ES2906335T3 - Aminotiazoles sustituidos como inhibidores de nucleasas - Google Patents
Aminotiazoles sustituidos como inhibidores de nucleasas Download PDFInfo
- Publication number
- ES2906335T3 ES2906335T3 ES18167651T ES18167651T ES2906335T3 ES 2906335 T3 ES2906335 T3 ES 2906335T3 ES 18167651 T ES18167651 T ES 18167651T ES 18167651 T ES18167651 T ES 18167651T ES 2906335 T3 ES2906335 T3 ES 2906335T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- mmol
- nmr
- mhz
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical class NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 title description 24
- 239000003112 inhibitor Substances 0.000 title description 15
- 101710163270 Nuclease Proteins 0.000 title description 9
- 238000000034 method Methods 0.000 claims abstract description 294
- 150000001875 compounds Chemical class 0.000 claims abstract description 192
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 146
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 135
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 54
- 125000003118 aryl group Chemical group 0.000 claims abstract description 53
- 229910052794 bromium Inorganic materials 0.000 claims abstract description 51
- 229910052801 chlorine Inorganic materials 0.000 claims abstract description 48
- 229910052731 fluorine Inorganic materials 0.000 claims abstract description 45
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 42
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims abstract description 30
- -1 O-phenyl Chemical group 0.000 claims abstract description 29
- 125000001424 substituent group Chemical group 0.000 claims abstract description 28
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 26
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 22
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims abstract description 20
- 201000011510 cancer Diseases 0.000 claims abstract description 19
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 19
- 125000000896 monocarboxylic acid group Chemical group 0.000 claims abstract description 19
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims abstract description 18
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 18
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims abstract description 18
- 208000031448 Genomic Instability Diseases 0.000 claims abstract description 13
- 208000012902 Nervous system disease Diseases 0.000 claims abstract description 13
- 208000025966 Neurological disease Diseases 0.000 claims abstract description 13
- 125000006367 bivalent amino carbonyl group Chemical group [H]N([*:1])C([*:2])=O 0.000 claims abstract description 13
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 13
- 206010063493 Premature ageing Diseases 0.000 claims abstract description 12
- 208000032038 Premature aging Diseases 0.000 claims abstract description 12
- 229910052740 iodine Inorganic materials 0.000 claims abstract description 10
- 125000006413 ring segment Chemical group 0.000 claims abstract description 9
- 150000003839 salts Chemical class 0.000 claims abstract description 8
- MDFFNEOEWAXZRQ-UHFFFAOYSA-N aminyl Chemical compound [NH2] MDFFNEOEWAXZRQ-UHFFFAOYSA-N 0.000 claims abstract description 6
- 239000012453 solvate Substances 0.000 claims abstract description 6
- 101100054666 Streptomyces halstedii sch3 gene Proteins 0.000 claims abstract description 5
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 4
- 150000002367 halogens Chemical class 0.000 claims abstract description 4
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims abstract 8
- 125000005004 perfluoroethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 claims abstract 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 9
- 125000002950 monocyclic group Chemical group 0.000 claims description 9
- 208000013414 ataxia-telangiectasia-like disease Diseases 0.000 claims description 8
- 208000011580 syndromic disease Diseases 0.000 claims description 8
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 7
- 239000008194 pharmaceutical composition Substances 0.000 claims description 7
- 206010003594 Ataxia telangiectasia Diseases 0.000 claims description 5
- 125000002619 bicyclic group Chemical group 0.000 claims description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 5
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 5
- 125000001624 naphthyl group Chemical group 0.000 claims description 5
- 229910052757 nitrogen Inorganic materials 0.000 claims description 5
- 206010053138 Congenital aplastic anaemia Diseases 0.000 claims description 4
- 201000004939 Fanconi anemia Diseases 0.000 claims description 4
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims description 4
- 210000000481 breast Anatomy 0.000 claims description 4
- 239000000872 buffer Substances 0.000 claims description 4
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 4
- 210000001072 colon Anatomy 0.000 claims description 4
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 4
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 4
- 230000002496 gastric effect Effects 0.000 claims description 4
- 210000003128 head Anatomy 0.000 claims description 4
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 4
- 208000032839 leukemia Diseases 0.000 claims description 4
- 210000004072 lung Anatomy 0.000 claims description 4
- 201000001441 melanoma Diseases 0.000 claims description 4
- 125000002757 morpholinyl group Chemical group 0.000 claims description 4
- 210000003739 neck Anatomy 0.000 claims description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 4
- 210000002307 prostate Anatomy 0.000 claims description 4
- 125000004076 pyridyl group Chemical group 0.000 claims description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 4
- 125000001544 thienyl group Chemical group 0.000 claims description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 3
- 210000004185 liver Anatomy 0.000 claims description 3
- 230000002611 ovarian Effects 0.000 claims description 3
- 125000003386 piperidinyl group Chemical group 0.000 claims description 3
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 3
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 3
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 3
- 239000011230 binding agent Substances 0.000 claims description 2
- 239000000969 carrier Substances 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 239000003995 emulsifying agent Substances 0.000 claims description 2
- 239000000945 filler Substances 0.000 claims description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 2
- 239000003755 preservative agent Substances 0.000 claims description 2
- 239000003381 stabilizer Substances 0.000 claims description 2
- 239000000080 wetting agent Substances 0.000 claims description 2
- 125000004860 4-ethylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])C([H])([H])[H] 0.000 claims 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims 1
- 229920001577 copolymer Chemical group 0.000 claims 1
- 102100033996 Double-strand break repair protein MRE11 Human genes 0.000 abstract description 25
- 101000591400 Homo sapiens Double-strand break repair protein MRE11 Proteins 0.000 abstract description 25
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 abstract 5
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 362
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 297
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 267
- 239000007787 solid Substances 0.000 description 260
- 239000000047 product Substances 0.000 description 234
- 238000005160 1H NMR spectroscopy Methods 0.000 description 214
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 204
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 201
- 235000019439 ethyl acetate Nutrition 0.000 description 180
- 239000000203 mixture Substances 0.000 description 155
- 238000004440 column chromatography Methods 0.000 description 142
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 110
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 94
- 230000035484 reaction time Effects 0.000 description 82
- 239000000243 solution Substances 0.000 description 82
- 239000002904 solvent Substances 0.000 description 80
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 77
- IBGBGRVKPALMCQ-UHFFFAOYSA-N 3,4-dihydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1O IBGBGRVKPALMCQ-UHFFFAOYSA-N 0.000 description 76
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 54
- 238000010626 work up procedure Methods 0.000 description 51
- ZIUSEGSNTOUIPT-UHFFFAOYSA-N ethyl 2-cyanoacetate Chemical compound CCOC(=O)CC#N ZIUSEGSNTOUIPT-UHFFFAOYSA-N 0.000 description 50
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 47
- 238000010992 reflux Methods 0.000 description 44
- 229920006395 saturated elastomer Polymers 0.000 description 44
- 238000001914 filtration Methods 0.000 description 43
- 239000012071 phase Substances 0.000 description 43
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 42
- PCYGLFXKCBFGPC-UHFFFAOYSA-N 3,4-Dihydroxy hydroxymethyl benzene Natural products OCC1=CC=C(O)C(O)=C1 PCYGLFXKCBFGPC-UHFFFAOYSA-N 0.000 description 38
- 239000002244 precipitate Substances 0.000 description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 30
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 29
- 239000012267 brine Substances 0.000 description 29
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 29
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 29
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 28
- 238000011282 treatment Methods 0.000 description 28
- 239000000284 extract Substances 0.000 description 27
- UBFLHMNQIQMLKX-UHFFFAOYSA-N 2-cyano-n-(4-phenyl-1,3-thiazol-2-yl)acetamide Chemical compound S1C(NC(CC#N)=O)=NC(C=2C=CC=CC=2)=C1 UBFLHMNQIQMLKX-UHFFFAOYSA-N 0.000 description 26
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 26
- 238000005481 NMR spectroscopy Methods 0.000 description 25
- 239000011541 reaction mixture Substances 0.000 description 24
- DLFVBJFMPXGRIB-UHFFFAOYSA-N thioacetamide Natural products CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 22
- 238000000746 purification Methods 0.000 description 21
- 239000000758 substrate Substances 0.000 description 21
- 150000001299 aldehydes Chemical class 0.000 description 19
- 239000000543 intermediate Substances 0.000 description 19
- 239000000741 silica gel Substances 0.000 description 19
- 229910002027 silica gel Inorganic materials 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 16
- 229950003476 aminothiazole Drugs 0.000 description 16
- 239000002480 mineral oil Substances 0.000 description 16
- 235000010446 mineral oil Nutrition 0.000 description 16
- 108020004414 DNA Proteins 0.000 description 15
- 208000004485 Nijmegen breakage syndrome Diseases 0.000 description 15
- MOIPGXQKZSZOQX-UHFFFAOYSA-N carbonyl bromide Chemical compound BrC(Br)=O MOIPGXQKZSZOQX-UHFFFAOYSA-N 0.000 description 15
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 14
- 210000004027 cell Anatomy 0.000 description 14
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 12
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 12
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 12
- 239000012074 organic phase Substances 0.000 description 12
- 235000019270 ammonium chloride Nutrition 0.000 description 11
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 238000006243 chemical reaction Methods 0.000 description 10
- VXIVSQZSERGHQP-UHFFFAOYSA-N chloroacetamide Chemical compound NC(=O)CCl VXIVSQZSERGHQP-UHFFFAOYSA-N 0.000 description 10
- 239000012230 colorless oil Substances 0.000 description 10
- 239000013067 intermediate product Substances 0.000 description 10
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 8
- 238000010898 silica gel chromatography Methods 0.000 description 8
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- 230000005764 inhibitory process Effects 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- 235000019341 magnesium sulphate Nutrition 0.000 description 7
- LLFNEIKPGDKEPC-UHFFFAOYSA-N 2-cyano-N-[4-(6-methoxypyridin-2-yl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=NC(=CC=C1)OC LLFNEIKPGDKEPC-UHFFFAOYSA-N 0.000 description 6
- PYSJLPAOBIGQPK-UHFFFAOYSA-N 4-phenyl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC=CC=2)=C1 PYSJLPAOBIGQPK-UHFFFAOYSA-N 0.000 description 6
- 230000033616 DNA repair Effects 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 6
- 150000008062 acetophenones Chemical class 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- FKEIGGWJRUYGHR-UHFFFAOYSA-N methyl 4-(2-amino-1,3-thiazol-4-yl)benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1C1=CSC(N)=N1 FKEIGGWJRUYGHR-UHFFFAOYSA-N 0.000 description 6
- CHEPDPSMYKFNAN-UHFFFAOYSA-N methyl 4-(2-bromoacetyl)benzoate Chemical compound COC(=O)C1=CC=C(C(=O)CBr)C=C1 CHEPDPSMYKFNAN-UHFFFAOYSA-N 0.000 description 6
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 6
- LIYGCLJYTHRBQV-UHFFFAOYSA-N 3,5-dichloro-4-hydroxybenzaldehyde Chemical compound OC1=C(Cl)C=C(C=O)C=C1Cl LIYGCLJYTHRBQV-UHFFFAOYSA-N 0.000 description 5
- 108091034117 Oligonucleotide Proteins 0.000 description 5
- 125000001931 aliphatic group Chemical group 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 238000009833 condensation Methods 0.000 description 5
- 230000005494 condensation Effects 0.000 description 5
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- QUKYVVGHLJGVEQ-UHFFFAOYSA-N 2-bromo-1-[3-(trifluoromethoxy)phenyl]ethanone Chemical compound FC(F)(F)OC1=CC=CC(C(=O)CBr)=C1 QUKYVVGHLJGVEQ-UHFFFAOYSA-N 0.000 description 4
- UOTMHAOCAJROQF-UHFFFAOYSA-N 3-bromo-4-hydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1Br UOTMHAOCAJROQF-UHFFFAOYSA-N 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 4
- KWOLFJPFCHCOCG-UHFFFAOYSA-N Acetophenone Chemical class CC(=O)C1=CC=CC=C1 KWOLFJPFCHCOCG-UHFFFAOYSA-N 0.000 description 4
- 101150065749 Churc1 gene Proteins 0.000 description 4
- 230000031709 bromination Effects 0.000 description 4
- 238000005893 bromination reaction Methods 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- DGJMPUGMZIKDRO-UHFFFAOYSA-N cyanoacetamide Chemical compound NC(=O)CC#N DGJMPUGMZIKDRO-UHFFFAOYSA-N 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 231100000518 lethal Toxicity 0.000 description 4
- 230000001665 lethal effect Effects 0.000 description 4
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 239000002245 particle Substances 0.000 description 4
- 238000012746 preparative thin layer chromatography Methods 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 3
- LAXPJIJQTHJGCK-UHFFFAOYSA-N 2-bromo-1-(2-bromophenyl)ethanone Chemical compound BrCC(=O)C1=CC=CC=C1Br LAXPJIJQTHJGCK-UHFFFAOYSA-N 0.000 description 3
- CJEGSZFRYLCTLK-UHFFFAOYSA-N 2-bromo-1-(3,5-difluorophenyl)ethanone Chemical compound FC1=CC(F)=CC(C(=O)CBr)=C1 CJEGSZFRYLCTLK-UHFFFAOYSA-N 0.000 description 3
- MZBXSQBQPJWECM-UHFFFAOYSA-N 2-bromo-1-(3-bromophenyl)ethanone Chemical compound BrCC(=O)C1=CC=CC(Br)=C1 MZBXSQBQPJWECM-UHFFFAOYSA-N 0.000 description 3
- CPXKTRDFYCGOSI-UHFFFAOYSA-N 2-bromo-1-(3-hydroxy-4-methoxyphenyl)ethanone Chemical compound COC1=CC=C(C(=O)CBr)C=C1O CPXKTRDFYCGOSI-UHFFFAOYSA-N 0.000 description 3
- JOCMYOUZIDSYFO-UHFFFAOYSA-N 2-bromo-1-(4-methylsulfonylphenyl)ethanone Chemical compound CS(=O)(=O)C1=CC=C(C(=O)CBr)C=C1 JOCMYOUZIDSYFO-UHFFFAOYSA-N 0.000 description 3
- RAXTYMXDSNWNJS-UHFFFAOYSA-N 2-bromo-1-(4-phenoxyphenyl)ethanone Chemical compound C1=CC(C(=O)CBr)=CC=C1OC1=CC=CC=C1 RAXTYMXDSNWNJS-UHFFFAOYSA-N 0.000 description 3
- OZBXRAVUJGCRFN-UHFFFAOYSA-N 2-bromo-1-(5-bromothiophen-2-yl)ethanone Chemical compound BrCC(=O)C1=CC=C(Br)S1 OZBXRAVUJGCRFN-UHFFFAOYSA-N 0.000 description 3
- UPWFYUIFTMKPFE-UHFFFAOYSA-N 2-bromo-1-(6-methylpyridin-3-yl)ethanone Chemical compound CC1=CC=C(C(=O)CBr)C=N1 UPWFYUIFTMKPFE-UHFFFAOYSA-N 0.000 description 3
- YWIVOTINWZTTTF-UHFFFAOYSA-N 2-bromo-1-pyridazin-3-ylethanone Chemical compound BrCC(=O)C1=CC=CN=N1 YWIVOTINWZTTTF-UHFFFAOYSA-N 0.000 description 3
- KWTHLSXHFFDZMK-UHFFFAOYSA-N 2-cyano-N-[4-[4-(trifluoromethoxy)phenyl]-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC=C(C=C1)OC(F)(F)F KWTHLSXHFFDZMK-UHFFFAOYSA-N 0.000 description 3
- DOYWPBYXBSSALL-UHFFFAOYSA-N 2-cyano-N-[5-cyclohexyl-4-[4-(trifluoromethoxy)phenyl]-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC(=C(N=1)C1=CC=C(C=C1)OC(F)(F)F)C1CCCCC1 DOYWPBYXBSSALL-UHFFFAOYSA-N 0.000 description 3
- YRRQAALAOMOXJW-UHFFFAOYSA-N 3-(2-amino-1,3-thiazol-4-yl)benzonitrile Chemical compound S1C(N)=NC(C=2C=C(C=CC=2)C#N)=C1 YRRQAALAOMOXJW-UHFFFAOYSA-N 0.000 description 3
- XWCGNFLHRINYCE-UHFFFAOYSA-N 3-(2-bromoacetyl)benzonitrile Chemical compound BrCC(=O)C1=CC=CC(C#N)=C1 XWCGNFLHRINYCE-UHFFFAOYSA-N 0.000 description 3
- WOCZNDZQTZMEAD-UHFFFAOYSA-N 4-(2-amino-1,3-thiazol-4-yl)-N,N-dimethylbenzamide Chemical compound NC=1SC=C(N=1)C1=CC=C(C(=O)N(C)C)C=C1 WOCZNDZQTZMEAD-UHFFFAOYSA-N 0.000 description 3
- HBAOQMFTWSDKDV-UHFFFAOYSA-N 4-(2-bromophenyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C(=CC=CC=2)Br)=C1 HBAOQMFTWSDKDV-UHFFFAOYSA-N 0.000 description 3
- DJGBSHUJYGXFQN-UHFFFAOYSA-N 4-(3,5-difluorophenyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=C(F)C=C(F)C=2)=C1 DJGBSHUJYGXFQN-UHFFFAOYSA-N 0.000 description 3
- PDNKBFMOKUBDDR-UHFFFAOYSA-N 4-(3-bromophenyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=C(Br)C=CC=2)=C1 PDNKBFMOKUBDDR-UHFFFAOYSA-N 0.000 description 3
- QQKMBIIGIPEUSO-UHFFFAOYSA-N 4-(3-cyclopropyl-4-methoxyphenyl)-1,3-thiazol-2-amine Chemical compound C1(CC1)C=1C=C(C=CC=1OC)C=1N=C(SC=1)N QQKMBIIGIPEUSO-UHFFFAOYSA-N 0.000 description 3
- TYRIFLPFJRVZMS-UHFFFAOYSA-N 4-(3-methoxypyridin-2-yl)-1,3-thiazol-2-amine Chemical compound COC=1C(=NC=CC=1)C=1N=C(SC=1)N TYRIFLPFJRVZMS-UHFFFAOYSA-N 0.000 description 3
- DEHWWMVRRPVEMH-UHFFFAOYSA-N 4-(3-methylpyridin-2-yl)-1,3-thiazol-2-amine Chemical compound CC1=CC=CN=C1C1=CSC(N)=N1 DEHWWMVRRPVEMH-UHFFFAOYSA-N 0.000 description 3
- ZBRNKOLWXWMLTA-UHFFFAOYSA-N 4-(4-bromophenyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC(Br)=CC=2)=C1 ZBRNKOLWXWMLTA-UHFFFAOYSA-N 0.000 description 3
- WSOKJBHBMAGBIP-UHFFFAOYSA-N 4-(4-fluorophenyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC(F)=CC=2)=C1 WSOKJBHBMAGBIP-UHFFFAOYSA-N 0.000 description 3
- VQEMDSRIOVZAOM-UHFFFAOYSA-N 4-(4-methylsulfonylphenyl)-1,3-thiazol-2-amine Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=CSC(N)=N1 VQEMDSRIOVZAOM-UHFFFAOYSA-N 0.000 description 3
- NFAIOGXJIPWLAS-UHFFFAOYSA-N 4-(4-methylthiophen-2-yl)-1,3-thiazol-2-amine Chemical compound CC1=CSC(C=2N=C(N)SC=2)=C1 NFAIOGXJIPWLAS-UHFFFAOYSA-N 0.000 description 3
- SSDOVEORQGFDOP-UHFFFAOYSA-N 4-(4-morpholin-4-ylphenyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC(=CC=2)N2CCOCC2)=C1 SSDOVEORQGFDOP-UHFFFAOYSA-N 0.000 description 3
- AZZJNMYKYSAYPZ-UHFFFAOYSA-N 4-(4-phenoxyphenyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC(OC=3C=CC=CC=3)=CC=2)=C1 AZZJNMYKYSAYPZ-UHFFFAOYSA-N 0.000 description 3
- MMZHPDCFNLBMBY-UHFFFAOYSA-N 4-(5-bromothiophen-2-yl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2SC(Br)=CC=2)=C1 MMZHPDCFNLBMBY-UHFFFAOYSA-N 0.000 description 3
- UXNSNCTVNSWUGZ-UHFFFAOYSA-N 4-(5-methylthiophen-2-yl)-1,3-thiazol-2-amine Chemical compound S1C(C)=CC=C1C1=CSC(N)=N1 UXNSNCTVNSWUGZ-UHFFFAOYSA-N 0.000 description 3
- SYIFPUGLNKNPJE-UHFFFAOYSA-N 4-(6-methoxypyridin-3-yl)-1,3-thiazol-2-amine Chemical compound NC=1SC=C(N=1)C=1C=NC(=CC=1)OC SYIFPUGLNKNPJE-UHFFFAOYSA-N 0.000 description 3
- VYRFEJAFSZHYEI-UHFFFAOYSA-N 4-[3-(trifluoromethoxy)phenyl]-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=C(OC(F)(F)F)C=CC=2)=C1 VYRFEJAFSZHYEI-UHFFFAOYSA-N 0.000 description 3
- KPLWOYYOIKYZHR-UHFFFAOYSA-N 4-[4-(2-trimethylsilylethynyl)phenyl]-1,3-thiazol-2-amine Chemical compound C[Si](C)(C)C#CC1=CC=C(C=C1)C=1N=C(SC=1)N KPLWOYYOIKYZHR-UHFFFAOYSA-N 0.000 description 3
- CRUWLPBETLMFFA-UHFFFAOYSA-N 4-phenyl-5-pyrazin-2-yl-1,3-thiazol-2-amine Chemical compound C1(=CC=CC=C1)C=1N=C(SC=1C1=NC=CN=C1)N CRUWLPBETLMFFA-UHFFFAOYSA-N 0.000 description 3
- OKUWMUDCFVWSLE-UHFFFAOYSA-N 4-pyridazin-3-yl-1,3-thiazol-2-amine Chemical compound N1=NC(=CC=C1)C=1N=C(SC=1)N OKUWMUDCFVWSLE-UHFFFAOYSA-N 0.000 description 3
- BLKHMTAXNXLDJP-UHFFFAOYSA-N 4-pyridin-2-yl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2N=CC=CC=2)=C1 BLKHMTAXNXLDJP-UHFFFAOYSA-N 0.000 description 3
- CUSJGENRTWNHPV-UHFFFAOYSA-N 4-pyridin-4-yl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CN=CC=2)=C1 CUSJGENRTWNHPV-UHFFFAOYSA-N 0.000 description 3
- JCXLOPXBXXSCEO-UHFFFAOYSA-N 5-(2-amino-1,3-thiazol-4-yl)-2-methoxyphenol Chemical compound NC=1SC=C(N=1)C=1C=CC(=C(C=1)O)OC JCXLOPXBXXSCEO-UHFFFAOYSA-N 0.000 description 3
- JVOJRKACQXYOAS-UHFFFAOYSA-N 5-(2-amino-1,3-thiazol-4-yl)thiophene-2-carbonitrile Chemical compound S1C(N)=NC(C=2SC(=CC=2)C#N)=C1 JVOJRKACQXYOAS-UHFFFAOYSA-N 0.000 description 3
- ONHYSHQEAKQAFB-UHFFFAOYSA-N 5-bromo-4-phenyl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC=CC=2)=C1Br ONHYSHQEAKQAFB-UHFFFAOYSA-N 0.000 description 3
- NVBRGMQJTDNHNS-UHFFFAOYSA-N 5-cyclohexyl-4-[4-(trifluoromethoxy)phenyl]-1,3-thiazol-2-amine Chemical compound C1(CCCCC1)C1=C(N=C(S1)N)C1=CC=C(C=C1)OC(F)(F)F NVBRGMQJTDNHNS-UHFFFAOYSA-N 0.000 description 3
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- 230000005778 DNA damage Effects 0.000 description 3
- 231100000277 DNA damage Toxicity 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- FGUAMQFRPZRZFE-UHFFFAOYSA-N FC(C=1C=CC(=NC=1)C=1N=C(SC=1)N)(F)F Chemical compound FC(C=1C=CC(=NC=1)C=1N=C(SC=1)N)(F)F FGUAMQFRPZRZFE-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 208000014766 Nijmegen breakage syndrome-like disease Diseases 0.000 description 3
- 102000012338 Poly(ADP-ribose) Polymerases Human genes 0.000 description 3
- 108010061844 Poly(ADP-ribose) Polymerases Proteins 0.000 description 3
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 238000010640 amide synthesis reaction Methods 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 235000010290 biphenyl Nutrition 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 3
- 230000028617 response to DNA damage stimulus Effects 0.000 description 3
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 2
- JYPGOBDETCKKKV-UHFFFAOYSA-N 1-(3-bromo-4-methoxyphenyl)ethanone Chemical compound COC1=CC=C(C(C)=O)C=C1Br JYPGOBDETCKKKV-UHFFFAOYSA-N 0.000 description 2
- VIFGSQVZWBYVJM-UHFFFAOYSA-N 1-(3-cyclopropyl-4-methoxyphenyl)ethanone Chemical compound C1(CC1)C=1C=C(C=CC=1OC)C(C)=O VIFGSQVZWBYVJM-UHFFFAOYSA-N 0.000 description 2
- ZMXWZZHFPDJCRB-UHFFFAOYSA-N 1-[4-(2-trimethylsilylethynyl)phenyl]ethanone Chemical compound CC(=O)C1=CC=C(C#C[Si](C)(C)C)C=C1 ZMXWZZHFPDJCRB-UHFFFAOYSA-N 0.000 description 2
- 238000004293 19F NMR spectroscopy Methods 0.000 description 2
- IXWOUPGDGMCKGT-UHFFFAOYSA-N 2,3-dihydroxybenzaldehyde Chemical compound OC1=CC=CC(C=O)=C1O IXWOUPGDGMCKGT-UHFFFAOYSA-N 0.000 description 2
- IUNJCFABHJZSKB-UHFFFAOYSA-N 2,4-dihydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C(O)=C1 IUNJCFABHJZSKB-UHFFFAOYSA-N 0.000 description 2
- NRQGTTQZNNQWJY-UHFFFAOYSA-N 2-(2-amino-1,3-thiazol-4-yl)propan-2-ol Chemical compound CC(C)(O)C1=CSC(N)=N1 NRQGTTQZNNQWJY-UHFFFAOYSA-N 0.000 description 2
- NRPWUDNMAYNCCH-UHFFFAOYSA-N 2-(oxan-4-yl)-1-phenylethanone Chemical compound C=1C=CC=CC=1C(=O)CC1CCOCC1 NRPWUDNMAYNCCH-UHFFFAOYSA-N 0.000 description 2
- KYSMSMOTZYPBPN-UHFFFAOYSA-N 2-[4-(2-amino-1,3-thiazol-4-yl)phenyl]propan-2-ol Chemical compound NC=1SC=C(N=1)C1=CC=C(C=C1)C(C)(C)O KYSMSMOTZYPBPN-UHFFFAOYSA-N 0.000 description 2
- OSELWQMHRSIZJF-UHFFFAOYSA-N 2-bromo-1-(3-cyclopropyl-4-methoxyphenyl)ethanone Chemical compound BrCC(=O)C1=CC(=C(C=C1)OC)C1CC1 OSELWQMHRSIZJF-UHFFFAOYSA-N 0.000 description 2
- CYANWVCVJQOGOK-UHFFFAOYSA-N 2-bromo-1-(3-methoxypyridin-2-yl)ethanone Chemical compound COC1=CC=CN=C1C(=O)CBr CYANWVCVJQOGOK-UHFFFAOYSA-N 0.000 description 2
- MFKZMURKFNOBQN-UHFFFAOYSA-N 2-bromo-1-(3-methylpyridin-2-yl)ethanone Chemical compound CC1=CC=CN=C1C(=O)CBr MFKZMURKFNOBQN-UHFFFAOYSA-N 0.000 description 2
- ZEDMENALRQNCBA-UHFFFAOYSA-N 2-bromo-1-(3-tert-butylphenyl)ethanone Chemical compound CC(C)(C)C1=CC=CC(C(=O)CBr)=C1 ZEDMENALRQNCBA-UHFFFAOYSA-N 0.000 description 2
- OUGMZFJPRSTGMJ-UHFFFAOYSA-N 2-bromo-1-(4-morpholin-4-ylphenyl)ethanone Chemical compound C1=CC(C(=O)CBr)=CC=C1N1CCOCC1 OUGMZFJPRSTGMJ-UHFFFAOYSA-N 0.000 description 2
- WIEZPKHDQRZMCI-UHFFFAOYSA-N 2-bromo-1-(5-methylthiophen-2-yl)ethanone Chemical compound CC1=CC=C(C(=O)CBr)S1 WIEZPKHDQRZMCI-UHFFFAOYSA-N 0.000 description 2
- CMOXUHAYWKQNPC-UHFFFAOYSA-N 2-bromo-1-(6-methoxypyridin-3-yl)ethanone Chemical compound COC1=CC=C(C(=O)CBr)C=N1 CMOXUHAYWKQNPC-UHFFFAOYSA-N 0.000 description 2
- ZYMAKPDWZDXVHC-UHFFFAOYSA-N 2-bromo-1-[4-(2-trimethylsilylethynyl)phenyl]ethanone Chemical compound BrCC(=O)C1=CC=C(C=C1)C#C[Si](C)(C)C ZYMAKPDWZDXVHC-UHFFFAOYSA-N 0.000 description 2
- HOUQSRLDJIVIOG-UHFFFAOYSA-N 2-bromo-1-[5-(trifluoromethyl)pyridin-2-yl]ethanone Chemical compound FC(F)(F)C1=CC=C(C(=O)CBr)N=C1 HOUQSRLDJIVIOG-UHFFFAOYSA-N 0.000 description 2
- SEUZMBMWEJRWNU-UHFFFAOYSA-N 2-bromo-1-phenyl-2-pyrazin-2-ylethanone Chemical compound BrC(C(=O)C1=CC=CC=C1)C1=NC=CN=C1 SEUZMBMWEJRWNU-UHFFFAOYSA-N 0.000 description 2
- WPDWOCRJBPXJFM-UHFFFAOYSA-N 2-bromo-1-phenylpropan-1-one Chemical compound CC(Br)C(=O)C1=CC=CC=C1 WPDWOCRJBPXJFM-UHFFFAOYSA-N 0.000 description 2
- DNPMOGQMEOPVNT-UHFFFAOYSA-N 2-bromo-1-pyridin-2-ylethanone Chemical compound BrCC(=O)C1=CC=CC=N1 DNPMOGQMEOPVNT-UHFFFAOYSA-N 0.000 description 2
- RGALBQILADNMKA-UHFFFAOYSA-N 2-bromo-1-pyridin-4-ylethanone;hydron;bromide Chemical compound Br.BrCC(=O)C1=CC=NC=C1 RGALBQILADNMKA-UHFFFAOYSA-N 0.000 description 2
- KBHQXNZCUIXQNH-UHFFFAOYSA-N 2-bromo-2-cyclohexyl-1-[4-(trifluoromethoxy)phenyl]ethanone Chemical compound BrC(C(=O)C1=CC=C(C=C1)OC(F)(F)F)C1CCCCC1 KBHQXNZCUIXQNH-UHFFFAOYSA-N 0.000 description 2
- KDXYEWRAWRZXFT-UHFFFAOYSA-N 2-bromocyclohexan-1-one Chemical compound BrC1CCCCC1=O KDXYEWRAWRZXFT-UHFFFAOYSA-N 0.000 description 2
- DPHLBGAHPBXFQL-UHFFFAOYSA-N 2-cyano-N-(4,5-diphenyl-1,3-thiazol-2-yl)acetamide Chemical compound C(#N)CC(=O)NC=1SC(=C(N=1)C1=CC=CC=C1)C1=CC=CC=C1 DPHLBGAHPBXFQL-UHFFFAOYSA-N 0.000 description 2
- GTWPKROVOGGMFN-UHFFFAOYSA-N 2-cyano-N-(4-phenyl-5-pyrazin-2-yl-1,3-thiazol-2-yl)acetamide Chemical compound C(#N)CC(=O)NC=1SC(=C(N=1)C1=CC=CC=C1)C1=NC=CN=C1 GTWPKROVOGGMFN-UHFFFAOYSA-N 0.000 description 2
- CIOBZAABVWDBDH-UHFFFAOYSA-N 2-cyano-N-(4-pyridazin-3-yl-1,3-thiazol-2-yl)acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C=1N=NC=CC=1 CIOBZAABVWDBDH-UHFFFAOYSA-N 0.000 description 2
- NYKHAGAXQJJHJN-UHFFFAOYSA-N 2-cyano-N-(5-phenyl-1,3-thiazol-2-yl)acetamide Chemical compound C(#N)CC(=O)NC=1SC(=CN=1)C1=CC=CC=C1 NYKHAGAXQJJHJN-UHFFFAOYSA-N 0.000 description 2
- OUDTXQTXYDAZCO-UHFFFAOYSA-N 2-cyano-N-[4-(2-hydroxypropan-2-yl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C(C)(C)O OUDTXQTXYDAZCO-UHFFFAOYSA-N 0.000 description 2
- BLCLHHOPJLGOQX-UHFFFAOYSA-N 2-cyano-N-[4-(3,5-difluorophenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC(=CC(=C1)F)F BLCLHHOPJLGOQX-UHFFFAOYSA-N 0.000 description 2
- SZHHQCVJLZYQSX-UHFFFAOYSA-N 2-cyano-N-[4-(3-cyanophenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC(=CC=C1)C#N SZHHQCVJLZYQSX-UHFFFAOYSA-N 0.000 description 2
- RTOCJXTZPUFYEJ-UHFFFAOYSA-N 2-cyano-N-[4-(3-fluorophenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC(=CC=C1)F RTOCJXTZPUFYEJ-UHFFFAOYSA-N 0.000 description 2
- LUMNYBKIFSPULX-UHFFFAOYSA-N 2-cyano-N-[4-(3-hydroxy-4-methoxyphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC(=C(C=C1)OC)O LUMNYBKIFSPULX-UHFFFAOYSA-N 0.000 description 2
- MACOKWWHAZLBNG-UHFFFAOYSA-N 2-cyano-N-[4-(3-methoxypyridin-2-yl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=NC=CC=C1OC MACOKWWHAZLBNG-UHFFFAOYSA-N 0.000 description 2
- ZREXZOHAHGMOHJ-UHFFFAOYSA-N 2-cyano-N-[4-(3-methylpyridin-2-yl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=NC=CC=C1C ZREXZOHAHGMOHJ-UHFFFAOYSA-N 0.000 description 2
- VZMHFTHXFFPXJW-UHFFFAOYSA-N 2-cyano-N-[4-(4-ethynylphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC=C(C=C1)C#C VZMHFTHXFFPXJW-UHFFFAOYSA-N 0.000 description 2
- RLDRXPAREARWDK-UHFFFAOYSA-N 2-cyano-N-[4-(4-fluorophenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC=C(C=C1)F RLDRXPAREARWDK-UHFFFAOYSA-N 0.000 description 2
- YWSMPVSSOZPRAN-UHFFFAOYSA-N 2-cyano-N-[4-(4-morpholin-4-ylphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC=C(C=C1)N1CCOCC1 YWSMPVSSOZPRAN-UHFFFAOYSA-N 0.000 description 2
- NXGZMLAPOMZNEO-UHFFFAOYSA-N 2-cyano-N-[4-(5-cyanothiophen-2-yl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C=1SC(=CC=1)C#N NXGZMLAPOMZNEO-UHFFFAOYSA-N 0.000 description 2
- RPFCWGBXOBOZJP-UHFFFAOYSA-N 2-cyano-N-[4-(5-methylthiophen-2-yl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C=1SC(=CC=1)C RPFCWGBXOBOZJP-UHFFFAOYSA-N 0.000 description 2
- ZAALSOMLZYNRDM-UHFFFAOYSA-N 2-cyano-N-[4-(6-methoxypyridin-3-yl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C=1C=NC(=CC=1)OC ZAALSOMLZYNRDM-UHFFFAOYSA-N 0.000 description 2
- FXMRKRWPIRTRCF-UHFFFAOYSA-N 2-cyano-N-[4-(6-methylpyridin-3-yl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C=1C=NC(=CC=1)C FXMRKRWPIRTRCF-UHFFFAOYSA-N 0.000 description 2
- QADLQDHCOMWTIG-UHFFFAOYSA-N 2-cyano-N-[4-[3-(trifluoromethoxy)phenyl]-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC(=CC=C1)OC(F)(F)F QADLQDHCOMWTIG-UHFFFAOYSA-N 0.000 description 2
- WMNMLZGUAXQJQW-UHFFFAOYSA-N 2-cyano-N-[4-[4-(2-hydroxypropan-2-yl)phenyl]-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC=C(C=C1)C(C)(C)O WMNMLZGUAXQJQW-UHFFFAOYSA-N 0.000 description 2
- AYULZMAQQNKQNC-UHFFFAOYSA-N 2-cyano-N-[4-[4-(2-trimethylsilylethynyl)phenyl]-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC=C(C=C1)C#C[Si](C)(C)C AYULZMAQQNKQNC-UHFFFAOYSA-N 0.000 description 2
- CFPMQDHOFUKINN-UHFFFAOYSA-N 2-cyano-N-[4-[5-(trifluoromethyl)pyridin-2-yl]-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=NC=C(C=C1)C(F)(F)F CFPMQDHOFUKINN-UHFFFAOYSA-N 0.000 description 2
- MHENNTXOHQSCCY-UHFFFAOYSA-N 2-cyano-N-[5-(oxan-4-yl)-4-phenyl-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC(=C(N=1)C1=CC=CC=C1)C1CCOCC1 MHENNTXOHQSCCY-UHFFFAOYSA-N 0.000 description 2
- NQTRGNVPQFOTJT-UHFFFAOYSA-N 2-cyano-N-methyl-N-(4-phenyl-1,3-thiazol-2-yl)acetamide Chemical compound C(#N)CC(=O)N(C=1SC=C(N=1)C1=CC=CC=C1)C NQTRGNVPQFOTJT-UHFFFAOYSA-N 0.000 description 2
- OFQQTJKSWPXQFP-UHFFFAOYSA-N 2-cyano-n-(4-naphthalen-2-yl-1,3-thiazol-2-yl)acetamide Chemical compound S1C(NC(CC#N)=O)=NC(C=2C=C3C=CC=CC3=CC=2)=C1 OFQQTJKSWPXQFP-UHFFFAOYSA-N 0.000 description 2
- QAUZAUFCZQAPNA-UHFFFAOYSA-N 2-cyano-n-(4-pyridin-4-yl-1,3-thiazol-2-yl)acetamide Chemical compound S1C(NC(CC#N)=O)=NC(C=2C=CN=CC=2)=C1 QAUZAUFCZQAPNA-UHFFFAOYSA-N 0.000 description 2
- VJSCTANFLGEHDX-UHFFFAOYSA-N 2-cyano-n-(4-thiophen-2-yl-1,3-thiazol-2-yl)acetamide Chemical compound S1C(NC(CC#N)=O)=NC(C=2SC=CC=2)=C1 VJSCTANFLGEHDX-UHFFFAOYSA-N 0.000 description 2
- GVIYDEOZDPLBST-UHFFFAOYSA-N 2-cyano-n-(5-methyl-4-phenyl-1,3-thiazol-2-yl)acetamide Chemical compound S1C(NC(=O)CC#N)=NC(C=2C=CC=CC=2)=C1C GVIYDEOZDPLBST-UHFFFAOYSA-N 0.000 description 2
- CHKDWZCJTXYBBP-UHFFFAOYSA-N 2-cyano-n-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C1=CC(OC)=CC=C1C1=CSC(NC(=O)CC#N)=N1 CHKDWZCJTXYBBP-UHFFFAOYSA-N 0.000 description 2
- YPCGNDIIQZOEHO-UHFFFAOYSA-N 2-cyano-n-[4-(4-methylphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C1=CC(C)=CC=C1C1=CSC(NC(=O)CC#N)=N1 YPCGNDIIQZOEHO-UHFFFAOYSA-N 0.000 description 2
- HHEHMLWJLZAKHM-UHFFFAOYSA-N 2-cyano-n-[4-(4-phenoxyphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound S1C(NC(CC#N)=O)=NC(C=2C=CC(OC=3C=CC=CC=3)=CC=2)=C1 HHEHMLWJLZAKHM-UHFFFAOYSA-N 0.000 description 2
- JJMDTERTPNYIGZ-UHFFFAOYSA-N 2-cyclohexylacetaldehyde Chemical compound O=CCC1CCCCC1 JJMDTERTPNYIGZ-UHFFFAOYSA-N 0.000 description 2
- SMWAOXCEPHEGFV-UHFFFAOYSA-N 4,5,6,7-tetrahydro-1,3-benzothiazol-2-amine Chemical compound C1CCCC2=C1N=C(N)S2 SMWAOXCEPHEGFV-UHFFFAOYSA-N 0.000 description 2
- XDWGEGZDMFHZBL-UHFFFAOYSA-N 4,5-diphenyl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 XDWGEGZDMFHZBL-UHFFFAOYSA-N 0.000 description 2
- JVJWJUQLRDIVJV-UHFFFAOYSA-N 4-(1-adamantyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C23CC4CC(CC(C4)C2)C3)=C1 JVJWJUQLRDIVJV-UHFFFAOYSA-N 0.000 description 2
- IQIAVCUUQIGJPO-UHFFFAOYSA-N 4-(1-benzofuran-2-yl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2OC3=CC=CC=C3C=2)=C1 IQIAVCUUQIGJPO-UHFFFAOYSA-N 0.000 description 2
- XBHHILITQUEDDC-UHFFFAOYSA-N 4-(3-fluorophenyl)-2-thiazolamine Chemical compound S1C(N)=NC(C=2C=C(F)C=CC=2)=C1 XBHHILITQUEDDC-UHFFFAOYSA-N 0.000 description 2
- PNKVOIPQWYPICP-UHFFFAOYSA-N 4-(3-tert-butylphenyl)-1,3-thiazol-2-amine Chemical compound CC(C)(C)C1=CC=CC(C=2N=C(N)SC=2)=C1 PNKVOIPQWYPICP-UHFFFAOYSA-N 0.000 description 2
- YPVVEXKDPBRGIK-UHFFFAOYSA-N 4-(4-methoxyphenyl)-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1C1=CSC(N)=N1 YPVVEXKDPBRGIK-UHFFFAOYSA-N 0.000 description 2
- ARLHWYFAPHJCJT-UHFFFAOYSA-N 4-(4-methylphenyl)-1,3-thiazol-2-amine Chemical compound C1=CC(C)=CC=C1C1=CSC(N)=N1 ARLHWYFAPHJCJT-UHFFFAOYSA-N 0.000 description 2
- WGNFXMXPBURHSR-UHFFFAOYSA-N 4-(4-tert-butyl-2,6-dimethylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(C(C)(C)C)=CC(C)=C1C1=CSC(N)=N1 WGNFXMXPBURHSR-UHFFFAOYSA-N 0.000 description 2
- ABCNUVGFUGMTOA-UHFFFAOYSA-N 4-(4-tert-butylphenyl)-1,3-thiazol-2-amine Chemical compound C1=CC(C(C)(C)C)=CC=C1C1=CSC(N)=N1 ABCNUVGFUGMTOA-UHFFFAOYSA-N 0.000 description 2
- NAFKXHMLGSTKAQ-UHFFFAOYSA-N 4-(6-methylpyridin-3-yl)-1,3-thiazol-2-amine Chemical compound C1=NC(C)=CC=C1C1=CSC(N)=N1 NAFKXHMLGSTKAQ-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- PWVLBESTTRJTFT-UHFFFAOYSA-N 4-[4-(trifluoromethoxy)phenyl]-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC(OC(F)(F)F)=CC=2)=C1 PWVLBESTTRJTFT-UHFFFAOYSA-N 0.000 description 2
- GOUHYARYYWKXHS-UHFFFAOYSA-N 4-formylbenzoic acid Chemical compound OC(=O)C1=CC=C(C=O)C=C1 GOUHYARYYWKXHS-UHFFFAOYSA-N 0.000 description 2
- UYGBSRJODQHNLQ-UHFFFAOYSA-N 4-hydroxy-3,5-dimethylbenzaldehyde Chemical compound CC1=CC(C=O)=CC(C)=C1O UYGBSRJODQHNLQ-UHFFFAOYSA-N 0.000 description 2
- XIMCLEAEWWYKLY-UHFFFAOYSA-N 4-hydroxy-3-phenylbenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1C1=CC=CC=C1 XIMCLEAEWWYKLY-UHFFFAOYSA-N 0.000 description 2
- GWDNDNTTXIIXRS-UHFFFAOYSA-N 4-naphthalen-2-yl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=C3C=CC=CC3=CC=2)=C1 GWDNDNTTXIIXRS-UHFFFAOYSA-N 0.000 description 2
- XRNPDKQNHIKARG-UHFFFAOYSA-N 4-thiophen-2-yl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2SC=CC=2)=C1 XRNPDKQNHIKARG-UHFFFAOYSA-N 0.000 description 2
- LUKVPYYHLIYJSW-UHFFFAOYSA-N 5-(2-bromoacetyl)thiophene-2-carbonitrile Chemical compound BrCC(=O)C1=CC=C(C#N)S1 LUKVPYYHLIYJSW-UHFFFAOYSA-N 0.000 description 2
- OUSQZOMOOSOTML-UHFFFAOYSA-N 5-hydroxy-6-oxo-1H-pyridine-3-carbaldehyde Chemical compound OC=1C(=NC=C(C=O)C=1)O OUSQZOMOOSOTML-UHFFFAOYSA-N 0.000 description 2
- GWVPOHCHOSLYSA-UHFFFAOYSA-N 7-(hydroxymethyl)-4h-1,4-benzoxazin-3-one Chemical compound N1C(=O)COC2=CC(CO)=CC=C21 GWVPOHCHOSLYSA-UHFFFAOYSA-N 0.000 description 2
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- XCTPNCOQDPDDME-UHFFFAOYSA-N FC(F)(F)Oc1ccc(cc1)C(=O)CC1CCCCC1 Chemical compound FC(F)(F)Oc1ccc(cc1)C(=O)CC1CCCCC1 XCTPNCOQDPDDME-UHFFFAOYSA-N 0.000 description 2
- 101001128138 Homo sapiens NACHT, LRR and PYD domains-containing protein 2 Proteins 0.000 description 2
- 101000981336 Homo sapiens Nibrin Proteins 0.000 description 2
- 229910010082 LiAlH Inorganic materials 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- RTBGNXAZTSRUHT-UHFFFAOYSA-N N-(5-bromo-4-phenyl-1,3-thiazol-2-yl)-2-cyanoacetamide Chemical compound BrC1=C(N=C(S1)NC(CC#N)=O)C1=CC=CC=C1 RTBGNXAZTSRUHT-UHFFFAOYSA-N 0.000 description 2
- MFXOGUHOPSUQID-UHFFFAOYSA-N N-(5-chloro-4-phenyl-1,3-thiazol-2-yl)-2-cyanoacetamide Chemical compound ClC1=C(N=C(S1)NC(CC#N)=O)C1=CC=CC=C1 MFXOGUHOPSUQID-UHFFFAOYSA-N 0.000 description 2
- PKCJXDYLBCNLSP-UHFFFAOYSA-N N-[4-(1-benzofuran-2-yl)-1,3-thiazol-2-yl]-2-cyanoacetamide Chemical compound O1C(=CC2=C1C=CC=C2)C=1N=C(SC=1)NC(CC#N)=O PKCJXDYLBCNLSP-UHFFFAOYSA-N 0.000 description 2
- PZGJIMCRPQJKIS-UHFFFAOYSA-N N-[4-(2-bromophenyl)-1,3-thiazol-2-yl]-2-cyanoacetamide Chemical compound BrC1=C(C=CC=C1)C=1N=C(SC=1)NC(CC#N)=O PZGJIMCRPQJKIS-UHFFFAOYSA-N 0.000 description 2
- COTDLXRITBAYLL-UHFFFAOYSA-N N-[4-(3-bromophenyl)-1,3-thiazol-2-yl]-2-cyanoacetamide Chemical compound BrC=1C=C(C=CC=1)C=1N=C(SC=1)NC(CC#N)=O COTDLXRITBAYLL-UHFFFAOYSA-N 0.000 description 2
- QQNKBGXFQXMZPX-UHFFFAOYSA-N N-[4-(4-tert-butyl-2,6-dimethylphenyl)-1,3-thiazol-2-yl]-2-cyanoacetamide Chemical compound C(C)(C)(C)C1=CC(=C(C(=C1)C)C=1N=C(SC=1)NC(CC#N)=O)C QQNKBGXFQXMZPX-UHFFFAOYSA-N 0.000 description 2
- RXDLIYWHMKMFQE-UHFFFAOYSA-N N-[4-(5-bromothiophen-2-yl)-1,3-thiazol-2-yl]-2-cyanoacetamide Chemical compound BrC1=CC=C(S1)C=1N=C(SC=1)NC(CC#N)=O RXDLIYWHMKMFQE-UHFFFAOYSA-N 0.000 description 2
- 102100031897 NACHT, LRR and PYD domains-containing protein 2 Human genes 0.000 description 2
- OYEXAVVDHBTMTP-UHFFFAOYSA-N O=C(CC#N)Nc1nc(cs1)-c1ccccn1 Chemical compound O=C(CC#N)Nc1nc(cs1)-c1ccccn1 OYEXAVVDHBTMTP-UHFFFAOYSA-N 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- 241000394605 Viola striata Species 0.000 description 2
- PZOTXXRWCKDMBC-UHFFFAOYSA-N [3-(cyclohexylcarbamoyl)phenyl]boronic acid Chemical compound OB(O)C1=CC=CC(C(=O)NC2CCCCC2)=C1 PZOTXXRWCKDMBC-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- 230000031018 biological processes and functions Effects 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 2
- 230000025084 cell cycle arrest Effects 0.000 description 2
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 2
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 230000005782 double-strand break Effects 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- CCERQOYLJJULMD-UHFFFAOYSA-M magnesium;carbanide;chloride Chemical compound [CH3-].[Mg+2].[Cl-] CCERQOYLJJULMD-UHFFFAOYSA-M 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- UEGCRFNWTGYVKX-UHFFFAOYSA-N methyl 3-hydroxy-4-nitrobenzoate Chemical compound COC(=O)C1=CC=C([N+]([O-])=O)C(O)=C1 UEGCRFNWTGYVKX-UHFFFAOYSA-N 0.000 description 2
- ICWZOGODOGTHMN-UHFFFAOYSA-N methyl 4-[2-[(2-cyanoacetyl)amino]-1,3-thiazol-4-yl]benzoate Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC=C(C(=O)OC)C=C1 ICWZOGODOGTHMN-UHFFFAOYSA-N 0.000 description 2
- YHEUXOONADIACE-UHFFFAOYSA-N methyl 4-acetamido-3-[tert-butyl(dimethyl)silyl]oxybenzoate Chemical compound C(C)(=O)NC1=C(C=C(C(=O)OC)C=C1)O[Si](C)(C)C(C)(C)C YHEUXOONADIACE-UHFFFAOYSA-N 0.000 description 2
- QPZCIKBBXHQUMD-UHFFFAOYSA-N methyl 4-acetamido-3-hydroxybenzoate Chemical compound COC(=O)C1=CC=C(NC(C)=O)C(O)=C1 QPZCIKBBXHQUMD-UHFFFAOYSA-N 0.000 description 2
- 229940095102 methyl benzoate Drugs 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- IMNDHOCGZLYMRO-UHFFFAOYSA-N n,n-dimethylbenzamide Chemical compound CN(C)C(=O)C1=CC=CC=C1 IMNDHOCGZLYMRO-UHFFFAOYSA-N 0.000 description 2
- KBKWZUVMKBNTFP-UHFFFAOYSA-N n-(3-formylphenyl)acetamide Chemical compound CC(=O)NC1=CC=CC(C=O)=C1 KBKWZUVMKBNTFP-UHFFFAOYSA-N 0.000 description 2
- SVISPFRZFMOMNM-UHFFFAOYSA-N n-(4-phenyl-1,3-thiazol-2-yl)formamide Chemical compound S1C(NC=O)=NC(C=2C=CC=CC=2)=C1 SVISPFRZFMOMNM-UHFFFAOYSA-N 0.000 description 2
- GQDQQKZXVSSDRH-UHFFFAOYSA-N n-[4-(4-bromophenyl)-1,3-thiazol-2-yl]-2-cyanoacetamide Chemical compound C1=CC(Br)=CC=C1C1=CSC(NC(=O)CC#N)=N1 GQDQQKZXVSSDRH-UHFFFAOYSA-N 0.000 description 2
- CISSQWUNYCOLRA-UHFFFAOYSA-N n-[4-(4-tert-butylphenyl)-1,3-thiazol-2-yl]-2-cyanoacetamide Chemical compound C1=CC(C(C)(C)C)=CC=C1C1=CSC(NC(=O)CC#N)=N1 CISSQWUNYCOLRA-UHFFFAOYSA-N 0.000 description 2
- BBKDLVZZNDOEET-UHFFFAOYSA-N n-methoxy-n-methyl-2-(oxan-4-yl)acetamide Chemical compound CON(C)C(=O)CC1CCOCC1 BBKDLVZZNDOEET-UHFFFAOYSA-N 0.000 description 2
- QPHATFGERAPTTA-UHFFFAOYSA-N n-methyl-4-phenyl-1,3-thiazol-2-amine Chemical compound S1C(NC)=NC(C=2C=CC=CC=2)=C1 QPHATFGERAPTTA-UHFFFAOYSA-N 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 230000006780 non-homologous end joining Effects 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 239000001301 oxygen Chemical group 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- LIGACIXOYTUXAW-UHFFFAOYSA-N phenacyl bromide Chemical compound BrCC(=O)C1=CC=CC=C1 LIGACIXOYTUXAW-UHFFFAOYSA-N 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- 235000015320 potassium carbonate Nutrition 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Chemical compound [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 description 2
- 230000008439 repair process Effects 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 229910052717 sulfur Chemical group 0.000 description 2
- 239000011593 sulfur Chemical group 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- ARWCZKJISXFBGI-UHFFFAOYSA-N (3,4-dihydroxyphenyl)-phenylmethanone Chemical compound C1=C(O)C(O)=CC=C1C(=O)C1=CC=CC=C1 ARWCZKJISXFBGI-UHFFFAOYSA-N 0.000 description 1
- MAYZWDRUFKUGGP-VIFPVBQESA-N (3s)-1-[5-tert-butyl-3-[(1-methyltetrazol-5-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol Chemical compound CN1N=NN=C1CN1C2=NC(C(C)(C)C)=NC(N3C[C@@H](O)CC3)=C2N=N1 MAYZWDRUFKUGGP-VIFPVBQESA-N 0.000 description 1
- GPURHDUTZUYAFI-GHXNOFRVSA-N (5z)-5-[(4-hydroxyphenyl)methylidene]-3-(2-methylpropyl)-2-sulfanylidene-1,3-thiazolidin-4-one Chemical compound O=C1N(CC(C)C)C(=S)S\C1=C/C1=CC=C(O)C=C1 GPURHDUTZUYAFI-GHXNOFRVSA-N 0.000 description 1
- WQWGKCNSOHRYLG-NTEUORMPSA-N (E)-2-cyano-3-(2,3-dihydroxyphenyl)-N-(4-phenyl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=CC=CC=C1)=C\C1=C(C(=CC=C1)O)O WQWGKCNSOHRYLG-NTEUORMPSA-N 0.000 description 1
- RTVMKWLJNCKONC-XNTDXEJSSA-N (E)-2-cyano-3-(2-fluorophenyl)-N-(4-phenyl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=CC=CC=C1)=C\C1=C(C=CC=C1)F RTVMKWLJNCKONC-XNTDXEJSSA-N 0.000 description 1
- NSXYLYMTBKULKT-RIYZIHGNSA-N (E)-2-cyano-3-(3,4-difluorophenyl)-N-(4-phenyl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=CC=CC=C1)=C\C1=CC(=C(C=C1)F)F NSXYLYMTBKULKT-RIYZIHGNSA-N 0.000 description 1
- FGUHOVCADLOZGR-GIJQJNRQSA-N (E)-2-cyano-3-(3,4-dihydroxyphenyl)-N-(4-naphthalen-2-yl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=CC2=CC=CC=C2C=C1)=C\C1=CC(=C(C=C1)O)O FGUHOVCADLOZGR-GIJQJNRQSA-N 0.000 description 1
- OJNIGPAIVNZDEC-KPKJPENVSA-N (E)-2-cyano-3-(3,4-dihydroxyphenyl)-N-[4-(3-methoxypyridin-2-yl)-1,3-thiazol-2-yl]prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=NC=CC=C1OC)=C\C1=CC(=C(C=C1)O)O OJNIGPAIVNZDEC-KPKJPENVSA-N 0.000 description 1
- UNDVFUPFAGDEOW-NTUHNPAUSA-N (E)-2-cyano-3-(3,4-dihydroxyphenyl)-N-[4-(4-fluorophenyl)-1,3-thiazol-2-yl]prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=CC=C(C=C1)F)=C\C1=CC(=C(C=C1)O)O UNDVFUPFAGDEOW-NTUHNPAUSA-N 0.000 description 1
- RZKNWDPJJYFNKD-LDADJPATSA-N (E)-2-cyano-3-(3,4-dihydroxyphenyl)-N-[4-(4-phenoxyphenyl)-1,3-thiazol-2-yl]prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=CC=C(C=C1)OC1=CC=CC=C1)=C\C1=CC(=C(C=C1)O)O RZKNWDPJJYFNKD-LDADJPATSA-N 0.000 description 1
- WQTFXIDWKJJMRD-AWNIVKPZSA-N (E)-2-cyano-3-(3,4-dihydroxyphenyl)-N-[4-(6-methoxypyridin-3-yl)-1,3-thiazol-2-yl]prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C=1C=NC(=CC=1)OC)=C\C1=CC(=C(C=C1)O)O WQTFXIDWKJJMRD-AWNIVKPZSA-N 0.000 description 1
- VCWWZZCTWQJMMG-VZUCSPMQSA-N (E)-2-cyano-3-(3,4-dihydroxyphenyl)-N-[4-[5-(trifluoromethyl)pyridin-2-yl]-1,3-thiazol-2-yl]prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=NC=C(C=C1)C(F)(F)F)=C\C1=CC(=C(C=C1)O)O VCWWZZCTWQJMMG-VZUCSPMQSA-N 0.000 description 1
- GBRRZNRQPQTQAN-VZUCSPMQSA-N (E)-2-cyano-3-(3,5-dichloro-4-hydroxyphenyl)-N-(4-pyridin-2-yl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=NC=CC=C1)=C\C1=CC(=C(C(=C1)Cl)O)Cl GBRRZNRQPQTQAN-VZUCSPMQSA-N 0.000 description 1
- OTOJKDCPJUCWIR-AWNIVKPZSA-N (E)-2-cyano-3-(5-hydroxy-6-oxo-1H-pyridin-3-yl)-N-(4-phenyl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=CC=CC=C1)=C\C=1C=NC(=C(C=1)O)O OTOJKDCPJUCWIR-AWNIVKPZSA-N 0.000 description 1
- WXEVUZHIJWWWRQ-VXLYETTFSA-N (E)-2-cyano-3-[4-hydroxy-3,5-di(propan-2-yl)phenyl]-N-(4-phenyl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=CC=CC=C1)=C\C1=CC(=C(C(=C1)C(C)C)O)C(C)C WXEVUZHIJWWWRQ-VXLYETTFSA-N 0.000 description 1
- LNWALUTYFWAOGC-VZUCSPMQSA-N (E)-2-cyano-N-[4-(5-cyanothiophen-2-yl)-1,3-thiazol-2-yl]-3-(3,4-dihydroxyphenyl)prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C=1SC(=CC=1)C#N)=C\C1=CC(=C(C=C1)O)O LNWALUTYFWAOGC-VZUCSPMQSA-N 0.000 description 1
- AENNFNADJNGDSY-GZTJUZNOSA-N (E)-3-(3-tert-butyl-4-hydroxyphenyl)-2-cyano-N-(4-phenyl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(C)(C)(C)C=1C=C(C=CC=1O)/C=C(/C(=O)NC=1SC=C(N=1)C1=CC=CC=C1)\C#N AENNFNADJNGDSY-GZTJUZNOSA-N 0.000 description 1
- AKNCUTYWJSUACI-IZZDOVSWSA-N (E)-N-(4-tert-butyl-1,3-thiazol-2-yl)-2-cyano-3-(3,4-dihydroxyphenyl)prop-2-enamide Chemical compound C(C)(C)(C)C=1N=C(SC=1)NC(\C(=C\C1=CC(=C(C=C1)O)O)\C#N)=O AKNCUTYWJSUACI-IZZDOVSWSA-N 0.000 description 1
- DFVVUVBHXQLTGD-VGOFMYFVSA-N (E)-N-[4-(1-benzofuran-2-yl)-1,3-thiazol-2-yl]-2-cyano-3-(3,4-dihydroxyphenyl)prop-2-enamide Chemical compound O1C(=CC2=C1C=CC=C2)C=1N=C(SC=1)NC(\C(=C\C1=CC(=C(C=C1)O)O)\C#N)=O DFVVUVBHXQLTGD-VGOFMYFVSA-N 0.000 description 1
- CDQYXQUEENZLOS-CXUHLZMHSA-N (E)-N-[4-(3-tert-butylphenyl)-1,3-thiazol-2-yl]-2-cyano-3-(3,4-dihydroxyphenyl)prop-2-enamide Chemical compound C(C)(C)(C)C=1C=C(C=CC=1)C=1N=C(SC=1)NC(\C(=C\C1=CC(=C(C=C1)O)O)\C#N)=O CDQYXQUEENZLOS-CXUHLZMHSA-N 0.000 description 1
- VKCPYJAVNVVFGD-OVCLIPMQSA-N (E)-N-[4-(4-tert-butylphenyl)-1,3-thiazol-2-yl]-2-cyano-3-(3,5-dichloro-4-hydroxyphenyl)prop-2-enamide Chemical compound C(C)(C)(C)C1=CC=C(C=C1)C=1N=C(SC=1)NC(\C(=C\C1=CC(=C(C(=C1)Cl)O)Cl)\C#N)=O VKCPYJAVNVVFGD-OVCLIPMQSA-N 0.000 description 1
- AINUZWOJVZDYRA-SDQBBNPISA-N (Z)-2-cyano-3-(1H-imidazol-5-yl)-N-(4-phenyl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=CC=CC=C1)=C/C=1N=CNC=1 AINUZWOJVZDYRA-SDQBBNPISA-N 0.000 description 1
- NTCBUXIQMLORSI-GIDUJCDVSA-N (e)-1-[4-(4-bromophenyl)phenyl]-3-phenylprop-2-en-1-one Chemical compound C1=CC(Br)=CC=C1C1=CC=C(C(=O)\C=C\C=2C=CC=CC=2)C=C1 NTCBUXIQMLORSI-GIDUJCDVSA-N 0.000 description 1
- ZGYIXVSQHOKQRZ-COIATFDQSA-N (e)-n-[4-[3-chloro-4-(pyridin-2-ylmethoxy)anilino]-3-cyano-7-[(3s)-oxolan-3-yl]oxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound N#CC1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZGYIXVSQHOKQRZ-COIATFDQSA-N 0.000 description 1
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 1
- KWCDIRFSULAMOC-UHFFFAOYSA-N 1-(1-adamantyl)-2-bromoethanone Chemical compound C1C(C2)CC3CC2CC1(C(=O)CBr)C3 KWCDIRFSULAMOC-UHFFFAOYSA-N 0.000 description 1
- NVRNCBWTEDOAQA-UHFFFAOYSA-N 1-(1-benzofuran-2-yl)-2-bromoethanone Chemical compound C1=CC=C2OC(C(=O)CBr)=CC2=C1 NVRNCBWTEDOAQA-UHFFFAOYSA-N 0.000 description 1
- APWRZPQBPCAXFP-UHFFFAOYSA-N 1-(1-oxo-2H-isoquinolin-5-yl)-5-(trifluoromethyl)-N-[2-(trifluoromethyl)pyridin-4-yl]pyrazole-4-carboxamide Chemical compound O=C1NC=CC2=C(C=CC=C12)N1N=CC(=C1C(F)(F)F)C(=O)NC1=CC(=NC=C1)C(F)(F)F APWRZPQBPCAXFP-UHFFFAOYSA-N 0.000 description 1
- PIMNFNXBTGPCIL-UHFFFAOYSA-N 1-(2-bromophenyl)ethanone Chemical compound CC(=O)C1=CC=CC=C1Br PIMNFNXBTGPCIL-UHFFFAOYSA-N 0.000 description 1
- OXJLDNSPGPBDCP-UHFFFAOYSA-N 1-(3,5-difluorophenyl)ethanone Chemical compound CC(=O)C1=CC(F)=CC(F)=C1 OXJLDNSPGPBDCP-UHFFFAOYSA-N 0.000 description 1
- JYAQYXOVOHJRCS-UHFFFAOYSA-N 1-(3-bromophenyl)ethanone Chemical compound CC(=O)C1=CC=CC(Br)=C1 JYAQYXOVOHJRCS-UHFFFAOYSA-N 0.000 description 1
- IGYMEVBVNSGNSD-UHFFFAOYSA-N 1-(3-methoxypyridin-2-yl)ethanone Chemical compound COC1=CC=CN=C1C(C)=O IGYMEVBVNSGNSD-UHFFFAOYSA-N 0.000 description 1
- SYISHRLXIIZBHJ-UHFFFAOYSA-N 1-(3-methylpyridin-2-yl)ethanone Chemical compound CC(=O)C1=NC=CC=C1C SYISHRLXIIZBHJ-UHFFFAOYSA-N 0.000 description 1
- TVEGMYYTIFLVAP-UHFFFAOYSA-N 1-(3-tert-butylphenyl)ethanone Chemical compound CC(=O)C1=CC=CC(C(C)(C)C)=C1 TVEGMYYTIFLVAP-UHFFFAOYSA-N 0.000 description 1
- WYECURVXVYPVAT-UHFFFAOYSA-N 1-(4-bromophenyl)ethanone Chemical compound CC(=O)C1=CC=C(Br)C=C1 WYECURVXVYPVAT-UHFFFAOYSA-N 0.000 description 1
- KAVZYDHKJNABPC-UHFFFAOYSA-N 1-(4-methylsulfonylphenyl)ethanone Chemical compound CC(=O)C1=CC=C(S(C)(=O)=O)C=C1 KAVZYDHKJNABPC-UHFFFAOYSA-N 0.000 description 1
- AKQWEDMTPCAESO-UHFFFAOYSA-N 1-(4-morpholin-4-ylphenyl)ethanone Chemical compound C1=CC(C(=O)C)=CC=C1N1CCOCC1 AKQWEDMTPCAESO-UHFFFAOYSA-N 0.000 description 1
- DJNIFZYQFLFGDT-UHFFFAOYSA-N 1-(4-phenoxyphenyl)ethanone Chemical compound C1=CC(C(=O)C)=CC=C1OC1=CC=CC=C1 DJNIFZYQFLFGDT-UHFFFAOYSA-N 0.000 description 1
- JNHLHPMTMTYLCP-UHFFFAOYSA-N 1-(4-tert-butyl-2,6-dimethylphenyl)ethanone Chemical compound CC(=O)C1=C(C)C=C(C(C)(C)C)C=C1C JNHLHPMTMTYLCP-UHFFFAOYSA-N 0.000 description 1
- LTHPBRNHHJIQME-UHFFFAOYSA-N 1-(4-tert-butylphenyl)-2-chloroethanone Chemical compound CC(C)(C)C1=CC=C(C(=O)CCl)C=C1 LTHPBRNHHJIQME-UHFFFAOYSA-N 0.000 description 1
- IGBZCOWXSCWSHO-UHFFFAOYSA-N 1-(5-bromothiophen-2-yl)ethanone Chemical compound CC(=O)C1=CC=C(Br)S1 IGBZCOWXSCWSHO-UHFFFAOYSA-N 0.000 description 1
- RYOQZXOVBJIUSX-UHFFFAOYSA-N 1-(6-methoxypyridin-3-yl)ethanone Chemical compound COC1=CC=C(C(C)=O)C=N1 RYOQZXOVBJIUSX-UHFFFAOYSA-N 0.000 description 1
- PVRYOKQFLBSILA-UHFFFAOYSA-N 1-(6-methylpyridin-3-yl)ethanone Chemical compound CC(=O)C1=CC=C(C)N=C1 PVRYOKQFLBSILA-UHFFFAOYSA-N 0.000 description 1
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 1
- UYHTUQHYGKAYJM-UHFFFAOYSA-N 1-[3-(trifluoromethoxy)phenyl]ethanone Chemical compound CC(=O)C1=CC=CC(OC(F)(F)F)=C1 UYHTUQHYGKAYJM-UHFFFAOYSA-N 0.000 description 1
- SEAOBYFQWJFORM-UHFFFAOYSA-N 1-bromo-4-(trifluoromethoxy)benzene Chemical compound FC(F)(F)OC1=CC=C(Br)C=C1 SEAOBYFQWJFORM-UHFFFAOYSA-N 0.000 description 1
- WUYTZBFFXRNJSB-UHFFFAOYSA-N 1-phenyl-2-pyrazin-2-ylethanone Chemical compound C=1C=CC=CC=1C(=O)CC1=CN=CC=N1 WUYTZBFFXRNJSB-UHFFFAOYSA-N 0.000 description 1
- FTSKLNSVINAQGJ-UHFFFAOYSA-N 1-pyridazin-3-ylethanone Chemical compound CC(=O)C1=CC=CN=N1 FTSKLNSVINAQGJ-UHFFFAOYSA-N 0.000 description 1
- WMQUKDQWMMOHSA-UHFFFAOYSA-N 1-pyridin-4-ylethanone Chemical compound CC(=O)C1=CC=NC=C1 WMQUKDQWMMOHSA-UHFFFAOYSA-N 0.000 description 1
- ZQEXIXXJFSQPNA-UHFFFAOYSA-N 1h-imidazole-5-carbaldehyde Chemical compound O=CC1=CNC=N1 ZQEXIXXJFSQPNA-UHFFFAOYSA-N 0.000 description 1
- JTWYTTXTJFDYAG-UHFFFAOYSA-N 1h-indazole-6-carbaldehyde Chemical compound O=CC1=CC=C2C=NNC2=C1 JTWYTTXTJFDYAG-UHFFFAOYSA-N 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- AJKVQEKCUACUMD-UHFFFAOYSA-N 2-Acetylpyridine Chemical compound CC(=O)C1=CC=CC=N1 AJKVQEKCUACUMD-UHFFFAOYSA-N 0.000 description 1
- APLNAFMUEHKRLM-UHFFFAOYSA-N 2-[5-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]-1-(3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NN=C(O1)CC(=O)N1CC2=C(CC1)N=CN2 APLNAFMUEHKRLM-UHFFFAOYSA-N 0.000 description 1
- YOSDTJYMDAEEAZ-UHFFFAOYSA-N 2-acetyl-5-methylthiophene Chemical compound CC(=O)C1=CC=C(C)S1 YOSDTJYMDAEEAZ-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- AXAVXPMQTGXXJZ-UHFFFAOYSA-N 2-aminoacetic acid;2-amino-2-(hydroxymethyl)propane-1,3-diol Chemical compound NCC(O)=O.OCC(N)(CO)CO AXAVXPMQTGXXJZ-UHFFFAOYSA-N 0.000 description 1
- ITAQNNGDCNFGID-UHFFFAOYSA-N 2-bromo-1-(3-fluorophenyl)ethanone Chemical compound FC1=CC=CC(C(=O)CBr)=C1 ITAQNNGDCNFGID-UHFFFAOYSA-N 0.000 description 1
- FKJSFKCZZIXQIP-UHFFFAOYSA-N 2-bromo-1-(4-bromophenyl)ethanone Chemical compound BrCC(=O)C1=CC=C(Br)C=C1 FKJSFKCZZIXQIP-UHFFFAOYSA-N 0.000 description 1
- ZJFWCELATJMDNO-UHFFFAOYSA-N 2-bromo-1-(4-fluorophenyl)ethanone Chemical compound FC1=CC=C(C(=O)CBr)C=C1 ZJFWCELATJMDNO-UHFFFAOYSA-N 0.000 description 1
- XQJAHBHCLXUGEP-UHFFFAOYSA-N 2-bromo-1-(4-methoxyphenyl)ethanone Chemical compound COC1=CC=C(C(=O)CBr)C=C1 XQJAHBHCLXUGEP-UHFFFAOYSA-N 0.000 description 1
- KRVGXFREOJHJAX-UHFFFAOYSA-N 2-bromo-1-(4-methylphenyl)ethanone Chemical compound CC1=CC=C(C(=O)CBr)C=C1 KRVGXFREOJHJAX-UHFFFAOYSA-N 0.000 description 1
- MOXZEXOTBFOOLT-UHFFFAOYSA-N 2-bromo-1-(4-methylthiophen-2-yl)ethanone Chemical compound CC1=CSC(C(=O)CBr)=C1 MOXZEXOTBFOOLT-UHFFFAOYSA-N 0.000 description 1
- AOAGGWLQIILIIV-UHFFFAOYSA-N 2-bromo-1-[4-(trifluoromethoxy)phenyl]ethanone Chemical compound FC(F)(F)OC1=CC=C(C(=O)CBr)C=C1 AOAGGWLQIILIIV-UHFFFAOYSA-N 0.000 description 1
- YHXHHGDUANVQHE-UHFFFAOYSA-N 2-bromo-1-naphthalen-2-ylethanone Chemical compound C1=CC=CC2=CC(C(=O)CBr)=CC=C21 YHXHHGDUANVQHE-UHFFFAOYSA-N 0.000 description 1
- UHWNENCHFSDZQP-UHFFFAOYSA-N 2-bromo-1-thiophen-2-ylethanone Chemical compound BrCC(=O)C1=CC=CS1 UHWNENCHFSDZQP-UHFFFAOYSA-N 0.000 description 1
- APOHNICSBWBQOM-UHFFFAOYSA-N 2-bromo-2-(oxan-4-yl)-1-phenylethanone Chemical compound BrC(C(=O)C1=CC=CC=C1)C1CCOCC1 APOHNICSBWBQOM-UHFFFAOYSA-N 0.000 description 1
- JVUMAEKJUGYQRX-UHFFFAOYSA-N 2-cyano-3-(3-cyano-4-hydroxyphenyl)-N-(4-phenyl-1,3-thiazol-2-yl)propanamide Chemical compound C(#N)C(C(=O)NC=1SC=C(N=1)C1=CC=CC=C1)CC1=CC(=C(C=C1)O)C#N JVUMAEKJUGYQRX-UHFFFAOYSA-N 0.000 description 1
- XNSWRRFTUXTRFM-UHFFFAOYSA-N 2-cyano-N-[4-(2-fluoro-4-methoxyphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=C(C=C(C=C1)OC)F XNSWRRFTUXTRFM-UHFFFAOYSA-N 0.000 description 1
- OHXGJJQQZQQYBO-UHFFFAOYSA-N 2-cyano-N-[4-(3-cyclopropyl-4-methoxyphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC(=C(C=C1)OC)C1CC1 OHXGJJQQZQQYBO-UHFFFAOYSA-N 0.000 description 1
- GYXVXMIPPGRWON-UHFFFAOYSA-N 2-cyano-N-[4-(4-methylsulfonylphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC=C(C=C1)S(=O)(=O)C GYXVXMIPPGRWON-UHFFFAOYSA-N 0.000 description 1
- WUPNEYCHEWSZGS-UHFFFAOYSA-N 2-cyano-N-[4-(4-phenylphenyl)-1,3-thiazol-2-yl]acetamide Chemical compound C1(=CC=C(C=C1)C=1N=C(SC=1)NC(CC#N)=O)C1=CC=CC=C1 WUPNEYCHEWSZGS-UHFFFAOYSA-N 0.000 description 1
- ZFFIOUNQFCBEJX-UHFFFAOYSA-N 2-cyclohexyl-1-[4-(trifluoromethoxy)phenyl]ethanol Chemical compound C=1C=C(OC(F)(F)F)C=CC=1C(O)CC1CCCCC1 ZFFIOUNQFCBEJX-UHFFFAOYSA-N 0.000 description 1
- ONRPXRPUBXXCCM-UHFFFAOYSA-N 2-fluoro-4-hydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C(F)=C1 ONRPXRPUBXXCCM-UHFFFAOYSA-N 0.000 description 1
- ZWDVQMVZZYIAHO-UHFFFAOYSA-N 2-fluorobenzaldehyde Chemical compound FC1=CC=CC=C1C=O ZWDVQMVZZYIAHO-UHFFFAOYSA-N 0.000 description 1
- JPHKMYXKNKLNDF-UHFFFAOYSA-N 3,4-difluorobenzaldehyde Chemical compound FC1=CC=C(C=O)C=C1F JPHKMYXKNKLNDF-UHFFFAOYSA-N 0.000 description 1
- SKOYTQILPMNZQO-UHFFFAOYSA-N 3,5-difluoro-4-hydroxybenzaldehyde Chemical compound OC1=C(F)C=C(C=O)C=C1F SKOYTQILPMNZQO-UHFFFAOYSA-N 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- QZBKFDMKAXUEFY-UHFFFAOYSA-N 3-(3-acetamidophenyl)-2-cyano-N-(4-phenyl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(C)(=O)NC=1C=C(C=CC=1)C=C(C(=O)NC=1SC=C(N=1)C1=CC=CC=C1)C#N QZBKFDMKAXUEFY-UHFFFAOYSA-N 0.000 description 1
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 1
- NMTUHPSKJJYGML-UHFFFAOYSA-N 3-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC=CC(C=O)=C1 NMTUHPSKJJYGML-UHFFFAOYSA-N 0.000 description 1
- UHDNUPHSDMOGCR-UHFFFAOYSA-N 3-Formylbenzoic acid Chemical compound OC(=O)C1=CC=CC(C=O)=C1 UHDNUPHSDMOGCR-UHFFFAOYSA-N 0.000 description 1
- UXPSCOZXPPIZNM-MHWRWJLKSA-N 3-[(E)-2-cyano-3-oxo-3-[(4-phenyl-1,3-thiazol-2-yl)amino]prop-1-enyl]benzoic acid Chemical compound C(#N)\C(=C/C=1C=C(C(=O)O)C=CC=1)\C(NC=1SC=C(N=1)C1=CC=CC=C1)=O UXPSCOZXPPIZNM-MHWRWJLKSA-N 0.000 description 1
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 1
- SRVXSISGYBMIHR-UHFFFAOYSA-N 3-[3-[3-(2-amino-2-oxoethyl)phenyl]-5-chlorophenyl]-3-(5-methyl-1,3-thiazol-2-yl)propanoic acid Chemical compound S1C(C)=CN=C1C(CC(O)=O)C1=CC(Cl)=CC(C=2C=C(CC(N)=O)C=CC=2)=C1 SRVXSISGYBMIHR-UHFFFAOYSA-N 0.000 description 1
- VFLZDVVSXIEMPZ-UHFFFAOYSA-N 3-[4-acetamido-3-[tert-butyl(dimethyl)silyl]oxyphenyl]-2-cyano-N-(4-phenyl-1,3-thiazol-2-yl)prop-2-enamide Chemical compound C(C)(=O)NC1=C(C=C(C=C1)C=C(C(=O)NC=1SC=C(N=1)C1=CC=CC=C1)C#N)O[Si](C)(C)C(C)(C)C VFLZDVVSXIEMPZ-UHFFFAOYSA-N 0.000 description 1
- SBCFGFDAZCTSRH-UHFFFAOYSA-N 3-acetylbenzonitrile Chemical compound CC(=O)C1=CC=CC(C#N)=C1 SBCFGFDAZCTSRH-UHFFFAOYSA-N 0.000 description 1
- RKFNAZGRJVNWEW-UHFFFAOYSA-N 3-cyclohexylpropionaldehyde Natural products O=CCCC1CCCCC1 RKFNAZGRJVNWEW-UHFFFAOYSA-N 0.000 description 1
- OOGOFUKAJDPHDJ-UHFFFAOYSA-N 3-fluoro-4-hydroxy-5-methoxybenzaldehyde Chemical compound COC1=CC(C=O)=CC(F)=C1O OOGOFUKAJDPHDJ-UHFFFAOYSA-N 0.000 description 1
- CWNPOQFCIIFQDM-UHFFFAOYSA-N 3-nitrobenzyl alcohol Chemical compound OCC1=CC=CC([N+]([O-])=O)=C1 CWNPOQFCIIFQDM-UHFFFAOYSA-N 0.000 description 1
- HTAUVJPDFDVVHV-UHFFFAOYSA-N 4-(4-phenylphenyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC(=CC=2)C=2C=CC=CC=2)=C1 HTAUVJPDFDVVHV-UHFFFAOYSA-N 0.000 description 1
- BAKYASSDAXQKKY-UHFFFAOYSA-N 4-Hydroxy-3-methylbenzaldehyde Chemical compound CC1=CC(C=O)=CC=C1O BAKYASSDAXQKKY-UHFFFAOYSA-N 0.000 description 1
- QJNMYPYXNCYQQT-MHWRWJLKSA-N 4-[(E)-2-cyano-3-oxo-3-[(4-phenyl-1,3-thiazol-2-yl)amino]prop-1-enyl]benzoic acid Chemical compound C(#N)\C(=C/C1=CC=C(C(=O)O)C=C1)\C(NC=1SC=C(N=1)C1=CC=CC=C1)=O QJNMYPYXNCYQQT-MHWRWJLKSA-N 0.000 description 1
- BONZNDGHGITCAY-UHFFFAOYSA-N 4-[2-[(2-cyanoacetyl)amino]-1,3-thiazol-4-yl]-N,N-dimethylbenzamide Chemical compound C(#N)CC(=O)NC=1SC=C(N=1)C1=CC=C(C(=O)N(C)C)C=C1 BONZNDGHGITCAY-UHFFFAOYSA-N 0.000 description 1
- YFCIFWOJYYFDQP-PTWZRHHISA-N 4-[3-amino-6-[(1S,3S,4S)-3-fluoro-4-hydroxycyclohexyl]pyrazin-2-yl]-N-[(1S)-1-(3-bromo-5-fluorophenyl)-2-(methylamino)ethyl]-2-fluorobenzamide Chemical compound CNC[C@@H](NC(=O)c1ccc(cc1F)-c1nc(cnc1N)[C@H]1CC[C@H](O)[C@@H](F)C1)c1cc(F)cc(Br)c1 YFCIFWOJYYFDQP-PTWZRHHISA-N 0.000 description 1
- KADXGLUHGAXACG-UHFFFAOYSA-N 4-acetyl-n,n-dimethylbenzamide Chemical compound CN(C)C(=O)C1=CC=C(C(C)=O)C=C1 KADXGLUHGAXACG-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- JDWWIEFMFPWBST-UHFFFAOYSA-N 4-hydroxy-2-methylbenzaldehyde Chemical compound CC1=CC(O)=CC=C1C=O JDWWIEFMFPWBST-UHFFFAOYSA-N 0.000 description 1
- WVGDLTQPAQUBMO-UHFFFAOYSA-N 4-hydroxy-3,5-di(propan-2-yl)benzaldehyde Chemical compound CC(C)C1=CC(C=O)=CC(C(C)C)=C1O WVGDLTQPAQUBMO-UHFFFAOYSA-N 0.000 description 1
- ZLMUFCNWTPSOSN-UHFFFAOYSA-N 4-hydroxy-3-(trifluoromethyl)benzaldehyde Chemical compound OC1=CC=C(C=O)C=C1C(F)(F)F ZLMUFCNWTPSOSN-UHFFFAOYSA-N 0.000 description 1
- YTHJCZRFJGXPTL-UHFFFAOYSA-N 4-hydroxy-3-nitrobenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1[N+]([O-])=O YTHJCZRFJGXPTL-UHFFFAOYSA-N 0.000 description 1
- LORPDGZOLAPNHP-UHFFFAOYSA-N 4-hydroxynaphthalene-1-carbaldehyde Chemical compound C1=CC=C2C(O)=CC=C(C=O)C2=C1 LORPDGZOLAPNHP-UHFFFAOYSA-N 0.000 description 1
- NTPLXRHDUXRPNE-UHFFFAOYSA-N 4-methoxyacetophenone Chemical compound COC1=CC=C(C(C)=O)C=C1 NTPLXRHDUXRPNE-UHFFFAOYSA-N 0.000 description 1
- BXRFQSNOROATLV-UHFFFAOYSA-N 4-nitrobenzaldehyde Chemical compound [O-][N+](=O)C1=CC=C(C=O)C=C1 BXRFQSNOROATLV-UHFFFAOYSA-N 0.000 description 1
- CUWZBHVYLVGOAB-UHFFFAOYSA-N 4-tert-butyl-1,3-thiazol-2-amine Chemical compound CC(C)(C)C1=CSC(N)=N1 CUWZBHVYLVGOAB-UHFFFAOYSA-N 0.000 description 1
- BVQOHVMOURVZMO-UHFFFAOYSA-N 5,6-dimethoxypyridine-3-carbaldehyde Chemical compound COC1=CC(C=O)=CN=C1OC BVQOHVMOURVZMO-UHFFFAOYSA-N 0.000 description 1
- JHAHYQCNCGGYDT-UHFFFAOYSA-N 5-(oxan-4-yl)-4-phenyl-1,3-thiazol-2-amine Chemical compound C1(=CC=CC=C1)C=1N=C(SC=1C1CCOCC1)N JHAHYQCNCGGYDT-UHFFFAOYSA-N 0.000 description 1
- IRPVABHDSJVBNZ-RTHVDDQRSA-N 5-[1-(cyclopropylmethyl)-5-[(1R,5S)-3-(oxetan-3-yl)-3-azabicyclo[3.1.0]hexan-6-yl]pyrazol-3-yl]-3-(trifluoromethyl)pyridin-2-amine Chemical compound C1=C(C(F)(F)F)C(N)=NC=C1C1=NN(CC2CC2)C(C2[C@@H]3CN(C[C@@H]32)C2COC2)=C1 IRPVABHDSJVBNZ-RTHVDDQRSA-N 0.000 description 1
- VSHPLUBHIUFLES-UHFFFAOYSA-N 5-acetylthiophene-2-carbonitrile Chemical compound CC(=O)C1=CC=C(C#N)S1 VSHPLUBHIUFLES-UHFFFAOYSA-N 0.000 description 1
- GHOLBYGCJQUZBQ-UHFFFAOYSA-N 5-chloro-4-phenyl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC=CC=2)=C1Cl GHOLBYGCJQUZBQ-UHFFFAOYSA-N 0.000 description 1
- HTXQOROHFFYFMC-UHFFFAOYSA-N 5-methyl-4-phenyl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC=CC=2)=C1C HTXQOROHFFYFMC-UHFFFAOYSA-N 0.000 description 1
- LSLUWQIENURREM-UHFFFAOYSA-N 5-phenyl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC=C1C1=CC=CC=C1 LSLUWQIENURREM-UHFFFAOYSA-N 0.000 description 1
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 101150065175 Atm gene Proteins 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- MTZMILDKMGKZEV-UHFFFAOYSA-N BrCC(=O)C1=CC=C(C(=O)N(C)C)C=C1 Chemical compound BrCC(=O)C1=CC=C(C(=O)N(C)C)C=C1 MTZMILDKMGKZEV-UHFFFAOYSA-N 0.000 description 1
- 208000017843 C syndrome Diseases 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- 208000031640 Chromosome Fragility Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 102000003844 DNA helicases Human genes 0.000 description 1
- 108090000133 DNA helicases Proteins 0.000 description 1
- 102100039116 DNA repair protein RAD50 Human genes 0.000 description 1
- 102100024607 DNA topoisomerase 1 Human genes 0.000 description 1
- 102100033587 DNA topoisomerase 2-alpha Human genes 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 108010042407 Endonucleases Proteins 0.000 description 1
- 102000004533 Endonucleases Human genes 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 108060002716 Exonuclease Proteins 0.000 description 1
- GISRWBROCYNDME-PELMWDNLSA-N F[C@H]1[C@H]([C@H](NC1=O)COC1=NC=CC2=CC(=C(C=C12)OC)C(=O)N)C Chemical compound F[C@H]1[C@H]([C@H](NC1=O)COC1=NC=CC2=CC(=C(C=C12)OC)C(=O)N)C GISRWBROCYNDME-PELMWDNLSA-N 0.000 description 1
- 101000743929 Homo sapiens DNA repair protein RAD50 Proteins 0.000 description 1
- 101000830681 Homo sapiens DNA topoisomerase 1 Proteins 0.000 description 1
- 101000801505 Homo sapiens DNA topoisomerase 2-alpha Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 206010028400 Mutagenic effect Diseases 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- KCRZZUWUHXTMTA-UHFFFAOYSA-N N-[4-(2-tert-butylphenyl)-1,3-thiazol-2-yl]-2-cyanoacetamide Chemical compound C(C)(C)(C)C1=C(C=CC=C1)C=1N=C(SC=1)NC(CC#N)=O KCRZZUWUHXTMTA-UHFFFAOYSA-N 0.000 description 1
- CBCDUXSBZAHVGG-UHFFFAOYSA-N N-[4-(3-tert-butylphenyl)-1,3-thiazol-2-yl]-2-cyanoacetamide Chemical compound C(C)(C)(C)C=1C=C(C=CC=1)C=1N=C(SC=1)NC(CC#N)=O CBCDUXSBZAHVGG-UHFFFAOYSA-N 0.000 description 1
- IDRGFNPZDVBSSE-UHFFFAOYSA-N OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F Chemical compound OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F IDRGFNPZDVBSSE-UHFFFAOYSA-N 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 101150003085 Pdcl gene Proteins 0.000 description 1
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- 101150085800 RPA2 gene Proteins 0.000 description 1
- 102100035525 Replication protein A 32 kDa subunit Human genes 0.000 description 1
- 206010070834 Sensitisation Diseases 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 101710183280 Topoisomerase Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 101100033868 Xenopus laevis rpa2-a gene Proteins 0.000 description 1
- 101100033871 Xenopus laevis rpa2-b gene Proteins 0.000 description 1
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 1
- XMYKNCNAZKMVQN-NYYWCZLTSA-N [(e)-(3-aminopyridin-2-yl)methylideneamino]thiourea Chemical compound NC(=S)N\N=C\C1=NC=CC=C1N XMYKNCNAZKMVQN-NYYWCZLTSA-N 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000000118 anti-neoplastic effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000004622 benzoxazinyl group Chemical group O1NC(=CC2=C1C=CC=C2)* 0.000 description 1
- 230000008236 biological pathway Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 235000019504 cigarettes Nutrition 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 238000011157 data evaluation Methods 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 239000011903 deuterated solvents Substances 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 125000005434 dihydrobenzoxazinyl group Chemical group O1N(CCC2=C1C=CC=C2)* 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- JLMMMEDWRUVCEW-UHFFFAOYSA-N ethyl 2-(oxan-4-yl)acetate Chemical compound CCOC(=O)CC1CCOCC1 JLMMMEDWRUVCEW-UHFFFAOYSA-N 0.000 description 1
- 102000013165 exonuclease Human genes 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- YLTGFGDODHXMFB-UHFFFAOYSA-N isoacetovanillone Chemical compound COC1=CC=C(C(C)=O)C=C1O YLTGFGDODHXMFB-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- DBYQHFPBWKKZAT-UHFFFAOYSA-N lithium;benzene Chemical compound [Li+].C1=CC=[C-]C=C1 DBYQHFPBWKKZAT-UHFFFAOYSA-N 0.000 description 1
- 239000012160 loading buffer Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- IMXBRVLCKXGWSS-UHFFFAOYSA-N methyl 2-cyclohexylacetate Chemical compound COC(=O)CC1CCCCC1 IMXBRVLCKXGWSS-UHFFFAOYSA-N 0.000 description 1
- HBGXBRNLTSFTIG-UHFFFAOYSA-N methyl 3-oxo-4h-1,4-benzoxazine-7-carboxylate Chemical compound N1C(=O)COC2=CC(C(=O)OC)=CC=C21 HBGXBRNLTSFTIG-UHFFFAOYSA-N 0.000 description 1
- ZCSQXAIOMRFZAN-OVCLIPMQSA-N methyl 4-[2-[[(E)-2-cyano-3-(3,4-dihydroxyphenyl)prop-2-enoyl]amino]-1,3-thiazol-4-yl]benzoate Chemical compound C(#N)/C(/C(=O)NC=1SC=C(N=1)C1=CC=C(C(=O)OC)C=C1)=C\C1=CC(=C(C=C1)O)O ZCSQXAIOMRFZAN-OVCLIPMQSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 231100000243 mutagenic effect Toxicity 0.000 description 1
- KELKOCKBZGULER-UHFFFAOYSA-N n-(4-tert-butyl-1,3-thiazol-2-yl)-2-cyanoacetamide Chemical compound CC(C)(C)C1=CSC(NC(=O)CC#N)=N1 KELKOCKBZGULER-UHFFFAOYSA-N 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- 230000013271 negative regulation by symbiont of host defense-related programmed cell death Effects 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229940072033 potash Drugs 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 150000003140 primary amides Chemical class 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- KRIOVPPHQSLHCZ-UHFFFAOYSA-N propiophenone Chemical compound CCC(=O)C1=CC=CC=C1 KRIOVPPHQSLHCZ-UHFFFAOYSA-N 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 150000003334 secondary amides Chemical class 0.000 description 1
- XIIOFHFUYBLOLW-UHFFFAOYSA-N selpercatinib Chemical compound OC(COC=1C=C(C=2N(C=1)N=CC=2C#N)C=1C=NC(=CC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC)(C)C XIIOFHFUYBLOLW-UHFFFAOYSA-N 0.000 description 1
- 230000008313 sensitization Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 150000003511 tertiary amides Chemical class 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 229960005526 triapine Drugs 0.000 description 1
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 238000009281 ultraviolet germicidal irradiation Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/46—Acylated amino or imino radicals by carboxylic acids, or sulfur or nitrogen analogues thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
Compuesto de fórmula general (1): **(Ver fórmula)** o una sal farmacéuticamente aceptable, o un solvato del mismo, en la que: R1 se selecciona del grupo que consiste en arilo; heteroarilo; en el que cada uno del arilo, heteroarilo puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, seleccionándose cada resto independientemente del grupo que consiste en F, Cl, Br, I, OH, CN, N3, =O, O(alquilo C1-C6), =S, SH, S(alquilo C1-C6), S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3, C2F5, OCF3, OC2F5, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), =N-OH, =N-O(alquilo C1-C6), NO2, COOH, COO(alquilo C1-C6), CO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, (alquil C1-C6)-S(O)2-NH-, (alquil C1-C6)-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-(SO)2-, (alquil C1-C6)2N-(SO)2-, (alquil C1-C6)-CO-NH-, (alquil C1-C6)-CO-N(alquil C1-C6)-, (alquil C1-C6)-OCO-NH-, (alquil C1-C6)-OCO-N(alquil C1-C6)-, (alquil C1-C6)-CO-NH-CO-, (alquil C1-C6)-CO- N(alquil C1-C6)-CO-, NH2-CO-NH-, (alquil C1-C6)-NH-CO-NH-, (alquil C1-C6)2N-CO-NH-, NH2-CO-N(alquil C1-C6)-, (alquil C1-C6)-NH-CO-N(alquil C1-C6)-, (alquil C1-C6)2N-CO-N(alquil C1-C6)-, NH2-S(O)2-NH-, (alquil C1-C6)-NH-S(O)2-NH-, (alquil C1-C6)2N-S(O)2-NH-, NH2-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-S(O)2- N(alquil C1-C6)-, (alquil C1-C6)2N-S(O)2-N(alquil C1-C6)-, alquilo C1-C6, O-alquilo C1-C6, O-fenilo, fenilo; mientras que el alquilo C1-C6, O-fenilo, fenilo en estos restos puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes independientemente seleccionados de: Cl, Br, I, alquilo C1-C6, OH, O-alquilo C1-C6, SH, SCH3, S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3, OCF3, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), COOH, COO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, NHC(O)-alquilo C1-C6 o NHC(O)NH2; R2 se selecciona de arilo C6-C12 y heteroarilo que tiene de 5 a 12 átomos de anillos, que puede estar opcionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, OH, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5, O(alquilo C1-C4), fenilo, O- fenilo, NH2, N(alquilo C1-C4)2, NO2, NHCO(alquilo C1-C4), CF3, OCF3, CN, S(O)2-alquilo C1-C6, SO2NH(alquilo C1-C6), SO2N(alquilo C1-C6)2; en particular seleccionados de F, Cl, Br, alquilo C1-C6, O(alquilo C1-C4), CF3, OCF3; R6 se selecciona del grupo que consiste en H; heterociclilo; cicloalquilo; heteroarilo; arilo; heteroarilo; en el que cada uno del arilo, cicloalquilo, heterociclilo, heteroarilo, puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, alquilo C1-C6, OH, O-alquilo C1-C6, SH, SCH3, S(O)- alquilo C1-C6, S(O)2-alquilo C1-C6, CF3, OCF3, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), NO2, COOH, COO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, NHC(O)-alquilo C1-C6 o NHC(O)NH2; R3 se selecciona del grupo que consiste en H; arilo; heteroarilo; heterociclilo; cicloalquilo; alquilo; y halógeno; en el que cada uno del arilo, cicloalquilo, alquilo o heteroarilo puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, seleccionándose cada resto independientemente del grupo que consiste en F, Cl, Br, I, OH, CN, N3, =O, O(alquilo C1-C6), =S, SH, S(alquilo C1-C6), S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3, C2F5, OCF3, OC2F5, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), =N-OH, =N-O(alquilo C1-C6), NO2, COOH, COO(alquilo C1-C6), CO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, (alquil C1-C6)-S(O)2-NH-, (alquil C1- C6)-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-(SO)2-, (alquil C1-C6)2N-(SO)2-, (alquil C1-C6)-CO-NH-, (alquil C1-C6)-CO-N(alquil C1-C6)-, (alquil C1-C6)-OCO-NH-, (alquil C1-C6)-OCO-N(alquil C1-C6)-, (alquil C1-C6)-CO- NH-CO-, (alquil C1-C6)-CO-N(alquil C1-C6)-CO-, NH2-CO-NH-, (alquil C1-C6)-NH-CO-NH-, (alquil C1-C6)2N- CO-NH-, NH2-CO-N(alquil C1-C6)-, (alquil C1-C6)-NH-CO-N(alquil C1-C6)-, (alquil C1-C6)2N-CO-N(alquil C1- C6)-, NH2-S(O)2-NH-, (alquil C1-C6)-NH-S(O)2-NH-, (alquil C1-C6)2N-S(O)2-NH-, NH2-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)2N-S(O)2-N(alquil C1-C6)-, alquilo C1-C6, O-alquilo C1- C6, O-fenilo, fenilo; mientras que el alquilo C1-C6, O-fenilo, fenilo en estos restos puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes independientemente seleccionados de: F, Cl, Br, alquilo C1-C6, OH, O-alquilo C1-C6, SH, SCH3, S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3, OCF3, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), NO2, COOH, COO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, NHC(O)-alquilo C1-C6 o NHC(O)NH2, R4 se selecciona del grupo que consiste en H y alquilo C1-C6; R5 se selecciona del grupo que consiste en H y arilo; para su uso en un método de tratamiento de cáncer, envejecimiento prematuro o enfermedades neurológicas, más específicamente de cáncer relacionado con la inestabilidad genómica, envejecimiento prematuro relacionado con la inestabilidad genómica y/o enfermedades neurológicas relacionadas con la inestabilidad genómica, en particular cáncer relacionado con MRE11, envejecimiento prematuro relacionado con MRE11 y/o enfermedades neurológicas relacionadas con MRE11.
Description
DESCRIPCIÓN
Aminotiazoles sustituidos como inhibidores de nucleasas
Campo de la invención
La presente invención se refiere a aminotiazoles sustituidos como inhibidores de nucleasas, especialmente nucleasa MRE11 y complejos que contienen MRE11, a composiciones farmacéuticas que contienen los compuestos y a métodos de tratamiento que usan los compuestos y las composiciones para tratar enfermedades tales como el cáncer y otras enfermedades asociadas con la inestabilidad genómica.
Técnica anterior
A pesar del intenso desarrollo de nuevas sustancias antineoplásicas, es necesario mejorar el tratamiento clínico de los tumores sólidos diagnosticados con mayor frecuencia, y para algunas neoplasias malignas es necesario desarrollar terapias razonablemente eficientes, ya que son prácticamente inexistentes. La detección temprana seguida de la cirugía sigue siendo la principal herramienta que permite una expansión significativa de la esperanza de vida para la mayoría de los pacientes. En la mayoría de las neoplasias malignas, puede ser necesario modular (preferiblemente de manera sinérgica) varias rutas biológicas relevantes. Por consiguiente, puede provocarse el fenotipo requerido (muerte de células tumorales) mediante la modulación letal sintética de procesos biológicos elegidos de manera apropiada. Las interacciones letales sintéticas tienden a formar agrupaciones; una red significativa de tales interacciones abarca los procesos biológicos implicados en el daño/reparación del ADN. Por tanto, la modulación selectiva y eficiente de la actividad de procesos seleccionados tiene una importancia significativa y puede conducir a una nueva generación de fármacos antineoplásicos modernos.
El mantenimiento de la integridad genómica garantizada por la respuesta multifacética al daño del ADN celular (DDR) es un fenómeno biológico fundamental que comparten todos los organismos. Por un lado, la red de DDR de las rutas de vigilancia del genoma, punto de control y reparación contrarresta los efectos potencialmente mutagénicos de ataques endógenos (lesiones oxidativas y replicativas) y exógenos (por ejemplo, ionización o irradiación UV, humo de cigarrillos) que dañan el ADN. Por otro lado, puede aprovecharse la modulación de componentes seleccionados en el tratamiento eficiente de enfermedades malignas. Es probable que los tratamientos letales sintéticos óptimos sean diferentes para las subpoblaciones tumorales particulares; por tanto, este enfoque es compatible con el concepto de medicina personalizada.
Entre las enzimas de procesamiento de la reparación del ADN, el complejo MRE11-RAD50-NBS1 (MRN) desempeña un papel importante en conservar la integridad genómica actuando como sensor de daño del ADN de roturas de doble hebra (DSB) y fomentando la reparación a través de unión de extremos no homólogos (NHEJ) o recombinación homóloga (Nature Reviews 2002, 3, 317; Trends Biochem. Sciences 2002, 27, 410). En respuesta a DSB, MRN activa y recluta ATM (perteneciente a la familia de cinasas relacionada con fosfatidilinositol-3' cinasa (PIKK)) a sitios de ADN dañados. ATM inicia una cascada de señales que conduce a la detención del ciclo celular y a la reparación del ADN. MRE11 es el núcleo de la subunidad del complejo MRN y presenta actividad 3'-5' exonucleasa, actividad endonucleasa monocatenaria y de horquilla de ADN. Las funciones del complejo MRE11-RAD50 incluyen unión al ADN, formación de puentes entre los extremos de DSB y su procesamiento. NBS1 no posee ninguna actividad enzimática; su papel se basa en señalizar e interactuar con otras proteínas (DNA Repair 2010, 9, 1299; Cell 2008, 135, 97). La importancia del complejo MRN se destaca por el hecho de que las mutaciones de la línea germinal de MRE11, NBS1 y RAD50 provocan enfermedad de tipo ataxia-telangiectasia (ATLD), síndrome de rotura de Nimega (NBS) y trastorno de tipo NBS (NBSLD), respectivamente (Cell 1998, 93, 477; Cell, 1999, 99, 577; Am. J. Hum. Genet. 2009, 84, 605). ATLD, NBS y NBSLD tienen características similares a las de la ataxia-telangiectasia (AT), provocada por mutaciones en el gen ATM, que incluyen hipersensibilidad a agentes inductores de DSB, fragilidad cromosómica, detención del ciclo celular dependiendo del daño del ADN y alta predisposición al cáncer (Cell 1998, 93, 477; Oncogene 2007, 26, 7749; Cell 1999, 99, 577; Am. J. Hum. Genet.
2009, 84, 605). Además, el agotamiento de MRE11 conduce a la sensibilización a la inhibición de la poli(ADP-ribosa) polimerasa (PARP) (Cancer Res. 2011, 71, 2632). Además, las células deficientes en MRE11 también son sensibles a los venenos de topoisomerasa, lo que sugiere un papel de MRE11 en la eliminación de las lesiones por parte de TOP1/TOP2 y en la estimulación de un efecto de topo-inhibidores (Mol. Cell. Biol. 2004, 24, 9682). De hecho, recientemente se ha demostrado que la triapina (inhibidor de RNR) bloquea la recombinación mediada por MRN y sensibiliza células de cáncer de ovario a PARP y topo-inhibidores (Mol. Cancer Res. 2014, 12, 381; Cancer Res.
2012, 72, 2814). La importancia terapéutica de los inhibidores de MRE11 en la oncología moderna se ve respaldada adicionalmente por las interacciones genéticas sintéticamente letales recientemente notificadas para MRE11-FEN1 (PLoS Genet. 2013, 9, 1, e1003254) y MRE11-BRCA2 (Cancer Res., 2012, 72, 2814). Los defectos en algunos procesos de reparación del ADN también se manifiestan por sí solos en tejidos neuronales y, por tanto, pueden asociarse con trastornos neurológicos humanos (Cell 2007, 130, 991). La mirina y sus dos análogos estrechamente relacionados a nivel estructural, PFM01 y PfM03 (Mol. Cell 2014, 53, 1; documento WO 2010/075372), son esencialmente los únicos ejemplos notificados de inhibidores (relativamente débiles) de la nucleasa MRE11.
Divulgación de la invención
La presente invención proporciona novedosos compuestos de aminotiazol sustituido, métodos de preparación de tales compuestos, composiciones farmacéuticas que comprenden uno o más de tales compuestos, métodos de preparación de formulaciones farmacéuticas que comprenden uno o más de tales compuestos y métodos de tratamiento, prevención, inhibición o mejora de una o más enfermedades, preferiblemente enfermedades asociadas con la nucleasa MRE11 y/o rutas de reparación del ADN relacionadas con MRE11 usando tales compuestos o composiciones farmacéuticas.
La presente invención se refiere a compuestos representados por la fórmula general (1):

