ES2898705T3 - Moduladores de los receptores farnesoides x - Google Patents
Moduladores de los receptores farnesoides x Download PDFInfo
- Publication number
- ES2898705T3 ES2898705T3 ES16854420T ES16854420T ES2898705T3 ES 2898705 T3 ES2898705 T3 ES 2898705T3 ES 16854420 T ES16854420 T ES 16854420T ES 16854420 T ES16854420 T ES 16854420T ES 2898705 T3 ES2898705 T3 ES 2898705T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- disease
- compounds
- mmol
- liver
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J9/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane
- C07J9/005—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane containing a carboxylic function directly attached or attached by a chain containing only carbon atoms to the cyclopenta[a]hydrophenanthrene skeleton
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/575—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of three or more carbon atoms, e.g. cholane, cholestane, ergosterol, sitosterol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/58—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids containing heterocyclic rings, e.g. danazol, stanozolol, pancuronium or digitogenin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J31/00—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring
- C07J31/006—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring not covered by C07J31/003
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J41/00—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring
- C07J41/0033—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005
- C07J41/0055—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 the 17-beta position being substituted by an uninterrupted chain of at least three carbon atoms which may or may not be branched, e.g. cholane or cholestane derivatives, optionally cyclised, e.g. 17-beta-phenyl or 17-beta-furyl derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J41/00—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring
- C07J41/0033—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005
- C07J41/0055—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 the 17-beta position being substituted by an uninterrupted chain of at least three carbon atoms which may or may not be branched, e.g. cholane or cholestane derivatives, optionally cyclised, e.g. 17-beta-phenyl or 17-beta-furyl derivatives
- C07J41/0061—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 the 17-beta position being substituted by an uninterrupted chain of at least three carbon atoms which may or may not be branched, e.g. cholane or cholestane derivatives, optionally cyclised, e.g. 17-beta-phenyl or 17-beta-furyl derivatives one of the carbon atoms being part of an amide group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J41/00—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring
- C07J41/0033—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005
- C07J41/0077—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 substituted in position 11-beta by a carbon atom, further substituted by a group comprising at least one further carbon atom
- C07J41/0083—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 substituted in position 11-beta by a carbon atom, further substituted by a group comprising at least one further carbon atom substituted in position 11-beta by an optionally substituted phenyl group not further condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J41/00—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring
- C07J41/0033—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005
- C07J41/0094—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 containing nitrile radicals, including thiocyanide radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J71/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton is condensed with a heterocyclic ring
- C07J71/0005—Oxygen-containing hetero ring
- C07J71/001—Oxiranes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J9/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Immunology (AREA)
- Heart & Thoracic Surgery (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Toxicology (AREA)
- Emergency Medicine (AREA)
- Transplantation (AREA)
- Vascular Medicine (AREA)
- Epidemiology (AREA)
- Child & Adolescent Psychology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Gastroenterology & Hepatology (AREA)
- Endocrinology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Steroid Compounds (AREA)
- Peptides Or Proteins (AREA)
Abstract
Un compuesto de fórmula Ie: **(Ver fórmula)** o una sal o un solvato farmacéuticamente aceptable del mismo, deonde R2 y R5 son OH; R4 es C1-C6 alquilo opcionalmente sustituido con uno o más halógenos u OH; R7 es OH, OSO3H, SO3H, tetrazolilo, oxadiazolilo, tiadiazolilo, 5-oxo-1,2,4-oxadiazolilo, 5-oxo-1,2,4-tiadiazolilo, oxazolidinadionilo, tiazolidinadionilo, 3-hidroxiisoxazolilo, 3-hidroxiisotiazolilo o 2,4-difluoro-3-hidroxifenilo; R8, R9 y R10 son cada uno independientemente H, OH, halógeno o alquilo opcionalmente sustituido con uno o más halógenos u OH, o R8 y R9 tomados junto con los átomos de carbono a los que están unidos forman un anillo carbocíclico o heterocíclico de 3 a 6 miembros que comprende 1 o 2 heteroátomos seleccionados entre N O, y S, o R9 y R10 tomados junto con los átomos de carbono a los que están unidos forman un anillo carbocíclico o heterocíclico de 3 a 6 miembros que comprende 1 o 2 heteroátomos seleccionados entre N, O, y S; m es 0, 1 o 2; n es 0 o 1; p es 0 o 1; y es un enlace simple o doble.
Description
DESCRIPCIÓN
Moduladores de los receptores farnesoides x
Antecedentes de la invención
El receptor farnesoide X (FXR) es un receptor nuclear que funciona como un sensor de ácidos biliares que controla la homeostasis de los ácidos biliares. El FXR se expresa en varios órganos y se ha demostrado que está implicado en muchas enfermedades y afecciones, como enfermedades hepáticas, pulmonares, renales, intestinales y cardíacas, y en procesos biológicos, como el metabolismo de la glucosa, de la insulina y de los lípidos. Varios ácidos biliares naturales son moduladores del FXR, y son capaces de regular las enfermedades y condiciones mediadas por el FXR (Gioiello, et al., 2014 Curr. Top. Med. Chem. 14, 2159). Por ejemplo, los ácidos biliares naturales como el ácido quenodesoxicólico (CDCA), el ácido desoxicólico (DCA), el ácido litocólico (LCA) y los conjugados de taurina y glicina de los mismos sirven como ligandos del FXR.
También se han descrito derivados de ácidos biliares naturales como moduladores del FXR. La patente europea No.
0312867 describe derivados 6-metilados de ácidos biliares naturales como el ácido ursodesoxicólico, el ácido ursocólico, el ácido quenodesoxicólico y el ácido cólico. El documento WO 2002/75298 divulga el ácido 3, 7-dihidroxi-6-etil-5-colan-24-oico (en lo sucesivo también denominado ácido 6-etil-chenodeoxicólico o 6-ECDCA), sales, solvatos y conjugados de aminoácidos del mismo como moduladores del FXR, que pueden utilizarse para prevenir o tratar enfermedades o afecciones mediadas por el FXR.
Sin embargo, es bien sabido que los ácidos biliares naturales y los derivados de ácidos biliares modulan no sólo otros receptores de hormonas nucleares, sino que también son moduladores del receptor acoplado a proteínas G (GPCR) TGR5. La selectividad del receptor es un problema en relación con el desarrollo de un compuesto terapéutico dirigido a modular un receptor hormonal nuclear como el FXR. Un compuesto terapéutico no selectivo puede conllevar un mayor riesgo de efectos secundarios. Otros obstáculos que hay que superar en el desarrollo de un compuesto terapéutico son un perfil farmacocinético no adecuado, problemas de seguridad como la toxicidad (por ejemplo, hepática) e interacciones farmacológicas indeseables. El documento WO-A-2014/184271 divulga el derivado 11.beta-hidróxido del 6-EDCA (denominado en la presente invención compuesto L1 y referido como compuesto 100 en la presente invención) como modulador del FXR con efectos reducidos del TGR5.
Por lo tanto, sigue habiendo una necesidad de moduladores selectivos adicionales de FXR adecuados para el desarrollo de fármacos, por ejemplo, un compuesto que sea selectivo contra otros receptores nucleares y/o que no active significativamente el GPCR de ácidos biliares TGR5.
Breve descripción de la invención
Un objetivo de la presente invención es proporcionar compuestos que modulen el FXR. En un aspecto, la presente invención proporciona un compuesto de fórmula Ie:

o una sal o un solvato farmacéuticamente aceptable del mismo, en el que R2 y R5 son OH;
R4 es C1-C6 alquilo opcionalmente sustituido con uno o más halógenos u OH;
R7 es OH, OSO3H, SO3H, tetrazolilo, oxadiazolilo, tiadiazolilo, 5-oxo-1,2,4-oxadiazolilo, 5-oxo-1,2,4-tiadiazolilo, oxazolidina-dionilo, tiazolidina-dionilo, 3-hidroxiisoxazolilo, 3-hidroxiisotiazolilo o 2,4-difluoro-3-hidroxifenilo; R8 , R9 y R10 son cada uno independientemente H, OH, halógeno o alquilo opcionalmente sustituido con uno o más halógenos u OH, o R8 y R9 tomados junto con los átomos de carbono a los que están unidos forman un anillo carbocíclico o heterocíclico de 3 a 6 miembros que comprende 1 o 2 heteroátomos seleccionados entre N O, y S, o R9 y R10 tomados junto con los átomos de carbono a los que están unidos forman un anillo carbocíclico o heterocíclico de 3 a 6 miembros que comprende 1 o 2 heteroátomos seleccionados entre N, O, y S;
m es 0, 1 o 2;
n es 0 o 1;
p es 0 o 1; y
= es un enlace simple o doble.
La presente invención proporciona además una composición farmacéutica que comprende un compuesto de fórmula le o una sal o solvato farmacéuticamente aceptable del mismo, y un portador o excipiente farmacéuticamente aceptable. La presente invención también proporciona los compuestos de fórmula Ie para su uso en un método para tratar o prevenir una enfermedad o condición mediada por FXR, que comprende administrar a un sujeto que lo necesita una cantidad eficaz de un compuesto de fórmula Ie o una sal o solvato farmacéuticamente aceptable del mismo.
La presente invención también proporciona la fabricación de un medicamento para tratar o prevenir una enfermedad o condición mediada por FXR, en el que el medicamento comprende un compuesto de fórmula Ie o una sal o solvato farmacéuticamente aceptable del mismo.
La presente invención proporciona además composiciones, incluyendo composiciones farmacéuticas, para su uso en el tratamiento o la prevención de una enfermedad o condición mediada por FXR, donde la composición comprende un compuesto de fórmula Ie o una sal o solvato farmacéuticamente aceptable del mismo.
A menos que se defina lo contrario, todos los términos técnicos y científicos utilizados en la presente invención tienen el mismo significado que comúnmente entiende una persona con conocimientos ordinarios en el arte al que pertenece esta invención. Aunque se pueden utilizar métodos y materiales similares o equivalentes a los descritos en la presente invención en la práctica o prueba de la presente invención, a continuación, se describen métodos y materiales adecuados. Los materiales, métodos y ejemplos son sólo ilustrativos y no pretenden ser limitantes.
Otras características y ventajas de la invención serán evidentes a partir de la siguiente descripción detallada.
Descripción detallada de la invención
Definiciones
Algunos términos utilizados en la descripción y las reivindicaciones se recogen aquí.
Tal como se utiliza aquí, la frase "un compuesto de la invención" se refiere a un compuesto de fórmula Ie.
Tal como se utiliza en la presente invención, el término "alquilo" se refiere a una fracción de hidrocarburo saturado de cadena recta o ramificada. El término "alquilo C1-C6" se refiere a una fracción de hidrocarburo de cadena recta o ramificada que tiene 1, 2, 3, 4, 5 o 6 átomos de carbono. El término "alquilo C1-C4" se refiere a una fracción de hidrocarburo de cadena recta o ramificada que tiene 1, 2, 3 o 4 átomos de carbono, incluyendo el metilo, el etilo, el npropilo, el isopropilo, el n-butilo, el isobutilo, el sec-butilo y el t-butilo.
El término "alquenilo" se refiere a una fracción de hidrocarburo de cadena recta o ramificada que contiene al menos un doble enlace carbono-carbono. Tanto los isómeros trans como cis del doble enlace carbono-carbono se engloban bajo el término "alquenilo". Los ejemplos de moléculas de alquenilo incluyen, pero no se limitan a, vinilo, alilo, 1 -butenilo, 2-butenilo, 3-butenilo y 2- hexenilo.
Tal como se utiliza en la presente invención, "alquinilo" se refiere a una fracción de hidrocarburo de cadena recta o ramificada que contiene al menos un triple enlace carbono-carbono. Los ejemplos de moléculas de alquilo incluyen, pero no se limitan a, etilo, 2-propinilo, 5-but-1-en- 3-quinilo y 3-hexinilo.
El término "alcoxi" se refiere a un hidrocarburo saturado de cadena recta o ramificado unido covalentemente a un átomo de oxígeno. Los ejemplos de moléculas alcoxi incluyen, pero no se limitan a, metoxi, etoxi, isopropiloxi, n-propoxi, nbutoxi, t-butoxiy pentoxi.
Tal como se utiliza aquí, el término "halógeno" se refiere al flúor, el bromo, el cloro y el yodo.
El término "opcionalmente sustituido" se refiere a la fracción indicada que puede o no estar sustituida, y cuando está sustituida es mono-, di-, o tri-sustituida, como por ejemplo con 1, 2, o 3 sustituyentes. En algunos casos, el sustituyente es halógeno u OH.
Tal como se utiliza aquí, "carbociclo", "carbocíclico" o "anillo carbocíclico" pretende incluir cualquier anillo monocíclico o bicíclico estable que tenga el número especificado de carbonos, cualquiera de los cuales puede ser saturado, insaturado o aromático. El anillo carbocíclico incluye el cicloalquilo y el arilo. Por ejemplo, un anillo carbocíclico C3-C8 incluye un anillo monocíclico o bicíclico con 3, 4, 5, 6, 7 u 8 átomos de carbono. Los ejemplos de carbocíclicos incluyen, pero no se limitan a, ciclopropilo, ciclobutilo, ciclobutenilo, ciclopentilo, ciclopentenilo, ciclohexilo, cicloheptenilo, cicloheptilo, cicloheptenilo, adamantilo, ciclooctilo, ciclooctenilo y fenilo.
Tal como se utiliza aquí, "heterocido", "heterocíclico" o "grupo heterocíclico" incluye cualquier estructura de anillo (saturado, insaturado o aromático) que contenga al menos un heteroátomo de anillo (por ejemplo, N, O o S). El heterociclo incluye el heterocicloalquilo y el heteroarilo. Los ejemplos de heterociclos incluyen, entre otros, morfolina, pirrolidina, tetrahidrotiofeno, piperidina, piperazina, oxetano, pirano, tetrahidropirano, azetidina y tetrahidrofurano. Los ejemplos de grupos heterocíclicos incluyen, pero no se limitan a, benzimidazolilo, benzofuranilo, benzotiofuranilo, tetrahidrofurano, furanilo, furazanilo, imidazolidinilo, imidazolinilo, imidazolilo, 1 H-indazolilo, indolenilo indolinilo, indolizinilo, indolilo, 3H-indolilo, isatinoilo, isobenzofuranoilo, isoindazolilo, isoindolinilo, isoquinolinilo, isotiazolilo, isoxazolilo, metilendioxifenilo, morfolinilo, piridinilo, piridilo y pirimidinilo.
Tal como se utiliza en la presente invención, el término "cicloalquilo" se refiere a un sistema de hidrocarburos no aromáticos saturados o insaturados de uno o varios anillos (por ejemplo, anillos fusionados, en puente o espiro) que tienen de 3 a 10 átomos de carbono (por ejemplo, C3-C6). Los ejemplos de cicloalquilo incluyen, pero no se limitan a, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, ciclooctilo, ciclopentenilo, ciclohexenilo y cicloheptenilo. El término "heterocicloalquilo" se refiere a un grupo monocíclico o bicíclico saturado o insaturado no aromático de 3-8 miembros (anillos fusionados, en puente o espiro) que tiene uno o más heteroátomos (como O, N o S), a menos que se especifique lo contrario. Los ejemplos de grupos heterocicloalquilo incluyen, pero no se limitan a, piperidinilo, piperazinilo, pirrolidinilo, dioxanilo, tetrahidrofuranoilo, isoindolinilo, indolinilo, imidazolidinilo, pirazolidinilo, oxazolidinilo isoxazolidinilo, triazolidinilo, tetrahidrofuranoilo, oxiranilo, azetidinilo, oxetanilo, tietanilo, 1,2,3,6-tetrahidropiridinilo, tetrahidropiranilo, dihidropiranilo, piranilo, morfolinilo y tetrahidrotiopiranilo y similares.
Tal como se utiliza en la presente invención, cualquier fracción recitada que incluya, pero no se limite a, alquilo, alquenilo, alquino, alcoxi, anillo carbocíclico, anillo heterocíclico, cicloalquilo, heterocicloalquilo, etc., puede estar opcionalmente sustituida.
El término "modulador de FXR" se refiere a cualquier compuesto que interactúa con el receptor FXR. La interacción no se limita a un compuesto que actúa como antagonista, agonista, agonista parcial o agonista inverso del receptor FXR. En una realización, el compuesto de la invención actúa como antagonista del receptor FXR. En otro aspecto, el compuesto de la invención actúa como un agonista del receptor FXR. En otro aspecto, el compuesto de la invención actúa como un agonista parcial del receptor FXR. En otro aspecto, el compuesto de la invención actúa como un agonista inverso del receptor FXR.
"Solvato", tal como se utiliza aquí, se refiere a una forma de adición de disolvente de un compuesto de la invención que contiene cantidades estequiométricas o no estequiométricas de disolvente. Algunos compuestos tienen tendencia a atrapar una proporción molar fija de moléculas de disolvente en el estado sólido cristalino, formando así un solvato. Si el disolvente es agua, el solvato formado es un hidrato, y cuando el disolvente es alcohol, el solvato formado es un alcoholato. Los hidratos se forman por la combinación de una o más moléculas de agua con una de las sustancias en las que el agua conserva su estado molecular como H2O, siendo dicha combinación capaz de formar uno o más hidratos.
Tal como se utiliza en la presente invención, el término "conjugados de aminoácidos" se refiere a conjugados de un compuesto de la invención con cualquier aminoácido adecuado. La taurina (-NH(CH2)2SO3H), la glicina (-NHCH2CO2H) y la sarcosina (-N(CH3)CH2CO2H) son ejemplos de conjugados de aminoácidos. Los conjugados de aminoácidos adecuados de los compuestos tienen la ventaja añadida de mejorar su integridad en los fluidos biliares o intestinales. Los aminoácidos adecuados no se limitan a la taurina, la glicina y la sarcosina.
Tal como se define en la presente invención, el término "metabolito" se refiere a los derivados glucuronidados y sulfatados de los compuestos descritos en la presente invención, en los que una o más moléculas de ácido glucurónico o sulfato están unidos al compuesto de la invención. Las moléculas de ácido glucurónico pueden unirse a los compuestos a través de enlaces glucosídicos con los grupos hidroxilo de los compuestos (por ejemplo, 3- hidroxilo, 7-hidroxilo, 11-hidroxilo, y/o el hidroxilo del grupo R7). Los derivados sulfatados de los compuestos pueden formarse a través de la sulfatación de los grupos hidroxilo (por ejemplo, 3-hidroxilo, 7-hidroxilo, 11-hidroxilo, y/o el hidroxilo del grupo R7). Los ejemplos de metabolitos incluyen, entre otros, 3-O-glucurónido, 7-O-glucurónido, 11-O- glucurónido, 3-O-7-O-diglucurónido, 3-O-11-O-triglucurónido, 7-O-11-O-triglucurónido y 3-O-7-O-11-O-triglucurónido, de los compuestos aquí descritos, y 3-sulfato, 7-sulfato, 11-sulfato, 3,7-bisulfato, 3,11 -bisulfato, 7,11 -bisulfato, y 3,7,11- trisulfato, de los compuestos aquí descritos.
Tal como se utiliza en la presente invención, las "sales farmacéuticamente aceptables" se refieren a los derivados de un compuesto de la invención en los que el compuesto original se modifica formando sales ácidas o básicas del mismo. Los ejemplos de sales farmacéuticamente aceptables incluyen, pero no se limitan a, sales minerales u orgánicas de residuos básicos como las aminas; sales alcalinas u orgánicas de residuos ácidos como los ácidos carboxílicos; y similares. Las sales farmacéuticamente aceptables incluyen las sales convencionales no tóxicas o las sales de amonio cuaternario del compuesto madre formadas, por ejemplo, a partir de ácidos inorgánicos u orgánicos no tóxicos. Por ejemplo, tales sales convencionales no tóxicas incluyen, pero no se limitan a, las derivadas de ácidos inorgánicos y orgánicos seleccionados de 2-acetoxibenzoico, 2-hidroxietano sulfónico, acético ascórbico, benceno sulfónico, benzoico, bicarbónico, carbónico, cítrico, edético, etano disulfónico, fumárico, glucoheptónico, glucónico, glutámico, glicólico,
glicolarsanílico, hexilresorcínico, hidrábico, bromhídrico, clorhídrico, hidroyódico, hidroximaleico, hidroxinaftoico, isetiónico, láctico, lactobiónico, lauril sulfónico, maleico, málico, mandélico, metanosulfónico, napsílico, nítrico, oxálico, pamoico, pantoténico, fenilacético, fosfórico, poligalacturónico, propiónico, salicílico, esteárico, subacético, succínico, sulfámico, sulfanílico, sulfúrico, tánico, tartárico y tolueno sulfónico.
La frase "portador farmacéuticamente aceptable" está reconocida en el arte, e incluye, por ejemplo, materiales, composiciones o vehículos farmacéuticamente aceptables, como un relleno líquido o sólido, diluyente, excipiente, disolvente o material de encapsulación, que participan en el transporte de cualquier composición temática desde un órgano, o porción del cuerpo, a otro órgano, o porción del cuerpo. Cada portador es "aceptable" en el sentido de que es compatible con los demás ingredientes de una composición temática y no es perjudicial para el paciente. En ciertas realizaciones, un portador farmacéuticamente aceptable no es pirogénico. Algunos ejemplos de materiales que pueden servir como portadores farmacéuticamente aceptables incluyen (1) azúcares, como la lactosa, la glucosa y la sacarosa; (2) almidones, como el almidón de maíz y la fécula de patata; (3) celulosa, y sus derivados, como la carboximetilcelulosa sódica, la etilcelulosa y el acetato de celulosa; (4) tragacanto en polvo; (5) malta; (6) gelatina; (7) talco; (8) excipientes, como la manteca de cacao y las ceras para supositorios; (9) aceites, como el aceite de cacahuete, el aceite de semilla de algodón, el aceite de girasol, el aceite de sésamo, el aceite de oliva, el aceite de maíz y el aceite de soja; (10) glicoles, como el propilenglicol; (11) polioles, como la glicerina, el sorbitol, el manitol y el polietilenglicol; (12) ésteres, como el oleato de etilo y el laurato de etilo; (13) agar; (14) agentes amortiguadores, como el hidróxido de magnesio y el hidróxido de aluminio; (15) ácido algínico; (16) agua libre de pirógenos; (17) solución salina isotónica; (18) solución de Ringer; (19) alcohol etílico; (20) soluciones tampón de fosfato; y (21) otras sustancias compatibles no tóxicas empleadas en formulaciones farmacéuticas.
Una "composición" o "composición farmacéutica" es una formulación que contiene un compuesto de la invención o una sal, un solvato o un conjugado de aminoácidos del mismo. En una realización, la composición farmacéutica es a granel o en forma de dosificación unitaria. La forma de dosificación unitaria es cualquiera de una variedad de formas, incluyendo, por ejemplo, una cápsula, una bolsa intravenosa, un comprimido, una bomba única en un inhalador de aerosol o un vial. La cantidad de ingrediente activo (por ejemplo, una formulación de un compuesto de la invención o sales del mismo) en una dosis unitaria de la composición es una cantidad efectiva y se varía según el tratamiento particular de que se trate. Un experto en la materia apreciará que puede ser necesario realizar variaciones rutinarias de la dosis en función de la edad y el estado del paciente. La dosis también dependerá de la vía de administración. Se contempla una variedad de vías, incluyendo la oral, ocular, oftálmica, pulmonar, rectal, parenteral, transdérmica, subcutánea, intravenosa, intramuscular, intraperitoneal, intranasal y similares. Las formas de dosificación para la administración tópica o transdérmica de un compuesto de esta aplicación incluyen polvos, aerosoles, ungüentos, pastas, cremas, lociones, geles, soluciones, parches e inhalantes. En otra realización, el compuesto activo se mezcla en condiciones estériles con un portador farmacéuticamente aceptable, y con los conservantes, tampones o propulsores que sean necesarios.
El término "tratar", tal y como se utiliza aquí, se refiere a aliviar, disminuir, reducir, eliminar, modular o mejorar, es decir, provocar la regresión del estado de la enfermedad o la condición.
El término "prevenir", tal y como se utiliza en este documento, se refiere a impedir por completo o casi por completo que se produzca un estado o condición de enfermedad en un paciente o sujeto, especialmente cuando el paciente o sujeto está predispuesto a ello o corre el riesgo de contraer un estado o condición de enfermedad. La prevención también puede incluir la inhibición, es decir, la detención del desarrollo, de un estado o condición de enfermedad, y el alivio o la mejora, es decir, la regresión del estado o condición de enfermedad, por ejemplo, cuando el estado o condición de enfermedad puede estar ya presente.
La frase "reducir el riesgo de", tal y como se utiliza aquí, se refiere a reducir la probabilidad de que una enfermedad del sistema nervioso central, una enfermedad inflamatoria y/o una enfermedad metabólica se produzca en un paciente, especialmente cuando el sujeto está predispuesto a que se produzca.
La "terapia de combinación" (o "co-terapia") se refiere a la administración de un compuesto de la invención y al menos un segundo agente como parte de un régimen de tratamiento específico destinado a proporcionar el efecto beneficioso de la co-acción de estos agentes terapéuticos (es decir, el compuesto de la invención y al menos un segundo agente). El efecto beneficioso de la combinación incluye, pero no se limita a, la coacción farmacocinética o farmacodinámica resultante de la combinación de agentes terapéuticos. La administración de estos agentes terapéuticos en combinación suele llevarse a cabo durante un periodo de tiempo definido (normalmente minutos, horas, días o semanas, dependiendo de la combinación seleccionada). La "terapia de combinación" puede, pero generalmente no se pretende que abarque la administración de dos o más de estos agentes terapéuticos como parte de regímenes de monoterapia separados que incidental y arbitrariamente dan lugar a las combinaciones de la presente solicitud. La "terapia combinada" pretende abarcar la administración de estos agentes terapéuticos de forma secuencial, es decir, en la que cada agente terapéutico se administra en un momento diferente, así como la administración de estos agentes terapéuticos, o al menos dos de los agentes terapéuticos, de forma sustancialmente simultánea. La administración sustancialmente simultánea puede lograrse, por ejemplo, administrando al sujeto una única cápsula con una proporción fija de cada agente terapéutico o en múltiples cápsulas individuales para cada uno de los agentes terapéuticos. La administración secuencial o sustancialmente simultánea de cada agente terapéutico puede efectuarse por cualquier vía
apropiada, incluyendo, pero sin limitarse a ello, las vías orales, intravenosas, intramusculares y la absorción directa a través de los tejidos de las membranas mucosas. Los agentes terapéuticos pueden administrarse por la misma vía o por vías diferentes. Por ejemplo, un primer agente terapéutico de la combinación seleccionada puede administrarse por inyección intravenosa, mientras que los demás agentes terapéuticos de la combinación pueden administrarse por vía oral. Alternativamente, por ejemplo, todos los agentes terapéuticos pueden administrarse por vía oral o todos los agentes terapéuticos pueden administrarse por inyección intravenosa. La secuencia en la que se administran los agentes terapéuticos no es crítica.
La "terapia combinada" también abarca la administración de los agentes terapéuticos descritos anteriormente en combinación con otros ingredientes biológicamente activos y terapias no farmacológicas (por ejemplo, cirugía o tratamientos mecánicos). Cuando la terapia combinada comprende además un tratamiento no farmacológico, el tratamiento no farmacológico puede llevarse a cabo en cualquier momento adecuado siempre que se consiga un efecto beneficioso de la coacción de la combinación de los agentes terapéuticos y el tratamiento no farmacológico. Por ejemplo, en casos apropiados, el efecto beneficioso se sigue logrando cuando el tratamiento no farmacológico se aleja temporalmente de la administración de los agentes terapéuticos, tal vez por días o incluso semanas.
Una "cantidad efectiva" de un compuesto de la invención, o una combinación de compuestos es una cantidad (cantidad o concentración) de compuesto o compuestos. En una realización, cuando se administra una cantidad terapéuticamente eficaz de un compuesto a un sujeto que necesita tratamiento, los síntomas derivados de la enfermedad mejoran inmediatamente o después de la administración del compuesto una o más veces. La cantidad del compuesto que debe administrarse a un sujeto dependerá del trastorno concreto, el modo de administración, los compuestos coadministrados, si los hay, y las características del sujeto, como el estado de salud general, otras enfermedades, la edad, el sexo, el genotipo, el peso corporal y la tolerancia a los fármacos. El artesano experto podrá determinar las dosis adecuadas en función de estos y otros factores.
El término "cantidad profilácticamente eficaz" significa una cantidad (cantidad o concentración) de un compuesto de la presente invención, o una combinación de compuestos, que se administra para prevenir o reducir el riesgo de una enfermedad -en otras palabras, una cantidad necesaria para proporcionar un efecto preventivo o profiláctico. La cantidad del presente compuesto que debe administrarse a un sujeto dependerá del trastorno concreto, del modo de administración, de los compuestos coadministrados, si los hay, y de las características del sujeto, como el estado de salud general, otras enfermedades, la edad, el sexo, el genotipo, el peso corporal y la tolerancia a los fármacos. El artesano experto podrá determinar las dosis adecuadas en función de estos y otros factores. Un "sujeto" incluye mamíferos, por ejemplo, seres humanos, animales de compañía (por ejemplo, perros, gatos, pájaros y similares), animales de granja (por ejemplo, vacas, ovejas, cerdos, caballos y similares) y animales de laboratorio (por ejemplo, ratas, ratones, cobayas y similares). Normalmente, el sujeto es humano.
Tal como se utiliza en la presente invención, el receptor X farnesoide o FXR se refiere a todas las formas de mamíferos de dicho receptor, incluyendo, por ejemplo, las isoformas de empalme alternativas y las isoformas naturales (véase, por ejemplo, Huber et al., Gene 290:35-43 (2002)). Las especies de FXR representativas incluyen, sin limitación, el FXR de rata (número de acceso del GenBank Nm 021745), el FXR de ratón (número de acceso del GenBank NM 009108) y el FXR humano (número de acceso del GenBank NM 005123).
Compuestos de la invención
En un aspecto, la presente divulgación proporciona un compuesto de fórmula Ie:

o una sal o un solvato farmacéuticamente aceptable del mismo, en el que R2 y R5 son OH; R4 es C1-C6 alquilo opcionalmente sustituido con uno o más halógenos u OH;
R7 es OH, OSO3H, SO3H, tetrazolilo, oxadiazolilo, tiadiazolilo, 5-oxo-1,2,4-oxadiazolilo, 5-oxo-1,2,4-tiadiazolilo, oxazolidina-dionilo, tiazolidina-dionilo, 3-hidroxiisoxazolilo, 3-hidroxiisotiazolilo o 2,4-difluoro-3-hidroxifenilo; R8 , R9 y R10 son cada uno independientemente H, OH, halógeno o alquilo opcionalmente sustituido con uno o más halógenos u OH, o R8 y R9 tomados junto con los átomos de carbono a los que están unidos forman un anillo carbocíclico o heterocíclico de 3 a 6 miembros que comprende 1 o 2 heteroátomos seleccionados entre N O, y S, o R9 y R10 tomados junto con los átomos de carbono a los que están unidos forman un anillo carbocíclico o heterocíclico de 3 a 6 miembros que
comprende 1 o 2 heteroátomos seleccionados entre N, O, y S;
m es 0, 1 o 2;
n es 0 o 1;
p es 0 o 1; y
= es un enlace simple o doble.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que R7 es OH, OSO3H o SO3H.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que R7 es OH.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que R7 es OSO3H. En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que R7 es SO3H. En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que R7 es tetrazolilo, oxadiazolilo, tiadiazolilo, 5-oxo-1,2,4-oxadiazolilo, 5-oxo-1,2,4-tiadiazolilo, oxazolidina-dionilo, tiazolidina- dionilo, 3-hidroxiisoxazolilo, 3-hidroxiisotiazolilo o 2,4-difluoro-3-hidroxifenilo.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que R7 es OH, OSO3H, tetrazolilo, oxadiazolilo, tiadiazolilo, 5-oxo-1,2,4-oxadiazolilo, 5-oxo-1,2,4-tiadiazolilo, oxazolidina-dionilo, tiazolidina-dionilo, 3-hidroxiisoxazolilo, 3-hidroxiisotiazolilo, o 2,4-difluoro-3-hidroxifenilo.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que R4 es metilo, etilo o propilo.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que m es 0.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que m es 1.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que m es 2.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, donde n es 1.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que p es 0.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que el compuesto se selecciona entre:

En una de las realizaciones, la presente divulgación proporciona sales de compuestos de fórmula Ie.
En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que R7 es OSO3. En una de las realizaciones, la presente divulgación proporciona compuestos de fórmula Ie, en los que R7 es OSO3 Na+. En una de las realizaciones, la presente divulgación proporciona un compuesto de fórmula Ie, en el que el compuesto se
selecciona de:

En una realización, R4 es metilo, etilo o propilo. En una realización, R4 es etilo.
En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es OH. En una En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es OSO3H. En otra realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es SO3H.
En otra realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es tetrazolilo, oxadiazolilo, tiadiazolilo, 5-oxo-1,2,4-oxadiazolilo, 5-oxo-1,2,4-tiadiazolilo, oxazolidina-dionilo, tiazolidina-dionilo, 3-hidroxiisoxazolilo, 3-hidroxiisotiazolilo, o 2,4-difluoro-3-hidroxifenilo. En otra realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es tetrazolilo. En una realización separada, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es oxadiazolilo. En una realización separada, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es tiadiazolilo. En otra realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es 5-oxo-1,2,4-oxadiazolilo. En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es oxazolidina-dionilo. En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es tiazolidina-dionilo. En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R7 es 2,4-difluoro-3-hidroxifenilo.
En una realización, la presente divulgación se refiere a un compuesto de Ie, en el que R8 es H. En una realización, R8 es independientemente H u OH. En una realización, R8 es independientemente H o halógeno. En una realización, R8 es independientemente H o alquilo. En una realización, R8 es independientemente H o alquilo sustituido con uno o más halógenos u OH.
En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R9 es H. En una realización, R9 es H u OH. En una realización, R9 es H o halógeno. En una realización, R9 es H o alquilo. En una realización, R9 es H o alquilo sustituido con uno o más halógenos u OH.
En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R8 y R9 son alquilos y tomados junto con los carbonos a los que están unidos forman un anillo de tamaño 3, 4, 5 o 6 átomos.
En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R10 es H. En una realización, R10 es H u OH. En una realización, R10 es H o halógeno. En una realización, R10 es H o alquilo. En una realización, R10 es H o alquilo sustituido con uno o más halógenos u OH.
En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que R9 y R10 son alquilos y tomados junto con los carbonos a los que están unidos forman un anillo de tamaño 3, 4, 5 o 6 átomos.
En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que m es 0. En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que m es 1. En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que m es 2.
En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que n es 0. En una realización, n es 1.
En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que p es 0. En una realización, p es 1.
En una realización, la presente divulgación se refiere a un compuesto de fórmula Ie, en el que el compuesto se selecciona entre:

En una de las realizaciones, la presente divulgación se refiere a un compuesto de fórmula le, en el que el compuesto se selecciona entre:

Los compuestos de la invención tienen centros asimétricos y pueden aislarse en formas ópticamente activas o racémicas. Es bien conocido en el arte cómo preparar formas ópticamente activas, como por resolución de formas racémicas o por síntesis a partir de materiales de partida ópticamente activos. Muchos isómeros geométricos de olefinas, y similares, también pueden estar presentes en los compuestos descritos aquí, y todos esos isómeros estables se contemplan en la presente invención. Los isómeros geométricos cis y trans de los compuestos de la invención y pueden aislarse como una mezcla de isómeros o como formas isoméricas separadas. Se contemplan todas las formas isoméricas quirales, diastereoméricas, racémicas y geométricas de una estructura, a menos que se indique específicamente la estereoquímica o la forma isomérica. Todos los procesos utilizados para preparar los compuestos de la presente invención y los intermedios realizados en ellos se consideran parte de la presente invención. Todos los tautómeros de los compuestos mostrados o descritos también se consideran parte de la presente invención.
La invención también comprende compuestos de la invención marcados isotópicamente, o sales o solvatos farmacéuticamente aceptables de los mismos, que son idénticos a los recitados en las fórmulas de la solicitud y siguientes, pero por el hecho de que uno o más átomos se sustituyen por un átomo que tiene una masa atómica o número de masa diferente de la masa atómica o número de masa que se encuentra más comúnmente en la naturaleza. Los ejemplos de isótopos que pueden incorporarse a los compuestos de la invención, o a las sales o solvatos farmacéuticamente aceptables de los mismos incluyen isótopos de hidrógeno, carbono, nitrógeno, flúor, como 3H, 11C, 14c 18F
Los isótopos tritiados, es decir, 3H, y de carbono-14, es decir, 14C, pueden utilizarse por su facilidad de preparación y detectabilidad. Además, la sustitución con isótopos más pesados como el deuterio, es decir, el 2H, puede ofrecer ciertas ventajas terapéuticas derivadas de una mayor estabilidad metabólica, por ejemplo, una mayor vida media in vivo o una reducción de las necesidades de dosificación y, por tanto, puede utilizarse en algunas circunstancias. Los compuestos de la invención etiquetados isotópicamente, o las sales farmacéuticas o solvatos de los mismos, pueden prepararse generalmente llevando a cabo los procedimientos descritos en los Esquemas y/o en los Ejemplos, sustituyendo un reactivo etiquetado isotópicamente disponible por un reactivo no etiquetado isotópicamente. Sin embargo, un experto en la materia reconocerá que no todos los isótopos pueden incluirse mediante la sustitución del reactivo no marcado isotópicamente. En una realización, los compuestos de la invención, o las sales o solvatos farmacéuticamente aceptables de los mismos no están etiquetados isotópicamente. En una realización, los compuestos deuterados de la invención son útiles para ensayos bioanalíticos. En otra realización, los compuestos de la invención, o las sales o solvatos farmacéuticamente aceptables de los mismos están radiomarcados.
Composiciones farmacéuticas
Una "composición farmacéutica" es una formulación que contiene uno o más compuestos de la invención en una forma adecuada para su administración a un sujeto. En una realización, la composición farmacéutica es a granel o en forma de unidad de dosificación. Puede ser ventajoso formular composiciones en forma de unidad de dosificación para facilitar
la administración y la uniformidad de la dosis. La forma de unidad de dosificación, tal como se utiliza aquí, se refiere a unidades físicamente discretas adecuadas como dosis unitarias para el sujeto a tratar; cada unidad contiene una cantidad predeterminada de reactivo activo calculada para producir el efecto terapéutico deseado en asociación con el portador farmacéutico requerido. Las especificaciones para las formas de unidades de dosificación están dictadas por y dependen directamente de las características únicas del reactivo activo y el efecto terapéutico particular que debe lograrse, y las limitaciones inherentes al arte de componer tal agente activo para el tratamiento de individuos.
Las posibles formulaciones incluyen las adecuadas para la administración oral, sublingual, bucal, parenteral (por ejemplo, subcutánea, intramuscular o intravenosa), rectal, tópica, incluyendo la transdérmica, intranasal y por inhalación. Los medios de administración más adecuados para un paciente en particular dependerán de la naturaleza y la gravedad de la enfermedad que se esté tratando o de la naturaleza de la terapia que se esté utilizando y de la naturaleza del compuesto activo, pero cuando sea posible, se puede utilizar la administración oral para la prevención y el tratamiento de enfermedades y afecciones mediadas por FXR. Las formulaciones adecuadas para la administración oral pueden suministrarse como unidades discretas, como comprimidos, cápsulas, cachets, pastillas, cada una de las cuales contiene una cantidad predeterminada del compuesto activo; como polvos o gránulos; como soluciones o suspensiones en líquidos acuosos o no acuosos; o como emulsiones de aceite en agua o agua en aceite. Las formulaciones adecuadas para la administración sublingual o bucal incluyen pastillas que comprenden el compuesto activo y, por lo general, una base aromatizada, como azúcar y acacia o tragacanto, y pastillas que comprenden el compuesto activo en una base inerte, como gelatina y glicerina o sacarosa acacia.
Las formulaciones adecuadas para la administración parenteral suelen comprender soluciones acuosas estériles que contienen una concentración predeterminada del compuesto activo; la solución puede ser isotónica con la sangre del receptor previsto. Otras formulaciones adecuadas para la administración parenteral incluyen formulaciones que contienen codisolventes fisiológicamente adecuados y/o agentes complejantes como tensioactivos y ciclodextrinas. Las emulsiones de aceite en agua también son formulaciones adecuadas para la administración parenteral. Aunque dichas soluciones pueden administrarse por vía intravenosa, también pueden administrarse por inyección subcutánea o intramuscular.
Las formulaciones adecuadas para la administración rectal pueden proporcionarse como supositorios de dosis unitarias que comprenden el ingrediente activo en uno o más portadores sólidos que forman la base del supositorio, por ejemplo, la manteca de cacao.
Las formulaciones adecuadas para la aplicación tópica o intranasal incluyen pomadas, cremas, lociones, pastas, geles, aerosoles y aceites. Los portadores adecuados para tales formulaciones incluyen vaselina, lanolina, polietilenglicoles, alcoholes y combinaciones de los mismos.
Las formulaciones de la invención pueden ser preparadas por cualquier método adecuado, típicamente mezclando uniforme e íntimamente el compuesto activo con líquidos o portadores sólidos finamente divididos o ambos, en las proporciones requeridas y luego, si es necesario, moldeando la mezcla resultante en la forma deseada.
Por ejemplo, un comprimido puede prepararse comprimiendo una mezcla íntima que comprenda un polvo o gránulos del ingrediente activo y uno o más ingredientes opcionales, como un aglutinante, un lubricante, un diluyente inerte o un agente dispersante tensioactivo, o moldeando una mezcla íntima de ingrediente activo en polvo y diluyente líquido inerte. Las formulaciones adecuadas para la administración por inhalación incluyen polvos o nieblas de partículas finas que pueden generarse mediante diversos tipos de aerosoles presurizados de dosis medida, nebulizadores o insufladores.
Para la administración pulmonar a través de la boca, el tamaño de las partículas del polvo o de las gotitas se encuentra típicamente en el rango de 0,5-10 mm, o puede ser de aproximadamente 1-5 mm, para asegurar la entrega en el árbol bronquial. Para la administración nasal, puede utilizarse un tamaño de partícula del orden de 10-500 mm para asegurar la retención en la cavidad nasal.
Los inhaladores de dosis medida son dispensadores de aerosol presurizados, que suelen contener una formulación de suspensión o solución del ingrediente activo en un propulsor licuado. Durante el uso, estos dispositivos descargan la formulación a través de una válvula adaptada para suministrar un volumen medido, típicamente de 10 a 150 mm, para producir un aerosol de partículas finas que contiene el ingrediente activo. Los propulsores adecuados incluyen ciertos compuestos de clorofluorocarbono, por ejemplo, diclorodifluorometano, triclorofluorometano, diclorotetrafluoroetano y mezclas de los mismos. La formulación puede contener además uno o más co-solventes, por ejemplo, tensioactivos de etanol, como el ácido oleico o el trioleato de sorbitán, antioxidantes y agentes aromatizantes adecuados.
Los nebulizadores son dispositivos disponibles en el mercado que transforman las soluciones o suspensiones del principio activo en una niebla de aerosol terapéutico, ya sea mediante la aceleración de un gas comprimido, típicamente aire u oxígeno, a través de un orificio venturi estrecho, o mediante agitación ultrasónica. Las formulaciones adecuadas para su uso en nebulizadores consisten en el ingrediente activo en un portador líquido y comprenden hasta el 40% p/p de la formulación, preferiblemente menos del 20% p/p. El portador suele ser agua o una solución alcohólica acuosa diluida, preferentemente isotónica con los fluidos corporales mediante la adición de, por ejemplo, cloruro de sodio. Los
aditivos opcionales incluyen conservantes si la formulación no se prepara de forma estéril, por ejemplo, hidroxibenzoato de metilo, antioxidantes, agentes aromatizantes, aceites volátiles, agentes amortiguadores y tensioactivos.
Las formulaciones adecuadas para la administración por insuflación incluyen polvos finamente triturados que pueden administrarse por medio de un insuflador o introducirse en la cavidad nasal a la manera de un rapé. En el insuflador, el polvo está contenido en cápsulas o cartuchos, normalmente de gelatina o plástico, que se perforan o se abren in situ y el polvo se suministra por medio de aire aspirado a través del dispositivo al inhalar o por medio de una bomba de accionamiento manual. El polvo empleado en el insuflador está compuesto únicamente por el principio activo o por una mezcla de polvo que comprende el principio activo, un diluyente en polvo adecuado, como la lactosa, y un tensioactivo opcional. El principio activo suele comprender entre el 0,1 y el 100 % en peso de la formulación.
En otra realización, la presente invención proporciona una composición farmacéutica que comprende, como ingrediente activo, un compuesto de la invención junto, y/o en mezcla, con al menos un portador o diluyente farmacéutico. Estas composiciones farmacéuticas pueden utilizarse en la prevención o el tratamiento de las enfermedades o condiciones anteriores.
El portador es farmacéuticamente aceptable y debe ser compatible con los demás ingredientes de la composición, es decir, no debe tener un efecto perjudicial sobre ellos. El portador puede ser un sólido o un líquido y se formula preferentemente como una formulación de dosis unitaria, por ejemplo, un comprimido que puede contener de 0,05 a 95% en peso del principio activo. Si se desea, también pueden incorporarse otros ingredientes fisiológicamente activos en las composiciones farmacéuticas de la invención.
Además de los ingredientes específicamente mencionados, las formulaciones de la presente invención pueden incluir otros agentes conocidos por los expertos en el arte de la farmacia, teniendo en cuenta el tipo de formulación en cuestión. Por ejemplo, las formulaciones adecuadas para la administración oral pueden incluir agentes aromatizantes y las formulaciones adecuadas para la administración intranasal pueden incluir perfumes.
En una de las realizaciones, la presente divulgación proporciona una composición farmacéutica que comprende los compuestos de fórmula Ie y un portador o excipiente farmacéuticamente aceptable.
Indicaciones medicinales
Los compuestos de la invención son útiles para la terapia en sujetos como los mamíferos, incluidos los humanos. En particular, los compuestos de la invención son útiles en un método de tratamiento o prevención de una enfermedad o condición en un sujeto que comprende administrar al sujeto que lo necesita una cantidad efectiva de un compuesto de la invención o una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la enfermedad o afección está mediada por el FXR (por ejemplo, el FXR desempeña un papel en el inicio o el progreso de la enfermedad o afección). En una realización, la enfermedad o afección está mediada por la disminución de la actividad del FXR. En una realización, la enfermedad o afección se selecciona entre enfermedades cardiovasculares, enfermedades hepáticas crónicas, trastornos lipídicos, enfermedades gastrointestinales, enfermedades renales, enfermedades metabólicas, cáncer y enfermedades neurológicas.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de enfermedades cardiovasculares en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de enfermedades cardiovasculares. En una realización, la enfermedad cardiovascular se selecciona entre aterosclerosis, arteriosclerosis, dislipidemia, hipercolesteremia, hiperlipidemia, hiperlipoproteinemia e hipertrigliceridemia.
El término "hiperlipidemia" se refiere a la presencia de un nivel anormalmente elevado de lípidos en la sangre. La hiperlipidemia puede aparecer en al menos tres formas: (1) hipercolesterolemia, es decir, un nivel elevado de colesterol; (2) hipertrigliceridemia, es decir, un nivel elevado de triglicéridos; y (3) hiperlipidemia combinada, es decir, una combinación de hipercolesterolemia e hipertrigliceridemia.
El término "dislipidemia" se refiere a niveles anormales de lipoproteínas en el plasma sanguíneo, incluyendo niveles deprimidos y/o elevados de lipoproteínas (por ejemplo, niveles elevados de LDL, VLDL y niveles deprimidos de HDL). En una realización, la invención se refiere a los compuestos de la invención para su uso en un método seleccionado de reducción de los niveles de colesterol o de modulación del metabolismo del colesterol, del catabolismo, de la absorción del colesterol de la dieta y del transporte inverso del colesterol en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal farmacéuticamente aceptable o de un solvato del mismo.
En otra realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de una enfermedad que afecta a los niveles de colesterol, triglicéridos o ácidos biliares en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una
sal o solvato farmacéuticamente aceptable del mismo.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método para reducir los triglicéridos en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal farmacéuticamente aceptable o un solvato del mismo.
En una realización, la invención se relaciona con los compuestos de la invención para su uso en un método de tratamiento o prevención de un estado de enfermedad asociado con un nivel elevado de colesterol en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de un estado de enfermedad asociado con un nivel elevado de colesterol en un sujeto. En una realización, la invención relaciona a los compuestos de la invención para el uso en un método de prevenir un estado de enfermedad asociado con un nivel de colesterol elevado en un sujeto. En una realización, el estado de la enfermedad se selecciona entre la enfermedad de las arterias coronarias, la angina de pecho, la enfermedad de las arterias carótidas, los accidentes cerebrovasculares, la arteriosclerosis cerebral y el xantoma.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de un trastorno lipídico en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de un trastorno lipídico. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de un trastorno lipídico.
Los trastornos lipídicos son el término que designa las anomalías del colesterol y los triglicéridos. Las anomalías lipídicas se asocian a un mayor riesgo de enfermedad vascular, y especialmente de ataques cardíacos y accidentes cerebrovasculares. Las anomalías en los trastornos lipídicos son una combinación de la predisposición genética, así como de la naturaleza de la ingesta dietética. Muchos trastornos lipídicos están asociados al sobrepeso. Los trastornos lipídicos también pueden estar asociados a otras enfermedades, como la diabetes, el síndrome metabólico (a veces denominado síndrome de resistencia a la insulina), la hipoactividad del tiroides o el resultado de ciertos medicamentos (como los utilizados para los regímenes antirrechazo en personas que han recibido trasplantes).
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de uno o más síntomas de una enfermedad que afecta al metabolismo de los lípidos (es decir, lipodistrofia) en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de uno o más síntomas de una enfermedad que afecta al metabolismo de los lípidos. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de uno o más síntomas de una enfermedad que afecta al metabolismo de los lípidos.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de disminución de la acumulación de lípidos en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de la enfermedad hepática crónica en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de la enfermedad hepática crónica. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de la enfermedad hepática crónica. En una realización, la enfermedad hepática crónica se selecciona entre la cirrosis biliar primaria (CBP), la xantomatosis cerebrotendinosa (CTX), la colangitis esclerosante primaria (CEP), la colestasis inducida por fármacos, la colestasis intrahepática del embarazo, la colestasis asociada a la nutrición parenteral (CPN), el sobrecrecimiento bacteriano o la colestasis asociada a la sepsis, la hepatitis autoinmune, la hepatitis viral crónica, la enfermedad hepática alcohólica, la enfermedad del hígado graso no alcohólico (NAFLD), la esteatohepatitis no alcohólica (NASH), la enfermedad de injerto contra huésped asociada al trasplante de hígado, la regeneración del hígado en el trasplante de donante vivo, la fibrosis hepática congénita, la coledocolitiasis, la enfermedad hepática granulomatosa, las neoplasias intra o extrahepáticas, el síndrome de Sjogren, la sarcoidosis, la enfermedad de Wilson, la enfermedad de Gaucher, la hemocromatosis y el déficit de alfa-1-antitripsina.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de uno o más síntomas de colestasis, incluyendo las complicaciones de la colestasis en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de uno o más síntomas de colestasis. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de uno o más síntomas
de colestasis.
La colestasis es causada típicamente por factores dentro del hígado (intrahepáticos) o fuera del hígado (extrahepáticos) y conduce a la acumulación de sales biliares, el pigmento biliar bilirrubina y lípidos en el torrente sanguíneo en lugar de ser eliminados normalmente. La colestasis intrahepática se caracteriza por la obstrucción generalizada de los pequeños conductos o por trastornos, como la hepatitis, que alteran la capacidad del organismo para eliminar la bilis. La colestasis intrahepática también puede estar causada por una enfermedad hepática alcohólica, una cirrosis biliar primaria, un cáncer que se ha extendido (metastatizado) desde otra parte del cuerpo, una colangitis esclerosante primaria, cálculos biliares, un cólico biliar y una colecistitis aguda. También puede producirse como complicación de una intervención quirúrgica, una lesión grave, la fibrosis quística, una infección o la alimentación intravenosa, o ser inducida por fármacos. La colestasis también puede producirse como complicación del embarazo y suele desarrollarse durante el segundo y tercer trimestre. La colestasis extrahepática está causada con mayor frecuencia por la coledocolitiasis (cálculos del conducto biliar), las estenosis biliares benignas (estrechamiento no canceroso del conducto común), el colangiocarcinoma (carcinoma ductal) y el carcinoma pancreático. La colestasis extrahepática puede producirse como efecto secundario de muchos medicamentos.
Un compuesto de la invención puede usarse para tratar o prevenir uno o más síntomas de colestasis intrahepática o extrahepática, incluyendo, sin limitación, atresia biliar, colestasis obstétrica, colestasis neonatal, colestasis inducida por fármacos, colestasis derivada de la infección por hepatitis C, enfermedad hepática colestática crónica como cirrosis biliar primaria (CBP) y colangitis esclerosante primaria (CEP).
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método para mejorar la regeneración del hígado en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o una sal o un solvato farmacéuticamente aceptable del mismo. En una realización, el método mejora la regeneración del hígado para el trasplante de hígado.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de la fibrosis en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de la fibrosis. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de la fibrosis.
En consecuencia, tal como se utiliza en la presente invención, el término fibrosis se refiere a todos los trastornos fibróticos reconocidos, incluida la fibrosis debida a condiciones o enfermedades patológicas, la fibrosis debida a un traumatismo físico ("fibrosis traumática"), la fibrosis debida a daños por radiación y la fibrosis debida a la exposición a quimioterapéuticos. Tal y como se utiliza en la presente invención, el término "fibrosis de órgano" incluye, entre otros, la fibrosis hepática, la fibrosis renal, la fibrosis pulmonar y la fibrosis intestinal. El término "fibrosis traumática" incluye, pero no se limita a, la fibrosis secundaria a la cirugía (cicatrización quirúrgica), el trauma físico accidental, las quemaduras y la cicatrización hipertrófica.
Tal como se utiliza en la presente invención, la "fibrosis hepática" incluye la fibrosis hepática debida a cualquier causa, incluyendo pero no limitándose a la fibrosis hepática inducida por virus, como la debida al virus de la hepatitis B o C; la exposición al alcohol (enfermedad hepática alcohólica), ciertos compuestos farmacéuticos, incluyendo pero no limitándose al metotrexato, algunos agentes quimioterapéuticos y la ingestión crónica de arsenicales o vitamina A en megadosis, el estrés oxidativo, la radioterapia contra el cáncer o ciertos productos químicos industriales, incluyendo pero no limitándose al tetracloruro de carbono y la dimetilnitrosamina; y enfermedades como la cirrosis biliar primaria, la colangitis esclerosante primaria, el hígado graso, la obesidad, la esteatohepatitis no alcohólica, la fibrosis quística, la hemocromatosis, la hepatitis autoinmune y la esteatohepatitis. El tratamiento actual de la fibrosis hepática se dirige principalmente a la eliminación del agente causal, por ejemplo, la eliminación del exceso de hierro (en el caso de la hemocromatosis), la disminución de la carga vírica (en el caso de la hepatitis vírica crónica) o la eliminación o disminución de la exposición a las toxinas (en el caso de la enfermedad hepática alcohólica). También se conoce el uso de fármacos antiinflamatorios, como los corticosteroides y la colchicina, para tratar la inflamación que puede conducir a la fibrosis hepática. Como se sabe en la técnica, la fibrosis hepática puede clasificarse clínicamente en cinco estadios de gravedad (S0, S1, S2, S3 y S4), normalmente basados en el examen histológico de una muestra de biopsia. S0 indica ausencia de fibrosis, mientras que S4 indica cirrosis. Aunque existen varios criterios para clasificar la gravedad de la fibrosis hepática, en general los estadios iniciales de la fibrosis se identifican por áreas discretas y localizadas de cicatrización en un portal (zona) del hígado, mientras que los estadios posteriores de la fibrosis se identifican por la fibrosis en puente (cicatrización que atraviesa zonas del hígado).
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de la fibrosis de órganos en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la fibrosis es una fibrosis hepática.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o
prevención de enfermedades gastrointestinales en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de enfermedades gastrointestinales. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de enfermedades gastrointestinales. En una realización, la enfermedad gastrointestinal se selecciona entre la enfermedad inflamatoria del intestino (EII), el síndrome del intestino irritable (SII), el sobrecrecimiento bacteriano, la malabsorción, la colitis post-radiación y la colitis microscópica. En una realización, la enfermedad inflamatoria intestinal se selecciona entre la enfermedad de Crohn y la colitis ulcerosa.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de la enfermedad renal en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de la enfermedad renal. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de la enfermedad renal. En una realización, la enfermedad renal se selecciona entre nefropatía diabética, glomeruloesclerosis segmentaria focal (FSGS), nefroesclerosis hipertensiva, glomerulonefritis crónica, glomerulopatía crónica por trasplante, nefritis intersticial crónica y enfermedad renal poliquística.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de enfermedades metabólicas en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de la enfermedad renal. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de la enfermedad renal. En una realización, la enfermedad metabólica se selecciona entre resistencia a la insulina, hiperglucemia, diabetes mellitus, diabesidad y obesidad. En una realización, la diabetes mellitus es de tipo I. En una realización, la diabetes mellitus es de tipo II.
La diabetes mellitus, comúnmente llamada diabetes, se refiere a una enfermedad o condición que se caracteriza generalmente por defectos metabólicos en la producción y utilización de la glucosa que resultan en la incapacidad de mantener niveles adecuados de azúcar en la sangre en el cuerpo.
En el caso de la diabetes de tipo II, la enfermedad se caracteriza por la resistencia a la insulina, en la que la insulina pierde su capacidad de ejercer sus efectos biológicos en una amplia gama de concentraciones. Esta resistencia a la capacidad de respuesta de la insulina da lugar a una activación insuficiente por parte de la insulina de la captación, oxidación y almacenamiento de la glucosa en el músculo y a una represión inadecuada por parte de la insulina de la lipólisis en el tejido adiposo y de la producción y secreción de glucosa en el hígado. La condición resultante es la elevación de la glucosa en sangre, que se denomina "hiperglucemia". La hiperglucemia no controlada se asocia a una mortalidad mayor y prematura debido a un mayor riesgo de enfermedades microvasculares y macrovasculares, como la retinopatía (el deterioro o la pérdida de la visión debido al daño de los vasos sanguíneos en los ojos); la neuropatía (daños en los nervios y problemas en los pies debido al daño de los vasos sanguíneos en el sistema nervioso); y la nefropatía (enfermedad renal debido al daño de los vasos sanguíneos en los riñones), la hipertensión, la enfermedad cerebrovascular y la enfermedad coronaria. Por lo tanto, el control de la homeostasis de la glucosa es un enfoque de importancia crítica para el tratamiento de la diabetes.
Se ha planteado la hipótesis de que la resistencia a la insulina unifica la agrupación de la hipertensión, la intolerancia a la glucosa, la hiperinsulinemia, el aumento de los niveles de triglicéridos y la disminución del colestero1HDL, y la obesidad central y general. La asociación de la resistencia a la insulina con la intolerancia a la glucosa, el aumento de los triglicéridos plasmáticos y la disminución de las concentraciones de colesterol de las lipoproteínas de alta densidad, la hipertensión, la hiperuricemia, las partículas de lipoproteínas de baja densidad más pequeñas y los niveles circulantes más elevados del inhibidor del activador del plasminógeno-1, se ha denominado "síndrome X". En consecuencia, se proporcionan métodos para tratar o prevenir cualquier trastorno relacionado con la resistencia a la insulina, incluyendo el grupo de estados de enfermedad, condiciones o trastornos que conforman el "Síndrome X". En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención del síndrome metabólico en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento del síndrome metabólico. En otra realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención del síndrome metabólico.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención del cáncer en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento del cáncer. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención del cáncer. En una realización, el cáncer se selecciona entre el carcinoma hepatocelular, el cáncer colorrectal, el cáncer gástrico, el cáncer renal, el cáncer de próstata, el cáncer suprarrenal, el cáncer de páncreas, el cáncer de mama, el
cáncer de vejiga, el cáncer de glándulas salivales, el cáncer de ovario, el cáncer de cuerpo uterino y el cáncer de pulmón. En una realización, el cáncer es un carcinoma hepatocelular. En una realización, el cáncer es un cáncer colorrectal. En una realización, el cáncer es un cáncer gástrico. En una realización, el cáncer es un cáncer renal. En una realización, el cáncer es de próstata. En una realización, el cáncer es un cáncer suprarrenal. En una realización, el cáncer es de páncreas. En una realización, el cáncer es de mama. En una realización, el cáncer es de vejiga. En una realización, el cáncer es de glándulas salivales. En una realización, el cáncer es de ovario. En una realización, el cáncer es de cuerpo uterino. En una realización, el cáncer es de pulmón.
En otra realización, al menos uno de los agentes seleccionados entre Sorafenib, Sunitinib, Erlotinib o Imatinib se coadministra con el compuesto de la invención para tratar el cáncer. En una realización, al menos uno de los agentes seleccionados entre abarelix, aldeleukina, alopurinol, altretamina, amifostina, anastozol, bevacizumab, capecitabina, carboplatino, cisplatino, docetaxel, doxorrubicina, erlotinib, exemestano, 5-fluorouracilo, fulvestrant, gemcitabina, acetato de goserelina, irinotecán, ditosilato de lapatinib, letozol, leucovorina, levamisol, oxaliplatino, paclitaxel, panitumumab, pemetrexed disódico, profimer sódico, tamoxifeno, topotecán y trastuzumab se coadministra con el compuesto de la invención para tratar el cáncer.
El tratamiento adecuado para los cánceres depende del tipo de célula de la que deriva el tumor, el estadio y la gravedad de la malignidad, y la anomalía genética que contribuye al tumor.
Los sistemas de estadificación del cáncer describen el grado de progresión del cáncer. En general, los sistemas de estadificación describen hasta dónde se ha extendido el tumor y colocan a los pacientes con un pronóstico y un tratamiento similares en el mismo grupo de estadificación. En general, hay peores pronósticos para los tumores que se han vuelto invasivos o han hecho metástasis.
En un tipo de sistema de estadificación, los casos se agrupan en cuatro estadios, denotados por los números romanos I a IV. En el estadio I, los cánceres suelen estar localizados y suelen ser curables. Los cánceres en estadio II y IIIA suelen estar más avanzados y pueden haber invadido los tejidos circundantes y haberse extendido a los ganglios linfáticos. Los cánceres en estadio IV incluyen los cánceres metastásicos que se han extendido a lugares fuera de los ganglios linfáticos.
Otro sistema de estadificación es la estadificación TNM que significa las categorías: Tumor, Ganglios y Metástasis. En este sistema, los tumores malignos se describen según la gravedad de las categorías individuales. Por ejemplo, T clasifica la extensión de un tumor primario de 0 a 4, donde 0 representa una neoplasia que no tiene actividad invasiva y 4 representa una neoplasia que ha invadido otros órganos por extensión desde el sitio original. N clasifica la extensión de la afectación de los ganglios linfáticos: 0 representa una neoplasia sin afectación de los ganglios linfáticos y 4 representa una neoplasia con una amplia afectación de los ganglios linfáticos. La M clasifica la extensión de la metástasis de 0 a 1, representando el 0 una neoplasia sin metástasis y el 1 una neoplasia con metástasis.
Estos sistemas de estadificación o variaciones de estos sistemas de estadificación u otros sistemas de estadificación adecuados pueden utilizarse para describir un tumor como el carcinoma hepatocelular. Para el tratamiento del cáncer hepatocelular sólo se dispone de unas pocas opciones en función del estadio y las características del cáncer. Los tratamientos incluyen la cirugía, el tratamiento con Sorafenib y las terapias dirigidas. En general, la cirugía es la primera línea de tratamiento para el cáncer hepatocelular localizado en estadio temprano. Pueden utilizarse tratamientos sistémicos adicionales para tratar los tumores invasivos y metastásicos.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de cálculos biliares en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de cálculos biliares. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de cálculos biliares.
Un cálculo biliar es una concreción cristalina formada dentro de la vesícula biliar por acumulación de componentes biliares. Estos cálculos se forman en la vesícula biliar, pero pueden pasar distalmente a otras partes del tracto biliar, como el conducto cístico, el conducto biliar común, el conducto pancreático o la ampolla de Vater. En raras ocasiones, en casos de inflamación grave, los cálculos biliares pueden erosionarse a través de la vesícula biliar hacia el intestino adherido, lo que puede causar una obstrucción denominada íleo biliar. La presencia de cálculos biliares en la vesícula puede dar lugar a una colecistitis aguda, una afección inflamatoria caracterizada por la retención de bilis en la vesícula y, a menudo, una infección secundaria por microorganismos intestinales, predominantemente Escherichia coli y especies de Bacteroides.
La presencia de cálculos biliares en otras partes del tracto biliar puede causar la obstrucción de los conductos biliares, lo que puede conducir a condiciones graves como la colangitis ascendente o la pancreatitis.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de la enfermedad de cálculos biliares de colesterol en un sujeto, que comprende la administración al sujeto
que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de la enfermedad de cálculos biliares de colesterol. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de la enfermedad de cálculos biliares de colesterol.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento o prevención de enfermedades neurológicas en un sujeto, que comprende la administración al sujeto que lo necesita de una cantidad eficaz de un compuesto de la invención o de una sal o solvato farmacéuticamente aceptable del mismo. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de tratamiento de enfermedades neurológicas. En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de prevención de enfermedades neurológicas. En una realización, la enfermedad neurológica es un accidente cerebrovascular.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método como el descrito en la presente invención y, además, en el que el compuesto se administra por una ruta seleccionada entre oral, parenteral, intramuscular, intranasal, sublingual, intratraqueal, inhalación, ocular, vaginal, rectal e intracerebroventricular. En una realización, la vía es oral.
En una realización, el compuesto utilizado en uno o más de los métodos descritos en la presente invención es un agonista del FXR. En una realización, el compuesto es un agonista FXR selectivo. En otra realización, el compuesto no activa TGR5. En una realización, el compuesto no activa otros receptores nucleares implicados en las vías metabólicas (p. ej., según las mediciones de un ensayo AlphaScreen). En una realización, dichos otros receptores nucleares implicados en las vías metabólicas se seleccionan entre LXR, PXR, CAR, PPARL PPAR, RAR, Ra R, VDR, TR, PR, RXR, GR y ER. En una realización, el compuesto induce la apoptosis.
En una realización, la invención se refiere a los compuestos de la invención para su uso en un método de regulación del nivel de expresión de uno o más genes implicados en la homeostasis del ácido biliar.
En una realización, la invención se refiere a un método no terapéutico de regulación descendente del nivel de expresión de uno o más genes seleccionados de CYP7al y SREBP-IC en una célula administrando a la célula un compuesto de la invención. En una realización, la invención se refiere a un método no terapéutico de regulación ascendente del nivel de expresión de uno o más genes seleccionados de OST, OST, BSEP, SHP, UGT2B4, MRP2, FGF-19, PPAR, PLTP, APOCII, y PEPCK en una célula administrando a la célula un compuesto de la invención.
La invención también se refiere a la fabricación de un medicamento para tratar o prevenir una enfermedad o afección (por ejemplo, una enfermedad o afección mediada por FXR), en el que el medicamento comprende un compuesto de la invención o una sal farmacéuticamente aceptable, un solvato o un conjugado de aminoácidos del mismo. En una realización, la invención se refiere a la fabricación de un medicamento para el tratamiento o la prevención de cualquiera de las enfermedades o afecciones descritas en la presente invención, en el que el medicamento comprende un compuesto de la invención o una sal, un solvato o un conjugado de aminoácidos farmacéuticamente aceptable del mismo.
La invención también se refiere a una composición para su uso en un método para tratar o prevenir una enfermedad o afección (por ejemplo, una enfermedad o afección mediada por el FXR), en la que la composición comprende un compuesto de la invención o una sal farmacéuticamente aceptable, un solvato o un conjugado de aminoácidos del mismo. En una realización, la invención se refiere a una composición para su uso en un método para tratar o prevenir cualquiera de las enfermedades o afecciones descritas en la presente invención, en la que la composición comprende un compuesto de la invención o una sal, un solvato o un conjugado de aminoácidos farmacéuticamente aceptable del mismo.
Los métodos para los que se indican los compuestos de la invención comprenden el paso de administrar una cantidad eficaz de un compuesto de la invención. Tal como se utiliza en la presente invención, el término "cantidad eficaz" se refiere a una cantidad de un compuesto de la invención que es suficiente para lograr el efecto indicado.
En consecuencia, una cantidad eficaz de un compuesto de la invención utilizado en un método para la prevención o el tratamiento de enfermedades o afecciones mediadas por FXR será una cantidad suficiente para prevenir o tratar la enfermedad o afección mediada por FXR.
Del mismo modo, una cantidad eficaz de un compuesto de la invención para su uso en un método para la prevención o el tratamiento de una enfermedad hepática colestásica o el aumento del flujo biliar será una cantidad suficiente para aumentar el flujo biliar al intestino.
La cantidad del compuesto de la invención que se requiere para lograr el efecto biológico deseado dependerá de una serie de factores tales como el uso para el que se destina, los medios de administración y el receptor, y será en última instancia a discreción del médico o veterinario asistente. En general, una dosis diaria típica para el tratamiento de una
enfermedad y afección mediada por FXR, por ejemplo, puede esperarse que se encuentre en el rango de aproximadamente 0,01 mg/kg a aproximadamente 100 mg/kg. Esta dosis puede administrarse como una dosis unitaria única o como varias dosis unitarias separadas o como una infusión continua. Dosis similares serían aplicables para el tratamiento de otras enfermedades, condiciones y terapias, incluyendo la prevención y el tratamiento de enfermedades hepáticas colestásicas.
En una de las realizaciones, la presente divulgación proporciona los compuestos de la invención para su uso en un método de tratamiento o prevención de una enfermedad o afección en un sujeto que lo necesita, que comprende la administración de una cantidad eficaz del compuesto de fórmula Ie o una sal o solvato farmacéuticamente aceptable del mismo, y en el que la enfermedad o afección está mediada por el FXR.
En una de las realizaciones, la presente divulgación proporciona los compuestos de la invención para su uso en un método de tratamiento o prevención de una enfermedad o afección en un sujeto que lo necesita, que comprende la administración de una cantidad eficaz del compuesto de fórmula Ie, en el que la enfermedad se selecciona entre enfermedad cardiovascular, enfermedad hepática crónica, trastorno lipídico, enfermedad gastrointestinal, enfermedad renal, enfermedad metabólica, cáncer y enfermedad neurológica.
En una de las realizaciones, la presente divulgación proporciona los compuestos de la invención para su uso en un método de tratamiento o prevención de una enfermedad o afección en un sujeto que la necesita, que comprende la administración de una cantidad eficaz del compuesto de fórmula Ie, donde la enfermedad es una enfermedad cardiovascular seleccionada entre aterosclerosis, arteriosclerosis, dislipidemia, hipercolesteremia, hiperlipidemia, hiperlipoproteinemia e hipertrigliceridemia.
En una de las realizaciones, la presente divulgación proporciona los compuestos de la invención para su uso en un método de tratamiento o prevención de una enfermedad o afección en un sujeto que la necesita, comprendiendo la administración de una cantidad eficaz del compuesto de fórmula Ie, donde la enfermedad es una enfermedad hepática crónica seleccionada entre cirrosis biliar primaria (CBP), xantomatosis cerebrotendinosa (CTX), colangitis esclerosante primaria (CEP), colestasis inducida por fármacos, colestasis intrahepática del embarazo, colestasis asociada a nutrición parenteral (CPN) sobrecrecimiento bacteriano o colestasis asociada a la sepsis, hepatitis autoinmune, hepatitis vírica crónica, enfermedad hepática alcohólica, enfermedad del hígado graso no alcohólico (HGNA), esteatohepatitis no alcohólica (EHNA), enfermedad de injerto contra huésped asociada al trasplante de hígado, regeneración hepática por trasplante de donante vivo, fibrosis hepática congénita, coledocolitiasis, enfermedad hepática granulomatosa, neoplasia intra o extrahepática, síndrome de Sjogren, sarcoidosis, enfermedad de Wilson, enfermedad de Gaucher, hemocromatosis y deficiencia de alfa-1 antitripsina.
En una de las realizaciones, la presente divulgación proporciona los compuestos de la invención para su uso en un método de tratamiento o prevención de una enfermedad o afección en un sujeto que la necesita, que comprende la administración de una cantidad eficaz del compuesto de fórmula Ie, donde la enfermedad es una enfermedad gastrointestinal seleccionada entre la enfermedad inflamatoria intestinal (EII), el síndrome del intestino irritable (SII), el sobrecrecimiento bacteriano, la mala absorción, la colitis post-radiación y la colitis microscópica.
En una de las realizaciones, la presente divulgación proporciona los compuestos de la invención para su uso en un método de tratamiento o prevención de una enfermedad o afección en un sujeto que lo necesita, que comprende la administración de una cantidad eficaz del compuesto de fórmula Ie, en el que la enfermedad inflamatoria intestinal es la enfermedad de Crohn o la colitis ulcerosa.
En una de las realizaciones, la presente divulgación proporciona los compuestos de la invención para su uso en un método de tratamiento o prevención de una enfermedad o afección en un sujeto que la necesita, que comprende la administración de una cantidad eficaz del compuesto de fórmula Ie, donde la enfermedad es una enfermedad renal seleccionada entre nefropatía diabética, glomeruloesclerosis segmentaria focal (FSGS), nefroesclerosis hipertensiva, glomerulonefritis crónica, glomerulopatía crónica por trasplante, nefritis intersticial crónica y enfermedad renal poliquística.
En una de las realizaciones, la presente divulgación proporciona los compuestos de la invención para su uso en un método de tratamiento o prevención de una enfermedad o afección en un sujeto que la necesita, que comprende la administración de una cantidad eficaz del compuesto de fórmula Ie, donde la enfermedad es una enfermedad metabólica seleccionada entre resistencia a la insulina, hiperglucemia, diabetes mellitus, diabesidad y obesidad.
En una de las realizaciones, la presente divulgación proporciona los compuestos de la invención para su uso en un método de tratamiento o prevención de una enfermedad o afección en un sujeto que la necesita, que comprende la administración de una cantidad eficaz del compuesto de fórmula Ie, donde la enfermedad es un cáncer seleccionado entre carcinoma hepatocelular, cáncer colorrectal, cáncer gástrico, cáncer renal, cáncer de próstata, cáncer suprarrenal, cáncer de páncreas, cáncer de mama, cáncer de vejiga, cáncer de glándulas salivales, cáncer de ovario, cáncer de cuerpo uterino y cáncer de pulmón.
Síntesis de los compuestos de la invención
Los siguientes esquemas y ejemplos son ilustrativos y no deben interpretarse de ninguna manera para limitar el alcance de la invención.
La presente invención proporciona un método de síntesis de compuestos de Fórmula Ie, como se define en la presente invención.
Los procesos sintéticos de la invención pueden tolerar una amplia variedad de grupos funcionales, por lo que pueden utilizarse diversos materiales de partida sustituidos. Los procesos generalmente proporcionan el compuesto final deseado en o cerca del final del proceso global, aunque puede ser deseable en ciertos casos convertir el compuesto en una sal farmacéuticamente aceptable, un solvato o un conjugado de aminoácidos del mismo.
Los compuestos de la invención pueden prepararse de diversas maneras utilizando materiales de partida disponibles en el mercado, compuestos conocidos en la bibliografía, o a partir de productos intermedios fácilmente preparados, empleando métodos y procedimientos sintéticos estándar conocidos por los expertos en la materia, o que serán evidentes para el artesano experto a la luz de las enseñanzas del presente documento. Los métodos y procedimientos sintéticos estándar para la preparación de moléculas orgánicas y transformaciones y manipulaciones de grupos funcionales pueden obtenerse de la literatura científica pertinente o de libros de texto estándar en la materia. Aunque no se limita a una o varias fuentes, los textos clásicos como Smith, M. B., March, J., March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5a edición, John Wiley & Sons: Nueva York, 2001; y Greene, T.W., Wuts, P.G. M., Protective Groups in Organic Synthesis, 3a edición, John Wiley & Sons: Nueva York, 1999, son útiles y reconocidos libros de texto de referencia de síntesis orgánica conocidos por los expertos. Las siguientes descripciones de los métodos sintéticos están diseñadas para ilustrar, pero no para limitar, los procedimientos generales para la preparación de los compuestos de la presente invención.
Todas las abreviaturas utilizadas en esta solicitud se encuentran en "Protective Groups in Organic Synthesis" de John Wiley & Sons, Inc. o en el MERCK INDEX de MERCK & Co., Inc. u otros libros de química o catálogos de productos químicos de proveedores de productos químicos como Aldrich, o según el uso conocido en la técnica.
El proceso sintético para obtener los compuestos de la invención puede utilizarse de acuerdo con los procedimientos expuestos a continuación en los esquemas 1-6.
Farmacología de los compuestos de la invención
En general, el potencial de un compuesto de la invención como candidato a fármaco puede probarse utilizando diversos ensayos conocidos en la técnica. Por ejemplo, para la validación in vitro de FXR, su actividad y selectividad pueden evaluarse utilizando AlphaScreen (ensayo bioquímico); la expresión génica puede evaluarse utilizando RT-PCR (gen diana de FXR); y la citotoxicidad (por ejemplo, HepG2) puede evaluarse utilizando el contenido de ATP, la liberación de LDH y la activación de Caspasa-3. Para la validación in vitro de TGR5, su actividad y selectividad pueden evaluarse mediante HTR-FRET (ensayo basado en células); la expresión génica puede evaluarse mediante rT-PCR (gen diana de TGR5 (es decir, cFOS)); y la citotoxicidad (por ejemplo, HepG2) puede evaluarse mediante el contenido de ATP, la liberación de LDH y la activación de la Caspasa-3. Los siguientes compuestos pueden utilizarse como controles en los ejemplos siguientes.
Como se usa aquí, el compuesto A es

