropiado de trihidrato de fosfato dipotásico y de dihidrogenofosfato de potasio en agua desionizada. Colocar en un agitador magnético y una vez disuelto completar el volumen requerido, verificar que el pH de la solución sea de 8,0 /- 0,1. Filtrar a través de un filtro de 0,2 pm.
por ejemplo, solución madre 3x de Fosfato de potasio (2,4 M) [50 ml]
23,963 g Trihidrato de fosfato dipotásico
2,041 g Dihidrogenofosfato de potasio
Completar hasta 50 ml Agua desionizada
Almacenar a 2°-8 °C durante hasta 1 mes.
o Preparar un tampón de desorción de fuerza de trabajo (tampón de fosfato de potasio 0,8 M que contiene BSA al 1 % y Tween20 al 0,05 %) añadiendo el volumen apropiado de solución madre de fosfato de potasio (2,4 M), de Tween20 y de BSA al volumen requerido de agua desionizada. Mezclar bien y usar el día de la preparación. por ejemplo, tampón de desorción de fuerza de trabajo [15 ml]
5 ml Fosfato de potasio (2,4 M)
7,5 pl Tween20
0,15 g BSA
10 ml Agua desionizada
Mantener el tampón de desorción de fuerza de trabajo a TA durante el día de uso.
1.5. Muestras de ensayo y anticuerpos
Muestras de ensayo:
o Principio activo y/o NIV (diversos lotes)
o Muestras de vacuna contra el JEV (vacuna final a granel y lote de vacuna final) Patrón de referencia de JEV inactivado (virus inactivado neutralizado - NIV) (Intercell Biomedical Ltd.)
Anticuerpos policlonales:
o Anticuerpo de recubrimiento: Anti-JEV de oveja purificado (Intercell Biomedical Ltd.)
o Anticuerpo de detección primaria: Anti-JEV de conejo purificado (Intercell Biomedical Ltd.)
Anticuerpo conjugado secundario: Conjugado de HRP de burro anti-conejo (Jackson Immuno Research N.° de Cat.
711-035-152)
2 Procedimiento
2.1. Recubrimiento de placas
o Etiquetar la placa con el número de placa, la fecha y el analista.
o Preparar un tampón de carbonato 0,05 M nuevo (pH 9,6) el día del recubrimiento de placas. Permitir aproximadamente 12 ml para cada placa recubierta.
o Retirar el número requerido de alícuotas del anticuerpo de recubrimiento del congelador y dejar que se descongele a TA. Preparar una dilución de anticuerpo anti-JEV de oveja purificado en tampón de carbonato. Mezclar bien por inversión del tubo.
o Usando la pipeta multicanal, aplicar 100 pl/pocillo a una placa Maxisorp de 96 pocillos a los 15 min de la preparación de la dilución de anticuerpo.
o Cubrir con cinta de sellado de microtitulación e incubar de 17 a 72 horas a 2-8 °C.
2.2. Lavado
o Retirar la placa del refrigerador y dejar que se caliente a temperatura ambiente.
o Lavar la placa o placas con el lavador de placas Microtiter 3 veces utilizando el programa de lavado respectivo (300 pl por pocillo, tres veces, dispensación final). Después de eso: retirar cualquier resto de tampón de lavado por decantación. Invertir la placa y golpearla contra una toalla de papel limpia. No permitir que la placa de microtitulación se seque entre las etapas de lavado y la adición del reactivo.
2.3. Bloqueo
o Preparar una solución de bloqueo al 5 % (p/v) de BSA en PBS-T como anteriormente.
o Aplicar 200 pl de solución de bloqueo por pocillo, cubrir la placa o placas con una placa de cubierta e incubar a 37 °C durante 1 hora /- 10 min.
2.4. Preparación de diluciones de curva patrón
o Retirar el patrón de referencia de NIV del congelador, dejar que se descongele a TA y mezclar bien. Preparar una dilución madre de 1 UA/ml del patrón de referencia actual; utilizar al menos 20 pl de patrón de referencia de NIV para la dilución.
por ejemplo, patrón de referencia de NIV Predilución:
Concentración: 235 UA/ml (lote N.° 03/2009)
Para preparar una solución patrón de trabajo de 1 UA/ml, diluirla de 1 a 235 en diluyente de muestra:
4680 pl diluyente de muestra
20 pl patrón de referencia de NIV
o Después, preparar las siguientes soluciones patrón de trabajo a partir de la predilución de 1 UA/ml:
0,8 UA/ml, 0,6 UA/ml, 0,4 UA/ml, 0,2 UA/ml, 0,1 UA/ml y 0,05 UA/ml en diluyente de muestra.
2.5. Muestras de control de calidad
a) Las muestras de control de calidad (QC) (por ejemplo, a 0,75, 0,30 y 0,18 UA/ml) se deben preparar a partir de la predilución del patrón de referencia de NIV nuevo en el momento del ensayo y después se desechan una vez utilizadas.
b) Estos controles son parte de los criterios de idoneidad del sistema y permiten controlar el rendimiento del ensayo a lo largo del tiempo.
2.6. Preparación de muestras de ensayo
Preparación de principio activo
Se reciben muestras de ensayo de principios activos para realizar ensayos en concentraciones desconocidas. Éstas se someterán a ensayo en seis diluciones por triplicado. Las diluciones se realizarán independientemente dentro del intervalo de la curva patrón, por ejemplo, una dilución previa de 1 en 15 u otra dilución adecuada y después seis diluciones con tampón de muestra.
Preparación de muestras de NIV
Las muestras de NIV se recibirán para su análisis en concentraciones desconocidas y se prediluirán en el intervalo de la curva patrón (por ejemplo, 1 en 30 u otra dilución adecuada) y después se diluirán seis veces de la misma manera que las muestras de DS (principio activo, del inglés drug substance).
Preparación de sobrenadante de medicamento
a) Para el análisis de muestras de DP (medicamento, del inglés drug product) a granel mezclar bien la muestra mediante agitación con formación de vórtice. Transferir exactamente 1 ml a un tubo Eppendorf LoBind de 1,5 ml.
Para el análisis de muestras de recipientes de productos finales, transferir el contenido de 2 jeringas (0,6 ml por jeringa) del mismo lote a un tubo Eppendorf LoBind de 1,5 ml. Mezclar bien el contenido del tubo por inversión para garantizar la homogeneidad del DP y transferir exactamente 1 ml a un tubo Eppendorf LoBind de 1,5 ml nuevo. b) Centrifugar tubos que contengan 1 ml de DP cada uno a 3300xg durante 5 minutos.
c) Para cada muestra, pipetear 25 pl de tampón de ELISA 20x en un tubo Eppendorf LoBind nuevo.
d) Retirar con cuidado 475 pl del sobrenadante sin alterar el sedimento de alumbre y transferirlo al tubo que contiene el tampón de ELISA 20x. Mezclar suavemente por inversión. Volver a centrifugar 2 min a 16.000xg. Almacenar la muestra a 2-8 °C antes del análisis.
NOTA: Las muestras de sobrenadante de DP preparadas de esta manera deben medirse puras por triplicado en el ELISA de JEV inactivado.
e) Retirar con cuidado la mayor cantidad posible de sobrenadante residual del tubo centrifugado utilizando una pipeta de 10-200 pl sin alterar el sedimento de alumbre y desechar el sobrenadante.
f) El sedimento obtenido se somete al procedimiento de Desorción como se describe a continuación.
Procedimiento de desorción del medicamento
a) Añadir 158 pl de tampón de desorción de fuerza de trabajo a cada sedimento que quede en el tubo LoBind. b) Volver a suspender el sedimento pipeteando hacia arriba y hacia abajo varias veces para garantizar la resuspensión completa del sedimento y la homogeneización de la muestra.
c) Incubar las muestras durante 10 min a TA en un agitador orbital a 500 rpm.
