ES2868973T3 - Derivados de cromano, isocromano y dihidroisobenzofurano como moduladores alostéricos negativos para mGluR2, composiciones y su uso - Google Patents
Derivados de cromano, isocromano y dihidroisobenzofurano como moduladores alostéricos negativos para mGluR2, composiciones y su uso Download PDFInfo
- Publication number
- ES2868973T3 ES2868973T3 ES17781253T ES17781253T ES2868973T3 ES 2868973 T3 ES2868973 T3 ES 2868973T3 ES 17781253 T ES17781253 T ES 17781253T ES 17781253 T ES17781253 T ES 17781253T ES 2868973 T3 ES2868973 T3 ES 2868973T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- mmol
- pharmaceutically acceptable
- methyl
- acceptable salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 COC(C=CC(*1)=CC=C1C(OC)=O)=O Chemical compound COC(C=CC(*1)=CC=C1C(OC)=O)=O 0.000 description 11
- DBSSGNLSYIUMOU-UHFFFAOYSA-N CC(C)(Cc(c(Cl)nc(Cl)c1)c1-c1ccc(C)cc1F)C(OC)=S Chemical compound CC(C)(Cc(c(Cl)nc(Cl)c1)c1-c1ccc(C)cc1F)C(OC)=S DBSSGNLSYIUMOU-UHFFFAOYSA-N 0.000 description 1
- CFABKBDXTPTREX-UHFFFAOYSA-N CC1(C)Oc2nc(C(N)=O)cc(-c3ccc(C)cc3F)c2CC1 Chemical compound CC1(C)Oc2nc(C(N)=O)cc(-c3ccc(C)cc3F)c2CC1 CFABKBDXTPTREX-UHFFFAOYSA-N 0.000 description 1
- CKJDSBFSYISFEF-UHFFFAOYSA-N CCC(CCC1C(C2=CCC(C)C=C2F)=C2)OC1N=C2C(OCC)=O Chemical compound CCC(CCC1C(C2=CCC(C)C=C2F)=C2)OC1N=C2C(OCC)=O CKJDSBFSYISFEF-UHFFFAOYSA-N 0.000 description 1
- DYCVWGSPDDQCNY-UHFFFAOYSA-N CCCC(c1cc(-c2ccc(C)cc2F)c(CCC(CO)O2)c2n1)O Chemical compound CCCC(c1cc(-c2ccc(C)cc2F)c(CCC(CO)O2)c2n1)O DYCVWGSPDDQCNY-UHFFFAOYSA-N 0.000 description 1
- XFPPFEQSDYZIFV-AWEZNQCLSA-N CCOC(c1cc(-c(c(F)c2)ccc2C(C)=C)c(CC[C@@H](COC(F)F)O2)c2n1)=O Chemical compound CCOC(c1cc(-c(c(F)c2)ccc2C(C)=C)c(CC[C@@H](COC(F)F)O2)c2n1)=O XFPPFEQSDYZIFV-AWEZNQCLSA-N 0.000 description 1
- XFPPFEQSDYZIFV-CQSZACIVSA-N CCOC(c1cc(-c(c(F)c2)ccc2C(C)=C)c(CC[C@H](COC(F)F)O2)c2n1)=O Chemical compound CCOC(c1cc(-c(c(F)c2)ccc2C(C)=C)c(CC[C@H](COC(F)F)O2)c2n1)=O XFPPFEQSDYZIFV-CQSZACIVSA-N 0.000 description 1
- CKSMVDUGKQFNKG-LBPRGKRZSA-N CCOC(c1cc(-c(ccc(C)c2)c2F)c(CC[C@@H](CF)O2)c2n1)=O Chemical compound CCOC(c1cc(-c(ccc(C)c2)c2F)c(CC[C@@H](CF)O2)c2n1)=O CKSMVDUGKQFNKG-LBPRGKRZSA-N 0.000 description 1
- HOSIDUMEDHWUNL-LJQANCHMSA-N CCOC(c1cc(-c(ccc(C)c2)c2F)c(CC[C@H](C(C)C)O2)c2n1)=O Chemical compound CCOC(c1cc(-c(ccc(C)c2)c2F)c(CC[C@H](C(C)C)O2)c2n1)=O HOSIDUMEDHWUNL-LJQANCHMSA-N 0.000 description 1
- HCJPZVDTYQAPRN-UHFFFAOYSA-N CCOC(c1cc(-c2ccc(C)cc2F)c(CC2OC3CC2)c3n1)=O Chemical compound CCOC(c1cc(-c2ccc(C)cc2F)c(CC2OC3CC2)c3n1)=O HCJPZVDTYQAPRN-UHFFFAOYSA-N 0.000 description 1
- HOSIDUMEDHWUNL-UHFFFAOYSA-N CCOC(c1cc(-c2ccc(C)cc2F)c(CCC(C(C)C)O2)c2n1)=O Chemical compound CCOC(c1cc(-c2ccc(C)cc2F)c(CCC(C(C)C)O2)c2n1)=O HOSIDUMEDHWUNL-UHFFFAOYSA-N 0.000 description 1
- CKSMVDUGKQFNKG-UHFFFAOYSA-N CCOC(c1cc(-c2ccc(C)cc2F)c(CCC(CF)O2)c2n1)=O Chemical compound CCOC(c1cc(-c2ccc(C)cc2F)c(CCC(CF)O2)c2n1)=O CKSMVDUGKQFNKG-UHFFFAOYSA-N 0.000 description 1
- KUBQWBDVPGJCSH-UHFFFAOYSA-N CCOC(c1cc(-c2ccc(C)cc2F)c(CCOC2)c2n1)=O Chemical compound CCOC(c1cc(-c2ccc(C)cc2F)c(CCOC2)c2n1)=O KUBQWBDVPGJCSH-UHFFFAOYSA-N 0.000 description 1
- HOSIDUMEDHWUNL-IBGZPJMESA-N CCOC(c1cc(-c2ccc(C)cc2F)c(CC[C@@H](C(C)C)O2)c2n1)=O Chemical compound CCOC(c1cc(-c2ccc(C)cc2F)c(CC[C@@H](C(C)C)O2)c2n1)=O HOSIDUMEDHWUNL-IBGZPJMESA-N 0.000 description 1
- SEYXBXCGOZPXEU-AWEZNQCLSA-N CCOC(c1cc(-c2ccc(C)cc2F)c(CC[C@@H](CC(C=C)(F)F)O2)c2n1)=O Chemical compound CCOC(c1cc(-c2ccc(C)cc2F)c(CC[C@@H](CC(C=C)(F)F)O2)c2n1)=O SEYXBXCGOZPXEU-AWEZNQCLSA-N 0.000 description 1
- SEYXBXCGOZPXEU-CQSZACIVSA-N CCOC(c1cc(-c2ccc(C)cc2F)c(CC[C@H](CC(C=C)(F)F)O2)c2n1)=O Chemical compound CCOC(c1cc(-c2ccc(C)cc2F)c(CC[C@H](CC(C=C)(F)F)O2)c2n1)=O SEYXBXCGOZPXEU-CQSZACIVSA-N 0.000 description 1
- CKSMVDUGKQFNKG-GFCCVEGCSA-N CCOC(c1cc(-c2ccc(C)cc2F)c(CC[C@H](CF)O2)c2n1)=O Chemical compound CCOC(c1cc(-c2ccc(C)cc2F)c(CC[C@H](CF)O2)c2n1)=O CKSMVDUGKQFNKG-GFCCVEGCSA-N 0.000 description 1
- SNALJBBCQBAUMG-UHFFFAOYSA-N CCOC(c1cc(Cl)c(CC2OC3CC2)c3n1)=O Chemical compound CCOC(c1cc(Cl)c(CC2OC3CC2)c3n1)=O SNALJBBCQBAUMG-UHFFFAOYSA-N 0.000 description 1
- NFGUKPADLCRMFA-UHFFFAOYSA-N CCOC(c1cc(Cl)c(CCOC2)c2n1)=O Chemical compound CCOC(c1cc(Cl)c(CCOC2)c2n1)=O NFGUKPADLCRMFA-UHFFFAOYSA-N 0.000 description 1
- WCZGYNLJAJNBCP-UHFFFAOYSA-N CC[O](C)C(C(C=[O](c1ccccc1)(c1ccccc1)c1ccccc1)=O)=O Chemical compound CC[O](C)C(C(C=[O](c1ccccc1)(c1ccccc1)c1ccccc1)=O)=O WCZGYNLJAJNBCP-UHFFFAOYSA-N 0.000 description 1
- VVFAQTMPKQNIGL-UHFFFAOYSA-N CCc1cc(N)c(C)cc1 Chemical compound CCc1cc(N)c(C)cc1 VVFAQTMPKQNIGL-UHFFFAOYSA-N 0.000 description 1
- WPTCQEANIFJPSA-NSHDSACASA-N CCc1ccc(-c2c(CC[C@@H](COC(F)F)O3)c3nc(C(N)=O)c2)c(F)c1 Chemical compound CCc1ccc(-c2c(CC[C@@H](COC(F)F)O3)c3nc(C(N)=O)c2)c(F)c1 WPTCQEANIFJPSA-NSHDSACASA-N 0.000 description 1
- WPTCQEANIFJPSA-LLVKDONJSA-N CCc1ccc(-c2c(CC[C@H](COC(F)F)O3)c3nc(C(N)=O)c2)c(F)c1 Chemical compound CCc1ccc(-c2c(CC[C@H](COC(F)F)O3)c3nc(C(N)=O)c2)c(F)c1 WPTCQEANIFJPSA-LLVKDONJSA-N 0.000 description 1
- FBPIDMAELBIRLE-UHFFFAOYSA-N COC(c([o]1)ccc1Br)=O Chemical compound COC(c([o]1)ccc1Br)=O FBPIDMAELBIRLE-UHFFFAOYSA-N 0.000 description 1
- YRSVVIITZJGBMH-LBPRGKRZSA-N Cc1ccc(-c2c(CC[C@@H](CC(C=C)(F)F)O3)c3nc(C(N)=O)c2)c(F)c1 Chemical compound Cc1ccc(-c2c(CC[C@@H](CC(C=C)(F)F)O3)c3nc(C(N)=O)c2)c(F)c1 YRSVVIITZJGBMH-LBPRGKRZSA-N 0.000 description 1
- YRSVVIITZJGBMH-GFCCVEGCSA-N Cc1ccc(-c2c(CC[C@H](CC(C=C)(F)F)O3)c3nc(C(N)=O)c2)c(F)c1 Chemical compound Cc1ccc(-c2c(CC[C@H](CC(C=C)(F)F)O3)c3nc(C(N)=O)c2)c(F)c1 YRSVVIITZJGBMH-GFCCVEGCSA-N 0.000 description 1
- LIXXGOMAGHXIMP-UHFFFAOYSA-N Cc1ccc(B(O)O)c(F)c1 Chemical compound Cc1ccc(B(O)O)c(F)c1 LIXXGOMAGHXIMP-UHFFFAOYSA-N 0.000 description 1
- RGCKJSPKMTWLLX-UHFFFAOYSA-N ICCCc1ccccc1 Chemical compound ICCCc1ccccc1 RGCKJSPKMTWLLX-UHFFFAOYSA-N 0.000 description 1
- ORCJOJBWARLEKN-ZDUSSCGKSA-N NC(c1cc(-c(c(F)c2)ccc2F)c(CC[C@@H](C(F)F)O2)c2n1)=O Chemical compound NC(c1cc(-c(c(F)c2)ccc2F)c(CC[C@@H](C(F)F)O2)c2n1)=O ORCJOJBWARLEKN-ZDUSSCGKSA-N 0.000 description 1
- ORCJOJBWARLEKN-CYBMUJFWSA-N NC(c1cc(-c(ccc(F)c2)c2F)c(CC[C@H](C(F)F)O2)c2n1)=O Chemical compound NC(c1cc(-c(ccc(F)c2)c2F)c(CC[C@H](C(F)F)O2)c2n1)=O ORCJOJBWARLEKN-CYBMUJFWSA-N 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N O=C1NCCC1 Chemical compound O=C1NCCC1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- LKYPXFBYDCHHFN-UHFFFAOYSA-N O=Cc(c(Cl)nc(Cl)c1)c1-c(ccc(F)c1)c1F Chemical compound O=Cc(c(Cl)nc(Cl)c1)c1-c(ccc(F)c1)c1F LKYPXFBYDCHHFN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/18—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/01—Hydrocarbons
- A61K31/015—Hydrocarbons carbocyclic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/02—Halogenated hydrocarbons
- A61K31/025—Halogenated hydrocarbons carbocyclic
- A61K31/03—Halogenated hydrocarbons carbocyclic aromatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/166—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the carbon of a carboxamide group directly attached to the aromatic ring, e.g. procainamide, procarbazine, metoclopramide, labetalol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/439—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom the ring forming part of a bridged ring system, e.g. quinuclidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/22—Bridged ring systems
- C07D221/24—Camphidines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Psychiatry (AREA)
- Hospice & Palliative Care (AREA)
- Pain & Pain Management (AREA)
- Anesthesiology (AREA)
- Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Medicines Containing Plant Substances (AREA)
Abstract
Un compuesto de Fórmula (I): **(Ver fórmula)** o un estereoisómero del mismo o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde: el anillo A es un resto seleccionado entre: **(Ver fórmula)** en donde: R2 se selecciona entre H, ciclopropilo, alquilo (C1-C4), -alquil (C1-C4)-OH, -alquil (C1-C4)-OCH3, -haloalquilo (C1C4), -alquil (C1-C4)-O-haloalquilo (C1-C4), -CH(CH3)2, -CH2-O-haloalquilo (C1-C4), -CH(CH3)-O-haloalquilo (C1-C4), -CH2- NH-haloalquilo (C1-C4) y -CH2-N(CH3)-haloalquilo (C1-C4), R2A se selecciona entre H y metilo; R3 se selecciona entre H y metilo; R3A se selecciona entre H y metilo; el anillo B es un resto seleccionado entre el grupo que consiste en fenilo, heteroarilo, -cicloalquilo (C5-C6) y - cicloalquenilo (C5-C6); n es 0, 1, 2 o 3, siempre que el valor de n no exceda el número máximo de átomos de hidrógeno sustituibles en el anillo B; y cada R1, (cuando está presente) se selecciona independientemente del grupo que consiste en halógeno, -CN, - OH, -alquilo (C1-C6), -O-alquilo (C1-C6), -haloalquilo (C1-C6), -O-haloalquilo (C1-C6), ciclopropilo, ciclobutilo, -NH2, - NH-alquilo (C1-C6), -N(alquilo C1-C6)2, -C(O)Oalquilo (C1-C6) y fenilo.
Description
DESCRIPCIÓN
Derivados de cromarlo, isocromano y dihidroisobenzofurano como moduladores alostéricos negativos para mGluR2, composiciones y su uso
Campo de la invención
La invención se dirige a determinados derivados de cromanos, isocromano y dihidroisobenzofurano, sus sales, a composiciones farmacéuticas que los comprenden y a su uso en terapia del cuerpo humano. Se ha descubierto que los compuestos de la invención modulan el receptor 2 de glutamato metabotrópico (mGluR2) y, por tanto, se espera que sean útiles en el tratamiento de la enfermedad de Alzheimer y otras enfermedades mediadas por el receptor mGluR2.
Antecedentes de la invención
Se sabe que los receptores metabotrópicos de glutamato contienen uno o más sitios alostéricos, que pueden alterar la afinidad con la que el glutamato y otros ligandos de glutamato metabotrópicos (mGluR) se unen a los sitios de unión primaria u ortostéricos. Como el sitio de unión ortostérico está altamente conservado entre todos los receptores de glutamato metabotrópicos conocidos, la selectividad funcional puede lograrse mejor a través de interacción alostérica con el receptor.
La modulación del receptor de glutamato metabotrópico 2 (mGluR2), que es prevalente en las terminales nerviosas presinápticas en la corteza y el hipocampo y regula la liberación del principal neurotransmisor excitador del cerebro, glutamato, en sinapsis neuronales clave, se ha demostrado que tiene un papel principal en el procesamiento cognitivo. Se cree que las enfermedades neurodegenerativas y los trastornos que afectan a la cognición están influenciados por la señalización del glutamato. Tales enfermedades y trastornos neurodegenerativos que afectan la cognición incluyen (pero no se limitan a) diversas formas de demencia, incluyendo demencia del tipo Alzheimer (enfermedad de Alzheimer), incluyendo enfermedad de Alzheimer leve, moderada y grave, deterioro cognitivo leve y otros. Tales enfermedades y trastornos pueden dar como resultado, o identificarse por manifestaciones tales como deterioro progresivo de la memoria, pérdida de lenguaje y habilidades visioespaciales, déficits de comportamiento y otros. El potencial de inhibición de mGluR2 para mejorar el rendimiento cognitivo se ha demostrado genética y farmacológicamente en especies preclínicas (Higgins et al. [2004], Neuropharmacology 46, 907-917). Además, la inhibición de mGluR2/3 con un modulador alostérico negativo muestra efectos precognitivos en primates no humanos (Goeldner et al., [2013], Neuropharmacology 64, 337-346). De manera similar, se espera que la inhibición de mGluR2 con moduladores alostéricos negativos mejore la cognición y revierta la demencia asociada a otros trastornos, tales como esquizofrenia (Marek [2010], Eur J Pharmacol 639, 81-90) y deterioro cognitivo leve general, dado que se ha demostrado que la mejora de la señalización glutamatérgica posterior mejora la cognición clínicamente (Lynch et al.
[1997], Exp Neurol 145, 89-92). Por estas razones, se cree que los inhibidores de mGluR2 son útiles para mejorar el rendimiento cognitivo asociado a diversas formas de demencia, incluyendo enfermedad de Alzheimer, deterioro cognitivo asociado a esquizofrenia y otras enfermedades y trastornos. Se han presentado patentes que desvelan inhibidores de mGluR2/3 para estas (y otras) indicaciones (Celanire et al. [2015], Expert Opin Ther Patents 25, 69-90).
Dada la capacidad del mGluR2 presináptico para modular la liberación de glutamato, la inhibición farmacológica de mGluR2 con moduladores alostéricos negativos tiene la capacidad de mejorar la señalización del glutamato para aliviar otros trastornos que implican la señalización del glutamato. Entre estos se encuentran los trastornos del estado de ánimo incluyendo trastorno depresivo mayor (MDD), depresión asociada a trastorno bipolar y ansiedad. La inhibición de mGluR2 y mGluR3 por antagonistas ortostéricos ha demostrado efectividad en modelos de depresión en roedores (Chaki et al. [2004], Neuropharmacology 46, 457-67) ya que tienen moduladores alostéricos negativos (Campo et al.
[2011], J Neurogenet 25, 152-66). Los antagonistas de mGluR2 y mGluR3 también han demostrado efectividad en modelos de ansiedad en roedores (Shimazaki et al. [2004], Eur J Pharmacol 501, 121-5; Iijima et al. [2007], Psychopharmacology (Berl) 190, 233-9) que ha dado lugar a la presentación de patentes de inhibidores de mGluR2/3 para estas (y otras) indicaciones (Celanire et al. [2015], Expert Opin Ther Patents 25, 69-90).
También se espera que la inhibición de los receptores mGluR2 con moduladores alostéricos negativos module el sueño y la excitación y la sincronización circadiana de los ciclos de sueño y vigilia. La activación de mGluR2 con un modulador alostérico positivo da como resultado un sueño profundo en ratas y clínicamente en voluntarios humanos sanos (Ahnaou et al. [2016], Neuropharmacology 103, 290-305), de modo que se espera que la inhibición con un modulador alostérico negativo promueva la excitación coincidente con la cognición mejorada. La señalización del glutamato modulada por los mGluR del grupo II (mGluR2, mGluR3) también está implicada en la sincronización circadiana de los ciclos de sueño / vigilia de tal manera que puede esperarse que la inhibición de mGluR2 mejore la coordinación de la actividad con los ciclos ambientales de luz / oscuridad. La pérdida genética de mGluR2 y mGluR3, así como la inhibición farmacológica con moduladores alostéricos negativos a estos receptores, da como resultado respuestas mejoradas a las señales de arrastre de luz (Pritchett et al. [2015], PLoS One 10, e0125523).
También se espera que la inhibición de mGluR2 con compuestos moduladores alostéricos negativos module la sensación de dolor y las respuestas al dolor. La señalización del glutamato media tanto en la transmisión de
información sobre el dolor como en los mecanismos periféricos y centrales de hipersensibilidad al dolor, de modo que la modulación de esta señalización a través de la inhibición de mGluR2 tiene el potencial de afectar la nocicepción, así como la percepción central de la memoria del dolor (Chiechio [2016], Adv Pharmacol 75, 63-89).
Determinadas carboxamidas de quinolina sustituidas, carbonitrilos de quinolina, tetrahidronaftiridinas y otros, se conocen en la técnica como inhibidores de mGluR2 o para otros usos. Véase, por ejemplo, el documento WO2016/032921, el documento WO2013/066736, la Solicitud de Patente de EE.UU. N.° 2008/0188521, el documento WO2007/038865, el documento WO 1996/13500, cada uno de los cuales describe compuestos como inhibidores de leucotrienos y la Solicitud de Patente canadiense N.° 2169231, que desvela compuestos como leucotrienos e inhibidores de SRS-A. Sigue existiendo una necesidad en la técnica de compuestos novedosos que sean eficaces como moduladores de mGluR2 no competitivos y/o moduladores alostéricos negativos de mGluR2 (NAM).
Sumario de la invención
En una realización, la presente invención se dirige a determinados derivados de cromano, isocromano y dihidroisobenzofurano sustituidos novedosos, que se denominan colectiva o individualmente en el presente documento "compuesto o compuestos de la invención". Los compuestos de la invención, descritos a continuación, son moduladores alostéricos negativos no competitivos del receptor metabotrópico de glutamato 2 (mGluR2 NAM) y pueden ser útiles en el tratamiento de enfermedades o trastornos en los que es útil la inhibición del receptor mGluR2. Dichas enfermedades o trastornos incluyen, pero pueden no limitarse a, enfermedad de Alzheimer, deterioro cognitivo, esquizofrenia y otros trastornos del estado de ánimo, trastornos de dolor y trastornos del sueño. En otra realización, la presente invención también se dirige a composiciones farmacéuticas que comprenden una cantidad eficaz de un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, y un vehículo farmacéuticamente aceptable y, en aún otra realización, al uso de los compuestos y composiciones farmacéuticas de la invención en el tratamiento de tales enfermedades o trastornos. En otras realizaciones, la presente invención también se dirige a una combinación que comprende un compuesto de la invención y uno, dos, tres o más de otros agentes terapéuticos, y al uso de dicha combinación en el tratamiento de las enfermedades o trastornos descritos en el presente documento. Estas y otras realizaciones se describen en detalle a continuación en el presente documento.
Descripción detallada de la invención
En una realización, los compuestos de la invención tienen la Fórmula estructural (I):

o un estereoisómero de los mismos o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
el anillo A es un resto seleccionado entre:

en donde:
R2 se selecciona entre H, ciclopropilo, alquilo (C1-C4), -alquil (C1-C4)-OH, -alquil (C1-C4)-OCH3, -haloalquilo (C1C4), -alquil (C1-C4)-O-haloalquilo (C1-C4), -CH2-O-haloalquilo (C1-C4), -CH(cHa)-O-haloalquilo (C1-C4), -CH2-NHhaloalquilo (C1-C4) y -CH2-N(CH3)-haloalquilo (C1-C4),
R2A se selecciona entre H y metilo;
R3 se selecciona entre H y metilo;
R3A se selecciona entre H y metilo;
el anillo B es un resto seleccionado entre el grupo que consiste en fenilo, heteroarilo, -cicloalquilo (C5-C6) y -cicloalquenilo (C5-C6);
n es 0, 1, 2 o 3, siempre que el valor de n no exceda el número máximo de átomos de hidrógeno sustituibles en el anillo B; y
cada R1, (cuando está presente) se selecciona independientemente del grupo que consiste en halógeno, -CN, -OH, -alquilo (C1-C6), -O-alquilo (C1-C6), -haloalquilo (C1-C6), -O-haloalquilo (C1-C6), ciclopropilo, ciclobutilo, -NH2 , -NH-alquilo (C1-C6), -N(alquilo C1-C6)2 , -C(O)Oalquilo (C1-C6) y fenilo.
En realizaciones en donde el anillo A es el resto:

en donde el anillo B, n, y cada R1 son como se definen en la Fórmula (I).
En una realización, en la Fórmula (IA):
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3 , -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R2A se selecciona entre H y metilo;
R3 se selecciona entre H y metilo;
R3A se selecciona entre H y metilo;
y el anillo B, n, y cada R1 es como se define en la Fórmula I.
En otra realización, en la Fórmula (IA):
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3 , -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R2A se selecciona entre H y metilo;
R3 se selecciona entre H y metilo;
R3A es H;
y el anillo B, n, y cada R1 es como se define en la Fórmula I.
En otra realización, en la Fórmula (IA):
R2 y R2A son ambos metilo;
R3 y R3A son ambos H;
y el anillo B, n, y cada R1 es como se define en la Fórmula I.
En otra alternativa de la realización inmediatamente anterior, En realizaciones en donde el anillo A es el resto:

la Formula (I) toma la forma de la Fórmula (IB):

