-
Die Erfindung betrifft das Gebiet der Pharmazeutika und Impfstoffe. Genauer gesagt betrifft die Erfindung das Gebiet der Verbindungen und Zusammensetzungen, die zusammen mit dem Medikament und/oder Antigen verabreicht werden.
-
Aluminium-Verbindungen (hier auch als „Aluminium” bezeichnet), einschließlich Aluminiumphosphat (AlPO4), Aluminiumhydroxid (Al(OH)3) und andere Aluminiumpräzipitierte Impfstoffe sind derzeit die am häufigsten verwendeten Adjuvantien mit menschlichen und veterinärmedizinischen Impfstoffen. Die Adjuvantien werden in der Literatur oft als „Alaun” bezeichnet.
-
Aluminium-Adjuvantien werden in der Praxis der Impfung seit mehr als einem halben Jahrhundert verwendet. Sie induzieren frühe, hochtitrige, lang anhaltende schützende Immunität. Milliarden von Dosen von Aluminium-adjuvantierten Impfstoffen wurden über die Jahre verabreicht. Ihre Sicherheit und Wirksamkeit haben sie zu den heutzutage beliebtesten Adjuvantien in Impfstoffen gemacht. Im Allgemeinen werden Aluminium-Adjuvantien als sicher angesehen, wenn sie in Übereinstimmung mit den geltenden Impfplänen verwendet werden.
-
Bei der Impfung von Menschen wurden Aluminium-Adjuvantien von jeher in Tetanus-, Diphtherie-, Pertussis- und Poliomyelitis-Impfstoffen als Teil der Standard-Impfprogramme von Kindern verwendet. Aluminium-Adjuvantien wurden ebenfalls in Hepatitis-A- und Hepatitis-B-Virus-Impfstoffe und Japanisches-Enzephalitis-Virus-(hier auch als „JEV” bezeichnet)-Impfstoffe eingeführt. Andere Aluminium-adsorbierten Impfstoffe gegen beispielsweise Anthrax stehen speziellen Risikogruppen zur Verfügung. In der Veterinärmedizin wurden Aluminium-Adjuvantien in einer großen Anzahl von Impfstoffformulierungen gegen virale und bakterielle Erkrankungen verwendet sowie bei Versuchen, Anti-Parasit-Impfstoffe herzustellen.
-
Adjuvantien dienen typischerweise dazu, das Antigen, die Substanz, welche die spezifische schützende Immunreaktion stimuliert, mit dem Immunsystem in Kontakt zu bringen und Einfluss auf die Art der erzeugten Immunität sowie die Qualität der Immunantwort (Ausmaß oder Dauer) zu nehmen; Adjuvantien können zudem die Toxizität bestimmter Antigene verringern; und einigen Impfstoffkomponenten zu Löslichkeit verhelfen. Studien haben gezeigt, dass viele aluminiumhaltige Impfstoffe höhere und länger andauernde Antikörperantworten als vergleichbare Impfstoffe ohne das Adjuvans hervorrufen. Der Vorteil von Adjuvantien wurde in der Regel während der Erstimmunisierung und nicht bei den Booster-Dosen beobachtet.
-
Es gibt drei Arten von aluminiumhaltigen Adjuvantien:
Aluminiumhydroxid, Aluminiumphosphat und Kaliumaluminiumsulfat (zusammen oft als „Alaun” bezeichnet) Die Wirksamkeit der einzelnen Salze als Adjuvans hängt von den Charakteristika des konkreten Impfstoffs ab und wie der Hersteller den Impfstoff herstellt. Um als Adjuvans zu wirken, wird das Antigen in der Regel am Aluminium adsorbiert; das heißt, es wird mit dem Aluminiumsalz verklumpt, um das Antigen an der Injektionsstelle zu halten.
-
Nicht alle Impfstoffe enthalten Aluminiumsalze. Manchmal kann ein Adjuvans nicht erforderlich gewesen sein, oder es wurde ein anderes Adjuvans ausgewählt. Beispiele für kommerzielle Impfstoffe, die keine Aluminiumsalze enthalten, sind der Impfstoff gegen inaktiviertes Polio-Virus (IPV), Masern-Mumps-Röteln-Impfstoff (MMR), Varizellen-Impfstoff, Meningokokken-Konjugat-(MCV4)-Impfstoff und Influenza-Impfstoffe. Dass die kommerziellen Impfstoffe keine Aluminiumsalze enthalten, bedeutet typischerweise nicht, dass ein Aluminium-Salz nicht funktionieren würde. Es bedeutet nur, dass aus irgendeinem Grund ein anderes Adjuvans ausgewählt wurde.
-
Beispiele für in den USA zugelassene Impfstoffe für Kinder, die Aluminium-Adjuvantien enthalten, sind: DTP (Diphtherie-Tetanus-Pertussis-Impfstoff); DTaP (Diphtherie-Tetanus-azellulärer Pertussis-Impfstoff); einige aber nicht alle Hib-(Haemophilus influenzae Typ b)-Konjugat-Impfstoffe; Pneumokokken-Konjugat-Impfstoff; Hepatitis-B-Impfstoffe; Hepatitis-A-Impfstoffe; Impfstoff gegen menschliches Papillomvirus; Anthrax-Impfstoff und Tollwut-Impfstoff.
-
Aluminium ist ein in unserer Umwelt sehr reichlich vorkommendes Element. Es kommt in vielen Lebensmitteln vor, die wir essen, in vielen Körperpflegeprodukten, die wir auf unsere Haut auftragen (zum Beispiel Deodorants) und in vielen Medikamenten, die wir einnehmen. Verschiedene Regierungsbehörden legen Leitlinien hinsichtlich der Exposition gegenüber potenziell toxischen Substanzen fest. Diese Leitlinien werden als „minimale Risikowerte” bezeichnet – die maximale Menge, der man im Laufe der Zeit – in der Regel täglich – ohne Schaden zu erwarten, ausgesetzt sein kann.
-
Die US-amerikanische Agency for Toxic Substances and Disease Registry (ATSDR) schätzte diese Werte für Säuglinge unter Berücksichtigung der Menge Aluminium (z. B. in Form eines Salzes), die ein Kind essen sowie durch Injektion von Impfstoffen erhalten würde. Die Belastung des Körpers durch Aluminium aus beiden Quellen liegt unterhalb des minimalen Risikowerts, außer vorübergehend nach Impfungen; da 50–70% des injizierten Aluminiums innerhalb von 24 Stunden ausgeschieden wird, wird angenommen, dass dies keine negativen Auswirkungen hat.
-
Aluminiumhydroxid- und Aluminiumphosphat-Adjuvantien werden im Allgemeinen hergestellt, indem wässrige Lösungen von Aluminium-Ionen in der Regel leicht alkalischen Bedingungen in einer gut definierten und kontrollierten chemischen Umgebung ausgesetzt werden. Verschiedene lösliche Aluminiumsalze können zur Herstellung von Aluminiumhydroxid verwendet werden. Zum Zeitpunkt der Präzipitation vorhandene Anionen können copräzipitieren (für eine Übersicht siehe, Lindblad, EB (2004) Immunol. and Cell Biol. B. 82: 497–505).
-
Aluminiumsalz wird auch bei der Herstellung und Zusammensetzung von Arzneimitteln verwendet. Zum Beispiel wird Faktor VIII aus Plasma-Kryopräzipitat aufgereinigt. Das Präzipitat wird solubilisiert, auf Aluminiumhydroxid absorbiert und dann behandelt, um lipidumhüllte Viren zu inaktivieren. Nach mehreren weiteren Verarbeitungsschritten wird das Konzentrat eingesetzt, um Patienten mit Hämophilie A to behandeln (Burnouf T, (1991) Vox Sang. B. 60: S. 8–15).
-
Bei der vorliegenden Erfindung hat es sich gezeigt, dass die Stabilität eines Biologikums in einer Zusammensetzung, die auch ein Aluminiumsalz umfasst, nicht immer gleich ist. Die vorliegende Erfindung zeigt zum Beispiel, dass die Stabilität einer Proteinkomponente (z. B. als solche oder in einem Komplex, wie z. B. einem Virus oder anderen Krankheitserreger) im Rahmen einer wässrigen Zusammensetzung, die ferner Aluminiumsalz umfasst, vom Schwermetallgehalt abhängt. Um von vornherein abzuschätzen, ob das Protein in dieser Zusammensetzung stabil sein wird, sieht die vorliegende Erfindung vor, dass es erforderlich ist, den Restgehalt an Schwermetallen in der Zusammensetzung zu bestimmen (sonst läuft die wässrige Zusammensetzung, die ein Protein umfasst, Gefahr, im Laufe der Zeit abgebaut zu werden, insbesondere sieht die Erfindung vor, dass diese Gefahr beträchtlich ist, wenn der Restgehalt an Schwermetallen in der wässrigen Zusammensetzung über 350 ppb (d. h. etwa 350 ng pro ml) beträgt). Ferner zeigt die Erfindung zudem, dass dieser Restgehalt an Schwermetallen nicht leicht aus der Aluminium-Verbindung entfernt werden kann. Zu diesem Zweck stellt die Erfindung ein Verfahren zum Herstellen einer wässrigen Zusammensetzung, die Aluminium und ein Protein umfasst, bereit, wobei das Verfahren umfasst:
- – das Kombinieren eines Aluminium-Salzes, eines Proteins und Wassers zur Herstellung der wässrigen Zusammensetzung, und
- – Bestimmen des Gehalts eines Schwermetalls in der wässrigen Zusammensetzung und/oder des Aluminium-Salzes. Zusammensetzungen, die weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, enthalten, können in einer Flüssigphase bei einer Temperatur zwischen 0 und 30 Grad Celsius mindestens 1 Monat lang, wie z. B. 20 Monate bei 2–8°C, gelagert werden. Die Proteinkomponente in der Zusammensetzung ist in der Flüssigphase mindestens 1 Monat lang stabil. Zusammensetzungen, die mehr als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, umfassen, können unter solchen Bedingungen nicht über einen längeren Zeitraum gelagert werden, da die Proteinkomponente in der Zusammensetzung über den angegebenen Zeitraum eine Änderung in mindestens einem Aspekt erfährt. Ein Milliliter oder ein Gramm wässrige Zusammensetzung umfasst somit vorzugsweise nicht mehr als 350 Nanogramm Schwermetall. Die wässrige Zusammensetzung umfasst vorzugsweise zwischen 0,1 mg/ml und 2,5 mg/ml Aluminium. Die durchschnittliche Aluminium-Dosis pro Verabreichung beträgt vorzugsweise nicht mehr als 1,25 Milligramm (mg). In einer besonders bevorzugten Ausführungsform beträgt die Aluminium-Dosis pro Verabreichung nicht mehr als 0,25 mg Aluminium. Eine Dosis umfasst typischerweise zwischen 0,5 und 1 ml der wässrigen Zusammensetzung.
-
In einem weiteren Aspekt der vorliegenden Erfindung hat es sich gezeigt, dass die Stabilität eines Biologikums in einer Zusammensetzung, die ein Aluminiumsalz und eine reaktive Verbindung umfasst, nicht immer gleich ist. Die vorliegende Erfindung zeigt zum Beispiel, dass die Stabilität einer Proteinkomponente (z. B. als solche oder in einem Komplex, wie z. B. einem Virus oder anderen Krankheitserreger) im Rahmen einer wässrigen Zusammensetzung, die ferner Aluminiumsalz und eine reaktive Komponente, wie z. B. Sulfit, umfasst, ganz entscheidend vom Schwermetallgehalt abhängt. Um von vornherein abzuschätzen, ob die Proteinkomponente (wie beispielsweise eine Proteinkomponente in einem Komplex, wie z. B. einem Viruspartikel; hier auch einfach als Protein bezeichnet) in dieser Zusammensetzung stabil sein wird, ist es erforderlich, den Schwermetallgehalt in der Zusammensetzung zu bestimmen. Eine wässrige Zusammensetzung wird beschrieben, die Aluminium und ein Protein umfasst, wobei ein Aluminiumsalz, das Protein und Wasser zur Herstellung der wässrigen Zusammensetzung kombiniert werden, und
- – Bestimmen des Gehalts eines Schwermetalls in der wässrigen Zusammensetzung und/oder dem Aluminium-Salz. Zusammensetzungen, die weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, umfassen, können in einer Flüssigphase bei einer Temperatur zwischen 0 und 30 Grad Celsius mindestens 1 Monat lang, wie z. B. 20 Monate bei 2–8°C, gelagert werden. Die Proteinkomponente in der Zusammensetzung ist in der Flüssigphase mindestens 1 Monat lang stabil, wie z. B. 20 Monate bei 2–8 °C. Zusammensetzungen, die mehr als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, umfassen, können unter solchen Bedingungen nicht über einen längeren Zeitraum gelagert werden, da die Proteinkomponente in der Zusammensetzung über den angegebenen Zeitraum eine Änderung in mindestens einem Aspekt erfährt. Ein Milliliter oder ein Gramm wässrige Zusammensetzung umfasst somit vorzugsweise nicht mehr als 350 Nanogramm Schwermetall. Die wässrige Zusammensetzung umfasst vorzugsweise zwischen 0,1 mg/ml (Milligramm pro Milliliter) und 2,5 mg/ml Aluminium. Die durchschnittliche Aluminium-Dosis pro Verabreichung beträgt vorzugsweise nicht mehr als 1,25 Milligramm (mg). In einer besonders bevorzugten Ausführungsform beträgt die Aluminium-Dosis pro Verabreichung nicht mehr als 0,25 mg Aluminium. Eine Dosis umfasst typischerweise zwischen 0,5 und 1 ml der wässrigen Zusammensetzung. Eine wässrige Zusammensetzung, die ein Protein, ein Aluminium-Salz und gegebenenfalls eine reaktive Verbindung umfasst, wobei die Zusammensetzung weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, wird hier auch als „eine erfindungsgemäße wässrige Zusammensetzung, die ein Protein umfasst” oder „eine erfindungsgemäße Zusammensetzung, die ein Protein umfasst” bezeichnet.
-
Der Aluminium-Gehalt der wässrigen Zusammensetzung beträgt in der Regel etwa 0,1 mg/ml bis 2,5 mg/ml, wobei der durchschnittliche Impfstoff etwa 0,5 bis 1,5 mg/ml Aluminium-Adjuvans umfasst. Zu diesem Zweck stellt die Erfindung eine wässrige Zusammensetzung, eine wässrige pharmazeutische oder Impfstoffzusammensetzung, bereit, die Aluminium, eine reaktive Verbindung und ein Protein umfasst, zwischen 5 μg/ml und 50 mg/ml Aluminium umfasst und verglichen mit dem Aluminium-Gehalt nicht mehr als 700 ppm eines Schwermetalls (Gramm/Gramm) umfasst. In diesem Zusammenhang entsprechen 700 ppm eines Schwermetalls 700 μg Schwermetall pro Gramm Aluminium.
-
In einer bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung, die wässrige pharmazeutische oder Impfstoffzusammensetzung, die Aluminium, eine reaktive Verbindung und ein Protein umfasst, zwischen 5 μg/ml und 50 mg/ml Aluminium und umfasst nicht mehr als 450 ppm eines Schwermetalls, bezogen auf den Aluminiumgehalt (Gramm/Gramm). In diesem Zusammenhang entsprechen 450 ppm eines Schwermetalls 450 μg Schwermetall pro Gramm Aluminium.
-
In einer bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung, die wässrige pharmazeutische oder Impfstoffzusammensetzung, die Aluminium, eine reaktive Verbindung und ein Protein umfasst, zwischen 5 μg/ml und 50 mg/ml Aluminium und umfasst nicht mehr als 700 ppm Fe, bezogen auf den Aluminiumgehalt (Gramm/Gramm). Vorzugsweise beträgt der Fe-Gehalt weniger als 420 ppm Fe, bezogen auf den Aluminium-Gehalt. Vorzugsweise beträgt der Fe-Gehalt weniger als 350 ppm, bevorzugt weniger als 100 und besonders bevorzugt weniger als 50 ppm, bezogen auf den Aluminium-Gehalt (1 ppm = 1 μg/Gramm Al). In einer besonders bevorzugten Ausführungsform beträgt der Fe-Gehalt der wässrigen Zusammensetzung, des wässrigen Pharmazeutikums oder Impfstoffs weniger als 420 ppm, bezogen auf den Aluminium-Gehalt. In einer bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung, das wässrige Pharmazeutikum oder der wässrige Impfstoff nicht mehr als 10 ppm Fe, bezogen auf den Aluminium-Gehalt.
-
In einer bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung, die wässrige pharmazeutische oder Impfstoffzusammensetzung, die Aluminium, eine reaktive Verbindung und ein Protein umfasst, zwischen 5 μg/ml und 50 mg/ml Aluminium und umfasst nicht mehr als 35 ppm Ni, bezogen auf den Aluminium-Gehalt (Gramm/Gramm). Vorzugsweise beträgt der Ni-Gehalt weniger als 18 ppm Ni, bezogen auf den Aluminium-Gehalt. Vorzugsweise beträgt der Ni-Gehalt weniger als 9 ppm, vorzugsweise weniger als 3 und besonders bevorzugt weniger als 1 ppm, bezogen auf den Aluminium-Gehalt (1 ppm = 1 μg/Gramm Al). In einer besonders bevorzugten Ausführungsform beträgt der Ni-Gehalt der wässrigen Zusammensetzung, des wässrigen Pharmazeutikums oder Impfstoffs weniger als 18 ppm, bezogen auf den Aluminium-Gehalt. In einer bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung, eine wässrige pharmazeutische oder Impfstoffzusammensetzung mindestens 200 ppb Ni, bezogen auf den Aluminium-Gehalt.
-
In einer bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung, die wässrige pharmazeutische oder Impfstoffzusammensetzung, die Aluminium, eine reaktive Verbindung und ein Protein umfasst, zwischen 5 μg/ml und 50 mg/ml Aluminium und umfasst nicht mehr als 5 ppm Cu, bezogen auf den Aluminium-Gehalt (Gramm/Gramm). Vorzugsweise beträgt der Cu-Gehalt weniger als 2,5 ppm Cu, bezogen auf den Aluminium-Gehalt. Vorzugsweise beträgt der Cu-Gehalt weniger als 1 ppm, bevorzugt weniger als 0,5 und besonders bevorzugt weniger als 0,25 ppm, bezogen auf den Aluminium-Gehalt (1 ppm = 1 μg/Gramm Al). In einer besonders bevorzugten Ausführungsform beträgt der Cu-Gehalt der wässrigen Zusammensetzung, des wässrigen Pharmazeutikums oder Impfstoffs weniger als 2,5 ppm, bezogen auf den Aluminium-Gehalt. In einer bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung, die wässrige pharmazeutische oder Impfstoffzusammensetzung mindestens 50 ppb Cu, bezogen auf den Aluminium-Gehalt.
-
In einer besonders bevorzugten Ausführungsform beträgt der Fe-Gehalt der wässrigen Zusammensetzung, des wässrigen Pharmazeutikums oder Impfstoffs weniger als 420 ppm, bezogen auf den Aluminium-Gehalt, der Ni-Gehalt der wässrigen Zusammensetzung, des wässrigen Pharmazeutikums oder Impfstoffs weniger als 18 ppm, bezogen auf den Aluminium-Gehalt, und der Cu-Gehalt der wässrigen Zusammensetzung, des wässrigen Pharmazeutikums oder Impfstoffs weniger als 2,5 ppm, bezogen auf den Aluminium-Gehalt. In einer bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung, das wässrige Pharmazeutikum oder der wässrige Impfstoff mehr als 10 ppm Fe, bezogen auf den Aluminium-Gehalt, mindestens 200 ppb Ni, bezogen auf den Aluminium-Gehalt, und mindestens 50 ppb Cu, bezogen auf den Aluminium-Gehalt.
-
Bei einem Verfahren zur Herstellung einer erfindungsgemäßen wässrigen Zusammensetzung, wässrigen pharmazeutischen oder Impfstoffzusammensetzung, die Aluminium, eine reaktive Verbindung und ein Protein umfasst, oder ein Verfahren zur Herstellung oder Auswahl eines erfindungsgemäßen Aluminiumsalzes, oder einem Verfahren zur Herstellung eines erfindungsgemäßes Aluminium-Salz-Präzipitats klinischer Qualität ist es bevorzugt, dass der Schwermetallgehalt, bezogen auf den Aluminium-Gehalt, wie vorstehend angegeben ist. Ein Verfahren wird bereitgestellt zur Herstellung einer wässrigen Zusammensetzung, die Aluminium, eine reaktive Verbindung und ein Protein umfasst bereit, wobei das Verfahren Folgendes umfasst:
- – Herstellen oder Auswählen eines Aluminium-Salzes, das eine wässrige Zusammensetzung bereitstellen kann, die weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, aufweist, und
- – Kombinieren des Aluminiumsalzes, der reaktiven Verbindung, des Proteins und Wassers zur Herstellung der wässrigen Zusammensetzung,
wobei die wässrige Zusammensetzung zwischen 5 μg/ml und 50 mg/ml Aluminium, vorzugsweise zwischen 0,1 mg/ml und 2,5 mg/ml, umfasst; und nicht mehr als 700 ppm Schwermetall und vorzugsweise nicht mehr als 450 ppm Schwermetall, bezogen auf den Aluminium-Gehalt; wobei die wässrige Zusammensetzung nicht mehr als 420 ppm Fe, vorzugsweise nicht mehr als 100 ppm, bezogen auf den Aluminium-Gehalt, umfasst; nicht mehr als 18 ppm, vorzugsweise nicht mehr als 3 ppm Ni, bezogen auf den Aluminium-Gehalt; und/oder nicht mehr als 2,5 ppm Cu, vorzugsweise nicht mehr als 0,5 ppm, bezogen auf den Aluminium-Gehalt. In einer bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung nicht mehr als 700 ppm Schwermetall und vorzugsweise nicht mehr als 450 ppm Schwermetall, bezogen auf den Aluminium-Gehalt; nicht mehr als 420 ppm Fe, vorzugsweise nicht mehr als 100 ppm, bezogen auf den Aluminium-Gehalt; nicht mehr als 18 ppm, vorzugsweise nicht mehr als 3 ppm Ni, bezogen auf den Aluminium-Gehalt; und nicht mehr als 2,5 ppm Cu, vorzugsweise nicht mehr als 0,5 ppm, bezogen auf den Aluminium-Gehalt.
-
Es wurde festgestellt, dass die Aluminium-Komponente eine wichtige Quelle für die Schwermetalle in der wässrigen Zusammensetzung darstellt. Somit besteht eine Möglichkeit, die Menge an Schwermetall in der wässrigen Zusammensetzung zu kontrollieren, darin, die Menge an Schwermetall in der Aluminiumquelle, die zur Erzeugung der wässrigen Zusammensetzung verwendet wurde, zu kontrollieren. Ferner wird ein Verfahren zur Herstellung einer wässrigen Zusammensetzung beschreiben, die Aluminium und ein Protein umfasst, bereit, wobei das Verfahren Folgendes umfasst:
- – Herstellen oder Auswählen einer Aluminium-Salz-Lösung (wie z. B. 10 mg/ml Aluminiumhydroxid-Flüssigkeit (wie z. B. Alhydrogel® 2% von Brenntag Biosector, Katalog-Nummer 843261), die in der fertigen Proteinformulierung nicht mehr als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, umfasst (z. B. sollte für das Alhydrogel® 2% und die Endmenge von 0,25 mg Aluminiumhydroxid in der wässrigen Zusammensetzung von 0,5 ml (= Dosis) die ausgewählte oder hergestellte Aluminium-Salz-Lösung nicht mehr als 7 Mikrogramm Schwermetall pro Milliliter der Alhydrogel® 2%-Lösung, einen Schwermetallgehalt von etwa 7 ppm, enthalten) und
- – Kombinieren der Aluminium-Salz-Lösung, des Proteins, Wassers und gegebenenfalls einer reaktiven Verbindung zur Herstellung der wässrigen Zusammensetzung (mit nicht mehr als 350 ppb Schwermetall in der wässrigen Zusammensetzung).
-
Zum Beispiel sollte die Aluminium-Salzlösung, z. B. die Aluminiumhydroxid-Flüssigkeit (verwendet als eine Komponente, die zum Ergeben der fertigen wässrigen Zusammensetzung beigemischt werden sollte), keinen Schwermetallgehalt höher als 7 ppm (hier als Beispiel angegeben, wobei die 10 mg/ml Aluminiumhydroxid-Flüssigkeit auf 0,5 mg/ml verdünnt werden, um einen Schwermetallgehalt von etwa 350 ppb, bezogen auf die wässrige Zusammensetzung, zu ergeben, unter der Annahme 1 ppm = etwa mg/ml), bezogen auf das Gewicht der Aluminiumhydroxid-Flüssigkeit aufweisen. Der Grenzwert des zulässigen Schwermetallgehalts kann auch bezogen auf das Gewicht des Aluminium-Salzes, wie beispielsweise des Aluminiumhydroxyds in Lösung (auch als „Aluminium-Ausgangsverbindung” bezeichnet), ausgedrückt werden. Der zulässige Schwermetallgehalt in diesem Beispiel darf 7 Mikrogramm Schwermetall für jedes Gramm Aluminiumhydroxid-Lösung (d. h. etwa 7 ppm), d. h. der Alhydrogel® 2%-Lösung, nicht überschreiten. Wie hier vorstehend angegeben umfasst die wässrige Zusammensetzung, die das Protein umfasst, vorzugsweise (wie z. B. bei Verwendung als ein Impfstoff) zwischen 0,1 bis 2,5 mg/ml Aluminium-Verbindung. Erfindungsgemäß sollte die Konzentration der Schwermetalle in der wässrigen Zusammensetzung jedoch 350 ppb, d. h. etwa 350 ng pro ml der fertigen Zusammensetzung, nicht überschreiten, und damit muss die Auswahl oder die Herstellung der Aluminium-Ausgangsverbindung entsprechend vorgenommen werden. Zur weiteren Erläuterung der Auswahl einer geeigneten Lösung einer Aluminium-Ausgangsverbindung (z. B. in Form einer konzentrierten Lösung (siehe vorstehendes Alhydrogel® 2% = 10 mg/ml) wird gezeigt, dass eine Lösung einer Aluminium-Verbindung von 10 mg/ml Aluminiumhydroxid, die etwa 7 ppm Schwermetallverunreinigungen aufweist, einer Schwermetallkonzentration in der Proteinzusammensetzung von etwa 70 Nanogramm/ml oder 70 ppb entspricht (also weit unter der von der Erfindung gelehrten Grenze des Schwermetallgehalts von 350 ppb), wenn die Aluminiumkonzentration etwa 0,1 mg/ml beträgt. Eine Konzentration von 2,5 mg/ml des Aluminiumhydroxids in der fertigen wässrigen Zusammensetzung ausgehend von einer Aluminium-Lösung von 10 mg/ml Aluminiumhydroxid, das etwa 7 ppm Schwermetallverunreinigungen aufweist, resultiert in einem Schwermetallgehalt in der wässrigen Proteinzusammensetzung, der einer Schwermetallkonzentration von etwa 1,75 Mikrogramm/ml oder etwa 1.750 ppm entspricht (also weit unter der von der Erfindung gelehrten Grenze des Schwermetallgehalts von 350 ppb). Somit sollte in diesem Fall die Aluminium-Lösung von 10 mg/ml Aluminiumhydroxid (der endgültige Aluminiumhydroxid-Gehalt beträgt 2,5 mg/ml) nicht mehr als 1,4 ppm Schwermetallverunreinigungen aufweisen.
-
Ein Verfahren zur Herstellung einer wässrigen Zusammensetzung, die ein Protein umfasst, umfasst zudem vorzugsweise Verpackungsaliquote der wässrigen Zusammensetzung, die weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, aufweisen, in separaten luftdichten Lagerbehältern. Das Protein in den luftdichten Lagerbehältern ist stabil und kann für mindestens drei Monate, wie z. B. 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 Monate, vorzugsweise 20 oder 24 Monate, besonders bevorzugt 20 Monate bei einer Temperatur von 2 bis 8°C Grad Celsius gelagert werden.
-
Ohne an eine Theorie gebunden zu sein, könnte der Antigenabbau in wässrigen Zusammensetzungen, wie beispielsweise immunogenen Zusammensetzungen, die in einem Aluminiumsalz, wie beispielsweise Aluminiumhydroxid, vorhandene Schwermetallionen umfassen, mit einem zugrunde liegenden Abbauweg erklärt werden, der freie Radikale voraussetzt, wie z. B. freie Sulfit-Radikale. Die Bildung von freien Radikalen kann durch Schwermetallionen katalysiert werden, die z. B. in Aluminiumhydroxid vorhanden sind, und dieser Effekt (bei einer bestimmten Menge an Schwermetall, wie erfindungsgemäß angegeben) könnte der Mechanismus der zugrunde liegenden Ursache der Stabilitätsprobleme sein, wie als Teil des erfinderischen Beitrags identifiziert. Der experimentelle Teil dieser Anmeldung zeigt in großer Ausführlichkeit die Hinweise auf diese Ursache für den Impfstoff gegen Japanische Enzephalitis (auch als „JEV” bezeichnet) und zeigt ein ähnliches Ergebnis für eine einfache Aluminium-adjuvantierte Polypeptidzusammensetzung, die eine reaktive Verbindung, wie Sulfit, umfasst. Somit ist ersichtlich, dass eine ähnliche Umsetzung auch in einer anderen wässrigen Zusammensetzung stattfinden kann, die Aluminium (mit einem hohen Schwermetallgehalt, z. B. über 350 ppb, bezogen auf das Gewicht der Zusammensetzung), Protein und möglicherweise eine reaktive Komponente, wie Sulfit und/oder andere radikalbildende Partikeln, umfasst. Schwermetallkatalysierte Oxidation ist ein Abbauweg, der in kovalenter Modifikation von Proteinen resultiert. Die modifizierten physikalisch-chemischen Eigenschaften des oxidierten/modifizierten Proteins oder Antigens können in einem Verlust der biologischen Aktivität resultieren (Li et al., 1995; Mayo et al., 2003; Stadtman, 1990).
-
Die folgenden Reaktionsschemata (wie sie möglicherweise im JEV-Produkt auftreten, wie im experimentellen Teil beschrieben) stehen für die erfindungsgemäßen Zusammensetzungen, die zusätzlich zu dem Protein und dem Schwermetall auch Sulfit und/oder eine andere reaktive Verbindung, wie z. B. Sulfit und/oder Formaldehyd, umfassen.
-
Natriummetabisulfit (Na2S2O5) löst sich in Lösung als Bisulfit (S2O5 2–), in einer alkalischen Lösung ist das Bisulfit-Gleichgewicht in Richtung Sulfit (SO3 2–) und in einer sauren Lösung in Richtung H2SO3/SO2 verschoben. Nach Auflösung von Na2S2O5 in Wasser wird es zu NaHSO3 wie folgt hydrolysiert: Na2S2O5 ↔ 2NaHSO3
-
Bei neutralem pH kann das folgende Gleichgewicht angenommen werden: HSO3 – ↔ H+ + SO3 2– pKa = 7,2
-
Dies bedeutet, dass bei pH 7 das Gleichgewicht in Richtung HSO3– und unter mehr basischen Bedingungen (z. B. pH 8) in Richtung SO3 2– verschoben ist.
-
Formaldehyd bildet ein Bisulfit-Addukt bei der Neutralisation gemäß der folgenden Gleichung: CH2O + HSO3 – → CH2(OH)(SO3)–
-
Nachdem das Bisulfat aufgebraucht ist, verläuft die Reaktion bis zum Erreichen des Gleichgewichts wie folgt: CH2O + SO3 2– + H2O → CH2(OH)(SO3)– + OH–
-
Formaldehyd und Sulfit reagieren miteinander, es wurde jedoch festgestellt, dass im JEV-Impfstoff Formaldehyd und Sulfit noch im Gleichgewicht vorliegen und im folgenden Bereich nachgewiesen werden können (n = 49):
| Freisetzungsergebnisse im Wirkstoff | Freies Sulfit | Freier Formaldehyd |
| Durchschnitt (ppm)
Durchschnitt (mM) | 113,9
1,41 | 41,9
1,36 |
| Standardabweichung (ppm) | 24 | 14,3 |
| Min. (ppm) | 66 | 10,6 |
| Max. (ppm) | 174 | 78,7 |
-
Nach Literaturangaben (Ranguelova et al., 2010) katalysieren Übergangsmetallionen die Autoxidation von (Bi)sulfit über Schwefeltrioxid-Radikalanion (•SO3 –)-Bildung: Mn+ + SO3 2– → M(n-1)+ + •SO3 – wobei M Kupfer (Cu2 +), Eisen (Fe3+), Oxovanadium (VO2 +), Mangan (Mn2 +), Nickel (Ni2+) oder Chromat-Anion (CrO4 2–) sein kann (Alipazaga et al. 2004; Berglund et al. 1993; Brandt und Elding 1998; Lima et al. 2002; Shi 1994).
-
Es wurde gezeigt, dass solche Sulfit-Radikale höchst reaktiv sind und verschiedene Substanzen, wie zum Beispiel Ascorbat, Hydrochinon und Histidin, oxidieren können (Huie et al., 1985). Eine Übersicht der Chemie freier Radikale von Sulfit wurde von Neta & Huie, 1985 veröffentlicht. Die Autoren zeigen auch, dass die Radikalbildung auch durch Photoionisation von Sulfit wie folgt katalysiert werden kann: SO3 2– + hv → •SO3 – + e–
-
Die durch Licht katalysierte Radikalbildung könnte auch Unterschiede, die bei der Wirkstärke und bei ELISA-Ergebnissen von unbeschrifteten nackten Spritzen (für Freigabetests und zu Referenzzwecken eingesetzt) und Proben von komplett verpackten Fertigimpfstofflosen für das JEV-Produkt beobachtet werden. Ganz eingepackte Proben sind vollständig vor Licht geschützt, während unbeschriftete Spritzen bei der Lagerung und Handhabung Licht ausgesetzt werden könnten.
-
Eine wichtige Reaktion des Sulfit-Radikals in Autoxidationssystemen findet mit molekularem Sauerstoff zu einem Peroxyl-Radikal statt, das sehr viel reaktiver ist: •SO3 –+ O2 → •SO5 –
-
Die Löslichkeit von 02 in Wasser bei 0°C und 20°C beträgt 0,4 mM bzw. 0,25 mM. Unter der Annahme, dass die O2-Löslichkeit bei einer erfindungsgemäßen Zusammensetzung in einem ähnlichen Bereich liegt, ist eine beträchtliche Menge an Sauerstoff vorhanden, um das Peroxyl-Radikal zu bilden. Dieses Radikal ist ein viel stärkeres Oxidationsmittel verglichen mit •SO3 – und kann bestimmte Substrate oxidieren, die von •SO3 – überhaupt nicht gebunden werden und die in der Tat Radikale bilden können, die Sulfit-Ionen oxidieren können. In solchen Fällen, bei denen das Redoxpotenzial des Substrats zwischen dem von •SO3 – und •SO5 – liegt, ist im Vorhandensein von O2 die Entwicklung einer Reaktionskette wahrscheinlich, die dem allgemeinen Muster folgt: •SO3 – + O2 → •SO5 – •SO5 – + X → SO5 2– + •X+ •X+ + SO3 2– → X + •SO3 –
-
Das Dazwischenliegen eines Substrats X kann den Kettenprozess der Sulfit-Oxidation durch Sauerstoff verstärken. Die Ein-Elektronen-Reduktion von •SO5 – ergibt HSO5 – (Peroxymonosulfat), ein sehr starkes Oxidationsmittel, das fähig ist, viele organische Verbindungen zu oxidieren (Lambeth et al., 1973; Ito & Kawanashi, 1991).
-
Peroxymonosulfat ist auch ein Vorläufer des Sulfat-Anionradikals •SO4 –. •SO5 – + HSO3 – → •SO4 – + HSO4 –
-
Das •SO4 –-Radikal ist ein sehr starkes Oxidationsmittel, fast so stark wie das Hydroxyl-Radikal (•OH), und oxidiert sehr wahrscheinlich andere Biomoleküle durch Ein-Elektronen-Oxidation.