d) Después de la incubación, centrifugar las muestras a 3300xg durante 5 minutos.
e) Para cada muestra, pipetear 250 pl de tampón de ELISA 2x en un tubo Eppendorf LoBind nuevo.
f) Retirar con cuidado 83,3 pl de la parte superior del sobrenadante que contiene el producto desorbido sin alterar el precipitado y transferirla al tubo que contiene el tampón de ELISA 2x. Retirar el sobrenadante restante utilizando una pipeta de 20-200 pl sin alterar el precipitado y desechar el sobrenadante.
g) Añadir otros 158 pl de tampón de desorción de fuerza de trabajo a cada sedimento.
h) Realizar 2 ciclos de desorción más (3 en total) agrupando los 3x 83,3 pl del material desorbido 250 pl de tampón de ELISA en el tubo apropiado. Después de la última etapa, el sedimento restante se puede desechar. i) La concentración final del antígeno vírico en la mezcla o mezclas desorbidas debe ser ahora la misma que el 1 ml original de DP del que se desorbió. Por lo tanto, la concentración del contenido de antígeno de JEV inactivado medido en el grupo desorbido puede estar directamente relacionada con la del DP original.
Nota: Analizar las muestras desorbidas mediante ELISA el día de la desorción.
j) Dilución de muestras de DP desorbidas:
Éstas se someterán a ensayo en seis diluciones por triplicado. Se realizará una predilución apropiada en el intervalo de la curva patrón, por ejemplo, 1 en 15 (100 pl a 1400 pl de diluyente) u otra dilución adecuada, después se realizarán seis diluciones de 1 en 15 independientemente utilizando diluyente de muestra.
2.7. Carga de muestra y plan de placa
Preparar muestras y patrones antes del análisis.
Después de bloquear, lavar la placa utilizando el lavador de placas empleando el programa de ELISA de JEV. Después de eso, retirar cualquier tampón de lavado restante por decantación. Invertir la placa y golpearla contra una toalla de papel limpia. No permitir que la placa de microtitulación se seque entre las etapas de lavado y la adición del reactivo.
Añadir 100 pl/pocillo de patrones/controles/muestras y cubrir con una placa de cubierta e incubar durante 1 hora /- 10 min a 37 °C.
Añadir 100 pl de diluyente de muestra a todos los pocillos que no sean necesarios para el ensayo.
2.8. Preparación del anticuerpo primario
Retirar la cantidad requerida de alícuotas del anticuerpo primario del congelador y dejar que se descongele a TA. Preparar máximo 15 min antes de su uso anticuerpo anti-JEV de Conejo en diluyente de muestra a una dilución adecuada. Después de la incubación de la muestra, lavar la placa utilizando el lavador de placas empleando el programa de ELISA de JEV. Después de eso, retirar cualquier tampón de lavado restante por decantación. Invertir la placa y golpearla contra una toalla de papel limpia. No permitir que la placa de microtitulación se seque entre las etapas de lavado y la adición del reactivo. Añadir 100 pl/pocillo de anticuerpo primario diluido, cubrir con una placa de cubierta e incubar durante 1 hora /-10 min a 37 °C.
2.9. Preparación de conjugado de anticuerpo secundario
Retirar la cantidad requerida de alícuotas del conjugado de anticuerpo secundario del congelador y dejar que se descongele a TA. Preparar máximo 15 min antes de su uso una dilución de anticuerpo de Burro anti-HRP de Conejo en diluyente de muestra; por ejemplo, para una dilución de 1 en 10.000 para preparar una predilución de 1 en 100 y después preparar una segunda dilución de 1 en 100.
Después de la incubación del anticuerpo primero, lavar la placa utilizando el lavador de placas empleando el programa de ELISA de JEV. Después de eso, retirar cualquier tampón de lavado restante por decantación. Invertir la placa y golpearla contra una toalla de papel limpia. No permitir que la placa de microtitulación se seque entre las etapas de lavado y la adición del reactivo. Añadir 100 pl/pocillo de conjugado de anticuerpo secundario diluido, cubrir con una placa de cubierta e incubar durante 1 hora /- 10 min a 37 °C.
2.10. Incubación de sustrato
Cuando se haya añadido el conjugado, retirar el TMB del refrigerador a 2-8 °C. Pipetear el volumen requerido (12 ml de TMB por placa) en un tubo de centrífuga de 50 ml, utilizando una pipeta serológica. Dejar que el TMB alcance la temperatura ambiente en la oscuridad. Después de la incubación del conjugado, lavar la placa 3 veces con el lavador de placas empleando el programa de ELISA de JEV. Después de eso: retirar cualquier resto de tampón de lavado por decantación. Invertir la placa y golpearla contra una toalla de papel limpia. No permitir que la placa de microtitulación se seque entre las etapas de lavado y la adición del reactivo. Añadir 100 pl/pocillo de TMB y revelar la placa en la oscuridad a temperatura ambiente durante 10 minutos.
2.11. Detención y lectura
Después de 10 minutos de incubación con TMB, detener el revelado añadiendo 100 pl/pocillo de ácido sulfúrico 2 M. Leer la placa a 450 nm (filtro de referencia 630 nm) dentro de los 10 minutos posteriores a la detención utilizando el lector BioTek y el software Gen5 Secure.
2.12. Análisis de datos
Análisis de datos de NIV/DS:
El software Gen5Secure se utilizará para calcular el % de recuperación de los QC, concentración x diluciones, concentración media de las muestras corregidas por diluciones y este valor multiplicado por 1,05 para corregir la adición de tampón de ELISA.
Análisis de datos de DP:
El software Gen5Secure se utilizará para calcular el % de recuperación de los QC, la concentración x diluciones y la concentración media de las diluciones de las muestras.
Para muestras de sobrenadante de DP:
Si la concentración de la muestra sobrenadante está por debajo del LIDC del ensayo (es decir, 0,05 UA/ml), entonces la muestra de sobrenadante debe registrarse como <0,05 UA/ml
Si la concentración de la muestra de sobrenadante está dentro de los LDC del ensayo (es decir, de 0,05 UA/ml a 1,25 UA/ml), se registra el valor de concentración para la muestra sobrenadante.
Si la concentración de la muestra de sobrenadante está por encima del LSDC del ensayo (es decir, 1,25 UA/ml), se debe repetir la preparación del sobrenadante de medicamento. La muestra de sobrenadante se volverá a someter a ensayo realizando una predilución adecuada en el intervalo de la curva patrón seguida de 6 diluciones de muestra. (No es necesario repetir la muestra del medicamento desorbido). La concentración media para las diluciones que están dentro de los LDC del ensayo (LIDC de 0,05 UA/ml a 1,25 UA/ml) será el valor de concentración registrado para la muestra de sobrenadante, siempre que se cumpla con la idoneidad del sistema y al menos 4 de las 6 diluciones de muestra estén dentro de los LDC.
2.13. Criterios de aceptación del ensayo
a) El coeficiente de correlación de la curva de calibración debe ser > 0,980.
b) % de CV <15 % para patrones y muestras (excepto sobrenadante de DP) para las cuatro concentraciones más altas de las diluciones, % de CV <15 % para controles
c) Las DO individuales en blanco deben ser < 0,2.