En realizaciones en donde el anillo A es el resto:

la Formula (I) toma la forma de la Fórmula (IC):

en donde el anillo B, n y cada R1 son 
En realizaciones en donde el anillo A es el resto:
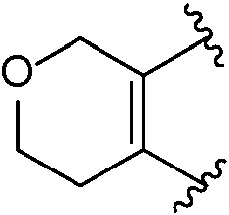
la Formula (I) toma la forma de la Fórmula (ID):

en donde el anillo B, n y cada R1 son como se definen en la Fórmula (I).
Las siguientes realizaciones alternativas del anillo B, n y R1 se aplican a cada una de las realizaciones descritas anteriormente.
En una realización, en cada una de las Fórmulas (I), (IA), (IB), (IC) y (ID):
el anillo B es un resto seleccionado entre el grupo que consiste en fenilo, ciclopentilo, ciclohexilo, piridinilo, pirimidinilo, pirazolilo, tienilo, tiazolilo, tiadiazolilo, isoxazolilo, oxadiazolilo y oxazolilo;
n es 0, 1, 2 o 3, siempre que el valor de n no exceda el número máximo de átomos de hidrógeno sustituibles en el anillo B; y
cada R1, (cuando está presente) se selecciona independientemente del grupo que consiste en halógeno, -CN, -OH, -alquilo (C1-C6 ), -O-alquilo (C1-C6), -haloalquilo (C1-C6), -O-haloalquilo (C1-C6), ciclopropilo, ciclobutilo, -NH2 , -NH-alquilo (C1-C6), -N(alquilo C1-C6)2 , -C(O)Oalquilo (C1-C6) y fenilo.
En una alternativa de la realización inmediatamente anterior, n es 0, 1 o 2; y
cada R1, (cuando está presente) se selecciona independientemente del grupo que consiste en flúor, cloro, -CN, -OH, -alquilo (C1-C6), -O-alquilo (C1-C6), -haloalquilo (C1-C6), -O-haloalquilo (C1-C6), ciclopropilo, ciclobutilo, -NH2 , -NH-alquilo (C1-C6), -N(alquilo C1-C6)2, -C(O)Oalquilo (C1-C6) y fenilo.
En otra realización, en cada una de las Fórmulas (I), (IA), (IB), (IC) y (ID):
el anillo B es un resto seleccionado entre el grupo que consiste en: fenilo, pirazolilo, piridinilo, tienilo, isoxazolilo, oxadiazolilo y oxazolilo;
n es 0, 1, 2 o 3; y
cada R1, (cuando está presente) se selecciona independientemente del grupo que consiste en flúor, cloro, -CN, -OH, -alquilo (C1-C6 ), -O-alquilo (C1-C6), -haloalquilo (C1-C6), -O-haloalquilo (C1-C6), ciclopropilo, ciclobutilo, -NH2 , -NH-alquilo (C1-C6), -N(alquilo C1-C6)2 , -C(O)Oalquilo (C1-C6) y fenilo.
En una alternativa de la realización inmediatamente anterior, n es 0, 1 o 2.
En otra realización, en cada una de las Fórmulas (I), (IA), (IB), (IC) y (ID):
el anillo B es un resto seleccionado entre el grupo que consiste en: fenilo, pirazolilo, piridinilo, tienilo, isoxazolilo, oxadiazolilo y oxazolilo;
n es 0, 1, 2 o 3; y
cada R1, (cuando está presente) se selecciona independientemente del grupo que consiste en flúor, cloro, -CH3 y -CHCF2.
En una alternativa de la realización inmediatamente anterior, n es 0, 1 o 2.
Los ejemplos no limitantes del anillo B, n y R1 se muestran en la posición correspondiente de cada uno de los compuestos de ejemplo de la invención como se muestra en los ejemplos preparativos y las reivindicaciones adjuntas. Como se describe en las Fórmulas (I), (IA), (IB), (IC) y (ID), y en cada una de las realizaciones alternativas del anillo B, n y R1 citados en el presente documento, el anillo B se puede sustituir con 0, 1, 2 o 3 grupos R1 seleccionados independientemente, siempre que el valor de n no exceda el número máximo de átomos de hidrógeno sustituibles en el anillo B. Por lo tanto, en realizaciones en donde el anillo B es fenilo, -cicloalquilo (C5-C6), -cicloalquenilo (C5-C6), piridinilo, pirimidinilo o tienilo, n es 0, 1, 2 o 3. En una alternativa de cada una de estas formas de realización, n es 0, 1 o 2. En otra alternativa de cada una de estas formas de realización, n es 0 o 1. Cuando el anillo B es pirazolilo, tiazolilo, isoxazolilo, oxadiazolilo u oxazolilo, n es 0, 1 o 2. En una alternativa de cada una de estas formas de realización, n es 0 o 1. Y cuando el anillo B es tiadiazolilo, n es 0 o 1.
Otra realización es un compuesto de Fórmula (IA-1):

o un estereoisómero del mismo o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3 , -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R2A se selecciona entre H y CH3 ;
R3 se selecciona entre H y CH3 ; y
R3A se selecciona entre H y CH3.
Otra realización es un compuesto de Fórmula (IA-1), o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3, -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R2A se selecciona entre H y CH3;
R3 se selecciona entre H y CH3; y
R3A es H.
Otra realización es un compuesto de Fórmula (IA-1), o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3, -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R2A es H;
R3 se selecciona entre H y CH3; y
R3A es H.
Otra realización es un compuesto de Fórmula (IA-1), o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3 , -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R2A es H;
R3 es H; y
R3A es H.
Otra realización es un compuesto de Fórmula (IA-1a):

o una sal farmacéuticamente 
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3, -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R3 es H; y
R3A es CH3.
Otra realización es un compuesto de Fórmula (IA-1a), o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3, -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R3 es CH3; y
R3A es H.
Otra realización es un compuesto de Fórmula (IA-1a), o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3, -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R3 es H; y
R3A es H.
Otra realización es un compuesto de Fórmula (IA-1 b):

o una sal farmacéuticamente 
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3 , -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R3 es H; y
R3A es CH3.
Otra realización es un compuesto de Fórmula (IA-1 b), o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3, -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R3 es CH3; y
R3A es H.
Otra realización es un compuesto de Fórmula (IA-1 b), o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3, -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R3 es H; y
R3A es H.
En una realización, los compuestos de la invención comprenden, colectiva e individualmente, cada uno de los compuestos de ejemplo mostrados en las tablas siguientes, y sus sales farmacéuticamente aceptables. Las sales farmacéuticamente aceptables adecuadas de cada uno de estos compuestos incluyen las discutidas en el presente documento a continuación.
Definiciones
Los términos utilizados en el presente documento tienen su significado habitual y el significado de dichos términos es independiente en cada aparición de los mismos. No obstante y excepto cuando se indique otra cosa, las siguientes definiciones se aplican en toda la memoria descriptiva y las reivindicaciones. Pueden usarse nombres químicos, nombres comunes y estructuras químicas indistintamente para describir esa misma estructura. Estas definiciones se aplican independientemente de si un término se usa por sí mismo o en combinación con otros términos, a menos que se indique otra cosa. Por tanto, la definición de "alquilo" se aplica tanto a "alquilo" como a la parte "alquilo" de "hidroxialquilo", "haloalquilo", arilalquil-, alquilaril-, "alcoxi" etc.
Se entenderá que, en las diversas realizaciones de la invención descritas en el presente documento, cualquier variable no definida explícitamente en el contexto de la realización es como se define en la Fórmula (I). Se supone que todas las valencias que no se llenan explícitamente están llenas de hidrógeno.
"Paciente" significa un ser humano que necesita un tratamiento descrito en el presente documento, como se determina
por un médico tratante u otro profesional de la salud o por cualquier otro método adecuado conocido por los expertos en la materia. Aunque el sujeto o paciente al que se administran los compuestos y composiciones de la presente invención es generalmente un ser humano, tales sujetos también pueden incluir mamíferos no humanos, incluyendo perros, gatos, ratones, ratas, ganado bovino, caballos, oveja, conejos, monos, chimpancés u otros monos o primates, para los cuales se desea el tratamiento de las enfermedades y trastornos anteriormente indicados o el estudio de la actividad biológica de los compuestos objeto.
"Composición farmacéutica" (o "composición farmacéuticamente aceptable") significa una composición adecuada para la administración a un paciente. Tales composiciones pueden contener el compuesto (o compuestos) de la invención o mezclas de los mismos, o sales, solvatos, profármacos, isómeros o tautómeros de los mismos, solos u opcionalmente junto con uno o más vehículos o diluyentes farmacéuticamente aceptables. La expresión "composición farmacéutica" también pretende abarcar tanto la composición en masa como las unidades farmacéuticas individuales que comprenden más de uno (por ejemplo, dos) agentes farmacéuticamente activos tal como, por ejemplo, un compuesto de la presente invención y un agente adicional seleccionado de entre las listas de los agentes adicionales descritos en el presente documento, junto con cualquier excipiente farmacéuticamente inactivo. La composición en masa y cada unidad farmacéutica individual puede contener cantidades fijas de los "más de un agente farmacéuticamente activo" anteriormente mencionados. La composición en masa es un material que aún no se ha formado en unidades farmacéuticas individuales. Una unidad farmacéutica ilustrativa es una unidad farmacéutica oral tal como comprimidos, píldoras y similares. De forma similar, el método descrito en el presente documento para tratar a un paciente mediante la administración de una composición farmacéutica de la presente invención también pretende abarcar la administración de la composición a granel mencionada anteriormente y las unidades de dosificación individuales.
"Halógeno" (o "halo") significa flúor (F), cloro (Cl), bromo (Br) o yodo (I). Se prefieren flúor, cloro y bromo. Son más preferidos, flúor y cloro.
"Alquilo" significa un grupo hidrocarburo alifático, que puede ser lineal o ramificado, que comprende de 1 a 6 átomos de carbono. "Alquilo inferior" significa un grupo alquilo lineal o ramificado que comprende de 1 a 4 átomos de carbono. Ramificado significa que uno o más grupos alquilo inferior tales como metilo, etilo o propilo, están unidos a una cadena alquilo lineal. Los ejemplos no limitantes de grupos alquilo adecuados incluyen metilo (Me o CH3), etilo (Et), n-propilo, isopropilo, n-butilo, i-butilo y t-butilo.
"Alquenilo" significa un grupo hidrocarburo alifático que contiene al menos un doble enlace carbono-carbono y que puede ser lineal o ramificado y que comprende de 2 a 10 átomos de carbono en la cadena lineal o ramificada. Ramificado significa que uno o más grupos alquilo inferior tales como metilo, etilo, propilo, etenilo o propenilo están unidos a una cadena de alquenilo lineal o ramificada. "Alquenilo inferior" significa de 2 a 4 átomos de carbono en la cadena que puede ser lineal o ramificada. Los ejemplos no limitantes de grupos alquinilo adecuados incluyen etenilo, propenilo, n-butenilo, 3-metilbut-2-enilo, n-pentenilo, octenilo y decenilo.
"Heteroarilo" significa un sistema de anillo aromático monocíclico o multicíclico que comprende de 5 a 14 átomos de anillo, preferentemente de 5 a 10 átomos en el anillo, en el que uno o más de los átomos en el anillo es un elemento diferente del carbono, por ejemplo nitrógeno, oxígeno o azufre, solos o en combinación. Los heteroarilos preferidos contienen de 5 a 6 átomos en el anillo. El "heteroarilo" puede estar opcionalmente sustituido con uno o más sustituyentes, que pueden ser iguales o diferentes, como se define en el presente documento. El prefijo aza, oxa o tia antes del nombre de la raíz de heteroarilo significa que al menos un átomo de nitrógeno, oxígeno o azufre, respectivamente, está presente como un átomo del anillo. Un átomo de nitrógeno de un heteroarilo se puede oxidar opcionalmente al correspondiente N-óxido sin caer fuera de la definición de heteroarilo. "Heteroarilo" también puede incluir un heteroarilo como se definió anteriormente fusionado a un arilo como se definió anteriormente. Los ejemplos no limitantes de heteroarilos adecuados incluyen piridilo, pirazinilo, furanilo, tienilo (que alternativamente puede denominarse tiofenilo), pirimidinilo, piridona (incluyendo piridonas N-sustituidas), isoxazolilo, isotiazolilo, oxazolilo, oxadiazolilo, tiazolilo, tiadiazolilo, pirazolilo, furazanilo, pirrolilo, triazolilo, 1,2,4-tiadiazolilo, pirazinilo, piridazinilo, quinoxalinilo, ftalazinilo, oxindolilo, imidazo[1,2-a]piridinilo, imidazo[2,1-b]tiazolilo, benzofurazanilo, indolilo, azaindolilo, benzoimidazolilo, benzotienilo, quinolinilo, imidazolilo, tienopiridilo, quinazolinilo, tienopirimidilo, pirrolopiridilo, imidazopiridilo, isoquinolinilo, benzoazaindolilo, 1,2,4-triazinilo, benzotiazolilo y similares. El término "heteroarilo" también se refiere a restos heteroarilo parcialmente saturados tales como, por ejemplo, tetrahidroisoquinolilo, tetrahidroquinolilo y similares. La expresión "heteroarilo monocíclico" se refiere a versiones monocíclicas de heteroarilo como se ha descrito anteriormente e incluye grupos heteroarilo monocíclicos de 4 a 7 miembros que comprenden de 1 a 4 heteroátomos en el anillo, seleccionándose dichos heteroátomos en el anillo independientemente de N, O y S y óxidos del mismo. El punto de unión al resto parental es cualquier carbono o heteroátomo del anillo disponible. Los ejemplos no limitantes de restos heteroarilo monocíclicos incluyen piridilo (o piridinilo), pirazinilo, furanilo, tienilo, pirimidinilo, piridazinilo, piridonailo, tiazolilo, isotiazolilo, oxazolilo, oxadiazolilo, isoxazolilo, pirazolilo, furazanilo, pirrolilo, triazolilo, tiadiazolilo (por ejemplo, 1,2,4-tiadiazolilo), imidazolilo y triazinilo (por ejemplo, 1,2,4-triazinilo) y óxidos de los mismos.
"Cicloalquilo" significa un sistema de anillo monocíclico o multicíclico no aromático que comprende de 3 a 10 átomos de carbono, preferiblemente de 3 a 6 átomos de carbono. El cicloalquilo puede estar opcionalmente sustituido con uno o más sustituyentes, que pueden ser iguales o diferentes, como se describe en el presente documento. Cicloalquilo
monocíclico se refiere a versiones monocíclicas de los restos cicloalquilo descritos en el presente documento. Los ejemplos no limitantes de cicloalquilos monocíclicos adecuados incluyen ciclopropilo, ciclopentilo, ciclohexilo, cicloheptilo y similares. Los ejemplos no limitantes de cicloalquilos multicíclicos incluyen [1.1.1]-biciclopentano, 1-decalinilo, norbornilo, adamantilo y similares.
"Cicloalquenilo" significa un sistema de anillo mono o multicíclico no aromático que comprende de 3 a 10 átomos de carbono, preferentemente de 4 a 6 átomos de carbono que contienen al menos un doble enlace carbono-carbono. Los anillos de cicloalquenilo preferidos contienen de 5 a 6 átomos en el anillo. La expresión "cicloalquenilo monocíclico" se refiere a versiones monocíclicas de grupos cicloalquenilo descritos en el presente documento e incluye grupos cicloalquilo monocíclicos de 3 a 7 miembros no aromáticos que contienen uno o más dobles enlaces carbono-carbono. Los ejemplos no limitantes incluyen ciclopropenilo, ciclobutenilo, ciclopentenilo, ciclohexenilo, ciclohet-penilo, ciclohepta-1,3-dienilo y similares. Un ejemplo no limitante de un cicloalquenilo multicíclico adecuado es norbornilenilo. Cualquiera de los grupos funcionales anteriores puede estar sin sustituir o sustituido como se describe en el presente documento. El término "sustituido" significa que uno o más hidrógenos en el átomo designado se reemplazan por una selección de entre el grupo indicado, a condición de que no se exceda la valencia normal del átomo designado en las circunstancias existentes y de que la sustitución dé como resultado un compuesto estable. Solo se permiten las combinaciones de sustituyentes y/o variables en caso de que dichas combinaciones den como resultado compuestos estables. Por "compuesto estable" o "estructura estable" se entiende un compuesto que es suficientemente robusto como para sobrevivir al aislamiento hasta un grado útil de pureza a partir de una mezcla de reacción y a su formulación en un agente terapéutico eficaz.
La expresión "opcionalmente sustituido" significa sustitución opcional con los grupos, radicales o restos especificados. Cuando una variable aparece más de una vez en un grupo, por ejemplo, R6 en -N(R6)2, o una variable aparece más de una vez en una estructura presentada en el presente documento, las variables pueden ser iguales o diferentes. La línea -, como enlace, generalmente indica una mezcla de, o cualquiera de, los posibles isómeros, por ejemplo, que contiene estereoquímica (R) y (S). Por ejemplo:

significa que contiene ambos

La línea ondulada
J\AA/\
como se usa en el presente documento, indica un punto de unión al resto del compuesto. Líneas dibujadas en los sistemas de anillos, tales como, por ejemplo:

indican que la línea indicada (enlace) puede estar unida a cualquiera de los átomos de carbono del anillo sustituible. "Oxo" significa un átomo de oxígeno que está doblemente unido a un anillo de carbono en un cicloalquilo, cicloalquenilo, heterociclilo, heterociclenilo, o tales otros anillos como se describen en este documento, por ejemplo,

En la presente memoria descriptiva, cuando hay múltiples átomos de oxígeno y/o azufre en un sistema de anillos, no puede haber oxígeno y/o azufre adyacentes presentes en dicho sistema de anillos.
Como es bien sabido en la técnica, un enlace extraído de un átomo particular en donde no se representa ningún resto en el extremo terminal del enlace indica un grupo metilo unido a través de ese enlace al átomo, a menos que se indique lo contrario. Por ejemplo:

representa

En otra realización, los compuestos de la invención, y/o las composiciones que los comprenden, están presentes en forma aislada y/o purificada. Las expresiones "purificado", "en forma purificada" o "en forma aislada y purificada" para un compuesto se refiere al estado físico de dicho compuesto después de haber sido aislado de un proceso de síntesiso (por ejemplo, de una mezcla de reacción), fuente natural o combinación del mismo. Por tanto, las expresiones "purificado", "en forma purificada" o "en forma aislada y purificada" para un compuesto se refiere al estado físico de dicho compuesto (o un tautómero del mismo, o sal farmacéuticamente aceptable de dicho compuesto o de dicho tautómero) después de ser obtenido de un proceso de purificación o procesos descritos en este documento o bien conocidos por el experto en la materia (por ejemplo, cromatografía, recristalización y similares), con suficiente pureza para ser adecuado para uso in vivo o medicinal y/o caracterizable mediante técnicas analíticas estándar descritas en el presente documento o bien conocidas por el experto en la materia.
Cuando un grupo funcional en un compuesto se denomina "protegido", esto significa que el grupo se encuentra en una forma modificada para impedir reacciones secundarias no deseadas en el sitio protegido cuando el compuesto se somete a una reacción. Los expertos en la materia reconocerán los grupos protectores adecuados, así como también pueden dirigirse a libros de texto convencionales tales como, por ejemplo, T. W. Greene et al, Protective Groups in organic Synthesis (1991), Wiley, Nueva York.
Los expertos en la materia reconocerán aquellos casos en los que los compuestos de la invención pueden convertirse en profármacos y/o solvatos, otra realización de la presente divulgación. Se proporciona una discusión sobre profármacos en T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 de la serie del Simposio A.C.S. y en Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press. El término "profármaco" significa un compuesto (por ejemplo, un precursor de un fármaco) que se transforma in vivo para producir un compuesto de la invención o una sal, hidrato o solvato farmacéuticamente aceptable del compuesto. La transformación puede tener lugar por varios mecanismos (por ejemplo, por procesos metabólicos o químicos), tales como, por ejemplo, a través de hidrólisis en la sangre. Un análisis del uso de profármacos se proporciona por T. Higuchi y W. Stella, "Pro-drugs as Novel Delivery Systems", Vol. 14 de la serie del Simposio A.C.S. y en Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
Uno o más compuestos de la invención pueden existir en formas no solvatadas así como solvatadas con disolventes farmacéuticamente aceptables tales como agua, etanol y similares, y se pretende que la invención abarque formas tanto solvatadas como no solvatadas cuando existan. "Solvato" significa una asociación física de un compuesto de la invención con una o más moléculas de disolvente. Esta asociación física implica grados variables de enlace iónico y covalente, incluyendo enlace de hidrógeno. En ciertos casos, el solvato podrá aislarse, por ejemplo, cuando se incorporan una o más moléculas de disolvente a la red cristalina del sólido cristalino. "Solvato" abarca solvatos tanto en fase de solución como aislables. Ejemplos no limitativos de solvatos adecuados incluyen etanolatos, metanolatos
y similares. "Hidrato" es un solvato en donde la molécula de disolvente es H2O.
"Cantidad eficaz" o "cantidad terapéuticamente eficaz" significa una cantidad de compuesto o una composición de la presente invención que provocará la respuesta biológica o médica de un tejido, sistema, animal o humano que está siendo buscado por el investigador, veterinario, doctor u otro médico o practicante. En las terapias de combinación de la presente invención, una cantidad eficaz puede referirse a cada agente individual o a la combinación como un todo, en donde las cantidades de todos los agentes administrados son conjuntamente eficaces. En algunas realizaciones, uno o más agentes componentes de la combinación pueden estar presentes individualmente en una cantidad menor que la cantidad necesaria para ser eficaz cuando se administra solo, como se describe adicionalmente a continuación. Una "cantidad terapéuticamente eficaz" puede variar dependiendo de, entre otros, el compuesto, de la enfermedad y su gravedad, y de la edad, peso, etc., del sujeto que se va a tratar.
El término "tratamiento" o "tratar" significa cualquier administración de un compuesto o composición de la presente invención a un sujeto que lo necesite, solo o en combinación con uno o más agentes terapéuticos adicionales, e incluye (1) inhibir o mejorar una patología y/o sintamotología de la enfermedad o trastorno en dicho sujeto, por ejemplo, animal, persona o paciente u otro sujeto que está experimentando o mostrando la patología o sintomatología de la enfermedad o trastorno. El término "prevención" o "profilaxis" significa cualquier administración de un compuesto o composición de la presente invención a un sujeto que lo necesite, solo o en combinación con uno o más agentes terapéuticos adicionales, e incluye (1) inhibir o mejorar un patología y/o sintomatología de la enfermedad o trastorno en dicho sujeto, por ejemplo, animal, persona o paciente u otro sujeto antes del inicio o manifestación de la patología o sintomatología de la enfermedad o trastorno. El término "control" incluye prevenir, tratar, erradicar, mejorar o reducir de otro modo la gravedad de la enfermedad o trastorno o síntoma o síntomas del mismo.
Los compuestos y composiciones de la presente invención, solos o en combinación con uno o más agentes terapéuticos adicionales, también pueden ser útiles en el tratamiento o prevención de las enfermedades o trastornos mencionados, o uno o más síntomas de los mismos, cuyos tratamientos y prevención también son contempladas como realizaciones adicionales de la presente invención.
Los expertos en la materia reconocerán aquellos casos en los que los compuestos de la invención pueden formar sales. En tales casos, otra realización proporciona sales farmacéuticamente aceptables de los compuestos de la invención. El término "sal" o "sales", como se emplea en el presente documento, representa cualquiera de las siguientes: sales ácidas formadas con ácidos inorgánicos y/u orgánicos, así como sales básicas formadas con bases inorgánicas y/u orgánicas. Además, cuando un compuesto de la invención contiene un resto básico, tales como, pero sin limitación, una piridina o imidazol, y un resto ácido, tales como, pero sin limitación, un ácido carboxílico, pueden formarse zwitteriones ("sales internas") y estos se incluyen dentro del término "sal" o "sales" como se usa en el presente documento. Se prefieren las sales farmacéuticamente aceptables (es decir, no tóxicas, fisiológicamente aceptables), aunque también son potencialmente útiles otras sales. Las sales de los compuestos de la invención se pueden formar por métodos conocidos por los expertos habituales en la materia, por ejemplo, haciendo reaccionar un compuesto de la invención con una cantidad de un ácido o base, tal como una cantidad equivalente, en un medio tal como uno en el que la sal precipita o en un medio acuoso seguido de liofilización.
Ejemplos de sales de adición de ácido que pueden ser útiles incluyen acetatos, ascorbatos, benzoatos, bencenosulfonatos, bisulfatos, boratos, butiratos, citratos, alcanforatos, alcanforsulfonatos, fumaratos, clorhidratos, bromhidratos, yodhidratos, lactatos, maleatos, metanosulfonatos, naftalenosulfonatos, nitratos, oxalatos, fosfatos, propionatos, salicilatos, succinatos, sulfatos, tartaratos, tiocianatos, toluenosulfonatos (también conocidos como tosilatos) y similares. Adicionalmente, los ácidos que, en general, se consideran adecuados para la formación de sales farmacéuticamente útiles a partir de compuestos farmacéuticos básicos se analizan, por ejemplo, de P. Stahl et al, Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P. Gould, International J. of Pharmaceutics (1986) 33 201-217; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, Nueva York; y en The Orange Book (Food & Drug Administration, Washington, D. C. en su sitio web). Estas divulgaciones se incorporan en el presente documento por referencia a las mismas.
Las sales básicas de ejemplo incluyen sales de amonio, sales de metales alcalinos, tales como sales de sodio, litio y sales de potasio, sales de metales alcalinotérreos, tales como sales de calcio y de magnesio, sales con bases orgánicas (por ejemplo, aminas orgánicas), tales como diciclohexilamina, t-butil aminas, y sales con aminoácidos, tales como arginina, lisina y similares. Los grupos que contienen nitrógeno básico se pueden cuaternizar con agentes tales como haluros de alquilo inferior (por ejemplo, cloruros, bromuros y yoduros de metilo, etilo y butilo), dialquilo sulfatos (por ejemplo, sulfatos de dimetilo, dietilo y dibutilo), haluros de cadena larga (por ejemplo, cloruros, bromuros y yoduros de decilo, laurilo y estearilo), haluros de aralquilo (por ejemplo, bromuros de bencilo y fenetilo) y otros.
Se pretende que todas estas sales ácidas y sales básicas sean sales farmacéuticamente aceptables dentro del alcance de la invención y todas las sales ácidas y básicas se consideran alternativas potencialmente útiles a las formas libres de los compuestos correspondientes para los fines de la invención.
Otra realización que puede ser útil incluye ésteres farmacéuticamente aceptables de los compuestos de la invención.
Tales ásteres pueden incluir los siguientes grupos: (1) ásteres de ácido carboxílico obtenidos por esterificación de los grupos hidroxi, en el que el resto no carbonilo de la porción de ácido carboxílico del grupo áster se selecciona de alquilo de cadena lineal o ramificada (por ejemplo, acetilo, n-propilo, t-butilo o n-butilo), alcoxialquilo (por ejemplo, metoximetilo), aralquilo (por ejemplo, bencilo), ariloxialquilo (por ejemplo, fenoximetilo), arilo (por ejemplo, fenilo opcionalmente sustituido con, por ejemplo, halógeno, alquilo C1-4 o alcoxi C1-4 o amino); (2) ásteres de sulfonato, tales como alquil- o aralquilsulfonilo (por ejemplo, metanosulfonilo); (3) ásteres de aminoácidos (por ejemplo, L-valilo o L-isoleucilo); (4) ásteres de aminoácidos y (5) ásteres de mono, di o trifosfato. Los ásteres de fosfato pueden esterificarse adicionalmente mediante, por ejemplo, un alcohol C1-20 o un derivado reactivo del mismo, o mediante un 2,3-di-acil (C6--24) glicerol.
Los compuestos de la invención pueden tener uno o más centros quirales (asimátricos). La presente invención abarca todas las formas estereoisomáricas de los compuestos de fórmula I. Los centros de asimetría que están presentes en los compuestos de fórmula I pueden tener todos independientemente entre sí una configuración (R) o (S). Como se indica anteriormente, cuando los enlaces a un carbono quiral se representan como líneas rectas en las fórmulas estructurales de la invención, o cuando el nombre de un compuesto se recita sin una designación quiral (R) o (S) para un carbono quiral, se entiende que tanto el (R) ) y (S) configuraciones de cada carbono quiral, y por lo tanto cada enantiómero o diastereómero y mezclas de los mismos, están abarcados dentro de la Fórmula o por el nombre.
Cuando son posibles varios estereoisómeros de los compuestos de la invención, otra realización proporciona mezclas diastereomáricas y enantiómeros individuales de los compuestos de la invención. Las mezclas diastereomáricas se pueden separar en sus diastereómeros individuales basándose en sus diferencias químicas mediante mátodos bien conocidos en la tácnica, tales como, por ejemplo, mediante cromatografía y/o cristalización fraccionada. Los enantiómeros se pueden separar convirtiendo la mezcla enantiomárica en una mezcla diastereoisomárica por reacción con un compuesto ópticamente activo apropiado (por ejemplo, un auxiliar quiral como un alcohol quiral o cloruro de ácido de Mosher), separando los diastereoisómeros y convirtiendo (por ejemplo, mediante hidrólisis) los diastereoisómeros individuales en los correspondientes enantiómeros puros. Tambián pueden separarse los enantiómeros mediante el uso de una columna de HPLC quiral.
Todos los estereoisómeros (por ejemplo, isómeros geomátricos, isómeros ópticos y similares) de los compuestos de la invención (incluidos los de las sales, solvatos, ásteres y profármacos de los compuestos así como las sales, solvatos y ásteres de los profármacos), tales como los que pueden existir debido a la presencia de carbonos asimátricos en diversos sustituyentes, incluyendo las formas enantiomáricas (que pueden existir incluso en ausencia de átomos de carbono asimátricos), formas rotamáricas, atropisómeros y formas diastereomáricas, se contemplan como realizaciones dentro del alcance de esta invención. (Por ejemplo, si un compuesto de la invención incorpora un doble enlace o un anillo condensado, tanto la forma cis como la forma trans, así como las mezclas, están incluidas dentro del alcance de la invención. Tambián, por ejemplo, todas las formas cetoenólicas e imina-enamina de los compuestos están incluidas en la invención).
Los estereoisómeros individuales de los compuestos de la invención, por ejemplo, pueden estar sustancialmente exentos de otros isómeros o pueden estar mezclados, por ejemplo, como compañeros de carrera o con todos los demás, u otros seleccionados, estereoisómeros. Los centros quirales de la presente invención pueden tener una configuración S o R como se define en las recomendaciones de la IUPAC de 1974. El uso de los tárminos "sal", "solvato", "áster", "profármaco" y similares, se pretende que se aplique igualmente a la sal, solvato, áster y profármaco de enantiómeros, estereoisómeros, rotámeros, tautómeros, racematos o profármacos de los compuestos de la invención.
Otra realización que puede ser útil incluye compuestos de la invención marcados isotópicamente. Tales compuestos son idánticos a los enumerados en el presente documento, salvo por el hecho de que uno o más átomos se reemplazan por un átomo que tiene una masa atómica o número másico diferente de la masa atómica o número másico que se encuentra normalmente en la naturaleza. Los ejemplos de isótopos que pueden incorporarse en los compuestos de la invención incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro, tal como 2H, 3H, 11C, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F y 36Cl, respectivamente.
En los compuestos de la invención, los átomos pueden mostrar sus abundancias isotópicas naturales o uno o más de los átomos pueden estar artificialmente enriquecidos en un isótopo particular que tiene el mismo número atómico, pero una masa atómica o número másico diferente de la masa atómica o número másico que se encuentra predominantemente en la naturaleza. La presente invención pretende incluir todas las variaciones isotópicas adecuadas de los compuestos de la invención. Por ejemplo, las diferentes formas isotópicas del hidrógeno (H) incluyen protio (1H) y deuterio (2H). El protio es el isótopo del hidrógeno predominante en la naturaleza. El enriquecimiento en deuterio puede proporcionar como resultado determinadas ventajas terapáuticas, tales como aumentar la vida media in vivo o reducir los requisitos de dosificación, o puede proporcionar un compuesto útil como estándar para la caracterización de muestras biológicas. Los compuestos enriquecidos isotópicamente de la invención se pueden preparar sin una experimentación excesiva mediante tácnicas convencionales bien conocidas por los expertos en la materia o mediante procesos análogos a los descritos en los esquemas y ejemplos del presente documento usando reactivos y/o intermedios con isótopos de manera apropiada.
Se pretende que las formas polimórficas de los compuestos de la invención y de las sales, solvatos, ásteres y profármacos de los compuestos de la invención están incluidas en la presente divulgación.
COMPOSICIONES Y ADMINISTRACIÓN
Otra realización proporciona composiciones farmacéuticas que comprenden una cantidad terapéuticamente eficaz de un compuesto de la invención, o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, y un vehículo farmacéuticamente aceptable.
Una dosificación representativa es de aproximadamente 0,001 a 100 mg/kg de peso corporal/día del compuesto de la invención. Una dosificación preferida es de aproximadamente 0,01 a 10 mg/kg de peso corporal/día de un compuesto de la invención o una sal farmacéuticamente aceptable de dicho compuesto.
La expresión "composición farmacéutica" también pretende abarcar tanto la composición en masa como las unidades farmacéuticas individuales que comprenden más de uno (por ejemplo, dos) agentes farmacéuticamente activos tal como, por ejemplo, un compuesto de la presente invención y un agente terapéutico adicional seleccionado de las listas de los agentes adicionales descritos en el presente documento a continuación, junto con cualquier excipiente farmacéuticamente inactivo. La composición en masa y cada unidad farmacéutica individual puede contener cantidades fijas de los "más de un agente farmacéuticamente activo" anteriormente mencionados. La composición en masa es un material que aún no se ha formado en unidades de dosificación individuales. Una unidad de dosificación ilustrativa es una unidad de dosificación oral tales como comprimidos, píldoras y similares. De manera similar, el método descrito en el presente documento para tratar a un paciente administrándole una composición farmacéutica de la presente invención también pretende abarcar la administración de la composición en masa anteriormente mencionada y unidades farmacéuticas individuales.
Para preparar composiciones farmacéuticas a partir de los compuestos descritos por esta invención, los vehículos inertes farmacéuticamente aceptables pueden ser sólidos o líquidos. Las preparaciones en forma sólida incluyen polvos, comprimidos, gránulos dispersables, cápsulas, obleas y supositorios. Los polvos y comprimidos pueden comprender de aproximadamente el 5 a aproximadamente el 95 por ciento de principio activo. Se conocen en la técnica vehículos sólidos adecuados, por ejemplo, carbonato de magnesio, estearato de magnesio, talco, azúcar o lactosa. Los comprimidos, polvos, obleas y cápsulas pueden usarse como formas de dosificación sólidas adecuadas para administración oral. Algunos ejemplos de vehículos farmacéuticamente aceptables y métodos de fabricación para diversas composiciones pueden encontrarse en A. Gennaro (ed.), Remington's Pharmaceutical Sciences, 18a edición, (1990), Mack Publishing Co., Easton, Pensilvania.
Las preparaciones en forma líquida incluyen, por ejemplo, soluciones, suspensiones y emulsiones. Los ejemplos de materiales útiles para formar tales preparaciones en forma líquida incluyen agua o soluciones de agua-propilenglicol para inyección parenteral, o edulcorantes y opacificantes para soluciones orales, suspensiones y emulsiones. Las preparaciones en forma líquida también pueden incluir soluciones o suspensiones para la administración intranasal. Las preparaciones en aerosol adecuadas para inhalación pueden incluir soluciones y sólidos en forma de polvo, que pueden combinarse con un vehículo farmacéuticamente aceptable, tal como un gas inerte comprimido, por ejemplo, nitrógeno.
También se incluyen preparaciones en forma sólida que se pretende que se conviertan, poco antes de su uso, en preparaciones en forma líquida para la administración oral o parenteral. Dichas formas líquidas incluyen soluciones, suspensiones y emulsiones.
Los compuestos de la invención también pueden administrarse por vía transdérmica. Las composiciones transdérmicas pueden tomar la forma de soluciones líquidas, cremas, lociones, aerosoles y/o emulsiones y pueden incluirse en un parche transdérmico del tipo matriz o depósito como son convencionales en la técnica para este fin. Los compuestos de esta invención también pueden administrarse por vía subcutánea.
Preferentemente el compuesto se administra por vía oral.
Preferentemente, la preparación farmacéutica está en una forma de dosificación unitaria. En dicha forma, la preparación se subdivide en dosis unitarias de tamaño adecuado que contienen cantidades apropiadas del componente activo, por ejemplo, una cantidad eficaz para conseguir el fin deseado.
La cantidad de compuesto activo en una dosis unitaria de preparación puede variarse o ajustarse de aproximadamente 0,001 mg a aproximadamente 100 mg por kg de peso corporal de un mamífero, preferentemente de aproximadamente 0,01 mg a aproximadamente 10 mg por kg. La dosificación real empleada puede variar dependiendo de los requisitos del paciente y la gravedad de la afección que se está tratando. La determinación del régimen de dosificación adecuado para una situación particular está dentro de los conocimientos de la técnica. Por conveniencia, la dosificación diaria total puede dividirse y administrarse en porciones durante el día según se requiera.
Las composiciones de la invención pueden comprender además uno o más agentes terapéuticos adicionales, como se analiza con más detalle a continuación. En consecuencia, en una realización, la presente invención proporciona composiciones que comprenden: (i) un compuesto de la invención, o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero; (ii) uno o más agentes terapéuticos adicionales, que no son compuestos de la invención; y (iii) un vehículo farmacéuticamente aceptable, en donde las cantidades en la composición son eficaces juntas para tratar una de las enfermedades o afecciones analizadas en el presente documento.
USOS DE LOS COMPUESTOS DE LA INVENCIÓN
Otra realización de la divulgación proporciona un método para tratar a un paciente (por ejemplo, un paciente humano o un animal de investigación) por enfermedades o trastornos en los que está implicado el receptor mGluR2. Estos métodos comprenden administrar una cantidad eficaz de un compuesto de la invención, o una composición que comprende un compuesto de la invención (o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero), a un paciente que lo necesite, para tratar una enfermedad o trastorno en el cual está implicado el receptor mGluR2.
Otra realización proporciona un compuesto de la invención para su uso en el tratamiento de una enfermedad o trastorno en el cual está implicado el receptor mGluR2, administrando una cantidad eficaz de un compuesto de la invención o una sal farmacéuticamente aceptable del mismo, a un paciente que lo necesite. Otra realización proporciona el uso de un compuesto de la invención para la fabricación de un medicamento para tratar una enfermedad o trastorno en el que está implicado el receptor mGluR2, administrando una cantidad eficaz de un compuesto de la invención o una sal farmacéuticamente aceptable del mismo, a un paciente que lo necesite. En el presente documento se describen ejemplos de tales enfermedades y trastornos.
En una realización, los compuestos de la invención útiles en dichos métodos o dichos usos comprenden un compuesto de acuerdo con una cualquiera de las Fórmulas (I), (IA), (IA-1), (IA-1a), (IA-1 b), (IB), (IC) y (ID) como se describió anteriormente o de acuerdo con cualquiera de las diversas realizaciones descritas en el presente documento o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, o una composición farmacéuticamente aceptable del mismo. En otra realización, los compuestos de la invención útiles en dichos métodos o dichos usos comprenden el compuesto del ejemplo 2-5, o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichos métodos o dichos usos comprende el compuesto del ejemplo 2-3A o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichos métodos o dichos usos comprende el compuesto del ejemplo 2-3B o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichos métodos o dichos usos comprende el compuesto del ejemplo 3-1A o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichos métodos o dichos usos comprende el compuesto del ejemplo 3-1B o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichos métodos o dichos usos comprende el compuesto del ejemplo 3-7A o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichos métodos o dichos usos comprende el compuesto del ejemplo 3-7B o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo.
En una realización, la presente divulgación se dirige a un método para tratar una enfermedad o trastornos neurodegenerativos que afectan a la cognición, comprendiendo dicho método administrar un compuesto de la invención, o un estereoisómero del mismo, o una sal farmacéuticamente aceptable del mismo, a un sujeto que lo necesite. Tales enfermedades o trastornos que afectan la cognición incluyen, pero no se limitan a, enfermedad de Alzheimer, deterioro cognitivo, cognición asociada a enfermedad de Parkinson, esquizofrenia, trastornos del estado de ánimo, incluyendo depresión y ansiedad, trastornos gastrointestinales, trastornos de dolor y trastornos del sueño y otros como se describen en el presente documento.
Los ejemplos adicionales de trastornos por dolor incluyen dolor agudo, dolor inflamatorio y dolor neuropático. El dolor neuropático incluye, pero no se limita a, neuralgia posherpética, daños neurales, las "dinias", por ejemplo, vulvodinia, dolor del miembro fantasma, avulsiones radiculares, neuropatía diabética dolorosa, mononeuropatía traumática dolorosa, polineuropatía dolorosa. Los ejemplos adicionales de trastornos de dolor incluyen síndromes de dolor central (potencialmente provocados por prácticamente cualquier lesión en cualquier nivel del sistema nervioso); síndromes de dolor posquirúrgico (por ejemplo, síndrome de posmastectomía, síndrome de postoracotomía, dolor de muñón); dolor de huesos y articulaciones (osteoartritis), dolor de movimiento repetitivo, dolor dental, dolor por cáncer, dolor miofascial (lesión muscular, fibromialgia); dolor perioperatorio (cirugía general, ginecológica), dolor crónico, dismennorrea, así como el dolor asociado a angina y dolor inflamatorio de orígenes variados (por ejemplo, osteoartritis, artritis reumatoide, enfermedad reumática, tenosinovitis y gota), dolor de cabeza, migraña y cefalea en racimos, dolor de cabeza, hiperalgesia primaria, hiperalgesia secundaria, alodinia primaria, alodinia secundaria u otro dolor
provocado por la sensibilización central.
Algunos ejemplos adicionales de trastornos cognitivos incluyen deterioro cognitivo leve. Otras afecciones que pueden tratarse mediante los compuestos y composiciones de la invención incluyen enfermedad de Parkinson, hipertensión pulmonar, enfermedad pulmonar obstructiva crónica (EPOC), asma, incontinencia urinaria, glaucoma, trisomía 21 (síndrome de Down), angiopatía amiloide cerebral, demencia degenerativa, hemorragia cerebral hereditaria con amiloidosis de tipo holandés (HCHWA-D), enfermedad de Creutzfeld-Jakob, trastornos priónicos, esclerosis lateral amiotrófica, parálisis supranuclear progresiva, traumatismo craneal, ictus, pancreatitis, miositis por cuerpos de inclusión, otras amiloidosis periféricas, diabetes, autismo y aterosclerosis.
En realizaciones preferidas, los compuestos de la invención pueden ser útiles en el tratamiento de enfermedad de Alzheimer, trastornos cognitivos, esquizofrenia, trastornos de dolor y trastornos del sueño. Por ejemplo, los compuestos pueden ser útiles para la prevención de demencia de tipo Alzheimer, así como para el tratamiento de la fase temprana, fase intermedia o fase tardía de demencia de tipo Alzheimer.
Las afecciones o trastornos de esquizofrenia potenciales para los que los compuestos de la invención pueden ser útiles incluyen una o más de las siguientes afecciones o enfermedades: esquizofrenia o psicosis incluyendo esquizofrenia (paranoica, desestructurada, catatónica o indiferenciada), trastorno esquizofreniforme, trastorno esquizoafectivo, trastorno delirante, trastorno psicótico breve, trastorno psicótico compartido, trastorno psicótico debido a una afección médica general e inducida por sustancias o fármacos (fenciclidina, ketanina y otros anestésicos disociativos, anfetamina y otros psicoestimulantes y cocaína) trastorno psicosispsicótico, psicosis asociada a trastornos afectivos, psicosis reactiva breve, psicosis esquizoafectiva, trastornos del "espectro de esquizofrenia" tales como trastornos esquizoides o esquizotípicos de la personalidad, o enfermedades asociadas a psicosis (tales como depresión mayor, trastorno maníaco depresivo (bipolar), Enfermedad de Alzheimer y síndrome de estrés postraumático), incluyendo tanto síntomas positivos como negativos de la esquizofrenia y otras psicosis; trastornos cognitivos incluyendo demencia (asociada a enfermedad de Alzheimer, isquemia, demencia multiinfarto, traumatismo, problemas vasculares o ictus, enfermedad del VIH, enfermedad de Parkinson, enfermedad de Huntington, enfermedad de Pick, enfermedad de Creutzfeldt-Jacob, hipoxia perinatal, otras afecciones médicas generales o abuso de sustancias); delirios, trastornos amnésicos o deterioro cognitivo relacionado con la edad.
En otra realización, la presente divulgación proporciona un método para tratar esquizofrenia o psicosis que comprende administrar a un paciente que lo necesite una cantidad eficaz de un compuesto (o composición que proporciona un compuesto) de la invención, o un estereoisómero del mismo.
Las posibles condiciones o trastornos del sueño para los que los compuestos de la invención pueden ser útiles incluyen potenciar la calidad del sueño; mejorar la calidad del sueño; aumentar el mantenimiento del sueño; aumentar el valor que se calcula a partir del tiempo que un sujeto duerme dividido por el tiempo que un sujeto intenta dormir; disminuir la latencia o inicio del sueño (el tiempo que lleva dormirse); disminuir las dificultades para quedarse dormido; aumentar de la continuidad del sueño; disminuir el número de despertares durante el sueño; disminuir las excitaciones nocturnas; disminuir el tiempo que pasa despierto después de la aparición inicial del sueño; aumentar la cantidad total de sueño; reducir la fragmentación del sueño; alterar el tiempo, la frecuencia o la duración de los episodios de sueño REM; alterar el tiempo, la frecuencia o la duración de los episodios de sueño de onda lenta (es decir, fases 3 o 4); aumentar la cantidad y el porcentaje de sueño en la fase 2; promover el sueño de onda lenta; mejorar la actividad EEG-delta durante el sueño; aumentar el estado de alerta durante el día; reducir la somnolencia diurna; tratar o reducir la somnolencia diurna excesiva; insomnio; hipersomnio; narcolepsia; sueño interrumpido; apnea del sueño; desvelo; mioclono nocturno; interrupciones del sueño REM; descompensación horaria; alteraciones del sueño de los trabajadores por turnos; disomnios; noche de terror; insomnios asociados a la depresión, trastornos emocionales/anímicos, así como caminar dormido enuresis y trastornos del sueño que acompañan al envejecimiento; ocaso del Alzheimer; afecciones asociadas a la ritmicidad circadiana, así como trastornos mentales y físicos asociados a los viajes a través de zonas horarias y con horarios rotativos de trabajo por turnos; afecciones debidas a medicamentos que provocan reducciones en el sueño REM como un efecto secundario; síndromes que se manifiestan por sueño no reparador y dolor muscular o apnea del sueño que se asocia a perturbaciones respiratorias durante el sueño; y afecciones que resultan de una disminución de la calidad del sueño.
Los compuestos de la invención también pueden usarse para tratar o prevenir discinesias. Adicionalmente, los compuestos de la invención pueden usarse para disminuir la tolerancia y/o dependencia al tratamiento del dolor con opioides y para el tratamiento del síndrome de abstinencia de, por ejemplo, alcohol, opioides y cocaína.
El sujeto o paciente al cual se administran los compuestos de la presente invención es generalmente un ser humano, de sexo masculino o femenino, en quienes se desea la inhibición del receptor mGluR2, pero también puede abarcar otros mamíferos tales como aquellos enumerados anteriormente, incluyendo perros, gatos, ratones, ratas, ganado bovino, caballos, oveja, conejos, monos, chimpancés u otros monos o primates, para los cuales se desea el tratamiento de los trastornos anteriormente indicados o el estudio de mGluR2.
Otra realización proporciona un medicamento o composición farmacéutica para la modulación alostérica negativa de un receptor mGluR2 y/o para el tratamiento de cualquiera de las enfermedades o trastornos enumerados anteriormente
a un paciente (preferentemente un ser humano) que necesita dicho tratamiento, que comprende un compuesto (o composición que comprende un compuesto) de la invención, o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, y un vehículo farmacéuticamente aceptable.
Otra realización proporciona un método para la fabricación de un medicamento o una composición farmacéutica para la modulación alostérica negativa de un receptor mGluR2 y/o para tratar una o más enfermedades o afecciones enumeradas anteriormente, que comprende combinar un compuesto (o composición que comprende un compuesto) de la invención, o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, con un vehículo farmacéuticamente aceptable.
TERAPIA DE COMBINACIÓN
Los compuestos y composiciones de la presente invención pueden usarse en combinación con uno o más fármacos distintos en el tratamiento de enfermedades o afecciones para las cuales los compuestos de la presente tienen utilidad, donde se desea la combinación de fármacos, por ejemplo, donde la combinación es más segura o más eficaz que cualquier fármaco solo. Los compuestos de la invención pueden usarse junto con uno o más de otros fármacos que tratan, previenen, controlan, mejoran o reducen el riesgo de efectos secundarios o toxicidad de los compuestos de la invención. Tales otros fármacos pueden administrarse por una vía y en una cantidad usada comúnmente para ello, de manera simultánea o secuencial con el compuesto de la presente invención. En consecuencia, las composiciones farmacéuticas de la presente invención incluyen aquellas que contienen uno o más principios activos diferentes, además de los compuestos de la presente invención. Las combinaciones pueden administrarse opcionalmente como parte de un producto de combinación de forma de dosificación unitaria, o como un kit o protocolo de tratamiento en donde uno o más fármacos adicionales se administran en formas de dosificación separadas como parte de un régimen de tratamiento. En una realización, los compuestos de la invención útiles en dichas combinaciones comprenden un compuesto de acuerdo con una cualquiera de las Fórmulas (I), (IA), (IA-1), (IA-1a), (IA-1 b), (IB), (IC) y (ID) como se describió en el presente documento o de acuerdo con cualquiera de las diversas realizaciones descritas en el presente documento o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, o una composición farmacéuticamente aceptable del mismo. En otra realización, los compuestos de la invención útiles en dichas combinaciones comprenden los compuestos de los ejemplos, por ejemplo, como se expone como compuestos de ejemplo de la invención. En otra realización, los compuestos de la invención útiles en dichas combinaciones comprenden el compuesto del ejemplo 2-5, o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichas combinaciones comprende el compuesto del ejemplo 2-3A o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichas combinaciones comprende el compuesto del ejemplo 2- 3B o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichas combinaciones comprende el compuesto del ejemplo 3-1A, o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichas combinaciones comprende el compuesto del ejemplo 3-1B o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichas combinaciones comprende el compuesto del ejemplo 3-7A o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo. En otra realización, el compuesto de la invención útil en dichas combinaciones comprende el compuesto del ejemplo 3-7B o una sal farmacéuticamente aceptable de dicho compuesto, o una composición farmacéuticamente aceptable del mismo.
En otra realización, puede emplearse un compuesto o composición de la invención en combinación con inhibidores de acetilcolinesterasa tales como donepezilo y rivastigmina, antagonistas de NMDA tales como memantina, moduladores del receptor muscarínico, moduladores del receptor de AMPA, moduladores de los receptores de mGluR3, moduladores de los receptores nicotínicos alfa-7 y alfa-4-beta 2, moduladores de los receptores 5-HT6 y 5-HT4, moduladores de fosfodiesterasas (PDE), antagonistas del receptor alfa 2c, histonas desacetilasas y terapias antioxidantes.
En otra realización, puede emplearse un compuesto o composición de la invención en combinación con terapias que pueden alterar o modificar el transcurso de la progresión de la enfermedad, incluyendo las terapias moduladoras de beta-amiloide tales como inhibidores de BACE, anticuerpos contra BACE, moduladores de gamma-secretasa, moduladores de tau y/o fósforo-tau, y terapias biológicas que modulan placas asociadas a trastornos neurológicos incluyendo anticuerpos, ARNi, miARN y terapias celulares. Los inhibidores de BACE adecuados incluyen pero no se limitan a verubecestat (Merck & Co., Inc.), AZD2392 (Eli Lilly & Co./Astra Zeneca), CTS-21166 (CoMentis), E2609 (Biogen I dec, Inc./Esai Co., Ltd.), and BAN2401 (Biogen Idec, Inc./Esai Co., Ltd.).
En otra realización, un compuesto o composición de la invención puede emplearse en combinación con levodopa (con o sin un inhibidor selectivo de la descarboxilasa extracerebral, tal como carbidopa o benserazida), anticolinérgicos, tales como biperideno (opcionalmente como su clorhidrato o sal de lactato) y clorhidrato de trihexifenidilo (benzexol), inhibidores de la COMT tales como entacapona, inhibidores de la MAO-B, antioxidantes, antagonistas de receptores
de adenosina A2a, agonistas colinérgicos, antagonistas de receptor NMDA, antagonistas de receptores de serotonina y agonistas de receptores de dopamina, tales como alentemol, bromocriptina, fenoldopam, lisurida, naxagolida, pergolida o pramipexol. Se apreciará que el agonista de la dopamina puede estar en forma de una sal farmacéuticamente aceptable, por ejemplo, bromhidrato de alentemol, mesilato de bromocriptina, mesilato de fenoldopam, clorhidrato de naxagolida y mesilato de pergolida.
Los ejemplos adicionales de combinaciones de los compuestos incluyen combinaciones con agentes para el tratamiento del dolor, por ejemplo agentes antiinflamatorios no esteroideos, tales como aspirina, diclofenaco, duflunisal, fenoprofeno, flurbiprofeno, ibuprofeno, indometacina, cetoprofeno, cetorolaco, naproxeno, oxaprozina, piroxicam, sulindaco y tolmetina; inhibidores de COX-2, tales como celecoxib, rofecoxib, valdecoxib, 406381 y 644784; agonistas de CB-2, tales como 842166 y SAB378; antagonistas de VR-1, tales como AMG517, 705498, 782443, PAC20030, V114380 y A425619; antagonistas del receptor de bradiquinina B1, tales como SSR240612 y NVPSAA164; bloqueantes y antagonistas de los canales de sodio, tales como VX409 y SPI860; inhibidores de la óxido nítrico sintasa (NOS) (incluyendo inhibidores de iNOS y nNOS), tales como SD6010 y 274150; antagonistas del sitio de glicina, incluyendo lacosamida; agonistas nicotínicos neuronales, tales como ABT 894; antagonistas de NMDA, tales como AZD4282; abridores del canal de potasio; antagonistas de los receptores de AMPA/kainato; bloqueantes de los canales de calcio, tales como ziconotida y NMED160; moduladores de IO del receptor GABA-A (por ejemplo, un agonista del receptor GABA-A); inhibidores de la metaloproteasa de matriz (MMP); agentes trombolíticos; analgésicos opioides tales como la codeína, fentanilo, hidromorfona, levofanol, meperidina, metadona, morfina, oxicodona, oximorfona, pentazocina, propoxifeno; factor inhibidor de neutrófilos (NIF); pramipexol, ropinirol; anticolinérgicos; amantadina; inhibidores de la monoaminooxidasa B15 ("MAO-B"); agonistas o antagonistas del receptor 5HT; antagonistas de mGlu5, tales como AZD9272; alfa agonistas, tales como AGNXX/YY; agonistas nicotínicos neuronales, tales como ABT894; agonistas o antagonistas del receptor NMDA, tales como AZD4282; antagonistas de NKI; inhibidores selectivos de la recaptación de serotonina ("SSRI") y/o inhibidores selectivos de la recaptación de serotonina y norepinefrina ("SSNRI"), tales como duloxetina; fármacos antidepresivos tricíclicos, moduladores de norepinefrina; litio; valproato; gabapentina; pregabalina; rizatriptán; zolmitriptán; naratriptán y sumatriptán.
En otra realización, los compuestos y composiciones de la invención pueden administrarse en combinación con compuestos útiles para el tratamiento de la esquizofrenia o para mejorar la calidad del sueño y prevenir y tratar los trastornos del sueño y las alteraciones del sueño, incluyendo, por ejemplo, sedantes, hipnóticos, ansiolíticos, antipsicóticos, agentes ansiolíticos, antihistamínicos, benzodiazepinas, barbitúricos, ciclopirrolonas, antagonistas de la orexina (tales como suvorexant), agonistas de orexina, antagonistas alfa-1, agonistas de GABA, antagonistas de 5HT-2 incluyendo antagonistas de 5HT-2A y antagonistas de 5HT-2A/2C, antagonistas de histamina incluyendo los antagonistas de histamina H3, agonistas inversos de histamina H3, imidazopiridinas, tranquilizantes leves, agonistas y antagonistas de melatonina, agentes melatonérgicos, agonistas y antagonistas de procineticina, pirazolopirimidinas, antagonistas de los canales de calcio tipo T, triazolopiridinas y similares, o el compuesto de la presente invención puede administrarse junto con el uso de métodos físicos tales como fototerapia o estimulación eléctrica.
Cuando se administra una terapia de combinación a un paciente que necesita dicha administración, los agentes terapéuticos en la combinación, o una composición o composiciones farmacéuticas que comprenden los agentes terapéuticos, pueden administrarse en cualquier orden tal como, por ejemplo, secuencialmente, simultáneamente, conjuntamente, simultáneamente y similares.
En una realización, el compuesto de la invención se administra durante un tiempo en el que el agente o agentes terapéuticos adicionales ejercen su efecto profiláctico o terapéutico, o viceversa.
En otra realización, el compuesto de la invención y el agente o agentes terapéuticos adicionales se administran en las dosis comúnmente empleadas cuando dichos agentes se usan como monoterapia para tratar el trastorno.
En otra realización, el compuesto de la invención y el agente o agentes terapéuticos adicionales se administran en dosis menores que las dosis comúnmente empleadas cuando dichos agentes se usan como monoterapia para tratar el trastorno.
En una realización, el compuesto de la invención y el agente o agentes terapéuticos adicionales están presentes en la misma composición, que es adecuada para la administración oral.
En algunas realizaciones, el compuesto de la invención y el agente o agentes terapéuticos adicionales pueden actuar de forma aditiva o sinérgica. Una combinación sinérgica puede permitir el uso de dosificaciones menores de uno o más agentes y/o la administración menos frecuente de uno o más agentes de una terapia de combinación. Una dosificación inferior o una administración menos frecuente de uno o más agentes pueden disminuir la toxicidad de la terapia sin reducir la efectividad de la terapia.
Las dosis y el régimen de dosificación del agente o agentes terapéuticos adicionales usados en las terapias de combinación de la presente invención para el tratamiento o la prevención de una enfermedad o trastorno pueden determinarse por el médico encargado, teniendo en cuenta las dosis y el régimen de dosificación aprobados en el
prospecto; la edad, el sexo y el estado de salud general del paciente; y el tipo y la gravedad de la infección vírica o la enfermedad o el trastorno relacionados.
Otra realización proporciona un kit que comprende una cantidad terapéuticamente eficaz del compuesto (o una composición que comprende un compuesto) de la invención, o un estereoisómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, opcionalmente junto con al menos un agente terapéutico adicional enumerado anteriormente, y un portador, vehículo o diluyente farmacéuticamente aceptable.
Cuando se administra una terapia de combinación a un paciente que necesita dicha administración, los agentes terapéuticos en la combinación, o una composición o composiciones farmacéuticas que comprenden los agentes terapéuticos, pueden administrarse en cualquier orden tal como, por ejemplo, secuencialmente, simultáneamente, conjuntamente, simultáneamente y similares.
En una realización, el compuesto de la invención se administra durante un tiempo en el que el agente o agentes terapéuticos adicionales ejercen su efecto profiláctico o terapéutico, o viceversa.
En una realización, el compuesto de la invención y el agente o agentes terapéuticos adicionales están presentes en la misma composición, que es adecuada para la administración oral.
EJEMPLOS PREPARATIVOS
En general, los compuestos de la invención pueden producirse mediante varios procesos conocidos por los expertos en la técnica y mediante procesos conocidos análogos a los mismos. La invención desvelada en el presente documento se ejemplifica mediante las siguientes preparaciones y ejemplos que no deben interpretarse como limitantes del alcance de la divulgación. Las vías mecánicas alternativas y las estructuras análogas resultarán evidentes para los expertos en la materia. El practicante no se limita a estos métodos.
Un experto en la materia reconocerá que una ruta se optimizará dependiendo de la elección de sustituyentes de apéndice. Adicionalmente, un experto en la materia reconocerá que, en algunos casos, el orden de los pasos debe controlarse para evitar la incompatibilidad del grupo funcional.
Los compuestos preparados pueden analizarse para determinar su composición y pureza, así como caracterizarse mediante técnicas analíticas estándar tales como, por ejemplo, análisis elemental, RMN, espectroscopía de masas y espectros de IR.
Un experto en la materia reconocerá que los reactivos y disolventes realmente usados pueden seleccionarse entre varios reactivos y disolventes bien conocidos en la técnica como equivalentes eficaces. Por tanto, cuando se menciona un disolvente o reactivo específico, se pretende que sea un ejemplo ilustrativo de las condiciones deseables para ese esquema de reacción particular o para la preparación descrita a continuación.
Cuando se presenten datos de RMN, los espectros de 1H se obtuvieron en un Varian 400, AVANCE III 400 o Varian AS500, y los cambios químicos se informan como ppm con el número de protones y multiplicidades indicados entre paréntesis. Cuando se presentan los datos de CL/Em , los análisis se realizaron usando un Waters Acquity UPLC (columna BEH C18, 1,0 x 50 mm, 1,7 um, UV 254 nm, 2 min MeCN al 10-99 %/agua gradiente de TFA al 0,05 %, IEN positivo) o un Agilent 1200 o Shimadzu 20AB series (con una columna Xtimate C18, 2,1 x 30 mm, 3 um, UV 220 o 254 nm, 1,5 ml/4 l de TFA en agua (disolvente A) y 0,75 ml/4 l de TFA en acetonitrilo (disolvente B), usando el gradiente de elución al 10 %-80 % (disolvente B) durante 0,9 minutos y manteniéndolo al 80 % durante 0,6 minutos a un caudal de 1,2 ml/min, IEN positivo).
Las separaciones por HPLC quiral preparativa se llevaron a cabo generalmente usando cromatografía de fluidos supercríticos eluyendo una columna quiral como OJ-H, (4,6 x 250 mm, Chiral Technologies, Inc., West Chester, Pensilvania) con una fase móvil de isopropanol y CO2 supercrítico.
Se pueden usar las siguientes abreviaturas a lo largo del texto:
Ac = acetilo; ac. = acuoso; CO = monóxido de carbono; Me = metilo; Et = etilo; f-Bu: = ferc-butilo; Ar: = arilo; Ph = fenilo; Bn = bencilo; EtOH = alcohol etílico; IPA = alcohol isopropílico; AIBN = Azobisisobutironitrilo; ACN = acetonitrilo; AcOK = acetato potásico; Boc = ferc-butiloxicarbonilo; BOP:= hexafluorofosfato de benzotriazoliloxitris(dimetilamino)fosfonio; calc. (o calcd.) = calculado; SFC quiral = cromatografía de fluidos supercríticos en columna quiral; CIZn = Clorocinc; DAST = trifluororo de dietilaminoazufre; DCE = dicloroetano; DCM = diclorometano; DCM/i-PrOH = diclorometano/iso-propanol; DEA = dietilamina; DIBAL-H = hidruro de diisobutilaluminio; DIPEA = N,N-diisopropiletilamina; DMA = dimetilacetamida; DMEM = medio Eagle modificado de Dulbecco (alto en glucosa); DMF:= dimetilformamida; DMFDMA = W,W-dimetilformamida dimetilacetal; DMP = peryodinano de dess-martin; DMSO = dimetilsulfóxido; dppf = fosforoferrocenilo de difenilo; FBS = suero bovino fetal; HATu = hexafluorofosfato de N,N,N',N'-tetrametil-O-(7-azabenzotriazol-1-il)uronio; (HCHO)n = paraformaldehído; HMDS = hexametildisilazano; HPLC = cromatografía líquida de alta resolución; m-CPBA = ácido metacloroperoxibenzoico; M = molar; m-CPBA (o mCPBA) = ácido meta-cloroperoxibenzoico; EM = espectrometría de masas; EM (IEN) calc. = espec. de masas (Ionización por electronebulización); Ms = mesilo; NMO = N-óxido de N-metil morfolina; Pd(dtbpf)Cl2 = 1,1'-Bis(di-terc-butilfosfino)ferroceno]dicloropaladio (N); PMB = p-metoxibencilo; TLC prep. = cromatografía preparativa de capa fina; ta = temperatura ambiente; SFC =cromatografía de fluidos supercríticos; TEA = trietilamina; TFA = ácido trifluoroacético; THF = tetrahidrofurano; TPAP = perrutenato de tetra-npropil amonio.
Los siguientes compuestos intermedios se prepararon como se describe a continuación para su uso en la preparación de compuestos de la invención.
INTERMEDIO 2-1