que también se conoce como ácido obetichólico, INT-747, 6-ECDCA, ácido 6-alfa-etil quenodesoxicólico o ácido 6 a -etil-3a,7a-dihidroxi-5p-colan-24-oico.
Tal como se utiliza en la presente invención, el compuesto B es
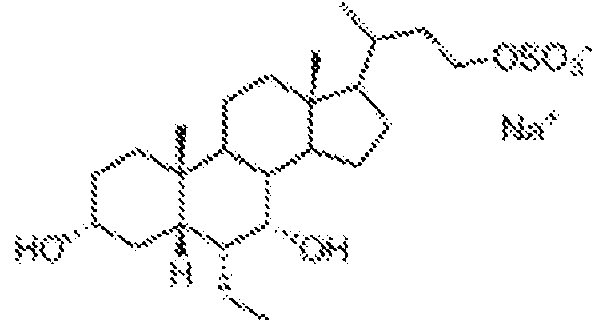
que también se conoce como INT-767 o sal sódica de 6a-etil-3a,7a,23-trihidroxi-24-nor-5p-colan-23-sulfato. Tal como se utiliza en la presente invención, el compuesto C es

que también se conoce como INT-777 o ácido 6a-etil-23(S)-metil-3a,7a,12a trihidroxi-5p-colan-24-oico. Tal como se utiliza en la presente invención, el compuesto D es

que también se conoce como ácido 6a-etil-23(R)-metil quenodesoxicólico, y S-EMCDCA.
Tal como se utiliza en la presente invención, el compuesto E es