-
In einer bevorzugten Ausführungsform ist die erfindungsgemäße Zusammensetzung, die ein Protein umfasst, eine therapeutische Zusammensetzung oder eine immunogene Zusammensetzung, wie beispielsweise ein Impfstoff. Therapeutische Zusammensetzungen werden Individuen verabreicht, wie beispielsweise einem Menschen oder einem Tier. Insbesondere ist es für solche Zusammensetzungen wichtig, dass das Protein in der Zusammensetzung zum Zeitpunkt der Verabreichung an das Individuum noch seine therapeutische Wirkung besitzt. Ein Abbau des Proteins oder Veränderungen des Proteins in seiner Struktur können zu einem Verlust der therapeutischen Wirksamkeit des Proteins führen. Ebenso führt ein Abbau oder führen strukturelle Veränderungen der immunogenen Zusammensetzung ebenfalls zu einer Verringerung der Wirksamkeit der Zusammensetzung bei der Induktion und/oder Steigerung einer Immunantwort in einem Individuum. Eine immunogene Zusammensetzung wird einem Individuum vorzugsweise verabreicht, um einer viralen oder bakteriellen Infektion entgegenzuwirken oder diese zu verhindern. In der wässrigen immunogenen Zusammensetzung umfassenes Protein kann ein einzelnes Protein oder ein multimeres Protein oder ein Teil eines Komplexes sein, der das Protein umfasst (z. B. als Teil eines Virus oder einer Zelle, z. B. einer Bakterienzelle).
-
In einer bevorzugten Ausführungsform umfasst das Protein oder der Proteinkomplex ein attenuiertes, inaktiviertes oder mutiertes Virus oder Bakterium oder ein immunogenes virales oder bakterielles Protein oder einen immunogenen Teil eines solchen Proteins (z. B. ein immunogenes Peptid), ein Spaltirus oder -bakterium oder ganze Bakterienzellen. Wenn die immunogene Zusammensetzung verabreicht wird, um Schutz gegen eine virale oder bakterielle Infektion zu bieten, kann ein Proteinabbau zum Verlust der Schutzfähigkeit der immunogenen Zusammensetzung führen. Im hier verwendeten Sinne ist ein „lebendes attenuiertes” Virus oder Bakterium ein Virus oder ein Bakterium, das weniger pathogen als das Wildtyp-Virus oder -Bakterium ist, aber immunogene Eigenschaften erhalten hat. Im hier verwendeten Sinne bezieht sich ein „inaktiviertes” Virus oder Bakterium auf ein Virus oder Bakterium, das inaktiviert wurde, so dass es nicht mehr infektiös ist, während immunogene Eigenschaften zumindest teilweise erhalten wurden. Ein inaktiviertes Virus oder Bakterium kann in Form von inaktivierten ganzen Virus- oder bakteriellen Zellen vorliegen. Allerdings kann eine Inaktivierung in der Spaltung von Viren oder bakteriellen Zellen resultieren. Daher kann ein inaktiviertes Virus oder Bakterium auch in gespaltener Form vorliegen. Im hier verwendeten Sinne bezieht sich „Spalt”-Virus oder -Bakterium auf ein Virus oder Bakterium, das beispielsweise unter Verwendung eines Detergens gespalten wird.
-
Die erfindungsgemäße wässrige Zusammensetzung kann eine reaktive Verbindung, wie beispielsweise Formaldehyd, umfassen, die im Allgemeinen vorhanden ist, um ein Virus oder Bakterium zu inaktivieren. Eine zusätzliche oder alternative reaktive Verbindung kann zudem vorhanden sein, um etwaigen restlichen Formaldehyd in der wässrigen Lösung zu inaktivieren, wie beispielsweise die reaktive Verbindung Sulfit. Wie vorstehend dargelegt wird angenommen, dass Schwermetalle die Oxidation von (Bi)sulfit und die Bildung von Sulfit-Radikalen, die in der wässrigen Zusammensetzung vorhanden sind, katalysieren, was wiederum einen Abbau des Proteins, das in der Zusammensetzung vorhanden ist, induziert. Daher ist es wahrscheinlich, dass Zusammensetzungen, die inaktivierte Viren oder Bakterien oder immunogene Teile davon enthalten, eine oder mehrere reaktive Verbindungen umfassen. Daher ist die Abwesenheit von Schwermetallen oder das Vorhandensein unterhalb der durch die vorliegende Erfindung identifizierten Gehalte von besonderer Bedeutung, wenn die erfindungsgemäße wässrige Zusammensetzung ein inaktiviertes Virus oder Bakterium oder einen immunogenen Teil davon umfasst. In einer besonders bevorzugten Ausführungsform umfasst eine erfindungsgemäße immunogene Zusammensetzung daher ein inaktiviertes Virus. In einer bevorzugten Ausführungsform umfasst die erfindungsgemäße wässrige Zusammensetzung, vorzugsweise immunogene Zusammensetzung inaktiviertes Virus oder Bakterium. Ein solches Virus oder Bakterium ist zum Beispiel durch eine reaktive Verbindung, wie beispielsweise Formaldehyd, inaktiviert, wie in der vorliegenden Erfindung beschrieben.
-
In einer weiteren bevorzugten Ausführungsform umfasst die erfindungsgemäße wässrige Zusammensetzung, immunogene Zusammensetzung oder der erfindungsgemäße Impfstoff ein Toxoid. Im hier verwendeten Sinne bezieht sich ein „Toxoid” auf ein inaktiviertes bakterielles Toxin, wie beispielsweise ein Exotoxin, auf Grund dessen die Toxizität reduziert oder aufgehoben wird, während immunogene Eigenschaften zumindest zum Teil erhalten bleiben. Ein solches Toxin wird zum Beispiel durch eine reaktive Verbindung, wie beispielsweise Formaldehyd, inaktiviert, wie in der vorliegenden Erfindung beschrieben. Beispiele für Toxoide, die in einer erfindungsgemäßen immunogenen Zusammensetzung vorhanden sind, umfassen, ohne darauf beschränkt zu sein, Diphtherie-, Tetanus- und Botulismus-Toxine.
-
Der Begriff „immunogene virales oder bakterielles Protein” bezieht sich auf ein virales oder bakterielles Protein, das fähig ist, eine Immunantwort hervorzurufen. Wie hier verwendet, bezieht sich der Begriff „immunogener Teil” auf einen Teil eines viralen oder bakteriellen Proteins, das fähig ist, eine Immunantwort hervorzurufen. Vorzugsweise erkennt die hervorgerufene Immunantwort sowohl den Teil des Proteins als auch das gesamte Protein. Daher ist in einer Ausführungsform eine erfindungsgemäße wässrige Zusammensetzung, die ein Protein umfasst, eine immunogene Zusammensetzung. Die Zusammensetzung ist vorzugsweise eine therapeutische Zusammensetzung und/oder eine prophylaktische Zusammensetzung, wie beispielsweise ein Impfstoff. Ferner wird ein Impfstoff bereitgestellt, der eine erfindungsgemäße wässrige Zusammensetzung ist, die ein Protein umfasst.
-
Eine „immunogene Zusammensetzung” ist hierin als eine Zusammensetzung definiert, die fähig ist, eine Immunantwort hervorzurufen, wenn sie einem Individuum verabreicht wird. Die hervorgerufene Immunantwort kann humoral, zellulär oder eine Kombination davon sein und umfasst, ohne darauf beschränkt zu sein, die Produktion von Antikörpern, B-Zellen, wie beispielsweise aktivierten B-Zellen, und T-Zellen, wie beispielsweise aktivierten T-Zellen. Im hier verwendeten Sinne ist eine Immunantwort vorzugsweise spezifisch auf ein oder mehrere immunogene in einer erfindungsgemäßen Zusammensetzung, die ein Protein umfasst, gerichtet. Eine erfindungsgemäße immunogene Zusammensetzung kann einem Individuum durch jede in der Technik bekannte Technik verabreicht werden, einschließlich, ohne darauf beschränkt zu sein, intramuskuläre (IM), intradermale (ID), subkutane (SC), intrakranielle (IC), intraperitoneale (IP) oder intravenöse (IV) Injektion, transdermale, orale, intranasale oder rektale Verabreichung, und Kombinationen davon, bevorzugt sind intramuskuläre (IM), intradermale (ID), subkutane (SC), intrakranielle (IC), intraperitoneale (IP) oder intravenöse (IV) Injektion. In einer bevorzugten Ausführungsform wird eine erfindungsgemäße immunogene Zusammensetzung, die ein Protein umfasst, zum Hervorrufen einer Immunantwort verwendet, die bei chronischer Behandlung (wie beispielsweise Krebsbehandlung) oder prophylaktischer Behandlung (wie beispielsweise einem typischen Impfstoff) nützlich sein kann. Es ist bevorzugt, dass die immunogene Zusammensetzung als ein Impfstoff verwendet wird, d. h. eine prophylaktische Anwendung. In dieser Ausführungsform ist das Aluminium typischerweise als Adjuvans vorhanden.
-
Im hier verwendeten Sinne bezieht sich ein „Adjuvans” auf ein pharmakologisches oder immunologisches Mittel, das die Wirkung von anderen Mitteln modifiziert, wie beispielsweise ein immunologisches Mittel, das die Antigenantwort erhöht. Adjuvantien dienen typischerweise dazu, das Antigen – die Substanz, welche die spezifische schützende Immunreaktion stimuliert – mit dem Immunsystem in Kontakt zu bringen und Einfluss auf die Art der erzeugten Immunität sowie die Qualität der Immunantwort (Ausmaß oder Dauer) zu nehmen; die Toxizität bestimmter Antigene zu verringern; und einigen Impfstoffkomponenten zu Löslichkeit zu verhelfen.
-
Ein „Individuum” ist hierin als ein Mensch oder ein Tier definiert. Individuen umfassen, ohne darauf beschränkt zu sein, Hühner, Enten, Gänse, Puten, Schwäne, Emus, Perlhühner und Fasane, Menschen, Schweine, Frettchen, Seehunde, Kaninchen, Katzen, Hunde und Pferde. In einer bevorzugten Ausführungsform der Erfindung ist ein Individuum ein Säuger, vorzugsweise ein Mensch.
-
Das „Mikro” in Mikrogramm oder Mikroliter oder einer anderen Einheit wird manchmal als mc, das Symbol μ oder der Buchstaben u bezeichnet. Ein Wert von 3 μg ist somit 3 Mikrogramm, 3 μl ist 3 Mikroliter und 3 uGramm ist 3 Mikrogramm.
-
Ein Aluminium-Adjuvans wird häufig hergestellt, indem eine wässrigen Lösung von Aluminium-Ionen kontrolliert alkalischen Bedingungen ausgesetzt wird (für eine Übersicht siehe Lindblad, EB (2004) Immunol. and Cell Biol. B. 82: 497–505). In der vorliegenden Erfindung wurde für das JEV-Produkt festgestellt (siehe experimentellen Teil), dass eine große Menge des Schwermetalls in dieser wässrigen Lösung von Aluminium-Ionen im Aluminiumsalz-Präzipitat für das Aluminium-Adjuvans anfällt. Es wurde ferner festgestellt, dass die Menge an Schwermetall im Aluminium-Präzipitat die Stabilität des Impfstoffs während der Lagerung des Impfstoffs beeinflusst. Die Menge an Schwermetall, die im Aluminium-Salz vorhanden ist, kann somit durch Festlegen der Menge an Schwermetall im Salz kontrolliert werden, aber auch und vorzugsweise durch Kontrollieren der Menge an Schwermetall in der wässrigen Lösung von Aluminium-Ionen. Ferner wird ein Verfahren beschrieben zur Herstellung eines Aluminium-Salzpräzipitats klinischer Qualität zur Aufnahme in einem Arzneimittel und/oder einem Impfstoff, wobei das Verfahren Herstellen einer wässrigen Lösung von Aluminium-Ionen und Präzipitieren der Aluminium-Ionen aus der Lösung und Bestimmen des Gehalts eines Schwermetalls in der Lösung und/oder im Aluminium-Salzpräzipitat umfasst, wobei vorzugsweise festgelegt wird, dass die Lösung und/oder das Aluminium-Salzpräzipitat eine Menge umfasst, die in weniger abs 350 ppb Schwermetall in der fertigen Zusammensetzung resultiert, z. B. beim Resuspendieren in der endgültigen Zusammensetzung. In einer bevorzugten Ausführungsform stellt die Erfindung ein Verfahren zur Herstellung eines Aluminium-Salzpräzipitats klinischer Qualität zur Aufnahme in einem Arzneimittel und/oder einem Impfstoff bereit, wobei das Verfahren Herstellen einer wässrigen Lösung von Aluminiumionen und Präzipitieren der Aluminium-Ionen aus der Lösung, und Bestimmen des Gehalts eines Schwermetalls in der Lösung und/oder im Aluminium-Salzpräzipitat umfasst, wobei das Präzipitat ausgewählt wird, das eine wässrige Zusammensetzung bereitstellen kann, die zwischen 5 μg/ml und 50 mg/ml Aluminium, vorzugsweise zwischen 0,1 mg/ml und 2,5 mg/ml umfasst, und das nicht mehr als 700 ppm Schwermetall umfasst und vorzugsweise nicht mehr als 450 ppm Schwermetall bezogen auf den Aluminium-Gehalt, wobei die Zusammensetzung nicht mehr als 420 ppm Fe umfasst, vorzugsweise nicht mehr als 100 ppm, bezogen auf den Aluminium-Gehalt, nicht mehr als 18 ppm, vorzugsweise nicht mehr als 3 ppm Ni, bezogen auf den Aluminium-Gehalt, und/oder nicht mehr als 2,5 ppm Cu, vorzugsweise nicht mehr als 0,5 ppm, bezogen auf den Aluminium-Gehalt. In einer bevorzugten Ausführungsform umfasst das Präzipitat nicht mehr als 700 ppm Schwermetall und vorzugsweise nicht mehr als 450 ppm Schwermetall, bezogen auf den Aluminium-Gehalt, nicht mehr als 420 ppm Fe, vorzugsweise nicht mehr als 100 ppm, bezogen auf den Aluminium-Gehalt, nicht mehr als 18 ppm, vorzugsweise nicht mehr als 3 ppm Ni, bezogen auf den Aluminium-Gehalt, und nicht mehr als 2,5 ppm Cu, vorzugsweise nicht mehr als 0,5 ppm, bezogen auf den Aluminium-Gehalt.
-
Es wird zudem eine erfindungsgemäße pharmazeutische Zusammensetzung, die ein Protein umfasst, bereitgestellt, die gegebenenfalls ferner einen pharmazeutisch verträglichen Träger und/oder ein pharmazeutisch verträgliches Verdünnungsmittel umfasst. Im hier verwendeten Sinne ist „ ein pharmazeutisch annehmbares Verdünnungsmittel” als eine beliebige Lösung, Substanz oder eine Kombination davon definiert, die keine biologische oder anderweitig unerwünschte Aktivität aufweist, was bedeutet, dass sie an ein Individuum zusammen mit anderen Komponenten einer immunologischen Zusammensetzung verabreicht werden kann, ohne dass eine wesentliche unerwünschte Reaktion hervorgerufen wird. Beispiele für geeignete Träger umfassen zum Beispiel Keyhole Limpet Hämocyanin (KLH), Serumalbumin (z. B. BSA oder RSA) und Ovalbumin. In einer bevorzugten Ausführungsform umfasst der geeignete Träger eine Lösung, wie zum Beispiel Kochsalzlösung.
-
In einem Aspekt wird die Erfindung für eine Verlängerung der Lagerbeständigkeit oder Haltbarkeit einer erfindungsgemäßen wässrigen Zusammensetzung, die ein Protein umfasst, benutzt. Im hier verwendeten Sinne wird der Begriff „Haltbarkeit” als der Zeitraum definiert, in der eine erfindungsgemäße Zusammensetzung, die ein Protein umfasst, gelagert werden kann, ohne dass sie zur Verwendung ungeeignet wird, z. B. aufgrund des Abbaus des Proteins (z. B. innerhalb der Spezifikation der Wirkstärke der Zusammensetzung, z. B. des Impfstoffs liegt, wie von der Zulassungsbehörde, die den Impfstoff zugelassen hat oder zulassen wird, gefordert). Während der Lagerung von wässrigen Zusammensetzungen wie hier beschrieben kann es zum Abbau des Proteins kommen, insbesondere wenn ein bestimmter Gehalt (wie hier beschrieben) an Schwermetallen überschritten wird. Bei der Lagerung solcher wässrigen Zusammensetzungen nimmt der Abbau in der Regel mit der Zeit zu. Da nun festgestellt wurde, dass der Abbau von Protein in einer wässrigen Zusammensetzung, die zusätzlich zu dem Protein ein Aluminium-Salz umfasst, verringert wird, wenn die Zusammensetzung weniger als 350 ppb Schwermetall bezogen auf das Gewicht der wässrigen Zusammensetzung umfasst, ist es möglich geworden, dem Proteinabbau in wässrigen Zusammensetzungen entgegenzuwirken. Indem dem Proteinabbau erfindungsgemäß entgegengewirkt wird, wird die Stabilität des Proteins in der Zusammensetzung erhöht und die Haltbarkeit einer wässrigen Zusammensetzung, die das Protein umfasst, verlängert. Eine erfindungsgemäße wässrige Zusammensetzung bietet den Vorteil, dass sie stabil ist, und dass kein Proteinabbau über einen längeren Zeitraum stattfindet. Eine solche erfindungsgemäße wässrige Zusammensetzung, die ein Protein umfasst, ist mindestens einen Monat lang bei erhöhter Temperatur, wie z. B. 20 oder 37°C stabil, vorzugsweise mindestens drei Monate lang bei erhöhter Temperatur, wie z. B. 20 oder 37°C. Um zu einer erhöhten Haltbarkeit beizutragen, wird eine erfindungsgemäße wässrige Zusammensetzung, die ein Protein umfasst, vorzugsweise bei einer Temperatur zwischen 0°C und 20°C gelagert, besonders bevorzugt zwischen 2°C und 15°C, besonders bevorzugt zwischen 2°C und 10°C, ganz besonders bevorzugt zwischen 2°C und 8°C. Die Haltbarkeit bei einer Temperatur zwischen 2°C und 8°C ist vorzugsweise mindestens drei Monate lang stabil, wie z. B. 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 bis 24 Monate, vorzugsweise 20 oder 24 Monate, besonders bevorzugt 20 Monate bei einer Temperatur von 2 bis 8°C Grad Celsius. Es wird somit eine erfindungsgemäße wässrige Zusammensetzung, die ein Protein umfasst, mit einer Haltbarkeit von etwa 12 bis 24 Monaten bereitgestellt. Die Erfindung stellt ferner eine erfindungsgemäße wässrige Lösung bereit, die mindestens einen Monat lang, gelagert wurde, vorzugsweise mindestens zwei Monate lang, besonders bevorzugt mindestens drei Monate lang, besonders bevorzugt mindestens sechs Monate lang. Eine wässrige Zusammensetzung umfasst vorzugsweise ein Protein und ein Aluminiumsalz, wobei die Zusammensetzung weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, zwischen 5 μg/ml und 50 mg/ml Aluminium, vorzugsweise zwischen 0,1 mg/ml und 2,5 mg/ml und nicht mehr als 700 ppm Schwermetall und vorzugsweise nicht mehr als 450 ppm Schwermetall bezogen auf den Aluminium-Gehalt umfasst, wobei die Zusammensetzung nicht mehr als 420 ppm Fe, vorzugsweise nicht mehr als 100 ppm bezogen auf den Aluminium-Gehalt, nicht mehr als 18 ppm, vorzugsweise nicht mehr als 3 ppm Ni bezogen auf den Aluminium-Gehalt, und/oder nicht mehr als 2,5 ppm Cu, vorzugsweise nicht mehr als 0,5 ppm bezogen auf den Aluminium-Gehalt umfasst. In einer bevorzugten Ausführungsform umfasst die Zusammensetzung nicht mehr als 700 ppm Schwermetall und vorzugsweise nicht mehr als 450 ppm Schwermetall bezogen auf den Aluminium-Gehalt, nicht mehr als 420 ppm Fe, vorzugsweise nicht mehr als 100 ppm bezogen auf den Aluminium-Gehalt, nicht mehr als 18 ppm, vorzugsweise nicht mehr als 3 ppm Ni bezogen auf den Aluminium-Gehalt, und nicht mehr als 2,5 ppm Cu, vorzugsweise nicht mehr als 0,5 ppm bezogen auf den Aluminium-Gehalt.
-
Im hier verwendeten Sinne bedeutet „eine stabile Proteinzusammensetzung”, dass bezogen auf eine Ausgangszusammensetzung nicht mehr als 50%, vorzugsweise nicht mehr als 40%, besonders bevorzugt nicht mehr als 30%, besonders bevorzugt nicht mehr als 20%, besonders bevorzugt nicht mehr als 10, besonders bevorzugt nicht mehr als 5% des Proteins in der Zusammensetzung abgebaut sind. In diesem Zusammenhang bedeutet „abgebaut” eine nachweisbare Modifikation des Proteins, verglichen mit dem Protein in der Ausgangszusammensetzung. Geeignet ist zum Beispiel eine Abnahme bei der Detektion des Proteins mit einem monoklonalen Antikörper, wie z. B. einem Antikörper, der ein neutralisierendes Epitop erkennt, und kann mit jedem in der Technik bekannten Verfahren, wie z. B. ELISA (siehe Beispiel 4), gemessen werden. Der detektierte Gehalt kann zum Beispiel mit dem Gehalt, der mit einem Polyklonalen, der für das gleiche Protein spezifisch ist, detektiert wird und die Gesamtmenge an Protein darstellt (verändert oder unverändert), verglichen werden. Zudem wird ein Verfahren zur Verlängerung der Haltbarkeit von einer wässrigen Zusammensetzung, die ein Protein und ein Aluminium-Salz umfasst, bereitgestellt, wobei das Verfahren Auswählen und/oder Herstellen eines Aluminium-Salzes umfasst, was in einer wässrigen Zusammensetzung mit einem Schwermetallgehalt von weniger als 350 ppb bezogen auf die Basis des Gewichts der wässrigen Zusammensetzung resultiert, und Kombinieren des Aluminiumsalzes, des Proteins und Wassers zur Herstellung der wässrigen Zusammensetzung. Es wird ferner ein Verfahren zur Verbesserung der Reproduzierbarkeit der Haltbarkeit von Präparaten von wässrigen Zusammensetzungen, die ein Protein und ein Aluminium-Salz enthalten, bereitgestellt, wobei das Verfahren Erlangen von mindestens zwei unterschiedlichen Aluminium-Salz-Präparaten, Bestimmen der Menge von mindestens einem Schwermetall in den Aluminium-Salz-Präparaten, Auswählen aus den Aluminium-Salz-Präparaten von Aluminium-Salz-Präparaten, die weniger als 350 ppb des mindestens einen Schwermetalls umfassen; und Kombinieren des Aluminiumsalzes der ausgewählten Präparate mit dem Protein und Wasser zur Herstellung der wässrigen Zusammensetzungen, umfasst. Vorzugsweise wird eine Zusammensetzung ausgewählt, die zwischen 5 μg/ml und 50 mg/ml Aluminium, vorzugsweise zwischen 0,1 mg/ml und 2,5 mg/ml umfasst, und nicht mehr als 700 ppm Schwermetall und vorzugsweise nicht mehr als 450 ppm Schwermetall bezogen auf den Aluminium-Gehalt umfasst, wobei die Zusammensetzung nicht mehr als 420 ppm Fe, vorzugsweise nicht mehr als 100 ppm bezogen auf den Aluminium-Gehalt; nicht mehr als 18 ppm, vorzugsweise nicht mehr als 3 ppm Ni, bezogen auf den Aluminium-Gehalt; und/oder nicht mehr 2,5 ppm Cu, vorzugsweise nicht mehr als 0,5 ppm bezogen auf den Aluminium-Gehalt umfasst. Besonders bevorzugt umfasst die Zusammensetzung nicht mehr als 700 ppm Schwermetall und vorzugsweise nicht mehr als 450 ppm Schwermetall bezogen auf den Aluminium-Gehalt, wobei die Zusammensetzung nicht mehr als 420 ppm Fe, vorzugsweise nicht mehr als 100 ppm bezogen auf den Aluminium-Gehalt; nicht mehr als 18 ppm, vorzugsweise nicht mehr als 3 ppm Ni bezogen auf den Aluminium-Gehalt; und nicht mehr als 2,5 ppm Cu, vorzugsweise nicht mehr als 0,5 ppm bezogen auf den Aluminium-Gehalt umfasst.
-
In einem weiteren Aspekt stellt die Erfindung ein Verfahren zur Analyse der Lagerstabilität einer Zusammensetzung, die Aluminium und ein Therapeutikum oder eine prophylaktische Verbindung umfasst, bereit, wobei das Verfahren Kombinieren einer vorbestimmten Menge an Therapeutikum oder Impfstoff und einer vorbestimmten Menge eines Aluminium-Salzes zu einer Zusammensetzung umfasst, wobei das Verfahren ferner Lagern der Zusammensetzung für mindestens 2 Wochen, vorzugsweise mindestens 4 Wochen und vorzugsweise für mindestens einen Monat bei einer Temperatur von über 20°C, vorzugsweise bei einer Temperatur von etwa 22°C und Bestimmen der Stabilität und/oder der Menge an Protein, vorzugsweise Therapeutikum oder prophylaktischer Verbindung, in der Zusammensetzung umfasst. Wie in den Beispielen 1 und 2 gezeigt, resultiert eine Temperatur von 22°C, welche eine höhere Temperatur verglichen mit normalen Lagerbedingungen von ungefähr 2–8°C eine höhere Temperatur ist, in einem beschleunigten Proteinabbau in einer wässrigen Zusammensetzung, die Protein, ein Aluminium-Salz und mehr als 350 ppb des Schwermetalls, bezogen auf das Gewicht der Zusammensetzung, umfasst. Somit sind eine Temperatur von etwa 22°C und eine Lagerdauer von mindestens 2 Wochen, vorzugsweise mindestens 4 Wochen, geeignet, um die Lagerstabilität von erfindungsgemäßen wässrigen Zusammensetzungen, die ein Protein enthalten, zu bestimmen. Die Lagerstabilität kann durch jedes in der Technik bekannte Verfahren bestimmt werden. Die Lagerstabilität einer erfindungsgemäßen wässrigen Zusammensetzung, die ein Protein umfasst, wird vorzugsweise durch Bestimmung der Lagerstabilität des Proteins analysiert, vorzugsweise durch Bestimmung eines gegenüber Lagerung empfindlichen Epitops des Proteins. Wie in Beispiel 1 und 2 beschrieben, wird die Stabilität eines Proteins, vorzugsweise eines Antigens, beispielsweise bestimmt, indem das Verhältnis von intaktem, gegenüber Lagerung empfindlichem Epitop, wie beispielsweise Gehalt an intaktem antigenen Epitop (z. B. Epitop eines neutralisierenden Epitops), zum Gesamtproteingehalt bestimmt wird. Im hier verwendeten Sinne bedeutet „intaktes gegenüber Lagerung empfindliches Epitop” oder „intaktes antigenes Epitop”, dass innerhalb des Epitops kein Abbau stattgefunden hat. Der Gehalt an intaktem antigenen Epitop wird beispielsweise durch Bestimmung von Protein, das an einen monoklonalen Antikörper gebunden ist, der spezifisch gegen das Epitop gerichtet ist, beispielsweise in einem ELISA, gemessen. Der Gesamtproteingehalt wird beispielsweise durch Bestimmung von Protein, das an polyklonalen Antikörper gebunden ist, der gegen verschiedene Epitope innerhalb des Proteins gerichtet ist, beispielsweise durch ELISA, gemessen. Der relative Gehalt an spezifischem Epitop kann dann als das Verhältnis des gesamten Antigengehalts, bestimmt durch Bindung an monoklonalen Antikörper, dividiert durch gesamten Antigengehalt, bestimmt durch Bindung an polyklonalen Antikörper, ausgedrückt werden. Ein hohes Verhältnis zeigt einen hohen Gehalt an antigenem Epitop an, und ein niedriges Verhältnis zeigt einen niedrigen Gehalt an antigenem Epitop an. Ein niedriges Verhältnis, das für eine wässrige Zusammensetzung nach Lagerung bei mindestens 20°C, vorzugsweise 22°C, für mindestens 2 Wochen, vorzugsweise 4 Wochen, gemessen wurde, verglichen mit dem Verhältnis der wässrigen Zusammensetzung, das vor der Lagerung gemessen wurde, zeigt an, dass innerhalb des antigenen Epitops strukturelle Veränderungen stattgefunden haben. Strukturelle Veränderungen innerhalb des antigenen Epitops zeigen eine verringerte Lagerstabilität der wässrigen Zusammensetzung an.
-
Der Schwermetallgehalt einer wässrigen Zusammensetzung, die gemäß der Erfindung hergestellt wird, beträgt weniger als 350 ppb, bezogen auf das Gewicht der wässrigen Zusammensetzung. Im Allgemeinen findet umso weniger Abbau des Proteins statt, je weniger Schwermetall eine solche wässrige Zusammensetzung umfasst. Vorzugsweise beträgt daher der Schwermetallgehalt weniger als 325 ppb, bezogen auf das Gewicht der wässrigen Zusammensetzung, besonders bevorzugt weniger als 300 ppb, besonders bevorzugt weniger als 275, besonders bevorzugt weniger als 250 ppb und besonders bevorzugt weniger als 235 ppb, bezogen auf das Gewicht der wässrigen Zusammensetzung.
-
Im hier verwendeten Sinne bezieht sich der Begriff „Schwermetall” auf die Gesamtmenge der Elemente, die metallische Eigenschaften aufweisen, und umfasst die Übergangsmetalle, Metalloide, Lanthanoide und Actinoide. Übergangsmetalle sind Elemente, deren Atom eine unvollständige d-Unterschale aufweist, oder die zu Kationen mit einer unvollständigen d-Unterschale führen können, und umfassen Zink, Molybdän, Cadmium, Scandium, Titan, Technetium, Palladium, Vanadium, Chrom, Mangan, Eisen, Kobalt, Rhodium, Hafnium, Kupfer, Nickel, Yttrium, Niob, Zirkon, Ruthenium, Silber, Tantal, Rhenium, Wolfram, Osmium, Meitnerium, Platin, Iridium, Quecksilber, Bohrium, Seaborgium, Hassium. Metalloide sind Bor (B), Silizium (Si), Germanium (Ge), Arsen (As), Antimon (Sb), Tellur (Te), Polonium (Po). Die Lanthanoiden sind die fünfzehn metallischen chemischen Elemente mit den Ordnungszahlen 57 bis 71, d. h. Lanthan, Cer, Praseodym, Neodym, Promethium, Samarium, Europium, Gadolinium, Terbium, Dysprosium, Holmium, Erbium, Thulium, Ytterbium und Lutetium. Die Actinoiden sind die fünfzehn metallischen chemischen Elemente mit den Ordnungszahlen 89 bis 103, Actinium, Thorium, Protactinium, Uran, Neptunium, Plutonium, Americium, Curium, Berkelium, Californium, Einsteinium, Fermium, Mendelevium, Nobelium und Lawrencium. Das Schwermetall ist vorzugsweise ein Metall des D-Blocks des Periodensystems. Das Schwermetall ist vorzugsweise ein Metall der Gruppe 3–12 des Periodensystems einschließlich der Lanthanoiden und der Actinoiden. In einer besonders bevorzugten Ausführungsform ist das Schwermetall ein Metall des D-Blocks oder der Gruppe 3–12 des Periodensystems und schließt die Lanthanoiden und die Actinoiden nicht ein. Vorzugsweise ist das Schwermetall ein Element, das aus den Übergangsmetallen ausgewählt ist. In einer weiteren bevorzugten Ausführungsform ist das Schwermetall aus Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, Cr, Pb, Rb und Mo ausgewählt. In einer weiteren bevorzugten Ausführungsform ist das Schwermetall ein Metall mit einer Molmasse zwischen 21 und 83, vorzugsweise ist das Schwermetall ein Metall des D-Blocks des Periodensystems mit einer Molmasse zwischen 21 und 83, besonders bevorzugt Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, Cr und Mo. Bevorzugt ist das Schwermetall ein Metall aus dem D-Block des Periodensystems mit einer Molmasse zwischen 21–30 und 39–48, besonders bevorzugt Cu, Ni, Co, Ru, Cd, Ag, Fe, V, Cr und Mo. In einem besonders bevorzugten Aspekt wird das Schwermetall aus den Schwermetallen Cu, Ni und Fe ausgewählt.
-
Wie in den Beispielen gezeigt, wurde festgestellt, dass Aluminiumhydroxid(Alaun)-Los 4230 wesentlich zum Antigenabbau in JEV-Impfstoff FVL09L37 beiträgt. In Beispiel 3 wird gezeigt, dass dieses Alaun-Los mindestens die folgenden Metalle umfasst: Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V. Verglichen mit anderen untersuchten Losen wurden in Alaun-Los 4230 höhere Gehalte an Fe-, Ni- und Cu-Ionen festgestellt. Los 4230 war das einzige, bei dem Rest-Cu-Ionen nachgewiesen wurden. Daher wird das Schwermetall bevorzugt aus der Gruppe bestehend aus Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, besonders bevorzugt Fe, Ni und Cu, ausgewählt.
-
Die Menge an Schwermetall in der wässrigen Zusammensetzung beträgt vorzugsweise weniger als 350 ppb, bezogen auf das Gewicht der wässrigen Zusammensetzung. Vorzugsweise beträgt die Menge an Schwermetall in der wässrigen Zusammensetzung weniger als 250 ppb, vorzugsweise weniger als 225, besonders bevorzugt weniger als 200, besonders bevorzugt weniger als 150, besonders bevorzugt weniger als 100, besonders bevorzugt weniger als 50, besonders bevorzugt weniger als 25 ppb, bezogen auf das Gewicht der wässrigen Zusammensetzung. Die Menge an Schwermetall in einer erfindungsgemäßen wässrigen Zusammensetzung wurde hierin vorstehend definiert. Diese Menge stellt in der Regel die Summe der festgelegten Schwermetalle oder der Schwermetalle Fe, Cr und Ni oder einer Kombination davon dar, welche die Hauptschwermetalle nach Gewicht in der erfindungsgemäßen wässrigen Zusammensetzung darstellen. Für bestimmte Schwermetalle können unterschiedliche Maximalmengen bevorzugt sein. Zum Beispiel ist es bevorzugt, dass die Menge an Fe in der erfindungsgemäßen wässrigen Zusammensetzung weniger als 350 ppb, bezogen auf das Gewicht der wässrigen Zusammensetzung, beträgt. In einer bevorzugten Ausführungsform beträgt die Menge an Fe weniger als 250 ppb, vorzugsweise weniger als 210 ppb Fe, bezogen auf das Gewicht der wässrigen Zusammensetzung.
-
Es gibt deutliche Hinweise darauf, dass viele der proinflammatorischen Wirkungen von Aluminium-Adjuvantien über die Bildung von reaktiven Sauerstoffspezies (ROS) vermittelt werden. Aluminium kann unter physiologischen Bedingungen die Reduktion von Fe(III) zu Fe(II) und die Oxidation des letzteren fördern. Somit potenziert die Kombination von Fe und Al im Adjuvans die Bildung und die Aktivitäten der ROS (Exley, C (2010). Trends in Immunol. B. 31: S. 103–109). In der vorliegenden Erfindung wurde festgestellt, dass Fe in einer erfindungsgemäßen wässrigen Zusammensetzung vorhanden sein kann, ohne die Lagerstabilität der Zusammensetzung wesentlich zu beeinträchtigen. In dieser Ausführungsform der Erfindung ist es bevorzugt, dass die erfindungsgemäße wässrige Zusammensetzung zwischen 5 ppb und 250 ppb Fe, bezogen auf das Gewicht der wässrigen Zusammensetzung, umfasst. Bei diesen Mengen beeinträchtigt die Bildung von ROS während der Lagerung aufgrund des Vorhandenseins einer solchen Menge an Fe (wenn überhaupt ROS) die Lagerstabilität der wässrigen Zusammensetzung, wie an anderer Stelle hier definiert, nicht wesentlich. Allerdings sind die Mengen ausreichend, um proinflammatorische Wirkungen nach der Verabreichung des Impfstoffs in vivo zu ermöglichen.