d) Los controles de ensayo deben estar dentro de los límites definidos especificados (para controles recién preparados, 2 de 3 QC deben tener concentraciones observadas dentro de ± 30 % de los valores nominales; O los niveles establecidos durante la calificación de QC) para que pase el ensayo.
e) La validez del ensayo se registrará en la copia impresa de Gen5. Si la placa no cumple con los criterios de aceptación definidos, el ensayo se considera inválido.
2.14. Notificación de datos
NIV
a) Contenido de antígeno
El valor notificado para UA/ml inactivado es la media de las concentraciones (que están dentro de los LDC de 0,04 a 1.25 UA/ml) calculadas para las diluciones de muestra única corregidas por los factores de dilución respectivos, y la media multiplicada por un factor de corrección de 1,05 para dar cuenta del 5 % del volumen de tampón de ELISA 20x que se añadió a cada muestra cuando se tomó. El contenido de antígeno se registrará en la copia impresa de Gen5. DS:
a) Identidad
Si las absorbancias de las muestras en la dilución más baja (concentración más alta) son superiores a 3 desviaciones estándar por encima del valor medio del blanco, el resultado se notificará como positivo.
b) Contenido de antígeno
El valor notificado para UA/ml inactivado es la media de las concentraciones (que están dentro de los LDC de 0,04 a 1.25 UA/ml) calculadas para las diluciones de muestra única corregidas por los factores de dilución respectivos, y la media multiplicada por un factor de corrección de 1,05 para dar cuenta del 5 % del volumen de tampón de ELISA 20x que se añadió a cada muestra cuando se tomó. El contenido de antígeno se registrará en la copia impresa de Gen5. DP desorbido:
a) Identidad:
Si las absorbancias de las muestras en la dilución más baja (concentración más alta) son superiores a 3 desviaciones estándar por encima del valor medio del blanco, el resultado se notificará como positivo.
b) Contenido de antígeno
El valor notificado para UA/ml inactivado es la media de las concentraciones (que están dentro de los LDC de 0,05 a 0,8 UA/ml) calculadas para las diluciones de muestra única corregidas por los factores de dilución respectivos. El contenido de antígeno se registrará en la copia impresa de Gen5.
Sobrenadante de DP (grado de adsorción/grado de no adsorción):
El grado de adsorción se notifica con respecto a la sustancia farmacológica formulada con hidróxido de aluminio después de la filtración.
a) Para el cálculo del valor notificado, el contenido de antígeno notificado (UA/ml) para la muestra de DS respectiva (postfiltración) corregido por la dilución con hidróxido de aluminio (5 %) se establecerá en 100 % y el porcentaje de la concentración medida en el sobrenadante (corregido por la adición de tampón de ELISA al 5 %) calculado con respecto a eso. El valor notificable será la diferencia entre el 100 % y el porcentaje calculado para el sobrenadante. Los resultados se notificarán con 2 decimales.
Sobrenadante de DP (UA/ml) * 1,05
Grado de adsorción (%) = 100 % - ------------------------------------------------- * 100 %
DS (UA/ml) * 0,95
El grado de no adsorción se calculará como se detalla a continuación y los resultados se notificarán con 2 decimales:
Sobrenadante de DP (UA/ml) * 1,05
Grado de no adsorción (%) = .......................................................... * 100 %
DS (UA/ml) *0,95
b) En caso de que el sobrenadante puro no contenga ningún antígeno medible (es decir, concentración de sobrenadante observada inferior al LIDC, donde LIDC = 0,05 UA/ml), se utilizará el LIDC para el cálculo del resultado. El resultado en este caso se notifica como “ mayor que x %” . Por ejemplo, si la muestra de DS se mide como 12,00 UA/ml y no se midió ninguna señal en el sobrenadante; con el LIDC de 0,05 UA/ml, la cantidad en el sobrenadante es <0,05*1,05 = < 0,0525 UA/ml. La cantidad de DS después de la corrección del tampón es de 12,00*0,95 = 11,40 UA/ml, y el resultado notificado para el grado de adsorción es < 100-0,0525/11,4*100 = > 99,54 %. También se notificará el grado de no adsorción (es decir, 100 - el % de grado de adsorción).
Ejemplo 5
Introducción:
Con el fin de investigar adicionalmente el modo de acción que conduce a la inestabilidad del producto/pérdida de potencia de la vacuna contra el JEV, se utilizó Ala-(His)6-OprF190-342-OprI21-83 (SEQ ID n O: 1, Figura 14) - en el presente documento también denominada “ proteína A” en un ensayo de detección preliminar que incorporaba lotes de alhidrogel con diferentes contenidos de metales y adiciones con iones de cobre y sulfito.
Material:
• Dihidrato de cloruro de cobre (II) (Sigma, N.° de pedido 807483)
• 10xPBS (Gibco, número de pedido 14200-091)
• Tubos Falcon de 15 ml (Greiner, N.° de Cat. 188724)
• Incubadora Infors HT Incubator Multitron Standard (InforsAG)
• Agua bidest. (Fresenius Kabi, N.° de Art. 0712221/01 A)
Preparación de soluciones madre:
• Solución madre de cobre (II)
• Se preparó una solución madre de cobre (II) 20 mM disolviendo 341 mg de dihidrato de cloruro de cobre (II) en 100 ml de agua bidestilada.
• Solución madre de metabisulfito de sodio
Se preparó una solución madre de metabisulfito de sodio 200 mM disolviendo 1,52 g de metabisulfito de sodio en 35 ml de PBS. A esta solución se le ajustó el pH a 7,3 con NaOH y se llenó hasta un volumen de 40 ml con PBS. La solución se filtró después a través de un filtro de jeringa de 0,2 p.
Preparación de soluciones de trabajo
Las soluciones de trabajo se prepararon mediante dilución de soluciones madre de metal con agua bidestilada y filtración estéril a través de un filtro de jeringa de 0,2 p. (Mini Kleenpak 25 mm-Pall)
Preparación de soluciones tampón
• / PBS NaCI al 0,9 %
Se preparó solución tampón de PBS 1 x mediante una dilución 1:10 de PBS 10 x con agua bidestilada. El pH de esta solución tampón fue de 7,5. Se prepararon soluciones tampón de PBS ajustadas a pH 7,3 y 8,0 ajustando el pH con HCl o NaOH, respectivamente.
Se disolvieron 9 g de NaCl en 333 ml de solución tampón de pH 7,3 o pH 8,0 y después se llevaron a 1000 ml con agua bidestilada, seguido de filtración a través de un filtro de botella de 0,2 p.
Preparación de la muestra:
Se prepararon formulaciones de Proteína A y diferentes lotes de alhidrogel (Lote 4230 y Lote 4074) en / PBS NaCl al 0,9 % a dos valores de pH diferentes y se adicionaron con sulfito según el siguiente esquema:
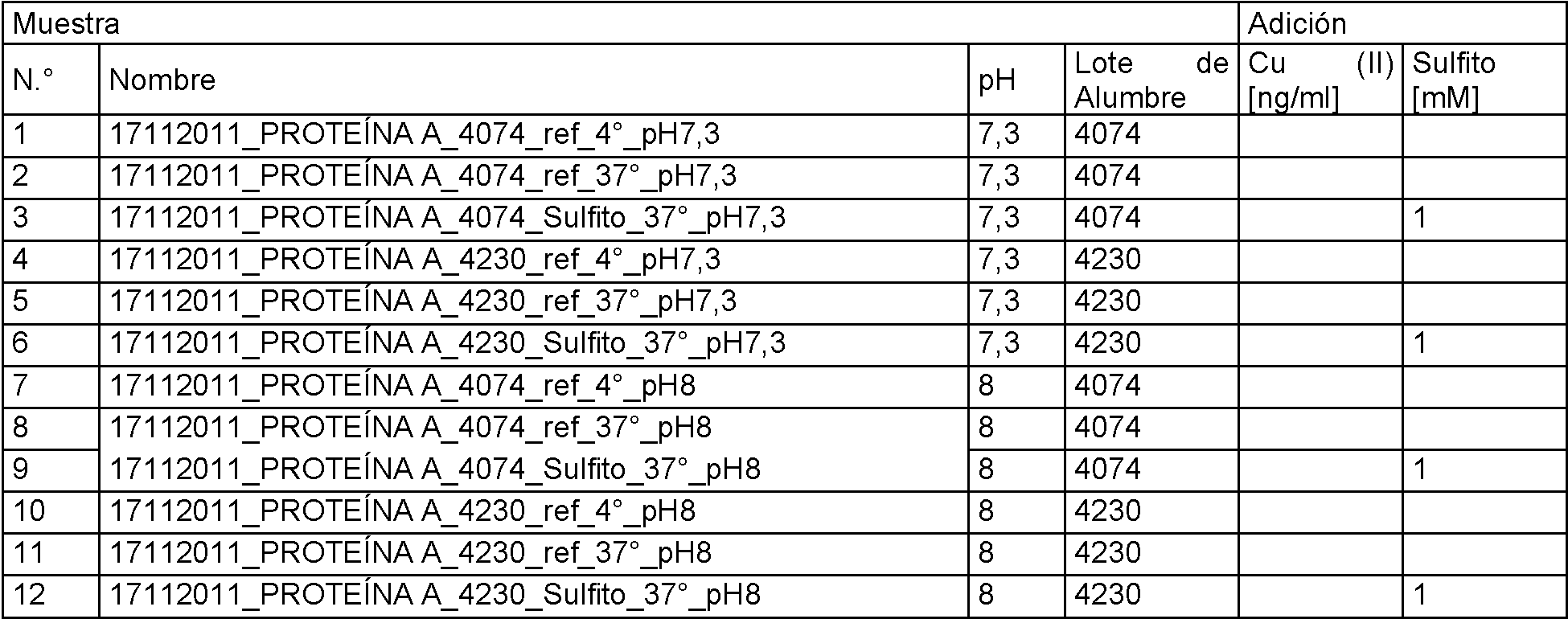
Las muestras 1, 4, 7 y 0 se almacenaron a 4 °C (muestras de referencia). Todas las demás muestras se incubaron a 37 °C durante 96 horas.
Después de 96 horas, todas las muestras se sometieron a un procedimiento de desorción para separar el antígeno del alhidrogel. El antígeno desorbido se analizó mediante RPC.
Resultados:
Los resultados mostraron una severa degradación del antígeno Proteína A en presencia de sulfito. La degradación fue más pronunciada en las muestras formuladas con alhidrogel de mayor contenido de impurezas metálicas. Tabla 1. Contenido de iones metálicos en los lotes 4230 y 4074 de Hidróxido de aluminio analizado mediante ICP-MS.