o una sal farmacéuticamente aceptable, o un solvato del mismo, que son para su uso en un método de tratamiento de cáncer, envejecimiento prematuro y/o enfermedades neurológicas, más específicamente de cáncer relacionado con la inestabilidad genómica, envejecimiento prematuro relacionado con la inestabilidad genómica y/o enfermedades neurológicas relacionadas con la inestabilidad genómica, en particular cáncer relacionado con MRE11, envejecimiento prematuro relacionado con MRE11 y/o enfermedades neurológicas relacionadas con MRE11, en el que:
R1 se selecciona del grupo que consiste en arilo; heteroarilo;
en el que cada uno del arilo, heteroarilo puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, seleccionándose cada resto independientemente del grupo que consiste en F, Cl, Br, I, OH, CN, N3, =O, O(alquilo C1-C6), =S, SH, S(alquilo C1-C6), S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6,
CFs, C2F5, OCF3, OC2F5, NH2 , NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), =N-OH, =N-O(alquilo C1-C6),
NO2 , COOH, COO(alquilo C1-C6), CO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, (alquil C1-C6)-S(O)2-NH-, (alquil C1-C6)-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-(SO)2-, (alquil C1-C6)2N-(SO)2-, (alquil C1-C6)-c O-NH-, (alquil C1-C6)-CO-N(alquil C1-C6)-, (alquil C1-C6)-OCO-NH-, (alquil C1-C6)-OCO-N(alquil C1-C6)-, (alquil C1-C6)-CO-NH-CO-, (alquil C1-C6)-CO-N(alquil C1-C6)-CO-, NH2-CO-NH-, (alquil C1-C6)-NH-CO-NH-, (alquil C1-C6)2N-CO-NH-, NH2-CO-N(alquil C1-C6)-, (alquil C1-C6)-NH-CO-N(alquil C1-C6)-, (alquil C1-C6)2 N-CO-N(alquil C1-C6)-, NH2-S(O)2-NH-, (alquil C1-C6)-NH-S(O)2-NH-, (alquil C1-C6)2N-S(O)2-NH-, NH2-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)2N-S(O)2-N(alquil C1-C6)-, alquilo C1-C6 , O-alquilo C1-C6 , O-fenilo, fenilo;
mientras que el alquilo C1-C6, O-fenilo, fenilo en estos restos puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes independientemente seleccionados de: Cl, Br, I, alquilo C1-C6, OH, O-alquilo
C1-C6, SH, SCH3, S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3, OCF3, NH2, NH(alquilo C1-C6), N(alquilo C1-C como N(CH3)2), COOH, COO(alquilo C1-C6), CONH2 , CONH(alquilo C1-C6), CON(alquilo C ^ h , NHC(O)-alquilo C1-C6 o NHC(O)NH2 ;
R2 se selecciona de arilo C6-C12 y heteroarilo que tiene de 5 a 12 átomos de anillos, que puede estar opcionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, OH, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5, O(alquilo C1-C4), fenilo, O-fenilo, NH2, N(alquilo C1-C4)2, NO2, NHCO(alquilo C1-C4), CF3, OCF3, CN, S(O)2-alquilo C1-C6, SO2NH(alquilo C1-C6), SO2N(alquilo C1-C6)2; en particular seleccionados de F, Cl, Br, alquilo C1-C6, O(alquilo C1-C4), CF3, OCF3;
R6 se selecciona del grupo que consiste en H; heterociclilo; cicloalquilo; heteroarilo; arilo; heteroarilo; en el que cada uno del arilo, cicloalquilo, heterociclilo, heteroarilo, puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, independientemente seleccionados del grupo que consiste en
F, Cl, Br, alquilo C1-C6 , OH, O-alquilo C1-C6, SH, SCH3, S(O)-alquilo C1-C6,S(O)2-alquilo C1-C6 , CF3, OCF3 , NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), NO2 , COOH, COO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, NHC(O)-alquilo C1-C6 o NHC(O)NH2 ;
R3 se selecciona del grupo que consiste en H; arilo; heteroarilo; heterociclilo; cicloalquilo; alquilo; y halógeno;
en el que cada uno del arilo, cicloalquilo, alquilo o heteroarilo puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, seleccionándose cada resto independientemente del grupo que consiste en F, Cl, Br, I, OH, CN, N3, =O, O(alquilo C1-C6), =S, SH, S(alquilo C1
Ce), S(O)-alquilo Ci-Ca, S(O)2-alquilo Ci-Ca, CF3, C2F5 , OCF3 , OC2F5, NH2 , NH(alquilo Ci-Ce), N(alquilo Ci-Ce)2 (tal como N(CH3)2), =N-OH, =N-O(alquilo Ci-Ca), NO2, COOH, COO(alquilo Ci-Ca), CO(alquilo Ci-Ca), CONH2 , CONH(alquilo Ci-Ca), CON(alquilo Ci-Ca)2, (alquil Ci-Ca)-S(O)2-NH-, (alquil Ci-Ca)-S(O)2-N(alquil Ci-Ca)-, (alquil Ci-Ca)-NH-(SO)2-, (alquil Ci-Ca)2N-(SO)2-, (alquil Ci-Ca)-CO-NH-, (alquil Ci-Ca)-CO-N(alquil Ci-Ca)-, (alquil Ci-Ca)-OCO-NH-, (alquil Ci-Ca)-OCO-N(alquil Ci-Ca)-, (alquil Ci-Ca)-CO-NH-CO-, (alquil Ci-Ca)-CO-N(alquil Ci-Ca)-CO-, NH2-CO-NH-, (alquil Ci-Ca)-NH-CO-NH-, (alquil Ci-Ca)2N-CO-NH-, NH2-CO-N(alquil Ci-Ca)-, (alquil Ci-Ca)-NH-CO-N(alquil Ci-Ca)-, (alquil Ci-Ca)2N-CO-N(alquil Ci-Ca)-, NH2-S(O)2-NH-, (alquil Ci-Ca)-NH-S(O)2-NH-, (alquil Ci-Ca)2N-S(O)2-NH-, NH2-S(O)2-N(alquil Ci-Ca)-, (alquil Ci-Ca)-NH-S(O)2-N(alquil Ci-Ca)-, (alquil Ci-Ca)2N-S(O)2-N(alquil Ci-Ca)-, alquilo Ci-Ca, O-alquilo Ci-Ca, O-fenilo, fenilo;
mientras que el alquilo Ci-Ca, O-fenilo, fenilo en estos restos puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes independientemente seleccionados de: F, Cl, Br, alquilo Ci-Ca, OH, O-alquilo Ci-Ca, SH, SCH3, S(O)-alquilo Ci-Ca,S(O)2-alquilo Ci-Ca, CF3 , OCF3 , NH2 , NH(alquilo Ci-Ca), N(alquilo Ci-Ca)2 (tal como N(CH3)2), NO2 , COOH, COO(alquilo Ci-Ca), CONH2 , CONH(alquilo Ci-Ca), CON(alquilo Ci-Ca)2, NHC(O)-alquilo Ci-Ca o NHC(O)NH2 ,
R4 se selecciona del grupo que consiste en H y alquilo Ci-Ca;
R5 se selecciona del grupo que consiste en H y arilo.
Los compuestos de fórmula ( i) se proporcionan para su uso en un método de tratamiento según la reivindicación i. Además, la invención proporciona un subgrupo formado por compuestos de fórmula (ia) según la reivindicación 9. En esta descripción y a menos que se indique lo contrario, los grupos sustituyentes genéricos tienen los siguientes significados:
“alquilo” significa un grupo hidrocarbonado alifático que puede ser lineal o ramificado y contiene de i a 8 átomos de carbono, preferiblemente de i a a átomos de carbono, incluso más preferiblemente de i a 4 átomos de carbono, en la cadena. Ejemplos de alquilos adecuados son metilo, etilo, propilo, isopropilo, butilo, isobutilo, ferc-butilo, pentilo, isopentilo, hexilo;
“arilo” significa un sistema de anillos aromático, monocíclico o policíclico, que contiene de a a i4 átomos de carbono, preferiblemente de a a i0 átomos de carbono. Los arilos pueden ser preferiblemente monocíclicos o bicíclicos o tricíclicos, condensados o no condensados. Ejemplos de arilos adecuados son fenilo, naftilo, bifenilo;
“cicloalquilo” significa un sistema de anillos alifático, monocíclico o policíclico, que comprende de 3 a i0 átomos de carbono, preferiblemente de 5 a 7 átomos de carbono. Los ejemplos adecuados incluyen ciclopentilo, ciclohexilo, cicloheptilo, i-decalinilo, norbornilo, adamantilo;
“heterociclilo” significa un sistema de anillos alifático, monocíclico o policíclico, que contiene de 3 a i0 átomos de carbono, preferiblemente de 4 a 8 átomos de carbono, y al menos un heteroátomo seleccionado del grupo que consiste en nitrógeno, oxígeno y azufre. Los ejemplos adecuados incluyen piperazinilo y morfolinilo;
“heteroarilo” significa un sistema de anillos aromático, monocíclico o policíclico, que contiene de 3 a i4 átomos de carbono, preferiblemente de 3 a 7 átomos de carbono, y al menos un heteroátomo seleccionado del grupo que consiste en nitrógeno, oxígeno y azufre. Los heteroarilos pueden ser preferiblemente monocíclicos o bicíclicos o tricíclicos, condensados o no condensados. Ejemplos de heteroarilos adecuados son piridilo, pirimidinilo, pirazinilo, furanilo, tienilo, pirazolilo, oxazolilo, tiazolilo, isotiazolilo, isoxazolilo, pirrolilo, imidazolilo, bencimidazolilo, dihidrobencimidazolilo, indolilo, indolinolilo, imidazopiridazinilo, benzoxazinilo, dihidrobenzoxazinilo, benzofuranilo. Especialmente preferidos son heteroarilos que contienen al menos un átomo de nitrógeno.
El sustituyente =O puede estar presente únicamente en restos alifáticos o en partes alifáticas de los restos.
Sales farmacéuticamente aceptables son sales con ácidos o bases, o sales de adición de ácido. Los ácidos y las bases pueden ser ácidos y bases inorgánicos u orgánicos habitualmente usados en la técnica de formulación, tales como clorhidrato, bromhidrato, sulfato, bisulfato, fosfato, hidrogenofosfato, acetato, benzoato, succinato, fumarato, maleato, lactato, citrato, tartrato, gluconato, metanosulfonato, bencenosulfonato, para-toluenosulfonato, amidas primarias, secundarias y terciarias, amoniaco. Los solvatos son estructuras que contienen moléculas de un disolvente, tal como agua (hidratos) o cualquier otra molécula de disolvente farmacéuticamente aceptable.
R1 se selecciona preferiblemente de arilo Ca-Ci2 y heteroarilo que tiene de 5 a i2 átomos de anillos, en el que dicho arilo o heteroarilo es monocíclico, bicíclico o tricíclico, y los anillos pueden estar condensados o no condensados. Los arilos y heteroarilos incluyen anillos condensados en los que uno o más anillos son aromáticos y uno o más anillos son alifáticos. El arilo o heteroarilo puede estar opcionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, OH, alquilo Ci-Ca, O(alquilo C1-C4), fenilo, O-fenilo, NH2, N(alquilo Ci-C4)2, NO2, NHCO(alquilo C1-C4), CF3 , OCF3 , CN, S(O)2-alquilo Ci-Ca, SO2NH(alquilo Ci-Ca),
SO2N(alquilo Ci-Ca)2.
Más preferiblemente, el arilo en R1 es fenilo, bifenilo o naftilo.
Más preferiblemente, los sustituyentes unidos al arilo o heteroarilo en R1 se seleccionan de F, Cl, Br, alquilo C1-Ca, O(alquilo C1-C4), NO2 , NHCO(alquilo C1-C4), CF3, CN, fenilo, OH; o los sustituyentes pueden seleccionarse de S(O)2-alquilo C1-C6 , SO2NH(alquilo C1-Ca), SO2N(alquilo C1-Ca)2.
Más preferiblemente, R1 se selecciona de arilo Ca-C12 (preferiblemente fenilo) y heteroarilo que tiene de 5 a
12 átomos de anillos, dicho arilo o heteroarilo es monocíclico, bicíclico o tricíclico, y los anillos pueden estar condensados o no condensados. Los arilos y heteroarilos incluyen anillos condensados en los que uno o más anillos son aromáticos y uno o más anillos son alifáticos. El arilo o heteroarilo está sustituido con de uno a tres grupos OH, preferiblemente con dos grupos OH o un grupo OH y un grupo seleccionado de CN, Cl, Br, F. Preferiblemente, los grupos OH están en posiciones vecinales. El arilo o heteroarilo puede estar opcionalmente sustituido con uno o más sustituyentes adicionales, independientemente seleccionados del grupo que consiste en F, Cl, Br, alquilo C1-Ca, O(alquilo C1-C4), fenilo, O-fenilo, NH2, N(alquilo C1-C4)2, NO2, NHCO(alquilo C1-C4), CF3 , OCF3, CN, S(O)2-alquilo
C1-C6 , SO2NH(alquilo C1-Ca), SO2N(alquilo CrCa)2; en particular seleccionados de F, Cl, Br, alquilo C1-Ca, O(alquilo
C1-C4), CF3, OCF3.
Lo más preferiblemente, R1 es 3,4-dihidroxifenilo.
Más preferiblemente, R2 se selecciona de fenilo, naftilo, benzofuranilo, piridilo, tiofenilo, piridazinilo, que no están sustituidos o que están sustituidos con de uno a dos sustituyentes independientemente seleccionados de O(alquilo
C1-C4), OH, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5 , F, Br, Cl, CF3, OCF3.
Ra se selecciona preferiblemente de fenilo, morfolinilo, opcionalmente sustituidos tal como se describió anteriormente en el presente documento.
R3 se selecciona preferiblemente de H, fenilo, metilo, etilo, propilo, isopropilo, butilo, isobutilo, ferc-butilo, F, Cl, Br, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, tetrahidropiranilo, tetrahidrofuranilo, piperidinilo, que no están sustituidos o que están sustituidos con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, OH, alquilo C1-Ca, fenilo, NH2, N(alquilo C1-C4)2, NO2, NHCO(alquilo C1-C4), CF3, OCF3 , Cn , SO2NH(alquilo CrCa), SO2N(alquilo C1-Ca)2, S(O)2-alquilo CrCa.
R4 se selecciona preferiblemente de H, metilo, etilo, isopropilo. Por ejemplo, R4 es H.
R5 se selecciona preferiblemente de H, fenilo. Más preferiblemente, R5 es H.
En todas las realizaciones preferidas, los alquilos, O-fenilos y fenilos pueden estar, de manera opcional, adicionalmente sustituidos con uno o más sustituyentes seleccionados de F, Cl, Br, alquilo CrCa, OH, O-alquilo C1-Ca, SH, SCH3, S(O)-alquilo CrCa, S(O)2-alquilo CrCa, CF3 , OCF3 , NH2, NH(alquilo CrCa), N(al como N(CH3)2), NO2 , COOH, COO(alquilo CrCa), CONH2 , CONH(alquilo CrCa), CON(alquilo CrCa)2, NHC(O)-alquilo CrCa o NHC(O)NH2.
Los compuestos de la presente invención pueden estar en configuración (E) o (Z) en el doble enlace C-C. Se prefiere la configuración (E).
Debe entenderse que las realizaciones preferidas y/o específicas pueden combinarse en cualquier combinación, y pueden combinarse en cualquier combinación con las realizaciones más generales de los grupos sustituyentes.
Los compuestos de la invención incluyen, por ejemplo:



En general, los compuestos descr¡tos en esta ¡nvenc¡ón pueden prepararse a través de las rutas generales descr¡tas a cont¡nuac¡ón en el esquema 1.
En resumen, la condensac¡ón de la bromocetona (2) aprop¡ada con t¡ourea proporc¡ona el am¡not¡azol (3) correspondente, que reacc¡ona con c¡anoacetato de et¡lo en cond¡c¡ones bás¡cas para proporc¡onar la am¡da (4) correspondente. Alternativamente, puede prepararse la am¡da (4) en dos etapas a part¡r de am¡not¡azol (3) med¡ante reacc¡ón con cloruro de cloroacet¡lo segu¡do de reacc¡ón con c¡anuro de potas¡o. La poster¡or condensac¡ón con el aldehido o la cetona aprop¡ado proporc¡ona el compuesto objetivo (1 ).
Esquema 1

Los compuestos de fórmula (1) actúan como inhibidores de nucleasa MRE11, y son útiles en el tratamiento y la prevención de enfermedades asociadas con la inestabilidad genómica, por ejemplo, cáncer (en particular, cánceres de mama, de colon, de próstata, de pulmón, de cabeza y cuello, hepático, de ovario, colorrectal, gástrico, melanoma, leucemias, síndrome de rotura de Nimega y síndrome de tipo rotura de Nimega, ataxia-telangiectasia y trastorno de tipo ataxia-telangiectasia, y anemia de Fanconi), envejecimiento prematuro y enfermedades neurológicas.
Por tanto, la presente invención proporciona los compuestos de fórmula (1) para su uso como medicamentos. Más específicamente, proporciona los compuestos de fórmula (1 ) para su uso en el tratamiento y la prevención de estados seleccionados de enfermedades asociadas con la inestabilidad genómica, por ejemplo, cáncer (en particular, cánceres de mama, de colon, de próstata, de pulmón, de cabeza y cuello, hepático, de ovario, colorrectal, gástrico, melanoma, leucemias, síndrome de rotura de Nimega y síndrome de tipo rotura de Nimega, ataxiatelangiectasia y trastorno de tipo ataxia-telangiectasia, y anemia de Fanconi), envejecimiento prematuro y enfermedades neurológicas. En una realización, la presente invención proporciona los compuestos de fórmula (1) para su uso en el tratamiento de tumores sólidos con BRCA-2 mutado. Hay pruebas comercialmente disponibles para diagnosticar mutaciones de BRCA-2. La presente invención también proporciona un método para el tratamiento, la inhibición, mejora o prevención de un estado seleccionado de enfermedades asociadas con la inestabilidad genómica, por ejemplo, cáncer (en particular, cánceres de mama, de colon, de próstata, de pulmón, de cabeza y cuello, hepático, de ovario, colorrectal, gástrico, melanoma, leucemias, síndrome de rotura de Nimega y síndrome de tipo rotura de Nimega, ataxia-telangiectasia y trastorno de tipo ataxia-telangiectasia, y anemia de Fanconi), envejecimiento prematuro y enfermedades neurológicas en un paciente que padece tal estado, que comprende la etapa de administrar al menos un compuesto de fórmula (1 ) a dicho paciente.
La presente invención incluye además composiciones farmacéuticas que comprenden al menos un compuesto de fórmula (1) y al menos un compuesto auxiliar farmacéuticamente aceptable. Los compuestos auxiliares pueden incluir, por ejemplo, portadores, diluyentes, cargas, conservantes, estabilizadores, aglutinantes, agentes humectantes, emulsionantes, tampones, etc. Los expertos en la técnica de formulación conocen bien compuestos auxiliares adecuados. Las composiciones farmacéuticas se preparan mediante métodos conocidos, por ejemplo, mezclado, disolución, etc.
Ejemplos para llevara cabo la invención
La presente invención proporciona aminotiazoles sustituidos que están representados por la fórmula estructural (1), o sales farmacéuticamente aceptables, solvatos, ésteres de los mismos, en los que los diversos restos son tal como se describió anteriormente.
Ejemplos preparativos
Materiales y métodos
Todos los reactivos comercialmente disponibles se usaron tal cual se suministraron sin purificación adicional. Los disolventes de reacción se adquirieron anhidros y se almacenaron bajo nitrógeno. A menos que se indique lo contrario, las reacciones se llevaron a cabo en material de vidrio secado al horno bajo una atmósfera de nitrógeno. La cromatografía en columna se llevó a cabo usando gel de sílice (tamaño de poro de 60 Á, tamaño de partícula de
malla de 230-400, tamaño de partícula de 40-63 |im). La purificación mediante cromatografía en capa fina preparativa se realizó usando placas de Merck (PLC Silica gel 60 F254, 1 mm). La cromatografía en columna de fase inversa se llevó a cabo usando gel de sílice con fase inversa C18 (tamaño de poro de 90 A, tamaño de partícula de malla de 230-400, tamaño de partícula de 40-63 |im). Los espectros de RMN se obtuvieron en los disolventes deuterados indicados; los desplazamientos químicos se dan en partes por millón (S) referenciados al disolvente deuterado indicado empleado. Las multiplicidades se indican mediante s (singlete), d (doblete), t (triplete), q (cuartete), p (pentete), sept (septete), m (multiplete) o (a) ancho, o combinaciones de los mismos. Los valores de constante de acoplamiento se dan en Hz.
Procedimiento general A1: Bromación de acetofenonas con bromo
A una disolución fría (0°C) del sustrato (es decir, derivado de acetofenona apropiado) en CH2Ch (14 ml por 1 mmol del sustrato, a menos que se indique lo contrario) se le añadió Br2 (1 eq), se permitió calentar la mezcla resultante hasta 25°C y se agitó durante 1 h (a menos que se indique lo contrario). Se añadió una disolución acuosa saturada de NaHCO3 (3 ml por 1 mmol del sustrato) y se extrajo la mezcla con CH2Ch (3 ml por 1 mmol del sustrato). Se lavaron los extractos orgánicos combinados con una disolución acuosa al 10 % de Na2S2O3 ( 2 ml por 1 mmol del sustrato), salmuera ( 2 ml por 1 mmol del sustrato), se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se usó el producto resultante, la bromocetona deseada, directamente sin purificación adicional en la siguiente etapa.
Procedimiento general A2: Bromación de acetofenonas con CuBr2
A una disolución del derivado de acetofenona apropiado en una mezcla de CHCh y acetato de etilo (1:1) (2 ml por 1 mmol del sustrato, a menos que se indique lo contrario), se le añadió CuBr2 ( 2 eq.) y se sometió a reflujo la mezcla resultante durante 2 h. Se filtró la mezcla a través de un filtro de HPLC y se evaporó a vacío. Se secó el producto a vacío y se usó en la siguiente etapa sin purificación adicional.
Procedimiento general A3: Bromación de acetofenonas con TMSOTf y NBS:
A una disolución del derivado de acetofenona apropiado en CH2Ch (2 ml por 1 mmol del sustrato, a menos que se indique lo contrario), se le añadieron NEt3 (1,2 eq) y TMSOTf (1,1 eq.) a 0°C. Se permitió calentar la mezcla hasta 25°C y se agitó durante 16 h. Luego se enfrió de nuevo la mezcla de reacción hasta 0°C y se le añadió NBS (1,1 eq.). Se agitó la mezcla durante 15-30 min a 0°C. Se absorbió la mezcla en bruto sobre sílice y se filtró rápidamente a través de un lecho de gel de sílice (hexano:EtOAc; 1:1, a menos que se indique lo contrario) para proporcionar la bromoacetofenona deseada, que se usó directamente en la siguiente etapa.
Procedimiento general A4: Bromación de acetofenonas con Br2 y HBr (al 47% en H2O):
Se añadió la acetofenona apropiada (1 eq) a una disolución de HBr (al 47% en H2O, 3 eq) y ácido acético (3 ml por 1 mmol del sustrato, a menos que se indique lo contrario) a 25°C. Luego se le añadió Br2 (1,1 eq) gota a gota a 25°C y se agitó la mezcla de reacción resultante durante 16 h a 25°C.
Se añadió una disolución acuosa saturada de NaHCO3 hasta que el pH fue neutro y se extrajo la mezcla con EtOAc (3 x 10 ml por 1 mmol del sustrato). Se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se usó el producto resultante directamente sin purificación adicional en la siguiente etapa.
Procedimiento general B: Condensación de bromocetonas con tiourea
Se sometió a reflujo una mezcla del sustrato (es decir, la bromocetona apropiada) (1 eq) y tiourea (1,5 eq) en EtOH (3 ml por 1 mmol de bromocetona, a menos que se indique lo contrario) durante 2 h (a menos que se indique lo contrario).
Tratamiento final 1:
Se añadió una disolución acuosa saturada de NaHCO3 (3 ml por 1 mmol de bromocetona) a la mezcla, que luego se extrajo con EtOAc (3 x 5 ml por 1 mmol de bromocetona). Se lavaron los extractos orgánicos combinados con salmuera (3 ml por 1 mmol de bromocetona), se secaron sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. Si era necesario, se purificó el producto mediante cromatografía en columna sobre gel de sílice (a menos que se indique lo contrario) para proporcionar el aminotiazol deseado.
Tratamiento final 2:
Se evaporó el disolvente a vacío. Se trituró el sólido resultante con EtOAc (1 ml por 1 mmol de bromocetona) y se filtró la mezcla. Se disolvió el residuo sólido en MeOH (1 ml por 1 mmol de bromocetona). Se añadió una disolución acuosa saturada de NaHCO3(3 ml por 1 mmol de bromocetona) y se extrajo la mezcla con EtOAc (3 x 5 ml por
1 mmol de bromocetona). Se lavaron los extractos orgánicos combinados con salmuera (3 ml por 1 mmol de bromocetona), se secaron sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. El producto, el aminotiazol deseado, habitualmente era suficientemente puro y se usó directamente sin purificación adicional (a menos que se indique lo contrario) en la siguiente etapa.
Procedimiento general C1: Formación de amida usando NaOEt o NaOMe
A una mezcla del aminotiazol apropiado (1 eq) y cianoacetato de etilo (1,5 eq) en MeOH o EtOH anhidro (2 ml por 1 mmol de aminotiazol, a menos que se indique lo contrario) se le añadió una disolución de NaOEt (al 21% en EtOH) (1,5 eq, a menos que se indique lo contrario) o NaOEt (1 mM en EtOH, recientemente preparado a partir de Na y EtOH anhidro) a 25°C. Se calentó la mezcla hasta 55°C durante 5 h (a menos que se indique lo contrario). Se añadió una disolución acuosa saturada de NH4Cl (10 ml por 1 mmol de aminotiazol) y se extrajo la mezcla con EtOAc (3 * 15 ml por 1 mmol de aminotiazol). Se lavaron los extractos orgánicos con agua (10 ml por 1 mmol de aminotiazol), salmuera ( 10 ml por 1 mmol de aminotiazol), se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna sobre gel de sílice (a menos que se indique lo contrario) para proporcionar la amida deseada.
Procedimiento general C2: Formación de amida con cloruro de cloroacetilo en dos etapas
Etapa 1: A una disolución del aminotiazol apropiado (1 eq) en acetonitrilo anhidro (1 ml por 1 mmol) se le añadió NEt3 (1 eq, a menos que se indique lo contrario) a 25°C. Se calentó la mezcla hasta 80°C y se añadió una disolución de cloruro de cloroacetilo (1,5 eq.) en acetonitrilo seco (0,5 ml por 1 mmol). Se agitó la mezcla de reacción a 80°C durante 2 h. Se añadió una disolución acuosa saturada de NH4Cl (10 ml por 1 mmol de aminotiazol) y se extrajo la mezcla con EtOAc (3 * 15 ml por 1 mmol de aminotiazol). Se combinaron las fases orgánicas y se lavaron con salmuera ( 10 ml por 1 mmol de aminotiazol), se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se filtró el residuo rápidamente a través de un lecho de gel de sílice (hexano:EtOAc; 1:1, a menos que se indique lo contrario) para proporcionar la cloroacetamida correspondiente.
Etapa 2: Se disolvió la cloroacetamida en DMF anhidra (1 ml por 1 mmol) y se añadió KCN (1 eq, a menos que se indique lo contrario) y se agitó a 25°C durante 6 h. Se añadió agua (5 ml por 1 mmol) y se extrajo la mezcla con EtOAc (3 * 15 ml por 1 mmol). Se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna para proporcionar la cianoacetamida deseada.
Procedimiento general C3: Formación de amida usando NaH
A una disolución del aminotiazol apropiado (1 eq) en THF anhidro:MeOH (5:1) ( 6 ml por 1 mmol, a menos que se indique lo contrario), se le añadió NaH (1,1 eq, al 60% en aceite mineral) a 0°C. Se agitó la mezcla a 55°C y se añadió cianoacetato de etilo (1,5 eq). Después de 4 h, se añadió cianoacetato de etilo adicional (1,5 eq) y se sometió a reflujo la mezcla durante 14 h. Se enfrió la mezcla hasta 25°C, se añadió una disolución acuosa saturada de NH4Cl (20 ml por 1 mmol de aminotiazol) y se extrajo la mezcla con EtOAc (3 * 20 ml por 1 mmol de aminotiazol). Se combinaron las fases orgánicas y se lavaron con salmuera ( 20 ml por 1 mmol de aminotiazol), se secaron sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna para proporcionar la cianoacetamida deseada.
Procedimiento general D1: Condensación con trietilamina como base
A una mezcla del aldehído apropiado (0,95 eq) y cianoacetamida (1 eq) en EtOH absoluto (17 ml por 1 mmol de aldehído, a menos que se indique lo contrario) se le añadió trietilamina (1 eq) y se agitó la mezcla a 50°C durante 2 h (a menos que se indique lo contrario).
Tratamiento final 1:
Cuando apareció un precipitado, se eliminó el disolvente mediante filtración. Se disolvió el residuo sólido en EtOAc (5 ml por 0,05 mmol de aldehído) y se añadió una disolución acuosa saturada de NH4Cl (5 ml por 0,05 mmol de aldehído). Se extrajo la mezcla con EtOAc (3 * 5 ml por 0,05 mmol de aldehído). Se lavaron los extractos orgánicos combinados con agua (aprox. 4 ml por 0,05 mmol de aldehído), salmuera (aprox. 4 ml por 0,05 mmol de aldehído), se secaron sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. El producto resultante, la acrilamida deseada, habitualmente era suficientemente pura y no requirió purificación adicional (a menos que se indique lo contrario).
Tratamiento final 2:
Cuando no apareció un precipitado, se añadió una disolución acuosa saturada de NH4Cl (5 ml por 0,05 mmol de aldehído) a la mezcla de reacción, que luego se extrajo con EtOAc (3 * 5 ml por 0,05 mmol de aldehído). Se lavaron los extractos orgánicos con agua (4 ml por 0,05 mmol de aldehído), salmuera (4 ml por 0,05 mmol de aldehído), se
secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna sobre gel de sílice y/o mediante cromatografía en columna de fase inversa sobre gel de sílice C18 para proporcionar la acrilamida deseada.
Procedimiento general D2: Condensación con piperidina como base
A una mezcla del aldehído apropiado (1 eq) y cianoacetamida (1 eq) en CH2Cl2 o CH3CN (10 ml por 1 mmol de aldehído, a menos que se indique lo contrario) se le añadió piperidina (0,1 eq). Se sometió a reflujo la mezcla durante 2 h (a menos que se indique lo contrario). Se añadió una disolución acuosa saturada de NH4Cl (5 ml por 0,1 mmol de aldehído) y se extrajo la mezcla con EtOAc (3 x 5 ml por 0,1 mmol de aldehído). Se lavaron los extractos orgánicos con agua (5 ml por 0,1 mmol de aldehído), salmuera (5 ml por 0,1 mmol de aldehído), se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante CCF preparativa y/o mediante cromatografía en columna sobre gel de sílice y/o mediante cromatografía en columna de fase inversa sobre gel de sílice C18 (a menos que se indique lo contrario) para proporcionar la acrilamida deseada. Preparación de productos intermedios individuales y compuestos objetivo como ejemplos:
Ejemplo preparativo 1
2-bromo-1-fenilpropan-1-ona

Se preparó según el procedimiento general A1 a partir de propiofenona (2,5 ml, 18,8 mmol) y bromo (964 |al, 18,8 mmol) en CH2Cl2 (25 ml); tiempo de reacción de 1 h. Se obtuvo el producto como un aceite incoloro (3,873 g, 97%).
1H-RMN (500 MHz, CDCl3) 5 (ppm) 8,07 - 7,98 (m, 2H), 7,64 - 7,56 (m, 1H), 7,53 - 7,45 (m, 2H), 5,29 (q, J = 6,6 Hz, 1H), 1,91 (d, J = 6,6 Hz, 3H);
EMAR calc. para C ^^B rO [M+H]+ 212,9910, hallado 212,9913.
Ejemplo preparativo 2
1-(4-((trimetilsilil)etinil)fenil)etan-1-ona

A una disolución de 4-bromoacetofenona (1 g, 5 mmol) en THF anhidro (6 ml) se le añadieron Cul (38 mg, 0,2 mmol), Et3N (0,75 g, 1 ml, 7,5 mmol) y Pd(PPh3)2Cl2 (70 mg, 0,1 mmol). Se añadió una disolución de trimetilsililacetileno (0,52 g, 0,75 ml, 5,3 mmol) en THF anhidro (2 ml) gota a gota a lo largo de 1 h, luego se agitó la mezcla a 25°C durante 4 h. Se adsorbió previamente la mezcla en bruto sobre gel de sílice y se purificó mediante cromatografía en columna (hexano:EtOAc; 10:1). Se aisló el producto como un aceite incoloro (0,91 g, 85%).
1H-RMN (500 MHz, CDCl3) 5 (ppm) 7,91 - 7,82 (m, 2H), 7,53 - 7,47 (m, 2H), 2,56 (s, 3H), 0,23 (s, 9H);
13C-RMN (126 MHz, CDCl3) 5 (ppm) 197,5, 136,7, 132,3, 128,3, 128,2, 104,3, 98,3, 26,8, 0,1;
EMAR calc. para C^H^OSi [M+H]+ 217,1043, hallado 217,1042.
Ejemplo preparativo 3
1-(3-bromo-4-metoxifenil)etan-1-ona
Se añadieron 1-(4-metoxifenil)etan-1-ona (0,98 g, 6,5 mmol) y NBS (1,16 g, 6,5 mmol) a 20 ml de agua. Se agitó la mezcla a 60°C y se añadió H2SO4 al 96% (0,7 ml, 13 mmol) gota a gota. Se agitó la mezcla resultante a 60°C durante 5 h, luego se enfrió hasta 25°C y se extrajo con EtOAc (3 * 15 ml). Se lavaron los extractos orgánicos combinados con una disolución acuosa saturada de NaHCO3 (50 ml), se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se obtuvo el producto (670 mg, 45%) como un sólido blanco.
1H-RMN (500 MHz, CDCh) S (ppm) 8,18 (d, J = 2,2 Hz, 1H), 7,93 (dd, J = 8 ,6 , 2,2 Hz, 1H), 6,95 (d, J= 8 , 6 Hz, 1H), 3,98 (s, 3H), 2,57 (s, 3H);
13C-RMN (126 MHz, CDCh) S (ppm) 195,8, 159,8, 134,1, 131,6, 129,7, 112,1, 111,3, 56,7, 26,5;
EMAR calc. para C a H ^B ^ [M+H]+ 228,9859, hallado 228,9858.
Ejemplo preparativo 4
1-(3-cicloprop¡l-4-metox¡fen¡l)etan-1-ona

Se agitó una mezcla de 1-(3-bromo-4-metoxifenil)etan-1-ona (250 mg, 1,1 mmol), K3PO4 (250 mg, 1,2 mmol), ácido ciclopropilborónico (103 mg, 1,2 mmol) y Pd(dppf)Ch (36 mg, 0,05 mmol) en dioxano (5 ml) y H2O (1 ml) a 90°C durante 16 h. Se adsorbió previamente la mezcla sobre gel de sílice y se purificó mediante cromatografía en columna (hexano:EtOAc; 5:1) para proporcionar el producto como un aceite incoloro (0,13 g, 60%).
1H-RMN (500 MHz, CDCla) S (ppm) 7,79 (dd, J = 8,5, 2,3 Hz, 1H), 7,51 (d, J = 2,3 Hz, 1H), 6,87 (d, J = 8,5 Hz, 1H), 3,94 (s, 3H), 2,54 (s, 3H), 2,21 - 2,12 (m, 1H), 0,99 - 0,93 (m, 2H), 0,74 - 0,68 (m, 2H);
13C-RMN (126 MHz, CDCh) S (ppm) 197,2, 162,5, 132,5, 130,3, 128,2, 125,6, 109,5, 56,0, 26,5, 9,7, 7,9;
EMAR calc. para C12H15O2 [M+H]+ 191,1067, hallado 191,1070.
Ejemplo preparativo 5
N-(3-formilfenil)acetamida
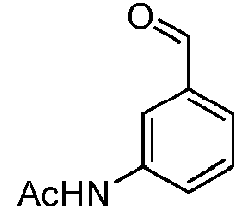
Se disolvió (3-nitrofenil)metanol (0,67 g, 4,38 mmol) en THF (5 ml), se añadió Pd/C (5 mg) y se agitó la mezcla a 25°C bajo H2 durante 4 h. Se filtró la suspensión a través de Celite y se evaporó el disolvente a vacío. Al residuo se le añadieron THF (5 ml), Et3N (1,8 g, 2,5 ml, 18 mmol) y Ac2O (1,3 g, 1,25 ml, 13 mmol) y se agitó la mezcla a 25°C durante 16 h. Se vertió la mezcla en una disolución acuosa saturada de NH4Cl (10 ml) y se extrajo con EtOAc (3 * 10 ml). Se combinaron las fracciones orgánicas, se secaron sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. Se disolvió el producto diacetilado en THF/H2O (4,5/0,5 ml) y se añadió LiOH (190 mg, 7,8 mmol). Se agitó la disolución a 25°C durante 16 h, se añadió H2O (20 ml) y se extrajo la mezcla con EtOAc (3 * 10 ml). Se combinaron las fases orgánicas, se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se obtuvo el producto intermedio deseado (es decir, W-(3-(hidroximetil)fenil)acetamida), purificado mediante cromatografía en columna (hexano:EtOAc; 1:2), como un sólido blanco (490 mg, 3 mmol, 70%).
EMAR calc. para C9H12NO2 [M+H]+ 166,0863, hallado 166,0865.
Se suspendieron W-(3-(hidroximetil)fenil)acetamida (100 mg, 0,6 mmol), PCC (390 mg, 1,8 mmol) y Celite (100 mg) en una mezcla de CH2Ch ( 6 ml) y DMF (0,5 ml). Se agitó la mezcla a 25°C durante 2 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; 1:2). Se obtuvo el producto como un sólido blanco (95 mg, 95%).
1H-RMN (500 MHz, CDCla) 5 (ppm) 9,99 (s, 1H), 8,02 - 7,98 (m, 1H), 7,92 - 7,86 (m, 1H), 7,70 (s, 1H), 7,63 (d, J = 7,6 Hz, 1H), 7,54 - 7,45 (m, 1H), 2,23 (s, 3H);
13C-RMN (126 MHz, CDCI3) 5 (ppm) 192,2, 168,9, 139,1, 137,3, 130,0, 125,9, 125,8, 120,5, 24,8;
EMAR calc. para C9 H8NO2 [M-H]- 162,0561, hallado 162,0560.
Ejemplo preparativo 6
6-hidroxi-[1,1’-bifenil]-3-carbaldehído

Se agitó una mezcla de 3-bromo-4-hidroxibenzaldehído (1 g, 5 mmol), K2CO3 (1,38 mg, 10mmol), ácido fenilborónico (0,66 g, 5,5 mmol) y Pd(dppf)Cl2 (36 mg, 0,05 mmol) en dioxano desgasificado (8 ml) y agua (2 ml) a 90°C durante 16 h. Se vertió la mezcla en una disolución acuosa saturada de NH4Cl (30 ml) y se extrajo con EtOAc (3 x 10 ml). Se lavaron las fracciones orgánicas combinadas con salmuera (20 ml), se secaron sobre MgSO4, se filtraron y se adsorbieron previamente sobre gel de sílice. Se aisló el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 5:1), como un aceite incoloro (0,82 g, 85%).
1H-RMN (500 MHz, CDCla) 5 (ppm) 59,92 (s, 1H), 7,85 - 7,81 (m, 2H), 7,57 - 7,52 (m, 2H), 7,51 - 7,44 (m, 3H), 7,13 (d, J = 8,2 Hz, 1H), 5,94 (s, 1H);
13C-RMN (126 MHz, CDCh) 5 (ppm) 191,1, 158,3, 135,7, 132,8, 131,7, 130,5, 129,9, 129,3, 129,1, 128,9, 116,8; EMAR calc. para C13H11O2 [M+H]+ 199,0754, hallado 199,0750.
Ejemplo preparativo 7
3-oxo-3,4-dihidro-2H-benzo[b][1,41oxazin-7-carboxilato de metilo
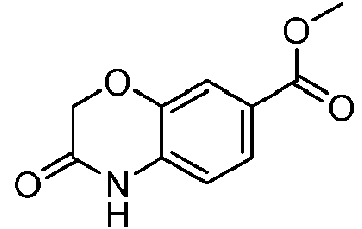
Se disolvió 3-hidroxi-4-nitrobenzoato de metilo (1,00 g, 5,0 mmol) en THF (15 ml), se añadió Pd/C (10 mg) y se agitó la mezcla bajo H2 a 50°C durante 3 h. Se filtró la mezcla y se concentró el filtrado a vacío. Se disolvió el residuo en DMF (15 ml), se añadieron cloruro de cloroacetilo (0,70 g, 0,50 ml, 6,0 mmol) y K2CO3 (1,80 g, 13,0 mmol) y se agitó la mezcla a 25°C durante 2 h. Luego se vertió la mezcla en agua (100 ml). Se filtró el precipitado, se lavó con agua (10 ml) y Et2O (15 ml) y se secó a vacío. Se obtuvo el producto como un sólido blanco (0,65 g, 65%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 11,04 (s, 1H), 7,58 (dd, J = 8,2, 1,8 Hz, 1H), 7,43 (d, J = 1,8 Hz, 1H), 6,98 (d, J = 8,1 Hz, 1H), 4,64 (s, 2H), 3,81 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 165,5, 164,9, 142,8, 131,8, 124,1, 124,0, 116,6, 115,7, 66,6, 52,0;
EMAR calc. para C10H10NO4 [M+H]+ 208,0604, hallado 208,0606.
Ejemplo preparativo 8
7-(hidroximetil)-2H-benzo[b][1,4]oxazin-3(4H)-ona

Se disolvió 3-oxo-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxilato de metilo (98 mg, 0,473 mmol) en THF (5 ml). Se enfrió la disolución hasta -78°C y se añadió DIBAL (disolución 1 M en Et2O, 2 ml, 2 mmol). Luego se agitó la mezcla
a 25°C durante 18 h. Se vertió la mezcla en agua (25 ml), se añadió NaHSO4 (1 g, 8,3 mmol) y se extrajo la disolución resultante con EtOAc (3 x 10 ml). Se lavaron las fracciones orgánicas combinadas con salmuera (20 ml), se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:1 a 0:1). Se obtuvo el producto como un sólido blanco (47 mg, 55%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 10,62 (s, 1H), 6,89 - 6 , 8 6 (m, 2H), 6,83 (d, J = 8,4 Hz, 1H), 4,53 (s, 2H), 4,39 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 164,7, 143,0, 137,8, 125,7, 120,3, 115,4, 114,3, 66,7, 62,3;
EMAR calc. para C10H8NO3 [M-H]- 178,051, hallado 178,051.
Ejemplo preparativo 9
3-oxo-3,4-dihidro-2H-benzo[b][1,41oxazin-7-carbaldehído

A una suspensión de 7-(hidroximetil)-2H-benzo[b][1,4]oxazin-3(4H)-ona (43 mg, 0,24 mmol) en CH2Cl2 (2 ml) y DMF (0,2 ml) se le añadieron Celite (103 mg) y PCC (103 mg, 0,48 mmol). Se agitó la mezcla de reacción a 25°C durante 2 h. Se eliminó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (EtOAc). Se obtuvo el producto como un sólido blanco (37 mg, 80%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 11,12 (s, 1H), 9,83 (s, 1H), 7,55 (dd, J = 8,0, 1,7 Hz, 1H), 7,42 (d, J = 1,7 Hz, 1H), 7,06 (d, J = 8,0 Hz, 1H), 4,67 (s, 2H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 191,3, 164,9, 143,3, 133,0, 131,7, 125,1, 116,1, 116,0, 6 6 ,6 ;
EMAR calc. para C9 H6NO [M-H]- 176,0353, hallado 176,0355.
Ejemplo preparativo 10
4-acetamido-3-hidroxibenzoato de metilo

Se disolvió 3-hidroxi-4-nitrobenzoato de metilo (0,7 g, 3,5 mmol) en THF (10 ml). Se añadió Pd/C (10 mg) y se agitó la mezcla bajo H2 a 50°C durante 3 h, luego se filtró a través de Celite y se concentró el filtrado a vacío. Se disolvió el residuo en THF (10 ml), se añadieron cloruro de acetilo (0,8 g, 0,75 ml, 10,5 mmol) y Et3N (1,4 g, 2 ml, 14 mmol) y se agitó la mezcla a 25°C durante 16 h. Se vertió la mezcla en agua (15 ml) y se extrajo con EtOAc (3 x 10 ml). Se combinaron las fases orgánicas, se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se disolvió el producto en MeOH (5 ml), se añadió EfeN (1,5 g, 2 ml, 14 mmol) y se agitó la disolución a 25°C durante 16 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; 1:1) para proporcionar el producto como un sólido amarillo (0,52 g, 70%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 10,28 (s, 1H), 9,31 (s, 1H), 8,07 (d, J = 8,4 Hz, 1H), 7,45 (d, J= 1,9 Hz, 1H), 7,40 (dd, J = 8,5, 1,9 Hz, 1H), 3,80 (s, 3H), 2,13 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 169,2, 165,9, 146,6, 131,3, 124,5, 120,6, 120,4, 115,4, 51,8, 23,9;
EMAR calc. para C10H12NO4 [M+H]+ 210,0761, hallado 210,0762.
Ejemplo preparativo 11
4-acetamido-3-((ferc-butildimetilsilil)oxi)benzoato de metilo

Se disolvió 4-acetamido-3-hidroxibenzoato de metilo (0,5 g, 2,6 mmol) en CH2CI2. EtaN (0,5 g, 0,73 ml, 5,2 mmol), se añadieron DMAP (31 mg, 0,26 mmol) y TBSCl (0,43 g, 2,8 mmol) y se agitó la mezcla a 25°C durante 16 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; 7:3). Se obtuvo el producto como un sólido blanco (0,6 g, 75%).
1H-RMN (500 MHz, CDCh) 5 (ppm) 8,42 (d, J = 8,5 Hz, 1H), 7,86 (s, 1H), 7,68 (dd, J = 8,5, 1,9 Hz, 1H), 7,50 (d, J = 1,8 Hz, 1H), 3,90 (s, 3H), 2,20 (s, 3H), 1,06 (s, 9H), 0,32 (s, 6H);
13C-RMN (126 MHz, CDCh) 5 (ppm) 168,2, 166,8, 143,5, 134,3, 125,2, 124,1, 119,2, 118,5, 52,3, 26,0, 25,1, 18,4, -4 1;
EMAR calc. para C16H*NO4Si [M+H]+ 324,1626, hallado 324,1630.
Ejemplo preparativo 12
N-(2-((ferc-butildimetilsili
r
)oxi)-4-(hidroximetil)fen
N
)acetamida

Se disolvió 4-acetamido-3-((ferc-butildimetilsilil)oxi)benzoato de metilo (180 mg, 0,55 mmol) en THF (3 ml) y se enfrió la disolución hasta 0°C. Se añadió DIBAL (1 M en THF, 1,95 ml, 1,95 mmol) y se agitó la mezcla a 25°C durante 16 h. Se vertió la mezcla en una disolución acuosa saturada de NH4Cl (10 ml) y se extrajo con EtOAc (3 x 10 ml). Se combinaron los extractos orgánicos, se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 7:3), como un sólido pardusco (70 mg, 45%).
1H-RMN (500 MHz, CDCh) 5 (ppm) 8,28 (d, J = 8,3 Hz, 1H), 7,67 (s, 1H), 6,94 (dd, J = 8,3, 1,8 Hz, 1H), 6,87 (d, J = 1,9 Hz, 1H), 4,60 (d, J = 5,4 Hz, 2H), 2,17 (s, 3H), 1,68 - 1,62 (m, 1H), 1,06 (s, 9H), 0,28 (s, 6H);
13C-RMN (126 MHz, CDCh) 5 (ppm) 168,0, 144,2, 136,7, 129,4, 120,6, 120,3, 116,6, 65,3, 26,0, 25,0, 18,4, -4,1; EMAR calc. para C15H26NO3Si [M+H]+ 296,1676, hallado 296,1679.
Ejemplo preparativo 13
N-(2-((ferc-butildimetilsilil)oxi)-4-formilfenil)acetamida

A una disolución de N-(2-((ferc-butildimetilsilil)oxi)-4-(hidroximetil)fenil)acetamida (65 mg, 0,22 mmol) en CH2Ch (2 ml) se le añadieron Celite (95 mg) y PCC (95 mg, 0,44 mmol). Se agitó la mezcla de reacción a 25°C durante 2 h. Se filtró la mezcla a través de un lecho de gel de sílice y se eluyó el producto con CH2Ch. Se evaporó el disolvente a vacío y se secó el producto a vacío. Se obtuvo el producto como un sólido amarillo pálido (65 mg, 100%).
1H-RMN (500 MHz, CDCh) 5 (ppm) 9,86 (s, 1H), 8,55 (d, J = 8,3 Hz, 1H), 7,94 (s, 1H), 7,49 (dd, J = 8,3, 1,8 Hz, 1H), 7,34 (d, J = 1,7 Hz, 1H), 2,22 (s, 3H), 1,07 (s, 9H), 0,33 (s, 6H);
13C-RMN (126 MHz, CDCh) 5 (ppm) 191,1, 168,3, 144,3, 135,9, 132,1, 126,8, 119,4, 115,8, 26,0, 25,2, 18,4, -4,1; EMAR calc. para C15H24NO3Si [M+H]+ 294,152, hallado 294,1524.
Ejemplo preparativo 14
N-metoxi-N-metil-2-(tetrahidro-2H-piran-4-il)acetamida

A una disolución de 2-(tetrahidro-2H-piran-4-il)acetato de etilo (300 mg, 1,74 mmol) y clorhidrato de N,O-dimetilhidroxilamina (200 mg, 2,1 mmol) en THF (2 ml) se le añadió /PrMgClLiCl (1,3 M en Th F, 3,4 ml, 4,95 mmol) a -10°C. Se agitó la mezcla de reacción a -5°C durante 1 h, luego se vertió en una disolución acuosa saturada de NH4Cl (10 ml) y se extrajo con EtOAc (3 * 10 ml). Se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; 1 0 :1 ). Se obtuvo el producto como un aceite incoloro (225 mg, 70%).
1H-RMN (500 MHz, CDCls) 5 (ppm) 4,00 - 3,91 (m, 2H), 3,69 (s, 3H), 3,43 (td, J = 11,8, 2,1 Hz, 2H), 3,19 (s, 3H), 2,37 (d, J = 7,0 Hz, 2H), 2,17 -2,06 (m, 1H), 1,73 - 1,64 (m, 2H), 1,42 - 1,30 (m, 2H);
13C-RMN (126 MHz, CDCls) 5 (ppm) 174,4, 68,1, 61,4, 39,0, 33,3, 32,3, 31,8.
Ejemplo preparativo 15
1 -fenil-2-(tetrahidro-2H-piran-4-il)etan-1-ona

A una disolución de N-metoxi-N-metil-2-(tetrahidro-2H-piran-4-il)acetamida (250 mg, 1,34 mmol) en THF (2 ml) se le añadió PhLi (1,8 M en THF, 0,74 ml, 1,34 mmol) a -78°C. Se agitó la mezcla de reacción a -78°C durante 30 min, luego se vertió en una disolución acuosa saturada de NH4Cl (10 ml) y se extrajo con EtOAc (3 * 10 ml). Se secaron los extractos orgánicos combinados sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1:1), como un sólido blanco (235 mg, 85%).
1H-RMN (500 MHz, CDCh) 5 (ppm) 7,98 - 7,94 (m, 2H), 7,61 - 7,53 (m, 1H), 7,50 - 7,44 (m, 2H), 4,02 - 3,91 (m, 2H), 3,45 (td, J = 11,8, 2,2 Hz, 2H), 2,90 (dd, J = 6,7, 2,3 Hz, 2H), 2,34 - 2,20 (m, 1H), 1,76 - 1, 66 (m, 2H), 1,48 - 1,33 (m, 2H);
13C-RMN (126 MHz, CDCh) 5 (ppm) 199,4, 137,5, 133,3, 128,8, 128,3, 68,1, 45,6, 33,3, 31,6;
EMAR calc. para C13H17O2 [M+H]+ 205,1223, hallado 205,1221.
Ejemplo preparativo 16
Benzoato de (2,6-di-ferc-butilpiridin-4-il)metilo

A una mezcla de 2,6-di-fe/'c-butil-4-metilpiridina (577 mg, 2,81 mmol), NBS (525 mg, 2,95 mmol) y AIBN (138 mg, 0,843 mmol) se le añadió CCl4 (25 ml). Se calentó la mezcla hasta reflujo. Después de 1 h, se añadió AIBN adicional (138 mg, 0,843 mmol) a la mezcla y se repitió dos veces la adición de la misma cantidad de AIBN durante 2 h. Se enfrió la mezcla hasta 25°C, se filtró y se concentró el filtrado a vacío. Se purificó el residuo mediante cromatografía en columna sobre gel de sílice (hexano), proporcionando una mezcla del producto (4-(bromometil)-2,6-di-fercbutilpiridina) con el material de partida (aprox. 1/1,3) como un sólido amorfo (673 mg). Se usó este material como tal en la siguiente etapa.
Se disolvió la mezcla que contenía 4-(bromometil)-2,6-di-/erc-butilpiridina (320 mg) en DMF (10 ml). Se añadieron ácido benzoico (275 mg, 2,252 mmol) y K2CO3 (342 mg, 2,48 mmol) y se agitó la mezcla a 50°C durante la noche.
Se evaporó el disolvente a vacío y se diluyó el residuo con EtOAc (25 ml). Se lavó la fase orgánica con H2O (3 x 20 ml), luego con salmuera (3 x 20 ml), se secó sobre MgSO4, se filtró y se concentró a vacío. Se purificó el residuo mediante cromatografía en columna sobre gel de sílice (hexano:EtOAc; de 1:0 a 10:1). Se obtuvo el producto como un aceite incoloro (364 mg).
1H-RMN (300 MHz, CDCls) 5 (ppm) 8,13 - 8,08 (m, 2H), 7,62 - 7,56 (m, 1H), 7,50 - 7,44 (m, 2H), 7,14 (s, 2H), 5,33 (s, 2H), 1,36 (s, 18H);
13C-RMN (126 MHz, CDCls) 5 (ppm) 168,4, 166,5, 144,7, 133,3, 130,1, 129,9, 128,6, 114,2, 66,0, 37,8, 30,3.
Ejemplo preparativo 17
(2,6-di-ferc-butilpiridin-4-il)metanol

Se disolvió benzoato de (2,6-di-ferc-butilpiridin-4-il)metilo (359 mg, 1,103 mmol) en MeOH (10 ml) y se enfrió la disolución hasta 0°C. Se añadió NaH (al 60% en aceite mineral, 27 mg, 1,103 mmol) y se agitó la mezcla y se permitió calentar hasta 25°C. Después de 1 h, se añadió una disolución acuosa saturada de cloruro de amonio (20 ml) y se extrajo la mezcla con EtOAc (3 x 30 ml). Se combinaron los extractos orgánicos, se lavaron con salmuera (40 ml), se secaron sobre MgSO4, se filtraron y se concentraron a vacío. Se purificó el residuo mediante cromatografía en columna sobre gel de sílice (hexano:EtOAc; de 1:0 a 5:1 a 4:1). Se obtuvo el producto como un sólido blanco (199 mg, 82%).
1H-RMN (300 MHz, CDCh) 5 (ppm) 7,08 (s, 2H), 4,69 (s, 2H), 1,35 (s, 18H);
13C-RMN (126 MHz, CDCh) 5 (ppm) 168,2, 149,4, 113,2, 64,9, 37,8, 30,3;
EMAR calc. para C14H24NO [M+H]+ 222,1852, hallado 222,1856.
Ejemplo preparativo 18
2 ,6 -di-ferc-butilisonicotinaldehído

Se agitó una mezcla de (2,6-di-ferc-butilpiridin-4-il)metanol (55 mg, 0,248 mmol), clorocromato de piridinio (107 mg, 0,497 mmol) y Celite (110 mg) en CH2Cl2 (2 ml) a 25°C durante 4 h. Se filtró la mezcla a través de un lecho de gel de sílice para proporcionar el producto como un aceite incoloro (40 mg, 73%).
1H-RMN (300 MHz, CDCh) 5 (ppm) 10,05 (s, 1H), 7,51 (s, 2H), 1,39 (s, 18H);
13C-RMN (126 MHz, CDCh) 5 (ppm) 193,2, 169,9, 142,7, 114,7, 38,1, 30,2.
Ejemplo preparativo 19
2 -ciclohexilacetaldehído

Se disolvió 2-ciclohexilacetato de metilo (1,04 g, 6 , 6 6 mmol) en THF (5 ml). Se enfrió la disolución hasta 0°C y se
añadió una disolución de LiAlH4 (1 M en Et2O, 13,3 ml, 13,3 mmol) y se agitó la mezcla a 25°C durante 16 h. Se vertió la mezcla resultante en agua (20 ml) con H2SO4 (4 ml) y se extrajo con EtOAc (3 x 10 ml). Se combinaron las fracciones orgánicas, se lavaron con salmuera (20 ml), se secaron sobre MgSO4 y se concentraron a vacío. Se disolvió el alcohol en bruto resultante en CH2Cl2 (1o ml), luego se añadieron PCC (2,9 g, 13,32 mmol) y Celite (2,9 g) y se agitó la mezcla a 25°C durante 2 h. Luego se cargó la mezcla sobre una columna corta de gel de sílice (50 ml) y se eluyó el producto con CH2Cl2. Se obtuvo el producto como un aceite incoloro (588 mg, 70%).
1H-RMN (300 MHz, CDCls) 59,78 (t, J = 2,4 Hz, 1H), 2,31 (dd, J = 6 ,8 , 2,4 Hz, 2H), 2,00 - 1,83 (m, 1H), 1,81 - 1,64 (m, 6 H), 1,42 -1,13 (m, 2H), 1,11 -0,95 (m, 2H);
13C-RMN (75 MHz, CDCls) 5203,1, 51,6, 33,5, 32,9, 26,3, 26,3.
Ejemplo preparativo 20
2-ciclohexil-1-(4-(trifluorometoxi)fenil)etan-1-ol

A una disolución de 1-bromo-4-(trifluorometoxi)benceno (486 mg, 0,3 ml, 2,0 mmol) en THF (3 ml) se le añadió n-BuLi (2,5 M en hexano, 435 |l, 6 mmol) a -78°C y se agitó la mezcla de reacción a -78°C durante 1 h. Se añadió una disolución de 2-ciclohexilacetaldehído (280 mg, 2,2 mmol) en THF (1 ml) y se agitó la mezcla a 25°C durante 5 h. Se vertió la mezcla de reacción en una disolución acuosa saturada de NH4Cl (20 ml) y se extrajo con EtOAc ( 3 x 15 ml). Se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; 10:1). Se obtuvo el producto como un aceite incoloro (450 mg, 80%).
1H-RMN (500 MHz, CDCh) 5 (ppm) 7,41 - 7,35 (m, 2H), 7,22 - 7,18 (m, 2H), 4,82 (dd, J = 8 ,8 , 4,9 Hz, 1H), 1,87 -1,79 (m, 1H), 1,79 -1,61 (m, 4H), 1,56 - 1,47 (m, 2H), 1,47 - 1,38 (m, 1H), 1,32 -1,14 (m, 3H), 1,07 -0,89 (m, 2H);
13C-RMN (126 MHz, CDCh) 5 (ppm) 148,7, 144,3, 127,4, 121,2, 120,7 (q, J = 256,9 Hz), 71,6, 47,4, 34,5, 26,8, 26,5, 26,3;
19F-RMN (471 MHz, CDCh) 5 (ppm) -57,89.
Ejemplo preparativo 21
2-ciclohexil-1-(4-(trifluorometoxi)fenil)etan-1-ona

A una disolución de 2-ciclohexil-1-(4-(trifluorometoxi)fenil)etan-1-ol (300 mg, 1,04 mmol) en CH2Ch (3 ml) se le añadieron Celite (450 mg) y PCC (450 mg, 2,08 mmol) y se agitó la mezcla de reacción a 25°C durante 2 h. Se cargó la mezcla sobre un tapón corto de gel de sílice y se eluyó el producto con CH2Cl2. Se obtuvo el producto como un sólido blanco (250 mg, 80%).
1H-RMN (500 MHz, CDCh) 5 (ppm) 8,06 - 7,96 (m, 2H), 7,31 -7,27 (m, 2H), 2,82 (d, J = 6,7 Hz, 2H), 2,06 - 1,92 (m, 1H), 1,82 - 1,62 (m, 4H), 1,36 - 1,25 (m, 3H), 1,23 -1,11 (m, 1H), 1,10 -0,96 (m, 2H);
13C-RMN (126 MHz, CDCh) 5 (ppm) 198,9, 152,7, 136,0, 130,4, 120,6, 120,6 (q, J = 258,5 Hz), 46,5, 34,8, 33,7, 26,5, 26,45;
19F-RMN (471 MHz, CDCh) 5 (ppm) -57,62;
EMAR calc. para C15H18F3O2 [M+H]+ 287,1253, hallado 287,1256.
Ejemplo preparativo 22
4-feniltiazol-2-amina

Se preparó el compuesto según el procedimiento general B a partir de 2-bromoacetofenona (3,21 g, 16,15 mmol) y tiourea (1,84 g, 24,22 mmol) en EtOH (20 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (2,82 g, 99%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 7,79 (d, J = 7,3 Hz, 2H), 7,40 - 7,31 (m, 2H), 7,29 - 7,21 (m, 1H), 7,01 (s, 2H), 6,99 (s, 1H).
Ejemplo preparativo 23
4-(4-bromofenil)tiazol-2-amina

Se preparó el compuesto según el procedimiento general B a partir de 2,4’-dibromoacetofenona (2,07 g, 7,45 mmol) y tiourea (0,85 g, 11,17 mmol) en EtOH (20 ml). Tratamiento final 1 del procedimiento general B. El producto, obtenido como un sólido amarillo (1,895 g, 100%), no requirió ninguna purificación adicional.
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 7,78 - 7,70 (m, 2H), 7,58 - 7,50 (m, 2H), 7,07 (s, 1H), 7,06 (s, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,3, 148,6, 134,1, 131,3, 127,5, 120,0, 102,4;
EMAR calc. para CgHsBr^S [M+H]+ 256,9565, hallado 256,9565.
Ejemplo preparativo 24
4-(4-metoxifenil)tiazol-2-amina

Se preparó el compuesto según el procedimiento general B a partir de 2-bromo-1-(4-metoxifenil)etanona (0,521 g, 2,27 mmol) y tiourea (0,26 g, 3,41 mmol) en EtOH (5 ml). Tratamiento final 1 del procedimiento general B. El producto, obtenido como un sólido amarillo pálido (0,460 g, 98%), no requirió ninguna purificación adicional.
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 7,76 - 7,67 (m, 2H), 6,96 (s, 2H), 6,95 - 6,88 (m, 2H), 6,81 (s, 1H), 3,76 (s, 3H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 168,0, 158,5, 149,7, 127,8, 126,8, 113,8, 99,3, 55,0;
EMAR calc. para C10H11N2OS [M+H]+ 207,0587, hallado 207,0585.
Ejemplo preparativo 25
4-(naftalen-2-il)tiazol-2-amina

Se preparó el compuesto según el procedimiento general B a partir de 2-bromo-2’-acetonaftona (0,967 g, 3,88 mmol) y tiourea (0,443 g, 5,82 mmol) en EtOH (5 ml). Tratamiento final 1 del procedimiento general B. El producto, obtenido como un sólido rosa pálido (0,747 g, 85%), no requirió ninguna purificación adicional.
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 8,32 (d, J = 1,8 Hz, 1H), 7,99 - 7,84 (m, 4H), 7,55 - 7,43 (m, 2H), 7,16 (s, 1H), 7,09 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,2, 149,8, 133,2, 132,3, 132,2, 128,0, 127,8, 127,5, 126,3, 125,7, 124,0, 124,0, 102,4;
EMAR calc. para C13H11N2S [M+H]+ 227,0637, hallado 227,0636.
Ejemplo preparativo 26
4-([1,1’-bifenil1-4-il)tiazol-2-amina

Se añadió una mezcla de dioxano y agua (2,0 0,5 ml) a una mezcla de 4-(4-bromofenil)tiazol-2-amina (0,127 g, 0,5 mmol), ácido fenilborónico (0,076 g, 0,62 mmol), K2CO3 (0,275 g, 1,99 mmol) y PdCl2(PPh3) 2 (0,035 g, 0,05 mmol). Se purgó la mezcla de reacción con N2 durante 5 min, luego se agitó a 80°C durante 16 h. Se enfrió la mezcla hasta 25°C, se diluyó con EtOAc (5 ml), se vertió en una disolución acuosa saturada de NH4Cl (20 ml) y se extrajo con EtOAc (3 * 25 ml). Se lavaron los extractos orgánicos combinados con agua (30 ml) y salmuera (30 ml), se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 2:1). Se obtuvo el producto como un sólido blanco (0,087 g, 55%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 7,9 (d, J = 8,4 Hz, 2H), 7,7 - 7,6 (m, 4H), 7,5 - 7,4 (m, 2H), 7,4 - 7,3 (m, 1H), 7,1 (s, 1H), 7,0 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,2, 149,4, 139,7, 138,6, 128,9, 127,3, 126,6, 126,4, 126,0, 101,7;
EMAR calc. para C ^ H ^ S [M+H]+ 253,0794, hallado 253,0796.
Ejemplo preparativo 27
4-(4-(trifuorometoxi)fenil)tiazol-2-amina

Se preparó el compuesto según el procedimiento general B a partir de 2-bromo-4’-(trifluorometoxi)acetofenona
(0,260 g, 0,918 mmol) y tiourea (0,105 g, 1,38 mmol) en EtOH (6 ml). Tratamiento final 1 del procedimiento general B. El producto, obtenido como un sólido blanco (0,227 g, 95%), no requirió ninguna purificación adicional.
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 7,95 - 7,85 (m, 2H), 7,38 - 7,30 (m, 2H), 7,12 - 7,03 (m, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,4, 148,4, 147,2, 134,2, 127,2, 121,0, 120,1 (q, J = 255,7 Hz), 102,5;
19F-RMN (471 MHz, DMSO-^) 5 (ppm) -56,74;
EMAR calc. para C10H8F3 N2OS [M+H]+ 261,0304, hallado 261,0303.
Ejemplo preparativo 28
4-(p-tolil)tiazol-2-amina

