ES2902136T3 - Composiciones farmacéuticas que comprenden un fármaco antirretrovírico y un potenciador farmacocinético - Google Patents
Composiciones farmacéuticas que comprenden un fármaco antirretrovírico y un potenciador farmacocinético Download PDFInfo
- Publication number
- ES2902136T3 ES2902136T3 ES17711359T ES17711359T ES2902136T3 ES 2902136 T3 ES2902136 T3 ES 2902136T3 ES 17711359 T ES17711359 T ES 17711359T ES 17711359 T ES17711359 T ES 17711359T ES 2902136 T3 ES2902136 T3 ES 2902136T3
- Authority
- ES
- Spain
- Prior art keywords
- piperine
- enhancer
- trans
- combination
- inhibitor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/683—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols
- A61K31/685—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols one of the hydroxy compounds having nitrogen atoms, e.g. phosphatidylserine, lecithin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/34—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4525—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/63—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide
- A61K31/635—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide having a heterocyclic ring, e.g. sulfadiazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/22—Heterocyclic compounds, e.g. ascorbic acid, tocopherol or pyrrolidones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
- A61K9/209—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat containing drug in at least two layers or in the core and in at least one outer layer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biophysics (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Biotechnology (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Una composición farmacéutica oral o inyectable que comprende una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico, que comprende (i) un inhibidor análogo de nucleótido de la transcriptasa inversa (NRTI) seleccionado del grupo que comprende: tenofovir alafenamida fumarato, tenofovir disoproxil fumarato, adefovir y una combinación de los mismos; o (ii) un inhibidor de la proteasa (IP) que comprende darunavir, y una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético que comprende piperina, tetrahidropiperina, cis-piperina, trans-piperina, cis,trans-piperina, trans,cis-piperina, cis,cis-piperina, trans,trans- piperina o una combinación de las mismas.
Description
DESCRIPCIÓN
Composiciones farmacéuticas que comprenden un fármaco antirretrovírico y un potenciador farmacocinético
Referencia cruzada a solicitudes relacionadas
Campo
La presente invención se refiere a composiciones farmacéuticas que comprenden al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético. La presente invención también proporciona el proceso de fabricación de las mismas y el uso de dichas composiciones para la prevención, tratamiento o profilaxis de enfermedades provocadas por virus, de manera específica provocadas por retrovirus o el virus de la hepatitis B.
Antecedentes
El virus de la inmunodeficiencia humana (VIH), el virus que provoca el síndrome de la inmunodeficiencia adquirida (SIDA), se ha convertido en uno de los problemas de salud más graves del mundo. El VIH pertenece a una clase de virus llamados retrovirus. Los retrovirus son virus de ARN (ácido ribonucleico) y para replicarse (duplicarse), los virus deben hacer una copia de ADN (ácido desoxirribonucleico) de su ARN. Son los genes del ADN los que permiten que el virus se replique. Como todos los virus, el VIH puede replicarse solo dentro de las células, apoderándose de la maquinaria de la célula para reproducirse. Sin embargo, solo el VIH y otros retrovirus, una vez dentro de una célula, utilizan una enzima llamada transcriptasa inversa para convertir su ARN en ADN, que se puede incorporar a los genes de la célula hospedadora.
El VIH destruye los linfocitos T positivos para CD4 (CD4), que son glóbulos blancos esenciales para mantener la función del sistema inmunitario humano. La destrucción de estas células deja a las personas infectadas con el VIH vulnerables a otras infecciones, enfermedades y otras complicaciones. Estas células, a veces llamadas "linfocitos T cooperadores", juegan un papel central en la respuesta inmunitaria, enviando señales a otras células del sistema inmunitario para que realicen sus funciones especiales. A medida que el VIH ataca a estas células, la persona infectada con el virus está menos preparada para luchas contra infecciones y enfermedades, lo que da como resultado, en última instancia, el desarrollo del SIDA.
Una persona sana no infectada suele tener de 800 a 1.200 linfocitos T CD4 por milímetro cúbico (mm3) de sangre. El VIH parece tener una afinidad particular por la célula de linfocito T-4 humano, que juega un papel vital en el sistema inmunitario del organismo. Los glóbulos blancos (GB) infectados por el VIH conducen a una disminución en la población de GB. Finalmente, el sistema inmunitario se vuelve inoperativo e ineficaz contra diversas enfermedades oportunistas. Durante la infección por VIH no tratada, el número de estas células en la sangre de una persona disminuye de manera progresiva. Cuando el recuento de linfocitos T CD4 cae por debajo de 200/mm3, una persona se vuelve especialmente vulnerable a las infecciones oportunistas y los cánceres que caracterizan al SIDA, la etapa final de la enfermedad del VIH. Las personas con SIDA a menudo sufren infecciones de los pulmones, tubo digestivo, cerebro, ojos y otros órganos, así como una pérdida de peso debilitante, diarrea, afecciones neurológicas y cánceres tales como el sarcoma de Kaposi y determinados tipos de linfomas.
El primer caso se informó en 1981 y en la actualidad hay aproximadamente 36,9 millones de personas que viven con el VIH y decenas de millones de personas han muerto por causas relacionadas con el sida desde el comienzo de la epidemia. Si bien se han notificado nuevos casos en todas las regiones del mundo, aproximadamente el 70 % se encuentran en África subsahariana. Además, según la hoja de datos de 2016 de Onusida, en 2015, había 36,7 millones de personas que vivían con el VIH. En diciembre de 2015, 17 millones de personas que vivían con el VIH estaban accediendo a la terapia antirretrovírica. En 2015, 1,1 millones de personas murieron por causas relacionadas con el sida en todo el mundo.
El VIH es el agente causante del SIDA que ha creado un importante problema de salud no solo en India sino a nivel mundial. El SIDA provoca una degradación gradual del sistema inmunitario del organismo, así como un deterioro progresivo de los sistemas nerviosos central y periférico. Desde su reconocimiento inicial a principios de la década de 1980, el SIDA se ha propagado rápidamente y ahora ha alcanzado proporciones epidémicas en un segmento relativamente limitado de la población. La investigación intensiva ha llevado al descubrimiento del agente responsable, el retrovirus linfotrópico T humano 111 (HTLV-111, del inglés "human T-lymphotropic retrovirus 111") comúnmente denominado virus de la inmunodeficiencia humana o VIH.
Los fármacos antirretrovíricos disponibles actualmente para el tratamiento del VIH incluyen: zidovudina o AZT (Retrovir®), didanosina o DDI (Videx®), estavudina o D4T (Zenith®), lamivudina o 3TC (Epivir®), zalcitabina o DDC (Hivid®), sulfato de abacavir (Ziagen®), tenofovir disoproxil fumarato (Viread®), emtricitabina (Emtriva®), Combivir® (contiene 3TC y AZT), Trizivir® (contiene abacavir, 3TC y AZT), Epzicom® (contiene abacavir y lamivudina); nevirapina (Viramune®), delavirdina (Rescriptor®), efavirenz (Sustiva®), saquinavir (Invirase®, Fortovase®), indinavir (Crixivan®), ritonavir (Norvir®), nelfinavir (Viracept®), amprenavir (Agenerase®), atazanavir (Reyataz®), Evotaz® (contiene atazanavir y cobicistat), fosamprenavir (Lexiva®), Kaletra® (contiene lopinavir y ritonavir), enfuvirtida (T-20,
Fuzeon®), Tolvada® (contiene tenofovir y emtricitabina), darunavir (Prezista®), Prezcobix® (contiene darunavir y cobicistat), dolutegravir (Tivicay®), Triumeq® (contiene dolutegravir, abacavir y lamivudina), elvitegravir (Vitekta®), Genvoya® (contiene elvitegravir, cobicistat, tenofovir alafenamida fumarato y emtricitabina), Stribild® (contiene elvitegravir, cobicistat, tenofovir disoproxil fumarato y emtricitabina), raltegravir (Isentress®), Complera® (contiene emtricitabina, tenofovir disoproxil fumarato, rilpivirina) y Atripla® (contiene una combinación triple de dosis fija de tenofovir, emtricitabina y efavirenz).
Entre el 5 y el 10 % de las personas con VIH también están infectadas con el virus de la hepatitis B (a menudo llamado infección conjunta). Las personas con VIH tienen menos probabilidades de eliminar la hepatitis B de manera natural sin tratamiento. Las personas con infección conjunta por el VIH y la hepatitis pueden tener una progresión más rápida de la enfermedad hepática y es posible que no respondan tan bien al tratamiento de la hepatitis B. Sin embargo, tener hepatitis B no parece empeorar la enfermedad del VIH. La infección por el virus de la hepatitis B (VHB) es la infección vírica crónica más común en el mundo. Se estima que 2 mil millones de personas se han infectado y más de 350 millones son portadores crónicos del virus. El VHB se transmite a través del contacto con sangre o semen infectados.
Además, los virus del SIDA (VIH) y de la hepatitis B son notablemente similares en cuanto a que comparten la transcripción inversa, en sus orígenes ancestrales y elementos genéticos comunes, y en sus modos de transmisión. Ambos son hipermutables y existen como cuasiespecies debido principalmente a errores en la transcripción inversa, aunque existe una restricción intensa en la competencia replicativa de la mayoría de los mutantes de la hepatitis B. Se diferencian por la falta de una integrasa en el virus de la hepatitis B y por su patogenia en el hospedador infectado. El VIH sobrevive principalmente por la variabilidad antigénica, la evasión inmunitaria y el deterioro de la función inmunitaria a través de elementos de control reguladores víricos que buscan restringir el daño fatal al hospedador. El virus de la hepatitis B sobrevive principalmente por la mutación del antígeno e/genes centrales que obvian directamente la destrucción de linfocitos T citotóxicos de las células hepáticas infectadas o limitan indirectamente la destrucción de las células infectadas mediante la inducción de anergia en la respuesta de los linfocitos T citotóxicos.
Además, los fármacos antirretrovíricos tales como lamivudina, adefovir, entecavir y tenofovir se han aprobado para el tratamiento de la infección crónica por el VHB.
Los reforzadores o potenciadores farmacocinéticos se utilizan para reforzar la eficacia de los fármacos antirretrovíricos. Cuando se administra de manera conjunta un reforzador o potenciador farmacocinético con un fármaco antirretrovírico, el potenciador farmacocinético interfiere con la degradación del fármaco antirretrovírico, lo que hace que el fármaco antirretrovírico permanezca en el organismo durante más tiempo y en una concentración más elevada. Los reforzadores o potenciadores farmacocinéticos provocan de manera específica la inhibición del sistema enzimático del citocromo P450 3A4 que conduce a un aumento de las concentraciones en plasma de los fármacos antirretrovíricos administrados de manera conjunta. Los inhibidores de la proteasa son una clase de fármacos antirretrovíricos que generalmente presentan una barrera genética elevada para la resistencia a los fármacos y, por lo tanto, requieren un reforzador o potenciador farmacocinético para ser administrado de manera conjunta. De todos los medicamentos aprobados para el tratamiento del VIH, el ritonavir y el cobicistat se denominan "reforzadores" o "potenciadores" farmacocinéticos. El ritonavir se utiliza debido a su capacidad para inhibir la enzima metabolizadora de fármacos citocromo P450 (CYP) 3A4. Administrado en una dosis baja, el ritonavir reduce el metabolismo de los inhibidores de la proteasa tales como lopinavir y atazanavir, que son ampliamente metabolizados por CYP3A4, mejorando así la exposición al fármaco. Cobicistat también es un potente inhibidor de las isoenzimas CYP3A y aumenta las concentraciones en plasma de fármacos que se metabolizan mediante CYP3A, tales como los inhibidores de la proteasa, esto es, atazanavir y darunavir.
Además de ritonavir y cobicistat, hay muchas sustancias de origen natural que se describen en la bibliografía y pueden explorarse para mejorar la actividad farmacocinética de determinados fármacos.
Estas sustancias de origen natural que actúan como biopotenciadores son entidades químicas que promueven y aumentan la biodisponibilidad de los fármacos que se mezclan con ellos y no presentan un efecto sinérgico con el fármaco. Los ejemplos de estos biopotenciadores incluyen piperina, ajo, Carum carvi, Cuminum cyminum, lisergol, naringina, quercetina, niaziridina, glicirricina, estevia, orina de vaca, jengibre destilado, etc.
Estos "reforzadores" o "potenciadores" farmacocinéticos podrían reducir el coste de la terapia antivírica, reducir la carga de píldoras para los pacientes y/o reducir el riesgo de concentraciones antivíricas subterapéuticas (por ejemplo, desarrollo de resistencia así como mejora de la adherencia a la terapia antivírica).
Sin embargo, esta mejora farmacocinética puede estar asociada a sus propios riesgos. El fármaco precipitante, por ejemplo, el reforzador o potenciador, puede tener que administrarse en una dosis que inhiba la eliminación del fármaco objeto y que no produzca sus propios efectos secundarios.
Por consiguiente, el potenciador, que normalmente es un inhibidor potente, puede inhibir involuntariamente la eliminación de otros fármacos, dando lugar a efectos adversos no deseados. Asimismo, si la dosis del potenciador no se ajusta de manera cuidadosa, o es inadecuada o en exceso, en última instancia, puede provocar una disminución o un aumento de la concentración del fármaco objeto. Por lo tanto, considerando estos aspectos, la selección de la dosis
adecuada del potenciador juega un papel fundamental.
Por ejemplo, aunque ritonavir tiene actividad antivírica, provoca efectos secundarios indeseables, que incluyen problemas gastrointestinales, especialmente diarrea crónica y anomalías de los lípidos. Después, se desarrolló cobicistat para producir aproximadamente el mismo grado de efecto que ritonavir, pero sin actividad antivírica ni ningún otro efecto secundario problemático.
Cobicistat es un sustrato de CYP3A4 (CYP2D6 es una vía secundaria de metabolismo) e inhibe su propio metabolismo. Además, cobicistat también inhibe la glucoproteína P (gp-P) y CYP2D6 y, por lo tanto, existen varias posibles interacciones que pueden ocurrir con cobicistat.
Además, los pacientes que reciben tratamiento para el VIH siempre corren el riesgo de interacciones con otros medicamentos no relacionados con el VIH y se sabe que cobicistat presenta interacciones farmacológicas clave con antiácidos, benzodiazepams, p-bloqueantes, bloqueantes de los canales de calcio, fármacos para la disfunción eréctil, corticosteroides inhalados/inyectables, estatinas, anticonceptivos gestagénico, rifampicina y maraviroc.
Los reforzadores o potenciadores farmacocinéticos que se utilizan actualmente inhiben involuntariamente la eliminación de otros fármacos, dando lugar a efectos adversos no deseados. Asimismo, no se sabe que el uso de piperina y/o sus análogos estructurales tales como tetrahidropiperina, cis,trans-piperina, trans,cis-piperina, cis,cis-piperina y trans,trans, mejore la biodisponibilidad de dichos fármacos antirretrovíricos.
Ravisekhar Kasibhatta et al., "Influence of piperine on the pharmacokinetics of nevirapine under fasting conditions: A Randomised, Crossover, Placebo-Controlled Study, Department of Clinical Pharmacology and Therapeutics, Nizam's Institute of Medical Sciences, Hyderabad, India, Drugs R D 2007; 8 (6): 383-391, se refiere a un estudio piloto para evaluar la biodisponibilidad de nevirapina cuando se administra con piperina.
J. VAN GELDER et al., "Intestinal absorption enhancement of the ester prodrug tenofovir disoproxil fumarate through modulation of the biochemical barrier by defined ester mixtures, drug metabolism and disposition", Vol. 30, N.° 8, 2002 de The American Society for Pharmacology and Experimental Therapeutics analiza el efecto de ésteres aislados y mezclas de ésteres en la estabilidad intestinal y la absorción de tenofovir disoproxil en comparación con el efecto del extracto de fresa, que se ha demostrado que mejora la absorción del profármaco a través de las monocapas de Caco-2 y en el íleon de rata.
Durg Vijay Singh et al., "A plausible explanation for enhanced bioavailability of P-Gp substrates in presence of piperine: simulation for next generation of P-Gp inhibitors", artículo en el Journal of Molecular Modeling • Agosto de 2012, analiza el papel de la gp-P en la biodisponibilidad de varios fármacos, principalmente fármacos citotóxicos hidrófobos y anticancerosos.
Por lo tanto, sigue existiendo una necesidad de proporcionar una terapia de combinación de un reforzador o potenciador farmacocinético con dichos fármacos antirretrovíricos para el tratamiento del VIH que reduzca la dosis de dichos fármacos antirretrovíricos, los efectos secundarios que presentan estos fármacos así como que mantenga la concentración óptima de los mismos. Además, el uso de un reforzador o potenciador farmacocinético de origen natural eliminaría o reduciría las interacciones con otros medicamentos distintos del VIH que se administrarían de manera simultánea.
Sumario
Todas las realizaciones definidas en el presente documento están destinadas a estar restringidas al fármaco(s) antirretrovírico(s) y al reforzador(es) o potenciador(es) farmacocinético(s) como se establece en las reivindicaciones adjuntas.
Por consiguiente, un objetivo de la presente invención es proporcionar una composición que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético como se establece en la reivindicación 1.
Otro objetivo de la presente invención es proporcionar una composición que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético con efectos secundarios reducidos.
Otro objetivo más de la presente invención es proporcionar una composición que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético con interacciones farmacológicas reducidas.
Otro objetivo de la presente invención es proporcionar una composición que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético para una administración una o dos veces al día.
Otro objetivo de la presente invención es proporcionar una composición que comprende al menos un fármaco
antirretrovírico y al menos un reforzador o potenciador farmacocinético con una dosis reducida.
Otro objetivo más de la presente invención es proporcionar una composición que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético en forma de un kit.
Otro objetivo más de la presente invención es proporcionar una composición que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético para su uso en la prevención, tratamiento o profilaxis de enfermedades provocadas por virus, de manera específica provocadas por retrovirus, de manera específica el síndrome de la inmunodeficiencia adquirida o una infección por el VIH, mediante la administración de la composición que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético.
Otro objetivo más de la presente invención es proporcionar una composición que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético para su uso en el tratamiento de enfermedades provocadas por virus, de manera específica provocadas por el virus de la hepatitis B, mediante la administración de la composición que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético.
Otro objetivo más de la presente invención es proporcionar el uso de una composición farmacéutica que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético para el tratamiento o profilaxis de enfermedades provocadas por virus, de manera específica provocadas por retrovirus, de manera específica el síndrome de la inmunodeficiencia adquirida o una infección por VIH.
Otro objetivo más de la presente invención es proporcionar el uso de una composición farmacéutica que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético para el tratamiento de enfermedades provocadas por virus, de manera específica el virus de la hepatitis B.
De acuerdo con un aspecto de la presente invención, se proporciona una composición farmacéutica que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético y uno o más excipientes farmacéuticamente aceptables.