*Contenido de metal residual calculado
Tabla 2: Esquema de pipeteo de DOE

Tabla 2 continuación

Tabla 2 continuación


Tabla 2 continuación

Tabla 3: Comparación de lixiviables de extracto de tapón y JEV09L37 SN. Todos los picos con un área de > O.lmUA.min están incluidos en esta tabla.


Tabla 4: Resultados del DOE obtenido después de 4 y 8 semanas a 22 °C. Se desorbió antígeno a partir de Alumbre y se analizó mediante ELISA (monoclonal y policlonal). * (aumento de x veces en comparación con FVL)

Tabla 4 continuación

Tabla 5: Análisis de varianza para la relación de ELISA monoclonal/policlonal después de un almacenamiento de 4 semanas a 22 °C
Análisis de Varianza para la Relación de 4 semanas
Fuente Suma de cuadrados Gl Cuadrado medio Relación F Valor P A: pH 0,02205 1 0,02205 5,48 0,0326
B: Alumbre 0,117612 1 0,117612 29,20 0,0001
C: Fragmentos de PS 0,0008 1 0,0008 0,20 0,6618
D: Extraíbles 0,0008 1 0,0008 0,20 0,6618
E: Formol 0,0242 1 0,0242 6,01 0,0261
AB 0,0001125 1 0,0001125 0,03 0,8694
AC 0,00045 1 0,00045 0,11 0,7425
AD 0,01125 1 0,01125 2,79 0,1141
AE 0,00405 1 0,00405 1,01 0,3309
BC 0,0000125 1 0,0000125 0,00 0,9563
BD 0,0010125 1 0,0010125 0,25 0,6229
BE 0,0006125 1 0,0006125 0,15 0,7017
CD 0,0008 1 0,0008 0,20 0,6618
CE 0,0008 1 0,0008 0,20 0,6618
DE 0,0072 1 0,0072 1,79 0,1999 Error total 0,0644375 16 0,00402734
Total (corr.) 0,2562 31
R cuadrado = 74,8488 por ciento
R cuadrado (ajustado para g.l.)= 51,2695 por ciento
Error estándar de Est. = 0,0634614
Error absoluto medio = 0,0352734
Estadístico de Durbin-Watson = 1,47556
Tabla 6: Análisis de varianza para la relación de ELISA monoclonal/policlonal después de un almacenamiento a 22 °C durante 8 semanas.
Análisis de Varianza para la Relación de 8 semanas
Fuente Suma de cuadrados Gl Cuadrado medio Relación F Valor P A: pH 0,102378 1 0,102378 11,19 0,0041
B: Alumbre 0,275653 1 0,275653 30,13 0,0000
C: Fragmentos de PS 0,00300312 1 0,00300312 0,33 0,5747
D: Extraíbles 0,0108781 1 0,0108781 1,19 0,2917
E: Formol 0,0318781 1 0,0318781 3,48 0,0804
AB 0,00137813 1 0,00137813 0,15 0,7031
AC 0,00382812 1 0,00382812 0,42 0,5269
AD 0,00137813 1 0,00137813 0,15 0,7031
AE 0,0166531 1 0,0166531 1,82 0,1961
BC 0,000253125 1 0,000253125 0,03 0,8700
BD 0,00382813 1 0,00382813 0,42 0,5269
BE 0,00195313 1 0,00195313 0,21 0,6503
CD 0,00195313 1 0,00195313 0,21 0,6503
CE 0,000153125 1 0,000153125 0,02 0,8987
DE 0,00525313 1 0,00525313 0,57 0,4596 Error total 0,1464 16 0,00915
Total (corr.) 0,606822 31
R cuadrado = 75,8743 por ciento
R cuadrado (ajustado para g.l.) = 53,2565 por ciento
Error estándar de Est. = 0,0956556
Error absoluto medio = 0,0571484
Estadístico de Durbin-Watson = 0,888586
Tabla 7: Análisis de regresión para “ Relación de 8 sem anas” incluyendo pH, A lum bre y Form aldehído.
Coeficientes de regresión para la Relación de 8 sem anas
Constante = 0,0228125
A: pH = 0,113125
B: A lum bre = -0,00185625
E: Formol = 0,0315625
Tabla 8: Estim ación de resultados de “ Relación de 8 sem anas” generados utilizando el m odelo ajustado.
Estim ación Resultados para la Relación de 8 sem anas
O bservado A justado Inferior LC del 95,0 % Superio r LC del 95,0 % Fila V alor V alor para la M edia para la M edia 1 0,58 0,5975 0,536766 0,658234
2 0,81 0,783125 0,722391 0,843859
3 0,9 0,84625 0,785516 0,906984
4 0,92 0,89625 0,835516 0,956984
6 0,99 0,84625 0,785516 0,906984
7 0,67 0,5975 0,536766 0,658234
8 0,99 0,89625 0,835516 0,956984
9 0,84 0,710625 0,649891 0,771359
10 0,78 0,710625 0,649891 0,771359
11 0,98 0,959375 0,898641 1,02011
12 0,59 0,5975 0,536766 0,658234
13 1,03 0,959375 0,898641 1,02011
14 0,78 0,660625 0,599891 0,721359
15 0,94 0,89625 0,835516 0,956984
16 0,81 0,77375 0,713016 0,834484
17 0,82 0,783125 0,722391 0,843859
18 0,88 0,84625 0,785516 0,906984
19 0,82 0,77375 0,713016 0,834484
20 0,82 0,959375 0,898641 1,02011
21 0,82 0,959375 0,898641 1,02011
22 0,63 0,710625 0,649891 0,771359
23 0,74 0,84625 0,785516 0,906984
24 0,66 0,660625 0,599891 0,721359
25 0,67 0,783125 0,722391 0,843859
26 0,76 0,783125 0,722391 0,843859
27 0,64 0,710625 0,649891 0,771359
28 0,74 0,77375 0,713016 0,834484
29 0,73 0,77375 0,713016 0,834484
30 0,44 0,5975 0,536766 0,658234
32 0,87 0,89625 0,835516 0,956984
33 0,67 0,660625 0,599891 0,721359
34 0,59 0,660625 0,599891 0,721359
Tabla 9: Plan para el experim ento 20110913 (NIV)
Plan de pipeteo
Sol madre [mM]
Volumen total: 2000 M Ni(II)SO4 1
Material de NIV: 11A74 Cu(II)Cl2 1


Tabla 9 continuación

Tabla 9 continuación
PM [g/mol] otros aditivos:
Ni 58,7 Sol madre:
C 63,6 PS frag 2 mg/ml Cr 52,0


Tabla 9 continuación

Tabla 10: Plan para el experimento 20110913(DP)
Plan de pipeteo
Sol madre [mM] Volumen total: 10000 Hl Ni(II)SO4 1
Cu(II)Cl2 1 DP: 11D87 a granel Cr(III)Cl3 1


