ES2928961T3 - Compuestos novedosos para controlar artrópodos - Google Patents
Compuestos novedosos para controlar artrópodos Download PDFInfo
- Publication number
- ES2928961T3 ES2928961T3 ES19733494T ES19733494T ES2928961T3 ES 2928961 T3 ES2928961 T3 ES 2928961T3 ES 19733494 T ES19733494 T ES 19733494T ES 19733494 T ES19733494 T ES 19733494T ES 2928961 T3 ES2928961 T3 ES 2928961T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- halogen
- chloro
- substituted
- butyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/14—Ectoparasiticides, e.g. scabicides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/96—Esters of carbonic or haloformic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Tropical Medicine & Parasitology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
Abstract
La presente invención se refiere a nuevos compuestos sustituidos con halógeno, a procesos para su preparación ya su uso para controlar plagas animales, en particular artrópodos y especialmente insectos y arácnidos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Compuestos novedosos para controlar artrópodos
Introducción y antecedentes de la invención
La presente invención se refiere a compuestos sustituidos con halógeno novedosos, a procesos para su preparación y a su uso para controlar plagas en animales, en particular artrópodos y especialmente insectos y arácnidos.
Estado de la técnica
Ectoparasiticidas
Carboxamidas halogenados con actividad insecticida y ectoparasiticidas se describen en EP 1911751, WO 2012/069366, WO 2012/080376, WO 2012/107434, WO 2012/175474, WO2014/122083 y WO2015/067647. Combinaciones de carboxamidas halogenadas para el tratamiento de plagas de animales se describen en WO 2016/174052.
Además de las clases antes mencionadas también se conocen en la técnica otros compuestos con actividad ectoparasiticida. Lo común en estas moléculas de una cantidad de modos moleculares de acción diferentes es que ninguna ejerce un efecto artropodicida amplio por un período de más de tres meses. El uso comercial de actividad artropodicida prolongada solo se puede encontrar en enfoques de liberación constante como se usa en collares (por ejemplo deltametrina en Scalibor® o flumetrina/imidacloprid en Seresto®). Una administración parenteral única conveniente con amplio control de artrópodos no se comercializa ni se demuestra que funciona en la práctica hasta el momento. Las razones para fallar en esa área se pueden encontrar en corta semivida, baja solubilidad, baja biodisponibilidad, estabilidad insuficiente, espectro de parásitos artrópodos incompleto, efectos secundarios sistémicos y locales. Una multitud de estos parámetros deben ser mejorados para llevar a un enfoque exitoso para cumplir con la necesidad de un tratamiento sistémico de ectoparásitos con un período amplio de eficacia.
Profármacos
En los casos en que la eficacia de un fármaco está limitada por sus propiedades fisicoquímicas, se puede usar un concepto de profármaco. Los profármacos se definen como derivados biorreversibles de los fármacos originales correspondientes. Esto significa que el profármaco transporta un grupo escindible, un denominado prorresto. Este grupo facilita la administración, absorción en el cuerpo y distribución en el animal o humano tratado. El prorresto se escinde mediante transformaciones biológicas o químicas en el cuerpo de paciente liberando el fármaco original una vez que se absorbió el profármaco. Se puede encontrar una descripción general de los conceptos de profármaco por ejemplo en un artículo de revisión de J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Jarvinen, J. Savolainen, Nature Reviews Drug Discovery 2008, 255-270. En muchos casos, la administración de un fármaco se limita por su poca solubilidad acuosa que puede impactar en la administración oral, aún más en la parenteral. Se han descrito los profármacos para aplicaciones orales e intravenosas de fármacos mientras que poco se sabe acerca de profármacos para uso subcutáneo (véase V.J. Stella, K.W. Nti-Addae, Advanced Drug Delivery Reviews 2007, 59, 677-694). Muchos profármacos usan un grupo de unión del fármaco original para enlazar el prorresto mediante un grupo enlazador y opcionalmente grupos espaciadores. Los puntos de unión son normalmente grupos funcionales del fármaco original que permiten la modificación química biorreversible, por ejemplo grupos hidroxilo, grupos de ácido carboxílico, grupos amino, amidas u otros. Un ejemplo reciente de enlazadores carbamato unidos con un grupo amida se puede encontrar en C. Liu, J. Lin, G. Everlof, C. Gesenberg, H. Zhang, P.H. Marathe, M. Malley, M.A. Gallella, M. McKinnon, J.H. Dodd, J.C. Barrish, G.L. Schieven, K. Leftheris, Bioorg. Med. Chem. Lett. 2013, 23, 3028-3033.
Además de los carbamatos, también los ésteres carbamato se describen ocasionalmente como enlazadores en la bibliografía de profármacos, por ejemplo, para la modificación de un grupo piridilo (véase E. Binderup, F. Bjorkling, P.V. Hjranaa, S. Latini, B. Balther, M. Carlsen, L. Binderup, Bioorg. Med. Chem. Lett. 2005, 15, 2491-2494).
Entonces era un objetivo de la presente invención proporcionar compuestos novedosos con alta actividad sistémica insecticida y ectoparasiticida y solubilidad potenciada. Otro objetivo de la presente invención era proporcionar compuestos novedosos con alta actividad sistémica ectoparasiticida, en particular insecticida y/o acaricida, y biodisponibilidad mejorada en comparación con compuestos conocidos. Otro objetivo de la presente invención era proporcionar compuestos novedosos con alta actividad sistémica ectoparasiticida, en particular insecticida y/o acaricida, con estabilidad suficiente en formulaciones para administración farmacéutica. Otro objetivo de la presente invención era proporcionar compuestos novedosos con actividad sistémica ectoparasiticida e insecticida con balance óptimo de estabilidad en formulaciones para administración farmacéutica y propiedades de liberación sistémica para liberar el principio activo de los compuestos novedosos bajo condiciones sistémicas en el cuerpo. En otro objetivo de la presente invención la liberación sistémica del principio activo de los compuestos novedoso debería ocurrir en un punto adecuado de tiempo o en un período de tiempo adecuado para alcanzar actividad sistémica mejorada en el tratamiento. Otro objetivo de la presente invención era proporcionar compuestos novedosos con alta actividad
sistémica insecticida y ectoparasiticida y solubilidad potenciada en formulaciones para administración subcutánea. Los compuestos novedosos no deberían ser tóxicos ni liberar grupos tóxicos tras la administración.
Los inventores de la presente invención han descubierto ahora de forma sorprendente que con los nuevos compuestos de la presente invención las desventajas de la técnica previa se pueden evitar y se pueden conseguir los objetivos descritos anteriormente.
Por lo tanto, los nuevos compuestos además muestran y por lo tanto se pueden emplear particularmente en el sector de salud animal.
Descripción Detallada de la Invención
La presente invención se puede describir en particular con las siguientes modalidades:
[1] En un primer aspecto, la invención se refiere a compuestos de fórmula (I)

(I)
Donde
Q es O o está ausente;
L1 es alcanodiilo C2-C4 lineal, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de halógeno, alquilo C1-C4 y cicloalquilo C3-C6, donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos; o es un resto (CH2)n-X-(CH2)m donde n y m son independientemente 0, 1 o 2 y X es cicloalcanodiilo C3-C7, que está opcionalmente sustituido con alquilo C1-C4; o L1 está ausente; con la condición de que en caso de que L1 esté ausente, Q también esté ausente;
L2 es C=O o está ausente,
L3 es alcanodiilo C1-C4 lineal, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C6 y halógeno, donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos; o es un grupo (CH2)n-(CH=CH)-CH2)m, que está opcionalmente sustituido con hasta 2 grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C5 y halógeno, donde n y m son independientemente 0, 1 o 2; o es cicloalcanodiilo C3-C7, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4 y halógeno;
Y se selecciona de un grupo T1
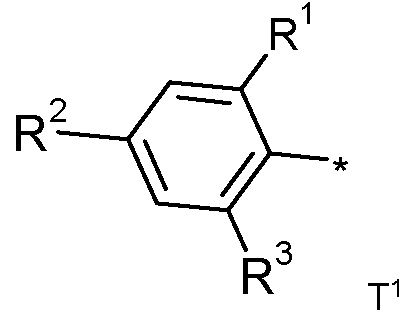
donde
cada uno de R1, R2 y R3 se selecciona independientemente de hidrógeno, halógeno, ciano, nitro, alquilo C1-C6 lineal o ramificado, cicloalquilo C3-C6, alcoxi C1-C6, alquilo C1-C6 sustituido con halógeno lineal o ramificado, alcoxi C1-C6 sustituido con halógeno, cicloalquilo C3-C6 sustituido con halógeno, alquilsulfanilo C1-C6, alquilsulfinilo C1-C6, alquilsulfonilo C1-C6, W-alquilamino C1-C6, W,A/-dialquilamino C1-C6, W-alcoxiC1-C3-alquilamino C1-C4 y 1 -pirrolidinilo
o un grupo T2

donde
cada uno de Z1yZ 2 se selecciona independientemente de hidrógeno, halógeno, ciano, nitro, alquilo C1-C6 lineal o ramificado, cicloalquilo C3-C6, alcoxi C1-C6, alquilcarbonilo C1-C6, alquilo C1-C6 lineal o ramificado sustituido con halógeno, alcoxi C1-C6 sustituido con halógeno, cicloalquilo C3-C6 sustituido con halógeno, alquilsulfanilo C1-C6, alquilsulfinilo C1-C6, alquilsulfonilo C1-C6, W-alquilamino C1-C6, W,W-dialquilamino C1-C6, W-alcoxiC1-C3-alquilamino C1-C4 y 1-pirrolidinilo; y
Z3 representa hidrógeno o alquilo C1-C6 lineal o ramificado, cicloalquilo C3-C6, alquenilo C2-Ca, alquinilo C2-C6, arilo o hetarilo, que pueden estar sustituidos independientemente unos de los otros con 1 a 5 sustituyentes que se seleccionan de hidroxi, halógeno, ciano, nitro, amino, alquilo C1-C3, alcoxi C1-C3, hidroxicarbonilo, alcoxicarbonilo, alquilcarbamoilo, cicloalquilcarbamoilo y fenilo
y las sales de estos.
[2] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en [1] donde
Q es O;
L2 es C=O;
L3 es un grupo (CH2)n-(CH=CH)-CH2)m, que está opcionalmente sustituido con hasta 2 grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C5 y halógeno, donde n y m son independientemente 0, 1 o 2; y
L1 tiene el significado como se define en [1];
y las sales de estos.
[3] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en [1] o [2] donde Q es O;
L2 es C=O;
L3 es un grupo (CH2)n-(CH=CH)-CH2)m, donde n y m son 0; y
L1 tiene el significado como se define en [1];
y las sales de estos.
[4] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en [1] donde
Q es O
L2 es C=O
L3 es alcanodiilo C1-C4 lineal, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-Ca y halógeno, donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos; y
L1 tiene el significado como se define en [1];
y las sales de estos.
[5] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en [1] donde
Q es O;
L2 está ausente;
L3 es alcanodiilo C1-C4 lineal, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C6 y halógeno, donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos; y
L1 tiene el significado como se define en [1];
y las sales de estos.
[6] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en [1] donde
L1, L2 y Q están ausentes; y
L3 tiene el significado como se define en [1];
y las sales de estos.
[7] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [6]
donde
Q es O o está ausente;
L1 es alcanodiilo C2-C4 lineal, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4 y cicloalquilo C3-C6, donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos; o es un resto (CH2)n-X-(CH2)m donde n y m son 0 y X es cicloalcanodiilo C5 o C6, que está opcionalmente sustituido con alquilo C1-C4; o L1 está ausente; con la condición de que en caso de que L1 esté ausente, Q también esté ausente;
L2 es C=O o está ausente,
L3 es alcanodiilo C1-C4 lineal, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C6 y halógeno, donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos; o es un grupo (CH2)n-(CH=CH)-CH2)m, que está opcionalmente sustituido con hasta 2 grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C5 y halógeno, donde n y m son 0; oes cicloalcanodiilo C3-C7 , que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4 y halógeno;
y las sales de estos.
[8] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [7]
donde
Q es O o está ausente;
L1 se selecciona de 1,2-etanodiilo, 1,3-propanodiilo, dimetil-propanodiilo, ciclopentanodiilo, ciclohexanodiilo; o
L1 está ausente; con la condición de que en caso de que L1 esté ausente, Q también esté ausente;
L2 es C=O o está ausente,
L3 tiene el significado como se define en [1];
y las sales de estos.
[9] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [8]
donde
Q es O o está ausente;
L2 es C=O o está ausente;
L3 se selecciona de metileno, 1,2-etanodiilo, 1,3-propanodiilo, dimetil-etanodiilo, eteno-1,2-diilo, y ciclohexanodiilo

o un grupo ,
donde n y m son independientemente 0, 1, 2;
L1 tiene el significado como se define en [1];
y las sales de estos.
[10] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [9]
donde
Q es O;
y las sales de estos.
[11] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [10]
donde
L1 es alcanodiilo C2-C4 lineal, que está opcionalmente sustituido con uno o más grupos alquilo C1-C4, donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos y las sales de estos.
[12] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [11]
donde
L2 es C=O
y las sales de estos.
[13] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [12]
donde
L3 es un grupo (CH2)n-(CH=CH)-CH2)m, que está opcionalmente sustituido con hasta 2 grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C5 y halógeno, donde n y m son independientemente 0, 1 o 2, preferentemente n y m son 0;
y las sales de estos.
[14] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [13]
donde
Y se selecciona de un grupo T1

donde
cada uno de R1, R2y R3 se selecciona independientemente de hidrógeno, halógeno, alquilo C1-C3 lineal o ramificado, cicloalquilo C3-C6, alcoxi C1-C3, alquilo C1-C3 sustituido con halógeno lineal o ramificado, alcoxi C1-C3 sustituido con halógeno, cicloalquilo C3-C6 sustituido con halógeno, y 1-pirrolidinilo
o un grupo T2

donde
Z1 representa alquilo C1-C3 lineal o ramificado, cicloalquilo C3-C6, alcoxi C1-C3,
que pueden estar sustituidos independientemente unos de los otros con 1 a 5 sustituyentes que se seleccionan de A hidroxi, halógeno, ciano, nitro, alquilo C1-C3, y alcoxi C1-C3,
Z2 representa halógeno, ciano, nitro, amino o alquilo C1-C6 lineal o ramificado, alquilcarbonilo C1-C6, alquilsulfanilo C1-C6, alquilsulfinilo C1-C6, alquilsulfonilo C1-C6, que pueden estar sustituidos independientemente unos de los otros con 1 a 5 sustituyentes que se seleccionan de hidroxi, halógeno, ciano, nitro, alquilo C1-C3, y alcoxi C1-C3; y
Z3 representa hidrógeno o alquilo C1-C6 lineal o ramificado, cicloalquilo C3-C6, alquenilo C2-C6, alquinilo C2-C6, arilo o hetarilo, que pueden estar sustituidos independientemente unos de los otros con 1 a 5 sustituyentes que se seleccionan de hidroxi, halógeno, ciano, nitro, alquilo C1-C3, y alcoxi C1-C3;
y las sales de estos.
[15] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [14]
donde
Y se selecciona de un grupo T1

donde
cada uno de R1, R2y R3 se seleccionan independientemente de halógeno, alquilo C1-C3 sustituido con halógeno lineal o ramificado, y alcoxi C1-C3 sustituido con halógeno;
o un grupo T2

donde
Z1 representa alquilo C1-C3 lineal o ramificado o cicloalquilo C3-C6, que pueden estar sustituidos independientemente unos de los otros con 1 a 5 sustituyentes de halógeno,
Z2 representa alquilo C1-C3 lineal o ramificado, que puede estar sustituido con 1 a 5 sustituyentes de halógeno, preferentemente con 1 a 3 sustituyentes de halógeno, más preferentemente trifluorometilo, o Z2 representa nitro, metilsulfanilo, metilsulfinilo, metilsulfonilo, flúor, cloro, bromo, yodo; y
Z3 representa hidrógeno o alquilo C1-C6 lineal o ramificado que puede estar sustituido con 1 a 5 sustituyentes que se seleccionan de hidroxi, halógeno, alquilo C1-C3, y alcoxi C1-C3;
y las sales de estos.
[16] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [15]
donde
Y se selecciona de un grupo T1

donde
R1 es halógeno;
R2 es alquilo C1-C3 lineal o ramificado sustituido con 1 a 7 halógenos, y
R3 es alcoxi C1-C3 sustituido con 1 a 3 halógenos;
o un grupo T2

donde
Z1 representa alquilo C1-C3 lineal o ramificado o cicloalquilo C3-C6, sustituido con 1 a 5 sustituyentes de halógeno,
Z2 representa alquilo C1-C3 lineal o ramificado sustituido con 1 a 3 sustituyentes de halógeno o
Z2 representa nitro, metilsulfanilo, metilsulfinilo, metilsulfonilo, flúor, cloro, bromo, yodo; y
Z3 representa hidrógeno o alquilo C1-C6 lineal o ramificado;
y las sales de estos.
[17] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [16]
donde
Y se selecciona de un grupo T1

donde
R1 es flúor, bromo o cloro, preferentemente cloro;
R2 es alquilo C1-C3 lineal o ramificado sustituido con 1 a 7 flúor, y
R3 es alcoxi C1-C3 sustituido con 1 a 3 flúor;
o un grupo T2
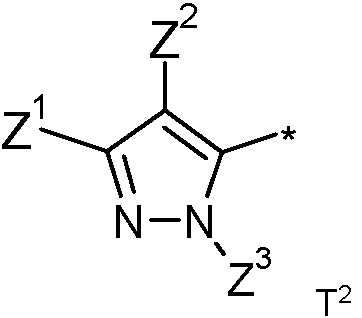
donde
Z1 representa trifluorometilo, 1-clorociclopropilo, 1-fluorociclopropilo o pentafluoroetilo;
Z2 representa alquilo C1-C3 lineal o ramificado sustituido con 1 a 3 flúor, y
Z3 representa alquilo C1-C6 lineal o ramificado;
y las sales de estos.
[18] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [17]
donde
Y se selecciona de un grupo T1

donde
R1 es cloro;
R2 es CF3, C2F5 o C3F7 y
R3 es OCF3, OC2F5 o OC3F7 ;
o un grupo T2

donde
Z1 representa trifluorometilo o pentafluoroetilo;
Z2 representa trifluorometilo; y
R3 representa hidrógeno, metilo, etilo o n-propilo;
y las sales de estos.
[19] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [18]
donde T1 está representado por el siguiente grupo T1-1:

T1-1;
y las sales de estos.
[20] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [18]
donde T1 está representado por el siguiente grupo T1-2:

y las sales de estos.
[21] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [18]
donde T1 está representado por el siguiente grupo T2-1:

y las sales de estos.
[22] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores donde Y es T1 como se define en cualquiera de los aspectos [1] a [20]
y las sales de estos.
[23] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [22], que están en la forma de sales, solvatos, N-óxidos y formas tautoméricas de estos.
[24] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [23], que se seleccionan de
Ácido (11E)-1-(2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-1,5,10-trioxo-4,6,9-trioxa-2-azatridec-11-en-13-oico,
Ácido (12E)-1-(2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-1,5,11-trioxo-4,6,10-trioxa-2-azatetradec-12-en-14-oico,
Ácido 1-(2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-1,5,10-trioxo-4,6,9-trioxa-2-azatridecan-13-oico,
Ácido (12E)-1-(2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-8,8-dimetil-1,5,11-trioxo-4,6,10-trioxa-2-azatetradec-12-en-14-oico, Ácido 1-(2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-8,8-dimetil-1,5,11-trioxo-4,6,10-trioxa-2-azatetradecan-14-oico,
Ácido (2E)-4-({(rel 1R,2S)-2-[({[(2-cloro-5-(H2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclopentil}oxi)-4-oxobut-2-enoico,
Ácido 4-({(rel 1R,2S)-2-[({[(2-cloro-5-(H2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclopentil}oxi)-4-oxobutanoico,
Ácido 1-(2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-11,11-dimetil-1,5,-dioxo-4,6,9-trioxa-2-azadodecan-12-oico,
Ácido 1-(2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-12,12-dimetil-1,5,10-trioxo-4,6,9-trioxa-2-azatridecan-13-oico,
Ácido (2E)-4-({(1S,2S)-2-[({[(2-doro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclopentil}oxi)-4-oxobut-2-enoico,
Ácido 4-({(1S,2S)-2-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclopentil}oxi)-4-oxobutanoico, Ácido 1-(2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-1,5,10-trioxo-4,6,9-trioxa-2-azatetradecan-14-oico,
Ácido 1-[10-(2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-9-(1-cianociclopropil)-6,10-dioxo-2,5,7-trioxa-9-azadecanan-1-oil]ciclopropano-1-carboxílico, Ácido (2E)-4-({cis-4-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexil}oxi)-4-oxobut-2-enoico, Ácido 4-({cis-4-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexil}oxi)-4-oxobutanoico,
Ácido (2E)-4-({trans-4-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexil}oxi)-4-oxobut-2-enoico, Ácido cis-4-[({[(2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexano-1-carboxílico,
Ácido Trans-4-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]cidohexano-1-carboxílico,
Ácido [({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]acético,
y las sales, solvatos, N-óxidos y formas tautoméricas de estos.
[25] En otro aspecto, la invención se refiere a compuestos de fórmula (I) como se define en cualquiera de los aspectos anteriores [1] a [24], que se usan como medicamentos.
[26] En otro aspecto, la invención se refiere a composiciones farmacéuticas que comprenden al menos un compuesto de acuerdo con cualquiera de los aspectos anteriores [1] a [25].
[27] En otro aspecto, la invención se refiere a composiciones farmacéuticas de acuerdo con el aspecto [26], que comprenden al menos un componente adicional que se selecciona de adyuvantes, excipientes y/o solventes.
[28] En un aspecto adicional, la invención se refiere a composiciones farmacéuticas de acuerdo con el aspecto [26] o [27], que comprenden al menos un principio adicional farmacéuticamente activo.
[29] En un aspecto adicional, la invención se refiere a composiciones farmacéuticas de acuerdo con el aspecto [28], donde el al menos un principio adicional farmacéuticamente activo se selecciona del grupo de principios activos con actividad ectoparasiticida, en particular con actividad insecticida y/o acaricida, o del grupo de antígenos con fines de vacunación.
[30] En otro aspecto, la invención se refiere a composiciones farmacéuticas de acuerdo con cualquiera de los
aspectos anteriores [26] a [29], que están en la forma de una formulación inyectable.
[31] En otro aspecto, la invención se refiere a composiciones farmacéuticas de acuerdo con cualquiera de los aspectos anteriores [26] a [29], que están en la forma de una formulación para administración oral.
[32] En otro aspecto, la invención se refiere a los compuestos o las composiciones farmacéuticas de acuerdo con cualquiera de los aspectos anteriores para administración subcutánea.
[33] En otro aspecto, la invención se refiere a los compuestos o las composiciones farmacéuticas de acuerdo con cualquiera de los aspectos anteriores para administración oral.
[34] En otro aspecto, la invención se refiere a los compuestos o las composiciones farmacéuticas de acuerdo con cualquiera de los aspectos anteriores para el tratamiento de animales.
[35] En otro aspecto, la invención se refiere a los compuestos o las composiciones farmacéuticas de acuerdo con el aspecto [34], donde los animales a ser tratados se seleccionan de animales de compañía.
[36] En otro aspecto, la invención se refiere a los compuestos o las composiciones farmacéuticas de acuerdo con el aspecto [34] o [35], donde los animales de compañía se seleccionan de gatos y perros, preferentemente perros.
[37] En otro aspecto, la invención se refiere al uso de los compuestos o las composiciones farmacéuticas de acuerdo con cualquiera de los aspectos anteriores para el control de insectos y arácnidos.
[38] En otro aspecto, la invención se refiere al uso de acuerdo con el aspecto [37], donde los insectos y arácnidos se selecciona del grupo de Chelicerata.
[39] En otro aspecto, la invención se refiere al uso de acuerdo con el aspecto [37] o [38], donde los insectos y arácnidos se selecciona del grupo que consiste en garrapatas, piojos, mosquitos, moscas, pulgas, Acari y ácaros.
[40] En otro aspecto, la invención se refiere al uso de los compuestos como se define en cualquiera de los aspectos anteriores para preparar composiciones farmacéuticas para controlar parásitos en animales.
[41] En otro aspecto, la invención se refiere al uso de los compuestos o las composiciones farmacéuticas de acuerdo con cualquiera de los aspectos anteriores con intervalos de tratamiento de 3 meses a dos años, preferentemente 4 meses a un año.
[42] En otro aspecto, la invención se refiere al uso de los compuestos o las composiciones farmacéuticas de acuerdo con el aspecto [41], donde los intervalos de tratamiento son 6 meses a un año, preferentemente 9 meses a un año.
[43] En otro aspecto, la invención se refiere al uso de los compuestos o las composiciones farmacéuticas de acuerdo con cualquiera de los aspectos anteriores, donde la cantidad total de los compuestos como se define en cualquiera de los aspectos anteriores a ser administrada se encuentra en el intervalo de 0,01 a 200 mg/kg de peso corporal por administración, preferentemente en el intervalo de 0,1 a 100 mg/kg de peso corporal por administración, más preferentemente de 0,5 a 75 mg/kg de peso corporal por administración, más preferentemente en el intervalo de 1,0 a 50 mg/kg de peso corporal por administración, más preferentemente en el intervalo de 2,0 a 20 mg/kg de peso corporal por administración.
[44] En otro aspecto, la invención se refiere a un proceso para preparar los compuestos de acuerdo con cualquiera de los aspectos anteriores que comprende el paso de hacer reaccionar un compuesto (A)

(A)
con un grupo (B)

(B)
donde
Y, Q, L1, L2 y L3 tienen el significado como se define en cualquiera de los aspectos anteriores y donde PG1 representa un grupo protector o hidrógeno para formar los compuestos de acuerdo con la fórmula (I), y donde en los casos donde PG1 no sea hidrógeno, la desprotección se lleva a cabo para formar los compuestos (I).
[45] En otro aspecto, la invención se refiere al proceso de acuerdo con el aspecto [44], que comprende además el paso preliminar de preparar el grupo (B):
i) haciendo reaccionar el compuesto (IIa) con un compuesto (IIIa) para formar el compuesto (Va), donde PG2 es un grupo protector o hidrógeno, y donde en los casos donde PG2 no sea hidrógeno, se obtiene (Va) haciendo escindir selectivamente PG2:
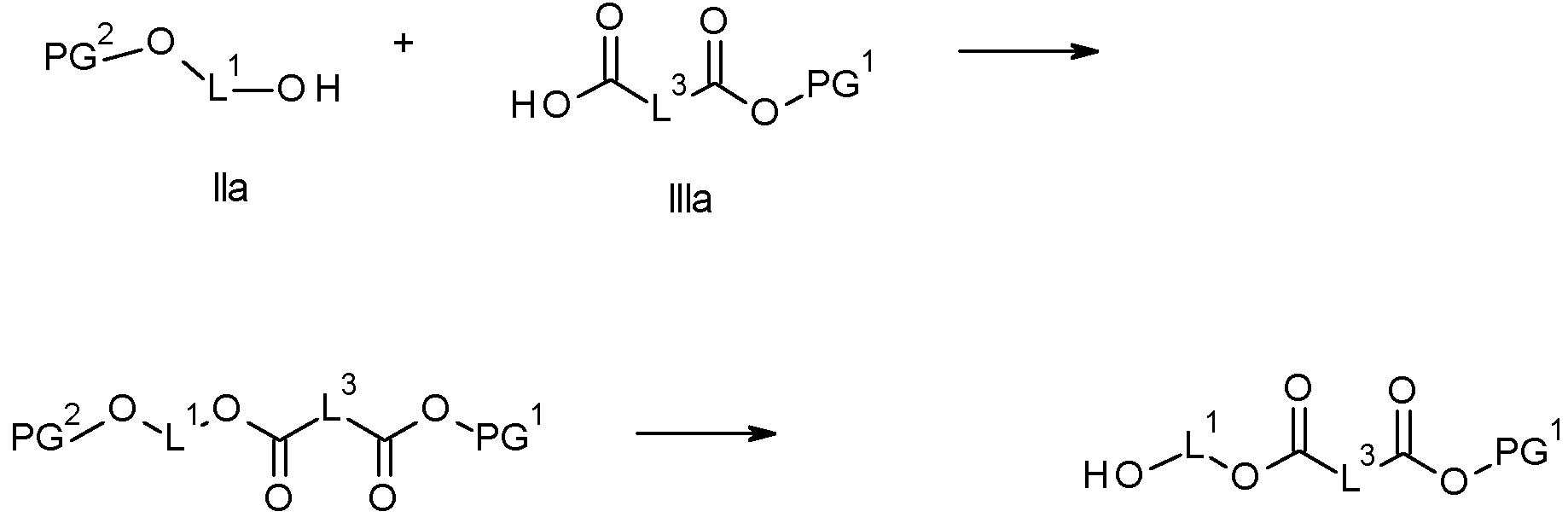
IVa Va donde L1 y L3 tienen el significado como se define en cualquiera de los aspectos anteriores y donde PG1 representa un grupo protector o hidrógeno;
o
ii) haciendo reaccionar el compuesto (VI) con un compuesto (IIIb) para formar el compuesto (Vb), donde PG2 es un grupo protector o hidrógeno, y donde en los casos donde PG2 no sea hidrógeno, se obtiene (Vb) haciendo escindir selectivamente PG2:

IVb Vb
donde L1, L2 y L3 tienen el significado como se define en cualquiera de los aspectos anteriores y donde PG1 representa un grupo protector o hidrógeno.
[46] En otro aspecto, la invención se refiere a compuestos intermedios de acuerdo con la fórmula (B),

(B)
donde
Q es O;
L2 es C=O;
L1 y L3 tienen el significado como se define en cualquiera de los aspectos anteriores y
PG1 representa un grupo terc-butilo.
[47] En otro aspecto, la invención se refiere a compuestos intermedios de acuerdo con la fórmula (C),

(C)
donde Y, Q, L1, L2 y L3 tienen el significado como se define en cualquiera de los aspectos anteriores y donde PG1 representa un grupo terc-butilo.
[48] En otro aspecto, la invención se refiere a compuestos intermedios que se seleccionan de ferc-Butil-2-{[(ciorometoxi)carbonil]oxi}etil-(2E)-but-2-enodioato (Intermedio 3A);
ferc-Butil-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropano-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil-(2É)-but-2-enodioato (Intermedio 4A);
terc-Butil 3-{[(clorometoxi)carbonil]oxi}propil-(2E)-but-2-enodioato (Intermedio 6A);
ferc-Butil 3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]propil (2£)-but-2-enodioato (Intermedio 7A);
ferc-Butil 2-{[(clorometoxi)carbonil]oxi}etilbutanodioato (Intermedio 9A);
ferc-Butil 2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil butanodioato (Intermedio 10A);
terc-Butil 3-{[(clorometoxi)carbonil]oxi}-2,2-dimetilpropil (2£)-but-2-enodioato (Intermedio 12A);
ferc-Butil 3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropil (2£)-but-2-enodioato (Intermedio 13A); ferc-Butil 3-{[(clorometoxi)carbonil]oxi}-2,2-dimetilpropil butanodiato (Intermedio 14A);
ferc-Butil 3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]-2,2-dimetilpropil butanodioato (Intermedio 15A); ferc-Butil (rel 1S,2R)-2-{[(clorometoxi)carbonil]oxi}ciclopentil (2£)-but-2-enodioato (Intermedio 17A);
ferc-Butil (rel 1R,2S)-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(tr¡fluorometox¡)fen¡l]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi} carbonil)oxi]ciclopentil (2£)-but-2-enodioato (Intermedio 18A);
ferc-Butil (rel 1S,2R)-2-{[(clorometoxi)carbonil]oxi}ciclopentil butanodioato (Intermedio 19A);
ferc-Butil (rel 1R,2S)-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(tr¡fluorometox¡)fen¡l]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]ciclopentil butanodioato (Intermedio 20A); ferc-Butil 3-[2-(benciloxi)etoxi]-2,2-dimetilpropanoato (Intermedio 21A);
ferc-Butil 3-(2-hidroxietoxi)-2,2-dimetilpropanoato (Intermedio 22A);
ferc-Butil 3-(2-{[(clorometoxi)carbonil]oxi}etoxi)-2,2-dimetilpropanoato (Intermedio 23A);
ferc-Butil 1-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-11,11-dimetil-1,5-dioxo-4,6,9-trioxa-2-azadodecan-12-oato (Intermedio 24A);
1 -ferc-Butil 4-(2-hidroxietil)2,2-dimetilsuccinato (Intermedio 28A)
1 -ferc-Butil 4-(2-{[(clorometoxi)carbonil]oxi}etil)2,2-dimetilbutanodioato (Intermedio 29A);
1 -ferc-Butil 4-{2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil} 2,2-dimetilbutanodioato (Intermedio 30A); ferc-Butil (1S,2S)-2-hidroxiciclopentil (2£)-but-2-enodioato (Intermedio 31A);
ferc-Butil (1S,2S)-2-{[(clorometoxi)carbonil]oxi}ciclopentil (2£)-but-2-enodioato (Intermedio 32A);
ferc-Butil (1S,2S)-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianocidopropil)amino]metoxi} carbonil)oxi]cidopentil (2£)-but-2-enodioato (Intermedio 33A);
ferc-Butil (1S,2S)-2-{[(clorometoxi)carbonil]oxi}ciclopentil butanodioato (Intermedio 34A);
ferc-Butil (1S,2S)-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]ciclopentil butanodioato (Intermedio 35A); ferc-Butil 2-hidroxietil pentanodioato (Intermedio 36A);
ferc-Butil 2-{[(clorometoxi)carbonil]oxi}etilpentanodioato (Intermedio 37A);
ferc-Butil 2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil pentanodioato (Intermedio 38A);
1 -ferc-Butil 1-(2-hidroxietil)ciclopropano-1,1-dicarboxilato (Intermedio 39A);
1 -ferc-Butil 1-(2-{[(clorometoxi)carbonil]oxi}etil)ciclopropano-1,1-dicarboxilato (Intermedio 40A);
1 -ferc-Butil 1-{2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil} ciclopropano-1,1-dicarboxilato (Intermedio 41A);
ferc-Butil-4-hidroxiciclohexil (2£)-but-2-enodioato (Mezcla de diasterómeros) (Intermedio 42A);
ferc-Butil c/s-4-hidroxiciclohexil (2£)-but-2-enodioato Intermedio 43A);
ferc-Butil frans-4-hidroxiciclohexil (2£)-but-2-enodioato (Intermedio 44A);
ferc-Butil c/s-4-{[(dorometoxi)carbonil]oxi}cidohexil (2£)-but-2-enodioato (Intermedio 45A);
ferc-Butil c/s-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianocidopropil)amino]metoxi}carbonil)oxi]cidohexil (2£)-but-2-enodioato (Intermedio 46A); ferc-Butil c/s-4-hidroxiciclohexil butanodioato (Intermedio 47A);
ferc-Butil c/s-4-{[(dorometoxi)carbonil]oxi}cidohexil butanodioato (Intermedio 48A);
ferc-Butil c/s-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianocidopropil)amino] metoxi}carbonil)oxi]cidohexil butanodioato (Intermedio 49A);
ferc-Butil frans-4-{[(clorometoxi)carbonil]oxi}ciclohexil (2£)-but-2-enodioato (Intermedio 50A);
terc-Butil frans-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianocidopropil)amino]metoxi}carbonil)oxi]cidohexil (2E)-but-2-enodioato (Intermedio 51A); ferc-Butil c/s-4-{[(dorometoxi)carbonil]oxi}cidohexano-1-carboxilato (Intermedio 52A);
ferc-Butil c/s-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianocidopropil)amino] metoxi}carbonil)oxi]cidohexano-1-carboxilato (Intermedio 53A);
ferc-Butil Trans-4-{[(clorometoxi)carbonil]oxi}ciclohexano-1-carboxilato (Intermedio 54A);
ferc-Butil frans-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianocidopropil)amino] metoxi}carbonil)oxi]cidohexano-1-carboxilato (Intermedio 55A); ferc-Butil [({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianocidopropil)amino] metoxi}carbonil)oxi]acetato (Intermedio 57A);
Definiciones
“Arácnidos” son una clase (Arachnida) de animales invertebrados de patas articuladas (artrópodos), en el subfilo Chelicerata. Una subclase preferida de arácnidos son los ácaros (oacarina) que comprende en particular ácaros y garrapatas.
El término “sustituido” significa que uno o más átomos de hidrógeno en el átomo o grupo designado se remplazan por una selección del grupo indicado, con la condición de que no se exceda la valencia normal del átomo designado bajo las circunstancias existentes. Se permiten combinaciones de sustituyentes y/o variables.
El término “opcionalmente sustituido” se refiere a que la cantidad de sustituyentes puede ser igual o diferente a cero. Salvo que se indique lo contrario, es posible que los grupos opcionalmente sustituidos estén sustituidos con todos los sustituyentes opcionales que se puedan alojar reemplazando el átomo de hidrógeno con un sustituyente diferente de hidrógeno en cualquier átomo de carbono o nitrógeno disponible. Comúnmente, es posible que la cantidad de sustituyentes opciones, cuando está presente, sea 1, 2, 3, 4 o 5, en particular 1, 2 o 3.
Como se usa en la presente, el término “uno o más”, por ejemplo en la definición de los sustituyentes de los compuestos de la fórmula general (I) de la presente invención, se refiere a “1, 2, 3, 4 o 5, particularmente 1, 2, 3 o 4, más particularmente 1, 2 o 3, aún más particularmente 1 o 2”.
Como se usa en la presente, la posición mediante la cual un sustituyente correspondiente está conectado con el resto de la molécula se puede representar en una estructura dibujada con un asterisco [*] en dicho sustituyente. El término “que comprende” cuando se usa en la memoria descriptiva incluye “que consiste en”.
Si dentro del presente texto cualquier ítem se denomina “como se menciona en la presente”, significa que puede mencionarse en cualquier lugar del presente texto.
Los términos como se mencionan en el presente texto tienen los significados a continuación:
El término “átomo de halógeno” se refiere a un átomo de flúor, cloro, bromo o yodo, particularmente un átomo de flúor, cloro o bromo, más particularmente cloro y/o flúor.
El término “alcanodiilo C1-C4” representa un radical alcanodiilo de cadena ramificada o (lineal) divalente con 1 a 4, preferentemente 1, 2 o 3, más preferentemente 2 o 3, átomos de carbono. El término “alcanodiilo C2-C4” representa un radical alcanodiilo de cadena ramificada o (lineal) divalente con 2 a 4, preferentemente 2 o 3 átomos de carbono. Los siguientes se pueden mencionar como ejemplos preferidos: metileno, 1,2-etanodiilo, etano-1,1-diilo, 1,3-propileno (1,3-propanodiilo), propano-1,1-diilo, propano-1,2-diilo, propano-2,2-diilo, 1,4-butileno (1,4-butanodiilo), butano-1,2-diilo, butano-1,3-diilo, butano-2,3-diilo. Se prefiere metileno, 1,2-etanodiilo y 1,3-propileno (1,3-propanodiilo), más preferentemente 1,2-etanodiilo y 1,3-propileno (1,3-propanodiilo).
En la presente invención un grupo alcanodiilo C1-C4 o un grupo alcanodiilo C2-C4 lineal puede estar sustituido con uno o más grupos que se seleccionan independientemente de halógeno (como se define anteriormente), alquilo C1-C4 y cicloalquilo C3-C6. En la presente invención un grupo alcanodiilo C1-C4 lineal o un grupo alcanodiilo C2-C4 que está sustituido con uno o más grupos que se seleccionan de alquilo C1-C4 comprende en particular un grupo 1,2-dimetil-etanodiilo y un grupo 2,2-dimetil-1,3-propanodiilo.
En el caso de un grupo a C1-C4 alcanodiilo o un grupo alcanodiilo C2-C4 que puede estar sustituido con uno o más sustituyentes alquilo C1-C4, también es posible que dos sustituyentes alquilo C1-C4 formen un anillo junto con el átomo de carbono con el que están unidos. Los grupos correspondientes comprenden en particular los siguientes grupos:

Preferentemente un grupo correspondiente es

Allí, n y m son independientemente 0, 1, 2 o 3. Preferentemente, n y m son independientemente 0, 1 o 2. Más preferentemente uno de n y m es 0 y el otro es 1.
El término “alquilo C1-C6-”, comprende grupos hidrocarburos monovalentes, saturados, lineales o ramificados con 1, 2, 3, 4, 5 o 6 átomos de carbono. El término “alquilo C1-C4-”, comprende grupos hidrocarburos monovalentes, saturados, lineales o ramificados con 1,2, 3, o 4 átomos de carbono. El término “alquilo C1-C3”, comprende grupos hidrocarburos monovalentes, saturados, lineales o ramificados con, 1, 2 o 3 átomos de carbono. Ejemplos de los grupos alquilo correspondientes son un grupo metilo, etilo, n-propilo, isopropilo, n-butilo, sec-butilo, isobutilo, terebutilo etc., o un isómero de estos. Los más preferidos son metilo, etilo, y n-propilo
El término “cicloalquilo C3-C7-”, se refiere a un anillo hidrocarburo monocíclico, monovalente, saturado, que contiene 3, 4, 5, 6 o 7 átomos de carbono. El término “cicloalquilo C3-C6-”, se refiere a un anillo hidrocarburo monocíclico, monovalente, saturado, que contiene 3, 4, 5 o 6 átomos de carbono. El término “cicloalquilo C3-C5-”, se refiere a un anillo hidrocarburo monocíclico, monovalente, saturado, que contiene 3, 4, o 5 átomos de carbono. Dichos grupos cicloalquilo C3-C7 son por ejemplo, un anillo hidrocarburo monocíclico, por ejemplo, un grupo ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo o cicloheptilo. Particularmente, dicho grupo cicloalquilo contiene 3, 5 o 6 átomos de carbono y es por ejemplo ciclopropilo, ciclopentilo o ciclohexilo.
El término “cicloalcanodiilo C3-C7” representa un radical hidrocarburo monocíclico divalente con 3 a 7, preferentemente 3 a 6 átomos de carbono. Los siguientes grupos son ejemplos:

y

Allí, se prefieren los siguientes grupos:

y
Dichos grupos cicloalcanodiilo C3-C7 , pueden estar sustituidos con uno o más grupos que se seleccionan independientemente de alquilo C1-C4 y halógeno, cada uno como se define anteriormente.
En la presente invención el sustituyente L1 puede ser un resto (CH2)n-X-(CH2)m donde n y m son independientemente 0, 1 o 2 y donde X es un grupo cicloalcanodiilo C3-C7 como se define anteriormente. Allí, los grupos (CH2)n y (CH2)m están unidos con los grupos cicloalcanodiilo C3-C7 en las posiciones indicadas con * en las fórmulas anteriores. En el caso de n y m, siendo ambos 0, el resto correspondiente corresponde a un grupo cicloalcanodiilo C3-C7 como se define anteriormente. Ejemplos particulares de dicho resto comprenden:
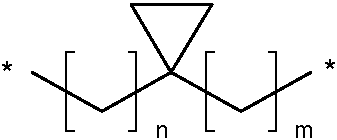
siendo uno de n o m 1 y el otro siendo 0 y

y
donde en cada caso n y m son ambos 0.
En la presente invención el sustituyente L3 puede ser un resto (CH2)n-(CH=CH)-CH2)m donde n y m son independientemente 0, 1 o 2. Los ejemplos comprenden eteno-1,1 -diilo, eteno-1,2-diilo, propeno-1,1-diilo, propeno-1,2-diilo, propeno-1,3-diilo, but-1-eno-1,4-diilo, but-1 -eno-1,3-diilo, but-2-eno-1,4-diilo, buta-1,3-dieno-1,4-diilo, pent-2-eno-1,5-diilo, hex-3-eno-1,6-diilo y hexa-2,4-dieno-1,6-diilo. Preferentemente n y m son ambos 0, es decir un grupo eteno-1,1 -diilo.
Dichos grupos (CH2)n-(CH=CH)-CH2)m pueden estar sustituidos con uno o más grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C5- y halógeno, cada uno como se define anteriormente.
El término "alcoxi C1-C6" representa un O-alquilo de cadena recta o ramificada, que tiene de 1 a 6 átomos de carbono, por ejemplo, metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, isobutoxi, s-butoxi y t-butoxi. También se da preferencia a los grupos alcoxi que tienen de 1 a 4 átomos de carbono. Los grupos alcoxi inventivos pueden estar sustituidos con uno o más radicales idénticos o diferentes.
El término "alquilsulfanilo C1-C6" representa S-alquilo de cadena recta o ramificada, con 1 a 6 átomos de carbono, por ejemplo, metiltio, etiltio, n-propiltio, isopropiltio, n-butiltio, isobutiltio, s-butiltio y t-butiltio. También se da preferencia a los grupos alquilsulfanilo que tienen de 1 a 4 átomos de carbono. Los grupos alquilsulfanilo inventivos pueden estar sustituidos con uno o más radicales idénticos o diferentes.
El término "alquilsulfinilo C1-C6" representa alquilsulfinilo de cadena recta o ramificada, con 1 a 6 átomos de carbono, por ejemplo, metilsulfinilo, etilsulfinilo, n-propilsulfinilo, isopropilsulfinilo, n-butilsulfinilo, isobutilsulfinilo, s-butilsulfinilo y t-butilsulfinilo. También se da preferencia a los grupos alquilsulfinilo que tienen de 1 a 4 átomos de carbono. Los grupos alquilsulfinilo inventivos pueden estar sustituidos con uno o más radicales idénticos o diferentes.
El término "alquilsulfonilo" representa alquilsulfonilo de cadena recta o ramificada, con 1 a 6 átomos de carbono, por ejemplo, metilsulfonilo, etilsulfonilo, n-propilsulfonilo, isopropilsulfonilo, n-butilsulfonilo, isobutilsulfonilo, sbutilsulfonilo y t-butilsulfonilo. También se da preferencia a los grupos alquilsulfonilo que tienen de 1 a 4 átomos de
carbono. Los grupos alquilsulfonilo inventivos pueden estar sustituidos con uno o más radicales idénticos o diferentes.
Los términos “alquilo C1-C6 sustituido con halógeno”, “alcoxi C1-C6 sustituido con halógeno” y “cicloalquilo C3-C6 sustituido con halógeno” representan grupos alquilo C1-C6, alcoxi C1-C6 y cicloalquilo C3-C6 como se define anteriormente, que están mono o poli sustituidos con halógeno hasta con la máxima cantidad posible de sustituyentes. Tales grupos también se refieren como grupos halo (por ejemplo, haloalquilo). En el caso de polisustitución por halógeno, los átomos de halógeno pueden ser los mismos o diferentes y todos pueden estar unidos a un átomo de carbono o pueden estar unidos a una pluralidad de átomos de carbono. Halógeno es especialmente flúor, cloro, bromo o yodo, preferentemente flúor, cloro o bromo y más preferentemente flúor. Más particularmente, los grupos sustituidos con halógeno son monohalocicloalquilo, tales como, 1-fluorociclopropilo, 2-fluorociclopropilo o 1 -fluorociclobutilo, monohaloalquilo tales como 2-cloroetilo, 2-fluoroetilo, 1 -cloroetilo, 1 -fluoroetilo, clorometilo, o fluorometilo; perhaloalquilo tales como triclorometilo o trifluorometilo o CF2CF3, polihaloalquilo tales como difluorometilo, 2-fluoro-2-cloroetilo, diclorometilo, 1,1,2,2-tetrafluoroetilo o 2,2,2-trifluoroetilo. Ejemplos adicionales de haloalquilos son triclorometilo, clorodifluorometilo, diclorofluorometilo, clorometilo, bromometilo, 1-fluoroetilo, 2-fluoroetilo, 2,2-difluoroetilo, 2,2,2-trifluoroetilo, 2,2,2-tricloroetilo, 2-cloro-2,2-difluoroetilo, pentafluoroetilo, 3,3,3-trifluoropropilo y pentafluoro-t-butilo. Se da preferencia a los haloalquilos que tienen 1 a 4 átomos de carbono y 1 a 9, preferentemente 1 a 5, átomos de halógeno idénticos o diferentes seleccionados de flúor, cloro y bromo, preferentemente de flúor. Se da particular preferencia a los haloalquilos con 1 o 2 átomos de carbono y 1 a 5 átomos de halógeno idénticos o diferentes seleccionados de flúor y cloro, tal como, entre otros, difluorometilo, trifluorometilo o 2,2-difluoroetilo. Ejemplos adicionales de compuestos sustituidos con halógeno son haloalcoxi tales como OCF3, OCHF2, OCH2F, OCF2CF3, OCH2CF3, OCH2CHF2 y OCH2CH2Cl, haloalquilsulfanilos tales como difluorometiltio, trifluorometiltio, triclorometiltio, clorodifluorometiltio, 1 -fluoroetiltio, 2-fluoroetiltio, 2,2-difluoroetiltio, 1,1,2,2-tetrafluoroetiltio, 2,2,2-trifluoroetiltio o 2-cloro-1,1,2-trifluoroetiltio, haloalquilsulfinilos tales como difluorometilsulfinilo, trifluorometilsulfinilo, triclorometilsulfinilo, clorodifluorometilsulfinilo, 1-fluoroetilsulfinilo, 2-fluoroetilsulfinilo, 2,2-difluoroetilsulfinilo, 1,1,2,2-tetrafluoroetilsulfinilo, 2,2,2-trifluoroetilsulfinilo y 2-cloro-1,1,2-trifluoroetilsulfinilo, haloalquilsulfinilos tales como difluorometilsulfinilo, trifluorometilsulfinilo, triclorometilsulfinilo, clorodifluorometilsulfinilo, 1-fluoroetilsulfinilo, 2-fluoroetilsulfinilo, 2,2-difluoroetilsulfinilo, 1,1,2,2-tetrafluoroetilsulfinilo, 2.2.2- trifluoroetilsulfinilo y 2-cloro-1,1,2-trifluoroetilsulfinilo, grupos haloalquilsulfonilo tales como difluorometilsulfonilo, trifluorometilsulfonilo, triclorometilsulfonilo, clorodifluorometilsulfonilo, 1-fluoroetilsulfonilo, 2-fluoroetilsulfonilo, 2,2-difluoroetilsulfonilo, 1,1,2,2-tetrafluoroetilsulfonilo, 2,2,2-trifluoroetilsulfonilo y 2-cloro-1,1,2-trifluoroetilsulfonilo. Los grupos preferidos de alquilo fluorados son CF3, C2F5 y C3F7 y los grupos alcoxi fluorados más preferidos son OCF3, OC2F5 y OC3F7.
Los términos “alquilamino N-C1-C6”, “N,N-dialquilamino C1-C6”, y “N-alcoxi C1-C3-alquilamino C1-C4” representa un grupo amino sustituido con uno o dos grupos C1-C6 o un grupo amino sustituido con un grupo alcoxi C1-C3 y un grupo alquilo C1-C4, cada uno como se define anteriormente.
El término "alquilcarbonilo C1-C6" representa alquilo-C(=O) de cadena recta o ramificada, con 2 a 7 átomos de carbono tales como metilcarbonilo, etilcarbonilo, n-propilcarbonilo, isopropilcarbonilo, s-butilcarbonilo y tbutilcarbonilo. También se le da preferencia a los alquilcarbonilos que tienen de 1 a 4 átomos de carbono. Los alquilcarbonilos inventivos pueden estar sustituidos con uno o más radicales idénticos o diferentes.
El término "alquenilo C2-C6" representa hidrocarburos de cadena recta o ramificada, preferentemente con 2 a 6 átomos de carbono y al menos un enlace doble, por ejemplo, vinilo, 2-propenilo, 2-butenilo, 3-butenilo, 1-metil-2-propenilo, 2-metil-2-propenilo, 2-pentenilo, 3-pentenilo, 4-pentenilo, 1-metil-2-butenilo, 2-metil-2-butenilo, 3-metil-2-butenilo, 1-metil-3-butenilo, 2-metil-3-butenilo, 3-metil-3-butenilo, 1,1 -dimetil-2-propenilo, 1,2-dimetil-2-propenilo, 1-etil-2-propenilo, 2-hexenilo, 3-hexenilo, 4-hexenilo, 5-hexenilo, 1-metil-2-pentenilo, 2-metil-2-pentenilo, 3-metil-2-pentenilo, 4-metil-2-pentenilo, 3-metil-3-pentenilo, 4-metil-3-pentenilo, 1-metil-4-pentenilo, 2-metil-4-pentenilo, 3-metil-4-pentenilo, 4-metil-4-pentenilo, 1,1-dimetil-2-butenilo, 1,1 -dimetil-3-butenilo, 1,2-dimetil-2-butenilo, 1,2-dimetil-3-butenilo, 1,3-dimetil-2-butenilo, 2,2-dimetil-3-butenilo, 2,3-dimetil-2-butenilo, 2,3-dimetil-3-butenilo, 1 -etil-2-butenilo, 1 -etil-3-butenilo, 2-etil-2-butenilo, 2-etil-3-butenilo, 1,1,2-trimetil-2-propenilo, 1 -etil-1 -metil-2-propenilo y 1 -etil-2-metil-2-propenilo. También se da preferencia a los alquenilos que tienen de 2 a 4 átomos de carbono tales como, entre otros, 2-propenilo, 2-butenilo o 1-metil-2-propenilo. Los alquenilos inventivos pueden estar sustituidos con uno o más radicales idénticos o diferentes.
El término "alquinilo C2-C6" representa hidrocarburos de cadena recta o ramificada, preferentemente con de 2 a 6 átomos de carbono y al menos un enlace triple, por ejemplo, 2-propinilo, 2-butinilo, 3-butinilo, 1 -metil-2-propinilo, 2-pentinilo, 3-pentinilo, 4-pentinilo, 1 -metil-3-butinilo, 2-metil-3-butinilo, 1 -metil-2-butinilo, 1,1 -dimetil-2-propinilo, 1-etil-2-propinilo, 2-hexinilo, 3-hexinilo, 4-hexinilo, 5-hexinilo, 1 -metil-2-pentinilo, 1 -metil-3-pentinilo, 1 -metil-4-pentinilo, 2-metil-3-pentinilo, 2-metil-4-pentinilo, 3-metil-4-pentinilo, 4-metil-2-pentinilo, 1,1 -dimetil-3-butinilo, 1,2-dimetil-3-butinilo, 2.2- dimetil-3-butinilo, 1 -etil-3-butinilo, 2-etil-3-butinilo, 1-etil-1-metil-2-propinilo y 2,5-hexadiinilo. También se da preferencia a los alquinilos que tienen de 2 a 4 átomos de carbono tales como, entre otros, etinilo, 2-propinilo o 2-butinilo-2-propenilo. Los alquinilos inventivos pueden estar sustituidos con uno o más radicales idénticos o diferentes.
El término "arilo" representa un sistema aromático mono, bi o policíclico con preferentemente de 6 a 14, especialmente de 6 a 10 átomos de carbono de anillo, por ejemplo, fenilo, naftilo, antrilo, fenantrenilo, preferentemente fenilo. Además, arilo también representa sistemas policíclicos tales como tetrahidronaftilo, indenilo, indanilo, fluorenilo, bifenilo, donde el sitio de enlace se encuentra en el sistema aromático. Los grupos arilo inventivos pueden estar sustituidos con uno o más radicales idénticos o diferentes.
El término “hetarilo” o "heteroarilo” representa compuestos heteroaromáticos, es decir compuestos heterocíclicos aromáticos completamente insaturados con al menos un anillo donde al menos un átomo de carbono se remplaza con un heteroátomo, preferentemente por un heteroátomo del grupo que consiste en N, O, S, P, B, Si, Se y que puede estar insustituido o sustituido, donde el sitio de enlace es sobre un átomo de anillo. A menos que se defina de forma diferente, el anillo heteroarilo contiene preferentemente de 3 a 9 átomos de anillo, especialmente de 3 a 6 átomos de anillo, especialmente 5 a 7 átomos de anillo y uno o más, preferentemente de 1 a 4, especialmente 1,2, o 3 heteroátomos en el anillo heteroarilo, preferentemente del grupo que consiste en N, O y S, aunque no debe haber juntos directamente dos átomos de oxígeno. Los anillos heteroarilo en general contienen no más de 4 átomos de nitrógeno y/o no más de 2 átomos de oxígeno y/o no más de 2 átomos de azufre. Se da preferencia particular a los anillos de 5 a 7 miembros con 1 a 3, preferentemente 1 o 2, heteroátomos idénticos o diferentes del grupo anterior. Los heteroarilos inventivos son, por ejemplo, urilo, tienilo, pirazolilo, imidazolilo, 1,2,3-y 1,2,4-triazolilo, isoxazolilo, tiazolilo, isotiazolilo, 1,2,3-, 1,3,4-, 1,2,4- y 1,2,5-oxadiazolilo, azepinilo, pirrolilo, piridilo, piridazinilo, pirimidinilo, pirazinilo, 1,3,5-, 1,2,4-y 1,2,3-triazinilo, 1,2,4-, 1,3,2-, 1,3,6-y 1,2,6-oxazinilo, oxepinilo, tiepinilo, 1,2,4-triazolonilo y 1,2,4-diazepinilo. Los grupos heteroarilo inventivos pueden estar sustituidos por uno o más radicales idénticos o diferentes.
Sustituyentes preferidos de los grupos arilo y heteroarilo se seleccionan de hidroxi, halógeno, ciano, nitro, amino, alquilo C1-C3, alcoxi C1-C3, hidroxicarbonilo, alcoxicarbonilo, alquilcarbamoilo, cicloalquilcarbamoilo y fenilo.
Preferentemente, los sustituyentes Y, Q, L1, L2 y L3 tienen el significado como se define anteriormente en los diferentes aspectos de la presente invención.
En un aspecto particularmente preferido de la invención el sustituyente Y en los compuestos de fórmula (I) como se define en otra parte de la presente se selecciona de un grupo T1 como se define en la presente, en particular un grupo T1-1 o T1-2, cada uno como se define en otra parte de la presente.
Ejemplos particularmente preferidos de la presente invención se enumeran en el aspecto [24] anterior y como se muestra en los Ejemplos a continuación. Compuestos preferidos de acuerdo con la fórmula (I) de la presente invención son los de los Ejemplos 1, 2, 3, 4, 5, 6, 7, 9, 10, 11, 12, 13, 14, 15, y 16. Compuestos más preferidos de acuerdo con la fórmula (I) de la presente invención son los de los Ejemplos 1, 2, 4, 6, 10, 14, y 16.
Sales de los compuestos de la invención que son adecuados de acuerdo con la invención, por ejemplo, sales con bases, son todas sales no tóxicas habituales, preferentemente sales aceptables desde el punto de vista agrícola y/o fisiológico. Se prefieren sales con bases inorgánicas, por ejemplo sales de metales alcalinos (por ejemplo sales de sodio, potasio o cesio), sales de metales alcalinotérreos (por ejemplo sales de calcio o magnesio), sales de amonio o sales con bases orgánicas, en particular con aminas orgánicas, por ejemplo sales de trietilamonio, diciclohexilamonio, W,W-dibenciletilendiamonio, piridinio, picolinio o etanolamonio. Preferentemente, las sales de los compuestos de la presente invención son sales farmacéuticamente aceptables.
Además, la presente invención incluye todas las formas cristalinas posibles o polimorfos de los compuestos de la presente invención como polimorfo simple o como mezcla de más de un polimorfo, en cualquier proporción.
Los compuestos de la presente invención y sus sales, en la medida de lo posible, pueden existir como un hidrato o como un solvato donde los compuestos de la presente invención contienen agua o solventes polares (por ejemplo metanol o etanol), respectivamente, como elemento estructural de la estructura cristalina de los compuestos. Es posible que la cantidad de solventes polares, en particular agua, exista en una relación estequimétrica o no estequimétrica. En el caso de solvatos estequimétricos, por ejemplo un hidrato, hemi-, (semi-), mono-, sesqui-, di-, tri-, tetra-, penta- etc. son posibles solvatos o hidratos, respectivamente. La presente invención incluye todos dichos hidratos o solvatos de los compuestos y sus sales, en la medida de lo posible.
Dependiendo de la naturaleza de los sustituyentes, los compuestos de la fórmula (I) pueden estar en la forma de isómeros geométricos y/u ópticamente activos o mezclas de isómeros correspondientes en diferentes composiciones. Estos estereoisómeros son, por ejemplo, enantiómeros, diastereómeros, atropisómeros, tautómeros o isómeros geométricos. Por consiguiente, la invención abarca estereoisómeros puros y cualquier mezcla de estos isómeros.
Los compuestos novedosos de acuerdo con la presente invención son particularmente adecuados para el uso como medicamentos, en particular para el uso como medicamentos para el tratamiento de animales. Los compuestos novedosos de acuerdo con la presente invención son particularmente adecuados para el uso como medicamentos para actuar contra parásitos de animales, especialmente ectoparásitos, tales como insectos y arácnidos u otros. Los ectoparásitos son normalmente y preferentemente artrópodos, especialmente insectos tales como moscas (que
muerden y lamen), larvas de moscas parasíticas, piojos que lamen, piojos que muerden, pulgas y similares; o acáridos como garrapatas, por ejemplo garrapatas duras o garrapatas suaves, o ácaros tales como ácaros de roña, ácaros de aves y similares y también los ectoparásitos acuáticos tales como los copepodos. Los compuestos novedosos de la presente invención son particularmente adecuados para actuar contra garrapatas, pulgas, piojos, moscas y ácaros.
Los compuestos novedosos de la fórmula (I) con toxicidad homeotérmica favorable son adecuados para controlar parásitos que ocurren en la cría de animales y reproducción de animales en ganadería, incluido acuicultura, animales de cría, animales de zoológico, animales de laboratorio, animales experimentales y animales domésticos. Son activos contra todas las etapas o etapas específicas de desarrollo de los parásitos.
El ganado agrícola incluye, por ejemplo, mamíferos, tales como ovejas, cabras, caballos, burros, camellos, búfalos, conejos, renos, gamos, y en particular ganado y cerdos; o aves de corral tales como pavos, patos, gansos y en particular gallinas; peces y crustáceos, por ejemplo, en cultivo acuático; y también insectos tales como abejas. Los animales domésticos (también indicados como animales de compañía) incluyen, por ejemplo, mamíferos, tales como hámster, cobayos, ratas, ratones, chinchillas, hurones y particularmente perros, gatos, aves enjauladas, reptiles, anfibios y peces de acuario. Los animales de compañía preferidos son gatos y perros.
En una modalidad preferida, los compuestos de la fórmula (I) se administran a mamíferos.
En una modalidad preferida, los compuestos de la fórmula (I) se administran a gatos.
En una modalidad preferida los compuestos de la fórmula (I) se administran a perros.
Se pretende que el uso de los compuestos de la fórmula (I) para controlar parásitos de animales reduzca o evite enfermedades, casos de muerte y reducciones de rendimiento (en el caso de carne, leche, lana, pieles, huevos, miel y similares), de modo de posibilitar el mantenimiento de animales más económica y simple y que se logre un mejor bienestar animal.
Con relación al campo de la salud animal, el término "control" o "controlar" significa que los compuestos de la fórmula (I) son eficaces para reducir la incidencia del parásito particular en un animal infectado con tales parásitos a un grado inocuo. Más específicamente, "controlar" en el presente contexto significa que los compuestos de la fórmula (I) matan el respectivo parásito, inhiben su crecimiento o inhiben su proliferación.
Estos parásitos incluyen:
Del orden de los anopluros, por ejemplo, Haematopinus spp., Linognathus spp., Pediculus spp., Phthirus spp., Solenopotes spp., los ejemplos específicos son: Linognathus setosus, Linognathus vituli, Linognathus ovillus, Linognathus oviformis, Linognathus pedalis, Linognathus stenopsis, Haematopinus asini macrocephalus, Haematopinus eurysternus, Haematopinus suis, Pediculus humanus capitis, Pediculus humanus corporis, Phylloxera vastatrix, Phthirus pubis, Solenopotes capillatus;
Del orden de los Mallophaga y los subórdenes Amblicerina e Ischnocera por ejemplo, Trimenopon spp., Menopon spp., Trinoton spp., Bovicola spp., Werneckiella spp., Lepikentron spp., Damalina spp., Trichodectes spp. y Felicola spp.; los ejemplos específicos son: Bovicola bovis, Bovicola ovis, Bovicola limbata, Damalina bovis, Trichodectes canis, Felicola subrostratus, Bovicola caprae, Lepikentron ovis, Werneckiella equi;
Del orden de los dípteros y los subórdenes Nematocerina y Branchycerina, por ejemplo, Aedes spp., Anopheles spp., Culex spp., Simulium spp., Eusimulium spp., Phlebotomus spp., Lutzomyia spp., Culicoides spp., Chrysops spp., Odagmia spp., Wilhelmia spp., Hybomitra spp., Atylotus spp., Tabanus spp., Haematopota spp., Philipomyia spp., Braula spp., Musca spp., Hydrotaea spp., Stomoxys spp., Haematobia spp., Morellia spp., Fannia spp., Glossina spp., Calliphora spp., Lucilia spp., Chrysomyia spp., Wohlfahrtia spp., Sarcophaga spp., Oestrus spp., Hypoderma spp., Gasterophilus spp., Hippobosca spp., Lipoptena spp., Melophagus spp., Rhinoestrus spp., Tipula spp.; los ejemplos específicos son: Aedes aegypti, Aedes albopictus, Aedes taeniorhynchus, Anopheles gambiae, Anopheles maculipennis, Calliphora erythrocephala, Chrysozona pluvialis, Culex quinquefasciatus, Culex pipiens, Culex tarsalis, Fannia canicularis, Sarcophaga carnaria, Stomoxys calcitrans, Tipula paludosa, Lucilia cuprina, Lucilia sericata, Simulium reptans, Phlebotomus papatasi, Phlebotomus longipalpis, Odagmia ornata, Wilhelmia equina, Boophthora erythrocephala, Tabanus bromius, Tabanus spodopterus, Tabanus atratus, Tabanus sudeticus, Hybomitra ciurea, Chrysops caecutiens, Chrysops relictus, Haematopota pluvialis, Haematopota italica, Musca autumnalis, Musca domestica, Haematobia irritans irritans, Haematobia irritans exigua, Haematobia stimulans, Hydrotaea irritans, Hydrotaea albipuncta, Chrysomya chloropyga, Chrysomya bezziana, Oestrus ovis, Hypoderma bovis, Hypoderma lineatum, Przhevalskiana silenus, Dermatobia hominis, Melophagus ovinus, Lipoptena capreoli, Lipoptena cervi, Hippobosca variegata, Hippobosca equina, Gasterophilus intestinalis, Gasterophilus haemorroidalis, Gasterophilus inermis, Gasterophilus nasalis, Gasterophilus nigricornis, Gasterophilus pecorum, Braula coeca;
Del orden de los sifonápteros, por ejemplo, Pulex spp., Ctenocephalides spp., Tunga spp., Xenopsylla spp., Ceratophyllus spp.; los ejemplos específicos son: Ctenocephalides canis, Ctenocephalides felis, Pulex irritans,
Tunga penetrans, Xenopsylla cheopis;
Del orden de los heterópteros, por ejemplo, Cimex spp., Triatoma spp., Rhodnius spp., Panstrongylus spp.
Del orden de los Blattarida, por ejemplo, Blatta orientalis, Periplaneta americana, Blattela germanica y Supella spp. (p.ej. Suppella longipalpa);
De la subclase de los Acari (Acarina) y los órdenes de los Meta y Mesostigmata, por ejemplo, Argas spp., Ornithodorus spp., Otobius spp., Ixodes spp., Amblyomma spp., Rhipicephalus (Boophilus) spp., Dermacentor spp., Haemophysalis spp., Hyalomma spp., Dermanyssus spp., Rhipicephalus spp., (el género original de las garrapatas de varios huéspedes), Ornithonyssus spp., Pneumonyssus spp., Raillietia spp., Pneumonyssus spp., Sternostoma spp., Varroa spp., Acarapis spp.; los ejemplos específicos son: Argas persicus, Argas reflexus, Ornithodorus moubata, Otobius megnini, Rhipicephalus (Boophilus) microplus, Rhipicephalus (Boophilus) decoloratus, Rhipicephalus (Boophilus) annulatus, Rhipicephalus (Boophilus) calceratus, Hyalomma anatolicum, Hyalomma aegypticum, Hyalomma marginatum, Hyalomma transiens, Rhipicephalus evertsi, Ixodes ricinus, Ixodes hexagonus, Ixodes canisuga, Ixodes pilosus, Ixodes rubicundus, Ixodes scapularis, Ixodes holocyclus, Haemaphysalis concinna, Haemaphysalis punctata, Haemaphysalis cinnabarina, Haemaphysalis otophila, Haemaphysalis leachi, Haemaphysalis longicorni, Dermacentor marginatus, Dermacentor reticulatus, Dermacentor pictus, Dermacentor albipictus, Dermacentor andersoni, Dermacentor variabilis, Hyalomma mauritanicum, Rhipicephalus sanguineus, Rhipicephalus bursa, Rhipicephalus appendiculatus, Rhipicephalus capensis, Rhipicephalus turanicus, Rhipicephalus zambeziensis, Amblyomma americanum, Amblyomma variegatum, Amblyomma maculatum, Amblyomma hebraeum, Amblyomma cajennense, Dermanyssus gallinae, Ornithonyssus bursa, Ornithonyssus sylviarum, Varroa jacobsoni;
De los órdenes de los Actinedida (Prostigmata) y Acaridida (Astigmata) por ejemplo, Acarapis spp., Cheyletiella spp., Ornithocheyletia spp., Myobia spp., Psorergates spp., Demodex spp., Trombicula spp., Listrophorus spp., Acarus spp., Tyrophagus spp., Caloglyphus spp., Hypodectes spp., Pterolichus spp., Psoroptes spp., Chorioptes spp., Otodectes spp., Sarcoptes spp., Notoedres spp., Knemidocoptes spp., Cytodites spp., y Laminosioptes spp.; los ejemplos específicos son: Cheyletiella yasguri, Cheyletiella blakei, Demodex canis, Demodex bovis, Demodex ovis, Demodex caprae, Demodex equi, Demodex caballi, Demodex suis, Neotrombicula autumnalis, Neotrombicula desaleri, Neoschongastia xerothermobia, Trombicula akamushi, Otodectes cynotis, Notoedres cati, Sarcoptis canis, Sarcoptes bovis, Sarcoptes ovis, Sarcoptes rupicaprae (=S. caprae), Sarcoptes equi, Sarcoptes suis, Psoroptes ovis, Psoroptes cuniculi, Psoroptes equi, Chorioptes bovis, Psoergates ovis, Pneumonyssoidic mange, Pneumonyssoides caninum, Acarapis woodi.
De la subclase de los copépodos con el orden Siphonostomatoida en particular los géneros Lepeophtheirus y Caligus; las especies Lepeophtheirus salmonis, Caligus elongatus y Caligus clemensi pueden mencionarse mediante ejemplos y con particular preferencia.
De acuerdo con una modalidad preferida los parásitos se seleccionan del siguiente grupo de especie de ectoparásito:
Pulgas: Ctenocephalides spp.;
Garrapatas: Amblyomma spp., Dermacentor spp., Rhipicephalus spp., Ixodes spp., Haemaphysalis spp., Hyalomma spp.;
Ácaros: Demodex spp., Otodectes spp., Sarcoptes spp.; y
Piojos: Linognathus spp.
Más particularmente los parásitos se seleccionan de:
Pulgas: Ctenocephalides felis, Ctenocephalides canis;
Garrapatas: Ixodes scapularis, Ixodes ricinus, Dermacentor variabilis, Amblyomma americanum, Rhipicephalus sanguineus, Dermacentor reticulatus, Ixodes holocyclus, Ixodes hexagonus, Haemaphysalis longicornis;
Ácaros: Otodectes cynotis, Sarcoptes scabiei, Demodex canis; y
Piojos: Linognathus setosus.
Generalmente, los ingredientes activos de la invención pueden utilizarse directamente cuando se utilizan para tratamiento de animales. Se utilizan (administran) preferentemente en la forma de composiciones farmacéuticas que pueden comprender excipientes, solventes y/o adyuvantes farmacéuticamente aceptables conocidos en la técnica previa.
Los compuestos activos novedosos de la presente invención se puede administrar de una manera conocida, por administración enteral en forma de, por ejemplo, comprimidos, cápsulas, pociones, emulsiones, gránulos, pastas, bolos, proceso de alimentación directa y supositorios, por administración parenteral, por ejemplo por inyección (intramuscular, subcutánea, intravenosa, intraperitoneal entre otras), implantes, por administración nasal, por administración dérmica en la forma, por ejemplo, de inmersión o baño, pulverización, vertiendo, tocando, lavando y espolvoreando, y también con la ayuda de cuerpos moldeados que contienen el compuesto activo, como collares, marcas para las oreja, marcas para el rabo, bandas para las extremidades, cabestros, marquiradores, etc. Preferentemente, los compuestos novedosos de la presente invención se administran por administración oral, más preferentemente por administración subcutánea (inyección).
Los compuestos activos novedosos de la presente invención se pueden formular en cualquier forma de administración adecuada para administración oral y subcutánea (inyectable) conocida en la técnica anterior.
En función de técnicas de laboratorio estándar conocidas para evaluar compuestos útiles para controlar parásitos en animales, mediante pruebas de toxicidad estándar y mediante ensayos farmacológicos estándar para la determinación de tratamiento de las afecciones antes identificadas en animales, y mediante comparación de estos resultados con los resultados de ingredientes activos conocidos o medicamentos que se usan para tratar estas afecciones, la dosificación eficaz de los compuestos de la presente invención se puede determinar fácilmente para tratamiento de cada indicación deseada. La cantidad del ingrediente activo a ser administrado en el tratamiento de una de estas afecciones puede variar ampliamente de acuerdo con dichas consideraciones como el compuesto particular y la unidad de dosificación empleada, el modo de administración, el período de tratamiento, la edad y sexo del sujeto tratado y la naturaleza y el alcance de la afección tratada.
El contenido de los compuestos activos novedosos de la presente invención en formulaciones para el uso (dosificación unitaria) de acuerdo con la presente invención puede variar dentro de límites amplios. La concentración de compuesto activo de las formas de administración puede ser de 0,00000001 a 98% en peso del compuesto activo, preferentemente de 0,00001 a 98 % en peso, más preferentemente de 0,001 a 98 % en peso. Más preferentemente, las composiciones farmacéuticas de la presente invención pueden comprender los compuestos novedosos de la invención en cantidades de 0,01 a 98% en peso del compuesto activo, preferentemente de 0,1 a 98 % en peso, más preferentemente de 0,5 a 90 % en peso. Las composiciones farmacéuticas de la presente invención pueden comprender los compuestos novedosos de la invención en cantidades de 0,001 a 95 % en peso del compuesto activo, preferentemente de 0,01 a 95% en peso, preferentemente de 0,1 a 50 % en peso, más preferentemente de 5 a 30 % en peso.
La cantidad total del ingrediente activo a ser administrado generalmente varía de 0,01 a 200 mg/kg de peso corporal por administración, preferentemente en el intervalo de 0,1 a 100 mg/kg de peso corporal por administración, más preferentemente de 0,5 a 75 mg/kg de peso corporal por administración, más preferentemente en el intervalo de 1,0 a 50 mg/kg de peso corporal, y más preferentemente en el intervalo de 2,0 a 20 mg/kg de peso corporal por administración.
En particular, la dosificación promedio para administración por técnicas de infusión o por inyección, incluida intravenosa, intramuscular y en particular inyecciones subcutáneas, preferentemente se encuentra dentro de los intervalos anteriores.
La dosificación promedio para administración oral preferentemente se encuentra dentro de los intervalos antes mencionados.
Los intervalos de administración o dosificación clínicamente útiles varían de una administración por mes a una administración cada dos años, preferentemente el intervalo del tratamiento es de una administración cada tres meses a una administración cada dos años, más preferentemente los intervalos de tratamiento son de una administración cada seis meses a una administración cada dos años. Otros intervalos de administración o dosificación preferidos varían de una administración por mes a una administración por año, preferentemente una administración cada tres meses a una administración por año, más preferentemente una administración cada cuatro meses a una administración por año, en particular una administración cada seis meses a una administración por año. De acuerdo con otra modalidad el intervalo de administración puede ser de una administración cada nueve meses a una administración por año.
Además, es posible “días libres de fármacos” en los cuales al sujeto no se le dosifica un fármaco durante un período de tiempo lo que es beneficioso en el balance global entre la tolerabilidad y el efecto farmacológico.
Claro que el régimen de dosificación continuo e inicial específico, la cantidad administrada de compuesto activo y el intervalo de dosificación particular varían para cada sujeto de acuerdo con la naturaleza y gravedad de la afección según se determine cada Los sujetos por el médico tratante, la actividad del compuesto específico empleado, la edad y estado general del sujeto, el tiempo de administración, la vía de administración, la velocidad de excreción del fármaco, las combinaciones de fármacos y similares. El modo de tratamiento deseado y la cantidad de dosis de un compuesto de la presente invención o una sal o composición farmacéuticamente aceptable de este puede ser determinado por aquellos expertos en la técnica usando pruebas de tratamiento convencionales.
Para ampliar el espectro de actividad, los compuestos activos novedosos de la presente invención se pueden usar en combinación con sinergistas, repelentes u otros ingredientes activos adecuados, por ejemplo acaricidas, insecticidas, antelmínticos y agentes antiprotozoarios.
Por ejemplo los compuestos de la presente invención se pueden usar en combinación con activadores del canal de cloro o moduladores de la clase de lactonas macrocíclicas, en particular avermectinas/milbemicinas, por ejemplo abamectina, doramectina, emamectina benzoato, eprinomectina, ivermectina, latidectina, lepimectina, milbemicina
oxima, milbemectina, moxidectina y selamectina, se prefiere particularmente aplicaciones contra ectoparásitos, doramectina, eprinomectina, ivermectina, milbemicina oxima, moxidectina o selamectina.
Además, los compuestos de la presente invención pueden utilizarse combinados con antígenos con fines de vacunación. Ejemplos de vacunas que se pueden combinar con los compuestos de la presente invención son contra leptospirosis, tracqueobronquitis infecciosa, leishmaniasis o lyme borreliosis (enfermedad de lyme).
Proceso para preparar los compuestos novedosos de Fórmula (I)
Los compuestos novedosos (I) de acuerdo con la presente invención se pueden sintetizar de acuerdo con los Esquemas generales 1 a 3 como se muestra a continuación. En general, la síntesis se puede dividir en dos etapas. En la primera etapa, se acumula un prorresto protegido con una función de alcohol terminal que formará parte del enlazador de carbonato éster en los compuestos novedosos de la invención.
En la segunda etapa, el grupo de alcohol terminal del prorresto se convierte en un clorometil carbonato éster que luego está acoplado con un grupo amida halogenado de un resto de compuesto activo y finalmente se desprotege para dar un ácido carboxílico libre.
La primera etapa de la síntesis difiere dependiendo del significado de los sustituyentes L1, L2 y Q en los compuestos de la fórmula (I) mientras que la segunda etapa es común para todos los compuestos de la presente invención. Primera etapa del proceso-acumulación del prorresto
Ruta de síntesis (a) para Compuestos (I) donde L2 es C=O y Q es O
El siguiente Esquema 1 describe la síntesis del prorresto protegido de la antes mencionada primera etapa del proceso de la presente invención para preparar compuestos (I) donde L1 y L3 tienen el significado como se define en cualquier parte de la presente y donde L2 se selecciona como C=O y Q se selecciona como O.
Esquema 1