Tal como se utiliza en la presente invención, el ácido cólico es

Tal como se utiliza en la presente invención, el ácido quenodesoxicólico es

que también se conoce como CDCA.
Tal como se utiliza en la presente invención, el ácido ursodesoxicólico es

que también se conoce como UDCA.
Tal como se utiliza en la presente invención, el ácido tauroconodesoxicólico es

que también se conoce como TCDCA.
Tal como se utiliza en la presente invención, el ácido tauroursodesoxicólico es

que también se conoce como TUDCA.
Tal como se utiliza en la presente invención, el ácido litochólico es

que también se conoce como LCA.
De los siguientes ejemplos, los ejemplos 2-7 se refieren a compuestos de la presente invención y el compuesto del ejemplo 1 es a efectos de comparación. Los ejemplos 8-14 son ejemplos de la presente invención en la medida en que se refieren a ensayos realizados con los compuestos de los ejemplos 2-7.
EJEMPLOS
Ejemplo 1. Síntesis del ácido 3a,7a,11p-trihidroxi-6a-etil-24-nor-5p-colan-23-oico (Compuesto 1)

El compuesto 1 se preparó según los procedimientos descritos en el esquema 1 y a partir de ácido 6-etil-cólico (6-ECA, compuesto A1) como material de partida. El compuesto A1 se preparó por métodos conocidos en la técnica. Por ejemplo, el compuesto A1 puede prepararse por los procedimientos descritos en Pellicciari, R., et al., J. Med. Chem.
2009, 52, 7958-7961.

Esquema 1. Reactivos y condiciones: a) MeOH, p-TSA, ultrasonido; b) PhMgBr en Et2O, THF, reflujo; HCl, EtOH, reflujo y luego temperatura ambiente; c) MeOAc, p-TSA, reflujo; d) PCC, CH2O 2; e) Ac2O, Bi (OTf)3 , CH2O 2; f) NaIO4 , H2O, H2SO4, RuCl3, MeCN, EtOAc; g) MeOH, p-TSA, reflujo; h) Br2, benceno, 30 °C; i) NaBH4 , NaOAc, piridina, 25 °C; j) HI, AcOH; k) CrO3 , AcOH; 1) polvo de Zn, NaOAc, AcOH, reflujo; m) NaOH, MeOH, H2O, reflujo; y n) NaBH4, THF, H2O. 3a,7a,12a-Trihidroxi-6a-etil-5p-bisnorcholanildifeniletileno (Compuesto B1):
Una solución del compuesto A1 (8 g, 18,32 mmol) y ácido para-toluenosulfónico (p-TSA) (352 mg, 1,83 mmol) en MeOH (200 mL) se trató bajo ultrasonidos durante 3 h. La mezcla se concentró al vacío, se diluyó con CHCb y se lavó con una solución saturada de NaHCO3. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para obtener 7,96 g de derivado de 6-etilcolato de metilo. El éster metílico así formado (17,66 mmol) se disolvió en THF recién destilado (80 mL) y la mezcla se calentó hasta 50 °C bajo agitación magnética y atmósfera de argón. A continuación, se añadió gota a gota PhMgBr en Et2O (176,6 mmol) y la mezcla resultante se sometió a reflujo durante 14 h. La suspensión se trató con HCl acuoso (50 mL) y se extrajo con EtOAc (3 x 120 mL). Las capas orgánicas recogidas se lavaron con una solución saturada de NaHCO3 , salmuera, se secaron sobre Na2SO4 anhidro y se
concentraron a presión reducida. El aceite resultante se trató con 160 mL de HCl:EtOH (3:1, v/v), se puso a reflujo durante 3 h y se agitó a temperatura ambiente durante la noche. El EtOH se eliminó al vacío y la mezcla se extrajo con EtOAc (3 x 100 mL). Las fases orgánicas combinadas se lavaron con una solución saturada de NaHCO3, salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El crudo se purificó por cromatografía flash en gel de sílice para obtener el producto deseado B1 en un 76% de rendimiento (7,7 g, 13,85 mmol).
3a,7a-Diacetoxi-12-oxo-6a-etil-5p- bisnorcholanildifeniletileno (Compuesto C1):
Una solución del compuesto B1 (7,7 g, 13,85 mmol) y p-TSA (266 mg, 1,38 mmol) en MeOAc (70 mL) se sometió a reflujo durante 2 d. La mezcla se lavó con una solución saturada de NaHCO3 , salmuera, se secó sobre Na2SO4 anhidro y se concentró al vacío para dar 8,15 g de 3-acetoxi-7,12-dihidroxi-6-etil-5-bisnorcholanyldiphenyletilene. El crudo (8,18 g) se disolvió en CH2O 2 seco (270 mL). Se añadió clorocromato de piridinio (PCC) (2,95 g) y la mezcla se agitó durante 4 h. La suspensión marrón resultante se filtró, se trató con HCl acuoso y la capa orgánica se lavó con H2O y salmuera. Tras secarse sobre Na2SO4 anhidro y concentrarse a presión reducida, el crudo se purificó mediante cromatografía flash en gel de sílice para obtener 5,6 g (9,39 mmol) del derivado 12-oxo deseado. El intermedio se disolvió en CH2O 2 (80 mL), se trató con Ac2O (4,5 mL, 46,95 mmol), Bi(OTf)3 (306 mg, 0,469 mmol) y se agitó durante 40 min. La suspensión así obtenida se filtró y se acidificó con HCl acuoso. La fase orgánica se lavó con H2O y salmuera, se secó sobre Na2SO4 anhidro y se concentró al vacío. El crudo se filtró sobre almohadilla de gel de sílice para obtener 5,3 g (8,29 mmol) del compuesto C1 en un rendimiento del 60%.
3a,7a-diacetoxi-12-oxo-6a-etil-24-nor-5p-colan-23-oato de metilo (compuesto D1):
Se agitó NaIO4 (15,97 g, 74,66 mmol) en 15 mL de H2O y 2 N H2SO4 (2,4 mL). Después de 1 h la solución se enfrió a 0 °C, se añadió RuCb (85,9 mg, 0,415 mmol) y la mezcla se agitó magnéticamente durante 1 h. Se añadió MeCN (23,5 mL) como transferencia de fase y después de 5 min se dejó caer una solución del compuesto C1 (5,3 g, 8,29 mmol) en EtOAc (32,5 mL) y se dejó reaccionar durante 1 h. La mezcla se filtró, se vertió en H2O y se extrajo con EtOAc (3 x 100 mL). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron a presión reducida. El residuo resultante se filtró sobre una almohadilla de gel de sílice para dar 5,33 g de derivado de ácido 6aetil-24-nor-cólico que se disolvió en MeOH (90 mL), se trató bajo ultrasonidos en presencia de p-TSA (160 mg, 0,829 mmol) durante 3 h y se reflujo durante 1 h. La mezcla se concentró al vacío, se diluyó con CHCb y se lavó con una solución saturada de NaHCO3. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró al vacío. El crudo se purificó mediante cromatografía flash en gel de sílice para dar el compuesto D1 en un rendimiento del 86% (3,73 g, 7,19 mmol).
11a-bromina-3a,7a-diacetoxi-12-oxo-6a-etil-24-nor-5p-colan-23-oato de metilo (compuesto E1):
Se añadió gota a gota una solución de Br2 en benceno anhidro (2 M, 4,67 mL) a una solución del compuesto D1 (3,73 g, 7,19 mmol) en benceno (156 mL). La solución roja resultante se dejó reaccionar a 30 °C bajo atmósfera de argón durante 3 días. La mezcla se vertió en una solución acuosa de Na2S2O3 y la suspensión amarilla se extrajo con EtOAc (3 x 100 mL). Las capas orgánicas recogidas se lavaron con H2O y salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El crudo se purificó por cromatografía flash en gel de sílice para obtener el compuesto E1 como sólido blanco-amarillo (2,65 g, 4,43 mmol).
3a,7a-diacetoxi-11p,12p-oxo-6a-etil-24-nor-5p-colan-23-oato de metilo (compuesto F1):
Se añadieron NaOAc (2,65 g, 32,81 mmol) y NaBH4 (808 mg, 21,27 mmol) a una solución del compuesto E1 (2,65 g, 4,43 mmol) en piridina recién destilada (27,5 mL) y la suspensión se dejó reaccionar a 25 °C bajo atmósfera de N2 durante 14 h. La mezcla se trató con HCl acuoso y se extrajo con EtOAc (3 x 80 mL). Las fases orgánicas combinadas se lavaron con H2O, salmuera y se secaron al vacío. El aceite crudo se purificó mediante cromatografía flash en gel de sílice para obtener 1,52 g (2,85 mmol) del compuesto F1 en un rendimiento del 64%.
12a-yodo-3a,7a-diacetoxi-11-oxo-6a-etil-24-nor-5p-colan-23-oato de metilo (compuesto G1):
A una solución del compuesto F1 (1,52 g, 2,85 mmol) en AcOH (40 mL), se añadió gota a gota HI 57% (3,6 g, 28,5 mmol) y la mezcla se dejó reaccionar a temperatura ambiente durante 30 min. La mezcla se trató con una solución acuosa de NaHSO3 , se vertió en H2O helado, se filtró y el sólido resultante se disolvió en AcOH (35 mL). Se añadió gota a gota una solución de CrO3 (1,4 g, 14,3 mmol) en AcOH (40 mL) y H2O (8 mL) y se agitó la mezcla durante 45 min. La reacción se apagó con una solución acuosa de NaHSO3 y se vertió en agua helada. La suspensión se filtró y el sólido se disolvió en CHCb. La solución se lavó con H2O, salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El crudo se purificó mediante cromatografía flash en gel de sílice para dar el compuesto G1 como producto puro (1 g, 1,67 mmol).
Ácido 3a,7a-dihidroxi-11-oxo-6a-etil-24-nor-5p-colan-23-oico (Compuesto H1):
Se añadieron NaOAc (3,8 g, 46,76 mmol) y polvo de Zn (3,8 g, 58,45 mmol) a una solución del compuesto G1 (1 g, 1,69
mmol) en AcOH (30 mL) y la suspensión resultante se sometió a reflujo durante 2 h. La mezcla se filtró y el filtrado se trató con H2O a 0 °C hasta la precipitación. El precipitado se disolvió en CHCb y la fase acuosa se extrajo con CHCb (3 x 50 mL). Las capas orgánicas recogidas se trataron con una solución saturada de NaHCO3, se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El crudo (880 mg) se disolvió en MeOH y H2O, se añadió NaOH (25,45 mmol) y la mezcla se sometió a reflujo durante 36 h. La solución resultante se concentró a presión reducida, se diluyó con H2O y se trató con HCl acuoso. Se extrajo con CHCb (3 x 50 mL) y las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío para dar el compuesto HI.
Ácido 3a,7a,11p-Trihidroxi-6a-etil-24-nor-5p-colan-23-oico (Compuesto 1):
A una solución del Compuesto H1 (650 mg, 1,54 mmol) en THF:H2O (33 mL, 4:1 v/v), se añadió porciones de NaBH4 (407 mg, 10,78 mmol) a 0 °C y la suspensión resultante se dejó reaccionar a temperatura ambiente durante 5 h. Después de tratarla con H2O y HCl acuoso, la mezcla de reacción cruda se extrajo con CHCb (3 x 50 mL). Las capas orgánicas recogidas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío para dar 650 mg del compuesto 1 (1,69 mmol, rendimiento cuantitativo) (9% de rendimiento global de 1).
Compuesto 1: 1H-NMR (400 MHz, CD3OD): 80,89 (3H, t, J= 7,33 Hz, CH3-25), 0,94 (3H, s, CH3-18), 1,04 (3H, d, J = 5,46 Hz,CH3-21), 1,13 (3H, s, CH3-19), 3,30-3,35 (1H, m, CH-3), 3,71 (1H, s, CH-7), 4,19 (1H, s, CH-11). 13C-NMR (400 MHz, CD3OD): 12,0,14,6, 19,9, 23,5, 24,6, 27,7, 29,1, 31,9, 34,7, 35,2, 36,4, 36,9, 38,3 (2 x), 42,6 (2 x), 42.8, 49.5, 49.9, 52.2, 57.9, 69.0, 71.4, 73.3, 177.7.
Ejemplo 2. Síntesis de 3a,7a,11p-trihidroxi-6a-etil-24-nor-5p-colan-23-ol (Compuesto 2)

El compuesto 2 se preparó de acuerdo con los procedimientos establecidos en el Esquema 2. El compuesto 2 se preparó a partir del compuesto 1 como material de partida.
Ejemplo 3. Síntesis de la sal sódica de 3o,7a,1ip,23-tetrahidroxi-6a-etil-24-nor-5p-colan-23-O-sulfato (Compuesto 3)

El compuesto 3 se preparó de acuerdo con los procedimientos establecidos en el Esquema 2. El compuesto 3 se preparó a partir del compuesto 1 como material de partida.

Esquema 2. Reactivos y condiciones: o) AC2O, THF, reflujo; p) EtCOCI, Et3N, THF; NaBH4, THF, H2O; y q) PyrSO3 , piridina; NaOH, MeOH, H2O, reflujo.
Ácido 3a-acetoxi-7a,11p-dihidroxi-6a-etil-24-nor-5p-colan-23-oico (Compuesto J1):
Se añadió AC2O (2,08 mL, 21,6 mmol) a una solución del compuesto 1 (460 mg, 1,08 mmol) en THF (35 mL) y la mezcla se sometió a reflujo durante 18 h. La solución resultante se trató con HCl acuoso y se extrajo con EtOAc (3 x 30 mL). Las fases orgánicas combinadas se lavaron con H2O, salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El crudo se purificó por cromatografía flash en gel de sílice para obtener el compuesto J1 (255 mg, 0,548 mmol).
3a-Acetoxy-7a, 11 p-dihidroxi-6-etil-24-nor-5p-colan-23-ol (Compuesto K1):
Una solución del compuesto J1 (250 mg, 0,538 mmol), EtCOCl (0,51 mL, 5,326 mmol) y Et3N (0,81 mL, 5,649 mmol) en THF (7,5 mL) se dejó reaccionar durante 14 h a temperatura ambiente. La mezcla de reacción se filtró, se trató con una suspensión de NaBH4 (306 mg, 8,07 mmol) en H2O (2,5 mL) y se agitó durante 2 h. La mezcla se acidificó con HCl acuoso y se extrajo con EtOAc (3 x 30 mL). Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron al vacío. El crudo se filtró sobre una almohadilla de gel de sílice para dar el compuesto K1 (150 mg, 0,333 mmol).
3a,7a,11p-trihidroxi-6a-etil-24-nor-5p-colan-23-ol (2) y sal sódica de 3a,7a,11p,23-tetrahidroxi-6a-etil-24-nor-5p-colan-23-O-sulfato (compuesto 3):
A una solución del compuesto K1 (150 mg, 0,333 mmol) en piridina (6 mL), se añadió PyrSO3 (133 mg, 0,832 mmol) y la mezcla resultante se agitó bajo atmósfera de argón durante 24 h. La mezcla de reacción se diluyó con H2O (2 mL) y se concentró a presión reducida para eliminar la piridina. El residuo se trató con una solución de NaOH (200 mg, 4,995 mmol) en MeOH:H2O (10 mL) y se sometió a reflujo durante la noche. La mezcla se secó al vacío para eliminar el MeOH, se diluyó con H2O (2 mL) y se lavó con Et2O (3 x 20 mL): las fases etéreas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se purificaron por cromatografía flash para dar el 3a,7a,11p-trihidroxi-6aetil-24-nor-5p-colan-23-ol (2) como sólido blanco puro (55 mg, 0,134 mmol). La fase acuosa alcalina se filtró en una almohadilla RP-18 de fase inversa para obtener el Compuesto 3 como sólido blanco puro (60 mg, 0,117 mmol).
Compuesto 2: 1H-NMR (400 MHz, CDCla): 80,88-0,92 (6H, m, CH3-25, CH3- I 8), 0,97 (3H, d, J= 6,5 Hz, CH3-21), 114 (3H, s, CH3-19), 3,40-3,47 (1H, m, CH-3), 3,62-3,72 (2H, m, CH2-23), 3,80 (1H, s, CH-7), 4,25 (1H, d, J = 2,72 Hz, CH-11). 13C-NMR (400 MHz, CDCb): 11,6, 14,4, 18,8, 22,2, 23,8, 27,0, 28,0, 31,1, 32,9, 34,1, 35,3, 35,7, 36,4, 37.1, 38.8, 40.6, 41.6, 47.7, 48.8, 50.9, 56.8, 60.7, 68.8, 71.0, 72.3.
Compuesto 3: 1H-NMR (400 MHz, CD3OD): 80,90-0,94 (6H, m, CH3-25, CH3-18 ), 1,04 (3H, d, J= 6,4 Hz, CH3-21), 115 (3H, s, CH3-19), 3,32-3,40 (1H, m, CH-3), 3,74 (1H, s, CH-7), 4,02-4,08 (2H, m, CH2-23), 4,21 (1H, s, CH-11). 13C-NMR (400 MHz, CD3OD): 12,0, 14,6, 19,1, 23,5, 24,7, 27,7, 29,1, 31,9, 34,3, 34,8, 36,4, 36,5, 36,9, 38,3 (x2), 42,6, 42.8, 49.5, 50.0, 52.2, 58.2, 67.2, 69.0, 71.4, 73.3.
Ejemplo 4. Síntesis de 3a,7a,11p-trihidroxi-6a-etil-5p-colan-24-ol (Compuesto 4)

4 se preparó de acuerdo con los procedimientos establecidos en el Esquema 3. La síntesis de 4 se preparó a partir del compuesto L1 como material de partida. El compuesto L1 se preparó por métodos conocidos en la técnica. Por ejemplo, el Compuesto L1 puede prepararse mediante los procedimientos descritos en la Publicación de Estados Unidos No.
2014/0371190.
Esquema 3.