De acuerdo con otro aspecto de la invención, se proporciona un proceso para preparar una composición farmacéutica que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético con al menos uno o más excipientes farmacéuticamente aceptables.
De acuerdo con otro aspecto de la presente invención, se proporciona una composición farmacéutica que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético para su uso en el tratamiento de enfermedades provocadas por virus, de manera específica provocadas por retrovirus, de manera especial el SIDA o una infección por el VIH, mediante la administración de una cantidad terapéuticamente eficaz de la composición farmacéutica a un sujeto que lo necesite.
De acuerdo con otro aspecto de la presente invención, se proporciona una composición farmacéutica que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético para su uso en el tratamiento de enfermedades provocadas por virus, de manera específica provocadas por el virus de la hepatitis B, mediante la administración de una cantidad terapéuticamente eficaz de la composición farmacéutica a un sujeto que lo necesite.
De acuerdo con otro aspecto de la presente invención, se proporciona el uso de una composición farmacéutica que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético de acuerdo con la presente invención en la fabricación de un medicamento para el tratamiento de enfermedades provocadas por virus, de manera específica provocadas por retrovirus, de manera especial el SIDA o una infección por el VIH.
De acuerdo con otro aspecto de la presente invención, se proporciona el uso de una composición farmacéutica que comprende al menos un fármaco antirretrovírico y al menos un reforzador o potenciador farmacocinético de acuerdo con la presente invención en la fabricación de un medicamento para el tratamiento de enfermedades provocadas por virus, de manera específica provocadas por el virus de la hepatitis B.
En algunas realizaciones, se proporciona una composición farmacéutica oral o inyectable que comprende una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico y una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético o derivado del mismo.
En algunas realizaciones, se proporciona una composición farmacéutica oral o inyectable que comprende una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico; una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético o derivado del mismo; y uno o más excipientes farmacéuticamente aceptables que comprenden vehículos, diluyentes, cargas, aglutinantes, lubricantes, deslizantes, disgregantes, agentes de carga, aromatizantes o cualquier combinación de los mismos.
En algunas realizaciones, la composición farmacéutica que comprende (i) una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico o un fármaco antivírico; (ii) una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético o derivado del mismo; y (iii) uno o más excipientes farmacéuticamente aceptables que comprenden vehículos, diluyentes, cargas, aglutinantes, lubricantes, deslizantes, disgregantes, agentes de carga, aromatizantes o cualquier combinación de los mismos se proporciona para su uso en el tratamiento de enfermedades provocadas por retrovirus o el virus de la hepatitis B en un paciente que necesita dicho tratamiento mediante la administración de la composición.
En algunas realizaciones, se proporciona un método para preparar una composición farmacéutica que mejora la biodisponibilidad de un fármaco antirretrovírico, comprendiendo el método: mezclar una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico y una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético o derivado del mismo con uno o más excipientes farmacéuticamente aceptables para preparar la composición farmacéutica.
En algunas realizaciones, se proporciona un kit para el tratamiento de enfermedades provocadas por retrovirus o el virus de la hepatitis B, comprendiendo el kit una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico y una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético o derivado del mismo, en donde el al menos un fármaco antirretrovírico está en una composición separada del al menos un reforzador o potenciador farmacocinético o derivado del mismo.
En algunas realizaciones, se proporciona una composición que comprende una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico y que proporciona una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético o derivado del mismo para mejorar la biodisponibilidad de un fármaco antirretrovírico oral.
Breve descripción de las figuras
La figura 1 representa un gráfico de barras de los resultados de un ensayo unidireccional que muestra la permeabilidad de tenofovir alafenamida fumarato (TAF) y de tenofovir disoproxil fumarato (TDF). Se observó que TDF y TAF eran fármacos de permeabilidad baja a moderada.
La figura 2 muestra un gráfico de barras de los resultados de un ensayo bidireccional de digoxina 10 pM, digoxina 10 pM (A-B) piperina 10 pM, TAF 100 pM (VRD-1063/16/187), TAF 100 pM (VRD-1063/16/187) piperina 0,1 pM y TAF 100 pM (VRD-1063/16/187) piperina 10 pM. Los resultados mostraron que la absorción de TAF aumenta con piperina al disminuir la relación de flujo de salida de TAF.
La figura 3 muestra un gráfico de barras de los resultados de un ensayo bidireccional de digoxina 10 pM, dolutegravir 5 pM, dolutegravir 5 pM piperina 1 pM, dolutegravir 5 pM piperina 10 pM, dolutegravir 5 pM verapamilo 1 pM y dolutegravir 5 pM verapamilo 10 pM.
La figura 4 muestra un gráfico de barras de los resultados de un ensayo bidireccional de digoxina 10 pM, darunavir 40 pM, darunavir 40 pM piperina 1 pM, darunavir 40 pM piperina 10 pM, darunavir 40 pM cobicistat 10 pM y darunavir 40 pM cobicistat 100 pM. Los resultados mostraron que la absorción de darunavir aumenta con piperina al disminuir la relación de flujo de salida de TAF.
La figura 5 representa un gráfico de barras de los resultados de un ensayo bidireccional de digoxina 10 pM, TDF 200 pM, TDF 100 pM, TDF 100 pM piperina 10 pM y TDF 100 pM tetrahidropiperina 10 pM. Los resultados mostraron que la absorción de TDF aumenta con piperina al disminuir la relación de flujo de salida, la absorción de TDF aumenta con la tetrahidropiperina al disminuir la relación de flujo de salida, y se observó una mejora comparable en la permeabilidad de TDF tanto con la piperina como la tetrahidropiperina.
La figura 6 muestra un gráfico de barras de las concentraciones en plasma de tenofovir para 300 mg de TDF y 300 mg de TDF 20 mg de piperina en diferentes momentos.
La figura 7 muestra un gráfico de barras de las concentraciones en plasma de tenofovir para 300 mg de TDF y 300 mg de TDF 20 mg de piperina en diferentes momentos.
La figura 8 representa las concentraciones en plasma de tenofovir dependientes del tiempo para 300 mg de TDF, 300 mg de t Df 20 mg de piperina y 150 mg de TDF 20 mg de piperina.
Debe entenderse que las figuras no están dibujadas ni fotografiadas a escala. Además, la relación entre los objetos de una figura puede no estar a escala y, de hecho, puede tener una relación inversa en cuanto al tamaño. Las figuras están destinadas a aportar comprensión y claridad a la estructura de cada objeto mostrado y, por lo tanto, algunas características pueden exagerarse para ilustrar una característica específica de una estructura.
Descripción detallada
Para el tratamiento de enfermedades provocadas por retrovirus o el virus de la hepatitis B, especialmente el SIDA, una infección por el VIH o la hepatitis B, es fundamental que la cantidad máxima de fármaco llegue al lugar de acción. La mayoría de los fármacos antirretrovíricos tienen poca solubilidad y/o poca permeabilidad, lo que deteriora la biodisponibilidad del fármaco en gran medida.
Los inventores de la presente invención han encontrado formas de abordar los problemas de biodisponibilidad de dichos fármacos antirretrovíricos. En particular, los inventores han descubierto que, las propiedades de biodisponibilidad de estos fármacos se pueden mejorar mediante el uso de un reforzador o potenciador farmacocinético.
La biodisponibilidad mejorada de un fármaco antivírico se divulga en varias referencias. Role of Piperine As A Bioavailability Enhancer, UMESH K PATIL et al., International Journal of Recent Advances in Pharmaceutical Research, octubre de 2011; 4: 16-23 divulga la piperina como potenciador de la biodisponibilidad.
El documento WO2004067018 divulga el uso de extractos de Carum carvi como biopotenciadores, ya sea solo o en combinación con piperina o con extracto de Zinzeber officinale para mejorar la biodisponibilidad de zidovudina.
Natural Bioenhancers: An overview, Deepthi V. Tatiraju et al., Journal of Pharmacognosy and Phytochemistry 2013; 2 (3): 55-60. Este artículo divulga la combinación de piperina con nevirapina, en donde la piperina mejora la biodisponibilidad de la nevirapina.
Oral bioavailability enhancement of an anti-viral drug using an herbal bio-enhancer, Mohammad Asif, una tesis presentada a la Universidad de Ganpat. Este artículo divulga la combinación de piperina con efavirenz, en donde la piperina mejora la biodisponibilidad de efavirenz.
Bioenhancement effect of piperine and ginger oleo resin on the bioavailability of atazanavir, Swati Prakash et al., International Journal of Pharmacy and Pharmaceutical Sciences Vol 7, fascículo 10, 2015. Este artículo divulga la combinación de piperina con atazanvir, en donde la piperina mejoró la biodisponibilidad de atazanvir.
El documento WO03084462 divulga el proceso para fabricar una composición farmacéutica que contiene un inhibidor de la proteasa antirretrovírico tal como indinavir, saquinavir, amprenavir, nelfinavir, lopinavir y piperina en una sola composición farmacéutica.
En algunas realizaciones, los fármacos antirretrovíricos, de acuerdo con la presente invención, incluyen, pero sin limitación, inhibidores nucleosídicos de la transcriptasa inversa (NRTI, del inglés "Nucleoside Reverse Transcriptase Inhibitors"), inhibidores no nucleosídicos de la transcriptasa inversa (NNRTI, del inglés "Non - Nucleoside Reverse Transcriptase Inhibitors"), inhibidores análogos de nucleótidos de la transcriptasa inversa, inhibidores de la proteasa (IP), inhibidores de la integrasa, inhibidores de fusión, inhibidores de CCR5, anticuerpos monoclonales, inhibidores de glucoproteínas y cualquier combinación de los mismos.
En una realización, los inhibidores nucleosídicos de la transcriptasa inversa (NRTI) y los inhibidores no nucleosídicos de la transcriptasa inversa (NNRTI) incluyen, pero sin limitación, lamivudina, abacavir, zidovudina, emtricitabina, didanosina, estavudina, lobucavir, entecavir, apricitabina, censavudina, zalcitabina, dexelvucitabina, alovudina, efavirenz, amdoxovir, elvucitabina, festinavir, racivir, lersivirina, rilpivirina, etravirina, estampidina, doravirina y dapivirina.
En algunas realizaciones, preferentemente, los inhibidores nucleosídicos de la transcriptasa inversa (NRTI) y los inhibidores no nucleosídicos de la transcriptasa inversa (NNRTI) son abacavir y didanosina. Preferentemente, la dosis de abacavir varía de aproximadamente 3 mg a aproximadamente 300 mg, y la de didanosina varía de aproximadamente 2 mg a aproximadamente 200 mg para una administración dos veces al día.
El inhibidor de la proteasa de la presente invención comprende darunavir. En otra realización, los inhibidores de la proteasa incluyen, pero sin limitación, lopinavir, ritonavir, saquinavir, nelfinavir, amprenavir, indinavir, nelfinavir, atazanavir, lasinavir, palinavir, tirpranavir, fosamprenavir o tipranavir. Preferentemente, el inhibidor de la proteasa es tirpranavir. Preferentemente, la dosis de tipranavir varía de aproximadamente 5 mg a aproximadamente 500 mg, y la de darunavir varía de aproximadamente 1 mg a aproximadamente 800 mg para una administración dos veces al día. En algunas realizaciones, la dosis de darunavir varía de aproximadamente 1 mg a aproximadamente 500 mg, de aproximadamente 20 mg a aproximadamente 500 mg, de aproximadamente 25 mg a aproximadamente 500 mg, de aproximadamente 30 mg a aproximadamente 500 mg, de aproximadamente 35 mg a aproximadamente 500 mg, de aproximadamente 25 mg a aproximadamente 35 mg, de aproximadamente 50 mg a aproximadamente 400 mg o de aproximadamente 100 mg a aproximadamente 300 mg para una administración dos veces al día. En algunas realizaciones, la dosis de darunavir varía de aproximadamente 1 mg, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490, 500, 510, 520, 530, 540, 550, 560, 570, 580, 590, 600, 610, 620, 630, 640, 650, 660, 670, 680, 690, 700, 710, 720, 730, 740, 750, 760, 770, 780, 790 hasta aproximadamente 800 mg para administración una o dos veces al día. Cada dosis puede estar en una o más formas farmacéuticas unitarias, como se describe en el presente documento.
En otra realización, los inhibidores de la integrasa incluyen, pero sin limitación, dolutegravir, elvitegravir, raltegravir, bictegravir y cabotegravir. Preferentemente, los inhibidores de la integrasa son elvitegravir, dolutegravir y raltegravir. Preferentemente, la dosis de dolutegravir varía de aproximadamente 1 mg a aproximadamente 50 mg, la de
elvitegravir varía de aproximadamente 1 mg a aproximadamente 150 mg para una administración una vez al día y la de raltegravir varía de aproximadamente 4 mg a aproximadamente 400 mg para una administración una vez al día. En algunas realizaciones, la dosis de dolutegravir varía de aproximadamente 5 mg a aproximadamente 50 mg, de aproximadamente 20 mg a aproximadamente 50 mg, de aproximadamente 25 mg a aproximadamente 50 mg, de aproximadamente 25 mg a aproximadamente 45 mg, de aproximadamente 30 mg a aproximadamente 50 mg, de aproximadamente 30 mg a aproximadamente 40 mg o de aproximadamente 35 mg a aproximadamente 50 mg para una administración dos veces al día. En algunas realizaciones, la dosis de dolutegravir varía de aproximadamente 1 mg, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 a aproximadamente 50 mg para una administración una o dos veces al día. Cada dosis puede estar en una o más formas farmacéuticas unitarias, como se describe en el presente documento.
En otra realización, los inhibidores de fusión incluyen, pero sin limitación, maraviroc, enfuvirtida, griffithsin, aplaviroc, vicriviroc, plerixafor, fostemsavir y albuvirtida.
En otra realización, Los inhibidores de CCR5 incluyen, pero sin limitación, aplaviroc, vicriviroc, maraviroc y cenicriviroc.
En otra realización, los anticuerpos monoclonales incluyen, pero sin limitación, ibalizumab.
En otra realización, los inhibidores de glucoproteínas incluyen, pero sin limitación, sifuvirtida.
Los inhibidores análogos de nucleótidos de la transcriptasa inversa incluyen tenofovir alafenamida fumarato, tenofovir disoproxil fumarato y adefovir. Los inhibidores análogos de nucleótidos de la transcriptasa inversa son tenofovir alafenamida fumarato y tenofovir disoproxil fumarato. En algunas realizaciones, la dosis de tenofovir alafenamida fumarato varía de aproximadamente 1 mg a aproximadamente 25 mg, de aproximadamente 2,5 mg a aproximadamente 25 mg, de aproximadamente 5 mg a aproximadamente 20 mg o de aproximadamente 5 mg a aproximadamente 15 mg para una administración dos veces al día. En algunas realizaciones, la dosis de tenofovir alafenamida fumarato oscila de aproximadamente 1 mg, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 a aproximadamente 25 mg para una administración una vez al día o dos veces al día. En algunas realizaciones, la dosis de tenofovir disoproxil fumarato varía de aproximadamente 1 mg a aproximadamente 300 mg, de aproximadamente 1 mg a aproximadamente 150 mg, de aproximadamente 75 mg a aproximadamente 250 mg, de aproximadamente 100 mg a aproximadamente 200 mg o de aproximadamente 120 a aproximadamente 180 mg para una administración dos veces al día. En algunas realizaciones, la dosis de tenofovir disoproxil fumarato oscila entre aproximadamente 1 mg, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290 hasta aproximadamente 300 mg para una administración una o dos veces al día. Cada dosis puede estar en una o más formas farmacéuticas unitarias, como se describe en el presente documento.
La expresión "fármaco antirretrovírico" y "reforzador o potenciador farmacocinético" se utiliza en un sentido amplio para incluir no solo un "fármaco antirretrovírico" per se y un "reforzador o potenciador farmacocinético" per se sino también derivados farmacéuticamente aceptables de los mismos. Los derivados farmacéuticamente aceptables adecuados incluyen sales farmacéuticamente aceptables, solvatos farmacéuticamente aceptables, hidratos farmacéuticamente aceptables, anhídridos farmacéuticamente aceptables, enantiómeros farmacéuticamente aceptables, ésteres farmacéuticamente aceptables, isómeros farmacéuticamente aceptables, polimorfos farmacéuticamente aceptables, profármacos farmacéuticamente aceptables, tautómeros farmacéuticamente aceptables, complejos farmacéuticamente aceptables, etc.
La expresión "reforzador o potenciador farmacocinético" es un alcaloide. El reforzador o potenciador farmacocinético comprende piperina, tetrahidropiperina, c/s-piperina, trans-piperina, cis-trans piperina, trans,cis-piperina, cis,cis-piperina, frans,frans-piperina o una combinación de las mismas. El reforzador o potenciador farmacocinético es piperina o tetrahidropiperina y sus análogos o derivados. En algunas realizaciones, el reforzador o potenciador farmacocinético aumenta las concentraciones en plasma del fármaco antirretrovírico en un 10 %, 20, 30, 40, 50, 60, 70, 80, 90, 100 % o más en comparación con cuando no se utiliza el reforzador o potenciador farmacocinético.
El término "inyectable" es un modo de administración de la composición farmacéutica. La composición farmacéutica se puede administrar de diversas formas. En los seres humanos, las composiciones farmacéuticas se pueden administrar mediante vía parenteral. Por ejemplo, la composición farmacéutica se puede administrar mediante vía intravenosa (por ejemplo, inyección intravenosa), subcutánea, intradérmica o mediante inyección intramuscular. La administración intravenosa se puede lograr mediante la mezcla de la composición farmacéutica en un portador (vehículo) o excipiente farmacéutico adecuado como lo entienden los profesionales en la materia. Las formulaciones adecuadas para la administración parenteral comprenden de manera conveniente una preparación acuosa estéril de la composición farmacéutica, que se puede formular para ser isotónica con la sangre del paciente.
La expresión "cantidad terapéuticamente eficaz" o "cantidad eficaz" es tal que cuando se administra, la composición farmacéutica da como resultado la inhibición de un virus o enfermedad. La dosis administrada a un paciente puede ser en dosis únicas o múltiples dependiendo de varios factores, que incluyen las propiedades farmacocinéticas
administradas del fármaco, la vía de administración, las condiciones y características del paciente (sexo, edad, peso corporal, salud, talla, etc.) y el alcance de los síntomas, los tratamientos simultáneos, la frecuencia de tratamiento y el efecto deseado.
El término "tratamiento" o "tratar" una enfermedad, virus o afección se refiere a la ejecución de un protocolo que puede incluir la administración de uno o más fármacos a un paciente, en un esfuerzo por aliviar los signos o síntomas de la enfermedad, virus o afección. El alivio puede ocurrir antes de la aparición de los signos o síntomas de la enfermedad, virus o afección, así como después de su aparición. Por lo tanto, tratar o tratamiento incluye reducir, prevenir o la prevención de la enfermedad, virus o afección. Además, tratar o el tratamiento no requiere el alivio completo de los signos o síntomas, no requiere cura y, de manera específica, incluye protocolos que solo tienen un efecto marginal en el paciente.
Los frutos de la pimienta negra (Piper nigrum L.) y pimienta larga (Piper longum L.) son hierbas medicinales importantes en los sistemas de medicina ayurvédica y unani (tradicional de la India), en donde el remedio generalmente consiste en mezclas de hierbas. Se conoce y se ha documentado una amplia gama de usos medicinales de la pimienta negra, incluido su uso en el tratamiento de la leucodermia.
La piperina, puede ser el reforzador o potenciador farmacocinético. La piperina, el alcaloide principal que se encuentra en el fruto de la pimienta negra (Piper nigrum L.; Piperaceae), estimula la replicación de los melanocitos e induce la formación de dendritas melanocíticas. Se espera que la piperina provoque la repoblación de los parches de vitiligo a través de un efecto estimulante sobre los melanocitos perilesionales y foliculares.
La piperina se conoce químicamente como (1-2E, 4E-piperinoil-piperidina) y se representa de manera estructural como se muestra a continuación.