IVa Va Allí, PG1 es un grupo protector. Los grupos protectores adecuados para la protección de ácidos carboxílicos junto con métodos para desprotección, se describen en P.G. Wuts, Greene's Protective Groups in Organic Synthesis, Wiley 2014. En un caso preferido de la presente invención, PG1 es un grupo terc-butilo. PG2 es un grupo protector adecuado para la protección de alcoholes o PG2 es hidrógeno. En el caso de que PG2 sea hidrógeno, el paso (IVa) en el Esquema 1 que se muestra anteriormente, es obsoleto y entonces el compuesto (IIIa) se hace reaccionar directamente para formar el compuesto (Va).
En un caso preferido de la presente invención, PG2 es hidrógeno y no es necesario un paso de desprotección (IVa ^ Va).
En el Esquema 1 que se muestra anteriormente para preparar prorresto protegido en la primera etapa del proceso de la presente invención, se hace reaccionar un compuesto de fórmula (IIa) con un ácido de fórmula (IIIa). Con este fin, el ácido (IIIa) primero se activa convirtiéndolo en un cloruro ácido o en un éster activado usando un agente de acoplamiento tal como HATU, TBTU. Se conocen métodos de acoplamiento adecuados, por ejemplo en C.A.G.N. Montalbetti, V. Falque, Tetrahedron 2005, 61, 10827-10852. En un segundo paso, el ácido activado se hace
reaccionar con el compuesto (IIa) en presencia de una base.
En un caso preferido, el ácido (IIIa) se activa convirtiéndolo en el cloruro ácido usando cloruro oxálico en presencia de una cantidad catalítica de DMF en diclorometano y luego se hace reaccionar con (IIa) en presencia de una base amina, por ejemplo DIPEA, TEA en un solvente aprótico polar, por ejemplo DMF, THF, acetonitrilo. Opcionalmente, se puede usar una cantidad catalítica de DMAP para facilitar esta reacción.
En los casos en los que PG2 no sea hidrógeno, el compuesto (Va) se obtiene escindiendo selectivamente PG2 del compuesto (IVa) en presencia de PG1.
Ruta de síntesis (b) para Compuestos (I) donde L2 está ausente y Q es O
El siguiente Esquema 2 describe la síntesis del prorresto protegido de la antes mencionada primera etapa del proceso de la presente invención para preparar compuestos (I) donde L1 y L3 tienen el significado como se define en cualquier parte de la presente y donde L2 está ausente y Q se selecciona como O.
Esquema 2
O
PG'2 0
V - X
l2
PG
H O ^
3J
^
L
.
O '
VI IIIb
2 O 1,0
P G ^ ' 
IVb Vb
Entonces, X es un grupo saliente y puede ser Cl, Br, I, o un éster sulfonato tal como éster triflato, mesilato o paratoluenosulfanto. X también puede ser OH. En un caso preferido, X es halógeno seleccionado de Cl, Br o I. PG2 es un grupo protector adecuado para la protección de alcoholes o PG2 es hidrógeno. En el caso de que PG2 sea hidrógeno, el paso (IVb) en el Esquema 2 que se muestra anteriormente, es obsoleto y entonces el compuesto (IIIb) se hace reaccionar directamente para formar el compuesto (Vb).
En un caso preferido de la presente invención, PG2 es un grupo de éter bencilo o un grupo de éter bencilo sustituido. En el Esquema 2 que se muestra anteriormente para preparar el prorresto protegido en la primera etapa del proceso de la presente invención, en un primer paso, el compuesto (VI) se hace reaccionar con el compuesto (IIIb) en condiciones de una síntesis de éter Williamson. Esto requiere tratar una solución de (IIIb) con una base, luego agregar el compuesto (VI) y dejar que la reacción proceda a temperatura ambiente o más. Se pueden usar bases fuertes, por ejemplo bases hidruro, bases bis(trimetildisilil)amida (HMDS), bases amidina o bases fosfaceno.
En un caso preferido, se usa hidruro de sodio o bis(trimetildisilil)amida de sodio. Los solventes preferidos son solventes apróticos polares, por ejemplo THF o DMF.
Los productos éter (IVb) se convierten en alcoholes (Vb) por remoción del grupo protector PG2.
En un caso preferido, PG2 es un grupo bencilo y la remoción se logra por hidrogenación en presencia de paladio sobre carbón en un solvente prótico tal como metanol, etanol, ácido acético o mezclas de estos solventes.
Ruta de síntesis (c) para Compuestos (I) donde Q está ausente
El siguiente Esquema 3 describe la síntesis del prorresto protegido de la antes mencionada primera etapa del proceso de la presente invención para preparar compuestos (I) donde L1, L2 y L3 tienen el significado como se define en cualquier parte de la presente y donde Q está ausente y entonces, no se necesita formar un enlace entre L1-Q o L2-Q o L3-Q.
Esquema 3

Vc
PG1 es un grupo protector adecuado para la protección de ácidos carboxílicos. Preferentemente, PG1 es un grupo terc-butilo. Los compuestos de fórmula (Vc) se pueden obtener comercialmente o sintetizarse de acuerdo con los métodos descritos en la bibliografía conocida.
Segunda etapa del proceso- preparación de los Compuestos (I)
La segunda etapa del proceso de la presente invención se muestra en el Esquema 4.
Esquema 4

X I
Allí, L1, L2, L3, Q, R y PG1 se definen como se describe anteriormente.
Esta segunda etapa de la síntesis de la invención se puede llevar a cabo comenzado con cualquier intermedio (Va), (Vb) y (Vc), que resulte de la primera etapa del proceso como se describe anteriormente. Dichos intermedios de partida se denominan generalmente en la presente como intermedio (V).
En un primer paso de la segunda etapa del proceso, los compuestos intermedio (V) se tratan con carbonocloridato de clorometilo que lleva a ésteres de clorometilo (VII). La reacción se lleva a cabo en un solvente en presencia de una base. Se pueden usar solventes apróticos dipolares tales como DMF, THF, acetonitrilo o piridina. La base puede ser una base amina, por ejemplo DIPEa , TEA, DMAP. En un caso preferido, la piridina sirve como solvente y como base.
En el próximo paso de la segunda etapa del proceso, se trata una amida de fórmula (VIII) con una base y luego se hace reaccionar con un éster clorocarbonato de fórmula (VII). Se pueden usar bases fuertes, por ejemplo terc-butilatos, bases hidruro, bases bis(trimetildisilil)amida (HMDS), bases amidina o bases fosfaceno. En un caso preferido, se usa hidruro de sodio o NaHMDS. La reacción generalmente se lleva a cabo en un solvente, normalmente en un solvente aprótico polar tal como THF, éter dietílico, DMF o una mezcla de solvente aprótico polar
y otros solventes.
El grupo protector PG1 se puede escindir para dar el compuesto (I) de la presente invención. En un caso preferido, PG1 es un grupo terc-butilo y se remueve tratando el compuesto (IX) con una solución de ácido clorhídrico en 1,4 dioxano o con una solución de TFA en diclorometano.
Ejemplos
Lista de abreviaciones
AA Amblyomma americanus (parasitología)
abs absoluto
Ac Acetilo
ac. Acuoso, solución acuosa
AUC Área bajo la curva (en farmacocinética)
cat. Catalítico
CF Ctenocephalides felis (parasitología)
CI Ionización (espectroscopía de masas)
Conc. Concentrado
d Doblete (NMR)
d Día(s)
dd Doblete de doblete (NMR)
DCM Diclorometano
DIPEA N,N,-diisopropiletilamina (base de Hünig)
DMAP 4-Dimetilaminopiridina
DMF N,N-dimetilformamida
DMSO Dimetilsulfóxido
dt Doblete de triplete (NMR)
DV Dermacentor variabilis (parasitología)
ent enantiomérico
eq. equivalente(s)
ESI Ionización por electropulverización (espectroscopía de masas)
F Biodisponibilidad (en farmacocinética)
GC cromatografía de gas
GC/MS espectroscopía de masas acoplada con cromatografía de Gas
h Hora/s
HATU 0-(7-Azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio-hexafluorofosfato
HMDS Bis(trimetildisilil)amida antes denominada también hexametildisilazida;
contraión en sales fuertemente básicas tales como LiHMDS, NaHMDS
KHMDS HPLC Cromatografía líquida de alta presión
iPr Isopropilo
IR Ixodes rhicinus (parasitología)
iv Intravenosa (PK)
KHMDS Bis(trimetildisilil)amida de potasio
LC Cromatografía líquida
LC-MS espectroscopía de masa acoplada con cromatografía líquida
LiHMDS Bis(trimetildisilil)amida de litio
Lit. Bibliografía
m Multiplete (NMR)
Me Metilo
min Minuto/s
EM Espectroscopía de masas
MTBE Terc-butil metil éter
NaHMDS Bis(trimetildisilil)amida de sodio
NMP N-Metil-2-pirrolidona
NMR Espectroscopía de resonancia magnética nuclear
PBS Solución salina tamponada con fosfato
PEG Polietilénglicol
Parámetro Farmacocinética
Pr Propilo
q (o quart) Cuartete (NMR)
qd Cuarteto de doblete (NMR)
cuant. Cuantitativo (se refiere a rendimiento químico)
quint Quinteto (NMR)
rac racémico
RP fase inversa (para cromatografía líquida)
RS Rhipicephalus sanguineus (parasitología)
Rt Tiempo de retención (cromatografía)
s Singlete (NMR)
sc Subcutáneo (PK y farmacología)
SD Día de ensayo
sept Septeto (NMR)
t Triplete (NMR)
t período de tiempo (durante un experimento)
t0 período de tiempo al comienzo de un experimento
TBTU Tetrafluoroborato de 0-(benzotriazol-1-il)-A/,W,W'W-tetrametiluronio
tBu ferc-Butilo
td Triplete de doblete (NMR)
TEA Trietilamina
TFA Ácido trifluoroacético
THF Tetrahidrofurano
UV Espectroscopía ultravioleta
Métodos analíticos
Método 1 (HPLC-MS)
Instrumento: Agilent MS Quad 6150;HPLC: Agilent 1290; Columna: Waters Acquity UPLC HSS T3 1,8 |jm 50 x 2,1 mm; Eluyente A: 1 l de agua 0,25 ml de 99% ácido fórmico, Eluyente B: 1 l de acetonitrilo 0,25 ml 99% ácido fórmico; Gradiente: 0,0 min 90% A ^ 0,3 min 90% A ^ 1,7 min 5% A ^ 3,0 min 5% A temperatura de columna: 50°C; Flujo: 1,20 ml/min; detección UV: 205 -305 nm.
Método 2 (HPLC-MS)
Instrumento: sistema Waters Acquity SQD UPLC; columna: Waters Acquity UPLC HSS T3 1,8 jm 50 x 1 mm; Eluyente A: 1 l de agua 0,25 ml de 99% ácido fórmico, Eluyente B: 1 l acetonitrilo 0,25 ml 99% ácido fórmico, gradiente: 0.0 min 90% A ^ 1.2 min 5% A ^ 2.0 min 5% A temperatura de columna: 50°C; flujo: 0,40 ml/min; detección UV: 210 nm.
Método 3 (HPLC-MS)
Instrumento MS: Thermo Scientific FT-MS; instrumento UHPLC+: Thermo Scientific UltiMate 3000; Columna: Waters, HSST3, 2,1 x 75 mm, C18 1,8 jm; eluyente A: 1 l agua+ 0,01% ácido fórmico, eluyente B: 1 l acetonitrilo 0,01% ácido fórmico, gradiente: 0,0 min 10% B ^ 2,5 min 95% B ^ 3,5 min 95% B; temperatura de columna: 50°C; flujo: 0,90 ml/min; detección UV: 210 nm/ senda de integración óptima 210-300 nm
Método 4 (HPLC-MS)
Instrumento: sistema Waters Acquity SQD UPLC; columna: Waters Acquity UPLC HSS T3 1,8 jm 50 x 1 mm; Eluyente A: 1 l agua+ 0,25 ml 99% ácido fórmico, eluyente B: 1 l acetonitrilo 0,25 ml 99% ácido fórmico, gradiente: 0,0 min 95% A ^ 6,0 min 5% A ^ 7,5 min 5% A temperatura de columna: 50°C; flujo: 0,35 ml/min; detección UV: 2 10 nm.
Método 5 (HPLC-MS)
La columna utilizada fue Shim-pack XR-ODS, 2,2 jm, 3,0 * 50 mm. Se aplicó un gradiente lineal, comenzando a partir de 95 % de A (A: TFA al 0,05% en agua) y finalizando en 100% de B (B: TFA al 0,05% en MeCN) durante 4,70 min con un tiempo de procesamiento total de 5,00 min. La temperatura de la columna se ubicó en 45 °C, con una velocidad de flujo de 1 , 20 ml/min.
Método 6 (HPLC-MS)
La columna utilizada fue Shim-pack XR-ODS, 2,2 jm, 3,0 * 50 mm. Se aplicó un gradiente lineal, comenzando a partir de 95 % de A (A: TFA al 0,05% en agua) y finalizando en 100% de B (B: TFA al 0,05% en MeCN) durante 1,70 min con un tiempo de procesamiento total de 2,00 min. La temperatura de la columna se ubicó en 40 °C, con una velocidad de flujo de 1 , 20 ml/min.
Método 7 (HPLC-MS)
La columna utilizada fue Kinetex EVO C18100A, 2,6 jm, 3,0 * 50 mm. Se aplicó un gradiente lineal, comenzando a partir de 90% de A (A: NH3H2O al 0,03% en agua) finalizando en 95% B (B: acetonitrilo) durante 1,70 min con un
tiempo de procesamiento total de 2,00 min. La temperatura de la columna se ubicó en 40 °C, con una velocidad de flujo de 1,20 ml/min.
Método 8 (HPLC)
Instrumento: HP 1260 Infinity HPLC System con detector de matriz de diodos G4212B; columna: Kromasil 100 C18ec 5 |jm 250 x 4 mm; eluyente A: 1 l agua+ 1,0 ml TFA, eluyente B: 1 l acetonitrilo 1,0 ml TFA; gradiente: 0,0 min 98% A ^ 1,0 min 98% A ^ 8,0 min 30% A ^ 16,0 min 30% A ^ 19,0 min 2% A ^ 20,0 min 2% A ^ 23,0 min 98% A ^ 25,0 min 30% A, temperatura de columna: 37°C; flujo: 1,5 ml/min; detección UV: 214 nm, volumen de inyección 10 jL.
Método 9 (HPLC-MS)
Instrumento: HP 1200 Infinity HPLC System con detector de matriz de diodos G1315B; columna Waters MS QuattroMicro (ESI+/ESI-): Kromasil 100 C18ec 5 jm 250 x 4 mm; eluyente A: 1 l agua+ 0,5 ml 50% HCOOH, eluyente B: 1 l acetonitrilo 0,5 ml 50% HCOOH; gradiente: 0,0 min 98% A ^ 1,0 min 98% A ^ 8,0 min 30% A ^ 16,0 min 30% A ^ 19,0 min 2% A ^ 20,0 min 2% A ^ 23,0 min 98% A ^ 25,0 min 30% A, temperatura de columna: 37°C; flujo: 1,5 ml/min; detección UV: 214 nm, volumen de inyección 10 jL
Síntesis de los materiales de partida
2-Clor-5-{1-[2-clor-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-W-(1-cianociclopropil)benzamida (CAS-RN 1771742-44-9) se sintetizó como se describe en WO 2015/067646 A1.
Ácido (2E)-4-terc-Butoxi-4-oxobut-2-énico (CAS RN 135355-96-3) se sintetizó de acuerdo con el método publicado por P.A. Clarke, R.l. Davie S. Peace, Tetrahedron Lett. 2002, 43, 2753-2756. terc-Butil 3-hidroxi-2,2-dimetilpropanoato (CAS RN 25307-76-0) se preparó como se describe en WO2007/116922 (página 51).
terc-Butil c/s-4-hidroxiciclohexanocarboxilato (CAS-RN 931110-79-1) se preparó de acuerdo con un procedimiento descrito en WO 2009/081195.
terc-Butil trans-4-hidroxiciclohexanocarboxilato (CAS-RN 869193-57-7) se preparó de acuerdo con un procedimiento descrito en 2010126030.
2-Cloro-N-(1-cianociclopropil)-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzamida (CAS-RN 1621436-41-6) se preparó de acuerdo con los procedimientos descritos en WO 2014/122083 A1, WO 2015/078846 A1 y WO 2015/181139 A1.
Síntesis de intermedios
Intermedio 1A.
íerc-Butil-(2E)-4-cloro-4-oxobut-2-enoato

Ácido (2£)-4-terc-Butoxi-4-oxobut-2-énico (172 mg, 1,00 mmol) y DMF (5 jL ) se disolvieron en DCM (2,0 ml) y se enfriaron hasta 0 °C. Se agregó cloruro oxálico (1,0 ml, 2,0 M, 2,0 mmol) gota a gota bajo agitación. Se continuó agitando hasta que terminó la evolución de gas. Luego el solvente se destiló y el residuo se usó sin purificación en el próximo paso. Rendimiento: 191 mg (cuant.).
Intermedio 2A.
ferc-Butil-2-hidroxietil-(2£)-but-2-enodioato

Se disolvió (1,1 ml, 20 mmol) y DIPEA (520 pl, 3,0 mmol) en DCM (2.0 ml) y una solución de ferc-Butil-(2E)-4-cloro-4-oxobut-2-enoato (Intermedio 1A, 191 mg, 1,00 mmol) en DCM (3,0 ml) se agregó lentamente. La mezcla se agitó a temperatura ambiente durante 16 h. Luego, se agregó agua, la mezcla se extrajo con 3 porciones de DCM y los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. La solución se purificó mediante cromatografía en gel de sílice (ciclohexano/acetato de etilo 1:1). Se obtuvo el compuesto del título (166 mg, 77 % de teoría). LC-MS (método 1): Rt = 1,02 min; MS (ESlpos): m/z = 217 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,467 (16,00), 3,607 (0,69), 3,618 (0,75), 4,144 (0,83), 4,157 (0,86), 4,169 (0,75), 4,875 (0,72), 6,690 (1,30), 6,698 (1,32).
Intermedio 3A.
íerc-Butil-2-{[(clorometoxi)carbonil]oxi}etil-(2E)-but-2-enodioato
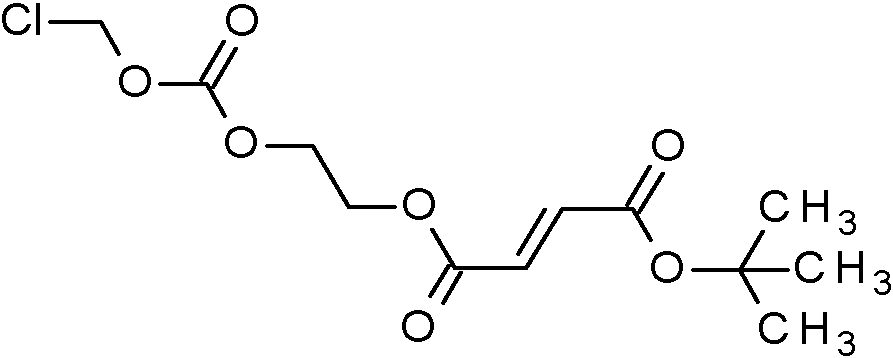
terc-But5l-2-hidroxietil-(2E)-but-2-enodioato (intermedio 2A, 164 mg, 758 pmol) se disolvió en piridina (3,0 ml) y la solución se enfrío hasta 0°C. Se agregó carbonoclorhidrato de clorometilo (100 pl, 1,2 mmol) de una vez y la mezcla se agitó a temperatura ambiente durante 45 min. El solvente se evaporó. El residuo se volvió a disolver en acetato de etilo (100 ml) se lavó con agua (30 ml) y salmuera (30 ml) se secó sobre sulfato de sodio anhidro y se evaporó. El producto bruto se purificó por cromatografía en gel de sílice (ciclohexano / acetato de etilo 3:1) para dar 174 mg (74 % de teoría) del compuesto del título.
LC-MS (método 2): Rt = 0,98 min; MS (ESlpos): m/z = 143 [M+H^H^ClOy]*
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,461 (16,00), 4,402 (0,63), 4,411 (0,64), 4,416 (0,79), 4,452 (0,80), 4,456 (0,64), 4,466 (0,64), 5,806 (3,88), 6,674 (3,58).
Intermedio 4A.
ferc-Butil-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropano-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil-(2E)-but-2-enodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluonTietox¡)feml]-1H-p¡razol-4-¡l}-A/-(1-cianocidopropil)benzamida (162 mg, 250 pmol) se disolvió en THF seco (8,0 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis^rimetilsNi âmida de potasio (800 pl, solución de 0,50 M en tolueno, 400 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (19 mg, 125 pmol) y una solución de terc-buiyl^-^clormetoxOcarboni^ox^etN-^E^but^-enodioato (intermedio 3A, 116 mg, 375 pmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 60 min. La mezcla de reacción se vertió en agua (30 ml) y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10pm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 210 mg (91 % de teoría) del compuesto del título.
LC-MS (método 3): Rt = 2,70 min; MS (ESlpos): m/z = 943 [M+Na]
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,76), 0,008 (0,80), 1,356 (0,69), 1,443 (16,00), 4,308 (0,69), 6,647 (3,15), 7,801 (0,59), 7,827 (0,68), 8,201 (0,73), 8,436 (0,70), 8,765 (0,47).
Intermedio 5A.
íerc-Butil 3-hidroxipropil (2E)-but-2-enodioato

Se disolvió propano-1,3-diol (7,2 ml, 100 mmol) y DIPEA (2,6 ml, 15,0 mmol) en DCM (25 ml) y una solución de terc-Butil-(2E)-4-cloro-4-oxobut-2-enoato (Intermedio 1A, 953 mg, 5,0 mmol) en DCM (15 ml) se agregó lentamente. La mezcla se agitó durante unos pocos minutos y luego se inactivó con agua. La mezcla se extrajo con 3 porciones de DCM y los extractos orgánicos combinados se secaron en sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó mediante cromatografía en gel de sílice (ciclohexano / acetato de etilo 1:1) para dar compuesto del título (800 mg, 69 % de teoría).
LC-MS (método 3): Rt = 1,51 min; MS (ESlpos): m/z = 175 [M+H^Hs]*
iH-NMR (500 MHz, DMSO-d6) 8 [ppm]: 1,464 (16,00), 1,755 (0,65), 1,768 (1,00), 1,781 (0,66), 3,476 (0,80), 3,486 (0,81), 4,197 (0,69), 4,210 (1,44), 4,223 (0,66), 4,529 (0,43), 4,539 (0,89), 4,550 (0,40), 6,658 (2,00), 6,665 (1,89). Intermedio 6A.
terc-Butil 3-{[(clorometoxi)carbonil]oxi}propil-(2E)-but-2-enodioato

terc-Butil 3 hidroxipropil (2E)-but-2-enodioato (intermedio 5A, 800 mg, 3,47 mmol) se disolvió en piridina (13 ml) y la solución se enfrío hasta 0°C. Se agregó carbonoclorhidrato de clorometilo (460 pl, 5,2 mmol) de una vez y la mezcla se agitó a temperatura ambiente durante 45 min. El solvente se evaporó. El residuo se volvió a disolver en acetato de etilo (250 ml) se lavó con agua (30 ml) y salmuera (30 ml) se secó sobre sulfato de sodio anhidro y se evaporó. El producto bruto se purificó por cromatografía en gel de sílice (ciclohexano -> ciclohexano / acetato de etilo 85:15) para dar 1,13 g (cuant.) del compuesto del título.
1H-NMR (500 MHz, DMSO-d6) 8 [ppm]: 1,465 (16,00), 1,761 (1,01), 2,007 (0,54), 2,020 (0,80), 2,032 (0,55), 3,602 (0,83), 4,213 (0,61), 4,225 (1,23), 4,238 (0,58), 4,283 (0,64), 4,296 (1,34), 4,308 (0,61), 5,889 (4,18), 6,673 (4,59). Intermedio 7A.
íerc-Butil 3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]propil (2E)-but-2-enodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (195 mg, 300 pmol) se disolvió en THF seco (9,6 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (960 pl, solución de 0,50 M en tolueno, 480 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (23 mg, 150 pmol) y una solución de terc-butil 3-{[(clorometoxi)carbonil]oxi}propil-(2E)-but-2-enodioato (intermedio 6A, 145 mg, 450 pmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 60 min. La mezcla de reacción se vertió en agua (30 ml) y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 pm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 195 mg (69 % de rendimiento) del compuesto del título.
LC-MS (método 2): Rt = 1,51 min; MS (ESlpos): m/z = 935 [M+H]+
iH-NMR (500 MHz, DMSO-d6) 8 [ppm]: -0,007 (0,89), 0,006 (0,67), 1,356 (0,48), 1,446 (16,00), 6,643 (1,46), 7,825 (0,48), 8,200 (0,54), 8,203 (0,52).
Intermedio 8A.
ferc-Butil 2-hidroxietil butanodioato

2-Hidroxietil (2£)-but-2-enodioato de ferc-butilo (intermedio 2A, 345 mg, 1,60 mmol) se disolvió en etanol (35 ml) bajo una atmósfera de argón. Se agregó paladio sobre carbón al 10 % (84,9 mg, 79,8 |jmol) y la mezcla se hidrogenó durante 30 min a presión ambiente y temperatura ambiente. Luego el catalizador se retiró por filtración sobre una capa de tierra diatomácea y el solvente se destiló. Se obtuvieron 347 mg (cuant.) del compuesto del título.
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,385 (16,00), 2,450 (0,68), 2,455 (0,60), 2,465 (0,86), 2,467 (0,83), 3,532 (0,41), 3,545 (0,87), 3,558 (0,88), 3,571 (0,47), 4,003 (0,86), 4,016 (1,07), 4,029 (0,77), 4,778 (0,68).
Intermedio 9A.
2-{[(Clorometoxi)carbonil]oxi}etil butanodioato de ferc-butilo

terc-Butil 2-hidroxietil butanodioato (intermedio 8A, 170 mg, 779 jmol) se disolvió en piridina (5,0 ml) y la solución se enfrío hasta 0 °C. Se agregó carbonoclorhidrato de clorometilo (100 jl, 1,2 mmol) de una vez y la mezcla se agitó a temperatura ambiente durante 45 min. El solvente se evaporó. El residuo se volvió a disolver en acetato de etilo (100 ml) se lavó con agua (30 ml) y salmuera (30 ml) se secó sobre sulfato de sodio anhidro y se evaporó. El producto bruto se purificó por cromatografía en gel de sílice (gradiente ciclohexano - ciclohexano / acetato de etilo 7:3) para dar 189 mg (78 % de teoría) del compuesto del título.
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,381 (16,00), 2,449 (0,71), 2,455 (0,58), 2,465 (0,79), 2,467 (0,83), 2,516 (0,86), 4,252 (0,57), 4,263 (0,73), 4,269 (0,55), 4,275 (0,87), 4,373 (0,80), 4,378 (0,56), 4,385 (0,74), 4,388 (0,54), 4,395 (0,59), 5,805 (3,70).
Intermedio 10A.
ferc-Butil 2-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil butanodioato

2-Clor-5-{1-[2-dor-4-(1,1,1,2,3,3,3-heptafluorpropan-2-¡l)-6-(tnfluonrietox¡)feml]-1H-p¡razol-4-¡l}-W-(1-cianocidopropil)benzamida (258 mg, 397 jmol) se disolvió en THF seco (13 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis^rimetilsiN âmida de potasio (1,3 ml, solución de 0,50 M en tolueno, 640 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (30 mg, 198 |jmol) y una solución de ferc-butil 2-{[(clorometox¡)carbon¡l]ox¡}et¡l butanodioato (intermedio 9A, 185 mg, 595 jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se vertió en agua (30 ml) y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 272 mg (100 % de pureza, 74 % de rendimiento) del compuesto del título. LC-MS (método 3): Rt = 2,67 min; MS (ESlpos): m/z = 923 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (1,00), 0,008 (0,86), 1,353 (16,00), 1,758 (0,64), 2,073 (0,70), 2,414 (0,82), 2,430 (1,07), 2,459 (0,86), 2,475 (0,81), 2,524 (0,56), 4,184 (0,80), 7,612 (0,44), 7,798 (0,77), 7,826 (0,46), 7,835 (1,11), 8,208 (1,25), 8,444 (1,01), 8,770 (0,84).
Intermedio 11A.
ferc-ButM 3-Hidroxi-2,2-dimetilpropil (2E)-but-2-enodioato

Se disolvió 2,2-dimetilpropano-1,3-diol (3,12 g, 30,0 mmol) y WA-diisopropiletNamina (3,1 ml, 18 mmol) en DCM (30 ml) y una solución de ferc-ButN-(2E)-4-cloro-4-oxobut-2-enoato (Intermedio 1A, 1,14 g, 6,00 mmol) en DCM (15 ml) se agregó lentamente. La mezcla se agitó a temperatura ambiente durante 30 min. Luego, se agregó agua, la mezcla se extrajo con 3 porciones de DCM y los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. La solución se purificó mediante cromatografía en gel de sílice (gradiente ciclohexano -ciclohexano/acetato de etilo 3:1). Se obtuvieron 1,35 g (98 % de pureza, 85 % de rendimiento) del compuesto del título.
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 0,851 (9,22), 1,469 (16,00), 3,189 (1,31), 3,202 (1,33), 3,829 (2,83), 4,641 (0,79), 6,663 (1,87), 6,676 (1,85).
Intermedio 12A.
terc-Butil 3-{[(clorometoxi)carbonil]oxi}-2,2-dimetilpropil (2E)-but-2-enodioato

terc-Butil 3-Hidroxi-2,2-dimetilpropil (2E)-but-2-enodioato (intermedio 11A, 675 mg, 2,61 mmol) se disolvió en piridina (30 ml) y la solución se enfrío hasta 0 °C. Se agregó carbonoclorhidrato de clorometilo (350 pl, 3,9 mmol) de una vez y la mezcla se agitó a temperatura ambiente durante 60 min. El solvente se evaporó. El residuo se volvió a disolver en acetato de etilo (250 ml) se lavó con agua (20 ml) y salmuera (20 ml) se secó sobre sulfato de sodio anhidro y se evaporó. El producto bruto se purificó por cromatografía en gel de sílice (gradiente ciclohexano - ciclohexano / acetato de etilo 8:2). Se aislaron 782 mg (98 % de pureza, 84 % de rendimiento) del compuesto del título.
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 0,862 (8,32), 1,470 (16,00), 3,879 (2,70), 4,073 (2,62), 5,891 (3,77), 6,692 (3,31).
Intermedio 13A.
íerc-Butil 3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropil (2E)-but-2-enodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (301 mg, 463 pmol) se disolvió en THF seco (8 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,2 ml, solución de 0,50 M en tolueno, 600 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (35 mg, 232 pmol) y una solución de terc-butil 3-{[(clorometoxi)carbonil]oxi}-2,2-dimetilpropil (2£)-but-2-enodioato (intermedio 12A, 195 mg, 556 pmol) en THF (4,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se vertió en agua (30 ml) y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-1810 pm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 303 mg (68 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 5,54 min; MS (ESlpos): m/z = 963 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 0,883 (2,25), 1,448 (16,00), 1,464 (0,51), 2,524 (0,83), 3,811 (1,10), 6,656 (1,25), 7,800 (0,60), 7,829 (0,60), 8,203 (0,70), 8,436 (0,53), 8,753 (0,43).
Intermedio 14A.
íerc-Butil 3-{[(clorometoxi)carbonil]oxi}-2,2-dimetilpropil butanodiato

3-{[(dorometoxi)carbonil]oxi}-2,2-dimetilpropil (2£)-but-2-enodioato de ferc-butilo (intermedio 12A, 390 mg, 1,11 mmol) se disolvió en THF (20 ml) bajo una atmósfera de argón. Se agregó paladio sobre carbón al 10 % (87,9 mg, 111 |jmol) y la mezcla se hidrogenó durante 40 min a presión ambiente y temperatura ambiente. Luego el catalizador se retiró por filtración sobre una capa de tierra diatomácea y el solvente se destiló. Se obtuvieron 385 mg (93 % de pureza, 92 % de rendimiento) del compuesto del título.
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 0,824 (8,41), 1,355 (0,62), 1,378 (16,00), 2,459 (0,44), 2,463 (0,69), 2,470 (0,79), 2,478 (1,07), 2,480 (1,14), 2,515 (0,83), 2,523 (0,76), 3,848 (2,59), 4,021 (2,73), 5,896 (3,77).
Intermedio 15A.
íerc-Butil 3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]-2,2-dimetilpropil butanodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (301 mg, 463 jmol) se disolvió en THF seco (8 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,2 ml, solución de 0,50 M en tolueno, 600 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (35 mg, 232 jmol) y ferc-butil 3-{[(clorometoxi)carbonil]oxi}-2,2-dimetilpropil butanodioato (intermedio 14A, 196 mg, 556 jmol) en THF (4,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 90 min. La mezcla de reacción se vertió en agua (30 ml) y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-1810 jm , gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 303 mg (68 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 5,41 min; MS (ESlpos): m/z = 965 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 0,008 (0,62), 0,849 (3,68), 1,352 (16,00), 1,753 (0,56), 2,327 (0,54), 2,442 (0,89), 2,670 (0,56), 3,784 (1,09), 3,866 (0,68), 7,803 (1,05), 7,836 (0,86), 8,213 (1,17), 8,442 (0,88), 8,758 (0,80).
Intermedio 16A.
íerc-Butil (re l 1S,2R)-2-hidrox¡c¡clopentil (2E)-but-2-enodioato

Se disolvió c/s-Ciclopentano-1,2-diol (2,68 g, 26,2 mmol) y A/^-diisopropiletilamina (2,7 ml, 16 mmol) en DCM (25ml). Luego, se agregó gota a gota una solución de ferc-butil (2E)-4-cloro-4-oxobut-2-enoato (intermedio 1A, 1,00 g, 5,25 mmol) en DCM (5 ml) a temperatura ambiente. La mezcla se agitó durante 16 h. La reacción se inactivó con cloruro de amonio acuoso concentrado y se extrajo con 3 porciones de MTBE. Se separó agua por filtración en un dispositivo Chromabond PTS y el solvente se destiló. El residuo se purificó por cromatografía ultrarrápida en gel de sílice (gradiente ciclohexano / acetato de etilo 9:1 - 1:1) y 1,09 g (81 % de rendimiento) del compuesto del título se aisló.
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,469 (16,00), 4,746 (0,81), 4,760 (0,88), 6,680 (1,81), 6,694 (1,82).
Intermedio 17A.
íerc-Butil (rel 1S,2R)-2-{[(clorometoxi)carbonil]oxi}ciclopentil (2E)-but-2-enodioato
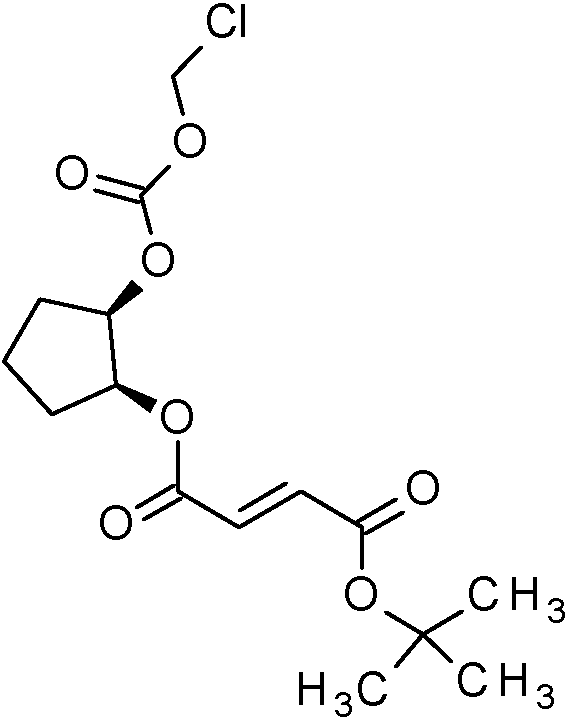
Se disolvió ferc-Butil (rel 1S,2R)-2-h¡drox¡c¡clopent¡l (2£)-but-2-enodioato (intermedio 16A, 700 mg, 2,73 mmol) en piridina (30 ml) y la solución se enfrió hasta 0 °C. S agregó carbonocloridato de clorometilo (364 pl, 4,1 mmol) de una vez y la mezcla se agitó a temperatura ambiente durante 90 min. Luego, la mezcla se vertió en agua, el pH se ajustó en 4 por adición de 1M ácido clorhídrico y la mezcla se extrajo con 3 porciones de DCM. Las capas orgánicas combinadas se lavaron con 1M ácido clorhídrico, agua y salmuera. Se separó agua residual por filtración en dispositivo Chromabond PTS, se destiló el solvente. El compuesto del título (900 mg, 94% de rendimiento) se utilizó sin purificación adicional.
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,453 (16,00), 1,465 (0,48), 1,820 (0,42), 5,220 (0,43), 5,833 (0,76), 5,849 (1,28), 5,893 (1,30), 5,810 (0,77), 6,626 (3,81).
Intermedio 18A.
íerc-Butil (rel 1R,2S)-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi} carbonil)oxi]ciclopentil (2E)-but-2-enodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluonTietox¡)feml]-1H-p¡razol-4-¡l}-A/-(1-cianocidopropil)benzamida (475 mg, 732 jmol) se disolvió en THF seco (8 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis^rimetNsili âmida de potasio (2,3 ml, solución de 0,50 M en tolueno, 1,2 mmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (55 mg, 366 jmol) y fere-butil (rel IS^R^-^clorometoxOcarbom^ox^dclopentN (2£)-but-2-enodioato (intermedio 17A, 383 mg, 1,10 mmol) en THF (4,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 4 h. La mezcla de reacción se vertió en solución de cloruro de amonio acuoso concentrado y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron pasándolos en dispositivo Chromabond PTD y se evaporaron. El producto bruto se purificó por cromatografía ultrarrápida (gel de sílice, gradiente DCM - DCM/ acetato de etilo 1:1) y posteriormente HPLC preparativa (RP C-18 10|jt , gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 460 mg (100 % de pureza, 65 % de rendimiento) del compuesto del título.
LC-MS (método 3): Rt = 2,79 min; MS (ESlneg): m/z = 1005 [M-H+CHOOH]-1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,66), 0,008 (0,58), 1,437 (16,00), 1,553 (0,48), 1,740 (1,41), 1,858 (0,49), 5,117 (0,49), 6,608 (2,79), 7,590 (0,53), 7,788 (1,06), 7,818 (0,61), 7,835 (1,45), 8,210 (1,58), 8,427 (1,74), 8,739 (0,89).
Intermedio 19A.
íerc-Butil (rel 1S,2R)-2-{[(clorometoxi)carbonil]oxi}cidopentil butanodioato

(rel 1S,2R)-2-{[(doroTetox¡)carbon¡l]ox¡}-ddopent¡l (2£)-but-2-enodioato de tere-butilo (intermedio 17A, 450 mg, 1,29 mmol) se disolvió en THF (20 ml) bajo una atmósfera de argón. Se agregó paladio sobre carbón al 10 % (28 mg, 26 jmol) y la mezcla se hidrogenó durante 16 h a presión ambiente y temperatura ambiente. Luego el catalizador se retiró por filtración sobre una capa de tierra diatomácea y el solvente se destiló. Se aislaron 430 mg (95 % de rendimiento) del compuesto del título.
1H-NMR (400 MHz, DMSO-d6) 5 [ppm]: 1,381 (16,00), 2,474 (0,49), 5,071 (0,45), 5,883 (1,66), 5,891 (1,67).
Intermedio 20A.
íerc-Butil (rel 1R,2S)-2-[({[(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]ciclopentil butanodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianocidopropil)benzamida (531 mg, 817 |jmol) se disolvió en THF seco (8 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (2,6 ml, solución de 0,50 M en tolueno, 1,3 mmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (61 mg, 409 pmol) y ferc-butil (rel 1S,2R)-2-{[(clorometoxi)carbonil]oxi}ciclopentil butenodioato (intermedio 19A, 430 mg, 1,23 mmol) en THF (4,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 4 h. La mezcla de reacción se vertió en solución de cloruro de amonio acuoso concentrado y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron pasándolos en dispositivo Chromabond PTD y se evaporaron. El producto bruto se purificó por cromatografía ultrarrápida (gel de sílice, gradiente DCM - DCM/ acetato de etilo 1:1) y posteriormente HPLC preparativa (RP C-1810pm, gradiente de aguaacetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 520 mg (98 % de pureza, 64 % de rendimiento) del compuesto del título.
LC-MS (método 3): Rt = 2,76 min; MS (ESlpos): m/z = 963 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 5 [ppm]: 1,333 (16,00), 1,356 (4,31), 1,507 (0,70), 1,757 (1,39), 1,898 (0,71), 2,184 (0,44), 2,367 (2,00), 4,885 (0,53), 4,898 (0,61), 7,587 (0,66), 7,608 (0,79), 7,805 (2,03), 7,824 (1,01), 7,837 (2,06), 8,212 (2,29), 8,445 (1,64), 8,756 (1,57).
Intermedio 21A.
íerc-Butil 3-[2-(benciloxi)etoxi]-2,2-dimetilpropanoato