Se preparó el compuesto según el procedimiento general B a partir de 2-bromo-4’-metilacetofenona (0,598 g, 2,8 mmol) y tiourea (0,320 g, 4,21 mmol) en EtOH (6 ml). Tratamiento final 1 del procedimiento general B. El producto, obtenido como un sólido amarillo pálido (0,534 g, 100%), no requirió ninguna purificación adicional.
1H-RMN (500 MHz, DMSO-d6) 5 (ppm) 7,68 (d, J = 8,3 Hz, 2H), 7,16 (d, J = 8,2 Hz, 2H), 6,98 (s, 2H), 6,90 (s, 1H), 2,30 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,0, 149,9, 136,3, 132,3, 129,0, 125,4, 101,0, 20,7;
EMAR calc. para C10H11N2S [M+H]+ 191,0637, hallado 191,0636.
Ejemplo preparativo 29
5-(2-aminotiazol-4-il)-2-metoxifenol

Se preparó 2-bromo-1-(3-hidroxi-4-metoxifenil)etan-1-ona según el procedimiento general A1 a partir de 1-(3-hidroxi-4-metoxifenil)etan-1-ona (0,30 g, 1,8 mmol) y Br2 (287,0 mg, 93 ^l, 1,8 mmol) en CH2Cl2 (5 ml). Se obtuvo el producto intermedio en bruto (2-bromo-1-(3-hidroxi-4-metoxifenil)etan-1-ona) como un sólido blanco (0,44 g, 100%) y se usó como tal en la siguiente etapa:
Se preparó 5-(2-aminotiazol-4-il)-2-metoxifenol según el procedimiento general B a partir de 2-bromo-1-(3-hidroxi-4-metoxifenil)etan-1-ona (200 mg, 0,8 mmol) y tiourea (90 mg, 1,2 mmol) en EtOH (3 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (110 mg, 30%).
1H-RMN (500 MHz, DMSO-CÍ6) 58,88 (ppm) (s, 1H), 7,22 (d, J = 2,1 Hz, 1H), 7,21 - 7,17 (m, 1H), 6,93 (s, 2H), 6,88 (d, J = 8,4 Hz, 1H), 6,72 (s, 1H), 3,77 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 167,8, 149,9, 147,1, 146,2, 128,3, 116,6, 113,1, 112,1, 99,2, 55,6;
EMAR calc. para C10H11N2O2S [M+H]+ 223,0536, hallado 223,0535.
Ejemplo preparativo 30
2-(2-aminotiazol-4-il)propan-2-ol

A una disolución de 2-aminotiazol-4-carboxilato de etilo en THF anhidro (5 ml) se le añadió lentamente MeMgCl (3 M en THF, 2,9 ml, 8,7 mmol) a 0°C. Se permitió calentar la mezcla hasta 25°C y se agitó durante 2 h. Se vertió la mezcla de reacción en una disolución acuosa saturada de NH4Cl (25 ml) y se extrajo con EtOAc (3 x 25 ml). Se secaron los extractos orgánicos combinados sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna (EtOAc). Se obtuvo el producto como un sólido blanco (160 mg, 60%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 6,76 (s, 2H), 6,21 (s, 1H), 4,72 (s, 1H), 1,34 (s, 6H);
13C-RMN (126 MHz, DMSO-cfe) 5 (ppm) 168,0, 160,5, 98,2, 70,1, 30,3;
EMAR calc. para C6 H11N2OS [M+H]+ 159,0587, hallado 159,0588.
Ejemplo preparativo 31
4-(3-bromofenil)tiazol-2-amina

Se preparó 2-bromo-1-(3-bromofenil)etan-1-ona según el procedimiento general A1 a partir de 1-(3-bromofenil)etan-1-ona (1,50 g, 6,0 mmol) y Br2 (960 mg, 310 |l, 6,0 mmol) en CH2Cl2 (5 ml). Se obtuvo el producto intermedio en bruto (2-bromo-1-(3-bromofenil)etan-1-ona) como un sólido blanco (1,80 g, 90%) y se usó como tal en la siguiente etapa:
Se preparó 4-(3-bromofenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(3-bromofenil)etan-1-ona (1,60 g, 6 mmol) y tiourea (690 mg, 9 mmol) en EtOH (10 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (0,90 g, 60%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 fppm) 7,99 - 7,97 (m, 1H), 7,82 - 7,76 (m, 1H), 7,47 - 7,40 (m, 1H), 7,34 - 7,30 (m, 1H), 7,15 (s, 1H), 7,09 (s, 2H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 fppm) 168,3, 148,0, 137,1, 130,6, 129,7, 128,1, 124,3, 121,9, 103,1;
EMAR calc. para CgHsBr^S [M+H]+ 256,9586, hallado 256,9585.
Ejemplo preparativo 32
4-(2-bromofenil)tiazol-2-amina

Se preparó 2-bromo-1-(2-bromofenil)etan-1-ona según el procedimiento general A1 a partir de 1-(2-bromofenil)etan-1-ona (1,47 g, 7,41 mmol) y Br2 (1,18 g, 380 |l, 7,41 mmol) en CH2Cl2 (10 ml). Se obtuvo el producto intermedio en bruto (2-bromo-1-(2-bromofenil)etan-1-ona) como un sólido blanco (1,59 g, 80%) y se usó como tal en la siguiente etapa:
Se preparó 4-(2-bromofenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(2-bromofenil)etan-1-ona (1,10 g, 4,1 mmol) y tiourea (850 mg, 11,11 mmol) en EtOH (10 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (1,30 g, 70%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 7,70 (dd, J = 7,8, 1,8 Hz, 1H), 7,66 (dd, J = 8,0, 1,2 Hz, 1H), 7,40 - 7,37 (m, 1H), 7,24 - 7,20 (m, 1H), 7,02 (s, 2H), 6,94 (s, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 167,3, 148,0, 135,8, 133,4, 131,4, 129,0, 127,5, 120,6, 105,7;
EMAR calc. para CgHsBr^S [M+H]+ 256,9565, hallado 256,9564.
Ejemplo preparativo 33
4-(3-(trifluorometoxi)fenil)tiazol-2-amina

Se preparó 2-bromo-1-(3-(trifluorometoxi)fenil)etan-1-ona según el procedimiento general A1 a partir de 1-(3-(trifluorometoxi)fenil)etan-1-ona (0,30 g, 1,46 mmol) y Br2 (0,24 g, 75 |l, 1,46 mmol) en CH2Cl2 (5 ml). Se obtuvo el producto intermedio en bruto (2-bromo-1-(3-(trifluorometoxi)fenil)etan-1-ona) como un sólido blanco (370 mg, 90%) y se usó como tal en la siguiente etapa:
Se preparó 4-(3-(trifluorometoxi)fenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(3-(trifluorometoxi)fenil)etan-1-ona (370 mg, 1,3 mmol) y tiourea (166 mg, 2,19 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (170 mg, 45%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 7,85 - 7,79 (m, 1H), 7,75 - 7,69 (m, 1H), 7,51 - 7,48 (m, 1H), 7,27 - 7,20 (m, 1H), 7,19 (s, 1H), 7,12 (s, 2H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 168,4, 148,7, 148,0, 137,1, 130,4, 124,2, 120,1 (q, J = 256,1 Hz), 119,3, 117,7, 103,4;
19F-RMN (471 MHz, DMSO-CÍ6) 5(ppm) -56,61;
EMAR calc. para C10H8F3N2OS [M+H]+ 261,0304, hallado 261,0302.
Ejemplo preparativo 34
4-(4-fenoxifenil)tiazol-2-amina

Se preparó 2-bromo-1-(4-fenoxifenil)etan-1-ona según el procedimiento general A1 a partir de 1-(4-fenoxifenil)etan-1-ona (0,90 g, 4,24 mmol) y Br2 (0,67 g, 220 |l, 4,24 mmol) en CH2Cl2 (10 ml). Se obtuvo el producto intermedio en bruto (2-bromo-1-(4-fenoxifenil)etan-1-ona) como un sólido blanco (1,20 g, 100%) y se usó como tal en la siguiente etapa:
Se preparó 4-(4-fenoxifenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(4-fenoxifenil)etan-1-ona (1,20 g, 4,24 mmol) y tiourea (480 mg, 6,36 mmol) en EtOH (10 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (0,60 g, 55%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 7,86 - 7,74 (m, 2H), 7,45 - 7,36 (m, 2H), 7,18 - 7,12 (m, 1H), 7,07 - 6,97 (m,
4H), 6,94 (s, 1H), 6,92 (s, 2H);
13C-RMN (126 MHz, CDCla) 5 (ppm) 168,2, 155,8, 155,2, 149,2, 132,8, 130,0, 127,2, 123,5, 120,7, 118,4, 100,6; EMAR calc. para C15H13N2OS [M+H]+ 269,0743, hallado 269,0743.
Ejemplo preparativo 35
4-(benzofuran-2-il)tiazol-2-amina

Se preparó el compuesto según el procedimiento general B a partir de 2-(bromoacetil)benzofurano (0,30 g, 1,25 mmol) y tiourea (0,143 g, 1,88 mmol) en EtOH (5 ml). Tratamiento final 1 del procedimiento general B. El producto (sólido blanquecino, 0,271 g, 100%) no requirió ninguna purificación adicional.
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 7,65 - 7,60 (m, 1H), 7,60 - 7,52 (m, 1H), 7,33 - 7,26 (m, 1H), 7,26 - 7,17 (m, 3H), 7,04 (s, 1H), 6,96 (s, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,8, 153,9, 152,2, 141,2, 128,5, 124,4, 123,1, 121,2, 110,8, 104,1, 101,9; EMAR calc. para C11H9 N2OS [M+H]+ 217,0430, hallado 217,0432.
Ejemplo preparativo 36
4-(adamantan-1-il)tiazol-2-amina (no se encuentra dentro del alcance de las reivindicaciones)
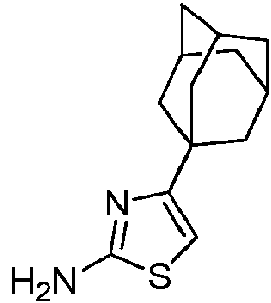
Se preparó el compuesto según el procedimiento general B a partir de 1-(adamantan-1-il)-2-bromoetanona (0,30 g, 1 , 16 6 mmol) y tiourea (0,133 g, 1,75 mmol) en EtOH (3 ml). Tratamiento final 2 del procedimiento general B. Se usó el producto (sólido blanquecino, 0,273 g, 100%) como tal en la siguiente etapa.
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 6,72 (s, 2H), 6,00 (s, 1H), 2,02 - 1,94 (m, 3H), 1,81 (d, J = 3,1 Hz, 6 H), 1,75 -1,63 (m, 3H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 167,6, 161,8, 97,3, 41,5, 36,4, 35,9, 27,9;
EMAR calc. para C13H18N2S [M+H]+ 235,1263, hallado 235,1262.
Ejemplo preparativo 37
5-ciclohexil-4-(4-(trifluorometoxi)fenil)tiazol-2-amina
Se preparó 2-bromo-2-ciclohexil-1-(4-(trifluorometoxi)fenil)etan-1-ona según el procedimiento general A1 a partir de 2-ciclohexil-1-(4-(trifluorometoxi)fenil)etan-1-ona (0,20 g, 0,7 mmol) y Br2 (0,11 g, 36 |al, 0,7 mmol) en CH2Cl2 (5 ml). Se obtuvo el producto intermedio en bruto (2-bromo-1-(3-(trifluorometoxi)fenil)etan-1-ona) como un aceite incoloro (230 mg, 90%) y se usó como tal en la siguiente etapa:
Se preparó 5-ciclohexil-4-(4-(trifluorometoxi)fenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-2-ciclohexil-1-(4-(trifluorometoxi)fenil)etan-1-ona (230 mg, 0,63 mmol) y tiourea (72,0 mg, 0,95 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (121 mg, 55%) y no requirió ninguna purificación adicional.
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 7,60 - 7,55 (m, 2H), 7,45 - 7,34 (m, 2H), 6,79 (s, 2H), 2,91 - 2,78 (m, 1H), 1,92 - 1, 88 (m, 2H), 1,78 - 1,69 (m, 2H), 1,68 - 1,61 (m, 1H), 1,37 -1,13 (m, 5H);
13C-RMN (126 MHz, DMSO-cfe) 5 (ppm) 164,7, 147,0, 142,1, 135,1, 129,8, 128,9, 120,7, 120,1 (q, J = 256,2 Hz), 36,5, 35,9, 26,0, 25,2;
EMAR calc. para C16H18F3N2OS [M+H]+ 343,1086, hallado 343,1080.
Ejemplo preparativo 38
5-bromo-4-feniltiazol-2-amina

Se añadió una disolución de NBS (1,062 g, 5,97 mmol) en CH2Cl2 (15 ml) a una disolución de 4-feniltiazol-2-amina (1,052 g, 5,97 mmol) en CH2Cl2 (50 ml) y se agitó la mezcla a 25°C durante 2 h. Se añadió una disolución acuosa saturada de NH4Cl (25 ml) a la mezcla, se separó la fase orgánica, se lavó con agua (25 ml), salmuera (25 ml), se secó sobre MgSO4, se filtró y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1). Se obtuvo el producto como un sólido violeta pálido (1,211 g, 80%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 7,83 - 7,78 (m, 2H), 7,45 - 7,39 (m, 2H), 7,38 - 7,32 (m, 1H), 7,28 (s, 2H);
13C-RMN (126 MHz, DMSO-cfe) 5 (ppm) 166,9, 147,2, 133,7, 128,1, 127,9, 87,0;
EMAR calc. para CgHsBr^S [M+H]+ 256,9565, hallado 256,9565.
Ejemplo preparativo 39
4,5-difeniltiazol-2-amina
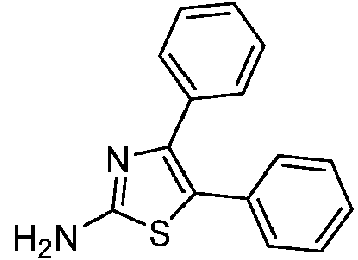
Se añadió una mezcla de dioxano y agua (2,0 ml 0,5 ml) a una mezcla de 5-bromo-4-feniltiazol-2-amina (0,116 g, 0,455 mmol), ácido fenilborónico (0,069 g, 0,568 mmol), K2CO3 (0,251 g, 1,82 mmol) y Pd(PPh3)4 (0,026 g, 0,023 mmol). Se desgasificó la mezcla de reacción burbujeando N2 durante 5 min, luego se agitó a 55°C durante 5 h y luego a 75°C durante 16 h. Se enfrió la mezcla hasta 25°C, se diluyó con EtOAc (5 ml), se vertió en una disolución acuosa saturada de NH4Cl (20 ml) y se extrajo con EtOAc (3 x 25 ml). Se lavaron los extractos orgánicos combinados con agua (20 ml) y salmuera (20 ml), se secaron sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1). Se obtuvo el producto como un sólido blanco (0,035 g, 30%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 7,39 - 7,35 (m, 2H), 7,31 - 7,18 (m, 8 H), 7,09 (s, 2H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 166,0, 144,9, 135,4, 132,8, 128,9, 128,6, 128,4, 128,0, 127,2, 127,0, 119,0;
EMAR calc. para C15H13N2S [M+H]+ 253,0794, hallado 253,0793.
Ejemplo preparativo 40
4-(4-morfolinofenil)tiazol-2-amina

Se preparó 2-bromo-1-(4-morfolinofenil)etan-1-ona según el procedimiento general A2 a partir de 1-(4-morfolinofenil)etanona (0,29 g, 1,41 mmol), CuBr2 (0,789 g, 3,53 mmol) en CHCl3 (2 ml) y EtOAc (2 ml). Se usó el producto en bruto como tal en la siguiente etapa.
Se preparó 4-(4-morfolinofenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(4-morfolinofenil)etan-1-ona (400 mg, 1,41 mmol) y tiourea (215 mg, 2,826 mmol) en EtOH (10 ml). Tratamiento final 1 del procedimiento general B. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:4), como un sólido rojo anaranjado (76 mg, 21%).
1H-RMN (300 MHz, CDCls) 5 (ppm) 7,65 (d, J = 8,9 Hz, 2H), 6,97 - 6 , 8 6 (m, 4H), 6,75 (s, 1H), 3,78 - 3,68 (m, 4H), 3,15 -3,08 (m, 4H);
13C-RMN (126 MHz, CDCls) 5 (ppm) 167,9, 150,1, 126,3, 114,7, 98,6, 66,0, 48,2;
EMAR calc. para C13H16N3OS [M+H]+ 262,1009, hallado 262,1008.
Ejemplo preparativo 41
4-(5-(trifluorometil)piridin-2-il)tiazol-2-amina

Se preparó 2-bromo-1-(5-(trifluorometil)piridin-2-il)etan-1-ona según el procedimiento general A4 a partir de 1-(5-trifluorometil)piridina-2-il]etanona (0,369 g, 1,95 mmol) y Br2 (0,343 g, 100 |al, 2,146 mmol) en AcOH (5 ml). Se obtuvo el producto intermedio en bruto como un sólido blanquecino en un rendimiento cuantitativo y se usó como tal en la siguiente etapa.
Se preparó 4-(5-(trifluorometil)piridin-2-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(5-(trifluorometil)piridin-2-il)etan-1-ona (0,52 g, 1,94 mmol) y tiourea (223 mg, 2,93 mmol) en EtOH (5 ml). Tratamiento final 1 del procedimiento general B. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 2:1 a 0:1), como un sólido blanquecino (359 mg, 75%).
1H-RMN (300 MHz, DMSO-CÍ6) 5 (ppm) 8,91 - 8,87 (m, 1H), 8,21 (dd, J = 8,4, 2,6 Hz, 1H), 7,99 (d, J = 8,2 Hz, 1H), 7,46 (s, 1H), 7,23 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,8 , 155,6, 148,6, 146,0 (q, J = 4,1 Hz), 134,7 (q, J = 3,7 Hz), 124,0 (q, J = 271,8 Hz); 123,0 (q, J = 32,2 Hz), 119,9, 108,5;
19F-RMN (471 MHz, DMSO-^) 5 (ppm) -60,68;
EMAR calc. para C9H7F3N3S [M+H]+ 246,0307, hallado 246,0309.
Ejemplo preparativo 42
3-(2-aminotiazol-4-il)benzonitrilo

Se preparó 3-(2-bromoacetil)benzonitrilo según el procedimiento general A1 a partir de 3-acetilbenzonitrilo (1,00 g, 7,08 mmol) y Br2 (1,13 g, 360 |l, 7,08 mmol) en CH2Cl2 (10 ml). Se obtuvo el producto intermedio en bruto (3-(2-bromoacetil)benzonitrilo) como un sólido blanco en rendimiento cuantitativo y se usó como tal en la siguiente etapa: Se preparó 3-(2-aminotiazol-4-il)benzonitrilo según el procedimiento general B a partir de 3-(2-bromoacetil)benzonitrilo (1,6 g, 7,08 mmol) y tiourea (810 mg, 10,6 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (950 mg, 70%).
1H-RMN (500 MHz, DMSO-^) 5 fppm) 8,21 - 8,18 (m, 1H), 8,14 - 8,09 (m, 1H), 7,72 - 7,69 (m, 1H), 7,61 - 7,55 (m, 1H), 7,25 (s, 1H), 7,13 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 fppm) 168,5, 147,6, 135,9, 130,4, 129,9, 129,8, 128,8, 118,8, 111,6, 103,8;
EMAR calc. para C10H8N3S [M+H]+ 202,0433, hallado 202,0437.
Ejemplo preparativo 43
4-(2-aminotiazol-4-il)-N,N-dimetilbenzamida

Se preparó 4-(2-bromoacetil)-N,N-dimetilbenzamida según el procedimiento general A2 a partir de 4-acetil-N,N-dimetilbenzamida (0,335 g, 1,75 mmol) y CuBr2 (0,78 g, 3,5 mmol) en CHCl3 (3 ml) y EtOAc (3 ml). Se usó el producto intermedio en bruto como tal en la siguiente etapa.
Se preparó 4-(2-aminotiazol-4-il)-N,N-dimetilbenzamida según el procedimiento general B a partir de 4-(2-bromoacetil)-N,Ñ-dimetilbenzamida (473 mg, 1,75 mmol) y tiourea (200 mg, 2,6 mmol) en EtOH (3 ml). Tratamiento final 1 del procedimiento general B. Se obtuvo el producto, purificado mediante cromatografía en columna (CH2Cl2 :EtOAc; de 7:3 a 0 :1 ), como un sólido blanco (220 mg, 50%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 fppm) 7,86 - 7,81 (m, 2H), 7,43 - 7,36 (m, 2H), 7,05 (s, 1H), 6,91 (s, 2H), 2,97 (s, 6 H);
13C-RMN (126 MHz, DMSO-^) 5 fppm) 169,8, 168,0, 149,0, 135,5, 134,8, 126,9, 125,0, 102,4;
EMAR calc. para C12H14N3OS [M+H]+ 248,0852, hallado 248,0850.
Ejemplo preparativo 44
4-(2-aminotiazol-4-il)benzoato de metilo

Se preparó 4-(2-bromoacetil)benzoato de metilo según el procedimiento general A1 a partir de 4-acetilbenzoato de metilo (1,00 g, 5,6 mmol) y Br2 (0,90 g, 290 |al, 5,6 mmol) en CH2Cl2 (10 ml). Se obtuvo el producto intermedio en bruto (4-(2-bromoacetil)benzoato de metilo) como un sólido blanco y se usó como tal en la siguiente etapa.
Se preparó 4-(2-aminotiazol-4-il)benzoato de metilo según el procedimiento general B a partir de 4-(2-bromoacetil)benzoato de metilo (1,40 g, 5,6 mmol) y tiourea (640 mg, 8,4 mmol) en EtOH (10 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (1,06 g, 82%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 7,99 - 7,89 (m, 4H), 7,23 (s, 1H), 7,12 (s, 2H), 3,85 (s, 3H);
13C-RMN (126 MHz, DMSO-cfe) 5 (ppm) 168,3, 166,0, 148,7, 139,1, 129,5, 127,8, 125,5, 104,4, 52,0;
EMAR calc. para C11H11N2O2S [M+H]+ 235,0536, hallado 235,0533.
Ejemplo preparativo 45
2-(4-(2-aminotiazol-4-il)fenil)propan-2-ol

A una disolución de 4-(2-aminotiazol-4-il)benzoato de metilo (300 mg, 1,28 mmol) en THF (5 ml) se le añadió una disolución de MeMgCl (3 M en THF, 2,1 ml, 6,4 mmol) a -78°C y se agitó la mezcla de reacción a 25°C durante 1 h. Se vertió la mezcla en una disolución acuosa saturada de NH4Cl (20 ml) y se extrajo con EtOAc (3 x 10 ml). Se lavaron los extractos orgánicos combinados con salmuera (20 ml), se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; 7:3). Se obtuvo el producto como un sólido blanco (160 mg, 55%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 7,71 (d, J = 8,4 Hz, 2H), 7,44 (d, J = 8,3 Hz, 2H), 6,99 (s, 2H), 6,92 (s, 1H), 4,95 (s, 1H), 1,43 (s, 6H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 168,0, 149,9, 149,4, 132,7, 124,9, 124,6, 100,7, 70,5, 31,8;
EMAR calc. para C12H15N2OS [M+H]+ 235,0900, hallado 235,0902.
Ejemplo preparativo 46
5-(2-aminotiazol-4-il)tiofen-2-carbonitrilo

Se preparó 5-(2-bromoacetil)tiofen-2-carbonitrilo según el procedimiento general A2 a partir de 5-acetiltiofen-2-carbonitrilo (0,25 g, 1,65 mmol) y CuBr2 (0,74 g, 3,3 mmol) en CHCh (3 ml) y EtOAc (3 ml). Se usó el producto
intermedio en bruto como tal en la siguiente etapa.
Se preparó 5-(2-aminotiazol-4-il)tiofen-2-carbonitrilo según el procedimiento general B a partir de 5-(2-bromoacetil)tiofen-2-carbonitrilo (380 mg, 1,65 mmol) y tiourea (188 mg, 2,5 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (243 mg, 71%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 7,87 (d, J = 4,0 Hz, 1H), 7,54 (d, J = 4,0 Hz, 1H), 7,32 (s, 2H), 7,25 (s, 1H); 13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,7, 146,6, 142,5, 139,7, 122,8, 114,9, 105,4, 104,3;
EMAR calc. para C8 H6N3S2 [M+H]+ 207,9998, hallado 207,9995.
Ejemplo preparativo 47
4-(tiofen-2-il)tiazol-2-amina

Se preparó el compuesto según el procedimiento general B a partir de 2-bromo-1-(2-tienil)etanona (0,506 g, 2,47 mmol) y tiourea (0,282 g, 3,70 mmol) en EtOH (7 ml). Tratamiento final 1 del procedimiento general B. El producto, obtenido como un sólido amarillo pálido (0,447 g, 99%), no requirió ninguna purificación adicional.
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 7,39 (dd, J = 5,0, 1,4 Hz, 1H), 7,36 (dd, J = 3,7, 1,4 Hz, 1H), 7,11 (s, 2H), 7,03 (dd, J = 5,1, 3,6 Hz, 1H), 6,83 (s, 1H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 168,2, 144,5, 139,2, 127,7, 124,6, 122,7, 99,7;
EMAR calc. para C7H7N2S2 [M+H]+ 183,0045, hallado 183,0045.
Ejemplo preparativo 48
4-(3,5-difluorofenil)tiazol-2-amina

Se preparó 2-bromo-1-(3,5-difluorofenil)etan-1-ona según el procedimiento general A1 a partir de 3’,5’-difluoroacetofenona (0,309 g, 1,98 mmol) y Br2 (0,316 g, 101 ^l, 1,98 mmol) en CH2Cl2 (16 ml). Se obtuvo el producto intermedio en bruto (2-bromo-1-(3,5-difluorofenil)etan-1-ona) como un aceite amarillo pálido en rendimiento cuantitativo y se usó como tal en la siguiente etapa.
Se preparó 4-(3,5-difluorofenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(3,5-difluorofenil)etan-1-ona (449 mg, 1,91 mmol) y tiourea (226 mg, 2,97 mmol) en EtOH (6 ml). Tratamiento final 1 del procedimiento general B. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 2:1), como un sólido blanco (334 mg, 79%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 7,52 - 7,44 (m, 2H), 7,27 (s, 1H), 7,14 (s, 2H), 7,09 (tt, J = 9,1,2,3 Hz, 1H); 13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,8, 163,1 (dd, J = 244,3, 13,6 Hz), 148,0, 138,9 (dd, J = 10,0 Hz), 108,8 (d, J = 26,3 Hz), 105,0, 102,7 (dd, J = 25,9 Hz);
19F-RMN (471 MHz, DMSO-^) 5 (ppm)-110,03;
EMAR calc. para C9H7F2N2S [M+H]+ 213,0293, hallado 213,0294.
Ejemplo preparativo 49
4-(piridazin-3-il)tiazol-2-amina

Se preparó 2-bromo-1-(piridazin-3-il)etan-1-ona según el procedimiento general A4 a partir de 1 -(piridazin-3-il)etanona (0,244 g, 2,0 mmol) y Br2 (0,319 g, 100 |al, 2,0 mmol) en AcOH (5 ml) y HBr (al 47% en H2O, 0,69 ml, 6.0 mmol). Se obtuvo 2-bromo-1-(piridazin-3-il)etan-1-ona en bruto como un sólido naranja y se usó como tal en la siguiente etapa.
Se preparó 4-(piridazin-3-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(piridazin-3-il)etan-1-ona (0,155 g, 0,771 mmol) y tiourea (230 mg, 3,0 mmol) en EtOH ( 8 ml). Tratamiento final 1 del procedimiento general B. Se obtuvo el producto, purificado mediante cromatografía en columna (EtOAc:MeOH; de 1:0 a 5:1), como un sólido rojo anaranjado ( 66 mg, 19%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 9,11 (dd, J = 4,8, 1,9 Hz, 1H), 7,98 (dd, J = 8,5, 2,1 Hz, 1H), 7,71 (dd, J = 8,5, 5.0 Hz, 1H), 7,53 (s, 1H), 7,25 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 fppm) 149,8, 127,2, 123,3, 106,5;
EMAR calc. para C9 H7N4S [M+H]+ 179,0386, hallado 179,0388.
Ejemplo preparativo 50
4-(4-fluorofenil)tiazol-2-amina

Se preparó 4-(4-fluorofenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-4’-fluoroacetofenona (586 mg, 2,70 mmol) y tiourea (308 mg, 4,05 mmol) en EtOH ( 8 ml). Tratamiento final 1 del procedimiento general B. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1 : 0 a 1 :1 ), como un sólido blanco (502 mg, 96%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 fppm) 7,86 - 7,78 (m, 2H), 7,23 -7,13 (m, 2H), 7,03 (s, 2H), 6,96 (s, 1H);
13C-RMN (126 MHz, DMSO-^) 5 fppm) 168,3, 161,3 (d, J = 244,3 Hz), 148,8, 131,5, 127,4 (d, J = 8,2 Hz), 115,2 (d, J = 20,9 Hz), 101,2;
19F-RMN (471 MHz, DMSO-CÍ6) 5 fppm)-115,16;
EMAR calc. para C9 H8FN2S [M+H]+ 195,0387, hallado 195,0388.
Ejemplo preparativo 51
4-(piridin-4-il)tiazol-2-amina
Se añadió 4-acetilpiridina (1,00 ml, 9,04 mmol) a una disolución de HBr (al 47% en H2O, 4,56 ml, 27,12 mmol) en AcOH (20 ml) a 25°C. Se añadió bromo (1,59 g, 0,51 ml, 9,94 mmol) gota a gota a la disolución, luego apareció lentamente un precipitado blanco. Se agitó la mezcla durante 20 h. Se añadió dietil éter (20 ml), se filtró el sólido y se lavó con dietil éter (2 * 5 ml). Después de secar a vacío, se obtuvo bromuro de 4-(2-bromoacetil)piridin-1-io como un sólido blanquecino (2,529 g) que se usó sin purificación adicional.
Se preparó 4-(piridin-4-il)tiazol-2-amina según el procedimiento general B a partir de bromuro de 4-(2-bromoacetil)piridin-1-io (1,483 g, 5,28 mmol) y tiourea (0,603 g, 7,92 mmol) en EtOH (15 ml). Se evaporó el disolvente a vacío. Se sometió a sonicación el residuo en dietil éter (10 ml) y se recogió el sólido naranja mediante filtración. Se mezcló el sólido con NaOH (2 M en H2O, 20 ml) y EtOAc (50 ml). Se lavó el extracto orgánico con agua (20 ml), salmuera (20 ml), se secó sobre MgSO4, se filtró y se evaporó el disolvente a vacío. Se obtuvo el producto como un sólido naranja (0,889 g, 95%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 8,57 - 8,50 (m, 2H), 7,77 -7,67 (m, 2H), 7,37 (s, 1H), 7,17 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,6, 150,0, 147,5, 141,4, 119,8, 106,1;
EMAR calc. para CsHsNsS [M+H]+ 178,0433, hallado 178,0432.
Ejemplo preparativo 52
4-(2-aminotiazol-4-il)benzoato de metilo

Se preparó 4-(2-bromoacetil)benzoato de metilo según el procedimiento general A1 a partir de 4-acetilbenzoato de metilo (1,00 g, 5,6 mmol) y Br2 (0,90 g, 290 |al, 5,6 mmol) en CH2Cl2 (10 ml). Se obtuvo el producto intermedio en bruto (4-(2-bromoacetil)benzoato de metilo) como un sólido blanco y se usó como tal en la siguiente etapa.
Se preparó 4-(2-aminotiazol-4-il)benzoato de metilo según el procedimiento general B a partir de 4-(2-bromoacetil)benzoato de metilo (1,40 g, 5,6 mmol) y tiourea (640 mg, 8,4 mmol) en EtOH (10 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (1,06 g, 82%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 7,99 - 7,89 (m, 4H), 7,23 (s, 1H), 7,12 (s, 2H), 3,85 (s, 3H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 168,3, 166,0, 148,7, 139,1, 129,5, 127,8, 125,5, 104,4, 52,0;
EMAR calc. para C11H11N2O2S [M+H]+ 235,0536, hallado 235,0533.
Ejemplo preparativo 53
4,5,6,7-tetrahidrobenzo[d1tiazol-2-amina (no se encuentra dentro del alcance de las reivindicaciones)

A ciclohexanona (4,70 g, 5,0 ml, 48,0 mmol) en CH2Cl2 (10 ml) a 0°C se le añadió una disolución de NBS (10,23 g, 57 mmol) y TsOH (0,90 g, 4,8 mmol) en CH2Cl2 (40 ml). Se agitó la mezcla a 25°C durante 3 h. Se añadió agua (30 ml) a la mezcla, se separó la fase orgánica, se secó sobre MgSO4, se filtró y se evaporó el disolvente a vacío. Se usó 2-bromociclohexan-1-ona, obtenida como un sólido blanco (7,16 g, 85%), en la siguiente etapa sin purificación adicional.
Se preparó 4,5,6,7-tetrahidrobenzo[d]tiazol-2-amina según el procedimiento general B con 2-bromociclohexan-1-ona (0,51 g, 2,9 mmol) y tiourea (330 mg, 4,35 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (350 mg, 80%).
1H-RMN (300 MHz, DMSO-da) 5 (ppm) 6,54 (s, 2H), 2,48 - 2,31 (m, 4H), 1,75 - 1,63 (m, 4H);
13C-RMN (75 MHz, DMSO-d6) 5 (ppm) 165,3, 144,7, 114,5, 26,2, 23,2, 22,6, 22,5;
EMAR calc. para C7H11N2S [M+H]+ 155,0598, hallado 155,0596.
Ejemplo preparativo 54
5-metil-4-fenilt¡azol-2-am¡na

Se preparó el compuesto según el procedimiento general B a partir de 2-bromo-1-fenilpropan-1-ona (1,555 g, 7,3 mmol) y tiourea (0,839 g, 11,02 mmol) en EtOH (15 ml). Tratamiento final 1 del procedimiento general B. Se obtuvo el producto como un sólido amarillo (1,361 g, 98%).
1H-RMN (500 MHz, DMSO-d6) 5 (ppm) 7,55 (d, J = 7,0 Hz, 2H), 7,38 (dd, J = 7,7 Hz, 2H), 7,31 - 7,23 (m, 1H), 6,74 (s, 2H), 2,32 (s, 3H);
13C-RMN (126 MHz, DMSO-d6) 5 (ppm) 164,2, 144,9, 135,4, 128,0, 127,9, 126,6, 114,6, 12,1;
EMAR calc. para C10H11N2S [M+H]+ 191,0637, hallado 191,0637.
Ejemplo preparativo 55
5-cloro-4-fen¡lt¡azol-2-am¡na
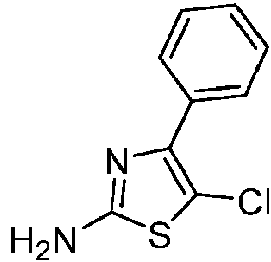
A una disolución de 4-feniltiazol-2-amina (150 mg, 0,84 mmol) en CH2Ch (5 ml) se le añadió lentamente una disolución de NCS (124 mg, 0,93 mmol) en CH2Cl2 (5 ml) a 0°C. Se agitó la mezcla de reacción a 25°C durante 4 h. Se añadió agua (20 ml) y se extrajo la mezcla con EtOAc (3 * 20 ml). Se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se obtuvo el producto, purificado mediante cromatografía en columna sobre gel de sílice (hexano:EtOAc; 1:1), como un sólido blanco (160 mg, 91%).
1H-RMN (500 MHz, CDCla) 5 (ppm) 7,86 (dd, J = 8,4, 1,3 Hz, 2H), 7,42 (d, J = 7,8 Hz, 2H), 7,35 (t, J = 7,4 Hz, 1H), 5,08 (s, 2H);
13C-RMN (126 MHz, CDCla) 5 (ppm) 163,2, 145,6, 133,1, 128,5, 128,4, 128,3, 108,7;
EMAR calc. para CgHaCl^S [M+H]+211,0091, hallado 211,0090.
Ejemplo preparativo 56
5-bromo-4-fen¡lt¡azol-2-am¡na

Se añadió una disolución de NBS (1,062 g, 5,97 mmol) en CH2Ch (15 ml) a una disolución de 2-amino-4-feniltiazol (1,052 g, 5,97 mmol) en CH2Ch (50 ml) y se agitó la mezcla a 25°C durante 2 h. Se añadió una disolución acuosa
saturada de NH4CI (25 ml) a la mezcla. Se separó la parte orgánica , se lavó con agua (25 ml), salmuera (25 ml), se secó sobre MgSO4, se filtró y se evaporó el disolvente a vacío. Se obtuvo el producto, purificado mediante cromatografía en columna sobre gel de sílice (hexano:EtOAc; de 1:0 a 1:1), como un sólido violeta pálido (1,211 mg, 80%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 7,83 - 7,78 (m, 2H), 7,45 - 7,39 (m, 2H), 7,38 - 7,32 (m, 1H), 7,28 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 166,9, 147,2, 133,7, 128,1, 127,9, 87,0;
EMAR calc. para CgHsBr^S [M+H]+ 256,9565, hallado 256,9565.
Ejemplo preparativo 57
4-(4-((trimetilsilil)etinil)fenil)tiazol-2-amina

Se preparó 2-bromo-1-(4-((trimetilsilil)etinil)fenil)etan-1-ona según el procedimiento general A2 a partir de 1-(4-((trimetilsilil)etinil)fenil)etan-1 -ona (0,25 g, 1,15 mmol) y CuBr2 (0,52 g, 2,3 mmol) en CHCl3 (4 ml) y EtOAc (4 ml). Se usó el producto intermedio en bruto como tal en la siguiente etapa.
Se preparó 4-(4-((trimetilsilil)etinil)fenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(4-((trimetilsilil)etinil)fenil)etan-1-ona (340 mg, 1,15 mmol) y tiourea (133 mg, 1,73 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (100 mg, 30%) y no requirió ninguna purificación adicional.
1H-RMN (500 MHz, CDCls) 5 (ppm) 7,75 - 7,70 (m, 2H), 7,50 - 7,45 (m, 2H), 6,77 (s, 1H), 5,04 (s, 2H), 0,27 (s, 9H);
13C-RMN (126 MHz, CDCls) 5 (ppm) 167,3, 150,9, 134,8, 132,5, 125,9, 122,5, 105,4, 104,1, 95,1, 0,2;
EMAR calc. para C ^ H ^ S S i [M+H]+ 273,0876, hallado 273,0873.
Ejemplo preparativo 58
4-(3-ciclopropil-4-metoxifenil)tiazol-2-amina

Se preparó 2-bromo-1-(3-ciclopropil-4-metoxifenil)etan-1-ona según el procedimiento general A2 a partir de 1-(3-ciclopropil-4-metoxifenil)etan-1-ona (0 , 1 2 g, 0,7 mmol) y CuBr2 (0,31 g, 1 ,4 mmol) en CHCl3 (2 ml) y EtOAc ( 2 ml). Se usó el producto intermedio en bruto como tal en la siguiente etapa.
Se preparó 4-(3-ciclopropil-4-metoxifenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(3-ciclopropil-4-metoxifenil)etan-1-ona (188 mg, 0,7 mmol) y tiourea (75 mg, 0,95 mmol) en EtOH (3 ml). Tratamiento final 1 del procedimiento general B. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 7:3), como un sólido blanco (100 mg, 60%).
1H-RMN (500 MHz, CDCh) 5 (ppm) 7,55 (dd, J = 8,5, 2,3 Hz, 1H), 7,28 (d, J = 2,3 Hz, 1H), 6,85 (d, J= 8,4 Hz, 1H), 6,56 (s, 1H), 5,12 (s, 2H), 3,90 (s, 3H), 2,18 (tt, J = 8,5, 5,4 Hz, 1H), 0,99 - 0,90 (m, 2H), 0,79 - 0,70 (m, 2H);
13C-RMN (126 MHz, CDCla) 5 (ppm) 167,3, 158,4, 151,6, 132,3, 127,6, 124,3, 123,2, 110,5, 101,1, 55,9, 9,8, 7,8; EMAR calc. para C13H17N2OS [M+2H+H]+ 249,1056, hallado 249,1060.
Ejemplo preparativo 59
4-(5-bromotiofen-2-il)tiazol-2-amina

Se preparó 2-bromo-1-(5-bromotiofen-2-il)etan-1-ona según el procedimiento general A2 a partir de 2-acetil-5-bromotiofene (0,53 g, 2,58 mmol) y CuBr2 (1,15 g, 5,16 mmol) en cloroformo (10 ml) y acetato de etilo (10 ml); el tiempo de reacción fue de 2 h. Se obtuvo el producto intermedio en bruto (2-bromo-1-(5-bromotiofen-2-il)etan-1-ona) como un sólido amarillo (630 mg) que se usó como tal en la siguiente etapa.
Se preparó 4-(5-bromotiofen-2-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(5-bromotiofen-2-il)etan-1-ona (630 mg, 2,21 mmol) y tiourea (253 mg, 3,32 mmol) en EtOH (15 ml). Tratamiento final 1 del procedimiento general B. Se obtuvo el producto, purificado mediante cromatografía en columna sobre gel de sílice (hexano:EtOAc; de 1:0 a 1:1), como un sólido amarillo pálido (250 mg, 38%).
1H-RMN (500 MHz, CDCh) 5 (ppm) 7,07 (d, J = 3,91 Hz, 1H), 6,97 (d, J = 3,90 Hz, 1H), 6,56 (s, 1H), 5,17 (s a, 2H); 13C-RMN (126 MHz, CDCh) 5 (ppm) 167,5, 144,7, 140,0, 130,6, 123,6, 111,8, 101,9;
EMAR calc. para C7H6BrN2S2 [M+H]+ 262,9128, hallado 262,9131.
Ejemplo preparativo 60
4-(4-(terc-butil)fenil)tiazol-2-amina

Se preparó 4-(4-(terc-butil)fenil)tiazol-2-amina según el procedimiento general B a partir de 1-(4-(terc-butil)fenil)-2-cloroetanona (570 mg, 2,705 mmol) y tiourea (309 mg, 4,058 mmol) en EtOH (15 ml). Se agitó la mezcla de reacción a 90°C durante 3 h. Tratamiento final 1 del procedimiento general B. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc, de 10:1 a 1:1), como un sólido blanco (616 mg, 98%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 7,73 - 7,68 (m, 2H), 7,40 - 7,34 (m, 2H), 6,99 (s, 2H), 6,90 (s, 1H), 1,29 (s, 9H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 168,1, 149,9, 149,5, 132,3, 125,2, 125,1, 100,6, 34,2, 31,1;
EMAR calc. para C13H17N2S [M+H]+ 233,1107, hallado 233,1107.
Ejemplo preparativo 61
4-(5-metiltiofen-2-il)tiazol-2-amina

Se preparó 2-bromo-1-(5-metiltiofen-2-il)etan-1-ona según el procedimiento general A2 a partir de 1-(5-metiltiofen-2-il)etan-1 -ona (0,37 g, 2 , 6 6 mmol) y CuBr2 (1 , 20 g, 5,3 mmol) en CHCl3 ( 2 ml) y EtOAc ( 2 ml). Se usó el producto intermedio en bruto como tal en la siguiente etapa.
Se preparó 4-(5-metiltiofen-2-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(5-metiltiofen-2-il)etan-1-ona (580 mg, 2,66 mmol) y tiourea (300 mg, 4 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (450 mg, 85%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 7,13 (d, J = 3,5 Hz, 1H), 7,06 (s, 2H), 6,71 (dd, J = 3,6, 1,2 Hz, 1H), 6,70 (s, 1H), 2,41 (d, J = 1,1 Hz, 3H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 168,6, 145,2, 138,4, 137,4, 126,4, 123,1, 99,3, 15,4;
EMAR calc. para CsHsN2S2 [M+H]+ 197,0202, hallado 197,0199.
Ejemplo preparativo 62
N-(4-feniltiazol-2-il)formamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-feniltiazol-2-amina (1,23 g, 6,9 mmol), formiato de metilo (0,70 g, 0,85 ml, 10 mmol) (en lugar de cianoacetato de etilo) y NaOEt (al 21% en EtOH, 2,60 ml, 6,9 mmol) en EtOH (2 ml). Se agitó la mezcla a 50°C durante 21 h. Se obtuvo el producto, purificado mediante cromatografía en columna (tolueno:EtOAc; 1:1), como un sólido blanco (1,26 g, 90%).
1H-RMN (500 MHz, DMSO-^) 5 fppm) 12,37 (s, 1H), 8,52 (s, 1H), 7,93 - 7,87 (m, 2H), 7,66 (d, J = 1,0 Hz, 1H), 7,43 (dd, J = 8,4, 7,0 Hz, 2H), 7,37 - 7,29 (m, 1H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 fppm) 159,7, 156,2, 148,9, 134,1, 128,7, 127,8, 125,7, 108,4;
EMAR calc. para C10H9 N2OS [M+H]+ 205,0430, hallado 205,0433.
Ejemplo preparativo 63
N-metil-4-feniltiazol-2-amina

Se añadió una disolución de LiAlH4 (1 M en THF, 9,80 ml, 9,8 mmol) lentamente a una disolución de N-(4-feniltiazol-2-il)formamida (1,00 g, 4,9 mmol) en THF (5 ml) a 0°C. Se permitió calentar la mezcla hasta 25°C y se agitó durante 16 h. Se añadió una disolución acuosa saturada de NH4Cl (15 ml) a 0°C y se extrajo la mezcla con EtOAc (3 * 10 ml). Se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; 1:1). Se obtuvo el producto como un sólido blanco (650 mg, 70%).
1H-RMN (500 MHz, CDCls) 5 fppm) 7,82 - 7,77 (m, 2H), 7,42 - 7,38 (m, 2H), 7,34 - 7,30 (m, 1H), 6,71 (s, 1H), 6,08 (s, 1H), 3,05 (d, J = 3,6 Hz, 3H);
13C-RMN (126 MHz, CDCla) 5 (ppm) 171,0, 150,1, 133,8, 129,0, 128,4, 126,2, 100,5, 32,6;
EMAR calc. para C10H11N2S [M+H]+ 191,0637, hallado 191,0639.
Ejemplo preparativo 64
4-(6-metilpiridin-3-il)tiazol-2-amina

Se preparó 2-bromo-1-(6-metilpiridin-3-il)etan-1-ona a partir de 1-(6-metilpiridin-3-il)etan-1-ona (0,5 g, 3,7 mmol), que se disolvió en HBr (al 47% en H2O, 0,67ml, 11,1 mmol) y CH3c Oo H (3 ml), MeOH (3 ml). Se añadió Br2 (448 mg, 0,14 ml, 3,7 mmol) y se agitó la mezcla a 25°C durante 16 h. Se evaporó el disolvente a vacío, se vertió el residuo en una disolución acuosa saturada de NaHCO3 (50 ml) y se extrajo la mezcla con EtOAc (3 x 10 ml). Se lavaron las fracciones orgánicas combinadas con salmuera (10 ml), se secaron sobre MgSO4 y se evaporó el disolvente a vacío. Se usó el producto intermedio en bruto (2-bromo-1-(6-metilpiridin-3-il)etan-1-ona) como tal en la siguiente etapa: Se preparó 4-(6-metilpiridin-3-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(6-metilpiridin-3- il)etan-1-ona (790 mg, 3,7 mmol) y tiourea (420 mg, 5,5 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (350 mg, 65%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 8,87 (d, J = 2,2 Hz, 1H), 8,00 (dd, J = 8,0, 2,3 Hz, 1H), 7,23 (d, J = 8,1 Hz, 1H), 7,10 (s, 2H), 7,08 (s, 1H), 2,46 (s, 3H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 168,5, 156,3, 147,1, 146,1, 132,9, 127,8, 122,8, 102,0, 23,7;
EMAR calc. para CgH10N3S [M+H]+ 192,0590, hallado 192,0588.
Ejemplo preparativo 65
4- fenil-5-(tetrahidro-2H-piran-4-il)tiazol-2-amina

Se preparó 2-bromo-1-fenil-2-(tetrahidro-2H-piran-4-il)etan-1-ona según el procedimiento general A2 a partir de 1-fenil-2-(tetrahidro-2H-piran-4-il)etan-1-ona (235 mg, 1,15 mmol) y CuBr2 (0,51 g, 2,3 mmol, 36 |al) en CHCl3 (2 ml) y EtOAc (2 ml). Se usó el producto intermedio en bruto como tal en la siguiente etapa.
Se preparó 4-(4-metiltiofen-2-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(4-metiltiofen-2-il)etan-1-ona (650 mg, 2,3 mmol) y tiourea (260 mg, 3,45 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco ( 66 mg, 45%).
1H-RMN (500 MHz, CDCl3) 5 (ppm) 7,51 - 7,46 (m, 2H), 7,45 - 7,40 (m, 2H), 7,38 - 7,32 (m, 1H), 5,10 (s, 2H), 4,01 (dd, J = 11,8, 4,6, 1,1 Hz, 2H), 3,43 (td, J = 11,8, 2,1 Hz, 2H), 3,19 (tt, J = 11,7, 4,0 Hz, 1H), 1,89 -1,81 (m, 2H), 1,81 - 1,70 (m, 2H);
13C-RMN (126 MHz, CDCl3) 5 (ppm) 164,5, 145,3, 135,4, 129,4, 128,7, 128,7, 128,0, 68,2, 36,1, 34,5;
EMAR calc. para C14H17N2O [M+H]+ 261,1056, hallado 261,1053.
Ejemplo preparativo 6 6
4-(3-fluorofenil)tiazol-2-amina

Se preparó 4-(4-metiltiofen-2-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(3-fluorofenil)etanona (300 mg, 1,382 mmol) y tiourea (158 mg, 2,073 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanquecino (242 mg, 90%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 7,64 (d, J = 7,9 Hz, 1H), 7,57 (ddd, J = 10,8, 2,7, 1,5 Hz, 1H), 7,43 - 7,36 (m, 1H), 7,13 (s, 1H), 7,11 -7,02 (m, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,2, 162,4 (d, J = 242,5 Hz), 148,5, 137,3 (d, J = 8,2 Hz), 130,4 (d, J = 9,1 Hz), 121,5, 113,7 (d, J = 20,9 Hz), 112,0 (d, J = 22,7 Hz), 103,0;
19F-RMN (471 MHz, DMSO-^) 5 (ppm) -113,4;
EMAR calc. para C9 H8FN2S [M+H]+ 195,0387, hallado 195,0385.
Ejemplo preparativo 67
4-fenil-5-(pirazin-2-il)tiazol-2-amina

Se preparó 2-bromo-1-fenil-2-(pirazin-2-il)etan-1-ona según el procedimiento general A3 a partir de 1-fenil-2-(pirazin-2-il)etan-1-ona (0,2 g, 1 mmol), EtaN (0,12 g, 0,17 ml, 1,2 mmol), TMSOTf (0,245 g, 0,2 ml, 1,1 mmol) y NBS (0,2 g, 1,1 mmol) en t Hf (3 ml). Se usó el producto intermedio en bruto como tal en la siguiente etapa.
Se preparó 4-fenil-5-(pirazin-2-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-fenil-2-(pirazin-2-il)etan-1-ona (277 mg, 1 mmol) y tiourea (115 mg, 1,5 mmol) en EtOH (3 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido amarillo (150 mg, 60%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 8,50 - 8,47 (m, 1H), 8,25 (d, J = 2,6 Hz, 1H), 8,10 (d, J = 1,6 Hz, 1H), 7,52 (s, 2H), 7,50 - 7,46 (m, 2H), 7,43 (dt, J = 4,5, 2,7 Hz, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,8, 150,5, 148,4, 143,9, 141,2, 140,3, 135,7, 128,7, 128,6, 128,6, 117,8; EMAR calc. para C13H11N4S [M+H]+ 255,0699, hallado 255,0695.
Ejemplo preparativo 6 8
4-(3-metilpiridin-2-il)tiazol-2-amina
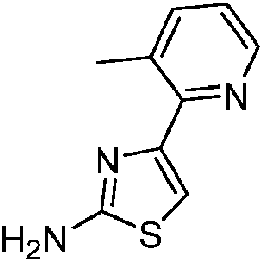
Se preparó 2-bromo-1-(3-metilpiridin-2-il)etan-1-ona según el procedimiento general A3 a partir de 1-(3-metilpiridin-2-il)etan-1-ona (0,34 g, 2,5 mmol), Et3N (0,28 g, 0,39 ml, 2,76 mmol), TMSOTf (0,61 g, 0,5 ml, 2,76 mmol) y NBS (0,49 g, 2,76 mmol) en CH2Cl2 (3 ml). Se usó el producto en bruto como tal en la siguiente etapa.
Se preparó 4-(3-metilpiridin-2-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(3-metilpiridin
2-il)etan-1-ona (0,54 g, 2,5 mmol) y tiourea (285 mg, 3,75 mmol) en EtOH (3 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (340 mg, 70%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 8,37 (dd, J = 4,8, 1,6 Hz, 1H), 7,62 - 7,57 (m, 1H), 7,22 - 7,17 (m, 1H), 6,99 (s, 1H), 6,95 (s, 2H), 2,52 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 167,4, 151,8, 151,3, 146,2, 139,0, 130,7, 122,1, 107,1,20,2;
EMAR calc. para C9 H10N3S [M+H]+ 192,0590, hallado 192,0592.
Ejemplo preparativo 69
4-(6-metoxipiridin-3-il)tiazol-2-amina

Se preparó 2-bromo-1-(6-metoxipiridin-3-il)etan-1-ona según el procedimiento general A3 a partir de 1-(6-metoxipiridin-3-il)etan-1-ona (0,3 g, 2 mmol), Et3N (0,25 g, 0,35 ml, 2,51 mmol), TMSOTf (0,51 g, 0,42 ml, 2,31 mmol) y NBS (0,45 g, 2,51 mmol) en CH2Cl2 (3 ml). Se usó el producto intermedio en bruto como tal en la siguiente etapa. Se preparó 4-(6-metoxipiridin-3-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(6-metoxipiridin-3-il)etan-1-ona (460 mg, 2 mmol) y tiourea (270 mg, 3,43 mmol) en EtOH (3 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido amarillo pálido (320 mg, 80%).
1H-RMN (500 MHz, CDCls) 5 (ppm) 8,60 - 8,56 (m, 1H), 7,94 (dd, J = 8 ,6 , 2,5 Hz, 1H), 6,76 (dd, J = 8,7, 0,7 Hz, 1H), 6,62 (s, 1H), 5,25 (s, 2H), 3,97 (s, 3H);
13C-RMN (126 MHz, CDCls) 5 (ppm) 167,9, 163,9, 148,7, 144,8, 136,7, 124,6, 110,8, 102,0, 53,8;
EMAR calc. para C9 H10N3OS [M+H]+ 208,0539, hallado 208,0536.
Ejemplo preparativo 70
4-(3-metoxipiridin-2-il)tiazol-2-amina

Se preparó 2-bromo-1-(3-metoxipiridin-2-il)etan-1-ona según el procedimiento general A3 a partir de 1-(3-metoxipiridin-2-il)etan-1-ona (0,4 g, 2,6 mmol), Et¿N (0,32 g, 440 |al, 3,13 mmol), TMSOTf (0,7 g, 0,57 ml, 3,13 mmol) y NBS (0,55 g, 3,1 mmol) en CH2Cl2 (3 ml). Se usó el producto intermedio en bruto como tal en la siguiente etapa. Se preparó 4-(3-metoxipiridin-2-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(3-metoxipiridin-2-il)etan-1-ona (0,6 g, 2,6 mmol) y tiourea (300 mg, 3,9 mmol) en EtOH (3 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (265 mg, 50%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 8,15 (dd, J = 4,5, 1,4 Hz, 1H), 7,47 (dd, J = 8,3, 1,3 Hz, 1H), 7,27 (dd, J = 8,3, 4,5 Hz, 1H), 7,15 (s, 1H), 6,97 (s, 2H), 3,86 (s, 3H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 166,5, 153,2, 147,5, 142,1, 140,3, 122,8, 118,8, 108,6, 55,5;
EMAR calc. para C9 H10N3OS [M+H]+ 208,0539, hallado 208,0536.
Ejemplo preparativo 71
4-(piridin-2-il)tiazol-2-amina

A una disolución de 1-(piridin-2-il)etan-1-ona (1,21 g, 10 mmol) en THF anhidro (2 ml) a -78°C se le añadió NaHMDS (1 M en THF, 11 ml, 11 mmol). Se permitió calentar la mezcla hasta 25°C y se agitó durante 1 h. Luego se enfrió la mezcla hasta -78°C, se añadió TMSCI (1,30 g, 1,50 ml, 12 mmol) y se agitó la mezcla a 25°C durante 16 h. Se enfrió la mezcla hasta 0°C, se añadió NBS (1,96 g, 11 mmol) y se agitó la mezcla a 0°C durante 30 min. Se adsorbió previamente la mezcla en bruto sobre un tapón de gel de sílice y se eluyó rápidamente el producto con hexano:EtOAc (1:1). Se obtuvo el producto intermedio en bruto (2-bromo-1-(piridin-2-il)etan-1-ona) como un sólido amarillo pálido, que se usó como tal en la siguiente etapa.
Se preparó 4-(piridin-2-il)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(piridin-2-il)etan-1-ona (1,47 g, 7,4 mmol) y tiourea (840 mg, 11 mmol) en EtOH (5 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (1,2 g, 6 8 %).
1H-RMN (500 MHz, DMSO-da) 5 fppm) 8,53 (dt, J = 4,7, 1,4 Hz, 1H), 7,84 -7,77 (m, 2H), 7,27 -7,22 (m, 2H), 7,07 (s, 2H);
13C-RMN (126 MHz, DMSO-da) 5 fppm) 168,4, 152,4, 150,1, 149,2, 136,9, 122,2, 120,1, 105,3;
EMAR calc. para CsHsNsS [M+H]+ 178,0433, hallado 178,0435.
Ejemplo preparativo 72
4-(4-(metilsulfonil)fenil)tiazol-2-amina