Tabla 10 Continuación
PM [g/mol] otros aditivos:
Ni 58,7 Sol madre:
C 63,6 PS frag 2 mg/ml
Cr 52,0
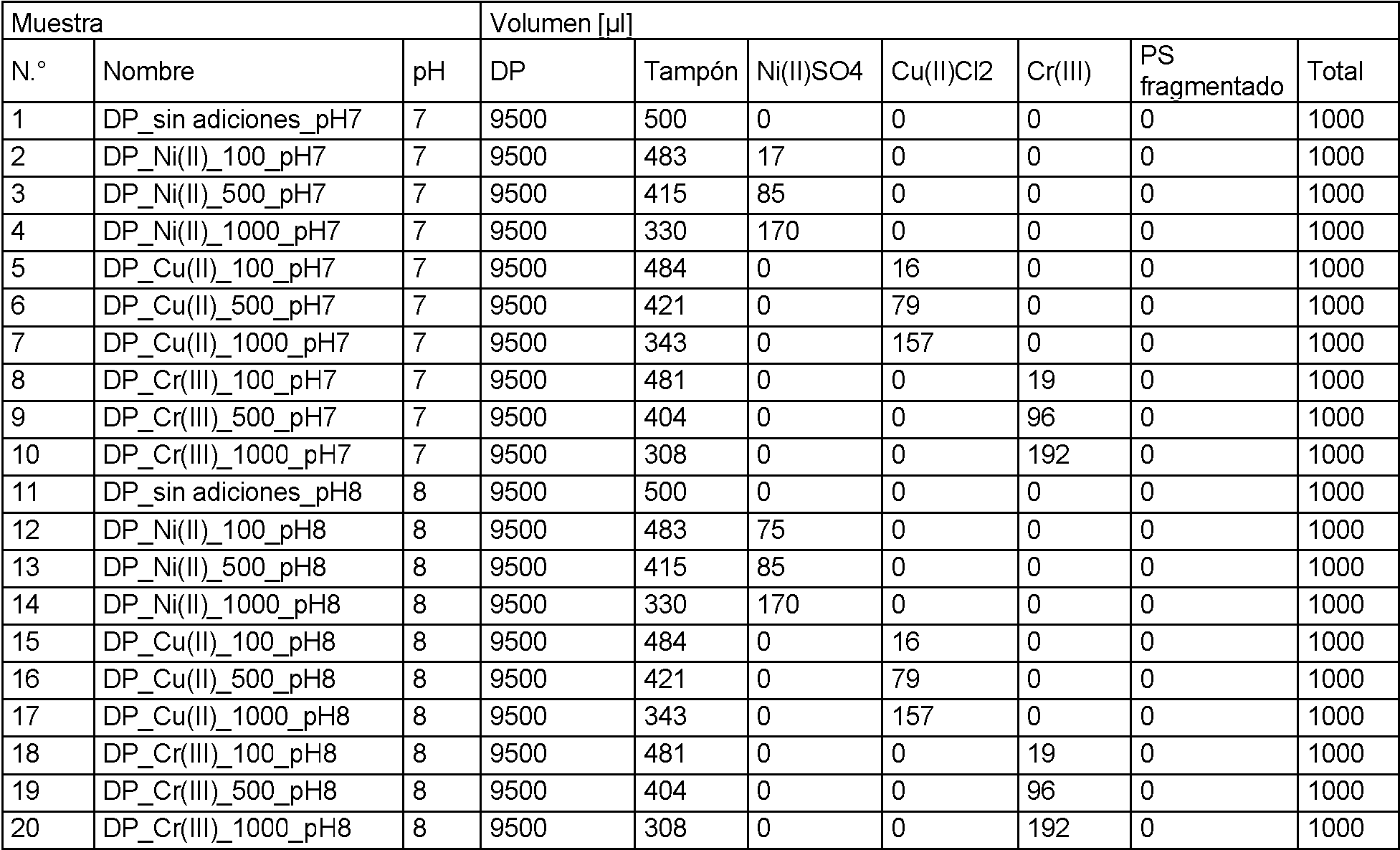
Tabla 11: Plan para el experimento 20110812-DP adicionado con iones metálicos
Plan de pipeteo
Sol madre [mM] PM [g/mol] FVL ng/ml pM Volumen total: 30000 pl Fe(II)Cl3 1 Fe 55,9 282 5,049 DP: 11D87 a granel Fe(III)Cl3 1 Fe 55,9 282 5,045
Ni(II)SO4 1 Ni 58,7 41 0,698
Co(II)Cl2 1 Co 58,9 0,33 0,006 Cu(II)Cl2 1 Cu 63,6 3 0,047 Zn(II)SO4 1 Zn 65,4
Cr(III)Cl3 1 Cr 52,0

Tabla 11 continuación

Muestra Volumen [pl] 
N.° Nombre NIV dii Tampón Fe(II)CI3 Fe(III)CI3 Ni(II)SO4 Co(II)CI2 Cu(II)CI2 Zn(II)SO4 Total 1 DP_Fe(II) 27750 1981 269 0 0 0 0 0 30000 2 DP_Fe(III 27750 1982 0 26 0 0 0 0 30000 3 DP_Ni(II) 27750 1981 0 0 26 0 0 0 30000 4 DP_Co(II) 27750 1994 0 0 0 25 0 0 30000 5 DP_Cu(II) 27750 1995 0 0 0 0 25 0 30000 6 DP_Zn_p 27750 2014 0 0 0 0 0 23 30000 7 DP_mezcl 27750 698 26 26 26 25 25 23 30000 8 DP_sin adiciones_pH7 7 27750 2250 0 0 0 0 0 0 30000 9 DP_Fe(II)_pH7,4 7,4 27750 1981 26 0 0 0 0 0 30000 10 DP_Fe(III)_pH7,4 7,4 27750 1982 0 26 0 0 0 0 30000 11 DP_Ni(II)_pH7,4 7,4 27750 1981 0 0 26 0 0 0 30000 12 DP_Co(II)_pH7,4
27750 698 26 26 26 25 25 23 30000 8 DP_sin adiciones_pH7 7 27750 2250 0 0 0 0 0 0 30000 9 DP_Fe(II)_pH7,4 7,4 27750 1981 26 0 0 0 0 0 30000 10 DP_Fe(III)_pH7,4 7,4 27750 1982 0 26 0 0 0 0 30000 11 DP_Ni(II)_pH7,4 7,4 27750 1981 0 0 26 0 0 0 30000 12 DP_Co(II)_pH7,4  7,4 27750 1994 0
7,4 27750 1994 0  0
0  0
0  25
25 0
0  0
0  30000 13 DP_Cu(II)_pH7,4 7,4 27750 1995 0 0 0 0 255 0 30000
30000 13 DP_Cu(II)_pH7,4 7,4 27750 1995 0 0 0 0 255 0 30000

Tabla 11 continuación
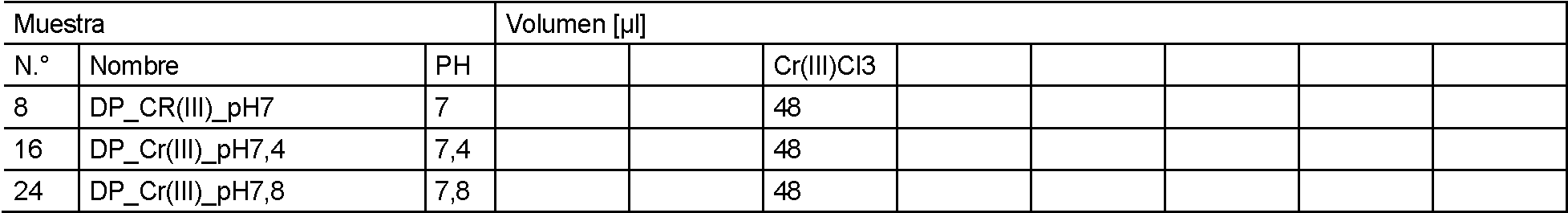
Tabla 12: Recuperación de antígeno determinada mediante SEC-HPLC del exp. 20110913(NIV). Para el pH 7 y el pH 8, las recuperaciones se basan en muestras de control de NIV sin adiciones n.° 1 y n.° 21, respectivamente. Las muestras se almacenaron a 22 °C durante 3 semanas. Las muestras marcadas con “ n.a.” no se analizaron debido a la priorización de muestras
Resultados

28 NIV_CR(III)_100_pH8_22 °C 4,213 100,0

Tabla 13: Resultados de ELISA del exp. 20110913(NIV) obtenidos después de 3 semanas a pH 8 (muestra 21-40) y 7 semanas a pH 7 (muestra 1-20) a 22 °C. Las muestras marcadas con “ n.a.” no se analizaron debido a la priorización de muestras.

20110913 adicionado con metal NIV 22 °C 7 semanas a pH 8 
1er análisis 2do análisis

32 NIV_Ni(II)_100_adición de PS_pH8_22 °C 18,729 19,250 1,028 n.a. n.a. n.a.

Tabla 14: Recuperaciones de antígeno después de 5 semanas a 22 °C de JEV desorbido obtenidas mediante SEC-HPLC. Las recuperaciones se basaron en muestras de control de DP sin adiciones almacenadas a pH 7 o pH 8

Tabla 15: Resultados de ELISA de antígeno de JEV desorbido después de 5 semanas a 22 °C


Tabla 16: Resultados de ELISA de antígeno de JEV desorbido después de 4 semanas y 7 semanas almacenado a 22 °C. Las muestras marcadas con “ n.a.” no se analizaron debido a la priorización de muestras