2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)n¡cot¡naldehído
Etapa 1: ácido 4-bromo-2.6-d¡cloron¡cotín¡co

Se añadió gota a gota n-butillitio (38,8 ml, 97 mmol) a diisopropilamina (14,91 ml, 106 mmol) en THF (50 ml) a -78 °C. La solución se agitó a -78 °C durante 0,5 h y a 0 °C durante 0,5 h. La solución en bruto de color amarillo pálido de diisopropilamida de litio se usó en la siguiente etapa sin purificación adicional.
A una solución en agitación y enfriada (-78 °C) de 4-bromo-2,6-dicloropiridina (20 g, 88 mmol) en THF (550 ml) se le añadió gota a gota la solución preformada de diisopropilamida de litio en atmósfera de N2. La mezcla se agitó a -78 °C durante 1 h, se añadió dióxido de carbono (hielo seco) (50 g, 1136 mmol) a la mezcla de reacción a -78 °C y se agitó durante 1 h. Se añadió Na2CO3 acuoso 1 M (1 l) para interrumpir la reacción. La mezcla se extrajo con acetato de etilo (1 l), la solución acuosa se acidificó con HCl acuoso 1 M a pH~4. La mezcla se extrajo con acetato de etilo (2 x 1 l). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar el producto que se usó en etapas posteriores sin purificación adicional. EM (IEN) calc. para (C6H3BrCbNO2)[M+H]+, [271,86], encontrado, [271,7].
RMN 1H (400 MHz, DMSO-d6) 68,48 - 7,66 (m, 1H)
Etapa 2: 4-bromo-2.6-d¡cloron¡cot¡nato de (trimetilsilil)metilo y 4-bromo-2.6-d¡cloron¡cot¡nato de metilo

A una solución de ácido 4-bromo-2,6-dicloronicotínico (19,5 g, 72,0 mmol) en THF (500 ml) se le añadió gota a gota (trimetilsilil)diazometano (54,0 ml, 108 mmol) a 15 °C. La mezcla se agitó a 15 °C durante 8 h. La mezcla se inactivó con HCl (2M) (20 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El producto en bruto se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 120 g SepaFlash®, eluyendo con acetato de etilo del 0 al 5 %/éter de petróleo) para dar una mezcla de los productos. EM (IEN) calc. para (C10H13BrCl2NO2Si)[M+H]+, 357,9, encontrado, 357,8. EM (IEN) calc. para (CyH5BrCbNO2) [M+H]+, 285,9, encontrado, 285,8.
Etapa 3: 4-bromo-2.6-d¡cloron¡cot¡naldehído

A una solución de 4-bromo-2,6-dicloronicotinato de (trimetilsilil)metilo y 4-bromo-2,6-dicloronicotinato de metilo (17 g, ~60 mmol) en tolueno (400 ml) se le añadió gota a gota DIBAL-H (65,6 ml, 65,6 mmol) a -78 °C. La mezcla se agitó a -78 °C durante 2 h. La mezcla se inactivó con NH4Cl acuoso saturado (300 ml). Se añadió HCl acuoso (2 M,150 ml) y la mezcla se extrajo con acetato de etilo (2 x 500 ml), los extractos orgánicos combinados se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 120 g SepaFlash®, eluyendo con acetato de etilo del 0 al 3 %/éter de petróleo) para dar el producto. RMN 1H (400 MHz, CDCfj) ó 10,36 (s, 1H), 7,77 - 7,63 (m, 1H)
Etapa 4: 2.6-d¡cloro-4-(2.4-difluorofen¡l)n¡cot¡naldehído

Una mezcla de ácido (2,4-difluorofenil)borónico (4,52 g, 28,6 mmol), 4-bromo-2,6-dicloronicotinaldehído (7,3 g, 28,6 mmol) y Na2CO3 (6,07 g, 57,3 mmol) en 1,4-dioxano (120 ml) y agua (12 ml) se añadió PdCh(dppf) (1,0 g, 1,4 mmol) en atmósfera de N2. La mezcla se calentó a 100 °C durante 2 h. Después de enfriar a 20 °C, la mezcla se vertió en agua (500 ml) y se extrajo con acetato de etilo (2 x 500 ml), las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 80 g SepaFlash®, eluyendo con acetato de etilo del 0 al 3 %/éter de petróleo) para dar el compuesto del título. RMN 1H (400 MHz, CDCla) ó 10,34 (s, 1H), 7,29 - 7,19 (m, 2H), 7,01 (dt, J = 1,5, 8,3 Hz, 1H), 6,95 - 6,86 (m, 1H)
INTERMEDIO 2-2

(E)-4-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-2-oxobut-3-enoato de etilo
Véase procedimiento para el Ejemplo 2-1A y el 2-1B, Etapa 1
INTERMEDIO 2-3

etil 4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-2-h¡drox¡butanoato
Véase procedimiento para el Ejemplo 2-1A y el 2-1B, Etapa 2
INTERMEDIO 2-4

5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-blp¡r¡d¡n-7-carbox¡lato de etilo
Véase proced¡m¡ento para el Ejemplo 2-1A y el 2-1B. Etapa 5
INTERMEDIOS 2-5A. 2-5B. 2-5C y 2-5D

(2S.3R)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-blp¡r¡d¡n-7-carbox¡lato de etilo. (2S.3S)-5-í2.4-d¡fluorofen¡l)-2-íh¡drox¡met¡h-3-met¡l-3.4-d¡h¡dro-2H-p¡ranor2.3-blp¡r¡d¡n-7-carbox¡lato de et¡lo. Í2R.3R1-5-Í2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-blp¡r¡d¡n-7-carbox¡lato de et¡lo y (2R.3S)-5-(2.4-d¡fluorofen¡l)-2-íh¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡ranor2.3-blp¡r¡d¡n-7-carbox¡lato de etilo
Véase proced¡m¡ento para el Ejemplo 2-2A. 2-2B. 2-2C y 2-2D. Etapa 8
INTERMEDIO 2-6

3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)propanoato de metilo Véase procedimiento para el Ejemplo 2-3A y el 2-3B, Etapa 3
INTERMEDIO 2-7

3-í2.6-d¡cloro-4-í2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)propanal
Véase procedimiento para el Ejemplo 2-3A y el 2-3B, Etapa 5
INTERMEDIO 3-1

5-(2.4-d¡fluorofen¡l)-2-form¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo Véase procedimiento para el Ejemplo 3-7A y el 3-7B, Etapa 1
INTERMEDIO 3-2

ácido 7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-carboxíl¡co Véase procedimiento para el Ejemplo 2-9A, 2-9B, 2-9C y 2-9D, Etapa 1

Los compuestos de Fórmula I (en donde en el Esquema 1 X es un puente de dos metilenos o no está presente, el anillo B, n, y cada R1 son como se definen en la Fórmula I, y R y R' son H o metilo) se pueden preparar de acuerdo con al Esquema 1 mediante la aminación del aldehído 1-1 seguido de acilación y ciclación de 1-3 para formar piridina 1-4. La piridina 1-4 se desacila, se clora y se carbonila secuencialmente para generar cloropiridina 1-7. La cloropiridina 1-7 puede acoplarse directamente con un ácido borónico aromático, o convertirse él mismo en un ácido borónico 1-7a y acoplarse a un bromuro aromático, para formar 1-8 que se trata con amoniaco para proporcionar 1-9.
EJEMPLO 1-1

4-(2.4-d¡fluorofen¡l)-5.8-d¡h¡dro-6H-p¡rano[3.4-b1p¡r¡d¡n-2-carboxam¡da
Etapa 1: 5-Am¡no-3.6-d¡h¡dro-2H-piran-4-carbox¡lato de etilo

Una mezcla de 5-(3-metox¡-3-oxoprop¡l)tetrah¡drofurano-2-carbox¡lato de met¡lo (3 g. 17.4 mmol) y NH4OCONH2 (2.65 g. 34.9 mmol) en MeOH (20 ml) se ag¡tó a 25 °C durante 2 h. La mezcla se concentró a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (eluyendo con 5:1 de éter de petróleo/acetato de et¡lo) para dar el producto. RMN 1H (400 MHz. CD3OD) ó 4.17 - 4.03 (m. 4H). 3.73 (t. J = 5.5 Hz. 2H). 2.37 - 2.19 (m. 2H). 1.24 (t. J = 7.0 Hz. 3H).
Etapa 2: 5-(3-metox¡-3-oxopropanam¡do)-3.6-d¡h¡dro-2H-p¡ran-4-carbox¡lato de etilo

Se d¡solv¡eron 5-am¡no-3.6-d¡h¡dro-2H-p¡ran-4-carbox¡lato de et¡lo (2.6 g. 15 mmol) y Et3N (6 ml. 45 mmol) en DCM (50 ml). se añad¡ó 3-cloro-3-oxopropanoato de met¡lo (2.26 g. 16 mmol) y la soluc¡ón resultante se ag¡tó durante 2 h a 0 °C. La mezcla se ¡nact¡vó con agua (30 ml) y se extrajo con DCM (3 x 40 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (30 ml). se secaron sobre Na2SO4. se f¡ltraron y se concentraron. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (eluyendo con 10:1 de éter de petróleo/acetato de et¡lo) para dar el producto. EM (IEN) calc. para (C12H18NO6) [M+H]+.272.1. encontrado. 271.9. RMN 1H (400 MHz. CDCla) ó 4.77 (s. 1H). 4.17 (c. J = 7.0 Hz. 1H). 3.79 - 3.63 (m. 3H). 3.35 (s. 1H). 2.38 (t. J = 5.3 Hz. 1H). 1.24 (t. J = 7.0 Hz. 3H).
Etapa 3: 2.4-D¡h¡drox¡-6.8-d¡h¡dro-5H-p¡rano[3.4-b1p¡r¡d¡n-3-carbox¡lato de met¡lo

Se d¡solv¡ó 5-(3-metox¡-3-oxopropanam¡do)-3.6-d¡h¡dro-2H-p¡ran-4-carbox¡lato de et¡lo (1.15 g. 4 mmol) en MeOH (15 ml). se añad¡ó metanolato sód¡co (0.25 g. 4.6 mmol) y la soluc¡ón resultante se ag¡tó durante 1 h a 80 °C. La mezcla se concentró a pres¡ón reduc¡da para proporc¡onar el producto que se usó en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón ad¡c¡onal. EM (IEN) calc. para (C10H12NO5)[M+H]+. 225.0. encontrado. 225.9. RMN 1H (400 MHz. CD3OD) ó 4.18 - 4.03 (m. 3H). 3.73 (t. J = 5.7 Hz. 2H). 2.27 (t. J = 5.7 Hz. 2H). 1.24 (t. J = 7.0 Hz. 2H).
Etapa 4: 6.8-d¡h¡dro-5H-p¡rano[3.4-b1p¡r¡d¡n-2.4-d¡ol

Se añadió HCl acuoso (6 M, 20 ml, 120 mmol) a 2,4-dihidroxi-6,8-dihidro-5H-pirano[3,4-b]piridin-3-carboxilato de metilo (1 g, en bruto) y la mezcla se agitó a 100 °C durante 4 h. El disolvente se retiró a presión reducida para dar el producto que se usó en la siguiente etapa sin purificación adicional. EM (IEN) calc. para (CsH10N03)[M+H]+, 168,0, encontrado, 167,8.
Etapa 5: 2.4-d¡cloro-6.8-d¡h¡dro-5H-p¡rano[3.4-b]p¡r¡d¡na

Una mezcla de 6,8-dihidro-5H-pirano[3,4-b]piridin-2,4-diol (0,9 g, en bruto) y DMF (5 ml, 64 mmol) en tricloruro de fosforilo (4,3 ml, 48,5 mmol) se agitó a 80 °C durante 12 h. La mezcla se dejó enfriar a temperatura ambiente y se vertió en agua enfriada con hielo y la mezcla se ajustó a pH 9 con Na2CO3 acuoso saturado. La mezcla se extrajo con EtOAc (3 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (45 ml), se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 100:0 a 20:1 de éter de petróleo/acetato de etilo) para dar el producto. EM (IEN) calc. para (C8HsC12No )[M+H]+, 204,0, encontrado, 203,9. RMN 1H (400 MHz, CDCla) ó 7,29 (s, 1H), 5,10 (d, J = 6,4 Hz, 1H), 4,86 (s a, 1H), 3,24 - 3,12 (m, 2H), 2,52 (d, J = 17,4 Hz, 2H), 2,11 (t, J = 9,6 Hz, 2H).
Etapa 6: 4-cloro-6.8-d¡h¡dro-5H-p¡rano[3.4-b1p¡r¡d¡n-2-carbox¡lato de etilo

A una solución de 2,4-dicloro-6,8-dihidro-5H-pirano[3,4-b]piridina (100 mg, 0,49 mmol) en EtOH (20 ml) se le añadieron acetato potásico (96 mg, 0,98 mmol) y PdCL(dppf) (36 mg, 0,049 mmol) en atmósfera de N2. La mezcla se desgasificó y se llenó de nuevo con CO (tres veces). La mezcla resultante se agitó a 0,34 MPa (50 psi) de CO a 60 °C durante 1,5 h. La mezcla se concentró a presión reducida para dar el producto en bruto, que se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 40 g SepaFlash®, eluyendo con acetato de etilo del 0 al 20 %/éter de petróleo at 40 ml/min) para formar el producto. Em (IEN) calc. para (CnH12ClNO3) [M+H]+,242,0, encontrado, 241,9. RMN 1H (400 MHz, CD3OD) ó 8,00 (s, 1H), 4,75 (s, 2H), 4,41 (c, J = 7,2 Hz, 2H), 4,01 (t, J = 5,9 Hz, 2H), 2,93 (t, J = 5,7 Hz, 2H), 1,39 (t, J = 7,2 Hz, 3H).
Etapa 7: 4-(2.4-D¡fluorofen¡l)-6.8-d¡h¡dro-5H-p¡rano[3.4-b1p¡r¡d¡n-2-carbox¡lato de etilo
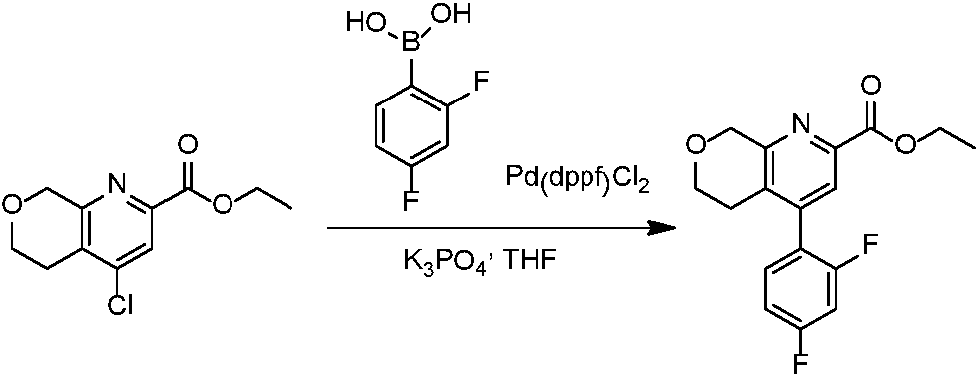
A una mezcla de 4-cloro-6,8-dihidro-5H-pirano[3,4-b]piridin-2-carboxilato de etilo (100 mg, 0,613 mmol), ácido (2,4-difluorofenil)borónico (260 mg, 1,65 mmol) y K3PO4 (263 mg, 1,24 mmol) en THF (4,5 ml) y agua (0,5 ml) se le añadió PdCh(dppf) (51 mg, 0,082 mmol), y la mezcla se desgasificó y se volvió a llenar con N2 (tres veces). La mezcla se calentó a 80 °C durante 2 h. La mezcla se concentró a presión reducida para dar el producto en bruto, que se purificó por TLC prep. (gel de sílice, eluyendo con 1:1 de acetato de etilo/éter de petróleo) para dar el producto. Em (IEN) calc. para (C17H16F2NO3) [M+H]+,320,1, encontrado, 319,9. RMN 1H (400 MHz, CD3OD) ó 7,85 (s, 1H), 7,46 - 7,34 (m, 1H), 7,13 (c, J = 7,7 Hz, 2H), 4,41 (c, J = 7,0 Hz, 2H), 4,03 - 3,85 (m, 2H), 2,80 - 2,66 (m, 2H), 1,39 (t, J = 7,2 Hz, 3H).
Etapa 8: 4-(2.4-d¡fluorofen¡l)-6.8-d¡h¡dro-5H-p¡rano[3.4-b1p¡r¡d¡n-2-carboxam¡da