-
In der vorliegenden Erfindung wurde festgestellt, dass insbesondere das Vorhandensein des Schwermetalls Cu die Lagerstabilität der erfindungsgemäßen Zusammensetzung ernsthaft beeinträchtigt. Die Menge an Cu in einer proteinhaltigen Zusammensetzung oder einem Verfahren zur Herstellung der erfindungsgemäßen Proteinzusammensetzung beträgt vorzugsweise weniger als 25 ppb. Vorzugsweise weniger als 5 ppb, besonders bevorzugt weniger als 1 ppb, besonders bevorzugt weniger als 0,2 ppb, bezogen auf das Gewicht der wässrigen Zusammensetzung. In einer besonders bevorzugten Ausführungsform umfasst eine erfindungsgemäße wässrige Zusammensetzung daher weniger als 3 ppb Cu, bezogen auf das Gewicht der wässrigen Zusammensetzung. Vorzugsweise weniger als 2,5 ppb. Die Menge an Cu in einer erfindungsgemäßen Zusammensetzung oder einem erfindungsgemäßen Verfahren beträgt in einer besonders bevorzugten Ausführungsform weniger als 1,25 ppb, bezogen auf das Gewicht der wässrigen Zusammensetzung. In einer besonders bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung Cu in einem Gehalt, der unterhalb der Nachweisgrenze des Verfahrens zum Nachweis von Kupfer liegt, wie in den Beispielen beschrieben.
-
In der vorliegenden Erfindung wurde festgestellt, dass insbesondere das Schwermetall Ni die Lagerstabilität der erfindungsgemäßen Zusammensetzung beeinträchtigt. Die Menge an Ni in einer erfindungsgemäßen Zusammensetzung oder einem erfindungsgemäßen Verfahren beträgt vorzugsweise weniger als 200 ppb. Vorzugsweise weniger als 40 ppb, vorzugsweise weniger als 9 ppb und vorzugsweise weniger als 2 ppb, bezogen auf das Gewicht der wässrigen Zusammensetzung. In einer besonders bevorzugten Ausführungsform umfasst eine erfindungsgemäße wässrige Zusammensetzung daher weniger als 40 ppb Ni, bezogen auf das Gewicht der wässrigen Zusammensetzung. Vorzugsweise weniger als 30 ppb, besonders bevorzugt weniger als 20 ppb und besonders bevorzugt weniger als 15 ppb Ni, bezogen auf das Gewicht der wässrigen Zusammensetzung. In einer besonders bevorzugten Ausführungsform umfasst die wässrige Zusammensetzung Ni in einem Gehalt, der unterhalb der Nachweisgrenze des Verfahrens zum Nachweis von Nickel liegt, wie in den Beispielen beschrieben.
-
Das Schwermetall kann in elektronisch neutraler Form vorliegen oder es kann ionisiert sein. Typischerweise und vorzugsweise liegt das Schwermetall in einer erfindungsgemäßen wässrigen Zusammensetzung in ionischer Form vor.
-
Metallgehalt einer Zusammensetzung kann auf verschiedene Weise bestimmt werden. In einem Aspekt umfasst ein erfindungsgemäßes Verfahren Bestimmen des Gehalts eines Schwermetalls in einer wässrigen Zusammensetzung und/oder des Aluminium-Salzes, das in der wässrigen Zusammensetzung vorhanden ist. Verfahren zur Messung des Gehalts von einem oder mehreren Schwermetallen in einer wässrigen Lösung sind in der Technik bekannt. Beispiele für solche Verfahren umfassen Massenspektrometrie, wie beispielsweise induktiv gekoppelte Plasmamassenspektrometrie (ICP-MS), Flammenatomabsorptionsspektrometrie (F-AAS) und/oder Graphitrohratomabsorptionsspektrometrie (GF-AAS).
-
Ein Beispiel für einen Assay, der verwendet werden kann, um den Gehalt an Schwermetallen zu bestimmen, wird in Beispiel 3 beschrieben. Der Assay beinhaltet Behandeln einer Probe einer wässrigen Lösung, die ein Aluminium-Salz umfasst, mit konzentrierter HNO3 in der Hitze, bis eine klare Lösung erhalten wird. Die klare Lösung kann dann weiter verdünnt und zum Beispiel durch ICP-MS, F-AAS und/oder GF-AAS auf das Vorhandensein und den Gehalt an Metallionen wie Pb, Cd, Cr, Co, Fe, Cu, Ni, Ag, W und Al analysiert werden.
-
Beispiele 1 und 2 zeigen, dass das JEV-Antigen höhere Stabilität bei pH 7,5–8 als bei pH 7 zeigt. In den Beispielen wird Antigenstabilität als das Verhältnis von monoklonalem/polyklonalem ELISA ausgedrückt. Es wurde gezeigt, dass der eingesetzte monoklonale Antikörper (Klon 52-2-5) ein neutralisierendes Epitop in dem Impfstoff gegen Japanische Enzephalitis (JEV) erkennt. Der relative Gehalt an spezifischem Epitop kann als das Verhältnis des gesamten Antigengehalts, bestimmt durch „monoklonalen ELISA”, dividiert durch gesamten Antigengehalt, bestimmt durch „polyklonalen ELISA”, ausgedrückt werden. Ohne an eine Theorie gebunden zu sein, kann der Effekt einer höheren Antigenstabilität bei pH 7,5–8 in Bezug auf die zugrunde liegende vermutete komplexe Reaktionschemie der Sulfite erklärt werden. pH beeinflusst möglicherweise die Reaktionsbedingungen im Zusammenhang mit dem Gleichgewicht der Sulfit/Formaldehyd-Reaktion und die Oberflächenladung bestimmter Protein-/Aminosäureseitenketten, die für eine Modifikation zugänglich sind. Der pH-Wert kann Oxidation durch direkten Einfluss auf Redoxpotenziale der Aminosäurereste und der Oxidationsmittel, z. B. freier Radikale, beeinflussen. Daher umfasst in einer Ausführungsform die wässrigen Zusammensetzung gepuffert bei einem pH zwischen 7,5 und 8,5.
-
In Zusammensetzungen zur Verabreichung an ein Individuum werden verschiedene Aluminium-Salze verwendet. Aluminium-Adjuvans umfasst typischerweise ein Aluminiumoxid oder -sulfat oder eine Kombination davon. In einer bevorzugten Ausführungsform umfasst das Aluminium-Salz Aluminiumoxid (Al2O3), Aluminiumhydroxid (Al(OH)3) oder Aluminiumphosphat (AlPO4).
-
In einer bevorzugten Ausführungsform umfasst die erfindungsgemäße wässrige Zusammensetzung ferner eine reaktive Verbindung. Typischerweise, wenn auch nicht notwendigerweise ist die reaktive Verbindung als Ergebnis einer Manipulation der wässrigen Zusammensetzung vorhanden, beispielsweise um ein etwaiges infektiöses Mittel in der Zusammensetzung zu behandeln oder zu inaktivieren. Die reaktive Verbindung kann auch aus einem anderen Grund vorhanden sein. Sulfit ist beispielsweise manchmal vorhanden, um etwaigen restlichen Formaldehyd in der wässrigen Lösung zu inaktivieren. Formaldehyd ist in der Regel eine Chemikalie, die häufig verwendet wird, um etwaiges infektiöses Mittel zu inaktivieren.
-
Die reaktive Verbindung ist vorzugsweise eine redoxaktive Verbindung, eine radikalbildende Verbindung und/oder eine stabilisierende Verbindung. In einer bevorzugten Ausführungsform umfasst die erfindungsgemäße wässrige Zusammensetzung Formaldehyd, Ethanol, Chloroform, Trichlorethylen, Aceton, Triton-X-100, Desoxycholat, Diethylpyrocarbonat, Sulfit, Na2S2O5, beta-Propriolacton, Polysorbat, wie beispielsweise Tween 20®, Tween 80®, O2, Phenol, Copolymere vom Pluronic-Typ oder eine Kombination von beliebigen davon.
-
Sulfit ist vorzugsweise in einer Menge von zwischen 0,1 mM und 5 mM oder vorzugsweise zwischen 0,5 bis 2 mM vorhanden. Formalin ist vorzugsweise in einer Menge zwischen 0,1 mM und 5 mM, besonders bevorzugt zwischen 0,5 mM und 2 mM vorhanden. Sauerstoff ist vorzugsweise in einer Menge vorhanden, die der Löslichkeit von O2 bei der gemessenen Temperatur äquivalent ist, O2 ist vorzugsweise in einer Menge zwischen 10 und 250 μM vorhanden, wenn bei 20°C gemessen. Wenn bei einer Temperatur von 0 Grad Celsius gemessen, ist O2 vorzugsweise in einer Menge zwischen 10 und 400 μM vorhanden. Eine stabilisierende Verbindung ist vorzugsweise in einer Menge zwischen 10 und 400 μM vorhanden. Ebenso ist eine redoxaktive Verbindung in einer Menge zwischen 0,1 mM und 5 mM oder vorzugsweise zwischen 0,5–2 mM vorhanden. Eine radikalbildende Verbindung ist vorzugsweise in einer Menge zwischen 0,1 mM und 5 mM oder vorzugsweise zwischen 0,5–2 mM vorhanden. In diesem Zusammenhang und im Interesse der Klarheit ist es wichtig zu beachten, dass die redoxaktive Verbindung, die radikalbildende Verbindung und/oder die stabilisierende Verbindung bei der Herstellung eines Radikals verbraucht werden, während das vorstehend angegebene Schwermetall ein Katalysator bei der Herstellung eines Radikals ist und als solches nicht verbraucht wird. Die redoxaktive Verbindung, die radikalbildende Verbindung und/oder die stabilisierende Verbindung ist daher kein Schwermetall.
-
Die Gesamtmenge an redoxaktiver Verbindung, der radikalbildenden Verbindung und/oder der stabilisierenden Verbindung, obwohl klein in absoluten Mengen, kann dennoch in Bezug auf das Antigen oder Protein in der erfindungsgemäßen wässrigen Zusammensetzung wesentlich sein. Das Antigen/Protein ist vorzugsweise in einer Menge zwischen 0,1 nmol und 1 μmol, besonders bevorzugt zwischen 1 nmol und 100 nmol, vorhanden.
-
Die Konzentration des Proteins, vorzugsweise eines Therapeutikums oder Impfstoffproteins in einer erfindungsgemäßen wässrigen Zusammensetzung, die ein Protein umfasst, beträgt vorzugsweise zwischen 1 ng/ml und 10 mg/ml, vorzugsweise zwischen 10 ng/ml und 1 mg/ml, besonders bevorzugt zwischen 100 ng/ml und 100 μg/ml, wie zum Beispiel zwischen 1 μg/ml und 100 μg/ml. Die Konzentration beträgt bevorzugt mindestens 1 ng/ml, um sicherzustellen, dass das Therapeutikum oder das Impfstoffprotein in einer Konzentration vorliegt, die ausreicht, um seine therapeutische Wirkung auszuüben, wenn es einem Individuum verabreicht wird. Die Konzentration sollte jedoch vorzugsweise 10 mg/ml nicht überschreiten, um das Auftreten von möglichen Nebenwirkungen, die mit der Verabreichung des Proteins an ein Individuum verbunden sind, zu verhindern oder zu reduzieren. Insbesondere beträgt die Konzentration des viralen Proteins in einer erfindungsgemäßen wässrigen Zusammensetzung, die JEV umfasst, vorzugsweise zwischen 0,01 μg/ml und 1 mg/ml, besonders bevorzugt zwischen 0,1 μg/ml und 100 μg/ml. In einem erfindungsgemäßen Ausführungsbeispiel umfasst eine erfindungsgemäße wässrige Zusammensetzung etwa 10 μg/ml JEV. Die Dosis einer einzelnen Verabreichung einer erfindungsgemäßen wässrigen Zusammensetzung, die ein Protein, vorzugsweise ein Therapeutikum oder Impfstoffprotein, umfasst, beträgt vorzugsweise zwischen 0,1 ml und 10 ml, bevorzugt zwischen 0,5 ml und 5 ml, wie beispielsweise 0,5 ml, 1 ml, 1,5 ml, 2 ml, 2,5 ml, da eine solche Dosis eine bequeme Verabreichung an ein Individuum, z. B. einen Menschen, ermöglicht.
-
Die erfindungsgemäße wässrige Zusammensetzung kann ferner ein Nukleinsäuremolekül enthalten. Die Nukleinsäure kann zu therapeutischen Zwecken verabreicht werden. In diesem Fall umfasst die erfindungsgemäße wässrige Zusammensetzung vorzugsweise ein Nukleinsäuremolekül, wie beispielsweise ein Plasmid, das die Nukleinsäuresequenz umfasst, die für ein Antigen oder Antigene oder Virus oder Bakterium oder immunogenen Teil davon codiert, gegen das bzw. die eine Immunantwort gesucht wird. Der Einbau eines solchen Nukleinsäuremoleküls stützt sich auf die In-situ-Erzeugung des Zielantigens, Virus, Bakteriums oder immunogenen Teils davon. In einer weiteren bevorzugten Ausführungsform umfasst die erfindungsgemäße wässrige Zusammensetzung eine Nukleinsäure in Form einer Antisense-RNA, RNAi oder eines Mimetikums davon. In einer weiteren Ausführungsform umfasst die erfindungsgemäße wässrige Zusammensetzung die Nukleinsäure in der Form eines infektiösen Mittels als solches, eines Virus oder eines modifizierten Virus, wie es bei vielen Gentherapieansätzen der Fall ist. Die Konzentration der Nukleinsäure in der erfindungsgemäßen wässrigen Zusammensetzung, immunogenen Zusammensetzung oder dem erfindungsgemäßen Impfstoff liegt vorzugsweise im Bereich von etwa 1 ng/ml bis etwa 10 mg/ml, vorzugsweise etwa 0,1 μg/ml bis etwa 1 mg/ml, besonders bevorzugt etwa 1 μg/ml bis etwa 100 μg/ml. Die geeignete Dosierung hängt von dem Individuum ab, dem die Zusammensetzung verabreicht wird, und der Größe der Nukleinsäuresequenzen, die in der Zusammensetzung vorhanden sind.
-
Eine erfindungsgemäße wässrige Zusammensetzung kann ferner ein Polysaccharid und/oder ein Oligosaccharid umfassen, vorzugsweise die Polysaccharid- und/oder Oligosaccharidkapsel von verkapselten Bakterien, gegen die eine Immunantwort gesucht wird. Beispiele für Polysaccharide und Oligosaccharide, die in einer erfindungsgemäßen wässrigen Zusammensetzung vorhanden sein können, umfassen, ohne darauf beschränkt zu sein, Pneumokokken-Polysaccharide, Meningokokken-Polysaccharide, Haemophilus influenzae Typ b-Polysaccharid, Streptococcus Gruppe B-Polysaccharide, Salmonella typhi-Vi-Poiysaccharid, Polysaccharide oder Oligosaccharide. die von Streptokokken der Gruppe A, Staphylococci, Neisseria meningitidis, Klebsiella pneumoniae, Enterococci, E. coli, Pseudomonas aeruginosa und Bacillus anthracis abgeleitet sind. Ein solches Polysaccharid und/oder ein Oligosaccharid kann in der erfindungsgemäßen wässrigen Zusammensetzung als solches enthalten sein, oder alternativ kann das Polysaccharid und/oder Oligosaccharid an ein Protein konjugiert sein. Die Polysaccharid- und/oder Oligosaccharid-Konzentration in der erfindungsgemäßen wässrigen Zusammensetzung, immunogenen Zusammensetzung oder dem erfindungsgemäßen Impfstoff liegt vorzugsweise im Bereich von etwa 10 ng/ml bis etwa 500 μg/ml, besonders bevorzugt etwa 0,1 μg/ml bis etwa 500 μg/ml, besonders bevorzugt etwa 1 μg/ml bis etwa 50 μg/ml. Die geeignete Dosierung hängt von dem Individuum ab, dem die Zusammensetzung verabreicht wird.
-
Ein erfindungsgemäßes Verfahren wird vorzugsweise verwendet, um die Stabilität einer immunogenen Zusammensetzung zu erhöhen, vorzugsweise eines Impfstoffs, die bzw. der ein Aluminium-Salz-Adjuvans umfasst. Nicht beschränkende Beispiele für solche Impfstoffe sind solche, die gegen eine Infektion mit Bacillus anthracis (ruft Anthrax hervor), Corynebacterium diphtheriae (ruft Diphtherie hervor), Clostridium tetani (ruft Tetanus hervor), Pseudomonas, wie beispielsweise Pseudomonas aeruginosa, Staphylococcus, wie beispielsweise Staphylococcus aureus oder Staphylococcus epidermidis, Haemophilus influenzae Typ B-Bakterien (Hib), Poliovirus, Hepatitis-A-Virus, Hepatitis-B-Virus, humanes Papillomavirus, Influenzavirus, Japanische-Enzephalitis-Virus, Rotavirus, Rickettsiae-Bakterien (rufen Typhus hervor), Gelbfiebervirus, Varicella-Zoster-Virus, Meningococcus oder Kombinationen davon gerichtet sind, wie beispielsweise, ohne darauf beschränkt sein, DTP (Diphtherie, Tetanus, Polio). Eine erfindungsgemäße wässrige Zusammensetzung, die ein Protein umfasst, umfasst daher vorzugsweise ein Protein, das ein virales oder bakterielles Protein ist, vorzugsweise ein Protein von Bacillus anthracis, Corynebacterium diphtheriae, Clostridium tetani, Haemophilus influenzae Typ B-Bakterien (Hib), Poliovirus, Hepatitis A-Virus, Hepatitis-B-Virus, humanem Papillomavirus, Influenzavirus, Japanische-Enzephalitis-Virus, Rotavirus, Rickettsiae-Bakterien, Gelbfiebervirus, Varicella-Zoster-Virus und/oder Meningokokken. In einer bevorzugten Ausführungsform ist das Protein, das in einer erfindungsgemäßen Zusammensetzung, die ein Protein umfasst, enthalten ist, ein virales Protein von einem Virus der Familie der Flaviviridae, vorzugsweise von einem Japanische-Enzephalitis-Virus (JEV). Wie in den Beispielen gezeigt, ist die Stabilität der wässrigen Zusammensetzungen, die ein JEV-Protein, ein Aluminium-Salz umfassen und weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, enthalten, und insbesondere wobei die Menge an Cu weniger als 3 ppb, bezogen auf das Gewicht der wässrigen Zusammensetzung, beträgt, verglichen mit der wässrigen Zusammensetzung, die mehr als 350 ppb Schwermetall und mehr als 3 ppb Cu umfasst, erhöht. Somit ist die Erfindung besonders geeignet, um die Stabilität einer wässrigen Zusammensetzung, die ein JEV-Protein JEV umfasst, zu erhöhen. In den Tabellen 30 und 31 wird eine nicht beschränkende Liste von Beispielen von Aluminium-basierten Impfstoffen sowohl für Human- als auch Veterinärmedizin bereitgestellt.
-
In einer weiteren bevorzugten Ausführungsform oder einem Aspekt der Erfindung ist das Protein, das in einer erfindungsgemäßen Zusammensetzung, die ein Protein umfasst, umfasst ist, ein bakterielles Protein von einem Bakterium der Familie Pseudomonas, vorzugsweise von Pseudomonas aeruginosa. Wie in den Beispielen gezeigt, wird die Stabilität von wässrigen Zusammensetzungen, die Pseudomonas aeruginosa-Fusionsprotein (SEQ ID NO: 1) und ein Aluminium-Salz umfassen, reduziert,, wenn mehr als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, vorhanden sind.
-
Eine erfindungsgemäße wässrige Zusammensetzung, die ein Protein umfasst, ist besonders zur Verwendung als immunogene Zusammensetzung oder Impfstoff geeignet. Zum Beispiel sind solche Zusammensetzungen besonders nützlich für die Immunisierung eines Individuums zur Behandlung oder Prävention einer viralen oder bakteriellen Infektion. In einer Ausführungsform beschreibt die Erfindung daher eine immunogene wässrigen Zusammensetzung, die ein Protein und ein Aluminium-Salz umfasst, wobei das Aluminium-Salz weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, aufweist, vorzugsweise weniger als 3 ppb Cu für die Behandlung eines Individuum umfasst. Auch offenbart ist eine immunogene wässrige Zusammensetzung umfassend ein Protein und ein Aluminium-Salz, wobei das Aluminium-Salz weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, aufweist, vorzugsweise weniger als 3 ppb Cu, für die Herstellung eines Medikaments zur prophylaktischen Behandlung eines Individuums. Ferner wird ein wird eine wässrigen Zusammensetzung beschrieben, die ein Protein, welches das Antigen umfasst, und ein Aluminium-Salz umfasst, wobei das Aluminium-Salz weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung aufweist, vorzugsweise weniger als 3 ppb Cu, für die Herstellung eines Medikaments zum Induzieren und/oder Verstärken einer Immunantwort gegenüber einem Antigen in einem Individuum. In einem weiteren Aspekt offenbart die Erfindung mindestens zwei immunogene Zusammensetzungen, die im Abstand von mindestens zwei Wochen zwischen den Verabreichungenerabreicht werden, und wobei jede der mindestens zwei immunogenen Zusammensetzungen das gleiche Antigen umfasst, und wobei mindestens eine der immunogenen Zusammensetzungen ferner ein Aluminiumsalz umfasst, wobei das Aluminium-Salz weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, aufweist, vorzugsweise weniger als 3 ppb Cu. Nukleinsäurezusammensetzungen werden manchmal auch zusammen mit Aluminium verabreicht. Somit ist es für die vorliegende Erfindung möglich, „Protein” in einer erfindungsgemäßen wässrigen Zusammensetzung mit Nukleinsäure zu ersetzen. Somit beschreibt die Erfindung in einer Ausführungsform eine wässrige Zusammensetzung, die Aluminium und eine Nukleinsäure umfasst, wobei:
- – Aluminium-Salz, die Nukleinsäure und Wassers wird kombiniert, um die wässrige Zusammensetzung herzustellen, und
- – Bestimmen des Gehalts eines Schwermetalls in der wässrigen Zusammensetzung und/oder dem Aluminium-Salz.
-
Die Erfindung offenbart auch eine wässrigen Zusammensetzung, die Aluminium und eine Nukleinsäure umfasst, wobei:
- – Aluminium-Salzes mit weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der fertigen wässrigen Zusammensetzung, vorzugsweise weniger als 3 ppb Cu, wird hergestellt oder ausgewählt, und die immunogene wässrige Zusammensetzung an ein Individuum, das derer bedarf, verabreicht, und
- – das Aluminiumsalzes, die Nukleinsäure und Wassers, werden kombiniert, um die wässrige Zusammensetzung herzustellen.
-
In einer bevorzugten Ausführungsform ist die wässrigen Zusammensetzung bei einem pH zwischen 7,5 und 8,5 gepuffert. In einer besonders bevorzugten Ausführungsform wird das Verpacken von Aliquoten der wässrigen Zusammensetzung beschrieben, die weniger als 350 ppb Schwermetalle, bezogen auf das Gewicht der wässrigen Zusammensetzung, aufweist, in separaten luftdichten Lagerbehältern. Die Nukleinsäure kann zu therapeutischen Zwecken verabreicht werden. Zum Beispiel in der Form einer Antisense-RNA, RNAi oder eines Mimetikums davon. Die Nukleinsäure kann auch in der Form eines infektiösen Mittels, typischerweise eines Virus oder eines modifizierten Virus, verabreicht werden, wie es bei vielen Gentherapieansätzen der Fall ist. In diesem Fall wird die Nukleinsäure in ein Partikel eingeschlossen, das Protein umfasst. Die Erfindung stellt daher ferner eine wässrige Zusammensetzung, die eine Nukleinsäure und ein Aluminium-Salz umfasst, bereit, wobei die Zusammensetzung weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, umfasst. Die Nukleinsäurekonzentration in der erfindungsgemäßen wässrigen Zusammensetzung, immunogenen Zusammensetzung oder dem erfindungsgemäßen Impfstoff liegt vorzugsweise im Bereich von etwa 1 ng/ml bis etwa 10 mg/ml, vorzugsweise etwa 0,1 μg/ml bis etwa 1 mg/ml, besonders bevorzugt etwa 1 μg/ml bis etwa 100 μg/ml. Die geeignete Dosierung hängt von dem Probanden ab, dem die Zusammensetzung verabreicht wird, und der Größe der Nukleinsäuresequenzen, die in der Zusammensetzung vorhanden sind.
-
Zusammensetzungen, die Polysaccharid, Oligosaccharid oder Polysaccharid-Polypeptid-Konjugat umfassen, werden manchmal auch zusammen mit Aluminium verabreicht. Für die vorliegende Erfindung ist es somit möglich, „Protein” in einer erfindungsgemäßen wässrigen Zusammensetzung durch Polysaccharid, Oligosaccharid oder Polysaccharid-Polypeptid-Konjugat oder eine Kombination davon zu ersetzen. Solches Polysaccharid oder Oligosaccharid ist vorzugsweise das Polysaccharid oder Oligosaccharid der Kapsel von verkapselten Bakterien, gegen die eine Immunantwort gesucht wird. Beispiele für Polysaccharide und Oligosaccharide, die in einer erfindungsgemäßen wässrigen Zusammensetzung vorhanden sein können, umfassen, ohne darauf beschränkt zu sein, Pneumokokken-Polysaccharide, Meningokokken-Polysaccharide, Haemophilus influenzae Typ b-Polysaccharid, Streptokokken Gruppe B-Polysaccharide, Salmonella typhi-Vi-Polysaccharid, Polysaccharide oder Oligosaccharide, die von Streptokokken der Gruppe A abgeleitet sind, Staphylokokken, Neisseria meningitidis, Klebsiella pneumoniae, Enterococci, E. coli, Pseudomonas aeruginosa und Bacillus anthracis. Solches Polysaccharid und/oder ein Oligosaccharid können in der erfindungsgemäßen wässrigen Zusammensetzung als solches bzw. als solche vorhanden sein, oder alternativ können das Polysaccharid und/oder ein Oligosaccharid an ein Protein konjugiert sein. Somit beschreibt die Erfindung in einer Ausführungsform eine wässrige Zusammensetzung, die Aluminium und ein Polysaccharid oder Oligosaccharid umfasst, wobei:
- – ein Aluminium-Salzes, das Polysaccharid oder Oligosaccharid und Wasser, kombiniert werden, um die wässrige Zusammensetzung herzustellen, und
- – der Gehalt eines Schwermetalls in der wässrigen Zusammensetzung und/oder im Aluminium-Salzbestimmt werden.
-
Die Erfindung offenbart auch eine wässrigen Zusammensetzung, die Aluminium und ein Polysaccharid oder Oligosaccharid umfasst, wobei:
- – ein Aluminium-Salze mit weniger 350 ppb Schwermetall, bezogen auf das Gewicht der fertigen wässrigen Zusammensetzung, vorzugsweise weniger als 3 ppb Cu, ergestellt oder ausgewählt wird, und die immunogene wässrige Zusammensetzung an ein Individuumverabreicht wird, das derer bedarf, und
- – das Aluminiumsalz, das Polysaccharid oder Oligosaccharid und Wasser kombiniert wird, um die wässrige Zusammensetzung herzustellen.
-
Die Polysaccharid- und/oder Oligosaccharidkonzentration in der erfindungsgemäßen wässrigen Zusammensetzung, immunogenen Zusammensetzung oder dem erfindungsgemäßen Impfstoff liegt vorzugsweise im Bereich von etwa 10 ng/ml bis etwa 500 μg ml, besonders bevorzugt etwa 0,1 μg/ml bis etwa 500 μg/ml, besonders bevorzugt etwa 1 μg/ml bis etwa 50 μg/ml. Die geeignete Dosierung hängt von dem Individuum ab, dem die Zusammensetzung verabreicht wird.
-
Das Polysaccharid-Polypeptid-Konjugat gemäß der vorliegenden Erfindung umfasst mindestens ein Polysaccharid und mindestens ein Polypeptid. Das erfindungsgemäße Polysaccharid ist vorzugsweise ein bakterielles Kapselpolysaccharid. Kapselpolysaccharide können mit dem Fachmann auf dem Gebiet bekannten Standardtechniken hergestellt werden.
-
In einer Ausführungsform ist das Polysaccharid ein Kapselpolysaccharid von S. pneumoniae. In einer weiteren Ausführungsform ist das Kapselpolysaccharid von S. pneumoniae aus der Gruppe bestehend aus Serotyp 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, HA, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F und 33F ausgewählt, welche die 23 Pneumokokken-Serotypen, welche die überwiegende Mehrheit der Pneumokokken-Krankheiten in allen Altersgruppen hervorrufen, aus den mehr als 90 bisher bekannten Serotypen repräsentieren.
-
Im hier verwendeten Sinne bezieht sich der Begriff „Polysaccharid” auf Polysaccharide und/oder Oligosaccharide. Polysaccharide werden aus Bakterien isoliert und können mit bekannten Verfahren auf einen bevorzugten Größenbereich depolymerisiert werden (siehe zum Beispiel
EP 497524 und
EP 497525 ). Oligosaccharide weisen eine niedrige Anzahl von Wiederholungseinheiten auf (typischerweise 5–30 Wiederholungseinheiten) und sind in der Regel hydrolysierte Polysaccharide.
-
Kapselpolysaccharide von Streptococcus pneumoniae umfassen Oligosaccharid-Wiederholungseinheiten, die bis zu 8 Zuckerreste umfassen können. Für eine Übersicht der Oligosaccharideinheiten für die wichtigsten Streptococcus pneumoniae-Serotypen siehe Jones et al., An. Acad. Bras. Cienc 2005, 77(2):293–324. In einer Ausführungsform kann ein Kapselsaccharid-Antigen ein Polysaccharid voller Länge sein, in anderen jedoch kann es eine Oligosaccharid-Einheit sein oder eine Saccharid-Kette von Oligosaccharid-Wiederholungseinheiten mit einer kürzeren als die native Länge. Polysaccharide voller Länge können „verkleinert” oder „depolymerisiert” werden, d. h. ihre Größe kann durch verschiedene in der Technik bekannte Verfahren reduziert werden (siehe oben). Der Begriff „Depolymerisation” umfasst Teildepolymerisation.
-
Vor der Konjugation an ein Trägerpolypeptid kann der Depolymerisation des Polysaccharides ein Aktivierungsschritt folgen. „Aktivierung” bedeutet chemische Behandlung des Polysaccharids, um chemische Gruppen bereitzustellen, die fähig sind, mit dem Trägerpolypeptid zu reagieren. Entsprechende Verfahren sind in der Technik bekannt.
-
„Polypeptid” oder „Protein” bedeutet eine Kette von Aminosäuren, die mindestens 10 und vorzugsweise mindestens 100 Aminosäuren in Peptidbindung miteinander, unabhängig von posttranslationaler Modifikation, aufweisen. Geeignete Polypeptidträger umfassen, ohne darauf beschränkt zu sein, Diphtherie-Toxin, Diphterie-Toxoid, CRM 197, Tetanus-Toxoid, Pertussis-Toxoid, E. coli-LT, E. coli-ST, Exotoxin A, Außenmembrankomplex C (OMPC), Porin, Transferrin-Bindungsprotein, Pneumolyse, Pneumokokken-Oberflächenprotein A (PspA), Pneumokokken-Adhäsin-Protein (PsaA), Ovalbumin, Keyhole Limpet Hämocyanin (KLH), Rinderserumalbumin (BSA) oder gereinigtes Proteinderivat von Tuberkulin (PPD) und dergleichen. Trägerpolypeptide sind vorzugsweise Polypeptide, die nicht toxisch und nicht reaktogen und in ausreichender Menge und Reinheit erhältlich sind. In einer besonders bevorzugten Ausführungsform umfasst das Trägerpolypeptid Tetanustoxoid. In einer weiteren bevorzugten Ausführungsform umfasst das Trägerpolypeptid ein Derivat von einem der oben genannten Trägerpolypeptide, beispielsweise eine Untereinheit oder eine mutierte Version von E. coli-LT, wie beispielsweise LT oder die A-Untereinheit von LT (LTA) mit einer Aminosäuresubstitution an der Position von AS 192 (z. B. LTG 192, LTT 192, LTS 192, LTA 192), LTK 63 LTR 72 oder andere Mutanten, wie z. B. in
WO 98/42375 ,
WO 02/64162 ,
US 4,761,372 ,
US 5,308,835 beschrieben. Die Polypeptide können auch Verlängerungen entweder am Carboxy- oder Amino-Terminus des Polypeptids zur Erleichterung der Interaktion mit der (den) polykationischen Verbindung(en) oder der (den) immunstimulierenden Verbindung(en) umfassen.
-
Darüber hinaus können die Polypeptide auch derivatisiert sein, um Moleküle zu umfassen, welche die Antigenpräsentation und das Antigen-Targeting von Antigenpräsentierenden Zellen zu verbessern.
-
Das Polypeptid kann vor der Konjugation aktiviert werden.
-
Die Art und Größe des Saccharids, die Art und Größe des Proteins oder Polypeptids, das Verhältnis des Saccharids zu Protein/Polypeptid sowie andere Faktoren und Bedingungen zur Herstellung eines Konjugats gemäß der vorliegenden Erfindung können von einem Fachmann ermittelt werden, wie z. B. in Robbins et al., JAMA, 1996, 276 (14): 1181–5 beschrieben. Zum Beispiel beträgt ein bevorzugtes Verhältnis für ein Pneumokokken-Polysaccharid zu einem Tetanus-Toxoid etwa 2:1.
-
„Konjugat” bedeutet eine Verbindung, bei der das Polysaccharid mit einem Trägerpolypeptid kovalent verbunden ist. Im Stand der Technik sind viele Konjugationsreaktionen bekannt, die zum kovalenten Verbinden von Polysacchariden an Polypeptide eingesetzt wurden, um ein Polysaccharid-Polypeptid-Konjugat zu erzeugen. Drei der häufiger eingesetzten Verfahren umfassen: 1) reduktive Aminierung, wobei die Aldehyd- oder Ketongruppe an einer Komponente der Reaktion mit der Amino- oder Hydrazidgruppe an der anderen Komponente reagiert, und die gebildete C-N-Doppelbindung anschließend mit einem Reduktionsmittel zu einer C-N-Einfachbindung reduziert wird; 2) Cyanylierung-Konjugation, wobei das Polysaccharid entweder durch Bromcyan (CNBr) oder durch 1-Cyano-4-dimethylammoniumpyridiniumtetrafluorborat (CDAP) aktiviert wird, um eine Cyanatgruppe an der Hydroxylgruppe einzuführen, die nach der Zugabe der Proteinkomponente eine kovalente Bindung mit der Amino- oder Hydrazidgruppe bildet; und 3) eine Carbodiimid-Reaktion, wobei Carbodiimid die Carboxylgruppe an einer Komponente der Konjugationsreaktion aktiviert, und die aktivierte Carbonylgruppe mit der Amino- oder Hydrazidgruppe an der anderen Komponente reagiert. Diese Reaktionen werden auch häufig verwendet, um die Komponenten des Konjugats vor der Konjugationsreaktion zu aktivieren. Das Polysaccharid kann an das Polypeptid direkt oder über einen Linker konjugiert werden. Die Verbindung über eine Linkergruppe kann mit einem beliebigen bekannten Verfahren vorgenommen werden, beispielsweise mit Verfahren, die in
US 4,882,317 und
US 4,695,624 beschrieben werden. Geeignete Linker umfassen Carbonyl, Adipinsäure, B-propionamido (
WO 00/10599 ), Nitrophenylethylamin (
Gever et al., Med. Microbiol. Immunol, 1979, 165:171–288), Halogenacylhalogenide (
US 4,057,685 ), Glycosidbindungen (
US 4,673,574 ;
US 4,761,283 ;
US 4,808,700 ), 6-Aminocapronsäure (
US 4,459,286 ), ADH (
US 4,965,338 ), C4- bis Ci2-Einheiten (
US 4,663,160 ) usw..