Se disolvió ferc-Butil 3-hidroxi-2,2-dimetilpropanoato (CAS RN 25307-76-0, 871 mg, 5,00 mmol) en THF (25 ml) y se enfrió hasta -20 °C. Se agregó en pequeñas porciones bis(trimetilsilil)amida de sodio (3,0 ml, 2,0 M en THF, 6,0 mmol) y la mezcla se agitó a -20 °C durante 30 min. Luego, se agregó [(2-yodoetoxi)metil]benceno (3,93 g, 15,0 mmol) y la mezcla primero se agitó a temperatura ambiente durante la noche y luego se calentó hasta reflujo durante 16 h. La mezcla de reacción se vertió en agua, se extrajo con acetato de etilo y la capa orgánica se secó sobre sulfato de sodio anhidro. El solvente se destiló y el residuo se purificó mediante cromatografía en gel de sílice (gradiente DCM - DCM /acetato de etilo 9:1). Se obtuvieron 305 mg (20 % de rendimiento) del compuesto del título.
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,054 (9,88), 1,361 (16,00), 3,378 (3,21), 3,541 (3,84), 4,490 (2,46), 7,316 (1,28), 7,321 (1,60), 7,337 (0,52), 7,339 (0,59).
Intermedio 22A.
ferc-Butil 3-(2-hidroxietoxi)-2,2-dimetilpropanoato

Se disolvió ferc-Butil 3-[2-(benciloxi)etoxi]-2,2-dimetilpropanoato (intermedio 21A, 303 mg, 982 jmol) en ácido acético glaciar (15 ml), se agregó 10% paladio sobre carbón (105 mg, 98,2 |jmol) y la mezcla se hidrogenó a presión ambiente y temperatura ambiente durante 3 h. Se agregó otra porción de 10% paladio sobre carbón (105 mg, 98,2 jmol) y la hidrogenación continuó durante otras 3 h. El catalizador se retiró por filtración y el solvente se destiló. El producto bruto se disolvió en DCM, se lavó con solución concentrada de carbonato de sodio acuoso, se secó sobre sulfato de sodio anhidro y se evaporó nuevamente. Se obtuvieron 198 mg (92 % de rendimiento) del compuesto del título.
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,047 (9,81), 1,380 (16,00), 3,362 (3,22), 3,379 (0,40), 3,392 (1,07), 3,405 (0,84), 3,449 (0,48), 3,461 (0,85), 3,475 (0,70), 4,483 (0,45), 4,496 (0,80), 4,509 (0,41).
Intermedio 23A.
ferc-Butil 3-(2-{[(clorometoxi)carbonil]oxi}etoxi)-2,2-dimetilpropanoato

Se disolvió ferc-Butil 3-(2-hidroxietoxi)-2,2-dimetilpropanoato (intermedio 22A, 95 mg, 435 jmol) en piridina (5,0 ml) bajo una atmósfera de argón y se enfrió hasta 0 °C. Se agregó carbonocloridato de clorometilo (58 jl, 650 jmol) de una vez y la mezcla se agitó a temperatura ambiente durante 45 min. El solvente se destiló, el residuo se absorbió en acetato de etilo (50 ml), se lavó consecutivamente con agua, y salmuera, se secó sobre sulfato de sodio anhidro y se evaporó. El producto bruto se purificó mediante cromatografía en gel de sílice (gradiente ciclohexano -ciclohexano acetato ciclohexano 82,5:17,5) para dar 103 mg (76 % de teoría) del compuesto del título.
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,040 (9,89), 1,374 (16,00), 3,377 (3,16), 3,607 (0,73), 3,615 (0,58), 3,619 (0,81), 3,623 (0,61), 3,630 (0,79), 4,273 (0,79), 4,279 (0,60), 4,284 (0,81), 4,288 (0,62), 4,295 (0,75), 5,893 (4,01). Intermedio 24A.
ferc-Butil 1-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-11,11-dimetil-1,5-dioxo-4,6,9-trioxa-2-azadodecan-12-oato

2-Cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-N-(1-cianocidopropil)benzamida (179 mg, 276 |jmol) se disolvió en THF seco (8 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (720 jL, solución de 0,50 M en tolueno, 331 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (21 mg, 138 jmol) y ferc-butil 3-(2-{[(clorometoxi)carbonil]oxi}etoxi)-2,2-dimetilpropanoato (intermedio 23A, 103 mg, 331 jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 90 min. La mezcla de reacción se vertió en agua y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron en sulfato de sodio anhidro. El producto bruto se purificó por cromatografía ultrarrápida (gel de sílice, gradiente DCM - DCM/ acetato de etilo 1:1) y posteriormente HPLC preparativa (RP C-18 10jm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 200 mg (78 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 5,47 min; MS (ESlpos): m/z = 923 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (1,22), 0,008 (1,08), 0,897 (11,12), 1,235 (0,47), 1,331 (16,00), 1,754 (1,15), 2,328 (0,74), 2,332 (0,55), 2,366 (0,48), 2,523 (1,67), 2,670 (0,78), 2,710 (0,50), 3,326 (2,89), 3,535 (1,49), 4,113 (0,84), 7,590 (0,68), 7,612 (0,81), 7,780 (1,11), 7,804 (0,81), 7,825 (0,81), 7,836 (1,89), 8,210 (2,27), 8,440 (1,72), 8,763 (1,49).
Intermedio 25A.
Ácido 4-Etoxi-2,2-dimetol-4-oxobutanoico
Una solución de 3,3-dimetil-dihidrofuran-2,5-diona, 70,0 g (0,55 mol) en etanol (400 ml) se agitó a 50 °C durante la noche. Después de enfriarse hasta temperatura ambiente, el solvente se retiró al vacío y el residuo se trituró con hexano a -50 °C. El sólido precipitado se recogió para dar el compuesto del título (60,0 g, 63% de teoría) como un sólido blanco.
LC-MS (método 6): Rt = 0,89 min, MS (ESlpos): m/z = 175 [M+H]+.
Intermedio 26A.
1-íerc-Butil 4-etil 2,2-dimetilsuccinato

Se disolvió ácido 4-Etoxi-2,2-dimetil-4-oxobutanoico (intermedio 25A,110 g, 0,63 mol), sulfato de magnesio (304 g, 2,53 mol), y ferc-butanol (330 ml) en DCM (2,6 l) Se agregó lentamente gota a gota ácido sulfúrico (93,0 g (0,95 mol), a 0 °C. La mezcla resultante se agitó a temperatura ambiente durante la noche. La mezcla de reacción se vertió en una solución saturada acuosa de bicarbonato de sodio. Se agregó DCM para extraer el producto deseado y la fase orgánica se lavó con salmuera, se secó sobre sulfato de sodio anhidro, y se concentró para dar 70,0 g (37%) del producto como aceite incoloro.
LC-MS (método 7): Rt = 1,26 min, MS (ESIpos): m/z = 253 [M+Na]+.
Intermedio 27A.
Ácido 4-íerc-butoxi-3,3-dimetil-4-oxobutanoico

A una solución de 1-ferc-butil 4-etil 2,2-dimetilsuccinato, (intermedio 26A, 70,0 g, 0,3 mol) en etanol (1,4 l) y agua (700 ml), se agregó hidróxido de potasio (85,3 g, 1,52 mol) a temperatura ambiente. Se agitó la mezcla de reacción a temperatura ambiente durante 2 horas. El pH de la mezcla de reacción se ajustó hasta 3-4 agregando ácido clorhídrico 1M. La solución resultante se extrajo con éter 3 veces, y las fases de éter combinadas se secaron sobre sulfato de sodio y se concentraron para dar el compuesto del título (37,0 g, 68% de teoría) como un sólido amarillo. LC-MS (método 7): Rt =0,36 min, MS (ESIpos): m/z = 201 [M-H]
Intermedio 28A.
1-íerc-Butil 4-(2-hidroxietil)2,2-dimetilsuccinato;

Una mezcla de ácido 4-terc-butoxi-3,3-dimetil-4-oxobutanoico, (intermedio 27A, 35,0 g 173,1 mmol) y carbonato de potasio, 120 g (0,87 mol) en DMF (1,0 l) se agitó a temperatura ambiente durante 1 hora. Se agregó 2-bromoetanol, 42,90 g (346,1 mmol), y la mezcla resultante se agitó durante la noche. El solvente se retiró al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice (hexano/ferc-butil metil éter 1:1) para proporcionar el compuesto del título (22,6 g, 52% de teoría) como un aceite amarillo.
LC-MS (método 5): Rt = 1,39 min, MS (ESIpos): m/z = 269 [M+Na]+.
1H-NMR (400 MHz, CD3OD): 8 [ppm] = 1,21 (s, 6H), 1,43 (s, 9H), 2,59 (s, 2H), 3,73 (t, 2H), 4,11 (t, 2H).
Intermedio 29A.
1-íerc-Butil 4-(2-{[(clorometoxi)carbonil]oxi}etil)2,2-dimetilbutanodioato

Se disolvió 1-terc-Butil 4-(2-hidroxietil)2,2-dimetilbutanodioato (intermedio 28A, 246 mg, 1,00 mmol) en piridina (5,0 ml) bajo una atmósfera de argón y se enfrió hasta 0 °C. Se agregó carbonocloridato de clorometilo (130 pl, 1,5 mmol) de una vez y la mezcla se agitó a temperatura ambiente durante 45 min. El solvente se destiló, el residuo se absorbió en acetato de etilo (50 ml), se lavó consecutivamente con agua, y salmuera, se secó sobre sulfato de sodio anhidro y se evaporó. El producto bruto se purificó mediante cromatografía en gel de sílice (gradiente ciclohexano -ciclohexano acetato ciclohexano 3:1) para dar 228 mg (66 % de rendimiento) del compuesto del título.
Intermedio 30A.
1-íerc-Butil 4-{2-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil} 2,2-dimetilbutanodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-N-(1-cianociclopropil)benzamida (285 mg, 439 pmol) se disolvió en THF seco (10 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,4 ml, solución de 0,50 M en tolueno, 700 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (33 mg, 220 pmol) y 1-íerc-butil 4-(2-{[(clorometoxi)carbonil]oxi}etil)2,2-dimetilbutanodiato (intermedio 29A, 227 mg, 98% de pureza, 659 pmol) en THF (4,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 90 min. La mezcla de reacción se vertió en agua y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron en sulfato de sodio anhidro. El producto bruto se purificó por HPLC preparativa (RP C-18 10 pm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 318 mg (100 % de pureza, 76 % de rendimiento) del compuesto del título.
LC-MS (método 3): Rt = 2,74 min; MS (ESlpos): m/z = 951 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,48), 0,008 (0,44), 1,098 (10,35), 1,326 (16,00), 1,356 (3,74), 1,756 (0,89), 2,073 (0,53), 2,183 (0,41), 4,168 (1,32), 4,197 (0,89), 7,592 (0,58), 7,611 (0,67), 7,788 (1,00), 7,806 (0,85), 7,827 (0,69), 7,835 (1,73), 8,208 (1,88), 8,438 (1,58), 8,767 (1,29).
Intermedio 31A.
íerc-Butil (1S,2S)-2-hidroxiciclopentil (2£)-but-2-enodioato

Se disolvió (1S,2S)-Ciclopentano-1,2-diol (2,68 g, 26,2 mmol) y W,A/-dMsoprop¡letilam¡na (2,7 ml, 16 mmol) en DCM (25ml). Luego, se agregó gota a gota una solución de ferc-butil (2E)-4-cloro-4-oxobut-2-enoato (intermedio 1A, 1,00 g, 5,25 mmol) en DCM (5 ml) a temperatura ambiente. La mezcla se dejó agitando durante 16 h. La reacción se inactivó con cloruro de amonio acuoso concentrado y se extrajo con 3 porciones de MTBE. Se separó agua por filtración en un dispositivo Chromabond PTS y el solvente se destiló. El residuo se purificó por cromatografía ultrarrápida en gel de sílice (gradiente ciclohexano / acetato de etilo 9:1 - 1:1) y 270 mg (20 % de rendimiento) del compuesto del título se aisló.
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,460 (16,00), 4,857 (0,84), 4,867 (0,81), 6,638 (4,27).
Intermedio 32A.
íerc-Butil (1S,2S)-2-{[(clorometoxi)carbonil]oxi}ciclopentil (2E)-but-2-enodioato

terc-Butil (1S,2S)-2-h¡drox¡c¡clopent¡l (2£)-but-2-enodioato (intermedio 31A, 275 mg, 1,07 mmol) se disolvió en piridina (10 ml) y la solución se enfrío hasta 0°C. Se agregó carbonoclorhidrato de clorometilo (190 pl, 2,1 mmol) gota a gota y la mezcla se agitó a temperatura ambiente durante la noche. Luego, la mezcla se vertió en agua, el pH se ajustó a 4 agregando 1M ácido clorhídrico y la mezcla se extrajo con 3 porciones de DCM. Las capas orgánicas combinadas se lavaron con 1M ácido clorhídrico, agua y salmuera. Se separó agua residual por filtración en dispositivo Chromabond PTS, se destiló el solvente. El compuesto del título (244 mg, 65 % de rendimiento) se utilizó sin purificación adicional.
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,464 (16,00), 1,734 (0,55), 1,744 (0,63), 1,753 (0,41), 5,890 (3,40), 6,666 (1,88), 6,677 (1,86).
Intermedio 33A.
íerc-Butil (1S,2S)-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi} carbonil)oxi]ciclopentil (2£)-but-2-enodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluonTietox¡)feml]-1H-p¡razol-4-¡l}-A/-(1-cianocidopropil)benzamida (199 mg, 308 jmol) se disolvió en THF seco (8 ml). Esta solución se enfrió a -45°C en un baño de acetona - hielo seco. Bis^nmetilsNi âmida de potasio (800 jL, solución de 0,50 M en tolueno, 400 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (23 mg, 154 jmol) y tere-butil (IS^S^-^clorometoxOcarboni^ox^ddopentN (2£)-but-2-enodioato (intermedio 32A, 118 mg, 338 jmol) en THF (4,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 4 h. La mezcla de reacción se vertió en solución de cloruro de amonio acuoso concentrado y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron pasándolos en dispositivo Chromabond PTD y se evaporaron. El producto bruto se purificó por cromatografía ultrarrápida (gel de sílice, gradiente DCM - DCM/ acetato de etilo 1:1) y posteriormente HPLC preparativa (RP C-1810jm, gradiente de aguaacetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 120 mg (41% de rendimiento) del compuesto del título.
LC-MS (método 3): Rt = 2,83 min; MS (ESlpos): m/z = 961 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,235 (0,66), 1,443 (16,00), 1,681 (0,41), 1,751 (0,43), 2,521 (0,65), 6,621 (1,79), 6,631 (1,85), 7,798 (0,73), 7,829 (0,62), 8,203 (0,66), 8,436 (0,70), 8,761 (0,47).
Intermedio 34A.
íerc-Butil (1S,2S)-2-{[(dorometoxi)carbonil]oxi}cidopentM butanodioato

(1S,2S)-2-{[(doroTetox¡)carbon¡l]ox¡}-ddopent¡l (2E)-but-2-enodioato de tere-butilo (intermedio 32A, 130 mg, 373 jmol) se disolvió en THF (5 ml) y se agregó 10% paladio sobre carbón (39,7 mg, 10 % de pureza, 37,3 jmol). La mezcla se hidrogenó a temperatura ambiente a presión ambiente durante la noche. Luego, el catalizador se retiró por filtración y el filtrado se evaporó. El compuesto del título (124 mg, 95 % de rendimiento) se utilizó sin purificación adicional.
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,356 (0,45), 1,383 (16,00), 1,700 (0,43), 1,714 (0,69), 1,721 (0,41), 1,727 (0,58), 2,452 (0,51), 2,456 (0,55), 2,463 (0,86), 2,468 (0,72), 2,483 (0,82), 5,886 (3,56).
Intermedio 35A.
íerc-Butil (1S,2S)-2-[({[(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]ciclopentil butanodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluonTietox¡)feml]-1H-p¡razol-4-¡l}-A/-(1-cianocidopropil)benzamida (203 mg, 313 jmol) se disolvió en THF seco (8 ml). Esta solución se enfrió a -45°C en un baño de acetona - hielo seco. Bis^rimetNsiNOamida de potasio (813 jL, solución de 0,50 M en tolueno, 407 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (23 mg, 154 jmol) y ferc-butil (1S,2S)-2-{[(cloroTetox¡)carbon¡l]ox¡}c¡clopent¡l butenodioato (intermedio 34A, 120 mg, 344 jmol) en THF (4,0 ml) en THF (3 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 4 h. La mezcla de reacción se vertió en solución de cloruro de amonio acuoso concentrado y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron pasándolos en dispositivo Chromabond PTD y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 100 mg (33% de rendimiento) del compuesto del título.
LC-MS (método 3): Rt = 2,78 min; MS (ESlpos): m/z = 963 [M+H]+
1H-NMR (500 MHz, DMSO-d6) 8 [ppm]: -0,022 (0,79), 1,235 (1,58), 1,354 (16,00), 1,650 (0,71), 1,753 (0,79), 1,863 (0,39), 2,424 (1,58), 2,435 (1,50), 4,855 (0,47), 7,586 (0,47), 7,603 (0,55), 7,799 (1,10), 7,819 (0,55), 7,834 (1,34), 8,208 (1,50), 8,211 (1,50), 8,442 (1,10), 8,760 (0,85).
Intermedio 36A.
íerc-Butil 2-hidroxietil pentanodioato

Se disolvió etano-1,2-diol (5,3 ml, 96 mmol) en DCM (10ml) y se agregó WA-diisopropiletilamina (5,0 ml, 29 mmol). Luego, una solución de ferc-butil 5-cloro-5-oxopentanoato (CAS RN 2011712-15-3, 1,98 g, 9,58 mmol) en DCM (5 ml) se agregó bajo enfriamiento con un baño de hielo. La reacción se dejó agitar durante la noche, luego se vertió en agua y el pH se ajustó a 5-6 con 1M ácido clorhídrico. La mezcla se extrajo con 2 porciones de DCM, los extractos orgánicos combinados se lavaron brevemente con carbonato de hidrógeno sódico acuoso concentrado, se secaron pasándolos por un dispositivo Chromabond PTS y se evaporaron. El residuo se purificó mediante cromatografía en gel de sílice (gradiente ciclohexano / acetato de etilo 80:20 - 50:50) para dar 670 mg (30 % de rendimiento) del compuesto del título.
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,395 (16,00), 1,706 (0,61), 1,725 (0,82), 1,743 (0,66), 2,215 (0,78), 2,234 (1,37), 2,252 (0,67), 2,308 (0,75), 2,327 (1,33), 2,345 (0,67), 3,546 (0,79), 3,559 (0,80), 3,572 (0,46), 4,008 (0,83), 4,021 (0,87), 4,034 (0,75), 4,772 (0,78).
Intermedio 37A.
2-{[(Clorometoxi)carbonil]oxi}etil pentanodioato de ferc-butilo

tere-Butil 2-hidroxietil pentanodioato (intermedio 36A, 640 mg, 2,76 mmol) se disolvió en piridina (10 ml), se agregó carbonoclorhidrato de clorometilo (490 pl, 5,5 mmol) gota a gota y la mezcla se agitó a temperatura ambiente durante la noche. Luego, la mezcla se vertió en 1M ácido clorhídrico y la mezcla se extrajo con 3 porciones de DCM. Las capas orgánicas combinadas se lavaron con 1M ácido clorhídrico, agua y solución de carbonato de hidrógeno sódico acuosa concentrada, se secaron sobre sulfato de magnesio y se evaporaron. El compuesto del título (648 mg, 72 % de rendimiento) se utilizó sin purificación adicional.
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,392 (16,00), 1,701 (0,62), 1,720 (1,00), 1,738 (0,77), 2,210 (0,76), 2,229 (1,45), 2,247 (0,80), 2,322 (0,72), 2,340 (1,29), 2,358 (0,66), 4,256 (0,53), 4,267 (0,76), 4,272 (0,57), 4,278 (0,86), 4,383 (0,78), 4,389 (0,50), 4,394 (0,67), 4,405 (0,52), 5,801 (3,42).
Intermedio 38A.
íerc-Butil 2-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil pentanodioato
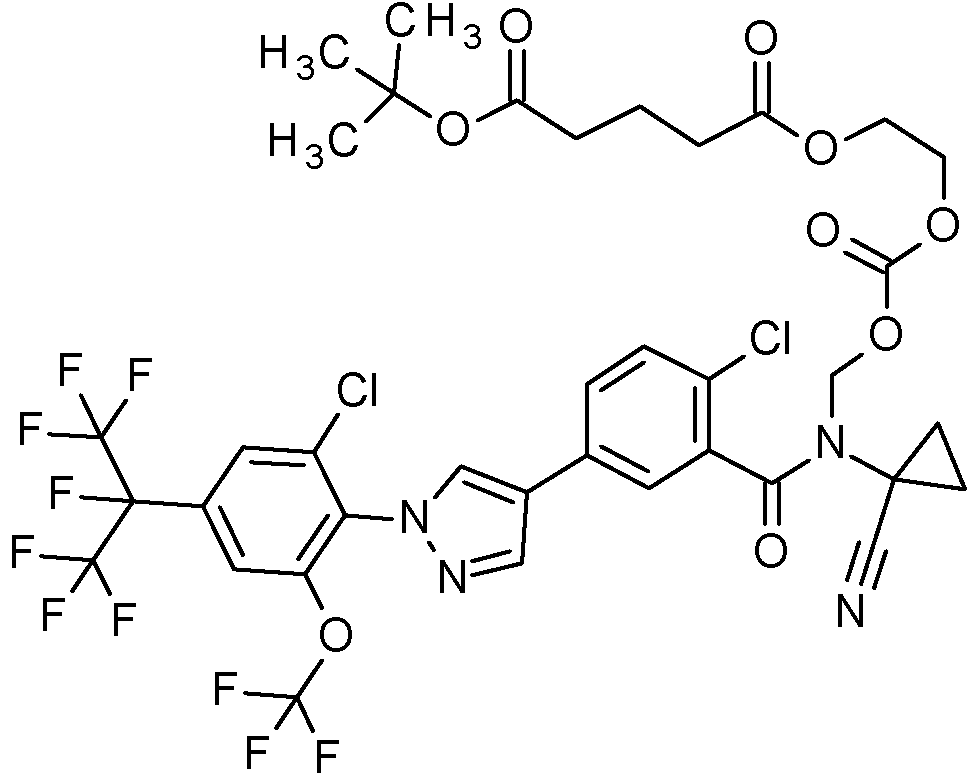
2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (300 mg, 462 pmol) se disolvió en THF seco (5 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,2 ml, solución de 0,50 M en tolueno, 600 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (35 mg, 231 pmol) y 2-{[(clorometoxi)carbonil]oxi}etil pentanodioato de tere-butilo (intermedio 37A, 180 mg, 554 pmol) en THF (3 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante la noche. La mezcla de reacción se vertió en solución de cloruro de amonio acuoso concentrado y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron pasándolos en dispositivo Chromabond PTD y se evaporaron. El producto bruto se purificó por HPLC preparativa (Rp C-18 10 pm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvo 155 mg (36 % de rendimiento).
LC-MS (método 3): Rt = 2,69 min; MS (ESlpos): m/z =937 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,356 (0,65), 1,369 (16,00), 1,669 (0,42), 1,687 (0,60), 1,705 (0,46), 2,178 (0,46), 2,196 (0,83), 2,215 (0,41), 2,296 (0,54), 4,180 (0,46), 7,834 (0,61), 8,207 (0,68), 8,440 (0,54), 8,767 (0,43).
Intermedio 39A.
1-íerc-Butil 1-(2-hidroxietil)ciclopropano-1,1-dicarboxilato

Se disolvió ácido 1-(ferc-Butoxicarbonil)ciclopropano-1-carboxílico (600 mg, 3,22 mmol) en DCM (6,0 ml) y se agregó una gota de DMF. La solución se enfrió hasta 0°C y se agregó gota a gota dicloruro oxálico (560 jL, 6,4 mmol). El baño de enfriamiento se retiró y la reacción se dejó agitar a temperatura ambiente hasta que cesó la evolución de gas. Las sustancias volátiles se retiraron a temperatura ambiente a presión reducida. El cloruro ácido resultante se disolvió en DCM (5 ml) y se agregó a una solución de etano-1,2-diol (1,8 ml, 32 mmol) y A/,W-diisopropiletilamina (1,7 ml, 9,7 mmol) en DCM. La mezcla se dejó agitar durante la noche a temperatura ambiente y luego se vertió en agua y el pH se ajustó a 6-6 con adición de 1M ácido clorhídrico. Esta mezcla se extrajo con 2 porciones de DCM, los extractos orgánicos combinados se lavaron con solución carbonato de hidrógeno sódico acuoso concentrado, se pasaron por un dispositivo Chromabond PTS y se evaporaron. El producto bruto se purificó mediante cromatografía en gel de sílice (gradiente ciclohexano / acetato de etilo 80:20 - 50:50) para dar 450 mg (61 % de rendimiento) del compuesto del título.
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,252 (0,80), 1,260 (1,68), 1,266 (0,81), 1,281 (0,85), 1,286 (1,65), 1,295 (0,71), 1,408 (16,00), 3,554 (0,44), 3,567 (0,85), 3,580 (1,02), 3,594 (0,46), 4,053 (0,84), 4,066 (1,13), 4,079 (0,81), 4,768 (0,43), 4,781 (0,85).
Intermedio 40A.
1-íerc-Butil 1-(2-{[(clorometoxi)carbonil]oxi}etil)cidopropano-1,1-dicarboxilato

1-terc-Butil 1 -(2-hidroxietil) ciclopropano-1,1-dicarboxilato (intermedio 39A, 445 mg, 1,93 mmol) se disolvió en piridina (5,0 ml), se agregó carbonoclorhidrato de clorometilo (340 jl, 3,9 mmol) gota a gota y la mezcla se agitó a temperatura ambiente durante la noche. Luego, la mezcla se vertió en 1M ácido clorhídrico y la mezcla resultante se extrajo con 3 porciones de DCM. Las capas orgánicas combinadas se lavaron con 1M ácido clorhídrico, agua y solución de carbonato de hidrógeno sódico acuosa concentrada, se secaron sobre sulfato de magnesio y se evaporaron. El compuesto del título (648 mg, 72 % de rendimiento) se utilizó sin purificación adicional.
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,284 (3,08), 1,288 (2,88), 1,297 (0,63), 1,397 (16,00), 1,420 (1,60), 4,312 (0,60), 4,319 (0,70), 4,322 (0,77), 4,333 (0,87), 4,394 (0,84), 4,398 (0,71), 4,405 (0,74), 4,415 (0,53), 5,811 (3,59). Intermedio 41A.
1-íerc-Butil 1-{2-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil} ciclopropano -1,1-dicarboxilato

2-Cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometox¡)feml]-1H-p¡razol-4-¡l}-A/-(1-cianocidopropil)benzamida (630 mg, 971 pmol) se disolvió en THF seco (20 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis^nmetilsiliOamida de potasio (2,5 ml, solución de 0,50 M en tolueno, 1,3 mmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (73 mg, 485 pmol) y ^^-{[(clorometoxOcarbom^ox êtN) c¡dopropano-1,1-d¡carbox¡lato de 1-terc-butilo (intermedio 40A, 470 mg, 1,46 mmol) en THF (3 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante la noche. La mezcla de reacción se vertió en agua y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de magnesio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 pm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 570 mg (63 % de rendimiento) del compuesto del título.
LC-MS (método 3): Rt = 2,70 min; MS (ESlpos): m/z = 935 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,245 (5,40), 1,324 (0,81), 1,356 (16,00), 1,758 (1,11), 4,235 (2,81), 7,592 (0,58), 7,612 (0,73), 7,791 (1,07), 7,807 (0,85), 7,829 (0,76), 7,835 (1,80), 8,209 (2,10), 8,213 (2,09), 8,443 (1,47), 8,766 (1,30).
Intermedio 42A.
ferc-ButN-4-hidroxicidohexN (2E)-but-2-enodioato (Mezcla de diasterómeros)

Se suspendió Ciclohexano-1,4-diol (mezcla de diastereómeros cis y trans, 23,23 g, 200 mmol) y N,N-diisopropiletilamina (5,23 ml, 30 mmol) en THF (100 ml). Se agregó lentamente una solución de terc-butil (2E)-4-cloro-4-oxobut-2-enoato (intermedio 1A, 1,91 g, 10,0 mmol) en THF (20 ml). Finalmente, se inactivó la reacción mediante adición de agua y se extrajo con 3 porciones de DCM. Los extractos orgánicos combinado se secaron sobre sulfato de sodio, se evaporaron y purificaron por cromatografía ultrarrápida sobre gel de sílice (gradiente ciclohexano - ciclohexano / acetato de etilo 3:2). El producto resultante (1,60 g, 59% de teoría) se obtuvo como una mezcla de isómeros.
LC-MS (método 2): Rt = 0,89 min (58 % de área) y 0,91 min (42 % de área); MS (ESlpos): m/z = 288 [M+NH4]+ Intermedio 43A.
íerc-Butil c/s-4-hidroxiciclohexil (2£)-but-2-enodioato

La mezcla del producto de intermedio 42A (1,60 g, 5,92 mmol) se presentó a SFC preparativa para separar los
diastereómeros. Condiciones de SFC: columna Chirapak AD-H (SFC) 5 |jm, 250x30 mm; eluyente: dióxido de carbono / etanol 7:3; presión: 135 bar; temperatura del eluyente: 38 °C, temperatura de ciclón 40 °C; flujo: 125 ml/min; detección UV a 210 nm.
El primer pico (Rt = 1,02 min) se recogió y evaporó. Este material se disolvió en DCM, se filtró sobre una capa de tierra diatomácea y finalmente se evaporó. Rendimiento: 845 mg del compuesto del título. La estereoquímica se asignó por comparación con el (1r,4r)-isómero auténtico preparado a partir de trans-ciclohexano-1,4-diol puro (intermedio 44A).
LC-MS (método 4): Rt = 2,68 min; MS (ESlpos): m/z = 288 [M+NH4]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,467 (16,00), 1,549 (0,57), 1,559 (0,73), 1,580 (0,45), 1,590 (0,58), 4,513 (0,84), 4,523 (0,83), 6,650 (2,12), 6,655 (2,11).
Intermedio 44A.
ferc-ButM trans-4-hidroxiddohexil (2E)-but-2-enodioato

De la separación por SFC preparativa descrita en el intermedio 43A, se recogió el segundo pico (Rt = 2,09 min). Este material se disolvió en DCM, se filtró sobre una capa de tierra diatomácea y finalmente se evaporó. Rendimiento: 365 mg del compuesto del título.
La estereoquímica se asignó por comparación con el material preparado independientemente usando trans-1,4-ciclohexanediol comercialmente disponible (CAS RN 6995-79-5):
Se disolvió trans-ciclohexno-1,4-diol (CAS Rn 6995-79-5, 1,00 g, 8,61 mmol) y W,W-diisopropiletilamina (900 jL, 5,2 mmol) en THF (20 ml) y una solución de terc-Butil-(2E)-4-cloro-4-oxobut-2-enoato (Intermedio 1A, 328 mg, 1,72 mmol) en THF (5 ml) se agregó lentamente. Finalmente, se inactivó la reacción mediante adición de agua y se extrajo con 3 porciones de DCM. Los extractos orgánicos combinado se secaron sobre sulfato de sodio, se evaporaron y purificaron por cromatografía ultrarrápida sobre gel de sílice (gradiente ciclohexano - ciclohexano / acetato de etilo 1:1). Se aislaron 214 mg (46 % de rendimiento).
LC-MS (método 4): Rt = 2,63 min; MS (ESlpos): m/z = 288 [M+NH4]+
iH-NMR (500 MHz, DMSO-d6) 8 [ppm]: 1,280 (0,56), 1,303 (0,62), 1,417 (0,73), 1,458 (16,00), 1,788 (0,54), 1,795 (0,57), 1,814 (0,49), 1,821 (0,46), 1,879 (0,57), 1,886 (0,55), 1,804 (0,50), 4,567 (1,04), 4,575 (0,81), 4,732 (0,42), 5,750 (0,62), 6,633 (2,89).
Intermedio 45A.
íerc-Butil c/s-4-{[(dorometoxi)carbonN]oxi}ddohexN (2E)-but-2-enodioato

C/s-4-hidroxiciclohexil (2E)-but-2-enodioato de terc-butilo (Intermedio 43A, 210 mg, 777 jmol) se disolvió en piridina (5,0 ml), se agregó carbonoclorhidrato de clorometilo (100 jl, 1,2 mmol) y la mezcla se agitó a temperatura ambiente durante 45 min. Luego, el solvente se destiló, el residuo se disolvió en acetato de etilo (100 ml) y posteriormente se lavó con agua y 3 porciones de salmuera. La fase orgánica se secó en sulfato de sodio y se evaporó. El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 4:1). Se aislaron 198 mg (100 % de pureza, 70 % de rendimiento) del compuesto del título.
1H-NMR (400 MHz, DMSO-d6) 5 [ppm]: 1,466 (16,00), 1,736 (0,43), 1,749 (0,66), 1,758 (0,83), 1,767 (0,71), 1,785 (0,78), 1,796 (0,83), 1,811 (0,60), 5,801 (3,59), 6,675 (4,24).
Intermedio 46A.
íerc-Butil c/s-4-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]cidohexil (2£)-but-2-enodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianocidopropil)benzamida (275 mg, 424 jmol) se disolvió en THF seco (8 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,4 ml, solución de 0,50 M en tolueno, 680 |jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (32 mg, 212 jmol) y c/s-4-{[(dorometoxi)carbonil]oxi}ddohexilo de ferc-butilo (2E)-but-2-enodioato (intermedio 45A, 231 mg, 636 jmol) en THF (3 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se vertió en agua y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 325 mg (100 % de pureza, 79 % de rendimiento) del compuesto del título. LC-MS (método 2): Rt = 1,55 min; MS (ESlpos): m/z = 975 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 5 [ppm]: 1,452 (16,00), 1,691 (0,88), 1,762 (0,44), 3,518 (0,87), 6,653 (2,33), 7,802 (0,68), 7,823 (0,57), 8,196 (0,66), 8,453 (0,46), 8,776 (0,42).
Intermedio 47A.
íerc-Butil c/s-4-hidroxiciclohexil butanodioato

Se disolvió ferc-Butil c/s-4-hidroxiciclohexil (2E)-but-2-enodioato (intermedio 43A, 210 mg, 777 jmol) en etanol (10 ml) y se hidrogenó a presión ambiente y temperatura ambiente en presencia de 10% de paladio sobre carbón activado (41 mg, 39 jmol) durante 90 min. La mezcla se filtró sobre una almohadilla de tierra diatomácea y se evaporó. El compuesto del título (167 mg, 79 % de rendimiento) se utilizó sin purificación adicional.
iH-NMR (400 MHz, DMSO-d6) 5 [ppm]: 1,384 (16,00), 1,504 (0,69), 1,522 (1,45), 2,444 (0,40), 4,477 (0,80), 4,487 (0,80).
Intermedio 48A.
íerc-Butil c/s-4-{[(clorometoxi)carbonil]oxi}ciclohexil butanodioato

C/s-4-hidroxicidohexil butanodioato de ferc-butilo (Intermedio 47A, 165 mg, 606 pmol) se disolvió en piridina (7,0 ml), se agregó carbonoclorhidrato de clorometilo (58 pl, 0,9 mmol) y la mezcla se agitó a temperatura ambiente durante 45 min. Luego, el solvente se destiló, el residuo se disolvió en acetato de etilo (100 ml) y posteriormente se lavó con agua y salmuera. La fase orgánica se secó en sulfato de sodio y se evaporó. El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 3:1). Se aislaron 180 mg (100 % de pureza, 81 % de rendimiento) del compuesto del título.
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,382 (16,00), 1,664 (0,50), 1,679 (0,78), 1,691 (0,67), 1,702 (0,47), 1,743 (0,47), 1,760 (0,73), 1,772 (0,70), 2,454 (0,42), 2,457 (0,40), 2,461 (0,65), 2,469 (1,10), 2,479 (1,14), 5,894 (3,54).
Intermedio 49A.
íerc-Butil c/s-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]ciclohexil butanodioato

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (267 mg, 411 pmol) se disolvió en THF seco (8 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,1 ml, solución de 0,50 M en tolueno, 530 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (31 mg, 206 pmol) y c/s-4-{[(clorometoxi)carbonil]oxi}ciclohexilo de ferc-butilo (2£)-but-2-enodioato (intermedio 48A, 180 mg, 493 pmol) en THF (3 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 45 min. La mezcla de reacción se vertió en agua y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 pm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 277 mg (100 % de pureza, 69 % de rendimiento) del compuesto del título. LC-MS (método 4): Rt = 5,30 min; MS (ESlpos): m/z = 977 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,356 (1,04), 1,369 (16,00), 1,633 (0,73), 1,662 (0,71), 1,761 (0,41), 2,442 (0,57), 2,449 (0,80), 2,462 (0,85), 2,470 (0,60), 2,476 (0,50), 7,800 (0,68), 7,834 (0,56), 8,208 (0,62), 8,212 (0,61), 8,448 (0,44), 8,772 (0,42).
Intermedio 50A.
íerc-Butil írans-4-{[(clorometoxi)carbonil]oxi}ciclohexil (2£)-but-2-enodioato

frans-4-hidroxiciclohexil (2E)-but-2-enodioato de ferc-butilo (Intermedio 44A, 210 mg, 777 |jmol) se disolvió en piridina (3,0 ml), se agregó carbonoclorhidrato de clorometilo (120 jl, 1,2 mmol) y la mezcla se agitó a temperatura ambiente durante 1 h. Luego, el solvente se destiló, el residuo se disolvió en acetato de etilo (50 ml) y posteriormente se lavó con agua y salmuera. La fase orgánica se secó en sulfato de sodio y se evaporó. El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 4:1). Se aislaron 237 mg (84 % de rendimiento) del compuesto del título.
1H-NMR (500 MHz, DMSO-d6) 8 [ppm]: 1,463 (16,00), 1,599 (0,86), 1,615 (0,87), 1,881 (0,42), 5,896 (3,73), 6,663 (4,43).
Intermedio 51A.
trans-4-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-M}benzoM)(1-cianocidopropM)amino]metoxi}carbonM)oxi]cidohexM (2E)-but-2-enodioato de terc-butilo