Se preparó 2-bromo-1-(4-(metilsulfonil)fenil)etan-1-ona según el procedimiento general A2 a partir de 1-(4-(metilsulfonil)fenil)etan-1-ona (0,4 g, 2 mmol) y CuBr2 (0,89 g, 4 mmol) en CHCl3 :EtOAc (1:1, 6 ml). Se obtuvo el producto intermedio en bruto (2-bromo-1-(4-(metilsulfonil)fenil)etan-1-ona) como un sólido blanco, que se usó como tal en la siguiente etapa.
Se preparó 4-(4-(metilsulfonil)fenil)tiazol-2-amina según el procedimiento general B a partir de 2-bromo-1-(4-(metilsulfonil)fenil)etan-1-ona (0,55 g, 2 mmol) y tiourea (230 mg, 3 mmol) en EtOH (3 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (390 mg, 75%).
1H-RMN (500 MHz, DMSO-^) 5 fppm) 8,05 - 8,02 (m, 2H), 7,93 - 7,88 (m, 2H), 7,29 (s, 1H), 7,15 (s, 2H), 3,21 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 fppm) 168,5, 148,2, 139,4, 138,8, 127,3, 126,0, 105,2, 43,6;
EMAR calc. para C10H11N2O2S2 [M+H]+ 255,0256, hallado 255,0259.
Ejemplo preparativo 73
4-(4-(ferc-butil)-2,6-dimetilfenil)tiazol-2-amina

Se preparó 2-bromo-1-(4-(ferc-but¡l)-2,6-d¡met¡lfeml)etanona según el procedimiento general A2 a partir de 1-(4-(tercbutil)-2,6-dimetilfenil)etanona (0,110 g, 5,38 mmol) y CuBr2 (0,240 g, 10,08 mmol) en CHCh (2 ml) y EtOAc (2 ml). Se usó el producto intermedio en bruto como tal en la siguiente etapa.
Se preparó 4-(4-(ferc-but¡l)-2,6-d¡met¡lfen¡l)t¡azol-2-am¡na según el procedimiento general B a partir de 2-bromo-1-(4-(ferc-butil^^-dimetilfeniOetanona (133 mg, 5,38 mmol) y tiourea (62 mg, 8,07 mmol) en EtOH (2 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (85 mg, 71%).
1H-RMN (500 MHz, CDCls) 5 (ppm) 7,07 (s, 2H), 6,29 (s, 1H), 2,18 (s, 6 H), 1,31 (s, 9H);
13C-RMN (126 MHz, CDCls) 5 fppm) 166,9, 151,0, 149,8, 137,1, 132,2, 124,5, 105,9, 34,5, 31,5, 20,8;
EMAR calc. para C15H21N2S [M+H]+ 261,1420, hallado 261,1423.
Ejemplo preparativo 74
3-(ferc-butil)feniDtiazol-2-amina

Se preparó 2-bromo-1-(3-(ferc-butil)fenil)etanona según el procedimiento general A2 a partir de 1-(3-(fercbutil)fenil)etanona (0,250 g, 1,41 mmol) y CuBr2 (0,633 g, 2,83 mmol) en CHCh (4 ml) y EtoAc (4 ml). Se usó el producto intermedio en bruto como tal en la siguiente etapa.
Se preparó 4-(3-(ferc-but¡l)fen¡l)t¡azol-2-am¡na según el procedimiento general B a partir de 2-bromo-1-(3-(fercbutil)fenil)etanona (359 mg, 1,41 mmol) y tiourea (160 mg, 2,11 mmol) en EtOH (4 ml). Tratamiento final 2 del procedimiento general B. Se obtuvo el producto como un sólido blanco (85 mg, 71%).
1H-RMN (500 MHz, CDCh) 5 fppm) 7,83 (m, 1H), 7,57 (dt, J = 6,5, 1,9 Hz, 1H), 7,38-7,28 (m, 2H), 6,71 (s, 1H), 5,06 (s a, 2H), 1,36 (s, 9H).
Ejemplo preparativo 75
2-ciano-N-(4-feniltiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 2-amino-4-feniltiazol (0,5 g, 2,837 mmol), cianoacetato de etilo (0,453 ml, 4,255 mmol) y NaOEt (al 21% en EtOH, 1,59 ml, 4,255 mmol) en EtOH (10 ml); el tiempo de reacción fue de 3 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna (tolueno:EtOAc; de 5:1 a 1:1), como un sólido blanquecino (0,586 g, 8 5 %).
IR (cm-1) 3207, 3081,2944, 2913, 2211, 1660, 1554, 1480, 1445, 1389, 1265, 1180, 945, 781,733;
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 12,59 (s, 1H), 7,93 -7,85 (m, 2H), 7,69 (s, 1H), 7,43 (dd, J = 8,3, 7,0 Hz, 2H), 7,36 -7,29 (m, 1H), 4,06 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,8, 157,3, 149,0, 134,0, 128,7, 127,9, 125,6, 115,1, 108,6, 25,9;
EMAR calc. para C12H10N3OS [M+H]+ 244,0539, hallado 244,0538.
Ejemplo preparativo 76
2-ciano-N-(4-(4-metoxifenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(4-metoxifenil)tiazol-2-amina (0,46 g, 2,23 mmol), cianoacetato de etilo (356 |al, 3,345 mmol) y NaOEt (al 21% en EtOH, 1,25 ml, 3,345 mmol) en MeOH (15 ml); se sometió a reflujo durante 3 h. Se purificó el producto mediante cromatografía en columna (tolueno:EtOAc; de 1:0 a 1:1) y luego mediante CCF preparativa (tolueno:EtOAc; 1:1). Se obtuvo el producto como un sólido amarillo pálido (0,312 g, 51%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 12,54 (s, 1H), 7,82 (d, J = 8,9 Hz, 2H), 7,51 (s, 1H), 6,99 (d, J = 8,9 Hz, 2H), 4,05 (s, 2H), 3,79 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,7, 159,0, 157,1, 148,9, 127,0, 115,2, 114,1, 106,5, 55,1,25,9;
EMAR calc. para C13H12N3O2S [M+H]+ 274,0645, hallado 274,0646.
Ejemplo preparativo 77
2-ciano-N-(4-(naftalen-2-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(naftalen-2-il)tiazol-2-amina (0,207 g, 0,915 mmol), cianoacetato de etilo (146 |al, 1,37 mmol) y NaOEt (al 21% en EtOH, 512 |al, 1,37 mmol) en EtOH (2 ml). Se agitó la mezcla a 50°C durante 21 h. Se evaporó el disolvente a vacío, se mezcló el residuo sólido con una disolución acuosa saturada de NH4Cl (20 ml) y se extrajo la mezcla con EtOAc (3 * 25 ml). Se lavaron los extractos orgánicos combinados con salmuera (20 ml), se secaron sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. El producto, obtenido como un sólido rosa pálido (0,161 g, 60%), no requirió ninguna otra purificación.
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 12,68 (s, 1H), 8,42 (s, 1H), 8,05 (d, J = 8,5, 1,7 Hz, 1H), 8,00 - 7,90 (m, 3H), 7,83 (d, J = 1,5 Hz, 1H), 7,59 - 7,47 (m, 2H), 4,08 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,9, 157,4, 148,9, 133,1, 132,5, 131,5, 128,3, 128,1, 127,6, 126,5, 126,2, 124,3, 123,9, 115,1, 109,2, 25,9;
EMAR calc. para C16H12N3OS [M+H]+ 294,0696, hallado 294,0695.
Ejemplo preparativo 78
2-ciano-N-(4-(4-(trifluorometoxi)fenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(4-(trifluorometoxi)fenil)tiazol-2-amina (0,20 g, 0,77 mmol), cianoacetato de etilo (123 |l, 1,15 mmol) y NaOEt (al 21% en EtOH, 430 |l, 1,15 mmol) en EtOH (2 ml). Se calentó la mezcla a 55°C durante 6 h. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1), como un sólido blanquecino (0,216 g, 85%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,62 (s, 1H), 8,00 (d, J = 8,7 Hz, 2H), 7,77 (s, 1H), 7,43 (d, J = 8,7 Hz, 2H), 4,06 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,5, 158,1, 148,3, 148,0, 133,8, 128,0, 121,8, 120,6 (q, J = 256,2 Hz), 115,6, 110,1,26,4;
19F-RMN (471 MHz, DMSO-d6) 8 (ppm) -56,73;
EMAR calc. para C13H9F3 N3O2S [M+H]+ 328,0362, hallado 328,0360.
Ejemplo preparativo 79
2-ciano-N-(4-(p-tolil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(p-tolil)tiazol-2-amina (0,259 g, 1,36 mmol), cianoacetato de etilo (217 |l, 2,04 mmol) y NaOEt (al 21% en EtOH, 763 |l, 2,04 mmol) en EtOH (2 ml). Se agitó la mezcla a 55°C durante 6 h. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1), como un sólido amarillo pálido (0,284 g, 81%).
1H-RMN (500 MHz, DMSO-CÍ6) 8 (ppm) 12,56 (s, 1H), 7,78 (d, J = 8,4 Hz, 2H), 7,60 (s, 1H), 7,24 (d, J = 8,1 Hz, 2H), 4,05 (s, 2H), 2,32 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,8, 157,2, 149,1, 137,2, 131,4, 129,3, 125,6, 115,1, 107,7, 25,9, 20,8; EMAR calc. para C13H12N3OS [M+H]+ 258,0696 hallado 258,0696.
Ejemplo preparativo 80
N-(4-([1,1’-bifenil1-4-il)tiazol-2-il)-2-cianoacetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-([1,1’-bifenil]-4-il)tiazol-2-amina (197 mg, 0,78 mmol), cianoacetato de etilo (125 |al, 1,17 mmol) y NaOEt (al 21% en EtOH, 0,437 ml, 1,17 mmol) en EtOH (2 ml). Se agitó la mezcla a 55°C durante 6 h. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1), como un sólido blanco (210 mg, 84%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,41 (s, 1H), 6,74 (s, 1H), 3,97 (s, 2H), 2,02 (s, 3H), 1,87 (d, J = 3,1 Hz, 6 H), 1,77 - 1,66 (m, 6 H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,3, 160,7, 156,5, 115,2, 105,1,41,6, 36,3, 35,8, 27,9, 25,7;
EMAR calc. para C18H12N3OS [M-H]- 318,0707, hallado 3180704.
Ejemplo preparativo 81
N-(4-(4-bromofenil)tiazol-2-il)-2-cianoacetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(4-bromofenil)tiazol-2-amina (540 mg, 2,12 mmol), cianoacetato de etilo (250 |al, 2,33 mmol) y NaOEt (al 21% en EtOH, 0,80 ml, 2,12 mmol). Se obtuvo el producto, purificado mediante cromatografía en columna (tolueno:EtOAc; 1:1), como un sólido blanco (425 mg, 65%).
1H-RMN (500 MHz, DMSO-CÍ6) 8 (ppm) 12,60 (s, 1H), 7,87 - 7,81 (m, 2H), 7,76 (s, 1H), 7,66 - 7,60 (m, 2H), 4,06 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,9, 157,5, 147,8, 133,2, 131,7, 127,6, 120,9, 115,1, 109,4, 25,9;
EMAR calc. para C^^BrNsOS [M-H]- 321,9478, hallado 321,9479.
Ejemplo preparativo 82
2-ciano-N-(4-(3-hidroxi-4-metoxifenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 5-(2-aminotiazol-4-il)-2-metoxifenol (90 mg, 0,4 mmol), cianoacetato de etilo (65 |al, 0,6 mmol) y NaOEt (al 21% en EtOH, 225 |al, 0,6 mmol). Se obtuvo el producto, purificado mediante cromatografía en columna (EtOAc:tolueno; 1:1), como un sólido blanco (43 mg, 40%).
1H-RMN (500 MHz, DMSO-cfe) 8 (ppm) 12,51 (s, 1H), 9,03 (s, 1H), 7,42 (s, 1H), 7,31 - 7,28 (m, 2H), 6,95 (d, 1H), 4,04 (s, 2H), 3,79 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,7, 156,9, 149,1, 147,7, 146,5, 127,3, 116,9, 115,1, 113,1, 112,3, 106,4, 55,6, 25,8;
EMAR calc. para C13H10N3O3S [M-H]- 288,0448, hallado 288,0447.
Ejemplo preparativo 83
2-ciano-N-(4-(2-hidroxipropan-2-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 2-(2-aminotiazol-4-il)propan-2-ol (126 mg, 0,8 mmol), cianoacetato de etilo (125 |al, 1,2 mmol) y NaOEt (al 21% en EtOH, 300 |al, 0,8 mmol). Se recogió el precipitado mediante filtración y se lavó con EtOAc (2 ml). Se mezcló el sólido con una disolución acuosa saturada de NH4Cl y EtOAc (25 ml 25 ml) y se separaron las fases. Se extrajo la fase acuosa con EtOAc (2 * 25 ml). Se secaron los extractos orgánicos combinados sobre MgSO4 y se evaporó el disolvente a vacío. Se obtuvo el producto como un sólido blanco (115 mg, 65%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,41 (s, 1H), 6,93 (s, 1H), 5,04 (s, 1H), 3,98 (s, 2H), 1,41 (s, 6 H);
13C-RMN (126 MHz, DMSO-CÍ6) 8 (ppm) 161,9, 160,3, 157,1, 115,7, 106,7, 70,6, 30,9, 26,2;
EMAR calc. para C9 H12N3O2S [M+H]+ 226,0645, hallado 226,0646.
Ejemplo preparativo 84
N-(4-(3-bromofenil)tiazol-2-il)-2-cianoacetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(3-bromofenil)tiazol-2-amina (0,50 g, 1,98 mmol) en EtOH (5 ml), cianoacetato de etilo (320 |al, 3,0 mmol) y NaOEt (al 21% en EtOH, 750 |al, 1,98 mmol). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1:1), como un sólido amarillo
(455 mg, 70%).
1H-RMN (500 MHz, DMSO-cfe) 8 (ppm) 12,61 (s, 1H), 8,11 - 8,08 (m, 1H), 7,93 - 7,87 (m, 1H), 7,83 (d, J = 1,2 Hz, 1H), 7,56 - 7,50 (m, 1H), 7,40 (t, J = 7,9 Hz, 1H), 4,06 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,5, 158,0, 147,7, 136,8, 131,5, 131,0, 128,8, 125,0, 122,7, 115,6, 110,6, 26,4;
EMAR calc. para C^^BrNsOS [M-H]- 321,9478, hallado 321,9478.
Ejemplo preparativo 85
N-(4-(2-bromofenil)tiazol-2-il)-2-cianoacetamida
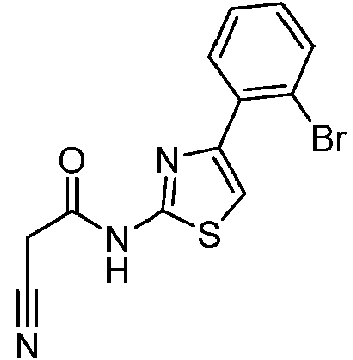
Se preparó el compuesto según el procedimiento general C1 a partir de 4-(2-bromofenil)tiazol-2-amina (0,30 g, 1,18 mmol) en EtOH (5 ml), cianoacetato de etilo (190 |al, 1,78 mmol) y NaOEt (al 21% en EtOH, 440 |al, 1,18 mmol). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1:1), como un sólido blanco (100 mg, 30%).
1H-RMN (500 MHz, DMSO-CÍ6) 8 (ppm) 12,60 (s, 1H), 7,73 (dd, J = 8,0, 1,2 Hz, 1H), 7,68 (dd, J = 7,8, 1,7 Hz, 1H), 7,58 (s, 1H), 7,48 - 7,45 (m, 1H), 7,32 - 7,30 (m, 1H), 4,06 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,9, 156,5, 147,4, 135,2, 133,5, 131,4, 129,7, 127,7, 121,1, 115,1, 113,0, 25,9;
EMAR calc. para C^^BrNsOS [M-H]- 321,9478, hallado 321,9478.
Ejemplo preparativo 86
2-ciano-N-(4-(3-(trifluorometoxi)fenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(3-(trifluorometoxi)fenil)tiazol-2-amina (0,10 g, 0,38 mmol), cianoacetato de etilo (62 |al, 0,6 mmol) y NaOEt (al 21% en EtOH, 140 |al, 0,38 mmol) en EtOH (3 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 3:2), como un sólido blanco (75 mg, 60%).
1H-RMN (500 MHz, DMSO-CÍ6) 8 (ppm) 12,64 (s, 1H), 7,96 - 7,91 (m, 1H), 7,88 (s, 1H), 7,86 - 7,84 (m, 1H), 7,59 -7,56 (m, 1H), 7,35 - 7,30 (m, 1H), 4,07 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,0, 157,6, 148,9, 147,2, 136,3, 130,8, 124,5, 120,2, 120,1 (q, J = 256,4 Hz), 117,8, 115,1, 110,4, 25,9;
19F-RMN (471 MHz, DMSO-CÍ6) 8 (ppm) -56,63;
EMAR calc. para C13H7F3 N3O2S [M-H]- 326,0217, hallado 326,0217.
Ejemplo preparativo 87
2-ciano-N-(4-(4-fenoxifenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(4-fenoxifenil)tiazol-2-amina (270 mg, 1,0 mmol) en EtOH (3 ml), cianoacetato de etilo (130 |l, 1,5 mmol) y NaOEt (al 21% en EtOH, 370 |l, 1,0 mmol). Se obtuvo el producto, purificado mediante cromatografía en columna (tolueno:EtOAc; 3:2), como un sólido blanco (75 mg, 25%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,59 (s, 1H), 7,96 - 7,87 (m, 2H), 7,65 - 7,56 (m, 1H), 7,45 - 7,38 (m, 2H), 7,20 -7,15 (m, 1H), 7,11 -6,99 (m, 4H), 4,05 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,8, 157,3, 156,6, 156,3, 148,4, 132,8, 130,1, 127,4, 123,7, 120,9, 118,9, 118,6, 107,7, 25,9;
EMAR calc. para C18H14N3O2S [M+H]+ 336,0801, hallado 336,0801.
Ejemplo preparativo 88
N-(4-(benzofuran-2-il)tiazol-2-il)-2-cianoacetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(benzofuran-2-il)tiazol-2-amina (0,148 g, 0,684 mmol), cianoacetato de etilo (109 |l, 1,026 mmol) y NaOEt (al 21% en EtOH, 383 |l, 0,47 mmol) en EtOH (3 ml). Se calentó la mezcla a 55°C durante 3 h, luego se enfrió hasta 25°C, se recogió el precipitado mediante filtración y se lavó con EtOH frío (1 ml). Se obtuvo el producto como un sólido blanco (0,129 g, 67%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,77 (s, 1H), 7,70 (s, 1H), 7,70 - 7,66 (m, 1H), 7,61 (dd, J = 8,2, 1,0 Hz, 1H), 7,34 (ddd, J = 8,4, 7,2, 1,4 Hz, 1H), 7,27 (ddd, J = 7,6, 7,6, 1,2 Hz, 1H), 7,14 (d, J = 1,1 Hz, 1H), 4,07 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,1, 158,3, 154,1, 151,5, 140,4, 128,3, 124,8, 123,3, 121,5, 115,1, 111,0, 110,8, 102,6, 25,9;
EMAR calc. para C14H8N3O2S [M-H]- 282,0343, hallado 282,0342.
Ejemplo preparativo 89
N-(4-adamantan-1-il)tiazol-2-il)-2-cianoacetamida (no se encuentra dentro del alcance de las reivindicaciones)

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(adamantan-1-il)tiazol-2-amina (0,253 g, 1,079 mmol), cianoacetato de etilo (172 |al, 1,62 mmol) y NaOEt (al 21% en EtOH, 0,403 ml, 1,079 mmol) en EtOH (2 ml). Se calentó la mezcla a 55°C durante 3 h. Se evaporó el disolvente a vacío. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1), como un sólido blanquecino (0,223 g, 69%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,41 (s, 1H), 6,74 (s, 1H), 3,97 (s, 2H), 2,02 (s, 3H), 1,87 (d, J = 3,1 Hz, 6 H), 1,77 - 1,66 (m, 6 H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,3, 160,7, 156,5, 115,2, 105,1,41,6, 36,3, 35,8, 27,9, 25,7;
EMARcalc. para C16H20N3OS [M+H]+ 302,1322, hallado 302,1321.
Ejemplo preparativo 90
2-ciano-N-(5-ciclohexil-4-(4-(trifluorometoxi)fenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 5-ciclohexil-4-(4-(trifluorometoxi)fenil)tiazol-2-amina (0,1 g, 0,3 mmol), cianoacetato de etilo (50 ^l, 0,45 mmol) y NaOEt (al 21% en EtOH, 110 |al, 0,3 mmol) en EtOH (2 ml). Se agitó la mezcla a 50°C durante 5 h. Se evaporó el disolvente a vacío, se mezcló el residuo con una disolución acuosa saturada de NH4Cl (10 ml) y se extrajo la mezcla con EtOAc (3 * 10 ml). Se lavaron los extractos orgánicos combinados con salmuera (10 ml), se secaron sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1:1), como un semisólido incoloro (0,110 g, 90%).
1H-RMN (500 MHz, CDCls) 8 11,39 (s, 1H), 7,59 - 7,54 (m, 2H), 7,38 -7,31 (m, 2H), 3,02 -2,97 (m, 1H), 2,94 (s, 2H), 2,05 - 1,98 (m, 2H), 1,89 - 1,82 (m, 2H), 1,80 - 1,72 (m, 1H), 1,50 (qd, J = 12,2, 3,1 Hz, 2H), 1,41 - 1,24 (m, 3H);
13C-RMN (126 MHz, CDCls) 8 159,7, 155,7, 149,3, 141,8, 137,6, 133,7, 130,4, 121,5, 120,3 (q, J = 258,0 Hz), 112,8, 37,2, 36,6, 26,7, 25,8, 25,4;
19F-RMN (471 MHz, CDCh) 8 -57,72;
EMAR calc. para C19H19F3N3O2S [M+H]+410,1145, hallado 410,1149.
Ejemplo preparativo 91
2-ciano-N-(4,5-difeniltiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4,5-difeniltiazol-2-amina (0,079 g, 0,313 mmol), cianoacetato de etilo (50 |al, 0,47 mmol) y NaOEt (al 21% en EtOH, 175 |al, 0,47 mmol) en EtOH (2 ml). Se agitó la mezcla de reacción a 55°C durante 3 h. Se trituró el producto en bruto con CH2Cl2 :EtOAc (1:1,2 ml) y se recogió el sólido mediante filtración. Se obtuvo el producto como un sólido blanco (0,059 g, 59%).
1H-RMN (500 MHz, DMSO-cfe) 8 (ppm) 12,66 (s, 1H), 7,47 - 7,36 (m, 5H), 7,36 - 7,27 (m, 5H), 4,08 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,0, 155,3, 143,9, 134,4, 131,6, 129,3, 128,9, 128,4, 128,3, 128,1, 127,8, 125,9, 115,1,25,9;
EMAR calc. para C18H12N3OS [M-H]- 318,0707, hallado 318,0709.
Ejemplo preparativo 92
2-ciano-N-(4-(4-morfolinofenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C2:
Etapa 1: a partir de 4-(4-morfolinofenil)tiazol-2-amina (72 mg, 0,276 mmol), Et3N (28 mg, 38 ^l, 0,276 mmol) y cloruro de cloroacetilo (47 mg, 33 ^l, 0,413 mmol) en CH3CN anhidro (3 ml). Después del tratamiento final, se filtró el residuo rápidamente a través de un lecho de gel de sílice (hexano:EtOAc; 1:1) para proporcionar la cloroacetamida correspondiente como un sólido blanquecino (62 mg, 0,184 mmol), que se usó en la etapa 2.
Etapa 2: a partir de KCN (13 mg, 0,193 mmol) en DMF (1 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:2), como un sólido blanquecino (27 mg, 30%).
1H-RMN (500 MHz, DMSO-CÍ6) 8 (ppm) 12,52 (s, 1H), 7,75 (d, J = 9,0 Hz, 2H), 7,45 (s, 1H), 6,98 (d, J = 8,9 Hz, 2H), 4,04 (s, 2H), 3,79 - 3,68 (m, 4H), 3,15 (dd, J = 6,0, 3,9 Hz, 4H);
13C-RMN (126 MHz, DMSO-CÍ6) 8 (ppm) 161,7, 157,0, 150,6, 149,2, 126,5, 125,1, 115,2, 114,8, 105,7, 66,0, 48,0, 25,8;
EMAR calc. para C16H17N4O2S [M+H]+ 329,1067, hallado 329,1070.
Ejemplo preparativo 93
2-ciano-N-(4-(3-cianofenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 3-(2-aminotiazol-4-il)benzonitrilo (300 mg, 1,5 mmol), cianoacetato de etilo (250 mg, 250 ^l, 2,25 mmol) y Na (35 mg, 1,5 mmol) en EtOH (3,5 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1:1), como un sólido blanco (125 mg, 30%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,64 (s, 1H), 8,31 (s, 1H), 8,23 - 8,19 (m, 1H), 7,92 (s, 1H), 7,81 - 7,77 (m, 1H), 7,71 - 7,63 (m, 1H), 4,07 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,1, 157,7, 146,8, 135,1, 131,2, 130,1, 129,0, 118,6, 115,1, 111,9, 110,7, 25,9;
EMAR calc. para C13H9 N4OS [M+H]+ 269,0492, hallado 269,0495.
Ejemplo preparativo 94
4-(2-(2-cianoacetamido)tiazol-4-il)-N.N-dimetilbenzamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(2-aminotiazol-4-il)-N,N-dimetilbenzamida (85 mg, 0,34 mmol), cianoacetato de etilo (58 mg, 55 |al, 0,51 mmol) y NaH (al 60% en aceite mineral, 14 mg, 0,34 mmol) en THF (2 ml) y MeOH (0,1 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1:1), como un sólido blanco (80 mg, 75%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,61 (s, 1H), 7,97 - 7,91 (m, 2H), 7,79 (s, 1H), 7,50 - 7,45 (m, 2H), 4,07 (s, 2H), 2,96 (s, 6 H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 169,7, 161,9, 157,4, 148,2, 135,7, 134,8, 127,6, 125,4, 115,1, 109,6, 34,8, 25,9;
EMAR calc. para C15H13N4O2S [M-H]- 313,0765, hallado 313,0766.
Ejemplo preparativo 95
2-ciano-N-(4-(4-(2-hidroxipropan-2-il)fenil)tiazol-2-il)acetamida
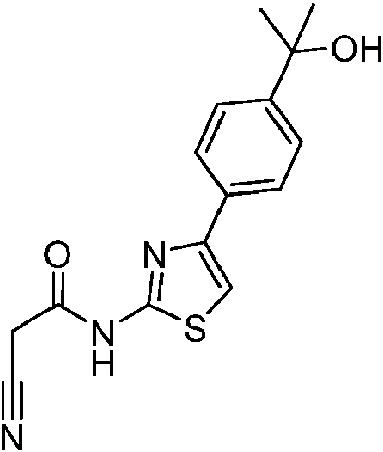
Se preparó el compuesto según el procedimiento general C1 a partir de 2-(4-(2-aminotiazol-4-il)fenil)propan-2-ol (150 mg, 0,64 mmol), cianoacetato de etilo (110 mg, 100 |l, 0,96 mmol) y Na (15 mg, 0,64 mmol) en EtOH (2 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 7:3), como un sólido blanco (55 mg, 30%).
1H-RMN (500 MHz, DMSO-cfe) 8 (ppm) 12,58 (s, 1H), 7,85 - 7,79 (m, 2H), 7,61 (s, 1H), 7,54 - 7,49 (m, 2H), 5,00 (s, 1H), 4,05 (s, 2H), 1,44 (s, 6 H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,8, 157,2, 150,26, 149,04, 131,82, 125,10, 124,89, 115,13, 107,82, 70,52, 31,79, 25,85;
EMAR calc. para C15H16N3O2S [M+H]+ 302,0958, hallado 302,0960.
Ejemplo preparativo 96
2-ciano-N-(4-(5-cianotiofen-2-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C2:
Etapa 1: a partir de 5-(2-aminotiazol-4-il)tiofen-2-carbonitrilo (230 mg, 1,11 mmol), cloruro de cloroacetilo (188 mg, 133 |l, 1,67 mmol) y Et3N (168 mg, 230 |l, 1,67 mmol) en CH3CN (2 ml). Después del tratamiento final, se purificó la mezcla en bruto mediante cromatografía en columna (hexano:EtOAc; 7:3), se obtuvo la 2 -cloroacetamida correspondiente como un sólido blanco (314 mg, 1,11 mmol), que se usó en la etapa 2.
Etapa 2: a partir de KCN (150 mg, 2,22 mmol) en DMF (2 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 7:3), como un sólido blanquecino (70 mg, 23%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,81 (s, 1H), 7,95 (d, J = 4,0 Hz, 1H), 7,91 (s, 1H), 7,71 (d, J = 4,0 Hz, 1H), 4,05 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,2, 158,1, 145,5, 141,8, 140,1, 124,2, 115,0, 114,6, 111,1, 106,5, 25,9;
EMAR calc. para C11H5 N4OS2 [M-H]- 272,991, hallado 272,9906.
Ejemplo preparativo 97
2-ciano-N-(4-(tiofen-2-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(tiofen-2-il)tiazol-2-amina (0,173 g, 0,949 mmol), cianoacetato de etilo (151 |al, 1,42 mmol) y NaOEt (al 21% en EtOH, 532 |al, 1,42 mmol) en MeOH (5 ml); tiempo de reacción 3 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1 :0 a 1 :1 ), como un sólido amarillo (0,081 g, 34%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 12,68 (s, 1H), 7,55 -7,51 (m, 2H), 7,50 (dd, J = 5,0, 1,2 Hz, 1H), 7,11 (dd, J = 5,0, 3,7 Hz, 1H), 4,04 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,9, 157,4, 143,8, 138,1, 128,1, 125,6, 123,9, 115,1, 106,9, 25,8;
EMAR calc. para C10H8 N3OS2 [M+H]+ 250,0103, hallado 250,0103.
Ejemplo preparativo 98
2-ciano-N-(4-(3,5-difluorofenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(3,5-difluorofenil)tiazol-2-amina (297 mg, 1,40 mmol), cianoacetato de etilo (237 mg, 0,223 ml, 2 , 1 0 mmol) y NaH (al 60% en aceite mineral, 62 mg, 1,54 mmol) en THF (5 ml) y MeOH (1 ml). Después de completarse la reacción, se recogió el precipitado mediante filtración, se lavó con hexano (5 ml) y se mezcló con una disolución acuosa saturada de NaHCO3 (15 ml) y EtOAc (25 ml). Se separaron las fases, se lavó la fase orgánica con salmuera (15 ml), se secó sobre MgSO4, se filtró y se evaporó el disolvente a vacío. Se obtuvo el producto como un sólido rosa pálido (312 mg, 80%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,64 (s, 1H), 7,93 (s, 1H), 7,62 - 7,54 (m, 2H), 7,24 - 7,16 (m, 1H), 4,07 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 162,8 (dd, J = 245,2, 13,6 Hz), 162,1, 157,7, 146,6, 137,5 (d, J = 10,2 Hz), 115,1, 111,3, 109,2- 107,9 (m), 103,1 (dd, J = 26,3 Hz), 25,9;
19F-RMN (471 MHz, DMSO-^) 5 (ppm) -109,41;
EMAR calc. para C12H8F2 N3OS [M+H]+ 280,0351, hallado 280,0349.
Ejemplo preparativo 99
2-ciano-N-(4-(piridazin-3-il)tiazol-2-il)acetamida
Se preparó el compuesto según el procedimiento general C2:
Etapa 1: a partir de 4-(piridazin-3-il)tiazol-2-amina (64 mg, 0,363 mmol), Et3N (37 mg, 51 ^l, 0,363 mmol) y cloruro de cloroacetilo (43 ^l, 62 mg, 0,545 mmol) en CH3CN (3 ml). Después del tratamiento final, se cargó rápidamente el residuo sobre un lecho de gel de sílice y se eluyó con EtOAc para proporcionar la cloroacetamida correspondiente como un sólido naranja (50 mg, 0,196 mmol), que se usó en la etapa 2.
Etapa 2: a partir de KCN (13 mg, 0,206 mmol) en DMF anhidra (1 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:1 a 0:1), como un sólido naranja rosáceo pálido ( 8 mg, 50%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,72 (s, 1H), 9,20 (dd, J = 5,0, 1,6 Hz, 1H), 8,17 (s, 1H), 8,09 (dd, J = 8,5, 1,7 Hz, 1H), 7,80 (dd, J = 8,5, 5,0 Hz, 1H), 4,08 (s, 2H);
13C-RMN (126 MHz, DMSO-CÍ6) 8 (ppm) 162,2, 158,1, 154,6, 150,6, 146,2, 127,8, 123,6, 115,1, 113,7, 26,0;
EMAR calc. para C10H8 N5OS [M+H]+ 246,0444, hallado 246,0447.
Ejemplo preparativo 100
2-ciano-N-(4-(4-fluorofenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(4-fluorofenil)tiazol-2-amina (438 mg, 2,25 mmol), cianoacetato de etilo (383 mg, 0,360 ml, 3,38 mmol) y NaH (al 60% en aceite mineral, 99 mg, 2,48 mmol) en THF (5 ml) y MeOH (1 ml). Después de completarse la reacción, se recogió el precipitado mediante filtración y se lavó con hexano (5 ml). Se mezcló el sólido con una disolución acuosa saturada de NaHCO3 (15 ml) y EtOAc (25 ml). Se separaron las fases, se lavó la fase orgánica con salmuera (15 ml), se secó sobre MgSO4, se filtró y se evaporó el disolvente a vacío. Se obtuvo el producto como un sólido rosa pálido (499 mg, 85%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,58 (s, 1H), 8,01 -7,83 (m, 2H), 7,66 (s, 1H), 7,31 - 7,18 (m, 2H), 4,06 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,9, 161,8 (d, J = 244,3 Hz), 157,4, 148,0, 130,7, 127,7 (d, J = 8,2 Hz), 115,6 (d, J = 20,9 Hz), 115,1, 108,3, 25,9;
19F-RMN (471 MHz, DMSO-^) 8 (ppm) -114,14;
EMAR calc. para C12H9FN3 OS [M+H]+ 262,0445, hallado 262,0447.
Ejemplo preparativo 101
2-ciano-N-(4-(piridin-4-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(piridin-4-il)tiazol-2-amina (0,05 g, 0,282 mmol), cianoacetato de etilo (45 |al, 0,423 mmol) y NaOEt (al 21% en EtOH, 158 |al, 0,423 mmol) en MeOH
(2 ml); el tiempo de reacción fue de 3 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna (tolueno:EtOAc; de 5:1 a 1:1), como un sólido blanquecino (0,033 g, 48%).
1H-RMN (500 MHz, DMSO-cfe) 8 (ppm) 12,69 (s, 1H), 8 , 6 6 - 8,59 (m, 2H), 8,05 (s, 1H), 7,87 - 7,78 (m, 2H), 4,07 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,2, 157,9, 150,3, 146,5, 140,7, 119,9, 115,1, 112,8, 25,9;
EMAR calc. para C11H9 N4OS [M+H]+ 245,0492, hallado 245,0491.
Ejemplo preparativo 102
4-(2-(2-cianoacetamido)tiazol-4-il)benzoato de metilo

Se preparó el compuesto según el procedimiento general C2:
Etapa 1: a partir de 4-(2-aminotiazol-4-il)benzoato de metilo (200 mg, 0,854 mmol), cloruro de cloroacetilo (130 mg, 95 |al, 0,1,18 mmol) y Et3N (85 mg, 120 |al, 0,85 mmol) en CH3CN (2 0,5 ml). Después del tratamiento final, se purificó la mezcla en bruto mediante cromatografía en columna (hexano:EtOAc; 7:3) para proporcionar la cloroacetamida correspondiente como un sólido blanco (263 mg, 0,85 mmol), que se usó en la etapa 2.
Etapa 2: a partir de KCN (55 mg, 0,85 mmol) en DMF (1 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 7:3), como un sólido blanquecino (30 mg, 12%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,69 (s, 1H), 8,09 - 8,00 (m, 4H), 7,90 (s, 1H), 4,42 (s, 2H), 3,87 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 165,9, 162,0, 157,6, 147,8, 138,3, 129,7, 128,6, 125,8, 115,1, 111,2, 52,1, 25,9;
EMAR calc. para C14H10N3O3S [M-H]- 300,0448, hallado 300,0443.
Ejemplo preparativo 103
2-ciano-N-(4,5,6,7-tetrahidrobenzo[d1tiazol-2-il)acetamida (no se encuentra dentro del alcance de las reivindicaciones)
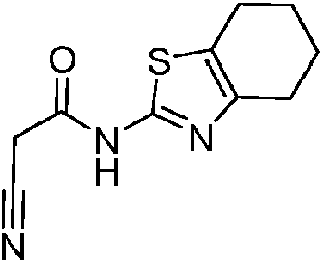
Se preparó el compuesto según el procedimiento general C1 a partir de 4,5,6,7-tetrahidrobenzo[d]tiazol-2-amina (200 mg, 1,3 mmol), cianoacetato de etilo (240 |al, 2,25 mmol) y NaOEt (al 21% en EtOH, 510 |al, 1,3 mmol). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1:1), como un sólido blanco (55 mg, 0,24 mmol, 2 0 %).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,18 (s, 1H), 3,98 (s, 2H), 2,67 -2,52 (m, 4H), 1,77 (q, J = 3,6 Hz, 4H);
13C-RMN (126 MHz, DMSO-CÍ6) 8 (ppm) 161,2, 144,3, 121,9, 115,3, 25,8, 22,8, 22,2;
EMAR calc. para C10H1qN3OS [M-H]- 220,0623, hallado 220,0624.
Ejemplo preparativo 104
2-ciano-N-(5-metil-4-feniltiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 con 5-metil-4-feniltiazol-2-amina (0,686 g, 3,60 mmol), cianoacetato de etilo (575 |al, 5,41 mmol) y NaOEt (al 21% en EtOH, 1,35 ml, 3,60 mmol) en EtOH (3 ml). Se calentó la mezcla a 55°C durante 3 h, luego se enfrió hasta 25°C, se recogió el precipitado mediante filtración y se lavó con EtOH frío (1 ml). Se obtuvo el producto como un sólido marrón pálido (0,736 g, 79%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,42 (s, 1H), 7,67 - 7,60 (m, 2H), 7,45 (dd, J = 7,7 Hz, 2H), 7,38 - 7,32 (m, 1H), 4,02 (s, 2H), 2,48 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,5, 153,3, 144,3, 134,6, 128,4, 127,9, 127,3, 121,7, 115,2, 25,8, 11,8; EMAR calc. para C13H10N3OS [M-H]- 256,0550, hallado 256,0552.
Ejemplo preparativo 105
N-(5-cloro-4-feniltiazol-2-il)-2-cianoacetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 5-cloro-4-feniltiazol-2-amina (100 mg, 0,48 mmol), cianoacetato de etilo (77 |al, 0,73 mmol) y NaOEt (al 21% en EtOH, 180 |al, 0,48 mmol). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1:1), como un sólido blanco (50 mg, 40%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,86 (s, 1H), 7,91 - 7,84 (m, 2H), 7,55 - 7,47 (m, 2H), 7,46 - 7,39 (m, 1H), 4,08 (s, 2H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 162,6, 153,5, 143,5, 132,3, 128,5, 128,5, 127,6, 114,9, 113,2, 25,8;
EMAR calc. para C12H7ClN3O3S [M-H]- 276,0004, hallado 276,0005.
Ejemplo preparativo 106
N-(5-bromo-4-feniltiazol-2-il)-2-cianoacetamida
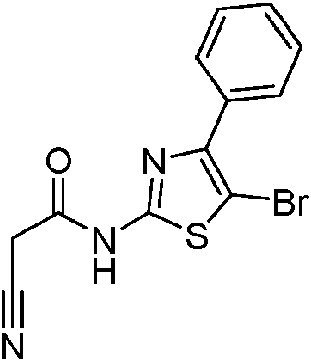
Se preparó el compuesto según el procedimiento general C1 a partir de 5-bromo-4-feniltiazol-2-amina (0,253 g,
0,99 mmol), cianoacetato de etilo (158 |l, 1,49 mmol) y NaOEt (al 21% en EtOH, 555 |l, 1,49 mmol) en EtOH (2 ml). Se calentó la mezcla a 55°C durante 6 h. Después de dos purificaciones mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1), se obtuvo el producto como un sólido amarillo pardo (0,059 g, 18%).
1H-RMN (500 MHz, DMSO-cfe) 8 (ppm) 7,84 - 7,79 (m, 2H), 7,54 - 7,46 (m, 3H), 2,80 (s, 2H);
13C-RMN (126 MHz, DMSO-cfe) 8 (ppm) 160,2, 158,0, 146,7, 133,0, 129,6, 129,1, 128,9, 126,7, 1 1 2 ,6 , 25,0;
EMAR calc. para C^HgBr^OS [M+H]+ 321,9644, hallado 321,9645.
Ejemplo preparativo 107
2-ciano-N-(4-(4-((trimetilsilil)etinil)fenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C2:
Etapa 1: a partir de 4-(4-((trimetilsilil)etinil)fenil)tiazol-2-amina (87 mg, 0,32 mmol), cloruro de cloroacetilo (54 mg, 38 |l, 0,48 mmol) y EtaN (50 mg, 6 8 |l, 0,48 mmol) en CH3CN (1,5 ml). Después del tratamiento final, se purificó la mezcla en bruto mediante cromatografía en columna (hexano:EtOAc; 7:3), se obtuvo la cloroacetamida correspondiente como un sólido blanco, que se usó en la etapa 2.
Etapa 2: a partir de KCN (33 mg, 0,48 mmol) en DMF anhidra (1 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 7:3), como un sólido blanquecino (40 mg, 40%).
1H-RMN (500 MHz, CDCh) 8 (ppm) 11,04 (s, 1H), 7,77 - 7,73 (m, 2H), 7,57 - 7,53 (m, 2H), 7,25 (s, 1H), 3,30 (s, 2H), 0,28 (s, 9H);
13C-RMN (126 MHz, CDCh) 8 (ppm) 159,6, 158,2, 149,5, 133,9, 132,9, 126,2, 123,7, 112,9, 109,9, 104,6, 96,3, 26,0, 0 ,1 ;
EMAR calc. para C17H1sN3OSS¡ [M+H]+ 340,0934, hallado 340,0928.
Ejemplo preparativo 108
2-ciano-N-(4-(4-etinilfenil)tiazol-2-il)acetamida

A una disolución de 2-ciano-N-(4-(4-((trimetilsilil)etinil)fenil)tiazol-2-il)acetamida (40 mg, 0,11 mmol) disuelta en MeOH (1 ml) se le añadió K2CO3 (65 mg, 0,44 mmol) y se agitó la mezcla a 25°C durante 48 h. Se adsorbió previamente la mezcla sobre gel de sílice y se purificó mediante cromatografía en columna (hexano:EtOAc; 2:1). Se aisló el producto como un sólido blanco (25 mg, 85%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,61 (s, 1H), 7,93 - 7,88 (m, 2H), 7,79 (s, 1H), 7,56 - 7,52 (m, 2H), 4,24 (s, 1H), 4,06 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,9, 157,5, 148,0, 134,3, 132,1, 125,8, 120,9, 115,1, 109,9, 83,3, 81,5, 25,9;
EMAR calc. para C14H10N3OS [M+H]+ 268,0539, hallado 268,0537.
Ejemplo preparativo 109
2-ciano-N-(4-(3-ciclopropil-4-metoxifenN)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(3-ciclopropil-4-metoxifenil)tiazol-2-amina (100 mg, 0,4 mmol), cianoacetato de etilo ( 68 mg, 65 ^l, 0,6 mmol) y Na (9 mg, 0,4 mmol) en EtOH (2 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 7:3), como un sólido blanco (70 mg, 55%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,53 (s, 1H), 7,65 (dd, J = 8,5, 2,2 Hz, 1H), 7,53 (s, 1H), 7,33 (d, J = 2,2 Hz, 1H), 6,99 (d, J = 8,5 Hz, 1H), 4,04 (s, 2H), 3,84 (s, 3H), 2,19 - 2,07 (m, 1H), 0,98 - 0,86 (m, 2H), 0,72 - 0,60 (m, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,7, 157,6, 157,0, 149,1, 131,3, 126,7, 123,8, 121,8, 115,1, 110,6, 106,4, 55,5, 25,8, 9,2, 7,8;
EMAR calc. para C16H16N3O3S [M+H]+ 314,0958, hallado 314,0955.
Ejemplo preparativo 110
N-(4-(ferc-butil)tiazol-2-iD-2-cianoacetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-terc-butiltiazol-2-ilamina (0,228 g, 1,46 mmol), cianoacetato de etilo (233 |al, 2,19 mmol) y NaOEt (al 21% en EtOH, 817 |al, 2,19 mmol) en EtOH (2 ml). Se calentó la mezcla a 50°C durante 21 h. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 2:1), como un sólido rosa pálido (0,242 g, 74%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,39 (s, 1H), 6,81 (s, 1H), 3,98 (s, 2H), 1,26 (s, 9H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 161,4, 160,3, 156,5, 115,2, 105,2, 34,1,29,7, 25,7;
EMAR calc. para C10H14N3OS [M+H]+ 224,0852, hallado 224,0852.
Ejemplo preparativo 111
N-(4-(5-bromotiofen-2-il)tiazol-2-il)-2-cianoacetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(5-bromotiofen-2-il)tiazol-2-amina (240 mg, 0,9l9mmol) en MeOH ( 6 ml), cianoacetato de etilo (0,147 ml, 1,37 mmol) y NaH (al 6 0 % en aceite mineral, 0,040 mg, 1,01 mmol); tiempo de reacción de 14 h a 60°C. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:1 a 0:1), como un sólido amarillo (300 mg, 0,914 mmol, 99%).
1H-RMN (300 MHz, DMSO-^) 5 (ppm) 12,7 (s, 1H), 7,6 (s, 1H), 7,4 (d, J = 3,9 Hz, 1H), 7,2 (d, J = 3,9 Hz, 1H), 4,0 (s, 2H);
EMAR calc. para C10H7BrNsOS2 [M+H]+ 329,9187, hallado 329,9191.
Ejemplo preparativo 112
N-(4-(4-(terc-butil)fenil)tiazol-2-il)-2-cianoacetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(4-(fe/'c-butil)fenil)tiazol-2-amina (626 mg, 2,694 mmol), cianoacetato de etilo (0,430 ml, 4,041 mmol) y NaOEt (al 21% en EtOH, 1,509 ml, 4,041 mmol) en EtOH ( 6 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (tolueno:EtOAc; de 10:1 a 4:1), como un sólido blanco (575 mg, 71%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,58 (s, 1H), 7,82 (d, J = 8,5 Hz, 2H), 7,60 (s, 1H), 7,45 (d, J = 8 , 6 Hz, 2H), 4,05 (s, 2H), 1,30 (s, 9H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,8, 157,2, 150,4, 149,0, 131,4, 125,5, 125,4, 115,1, 107,8, 34,3, 31,0, 25,9;
EMAR calc. para C16H18N3OS [M+H]+ 300,1165, hallado 300,1168.
Ejemplo preparativo 113
2-ciano-N-(4-(5-metiltiofen-2-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C2:
Etapa 1: a partir de 4-(5-metiltiofen-2-il)tiazol-2-amina (240 mg, 1,22 mmol), cloruro de cloroacetilo (205 mg, 145 |l, 1,84 mmol) y Et3N (186 mg, 255 |l, 1,84 mmol) en CH3CN (2,5 ml). Después del tratamiento final, se purificó la mezcla en bruto mediante cromatografía en columna (hexano:EtOAc; 7:3), la cloroacetamida correspondiente se obtuvo como un sólido blanco (331 mg), que se usó en la etapa 2.
Etapa 2: a partir de KCN (165 mg, 2,44 mmol) en DMF (2 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 7:3), como un sólido blanquecino (150 mg, 50%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,64 (s, 1H), 7,41 (s, 1H), 7,29 (d, J = 3,5 Hz, 1H), 6,79 (dd, J = 3,5, 1,3 Hz, 1H), 4,03 (s, 2H), 2,45 (d, J = 1,1 Hz, 3H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,3, 157,7, 144,5, 139,6, 136,2, 126,8, 124,3, 115,6, 106,5, 26,3, 15,5; EMAR calc. para C11H10N3 OS2 [M+H]+ 264,0260, hallado 264,0256.
Ejemplo preparativo 114
2-ciano-N-metil-N-(4-feniltiazol-2-il)acetamida
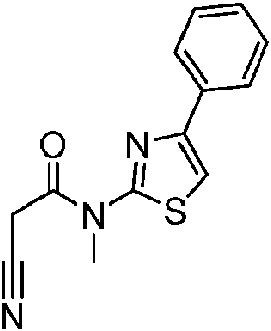
Se preparó el compuesto según el procedimiento general C2:
Etapa 1: a partir de N-metil-4-feniltiazol-2-amina (130 mg, 0,70 mmol), Et3N (70 mg, 0,1 ml, 0,70 mmol) y cloruro de cloroacetilo (120 mg, 85 |l, 1,05 mmol) en CH3CN (1,5 ml). Después del tratamiento final, se purificó la mezcla en bruto mediante cromatografía en columna (hexano:EtOAc; 7:3), la cloroacetamida correspondiente se obtuvo como un sólido blanco (187 mg, 0,70 mmol), que se usó en la etapa 2.
Etapa 2: a partir de KCN (90 mg, 1,4mmol) en DMF anhidra (1 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 7:3), como un sólido blanco (90 mg, 50%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 7,98 - 7,92 (m, 2H), 7,75 (s, 1H), 7,48 - 7,39 (m, 2H), 7,37 - 7,30 (m, 1H), 4,49 (s, 2H), 3,70 (s, 3H);
13C-RMN (126 MHz, DMSO-CÍ6) 8 (ppm) 164,2, 158,9, 148,0, 134,0, 128,7, 127,8, 125,7, 115,1, 110,1, 34,7, 26,7; EMAR calc. para C13H12N3OS [M+H]+ 258,0696, hallado 258,0700.
Ejemplo preparativo 115
2-ciano-N-(4-(6-metilpiridin-3-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(6-metilpiridin-3-il)tiazol-2-amina (320 mg, 1,67 mmol), cianoacetato de etilo (280 mg, 270 |l, 2,5 mmol) y NaH (al 60% en aceite mineral, 40 mg, 1,67 mmol) en THF (3 ml) y MeOH (0,1 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 7:3 a 0 :1 ), como un sólido blanco (320 mg, 75%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,64 (s, 1H), 8,97 (d, J = 2,3 Hz, 1H), 8,11 (dd, J = 8,1,2,3 Hz, 1H), 7,77 (s,
1H), 7,32 (d, J = 8,2 Hz, 1H), 4,06 (s, 2H), 2,49 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 162,5, 158,3, 157,7, 146,9, 146,7, 133,7, 127,6, 123,6, 115,6, 109,7, 26,4, 24,3;
EMAR calc. para C12H11N4OS [M+H]+ 259,0648, hallado 259,0644.
Ejemplo preparativo 116
2-ciano-N-(4-fenil-5-(tetrahidro-2H-piran-4-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C2:
Etapa 1: 4-(4-metiltiofen-2-il)tiazol-2-amina (65 mg, 0,24 mmol), cloruro de cloroacetilo (40 mg, 30 |l, 0,5 mmol) y Et3N (24 mg, 34 |l, 0,24 mmol) en CH3CN (2,5 ml). Después del tratamiento final, se purificó la mezcla en bruto mediante cromatografía en columna (hexano:EtOAc; 7:3), la cloroacetamida correspondiente se obtuvo como un sólido blanco (80 mg), que se usó en la etapa 2.
Etapa 2: a partir de KCN (31 mg, 0,48 mmol) en DMF (2 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 2:3), como un sólido blanquecino (43 mg, 55%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,46 (s, 1H), 7,57 - 7,53 (m, 2H), 7,49 - 7,44 (m, 2H), 7,41 - 7,36 (m, 1H), 4,02 (s, 2H), 3,96 - 3,85 (m, 2H), 3,37 (td, J = 11,8, 1,9 Hz, 2H), 3,33 -3,21 (m, 1H), 1,90 - 1,83 (m, 2H), 1,74 - 1,57 (m, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,5, 153,9, 143,9, 134,9, 132,7, 128,5, 128,3, 127,7, 115,1, 67,0, 35,4, 33,7, 25,8;
EMAR calc. para C ^ H ^ N ^ S [M+H]+ 328,1114, hallado 328,1109.
Ejemplo preparativo 117
2-ciano-N-(4-(3-fluorofenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C1 a partir de 4-(3-fluorofenil)tiazol-2-amina (150 mg, 0,772 mmol), cianoacetato de etilo (174 |l, 1,544 mmol) y NaH (al 60% en aceite mineral, 34 mg, 0,849 mmol) en MeOH (1 ml) y THF (3 ml). Se agitó la mezcla a 50°C durante 24 h. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 10:1 a 2:1), como un sólido blanco (126 mg, 63%).
1H-RMN (500 MHz, DMSO-CÍ6) 8 (ppm) 12,61 (s, 1H), 7,82 (s, 1H), 7,74 (d, J = 7,9 Hz, 1H), 7,68 (ddd, J = 10,7, 2,7, 1,5 Hz, 1H), 7,53 - 7,42 (m, 1H), 7,22 - 7,07 (m, 1H), 4,06 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,5 (d, J = 243,0), 162,0, 157,4, 147,7, 136,4 (d, J = 8,2 Hz), 130,8 (d, J = 8 , 8 Hz), 121,7, 115,1, 114,6 (d, J = 21,2 Hz), 112,2 (d, J = 23,4 Hz), 110,0, 25,9;
19F-RMN (471 MHz, DMSO-^) 8 (ppm) -112,94;
EMAR calc. para C12H9FN3OS [M+H]+ 262,0445, hallado 262,0444.
Ejemplo preparativo 118
2-ciano-N-(4-fenil-5-(pirazin-2-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-fenil-5-(pirazin-2-il)tiazol-2-amina (140 mg, 0,55 mmol), cianoacetato de etilo (95 mg, 90 |l, 0,83 mmol) y NaH (al 60% en aceite mineral, 22 mg, 0,55 mmol) en THF (1 ml) y MeOH (0,1 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (CH2Cl2:EtOAc; 2:1), como un sólido amarillo (77 mg, 45%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 12,78 (s, 1H), 8 , 6 8 - 8,61 (m, 1H), 8,46 (d, J = 2,5 Hz, 1H), 8,29 (d, J = 1,6 Hz, 1H), 7,55 - 7,50 (m, 2H), 7,47 (q, J = 3,2, 2,8 Hz, 3H), 4,10 (s, 2H);
13C-RMN (126 MHz, DMSO-cfe) 5 (ppm) 162,9, 158,4, 148,6, 148,0, 144,9, 142,9, 142,8, 135,1, 129,5, 129,4, 129,3, 124,8, 115,6, 26,5;
EMAR calc. para C16H12N5OS [M+H]+ 322,0757, hallado 322,0759.
Ejemplo preparativo 119
2-ciano-N-(4-(3-metilpiridin-2-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(3-metilpiridin-2-il)tiazol-2-amina (290 mg, 1,52 mmol), cianoacetato de etilo (270 mg, 250 |l, 2,3 mmol) y NaH (al 60% en aceite mineral, 61 mg, 1,52 mmol) en THF (3 ml) y MeOH (0,1 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (CH2Cl2:MeOH; 10:1), como un sólido naranja (275 mg, 70%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 12,48 (s, 1H), 8,44 (dd, J = 4,7, 1,6 Hz, 1H), 7,69 - 7,68 (m, 1H), 7,67 (s, 1H), 7,27 (dd, J = 7,7, 4,7 Hz, 1H), 4,07 (s, 2H), 2,55 (s, 3H);
13C-RMN (126 MHz, DMSO-cfe) 5 (ppm) 161,9, 156,3, 150,9, 150,2, 146,6, 139,3, 130,9, 122,7, 115,2, 114,1, 25,9, 2 0 ,1 ;
EMAR calc. para C12H11N4OS [M+H]+ 259,0648, hallado 259,0651.
Ejemplo preparativo 120
2-ciano-N-(4-(6-metoxipiridin-3-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(6-metoxipiridin-3-il)tiazol-2-amina (270 mg, 1,3mmol), cianoacetato de etilo (220 mg, 200 |l, 1,95 mmol) y NaH (al 60% en aceite mineral, 60 mg, 1,43 mmol) en THF (2 ml) y MeOH (0,1 ml). Se vertió la mezcla de reacción en una disolución acuosa saturada de NH4Cl (10 ml). Se recogió el precipitado mediante filtración, se lavó con agua (10 ml) y Et2O (5 ml). Se obtuvo el producto, secado a vacío, como un sólido blanco (270 mg, 75%).
1H-RMN (500 MHz, DMSO-cfe) 8 (ppm) 12,61 (s, 1H), 8,69 (d, J = 2,5 Hz, 1H), 8,16 (dd, J = 8 ,6 , 2,5 Hz, 1H), 7,66 (s, 1H), 6,90 (d, J = 8 , 6 Hz, 1H), 4,06 (s, 2H), 3,89 (s, 3H);
13C-RMN (126 MHz, DMSO-CÍ6) 8 (ppm) 163,1, 161,9, 157,6, 146,2, 144,1, 136,5, 123,9, 115,1, 110,6, 107,8, 53,3, 25,9;
EMAR calc. para C12H11N4O2S [M+H]+ 275,0597, hallado 275,0594.
Ejemplo preparativo 121
2-ciano-N-(4-(3-metoxipiridin-2-il)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(3-metoxipiridin-2-il)tiazol-2-amina (235 mg, 1,13 mmol), cianoacetato de etilo (190 mg, 180 |l, 1,7 mmol) y NaH (al 60% en aceite mineral, 45 mg, 1,13 mmol) en THF (3 ml) y MeOH (0,1 ml). Se vertió la mezcla en una disolución acuosa saturada de NH4Cl (10 ml). Se recogió el precipitado mediante filtración, se lavó con agua (10 ml) y Et2O (5 ml). Se secó el producto a vacío y se obtuvo como un sólido blanco (190 mg, 60%).
1H-RMN (500 MHz, DMSO-CÍ6) 8 (ppm) 12,72 (s, 1H), 8,21 (dd, J = 4,5, 1,3 Hz, 1H), 7,78 (s, 1H), 7,56 (dd, J = 8,4, 1,3 Hz, 1H), 7,36 (dd, J = 8,4, 4,5 Hz, 1H), 4,03 (s, 2H), 3,91 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,4, 156,5, 154,0, 150,0, 146,9, 141,1, 128,1, 124,1, 119,8, 115,5, 56,1, 26,4;
EMAR calc. para C12H11N4O2S [M+H]+ 275,0597, hallado 275,0594.
Ejemplo preparativo 122
2-ciano-N-(4-(piridin-2-il)tiazol-2-il)acetamida
Se preparó el compuesto según el procedimiento general C3 a partir de 4-(piridin-2-il)tiazol-2-amina (440 mg, 2,2 mmol), cianoacetato de etilo (370 mg, 350 ^l, 3,3 mmol) y NaH (al 60% en aceite mineral, 90 mg, 2,2 mmol) en THF (3 ml) y MeOH (0,1 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1:1), como un sólido blanco (385 mg, 70%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 12,63 (s, 1H), 8,61 (dt, J = 4,66, 1,47 Hz, 1H), 7,95 - 7,86 (m, 3H), 7,36 - 7,32 (m, 1H), 4,07 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 161,9, 157,5, 151,8, 149,5, 149,1, 137,3, 122,9, 119,9, 115,1, 112,1,25,9; EMAR calc. para C11H9 N4OS [M+H]+ 245,0492, hallado 245,0497.
Ejemplo preparativo 123
2-ciano-N-(4-(2-fluoro-4-metoxifenil)tiazol-2-il)acetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(4-(metilsulfonil)fenil)tiazol-2-amina (300 mg, 1,18 mmol), cianoacetato de etilo (200 mg, 200 ^l, 1,77 mmol) y NaH (al 60% en aceite mineral, 52 mg, 1,3 mmol) en THF (3 ml) y MeOH (0,1 ml). Se vertió la mezcla de reacción en una disolución acuosa saturada de NH4Cl (10 ml). Se recogió el precipitado mediante filtración y se lavó con agua (10 ml) y Et2O (5 ml) y se secó a vacío. Se obtuvo el producto como un sólido blanco (320 mg, 85%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 2,61 (s, 1H), 8,16 - 8,13 (m, 2H), 8,00 - 7,97 (m, 2H), 7,97 (s, 1H), 4,08 (s, 2H), 3,24 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 8 (ppm) 162,1, 157,8, 147,2, 139,6, 138,5, 127,6, 126,2, 115,1, 111,8, 43,5, 25,9; EMAR calc. para C13H12N3O3S2 [M+H]+ 322,0315, hallado 322,0318.
Ejemplo preparativo 124
2-ciano-N-(5-feniltiazol-2-il)acetamida (no se encuentra dentro del alcance de las reivindicaciones)