Tabla 17: ANOVA para muestras de estabilidad almacenadas a 22 °C durante 7 semanas.
Análisis de Varianza para Relación - Sumas de Cuadrados de Tipo III
Fuente Suma de cuadrados Gl Cuadrado medio Relación F Valor P EFECTOS PRINCIPALES
R: tipo de metal 0,139643 8 0,0174554 3,00 0,0293 B: pH 0,0462028 2 0,0231014 3,97 0,0398 RESIDUAL 0,0931046 16 0,00581904
TOTAL (CORREGIDO) 0,27895 26
Todas relaciones F se basan en el error cuadrado medio residual.
Tabla 18: Ensayo de intervalo múltiple para la relación por tipo de ion metálico. l=Fe(II); 2=Fe(III); 3=Ni(II); 4=Co(II); 5=Cu(II); 6=Zn(II); 7=Cr(III); 8=Mezcla[l-6]; 9=control no adicionado
Ensayos de intervalo múltiple para la Relación por tipo de metal
Método: LSD 95,0 por ciento
Tipo de metal Recuento Media de LS Grupos Homogéneos
8 3 0,666087 X
5 3 0,770168 XX
2 3 0,793725 XXX
7 3 0,807248 XX
4 3 0,844388 XX
6 3 0,853867 XX
1 3 0,878563 XX
3 3 0,887505 XX
9 3 0,919926 X
Contraste Diferencia /- Límites
1-2 0,084838 0,132037
1-3 -0,0089421 0,132037
1-4 0,0341754 0,132037
1-5 0,108395 0,132037
1-6 0,0246961 0,132037
1-7 0,0713155 0,132037
1-8 *0,212476 0,132037
1-9 -0,0413627 0,132037
2-3 -0,0937801 0,132037
2-4 -0,0506626 0,132037
2-5 0,0235569 0,132037
2-6 -0,0601419 0,132037
2-7 -0,0135225 0,132037
2-8 0,127638 0,132037
2-9 -0,126201 0,132037
3-4 0,0431175 0,132037
3-5 0,117337 0,132037
3-6 0,0336382 0,132037
3-7 0,0802576 0,132037
3-8 *0,221418 0,132037
3-9 -0,0324206 0,132037
4-5 0,0742195 0,132037
4-6 -0,00947935 0,132037
4-7 0,0371401 0,132037
4-8 *0,1783 0,132037
4-9 -0,0755381 0,132037
5-6 -0,0836988 0,132037
5-7 -0,0370794 0,132037
5-8 0,104081 0,132037
5-9 *-0,149758 0,132037
6-7 0,0466195 0,132037
6-8 *0,18778 0,132037
6-9 -0,0660588 0,132037
7-8 *0,14116 0,132037
7-9 -0,112678 0,132037
8-9 *-0,253838 0,132037
* indica una diferencia estadísticamente significativa
Tabla 19: Resultados de ICP-MS de impurezas de iones metálicos residuales presentes en diversos lotes de Alumbre (2 %)
Contenido de metal residual (ng/ml)
Lote de Alumbre (2 %) Cr Fe Ni Cu V Co
4074 19,8 266 14,8 <25 <5 <5 
*Contenido calculado de metales residuales basado en los lotes 4230 y 4074 de Alumbre
**no GI: no irradiado con rayos gamma
***GI: irradiado con rayos gamma
Tabla 20: Resumen del contenido de iones metálicos y análisis de muestras de DP formuladas con diversos lotes de Alumbre. Las muestras se analizaron por duplicado mediante ELISA y se notifica la relación de ELISA monoclonal/policlonal. Las formulaciones se almacenaron a 22 °C durante 6 semanas.

*El intervalo es la diferencia absoluta entre el 1er y el 2do análisis.
Tabla 21: Resultados del análisis PSD de la solución madre de Alhydrogel® (2 %) en agua.

d(0,1): El 10 % de todas las partículas medidas tienen un diámetro por debajo de este valor
d(0,5): El 50 % de todas las partículas medidas tienen un diámetro por debajo de este valor
d(0,9): El 90 % de todas las partículas medidas tienen un diámetro por debajo de este valor
Oscurecimiento: cantidad de reducción de luz láser por muestra; corresponde a la concentración de la muestra en la cámara de medición
Tabla 22: Resultados de las curvas de titulación de Alhydrogel® para la determinación de POZ. Las muestras se analizaron en PBS (dilución 1:20).

Tabla 23: Sumario del análisis de iones metálicos para diversas soluciones madre de hidróxido de aluminio; Nota: Una solución madre al 2 %


*los resultados por debajo del LDC no se utilizaron para el cálculo del promedio; **no es posible calcular el promedio debido a los resultados por debajo del LDC
Tabla 24: El análisis de la fracción de gel de hidróxido de aluminio y sobrenadante (Lote 4230) para detectar iones metálicos contaminantes muestra que los iones metálicos se encuentran en el gel, no en el sobrenadante


