ES2911888T3 - Compuestos de heteroarilo como inhibidores de IRAK y usos de los mismos - Google Patents
Compuestos de heteroarilo como inhibidores de IRAK y usos de los mismos Download PDFInfo
- Publication number
- ES2911888T3 ES2911888T3 ES16770654T ES16770654T ES2911888T3 ES 2911888 T3 ES2911888 T3 ES 2911888T3 ES 16770654 T ES16770654 T ES 16770654T ES 16770654 T ES16770654 T ES 16770654T ES 2911888 T3 ES2911888 T3 ES 2911888T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- mmol
- atom
- nitrogen
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/24—Benzimidazoles; Hydrogenated benzimidazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D235/30—Nitrogen atoms not forming part of a nitro radical
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Immunology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Dermatology (AREA)
- Emergency Medicine (AREA)
- Communicable Diseases (AREA)
- Endocrinology (AREA)
- Oncology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Vascular Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Un compuesto de fórmula I, **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, donde: **(Ver fórmula)** se selecciona entre **(Ver fórmula)** el anillo X es **(Ver fórmula)** R3 es -R o -haloalquilo; R4 es -R, halógeno, -haloalquilo, -OR, -SR, -CN, -NO2, -SO2R, -SOR, -C(O)R, -CO2R, -C(O)N(R)2, -NRC(O)R, -NRC(O)N(R)2, -NRSO2R o -N(R)2; y cada R es independientemente hidrógeno, grupo alifático C1-6, arilo C3-10, un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado, un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico de 5-6 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido; o se toman dos grupos R del mismo átomo junto con el átomo al que están unidos para formar un arilo C3-10, un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado, un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico de 5-6 átomos que tiene 1-4 heteroátomos, seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido.
Description
DESCRIPCIÓN
Compuestos de heteroarilo como inhibidores de IRAK y usos de los mismos
Solicitudes relacionadas
La presente solicitud reivindica el beneficio de la solicitud provisional de EE. UU. 62/220.358, presentada el 18 de septiembre de 2015.
Campo técnico de la invención
La presente invención proporciona compuestos de fórmula (I) según se define adicionalmente en las reivindicaciones como inhibidores de IRAK y su uso en el tratamiento del cáncer y otras enfermedades relacionadas con las sobreexpresión de IRAK, incluyendo artritis reumatoide, lupus eritematoso sistémico o nefritis lúpica.
Antecedentes de la invención
Las quinasas catalizan la fosforilación de proteínas, lípidos, azúcares, nucleósidos y otros metabolitos celulares y desempeñan una función importante en todos los aspectos de la fisiología de las células eucariotas. En especial, las proteína quinasas y las lípido quinasas participan en la señalización que controla la activación, crecimiento, diferenciación y supervivencia de las células en respuesta a mediadores extracelulares o estímulos como factores de crecimiento, citoquinas o quimioquinas. En general, las proteína quinasas se clasifican en dos grupos, las que fosforilan preferentemente restos tirosina y las que fosforilan preferentemente restos serina y/o treonina.
Las quinasas son objetivos terapéuticos importantes para el desarrollo de fármacos antiinflamatorios (Cohen, 2009. Current Opinion in Cell Biology 21, 1-8), como las quinasas que están implicadas en la organización de las respuestas inmunitarias innata y adquirida. Son quinasas objetivo de especial interés los miembros de la familia IRAK.
Las quinasas asociadas al receptor de interleuquina-1 (IRAK, por sus siglas en inglés) están críticamente implicadas en la regulación de redes de señalización intracelular que controlan la inflamación (Ringwood y Li, 2008. Cytokine 42, 1-7). Las IRAK se expresan en muchos tipos celulares y pueden mediar señales desde diferentes receptores celulares, como los receptores de tipo toll (TLR). Se cree que IRAK4 es la proteína quinasa inicial activada después del receptor de interleuquina-1 (IL-1) y de todos los receptores similares a toll (TLR), excepto TLR3, y que inicia la señalización en el sistema inmunitario innato a través de la rápida activación de IRAK1 y la activación más lenta de IRAK2. IRAK1 fue la primera en identificarse mediante purificación bioquímica de la actividad quinasa dependiente de IL-1 que coinmunoprecipita con el receptor de tipo 1 de la IL-1 (Cao y cols., 1996. Science 271[5252]: 1128-31). IRAK2 se identificó mediante la investigación de secuencias homólogas a IRAKI en la base de datos de etiquetas de secuencias expresadas (EST) en humanos (Muzio y cols., 1997. Science 278[5343]: 1612-5). IRAK3 (también denominada IRAKM) se identificó usando una secuencia EST murina que codifica un polipéptido con homología significativa con IRAK1 para hacer un cribado de una biblioteca de ADNc de leucocitos de sangre periférica activados con fitohemaglutinina (Wesche y cols., 1999. J. Biol. Chem. 274[27]: 19403-10). IRAK4 se identificó buscando en una base de datos secuencias similares a IRAK y mediante PCR de una biblioteca de ADNc universal (Li y cols., 2002. Proc. Natl. Acad. Sci. USA 99[8]: 5567-5572).
En el documento WO 03/030902 se describen derivados de imidazol específicos como fármacos antiinflamatorios y composiciones farmacéuticas de los mismos que son útiles para el tratamiento de afecciones o trastornos relacionados con el sistema inmunitario. En particular, los compuestos se describen para modular la expresión y/o función de proteínas implicadas en la inflamación, la regulación de la respuesta inmunitaria y la proliferación celular.
Los ratones que expresan un mutante catalíticamente inactivo de IRAK4 en lugar de la quinasa natural son completamente resistentes al choque septicémico desencadenado por varios agonistas de TLR y tienen alterada su respuesta a IL-1. Los niños que carecen de actividad IRAK4 debido a un defecto genético sufren infección recurrente por bacterias piógenas. Parece que los TLR y los IL-1R dependientes de IRAK son vitales para la inmunidad infantil frente a algunas bacterias piógenas, pero en adultos tienen una función redundante en la inmunidad protectora frente a la mayoría de las infecciones. Por tanto, los inhibidores de IRAK4 pueden ser útiles para el tratamiento de enfermedades inflamatorias crónicas en adultos sin hacerles demasiado susceptibles a infecciones bacterianas y víricas (Cohen, 2009. Current Opinion in Cell Biology 21, 1-8). Se han desarrollado inhibidores potentes de IRAK4 (Buckley y cols., 2008. Bioorg Med Chem Lett. 18[12]: 3656-60). IRAK1 es esencial para la activación mediada por TLR7 y por TLR9 de IRF7 y la producción de interferón alfa (IFN-a), lo que sugiere que los inhibidores de IRAK1 pueden ser útiles para el tratamiento del lupus eritematoso sistémico (LES). IRAK2 se activa después de IRAK4 y está implicada en la producción de citoquinas proinflamatorias. Por tanto, los inhibidores de IRAK2 pueden ser útiles para enfermedades inflamatorias.
Resumen de la invención
La invención se define en las reivindicaciones. Cualquier tema que quede fuera del alcance de las reivindicaciones se proporciona exclusivamente con fines informativos. En un aspecto, la presente memoria descriptiva proporciona compuestos de fórmula (I):

y derivados, solvatos, sales, hidratos y estereoisómeros farmacéuticamente aceptables de los mismos, donde el anillo X, R3, R4 y Rb es como se define a continuación y como se describe en la memoria descriptiva adicional.
En otro aspecto, en la presente memoria descriptiva se proporcionan compuestos de fórmula (I) que son adecuados para el tratamiento y/o prevención de trastornos relacionados con IRAK. En otro aspecto, la invención proporciona dichos compuestos que son capaces de modular, especialmente inhibir, la actividad o función de IRAK en estados patológicos en mamíferos, especialmente en seres humanos.
Según otro aspecto de la presente memoria descriptiva se proporcionan métodos para el tratamiento y/o prevención de trastornos seleccionados entre trastornos autoinmunitarios, trastornos inflamatorios, enfermedades cardiovasculares, trastornos neurodegenerativos, infecciones bacterianas y víricas, alergia, asma, pancreatitis, fallo multiorgánico, enfermedades renales, agregación plaquetaria, cáncer, trasplante, movilidad espermática, deficiencia de eritrocitos, rechazo de injerto, lesiones pulmonares, enfermedades respiratorias y afecciones isquémicas.
Según otro aspecto, la presente memoria descriptiva proporciona compuestos de fórmula (I) que son selectivos de IRAK-4 y/o IRAK-1.
Según otro aspecto, la presente memoria descriptiva proporciona compuestos de fórmula (I) que son selectivos de IRAK-4 e IRAK-1.
Descripción detallada de determinadas realizaciones o aspectos
1. Descripción general de los compuestos de la invención
En determinados aspectos, en la presente memoria descriptiva se proporcionan inhibidores de IRAK. En algunas realizaciones, estos compuestos incluyen aquellos de las fórmulas descritas en este documento, o una sal farmacéuticamente aceptable de los mismos, donde cada variable es como se define y se describe en este documento.
2. Compuestos y definiciones
Entre los compuestos de la presente memoria descriptiva se incluyen aquellos descritos en general anteriormente, y se ilustran adicionalmente mediante las clases, subclases y especies descritas en este documento. Según se utiliza en este documento, se aplicarán las siguientes definiciones siempre que no se indique otra cosa. A los fines de la presente memoria descriptiva, los elementos químicos se identifican según la tabla periódica de los elementos, versión CAS, Handbook of Chemistry and Physics, 75.a Ed. Adicionalmente, los principios generales de química orgánica se describen en «Organic Chemistry», Thomas Sorrel, University Science Books, Sausalito: 1999, y «March’s Advanced Organic Chemistry», 5.a Ed., Ed.: Smith, M.B. y March, J., John Wiley & Sons, Nueva York: 2001.
El término «alifático» o «grupo alifático», según se utiliza en este documento, significa una cadena de hidrocarburo lineal (es decir, no ramificada) o ramificada, sustituida o no sustituida que está completamente saturada o que contiene una o más unidades de insaturación, o un hidrocarburo monocíclico o hidrocarburo bicíclico que está completamente saturado o contiene una o más unidades de insaturación, pero que no es aromático (también denominado en este documento «carbociclo», «cicloalifático» o «cicloalquilo») que tiene un único punto de unión al resto de la molécula. Siempre que no se especifique otra cosa, los grupos alifáticos contienen 1-6 átomos de carbono alifáticos. En algunos aspectos, los grupos alifáticos contienen 1-5 átomos de carbono alifáticos. En otros aspectos, los grupos alifáticos contienen 1-4 átomos de carbono alifáticos. Aun en otros aspectos, los grupos alifáticos contienen 1-3 átomos de carbono alifáticos, y todavía en otros aspectos, los grupos alifáticos contienen 1-2 átomos de carbono alifáticos. En algunos aspectos, «cicloalifático» (o «carbociclo» o «cicloalquilo») se refiere a un hidrocarburo C3-C6 monocíclico que está completamente saturado o que contiene una o más unidades de insaturación, pero que no es aromático, y tiene un único punto de unión al resto de la molécula. Son ejemplos de grupos alifáticos grupos alquilo C1-C8 , alquenilo C2-C8 , alquinilo C2-C8 lineales o ramificados, sustituidos o no sustituidos e híbridos de los mismos, tales como (cicloalquil)alquilo, (cicloalquenil)alquilo o (cicloalquil)alquenilo.
El término «alquilo inferior» se refiere a un grupo alquilo C1-4 lineal o ramificado. Son ejemplos de grupos alquilo inferiores metilo, etilo, propilo, isopropilo, butilo, isobutilo y tere-butilo.
El término «haloalquilo inferior» se refiere a un grupo alquilo C1-4 lineal o ramificado que está sustituido con uno o más átomos de halógeno.
El término «heteroátomo» significa uno o más de oxígeno, azufre, nitrógeno o fósforo (incluida cualquier forma oxidada de nitrógeno, azufre o fósforo; la forma cuaternizada de cualquier nitrógeno básico o un nitrógeno sustituible de un anillo heterocíclico, por ejemplo N (como en 3,4-dihidro-2H-pirrolilo), NH (como en pirrolidinilo) o NR+ (como en pirrolidinilo N-sustituido).
El término «insaturado», según se usa en este documento, significa que un resto tiene una o más unidades de insaturación.
Según se usa en este documento, el término «cadena de hidrocarburo C1-8 (o Ci-a) bivalente lineal o ramificada, saturada o insaturada» se refiere a cadenas alquileno, alquenileno y alquinileno bivalentes que son lineales o ramificadas según se define en este documento.
El término «alquileno» se refiere a un grupo alquilo bivalente. Una «cadena alquileno» es un grupo polimetileno, es decir, -(CH 2)n-, donde n es un número entero positivo, preferiblemente de 1 a 6, de 1 a 4, de 1 a 3, de 1 a 2 o de 2 a 3. Una cadena alquileno sustituida es un grupo polimetileno en el que uno o más átomos de hidrógeno del metileno se han sustituido con un sustituyente. Entre los sustituyentes adecuados se incluyen aquellos que se describen a continuación para un grupo alifático sustituido.
El término «alquenileno» se refiere a un grupo alquenilo bivalente. Una cadena alquenileno sustituida es un grupo polimetileno que contiene al menos un enlace doble en el que uno o más átomos de hidrógeno se han sustituido con un sustituyente. Entre los sustituyentes adecuados se incluyen aquellos que se describen a continuación para un grupo alifático sustituido.
El término «halógeno» significa F, Cl, Br o I.
El término «arilo» usado solo o como parte de un resto mayor como en «aralquilo», «aralcoxi» o «ariloxialquilo», se refiere a sistemas de anillo monocíclico o bicíclico que tiene un total de cinco a catorce átomos del anillo, donde al menos un anillo del sistema es aromático y donde cada anillo del sistema contiene de tres a siete átomos del anillo. El término «arilo» se utiliza indistintamente con el término «anillo arilo». En determinadas realizaciones de la presente invención, «arilo» se refiere a un sistema de anillo aromático. Son ejemplos de grupos arilo fenilo, bifenilo, naftilo, antracilo y similares, que opcionalmente incluyen uno o más sustituyentes. También se incluyen dentro del alcance del término «arilo», según se usa en este documento, un grupo en el que un anillo aromático se fusiona con uno o más anillos no aromáticos, como indanilo, ftalimidilo, naftimidilo, fenantridinilo o tetrahidronaftilo, y similares.
Los términos «heteroarilo» y «heteroar-», utilizados solos o como parte un resto más grande, por ejemplo, «heteroaralquilo» o «heteroaralcoxi», se refieren a grupos con 5 a 10 átomos del anillo, preferiblemente 5, 6 o 9 átomos del anillo; con 6, 10 o 14 electrones n compartidos en una disposición cíclica; y tienen, además de los átomos de carbono, de uno a cinco heteroátomos. El término «heteroátomo» se refiere a nitrógeno, oxígeno o azufre, e incluye cualquier forma oxidada de nitrógeno o azufre, y cualquier forma cuaternizada de un nitrógeno básico. Entre los grupos heteroarilo se incluyen, sin limitaciones, tienilo, furanilo, pirrolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, oxazolilo, isoxazolilo, oxadiazolilo, tiazolilo, isotiazolilo, tiadiazolilo, piridilo, piridazinilo, pirimidinilo, pirazinilo, indolizinilo, purinilo, naftiridinilo y pteridinilo. Los términos «heteroarilo» y «heteroar-», según se usa en este documento, también incluyen grupos en los que un anillo heteroaromático se fusiona con uno o más anillos arilo, cicloalifático o heterociclilo, donde el radical o punto de unión está en el anillo heteroaromático. Entre los ejemplos no limitantes se incluyen indolilo, isoindolilo, benzotienilo, benzofuranilo, dibenzofuranilo, indazolilo, bencimidazolilo, benzotiazolilo, quinolilo, isoquinolilo, cinolinilo, ftalazinilo, quinazolinilo, quinoxalinilo, 4H-quinolizinilo, carbazolilo, acridinilo, fenazinilo, fenotiazinilo, fenoxazinilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo y pirido[2,3-b]-1,4-oxazin-3(4H)-ona. Un grupo heteroarilo es opcionalmente mono o bicíclico. El término «heteroarilo» se utiliza indistintamente con los términos «anillo heteroarilo», «grupo heteroarilo» o «heteroaromático», incluyendo cualquiera de los términos anillos que están opcionalmente sustituidos. El término «heteroaralquilo» se refiere a un grupo alquilo sustituido por un heteroarilo, donde las porciones alquilo y heteroarilo independientemente están opcionalmente sustituidas.
Según se usa en este documento, los términos «heterociclo», «heterociclilo», «radial heterocíclico» y «anillo heterocíclico» se usan indistintamente y se refieren a un resto estable monocíclico de 5 a 7 átomos o heterocíclico bicíclico de 7 a 10 átomos que está saturado o parcialmente insaturado y tiene, además de los átomos de carbono, uno o más, preferiblemente de uno a cuatro, heteroátomos, como se define anteriormente. Cuando se utiliza en referencia a un átomo de anillo de un heterociclo, el término «nitrógeno» incluye un nitrógeno sustituido. Como ejemplo, en un anillo saturado o parcialmente insaturado con 0-3 heteroátomos seleccionados entre oxígeno, azufre o nitrógeno, el nitrógeno es N (como en 3,4-dihidro-2H-pirrolilo), NH (como en pirrolidinilo) o NR (como en pirrolidinilo W-sustituido).
Un anillo heterocíclico puede estar unido a su grupo pendiente en cualquier heteroátomo o átomo de carbono, lo que da lugar a una estructura estable y cualquiera de los átomos del anillo puede estar opcionalmente sustituido. Entre los ejemplos de dichos radicales heterocíclicos saturados o parcialmente insaturados se incluyen, sin limitaciones, tetrahidrofuranilo, tetrahidrotiofenilo, pirrolidinilo, piperidinilo, pirrolinilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo, decahidroquinolinilo, oxazolidinilo, piperazinilo, dioxanilo, dioxolanilo, diazepinilo, oxazepinilo, tiazepinilo, morfolinilo y
quinudidinilo. Los términos «heterocido», «heterocidilo», «anillo heterociclilo», «grupo heterocíclico», «resto heterocídico» y «radical heterocíclico» se utilizan indistintamente en este documento, y también incluyen grupos en los que un anillo heterociclilo se fusiona con uno o más anillos arilo, heteroarilo o cicloalifático, como indolinilo, 3H-indolilo, cromanilo, fenantridinilo o tetrahidroquinolinilo, donde el radical o punto de unión está en el anillo heterociclilo. Un grupo heterociclilo es opcionalmente mono o bicíclico. El término «heterociclilalquilo» se refiere a un grupo alquilo sustituido por un heterociclilo, donde las porciones alquilo y heterociclilo independientemente están opcionalmente sustituidas.
Según se usa en este documento, el término «parcialmente insaturado» se refiere a un resto del anillo que incluye al menos uno enlace doble o triple. El término «parcialmente insaturado» pretende abarcar los anillos con sitios múltiples de insaturación, pero no pretende incluir restos arilo o heteroarilo, según se define en este documento.
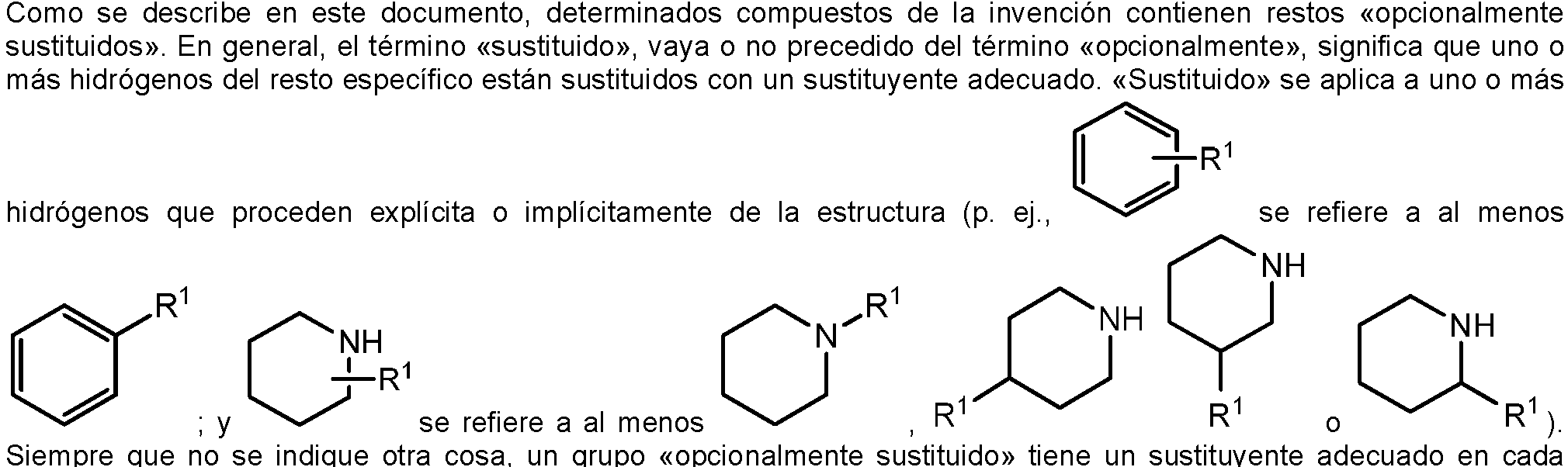
posición sustituible del grupo, y cuando más de una posición en una estructura determinada está sustituida con más de un sustituyente seleccionado entre un grupo específico, el sustituyente es el mismo o diferente en cada posición. Las combinaciones de sustituyentes contempladas en esta invención son preferiblemente aquellas que tiene como resultado la formación de compuestos estables o químicamente viables. El término «estable», según se utiliza en este documento, se refiere a compuestos que no se alteran sustancialmente cuando se someten a condiciones que permitan su producción, detección y, en determinadas realizaciones, su recuperación, purificación y uso para uno o más de los objetivos descritos en este documento.
Los sustituyentes monovalentes adecuados en un átomo de carbono sustituible de un grupo «opcionalmente sustituido» son independientemente deuterio; halógeno; -(CH2)o-4R°; -(CH2)o-4OR°; -O(CH2)o-4R°, -O-(CH2)o-4C(O)OR°; -(CH 2)o-4CH(OR°)2; -(CH2)o-4SR°; -(CH2)o-4Ph, que están opcionalmente sustituidos con R°; -(CH2)o-4O(CH2)o-iPh que está opcionalmente sustituido con R°; -CH=CHPh, que están opcionalmente sustituidos con R°; -(CH2)o-4O(CH2)o-i-piridilo que está opcionalmente sustituido con R°; -N O 2 ; -CN; -N 3 ; -(CH2)o-4N(R°)2; -(CH2)o-4N(R°)C(O)R°; -N(R°)C(S)R°; -(CH2)o-4N(R°)C(O)NR°2; -N(R°)C(S)NR°2 ; -(CH2)o-4N(R°)C(O)OR°; -N(R°)N(R°)C(O)R°; -N(R°)N(R°)C(O)NR°2 ; -N(R°)N(R°)C(O)OR°; -(CH2)o-4C(O)R°; -C(S)R°; -(CH2)o-4C(O)OR°; -(CH2)o-4C(O)SR°; -(CH2)o-4C(O)OSiR°3; -(CH2)o-4OC(O)R°; -OC(O)(CH2)o-4SR°, SC(S)SR°; -(CH2)o-4SC(O)R°; -(CH 2)o-4C(O)NR°2; -C(S)NR°2; -C(S)SR°; -SC(S)SR°, -(CH2)o-4OC(O)NR°2; -C(O)N(OR°)R°; -C(O)C(O)R°; -C(O)CH2C(O)R°; -C(NOR°)R°; -(CH2)o-4SSR°; -(CH2)o--4S(O)2R°; -(CH2)o-4S(O)2OR°; -(CH2)o-4OS(O)2R°; -S(O)2NR°2 ; -(CH2)o-4S(O)R°; -N(R°)S(O)2NR°2; -N(R°)S(O)2R°; -N(OR°)R°; -C(NH)NR°2; -P(O)2R°; -P(O)R°2; -OP(O)R°2; -OP(O)(OR°)2; SiR°3; -(C 1-4 alquileno lineal o ramificado)O-N(R°)2 ; o -(C 1-4 alquileno lineal o ramificado)C(O)O-N(R°)2 , donde cada R° está opcionalmente sustituido como se define a continuación y es independientemente hidrógeno, grupo alifático C1 -6 , -CH2Ph, -O(CH 2)o-iPh, -CH2-(anillo heteroarilo de 5-6 átomos), o un anillo de 5-6 átomos saturado, parcialmente insaturado o arilo que tiene o-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o, a pesar de la definición anterior, dos apariciones independientes de R°, tomados junto con sus átomos intermedios, forman un anillo mono o bicíclico saturado, parcialmente insaturado o arilo de 3-12 átomos que tiene o-4 heteroátomos que se seleccionan independientemente entre nitrógeno, oxígeno o azufre, que está opcionalmente sustituido como se define a continuación.
Los sustituyentes monovalentes adecuados en R° (o el anillo formado tomando las dos apariciones independiente de R° junto con sus átomos intermedios) son independientemente deuterio, halógeno, -(CH 2)o- 2 R*, -(haloR*), -(CH 2)o- 2 OH, -(CH2)o- 2 OR*, -(CH 2)o- 2 CH(OR*)2 ; -O(haloR*), -CN, -N 3 , -(CH 2)o- 2 C(O)R*, -(CH 2)o-2C(O)OH, -(CH 2)o- 2 C(O)OR*, -(CH2)o- 2 SR*, -(CH 2)o- 2 SH, -(CH 2)o- 2 NH2 , -(CH 2)o- 2 NHR*, -(CH 2)o- 2 NR*2 , -N O 2 , -SiR*3, -OSiR*3, -C(O)SR* -(C 1-4 alquileno lineal o ramificado)C(O)OR* o -SSR* donde cada R* no está sustituido o cuando va precedido por «halo» está sustituido solo con uno o más halógenos, y se selecciona independientemente entre alifático C1 -4 , -C H 2Ph, -O(CH 2)o-1Ph, o un anillo de 5-6 átomos saturado, parcialmente insaturado o arilo que tiene o-4 heteroátomos independientemente seleccionados entre nitrógeno, oxígeno o azufre. Los sustituyentes divalentes adecuados en un átomo de carbono saturado de R° son =O y =S.
Los sustituyentes divalentes adecuados en un átomo de carbono saturado de un grupo «opcionalmente sustituido» son los siguientes: =O, =S, =NNR*2, =NNHC(O)R*, =NNHC(O)OR*, =NNHS(O)2R*, =NR*, =NOR*, -O(C(R *2))2-aO- o -S(C(R*2))2-3S-, donde cada aparición independiente de R* se selecciona entre hidrógeno, grupo alifático C1-6 que está
sustituido como se define a continuación, o un anillo no sustituido de 5-6 átomos saturado, parcialmente insaturado o arilo que tiene 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. Los sustituyentes divalentes adecuados que se unen a los carbonos vecinos sustituibles de un grupo «opcionalmente sustituido» son: -O(CR*2)2-3O-, donde cada aparición independiente de R* se selecciona entre hidrógeno, grupo alifático Ci-6 que está opcionalmente sustituido como se define a continuación, o un anillo no sustituido de 5-6 átomos saturado, parcialmente insaturado o arilo que tiene 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
Los sustituyentes adecuados en el grupo alifático de R* son halógeno, -R*, -(haloR*), -OH, -OR*, -O(haloR*), -CN, -C(O)OH, -C(O)OR*, -N H 2 , -NHR*, -N R *2 o -N O 2 , donde cada R* no está sustituido o cuando va precedido por «halo» está sustituido solo con uno o más halógenos y es independientemente alifático C1-4 , -CH 2Ph, -O(CH 2)o-iPh o un anillo de 5-6 átomos saturado, parcialmente insaturado o arilo que tiene 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
Los sustituyentes adecuados en un átomo de nitrógeno sustituible de un grupo «opcionalmente sustituido» son -R t -NRt>, -C(O)Rt -C(O)ORt -C(O)C(O)Rt -C(O)CH2C(O)Rt -S (O ^R f , -S(O)2NR^, -C(S)NR^, -C(NH)NR^ o -N(Rt )S(O)2Rt ; donde cada Rf es independientemente hidrógeno, grupo alifático C1-6 que está opcionalmente sustituido como se define a continuación, -OPh no sustituido, o un anillo no sustituido de 5-6 átomos saturado, parcialmente insaturado o arilo que tiene 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o a pesar de la definición anterior, dos apariciones independientes de Rt , junto con sus átomos intermedios forman un anillo mono o bicíclico no sustituido de 3-12 átomos saturado, parcialmente insaturado o arilo que tiene 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
Los sustituyentes adecuados en el grupo alifático de Rt son independientemente halógeno, -R*, -(haloR*), -OH, -OR*, -O(haloR*), -CN, -C(O)OH, -C(O)OR*, -NH 2 , -NHR*, -N R *2 o -N O 2 , donde cada R* no está sustituido o cuando va precedido por «halo» está sustituido solo con uno o más halógenos y es independientemente alifático C1-4 , -C H 2Ph, -O(CH2)0-iPh o un anillo de 5-6 átomos saturado, parcialmente insaturado o arilo que tiene 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
En determinadas realizaciones, los términos «opcionalmente sustituido», «alquilo opcionalmente sustituido», «opcionalmente sustituido», «alquenilo opcionalmente sustituido», «alquinilo opcionalmente sustituido», «carbocíclico opcionalmente sustituido», «arilo opcionalmente sustituido», «heteroarilo opcionalmente sustituido», «heterocíclico opcionalmente sustituido» y cualquier otro grupo opcionalmente sustituido según se utiliza en este documento, se refiere a grupos que están sustituidos o no sustituidos mediante sustitución independiente de uno, dos , tres o más de los átomos de hidrógeno en los mismos con los sustituyentes típicos que incluyen, entre otros:
-F, -Cl, -Br, -I, deuterio,
-OH, hidroxi protegido, alcoxi, oxo, tiooxo,
-NO2 , -CN, CF3 , N3 ,
-NH2 , amino protegido, -NH-alquilo, -NH-alquenilo, -NH-alquinilo, -NH-cicloalquilo, -NH-arilo, -NH-heteroarilo, -NH-heterocíclico, -dialquilamino, -diarilamino, -diheteroarilamino,
-O-alquilo, -O-alquenilo, -O-alquinilo, -O-cicloalquilo, -O-arilo, -O-heteroarilo, -O-heterocíclico,
-C(O)-alquilo, -C(O)-alquenilo, -C(O)-alquinilo, -C(O)-carbociclilo, -C(O)-arilo, -C(O)-heteroarilo, -C(O)-heterociclilo,
-CONH2 , -CONH-alquilo, -CONH-alquenilo, -CONH-alquinilo, -CONH-carbociclilo, -CONH-arilo, -CONH-heteroarilo, -CONH-heterociclilo,
-OCO2-alquilo, -OCO2-alquenilo -OCO2-alquinilo, -OCO2-carbociclilo, -OCO2-arilo, -OCO2-heteroarilo, -OCO2-heterociclilo, -OCONH2 , -OCONH-alquilo, -OCONH-alquenilo, -OCONH-alquinilo, -OCONH-carbociclilo, -OCONH-arilo, -OCONH-heteroarilo, -OCONH-heterociclilo,
-NHC(O)-alquilo, -NHC(O)-alquenilo, -NHC(O)-alquinilo, -NHC(O)-carbociclilo, -NHC(O)-arilo, -NHC(O)-heteroarilo, -NHC(O)-heterociclilo, -NHCO2-alquilo, -NHCO2-alquenilo, -NHCO2-alquinilo, -NHCO2-carbociclilo, -NHCO2-arilo, -NHCO2-heteroarilo, -NHCO2-heterociclilo, -NHC(O)NH2 , -NHC(O)NH-alquilo, -NHC(O)NH-alquenilo, -NHC(O)NH-alquenilo, -NHC(O)NH-carbociclilo, -NHC(O)NH-arilo, -NHC(O)NH-heteroarilo, -NHC(O)NH-heterociclilo, NHC(S)NH2, -NHC(S)NH-alquilo, -NHC(S)NH-alquenilo, -NHC(S)NH-alquinilo, -NHC(S)NH-carbociclilo, -NHC(S)NH-arilo, -NHC(S)NH-heteroarilo, -NHC(S)NH-heterociclilo, -NHC(NH)NH2 , -NHC(NH)NH-alquilo, -NHC(NH)NH-alquenilo, -NHC(NH)NH-alquenilo, -NHC(NH)NH-carbociclilo, -NHC(NH)NH-arilo, -NHC(NH)NH-heteroarilo, -NHC(NH)NH-heterociclilo, -NHC(NH)-alquilo, -NHC(NH)-alquenilo, -NHC(NH)-alquenilo, -NHC(NH)-carbociclilo, -NHC(NH)-arilo, -NHC(NH)-heteroarilo, -NHC(NH)-heterociclilo,
-C(NH)NH-alquilo, -C(NH)NH-alquenilo, -C(NH)NH-alquinilo, -C(NH)NH-carbociclilo, -C(NH)NH-arilo, -C(NH)NH-heteroarilo, -C(NH)NH-heterociclilo,
-S(O)-alquilo, -S(O)-alquenilo, -S(O)-alquinilo, -S(O)-carbocid ilo, -S(O)-arilo, -S(O)-heteroarilo, -S(O)-heterocid ilo, -SO2NH2, -SO2NH-alquilo, -sO2NH-alquenilo, -SO2NH-alquinilo, -SO2NH-carbod d ilo, -SO2NH-arilo, -SO2NH-heteroarilo, -SO2NH-heterocid ilo,
-NHSO2-alquilo, -NHSO2-alquenilo, -NHSO2-alquinilo, -NHSO2-carbociclilo, -NHSO2-arilo, -NHSO2-heteroarilo, -NHSO2-heteroddilo,
-CH2NH2 , -CH2SO2CH3 ,
-mono-, di- o tri-alquil sililo,
-alquilo, -alquenilo, -alquinilo, -arilo, -arilalquilo, -heteroarilo, -heteroarilalquilo, -heterocicloalquilo, -cicloalquilo, -carbocíclico, -heterocíclico, polialcoxialquilo, polialcoxi, -metoximetoxi, -metoxietoxi, -SH, -S-alquilo, -S-alquenilo, -S-alquinilo, -S-carbociclilo, -S-arilo, -S-heteroarilo, -S-heterociclilo o metiltiometilo.
Según se usa en este documento, el término «sal farmacéuticamente aceptable» se refiere a aquellas sales que son, dentro del alcance del buen criterio médico, adecuadas para su uso en contacto con los tejidos de seres humanos y animales inferiores sin toxicidad indebida, irritación, respuesta alérgica y similares, y son proporcionales con una relación beneficio/riesgo razonable. Las sales farmacéuticamente aceptables son bien conocidas en la técnica. Por ejemplo, S. M. Berge y cols. describen sales farmacéuticamente aceptables en detalle en J. Pharmaceutical Sciences, 1977, 66, 1-19. Las sales farmacéuticamente aceptables de los compuestos de esta invención incluyen aquellas derivadas de bases y ácidos orgánicos e inorgánicos aceptables. Son ejemplos de sales de adición de ácido no tóxicas farmacéuticamente aceptables las sales de un grupo amino formado con ácidos inorgánicos como ácido clorhídrico, ácido bromhídrico, ácido fosfórico, ácido sulfúrico y ácido perclórico o con ácidos orgánicos como ácido acético, ácido oxálico, ácido maleico, ácido tartárico, ácido cítrico, ácido succínico o ácido malónico o utilizando otros métodos usados en la técnica como intercambio iónico. Otras sales farmacéuticamente aceptables incluyen sales de adipato, alginato, ascorbato, aspartato, bencenosulfonato, benzoato, bisulfato, borato, butirato, alcanforato, alcanforsulfonato, citrato, ciclopentanopropionato, digluconato, dodecilsulfato, etanosulfonato, formato, fumarato, glucoheptonato, glicerofosfato, gluconato, hemisulfato, heptanoato, hexanoato, yodhidrato, 2-hidroxi-etanosulfonato, lactobionato, lactato, laurato, laurilsulfato, malato, maleato, malonato, metanosulfonato, 2-naftalenosulfonato, nicotinato, nitrato, oleato, oxalato, palmitato, pamoato, pectinato, persulfato, 3-fenilpropionato, fosfato, pivalato, propionato, estearato, succinato, sulfato, tartrato, tiocianato, ptoluenosulfonato, undecanoato, valerato y similares.
Entre las sales derivadas de bases adecuadas se incluyen sales de metales alcalinos, metales alcalinotérreos, amonio y N+(alquilo C1-4)4. Entre las sales alcalinas o alcalinotérreas representativas se incluyen sodio, litio, potasio, calcio, magnesio y similares. Las sales farmacéuticamente aceptables adicionales incluyen, cuando es adecuado, cationes no tóxicos de amonio, amonio cuaternario y amina formados utilizando contraiones como haluro, hidróxido, carboxilato, sulfato, fosfato, nitrato, sulfonato de alquilo y sulfonato de arilo inferiores.
Siempre que no se indique otra cosa, también se pretende que las estructuras descritas en este documento incluyan todas las formas isoméricas (p. ej., enantioméricas, diastereoméricas y geométricas [o conformacionales]) de la estructura; por ejemplo, las configuraciones R y S para cada centro asimétrico, isómeros Z y E con dobles enlaces e isómeros conformacionales Z y E. Por tanto, los isómeros estereoquímicos sencillos, así como las mezclas enantioméricas, diastereoméricas y geométricas (o conformacionales) de los presentes compuestos están dentro del alcance de la invención. Siempre que no se indique otra cosa, todas las formas tautoméricas de los compuestos de la invención están dentro del alcance de la invención. Por ejemplo, la invención contempla tautómeros de las siguientes fórmulas, siempre que la valencia y/u otros requisitos químicos se cumplan:

Adicionalmente, siempre que no se indique otra cosa, también se pretende que las estructuras descritas en este documento incluyan compuestos que difieren únicamente en la presencia de uno o más átomos isotópicamente enriquecidos. Por ejemplo, los compuestos que tienen las estructuras presentes que incluyen la sustitución de hidrógeno
por deuterio o tritio, o la sustitución de un carbono por un carbono enriquecido en 13C o 14C están dentro del alcance de esta invención. En algunas realizaciones, el grupo comprende uno o más átomos de deuterio.
Se pretende además que un compuesto de fórmula I incluya sus formas marcadas con isótopos. Una forma marcada con isótopo de un compuesto de fórmula I es idéntica a este compuesto, excepto por el hecho de que uno o más átomos del compuesto se ha sustituido por un átomo o átomos con una masa atómica o un número másico que difiere de la masa atómica o el número másico del átomo natural. Entre los ejemplos de isótopos que se encuentran fácilmente en el mercado y que pueden incorporarse a un compuesto de fórmula I mediante métodos bien conocidos se incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro, por ejemplo, 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F y 36Cl, respectivamente. Se pretende que un compuesto de la fórmula I, un profármaco del mismo o una sal farmacéuticamente aceptable de cualquiera de los dos que contenga uno o más de los isótopos mencionados anteriormente y/u otros isótopos de otros átomos sea parte de la presente invención. Se puede usar un compuesto de fórmula I marcado con isótopo de diversas formas beneficiosas. Por ejemplo, un compuesto de fórmula I marcado con isótopo dentro del cual se ha incorporado, por ejemplo, un radioisótopo, como 3H o 14C, es adecuado para ensayos de distribución en tejidos de medicamentos y/o sustratos. Estos radioisótopos, es decir, tritio (3H) y carbono 14 (14C), son especialmente preferidos debido a su sencilla preparación y a la excelente capacidad de detección. La incorporación de isótopos más pesados, por ejemplo deuterio (2H), a un compuesto de fórmula I tiene ventajas terapéuticas debido a la mayor estabilidad metabólica de este compuesto marcado con el isótopo. Una mayor estabilidad metabólica se traduce directamente en dosis más bajas o un aumento de la semivida in vivo, lo que en la mayoría de las circunstancias representaría una realización preferida de la presente invención. Un compuesto de fórmula I marcado con isótopo puede prepararse habitualmente realizando los procedimientos descritos en los esquemas de síntesis y la descripción relacionada, en la parte de ejemplos y en la parte de preparación del presente texto, sustituyendo un reactivo no marcado con isótopo por un reactivo marcado con isótopo fácilmente disponible.
También se puede incorporar deuterio (2H) en un compuesto de fórmula I con el fin de manipular el metabolismo oxidativo del compuesto por medio del efecto isotópico cinético primario. El efecto isotópico cinético primario es un cambio de la velocidad de una reacción química que es consecuencia del intercambio de núcleos isotópicos, lo que a su vez está causado por el cambio en las energías del estado fundamental necesarias para la formación de enlaces covalentes después de este intercambio isotópico. El intercambio de un isótopo más pesado normalmente tiene como consecuencia una reducción de la energía del estado fundamental para un enlace químico y causa, por tanto, una reducción de la velocidad de rotura de enlaces limitantes de la velocidad. Si la rotura de enlaces se produce en o en las proximidades de una región de punto de silla a lo largo de la coordenada de una reacción multiproducto, los cocientes de distribución de productos se pueden alterar de forma sustancial. Como explicación: si el deuterio se une a un átomo de carbono en una posición no intercambiable, son típicas las diferencias de velocidad de kM/kD = 2-7. Si esta diferencia de velocidad se aplica con éxito a un compuesto de fórmula I que es susceptible a la oxidación, el perfil de este compuesto in vivo se puede modificar considerablemente y tiene como resultado una mejora de las propiedades farmacocinéticas.
Cuando se descubren y desarrollan agentes terapéuticos, la persona experta en la materia es capaz de optimizar los parámetros farmacocinéticos, conservando a la vez las propiedades in vitro deseables. Es razonable asumir que muchos compuestos con malos perfiles farmacocinéticos son susceptibles del metabolismo oxidativo. Los ensayos de microsomas hepáticos in vitro actualmente disponibles proporcionan información valiosa sobre el curso del metabolismo oxidativo de este tipo, lo que a su vez permite el diseño racional de compuestos deuterados de fórmula I con mejor estabilidad a través de la resistencia a dicho metabolismo oxidativo. Se obtienen de este modo mejoras significativas en los perfiles farmacocinéticos de compuestos de fórmula I, y se pueden expresar cuantitativamente en términos de aumentos en la semivida (t-io) in vivo, concentración en el efecto terapéutico máximo (Cmáx), área bajo la curva de dosis-respuesta (AUC) y F; y en términos de reducción del aclaramiento, dosis y costes de materiales.
La siguiente explicación está destinada a ilustrar lo anterior: un compuesto de fórmula I que tiene múltiples sitios potenciales de ataque para el metabolismo oxidativo, por ejemplo, átomos de hidrógeno bencílico y átomos de hidrógeno unidos a un átomo de nitrógeno, se prepara como una serie de análogos en los que diversas combinaciones de átomos de hidrógeno se sustituyen por átomos de deuterio, de modo que algunos, la mayoría o todos estos átomos de hidrógeno se han sustituido por átomos de deuterio. Las determinaciones de la semivida permiten la determinación favorable y precisa del grado al cual ha mejorado la resistencia al metabolismo oxidativo. De este modo, se determina que la semivida del compuesto original se puede extender hasta el 100 % como consecuencia de un intercambio deuterio-hidrógeno de este tipo.
El intercambio deuterio-hidrógeno en un compuesto de fórmula I también se puede usar para conseguir una modificación favorable del espectro de metabolitos del compuesto de partida con el fin de disminuir o eliminar los metabolitos tóxicos no deseados. Por ejemplo, si aparece un metabolito tóxico por escisión oxidativa del enlace carbono-hidrógeno (C-H), puede ser razonable asumir que el análogo deuterado disminuirá o eliminará en gran medida la producción del metabolito no deseado, incluso si la oxidación en particular no es un paso determinante de la velocidad. Se puede encontrar más información sobre el estado de la técnica con respecto al intercambio deuterio-hidrógeno por ejemplo en Hanzlik y cols., J. Org. Chem. 55, 3992-3997, 1990, Reider y cols., J. Org. Chem. 52, 3326-3334, 1987, Foster, Adv. Drug Res. 14, 1-40, 1985, Gillette y cols., Biochemistry 33(10) 2927-2937, 1994, y Jarman y cols. Carcinogenesis 16(4), 683-688, 1993.
Según se usa en este documento, el término «modulador» se define como un compuesto que se une y/o inhibe la diana con una afinidad medible. En determinadas realizaciones, un modulador tiene una IC50 y/o constante de unión de menos
de aproximadamente 50 ^M, menos de aproximadamente 1 ^M, menos de aproximadamente 500 nM, menos de aproximadamente 100 nM o menos de aproximadamente 10 nM.
Los términos «afinidad medible» e «inhibir de forma medible» , según se usa en este documento, significa un cambio medible en la actividad IRAK entre una muestra que comprende un compuesto de la presente invención, o composición del mismo, e IRAK, y una muestra equivalente que comprende IRAK, en ausencia de dicho compuesto, o composición del mismo.
Las combinaciones de sustituyentes y variables contempladas por esta invención son únicamente aquellas que tienen como resultado la formación de compuestos estables. El término «estable», según se usa en este documento, se refiere a compuestos que poseen una estabilidad suficiente que permite su fabricación y que mantiene la integridad del compuesto durante un periodo de tiempo suficiente para que sea útil a los fines descritos en este documento (p. ej., administración terapéutica o profiláctica a un sujeto).
La relación de una lista de grupos químicos en cualquier definición de una variable de este documento incluye definiciones de esa variable como cualquier grupo único o combinación de los grupos enumerados. La relación de una realización de una variable en este documento incluye esa realización, así como cualquier realización sola o en combinación con cualquier otra realización o partes de la misma.
3. Descripción de los compuestos según la invención
Según un aspecto, la presente descripción proporciona un compuesto de fórmula I,

o una sal farmacéuticamente aceptable del mismo, donde:

se selecciona entre

y

el anillo X es

R3 es -R o -haloalquilo;
R4 es -R , halógeno, -haloalquilo, -OR, -SR, -CN, -N O 2 , -SO2R, -SOR, -C(O)R, -CO2R, -C(O)N(R)2 , -NRC(O)R, -NRC(O)N(R)2 , -NRSO2 R, o -N(R)2 ; y
cada R es independientemente hidrógeno, grupo alifático C1 -6 , arilo C3 -10 , un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado, un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico de 5-6 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido; o
se toman dos grupos R del mismo átomo junto con el átomo al que están unidos para formar un arilo C3-10, un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado, un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico de 5-6 átomos que tiene 1-4 heteroátomos, seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido.
Se prefieren los compuestos de fórmula I, donde R3 es un grupo alifático C1-6, un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado o un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido.
Se prefieren los compuestos de fórmula I, donde R3 es metilo, etilo, propilo, i-propilo, n-butilo, s-butilo, t-butilo, un pentilo de cadena lineal o ramificada, o un hexilo de cadena lineal o ramificada; cada uno de ellos opcionalmente sustituido. Se prefieren compuestos de fórmula I, donde R3 se selecciona a partir de:

Se prefieren compuestos de fórmula I, seleccionados a partir de





Se prefiere una composición farmacéutica que comprende un compuesto de fórmula I y un adyuvante, transportador o vehículo farmacéuticamente aceptable.
Según un aspecto, en la presente memoria descriptiva se proporciona un compuesto de fórmula I,

o una sal farmacéuticamente aceptable del mismo, donde:

es un arilo C3-10, un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado, un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico de 5-6 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
Rb es -R, halógeno, -haloalquilo, -OR, -SR, -CN, -NO 2 , -SO 2R, -SOR, -C(O)R, -CO2R, -C(O)N(R)2 , -NRC(O)R, -NRC(O)N(R)2 , -NRSO2R o -N(R)2 ; o Rb está ausente;
el anillo X es un arilo C3-10; un anillo heteroarilo monocíclico de 5-6 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; un arilo C3-10 fusionado, un anillo carbocíclico fusionado de 5-10 átomos saturado o parcialmente insaturado, un anillo heterocíclico fusionado de 5-10 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico fusionado de 5 10 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido;
R3 es -R o -haloalquilo;
R4 es -R, halógeno, -haloalquilo, -OR, - SR, -CN, -N O 2 , -SO2R, -SOR, -C(O)R, -CO2R, -C(O)N(R)2 , -NRC(O)R, -NRC(O)N(R)2 , -NRSO2R o -N(R)2 ;
cada R es independientemente hidrógeno, grupo alifático C1 -6 , arilo C3 -10 , un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado, un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico de 5-6 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido; o
se toman dos grupos R del mismo átomo junto con el átomo al que están unidos para formar un arilo C3-10, un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado, un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico de 5-6 átomos que tiene 1-4 heteroátomos, seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido.
En determinados aspectos,

es fenilo, naftilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, adamantilo, ciclooctilo, [3.3.0]biciclooctanilo, [4.3.0]biciclononanilo, [4.4.0]biciclodecanilo, [2.2.2]biciclooctanilo, fluorenilo, indanilo, tetrahidronaftilo, acridinilo, azocinilo, bencimidazolilo, benzofuranilo, benzotiofuranilo, benzotiofenilo, benzoxazolilo, benctiazolilo, benctriazolilo, benctetrazolilo, bencisoxazolilo, bencisotiazolilo, bencimidazolinilo, carbazolilo, NH-carbazolilo, carbolinilo, cromanilo, cromenilo, cinnolinilo, decahidroquinolinilo, 2H,6H-1,5,2-ditiazinilo, dihidrofuro [2,3-b] tetrahidrofurano, furanilo, furazanilo, imidazolidinilo, imidazolinilo, imidazolilo, 1 H-indazolilo, indolenilo, indolinilo, indolizinilo, indolilo, 3H-indolilo, isoindolinilo, isoindolenilo, isobenzofuranilo, isocromanilo, isoindazolilo, isoindolinilo, isoindolilo, isoquinolinilo, isotiazolilo, isoxazolilo, morfolinilo, naftiridinilo, octahidroisoquinolinilo, oxadiazolilo, 1,2,3-oxadiazolilo, 1,2,4-oxadiazolilo; 1,2,5-oxadiazolilo, 1.3.4- oxadiazolilo, oxazolidinilo, oxazolilo, oxazolidinilo, pirimidinilo, fenantridinilo, fenantrolinilo, fenazinilo, fenotiazinilo, fenoxatiinilo, fenoxazinilo, ftalazinilo, piperazinilo, piperidinilo, pteridinilo, purinilo, piranilo, pirazinilo, pirazolidinilo, pirazolinilo, pirazolilo, piridazinilo, piridooxazol, piridoimidazol, piridotiazol, piridinilo, pridilo, pirimidinilo, pirrolidinilo, pirrolinilo, 2H-pirrolilo, pirrolilo, quinazolinilo, quinolinilo, 4H-quinolizinilo, quinoxalinilo, quinuclidinilo, tetrahidrofuranilo, tetrahidroisoquinolinilo, tetrahidroquinolinilo, 6H-1,2,5-tiadiazinilo, 1,2,3-tiadiazolilo, 1,2,4-tiadiazolilo, 1,2,5-tiadiazolilo, 1.3.4- tiadiazolilo, tiantrenilo, tiazolilo, tienilo, tienotiazolilo, tienooxazolilo, tienoimidazolilo, tiofenilo, triazinilo, 1,2,3-triazolilo, 1,2,4-triazolilo, 1,2,5-triazolilo, 1,3,4-triazolilo, oxetanilo, azetidinilo o xantenilo.
En determinados aspectos,

es ciclohexilo, furanilo, dihidrofuranilo, piperidinilo, piridinilo, dihidropiridinilo, piridilo, pirimidinilo, dihidropirimidina, pirrolidinilo u oxetanilo.
En determinados aspectos,

es ciclohexanona, furanona, piperidinona, piridinona, pirimidinona, pirrolidinona, u oxetanilo.
En determinados aspectos, el anillo
se selecciona entre:

En determinados aspectos, el anillo
se selecciona entre:

En determinados aspectos, el anillo X es un arilo C3-10 opcionalmente sustituido. En determinados aspectos, el anillo X es un anillo heteroarilo monocíclico de 5-6 átomos opcionalmente sustituido que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En determinados aspectos, el anillo X es un arilo C3-10 fusionado opcionalmente sustituido. En determinados aspectos, el anillo X es un anillo carbocíclico fusionado de 5-10 átomos saturado o parcialmente insaturado opcionalmente sustituido. En determinados aspectos, el anillo X es un anillo heterocíclico fusionado de 5-10 átomos opcionalmente sustituido que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En determinados aspectos, el anillo X es un anillo heteroarilo
monocíclico fusionado de 5-10 átomos opcionalmente sustituido que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
En determinados aspectos, el anillo X es fenilo, naftilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, adamantilo, ciclooctilo, [3.3.0]biciclooctanilo, [4.3.0]biciclononanilo, [4.4.0]biciclodecanilo, [2.2.2]biciclooctanilo, fluorenilo, indanilo, tetrahidronaftilo, acridinilo, azocinilo, bencimidazolilo, benzofuranilo, benzotiofuranilo, benzotiofenilo, benzoxazolilo, benctiazolilo, benctriazolilo, benctetrazolilo, bencisoxazolilo, bencisotiazolilo, bencimidazolinilo, carbazolilo, NH-carbazolilo, carbolinilo, cromanilo, cromenilo, cinnolinilo, decahidroquinolinilo, 2H,6H-1,5,2-ditiazinilo, dihidrofuro [2,3-b] tetrahidrofurano, furanilo, furazanilo, imidazolidinilo, imidazolinilo, imidazolilo, 1 H-indazolilo, indolenilo, indolinilo, indolizinilo, indolilo, 3H-indolilo, isoindolinilo, isoindolenilo, isobenzofuranilo, isocromanilo, isoindazolilo, isoindolinilo, isoindolilo, isoquinolinilo, isotiazolilo, isoxazolilo, morfolinilo, naftiridinilo, octahidroisoquinolinilo, oxadiazolilo, 1.2.3- oxadiazolilo, 1,2,4-oxadiazolilo; 1,2,5-oxadiazolilo, 1,3,4-oxadiazolilo, oxazolidinilo, oxazolilo, oxazolidinilo, pirimidinilo, fenantridinilo, fenantrolinilo, fenazinilo, fenotiazinilo, fenoxatiinilo, fenoxazinilo, ftalazinilo, piperazinilo, piperidinilo, pteridinilo, purinilo, piranilo, pirazinilo, pirazolidinilo, pirazolinilo, pirazolilo, piridazinilo, piridooxazol, piridoimidazol, piridotiazol, piridinilo, piridilo, pirimidinilo, pirrolidinilo, pirrolinilo, 2H-pirrolilo, pirrolilo, quinazolinilo, quinolinilo, 4H-quinolizinilo, quinoxalinilo, quinuclidinilo, tetrahidrofuranilo, tetrahidroisoquinolinilo, tetrahidroquinolinilo, 6H-1,2,5-tiadiazinilo, 1,2,3-tiadiazolilo, 1,2,4-tiadiazolilo, 1,2,5-tiadiazolilo, 1,3,4-tiadiazolilo, tiantrenilo, tiazolilo, tienilo, tienotiazolilo, tienooxazolilo, tienoimidazolilo, tiofenilo, triazinilo, 1,2,3-triazolilo, 1,2,4-triazolilo, 1,2,5-triazolilo, 1.3.4- triazolilo, oxetanilo, azetidinilo, o xantenilo; cada uno de los cuales está opcionalmente sustituido.
En determinados aspectos, el anillo X es un arilo C3-10 opcionalmente sustituido; un anillo heteroarilo monocíclico de 5-6 átomos opcionalmente sustituido que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; un arilo C3-10 fusionado opcionalmente sustituido; o un anillo heteroarilo monocíclico fusionado de 5-10 átomos opcionalmente sustituido que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
En determinados aspectos, el anillo X es fenilo, bencimidazolilo, benzofuranilo, benzotiofuranilo, benzotiofenilo, benzoxazolilo, benctiazolilo, benctriazolilo, benctetrazolilo, bencisoxazolilo, bencisotiazolilo, bencimidazolinilo, furanilo, furazanilo, imidazolidinilo, imidazolinilo, imidazolilo, 1 H-indazolilo, indolenilo, indolinilo, indolizinilo, indolilo, 3H-indolilo, isoindolinilo, isoindolenilo, isobenzofuranilo, isoindazolilo, isoindolinilo, isoindolilo, isoquinolinilo, isotiazolilo, isoxazolilo, oxadiazolilo, 1,2,3-oxadiazolilo, 1,2,4-oxadiazolilo; 1,2,5-oxadiazolilo, 1,3,4-oxadiazolilo, oxazolidinilo, oxazolilo, oxazolidinilo, pirimidinilo, piranilo, pirazinilo, pirazolidinilo, pirazolinilo, pirazolilo, piridazinilo, piridooxazol, piridoimidazol, piridotiazol, piridinilo, piridilo, pirimidinilo, pirrolidinilo, pirrolinilo, 2H-pirrolilo, pirrolilo, quinazolinilo, quinolinilo, 4H-quinolizinilo, quinoxalinilo, tetrazol, triazinilo, 1,2,3-triazolilo, 1,2,4-triazolilo, 1,2,5-triazolilo o 1,3,4-triazolilo; cada uno de los cuales está opcionalmente sustituido.
En determinados aspectos, el anillo X es fenilo, bencimidazolilo, benzofuranilo, benzotiofuranilo, benzotiofenilo, 1 H-indazolilo, isobenzofuranilo, isoxazolilo, pirazinilo, pirazolilo, piridinilo, piridilo o tetrazol; cada uno de los cuales está opcionalmente sustituido.
En determinados aspectos, el anillo X se selecciona entre:

En determinados aspectos, el anillo X es

En determinados aspectos, el anillo X se selecciona entre:

En determinados aspectos, el anillo X es:

En determinados aspectos, R3 es -R.
En determinados aspectos, R3 es un grupo alifático Ci-6, arilo C3 -10 , un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado, un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico de 5-6 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido.
En determinados aspectos, R3 es un grupo alifático C1-6, un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado o un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido.
En determinados aspectos, R3 es metilo, etilo, propilo, i-propilo, n-butilo, s-butilo, t-butilo, un pentilo de cadena lineal o ramificada, o un hexilo de cadena lineal o ramificada; cada uno de los cuales está opcionalmente sustituido.
En determinados aspectos, R3 se selecciona entre:

En determinados aspectos, R3 es

En determinados aspectos, R3 es
a J x ^ oh
En determinados aspectos, R3 es

En determinados aspectos, R3 es

En determinados aspectos, R3 es

En determinados aspectos, R3 es

En determinados aspectos, R3 es

En determinados aspectos, R3 es

En determinados aspectos, cada uno de anillo X, R, R3, R4 y Rb, es como se define anteriormente y se describe en los aspectos, clases y subclases anteriores y en este documento, individualmente o en combinación.
En determinados aspectos, la presente memoria descriptiva proporciona un compuesto de fórmula I-a,

o una sal farmacéuticamente aceptable del mismo, donde cada uno de anillo X, R3 y Rb, es como se define anteriormente y se describe en los aspectos, clases y subclases anteriores y en este documento, de manera individual o en combinación. En determinados aspectos, la presente memoria descriptiva proporciona un compuesto de fórmula I-b,

o una sal farmacéuticamente aceptable del mismo, donde cada uno de R3 y Rb, es como se define anteriormente y se describe en los aspectos, clases y subclases anteriores y en este documento, de manera individual o en combinación. En determinados aspectos, la presente memoria descriptiva proporciona un compuesto de fórmula I-c,

I-c;
o una sal farmacéuticamente aceptable del mismo, donde cada uno de anillo X y R3, es como se define anteriormente y se describe en los aspectos, clases y subclases anteriores y en este documento, de manera individual o en combinación. En determinados aspectos, la invención proporciona un compuesto seleccionado a partir de la tabla 1:
Tabla 1