Una mezcla de 4-(2,4-difluorofenil)-6,8-dihidro-5H-pirano[3,4-b]piridin-2-carboxilato de etilo (80 mg, 0,25 mmol) en NH3 en MeOH (15 ml, 150 mmol) se agitó a 25 °C durante 12 h. La mezcla se concentró a presión reducida y se purificó por HPLC prep. (Columna: Agela ASB 150x25 mm, 5 um; Fase móvil: agua del 30 % al 60 % (que contenía TFA al 0,1 %)-ACN; Caudal: 25 ml/min) para dar el compuesto del título. EM (IEN) calc. para (C15H14F2N2O2) [M+H]+,291,1, encontrado, 291,0. RMN 1H (400 MHz, CD3OD) 67,82 (s, 1H), 7,44 - 7,32 (m, 1H), 7,18 - 7,05 (m, 2H), 3,93 (t, J = 5,7 Hz, 2H), 2,70 (t, J = 5,3 Hz, 2H).
EJEMPLOS 1-2A y 1-2B

(6S.9R)-4-(2.4-d¡fluorofen¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epoxic¡clohepta[b1p¡r¡d¡n-2-carboxam¡da y (6R,9S)-4-(2,4-d¡fluorofen¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epoxic¡clohepta[b1p¡r¡d¡n-2-carboxam¡da
Etapa 1: 5-(3-metoxi-3-oxoprop¡l)furan-2-carbox¡lato de metilo

Una mezcla de 5-bromofuran-2-carboxilato de metilo (10 g, 64 mmol), acrilato de metilo (16,5 g, 192 mmol), (MeO)3P (0,4 g, 0,32 mmol), Et3N(17 ml, 128 mmol) y Pd(OAc)2 (0,36 g, 0,16 mmol) en DMF (120 ml) se desgasificó y se volvió a llenar con N2 (tres veces). La mezcla se calentó a 110 °C durante 3 h. La mezcla enfriada se retiró por filtración y el filtrado se concentró a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 5:1 de éter de petróleo/acetato de etilo) para dar el producto. RMN 1H (400 MHz, CDCb) 67,45 (d, J = 15,9 Hz, 1H), 7,20 (d, J = 3,5 Hz, 1H), 6,68 (d, J = 3,5 Hz, 1H), 6,57 (d, J = 15,9 Hz, 1H), 3,92 (s, 3H), 3,81 (s, 3H).
Etapa 2: 5-(3-metox¡-3-oxoprop¡l)tetrahidrofurano-2-carbox¡lato de metilo

A una solución de 5-(3-metoxi-3-oxopropil)furan-2-carboxilato de metilo (4,7 g, 22 mmol) en acetato de etilo (100 ml) se le añadió Pd/C (1 g) (10 % en peso) en atmósfera de N2. La mezcla se desgasificó y se llenó de nuevo con H2 (tres veces). La mezcla resultante se agitó a 0,28 MPa (40 psi) de H2 a 50 °C durante 15 h. La mezcla se filtró y el filtrado se concentró a presión reducida para dar el producto que se usó en etapas posteriores sin purificación adicional. RMN 1H (400 MHz, CDCl3) 64,42 (dd, J = 5,0, 8,5 Hz, 1H), 4,07 - 3,95 (m, 1H), 3,74 - 3,67 (m, 3H), 3,64 (s, 3H), 2,57 - 2,36 (m, 2H), 2,20 (cd, J = 8,4, 12,6 Hz, 1H), 2,11 - 1,81 (m, 5H), 1,58 (cd, J = 8,2, 11,9 Hz, 1H).
Etapa 3: 2-oxo-8-oxab¡ciclo[3.2.11octano-3-carbox¡lato de metilo

A una mezcla de 5-(3-metoxi-3-oxopropil)tetrahidrofurano-2-carboxilato de metilo (4,6 g, 21 mmol) en THF (60 ml) se le añadió NaH (840 mg, 60 % en aceite) lentamente en atmósfera de N2 y la mezcla se calentó a reflujo durante 6 h. La reacción se interrumpió con NH4Cl (50 ml) y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (60 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 5:1 de éter de petróleo/acetato de etilo) para dar el producto. RMN 1H (400 MHz, CDCla) 64,65 - 4,48 (m, 1H), 4,41 - 4,22 (m, 1H), 3,75 - 3,64 (m, 1H), 2,69 (dd, J = 4,9, 15,7 Hz, 1H), 2,40 - 2,29 (m, 1H), 2,18 - 2,01 (m, 3H), 1,94 - 1,72 (m, 2H).
Etapa 4: 2-am¡no-8-oxabic¡clo[3.2.11oct-2-eno-3-carbox¡lato de metilo

Una mezcla de 2-oxo-8-oxabiciclo[3.2.1]octano-3-carboxilato de metilo (200 mg, 1,08 mmol) y NH4OAc (770 mg, 10 mmol) en MeOH se agitó en un tubo cerrado herméticamente a 90 °C durante 15 h. La mezcla se concentró a presión reducida. El residuo se disolvió en agua (10 ml) y se extrajo con acetato de etilo (5 x 15 ml). Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 5:1 de éter de petróleo/acetato de etilo) para dar el producto. RMN 1H (400 MHz, CDCls) 64,52 (t, J = 5,0 Hz, 1H), 4,38 (s a, 1H), 3,52 (s, 3H), 2,56 (d, J = 5,3 Hz, 1H), 2,00 - 1,82 (m, 5H), 1,58 (t, J = 8,2 Hz, 1H).
Etapa 5: 2-(3-Etox¡-3-oxopropanam¡do)-8-oxab¡c¡clo[3.2.11oct-2-eno-3-carbox¡lato de metilo

Se disolvieron 2-amino-8-oxabiciclo[3.2.1]oct-2-eno-3-carboxilato de metilo (490 mg, 2,67 mmol) y Et3N (0,7 ml, 5,3 mmol) en DCM (20 ml), se añadió etil-3-cloro-3-oxopropanoato (481 mg, 3,1 mmol) y la solución resultante se agitó durante 2 h a 20 °C. La mezcla se inactivó con agua (15 ml) y se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 4:1 de éter de petróleo/acetato de etilo) para dar el producto. EM (IEN) calc. para (C14H20NO6) [M+H]+, 298,1, encontrado, 298,1.
Etapa 6: 2.4-d¡h¡drox¡-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-3-carbox¡lato de metilo

Se disolvió 2-(3-etoxi-3-oxopropanamido)-8-oxabiciclo[3.2.1]oct-2-eno-3-carboxilato de metilo (4,8 g, 16 mmol) en MeOH (80 ml), se añadió metanolato sódico (368 mg, 16,1 mmol) y la solución resultante se agitó durante 1,5 h a 80 °C. La mezcla enfriada se filtró y se concentró a presión reducida para proporcionar el producto que se usó en la siguiente etapa sin purificación adicional. EM (IEN) calc. para (C12H14NO5) [M+H]+, 252,1, encontrado, 252,0.
Etapa 7: 6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2.4-d¡ol

Se añadió HCl acuoso (20 ml, 120 mmol, 6 M) a 2,4-dihidroxi-6,7,8,9-tetrahidro-5H-6,9-epoxiciclohepta[b]piridin-3-carboxilato de metilo (1,2 g, 4,8 mmol) y la mezcla se agitó a 100 °C durante 20 h. La mezcla se extrajo con 3:1 de DCM/i-PrOH (4 x 100 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el producto que se usó en la siguiente etapa sin purificación adicional. EM (IEN) calc. para (C10H11NO3) [M+H]+, 194,1, encontrado, 193,9.
Etapa 8: 2.4-d¡cloro-6.7.8.9-tetrah¡dro-5H-6,9-epox¡c¡clohepta[b1p¡r¡d¡na

Una mezcla de 6,7,8,9-tetrahidro-5H-6,9-epoxiciclohepta[b]piridin-2,4-diol (1,2 g, 6,21 mmol) y DMF (6,5 ml, 6,21 mmol) en tricloruro de fosforilo (8,48 g, 55,3 mmol) se agitó a 90 °C durante 15 h. Se añadió Dm F (0,2 ml) y la mezcla se agitó a 90 °C durante 15 h. La mezcla enfriada se vertió en agua enfriada con hielo y el pH se ajustó a 7 con NaHCO3 sat. La mezcla se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (45 ml), se secaron sobre Na2SO4, se filtraron y se concentraron, el residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 4:1 de éter de petróleo/acetato de etilo) para dar el producto. e M (IEN) calc. para (C10H10ChNO) [M+H]+, 230,0, encontrado, 230,1. RMN 1H (400 MHz, CDCla) ó 7,29 (s, 1H), 5,10 (d, J = 6,4 Hz, 1H), 4,86 (s a, 1H), 3,24 - 3,12 (m, 2H), 2,52 (d, J = 17,4 Hz, 2H), 2,11 (t, J = 9,6 Hz, 2H).
Etapa 9: 4-cloro-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carbox¡lato de etilo

A una solución de 2,4-dicloro-6,7,8,9-tetrahidro-5H-6,9-epoxiciclohepta[b]piridina (330 mg, 1,434 mmol) en EtOH (10 ml) se le añadieron acetato potásico (282 mg, 2,87 mmol) y PdCb(dppf) (105 mg, 0,143 mmol) en atmósfera de N2. La mezcla se desgasificó y se llenó de nuevo con CO (tres veces). La mezcla resultante se agitó a 0,34 MPa (50 psi) de CO a 80 °C durante 2 h. La mezcla se concentró a presión reducida para dar el producto en bruto, que se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 40 g SepaFlash®, eluyendo con acetato de etilo del 0 al 20 % / éter de petróleo) para formar el producto. EM (IEN) calc. para (C13H15ClNO3) [M+H]+, 268,1, encontrado, 268,1.
Etapa 10: 4-(2.4-d¡fluorofen¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carbox¡lato de etilo
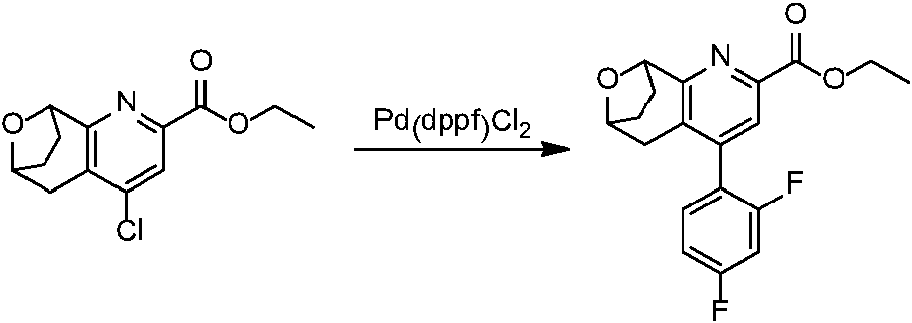
A una mezcla de 4-cloro-6,7,8,9-tetrahidro-5H-6,9-epoxiciclohepta[b]piridin-2-carboxilato de etilo (140 mg, 0,523 mmol), ácido (2,4-difluorofenil)borónico (165 mg, 1,046 mmol) y K2CO3 (145 mg, 1,046 mmol) en 1,4-dioxano (5 ml) y agua (0,5 ml) se le añadió PdCb(dppf) (38,3 mg, 0,052 mmol). La mezcla se desgasificó y se llenó de nuevo con N2 (tres veces), después se calentó a 100 °C durante 2 h. La mezcla se concentró a presión reducida para dar el producto en bruto, que se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 40 g SepaFlash®, eluyendo con acetato de etilo del 0 al 30 % / éter de petróleo) para dar el producto. EM (IEN) calc. para (C19H18F2NO3) [M+H]+, 346,1, encontrado, 346,0. RMN 1H (400 MHz, CDCla) ó 7,86 (s, 1 H), 7,21 (d, J = 6,26 Hz, 1 H), 6,91 - 7,06 (m, 2 H), 5,37 (d, J = 6,26 Hz, 1 H), 4,73 - 4,80 (m, 1 H), 4,49 (d, J = 7,04 Hz, 2 H), 3,12 - 3,22 (m, 1 H), 2,20 - 2,36 (m, 4 H), 1,57 - 1,76 (m, 2 H), 1,43 (t, J = 7,04 Hz, 3H).
Etapa 11: (6S.9R)-4-(2.4-d¡fluorofen¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carbox¡lato de etilo y (6R.9S)-4-(2.4-d¡fluorofen¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carbox¡lato de etilo

Se resolvió 4-(2,4-difluorofenil)-6,7,8,9-tetrahidro-5H-6,9-epoxiciclohepta[b]piridin-2-carboxilato de etilo racémico (100 mg, 0,290 mmol) a través de SFC quiral (Columna: Chiralpak AD 250x30 mm, 5 um; Fase móvil: EtOH del 15 % al 15 % (que contenía DEA al 0,05 %) en CO2; Caudal: 60 ml/min) para dar los dos enantiómeros.
Etapa 12: (6S.9R)-4-(2.4-d¡fluorofen¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epoxic¡clohepta[b1p¡r¡d¡n-2-carboxam¡da y (6R,9S)-4-(2.4-d¡fluorofen¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epoxic¡clohepta[b1p¡r¡d¡n-2-carboxam¡da

Un enantiómero de 4-(2,4-difluorofenil)-6,7,8,9-tetrahidro-5H-6,9-epoxiciclohepta[b]piridin-2-carboxilato de etilo (40 mg, 0,116 mmol) se mezcló con amoniaco en MeOH (15 ml, 150 mmol) y se agitó a 20 °C durante 17 h. La mezcla se concentró a presión reducida para dar un enantiómero del compuesto del título. EM (IEN) calc. para (C17H15F2 N2O2) [M+H]+, 317,1, encontrado, 317,0. RMN 1H (400 MHz, CD3OD) ó 7,82 (s, 1 H), 7,31 - 7,41 (m, 1 H), 7,07 - 7,18 (m, 2 H), 5,17 (d, J = 5,95 Hz, 1 H), 4,75 (t, J = 5,84 Hz, 1 H), 3,11 (s a, 1 H), 2,03 - 2,39 (m, 5 H), 1,69 (s a, 1 H).
El similar tratamiento del otro enantiómero de 4-(2,4-difluorofenil)-6,7,8,9-tetrahidro-5H-6,9-epoxiciclohepta[b]piridin-2-carboxilato de etilo proporcionó el otro enantiómero del compuesto del título. EM (IEN) calc. para (C17H15F2N2O2) [M+H]+, 317,1, encontrado, 317,0. RMN 1H (400 MHz, CD3OD) ó 7,81 (s, 1 H), 7,36 (d, J = 6,39 Hz, 1 H), 7,07 - 7,18 (m, 2 H), 5,17 (d, J = 5,95 Hz, 1 H), 4,74 (s a, 1 H), 3,12 (dd, J = 17,42, 5,29 Hz, 1 H), 2,03 - 2,39 (m, 4 H), 1,62 - 1,72 (m, 1 H).
Los siguientes compuestos se prepararon de acuerdo con los procedimientos descritos anteriormente.

continuación

EJEMPLOS 1-6A y 1-6B

(6S.9R)-4-(3.5-d¡fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carboxam¡da y (6R.9S)-4-(3.5-d¡fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carboxam¡da
Etapa 1: ác¡do (2-(etox¡carbon¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-4-¡l)borón¡co

Una mezcla de 4-cloro-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carbox¡lato de et¡lo (Ejemplo 1-2 Etapa 9) (200 mg. 0.75 mmol). 4.4.4'.4'.5.5.5'.5'-octamet¡l-2.2'-b¡(1.3.2-d¡oxaborolano) (1900 mg. 7.5 mmol). Pd(dtbpf)Cl2 (48.7 mg. 0.08 mmol) y AcOK (220 mg. 2.2 mmol) en 1.4-d¡oxano (5 ml) se ag¡tó a 100 °C en atmósfera de N2 durante 2 h. La mezcla se concentró a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por HPLC prep. (Columna: Waters Xbr¡dge Prep OBD C18 100x19 mm. 5 um; Fase móv¡l: agua del 28 % al 58 % (que contenía h¡dróx¡do de amon¡aco al 0.05 % v/v)-ACN; Caudal: 25 ml/m¡n) para dar el producto. EM (IEN) calc. para (C13H17BNO5) [M+H]+. 278.1. encontrado. 277.9. Etapa 2: 4-(3.5-D¡fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carbox¡lato de etilo

Una mezcla de ác¡do (2-(etox¡carbon¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-4-¡l)borón¡co (100 mg.
0.361 mmol). 2-bromo-3.5-d¡fluorop¡r¡d¡na (140 mg. 0.722 mmol). Pd(dtbpf)Cl2 (23.5 mg. 0.04 mmol) y K3PO4 (230 mg.
1.08 mmol) en THF (5 ml) y H2O (1 ml) se ag¡tó a 80 °C en atmósfera de N2 durante 2 h. La mezcla se concentró a pres¡ón reduc¡da y se pur¡f¡có por HPLC prep. (Columna: Phenomenex Synerg¡ C18 150x30 mm. 4 um; Fase móv¡l: agua del 41 % al 61 % (que contenía TFA al 0.1 %)-ACN; Caudal: 25 ml/m¡n) para dar el producto. EM (IEN) calc. para (C18H17F2N2O3) [M+H1+. 347.1. encontrado. 346.9.
Etapa 3: (6R.9S)-4-(3.5-d¡fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carbox¡lato de et¡lo y (6S.9R)-4-(3.5-d¡fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carbox¡lato de etilo

Se resolv¡ó 4-(3.5-d¡fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carbox¡lato de et¡lo racém¡co (30 mg. 0.087 mmol) por SFC qu¡ral (Columna: Ch¡ralpak AD 250x30 mm. 10 um; Fase móv¡l: EtOH al 45 % (que contenía DEA al 0.05 %) en CO2 ; Caudal: 80 ml/m¡n) para dar los dos enant¡ómeros.
Etapa 4: (6R.9S)-4-(3.5-d¡fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b]p¡r¡d¡n-2-carboxaiT i¡da y (6S.9R)-4-(3.5-d¡fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b]p¡r¡d¡n-2-carboxam¡da

Una soluc¡ón de un enant¡ómero de 4-(3.5-d¡fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b]p¡r¡d¡n-2-carbox¡lato de et¡lo (12 mg. 0.035 mmol) en amon¡aco (10 M en MeOH) (20 ml) se ag¡tó a 26 °C durante 12 h. La mezcla se concentró a pres¡ón reduc¡da y se pur¡f¡có por HPLC prep. (Columna: Phenomenex Synerg¡ C18 150x30 mm. 4 um; Fase móv¡l: agua del 25 % al 45 % (que contenía TFA al 0.1 %)-ACN; Caudal: 25 ml/m¡n) para dar un enant¡ómero del compuesto del título. EM (IEN) calc. para (C16H14F2N3O2 ) [M+H]+.318.1. encontrado. 317.9. RMN 1H (400 MHz. CD3OD) 68.53 (s. 1H). 7.96 (s. 1H). 7.79 (t. J = 8.38 Hz. 1H). 5.19 (d. J = 5.95 Hz. 1H). 4.68 - 4.79 (m.
1H). 3.25 (dd. J = 5.40; 17.75 Hz. 1H). 2.48 (d. J = 17.86 Hz. 1H). 2.07 - 2.32 (m. 4H). 1.71 (s a. 1H).
El s¡m¡lar tratam¡ento del otro enant¡ómero de 4-(3.5-d¡fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b]p¡r¡d¡n-2-carbox¡lato de et¡lo proporc¡onó el otro enant¡ómero del compuesto del título. EM (IEN) calc. para (C16H14F2N3O2) [M+H]+. 318.1. encontrado. 317.9. RMN 1H (400 MHz. CD3OD)6 8.53 (s. 1H). 7.97 (s. 1H). 7.79 (t. J = 8.16 Hz. 1H). 5.19 (d. J = 5.51 Hz. 1H). 4.65 - 4.80 (m. 1H). 3.20 - 3.27 (m. 1H). 2.48 (d. J = 17.42 Hz. 1H). 2.16 - 2.31 (m. 2H). 2.05 - 2.16 (m. 1H). 1.71 (s a. 1H).
EJEMPLOS 1-7A y 1-7B

(6S.9R)-4-(5-cloro-3-fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carboxam¡da y (6R.9S)-4-(5-cloro-3-fluorop¡r¡d¡n-2-¡l)-6.7.8.9-tetrah¡dro-5H-6.9-epox¡c¡clohepta[b1p¡r¡d¡n-2-carboxam¡da
Los compuestos del título se prepararon de acuerdo con el m¡smo proced¡m¡ento como el Ejemplo 1-6A y 1-6B. sust¡tuyendo 2-bromo-3.5-d¡fluorop¡r¡d¡na por 2-bromo-5-cloro-3-fluorop¡r¡d¡na.
Enant¡ómero 1: EM (IEN) calc. para (C16H13CFN3O2) [M+H]+. 334.07. encontrado. 333.8. RMN 1H (400 MHz. CD3OD) 68.56 (s. 1H). 7.94 - 8.00 (m. 2H). 5.18 (d. J = 5.87 Hz. 1H). 4.73 (t. J = 5.67 Hz. 1H). 3.20 - 3.26 (m. 1H).
2.48 (d. J= 17.61 Hz. 1H). 2.17 - 2.27 (m. 2H). 2.08 (t. J = 9.78 Hz. 1H). 1.63 - 1.75 (m. 1H).
Enant¡ómero 2: EM (IEN) calc. para (C16H13CFN3O2) [M+H]+. 334.07. encontrado. 333.8. RMN 1H (400 MHz. CD3OD) 68.56 (s. 1H). 7.93 - 8.00 (m. 2H). 5.17 (d. J = 6.26 Hz. 1H). 4.73 (t. J = 5.87 Hz. 1H). 3.24 (dd. J = 17.80; 5.28 Hz. 1H). 2.47 (d. J = 18.00 Hz. 1H). 2.12 - 2.30 (m. 2H). 2.04 - 2.12 (m. 1H). 1.63 - 1.75 (m. 1H).

Los compuestos de Fórmula I (en donde, en el esquema 2, R2, R2A, R3, R3A, el anillo B, n y cada R1 son como se describen en la Fórmula I, y en donde R2x, R2y R3x, R3y corresponde a R2, R2A, R3, R3A, o pueden convertirse por métodos conocidos por los expertos en la materia a R2, R2A R3, R3A, respectivamente) se puede preparar de acuerdo con el Esquema 2 mediante una reacción de Wittig con aldehído 2-1 seguida de reducción (y/o adición según sea apropiado) y ciclación de 2-3 para formar el biciclo 2-4. Como alternativa, se pueden formar 2-3 a través de una alquilación del aldehído 2-1 seguida de desoxigenación radical y posterior reducción (y/o adición según sea apropiado). El biciclo 2-4 se carbonila secuencialmente y se trata con amoniaco para producir 2-6. Las reacciones en la cadena de carbono de 2-2 y 2-3 y/o en el grupo R2x, R2y R3x, R3y de 2-2, 2-2a, 2-3 o 2-4 se pueden usar para realizar cambios adicionales en el núcleo bicíclico o sustituyentes de los mismos.
EJEMPLOS 2-1A y 2-1B

(S)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da y (R)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: (E)-4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-2-oxobut-3-enoato de et¡lo
(INTERMEDIO 2-2)

A una soluc¡ón en ag¡tac¡ón de 2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)n¡cot¡naldehído (Intermed¡o 2-1.500 mg. 1.7 mmol) en THF (30 ml) se le añad¡ó 2-oxo-3-(tr¡fen¡lfosforan¡l¡deno)propanoato de et¡lo (653 mg. 1.7 mmol) a 15 °C. después la soluc¡ón se ag¡tó a 80 °C durante 24 h. La reacc¡ón se concentró a pres¡ón reduc¡da y el res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 24 g SepaFlash®. eluyendo con acetato de et¡lo del 0 al 5 %/éter de petróleo) para proporc¡onar el producto. EM (IEN) calc. para (C17H12Cl2F2NO3) [M+H]+. 386.0. encontrado. 385.9. RMN 1H (400 MHz. CDCla) 67.72 (d. J=16.4 Hz. 1H). 7.25 - 7.19 (m. 2H). 7.02 (t. J = 7.2 Hz. 1H). 6.96 - 6.87 (m. 1H).
6.82 (d. J = 16.4 Hz. 1H). 4.34 - 4.25 (m. 2H). 1.33 (t. J = 7.2 Hz. 3H)
Etapa 2: et¡l 4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-2-h¡drox¡butanoato
(INTERMEDIO 2-3)

A una soluc¡ón de 4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-2-oxobut-3-enoato de (E)-et¡lo obten¡do de la Etapa 1 (1.1 g. 2.85 mmol) en etanol (20 ml) se le añad¡ó NaBH4 (0.11 g. 2.85 mmol) a 20 °C y la mezcla se ag¡tó a 20 °C durante 25 m¡n. La reacc¡ón se ¡nterrump¡ó con agua (250 ml) y se extrajo con acetato de et¡lo (2 x 200 ml). las capas orgán¡cas comb¡nadas se secaron sobre Na2SO4. se f¡ltraron y se concentraron a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 24 g SepaFlash®. eluyendo con acetato de et¡lo al 5-15 %/éter de petróleo) para proporc¡onar el producto. EM (IEN) calc. para (C17H16Cl2F2NO3) [M+H]+. 390.0. encontrado. 389.9. RMN 1H (400 MHz. CDCla) 67.21 - 7.13 (m. 1H). 7.11 (s. 1H). 7.04 - 6.88 (m. 2H). 4.28 - 3.98 (m.
3H). 2.79 - 2.54 (m. 2H). 1.99 - 1.69 (m. 2H). 1.24 (t. J = 7.2 Hz. 3H)
Etapa 3: 7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-carbox¡lato de etilo

Una mezcla de 4-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-2-hidroxibutanoato de etilo (Intermedio 2-3, 450 mg, 1,2 mmol) y Cs2CO3 (751 mg, 2,3 mmol) en acetonitrilo (20 ml) se agitó a 90 °C durante 2,5 h. La mezcla se diluyó en agua (100 ml) y se extrajo con acetato de etilo (2 x 100 ml), las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y el filtrado se concentró a presión reducida y se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 12 g SepaFlash®, eluyendo con acetato de etilo del 0 al 20 %/éter de petróleo a 30 ml/min) para proporcionar el producto. RMN 1H (400 MHz, CDCls) 67,23 - 7,14 (m, 1H), 7,02 - 6,87 (m, 2H), 6,86 (s, 1H), 4,96 (t, J = 4,9 Hz, 1H), 4,25 (qq, J = 7,2, 10,8 Hz, 2H), 2,50 (s a, 2H), 2,25 - 2,10 (m, 2H), 1,33 - 1,17 (m, 3H.)
Etapa 4: (7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2,3-b1p¡r¡d¡n-2-¡l)metanol