La piperina puede mejorar la biodisponibilidad del fármaco al promover la rápida absorción de fármacos y nutrientes mediante el aumento del suministro de sangre al tracto gastrointestinal, la disminución de la secreción de ácido clorhídrico para prevenir la degradación de algunos medicamentos, el aumento del contenido emulsionante del intestino, el aumento de enzimas como la Y-glutamil transpeptidasa que participan en el transporte activo y pasivo de nutrientes a las células intestinales.
La piperina puede aumentar la biodisponibilidad del fármaco mediante la inhibición de las enzimas que participan en la biotransformación de los fármacos y evitando así su inactivación y eliminación. También inhibe la p-glucoproteína, la proteína 'bomba' que elimina sustancias de las células y puede disminuir la producción intestinal de ácido glucurónico, permitiendo así que más sustancias entren al organismo en forma activa.
También se ha informado de la presencia de piperina en otras especies Piper, es decir P. acutisleginum, album, argyrophylum, attenuatum, aurantiacum, betle, callosum, chaba, cubeba, guineense, hancei, khasiana, longum, macropodum, nepalense, novae hollandiae, peepuloides, retrokacturn y sylvaticum.
La tetrahidropiperina es un análogo estructural de la piperina. Los dos enlaces dobles en las posiciones 2 y 4 están saturados para dar un análogo tetrahidro. La tetrahidropiperina se conoce químicamente como 5-(1,3-benzodioxol-5-il)-1-piperidin-1-ilpentan-1-ona y se representa de manera estructural como se muestra a continuación.