Ċ





En algunos aspectos, la presente memoria descriptiva proporciona un compuesto seleccionado entre aquellos descritos anteriormente, o una sal farmacéuticamente aceptable del mismo.
Varias representaciones estructurales pueden mostrar un heteroátomo sin un grupo, radical, carga o contraión unidos. Los expertos en la materia son conscientes de que dichas representaciones pretenden indicar que el heteroátomo está \ / 0
unido al hidrógeno (p. ej., ^ se entiende que es
En determinadas realizaciones, los compuestos de la invención se sintetizaron según los esquemas que se proporcionan en los ejemplos que aparece a continuación.
4. Usos, formulación y administración
Composiciones farmacéuticamente aceptables
Según otra realización, la invención proporciona una composición que comprende un compuesto de esta invención o un derivado farmacéuticamente aceptable del mismo y un transportador, adyuvante o vehículo farmacéuticamente aceptable. La cantidad de compuesto en las composiciones de esta invención es aquella que resulta eficaz para inhibir de forma medible la actividad IRAK, o una forma mutada de la misma, en una muestra biológica o en un paciente. En determinadas realizaciones, la cantidad de compuesto en las composiciones de esta invención es aquella que resulta eficaz para inhibir de forma medible la actividad IrA k , o una forma mutada de la misma, en una muestra biológica o en un paciente. En determinadas realizaciones, una composición de esta invención se formula para la administración a un paciente que necesita dicha composición.
El término «paciente» o «sujeto», según se usa en este documento, significa un animal, preferiblemente un mamífero y más preferiblemente un ser humano.
El término «transportador, adyuvante o vehículo farmacéuticamente aceptable» se refiere a un transportador, adyuvante o vehículo no tóxico que no destruye la actividad farmacológica del compuesto en el que está formulado. Los transportadores, adyuvantes o vehículos farmacéuticamente aceptables que se utilizan en las composiciones de esta invención incluyen, pero sin limitaciones, intercambiadores de iones, alúmina, estearato de aluminio, lecitina, proteínas séricas, como albúmina sérica humana, sustancias tampón como fosfatos, glicina, ácido sórbico, sorbato de potasio, mezclas parciales de glicéridos de ácidos grasos vegetales saturados, agua, sales o electrolitos, como sulfato de protamina, hidrogenofosfato disódico, hidrogenofosfato de potasio, cloruro sódico, sales de cinc, sílice coloidal, trisilicato de magnesio, polivinilpirrolidona, sustancias a base de celulosa, polietilenglicol, carboximetilcelulosa sódica, poliacrilatos, ceras, polímeros de bloque de polietileno-polioxipropileno, polietilenglicol y lanolina.
Un «derivado farmacéuticamente aceptable» significa cualquier sal, éster, sal de un éster u otro derivado no tóxico de un compuesto de esta invención que, tras la administración a un receptor, es capaz de proporcionar, directa o indirectamente, un compuesto de esta invención o un metabolito o resto inhibitoriamente activo del mismo.
Las composiciones de la presente invención se administran por vía oral, parenteral, mediante inhalación con un pulverizador, por vía tópica, rectal, nasal, bucal, vaginal o mediante un reservorio implantado. El término «parenteral» según se usa en este documento incluye técnicas de infusión o inyección subcutánea, intravenosa, intramuscular, intraarticular, intrasinovial, intraesternal, intratecal, intrahepática, intralesión e intracraneal. Preferiblemente, las composiciones se administran por vía oral, intraperitoneal o intravenosa. Las formas inyectables estériles de las composiciones de esta invención incluyen suspensiones acuosas u oleaginosas. Estas suspensiones se formulan según técnicas conocidas en la materia usando agentes dispersantes o humectantes y agentes de suspensión adecuados. La preparación estéril inyectable también puede ser una solución o suspensión inyectable estéril en un diluyente o solvente no tóxico aceptable por vía parenteral, por ejemplo, como una solución en 1,3-butanediol. Entre los vehículos y solventes aceptables que se emplean se encuentran agua, solución de Ringer y solución isotónica de cloruro sódico. Además, convencionalmente se emplean como solvente o medio de suspensión aceites fijados estériles.
Con este objetivo, cualquier aceite fijado insípido incluye mono o diglicéridos sintéticos. Los ácidos grasos, como ácido oleico y sus derivados glicéridos, son útiles para la preparación de inyectables, así como los aceites naturales farmacéuticamente aceptables, como aceite de oliva o aceite de ricino, especialmente en sus versiones polioxietiladas. Estas soluciones o suspensiones oleaginosas también contienen un diluyente o dispersante alcohol de cadena larga, como carboximetilcelulosa o agentes dispersantes similares que normalmente se utilizan en la formulación de formas farmacéuticas farmacéuticamente aceptables incluidas emulsiones y suspensiones. También pueden utilizarse a los fines de formulación otros tensioactivos utilizados con frecuencia, como Tweens, Spans y otros agentes emulsionantes o potenciadores de la biodisponibilidad que normalmente se utilizan en la fabricación de formas farmacéuticas sólidas, líquidas o de otro tipo farmacéuticamente aceptables.
Las composiciones farmacéuticamente aceptables de esta invención se administran por vía oral en cualquier forma farmacéutica aceptable por vía oral. Son ejemplos de formas farmacéuticas orales cápsulas, comprimidos, suspensiones o soluciones acuosas. En el caso de comprimidos para uso oral, entre los vehículos utilizados normalmente se incluyen lactosa y almidón de maíz. También se añaden típicamente agentes lubricantes como estearato de magnesio. Para la administración oral en forma de cápsula, son diluyentes útiles la lactosa y el almidón de maíz seco. Cuando se requieren suspensiones acuosas para uso oral, el principio activo se combina con agentes emulsionantes y de suspensión. Si se desea, también se añaden opcionalmente determinados agentes edulcorantes, aromatizantes o colorantes.
Alternativamente, las composiciones farmacéuticamente aceptables de esta invención se administran en forma de supositorios para administración rectal. Estos pueden preparase mezclando el agente con un excipiente adecuado no irritante que sea sólido a temperatura ambiente pero líquido a temperatura rectal y, por tanto, se funda en el recto para que se libere el fármaco. Entre estos materiales se incluyen manteca de cacao, cera de abeja y polietilenglicoles.
Las composiciones farmacéuticamente aceptables de esta invención también pueden administrarse por vía tópica, especialmente cuando el objetivo de tratamiento son zonas u órganos fácilmente accesibles mediante aplicación tópica, como enfermedades oculares, cutáneas o del aparato digestivo inferior. Se preparan fácilmente formulaciones tópicas adecuadas para cada una de estas zonas u órganos.
La aplicación tópica para el aparato digestivo inferior puede realizarse en una formulación de supositorio rectal (véase anteriormente) o en una formulación de enema adecuada. También se utilizan parches transdérmicos de aplicación tópica.
Para aplicaciones tópicas, las composiciones farmacéuticamente aceptables se formulan en una pomada adecuada que contiene el componente activo suspendido o disuelto en uno o más vehículos. Son ejemplos de vehículos para la administración tópica de los compuestos de esta: aceite mineral, vaselina líquida, vaselina filante, propilenglicol, polioxietileno, compuesto polioxipropileno, cera emulsionante y agua. Alternativamente, las composiciones farmacéuticamente aceptables proporcionadas pueden formularse en una loción o crema adecuada que contenga los componentes activos resuspendidos o disueltos en uno o más vehículos farmacéuticamente aceptables. Entre los
vehículos adecuados se incluyen, pero sin limitaciones, aceite mineral, monoestearato de sorbitán, polisorbato 60, cera de ésteres cetílicos, alcohol cetearílico, 2-octildodecanol, alcohol bencílico y agua.
Las composiciones farmacéuticamente aceptables de esta invención se administran opcionalmente mediante aerosol nasal o inhalación. Estas composiciones se preparan según técnicas de formulación farmacéutica bien conocidas en la técnica y se preparan como soluciones en solución salina, empleando alcohol bencílico u otros conservantes adecuados, promotores de absorción para aumentar la biodisponibilidad, fluorocarbonos y/u otros agentes solubilizantes o dispersantes convencionales.
Más preferiblemente, las composiciones farmacéuticamente aceptables de esta invención se formulan para administración oral. Estas formulaciones pueden administrase con o sin alimentos. En algunas realizaciones, las composiciones farmacéuticamente aceptables de esta invención se administran sin alimentos. En otras realizaciones, las composiciones farmacéuticamente aceptables de esta invención se administran con alimentos.
La cantidad de compuestos de la presente invención que se combina opcionalmente con los materiales vehículo para producir una composición en una forma farmacéutica única variará dependiendo del huésped tratado y del modo particular de administración. Preferiblemente, las composiciones proporcionadas deben formularse de modo que pueda administrarse a un paciente que recibe estas composiciones una dosis de entre 0,01 y 100 mg/kg de peso corporal/día del compuesto.
También debe entenderse que una dosificación y pauta de tratamiento específicas para cualquier paciente en particular dependerá de diversos factores, como la actividad del compuesto específico empleado, la edad, el peso corporal, la salud general, el sexo, la dieta, el tiempo de administración, la tasa de excreción, la combinación de fármacos y el criterio del médico responsable del tratamiento, así como la gravedad de la enfermedad en concreto que se esté tratando. La cantidad de un compuesto de la presente invención en la composición también dependerá del compuesto en particular en la composición.
Usos de compuestos y composiciones farmacéuticamente aceptables
La presente invención además se refiere a un método para tratar a un sujeto que padece un trastorno relacionado con IRAK, que comprende la administración a dicho sujeto de una cantidad eficaz de un compuesto de fórmula I y fórmulas relacionadas.
La presente invención preferiblemente se refiere a un método en el que el trastorno asociado con IRAK es un trastorno o afección autoinmune asociado con una respuesta inmunitaria hiperactiva o cáncer. La presente invención además se refiere a un método para tratar a un sujeto que padece una anomalía inmunorreguladora, que comprende la administración a dicho sujeto de un compuesto de fórmula (I), y formas relacionadas en una cantidad que es eficaz para tratar dicha anomalía inmunorreguladora.
La presente invención se refiere preferiblemente a un método en el que la anomalía inmunorreguladora es una enfermedad autoinmune o inflamatoria crónica seleccionada entre el grupo compuesto por: enfermedades alérgicas, esclerosis lateral amiotrófica (ELA), lupus eritematoso sistémico, artritis reumatoide crónica, diabetes mellitus de tipo I, enfermedad inflamatoria intestinal, cirrosis biliar, uveítis, esclerosis múltiple, enfermedad de Crohn, colitis ulcerosa, penfigoide ampolloso, sarcoidosis, psoriasis, miositis autoinmune, granulomatosis de Wegener, ictiosis, oftalmopatía de Graves y asma.
La presente invención además se refiere a un método en el que la anomalía inmunorreguladora es un rechazo de trasplante de médula ósea o de órgano, o una enfermedad de injerto contra huésped.
La presente invención además se refiere a un método en el que la anomalía inmunorreguladora se selecciona entre el grupo compuesto por: trasplante de órganos o tejidos, enfermedades de injerto contra huésped causadas por trasplante, síndromes autoinmunes como artritis reumatoide, lupus eritematoso sistémico, tiroiditis de Hashimoto, esclerosis múltiple, esclerosis sistémica, miastenia gravis, diabetes de tipo I, uveítis, uveítis posterior, encefalomielitis alérgica, glomerulonefritis, enfermedades autoinmunes posinfecciosas como fiebre reumática y glomerulonefritis posinfecciosa, enfermedades cutáneas inflamatorias e hiperproliferativas, psoriasis, dermatitis atópica, dermatitis de contacto, dermatitis eccematosa, dermatitis seborreica, liquen plano, pénfigo, penfigoide ampolloso, epidermólisis ampollosa, urticaria, angioedemas, vasculitis, eritema, eosinofilia cutánea, lupus eritematoso, acné, alopecia areata, queratoconjuntivitis, oftalmía primaveral, uveítis asociada a la enfermedad de Behcet, queratitis, queratitis herpética, queratocono, distrofia epitelial de la córnea, leucoma corneal, pénfigo ocular, úlcera de Mooren, escleritis, oftalmopatía de Graves, síndrome de Vogt-Koyanagi-Harada, sarcoidosis, alergias al polen, enfermedad obstructiva de las vías respiratorias reversible, asma bronquial, asma alérgica, asma intrínseca, asma extrínseca, asma por polvo, asma crónica o inveterada, asma tardía e hipersensibilidad de las vías respiratorias, bronquitis, úlceras gástricas, daño vascular causado por enfermedades isquémicas y trombosis, enfermedades intestinales isquémicas, enfermedades inflamatorias intestinales, enterocolitis necrosante, lesiones intestinales asociadas a quemaduras térmicas, enfermedades celíacas, proctitis, gastroenteritis eosinofílica, mastocitosis, enfermedad de Crohn, colitis ulcerosa, migrañas, rinitis, eccema, nefritis intersticial, síndrome de Goodpasture, síndrome urémico hemolítico, nefropatía diabética, miositis múltiple, síndrome de Guillain-Barre, enfermedad de Meniere, polineuritis, neuritis múltiple, mononeuritis, radiculopatía, hipertiroidismo, enfermedad de Basedow, aplasia eritrocitaria pura, anemia aplásica, anemia hipoplásica, púrpura trombocitopénica idiopática, anemia
hemolítica autoinmune, agranulocitosis, anemia perniciosa, anemia megaloblástica, aneritroplasia, osteoporosis, sarcoidosis, fibrosis pulmonar, neumonía intersticial idiopática, dermatomiositis, leucodermia vulgar, ictiosis vulgar, sensibilidad fotoalérgica, linfoma de células T cutáneo, leucemia linfocítica crónica, arteriesclerosis, ateroesclerosis, síndrome de aortitis, poliarteritis nodosa, miocardosis, escleroderma, granuloma de Wegener, síndrome de Sjogren, adiposis, fascitis eosinofílica, lesiones de la encía, periodontio, hueso alveolar y sustancia ósea dentaria, glomerulonefritis, alopecia de patrón masculino o alopecia senil previniendo la depilación o proporcionando germinación de pelo y/o favoreciendo la generación y el crecimiento del pelo, distrofia muscular, pioderma y síndrome de Sezary, enfermedad de Addison, lesión de órganos por isquemia-reperfusión que se produce tras su conservación, trasplante o enfermedad isquémica, choque endotóxico, colitis pseudomembranosa, colitis causada por fármaco o radiación, insuficiencia renal aguda isquémica, insuficiencia renal crónica, toxinosis causada por el oxígeno pulmonar o fármacos, cáncer de pulmón, enfisema pulmonar, catarata, siderosis, retinitis pigmentosa, degeneración macular senil, cicatrización del vítreo, quemadura de la córnea por álcalis, dermatitis, eritema multiforme, dermatitis ampollosa por IgA lineal y dermatitis por cemento, gingivitis, periodontitis, septicemia, pancreatitis, enfermedades causadas por la contaminación ambiental, envejecimiento, carcinogénesis, metástasis de carcinoma e hipobaropatía, enfermedad causada por la liberación de histamina o leucotrieno-C4, enfermedad de Behcet, hepatitis autoinmune, cirrosis biliar primaria, colangitis esclerosante, resección hepática parcial, necrosis hepática aguda, necrosis causada por toxina, hepatitis vírica, shock, anorexia, hepatitis B, hepatitis no A/no B, cirrosis, cirrosis alcohólica, insuficiencia hepática, insuficiencia hepática fulminante, insuficiencia hepática de aparición tardía, insuficiencia hepática crónica reagudizada, aumento del efecto quimioterapéutico, infección por citomegalovirus, infección por HCMV, SIDA, cáncer, demencia senil, enfermedades parkinsonianas, traumatismo e infección bacteriana crónica.
En determinadas realizaciones, las enfermedades asociadas con IRAK se seleccionan entre artritis reumatoide, artritis psoriásica, artrosis, lupus eritematoso sistémico, nefritis lúpica, espondilitis anquilosante, osteoporosis, esclerosis sistémica, esclerosis múltiple, psoriasis, diabetes de tipo I, diabetes de tipo II, enfermedad inflamatoria intestinal (enfermedad de Crohn y colitis ulcerosa), hiperinmunoglobulinemia D y síndrome de fiebre periódica, síndromes periódicos asociados con criopirina, síndrome de Schnitzler, artritis idiopática juvenil sistémica, enfermedad de Still del adulto, gota, pseudogota, síndrome SAPHO, enfermedad de Castleman, septicemia, ictus, ateroesclerosis, enfermedad celíaca, DIRA (deficiencia del antagonista del receptor de IL-1), enfermedad de Alzheimer, enfermedad de Parkinson y cáncer.
En determinadas realizaciones, el cáncer se selecciona entre carcinoma, linfoma, blastoma (incluidos meduloblastoma y retinoblastoma), sarcoma (incluidos liposarcoma y sarcoma sinovial), tumores neuroendocrinos (incluidos tumores carcinoides, gastrinoma y cáncer de células de los islotes), mesotelioma, schwannoma (incluido neuroma acústico), meningioma, adenocarcinoma, melanoma y leucemia o neoplasias malignas linfoides. Ejemplos más en particular de estos cánceres son carcinoma de células escamosas (p. ej., carcinoma espinocelular), cáncer de pulmón incluido el cáncer de pulmón microcítico (CPM), cáncer de pulmón no microcítico (CPNM), adenocarcinoma de pulmón y carcinoma epidermoide de pulmón, cáncer del peritoneo, cáncer hepatocelular, cáncer gástrico o de estómago incluido cáncer gastrointestinal, cáncer pancreático, gliobastoma, cáncer de cuello uterino, cáncer de ovario, cáncer hepático, cáncer de vejiga, hepatoma, cáncer de mama (incluido cáncer de mama metastásico), cáncer de colon, cáncer rectal, cáncer colorrectal, carcinoma endometrial o uterino, carcinoma de las glándulas salivares, cáncer de riñón o renal, cáncer de próstata, cáncer de vulva, cáncer de tiroides, carcinoma hepático, carcinoma anal, carcinoma de pene, cáncer de testículo, cáncer de esófago, tumores del conducto biliar, así como cáncer de cabeza y cuello.
En determinadas realizaciones, el cáncer es cáncer de cerebro, pulmón, colon, epidermoide, de células escamosas, de vejiga, gástrico, pancreático, de mama, cabeza, cuello, renal, de riñón, hígado, ovario, próstata, colorrectal, uterino, rectal, esófago, testicular, ginecológico, de tiroides, melanoma, neoplasias malignas hematológicas como leucemia mielógena aguda, mieloma múltiple, leucemia mielógena crónica, leucemia de células mieloides, glioma, sarcoma de Kaposi o cualquier otro tipo de tumores sólidos o líquidos. En algunas realizaciones, el cáncer es cáncer metastásico. En algunas realizaciones, el cáncer es cáncer colorrectal. En algunas realizaciones, el cáncer es cáncer de colon.
En diversas realizaciones, los compuestos de fórmula (I) y fórmulas relacionadas muestran una IC50 para la unión a IRAK de menos de aproximadamente 5 |j M, preferiblemente menos de aproximadamente 1 |j M e incluso más preferiblemente menos de aproximadamente 0,100 j M.
El método de la invención puede realizarse in vitro o in vivo. La susceptibilidad de una célula en particular al tratamiento con los compuestos según la invención puede determinarse especialmente mediante pruebas in vitro, ya sea en el curso de una investigación o en la aplicación clínica. Normalmente, se combina un cultivo de la célula con un compuesto según la invención a diversas concentraciones durante un periodo de tiempo que es suficiente como para permitir que los principios activos inhiban la actividad IRAK, generalmente entre aproximadamente una hora y una semana. El tratamiento in vitro se puede realizar usando células en cultivo procedentes de una muestra de biopsia o de una línea celular.
El huésped o paciente puede pertenecer a cualquier especie de mamífero, por ejemplo, una especie de primate, especialmente humanos; roedores, como ratones, ratas y hámsteres; conejos; caballos, vacas, perros, gatos, etc. Los modelos de animales son interesantes para las investigaciones experimentales, proporcionando un modelo para el tratamiento de una enfermedad humana.
Para la identificación de una vía de transducción de señales y para la detección de interacciones entre diversas vías de transducción de señales, varios científicos han desarrollado modelos o sistemas de modelos idóneos, por ejemplo
modelos de cultivo celular y modelos de animales transgénicos. Para la determinación de ciertas etapas de la cascada de transdu cción de señales pueden utilizarse compuestos que interaccionan para modular la señal. Los compuestos según la invención también pueden ser útiles como reactivos para el análisis de vías de transducción de señales dependientes de IRAK en modelos animales y/o de cultivo celular, o en las enfermedades mencionadas en esta solicitud.
Asimismo, las consecuentes explicaciones de la presente memoria descriptiva referentes al uso de los compuestos según la fórmula (I) y sus derivados para la producción de un medicamento para el control y/o el tratamiento profiláctico o terapéutico se consideran válidas y aplicables sin restricciones al uso del compuesto para la inhibición de la actividad IRAK si es conveniente.
La invención también se refiere al uso de los compuestos según la fórmula (I) y/o las sales fisiológicamente aceptables de los mismos para el control y/o tratamiento profiláctico o terapéutico de enfermedades que están causadas, mediadas y/o se propagan por la actividad IRAK. Adicionalmente, la invención se refiere al uso de los compuestos según la fórmula (I) y/o las sales fisiológicamente aceptables de los mismos para la producción de un medicamento para el control y/o tratamiento profiláctico o terapéutico de enfermedades que están causadas, mediadas y/o se propagan por la actividad IRAK. En determinadas realizaciones, la invención proporciona el uso de un compuesto según la fórmula I o las sales fisiológicamente aceptables de los mismos, para la producción de un medicamento para el tratamiento profiláctico o terapéutico de un trastorno mediado por IRAK.
Pueden además emplearse compuestos de fórmula (I) y/o una sal fisiológicamente aceptable de los mismos como compuestos intermedios para la preparación de principios activos de medicamentos adicionales. El medicamento se prepara preferiblemente de forma no química, por ejemplo, combinando el principio activo con al menos un vehículo o excipiente sólido, fluido y/o semifluido y, opcionalmente, junto con uno o más principios activos adicionales en una forma farmacéutica apropiada.
Los compuestos de fórmula (I) según la invención se pueden administrar antes o después de la aparición de una enfermedad una o varias veces actuando como terapia. Los compuestos y medicamentos de la invención mencionados anteriormente se usan especialmente para el tratamiento terapéutico. Un efecto terapéuticamente relevante alivia en cierto grado uno o más síntomas de un trastorno, o recupera la normalidad parcial o completamente de uno o más parámetros fisiológicos o bioquímicos asociados o causantes de una enfermedad o patología. El control se considera una clase de tratamiento siempre que los compuestos se administren en distintos intervalos, por ejemplo, para reforzar la respuesta y erradicar completamente los patógenos y/o síntomas de la enfermedad. Se puede aplicar el compuesto idéntico o compuestos diferentes. Los métodos de la invención también se pueden usar para reducir la probabilidad de desarrollar un trastorno o incluso prevenir el inicio de enfermedades asociadas con la actividad IRAK por anticipado o para tratar los síntomas iniciales y continuos.
En el significado de la invención, el tratamiento profiláctico es aconsejable si el sujeto tiene alguna condición previa para las afecciones fisiológicas o patológicas mencionadas anteriormente, como una predisposición familiar, un defecto genético o una enfermedad previamente contraída.
La invención además se refiere a un medicamento que comprende al menos un compuesto según la invención, y/o derivados, sales, solvatos y estereoisómeros del mismo farmacéuticamente útiles, incluyendo sus mezclas en todas las proporciones. En determinados realizaciones, la invención se refiere a un medicamento que comprende al menos un compuesto según la invención y/o las sales fisiológicamente aceptables del mismo.
Un «medicamento» en el significado de la invención es cualquier principio en el campo de la medicina, que comprende uno o más compuestos de fórmula (I) o preparaciones de los mismos (p. ej., una composición farmacéutica o formulación farmacéutica) que se pueden usar en la profilaxis, terapia, seguimiento o tratamiento posoperatorio de pacientes que sufren enfermedades, las cuales están asociadas con la actividad IRAK, de tal forma que se pueda establecer, al menos temporalmente, una modificación patogénica de su afección general o de la afección de regiones en particular.
En diversas realizaciones, el principio activo puede administrarse solo o en combinación con otros tratamientos. Se puede lograr un efecto sinérgico mediante el uso de más de un compuesto en la composición farmacéutica, es decir, el compuesto de fórmula (I) se combina con al menos otro fármaco como principio activo, que es otro compuesto de fórmula (I) o un compuesto con un esqueleto estructural diferente. Los principios activos se pueden usar simultánea o secuencialmente.
En este documento se incluyen métodos de tratamiento en los que al menos se administra una entidad química proporcionada en este documento en combinación con un antiinflamatorio. Entre los antiinflamatorios se incluyen, pero sin limitaciones, los AINE, inhibidores de la enzima ciclooxigenasa inespecíficos y específicos de COX-2, compuestos de oro, corticoesteroides, metotrexato, antagonistas del factor de necrosis tumoral (TNF), inmunodepresores y metotrexato.
Entre los ejemplos de AINE se incluyen, pero sin limitaciones, ibuprofeno, flurbiprofeno, naproxeno y naproxeno sódico, diclofenaco, combinaciones de diclofenaco sódico y misoprostol, sulindaco, oxaprozina, diflunisal, piroxicam, indometacina, etodolaco, fenoprofeno de calcio, ketoprofeno, nabumetona sódica, sulfasalazina, tolmetina sódica e hidroxicloroquina. Los ejemplos de AINE también incluyen inhibidores específicos de COX-2 como celecoxib, valdecoxib, lumiracoxib y/o etoricoxib.
En algu nas realizaciones, el antiinflamatorio es un salicilato. Entre los salicilatos se incluyen, pero sin limitaciones, ácido acetilsa licílico o aspirina, salicilato sódico y salicilatos de colina y magnesio.
El antiinflamatorio también puede ser un corticoesteroide. Por ejemplo, el corticoesteroide puede ser cortisona, dexametasona, metilprednisolona, prednisolona, fosfato sódico de prednisolona o prednisona.
En realizaciones adicionales el antiinflamatorio es un compuesto de oro como triomalato sódico de oro o auranofina.
La invención también incluye realizaciones en las que el antiinflamatorio es un inhibidor metabólico como un inhibidor de la dihidrofolato reductasa, como metotrexato, o un inhibidor de la dihidroorotato deshidrogenasa, como leflunomida.
Otras realizaciones de la invención se relacionan con combinaciones en las que al menos un compuesto antiinflamatorio es un anticuerpo monoclonal (como eculizumab o pexelizumab), un agonista de TNF, como entanercept, o infliximab, que es un anticuerpo monoclonal anti-TNF alfa.
Aún otras realizaciones de la invención se refieren a combinaciones en las que al menos un principio activo es un compuesto inmunodepresor como un compuesto inmunodepresor elegido entre metotrexato, leflunomida, ciclosporina, tacrolimús, azatioprina y micofenolato de mofetilo.
Los compuestos descritos de fórmula I se pueden administrar en combinación con otros agentes terapéuticos conocidos, incluidos agentes antineoplásicos. Como se usa aquí, el término «agente antineoplásico» se refiere a cualquier agente que se administre a un paciente con cáncer para los fines de tratamiento del cáncer.
El tratamiento antineoplásico definido anteriormente se puede aplicar como monoterapia o puede implicar, además de los compuestos de fórmula I descritos en este documento, cirugía convencional o radioterapia o tratamiento farmacológico. Dicho tratamiento farmacológico, por ejemplo, una quimioterapia o una terapia dirigida, puede incluir uno o más, pero preferiblemente uno, de los siguientes agentes antitumorales:
Agentes alquilantes: como altretamina, bendamustina, busulfán, carmustina, clorambucilo, clormetina, ciclofosfamida, dacarbazina, ifosfamida, improsulfán, tosilato, lomustina, melfalán, mitobronitol, mitolactol, nimustina, ranimustina, temozolomida, tiotepa, treosulfán, mecloretamina, carboquona; apazicuona, fotemustina, glufosfamida, palifosfamida, pipobromán, trofosfamida, uramustina, TH-3024, VAL-0834;
Compuestos de platino: como carboplatino, cisplatino, eptaplatino, hidrato de miriplatino, oxaliplatino, lobaplatino, nedaplatino, picoplatino, satraplatino; lobaplatino, nedaplatino, picoplatino, satraplatino;
Agentes que alteran el ADN: como amrubicina, bisantreno, decitabina, mitoxantrona, procarbazina, trabectedina, clofarabina; amsacrina, brostalicina, pixantrona, laromustina13;
Inhibidores de la topoisomerasa: como etopósido, irinotecán, razoxano, sobuzoxano, tenipósido, topotecán; amonafida, belotecán, acetato de eliptinio, voreloxina;
Modificadores de microtúbulos: como cabazitaxel, docetaxel, eribulina, ixabepilona, paclitaxel, vinblastina, vincristina, vinorelbina, vindesina, vinflunina; fosbretabulina, tesetaxel;
Antimetabolitos: como asparaginasa3, azacitidina, levofolinato de calcio, capecitabina, cladribina, citarabina, enocitabina, floxuridina, fludarabina, fluorouracilo, gemcitabina, mercaptopurina, metotrexato, nelarabina, pemetrexed, pralatrexato, azatioprina, tioguanina, carmofur, doxifluridina, elacitarabina, raltitrexed, sapacitabina, tegafur23, trimetrexato;
Antibióticos antineoplásicos: como bleomicina, dactinomicina, doxorubicina, epirubicina, idarubicina, levamisol, miltefosina, mitomicina C, romidepsina, estreptozocina, valrubicina, zinostatina, zorubicina, daunorubicina, plicamicina, aclarubicina, peplomicina, pirarubicina;
Hormonas/antagonistas: como abarelix, abiraterona, bicalutamida, buserelina, calusterona, clorotrianiseno, degarelix, dexametasona, estradiol, fluocortolona, fluoximesterona, flutamida, fulvestrant, goserelina, histrelina, leuprorelina, megestrol, mitotano, nafarelina, nandrolona, nilutamida, octreotida, prednisolona, raloxifeno, tamoxifeno, tirotropina alfa, toremifeno, trilostano, triptorelina, dietilestilbestrol, acolbifeno, danazol, deslorelina, epitiostanol, orteronel, enzalutamida13;
Inhibidores de la aromatasa: como aminoglutetimida, anastrozol, exemestano, fadrozol, letrozol, testolactona, formestano;
Moléculas pequeñas inhibidoras de quinasas: como crizotinib, dasatinib, erlotinib, imatinib, lapatinib, nilotinib, pazopanib, regorafenib, ruxolitinib, sorafenib, sunitinib, vandetanib, vemurafenib, bosutinib, gefitinib, axitinib, afatinib, alisertib, dabrafenib, dacomitinib, dinaciclib, dovitinib, enzastaurina, nintedanib, lenvatinib, linifanib, linsitinib, masitinib, midostaurina, motesanib, neratinib, orantinib, perifosina, ponatinib, radotinib, rigosertib, tipifarnib, tivantinib, tivozanib, trametinib, pimasertib, alaninato de brivanib, cediranib, apatinib4, S-malato de cabozantinib13 ibrutinib13 icotinib4, buparlisib2, cipatinib4, cobimetinib13, idelalisib13, fedratinib1, XL-6474;
Fotosensibilizadores: como metoxaleno3, porfímero de sodio, talaporfina, temoporfina;
Anticue rpos: como alemtuzumab, besilesomab, brentuximab vedotina, cetuximab, denosumab, ipilimumab, ofatumumab, panitumumab, rituximab, tositumomab; trastuzumab, bevacizumab, pertuzumab23, catumaxomab, elotuzumab, epratuzumab, farletuzumab, mogamulizumab, necitumumab, nimotuzumab, obinutuzumab, ocaratuzumab, oregovomab, ramucirumab, rilotumumab, siltuximab, tocilizumab, zalutumumab, zanolimumab, matuzumab, dalotuzumab123, onartuzumab13 racotumomab1, tabalumab13, EMD-5257974, nivolumab13;
Citoquinas: como aldesleuquina, interferón alfa2, interferón alfa2a3, interferón alfa2b23, celmoleuquina, tasonermina, teceleuquina, oprelvequina13 interferón beta-1a recombinante4;
Fármacos conjugados: como denileuquina diftitox, ibritumomab tiuxetán, iobenguano I123, prednimustina, trastuzumab emtansina, estramustina, gemtuzumab, ozogamicina, aflibercept, cintredequina besudotox, edotreotida, inotuzumab ozogamicina, naptumomab estafenatox, oportuzumab monatox, tecnecio (99mTc) arcitumomab13, vintafolida13;
Vacunas: como sipuleucel3, vitespen3, emepepimut-S3, oncoVAX4, rindopepimut3, troVax4, MGN-16014, MGN-17034; y
Varios: alitretinoína, bexaroteno, bortezomib, everolimús, ácido ibandrónico, imiquimod, lenalidomida, lentinano, metirosina, mifamurtida, ácido pamidrónico, pegaspargasa, pentostatina, sipuleucel3, sizofirano, tamibaroteno, temsirolimús, talidomida, tretinoína, vismodegib, ácido zoledrónico, vorinostat; celecoxib, cilengitida, entinostat, etanidazol, ganetespib, idronoxilo, iniparib, ixazomib, lonidamina, nimorazol, panobinostat, peretinoína, plitidepsina, pomalidomida, procodazol, ridaforolimús, tasquinimod, telotristat, timalfasina, tirapazamina, tosedostat, trabedersen, ubenimex, valspodar, gendicina4, picibanil4, reolisina4, clorhidrato de retaspimicina13, trebananib23, virulizina4, carfilzomib13, endostatina4, immucothel4, belinostat3, MGN-17034.
(1 DCI prop. [denominación común internacional propuesta]; 2 DCI rec. [denominación común internacional recomendada]; 3 USAN [nombre adoptado en Estados Unidos]; 4 no DCI).
En otro aspecto, la invención proporciona un kit compuesto por envases independientes de una cantidad eficaz de un compuesto según la invención y/o sales, derivados, solvatos y estereoisómeros del mismo farmacéuticamente aceptables, incluidas sus mezclas en todas las proporciones, y opcionalmente, una cantidad eficaz de un principio activo adicional. El kit comprende recipientes adecuados, como cajas, frascos individuales, bolsas o ampollas. El kit puede comprender, por ejemplo, ampollas independientes, cada una con una cantidad eficaz de un compuesto según la invención y/o sales, derivados, solvatos y estereoisómeros del mismo farmacéuticamente aceptables, incluidas sus mezclas en todas las proporciones y una cantidad eficaz de un principio activo adicional en forma disuelta o liofilizada.
Según se usa en este documento, los términos «tratamiento», «tratar» y «tratando» se refieren a revertir, aliviar, retrasar la aparición o inhibir el progreso de una enfermedad o trastorno, o uno o más síntomas del mismo, como se describe en este documento. En algunas realizaciones, el tratamiento se administra tras el desarrollo de uno o más síntomas. En otras realizaciones, el tratamiento se administra en ausencia de síntomas. Por ejemplo, el tratamiento se administra a un individuo susceptible antes de la aparición de los síntomas (p. ej., a tenor de antecedentes de síntomas y/o a tenor de factores genéticos u otros factores de susceptibilidad). El tratamiento también se continúa tras la resolución de los síntomas, por ejemplo para prevenir o retrasar su reaparición.
Los compuestos y composiciones, según el método de la presente invención, se administran usando cualquier cantidad y vía de administración eficaz para tratar o reducir la gravedad de un trastorno mencionado anteriormente. La cantidad exacta necesaria variará de un sujeto a otro, dependiendo de la especie, edad y estado general del sujeto, la gravedad de la infección, el fármaco en particular utilizado, su vía de administración y similares. Los compuestos de la invención se formulan preferiblemente en forma de unidades de dosis para una fácil administración y uniformidad de dosis. La expresión «forma de unidad de dosis» según se usa en este documento se refiere a una unidad físicamente discreta de un fármaco apropiado para el paciente que se va a tratar. No obstante, se entenderá que el médico responsable del tratamiento decidirá sobre el uso diario total de los compuestos y composiciones de la presente invención dentro del alcance del buen criterio médico. El nivel de dosis eficaz específico para cualquier paciente u organismo en particular dependerá de diversos factores que incluyen el trastorno que se está tratando y la gravedad del trastorno; la actividad del compuesto específico empleado; la composición específica empleada; la edad, peso corporal, estado de salud general, sexo y dieta del paciente; el tiempo de administración, la vía de administración y la velocidad de excreción del compuesto específico empleado; la duración del tratamiento; fármacos utilizados en combinación o coincidencia con el compuesto específico empleado, y factores similares bien conocidos en la técnica médica.
Las composiciones farmacéuticamente aceptables de esta invención pueden administrarse a humanos y a otros animales por vía oral, rectal, parenteral, intracisternal, intravaginal, intraperitoneal, tópica (mediante polvos, pomadas o gotas), bucal, como un spray bucal o nasal, o similar, dependiendo de la gravedad de la infección que se esté tratando. En determinadas realizaciones, los compuestos de la invención se administran por vía oral o parenteral a niveles de dosis de aproximadamente 0,01 mg/kg a aproximadamente 100 mg/kg y preferiblemente de aproximadamente 1 mg/kg a aproximadamente 50 mg/kg de peso corporal del sujeto al día, una o más veces al día, para obtener el efecto terapéutico deseado.
En determinadas realizaciones, la cantidad terapéuticamente eficaz de un compuesto de fórmula (I) y fórmulas relacionadas y del otro principio activo depende de varios factores, como por ejemplo, la edad y el peso del animal, la enfermedad precisa que requiere tratamiento y su gravedad, la naturaleza de la formulación y el método de administración,
y la determina finalmente el médico o veterinario responsable del tratamiento. Sin embargo, una cantidad eficaz de un compuesto generalmente está en el intervalo de 0,1 a 100 mg/kg de peso corporal del receptor (mamífero) al día y en particular, típicamente en el intervalo de 1 a 10 mg/kg de peso corporal al día. Por tanto, la cantidad real al día para un mamífero adulto que pesa 70 kg normalmente está entre 70 y 700 mg, donde esta cantidad puede administrarse como una dosis individual al día o normalmente en una serie de dosis divididas (como, por ejemplo, dos, tres, cuatro, cinco o seis) al día, de modo que la dosis total diaria sea la misma. Una cantidad eficaz de una sal o solvato, o de un derivado fisiológicamente funcional del mismo puede determinarse como la fracción de la cantidad eficaz de compuesto per se.
En determinadas realizaciones, las formulaciones farmacéuticas pueden administrarse en forma de unidades de dosis que comprenden una cantidad predeterminada de principio activo por unidad de dosis. Esta unidad puede comprender, por ejemplo, de 0,5 mg a 1 g, preferiblemente de 1 mg a 700 mg, en especial, preferiblemente de 5 mg a 100 mg, de un compuesto según la invención, dependiendo de la enfermedad tratada, el método de administración y la edad, peso y estado del paciente, o las formulaciones farmacéuticas pueden administrarse en forma de unidades de dosis que comprenden una cantidad predeterminada del principio activo por unidad de dosis. Las formulaciones de unidad de dosis preferidas son aquellas que comprenden una dosis, o parte de una dosis, diaria como se indica anteriormente, o una fracción correspondiente de la misma de un principio activo. Además, las formulaciones farmacéuticas de este tipo pueden prepararse usando un proceso, que generalmente es conocido en la técnica farmacéutica.
Entre las formas farmacéuticas líquidas para administración oral se incluyen, pero sin limitaciones, emulsiones, microemulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables. Además de los compuestos activos, las formas farmacéuticas líquidas opcionalmente contienen diluyentes inertes normalmente utilizados en la técnica, como por ejemplo, agua u otros solventes, agentes solubilizantes y emulsionantes como alcohol etílico, alcohol isopropílico, carbonato de etilo, acetato de etilo, alcohol bencílico, benzoato de bencilo, propilenglicol, 1,3-butilenglicol, dimetilformamida, aceites (en particular, aceites de semillas de algodón, cacahuete, maíz, germen de trigo, oliva, ricino y sésamo), glicerol, alcohol tetrahidrofurfurilo, polietilenglicoles y ésteres de ácidos grasos de sorbitán, y mezclas de los mismos. Además de los diluyentes inertes, las composiciones orales también pueden incluir adyuvantes como agentes humectantes, emulsionantes y agentes de suspensión, edulcorantes, aromatizantes y perfumantes.
Las preparaciones inyectables, por ejemplo, suspensiones acuosas u oleaginosas inyectables estériles, se formulan según la técnica conocida utilizando agentes de dispersión o agentes humectantes adecuados. La preparación inyectable estéril también es una solución, suspensión o emulsión inyectable estéril en un diluyente o solvente no tóxico aceptable por vía parenteral, por ejemplo, como una solución en 1,3-butanediol. Entre los vehículos y solventes aceptables que pueden emplearse figuran agua, solución de Ringer y solución de cloruro sódico isotónica y según la farmacopea estadounidense (U.S.P.). Además, convencionalmente se emplean como solvente o medio de suspensión aceites fijados estériles. Con este objetivo, puede emplearse cualquier aceite fijado insípido, como mono o diglicéridos sintéticos. Además, se usan ácidos grasos como ácido oleico en la preparación de inyectables.
Las formulaciones inyectables pueden esterilizarse, por ejemplo, mediante filtración a través de un filtro que retiene bacterias, o incorporando agentes esterilizantes en la forma de composiciones sólidas estériles que pueden disolverse o dispersarse en agua estéril u otro medio inyectable estéril antes de su uso.
Para prolongar el efecto de un compuesto de la presente invención, a menudo es deseable ralentizar la absorción del compuesto a partir de la inyección subcutánea o intramuscular. Esto se consigue mediante el uso de una suspensión líquida de material cristalino o amorfo con baja solubilidad en agua. La velocidad de absorción del compuesto depende por tanto de su velocidad de disolución que, a su vez, puede depender del tamaño del cristal y de la forma cristalina. Alternativamente, el retraso en la absorción de una forma del compuesto administrada por vía parenteral se consigue disolviendo o resuspendiendo el compuesto en un vehículo oleoso. Las formas de liberación lenta inyectables se hacen formando matrices microcapsulares del compuesto en polímeros biodegradables como polilactida-poliglicólido. Dependiendo de la relación entre compuesto y polímero y de la naturaleza del polímero empleado en particular, puede controlarse la velocidad de liberación del compuesto. Entre los ejemplos de otros polímeros biodegradables se incluyen poli(ortoésteres) y poli(anhídridos). Las formulaciones de liberación lenta inyectables también se preparan embebiendo el compuesto en liposomas o microemulsiones que son compatibles con los tejidos del organismo.
Las composiciones para administración rectal o vaginal son preferiblemente supositorios que pueden prepararse mezclando los compuestos de esta invención con excipientes o vehículos adecuados no irritantes como manteca de cacao, polietilenglicol o cera para supositorios que son sólidos a temperatura ambiente pero líquidos a temperatura corporal y, por tanto, se funden en el recto o en la cavidad vaginal y liberan el compuesto activo.
Entre las formas farmacéuticas sólidas para administración oral se incluyen cápsulas, comprimidos, píldoras, polvos y gránulos. En dichas formas farmacéuticas sólidas, el compuesto activo se mezcla con al menos un excipiente o vehículo inerte, farmacéuticamente aceptables como citrato sódico o fosfato dicálcico y/o a) cargas o extensores como almidones, lactosa, sacarosa, glucosa, manitol y ácido silícico, b) aglutinantes, como por ejemplo, carboximetilcelulosa, alginatos, gelatina, polivinilpirrolidona, sacarosa y goma de acacia, c) humectantes como glicerol, d) agentes desintegrantes como agar-agar, carbonato de calcio, almidón de patata o tapioca, ácido algínico, determinados silicatos y carbonato sódico, e) solución de agentes retardantes como parafina, f) aceleradores de absorción como compuestos de amonio cuaternario, g) agentes humectantes, como por ejemplo, alcohol cetílico y monoestearato de glicerol, h) absorbentes como kaolina y arcilla bentonita e i) lubricantes como talco, estearato cálcico, estearato de magnesio, polietilenglicoles sólidos, lauril
sulfato sódico y mezclas de los mismos. En el caso de cápsulas, comprimidos y píldoras, la forma farmacéutica también compre nde opcionalmente agentes tamponadores.
Las composiciones sólidas de un tipo similar también se emplean como cargas en cápsulas de gelatina cargadas blandas y duras usando excipientes como lactosa o azúcar de la leche, así como polietilenglicoles de alto peso molecular y similares. Las formas farmacéuticas sólidas de comprimidos, grageas, cápsulas, píldoras y gránulos pueden prepararse con recubrimientos y envoltorios como recubrimientos entéricos y otros recubrimientos bien conocidos en la técnica de formulación farmacéutica. Opcionalmente contienen agentes opacificantes y también pueden formar parte de una composición que libere solo el o los principios activos, o preferiblemente, en una parte determinada del tubo digestivo, opcionalmente, de forma retardada. Entre los ejemplos de composiciones de inclusión que pueden usarse pueden incluirse sustancias poliméricas y ceras. Las composiciones sólidas de un tipo similar también se emplean como cargas en cápsulas de gelatina cargadas blandas y duras usando excipientes como lactosa o azúcar de la leche, así como polietilenglicoles de alto peso molecular y similares.
Los compuestos activos también pueden estar en forma microencapsulada con uno o más excipientes como se ha indicado anteriormente. Las formas farmacéuticas sólidas de comprimidos, grageas, cápsulas, píldoras y gránulos pueden prepararse con recubrimientos y envoltorios como recubrimientos entéricos, recubrimientos de liberación controlada u otros recubrimientos bien conocidos en la técnica de formulación farmacéutica. En dichas formas farmacéuticas sólidas, el compuesto activo puede mezclarse con al menos un diluyente inerte como sacarosa, lactosa o almidón. Dichas formas farmacéuticas también comprenden, como en la práctica normal, sustancias adicionales distintas de los diluyentes inertes, por ejemplo, lubricantes para comprimidos y otros excipientes para comprimidos como estearato de magnesio y celulosa microcristalina. En el caso de cápsulas, comprimidos y píldoras, las formas farmacéuticas también comprenden opcionalmente agentes tamponadores. Opcionalmente contienen agentes opacificantes y también pueden formar parte de una composición que libere solo el o los principios activos, o preferiblemente, en una parte determinada del tubo digestivo, opcionalmente, de forma retardada. Entre los ejemplos de composiciones de inclusión que pueden usarse pueden incluirse sustancias poliméricas y ceras.
Entre las formas farmacéuticas para administración tópica o transdérmica de un compuesto de esta invención se incluyen pomadas, pastas, cremas, lociones, geles, polvos, soluciones, sprays, inhaladores o parches. El componente activo se mezcla en condiciones estériles con un vehículo farmacéuticamente aceptable y cualquier conservante o tampón necesario según se requiera. También se contemplan dentro del alcance de esta invención formulaciones oftálmicas, gotas para los oídos y colirios. Adicionalmente, la presente invención contempla el uso de parches transdérmicos, que tienen la ventaja añadida de proporcionar la administración controlada de un compuesto al organismo. Dichas formas farmacéuticas puede prepararse disolviendo o dispersando el compuesto en el medio adecuado. También pueden usarse potenciadores de la absorción para aumentar el flujo del compuesto a través de la piel. La velocidad puede controlarse proporcionando una membrana que controle la velocidad o dispersando el compuesto en una matriz polimérica o gel.
Según una realización, la invención se refiere a un método de inhibición de la actividad IRAK en una muestra biológica que comprende los pasos de poner en contacto dicha muestra biológica con un compuesto de esta invención, o una composición que comprende dicho compuesto.
Según otra realización, la invención se refiere a un método de inhibición de la actividad IRAK, o una forma mutada de la misma, en una muestra biológica de forma positiva, que comprende el paso de poner en contacto dicha muestra biológica con un compuesto de esta invención, o una composición que comprende dicho compuesto.
Los compuestos de la invención son útiles in vitro como herramientas exclusivas para entender la función biológica de IRAK, incluida la evaluación de los numerosos factores que se cree influyen, y están influenciados, por la producción de IRAK, así como su interacción. Los presentes compuestos también son útiles en el desarrollo de otros compuestos que interaccionan con IRAK, ya que los presentes compuestos proporcionan información importante sobre la relación estructura-actividad que facilita este desarrollo. Los compuestos de la presente invención que se unen a IRAK pueden usarse como reactivos para detectar IRAK en células vivas, células fijadas, en líquidos biológicos, en homogeneizados de tejido, en materiales biológicos naturales purificados, etc. Por ejemplo, marcando estos compuestos, pueden identificarse las células que expresan IRAK. Además, en función de su capacidad para unirse a IRAK, los compuestos de la presente invención pueden usarse en la tinción in situ, FACS (clasificación de células activada por fluorescencia), electroforesis en gel de poliacrilamida con dodecil sulfato sódico (SDS-PAGE), ELISA (ensayo de inmunoadsorción ligado a enzima), etc., en la purificación de enzimas o en la purificación de células que expresan IRAK dentro de células permeabilizadas. Los compuestos de la invención también pueden utilizarse como reactivos comerciales para diversos usos en investigación médica y diagnóstico. Estos usos pueden incluir, pero sin limitaciones: uso como patrón de calibración para cuantificar las actividades de candidatos a inhibidores de IRAK en diversos ensayos funcionales; uso como reactivos de bloqueo en el cribado aleatorio de compuestos, es decir, para buscar nuevas familias de ligandos de IRAK, los compuestos pueden usarse para bloquear la recuperación de los compuestos de IRAK actualmente reivindicados; uso en la cocristalización con la enzima IRAK, es decir, los compuestos de la presente invención permitirán la formación de cristales del compuesto unidos a IRAK, lo que permite la determinación de la estructura enzima/compuesto mediante cristalografía de rayos X; otras aplicaciones en investigación y diagnóstico, donde IRAK está preferiblemente activada o dicha activación se calibra convenientemente frente a una cantidad conocida de un inhibidor de IRAK, etc.; uso en ensayos como sondas para determinar la expresión de IRAK en células; y desarrollo de ensayos para detectar compuestos que se unen al mismo sitio que los ligandos de unión a IRAK.
Los compuestos de la invención pueden aplicarse por sí mismos y/o en combinación con medidas físicas para el diagnóstico de la eficacia del tratamiento. Las composiciones farmacéuticas que contienen dichos compuestos y el uso de dichos compuestos para tratar afecciones mediadas por IRAK es una nueva estrategia prometedora para un amplio espectro de terapias que causan una mejora directa e inmediata del estado de salud, ya sea en seres humanos o en animales. Las nuevas entidades químicas biodisponibles por vía oral y activas de la invención mejoran la conveniencia para los pacientes y el cumplimiento terapéutico para los médicos.
Los compuestos de fórmula (I), sus sales, isómeros, tautómeros, formas enantioméricas, diastereómeros, racematos, derivados, profármacos y/o metabolitos se caracterizan por una alta especificidad y estabilidad, bajo coste de fabricación y manejo conveniente. Estas características constituyen la base de una acción reproducible, en la que se incluye la falta de reactividad cruzada, y de una interacción fiable y segura con la estructura diana.
El término «muestra biológica», según se usa en este documento, incluye, sin limitaciones, cultivos celulares o extractos de los mismos; material biopsiado obtenido a partir de un mamífero o extractos del mismo; y sangre, saliva, orina, heces, semen, lágrimas u otros líquidos corporales o extractos de los mismos.
La modulación de la actividad IRAK, o una forma mutada de la misma, en una muestra biológica es útil para diversos fines que son conocidos por un experto en la materia. Entre los ejemplos de dichos fines se incluyen, pero sin limitaciones, transfusión de sangre, trasplante de órganos, conservación de muestras biológicas y ensayos biológicos.
Ejemplos
Como se muestra en los ejemplos a continuación, en determinados ejemplos de realizaciones, los compuestos se preparan según los siguientes procedimientos generales. Se apreciará que, aunque los métodos generales describen la síntesis de determinados compuestos de la presente invención, los siguientes métodos generales y otros métodos conocidos por un experto en la materia, pueden aplicarse a todos los compuestos y subclases y especies de cada uno de estos compuestos, como se describe en este documento.
Los símbolos y convenciones utilizados en las siguientes descripciones de procesos, esquemas y ejemplos coinciden con los utilizados en la literatura científica contemporánea, por ejemplo, en el Journal of the American Chemical Society o en el Journal of Biological Chemistry.
Siempre que no se indique otra cosa, todas las temperaturas se expresan en °C (grados centígrados).
Todas las reacciones se llevaron a cabo a temperatura ambiente, siempre que no se indique otra cosa. Todos los compuestos de la presente invención se sintetizaron mediante procesos desarrollados por los inventores.
Los espectros de RMN 1H se adquirieron en un instrumento Bruker Fourier-300MHz. Los desplazamientos químicos se expresan en partes por millón (ppm, unidades 5). Las constantes de acoplamiento se expresan en hercios (Hz). Los patrones de división describen multiplicidades aparentes y se designan como s (singlete), d (doblete), t (triplete), c (cuadruplete), m (multiplete), q (quintuplete) o a (ancho).
Los espectros de masas se obtuvieron en espectrómetros de masas de la serie LC-MS-2020 de Shimadzu, usando ionización por electropulverización (ESI). Columna: Luna C18, 5 |jm, 2,0 * 50 mm; solvente A: agua ácido fórmico al 0,1 %; solvente B: MeCN ácido fórmico al 0,1%; flujo: 0,7 ml/min; gradiente: 0 min: 5 % de B; 5 min: 100 % de B; 6,5 min: 100 % de B; 6,51 min: 5 % de B; 7 min 5 % de B.
Los números de compuesto utilizados en los siguientes ejemplos se corresponden con los números indicados anteriormente.
Las siguientes abreviaturas se refieren a las abreviaturas que se utilizan a continuación:
Ac (acetilo), BINAP (2,2'-bis(disfenilfosfino)-1,1'-binaftaleno), dba (dibencilidenacetona), Bu (butilo), fBu (ferc-butilo), DCE (dicloroetano), DCM (diclorometano), 5 (desplazamiento químico), DIEA (di-isopropil etilamina), DMA (dimetil acetamida), DMSO (dimetilsulfóxido), DMF (N,N-dimetilformamida), Dppf (1,1 '-bis (difenil fosfina ferroceno)), EtOAc (acetato de etilo), EtOH (etanol), eq. (equivalente), g (gramo), Hex (hexano), cHex (ciclohexano), HATU (fosfato de N-[(dimetilamino)(3H-[1,2,3]triazolo[4,5-b]piridin-3-iloxi)metilen]-N-metilmetanaminohexafluoro), HPLC (cromatografía líquida de alta resolución), h (hora), LDA (diisopropilamina de litio), LiHMDS (bis(trimetilsilil)amida de litio), MHz (megahercio), MeOH (metanol), min (minuto), ml (mililitro), mmol (milimol), mM (milimolar), EM (espectrometría de masas), RMN (resonancia magnética nuclear), T/N (toda la noche), PBS (solución salina tamponada con fosfato), TA (temperatura ambiente), TEA (trietilamina), TFA (ácido trifluoroacético), THF (tetrahidrofurano), TLC (cromatografía en capa fina).
En general, los compuestos según la fórmula (I) y fórmulas relacionadas de esta invención pueden prepararse a partir de materiales de partida fácilmente disponibles. Si estos materiales de partida no están disponibles en el mercado, pueden prepararse mediante técnicas de síntesis convencionales. En general, la ruta de síntesis para cualquier compuesto individual de fórmula (I) y fórmulas relacionadas dependerá de los sustituyentes específicos de cada molécula, siendo apreciados estos factores por los expertos en la materia. Los siguientes métodos y procedimientos generales descritos a continuación en este documento en los ejemplos se pueden emplear para preparar compuestos de fórmula (I) y fórmulas
relacionadas. Las condiciones de reacción mostradas en los esquemas siguientes, como las temperaturas, solventes o correactivos, se proporcionan solo como ejemplos y no son restrictivos. Se apreciará que cuando se proporcionan condiciones experimentales típicas o preferidas (es decir, temperaturas de reacción, tiempo, moles de reactivos, solventes, etc.), también pueden usarse otras condiciones experimentales siempre que no se establezca otra cosa. Las condiciones óptimas de reacción pueden variar con los reactivos o solventes utilizados en particular, aunque estas condiciones pueden ser determinadas por un experto en la materia mediante procedimientos rutinarios de optimización. Para obtener información sobre todos los métodos de protección y desprotección, consulte Philip J. Kocienski, en «Protecting Groups», Georg Thieme Verlag Stuttgart, Nueva York, 1994 y Theodora W. Greene y Peter G. M. Wuts en «Protective Groups in Organic Synthesis», Wiley Interscience, 3.a edición 1999.
Compuesto intermedio ácido 1
O O
H 0 ^ 0Et
F Me
Etapa 1: 2-Fluoro-2-metilmalonato de dietilo. A una solución en THF (0,5 M) de metilmalonato de dietilo (1 eq.) se añadió hidruro de sodio (1,4 eq., dispersión del 60 % [p/p] en aceite de parafina) en cuatro porciones iguales, con tres a cinco minutos de diferencia. La reacción se mantuvo a 0 °C durante 15 min, antes se dejó atemperar a TA durante 30 min. Después de otros 30 min de agitación a TA, la mezcla se volvió a enfriar a 0 °C y posteriormente se añadió N-fluorobencenosulfonamida (1,1 eq.) en cuatro porciones iguales. Se continuó con la agitación a 0 °C durante 30 min y luego a TA durante 4 h, en cuyo momento se determinó que se había completado >95 % de la reacción mediante RMN 1H. La reacción se diluyó a continuación con hexanos y se filtró al vacío. La torta de filtración se lavó adicionalmente con hexanos y se concentró el filtrado que contenía el producto. Se añadieron más hexanos para inducir la precipitación adicional de subproductos no deseados y la suspensión resultante se filtró de nuevo. A continuación, el filtrado obtenido de este modo se concentró al vacío para obtener un aceite bifásico. La capa superior se determinó que era aceite de parafina y se desechó. La capa inferior era el producto deseado (rendimiento del 82 %).
Etapa 2: Ácido (S)-3-etoxi-2-fluoro-2-metil-3-oxopropanoico. Se recogió 2-fluoro-2-metilmalonato de dietilo (1 eq.) procedente de la etapa previa en un tampón fosfato acuoso a pH = 7,3 (0,14 M, preparado disolviendo 7,3 g de NaHPO4 y 2,1 g de KH2PO4 por litro de agua). A continuación, se añadió lipasa de Candida rugosa (70 mg por mmol de sustrato, 847 U/mg, Sigma n.° cat. L1754) y la mezcla heterogénea resultante se agitó vigorosamente a TA durante 18 h. Dependiendo de la escala, fue necesaria la adición ocasional de NaOH 1 N para mantener el pH de la mezcla de reacción en ~7,3 para garantizar una actividad óptima. A continuación, se añadió celite a la mezcla de reacción, se agitó a TA durante 1 h y se filtró. El filtrado se extrajo con EtOAc, se secó sobre MgSO4 y se filtró. La concentración del filtrado al vacío permitió obtener el producto deseado como un sólido cristalino de color blanco (rendimiento del 71 %, exceso enantiomérico del 93 %).
Compuesto intermedio ácido 2