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (162 mg, 250 jmol) se disolvió en THF seco (8 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (800 jL, solución de 0,50 M en tolueno, 400 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (19 mg, 125 jmol) y frans-4-{[(clorometoxi)carbonil]oxi}ciclohexilo de ferc-butilo (2£)-but-2-enodioato (intermedio 50A, 136 mg, 375 jmol) en t Hf (3 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se vertió en agua y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 197 mg (81 % de teoría) del compuesto del título.
LC-MS (método 4): Rt = 5,49 min; MS (ESlpos): m/z = 975 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,459 (16,00), 1,502 (0,48), 3,392 (0,44), 5,757 (0,95), 6,641 (1,72), 7,804 (0,56), 7,929 (0,58), 8,206 (0,69), 8,461 (0,55), 8,779 (0,53).
Intermedio 52A.
C/s-4-{[(clorometoxi)carbonN]oxi}ciclohexano-1-carboxMatode terc-butilo
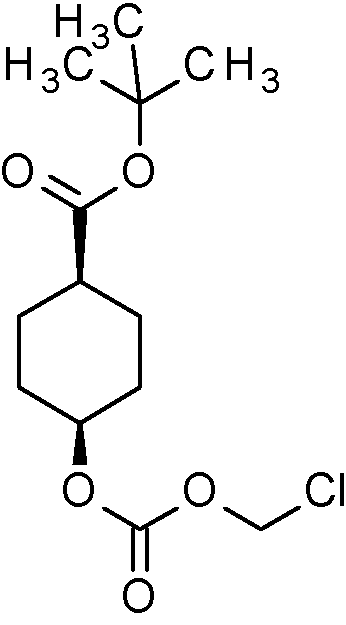
C/s-4-hidroxicidohexano-1-carboxilato de ferc-butilo (CAS-RN 931110-79-1, 401 mg, 2,00 mmol) se disolvió en piridina (6,5 ml), se agregó carbonoclorhidrato de clorometilo (300 pl, 3,0 mmol) y la mezcla se agitó a temperatura ambiente durante 1 h. Luego, el solvente se destiló, el residuo se disolvió en acetato de etilo (50 ml) y posteriormente se lavó con agua y salmuera. La fase orgánica se secó en sulfato de sodio y se evaporó. El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 4:1). Se aislaron 478 mg (82 % de rendimiento) del compuesto del título.
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,397 (16,00), 1,639 (0,63), 1,646 (0,61), 1,654 (0,51), 5,887 (3,63).
Intermedio 53A.
C/s-4-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-M}benzoM)(1-cianocidopropM)amino]metoxi}carbonM)oxi]cidohexano-1-carboxMato de terc-butílo
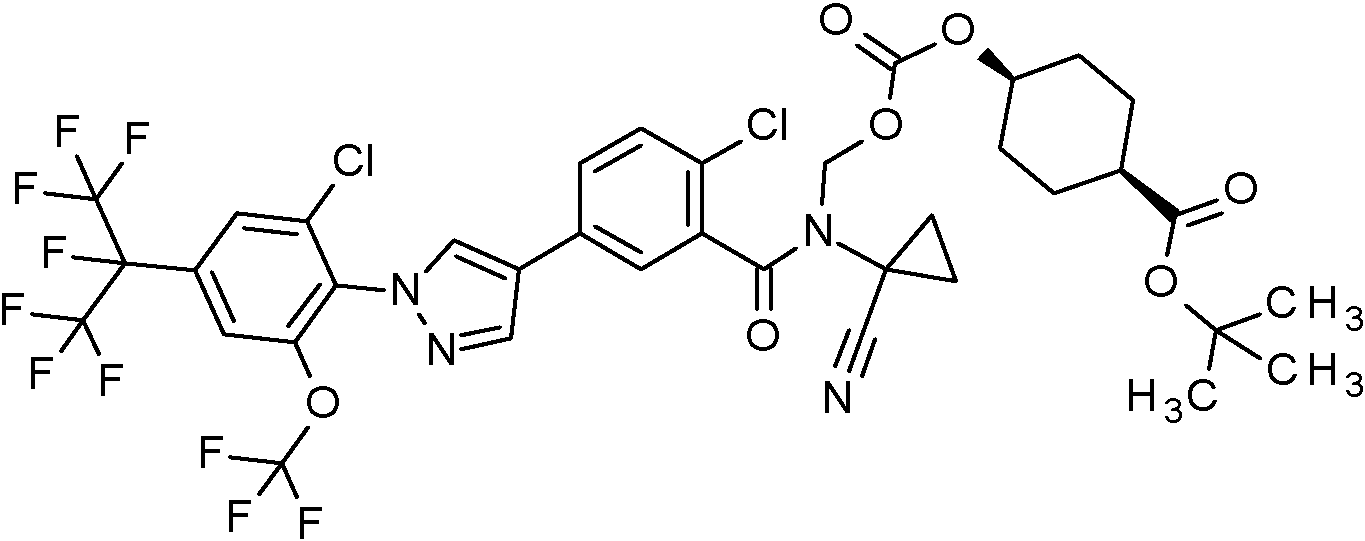
2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (162 mg, 250 pmol) se disolvió en THF seco (8 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (800 pL, solución de 0,50 M en tolueno, 400 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (19 mg, 125 pmol) y c/s-4-{[(clorometoxi)carbonil]oxi}ciclohexano-1-carboxilato de ferc-butilo (2E)-but-1-enodioato (intermedio 52A, 110 mg, 375 pmol) en THF (3 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se vertió en agua y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 pm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 196 mg (87 % de teoría) del compuesto del título.
LC-MS (método 4): Rt = 5,41 min; MS (ESlpos): m/z = 905 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,234 (0,49), 1,358 (0,50), 1,375 (16,00), 1,395 (0,47), 1,400 (0,55), 1,591 (0,91), 1,605 (0,84), 1,660 (0,56), 1,762 (0,59), 7,813 (0,87), 7,932 (1,04), 8,209 (1,16), 8,452 (0,78), 8,777 (0,76).
Intermedio 54A.
7rans-4-{[(clorometoxi)carbonN]oxi}ciclohexano-1-carboxNato de terc-butílo

Trans-4-hidroxiciclohexano-1-carboxilato de ferc-butilo (CAS-RN 869193-57-7, 401 mg, 2,00 mmol) se disolvió en piridina (6,5 ml), se agregó carbonoclorhidrato de clorometilo (300 pl, 3,0 mmol) y la mezcla se agitó a temperatura ambiente durante 1 h. Luego, el solvente se destiló, el residuo se disolvió en acetato de etilo (50 ml) y posteriormente se lavó con agua y salmuera. La fase orgánica se secó en sulfato de sodio y se evaporó. El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 4:1). Se aislaron 500 mg (85 % de rendimiento) del compuesto del título.
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,389 (16,00), 1,413 (0,58), 1,429 (1,06), 1,446 (0,66), 1,984 (0,44), 5,881 (3,82).
Intermedio 55A.
7rans-4-[({[(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-M}benzoM)(1-cianoddopropM)amino]metoxi}carbonM)oxi]ddohexano-1-carboxMato de tere-butilo

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (162 mg, 250 pmol) se disolvió en THF seco (8 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (800 pL, solución de 0,50 M en tolueno, 400 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (19 mg, 125 pmol) y frans-4-{[(clorometoxi)carbonil]oxi}ciclohexano-1-carboxilato de ferc-butilo (2E)-but-1-enodioato (intermedio 54A, 110 mg, 375 pmol) en THF (3 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se vertió en agua y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 pm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95). Se obtuvieron 191 mg (98 % de pureza, 82 % de teoría) del compuesto del título. LC-MS (método 4): Rt = 5,44 min; MS (ESlpos): m/z = 905 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,129 (0,53), 1,257 (0,72), 1,391 (13,61), 1,407 (2,06), 1,780 (0,74), 1,847 (0,61), 2,564 (16,00), 7,620 (0,48), 7,634 (0,53), 7,806 (0,79), 7,827 (0,56), 7,842 (0,50), 7,957 (1,20), 8,234 (1,26), 8,478 (0,93), 8,801 (0,92).
Intermedio 56A.
{[(Clorometoxi)carbonil]oxi}acetato de tere-butilo

Hidroxiacetato de ferc-butilo (CAS-RN 50595-15-8, 264 mg, 2,00 mmol) se disolvió en piridina (6,5 ml), se agregó carbonoclorhidrato de clorometilo (300 pl, 3,0 mmol) y la mezcla se agitó a temperatura ambiente durante 1 h. Luego, el solvente se destiló, el residuo se disolvió en acetato de etilo (50 ml) y posteriormente se lavó con agua y salmuera. La fase orgánica se secó en sulfato de sodio y se evaporó. El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 4:1). Se aislaron 430 mg (89 % de pureza, 85 % de rendimiento) del compuesto del título.
1H-NMR (500 MHz, DMSO-d6) 8 [ppm]: 1,426 (2,11), 1,431 (16,00), 4,688 (3,87), 5,946 (4,12).
Intermedio 57A.
[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]acetato de terc-butilo

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}-N-(1-cianociclopropil)benzamida (162 mg, 250 pmol) se disolvió en THF seco (8 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (800 pL, solución de 0,50 M en tolueno, 400 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (19 mg, 125 pmol) y {[(clorometoxi)carbonil]oxi}acetato de ferc-butilo (2E)-but-1-enodioato (intermedio 56A, 84,2 mg, 375 pmol) en t Hf (3 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se vertió en agua y se extrajo con 3 porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 pm, gradiente de agua-acetonitrilo con TFA al 0,01 % en ambos eluyentes, 90:10->5:95) del compuesto del título se obtuvieron. Se aislaron 120 mg (57 % de rendimiento).
LC-MS (método 4): Rt = 4,99 min; MS (ESlpos): m/z = 837 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: -0,022 (0,77), 1,235 (1,70), 1,357 (10,57), 1,364 (16,00), 1,417 (1,30), 1,504 (0,42), 1,767 (1,65), 2,184 (0,68), 4,491 (3,15), 6,872 (0,44), 7,604 (0,89), 7,618 (0,98), 7,782 (1,45), 7,813 (1,01), 7,826 (0,95), 7,939 (2,92), 8,214 (3,08), 8,440 (5,00), 8,760 (1,98).
Intermedio 58A
4-{[(Clorometoxi)carbonil]oxi}butanoato de tere-butilo

4-Hidroxibutanoato de ferc-butilo (CAS RN 59854-12-5, 250 mg, 1,56 mmol) se disolvió en piridina (5,1 ml). Esta solución se enfrió a 0 °C, se agregó carbonoclorhidrato de clorometilo (240 pL, 2,4 mmol) y la mezcla se agitó a temperatura ambiente durante la noche. Luego el solvente se destiló, el residuo se disolvió en acetato de etilo y se lavó posteriormente con agua y salmuera. La fase orgánica se secó en sulfato de sodio y se evaporó. El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 4:1). Se aislaron 263 mg (67 % de rendimiento) del compuesto del título. 1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,400 (16,00), 1,836 (0,69), 1,847 (1,06), 1,859 (0,73), 2,271 (0,87), 2,283 (1,65), 2,295 (0,78), 4,185 (0,85), 4,196 (1,73), 4,207 (0,83), 5,890 (3,93).
Intermedio 59A.
4-[({K2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]butanoato de tere-butilo

2-Cloro-W-(1-cianociclopropil)-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzamida (292 mg, 528 pmol) se disolvió en THF seco (16 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,6 ml, solución de 0,50 M en tolueno, 840 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (39,5 mg, 264 pmol) y 4-{[(clorometoxi)carbonil]oxi}butanoato de terc-butilo (intermedio 58A, 160 mg, 633 pmol) en THF (5,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante la noche. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 pm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 231 mg (100 % de pureza, 57 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,80 min; MS (ESlpos): m/z = 769 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,367 (16,00), 1,754 (1,69), 1,764 (1,54), 2,074 (1,78), 2,204 (0,77), 2,216 (1,32), 2,228 (0,72), 3,824 (7,66), 4,021 (0,65), 7,616 (0,65), 7,629 (0,72), 7,825 (1,06), 7,849 (0,68), 7,863 (0,64), 8,566 (1,34), 8,842 (1,30).
Intermedio 60A.
3-{[(Clorometoxi)carbonil]oxi}-2,2-dimetilpropanoato de tere-butilo

3-Hidroxi-2,2-dimetilpropanoato de ferc-butilo (CAS RN 25307-76-0, 348 mg, 2,00 mmol) se disolvió en piridina (7,0 ml). Esta solución se enfrió a 0 °C, se agregó carbonoclorhidrato de clorometilo (240 pL, 3,0 mmol) y la mezcla se agitó a temperatura ambiente durante la noche. Luego el solvente se destiló, el residuo se disolvió en acetato de etilo y se lavó posteriormente con agua y salmuera. La fase orgánica se secó en sulfato de sodio y se evaporó. El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano -ciclohexano acetato de etilo). Se aislaron 258 mg (48 % de rendimiento) del compuesto del título.
1H-NMR (400 MHz, DMSO-d6) 6 [ppm]: 1,118 (9,71), 1,371 (0,81), 1,378 (16,00), 4,194
Intermedio 61A.
3-[({K2-cloro-5-[2'-metN-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-N]benzoM}(1-cianociclopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropanoato de tere-butilo

2- Cloro-W-(1-cianociclopropil)-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzamida (262 mg, 475 pmol) se disolvió en THF seco (15 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,5 ml, solución de 0,50 M en tolueno, 760 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (35,6 mg, 237 pmol) y 3- {[(clorometoxi)carbonil]oxi}-2,2-dimetilpropanoato de terc-butilo (intermedio 60A, 190 mg, 712 pmol) en THF (5,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante el fin de semana. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-1810 pm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 166 mg (91 % de pureza, 41 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 5,02 min; MS (ESlpos): m/z = 727 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 6 [ppm]: -0,022 (0,59), -0,008 (3,30), 0,008 (3,41), 1,044 (9,00), 1,095 (2,59), 1,109 (0,96), 1,235 (0,98), 1,306 (1,20), 1,329 (16,00), 1,370 (5,59), 1,379 (2,07), 1,484 (0,42), 1,758 (1,21), 2,524 (1,26), 3,824 (11,37), 4,024 (1,01), 4,069 (0,85), 7,613 (0,75), 7,633 (0,91), 7,826 (1,30), 7,846 (1,05), 7,868 (0,89), 8,560 (1,75), 8,815 (1,78).
Intermedio 62A.
7r3ns-4-[({[{2-cloro-5-[2'-metM-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-N]benzoM}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexil (2E)-but-2-enodioato de tere-butilo

2-Cloro-W-(1-c¡anoc¡cloprop¡l)-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzam¡da (203 mg, 368 jmol) se d¡solv¡ó en THF seco (9 ml). La mezcla se enfr¡ó hasta -45 °C en un baño de acetona - h¡elo seco. B¡s(tr¡met¡ls¡l¡l)am¡da de potas¡o (1,1 ml, soluc¡ón de 0,50 M en tolueno, 590 jmol) se agregó gota a gota y la mezcla se ag¡tó durante 30 m¡n. a -45 °C. Luego, se agregó una soluc¡ón de yoduro de sod¡o (27,5 mg, 184 jmol) y (1r,4r)-4-{[(clorometox¡)carbon¡l]ox¡}c¡clohex¡lo (2£)-but-2-enod¡oato de terc-but¡lo (¡ntermed¡o 50A, 200 mg, 551 jmol) en THF (4,0 ml), se ret¡ró el baño de h¡elo seco y la mezcla se ag¡tó a temperatura amb¡ente durante la noche. La mezcla de reacc¡ón se vert¡ó en agua y se extrajo con tres porc¡ones de acetato de et¡lo. Los extractos orgán¡cos comb¡nados se secaron sobre sulfato de sod¡o anh¡dro y se evaporaron. El producto bruto se pur¡f¡có por HPLC preparat¡va (RP C-1810 |jm, grad¡ente de agua-aceton¡tr¡lo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuv¡eron 206 mg (100 % de pureza, 64 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 5,12 m¡n; MS (ESlpos): m/z = 879 [M+H]+ 1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,60), 0,008 (0,70), 1,157 (0,55), 1,175 (1,11), 1,193 (0,57), 1,234 (0,40), 1,458 (16,00), 1,504 (0,55), 1,767 (0,41), 1,856 (0,44), 1,988 (2,03), 3,823 (3,34), 4,021 (0,47), 4,039 (0,47), 6,638 (1,73), 7,840 (0,56), 8,579 (0,55), 8,844 (0,53).
Intermedio 63A.
73ns-4-[({[{2-doro-5-[2'-metN-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-N]benzoM}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexano-1-carboxilato de tere-butilo

2-Cloro-W-(1-c¡anoc¡cloprop¡l)-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzam¡da (315 mg, 569 pmol) se d¡solv¡ó en THF seco (16 ml). La mezcla se enfr¡ó hasta -45 °C en un baño de acetona - h¡elo seco. B¡s(tr¡met¡ls¡l¡l)am¡da de potas¡o (1,8 ml, soluc¡ón de 0,50 M en tolueno, 910 pmol) se agregó gota a gota y la mezcla se ag¡tó durante 30 m¡n. a -45 °C. Luego, se agregó una soluc¡ón de yoduro de sod¡o (42,7mg, 285 pmol) y (1s,4s)-4-{[(clorometox¡)carbon¡l]ox¡}c¡clohexano-1-carbox¡lato de ferc-but¡lo (2E)-but-1-enod¡oato (¡ntermed¡o 52A, 250 mg, 854 pmol) en THF (5,0 ml), se ret¡ró el baño de h¡elo seco y la mezcla se ag¡tó a temperatura amb¡ente durante 2 h. La mezcla de reacc¡ón se vert¡ó en agua y se extrajo con tres porc¡ones de acetato de et¡lo. Los extractos orgán¡cos comb¡nados se secaron sobre sulfato de sod¡o anh¡dro y se evaporaron. El producto bruto se pur¡f¡có por HPLC preparat¡va (RP C-18 10 pm, grad¡ente de agua-aceton¡tr¡lo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuv¡eron 311 mg (98 % de pureza, 66 % de rend¡m¡ento) del compuesto del título. LC-MS (método 4): Rt = 5,08 m¡n; MS (ESlpos): m/z = 809 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,357 (1,18), 1,379 (16,00), 1,590 (0,86), 1,767 (0,56), 2,075 (1,06), 3,826 (5,33), 7,612 (0,43), 7,627 (0,48), 7,851 (1,16), 7,861 (0,58), 8,574 (0,95), 8,848 (0,92).
Intermedio 64A.
7r3ns-4-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexano-1-carboxilato de tere-butilo

2-Cloro-W-(1-c¡anoc¡cloprop¡l)-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzam¡da (189 mg, 342 pmol) se d¡solv¡ó en THF seco (11 ml). La mezcla se enfr¡ó hasta -45 °C en un baño de acetona - h¡elo seco. B¡s(tr¡met¡ls¡l¡l)am¡da de potas¡o (1,1 ml, soluc¡ón de 0,50 M en tolueno, 550 pmol) se agregó gota a gota y la mezcla se ag¡tó durante 30 m¡n. a -45 °C. Luego, se agregó una soluc¡ón de yoduro de sod¡o (25,6 mg, 171 pmol) y (1r,4r)-4-{[(clorometox¡)carbon¡l]ox¡}c¡clohexano-1-carbox¡lato de ferc-but¡lo (¡ntermed¡o 54A, 150 mg, 512 pmol) en THF (3,0 ml), se ret¡ró el baño de h¡elo seco y la mezcla se ag¡tó a temperatura amb¡ente durante la noche. La mezcla de reacc¡ón se vert¡ó en agua y se extrajo con tres porc¡ones de acetato de et¡lo. Los extractos orgán¡cos comb¡nados se secaron sobre sulfato de sod¡o anh¡dro y se evaporaron. El producto bruto se pur¡f¡có por HPLC preparativa (RP C-1810 pm, grad¡ente de agua-aceton¡tr¡lo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuv¡eron 201 mg (100 % de pureza, 73 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 5,07 m¡n; MS (ESlpos): m/z = 809 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,234 (0,53), 1,335 (0,85), 1,358 (1,57), 1,370 (16,00), 1,765 (0,75), 1,824 (0,66), 2,075 (6,04), 3,370 (0,40), 3,828 (6,58), 7,623 (0,61), 7,637 (0,71), 7,827 (0,96), 7,850 (0,62), 7,865 (0,59), 8,579 (1,25), 8,847 (1,22).
Intermedio 65A.
3-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]propil (2E)-but-2-enodioato de tere-butilo

2- Cloro-W-(1-cianociclopropil)-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzamida (228 mg, 413 jmol) se disolvió en THF seco (16 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,3 ml, solución de 0,50 M en tolueno, 660 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (31,0 mg, 207 jmol) y 3- {[(clorometoxi)carbonil]oxi}propil (2E)-but-2-enodioato de terc-butilo (intermedio 6A, 200 mg, 620 jmol) en THF (4,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 2 h. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-1810 |jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 202 mg (100 % de pureza, 58 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,85 min; MS (ESlpos): m/z = 783 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,450 (16,00), 1,760 (0,45), 1,926 (0,42), 2,073 (0,68), 3,820 (3,80), 4,138 (0,56), 4,154 (0,67), 6,643 (1,46), 7,830 (0,49), 7,842 (0,41), 8,559 (0,63), 8,835 (0,60).
Intermedio 66A.
3-hidroxipropil butanodioato de tere-butilo

3-hidroxipropil (2E)-but-2-enodioato de tere-butilo (intermedio 5A, 450 mg, 1,95 mmol) se disolvió en etanol (10 ml) bajo una atmósfera de argón. Se agregó paladio sobre carbón al 10 % (41,6 mg, 39,1 jmol) y la mezcla se hidrogenó durante la noche a presión ambiente y temperatura ambiente. Luego el catalizador se retiró por filtración sobre una capa de tierra diatomácea y el solvente se destiló. Se obtuvieron 430 mg (95 % de rendimiento) del compuesto del título.
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,384 (16,00), 1,685 (0,76), 1,701 (1,19), 1,717 (0,80), 2,438 (0,52), 2,440 (0,48), 2,445 (0,66), 2,453 (0,95), 2,454 (1,08), 2,467 (1,04), 2,477 (0,61), 2,484 (0,54), 3,427 (0,41), 3,443 (0,93), 3,456 (0,92), 4,049 (0,78), 4,065 (1,59), 4,082 (0,75), 4,484 (0,42), 4,496 (0,85).
Intermedio 67A.
3-{[(clorometoxi)carbonil]oxi}propil butanodioato de tere-butilo

3-hidroxipropil butanodioato de ferc-butilo (intermedio 66A,120 mg, 517 jmol) se disolvió en piridina (4,0 ml). Esta solución se enfrió a 0 °C, se agregó carbonoclorhidrato de clorometilo (69 jl, 770 |jmol) y la mezcla se agitó a temperatura ambiente durante 2 h. Luego el solvente se destiló, el residuo se disolvió en acetato de etilo y se lavó posteriormente con agua y salmuera. La fase orgánica se secó en sulfato de sodio y se evaporó. El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 4:1). Se aislaron 144 mg (100 % de pureza, 86 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 3,09 min; MS (ESlpos): m/z = 151 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,381 (16,00), 1,931 (0,71), 1,947 (1,08), 1,963 (0,73), 2,450 (0,62), 2,456 (0,65), 2,466 (1,08), 2,482 (1,11), 4,073 (0,74), 4,089 (1,51), 4,105 (0,72), 4,235 (0,76), 4,251 (1,58), 4,267 (0,75), 5,894 (3,71).
Intermedio 68A.
3-[({K2-cloro-5-[2'-metM-5'-(pentafluoroetN)-4'-(trifluorometN)-2'H-[1,3'-bipirazol]-4-M]benzoN}(1-cianociclopropil)amino]metoxi}carbonil)oxi]propil butanodioato de tere-butilo

2-Cloro-W-(1-cianociclopropil)-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzamida (159 mg, 287 jmol) se disolvió en THF seco (9 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (900 l, solución de 0,50 M en tolueno, 460 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (21,5 mg, 144 jmol) y 3-{[(clorometoxi)carbonil]oxi}propil butanodioato de ferc-butilo (intermedio 67A, 140 mg, 431 jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 196 mg (100 % de pureza, 81 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,71 min; MS (ESlpos): m/z = 841 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 5 [ppm]: 1,030 (6,03), 1,045 (6,10), 1,360 (16,00), 1,761 (0,67), 1,842 (0,45), 1,857 (0,64), 1,872 (0,48), 2,086 (2,40), 2,437 (1,36), 2,451 (1,24), 3,757 (0,42), 3,772 (0,56), 3,788 (0,43), 3,821 (5,60), 4,011 (0,50), 4,026 (0,88), 4,042 (0,56), 4,086 (0,65), 7,635 (0,47), 7,832 (0,72), 7,843 (0,60), 7,865 (0,43), 8,563 (0,91), 8,837 (0,91).
Intermedio 69A.
1-{2-[({K2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]etil} ciclopropano-1,1-dicarboxilato de 1-ferc-butilo

2-Cloro-W-(1-c¡anoc¡cloprop¡l)-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzam¡da (171 mg, 310 |jmol) se d¡solv¡ó en THF seco (8 ml). La mezcla se enfr¡ó hasta -45 °C en un baño de acetona - h¡elo seco. B¡s(tr¡met¡ls¡l¡l)am¡da de potas¡o (910 l, soluc¡ón de 0,50 M en tolueno, 500 jmol) se agregó gota a gota y la mezcla se ag¡tó durante 30 m¡n. a -45 °C. Luego, se agregó una soluc¡ón de yoduro de sod¡o (27,9 mg, 186 jmol) y 1-(2-{[(clorometox¡)carbon¡l]ox¡}et¡l) c¡clopropano-1,1-d¡carbox¡lato de 1-terc-but¡lo (¡ntermed¡o 40A, 150 mg, 465 jmol) en THF (3,0 ml), se ret¡ró el baño de h¡elo seco y la mezcla se ag¡tó a temperatura amb¡ente durante 3 h. La mezcla de reacc¡ón se vert¡ó en agua y se extrajo con tres porc¡ones de acetato de et¡lo. Los extractos orgán¡cos comb¡nados se secaron sobre sulfato de sod¡o anh¡dro y se evaporaron. El producto bruto se pur¡f¡có por HPLC preparat¡va (RP C-1810 jm , grad¡ente de agua-aceton¡tr¡lo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuv¡eron 180 mg (86 % de pureza, 60 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 4,77 m¡n; MS (ESlpos): m/z = 783 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 5 [ppm]: 1,157 (0,80), 1,175 (1,60), 1,193 (0,88), 1,249 (6,35), 1,358 (16,00), 1,760 (1,22), 1,988 (2,93), 2,073 (1,87), 3,820 (11,25), 4,021 (0,72), 4,038 (0,73), 4,236 (3,37), 7,614 (0,71), 7,636 (0,88), 7,831 (1,26), 7,849 (0,99), 7,871 (0,84), 8,561 (1,70), 8,834 (1,60).
Intermedio 70A.
2-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]etil pentanodioato de ferc-butilo

2-Cloro-W-(1-c¡anoc¡cloprop¡l)-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzam¡da (238 mg, 431 pmol) se d¡solv¡ó en THF seco (12 ml). La mezcla se enfr¡ó hasta -45 °C en un baño de acetona - h¡elo seco. B¡s(tr¡met¡ls¡l¡l)am¡da de potas¡o (1,4 ml, soluc¡ón de 0,50 M en tolueno, 690 pmol) se agregó gota a gota y la mezcla se ag¡tó durante 30 m¡n. a -45 °C. Luego, se agregó una soluc¡ón de yoduro de sod¡o (32,3 mg, 216 pmol) y 2-{[(clorometox¡)carbon¡l]ox¡}et¡l pentanod¡oato de terc-but¡lo (¡ntermed¡o 37A, 210 mg, 647 pmol) en THF (4,0 ml), se ret¡ró el baño de h¡elo seco y la mezcla se ag¡tó a temperatura amb¡ente durante la noche. La mezcla de reacc¡ón se vert¡ó en agua y se extrajo con tres porc¡ones de acetato de et¡lo. Los extractos orgán¡cos comb¡nados se secaron sobre sulfato de sod¡o anh¡dro y se evaporaron. El producto bruto se pur¡f¡có por HPLC preparat¡va (RP C-1810 pm, grad¡ente de agua-aceton¡tr¡lo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuv¡eron 201 mg (66 % de pureza, 37 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 4,75 m¡n; MS (ESlpos): m/z = 785 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,373 (16,00), 1,668 (0,43), 1,687 (0,62), 1,705 (0,49), 1,761 (0,42), 2,180 (0,44), 2,198 (0,82), 2,216 (0,44), 2,295 (0,59), 3,821 (3,64), 4,177 (0,53), 7,834 (0,44), 8,559 (0,60), 8,836 (0,55).
Intermedio 71A.
3-[({K2-cloro-5-[2'-metN-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-N]benzoM}(1-cianociclopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropil butanodioato de tere-butilo

2-Cloro-W-(1-c¡anoc¡cloprop¡l)-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzam¡da (261 mg, 472 pmol) se d¡solv¡ó en THF seco (12 ml). La mezcla se enfr¡ó hasta -45 °C en un baño de acetona - h¡elo seco. B¡s(tr¡met¡ls¡l¡l)am¡da de potas¡o (1,4 ml, soluc¡ón de 0,50 M en tolueno, 760 pmol) se agregó gota a gota y la
mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (35,4 mg, 236 |jmol) y 3-{[(dorometoxi)carbonil]oxi}-2,2-dimetilpropil butanodioato de ferc-butilo (intermedio 14A, 250 mg, 709 jmol) en THF (4,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 1 h. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-1810 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 349 mg (100 % de pureza, 85 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,98 min; MS (ESlpos): m/z = 868 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 0,851 (1,90), 1,175 (0,72), 1,355 (7,00), 1,988 (1,27), 2,444 (0,47), 2,466 (0,44), 2,523 (0,41), 3,318 (16,00), 3,321 (15,00), 3,785 (0,54), 3,822 (2,61), 7,842 (0,55).
Intermedio 72A.
[({[{2-doro-5-[2'-metN-5'-(pentafluoroetM)-4'-(trifluorometN)-2'H-[1,3'-bipirazol]-4-N]benzoM}(1-cianociclopropil)amino]metoxi}carbonil)oxi]acetato de tere-butilo

2-Cloro-N-(1-cianociclopropil)-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzamida (246 mg, 445 jmol) se disolvió en THF seco (13 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,8 ml, solución de 0,50 M en tolueno, 710 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (33,4 mg, 223 jmol) y {[(clorometoxi)carbonil]oxi}acetato de ferc-butilo (intermedio 56A, 150 mg, 668 jmol) en THF (4,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante la noche. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 230 mg (100 % de pureza, 70 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,65 min; MS (ESlpos): m/z = 685 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,365 (16,00), 1,430 (1,20), 1,772 (1,41), 2,075 (0,41), 3,295 (0,63), 3,823 (13,60), 4,502 (3,21), 7,628 (0,89), 7,642 (0,99), 7,825 (1,45), 7,855 (0,91), 7,868 (0,87), 8,558 (3,53), 8,829 (2,26).
Intermedio 73A.
1-metil ciclopropano-1,1-dicarboxilato de 1-tere-butilo

A una solución de metil malonato de ferc-butilo (CAS-RN: 42726-73-8, 35,00 g 200,92 mmol) en N,Ndimetilformamida (350 ml), se agregaron 1-bromo-2-cloroetano (40 ml, 401,84 mmol), carbonato de potasio (69,42 g, 502,30 mmol) y tetrafluoroborato de 1-butil-3-metil-1H-imidazol-3-io (11,15 g, 50,32 mmol) con agitación a temperatura ambiente. Después de agitar a durante 48 h, la mezcla de reacción se diluyó con agua (800 ml) y se extrajo con éter dietílico (800 ml, tres veces). Las capas orgánicas combinadas se lavaron con agua (800 ml) y salmuera (800 ml), se secaron sobre sulfato de sodio anhidro y se filtraron. El filtrado se evaporó a presión reducida para dar 40 g (60 % de pureza, 60 % de rendimiento) del compuesto del subtítulo como un aceite incoloro.
1H-NMR (300 MHz, DMSO-d6) 8 [ppm]: 1,175 (0,54), 1,260 (0,47), 1,271 (2,24), 1,278 (2,26), 1,289 (0,47), 1,369 (0,41), 1,379 (0,40), 1,398 (16,00), 1,408 (13,59), 1,989 (1,01), 2,891 (0,41), 3,387 (2,76), 3,639 (4,45), 3,652 (5,98).
Intermedio 74A.
Acido 1-(ferc-butoxicarbonil)ciclopropano-1-carboxíNco

A una solución de 1-metil ciclopropano-1,1-dicarboxilato de 1-terf-butilo (intermedio 73A, 40,00 g, 119,86 mmol) en una mezcla de tetrahidrofurano (200 ml), metanol (100 ml) y agua (100 ml), se agregó hidróxido de litio monohidrato (10,06 g, 239,72 mmol). La mezcla se agitó a temperatura ambiente durante 2 h, luego la mayoría de los solventes se retiraron por evaporación al vacío, el residuo se diluyó con agua (200 ml) y se extrajo con éter dietílico (200 ml, tres veces). La capa acuosa se acidificó hasta pH 2-3 con ácido clorhídrico 6 M helado y se extrajo con diclorometano (2,0 l, tres veces), se secó sobre sulfato de sodio anhidro y se filtró. El residuo se concentró a presión reducida para dar 12,00 g (95% de pureza, 51% de rendimiento) del compuesto del subtítulo como un aceite incoloro.
1H-NMR (300 MHz, DMSO-d6) 8 [ppm]: 1,202 (0,43), 1,213 (1,95), 1,223 (2,00), 1,233 (0,52), 1,397 (16,00).
Intermedio 75A.
1-(hidroximetil)ciclopropano-1-carboxilato de ferc-butilo

A una solución de ácido 1-(terc-butoxicarbonil)ciclopropano-1-carboxílico (Intermedio 74A, 12,80 g, 68,74 mmol) y trietilamina (13,91 g, 137,48 mmol) en tetrahidrofurano (160 ml), se agregó cloroformiato de isobutilo (11,74 g, 85,93 mmol) gota a gota con agitación a 0 °C. La agitación se mantuvo durante 1 h, luego la mezcla se filtró y lavó con tetrahidrofurano (20 ml). El filtrado se enfrió hasta 0 °C y luego se agregó una solución de borohidruro de sodio (5,20 g, 137,48 mmol) en 1 -metil-2-pirrolidinona (20 ml ). La mezcla de reacción se agitó a temperatura ambiente durante 2 h, luego se diluyó con éter (100 ml), se lavó con solución acuosa de bicarbonato de sodio saturado (200 ml), agua (200 ml) y salmuera (200 ml). Las capas orgánicas combinadas se secaron con sulfato de sodio anhidro y se filtraron. El filtrado se evaporó a presión reducida. El residuo se purificó mediante cromatografía en columna en gel de sílice (120 g, eluyente: éter de petróleo-acetato de etilo, 20:1) para dar 6,50 g (95 % de pureza, 52 % de rendimiento) del compuesto del título como un aceite incoloro.
1H-NMR (300 MHz, DMSO-d6) 8 [ppm]: 0,785 (0,92), 0,794 (1,35), 0,805 (0,61), 0,911 (0,57), 0,922 (1,34), 0,931 (0,88), 0,945 (0,40), 1,375 (16,00), 1,398 (0,50), 3,502 (1,55), 3,521 (1,64), 4,499 (0,48), 4,518 (1,00), 4,538 (0,48), 5,760 (0,80).
Intermedio 76A.
2-({[(Clorometoxi)carbonil]oxi}metil)-2-metilbutanoato de ferc-butilo

2-(hidroximetil)-2-metilbutanoato de ferc-butilo (intermedio 75A, 377 mg, 2,00 mmol) se disolvió en piridina (6,5 ml) y la solución se enfrío hasta 0 °C. Se agregó carbonoclorhidrato de clorometilo (300 pL, 3,0 mmol) de una vez y la mezcla se agitó a temperatura ambiente durante 2 horas. El solvente se evaporó. El residuo se volvió a disolver en acetato de etilo (50 ml) se lavó con agua (20 ml) y salmuera (20 ml) se secó sobre sulfato de sodio anhidro y se evaporó. El producto bruto se purificó por cromatografía en gel de sílice (ciclohexano / acetato de etilo 4:1) para dar 440 mg (100 % de pureza, 78 % de rendimiento) del compuesto del título.
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 0,985 (0,51), 0,992 (1,36), 0,997 (1,36), 1,004 (0,54), 1,129 (0,63), 1,135 (1,42), 1,140 (1,22), 1,147 (0,40), 1,374 (16,00), 4,290 (3,28), 5,905 (3,83)
Intermedio 77A.
1-{[({K2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]metil}ciclopropano-1-carboxilato de tere-butilo

2-Cloro-W-(1-cianociclopropil)-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzamida (278 mg, 504 pmol) se disolvió en THF seco (16 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (1,6 ml, solución de 0,50 M en tolueno, 810 pmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (37,7 mg, 236 pmol) y 2-({[(clorometoxi)carbonil]oxi}metil)-2-metilbutanoato de terc-butilo (intermedio 76A, 200 mg, 756 pmol) en THF (5,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante la noche. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-1810 pm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 213 mg (100 % de pureza, 54 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,87 min; MS (ESlpos): m/z = 781 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: 0,913 (1,97), 1,085 (2,09), 1,235 (0,58), 1,318 (16,00), 1,368 (1,19), 1,761 (1,15), 2,075 (1,55), 3,374 (1,26), 3,824 (11,32), 4,118 (0,50), 4,154 (0,54), 7,619 (0,89), 7,633 (1,03), 7,844 (1,57), 7,852 (1,19), 7,866 (0,95), 8,568 (1,94), 8,838 (1,81).
Intermedio 78A.
7r3ns-4-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexil (2E)-but-2-enodioato de tere-butilo

2-Cloro-W-(1-c¡anoc¡cloprop¡l)-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzam¡da (120 mg, 217 jmol) se d¡solv¡ó en THF seco (7 ml). La mezcla se enfr¡ó hasta -45 °C en un baño de acetona - h¡elo seco. B¡s(tr¡met¡ls¡l¡l)am¡da de potas¡o (800 jL, soluc¡ón de 0,50 M en tolueno, 350 jmol) se agregó gota a gota y la mezcla se ag¡tó durante 30 m¡n. a -45 °C. Luego, se agregó una soluc¡ón de yoduro de sod¡o (19,5 mg, 130 jmol) y (1S,4S)-4-{[(clorometox¡)carbon¡l]ox¡}c¡clohex¡lo (2£)-but-2-enod¡oato de terc-but¡lo (¡ntermed¡o 45A, 118 mg, 326 jmol) en THF (2,0 ml), se ret¡ró el baño de h¡elo seco y la mezcla se ag¡tó a temperatura amb¡ente durante la noche. La mezcla de reacc¡ón se vert¡ó en agua y se extrajo con tres porc¡ones de acetato de et¡lo. Los extractos orgán¡cos comb¡nados se secaron sobre sulfato de sod¡o anh¡dro y se evaporaron. El producto bruto se pur¡f¡có por HPLC preparat¡va (RP C-1810 jm, grad¡ente de agua-aceton¡tr¡lo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuv¡eron 120 mg (100 % de pureza, 63 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 5,07 m¡n; MS (ESlpos): m/z = 879 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,022 (0,60), -0,008 (1,01), 0,008 (1,09), 1,234 (1,46), 1,356 (0,79), 1,457 (16,00), 1,685 (1,04), 1,767 (0,57), 2,073 (4,31), 2,086 (0,45), 3,816 (3,69), 6,652 (1,84), 7,843 (0,62), 8,572 (0,58), 8,847 (0,56).
Intermedio 79A.
2-[({K2-cloro-5-[2'-metM-5'-(pentafluoroetN)-4'-(trifluorometN)-2'H-[1,3'-bipirazol]-4-M]benzoN}(1-cianociclopropil)amino]metoxi}carbonil)oxi]etil butanodioato de tere-butilo

2-Cloro-W-(1-c¡anoc¡cloprop¡l)-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzam¡da (111 mg, 200 jmol) se d¡solv¡ó en THF seco (6 ml). La mezcla se enfr¡ó hasta -45 °C en un baño de acetona - h¡elo seco. B¡s(tr¡met¡ls¡l¡l)am¡da de potas¡o (640 l, soluc¡ón de 0,50 M en tolueno, 320 jmol) se agregó gota a gota y la mezcla se ag¡tó durante 30 m¡n. a -45 °C. Luego, se agregó una soluc¡ón de yoduro de sod¡o (18 mg, 120 jmol) y 2-{[(clorometox¡)carbon¡l]ox¡}et¡l butanod¡oato de terc-but¡lo (¡ntermed¡o 9A, 93 mg, 300 jmol)) en THF (2,0 ml), se
retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 45 min. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-1810 |jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 132 mg (100 % de pureza, 80 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,75 min; MS (ESlpos): m/z = 827 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,44), 0,008 (0,50), 1,235 (0,40), 1,356 (16,00), 1,385 (0,59), 1,763 (0,67), 2,416 (0,87), 2,431 (1,09), 2,459 (0,91), 2,474 (0,91), 3,441 (1,82), 4,184 (0,96), 4,212 (0,65), 7,615 (0,40), 7,636 (0,50), 7,838 (0,79), 7,868 (0,47), 8,562 (1,02), 8,837 (0,97).
Intermedio 80A.
2-[({K2-cloro-5-[2'-metN-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-N]benzoM}(1-cianociclopropil)amino]metoxi}carbonil)oxi]etil (2E)-but-2-enodioato de tere-butilo