Tabla 25: Vacunas a base de aluminio para su uso humano


Tabla 26: Vacunas a base de aluminio para su uso veterinario









Claims (18)
- REIVINDICACIONESi . Un método para preparar una composición acuosa que comprende aluminio, un compuesto reactivo y una proteína, en donde dicho método comprende:- seleccionar una sal de aluminio que sea capaz de proporcionar una composición acuosa que tenga menos de 350 ppb de metal pesado total y menos de 2,5 ppb de Cu basándose en el peso de la composición acuosa y- combinar dicha sal de aluminio, dicho compuesto reactivo, dicha proteína dentro de partículas de virus inactivadas con formaldehído y agua para producir dicha composición acuosa que tiene menos de 350 ppb de metal pesado total y menos de 2,5 ppb de Cu basándose en el peso de la composición acuosa; yen donde el compuesto reactivo es un compuesto activo redox, un compuesto formador de radicales y/o un compuesto estabilizante.
- 2. Un método según la reivindicación 1, que comprende además tamponar dicha composición acuosa a un pH de entre 6,5 y 8,5.
- 3. Un método según la reivindicación 1 o la reivindicación 2, en donde la composición acuosa comprende no más de 5 ppm de Cu en comparación con el contenido de aluminio.
- 4. Un método según cualquiera de las reivindicaciones 1 a 3, en donde la composición acuosa comprende menos de 1,25 ppb de Cu basándose en el peso de la composición acuosa.
- 5. Un método según cualquiera de las reivindicaciones 1 a 4, que comprende además almacenar la composición acuosa durante al menos tres meses a una temperatura de entre 2 °C y 8 °C.
- 6. Un método según la reivindicación 5, en donde se contrarresta la degradación de la proteína durante la etapa de almacenamiento de manera que:(i) la composición acuosa es estable durante al menos tres meses a una temperatura de entre 2 °C y 8 °C;(ii) la composición acuosa tiene una vida útil de 20 o 24 meses;(iii) la composición acuosa se puede almacenar durante 20 o 24 meses sin volverse inadecuada para su uso debido a la degradación de la proteína;(iv) una vacuna que comprende la composición acuosa permanece dentro de la especificación de potencia después de 20 o 24 meses de almacenamiento; o(v) no más del 30 % de la proteína en la composición se degrada después de 20 o 24 meses de almacenamiento.
- 7. Un método según cualquiera de las reivindicaciones 1 a 6, en donde la sal de aluminio que se selecciona comprende menos de 700 ppm de metal pesado, sobre la base del peso con respecto al contenido de aluminio.
- 8. Un método según cualquiera de las reivindicaciones 1 a 7, en donde la sal de aluminio es hidróxido de aluminio (Al(OH)3) o fosfato de aluminio (AlPO4), preferiblemente en donde la sal de aluminio es hidróxido de aluminio (Al(OH)3).
- 9. Un método según cualquiera de las reivindicaciones 1 a 8, que comprende entre 5 pg/ml y 50 mg/ml de aluminio, preferiblemente que comprende entre 50 pg/ml y 5 mg/ml de aluminio.
- 10. Un método según cualquiera de las reivindicaciones 1 a 9, en donde el compuesto reactivo se selecciona del grupo que consiste en formaldehído, etanol, cloroformo, tricloroetileno, acetona, 2-[4-(2,4,4-trimetilpentan-2-il) fenoxijetanol, desoxicolato, dietilpirocarbonato, sulfito, Na2S2O5, beta-propriolactona, polisorbato opcionalmente Polisorbato 20 o Polisorbato 80, O2 , fenol, copolímeros de tipo pluronic y una combinación de cualquiera de los mismos, preferiblemente en donde el compuesto reactivo comprende sulfito.
- 11. Un método según cualquiera de las reivindicaciones 1 a 10, en donde la proteína es una proteína vírica de un virus de la familia Flaviviridae, preferiblemente en donde dicho virus de la familia Flaviviridae es un virus de la encefalitis japonesa.
- 12. Un método según cualquiera de las reivindicaciones 1 a 11, que comprende además determinar el contenido de metales pesados en la composición acuosa y/o la sal de aluminio.
- 13. Uso de una sal de aluminio que comprende menos de 700 ppm de metal pesado sobre la base del peso con respecto al contenido de aluminio, para preparar una composición acuosa o vacuna, en donde la com posic ión acuosa o vacuna com prende una proteína, un com puesto reactivo y una sal de alum inio, com prendiendo dicha com posic ión m enos de 350 ppb de metal pesado y m enos de 2,5 ppb de Cu basándose en el peso de la com posic ión acuosa, en donde el com puesto reactivo es un com puesto activo redox, un com puesto fo rm ador de rad ica les y/o un com puesto estabilizante, y en donde dicha proteína es una proteína dentro de partícu las v íricas inactivadas con form aldehído.
- 14. Uso de una sal de aluminio que comprende menos de 700 ppm de metal pesado sobre la base del peso con respecto al contenido de aluminio, para (i) contrarrestar la degradación de una proteína en una composición acuosa o vacuna; y/o (ii) prolongar la vida de almacenamiento de una composición acuosa o vacuna; en donde la composición acuosa o vacuna se obtiene mediante el método de cualquiera de las reivindicaciones 2 a 12 o comprende una proteína, un compuesto reactivo y una sal de aluminio, comprendiendo dicha composición menos de 350 ppb de metal pesado y menos de 2,5 ppb de Cu basándose en el peso de la composición acuosa, en donde el compuesto reactivo es un compuesto activo redox, un compuesto formador de radicales y/o un compuesto estabilizante, y en donde dicha proteína es una proteína dentro de partículas víricas inactivadas con formaldehído.
- 15. Uso según la reivindicación 13 o la reivindicación 14, en donde la com posición acuosa com prende no más de 5 ppm de Cu en com paración con el contenido de aluminio.
- 16. Uso según la reivindicación 15, en donde la composición acuosa comprende no más de 2,5 ppm de Cu en comparación con el contenido de aluminio.
- 17. Uso según cualquiera de las reivindicaciones 13 a 16, en donde la sal de aluminio es hidróxido de aluminio (Al(OH)3).
- 18. Uso según cua lqu iera de las re iv ind icaciones 13 a 17, en donde la com posic ión acuosa o vacuna com prende entre 0,1 y 2,5 mg/m l de alum inio, p re ferib lem ente en donde la com posic ión acuosa o vacuna tiene un conten ido de a lum in io de 0,5 mg/ml.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11192230 | 2011-12-06 | ||
| PCT/EP2012/054387 WO2013135274A1 (en) | 2012-03-13 | 2012-03-13 | Aluminium compounds for use in therapeutics and vaccines |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2930458T3 true ES2930458T3 (es) | 2022-12-13 |
Family
ID=47294919
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16183076.5T Active ES2647882T3 (es) | 2011-12-06 | 2012-12-06 | Compuestos de aluminio para su uso en productos terapéuticos y vacunas |
| ES17185526T Active ES2930458T3 (es) | 2011-12-06 | 2012-12-06 | Compuestos de aluminio para su uso en agentes terapéuticos y vacunas |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16183076.5T Active ES2647882T3 (es) | 2011-12-06 | 2012-12-06 | Compuestos de aluminio para su uso en productos terapéuticos y vacunas |
Country Status (15)
| Country | Link |
|---|---|
| EP (5) | EP3106176B1 (es) |
| CY (1) | CY1119927T1 (es) |
| DE (1) | DE202012012768U1 (es) |
| DK (6) | DK3269386T3 (es) |
| ES (2) | ES2647882T3 (es) |
| FI (1) | FI3785730T3 (es) |
| HR (1) | HRP20171871T1 (es) |
| HU (1) | HUE037325T2 (es) |
| LT (1) | LT3106176T (es) |
| NO (1) | NO3106176T3 (es) |
| PL (2) | PL3106176T3 (es) |
| PT (2) | PT3269386T (es) |
| RS (1) | RS56709B1 (es) |
| SI (1) | SI3106176T1 (es) |
| WO (1) | WO2013083726A1 (es) |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK3269386T3 (da) | 2011-12-06 | 2022-10-24 | Valneva Austria Gmbh | Aluminiumforbindelser til anvendelse inden for behandling og i vacciner |
| US9895437B2 (en) | 2012-04-18 | 2018-02-20 | Valneva Austria Gmbh | Aluminum compounds for use in therapeutics and vaccines |
| US10434168B2 (en) | 2014-09-03 | 2019-10-08 | Intervet Inc. | Attenuated bovine coronavirus and related vaccines |
| CN108697785B (zh) | 2015-12-23 | 2022-08-02 | 瓦尔尼瓦奥地利有限责任公司 | Zika病毒疫苗 |
| EP3562503A2 (en) | 2016-12-30 | 2019-11-06 | Sutrovax, Inc. | Polypeptide-antigen conjugates with non-natural amino acids |
| US11951165B2 (en) | 2016-12-30 | 2024-04-09 | Vaxcyte, Inc. | Conjugated vaccine carrier proteins |
| US12018054B2 (en) | 2017-04-13 | 2024-06-25 | Valneva Austria Gmbh | Multivalent OspA polypeptides and methods and uses relating thereto |
| SG11202001161YA (en) | 2017-09-21 | 2020-03-30 | Valneva Se | Method of producing pharmaceutical compositions comprising immunogenic chikungunya virus chikv-delta5nsp3 |
| CA3081578A1 (en) | 2017-11-03 | 2019-05-09 | Takeda Vaccines, Inc. | Zika vaccines and immunogenic compositions, and methods of using the same |
| CN111601885A (zh) | 2017-11-30 | 2020-08-28 | 武田疫苗股份有限公司 | 用于将寨卡病毒灭活的方法和相关方法 |
| US12053518B2 (en) | 2018-07-13 | 2024-08-06 | Valneva Se | Method for rescuing and producing a virus in avian cells |
| MX2021013111A (es) | 2019-05-20 | 2021-11-17 | Valneva Se | Una vacuna de subunidad para el tratamiento o la prevencion de una infeccion del tracto respiratorio. |
| WO2021178306A1 (en) * | 2020-03-01 | 2021-09-10 | Dynavax Technologies Corporation | Coronavirus vaccines comprising a tlr9 agonist |
| US20230218740A1 (en) * | 2020-03-01 | 2023-07-13 | Dynavax Technologies Corporation | Coronavirus vaccines comprising a tlr9 agonist |