Etapa 1 :3-Etil-2-fluoro-2-metilmalonato de (R)-1-terc-butilo. Se disolvió el compuesto intermedio ácido 1 (1 eq.) en cloruro de tionilo (2,3 M) a TA. A continuación, a esta mezcla se añadieron unas gotas de DMF puro y la mezcla resultante se calentó a 75 °C durante 2 h. Los compuestos volátiles se eliminaron después al vacío y el compuesto intermedio ácido clorhídrico se recogió en diclorometano (0,12 M). A continuación, se añadió a 0 °C N,N-diisopropiletilamina (3 eq.) y terc-butanol (1,5 eq.) secuencialmente a la solución anterior de diclorometano y la mezcla de reacción resultante se dejó atemperar lentamente a TA durante 16 h. La reacción se detuvo después cuidadosamente con la adición de solución acuosa de HCl 1 N y se extrajo con diclorometano. Los extractos orgánicos combinados se lavaron adicionalmente con solución acuosa de NaHCO3 saturada, agua y salmuera, se secaron sobre MgSO4 y se filtraron. La concentración del filtrado al vacío permitió obtener el producto deseado como un aceite incoloro (rendimiento del 65 %).
Etapa 2: Ácido (R)-3-(terc-butoxi)-2-fluoro-2-metil-3-oxopropanoico. A una solución en THF (0,16 M) de 3-etil-2-fluoro-2-metilmalonato de (R)-1-terc-butilo (1 eq.) procedente de la etapa previa se añadió solución acuosa de NaOH 1 N (2 eq.). La mezcla bifásica resultante se agitó vigorosamente a TA durante 2 h. A continuación, la reacción se detuvo cuidadosamente con la adición de solución acuosa de HCl 1 N (hasta un pH de ~4) y se extrajo con EtOAc. Los extractos orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre MgSO4 y se filtraron. La concentración del filtrado al vacío permitió obtener el producto deseado como un sólido de color blanco (rendimiento del 52 %).
Compuesto intermedio amina 1:

Etapa 1: 2-Fluoro-3-((4-metoxibencil)amino)-2-metil-3-oxopropanoato de (S)-etilo. El compuesto intermedio ácido 1 (1 eq.), clorhidrato de 4-metoxibencil amina (1,1 eq.) y N,N-diisopropiletilamina (3,5 eq.) se combinaron en diclorometano (0,15 M). A continuación, a esta mezcla se añadió HATU (1,05 eq.) y la mezcla resultante se agitó a TA durante 18 h. Después la reacción se detuvo con la adición de solución acuosa de HCl 1 N y se extrajo con diclorometano. Los extractos orgánicos combinados se lavaron adicionalmente con agua, solución acuosa de NaHCO3 al 10 % y salmuera, se secaron sobre MgSO4, se filtraron y el filtrado se concentró al vacío. La purificación adicional del producto sin procesar obtenido de este modo mediante cromatografía en columna (SiO2 , elución en gradiente, Hex ^ 7:3 (v/v) Hex: EtOAc) permitió obtener el producto deseado como un aceite de color amarillo dorado (rendimiento del 70 %).
Etapa 2: (R)-2-Fluoro-3-hidroxi-N-(4-metoxibencil)-2-metilpropanamida. A una solución en metanol (0,23 M) de 2-fluoro-3-((4-metoxibencil)amino)-2-metil-3-oxopropanoato de (S)-etilo: (1 eq.) procedente de la etapa previa se añadió a 0 °C yoduro de calcio (1 eq.) y borohidruro de sodio (2 eq.). Tras el desarrollo vigoroso de gas, se retiró el baño frío y la mezcla de reacción se agitó a TA durante 4 h. A continuación, la reacción se detuvo con la adición de solución acuosa de HCl al 10 % y se extrajo con EtOAc. Los extractos orgánicos combinados se lavaron adicionalmente con agua, solución acuosa de NaOH 1 N y salmuera, se secaron sobre MgSO4, se filtraron y el filtrado se concentró al vacío. La recristalización del producto sin procesar obtenido de este modo a partir de diclorometano, éter y hexanos permitió obtener el producto deseado como un sólido cristalino de color blanco (rendimiento del 86 %).
Etapa 3: (R)-2-Fluom-N-(4-metoxibendl)-2-metil-3-((triisopmpilsWl)oxi)-pmpanamida. A una solución en diclorometano (0,31 M) de (R)-2-fluoro-3-hidroxi-N-(4-metoxibencil)-2-metilpropanamida (1 eq.) procedente de la etapa previa se añadió a 0 °C 2,6-lutidina (1,5 eq.) y, a continuación, triisopropilsilil trifluorometanosulfonato (1,2 eq.). La solución resultante se agitó a 0 °C durante 10 min y después a TA durante 30 min. A continuación, la reacción se detuvo cuidadosamente con la adición de solución acuosa de HCl 0,1 N y se extrajo con diclorometano. Los extractos orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre MgSO4 y se filtraron. La concentración del filtrado al vacío permitió obtener el producto deseado como un aceite incoloro (rendimiento del 71 %).
Etapa 4: (S)-2-Fluom-N-(4-metoxibendl)-2-metil-3-((triisopropilsiW)oxi)pmpan-1-amina. A una solución en THF (0,25 M) de (R)-2-fluoro-N-(4-metoxibencil)-2-metil-3-((triisopropilsilil)oxi)-propanamida (1 eq.) procedente de la etapa previa se añadió a TA borano (6 eq., solución 1 M en THF) gota a gota durante un periodo de 30 min. Tras completar la adición, se acopló un condensador de reflujo y la mezcla de reacción se calentó a reflujo durante 18 h. Cuando se estimó que se había completado la reacción, esta se detuvo lenta y cuidadosamente a 0 °C con la adición gota a gota de metanol. A continuación, los compuestos volátiles se eliminaron al vacío y el residuo resultante se coevaporó con tolueno (3x). El producto sin procesar obtenido de este modo se utilizó inmediatamente en la siguiente etapa.
Etapa 5: (2-Fluom-2-metil-3-((triisopmpilsilil)oxi)pmpil)(4-metoxibendl)carbamato de (S)-terc-butilo. A una solución en diclorometano (0,29 M) de (S)-2-fluoro-N-(4-metoxibencil)-2-metil-3-((triisopropilsilil)oxi)propan-1-amina (1 eq.) procedente de la etapa previa se añadió N,N-diisopropiletilamina (3 eq.) y, a continuación, dicarbonato de di-terc-butilo (1,5 eq.). La solución resultante se agitó a TA durante 5 h. A continuación, la reacción se detuvo cuidadosamente con la adición de solución acuosa de HCl 1 N y se extrajo con diclorometano. Los extractos orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre MgSO4 y se filtraron. La concentración del filtrado al vacío permitió obtener el producto deseado como un aceite incoloro (rendimiento del 62 % en dos etapas).
Etapa 6: (2-Fluoro-3-hidroxi-2-metilpropil)carbamato de (S)-terc-butilo. A una solución 3:1 (v/v) de acetonitrilo:agua (0,1 M) de (2-fluoro-2-metil-3-((triisopropilsilil)-oxi)propil)(4-metoxibencil)carbamato de (S)-terc-butilo (1 eq.) procedente de la etapa previa se añadió nitrato de amonio y cerio (2 eq.) y la mezcla resultante se agitó a TA durante 18 h. A continuación, la solución de reacción se diluyó con agua y se extrajo con EtOAc. Los extractos orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre MgSO4, se filtraron y el filtrado se concentró al vacío. La purificación adicional del producto sin procesar obtenido de este modo mediante cromatografía en columna (SiO2 , elución en gradiente, Hex ^ EtOAc) permitió obtener el producto deseado como un sólido cristalino de color blanco (rendimiento del 70 %).
Etapa 7: (3-((terc-Butildifenilsilil)oxi)-2-fluoro-2-metilpropil)carbamato de (S)-terc-butilo. A una solución en diclorometano (0,1 M) de (2-fluoro-3-hidroxi-2-metilpropil)carbamato de (S)-terc-butilo (1 eq.) procedente de la etapa previa se añadió secuencialmente N,N-diisopropiletilamina (2,5 eq.), DMAP (0,05 eq.) y terc-butildifenilclorosilano (1,3 eq.). La solución resultante se agitó a TA durante 72 h. A continuación, la reacción se detuvo cuidadosamente con la adición de solución acuosa de HCl 1 N y se extrajo con diclorometano. Los extractos orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre MgSO4, se filtraron y el filtrado se concentró al vacío. La purificación adicional del producto sin procesar obtenido de este modo mediante cromatografía en columna (SiO2 , elución en gradiente, Hex ^ 1:1 (v/v) Hex: EtOAc) permitió obtener el producto deseado como un aceite incoloro (rendimiento del 88 %).
Etapa 8: Clorhidrato de (S)-3-((terc-butildifenilsilil)oxi)-2-fluoro-2-metilpropan-1-amina. A una solución en diclorometano (0,1 M) de (3-((terc-butildifenilsilil)oxi)-2-fluoro-2-metilpropil)carbamato de (S)-terc-butilo (1 eq.) procedente de la etapa previa se añadió HCl (5 eq., solución 4 M en 1,4-dioxano) y la solución resultante se agitó a TA durante 2 h. Los compuestos volátiles se eliminaron a continuación al vacío y el residuo resultante se recristalizó a partir de éter y hexanos. El compuesto del título se aisló como un sólido cristalino de color blanco (rendimiento del 86 %).
Compuesto intermedio amina 2:
Q ív
Si Me
H2N X 0 HCl
F* Me / / "%
Se preparó de forma análoga al compuesto intermedio amina 1, pero utilizando el compuesto intermedio ácido 2 (1 eq.) en lugar del compuesto intermedio ácido 1 en la etapa 1.
Compuesto intermedio amina 3:

Etapa 1: 1-Azido-3-(benciloxi)-2-metilpropan-2-ol. Una solución de 2-[(benciloxi)metil]-2-metiloxirano (Matrix Scientifics, 4,1 g; 20,7 mmol; 1,00 eq.), NH4Cl (2,3 g; 42,14 mmol; 2,04 eq.) y NaNa (4,2 g; 63,31 mmol; 3,06 eq.; 98 %) en DMF (30 ml) se agitó durante 16 h a 60 °C. La mezcla de reacción se diluyó con agua (300 ml) y se extrajo con DCM (3 * 50 ml). Las fases orgánicas combinadas se secaron sobre sulfato de sodio, se filtraron y se concentraron. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (éter de petróleo:EtOAc, gradiente de 50:1 a 20:1) proporcionó el compuesto del título como un aceite de color amarillo (4 g, 69 %).
Etapa 2 :[(3-Azido-2-fluoro-2-metilpropoxi)metil]benceno. Se añadió DAST (5,3 g; 32,22 mmol; 1,12 eq.; 98 %) gota a gota a una solución de 1-azido-3-(benciloxi)-2-metilpropan-2-ol (7,1 g; 28,9 mmol; 1,00 eq.) in DCM (100 ml) mantenido bajo atmósfera de nitrógeno a °C. La mezcla de reacción se atemperó a TA y se agitó durante 4 h. A continuación se detuvo con agua (30 ml) y se extrajo con DCM (3 * 30 ml). Las fases orgánicas combinadas se concentraron a presión reducida y se purificaron mediante cromatografía ultrarrápida sobre sílice (éter de petróleo:EtOAc, gradiente de 15:1 a 10:1) para obtener el compuesto del título como un aceite de color amarillo (6 g; 84 %).
Etapa 3: 3-(Benciloxi)-2-fluoro-2-metilpropan-1-amina. Se añadió trifenilfosfano (3,76 g; 13,62 mmol; 1,30 eq.) lentamente a una solución de [(3-azido-2-fluoro-2-metilpropoxi)metil]benceno (2,60 g; 10,48 mmol; 1,00 eq.) en THF (40 ml) y agua (10 ml) previamente purgado con nitrógeno y mantenido bajo atmósfera de nitrógeno y a °C. A continuación, la mezcla de reacción se atemperó a TA y se agitó durante la noche antes de su concentración al vacío. El residuo resultante se trituró en éter y los sólidos se recogieron mediante filtración. El filtrado se concentró y se purificó mediante cromatografía ultrarrápida sobre gel de sílice (DCM:MeOH, 20:1) para obtener el compuesto del título como un sólido de color amarillo (2 g, 67 %).
Compuesto intermedio 1 -CF3 :

Etapa 1: 3-(4-Yodo-2-nitro-fenilamino)-propan-1-ol. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvió 1-fluoro-4-yodo-2-nitro-benceno (1 eq.) en DMF (0,3 M). A continuación, a esta mezcla se añadió 3-amino-propan-1-ol (1,2 eq.) y carbonato de potasio (3 eq.). La solución resultante se calentó a 65 °C durante 18 h. A continuación, la mezcla de reacción sin procesar se diluyó con EtOAc y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó a continuación sobre MgSO4, se filtró y el filtrado se concentró al vacío. La purificación del producto sin procesar obtenido de este modo mediante cromatografía en columna (SiO2 , elución en gradiente, 8:1 (v/v) Hex: EtOAc ^ EtOAc) permitió obtener el producto deseado (rendimiento del 79 %).
Etapa 2: Acetato de 3-((4-yodo-2-nitrofenil)amino)propilo. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvió 3-(4-yodo-2-nitro-fenilamino)-propan-1-ol (1 eq.) procedente de la etapa previa en piridina
(0,1 M). A continuación, a esta mezcla se añadió anhídrido acético (1,8 eq.) gota a gota y puro durante 10 min. Tras 18 h de agitación a TA, los compuestos volátiles se eliminaron al vacío y el residuo resultante se repartió entre EtOAc y solución acuosa de NH4Cl saturada. La capa acuosa se separó y se extrajo de nuevo con EtOAc. Los extractos orgánicos combinados se secaron después sobre MgSO4, se filtraron y el filtrado se concentró al vacío. La purificación del producto sin procesar obtenido de este modo mediante cromatografía en columna (SiO2 , elución en gradiente, 8:1 (v/v) Hex: EtOAc ^ EtOAc) permitió obtener el producto deseado (rendimiento del 91 %).
Etapa 3: Acetato de 3-((2-amino-4-yodofenil)amino)propilo. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se resuspendió tricloruro de hierro (0,25 eq.), carbón activado (100 mg por mmol de sustrato) y acetato de 3-((4-yodo-2-nitrofenil)amino)propilo (1 eq.) procedente de la etapa previa en metanol (0,15 M). A continuación, a esta mezcla se añadió N,N-dimetil hidrazina (5 eq.) gota a gota y puro durante un periodo de 10 min. Después, la suspensión resultante se calentó a 65 °C durante 4 h. El material insoluble se eliminó mediante filtración a través de un lecho de celite humedecido con MeOH y el filtrado obtenido de este modo se concentró al vacío. El residuo resultante se recogió en EtOAc, se lavó secuencialmente con agua y salmuera, se secó sobre MgSO4, se filtró y el filtrado se concentró al vacío. La purificación del producto sin procesar obtenido de este modo mediante cromatografía en columna (SiO2 , elución en gradiente, 1:1 (v/v) Hex: EtOAc ^ EtOAc) permitió obtener el producto deseado (rendimiento del 89 %).
Etapa 4: Acetato de 3-(2-amino-5-yodo-1H-benzo[d]imidazol-1-il)propilo. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se añadió lentamente bromuro de cianógeno (2 eq., solución 5 M en acetonitrilo) en etanol (0,5 M) durante un periodo de 10 min. A continuación, a esta solución se añadió una solución en etanol (0,25 M) de acetato de 3-((2-amino-4-yodofenil)amino)propilo (1 eq.) procedente de la etapa previa gota a gota durante un periodo de 10 min. La mezcla resultante se agitó a Ta durante 3 h. Después, los compuestos volátiles se eliminaron al vacío y el residuo resultante se recogió en EtOAc y se lavó secuencialmente con solución acuosa de NaHCO3 al 10 %, agua y salmuera. El extracto orgánico obtenido de este modo se secó a continuación sobre Na2SO4, se filtró y el filtrado se concentró al vacío. La purificación del producto sin procesar obtenido de este modo mediante cromatografía en columna (SiO2 , elución en gradiente, 1:1 (v/v) Hex: EtOAc ^ EtOAc ^ 10:1 (v/v) EtOAc: MeOH) permitió obtener el producto deseado (rendimiento del 81 %).
Etapa 5: Acetato de (E)-3-(5-yodo-2-((3-(trifluorometil)benzoil)imino)-2,3-dihidro-1H-benzo[d]imidazol-1-il)propilo. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvió acetato de 3-(2-amino-5-yodo-1H-benzo[d]imidazol-1-il)propilo (1 eq.) procedente de la etapa previa, ácido 3-trifluorometil-benzoico (1 eq.) y HATU (1,2 eq.) en DMF (0,1 M). A continuación, a esta mezcla se añadió etil-diisopropil-amina (2,5 eq.) y la solución amarilla resultante se agitó a TA durante 14 h. La mezcla de reacción sin procesar se diluyó con EtOAc y se lavó secuencialmente con agua, solución acuosa de HCl al 10 %, agua y salmuera. El extracto orgánico se secó a continuación sobre MgSO4, se filtró y el filtrado se concentró al vacío. La purificación adicional mediante cromatografía en columna (SiO2 , elución en gradiente, 9:1 (v/v) Hex: EtOAc ^ EtOAc) permitió obtener el producto deseado como un sólido blanquecino (rendimiento del 77 %).
Etapa 6: Acetato de (Z)-3-(5-yodo-2-((3-(trifluorometil)benzoil)imino)-3-((2-(trimetilsilil)etoxi)metil)-2,3-dihidro-1H-benzo[d]imidazol-1-il)propilo. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se resuspendió hidruro de sodio (1,6 eq., dispersión del 60 % [p/p] en aceite de parafina) en DMF (0,09 M). A continuación, a esta mezcla se añadió, a 0 °C, acetato de (E)-3-(5-yodo-2-((3-(trifluorometil)benzoil)imino)-2,3-dihidro-1H-benzo[d]imidazol-1-il)propilo (1 eq.) procedente de la etapa previa. La suspensión resultante se agitó a 0 °C durante 30 min antes de añadir (2-(clorometoxi)etil)trimetilsilano (2 eq.) gota a gota y puro durante un periodo de 5 min. Tras otra hora de agitación a 0 °C, la reacción se detuvo con la adición de agua y se extrajo con EtOAc. Los extractos orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre MgSO4, se filtraron y el filtrado se concentró al vacío. La purificación adicional mediante cromatografía en columna (SiO2 , elución en gradiente, 9:1 (v/v) Hex:acetona ^ acetona) permitió obtener el producto deseado como un aceite viscoso (rendimiento del 78 %).
Compuesto intermedio 2:

Etapa 1: 3-(4-Fluoro-3-nitro-fenil)-2-metoxipiridina. En un recipiente de reacción de vidrio equipado con un tapón de rosca recubierto de Teflón se combinaron ácido (4-fluoro-3-nitro-fenil)borónico (1,5 eq.), acetato de paladio(II) (0,05 eq.), diciclohexil-(2'6'-dimetoxi-bifenil-2-il)-fosfina (0,1 eq.) y fluoruro de potasio (3 eq.) en THF (0,11 M). A continuación, la suspensión de reacción se purgó subsuperficialmente con nitrógeno durante 15 min antes de que se añadiera 3-bromo-2-metoxi-piridina (1 eq.). A continuación, el recipiente de reacción se selló herméticamente y se calentó a 70 °C durante 16 h. La suspensión de reacción de color marrón resultante se diluyó con tBuOMe y se lavó secuencialmente con agua, solución acuosa de NaHCO3 al 10 %, agua y salmuera. El extracto orgánico se secó a continuación sobre MgSO4, se filtró y el filtrado se concentró al vacío para obtener un sólido de color marrón oscuro. La purificación del producto sin procesar
obtenido de este modo mediante cromatografía en columna (SÍO2, elución en gradiente, Hex ^ 3:7 (v/v) Hex:EtOAc) permitió obtener el producto deseado como un sólido de color blanco (rendimiento del 98 %).
Etapa 2: 3-(4-Fluoro-3-nitro-fenil)-piridin-2-ol. En un recipiente de reacción de vidrio equipado con un tapón de rosca recubierto de Teflón se disolvió 3-(4-fluoro-3-nitro-fenil)-2-metoxipiridina (1 eq.) procedente de la etapa previa en ácido acético glacial (0,078 M). A este se añadió a continuación bromuro de hidrógeno (3 eq., solución acuosa al 48 % [p/v]) y la mezcla resultante se calentó a 70 °C durante 16 h. La solución ahora de color amarillo se enfrió a TA y los compuestos volátiles se eliminaron al vacío. Después, el semisólido resultante se trituró en diclorometano y éter durante 2 h. El producto deseado se aisló mediante filtración al vacío como un sólido cristalino de color blanco (rendimiento del 98 %).
Etapa 3 :3-(4-Fluoro-3-nitrofenil)-1-metilpiridin-2(1H)-ona. En un recipiente de reacción de vidrio equipado con un agitador magnético recubierto de Teflón se resuspendió 3-(4-fluoro-3-nitro-fenil)-piridin-2-ol (1 eq.) procedente de la etapa previa, carbonato de cesio (1,5 eq.) y yodometano (3 eq.) en DMF (0,12 M). A continuación, la mezcla resultante se agitó a TA durante 48 h. La reacción se detuvo cuidadosamente con la adición de solución acuosa de HCl al 10 % y se extrajo con EtOAc. Después, los extractos orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre MgSO4 y se filtraron. La concentración del filtrado al vacío permitió obtener el producto deseado, tras una trituración adicional en éter y hexanos, como un sólido cristalino de color amarillo (rendimiento del 65 %).
Etapa 4: 3-(4-((3-Hidroxipropil)amino)-3-nitrofenil)-1-metilpiridin-2(1H)-ona. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvió 3-(4-fluoro-3-nitrofenil)-1-metilpiridin-2(1H)-ona (1 eq.) procedente de la etapa previa en acetonitrilo (0,1 M). A continuación, a esta mezcla se añadió 3-aminopropan-1-ol (1,5 eq.) y carbonato de potasio (2 eq.). La suspensión resultante se calentó a 80 °C durante 16 h. Los compuestos volátiles se eliminaron al vacío y el residuo resultante se repartió entre EtOAc y una solución acuosa de HCl al 10 %. Después, el extracto orgánico se separó y se lavó adicionalmente con agua y salmuera, se secó sobre MgSO4, se filtró y el filtrado se concentró al vacío. La purificación adicional del producto sin procesar obtenido de este modo mediante cromatografía en columna (SiO2 , elución en gradiente, 4:1 (v/v) Hex: EtOAc ^ EtOAc ^ 10:1 (v/v) EtOAc: MeOH) proporciona el producto deseado como un aceite de color naranja que solidificó tras dejarlo reposar (rendimiento del 85 %).
Etapa 5: 1-Metil-3-(3-nitro-4-((3-((triisopropilsilil)oxi)propil)amino)fenil)-piridin-2(1H)-ona. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvió 3-(4-((3-hidroxipropil)amino)-3-nitrofenil)-1-metilpiridin-2(1H)-ona (1 eq.) procedente de la etapa previa en DMF (0,11 M). A continuación, a esta mezcla se añadió cloro-triisopropil-silano (6 eq.), imidazol (6 eq.) y algunos cristales de DMAP. La solución resultante se agitó a TA durante 18 h. La mezcla de reacción sin procesar se diluyó con EtOAc y se lavó secuencialmente con agua, solución acuosa de NaHCO3 al 10 %, agua y salmuera. El extracto orgánico se secó a continuación sobre Na2SO4, se filtró y el filtrado se concentró al vacío. La purificación adicional mediante cromatografía en columna (SiO2 , elución en gradiente, 1:1 (v/v) Hex: EtOAc ^ EtOAc) proporcionó el producto deseado como un aceite de color naranja que solidificó tras dejarlo reposar (rendimiento del 61 %).
Etapa 6: 3-(3-Amino-4-((3-((tríisopmpilsilil)oxi)propil)amino)fenil)-1-metilpiperídin-2-ona. En un matraz con agitador Parr se resuspendió 1-metil-3-(3-nitro-4-((3-((triisopropilsilil)oxi)propil)amino)fenil)-piridin-2(1H)-ona (1 eq.) procedente de la etapa previa y negro de paladio (seco, 10 % p/p sobre carbono, 0,4 eq.) en metanol (0,12 M). A continuación, el recipiente se vació repetidamente y se volvió a llenar con nitrógeno (3 veces) y luego con hidrógeno (3 veces). La suspensión resultante se agitó bajo una presión de hidrógeno de 50 psi durante 16 h. La reacción se detuvo a continuación con CH2O 2 y se filtró a través de un lecho de celite humedecido con CH2O 2. El lecho insoluble se lavó adicionalmente con MeOH y CH2O 2. La concentración del filtrado obtenido de este modo al vacío proporcionó un aceite de color marrón. La purificación adicional del producto sin procesar obtenido de este modo mediante cromatografía en columna (SiO2 , elución en gradiente, CH2O 2 ^ 10:1 (v/v) CH2O 2 : MeOH) proporcionó el producto deseado como un sólido de color marrón claro que se oscureció tras dejarlo reposar (rendimiento del 75 %).
Etapa 7: 3-(2-Amino-1-(3-((triisopropilsilil)oxi)propil)-1H-benzo[d]imidazol-5-il)-1-metilpiperidin-2-ona. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvió 3-(3-amino-4-((3-((triisopropilsilil)oxi)propil)amino)fenil)-1-metilpiperidin-2-ona (1 eq.) procedente de la etapa previa en etanol (0,05 M). A continuación, a esta mezcla se añadió bromuro de cianógeno (solución 5 M en acetonitrilo, 1,5 eq) gota a gota durante un periodo de 10 min. La mezcla resultante se agitó a TA durante 12 h. La mezcla de reacción sin procesar se diluyó con EtOAc y se lavó secuencialmente con una solución acuosa de NaHCO3 al 10 %, agua y salmuera. El extracto orgánico se secó a continuación sobre Na2SO4 y se filtró. La concentración del filtrado obtenido de este modo al vacío permitió obtener el producto deseado como un sólido blanquecino (rendimiento del 82 %).
Compuesto intermedio 3:
Se preparó de forma análoga al compuesto intermedio 2, pero utilizando tricloruro de hierro (0,25 eq.), carbón activado (100 mg por mmol de sustrato) y N,N-dimetil hidrazina (5 eq.) en lugar de negro de paladio y gas de hidrógeno en la etapa 6.
Compuesto intermedio 4:

Se preparó de forma análoga al compuesto intermedio 2, pero utilizando bromometil-ciclopropano (1,5 eq.) en lugar de yodometano en la etapa 3.
Compuesto intermedio 5:

Se preparó de forma análoga al compuesto intermedio 2, pero utilizando 5-bromo-4-metoxi-pirimidina (1 eq.) en lugar de 3-bromo-2-metoxi-piridina en la etapa 1. Además, se utilizaron tricloruro de hierro (0,25 eq.), carbón activado (100 mg por mmol de sustrato) y N,N-dimetil hidrazina (5 eq.) en lugar de negro de paladio y gas de hidrógeno en la etapa 6.
Compuesto intermedio 6:

Se preparó de forma análoga al compuesto intermedio 2, pero utilizando 3-bromo-2-metoxi-4-metilpiridina (1 eq.) en lugar de 3-bromo-2-metoxi-piridina en la etapa 1. Además, se utilizaron tricloruro de hierro (0,25 eq.), carbón activado (100 mg por mmol de sustrato) y N,N-dimetil hidrazina (5 eq.) en lugar de negro de paladio y gas de hidrógeno en la etapa 6. Compuesto intermedio 7:

Se preparó de forma análoga al compuesto intermedio 2, pero utilizando 4-bromo-5H-furan-2-ona (1 eq.) en lugar de 3-bromo-2-metoxi-piridina en la etapa 1. Adicionalmente, se omitieron la etapa 2, la etapa 3 y la etapa 5.
Compuesto intermedio 8 -CF3 :

En un recipiente de reacción de vidrio equipado con un tapón de rosca recubierto de Teflón se resuspendió el compuesto intermedio 1 -CF3 (1 eq.), 4,4,4',4',5,5,5',5'-octametil-2,2'-bi(1,3,2-dioxaborolano) (1,5 eq.) y carbonato de cesio (1,5 eq.) en acetonitrilo (0,1 M). A continuación, la mezcla de reacción se desoxigenó a fondo mediante purgado subsuperficial con nitrógeno durante 30 min. Finalmente, se añadieron acetato de paladio(II) (0,1 eq.), trifenilfosfina (0,1 eq.) y yoduro de cobre (0,4 eq.) en una porción rápida. La mezcla resultante se agitó a TA durante 18 h. Después, la mezcla de reacción sin procesar se diluyó con EtOAc y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó a continuación sobre MgSO4, se filtró y el filtrado se concentró al vacío. La purificación del producto sin procesar obtenido de este modo mediante cromatografía en columna (SiO2 , elución en gradiente, Hex ^ 2:3 (v/v) Hex: EtOAc) permitió obtener el producto deseado como un aceite viscoso de color amarillo (rendimiento del 91 %).
Compuesto intermedio 9:

Se preparó de forma análoga al compuesto intermedio 2, pero utilizando amina 1 (1,0 eq.) en lugar de 3-aminopropan-1-ol en la etapa 4.
Compuesto intermedio 10:

Se preparó de forma análoga al compuesto intermedio 2, pero utilizando amina 2 (1,0 eq.) en lugar de 3-aminopropan-1-ol en la etapa 4.
Compuesto intermedio 11: Bromhidrato de 3-(2-amino-1-but-3-enil-1H-benzoimidazol-5-il)-1-metil-pirrolidin-2-ona