La tetrahidropiperina se produce como la piperina de forma natural en la pimienta negra (aproximadamente un 0,7 %
en la oleorresina de la pimienta negra). La tetrahidropiperina se puede sintetizar a partir de piperina que se extrae previamente de la oleorresina de la pimienta negra.
En algunas realizaciones, preferentemente, la dosis de piperina varía de aproximadamente 0,5 mg a aproximadamente 400 mg y la dosis de tetrahidropiperina varía de aproximadamente 0,5 mg a aproximadamente 400 mg. En algunas realizaciones, la dosis de piperina y/o de la tetrahidropiperina varía de aproximadamente 0,5 mg, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, a aproximadamente 400 mg. En algunas realizaciones, la relación del al menos un fármaco antirretrovírico con el al menos un reforzador o potenciador farmacocinético es de aproximadamente 100:1 a aproximadamente 1:1 en peso.
Preferentemente, la composición farmacéutica se puede proporcionar en formas farmacéuticas tales como, pero sin limitación, formas farmacéuticas unitarias que incluyen comprimidos, cápsulas (llenas de polvos, miniesferas, perlas, minicomprimidos, píldoras, microgránulos, unidades de comprimidos pequeños, sistemas de microgránulos de unidades múltiples (MUPS), comprimidos que se desintegran, comprimidos dispersables, gránulos y microesferas, multiparticulados), sobres (llenos de polvos, miniesferas, perlas, minicomprimidos, píldoras, microgránulos, unidades de comprimidos pequeños, MUPS, comprimidos que se desintegran, comprimidos dispersables, gránulos y microesferas, multiparticulados), polvos para reconstituir, parches transdérmicos y rociadores, sin embargo, otras formas farmacéuticas tales como formulaciones de liberación controlada, formulaciones liofilizadas, polvo liofilizado, formulaciones de liberación modificada, formulaciones de liberación retardada, formulaciones de liberación prolongada, formulaciones de liberación pulsátil, formulaciones de liberación dual y similares. También se pueden contemplar en el ámbito de la invención formas farmacéuticas líquidas, líquidas inyectables y semisólidas (líquidos, suspensiones, soluciones, dispersiones, pomadas, cremas, emulsiones, microemulsiones, pulverizadores, parches, unción dorsal puntual), preparaciones para inyección, parenteral, tópica, inhalaciones, bucal, nasal, etc. En algunas realizaciones, la composición farmacéutica se administra mediante un jarabe. Se puede preparar un jarabe mediante la adición del compuesto activo a una solución acuosa concentrada de un azúcar, por ejemplo, sacarosa, a la que también se le puede añadir cualquier ingrediente(s) accesorio. Dichos ingredientes accesorios pueden incluir aromatizantes, conservantes adecuados, un agente para retardar la cristalización del azúcar y un agente para aumentar la solubilidad de cualquier otro ingrediente, tal como alcohol polihídrico, por ejemplo, glicerol o sorbitol.
En algunas realizaciones, una dosis unitaria de, por ejemplo, un comprimido, se puede preparar mediante compresión o moldeo, opcionalmente con uno o más ingredientes accesorios. Los comprimidos de compresión pueden prepararse mediante la compresión en una máquina adecuada, con el compuesto activo en forma suelta, tal como un polvo o gránulos, que opcionalmente se mezcla con un aglutinante, disgregante, lubricante, diluyente inerte, agente tensioactivo o agente dispersante. Los comprimidos moldeados comprendidos con un vehículo adecuado se pueden fabricar mediante el moldeo en una máquina adecuada.
Las composiciones farmacéuticas de la presente invención comprenden al menos un fármaco antirretrovírico y piperina o tetrahidropiperina. Estos principios activos están formulados para una administración simultánea, separada o secuencial. Cuando los principios activos se administran de manera secuencial, al menos un fármaco antirretrovírico o la piperina/tetrahidropiperina, puede administrarse primero. Cuando la administración es simultánea, los principios activos se pueden administrar en la misma composición farmacéutica o en una diferente. El tratamiento complementario, por ejemplo, donde un principio activo se utiliza como tratamiento primario y los otros principios activos se utilizan para ayudar a ese tratamiento primario también es una realización de la presente invención.
Por consiguiente, se proporciona un producto que comprende al menos un fármaco antirretrovírico y piperina o tetrahidropiperina como preparación combinada para uso simultáneo, separado o secuencial para el tratamiento de enfermedades provocadas por retrovirus o el virus de la hepatitis B, de manera especial el SIDA o una infección por el VIH o hepatitis B.
En algunas realizaciones, las composiciones farmacéuticas de la presente invención comprenden tenofovir disproxil fumarato y piperina para el tratamiento de enfermedades provocadas por retrovirus, de manera especial el síndrome de la inmunodeficiencia adquirida o una infección por el VIH.
De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden tenofovir disproxil fumarato y piperina en una proporción de aproximadamente 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas de la presente invención comprenden tenofovir alafenamida fumarato y piperina para el tratamiento de enfermedades provocadas por retrovirus, de manera especial el SIDA o una infección por el VIH.
De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden tenofovir alafenamida fumarato y piperina en una proporción de aproximadamente 100:1, 50:1,40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1,2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas divulgadas en el presente documento comprenden dolutegravir y piperina para el tratamiento de enfermedades provocadas por retrovirus, de manera especial el síndrome de la inmunodeficiencia adquirida o una infección por el VIH.
De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden dolutegravir y piperina en una proporción de aproximadamente 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas de la presente invención comprenden darunavir y piperina para el tratamiento de enfermedades provocadas por retrovirus, de manera especial el síndrome de la inmunodeficiencia adquirida o una infección por el VIH.
De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden darunavir y piperina en una proporción de aproximadamente 100:1, 50:1,40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas de la presente invención comprenden tenofovir disproxil fumarato y piperina para el tratamiento de enfermedades provocadas por el virus de la hepatitis B.
De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden tenofovir disproxil fumarato y piperina en una proporción de aproximadamente 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1,2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas de la presente invención comprenden tenofovir alafenamida fumarato y piperina para el tratamiento de enfermedades provocadas por el virus de la hepatitis B. De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden tenofovir alafenamida fumarato y piperina en una proporción de aproximadamente 100:1, 50:1,40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1,2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas de la presente invención comprenden tenofovir disproxil fumarato y tetrahidropiperina para el tratamiento de enfermedades provocadas por retrovirus, de manera especial el síndrome de la inmunodeficiencia adquirida o una infección por el VIH.
De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden tenofovir disproxil fumarato y tetrahidropiperina en una proporción de aproximadamente 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1,2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas de la presente invención comprenden tenofovir alafenamida fumarato y tetrahidropiperina para el tratamiento de enfermedades provocadas por retrovirus, de manera especial el síndrome de la inmunodeficiencia adquirida o una infección por el VIH.
De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden tenofovir alafenamida fumarato y tetrahidropiperina en una proporción de aproximadamente 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1,6:1, 5:1, 4:1, 3:1, 2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas divulgadas en el presente documento comprenden dolutegravir y tetrahidropiperina para el tratamiento de enfermedades provocadas por retrovirus, de manera especial el síndrome de la inmunodeficiencia adquirida o una infección por el VIH.
De acuerdo con una realización preferida, las composiciones farmacéuticas divulgadas en el presente documento comprenden dolutegravir y tetrahidropiperina en una proporción de aproximadamente 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1,2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas de la presente invención comprenden darunavir y tetrahidropiperina para el tratamiento de enfermedades provocadas por retrovirus, de manera especial el síndrome de la inmunodeficiencia adquirida o una infección por el VIH.
De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden darunavir y tetrahidropiperina en una proporción de aproximadamente 100:1, 50:1,40:1, 30:1,20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas de la presente invención comprenden tenofovir disproxil fumarato y tetrahidropiperina para el tratamiento de enfermedades provocadas por el virus de la hepatitis B.
De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden
tenofovir disproxil fumarato y tetrahidropiperina en una proporción de aproximadamente 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1,2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, las composiciones farmacéuticas de la presente invención comprenden tenofovir alafenamida fumarato y tetrahidropiperina para el tratamiento de enfermedades provocadas por el virus de la hepatitis B.
De acuerdo con una realización preferida, las composiciones farmacéuticas de la presente invención comprenden tenofovir alafenamida fumarato y tetrahidropiperina en una proporción de aproximadamente 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1,6:1, 5:1, 4:1, 3:1, 2:1, a aproximadamente 1:1 en peso.
En algunas realizaciones, cuando el reforzador o potenciador farmacocinético o derivado del mismo se administra con el fármaco antirretrovírico en la composición farmacéutica, se reduce la frecuencia de dosificación del al menos un fármaco antirretrovírico que se administra a un paciente. En algunas realizaciones, el al menos un reforzador o potenciador farmacocinético o derivado del mismo aumenta la biodisponibilidad del al menos un fármaco antirretrovírico de aproximadamente un 10% a aproximadamente el 100%, de aproximadamente un 10% a aproximadamente un 70 %, de aproximadamente un 10 % a aproximadamente un 50 %, de aproximadamente un 10 % a aproximadamente un 30 % o de aproximadamente un 10 % a aproximadamente un 20 %. En algunas realizaciones, el al menos un reforzador o potenciador farmacocinético o derivado del mismo aumenta la biodisponibilidad del al menos un fármaco antirretrovírico de aproximadamente un 10 %, 15, 16, 17, 18, 19, 20, 21,22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 o 100 %.
Los inventores de la presente invención también han descubierto que las propiedades de biodisponibilidad de los fármacos antirretrovíricos también pueden mejorarse mediante el nanoescalado. En algunas realizaciones, la composición farmacéutica se administra mediante nanopartículas que tienen un tamaño de aproximadamente 1 nanómetro (nm) a aproximadamente 50 nm. En algunas realizaciones, las nanopartículas tienen un tamaño de aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 o 50 nm.
En algunas realizaciones, se pueden utilizar excipientes adecuados para formular las formas farmacéuticas de acuerdo con la presente invención, tales como, pero sin limitación, estabilizadores de superficie o tensioactivos, agentes modificadores de la viscosidad, polímeros
que incluyen polímeros de liberación prolongada, estabilizadores, desintegrantes o superdesintegrantes, diluyentes, plastificantes, aglutinantes, deslizantes, lubricantes, edulcorantes, agentes aromatizantes, agentes antiaglomerantes, opacificantes, agentes antimicrobianos, agentes antiespumantes, emulsionantes, agentes tamponantes, agentes colorantes, portadores, cargas, antiadherentes, disolventes, agentes de enmascaramiento del sabor, conservantes, antioxidantes, potenciadores de textura, agentes canalizadores, agentes de recubrimiento o combinaciones de los mismos.
En algunas realizaciones, cuando la composición farmacéutica se proporciona en formas farmacéuticas unitarias, como se ha indicado anteriormente, la forma farmacéutica unitaria puede estar revestida o sin revestir.
Estos y otros aspectos de la presente solicitud se apreciarán adicionalmente al considerar los siguientes ejemplos, que pretenden ilustrar determinadas realizaciones particulares de la solicitud pero no pretenden limitar su alcance, como se define mediante las reivindicaciones.
Ejemplos
Ejemplo 1
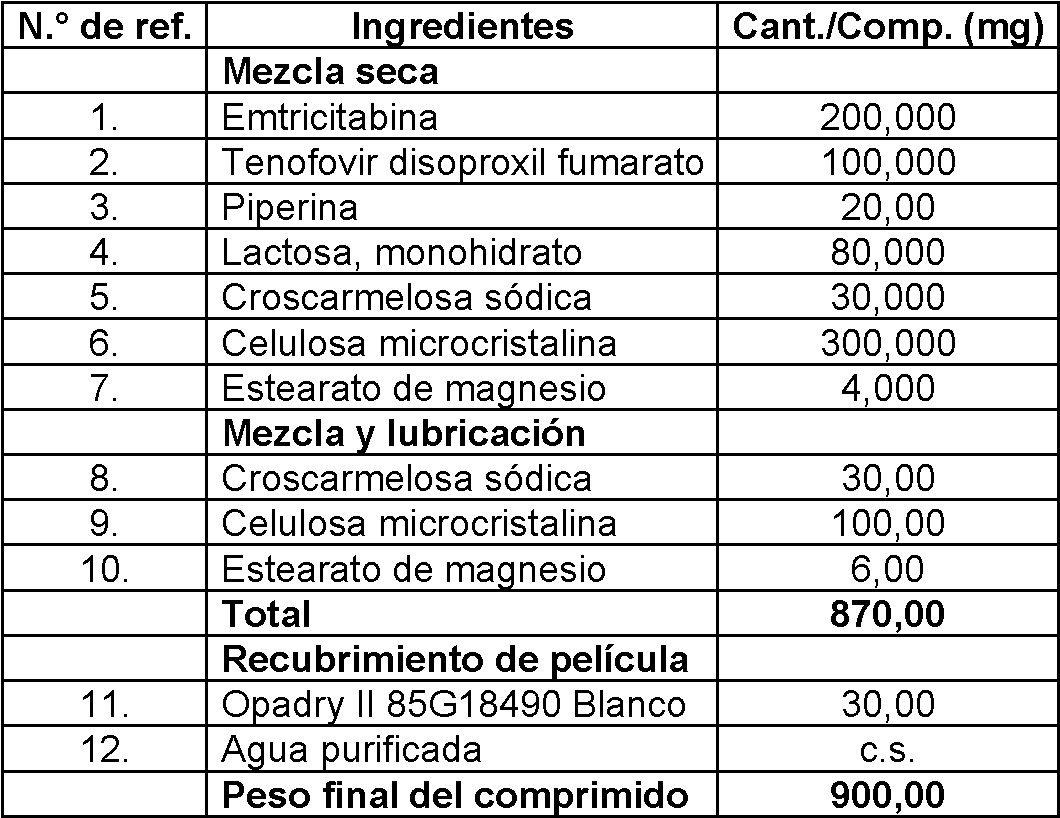
Tabla 1