Se preparó el compuesto según el procedimiento general C3 a partir de 5-feniltiazol-2-amina (250 mg, 1,41 mmol), cianoacetato de etilo (191 mg, 0,18 ml, 1,69 mmol) y NaH (al 60% en aceite mineral, 56 mg, 1,74 mmol) en THF (3 ml) y MeOH (7 ml). Se vertió la mezcla de reacción en una disolución acuosa saturada de NH4Cl (20 ml), se recogió el precipitado mediante filtración, se lavó con agua (15 ml) y EtOAc (25 ml) y se secó a vacío. Se obtuvo el producto como un sólido blanco (205 mg, 60%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 7,89 (s, 1H), 7,72-7,53 (m, 2H), 7,48-7,35 (m, 2H), 7,38-7,26 (m, 1H), 4,05 (s, 2H);
EMAR calc. para C12H8 N3OS [M-H]- 242,0394, hallado [M+H]+ 242,0395.
Ejemplo preparativo 125
N-(4-(4-(ferc-butil)-2,6-dimetilfenil)tiazol-2-il)-2-cianoacetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(4-(ferc-butil)-2,6-dimetilfenil)tiazol-2-amina ( 8 8 mg, 0,34 mmol), cianoacetato de etilo (38 mg, 0,04 ml, 0,34 mmol) y NaH (13 mg, 0,34 mmol) en THF (1,5 ml) y MeOH (0,1 ml). Se vertió la mezcla de reacción en una disolución acuosa saturada de NH4Cl (10 ml), se recogió el precipitado mediante filtración, se lavó con agua (3 ml) y EtOAc (5 ml) y se secó a vacío. Se obtuvo el producto como un sólido blanco (82 mg, 75%).
1H-RMN (500 MHz, DMSO-^) 8 (ppm) 7,22 (s, 2H), 6 , 8 6 (s, 1H), 2,44 (s, 2H), 2,13 (s, 6 H), 1,36 (s, 9H);
13C-RMN (126 MHz, CDCls) 8 (ppm) 160,6, 159,2, 153,0, 147,5, 137,6, 131,2, 125,4, 112,8, 112,4, 34,8, 31,4, 24,3, 20,9, 20,8;
EMAR calc. para C18H22N3SO [M+H]+ 328,1478, hallado 328,1481.
Ejemplo preparativo 126
N-(4-(3-(terc-butil)fenil)tiazol-2-il)-2-cianoacetamida

Se preparó el compuesto según el procedimiento general C3 a partir de 4-(3-(ferc-butil)fenil)tiazol-2-amina (265 mg, 1,13 mmol, cianoacetato de etilo (128 mg, 0,12 ml, 1,13 mmol) y NaH (al 60% en aceite mineral, 45 mg, 1,13 mmol) en THF (3 ml) y MeOH (3 ml). Se vertió la mezcla de reacción en una disolución acuosa saturada de NH4Cl (20 ml), se recogió el precipitado mediante filtración, se lavó con agua (5 ml) y EtOAc (10 ml) y se secó a vacío. Se obtuvo el producto como un sólido blanco (277 mg, 82%).
1H-RMN (500 MHz, CDCls) 8 (ppm) 7,81 (t, J = 1,9 Hz, 1H), 7,58 (dt, J = 7,2, 1,6 Hz, 1H), 7,49-7,36 (m, 2H), 7,19 (s, 1H), 3,10 (s, 2H), 1,37 (s, 9H);
Ejemplo preparativo 127
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(4-metoxifenil)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-(4-metoxifenil)tiazol-2-il)acetamida (41 mg, 0,15 mmol), 3,4-dihidroxibenzaldehído (20 mg, 0,143 mmol) y NEt3 (21 |l, 0,15 mmol) en EtOH
(1 ml); el tiempo de reacción fue de 3 h. Se evaporó el disolvente a vacío y se agitó el sólido resultante en CH3CN (2 ml) a 25°C durante 48 h. Se recogió el sólido mediante filtración, se lavó con dietil éter (3 x 1 ml) y se secó a vacío. Se obtuvo el producto como un sólido naranja (30 mg, 51%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 8,28 (s, 1H), 7,86 (d, J = 8,7 Hz, 2H), 7,65 - 7,56 (m, 1H), 7,49 (s, 1H), 7,37 (d, J = 8,4 Hz, 1H), 7,00 (d, J = 8,7 Hz, 2H), 6,92 (d, J = 8,4 Hz, 1H), 3,80 (s, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,4, 159,0, 151,7, 148,3, 145,8, 127,1, 126,8, 125,9, 123,1, 116,7, 116,4, 116,1, 114,1, 106,5, 55,1;
EMAR calc. para C20H14N3O4S [M-H]- 392,0711, hallado 392,0712.
Ejemplo preparativo 128
(E)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(naftalen-2-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-(naftalen-2-il)tiazol-2-il)acetamida (56 mg, 0,191 mmol), 3,4-dihidroxibenzaldehído (25 mg, 0,181 mmol) y NEt3 (27 |al, 0,191 mmol) en EtOH (1 ml); el tiempo de reacción fue de 4 h. Se evaporó el disolvente a vacío y se agitó el residuo en una mezcla de CH2Cl2 y CH3CN (1,5 ml 1,5 ml) a 25°C durante 2 h. Se recogió el sólido mediante filtración y se secó a vacío. Se obtuvo el producto como un sólido marrón anaranjado (34 mg, 43%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 8,49 (s, 1H), 8,33 (s, 1H), 8,08 (d, J = 8,5, 1,7 Hz, 1H), 8,04 - 7,90 (m, 3H), 7,83 (s, 1H), 7,63 (d, J = 2,4 Hz, 1H), 7,58 - 7,49 (m, 1H), 7,40 (dd, J = 8,3, 2,4 Hz, 1H), 6,94 (d, J = 8,2 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,1, 152,0 , 151,6 ,149,0, 145,8, 133,1, 132,5, 128,3, 128,1, 127,6, 126,5, 126,2, 125,9, 124,3, 124,0, 123,1, 116,5, 116,1, 109,3;
EMAR calc. para C23H14N3O3S [M-H]- 412,0761, hallado 412,0761.
Ejemplo preparativo 129
(£)-N-(4-([1,1’-bifenil)-4-il)tiazol-2-il)-2-ciano-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de N-(4-([1,1’-bifenil]-4-il)tiazol-2-il)-2-cianoacetamida (58 mg, 0,182 mmol), 3,4-dihidroxibenzaldehído (24 mg, 0,173 mmol) y piperidina (3 |al, 0,027 mmol) en CH3CN (2 ml); el tiempo de reacción fue de 2 h. Tratamiento final 1 del procedimiento general D2. Se obtuvo el producto como un sólido amarillo (48 mg, 60%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 8,30 (s, 1H), 8,03 (d, J = 8,5 Hz, 3H), 7,80 - 7,67 (m, 5H), 7,62 (d, J = 2,3 Hz, 1H), 7,53 - 7,44 (m, 2H), 7,41 - 7,34 (m, 2H), 6,95 - 6,87 (m, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,5, 151,6, 145,9, 139,6, 139,3, 133,2, 128,9, 127,5, 126,9, 126,5, 126,3, 116,8, 116,2, 116,1, 108,6;
EMAR calc. para C25H16N3O3S [M-H]- 438,0918, hallado 438,0919.
Ejemplo preparativo 130
(£)-N-(4-(4-bromofenil)tiazol-2-il)-2-ciano-3-(3,5-dicloro-4-hidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de N-(4-(4-bromofenil)tiazol-2-il)-2-cianoacetamida (50 mg, 0,16 mmol), 3,5-dicloro-4-hidroxibenzaldehído (28 mg, 0,15 mmol) y NEt3 (20 |al, 0,15 mmol) en EtOH (3 ml); el tiempo de reacción fue de 4 h. Tratamiento final 1 del procedimiento general D1. Se obtuvo el producto como un sólido amarillo (15 mg, 20%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,39 (s, 1H), 8,18 (s, 1H), 7,96 (s, 2H), 7,91 - 7,86 (m, 2H), 7,73 (s, 1H), 7,67 -7,61 (m, 2H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,5, 158,8, 149,1, 147,1, 133,3, 131,6, 131,4, 127,7, 124,0, 120,9, 117,4, 109,3, 103,0;
EMAR calc. para C1gH14BrN3O3S [M-H]- 493,8958, hallado 493,8960.
Ejemplo preparativo 131
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(3-hidroxi-4-metoxifenil)tiazol-2-il)acrilamida
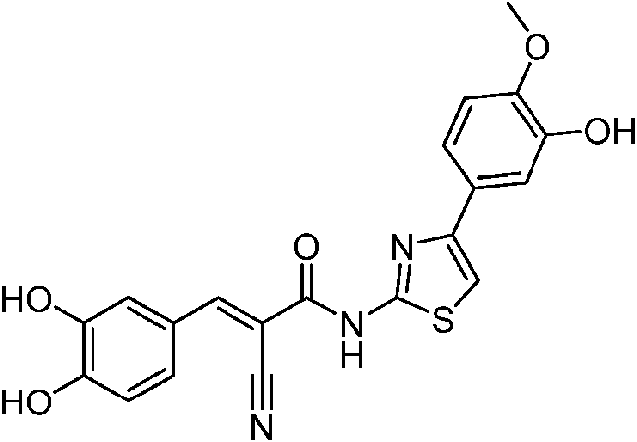
Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-(3-hidroxi-4-metoxifenil)tiazol-2-il)acetamida (41 mg, 0,14 mmol), 3,4-dihidroxibenzaldehído (18 mg, 0,13 mmol) y NEt3 (20 |al, 0,14 mmol) en EtOH (1,5 ml); el tiempo de reacción fue de 4 h. Tratamiento final 1 del procedimiento general D1. Se obtuvo el producto como un sólido amarillo (25 mg, 45%).
1H-RMN (500 MHz, DMSOd) 5 (ppm) 12,55 (s, 1H), 9,81 (s, 2H), 9,03 (s, 1H), 8,29 (s, 1H), 7,61 (d, J = 2,3 Hz, 1H), 7,41 (s, 1H), 7,38 (dd, J = 8,5, 2,2 Hz, 1H), 7,37 - 7,31 (m, 2H), 6,97 (d, J = 9,0 Hz, 1H), 6,93 (d, J = 8,3 Hz, 1H), 3,81 (s, 3H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 151,9, 151,5, 147,7, 146,5, 145,8, 125,8, 123,2, 117,0, 116,5, 116,1, 113,2, 112,2, 106,5, 55,6;
EMAR calc. para C20H14N3O5S [M-H]- 408,0660, hallado 408,0659.
Ejemplo preparativo 132
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(2-hidroxipropan-2-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-(2-hidroxipropan-2-il)tiazol-2-il)acetamida (59 mg, 0,26 mmol), 3,4-dihidroxibenzaldehído (32 mg, 0,23 mmol) y NEt3 (36 |l, 0,26 mmol) en EtOH (1,5 ml); el tiempo de reacción fue de 4 h. Tratamiento final 1 del procedimiento general D1. Se obtuvo el producto como un sólido amarillo (33 mg, 40%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,47 (s, 1H), 10,22 (s, 1H), 9,61 (s, 1H), 8,25 (s, 1H), 7,59 (d, J = 2,1 Hz, 1H), 7,35 (d, J = 8,4 Hz, 1H), 6,91 (d, J = 8,3 Hz, 2H), 5,05 (s, 1H), 1,45 (s, 6 H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 151,6, 151,2, 145,7, 125,6, 123,3, 116,5, 116,0, 70,0, 30,2;
EMAR calc. para C16H14N3O4S [M+H]+ 344,0711, hallado 344,0711.
Ejemplo preparativo 133
(£)-2-ciano-3-(3,5-dicloro-4-hidroxifenil)-N-(4-(p-tolil)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-(p-tolil)tiazol-2-il)acetamida (50 mg, 0,19 mmol), 3,5-dicloro-4-hidroxibenzaldehído (35 mg, 0,18 mmol) y NEt3 (26 |l, 0,19 mmol) en EtOH (3 ml); el tiempo de reacción fue de 4 h. Tratamiento final 1 del procedimiento general D1. Se obtuvo el producto como un sólido rojo pardo (35 mg, 43%).
1H-RMN (500 MHz, DMSO-de) 5 fppm) 12,24 (s, 1H), 8,14 (s, 1H), 7,95 (s, 2H), 7,86 - 7,77 (m, 2H), 7,55 (s, 1H), 7,25 (d, J = 7,9 Hz, 2H), 2,33 (s, 3H);
13C-RMN (126 MHz, DMSO-de) 5 fppm) 197,3, 162,4, 148,9, 137,1, 129,2, 125,6, 125,2, 124,2, 117,8, 107,4, 20,7; EMAR calc. para C20H12Cl2N3O2S [M-H]- 428,0033, hallado 428,0035.
Ejemplo preparativo 134
(£)-N-(4-(3-bromofenil)tiazol-2-il)-2-ciano-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de N-(4-(3-bromofenil)tiazol-2-il)-2-cianoacetamida (80 mg, 0,24 mmol), 3,4-dihidroxibenzaldehído (31 mg, 0,23 mmol) y NEt3 (33 |l, 0,24 mmol) en EtOH (3 ml); el tiempo de reacción fue de 4 h. Tratamiento final 2 del procedimiento general D1. Se purificó el
residuo mediante cromatografía en columna (hexano:EtOAc; 2:1). Se sometió a sonicación el producto semipuro así obtenido en CH3CN (4 ml) y se recogió el sólido mediante filtración. Se obtuvo el producto como un sólido amarillo (40 mg, 40%).
1H-RMN (500 MHz, DMSO-da) 5 (ppm) 12,67 (s, 1H), 10,29 (s, 1H), 9,66 (s, 1H), 8,32 (s, 1H), 8,17 - 8,15 (m, 1H), 7,97 - 7,92 (m, 1H), 7,85 (s, 1H), 7,62 (d, J = 2,3 Hz, 1H), 7,57 - 7,48 (m, 1H), 7,44 - 7,39 (m, 1H), 7,40 - 7,36 (m, 1H), 6,94 (d, J = 8,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-da) 5 (ppm) 161,9, 158,2, 152,2, 151,6, 147,4, 145,8, 136,4, 130,9, 130,4, 128,4, 125,9, 124,6, 123,1, 122,2, 116,5, 116,3, 116,1, 110,2, 99,5;
EMAR calc. para C1gH11BrN3OsS [M-H]- 441,9691, hallado 441,9689.
Ejemplo preparativo 135
(£)-N-(4-(2-bromofenil)tiazol-2-il)-2-ciano-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de N-(4-(2-bromofenil)tiazol-2-il)-2-cianoacetamida (100 mg, 0,3 mmol), 3,4-dihidroxibenzaldehído (41 mg, 0,28 mmol) y NEt3 (42 |l, 0,3 mmol) en EtOH (3 ml); el tiempo de reacción fue de 4 h. Tratamiento final 2 de procedimiento general D1. Se purificó el residuo mediante cromatografía en columna (tolueno:EtOAc; 1:1). Se sometió a sonicación el producto semipuro resultante en CH3CN (4 ml) y se recogió el sólido mediante filtración. Se obtuvo el producto como un sólido amarillo (40 mg, 30%).
1H-RMN (500 MHz, DMSO-da) 5 (ppm) 12,70 (s, 1H), 10,27 (s, 1H), 9,65 (s, 1H), 8,30 (s, 1H), 7,77 - 7,71 (m, 2H), 7,61 (d, J = 2,2 Hz, 2H), 7,49 - 7,46 (m, 1H), 7,37 (dd, J = 8,4, 2,3 Hz, 1H), 7,36 - 7,29 (m, 1H), 6,93 (d, J = 8,2 Hz, 1H);
13C-RMN (126 MHz, DMSO-da) 5 (ppm) 161,8, 152,1, 151,5, 145,8, 133,4, 131,5, 129,8, 127,7, 125,8, 123,1, 121,2, 116,5, 116,4, 116,1, 113,1;
EMAR calc. para C19 HnBrN3O3S [M-H]- 441,9691, hallado 441,9692.
Ejemplo preparativo 136
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(3-(trifluorometoxi)fenil)tiazol-2-il)acrilamida
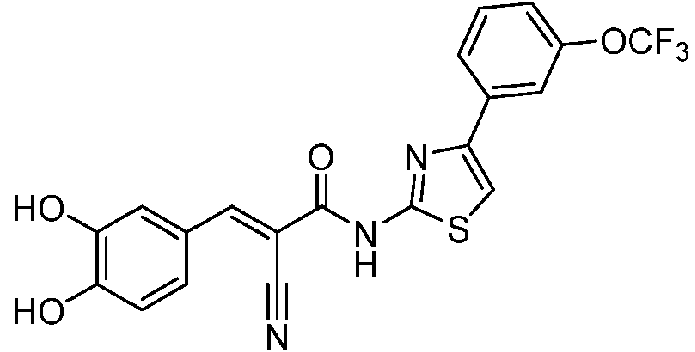
Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(3-(trifluorometoxi)fenil)tiazol-2-il)acetamida (70 mg, 0,22 mmol), 3,4-dihidroxibenzaldehído (30 mg, 0,22 mmol) y piperidina (2,0 |l, 0,02 mmol) en CH2Cl2 ( 2 ml); el tiempo de reacción fue de 4 h a reflujo y luego de 16 h a 25°C. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 45:55:0,05 a 10:90:0,05), como un sólido amarillo (60 mg, 60%).
1H-RMN (500 MHz, DMSO-da) 5 (ppm) 12,68 (s, 1H), 10,29 (s, 1H), 9,67 (s, 1H), 8,32 (s, 1H), 8,01 - 7,95 (m, 1H), 7.93 - 7,87 (m, 2H), 7,62 (d, J = 2,2 Hz, 1H), 7,60 - 7,58 (m, 1H), 7,39 (dd, J = 8,4, 2,2 Hz, 1H), 7,37 - 7,31 (m, 1H), 6.94 (d, J = 8,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-da) 5 (ppm) 162,0, 158,4, 152,2, 151,6, 148,9, 147,4, 145,8, 136,4, 130,8, 128,7, 125,9, 124,6, 123,1, 120,2, 120,1 (q, J = 256,4 Hz), 117,9, 116,6, 116,4, 116,9, 110,5;
19F-RMN (471 MHz, DMSO-da) 5 (ppm) -56,61;
EMAR calc. para C20H11F3N3O4S [M-H]- 446,0428, hallado 446,0428.
Ejemplo preparativo 137
(E)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(4-fenoxifenil)tiazol-2-il)acrilamida
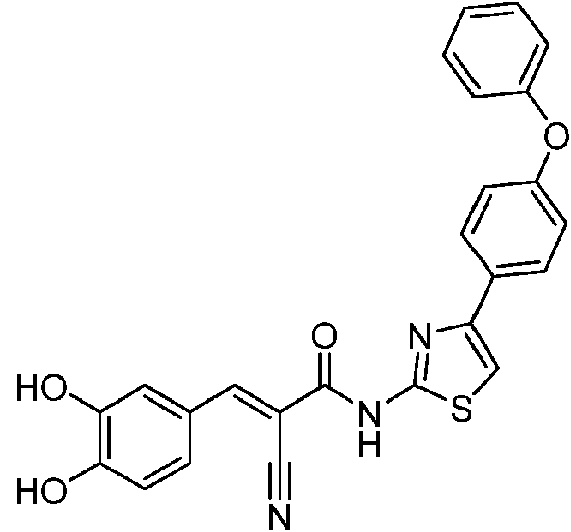
Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(4-fenoxifenil)tiazol-2-il)acetamida ( 68 mg, 0,2 mmol), 3,4-dihidroxibenzaldehído (28 mg, 0,2 mmol) y piperidina (2,0 |al, 0,02 mmol) en CH2Cl2 (2 ml); el tiempo de reacción fue de 4 h a reflujo y luego de 16 h a 25°C. Se recogió el precipitado mediante filtración y se lavó con CH2Cl2 (2 ml). Se mezcló el sólido con una disolución acuosa saturada de NH4Cl (15 ml) y se extrajo la mezcla con EtOAc (3 x 15 ml). Se secaron los extractos orgánicos combinados sobre MgSO4 , se filtraron y se evaporó el disolvente a vacío. Se obtuvo el producto como un sólido amarillo (45 mg, 40%).
1H-RMN (500 MHz, DMSO-da) 5 (ppm) 12,67 (s, 1H), 10,24 (s, 1H), 9,66 (s, 1H), 8,31 (s, 1H), 7,97 - 7,95 (m, 2H), 7,68 - 7,56 (m, 3H), 7,48-7,41 (m, 1H), 7,39 (dd, J = 8,5, 2,2 Hz, 1H), 7,22 - 7,16 (m, 1H), 7,14 -7,01 (m, 4H), 6,94 (d, J = 8,4 Hz, 1H);
13C-RMN (126 MHz, DMSO-da) 5 (ppm) 156,6, 156,3, 156,0, 155,9, 151,5, 145,8, 132,8, 130,1, 127,4, 125,7, 123,7, 123,1, 121,0, 119,0, 118,8, 118,5, 116,5, 116,9, 116,1, 115,3, 107,8;
EMAR calc. para C25H16N3O4S [M-H]-454,0867, hallado 454,0868.
Ejemplo preparativo 138
(E)-N-(4-(benzofuran-2-il)tiazol-2-il)-2-ciano-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de N-(4-(benzofuran-2-il)tiazol-2-il)-2-cianoacetamida (53 mg, 0,187 mmol), 3,4-dihidroxibenzaldehído (25 mg, 0,183 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (25 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:1 a 0:1) seguido de cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 10:90:0,05), como un sólido naranja (50 mg, 6 6 %).
1H-RMN (500 MHz, DMSO-da) 5 (ppm) 8,35 - 8,28 (m, 1H), 7,74 - 7,66 (m, 2H), 7,65 - 7,59 (m, 2H), 7,40 - 7,36 (m, 1H), 7,36 -7,31 (m, 1H), 7,31 -7,26 (m, 1H), 7,18 (s, 1H), 6,93 (d, J = 8,4 Hz, 1H);
13C-RMN (126 MHz, DMSO-da) 5 (ppm) 162,6, 154,1, 151,6, 145,8, 140,4, 128,4, 125,9, 124,7, 123,3, 123,1, 121,4,
116,6, 116,4, 116,1, 111,0, 110,8, 102,4;
EMAR calc. para C21H12N3O4S [M-H]- 402,0554, hallado 402,0552.
Ejemplo preparativo 139
(E)-N-(4-((1S,3s)-adamantan-1-iDtiazol-2-il)-2-ciano-3-(3,4-dihidroxifeni r )acrilamida (no se encuentra dentro del alcance de las reivindicaciones)

Se preparó el compuesto según el procedimiento general D1 a partir de N-(4-adamantan-1-il)tiazol-2-il)-2-cianoacetamida (65 mg, 0,216 mmol), 3,4-dihidroxibenzaldehído (28 mg, 0,205 mmol) y NEt3 (30 ^l, 0,216 mmol) en EtOH (2 ml). El tiempo de reacción fue de 5 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1) seguido de cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 10:90:0,05). Se obtuvo el producto como un sólido amarillo pardo (17 mg, 19%).
1H-RMN (500 MHz, DMSO-de) 5 fppm) 8,18 (s, 1H), 7,58 (d, J = 2,3 Hz, 1H), 7,32 (dd, J = 8,4, 2,3 Hz, 1H), 6,85 (s, 1H), 6,67 (s, 1H), 2,04 (s, 3H), 1,91 (d, J = 3,7 Hz, 6 H), 1,79 - 1, 68 (m, 6 H);
EMAR calc. para C23H22N3O3S [M-H]- 420,1387, hallado 420,1385.
Ejemplo preparativo 140
(£)-2-ciano-N-(5-ciclohexil-4-(4-(trifluorometoxi)fenil)tiazol-2-il)-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(5-ciclohexil-4-(4-(trifluorometoxi)fenil)tiazol-2-il)acetamida (70 mg, 0,17 mmol), 3,4-dihidroxibenzaldehído (25 mg, 0,17 mmol) y piperidina (2,0 |al, 0,02 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 4 h a reflujo y luego de 16 h a 25°C. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 45:55:0,05 a 10:90:0,05), como un sólido naranja (55 mg, 60%).
1H-RMN (500 MHz, DMSO-da) 512,52 (s, 1H), 10,20 (s, 1H), 9,97 - 9,47 (m, 1H), 8,25 (s, 1H), 7,71 - 7,66 (m, 2H), 7,59 (d, J = 2,2 Hz, 1H), 7,47 (d, J = 8,3 Hz, 2H), 7,38 - 7,33 (m, 1H), 6,92 (d, J = 8,3 Hz, 1H), 3,09 - 2,87 (m, 1H), 2,01 - 1,92 (m, 2H), 1,83 - 1,75 (m, 2H), 1,73 - 1,63 (m, 1H), 1,52 - 1,38 (m, 2H), 1,39 - 1,20 (m, 3H);
13C-RMN (126 MHz, DMSO-da) 5151,8, 151,5, 145,8, 142,0, 134,4, 130,2, 125,8, 123,1, 122,8, 122,7, 122,7, 120,9, 120,1 (q, J = 256,6 Hz) 117,0, 116,5, 116,0, 36,3, 35,7, 26,0, 25,1;
EMAR calc. para C26H23F3N3O4S [M+H]+ 530,1356, hallado 530,1350.
Ejemplo preparativo 141
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4,5-difeniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4,5-difeniltiazol-2-il)acetamida (53 mg, 0,166 mmol), 3,4-dihidroxibenzaldehído (22 mg, 0,158 mmol) y NEt3 (23 |al, 0,166 mmol) en EtOH (1 ml); el tiempo de reacción fue de 2 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 0:1) seguido de cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 70:30:0,05 a 10:90:0,05). Se obtuvo el producto como un sólido amarillo (21 mg, 29%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,70 (s, 1H), 10,19 (s, 1H), 9,67 (s, 1H), 8,31 (s, 1H), 7,62 (d, J = 2,3 Hz, 1H), 7,49 - 7,44 (m, 2H), 7,41 - 7,30 (m, 9H), 6,93 (d, J = 8,2 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 152,0, 151,6, 145,8, 131,6, 129,2, 128,9, 128,5, 128,3, 128,0, 125,9, 123,1, 116,5, 116,1;
EMAR calc. para C25H16N3O3S [M-H]- 438,0918, hallado 438,0920.
Ejemplo preparativo 142
(£)-2-ciano-3-(3,5-dimetil-4-hidroxifenil)-N-(4-(4-(trifluorometoxi)fenil)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(4-(trifluorometoxi)fenil)tiazol-2-il)acetamida (80 mg, 0,25 mmol), 3,5-dimetil-4-hidroxibenzaldehído (37 mg, 0,25 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 anhidro (3 ml). El tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (80 mg, 70%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,71 (s, 1H), 9,61 (s, 1H), 8,34 (s, 1H), 8,10 - 8,03 (m, 2H), 7,78 (s, 1H), 7,71 (s, 2H), 7,50 - 7,42 (m, 2H), 2,24 (s, 6 H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 161,8, 158,8, 152,0, 147,8, 133,4, 131,9, 127,5, 125,1, 122,7, 121,3, 120,1 (q, J = 256,2 Hz), 109,7, 16,6;
19F-RMN (471 MHz, DMSO-de) 5 (ppm) -56,68;
EMAR calc. para C22H17F3N3O3S [M+H]+ 460,0937, hallado 460,0940.
Ejemplo preparativo 143
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(4-morfolinofenil)tiazo1-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(4-morfolinofenil)tiazol-2-il)acetamida (26 mg, 0,079 mmol), 3,4-dihidroxibenzaldehído (12 mg, 0,087 mmol) y piperidina (1 |l, 0,009 mmol) en CH2CI2 (3 ml); el tiempo de reacción fue de 4 h a 55°C. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 70:30:0,05 a 0:100:0,05), como un sólido amarillo (5 mg, 0,011 mmol, 14%).
1H-RMN (500 MHz, DMSO-da) 5 (ppm) 12,57 (s, 1H), 10,26 (s, 1H), 9,63 (s, 1H), 8,29 (s, 1H), 7,79 (d, J = 8,9 Hz, 2H), 7,61 (d, J = 2,1 Hz, 1H), 7,45 (s, 1H), 7,38 (dd, J = 8,5, 2,4 Hz, 1H), 7,00 (d, J = 9,0 Hz, 2H), 6,93 (d, J = 8,2 Hz, 1H), 3,80 - 3,70 (m, 4H), 3,19 - 3,13 (m, 4H);
13C-RMN (126 MHz, DMSO-da) 5 (ppm) 151,7, 126,4, 125,5, 116,3, 115,8, 114,5, 65,7, 47,7;
EMAR calc. para C23H19N4O4S [M-H]- 447,1132, hallado 447,1135.
Ejemplo preparativo 144
(E)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(5-(trifluorometil)piridin-2-il)tiazol-2-il)acrilamida

Se preparó 2-ciano-N-(4-(5-(trifluorometil)piridin-2-il)tiazol-2-il)acetamida según el procedimiento general C3 a partir de 4-(5-(trifluorometil)piridin-2-il)tiazol-2-amina (321 mg, 1,31 mmol), NaH (35 mg, 1,44 mmol), cianoacetato de etilo (0,222 mg, 209 |l, 1,96 mmol) en THF (5 ml) y MeOH (1 ml) a reflujo durante 2 h. Se añadió cianoacetato de etilo (0,222 mg, 209 |l, 1,96 mmol) y se sometió a reflujo la mezcla durante 16 h adicionales. Después de la purificación mediante cromatografía en columna (hexano:EtOAc; de 1 :0 a 1 :2 ), se obtuvo una mezcla de la acetamida correspondiente con el material de partida (aprox. 5/3) como un sólido amarillo pálido (335 mg), que se usó como tal en la siguiente etapa.
Se preparó (£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(5-(trifluorometil)piridin-2-il)tiazol-2-il)acrilamida según el procedimiento general D2 a partir de la mezcla que contenía 2-ciano-N-(4-(5-(trifluorometil)piridin-2-il)tiazol-2-il)acetamida (113 mg de la mezcla correspondiente con respecto a 71 mg del producto intermedio de acetamida puro, 0,226 mmol), 3,4-dihidroxibenzaldehído (34 mg, 0,249 mmol) y piperidina (2 |l, 0,023 mmol) en CH2Cl2 anhidro (5 ml). Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 50:50:0,05 a 10:90:0,05), como un sólido naranja oscuro (80 mg, 82%).
1H-RMN (500 MHz, DMSO-da) 5 (ppm) 8,99 (s, 1H), 8,35 - 8,29 (m, 2H), 8,18 (d, J = 8,2 Hz, 1H), 8,08 (s, 1H), 7,62 (d, J = 2,3 Hz, 1H), 7,38 (dd, J = 8,4, 2,3 Hz, 1H), 6,94 (d, J = 8,2 Hz, 1H);
13C-RMN (126 MHz, DMSO-da) 5 (ppm) 162,9, 155,8, 152,5, 152,1, 151,9, 147,07 - 146,64 (m), 146,3, 135,5 (d, J = 3,6 Hz), 126,4, 124,1 (q, J = 31,8 Hz), 123,6, 123,3, 120,5, 117,0, 116,6, 115,6;
19F-RMN (471 MHz, DMSO-da) 5 (ppm) -60,07;
EMAR calc. para C19H12F3N4O3S [M+H]+ 433,0577, hallado 433,0578.
Ejemplo preparativo 145
(E)-2-ciano-N-(4-(3-cianofenN)tiazol-2-il)-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(3-cianofenil)tiazol-2-il)acetamida (100 mg, 0,37 mmol), 3,4-dihidroxibenzaldehído (51 mg, 0,37 mmol) y piperidina (3 |l, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se recogió el precipitado mediante filtración, se lavó con CH2Cl2 (3 ml), MeOH (3 ml) y se secó a vacío. Se obtuvo el producto como un sólido amarillo (80 mg, 55%).
1H-RMN (500 MHz, DMSOd) 5 fppm) 12,68 (s, 1H), 10,27 (s, 1H), 9,68 (s, 1H), 8,37 (d, J = 1,7 Hz, 1H), 8,32 (s, 1H), 8,29 - 8,24 (m, 1H), 7,94 (s, 1H), 7,80 (d, J = 7,6 Hz, 1H), 7,72 - 7,65 (m, 1H), 7,62 (d, J = 2,2 Hz, 1H), 7,39 (dd, J = 8,4, 2,3 Hz, 1H), 6,94 (d, J = 8,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 fppm) 162,3, 159,5, 152,0, 151,6, 146,7, 145,8, 135,2, 131,2, 130,1, 130,1, 129,1, 125,9, 123,1, 118,6, 116,5, 116,5, 116,1, 111,9, 110,7;
EMAR calc. para C20H13N4O3S [M+H]+ 389,0703, hallado 389,0704.
Ejemplo preparativo 146
(£)-4-(2-(2-ciano-3-(3,4-dihidroxifenil)acrilamido)tiazol-4-il)-N.N-dimetilbenzamida

Se preparó el compuesto según el procedimiento general D2 a partir de 4-(2-(2-cianoacetamido)tiazol-4-il)-N,N-dimetilbenzamida (70 mg, 0,22 mmol), 3,4-dihidroxibenzaldehído (31 mg, 0,22 mmol) y piperidina (3 |l, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se recogió el precipitado mediante filtración y se lavó con CH2Cl2 (3 ml). Se obtuvo el producto como un sólido amarillo (73 mg, 75%).
1H-RMN (500 MHz, DMSO-de) 5 fppm) 12,5 (s, 1H), 9,87 (s, 2H), 8,31 (s, 1H), 7,98 (d, J = 7,9 Hz, 2H), 7,78 (s, 1H), 7,62 (d, J = 2,2 Hz, 1H), 7,48 (d, 2H), 7,38 (dd, J = 8,4, 2,3 Hz, 1H), 6,94 (d, J = 8,3 Hz, 1H), 2,98 (s, 6 H);
13C-RMN (126 MHz, DMSO-d6) 5 fppm) 169,8, 162,1, 158,2, 152,0, 151,6, 148,2, 145,8, 135,6, 134,9, 127,6, 125,9, 125,4, 123,1, 116,5, 116,1, 109,7, 34,7;
EMAR calc. para C22H19N4O4S [M+H]+ 435,1122, hallado 435,1126.
Ejemplo preparativo 147
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(4-(2-hidroxipropan-2-il)fenil)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(4-(2-hidroxipropan-2-il)fenil)tiazol-2-il)acetamida (50 mg, 0,17 mmol), 3,4-dihidroxibenzaldehído (23 mg, 0,17 mmol) y piperidina (3 l^, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (45 mg, 60%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,62 (s, 1H), 9,82 (s, 2H), 8,31 (s, 1H), 7,86 (dd, J = 8,4, 1,9 Hz, 2H), 7,61 (d, J = 2,3 Hz, 2H), 7,56 - 7,50 (m, 2H), 7,38 (dd, J = 8,4, 2,3 Hz, 1H), 6,93 (d, J = 8,3 Hz, 1H), 5,01 (s, 1H), 1,45 (s, 6 H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,2, 151,9, 151,6, 150,3, 145,8, 131,7, 125,8, 125,2, 124,9, 123,1, 116,6, 116,5, 116,1, 107,8, 70,5, 31,8;
EMAR calc. para C22H20N3O4S [M+H]+ 422,1169, hallado 422,1167.
Ejemplo preparativo 148
(E)-2-ciano-N-(4-(5-cianotiofen-2-il)tiazol-2-il)-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(5-cianotiofen-2-il)tiazol-2-il)acetamida (50 mg, 0,18 mmol), 3,4-dihidroxibenzaldehído (25 mg, 0,18 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (2 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (40 mg, 55%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,87 (s, 1H), 10,30 (s, 1H), 9,66 (s, 1H), 8,33 (s, 1H), 7,96 (d, J = 4,0 Hz, 1H), 7,93 (s, 1H), 7,73 (d, J = 4,0 Hz, 1H), 7,61 (d, J = 2,3 Hz, 1H), 7,38 (dd, J = 8,4, 2,3 Hz, 1H), 6,94 (d, J = 8,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,1, 158,9, 152,4, 151,7, 145,8, 145,7, 142,0, 140,1, 126,0, 124,2, 123,0, 116,6, 116,3, 116,1, 114,6, 111,3, 106,4, 105,1;
EMAR calc. para C18H9 N4O3S2 [M-H]- 393,0122, hallado 393,0121.
Ejemplo preparativo 149
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(tiofen-2-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-(tiofen-2-il)tiazol-2-il)acetamida (41 mg, 0,164 mmol), 3,4-dihidroxibenzaldehído (22 mg, 0,156 mmol) y NEt3 (23 |al, 0,164 mmol) en EtOH (1,5 ml); el tiempo de reacción fue de 4 h. Se evaporó el disolvente a vacío y se trituró el sólido resultante con una mezcla de CH2Cl2 (2 ml) y EtOH (0,1 ml). Se recogió el sólido mediante filtración y se secó a vacío. Se obtuvo el producto como un sólido naranja (22 mg, 36%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 8,31 (s, 1H), 7,61 (s, 1H), 7,58 - 7,46 (m, 3H), 7,38 (d, J = 8,0 Hz, 1H), 7,19 -7,05 (m, 1H), 6,93 (d, J = 8,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,0, 152,1, 151,6, 145,8, 138,2, 128,0, 125,9, 125,6, 123,9, 123,1, 116,5, 116,4, 116,1, 107,1, 99,7;
EMAR calc. para C17H10N3O3S2 [M-H]- 368,0169, hallado 368,0169.
Ejemplo preparativo 150
(£)-2-ciano-N-(4-(3,5-difluorofenil)tiazol-2-il)-3-(3,4-dihidroxifenil)acrilamida
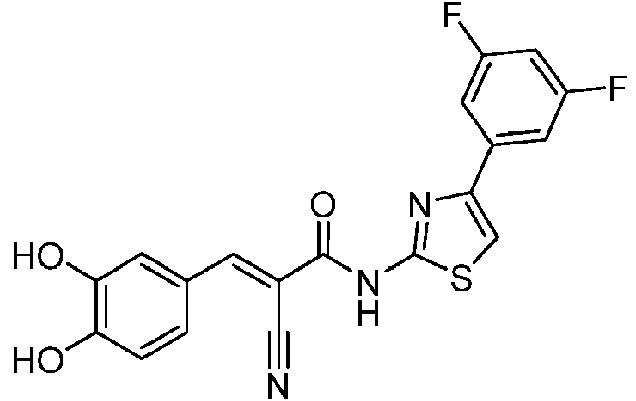
Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(3,5-difluorofenil)tiazol-2-il)acetamida (102 mg, 0,365 mmol), 3,4-dihidroxibenzaldehído (50 mg, 0,365 mmol) y piperidina (3,3 mg, 0,037 mmol, 4 |al) en CH2Cl2 ( 6 ml). Se recogió el precipitado mediante filtración y se lavó con CH2Cl2 (3 ml). Se mezcló el sólido con una disolución acuosa saturada de NaHCO3 (15 ml) y EtOAc (25 ml). Se separaron las fases, se lavó la fase orgánica con salmuera (15 ml), se secó sobre MgSO4, se filtró y se evaporó el disolvente a vacío. Se obtuvo el producto como un sólido naranja oscuro (79 mg, 50%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 8,31 (s, 1H), 7,93 (s, 1H), 7,68 - 7,62 (m, 2H), 7,61 (d, J = 2,3 Hz, 1H), 7,39 (dd, J = 8,4, 2,3 Hz, 1H), 7,24 -7,16 (m, 1H), 6,94 (d, J = 8,4 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 163,8 (d, J = 13,6 Hz), 162,1, 161,8 (d, J = 13,6 Hz), 158,5, 151,9 (d, J = 71,8 Hz), 146,7, 145,8, 137,6, 125,9, 123,1, 116,5, 116,1, 111,4, 108,8- 108,4 (m), 103,0 (dd, J = 25,9 Hz);
19F-RMN (471 MHz, DMSO-de) 5 (ppm) -109,5;
EMAR calc. para C19H12F2N3O3S [M+H]+ 400,0562, hallado 400,0559.
Ejemplo preparativo 151
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(piridazin-3-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(piridazin-3-il)tiazol-2-il)acetamida ( 8 mg, 0,033 mmol), 3,4-dihidroxibenzaldehído (5 mg, 0,036 mmol) y piperidina (0,3 |al, 0,004 mmol) en CH2CI2 (1 ml) y DMSO (0,5 ml); el tiempo de reacción fue de 4 h a 55°C. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; del 70:30:0,05% al 0:100:0,05%), como un sólido amarillo (3 mg, 25%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 9,23 - 9,16 (m, 1H), 8,28 (s, 1H), 8,20 - 8,08 (m, 2H), 7,80 (dd, J = 8,5, 4,9 Hz, 1H), 7,61 (d, J = 2,3 Hz, 1H), 7,37 (dd, J = 8,4, 2,3 Hz, 1H), 6,92 (d, J = 8,2 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 150,1, 127,6, 125,3, 123,6, 116,0, 115,7;
EMAR calc. para C17H10N5O3S [M-H]- 364,0510, hallado 364,0513.
Ejemplo preparativo 152
(E)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(4-fluorofenil)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(4-fluorofenil)tiazol-2-il)acetamida (108 mg, 0,413 mmol), 3,4-dihidroxibenzaldehído (57 mg, 0,413 mmol) y piperidina (4 ^l, 0,041 mmol) en CH2Cl2 ( 6 ml). Se recogió el precipitado mediante filtración y se lavó con CH2Cl2 (3 ml). Se mezcló el sólido con una disolución acuosa saturada de NaHCO3 (15 ml) y EtOAc (25 ml), se separaron las fases, se lavó la fase orgánica con salmuera (15 ml), se secó sobre MgSO4 , se filtró y se evaporó el disolvente a vacío. Se obtuvo el producto como un sólido naranja (143 mg, 91%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 8,31 (s, 1H), 8,01 - 7,93 (m, 2H), 7,66 (s, 1H), 7,61 (d, J = 2,1 Hz, 1H), 7,38 (dd, J = 8,5, 2,3 Hz, 1H), 7,32 - 7,24 (m, 2H), 6,94 (d, J = 8,2 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,8, 160,8, 151,8 (d, J = 63,6 Hz), 145,8, 130,6, 127,8 (d, J= 8,2 Hz), 125,9, 123,1, 116,5, 116,1, 115,6 (d, J = 20,9 Hz), 108,5;
19F-RMN (471 MHz, DMSO-de) 5 (ppm) -114,2;
EMAR calc. para C19H13FN3O3S [M+H]+ 382,0656, hallado 382,0656.
Ejemplo preparativo 153
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(piridin-4-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-(piridin-4-il)tiazol-2-il)acetamida (37 mg, 0,151 mmol), 3,4-dihidroxibenzaldehído (20 mg, 0,144 mmol) y NEt3 (21 |l, 0,151 mmol) en EtOH (2 ml); el tiempo de reacción fue de 4 h. Se recogió el precipitado mediante filtración, se lavó con etil éter (2 x 2 ml) y se secó a vacío. Se obtuvo el producto como un sólido amarillo (14 mg, 25%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,77 (sa, 1H), 10,29 (sa, 1H), 9,66 (s, 1H), 8,64 (d, J = 6,3 Hz, 2H), 8,32 (s, 1H), 8,06 (s, 1H), 7,88 (d, J = 6,3 Hz, 2H), 7,62 (d, J = 2,3 Hz, 1H), 7,39 (dd, J = 8,3, 2,4 Hz, 1H), 6,94 (d, J = 8,4 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,2, 158,9, 152,3, 151,6, 150,3, 146,5, 145,8, 140,8, 126,0, 123,1, 120,0, 116,6, 116,4, 116,1, 112,9, 99,6;
EMAR calc. para C18H11N4O3S [M-H]- 363,0557, hallado 363,0558.
Ejemplo preparativo 154
(E)-4-(2-(2-ciano-3-(3,4-dihidroxifenil)acrilamido)tiazol-4-il)benzoato de metilo

Se preparó el compuesto según el procedimiento general D2 a partir de 4-(2-(2-cianoacetamido)tiazol-4-il)benzoato de metilo (25 mg, 0,08 mmol), 3,4-dihidroxibenzaldehído (15 mg, 0,08 mmol) y piperidina (3 |l, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (22 mg, 70%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,74 (s, 1H), 10,29 (s, 1H), 9,66 (s, 1H), 8,33 (s, 1H), 8,12 - 8,02 (m, 4H), 7,91 (s, 1H), 7,62 (d, J = 2,2 Hz, 1H), 7,39 (dd, J = 8,4, 2,2 Hz, 1H), 6,94 (d, J = 8,3 Hz, 1H), 3,87 (s, 3H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 170,3, 165,9, 161,9, 158,4, 152,1, 151,6, 147,9, 145,8, 138,4, 129,7, 128,6, 125,9, 125,8, 123,1, 116,5, 116,3, 116,1, 111,3, 52,1;
EMAR calc. para C21H16N3O5S [M+H]+ 422,0805, hallado 422,0803.
Ejemplo preparativo 155
(E/Z)-2-ciano-3-(3,4-dihidroxifenil)-3-fenil-N-(4-feniltiazol-2-il)acrilamida
Se calentó una mezcla de 2-ciano-N-(4-femlt¡azol-2-¡l)acetam¡da (201 mg, 0,826 mmol), (3,4-dihidroxifenil)fenilmetanona (177 mg, 0,826 mmol) y NH4OAc (191 mg, 2,48 mmol) en tolueno ( 6 ml) a reflujo durante 24 h con eliminación azeotrópica de agua mediante una trampa de Dean-Stark. Se evaporó el disolvente a vacío, se disolvió el residuo en EtOAc (50 ml) y se lavó la disolución con una disolución acuosa saturada de NH4Cl (25 ml), H2O (25 ml) y salmuera (25 ml). Se secó la fase orgánica sobre MgSO4, se filtró y se evaporó el disolvente a vacío. Se purificó el residuo mediante cromatografía en columna (tolueno:EtOAc; de 1:0 a 2:1). Se concentraron las fracciones que contenían el producto hasta el volumen residual de 20 ml y se permitió reposar a la mezcla a 25°C. Se recogió el precipitado mediante filtración y se secó a vacío. Se obtuvo el producto (una mezcla 1:0,8 de isómeros E y Z) como un sólido amarillo (80 mg, 22%).
1H-RMN (500 MHz, DMSO-d6) S (ppm) 12,80 (s, 1H, isómero mayoritario), 12,72 (s, 0,8H, isómero minoritario), 9,70 (s, 0,8H, isómero minoritario), 9,59 (s, 1H, isómero mayoritario), 9,36 (s, 0,8H, isómero minoritario), 9,23 (s, 1H, isómero mayoritario), 7,90 - 7,78 (m, 3,6H), 7,70 (s, 1H, isómero mayoritario), 7,65 (s, 0,8H, isómero minoritario), 7,57 - 7,52 (m, 2,8H), 7,44 - 7,36 (m, 8 H), 7,35 - 7,29 (m, 1,8H), 7,23 - 7,18 (m, 1,8H), 6 , 8 8 - 6,82 (m, 1,8H), 6,77 (dd, J = 8 ,2 , 2,3 Hz, 0,8H, isómero minoritario), 6,69 (d, J = 8,7 Hz, 1 H, isómero mayoritario), 6,59 - 6,54 (m, 1 ,8 h);
13C-RMN (126 MHz, DMSO-da) S (ppm) 163,9, 163,8, 161,8, 161,6, 157,4, 157,2, 149,2, 149,2, 148,8, 148,6, 145,1, 138,6, 138,0, 134,0, 133,9, 130,5, 130,4, 129,8, 129,7, 128,7, 128,6, 128,5, 128,2, 127,9, 125,7, 122,5, 122,0, 117,4, 117,3, 117,1, 115,4, 115,3, 108,9, 108,9, 102,7, 102,7;
EMAR calc. para C25H18N3O3S [M+H]+ 440,1063, hallado 440,1061.
Ejemplo preparativo 156
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4,5,6,7-tetrahidrobenzo[d1tiazol-2-il)acrilamida (no se encuentra dentro del alcance de las reivindicaciones)

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4,5,6,7-tetrahidrobenzo[d1tiazol-2-il)acetamida (45 mg, 0,2 mmol) , 3,4-dihidroxibenzaldehído (26 mg, 0,2 mmol) y NEt3 (30 |al, 0,20 mmol) en EtOH (3 ml); el tiempo de reacción fue de 2,5 h a 50°C y luego de 16 h a 25°C. Tratamiento final 1 del procedimiento general D1. Se obtuvo el producto como un sólido amarillo (73 mg, 77%).
1H-RMN (500 MHz, DMSO-da) S (ppm) 12,81 (s, 1H), 10,19 (s, 1H), 9,63 (s, 1H), 8,22 (s, 1H), 7,67 (d, J = 2,2 Hz, 1H), 7,41 (dd, J = 8,4, 2,2 Hz, 1H), 6,97 (d, J = 8,2 Hz, 1H), 2,69 - 2,59 (m, 4H), 1, 8 6 (p, J = 3,3 Hz, 4H);
13C-RMN (126 MHz, DMSO-da) S (ppm) 151,1, 150,8, 145,7, 125,3, 123,6, 117,5, 116,4, 116,0, 23,6, 22,5, 22,2, 2 1 ,8 ;
EMAR calc. para C17H14N3O3S [M-H]- 340,0761, hallado 340,0760.
Ejemplo preparativo 157
(£)-2-ciano-3-(3,5-dibromo-4-hidroxifenil)-N-(4-feniltiazol-2-il)acrilamida
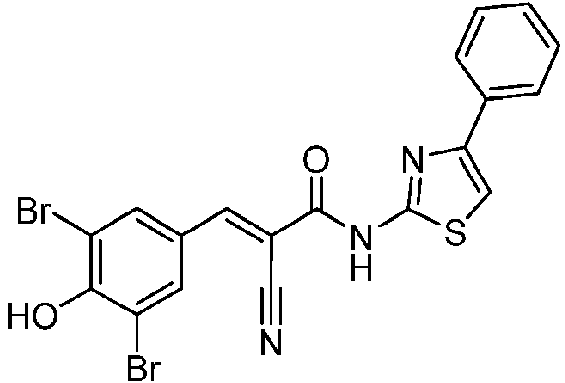
Se preparó el compuesto según el procedimiento general D1 a partir de 2-c¡ano-N-(4-fen¡lt¡azol-2-¡l)acetam¡da (60 mg, 0,247 mmol), 3,5-d¡bromo-4-h¡drox¡benzaldehído ( 66 mg, 0,234 mmol) y NEt3 (34 |al, 0,247 mmol) en EtOH (1 ml); el tiempo de reacción fue de 2 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante
cromatografía en columna (hexano:EtOAc; de 1:0 a 0:1). Se obtuvo el producto como un sólido amarillo (74 mg, 59%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,38 (s, 1H), 8,19 (s, 1H), 8,17 (s, 2H), 7,95 - 7,90 (m, 2H), 7,65 (s, 1H), 7,48 - 7,41 (m, 2H), 7,38 - 7,31 (m, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,5, 148,7, 135,1, 134,0, 128,7, 127,9, 125,7, 117,3, 114,1, 108,4;
EMAR calc. para C19H10Br2NsO2S [M-H]- 503,8846, hallado 503,8846.
Ejemplo preparativo 158
(£)-2-ciano-3-(3,5-dicloro-4-hidroxifenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (30 mg, 0,123 mmol), 3,5-dicloro-4-hidroxibenzaldehído (22 mg, 0,117 mmol) y NEt3 (17 |al, 0,123 mmol) en EtOH (2 ml); el tiempo de reacción fue de 4 h. Tratamiento final 2 del procedimiento general D1. Se obtuvo el producto, purificado mediante CCF preparativa (EtOAc), como un sólido marrón anaranjado (32 mg, 62%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,34 (s, 1H), 8,20 (s, 1H), 8,05 - 7,90 (m, 5H), 7,65 (s, 1H), 7,45 (t, J = 7,7 Hz, 2H), 7,37 -7,31 (m, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 151,6, 149,1, 131,4, 128,7, 127,9, 125,7, 123,9, 108,4;
EMAR calc. para C1gH10Cl2N3O2S [M-H]- 413,9876, hallado 413,9880.
Ejemplo preparativo 159
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(5-metil-4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(5-metil-4-feniltiazol-2-il)acetamida (63 mg, 0,229 mmol), 3,4-dihidroxibenzaldehído (30 mg, 0,217 mmol) y NEt3 (32 |al, 0,229 mmol) en EtOH (1 ml); el tiempo de reacción fue de 2 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 0:1) seguido de cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 70:30:0,05 a 10:90:0,05). Se obtuvo el producto como un sólido naranja (37 mg, 43%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,49 (s, 1H), 10,24 (s, 1H), 9,62 (s, 1H), 7,66 (d, J = 7,5 Hz, 2H), 7,60 (d, J = 2,3 Hz, 1H), 7,50 - 7,44 (m, 2H), 7,41 - 7,34 (m, 2H), 6,92 (d, J = 8,4 Hz, 1H), 2,47 (s, 3H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 151,8, 151,3, 145,8, 128,4, 128,1, 127,5, 125,7, 123,2, 116,5, 116,0, 11,8; EMAR calc. para C20H14N3O3S [M-H]- 376,0761, hallado 376,0760.
Ejemplo preparativo 160
(£)-N-(5-cloro-4-feniltiazol-2-il)-2-ciano-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de N-(5-cloro-4-feniltiazol-2-il)-2-cianoacetamida (56 mg, 0,2 mmol), 3,4-dihidroxibenzaldehído (27 mg, 0,2 mmol) y piperidina (2,0 ^l, 0,02 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 4 h a 50°C y luego de 16 h a 25°C. Se obtuvo el producto como un sólido amarillo (30 mg, 40%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,96 (s, 1H), 10,33 (s, 1H), 9,67 (s, 1H), 8,32 (s, 1H), 7,91 (d, J = 6,9 Hz, 2H), 7,61 (d, J = 2,2 Hz, 1H), 7,51 (t, J = 7,5 Hz, 2H), 7,47 - 7,40 (m, 1H), 7,38 (dd, J = 8,4, 2,2 Hz, 1H), 6,94 (d, J = 8,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,0, 152,5, 151,8, 145,8, 132,3, 128,5, 127,7, 126,1, 123,0, 116,6, 116,2, 116,1;
EMAR calc. para C19H11ClN3OsS [M-H]- 396,0215, hallado 396,0213.
Ejemplo preparativo 161
(£)-N-(5-bromo-4-feniltiazol-2-il)-2-ciano-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de N-(5-bromo-4-feniltiazol-2-il)-2-cianoacetamida (53 mg, 0,164 mmol), 3,4-dihidroxibenzaldehído (22 mg, 0,156 mmol) y NEt3 (23 ^l, 0,164 mmol) en EtOH (2 ml); el tiempo de reacción fue de 2 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 0:1) seguido de cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 50:50:0,05 a 0:100:0,05). Se obtuvo el producto como un sólido naranja amarillento (14 mg, 19%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,97 (s, 1H), 10,32 (s, 1H), 9,67 (s, 1H), 8,32 (s, 1H), 7,89 (d, J = 7,2 Hz, 2H), 7,61 (d, J = 2,3 Hz, 1H), 7,55 - 7,47 (m, 2H), 7,47 - 7,41 (m, 1H), 7,38 (dd, J = 8,4, 2,3 Hz, 1H), 6,94 (d, J = 8,4 Hz, 1H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 162,5, 152,5, 151,8, 145,8, 133,0, 128,5, 128,4, 128,0, 126,1, 123,1, 116,6, 116,1;
EMAR calc. para C1gH12BrN3OsS [M-H]- 439,9710, hallado 439,9709.
Ejemplo preparativo 162
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(4-etinilfenil)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(4-etinilfenil)tiazol-2-il)acetamida (80 mg, 0,25 mmol), 3,4-dihidroxibenzaldehído (13 mg, 0,09 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (21 mg, 60%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 12,69 (s, 1H), 10,27 (s, 1H), 9,68 (s, 1H), 8,31 (s, 1H), 7,98 - 7,92 (m, 2H), 7,79 (s, 1H), 7,61 (d, J = 2,3 Hz, 1H), 7,58 - 7,53 (m, 2H), 7,38 (dd, J = 8,4, 2,2 Hz, 1H), 6,94 (d, J = 8,3 Hz, 1H), 4,24 (s, 1H);
13C-RMN (126 MHz, DMSO-d6) 5 (ppm) 161,9, 158,4, 152,0, 151,6, 148,1, 145,8, 134,4, 132,1, 125,9, 125,8, 123,1, 120,9, 116,5, 116,1, 110,0, 83,4, 81,5;
EMAR calc. para C21H14N3O3S [M+H]+ 388,0750, hallado 388,0752.
Ejemplo preparativo 163
(£)-2-ciano-N-(4-(3-ciclopropil-4-metoxifenil)tiazol-2-il)-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(3-ciclopropil-4-metoxifenil)tiazol-2-il)acetamida (70 mg, 0,22 mmol), 3,4-dihidroxibenzaldehído (30 mg, 0,22 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (50 mg, 50%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 12,6 (s, 1H), 9,6 (s, 2H), 8,28 (s, 1H), 7,69 (dd, J = 8,5, 2,2 Hz, 1H), 7,61 (d, J = 2,3 Hz, 1H), 7,51 (s, 1H), 7,41 - 7,34 (m, 2H), 7,01 (d, J = 8,5 Hz, 1H), 6,92 (d, J = 8,3 Hz, 1H), 3,85 (s, 3H), 2,21 -2,10 (m, 1H), 0,99 -0,88 (m, 2H), 0,76 -0,61 (m, 2H);
13C-RMN (126 MHz, DMSO-cfe) 5 (ppm) 162,1, 157,7, 151,9, 151,4, 145,8, 131,4, 126,5, 125,8, 123,9, 123,2, 1 2 2 ,0 , 116,5, 116,0, 110,6, 55,5, 9,2, 7,9;
EMAR calc. para C23H20N3O4S [M+H]+ 434,1169, hallado 434,1172.
Ejemplo preparativo 164
(E)-N-(4-(ferc-butil)tiazol-2-il)-2-ciano-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de N-(4-(terc-butil)tiazol-2-il)-2-cianoacetamida (60 mg, 0,269 mmol), 3,4-dihidroxibenzaldehído (35 mg, 0,255 mmol) y NEt3 (38 |al, 0,269 mmol) en EtOH (1 ml); el tiempo de reacción fue de 3 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1) seguido de cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 70:30:0,05 a 10:90:0,05). Se obtuvo el producto como un sólido amarillo (14 mg, 15%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 8,22 (s, 1H), 7,59 (d, J = 2,3 Hz, 1H), 7,34 (dd, J = 8,2, 2,3 Hz, 1H), 6,89 (d, J = 8,4 Hz, 1H), 6,75 (s, 1H), 1,29 (s, 9H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 151,4, 145,8, 125,7, 123,2, 117,1, 116,3, 116,0, 104,6, 33,7, 29,4;
EMAR calc. para C17H16N3O3S [M-H]- 342,0918, hallado 342,0919.
Ejemplo preparativo 165
(E)-2-ciano-3-(5,6-dihidroxipiridin-3-il)-N-(4-feniltiazol-2-il)acrilamida