-
Nach Konjugation des Polysaccharids an das Trägerpolypeptid kann das Polysaccharid-Polypeptid-Konjugat durch eine Vielzahl in der Technik bekannter Techniken gereinigt (bezüglich der Menge an Polysaccharid-Polypeptid Konjugat angereichert) werden. Ein Ziel des Reinigungsschritts ist die Entfernung von ungebundenem Polysaccharid und/oder Polypeptid aus dem Polysaccharid-Polypeptid-Konjugat. Verfahren zur Reinigung umfassen z. B. Ultrafiltration im Vorhandensein von Ammoniumsulfat, Größenausschlusschromatografie, Dichtegradientenzentrifugierung und hydrophobe Interaktionschromatografie.
-
Aluminium-Adjuvans, wie beispielsweise Alumiuniumhydroxid- und Aluminiumphosphat-Adjuvantien, werden in der Regel hergestellt, indem wässrige Lösungen von Aluminium-Ionen in der Regel leicht alkalischen Bedingungen in einer gut definierten und kontrollierten chemischen Umgebung ausgesetzt werden. Verschiedene lösliche Aluminiumsalze können zur Herstellung von Aluminiumhydroxid verwendet werden. Zum Zeitpunkt der Präzipitation vorhandene Anionen können mit dem Aluminiumhydroxid oder -sulfat copräzipitieren. Nach der Präzipitation des Ammoniumsalzes durch pH-Verschiebung mit beispielsweise NaOH ist es nicht möglich, etwaiges in dem Aluminium-Adjuvans vorhandenes Schwermetall zu entfernen. Selbst gründliches Waschen resultiert nicht in einer ausreichenden Reduzierung des Schwermetallgehalts. Daher ist es wichtig, das Schwermetall vor der Präzipitation des Aluminium-Salzes zu kontrollieren, d. h. durch Auswahl geeigneter Rohmaterialien, Kontrolle der Prozessbedingungen, denn z. B. mit Ammoniak-Alaun und Aluminiumsulfat oder anderen Aluminium-Quellen in Lösung sollte kein Metall zugegeben werden, beispielsweise andere Salze, wie beispielsweise CuSO3. Alternativ oder zusätzlich werden etwaige in Lösung vorhandene Schwermetalle vor der Präzipitation des Aluminium-Adjuvans entfernt. Dies kann beispielsweise durch Kristallisation oder Ionen(Kationen)-Austausch erreicht werden, Verfahren, die in der Technik bekannt sind. Ionenaustausch bezieht sich auf einen Prozess, durch den (Metall)-Ionen in Lösung auf eine feste Matrix übertragen werden, welche wiederum Ionen eines unterschiedlichen Typs, aber mit der gleichen Polarität, in die Lösung freisetzt. Somit werden die Ionen in der Lösung durch verschiedene Ionen, die ursprünglich in der festen Matrix vorhanden sind, ersetzt. Kristallisation von Metallen bezieht sich auf die Präzipitation unlöslicher Metallkristalle von Metallionen in Lösung. Deshalb beschreibt die Erfindung in einer bevorzugten Ausführungsform eine wässrige Zusammensetzung, die Aluminium, eine reaktive Verbindung und ein Protein umfasst, wobei:
- – ein Aluminium-Salz, das eine wässrige Zusammensetzung bereitstellen kann, die weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, hergestellt wird, und
- – das Aluminiumsalzes, die reaktive Verbindung, das Protein und Wasser kombiniert werden, um die wässrige Zusammensetzung herzustellen,
wobei das Aluminium-Salz durch Herstellen einer wässrigen Lösung von Aluminium-Ionen, Entfernen von Schwermetallen aus der wässrigen Lösung, wie beispielsweise durch Kristallisation oder Ionenaustausch, bevorzugt Kationenaustausch, und Präzipitieren der Aluminium-Ionen aus der Lösung, vorzugsweise unter Verwendung einer Base, hergestellt wird.
-
Ferner wird ein Aluminium-Salzpräzipitats klinischer Qualität zum Einbau in ein Arzneimittel und/oder einen Impfstoff bereitgestellt, wobei Schwermetalle aus der wässrigen Lösung entfernt werden, wie beispielsweise durch Kristallisation oder Ionenaustausch, vorzugsweise Kationenaustausch, und Präzipitieren der Aluminium-Ionen aus der Lösung und Bestimmen des Gehalts eines Schwermetalls in der Lösung und/oder dem Aluminium-Salzpräzipitat umfasst, wobei das Präzipitat ausgewählt wird, das eine wässrige Zusammensetzung bereitstellen kann, die weniger als 350 ppb Schwermetall umfasst, bezogen auf das Gewicht der wässrigen Zusammensetzung.
-
Die Herstellung von Aluminiumhydroxid aus Rohmaterialien wird unter anderem in
CN101734698 und
WO98/14401 beschrieben, die durch Querverweis in den vorliegenden Gegenstand aufgenommen werden.
-
Die Erfindung offenbart ferner eine wässrige pharmazeutische oder Impfstoffzusammensetzung, die Aluminium, eine reaktive Verbindung und ein Protein umfasst, wobei:
- – ein Aluminium-Salz, das geeignet ist, eine wässrige Zusammensetzung bereitzustellen, die weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, ausgewählt wird,
- – Kombinieren des Aluminiumsalzes, der reaktiven Verbindung, des Proteins und Wassers, um die wässrige Zusammensetzung herzustellen, die (i) weniger als 350 ppb Schwermetalle, bezogen auf das Gewicht der wässrigen Zusammensetzung, aufweist und (ii) zwischen 5 μg/ml und 50 mg/ml Aluminium umfasst;
wobei die reaktive Verbindung aus der Gruppe bestehend aus einer redoxaktiven Verbindung, einer radikalbildenden Verbindung, einer stabilisierenden Verbindung und einer Kombination von beliebigen davon ausgewählt wird.
-
Die wässrigen Zusammensetzung ist bei einem pH-Wert zwischen 6,5 und 8,5 gepuffert.
-
Ferner wird eine wässrige Zusammensetzung, die weniger als 350 ppb Schwermetalle, bezogen auf das Gewicht der wässrigen Zusammensetzung, aufweist, in separaten luftdichten Lagerbehältern beschrieben.
-
Die Erfindung beschreibt ein Aluminium-Salzpräzipitat klinischer Qualität zum Einbau in ein Arzneimittel und/oder einen Impfstoff, wobei eine wässrigen Lösung von Aluminium-Ionen bereitet wird, und die Aluminium-Ionen aus der Lösung präzipitiert werden, und der Gehalts eines Schwermetalls in der Lösung und/oder des Aluminium-Salzpräzipitat bestimmt werden, wobei das Präzipitat ausgewählt wird, das eine wässrige Zusammensetzung bereitstellen kann, die (i) weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, und (ii) zwischen 5 μg/ml und 50 mg/ml Aluminium umfasst.
-
Ebenfalls wird eine wässrige pharmazeutische oder Impfstoffzusammensetzung, die ein Protein, eine reaktive Verbindung und ein Aluminium-Salz umfasst, bereitgestellt, wobei die Zusammensetzung (i) weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung, und (ii) zwischen 5 μg/ml und 50 mg/ml Aluminium umfasst, wobei die reaktive Verbindung aus der Gruppe bestehend aus einer redoxaktiven Verbindung, einer radikalbildenden Verbindung, einer stabilisierenden Verbindung und einer Kombination von beliebigen davon ausgewählt wird. In einer bevorzugten Ausführungsform wird das Schwermetall aus Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, Cr, Pb, Rb und Mo ausgewählt. Das Schwermetall wird vorzugsweise aus Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V ausgewählt. In einer besonders bevorzugten Ausführungsform wird das Schwermetall aus Cu oder Ni ausgewählt. Das Schwermetall liegt vorzugsweise in ionischer Form vor. Das Aluminium-Salz ist vorzugsweise Aluminiumhydroxid (Al(OH)3) oder Aluminiumphosphat (AlPO4). Das Aluminium-Salz ist vorzugsweise Aluminiumhydroxid (Al(OH)3). Die reaktive Verbindung wird vorzugsweise aus der Gruppe bestehend aus Formaldehyd, Ethanol, Chloroform, Trichlorethylen, Aceton, 4-(1,1,3,3-Tetramethylbutyl)phenylpolyethylenglykol, Desoxycholat, Diethylpyrocarbonat, Sulfit, Na2S2O5, beta-Propriolacton, Polysorbat, wie z. B. Polyethylenglykolsorbitanmonolaurat, Polyethylenglykolsorbitanmonooleat, O2, Phenol, Copolymeren vom Pluronic-Typ und einer beliebige Kombination davon ausgewählt. Die wässrige pharmazeutische oder Impfstoffzusammensetzung umfasst vorzugsweise zwischen 50 μg/ml und 5 mg/ml Aluminium. Die wässrige pharmazeutische oder Impfstoffzusammensetzung umfasst vorzugsweise zwischen 5 ppb und 250 ppb Fe, bezogen auf das Gewicht der wässrigen Zusammensetzung. Die wässrige pharmazeutische oder Impfstoffzusammensetzung umfasst vorzugsweise weniger als 3 ppb Cu, bezogen auf das Gewicht der wässrigen Zusammensetzung. Die wässrige pharmazeutische oder Impfstoffzusammensetzung umfasst vorzugsweise weniger als 40 ppb Ni, bezogen auf das Gewicht der wässrigen Zusammensetzung. Das Protein in der wässrigen pharmazeutischen oder Impfstoffzusammensetzung ist vorzugsweise ein Therapeutikum und/oder ein Impfstoff. Das Protein ist vorzugsweise ein virales oder bakterielles Protein. Das virale Protein ist vorzugsweise ein Protein des Japanische-Enzephalitis-Virus oder ein Protein des Bakteriums Pseudomonas aeruginosa. Das Protein ist vorzugsweise ein Protein in einem Formaldehyd-inaktivierten Viruspartikel. Die wässrige pharmazeutische oder Impfstoffzusammensetzung umfasst ferner vorzugsweise Sulfit. Die Erfindung stellt ferner einen Impfstoff bereit, der eine erfindungsgemäße wässrige Impfstoffzusammensetzung umfasst.
-
Wenn hier ein Bereich zwischen Wert X und Y angegeben ist, schließt der Bereich die Werte X und Y ein.
-
Die Erfindung offenbart ferner eine wässrigen Zusammensetzung, die Aluminium, eine reaktive Verbindung und ein Protein umfasst, wobei:
- – ein Aluminium-Salz, das eine wässrige Zusammensetzung bereitstellen kann, die weniger als 450 ppm Schwermetall, bezogen auf das Gewicht des Aluminiums (Gramm/Gramm) aufweist, hergestellt oder ausgewählt wird, und
- – das Aluminiumsalzes, die reaktive Verbindung, das Proteins und Wasser werden kombiniert, um die wässrige Zusammensetzung herzustellen. Vorzugsweise beträgt der Fe-Gehalt der wässrigen Zusammensetzung weniger als 700 ppm, bezogen auf das Gewicht des Aluminiums in der Zusammensetzung; der Ni-Gehalt beträgt weniger als 18 ppm, bezogen auf das Gewicht des Aluminiums in der Zusammensetzung; oder der Cu-Gehalt beträgt weniger als 2,5 ppm, bezogen auf das Gewicht des Aluminiums in der wässrigen Zusammensetzung, oder eine Kombination davon. Vorzugsweise umfasst das Verfahren ferner Puffern der wässrigen Zusammensetzung bei einem pH-Wert zwischen 6,5 und 8,5. Vorzugsweise umfasst das Verfahren ferner Verpacken von Aliquoten der wässrigen Zusammensetzung, die weniger 450 ppm Schwermetall, bezogen auf das Gewicht des Aluminiums in der Zusammensetzung, aufweist, in separate luftdichte Lagerbehälter.
-
Die Erfindung offenbart ferner ein Aluminium-Salzpräzipitat klinischer Qualität zum Einbau in ein Arzneimittel und/oder einen Impfstoff, wobei eine wässrigen Lösung von Aluminium-Ionen bereitet wird, und Aluminium-Ionen aus der Lösung präzipitiert werden, und der Gehalt eines Schwermetalls in der Lösung und/oder dem Aluminium-Salzpräzipitat bestimmt wird, wobei das Präzipitat ausgewählt wird, das eine wässrige Zusammensetzung bereitstellen kann, die weniger als 450 ppm Schwermetall, bezogen auf das Gewicht der Aluminium-Ionen (Gramm/Gramm) in der Lösung, umfasst.
-
Ferner wird eine wässrige Zusammensetzung bereitgestellt, die ein Protein und ein Aluminium-Salz umfasst, wobei die Zusammensetzung weniger als 450 ppm Schwermetall, bezogen auf das Gewicht des Aluminiums in der Zusammensetzung, umfasst. Die wässrige Zusammensetzung wurde vorzugsweise bei Temperaturen höher als 20°C mindestens 1 Monat lang gelagert. Das Schwermetall wird vorzugsweise aus Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, Cr und Mo ausgewählt. Vorzugsweise wird das Schwermetall aus Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V ausgewählt. In einer besonders bevorzugten Ausführungsform wird das Schwermetall aus Cu oder Ni ausgewählt. Vorzugsweise ist das Schwermetall Cu.
-
Die Erfindung wird weiter in den folgenden Beispielen erläutert. Diese Beispiele beschränken nicht den Umfang der Erfindung, sondern dienen lediglich dazu, die Erfindung zu verdeutlichen.
-
Literaturverweise
-
- Alipazaga MV, Moreno RGM, Coichev N. 2004. Synergistic effect of Ni(II) and Co(II) ions on the sulphite induced autoxidation of Cu(II)/tetraglycine complex. Dalton Trans 13:2036–2040.
-
Arunee Wittayanukulluk, Dongping Jiang, Fred E. Regnier, Stanley L. Hem, „Effect of microenvironment pH of aluminum hydroxide adjuvant on the chemical stability of adsorbiert antigen", Vaccine 22 (2004) 1172–1176
- Berglund J, Fronaeus S, Elding Ll. 1993. Kinetics and mechanism for manganese-catalyzed oxidation of sulfur(IV) by oxygen in aqueous solution. Inorg Chem 32:4527–4538.
- Brandt C, Elding Ll. 1998. Role of chromium and vanadium in the atmospheric oxidation of sulfur (IV). Atmos Environ 32(4):797–800.
- Exley, C (2010). Trends in Immunol. Vol. 31: pp 103–109.
-
Ito, Kimiko und Kawanashi, Shosuke. Site-specific fragmentation and modification of Albumin by sulphite in presence of metal ions or peroxidase/H2O2: Role of Sulphate radical. Biochem and Biophys Res Comm., 1991, 176, 1306–1312
-
Huie R. E., Neta P. One-electron redox reaction in aqueous solutions of sulphite with hydroquinone and other hydroxyphenols. J. Phys. Chem., 1985, 89 (18), 3918–3921
-
Kalina Ranguelova, Marcelo G. Bonini und Ronald P. Mason: (Bi)sulphite Oxidation by Copper, Zinc-Superoxide Dismutase: Sulphite-Derived, Radical-Initiated Protein Radical Formation. Environmental Health Perspectives 2010, 118 (7), 970–975
-
Lambeth D. O., Palmer G. The kinetics and mechanism of reduction of electron transfer proteins and other compounds of biological interest by dithionite. J. Biochem. Chem. 1973, 248, 6095–6103
-
Li S, Schöneich C, Borchardt RT. Chemical instability of protein pharmaceuticals: Mechanisms of oxidation and strategies for stabilization. Biotechnol Bioeng. 1995 Dez. 5; 48(5): 490–500
- Lindblad, EB (2004) Immunol. and Cell Biol. B. 82: 497–505.
- Lima S, Bonifacio RL, Azzellini GC, Coichev N. 2002. Ruthenium(II) tris(bipyridyl) ion as a luminescent probe for oxygen uptake on the catalyzed oxidation of HSO3–. Talanta 56: 547–556.
- Mayo JC, Tan DX, Sainz RM, Natarajan M, Lopez-Burillo S, Reiter RJ. Protection against oxidative protein damage induced by metal-catalyzed reaction or alkylperoxyl radicals: comparative effects of melatonin and other antioxidants. Biochim Biophys Acta. 2003 März 17;1620(1–3): 139–50.
-
Neta P., Huie R. E.: Free Radical Chemistry of Sulphite. Environmental Health Perspectives 1985, 64, 209–217
- Shi X. 1994. Generation of •SO3– and OH radicals in SO32– reactions with inorganic environmental pollutants and its implications to SO32– toxicity. J Inorg Biochem 56(3): 155–165.
- Stadtman ER. Metal ion-catalyzed oxidation of proteins: biochemical mechanism and biological consequences. Free Radic Biol Med. 1990; 9(4): 315–25.
-
Kurze Beschreibung der Zeichnungen
-
1: RP-HPLC-Elutionsprofile von Chlorbutylstopfenextrakt (1:4 verdünnt) und JEV09L37 SN.
-
2: SEC-HPLC-Elutionsprofile von PS (2 mg/ml) vor und nach der Spaltung durch Trypsin.
-
3: SEC-HPLC Elutionsprofile von Trypsin-behandeltem PS und abgebautem PS, wie es in NIV11A74 vorhanden ist. Man beachte, dass Elutionsprofile auf ähnliche Peakhöhe normiert wurden, um einen besseren Vergleich zu ermöglichen.
-
4: DOE-Auswertung des Verhältnisses monoklonaler/polyklonaler ELISA durch Pareto-Diagramm-Analyse und Haupteffektdiagramme (4 Wochen bei 22°C).
-
5: DOE-Auswertung des Verhältnisses monoklonaler/polyklonaler ELISA durch Pareto-Diagramm-Analyse und Haupteffektdiagramme (8 Wochen bei 22°C).
-
6: Kontourdiagramm der geschätzten Antwort
-
7: Residuendiagramm der geschätzten Antwort
-
8: ELISA-Verhältnis (monoklonal/polyklonal) für JEV-Formulierungen bei pH 7 im Vorhandensein von Ni, Cu und Cr. Die Proben wurden 5 Wochen lang bei 22°C gelagert.
-
9: Zusammenfassung der nach 7 Wochen bei 22°C erhaltenen Ergebnisse. Dargestellt sind die Rohdaten als Funktion des pH-Wertes und Metallionenart und kombinierte Ergebnisse für jeden Parameter.
-
10: Mittleres Verhältnis von DP-Formulierungen, die mit unterschiedlichen Alaun-Losen hergestellt wurden. Die Proben wurden 6 Wochen lang bei 22°C gelagert. Fehlerbalken stellen das 95%-Konfidenzintervall dar, basierend auf gepoolter Standardabweichung berechnet. Proben von links nach rechts: Alaun 3877, Alaun 4074, Alaun 4230 nonGl, Alaun 4230 GI, Alaun 4470, Alaun 4563, Alaun 4621, Alaun-Gemisch 4074_4230.
-
11: Partikelgrößenverteilung von Alhydrogel®-Proben.
-
12: Alhydrogel
®-Titrationskurven in PBS. ∎ nicht bestrahltes AlOH (RQCS0890),
Gl AlOH (RQCS1200), ♦ Gl AlOH (RQCS1342), + Gl AlOH (RQCS0448)
-
13: Übersicht der getesteten Alhydrogel®-Chargen. Es sind die Gesamtkonzentration von verunreinigenden Metallionen in ng/ml und der Anteil der Hauptmetallionen Fe, Cr und Ni gezeigt.
-
14: Vergleich der Metallionenverunreinigung in den analysierten Ammoniak-Alaun-Rohmaterialchargen; Lose 6-80578-28, AA0427 und 91480 wurden bei der JEV-Impfstoff-Produktion eingesetzt.
-
15: Aminosäuresequenz von Ala-(His) 6-OprF190-342-Opr121-83 (SEQ ID NO: 1) – hier auch als „Protein A” bezeichnet.
-
Beispiele
-
Beispiel 1
-
Es wurde bereits erkannt, dass Aluminiumhydroxid (Alaun) Los 4230 wesentlich zum Antigenabbau in FVL09L37 beiträgt. Bei diesem speziellen Alaun-Los wurde ein viel höherer Restmetallionengehalt im Vergleich zu anderen Alaun-Losen, die zur Formulierung des inaktivierten JEV-Antigens eingesetzt wurden, beobachtet. Dieses Beispiel zeigt zusätzliche Untersuchungen, die durchgeführt wurden, um den Mechanismus der zugrunde liegenden Ursache und den Einfluss von Metallionen auf den Abbauweg von JEV genauer zu ermitteln. Design of Experiments (DOE) wurde durchgeführt, um den Einfluss der einzelnen Parameter auf Antigenstabilität herauszufinden.
-
Die folgenden Parameter wurden in einem 25 vollfaktoriellen DOE getestet:
- – Aluminiumhydroxid-Los 4230 vs. Aluminiumhydroxid Los-4074
- – Vorhandensein von überschüssigen Protaminsulfat-Fragmenten
- – Vorhandensein von Leachables aus Chlorbutylgummistopfen
- – pH-Bereich von 7 bis 8
- – Resttormaldehydgehalt
-
Alaun-Los 4230 enthält verglichen mit anderen Alaun-Losen, die zur Formulierung von JEV eingesetzt wurden, viel höhere Gehalte an Restmetallionenverunreinigungen. Ein „Design-of-Experiment” (DOE) wurde gewählt, um den potenziellen Mechanismus der zugrunde liegenden Ursache und die Interaktion von Parametern, die schließlich zum Abbau des Produktes führen könnten, genauer zu untersuchen. Bei faktoriellen Designs werden während des Tests mehrere Faktoren gleichzeitig untersucht. Wie bei Ein-Faktor-Designs können qualitative und/oder quantitative Faktoren berücksichtigt werden. Das Ziel dieser Designs ist es, die Faktoren zu identifizieren, die einen signifikanten Einfluss auf die Antwort haben, sowie die Wirkung von Interaktionen zu untersuchen (je nach verwendetem Versuchsdesign). Es können auch Prognosen durchgeführt werden, wenn quantitative Faktoren vorhanden sind, aber man sollte vorsichtig sein, da bestimmte Designs in der Wahl des prädiktiven Modells sehr begrenzt sind. Für Informationen über DOE im Allgemeinen siehe (Siebertz, Karl; van Bebber, David, Hochkirchen, Thomas: Statistische Versuchsplanung: Design of Experiments (DoE). Verlag: Springer Berlin Heidelberg; 1. Auflage (2010), ISBN-10: 3642054927).
-
1.1 DOE-Studiendesign
-
1.1.1 Definition der Parameter und Stufen für das DOE-Design
-
Die folgenden Parameter und Stufen wurden für das Design eines geeigneten-DOE-Versuchs in Betracht gezogen:
- – Restmetallionengehalt von Alaun: Aluminiumhydroxid-Los 4230 und -Los 4074 wurden als Vertreter der beiden Extreme Qualität im Hinblick auf Restmetallionengehalt von Aluminiumhydroxid ausgewählt. Der Mittelpunkt war eine Mischung von 50/50% beider Alaun-Lose. Erste Analysen auf Restmetallionenverunreinigungen in 2% Aluminiumhydroxid-Stammlösung mittels ICP-MS zeigten signifikante Unterschiede in Cr-, Fe-, Ni- und Cu-Ionen-Gehalt zwischen diesen beiden Losen (siehe Tabelle 1).
- – Protaminsulfat-Fragmente: Protaminsulfat (PS) Fragmente sind in niedrigen Mengen (< 5 μg/ml) im Fertigimpfstoff vorhanden. Es wurde geprüft, ob PS-Fragmente zur Oberflächenmodifizierung des Virus (z. B. Interaktion mit/kovalente Bindung an Virus-Oberflächenproteine(n)) in Kombination mit Alaun und anderen in dieser Studie eingesetzten Faktoren beitragen könnten. Daher wurde eine Stammlösung von PS-Fragmenten durch Verdau mit Trypsin gefolgt von Hitzeinaktivierung und Ultrafiltration unter Verwendung einer 5-kDa-Membran zur Protease-Inaktivierung und Entfernung des Enzyms hergestellt. Diese Stammlösung wurde zum Versetzen von jeweiligen Formulierungen mit zusätzlichen PS-Fragmenten auf den hohen Gehalt von 50 μg/ml verwendet. Bei Proben mit niedrigem Gehalt wurden keine zusätzlichen PS-Fragmente zugesetzt und der tatsächliche Gehalt in Formulierungen betrug < 5 μg/ml gemäß HPLC-Analyse.
- – pH: Der untere und obere Wert des pH-Werts in Formulierungen betrug 7 und 8 mit dem Mittelpunkt bei pH 7,5.
- – Leachables/Extractables vom Spritzenstempel: Spritzenstempel (hergestellt aus Chlorbutyl PH701/50 schwarz), die derzeit im Verschlusssystem des Behältnisses verwendet werden. Es wurde geprüft, ob Leachables aus dem Chlorbutylgummi in der Formulierung zur Antigenmodifizierung beitragen könnten. Daher wurde eine Stammlösung von Leachables hergestellt und für Aufstockungsversuche verwendet. Es wurde geschätzt, dass der hohe Gehalt der aufgestockten Leachables in der Formulierung im Durchschnitt 1,4× höher im Vergleich zu dem kommerziellen Fertigimpfstofflos (FVL) ist. Aufgrund der harschen Extraktionsbedingungen wurden zusätzliche Peaks detektiert, die in FVL-Proben nicht vorhanden sind. Daher stellen die aufgestockten Formulierungen den „schlimmsten Fall” in Bezug auf Extractables und Leachables dar. Formulierungen mit dem niedrigen Gehalt enthielten keine Leachables aus Chlorbutylgummi.
- – Restformaldehyd: Bei Formulierungen mit niedrigem Gehalt wurde die Formulierung nicht zusätzlich mit Formaldehyd aufgestockt. Der niedrigere Gehalt war der Restformaldehyd, der in verdünnter NIV-Probe nach Inaktivierung/Neutralisation und 2-facher Verdünnung noch vorhanden war, und lag im Bereich von etwa 37 ppm (berechnet vom GMP-Analysezertifikat der kommerziellen DS-Freigabe). Für den hohen Gehalt wurde die entsprechende Formulierung mit weiteren 40 ppm Formaldehyd aufgestockt (Endgehalt insgesamt etwa 77 ppm). Es wurde geprüft, ob Restformaldehyd in Kombination mit einem höheren Gehalt an Metallionen, der in Alaun 4230 vorhanden ist, und möglicherweise anderen Faktoren weiter mit dem Virus reagieren und zu Hyper-Vernetzung der Oberflächenproteine und Verlust der relevanten Epitope führen könnten.
-
Bestimmung von anderen prozessbezogenen Restverunreinigungen:
-
Restformaldehyd, -sulfit und -saccharose in Endformulierungen wurden basierend auf GMP-Zertifikaten für den kommerziellen Wirkstoff JEV11A74 abgeschätzt. Die Ergebnisse wurden aufgrund der durchgeführten 2-fachen Verdünnung von NIV zu DS innerhalb der DOE-Versuche neu berechnet.
-
Restsulfit: Die Konzentration des Restsulfits war in allen Formulierungen konstant bei etwa 93 ppm.
-
Restsaccharose: Die Konzentration der Restsaccharose war in allen Formulierungen konstant bei etwa 1% Vol./Gew.
-
1.1.2 DOE-Design
-
Diese 5 Faktoren wurden in einem 25-DOE-Plan kombiniert, der in einer Gesamtzahl von 34 Versuchen, darunter 2 Mittelpunkte, mit dem folgenden Basis-Design resultierte. Die DOE-Planung und Auswertung wurden mit einer geeigneten Software durchgeführt (Statgraphic Plus 3.0)
- 1) Kontinuierlich bedeutet, dass das Studiendesign einen Mittelpunkt (Mittelwert aus hohem und niedrigem Gehalt) enthält. Kein Mittelpunkt bedeutet, dass im Studiendesign nur ein hoher und ein niedriger Gehalt vorhanden sind.
- 2) Niedriger Gehalt (0%) bedeutet, dass die Formulierung mit Alaun-Los 4074 hergestellt wurde. Hoher Gehalt (100%) bedeutet, dass die Formulierung mit Alaun-Los 4230 hergestellt wurde. Für Mittelpunkt-Formulierungen wurde eine Mischung aus gleichen Anteilen (50/50%) der beiden Alaun-Lose verwendet.
- 3) Da PS in NIV vorhanden ist, das zur Herstellung von Fertigarzneistoffproben verwendet wird, betrug die tatsächliche PS-Konzentration bei nicht aufgestockten Formulierungen < 5 μg/ml und bei aufgestockten Formulierungen ~50–55 μg/ml.
- 4) In nicht aufgestockten Formulierungen wurden keine Leachables angenommen, da Proben in LoBind-Eppendorf-Röhrchen hergestellt/gelagert wurden. Der Gesamtgehalt an Leachables aus Chlorbutylgummi-Spritzenstempel in aufgestockten Proben war verglichen mit dem FVL etwa 1,4-mal höher.
- 5) Ist-Formaldehyd-Konzentration in nicht aufgestockten DP-Proben betrug etwa 37 ppm, die Gesamtformaldehydkonzentration in aufgestockten Proben betrug etwa 77 ppm.
-
2 Begriffsbestimmungen und Abkürzungen
-
-
- AcCN
- Acetonitril
- DOE
- Design of Experiments
- DS
- Wirkstoff
- FVL
- Fertigimpfstofflos
- HPLC
- Hochdruckflüssigkeitschromatografie
- ICP-MS
- Induktiv gekoppelte Plasma-Massenspektroskopie
- PS
- Protaminsulfat
- RP
- Umkehrphase
- SEC
- Größenausschlusschromatografie
- SN
- Überstand
- TFA
- Trifluoressigsäure
- w/o
- ohne
-
3 Materialien und Methoden
-
3.1 DOE-Studien
-
3.1.1 Materialien
-
- – Spritzenkolbenstopfen: PH701/50/C schwarz Silo 7002-1051 (bezogen von West, Best.-Nr. 2116)
- – 100 ml-Glasflasche (Schott) + Teflon beschichteter Schraubverschluss
- – Aluminiumfolie
- – HQ-Wasser
- – Elektrisches Wasserbad (IKA, HBR 4 digital)
- – LoBind Eppis 2 ml (Eppendorf, Kat.-Nr. 0030 108.132)
- – Speed Vac (Christ, RVC-2-25)
- – HPLC-Gläschen, Klarglas, 900 μl, Chromacol (VWR, Kat.-Nr.: 548-1124)
- – HPLC-Gläschen, PP, 900 μl, (Agilent, Artikel-Nr. 5182-0567)
- – Kappen für HPLC-Gläschen, Pre-Cut (VWR, Kat.-Nr.: 548-1260)
- – 15-ml-Falcon-Röhrchen (Greiner, Kat.-Nr. 188724)
- – Alaun-Charge 4470 (RQCS 1342); Alaun-Los 4230 (RQCS 1200)
- – 10 × PBS (Gibco, Best.-Nr. 14200-091)
- – Parafilm
- – Waters Atlantis T3-Säule; 3 um Partikeldurchmesser; Säulendurchmesser/Länge 2,1 × 100 mm (Best.-Nr. 186003718; Los 0107372331)
- – Acetonitril (Merck, Kat.-Nr. 1.13358.2500)
- – TFA (Sigma, Best.-Nr. 302031
- – HPLC-System Dionex 3000
- – Solvent-Rack SR-3000
- – Pumpe UltiMate-3000, analytische Niederdruckgradientenpumpe
- – Autosampler WPS-3000 TSL, analytischer Autosampler – temperaturgesteuert
- – Säulenofen TCC-3200, temperaturgesteuert
- – PDA-Detektor PDA-3000
- – Formaldehyd-Lösung 37% (Merck, Kat.-Nr. 1.040031000)
- – Protaminsulfat (Intercell Biomedical Ltd., Chargen-Nr. 086056)
- – Ultrafiltrationsgerät (Amicon® Ultra 3 kDa) (Millipore, Kat.-Nr. UFC900324)
- – Inkubator Infors HT Incubator Multitron Standard (InforsAG)
- – Trypsin (Sigma, Bestell-Nr: T0303)
-
3.1.2 Vorschrift zur Herstellung von Extractables aus Spritzenstempel
-
Spritzenstempel (hergestellt aus Chlorbutyl PH701/50 schwarz), die derzeit im Verschlusssystem des Behältnisses für das FVL verwendet werden, wurden von West (Deutschland) bezogen. Daher wurde eine Stammlösung von Leachables durch Wärmebehandlung von Spritzenstempeln in Wasser hergestellt (90°C/2 h), gefolgt von Konzentration in einem Speed-Vac. Der relative Gehalt an Leachables in dieser Stammlösung wurde durch RP-HPLC unter Verwendung einer C18-Säule (Atlantis T3-Säule) abgeschätzt und mit dem FVL JEV09L37-Überstand verglichen.
-
Extraktionsverfahren
-
Eine 100-ml-Glasflasche von Schott mit einem Teflon beschichteten Schraubverschluss und ein Stück Aluminiumfolie wurden mit heißem Wasser gewaschen und gründlich mit HQ-Wasser gespült. 30 Stopfen wurden in die Flasche gefüllt, und 30 ml HQ-Wasser wurden zugegeben. Die Flasche wurde mit zwischen Flasche und Stopfen angebrachter Aluminiumfolie verschlossen und zusätzlich mit Parafilm versiegelt. Die Flasche wurde im Wasserbad 2 Stunden lang auf 90°C erwärmt und auf Raumtemperatur abkühlen gelassen. Der Extrakt wurde in 14 LoBind-Eppendorf-Röhrchen (insgesamt wurden 28 ml Extrakt gewonnen) überführt. Zwölf Gläschen (insgesamt 24 ml) wurden in einem Speed-Vac etwa 44 Stunden lang eingeengt, in einem Falcon-Röhrchen gepoolt, wobei 6 ml 4× konzentrierter Stopfenextrakt erlangt wurde. Eine Kontrollprobe mit 30 ml HQ-Wasser HQ ohne Stopfen wurde in der gleichen Art und Weise hergestellt, um mögliche Verunreinigungen zu bewerten.
-
C18-RP-HPLC-Methode
-
Leachables wurden durch eine RP-HPLC-C18-Säule (Atlantis T3), die bei 40°C und 0,25 ml/min betrieben wurde, getrennt. Lösungsmittel A war 0,1% TFA in H2O, Lösungsmittel B war 0,1% TFA in AcCN. Die Trennung wurde mittels eines linearen Gradienten von 0 bis 95% B in 30 min durchgeführt. Die Detektion erfolgte bei 214 nm, 254 nm und 280 nm. Es wurde geschätzt, dass verglichen mit Peaks im Überstand des Fertigimpfstoff loses (FVL-SN; erlangt durch Entfernung von Alaun-Partikeln durch Zentrifugieren bei 5000 g/5 min) gemäß Detektion bei 254 nm die gesamte relative Konzentration des konzentrierten Stopfenextrakts 80-fach höher war. Daher wurde der Stammlösung ein relativer Gesamtgehalt von 80 E/ml (willkürliche Einheiten E) zugeordnet, während die relative Gesamtkonzentration von Leachables in FVL-SN auf 1 E/ml gesetzt wurde. Für DOE-Studien wurde die Stammlösung 16-fach in den jeweiligen Formulierungen verdünnt, was etwa 5 E/ml Gesamt-Extractables ergab.
-
3.1.3 Herstellung von Protaminsulfat-Fragmenten
-
Eine Stammlösung von PS-Fragmenten wurde durch Verdauen einer PS-Lösung (2 mg/ml in PBS) mit Trypsin (200 ng/ml 60 min lang bei 37°C) hergestellt. Das Enzym wurde anschließend durch Hitze inaktiviert (10 min lang bei 90°C), gefolgt von Ultrafiltration mit einer 3-kDa-Membran (Zentrifugalfilter Amicon® Ultra). Aufgrund der Ausschlussgrenze der Membran blieb Trypsin im Retentat, während die PS-Fragmente im Permeat vorhanden waren. Die vollständige Inaktivierung des Enzyms wurde durch Aufstocken mit 500 μg/ml PS voller Länge in einem Aliquot des erhaltenen PS-Fragments gefolgt von 18-stündiger Inkubation bei 37°C beurteilt. Es wurde kein Abbau von PS voller Länge beobachtet, was die komplette Inaktivierung/Eliminierung von Trypsin anzeigte. Der Abbau wurde durch PS-SEC-HPLC verfolgt.