2-Cloro-W-(1-cianociclopropil)-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzamida (75,0 mg, 136 jmol) se disolvió en THF seco (4 ml). La mezcla se enfrió hasta -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (400 jL, solución de 0,50 M en tolueno, 220 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó una solución de yoduro de sodio (10,2 mg, 67,8 jmol) y 2-{[(clorometoxi)carbonil]oxi}etil (2E)-but-2-enodioato de terc-butilo (intermedio 3A, 62,8 mg, 204 jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 1 h. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-1810 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 75,7 mg (100 % de pureza, 68 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,85 min; MS (ESlpos): m/z = 825 [M+H]+
Intermedio 81A.
1-{[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]metil}ciclopropano-1-carboxilato de tere-butilo

2- Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluonTietox¡)feml]-1H-p¡razol-4-¡l}-A/-(1-cianocidopropil)benzamida (162 mg, 250 jmol) se disolvió en THF seco (6,0 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis^rimetNsiNOamida de potasio (800 jL, solución de 0,50 M en tolueno, 400 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (18,7 mg, 125 jmol) y una solución de 2-({[(doroTetox¡)carbon¡l]ox¡}Tet¡l)-2-Tet¡lbutanoato de ferc-butilo (intermedio 76A, 105 mg, 375 jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 60 min. La mezcla de reacción se evaporó. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 205 mg (100 % de pureza, 93 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 5,25 min; MS (ESlneg): m/z = 920 [M-H]-1H-NMR (500 MHz, DMSO-d6) 8 [ppm]: 0,913 (1,92), 1,078 (2,08), 1,162 (1,60), 1,176 (3,19), 1,190 (1,66), 1,234 (0,56), 1,313 (16,00), 1,357 (4,37), 1,757 (1,28), 1,990 (5,95), 2,185 (0,49), 4,009 (0,50), 4,023 (1,43), 4,038 (1,42), 4,052 (0,51), 4,120 (0,57), 4,150 (0,59), 7,596 (0,82), 7,612 (0,98), 7,809 (2,54), 7,826 (0,98), 7,943 (2,24), 8,220 (2,52), 8,454 (2,00), 8,772 (1,80).
Intermedio 82A.
3- {[(clorometoxi)carbonil]oxi}propanoato de tere-butilo

3-Hidroxipropanoato de ferc-butilo (CAS-RN: 59854-11-4, 590 jl, 4,0 mmol) se disolvió en piridina (13,0 ml) y la solución se enfrío hasta 0 °C. Se agregó carbonoclorhidrato de clorometilo (610 jL, 6,0 mmol) de una vez y la mezcla se agitó a temperatura ambiente durante 2 horas. El solvente se evaporó. El residuo se disolvió en acetato de etilo (50 ml), se lavó con agua (20 ml), salmuera (20 ml) y se secó en sulfato de sodio anhidro y se evaporó. El producto bruto se purificó mediante cromatografía en gel de sílice (ciclohexano / acetato de etilo 4:1) para dar 768 mg (80 % de teoría) del compuesto del título.
iH-NMR (500 MHz, DMSO-d6) 8 [ppm]: 1,402 (16,00), 2,619 (0,78), 2,631 (1,48), 2,643 (0,80), 4,337 (0,80), 4,349 (1,48), 4,361 (0,77), 5,897 (3,97).
Intermedio 83A.
3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]propanoato de tere-butilo

2-Cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-¡l)-6-(tr¡fluorTetox¡)fen¡l]-1H-p¡razol-4-¡l}-A/-(1-danodclopropi^benzamida (162 mg, 250 jmol) se disolvió en THF seco (6,0 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis^nmetNsiliOamida de potasio (800 jL, solución de 0,50 M en tolueno, 400 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (18,7 mg, 125 jmol) y una solución de 3-{[(doroTetox¡)carbon¡l]ox¡}propanoato de ferc-butilo (intermedio 82A, 89,5 mg, 375
|jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 60 min. La mezcla de reacción se evaporó. El producto bruto se purificó por HPLC preparativa (RP C-1810 jm , gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 198 mg (100 % de pureza, 93 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 5,10 min; MS (ESlneg): m/z = 849 [M-H]-1H-NMR (500 MHz, DMSO-d6)8[ppm]: 1,234 (0,54), 1,360 (16,00), 1,757 (1,05), 2,077 (0,83), 2,184 (0,45), 2,524 (0,86), 4,181 (0,62), 7,596 (0,68), 7,613 (0,79), 7,785 (1,08), 7,813 (0,76), 7,830 (0,70), 7,942 (1,84), 8,217 (2,09), 8,220 (2,05), 8,451 (1,54), 8,771 (1,42).
Intermedio 84A.
4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]butanoato de tere-butilo

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (162 mg, 250 jmol) se disolvió en THF seco (6,0 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (800 jL, solución de 0,50 M en tolueno, 400 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (18,7 mg, 125 jmol) y una solución de 4-{[(clorometoxi)carbonil]oxi}butanoato de ferc-butilo (intermedio 58A, 94,8 mg, 375 jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante 60 min. La mezcla de reacción se evaporó. El producto bruto se purificó por HPLC preparativa (RP C-1810 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 196 mg (100 % de pureza, 91 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 5,19 min; MS (ESlpos): m/z = 865 [M+H]+
1H-NMR (500 MHz, DMSO-d6) 8 [ppm]: 1,234 (0,53), 1,365 (16,00), 1,756 (1,66), 2,202 (0,65), 2,216 (1,11), 2,231 (0,61), 4,023 (0,62), 7,592 (0,54), 7,609 (0,63), 7,789 (0,91), 7,807 (0,65), 7,823 (0,56), 7,941 (1,34), 8,216 (1,50), 8,451 (1,18), 8,779 (1,11).
Intermedio 85A.
(1R,2R)-2-[({[{2-cloro-5-[2'-metN-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-N]benzoM}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclopentil butanodioato de tere-butilo

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluonTietox¡)feml]-1H-p¡razol-4-¡l}-A/-(1-cianocidopropil)benzamida (105 mg, 190 jmol) se disolvió en THF seco (6,0 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis^rimetilsiNOamida de potasio (600 jL, solución de 0,50 M en tolueno, 300 jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (14,2 mg, 95 jmol) y una solución de (IR^R^-^clorometoxOcarbomlJoxiJcidopentil butanodioato de tere-butilo (intermedio 34A, 100 mg, 285 jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente por una noche. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 87,1 mg (95 % de pureza, 50 % de rendimiento) del compuesto del título. LC-MS (método 4): Rt = 5,05 min; MS (ESlpos): m/z = 867 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,236 (0,45), 1,357 (16,00), 1,382 (0,41), 1,655 (0,49), 1,757 (0,55), 2,085 (4,91), 2,183 (0,77), 2,423 (1,20), 2,438 (0,96), 2,500 (11,24), 3,821 (5,47), 5,746 (3,10), 6,871 (0,49), 7,832 (0,60), 7,843 (0,47), 8,556 (0,70), 8,826 (0,67).
Intermedio 86A.
3-[({K2-cloro-5-[2'-metN-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-N]benzoM}(1-cianociclopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropil (2£)-but-2-enodioato de tere-butilo

2-Cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-¡l)-6-(tr¡fluorTetox¡)fen¡l]-1H-p¡razol-4-¡l}-A/-(1-cianodclopropi^benzamida (210 mg, 380 jmol) se disolvió en THF seco (10 ml). Esta solución se enfrió a -45 °C en
un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (900 jL, solución de 0,50 M en tolueno, 600 |jmol) se agregó gota a gota y la mezcla se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (28,5 mg, 190 jmol) y una solución de 3-{[(clorometoxi)carbonil]oxi}-2,2-dimetilpropil (2E)-but-2-enodioato de ferc-butilo (intermedio 12A, 200 mg, 570 jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante la noche. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:l0->5:95). Se obtuvieron 295 mg (100 % de pureza, 89 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 5,12 min; MS (ESlpos): m/z = 811 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,93), 0,008 (1,01), 0,887 (2,51), 1,356 (1,22), 1,453 (16,00), 2,073 (5,27), 3,438 (1,10), 3,821 (3,53), 3,911 (1,18), 6,654 (1,00), 7,840 (0,73), 8,556 (0,55), 8,828 (0,50).
Intermedio 87A.
4-{2-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]etil} 2,2-dimetilbutanodioato de 1-ferc-butilo

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (97,9 mg, 177 jmol) se disolvió en THF seco (5,0 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis(trimetilsilil)amida de potasio (560 jL, solución de 0,50 M en tolueno, 280 jmol) se agregó gota a gota y se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (13,3 mg, 88,6 jmol) y una solución de 4-(2-{[(clorometoxi)carbonil]oxi}etil) 2,2-dimetilbutanodioato de ferc-butilo (intermedio 29A, 90,0 mg, 266 jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante la noche. Se agregó 4-(2-{[(clorometoxi)carbonil]oxi}etil) 2,2-dimetilbutanodioato de 1-ferc-butilo (intermedio 29A, 30,0 mg, 88,7 jmol). La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 56,2 mg (100 % de pureza, 37 % de rendimiento) del compuesto del título. LC-MS (método 4): Rt = 4,93 min; MS (ESlpos): m/z = 855 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,143 (2,13), 1,228 (2,13), 1,239 (3,20), 1,912 (4,27), 2,392 (3,20), 2,430 (5,33), 2,619 (3,20), 2,660 (5,33), 3,338 (4,27), 3,353 (5,33), 3,362 (3,20), 3,388 (16,00), 3,494 (14,93), 3,512 (6,40), 7,023 (2,13), 7,108 (2,13), 7,193 (2,13).
Intermedio 88A.
3-{[(clorometoxi)carbonil]oxi}-2,2-dimetilpropanoato de bencilo

3-Hidroxi-2,2-dimetilpropanoato de bencilo (CAS-RN: 17701-61-0,417 mg, 2,00 mmol) se disolvió en piridina (7,5 ml) y la solución se enfrío hasta 0°C. Se agregó carbonoclorhidrato de clorometilo (270 pL, 3,0 mmol) de una vez y la mezcla se agitó a temperatura ambiente durante 16 horas. El solvente se evaporó. El residuo se volvió a disolver en acetato de etilo (50 ml) se lavó con agua (20 ml) y salmuera (20 ml) se secó sobre sulfato de sodio anhidro y se evaporó. El producto bruto se purificó por cromatografía en gel de sílice (ciclohexano / acetato de etilo 5:1) para dar 230 mg (100 % de pureza, 38 % de rendimiento) del compuesto del título.
LC-MS (método 2): Rt = 1,07 min; MS (ESlpos): m/z = 301 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,53), 0,008 (0,63), 1,195 (16,00), 4,263 (5,41), 5,122 (4,61), 5,875 (6,25), 7,322 (1,17), 7,340 (2,99), 7,360 (1,79), 7,375 (1,15).
Intermedio 89A.
3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)fenM]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropanoato de bencilo

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-A/-(1-cianociclopropil)benzamida (331 mg, 510 pmol) se disolvió en THF seco (15 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Se agregó bis(trimetilsilil)amida de potasio (1,6 ml, solución de 0,50 M en tolueno, 820 pmol) gota a gota y se agregó a 30 min. a -45 °C. Luego se agregó yoduro de sodio (38,2 mg, 255 pmol) y una solución de 3-{[(clorometoxi)carbonil]oxi}-2,2-dimetilpropanoato de bencilo (intermedio 88A,230 mg, 765 pmol) en THF (2 ml), el baño de hielo seco se retiró y la mezcla se agitó a temperatura ambiente durante 90 min. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-1810 pm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 435 mg (99 % de pureza, 92 % de rendimiento) del compuesto del título.
LC-MS (método 3): Rt = 2,72 min; MS (ESlpos): m/z = 913 [M+H]+ 1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008
(3,58), 0,008 (2,47), 1,122 (16,00), 1,157 (2,02), 1,175 (2,57), 1,193 (1,74), 1,235 (1,44), 1,356 (3,95), 1,490 (0,69), 1,644 (0,62), 1,754 (2,39), 1,988 (2,13), 2,184 (0,50), 2,328 (0,69), 2,524 (2,36), 2,670 (0,76), 4,021 (0,56), 4,039 (0,62), 4,098 (2,12), 5,070 (6,65), 5,240 (0,53), 5,374 (0,55), 7,301 (7,10), 7,313 (5,22), 7,328 (3,15), 7,579 (1,35), 7,600 (1,58), 7,772 (2,32), 7,800 (1,93), 7,820 (1,60), 7,934 (3,98), 8,206 (4,50), 8,422 (4,34), 8,704 (4,44).
Intermedio 90A.
(1S,2S)-2-[({[{2-doro-5-[2'-metM-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-N]benzoM}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclopentil (2E)-but-2-enodioato de tere-butilo

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluonTietox¡)feml]-1H-p¡razol-4-¡l}-A/-(1-cianocidopropil)benzamida (87,1 mg, 157 pmol) se disolvió en THF seco (6,0 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis^nmetilsiliOamida de potasio (600 pL, solución de 0,50 M en tolueno, 250 pmol) se agregó gota a gota y se agregó 30 min. a -45 °C. Luego, se agregó yoduro de sodio (11,8 mg, 78,7 pmol) y una solución de (1S,2S)-2-{[(doroTetox¡)carbon¡l]ox¡}c¡dopent¡l (2E)-but-2-enodioato de tere-butilo (intermedio 32A, 82,4 mg, 236 pmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante la noche. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-18 10 pm, gradiente de agua-acetonitrilo con t Fa al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 57,6 mg (42 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 5,19 min; MS (ESlpos): m/z = 809 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,41), 0,008 (0,46), 1,235 (0,76), 1,356 (3,04), 1,447 (16,00), 1,462 (1,49), 1,675 (0,48), 1,759 (0,52), 2,183 (0,41), 3,820 (4,01), 5,754 (4,57), 6,622 (0,82), 6,632 (0,97), 7,839 (0,85), 8,557 (0,59), 8,831 (0,57).
Intermedio 91A.
3-[({K2-cloro-5-[2'-metN-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-N]benzoN}(1-cianociclopropil)amino]metoxi}carbonil)oxi]propanoato de tere-butilo

2-Cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluonTietox¡)feml]-1H-p¡razol-4-¡l}-A/-(1-cianocidopropil)benzamida (276 mg, 500 jmol) se disolvió en THF seco (12,0 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis^rimetilsNi âmida de potasio (1,6 ml, solución de 0,50 M en tolueno, 800 |jmol) se agregó gota a gota y se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (37,5 mg, 250 |jmol) y una solución de 3-{[(doroTetox¡)carbon¡l]ox¡}propanoato de ferc-butilo (intermedio 82A, 179 mg, 750 jmol) en THF (4 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante la noche. La mezcla de reacción se vertió en agua y se extrajo con tres porciones de acetato de etilo. Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se evaporaron. El producto bruto se purificó por HPLC preparativa (RP C-1810 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 254 mg (100 % de pureza, 67 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,70 min; MS (ESlpos): m/z = 699 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,365 (16,00), 1,758 (1,31), 2,071 (4,24), 3,822 (11,81), 4,183 (0,88), 7,616 (0,74), 7,630 (0,85), 7,818 (1,23), 7,854 (0,94), 7,868 (0,93), 8,556 (1,65), 8,827 (1,62).
Intermedio 92A.
1-{2-doro-5-[2'-metN-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-N]fenM}-2-(1-cianocidopropN)-11,11-dimetiM,5-dioxo-4,6,9-trioxa-2-azadodecan-12-oato de tere-butilo

2-Cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluorpropan-2-¡l)-6-(t^fluo^Tetox¡)fen¡l]-1H-p¡razol-4-¡l}-A/-(1-danodclopropi^benzamida (138 mg, 250 jmol) se disolvió en THF seco (5,5 ml). Esta solución se enfrió a -45 °C en un baño de acetona - hielo seco. Bis^rimetNsiNOamida de potasio (650 jL, solución de 0,50 M en tolueno, 330 jmol) se agregó gota a gota y se agitó durante 30 min. a -45 °C. Luego, se agregó yoduro de sodio (18,7 mg, 125 jmol) y una solución de 3-(2-{[(doroTetox¡)carbon¡l]ox¡}etox¡)-2,2-d¡Tet¡lpropanoato de ferc-butilo (intermedio 23A, 93,2 mg, 300 jmol) en THF (2,0 ml), se retiró el baño de hielo seco y la mezcla se agitó a temperatura ambiente durante la noche. La mezcla de reacción se evaporó. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 110 mg (100 % de pureza, 53 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 5,15 min; MS (ESlpos): m/z = 771 [M+H-C4H8]++
1H-NMR (600 MHz, DMSO-d6) 5 [ppm]: 0,005 (0,68), 1,001 (10,40), 1,041 (2,96), 1,043 (3,10), 1,236 (0,57), 1,335 (16,00), 1,357 (6,04), 1,374 (4,82), 1,376 (3,88), 1,755 (1,20), 2,183 (0,59), 2,515 (0,76), 2,518 (0,74), 2,521 (0,68), 3,267 (0,76), 3,271 (0,48), 3,329 (3,16), 3,359 (0,65), 3,369 (0,78), 3,376 (0,59), 3,537 (1,72), 3,819 (14,82), 4,113 (0,87), 5,247 (0,59), 7,614 (0,74), 7,628 (0,87), 7,815 (1,24), 7,850 (0,87), 7,863 (0,83), 8,552 (1,72), 8,826 (1,63).
Síntesis de los ejemplos de compuestos de acuerdo con la presente invención
Compuesto de Ejemplo 1
Ácido (11E)-1-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-1,5,10-trioxo-4,6,9-trioxa-2-azatridec-11-en-13-oico

2-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometox¡)feml]-1H-p¡razol-4-il}benzo¡l)(1-cianocidopropil)amino]metoxi} carbonil)oxi]etil (2£)-but-2-enodioato de ferc-butilo (intermedio 4A, 105 mg, 114 jmol) se disolvió en TFA al 30 % en DCM (11 ml) y se agitó a temperatura ambiente durante 45 min. Los volátiles se evaporaron y el residuo se purificó por HPLC preparativa (RP C-18 10jm de gradiente de acetonitrilo-agua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5) y eventualmente por cromatografía ultrarrápida en gel de sílice eluida consecutivamente con ciclohexano, ciclohexano / acetato de etilo 4:1, acetato de etilo y por último acetato de etilo / metanol 10:1. El compuesto del título se aisló en un rendimiento de 56 mg (57% de teoría).
LC-MS (método 2): Rt = 1,26 min; MS (ESlpos): m/z = 865 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 5 [ppm]: -0,149 (1,74), -0,008 (16,00), 0,008 (11,78), 0,146 (1,48), 0,853 (0,63), 1,183 (0,78), 1,235 (3,59), 1,259 (1,56), 1,298 (0,78), 1,337 (0,81), 1,352 (0,85), 1,504 (1,96), 1,643 (1,89), 1,756 (6,78), 2,328 (1,41), 2,367 (1,07), 2,670 (1,44), 2,710 (0,96), 4,311 (11,48), 5,273 (1,44), 5,409 (1,48), 6,621 (1,48), 6,661 (10,11), 6,675 (13,67), 6,715 (2,04), 7,583 (3,30), 7,603 (4,07), 7,799 (9,78), 7,823 (4,70), 7,928 (11,37), 8,204 (11,96), 8,441 (10,78), 8,764 (7,52), 11,201 (0,52), 13,223 (1,63).
Compuesto de Ejemplo 2
Ácido (12E)-1-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-1,5,11-trioxo-4,6,10-trioxa-2-azatetradec-12-en-14-oico

3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-¡l}benzo¡l)(1-cianociclopropiOamino] metox^carboni^ox^propil (2£)-but-2-enodioato de ferc-butilo (intermedio 7A, 60,0 mg, 64,1 |jmol) se disolvió en TFA al 30 % en DCM (3,0 ml) y se agitó a temperatura ambiente durante 15 min. Los volátiles
se evaporaron y el residuo se purificó por HPLC preparativa (RP C-18 10|jm de gradiente de acetonitrilo-agua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuvieron 38,0 mg (64 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,34 min; MS (ESlpos): m/z = 879 [M+H]+
1H-NMR (500 MHz, DMSO-d6) 8 [ppm]: -0,120 (0,56), 1,235 (1,78), 1,490 (2,29), 1,655 (1,95), 1,756 (8,08), 1,930 (7,19), 2,363 (1,39), 2,637 (1,28), 4,123 (6,58), 4,159 (10,37), 5,254 (1,56), 5,405 (1,67), 6,638 (1,90), 6,670 (15,33), 6,679 (16,00), 6,710 (2,01), 7,589 (4,40), 7,605 (5,24), 7,790 (8,59), 7,803 (6,63), 7,820 (5,30), 7,927 (13,60), 8,201 (15,00), 8,443 (10,98), 8,766 (9,59), 13,209 (0,78).
Compuesto de Ejemplo 3
Ácido 1-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-1,5,10-trioxo-4,6,9-trioxa-2-azatridecan-13-oico

2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil butanodioato de ferc-butilo (intermedio 10A, 100 mg, 108 jmol) se disolvió en TFA al 30 % en DCM (5,0 ml) y se agitó a temperatura ambiente durante 15 min. Los volátiles se evaporaron y el residuo se purificó por HPLC preparativa (RP C-18 10jm de gradiente de acetonitrilo-agua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuvieron 78,0 mg (95 % de pureza, 79 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,20 min; MS (ESlpos): m/z = 867 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,149 (0,42), -0,022 (0,86), -0,008 (3,63), 0,008 (2,88), 1,235 (1,93), 1,513 (2,33), 1,647 (2,07), 1,761 (8,31), 2,323 (0,89), 2,328 (1,12), 2,367 (1,10), 2,441 (11,63), 2,454 (15,22), 2,473 (12,55), 2,670 (1,10), 2,710 (0,99), 3,540 (1,05), 3,552 (0,99), 3,566 (0,78), 4,018 (0,47), 4,030 (0,44), 4,183 (11,61), 4,363 (0,89), 5,279 (1,65), 5,403 (1,75), 7,556 (0,44), 7,592 (4,26), 7,612 (5,25), 7,758 (0,84), 7,801 (11,92), 7,824 (5,73), 7,936 (14,56), 8,209 (16,00), 8,213 (15,61), 8,446 (12,94), 8,770 (10,75), 12,198 (0,81).
Compuesto de Ejemplo 4
Ácido (12£)-1-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-8,8-dimetil-1,5,11-trioxo-4,6,10-trioxa-2-azatetradec-12-en-14-oico

3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-¡l}benzo¡l)(1cianocidopropil)amino] metoxi}carbonil)oxi]-2,2-dimetilpropil (2E)-but-2-enodioato de ferc-butilo (intermedio 13A, 298 mg, 309 |jmol) se disolvió en TFA al 30 % en DCM (15 ml) y se agitó a temperatura ambiente durante 25 min. Los volátiles se evaporaron y el residuo se purificó por HPLC preparativa (RP C-18 10jm de gradiente de acetonitriloagua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuvieron 204 mg (100 % de pureza, 73 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,60 min; MS (ESIpos): m/z = 907 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (2,25), 0,888 (16,00), 1,235 (0,89), 1,751 (2,55), 2,073 (13,35), 2,328 (1,35), 2,367 (0,82), 2,670 (1,53), 2,710 (0,97), 3,915 (7,86), 5,399 (0,64), 6,688 (12,63), 7,577 (1,40), 7,599 (1,79), 7,800 (3,96), 7,930 (4,29), 8,203 (5,10), 8,439 (4,13), 8,753 (3,27), 13,206 (1,89).
Compuesto de Ejemplo 5
Ácido 1-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-8,8-dimetil-1,5,11-trioxo-4,6,10-trioxa-2-azatetradecan-14-oico

3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]-2,2-dimetilpropil butanodioato de ferc-butilo (intermedio 15A, 325 mg, 337 jmol) se disolvió en TFA al 30 % en DCM (16 ml) y se agitó a temperatura ambiente durante 10 min. Los volátiles se evaporaron y el residuo se purificó por HPLC preparativa (RP C-18 10jm de gradiente de acetonitriloagua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuvieron 237 mg (77 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,54 min; MS (ESIpos): m/z = 909 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (2,76), 0,008 (1,95), 0,848 (16,00), 1,234 (0,49), 1,501 (0,70), 1,757 (2,59), 2,073 (8,11), 2,328 (1,65), 2,366 (1,08), 2,463 (4,81), 2,670 (1,68), 2,710 (1,05), 3,784 (4,59), 3,867 (2,84), 5,404 (0,59), 7,585 (1,35), 7,606 (1,62), 7,803 (4,27), 7,938 (4,38), 8,210 (5,05), 8,443 (3,68), 8,760 (3,19), 12,197 (2,14).
Compuesto de Ejemplo 6
Ácido (2E)-4-({(rel 1R,2S)-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclopentil}oxi)-4-oxobut-2-enoico

(rel 1R,2S)-2-[({[(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-il}benzoil)(1-cianocidopropil)amino] metox¡}carbon¡l)ox¡]c¡clopent¡l (2E)-but-2-enod¡oato de ferc-but¡lo (intermedio 18A, 455 mg, 473 pmol) se d¡solv¡ó en DCM (10 ml) y se agregó TFA (1,8 ml). La mezcla se ag¡tó a temperatura amb¡ente durante 1 h. Los volát¡les se evaporaron y el res¡duo se pur¡f¡có por HPLC preparat¡va (RP C-18 10|jm de grad¡ente de aceton¡tr¡lo-agua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuv¡eron 135 mg (100 % de pureza, 32 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 2): Rt = 1,34 m¡n; MS (ESlpos): m/z = 905 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,149 (0,57), -0,008 (4,30), 0,008 (3,38), 0,146 (0,39), 1,029 (0,75), 1,235 (1,80), 1,459 (3,38), 1,547 (4,12), 1,742 (13,68), 1,958 (4,30), 2,073 (1,10), 2,328 (1,36), 2,367 (1,71), 2,524 (4,08), 2,671 (1,36), 2,711 (1,58), 4,944 (3,59), 5,110 (4,82), 5,244 (1,75), 6,632 (10,43), 7,568 (4,34), 7,587 (5,35), 7,790 (15,30), 7,810 (6,44), 7,934 (14,73), 8,209 (16,00), 8,432 (15,39), 8,741 (9,60), 13,189 (2,81).
Compuesto de Ejemplo 7
Ácido 4-({(rel 1R,2S)-2-[({[(2-cloro-5-{H2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)ammo]metoxi}carboml)oxi]ciclopentil}oxi)-4-oxobutanoico

(rel 1R,2S)-2-[({[(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-¡l}benzo¡l)(1-danoddoprop¡l)am¡no] metox¡}carbon¡l)ox¡]c¡clopent¡l butanod¡oato de ferc-but¡lo (¡ntermed¡o 20A, 515 mg, 534 pmol) se d¡solv¡ó en DCM (10 ml) y se agregó TFA (2,1 ml). La mezcla se ag¡tó a temperatura amb¡ente durante 20 m¡n. Los volát¡les se evaporaron y el res¡duo se pur¡f¡có por HPLC preparat¡va (RP C-18 10pm de grad¡ente de aceton¡tr¡lo-agua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuv¡eron 300 mg (98 % de pureza, 61 % de rend¡m¡ento) del compuesto del título.
LC -M S (m é to d o 3): Rt = 2,41 m in; M S (E S lp o s ): m /z = 907 [M H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (1,23), 0,008 (1,36), 1,218 (1,44), 1,235 (0,94), 1,283 (3,94), 1,509 (4,09), 1,628 (4,47), 1,693 (5,07), 1,758 (8,98), 1,908 (4,09), 2,074 (2,64), 2,671 (0,76), 2,711 (0,65), 4,874 (2,93), 4,996 (3,70), 5,277 (1,31), 7,588 (4,05), 7,609 (5,11), 7,802 (12,66), 7,820 (6,24), 7,936 (13,33), 8,211 (14,70), 8,405 (0,96), 8,444 (11,48), 8,759 (9,61), 12,165 (16,00).
Compuesto de Ejemplo 8
Ácido 1-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-11,11-dimetil-1,5-dioxo-4,6,9-trioxa-2-azadodecan-12-oico

1-(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-danoddopropil)-11,11-dimetil-1,5-dioxo-4,6,9-trioxa-2-azadodecan-12-oato de tere-butilo (intermedio 24A, 195 mg, 211 |jmol) se disolvió en TFA al 30 % en DCM (10 ml) y se agitó por 5 min a temperatura ambiente. Los volátiles se evaporaron al vacío a temperatura ambiente. El residuo se purificó por HPLC preparativa (RP C-1810 jm , gradiente de acetonitrilo- agua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se aislaron 152 mg (83 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,57 min; MS (ESlpos): m/z = 867 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,027 (5,86), 1,758 (0,71), 3,340 (16,00), 3,360 (2,50), 3,546 (0,85), 4,115 (0,46), 7,611 (0,45), 7,794 (0,72), 7,802 (0,70), 7,824 (0,46), 7,937 (1,16), 8,213 (1,28), 8,444 (1,07), 8,768 (0,88). Compuesto de Ejemplo 9
Ácido 1-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-12,12-dimetil-1,5,10-trioxo-4,6,9-trioxa-2-azatridecan-13-oico

4-{2-[({[(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]etil} 2,2-dimetilbutanodioato de 1-tere-butilo (intermedio 30A, 310 mg, 100 % de pureza, 326 jmol) se disolvió en TFA al 30 % en DCM (16 ml) y se agitó por 10 min a temperatura ambiente. Los volátiles se evaporaron y el residuo se purificó por HPLC preparativa (RP C-1810 jm , gradiente de acetonitriloagua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuvieron 207 mg (71 % de rendimiento) del compuesto del título.
LC -M S (m é to d o 4): Rt = 4 ,43 m in; M S (E S lp o s ): m /z = 895 [M H]+
1H-NMR (500 MHz, DMSO-d6) 8 [ppm]: -0,120 (0,63), -0,007 (7,27), 0,006 (4,39), 0,116 (0,58), 1,127 (16,00), 1,168 (1,12), 1,512 (0,48), 1,660 (0,43), 1,760 (1,88), 2,362 (0,42), 2,636 (0,45), 3,541 (1,40), 4,157 (2,46), 4,200 (1,25), 7,593 (1,01), 7,609 (1,16), 7,793 (1,88), 7,805 (1,58), 7,822 (1,24), 7,935 (3,31), 8,208 (3,72), 8,441 (2,73), 8,769 (2,32).
Compuesto de Ejemplo 10
Acido (2E)-4-({(1S,2S)-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)feml]-1H-p¡razol-4-¡l}benzo¡l)(1-c¡anoc¡cloprop¡l)ammo]metox¡}carboml)ox¡]c¡clopent¡l}ox¡)-4-oxobut-2-enoico

(1S,2S)-2-[({[(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-p¡razol-4-il}benzoN)(1-danoddopropN)aiTiino] metoxi}carboml)oxi]ddopent¡l (2£)-but-2-enodioato de ferc-butilo (intermedio 33A, 120 mg, 125 pmol) se disolvió en DCM (5,0 ml) y se agregó TFA (480 pL). La mezcla se agitó a temperatura ambiente durante 1 hora. Luego, se agregaron otros 480 pL de TFA y se siguió agitando durante 45 min. El solvente se destiló a temperatura ambiente al vacío y el residuo se purificó por HPLC preparativa (RP C-18 10 pm, gradiente de acetonitrilo- agua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuvieron 58,0 mg (51 % de rendimiento) del compuesto del título.
LC-MS (método 3): Rt = 2,46 min; MS (ESlpos): m/z = 905 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,149 (1,27), -0,008 (9,64), 0,008 (10,30), 0,146 (1,15), 1,141 (1,33), 1,235 (2,85), 1,465 (2,61), 1,501 (2,30), 1,673 (8,42), 1,754 (9,70), 2,025 (4,55), 2,328 (1,70), 2,367 (2,36), 2,670 (1,64), 2,710 (2,36), 4,883 (3,33), 5,067 (4,30), 5,277 (1,45), 5,384 (1,39), 6,583 (5,09), 6,622 (13,39), 6,669 (16,00), 6,709 (6,00), 7,583 (4,00), 7,603 (4,97), 7,799 (14,30), 7,820 (5,82), 7,931 (12,73), 8,204 (13,94), 8,442 (13,33), 8,762 (9,27), 13,231 (1,45).
Compuesto de Ejemplo 11
Ac¡do 4-({(1S,2S)-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-¡l}benzo¡l)(1-c¡anoc¡cloprop¡l)ammo]metox¡}carboml)ox¡]c¡clopent¡l}ox¡)-4-oxobutano¡co

(1S,2S)-2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-il}benzoil)(1-cianocidopropil)amino] metox¡}carbon¡l)ox¡]c¡clopent¡l butanodioato de ferc-butilo (intermedio 35A, 100 mg, 104 |jmol) se disolvió en DCM (5,0 ml) y se agregó TFA (1,0 ml). La mezcla se agitó a temperatura ambiente durante 30 min, luego los volátiles se evaporaron a temperatura ambiente al vacío. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm, gradiente de acetonitrilo- agua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuvieron 66 mg (70 % de rendimiento) del compuesto del título.
LC-MS (método 3): Rt = 2,41 min; MS (ESlpos): m/z = 907 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (2,67), 0,008 (3,56), 1,235 (0,89), 1,567 (2,67), 1,646 (4,44), 1,755 (4 ,4 4 ), 1 , 95 3 (2,67), 2,330 (1,78), 2,368 (2,67), 2,445 (16,00), 2,672 (1,78), 2,712 (1,78), 3,071 (0,89), 3,105 (0,89), 3,229 (1,78), 3,489 (0,89), 3,643 (0,89), 3,664 (0,89), 4,279 (0,89), 4,791 (1,78), 4,955 (2,67), 5,389 (0,89), 7,585 (2,67), 7,606 (2,67), 7,799 (6,22), 7,823 (2,67), 7,936 (7,11), 8,209 (8,00), 8,442 (6,22), 8,759 (5,33).
Compuesto de Ejemplo 12
Ácido 1-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}fenil)-2-(1-cianociclopropil)-1,5,10-trioxo-4,6,9-trioxa-2-azatetradecan-l4-oico

2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-¡l}benzo¡l)(1-c¡anoc¡cloprop¡l)am¡no] metoxi}carbonil)ox¡]et¡l pentanodioato de ferc-butilo (intermedio 38A, 150 mg, 160 jmol) se disolvió en DCM (5,0 ml) y se agregó TFA (2,0 ml). La mezcla se agitó a temperatura ambiente durante 10 min, luego los volátiles se evaporaron a temperatura ambiente al vacío. El producto bruto se purificó por HPLC preparativa (RP C-18 10 jm , gradiente de acetonitrilo- agua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuvieron 79 mg (56 % de rendimiento) del compuesto del título.
LC-MS (método 3): Rt = 2,34 min; MS (ESlpos): m/z = 881 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 5 [ppm]: -0,149 (0,83), 0,008 (6,71), 0,146 (0,91), 1,044 (0,61), 1,235 (1,59), 1,508 (2,69), 1,681 (9,25), 1,699 (12,06), 1,718 (9,78), 1,756 (7,51), 2,073 (4,51), 2,203 (8,87), 2,221 (16,00), 2,240 (7,85), 2,293 (6,10), 2,311 (10,65), 2,328 (7,70), 2,366 (1,97), 2,670 (1,97), 2,709 (1,67), 3,409 (8,80), 4,177 (9,52), 4,216 (6,26), 4,590 (0,76), 5,175 (1,21), 5,272 (1,52), 5,404 (1,71), 7,557 (1,25), 7,588 (4,09), 7,608 (5,04), 7,757 (2,35), 7,794 (8,95), 7,823 (5,12), 7,935 (13,95), 8,209 (15,32), 8,442 (11,79), 8,767 (9,21), 12,058 (1,97).
Compuesto de Ejemplo 13
Ácido 1-[10-(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}fenil)-9-(1-cianociclopropil)-6,10-dioxo-2,5,7-trioxa-9-azadecanan-1-oil]ciclopropano-1-carboxilico