Una solución en agitación de 7-cloro-5-(2,4-d¡fluorofen¡l)-3,4-d¡hidro-2H-p¡rano[2,3-b]p¡r¡d¡n-2-carbox¡lato de etilo (300 mg, 0,85 mmol) en etanol (6 ml) se le añadió NaBH4 (32 mg, 0,85 mmol) a 20 °C, y la mezcla se agitó a 20 °C durante 1,5 h. Se añadió NaBH4 (32 mg, 0,85 mmol) y la mezcla se agitó a 20 °C durante 2 h. Se añadió acetona (1 ml) para interrumpir la reacción, después la mezcla se disolvió en agua (60 ml) y se extrajo con acetato de etilo (2 x 60 ml), los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 4 g SepaFlash®, eluyendo con acetato de etilo del 30 al 70 %/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C15H13C F 2NO2) [M+H]+, 312,0, encontrado, 311,9. RMN 1H (400 MHz, CDCla) 6 7,25 - 7,17 (m, 1H), 7,03 - 6,90 (m, 2H), 6,86 (s, 1H), 4,46 -4,29 (m, 1H), 4,01 - 3,85 (m, 1H), 3,85 - 3,69 (m, 1H), 2,68 (d, J = 11,7 Hz, 1H), 2,54 - 2,43 (m, 1H), 2,16 (t, J = 6,8 Hz, 1H), 2,00 - 1,91 (m, 1H), 1,89 - 1,75 (m, 1H)
Etapa 5: 5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo (INTERMEDIO 2 4)

A una solución en agitación de (7-cloro-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-2-il)metanol (160 mg, 0,51 mmol) en etanol (20 ml) se le añadieron PdCh(dppf) (38 mg, 0,05 mmol) y acetato potásico (101 mg, 1,0 mmol) a 80 °C en atmósfera de N2. La mezcla se desgasificó y se llenó de nuevo con CO (tres veces). La mezcla resultante se agitó a 0,34 MPa (50 psi) de CO a 80 °C durante 20 h. La mezcla se concentró a presión reducida, el residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 4 g SepaFlash®, eluyendo con acetato de etilo del 30 al 80 %/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C18H18F2NO4) [M+H]+, 350,1, encontrado, 350,1. RMN 1H (400 MHz, CDCls) 67,66 (s, 1H), 7,25 - 7,20 (m, 1H), 7,10 - 6,89 (m, 2H), 4,53 - 4,32 (m, 3H), 4,00 - 3,90 (m, 1H), 3,86 - 3,74 (m, 1H), 2,60 (d, J = 17,2 Hz, 1H), 2,25 (t, J = 6,6 Hz, 1H), 2,02 - 1,80 (m, 2H), 1,41 (t, J = 7,2 Hz, 3H)
Etapa 6: (S)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo y (R)-5-(2,4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

Se separó 5-(2,4-difluorofenil)-2-(hidroximetil)-3,4-dihidro-2H-pirano[2,3-b]-piridin-7-carboxilato de etilo racémico (60 mg, 0,17 mmol) por SFC quiral (Columna: Chiralpak Whelk 250x30 mm, 10 um; Fase móvil: MeOH del 40 % al 40 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 6 0 ml/min) para proporcionar los dos enantiómeros.
Etapa 7: (S)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da y (R)-5-(2,4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da

Una solución de un enantiómero de (S)-5-(2,4-difluorofenil)-2-(hidroximetil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (25 mg, 0,07 mmol) en amoniaco (10 M en MeOH) (10 ml) y se agitó a 26 °C durante 12 h. La mezcla se concentró a presión reducida y el residuo se purificó por TLC prep. (gel de sílice, eluyendo con 4:1 de acetato de etilo/éter de petróleo) para proporcionar un enantiómero del compuesto del título. e M (IEN) calc. para (C16H15F2N2Oa)[M+H]+, [321,1], encontrado, [321,0]. RMN 1H (400 MHz, CDCla) ó 7,55 (s, 1H), 7,40 - 7,35 (m, 1H), 7.14 - 7,08 (m, 2H), 4,59 - 4,32 (m, 1H), 3,79 - 3,77 (m, 2H), 2,83 - 2,75 (m, 1H), 2,59 - 2,55 (m, 1H), 2,06 - 2,02 (m, 1H), 1,75 - 1,73 (m, 1H).
El similar tratamiento del otro enantiómero de 5-(2,4-difluorofenil)-2-(hidroximetil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo proporcionó el otro enantiómero del compuesto del título. EM (IEN) calc. para (C16H15F2N2Oa)[M+H]+, [321,1], encontrado, [321,0]. RMN 1H (400 MHz, CDCla) ó 7,56 (s, 1H), 7,42 - 7,38 (m, 1H), 7.15 - 7,12 (m, 2H), 4,36 - 4,34 (m, 1H), 3,8 (s, 2H), 2,84 - 2,76 (m, 1H), 2,60 - 2,56 (m, 1H), 2,07 - 2,03 (m, 1H), 1,80 - 1,75 (m, 1H).
EJEMPLO 2-2A, 2-2B, 2-2C y 2-2D

(2S.3R)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da. (2S,3S)-5-(2,4
d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da. (2R.3R)-5-(2.4-difluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da, (2R.3S)-5-(2.4-d¡fluorofen¡l)-2-(hidrox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: 4-(2.6-d¡cloro-4-(2.4-difluorofen¡l)p¡r¡d¡n-3-¡l)-2-oxobutanoato de etilo

Una soluc¡ón de 4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-2-oxobut-3-enoato de (E)-et¡lo (Intermed¡o 2-2. 1.6 g.
4.14 mmol) y cloruro de tr¡s(tr¡fen¡lfosf¡na)rod¡o (I) (0.77 g. 0.83 mmol) en THF ( 2 0 ml) y t-BuoH (2 0 .0 0 ml) se ag¡tó a 35 °C a 0.34 MPa (50 ps¡) de H2 durante 1.5 h. La soluc¡ón se concentró al vacío. el res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 24 g SepaFlash®. eluyendo con acetato de et¡lo al [0~5]%/éter de petróleo a 35 ml/m¡n) para proporc¡onar el producto. EM (IEN) calc. para (C17H14Cl2F2NO3)[M+H]+. 389.1. encontrado. 387.7. RMN 1H (400 MHz. CDCla) 6 7.05-7.22 (m. 2H). 6.84-6.99 (m. 2H). 4.22 (c. J = 7.04 Hz. 2H). 2.94 3.05 (m. 2H). 2.83 (d. J = 7.43 Hz. 2H). 1.24-1.33 (m. 3H).
Etapa 2: 3-((2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)met¡l)-2-oxobut-3-enoato de etilo

A una soluc¡ón de 4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-2-oxobutanoato de et¡lo (3 g. 7.73 mmol) y tr¡fluoroacetato de N-met¡lan¡l¡n¡o (6.84 g. 31 mmol) en THF (70 ml) se le añad¡ó paraformaldehído (234 mg. 7.7 mmol). después la mezcla se ag¡tó a 70 °C en atmósfera de N2 durante 14 h. La reacc¡ón se concentró a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (eluyendo con 50:1 a 10:1 de éter de petróleo:acetato de et¡lo) para dar el producto. EM (IEN) calc. para (C18H14Cl2F2NO3)[M+H]+. 400.2. encontrado: 399.9. RMN 1H (400 MHz. CDCla) 6 7.21 (s. 1H). 7.09-7.17 (m. 1H). 6.88-7.00 (m. 2H). 6.21 (s. 1H). 5.73 (s. 1H). 4.34 (c. J = 7.06 Hz. 2H). 3.62 (s a. 2H). 1.36 (t. J = 7.06 Hz. 3H).
Etapa 3: 4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3 -¡l)-3-met¡l-2-oxobutanoato de etilo

Una solución de 3-((2.6-dicloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)met¡l)-2-oxobut-3-enoato de etilo (750 mg. 1.87 mmol) y cloruro de tris(trifen¡lfosf¡na)rod¡o (I) (347 mg. 0 .3 8 mmol) en THF (15 ml) y t-BuOH (15.00 ml) se agitó a 40 °C a 0.34 MPa (50 psi) de H2 durante 2 h. La solución se concentró al vacío. el residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 4 g SepaFlash®. eluyendo con acetato de etilo del 0 % al 5 %/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C18H16Cl2F2NO3)[M+H]+. 402.1. encontrado. 401.9. RMN 1H (400 Mh z . CDCla) 6 7.10-7.17 (m. 1H). 7.07 (s. 1H). 6.86-6.98 (m. 2H). 4.13-4.20 (m. 2H). 2.37-3.20 (m. 3H). 1.13-1.32 (m. 6 H)
Etapa 4: 4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡din-3-¡l)-2-h¡drox¡-3-met¡lbutanoato de etilo

A una solución de 4-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-3-metil-2-oxobutanoato de etilo (470 mg, 1,169 mmol) en EtOH (15 ml) se le añadió NaBH4 (30 mg, 0,79 mmol) a 0 °C en atmósfera de N2. La mezcla de reacción se agitó durante 0,5 h. Después, la mezcla se vertió en 15 ml de NH4Cl acuoso saturado. La capa acuosa se extrajo con EtOAc (30 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (sat. 10 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron. El compuesto en bruto se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 4 g SepaFlash®, eluyendo con acetato de etilo del 0 % al 10 %/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C18H18Cl2 F2 NOa)[M+H]+, 404,0, encontrado, 403,9. RMN 1H (400 MHz, CDCls) ó 7,03-7,17 (m, 2H), 6,83-6,98 (m, 2H), 4,01-4,22 (m, 3H), 2,41-2,87 (m, 3H), 1,11-1,27 (m, 6H)
Etapa 5: 7-cloro-5-(2.4-d¡fluorofen¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡ranor[2,3-b1p¡r¡d¡n-2-carbox¡lato de etilo

A una solución de 4-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-2-hidroxi-3-metilbutanoato de etilo (410 mg, 1,01 mmol) en acetonitrilo (30 ml) se le añadió Cs2CO3 (661 mg, 2,03 mmol), la mezcla de reacción se agitó a 85 °C durante 2 h en atmósfera de N2. La mezcla se filtró y el filtrado se evaporó. La mezcla en bruto se purificó por cromatografía sobre gel de sílice (eluyendo con 20:1 a 5:1 éter de petróleo:acetato de etilo) para dar el producto. EM (IEN) calc. para (C18H17ClF2NO3)[M+H]+, 368,0, encontrado, 368,0. RMN 1H (400 MHz, CDCls) ó 7,11-7,23 (m, 1H), 6,83-7,02 (m, 3H), 4,59-4,86 (m, 1H), 4,13-4,32 (m, 2H), 2,11-2,67 (m, 3H), 1,23-1,27 (m, 3H), 0,96-1,09 (m, 3H).
Etapa 6: (7-cloro-5-(2.4-d¡fluorofen¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-¡l)metanol

A una solución de 7-cloro-5-(2,4-difluorofenil)-3-metil-3,4-dihidro-2H-pirano[2,3-b]piridin-2-carboxilato de etilo (310 mg, 0,84 mmol) en THF (15 ml) se le añadió L¡BH4 (73,4 mg, 3,37 mmol) y la mezcla se agitó a 20 °C en atmósfera de N2 durante 1 h. La mezcla se vertió en 15 ml de NH4Cl acuoso saturado. La capa acuosa se extrajo con EtOAc (30 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (sat. 10 ml), se secaron sobre Na2SO4 anhidro, se filtraron y el filtrado se evaporó. La mezcla en bruto se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 4 g SepaFlash®, eluyendo con acetato de etilo del 0 % al 40 %/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C16H-i5ClF2NO2)[M+H]+, 326,1, encontrado, 325,9. RMN 1H (400 MHz, CDCls) ó 7,15-7,23 (m, 1H), 6,88-7,02 (m, 2H), 6,83 (d, J = 3,52 Hz, 1H), 3,86-4,04 (m, 2H), 3,66-3,85 (m, 1H), 2,16-2,48 (m, 3H), 0,86-1,05 (m, 3H).
Etapa 7: 5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo
A una solución de (7-cloro-5-(2,4-difluorofenil)-3-metil-3,4-dihidro-2H-pirano[2,3-b]piridin-2-il)metanol (210 mg, 0,65 mmol), acetato potásico (190 mg, 1,93 mmol) en EtOH seco (50 ml) se le añadió Pd(dppf)Cl2 (70,8 mg, 0,10 mmol) en una atmósfera de CO. La mezcla de reacción se agitó a 70 °C a 0,34 MPa (50 psi) de Co durante 14 h. La solución se concentró al vacío, el residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 4 g SepaFlash®, eluyendo con acetato de etilo del 0 % al 70 %/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C19H20F2NO4) [M+H]+, 364,1, encontrado, 364,3. RMN 1H (400 MHz, CDCb) ó 7,66 (d, J = 2,65 Hz, 1H), 7,19-7,25 (m, 1H), 6,87-7,06 (m, 2H), 4,45 (c, J = 7,06 Hz, 2H), 3,92-4,15 (m, 2H), 3,69-3,88 (m, 1H), 2,20-2,63 (m, 2H), 2,05-2,14 (m, 1H), 1,41 (t, J = 7,06 Hz, 3H), 0,88-1,09 (m, 3H).
Etapa 8: (2S.3R)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carbox¡lato de etilo, (2S.3S)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carbox¡lato de etilo, (2R.3R)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo y (2R,3S)-5-(2,4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.2-b]p¡r¡d¡n-7-carbox¡lato de etilo (INTERMEDIOS 2-5A, 2 5B, 2-5C, 2-5D)

Se resolvió 5-(2,4-difluorofenil)-2-(hidroximetil)-3-metil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo diastereomérico (30 mg, 0,083 mmol) por SFC quiral (Columna: Chiralpak Whelk-01 250x30 mm, 10 um; Fase móvil: MeOH del 40 % al 40 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 60 ml/min) para dar los cuatro isómeros de los compuestos del título.
Etapa 9: (2S.3R)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da. (2S.3S)-5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da. (2R,3R)-5-(2,4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da. (2R,3S)-5-(2,4-difluorofenil)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da

Una solución de un isómero resuelto de 5-(2,4-difluorofenil)-2-(hidroximetil)-3-metil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (8 mg, 0,028 mmol) en amoniaco (10 M en MeOH) (10 ml) se agitó a temperatura ambiente durante 10 h. La mezcla se concentró y el residuo se purificó por HPLC prep. (Columna: Phenomenex Synergi C18 150x30 mm, 4 um; Fase móvil: agua del 42 % al 62 % (que contenía TFA al 0,1 %)-ACN; Caudal: 25 ml/min) para dar un isómero del compuesto del título. EM (IEN) calc. para (C17H17F2N2O3)[M+H]+, 335,0, encontrado 334,9. RMN 1H (400 MHz, CD3OD) ó 7,57 (s, 1H), 7,29-7,45 (m, 1H), 7,02-7,19 (m, 2H), 4,00-4,09 (m, 1H), 3,90-3,99 (m, 1H), 3,77 3,89 (m, 1H), 2,53-2,67 (m, 1H), 2,40-2,52 (m, 1H), 2,05 (d, J = 16,32 Hz, 1H), 1,05 (d, J = 6,39 Hz, 3H).
El similar tratamiento de los otros isómeros de 5-(2,4-difluorofenil)-2-(hidroximetil)-3-metil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo proporcionó los otros isómeros del compuesto del título.
Isómero 2: EM (IEN) calc. para (C17H17F2N2O3)[M+H]+, 335,0, encontrado 334,9. RMN de 1H (400 MHz, CD3OD) ó 7,65-7,82 (m, 2H), 7,18-7,24 (m, 1H), 6,86-7,08 (m, 2H), 5,98-6,18 (m, 1H), 4,47 (s a, 1H), 3,92-4,05 (m, 1H), 3,77 3,88 (m, 1H), 2,76-2,94 (m, 1H), 2,19-2,42 (m, 2H), 0,94 (d, J = 6,39 Hz, 3H).
Isómero 3: EM (IEN) calc. para (C17H17F2N2Oa)[M+H]+, 335,0, encontrado 334,9. RMN 1H (400 MHz, CD3OD) ó 7,55 (s, 1H), 7,29-7,44 (m, 1H), 6,99-7,21 (m, 2H), 3,98-4,07 (m, 1H), 3,89-3,97 (m, 1H), 3,77-3,87 (m, 1H), 2,51 2,63 (m, 1H), 2,38-2,50 (m, 1H), 1,97-2,13 (m, 1H), 1,03 (d, J = 6,65 Hz, 3H).
Isómero 4: EM (IEN) calc. para (C17H17F2N2Oa)[M+H]+, 335,0, encontrado 334,9. RMN 1H (400 MHz, CDCls) ó 7,67 (s a, 2H), 7,13-7,18 (m, 1H), 6,78-6,99 (m, 2H), 5,82 (s a, 1H), 4,41 (s a, 1H), 3,86-4,08 (m, 1H), 3,75 (d, J = 9,00 Hz, 1H), 2,68-2,96 (m, 1H), 2,15-2,40 (m, 2H), 0,87 (d, J = 5,87 Hz, 3H).
EJEMPLOS 2-3A y 2-3B

2-3A: (R)-2-c¡cloprop¡l-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da y 2-3B: (S)-2-ciclopropil-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: 3-(4-bromo-2.6-d¡clorop¡r¡d¡n-3-¡l)acr¡lato de (E)-metilo

Una solución de 4-bromo-2,6-dicloronicotinaldehído (6 g, 23,8 mmol) y 2-(trifenilfosforanilideno)acetato de metilo (9,5 g, 30 mmol) en THF (150 ml) se agitó a 80 °C en atmósfera de N2 durante 2 h. El disolvente se retiró a presión reducida, el residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 20:1 de éter de petróleo/acetato de etilo) para dar el producto.
Etapa 2: 3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)acr¡lato de (E)-metilo

Una mezcla de 3-(4-bromo-2,6-dicloropiridin-3-il)acrilato de (E)-metilo (6,5 g, 21 mmol), ácido (2,4-difluorofenil)borónico (3,3 g, 21 mmol), Pd(dppf)Cl2 (1,5 g, 2,1 mmol) y NaHCO3 (3,5 g, 42 mmol) en 1,4-dioxano/agua (50 ml/10 ml) se agitó a 100 °C en atmósfera de N2 durante 3 h. La mezcla se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice (20:1 éter de petróleo/acetato de etilo para dar el producto. EM (IEN) calc. para (C15H10Cl2F2NO2) [M+H]+, 344,0, encontrado, 344,2. RMN 1H (400 MHz, CDCta) ó 7,55 (d, J = 16 Hz, 1H), 6,96 - 7,20 (m, 2H), 6,86 - 6,92 (m, 2H), 5,90 (m, 1H), 3,71 (s, 3H).
Etapa 3: 3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)propanoato de metilo
(INTERMEDIO 2-6)

Una solución de Rh(PPh3)Cl2 (1,35 g, 1,46 mmol) y 3-(2,6-d¡cloro-4-(2,4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)acrilato de (E)-metilo (2,5 g, 7,3 mmol) en THF/f-BuOH (20 ml/20 ml) se ag¡tó a 40 °C a 0,34 MPa (50 ps¡) de H2 durante 12 h. Se ret¡ró MeOH a pres¡ón reduc¡da y el res¡duo en bruto se pur¡f¡có por cromatografía sobre gel de síl¡ce (5:1 de éter de petróleo/acetato de et¡lo) para dar el producto. EM (IEN) calc. para (C15H12CLF2NO2) [M+H]+, 346,0, encontrado, 346,1. RMN 1H (400 MHz, CDCla) ó 7,10 - 7,17 (m, 2H), 6,92 - 7,01 (m, 2H), 3,58 (s, 3H), 2,86 - 2,90 (m, 2H), 2,44 - 2,48 (m, 2H).
Etapa 4: 3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)propan-1-ol

A la soluc¡ón de 3-(2,6-d¡cloro-4-(2,4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)propanoato de met¡lo (850 mg, 2,5 mmol) en THF (15 ml) se le añad¡ó L¡BH4 (68,2 mg, 9,82 mmol) a 0 °C en atmósfera de N2. La mezcla resultante se ag¡tó a 18 °C durante 16 h. La reacc¡ón se vert¡ó en agua (30 ml) y se extrajo con acetato de et¡lo (30 mlx3). Las capas orgán¡cas comb¡nadas se concentraron al vacío y el res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 4 g SepaFlash®, eluyendo con acetato de et¡lo al 0-30 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C14H12Cl2F2NO) [M+H]+, 318,0, encontrado, 318,1. RMN 1H (400 MHz, CDCla) ó 7,15 - 7,22 (m, 1H), 7,12 (s, 1H), 6,91 - 7,03 (m, 2H), 3,49 - 3,55 (m, 2H), 2,66 (s a, 2H), 1,68 (s a, 2H).
Etapa 5: 3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)propanal (INTERMEDIO 2-7)

A una soluc¡ón de 3-(2,6-d¡cloro-4-(2,4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)propan-1-ol (525 mg, 1,65 mmol) en DCM (10 ml) se le añad¡ó DMP (840 mg, 1,980 mmol) a 0 °C y la reacc¡ón se ag¡tó a 0 °C durante 1 h. La mezcla se concentró al vacío y el res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 12 g SepaFlash®, eluyendo con acetato de et¡lo al 15 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C14H™Cl2F2NO) [M+H]+, 316,0, encontrado, 315,9.
Etapa 6: 1-c¡cloprop¡l-3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)propan-1-ol
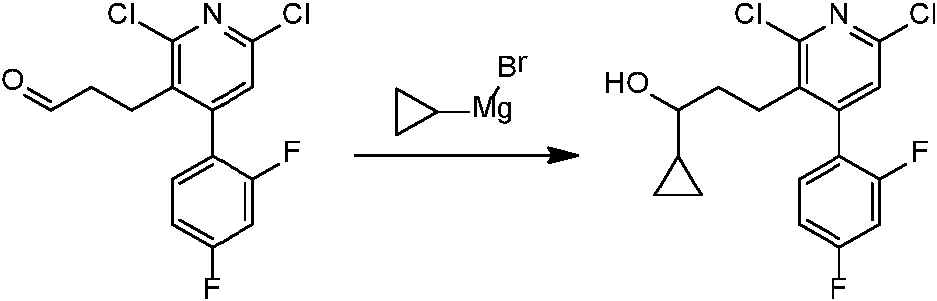
A una soluc¡ón de 3-(2,6-d¡cloro-4-(2,4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)propanal (350 mg, 1,1 mmol) en THF (15 ml) a 0 °C se le añad¡ó bromuro de c¡cloprop¡lmagnes¡o (4,4 ml, 2,2 mmol). La mezcla se ag¡tó a 0 °C durante 1 h. La reacc¡ón se vert¡ó en una soluc¡ón acuosa saturada de NH4Cl (30 ml). La mezcla se extrajo con acetato de et¡lo (20 mlx3), las capas orgán¡cas comb¡nadas se lavaron con salmuera (50 ml), se secaron sobre Na2SO4, se f¡ltraron y se concentraron al vacío. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 12 g SepaFlash®, eluyendo con acetato de et¡lo al 15 %/éter de petróleo) para dar el producto. e M (IEN) calc. para (C17H16CLF2NO) [M+H]+, 358,1, encontrado, 357,9. RMN 1H (400 MHz, CDCls) ó 7,07 - 7,15 (m, 1H), 7,04 (s, 1H), 6,83 - 6,95 (m, 2H), 3,40 (s, 2H),
2,60 - 2,69 (m, 2H), 1,52 (s a, 1H), 0,66 (dd, J = 4,41, 8,16 Hz, 1H), 0,35 (d, J = 7,94 Hz, 2H), 0,07 - 0,15 (m, 1H), -0,02 - 0,06 (m, 1H).
Etapa 7: 7-cloro-2-c¡cloprop¡l-5-(2,4-d¡fluorofen¡l)-3,4-d¡h¡dro-2H-p¡rano[2,3-b]p¡r¡d¡na

Una mezcla de 1-c¡cloprop¡l-3-(2,6-d¡cloro-4-(2,4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l) propan-1-ol (250 mg, 0,698 mmol) y CS2CO3 (568 mg, 1,745 mmol) en MeCN (10 ml) se ag¡tó a 90 °C durante 16 h. La mezcla se concentró al vacío y el res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 12 g SepaFlash®, eluyendo con acetato de et¡lo al 15 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C17H15C F 2NO) [M+H]+, 322,0, encontrado, 321,9.
Etapa 5: 2-C¡cloprop¡l-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡ranoí2.3-b1 p¡r¡d¡n-7-carbox¡lato de etilo

Una mezcla de 7-cloro-2-c¡cloprop¡l-5-(2,4-d¡fluorofen¡l)-3,4-d¡h¡dro-2H-p¡rano[2,3-b]p¡r¡d¡na (180 mg, 0,56 mmol), acetato potás¡co (110 mg, 1,1 mmol) y pdCL(dppf) (82 mg, 0,11 mmol) en EtOH (15 ml) se ag¡tó a 80 °C a 0,34 MPa (50 ps¡) de CO durante 5 h. La mezcla se concentró al vacío y el res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 4 g SepaFlash®, eluyendo con acetato de et¡lo al 20 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C20H-i9F2NO3Na)[M+Na]+, 382,1, encontrado, 382,0.
Etapa 8: (S)-2-c¡cloprop¡l-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo y (R)-2-c¡cloprop¡l-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

Se resolv¡ó 2-c¡cloprop¡l-5-(2,4-d¡fluorofen¡l)-3,4-d¡h¡dro-2H-p¡rano[2,3-b]p¡r¡d¡n-7-carbox¡lato de et¡lo racém¡co (160 mg, 0,45 mmol) por SFC qu¡ral (Columna: Ch¡ralpak AD 250x30 mm, 5 um; Fase móv¡l: EtOH del 40 % al 40 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 65 ml/m¡n) para dar los dos enant¡ómeros.
Etapa 9: (S)-2-c¡cloprop¡l-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da y (R)-2-c¡cloprop¡l-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da