Etapa 1: Éster metílico del ácido 4-fluorofenilacético. A una solución de ácido 4-fluorofenilacético en MeOH (400 ml) a 0 °C se añadió gota a gota SOCh (92 g, 773 mmol, 56 ml). La reacción se agitó a 60 °C bajo atmósfera de N2 durante 1 hora. La TLC (EtOAc:éter de petróleo, 1:1, Rf del material de partida = 0,1, Rf del producto = 0,9) mostró que la reacción había finalizado. La mezcla de reacción se concentró a presión reducida para eliminar el MeOH y el SOCh. El residuo se diluyó con solución de NaHCO3 (300 ml) saturada y se extrajo con EtOAc (2 * 300 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron para obtener el compuesto del título (45 g; sin procesar) como un aceite incoloro. RMN 1H (400 MHz, CDCla,) 57,29-7,25 (m, 2H), 7,06-7,01 (m 2H), 3,72 (s, 3H), 3,62 (s, 2H).
Etapa 2: Éster metílico del ácido 3-ciano-2-(4-fuorofenil)-propiónico. A una solución de éster metílico del ácido 4-fluorofe nilacético (20 g, 119 mmol) en THF (300 ml) a -70 °C se añadió gota a gota LDA (2 M, 81 ml). Tras agitar durante 1 h, se añadió una solución de 2-bromoacetonitrilo (14,4 g, 120 mmol, 8 ml) en THF anhidro (50 ml) y la reacción se agitó a -70 °C bajo atmósfera de N2 durante 1 h. La TLC (éter de petróleo:EtOAc, 3:1, Rf del material de partida = 0,7, Rf del producto = 0,4) mostró que la reacción había finalizado. La reacción se detuvo con solución de NH4Cl saturada y, a continuación, se extrajo con EtOAc (2 * 300 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 * 100 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron al vacío. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (éter de petróleo:EtOAc, gradiente de 20:1 a 3:1) permitió obtener el compuesto del título (19 g, 77 %) como un aceite incoloro. RMN 1H (400 MHz, CDCb) 57,30-7,03 (m, 2H), 7,12-7,07 (m, 2H), 3,98-3,93 (m, 1H) 3,75 (s, 3H), 3,09-3,00 (m, 1H) 2,86-2,80 (m, 1H).
Etapa 3: 3-(4-Fluorofenil)-pirrolidin-2-ona. A una solución de éster metílico del ácido 3-ciano-2-(4-fuorofenil)-propiónico (7 g, 33,8 mmol), NH3.H2O (10,9 g, 312 mmol, 12 ml) en EtOH (120 ml) se añadió Raney-Ni (1,2 g) bajo atmósfera de nitrógeno. La suspensión se desgaseó al vacío y se purgó con H2 varias veces. La mezcla se agitó con H2 (50 psi) a 40 °C durante 16 h. La TLC (DCM:MeOH, 20:1, Rf del material de partida = 0,9, Rf del producto = 0,3) mostró que la reacción había finalizado. La mezcla de reacción se filtró a través de un lecho de celite. El lecho se lavó con EtOH (3 * 20 ml)y el filtrado se concentró a presión reducida para obtener el compuesto del título (12 g, rendimiento del 49,6 %) como un sólido de color blanco. RMN 1H (400 MHz, D M S O d) 57,82 (s, 1H), 7,30-7,26 (m, 2H), 7,16-7,12 (m, 2H) 3,55 (t, J = 9,2 Hz, 1H), 3,32-3,27 (m, 2H), 2,49-2,46 (m, 1H), 2,09-2,02 (m, 1H).
Etapa 4: 3-(4-Fluorofenil)-1-metilpirrolidin-2-ona. A una solución de 3-(4-fluorofenil)-pirrolidin-2-ona (10 g; 55,8 mmol), KOH (3,44 g; 61,4 mmol) y bromuro de tetrabutilamonio (1,6 g; 5,6 mmol) en THF (300 ml) a 0 °C se añadió gota a gota Mel (8,48 g; 59,8 mmol; 3,72 ml) en THF (20 ml). La reacción se agitó a 28 °C bajo atmósfera de N2 durante 3 horas. La TLC (éter de petróleo, EtOAc, 1:1, Rf del material de partida = 0,1, Rf del producto = 0,2) mostró que la reacción había finalizado. La mezcla de reacción se concentró a presión reducida para eliminar el THF. A continuación, se diluyó con agua (200 ml) y se extrajo con EtOAc (2 * 400 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (éter de petróleo:EtOAc, gradiente de 20:1 a 3:1) proporcionó el compuesto del título (7,5 g, rendimiento del 70 %) como un sólido de color blanco. CL/EM: TR = 0,619 min, m/z = 194,1 (M+H)+, RMN 1H (400 MHz, CDCla) 57,25-7,22 (m, 2H), 7,06-7,02 (m, 2H), 3,66 (t, J = 9,2Hz, 1H), 3,49-3,44 (m, 2H), 2,96 (s, 3H), 2,55-2,53 (m, 1H), 2,14-2,06 (m, 1H).
Etapa 5: 3-(4-Fluoro-3-nitrofenil)-1-metilpirrolidin-2-ona. A una solución de 3-(4-fluorofenil)-1 -metilpirrolidin-2-ona (7,5 g; 38,8 mmol) en solución de H2SO4 conc. (80 ml) a -10 °C se añadió gota a gota ácido nítrico (4,2 g; 65 %; 43,3 mmol; 3 ml). La reacción se agitó a 0 °C durante 20 minutos. La TLC (éter de petróleo, EtOAc, 1:1, Rf del material de partida = 0,2, Rf del producto = 0,1) mostró que la reacción había finalizado. La reacción se detuvo mediante la adición lenta de agua con hielo y a continuación se extrajo con EtOAc (2 * 400 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron para obtener el compuesto del título (8,9 g; rendimiento del 96 %) como un sólido de color blanco. CL/EM: TR = 0,631 min, m/z = 239,1 (M+H)+, RMN 1H (400 MHz, CDCb) 58,00-7,98 (m, 1H), 7,62-7,59 (m, 1H), 7,30-7,25 (m, 1H), 3,73 (t, J = 5,6 Hz, 1H) 3,51-3,48 (m, 2H), 2,96 (s, 3H), 2,63-2,57 (m, 1H), 2,18-2,13 (m, 1H).
Etapa 6: 3-(4-But-3-enilamino-3-nitrofenil)-1-metilpirrolidin-2-ona. A una solución de clorhidrato de but-3-enilamina (2,4 g; 22,3 mmol) y K2CO3 (6 g; 43,4 mmol) en DMF (40 ml), se añadió el compuesto 3-(4-fluoro-3-nitrofenil)-1-metilpirrolidin-2-ona (3,5 g; 14,7 mmol). La reacción se agitó a 28 °C bajo atmósfera de nitrógeno durante 16 horas. La t Lc (éter de petróleo:EtOAc, 1:1, Rf del material de partida = 0,1, Rf del producto = 0,2) mostró que la reacción había finalizado. La reacción se diluyó con agua (200 ml) y se extrajo con EtOAc (2 * 200 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 * 200 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron para obtener el compuesto del título (5,5 g; sin procesar) como un aceite de color amarillo. RMN 1H (400 MHz, CDCb) 58,05-8,015 (m, 2H), 7,428 7,4 (m, 1H), 6,852 (d, J = 8,8, 1H), 5,874-5,788 (m, 1H) 5,194-5,154 (m, 2H) 3,58-3,348 (m, 5H) 2,927 (s, 3H) 2,508-2,426 (m, 3H) 2,148-2,116 (m, 1H), CL/EM: TR = 0,790 min, m/z = 290,1(+H)+.
Etapa 7: 3-(4-But-3-enilamino-3-aminofenil)-1-metilpirrolidin-2-ona. A una solución de 3-(4-but-3-enilamino-3-nitrofenil)-1-metilpirrolidin-2-ona (5,5 g; 19 mmol) en una solución acuosa de NH4Cl (40 ml) y MeOH (40 ml) a 50 °C se añadieron varias secciones de Fe (5,5 g; 98,5 mmol). La reacción se agitó a 90 °C bajo atmósfera de nitrógeno durante 3 horas. La TLC (DCM:MeOH, 20:1, Rf del material de partida = 0,9, Rf del producto = 0,5) mostró que la reacción había finalizado. La mezcla de reacción se filtró a través de un lecho de celite. El lecho se lavó con MeOH (3 * 150 ml) y el filtrado se concentró a presión reducida para eliminar el MeOH. El residuo se diluyó con agua (200 ml) y se extrajo con EtOAc (2 * 300 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (éter de petróleo:EtOAc, 1:1, a continuación DCM:MeOH, 100:1) proporcionó el compuesto del título (3,6 g, rendimiento del 73 %) como un aceite de color rojo. RMN 1H (400 MHz, CDCb) 5 6,695-6,70 (m, 1H), 6,614 (d, J = 8Hz, 2H), 5,905-5,863 (m, 1H) 5,185-5,095 (m, 2H), 3,540-3,374 (m, 3H) 3,16 (t, J = 6,4Hz 2H) 2,925 (s, 3H) 2,451-2,415 (m, 3H) 2,095-2,053 (m, 1H), CL/EM: TR = 0,561 min, m/z = 260,3(+H)+.
Etapa 8: Bromhidrato de 3-(2-amino-1-but-3-enil-1H-benzoimidazol-5-il)-1-metil-pirrolidin-2-ona. Una solución de 3-(4-but-3-enilamino-3-aminofenil)-1-metilpirrolidin-2-ona (3,6 g, 13,9 mmol) y BrCN (1,47 g, 13,9 mmol) en EtOH (35 ml) se agitó a 28 °C bajo atmósfera de nitrógeno durante 3 h. La TLC (DCM:MeOH, 10:1, Rf del material de partida = 0,7, Rf del producto = 0,1) mostró que la reacción había finalizado. La reacción se concentró a presión reducida para obtener el compuesto del título (5 g; rendimiento del 98,6 %) como un sólido de color amarillo. Rm N 1H (400 MHz, cD cb) 57,542 (t,
J = 7,2Hz 1H), 7,224 (t, J = 4Hz 2H), 5,710-5,642 (m, 1H) 4,972-4,924 (m 2H), 4,078-3,765 (m, 3H) 3,578-3,542 (m, 2H) 2,992 (s, 3H) 2,608-2,599 (m, 1H) 2,408-2,356 (m, 2H) 2,215-2,182 (m, 1H), CL/EM: TR = 1,767 min, m/z = 285(+H)+.
Compuesto intermedio 12: 3-(2-Amino-1-but-3-enil-1H-benzoimidazol-5-il)-1-metil-piperidin-2-ona

El compuesto del título se preparó de forma análoga al compuesto intermedio 11 utilizando 3-bromopropanonitrilo en lugar de bromoacetonitrilo en la etapa 2. CL/EM: TR = 1,187 min, m/z = 299,2 (M+H)+, RMN 1H (400 MHz, CDCb) 57,17 (s, 1H) 6,955 (d, J = 8,4Hz 1H) 6,864-6,839 (m, 1H) 5,837-5,734 (m, 1H) 5,112-5,056 (m, 2H) 3,808-3,391 (m, 5H) 3,055 (s 3H) 2,449-2,396 (m, 2H) 2,04-1,806 (s, 4H).
Compuesto intermedio 13: Bromhidrato de 3-(1-alil-2-amino-1H-benzoimidazol-5-il)-1-metil-pirrolidin-2-ona

El compuesto del título se preparó de forma análoga al compuesto intermedio 11 utilizando prop-2-en-1-amina en lugar de clorhidrato de but-3-enilamina en la etapa 6. CL/EM: TR = 1,187 min, m/z = 299,2 (M+H)+, RMN 1H (400 MHz, CDCla) 5 7,17 (s, 1H) 6,955 (d, J = 8,4Hz 1H) 6,864-6,839 (m, 1H) 5,837-5,734 (m, 1H) 5,112-5,056 (m, 2H) 3,808-3,391 (m, 5H) 3,055 (s 3H) 2,449-2,396 (m, 2H) 2,04-1,806 (s, 4H). CL/EM: TR = 0,510 min, m/z = 271,1(M+H)+, RMN 1H: 400 MHz, CDCl3, 57,548-7,527 (m, 3H) 7,32 (s,1H) 7,221-7,201 (m, 1H) 5,743-5,674 (m, 1H) 5,207 (t, J = 16Hz 2H) 4,635 (t, J = 5,2Hz 1H) 4,635 (t, J = 11,6Hz 1H) 3,823 (t, J = 9,6Hz 1H) 3,576-3,54 (m, 1H) 2,998 (s, 3H) 2,623-2,581 (m, 1H) 2,197 2,139 (m, 1H).
Compuesto intermedio 14: 3-(2-Amino-1-{3-[(terc-butildimetilsilil)oxi]propil}-1H-1,3-benzodiazol-5-il)-1-metilpirrolidin-2-ona

Etapa 1: 3-(4-Fluorofenil)-1-metilpirrolidin-2-ona. Se añadió terc-butóxido de sodio (1,597 g; 14,23 mmol; 1,50 eq.) a una solución de 3-(4-fluorofenil)pirrolidin-2-ona (Otava Building Blocks, 2 g; 9,49 mmol; 1,00 eq.; 85 %) en DMF (20 ml) en varios lotes a 0 °C. Después de 1 h, se añadió gota a gota yodometano (1,481 g; 10,43 mmol; 1,10 eq.). La mezcla de reacción se agitó durante 2 h a 0 °C y se detuvo mediante la adición de 100 ml de agua. La solución resultante se extrajo con EtOAc (3 * 100 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 * 50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (acetato de etilo:éter de petróleo; 3:1) permitió obtener el compuesto del título como un aceite de color amarillo (1,3 g; 64 %). ESI+: M+1: 193,9.
Etapa 2: 3-(4-Fluoro-3-nitrofenil)-1-metilpirrolidin-2-ona. Se añadió ácido nítrico (2 ml; 44,59 mmol; 7,36 eq.) gota a gota a una solución de 3-(4-fluorofenil)-1 -metilpirrolidin-2-ona (1,3 g; 6,06 mmol; 1,00 eq.; 90 %) en ácido sulfúrico (13 ml) a 0 °C. La solución resultante se agitó durante 2 h a 0 °C en un baño de agua con hielo y se detuvo mediante la adición de 50 ml de agua con hielo. A continuación, se extrajo con EtOAc (2 * 100 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 * 30 ml), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para obtener el compuesto del título como un sólido de color amarillo (1,2 g; 75 %). ESI+: M+1: 238,9.
Etapa 3: 3-{4-[(3-Hidroxipropil)amino]-3-nitrofenil}-1-metilpirrolidin-2-ona. Una solución de 3-(4-fluoro-3-nitrofenil)-1-metilpirrolidin-2-ona (1,2 g; 4,53 mmol; 1,00 eq.; 90 %), carbonato de potasio (1,88 g; 13,60 mmol; 3,00 eq.) y 3-aminopropan-1-ol (681 mg; 9,07 mmol; 2,00 eq.) en DMF (10 ml) se agitó durante 1 h a 70 °C. A continuación, la mezcla de reacción se detuvo mediante la adición de 100 ml de agua y se extrajo con EtOAc (2 * 100 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 * 50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron. La purificación mediante cromatografía ultrarrápida sobre sílice (DCM:MeOH, 98:2) permitió obtener el compuesto del título como un aceite de color rojo (1,25; 89 %). ESI+: M+1: 294,1.
Etapa 4: 3-{3-Amino-4-[(3-hidroxipropil)amino]fenil}-1-metilpirrolidin-2-ona. Se añadió Raney Ni (20 mg; 0,23 mmol; 2,40 eq.) a una solución purgada con nitrógeno de 3-[4-[(3-hidroxipropil)amino]-3-nitrofenil]-1-metilpirrolidin-2-ona (30 mg; 0,10 mmol; 1,00 eq.; 95 %) en MeOH (3 ml). A continuación, la mezcla de reacción se hidrogenó (1 atm) durante 3 h a TA. Los sólidos se recogieron por filtración y la mezcla resultante se concentró al vacío para obtener el compuesto del título como un aceite de color morado (880 mg, 86 %). ESI+: M+1: 264,1.
Etapa 5: 3-[2-Amino-1-(3-hidroxipropil)-1H-1,3-benzodiazol-5-il]-1-metilpirrolidin-2-ona. Una solución de 3-[3-amino-4-[(3-hidroxipropil)amino]fenil]-1 -metilpirrolidin-2-ona (830 mg; 2,63 mmol; 1,00 eq.) y BrCN (335 mg; 3,16 mmol; 1,20 eq.) en etanol (10 ml) se agitó durante 2 h a 50 °C. A continuación, se concentró al vacío para obtener el compuesto del título como un sólido de color morado (1,1 g; 100 %). ESI+: M+1: 289,1.
Etapa 6: 3-(2-Amino-1-{3-[(terc-butildimetilsilil)oxi]propil}-1H-1,3-benzodiazol-5-il)-1-metilpirrolidin-2-ona. Una solución de terc-butil(cloro)dimetilsilano (566 mg; 3,76 mmol; 1,20 eq.) en DCM (2 mi) se añadió gota a gota a otra solución de 3-[2-amino-1-(3-hidroxipropil)-1H-1,3-benzodiazol-5-il]-1-metilpirrolidin-2-ona (1,29 g; 3,13 mmol; 1,00 eq.) y 1H-imidazol (469 mg; 6,89 mmol; 2,20 eq.) en DCM (50 ml) mantenido a 0 °C. La solución resultante se agitó durante 4 h a temperatura ambiente. A continuación, los sólidos se recogieron por filtración y el filtrado se concentró al vacío. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (DCM:MeOH, 97,3:2,7) permitió obtener el compuesto del título como un sólido de color marrón (1,1 g; 84 %). ESI+: M+1: 403,3.
Compuesto intermedio 15: 4-[2-Amino-1-(3-hidroxipropil)-1H-1,3-benzodiazol-5-il]-1,3,3-trimetilpirrolidin-2-ona

Etapa 1: 3-(4-Fluorofenil)-2,2-dimetil-4-nitrobutanoato de metilo. Se añadió gota a gota n-BuLi (2,5 M en THF, 6,765 ml; 16,37 mmol; 1,60 eq.) a una solución de di-isopropilamina (1,744 g; 16,37 mmol; 1,60 eq.) en THF (40 ml) que se mantuvo bajo atmósfera de nitrógeno a -60 °C. La solución resultante se agitó durante 0,5 h a -30 °C antes de la adición de 2-metilpropanoato de metilo (1,65 g; 15,35 mmol; 1,50 eq.). A continuación, la mezcla de reacción se agitó durante 1 h a -40 °C y se añadió gota a gota una solución de 1-fluoro-4-[(E)-2-nitroetenil]benceno (1,900 g, 10,23 mmol, 1,00 eq.) en THF (10 ml) mientras se agitaba a -60 °C. La temperatura se llevó de nuevo a TA y la mezcla se agitó durante 1 h antes de la adición de una solución de NH4Cl saturada (50 ml). La solución resultante se extrajo con EtOAc (2 * 50 ml). Las fases orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (EtOAc:éter de petróleo; 1:10) permitió obtener el compuesto del título como un aceite de color amarillo (2 g; 65 %). RMN 1H (300 MHz, CDCl3, ppm) 5: 7,24 - 7,10 (m, 2H), 7,07 - 6,94 (m, 2H), 4,92 (dd, J = 13,1, 11,3 Hz, 1H), 4,77 (dd, J = 13,1, 4,2 Hz, 1H), 3,76 (dd, J = 11,3, 4,2 Hz, 1H), 3,69 (s, 3H), 1,18 (d, J = 13,5 Hz, 6H).
Etapa 2: 4-(4-Fluorofenil)-3,3-dimetilpirrolidin-2-ona. Una suspensión desgaseada de 3-(4-fluorofenil)-2,2-dimetil-4-nitrobutanoato de metilo (2,0 g; 6,68 mmol; 1,00 eq.) y Raney Ni (603 mg; 6,69 mmol; 1,00 eq.) en EtOH (60 ml) se hidrogenó a 50 °C a una presión de hidrógeno de 15 atm durante toda la noche. Después se filtró a través de un relleno de celite y el filtrado se concentró a presión reducida para obtener el compuesto del título como un sólido de color amarillo (2,0 g; cuantitativo). ESI+: M+1: 208,0.
Etapa 3: 4-(4-Fluorofenil)-1,3,3-trimetilpirrolidin-2-ona. Se añadió yodometano (1,557 g; 10,42 mmol; 1,50 eq.) a una suspensión de hidruro sódico (361 mg; 9,03 mmol; 1,30 eq.; 60 %) y 4-(4-fluorofenil)-3,3-dimetilpirrolidin-2-ona (1,6 g; 6,95 mmol; 1,00 eq.) en THF (50 ml) que se mantuvo bajo atmósfera de nitrógeno y a °C. La solución resultante se agitó durante 30 min a 0 °C, se dejó atemperar a TA y se agitó durante toda la noche. A continuación, la reacción se detuvo con agua (30 ml) y se extrajo con EtOAc (3 * 25 ml). Las fases orgánicas combinadas se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron al vacío. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (DCM:MeOH, 10:1) permitió obtener el compuesto del título como un aceite de color amarillo (1,5 g; 88 %). RMN 1H (300MHz,CDCl3, ppm) 57,22 - 7,11 (m, 2H), 7,09 - 6,96 (m, 2H), 3,65 - 3,46 (m, 2H), 3,22 (t, J = 8,1 Hz, 1H), 2,95 (s, 3H), 1,21 (s, 3H), 0,73 (s, 3H).
Etapa 4 :4-(4-Fluoro-3-nitrofenil)-1,3,3-trimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 2 del compuesto intermedio 14 pero empezando a partir de 4-(4-fluorofenil)-1,3,3-trimetilpirrolidin-2- ona (1,500 g; 6,10 mmol) como un sólido de color amarillo (1,8 g; 100 %). ESI+: M+1: 226,9.
Etapa 5: 4-{4-[(3-Hidroxipropil)amino]-3-nitrofenil}-1,3,3-trimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 3 del compuesto intermedio 14 pero empezando a partir de 4-(4-fluoro-3- nitrofenil)-1,3,3-trimetilpirrolidin-2-ona (1,70 g; 5,75 mmol; 1,00 eq.) como un sólido de color rojo (2 g; 97 %). e S|+: M+1: 322,4.
Etapa 6: 4-{3-Amino-4-[(3-hidroxipropil)amino]fenil}-1,3,3-trimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 4 del compuesto intermedio 14 pero empezando a partir de 4-[4-[(3-hidroxipropil)amino]-3-nitrofenil]-1,3,3-trimetilpirrolidin-2-ona (900 mg; 1,00 eq.) como un aceite de color marrón (800 mg; 98 %). ESI+: M+1: 292,4.
Etapa 7: 4-[2-Amino-1-(3-hidroxipropil)-1H-1,3-benzodiazol-5-il]-1,3,3-trimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 5 del compuesto intermedio 14 pero empezando a partir de 4-[3-amino-4-[(3-hidroxipropil)amino]fenil]-1,3,3-trimetilpirrolidin-2-ona (1,7 g; 5,25 mmol; 1,00 eq.) como un sólido de color marrón (1,5 g; 81 %). ESI+: M+1: 292,4.
Compuesto intermedio 16: 2-{2-Amino-1-[3-(benciloxi)-2-fluoro-2-metilpropil]-1H-1,3-benzodiazol-5-il}-4-metilmorfolin-3-ona

Etapa 1: Metil(2-{[tris(pmpan-2-il)sWl]oxi}etil)amina. Se añadió gota a gota 2,6-dimetilpiridina (14 g; 131 mmol; 1,00 eq.) a una solución de 2-(metilamino)etan-1-ol (9,8 g; 130,5 mmol; 1,00 eq.) y tris(propan-2-il)silil trifluorometansulfonato (100 g; 326,35 mmol; 2,50 eq.) en DCM (300 ml) que se mantuvo bajo atmósfera de nitrógeno a 0 °C. La mezcla de reacción se dejó atemperar a TA y se agitó durante 5 h. La reacción se detuvo mediante la adición de agua con hielo (100 mi) y se extrajo con DCM (3 * 30 ml). Las fases orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para obtener el compuesto del título como un sólido de color amarillo (27 g, 72 %).
Etapa 2: N-{2-[(terc-Butildimetilsilil)oxi]etil}-2-(4-fluorofenil)-2-hidroxi-N-metilacetamida. Una mezcla de [2-[(tercbutildimetilsilil)oxi]etil](metil)amina (3,4 g; 16,2 mmol; 1,00 eq.), ácido 2-(4-fluorofenil)-2-hidroxiacético (4,1 g; 24,10 mmol; 1,49 eq.), Ha Tu (7,4 g; 19,46 mmol; 1,20 eq.) y DIEA (3,2 g; 24,76 mmol; 1,53 eq.) en DMF (10 ml) se agitó durante toda la noche a TA. A continuación, la mezcla de reacción se detuvo mediante la adición de agua (50 ml) y se extrajo con EtOAc (3 * 30 ml). Las fases orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (EtOAc:éter de petróleo, gradiente de 1:10 a 7:3) proporcionó el compuesto del título como un aceite de color amarillo (2,0 g, 33 %).
Etapa 3: Metanosulfonato de ({2-[(terc-butildimetilsilil)oxi]etil}(metil)carbamoil)(4-fluorofenil)metilo. Se añadió gota a gota MsCl (740 mg; 6,46 mmol; 1,20 eq.) a una solución de N-[2-[(terc-butildimetilsilil)oxi]etil]-2-(4-fl uorofenil)-2-hidroxi-N-metilacetamida (2,3 g; 5,39 mmol; 1,00 eq.) y TEA (820 mg; 8,10 mmol; 1,50 eq.) en d Cm (60 ml) que se mantuvo bajo atmósfera de nitrógeno y a °C. La mezcla de reacción se agitó durante 1 h a °C y se detuvo mediante la adición de agua (100 ml). Se extrajo con EtOAc (3 * 40 ml) y las fases orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para obtener el compuesto del título como un aceite de color amarillo (2,5 g; 88 %).
Etapa 4: 2-(4-Fluorofenil)-4-metilmorfolin-3-ona. Una solución de metanosulfonato de ([2-[(tercbutildimetilsilil)oxi]etil](metil)carbamoil)(4-fluorofenil)metilo (2,5 g; 4,77 mmol; 1,00 eq.) y TBAF (5 g; 19,12 mmol; 4,01 eq.) en THF (60 ml) se agitó durante 16 h a TA. A continuación, la mezcla de reacción se detuvo mediante la adición de agua (60 ml) y se extrajo con EtOAc (3 * 40 ml). Las fases orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para proporcionar el compuesto del título como un sólido de color amarillo (1 g, 83 %). ESI+: M+1: 210,0.
Etapa 5: 2-(4-Fluoro-3-nitrofenil)-4-metilmorfolin-3-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 2 del compuesto intermedio 14 pero empezando a partir de 2-(4-fluorofenil)-4-metilmorfolin-3-ona (1 g; 3,93 mmol; 1,00 eq.) como un sólido de color amarillo (500 mg; 47 %). ESI+: M+1: 255,0.
Etapa 6: 2-(4-{[3-(Bendloxi)-2-fíuoro-2-metilpropil]amino}-3-nitrofenil)-4-metilmorfolin-3-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 3 del compuesto intermedio 14 pero empezando a partir de 2-(4-fluoro-3-nitrofenil)-4-metilmorfolin-3-ona (350 mg, 1,29 mmol, 1,00 eq.) y 3-(benciloxi)-2-fluoro-2-metilpropan-1-amina (amina intermedia 2, 306 mg; 1,55 mmol; 1,20 eq.) como un aceite de color amarillo (460 mg; 72 %). ESI+: M+1: 432,2.
Etapa 7: 2-(3-Amino-4-{[3-(benciloxi)-2-fluoro-2-metilpropil]amino}fenil)-4-metilmorfolin-3-ona. Una suspensión desgaseado de 2-(4-[[3-(benciloxi)-2-fluoro-2-metilpropil]amino]-3-nitrofenil)-4-metilmorfolin-3-ona (450 mg; 0,91 mmol; 1,00 eq.; 87,3 %) y paladio carbono (15 mg; 0,01 mmol; 0,01 eq.; 10 %) en MeOH (10 ml) se hidrogenó a temperatura ambiente con una presión de hidrógeno de 30 psi durante 1 h. A continuación se filtró a través de un relleno de celite y el filtrado se concentró para obtener el compuesto del título como un sólido de color rojo (340 mg, 83 %). ESI+: M+1: 402,2.
Etapa 8: 2-{2-Amino-1-[3-(benciloxi)-2-fluoro-2-metilpropil]-1H-1,3-benzodiazol-5-il}-4-metilmorfolin-3-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 5 del compuesto intermedio 14 pero empezando a partir de 2-(3-amino-4-[[3-(benciloxi)-2-fluoro-2-metilpropil]amino]fenil)-4-metilmorfolin-3-ona (330 mg; 0,73 mmol; 1,00 eq.) como un sólido de color rojo (255 mg; 55 %). ESI+: M+1: 427,1.
Compuesto intermedio 17: 3-(2-Amino-1 -{3-[(terc-butildimetilsilil)oxi]propil}-1 H-1,3-benzodiazol-5-il)-1,3-metilpirrolidi n-2-ona

Etapa 1: 3-(4-Fluorofenil)-1,3-dimetilpirrolidin-2-ona. Se añadió terc-butóxido de sodio (1,63 g; 14,24 mmol; 1,50 eq.) en varios lotes a una solución de 3-(4-fluorofenil)pirrolidin-2-ona (etapa 1 del compuesto intermedio 11,2 g; 9,49 mmol; 1,00 eq.) en DMF (20 ml) que se mantuvo a 0 °C. A continuación, la mezcla de reacción se agitó a 0 °C durante 60 min antes de la adición gota a gota de yodometano (1,5 g; 10,36 mmol; 1,09 eq.). La mezcla de reacción se agitó durante 2 h más a 0 °C y se detuvo con agua (100 ml). La solución resultante se extrajo con acetato de etilo (3 * 100 ml) y las fases orgánicas combinadas se lavaron con salmuera (2 * 50 ml), se secó sobre sulfato sódico anhidro, se filtró y se concentró. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (acetato de etilo:éter de petróleo; 3:1) permitió obtener el compuesto del título como un aceite de color amarillo (500 mg; 23 %). ESI+ (M+1): 208,2.
Etapa 2: 3-(4-Fluoro-3-nitrofenil)-1,3-dimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 2 del compuesto intermedio 14 pero empezando a partir de 3-(4-fluorofenil)-1,3-dimetilpi rrolidin-2-ona (480 mg; 2,08 mmol; 1,00 eq.) como un sólido de color amarillo (580 mg; 99 %). Es I+ (M+1): 253,2.
Etapa 3: 3-{4-[(3-Hidroxipropil)amino]-3-nitrofenil}-1,3-dimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 3 del compuesto intermedio 11 pero empezando a partir de 3-(4-fluoro-3-nitrofenil)-1,3-dimetilpirrolidin-2-ona (560 mg; 2,00 mmol; 1,00 eq.) y 3-aminopropan-1-ol (307 mg; 4,01 mmol; 2,00 eq.) como un sólido de color naranja (690 mg; 101 %). ESI+ (M+1): 308,2.
Etapa 4: 3-[4-({3-[(terc-Butildimetilsilil)oxi]propil}amino)-3-nitrofenil]-1,3-dimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 6 del compuesto intermedio 14 pero empezando a partir de 3-[4-[(3-hidroxipropil)amino]-3-nitrofenil]-1,3-dimetilpirrolidin-2-ona (670 mg; 1,96 mmol; 1,00 eq.) como un sólido de color marrón ( 9 80 mg, cuantitativo). ESI+ (M+1): 422,1.
Etapa 5: 3-[3-Amino-4-({3-[(terc-butildimetilsilil)oxi]propil}amino)fenil]-1,3-dimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 4 del compuesto intermedio 14 pero empezando a partir de 3-[4-([3-[(terc-butildimetilsilil)oxi]propil]amino)-3-nitrofenil]-1,3-dimetilpirrolidin-2-ona (950 mg; 2,03 mmol; 1,00 eq.) como un aceite de color amarillo (840 mg; 95 %). ESI+ (M+1): 392,3.
Etapa 6: 3-(2-Amino-1-{3-[(terc-butildimetilsilil)oxi]propil}-1H-1,3-benzodiazol-5-il)-1,3-dimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 6 del compuesto intermedio 14 pero empezando a partir de 3-[3-amino-4-([3-[(terc-butildimetilsilil)oxi]propil]amino)fenil]-1,3-dimetilpirrolidin-2-ona (820 mg; 1,88 mmol; 1,00 eq.) como un sólido de color beis (850 mg; 98 %). e S i+ (M+1): 417,2.
Compuesto intermedio 18: 5-[2-Amino-1-(3-hidroxipropil)-1H-1,3-benzodiazol-5-il]-1,5-dimetilpirrolidin-2-ona

Etapa 1: 4-(4-Fluorofenil)-4-nitropentanoato de metilo. Una solución de DBU (639 mg, 3,99 mmol, 0,50 eq.) en MeCN (2 ml) se añadió gota a gota a una solución de 1-fluoro-4-(1-nitroetil)benceno (Aurora Building Blocks, 1,5 g; 7,98 mmol; 1,00 eq.) y prop-2-enoato de metilo (1,446 g; 15,96 mmol; 2,00 eq.) en MeCN (40 ml) que se mantuvo bajo atmósfera de nitrógeno a 0 °C. La mezcla de reacción se agitó durante 3 h a TA, se detuvo con solución de NH4Cl saturada (50 ml) y se extrajo con EtOAc (3 * 50 ml). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (éter de petróleo:EtOAc, gradiente de 100:0 a 90:10) proporcionó el compuesto del título como un aceite de color amarillo (1,8 g; 80 %). RMN 1H (400MHz, CDCl3, ppm) 57,42-7,35 (m, 2H), 7,12-7,03 (m, 2H), 3,67 (s, 3H), 2,84-2,72 (m, 1H), 2,72 2,61 (m, 1H), 2,34-2,26 (m, 2H), 1,95 (s, 3H).
Etapa 2: 5-(4-Fluorofenil)-5-metilpirrolidin-2-ona. Una solución de 4-(4-fluorofenil)-4-nitropentanoato de metilo (8,0 g; 28,2 mmol; 1,00 eq.), en MeOH (150 ml) se purgó con nitrógeno antes de la adición de Raney-Ni (2,54 g; 28,2 mmol; 1,00 eq.). A continuación, la mezcla de reacción resultante se hidrogenó a presión atmosférica a Ta durante 16 h. Después se filtró a través de un lecho de celite y el filtrado se concentró a presión reducida. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (DCM:MeOH, gradiente de 100:0 a 90:10) proporcionó el compuesto del título como un sólido incoloro (5 g; 83 %). RMN 1H (300MHz,CDCl3, ppm) 57,37 - 7,31 (m, 2H), 7,09 - 7,01 (m, 2H), 6,94 (s, 1H), 2,51 -2.35 (m, 2H), 2,33 - 2,21 (m, 2H), 1,65 (s, 3H).
Etapa 3: 5-(4-Fluorofenil)-1,5-dimetilpirrolidin-2-ona. Se añadió yodometano (5,220 g; 34,94 mmol; 1,50 eq.) a una suspensión de hidruro sódico (1,21 g; 30,3 mmol; 1,30 eq.; 60 %) y 4-(4-fluorofenil)-3,3-dimetilpirrolidin-2-ona (5,0 g; 23,29 mmol; 1,3 eq.) en DMF (75 ml) que se mantuvo bajo atmósfera de nitrógeno y a 0 °C. La mezcla de reacción se dejó atemperar a TA y se agitó 1 h a Ta . A continuación, la reacción se detuvo con agua (30 ml) y se extrajo con EtOAc (3 * 20 ml). Las fases orgánicas combinadas se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron al vacío. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (DCM:MeOH, gradiente de 10:0 a 15:1) proporcionó el compuesto del título como un sólido de color amarillo (5 g, 93 %). RMN 1H (300 MHz,CDCb) 57,25 - 7,14 (m, 2h ), 7,12 - 6,99 (m, 2H), 2,66 (s, 3H), 2,53 - 2,39 (m, 2H), 2,19 - 2,01 (m, 2H), 1,67 (s, 3H). ESI+ (M+1): 208,0.
Etapa 4: 5-(4-Fluoro-3-nitrofenil)-1,5-dimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 2 del compuesto intermedio 14 pero empezando a partir de 5-(4-fluorofenil)-1,5-dimetilpi rrolidin-2-ona (5,500 g; 23,88 mmol; 1,00 eq.) como un aceite de color amarillo (6 g; 82 %). ESI+ (M+1): 253,0.
Etapa 5: 5-{4-[(3-Hidroxipropil)amino]-3-nitrofenil}-1,5-dimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 3 del compuesto intermedio 14 pero empezando a partir de 5-(4-fluoro-3-nitrofenil)-1,5-dimetilpirrolidin-2-ona (2,5 g; 8,92 mmol; 1,00 eq.) y 3-aminopropan-1-ol (1,06 g; 13,38 mmol; 1,50 eq.) como un aceite de color rojo (3 g; 98 %). ESI+ (M+1): 308,0.
Etapa 6: 5-[4-({3-[(terc-Butildimetilsilil)oxi]propil}amino)-3-nitrofenil]-1,5-dimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 6 del compuesto intermedio 14 pero empezando a partir de 5-[4-[(3-hidroxipropil)amino]-3-nitrofenil]-1,5-dimetilpirrolidin-2-ona (3,0 g; 8,78 mmol; 1,00 eq.) como un aceite de color rojo (4 g, 97 %). ESI+ (M+1): 422,1.
Etapa 7 :5-[3-Amino-4-({3-[(terc-butildimetilsilil)oxi]propil}amino)fenil]-1,5-dimetilpirrolidin-2-ona. El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 4 del compuesto intermedio 14 pero empezando a partir de 5-[4-([3-[(terc-butildimetilsilil)oxi]propil]amino)-3-nitrofenil]-1,5-dimetilpirrolidin-2-ona (3,5 g; 7,47 mmol; 1,00 eq.) como un aceite de color marrón (4 g; cuantitativo). ESI+ (M+1): 392,1.
Etapa 8: 5-[2-Amino-1-(3-hidroxipropil)-1H-1,3-benzodiazol-5-il]-1,5-dimetilpirrolidin-2-ona.
El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 5 del compuesto intermedio 14 pero empezando a partir de 5-[3-amino-4-([3-[(terc-butildimetilsilil)oxi]propil]amino)fenil]-1,5-dimetilpirrolidin-2-ona (3,200 g; 7.35 mmol; 1,00 eq.) como un aceite de color marrón (3 g; cuantitativo).
ESI+ (M+1): 303,2.
Compuesto intermedio 19: 3-{2-Amino-1-[3-(benciloxi)-2-fluoro-2-metilpropil]-1H-1,3-benzodiazol-5-il}-1-metilpiperidin-2-ona

El compuesto del título se obtuvo siguiendo un procedimiento similar al descrito para el compuesto intermedio 2 pero utilizando el compuesto intermedio amina 3 en lugar del compuesto intermedio amina 3-aminopropanol-1 de la etapa 4 como un sólido de color amarillo. ESI+ (M+1): 425,0.
Ejemplo 1: (E)-N-(1-(3-Hidroxipropil)-5-(1-metil-2-oxopiperidin-3-il)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)benzamida (1)

Etapa 1: (E)-N-(5-(1-Metil-2-oxopiperidin-3-il)-1-(3-((triisopropilsWl)oxi)propil)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)benzamida. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvieron el compuesto intermedio 2 (1 eq.), ácido 3-trifluorometil-benzoico (1,1 eq.) y HATU (1,2 eq.) en DMF (0,1 M). A continuación, a esta mezcla se añadió etil-diisopropil-amina (2,5 eq.) y la solución amarilla resultante se agitó a TA durante 14 h. La mezcla de reacción sin procesar se diluyó con EtoAc y se lavó secuencialmente con agua, solución acuosa de NaHCO3 al 10 %, solución acuosa de NH4Cl al 10 %, agua y salmuera. El extracto orgánico se secó a continuación sobre Na2SO4, se filtró y el filtrado se concentró al vacío. La purificación adicional mediante cromatografía en columna (SiO2 , elución en gradiente, 1:1 (v/v) Hex: EtOAc ^ EtOAc) permitió obtener el producto deseado como un aceite viscoso (rendimiento del 68 %).
Etapa 2: (E)-N-(1-(3-Hidroxipropil)-5-(1-metil-2-oxopiperidin-3-il)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)benzamida. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvió (E)-N-(5-(1-metil-2-oxopiperidin-3-il)-1-(3-((triisopropilsilil)oxi)propil)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)benzamida (1 eq.) procedente de la etapa previa en THF (0,1 M). A continuación, a esta mezcla se añadió fluoruro de tetrabutilamonio (3,5 eq., solución 1 M en THF) gota a gota durante un periodo de 1 min. La mezcla resultante se agitó a TA durante 3 h. La mezcla de reacción sin procesar se diluyó con EtOAc y se lavó secuencialmente con solución acuosa de NaHCO3 al 10 %, agua y salmuera. El extracto orgánico se secó a continuación sobre Na2SO4, se filtró y el filtrado se concentró al vacío. La trituración del producto sin procesar obtenido de este modo en éter y hexanos permitió obtener el producto del título como un sólido de color blanco (rendimiento del 42 %). ESI+: M+1: 475. Rm N 1H (300 MHz, CDCl3) 512,42 (s, 1H), 8,50 (d, J = 7,8 Hz, 1H), 8,46 - 8,34 (m, 1H), 7,74 (d, J = 7,8 Hz, 1H), 7,58 (t, J = 7,8 Hz, 1H), 7,31 - 7,12 (m, 3H), 4,77 - 4,70 (m, 1H), 4,46 (t, J = 6,0 Hz, 2H), 3,74 (dd, J = 8,6, 5,8 Hz, 1H), 3,57 - 3,33 (m, 4H), 3,05 (s, 3H), 2,31 - 2,14 (m, 1H), 2,10 - 1,82 (m, 5H).
Ejemplos 1a y 1b: (S,E)-N-(1-(3-Hidroxipropil)-5-(1-metil-2-oxopiperidin-3-il)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)benzamida (1a) y (R,E)-N-(1-(3-hidroxipropil)-5-(1-metil-2-oxopiperidin-3-il)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)benzamida (1b)
Los enantiómeros del ejemplo 1 se resolvieron con facilidad mediante cromatografía SFC quiral (ChiraCel OJ 10 * 250 mm, columna de 5 |jm, isocrático de MeOH al 55 % NH4OH acuosa 0,1 %, 10 mi/min, 100 Bar CO235 °C) en 1a (tiempo de retención: 2,74 min) y 1b (tiempo de retención: 7,56 min).
Ejemplo 2a: (E)-3-(Difluorometil)-N-(1-((S)-2-fluoro-3-hidroxi-2-metilpropil)-5-(1-metil-2-oxopiperidin-3-il)-1H-benzo[d]imidazol-2(3H)-iliden)benzamida (2a)