1-{2-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-¡l}benzo¡l)(1-cianocidopropil)amino] metox¡}carbon¡l)ox¡]et¡l} c¡clopropano-1,1-d¡carbox¡lato de 1-terc-but¡lo (¡ntermed¡o 41 A, 570 mg, 609 pmol) se d¡solv¡ó en DCM (10 ml) y se agregó TFA (2,0 ml). La mezcla se ag¡tó a temperatura amb¡ente durante 10 m¡n, luego los volát¡les se evaporaron a temperatura amb¡ente al vacío. El producto bruto se pur¡f¡có por HPLC preparat¡va (RP C-18 10 pm, grad¡ente de aceton¡tr¡lo-agua con TFA al 0,01 % en ambos eluyentes, 10:90->95:5). Se obtuv¡eron 365 mg (66 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 3): Rt = 2,39 m¡n; MS (ESlpos): m/z = 879 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 5 [ppm]: 1,280 (16,00), 1,505 (0,86), 1,648 (0,73), 1,762 (2,91), 2,073 (0,50), 2,328 (0,52), 2,670 (0,53), 4,226 (6,88), 5,277 (0,61), 5,395 (0,64), 7,591 (1,44), 7,611 (1,82), 7,802 (3,23), 7,826 (1,95), 7,936 (5,09), 8,212 (5,51), 8,446 (4,38), 8,767 (3,48), 12,780 (0,99).
Compuesto de Ejemplo 14
Ácido (2E)-4-({c/s-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexil}oxi)-4-oxobut-2-enoico

c/s-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-¡l}benzo¡l)(1-c¡anoc¡cloprop¡l)am¡no] metox¡}carbon¡l)ox¡]c¡clohex¡l (2E)-but-2-enod¡oato de terc-but¡lo (¡ntermed¡o 46A, 320 mg, 328 pmol) se d¡solv¡ó en TFA al 30 % en DCM (16 ml) y la mezcla se ag¡tó a temperatura amb¡ente durante 25 m¡n. El solvente se dest¡ló, el res¡duo se absorb¡ó en aceton¡tr¡lo y se pur¡f¡có por HPLC preparat¡va (RP C-18 10 pm de aceton¡tr¡lo:agua TFA al 0,01% 10:90->95:5). Las fracc¡ones que cont¡enen el producto deseado se comb¡naron y l¡of¡l¡zaron para dar 215 mg (71 % de rend¡m¡ento) del compuesto del título.
LC -M S (m é tod o 4): Rt = 4 ,57 m in; M S (E S lp os ): m /z = 919 [M H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (3,46), 0,008 (3,26), 1,234 (0,64), 1,697 (11,46), 1,764 (4,98), 2,073 (1,23), 2,328 (2,39), 2,366 (1,39), 2,670 (2,23), 2,710 (1,35), 3,472 (14,21), 4,595 (1,71), 4,850 (2,19), 5,261 (0,84), 6,643 (0,88), 6,683 (16,00), 6,688 (15,20), 6,727 (0,84), 7,593 (2,11), 7,614 (2,43), 7,802 (7,68), 7,821 (3,18), 7,924 (6,29), 8,196 (7,44), 8,452 (5,13), 8,775 (4,74), 13,181 (0,64).
Compuesto de Ejemplo 15
Ácido 4-({c/s-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexil}oxi)-4-oxobutanoico

c/s-4-[({[(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-p¡razol-4-N}benzoN)(1-danoddopropN)amino] metoxi}carboml)ox¡]ddohexil butanodioato de ferc-butilo (intermedio 49A, 273 mg, 279 pmol) se disolvió en TFA al 30 % en DCM (14 ml) y la mezcla se agitó a temperatura ambiente durante 15 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 pm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Las fracciones que contienen el producto deseado se combinaron y liofilizaron para dar 255 mg (99 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,50 min; MS (ESlpos): m/z = 921 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,149 (0,62), -0,008 (5,28), 0,008 (4,78), 0,146 (0,62), 1,234 (0,45), 1,505 (1,28), 1,663 (9,81), 1,762 (5,48), 2,073 (2,23), 2,323 (2,10), 2,327 (2,89), 2,332 (2,23), 2,366 (1,20), 2,443 (2,27), 2,467 (15,05), 2,473 (16,00), 2,523 (8,99), 2,665 (2,47), 2,670 (3,34), 2,674 (2,52), 2,710 (1,40), 4,566 (2,02), 4,725 (2,72), 5,262 (0,91), 5,410 (0,95), 7,592 (2,27), 7,614 (2,76), 7,801 (8,74), 7,820 (3,59), 7,937 (7,55), 8,210 (8,29), 8,214 (8,29), 8,451 (6,06), 8,773 (5,65), 12,184 (1,36).
Compuesto de Ejemplo 16
Ácido (2£)-4-({írans-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)ammo]metoxi}carboml)oxi]ciclohexil}oxi)-4-oxobut-2-enoico

frans-4-[({[(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-l^eptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-¡l}benzo¡l)(1-danoddoprop¡l)am¡no] metox^carboni^ox^ciclohexil (2£)-but-2-enodioato de ferc-butilo (intermedio 51A, 192 mg, 197 pmol) se disolvió en TFA al 30 % en DCM (19 ml) y la mezcla se agitó a temperatura ambiente durante 30 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-1810
|jm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Las fracciones que contienen el producto deseado se combinaron y liofilizaron para dar 133 mg (73 % de teoría) del compuesto del título.
LC-MS método 4): Rt = 4,56 min; MS (ESIpos): m/z = 919 [M+H]+
1H-NMR (500 MHz, DMSO-d6) 8 [ppm]: -0,007 (3,06), 0,006 (2,21), 1,234 (0,72), 1,511 (9,00), 1,654 (2,19), 1,766 (7,12), 1,860 (7,44), 2,365 (0,55), 2,639 (0,54), 4,561 (3,25), 4,806 (3,40), 5,262 (1,65), 5,408 (1,74), 6,636 (2,45), 6,668 (15,70), 6,679 (16,00), 6,710 (2,42), 7,601 (5,00), 7,617 (5,79), 7,805 (13,70), 7,823 (5,29), 7,941 (10,76), 8,214 (12,49), 8,464 (10,82), 8,704 (0,47), 8,784 (10,72).
Compuesto de Ejemplo 17
Ácido c/s-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexano-1-carboxílico

c/s-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]ciclohexano-1-carboxilato de ferc-butilo (intermedio 53A, 190 mg, 210 jmol) se disolvió en TFA al 30 % en DCM (19 ml) y la mezcla se agitó a temperatura ambiente durante 10 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 jm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Las fracciones que contienen el producto deseado se combinaron y liofilizaron para dar 110 mg (62 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,47 min; MS (ESIpos): m/z = 849 [M+H]+
iH-NMR (500 MHz, DMSO-d6) 8 [ppm]: -0,022 (0,83), 0,006 (1,66), 1,234 (1,39), 1,275 (0,53), 1,491 (2,58), 1,605 (16,00), 1,648 (8,81), 1,673 (8,69), 1,763 (9,17), 2,297 (3,65), 2,361 (0,73), 2,638 (0,56), 4,637 (4,73), 4,805 (0,50), 5,252 (1,82), 5,392 (1,89), 7,589 (5,24), 7,607 (5,98), 7,814 (13,64), 7,944 (13,72), 8,217 (15,39), 8,457 (12,23), 8,776 (11,73), 12,133 (0,48).
Compuesto de Ejemplo 18
frans-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)ammo]metoxi}carboml)oxi]ciclohexano-1-carboxílico

frans-4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino] metoxi}carbonil)oxi]ciclohexano-1-carboxilato de ferc-butilo (intermedio 55A, 189 mg, 209 jmol) se disolvió en TFA al 30 % en DCM (19 ml) y la mezcla se agitó a temperatura ambiente durante 10 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 jm
de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Las fracciones que contienen el producto deseado se combinaron y liofilizaron para dar 121 mg (94 % de pureza, 64 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,48 min; MS (ESIpos): m/z = 849 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: -0,022 (0,75), 0,005 (1,63), 1,190 (0,66), 1,234 (1,15), 1,360 (8,78), 1,454 (2,90), 1,659 (2,14), 1,759 (9,21), 1,863 (8,45), 2,033 (0,88), 2,075 (1,12), 2,167 (3,86), 2,387 (1,15), 2,426 (1,45), 2,518 (3,02), 2,521 (3,23), 2,524 (3,17), 2,615 (1,03), 2,655 (1,09), 4,398 (3,71), 4,573 (0,54), 5,241 (1,96), 5,389 (2,05), 7,563 (0,69), 7,577 (1,03), 7,599 (6,37), 7,613 (7,00), 7,760 (1,39), 7,785 (10,20), 7,806 (6,97), 7,820 (6,13), 7,940 (14,97), 8,215 (16,00), 8,454 (13,74), 8,778 (13,13), 12,109 (0,78).
Compuesto de Ejemplo 19
Ácido [({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]acético

[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)fenil]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]acetato de ferc-butilo (intermedio 57A, 115 mg, 137 pmol) se disolvió en
TFA al 30 % en DCM (19 ml) y la mezcla se agitó a temperatura ambiente durante 10 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-1810 pm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Las fracciones que contienen el producto deseado se combinaron y liofilizaron para dar 34,0 mg
(97 % de pureza, 31 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,18 min; MS (ESIpos): m/z = 781 [M+H]+
iH-NMR (500 MHz, DMSO-d6) 8 [ppm]: -0,120 (1,08), 0,117 (0,99), 1,170 (0,48), 1,184 (0,97), 1,198 (0,48), 1,234 (0,97), 1,516 (2,26), 1,658 (1,97), 1,767 (9,02), 4,204 (0,50), 4,218 (0,54), 4,475 (1,18), 4,524 (16,00), 4,688 (1,39), 5,210 (0,93), 5,312 (1,82), 5,444 (1,93), 7,539 (0,44), 7,603 (4,58), 7,619 (5,41), 7,689 (0,62), 7,780 (7,71), 7,808 (5,37), 7,824 (4,85), 7,947 (14,20), 8,223 (15,38), 8,321 (0,39), 8,444 (15,83), 8,771 (12,83), 13,240 (1,08).
Ejemplo 20
Ácido 4-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetM)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoM}(1-cianociclopropil)amino]metoxi}carbonil)oxi]butanoico

4-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianocidopropil)amino]metoxi}carbonil)oxi]butanoato de ferc-butilo (intermedio 59A, 230 mg, 299 |jmol) se disolvió en TFA al 30 % en DCM (10 ml) y la mezcla se agitó a temperatura ambiente durante 7 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-1810 jm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 2:1-acetato de etilo metanol 80:20). Se aislaron 60,9 mg (100 % de pureza, 29 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 3,89 min; MS (ESlpos): m/z = 713 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,500 (0,44), 1,760 (2,79), 2,242 (1,98), 3,820 (16,00), 4,037 (1,23), 7,613 (0,94), 7,627 (1,05), 7,825 (1,70), 7,843 (1,21), 7,858 (1,12), 8,557 (2,18), 8,830 (2,26).
Ejemplo 21
Ácido 3-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetM)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropanoico

3-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropanoato de ferc-butilo (intermedio 61A, 160 mg, 204 jmol) se disolvió en TFA al 30 % en DCM (8 ml) y la mezcla se agitó a temperatura ambiente durante 5 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-1810 jm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Se aislaron 78,3 mg (100 % de pureza, 53 % de rendimiento) del compuesto del título. LC-MS (método 4): Rt = 4,10 min; MS (ESlpos): m/z = 727 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,47), 0,008 (0,47), 1,034 (0,97), 1,064 (12,20), 1,145 (0,70), 1,493 (0,48), 1,644 (0,43), 1,762 (1,81), 2,524 (1,47), 3,823 (16,00), 4,035 (1,24), 7,612 (1,00), 7,632 (1,26), 7,841 (2,64), 7,864 (1,27), 8,564 (2,70), 8,828 (2,75).
Ejemplo 22
Ácido trans (2£)-4-({-4-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexil}oxi)-4-oxobut-2-enoico

trans-4-[({[{2-doro-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzo¡l}(1-cianocidopropil)amino]metoxi}carbonil)oxi]cidohexil (2E)-but-2-ened¡oato de ferc-butilo (¡ntermed¡o 62A, 188 mg, 214 |jmol) se disolvió en TFA al 30 % en DCM (15 ml) y la mezcla se agitó a temperatura ambiente durante 15 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 jm de aceton¡tr¡lo:agua TFA al 0,01% 10:90->95:5). Se aislaron 125 mg (100 % de pureza, 71 % de rendimiento).
LC-MS (método 4): Rt = 4,11 min; MS (ESlpos): m/z = 823 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,149 (1,71), 0,008 (16,00), 0,146 (1,67), 1,236 (1,47), 1,511 (6,79), 1,766 (5,25), 1,863 (5,63), 2,328 (0,85), 2,366 (1,19), 2,670 (1,02), 2,710 (1,26), 3,431 (8,97), 4,560 (2,15), 4,810 (2,39), 5,255 (1,16), 5,400 (1,19), 6,626 (1,16), 6,666 (10,85), 6,676 (11,39), 6,715 (1,23), 7,621 (3,17), 7,641 (3,92), 7,841 (7,23), 7,866 (3,72), 8,577 (7,03), 8,844 (7,03), 13,178 (0,89).
Ejemplo 23
Ácido c/s-4-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-M]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexano-1-carboxílico

c/s-4-[({[{2-Cloro-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzo¡l}(1-cianocidopropil)amino]metoxi}carbonil)oxi]cidohexano-1-carboxilato de terc-but¡lo (¡ntermed¡o 63A, 300 mg, 371 |jmol) se d¡solv¡ó en TFA al 30 % en DCM (15 ml) y la mezcla se ag¡tó a temperatura amb¡ente durante 15 m¡n. El solvente se dest¡ló, el res¡duo se absorb¡ó en aceton¡tr¡lo y se pur¡f¡có por HPLC preparat¡va (RP C-18 10 jm de aceton¡tr¡lo:agua TFA al 0,01% 10:90->95:5). Se obtuv¡eron 51,4 mg (100 % de pureza, 18 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 4,07 m¡n; MS (ESlpos): m/z = 753 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,489 (0,54), 1,609 (2,64), 1,643 (1,72), 1,677 (1,68), 1,767 (1,77), 2,301 (0,79), 3,824 (16,00), 4,636 (1,05), 5,399 (0,41), 7,613 (1,19), 7,628 (1,36), 7,849 (3,12), 8,573 (2,84), 8,842 (2,82).
Ejemplo 24
Ácido trans -4-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexano-1-carboxílico

frans-4-[({[{2-Cloro-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzo¡l}(1-danoddoprop¡l)am¡no]metox¡}carbon¡l)ox¡]ddohexano-1-carbox¡lato de terc-but¡lo (¡ntermed¡o 64A, 200 mg, 247 jmol) se d¡solv¡ó en TFA al 30 % en DCM (10 ml) y la mezcla se ag¡tó a temperatura amb¡ente durante 5 m¡n. El solvente se dest¡ló, el res¡duo se absorb¡ó en aceton¡tr¡lo y se pur¡f¡có por HPLC preparativa (RP C-18 10 jm de aceton¡tr¡lo:agua TFA al 0,01% 10:90->95:5). Se obtuv¡eron 53,1 mg (100 % de pureza, 29 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 4,04 m¡n; MS (ESlpos): m/z = 753 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,360 (1,47), 1,461 (0,56), 1,655 (0,41), 1,763 (1,63), 1,861 (1,32), 2,172 (0,75), 3,823 (16,00), 4,393 (0,69), 7,624 (1,30), 7,638 (1,48), 7,825 (2,02), 7,847 (1,26), 7,863 (1,15), 8,576 (2,69), 8,845 (2,67).
Ejemplo 25
Ácido (12£)-1-{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]fenil}-2-(1-cianociclopropil)-1,5,11-trioxo-4,6,10-trioxa-2-azatetradec-12-en-14-oico

3-[({[{2-doro-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzo¡l}(1-cianocidopropil)amino]metoxi}carbonil)oxi]propil (2E)-but-2-ened¡oato de terc-but¡lo (¡ntermed¡o 65A, 200 mg, 238 pmol) se d¡solv¡ó en TFA al 30 % en DCM (12 ml) y la mezcla se ag¡tó a temperatura amb¡ente durante 10 m¡n. El solvente se dest¡ló, el res¡duo se absorb¡ó en aceton¡tr¡lo y se pur¡f¡có por HPLC preparat¡va (RP C-18 10 pm de aceton¡tr¡lo:agua TFA al 0,01% 10:90->95:5). Se obtuv¡eron 8,10 mg (100 % de pureza, 4 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 4,00 m¡n; MS (ESlpos): m/z = 783 [M+H]+
1H-NMR (500 MHz, DMSO-d6) 8 [ppm]: -0,120 (0,72), -0,007 (7,75), 0,006 (5,08), 0,116 (0,66), 1,225 (1,22), 1,236 (1,36), 1,259 (0,63), 1,285 (0,81), 1,297 (0,83), 1,337 (0,74), 1,494 (0,46), 1,760 (1,58), 1,908 (0,98), 1,917 (1,05), 1,929 (1,49), 1,941 (1,10), 2,040 (1,53), 2,358 (0,42), 2,362 (0,61), 2,365 (0,42), 2,518 (0,85), 2,522 (0,59), 2,632 (0,42), 2,635 (0,61), 2,639 (0,41), 3,820 (16,00), 4,124 (1,36), 4,144 (1,64), 4,156 (2,17), 6,622 (0,46), 6,653 (1,90), 6,674 (2,34), 6,706 (0,68), 7,612 (0,98), 7,628 (1,14), 7,829 (1,68), 7,843 (1,31), 7,860 (1,03), 8,560 (2,30), 8,834 (2,17).
Ejemplo 26
Ácido 1-{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]fenil}-2-(1-cianociclopropil)-1,5,1l-trioxo-4,6,10-trioxa-2-azatetradecan-14-oico

3-[({[{2-cloro-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzo¡l}(1-c¡anoc¡doprop¡l)am¡no]metox¡}carbon¡l)ox¡]prop¡l butanod¡oato de terc-but¡lo (¡ntermed¡o 68A, 190 mg, 226 pmol) se d¡solv¡ó en t Fa al 30 % en DCM (10 ml) y la mezcla se ag¡tó a temperatura amb¡ente durante 5 m¡n. El solvente se dest¡ló, el res¡duo se absorb¡ó en aceton¡tr¡lo y se pur¡f¡có por HPLC preparativa (RP C-1810 pm de aceton¡tr¡lo:agua TFA al 0,01% 10:90->95:5). Se obtuv¡eron 36,1 mg (100 % de pureza, 20 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 3,96 m¡n; MS (ESlpos): m/z = 785 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 5 [ppm]: 1,397 (0,45), 1,491 (0,52), 1,664 (0,46), 1,765 (1,77), 1,847 (1,27), 1,857 (1,82), 1,867 (1,36), 2,426 (0,45), 2,456 (3,27), 2,467 (3,22), 2,575 (0,52), 3,822 (16,00), 4,027 (2,34), 4,036 (1,52), 4,090 (1,44), 5,249 (0,40), 5,405 (0,42), 7,620 (1,13), 7,634 (1,29), 7,832 (1,98), 7,847 (1,31), 7,861 (1,22), 8,568 (2,47), 8,839 (2,62).
Ejemplo 27
Ácido 1-[10-{2-cloro-5-[2'-metil-5'-(pentafluoroetM)-4'-(t
N fl
uorometil)-2'H-[1,3'-bipirazol]-4-il]feml}-9-(1-cianociclopropil)-6,10-dioxo-2,5,7-trioxa-9-azadecanan-1-oil]ciclopropano-1-carboxílico

2-[({[{2-cloro-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(trifluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzo¡l}(1-cianocidopropil)amino]metoxi}carbonil)oxi]etil et¡l(met¡l)propanod¡oato de terc-but¡lo (¡ntermed¡o 69A, 167 mg, 195 |jmol) se d¡solv¡ó en TFA al 30 % en DCM (15 ml) y la mezcla se ag¡tó a temperatura amb¡ente durante 5 m¡n. El solvente se dest¡ló, el res¡duo se absorb¡ó en aceton¡tr¡lo y se pur¡f¡có por HPLC preparat¡va (RP C-18 10 pm de aceton¡tr¡lo:agua TFA al 0,01% 10:90->95:5). Se obtuv¡eron 24,6 mg (100 % de pureza, 16 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 4,03 m¡n; MS (ESlpos): m/z = 783 [M+H]+
iH-NMR (500 MHz, DMSO-d6) 5 [ppm]: -0,120 (0,79), -0,006 (9,82), 0,006 (6,98), 0,117 (0,79), 1,280 (7,07), 1,397 (2,03), 1,492 (0,48), 1,646 (0,50), 1,765 (1,75), 2,362 (0,64), 2,635 (0,69), 3,545 (0,50), 3,820 (16,00), 4,227 (3,99), 5,886 (0,43), 7,616 (0,98), 7,633
Ejemplo 28
Ácido 1-{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]fenil}-2-(1-cianociclopropil)-1,5,10-trioxo-4,6,9-trioxa-2-azatetradecan-14-oico

2-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianocidopropil)amino]metoxi}carbonil)oxi]etil pentanodioato de ferc-butilo (intermedio 70A, 200 mg, 238 |jmol) se disolvió en TFA al 30 % en DCM (12 ml) y la mezcla se agitó a temperatura ambiente durante 5 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-1810 jm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Se obtuvieron 131 mg (100 % de pureza, 70 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 3,84 min; MS (ESlpos): m/z = 785 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,372 (0,41), 1,503 (0,58), 1,681 (1,89), 1,699 (2,52), 1,717 (1,98), 1,736 (1,17), 1,762 (1,78), 2,205 (1,76), 2,223 (3,23), 2,241 (1,68), 2,290 (1,30), 2,309 (2,32), 2,327 (1,62), 2,366 (0,46), 3,821 (16,00), 4,177 (2,15), 4,220 (1,47), 7,612 (0,96), 7,632 (1,15), 7,791 (0,40), 7,835 (2,02), 7,866 (1,14), 8,561 (2,51), 8,834 (2,41).
Ejemplo 29
Ácido 1-{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(t
N fl
uorometil)-2'H-[1,3'-bipirazol]-4-il]feml}-2-(1-cianociclopropil)-8,8-dimetil-1,5,11-trioxo-4,6,10-trioxa-2-azatetradecan-14-oico
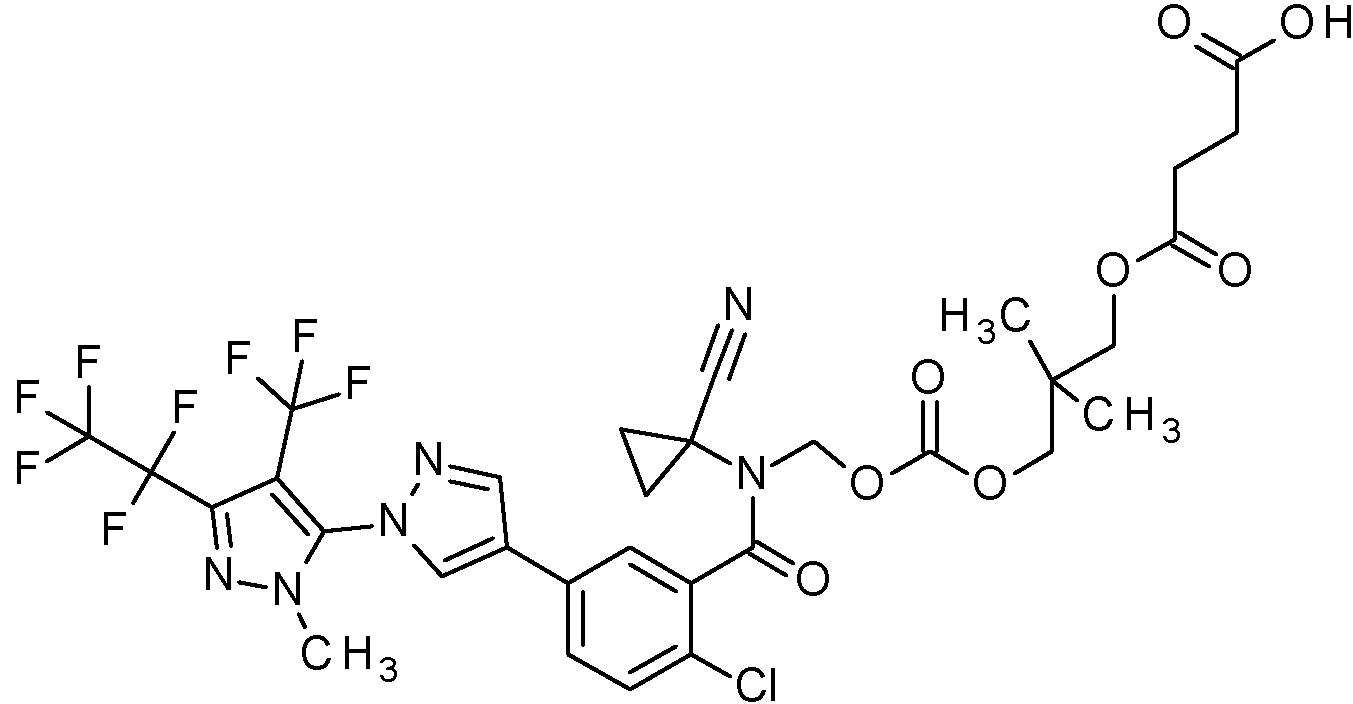
3-[({[{2-cloro-5-[2-metil-5'-(pentafluoroetil)-4-(trifluorometil)-2'H-[1,3-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropil butanodioato de ferc-butilo (intermedio 71A, 300 mg, 345 jmol) se disolvió en TFA al 30 % en DCM (16 ml) y la mezcla se agitó a temperatura ambiente durante 15 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 jm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Se obtuvieron 91,1 mg (100 % de pureza, 32 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,13 min; MS (ESlpos): m/z = 813 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,62), 0,008 (0,76), 0,851 (10,99), 0,934 (0,59), 1,500 (0,44), 1,762 (1,66), 2,463 (2,83), 3,785 (3,21), 3,822 (16,00), 3,866 (1,89), 7,610 (0,90), 7,631 (1,11), 7,842 (3,25), 7,860 (1,33), 8,563 (2,40), 8,831 (2,28).
Ejemplo 30
Ácido [({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]acético

[({[{2-doro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianocidopropil)amino]metoxi}carbonil)oxi]aceteato de ferc-butilo (intermedio 72a , 230 mg, 310 pmol) se disolvió en TFA al 30 % en DCM (15 ml) y la mezcla se agitó a temperatura ambiente durante 10 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-1810 pm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 4:1). Se aislaron 45,3 mg (100 % de pureza, 21 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 3,82 min; MS (ESlneg): m/z = 683 [M-H]-1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (1,75), 0,008 (1,76), 1,512 (0,46), 1,682 (0,44), 1,766 (1,61), 2,328 (0,41), 2,524 (1,93), 2,670 (0,45), 3,818 (16,00), 3,906 (0,42), 4,533 (2,86), 7,621 (0,78), 7,643 (0,97), 7,815 (1,45), 7,846 (1,05), 7,869 (0,96), 8,551 (4,10), 8,569 (0,45), 8,824 (4,02).
Ejemplo 31
Ácido 1-{[({K2-cloro-5-[2'-metil-5'-(pentafluoroetM)-4'-(trifluorometM)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]metil}ciclopropano-1-carboxílico

1-{[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]metil}ciclopropano-1-carboxilato de ferc-butilo (intermedio 77A, 210 mg, 269 pmol) se disolvió en TFA al 30 % en DCM (9,0 ml) y la mezcla se agitó a temperatura ambiente durante 7 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 pm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano - ciclohexano acetato de etilo 4:1). Se aislaron 62 mg (100 % de pureza, 32 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 3,99 min; MS (ESlpos): m/z = 725 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (0,54), 0,008 (0,58), 0,921 (2,34), 1,117 (2,55), 1,491 (0,54), 1,659 (0,45), 1,759 (1,82), 3,823 (16,00), 4,153 (0,70), 7,613 (1,00), 7,634 (1,26), 7,833 (2,10), 7,864 (1,19), 8,565 (2,97), 8,835 (2,83).
Ejemplo 32
Ácido c/s-(2E)-4-({-4-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(t
N fl
uorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclohexil}oxi)-4-oxobut-2-enoico

(1S,4S)-4-[({[{2-doro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2,H-[1,3'-bipirazol]-4-il]benzoil}(1-cianocidopropil)amino]metoxi}carbonil)oxi]cidohexil (2£)-but-2-enodioato de ferc-butilo (intermedio 78A, 73 mg, 83 pmol) se disolvió en TFA al 30 % en DCM (7,0 ml) y la mezcla se agitó a temperatura ambiente durante 10 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 pm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Se aislaron 39,5 mg (l00 % de pureza, 58 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,10 min; MS (ESlpos): m/z = 823 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,149 (0,40), -0,022 (0,42), -0,008 (3,14), 0,008 (3,57), 1,235 (0,86), 1,457 (0,99), 1,467 (0,55), 1,506 (0,53), 1,695 (4,74), 1,767 (2,39), 3,568 (0,44), 3,818 (16,00), 4,584 (0,83), 4,857 (0,94), 6,684 (5,76), 6,687 (5,79), 7,619 (1,04), 7,639 (1,25), 7,845 (3,70), 7,865 (1,33), 8,574 (2,46), 8,845 (2,38).
Ejemplo 33
Ácido 1-{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]fenil}-2-(1-cianociclopropil)-1,5,10-trioxo-4,6,9-trioxa-2-azatridecan-13-oico

2-[({[{2-doro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]etil butanodioato de ferc-butilo (intermedio 79a , 130 mg, 157 pmol) se disolvió en t Fa al 30 % en DCM (15,0 ml) y la mezcla se agitó a temperatura ambiente durante 10 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 pm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Se aislaron 109 mg (95 % de pureza, 86 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 3,96 min; MS (ESlpos): m/z = 771 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,501 (0,48), 1,669 (0,43), 1,767 (1,73), 2,444 (2,28), 2,455 (3,01), 2,473 (2,80), 3,822 (16,00), 4,184 (2,35), 4,213 (1,52), 7,617 (0,96), 7,637 (1,19), 7,843 (2,31), 7,868 (1,18), 8,564 (2,63), 8,837 (2,53).
Ejemplo 34
Ácido (11E)-1-{2-cloro-5-[2'-metM-5'-(pentafluoroetil)-4'-(t
N fl
uorometil)-2'H-[1,3'-bipirazol]-4-il]feml}-2-(1-cianociclopropil)-1,5,10-trioxo-4,6,9-trioxa-2-azatridec-11-en-l3-oico

2-[({[{2-doro-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡pirazol]-4-¡l]benzo¡l}(1-cianocidopropil)amino]metoxi}carbonil)oxi]etil (2E)-but-2-enod¡oato de ferc-butilo (¡ntermed¡o 80A, 74,0 mg, 89,7 |jmol) se disolvió en TFA al 30 % en DCM (7,4 ml) y la mezcla se agitó a temperatura ambiente durante 5 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 jm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Se aislaron 54,9 mg (94 % de pureza, 69 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,01 min; MS (ESlpos): m/z = 769 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: -0,008 (1,28), 0,008 (1,19), 1,496 (0,55), 1,683 (0,50), 1,758 (1,64), 2,323 (0,73), 2,327 (1,00), 2,332 (0,77), 2,366 (0,89), 2,523 (3,51), 2,665 (0,68), 2,670 (0,93), 2,674 (0,71), 2,710 (0,75), 3,820 (16,00), 4,309 (3,05), 6,665 (2,39), 6,672 (2,67), 7,603 (1,09), 7,625 (1,12), 7,839 (2,55), 7,861 (1,12), 8,556 (2,62), 8,833 (2,23), 13,218 (0,52).
Ejemplo 35
Ácido 1-{[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]metil}ciclopropano-1-carboxílico
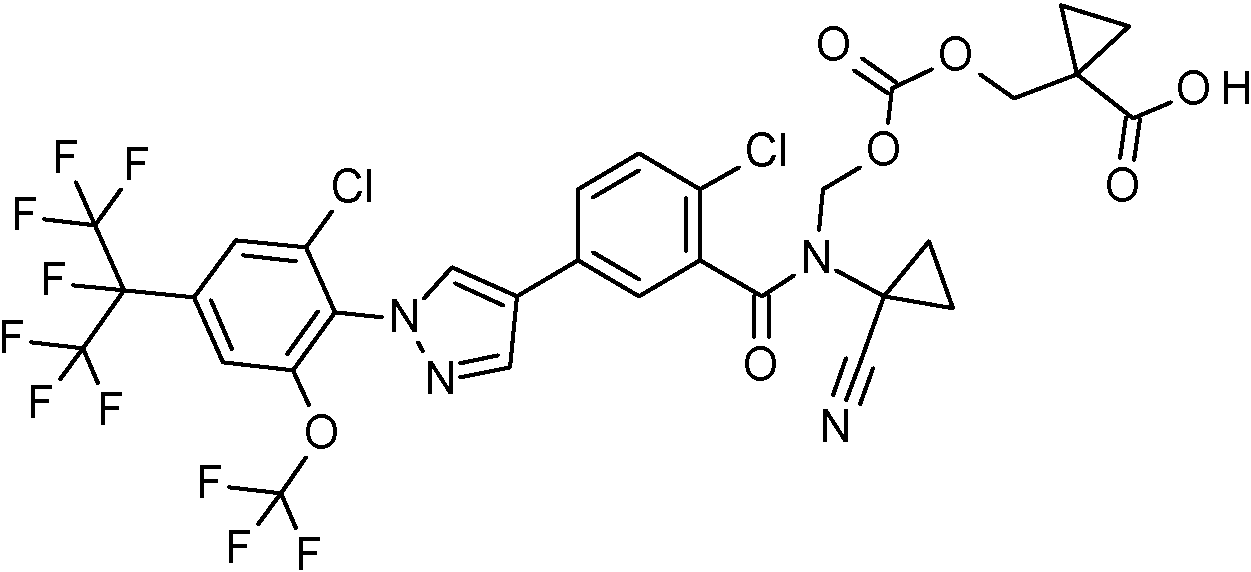
1-{[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(trifluorometox¡)fen¡l]-1H-p¡razol-4-¡l}benzo¡l)(1-danoddoprop¡l)amino]metox¡}carbon¡l)ox¡]met¡l}ddopropano-1-carbox¡lato de terc-butilo (intermedio 81A, 200 mg, 228 jmol) se disolvió en TFA al 30 % en DCM (20,0 ml) y la mezcla se agitó a temperatura ambiente durante 15 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 jm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Se aislaron 135 mg (95 % de pureza, 68 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,43 min; MS (ESlpos): m/z = 821 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 0,008 (1,67), 0,921 (10,12), 1,114 (11,51), 1,186 (1,08), 1,234 (1,08), 1,501 (2,38), 1,640 (1,98), 1,754 (8,17), 2,328 (0,96), 2,367 (1,08), 2,670 (0,99), 2,711 (1,15), 4,167 (2,60), 4,275 (1,08), 5,265 (1,67), 5,395 (1,73), 7,589 (4,33), 7,609 (5,32), 7,795 (10,58), 7,820 (5,45), 7,935 (14,42), 8,210 (16,00), 8,446 (13,80), 8,763 (11,91), 12,471 (1,33).
Ejemplo 36
Ácido 3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]propanoico

3-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometox¡)feml]-1H-p¡razol-4-il}benzo¡l)(1-danocidopropN)ammo]metoxi}carboml)oxi]propanoato de ferc-butilo (intermedio 83A, 192 mg, 225 pmol) se disolvió en TFA al 30 % en DCM (19 ml) y la mezcla se agitó a temperatura ambiente durante 15 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 pm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Se aislaron 134 mg (100 % de pureza, 75 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,28 min; MS (ESlpos): m/z = 795 [M+H]+
iH-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,235 (0,79), 1,505 (2,52), 1,638 (2,12), 1,756 (8,75), 2,073 (12,12), 2,328 (0,62), 2,367 (0,65), 2,671 (1,31), 2,711 (0,76), 4,191 (5,65), 4,345 (0,86), 5,163 (0,47), 5,271 (1,76), 5,386 (1,85), 7,558 (0,47), 7,594 (4,25), 7,614 (5,32), 7,783 (7,52), 7,805 (6,74), 7,826 (5,60), 7,935 (14,80), 8,208 (16,00), 8,211 (15,98), 8,446 (13,07), 8,768 (12,06), 12,416 (1,42).
Ejemplo 37
Ácido 4-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]butanoico

4-[({[(2-doro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-¡l)-6-(tr¡fluorometox¡)fen¡l]-1H-p¡razol-4-¡l}benzo¡l)(1-danodclopropi^amino^etox^carboni^ox^butanoato de ferc-butilo (intermedio 84A, 190 mg, 220 pmol) se disolvió en TFA al 30 % en DCM (19 ml) y la mezcla se agitó a temperatura ambiente durante 10 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-1810 pm de acetonitrilo:agua TFA al
0 ,01% 10 :90 -> 95 :5 ). S e a is la ro n 139 mg (96 % de pu reza , 75 % de re n d im ie n to ) de l co m p u e s to de l títu lo .
LC-MS (método 4): Rt = 4,33 min; MS (ESIpos): m/z = 809 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,234 (0,62), 1,508 (2,36), 1,639 (2,19), 1,755 (13,94), 1,768 (14,21), 2,226 (6,06), 2,244 (10,41), 2,261 (6,12), 2,329 (1,14), 2,367 (0,63), 2,671 (0,52), 2,711 (0,48), 4,036 (6,76), 4,185 (0,78), 5,257 (1,74), 5,380 (1,83), 7,590 (4,72), 7,610 (5,76), 7,793 (9,88), 7,822 (5,85), 7,935 (14,31), 8,209 (16,00), 8,446 (12,78), 8,770 (11,49), 12,122 (0,66).
Ejemplo 38
Ácido 4-({(1R,2R)-2-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclopentil}oxi)-4-oxobutanoico

(1R,2R)-2-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclopentil butanodioato de ferc-butilo (intermedio 85A, 87,0 mg, 100 pmol) se disolvió en TFA al 30 % en DCM (8,0 ml) y la mezcla se agitó a temperatura ambiente durante 5 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-18 10 pm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Se aislaron 32,3 mg (100 % de pureza, 40 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,16 min; MS (ESIpos): m/z = 811 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,236 (0,46), 1,320 (0,89), 1,651 (1,39), 1,756 (1,73), 1,966 (0,89), 2,085 (0,46), 2,384 (1,39), 2,422 (2,03), 2,449 (4,39), 2,517 (2,36), 2,611 (0,93), 2,652 (0,84), 3,266 (2,32), 3,820 (16,00), 4,791 (0,68), 4,950 (0,89), 7,611 (0,93), 7,626 (1,10), 7,831 (1,60), 7,859 (1,14), 8,557 (1,90), 8,826 (1,90), 12,182 (0,84).
Ejemplo 39
Ácido (12£)-1-{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]fenil}-2-(1-cianociclopropil)-8,8-dimetil-1,5,11-trioxo-4,6,10-trioxa-2-azatetradec-12-en-14-oico

3-[({[{2-doro-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzo¡l}(1-cianocidopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropil (2E)-but-2-enod¡oato de terc-but¡lo (¡ntermed¡o 86A, 280 mg, 323 pmol) se d¡solv¡ó en TFA al 30 % en DCM (16 ml) y la mezcla se ag¡tó a temperatura amb¡ente durante 15 m¡n. El solvente se dest¡ló, el res¡duo se absorb¡ó en aceton¡tr¡lo y se pur¡f¡có por HPLC preparat¡va (RP C-18 10 pm de aceton¡tr¡lo:agua TFA al 0,01% 10:90->95:5). Se a¡slaron 88,2 mg (100 % de pureza, 34 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 4,18 m¡n; MS (ESlpos): m/z = 811 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 0,892 (12,63), 0,925 (0,51), 0,982 (0,62), 1,379 (0,40), 1,483 (0,45), 1,668 (0,40), 1,759 (1,55), 2,521 (0,46), 3,823 (16,00), 3,915 (5,04), 6,687 (5,15), 7,604 (1,07), 7,619 (1,19), 7,843 (3,36), 7,854 (1,45), 8,561 (2,50), 8,830 (2,39).
Ejemplo 40
Ácido 1-{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(t
N fl
uorometil)-2'H-[1,3'-bipirazol]-4-il]feml}-2-(1-cianociclopropil)-12,12-dimetil-1,5,10-trioxo-4,6,9-trioxa-2-azatridecan-13-oico

4-{2-[({[{2-doro-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzo¡l}(1-danoddoprop¡l)am¡no]metox¡}carbon¡l)ox¡]et¡l} 2,2-d¡met¡lbutanod¡oato de terc-but¡lo (¡ntermed¡o 87A, 55,3 mg, 64,7 pmol) se d¡solv¡ó en TFA al 30 % en DCM (5 ml) y la mezcla se ag¡tó a temperatura amb¡ente durante 5 m¡n. El solvente se dest¡ló, el res¡duo se absorb¡ó en aceton¡tr¡lo y se pur¡f¡có por HPLC preparat¡va (RP C-18 10 pm de aceton¡tr¡lo:agua TFA al 0,01% 10:90->95:5). Se a¡slaron 10,9mg (100 % de pureza, 21 % de rend¡m¡ento) del compuesto del título.
LC-MS (método 4): Rt = 4,07 m¡n; MS (ESlpos): m/z = 799 [M+H]+
iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: -0,022 (0,51), 0,843 (0,46), 0,855 (0,88), 0,866 (0,41), 1,129 (14,61), 1,167 (0,78), 1,237 (3,81), 1,490 (0,53), 1,671 (0,42), 1,769 (1,56), 3,824 (16,00), 4,158 (2,44), 4,216 (0,82), 5,254 (0,41), 5,416 (0,41), 7,623 (1,28), 7,637 (1,49), 7,840 (2,01), 7,854 (1,28), 7,868 (1,12), 8,572 (2,76), 8,845 (2,61).
Ejemplo 41
Ácido 3-[({[(2-cloro-5-{1-[2-cloro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-M)-6-(trifluorometoxi)feml]-1H-pirazol-4-il}benzoil)(1-cianociclopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropanoico

3-[({[(2-cloro-5-{1-[2-doro-4-(1,1,1,2,3,3,3-heptafluoropropan-2-il)-6-(trifluorometox¡)feml]-1H-p¡razol-4-¡l}benzo¡l)(1-cianocidopropil)amino]metoxi}carbonil)oxi]-2,2-dimetilpropanoato de bencilo (intermedio 89A, 415 mg, 454 pmol) se disolvió en acetato de etilo (200 ml) en una atmósfera de argón. Se agregó paladio sobre carbón al 10 % (22,0 mg) y la mezcla se hidrogenó durante 6 h a presión ambiente y temperatura ambiente. Luego el catalizador se retiró por filtración sobre una capa de tierra diatomácea y el solvente se destiló. El producto bruto se purificó por HPLC preparativa (RP C-1810 pm, gradiente de agua-acetonitrilo con TFA al 0,01% en ambos eluyentes, 90:10->5:95). Se obtuvieron 299 mg (100 % de pureza, 80 % de rendimiento) del compuesto del título.
LC-MS (método 4): Rt = 4,54 min; MS (ESlpos): m/z = 823 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 8 [ppm]: 1,017 (4,00), 1,063 (16,00), 1,235 (0,82), 1,513 (0,87), 1,640 (0,77), 1,756 (3,11), 3,363 (1,74), 4,037 (1,81), 5,277 (0,68), 5,400 (0,59), 7,588 (1,42), 7,608 (1,78), 7,762 (0,76), 7,801 (4,81), 7,822 (2,28), 7,935 (4,90), 8,213 (5,67), 8,446 (4,01), 8,755 (4,30).
Ejemplo 42
Ácido (2E)-4-({(1S,2S)-2-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(t
N fl
uorometM)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]ciclopentil}oxi)-4-oxobut-2-enoico