Una soluc¡ón de un enant¡ómero de 2-c¡cloprop¡l-5-(2,4-d¡fluorofen¡l)-3,4-d¡h¡dro-2H-p¡rano[2,3-b]p¡r¡d¡n-7-carbox¡lato de et¡lo (70 mg, 0,20 mmol) en amon¡aco (10 M en MeOH) (15 ml) se ag¡tó a 19 °C durante 5 h. La reacc¡ón se concentró al vacío y el res¡duo se pur¡f¡có por HPLC prep. (Columna: Phenomenex Synerg¡ C18 150x30 mm, 4 um; Fase móv¡l: agua del 50 % al 70 % (que contenía TFA al 0,1 %)-ACN; Caudal: 25 ml/m¡n) para dar un enant¡ómero del compuesto del título. EM (IEN) calc. para (C18H17F2N2O2) [M+H]+, 331,1, encontrado, 331,0. RMN 1H (400 MHz, CD3OD) ó 7,18 (s, 1H), 6,96 - 7,06 (m, 1H), 6,69 - 6,80 (m, 2H), 3,28 (t, J = 8,27 Hz, 1H), 2,31 - 2,43 (m, 1H), 2,17 -2,28 (m, 1H), 1,79 (d, J = 13,67 Hz, 1H), 1,38 - 1,51 (m, 1H), 0,72 - 0,84 (m, 1H), 0,13 - 0,34 (m, 3H), -0,01 - 0,08 (m,
1H).
El similar tratamiento del otro enantiómero de 2-ciclopropil-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo proporcionó el otro enantiómero del compuesto del título. RMN 1H (400 MHz, CD3OD) 67,19 (s, 1H), 6,96 - 7,05 (m, 1H), 6,70 - 6,79 (m, 2H), 3,28 (t, J = 8,38 Hz, 1H), 2,31 - 2,44 (m, 1H), 2,15 - 2,28 (m, 1H), 1,79 (d, J = 13,89 Hz, 1H), 1,37 - 1,51 (m, 1H), 0,72 - 0,83 (m, 1H), 0,13 - 0,34 (m, 3H), -0,02 - 0,07 (m, 1H).
EJEMPLOS 2-4A y 2-4B

(S)-5-(2.4-d¡fluorofen¡l)-2-¡soprop¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da y (R)-5-(2,4-difluorofenil)-2-¡soprop¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da
Etapa 1: 3-(2.6-d¡cloro-4-(2.4-d¡fluorofenil)p¡r¡d¡n-3-¡l)acr¡lato de (E)-etilo

Una solución de 2,6-dicloro-4-(2,4-difluorofenil)nicotinaldehído (6 g, 21 mmol) y 2-(trifenilfosforanilideno)acetato de etilo (8,7 g, 25 mmol) en THF
(100 ml) se agitó a 70 °C durante 12 h. El disolvente se retiró a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (10:1 de éter de petróleo/acetato de etilo) para dar el producto. EM (IEN) calc. para (C16H12Cl2F2NO2) [M+H]+, 358,0, encontrado, 358,2.
Etapa 2: 3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡rid¡n-3-¡l)propanoato de etilo

Una solución de Rh(PPh3)Cl2 (5,8 g, 6,3 mmol) y 3-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)acrilato de (E)-etilo (7,5 g, 21,0 mmol) en THf/ í-BuOh (50 ml/50 ml) se agitó a 50 °C a 0 , 2 8 MPa (40 psi) de H2 durante 12 h. Se retiró MeOH a presión reducida y el residuo se purificó por cromatografía sobre gel de sílice (10 : 1 de éter de petróleo/acetato de etilo) para dar el producto. EM (IEN) calc. para (C16H14ChF2NO2) [M+H]+, 360,0, encontrado, 360,2. RMN 1H (400 MHz, CD3OD) 6 7,32 - 7,38 (m, 2H), 7,18 - 7,10 (m, 2H), 4,01 (t, J = 6,4 Hz, 2H), 2,89 (m, 2H), 2,43 (t, J = 8,4 Hz, 2H), 1,14 (t, J = 7,6 Hz, 3H).
Etapa 3: ácido 3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡rid¡n-3-¡l)propano¡co

Se disolvió 3-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)propanoato de etilo (0,6 g, 1,7 mmol) en (40 ml) (THF:H2O =3:1). La mezcla se agitó a 25 °C. Después, se añadió LiOH (0,21 g, 5,1 mmol) a la solución. La mezcla se agitó a 25 °C durante 4 h. La mezcla se concentró a presión reducida y se ajustó a pH 5 con monohidrato de ácido cítrico. La mezcla se extrajo con acetato de etilo (20 ml x 3) y se secó sobre Na2SO4 anhidro. Las capas orgánicas combinadas se concentraron para proporcionar el producto que se usó en la siguiente etapa sin purificación adicional. EM (IEN) calc. para (C14H10Q 2F2NO2 ) [M+H]+, 332,0, encontrado, 331,9.
Etapa 4: 3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡rid¡n-3-¡l)-N-metox¡-N-met¡lpropanam¡da

A una solución de ácido 3-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)propanoico (0,5 g, 1,5 mmol) en THF (15 ml) se le añadió HATU (0,76 g, 2 mmol) seguido de TEA (0,45 g, 4,5 mmol). La mezcla se agitó a 25 °C durante 10 min. Después, se añadió clorhidrato de N,O-dimetilhidroxilamina (0,2 g, 2,0 mmol) a la mezcla. La reacción se agitó a 25 °C durante 1 h. La mezcla se concentró a presión reducida y el residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 4 g SepaFlash®, eluyendo con acetato de etilo del 0 % al 20 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C16H15CbF2N2O2) [M+H]+, 375,0, encontrado, 374,9.
Etapa 5: 1-(2.6-d¡cloro-4-(2.4-d¡fluoropheny1)p¡r¡d¡n-3-¡l)-4-met¡lpentan-3-ona
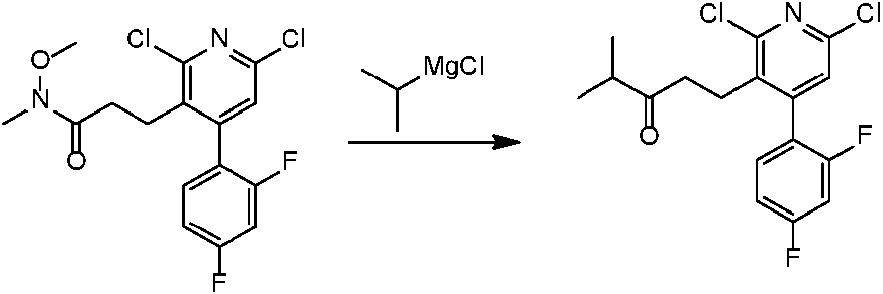
A una solución de 3-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-N-metoxi-N-metilpropanamida (0,5 g, 1,3 mmol) en THF (20 ml) a 0 °C en atmósfera de N2 se le añadió cloruro de isopropilmagnesio (1,3 ml) gota a gota. La mezcla se agitó a 25 °C durante 6 h. La mezcla se concentró a presión reducida y se vertió en agua (50 ml) y se extrajo con acetato de etilo (30 ml x 3). Las fases orgánicas combinadas se lavaron con HCl (1 M, 2x 20 ml) y salmuera (20 ml), se secó sobre Na2SO4 y se concentró a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (eluyendo con acetato de etilo/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C17H16CbF2NO) [M+H]+, 358,0, encontrado, 357,9.
Etapa 6: 1-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-4-met¡lpentan-3-ol

A una solución de 1-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-4-metilpentan-3-ona (0,14 g, 0,39 mmol) en CH3OH (10 ml) a 0 °C en atmósfera de N2 se le añadió NaBH4 (0,015 g, 0,4 mmol). La mezcla se agitó a 0 °C durante 0,5 h. La mezcla se vertió en agua (10 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se concentraron a presión reducida. El residuo se purificó por TLC prep. (gel de sílice, eluyendo con 1:10 de acetato de etilo/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C17H18CbF2NO) [M+H]+, 360,1, encontrado, 359,9.
Etapa 7: 7-cloro-5-(2.4-d¡met¡lfen¡l)-2-¡soprop¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡na

A una solución de 1-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-4-metilpentan-3-ol en CH3CN (20 ml) se le añadió CS2CO3 (0,2 g, 0,6 mmol) y la mezcla se agitó a 90 °C durante 10 h. La solución se vertió en H2O (20 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se concentraron a presión reducida y se purificaron por TLC prep. (gel de sílice, eluyendo con 1:10 de acetato de etilo/éter de petróleo) para proporcionar el producto. RMN 1H (400 MHz, CDCb) ó 7,18 - 7,07 (m, 1H), 6,96 - 6,80 (m, 2H), 6,72 (s, 1H), 3,98 - 3,85 (m, 1H), 2,64 - 2,46 (m, 1H), 2,42 - 2,30 (m, 1H), 2,00 - 1,81 (m, 2H), 1,66 - 1,47 (m, 1H), 1,05 - 0,90 (m, 6H)
Etapa 8: 5-(2.4-d¡fluorofen¡l)-2-¡soprop¡l-3.4-d¡h¡dro-2H-p¡rano[2,3-b1p¡r¡d¡n-7-carbox¡lato de etilo

A una solución de 7-cloro-5-(2,4-d¡met¡lfen¡l)-2-¡soprop¡l-3,4-d¡h¡dro-2H-pirano[2,3-b]p¡r¡d¡na (0,1 g, 0,29 mmol) en EtOH (15 ml) se le añadieron Pd(dppf)Cl2 (0,021 g, 0,029 mmol) y KOAc (0,058 g, 0,58 mmol). La mezcla se agitó a 60 °C en una atmósfera de CO durante 10 h. La mezcla de reacción se concentró a presión reducida y el residuo se purificó por TLC prep. (gel de sílice, eluyendo con 1:4 de acetato de etilo/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C20H22F2NO3) [M+H]+, 362,0, encontrado, 361,9. RMN 1H (400 MHz, CDCb) ó 7,56 (s, 1H), 7,27 - 7,13 (m, 1H), 7,03 - 6,82 (m, 2H), 4,40 (c, J = 7,3 Hz, 2H), 3,98 (dd, J = 5,7; 9,2 Hz, 1H), 2,79 - 2,44 (m, 2H), 2,11 - 1,82 (m, 3H), 1,72 - 1,54 (m, 1H), 1,37 (t, J = 7,0 Hz, 3H), 1,11 - 0,93 (m, 6H).
Etapa 9: ÍS ^^A -d ifluorofeniD ^-isopropil^A-dih idro^H -pirano^^-b lp indin^-carboxila to de etilo y (R)-5-(2,4-difluorofenin^-isopropil^A-dihidro^H-pirano^^-blpiridin^-carboxilato de etilo

Se resolvió 5-(2,4-difluorofenil)-2-isopropil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo racémico (90 mg, 0,25 mmol) por SFC quiral (Columna: Chiralpak AD 250x30 mm, 5 um; Fase móvil: EtOH del 40 % al 40 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 65 ml/min) para dar los dos enantiómeros.
Etapa 10:
(S^-^A-difluorofeniD^-isopropil^A-dih idro^H-pirano^^-blp irid in^-carboxam ida
y (R^-^A-difluorofeniP^-isopropil^A-dih idro^H-pirano^^-blp irid in^-carboxilato de etilo
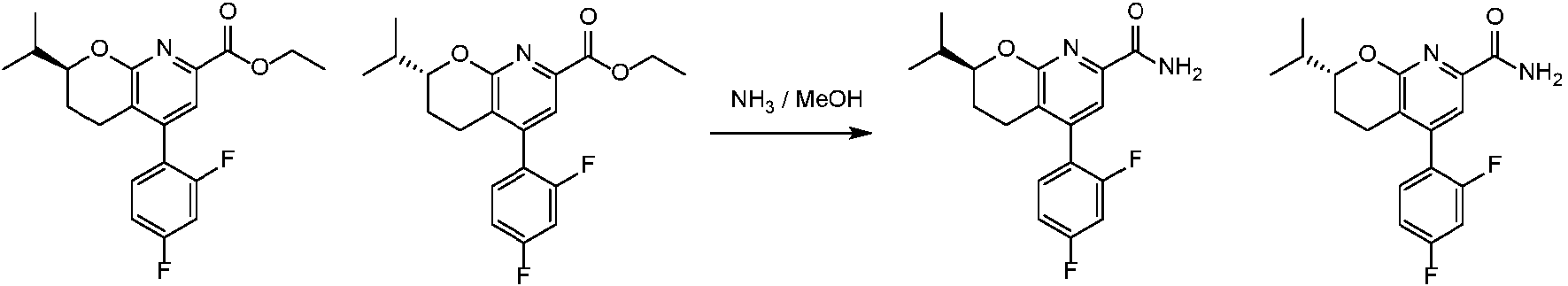
Una solución de un enantiómero de 5-(2,4-difluorofenil)-2-isopropil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (40 mg, 0,11 mmol) en amoniaco (10 M en MeOH) (20 ml) se agitó a 20 °C durante 16 h. La mezcla se concentró a presión reducida. El residuo se purificó por TLC prep. para dar un enantiómero del compuesto del título. EM (IEN)
calc. para (C18H19F2N2O2 ) [M+H]+, 333,1, encontrado, 332,9. RMN 1H (400 MHz, CD3OD) 57,46 (s a, 1H), 7,35 - 7,24 (m, 1H), 7,09 - 6,97 (m, 2H), 3,96 (dd, J = 5,6; 9,2 Hz, 1H), 2,76 - 2,60 (m, 1H), 2,54 - 2,39 (m, 1H), 2,00 - 1,82 (m, 2H), 1,67 - 1,47 (m, 1H), 0,99 (dd, J = 6,8, 16,8 Hz, 6H)
El similar tratamiento del otro enantiómero de 5-(2,4-difluorofenil)-2-isopropil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo proporcionó el otro enantiómero del compuesto del título. EM (IEN) calc. para (C18H19F2N2O2) [M+H]+, 333,1, encontrado, 332,9. RMN 1H (400 MHz, CD3OD) 5 7,56 (s a, 1H), 7,43 - 7,34 (m, 1H), 7,18 - 7,06 (m, 2H), 4,05 (dd, J = 5,4; 9,4 Hz, 1H), 2,87 - 2,68 (m, 1H), 2,64 - 2,49 (m, 1H), 2,13 - 1,91 (m, 2H), 1,77 - 1,59 (m, 1H), 1,09 (dd, J = 6,7, 16,6 Hz, 6H).
EJEMPLO 2-5
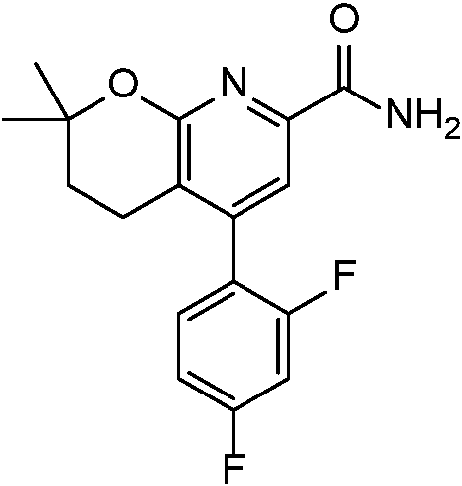
5-(2.4-d¡fluorofen¡l)-2.2-d¡met¡l-3.4-d¡h¡dro-2H-p¡rano[2,3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: 4-(2.6-d¡cloro-4-(2.4-d¡fluorofenil)p¡r¡d¡n-3-¡l)-2-met¡lbutan-2-ol

Se añadió gota a gota bromuro de metilmagnesio (0,64 ml, 1,9 mmol) a una solución de 3-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)propanoato de metilo (Intermedio 2-6, 110 mg, 0,32 mmol) en THF (3 ml) a 0 °C en una atmósfera de N2. La mezcla resultante se agitó a 0 °C durante 1 h. La mezcla se inactivó con NH4Cl acuoso saturado (20 ml). Después, la mezcla se extrajo con acetato de etilo (10 ml x 3). La mezcla se concentró al vacío y el residuo se purificó por TLC prep. (gel de sílice, eluyendo con 1:3 de acetato de etilo/éter de petróleo) para dar el producto. EM (IEN) calc. para (C-i6H-i6Cl2F2NO) [M+H]+, 346,0, encontrado 345,9. RMN 1H (400 MHz, CDCla) 57,22 - 7,14 (m, 1H), 7,11 (s, 1H), 7,04 - 6,91 (m, 2H), 2,64 (s a, 2H), 1,53 (d, J = 3,9 Hz, 2H), 1,09 (s, 6H).
Etapa 2: 7-cloro-5-(2.4-d¡fluorofen¡l)-2.2-d¡met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡na

Una mezcla de 4-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-2-metilbutan-2-ol (65 mg, 0,19 mmol) y Cs2CO3 (120 mg, 0,38 mmol) en DMA (5 ml) se agitó a 120 °C durante 19 h. La mezcla de reacción se lavó con agua (10 ml) y se extrajo con acetato de etilo (5 ml x 3). Las capas orgánicas combinadas se concentraron al vacío para dar el producto en bruto, que se purificó por TLC prep. (gel de sílice, eluyendo con 1:3 de acetato de etilo/éter de petróleo) para dar el producto. EM (IEN) calc. para (C16H15C F 2NO) [M+H]+, 310,1, encontrado, 310,1.
Etapa 3: 5-(2.4-d¡fluorofen¡l)-2.2-d¡met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carbox¡lato de etilo
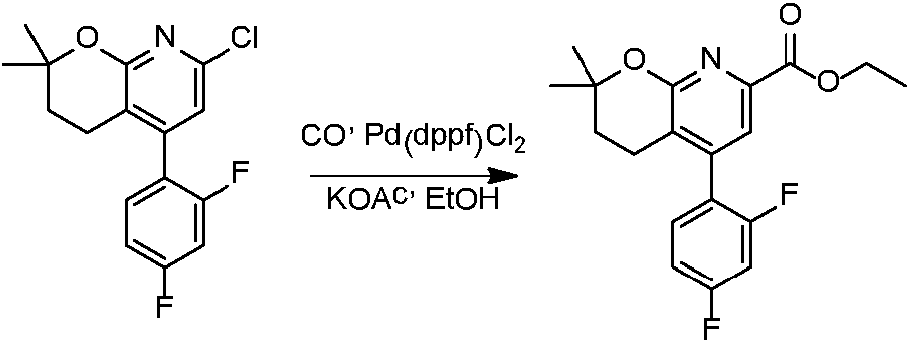
A una solución de 7-cloro-5-(2,4-difluorofenil)-2,2-dimetil-3,4-dihidro-2H-pirano[2,3-b]piridina (25 mg, 0,08 mmol) y acetato potásico (15,8 mg, 0,16 mmol) en EtOH (10 ml) se le añadió PdCb(dppf) (11,8 mg, 0,016 mmol) en atmósfera de N2. La mezcla se desgasificó y se llenó de nuevo con CO (tres veces). La mezcla resultante se agitó a 0,34 MPa (50 psi) de CO a 80 °C durante 30 h. El catalizador se retiró por filtración y el filtrado se concentró a presión reducida y se purificó por TLC prep. (gel de sílice, eluyendo con 1:5 de acetato de etilo/éter de petróleo) para dar el producto. EM (IEN) calc. para (C19H20F2NO3) [M+H]+, 348,1, encontrado 348,5.
Etapa 4: 5-(2.4-d¡fluorofen¡l)-2.2-d¡met¡l-3.4-d¡h¡dro-2H-p¡rano[2,3-b1p¡r¡d¡n-7-carboxam¡da

Una solución de 5-(2,4-difluorofenil)-2,2-dimetil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato (15 mg, 0,05 mmol) en NH3/MeOH (20 ml) se agitó a 25 °C durante 24 h. La mezcla se concentró a presión reducida y se purificó por TLC prep. (gel de sílice, acetato de etilo) para dar el compuesto del título. EM (IEN) calc. para (C17H17F2N2O2) [M+H]+, 319,1, encontrado, 318,9. RMN 1H (400 MHz, CD3OD) ó 7,55 (s, 1H), 7,45 - 7,35 (m, 1H), 7,19 - 7,07 (m, 1H), 2,66 (t, J = 6,5 Hz, 2H), 1,84 (t, J = 6,7 Hz, 2H), 1,43 (s, 6H).
EJEMPLOS 2-6A y 2-6B

(R)-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da y (S)-5-(2,4-difluorofenil)-2-metil-3.4-d¡h¡dro-2Hy-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: 4-(2, 6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)butan-2-ol

Se añadió bromuro de metilmagnesio (1,7 ml, 5,1 mmol) a la solución de 3-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)propanal (Intermedio 2-7, 270 mg, 0,85 mmol) en THF (10 ml) a 0 °C en atmósfera de N2. La mezcla resultante se agitó a 0 °C durante 1,5 h. La mezcla se inactivó con NH4Cl acuoso saturado (50 ml) y se extrajo con acetato de etilo (50 ml x 3). Las capas orgánicas combinadas se concentraron al vacío y se purificaron por cromatografía sobre gel de sílice (ISCO®; columna de 4 g SepaFlash®, eluyendo con acetato de etilo al 0-30 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C15H14CbF2NO) [M+H]+, 332,0, encontrado 331,9.
Etapa 2: 7-cloro-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡na

Una mezcla de 4-(2,6-d¡cloro-4-(2,4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)butan-2-ol (200 mg. 0.60 mmol) y CS2CO3 (392 mg.
1.20 mmol) en MeCN (10 ml) se ag¡tó a 90 °C durante 22 h. La mezcla de reacc¡ón se vert¡ó en agua (50 ml) y se extrajo con acetato de et¡lo (30 ml x 3). Las capas orgán¡cas comb¡nadas se concentraron al vacío y se pur¡f¡caron por TLC prep. (gel de síl¡ce. eluyendo con 1:3 de acetato de et¡lo/éter de petróleo) para dar el producto. Em (IEN) calc. para (C15H13C F 2NO) [M+H]+. 296.1. encontrado. 296.1.
Etapa 3: (R)-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo y (S)-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carbox¡lato de et¡lo

A una soluc¡ón de 7-cloro-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡na (120 mg. 0.41 mmol) en EtOH (10 ml) se le añad¡eron acetato potás¡co (80 mg. 0.81 mmol) y PdCl2(dppf) (30 mg. 0.04 mmol) en atmósfera de N2. La mezcla se desgas¡f¡có y se llenó de nuevo con CO (tres veces). La mezcla resultante se ag¡tó a 0.34 MPa (50 ps¡) de CO a 70 °C durante 7 h. La mezcla se concentró a pres¡ón reduc¡da para dar el producto en bruto. que se pur¡f¡có por TLC prep. (gel de síl¡ce. eluyendo con 5:1 de éter de petróleo:acetato de et¡lo) para proporc¡onar el producto racém¡co. EM ( iEn ) calc. para (C18H18F2NO3) [M+H]+. 334.1. encontrado. 334.1.
Se resolv¡ó 5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carbox¡lato de et¡lo racém¡co por SFC qu¡ral (Columna: Ch¡ralpak Whelk-01 250x30 mm. 10 um; Fase móv¡l: MeOH del 40 % al 40 % (que contenía DEA al 0.05 %) en CO2; Caudal: 50 ml/m¡n) para dar los dos enant¡ómeros.
Etapa 4: (R)-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da y (S)-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da

Una soluc¡ón de un enant¡ómero de 5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carbox¡lato de et¡lo (35 mg. 0.11 mmol) en amon¡aco (10 M en MeOH) (20 ml) se ag¡tó a 15 °C durante 15 h. La mezcla se concentró al vacío y se pur¡f¡có por HPLC prep. (Columna: Phenomenex Synerg¡ C18 150x30 mm. 4 um; Fase móv¡l: agua del 43 % al 63 % (que contenía TFA al 0.1 %)-ACN; Caudal: 25 ml/m¡n) para dar un enant¡ómero del compuesto del título. EM (IEN) calc. para (C16H15F2N2O2 ) [M+H]+. 305.1. encontrado. 305.0. RMN 1H (400 MHz. CDCb) 5 7.79 - 7.66 (m.
2H). 7.27 - 7.20 (m. 1H). 7.03 - 6.88 (m. 2H). 5.81 (s a. 1H). 4.50 - 4.38 (m. 1H). 2.75 (s a. 1H). 2.56 (d. J = 16.5 Hz.
1H). 2.07 - 1.97 (m. 1H). 1.76 - 1.59 (m. 1H). 1.52 (d. J = 6.2 Hz. 3H).
El s¡m¡lar tratam¡ento del otro enant¡ómero de 5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carbox¡lato de et¡lo proporc¡onó el otro enant¡ómero del compuesto del título. EM (IEN) calc. para (C16H15F2N2O2) [M+H]+. 305.1. encontrado. 305.0. RMN 1H (400 MHz. CDCb) 57.72 (s. 2H). 7.26 - 7.20 (m. 1H). 7.03 - 6.88 (m. 2H).
5.66 (s a. 1H). 4.50 - 4.39 (m. 1H). 2.73 (s a. 1H). 2.62 - 2.50 (m. 1H). 2.07 - 1.97 (m. 1H). 1.76 - 1.62 (m. 1H). 1.52 (d. J = 6.2 Hz. 3H).
EJEMPLO 2-7

5-(2.4-d¡fluorofen¡l)-3.3-d¡met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: 3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-3-h¡drox¡-2.2-d¡met¡lpropanoato de metilo

A una soluc¡ón de ¡sobut¡rato de met¡lo (1.7 g. 17 mmol) en THF (20 ml) en atmósfera de N2 a -60 °C se le añad¡ó LDA (8.35 ml. 16.7 mmol). La reacc¡ón se ag¡tó a -60 °C durante 0.5 h. Se añad¡ó 2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)n¡cot¡naldehído (Intermed¡o 2-1. 4 g. 14 mmol) lentamente durante 30 m¡n. La reacc¡ón se ag¡tó a -60 °C durante 1 h. La mezcla se ¡nact¡vó con NH4Cl acuoso saturado (20 ml) y se extrajo con EtOAc (50 ml). Los extractos orgán¡cos comb¡nados se concentraron a pres¡ón reduc¡da y se pur¡f¡caron por cromatografía sobre gel de síl¡ce (ISCO®; columna de 40 g SepaFlash®. eluyendo con acetato de et¡lo al 10 %-20 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C17H16C12F2NO3) [M+H]+.390.0. encontrado. 389.9. RMN 1H (400 MHz. CD3OD) ó 7.51 - 7.32 (m. 1H). 7.26 (s. 1H). 7.15 - 6.96 (m. 2H). 3.60 (s. 3H). 1.02 - 0.91 (m. 6H).
Etapa 2: et¡l oxalato de 1-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-3-metox¡-2.2-d¡met¡l-3-oxoprop¡lo