-
3.1.4 DOE-Plan
-
Die Proben wurden nach dem in Tabelle 2 gezeigten Pipettierschema hergestellt. Die NIV-Charge JEV11A74, die von einem kommerziellen Produktionslauf erlangt wurde, wurde als Ausgangsprobe verwendet. NIV wurde 2-fach als DS mit PBS-Puffer verdünnt, gefolgt von pH-Einstellung. 5-ml-Aliquote wurden entnommen und mit dem entsprechenden Alaun-Los 4230, 4074 oder einer 50/50%-Mischung von beiden adjuvantiert. Die endgültige Menge an hinzugefügter Alaun-Stammlösung (2% Al2O3) betrug 500 μg/ml Aluminium (0,1% Al2O3). Jede Formulierung (5 ml) wurde in zwei Teile (2 × 2,5 ml) unter Verwendung von LoBind-Eppendorf-Röhrchen aufgeteilt. Ein Aliquot wurde bei 2–8°C gelagert, ein weiteres Aliquot wurde bei 22 ± 1°C (Infors HT Inkubator) unter leichtem Schütteln (20 UpM) gelagert.
-
3.2 ELISA von inaktiviertem JEV (auf polyklonaler Basis)
-
Die Desorption des Antigens von Alaun und ELISA-Analyse wurden unter Verwendung von polyklonalen Schaf-anti-JEV-Antikörpern zur Beschichtung der 96-Well-ELISA-Platten durchgeführt, wie in Beispiel 4 beschrieben.
-
3.3 ELISA von inaktiviertem JEV (auf monoklonaler Basis)
-
Es wurde ein JEV-ELISA auf monoklonaler (mAb) Basis entwickelt. Der Assay basiert in erster Linie auf dem „polyklonalen JEV-ELISA”-Assayformat, wobei allerdings ein monoklonaler Anti-JEV-Antikörper (Klon 52-2-5) zur Beschichtung verwendet wird. Es wurde gezeigt, dass der eingesetzte mAb 52-2-5 spezifisch für JEV ist und ein neutralisierendes Epitop erkennt. Der mAb-Klon 52-2-5 wurde durch subkutane Immunisierung von BALB/c-Mäusen mit handelsüblichem Impfstofflos JEV08J14B erlangt. Milzzellen der Mäuse wurden mit Myelomzellen fusioniert. Aus den resultierenden Hybridomzellen wurden einzelne Klone ausgewählt und subkloniert. Die Klone wurden negativ gegen Rinderserumalbumin, Protaminsulfat und einen Extrakt von der Produktionszelllinie des JE-Impfstoffs (Vero-Zellen) gescreent. Ein positives Screening wurde gegen neutralisiertes inaktiviertes Virus (NIV) des Impfstoffloses JEV08M20 durchgeführt. Für das Screening wurden Mikrotiterplatten mit dem entsprechenden Antigen beschichtet und mit Überstand von Kulturen der ausgewählten Klone umgesetzt. Zur Detektion wurde ein polyklonaler Ziege-anti-Maus Antikörper, konjugiert mit alkalischer Phosphatase verwendet. Es wurde gezeigt, dass der mAb-Klon 52-2-5 ein neutralisierendes Epitop auf Domäne III des Hüll(E)-Proteins von JEV, das Ser331 und Asp332 enthält, erkennt (Lin C.-W. und Wu W.-C. J. Virol. 2003; 77(4):2600–6). Die Bindung des mAb an das angegebene neutralisierende Epitop wird beispielsweise, wie in Lin und Wu (2003) beschrieben, durch ortsspezifische Mutagenese der Domäne III an Position 331 (zum Beispiel: S → R), und/oder Alanin-Mutationen an oder nahe Position 331 der Domäne III, beispielsweise der Reste Ser 331 und Asp332, festgestellt, gefolgt von Immunblots zur Feststellung der Bindung des mAb an die mutierten Proteine.
-
Negative Bindungsergebnisse zeigen an, dass das Epitop des mAb das von Lin und Wu (2003) identifizierte neutralisierende Epitop ist. Das neutralisierende Charakteristikum des Epitops führt zu der Annahme, dass das Epitop für das Antigen von Bedeutung sein könnte, um eine schützende Immunantwort hervorzurufen.
-
JEV-Proben wurden mit beiden ELISA-Assays, polyklonal und monoklonal, analysiert. Der relative spezifische Epitopgehalt kann als das Verhältnis des gesamten Antigengehalts, bestimmt durch „monoklonalen ELISA” (Klon 52-2-5), dividiert durch gesamten Antigengehalt, bestimmt durch „polyklonalen ELISA”, ausgedrückt werden. Etwaige Unterschiede im Verhältnis können Unterschiede im spezifischen Epitopgehalt 52-2-5 anzeigen. Ergebnisse nahe 1 würden hohen Epitopgehalten entsprechen, und Ergebnisse nahe 0 entsprechen einem niedrigen relativen Epitopgehalt. Ein niedriges Verhältnis zeigt das Vorhandensein von strukturellen Veränderungen des neutralisierenden Epitops an.
-
Im Zuge der Entwicklung dieses „mAb-ELISA” wurden Unterschiede zwischen Impfstofflosen entdeckt, die mit den Wirkstärkeergebnissen dieser Lose korreliert werden konnten.
-
3.4 SEC-HPLC von Protaminsulfat
-
PS (voller Länge) und seine Fragmente wurden durch Größenausschluss-HPLC (SEC-HPLC) unter Verwendung einer Superdex Peptide 10/300 GL, 10 × 300 mm, 13 um (GE Healthcare) unter Verwendung von 0,1% (Vol./Vol.) Trifluoressigsäure (TFA) in 30% Acetonitril (ACN) als mobile Phase mit einer Flussrate von 0,6 ml/min analysiert. PS-haltige Proben wurden in zweifacher Ausführung hergestellt, d. h. mit der mobilen Phase vor der Injektion verdünnt.
-
4 Ergebnisse
-
4.1 Analyse der für Aufstockungsversuche verwendeten Stopfen-Leachables
-
1 zeigt einen Vergleich von RP-HPLC-Elutionsprofilen von konzentrierter Stammlösung, die durch Extraktion von Stopfen in der Hitze erhalten wurde, und von FVL-SN. Beide Proben zeigten ähnliche Peakmuster. Aufgrund der harschen Extraktionsbedingungen wurden im Konzentrat zusätzliche Peaks detektiert, die in FVL-Proben nicht oder nur in einem niedrigeren relativen Gehalt vorhanden waren. Daher stellen die aufgestockten Formulierungen den „schlimmsten Fall” in Bezug auf Extractables und Leachables dar. Der relative Gesamtgehalt der einzelnen Peaks in Extraktkonzentrat und aufgestockter Formulierung verglichen mit FVL-SN ist in Tabelle 3 zusammengefasst. Die Gesamtmenge an Leachables in der Stammlösung wurde als die Summe aller detektierten Peaks berechnet und in willkürlichen Einheiten als 67 E/ml ausgedrückt. Da die Stammlösung in der jeweiligen Formulierung 16-fach verdünnt war, wurde der resultierende Gesamtgehalt an Leachables zu 4,2 E/ml geschätzt. Dies entspricht im Durchschnitt einer 1,4-fachen Erhöhung verglichen mit dem Überstand von FVL JEV09L37 (3,0 E/ml).
-
4.2 Analyse der Protaminsulfat-Fragmente
-
2 zeigt PS-Fragmente, die nach der Spaltung von PS voller Länge durch Trypsin erlangt wurden. Mit HPLC wurden ähnliche Peakprofile von Trypsin-behandeltem PS und bereits abgebautem PS, das in NIV11A74 vorhanden ist, erlangt (siehe 3).
-
4.3 DOE-Auswertung
-
Für dieses DOE hergestellte Formulierungen wurden nach 4 Wochen und 8 Wochen Inkubation unter beschleunigten Bedingungen (22°C) analysiert. Es wurde davon ausgegangen, dass bei Lagerung bei höheren Temperaturen verglichen mit normalen Lagerbedingungen (2–8°C) eine Abbaureaktion beschleunigt werden würde. Proben werden allerdings auch noch bei 2–8°C gelagert und zu einem späteren Zeitpunkt (~4–6 Monate) analysiert. Tabelle 4 zeigt die erste Analyse von Proben, die 4 bis 8 Wochen lang bei 22°C gelagert wurden.
-
4.3.1 DOE-Auswertung nach 4 Wochen bei 22°C
-
Die statistische Auswertung der Ergebnisse der DOE-Matrix, die nach 4 Wochen bei 22°C erlangt wurden, zeigte, dass der spezifische Epitopgehalt 52-2-5 (ausgedrückt als das Verhältnis des desorbierten Antigens, wie durch monoklonalen/polyklonalen ELISA analysiert) statistisch signifikant (95% Konfidenzniveau, siehe Tabelle 5) durch die folgenden Faktoren beeinflusst wurde:
- – niedrigerer spezifischer Epitopgehalt 52-2-5 im Vorhandensein von Alaun-Los 4230
- – niedrigerer spezifischer Epitopgehalt 52-2-5 bei unter pH 7
- – höherer spezifischer Epitopgehalt 52-2-5 bei erhöhter Konzentration von Formaldehyd
-
Das Vorhandensein einer höheren Konzentration von PS-Fragmenten und Leachables aus Chlorbutylgummi zeigte keinen Einfluss auf den spezifischen Epitopgehalt. Es wurden keine Interaktionen 2. oder höherer Ordnung zwischen einzelnen Parametern festgestellt.
-
Die ANOVA-Tabelle partitioniert die Variabilität bei „Verhältnis 4 Wochen” in einzelne Teile für jeden der Effekte. Die statistische Signifikanz der einzelnen Effekte wird dann durch Vergleich des quadratischen Mittels mit einer Schätzung des experimentellen Fehlers geprüft. In diesem Fall weisen 3 Effekte (Alaun, pH, Formaldehyd) p-Werte von weniger als 0,05 auf, was anzeigt, dass sie auf dem Konfidenzniveau von 95,0% signifikant von Null verschieden sind. Die R-Quadrat-Statistik zeigt, dass das so angepasste Modell 74,85% der Variabilität bei Verhältnis 4 Wochen erklärt. Die angepasste R-Quadrat-Statistik, die für den Vergleich von Modellen mit unterschiedlichen Anzahlen von unabhängigen Variablen besser geeignet ist, beträgt 51,27%. Der Standardfehler der Schätzung zeigt, dass die Standardabweichung der Residuen 0,063 beträgt. Der mittlere absolute Fehler (MAE) von 0,0353 ist der Mittelwert der Residuen. Die Durbin-Watson(DW)-Statistik prüft die Residuen, um zu bestimmen, ob es irgendeine signifikante Korrelation basierend auf der Reihenfolge, in der sie in ihrer Datendatei auftreten, gibt. Da der DW-Wert größer als 1,4 ist, gibt es wahrscheinlich keine ernsthafte Autokorrelation bei den Residuen. Effekte werden auch durch standardisierte Pareto- und Haupteffekt-Diagramme dargestellt, wie in 4 gezeigt.
-
4.3.2 DOE-Auswertung nach 8 Wochen bei 22°C
-
Die statistische Auswertung der Ergebnisse der DOE-Matrix, die nach 8 Wochen bei 22 °C erlangt wurden, waren ähnlich den Ergebnissen, die nach 4 Wochen erlangt wurden. Die Auswertung zeigt, dass der spezifische Epitopgehalt 52-2-5 (ausgedrückt als das Verhältnis des desorbierten Antigens, wie durch monoklonalen/polyklonalen ELISA analysiert) statistisch signifikant (95% Konfidenzniveau, siehe Tabelle 6) durch die folgenden Faktoren beeinflusst wurde:
- – niedrigerer spezifischer Epitopgehalt 52-2-5 im Vorhandensein von Alaun-Los 4230
- – niedrigerer spezifischer Epitopgehalt 52-2-5 bei unter pH 7
-
Das Vorhandensein einer höheren Konzentration von PS-Fragmenten und Leachables aus Chlorbutylgummi und Formaldehyd zeigte keinen Einfluss auf den spezifischen Epitopgehalt. Man beachte, dass der p-Wert für Formaldehyd (p = 0,08) ganz nahe bei Signifikanz liegt. Es wurden keine Interaktionen 2. oder höherer Ordnung zwischen einzelnen Parametern festgestellt.
-
Die ANOVA-Tabelle partitioniert die Variabilität bei „Verhältnis 8 Wochen” in einzelne Teile für jeden der Effekte. Die statistische Signifikanz der einzelnen Effekte wird dann durch Vergleich des quadratischen Mittels mit einer Schätzung des experimentellen Fehlers geprüft. In diesem Fall weisen 2 Effekte (Alaun und pH) p-Werte von weniger als 0,05 auf, was anzeigt, dass sie auf dem Konfidenzniveau von 95,0% signifikant von Null verschieden sind. Die R-Quadrat-Statistik zeigt, dass das so angepasste Modell 75,9% der Variabilität bei „Verhältnis 8 Wochen” erklärt. Die angepasste R-Quadrat-Statistik, die für den Vergleich von Modellen mit unterschiedlichen Anzahlen von unabhängigen Variablen besser geeignet ist, beträgt 53,3%. Der Standardfehler der Schätzung zeigt, dass die Standardabweichung der Residuen 0,095 beträgt. Der mittlere absolute Fehler (MAE) von 0,057 ist der Mittelwert der Residuen. Effekte werden auch durch standardisierte Pareto- und Haupteffekt-Diagramme dargestellt, wie in 5 gezeigt.
-
Mit den angepassten Daten wurde auch eine Regressionsanalyse durchgeführt, und die berechneten Regressionskoeffizienten sind in Tabelle 7 gezeigt. Die nachfolgend angegebene Regressionsgleichung wurde den Daten angepasst, einschließlich pH-Wert, Alaun und Formaldehyd. Die Gleichung des angepassten Modells lautet „Verhältnis 8 Wochen” = 0,0228125 + 0,113125·pH – 0,00185625·Alaun + 0,0315625·Formaldehyd wobei die Werte der Variablen in ihren ursprünglichen Einheiten angegeben sind, mit Ausnahme der kategorischen Faktoren, die die Werte –1 für den niedrigen Gehalt und +1 für den hohen Gehalt annehmen. Die Kontur der geschätzten Antwort und das Residuendiagramm sind in 6 und 7 dargestellt. Das Verhältnis steigt, wenn der relative Gehalt an Alaun-Los 4230 abnimmt und der pH-Wert steigt.
-
Tabelle 8 enthält Informationen über Werte von „Verhältnis 8 Wochen”, die unter Verwendung des angepassten Modells erzeugt wurden. Die Tabelle enthält:
- (1) den beobachteten Wert von „Verhältnis 8 Wochen”
- (2) den vorhergesagten Wert von „Verhältnis 8 Wochen” unter Verwendung des angepassten Modells
- (3) 95,0% Konfidenzniveau für die mittlere Antwort
-
Wie gezeigt, werden die experimentellen Ergebnisse durch das Regressionsmodell gut vorhergesagt.
-
5. Zusammenfassung
-
Es wurde gezeigt, dass unter den getesteten Parametern Alaun-Los 4230 unter beschleunigten Bedingungen (22°C, Testzeitpunkte 4 und 8 Wochen) signifikant zum Antigenabbau beiträgt, wie die Analyse durch monoklonalen/polyklonalen ELISA ergab. DOE-Ergebnisse, die nach 4 und 8 Wochen bei 22°C erlangt wurden, zeigen, dass Alaun 4230 der Faktor mit der höchsten Signifikanz in Bezug auf Antigenabbau ist, wie durch das Verhältnis von monoklonalem/polyklonalem ELISA detektiert. Formulierungen, die mit Alaun 4074 (viel höhere Reinheit im Hinblick auf Restmetallionen) hergestellt wurden, zeigen in der Regel einen viel höheren spezifischen Epitopgehalt.
-
Formalaldehyd und pH trugen ebenfalls zu Antigenstabilität bei, allerdings in einem geringeren Ausmaß. Der Effekt einer erhöhten Antigenstabilität in Proben, die mit Alaun 4230 formuliert wurden, bei einem höheren Formaldehydgehalt wurde klar nachgewiesen (z. B. Proben Nr. 19 und 29). Allerdings war nach längerer Lagerzeit (8 Wochen bei 22 °C) der Einfluss von Formaldehyd weniger ausgeprägt.
-
Im Vergleich zu pH 7 wurde bei pH 8 bessere Stabilität des Antigens beobachtet. Protaminsulfat und Leachables aus Chlorbutylgummistopfen trugen nicht zum Antigenabbau bei.
-
Beispiel 2
-
In früheren Studien (siehe Beispiel 1) wurde Aluminiumhydroxid Los 4230 bereits als signifikant beitragender Faktor zu dem Antigenabbau, der bei FVL09L37 beobachtet wird, identifiziert. Bei diesem speziellen Aluminiumhydroxid(Alaun)-Los wurde ein viel höherer Restmetallionengehalt im Vergleich zu anderen Alaun-Losen, die zur Formulierung des inaktivierten JEV-Antigens eingesetzt wurden, beobachtet. Dieses Beispiel fasst zusätzliche Untersuchungen zusammen, die durchgeführt wurden, um den Einfluss von Metallionen auf die Stabilität von inaktiviertem JEV zu ermitteln. Aufstockungsstudien wurden mit dem Antigen durchgeführt, das entweder in einer Lösung des inaktivierten neutralisierten Virus (NIV) oder in einer Fertigarzneimittel(DP)-Suspension nach Formulierung des Antigens mit Aluminiumhydroxid vorhanden ist.
-
1 Beschreibung der Studie
-
Es wurde bereits gezeigt, dass Alaun-Los 4230 viel höhere Gehalte an Restmetallverunreinigungen im Vergleich zu anderen Alaun-Losen, die zur Formulierung von JEV eingesetzt werden, enthält (siehe auch Beispiel 3). Es wurden zusätzliche Studien durchgeführt, um den Einfluss von Metallionen auf die Stabilität und auf eine mögliche Oberflächenmodifizierung von JEV zu beurteilen. Das inaktivierte Antigen war entweder in einer Lösung des inaktivierten neutralisierten Virus (NIV) oder in einer Fertigarzneimittel(DP)-Suspension nach weiterer Verdünnung des NIV und Formulierung mit Aluminiumhydroxid vorhanden. In einer weiteren Versuchsreihe wurden verschiedene Alaun-Lose, die einen breiten Gehaltbereich an Restmetallionen abdecken, eingesetzt und mit einem einzigen definierten NIV-Los formuliert. All diese Formulierungen enthielten noch Restformaldehyd und -bisulfit in repräsentativen Konzentrationen verglichen mit dem kommerziellen Produkt. Metallsalz-Stammlösungen wurden in Wasser gelöst und Proben wurden damit bis zur gewünschten Endkonzentration aufgestockt.
-
2 Begriffsbestimmungen und Abkürzungen
-
-
- AcCN
- Acetonitril
- ANOVA
- Analyse der Varianz
- DOE
- Design of Experiments
- DP
- Fertigarzneimittel
- DS
- Wirkstoff
- FBV
- Fertigbulkimpfstoff
- FVL
- Fertigimpfstofflos
- GI
- gamma-bestrahlt
- HPLC
- Hochdruckflüssigkeitschromatografie
- LSD
- kleinster signifikanter Unterschied nach Fisher
- mAb
- monoklonaler Antikörper
- NIV
- neutralisiertes inaktiviertes Virus
- PS
- Protaminsulfat
- RP
- Umkehrphase
- SEC
- Größenausschlusschromatografie
- SN
- Überstand
- TFA
- Trifluoressigsäure
- w/o
- ohne
-
3 Materialien und Methoden
-
3.1 Materialien
-
- – Eisen(II)-chlorid-Tetrahydrat (Sigma, Bestell-Nr. 44939)
- – Eisen(III)-chlorid-Hexahydrat (Sigma, Bestell-Nr. 31232)
- – Nickel(II)-sulfat-Hexahydrat (Sigma, Bestell-Nr. N4882)
- – Kobalt(II)-chlorid-Hexahydrat (Sigma, Bestell-Nr. 31277)
- – Kupfer(II)-chlorid-Dihydrat (Sigma, Bestell-Nr. 807483)
- – Zinksulfat-Heptahydrat (Sigma, Bestell-Nr. 24750)
- – Crom(III)-chlorid-Hexahydrat (AlfaAesar, Bestell-Nr. 42114)
- – Ethylendiamintetraessigsäuredinatriumsalz-Dihydrat (EDTA) (Sigma, E5134)
- – doppeldestilliertes Wasser (Fresenius Kabi, Art.-Nr. 0712221/01 A)
- – 10 × PBS (Gibco, Best.-Nr. 14200-091)
- – Formaldehyd-Lösung 37% (Merck, Kat.-Nr. 1.040031000)
- – Protaminsulfat (Intercell Biomedical Ltd., Chargen-Nr. 086056)
- – LoBind Eppis 2 ml (Eppendorf, Kat.-Nr. 0030 108.132)
- – 15-ml-Falcon-Röhrchen (Greiner, Kat.-Nr. 188724)
- – Inkubator Infors HT Incubator Multitron Standard (InforsAG)
- – 0,2-μm-Filter Mini Kleenpak 25 mm (Pall)
- – NIV11A74 und Fertigbulkimpfstoff (FBV, mit Alaun-Los 4539 formuliert) JEV11D87 von kommerziellen Produktionsläufen wurde von Intercell Biomedical (Livingston, GB) bezogen und bei 2–8°C bis zur Weiterverarbeitung gelagert
- – Stammlösungen der Metallsalze in Wasser (Endkonzentration 1 mM) zur Verwendung bei Aufstockungsversuchen wurden hergestellt und bis zur Verwendung bei 2–8°C gelagert.
- – Aluminiumhydroxid-Proben (2% Al2O3, Brenntag Biosector) waren entweder Rückstellproben, die von Intercell Biomedical bezogen wurden, oder wurden direkt von Brenntag gekauft. Alaun-Proben wurden bei 2–8°C gelagert. In dieser Studie wurden die folgenden Alaun-Lose eingesetzt: 4470, 4563, 4621, 3877, 4230 (nicht gammabestrahlt und gammabestrahlt).
-
3.2 Herstellung von Metallstammlösungen
-
3.2.1 Eisen(II)-Stammlösung
-
20 mM Eisen(II)-Stammlösung wurden durch Lösen von 397 mg Eisen(II)-chlorid-Tetrahydrat in 100 ml doppeldestilliertem Wasser hergestellt.
-
3.2.2 Eisen(III)-Stammlösung
-
20 mM Eisen(III)-Stammlösung wurden durch Lösen von 540 mg Eisen(III)-chlorid-Hexahydrat in 100 ml doppeldestilliertem Wasser hergestellt.
-
3.2.3 Nickel(II)-Stammlösung
-
20 mM Nickel(II)-Stammlösung wurden durch Lösen von 525 mg Nickel(II)-sulfat-Hexahydrat in 100 ml doppeldestilliertem Wasser hergestellt.
-
3.2.4 Kobalt(II)-Stammlösung
-
20 mM Kobalt(II)-Stammlösung wurden durch Lösen von 476 mg Kobalt(II)-Hexahydrat in 100 ml doppeldestilliertem Wasser hergestellt.
-
3.2.5 Kupfer(II)-Stammlösung
-
20 mM Kupfer(II)-Stammlösung wurden durch Lösen von 341 mg Kupfer(II)-chlorid-Dihydrat in 100 ml doppeldestilliertem Wasser hergestellt.
-
3.2.6 Zink-Stammlösung
-
20 mM Zink-Stammlösung wurden durch Lösen von 575 mg Zinksulfat-Heptahydrate in 100 ml doppeldestilliertem Wasser hergestellt.
-
3.2.7 Crom(III)-Stammlösung
-
20 mM Crom(III)-Stammlösung wurden durch Lösen von 533 mg Crom(III)-chlorid-Hexahydrat in 100 ml doppeldestilliertem Wasser hergestellt.
-
3.3 Herstellung der Arbeitslösungen
-
Arbeitslösungen von Metallionen (1 mM Endkonzentration, sofern nicht anders angegeben) wurden durch Verdünnung von Metallionenstammlösungen mit doppeldestilliertem Wasser und Sterilfiltration mittels 0,2 um Spritzenfilter hergestellt.
-
3.4 Herstellung der Formulierung
-
Alle Formulierungen wurden unter sterilen Bedingungen hergestellt. NIV und FBV, die aus kommerziellen Produktionsläufen erlangt wurden, wurden auf den gewünschten pH-Wert eingestellt und mit Aliquoten von Metallstammlösung aufgestockt. Alle Proben wurden in Plastikröhrchen gelagert, sofern nicht anders angegeben. Bei allen Formulierungen unter Verwendung von Alaun betrug der Endgehalt an Al 500 μg/ml, was 0,1% Al2O3 entspricht. Man beachte, dass Metallionen, insbesondere Eisen(II), Eisen(III) und zu einem gewissen Ausmaß Cr(III), ein Präzipitat mit den im Puffer vorhandenen Phosphationen bildeten, was in einer teilweisen Copräzipitation des inaktivierten Virus resultierte, was sich an der geringen Rückgewinnung, wie durch Größenausschluss-HPLC (SEC-HPLC) bestimmt, zeigte.
-
3.4.1 Versuch 20110913 (NIV): NIV-Formulierung bei verschiedenen Metallionenkonzentration von Ni(II), Cu(II), Cr(III) im oder ohne Vorhandensein von PS-Fragmenten
-
NIV 11A74 wurde auf pH 7 und pH 8 eingestellt, gefolgt von Aufstocken mit Metallionen (Ni(II), Cu(II), Cr(III)) bei Endkonzentration von 100/500/1000 ng/ml. Alle Formulierungen wurden in LoBind-Eppendorf-Röhrchen bei 22°C gelagert. Aliquote von sämtlichen Formulierungen wurden auch im Vorhandensein von Protaminsulfat-Fragmenten hergestellt (50 μg/ml). Dies geschah zur Beurteilung eines etwaigen Effekts von PS-Fragmenten auf JEV-Stabilität im Vorhandensein von Metallen. Die Herstellung von Protaminsulfat(PS)-Fragmenten wird in Beispiel 1 beschrieben. Die Proben wurden am selben Tag hergestellt (siehe Tabelle 9) und drei Wochen später analysiert. Alle Proben wurden mit SEC-HPLC analysiert, allerdings wurden lediglich Proben bei pH 8 (Nr. 21–40) durch ELISA analysiert.
-
3.4.2 Experiment 20110913 (DP): DP-Formulierung mit verschiedener Metallionenkonzentration von Ni(II), Cu(II), Cr(III)
-
In dieser Studie wurde FBV 11D87 (formuliert mit Alaun-Los 4539) eingesetzt. FBV wurde auf pH 7 und pH 8 eingestellt und mit Ni(II)/Cu(II)/Cr(III) bei 100, 500 und 1000 ng/ml aufgestockt, um einen etwaigen Metallionenkonzentration/pH-abhängigen Effekt zu beurteilen. Tabelle 10 zeigt das experimentelle Design des Versuchs. Alle Formulierungen wurden in Falcon-Röhrchen bei 2–8°C und 22°C gelagert. Bei 22°C gelagerte Proben wurden nach 5 Wochen mit SEC-HPLC und ELISA analysiert.
-
3.4.3 Experiment 20110812 – Metallaufgestocktes DP
-
Fertigbulkimpfstoff 11D87 (formuliert mit Alaun-Los 4539) wurde aus einem kommerziellen Produktionslauf erlangt und in dieser Studie eingesetzt. Restformalin in DS wurde zu 28,1 ppm analysiert, Restsulfite betrugen 92,2 ppm. Der Ist-Gehalt in DP kann als in der gleichen Größenordnung liegend angenommen werden. FBV JEV11D87 wurde auf pH 7,0/7,4/7,8 eingestellt und mit 500 ng/ml (Endkonzentration) Fe(II), Fe(III), Ni(II), Co(II), Cu(II), Zn(II) aufgestockt. Eine Metallionenmischungsformulierung wurde ebenfalls hergestellt, die alle einzelnen Metallionen zusammen in Lösung enthielt. Formulierungen mit Cr(III) wurden später hergestellt und Cr(III) wurde nicht in die Metallionenmischung einbezogen. Kontrollformulierungen wurden lediglich auf den gewünschten pH eingestellt, aber nicht mit Metallen aufgestockt. Alle Formulierungen (Nr. 1–24) wurden am selben Tag hergestellt und in Falcon-Röhrchen bei 2–8°C und 22°C gelagert.
-
Zusätzliche mit Cr(III) aufgestockte Proben (Nr. 25–27) wurden durch Entnahme von Aliquoten der bei 2–8°C gelagerten Kontrollproben und Aufstockung mit Cr(III) zu einer Endkonzentration von 500 ng/ml hergestellt. Formulierungen wurden ausschließlich unter beschleunigten Bedingungen (22°C) gelagert. Tabelle 11 zeigt die Versuchsanordnung dieses Versuchs. Alle bei 22°C gelagerten Proben wurden nach 4 Wochen und 7 Wochen mit ELISA (monoklonal und polyklonal) analysiert.
-
3.4.4 Experiment 20110819: DP-Formulierung unter Verwendung verschiedener Alaun-Lose
-
Aufstockungsstudien, wie oben beschrieben, können erste Hinweise auf mögliche Instabilität des formulierten Antigens im Vorhandensein von bestimmten Metallen geben, sind aber möglicherweise nicht ganz repräsentativ für die tatsächlichen Bedingungen, bei denen in Aluminiumhydroxid vorhandene Metalle in der dreidimensionalen Struktur des Gels eingebaut sind, was in unterschiedlicher lokaler Konzentration und Orientierung/Zugänglichkeit resultiert. Um diese Einschränkungen zu überwinden, wurde eine erste Studie gestartet, um die tatsächlichen Bedingungen zu simulieren. Eine einzelne NIV-Charge (11A74), die von einem kommerziellen Produktionslauf erlangt wurde, wurde mit verschiedenen Alaun-Losen formuliert, die von Brenntag produziert wurden und ein breites Spektrum von Restmetallen abdecken. In Falcon-Röhrchen wurden 4,75 ml NIV mit 0,25 ml Alaun (2%) gemischt. Die Endkonzentration des Aluminiumhydroxids betrug 500 μg/ml (= 0,1% Al2O3). Formulierte Impfstoffproben wurden bei 2–8°C und unter beschleunigten Bedingungen bei 22°C gelagert. Alle diese Alaun-Lose enthielten Restmetallionen in verschiedenen Konzentrationen. Alaun-Los 4230 weist den höchsten Gehalt an Fe, Cu, Ni und V auf (siehe Beispiel 3). Man beachte, dass Metallionenwertigkeiten mit ICP-MS nicht angegeben werden können. Eine Alaun-Mischprobe, die gleiche Mengen an 4230 und 4074 enthielt, wurde ebenfalls hergestellt, um einen „mittleren” Gehalt für Ni(II) und Cu(II) zu erlangen. Die Proben wurden nach 6 Wochen Lagerung bei 22°C analysiert. Die Restmengen an Formaldehyd und Sulfit, die durch Neuberechnung aus den verfügbaren DS-Analyseergebnissen, korrigiert mit dem Verdünnungsfaktor von NIV zu DS, geschätzt wurden, betrugen 76 ppm Formaldehyd bzw. 192 ppm Sulfit.
-
3.1 Antigendesorption von Aluminiumhydroxid für SEC-MALLS-Analyse
-
Viruspartikel wurden von Aluminiumhydroxid desorbiert. ~625 μl DP wurden zentrifugiert (8°C, 5 min, 3300 × g) und der Überstand wurde entweder verworfen, sofern nicht anders angegeben, oder durch JEV-SEC-MALLS analysiert, um die ungebundene Antigen-Konzentration zu detektieren. Viruspartikel wurden durch Suspendieren der Aluminium-Partikel mit 62,5 μl 0,8 M Kaliumphosphatpuffer (pH 8), der BSA (50 μg/ml) enthielt, desorbiert. BSA wurde zu dem Desorptionspuffer für die SEC-MALLS-Analyse gegeben, um Verluste zu minimieren, die durch unspezifische Adsorption des Antigens verursacht werden. Nach Schütteln (500 UpM) der Aluminiumhydroxid-Partikel für 10 min bei Raumtemperatur wurden die Partikel abzentrifugiert und der Überstand wurde in LoBind-Eppendorf-Röhrchen gesammelt, und das Desorptionsverfahren wurde mit verbleibender Probe wiederholt. Das gepoolte desorbierte Antigen (~5× konzentrierte Probe; Endvolumen 125 μl; Startvolumen ~625 μl) wurde dann weiter mit SEC-MALLS analysiert.
-
3.2 SEC-HPLC-MALLS-Verfahren
-
Desorbiertes Antigen wurde mit SEC-MALLS analysiert. Kurz gesagt, nach der Desorption des Antigens von dem Aluminiumhydroxid wurden anschließend 100 μl des gepoolten desorbierten Materials (~5× konzentriert) auf eine Superose 6 10/300 GL SEC-Säule geladen. 1 × PBS + 250 mM NaCl wurde als mobile Phase verwendet. Ultraviolett(UV)-214-nm- und MALLS-Signale von Viruspartikeln wurden aufgenommen und unter Verwendung der Softwarepakete Chromeleon und ASTRA analysiert.
-
3.3 ELISA von inaktiviertem JEV (auf polyklonaler Basis)
-
Die Desorption des Antigens von Alaun und ELISA-Analyse wurden unter Verwendung von polyklonalen Schaf-anti-JEV-Antikörpern zur Beschichtung der 96-Well-ELISA-Platten durchgeführt, wie in Beispiel 4 beschrieben.
-
3.4 ELISA von inaktiviertem JEV (auf monoklonaler Basis)
-
Im Zuge dieser Untersuchungen wurde ein JEV-ELISA auf monoklonaler (mAb) Basis entwickelt. Der Assay basiert in erster Linie auf dem „polyklonalen JEV-ELISA”-Assayformat, wobei allerdings ein monoklonaler Anti-JEV-Antikörper (Klon 52-2-5) zur Beschichtung und der derzeitige polyklonale Antikörper zur Detektion verwendet werden. Es wurde gezeigt, dass der eingesetzte mAb 52-2-5 spezifisch für JEV ist und ein neutralisierendes Epitop erkennt. Der mAb-Klon 52-2-5 wurde durch subkutane Immunisierung von BALB/c-Mäusen mit handelsüblichem Impfstofflos JEV08J14B erlangt. Milzzellen der Mäuse wurden mit Myelomzellen fusioniert. Aus den resultierenden Hybridomzellen wurden einzelne Klone ausgewählt und subkloniert. Die Klone wurden negativ gegen Rinderserumalbumin, Protaminsulfat und einen Extrakt von der Produktionszelllinie des JE-Impfstoffs (Vero-Zellen) gescreent. Ein positives Screening wurde gegen neutralisiertes inaktiviertes Virus (NIV) des Impfstoffloses JEV08M20 durchgeführt. Für das Screening wurden Mikrotiterplatten mit dem entsprechenden Antigen beschichtet und mit Überstand von Kulturen der ausgewählten Klone umgesetzt. Zur Detektion wurde ein polyklonaler Ziege-anti-Maus Antikörper, konjugiert mit alkalischer Phosphatase, verwendet. Es wurde gezeigt, dass der mAb-Klon 52-2-5 ein neutralisierendes Epitop auf Domäne III des Hüll-(E)-Proteins von JEV, das Ser331 und Asp332 enthält, erkennt (Lin C.-W. und Wu W.-C. J. Virol. 2003; 77(4):2600–6). Die Bindung des mAb an das angegebene neutralisierende Epitop wird beispielsweise, wie in Lin und Wu (2003) beschrieben durch ortsspezifische Mutagenese der Domäne III an Position 331 (zum Beispiel: S → R) und/oder Alanin-Mutationen an oder nahe Position 331 der Domäne III, beispielsweise der Reste Ser 331 und Asp332, festgestellt, gefolgt von Immunblots zur Feststellung der Bindung des mAb an die mutierten Proteine. Negative Bindungsergebnisse zeigen an, dass das Epitop des mAb das von Lin und Wu (2003) identifizierte neutralisierende Epitop ist. Das neutralisierende Charakteristikum des Epitops führt zu der Annahme, dass das Epitop für das Antigen von Bedeutung sein könnte, um eine schützende Immunantwort hervorzurufen. JEV-Proben wurden mit beiden ELISA-Assays, polyklonal und monoklonal, analysiert. Der relative spezifische Epitopgehalt kann als das Verhältnis des gesamten Antigengehalts, bestimmt durch „monoklonalen ELISA” (Klon 52-2-5), dividiert durch gesamten Antigengehalt, bestimmt durch „polyklonalen ELISA”, ausgedrückt werden. Etwaige Unterschiede im Verhältnis können Unterschiede im spezifischen Epitopgehalt 52-2-5 anzeigen. Ergebnisse nahe 1 würden hohen Epitopgehalten entsprechen, und Ergebnisse nahe 0 entsprechen einem niedrigen relativen Epitopgehalt. Ein niedriges Verhältnis zeigt das Vorhandensein von strukturellen Veränderungen des neutralisierenden Epitops an.