| WO2021176434A1 (en) | 2020-03-01 | 2021-09-10 | Valneva Austria Gmbh | Cpg-adjuvanted sars-cov-2 virus vaccine |
| WO2021178318A1 (en) * | 2020-03-01 | 2021-09-10 | Dynavax Technologies Corporation | Coronavirus vaccines comprising a tlr9 agonist |
| EP3895729A1 (en) | 2020-03-01 | 2021-10-20 | Valneva Austria GmbH | Cpg-adjuvanted sars-cov-2 virus vaccine |
| WO2021204825A2 (en) | 2020-04-06 | 2021-10-14 | Valneva Austria Gmbh | INACTIVATED SARS-CoV-2 VIRUS VACCINE |
| WO2021205017A1 (en) | 2020-04-09 | 2021-10-14 | Valneva Austria Gmbh | Improvements in vaccine formulations for medical use |
| US20240358812A1 (en) | 2021-04-09 | 2024-10-31 | Valneva Se | Human metapneumovirus combination vaccine |
| WO2023148256A1 (en) | 2022-02-02 | 2023-08-10 | Valneva Austria Gmbh | Inactivated sars-cov-2 virus vaccine |
| WO2023232901A1 (en) | 2022-06-01 | 2023-12-07 | Valneva Austria Gmbh | Clostridium difficile vaccine |
| WO2024153686A1 (en) | 2023-01-18 | 2024-07-25 | Valneva Austria Gmbh | Process for virus production in a bioreactor |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4057685A (en) | 1972-02-02 | 1977-11-08 | Abbott Laboratories | Chemically modified endotoxin immunizing agent |
| US4761372A (en) | 1981-01-12 | 1988-08-02 | New York University | Mutant enterotoxin of E. coli |
| US4673574A (en) | 1981-08-31 | 1987-06-16 | Anderson Porter W | Immunogenic conjugates |
| US4459286A (en) | 1983-01-31 | 1984-07-10 | Merck & Co., Inc. | Coupled H. influenzae type B vaccine |
| US4663160A (en) | 1983-03-14 | 1987-05-05 | Miles Laboratories, Inc. | Vaccines for gram-negative bacteria |
| US4761283A (en) | 1983-07-05 | 1988-08-02 | The University Of Rochester | Immunogenic conjugates |
| US4695624A (en) | 1984-05-10 | 1987-09-22 | Merck & Co., Inc. | Covalently-modified polyanionic bacterial polysaccharides, stable covalent conjugates of such polysaccharides and immunogenic proteins with bigeneric spacers, and methods of preparing such polysaccharides and conjugates and of confirming covalency |
| US4882317A (en) | 1984-05-10 | 1989-11-21 | Merck & Co., Inc. | Covalently-modified bacterial polysaccharides, stable covalent conjugates of such polysaccharides and immunogenic proteins with bigeneric spacers and methods of preparing such polysaccharides and conjugataes and of confirming covalency |
| US5308835A (en) | 1984-07-09 | 1994-05-03 | Praxis Biologics, Inc. | Production of the E. coli LT-B enterotoxin subunit |
| US4808700A (en) | 1984-07-09 | 1989-02-28 | Praxis Biologics, Inc. | Immunogenic conjugates of non-toxic E. coli LT-B enterotoxin subunit and capsular polymers |
| NL8802046A (nl) | 1988-08-18 | 1990-03-16 | Gen Electric | Polymeermengsel met polyester en alkaansulfonaat, daaruit gevormde voorwerpen. |
| CA2059693C (en) | 1991-01-28 | 2003-08-19 | Peter J. Kniskern | Polysaccharide antigens from streptococcus pneumoniae |
| CA2059692C (en) | 1991-01-28 | 2004-11-16 | Peter J. Kniskern | Pneumoccoccal polysaccharide conjugate vaccine |
| AUPO264096A0 (en) | 1996-09-30 | 1996-10-24 | Mclaughlin Geosurveys Pty Ltd. | Value improvement of clays |
| US6818222B1 (en) | 1997-03-21 | 2004-11-16 | Chiron Corporation | Detoxified mutants of bacterial ADP-ribosylating toxins as parenteral adjuvants |
| DK1025209T3 (da) | 1997-08-28 | 2007-06-25 | Cheil Jedang Corp | Et attenueret japansk encephalitis-virus tilpasset til Vero-celle og en japansk encephalitis-vaccine |
| ATE446107T1 (de) | 1998-08-19 | 2009-11-15 | Baxter Healthcare Sa | Immunogenes beta-propionamido-gebundenes polysaccharid-protein konjugat geeignet als impfstoff und hergestellt bei verwendung von n- acryloyliertem polysaccharid |
| WO2002064162A2 (en) | 2001-02-13 | 2002-08-22 | Government Of The United States, As Represented By The Secretary Of The Army | Vaccine for transcutaneous immunization |
| GB0118249D0 (en) * | 2001-07-26 | 2001-09-19 | Chiron Spa | Histidine vaccines |
| US20110189217A1 (en) * | 2008-06-26 | 2011-08-04 | Barry Michael A | Methods and materials for producing immune responses against polypeptides involved in antibiotic resistance |
| CN101734698B (zh) | 2009-09-08 | 2013-01-09 | 东北大学 | 一种由含铝物料制备氧化铝的方法 |
| DK3269386T3 (da) | 2011-12-06 | 2022-10-24 | Valneva Austria Gmbh | Aluminiumforbindelser til anvendelse inden for behandling og i vacciner |
-
2012
- 2012-12-06 DK DK17185526.5T patent/DK3269386T3/da active
- 2012-12-06 ES ES16183076.5T patent/ES2647882T3/es active Active
- 2012-12-06 SI SI201231133T patent/SI3106176T1/en unknown
- 2012-12-06 PL PL16183076T patent/PL3106176T3/pl unknown
- 2012-12-06 EP EP16183076.5A patent/EP3106176B1/en not_active Revoked
- 2012-12-06 DK DK20184308.3T patent/DK3785730T3/da active
- 2012-12-06 EP EP17185526.5A patent/EP3269386B1/en active Active
- 2012-12-06 PT PT171855265T patent/PT3269386T/pt unknown
- 2012-12-06 RS RS20171241A patent/RS56709B1/sr unknown
- 2012-12-06 LT LTEP16183076.5T patent/LT3106176T/lt unknown
- 2012-12-06 EP EP24167413.4A patent/EP4400115A3/en active Pending
- 2012-12-06 FI FIEP20184308.3T patent/FI3785730T3/fi active
- 2012-12-06 DK DK16183076.5T patent/DK3106176T3/en active
- 2012-12-06 EP EP12795830.4A patent/EP2788023B1/en active Active
- 2012-12-06 EP EP20184308.3A patent/EP3785730B1/en active Active
- 2012-12-06 HU HUE16183076A patent/HUE037325T2/hu unknown
- 2012-12-06 PL PL17185526.5T patent/PL3269386T3/pl unknown
- 2012-12-06 DK DK12795830.4T patent/DK2788023T3/en active
- 2012-12-06 DE DE202012012768.3U patent/DE202012012768U1/de not_active Expired - Lifetime
- 2012-12-06 ES ES17185526T patent/ES2930458T3/es active Active
- 2012-12-06 PT PT161830765T patent/PT3106176T/pt unknown
- 2012-12-06 WO PCT/EP2012/074701 patent/WO2013083726A1/en active Application Filing
- 2012-12-06 NO NO16183076A patent/NO3106176T3/no unknown
-
2013
- 2013-12-09 DK DKBA201300194U patent/DK201300194U1/da not_active Application Discontinuation
- 2013-12-09 DK DKBA201300193U patent/DK201300193U1/da not_active IP Right Cessation
-
2017
- 2017-12-04 HR HRP20171871TT patent/HRP20171871T1/hr unknown
-
2018
- 2018-01-10 CY CY20181100033T patent/CY1119927T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| EP3106176B1 (en) | 2017-10-11 |
| EP3785730B1 (en) | 2024-04-24 |
| EP4400115A2 (en) | 2024-07-17 |
| NO3106176T3 (es) | 2018-03-10 |
| DE202012012768U1 (de) | 2014-01-21 |
| SI3106176T1 (en) | 2018-04-30 |
| EP4400115A3 (en) | 2024-10-23 |
| EP2788023B1 (en) | 2016-11-02 |
| DK3106176T3 (en) | 2018-01-08 |
| PL3269386T3 (pl) | 2022-12-05 |
| DK3785730T3 (da) | 2024-05-06 |
| DK201300194U1 (da) | 2014-01-17 |
| EP3269386A1 (en) | 2018-01-17 |
| ES2647882T3 (es) | 2017-12-27 |
| LT3106176T (lt) | 2018-01-10 |
| EP3106176A1 (en) | 2016-12-21 |
| CY1119927T1 (el) | 2018-12-12 |
| HUE037325T2 (hu) | 2018-08-28 |
| DK2788023T3 (en) | 2016-12-19 |
| EP3269386B1 (en) | 2022-09-14 |
| EP3785730A1 (en) | 2021-03-03 |
| EP2788023A1 (en) | 2014-10-15 |
| HRP20171871T1 (hr) | 2018-01-12 |
| DK3269386T3 (da) | 2022-10-24 |
| PT3269386T (pt) | 2022-12-05 |
| DK201300193U1 (da) | 2014-01-17 |
| FI3785730T3 (fi) | 2024-05-06 |
| PT3106176T (pt) | 2017-12-20 |
| PL3106176T3 (pl) | 2018-03-30 |
| WO2013083726A1 (en) | 2013-06-13 |
| RS56709B1 (sr) | 2018-03-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2930458T3 (es) | Compuestos de aluminio para su uso en agentes terapéuticos y vacunas | |
| US11110170B2 (en) | Aluminum compounds for use in therapeutics and vaccines | |
| ES2553385T3 (es) | Proceso para preparar una composición de inmunoglobulina | |
| ES2658851T3 (es) | Vacunas de matriz de proteína y métodos de preparación y administración de tales vacunas | |
| ES2797504T3 (es) | Proteína de enlace del factor H meningocócico utilizada como adyuvante | |
| Test et al. | Increased immunogenicity and induction of class switching by conjugation of complement C3d to pneumococcal serotype 14 capsular polysaccharide | |
| TW201925458A (zh) | 包含肺炎鏈球菌多醣-蛋白結合物之組合物及其使用方法 | |
| Ljutic et al. | Formulation, stability and immunogenicity of a trivalent pneumococcal protein vaccine formulated with aluminum salt adjuvants | |
| ES2803578T3 (es) | Métodos mejorados para la inactivación del enterovirus, adsorción de adjuvante y composiciones de vacuna de dosis reducida obtenidas de los mismos | |
| Khim et al. | Deimmunization of flagellin for repeated administration as a vaccine adjuvant | |
| WO2013135274A1 (en) | Aluminium compounds for use in therapeutics and vaccines | |
| US20100040625A1 (en) | Pharmaceutical Compositions Containing Monoclonal Anti-Idiotypic Anti-Ca-125 Antibody and Aluminium Derivatives | |
| GB2500204A (en) | Vaccine comprising aluminium adjuvant having low levels of contaminating heavy metal ions | |
| EP3111953A1 (en) | Whole-inactivated influenza virus as an adjuvant for peptide antigens | |
| EP3307315B1 (en) | Method for preparation of aluminium phosphate gel for application in vaccine formulations | |
| Mudholkar | Relationship between the degree of antigen adsorption in the vaccine or in interstitial fluid and the ensuing immune response | |
| TW201908481A (zh) | 使腸病毒去活化之改良方法、佐劑吸附及所獲得之劑量減少的疫苗組成物 | |
| Sandle | Tackling Safety Issues Of Adjuvanted Vaccines | |
| Osterholm | Current trivalent flu vaccine lacks efficacy; improved flu vaccine needed |