Se preparó de forma análoga al ejemplo 1, pero utilizando el compuesto intermedio 9 (1 eq.) en lugar del compuesto intermedio 2 y ácido 3-difluorometil-benzoico (1,2 eq.) en lugar de ácido 3-trifluorometil-benzoico en la etapa 1. ESI+: M+1: 489. RMN 1H (300 MHz, CDCla) 512,46 (s, 1H), 8,34 - 8,24 (m, 2H), 7,67 (d, J = 7,7 Hz, 1H), 7,56 (t, J = 7,7 Hz, 1H), 7,42 - 7,30 (m, 1H), 7,26 - 7,11 (m, 2H), 6,73 (t, J = 56,3 Hz, 1H), 5,78 - 5,72 (m, 1H), 4,70 (dd, J = 32,4, 15,3 Hz, 1H), 4,20 (dd, J = 15,2, 9,7 Hz, 1H), 3,75 (t, J = 7,3 Hz, 1H), 3,59 - 3,22 (m, 4H), 3,05 (s, 3H), 2,25 - 2,18 (m, 1H), 2,10 - 1,79 (m, 3H), 1,65 (d, J = 22,2 Hz, 3H).
Ejemplo 2b: (E)-3-(Difluorometil)-N-(1-((£)-2-fluoro-3-hidroxi-2-metilpropil)-5-(1-metil-2-oxopiperidin-3-il)-1H-benzo[d]imidazol-2(3H)-iliden)benzamida (2b)

Se preparó de forma análoga al ejemplo 1, pero utilizando el compuesto intermedio 10 (1 eq.) en lugar del compuesto intermedio 2 y ácido 3-difluorometil-benzoico (1,2 eq.) en lugar de ácido 3-trifluorometil-benzoico en la etapa 1. ESI+: M+1: 489. RMN 1H 300 MHz, CDCla) 512,48 (s, 1H), 8,34 - 8,24 (m, 2H), 7,68 (d, J = 7,7 Hz, 1H), 7,57 (t, J = 7,7 Hz, 1H), 7,43 - 7,30 (m, 1H), 7,21 - 7,15 (m, 2H), 6,73 (t, J = 56,3 Hz, 1H), 5,78 - 5,72 (m, 1H), 4,70 (dd, J = 32,1, 15,1 Hz, 1H), 4,29 -4,14 (m, 1H), 3,74 (t, J = 7,3 Hz, 1H), 3,58 - 3,30 (m, 4H), 3,06 (s, 3H), 2,30 - 2,13 (m, 1H), 2,08 - 1,77 (m, 3H), 1,70 (d, J = 22,2 Hz, 3H).
Ejemplos 3a , 3b, 3c y 3d: 3-(Difluorometil)-N-[(2E)-1-[(2R)-2-fluoro-3-hidroxi-2-metilpropil]-5-[(3R)-1-metil-2-oxopiperidin-3-il]-2,3-dihidro-1H-1,3-benzodiazol-2-iliden]benzamida (3a), 3-(difluorometil)-N-[(2E)-1-[(2s)-2-fluoro-3-hidroxi-2-metilpropil]-5-[(3S)-1-metil-2-oxopiperidin-3-il]-2,3-dihidro-1H-1,3-benzodiazol-2-iliden]benzamida (3b), 3 (difluorometil)-N-[(2E)-1-[(2S)-2-fluoro-3-hidroxi-2-metilpropil]-5-[(3R)-1-metil-2-oxopiperidin-3-il]-2,3-dihidro-1H-1,3-benzodiazol-2-iliden]benzamida (3c) y 3-(difluorometil)-N-[(2E)-1-[(2R)-2-fluoro-3-hidroxi-2-metilpropil]-5-[(3S)-1-metil-2-oxopiperidin-3-il]-2,3-di hidro-1 H-1,3-benzodiazol-2-iliden]benzamida (3d)
Etapa 1: N-{1-[3-(Bendloxi)-2-fíuoro-2-metilpropil]-5-(1-metil-2-oxopiperídin-3-il)-1H-1,3-benzodiazol-2-il}-3-(difluorometil)benzamida
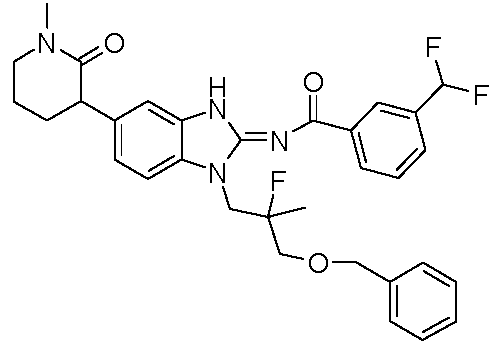
Una solución de 3-[2-amino-1-[3-(benciloxi)-2-fluoro-2-metilpropil]-1H-1,3-benzodiazol-5-il]-1-metilpiperidin-2-ona (compu esto intermedio 19, 501 mg; 1,06 mmol; 1,10 eq.), ácido 3-(difluorometil)benzoico (175 mg; 0,97 mmol; 1,00 eq.), HAt U (406 mg; 1,01 mmol; 1,05 eq.) y DIEA (328 mg; 2,41 mmol; 2,50 eq.) en Dm F (5 ml) se agitó durante toda la noche a TA. La mezcla de reacción se diluyó con EtOAc (100 ml), se lavó con salmuera (3 * 100 ml), se secó sobre sulfato sódico anhidro, se filtró y se concentró. La purificación mediante cromatografía ultrarrápida sobre gel de sílice (DCM:MeOH, 10:1) permitió obtener el compuesto del título como un aceite de color marrón (570 mg; 92 %). ESI+ (M+1): 579,4.
Etapa 2: 3-(Difluorometil)-N-[(2E)-1-[2-fluoro-3-hidroxi-2-metilpropil]-5-[1-metil-2-oxopiperidin-3-il]-2,3-dihidro-1H-1,3-benzodiazol-2-iliden]benzamida

Una solución de N-[(2E)-1-[3-(benciloxi)-2-fluoro-2-metilpropil]-5-(1-metil-2-oxopiperidin-3-il)-2,3-dihidro-1H-1,3-benzodiazol-2-iliden]-3-(difluorometil)benzamida (570 mg; 0,89 mmol; 1,00 eq.) y cloruro de hidrógeno (0,3 ml; 1,18 mmol; 11,14 eq.; 12 M) en MeOH (30 ml) se purgó con nitrógeno antes de la adición de paladio carbono (38 mg; 0,18 mmol; 0,20 eq.). A continuación, el matraz se conectó al vacío y se purgó con hidrógeno. La mezcla de reacción se hidrogenó a temperatura ambiente durante 1 día a presión atmosférica. Después se filtró a través de un relleno de celite y el filtrado se concentró y purificó mediante HPLC preparativa para obtener el compuesto del título como un sólido de color marrón (300 mg; 69 %). RMN 1H (400MHz,CDCl3) 5 12,51 (s, 1H), 8,30 (d, J = 11,2 Hz, 2H), 7,67 (d, J = 7,2 Hz, 1H), 7,57 (d, J = 7,1 Hz, 1H), 7,36 (s, 1H), 7,24 - 7,11 (m, 2H), 6,73 (m, 1H), 5,71 (m, 1H), 4,73 (m, 1H), 4,20 (m, 1H), 3,76 (m, 1H), 3,57 -3,33 (m, 4H), 3,05 (s, 3H), 2,21 (m, 1H), 2,09 - 1,81 (m, 3H), 1,65 (d, J = 22,1 Hz, 3H).
Etapa 3: Separación quiral
La mezcla obtenida en la etapa 2 se purificó en primer lugar mediante HPLC quiral preparativa utilizando una
columna Chiralpak IB, 2 * 25 cm, 5 |jm (hexano: EtOHOH, 50:50). La primera fracción de elución contenía dos picos solapados (tiempo de retención = 9,3-9,7 min).
Tercera fracción de elución: 3-(difluorometil)-N-[(2E)-1-[(2R)-2-fluoro-3-hidroxi-2-metilpropil]-5-[(3R)-1-metil-2-oxopiperidin-3-il]-2,3-di hidro-1 H-1,3-benzodiazol-2-iliden]benzamida (3a).
55 mg, sólido blanquecino, tiempo de retención = 19,7 min, p.f.: 2444-246 grados centígrados.
Cuarta fracción de elución: 3-(difluorometil)-N-[(2E)-1-[(2S)-2-fluoro-3-hidroxi-2-metilpropil]-5-[(3S)-1-metil-2-oxopiperidin-3-il]-2,3-dihidro-1 H-1,3-benzodiazol-2-iliden]benzamida (3b).
55 mg, sólido blanquecino, tiempo de retención = 33,3 min, p.f.: 102-104 °C.
A continuación, se separó la primera fracción utilizando una columna Chiralpak IA, 2 * 25 cm, 5 jm (DCM:MeOH, 20:80). Primera fracción de elución: 3-(difluorometil)-N-[(2E)-1-[(2S)-2-fluoro-3-hidroxi-2-metilpropil]-5-[(3R)-1-metil-2-oxopi peridin-3-il]-2,3-di hidro-1 H-1,3-benzodiazol-2-iliden]benzamida (3c).
45 mg, sólido de color marrón, tiempo de retención = 6,8 min, p.f.: 236-238 °C.
Segunda fracción de elución: 3-(difluorometil)-N-[(2E)-1-[(2R)-2-fluoro-3-hidroxi-2-metilpropil]-5-[(3S)-1-metil-2-oxopi peridin-3-il]-2,3-di hidro-1 H-1,3-benzodiazol-2-iliden]benzamida (3d).
45 mg, sólido de color marrón, tiempo de retención = 9,7 min, p.f.: 104-106 °C
emp o : - - - - ¡ rox¡prop¡ - - -met¡- -oxo- , - ¡ ¡ ropm m- -¡ - - enzo ¡m¡ azo- -¡¡ en - -(trifluorometil)benzamida (4)
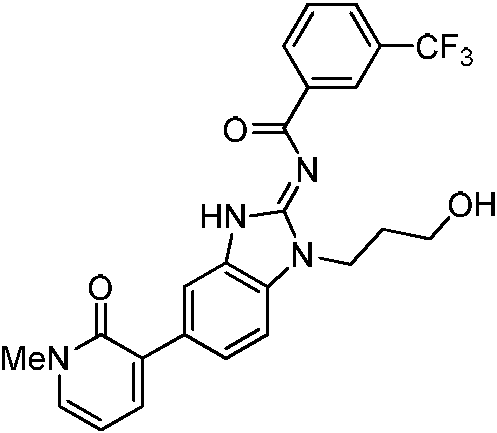
Se preparó de forma análoga al ejemplo 1, pero ut¡l¡zando el compuesto ¡ntermed¡o 3 (1 eq.) en lugar del compuesto ¡ntermed¡o 2 en la etapa 1. ESI+: M+1: 471. RMN 1H (300 MHz, CDCla) 5 12,46 (s, 1H), 8,55 - 8,39 (m, 2H), 7,83 - 7,45 (m, 5H), 7,42 - 7,22 (m, 2H), 6,36 - 6,23 (m, 1H), 4,70 (s, 1H), 4,52 - 4,48 (s, 2H), 3,65 (s, 3H), 3,48 - 3,44 (m, 2H), 2,05 -2,03 (m, 2H).
Ejemplo 5: (£)-N-(1-(3-H¡drox¡prop¡l)-5-(1-met¡l-6-oxo-1,6-d¡h¡dropmdm-5-¡l)-1H-benzo[d]¡m¡dazol-2(3H)-¡l¡den)-3-(tr¡fluoromet¡l)benzam¡da (5)
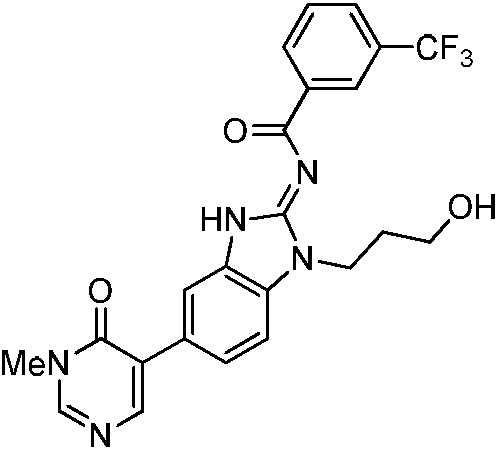
¡ntermed¡o 2 en la etapa 1. ESI+: M+1: 472. RMN 1H (300 MHz, CDCla) 5 12,46 (s, 1H), 8,54 - 8,39 (m, 2H), 8,26 (s, 1H), 8,12 (s, 1H), 7,82 - 7,79 (m, 2H), 7,62 - 7,59 (m, 2H), 7,46 (d, J = 7,6 Hz, 1H), 4,50 (s a, 2H), 3,66 (s, 3H), 3,55 (s a, 2H), 3,37 (s, 1H), 2,10 (s a, 2H).
Ejemplo 6: (£)-N-(5-(1,4-D¡met¡l-2-oxo-1,2-d¡h¡dropmdm-3-¡l)-1-(3-h¡drox¡prop¡l)-1H-benzo[d]¡m¡dazol-2(3H)-¡l¡den)-3-(tr¡fluoromet¡l)benzam¡da (6)
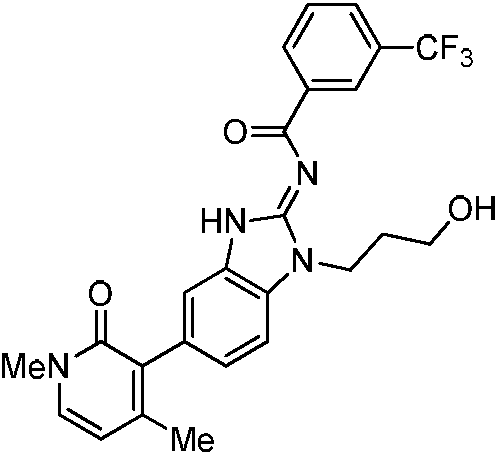
¡ntermed¡o 2 en la etapa 1. ESI+: M+1: 485. RMN 1H (300 MHz, DMSO-d6) 5 12,82 (s, 1H), 8,59 - 8,49 (m, 2H), 7,91 (d, J = 7,7 Hz, 1H), 7,74 (t, J = 7,7 Hz, 1H), 7,62 - 7,57 (m, 2H), 7,37 (d, J = 1,4 Hz, 1H), 7,09 (dd, J = 8,2, 1,6 Hz, 1H), 6,22 (d, J = 7,0 Hz, 1H), 4,72 (t, J = 5,1 Hz, 1H), 4,37 (t, J = 6,9 Hz, 2H), 3,51 (t, J = 5,8 Hz, 2H), 3,44 (s, 3H), 2,52 - 2,24 (m, 2H),
Ejemplo 7 : (E)-N-(5-(1-(C¡cloprop¡lmet¡l)-2-oxop¡per¡d¡n-3-¡l)-1-(3-h¡drox¡prop¡l)-1H-benzo[d]¡m¡dazol-2(3H)-¡l¡den)-3-(trifluorometil)benzamida (7)

Se preparó de forma análoga al ejemplo 1, pero ut¡l¡zando el compuesto ¡ntermed¡o 4 (1 eq.) en lugar del compuesto ¡ntermed¡o 2 en la etapa 1. ESI+: M+1: 515. RMN 1H (300 MHz, DMSO-da) 512,77 (s, 1H), 8,57 - 8,47 (m, 2H), 7,97 - 7,85 (m, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,46 (d, J = 8,3 Hz, 1H), 7,37 (d, J = 1,5 Hz, 1H), 7,10 (dd, J = 8,3, 1,6 Hz, 1H), 4,68 (t, J = 5,1 Hz, 1H), 4,33 (t, J = 6,8 Hz, 2H), 3,72 - 3,67 (m, 1H), 3,60 - 3,32 (m, 4H), 3,11 (dd, J = 13,6, 7,0 Hz, 1H), 2,10 (s, 1H), 1,96 (t, J = 7,7 Hz, 1H), 1,91 - 1,81 (m, 5H), 0,55 - 0,43 (m, 2H), 0,28 - 0,22 (m, 2H).
Ejemplo 8: (E)-N-(1-(3-H¡drox¡prop¡l)-5-(2-oxoc¡clohex¡l)-1H-benzo[d]¡m¡dazol-2(3H)-¡l¡den)-3-(tr¡fluoromet¡l)benzam¡da (8)

Etapa 1: Acetato de (Z)-3-(5-(2-oxoddohexil)-2-((3-(trifluommetil)benzoil)imino)-3-((2-(trimetilsilil)etoxi)metil)-2,3-dihidm-1H-benzo[d]imidazol-1-il)propilo. En un rec¡p¡ente de reacc¡ón de v¡dr¡o equ¡pado con un tapón de rosca recub¡erto de Teflón se d¡solv¡ó el compuesto ¡ntermed¡o 1 -CF3 (1 eq.), (c¡clohex-1-en-1-¡lox¡)tr¡met¡ls¡lano (2 eq.), tr¡but¡l(fluoro)estannano (2 eq.) y Pd(P(tBu)3)2 (0,06 eq.) en benceno (0,28 M). A cont¡nuac¡ón, la soluc¡ón resultante se desox¡genó med¡ante purgado subsuperf¡c¡al durante 5 m¡n antes de que el rec¡p¡ente se sellara hermét¡camente y se calentara a 90 °C durante 18 h. Los compuestos volát¡les se el¡m¡naron al vacío y el res¡duo resultante se repart¡ó entre agua y éter. La capa acuosa se separó y se retroextrajo con éter. Los extractos orgán¡cos comb¡nados se lavaron ad¡c¡onalmente con soluc¡ón acuosa de NaOH 1 N, agua y salmuera, se secaron sobre Na2SO4, se f¡ltraron y el f¡ltrado se concentró al vacío. La pur¡f¡cac¡ón med¡ante cromatografía en columna (S¡O2 , eluc¡ón en grad¡ente, 1:1 (v/v) Hex: EtOAc ^ EtOAc) perm¡t¡ó obtener el compuesto del producto deseado como un ace¡te v¡scoso (rend¡m¡ento del 28 %).
Etapa 2: Acetato de (E)-3-(5-(2-oxoddohexilo)-2-((3-(trifluorometil)benzoil)imino)-2,3-dihidro-1H-benzo[d]imidazol-1-il)propilo. En un matraz RBF de v¡dr¡o equ¡pado con un ag¡tador magnét¡co recub¡erto de Teflón se d¡solv¡ó acetato de (Z)-3-(5-(2-oxoc¡clohex¡l)-2-((3-(tr¡fluoromet¡l)benzo¡l)¡m¡no)-3-((2-(tr¡met¡ls¡l¡l)etox¡)met¡l)-2,3-d¡h¡dro-1H-benzo[d]¡m¡dazol-1-¡l)prop¡lo (1 eq.) procedente de la etapa prev¡a en d¡clorometano (0,1 M). A cont¡nuac¡ón, a esta mezcla se añad¡ó ác¡do tr¡fluoroacét¡co (50 eq.) puro gota a gota durante un per¡odo de 1 m¡n. La mezcla resultante se ag¡tó a TA durante 14 h. La mezcla de reacc¡ón s¡n procesar se d¡luyó con EtOAc y se lavó secuenc¡almente con soluc¡ón acuosa de NaHCO3 saturada, agua y salmuera. El extracto orgán¡co se secó a cont¡nuac¡ón sobre MgSO4, se f¡ltró y el f¡ltrado se concentró al vacío. La pur¡f¡cac¡ón med¡ante cromatografía en columna (S¡O2 , eluc¡ón en grad¡ente, 1:1 (v/v) Hex: EtOAc ^ EtOAc ^ 10:1 (v/v) EtOAc: MeOH) perm¡t¡ó obtener el compuesto del producto deseado (rend¡m¡ento del 76 %).
Etapa 3: (E)-N-(1-(3-Hidmxipropil)-5-(2-oxoddohexil)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifíuorometil)benzamida. En un matraz RBF de v¡dr¡o equ¡pado con un ag¡tador magnét¡co recub¡erto de Teflón se d¡solv¡ó acetato de (E)-3-(5-(2-oxoc¡clohex¡l)-2-((3-(tr¡fluoromet¡l)benzo¡l)¡m¡no)-2,3-d¡h¡dro-1H-benzo[d]¡m¡dazol-1-¡l)prop¡lo (1 eq.) procedente de la etapa prev¡a en metanol (0,021 M). A cont¡nuac¡ón, a esta mezcla se añad¡ó carbonato de potas¡o (2 eq.) y la soluc¡ón resultante se ag¡tó a TA durante 4 h. Los compuestos volát¡les se el¡m¡naron después al vacío y el res¡duo resultante se repart¡ó entre una soluc¡ón acuosa de NaHCO3 al 10 % y EtOAc. La capa acuosa se separó y se extrajo de nuevo con EtOAc. Los extractos orgán¡cos comb¡nados se lavaron ad¡c¡onalmente con agua y salmuera, se secaron sobre Na2SO4, se f¡ltraron y el f¡ltrado se concentró al vacío. La pur¡f¡cac¡ón med¡ante cromatografía líqu¡da a alta pres¡ón (C18, eluc¡ón en grad¡ente, 4:1 (v/v) H2O: MeCN TFA al 0,1 % ^ MeCN TFA al 0,1 %) perm¡t¡ó obtener el compuesto del título como un sól¡do de color blanco (rend¡m¡ento del 37 %). ESI+: M+1: 460. RMN 1H (300 MHz, CDCla) 512,54 (s, 1H), 8,52 (s, 1H), 8,44 (dd, J = 7,8 Hz, 1H), 7,75 (d, J = 7,7 Hz, 1H), 7,58 (t, J = 7,7 Hz, 1H), 7,33 - 7,23 (m, 1H), 7,17 - 7,04 (m, 2H), 4,53 -4,42 (m, 2H), 3,67 (dd, J = 12,1, 5,3 Hz, 1H), 3,48 (t, J = 5,5 Hz, 2H), 2,62 - 2,39 (m, 2H), 2,36 - 2,11 (m, 2H), 2,10 - 1,69 (m, 7H).
Ejemplo 9: (E)-N-(1-(3-H¡drox¡prop¡l)-5-(5-oxo-2,5-d¡h¡drofuran-3-¡l)-1H-benzo[d]¡m¡dazol-2(3H)-¡l¡den)-3-(trifluorometil)benzamida (9)

benzo¡co (1,5 eq.), HBTU (1,1 eq.) y et¡l-d¡¡soprop¡l-am¡na (3 eq.) en aceton¡tr¡lo (0,12 M). A cont¡nuac¡ón, a esta mezcla se añadió el compuesto intermedio 7 (1 eq.) como soluc¡ón (0,12 M) en 9:1 (v/v) DMF:MeCN y la soluc¡ón de color amar¡llo resultante se ag¡tó a TA durante 1 h. La mezcla de reacc¡ón s¡n procesar se somet¡ó d¡rectamente a pur¡f¡cac¡ón med¡ante cromatografía en columna (S¡O2 , eluc¡ón en grad¡ente, CH2O 2 ^ 9:1 (v/v) CH2Cl2 :MeOH). A cont¡nuac¡ón, el producto obten¡do de este modo se tr¡turó en metanol y éter para obtener el compuesto del título como un sól¡do de color blanco (rend¡m¡ento del 21 %). ESI+: M+1: 446. RMN 1H (300 MHz, DMSO-d6) 5 12,94 (s, 1H), 8,58 - 8,48 (m, 2H), 7,92 (d, J = 7,6 Hz, 1H), 7,79 - 7,66 (m, 4H), 6,66 (s, 1H), 5,43 (s, 2H), 4,71 - 4,65 (m, 1H), 4,40 - 4,34 (m, 2H), 3,51 - 3,44 (m, 2H), 2,01 - 1,94 (m, 2H).
Ejemplo 10: (E)-N-(1-(3-H¡drox¡prop¡l)-5-(1-met¡l-5-oxop¡rrol¡d¡n-3-¡l)-1H-benzo[d]¡m¡dazol-2(3H)-¡l¡den)-3-(tr¡-fluoromet¡l)benzam¡da (10)

Etapa 1: (E)-N-(1-(3-Hidroxipropil)-5-(1-metil-5-oxo-2,5-dihidro-1H-pirrol-3-il)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)benzamida. En un matraz RBF de v¡dr¡o equ¡pado con un ag¡tador magnét¡co recub¡erto de Teflón se d¡solv¡ó el ejemplo 9 (1 eq.) en met¡lam¡na (100 eq., soluc¡ón acuosa al 40 % [p/v]). La mezcla de reacc¡ón resultante se ag¡tó a TA durante 16 h. A cont¡nuac¡ón, los compuestos volát¡les se el¡m¡naron al vacío y el res¡duo resultante se somet¡ó d¡rectamente a cromatografía en columna (S¡O2 , eluc¡ón en grad¡ente, 4:1 (v/v) Hex: EtOAc ^ EtOAc ^ 5:1 (v/v) EtOAc: MeOH) para obtener el producto deseado como un sól¡do de color blanco (rend¡m¡ento del 12 %).
Etapa 2: (E)-N-(1-(3-Hidroxipropil)-5-(1-metil-5-oxopirrolidin-3-il)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)benzamida. En un matraz RBF de v¡dr¡o equ¡pado con un ag¡tador magnét¡co recub¡erto de Teflón se d¡solv¡ó (E)-N-(1-(3-h¡drox¡prop¡l)-5-(1-met¡l-5-oxo-2,5-d¡h¡dro-1H-p¡rrol-3-¡l)-1H-benzo[d]¡m¡dazol-2(3H)-¡l¡den)-3-(tr¡fluoromet¡l)benzam¡da (1 eq.) procedente de la etapa prev¡a y cloruro de palad¡o(II) (0,5 eq.) en MeOH (0,01 M). La suspens¡ón resultante se purgó subsuperf¡c¡almente con h¡drógeno durante 30 m¡n y, a cont¡nuac¡ón, se ag¡tó bajo una atmósfera de h¡drógeno con globo estát¡co durante 3 días. Después, el mater¡al ¡nsoluble se el¡m¡nó med¡ante f¡ltrac¡ón a través de un lecho de cel¡te humedec¡do con metanol. El mater¡al ¡nsoluble se lavó de nuevo con metanol y el f¡ltrado obten¡do de este modo se concentró al vacío. El res¡duo obten¡do de este modo se somet¡ó d¡rectamente a pur¡f¡cac¡ón med¡ante cromatografía en columna (S¡O2 , eluc¡ón en grad¡ente, 4:1 (v/v) Hex:EtOAc ^ EtOAc ^ 10:1 (v/v) EtOAc:MeOH) para obtener el producto deseado como un sól¡do de color blanco (rend¡m¡ento del 81 %). ESI+: M+1: 461. RMN 1H (300 MHz, DMSO-d6) 512,94 (s, 1H), 8,56 - 8,47 (m, 2H), 7,90 (d, J = 8,1 Hz, 1H), 7,74 (t, J = 7,7 Hz, 1H), 7,57 - 7,38 (m, 2H), 7,30 - 7,21 (m, 1H), 4,33 (t, J = 6,8 Hz, 2H), 3,84 - 3,72 (m, 1H), 3,72 - 3,59 (m, 1H), 3,47 (t, J = 6,2 Hz, 2H), 3,35 (dd, J = 9,2, 6,6 Hz, 1H), 2,80 (s, 3H), 2,76 - 2,65 (m, 1H), 2,41 - 2,28 (m, 1H), 2,01 - 1,90 (m, 2H).
benzamida (11)

dihidro-1H-benzo[d]imidazol-1-il)propilo. En un matraz RBF de v¡dr¡o equ¡pado con un ag¡tador magnét¡co recub¡erto de Teflón se d¡solv¡ó el compuesto ¡ntermed¡o I-CF3 (1 eq.) en THF (0,05 M). A cont¡nuac¡ón, a esta mezcla se añad¡ó a 0 °C, cloruro de ¡soprop¡lmagnes¡o (1 eq., soluc¡ón 2,9 M en 2-MeTHF) gota a gota durante un per¡odo de 5 m¡n. La mezcla de reacc¡ón resultante se ag¡tó a 0 °C durante 15 m¡n y después a TA durante 30 m¡n. Tras enfr¡ar de nuevo a 0 °C, se añad¡ó oxetan-3-ona (1,2 eq.) puro gota a gota durante 5 m¡n. Después, la mezcla de reacc¡ón se dejó atemperar lentamente a TA durante 1 h. La reacc¡ón se detuvo con la ad¡c¡ón de soluc¡ón acuosa de HCl 1 N y se extrajo con EtOAc. A cont¡nuac¡ón, los extractos orgán¡cos comb¡nados se lavaron ad¡c¡onalmente con soluc¡ón acuosa de NaHCO3 al 10 % y salmuera, se secaron sobre Na2SO4 , se f¡ltraron y el f¡ltrado se concentró al vacío. La pur¡f¡cac¡ón ad¡c¡onal med¡ante cromatografía en columna (S¡O2 , eluc¡ón en grad¡ente, EtOAc ^ 10:1 (v/v) EtOAc: MeOH) perm¡t¡ó obtener el producto deseado como un sól¡do de color blanco (rend¡m¡ento del 21 %).
Etapa 2: Acetato de (E)-3-(5-(3-hidroxioxetan-3-il)-2-((3-(trifluorometil)benzoil)imino)-2,3-dihidro-1H-benzo[d]imidazol-1-il)propilo. En un matraz RBF de v¡dr¡o equ¡pado con un ag¡tador magnét¡co recub¡erto de Teflón se d¡solv¡ó acetato de (Z)-3-(5-(3-h¡drox¡oxetan-3-¡l)-2-((3-(tr¡fluoromet¡l)benzo¡l)¡m¡no)-3-((2-(tr¡met¡ls¡l¡l)etox¡)met¡l)-2,3-d¡h¡dro-1H-benzo[d]¡m¡dazol-1-¡l)prop¡lo (1 eq.) procedente de la etapa prev¡a en d¡clorometano (0,02 M). A cont¡nuac¡ón, a esta mezcla se añad¡ó TFA (50 eq.) puro gota a gota durante un per¡odo de 1 m¡n. La mezcla resultante se ag¡tó a TA durante 18 h. Después, la mezcla de reacc¡ón s¡n procesar se d¡luyó con DCM y se lavó secuenc¡almente con soluc¡ón acuosa de NaHCO3 saturada, agua y salmuera. El extracto orgán¡co se secó a cont¡nuac¡ón sobre Na2SO4 y se f¡ltró. La concentrac¡ón del f¡ltrado al vacío perm¡t¡ó obtener el compuesto del producto deseado como un sól¡do blanquec¡no.
Etapa 3: (E)-N-(5-(3-Hidroxioxetan-3-il)-1-(3-hidroxipropil)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)benzamida. En un matraz RBF de v¡dr¡o equ¡pado con un ag¡tador magnét¡co recub¡erto de Teflón se d¡solv¡ó acetato de (E)-3-(5-(3-h¡drox¡oxetan-3-¡l)-2-((3-(tr¡fluoromet¡l)benzo¡l)¡m¡no)-2,3-d¡h¡dro-1H-benzo[d]¡m¡dazol-1-¡l)prop¡lo (1 eq.) procedente de la etapa prev¡a en metanol (0,02 M). A cont¡nuac¡ón, a esta se añad¡ó carbonato de potas¡o (2 eq.) y la suspens¡ón resultante se ag¡tó a TA durante 18 h. Los compuestos volát¡les se el¡m¡naron al vacío y el res¡duo resultante se somet¡ó d¡rectamente a pur¡f¡cac¡ón med¡ante cromatografía líqu¡da a alta pres¡ón (C18, eluc¡ón en grad¡ente, 9:1 (v/v) ^ 1:9 (v/v) H2O:MeCN TFA al 0,1 %). El compuesto del título se a¡sló como un sól¡do de color blanco (rend¡m¡ento del 49 % en 2 etapas). ESI+: M+1: 436. RMN 1H (300 MHz, DMSO-d6) 5 12,83 (s, 1H), 8,57 - 8,47 (m, 2H), 7,90 (d, J = 7,8 Hz, 1H), 7,81 (s, 1H), 7,73 (t, J = 7,5 Hz, 1H), 7,60 - 7,53 (m, 2H), 6,50 - 6,42 (m, 1H), 4,84 - 4,77 (m, 2H), 4,74 - 4,64 (m, 3H), 4,38 - 4,31 (m, 2H), 3,52 - 3,43 (m, 2H), 2,02 - 1,91 (m, 2H).
Ejemplo 12: (E)-N-(5-(1-Acet¡lp¡rrol¡d¡n-3-¡l)-1-(3-h¡drox¡prop¡l)-1H-benzo[d]¡m¡dazol-2(3H)-¡l¡den)-3-(tr¡fluoromet¡l)-benzam¡da (12)

Etapa 1: 4-(1-(3-Acetoxipropil)-2-((3-(trifluorometil)benzoil)imino)-3-((2-(trimetilsilil)etoxi)metil)-2,3-dihidro-1H-benzo[d]imidazol-5-il)-2,3-dihidro-1H-pirrol-1-carboxilato de (Z)-terc-butilo. En un matraz RBF de v¡dr¡o equ¡pado con un ag¡tador magnét¡co recub¡erto de Teflón se d¡solv¡ó el compuesto ¡ntermed¡o 8 -CF3 (1 eq.), 4-(((tr¡fluoromet¡l)sulfon¡l)ox¡)-2,3-d¡h¡dro-1H-p¡rrol-1-carbox¡lato de terc-but¡lo (1 eq.), Pd(dppf)Cl2 (0,2 eq.) y fosfato de potas¡o (3 eq.) en una soluc¡ón 10:1 (v/v) (0,075 M) de THF:agua. Después, la mezcla de reacc¡ón obten¡da de este modo se desox¡genó a fondo med¡ante purgado subsuperf¡c¡al con n¡trógeno durante 15 m¡n. El rec¡p¡ente se selló y ag¡tó a TA durante 18 h. La mezcla de reacc¡ón se detuvo con la ad¡c¡ón de agua y se extrajo con EtOAc. A cont¡nuac¡ón, los extractos orgán¡cos comb¡nados se lavaron ad¡c¡onalmente con soluc¡ón acuosa de NaHCO3 al 10 % y salmuera, se secaron sobre Na2SO4, se f¡ltraron y el f¡ltrado se concentró al vacío. La pur¡f¡cac¡ón ad¡c¡onal med¡ante cromatografía en columna (S¡O2 , eluc¡ón en grad¡ente, 10:1 (v/v) Hex:EtOAc ^ EtOAc ^ 10:1 (v/v) EtOAc:MeOH) perm¡t¡ó obtener el producto deseado (rend¡m¡ento del 58 %).
Etapa 2: 3-(1-(3-Acetoxipropil)-2-((3-(trifluorometil)benzoil)imino)-3-((2-(trimetilsilil)etoxi)metil)-2,3-dihidro-1H-benzo[d]imidazol-5-il)pirrolidin-1-carboxilato de (Z)-terc-butilo. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se resuspendió paladio (0,2 eq., seco, al 10 % p/p sobre carbono) y 4-(1-(3-acetoxipropil)-2- ((3-(trifluorometil)benzoil)imino)-3-((2-(trimetilsilil)etoxi)metil)-2,3-dihidro-1H-benzo[d]imidazol-5-il)-2,3-dihidro-1H-pirrol-1-carboxilato de (Z)-terc-butilo (1 eq.) procedente de la etapa previa en metanol (0,05 M). A continuación, la suspensión resultante se vació y se volvió a llenar con nitrógeno (3 veces) y luego con hidrógeno (3 veces). La suspensión de reacción se agitó bajo una atmósfera de hidrógeno mantenida con balón estático durante 18 h. La reacción se detuvo con diclorometano y la suspensión resultante se filtró a través de un relleno de celite humedecido con diclorometano. El filtrado obtenidos de este modo se concentró al vacío y se sometió a cromatografía en columna (SiO2 , elución en gradiente, 10:1 (v/v) Hex:EtOAc ^ EtOAc ^ 10:1 (v/v) EtOAc:MeOH). El producto deseado se aisló como un aceite incoloro (rendimiento del 88 %).
Etapa 3: Acetato de (E)-3-(5-(pirrolidin-3-il)-2-((3-(trifluorometil)benzoil)imino)-2,3-dihidro-1 H-benzo[d]imidazol-1-il)propilo. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvió 3-(1-(3-acetoxipropil)-2-((3-(trifluorometil)benzoil)imino)-3-((2-(trimetilsilil)etoxi)metil)-2,3-dihidro-1H-benzo[d]imidazol-5-il)pirrolidin-1-carboxilato de (Z)-terc-butilo (1 eq.) procedente de la etapa previa en diclorometano (0,1 M). A continuación, a esta solución se añadió TFA (30 eq.) puro gota a gota durante un periodo de 1 min. La mezcla resultante se agitó a TA durante 8 h. Después, la mezcla de reacción sin procesar se diluyó con DCM y se lavó secuencialmente con solución acuosa de NaHCO3 saturada, agua y salmuera. El extracto orgánico se secó a continuación sobre Na2SO4 y se filtró. La concentración del filtrado al vacío permitió obtener el compuesto del producto deseado como un aceite viscoso.
Etapa 4: Acetato de (E)-3-(5-(1-acetilpirrolidin-3-il)-2-((3-(trifluorometil)benzoil)imino)-2,3-dihidro-1H-benzo[d]imidazol-1-il)propilo. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvió acetato de (E)-3- (5-(pirrolidin-3-il)-2-((3-(trifluorometil)benzoil)imino)-2,3-dihidro-1H-benzo[d]imidazol-1-il)propilo (1 eq.) procedente de la etapa previa en piridina (0,1 M). A continuación, a esta solución se añadió anhídrido acético (1,8 eq.) puro gota a gota durante un periodo de 1 min. La mezcla resultante se agitó a TA durante 48 h. Después, la mezcla de reacción sin procesar se diluyó con EtOAc y se lavó secuencialmente con solución acuosa de NH4Cl saturada, agua y salmuera. El extracto orgánico se secó a continuación sobre Na2SO4 y se filtró. El filtrado obtenido de este modo se concentró al vacío y se sometió a cromatografía en columna (SiO2 , elución en gradiente, 10:1 (v/v) Hex:EtOAc ^ EtOAc ^ 10:1 (v/v) EtOAc:MeOH). El producto deseado se aisló como un aceite de color amarillo claro (rendimiento del 38 % en dos etapas).
Etapa 5: (E)-N-(5-(1-Acetilpirrolidin-3-il)-1-(3-hidroxipropil)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)benzamida. En un matraz RBF de vidrio equipado con un agitador magnético recubierto de Teflón se disolvió acetato de (E)-3-(5-(1-acetilpirrolidin-3-il)-2-((3-(trifluorometil)benzoil)imino)-2,3-dihidro-1 H-benzo[d]imidazol-1 -il)propilo (1 eq.) procedente de la etapa previa en metanol (0,02 M). A continuación, a esta se añadió carbonato de potasio (2 eq.) y la suspensión resultante se agitó a TA durante 8 h. Los compuestos volátiles se eliminaron al vacío y el residuo resultante se aplicó directamente en una columna cromatográfica (SiO2 , elución en gradiente, 10:1 (v/v) Hex:EtOAc ^ EtOAc ^ 10:1 (v/v) EtOAc:MeOH). El compuesto del título se aisló como un sólido de color blanco (rendimiento del 95 %). ESI+: M+1: 475. RMN 1H (300 MHz, CDCla) 5 12,61 (s, 1H), 8,53 (s, 1H), 8,45 (d, J = 7,8 Hz, 1H), 7,75 (d, J = 7,8 Hz, 1H), 7,58 (t, J = 7,8 Hz, 1H), 7,34 - 7,13 (m, 3H), 4,67 - 4,60 (m, 1H), 4,47 (t, J = 5,4 Hz, 2H), 4,06 - 3,71 (m, 2H), 3,70 - 3,27 (m, 2H), 3,70 - 3,27 (m, 5H), 2,38 - 2,21 (m, 1H), 2,15 - 1,78 (m, 6H).
Ejemplo 13: (E)-N-(5-(1-Acetilpiperidin-4-il)-1-(3-hidroxipropil)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)-benzamida (13)