-
Im Zuge der Entwicklung dieses „mAb-ELISA” wurden Unterschiede zwischen Impfstofflosen entdeckt, die mit den Wirkstärkeergebnissen dieser Lose korreliert werden konnten.
-
3.5 Statistische Auswertung
-
Statistische Auswertung erfolgte mit Statgraphic Plus 3.0.
-
4 Ergebnisse
-
4.1 Experiment 20110913 (NIV): NIV-Formulierung bei verschiedenen Metallionenkonzentration von Ni(II), Cu(II), Cr(III) im oder ohne Vorhandensein von PS-Fragmenten
-
Die SEC-HPLC-Ergebnisse von NIV-Formulierungen (pH 7 und pH 8), die Metallionen [Ni(II), Cu(II), Cr(III)] mit und ohne PS-Fragmente sind in Tabelle 12 zusammengefasst. SEC-HPLC-Ergebnisse zeigen, dass die Antigenrückgewinnung der meisten Proben bei > 80% lag. Einige Proben (Nr. 7, Nr. 36, Nr. 38) zeigten leicht reduzierte Rückgewinnungen im Bereich von 70–80%. Man beachte, dass der tatsächliche Virusgehalt ziemlich niedrig ist und die Genauigkeit der HPLC-Ergebnisse bei etwa ±20% geschätzt werden kann. Da für die Proben Nr. 36 und Nr. 38 die Rückgewinnungen für die folgenden Formulierungen (Nr. 37, Nr. 39) auf der nächsten Ebene des einzelnen Metallionengehalts wieder höher waren, könnten diese Unterschiede durch Assay-Variabilität verursacht worden sein und wurden nicht als signifikant angesehen. Basierend auf den erlangten Ergebnissen war es nicht möglich, den Einfluss von Metallionen in Bezug auf die Rückgewinnung von löslichem inaktiviertem JEV zu klären. SEC-HPLC liefert allerdings nur Informationen über den Gehalt an löslichem Virus, jedoch keine Informationen über etwaige potenzielle Oberflächenmodifizierung. Nur Formulierungen, die bei pH 8 hergestellt wurden, wurden auch durch ELISA (Analyse in doppelter Ausführung) analysiert. Das Verhältnis von monoklonalem/polyklonalem ELISA wurde berechnet und kann zu Vergleichszwecken von Ergebnissen verwendet werden. Die Analyse der Proben mit ELISA (siehe Tabelle 13) zeigen keinen signifikanten Einfluss der getesteten Metalle auf den Abbau von inaktiviertem JEV bei pH 8 nach drei Wochen bei 22°C. Es gibt möglicherweise einen Trend in Richtung verringertes Verhältnis im Vorhandensein von Cu(II), aber insgesamt scheint eine Inkubationszeit von drei Wochen bei 22°C nicht auszureichen, um einen signifikanten Abbau zu erkennen. Wie der DOE-Versuch ebenfalls zeigt (Beispiel 1), scheint inaktiviertes JEV bei Lagerung unter beschleunigten Bedingungen bei 22°C eine höhere Stabilität bei pH 8 zu haben, und dies würde auch dazu beitragen, dass signifikante Effekte nicht beobachtet wurden. In diesem Experiment wurde auch gezeigt, dass PS-Fragmente keinen Einfluss auf die JEV-Stabilität haben. Dies stimmt auch gut mit DOE-Ergebnissen überein. NIV-Proben 1–20, die bei pH 7 formuliert wurden, zeigten eine signifikante Reduktion im monoklonalen Epitopgehalt im Vorhandensein von Cu(II). Bei der höchsten getesteten Konzentration (1000 ng/ml) war das Verhältnis nahe Null, was signifikante strukturelle Veränderungen des Antigens anzeigt.
-
4.2 Experiment 20110913 (DP): DP-Formulierung mit verschiedener Metallionenkonzentration von Ni(II), Cu(II), Cr(III)
-
Die Analyse des desorbierten JEV-Antigens wird in Tabelle 14 (SEC-HPLC) und Tabelle 15 (ELISA) zusammengefasst. Antigenrückgewinnungen für alle Proben, bestimmt mit SEC-HPLC, betrugen > 80% nach 5 Wochen bei 22°C, was anzeigt, dass die getesteten Metallionen keinen signifikanten Einfluss auf die Rückgewinnung nach der Desorption haben. Wie in 8 gezeigt, gibt es einen Trend in Richtung verringertes Verhältnis, wie mit ELISA analysiert, im Vorhandensein von Cu(II) und Cr(III) bei pH 7. Formulierungen bei pH 8 scheinen stabiler zu sein.
-
4.3 Experiment 20110812 (DP): Mit Metallionen aufgestocktes DP
-
Bei diesem Versuch wurde FBV11D87 als Ausgangsmaterial eingesetzt. Der pH-Wert der Formulierung wurde in einem engeren Bereich eingestellt (pH 7,0, 7,4, 7,8) und zum Aufstocken wurden zusätzliche Metallionen verwendet, jeweils bei 500 ng/ml (Endkonzentration). Die Metallionenmischung enthielt alle einzelnen Metallionen mit Ausnahme von Cr(III) in einzelnen Formulierungen (jedes Metall bei 500 ng/ml). ELISA-Ergebnisse, die nach 4 Wochen und 7 Wochen bei 22°C erlangt wurden, sind in Tabelle 16 zusammengefasst. Die Ergebnisse werden auch als Diagramme in 9 dargestellt.
-
Es wurde eine statistische Auswertung der Stabilitätsproben, die 7 Wochen lang bei 22°C gelagert wurden, durchgeführt. ANOVA (Analyse der Varianz) zeigte signifikante Effekte von Parametern (pH und Metallart) auf Antigenstabilität, die als das Verhältnis von monoklonalem/polyklonalem ELISA ausgedrückt wird (siehe Tabelle 17). Die ANOVA-Tabelle zerlegt die Verhältnisvariabilität in Beiträge der verschiedenen Faktoren. Da Typ-III-Quadratsummen ausgewählt wurden, wird der Beitrag der einzelnen Faktoren gemessen, wobei die Effekte aller anderen Faktoren entfernt worden sind. Die p-Werte prüfen die statistische Signifikanz von jedem der Faktoren. Da die p-Werte für pH und Metallionenart kleiner als 0,05 sind, üben diese Faktoren einen statistisch signifikanten Effekt auf Verhältnis auf dem Konfidenzniveau von 95,0% aus.
-
In Tabelle 18 wird ein Verfahren mit mehrfachen Vergleichen angewendet, um die Signifikanz der beobachteten Unterschiede in Bezug auf die Mittel zu bestimmen. Ein signifikanter Effekt auf das Verhältnis wurde für Cu(II) und die Metallmischung verglichen mit den nicht aufgestockten Kontrollformulierungen gezeigt. Die untere Hälfte der Ergebnisse zeigt den geschätzten Unterschied zwischen jedem Paar von Mitteln. Ein Sternchen wurde neben 7 Paaren platziert, was anzeigt, dass diese Paare statistisch signifikante Unterschiede auf dem Konfidenzniveau von 95,0% zeigen. Am oberen Rand der Seite werden 3 homogene Gruppen mit Spalten von X identifiziert. Innerhalb jeder Spalte bilden die Gehalte, die X umfassen, eine Gruppe von Mitteln, in der es keine statistisch signifikanten Unterschiede gibt. Das Verfahren, das gegenwärtig verwendet wird, um zwischen den Mitteln zu unterscheiden, ist das Verfahren des kleinsten signifikanten Unterschieds (LSD) nach Fisher. Bei diesem Verfahren besteht ein 5,0%-iges Risiko, ein Paar von Mitteln als signifikant verschieden zu bezeichnen, wenn der Ist-Unterschied gleich 0 ist.
-
Ein signifikanter Effekt auf das Verhältnis wurde für Cu(II) und die Metallionenmischung gezeigt. Der Einfluss anderer Metallionen könnte bei längerer Lagerung signifikant werden. Die Metallionenmischung enthielt die höchste Gesamtkonzentration und könnte einen schlimmsten Fall darstellen. Es wurde jedoch die Schlussfolgerung gezogen, dass mehrere in Alaun vorhandene Metallionen zum Abbau beitragen könnten, jeweils in einem unterschiedlichen Ausmaß. Diese Ergebnisse bekräftigen ferner die vorgeschlagene Ursache des metallionenkatalysierten Antigenabbaus. Man beachte, dass Aufstockungsversuche möglicherweise die tatsächlichen Bedingungen der in Alaun 4230 vorhandenen Restmetallionenverunreinigungen nicht vollständig simulieren. Metallionen werden in die Alaun-Struktur eingebaut, und die lokale Konzentration und Orientierung könnte von Metallionen, die in Aufstockungsversuchen eingesetzt werden, verschieden sein. Es ist auch bekannt, dass Metallionen (z. B. Fe) im Vorhandensein von Phosphat-Ion (PO43–) eine geringe Löslichkeit aufweisen. Daher sind die tatsächliche Konzentration der löslichen Metallionen und der Beitrag der Metalle, die als Metall-Phosphat-Komplex vorhanden sind, auf den JEV-Abbau nicht bekannt.
-
4.4 Experiment 20110819: Herstellung von DP-Proben mit unterschiedlichen Alaun-Losen
-
Aufstockungsstudien, wie oben beschrieben, können erste Hinweise auf mögliche Instabilität des formulierten Antigens im Vorhandensein von bestimmten Metallen geben, sind aber möglicherweise nicht ganz repräsentativ für die tatsächlichen Bedingungen, unter denen zu erwarten ist, dass in Aluminiumhydroxid vorhandene Metalle in der dreidimensionalen Struktur des Gels eingebaut sind, was in unterschiedlicher lokaler Konzentration und Orientierung/Zugänglichkeit resultiert. Um diese Einschränkungen zu überwinden, wurde eine erste Studie gestartet, um die tatsächlichen Bedingungen zu simulieren. Eine einzelnes NIV (11A74) wurde mit verschiedenen Alaun-Losen formuliert, die von Brenntag bezogen wurden und ein breites Spektrum von Restmetallen abdecken. Formulierte Impfstoffproben wurden bei 2–8°C und unter beschleunigten Bedingungen bei 22°C gelagert. Alle diese Alaun-Lose enthielten Restmetallionen mit verschiedenen Gehalten. Los 4230 hatte den höchsten Gehalt an Fe, Cu, Ni und V (siehe Tabelle 19). Man beachte, dass der eigentliche Gehalt von Metallionen, die in dem formulierten Produkt vorhanden sind, nur 1/20 der Konzentration in der Alaun-(2%)-Stammlösung beträgt. Man beachte, dass Metallionenwertigkeiten mit ICP-MS nicht angegeben werden können. Die Analyse von desorbiertem Antigen mit ELISA von Proben, die 6 Wochen lang bei 22°C gelagert wurden, ist in Tabelle 20 gezeigt.
-
Als Maß für die experimentelle Unsicherheit wurde die gepoolte Standardabweichung aus allen Proben berechnet (Sgepoolt ~ 0,075). Mittelwerte für Verhältnis und Konfidenzniveaus von 95% (berechnet basierend auf Sgepoolt) wurden in Gegenüberstellung zu den einzelnen Formulierungen eingetragen (siehe 10). Proben, die mit Alaun 4230 formuliert wurden, zeigten einen Trend zu niedrigerem Verhältnis verglichen mit den anderen Proben. Allerdings waren die Unterschiede nicht so groß, um statistisch signifikante Unterschiede zwischen den verschiedenen Formulierungen zu zeigen.
-
5 Zusammenfassung
-
Es wurde gezeigt, dass Metallionen zum Abbau des inaktivierten JEV unter beschleunigten Lagerungsbedingungen (22°) beitragen. In Aufstockungsstudien wurde eine höhere Konzentration an Restmetallionen (Bereich 100–1000 ng/ml) verwendet, als in FVL vorhanden ist, das mit Alaun-Los 4230 formuliert wird (z. B. Fe ~310 ng/ml; Cr ~64 ng/ml; Ni ~52 ng/ml). Der Cu-Gehalt in FVL kann basierend auf ICP-MS-Daten von 2 Alaun-Stammlösung nur als 3 ng/ml geschätzt werden, da die LOD 25 ng/ml beträgt. Es wurden eine höhere Metallkonzentration und Lagertemperatur ausgewählt, um die Geschwindigkeit einer potenziellen Abbaureaktion zu erhöhen. Bei FVL JEV09L37 trat der Verlust der Wirkstärke sogar nach 11 Monaten Lagerung bei 2–8°C ein. Es wurde auch gezeigt, dass Metalle unlösliche Komplexe mit Phosphat-Ionen bilden können, was die Abschätzung der tatsächlichen Gehalte an vorhandenen Metallen erschwert. In Aufstockungsversuchen zeigten ELISA-Ergebnisse statistisch signifikante strukturelle Veränderungen der Virusoberfläche schon nach 4 Wochen bei 22°C im Vorhandensein von Metallionen. Es wurde gezeigt, dass die ELISA-Verhältnisse für Formulierungen, die Cu(II) und Metallionenmischung (die Fe(II), Fe(III), Co(II), Cu(II) und Zn(II) enthielt) enthielten, im Vergleich zur nicht aufgestockten Kontrollformulierung statistisch signifikant niedriger waren. Cu(II) wurde auch in Alaun-Los 4230 (2% Stammlösung) mit 64 ng/ml, was ~3 ng/ml in FVL entspricht, gefunden. In allen anderen Alaun-(2%)-Losen lag der Cu(II)-Gehalt bei < 25 ng/ml (unterhalb der Nachweisgrenze).
-
Für Formulierungsversuche des Antigens unter Verwendung verschiedener Alaun-Lose sind längere Lagerungszeiten (> 6 Wochen bei 22°C) unter beschleunigten Bedingungen erforderlich. Es gibt einen Trend, dass Formulierungen, die mit Alaun 4230 hergestellt wurden, verglichen mit anderen Losen niedrigere ELISA-Verhältnisse zeigen. Eine verglichen mit aufgestockter Formulierung langsamere Abbaurate könnte zu niedrigerem Metallionengehalt in kommerziellen Alaun-Losen beitragen. Es wurde auch beobachtet, dass das Antigen bei pH 7,5–8 verglichen mit pH 7 eine höhere Stabilität zeigt und dass PS-Fragmente zu keiner Abbaureaktion beitragen. Diese Ergebnisse stimmen gut mit in Beispiel 1 beschriebenen DOE-Ergebnissen überein.
-
Beispiel 3
-
Als Teil der Untersuchung bei von der Spezifikation abweichenden Ergebnissen (OOS-Untersuchung) in Bezug auf FVL JEV09L37 wurde festgestellt, dass das verwendete Aluminiumhydroxid-Los (Los 4230) die wahrscheinlichste Ursache für den beobachteten Verlust der Wirkstärke ist. Aluminiumhydroxid (wird während des Herstellungsprozesses von JE-PIV als Alaun bezeichnet) wird von Brenntag Biosector als autoklavierte Suspension mit der Bezeichnung „Alhydrogel® Aluminium Hydroxide Gel Adjuvans” käuflich erworben. Vor der Verwendung im JEV-Produktionsprozess wird jede Charge durch Strahlung sterilisiert.
-
Eine Reihe von verschiedenen Alhydrogel®-Chargen wurde hinsichtlich Aussehen, Metallionengehalt und physikalischen Eigenschaften analysiert.
-
1 Einführung
-
1.1 Aluminiumhydroxid
-
Das Alhydrogel® von Brenntag Biosector weist einen vorgegebenen Aluminium-Gehalt von 10 mg/ml auf, der 2% Al2O3 bzw. 3% Al(OH)3 entspricht. Weitere Spezifikationen umfassen Stickstoff (max. 0,005%), freies Sulfat (max. 0,05%), Gesamt-Sulfat (max. 0,1%) und pH-Wert (6,5 ± 0,5). Bei Lagerung bei Raumtemperatur hat es eine Haltbarkeit von 26 Monaten.
-
1.2 Erzeugung von Aluminiumhydroxid
-
Alhydrogel® 2% (als Aluminiumhydroxid bezeichnet) wird von Brenntag produziert (CAS-Nr. 21645-51-2).
-
Zu beachten ist, dass nach der ersten Fällung durch Ammoniak und Ammoniumsulfat alle verbleibenden nachgelagerten Verarbeitungsschritte nur wasserlösliche Verunreinigungen entfernen können und daher keine Metallionen entfernen, die entweder an den Aluminiumhydroxid-Partikeln adsorbiert sind oder mit diesen präzipitierten. Der Herstellungsprozess enthält keinen speziellen Reinigungsschritt zur Entfernung von verunreinigenden Metallionen.
-
Die Ammoniumsulfat-Lösung wird nach der Präzipitation und der Konzentration des Aluminiumhydroxid-Gels recycelt.
-
1.3 Verwendung von Aluminiumhydroxid-Losen bei der JEV-Produktion
-
Zur Herstellung von kommerziellen JEV-Impfstoff-Chargen wurden bisher insgesamt 5 verschiedenen Alhydrogel®-2%-Lose von Brenntag verwendet. Diese Lose unterschieden sich in der Herkunft der Ammoniak-Alaun-Rohmaterialquelle und der Anzahl der JEV-Chargen, zu deren Formulierung sie verwendet wurden. Tabelle 21 bietet eine Übersicht.
-
2 Begriffsbestimmungen und Abkürzungen
-
-
- Alhydrogel
- 2% Aluminiumhydroxid-Lösung (auch als Alaun bezeichnet)
- DS/DP
- Wirkstoff/Fertigarzneimittel
- ESG
- Environmental Scientifics Group
- F-AAS
- Flammenatomabsorptionsspektrometrie
- FVL
- Fertigimpfstofflos
- GF-AAS
- Graphitrohratomabsorptionsspektrometrie
- ICP-MS
- induktiv gekoppelte Plasmamassenspektrometrie
- JEV
- Japanisches Enzephalitis-Virus
- JE-PrV
- gereinigtes inaktiviertes japanisches Enzephalitis-Virus
- LOQ
- Bestimmungsgrenze
- P & TD
- Patch & Technische Entwicklung
- PSD
- Partikelgrößenverteilung
- PZC
- Nullpunktladung
- QCI
- Qualitätskontrolle Immunologie
-
3 Materialien und Methoden
-
3.1 Alhydrogel®-Chargen und Rohmaterialien
-
- Alhydrogel® 2%-Lose: 3877, 4074, 4187, 4230, 4414, 4470, 4539, 4563, 4587, 4621 (nicht alle aufgeführten Alaun-Lose wurden in der Formulierung von JE-PIV verwendet; siehe Tabelle 21)
- 7× gewaschene Alhydrogel® 2%-Lose: 4577, 4580, 4596 (von Brenntag bezogen, nicht typisch für den 2% Alaun, der zur Formulierung erlangt wird)
- Ammoniak-Alaun: AA0427, 6-80578-28, 10094, 10095, 91480 (stellt Ammoniak-Alaun dar, der zur Herstellung verschiedener Alaun-Lose verwendet wurde. AA0427 wurde zur Herstellung von Alaun-Los Nr. 4230 verwendet, das zur Formulierung von JEV09L37 verwendet wurde)
- Ammoniak-Lösung 24%, technische Qualität: 4140716; lediglich als Probe und nicht mit getesteten Alaun-Chargen verbunden.
-
3.2 PSD-Messungen von Alhydrogel®
-
Die Aluminiumhydroxid-Partikelgrößenverteilung (PSD) wurde auf einem Malvern Mastersizer 2000μP-System mit einer 20-ml-Probenküvette analysiert. Alhydrogel® 2%-Bulkware wurde 1:20 in Wasser verdünnt und 1 ml wurde in die Probenküvette gegeben. Die endgültige Verdünnung der Probe in der Probenküvette war daher 400-fach (0,005 Aluminiumhydroxid).
-
3.3 Zeta-Potenzial-Messungen von Alhydrogel®
-
Zeta-Potenzial und Nullpunktladung (PZC) wurden auf einem Malvern Zetasizer ZS-System, ausgestattet mit einem MPT-2-Autotitrator, gemessen. Alhydrogel® 2%-Bulkware wurde 1:20 in PBS verdünnt und über Nacht bei Raumtemperatur äquilibriert. Zur Aufnahme der Ladungstitrationskurve wurde der pH unter Verwendung von 100 mM HCl- und 100 mM NaOH-Lösungen eingestellt. PZC wurde durch Extrapolation der Null-Ladung in der Titrationskurve ermittelt (Schnittpunkt der Titrationskurve und x-Achse). Die Nullpunktladung entspricht dem pH-Wert, bei dem die Oberfläche der Probe keine Nettoladung aufweist.
-
3.4 Analyse von Metallionen in Aluminiumhydroxid und Rohmaterialien
-
Ausgewählte Metallionen wurden entweder durch induktiv gekoppelte Plasmamassenspektrometrie (ICP-MS), Flammenatomabsorptionsspektrometrie (F-AAS) oder Graphitrohratomabsorptionsspektrometrie (GF-AAS) am Medizinischen Labor Bremen (Deutschland) analysiert. Kurz gesagt, wurden Proben, die Aluminiumhydroxid enthielten, unter Erhitzen mit konzentrierter HNO3 behandelt, bis eine klare Lösung erlangt wird. Die klare Lösung wird dann weiter verdünnt und analysiert. Das Vorhandensein und der Gehalt der folgenden Metallionen wurden ermittelt: Pb, Cd, Cr, Co, Fe, Cu, Ni, Ag, W und Al. Abhängig von der Verdünnung der Probe betrug die Bestimmungsgrenze (LOQ) 5 bis 25 ng/ml. Kristalline Ammoniak-Alaun-Proben wurden vor der Analyse in HNO3 gelöst. Vor der Analyse musste Ammoniak auf einen sauren pH eingestellt werden.
-
Zusätzlich wurde ein semiquantitativer 70-Elemente-Scan von ESG (GB) unter Verwendung einer Kombination von ICP-MS (Agilent 7500ce) und ICP-AES (Perkin Elmer Optima 4300DV) durchgeführt, die unter Verwendung zertifizierter Standards kalibriert wurden. Der Element-Scan ist ein Screening-Verfahren und nicht so empfindlich wie die Spurenanalyse für ausgewählte Metalle, wie sie vom Medizinischen Labor Bremen durchgeführt wird. Allerdings gibt ein solches Screening einen guten Überblick über das Vorhandensein und die Gehalte von bestimmten Metallen.
-
4 Ergebnisse
-
4.1 Bestimmung der Partikelgrößenverteilung von Alhydrogel®
-
Tabelle 22 fasst PSD-Daten von zwei Unterlosen jeweils von Alhydrogel®-Losen 4230 und 4740 zusammen. Verteilungsergebnisse sind in 11 dargestellt. Mittlere Partikel betrugen ~2–4 um, wobei Populationen von kleineren (< 1 um]) und größeren (> 20 um) Partikeln in allen vier Proben vorhanden sind. Die vier getesteten Alhydrogel®-Proben zeigten keinen signifikanten Unterschied in der mittleren Partikelgrößenverteilung.
-
4.2 Zeta-Potenzial-Messungen
-
Zwei Unterlose jeweils von Alhydrogel®-Losen 4230 und 4740 (2% Stammlösung vor der Analyse 20-fach in PBS verdünnt und über Nacht bei RT äquilibriert) wurden auf Nullpunktladung analysiert. Tabelle 23 fasst die Ergebnisse von PZC für die vier Proben zusammen, die sehr ähnliche PZC in PBS-Puffer zeigen. Titrationskurven sind in 12 dargestellt. Zwischen den vier untersuchten Proben konnte kein Unterschied bei den Titrationskurven und PZC beobachtet werden.
-
4.3 Bestimmung des Restmetallionengehalts in Alhydrogel®-Chargen
-
Die aktuellen Grenzwerte für Fe in 2% Aluminiumhydroxid-Lösungen nach Ph. Eur. sind 15 ppm (= 15 μg/ml) und ein Gesamtmaximum von 20 ppm (= 20 μg/ml) für andere Schwermetalle (wie beispielsweise Pb). Eine Konzentration von 15 ppm Fe würde jedoch 0,27 mM Fe in Lösung entsprechen. Berücksichtigt man, dass auch nur Spuren von Restmetallionen eine Vielzahl von Abbaureaktionen für Proteine katalysieren können (z. B. Oxidation, Aktivierung von Proteasen usw.) und dass Metalle in Lösung stabil bleiben, könnten Unterschiede im Metallionengehalt zwischen Aluminiumhydroxid-Losen Unterschiede hinsichtlich der Antigenstabilität über die Zeit verursachen.
-
Die Konzentrationen einer Reihe von Metallionen in handelsüblichen Aluminiumhydroxid-Losen wurden unter Verwendung von ICP-MS analysiert. Die Ergebnisse dieser Analysen sind in Tabelle 24 zusammengefasst. Lose 4074, 4230, 4470, 4414 und 4539 wurden bei der Herstellung von kommerziellen JEV-Chargen eingesetzt. Da eine 2% Alhydrogel®-Stammlösung einer Al-Konzentration von 10 mg/ml entspricht, kann die Quantifizierung des Al-Gehalts in den verschiedenen Proben als Bezugspunkt für die Ergebnisse, die für die anderen Metallionen erlangt werden, verwendet werden. Tatsächlich konnte ein durchschnittlicher Aluminiumgehalt von 10,3 mg/ml gemessen werden, was die Genauigkeit und Reproduzierbarkeit des Verfahrens zeigt.
-
Bei einem Vergleich der verschiedenen Alhydrogel®-Lose konnten große Schwankungen in der Menge an verunreinigenden Metallionen beobachtet werden. Die hervorstechendsten verunreinigenden Metallionen sind Fe, Cr und Ni, die in allen Chargen detektiert wurden. Darüber hinaus enthielt Los 4230 detektierbare Mengen an Cu, welches bei allen anderen Chargen unter der LOQ lag.
-
Man beachte jedoch, dass keines dieser Metalle in Mengen nahe der vorstehend erwähnten Spezifikationen von Alhydrogel® detektiert wurde. Zum Beispiel betrug die höchste Konzentration von Eisen, die in Los 4230 gefunden wurde, 5,6 μg/ml oder etwa 40% der zulässigen Konzentration.
-
Da die Präzipitation von Aluminiumhydroxid während des Prozesses in erheblichen Mengen an Restammoniak im Endprodukt resultiert, bietet Brenntag Biosector eine zweite Qualität von Alhydrogel® an. Im Gegensatz zum Standardprodukt wurde dieses „verbesserte” Alhydrogel® während des Reinigungsschritts 7 Mal mit Wasser statt nur 4 Mal für standardmäßiges Alhydrogel® gewaschen. Um zu testen, ob diese zusätzlichen Waschschritte in reduzierter Metallionenverunreinigung resultieren, wurden drei verschiedene Lose (4580, 4596 und 4577) analysiert. Die Ergebnisse sind in Tabelle 24 umfassen. Hinsichtlich Metallionen konnte kein Unterschied verglichen mit der Standardqualität von Alhydrogel® beobachtet werden, was nahe legt, dass die Metallionen entweder fest an die Oberfläche der Aluminium-Partikel gebunden sind oder tatsächlich während des Herstellungsprozesses copräzipitieren.
-
13 zeigt einen Vergleich der verschiedenen Alhydrogel®-Lose, die analysiert wurden. Es wird der Gesamtgehalt der verunreinigenden Metallionen für alle getesteten verunreinigenden Elemente gezeigt, wobei die absoluten Anteile der drei Hauptmetalle Fe, Cr und Ni in verschiedenen Farben dargestellt sind. Wie zu sehen ist, weist Los 4074 verglichen mit der Mehrzahl der anderen analysierten Chargen sehr wenig verunreinigende Metallionen auf. Nur Los 3877 zeigte eine ähnliche Losverunreinigung, während Los 4230 mit Abstand die höchste Verunreinigung von allen Chargen zeigt, die während dieser Untersuchung analysiert wurden.
-
Bei den Tests im Rahmen der Untersuchung wurde eine große Schwankung bezüglich des Metallionengehalts zwischen verschiedenen Chargen von Alhydrogel® beobachtet (siehe 13). Um zu testen, ob sich diese verunreinigenden Metallionen in der Aluminiumhydroxid-Fraktion oder im Überstand befinden, wurde Los 4230 in eine Überstand- und eine Sedimentfraktion getrennt (siehe Tabelle 25). Wie zu sehen ist, konnten weniger als 2% der Metallionen im Überstand detektiert werden, was anzeigt, dass alle verunreinigenden Metallionen entweder an die Aluminiumhydroxid-Partikeloberfläche oder innerhalb der Partikelstrukturen gebunden sind. Die lokale Metallionenkonzentration ist daher schätzungsweise mindestens 50–100 Mal höher, da der feste Volumenanteil (Volumen des Alaun-Pellets nach Zentrifugieren) von 0,1% Al2O3 (entspricht 0,5 mg/ml Al), das bei JEV-Impfstoff-Formulierung verwendet wird, etwa 10–20 μl pro 1000 μl FVL beträgt.
-
4.4 Bestimmung des Restmetallionengehalts in Alhydrogel®-Rohmaterialien
-
Insgesamt wurden 5 Chargen Ammoniak-Alaun (Rohmaterial für Alhydrogel®) von Brenntag Biosector bezogen. Diese Chargen wurden zur Herstellung von 4 Alhydrogel®-Chargen verwendet (siehe Tabelle 26; man beachte, dass zur Herstellung von Alaun-Los Nr. 4563 2 unterschiedliche Chargen Ammoniak-Alaun verwendet wurden), von denen drei für JEV-Impfstoff-Lose verwendet wurden. Bei der Anlieferung wurde ein deutlicher Unterschied beim visuellen Aussehen zwischen den Chargen beobachtet. Während vier Proben eine kristalline Struktur ähnlich großen Salzkristallen zeigten, war ein Los eher sandkornartig. Während die Kristalle völlig farblos waren, war außerdem das Material von Ammoniak-Alaun AA0427 zinkweiß. Dieser Befund ist von Bedeutung, da Los AA0427 zur Herstellung von Alhydrogel®-Charge 4230 verwendet wurde, die anschließend bei der Herstellung von JEV09L37 verwendet wurde (siehe Tabelle 26). Allerdings kann nach Ph. Eur. das Aussehen von Alaun „körniges Pulver oder farblose, durchsichtige, kristalline Massen” sein (Europäisches Arzneibuch 7. Auflage. Monograf 1357ff). Folglich entsprechen alle fünf Rohmaterialien dieser Definition.
-
Die ICP-MS-Analyse dieser fünf Ammoniak-Alaun-Lose und eines Loses 24% Ammoniak-Lösung (technische Qualität) zeigte, dass das Los 6-80578-28 sehr geringe Mengen an verunreinigenden Metallionen aufweist, während Los AA0427 sehr hohe Mengen aufweist (siehe Tabelle 27). Dies korreliert mit der Analyse der entsprechenden Alhydrogel®-Chargen (siehe Tabelle 24), was zeigt, dass diese Metallionen ihren Ursprung im Rohmaterial haben und nicht prozessbedingt sind.
-
14 zeigt einen Vergleich der verschiedenen Ammoniak-Alaun-Lose, die analysiert wurden. Es wird der Gesamtgehalt der Metallionen für alle getesteten verunreinigenden Elemente gezeigt, wobei die absoluten Anteile der drei Hauptmetalle Fe, Cr und Ni in verschiedenen Farben dargestellt sind. Ammoniak-Alaun-Los 6-80578-28 weist verglichen mit Los AA0427 bei gleicher Al-Konzentration ~ 40 Mal weniger verunreinigende Metallionen auf, was in der beobachteten viel höheren Reinheit der entsprechenden fertigen Alhydrogel®-Chargen 4074 und 4230 resultiert.
-
Beim Vergleich der Ergebnisse für die drei Ammoniak-Alaun-Lose mit ihren entsprechenden Alhydrogel®-Losen kann ein ähnliches Verhältnis für die drei Hauptverunreinigungen Fe, Ni und Cr beobachtet werden (Tabelle 28).
-
Dies zeigt, dass während des gesamten Aluminiumhydroxid-Produktionsprozesses im Wesentlichen keine Veränderung bei der Metallionenzusammensetzung erfolgt. Folglich kann eine Sekundärverunreinigung während des Herstellungsprozesses (z. B. andere Chemikalien oder der Vorratsbehälter) ausgeschlossen werden.
-
Tabelle 29 fasst die Ergebnisse der semiquantitativen Element-Scan-Analyse, die von ESG durchgeführt wurde, zusammen.
-
5 Zusammenfassung
-
Alhydrogel® wird in einer Endkonzentration von 0,1% als Adjuvans in der derzeitigen JEV-Impfstoff-Formulierung verwendet. Während der Untersuchung eines von der Spezifikation abweichenden (OOS) Wirkstärkeergebnisses für Produktions-FVL JEV09L37 wurde eine Beurteilung des Alhydrogel®-Produktionsprozesses eingeleitet. Insgesamt wurden 13 verschiedene Alhydrogel®-Lose auf das Vorhandensein von verunreinigenden Metallionen, welche die Stabilität des Proteins reduzieren könnten, analysiert.
-
Bei den verschiedenen Alhydrogel®-Losen wurden große Schwankungen bei der Konzentration einer Reihe von Metallionen beobachtet. Bei der Analyse der Rohmaterialien zeigte sich, dass diese Verunreinigungen in der gleichen Konzentration vorhanden waren, wie sie im Alhydrogel gefunden wurde.
-
Verglichen mit den anderen untersuchten Losen wurden höhere Konzentrationen von Fe-, Ni- und Cu-Ionen in Alhydrogel®-Los 4230 festgestellt. Los 4230 war das einzige, in dem Rest-Cu-Ionen detektiert wurden. Dieses Los 4230 wurde für die Formulierung von FVL JEV09L37 verwendet.
-
Bei der Analyse von Überstand und unlöslicher Fraktion einer Alhydrogel®-Charge konnten diese verunreinigenden Metallionen nur im Präzipitat gefunden werden, was anzeigt, dass diese Ionen entweder an die Aluminiumhydroxid-Partikeloberfläche gebunden sind oder tatsächlich Teil des Partikels sind.
-
Das Rohmaterial, das für Charge 4230 verwendet wurde, zeigte einen optischen Unterschied zu den anderen Chargen, die bei der JEV-Impfstoff-Herstellung verwendet wurden. Obwohl makroskopisch und hinsichtlich der Zusammensetzung verschieden von anderen Alhydrogel®-Losen, die bei der JEV-Herstellung verwendet wurden, entsprach Los 4230 allen Anforderungen, die Ph. Eur. angibt. Zudem zeigte die physikalische Charakterisierung (Partikelgrößenverteilung und Nullpunktladung) keine Unterschiede zwischen Los 4230 und anderen Alhydrogel®-Losen, die diese hohen Metallionenverunreinigungen nicht zeigen.