Esquema 3. Reactivos y condiciones: r) LiAlH4, THF, de 0 °C a temperatura ambiente.
3a,7a,11p-Trihidroxi-6a-etil-5p-colan-24-ol (Compuesto 4):
Se añadió gota a gota una solución del compuesto L1 (25 mg, 0,057 mmol) en THF (2 mL) a una suspensión de LiAlH4 (21,8 mg, 0,572 mmol) en THF (1 mL) enfriada a 0 °C. La mezcla resultante se dejó reaccionar bajo atmósfera de argón y a temperatura ambiente durante 12 h. La suspensión se diluyó con EtOAc (5 mL) y se trató primero con H2O y luego con HCl acuoso. La mezcla resultante se dejó reaccionar bajo atmósfera de argón y a temperatura ambiente durante 12 h. La suspensión se diluyó con EtOAc (5 mL), se trató primero con H2O, luego con HCl acuoso y finalmente se extrajo con EtOAc (3 x 5 mL). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El crudo se purificó por cromatografía flash para dar el compuesto 4 como un sólido blanco puro (21 mg, 0,051 mmol, rendimiento del 90%).
Compuesto 4: 1H-NMR (400 MHz, CD3OD): 8 0,88-0,92 (6H, m, CH3-26, CH3- I 8), 1,00 (3H, d, J = 6,25 Hz, CH3-21), 114 (3H, s, CH3-19), 3,31-3,40 (1H, m, CH-3), 3,48-3,55 (2H, m, CHi -24), 3,73 (1H, s, CH-7), 4,19 (1H, s, CH-11). 13C-NMR (400 MHz, CDaOD):12,0, 14,6, 19,2, 23,5, 24,7, 27,7, 29,1, 30,3, 31,9, 33,2, 34,8, 36,4, 36,9, 37,2, 38,3 (x2), 42,6, 42,7, 49,5, 50,1, 52,2, 58,1, 63,6, 69,1, 71,4, 73,3.
Ejemplo 5. Síntesis de 3a,7a,11(3-r¡h¡drox¡-6a-et¡l-5(3-colan-24-0-sulfato, sal de sodio (Compuesto 5)

El compuesto 5 se preparó de acuerdo con los procedimientos establecidos en el esquema 4. La síntesis del compuesto 5 se preparó a partir del compuesto M1 como material de partida. El compuesto M1 se preparó por métodos conocidos en la técnica. Por ejemplo, el Compuesto M1 puede prepararse mediante los procedimientos descritos en la Publicación de Estados Unidos No. 2014/0371190.

Esquema 4. Reactivos y condiciones: s) AC2O, NaHCO3 , THF, reflujo; t) Et3N, ClCO2Et, THF, r.t.; NaBH4, THF, H2O; u) NaBH4, THF, H2O; y v) PyrSO3 , piridina; NaOH, MeOH, H2O.
3a-Acetoxy-7a-hidroxi-11p-oxo-6a-etil-5p-cholan-24-ol (Compuesto N1):
A una solución del compuesto M1 (120 mg, 0,27 mmol) en THF recién destilado (4 mL), se añadieron NaHCO3 (417 mg, 4,97 mmol) y AC2O (0,47 mL, 4,97 mmol) y la suspensión se sometió a reflujo durante 24 h bajo atmósfera de argón. La mezcla se enfrió a temperatura ambiente, se trató con HCl acuoso y se extrajo con EtOAc (3 x 10 mL). Las fases orgánicas recogidas se lavaron secuencialmente con HCl acuoso, agua, una solución saturada de NaHCO3, salmuera y se secaron sobre Na2SO4 anhidro. Tras concentrarse a presión reducida, el crudo se disolvió en THF recién destilado (3 mL), se trató con Et3N (0,22 mL, 1,54 mmol) y ClcO2Et (0,14 mL, 1,45 mmol) y se dejó reaccionar la mezcla a temperatura ambiente durante 2 h bajo atmósfera de argón. La suspensión se filtró y el filtrado se trató con una suspensión de NaBH4 (125 mg, 3,30 mmol) en H2O (1 mL) y se agitó durante 3 h. La mezcla se acidificó con HCl acuoso y se extrajo con EtOAc (3 x 10 mL). Las capas orgánicas combinadas se lavaron con H2O, salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El crudo se purificó por cromatografía flash obteniéndose el compuesto N1 (70 mg, 0,15 mmol).
3a-Acetoxi-7a, 11 p-dihidroxi-6a-etil-5p-colan-24-ol (Compuesto P1):
A una solución del compuesto N1 (0,15 mmol) en una mezcla binaria de THF y H2O, se añadió NaBH4 (3,75 mmol) y la mezcla se agitó a temperatura ambiente durante 24 h. La suspensión se trató con HCl acuoso y se extrajo con EtOAc (3 x 10 mL). Las fases orgánicas recogidas se lavaron con H2O, salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida dando el compuesto P1 en rendimiento cuantitativo.
3a,7a,11p-Trihidroxi-6a-etil-5p-colan-24-O-sulfato, sal de sodio (Compuesto 5):
Se añadió PirSO3 (48 mg, 0,30 mmol) a una solución del compuesto PI (70 mg, 0,15 mmol) en piridina (2,7 mL) y se dejó reaccionar a temperatura ambiente durante 30 h bajo atmósfera de argón. La piridina se eliminó al vacío y el residuo se agitó con una solución de NaOH (60 mg, 1,5 mmol) en una mezcla de MeOH y H2O durante 3 d. La mezcla se concentró a presión reducida para evaporar el MeOH, se diluyó con H2O (2 mL) y se lavó con Et2O (3 x 10 mL). La fase acuosa alcalina se filtró en una almohadilla RP-18 de fase inversa para obtener el Compuesto 5 (47 mg, 0,085 mmol, 57% de rendimiento) como sólido blanco puro.
Compuesto 5: 1H-NMR (400 MHz, CD3OD): 80,88-0,92 (6H, m, CH3-I8, CH3-26), 1,00 (3H, d, J = 6,3 Hz, CH3- 21), 1.14 (3H, s, CH3-19), 3,32-3,35 (1H, brm, CH-3), 3,72 (1H, brs, CH-7), 3,94-3,97 (2H, brm, CH2-24), 4,20 (1H, brs, CH-11).
13C-NMR (400 MHz, CD3OD): 12,1, 14,7, 19,1, 23,6, 24,7, 27,1, 27,7, 29,1, 31,9, 33,1, 34,8, 36,4, 36,9, 37,0, 38.3 (x 2), 42.6, 42.7, 49.5, 50.1, 52.2, 58.0, 69.1, 69.7, 71.4, 73.3.
Ejemplo 6. Síntesis del 3a,7a,11p-tr¡h¡droxi-6a-et¡l-22-(1,2,4-oxad¡azol-5-oxo-3-¡l)-23,24-b¡snor-5p-colano (Compuesto 6) Esquema 5.

Esquema 5. Reactivos y condiciones: a) Ac2O, Bi(OTf)3, CH2Ch ; b) TFAA, NaNO2, TFA; c) NaOH, MeOH; d) NH2OH-HCl, Na2CO3- 10H2O, EtOH; e) ClCO2Et, piridina, THF; f) piridina, tolueno.
Ác¡do 3a,7a,11p-Triacetox¡-6a-et¡l-5p-colan-24-o¡co (Compuesto Q1):
A una suspensión del compuesto L1 (660 mg, 1,5 mmol) en CH2Cl2 (15 mL), se añadieron AC2O (22,7 mmol) y Bi(OTf)3 (0,08 mmol) y la mezcla se agitó a temperatura ambiente durante 1 h. La mezcla de reacción se filtró y el filtrado se trató con HCl al 37%. La fase orgánica se lavó con H2O, con una solución saturada de NaHCO3 y con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida para obtener Q1 (820 mg, 1,46 mmol, 96% de rendimiento), que se utilizó para el siguiente paso sin más purificación.
Compuesto Q1: 1H-NMR (400 MHz, CDCl3): 80,76 (3H, s, CH3- I 8), 0,87-0,90 (6H, m, CH3-21, CH3-26), 1,04 (3H, s, CH3-19), 2.03-2.05 (6H, m, OCOCH3 x2), 2.08 (3H, s, OCOCH3), 4.52-4.61 (1H, m, CH-3), 5.20 (1H, s, CH-7), 5.25 (1H, s, CH-11).
3a,7a,11p-Tr¡acetoxi-6a-et¡l-24-nor-5p-colan-23-n¡tr¡lo (Compuesto R1):
Una suspensión del compuesto Q1 (820 mg, 1,46 mmol) en TFA (4,6 mL) a 0 °C se trató con TFAA (1,55 mL) y se agitó a 0 °C durante 45 min. Se añadió NaNO2 (4,4 mmol) y la mezcla se hizo reaccionar a 0 °C durante 45 min y a 50 °C durante 45 min adicionales. La mezcla de reacción se enfrió a temperatura ambiente y se vertió en hielo picado. La fase acuosa se filtró al vacío y el sólido amarillo anaranjado resultante se disolvió en EtOAc (30 mL), se lavó con una solución saturada de NaHCO3, H2O y salmuera. La capa orgánica se secó sobre Na2SO4 anhidro, se concentró a presión reducida y se obtuvo el compuesto R1 (770 mg) como crudo que se utilizó para el siguiente paso sin más purificación.
Compuesto R1: 1H-NMR (400 MHz, CDCb): 80,76 (3H, s, CH3- I 8), 0,84-0,87 (3H, m, CH3-25), 1,02 (3H, s, CH3-19), 1,08 (3H, d, J = 6.4 Hz, CH3-21), 2,01 (6H, brs, OCOCH3 x2), 2,07 (3H, s, OCOCH3), 4,52-4,61 (1H, m, CH- 3), 5,18 (1H, s, CH-7), 5,24 (1H, s, CH- 11).
3a,7a, 11 p-T r¡h¡drox¡-6a-et¡l-24-nor-5p-colan-23-nitr¡lo (Compuesto S1):
El compuesto R1 (770 mg) se disolvió en MeOH (10 mL) y se sometió a reflujo durante 3 d con NaOH (1,2 g). Tras eliminar el disolvente, el residuo se disolvió en CHCb (30 mL) y se trató con HCl 1 N. La fase acuosa se extrajo con CHCl3 y las capas orgánicas combinadas se lavaron con H2O y salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El crudo se purificó mediante cromatografía flash en gel de sílice utilizando CH2Cl2 y MeOH como disolventes de elución para obtener el compuesto S1 (180 mg, 0,445 mmol) en grado de alta pureza.
Compuesto S1: 1H-NMR (400 MHz, CDCla): 8 0,87-0,91 (6H, m, CH3- I 8, CH3-25), 1,13 (3H, s, CH3-19), 1,17 (3H, d, J = 6,5 Hz, CH3-21), 3,42-3,50 (1H, m, CH-3), 3,77 (1H, s, CH-7), 4,28 (1H, s, CH-11).
3a,7a, 11 p-T rihidroxi-6a-etil-24-nor-N-hidroxi-5p-colan-23-amidina (Compuesto T1):
Se añadieron NH2OH-HCI (557 mg) y Na2CO3-10H2O (2,30 g) a una solución del compuesto S1 (180 mg, 0,445 mmol) en EtOH (8 mL) y la mezcla resultante se sometió a reflujo durante 2 d. La suspensión se enfrió a temperatura ambiente y se filtró al vacío. El sólido se lavó con EtOAc y la fase orgánica se lavó con H2O, salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El crudo, que contenía el compuesto intermedio deseado T1, se utilizó para el siguiente paso sin más purificación.
Compuesto T1: 1H-NMR (400 MHz, CD3OD): 8 0,90 (3H, t, J = 7,3 Hz, CH3-25), 0,96-0,98 (6H, m, CH3- I 8, CH3- 2 I), 1,14 (3H, s,CH3-19), 3,31-3,40 (1H, m, CH-3), 3,72 (1H, s, CH-7), 4,20 (1H, s, CH-11).
3a,7a,11p-Trihidroxi-6a-etil-24-nor-N-[(etoxicarbonil)oxi]-5p-colan-23-amidina (Compuesto U1):
A una solución del compuesto T1, (180 mg) en THF (2 mL) y piridina (50 mL, 0,6 mmol) enfriada a 0 °C, se añadió gota a gota una solución de ClCO2Et (0,45 mmol) en THF (1 mL) y la suspensión resultante se agitó bajo atmósfera de argón durante 30 min. La mezcla se trató con H2O y se extrajo con EtOAc (3 x 10 mL). Las fases orgánicas recogidas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío para obtener el compuesto U1, que se utilizó para el siguiente paso sin más purificación.
Compuesto U1: 1H-NMR (400 MHz, CDCb): 8 0,86-0,92 (6H, m, CHs-25, C02CH2CH3), 1,00 (3H, d, J = 6,1 Hz, CH3-21), 111 (3H, s, CH3- I 8), 1,23 (3H, s, CH3-19), 3,35-3,44 (1H, m, CH-3), 3,77 (1H, s, CH-7), 4,13-4,39 (3H, m, CH-11, CO2CH2CH3), 4,90-5,05 (1H, m, NH).
3a,7a,11p-Trihidroxi-6a-etil-22-(1,2,4-oxadiazol-5-oxo-3-il)-23,24-bisnor-5p-colano (Compuesto 6):
El compuesto U1 (210 mg, 0,412 mmol) se disolvió en tolueno (6 mL) y piridina (0,6 mL) y se sometió a reflujo bajo atmósfera de argón durante 20 h. Tras enfriarse a temperatura ambiente, la mezcla se diluyó con EtOAc (10 mL) y se lavó con HCl 1 N, H2O, una solución saturada de NaHCO3 y salmuera. La capa orgánica se secó sobre Na2SO4 anhidro, se concentró a presión reducida y se purificó para obtener el compuesto 6 (35 mg, 0,076mmol).
Compuesto 6: 1H-NMR (400 MHz, CD3OD): 80,89-0,93 (3H, t, J = 7,3 Hz, CH3-24), 0,96-0,99 (6H, m, CH3-18, CH3-21), 115 (3H, s, CH3-19), 2,09 (1H, d, J = 14,1 Hz), 2,37 (1H, d, J = 12,3 Hz), 2,57 (1H, d, J = 13,4 Hz), 3,31-3,40 (1H, m, CH-3), 3,66 (1H, s, OH), 3,73 (1H, s, CH-7), 4,21 (1H, s, CH-11). 13C-NMR (100,6 MHz, CD3OD): 12,0, 14,7, 19.2, 23.5, 24.7, 27.6, 29.1, 31.9, 34.5, 34.7, 36.1, 36.4, 36.9, 38.2 (x2), 42.6, 43.0, 49.4, 49.9, 52.2, 58.4, 68.9, 71.3, 73.3, 169.3, 173.0.
Ejemplo 7. Síntesis del 3a,7a,11p-tr¡h¡droxi-6a-et¡l-23-(1,2,4-oxad¡azol-5-oxo-3-¡l)-23-nor-5p-colano (Compuesto 7 )