(1S,2S)-2-[({[{2-cloro-5-[2'-met¡l-5'-(pentafluoroet¡l)-4'-(tr¡fluoromet¡l)-2'H-[1,3'-b¡p¡razol]-4-¡l]benzo¡l}(1-cianociclopropi^amino^etox^carboni^ox^ciclopentil (2£)-but-2-enodioato de tere-butilo (intermedio 90A, 57,6 mg, 66,6 pmol) se disolvió en TFA al 30 % en DCM (6 ml) y la mezcla se agitó a temperatura ambiente durante 8 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLc preparativa (RP C-18 10 pm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). Se aislaron 27,1 mg (100 % de pureza, 50 % de rendimiento) del
c o m p u e s to de l títu lo .
LC-MS (método 4): Rt = 4,21 min; MS (ESlpos): m/z = 809 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,236 (0,46), 1,494 (0,43), 1,643 (1,35), 1,694 (1,37), 1,756 (1,86), 2,033 (0,85), 2,070 (0,66), 2,572 (0,40), 3,820 (16,00), 4,884 (0,65), 5,065 (0,80), 6,612 (0,70), 6,638 (1,67), 6,674 (1,68), 6,699 (0,77), 7,606 (0,90), 7,619 (1,06), 7,835 (1,83), 7,856 (1,23), 8,554 (2,09), 8,825 (1,88).
Ejemplo 43
Ácido 3-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianociclopropil)amino]metoxi}carbonil)oxi]propanoico

3-[({[{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzoil}(1-cianocidopropil)amino]metoxi}carbonil)oxi]propanoato de tere-butilo (intermedio 91A, 250 mg, 331 pmol) se disolvió en TFA al 30 % en DCM (12 ml) y la mezcla se agitó a temperatura ambiente durante 8 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-1810 pm de acetonitrilo:agua TFA al 0,01% 10:90->95:5). El producto bruto se purificó mediante cromatografía ultrarrápida en gel de sílice (gradiente de ciclohexano:acetato de etilo 2:1-acetato de etilo metanol 4:1). Se aislaron 42,9 mg (100 % de pureza, 19 % de rendimiento) del compuesto del título. LC-MS (método 4): Rt= 3,85 min; MS (ESlpos): m/z = 699 [M+H]+ iH-NMR (600 MHz, DMSO-d6) 8 [ppm]: 1,494 (1,33), 1,650 (1,10), 1,759 (4,94), 2,085 (16,00), 2,344 (0,66), 2,355 (0,41), 2,384 (0,45), 2,423 (0,73), 2,613 (0,57), 2,652 (1,15), 3,421 (6,26), 3,598 (0,49), 3,608 (0,83), 3,619 (0,50), 3,697 (0,41), 3,807 (2,81), 4,191 (2,93), 4,348 (0,56), 5,259 (0,89), 5,393 (0,98), 7,618 (2,64), 7,631 (3,04), 7,787 (0,65), 7,820 (4,69), 7,850 (3,42), 7,864 (3,28), 8,557 (6,79), 8,830 (7,19), 12,411 (0,42).
Ejemplo 44
Ácido 1-{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]fenil}-2-(1-cianociclopropil)-11,11 -dimetil-1,5-dioxo-4,6,9-trioxa-2-azadodecan-12-oico

1-{2-cloro-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]fenil}-2-(1-cianociclopropil)-11,11-dimetil-1,5-dioxo-4,6,9-trioxa-2-azadodecan-12-oato de tere-butilo (intermedio 92A, 108 mg, 131 pmol) se disolvió en TFA al 30 % en DCM (11 ml) y la mezcla se agitó a temperatura ambiente durante 10 min. El solvente se destiló, el residuo se absorbió en acetonitrilo y se purificó por HPLC preparativa (RP C-1810 pm de acetonitrilo:agua TFA al
0 ,01 % 10 :90 -> 95 :5 ). S e a is la ro n 80 ,0 mg (100 % de pu reza , 79 % de re n d im ie n to ) de l c o m p u e s to de l títu lo .
LC-MS (método 4): Rt = 4,24 min; MS (ESIpos): m/z = 771 [M+H]+
1H-NMR (600 MHz, DMSO-d6) 8 [ppm]: 0,005 (0,68), 1,029 (10,00), 1,076 (1,10), 1,760 (1,36), 2,515 (0,65), 2,518 (0,72), 2,521 (0,57), 2,572 (0,51), 3,391 (0,96), 3,548 (1,76), 3,819 (16,00), 4,119 (0,85), 7,615 (0,76), 7,628 (0,87), 7,826 (1,26), 7,847 (0,95), 7,861
Ejemplos fisicoquímicos y biológicos: evaluación de la estabilidad del profármaco y liberación del compuesto original
Preparación de la solución amortiguadora PBS pH 7,4: 90 g de cloruro de sodio, 13,61 g dihidrogenofosfato de potasio, 83,35 g de hidróxido de sodio acuoso 1M se disolvieron en agua. Se agregó más agua hasta un volumen total de 1 l. Esta solución se diluyó 1:10 con agua. Eventualmente, el pH se ajustó a pH 7,4 por la adición de ácido fosfórico.
Se usó solución amortiguadora de ácido cítrico pH 3,0: amortiguador de ácido cítrico comercialmente disponible (pH 3) de Fluka (art. No 31046) que contenía 8,47 g de ácido cítrico, 3,49 g de cloruro de sodio y 0,82 g de hidróxido de sodio.
Ejemplos fisicoquímicos y biológicos
Evaluación de la estabilidad del profármaco y liberación del compuesto original
Preparación de la solución amortiguadora PBS pH 7,4: 90 g de cloruro de sodio, 13,61 g dihidrogenofosfato de potasio, 83,35 g de hidróxido de sodio acuoso 1M se disolvieron en agua. Se agregó más agua hasta un volumen total de 1 l. Esta solución se diluyó 1:10 con agua. Eventualmente, el pH se ajustó a pH 7,4 por la adición de ácido fosfórico.
Se usó solución amortiguadora de ácido cítrico pH 3,0: amortiguador de ácido cítrico comercialmente disponible (pH 3) de Fluka (art. No 31046) que contenía 8,47 g de ácido cítrico, 3,49 g de cloruro de sodio y 0,82 g de hidróxido de sodio.
Ejemplo biológico B1
Medición de la solubilidad en amortiguador a pH 6,5
Se disuelven 2 - 4 mg del compuesto de prueba en DMSO para alcanzar una concentración de 50 g/l (solución A). A 10 |jl de esta solución, se le agregan 960 j l de amortiguador PBS pH 6,5 (concentración final: 515 jg/l); la mezcla se agita durante 24 h a ta en una placa de 96 pocillos. Una alícuota se centrifuga a 42000 rpm durante 30 min. El sobrenadante se diluye con acetonitrilo/agua (8:2) 1:10 y 1:1000 resp. Estas muestras diluidas se analizan por LC-MSMS. Los resultados se proveen en la Tabla 0, a continuación.
Calibración: 10 j l de la solución A se diluyen con 823 j l de DMSO (concentración final: 600 jg/ml), que se diluye adicionalmente con acetonitrilo/agua 8:2 por un factor de 100 (provee la solución B).
La curva de calibración se obtiene de la solución B por dilución adicional con acetonitrilo/agua 8:2 con concentraciones objetivo de 1,2 -12 -60 - 600 ng/ml e inyectando estas cuatro soluciones para medición de MS. Optimización del método MS: La solución B se utiliza para la optimización del método MS.
Amortiguador PBS: 6,18 g de cloruro de sodio y 3,96 g de dihidrógenofosfato de sodio se disuelven en 1L de agua destilada, el pH se ajusta a 6,5 con hidróxido de sodio 1N.
Condiciones de LC y MS:
Optimización de LC-MSMS: Las siguientes configuraciones se usaron para optimización.
AB Sciex TRIPLE QUAD 4500, Agilent 1260 Infinity (G1312B), desgasificador (G4225A), horno de columna (G1316C y G1316A), sistema de inyección CTC Analytics pAl HTS-xt
Eluyente A: 0,5 ml de ácido fórmico, (50 % de resistencia)/ l agua, Eluyente B: 0,5 ml de ácido fórmico, (50 % de resistencia) / l acetonitrilo
tiempo [min] flujo [pl/min] % B
0,00 200 70
0,08 200 70
0,09 25 70
0,60 25 70
0,65 200 70
1,10 200 70
Automuestreador: sin configuración de autoinyección adelante; columna: capilar acero inoxidable, temperatura del horno: 22 °C; velocidad de flujo: gradiente de flujo, volumen inyectado: 2 pL.
Waters Quattro Micro MS, Agilent 1100 (G1312A), desgasificador (G1322A), horno de columna (G1316A), sistema de inyección HTS CTC Analytics PAL, eluyentes como antecede
tiempo [min] flujo [pl/min] % B
0,00 250 70
1,50 250 70
Automuestreador con configuración de autoinyección adelante; columna: capilar acero inoxidable, temperatura del horno: 22 °C; velocidad de flujo: gradiente de flujo, volumen inyectado: 5 pL.
Método MS: Análisis de inyección de flujo (FIA) para optimización („MS-OPTI“); modo de ionización ABSciex-MS: ESl-pos/neg, Waters-MS: ESl-pos
Método HPLC para cuantificación MSMS:
Las siguientes condiciones se usaron para cuantificación:
Eluyente A, B como antes
ABSciex-MS
tiempo [min] % A % B
0 90 10
0,5 5 95
0,84 5 95
0,85 90 10
1,22 90 10
Automuestreador sin configuración de autoinyección adelante; columna: Waters OASIS HLB, 2,1 x 20 mm, 25 p, temperatura de la columna: 30 °C, velocidad de flujo: 2,5 ml/min; volumen inyectado: 2 pl, Divisor (antes de MS) 1:20.
Waters-MS
Gradiente como antes
Automuestreador con configuración de autoinyección adelante; columna: capilar acero inoxidable, columna: Waters OASIS HLB, 2,1 x 20 mm, 25 p, temperatura de la columna: 30 °C, velocidad de flujo: 2,5 ml/min; volumen inyectado: 5 pL, Divisor (antes de MS) 1:20, método MS: Monitoreo de reacción múltiple (MRM).
T l : l ili l m m l H

continuación

Ejemplo biológico B2
Medición de la estabilidad de profármacos en amortiguador a pH 7,4
Medición: 0,15 mg del compuesto de prueba se disolvieron en 0,1 ml de dimetilsulfóxido y 0,4 ml de acetonitrilo. Para una disolución completa, el recipiente de HPLC con la solución de muestra se agitó y se trató con ultrasonido. Luego se agregaron 1,0 ml de solución amortiguadora PBS pH 7,4 y la muestra se agitó en vórtex. La solución de muestra se analizó por HPLC (método 8) para determinar la cantidad de compuesto de prueba en un momento particular en un período de 24 h a 37 °C. Las áreas pico, dadas como porcentaje del área total, se usan para cuantificación. Además, la mezcla de reacción se analizó por el método 9 (HPLC-MS) en el punto de tiempo final.
T l 1: E ili l m ml H 74

continuación

Ejemplo biológico B3
Medición de liberación del compuesto original en plasma de rata
1 mg del compuesto de prueba se disolvió en 0,5 ml de acetonitrilo/dimetilsulfóxido 9:1. Para una disolución completa, el recipiente de HPLC se agitó y se trató con ultrasonido. Se agregaron 20 j l de esta solución a 1 ml de plasma de rata (plasma Li-heparina, rata Hannover-Janvier, RjHan macho) con agitación en vórtex a una temperatura de 37 °C. Se tomaron alícuotas (100 jL cada una) a 0,17, 0,5, 1, 1,5, 2 y 4 horas. Cada alícuota se transfirió a un recipiente que contenía 300 jL de amortiguador de ácido cítrico/ acetonitrilo/ a pH 3 8:2. Estas soluciones se centrifugaron a 5000 rpm durante 10 minutos. El sobrenadante se analizó por HPLC (Método 8) para determinar la cantidad de compuesto de prueba. Además, la mezcla de reacción se analizó por el método 9 (HpLC-MS) en el punto de tiempo final. Se monitorearon tanto la reducción de la concentración de profármaco como el aumento de la concentración del compuesto original. Todos los datos se dan como área porcentual de profármaco en tü.
El compuesto original A significa 2-Clor-5-{1-[2-clor-4-(1,1,1,2,3,3,3-heptafluorpropan-2-il)-6-(trifluormetoxi)fenil]-1H-pirazol-4-il}-W-(1-cianociclopropil)benzamida (Ca S-RN 1771742-44-9).

El compuesto original B significa 2-cloro-A/-(1-cianociclopropil)-5-[2'-metil-5'-(pentafluoroetil)-4'-(trifluorometil)-2'H-[1,3'-bipirazol]-4-il]benzamida (CAS-RN 1621436-41-6)

T l 2: D r i n r f rm n l m r li r i n f rm ri in l.

continuación

Ejemplo biológico B4
Medición de liberación del compuesto original en plasma de perro
1 mg del compuesto de prueba se disolvió en 0,5 ml de acetonitrilo/dimetilsulfóxido 9:1. Para una disolución completa, el recipiente de HPLC se agitó y se trató con ultrasonido. Se agregaron 20 j l de esta solución a 1 ml de plasma de perro (Beagle) con agitación en vórtex a una temperatura de 37 °C. Se tomaron alícuotas (100 jL cada una) a 0,17, 0,5, 1, 1,5, 2 y 4 horas. Cada alícuota se transfirió a un recipiente que contenía 300 jL de amortiguador de ácido cítrico/ acetonitrilo/ a pH 380:20. Estas soluciones se centrifugaron a 5000 rpm durante 10 minutos. El sobrenadante se analizó por HPLC (Método 8) para determinar la cantidad de compuesto de prueba. Además, la mezcla de reacción se analizó por el método 9 (HPLC-MS) en el punto de tiempo final.
Se monitorearon tanto la reducción de la concentración de profármaco como el aumento de la concentración del compuesto original. Todos los datos se dan como área porcentual de profármaco en to.
T l : D r i n r f rm n l m ^ rr

Ejemplo biológico B5
Evaluación de farmacocinética (in vivo)
Para evaluar la farmacocinética de las sustancias de prueba in vivo, estas sustancias de prueba se disuelven en vehículos de formulación apropiados (etanol, dimetil sulfóxido, PEG400, glicerol, formal, etc.) o mezclas de estos. Luego las sustancias de prueba se administran a ratas o perros por vía intravenosa, oral o subcutánea. La administración intravenosa se realiza en forma de bolo. Las dosis administradas habitualmente varían de 0,1 a 5 mg/kg, sin embargo, las dosis para administración subcutánea y oral pueden exceder este intervalo hasta dosis de 30 mg/kg. Las muestras de sangre se extraen mediante un catéter, desangrado o punción venosa en recipientes que contienen anticoagulantes apropiados, como heparinato de litio o EDTA de potasio. El plasma se genera a partir de la sangre mediante centrifugación. Se toman muestras de sangre en un intervalo de tiempo apropiado que habitualmente dura hasta 144 h después de la administración. De ser necesario, también se pueden tomar muestras de puntos de tiempo posteriores. De ser posible, los puntos de tiempo se eligen de modo de describir la fase de absorción inicial, la concentración máxima en plasma (Cmáx) y la exposición dentro de cierto intervalo de tiempo (AUC(0-t)). Además, también es posible recuperar muestras de órganos, tejidos y orina. La medición cuantitativa de la sustancias de prueba en las muestras se realiza usando curvas de calibración en los matrices correspondientes. El contenido de proteína de las muestras se precipita usando acetonitrilo o metanol. A partir de allí, las muestras se separan usando HPLC en combinación con columnas de cromatografía de fase inversa. El sistema de HPLC se acopla a un espectrómetro de masas triple cuadrupolo mediante una interfaz de electropulverización. La evaluación de los perfiles de concentración en plasma/tiempo se evalúan después usando un programa de evaluación farmacocinética validado.
La exposición del compuesto original A dentro de cierto intervalo de tiempo mostrado como AUC(0-t) en las tablas 4 6 se usó para evaluar la conversión del profármaco en fármaco y la absorción del profármaco en comparación con el compuesto original in vivo, respectivamente. La comparación de exposiciones después de la administración intravenosa del compuesto original A y los profármacos mostró la cantidad de profármaco que se convirtió en el original. La comparación de exposiciones después de la administración subcutánea u oral del compuesto original y los profármacos mostró además características de absorción alteradas de profármacos en comparación con el compuesto original. La biodisponibilidad relativa (Frel) se usó para describir la exposición del compuesto original después de la administración del profármaco en comparación con la exposición del compuesto original después de la administración directa del compuesto A que se normalizó a 100 %.
Tabla 4: AUC del compuesto original A después de la administración intravenosa de los ejemplos de profármaco y des ués de la administración intravenosa del com uesto ori inal A.

continuación

Tabla 5: AUC del compuesto original A después de la administración subcutánea de los ejemplos de profármaco y des ués de la administración subcutánea del com uesto ori inal A.

Tabla 6: AUC del compuesto original A después de la administración oral de los ejemplos de profármaco y después de la administración oral del com uesto ori inal A.

continuación

Ejemplo biológico B6
Medición de la actividad antiparasitaria después de la administración de un profármaco a las ratas
Los compuestos se disolvieron en el volumen apropiado de glicerol formal (p.a.) justo antes de la administración a los animales (el volumen de dosis se ajustó a 0,03 ml/kg de peso corporal). 5 ratas por grupo se inyectaron intraperitonealmente o subcutáneamente.
Las ratas se infectaron con 30 ninfas de Dermacentor variabilis en los días de estudio -2, 7, 14, 21, 28, 35 y 30 pulgas Ctenocephalides adultas los días de estudio -1, 8, 15, 22, 29, 36.
Se realizaron conteos de parásitos los días del estudio 2, 9, 16, 23, 30, 37.
El porcentaje de eficacia se calculó como cantidad de parásitos promedio aritmético como cantidad de pulgas vivas agarradas y cantidad de pulgas vivas en el grupo de estudio correspondiente en comparación con los conteos de parásitos en un grupo de control tratado con placebo. La infestación de grupos con una eficacia menor que 75 % en dos ocasiones de conteo consecutivas se terminará para el parásito correspondiente.
Ambos tratamientos de profármaco SC (ejemplo 1 y ejemplo 4) son superiores en su eficacia en pulgas y garrapatas en ratas respecto del fármaco original que también se inyectó subcutáneamente.
Tabla 7: Eficacia in vivo del compuesto original A y formulaciones de ejemplo de profármaco aplicadas i.p. o sc en ratas

Ejemplo biológico B7
Medición de la actividad antiparasitaria después de la administración de un profármaco a los perros Los compuestos se disolvieron en el volumen apropiado de glicerol formal (p.a.) justo antes de la administración a los animales (el volumen de dosis se ajustó a 0,1 ml/kg de peso corporal). Los perros recibieron una inyección intravenosa subcutánea respectiva (véase tabla 8 para más detalles).
Los perros se infectaron con ectoparásitos de acuerdo con la tabla el día 1 del estudio antes de continuar con pulgas, 2 días antes de continuar con pulgas de RS, IR, DV y 3 días antes de continuar con pulgas de AA. Con este objetivo, los perros se colocaron en una caja etiquetada individualmente con ventanas de red. Las garrapatas se liberaron en el lomo de los perros y se las dejó dispersarse y entrar en el pelo sin molestarlas. El perro permaneció en la caja por aproximadamente 120 minutos con la tapa cerrada y la luz apagada en la habitación. Para las infestaciones con pulgas, los recipientes de pulgas se abrieron en la jaula del animal y se aplicaron pulgas en el lomo del perro entre los hombros.
Los conteos de parásitos se realizaron como se indica en la Tabla 8. Los conteos de pulgas, inclusive la eliminación de I. ricinus, R. sanguineus y D. variabilis se realizaron 48(±4) horas después de la infestación. Los conteos de pulgas, inclusive la eliminación de A. americanum se realizaron 72(±4) horas después de la infestación. Los conteos de pulgas se realizaron 24(±4) horas después de la infestación.
Cada día de evaluación de parásitos, se calculó el porcentaje de eficacia como cantidad de parásitos promedio aritmético como cantidad de pulgas vivas agarradas y vivas libres y cantidad de pulgas vivas en el grupo de estudio correspondiente en comparación con conteos de parásitos en un grupo de control no tratado con placebo.
En función de los resultados de estas infestaciones, se puede alcanzar una eficacia de cinco meses del profármaco “1” contra D. variabilis como especies menos sensibles.
T l : L fi i in viv f rm l i n ri in l ^ r f rm li iv n rr

Claims (15)
1. Compuestos de la fórmula (I)

en la que
Q es O o está ausente;
L1 es alcanodiilo C2-C4 lineal, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de halógeno, alquilo C1-C4 y cicloalquilo C3-C6, en donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos; o
es un resto (CH2)n-X-(CH2)m en donde
n y m son independientemente 0, 1 o 2 y
X es cicloalcanodiilo C3-C7 , que está opcionalmente sustituido con alquilo C1-C4; o
L1 está ausente;
con la condición de que en caso de que L1 esté ausente, Q también esté ausente;
L2 es C=O o está ausente,
L3 es alcanodiilo C1-C4 lineal, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C6 y halógeno, en donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos; o
es un grupo (CH2)n-(CH=CH)-CH2)m, que está opcionalmente sustituido con hasta 2 grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C5 y halógeno, en donde
n y m son independientemente 0, 1 o 2; o
es cicloalcanodiilo C3-C7, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4 y halógeno;
Y se selecciona de T1

en donde
cada uno de R1, R2 y R3 se selecciona independientemente de hidrógeno, halógeno, ciano, nitro, alquilo C1-C6 lineal o ramificado, cicloalquilo C3-C6, alcoxi C1-C6, alquilo C1-C6 sustituido con halógeno lineal o ramificado, alcoxi C1-C6 sustituido con halógeno, cicloalquilo C3-C6 sustituido con halógeno, alquilsulfanilo C1-C6, alquilsulfinilo C1-C6, alquilsulfonilo C1-C6, N-alquilamino C1-C6, N,N-dialquilamino C1-C6, N-alcoxi C1-C3-alquilamino C1-C4 y 1 -pirrolidinilo
o T2

en donde
cada uno de Z1 y Z2 se selecciona independientemente de hidrógeno, halógeno, ciano, nitro, alquilo C1-C6 lineal o ramificado, cicloalquilo C3-C6, alcoxi C1-C6, alquilcarbonilo C1-C6, alquilo C1-C6 lineal o ramificado sustituido con halógeno, alcoxi C1-C6 sustituido con halógeno, cicloalquilo C3-C6 sustituido con halógeno, alquilsulfanilo C1-C6, alquilsulfinilo C1-C6, alquilsulfonilo C1-C6, N-alquilamino C1-C6, A/,W-dialquilamino C1-C6, W-alcoxi C1-C3-alquilamino C1-C4 y 1-pirrolidinilo; y
Z3 representa hidrógeno o alquilo C1-C6 lineal, cicloalquilo C3-C6, alquenilo C2-C6, alquinilo C2-C6, arilo o hetarilo, que pueden estar sustituidos independientemente unos de los otros con 1 a 5 sustituyentes que se seleccionan de hidroxi, halógeno, ciano, nitro, amino, alquilo C1-C3, alcoxi C1-C3, hidroxicarbonilo, alcoxicarbonilo, alquilcarbamoilo, cicloalquilcarbamoilo y fenilo
y las sales de estos.
2. Compuestos de acuerdo con la reivindicación 1,
en donde
Q es O;
L2 es C=O;
L3 es un grupo (CH2)n-(CH=CH)-CH2)m, que está opcionalmente sustituido con hasta 2 grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C5 y halógeno, en donde
n y m son independientemente 0, 1 o 2; y
L1 tiene el significado como se define en la reivindicación 1;
y las sales de estos.
3. Compuestos de acuerdo con las reivindicaciones 1 o 2,
en donde
Q es O;
L2 es C=O;
L3 es un grupo (CH2)n-(CH=CH)-CH2)m, en donde n y m son 0; y
L1 tiene el significado como se define en la reivindicación 1;
y las sales de estos.
4. Compuestos de acuerdo con la reivindicación 1,
en donde
Q es O
L2 es C=O
L3 es alcanodiilo C1-C4 lineal, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C6 y halógeno, en donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos; y
L1 tiene el significado como se define en la reivindicación 1;
y las sales de estos.
5. Compuestos de acuerdo con la reivindicación 1,
en donde
Q es O;
L2 está ausente;
L3 es alcanodiilo C1-C4 lineal, que está opcionalmente sustituido con uno o más grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C6 y halógeno, en donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos; y
L1 tiene el significado como se define en la reivindicación 1;
y las sales de estos.
6. Compuestos de acuerdo con la reivindicación 1,
en donde
L1, L2 y Q están ausentes; y
L3 tiene el significado como se define en la reivindicación 1;
y las sales de estos.
7. Compuestos de acuerdo con una cualquiera de las reivindicaciones anteriores,
en donde se aplican una o más de las siguientes condiciones
Q es O;
y/o
L1 es alcanodiilo C2-C4 lineal, que está opcionalmente sustituido con uno o más grupos alquilo C1-C4, en donde dos sustituyentes alquilo C1-C4 pueden formar un anillo junto con el átomo de carbono al que están unidos y/o
L2 es C=O
y/o
L3 es un grupo (CH2)n-(CH=CH)-CH2)m, que está opcionalmente sustituido con hasta 2 grupos que se seleccionan independientemente de alquilo C1-C4, cicloalquilo C3-C5 y halógeno, en donde
n y m son independientemente 0, 1 o 2, preferentemente n y m son 0;
y las sales de estos.
8. Compuestos de acuerdo con una cualquiera de las reivindicaciones anteriores,
en donde
Y se selecciona de T1

en donde
cada uno de R1, R2 y R3 se selecciona independientemente de hidrógeno, halógeno, alquilo C1-C3 lineal o ramificado, cicloalquilo C3-C6, alcoxi C1-C3, alquilo C1-C3 sustituido con halógeno, lineal o ramificado, alcoxi C1-C3 sustituido con halógeno, cicloalquilo C3-C6 sustituido con halógeno y 1-pirrolidinilo,
preferentemente cada uno de R1, R2 y R3 se seleccionan independientemente de halógeno, alquilo C1-C3 sustituido con halógeno, lineal o ramificado y alcoxi C1-C3 sustituido con halógeno;
o T2

en donde
Z1 representa alquilo C1-C3 lineal o ramificado, cicloalquilo C3-C6, alcoxi C1-C3,
que pueden estar sustituidos independientemente unos de los otros con 1 a 5 sustituyentes que se seleccionan de hidroxi, halógeno, ciano, nitro, alquilo C1-C3 y alcoxi C1-C3,
preferentemente Z1 representa alquilo C1-C3 lineal o ramificado o cicloalquilo C3-C6, que pueden estar sustituidos independientemente unos de los otros con 1 a 5 sustituyentes de halógeno;
Z2 representa halógeno, ciano, nitro, amino o alquilo C1-C6 lineal o ramificado, alquilcarbonilo C1-C6, alquilsulfanilo C1-C6, alquilsulfinilo C1-C6, alquilsulfonilo C1-C6, que independientemente unos de otros pueden estar sustituidos con 1 a 5 sustituyentes que se seleccionan de hidroxi, halógeno, ciano, nitro, alquilo C1-C3 y alcoxi C1-C3,
preferentemente Z2 representa alquilo C1-C3 lineal o ramificado, que puede estar sustituido con 1 a 5
sustituyentes de halógeno, preferentemente con 1 a 3 sustituyentes de halógeno, más preferentemente trifluorometilo, o Z2 representa nitro, metilsulfanilo, metilsulfinilo, metilsulfonilo, flúor, cloro, bromo, yodo; y Z3 representa hidrógeno o alquilo C1-C6 lineal o ramificado, cicloalquilo C3-C6, alquenilo C2-C6, alquinilo C2-C6, arilo o hetarilo, que pueden estar sustituidos independientemente unos de los otros con 1 a 5 sustituyentes que se seleccionan de hidroxi, halógeno, ciano, nitro, alquilo C1-C3 y alcoxi C1-C3,
preferentemente Z3 representa hidrógeno o alquilo C1-C6 lineal o ramificado que puede estar sustituido con 1 a 5 sustituyentes que se seleccionan de hidroxi, halógeno, alquilo C1-C3 y alcoxi C1-C3;
y las sales de estos.
9. Compuestos de acuerdo con una cualquiera de las reivindicaciones anteriores,
en donde
Y se selecciona de T1

en donde
R1 es halógeno, preferentemente flúor, bromo o cloro, más preferentemente cloro;
R2 es alquilo C1-C3 lineal o ramificado sustituido con 1 a 7 halógenos, preferentemente alquilo C1-C3 lineal o ramificado sustituido con 1 a 7 flúor, más preferentemente CF3, C2F5 o C3F7; y
R3 es alcoxi C1-C3 sustituido con 1 a 3 halógenos, preferentemente alcoxi C1-C3 sustituido con 1 a 3 flúor, más preferentemente OCF3, OC2F5 u OC3F7;
o T2

en donde
Z1 representa alquilo C1-C3 lineal o ramificado o cicloalquilo C3-C6, sustituido con 1 a 5 sustituyentes de halógeno, preferentemente trifluorometilo, 1-clorociclopropilo, 1 -fluorociclopropilo o pentafluoroetilo, más preferentemente trifluorometilo o pentafluoroetilo;
Z2 representa alquilo C1-C3 lineal o ramificado sustituido con 1 a 3 sustituyentes de halógeno o
Z2 representa nitro, metilsulfanilo, metilsulfinilo, metilsulfonilo, flúor, cloro, bromo, yodo; preferentemente Z2 representa alquilo C1-C3 lineal o ramificado sustituido con 1 a 3 flúor, más preferentemente trifluorometilo; y Z3 representa hidrógeno o alquilo C1-C6 lineal o ramificado; preferentemente alquilo C1-C6 lineal o ramificado; más preferentemente hidrógeno, metilo, etilo o n-propilo;
y las sales de estos.
10. Compuestos de acuerdo con una cualquiera de las reivindicaciones anteriores, donde T1 está representado por uno 
T1-1 T1-2 T2-1
11. Composición farmacéutica que comprende al menos un compuesto de acuerdo con cualquiera de las reivindicaciones anteriores, que opcionalmente comprende al menos un componente adicional que se selecciona de adyuvantes, excipientes, solventes y/o al menos un principio farmacéuticamente activo adicional.
12. Composición farmacéutica de acuerdo con la reivindicación 11, para su uso en administración subcutánea u oral.
13. Compuestos o las composiciones farmacéuticas de acuerdo con una cualquiera de las reivindicaciones anteriores para su uso en el control de parásitos en animales, preferentemente para controlar insectos y arácnidos, que preferentemente se seleccionan del grupo de Chelicerata, tal como preferentemente garrapatas, moscas, Acari y ácaros, preferentemente en animales de compañía tales como gatos y perros.
14. Proceso para preparar los compuestos de acuerdo con cualquiera de las reivindicaciones anteriores que comprende el paso de hacer reaccionar un compuesto (A)

con un grupo (B)

(B)
en donde
Y, Q, L1, L2 y L3 tienen el significado como se define en una cualquiera de las reivindicaciones anteriores y en donde PG1 representa un grupo protector o hidrógeno para formar los compuestos de acuerdo con la fórmula (I) y donde en los casos donde PG1 no sea hidrógeno, la desprotección se lleva a cabo para formar los compuestos (I);
en donde el proceso también puede comprender el paso preliminar de preparar el grupo (B)
i) haciendo reaccionar el compuesto (IIa) con un compuesto (IIIa) para formar el compuesto (Va), en donde PG2 es un grupo protector o hidrógeno, y en donde en los casos donde PG2 no sea hidrógeno, se obtiene (Va) haciendo escindir selectivamente PG2:

IVa Va
en donde L1 y L3 tienen el significado como se define en una cualquiera de las reivindicaciones anteriores y en donde PG1 representa un grupo protector o hidrógeno;
o
ii) haciendo reaccionar un compuesto (VI) con un compuesto (IIIb) para formar el compuesto (Vb), en donde PG2 es un grupo protector o hidrógeno, y en donde en los casos donde PG2 no sea hidrógeno, se obtiene (Vb) haciendo escindir selectivamente PG2:

IVb Vb
en donde L1, L2 y L3 tienen el significado como se define en una cualquiera de las reivindicaciones anteriores y donde PG1 representa un grupo protector o hidrógeno.
15. Compuestos intermedios de acuerdo con la fórmula (B),

en donde
Q es O;
L2 es C=O;
L1 y L3 tienen el significado como se define en una cualquiera de las reivindicaciones anteriores y PG1 representa un grupo terc-butilo
o
de acuerdo con la fórmula (C),

donde Y, Q, L1, L2 y L3 tienen el significado como se define en una cualquiera de las reivindicaciones anteriores y donde PG1 representa un grupo terc-butilo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18181950.9A EP3590927A1 (en) | 2018-07-05 | 2018-07-05 | Novel compounds for controlling arthropods |
| PCT/EP2019/067165 WO2020007704A1 (en) | 2018-07-05 | 2019-06-27 | Novel compounds for controlling arthropods |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2928961T3 true ES2928961T3 (es) | 2022-11-24 |
Family
ID=62874671
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19733494T Active ES2928961T3 (es) | 2018-07-05 | 2019-06-27 | Compuestos novedosos para controlar artrópodos |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US11357755B2 (es) |
| EP (2) | EP3590927A1 (es) |
| JP (1) | JP2021528477A (es) |
| KR (1) | KR20210029206A (es) |
| CN (1) | CN112424170A (es) |
| AU (1) | AU2019298306B2 (es) |
| BR (1) | BR112021000028A2 (es) |
| CA (1) | CA3105234A1 (es) |
| DK (1) | DK3818047T3 (es) |
| ES (1) | ES2928961T3 (es) |
| MX (1) | MX2020013770A (es) |
| TW (1) | TW202005532A (es) |
| UY (1) | UY38292A (es) |
| WO (1) | WO2020007704A1 (es) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020164994A1 (en) * | 2019-02-13 | 2020-08-20 | Syngenta Crop Protection Ag | Pesticidally active pyrazole derivatives |
| EP3771711A1 (en) | 2019-07-29 | 2021-02-03 | Bayer Animal Health GmbH | Pyrazole derivatives for controlling arthropods |
| WO2021242581A1 (en) | 2020-05-29 | 2021-12-02 | Boehringer Ingelheim Animal Health USA Inc. | Anthelmintic heterocyclic compounds |
| CN118221639A (zh) * | 2022-12-21 | 2024-06-21 | 顺毅股份有限公司 | 一种n-取代的苯基吡唑衍生物及其制备和应用 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU1494702A (en) * | 2000-11-21 | 2002-06-03 | Leo Pharma As | Cyanoguanidine prodrugs |
| MX295245B (es) | 2005-06-21 | 2012-01-26 | Mitsui Chemicals Inc | Derivado de amida e insecticida que contiene el mismo. |
| UY30244A1 (es) | 2006-03-30 | 2007-11-30 | Tanabe Seiyaku Co | Un proceso para preparar derivados de tetrahidroquinolina |
| AR069802A1 (es) | 2007-12-20 | 2010-02-17 | Astrazeneca Ab | Compuestos de carbamoilo como inhibidores de dgat1 190 |
| PT2433940E (pt) | 2009-04-28 | 2014-12-12 | Chugai Pharmaceutical Co Ltd | Derivado de espiroimidazolona |
| WO2012069366A1 (en) | 2010-11-23 | 2012-05-31 | Syngenta Participations Ag | Insecticidal compounds |
| US20130253011A1 (en) | 2010-12-17 | 2013-09-26 | Syngenta Participations Ag | Insecticidal compounds |
| EA201300894A1 (ru) | 2011-02-09 | 2014-01-30 | Зингента Партисипейшнс Аг | Инсектицидные соединения |
| WO2012175474A1 (en) | 2011-06-20 | 2012-12-27 | Syngenta Participations Ag | 1,2,3 triazole pesticides |
| LT2953942T (lt) | 2013-02-06 | 2018-02-12 | Bayer Cropscience Aktiengesellschaft | Halogenpakeistieji pirazolo dariniai kaip pesticidai |
| EP3066079B1 (de) | 2013-11-05 | 2018-05-09 | Bayer CropScience Aktiengesellschaft | Substituierte benzamide zur behandlung von arthropoden |
| KR102367319B1 (ko) | 2013-11-27 | 2022-02-23 | 바이엘 애니멀 헬스 게엠베하 | 5-플루오로-1h-피라졸의 제조 방법 |
| TWI694066B (zh) | 2014-05-27 | 2020-05-21 | 德商拜耳動物保健有限公司 | 始於六氟丙烯之製備5-氟-1h-吡唑的方法 |
| AR101448A1 (es) * | 2014-08-08 | 2016-12-21 | Bayer Cropscience Ag | Compuestos sustituidos por halógeno |
| AR104398A1 (es) | 2015-04-30 | 2017-07-19 | Bayer Animal Health Gmbh | Combinaciones antiparasíticas |
| WO2016174049A1 (en) * | 2015-04-30 | 2016-11-03 | Bayer Animal Health Gmbh | Anti-parasitic combinations including halogen-substituted compounds |
-
2018
- 2018-07-05 EP EP18181950.9A patent/EP3590927A1/en not_active Ceased
-
2019
- 2019-06-27 BR BR112021000028-3A patent/BR112021000028A2/pt not_active Application Discontinuation
- 2019-06-27 KR KR1020217001583A patent/KR20210029206A/ko not_active Application Discontinuation
- 2019-06-27 JP JP2020573343A patent/JP2021528477A/ja active Pending
- 2019-06-27 CA CA3105234A patent/CA3105234A1/en active Pending
- 2019-06-27 US US17/256,867 patent/US11357755B2/en active Active
- 2019-06-27 CN CN201980048783.1A patent/CN112424170A/zh active Pending
- 2019-06-27 EP EP19733494.9A patent/EP3818047B1/en active Active
- 2019-06-27 MX MX2020013770A patent/MX2020013770A/es unknown
- 2019-06-27 AU AU2019298306A patent/AU2019298306B2/en not_active Expired - Fee Related
- 2019-06-27 ES ES19733494T patent/ES2928961T3/es active Active
- 2019-06-27 WO PCT/EP2019/067165 patent/WO2020007704A1/en active Application Filing
- 2019-06-27 DK DK19733494.9T patent/DK3818047T3/da active
- 2019-07-03 TW TW108123357A patent/TW202005532A/zh unknown
- 2019-07-05 UY UY0001038292A patent/UY38292A/es not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| EP3818047A1 (en) | 2021-05-12 |
| KR20210029206A (ko) | 2021-03-15 |
| AU2019298306A1 (en) | 2021-01-21 |
| EP3818047B1 (en) | 2022-07-27 |
| DK3818047T3 (da) | 2022-10-24 |
| CN112424170A (zh) | 2021-02-26 |
| EP3590927A1 (en) | 2020-01-08 |
| US11357755B2 (en) | 2022-06-14 |
| JP2021528477A (ja) | 2021-10-21 |
| CA3105234A1 (en) | 2020-01-09 |
| TW202005532A (zh) | 2020-02-01 |
| US20220008391A1 (en) | 2022-01-13 |
| UY38292A (es) | 2020-01-31 |
| BR112021000028A2 (pt) | 2021-03-30 |
| MX2020013770A (es) | 2021-03-02 |
| WO2020007704A1 (en) | 2020-01-09 |
| AU2019298306B2 (en) | 2023-08-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2928961T3 (es) | Compuestos novedosos para controlar artrópodos | |
| ES2616230T3 (es) | Compuestos pesticidas de N-aril- o N-heteroaril-pirazolcarboxamida | |
| JP2011057661A (ja) | 殺虫性カルボキサミド類 | |
| EP2253617A1 (de) | Halogen-substituierte Verbindungen als Pestizide | |
| JP2011136928A (ja) | 殺虫性アリールピロリジン類 | |
| WO2021000867A1 (zh) | 一种异噁唑啉类化合物及其应用 | |
| WO2021036966A1 (zh) | 一种双酰胺类化合物及其应用 | |
| WO2021139370A1 (zh) | 一种酰胺类化合物及其应用 | |
| EP4003963B1 (en) | Pyrazole derivatives for controlling arthropods | |
| AU2022310062B2 (en) | Amide compound and use thereof | |
| US20230203013A1 (en) | Method for preparing 2-chloro-n-(1-cyanocyclopropyl)-5-[2'-methyl-5'-(pentafluoroethyl)-4'-(trifluoromethyl)-2'h-1,3'-bipyrazol-4-yl]benzamide | |
| JP2014521592A (ja) | 殺害虫性ジアリールヘテロ環誘導体 |