continuación

Proceso:
1) Se mezclaron en seco tenofovir alafenamida fumarato, emtricitabina, piperina, lactosa, dióxido de silicio coloidal, celulosa microcristalina y croscarmelosa sódica en un mezclador adecuado.
2) La mezcla obtenida en la etapa (1) se lubricó con estearato de magnesio y se compactó y granuló en seco. 3) Se mezclaron los gránulos obtenidos en la etapa (3), dióxido de silicio coloidal, celulosa microcristalina y croscarmelosa sódica para formar una mezcla.
4) La mezcla obtenida en la etapa (3) se comprimió para formar comprimidos y se revistieron con Opadry.
Ejemplo 2
Tabla 2

Proceso:
1) Se mezclaron en seco tenofovir alafenamida fumarato, piperina, lactosa, dióxido de silicio coloidal, celulosa microcristalina y croscarmelosa sódica en un mezclador adecuado.
2) La mezcla obtenida en la etapa (1) se lubricó con estearato de magnesio, se comprimió para formar comprimidos y se recubrieron con Opadry.
Ejemplo 3
Tabla 3

continuación

Proceso:
1) Se mezclaron en seco tenofovir alafenamida fumarato, emtricitabina, piperina, elvitegravir, lactosa, dióxido de silicio coloidal, croscarmelosa sódica y celulosa microcristalina para obtener una mezcla.
2) La mezcla obtenida en la etapa (1) se lubricó con estearato de magnesio, se compactó y se comprimió el tamaño para formar comprimidos.
Ejemplo 4
Tabla 4

Proceso:
Se mezclaron en seco tenofovir disoproxil fumarato, emtricitabina, piperina, elvitegravir, lactosa, dióxido de silicio coloidal, croscarmelosa sódica y celulosa microcristalina para obtener una mezcla.
2) La mezcla obtenida en la etapa (1) se lubricó con estearato de magnesio, se compactó y se comprimió el tamaño para formar comprimidos.
Ejemplo 5 - no es un ejemplo de la invención
Tabla 5

Proceso:
Se mezclaron en seco dolutegravir sódico, piperina y manitol y la povidona se disolvió en agua.
2) Se granuló la mezcla seca obtenida en la etapa (1) y se molieron los gránulos obtenidos.
3) Los gránulos obtenidos en la etapa (2) se mezclaron con manitol, glicolato sódico de almidón, celulosa microcristalina y dióxido de silicio coloidal.
4) La mezcla obtenida en la etapa (3) se lubricó con estearil fumarato de sodio, se comprimió y se recubrió.
Ejemplo 6
Tabla 6

Proceso:
1) Se tamizaron y mezclaron hidrato de darunavir, piperina, celulosa microcristalina, crospovidona y dióxido de silicio coloidal.
2) La mezcla seca obtenida en la etapa (1) se granuló y se lubricó con estearato de magnesio.
3) Los gránulos obtenidos en la etapa (2) se comprimieron y recubrieron con Opadry.
Ejemplo 7
Tabla 7

Proceso:
1) Se tamizaron y mezclaron hidrato de darunavir, piperina, povidona, celulosa microcristalina, crospovidona y dióxido de silicio coloidal.
2) La mezcla seca obtenida en la etapa (1) se granuló y se lubricó con estearato de magnesio.
3) Los gránulos obtenidos en la etapa (2) se comprimieron y recubrieron con Opadry.
Ejemplo 8
Tabla 8

continuación

Proceso:
Se tamizaron y mezclaron hidrato de darunavir, piperina, povidona, celulosa microcristalina silicificada, crospovidona y dióxido de silicio coloidal.
2) La mezcla seca obtenida en la etapa (1) se granuló y se lubricó con estearato de magnesio.
3) Los gránulos obtenidos en la etapa (2) se comprimieron y recubrieron con Opadry.
Ejemplo 9
Tabla 9

Proceso:
1) Se tamizaron y mezclaron etanolato de darunavir, piperina, hidroxipropilmetilcelulosa celulosa microcristalina silicificada, crospovidona y dióxido de silicio coloidal.
2) La mezcla seca obtenida en la etapa (1) se granuló y se lubricó con estearato de magnesio.
3) Los gránulos obtenidos en la etapa (2) se comprimieron y recubrieron con Opadry.
Ejemplo 10
Tabla 10

continuación

Proceso:
1) Se mezclaron lamivudina, celulosa microcristalina, croscarmelosa de sodio, almidón pregelatinizado y estearato de magnesio y se compactaron en una masa granular.
2) Se mezclaron tenofovir disoproxil fumarato, piperina, celulosa microcristalina, croscarmelosa de sodio y estearato de magnesio y se compactaron en una masa granular.
3) Se mezclaron celulosa microcristalina, croscarmelosa de sodio y estearato de magnesio con las mezclas obtenidas en la etapa (1) y en la etapa (2) y se comprimieron para formar comprimidos con recubrimiento de sellado seguido de recubrimiento de película.
Ejemplo 11
Tabla 11

Proceso:
1) Se mezclaron emtricitabina, tenofovir disoproxil fumarato, piperina, lactosa monohidrato, celulosa microcristalina, croscarmelosa de sodio y estearato de magnesio y se compactaron en una masa granular. 2) Se mezclaron celulosa microcristalina, croscarmelosa de sodio y estearato de magnesio y se compactaron en una masa granular.
3) Se disolvieron polisorbato 80, povidona y lactosa en agua.
4) Se añadió rilpivirina a la solución obtenida en la etapa (3) para formar una suspensión.
5) Se añadió la mezcla seca de lactosa monohidrato y crospovidona a la suspensión obtenida en la etapa (4). 6) Se añadieron celulosa microcristalina, crospovidona y estearato de magnesio a la mezcla seca obtenida en la etapa (5).
7) La mezcla obtenida en la etapa (1) se comprimió con la mezcla obtenida en la etapa (6) para formar un comprimido bicapa con recubrimiento de película.
Ejemplo 12
Tabla 12
Proceso:
1) Se mezclaron emtricitabina, tenofovir disoproxil fumarato, piperina, lactosa monohidrato, celulosa microcristalina, croscarmelosa de sodio y estearato de magnesio y se combinaron para formar una masa granular.
2) Se mezclaron y combinaron celulosa microcristalina, croscarmelosa de sodio y estearato de magnesio.
3) La mezcla obtenida en la etapa (1) y en la etapa (2) se comprimió y recubrió para formar comprimidos con recubrimiento de película.
Ejemplo 13
Tabla 13

continuación

Proceso:
1) Se mezclaron emtricitabina, tenofovir disoproxil fumarato, piperina, lactosa monohidrato, celulosa microcristalina, croscarmelosa de sodio y estearato de magnesio y se combinaron para formar una masa granular.
2) Se añadieron celulosa microcristalina, croscarmelosa de sodio y estearato de magnesio a la mezcla obtenida en la etapa (1) y se mezclaron de manera adicional.
3) Se añadieron efavirenz, celulosa microcristalina y croscarmelosa sódica a SLS seguido de hidroxipropilcelulosa para formar una solución y se granuló.
4) Se mezclaron y comprimieron lactosa monohidrato y estearato de magnesio para formar un comprimido bicapa con un recubrimiento de película.
Ejemplo 14
Tabla 14

continuación

Proceso:
1) Se mezclaron lamivudina, celulosa microcristalina, croscarmelosa de sodio, almidón pregelatinizado y estearato de magnesio para formar una masa granular.
2) Se mezclaron tenofovir disoproxil fumarato, piperina, celulosa microcristalina, croscarmelosa de sodio y estearato de magnesio para formar una masa granular.
3) Se mezclaron celulosa microcristalina, croscarmelosa de sodio y estearato de magnesio con las mezclas obtenidas en la etapa (1) y en la etapa (2).
4) Se añadieron efavirenz, celulosa microcristalina y croscarmelosa sódica a SLS seguido de hidroxipropilcelulosa para formar una solución y se granuló.
5) Se mezclaron lactosa monohidrato y estearato de magnesio y se comprimieron para formar un comprimido bicapa que tenía un recubrimiento de sellado seguido de un recubrimiento de película.
Con el fin de que la presente invención pueda comprenderse mejor, se establecen los siguientes métodos de preparación y de prueba. Estos métodos son solo para fines ilustrativos y no deben interpretarse como limitantes del alcance de la invención de ninguna manera.
MÉTODOS DE PREPARACIÓN Y DE PRUEBA
I) MATERIAL
Cafeína (marcador de permeabilidad elevada), atenolol (marcador de permeabilidad baja), digoxina (sustrato conocido de la gp-P), TDF (MD1431532), TAF (VRD-1063/16/187), tampón de HBSS, hidrato de MES, polvo de HEPES, suero fetal bovino (FBS), medio mínimo esencial (MEM), amarillo Lucifer, piperina (inhibidor de la gp-P)
MÉTODO
1) Cultivo de células Caco-2
Se cultivaron células Caco-2 en medio MEM con suero al 10 % y se sembraron a una densidad de 75000 células por ml y se cultivaron durante 21 días en una placa transwell de 24 pocillos a 37 °C, CO2 al 5 %. La integridad de la monocapa se comprobó de forma intermitente (día 0-21) usando resistencia eléctrica transepitelial (TEER, del inglés "Trans Epithelial Electric Resistance"). Las células se trataron con fármacos de la siguiente manera:
2) Ensayo unidireccional (A-B)
Preparaciones madre: Se prepararon madres 10 mM de todos los fármacos en DMSO. Las concentraciones de prueba se prepararon de manera adicional en tampón de HBSS que contenía hidrato de MES 10 mM a pH 6,8 según el plan de la placa. Asimismo, se preparó tampón de HBSS con HEPES 10 mM con pH 7,4.
Plan del estudio
Configuración de la placa
Tabla 15