Esquema 6. Reactivos y condiciones: a) MeOH, p-TSA, ultrasonidos; b) MOMCl, DIPEA, DMAP, CH2CI2; c) NaOH, MeOH; d) CICO2¡Bu, EtsN, THF, NH4OH 30%; e) CNCl, DMF; f) NH2OH-HCI, Na2COa 10 H2O, EtOH; g) ClCO2Et, piridina, THF; h) piridina, tolueno; ¡) HCl 3 N, acetona.
6a-et¡l-3a,7a,11p-trimetox¡metalox¡-5p-colan-24-oato de metilo (compuesto V1):
Una solución del compuesto L1 (730 mg, 1,7 mmol) y p-TSA (0,17 mmol) en MeOH (10 mL) se trató bajo ultrasonidos durante 3 h. El disolvente se eliminó, el residuo se disolvió en EtOAc (10 mL) y se lavó con una solución saturada de NaHCOa. La fase acuosa se extrajo con EtOAc (2 x 10 mL) y las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El crudo (700 mg, 1,55 mmol) se disolvió en CH2CI2 (20 mL) y se puso a reflujo con DIPEA (18,6 mmol), DMAP (0,16 mmol) y M o M C I (15,5 mmol) durante 3 d. La mezcla se enfrió a temperatura ambiente y se lavó secuencialmente con una solución saturada de NH4CI, H2O y salmuera. La fase orgánica se secó sobre Na2SO4 anhidro, se concentró a presión reducida y se obtuvo el Compuesto V1 crudo que se utilizó para el siguiente paso sin más purificación.
Compuesto VI: 1H-NMR (400 MHz, CDCI3): 80,81 (3H, s, CH3- I 8), 0,85-0,89 (3H, m, CH3-26), 0,93 (3H, d, J = 6,2 Hz, CH3-21),110 (3H, s, CH3-19), 3,34-3,40 (10H, m, CH-3, OCH2OCH3 x3), 3,53 (1H, s, CH-7), 3,65 (3H, s, CO2CH3), 3,93 (1H, s, CH-11), 4,55-4,70 (6H, m, OCH2OCH3 x3).
6a-Etil-3a,7a,11p-tr¡metox¡metox¡-5p-colan-24-am¡da (Compuesto W1):
A una solución del compuesto V1 (980 mg, 1,6 mmol) en MeOH (10 mL), se añadió NaOH (15,5 mmol) y la mezcla se dejó reaccionar a 50 °C. El disolvente se eliminó al vacío, el residuo se disolvió en H2O (5 mL) y se trató con HCl 1 N. La suspensión se extrajo con CHCI3 (3 x 10 mL) y las fases orgánicas combinadas se lavaron con H2O y salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El crudo (840 mg) se disolvió en THF recién destilado (18 mL), se enfrió a 0 °C y se agitó con Et3N (0,288 mL) y ClCO2¡Bu (0,250 mL) durante 20 min bajo atmósfera de argón. Se añadió NH4OH 30% (0,28 mL) y la suspensión resultante se hizo reaccionar durante 40 min a temperatura ambiente. La mezcla se trató con H2O y se extrajo con EtOAc (3 x 10 mL). Las fases orgánicas recogidas se lavaron con HCl 1 N, H2O, una solución saturada de NaHCO3 , H2O y salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para obtener el compuesto W1 (900 mg) que se utilizó para el siguiente paso sin más purificación.
Compuesto W1: 1H-NMR (400 MHz, CDCI3): 80,62-0,82 (9H, m, CH3-18 , CH3-21, CH3-26), 0,99 (3H, s, CH3-19), 3,22 3.30 (10H, m, CH-3, OCH2OCH3 x3), 3,42 (1H, s, CH-7), 3,82 (1H, s, CH-11), 4,46-4,57 (6H, m, OCH2OCH3 x3), 6,03 (1H, brs, CONH2), 6,27 (1H, brs, CONH2).
6a-Etil-3a,7a, 11 p-trimetoximetaloxi-5p-colan-24-nitrilo (Compuesto X1):
Una solución del compuesto W1 (890 mg) y CNCI (578 mg) en DMF (22 mL) se agitó a temperatura ambiente bajo atmósfera de argón durante 12 h. La suspensión resultante se diluyó con EtOAc (50 mL) y se lavó con H2O (3 x 15 mL). La capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El crudo se purificó por cromatografía flash sobre gel de sílice utilizando PET-EtOAc como sistema de disolvente de elución para obtener el compuesto X1 como un aceite amarillo pálido (260 mg, 0,473 mmol).
Compuesto X1: 1H-NMR (400 MHz, CDCl3): 80,83-0,88 (6H, m, CH3- I 8, CH3 -26), 0,95 (3H, d, J = 6,0 Hz, CH3-21), 1,05 (3H, s, CH3-19), 3.33-3.40 (10H, m, CH-3, OCH2OCH3 x3), 3.52 (1H, s, CH-7), 3.91 (1H, s, CH-11), 4.55-4.69 (6H, m, OCH2OCH3 x3).
6a-Etil-3a,7a,11p-trimetoximetaloxi-N-hidroxi-5p-colan-24-amidina (Compuesto Y1):
Se añadieron NH2OH-HQ (386 mg) y Na2CO3-(1,6 g) a una solución del compuesto X1 (170 mg, 0,309 mmol) en EtOH (6 mL) y se sometió a reflujo hasta el consumo del material de partida. La suspensión se enfrió a temperatura ambiente y se filtró al vacío. El sólido restante se lavó con EtOAc (15 mL) y la fase orgánica filtrada se lavó con H2O, salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El crudo, que contenía el compuesto intermedio deseado Y1, se utilizó para el siguiente paso sin más purificación.
Compuesto Y1: 1H-NMR (400 MHz, CDCb): 80,77 (3H, s, CH3- I 8), 0,81-0,84 (3H, t, J = 6,8 Hz, CH3-26), 0,90 (3H, d, J = 6,2 Hz, CH3-21), 1.05 (3H, s, CH3-19), 3,30-3,36 (10H, m, CH-3, OCH2OCH3 x3), 3,48 (1H, s, CH-7), 3,88 (1H, s, CH-11), 4,51-4,65 (6H, m, OCH2OCH3 x3), 4,76 (2H, brs, NH, NH-OH).
6a-Etil-3a,7a,11p-trimetoxiloxi-N-[(etoxicarbonil)oxi]-5p-colan-24-amidina (Compuesto Z1):
A una solución del compuesto Y1 (200 mg) en THF (2 mL) y piridina (0,46 mmol) enfriada a 0 °C, se añadió gota a gota una solución de ClCO2Et (0,34 mmol) en THF (1 mL) y la suspensión resultante se agitó bajo atmósfera de argón durante 30 min. La mezcla se trató con H2O y se extrajo con EtOAc (3 x 10 mL). Las fases orgánicas recogidas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío para proporcionar el compuesto Z1, que se utilizó para el siguiente paso sin más purificación.
Compuesto Z1: 1H-NMR (400 MHz, CDCl3): 80,77 (3H, s, CH3- I 8), 0,81-0,84 (3H, t, J = 6,8 Hz, CH3-26), 0,92 (3H, d, J = 6,1 Hz, CH3-21), 1,05 (3H, s, CH3-19), 1,27 (3H, t, J = 7.0 Hz, OCH2CH3), 3,30-3,36 (10H, m, CH-3, OCH2OCH3 x3), 3,48 (1H, s, CH-7), 3,88 (1H, s, CH-11), 4,21 (2H, q, J = 7,0 Hz, OCH2CH3), 4,51-4,65 (6H, m, OCH2OCH3 x3), 4,88 (2H, brm, NH, NH-OH).
3a,7a,11p-Trihidroxi-6a-etil-23-(1,2,4-oxadiazol-5-oxo-3-il)-24-nor-5p-colano (Compuesto 7):
El compuesto Z1 (200 mg) obtenido en el paso anterior se disolvió en tolueno (5 mL) y piridina (0,5 mL), se sometió a reflujo bajo atmósfera de argón durante 8 h y se agitó a temperatura ambiente durante 12 h adicionales. La mezcla se diluyó con EtOAc (10 mL) y se lavó secuencialmente con HCl 1 N, H2O, una solución saturada de NaHCO3 y salmuera. La capa orgánica se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El crudo resultante se disolvió en acetona (15 mL) y se agitó con HCl 3 N (1,5 mL) a 40 °C durante 6 h. La mezcla se diluyó con H2O y la capa orgánica se concentró a presión reducida. La fase acuosa se extrajo con EtOAc (3 x 10 mL) y las capas orgánicas combinadas se trataron con una solución saturada de NaHCO3 , se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El crudo se purificó por cromatografía flash para obtener el compuesto 7 (29,6 mg, 0,062 mmol) como un sólido blanco.
Compuesto 7: 1H-NMR (400 MHz, CD3OD): 8 0,89-0,93 (6H, m, CH3-18 , CH3-25), 1,05 (3H, d, J = 6,3 Hz, CH3- 21), 1.15 (3H, s, CH3-19), 2,07-2,11 (1H, m), 2,20-2,24 (1H, m), 2,45-2,50 (1H, m, CH-23), 2,58-2,68 (1H, m, CH-23), 3,32-3,36 (1H, m, CH-3), 3,73 (1H, s, CH-7), 4,20 (1H, s, CH-11). 13C-NMR (100,6 MHz, CD3OD): 12,0, 14,6, 18,7, 23.0, 23.5, 24.7, 27.7, 29.1, 30.8, 31.9, 33.1, 34.7, 36.4, 36.8, 36.9, 38.3 (x2), 42.6, 42.8, 50.0, 52.2, 57.6, 69.0, 71.4, 73.3, 162.2, 162.8.
Ejemplo 8. Actividad FXR / TGR5 de los compuestos 1-7
En el núcleo, los receptores nucleares (NR) unidos a ligandos modulan el inicio de la transcripción interactuando directamente con la maquinaria transcripcional basal o poniéndose en contacto con factores puente llamados coactivadores (Onate, et al., Science, 1995, 270, 1354-1357; Wang, et al., J Biol Chem, 1998, 273, 30847-30850; y Zhu, et al., Gene Expr, 1996, 6, 185-195). La interacción dependiente del ligando de los NR con sus coactivadores se produce entre la función de activación 2 (AF-2), situada en el dominio de unión al ligando del receptor (LBD) y las cajas del receptor nuclear (NR box), situadas en los coactivadores (Nolte, et al., Nature, 1998, 395, 137-143). Varias líneas de evidencia han demostrado que la secuencia peptídica LXXLL presente en la caja NR representa un motivo de firma que facilita la interacción de diferentes proteínas con la región AF-2 (Heery, et al., Nature, 1997, 387, 733-736; y Torchia, et al., Nature, 1997, 387, 677-684).
AlphaScreen se utilizó con el objetivo de identificar nuevos moduladores aprovechando la interacción bimolecular que prevalece entre el FXR y el motivo LXXLL presente en la caja NR del coactivador del receptor de esteroides 1 (SRC-1). El FXR-LBD-GST humano se incubó con concentraciones crecientes de los ligandos indicados en presencia del péptido LXXLL SRC-1 biotinilado. La señal de AlphaScreen aumenta cuando se forma el complejo receptor-coactivador. Los compuestos de esta invención son potentes agonistas del FXR. Los datos se proporcionan en las Tablas 1 y 2.
Los ácidos biliares (AB) no sólo modulan varios receptores hormonales nucleares, sino que también son agonistas del receptor acoplado a proteínas G (GPCR) TGR5 (Makishima, et al, Science, 1999, 284, 1362-1365; Parks, et al., Science, 1999, 284, 1365-1368; Maruyama, et al., Biochem Biophys Res Commun, 2002, 298, 714-719; y Kawamata, et al., J Biol Chem, 2003, 278, 9435- 9440). La señalización a través de FXR y TGR5 modula varias vías metabólicas, regulando no sólo la síntesis de BA y la recirculación enterohepática, sino también la homeostasis de los triglicéridos, el colesterol, la glucosa y la energía. Para evaluar la capacidad de un compuesto de la invención para activar la TGR5, se examinó el compuesto de la invención y otros compuestos de comparación para comprobar el aumento del AMPc intracelular como lectura de la activación de la TGR5. Las células humanas enteroendocrinas NCI-H716 que expresan constitutivamente TGR5 se expusieron a concentraciones crecientes de un compuesto de la invención, y los niveles intracelulares de AMPc se midieron por TR-FRET. El ácido litochólico (LCA) se utilizó como control positivo. Los compuestos de esta invención muestran una alta selectividad para FXR sobre TGR5. Los datos se proporcionan en Tabla 1. Actividad FXR / TGR5 de los compuestos 1-5
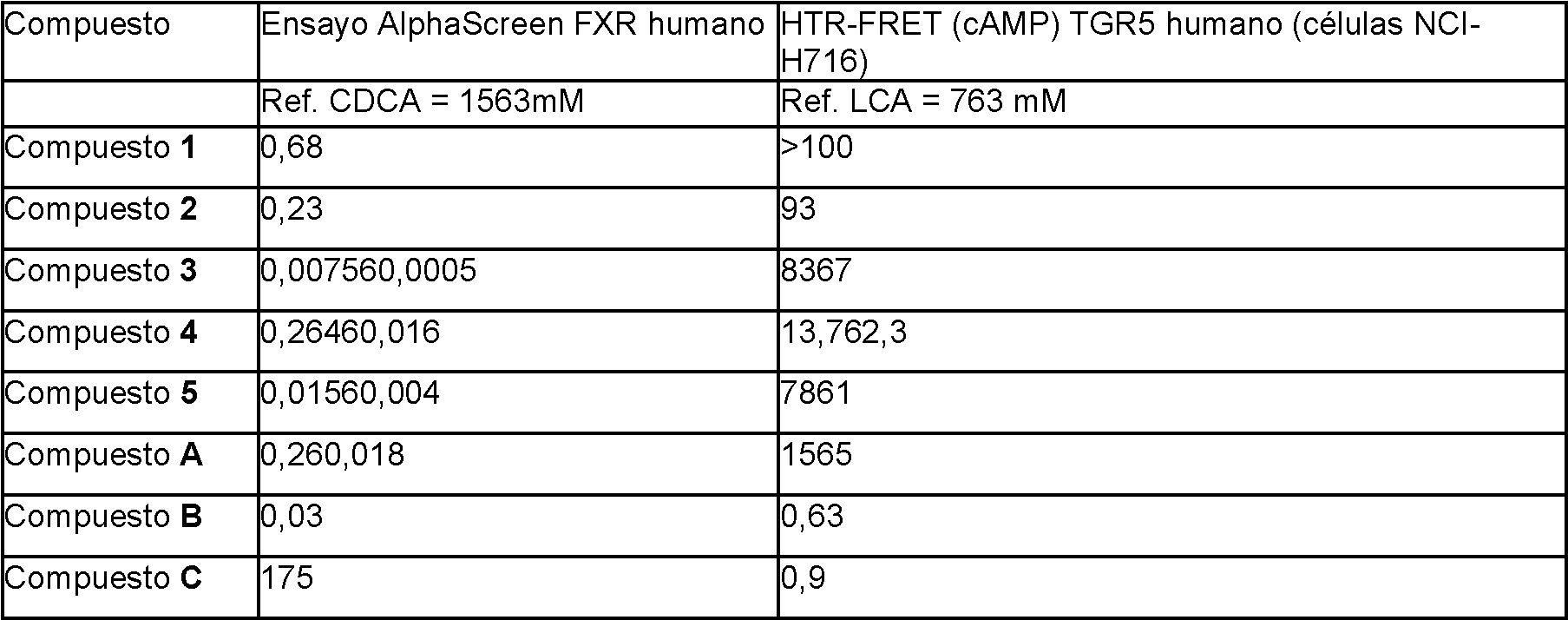
Tabla 2. Actividad FXR / TGR5 de los compuestos L1, 3, 5, 6 y 7

Tabla 3. Actividad agonista del FXR en los ortólogos humanos, de ratón, de rata y de perro


Tabla 4. Actividad TGR5 entre especies

Ejemplo 9. Perfil de selectividad de receptores nucleares
Utilizando el ensayo AlphaScreen, puede evaluarse la selectividad de un compuesto de la invención contra los siguientes receptores nucleares implicados en las vías metabólicas: LXRp, PXR, CAR, PPARa, PPAP8, PPAPy , RAR, RARa, VDR, TR, PR, RXR, GR y ER.
Los compuestos L1, 3, 5, 6 y 7 se probaron contra el panel de receptores nucleares disponibles tanto en modo agonista como antagonista. Ninguno de los compuestos activó ninguno de los receptores en modo agonista (respuesta de dosis a 200 mM) o antagonista (concentración fija a 10 mM).
Ejemplo 10. Panel de genes diana FXR
Para evaluar la capacidad de un compuesto de la invención para modular los genes diana del FXR, se realizan ensayos cuantitativos de RT-PCR. Se seleccionan células HepG2 como línea celular relevante para determinar si un compuesto de la invención puede regular la red genética FXR endógena. La capacidad de un compuesto de la invención para inducir los genes diana del FXR se evalúa aislando el ARN total de las células tratadas durante la noche con 1 mM de los compuestos A, B y un compuesto de la invención. El compuesto A se establece como un potente agonista selectivo de FXR y el compuesto B se establece como un potente agonista dual de FXR/TGR5.
El FXR regula la expresión de varios genes diana implicados en la homeostasis del BA. Brevemente, el FXR desempeña un papel central en varias vías metabólicas, incluyendo, por ejemplo, el metabolismo de los lípidos, el metabolismo de los ácidos biliares y el metabolismo de los carbohidratos. En lo que respecta al perfil de expresión génica, los genes que codifican las proteínas implicadas en el metabolismo de los lípidos incluyen, por ejemplo, APOCII, APOE, APOAI, SREBP-1C, VLDL-R, PLTP y LPL; los genes que codifican las proteínas implicadas en el metabolismo de los ácidos biliares incluyen, por ejemplo, OSTa /p, BSEP, MRP2, SHP, CYP7A1, FGF19, SULT2A1 y UGT2B4; y los genes que codifican proteínas implicadas en el metabolismo de los carbohidratos incluyen, por ejemplo, PGCla, PEPCK y GLUT2. Genes diana del FXR: BSEP, SHP, OSTa y CYP7A1 se evaluaron tras la estimulación de los compuestos 3, 5, 6 y 7 en células HepG2 durante 18 horas. El compuesto L1 se utilizó como control. Los compuestos 3, 5, 6 y 7 se unen significativamente al FXR en las células hepáticas modulando los genes diana del FXR.
Ejemplo 11. Citotoxicidad in vitro
Para evaluar la citotoxicidad in vitro de un compuesto de la invención, se emplean dos métodos de ensayo diferentes. Los ensayos evalúan la viabilidad celular midiendo los niveles de ATP y la citotoxicidad midiendo la liberación de LDH. El nucleótido trifosfato de adenosina (ATP) representa la fuente de energía a nivel molecular básico, ya que es una molécula multifuncional que se utiliza en todas las células como coenzima y forma parte integral del ADN mitocondrial (Kangas, et al., Medical Biology, 1984, 62,
338-343; Crouch, et al., J Immunol. Methods, 1993, 160, 81-88; y Petty, et al., J Biolumin. Chemilumin. 1995, 10, 29-34). Se le ha llamado la "unidad molecular de moneda" cuando se trata de la transferencia de energía intracelular. Esto es para asegurar el importante papel del ATP en el metabolismo y una caída en el contenido de ATP es el primer paso para revelar el daño celular (Storer, et al., Mutation Research, 1996, 368, 59-101; y Cree y Andreotti, Toxicology In-Vitro, 1997, 11, 553-556).
Un método adicional para determinar la viabilidad de las células es detectar la integridad de la membrana que define la
compartimentación celular. La medición de la fuga de componentes fuera del citoplasma, en membranas celulares dañadas, indica la pérdida de la integridad de la membrana, y la liberación de LDH es el método utilizado para determinar la toxicidad común en las células. Las células HepG2 se tratan con un compuesto de la invención y se realizan diluciones en serie. Se añaden diluciones de LCA a las células chapadas como controles del ensayo, junto con células sin tratar y células sin tratar. El ensayo se realiza por triplicado para cada concentración del compuesto de prueba.
La viabilidad celular se determinó como una medida de ATP intracelular relacionada con el tiempo de exposición y la concentración de los compuestos de prueba (Sussman, Promega Cell Notes, número 3, 2002). Los datos se proporcionan en las Tablas 5a y 5B.
Tabla 5A. Citotoxicidad in vitro de los compuestos 3 y 5

Tabla 5B. Citotoxicidad in vitro de los compuestos 6 y 7 (liberación de LDH y contenido de ATP en relación con los compuestos 6 y 7 estimulación en HepG2.