-
Beispiel 4
-
1.1. Materialien, Ausrüstung und Methoden
-
1.2. Ausrüstung
-
- Analysenwaage (Lesbarkeit 0,1 mg, z. B. Mettler Toledo XP205DR/M) Präzisionswaage (Lesbarkeit 0,1 g, z. B. Mettler Toledo, Model Nr. XS6002S Delta Range)
- Filtereinheiten 0,22-μm-(z. B. Stericup Kat.-Nr. SCGPV01RE) oder 0,2-μm-Filtersystem (z. B. 50 ml Millipore Steriflip)
- Gefrierschrank (–20°C) und Ultra-Gefrierschrank (–80°C)
- Kühlschrank (+ 2 bis 8°C)
- Magnetrührer (z. B. KIKA Labortechnik RCT basic) und Magnetrührstäbe
- Mikroplatten-Wäscher: z. B. BioTek ELx405
- Mikroplatten-Lesegerät: z. B. BioTek Synergy 2 und Gen5 Secure-Software
- Mikroplatten-Inkubator (37°C)
- Microtiter-Dichtband (z. B. Thermo Electron 9503130)
- Mehrkanalpipetten und Spitzen (z. B. Eppendorf Research Pro 50–1200 μl, Eppendorf Research, 10–100 μl)
- pH-Meter (z. B. WTW-Serie ino Lab, Terminal 740 und pH/Cond. 740)
- Pipetten und Spitzen (z. B. Eppendorf Research, 0,5–10 μl, 2–20 μl, 20–200 μl, 100–1000 μl, 500–5000 μl)
- Pipettierer (z. B. IBS Biosciences Pipetboy)
- PP-Röhrchen 15 ml (z. B. Sarstedt 62.515.006) oder PP-Röhrchen 50 ml (z. B. Greiner 227261)
- Reagenz-Reservoir 50 ml (z. B. Corning Incorporated 4870)
- Serologische Pipetten (z. B. Falcon, 2 ml, 5 ml, 10 ml, 25 ml, 50 ml)
- Titertube-Mikroröhrchen – Bulk (BioRad 223-9391)
- Vortex-Mischer (z. B. VWR Analog Vortex Mixer, Modell-Nr. 945304) 1,5-ml- oder 2,0-ml-Eppendorf-LoBind-Röhrchen (Kat.-Nr. 0030 108.116 bzw. Kat.-Nr. 0030 108.132)
- 96-Well-Mikroplatten (F96 Cert. Maxisorp Nunc-Immunplatten)
-
Zusätzlich für die Analyse der DP-Proben:
Tischzentrifuge (z. B. Beckman Coulter, Microfuge 16 Centrifuge, Kat.-Nr. A46473)
Orbitalschüttler (z. B. Eppendorfer Thermomixer compact)
50-ml-PP-Röhrchen (z. B. Greiner 227261)
-
1.3. Reagenzien
-
- PBS 10× (z. B. Gibco, Kat.-Nr. 14200-083)
- Tween 20 (z. B. Sigma Kat.-Nr. P7949)
- 2 M Schwefelsäure (volumetrische Lösung, z. B. Fisher, Kat.-Nr. J/8410/17)
- Entionisiertes Wasser, z. B. (Milli-Q, 18,2 Ω)
- Natriumcarbonat-Hydrogencarbonat-Kapseln (z. B. Sigma, Kat.-Nr. C3041)
- Salzsäure (HCl) 1 mol/l (z. B. Merck, Kat.-Nr. 1.09057.1000)
- Natriumhydroxid (NaOH) 1 mol/l (z. B. Merck, Kat.-Nr. 1.09132.1000)
- Glycerin (z. B. Sigma)
-
Zusätzlich für die Analyse der DP-Proben:
Dikaliumphosphat-Trihydrat (z. B. Sigma, Kat.-Nr. P5504)
Kaliumdihydrogenphosphat (z. B. VWR, AnalaR Normapur, Kat.-Nr. 26936.260)
Albumin, Rinderserum (BSA), ELISA-Qualität (z. B. Sigma, Kat.-Nr. A3059)
TMB-Substrat (z. B. BioFX, TMBW-1000-1001)
Esel-anti-Kaninchen-IgG-HRP-Konjugat (Jackson Immuno Research, Kat.-Nr. 711-035-152)
-
Rekonstitution:
-
Der Inhalt von 1 Gläschen (0,4 mg) wird in 0,5 ml entionisiertem Wasser rekonstituiert und bis zur vollständigen Auflösung gründlich gemischt. Zugabe von 0,5 ml Glycerin und weiteres Mischen bis zur Homogenität. Aliquote werden bis zur Verwendeung bei –20°C gelagert.
Referenzstandard von inaktiviertem JEV (Intercell Biomedical Ltd.)
Gereinigtes Schaf-anti-JEV (Intercell Biomedical Ltd.)
Gereinigtes Kaninchen-anti-JEV (Intercell Biomedical Ltd.)
-
1.4. Lösungen
-
a) 0,05 M Carbonat-Puffer bei pH 9,6 (zur Beschichtung von ELISA-Platten verwendet)
-
Für 100 ml Puffer eine Bicarbonat/Carbonat-Puffer-Kapsel in 100 ml entionisiertem Wasser lösen. Den pH prüfen und ihn gegebenenfalls mit HCl oder NaOH auf 9,6 ± 0,1 einstellen. Nur am Tag der Herstellung verwenden. Am Tag der Verwendung den ELISA-Beschichtungspuffer bei RT halten, dann verwerfen.
-
b) ELISA-Waschpuffer und Teil des Blockier-/Probenverdünnungsmittels (PBS-T)
-
Etwa 1 Liter für jede verwendete Platte herstellen. 10 × PBS-Stammlösung 1 + 9 in entionisiertem Wasser verdünnen, gut mischen und den pH prüfen (7,4 ± 0,1), gegebenenfalls mit 1 M HCl oder 1 M NaOH einstellen. 0,05% (Vol./Vol.) Tween 20 zugeben, gut mischen.
z. B. ELISA-Waschpuffer (PBS-T) [1 l]:
100 ml 10 × PBS
900 ml entionisiertes Wasser
Gut mischen, pH prüfen/einstellen (7,4 ± 0,1)
0,5 ml Tween20
Gut mischen.
-
Nur am Tag der Herstellung verwenden; am Tag der Verwendung den ELISA-Waschpuffer bei RT halten, dann verwerfen.
-
c) Blockierlösung: 5% BSA in PBS-T
-
Etwa 25 ml für jede Platte herstellen. Die erforderliche Menge PBS-T mit einer serologischen Pipette in eine saubere Glasflasche einmessen. Einen sauberen Magnetrührstab zugeben. Die erforderliche Menge BSA wiegen, an der Oberfläche von PBS-T zugeben und auf einem Magnetrührer behutsam mischen, bis das gesamte BSA in Lösung gegangen ist. Lösung mit einem 0,2-μm-Filter (entweder dem Steriflip-Filtersystem oder einem Spritzenfilter) filtrieren.
z. B. Blockierlösung [100 ml]
5 g BSA
100 ml PBS-T
-
Nur am Tag der Herstellung verwenden; am Tag der Verwendung die Blockierlösung bei RT halten, dann verwerfen.
-
d) Probenverdünnungsmittel: 1% BSA in PBS-T
-
Wie oben angegeben herstellen, jedoch mit 1 g BSA pro 100 ml PBS-T, es sind etwa 25 ml pro Platte erforderlich,
z. B. Probenverdünnungsmittel [100 ml]
1 g BSA
100 ml PBS-T
-
Nur am Tag der Herstellung verwenden; am Tag der Verwendung das Probenverdünnungsmittel bei RT halten, dann verwerfen.
-
Zusätzlich für die Analyse von DP-Proben:
-
e) 1 × PBS
-
1 Teil 10 × PBS mit 9 Teilen entionisiertem Wasser herstellen
z. B. 1 × PBS [100 ml]
10 ml 10 × PBS
90 ml entionisiertes Wasser
-
Nur am Tag der Herstellung verwenden; am Tag der Verwendung 1 × PBS bei RT halten, dann verwerfen.
-
f) 20 × ELISA-Puffer
-
Zur Herstellung einer 20 × Lösung eine entsprechende Menge BSA in einen geeigneten Behälter einwiegen. Das entsprechende Volumen 1 × PBS hinzugeben. Tween20 bis zu einer Endkonzentration von 0,05% zugeben. Auf dem Magnetrührer rühren, bis das BSA vollständig gelöst ist. Die Lösung durch einen 0,2-um-Filter (entweder das Steriflip-Filtersystem oder einen Spritzenfilter verwenden) in einen sterilen Behälter filtrieren (und nach Bedarf aliquotieren).
z. B. 20 × ELISA-Puffer [25 ml]
5 g BSA
25 ml 1 × PBS
12,5 μl Tween20
-
Die Lösung kann bei 2–8°C 1 Woche lang gelagert werden.
-
g) 2 × ELISA-Puffer
-
Er wird durch Verdünnung des 20 × ELISA-Puffers mit 1 × PBS (1 Teil 20 × ELISA-Puffer und 9 Teile 1 × PBS) hergestellt.
z. B. 2 × ELISA-Puffer [20 ml]
2 ml 20 × ELISA-Puffer
18 ml 1 × PBS
-
Nur am Tag der Herstellung verwenden; am Tag der Verwendung 2 × ELISA-Puffer bei RT halten, dann verwerten.
-
h) Desorptionspuffer
-
- • Kaliumphosphat-Stammlösung: Herstellung einer 3 × Stammlösung von Kaliumphosphat (2,4 M) durch Auflösen des entsprechenden Volumens Dikaliumphosphat-Trihydrat und von Kaliumdihydrogenphosphat in entionisiertem Wasser. Auf einem Magnetrührer platzieren und nach Auflösung auf das erforderliche Volumen auffüllen, prüfen, dass der pH der Lösung 8,0 ± 0,1 ist. Durch einen 0,2-um-Filter filtrieren.
z. B. 3 × Stammlösung von Kaliumphosphat (2,4 M) [50 ml]
23,963 g Dikaliumphosphat-Trihydrat
2,041 g Kaliumdihydrogenphosphat
Auf 50 ml auffüllen Entionisiertes Wasser
Bei +2°–8°C für bis zu 1 Monat lagern.
- • Herstellung von Desorptionspuffer in Arbeitsstärke (0,8 M Kaliumphosphatpuffer, der 1% BSA und 0,05% Tween20 enthält) durch Zugabe des entsprechenden Volumens Kaliumphosphat-Stammlösung (2,4 M), Tween20 und BSA zum erforderlichen Volumen entionisierten Wassers. Gründlich mischen und am Tag der Herstellung verwenden.
z. B. Desorptionspuffer in Arbeitsstärke [15 ml]
5 ml Kaliumphosphat (2,4 M)
7,5 μl Tween20
0,15 g BSA
10 ml Entionisiertes Wasser
Den Desorptionspuffer in Arbeitsstärke am Tage der Verwendung bei RT halten.
-
1.5. Testproben und Antikörper
-
Testproben:
-
- • Wirkstoff und/oder NIV (verschiedene Chargen)
- • JEV-Impfstoffproben (Fertigbulkimpfstoff und Fertigimpfstofflos) Referenzstandard von inaktiviertem JEV (Neutralisiertes Inaktiviertes Virus – NIV) (Intercell Biomedical Ltd.)
-
Polyklonale Antikörper:
-
- • Beschichtungsantikörper: Gereinigtes Schaf-anti-JEV (Intercell Biomedical Ltd.)
- • Primärer Detektionsantikörper: Gereinigtes Kaninchen-anti-JEV (Intercell Biomedical Ltd.)
-
Konjugierter Sekundärantikörper: Esel-anti-Kaninchen-HRP-Konjugat (Jackson Immuno Research Kat.-Nr. 711-035-152)
-
2. Verfahren
-
2.1. Plattenbeschichtung
-
- • Die Platte mit Plattennummer, Datum und Analyst beschriften.
- • Frischen 0,05 M Carbonatpuffer (pH 9,6) am Tag der Plattenbeschichtung herstellen. Etwa 12 ml für jede beschichtete Platte einkalkulieren.
- • Die erforderliche Anzahl von Aliquoten des Beschichtungsantikörpers aus dem Gefrierschrank nehmen und bei RT auftauen lassen. Eine Verdünnung des gereinigten Schaf-anti-JEV in Carbonat-Puffer herstellen. Durch Inversion des Röhrchens gut mischen.
- • Unter Verwendung der Mehrkanalpipette 100 μl/Vertiefung auf eine 96-Well-Maxisorp-Platte innerhalb von 15 min der Herstellung der Antikörperverdünnung aufgeben.
- • Mit Mikrotiter-Dichtband abdecken und 17 bis 72 Stunden bei +2–8°C inkubieren.
-
2.2. Waschen
-
- • Platte aus dem Kühlschrank nehmen und auf Raumtemperatur erwärmen lassen.
- • Die Platte(n) mit dem Mikrotiterplatten-Wäscher 3 Mal unter Verwendung des jeweiligen Waschprogramms waschen (300 μl pro Vertiefung, 3 Mal, finale Dispensierung). Danach: etwaigen verbleibenden Waschpuffer durch Dekantieren entfernen. Die Platte invertieren und mit einem sauberen Papiertuch abtupfen. Mikrotiterplatten zwischen Waschschritten und Zugabe von Reagenzien nicht trocknen lassen.
-
2.3. Blockierung
-
- • Eine Blockierungslösung 5% (Gew./Vol.) BSA in PBS-T wie oben herstellen.
- • 200 μl Blockierlösung pro Vertiefung aufgeben, die Platte(n) mit einer Abdeckplatte abdecken und bei 37°C 1 Stunde ± 10 min lang inkubieren.
-
2.4. Herstellung von Standardkurvenverdünnungen
-
- • Den NIV-Referenzstandard aus dem Gefrierschrank nehmen, bei RT auftauen lassen, gut mischen. Eine 1 AE/ml Stammverdünnung des derzeitigen Referenzstandards herstellen; mindestens 20 μL NIV-Referenzstandard zur Verdünnung verwenden.
z. B. Vorverdünnung des NIV-Referenzstandards:
Konzentration: 235 AE/ml (Los Nr. 03/2009)
Zur Herstellung einer 1 AE/ml Arbeitsstandardlösung 1 zu 235 in Probenverdünnungsmittel verdünnen:
4680 μl Probenverdünnungsmittel
20 μl NIV-Referenzstandard
- • Dann die folgenden Arbeitsstandardlösungen aus der 1 AE/ml-Vorverdünnung herstellen:
0,8 AE/ml, 0,6 AE/ml, 0,4 AE/ml, 0,2 AE/ml, 0,1 AE/ml und 0,05 AE/ml in Probenverdünnungsmittel.
-
2.5. Proben zur Qualitätskontrolle
-
- a) Proben zur Qualitätskontrolle (QC) (zum Beispiel bei 0,75, 0,30 und 0,18 AE/ml) sollten aus der Vorverdünnung des NIV-Referenzstandards frisch zum Zeitpunkt des Assays hergestellt werden, dann nach Verwendung verworfen werden.
- b) Diese Kontrollen sind Teil der Systemeignungskriterien und ermöglichen, dass die Performance des Assays über die Zeit überwacht werden kann.
-
2.6. Herstellung der Testproben
-
Herstellung des Wirkstoffs
-
Testproben des Wirkstoffs werden für die Tests mit unbekannten Konzentrationen empfangen. Sie werden bei sechs Verdünnungen in dreifacher Ausführung getestet. Die Verdünnungen werden unabhängig im Bereich der Standardkurve gemacht, z. B. Vorverdünnung von 1 zu 15 oder eine andere geeignete Verdünnung, dann sechs Verdünnungen mit Probenpuffer.
-
Herstellung der NIV-Proben
-
NIV-Proben werden für die Tests mit unbekannten Konzentrationen empfangen und im Bereich der Standardkurve vorverdünnt (z. B. 1 zu 30 oder eine andere geeignete Verdünnung), dann 6 Mal in der gleichen Weise wie die DS-Proben verdünnt.
-
Herstellung des Fertigarzneimittel-Überstands
-
- a) Für die Analyse der Bulk-DP-Proben die Probe durch Vortexen gut mischen. Genau 1 ml in ein 1,5-ml-LoBind-Eppendort-Röhrchen übertragen.
Für die Analyse der Endproduktbehälterproben den Inhalt von 2 Spritzen (0,6 ml je Spritze) vom gleichen Los in ein 1,5-ml-LoBind-Eppendort-Röhrchen übertragen. Den Inhalt des Röhrchens gründlich durch Inversion mischen, um Homogenität des DP zu gewährleisten und genau 1 ml in ein frisches 1,5-ml-LoBind-Eppendort-Röhrchen übertragen.
- b) Röhrchen, die 1 ml DP enthalten, jeweils bei 3300 × g 5 Minuten lang zentrifugieren.
- c) Für jede Probe 25 μl 20 × ELISA-Puffer in frische LoBind-Eppendorf-Röhrchen pipettieren.
- d) Vorsichtig 475 μl des Überstands entfernen, ohne das Alaun-Pellet zu stören, und in das Röhrchen, das den 20 × ELISA-Puffer enthält, übertragen. Durch Inversion behutsam mischen. Noch einmal 2 min bei 16.000 × g zentrifugieren. Vor der Analyse Probe bei +2–8°C lagern.
HINWEIS: Auf diese Weise hergestellte DP-Überstandsproben sollten ohne Verdünnung in dreifacher Ausfertigung in dem ELISA für inaktiviertes JEV gemessen werden.
- e) So viel wie möglich von dem restlichen Überstand von dem zentrifugierten Röhrchen unter Verwendung einer 10-200-ul-Pipette entfernen, ohne das Alaun-Pellet zu stören, und den Überstand verwerfen.
- f) Das erhaltene Pellet wird der Desorptionsvorschrift unterworfen, wie nachfolgend beschrieben.
-
Fertigarzneimittel-Desorptionsvorschrift
-
- a) 158 μl Desorptionspuffer in Arbeitsstärke zu jedem Pellet, das in einem LoBind-Röhrchen verblieben ist, geben.
- b) Das Pellet durch mehrmaliges Auf- und Ab-Pipettieren resuspendieren, um eine vollständige Resuspension des Pellets und Homogenisierung der Probe zu gewährleisten.
- c) Proben 10 min lang bei RT auf einem Orbitalschüttler bei 500 UpM inkubieren.
- d) Nach der Inkubation Proben bei 3300 × g 5 Minuten lang zentrifugieren.
- e) Für jede Probe 250 μl 2 × ELISA-Puffer in ein frisches LoBind-Eppendorf-Röhrchen Pipettieren.
- f) Vorsichtig 83,3 μl aus dem oberen Teil des Überstands, der das desorbierte Produkt enthält, entfernen, ohne das Pellet zu stören und in das Röhrchen mit dem 2 × ELISA-Puffer übertragen. Verbleibenden Überstand unter Verwendung einer 20–200 μl-Pipette entfernen, ohne das Pellet zu stören, und den Überstand verwerfen.
- g) Weitere 158 μl Desorptionspuffer in Arbeitsstärke zu jedem Pellet geben.
- h) 2 weitere Desorptionszyklen (insgesamt 3) durchführen, wobei die 3 × 83,3 μl des desorbierten Materials + 250 μl ELISA-Puffer in dem entsprechenden Röhrchen gepoolt werden. Nach dem letzten Schritt kann das verbleibende Pellet verworfen werden.
- i) Die Endkonzentration des viralen Antigens in dem (den) desorbierten Pool(en) sollte nun die gleiche sein, wie sie ursprünglich in 1 ml DP war, von dem sie desorbiert wurde. Daher kann die Konzentration des inaktivierten JEV-Antigen-Gehalts, der im desorbierten Pool gemessen wird, direkt auf das ursprüngliche DP bezogen werden. Hinweis: Die desorbierten Proben mittels ELISA am Tag der Desorption analysieren.
- j) Verdünnung der desorbierten DP-Proben:
Diese werden bei sechs Verdünnungen in dreifacher Ausführung getestet. Eine entsprechende Vorverdünnung wird im Bereich der Standardkurve gemacht, z. B. 1 zu 15 (100 μl bis 1400 μl Verdünnungsmittel) oder eine andere geeignete Verdünnung, dann werden sechs Verdünnungen von 1 zu 15 Vorverdünnung unabhängig unter Verwendung von Probenverdünnungsmittel gemacht.
-
2.7. Probenbeladung und Plattenplan
-
Proben und Standards vor der Analyse herstellen.
-
Nach der Blockierung die Platte unter Verwendung des Plattenwäschers unter Einsatz des JEV-ELISA-Programms waschen. Danach etwaigen verbleibenden Waschpuffer durch Dekantieren entfernen. Die Platte invertieren und mit einem sauberen Papiertuch abtupfen. Mikrotiterplatten zwischen Waschschritten und Zugabe von Reagenzien nicht trocknen lassen.
-
100 μl/Vertiefung von Standards/Kontrollen/Proben zugeben und mit Abdeckplatte abdecken und 1 Stunde ± 10 min lang bei 37°C inkubieren.
-
100 μl Probenverdünnungslösung in alle Vertiefungen geben, die zum Testen nicht erforderlich sind.
-
2.8. Herstellung von Primärantikörper
-
Die erforderliche Anzahl von Aliquoten des Primärantikörpers aus dem Gefrierschrank nehmen und bei RT auftauen lassen. Max. 15 min vor der Verwendung Kaninchen-anti-JEV in Probenverdünnungsmittel bei einer geeigneten Verdünnung herstellen. Nach der Probeninkubation die Platte unter Verwendung des Plattenwäschers unter Einsatz des JEV-ELISA-Programms waschen. Danach etwaigen verbleibenden Waschpuffer durch Dekantieren entfernen. Die Platte invertieren und mit einem sauberen Papiertuch abtupfen. Mikrotiterplatten zwischen Waschschritten und Zugabe von Reagenzien nicht trocknen lassen.
-
100 μl/Vertiefung des verdünnten Primärantikörper zugeben, mit Abdeckplatte abdecken und 1 Stunde ± 10 min lang bei 37°C inkubieren.
-
2.9. Herstellung von Sekundärantikörper-Konjugat
-
Die erforderliche Anzahl von Aliquoten des Sekundärantikörper-Konjugats aus dem Gefrierschrank nehmen und bei RT auftauen lassen. Max. 15 min vor der Verwendung eine Verdünnung von Esel-anti-Kaninchen-JEV in Probenverdünnungsmittel bei einer geeigneten Verdünnung herstellen; z. B. für eine 1-zu-10.000-Verdünnung eine 1-zu-100-Vorverdünnung machen, dann eine zweite Verdünnung von 1 zu 100 machen.
-
Nach der Primärantikörperinkubation die Platte unter Verwendung des Plattenwäschers unter Einsatz des JEV-ELISA-Programms waschen. Danach etwaigen verbleibenden Waschpuffer durch Dekantieren entfernen. Die Platte invertieren und mit einem sauberen Papiertuch abtupfen. Mikrotiterplatten zwischen Waschschritten und Zugabe von Reagenzien nicht trocknen lassen.
-
100 μl/Vertiefung des verdünnten Sekundärantikörper-Konjugats zugeben, mit Abdeckplatte abdecken und 1 Stunde ± 10 min lang bei 37°C inkubieren.
-
2.10. Substratinkubation
-
Nach Zugabe des Konjugats TMB aus dem 2–8°C-Kühlschrank nehmen. Das erforderliche Volumen (12 ml TMB pro Platte) unter Verwendung einer serologischen Pipette in ein 50-ml-Zentrifugenröhrchen pipettieren. Das TMB im Dunkeln Raumtemperatur erreichen lassen. Nach der Konjugatinkubation die Platte unter Verwendung des Plattenwäschers unter Einsatz des JEV-ELISA-Programms 3 Mal waschen. Danach etwaigen verbleibenden Waschpuffer durch Dekantieren entfernen. Die Platte invertieren und mit einem sauberen Papiertuch abtupfen. Mikrotiterplatten zwischen Waschschritten und Zugabe von Reagenzien nicht trocknen lassen. 100 μl/Vertiefung TMB zugeben und die Platte im Dunkeln bei Raumtemperatur 10 Minuten lang entwickeln.
-
2.11. Stoppen und Lesen
-
Nach 10-minütiger TMB-Inkubation die Entwicklung durch Zugabe von 100 μl/Vertiefung 2 M Schwefelsäure stoppen. Die Platte bei 450 nm (Referenzfilter 630 nm) innerhalb von 10 Minuten nach dem Stoppen unter Verwendung des BioTek-Lesers und der Gen5 Secure-Software lesen.
-
2.12. Datenanalyse
-
NIV/DS-Datenanalyse:
-
Gen5Secure-Software wird zur Berechnung der prozentualen Rückgewinnung der QCs, Konzentration × Verdünnungen, mittlere Konzentration der Proben korrigiert für Verdünnungen und diesen Wert mit 1,05 multipliziert, um für die Zugabe von ELISA-Puffer zu korrigieren, verwendet.
-
DP-Datenanalyse:
-
Gen5Secure-Software wird zur Berechnung der prozentualen Rückgewinnung der QCs, Konzentration × Verdünnungen und mittlere Konzentration der Verdünnungen für die Proben verwendet.
-
Für DP-Überstandsproben:
-
Wenn die Konzentration der Überstandsprobe unter der LLOQ des Assays liegt (d. h. 0,05 AE/ml), sollte die Überstandsprobe als < 0,05 AE/ml dokumentiert werden.
-
Wenn die Konzentration der Überstandsprobe innerhalb der LOQs des Assays liegt (d. h. 0,05 AE/ml bis 1,25 AE/ml), dann wird der Konzentrationswert für die Überstandsprobe dokumentiert.
-
Wenn die Konzentration der Überstandsprobe über der ULOQ des Assays liegt (d. h. 1,25 AE/ml), sollte die Herstellung des Fertigarzneimittelüberstands wiederholt werden. Die Überstandsprobe wird erneut getestet, indem eine geeignete Vorverdünnung im Bereich der Standardkurve, gefolgt von 6 Probenverdünnungen, durchgeführt wird. (Die desorbierte Fertigarzneimittelprobe muss nicht wiederholt werden.)
-
Die mittlere Konzentration für die Verdünnungen, die innerhalb der LOQs des Assays liegen (LLOQ 0,05 AE/ml bis 1,25 AE/ml), ist der dokumentierte Konzentrationswert für die Überstandsprobe, vorausgesetzt, dass die Systemeignung gegeben ist und mindestens 4 aus den 6 Probenverdünnungen innerhalb der LOQs liegen.
-
2.13. Assay-Annahmebedingungen
-
- a) Der Korrelationskoeffizient für die Eichkurve muss > 0,980 sein.
- b) proz. CVs ≤ 15% für Standards und Proben (außer DP-Überstand) für die vier höchsten Konzentrationen der Verdünnungen, proz. CVs ≤ 15% für Kontrollen
- c) Einzelne Leerproben-ODs müssen ≤ 0,2 sein.
- d) Assay-Kontrollen müssen innerhalb der angegebenen definierten Grenzen liegen (für frisch hergestellte Kontrollen sollten 2 von 3 QCs beobachtete Konzentrationen innerhalb ±30% der Nennwerte aufweisen; ODER der Gehalte, die während der QC-Qualifikation festgelegt wurden), um den Test zu bestehen.
- e) Die Gültigkeit des Assays wird auf dem Gen5-Ausdruck dokumentiert. Wenn die Platte die definierten Annahmekriterien nicht erfüllt, ist der Test ungültig.
-
2.14. Dokumentation von Daten
-
NIV
-
a) Antigengehalt
-
Der dokumentierte Wert für inaktivierte AE/ml ist das Mittel der Konzentrationen (die innerhalb der LOQs von 0,04 bis 1,25 AE/ml liegen), die für die einzelnen Probenverdünnungen berechnet wurden, korrigiert mit den entsprechenden Verdünnungsfaktoren, und wobei der Mittelwert mit einem Korrekturfaktor 1,05 multipliziert wird, um das 5% Volumen von 20 × ELISA-Puffer zu berücksichtigen, das zu jeder Probe zugegeben wurde, als sie entnommen wurde. Antigengehalt wird auf dem Gen5-Ausdruck dokumentiert.
-
DS:
-
a) Identität
-
Wenn die Absorptionen der Proben bei der niedrigsten Verdünnung (höchsten Konzentration) mehr als 3 Standardabweichungen über dem Mittelwert der Leerprobe liegen, wird das Ergebnis als positiv dokumentiert.
-
b) Antigengehalt
-
Der dokumentierte Wert für inaktivierte AE/ml ist das Mittel der Konzentrationen (die innerhalb der LOQs von 0,04 bis 1,25 AE/ml liegen), die für die einzelnen Probenverdünnungen berechnet wurden, korrigiert mit den entsprechenden Verdünnungsfaktoren, und wobei der Mittelwert mit einem Korrekturfaktor 1,05 multipliziert wird, um das 5% Volumen von 20 × ELISA-Puffer zu berücksichtigen, das zu jeder Probe zugegeben wurde, als sie entnommen wurde. Antigengehalt wird auf dem Gen5-Ausdruck dokumentiert.
-
Desorbiertes DP:
-
a) Identität:
-
Wenn die Absorptionen der Proben bei der niedrigsten Verdünnung (höchsten Konzentration) mehr als 3 Standardabweichungen über dem Mittelwert der Leerprobe liegen, wird das Ergebnis als positiv dokumentiert.
-
b) Antigengehalt
-
Der dokumentierte Wert für inaktivierte AE/ml ist das Mittel der Konzentrationen (die innerhalb der LOQs von 0,04 bis 0,8 AE/ml liegen), die für die einzelnen Probenverdünnungen berechnet wurden, korrigiert mit den entsprechenden Verdünnungsfaktoren. Antigengehalt wird auf dem Gen5-Ausdruck dokumentiert
-
DP-Überstand (Grad der Adsorption/Grad der Nicht-Adsorption):
-
Grad der Adsorption wird in Beziehung zu Aluminiumhydroxid-formulierten Wirkstoff nach Filtration dokumentiert.
- a) Für die Berechnung des dokumentierten Werts wird der dokumentierte Antigengehalt (AE/ml) für die jeweilige DS-Probe (nach Filtration), korrigiert für die Verdünnung mit Aluminiumhydroxid (5%), auf 100% gesetzt, und der Prozentsatz der gemessenen Konzentration im Überstand (korrigiert für die Zugabe von 5% ELISA-Puffer) wird in Bezug darauf berechnet. Der dokumentierbare Wert ist die Differenz zwischen 100% und dem für den Überstand berechneten Prozentsatz. Ergebnisse werden auf 2 Dezimalstellen dokumentiert.
DP-Überstand (AE/ml)·1,05 Der Grad der Nicht-Adsorption wird wie nachfolgend erläutert berechnet und Ergebnisse werden auf 2 Dezimalstellen dokumentiert:
DP-Überstand (AE/ml)· 1,05
- b) Im Fall, dass der unverdünnte Überstand kein messbares Antigen enthält (d. h. eine beobachtete Überstandskonzentration von weniger als LLOQ, wobei LLOQ = 0,05 AE/ml), wird die 1100 zur Berechnung des Ergebnisses verwendet. Das Ergebnis in diesem Fall wird als „größer als x%” dokumentiert. Wenn die DS-Probe beispielsweise als 12,00 AE/ml gemessen wird und im Überstand kein Signal gemessen wurde; mit der LLOQ von 0,05 AE/ml ist dann die Menge im Überstand < 0,05·1,05 = < 0,0525 AE/ml. Die Mange DS nach Pufferkorrektur ist 12,00·0,95 = 11,40 AE/ml, und das dokumentierte Ergebnis für Grad der Adsorption ist < 100 – 0,0525/11,4·100 => 99,54%. Der Grad der Nicht-Adsorption wird ebenfalls dokumentiert (d. h. 100 – der proz. Grad der Adsorption).
-
Beispiel 5
-
Einleitung:
-
Zur weiteren Untersuchung der Wirkungsweise, die zu Produktinstabilität/Wirkstärkeverlust des JEV-Impfstoffs führt, wurde Ala-(His)6 OprF190-342-OprI21-83 (SEQ ID NO: 1, 15) – hier auch als „Protein A” bezeichnet – in einem vorläufigen Screening-Assay unter Einbeziehung von Alhydrogel-Losen mit unterschiedlichem Metallgehalt und Aufstocken mit Kupferionen und Sulfit eingesetzt.
-
Material:
-
- – Kupfer(II)-chlorid-Dihydrat (Sigma, Bestell-Nr. 807483)
- – 10 × PBS (Gibco, Best.-Nr. 14200-091)
- – 15 ml-Falcon-Röhrchen (Greiner, Kat.-Nr. 188724)
- – Inkubator Infors HT Inkubator Multitron Standard (InforsAG)
- – Doppeldestilliertes Wasser (Fresenius Kabi, Art.-Nr. 0712221/01 A)
-
Herstellung der Stammlösungen:
-
- – Kupfer(II)-Stammlösung
20 mM Kupfer(II)-Stammlösung wurden durch Lösen von 341 mg Kupfer(II)-chlorid-Dihydrat in 100 ml doppeldestilliertem Wasser hergestellt.
- – Natriummetabisulfit-Stammlösung
200 mM Natriummetabisulfit-Stammlösung wurden durch Lösen von 1,52 g Natriummetabisulfit in 35 ml PBS hergestellt. Diese Lösung wurde mit NaOH auf pH 7,3 eingestellt und mit PBS auf ein Volumen von 40 ml aufgefüllt. Die Lösung wurde dann durch ein 0,2-μ-Spritzenfilter filtriert.
-
Herstellung von Arbeitslösungen
-
Arbeitslösungen wurden durch Verdünnung von Metallstammlösungen mit doppeldestilliertem Wasser und Sterilfiltration durch ein 0,2-μ-Spritzenfilter hergestellt.
(Mini Kleenpak 25 mm – Pall)
-
Herstellung von Pufferlösungen
-
- – 1/3 PBS + 0,9% NaCl
1 × PBS-Pufferlösung wurde durch 1:10-Verdünnung von 10 × PBS mit doppeldestilliertem Wasser hergestellt. Der pH dieser Pufferlösung betrug 7,5. Auf pH 7,3 und 8,0 eingestellte PBS-Pufferlösungen wurden durch Einstellen des pH mit NaOH bzw. HCl hergestellt.
9 g NaCl wurden in 333 ml entweder Puffer pH 7,3 oder Puffer pH 8,0 gelöst und mit doppeldestilliertem Wasser auf 1000 ml gebracht, gefolgt von Filtration durch 0,2-μ-Flaschenaufsatzfilter.
-
Probenherstellung:
-
Formulierungen von Protein A und verschiedenen Losen Alhydrogel (Los 4230 und Los 4074) wurden in 1/3 PBS + 0,9% NaCl bei zwei unterschiedlichen pH-Werten hergestellt und mit Sulfit nach folgendem Schema aufgestockt:
| Probe | Aufstockung |
| Nr. | Bezeichnung | PH | Alauncharge | Cu(II) [ng/ml] | Sulfit [mM] |
| 1 | 17112011_PROTEIN A_4074_ref_4°_pH7.3 | 7,3 | 4074 | | |
| 2 | 17112011_PROTEIN A_4074_ref_37°_pH7.3 | 7,3 | 4074 | | |
| 3 | 17112011_PROTEIN A_4074_Sulfit_37°_pH7.3 | 7,3 | 4074 | | 1 |
| 4 | 17112011_PROTEIN A_4230_ref_4°_pH7.3 | 7,3 | 4230 | | |
| 5 | 17112011_PROTEIN A_4230_ref_37°_pH7.3 | 7,3 | 4230 | | |
| 6 | 17112011_PROTEIN A_4230_Sulfit_37°_pH7.3 | 7,3 | 4230 | | 1 |
| 7 | 17112011_PROTEIN A_4074_ref_4°_pH8 | 8 | 4074 | | |
| 8 | 17112011_PROTEIN A_4074_ref_37°_pH8 | 8 | 4074 | | |
| 9 | 17112011_PROTEIN A_4074_Sulfit_37°_pH8 | 8 | 4074 | | 1 |
| 10 | 17112011_PROTEIN A_4230_ref_4°_pH8 | 8 | 4230 | | |
| 11 | 17112011_PROTEIN A_4230_ref_37°_pH8 | 8 | 4230 | | |
| 12 | 17112011_PROTEIN A_4230_Sulfit_37°_pH8 | 8 | 4230 | | 1 |
-
Beispiele 1, 6, 11 und 16 wurden bei 4°C (Referenzproben) gelagert. Alle anderen Proben wurden bei 37°C 96 Stunden lang inkubiert.