Protocolo del ensayo
Se añadieron 0400 |jl de muestras a los pocilios según la configuración de la placa en el lado apical (A) preparado en hidrato de MES 10 mM pH 6,8 por duplicado y se añadieron 800 j l de HBSS con HEPES 10 mM pH 7,4 a todos los pocillos basales (B). Las muestras se recogieron a los 60, 90 y 120 minutos del lado basal. Se recogieron muestras de equilibrio de masas a los 0 y 120 minutos del lado apical. Las muestras se analizaron en LCMS-MS.
4) Ensayo bidireccional (A-B y B-A) para estudiar el efecto de la piperina (inhibidor de la gp-P) sobre la permeabilidad Plan de la placa
Tabla 16

Protocolo del ensayo
Se añadieron 400 j l de muestras a los pocillos según la configuración de la placa en el lado apical por duplicado con 800 j l de HBSS pH 7,4 en los pocillos basales. Las muestras se recogieron a los 60, 90 y 120 minutos del lado basal. Se recogieron muestras de equilibrio de masas a los 0 y 120 minutos del lado apical.
Para B-A, se añadieron 800 j l de las diluciones respectivas al lado basal por duplicado con 400 j l de HBSS pH 7,4 en los pocillos apicales. Las muestras se recogieron a los 60, 90 y 120 minutos del lado apical. Se recogieron muestras de equilibrio de masas a los 0 y 120 minutos del lado basal. Las muestras se analizaron en LCMS-MS.
Al final del experimento, se comprobó la integridad de la monocapa utilizando amarillo Lucifer y calculando el % de rechazo del amarillo Lucifer mediante la incubación de las células con Lucifer 100 jg/ml.
5) Análisis de los datos:
Se calculó la Pap de la siguiente manera:
La permeabilidad aparente (Pap) en unidades por segundo se puede calcular con la siguiente ecuación, Para el método de un solo punto:
Pap = (V/(T x A)) x (C0/Ct)
Para el método multipunto:
Pap = (dQ/dt)/(A x C0)
% de equilibrio de masa = 100 -[CR120 x VR CD120 x VD/C0 x VD] Para amarillo Lucifer,
% de pasaje amarillo Lucifer = [RFU (prueba) - RFU (en blanco)/RFU (equilibrio) - RFU (en blanco)] x 100 Clasificación de la permeabilidad:
Tabla 17

Relación de flujo de salida = Pap B-A/Pap A-B
La relación de flujo de salida >2 indica que el fármaco es un sustrato de la gp-P
Resultados
Ensayo unidireccional (Figura 1)
Tabla 18

Ensayo bidireccional (Figura 2)
Tabla 19

Conclusiones
Se observa que el TDF y el TAF son fármacos de permeabilidad baja a moderada. Además, la absorción de TAF aumenta con piperina al disminuir la relación de flujo de salida de TAF. Los datos anteriores indican que el TAF es un sustrato del transportador de flujo de salida y, por tanto, su biodisponibilidad es baja. Como puede observarse en los datos, la Pap A-B fue de 34,49 nm/s y su relación de flujo de salida fue de 5,17. Mediante la adición de piperina, que es un inhibidor conocido de los transportadores de flujo de salida, la Pap A-B aumentó a más de 282,66 nm/s mientras que la relación de flujo de salida disminuyó a menos de 1,18. Por tanto, indicar que la adición de piperina mejora la permeabilidad. Por lo tanto, se puede concluir que el uso de piperina disminuye la relación de flujo de salida, lo que a su vez aumentaría su biodisponibilidad.
II) Material
digoxina (sustrato conocido de la gp-P), dolutegravir (KK1406229), tampón de HBSS, hidrato de MES, polvo de HEPES, suero fetal bovino (FBS), medio mínimo esencial (MEM), amarillo Lucifer, piperina (inhibidor de la gp-P), cobicistat (inhibidor de la gp-P).
Método
1) Cultivo de células Caco-2
Se cultivaron células Caco-2 en medio MEM con suero al 10 % y se sembraron a una densidad de 75000 células por ml y se cultivaron durante 21 días en una placa transwell de 24 pocillos a 37 °C, CO2 al 5 %. La integridad de la monocapa se comprobó de forma intermitente (día 0-21) usando resistencia eléctrica transepitelial (TEER, del inglés "Trans Epithelial Electric Resistance"). Las células se trataron con fármacos de la siguiente manera:
2) Ensayo bidireccional (A-B y B-A) para estudiar el efecto de la piperina (inhibidor de la gp-P) sobre la permeabilidad
Plan de la placa
Tabla 20

Protocolo del ensayo
Se añadieron 400 |jl de muestras a los pocilios según la configuración de la placa en el lado apical por duplicado con 800 j l de HBSS pH 7,4 en los pocillos basales. Las muestras se recogieron a los 60, 90 y 120 minutos del lado basal. Se recogieron muestras de equilibrio de masas a los 0 y 120 minutos del lado apical.
Para B-A, se añadieron 800 j l de las diluciones respectivas al lado basal por duplicado con 400 j l de HBSS pH 7,4 en los pocillos apicales. Las muestras se recogieron a los 60, 90 y 120 minutos del lado apical. Se recogieron muestras de equilibrio de masas a los 0 y 120 minutos del lado basal.
Las muestras se analizaron en LCMS-MS. Al final del experimento, se comprobó la integridad de la monocapa utilizando amarillo Lucifer y calculando el % de rechazo del amarillo Lucifer mediante la incubación de las células con Lucifer 100 jg/ml.
3) Análisis de los datos:
Se calculó la Pap de la siguiente manera:
La permeabilidad aparente (Pap) en unidades por segundo se puede calcular con la siguiente ecuación, Para el método de un solo punto:
Pap = (V/(T x A)) x (Cü/Ct)
Para el método multipunto:
Pap = (dQ/dt)/(A x C0)
% de equilibrio de masa = 100 -[CR120 x Vr CD120 x Vd/C0 x Vd]
Para amarillo Lucifer,
% de pasaje amarillo Lucifer = [RFU (prueba) - RFU (en blanco)/RFU (equilibrio) - RFU (en blanco)] x 100 Clasificación de la permeabilidad:
Tabla 21

Relación de flujo de salida = Pap B-A/Pap A-B
La relación de flujo de salida >2 indica que el fármaco es un sustrato de la gp-P
Resultados
Ensayo bidireccional (Figura 3)
Tabla 22

Conclusiones
Dolutegravir es un conocido sustrato de la gp-P. El dolutegravir es un fármaco de permeabilidad elevada y la piperina no afecta la permeabilidad del dolutegravir a través de la monocapa de caco-2. Por lo tanto, se puede concluir que el uso de piperina disminuye la relación de flujo de salida, lo que a su vez aumentaría su biodisponibilidad.
II) Material
digoxina (sustrato conocido de la gp-P), darunavir (DN0011215), tampón de HBSS, hidrato de MES, polvo de HEPES, suero fetal bovino (FBS), medio mínimo esencial (MEM), amarillo Lucifer, piperina (inhibidor de la gp-P), cobicistat (inhibidor de la gp-P)
Método
1. ) Cultivo de células Caco-2
Se cultivaron células Caco-2 en medio MEM con suero al 10 % y se sembraron a una densidad de 75000 células por ml y se cultivaron durante 21 días en una placa transwell de 24 pocillos a 37 °C, CO2 al 5 %. La integridad de la monocapa se comprobó de forma intermitente (día 0-21) usando resistencia eléctrica transepitelial (TEER, del inglés "Trans Epithelial Electric Resistance"). Las células se trataron con fármacos de la siguiente manera:
2. ) Ensayo bidireccional (A-B y B-A) para estudiar el efecto de la piperina (inhibidor de la gp-P) en la permeabilidad
Plan de la placa
Tabla 23

Protocolo del ensayo
Se añadieron 400 pl de muestras a los pocillos según la configuración de la placa en el lado apical por duplicado con 800 pl de HBSS pH 7,4 en los pocillos basales. Las muestras se recogieron a los 60, 90 y 120 minutos del lado basal. Se recogieron muestras de equilibrio de masas a los 0 y 120 minutos del lado apical.
Para B-A, se añadieron 800 pl de las diluciones respectivas al lado basal por duplicado con 400 pl de HBSS pH 7,4 en los pocillos apicales. Las muestras se recogieron a los 60, 90 y 120 minutos del lado apical. Se recogieron muestras de equilibrio de masas a los 0 y 120 minutos del lado basal.
Las muestras se analizaron en LCMS-MS. Al final del experimento, se comprobó la integridad de la monocapa utilizando amarillo Lucifer, calculando el % de rechazo de amarillo Lucifer mediante la incubación de células con Lucifer 100 pg/ml.
3.) Análisis de los datos:
Se calculó la Pap de la siguiente manera:
La permeabilidad aparente (Pap) en unidades por segundo se puede calcular con la siguiente ecuación,
Para el método de un solo punto:
Pap = (V/(T x A)) x (C0/Ct)
Para el método multipunto:
Pap = (dQ/dt)/(A x CO)
% de equilibrio de masa = 100 -[CR120 x VR CD120 x VD/C0 x VD] Para amarillo Lucifer,
% de pasaje amarillo Lucifer = [RFU (prueba) - RFU (en blanco)/RFU (equilibrio) - RFU (en blanco)] x 100 Clasificación de la permeabilidad:
Tabla 24

Relación de flujo de salida = Pap B-A/Pap A-B
La relación de flujo de salida >2 indica que el fármaco es un sustrato de la gp-P
Resultados
Ensayo bidireccional (Figura 4)
Tabla 25

Conclusiones
Darunavir es un sustrato conocido de la gp-P. La absorción de darunavir aumenta con piperina al disminuir la relación de flujo de salida del TAF. Por lo tanto, se puede concluir que el uso de piperina disminuye la relación de flujo de salida, lo que a su vez aumentaría su biodisponibilidad.
III) Material
digoxina (sustrato conocido de la gp-P), TDF, tampón de HBSS, hidrato de MES, polvo de HEPES, suero fetal bovino (FBS), medio mínimo esencial (MEM), amarillo Lucifer, piperina (inhibidor de la gp-P), cobicistat (inhibidor de la gp-P), tetrahidropiperina (inhibidor de la gp-P)
Método
1. ) Cultivo de células Caco-2
Se cultivaron células Caco-2 en medio MEM con suero al 10 % y se sembraron a una densidad de 75000 células por ml y se cultivaron durante 21 días en una placa transwell de 24 pocillos a 37 °C, CO2 al 5 %. La integridad de la monocapa se comprobó de forma intermitente (día 0-21) usando resistencia eléctrica transepitelial (TEER, del inglés "Trans Epithelial Electric Resistance"). Las células se trataron con fármacos de la siguiente manera:
2. ) Ensayo bidireccional (A-B y B-A) para estudiar el efecto de la piperina (inhibidor de la gp-P) y la tetrahidropiperina en la permeabilidad
Plan de la placa
Tabla 26

__________ ________ ________
Protocolo del ensayo
Se añadieron 400 |jl de muestras a los pocilios según la configuración de la placa en el lado apical por duplicado con 800 j l de HBSS pH 7,4 en los pocillos basales. Las muestras se recogieron a los 60, 90 y 120 minutos del lado basal. Se recogieron muestras de equilibrio de masas a los 0 y 120 minutos del lado apical.
Para B-A, se añadieron 800 j l de las diluciones respectivas al lado basal por duplicado con 400 j l de HBSS pH 7,4 en los pocillos apicales. Las muestras se recogieron a los 60, 90 y 120 minutos del lado apical. Se recogieron muestras de equilibrio de masas a los 0 y 120 minutos del lado basal.
Las muestras se analizaron en LCMS-MS. Al final del experimento, se comprobó la integridad de la monocapa utilizando amarillo Lucifer, calculando el % de rechazo de amarillo Lucifer mediante la incubación de células con Lucifer 100 jg/ml.
3.) Análisis de los datos:
Se calculó la Pap de la siguiente manera:
La permeabilidad aparente (Pap) en unidades por segundo se puede calcular con la siguiente ecuación, Para el método de un solo punto:
Pap = (V/(T x A)) x (C0/Ct)
Para el método multipunto:
Pap = (dQ/dt)/(A x C0)
% de equilibrio de masa = 100 -[CR120 x VR CD120 x VD/C0 x VD] Para amarillo Lucifer, % de pasaje amarillo Lucifer = [RFU (prueba) - RFU (en blanco)/RFU (equilibrio) - RFU (en blanco)] x 100
Clasificación de la permeabilidad:
Tabla 27