A 5,6-dimetoxinicotinaldehído (50 mg, 0,299 mmol) se le añadió AcOH (1 ml) y HBr (al 47% en H2O, 1 ml). Se sometió a irradiación microondas la mezcla durante 30 min a 150°C. Se diluyó el residuo con agua (5 ml) y se ajustó el pH a 7 con una disolución acuosa saturada de NaHCO3. Se extrajo la mezcla con EtOAc (3 * 50 ml). Se combinaron las fases orgánicas, se secaron sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se obtuvo 5,6-dihidroxinicotinaldehído, purificado mediante cromatografía en columna (EtOAc:MeOH:7M NH3 en MeOH; de 4:1:0,1 a 0:1:0,1), como un sólido blanquecino (10 mg, 24%) y se usó como tal en la siguiente etapa.
1H-RMN (300 MHz, DMSO-cfe) 59,54 (s, 1H), 7,80 (s, 1H), 6,91 (s, 1H).
Se preparó (£)-2-ciano-3-(5,6-dihidroxipiridin-3-il)-N-(4-feniltiazol-2-il)acrilamida según el procedimiento general D2 a partir de 2-ciano-N-(4-(6-metoxipiridin-2-il)tiazol-2-il)acetamida ( 80 mg, 0,3 mmol), 5,6-dihidroxinicotinaldehído (10 mg, 0,072 mmol) y piperidina ( 8 |al, 0,083 mmol) en CH2Cl2 (3 ml) y MeOH (2 ml); el tiempo de reacción fue de 5 h a 55°C. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 10:90:0,05), como un sólido amarillo (2 mg, 8 %).
1H-RMN (500 MHz, acetona-cfe) 5 (ppm) 8,23 (s, 1H), 7,97 - 7,90 (m, 2H), 7,82 (s, 1H), 7,55 (s, 1H), 7,47 - 7,39 (m, 2H), 7,37 -7,30 (m, 1H);
EMAR calc. para C18H11N4O3S [M-H]- 363,0557, hallado 363,0557.
Ejemplo preparativo 166
(£)-3-(3-cloro-4-hidroxifenil)-2-ciano-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (51 mg, 0,21 mmol), 3,4-dihidroxibenzaldehído (31 mg, 0,20 mmol) y NEt3 (29 ^l, 0,21 mmol) en EtOH (1 ml); el tiempo de reacción fue de 2 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1). Se obtuvo el producto como un sólido amarillo pálido (50 mg, 62%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,74 (sa, 1H), 11,53 (s, 1H), 8,39 (s, 1H), 8,09 (d, J = 2,3 Hz, 1H), 7,93 (d, J = 7,0 Hz, 2H), 7,89 (dd, J = 8,7, 2,3 Hz, 1H), 7,70 (s, 1H), 7,45 (dd, J = 7,7 Hz, 2H), 7,39 -7,31 (m, 1H), 7,18 (d, J = 8,5 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 157,6, 150,6, 132,4, 131,5, 128,7, 128,0, 125,7, 123,8, 120,7, 117,2, 108,8; EMAR calc. para C ^H u C lN ^S [M-H]- 380,0266, hallado 380,0266.
Ejemplo preparativo 167
(£)-3-(3-bromo-4-hidroxifenil)-2-ciano-N-(4-feniltiazol-2-il)acrilamida
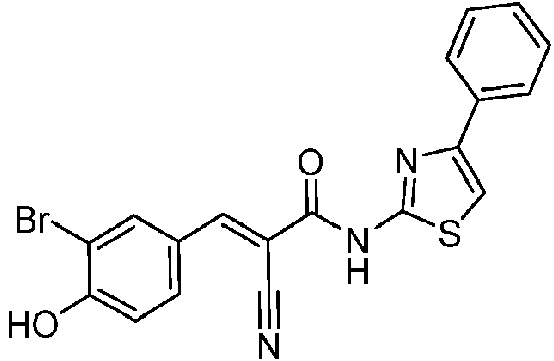
Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (75 mg, 0,308 mmol), 3-bromo-4-hidroxibenzaldehído (59 mg, 0,293 mmol) y NEt3 (43 |al, 0,308 mmol) en EtOH (5 ml); el tiempo de reacción fue de 4 h. Tratamiento final 2 del procedimiento general D1. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1), como un sólido amarillo (32 mg, 24%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,74 (s, 1H), 11,60 (s, 1H), 8,39 (s, 1H), 8,24 (d, J = 2,3 Hz, 1H), 8,00 - 7,87 (m, 3H), 7,70 (s, 1H), 7,45 (t, J = 7,7 Hz, 2H), 7,35 (t, J = 7,4 Hz, 1H), 7,17 (d, J = 8,7 Hz, 1H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 158,6, 150,4, 135,6, 132,0, 128,7, 128,0, 125,7, 124,2, 116,9, 110,1, 108,8; EMAR calc. para C ^H u B rN ^S [M-H]- 423,9761, hallado 423,9769.
Ejemplo preparativo 168
(£)-2-ciano-3-(3-fluoro-4-hidroxi-5-metoxifenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (80 mg, 0,33 mmol) en EtOH (3 ml), 3-fluoro-4-hidroxi-5-metoxibenzaldehído (53 mg, 0,31 mmol) y NEt3 (46 |al, 0,33 mmol); el tiempo de reacción fue de 4 h. Tratamiento final 1 del procedimiento general D1. Se obtuvo el
producto como un sólido amarillo (50 mg, 40%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 12,79 (s, 1H), 10,70 (s, 1H), 8,40 (s, 1H), 8,00 - 7,86 (m, 2H), 7,70 (s, 1H), 7,56 (dd, J = 9,7, 2,0 Hz, 2H), 7,45 (t, J = 7,7 Hz, 2H), 7,38 - 7,32 (m, 1H), 3,90 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,6, 151,8, 151,2, 149,9, 149,5 (d, J = 6,4 Hz), 139,8 (d, J = 14,4 Hz), 136,4, 133,7, 128,7, 128,0, 125,7, 121,7 (d, J = 9,5 Hz), 116,3, 111,7 (d, J = 20,1 Hz), 110,4, 108,7, 56,3;
19F-RMN (471 MHz, DMSO-^) 5 (ppm) -134,29;
EMAR calc. para C20H13FN3O3S [M-H]- 394,0667, hallado 394,0666.
Ejemplo preparativo 169
Ácido (E)-4-(2-ciano-3-oxo-3-((4-feniltiazol-2-il)amino)prop-1-en-1-il)benzoico

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (80 mg, 0,33 mmol), ácido 4-formilbenzoico (47 mg, 0,31 mmol) y NEt3 (92 |al, 0,66 mmol) en EtOH (3 ml); el tiempo de reacción fue de 4 h a 50°C y luego de 16 h a 25°C. Tratamiento final 2 del procedimiento general D1. Se purificó el producto en bruto mediante cromatografía en columna (hexano:EtOAc; 1:1). Se combinaron las fracciones que contenían el producto, se evaporó el disolvente a vacío y se trituró el producto sólido con una mezcla de tolueno (3 ml) y EtOAc (0,3 ml). Se recogió el sólido mediante filtración, se lavó con una mezcla de tolueno y EtOAc (10:1; 2 ml) y se secó a vacío. Se obtuvo el producto como un sólido amarillo (55 mg, 45%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 8,19 (s, 1H), 7,99 (d, J = 8,2 Hz, 2H), 7,97 - 7,89 (m, 4H), 7,37 (t, J = 7,6 Hz, 2H), 7,25 (s, 1H), 7,23 (t, J = 7,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 169,1, 167,9, 164,4, 148,2, 145,7, 138,4, 136,1, 135,0, 129,4, 129,0, 128,7, 128,3, 126,6, 125,6, 125,5, 118,4, 114,5, 106,5;
EMAR calc. para C20H12N3O3S [M-H]- 374,0605, hallado 374,0606.
Ejemplo preparativo 170
(£)-2-ciano-3-(1H-indazol-6-il)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (60 mg, 0,247 mmol), 1H-indazol-6-carboxaldehído (34 mg, 0,234 mmol) y NEt3 (34 |al, 0,247 mmol) en EtOH (1 ml); el tiempo de reacción fue de 3 h. Se evaporó el disolvente a vacío y se trituró el residuo con una mezcla de CH2Cl2 y CH3CN (1,5 ml 1,5 ml). Se recogió el sólido mediante filtración y se secó a vacío. Se obtuvo el producto como un sólido amarillo (74 mg, 8 1 %).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 13,55 (s, 1H), 12,90 (s, 1H), 8,67 (s, 1H), 8,28 (s, 1H), 8,23 (s, 1H), 7,98 (d, J = 8,4 Hz, 1H), 7,95 (d, J = 7,3 Hz, 2H), 7,76 (d, J = 8,5, 1,6 Hz, 1H), 7,73 (s, 1H), 7,46 (dd, J = 7,7 Hz, 2H), 7,36 (dd, J = 7,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,3, 152,8, 149,4, 139,6, 133,9, 129,2, 128,8, 128,0, 125,7, 124,9, 121,5, 121,5, 116,0, 113,4, 108,9;
EMAR calc. para C20H12N5OS [M-H]- 370,0768, hallado 370,0767.
Ejemplo preparativo 171
(E)-2-ciano-3-(2-fluorofenil)-N-(4-feniltiazol-2-il)acrilamida
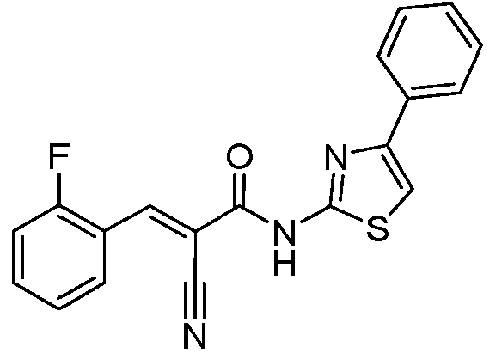
Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (50 mg, 0,206 mmol), 2-fluorobenzaldehído (24 mg, 0,195 mmol) y NEt3 (29 |al, 0,206 mmol) en EtOH (1 ml); el tiempo de reacción fue de 2 h. Tratamiento final 2 del procedimiento general D1. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 1:0 a 1:1), como un sólido amarillo (42 mg, 59%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 13,13 (s, 1H), 8,59 (s, 1H), 8,19 (t, J = 7,8 Hz, 1H), 7,94 (d, J = 7,8 Hz, 2H), 7,79 - 7,64 (m, 2H), 7,52 - 7,41 (m, 4H), 7,36 (t, J = 7,5 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,6, 159,6, 143,8, 135,0, 129,0, 128,8, 128,1, 125,8, 125,2, 125,2, 120,0, 119,9, 116,5, 116,3, 115,2, 108,9;
19F-RMN (471 MHz, DMSO-CÍ6) 5 (ppm) -111,17;
EMAR calc. para C19H13FN3OS [M+H]+ 350,0758, hallado 350,0759.
Ejemplo preparativo 172
3-(3-acetamidofenil)-2-ciano-N-(4-feniltiazol-2-il)acrilamida
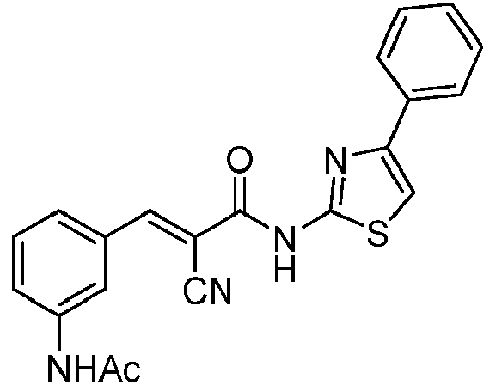
Se preparó el compuesto según el procedimiento general D2 a partir de N-(3-formilfenil)acetamida (76 mg, 0,3 mmol), 2-ciano-N-(4-feniltiazol-2-il)acetamida (50 mg, 0,3 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (5 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (40 mg, 35%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 12,93 (s, 1H), 10,21 (s, 1H), 8,49 (s, 1H), 8,30 (s, 1H), 7,99 - 7,92 (m, 2H), 7,80 - 7,69 (m, 3H), 7,56 - 7,51 (m, 1H), 7,49 - 7,43 (m, 2H), 7,35 (t, J = 7,4 Hz, 1H), 2,09 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 168,7, 161,1, 152,0, 147,2, 140,0, 133,4, 132,1, 129,6, 128,7, 128,0, 125,7, 124,5, 123,2, 120,7, 115,4, 108,8, 24,0;
EMAR calc. para C21H17N4O2S [M+H]+ 389,1067, hallado 389,1069.
Ejemplo preparativo 173
(£)-2-ciano-3-(4-hidroxi-3-nitrofenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (69 mg, 0,284 mmol), 4-hidroxi-3-nitrobenzaldehído (45 mg, 0,269 mmol) y NEt3 (40 |l, 0,284 mmol) en EtOH (1 ml); el tiempo de reacción fue de 3 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc:MeOH; de 1:1:0 a 0:9:1). Se obtuvo el producto como un sólido naranja (65 mg, 58%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,72 (s, 1H), 8,54 (d, J = 2,4 Hz, 1H), 8,44 (s, 1H), 8,13 (dd, J = 9,0, 2,4 Hz, 1H), 7,97 -7,91 (m, 2H), 7,69 (s, 1H), 7,45 (dd, J = 7,7, 7,7 Hz, 2H), 7,38 -7,32 (m, 1H), 7,15 (d, J = 8,9 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 162,0, 159,0, 150,1, 148,3, 137,5, 135,4, 133,8, 129,8, 128,7, 128,0, 125,7, 121,9, 120,0, 116,3, 108,7;
EMAR calc. para C19H11N4O4S [M-H]- 391,0506, hallado 391,0505.
Ejemplo preparativo 174
(£)-2-ciano-3-(4-nitrofenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (51 mg, 0,209 mmol), 4-nitrobenzaldehído (30 mg, 0,198 mmol) y NEt3 (29 |l, 0,209 mmol) en EtOH (2 ml); el tiempo de reacción fue de 2 h. Se evaporó el disolvente a vacío y se trituró el residuo con CH3CN (0,7 ml). Se recogió el sólido mediante filtración y se lavó con dietil éter (2 ml). Después de secar a vacío, se obtuvo el producto como un sólido naranja (47 mg, 63%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 13,14 (s, 1H), 8,61 (s, 1H), 8,43 (d, J = 8,7 Hz, 2H), 8,19 (d, J = 8,7 Hz, 2H), 7,94 (d, J = 7,8 Hz, 2H), 7,72 (s, 1H), 7,46 (t, J = 7,6 Hz, 2H), 7,36 (t, J = 7,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 160,8, 149,6, 149,0, 137,8, 131,1, 130,6, 128,8, 128,1, 125,8, 124,2, 115,1, 108,9;
EMAR calc. para C ^ H ^ O s S [M+H]+ 377,0703, hallado 377,0701.
Ejemplo preparativo 175
Ácido (E)-3-(2-ciano-3-oxo-3-((4-feniltiazol-2-il)amino)prop-1-en-1-il)benzoico

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida
(61 mg, 0,251 mmol), ácido 3-formilbenzoico (36 mg, 0,238 mmol) y NEt3 (35 |al, 0,251 mmol) en EtOH (1 ml); el tiempo de reacción fue de 3 h. Se evaporó el disolvente a vacío y se trituró el residuo con una mezcla de C ^C h y CH3CN (1,5 ml 1,5 ml). Se recogió el sólido mediante filtración y se secó a vacío. Se obtuvo el producto como un sólido amarillo (47 mg, 52%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 13,18 (s, 2H), 8,61 (s, 2H), 8,21 (d, J = 7,9 Hz, 1H), 8,16 (d, J = 7,8 Hz, 1H), 7,99 - 7,90 (m, 2H), 7,81 - 7,66 (m, 2H), 7,46 (dd, J = 7,7 Hz, 2H), 7,36 (dd, J = 7,4 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 166,4, 151,1, 134,1, 133,0, 132,1, 131,8, 130,8, 129,8, 128,8, 128,0, 125,7, 115,4, 108,9;
EMAR calc. para C20H12N3O3S [M-H]- 374,0605, hallado 374,0604.
Ejemplo preparativo 176
(£)-2-ciano-3-(6-hidroxi-[1,1’-bifenil1-3-il)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (62 mg, 0,25 mmol), 6-hidroxi-[1,1’-bifenil]-3-carbaldehído (50 mg, 0,25 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (70 mg, 65%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 11, 66 (s, 2H), 8,47 (s, 1H), 8,06 (d, J = 2,3 Hz, 1H), 7,96 - 7,90 (m, 3H), 7,67 (s, 1H), 7,62 - 7,57 (m, 2H), 7,49 - 7,41 (m, 4H), 7,40 - 7,31 (m, 2H), 7,17 (d, J = 8,5 Hz, 1H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 162,2, 159,4, 151,6, 148,7, 137,1, 134,0, 133,7, 132,1, 128,9, 128,7, 128,7, 128,1, 127,9,127,2, 125,7, 123,2, 117,0, 116,6, 108,6;
EMAR calc. para C25H18N3O2S [M+H]+ 424,1114, hallado 424,1112.
Ejemplo preparativo 177
(£)-2-ciano-3-(3,5-difluoro-4-hidroxifenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (80 mg, 0,33 mmol), 3,5-difluoro-4-hidroxibenzaldehído (50 mg, 0,31 mmol) y NEt3 (46 |al, 0,33 mmol) en EtOH (3 ml); el tiempo de reacción fue de 4 h. Tratamiento final 1 del procedimiento general D1. Se obtuvo el producto como un sólido amarillo (90 mg, 71%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 12,87 (s, 1H), 11, 66 (s, 1H), 8,37 (s, 1H), 7,97 - 7,90 (m, 2H), 7,79 - 7,71 (m, 2H), 7,71 (s, 1H), 7,46 (t, J = 7,7 Hz, 2H), 7,36 (t, J = 7,4 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 152,8 (d, J = 7,6 Hz), 150,9 (d, J = 7,5 Hz), 149,9, 138,8, 128,8, 128,0, 125,7, 121,5, 115,8, 115,2 - 113,3 (m), 108,8;
EMAR calc. para C19H10F2N3O2S [M-H]- 382,0467, hallado 382,0469.
Ejemplo preparativo 178
(E)-2-ciano-3-(3,4-difluorofenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (80 mg, 0,33 mmol), 3,4-difluorobenzaldehído (44 mg, 0,31 mmol) y NEt3 (46 |al, 0,33 mmol) en EtOH (3 ml); el tiempo de reacción fue de 4 h. Tratamiento final 1 del procedimiento general D1. Se obtuvo el producto como un sólido amarillo (65 mg, 55%).
1H-RMN (500 MHz, DMSO-da) 5 (ppm) 13,03 (s, 1H), 8,47 (s, 1H), 8,03 (ddd, J = 11,6, 7,8, 2,2 Hz, 1H), 7,95 - 7,91 (m, 2H), 7,90 - 7,86 (m, 1H), 7,77 - 7,65 (m, 2H), 7,45 (t, J = 7,7 Hz, 2H), 7,39 - 7,29 (m, 1H);
13C-RMN (126 MHz, DMSO-^) 5 fppm) 161,7, 153,2 (d, J = 12,4 Hz), 151,2 (d, J = 12,8 Hz), 151,0 (d, J = 13,5 Hz), 150,2, 149,0 (d, J = 13,4 Hz), 134,1, 129,9 (dd, J = 3,8 Hz), 129,3, 128,7 (dd, J = 7,4, 3,4 Hz), 128,6, 126,3, 119,3 (dd, J = 17,7, 15,8 Hz), 116,0, 109,4;
19F-RMN (471 MHz, DMSO-^) 5fppm) -136,40, -136,45;
EMAR calc. para C19H10F2N3OS [M-H]- 366,0518, hallado 366,0520.
Ejemplo preparativo 179
(E)- y (Z)-2-ciano-3-(1H-imidazol-4-il)-N-(4-feniltiazol-2-il)acrilamida

Se prepararon los compuestos según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (59 mg, 0,242 mmol), 4-imidazolcarboxaldehído (22 mg, 0,23 mmol) y NEt3 (34 |al, 0,242 mmol) en EtOH (1 ml); el tiempo de reacción fue de 3 h. Se evaporó el disolvente a vacío y se purificó el residuo mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 70:30:0,05 a 10:90:0,05). Se obtuvo el producto, una mezcla de isómeros (E) y (Z), como un sólido amarillo (14 mg, 18%).
1H-RMN (500 MHz, DMSO-^) 5 fppm) 2,92 (s, 1H), 12,67 (s, 1H), 8,46 (s, 0,15H), 8,37 (s, 0,85H), 8,09 (s, 0,15H), 8,05 (s, 0,85H), 8,03 - 7,91 (m, 3H), 7,70 (s, 0,15H), 7,68 (s, 0,85H), 7,50 - 7,41 (m, 2H), 7,34 (t, J = 7,3 Hz, 1H); EMAR calc. para C16H10N5OS [M-H]- 320,0612, hallado 320,0614.
Ejemplo preparativo 180
(E)-2-ciano-3-(2,3-dihidroxifenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(6-metoxipiridin-2-il)tiazol-2-il)acetamida (87 mg, 0,358 mmol), 2,3-dihidroxibenzaldehído (49 mg, 0,358 mmol) y piperidina (4 ^l, 0,036 mmol) en CH2Cl2 (3 ml). Se recogió el precipitado mediante filtración y se lavó con CH2Cl2 (3 ml). Se mezcló el sólido con una disolución acuosa saturada de NaHCO3 (15 ml) y EtOAc (25 ml). Se separaron las fases y se lavó la fase orgánica con salmuera (15 ml), se secó sobre MgSO4, se filtró y se evaporó el disolvente a vacío. Se obtuvo el producto como un sólido verde pálido (120 mg, 91%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 14,46 (s, 1H), 10,27 (s, 1H), 9,29 (s, 1H), 8,61 (s, 1H), 7,99 - 7,87 (m, 2H), 7,73 (s, 1H), 7,47 -7,39 (m, 2H), 7,36 -7,31 (m, 1H), 7,28 (dd, J = 7,3, 2,1 Hz, 1H), 7,18 -7,09 (m, 2H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 160,0, 156,9, 155,6, 149,4, 144,1, 143,4, 142,1, 134,0, 128,7, 127,9, 125,8, 124,3, 120,4, 120,3, 119,2, 117,8, 108,9;
EMAR calc. para C19H14N3O3S [M+H]+ 364,0750, hallado 364,0748.
Ejemplo preparativo 181
(£)-2-ciano-3-(2-oxo-2,3-dihidro-1H-benzo[d1imidazol-5-il)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (49 mg, 0,201 mmol), 2-oxo-2,3-dihidro-1H-benzodiazol-5-carbaldehído (31 mg, 0,191 mmol) y NEt3 (28 |al, 0,201 mmol) en EtoH ( 2 ml); el tiempo de reacción fue de 3 h. Se evaporó el disolvente a vacío, se mezcló el residuo con una mezcla de CH3CN y EtOH (1 ml 1 ml) y se agitó durante 16 h a 60°C. Se recogió el sólido mediante filtración y se secó a vacío. Se obtuvo el producto como un sólido amarillo (39 mg, 53%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,71 (sa, 1H), 11,21 (s, 1H), 11,02 (s, 1H), 8,48 (s, 1H), 7,94 (d, J = 7,5 Hz, 2H), 7,80 (s, 1H), 7,70 (s, 1H), 7,62 (dd, J = 8,2, 1,8 Hz, 1H), 7,45 (t, J = 7,7 Hz, 2H), 7,35 (t, J = 7,4 Hz, 1H), 7,15 (d, J = 8,2 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,7, 155,2, 152,7, 134,6, 134,1, 130,4, 128,7, 127,9, 127,0, 125,7, 124,1, 116,5, 109,0, 108,8;
EMAR calc. para C20H12N5O2S [M-H]- 386,0717, hallado 386,0716.
Ejemplo preparativo 182
(£)-2-ciano-3-(4-hidroxi-3-metilfenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D1 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (60 mg, 0,247 mmol), 4-hidroxi-3-metilbenzaldehído (32 mg, 0,234 mmol) y NEt3 (34 |al, 0,247 mmol) en EtOH (2 ml); el tiempo de reacción fue de 4 h. Se enfrió la mezcla de reacción hasta 25°C y se sometió a sonicación durante 10 min. Se recogió el precipitado mediante filtración, se lavó con EtOH frío (1 ml) y dietil éter (1 ml) y se secó a vacío. Se obtuvo el producto como un sólido amarillo (45 mg, 50%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,68 (s, 1H), 10,70 (s, 1H), 8,37 (s, 1H), 7,94 (d, J = 7,3 Hz, 2H), 7,84 (s, 1H), 7,81 (dd, J = 8,5, 2,4 Hz, 1H), 7,69 (s, 1H), 7,49 - 7,41 (m, 2H), 7,39 - 7,29 (m, 1H), 6,99 (d, J = 8,4 Hz, 1H), 2,19 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,8, 160,8, 151,9, 141,9, 134,2, 133,9, 131,1, 128,7, 127,9, 125,7, 125,2, 122,6, 115,4, 108,7, 15,9;
EMAR calc. para C20H14N3O2S [M-H]- 360,0812, hallado 360,0812.
Ejemplo preparativo 183
(£)-2-ciano-3-(2,4-dihidroxifenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (242 mg, 1 mmol), 2,4-dihidroxibenzaldehído (138 mg, 1 mmol) y piperidina (3 ^l, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se recogió el precipitado mediante filtración, se lavó con CH2Cl2 (5 ml) y se secó a vacío. Se obtuvo el producto como un sólido amarillo (150 mg, 40%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 14,32 (s, 1H), 10,91 (s, 1H), 9,24 - 9,02 (m, 1H), 8,57 (s, 1H), 7,96 - 7,89 (m, 2H), 7,71 - 7,65 (m, 2H), 7,47 - 7,40 (m, 2H), 7,37 - 7,29 (m, 1H), 6,76 (dd, J = 8,5, 2,3 Hz, 1H), 6,65 (d, J = 2,1 Hz, 1H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 163,4, 160,4, 157,0, 156,0, 155,7, 149,2, 143,4, 134,0, 132,1, 128,7, 127,8, 125,7, 113,2, 113,1, 110,7, 108,7, 101,4;
EMAR calc. para C19H14N3O3S [M+H]+ 364,0750, hallado 364,0757.
Ejemplo preparativo 184
(£)-N-(4-(5-bromotiofen-2-il)tiazol-2-il)-2-ciano-3-(3,4-dihidroxifenil)acrilamida
Se preparó el compuesto según el procedimiento general D2 a partir de N-(4-(5-bromotiofen-2-il)tiazol-2-il)-2-cianoacetamida (300 mg, 0,914 mmol), 3,4-dihidroxibenzaldehído (126 mg, 0,914 mmol) y piperidina (9,0 |al, 0,091 mmol) en Ch2CI2 ( 8 ml) y THF (3 ml); el tiempo de reacción fue de 3 h. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH: AcOH; de 50:50:0,05 a 10:90:0,05), como un sólido amarillo oscuro (50 mg, 12%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 12,72 (s a, 1H), 10,28 (s a, 1H), 9,67 (s a, 1H), 8,31 (s, 1H), 7,60 (d, J = 2,69 Hz, 2H), 7,42 - 7,36 (m, 2H), 7,24 (d, J = 3,88 Hz, 1H), 6,94 (d, J = 8,28 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 fppm) 152,2, 151,6, 145,8, 131,4, 125,9, 124,3, 123,0, 116,6, 116,4, 116,0, 110,9, 107,7;
EMAR calc. para C^HnBrN3O3S2 [M+H]+ 449,9399, hallado 449,9401.
Ejemplo preparativo 185
(£)-2-ciano-3-(3-oxo-3,4-dihidro-2H-benzo[b1[1,41oxazin-7-il)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (42 mg, 0,17 mmol), 3-oxo-3,4-dihidro-2H-benzo[b1[1,4]oxazin-7-carbaldehído (30 mg, 0,17 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se recogió el precipitado mediante filtración y se lavó con CH2Cl2 (2 ml). Se mezcló el sólido con una disolución acuosa saturada de NH4Cl (10 ml) y se extrajo la mezcla con EtOAc (3 * 10 ml). Se secaron los extractos orgánicos combinados sobre MgSO4, se filtraron y se evaporó el disolvente a vacío. Se obtuvo el producto como un sólido amarillo (60 mg, 85%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 fppm) 12,72 (s, 1H), 11,15 (s, 1H), 8,35 (s, 1H), 7,95 - 7,91 (m, 2H), 7,69 (d, J = 1,9 Hz, 1H), 7,63 (s, 1H), 7,61 (dd, J = 8,3, 2,0 Hz, 1H), 7,48 - 7,41 (m, 2H), 7,37 - 7,30 (m, 1H), 7,08 (d, J = 8,2 Hz, 1H), 4,70 (s, 2H);
13C-RMN (126 MHz, DMSO-^) 5 fppm) 164,8, 150,3, 143,1, 131,5, 128,7, 126,6, 126,5, 125,7, 116,9, 116,5, 116,2, 108,44, 6 6 ,6 ;
EMAR calc. para C21H15N4O3S [M+H]+ 403,0859, hallado 403,0863.
Ejemplo preparativo 186
(E)-2-ciano-3-(3,5-di-ferc-butil-4-hidroxifenil)-N-(4-(4-(trifluorometoxi)fenil)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(4-(trifluorometoxi)fenil)tiazol-2-il)acetamida (80 mg, 0,25 mmol), 3,5-di-ferc-butil-4-hidroxibenzaldehído (60 mg, 0,25 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo
(80 mg, 60%).
1H-RMN (500 MHz, DMSO-cfe) 5 (ppm) 12,71 (s, 1H), 8,50 (s, 1H), 8,17 (s, 1H), 8,09 - 8,02 (m, 2H), 7,94 (s, 2H), 7,79 (s, 1H), 7,45 (d, J = 8,3 Hz, 2H), 1,43 (s, 18H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,8, 159,2, 158,3, 153,0, 147,8, 139,0, 133,4, 128,7, 127,5, 122,9, 121,3, 120,1 (q, J = 256,3 Hz), 116,5, 109,7, 100,2, 34,7, 29,9;
19F-RMN (471 MHz, DMSO-^) 5 (ppm) -56,68;
EMAR calc. para C28H29F3N3O3S [M+H]+ 544,1876, hallado 544,1876.
Ejemplo preparativo 187
(E)-3-(4-acetamido-3-((ferc-butildimetilsilil)oxi)fenil)-2-ciano-N-(4-fenilmiazol-2-i
r
)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (40 mg, 0,22 mmol), N-(2-((ferc-butildimetilsilil)oxi)-4-formilfenil)acetamida (65 mg, 0,22 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1 :1 ), como un sólido amarillo (65 mg, 60%).
1H-RMN (500 MHz, CDCh) 5 (ppm) 9,61 (s, 1H), 8,53 (d, J = 8,5 Hz, 1H), 8,34 (s, 1H), 8,00 (s, 1H), 7,90 - 7,83 (m, 2H), 7,76 (d, J = 2,1 Hz, 1H), 7,49 - 7,40 (m, 3H), 7,38 - 7,32 (m, 1H), 7,23 (s, 1H), 2,24 (s, 3H), 1,09 (s, 9H), 0,40 (s, 6 H);
13C-RMN (126 MHz, CDCh) 5 (ppm) 168,4, 159,0, 157,0, 154,6, 150,8, 144,2, 135,6, 134,3, 129,1, 129,0, 128,5, 126,7, 126,3, 119,7, 117,3, 116,8, 108,9, 26,0, 25,2, 18,4, -4,2;
EMAR calc. para C27H31^ O 3SSi [M+H]+ 519,1881, hallado 519,1880.
Ejemplo preparativo 188
(£)-3-(4-acetamido-3-hidroxifenil)-2-ciano-N-(4-feniltiazol-2-il)acrilamida

Se disolvió 3-(4-acetamido-3-((ferc-butildimetilsilil)oxi)fenil)-2-ciano-N-(4-feniltiazol-2-il)acrilamida en THF anhidro (2 ml), se enfrió la disolución hasta 0°C y se añadió TBa F (1 M en THF, 0,15 ml, 0,15 mmol). Se agitó la mezcla durante 1 h, se evaporó el disolvente y se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 1:1 a 0:1). Se obtuvo el producto como un sólido amarillo (33 mg, 65%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,78 (s, 1H), 10,59 (s, 1H), 9,47 (s, 1H), 8,36 (s, 1H), 8,20 (d, J = 8,5 Hz, 1H), 7,98 - 7,90 (m, 2H), 7,76 - 7,67 (m, 2H), 7,48 - 7,43 (m, 2H), 7,41 (dd, J = 8,5, 2,1 Hz, 1H), 7,38 - 7,32 (m, 1H), 2,17 (s, 3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 169,4, 161,6, 151,6, 149,3, 146,9, 134,1, 131,9, 128,7, 127,9, 126,8, 125,7, 124,4, 120,5, 115,1, 108,8, 24,0;
EMAR calc. para C21H17N4O3S [M+H]+ 405,1016, hallado 405,1020.
Ejemplo preparativo 189
(E)-N-(4-(4-(terc-butil)fenil)tiazol-2-il)-2-ciano-3-(3,5-dicloro-4-hidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de N-(4-(4-(terc-butil)fenil)tiazol-2-il)-2-cianoacetamida ((50 mg, 0,167 mmol), 3,5-dicloro-4-hidroxibenzaldehído (32 mg, 0,167 mmol) y piperidina (2 l^, 0,017 mmol) en CH2Cl2 (4 ml); el tiempo de reacción fue de 3 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; 1 :1 ), como un sólido amarillo (52 mg, rendimiento del 6 6 %).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,80 (s, 1H), 8,35 (s, 1H), 8,05 (s, 2H), 7,85 (d, J = 8,5 Hz, 2H), 7,62 (s, 1H), 7,47 (d, J = 8 , 6 Hz, 2H), 1,31 (s, 9H);
13C-RMN (126 MHz, DMSO-^) 5153,4, 150,6, 149,2, 130,8, 125,5, 122,7, 116,0, 34,3, 31,0;
EMAR calc. para C23H20Cl2NsO2S [M+H]+ 472,0648, hallado 472,0646.
Ejemplo preparativo 190
(£)-2-ciano-3-(4-hidroxi-2-metilfenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (80 mg, 0,329 mmol), 4-hidroxi-2-metilbenzaldehído (45 mg, 0,329 mmol) y piperidina (3 |al, 0,033 mmol) en CH2Cl2 (4 ml); el tiempo de reacción fue de 3 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 10:1 a 1:2), como un sólido amarillo (112 mg, rendimiento del 94%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,87 (s, 1H), 10,48 (s, 1H), 8,51 (s, 1H), 8,11 (d, J = 8,4 Hz, 1H), 7,94 (d, J = 7,2 Hz, 2H), 7,70 (s, 1H), 7,45 (dd, J = 7,7 Hz, 2H), 7,34 (dd, J = 7,3 Hz, 1H), 6,83 - 6,77 (m, 2H), 2,45 (s, 3H);
13C-RMN (126 MHz, DMSO-CÍ6) 5 (ppm) 161,8, 149,4, 143,5, 134,2, 130,3, 128,7, 127,9, 125,8, 121,6, 117,8, 116,6, 113,8, 108,8, 19,7;
EMAR calc. para C20H16N3O2S [M+H]+ 362,0958, hallado 362,0955.
Ejemplo preparativo 191
(£)-2-ciano-3-(2-fluoro-4-hidroxifenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (80 mg, 0,329 mmol), 2-fluoro-4-hidroxibenzaldehído (46 mg, 0,329 mmol) y piperidina (3 |al, 0,033 mmol) en CH2Cl2 (4 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 10:1 a 1:4), como un sólido amarillo (108 mg, 90%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,94 (s, 1H), 11,19 (s, 1H), 8,46 (s, 1H), 8,23 (dd, J = 8,9 Hz, 1H), 7,93 (d, J = 7,0 Hz, 2H), 7,69 (s, 1H), 7,45 (dd, J = 7,7 Hz, 2H), 7,35 (dd, J = 7,4 Hz, 1H), 6,87 (dd, J = 8,9, 2,4 Hz, 1H), 6,78 (dd, J = 12,5, 2,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 164,9, 163,4 (d, J = 256,1 Hz), 143,5, 130,4, 129,3, 128,5, 126,3, 116,7, 113,7, 111,2 (d, J = 10,9 Hz), 109,3, 103,7 (d, J = 23,6 Hz);
19F-RMN (471 MHz, DMSO-^) 5 (ppm) -109,0;
EMAR calc. para C19H13FN3O2S [M+H]+ 366,0707, hallado 366,0704.
Ejemplo preparativo 192
(£)-2-ciano-3-(4-hidroxinaftalen-1-il)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (81 mg, 0,331 mmol), 4-hidroxi-1-naftaldehído (57 mg, 0,331 mmol) y piperidina (3 |al, 0,033 mmol) en CH2Cl2 (5 ml); el tiempo de reacción fue de 4 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 10:1 a 1:3), como un sólido naranja (118 mg, 90%).
1H-RMN (500 MHz, DMSO-CÍ6) 5 (ppm) 13,10 (s, 1H), 11,42 (s, 1H), 9,14 (s, 1H), 8,46 (d, J = 8,7 Hz, 1H), 8,33 (d, J = 8,2 Hz, 1H), 8,28 (dd, J = 8,4, 1,5 Hz, 1H), 7,97 (d, J = 7,8 Hz, 2H), 7,73 (ddd, J = 8,4, 5,0, 1,5 Hz, 2H), 7,65 - 7,58 (m, 1H), 7,46 (dd, J = 7,7 Hz, 2H), 7,35 (dd, J = 7,4 Hz, 1H), 7,08 (d, J = 8,2 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 161,6, 158,7, 158,1, 149,3, 148,8, 134,2, 133,4, 130,1, 128,7, 128,1, 127,9, 125,8, 125,7, 124,4, 123,7, 122,8, 119,1, 116,6, 108,7, 108,2;
EMAR calc. para C23H16N3O2S [M+H]+ 398,0958, hallado 398,0956.
Ejemplo preparativo 193
2-ciano-3-(3-ciano-4-hidroxifenil)-N-(4-feniltiazol-2-il)propanamida
Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (70 mg, 0,4 mmol), 4-hidroxi-3-(trifluorometil)benzaldehído (75 mg, 0,4 mmol) y piperidina (3 |l, 0,027 mmol) en Ch2CI2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (100 mg, 70%).
1H-RMN (500 MHz, DMSO-^) 5 (ppm) 12,80 (s, 1H), 11,90 (s, 1H), 8,50 (s, 1H), 8,28 - 8,23 (m, 1H), 8,18 (dd, J = 8 ,8 , 2,2 Hz, 1H), 7,96 - 7,90 (m, 2H), 7,70 (s, 1H), 7,49 - 7,42 (m, 2H), 7,39 - 7,32 (m, 1H), 7,24 (d, J = 8,7 Hz, 1H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 170,3, 160,1, 150,7, 136,0, 130,2 (q, J = 3,8 Hz), 128,7, 128,0, 125,7, 123,4 (q, J = 272,3 Hz), 122,2, 118,1, 116,3 (q, J = 30,3 Hz), 116,1, 108,7;
19F-RMN (471 MHz, DMSO-^) 5 (ppm) -61,76;
EMAR calc. para C20H13F3N3O2S [M+H]+ 416,0675, hallado 416,0678.
Ejemplo preparativo 194
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(5-metiltiofen-2-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(5-metiltiofen-2-il)tiazol-2-il)acetamida (60 mg, 0,23 mmol), 3,4-dihidroxibenzaldehído (32 mg, 0,23 mmol) y piperidina (3 |l, 0,027 mmol) en CH2Cl2 (2 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (70 mg, 80%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 10,01 (s, 2H), 8,30 (s, 1H), 7,60 (d, J = 2,3 Hz, 1H), 7,40 (s, 1H), 7,37 (dd, J = 8,5, 2,3 Hz, 1H), 7,32 (d, J = 3,5 Hz, 1H), 6,94 (d, J = 8,3 Hz, 1H), 6,79 (dd, J = 3,5, 1,3 Hz, 1H), 2,46 (s, 3H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,1, 158,4, 151,9, 151,6, 145,8, 144,0, 138,9, 135,9, 126,3, 125,8, 123,8, 123,1, 116,6, 116,5, 116,1, 106,2, 15,0;
EMAR calc. para C18H14N3O3S2 [M+H]+ 384,0471, hallado 384,0469.
Ejemplo preparativo 195
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-metil-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-metil-N-(4-feniltiazol-2-il)acetamida (53 mg, 0,28 mmol), 3,4-dihidroxibenzaldehído (38 mg, 0,28 mmol) y piperidina (2 |l, 0,02 mmol) en CH2Cl2 (2 ml). Se agitó la mezcla durante 4 h a reflujo y luego durante 16 h a 25°C. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (50 mg, 60%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 10,14 (s, 1H), 9,70 (s, 1H), 8,00 - 7,94 (m, 2H), 7,93 (s, 1H), 7,80 (s, 1H), 7,61 (d, J = 2,3 Hz, 1H), 7,48 - 7,41 (m, 2H), 7,40 - 7,30 (m, 2H), 6,91 (d, J = 8,3 Hz, 1H), 3,85 (s, 3H);
13C-RMN (126 MHz, DMSO-da) 5 (ppm) 164,2, 159,7, 153,3, 151,3, 148,6, 145,7, 134,1, 128,7, 127,9, 125,7, 125,5, 123,4, 116,3, 116,2, 115,9, 110,4, 99,0, 38,1;
EMAR calc. para C20H16N3O3S [M+H]+ 378,0907, hallado 378,0910.
Ejemplo preparativo 196
(£)-2-ciano-N-(4-feniltiazol-2-il)-3-(3-(trifluorometil)fenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-feniltiazol-2-il)acetamida (220 mg, 0,9 mmol), 3-(trifluorometil)benzaldehído (157 mg, 0,9 mmol) y piperidina (3 |l, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se purificó el residuo mediante cromatografía en columna (hexano:EtOAc; de 10:1 a 10:3). Se combinaron las fracciones que contenían el producto, se evaporó el disolvente y se suspendió el residuo en una mezcla de hexano (5 ml) y EtOAc (0,5 ml). Se sometió a sonicación la mezcla durante 10 min, se recogió el sólido mediante filtración y se secó a vacío. Se obtuvo el producto como un sólido amarillo (200 mg, 55%).
1H-RMN (500 MHz, DMSO-da) 5 (ppm) 12,97 (s, 1H), 8,62 (s, 1H), 8,32 (s, 1H), 8,28 (d, J = 8,0 Hz, 1H), 8,00 (d, J = 7,9 Hz, 1H), 7,94 (d, J = 7,7 Hz, 2H), 7,91 - 7,84 (m, 1H), 7,74 (s, 1H), 7,50 - 7,44 (m, 2H), 7,39 - 7,33 (m, 1H);
13C-RMN (126 MHz, DMSO-da) 5 (ppm) 160,7, 150,5, 149,2, 133,6, 132,7 (q, J = 4,7 Hz), 132,6, 130,6, 129,9 (q, J = 32,7 Hz), 128,8, 128,0, 126,5 (q, J = 4,7 Hz), 125,8, 123,7 (q, J = 273,2 Hz), 115,2;
19F-RMN (471 MHz, DMSO-da) 5 (ppm) -61,53;
EMAR calc. para C20H13F3N3OS [M+H]+ 400,0726, hallado 400,0722.
Ejemplo preparativo 197
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(6-metilpiridin-3-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(6-metilpiridin-3-il)tiazol-2-il)acetamida (100 mg, 0,39 mmol), 3,4-dihidroxibenzaldehído (55 mg, 0,39 mmol) y piperidina (3 |l, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (97 mg, 65%).
1H-RMN (500 MHz, DMSO-da) 5 (ppm) 12,51 (s, 1H), 10,82 -9,33 (m, 2H), 9,02 (d, J = 2,3 Hz, 1H), 8,31 (s, 1H), 8,15 (dd, J = 8,1, 2,4 Hz, 1H), 7,77 (s, 1H), 7,62 (d, J = 2,2 Hz, 1H), 7,38 (dd, J = 8,4, 2,2 Hz, 1H), 7,33 (d, J = 8,1 Hz, 1H), 6,94 (d, J = 8,3 Hz, 1H), 2,46 (s, 3H);
13C-RMN (126 MHz, DMSO-da) 5 (ppm) 157,2, 152,2, 151,6, 146,2, 145,8, 136,1, 133,3, 127,0, 125,9, 123,1, 123,1, 116,6, 116,4, 116,1, 109,3, 99,8, 23,7;
EMAR calc. para C19H15N4O3S [M+H]+ 379,0859, hallado 379,0854.
Ejemplo preparativo 198
(E)-2-ciano-3-(3,4-dihidroxifenir)-N-(4-fenil-5-(tetrahidro-2H-piran-4-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-fenil-5-(tetrahidro-2H-piran-4-il)tiazol-2-il)acetamida (37 mg, 0,11 mmol), 3,4-dihidroxibenzaldehído (15 mg, 0,11 mmol) y piperidina (3 ^l, 0,027 mmol) en CH2Cl2 (2 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (30 mg, 60%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,54 (s, 1H), 10,25 (s, 1H), 9,63 (s, 1H), 8,26 (s, 1H), 7,60 (d, J = 2,3 Hz, 1H), 7,59 - 7,54 (m, 2H), 7,52 - 7,46 (m, 2H), 7,44 - 7,38 (m, 1H), 7,36 (dd, J = 8,4, 2,2 Hz, 1H), 6,92 (d, J = 8,3 Hz, 1H), 3,97 -3,86 (m, 2H), 3,44 -3,34 (m, 2H), 3,30 - 3,33 (m, 1H), 1,86 (d, J= 12,9 Hz, 2H), 1,68 (qd, J = 12,2, 4,4 Hz, 2H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 151,9, 145,8, 144,0, 134,7, 128,5, 127,7, 125,7, 123,1, 116,5, 116,0, 67,0, 35,4, 33,7;
EMAR calc. para C24H22N3O4S [M+H]+ 448,1326, hallado 448,1321.
Ejemplo preparativo 199
(E-3-(3-(ferc-butil)-4-hidroxi-5-metilfenN)-2-ciano-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(6-metoxipiridin-2-il)tiazol-2-il)acetamida (96 mg, 0,395 mmol), 3-(íerc-butil)-4-hidroxi-5-metilbenzaldehído (76 mg, 0,395 mmol) y piperidina (4 ^l, 0,039 mmol) en CH2Cl2 (3 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 10 :1 a 1 :1 ), como un sólido amarillo (137 mg, 83%).
1H-RMN (500 MHz, DMSO-d6) 5 (ppm) 12,65 (s, 1H), 9,51 (s, 1H), 8,41 (s, 1H), 8,02 - 7,88 (m, 3H), 7,78 - 7,62 (m, 2H), 7,50 - 7,39 (m, 2H), 7,41 - 7,26 (m, 1H), 2,27 (s, 3H), 1,42 (s, 9H);
13C-RMN (126 MHz, DMSO-d6) 5 (ppm) 161,9, 159,2, 152,4, 149,1, 137,3, 134,1, 132,4, 128,7, 128,2, 127,9, 125,7, 122,6, 116,6, 108,7, 34,8, 29,3, 17,1;
EMAR calc. para C24H24N3O2S [M+H]+ 418,1584, hallado 418,1585.
Ejemplo preparativo 200
(E)-3-(3-(ferc-butil)-4-hidroxifenil)-2-ciano-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(6-metoxipiridin-2-il)tiazol-2-il)acetamida (102 mg, 0,421 mmol), 3-(ferc-butil)-4-hidroxibenzaldehído (75 mg, 0,421 mmol) y piperidina (4 |al, 0,041 mmol) en CH2Cl2 (4 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 10 : 1 a 1 :1 ), como un sólido amarillo (138 mg, 81%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,66 (s, 1H), 10,77 (s, 1H), 8,44 (s, 1H), 8,03 (d, J = 2,4 Hz, 1H), 7,94 (d, J = 7,6 Hz, 2H), 7,79 (dd, J = 8,5, 2,3 Hz, 1H), 7,70 (s, 1H), 7,48 - 7,42 (m, 2H), 7,38 - 7,32 (m, 1H), 7,00 (d, J = 8,4 Hz, 1H), 1,40 (s, 9H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 161,8, 161,4, 158,1, 152,4, 149,1, 136,4, 134,2, 131,2, 130,4, 128,7, 127,9, 125,7, 122,4, 117,1, 116,6, 108,7, 34,7, 29,0;
EMAR calc. para C23H22N3O2S [M+H]+ 404,1427, hallado 404,1429.
Ejemplo preparativo 201
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(3-fluorofenil)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(3-fluorofenil)tiazol-2-il)acetamida (106 mg, 0,406 mmol), 3,4-dihidroxibenzaldehído (56 mg, 0,406 mmol) y piperidina (3 |al, 0,041 mmol) en CH2Cl2 (4 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 10:1 a 1:2) seguido de cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 50:50:0,05 a 10:90:0,05), como un sólido naranja (132 mg, 85%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 8,20 (s, 1H), 7,82 -7,76 (m, 1H), 7,75 - 7,68 (m, 2H), 7,59 (d, J = 2,3 Hz, 1H), 7,51 -7,43 (m, 1H), 7,34 (dd, J = 8,4, 2,4 Hz, 1H), 7,14 (ddd, J = 8 ,8 , 3,5 Hz, 1H), 6,85 (d, J = 7,8 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 163,0, 162,57 (d, J = 243,4 Hz), 150,9, 147,3, 146,1, 137,0 (d, J = 8,1 Hz), 130,6 (d, J = 9,1 Hz), 121,7, 117,4, 116,2, 114,2 (d, J = 20,9 Hz), 112,2 (d, J = 22,7 Hz), 109,5;
19F-RMN (471 MHz, DMSO-de) 5 (ppm) -113,1;
EMAR calc. para C19H13FN3O3S [M+H]+ 382,0656, hallado 382,0653.
Ejemplo preparativo 202
(E)-2-ciano-3-(4-hidroxi-3,5-diisopropilfenil)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(6-metoxipiridin-2-il)tiazol-2-il)acetamida (96 mg, 0,395 mmol), 4-hidroxi-3,5-diisopropilbenzaldehído (81 mg, 0,395 mmol) y piperidina (4 l^, 0,039 mmol) en CH2Cl2 (3 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 10:1 a 1:1), como un sólido amarillo (121 mg, 71%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,65 (s, 1H), 9,47 (s, 1H), 8,47 (s, 1H), 7,94 (dd, J = 8,4, 1,4 Hz, 2H), 7,84 (s, 2H), 7,69 (s, 1H), 7,45 (t, J = 7,7 Hz, 2H), 7,39 - 7,31 (m, 1H), 3,36 (hept, J = 6 , 8 Hz, 2H), 1,21 (d, J = 6,7 Hz, 12H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 161,9, 158,0, 156,4, 152,6, 149,1, 135,8, 134,2, 128,7, 127,9, 127,3, 125,7, 123,2, 116,7, 108,6, 26,1, 22,7;
EMAR calc. para C25H26N3O2S [M+H]+ 432,1740, hallado 432,1739.
Ejemplo preparativo 203
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-fenil-5-(pirazin-2-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-fenil-5-(pirazin-2-il)tiazol-2-il)acetamida (75 mg, 0,23 mmol), 3,4-dihidroxibenzaldehído (33 mg, 0,23 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se recogió el precipitado mediante filtración, se lavó con CH2Cl2 (3 ml) y se secó a vacío. Se obtuvo el producto como un sólido amarillo (90 mg, 90%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,84 (s, 1H), 10,23 (s, 1H), 9,67 (s, 1H), 8 , 6 8 - 8,62 (m, 1H), 8,45 (d, J = 2,6 Hz, 1H), 8,33 (s, 1H), 8,27 (s, 1H), 7,62 (d, J = 2,2 Hz, 1H), 7,59 - 7,53 (m, 2H), 7,52 - 7,46 (m, 3H), 7,38 (dd, J = 8,4, 2,3 Hz, 1H), 6,93 (d, J = 8,3 Hz, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,2, 152,3, 151,6, 147,6, 145,8, 144,4, 142,3, 142,2, 134,6, 131,7, 128,9, 126,0, 123,1, 116,5, 116,1;
EMAR calc. para C23H16N5O3S [M+H]+ 442,0968, hallado 442,0967.
Ejemplo preparativo 204
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(3-metilpiridin-2-il)tiazol-2-il)acrilamida
Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(3-metilpiridin-2-il)tiazol-2-il)acetamida (74 mg, 0,29 mmol), 3,4-dihidroxibenzaldehído (40 mg, 0,29 mmol) y piperidina (3 |al, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se recogió el precipitado mediante filtración, se lavó con CH2Cl2 (5 ml) y se secó a vacío. Se obtuvo el producto como un sólido amarillo ( 88 mg, 80%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 8,46 (dd, J = 4,7, 1,6 Hz, 1H), 8,27 (s, 1H), 7,71 (d, J = 7,6 Hz, 1H), 7,65 -7,58 (m, 2H), 7,38 (dd, J = 8,4, 2,3 Hz, 1H), 7,29 (dd, J = 7,7, 4,7 Hz, 1H), 6,92 (d, J = 8,3 Hz, 1H), 2,57 (s, 3H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,7, 151,9, 151,6, 150,8, 146,6, 145,8, 139,2, 131,1, 125,9, 123,1, 122,8, 116,7, 116,4, 116,0, 113,8, 20,0;
EMAR calc. para C19H15N4O3S [M+H]+ 379,0859, hallado 379,0865.
Ejemplo preparativo 205
(£)-2-ciano-3-(2,6-di-ferc-butilpiridin-4-il)-N-(4-feniltiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(6-metoxipiridin-2-il)tiazol-2-il)acetamida (44 mg, 0,182 mmol), 2,6-di-ferc-butilisonicotinaldehído (40 mg, 0,182 mmol) y piperidina (2 ^l, 0,018 mmol) en CH2Cl2 (2 ml). Se obtuvo el producto, purificado mediante cromatografía en columna (hexano:EtOAc; de 10 : 1 a 1 :1 ), como un sólido amarillo (64 mg, 79%).
1H-RMN (500 MHz, CDCls) 5 (ppm) 8,45 (s, 1H), 7,88 - 7,82 (m, 2H), 7,57 (s, 2H), 7,47 - 7,41 (m, 2H), 7,38 - 7,33 (m, 1H), 1,41 (s, 18H);
13C-RMN (126 MHz, CDCls) 5 (ppm) 170,0, 157,8, 156,8, 155,0, 150,8, 138,3, 134,0, 129,0, 128,5, 126,3, 115,6, 109,1, 106,1, 38,2, 30,2;
EMAR calc. para C26H29N4OS [M+H]+ 445,2057, hallado 445,2060.
Ejemplo preparativo 206
(£)-2-ciano-N-(5-ciclohexil-4-(4-(trifluorometoxi)fenil)tiazol-2-il)-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(5-ciclohexil-4-(4-(trifluorometoxi)fenil)tiazol-2-il)acetamida (70 mg, 0,17 mmol), 3,4-dihidroxibenzaldehído (25 mg, 0,17 mmol) y piperidina (3 ^l, 0,027 mmol) en CH2Cl2 (5 ml); el tiempo de reacción fue de 2 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (55 mg, 60%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,52 (s, 1H), 10,20 (s, 1H), 9,68 (s, 1H), 8,25 (s, 1H), 7,71 - 7,66 (m, 2H), 7,59 (d, J = 2,2 Hz, 1H), 7,51 - 7,45 (m, 2H), 7,38 - 7,33 (m, 1H), 7,03 - 6 , 8 6 (m, 1H),
3,09 - 2,87 (m, 1H), 2,01 - 1,92 (m, 2H), 1,83 - 1,75 (m, 2H), 1,73 - 1,63 (m, 1H), 1,52 - 1,38 (m, 2H), 1,39 - 1,20 (m,
3H);
13C-RMN (126 MHz, DMSO-^) 5 (ppm) 151,8, 151,5, 145,8, 147,6, 142,0, 134,4, 130,2, 125,8, 123,1, 121,1, 120,1 (q, J = 256,6 Hz), 120,9, 116,5, 116,0, 36,3, 35,7, 26,0, 25,1;
EMAR calc. para C26H23F3N3O4S [M+H]+ 530,1356, hallado 530,1353.
Ejemplo preparativo 207
(E)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(6-metoxipiridin-3-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(6-metoxipiridin-3-il)tiazol-2-il)acetamida (150 mg, 0,54 mmol), 3,4-dihidroxibenzaldehído (76 mg, 0,56 mmol) y piperidina (3 |l, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 2 h a reflujo. Se recogió el precipitado mediante filtración, se suspendió en CH2Q 2 (5 ml) y se agitó la mezcla durante 2 h. Se recogió el sólido mediante filtración y se secó a vacío. Se obtuvo el producto como un sólido amarillo ( 120 mg, 60%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 10,65 (s, 3H), 8,73 (d, J = 2,4 Hz, 1H), 8,29 (s, 1H), 8,19 (dd, J = 8,7, 2,5 Hz, 1H), 7,62 (s, 1H), 7,61 (d, J = 2,3 Hz, 1H), 7,38 (dd, J = 8,4, 2,2 Hz, 1H), 6,96 - 6 , 8 8 (m, 2H), 3,90 (s, 3H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 163,1, 162,3, 159,5, 151,8, 151,7, 145,8, 144,2, 136,6, 125,9, 123,9, 123,1, 116,6, 116,4, 116,1, 110,5, 107,8, 53,3;
EMAR calc. para C19H15N4O4S [M+H]+ 395,0809, hallado 395,0810
Ejemplo preparativo 208
(E)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(3-metoxipiridin-2-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(3-metoxipiridin-2-il)tiazol-2-il)acetamida (76 mg, 0,27 mmol), 3,4-dihidroxibenzaldehído (38 mg, 0,27 mmol) y piperidina (3 |l, 0,027 mmol) en CH2Cl2 (5 ml); el tiempo de reacción fue de 2 h a reflujo. Se recogió el precipitado mediante filtración, se lavó con CH2Cl2 (5 ml) y se secó a vacío. Se obtuvo el producto como un sólido amarillo (94 mg, 90%).
1H-RMN (500 MHz, DMSOd) 5 (ppm) 10,14 (s, 2H), 8,31 (s, 1H), 8,24 (dd, J = 4,5, 1,2 Hz, 1H), 7,78 (s, 1H), 7,63 -7,56 (m, 2H), 7,39 (dd, J = 8,4, 4,6 Hz, 1H), 7,36 (dd, J = 8,4, 2,2 Hz, 1H), 6,92 (d, J = 8,3 Hz, 1H), 3,95 (s, 3H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 164,5, 160,3, 153,4, 151,6, 151,4, 145,8, 140,5, 139,4, 125,7, 123,9, 123,3, 119,3, 117,0, 116,4, 116,0, 114,2, 55,7;
EMAR calc. para C19H15N4O4S [M+H]+ 395,0809, hallado 395,0807.
Ejemplo preparativo 209
(E)-2-ciano-3-(3,5-dicloro-4-hidroxifenir)-N-(4-(piridin-2-il)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(piridin-2-il)tiazol-2-il)acetamida (80 mg, 0,33 mmol), 3,5-dicloro-4-hidroxibenzaldehído (63 mg, 0,33 mmol) y piperidina (3 ^l, 0,027 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 16 h a reflujo. Se recogió el precipitado mediante filtración, se lavó con CH2Cl2 (5 ml) y se secó a vacío. Se obtuvo el producto como un sólido amarillo (95 mg, 70%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 12,58 (s, 1H), 8,65 - 8,59 (m, 1H), 8,25 (s, 1H), 8,03 - 7,97 (m, 3H), 7,95 -7,86 (m, 2H), 7,38 -7,32 (m, 1H);
13C-RMN (126 MHz, DMSO-de) 5 (ppm) 162,2, 159,0, 158,3, 151,7, 149,5, 149,2, 137,3, 136,0, 131,2, 126,3, 123,5, 122,9, 120,0, 116,9, 112,2;
EMAR calc. para C18HnCl2N4O2S [M+H]+ 416,9974, hallado 416,9970.
Ejemplo preparativo 210
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(4-(4-(metilsulfonil)fenil)tiazol-2-il)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(4-(4-(metilsulfonil)fenil)tiazol-2-il)acetamida (100 mg, 0,311 mmol), 3,4-dihidroxibenzaldehído (43 mg, 0,31 mmol) y piperidina (3 ^l, 0,027 mmol) en CH2Cl2 (5 ml); el tiempo de reacción fue de 2 h a reflujo. Se recogió el precipitado mediante filtración, se lavó con una mezcla de CH2Cl2 y MeOH (5 ml 0,5 ml) y se secó a vacío. Se obtuvo el producto como un sólido amarillo (90 mg, 65%).
1H-RMN (500 MHz, DMSO-de) 5 (ppm) 10,39 (s, 3H), 8,27 (s, 1H), 8,19 (d, J = 8,1 Hz, 2H), 7,99 (d, J= 8,1 Hz, 2H), 7,91 (s, 1H), 7,61 (d, J = 2,3 Hz, 1H), 7,37 (dd, J = 8,4, 2,3 Hz, 1H), 6,92 (d, J = 8,3 Hz, 1H), 3,24 (s, 3H);
13C-RMN (126 MHz, DMSO-d6) 5 (ppm) 162,6, 160,2, 151,6, 151,5, 147,1, 145,8, 139,4, 138,9, 133,1, 127,6, 126,2, 125,8, 123,1, 116,8, 116,3, 116,0, 111,7, 43,6;
EMAR calc. para C20H16N3O5S2 [M+H]+ 442,0526, hallado 442,0528.
Ejemplo preparativo 211
(£)-2-ciano-3-(3,4-dihidroxifenil)-N-(5-feniltiazol-2-il)acrilamida (no se encuentra dentro del alcance de las reivindicaciones)
Se preparó el compuesto según el procedimiento general D2 a partir de 2-ciano-N-(5-feniltiazol-2-il)acetamida ( 66 mg, 0,271 mmol), 3,4-dihidroxibenzaldehído (0,244 mg, 33 mmol) y piperidina (2 ^l, 0,03 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 4 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo ( 66 mg, 67%).
1H-RMN (500 MHz, acetona-cfe) 5 (ppm) 8,29 (s, 1H), 7,82 (s, 1H), 7,79 (s, 1H), 7,66 (d, J = 7,9 Hz, 2H), 7,52-7,42 (m, 3H), 7,34 (dd, J = 14,8, 7,7 Hz, 1H), 7,03 (d, J = 8,3 Hz, 1H);
13C-RMN (126 MHz, acetona-^) 5 fppm) 153,4, 151,9, 146,6, 132,8, 130,19, 128,8, 127,7, 126,9, 125,4, 117,4, 117,2, 116,9, 79,7, 79,1,78,9;
EMAR calc. para C19H14N3O3S [M+H]+ 364,0750, hallado [M+H]+ 364,0752.
Ejemplo preparativo 212
(£)-N-(4-(4-(ferc-butil)-2,6-dimetilfenil)tiazol-2-il)-2-ciano-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de N-(4-(4-(terc-butil)-2,6-dimetilfenil)tiazol-2-il)-2-cianoacetamida (80 mg, 0,24 mmol), 3,4-dihidroxibenzaldehído (33 mg, 0,24 mmol) y piperidina (3 |al, 0,02 mmol) en CH2Cl2 (2 ml); el tiempo de reacción fue de 4 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (65 mg, 65%).
1H-RMN (500 MHz, acetona-^) 5 fppm) 8,25 (s, 1H), 7,77 (s, 1H), 7,47 (d, J = 8,2 Hz, 1H), 7,16 (s, 2H), 7,02 (d, J = 8,3 Hz, 1H), 6,95 (s, 1H), 2,13 (s, 6 H), 1,32 (s, 10H);
13C-RMN (126 MHz, acetona-^) 5 fppm) 163,3, 153,2, 151,9, 151,8, 146,5, 137,8, 132,7, 127,6, 125,5, 125,4, 117,5, 117,2, 116,8, 112,0, 102,3, 79,39, 79,1,35,1, 31,7, 20,9;
EMAR calc. para C25H26N3O3S [M+H]+ 448,1689, hallado 448,1691.
Ejemplo preparativo 213
(E)-N-(4-(3-(terc-butil)fenil)tiazol-2-il)-2-ciano-3-(3,4-dihidroxifenil)acrilamida