-
Nach 96 Stunden wurden alle Proben einer Desorptionsvorschrift unterworfen, um das Antigen von Alhydrogel zu trennen. Das desorbierte Antigen wurde durch RPC analysiert.
-
Ergebnisse:
-
Die Ergebnisse zeigten einen starken Abbau des Antigens Protein A im Vorhandensein von Sulfit. Der Abbau war in den Proben, die mit Alhydrogel mit einem höheren Gehalt an metallischen Verunreinigungen formuliert wurden, stärker ausgeprägt.
Tabelle 3: Vergleich von Leachables aus Stopfenextrakt und JEV09L37 SN. In dieser Tabelle sind alle Peaks mit einer Fläche von > 0,1 mAE.min enthalten.
| Retentionszeit | Stopfenextraktkonzentrat | Stopfenextraktkonzentrat 1:16 verdünnt in Formulierung | FVL L37 SN | Relative Konzentration verglichen mit FVL |
| (min) | Peakfläche (mAE.min) | (%) |
| 12,40 | 0,79 | 0,05 | 0,10 | 47 |
| 13,14 | 1,91 | 0,12 | n. d. | zusätzlicher Peak verglichen mit FVL |
| 13,48 | 0,81 | 0,05 | n. d. | zusätzlicher Peak verglichen mit FVL |
| 13,74 | 0,43 | 0,03 | n. d. | zusätzlicher Peak verglichen mit FVL |
| 14,10 | 0,75 | 0,05 | n. d. | zusätzlicher Peak verglichen mit FVL |
| 14,41 | 0,49 | 0,03 | 0,25 | 12 |
| 16,12 | 29,64 | 1,85 | 0,45 | 414 |
| 16.71 | 2,75 | 0,17 | n. d. | zusätzlicher Peak verglichen mit FVL |
| 17,15 | 14,68 | 0,92 | 0,11 | 815 |
| 18,68 | 0,90 | 0,06 | 0,57 | 10 |
| 20,08 | 0,70 | 0,04 | 0,25 | 18 |
| 20,75 | 0,42 | 0,03 | n. d. | zusätzlicher Peak verglichen mit FVL |
| 21,27 | 0,54 | 0,03 | n. d. | zusätzlicher Peak verglichen mit FVL |
| 22,13 | 2,84 | 0,18 | 0,15 | 122 |
| 22,74 | 0,55 | 0,03 | 0,17 | 20 |
| 23,74 | 1,74 | 0,11 | 0,27 | 41 |
| 24,88 | 0,98 | 0,06 | 0,12 | 51 |
| 27,63 | 0,84 | 0,05 | 0,16 | 32 |
| 29,46 | 0,72 | 0,04 | n. d. | zusätzlicher Peak verglichen mit FVL |
| 31,40 | 0,39 | 0,02 | n. d. | zusätzlicher Peak verglichen mit FVL |
| 35,23 | 4,70 | 0,29 | 0,41 | 72 |
| SUMME | 67,59 | 4,2 | 3,0 | 140 |




Tabelle 7: Regressionsanalyse für „Verhältnis 8 Wochen” einschließlich pH, Alaun und Formaldehyd.
Tabelle 8: Schätzung der Ergebnisse „Verhältnis 8 Wochen”, erzeugt mit dem angepassten Modell. Schätzergebnisse für Verhältnis 8 Wochen
| Reihe | Beobachteter Wert | Angepasster Wert | Untere 95,0% KG für das Mittel | obere 95,0% KG für das Mittel |
| 1 | 0,58 | 0,5975 | 0,536766 | 0,658234 |
| 2 | 0,81 | 0,783125 | 0,722391 | 0,843859 |
| 3 | 0,9 | 0,84625 | 0,785516 | 0,906984 |
| 4 | 0,92 | 0,89625 | 0,835516 | 0,956984 |
| 6 | 0,99 | 0,84625 | 0,785516 | 0,906984 |
| 7 | 0,67 | 0,5975 | 0,536766 | 0,658234 |
| 8 | 0,99 | 0,89625 | 0,835516 | 0,956984 |
| 9 | 0,84 | 0,710625 | 0,649891 | 0,771359 |
| 10 | 0,78 | 0,710625 | 0,649891 | 0,771359 |
| 11 | 0,98 | 0,959375 | 0,898641 | 1,02011 |
| 12 | 0,59 | 0,5975 | 0,536766 | 0,658234 |
| 13 | 1,03 | 0,959375 | 0‚898641 | 1,02011 |
| 14 | 0,78 | 0,660625 | 0,599891 | 0,721359 |
| 15 | 0,94 | 0,89625 | 0,835516 | 0,956984 |
| 16 | 0,31 | 0,77375 | 0,713016 | 0,834484 |
| 17 | 0,82 | 0,783125 | 0,722391 | 0,843859 |
| 18 | 0,88 | 0,84625 | 0,785516 | 0,906984 |
| 19 | 0,82 | 0,77375 | 0,713016 | 0,834484 |
| 20 | 0,82 | 0,959375 | 0,398641 | 1,02011 |
| 21 | 0,82 | 09959375 | 0,898641 | 1,02011 |
| 22 | 0,63 | 0,710625 | 0,649891 | 0,771359 |
| 23 | 0,74 | 0,84625 | 0,785516 | 0,906984 |
| 24 | 0,66 | 0,660625 | 0,599891 | 0,721359 |
| 25 | 0,67 | 0,783125 | 0,722391 | 0,843859 |
| 26 | 0,76 | 0,783125 | 0,722391 | 0,843859 |
| 27 | 0,64 | 0,710625 | 0,649891 | 0,771359 |
| 28 | 0,74 | 0,77375 | 0,713016 | 0,834484 |
| 29 | 0,73 | 0,77375 | 0,713016 | 0,834484 |
| 30 | 0,44 | 0,5975 | 0,536766 | 0,658234 |
| 32 | 0,87 | 0,89625 | 0,835516 | 0,956984 |
| 33 | 0,67 | 0,660625 | 0,599891 | 0,721359 |
| 34 | 0,59 | 0,660625 | 0,599891 | 0,721359 |

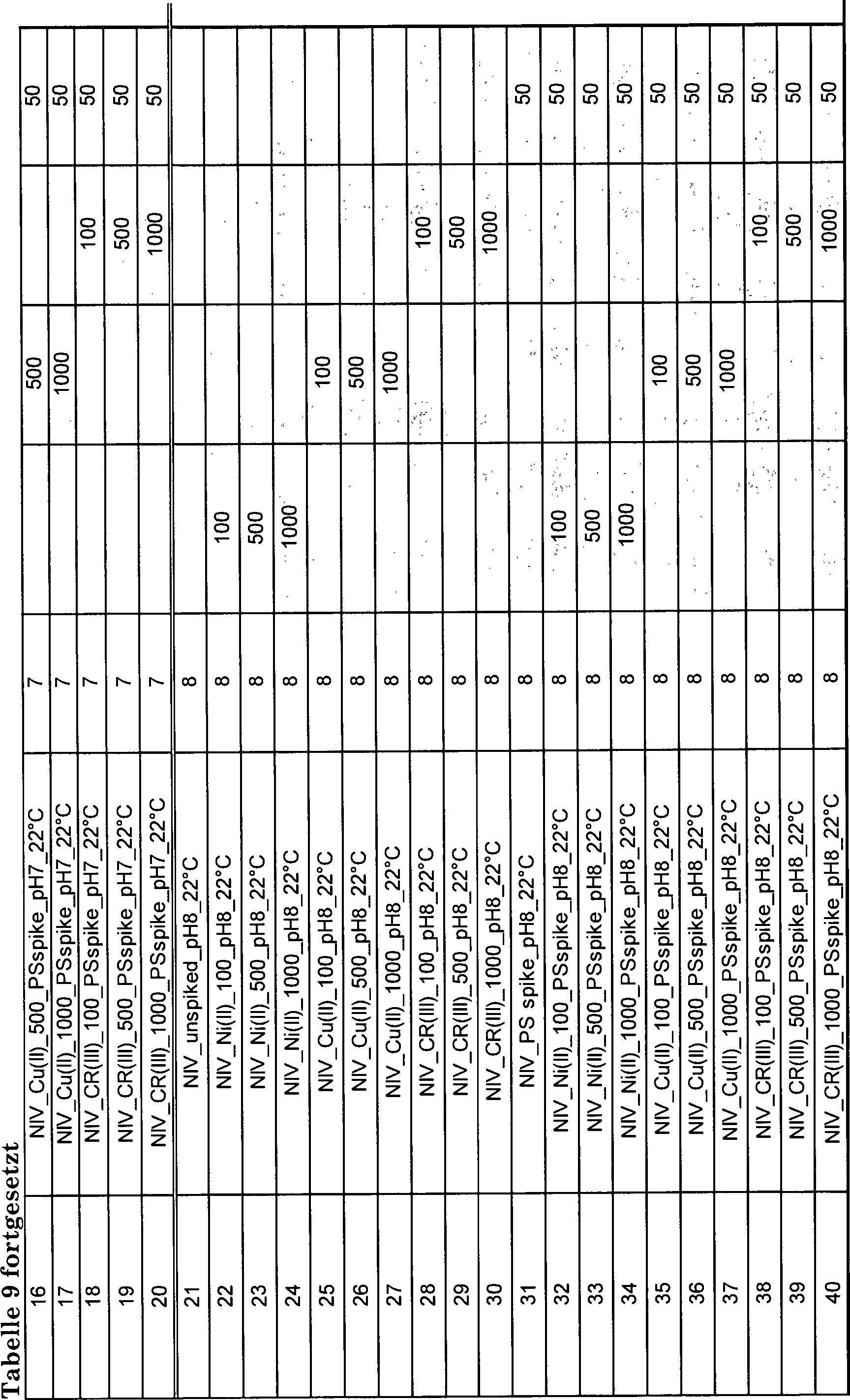
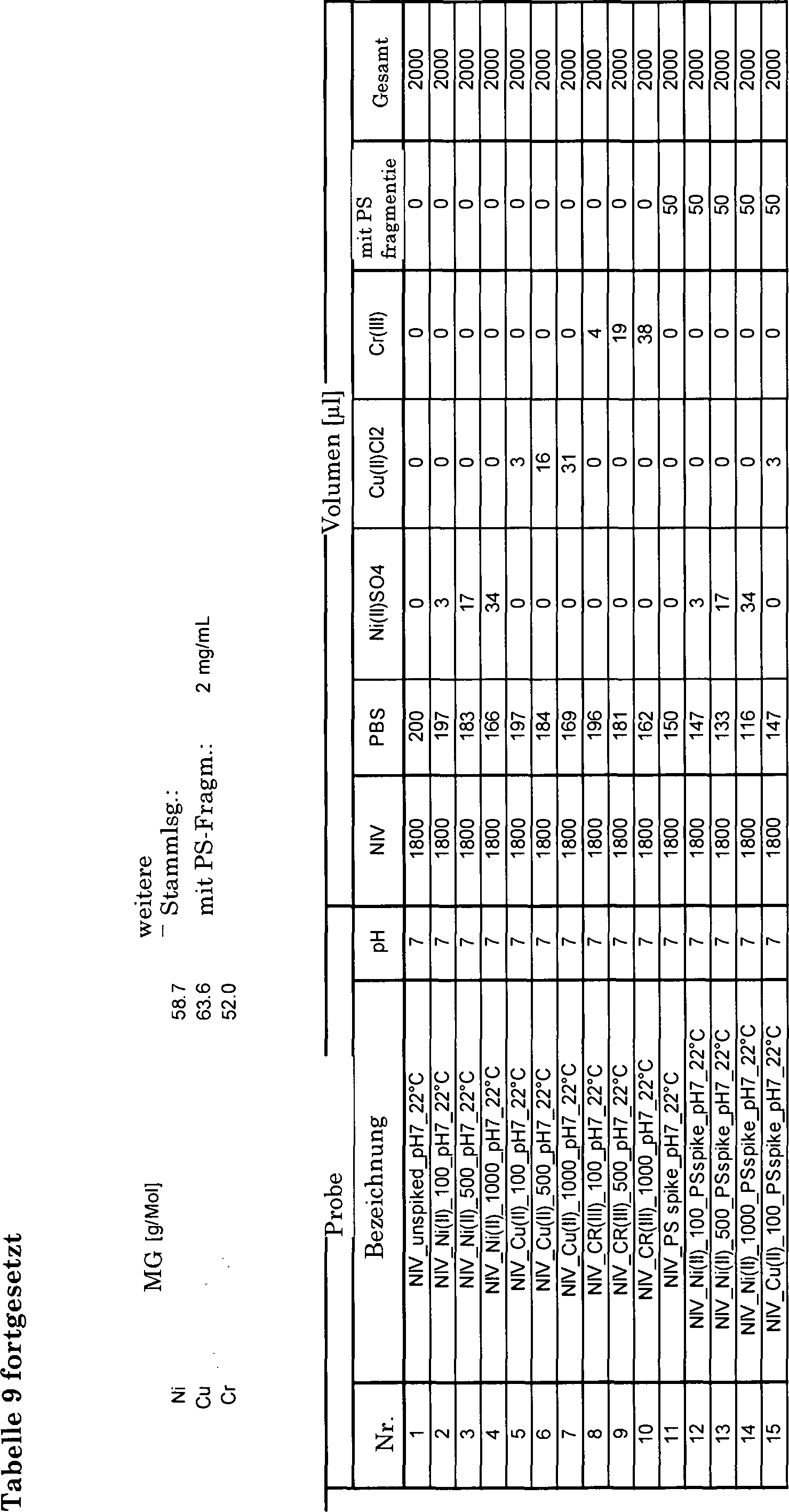






Tabelle 12: Antigenrückgewinnung von Vers. 20110913(NIV), durch SEC-HPLC bestimmt. Für pH 7 und pH 8 basieren die Rückgewinnungen auf NIV-Kontrollproben Nr. 1 bzw. Nr. 21 ohne Zusatz. Die Proben wurden bei 22°C 3 Wochen lang gelagert. Mit „n. a” gekennzeichnete Proben wurden aufgrund von Probenpriorisierung nicht analysiert.
Tabelle 13: ELISA-Ergebnisse von Vers. 20110913(NIV), erlangt nach 3 Wochen bei pH 8 (Probe 21–40) und 7 Wochen bei pH 7 (Probe 1–20) bei 22°C. Mit „n. a” gekennzeichnete Proben wurden aufgrund von Probenpriorisierung nicht analysiert.
Tabelle 14: Antigenrückgewinnung nach 5 Wochen bei 22°C von desorbiertem JEV, erlangt durch SEC-HPLC. Rückgewinnungen basierten auf DP-Kontrollproben ohne Zusatz, gelagert entweder bei pH 7 oder pH 8
Tabelle 15: ELISA-Ergebnisse von desorbiertem JEV-Antigen nach 5 Wochen bei 22°C.
Tabelle 16: ELISA-Ergebnisse von desorbiertem JEV-Antigen nach 4 Wochen und 7 Wochen Lagerung bei 22°C. Mit „n. a” gekennzeichnete Proben wurden aufgrund von Probenpriorisierung nicht analysiert.
Tabelle 17: ANOVA für Stabilitätsproben, die bei 22°C 7 Wochen lang gelagert wurden. Varianzanalyse für Verhältnis – Quadratsumme Typ III
Alle F-Verhältnisse basieren auf der mittleren quadratischen Tabelle 18: Mehrfachreihentest für Verhältnis nach Metallionenart. 1 = Fe(II); 2 = Fe(III); 3 = Ni(II); 4 = Co(II); 5 = Cu(II); 6 = Zn(II); 7 = Cr(III); 8 = Gemisch[1–6]; 9 = Kontrolle ohne Zusatz Mehrfachreihentest für Verhältnis nach Metallionenart
* bezeichnet einen statistisch signifikanten Unterschied Tabelle 19: ICP-MS-Ergebnisse von Restmetallionen-Verunreinigungen in verschiedenen Alaun(2%)-Losen
| | Restmetallgehalt (ng/ml) |
| Alaun-(2%)-Los | Cr | Fe | Ni | Cu | V | Co |
| 4074 | 19,8 | 266 | 14,8 | < 25 | < 5 | < 5 |
| 4470 | 1637 | 1179 | 17,5 | < 25 | < 5 | < 5 |
| 4563 | 1874 | 2485 | 8,9 | < 25 | < 5 | < 5 |
| 4621 | 1333 | 1183 | 7,6 | < 25 | < 5 | < 5 |
| 3877 | 48,2 | 183 | 12,2 | < 25 | < 5 | < 5 |
| 4230 (nonGI**, GI***) | 1139 | 5640 | 816 | 64 | 12,6 | 7 |
| Gemisch* 4074/4230 | 579,4 | 2953 | 4154 | < 44,5 | < 6 | < 6 |
*Auf Alaun-Losen 4230 und 4074 berechneter Restmetallgehalt
**nonGI: nicht gammabestrahlt
***GI: gammabestrahlt
Tabelle 21: Übersicht der Alhydrogel
®-Lose, die bei der JEV-Produktion eingesetzt wurden.
| Los Nr. | Brenntag-ID | Ammoniak-Alaun-Quelle | bei wie vielen JEV-Chargen eingesetzt |
| 1 | 4074 | BK Giulini | 17 |
| 2 | 4230 | Holland (USA) | 24 |
| 3 | 4414 | Canton India | 7 |
| 4 | 4470 | Canton India | 19 |
| 5 | 4539 | Holland (USA) | 4 |
Tabelle 22: Ergebnisse der PSD-Analyse von Alhydrogel
® (2%) Stammlösung in Wasser.
| Nr. | Probenbezeichnung | d (0,1) | d (0,5) | d (0,9) | Undurchlässigkeit (%) |
| 1 | Nicht bestrahltes AlOH RQCS0890 Lot 4230 | 0,70 | 2,13 | 46,53 | 1,51 |
| 2 | GI AlOH RQCS1200 Lot 4230 | 0,71 | 4,14 | 9,64 | 1,9 |
| 3 | GI AlOH RQCS1342 Lot 4740 | 0,78 | 2,23 | 5 ,44 | 2, |
| 4 | GI AlOH RQCS0448 Lot 4074 | 0,73 | 4,49 | 78,58 | 1,96 |
d (0,1): 10% von allen gemessenen Partikeln weisen einen Durchmesser unterhalb dieses Werts auf
d (0,5): 50% von allen gemessenen Partikeln weisen einen Durchmesser unterhalb dieses Werts auf
d (0,9): 90% von allen gemessenen Partikeln weisen einen Durchmesser unterhalb dieses Werts auf
Undurchlässigkeit: Menge des durch die Probe reduzierten Laserlichts; entspricht der Probenkonzentration in der Messzelle Tabelle 23: Ergebnisse der Alhydrogel
®-Titrationskurven zur Bestimmung von POZ. Die Proben wurden in PBS (Verdünnung 1:20) analysiert.
| Proben-ID | PZC (pH) |
| Nicht bestrahltes AlOH RQCS0890
Los 4230 | 4,58 |
| GI AlOH RQCS1200
Los 4230 | 4,62 |
| GI AlOH RQCS1342
Los 4740 | 4,49 |
| GI AlOH RQCSO448
Los 4074 | 4,48 |
Tabelle 25: Analyse des Überstands und der Aluminiumhydroxid-(Los 4230)-Gelfraktion auf verunreinigende Metallionen zeigt, dass sich die Metallionen im Gel befinden und nicht im Überstand
Tabelle 26: Übersicht über die Rohmaterialien, die zur Alhydrogel
®-Produktion eingesetzt wurden
| Ammoniak-Alaun-Los | Herkunft | Resultierende Alhydrogel-Charge | Anzahl an JEV – Produktionschargen |
| 6-80578-28 | BK Giulini | 4074 | 17 (z. B. JEV08A02) |
| AA0427 | | 4230 | 24 (z. B. JEV09L37) |
| 91480 | Canton India | 4470 | 19 (z. B. JEV10F54) |
| 10094 | unbekannt | 4563 | Nicht eingesetzt |
| 10095 |
Tabelle 28: Vergleich von Ammoniak-Alaun und entsprechenden Alhydrogel
®-Proben
| | Fe | Cr | Ni | Cu |
| rel.% |
| Ammoniak-Alaun-Los | 6-80578 | 88 | 7 | 2 | 3 |
| Alaun-Los | 4074 | 82 | 6 | 5 | 8 |
| Ammoniak-Alaun-Los | AA0427 | 72 | 18 | 10 | 1 |
| Alaun-Los | 4230 | 74 | 15 | 11 | 1 |
| Ammoniak-Alaun-Los | 91480 | 41 | 59 | 0 | 0 |
| Alaun-Los | 4470 | 41 | 57 | 1 | 1 |
Tabelle 29: Zusammenfassung eines bei ESG durchgeführten 70 Elemente umfassenden Scans. Es werden nur diejenigen Elemente gezeigt, bei denen Unterschiede zwischen verschiedenen Losen beobachtet wurden.
Tabelle 29 fortgesetzt
Tabelle 30: Aluminium-basierte Impfstoff zur Anwendung am Menschen
Tabelle 31: Aluminium-basierte Impfstoffe als Tierarzneimittel
-
Die folgenden Begriffe, die in dieser Beschreibung verwendet werden, sind anerkannte eingetragene Marken:
„Triton”, „Falcon”, „UltiMate”, „Infors”, „Multitron”, „LoBind”, „Kleenpak”, „Eppendorf”, „Superose”, „Millipore”, „Steriflip”, „BioTek” und „Tween”.
-
Bevorzugte Aspekte
-
- Aspekt 1. Wässrige Zusammensetzung, die ein Protein und ein Aluminium-Salz beinhaltet, wobei die Zusammensetzung weniger als 350 ppb Schwermetall, bezogen auf das Gewicht der wässrigen Zusammensetzung beinhaltet.
- Aspekt 2. Wässrige Zusammensetzung gemäß Aspekt 1, die bei Raumtemperatur von mehr als 20°C für mindestens 1 Monat gelagert wurde.
- Aspekt 3. Wässrige Zusammensetzung gemäß Aspekt 1, die eine Haltbarkeitsdauervon mindestens 20 Monaten aufweist.
- Aspekt 4. Wässrige Zusammensetzung gemäß Aspekt 1–3, wobei das Schwermetall aus Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V, Cr, Pb, Rb und Mo ausgewaehlt wird.
- Aspekt 5. Wässrige Zusammensetzung gemäß Aspekt 1–4, wobei das Schwermetall aus Cu, Ni, W, Co, Os, Ru, Cd, Ag, Fe, V ausgewählt wird.
- Aspekt 6. Wässrige Zusammensetzung gemäß Aspekt 1–5, wobei das Schwermetall aus Cu oder Ni ausgewählt wird.
- Aspekt 7. Wässrige Zusammensetzung gemäß Aspekt 1–6, wobei das Schwermetall in ionischer Form vorhanden ist.
- Aspekt 8. Wässrige Zusammensetzung gemäß Aspekt 1–7, wobei das Aluminium-Salz Aluminiumhydroxid (Al(OH)3) oder Aluminiumphosphat (Al(PO4) ist.
- Aspekt 9. Wässrige Zusammensetzung gemäß Aspekt 1–8, das ferner eine reaktive Verbindung umfasst.
- Aspekt 10. Wässrige Zusammensetzung gemäß Aspekt 9, wobei die reaktive Verbindung aus der Gruppe bestehend aus einer redoxaktiven Verbindung, einer radikalbildenden Verbindung, einer stabilisierenden Verbindung und einer Kombination von beliebigen davon ausgewählt ist.
- Aspekt 11. Wässrige Zusammensetzung gemäß Aspekt 9–10, wobei die reaktive Verbindung aus der Gruppe bestehend aus Formaldehyd, Ethanol, Chloroform, Trichlorethylen, Aceton, Triton-X-100, Desoxycholat, Diethylpyrocarbonat, Sulfit, Na2S2O5, beta-Propriolacton, Polysorbat, wie beispielsweise Tween 20®, Tween 80®, O2, Phenol, Copolymeren vom Pluronic-Typ und einer Kombination von beliebigen davon ausgewählt ist.
- Aspekt 12. Wässrige Zusammensetzung gemäß Aspekt 1–11, die zwischen 5 μg/ml und 50 mg/ml Aluminium umfasst.
- Aspekt 13. Wässrige Zusammensetzung gemäß Aspekt 1–12, die zwischen 50 μg/ml und 5 mg/ml Aluminium umfasst.
- Aspekt 14. Wässrige Zusammensetzung gemäß Aspekt 1–13, die zwischen 5 ppb und 250 ppb Fe, bezogen auf das Gewicht der wässrigen Zusammensetzung, umfasst.
- Aspekt 15. Wässrige Zusammensetzung gemäß Aspekt 1–14, die weniger als 3 ppb Cu, bezogen auf das Gewicht der wässrigen Zusammensetzung, umfasst.
- Aspekt 16. Wässrige Zusammensetzung gemäß Aspekt 1–15, die weniger als 40 ppb Ni, bezogen auf das Gewicht der wässrigen Zusammensetzung, umfasst.
- Aspekt 17. Wässrige Zusammensetzung gemäß Aspekt 1–16, wobei das Protein ein Therapeutikum und/oder ein Impfstoff ist.
- Aspekt 18. Wässrige Zusammensetzung gemäß Aspekt 1–17, wobei das Protein ein virales oder bakterielles Protein ist.
- Aspekt 19. Wässrige Zusammensetzung gemäß Aspekt 1–18, wobei das virale Protein ein Protein des japanischen Enzephalitis-Virus oder ein Protein des Bakteriums Pseudomonas aeruginosa ist.
- Aspekt 20. Wässrige Zusammensetzung gemäß Aspekt 1–19, wobei das Protein ein Protein in einem Formaldehyd-inaktivierten Viruspartikel ist.
- Aspekt 21. Wässrige Zusammensetzung gemäß Aspekt 1–20, die ferner Sulfit umfasst.
- Aspekt 22. Impfstoff, der eine wässrige Zusammensetzung gemäß Aspekt 1–21 umfasst.
- Aspekt 23. Aluminiumhydroxid-Konzentrat, das a) 10 mg/ml des Aluminiumhydroxids und b) weniger als 7 Mikrogramm Schwermetall umfasst, zur Verwendung bei der Herstellung eines Medikaments, vorzugsweise zur Verwendung bei der Herstellung eines Impfstoffs.
- Aspekt 24. Aluminiumhydroxid-Konzentrat, das 10 mg/ml Aluminiumhydroxid und weniger als 7 Mikrogramm Schwermetall umfasst, zur Verwendung bei der Herstellung eines Medikaments, vorzugsweise zur Verwendung bei der Herstellung eines Impfstoffs.
- Aspekt 25. Aluminiumsalz-Konzentrat, das weniger als 7 ppm Schwermetall, bezogen auf das Gewicht des Konzentrats umfasst, zur Verwendung bei der Herstellung eines Medikaments, bevorzugt zur Verwendung bei der Herstellung eines Impfstoffs.
- Aspekt 26. Aluminiumsalz-Konzentrat, das weniger als 700 ppm Schwermetall, bezogen auf das Gewicht des Aluminiumsalzes, umfasst, zur Verwendung bei der Herstellung eines Medikaments, vorzugsweise zur Verwendung bei der Herstellung eines Impfstoffs.
- Aspekt 27. Aluminiumhydroxid-Konzentrat, das weniger als 700 ppm Schwermetall, bezogen auf das Gewicht des Aluminiumhydroxids, umfasst, zur Verwendung bei der Herstellung eines Medikaments, bevorzugt zur Verwendung bei der Herstellung eines Impfstoffs.
- Aspekt 28. Aluminiumhydroxid-Konzentrat, das 10 weniger als 1,4 Mikrogramm Schwermetall umfasst, zur Verwendung bei der Herstellung eines Medikaments, bevorzugt zur Verwendung bei der Herstellung eines Impfstoffs.
- Aspekt 29. Aluminiumsalz-Konzentrat, das weniger als 1,4 ppm Schwermetall, bezogen auf das Gewicht des Konzentrats, umfasst, zur Verwendung bei der Herstellung eines Medikaments, bevorzugt zur Verwendung bei der Herstellung eines Impfstoffs.
- Aspekt 30. Aluminiumsalz-Konzentrat, das weniger als 140 ppm Schwermetall, bezogen auf das Gewicht des Aluminiumsalzes, umfasst, zur Verwendung bei der Herstellung eines Medikaments, bevorzugt zur Verwendung bei der Herstellung eines Impfstoffs.
- Aspekt 31. Aluminiumhydroxid-Konzentrat, das weniger als 140 ppm Schwermetall, bezogen auf das Gewicht des Aluminiumhydroxids, umfasst, zur Verwendung bei der Herstellung eines Medikaments, bevorzugt zur Verwendung bei der Herstellung eines Impfstoffs.
-
ZITATE ENTHALTEN IN DER BESCHREIBUNG
-
Diese Liste der vom Anmelder aufgeführten Dokumente wurde automatisiert erzeugt und ist ausschließlich zur besseren Information des Lesers aufgenommen. Die Liste ist nicht Bestandteil der deutschen Patent- bzw. Gebrauchsmusteranmeldung. Das DPMA übernimmt keinerlei Haftung für etwaige Fehler oder Auslassungen.
-
Zitierte Patentliteratur
-
- EP 497524 [0084]
- EP 497525 [0084]
- WO 98/42375 [0087]
- WO 02/64162 [0087]
- US 4761372 [0087]
- US 5308835 [0087]
- US 4882317 [0091]
- US 4695624 [0091]
- WO 00/10599 [0091]
- US 4057685 [0091]
- US 4673574 [0091]
- US 4761283 [0091]
- US 4808700 [0091]
- US 4459286 [0091]
- US 4965338 [0091]
- US 4663160 [0091]
- CN 101734698 [0095]
- WO 98/14401 [0095]
-
Zitierte Nicht-Patentliteratur
-
- Lindblad, EB (2004) Immunol. and Cell Biol. B. 82: 497–505 [0011]
- Burnouf T, (1991) Vox Sang. B. 60: S. 8–15 [0012]
- Li et al., 1995; Mayo et al., 2003; Stadtman, 1990 [0025]
- Ranguelova et al., 2010 [0033]
- Alipazaga et al. 2004 [0033]
- Berglund et al. 1993 [0033]
- Brandt und Elding 1998 [0033]
- Lima et al. 2002 [0033]
- Shi 1994 [0033]
- Huie et al., 1985 [0034]
- Neta & Huie, 1985 [0034]
- Lambeth et al., 1973 [0038]
- Ito & Kawanashi, 1991 [0038]
- Lindblad, EB (2004) Immunol. and Cell Biol. B. 82: 497–505 [0050]
- Exley, C (2010). Trends in Immunol. B. 31: S. 103–109 [0059]
- Jones et al., An. Acad. Bras. Cienc 2005, 77(2):293–324 [0085]
- Robbins et al., JAMA, 1996, 276 (14): 1181–5 [0090]
- Gever et al., Med. Microbiol. Immunol, 1979, 165:171–288 [0091]
- Siebertz, Karl; van Bebber, David, Hochkirchen, Thomas: Statistische Versuchsplanung: Design of Experiments (DoE). Verlag: Springer Berlin Heidelberg; 1. Auflage (2010), ISBN-10: 3642054927 [0123]
- Lin C.-W. und Wu W.-C. J. Virol. 2003; 77(4):2600–6 [0135]
- Lin und Wu (2003) [0136]
- Lin C.-W. und Wu W.-C. J. Virol. 2003; 77(4):2600–6 [0174]
- Lin und Wu (2003) [0174]























 Tabelle 3: Vergleich von Leachables aus Stopfenextrakt und JEV09L37 SN. In dieser Tabelle sind alle Peaks mit einer Fläche von > 0,1 mAE.min enthalten.
Tabelle 3: Vergleich von Leachables aus Stopfenextrakt und JEV09L37 SN. In dieser Tabelle sind alle Peaks mit einer Fläche von > 0,1 mAE.min enthalten.

 Tabelle 13: ELISA-Ergebnisse von Vers. 20110913(NIV), erlangt nach 3 Wochen bei pH 8 (Probe 21–40) und 7 Wochen bei pH 7 (Probe 1–20) bei 22°C. Mit „n. a” gekennzeichnete Proben wurden aufgrund von Probenpriorisierung nicht analysiert.
Tabelle 13: ELISA-Ergebnisse von Vers. 20110913(NIV), erlangt nach 3 Wochen bei pH 8 (Probe 21–40) und 7 Wochen bei pH 7 (Probe 1–20) bei 22°C. Mit „n. a” gekennzeichnete Proben wurden aufgrund von Probenpriorisierung nicht analysiert.

 Tabelle 14: Antigenrückgewinnung nach 5 Wochen bei 22°C von desorbiertem JEV, erlangt durch SEC-HPLC. Rückgewinnungen basierten auf DP-Kontrollproben ohne Zusatz, gelagert entweder bei pH 7 oder pH 8
Tabelle 14: Antigenrückgewinnung nach 5 Wochen bei 22°C von desorbiertem JEV, erlangt durch SEC-HPLC. Rückgewinnungen basierten auf DP-Kontrollproben ohne Zusatz, gelagert entweder bei pH 7 oder pH 8
 Tabelle 15: ELISA-Ergebnisse von desorbiertem JEV-Antigen nach 5 Wochen bei 22°C.
Tabelle 15: ELISA-Ergebnisse von desorbiertem JEV-Antigen nach 5 Wochen bei 22°C. Tabelle 16: ELISA-Ergebnisse von desorbiertem JEV-Antigen nach 4 Wochen und 7 Wochen Lagerung bei 22°C. Mit „n. a” gekennzeichnete Proben wurden aufgrund von Probenpriorisierung nicht analysiert.
Tabelle 16: ELISA-Ergebnisse von desorbiertem JEV-Antigen nach 4 Wochen und 7 Wochen Lagerung bei 22°C. Mit „n. a” gekennzeichnete Proben wurden aufgrund von Probenpriorisierung nicht analysiert. Tabelle 17: ANOVA für Stabilitätsproben, die bei 22°C 7 Wochen lang gelagert wurden. Varianzanalyse für Verhältnis – Quadratsumme Typ III
Tabelle 17: ANOVA für Stabilitätsproben, die bei 22°C 7 Wochen lang gelagert wurden. Varianzanalyse für Verhältnis – Quadratsumme Typ III Alle F-Verhältnisse basieren auf der mittleren quadratischen Tabelle 18: Mehrfachreihentest für Verhältnis nach Metallionenart. 1 = Fe(II); 2 = Fe(III); 3 = Ni(II); 4 = Co(II); 5 = Cu(II); 6 = Zn(II); 7 = Cr(III); 8 = Gemisch[1–6]; 9 = Kontrolle ohne Zusatz Mehrfachreihentest für Verhältnis nach Metallionenart
Alle F-Verhältnisse basieren auf der mittleren quadratischen Tabelle 18: Mehrfachreihentest für Verhältnis nach Metallionenart. 1 = Fe(II); 2 = Fe(III); 3 = Ni(II); 4 = Co(II); 5 = Cu(II); 6 = Zn(II); 7 = Cr(III); 8 = Gemisch[1–6]; 9 = Kontrolle ohne Zusatz Mehrfachreihentest für Verhältnis nach Metallionenart * bezeichnet einen statistisch signifikanten Unterschied Tabelle 19: ICP-MS-Ergebnisse von Restmetallionen-Verunreinigungen in verschiedenen Alaun(2%)-Losen
* bezeichnet einen statistisch signifikanten Unterschied Tabelle 19: ICP-MS-Ergebnisse von Restmetallionen-Verunreinigungen in verschiedenen Alaun(2%)-Losen

 Tabelle 26: Übersicht über die Rohmaterialien, die zur Alhydrogel®-Produktion eingesetzt wurden
Tabelle 26: Übersicht über die Rohmaterialien, die zur Alhydrogel®-Produktion eingesetzt wurden

 Tabelle 30: Aluminium-basierte Impfstoff zur Anwendung am Menschen
Tabelle 30: Aluminium-basierte Impfstoff zur Anwendung am Menschen

 Tabelle 31: Aluminium-basierte Impfstoffe als Tierarzneimittel
Tabelle 31: Aluminium-basierte Impfstoffe als Tierarzneimittel