Relación de flujo de salida = Pap B-A/Pap A-B
La relación de flujo de salida >2 indica que el fármaco es un sustrato de la gp-P
Resultados:
Ensayo bidireccional (Figura 5)
Tabla 28

continuación

Conclusiones
TDF es un sustrato conocida de la gp-P. La absorción de TDF aumenta con piperina al disminuir la relación de flujo de salida. Además, la absorción de TDF aumenta con tetrahidropiperina al disminuir la relación de flujo de salida. Tanto con la piperina como con la tetrahidropiperina se observó una mejora comparable en la permeabilidad del TDF. Por lo tanto, se puede concluir que el uso de piperina y tetrahidropiperina disminuye la relación de flujo de salida, lo que a su vez aumentaría su biodisponibilidad.
ESTUDIO EN ANIMALES
Estudio FC in vivo ratas
El objetivo de este estudio sin GLP fue determinar la farmacocinética del TDF solo y en combinaciones con piperina en diferentes grupos de ratas Wistar macho después de la administración intravenosa y oral de una dosis única. Este estudio se realizó con la aprobación del Institutional Animal Ethics Committee (IAEC) de acuerdo con el requisito del Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India.
Diseño del estudio
El estudio se realizó utilizando seis ratas Wistar macho en cada grupo, como se muestra en la tabla 30 a continuación.
Tabla 29

Preparación de la formulación
Las formulaciones en solución se prepararon de la siguiente manera:
Vía intravenosa:
Se pesó la cantidad necesaria de compuesto B (TDF) (23,03 mg) y a esto se le añadieron 10,94 ml de vehículo (solución salina normal), se agitó en vórtex y se sometió a ultrasonidos durante 2 minutos para obtener una formulación clara.
Por vía oral:
Para el grupo 2: Se pesó la cantidad necesaria de compuesto B (79,37 mg) y a esto se le añadieron 9,42 ml de vehículo (Carboximetilcelulosa de sodio), se agitó en vórtex y se sometió a ultrasonidos durante 2 minutos para obtener una formulación de 6,2 mg/ml de concentración.
Para el grupo 3: Se pesó la cantidad necesaria de compuesto B (46,14 mg) y a esto le añadieron 10,95 ml de vehículo (Carboximetilcelulosa de sodio), se agito en vórtex y se sometió a ultrasonidos durante 2 minutos para obtener una formulación uniforme.
Para el grupo 4: Se pesó la cantidad necesaria de compuesto B (79,56 mg) y a esto se le añadieron 4,72 ml de vehículo (Carboximetilcelulosa de sodio), se agitó en vórtex y se sometió a ultrasonidos durante 2 minutos para obtener una formulación clara. Se pesó la cantidad necesaria de compuesto B1 (piperina) (18,95 mg) y a esto se le añadieron 22,71 ml de vehículo (Carboximetilcelulosa de sodio), se agitó en vórtex y se sometió a ultrasonidos durante 2 minutos
para obtener una formulación clara. Se mezcló un volumen igual (4,72 ml) de cada formulación en frascos separados para obtener 5 ml/kg.
Para el grupo 5: Se pesó la cantidad necesaria de compuesto B (47,20 mg) y de compuesto B1 (18,95 mg) y a esto se le añadieron 5,61 ml de vehículo (Carboximetilcelulosa de sodio), se agitó en vórtex y se sometió a ultrasonidos durante 2 minutos para obtener una formulación clara. Se mezcló un volumen igual (5,61 ml) de cada formulación en frascos separados para obtener 5 ml/kg.
Bioanálisis
El bioanálisis se realizó utilizando el método LC-MS/MS adaptado para el propósito para la cuantificación de TDF y PMPA en muestras de plasma de rata. La curva de calibración (CC) para el método consistió en nueve patrones de calibración distintos de cero junto con un doble blanco y muestras patrones cero.
Análisis farmacocinético
Los parámetros farmacocinéticos plasmáticos se calcularon utilizando la herramienta de análisis no compartimental del programa informático Phoenix (Versión 6.3) y se determinaron a partir de animales individuales en cada grupo. La concentración en plasma máxima (Cmáx), el tiempo para alcanzar la concentración en plasma máxima (Tmáx), el área bajo la curva de la concentración en plasma-tiempo (ABC0-t y ABCinf), el ABC extra (%), la semivida de eliminación (T1/2), la eliminación (CL), el volumen de distribución Vd (l/kg) y el tiempo medio de residencia (MRT, del inglés "Mean residence time") se calcularon a partir del grupo de administración intravenosa. La concentración en plasma máxima (Cmáx), el tiempo para alcanzar la concentración en plasma máxima (Tmáx), el ABC0-t y el ABCinf, el ABC extra (%), el tiempo medio de residencia (MRT) y la biodisponibilidad oral absoluta (F) se calcularon a partir del grupo oral.
Resultados
Los datos de la concentración en plasma-tiempo y los parámetros farmacocinéticos plasmáticos de PMPA después de la administración intravenosa y oral de TDF en ratas Wistar macho se presentan en la tabla 31.
Tabla 30

Las figuras 6 y 7 muestran la concentración en plasma de tenofovir para 300 mg de TDF y 300 mg de TDF 20 mg de piperina en diferentes momentos.
Tabla 31: Valores de Cmáx Tmáx ABC de diferentes combinaciones