Se preparó el compuesto según el procedimiento general D2 a partir de N-(4-(2-(terc-butil)fenil)tiazol-2-il)-2-cianoacetamida (100 mg, 0,33 mmol), 3,4-dihidroxibenzaldehído (46 mg, 0,33 mmol) y piperidina (3 |al, 0,03 mmol) en CH2Cl2 (3 ml); el tiempo de reacción fue de 4 h a reflujo. Se obtuvo el producto, purificado mediante cromatografía en columna de fase inversa (H2O:MeOH:AcOH; de 60:40:0,05 a 5:95:0,05), como un sólido amarillo (83 mg, 60%).
1H-RMN (500 MHz, acetona-^) 5 fppm) 8,31 (s, 1H), 8,02 (s, 1H), 7,80 (s, 1H), 7,74 (d, J = 7,5 Hz, 1H), 7,56 (s, 1H), 7,51 (d, J = 8,1 Hz, 1H), 7,38 (dd, J = 24,3, 11,6 Hz, 2H), 7,04 (d, J = 8,3 Hz, 1H), 1,37 (s, 9H);
13C-RMN (126 MHz, acetona-da) 5 (ppm) 172,1, 162,1, 159,1, 153,7, 152,41, 152,1, 151,0, 146,6, 135,1, 129,4, 129,2, 127,9, 126,2, 125,9, 125,3, 124,2, 124,0, 117,2, 116,9, 109,1, 101,2, 31,8, 30,9;
EMAR calc. para C23H22N3O3S [M+H]+ 420,1376, hallado 420,1379.
ENSAYOS:
Ensayo HTS
Preparación de sustrato de ADN
Se prepara el sustrato de ADN mezclando los oligonucleótidos enumerados a continuación en una razón 1:1 para conseguir una concentración final de 6 |iM en un tampón que contenía Tris 50 mM pH 7,5, NaCl 100 mM y MgCh 8 mM.
Se calienta la mezcla durante 3 min a 65°C y se deja enfriar lentamente hasta TA. Luego se almacena a -20°C. Oligonucleótido 1: 5' CY5 - CTAAGTTCGTCAGGATTCCAGC
Oligonucleótido 2: 5' CTCTATCACTGTTACAATGCTGGAATCCTGACGAACTTAG - BBQ [650]
Este sustrato es una proyección en 5', que es un sustrato preferido de MRE11. La etiqueta fluorescente CY5 se inhibe mediante el inhibidor BBQ [650] y, por tanto, muestra muy poca o ninguna fluorescencia. La fluorescencia aumenta tras la escisión del sustrato por parte de MRE11, cuando se produce la separación de las dos etiquetas. Configuración
Las condiciones del ensayo son las siguientes:
Tipo de microplaca: 1536 pocillos, negra, de fondo redondo, de poliestireno, no tratada (n.° de cat. De Corning 3936) Volumen de reacción total: 5 |il
Concentración de MRE11 en la reacción: 18 nM
Concentración de sustrato de ADN en la reacción: 40 nM
Número de compuestos para someter a prueba: 257
Intervalo de concentración de inhibidor sometido a prueba: 7 nM - 50 |iM
Réplicas: 3
Puntos de concentración: 13
Etapa de dilución: 2,1
Cada placa contiene una serie de pocillos de control de señal alta y baja, en los que no se añade compuesto:
Señal alta: MRE11 sustrato de ADN
Señal baja: sustrato de ADN solamente
Estos se usan durante la evaluación de los datos.
Dos controles de ensayo adicionales:
1) Para comprobar si se produce el desenrollamiento del ADN por parte de los compuestos solos.
Se mezcla el sustrato de ADN [CY5+BBQ(650)] con los compuestos sin ninguna proteína presente. Esto se realiza como medición única a una concentración de inhibidor de 25 |iM.
2) Para comprobar si los compuestos pueden inhibir CY5.
Se mezcla el oligonucleótido de una sola hebra CY5 con los compuestos sin ninguna proteína presente. Esto se realiza como medición única a una concentración de inhibidor de 25 |iM.
Se crea la disposición de las placas mediante el software interno (CZ-Openscreen Prague) y se transfiere esta información a la estación robótica HTS.
Etapas del ensayo
1) Preparar 50 ml de mezcla maestra:
16,7 ml 5x tampón de reacción (Bis Tris 150 mM pH 7; DTT 5 mM)
1042 |il de MnCl2400 mM
32,3 ml de H2O
2) Llenar las placas con 3 |il de mezcla maestra por pocillo usando MultiDrop (Thermo Scientific)
3) Transferir los compuestos a las placas en la estación robótica con el dispensador inalámbrico Echo (Labcyte) 4) Medir la autofluorescencia con el lector EnVision (PerkinElmer)
5) Preparar 20 ml de MRE11 90 nM en tampón T+50 (Tris-HCl 25 mM pH 7,5, KCl 50 mM, glicerol al 8,7%, EDTA 0,5 mM)
6 ) Añadir 1 |il de MRE11 90 nM a los pocillos correspondientes usando MultiDrop
7) Incubar previamente a TA durante 30 min
8 ) Preparar 20 ml de una disolución 200 nM en el sustrato de ADN con proyección en 5':
480 |il de ADN 6 |iM 19,52 ml de H2O
9) Preparar 4 ml de una disolución 200 nM del ADN de una sola hebra (oligonucleótido 1):
8 |il de ADN 100 |iM 4 ml de H2O
10) Añadir 1 |il de cada disolución 200 nM de ADN a los pocillos correspondientes con MultiDrop
11) Medir la fluorescencia con el lector EnVision cada 45 minutos
Lectura de fluorescencia: CY5 Xex/em = 620/665 nm
Análisis
La reacción se inicia mediante la adición de ADN y se cuenta el tiempo de reacción a partir ese momento, incluyendo un retardo de 15 min. Se miden diez puntos de tiempo:

El análisis de los datos del ensayo se realizó a t=4 h. Esto corresponde al tiempo cuando la reacción está próxima a su máximo.
El análisis de los datos se realizó usando el software interno para obtener la CI50 para cada compuesto.
Ensayo HR
Se transfectaron células DR-GFP U2OS (Methods in Molecular Biology 2012, 920, 379) con 2,5 |ig de vector pCAGGS que expresa I-SceI y se trataron con los inhibidores a una concentración de 25 |iM. 72 horas después de la transfección, se sometieron a tripsinización las células y se resuspendieron en BSA al 3% en PBS. Se llevó a cabo la detección de la fluorescencia de GFP usando un citómetro de flujo BD FACSVerse y se analizaron los datos con el software FlowJo.
Ensayo RPA
Se trataron previamente células U2OS durante 1 h con inhibidores de MRE11 seguido de la adición de camptotecina 1 |iM durante 1 h. Se lisaron las células en tampón de carga de SDS-PAGE, se sometieron a sonicación y se sometieron a ebullición a 70°C durante 10 min. Se analizaron cantidades iguales de proteína (50-100 |ig) mediante electroforesis en gel de Tris-glicina. Se cuantificaron los niveles de expresión usando el software Multi Gauge y se expresaron en relación con el control de carga. Se usó una dilución 1:1000 de RPA32 S4/S8 fosforilado (A300-245A, Bethyl Laboratories).
Resultados
La tabla 1 resume las actividades inhibidoras de los compuestos indicados sometidos a prueba en el ensayo de nucleasas HTS in vitro (CI50), ensayo HR (inhibición de HR a 25 |iM) y ensayo RPA (% de inhibición de fosforilación de RPA a [10 |iM] o [25 |iM] (concentración del inhibidor)).
Ensayo de nucleasas HTS: (CI50)
A: CI50 < 2 jiM
B: 2 |iM < CI50 < 10 |iM
C: 10 |iM< CI50 < 90 |iM
Ensayo HR: inhibición de HR a 25 |iM
A: inhibición de HR > 75%
B: 75% > inhibición de HR > 50%
C: 50% > inhibición de HR
Tabla 1:



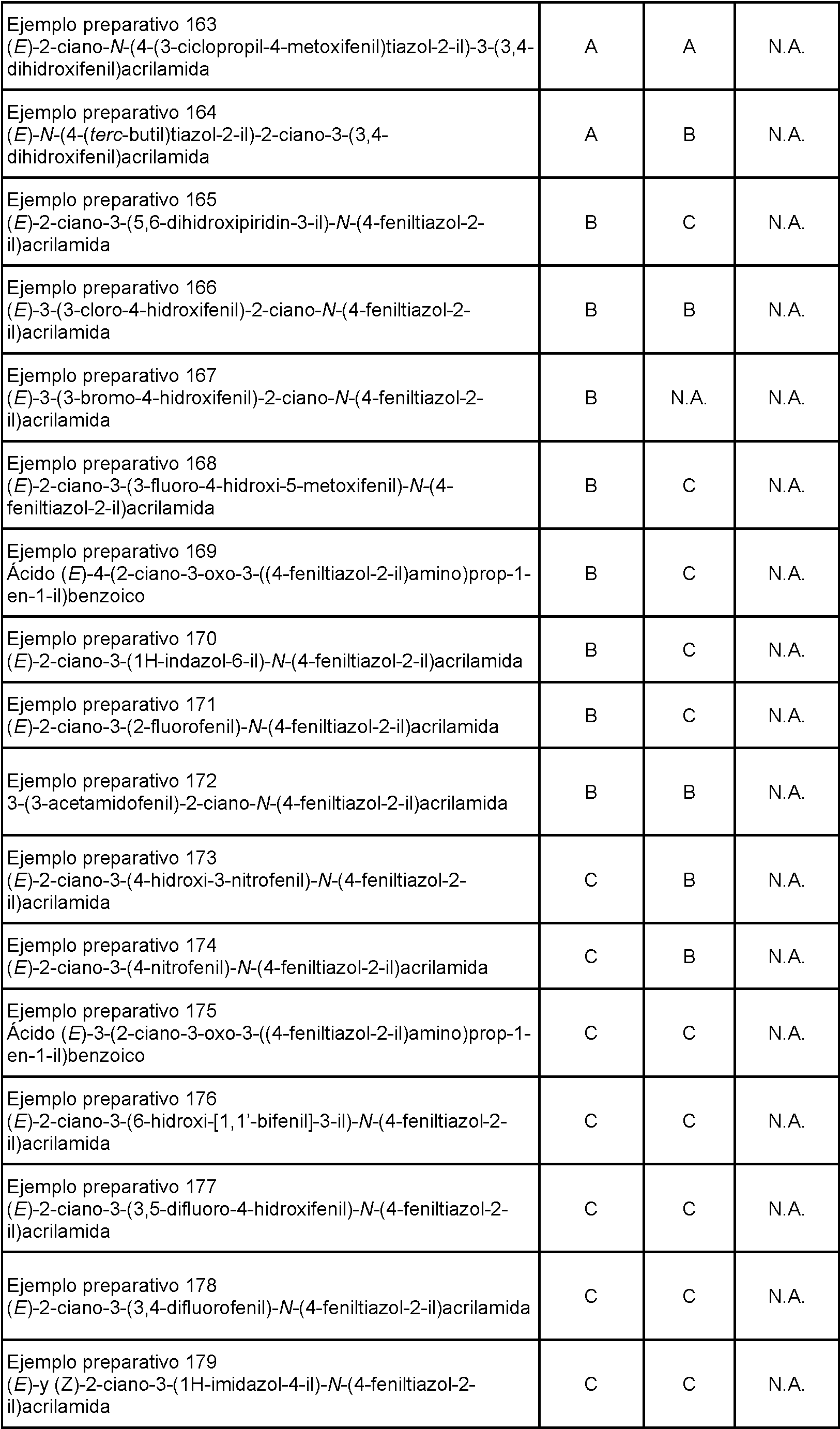


Claims (13)
- REIVINDICACIONESCompuesto de fórmula general (1):
 o una sal farmacéuticamente aceptable, o un solvato del mismo,en la que:R1 se selecciona del grupo que consiste en arilo; heteroarilo;en el que cada uno del arilo, heteroarilo puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, seleccionándose cada resto independientemente del grupo que consiste en F, Cl, Br, I, OH, CN, N3, =O, O(alquilo C1-C6), =S, SH, S(alquilo C1-C6), S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3, C2F5, OCF3, OC2F5, n H2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), =N-OH, =N-O(alquilo C1-C6), NO2 , COOH, COO(alquilo C1-C6), CO(alquilo C1-C6), CONH2 , CONH(alquilo C1-C6), CON(alquilo C1-C6)2 , (alquil C rC 6)-S(O)2-NH-, (alquil C1-C6)-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-(SO)2-, (alquil C1-C6)2N-(SO)2-, (alquil C1-C6)-CO-NH-, (alquil Cr C6)-CO-N(alquil C1-C6)-, (alquil C1-C6)-OCO-NH-, (alquil Cr C6)-OCO-N(alquil C1-C6)-, (alquil C1-C6)-CO-NH-CO-, (alquil CrC6)-CO-N(alquil C1-C6)-CO-, NH2-CO-NH-, (alquil CrC6)-NH-CO-NH-, (alquil C1-C6)2N-CO-NH-, NH2-CO-N(alquil C1-C6)-, (alquil C1-C6)-NH-CO-N(alquil C1-C6)-, (alquil C rC 6)2 N-CO-N(alquil C1-C6)-, NH2-S(O)2-NH-, (alquil C1-C6)-NH-S(O)2-NH-, (alquil C rC 6hN-S(O)2-NH-, NH2-S(O)2-N(alquil C1-C6)-, (alquil C rC 6)-NH-S(O)2-N(alquil C1-C6)-, (alquil Ci-C6)2N-S(O)2-N(alquil C1-C6)-, alquilo C1-C6, O-alquilo C1-C6, O-fenilo, fenilo; mientras que el alquilo C1-C6 , O-fenilo, fenilo en estos restos puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes independientemente seleccionados de: Cl, Br, I, alquilo C1-C6, OH, O-alquilo C1-C6 , SH, SCH3, S(O)-alquilo C1-C6 , S(O)2-alquilo C1-C6 , CF3 , OCF3, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), COOH, COO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2 , NHC(O)-alquilo C1-C6 o NHC(O)NH2 ;R2 se selecciona de arilo C6-C12 y heteroarilo que tiene de 5 a 12 átomos de anillos, que puede estar opcionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, OH, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5, O(alquilo C1-C4), fenilo, O-fenilo, NH2, N(alquilo C1-C4)2, NO2, NHCO(alquilo C1-C4), c F3, OCF3, CN, S(O)2-alquilo C1-C6, SO2NH(alquilo C1-C6), SO2N(alquilo C1-C6)2; en particular seleccionados de F, Cl, Br, alquilo C1-C6, O(alquilo C1-C4), CF3, OCF3;R6 se selecciona del grupo que consiste en H; heterociclilo; cicloalquilo; heteroarilo; arilo; heteroarilo; en el que cada uno del arilo, cicloalquilo, heterociclilo, heteroarilo, puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, alquilo C1-C6, OH, O-alquilo C1-C6, SH, SCH3, S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3, OCF3, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), NO2 , COOH, COO(alquilo C1-C6), CONH2 , CONH(alquilo C1-C6), CON(alquilo C1-C6)2, NHC(O)-alquilo C1-C6 o NHC(O)NH2;R3 se selecciona del grupo que consiste en H; arilo; heteroarilo; heterociclilo; cicloalquilo; alquilo; y halógeno;en el que cada uno del arilo, cicloalquilo, alquilo o heteroarilo puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, seleccionándose cada resto independientemente del grupo que consiste en F, Cl, Br, I, OH, CN, N3, =O, O(alquilo C1-C6), =S, SH, S(alquilo C1-C6), S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3 , C2 F5 , OCF3 , OC2F5, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), =N-OH, =N-O(alquilo C1-C6), NO2 , COOH, COO(alquilo C1-C6), CO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, (alquil C1-C6)-S(O)2-NH-, (alquil C1-C6)-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-(SO)2-, (alquil C1-C6)2N-(SO)2-, (alquil C1-C6)-CO-NH-, (alquil C1-C6)-CO-N(alquil C1-C6)-, (alquil C1-C6)-Oc O-NH-, (alquil C1-C6)-OCO-N(alquil C1-C6)-, (alquil C1-C6)-CONH-CO-, (alquil Ci-Ca)-CO-N(alquil Ci-Ca)-CO-, NH2-CO-NH-, (alquil Ci-Ca)-NH-CO-NH-, (alquil Ci-Ca)2N-CO-NH-, NH2-CO-N(alquil Ci-Ca)-, (alquil Ci-Ca)-NH-CO-N(alquil Ci-Ca)-, (alquil Ci-Ca)2N-CO-N(alquil CiCa)-, NH2-S(O)2-NH-, (alquil Ci-Ca)-NH-S(O)2-NH-, (alquil Ci-Ca)2N-S(O)2-NH-, NH2-S(O)2-N(alquil Ci-Ca)-, (alquil Ci-Ca)-NH-S(O)2-N(alquil Ci-Ca)-, (alquil Ci-Ca)2N-S(O)2-N(alquil Ci-Ca)-, alquilo Ci-Ca, O-alquilo CiCa, O-fenilo, fenilo;mientras que el alquilo Ci-Ca, O-fenilo, fenilo en estos restos puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes independientemente seleccionados de: F, Cl, Br, alquilo Ci-Ca, OH, O-alquilo Ci-Ca, SH, SCH3, S(O)-alquilo Ci-Ca, S(O)2-alquilo Ci-Ca, CF3 , OCF3, NH2, NH(alquilo Ci-Ca), N(alquilo Ci-Ca)2 (tal como N(CH3)2), NO2 , COOH, COO(alquilo Ci-Ca), CONH2 , CONH(alquilo Ci-Ca), CON(alquilo Ci-Ca)2, NHC(O)-alquilo Ci-Ca o NHC(O)NH2 ,R4 se selecciona del grupo que consiste en H y alquilo Ci-Ca;R5 se selecciona del grupo que consiste en H y arilo;para su uso en un método de tratamiento de cáncer, envejecimiento prematuro o enfermedades neurológicas, más específicamente de cáncer relacionado con la inestabilidad genómica, envejecimiento prematuro relacionado con la inestabilidad genómica y/o enfermedades neurológicas relacionadas con la inestabilidad genómica, en particular cáncer relacionado con MREii, envejecimiento prematuro relacionado con MREii y/o enfermedades neurológicas relacionadas con MREii.
o una sal farmacéuticamente aceptable, o un solvato del mismo,en la que:R1 se selecciona del grupo que consiste en arilo; heteroarilo;en el que cada uno del arilo, heteroarilo puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, seleccionándose cada resto independientemente del grupo que consiste en F, Cl, Br, I, OH, CN, N3, =O, O(alquilo C1-C6), =S, SH, S(alquilo C1-C6), S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3, C2F5, OCF3, OC2F5, n H2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), =N-OH, =N-O(alquilo C1-C6), NO2 , COOH, COO(alquilo C1-C6), CO(alquilo C1-C6), CONH2 , CONH(alquilo C1-C6), CON(alquilo C1-C6)2 , (alquil C rC 6)-S(O)2-NH-, (alquil C1-C6)-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-(SO)2-, (alquil C1-C6)2N-(SO)2-, (alquil C1-C6)-CO-NH-, (alquil Cr C6)-CO-N(alquil C1-C6)-, (alquil C1-C6)-OCO-NH-, (alquil Cr C6)-OCO-N(alquil C1-C6)-, (alquil C1-C6)-CO-NH-CO-, (alquil CrC6)-CO-N(alquil C1-C6)-CO-, NH2-CO-NH-, (alquil CrC6)-NH-CO-NH-, (alquil C1-C6)2N-CO-NH-, NH2-CO-N(alquil C1-C6)-, (alquil C1-C6)-NH-CO-N(alquil C1-C6)-, (alquil C rC 6)2 N-CO-N(alquil C1-C6)-, NH2-S(O)2-NH-, (alquil C1-C6)-NH-S(O)2-NH-, (alquil C rC 6hN-S(O)2-NH-, NH2-S(O)2-N(alquil C1-C6)-, (alquil C rC 6)-NH-S(O)2-N(alquil C1-C6)-, (alquil Ci-C6)2N-S(O)2-N(alquil C1-C6)-, alquilo C1-C6, O-alquilo C1-C6, O-fenilo, fenilo; mientras que el alquilo C1-C6 , O-fenilo, fenilo en estos restos puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes independientemente seleccionados de: Cl, Br, I, alquilo C1-C6, OH, O-alquilo C1-C6 , SH, SCH3, S(O)-alquilo C1-C6 , S(O)2-alquilo C1-C6 , CF3 , OCF3, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), COOH, COO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2 , NHC(O)-alquilo C1-C6 o NHC(O)NH2 ;R2 se selecciona de arilo C6-C12 y heteroarilo que tiene de 5 a 12 átomos de anillos, que puede estar opcionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, OH, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5, O(alquilo C1-C4), fenilo, O-fenilo, NH2, N(alquilo C1-C4)2, NO2, NHCO(alquilo C1-C4), c F3, OCF3, CN, S(O)2-alquilo C1-C6, SO2NH(alquilo C1-C6), SO2N(alquilo C1-C6)2; en particular seleccionados de F, Cl, Br, alquilo C1-C6, O(alquilo C1-C4), CF3, OCF3;R6 se selecciona del grupo que consiste en H; heterociclilo; cicloalquilo; heteroarilo; arilo; heteroarilo; en el que cada uno del arilo, cicloalquilo, heterociclilo, heteroarilo, puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, alquilo C1-C6, OH, O-alquilo C1-C6, SH, SCH3, S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3, OCF3, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), NO2 , COOH, COO(alquilo C1-C6), CONH2 , CONH(alquilo C1-C6), CON(alquilo C1-C6)2, NHC(O)-alquilo C1-C6 o NHC(O)NH2;R3 se selecciona del grupo que consiste en H; arilo; heteroarilo; heterociclilo; cicloalquilo; alquilo; y halógeno;en el que cada uno del arilo, cicloalquilo, alquilo o heteroarilo puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, seleccionándose cada resto independientemente del grupo que consiste en F, Cl, Br, I, OH, CN, N3, =O, O(alquilo C1-C6), =S, SH, S(alquilo C1-C6), S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, CF3 , C2 F5 , OCF3 , OC2F5, NH2, NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), =N-OH, =N-O(alquilo C1-C6), NO2 , COOH, COO(alquilo C1-C6), CO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, (alquil C1-C6)-S(O)2-NH-, (alquil C1-C6)-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-(SO)2-, (alquil C1-C6)2N-(SO)2-, (alquil C1-C6)-CO-NH-, (alquil C1-C6)-CO-N(alquil C1-C6)-, (alquil C1-C6)-Oc O-NH-, (alquil C1-C6)-OCO-N(alquil C1-C6)-, (alquil C1-C6)-CONH-CO-, (alquil Ci-Ca)-CO-N(alquil Ci-Ca)-CO-, NH2-CO-NH-, (alquil Ci-Ca)-NH-CO-NH-, (alquil Ci-Ca)2N-CO-NH-, NH2-CO-N(alquil Ci-Ca)-, (alquil Ci-Ca)-NH-CO-N(alquil Ci-Ca)-, (alquil Ci-Ca)2N-CO-N(alquil CiCa)-, NH2-S(O)2-NH-, (alquil Ci-Ca)-NH-S(O)2-NH-, (alquil Ci-Ca)2N-S(O)2-NH-, NH2-S(O)2-N(alquil Ci-Ca)-, (alquil Ci-Ca)-NH-S(O)2-N(alquil Ci-Ca)-, (alquil Ci-Ca)2N-S(O)2-N(alquil Ci-Ca)-, alquilo Ci-Ca, O-alquilo CiCa, O-fenilo, fenilo;mientras que el alquilo Ci-Ca, O-fenilo, fenilo en estos restos puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes independientemente seleccionados de: F, Cl, Br, alquilo Ci-Ca, OH, O-alquilo Ci-Ca, SH, SCH3, S(O)-alquilo Ci-Ca, S(O)2-alquilo Ci-Ca, CF3 , OCF3, NH2, NH(alquilo Ci-Ca), N(alquilo Ci-Ca)2 (tal como N(CH3)2), NO2 , COOH, COO(alquilo Ci-Ca), CONH2 , CONH(alquilo Ci-Ca), CON(alquilo Ci-Ca)2, NHC(O)-alquilo Ci-Ca o NHC(O)NH2 ,R4 se selecciona del grupo que consiste en H y alquilo Ci-Ca;R5 se selecciona del grupo que consiste en H y arilo;para su uso en un método de tratamiento de cáncer, envejecimiento prematuro o enfermedades neurológicas, más específicamente de cáncer relacionado con la inestabilidad genómica, envejecimiento prematuro relacionado con la inestabilidad genómica y/o enfermedades neurológicas relacionadas con la inestabilidad genómica, en particular cáncer relacionado con MREii, envejecimiento prematuro relacionado con MREii y/o enfermedades neurológicas relacionadas con MREii. - 2. Compuesto para su uso según la reivindicación i, en el que R1 se selecciona de arilo Ca-Ci2 y heteroariloque tiene de 5 a 12 átomos de anillos, siendo dicho arilo o heteroarilo monocíclico, bicíclico o tricíclico, en el que el arilo o heteroarilo está sustituido con de uno a tres grupos OH, y el arilo o heteroarilo puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, alquilo Ci-Ca, O(alquilo C1-C4), fenilo, O-fenilo, NH2, N(alquilo Ci-C4)2 , NO2 , NHCO(alquilo C1-C4), CF3, OCF3, CN, S(O)2-alquilo Ci-Ca, SO2NH(alquilo Ci-C SO2N(alquilo Ci-Ca)2 ; en particular seleccionados de F, Cl, Br, alquilo Ci-Ca, O(alquilo C1-C4), CF3, OCF3.
- 3. Compuesto para su uso según la reivindicación i, en el que Ri se selecciona de arilo Ca-Ci2 y heteroariloque tiene de 5 a i2 átomos de anillos, en el que el arilo o heteroarilo está sustituido con dos grupos OH o un grupo OH y un grupo seleccionado de CN, Cl, Br, F, y el arilo o heteroarilo puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, alquilo Ci-Ca, O(alquilo C1-C4), fenilo, O-fenilo, NH2 , N(alquilo Ci-C4)2, NO2 , NHCO(alquilo C1-C4), CF3 , OCF3, CN, S(O)2-alquilo Ci-Ca, SO2NH(alquilo Ci-Ca), SO2N(alquilo Ci-Ca)2; en particular seleccionados de F, Cl, Br, alquilo Ci-Ca, O(alquilo C1-C4), CF3 , OCF3; preferiblemente, R1 es 3,4-dihidroxifenilo.
- 4. Compuesto para su uso según una cualquiera de las reivindicaciones anteriores, en el que R2 se seleccionade fenilo, naftilo, benzofuranilo, piridilo, tiofenilo, piridazinilo, que no están sustituidos o que están sustituidos con de uno a dos sustituyentes independientemente seleccionados de O(alquilo C1-C4), OH, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5, F, Br, Cl, CF3 , OCF3.
- 5. Compuesto para su uso según una cualquiera de las reivindicaciones anteriores, en el que Ra se seleccionade H, fenilo, morfolinilo, opcionalmente sustituidos tal como se describió anteriormente en el presente documento.
- 6. Compuesto para su uso según una cualquiera de las reivindicaciones anteriores, en el que R3 se seleccionade H, fenilo, metilo, etilo, propilo, isopropilo, butilo, isobutilo, ferc-butilo, F, Cl, Br, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, tetrahidropiranilo, tetrahidrofuranilo, piperidinilo, que no están sustituidos o que están sustituidos con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, OH, alquilo Ci-Ca, fenilo, NH2 , N(alquilo Ci-C4)2 , NO2 , NHCO(alquilo C1-C4), CF3 , OCF3 , CN, SO2NH(alquilo Ci-Ca), SO2N(alquilo Ci-Ca)2, S(O)2-alquilo Ci-Ca.
- 7. Compuesto para su uso según una cualquiera de las reivindicaciones i a a, en el que el uso es en un método de tratamiento de cánceres de mama, de colon, de próstata, de pulmón, de cabeza y cuello, hepático, de ovario, colorrectal, gástrico, melanoma, leucemias, síndrome de rotura de Nimega y síndrome de tipo rotura de Nimega, ataxia-telangiectasia y trastorno de tipo ataxia-telangiectasia, y anemia de Fanconi.
- 8. Compuesto para su uso según una cualquiera de las reivindicaciones i a a, en el que el uso es en un método de tratamiento de tumores sólidos con BRCA-2 mutado.
- 9. Compuesto de fórmula general (la):i i 3
 o una sal farmacéuticamente aceptable, o un solvato del mismo,en la que:R1 se selecciona de arilo C6-C12 y heteroarilo que tiene de 5 a 12 átomos de anillos, en el que el arilo o heteroarilo está sustituido con de uno a tres grupos OH, preferiblemente con dos grupos OH, y el arilo o heteroarilo puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, alquilo C1-C6, O(alquilo C1-C4), fenilo, O-fenilo, NH2, N(alquilo C1-C4)2, NO2, NHCO(alquilo C1-C4), CF3, OCF3, CN, S(O)2-alquilo C1-C6, SO2NH(alquilo C1-C6), SO2N(alquilo C1-C6)2; en particular seleccionados de F, Cl, Br, alquilo C1-C6, O(alquilo C1-C4), CF3, OCF3; siempre que R1 no sea 4-hidroxi-3-metoxifenilo cuando R2 sea 4-etilfenilo y R3,R4, R5, R6 sean H;R2 se selecciona de arilo C6-C12 y heteroarilo que tiene de 5 a 12 átomos de anillos, que puede estar opcionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, OH, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5, O(alquilo C1-C4), fenilo, O-fenilo, NH2, N(alquilo C1-C4)2, NO2, NHCO(alquilo C1-C4), c F3, OCF3, CN, S(O)2-alquilo C1-C6, SO2NH(alquilo C1-C6), SO2N(alquilo C1-C6)2; en particular seleccionados de F, Cl, Br, alquilo C1-C6, O(alquilo C1-C4), CF3, OCF3;R6 se selecciona del grupo que consiste en H; heterociclilo; cicloalquilo; heteroarilo; arilo; heteroarilo; en el que cada uno del arilo, cicloalquilo, heterociclilo, heteroarilo, puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, alquilo C1-C6 , OH, O-alquilo C1-C6, SH, SCH3, S(O)-alquilo C1-C6 , S(O)2-alquilo C1-C6 , CF3 , OCF3 , NH2 , NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2),NO2 , COOH, COO(alquilo C1-C6), CONH2 , CONH(alquilo C1-C6), CON(alquilo C1-C6)2, NHC(O)-alquilo C1-C6 o NHC(O)NH2;R3 se selecciona del grupo que consiste en H; arilo; heteroarilo; heterociclilo; cicloalquilo; alquilo; y halógeno;en el que cada uno del arilo, cicloalquilo, alquilo o heteroarilo puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, seleccionándose cada resto independientemente del grupo que consiste en F, Cl, Br, I, OH, CN, N3, =O, O(alquilo C1-C6), =S, SH, S(alquilo C1-C6), S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, Cf3 , C2F5, Oc F3 , Oc 2F5, Nh2, NH( N(alquilo C1-C6)2 (tal como N(CH3)2), =N-OH, =N-O(alquilo C1-C6), NO2 , COOH, COO(alquilo C1-C6), CO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, (alquil C1-C6)-S(O)2-NH-, (alquil C1-C6)-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-(SO)2-, (alquil C1-C6)2N-(SO)2-, (alquil C1-C6)-CO-NH-, (alquilC1-C6)-CO-N(alquil C1-C6)-, (alquil C1-C6)-Oc O-NH-, (alquil C1-C6)-OCO-N(alquil C1-C6)-, (alquil C1-C6)-CO-NH-CO-, (alquil C1-C6)-CO-N(alquil Cr C6)-CO-, NH2-CO-NH-, (alquil C1-CO-NH-CO-NH-, (alquil C ^ fc N -CO-NH-, NH2-CO-N(alquil C1-C6)-, (alquil Cr C6)-NH-CO-N(alquil C1-C 5)-, (alquil C1-C6)2N-CO-N(alquil C1-C6)-, NH2-S(O)2-NH-, (alquil C1-C)-NH-S(O)2-NH-, (alquil C1-Cs)2N-S(O)2-NH-, NH2-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)2N-S(O)2-N(alquil C1-C6)-, alquilo C1-C6, O-alquilo C1-C6, O-fenilo, fenilo;mientras que el alquilo C1-C6 , O-fenilo, fenilo en estos restos puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes independientemente seleccionados de: F, Cl, Br, alquilo C1-C6, OH, O-alquilo C1-C6 , SH, SCH3, S(O)-alquilo C1-C6 , S(O)2-alquilo C1-C6 , CF3 , OCF NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), NO2 , COOH, COO(alquilo C1-C6), CONH2 , CONH(alquilo C1-C6), CON(alquilo C1-C6)2 , NHC(O)-alquilo C1-C6 o NHC(O)NH2 ,R4 se selecciona del grupo que consiste en H y alquilo C1-C6;R5 se selecciona del grupo que consiste en H y arilo.
o una sal farmacéuticamente aceptable, o un solvato del mismo,en la que:R1 se selecciona de arilo C6-C12 y heteroarilo que tiene de 5 a 12 átomos de anillos, en el que el arilo o heteroarilo está sustituido con de uno a tres grupos OH, preferiblemente con dos grupos OH, y el arilo o heteroarilo puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, alquilo C1-C6, O(alquilo C1-C4), fenilo, O-fenilo, NH2, N(alquilo C1-C4)2, NO2, NHCO(alquilo C1-C4), CF3, OCF3, CN, S(O)2-alquilo C1-C6, SO2NH(alquilo C1-C6), SO2N(alquilo C1-C6)2; en particular seleccionados de F, Cl, Br, alquilo C1-C6, O(alquilo C1-C4), CF3, OCF3; siempre que R1 no sea 4-hidroxi-3-metoxifenilo cuando R2 sea 4-etilfenilo y R3,R4, R5, R6 sean H;R2 se selecciona de arilo C6-C12 y heteroarilo que tiene de 5 a 12 átomos de anillos, que puede estar opcionalmente sustituido con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, OH, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5, O(alquilo C1-C4), fenilo, O-fenilo, NH2, N(alquilo C1-C4)2, NO2, NHCO(alquilo C1-C4), c F3, OCF3, CN, S(O)2-alquilo C1-C6, SO2NH(alquilo C1-C6), SO2N(alquilo C1-C6)2; en particular seleccionados de F, Cl, Br, alquilo C1-C6, O(alquilo C1-C4), CF3, OCF3;R6 se selecciona del grupo que consiste en H; heterociclilo; cicloalquilo; heteroarilo; arilo; heteroarilo; en el que cada uno del arilo, cicloalquilo, heterociclilo, heteroarilo, puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, alquilo C1-C6 , OH, O-alquilo C1-C6, SH, SCH3, S(O)-alquilo C1-C6 , S(O)2-alquilo C1-C6 , CF3 , OCF3 , NH2 , NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2),NO2 , COOH, COO(alquilo C1-C6), CONH2 , CONH(alquilo C1-C6), CON(alquilo C1-C6)2, NHC(O)-alquilo C1-C6 o NHC(O)NH2;R3 se selecciona del grupo que consiste en H; arilo; heteroarilo; heterociclilo; cicloalquilo; alquilo; y halógeno;en el que cada uno del arilo, cicloalquilo, alquilo o heteroarilo puede no estar sustituido o estar opcionalmente sustituido con uno o más restos que pueden ser iguales o diferentes, seleccionándose cada resto independientemente del grupo que consiste en F, Cl, Br, I, OH, CN, N3, =O, O(alquilo C1-C6), =S, SH, S(alquilo C1-C6), S(O)-alquilo C1-C6, S(O)2-alquilo C1-C6, Cf3 , C2F5, Oc F3 , Oc 2F5, Nh2, NH( N(alquilo C1-C6)2 (tal como N(CH3)2), =N-OH, =N-O(alquilo C1-C6), NO2 , COOH, COO(alquilo C1-C6), CO(alquilo C1-C6), CONH2, CONH(alquilo C1-C6), CON(alquilo C1-C6)2, (alquil C1-C6)-S(O)2-NH-, (alquil C1-C6)-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-(SO)2-, (alquil C1-C6)2N-(SO)2-, (alquil C1-C6)-CO-NH-, (alquilC1-C6)-CO-N(alquil C1-C6)-, (alquil C1-C6)-Oc O-NH-, (alquil C1-C6)-OCO-N(alquil C1-C6)-, (alquil C1-C6)-CO-NH-CO-, (alquil C1-C6)-CO-N(alquil Cr C6)-CO-, NH2-CO-NH-, (alquil C1-CO-NH-CO-NH-, (alquil C ^ fc N -CO-NH-, NH2-CO-N(alquil C1-C6)-, (alquil Cr C6)-NH-CO-N(alquil C1-C 5)-, (alquil C1-C6)2N-CO-N(alquil C1-C6)-, NH2-S(O)2-NH-, (alquil C1-C)-NH-S(O)2-NH-, (alquil C1-Cs)2N-S(O)2-NH-, NH2-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)-NH-S(O)2-N(alquil C1-C6)-, (alquil C1-C6)2N-S(O)2-N(alquil C1-C6)-, alquilo C1-C6, O-alquilo C1-C6, O-fenilo, fenilo;mientras que el alquilo C1-C6 , O-fenilo, fenilo en estos restos puede estar, de manera opcional, adicionalmente sustituido con uno o más sustituyentes independientemente seleccionados de: F, Cl, Br, alquilo C1-C6, OH, O-alquilo C1-C6 , SH, SCH3, S(O)-alquilo C1-C6 , S(O)2-alquilo C1-C6 , CF3 , OCF NH(alquilo C1-C6), N(alquilo C1-C6)2 (tal como N(CH3)2), NO2 , COOH, COO(alquilo C1-C6), CONH2 , CONH(alquilo C1-C6), CON(alquilo C1-C6)2 , NHC(O)-alquilo C1-C6 o NHC(O)NH2 ,R4 se selecciona del grupo que consiste en H y alquilo C1-C6;R5 se selecciona del grupo que consiste en H y arilo. - 10. Compuesto según la reivindicación 9, en el que R2 se selecciona de fenilo, naftilo, benzofuranilo, piridilo, tiofenilo, piridazinilo, que no están sustituidos o que están sustituidos con de uno a dos sustituyentes independientemente seleccionados de O(alquilo C1-C4), OH, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5 , F, Br, Cl, CF3 , OCF3.
- 11. Compuesto según una cualquiera de las reivindicaciones 9 y 10, en el que R6 se selecciona de H, fenilo, morfolinilo, opcionalmente sustituidos tal como se describió anteriormente en el presente documento.
- 12. Compuesto según una cualquiera de las reivindicaciones 9 a 11, en el que R3 se selecciona de H, fenilo, metilo, etilo, propilo, isopropilo, butilo, isobutilo, ferc-butilo, F, Cl, Br, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, tetrahidropiranilo, tetrahidrofuranilo, piperidinilo, que no están sustituidos o que están sustituidos con uno o más sustituyentes, independientemente seleccionados del grupo que consiste en F, Cl, Br, OH, alquilo C1-C6 , fenilo, NH2, N(alquilo C rC 4)2, NO2, NHCO(alquilo C1-C4), CF3 , OCF3 , CN, SO2NH(alquilo C1-C6), SO2N(alquilo C1-C6)2, S(O)2-alquilo C1-C6.
- 13. Composición farmacéutica que comprende al menos un compuesto de fórmula (1a) según una cualquiera de las reivindicaciones 9 a 12 y al menos un compuesto auxiliar farmacéuticamente aceptable seleccionado del grupo que consiste en portadores, diluyentes, cargas, conservantes, estabilizadores, aglutinantes, agentes humectantes, emulsionantes, tampones farmacéuticamente aceptables.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18167651.1A EP3556755B1 (en) | 2018-04-17 | 2018-04-17 | Substituted aminothiazoles as inhibitors of nucleases |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2906335T3 true ES2906335T3 (es) | 2022-04-18 |
Family
ID=62017184
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18167651T Active ES2906335T3 (es) | 2018-04-17 | 2018-04-17 | Aminotiazoles sustituidos como inhibidores de nucleasas |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US11584742B2 (es) |
| EP (1) | EP3556755B1 (es) |
| JP (1) | JP7175031B2 (es) |
| CN (1) | CN112313229A (es) |
| AU (1) | AU2019256670B2 (es) |
| CA (1) | CA3096935A1 (es) |
| ES (1) | ES2906335T3 (es) |
| IL (1) | IL277866B2 (es) |
| WO (1) | WO2019201865A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3556756B1 (en) * | 2018-04-17 | 2020-10-07 | Masarykova univerzita | Substituted propanamides as inhibitors of nucleases |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08248567A (ja) * | 1995-03-14 | 1996-09-27 | Konica Corp | ハロゲン化銀写真感光材料 |
| WO2005072731A1 (en) * | 2004-01-29 | 2005-08-11 | X-Ceptor Therapeutics, Inc. | 3-phenyl-n- ((1, 3, 4) thiadiazol-2-yl) -acrylamide derivatives and related compounds as modulators of estrogen-related receptors for the treatment of e.g. cancer, rheumatoid arthritis or neurological disorders |
| WO2009086303A2 (en) * | 2007-12-21 | 2009-07-09 | University Of Rochester | Method for altering the lifespan of eukaryotic organisms |
| WO2010075372A1 (en) | 2008-12-22 | 2010-07-01 | The Trustees Of Columbia University In The City Of New York | Inhibitors of mre11, rad50 and/or nbs1 |
| ES2763527T3 (es) * | 2014-04-04 | 2020-05-29 | Univ Michigan Regents | Inhibidores de molécula pequeña de mcl-1 y usos de los mismos |
| WO2018036501A1 (en) * | 2016-08-24 | 2018-03-01 | National Institute Of Biological Sciences, Beijing | Entacapone-related compounds to treat injury |
| CN108947931B (zh) * | 2017-05-27 | 2023-07-07 | 中国科学院上海有机化学研究所 | 一种噻唑酰胺类化合物、其制备方法、药物组合物及应用 |
| EP3556756B1 (en) * | 2018-04-17 | 2020-10-07 | Masarykova univerzita | Substituted propanamides as inhibitors of nucleases |
-
2018
- 2018-04-17 ES ES18167651T patent/ES2906335T3/es active Active
- 2018-04-17 EP EP18167651.1A patent/EP3556755B1/en not_active Not-in-force
-
2019
- 2019-04-15 WO PCT/EP2019/059689 patent/WO2019201865A1/en active Application Filing
- 2019-04-15 AU AU2019256670A patent/AU2019256670B2/en not_active Ceased
- 2019-04-15 CA CA3096935A patent/CA3096935A1/en active Pending
- 2019-04-15 IL IL277866A patent/IL277866B2/en unknown
- 2019-04-15 CN CN201980040558.3A patent/CN112313229A/zh active Pending
- 2019-04-15 JP JP2020556927A patent/JP7175031B2/ja active Active
- 2019-04-15 US US17/047,004 patent/US11584742B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| IL277866B2 (en) | 2024-01-01 |
| IL277866B1 (en) | 2023-09-01 |
| EP3556755A1 (en) | 2019-10-23 |
| US20220002282A1 (en) | 2022-01-06 |
| IL277866A (en) | 2020-11-30 |
| JP7175031B2 (ja) | 2022-11-18 |
| CA3096935A1 (en) | 2019-10-24 |
| CN112313229A (zh) | 2021-02-02 |
| EP3556755B1 (en) | 2021-11-17 |
| WO2019201865A1 (en) | 2019-10-24 |
| JP2021521235A (ja) | 2021-08-26 |
| AU2019256670A1 (en) | 2020-11-05 |
| US11584742B2 (en) | 2023-02-21 |
| AU2019256670B2 (en) | 2022-09-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5524173B2 (ja) | 抗増殖性化合物およびその治療的使用 | |
| CA2371158A1 (en) | Diaminothiazoles and their use for inhibiting protein kinases | |
| BRPI0709680B1 (pt) | Derivados de pirrol, tiofeno e furano substituídos por piridil e piridimil como inibidores de quinase | |
| KR20070073791A (ko) | 티아디아졸 화합물 및 이의 사용방법 | |
| EP2942349A1 (en) | Enzyme modulators and treatments | |
| WO2003062215A1 (en) | 4(hetero-) aryl substituted (thia-/oxa-/pyra) zoles for inhibition of tie-2 | |
| WO2004110351A2 (en) | Heterocyclic compounds for treating hepatitis c virus | |
| BRPI0809506A2 (pt) | Composto, e, uso de um composto | |
| CN114127049A (zh) | Acss2抑制剂和其使用方法 | |
| JP2021528454A (ja) | 化合物 | |
| KR102214225B1 (ko) | 5원 헤테로시클릭 아미드계 wnt 경로 억제제 | |
| WO2002090360A1 (en) | Compounds useful as kinase inhibitors for the treatment of hyperproliferative diseases | |
| ES2906335T3 (es) | Aminotiazoles sustituidos como inhibidores de nucleasas | |
| EP4089089A1 (en) | Polycyclic compounds for inhibiting rna helicase dhx33 and use thereof | |
| CN109843879B (zh) | 作为dyrk1抑制剂的苯并噻唑衍生物 | |
| ES2841951T3 (es) | Propanamidas sustituidas como inhibidores de nucleasas | |
| CN111247137A (zh) | 一种嘧啶类化合物、其制备方法及其医药用途 | |
| JP2002531500A (ja) | Myt1キナーゼ阻害剤 | |
| JP2002531503A (ja) | Myt1キナーゼ阻害剤 | |
| KR20230165833A (ko) | 치환된 피롤 카복스아미드, 이의 제조방법 및 이의 키나제 억제제로서의 용도 | |
| AU2003236875A1 (en) | 4(hetero-) aryl substituted (thia-/oxa-/pyra) zoles for inhibition of tie-2 |