Se preparó de forma análoga al ejemplo 12, pero utilizando 4-(((trifluorometil)sulfonil)oxi)-5,6-dihidropiridin-1(2H)-carboxilato de terc-butilo (1 eq.) en lugar de 4-(((trifluorometil)sulfonil)oxi)-2,3-dihidro-1 H-pirrol-1-carboxilato de terc-butilo en la etapa 1. ESI+: M+1: 489. RMN 1H NMR (300 MHz, CDCla) 512,51 (s, 1H), 8,51 (s, 1H), 8,43 (d, J = 7,7 Hz, 1H), 7,75 (d, J = 7,7 Hz, 1H), 7,58 (t, J = 7,8 Hz, 1H), 7,33 - 7,12 (m, 3H), 4,80 (d, J = 13,3 Hz, 1H), 4,72 - 4,65 (m, 1H), 4,47 (t, J = 5,8 Hz, 2H), 3,95 (t, J = 13,4 Hz, 1H), 3,51 - 3,45 (m, 2H), 3,18 (t, J = 12,8 Hz, 1H), 2,82 (s, 1H), 2,63 (t, J = 12,9 Hz, 1H), 2,15 (s, 3H), 2,05 (d, J = 5,9 Hz, 2H), 1,96 - 1,82 (m, 2H), 1,73 - 1,54 (m, 2H).
Ejemplo 14: (E)-N-(5-(1-Acetilpiperidin-3-il)-1-(3-hidroxipropil)-1H-benzo[d]imidazol-2(3H)-iliden)-3-(trifluorometil)-benzamida (14)
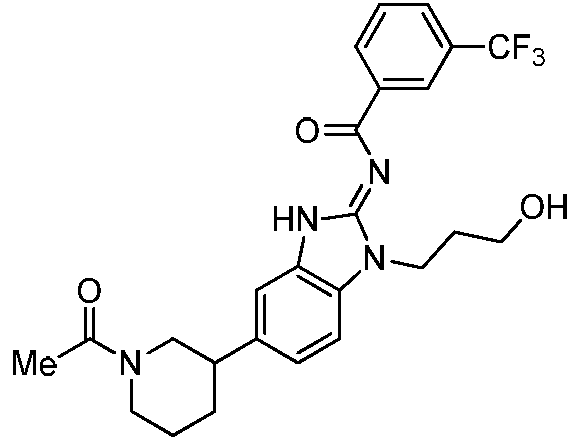
Se preparó de forma análoga al ejemplo 12, pero utilizando 5-(((tr¡fluoromet¡l)sulfon¡l)oxi)-3,4-d¡h¡drop¡r¡d¡n-1(2H)-carboxilato de tere-butilo (1 eq.) en lugar de 4-(((trifluoromet¡l)sulfon¡l)ox¡)-2,3-d¡h¡dro-1H-p¡rrol-1-carbox¡lato de tere-butilo en la etapa 1. ESI+: M+1: 489. RMN 1H (300 MHz, CDCb) 5 12,55 (s, 1H), 8,53 (s, 1H), 8,45 (d, J = 7,8 Hz, 1H), 7,75 (d, J = 7,8 Hz, 1H), 7,58 (t, J = 7,8 Hz, 1H), 7,34 - 7,16 (m, 3H), 4,79 - 4,54 (m, 2H), 4,48 (dd, J = 5,5, 4,8 Hz, 2H), 3,85 (t, J = 13,5 Hz, 1H), 3,51 - 3,45 (m, 2H), 3,08 - 2,99 (m, 1H), 2,78 - 2,71 (m, 1H), 2,56 - 2,49 (m, 1H), 2,18 - 1,98 (m, 5H), 1,92 - 1,54 (m, 4H).
Ejemplo 15: (£)-N-(5-(8-Acetil-8-azab¡ddo[3.2.1]octan-3-¡l)-1-(3-h¡drox¡prop¡l)-1H-benzo[cí]¡m¡dazol-2(3H)-¡l¡den)-3-(trifluorometil)benzamida (15)

Se preparó de forma análoga al ejemplo 12, pero utilizando 3-(((trifluoromet¡l)sulfon¡l)ox¡)-8-azab¡ddo[3.2.1]oct-3-ene-8-carboxilato de tere-butilo (1 eq.) en lugar de 4-(((trifluoromet¡l)sulfon¡l)ox¡)-2,3-d¡h¡dro-1H-p¡rrol-1-carbox¡lato de tere-butilo en la etapa 1. ESI+: M+1: 515. RMN 1H (300 MHz, CDCb) 5 12,50 (s, 1H), 8,51 (s, 1H), 8,44 (d, J = 7,7 Hz, 1H), 7,75 (d, J = 7,7 Hz, 1H), 7,58 (t, J = 7,8 Hz, 1H), 7,30 - 7,15 (m, 3H), 4,82 - 4,66 (m, 2H), 4,47 (t, J = 5,9 Hz, 2H), 4,26 - 4,16 (m, 1H), 3,52 - 3,45 (m, 2H), 2,80 - 2,40 (m, 2H), 2,20 - 1,44 (m, 12H).
Ejemplo 16: (£)-W-(1-((S)-2-Fluoro-3-h¡drox¡-2-metilprop¡l)-5-(1-met¡l-2-oxop¡per¡d¡n-3-¡l)-1H-benzo[d]¡m¡dazol-2(3H)-iliden)-3-(tr¡fluoromet¡l)benzam¡da (16)

Se preparó de forma análoga al ejemplo 1, pero utilizando el compuesto intermedio 9 (1 eq.) en lugar del compuesto intermedio 2 en la etapa 1. ESI+: M+1: 507. RMN 1H (300 MHz, CDCb) 5 12,45 (s, 1H), 8,45 (s, 1H), 8,35 (d, J = 7,9 Hz, 1H), 7,76 (d, J = 7,9 Hz, 1H), 7,59 (t, J = 7,7 Hz, 1H), 7,43 - 7,30 (m, 1H), 7,28 - 7,13 (m, 2H), 5,69 - 5,59 (m, 1H), 4,71 (dd, J = 32,5, 15,4 Hz, 1H), 4,28 - 4,13 (m, 1H), 3,76 (t, J = 7,3 Hz, 1H), 3,65 - 3,32 (m, 4H), 3,06 (s, 3H), 2,26 - 2,19 (m, 1H), 2,12 - 1,82 (m, 3H), 1,65 (d, J = 22,2 Hz, 3H).
Ejemplo 17: N-[1-But-3-en¡l-5-(1-met¡l-2-oxo-p¡rrol¡d¡n-3-il)-1,3-d¡h¡dro-benzo¡m¡dazol-(2E)-¡l¡den]-3-tr¡fluoromet¡lbenzamida (17)

Una mezcla de bromhidrato de 3-(2-amino-1-but-3-enil-1H-benzoimidazol-5-il)-1-metil-pirrolidin-2-ona (compuesto intermedio 11, 300 mg; 0,82 mmol; 1,0 eq.), ácido 3-(trifluorometil)benzoico (Aldrich, 163 mg; 0,86 mmol; 1,05 eq.), hexafluorofosfato de (benzotriazol-1-iloxi)tris(dimetilamino)fosfonio (449 mg; 0,99 mmol; 1,2 eq.) y diisopropiletilamina (358 |jl; 2,05 mmol; 2,5 eq.) en DMF (2 ml) se agitó a TA durante 1 h. La mezcla de reacción se vertió en agua. El precipitado se recogió mediante filtración y, a continuación, se lavó con agua para obtener el producto sin procesar que se purificó mediante HPLC preparativa para obtener el compuesto del título como un sólido de color blanco (rendimiento del 89 %). ESI+: M+1: 457. RMN 1H (400 MHz, DMSO-d6) 52,69 (s, 1H), 8,52 (dt, J = 4,3, 1,8 Hz, 2H), 7,90 (d, J = 7,8 Hz, 1H), 7,74 (t, J = 7,9 Hz, 1H), 7,51 (d, J = 8,3 Hz, 1H), 7,42 (d, J = 1,6 Hz, 1H), 7,14 (dd, J = 8,3, 1,7 Hz, 1H), 5,90 (ddt, J = 17,1, 10,2, 6,8 Hz, 1H), 5,13 - 4,90 (m, 2H), 4,35 (t, J = 7,0 Hz, 2H), 3,71 (t, J = 8,9 Hz, 1H), 3,53 - 3,39 (m, 2H), 2,83 (s, 3H), 2,62 (p, J = 6,8 Hz, 2H), 2,49 - 2,42 (m, 1H), 2,13 - 1,92 (m, 1H).
Ejemplo 18: N-[1-But-3-enil-5-(1-metil-2-oxo-piperidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometilbenzamida (18)

El compuesto del título se preparó de forma análoga al ejemplo 17 utilizando 3-(2-amino-1-but-3-enil-1H-benzoimidazol-5-il)-1-metil-piperidin-2-ona (compuesto intermedio 12) en lugar de bromhidrato de 3-(2-amino-1-but-3-enil-1H-benzoimidazol-5-il)-1-metil-pirrolidin-2-ona (rendimiento del 88 %). ESI+: M+1: 471. RMN 1H (400 MHz, DMSO-d6) 512,68 (s, 1H), 8,57 - 8,45 (m, 2H), 7,89 (d, J = 7,8 Hz, 1H), 7,74 (t, J = 7,7 Hz, 1H), 7,48 (d, J = 8,3 Hz, 1H), 7,36 (d, J = 1,6 Hz, 1H), 7,09 (dd, J = 8,3, 1,7 Hz, 1H), 5,90 (ddt, J = 17,0, 10,2, 6,9 Hz, 1H), 5,08 (dc, J = 17,2, 1,6 Hz, 1H), 5,00 (ddt, J = 10,3, 2,1, 1,1 Hz, 1H), 4,35 (t, J = 7,0 Hz, 2H), 3,67 (dd, J = 8,4, 5,9 Hz, 1H), 3,52 - 3,39 (m, 1H), 3,42 - 3,32 (m, 1H), 2,91 (s, 3H), 2,61 (c, J = 6,9 Hz, 2H), 2,13 - 2,08 (m, 1H), 1,96 - 1,76 (m, 3H).
Ejemplo 19: N-[1-Alil-5-(1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (19)

El compuesto del título se preparó de forma análoga al ejemplo 17 utilizando bromhidrato de 3-(1-alil-2-amino-1H-benzoimidazol-5-il)-1-metil-pirrolidin-2-ona (compuesto intermedio 13) en lugar de bromhidrato de 3-(2-amino-1-but-3-enil-1H-benzoimidazol-5-il)-1-metil-pirrolidin-2-ona (rendimiento del 83 %). ESI+: M+1: 443. RMN 1H (400 MHz, DMSO-d6) 5 12,77 (s, 1H), 8,52 (d, J = 7,8 Hz, 1H), 8,49 (s, 1H), 7,93 - 7,86 (m, 1H), 7,74 (t, J = 7,8 Hz, 1H), 7,44 (d, J = 1,7 Hz, 1H), 7,41 (d, J = 8,3 Hz, 1H), 7,13 (dd, J = 8,3, 1,7 Hz, 1H), 6,04 (ddt, J = 17,1, 10,5, 5,4 Hz, 1H), 5,29 - 5,13 (m, 2H), 5,01 -4,85 (m, 2H), 3,72 (t, J = 8,9 Hz, 1H), 3,55 - 3,36 (m, 2H), 2,83 (s, 3H), 2,55 - 2,46 (m, 1H), 2,02 (dtd, J = 12,7, 8,7, 7,6 Hz, 1H).
Ejemplo 20: N-[1-(2,3-Dihidroxi-propil)-5-(1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (20)
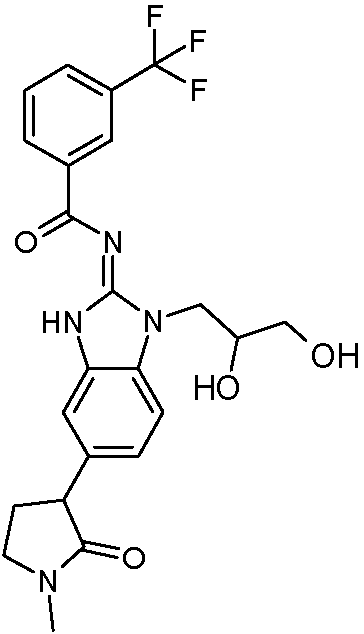
A una solución en agitación de N-[1-alil-5-(1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (ejemplo 19, 30 mg; 0,07 mmol; 1,0 eq.) en acetona (5 ml) y agua (0,6 ml), se añadió tetróxido de osmio (2,5 % en peso en t-BuOH, 0,13 ml; 0,01 mmol; 0,15 eq.) y n-óxido de 4-metilmorfolina (15 mg; 0,14 mmol; 2,0 eq.). La mezcla resultante se agitó durante toda la noche a TA. El producto sin procesar se purificó mediante HPLC preparativa obteniéndose el compuesto del título como un sólido de color blanco (rendimiento del 56 %). ESI+: M+1: 477. RMN 1H (400 MHz, DMSO-d6) 5 12,72 (s, 1H), 8,63 - 8,42 (m, 2H), 7,89 (d, J = 7,7 Hz, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,49 (d, J = 8,3 Hz, 1H), 7,42 (d, J = 1,7 Hz, 1H), 7,12 (dd, J = 8,3, 1,7 Hz, 1H), 5,08 (d, J = 5,2 Hz, 1H), 4,85 (t, J = 5,6 Hz, 1H), 4,41 (dd, J = 14,0, 4,1 Hz, 1H), 4,25 - 4,10 (m, 1H), 4,08 - 3,95 (m, 1H), 3,71 (t, J = 8,9 Hz, 1H), 3,53 - 3,38 (m, 4H), 2,83 (s, 3H), 2,55 - 2,48 (m, 1H), 2,12 - 1,94 (m, 1H).
Ejemplos 20a y 20b: N-[1-(2,3-Dihidroxi-propil)-5-((S)-1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (20a) y N-[1-(2,3-dihidroxi-propil)-5-((R)-1-metil-2-oxo-pi rrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (20b)
Los dos diastereoisómeros del ejemplo 20 se separaron mediante cromatografía SFC quiral (columna CHIRALPAK OJ-H, 4,6 x 250 mm, 5 |jm, gradiente isocrático MeOH al 55 %, 8 ml/min, 100 Bar CO240 °C).
Primer isómero eluido (20a): 21 mg de un sólido de color rosa, tiempo de retención: 3,44 min. ESI+ (M+1): 477. RMN 1H (400 MHz, DMSO-d6) 5 12,76 (s, 1H), 8,61 - 8,43 (m, 2H), 7,89 (d, J = 7,7 Hz, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,49 (d, J = 8,3 Hz, 1H), 7,42 (d, J = 1,6 Hz, 1H), 7,12 (dd, J = 8,4, 1,6 Hz, 1H), 5,07 (d, J = 5,3 Hz, 1H), 4,85 (t, J = 5,7 Hz, 1H), 4,41 (dd, J = 14,0, 4,0 Hz, 1H), 4,16 (dd, J = 14,0, 7,4 Hz, 1H), 4,08 - 3,92 (m, 1H), 3,71 (t, J = 8,9 Hz, 1H), 3,55 - 3,37 (m, 4H), 2,83 (s, 3H), 2,49 - 2,40 (m, 1H), 2,12 - 1,93 (m, 1H).
Segundo isómero eluido (20b): 20 mg de un sólido de color rosa, tiempo de retención: 5,42 min. ESI+ (M+1): 477. RMN 1H (400 MHz, DMSO-d6) 5 12,75 (s, 1H), 8,60 - 8,45 (m, 2H), 7,89 (d, J = 7,7 Hz, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,49 (d, J = 8,3 Hz, 1H), 7,42 (d, J = 1,6 Hz, 1H), 7,12 (dd, J = 8,4, 1,7 Hz, 1H), 5,08 (d, J = 5,3 Hz, 1H), 4,85 (t, J = 5,7 Hz, 1H), 4,41 (dd, J = 13,8, 4,0 Hz, 1H), 4,22 - 4,11 (m, 1H), 4,01 (c, J = 5,6 Hz, 1H), 3,71 (t, J = 8,9 Hz, 1H), 3,54 - 3,37 (m, 4H), 2,83 (s, 3H), 2,50 - 2,44 (m, 1H), 2,11 - 1,93 (m, 1H).
Ejemplo 21: N-[1-(3,4-Dihidroxi-butil)-5-(1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (21)

El compuesto del título se preparó de forma análoga al ejemplo 20 utilizando N-[1-but-3-enil-5-(1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida en lugar de N-[1-alil-5-(1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (rendimiento del 74 %). ESI+: M+1: 491. RMN 1H (400 MHz, DMSO-d6) 5 12,65 (sa, 1H), 8,58 - 8,46 (m, 2H), 7,94 - 7,85 (m, 1H), 7,74 (t, J = 7,7 Hz, 1H), 7,49 (d, J = 8,3 Hz, 1H), 7,43 (d, J = 1,6 Hz, 1H), 7,15 (dt, J = 8,4, 1,6 Hz, 1H), 4,76 (d, J = 4,9 Hz, 1H), 4,52 (t, J = 5,7 Hz, 1H), 4,42 - 4,32 (m, 2H), 3,72 (t, J = 8,9 Hz, 1H), 3,53 - 3,44 (m, 2H), 3,44 - 3,34 (m, 2H), 3,29 - 3,21 (m, 1H), 2,83 (s, 3H), 2,54 - 2,46 (m, 1H), 2,07 - 1,98 (m, 2H), 1,80 - 1,66 (m, 1H).
Ejemplos 21a, 21b, 21c y 21d: N-[1-((S)-3,4-Dihidroxi-butil)-5-((R)-1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidrobenzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (21a), N-[1-((R)-3,4-dihidroxi-butil)-5-((R)-1-metil-2-oxo-pi rrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (21b), N-[1-((R)-3,4-dihidroxi-butil)-5-((S)-1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (21c) y N-[1-((S)-3,4-dihidroxi-butil)-5-((S)-1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (21d)
Los cuatro diastereoisómeros del ejemplo 21 se separaron mediante cromatografía SFC quiral. Una primera purificación utilizando una columna CHIRALPAK OJ-H (columna de 4,6 * 250 mm, 5 |jm, gradiente isocrático, MeOH al 30 %, 8 ml/min, 100 Bar de CO240 °C) permitió la separación de tres fracciones:
Primera fracción de elución: 38 mg de polvo blanquecino. Contiene una mezcla racémica.
Segunda fracción de elución (21a): 18 mg de polvo blanquecino, tiempo de retención: 14,02 min ESI+ (M+1): 491. RMN 1H (400 MHz, DMSO-d6) 5 12,74 (s, 1H), 8,57 - 8,48 (m, 2H), 7,90 (d, J = 7,7 Hz, 1H), 7,74 (t, J = 7,7 Hz, 1H), 7,49 (d, J = 8,4 Hz, 1H), 7,44 (t, J = 1,6 Hz, 1H), 7,15 (dt, J = 8,4, 1,6 Hz, 1H), 4,76 (d, J = 4,9 Hz, 1H), 4,53 (t, J = 5,3 Hz, 1H), 4,45 - 4,29 (m, 2H), 3,72 (t, J = 8,9 Hz, 1H), 3,53 - 3,45 (m, 2H), 3,44 - 3,35 (m, 2H), 3,29 - 3,25 (m, 1H), 2,83 (s, 3H), 2,54 -2,50 (m, 1H), 2,12 - 1,95 (m, 2H), 1,80 - 1,66 (m, 1H).
Tercera fracción de elución (21b): 9 mg de polvo blanquecino, tiempo de retención: 17,01 min. ESI+ (M+1): 491. RMN 1H (400 MHz, DMSO-d6) 5 12,74 (s, 1H), 8,57 - 8,49 (m, 2H), 7,93 - 7,86 (m, 1H), 7,74 (t, J = 7,7 Hz, 1H), 7,49 (d, J = 8,3 Hz, 1H), 7,44 (d, J = 1,6 Hz, 1H), 7,15 (dd, J = 8,4, 1,7 Hz, 1H), 4,76 (d, J = 4,9 Hz, 1H), 4,53 (t, J = 5,6 Hz, 1H), 4,46 -4,29 (m, 2H), 3,72 (t, J = 8,9 Hz, 1H), 3,53 - 3,43 (m, 2H), 3,42 - 3,33 (m, 2H), 3,30 - 3,25 (m, 1H), 2,83 (s, 3H), 2,60 -2,46 (m, 1H), 2,10 - 1,97 (m, 2H), 1,79 - 1,67 (m, 1H).
La primera fracción de elución se separó utilizando una columna CHIRALPAK IA (columna de 10 * 250 mm, 5 jm , gradiente isocrático MeOH al 60 % con DMEA al 0,5 %, 8 ml/min, 100 Bar CO235 °C).
Primera fracción de elución (21c): 10 mg de un polvo de color blanco, tiempo de retención: 5,32 min ESI+ (M+1): 491. RMN 1H (400 MHz, DMSO-d6) 5 12,66 (s, 1H), 8,57 - 8,48 (m, 2H), 7,89 (d, J = 7,7 Hz, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,49 (d, J = 8,2 Hz, 1H), 7,43 (d, J = 1,5 Hz, 1H), 7,15 (dd, J = 8,3, 1,7 Hz, 1H), 4,76 (d, J = 4,8 Hz, 1H), 4,52 (t, J = 5,7 Hz, 1H), 4,44 - 4,29 (m, 2H), 3,72 (t, J = 8,9 Hz, 1H), 3,50 - 3,44 (m, 2H), 3,43 - 3,35 (m, 2H), 3,29- 3,17 (m, 1H), 2,83 (s, 3H), 2,57 - 2,43 (m, 1H), 2,11 - 1,94 (m, 2H), 1,81 - 1,65 (m, 1H).
Segunda fracción de elución (21d): 14 mg de un polvo de color blanco, tiempo de retención: 6,69 min ESI+ (M+1): 491. RMN 1H (400 MHz, DMSO-d6) 5 12,54 (s, 1H), 8,57 - 8,48 (m, 2H), 7,89 (d, J = 7,7 Hz, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,48
(d, J = 8,3 Hz, 1H), 7,43 (d, J = 1,5 Hz, 1H), 7,14 (dd, J = 8,3, 1,7 Hz, 1H), 4,77 (sa, 1H), 4,53 (sa, 1H), 4,45 - 4,28 (m, 2H), 3,72 (t, J = 8,9 Hz, 1H), 3,54 - 3,45 (m, 2H), 3,43 - 3,37 (m, 2H), 3,26 - 3,21 (m, 1H), 2,83 (s, 3H), 2,52 - 2,43 (m, 1H), 2,13 - 1,94 (m, 2H), 1,79 - 1,65 (m, 1H).
Ejemplo 22: N-[1-(3,4-Dihidroxi-butil)-5-(1-metil-2-oxo-piperidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (22)

El compuesto del título se preparó de forma análoga al ejemplo 20 utilizando N-[1-but-3-enil-5-(1-metil-2-oxo-piperidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida en lugar de N-[1-alil-5-(1-metil-2-oxo-pirrolidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (rendimiento del 80 %). ESI+: M+1: 505. RMN 1H (400 MHz, DMSO-d6) 5 12,65 (s, 1H), 8,59 - 8,47 (m, 2H), 7,89 (dd, J = 7,7, 1,7 Hz, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,45 (d, J = 8,3 Hz, 1H), 7,38 (d, J = 1,6 Hz, 1H), 7,10 (dt, J = 8,3, 1,5 Hz, 1H), 4,78 (sa, 1H), 4,52 (t, J = 5,7 Hz, 1H), 4,44 - 4,28 (m, 2H), 3,68 (dd, J = 8,4, 5,9 Hz, 1H), 3,55 - 3,42 (m, 2H), 3,42 - 3,35 (m, 2H), 3,29 - 3,23 (m, 1H), 2,91 (s, 3H), 2,19 - 1,99 (m, 2H), 1,99 - 1,79 (m, 3H), 1,79 - 1,64 (m, 1H).
Ejemplos 22a, 22b, 22c y 22d: N-[1-((R)-3,4-Dihidroxi-butil)-5-((S)-1-metil-2-oxo-piperidin-3-il)-1,3-dihidrobenzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (22a), N-[1-((S)-3,4-dihidroxi-butil)-5-((S)-1-metil-2-oxo-piperidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (22b), N-[1-((R)-3,4-dihidroxi-butil)-5-((R)-1-metil-2-oxo-piperidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (22c) y N-[1-((S)-3,4-dihidroxi-butil)-5-((R)-1-metil-2-oxo-piperidin-3-il)-1,3-dihidro-benzoimidazol-(2E)-iliden]-3-trifluorometil-benzamida (22d).
Los cuatro diastereoisómeros del ejemplo 22 se separaron mediante cromatografía SFC quiral (columna CHIRALPAK IA, 10 x 250 mm, 5|jm, gradiente isocrático MeOH al 60 % con DMEA al 0,5 %, 8 ml/min, 100 Bar CO235 °C).
Primera fracción de elución (22a): 12 mg de un sólido de color blanco, tiempo de retención: 4,97 min. ESI+ (M+1): 505. RMN 1H (400 MHz, DMSO-d6) 5 12,70 (s, 1H), 8,57 - 8,48 (m, 2H), 7,89 (d, J = 7,7 Hz, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,45 (d, J = 8,3 Hz, 1H), 7,38 (d, J = 1,6 Hz, 1H), 7,10 (dd, J = 8,3, 1,7 Hz, 1H), 4,77 (d, J = 5,0 Hz, 1H), 4,53 (t, J = 5,7 Hz, 1H), 4,45 - 4,28 (m, 2H), 3,67 (dd, J = 8,5, 5,8 Hz, 1H), 3,54 - 3,38 (m, 2H), 3,42 - 3,31 (m, 2H), 3,33 - 3,22 (m, 1H), 2,91 (s, 3H), 2,17 - 1,98 (m, 2H), 1,97 - 1,81 (m, 3H), 1,86 - 1,65 (m, 1H).
Segunda fracción de elución (22b): 14 mg de un polvo de color blanco, tiempo de retención: 6,41 min ESI+ (M+1): 505. RMN 1H (400 MHz, DMSO-d6) 5 12,70 (s, 1H), 8,57 - 8,48 (m, 2H), 7,92 - 7,85 (m, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,45 (d, J = 8,3 Hz, 1H), 7,38 (d, J = 1,6 Hz, 1H), 7,10 (dd, J = 8,3, 1,6 Hz, 1H), 4,78 (d, J = 4,9 Hz, 1H), 4,53 (t, J = 5,6 Hz, 1H), 4,45 - 4,28 (m, 2H), 3,67 (dd, J = 8,4, 5,8 Hz, 1H), 3,52 - 3,33 (m, 4H), 3,30 - 3,21 (m, 1H), 2,91 (s, 3H), 2,17 - 1,99 (m, 2H), 1,97 - 1,77 (m, 3H), 1,79 - 1,65 (m, 1H).
Tercera fracción de elución (22c): 10 mg de un polvo de color blanco, tiempo de retención: 6,98 min ESI+ (M+1): 505. RMN 1H (400 MHz, DMSO-d6) 5 12,70 (s, 1H), 8,57 - 8,48 (m, 2H), 7,89 (d, J = 7,7 Hz, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,45 (d, J = 8,3 Hz, 1H), 7,38 (d, J = 1,6 Hz, 1H), 7,10 (dd, J = 8,3, 1,7 Hz, 1H), 4,79 (sa, 1H), 4,53 (sa, 1H), 4,41 - 4,30 (m, 2H), 3,67 (dd, J = 8,4, 5,9 Hz, 1H), 3,53 - 3,42 (m, 2H), 3,47 - 3,34 (m, 2H), 3,28 - 3,17 (m, 1H), 2,91 (s, 3H), 2,17 - 1,98 (m, 2H), 1,97 - 1,65 (m, 3H), 1,33 - 1,14 (m, 1H).
Cuarta fracción de elución (22d): 12 mg de un polvo de color blanco, tiempo de retención: 8,15 min ESI+ (M+1): 505. RMN 1H (400 MHz, DMSO-d6) 5 12,71 (s, 1H), 8,57 - 8,48 (m, 2H), 7,89 (dt, J = 7,9, 1,3 Hz, 1H), 7,73 (t, J = 7,7 Hz, 1H), 7,45 (d, J = 8,3 Hz, 1H), 7,38 (d, J = 1,5 Hz, 1H), 7,10 (dd, J = 8,3, 1,7 Hz, 1H), 4,78 (sa, 1H), 4,54 (sa, 1H), 4,41 - 4,30 (m, 2H), 3,67 (dd, J = 8,5, 5,8 Hz, 1H), 3,53 - 3,34 (m, 4H), 3,27 - 3,18 (m, 1H), 2,91 (s, 3H), 2,14 - 2,00 (m, 2H), 1,97 - 1,65 (m, 4H).
Ejemplo 23: N-[1-(3-Hidroxipropil)-5-(1-metil-2-oxopirrolidin-3-il)-1H-1,3-benzodiazol-2-il]-3-(trifluorometil)benzamida (23)
Etapa 1: N-(1-{3-[(ferc-Butildimetilsilil)oxi]propil}-5-(1-metil-2-oxopirrolidin-3-il)-1H-1,3-benzodiazol-2-il)-3-(trifluorometil)benzamida

Una solución de ácido 3-(trifluorometil)benzoico (68 mg; 0,36 mmol; 1,00 eq.), HOBT (58 mg; 0,43 mmol; 1,20 eq.), EDCI (82 mg; 0,43 mmol; 1,20 eq.), DIEA (138 mg; 1,07 mmol; 3,00 eq.) y 3-(2-amino-1-[3-[(terc-butildimetilsilil)oxi]propil]-1H-1,3-benzodiazol-5-il)-1-metilpirrolidin-2-ona (compuesto intermedio 11, 150 mg; 0,36 mmol; 1,00 eq.) en DMF (2 ml) se agitó durante toda la noche a temperatura ambiente. A continuación, se diluyó con EtOAc y se lavó con una solución de NH4Cl (2 x 50 ml) saturada y salmuera (2 * 50 ml). La capa orgánica se secó sobre sulfato de sodio anhidro, se filtró y se concentró para obtener el compuesto del título como un sólido de color marrón (240 mg, 82 %). ESI+ (M+1): 575,3.
Etapa 2: N-[1-(3-Hidroxipropil)-5-(1-metil-2-oxopirrolidin-3-il)-1H-1,3-benzodiazol-2-il]-3-(trifluorometil)benzamida

Una solución de N-(1-[3-[(terc-butildimetilsilil)oxi]propil]-5-(1-metil-2-oxopirrolidin-3-il)-1H-1,3-benzodiazol-2-il)-3-(trifluorometil)benzamida (240 mg; 0,29 mmol; 1,00 eq.) y TbA f (306 mg; 1,17 mmol; 4,00 eq.) en THF (4 ml) se agitó durante 3 h a temperatura ambiente. Esta solución se diluyó con EtOAc (60 ml) y se lavó con solución de NH4Cl saturada (3 * 60 ml) y salmuera (60 ml). La fase orgánica se secó sobre sulfato sódico anhidro, se filtró y se concentró. La purificación mediante HPLC preparativa permitió obtener el compuesto del título como un sólido de color rosa (110 mg, 81 %). ESI+: (M+1): 461,2. RMN 1H (400 MHz, CDCla) 5 12,48 (s, 1H), 8,53(s, 1H), 8,46-8,44(d, J=8, 1H), 7,78-7,76 (d, J=8, 1H), 7,62-7,58 (m, 1H), 7,31-7,28 (m, 2H), 7,25-7,23 (m, 1H), 4,71 (s, 1H), 4,50-4,47 (m, 2H), 3,80-3,75 (m, 1H), 3,53 3,46 (m, 4H), 2,98 (s, 3H), 2,64-2,56 (m, 1H), 2,20-2,13 (m, 1H), 2,10-2,01 (m, 2H).
Ejemplos 23a y 23b: N-[1-(3-Hidroxipropil)-5-[(3S)-1-metil-2-oxopirrolidin-3-il]-1H-1,3-benzodiazol-2-il]-3-(trifluorometil)benzamida (23a) y N-[1-(3-hidroxipropil)-5-[(3R)-1-metil-2-oxopirrolidin-3-il]-1H-1,3-benzodiazol-2-il]-3-(trifluorometil)benzamida (23b)
Los dos enantiómeros de N-[1-(3-hidroxipropil)-5-(1-metil-2-oxopirrolidin-3-il)-1H-1,3-benzodiazol-2-il]-3-(trifluorometil)benzamida (60 mg) se separaron mediante una columna de HPLC preparativa quiral (columna CHIRALPAK-AD-H-SL002, 20 * 250 mm; hexano:EtOH, 50:50). Primera fracción de elución: 23a, 21 mg de un sólido blando, segundo fracción de elución: 23b, 21 mg de un sólido de color blanco. p.f.: 161-164 °C.
Ejemplo 24: 3-(Difluorometil)-N-[1-(3-hidroxipropil)-5-(1-metil-2-oxopirrolidin-3-il)-1H-1,3-benzodiazol-2-il]benzamida (24)

El compuesto del título se obtuvo siguiendo el procedimiento descrito para el ejemplo 23, pero usando ácido 3-(difluorometil)benzoico (enamina, 41 mg; 0,24 mmol; 1 eq.) en lugar de ácido 3-(trifluorometil)benzoico como un sólido de color blanco. ESI+: M+1: 443,2. RMN 1H (400 MHz, CDCla) 5 12,52 (s, 1H), 8,39-8,37 (m, 2H), 7,69-7,67 (d, J=8, 1H), 7,59-7,55 (m, 1H), 7,30-7,27 (m, 2H), 7,24-7,21 (m, 1H), 6,89-6,61 (m, 1H), 4,82 (s, 1H), 4,49-4,46 (m, 2H), 3,77-3,73 (m, 1H), 3,51-3,45 (m, 4H), 2,97 (s, 3H), 2,61-2,53 (m, 1H), 2,18-2,08 (m, 1H), 2,04-2,00 (m, 2H). p.f.: 166,0-168,0 °C.
Ejemplo 25: N-[1-(3-Hidroxi-propil)-5-(1,4,4-trimetil-5-oxo-pirrolidin-3-il)-1H-benzoimidazol-2-il]-3-trifluorometilbenzamida (25)

El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 1 del ejemplo 23 pero empezando a partir de 4-[2-amino-1-(3-hidroxipropil)-1H-1,3-benzodiazol-5-il]-1,3,3-trimetilpirrolidin-2-ona (100 mg, 0,28 mmol, 1,00 eq.) y ácido 3-(trifluorometil)benzoico (57 mg, 0,28 mmol, 1,00 eq.) como un (30 mg; 21 %). RMN 1H (300 MHz, CDCb) 5: 12,50 (s, 1H), 8,52 (s, 1H), 8,44 (d, J = 7,8 Hz, 1H), 7,77 (d, J = 7,7 Hz, 1H), 7,60 (t, J = 7,7 Hz, 1H), 7,30 (d, J = 8,3 Hz, 1H), 7,26 - 7,21 (m, 1H), 7,17 (dd, J = 8,4, 1,5 Hz, 1H), 4,50 (t, J = 6,0 Hz, 3H), 3,67 - 3,57 (m, 2H), 3,53 (t, J = 5,5 Hz, 2H), 3,35 (t, J = 7,9 Hz, 1H), 2,99 (s, 3H), 2,08 (p, J = 5,7 Hz, 2H), 1,27 (s, 3H), 0,77 (s, 3H). ESI+ (M+1): 489,3. p.f.: 104 106 °C.
Ejemplos 25a y 25b: N-[1-(3-Hidroxipropil)-5-[(3S)-1,4,4-trimetil-5-oxopirrolidin-3-il]-1H-1,3-benzodiazol-2-il]-3-(trifluorometil)benzamida (25a) y N-[1-(3-hidroxipropil)-5-[(3R)-1,4,4-trimetil-5-oxopirrolidin-3-il]-1H-1,3-benzodiazol-2-il]-3-(trifluorometil)benzamida (25b)
Los dos enantiómeros de N-[1-(3-hidroxi-propil)-5-(1,4,4-trimetil-5-oxo-pirrolidin-3-il)-1H-benzoimidazol-2-il]-3-trifluorometil-benzamida se separaron mediante HPLC preparativa quiral (columna IA, 21,2 * 250 mm,5 micras, hexano:EtOH, 50:50).
Primera fracción de elución: 25a, 34 mg de un sólido de color blanco, p.f.: 90-91 °C, segunda fracción de elución: 25b, 30 mg de un sólido de color blanco. p.f.: 88-89 °C.
Ejemplo 26: 3-Difluorometil-N-[1-(2-fluoro-3-hidroxi-2-metil-propil)-5-(4-metil-3-oxo-morfolin-2-il)-1H-benzoimidazol-2-il]-benzamida (26)
Etapa 1: N-{1-[3-(Benciloxi)-2-fluoro-2-metilpropil]-5-(4-metil-3-oxomorfolin-2-il)-1H-1,3-benzodiazol-2-il}-3-(difluorometil)benzamida
El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 1 del ejemplo 23 pero empezando a partir de ácido 3-(difluorometil)benzoico (83,5 mg; 0,49 mmol; 1,27 eq.) y 2-[2-amino-1-[3-(benciloxi)-2-fluoro-2-metilpropil]-1H-1,3-benzodiazol-5-il]-4-metilmorfolin-3-ona (compuesto intermedio 16, 245 mg; 0,38 mmol; 1,00 eq.) como un sólido de color rojo (200 mg; 86 %).
Etapa 2: 3-Difluorometil-N-[1-(2-fluoro-3-hidroxi-2-metil-propil)-5-(4-metil-3-oxo-morfolin-2-il)-1H-benzoimidazol-2-il]-benzamida