El tamoxifeno se utilizó como control positivo del ensayo y el LCA se utilizó como referencia.
Ejemplo 12. Cribado del CYP450
Para evaluar el potencial de un compuesto de la invención para las interacciones farmacológicas, se investigan las seis isoformas principales del CYP450 (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4) (Obach, et al., J Pharmacol. Exp. Ther, 2006, 316, 336-348).
Para determinar la interacción entre un compuesto de la invención y las enzimas del citocromo P450, el compuesto de la invención se analiza por su capacidad de inhibir (o no) la producción de una señal fluorescente, utilizando proteínas CYP450 recombinantes (baculosomas; Invitrogen), sustratos e inhibidores (Bidstrup, et al., Br J Clin. Pharmacol, 2003, 56, 305-14). Como control positivo, se prueba un inhibidor selectivo para cada isoforma del CYP450 en la misma placa. Tabla 6. Inhibición de CYP450s (prueba contra las 6 isozimas principales)


Ejemplo 13. Canal de potasio humano ERG
Para determinar la función del canal iónico, se emplea el ensayo Predictor™ hERG Fluorescence Polarization, ya que proporciona un método eficiente para una determinación inicial de la propensión de los compuestos de prueba a bloquear el canal hERG (Dorn, et al. J Biomol. Screen, 2005, 10, 339-347). El ensayo se basa en la suposición de que la actividad del canal de potasio hERG contribuye al potencial de membrana en reposo en las células transfectadas permanentemente y, por tanto, un bloqueo de los canales hERG debería dar lugar a una despolarización de la membrana celular. El ensayo está diseñado para identificar potenciales bloqueadores del canal hERG produciendo datos que se correlacionan con precisión con los estudios de electrofisiología patch-clamp. Los resultados del ensayo Predictor™ demuestran una alta correlación con los obtenidos mediante técnicas de patch clamp (Dorn, et al. J Biomol Screen, 2005, 10, 339-347).
Se utilizan preparaciones de membrana de células de ovario de hámster chino transfectadas de forma estable con el canal de potasio hERG para evaluar el efecto inhibidor potencial de un compuesto de la invención sobre este canal utilizando el ensayo de polarización de fluorescencia Predictor™. La reducción de la polarización de la membrana como resultado de la inhibición del canal de potasio hERG se correlaciona directamente con una reducción de la polarización de fluorescencia (PF).
El ensayo se realiza por triplicado utilizando una dosis-respuesta de 16 puntos del compuesto de prueba y los controles positivos E-4031 y Tamoxifeno. Se obtiene un IC50 de 15 nM (AmP = 163) para el E-4031 y 1,4 mM (AP = 183) para el Tamoxifeno. Una ventana de ensayo superior a 100 mP (milipolarización) se considera buena. Las curvas de regresión no lineal se obtienen mediante el análisis de GraphPad Prism (GraphPad Software Inc.), para calcular los valores IC50. Tabla 7. Inhibición del canal de potasio del ERG humano

Los compuestos L1, 3, 5, 6 y 7 no inhibieron el canal de potasio hERG.
Ejemplo 14. Propiedades fisioquímicas
Las propiedades fisioquímicas de un compuesto de la invención, como la solubilidad en agua, la concentración micelar crítica, la tensión superficial y el LogPA, se determinaron mediante métodos conocidos en la técnica. Los datos se proporcionan en la Tabla 8.
Tabla 8. Propiedades fisicoquímicas
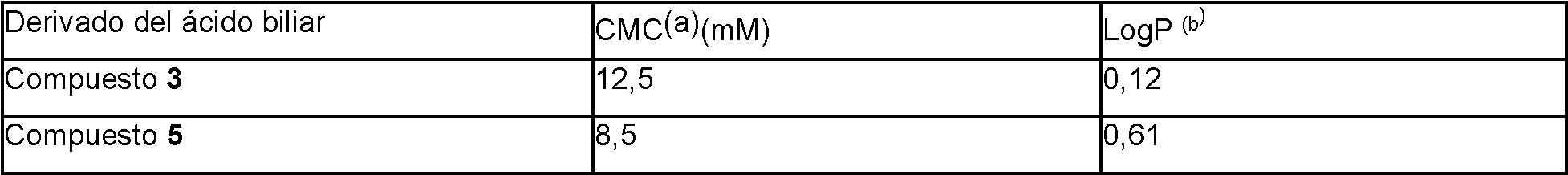

Claims (8)
1. Un compuesto de fórmula le:

o una sal o un solvato farmacéuticamente aceptable del mismo, deonde R2 y R5 son OH; R4 es C1-C6 alquilo opcionalmente sustituido con uno o más halógenos u OH;
R7 es OH, OSO3H, SO3H, tetrazolilo, oxadiazolilo, tiadiazolilo, 5-oxo-1,2,4-oxadiazolilo, 5-oxo-1,2,4-tiadiazolilo, oxazolidinadionilo, tiazolidinadionilo, 3-hidroxiisoxazolilo, 3-hidroxiisotiazolilo o 2,4-difluoro-3-hidroxifenilo; R8, R9 y R10 son cada uno independientemente H, OH, halógeno o alquilo opcionalmente sustituido con uno o más halógenos u OH, o R8 y R9 tomados junto con los átomos de carbono a los que están unidos forman un anillo carbocíclico o heterocíclico de 3 a 6 miembros que comprende 1 o 2 heteroátomos seleccionados entre N O, y S, o R9 y R10 tomados junto con los átomos de carbono a los que están unidos forman un anillo carbocíclico o heterocíclico de 3 a 6 miembros que comprende 1 o 2 heteroátomos seleccionados entre N, O, y S;
m es 0, 1 o 2;
n es 0 o 1;
p es 0 o 1; y
= es un enlace simple o doble.
2. El compuesto de la reivindicación 1, donde R7 es OH.
3. El compuesto de la reivindicación 1, donde R7 es tetrazolilo, oxadiazolilo, tiadiazolilo, 5-oxo-1,2,4-oxadiazolilo, 5-oxo- 1,2,4-tiadiazolilo, oxazolidina-dionilo, tiazolidina-dionilo, 3-hidroxiisoxazolilo, 3-hidroxiisotiazolilo o 2,4-difluoro-3-hidroxifenilo.
4. El compuesto de cualquiera de las reivindicaciones 1 a 3, donde R4 es metilo, etilo o propilo, y donde R4 está en la posición a.
5. El compuesto de la reivindicación 1, donde el compuesto se selecciona entre:

o en el que el compuesto se selecciona entre:
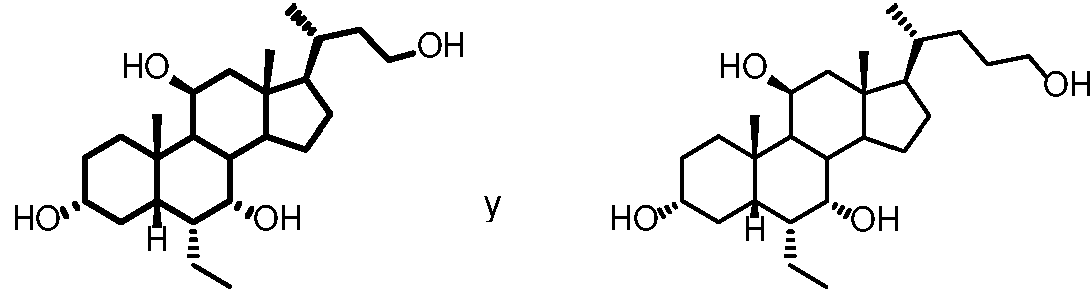
6. Una composición farmacéutica que comprende el compuesto de cualquiera de las reivindicaciones 1-5 y un portador o excipiente farmacéuticamente aceptable.
7. Un compuesto de cualquiera de las reivindicaciones 1-5 o una sal o solvato farmacéuticamente aceptable del mismo, para usarse en un método de tratamiento o prevención de una enfermedad o condición en un sujeto que lo necesite, donde la enfermedad o condición está mediada por el FXR.
8. El compuesto para usarse de la reivindicación 7, donde:
(i) la enfermedad se selecciona entre las enfermedades cardiovasculares, las enfermedades hepáticas crónicas, los trastornos lipídicos, las enfermedades gastrointestinales, las enfermedades renales, las enfermedades metabólicas, el cáncer y las enfermedades neurológicas; o
(ii) la enfermedad es una enfermedad hepática crónica seleccionada entre la cirrosis biliar primaria (CBP), la xantomatosis cerebrotendinosa (CTX), la colangitis esclerosante primaria (CEP), la colestasis inducida por fármacos, la colestasis intrahepática del embarazo, la colestasis asociada a la nutrición parenteral (CPN), el sobrecrecimiento bacteriano o la colestasis asociada a la sepsis, la hepatitis autoinmune, la hepatitis viral crónica, la enfermedad hepática alcohólica la enfermedad del hígado graso no alcohólico (NAFLD), la esteatohepatitis no alcohólica (NASH), la enfermedad de injerto contra huésped asociada al trasplante de hígado, la regeneración del hígado en el trasplante de donante vivo, la fibrosis hepática congénita, la coledocolitiasis, la enfermedad hepática granulomatosa, la neoplasia intra o extrahepática, el síndrome de Sjogren, la sarcoidosis, la enfermedad de Wilson, la enfermedad de Gaucher, la hemocromatosis y el déficit de alfa-1-antitripsina.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562238246P | 2015-10-07 | 2015-10-07 | |
| PCT/US2016/055980 WO2017062763A1 (en) | 2015-10-07 | 2016-10-07 | Farnesoid x receptor modulators |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2898705T3 true ES2898705T3 (es) | 2022-03-08 |
Family
ID=58488545
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16854420T Active ES2898705T3 (es) | 2015-10-07 | 2016-10-07 | Moduladores de los receptores farnesoides x |
Country Status (26)
| Country | Link |
|---|---|
| US (3) | US11034717B2 (es) |
| EP (2) | EP3981779A1 (es) |
| JP (2) | JP7021080B2 (es) |
| KR (2) | KR20180061351A (es) |
| CN (1) | CN108348530B (es) |
| AR (1) | AR106293A1 (es) |
| AU (1) | AU2016335765B2 (es) |
| BR (1) | BR112018006892B1 (es) |
| CA (1) | CA3000881C (es) |
| CL (1) | CL2018000883A1 (es) |
| CO (1) | CO2018003728A2 (es) |
| CR (1) | CR20180201A (es) |
| DK (1) | DK3359160T3 (es) |
| EA (1) | EA038665B1 (es) |
| EC (1) | ECSP18030284A (es) |
| ES (1) | ES2898705T3 (es) |
| IL (1) | IL258359B2 (es) |
| MA (2) | MA45761A (es) |
| MX (1) | MX387172B (es) |
| PH (1) | PH12018500777A1 (es) |
| SV (1) | SV2018005662A (es) |
| TN (1) | TN2018000106A1 (es) |
| TW (1) | TW201718621A (es) |
| UA (1) | UA123948C2 (es) |
| WO (1) | WO2017062763A1 (es) |
| ZA (1) | ZA201802901B (es) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016073767A1 (en) | 2014-11-06 | 2016-05-12 | Enanta Pharmaceuticals, Inc. | Bile acid analogs an fxr/tgr5 agonists and methods of use thereof |
| US11578097B2 (en) | 2014-11-26 | 2023-02-14 | Enanta Pharmaceuticals, Inc. | Tetrazole derivatives of bile acids as FXR/TGR5 agonists and methods of use thereof |
| JP2017535570A (ja) | 2014-11-26 | 2017-11-30 | エナンタ ファーマシューティカルズ インコーポレイテッド | Fxr/tgr5アゴニストとしての胆汁酸類似体およびその使用方法 |
| US10208081B2 (en) | 2014-11-26 | 2019-02-19 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as FXR/TGR5 agonists and methods of use thereof |
| MX2017010376A (es) | 2015-02-11 | 2017-12-20 | Enanta Pharm Inc | Análogos de ácido biliar como agonistas de fxr/tgr5 y métodos para el uso de los mismos. |
| CA2981503C (en) | 2015-03-31 | 2023-09-19 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as fxr/tgr5 agonists and methods of use thereof |
| EP3981779A1 (en) | 2015-10-07 | 2022-04-13 | Intercept Pharmaceuticals, Inc. | Farnesoid x receptor modulators |
| WO2017147159A1 (en) | 2016-02-23 | 2017-08-31 | Enanta Pharmaceuticals, Inc. | Deuterated bile acid derivatives as fxr/tgr5 agonists and methods of use thereof |
| WO2017147174A1 (en) | 2016-02-23 | 2017-08-31 | Enanta Pharmaceuticals, Inc. | Heteroaryl containing bile acid analogs as fxr/tgr5 agonists and methods of use thereof |
| US10323060B2 (en) | 2016-02-23 | 2019-06-18 | Enanta Pharmaceuticals, Inc. | Benzoic acid derivatives of bile acid as FXR/TGR5 agonists and methods of use thereof |
| CA3045023A1 (en) | 2016-11-29 | 2018-06-07 | Enanta Pharmaceuticals, Inc. | Process for preparation of sulfonylurea bile acid derivatives |
| US10472386B2 (en) | 2017-02-14 | 2019-11-12 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as FXR agonists and methods of use thereof |
| CA3058754A1 (en) | 2017-04-07 | 2018-10-11 | Enanta Pharmaceuticals, Inc. | Process for preparation of sulfonyl carbamate bile acid derivatives |
| JP7224307B2 (ja) * | 2017-06-23 | 2023-02-17 | インターセプト ファーマシューティカルズ, インコーポレイテッド | 胆汁酸誘導体の調製のための方法及び中間体 |
| WO2019023103A1 (en) | 2017-07-24 | 2019-01-31 | Intercept Pharmaceuticals, Inc. | BILIARY ACID DERIVATIVES WITH ISOTOPIC MARKING |
| US11028111B2 (en) | 2017-12-19 | 2021-06-08 | Xi' An Biocare Pharma Ltd. | Compound for treating metabolic diseases and preparation method and use thereof |
| GB201812382D0 (en) | 2018-07-30 | 2018-09-12 | Nzp Uk Ltd | Compounds |
| SG11202113155XA (en) | 2019-05-30 | 2021-12-30 | Intercept Pharmaceuticals Inc | Pharmaceutical compositions comprising a fxr agonist and a fibrate for use in the treatment of cholestatic liver disease |
| WO2023288123A1 (en) | 2021-07-16 | 2023-01-19 | Interecept Pharmaceuticals, Inc. | Methods and intermediates for the preparation of 3.alpha.,7.alpha.,11.beta.-trihydroxy-6.alpha.-ethyl-5.beta.-cholan-24-oic acid |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2599481A (en) | 1948-09-28 | 1952-06-03 | Ciba Pharm Prod Inc | Reduction of steroid epoxides |
| DE3366932D1 (en) * | 1982-07-29 | 1986-11-20 | Lehner Ag | New derivatives of biliary acids, process for the production thereof and pharmaceutical compositions containing the same |
| IT1223313B (it) | 1987-10-20 | 1990-09-19 | Gipharmex Spa | Derivati di acidi biliari loro preparazione e composizioni farmaceutiche che li contengono |
| DE10113257C1 (de) | 2001-03-19 | 2002-11-14 | Inst Mikrotechnik Mainz Gmbh | Elektrophoresevorrichtung und ihre Verwendung |
| US20070197484A1 (en) | 2001-05-03 | 2007-08-23 | Ching Song | Method of treating disorder related to high cholesterol concentration |
| PT2040713E (pt) * | 2006-06-27 | 2014-10-13 | Intercept Pharmaceuticals Inc | Para a prevenção ou o tratamento de doenças ou estados clínicos mediados por fxr |
| ES2663948T3 (es) * | 2008-11-19 | 2018-04-17 | Intercept Pharmaceuticals, Inc. | Moduladores de TGR5 y método de uso de los mismos |
| NZ734451A (en) | 2012-06-19 | 2018-12-21 | Intercept Pharmaceuticals Inc | Preparation, uses and solid forms of obeticholic acid |
| SG11201503697TA (en) * | 2012-11-28 | 2015-06-29 | Intercept Pharmaceuticals Inc | Treatment of pulmonary disease |
| CA2912139C (en) | 2013-05-14 | 2021-04-20 | Roberto Pellicciari | 11-hydroxyl-derivatives of bile acids and amino acid conjugates thereof as farnesoid x receptor modulators |
| US9351083B2 (en) | 2013-10-17 | 2016-05-24 | Turtle Beach Corporation | Transparent parametric emitter |
| JP2017535570A (ja) * | 2014-11-26 | 2017-11-30 | エナンタ ファーマシューティカルズ インコーポレイテッド | Fxr/tgr5アゴニストとしての胆汁酸類似体およびその使用方法 |
| US11578097B2 (en) * | 2014-11-26 | 2023-02-14 | Enanta Pharmaceuticals, Inc. | Tetrazole derivatives of bile acids as FXR/TGR5 agonists and methods of use thereof |
| EP3358955B1 (en) | 2015-10-07 | 2026-04-15 | Corteva Agriscience LLC | Managing ethylene in plants using an agricultural formula comprising diformyl urea and cobalt sulfate |
| EP3981779A1 (en) * | 2015-10-07 | 2022-04-13 | Intercept Pharmaceuticals, Inc. | Farnesoid x receptor modulators |
| EP3426348B1 (en) | 2016-03-11 | 2021-05-05 | Intercept Pharmaceuticals, Inc. | 3-desoxy derivative and pharmaceutical compositions thereof |
| JP7224307B2 (ja) | 2017-06-23 | 2023-02-17 | インターセプト ファーマシューティカルズ, インコーポレイテッド | 胆汁酸誘導体の調製のための方法及び中間体 |
| SG11202113155XA (en) | 2019-05-30 | 2021-12-30 | Intercept Pharmaceuticals Inc | Pharmaceutical compositions comprising a fxr agonist and a fibrate for use in the treatment of cholestatic liver disease |
-
2016
- 2016-10-07 EP EP21192609.2A patent/EP3981779A1/en not_active Withdrawn
- 2016-10-07 CA CA3000881A patent/CA3000881C/en active Active
- 2016-10-07 TW TW105132613A patent/TW201718621A/zh unknown
- 2016-10-07 TN TNP/2018/000106A patent/TN2018000106A1/en unknown
- 2016-10-07 WO PCT/US2016/055980 patent/WO2017062763A1/en not_active Ceased
- 2016-10-07 CR CR20180201A patent/CR20180201A/es unknown
- 2016-10-07 ES ES16854420T patent/ES2898705T3/es active Active
- 2016-10-07 UA UAA201804942A patent/UA123948C2/uk unknown
- 2016-10-07 EA EA201890899A patent/EA038665B1/ru unknown
- 2016-10-07 KR KR1020187012652A patent/KR20180061351A/ko not_active Ceased
- 2016-10-07 CN CN201680063616.0A patent/CN108348530B/zh active Active
- 2016-10-07 KR KR1020257025658A patent/KR20250120445A/ko active Pending
- 2016-10-07 BR BR112018006892-6A patent/BR112018006892B1/pt active IP Right Grant
- 2016-10-07 IL IL258359A patent/IL258359B2/en unknown
- 2016-10-07 US US15/288,390 patent/US11034717B2/en active Active
- 2016-10-07 AU AU2016335765A patent/AU2016335765B2/en active Active
- 2016-10-07 MX MX2018004186A patent/MX387172B/es unknown
- 2016-10-07 AR ARP160103074A patent/AR106293A1/es unknown
- 2016-10-07 MA MA045761A patent/MA45761A/fr unknown
- 2016-10-07 MA MA42339A patent/MA42339A1/fr unknown
- 2016-10-07 JP JP2018517509A patent/JP7021080B2/ja active Active
- 2016-10-07 EP EP16854420.3A patent/EP3359160B1/en active Active
- 2016-10-07 DK DK16854420.3T patent/DK3359160T3/da active
-
2018
- 2018-04-05 SV SV2018005662A patent/SV2018005662A/es unknown
- 2018-04-05 CL CL2018000883A patent/CL2018000883A1/es unknown
- 2018-04-06 CO CONC2018/0003728A patent/CO2018003728A2/es unknown
- 2018-04-10 PH PH12018500777A patent/PH12018500777A1/en unknown
- 2018-04-19 EC ECIEPI201830284A patent/ECSP18030284A/es unknown
- 2018-05-03 ZA ZA2018/02901A patent/ZA201802901B/en unknown
-
2019
- 2019-09-20 JP JP2019171488A patent/JP2019210297A/ja active Pending
-
2021
- 2021-05-14 US US17/320,286 patent/US12291549B2/en active Active
-
2025
- 2025-05-05 US US19/198,431 patent/US20250376488A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2898705T3 (es) | Moduladores de los receptores farnesoides x | |
| US11319337B2 (en) | 3-desoxy derivative and pharmaceutical compositions thereof | |
| ES2936638T3 (es) | Derivados sustituidos con 11-Hidroxil-6 de ácidos biliares y conjugados de aminoácidos de los mismos como moduladores del receptor X farnesoide | |
| HK1253558B (zh) | 法尼醇x受体调节剂 | |
| HK1259145B (zh) | 3-脱氧衍生物及其药物组合物 |