La figura 8 muestra la concentración en plasma dependiente del tiempo de tenofovir para 300 mg de TDF, 300 mg de TDF 20 mg de piperina y 150 mg de TDF 20 mg de piperina
Conclusiones
El estudio FC en ratas indica claramente que la concentración en plasma máxima de tenofovir aumentó de manera significativa cuando se administra TDF en combinación con piperina. Los resultados demuestran una mejora significativa de la biodisponibilidad cuando se administran 150 mg de TDF con 20 mg de piperina (52,48 ± 13,48) en comparación con 300 mg de TDF solo (34,39 ± 6,07).
Claims (15)
1. Una composición farmacéutica oral o inyectable que comprende una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico, que comprende (i) un inhibidor análogo de nucleótido de la transcriptasa inversa (NRTI) seleccionado del grupo que comprende: tenofovir alafenamida fumarato, tenofovir disoproxil fumarato, adefovir y una combinación de los mismos; o (ii) un inhibidor de la proteasa (IP) que comprende darunavir, y una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético que comprende piperina, tetrahidropiperina, c/s-piperina, trans-piperina, c/s,trans-piperina, trans,c/s-piperina, c/s,c/s-piperina, trans,trans-piperina o una combinación de las mismas.
2. La composición farmacéutica oral o inyectable de la reivindicación 1, en donde (i) el al menos un reforzador o potenciador farmacocinético reduce la frecuencia de dosificación del al menos un fármaco antirretrovírico que se administra a un paciente y (ii) el al menos un reforzador o potenciador farmacocinético aumenta la biodisponibilidad del al menos un fármaco antirretrovírico de aproximadamente un 10 % a aproximadamente un 70 %.
3. La composición farmacéutica oral o inyectable de la reivindicación 1, en donde (i) el al menos un fármaco antirretrovírico comprende además un inhibidor nucleosídico de la transcriptasa inversa (NRTI), un inhibidor no nucleosídico de la transcriptasa inversa (NNRTI), un inhibidor de la proteasa (IP), un inhibidor de la integrasa, un inhibidor de fusión, un inhibidor de CCR5, un anticuerpo monoclonal, un inhibidor de glucoproteínas o combinaciones de los mismos.
4. La composición farmacéutica oral o inyectable de la reivindicación 3, en donde el inhibidor nucleosídico de la transcriptasa inversa (NRTI) comprende lamivudina, abacavir, emtricitabina, didanosina, estavudina, entecavir, apricitabina, censavudina, zalcitabina, dexelvucitabina, amdoxovir, elvucitabina, festinavir, racivir, estampidina o una combinación de los mismos; el inhibidor no nucleosídico de la transcriptasa inversa (NNRTI) comprende lersivirina, rilpivirina, etravirina, doravirina, dapivirina o una combinación de los mismos; el inhibidor de la proteasa (IP) comprende, ritonavir, lasinavir, palinavir, tirpranavir, fosamprenavir o una combinación de los mismos; el inhibidor de la integrasa comprende dolutegravir, elvitegravir, raltegravir, bictegravir, cabotegravir o una combinación de los mismos; el inhibidor de fusión comprende maraviroc, enfuvirtida, griffithsin, aplaviroc, vicriviroc, plerixafor, fostemsavir, albuvirtida o una combinación de los mismos; el inhibidor de CCR5 comprende aplaviroc, vicriviroc, maraviroc, cenicriviroc o una combinación de los mismos; el anticuerpo monoclonal comprende ibalizumab; el inhibidor de glucoproteínas comprende sifuvirtida; o combinaciones de los mismos.
5. La composición farmacéutica oral o inyectable de la reivindicación 1, en donde la relación del al menos un fármaco antirretrovírico a al menos un reforzador o potenciador farmacocinético es de aproximadamente 100:1 a aproximadamente 1:1 en peso.
6. La composición farmacéutica oral o inyectable de las reivindicaciones 1 o 4, que comprende además las siguientes características:
- en donde el tenofovir disoproxil fumarato en la composición es de aproximadamente 1 mg a aproximadamente 300 mg; o
- en donde el tenofovir alafenamida fumarato en la composición es de aproximadamente 1 a aproximadamente 25 mg; o
- en donde el dolutegravir en la composición es de aproximadamente 1 mg a aproximadamente 50 mg; o - en donde el darunavir en la composición es de aproximadamente 1 mg a aproximadamente 800 mg; o - en donde el elvitegravir en la composición es de aproximadamente 1 mg a aproximadamente 150 mg; y el raltegravir en la composición es de aproximadamente 1 mg a aproximadamente 400 mg.
7. La composición farmacéutica oral o inyectable de la reivindicación 1, en donde la piperina está en la composición de aproximadamente 0,5 mg a aproximadamente 400 mg.
8. La composición farmacéutica oral o inyectable de la reivindicación 1, que además comprende uno o más excipientes farmacéuticamente aceptables que comprenden vehículos, diluyentes, cargas, aglutinantes, lubricantes, deslizantes, disgregantes, agentes de carga, aromatizantes o cualquier combinación de los mismos.
9. La composición farmacéutica oral o inyectable de la reivindicación 8, en donde la composición oral está en forma de comprimido, minicomprimido, gránulos, aspersiones, cápsulas, sobres, polvos, gránulos, y la composición inyectable está en forma de solución, suspensión, emulsión, polvo liofilizado o en forma de kit.
10. La composición farmacéutica oral o inyectable de la reivindicación 8, en donde la composición farmacéutica oral o inyectable es para su uso en el tratamiento o la profilaxis de enfermedades provocadas por retrovirus o en el tratamiento de enfermedades provocadas por el virus de la hepatitis B.
11. Una composición farmacéutica que comprende (i) una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico que comprende (a) un inhibidor análogo de nucleótido de la transcriptasa inversa seleccionado del
grupo que comprende: tenofovir alafenamida fumarato, tenofovir disoproxil fumarato, adefovir y una combinación de los mismos; o (b) un inhibidor de la proteasa (IP) que comprende darunavir; (ii) una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético que comprende piperina, tetrahidropiperina, c/s-piperina, trans-piperina, cis-trans piperina, trans,cis-piperina, cis,cis-piperina, trans,trans-piperina o una combinación de las mismas; y (iii) uno o más excipientes farmacéuticamente aceptables que comprenden vehículos, diluyentes, cargas, aglutinantes, lubricantes, deslizantes, disgregantes, agentes de carga, aromatizantes o cualquier combinación de los mismos, para su uso en el tratamiento o la profilaxis de enfermedades provocadas por retrovirus, opcionalmente, las enfermedades provocadas por retrovirus comprenden el síndrome de la inmunodeficiencia adquirida o una infección por el VIH.
12. Un método para preparar una composición farmacéutica que mejora la biodisponibilidad de un fármaco antirretrovírico, comprendiendo el método: mezclar una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico que comprende (i) un inhibidor análogo de nucleótido de la transcriptasa inversa seleccionado del grupo que comprende: tenofovir alafenamida fumarato, tenofovir disoproxil fumarato, adefovir y una combinación de los mismos; o (ii) un inhibidor de la proteasa (IP) que comprende darunavir, y una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético que comprende piperina, tetrahidropiperina, c/s-piperina, trans-piperina, cis-trans piperina, trans,cis-piperina, cis,cis-piperina, frans,frans-piperina o una combinación de las mismas, con uno o más excipientes farmacéuticamente aceptables para preparar la composición farmacéutica.
13. El método de acuerdo con la reivindicación 12, en donde el al menos un fármaco antirretrovírico comprende además un inhibidor nucleosídico de la transcriptasa inversa (NRTI) que comprende lamivudina, abacavir, emtricitabina, didanosina, estavudina, entecavir, apricitabina, censavudina, zalcitabina, dexelvucitabina, amdoxovir, elvucitabina, festinavir, racivir, estampidina o una combinación de los mismos; un inhibidor no nucleosídico de la transcriptasa inversa (NNRTI) que comprende lersivirina, rilpivirina, etravirina, doravirina, dapivirina o una combinación de los mismos; un inhibidor de la proteasa (IP) que comprende, ritonavir, lasinavir, palinavir, tirpranavir, fosamprenavir, darunavir, tipranavir o una combinación de los mismos; un inhibidor de la integrasa que comprende dolutegravir, elvitegravir, raltegravir, bictegravir, cabotegravir o una combinación de los mismos; un inhibidor de fusión que comprende maraviroc, enfuvirtida, griffithsin, aplaviroc, vicriviroc, plerixafor, fostemsavir, albuvirtida o una combinación de los mismos; un inhibidor de CCR5 que comprende aplaviroc, vicriviroc, maraviroc, cenicriviroc o una combinación de los mismos; un anticuerpo monoclonal que comprende ibalizumab; un inhibidor de glucoproteínas que comprende sifuvirtida; o combinaciones de los mismos.
14. Un kit para el tratamiento de enfermedades provocadas por retrovirus o por el virus de la hepatitis B, comprendiendo el kit una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico que comprende (i) un inhibidor análogo de nucleótido de la transcriptasa inversa seleccionado del grupo que comprende: tenofovir alafenamida fumarato, tenofovir disoproxil fumarato, adefovir y una combinación de los mismos; o (ii) un inhibidor de la proteasa (IP) que comprende darunavir, y una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético que comprende piperina, tetrahidropiperina, c/s-piperina, trans-piperina, cis-trans piperina, trans,cis-piperina, cis,cis-piperina, trans,trans-piperina o una combinación de las mismas, en donde el al menos un fármaco antirretrovírico está en una composición separada del al menos un reforzador o potenciador farmacocinético.
15. Uso de una combinación que comprende (i) una cantidad terapéuticamente eficaz de al menos un fármaco antirretrovírico que comprende (a) un inhibidor análogo de nucleótido de la transcriptasa inversa seleccionado del grupo que comprende: tenofovir alafenamida fumarato, tenofovir disoproxil fumarato, adefovir y una combinación de los mismos; o (b) un inhibidor de la proteasa (IP) que comprende darunavir, y (ii) una cantidad terapéuticamente eficaz de al menos un reforzador o potenciador farmacocinético que comprende piperina, tetrahidropiperina, c/s-piperina, trans-piperina, cis-trans piperina, trans,cis-piperina, cis,cis-piperina, trans,trans-piperina o una combinación de las mismas para mejorar la biodisponibilidad del fármaco antirretrovírico oral, opcionalmente en donde (I) el al menos un fármaco antirretrovírico está en una primera composición y el al menos un reforzador o potenciador farmacocinético está en una segunda composición; o (II) el al menos un fármaco antirretrovírico y el al menos un reforzador o potenciador farmacocinético se combinan en una composición.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201621005051 | 2016-02-12 | ||
| IN201621032504 | 2016-09-23 | ||
| IN201621040945 | 2016-11-30 | ||
| PCT/IN2017/050046 WO2017138022A1 (en) | 2016-02-12 | 2017-02-01 | Pharmaceutical compositions comprising an anti-retroviral drug and a pharmacokinetic enhancer |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2902136T3 true ES2902136T3 (es) | 2022-03-25 |
Family
ID=58347738
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17711359T Active ES2902136T3 (es) | 2016-02-12 | 2017-02-01 | Composiciones farmacéuticas que comprenden un fármaco antirretrovírico y un potenciador farmacocinético |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US20170232020A1 (es) |
| EP (1) | EP3413913B1 (es) |
| JP (1) | JP7085998B2 (es) |
| CN (1) | CN109195633A (es) |
| AU (2) | AU2017218800B2 (es) |
| BR (1) | BR112018016517B1 (es) |
| CA (1) | CA3014411A1 (es) |
| ES (1) | ES2902136T3 (es) |
| RU (1) | RU2745204C2 (es) |
| WO (1) | WO2017138022A1 (es) |
| ZA (1) | ZA201806061B (es) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3612226A1 (en) * | 2017-04-18 | 2020-02-26 | Cipla Limited | Combination therapy for use in treating retroviral infections |
| EP3700573A1 (en) * | 2017-10-24 | 2020-09-02 | Gilead Sciences, Inc. | Methods of treating patients co-infected with a virus and tuberculosis |
| CN108299500A (zh) * | 2018-04-04 | 2018-07-20 | 安徽安科恒益药业有限公司 | 一种富马酸替诺福韦艾拉酚胺原料药及其生产工艺 |
| CN112243383A (zh) * | 2018-04-05 | 2021-01-19 | 希普拉有限公司 | 药物制剂 |
| US11110123B2 (en) * | 2018-07-17 | 2021-09-07 | Triumvira Immunologics Usa, Inc. | T cell-antigen coupler with various construct optimizations |
| AU2020211600A1 (en) | 2019-01-25 | 2021-07-22 | Brown University | Compositions and methods for treating, preventing or reversing age-associated inflammation and disorders |
| MX2022006274A (es) * | 2019-11-26 | 2022-10-13 | Biotron Ltd | Metodos de tratamiento de la infeccion por vih-1. |
| IL293566A (en) * | 2019-12-09 | 2022-08-01 | Viiv Healthcare Co | Pharmaceutical preparations containing Cabotgravir |
| CN110946866B (zh) * | 2019-12-12 | 2020-12-15 | 中山大学 | 达匹维林在制备抑制鱼类病毒性出血性败血症病毒对细胞感染的药物中的应用 |
| WO2021116244A1 (en) * | 2019-12-12 | 2021-06-17 | Sandoz Ag | Modulation of drug release and bioavailability of compositions containing dolutegravir sodium and other anti hiv drugs |
| CN113133970A (zh) * | 2020-01-17 | 2021-07-20 | 美国琛蓝营养制品股份有限公司 | 一种姜黄素复合物及其制备方法和检测方法 |
| CN113662937B (zh) * | 2021-10-09 | 2022-08-23 | 江苏省人民医院(南京医科大学第一附属医院) | Plerixafor在上调EFTUD2表达和抑制HBV药物中的用途 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8178123B2 (en) * | 2001-08-29 | 2012-05-15 | Femina Pharma Incorporated | Method for augmentation of intraepithelial and systemic exposure of therapeutic agents having substrate activity for cytochrome P450 enzymes and membrane efflux systems following vaginal and oral cavity administration |
| DE60309582T2 (de) | 2002-04-04 | 2007-09-13 | Cadila Pharmaceuticals Ltd. | Verfahren zur herstellung einer einen antiretroviralen proteasehemmer enthaltenden pharmazeutischen zusammensetzung mit verbesserter bioverfügbarkeit |
| DE10215724A1 (de) * | 2002-04-10 | 2003-10-30 | Celanese Ventures Gmbh | Kovalent fixierte Ligandensysteme, Non-Metallocene und Katalysatorsysteme, Verfahren zur Herstellung von diesen und deren Verwendung zur Polymerisation von Olefinen |
| EP1587521A1 (en) * | 2003-01-30 | 2005-10-26 | Council of Scientific and Industrial Research | Bioavailability enhancing activity of carum carvi extracts and fractions thereof |
| WO2008043829A2 (en) * | 2006-10-14 | 2008-04-17 | Boehringer Ingelheim International Gmbh | Method for treating hiv-1 infection by the combined administration of nevirapine, tenofovir and emtricitabine |
| BR112014018918A8 (pt) * | 2012-02-03 | 2017-07-11 | Gilead Sciences Inc | Terapia de combinação compreendendo hemifumarato de tenofovir alafenamida e cobicistato para uso no tratamento de infecções virais |
| GB201401617D0 (en) | 2014-01-30 | 2014-03-19 | Helperby Therapeutics Ltd | Novel combination and use |
| EP3612226A1 (en) * | 2017-04-18 | 2020-02-26 | Cipla Limited | Combination therapy for use in treating retroviral infections |
| CN112243383A (zh) * | 2018-04-05 | 2021-01-19 | 希普拉有限公司 | 药物制剂 |
-
2017
- 2017-02-01 US US15/421,939 patent/US20170232020A1/en not_active Abandoned
- 2017-02-01 CA CA3014411A patent/CA3014411A1/en active Pending
- 2017-02-01 AU AU2017218800A patent/AU2017218800B2/en not_active Ceased
- 2017-02-01 WO PCT/IN2017/050046 patent/WO2017138022A1/en not_active Ceased
- 2017-02-01 BR BR112018016517-4A patent/BR112018016517B1/pt not_active IP Right Cessation
- 2017-02-01 JP JP2018543111A patent/JP7085998B2/ja active Active
- 2017-02-01 ES ES17711359T patent/ES2902136T3/es active Active
- 2017-02-01 EP EP17711359.4A patent/EP3413913B1/en active Active
- 2017-02-01 RU RU2018132408A patent/RU2745204C2/ru active
- 2017-02-01 CN CN201780019366.5A patent/CN109195633A/zh active Pending
-
2018
- 2018-09-10 ZA ZA2018/06061A patent/ZA201806061B/en unknown
-
2020
- 2020-10-15 US US17/071,538 patent/US11612612B2/en active Active
-
2022
- 2022-12-21 AU AU2022291490A patent/AU2022291490A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| NZ746125A (en) | 2024-08-30 |
| WO2017138022A1 (en) | 2017-08-17 |
| JP2019504869A (ja) | 2019-02-21 |
| AU2022291490A1 (en) | 2023-02-02 |
| RU2018132408A3 (es) | 2020-03-12 |
| US20170232020A1 (en) | 2017-08-17 |
| ZA201806061B (en) | 2022-04-28 |
| EP3413913A1 (en) | 2018-12-19 |
| CN109195633A (zh) | 2019-01-11 |
| RU2018132408A (ru) | 2020-03-12 |
| BR112018016517B1 (pt) | 2024-03-12 |
| RU2745204C2 (ru) | 2021-03-22 |
| BR112018016517A2 (pt) | 2019-03-19 |
| EP3413913B1 (en) | 2021-11-24 |
| AU2017218800B2 (en) | 2022-09-22 |
| US11612612B2 (en) | 2023-03-28 |
| US20210038619A1 (en) | 2021-02-11 |
| JP7085998B2 (ja) | 2022-06-17 |
| CA3014411A1 (en) | 2017-08-17 |
| AU2017218800A1 (en) | 2018-09-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2902136T3 (es) | Composiciones farmacéuticas que comprenden un fármaco antirretrovírico y un potenciador farmacocinético | |
| ES2552238T3 (es) | Combinación farmacéutica | |
| US20240285629A1 (en) | Combination Therapy for Use in Treating Retroviral Infections | |
| US20210100786A1 (en) | Pharmaceutical Formulations | |
| BR112019014712A2 (pt) | Tratamento médio que compreende a administração entérica de edaravona | |
| US11045423B2 (en) | Anti-retroviral compositions | |
| CN112957334A (zh) | 含alpelisib的药物组合物 | |
| ES3033621T3 (en) | Dispersible tablet formulations comprising dolutegravir | |
| KR20230027049A (ko) | 제제 | |
| RU2847035C1 (ru) | Композиции | |
| CN110087688B (zh) | 包含利福昔明的药物组合物 | |
| CN111565721A (zh) | 具有增强的生物利用度的包含依鲁替尼和生物碱的组合物 | |
| RU2543322C1 (ru) | Фармацевтическая композиция для лечения вич-инфекции, способ ее получения и способ лечения | |
| Archana et al. | FORMULATION AND INVITRO EVALUATION OF ATAZANAVIR ORAL DISINTEGRATING TABLETS |