Una suspensión desgaseada de N-[1-[3-(benciloxi)-2-fluoro-2-metilpropil]-5-(4-metil-3-oxomorfolin-2-il)-1H-1,3-benzodiazol-2-il]-3-(difluorometil)benzamida (80 mg; 0,13 mmol; 1,00 eq.) y paladio carbono (8,06 mg; 0,07 mmol; 0,55 eq.) en MeOH (8,00 ml) se hidrogenó bajo presión atmosférica de hidrógeno durante toda la noche a TA. Los sólidos se recogieron por filtración y el filtrado se concentró y purificó mediante HPLC preparativa para obtener el compuesto del título como un sólido de color blanco (20 mg; 30 %). RMN 1H (300 MHz, DMSO) 512,87 (d, J=7,9Hz,1H),8,40 (d, J=7,5Hz, 2H), 7,83-7,53 (m, 3H), 7,43 (d, J=7,5Hz, 1H), 7,36-6,93 (m, 2H), 5,37 (t, J = 6,1Hz,1 H), 5,19 (s, 1H), 4,72-4,39 (m, 2H), 4,04-3,84 (m, 2H), 3,61 (ddt, J=17,2, 10,6, 6,7Hz, 3H), 2,93 (s, 3H), 1,32 (d, J=22,2Hz, 3H). ESI+ (M+1): 491,3. p.f.: 118 120 °C.
Ejemplo 27: 3-Difluorometil-N-[1-(3-hidroxi-propil)-5-(1,4,4-trimetil-5-oxo-pirrolidin-3-il)-1H-benzoimidazol-2-il]-benzamida (27)

El compuesto del título se obtuvo siguiendo el procedimiento descrito para el ejemplo 23 pero empezando a partir de 4-[2-amino-1-(3-hidroxipropil)-1H-1,3-benzodiazol-5-il]-1,3,3-trimetilpirrolidin-2-ona (compuesto intermedio 15, 20 mg; 0,06 mmol; 1,00 eq.) y ácido 3-(difluorometil)benzoico (10,3 mg; 0,06 mmol; 1,00 eq.) como un sólido de color blanco (5 mg; 18 %). RMN 1H (300 MHz,CDCla) 5 12,55 (s, 1H), 8,42 - 8,32 (m, 2H), 7,67 (d, J = 7,6 Hz, 1H), 7,56 (t, J = 7,9 Hz, 1H), 7,29 (s, 1H), 7,23 - 7,10 (m, 2H), 4,69 (t, J = 7,1 Hz, 1H), 4,54 - 4,43 (m, 2H), 3,55 (dd, J = 22,7, 6,8 Hz, 4H), 3,32 (t, J = 7,9 Hz, 1H), 2,97 (s, 3H), 2,05 (td, J = 10,0, 8,4, 4,1 Hz, 2H), 1,24 (s, 3H), 0,74 (s, 3H). ESI+ (M+1): 471,3. p.f.: 95-96 °C.
Ejemplos 27a y 27b: 3-(Difluorometil)-N-[1-(3-hidroxipropil)-5-[(3R)-1,4,4-trimetil-5-oxopirrolidin-3-il]-1H-1,3-benzodiazol-2-il]benzamida (27a) y 3-(difluorometil)-N-[1-(3-hidroxipropil)-5-[(3S)-1,4,4-trimetil-5-oxopirrolidin-3-il]-1 H-1,3-benzodiazol-2-il]benzamida (27b)
Los dos enantiómeros de 3-difluorometil-N-[1-(3-hidroxi-propil)-5-(1,4,4-trimetil-5-oxo-pirrolidin-3-il)-1 H-benzoimidazol-2-il]-benzamida se separaron mediante HPLC preparativa quiral (columna IC, 21,2 * 250 mm, 5 |jm, hexano:EtOH, 50:50). Primera fracción de elución: 27a, 20 mg de un sólido de color blanco, p.f.: 94-95 °C, segunda fracción de elución: 27b, 20 mg de un sólido de color blanco. p.f.: 94-95 °C.
benzamida (28)

El compuesto del título se obtuvo siguiendo el procedimiento descrito para el ejemplo 26 pero empezando a partir de 2-[2-amino-1-[3-(benciloxi)-2-fluoro-2-metilpropil]-1H-1,3-benzodiazol-5-il]-4-metilmorfolin-3-ona (compuesto intermedio 16, 100 mg; 0,21 mmol; 1,00 eq.) y ácido 3-(trifluorometil)benzoico (50 mg; 0,26 mmol; 1,22 eq.) como un sólido de color blanco (80 mg; 67 %; 2 etapas). RMN 1H (300MHz, DMSO-d6) 5 12,87 (s,1H), 8,56-8,45 (m,2h), 7,90 (d, J=7,9Hz, 1H), 7,78-7,69 (m, 1H), 7,60-7,53 (m, 1H), 7,43 (dd, J=8,6, 2,2Hz, 1H), 7,25 (d, J=8,2Hz, 1H), 5,40 (s, 1H), 5,19 (s, 1H), 4,72 4,40 (m, 2H), 4,05-3,85 (m, 2H), 3,72-3,51 (m, 3H), 3,44-3,38 (m, 1H), 2,93 (s, 3H), 1,32 (d, J=22,2Hz, 3H). ESI+ (M+1): 509,2. p.f.: 121-124 °C.
Ejemplo 29: N-[5-(1,3-Dimetil-2-oxo-pirrolidin-3-il)-1-(3-hidroxi-propil)-1 H-benzoimidazol-2-il]-3-trifluorometil-benzamida (29)
Etapa 1: N-(1-{3-[(ferc-Butildimetilsilil)oxi]propil}-5-(1,3-dimetil-2-oxopirrolidin-3-il)-1 H-1,3-benzodiazol-2-il)-3-(trifluorometil)benzamida

Una mezcla de 3-(2-amino-1-[3-[(ferc-butildimetilsilil)oxi]propil]-1 H-1,3-benzodiazol-5-il)-1,3-dimetilpi rrolidin-2-ona (compuesto intermedio 17, 250 mg; 0,54 mmol; 1,00 eq.), ácido 3-(trifluorometil)benzoico (137 mg; 0,71 mmol; 1,31 eq.), HAt U (231 mg; 0,60 mmol; 1,10 eq.) y DIEA (107 mg; 0,81 mmol; 1,50 eq.) en DMF (3 ml) se agitó a TA durante 2 h. El solvente se eliminó a presión reducida y el producto sin procesar se purificó mediante cromatografía ultrarrápida sobre gel de sílice (DCM: MeOH, gradiente de 100:1 a 30:1) para obtener el compuesto del título como un sólido de color amarillo (270 mg, 76 %). ESI+ (M+1): 589,2.
Etapa 2: N-[5-(1,3-Dimetil-2-oxo-pirrolidin-3-il)-1-(3-hidroxi-propil)-1H-benzoimidazol-2-il]-3-trifluorometil-benzamida
Se introdujo cloruro de hidrógeno (250 mg, 6,72 mmol, 17,58 eq.) en una solución de N-(1-[3-[(fercbutildimetilsilil)oxi]propil]-5-(1,3-dimetil-2-oxopirrolidin-3-il)-1 H-1,3-benzodiazol-2-il)-3-(trifluorometil)benzamida (250 mg; 0,38 mmol; 1,00 eq.) en dioxano (3 ml). La mezcla de reacción se agitó durante 2 h a TA y se concentró al vacío. La purificación mediante HPLC preparativa permitió obtener el compuesto del título como un sólido de color blanco (35 mg, 19 %). RMN 1H (300MHz, DMSO-d6, ppm) 512,71 (s, 1H), 8,57-8,47 (m, 2H), 7,94-7,84 (m, 1H), 7,79-7,67 (m, 1H), 7,64 7,57 (m, 1H), 7,48 (d, J=8,5Hz, 1H), 7,29 (dd, J=8,5, 1,8Hz, 1H), 4,65 (t, J=5,1Hz, 1H), 4,32 (t, J=6,9Hz, 2H), 3,53-3,43 (m, 2H), 3,41-3,33 (m, 1H), 3,32-3,23 (m, 1H), 2,83 (s, 3H), 2,38-2,28 (m, 1H), 2,22-2,10 (m, 1H), 2,01-1,90 (m, 2H), 1,45 (s, 3H). ESI+ (M+1): 475,2. p.f.: 72-74 °C.
Ejemplos 29a y 29b: N-{5-[(3R)-1,3-Dimetil-2-oxopirrolidin-3-il]-1-(3-hidroxipropil)-1H-1,3-benzodiazol-2-il}-3-(trifluorometil)benzamida (29a) y N-{5-[(3S)-1,3-dimetil-2-oxopirrolidin-3-il]-1-(3-hidroxipropil)-1H-1,3-benzodiazol-2-il}-3-(trifluorometil)benzamida (29b)
Los dos enantiómeros de N-[5-(1,3-dimetil-2-oxo-pirrolidin-3-il)-1-(3-hidroxi-propil)-1H-benzoimidazol-2-il]-3-trifluorometilbenzamida se separaron mediante HPLC preparativa quiral (columna alfa Phenomenex Lux, 21,2 * 250 mm, 5 |jm, hexano:EtOH, 50:50). Primera fracción de elución: 29a, 30 mg de un sólido de color blanco, p.f.: 78-81 °C, segunda fracción de elución: 29b, 30 mg de un sólido de color blanco. p.f.: 78-81 °C.
Ejemplo 30: 3-Difluorometil-N-[5-(1,3-dimetil-2-oxo-pirrolidin-3-il)-1-(3-hidroxi-propil)-1H-benzoimidazol-2-il]-benzamida (30)

El compuesto del título se preparó siguiendo el procedimiento descrito para el ejemplo 29, pero partiendo de 3-(2-amino-1-[3-[(terc-butildimetilsilil)oxi]propil]-1H-1,3-benzodiazol-5-il)-1,3-dimetilpirrolidin-2-ona (compuesto intermedio 17, 250 mg; 0,54 mmol; 1,00 eq.) y ácido 3-(difluorometil)benzoico (124 mg; 0,71 mmol; 1,31 eq.) como un sólido de color blanco (30 mg, 13 %, 2 etapas). RMN 1H (300 MHz, DMSO-d6) 5 12,69 (s, 1H), 8,45-8,36 (m, 2H), 7,76-7,68 (m, 1H), 7,68-7,56 (m, 2H), 7,52-7,42 (m, 1H), 7,36-6,93 (m, 2H), 4,65 (t, J=5,1Hz, 1H), 4,31 (t, J=6,8Hz, 2H), 3,51-3,42 (m, 2H), 3,41-3,33 (m, 1H), 3,32-3,22 (m, 1H), 2,83 (s, 3H), 2,38-2,26 (m, 1H), 2,22-2,08 (m, 1H), 2,03-1,89 (m, 2H), 1,44 (s, 3H). ESI+ (M+1): 457,2. p.f.: 78-81 °C.
Ejemplos 30a y 30b: N-{5-[(3R)-1,3-Dimetil-2-oxopirrolidin-3-il]-1-(3-hidroxipropil)-1H-1,3-benzodiazol-2-il}-3-(difluorometil)benzamida (30a) y N-{5-[(3S)-1,3-dimetil-2-oxopirrolidin-3-il]-1 -(3-hidroxi propil)-1H-1,3-benzodiazol-2-il}-3-(difluorometil)benzamida (30b)
Los dos enantiómeros de N-[5-(1,3-dimetil-2-oxo-pirrolidin-3-il)-1-(3-hidroxi-propil)-1H-benzoimidazol-2-il]-3-difluorometilbenzamida se separaron mediante HPLC preparativa quiral (columna alfa Phenomenex Lux, 21,2 * 250 mm, 5 micras, hexano:EtOH, 50:50).
Primera fracción de elución: 30a, 30 mg de un sólido de color blanco, p.f.: 78-81 °C, segunda fracción de elución: 30b, 30 mg de un sólido de color blanco. p.f.: 78-81 °C.
Ejemplo 31: 3-Difluorometil-N-[5-(1,2-dimetil-5-oxo-pirrolidin-2-il)-1-(3-hidroxi-propil)-1H-benzoimidazol-2-il]-benzamida (31)

El compuesto del título se obtuvo siguiendo el procedimiento descrito para la etapa 1 del ejemplo 29 pero empezando a partir de 5-[2-amino-1-(3-hidroxipropil)-1H-1,3-benzodiazol-5-il]-1,5-dimetilpirrolidin-2-ona (compuesto intermedio 18, 200 mg; 0,60 mmol; 1,00 eq.) y ácido 3-(difluorometil)benzoico (108 mg; 0,06 mmol; 1,00 eq.) como un sólido de color blanco (90 mg; 33 %). RMN 1H (300 MHz,CDCl3) 5 12,52 (s, 1H), 8,41 - 8,28 (m, 2H), 7,70 - 7,60 (m, 1H), 7,56 - 7,51 (m, 1H), 7,29 - 7,25 (m, 2H), 7,16 - 7,13 (m, 1H), 6,70 (t, J = 56,4 Hz, 1H), 4,63 - 4,59 (m, 1H), 4,52 - 4,39 (m, 2H), 3,50 - 3,45 (m, 2H), 2,66 (s, 3H), 2,50 - 2,44 (m, 2H), 2,22 - 1,95 (m, 4H), 1,68 (s, 3H). ESI+ (M+1): 457,2. p.f.: 98-99 °C.
Ejemplos 31a y 31b: 3-(Difluorometil)-N-{5-[(2R)-1,2-dimetil-5-oxopirrolidin-2-il]-1-(3-hidroxipropil)-1H-1,3-benzodiazol-2-il}benzamida (31a) y 3-(difluorometil)-N-{5-[(2s)-1,2-dimetil-5-oxopirrolidin-2-il]-1-(3-hidroxipropil)-1H-1,3-benzodiazol-2-il}benzamida (31b)
Los dos enantiómeros de 3-difluorometil-N-[5-(1,2-dimetil-5-oxo-pirrolidin-2-il)-1-(3-hidroxi-propil)-1 H-benzoimidazol-2-il]-benzamida se separaron mediante HPLC preparativa quiral (columna CHIRAL ART celulosa SB Lux, 20 * 250 mm, hexano:EtOH, 65:35). Primera fracción de elución: 31a, 24 mg de un sólido de color marrón., p.f.: 98-99 °C, segunda fracción de elución: 31b, 21 mg de un sólido de color beis. p.f.: 98-99 °C.
Ejemplo 32: N-[5-(1,2-Dimetil-5-oxo-pirrolidin-2-il)-1-(3-hidroxi-propil)-1H-benzoimidazol-2-il]-3-trifluorometil-benzamida (32)

El compuesto del título se obtuvo siguiendo el procedimiento descrito para el ejemplo 31 comenzando a partir de 5-[2-amino-1-(3-hidroxipropil)-1H-1,3-benzodiazol-5-il]-1,5-dimetilpirrolidin-2-ona (compuesto intermedio 18, 300 mg, 0,89 mmol, 1,00 eq.) y ácido 3-(trifluorometil)benzoico (179 mg; 0,89 mmol; 1,00 eq.), como un sólido de color blanco (90 mg, 21 %). RMN 1H (300 MHz,CDCl3) 5 12,56 (s, 1H), 8,56 - 8,38 (m, 2H), 7,77 - 7,7 (m, 1H), 7,59 - 7,45 (m, 1H), 7,35 - 7,27 (m, 2H), 7,19 - 7,17 (m, 1H), 4,58 - 4,36 (m, 2H), 3,54 - 3,50 (m, 2H), 2,69 (s, 3H), 2,50 - 2,47 (m, 2H), 2,23 - 1,99 (m, 4H), 1,72 (s, 3H). ESI+ (M+1): 475,2. p.f.: 99-100 °C.
Ejemplos 32a y 32b: N-{5-[(2R)-1,2-Dimetil-5-oxopirrolidin-2-il]-1-(3-hidroxipropil)-1H-1,3-benzodiazol-2-il}-3-(trifluorometil)benzamida (32a) y N-{5-[(2S)-1,2-dimetil-5-oxopirrolidin-2-il]-1-(3-hidroxipropil)-1H-1,3-benzodiazol-2-il}-3-(trifluorometil)benzamida (32b)
Los dos enantiómeros de N-[5-(1,2-dimetil-5-oxo-pirrolidin-2-il)-1-(3-hidroxi-propil)-1H-benzoimidazol-2-il]-3-trifluorometilbenzamida se separaron mediante HPLC preparativa quiral (columna ChiralPak ID, 20 * 250 mm, 5 micras, MTBE:EtOH, 70:30).
Primera fracción de elución: 32a, 32 mg de un de color blanco, p.f.: 99-100 °C, segunda fracción de elución: 32b, 29 mg de un sólido de color blanco, p.f.: 99-100 °C.
Ejemplo 33: Ensayos enzimáticos
Ensayo enzimático para IRAK4:
IRAK4 es una enzima recombinante humana purificada (His-TEV-IRAK4 [1-460]).
En este ensayo, IRAK4 hidroliza el ATP, se autofosforila y fosforila un sustrato peptídico genérico serina/treonina (STK: 61ST1BLC de CisBio International). La medición de la inhibición de IRAK-4 se realiza en un formato de 384 pocillos basado en un ensayo de luminiscencia (ensayo de actividad quinasa ADP-Glo™ de Promega). IRAK4 recombinante humana purificada (0,3 |jg/ml) y los compuestos diluidos en serie en DMSO (intervalo de concentraciones de 10 |jM a 0,5 nM) o los controles (DMSO al 1 %) se incuban durante 15 minutos a TA en tampón de ensayo que contiene Hepes 50 mM pH 7,0, BSA libre de ácidos grasos al 0,1 %, ditiotreitol (DTT) 2 mM, MgCh 10 mM, EGTA 0,5 mM, Tritón X-100 al 0,01 % y MnCl2 5 mM. A continuación, la reacción quinasa se inicia mediante la adición de ATP (2 jM ) y el sustrato peptídico péptido STK1-biotina (300 mM). Después de 2 horas de incubación a TA, la reacción se detiene y el ATP no consumido se elimina mediante la adicción del reactivo ADP-Glo™ según las instrucciones del proveedor. Después de 40 minutos de incubación a TA, se añade a la placa de ensayo el reactivo de detección de quinasas según las instrucciones del proveedor. Después de 20 minutos de incubación a TA, se mide la señal de luminiscencia con un luminómetro de lectura de placas (PerkinElmer Envision o lector equivalente).
Ensayo enzimático para IRAK1:
IRAK1 es una enzima recombinante purificada humana (His-TEV-IRAK1 [194-712])
En este ensayo, IRAK1 hidroliza el ATP y se autofosforila. La medición de la inhibición de IRAK-1 se realiza en un formato de 384 pocillos basado en un ensayo de luminiscencia (ensayo de actividad quinasa ADP-Glo™ de Promega). IRAK1 recombinante humano purificado (0,3 jg/m l) y los compuestos diluidos en serie en DMSO (intervalo de concentraciones de 10 jM a 0,5 nM) o los controles (DMSO al 1 %) se incuban durante 15 minutos a TA en tampón de ensayo que contiene Hepes 50 mM pH 7,0, BSA libre de ácidos grasos al 0,1 %, ditiotreitol (DTT) 2 mM, MgCh 10 mM, EGTA 0,5 mM y Tritón X-100 al 0,01 %. A continuación, la reacción quinasa se inicia mediante la adición de ATP a una concentración de 1 jM . Después de 2 horas de incubación a TA, la reacción se detiene y el ATP no consumido se elimina mediante la adicción del reactivo ADP-Glo™ según las instrucciones del proveedor. Después de 40 minutos de incubación a TA, se añade a la placa de ensayo el reactivo de detección de quinasas según las instrucciones del proveedor. Después de 20 minutos de incubación a TA, se mide la señal de luminiscencia con un luminómetro (PerkinElmer Envision o lector equivalente).
IL-6 inducida por TLR7 en PBMC humanas
Los ensayos con células mononucleares de sangre periférica (PBMC) humanas se usaron como uno de los ensayos funcionales para monitorizar la actividad de moléculas pequeñas inhibidoras de IRAK1 e IRAK4 sobre la secreción de IL-6 inducida por TLR7 en PBMC humanas. Las PBMC humanas se prepararon a partir de las capas leucoplaquetarias (sangre completa enriquecida en leucocitos y plaquetas) obtenidas de voluntarios sanos utilizadas recién obtenidas o congeladas, se colocaron en placas con medio del ensayo (RPMI P/S/L-glu al 2 % HI-FBS al 10 %) y se trataron previamente con compuestos en DMSO/medios (intervalo de concentraciones de 25 |jM a 0,4 nM) o controles (DMSO al 0,25 %) durante 30 minutos a 37 °C en el medio del ensayo. Tras el tratamiento previo con los inhibidores de IRAK1 e IRAK4, las PBMC se estimularon con ligando específico de TLR7 (2 jM ) durante toda la noche (16-18 horas) a 37 °C. Tras la incubación, el sobrenadante se transfirió a placas AlphaPlate-384 PE de 384 pocillos (6005350) y se cuantificó la IL-6 usando un kit Alpha LISA para IL-6 de Perkin Elmer (AL223C). Las placas se leyeron en un lector de placas Envision® con Alpha Technology®.
Los resultados se resumen en la tabla siguiente.
Tabla 2



Ejemplo 34: Preparaciones farmacéuticas
(A) Viales para inyección: se ajusta el pH de una solución de 100 g de un principio activo según la invención y 5 g de hidrogenofosfato disódico en 3 litros de agua bidestilada a pH 6,5 usando ácido clorhídrico 2 N, se esteriliza mediante filtración, se transfiere a viales para inyección, se liofiliza en condiciones estériles y se sella en condiciones estériles. Cada vial para inyección contiene 5 mg del principio activo.
(B) Supositorios: se funde una mezcla de 20 g de un principio activo según la invención con 100 g de lecitina de soja y 1400 g de manteca de cacao, se vierte en los moldes y se deja enfriar. Cada supositorio contiene 20 mg de principio activo.
(C) Solución: se prepara una solución de 1 g de un principio activo según la invención, 9,38 g de NaH2PO4 • 2 H2O, 28,48 g de Na2HPO4, 12 H2O y 0,1 g de cloruro de benzalconio en 940 ml de agua bidestilada. El pH se ajusta a 6,8, la solución se lleva a 1 litro y se esteriliza mediante radiación. Esta solución podría usarse en forma de colirio.
(D) Pomada: se mezclan 500 mg de un principio activo según la invención con 99,5 g de vaselina en condiciones asépticas.
(E) Comprimidos: se prensa una mezcla de 1 kg de un principio activo según la invención, 4 kg de lactosa, 1,2 kg de almidón de patata, 0,2 kg de talco y 0,1 kg de estearato de magnesio para obtener comprimidos de la forma habitual, de manera que cada comprimido contiene 10 mg de principio activo.
(F) Comprimidos recubiertos: los comprimidos se prensan de forma análoga al ejemplo E y, posteriormente, se recubren de la forma habitual con un recubrimiento de sacarosa, almidón de patata, talco, goma de tragacanto y colorante.
(G) Cápsulas: se introducen 2 kg de un principio activo según la invención dentro de cápsulas de gelatina dura de la forma habitual, de modo que cada cápsula contiene 20 mg de principio activo.
(H) Ampollas: se esteriliza por filtración una solución de 1 kg de un principio activo según la invención en 60 litros de agua bidestilada, se transfiere a ampollas, se liofiliza en condiciones estériles y se sella en condiciones estériles. Cada ampolla contiene 10 mg del principio activo.
(I) Spray para inhalación: se disuelven 14 g de un principio activo según la invención en 10 litros de solución isotónica de NaCl y la solución se transfiere a recipientes para aerosoles disponibles en el mercado con un mecanismo de bombeo. La solución podría rociarse en la boca o la nariz. Cada descarga del inhalador (aproximadamente 0,1 ml) se corresponde con una dosis de aproximadamente 0,14 mg.
Aunque aquí se describen varias realizaciones de esta invención, es evidente que los ejemplos básicos pueden alterarse para proporcionar otras realizaciones que utilicen los compuestos y métodos de esta invención. Por tanto, se apreciará que el alcance de esta invención se define mediante las reivindicaciones adjuntas en lugar de por las realizaciones específicas que se ha presentado a modo de ejemplo.
Claims (8)
1. Un compuesto de fórmula I,

o una sal farmacéuticamente aceptable del mismo, donde:

R4 es -R , halógeno, -haloalquilo, -OR, -SR, -CN, -NO 2 , -SO2 R, -SOR, -C(O)R, -CO2 R, -C(O)N(R)2 , -NRC(O)R, -NRC(O)N(R)2 , -NRSO2R o -N(R)2 ; y
cada R es independientemente hidrógeno, grupo alifático C1 -6 , arilo C3 -10 , un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado, un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico de 5-6 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido; o
se toman dos grupos R del mismo átomo junto con el átomo al que están unidos para formar un arilo C3-10, un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado, un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroarilo monocíclico
de 5-6 átomos que tiene 1-4 heteroátomos, seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido.
2. El compuesto de la reivindicación 1, donde R3 es un grupo alifático C1-6 , un anillo carbocíclico de 3-8 átomos saturado o parcialmente insaturado o un anillo heterocíclico de 3-7 átomos que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada uno de los cuales está opcionalmente sustituido.
3. El compuesto de la reivindicación 2, donde R3 es metilo, etilo, propilo, i-propilo, n-butilo, s-butilo, t-butilo, un pentilo de cadena lineal o ramificada, o un hexilo de cadena lineal o ramificada; cada uno de ellos opcionalmente sustituido.
4. El compuesto la reivindicación 3, donde R3 se selecciona entre:
/ ......... /O H / 0


5. El compuesto de la reivindicación 1, seleccionado entre


6. Una composición farmacéutica que comprende un compuesto de la reivindicación 1 y un adyuvante, transportador o vehículo farmacéuticamente aceptable.
7. Un compuesto según la reivindicación 1 para su uso en el tratamiento de un trastorno mediado por IRAK, en el que el trastorno se selecciona entre artritis reumatoide, artritis psoriásica, artrosis, lupus eritematoso sistémico, nefritis lúpica, espondilitis anquilosante, osteoporosis, esclerosis sistémica, esclerosis múltiple, psoriasis, diabetes de tipo I, diabetes de tipo II, enfermedad inflamatoria intestinal (enfermedad de Crohn y colitis ulcerosa), hiperinmunoglobulinemia D y síndrome de fiebre periódica, síndromes periódicos asociados con criopirina, síndrome de Schnitzler, artritis idiopática juvenil sistémica, enfermedad de Still del adulto, gota, pseudogota, síndrome SAPHO, enfermedad de Castleman, septicemia, ictus, ateroesclerosis, enfermedad celíaca, DiRa (deficiencia del antagonista del receptor de IL-1), enfermedad de Alzheimer, enfermedad de Parkinson y cáncer.
8. El compuesto para su uso según la reivindicación 7, en el que el trastorno se selecciona entre artritis reumatoide, lupus eritematoso sistémico, nefritis lúpica y esclerosis múltiple.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562220358P | 2015-09-18 | 2015-09-18 | |
| PCT/US2016/052107 WO2017049069A1 (en) | 2015-09-18 | 2016-09-16 | Heteroaryl compounds as irak inhibitors and uses thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2911888T3 true ES2911888T3 (es) | 2022-05-23 |
Family
ID=56991011
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16770654T Active ES2911888T3 (es) | 2015-09-18 | 2016-09-16 | Compuestos de heteroarilo como inhibidores de IRAK y usos de los mismos |
Country Status (13)
| Country | Link |
|---|---|
| US (2) | US9969710B2 (es) |
| EP (1) | EP3350170B1 (es) |
| JP (1) | JP7083750B2 (es) |
| KR (1) | KR102650026B1 (es) |
| CN (1) | CN108602776B (es) |
| AU (1) | AU2016323613B2 (es) |
| CA (1) | CA2996318C (es) |
| ES (1) | ES2911888T3 (es) |
| IL (1) | IL258081B2 (es) |
| MX (1) | MX2018002986A (es) |
| RU (1) | RU2743360C2 (es) |
| WO (1) | WO2017049069A1 (es) |
| ZA (1) | ZA201801056B (es) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI808055B (zh) | 2016-05-11 | 2023-07-11 | 美商滬亞生物國際有限公司 | Hdac 抑制劑與 pd-1 抑制劑之組合治療 |
| TWI794171B (zh) | 2016-05-11 | 2023-03-01 | 美商滬亞生物國際有限公司 | Hdac抑制劑與pd-l1抑制劑之組合治療 |
| JOP20180011A1 (ar) | 2017-02-16 | 2019-01-30 | Gilead Sciences Inc | مشتقات بيرولو [1، 2-b]بيريدازين |
| WO2019060693A1 (en) | 2017-09-22 | 2019-03-28 | Kymera Therapeutics, Inc. | CRBN LIGANDS AND USES THEREOF |
| AU2018338314A1 (en) | 2017-09-22 | 2020-04-09 | Kymera Therapeutics, Inc | Protein degraders and uses thereof |
| WO2019111218A1 (en) | 2017-12-08 | 2019-06-13 | Cadila Healthcare Limited | Novel heterocyclic compounds as irak4 inhibitors |
| IL315310A (en) | 2017-12-26 | 2024-10-01 | Kymera Therapeutics Inc | IRAK joints and used in them |
| WO2019140380A1 (en) | 2018-01-12 | 2019-07-18 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US11512080B2 (en) | 2018-01-12 | 2022-11-29 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| JP7313364B2 (ja) | 2018-02-21 | 2023-07-24 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Egfr阻害剤としての新たなベンズイミダゾール化合物および誘導体 |
| US11292792B2 (en) | 2018-07-06 | 2022-04-05 | Kymera Therapeutics, Inc. | Tricyclic CRBN ligands and uses thereof |
| TWI721483B (zh) | 2018-07-13 | 2021-03-11 | 美商基利科學股份有限公司 | 吡咯并[1,2-b]嗒𠯤衍生物 |
| KR102612513B1 (ko) | 2018-07-31 | 2023-12-12 | 록쏘 온콜로지, 인코포레이티드 | (s)-5-아미노-3-(4-((5-플루오로-2-메톡시벤즈아미도)메틸)페닐)-1-(1,1,1-트리플루오로프로판-2-일)-1h-피라졸-4-카르복스아미드의 분무-건조된 분산물 및 제제 |
| MX2021006154A (es) | 2018-11-30 | 2021-08-24 | Kymera Therapeutics Inc | Degradadores de cinasas asociadas al receptor de interleucina 1 (irak) y usos de los mismos. |
| US11485750B1 (en) | 2019-04-05 | 2022-11-01 | Kymera Therapeutics, Inc. | STAT degraders and uses thereof |
| CN114007698B (zh) | 2019-06-24 | 2024-01-09 | 勃林格殷格翰国际有限公司 | 作为egfr抑制剂的大环化合物和衍生物 |
| EP4076520A4 (en) | 2019-12-17 | 2024-03-27 | Kymera Therapeutics, Inc. | IRAQ DEGRADERS AND USES THEREOF |
| US11591332B2 (en) | 2019-12-17 | 2023-02-28 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| TW202210483A (zh) | 2020-06-03 | 2022-03-16 | 美商凱麥拉醫療公司 | Irak降解劑之結晶型 |
| CN115108968A (zh) * | 2021-03-23 | 2022-09-27 | 上药康丽(常州)药业有限公司 | 一种3-(4-溴苯基)-哌啶的生产方法 |
| AU2022271290A1 (en) | 2021-05-07 | 2023-11-23 | Kymera Therapeutics, Inc. | Cdk2 degraders and uses thereof |
| IL314437A (en) | 2022-01-31 | 2024-09-01 | Kymera Therapeutics Inc | IRAK joints and their uses |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2367422T3 (es) * | 2001-10-09 | 2011-11-03 | Amgen Inc. | Derivados de imidazol como agentes antiinflamatorios. |
| WO2005076854A2 (en) * | 2004-02-04 | 2005-08-25 | Smithkline Beecham Corporation | Pyrimidinone compounds useful as kinase inhibitors |
| WO2005079791A1 (en) | 2004-02-12 | 2005-09-01 | Boehringer Ingelheim Pharmaceuticals, Inc. | Thiophene -2- carboxylic acid - (1h - benzimidazol - 2 yl) - amide derivatives and related compounds as inhibitors of the tec kinase itk (interleukin -2- inducible t cell kinase) for the treatment of inflammation, immunological and allergic disorders |
| EP1732919A1 (en) * | 2004-03-30 | 2006-12-20 | Chiron Corporation | Substituted thiophene derivatives as anti-cancer agents |
| BRPI0608581A2 (pt) * | 2005-03-14 | 2010-01-19 | Transtech Pharma Inc | derivados de benzazol, composiÇÕes e mÉtodos de uso como inibidores de b-secretase |
| JP2012525389A (ja) | 2009-04-27 | 2012-10-22 | ハイ ポイント ファーマシューティカルズ,リミティド ライアビリティ カンパニー | 置換イソキノリン誘導体、医薬組成物、及びβ−セクレターゼ阻害剤としての使用方法 |
| MX2011011396A (es) | 2009-04-27 | 2012-01-30 | High Point Pharmaceuticals Llc | DERIVADOS DE IMIDAZO[1,2-A]PIRIDINA SUSTITUIDA, COMPOSICIONES FARMACEUTICAS Y METODOS DE USO COMO INHIBIDORES DE LA ß-SECRETASA. |
| US8450354B2 (en) | 2010-03-23 | 2013-05-28 | High Point Pharmaceuticals, Llc | Substituted imidazo[1,2-b]pyridazine derivatives, pharmaceutical compositions, and methods of use as β-secretase inhibitors |
| RU2013109143A (ru) * | 2010-08-05 | 2014-09-10 | Амджен Инк. | Бензимидазол- и азабензимидазолсодержащие соединения, которые ингибируют киназу анапластической лимфомы |
| JO3300B1 (ar) * | 2012-06-06 | 2018-09-16 | Novartis Ag | مركبات وتركيبات لتعديل نشاط egfr |
-
2016
- 2016-09-16 RU RU2018113451A patent/RU2743360C2/ru active
- 2016-09-16 AU AU2016323613A patent/AU2016323613B2/en active Active
- 2016-09-16 CN CN201680065303.9A patent/CN108602776B/zh active Active
- 2016-09-16 US US15/267,606 patent/US9969710B2/en active Active
- 2016-09-16 ES ES16770654T patent/ES2911888T3/es active Active
- 2016-09-16 CA CA2996318A patent/CA2996318C/en active Active
- 2016-09-16 KR KR1020187010914A patent/KR102650026B1/ko active IP Right Grant
- 2016-09-16 JP JP2018513805A patent/JP7083750B2/ja active Active
- 2016-09-16 EP EP16770654.8A patent/EP3350170B1/en active Active
- 2016-09-16 MX MX2018002986A patent/MX2018002986A/es unknown
- 2016-09-16 WO PCT/US2016/052107 patent/WO2017049069A1/en active Application Filing
-
2018
- 2018-02-15 ZA ZA2018/01056A patent/ZA201801056B/en unknown
- 2018-03-13 IL IL258081A patent/IL258081B2/en unknown
- 2018-04-17 US US15/954,759 patent/US20180237413A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| AU2016323613A1 (en) | 2018-03-01 |
| CA2996318C (en) | 2023-10-10 |
| ZA201801056B (en) | 2020-05-27 |
| NZ739742A (en) | 2022-03-25 |
| JP2018531225A (ja) | 2018-10-25 |
| IL258081B (en) | 2022-10-01 |
| EP3350170B1 (en) | 2022-01-26 |
| RU2018113451A3 (es) | 2020-01-15 |
| IL258081B2 (en) | 2023-02-01 |
| MX2018002986A (es) | 2018-05-02 |
| RU2018113451A (ru) | 2019-10-18 |
| US20170081307A1 (en) | 2017-03-23 |
| WO2017049069A1 (en) | 2017-03-23 |
| AU2016323613B2 (en) | 2020-12-10 |
| CN108602776B (zh) | 2021-12-03 |
| EP3350170A1 (en) | 2018-07-25 |
| KR102650026B1 (ko) | 2024-03-22 |
| CA2996318A1 (en) | 2017-03-23 |
| JP7083750B2 (ja) | 2022-06-13 |
| IL258081A (en) | 2018-05-31 |
| US20180237413A1 (en) | 2018-08-23 |
| CN108602776A (zh) | 2018-09-28 |
| BR112018003343A2 (pt) | 2018-09-25 |
| RU2743360C2 (ru) | 2021-02-17 |
| KR20180063145A (ko) | 2018-06-11 |
| US9969710B2 (en) | 2018-05-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2911888T3 (es) | Compuestos de heteroarilo como inhibidores de IRAK y usos de los mismos | |
| ES2911524T3 (es) | Compuestos heteroarilo como inhibidores de IRAK y sus usos | |
| ES2772133T3 (es) | Compuestos heteroarilo como inhibidores de IRAK y sus usos | |
| ES2886577T3 (es) | Compuestos macrólidos como inhibidores de irak1/4 y sus usos | |
| AU2019249721B2 (en) | Heteroaryl compounds as type II IRAK inhibitors and uses hereof | |
| ES2841452T3 (es) | Macrociclos de piridazinona como inhibidores de IRAK y sus usos | |
| JP2018531225A6 (ja) | Irak阻害剤としてのヘテロアリール化合物及びその使用 | |
| BR112018003343B1 (pt) | Compostos heteroarila como inibidores de irak e composição farmacêutica compreendendo os mesmos | |
| NZ732527B2 (en) | Pyridazinone macrocycles as irak inhibitors and uses thereof |