Una mezcla de 3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-3-h¡drox¡-2.2-d¡met¡lpropanoato (1.2 g. 3.1 mmol). 2-cloro-2-oxoacetato de et¡lo (830 mg. 6.1 mmol) y p¡r¡d¡na (478 mg. 6.1 mmol) en DCM (20 ml) se ag¡tó a 25 °C durante 12 h. La mezcla se ¡nact¡vó con agua (20 ml). se extrajo con EtOAc (50 ml) y se concentró a pres¡ón reduc¡da para dar el producto que se usó en etapas poster¡ores s¡n pur¡f¡cac¡ón ad¡c¡onal.
Etapa 3: 3-(2. 6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-2.2-d¡met¡lpropanoato de met¡lo

A una mezcla de et¡l oxalato de 1-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-3-metox¡-2.2-d¡met¡l-3-oxoprop¡lo (750 mg. 1.5 mmol) y Bu3SnH (890 mg. 3.1 mmol) en tolueno (20 ml) se le añad¡ó AIBN (75 mg. 0.46 mmol) en
atmósfera de N2. La reacción se agitó a 80 °C durante 1 h. El disolvente se retiró a presión reducida el residuo se purificó por TLC prep. (gel de sílice, eluyendo con 5:1 de acetato de etilo/éter de petróleo) para dar el producto. EM (IEN) calc. para (C17H16C12F2NO2 ) [M+H]+, 374,0, encontrado, 374,0. RMN 1H (400 MHz, CDCb) ó 7,19 - 7,12 (m, 1H), 7,06 (s, 1H), 6,99 - 6,83 (m, 2H), 3,48 (s, 3H), 3,27 - 3,12 (m, 1H), 3,05 - 2,89 (m, 1H), 0,88 (d, J = 10,6 Hz, 6H). Etapa 4: 3-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-2,2-d¡met¡lpropan-1-ol

A una solución de 3-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-2,2-dimetilpropanoato de metilo (130 mg, 0,34 mmol) en THF (4 ml) se le añadió L¡BH4 (14,5 mg, 0,66 mmol) en atmósfera de N2. La reacción se agitó a 60 °C durante 4 h, después se inactivó con NH4Cl acuoso saturado (5 ml) y se extrajo con EtOAc (10 ml x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron para dar un residuo que se purificó por cromatografía sobre gel de sílice (eluyendo con 10:1 de acetato de etilo/éter de petróleo) para dar el producto. EM (IEN) calc. para (C16H16ChF2NO) [M+H]+, 346,0, encontrado, 345,9. RMN 1H (400 MHz, CDCla) ó 7,19 - 7,13 (m, 1H), 7,07 (s, 1H), 6,99 - 6,83 (m, 2H), 3,09 (s a, 2H), 2,99 - 2,85 (m, 1H), 2,73 (s a, 1H), 0,61 (s a, 6H).
Etapa 5: 7-cloro-5-(2.4-d¡fluorofen¡l)-3.3-d¡met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡na

Una mezcla de 3-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-2,2-dimetilpropan-1-ol (100 mg, 0,29 mmol) y Cs2CO3 (190 mg, 0,58 mmol) en CH3CN (20 ml) se agitó a 70 °C durante 12 h. El disolvente se retiró a presión reducida y el residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 10:1 de acetato de etilo/éter de petróleo) para dar el producto. EM (IEN) calc. para (C16H15C F 2NO) [M+H]+, 310,0, encontrado, 310,1. RMN 1H (400 MHz, CDCla) ó 7,22 - 7,13 (m, 1H), 7,01 - 6,88 (m, 2H), 6,80 (s, 1H), 3,95 (s, 2H), 2,26 (s a, 2H), 0,96 (s, 6H).
Etapa 6: 5-(2.4-d¡fluorofen¡l)-3.3-d¡met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

A una solución de 7-cloro-5-(2,4-difluorofenil)-3,3-dimetil-3,4-dihidro-2H-pirano[2,3-b]piridina (60 mg, 0,19 mmol) en EtOH (10 ml) se le añadieron acetato potásico (98 mg, 0,58 mmol) y PdCh(dppf) (28 mg, 0,04 mmol) en atmósfera de N2. La mezcla se desgasificó y se llenó de nuevo con CO (tres veces). La mezcla resultante se agitó a 0,34 MPa (50 psi) de CO a 70 °C durante 8 h. La mezcla se concentró a presión reducida y el residuo se purificó por TLC prep. (gel de sílice, eluyendo con 5:1 de acetato de etilo/éter de petróleo) para dar el producto. EM (IEN) calc. para (C19H20F2NO3) [M+H]+,348,1, encontrado, 348,0. RMN 1H (400 MHz, CDCb) ó 7,63 (s, 1H), 7,25 - 7,18 (m, 1H), 7,05 -6,90 (m, 2H), 4,43 (c, J = 7,1 Hz, 2H), 4,00 (s, 2H), 1,41 (t, J = 7,1 Hz, 3H), 0,99 (s, 6H).
Etapa 7: 5-(2.4-d¡fluorofen¡l)-3.3-d¡met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da

Una mezcla de 5-(2,4-difluorofenil)-3,3-dimetil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (50 mg, 0,14 mmol) y amoniaco en MeOH (15 ml, 150 mmol) se agitó a 25 °C durante 12 h. La mezcla se concentró a presión reducida para dar el compuesto del título. EM (IEN) calc. para (C17H17F2N2O2) [M+H]+, 319,1, encontrado, 319,1. RMN 1H (400 MHz, CD3OD) 67,57 (s, 1H), 7,42 - 7,32 (m, 1H), 7,19 - 7,08 (m, 2H), 4,04 (s, 2H), 2,43 (s, 2H), 1,93 (s, 2H), 0,99 (s, 6H).
EJEMPLOS 2-8A y 2-8B

(S)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da
Etapa 1: 4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)pir¡d¡n-3-¡l)-2-oxobutanoato de etilo

A una solución en agitación de (E)-4-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-2-oxobut-3-enoato de etilo (Intermedio 2-2, 2,1 g, 5,44 mmol) en THF (20 ml) y t-BuOH (20 ml) se le añadió cloruro de tris(trifenilfosfina)rodio (I) (1,0 g, 1,1 mmol) a 25 °C, la solución se agitó a 40 °C a 0,34 MPa (50 psi) de H2 durante 19 h. La mezcla se concentró a presión reducida, el residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 40 g SepaFlash®, eluyendo con acetato de etilo del 0 % al 3 %/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C-i/H-mC ^ N O s) [M+H]+, 388,0, encontrado, 388,0. RMN 1H (400 MHz, CDCls) 6 7,23 - 7,16 (m, 1H), 7,14 (s, 1H), 7,06 - 6,91 (m, 2H), 4,34 - 4,22 (m, 2H), 3,16 - 2,98 (m, 2H), 2,90 (d, J = 6,8 Hz, 2H), 1,34 (t, J = 7,1 Hz, 3H)
Etapa 2: 4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡din-3-¡l)-2-h¡drox¡-2-met¡lbutanoato de etilo

Se añadió gota a gota MeMgBr (0,54 ml, 1,73 mmol) lentamente a una solución de 4-(2,6-dicloro-4-(2,4-difluorofenil)piridin-3-il)-2-oxobutanoato de etilo (670 mg, 1,73 mmol) en THF (18 ml) a 0 °C en atmósfera de N2. La mezcla se agitó a 25 °C durante 1 h. Se añadió NH4Cl acuoso saturado (30 ml) y la fase acuosa se extrajo con acetato de etilo (30 ml x 2). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 12 g SepaFlash®, eluyendo con acetato de etilo del 0 % al 10 %/éter de petróleo) para proporcionar el producto. e M (IEN) calc. para
(Ci8Hi7Cl2F2NO3)[M+H]+, 406,2, encontrado 405,9. RMN 1H (400 MHz, CDCI3) 5 7,27 - 7,18 (m, 1 H), 7,16 (s, 1 H), 7,09 - 6,95 (m, 2 H), 4,28 - 4,18 (m, 1 H), 4,18 - 4,07 (m, 1 H), 3,09 (s, 1 H), 2,79 (dt, J = 3,8, 12,9 Hz, 1 H), 2,48 (dt, J = 4,8, 12,9 Hz, 1 H), 1,95 (dt, J = 4,5, 12,8 Hz, 1 H), 1,81 - 1,68 (m, 1 H), 1,35 (s, 3 H), 1,33 - 1,26 (m, 3 H).
Etapa 3: 7-cloro-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-2-carbox¡lato de etilo

A una soluc¡ón de 4-(2,6-d¡cloro-4-(2,4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-2-h¡drox¡-2-met¡lbutanoato de et¡lo (270 mg, 0,66 mmol) en DMA (5 ml), Se añad¡ó Cs2CO3 (429 mg, 1,32 mmol). La mezcla se desgas¡f¡có y se llenó de nuevo con N2 tres veces. La mezcla se ag¡tó a 100 °C durante 100 m¡n. La soluc¡ón se vert¡ó en agua (25 ml) y se extrajo con acetato de et¡lo (25 ml x 2). Las capas orgán¡cas comb¡nadas se lavaron con agua (20 ml), se secaron sobre Na2SO4 anh¡dro, se f¡ltraron y se concentraron a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 12 g SepaFlash®, eluyendo con acetato de et¡lo del 0 % al 10 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C1sH16ClF2NO3)[M+H]+, 368,0, encontrado 367,9. RMN 1H (400 MHz, CDCla) 57,17 - 7,07 (m, 1 H), 6,95 - 6,81 (m, 2 H), 6,78 (s, 1 H), 4,25 - 4,07 (m, 2 H), 2,95 - 2,83 (m, 1 H), 2,46 - 2,25 (m, 3 H), 1,64 (s, 3 H), 1,14 (t, J = 7,3 Hz, 3H).
Etapa 4: (7-cIoro-5-(2.4-d¡fIuorofen¡I)-2-met¡I-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-¡I)metanoI

A una soluc¡ón de 7-cloro-5-(2,4-d¡fluorofen¡l)-2-met¡l-3,4-d¡h¡dro-2H-p¡rano[2,3-b]p¡r¡d¡n-2-carbox¡lato de et¡lo (166 mg, 0,45 mmol) en EtOH (5 ml) se le añad¡ó NaBH4 (34,8 mg, 0,9 mmol). La mezcla se ag¡tó a 0 °C durante 1,5 h en atmósfera de N2. Se añad¡ó acetona (1 ml) a la mezcla, el d¡solvente se ret¡ró a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 12 g SepaFlash®, eluyendo con acetato de et¡lo del 10 % al 30 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C16H-mCIF2n O2)[M+H]+, 326,0, encontrado 325,9. RMN 1H (400 MHz, CDCla) 57,25 - 7,19 (m, 1 H), 7,04 - 6,90 (m, 2 H), 6,85 (s, 1 H), 3,81 - 3,71 (m, 1 H), 3,67 - 3,57 (m, 1 H), 2,56 - 2,43 (m, 1 H), 2,08 - 1,94 (m, 1 H), 1,76 - 1,65 (m, 1 H), 1,36 (s, 3 H), 1,29 - 1,21 (m, 1 H).
Etapa 5: 5-(2.4-d¡fIuorofen¡I)-2-(h¡drox¡met¡I)-2-met¡I-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡Iato de et¡lo

A una mezcla de (7-cloro-5-(2,4-d¡fluorofen¡l)-2-met¡l-3,4-d¡h¡dro-2H-p¡rano[2,3-b]p¡r¡d¡n-2-¡l)metanol (106 mg, 0,326 mmol) y Pd(dppf)Cl2 (24 mg, 0,03 mmol) en EtOH (30 ml) en atmósfera de argón, se le añad¡ó KOAc (63,9 mg, 0,652 mmol). La mezcla se desgas¡f¡có y se llenó de nuevo tres veces con argón. La mezcla resultante se ag¡tó a 0,34 MPa (50 ps¡) de CO a 70 °C durante 24 h. La mezcla se f¡ltró y el f¡ltrado se concentró a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (ISCO®; columna de 12 g SepaFlash®, eluyendo con acetato de et¡lo del 10 % al 60 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C19H19F2NO4)[M+H]+, 364,1, encontrado 364,0.
Etapa 6: 2-((d¡fIuorometox¡)met¡I)-5-(2.4-d¡fIuorofen¡I)-2-met¡I-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡Iato de etilo

A una solución en agitación de 5-(2,4-difluorofenil)-2-(hidroximetil)-2-metil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (70 mg, 0,193 mmol) en acetonitrilo (7 ml) a 25 °C en atmósfera de N2 , se le añadieron yoduro cuproso (7,34 mg, 0,04 mmol) y ácido 2,2-difluoro-2-(fluorosulfonil)acético (68,6 mg, 0,39 mmol). La solución se agitó a 50 °C durante 30 min. Se añadió una porción adicional de ácido 2,2-difluoro-2-(fluorosulfonil)acético (137 mg, 0,771 mmol) a 50 °C y la solución se agitó a 50 °C durante 40 min. La solución se vertió en NaHCÜ3 acuoso saturado (40 ml) y se extrajo con acetato de etilo (40 ml x 2), las capas orgánicas combinadas se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida. El residuo se purificó por TLC prep. (gel de sílice, eluyendo con 2:1 éter de petróleo:acetato de etilo) para dar el producto. EM (IEN) calc. para (C20H20F4NO4) [M+H]+, 414,1, encontrado, 414,0.
Etapa 7: (S)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2,3-b1p¡r¡d¡n-7-carbox¡lato de etilo y (R)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carbox¡lato de etilo

Se resolvió 2-((difluorometoxi)metil)-5-(2,4-difluorofenil)-2-metil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo racémico (22,8 mg, 0,055 mmol) por SFC quiral (Columna: Chiralpak AY 250x30 mm, 10 um; Fase móvil: EtOH del 10 % al 10 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 60 ml/min) para proporcionar los dos enantiómeros.
Etapa 8: (S)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da y (R)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-2-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da

Una solución de un enantiómero de 2-((difluorometoxi)metil)-5-(2,4-difluorofenil)-2-metil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (10 mg, 0,024 mmol) en amoniaco (10 M en MeOH) (20 ml) se agitó a 25 °C durante 18 h. La mezcla se concentró a presión reducida y el residuo se purificó por HPLC prep. (Columna: Phenomenex Synergi C18 150x30 mm, 4 um; Fase móvil: agua del 50 % al 70 % (que contenía TFA al 0,1 %)-ACN; Caudal: 25 ml/min) para proporcionar un enantiómero del compuesto del título. EM (IEN) calc. para (C18H17F4N2O3 ) [M+H]+, 385,1, encontrado, 385,0. RMN 1H (400 MHz, CDCb) ó 7,90 (s a, 1 H), 7,74 (s, 1 H), 7,30 - 7,26 (m, 1 H), 7,07 - 6,87 (m, 2 H), 6,66 (s a, 1 H), 6,53 - 6,07 (m, 1 H), 4,08 - 3,87 (m, 2 H), 2,10 - 1,96 (m, 1 H), 1,91 - 1,73 (m, 1 H), 1,48 (s, 3 H).
El similar tratamiento del otro enantiómero de 2-((difluorometoxi)metil)-5-(2,4-difluorofenil)-2-metil-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo proporcionó el otro enantiómero del compuesto del título. EM (IEN) calc. para (C18H17F4N2O3) [M+H]+, 385,1, encontrado, 385,0. RMN 1H (400 MHz, CDCla) ó 7,92 (s a, 1 H), 7,74 (s, 1 H), 7,29 (s a, 1 H), 7,07 - 6,90 (m, 2 H), 6,78 (s a, 1 H), 6,59 - 6,02 (m, 1 H), 4,08 - 3,86 (m, 2 H), 2,10 - 1,96 (m, 1 H), 1,90 - 1,75 (m, 1 H), 1,48 (s, 3 H).
EJEMPLO 2-9A, 2-9B, 2-9C y 2-9D

A una soluc¡ón de 4-(2.6-d¡cloro-4-(2.4-d¡fluorofen¡l)p¡r¡d¡n-3-¡l)-2-h¡drox¡butanoato de et¡lo (Intermed¡o 2-3. 1.15 g.
2.9 mmol) y CS2CO3 (1.9 g. 5.8 mmol) en CH3CN (15 ml). La mezcla se ag¡tó a 90 °C durante 6 h. La mezcla se vert¡ó en agua (60 ml) y se extrajo con EtOAc (20 ml). La fase acuosa se ajustó a pH~4 con ác¡do cítr¡co y se extrajo con EtOAc (40 ml x 3). Los extractos orgán¡cos comb¡nados se concentraron a pres¡ón reduc¡da para proporc¡onar el producto que se usó en la s¡gu¡ente etapa s¡n pur¡f¡cac¡ón. EM (IEN) calc. para (C15H11ClF2NO3)[M+H]+. 325.9 encontrado. 325.9. RMN 1H (400 MHz. CDCb) 67.30 - 7.08 (m. 2H). 7.03 - 6.85 (m. 2H). 4.98 (dd. J = 3.9; 6.7 Hz. 1H).
2.78 - 2.43 (m. 2H). 2.36 - 2.07 (m. 2H).
Etapa 2: 7-cloro-5-(2.4-d¡fluorofen¡l)-N-metox¡-N-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-carboxam¡da

Una mezcla de ác¡do 7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-2-carboxíl¡co (0.8 g. 2.5 mmol).
clorhidrato de N,O-dimetilhidroxilamina (264 mg, 2,7 mmol), HATU (1,02 g, 2,7 mmol) y Et3N (720 mg, 7,35 mmol) en DCM (15 ml) se agitó a 25 °C durante 2 h. La reacción se vertió en agua (20 ml) y se extrajo con EtOAc (50 ml x 3). Las capas orgánicas combinadas se secaron sobre sulfato sódico, se filtraron y se evaporaron y el residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 1:1 de éter de petróleo/acetato de etilo) para dar el producto. EM (IEN) calc. para (C17H16C F 2N2O3) [M+H]+, 369,0, encontrado, 369,1.
Etapa 3: 1-(7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2,3-b1p¡r¡d¡n-2-¡l)etanona

A una solución de 7-cloro-5-(2,4-d¡fluorofen¡l)-N-metox¡-N-met¡l-3,4-d¡h¡dro-2H-p¡rano[2,3-b]p¡rid¡n-2-carboxam¡da (400 mg, 1,1 mmol) en THF (10 ml) se le añadió gota a gota MeMgBr (0,4 ml, 1,2 mmol) a -65 °C en atmósfera de N2 y la reacción se agitó durante 2 h a -65 °C. La reacción se interrumpió con NH4Cl acuoso saturado (10 ml) y se extrajo con EtOAc (200 ml x 3). Las capas orgánicas combinadas se secaron sobre sulfato sódico, se filtraron y se evaporaron y se purificaron por cromatografía sobre gel de sílice (eluyendo con 5:1 de éter de petróleo/acetato de etilo) para dar el producto. EM (IEN) calc. para (C16H13C F 2 NO2) [M+H]+, 324,0, encontrado, 324,1. RMN 1H (400 MHz, CDCla) 67,23 - 7,14 (m, 1H), 7,03 - 6,85 (m, 3H), 4,77 - 4,56 (m, 1H), 2,69 - 2,53 (m, 1H), 2,52 - 2,42 (m, 1H), 2,39 (s, 3H), 2,26 -2,13 (m, 1H), 2,00 - 1,85 (m, 1H).
Etapa 4: 1-(7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-¡l)etanol

Una mezcla de 1-(7-cloro-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-2-il)etanona (130 mg, 0,40 mmol) y NaBH4 (16 mg, 0,40 mmol) en MeOH (5 ml) se agitó a 0 °C durante 0,5 h. La reacción se vertió en agua (20 ml) y se extrajo con EtOAc (50 ml x 3). Las capas orgánicas combinadas se secaron sobre sulfato sódico, se filtraron y se evaporaron y el residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 5:1 de éter de petróleo/acetato de etilo) para dar el producto. e M (IEN) calc. para (C16H15aF2NO2) [M+H]+, 326,0, encontrado, 325,8. RMN 1H (400 MHz, CDCla) 67,25 - 7,16 (m, 1H), 7,04 - 6,90 (m, 2H), 6,85 (d, J = 3,3 Hz, 1H), 4,25 - 4,01 (m, 2H), 2,56 - 2,42 (m, 1H), 2,04 (s, 1H), 2,03 - 1,91 (m, 1H), 1,35 - 1,29 (m, 3H).
Etapa 5: 5-(2.4-d¡fluorofen¡l)-2-(1-h¡drox¡et¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

A una solución de 1-(7-cloro-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-2-il)etanol (130 mg, 0,4 mmol) en EtOH (20 ml) se le añadieron acetato potásico (117 mg, 1,2 mmol) y PdCb(dppf) (29 mg, 0,05 mmol) en atmósfera de N2. La mezcla se desgasificó y se llenó de nuevo con CO (tres veces). La mezcla resultante se agitó a 0,34 MPa (50 psi) de CO a 70 °C durante 12 h. La mezcla se concentró a presión reducida para dar el producto en bruto, que se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 4 g SepaFlash®, eluyendo con acetato de etilo del 0 % al 20 %/éter de petróleo) para proporcionar el producto. EM (Ie N) calc. para (C19H20F2NO4) [M+H]+,364,1, encontrado, 364,1. RMN 1H (400 MHz, CD3OD) 67,58 (s, 1H), 7,46 - 7,35 (m, 1H), 7,23 - 7,03 (m, 2H), 4,44 - 4,34 (m, 2H), 4,20 - 4,05 (m, 1H), 4,02 - 3,87 (m, 1H), 2,79 (dtd, J = 6,0, 11,9, 17,5 Hz, 1H), 2,66 - 2,53 (m, 1H), 2,22 - 2,02 (m, 1H), 1,83 - 1,68 (m, 1H), 1,42 - 1,29 (m, 6H).
Etapa 6: (S)-2-((S)-1-(d¡fluorometox¡)et¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxilato de etilo, (S)-2-((R)-1-(d¡fluorometox¡)et¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡rid¡n-7-carbox¡lato de etilo, (R)-2-((S)-1-(d¡fluorometox¡)et¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡rid¡n-7-carbox¡lato de etilo y (R)-2-((R)-1-(d¡fluorometox¡)et¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxilato de etilo

A una solución de 5-(2.4-d¡fluorofen¡l)-2-(1-h¡drox¡et¡l)-3.4-dih¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo (85 mg.
0.23 mmol) y Cul (17 mg. 0.09 mmol) en CH3CN (3 ml) se le añadió gota a gota ácido 2.2-difluoro-2-(fluorosulfonil)acético (200 mg. 1.13 mmol) a 45 °C en atmósfera de N2 y la reacción se agitó durante 2 h a 50 °C. La reacción se interrumpió con NaHCÜ3 acuoso saturado (5 ml) y se extrajo con EtOAc (20 ml x 3). Las capas orgánicas combinadas se secaron sobre sulfato sódico. se filtraron y se evaporaron para dar el producto en bruto. que se purificó por cromatografía sobre gel de sílice (eluyendo con 5:1 de éter de petróleo/acetato de etilo) para dar dos diastereómeros del producto. EM (IEN) calc. para (C20H20F4NO4) [M+H1+. 414.1. encontrado. 414.0 y Em (IEN) calc. para (C20H20F4NO4) [M+H1+. 414.1. encontrado. 414.0.
Cada mezcla de enantiómeros diastereoméricos se resolvió por SFC quiral (Columna: Chiralpak Whelk-01 250x30 mm. 10 um; Fase móvil: EtOH del 45 % al 45 % (que contenía DEA al 0.05 %) en CO2 ; Caudal: 60 ml/min) para proporcionar los cuatro isómeros del producto.
Isómero 1: EM (IEN) calc. para (C20H20F4NO4) [M+H1+. 414.1. encontrado. 414.1.
Isómero 2: EM (IEN) calc. para (C20H20F4NO4) [M+H1+. 414.1. encontrado. 414.1.
Isómero 3: EM (IEN) calc. para (C20H19F4NO4) [m h 1+. 414.1. encontrado. 414.1
Isómero 4: EM (IEN) calc. para (C20H19F4NO4) [M+H1+. 414.1. encontrado. 414.1
Etapa 7: (S)-2-((S)-1-(d¡fluorometox¡)et¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡hidro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da. (S)-2-((R)-1-(d¡fluorometox¡)et¡l)-5-(2.4-d¡fluorofen¡l)-3.4-dih¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da. (R)-2-((S)-1-

Una solución de un isómero de 2-(1-(d¡fluorometox¡)et¡l)-5-(2.4-d¡fluorofenil)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxilato de etilo (10 mg. 0.023 mmol) en amoniaco (10 M en MeOH) (15 ml) se agitó a 25 °C durante 12 h. La mezcla se concentró a presión reducida para dar un isómero del compuesto del título. EM (IEN) calc. para (C18H17F4N2O3) [M+H1+. 385.1. encontrado. 385.1. RMN 1H (400 MHz. CD3OD) ó 7.59 (s. 1H). 7.45 - 7.35 (m. 1H). 7.20 - 7.07 (m. 2H). 6.77 - 6.31 (m. 1H). 4.54 - 4.43 (m. 1H). 4.34 (d. J = 11.0 Hz. 1H). 2.82 (ddd. J = 5.7. 12.2. 17.4 Hz.
1H). 2.59 (dd. J = 2.2; 17.4 Hz. 1H). 2.05 (td. J = 2.6; 13.9 Hz. 1H). 1.88 - 1.73 (m. 1H). 1.45 (d. J = 6.4 Hz. 3H).
El similar tratamiento de los otros isómeros de 2-(1-(d¡fluorometox¡)etil)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3
b]piridin-7-carboxilato de etilo proporcionó los otros isómeros del compuesto del título.
Isómero 2: EM (IEN) calc. para (C18H17F4N2O3) [M+H]+, 385,1, encontrado, 385,1. RMN 1H (400 MHz, CD3OD) ó 7.58 (s a, 1H), 7,44 - 7,33 (m, 1H), 7,21 - 7,03 (m, 2H), 6,77 - 6,26 (m, 1H), 4,45 (d, J = 5,5 Hz, 1H), 4,39 - 4,26 (m, 1H), 2,90 - 2,72 (m, 1H), 2,67 - 2,49 (m, 1H), 2,04 (d, J = 9,4 Hz, 1H), 1,85 - 1,69 (m, 1H), 1,44 (d, J = 6,3 Hz, 3H). Isómero 3: EM (IEN) calc. para (C18H17F4N2O3) [M+H]+, 385,1, encontrado, 385,1. RMN 1H (400 MHz, CD3OD) ó 7.58 (s, 1H), 7,46 - 7,31 (m, 1H), 7,12 (c, J = 7,7 Hz, 2H), 6,75 - 6,26 (m, 1H), 4,53 - 4,42 (m, 1H), 4,31 (d, J = 11,0 Hz, 1H), 2,79 (ddd, J = 5,7, 12,1, 17,4 Hz, 1H), 2,67 - 2,51 (m, 1H), 2,14 (dd, J = 2,7; 11,0 Hz, 1H), 1,84 - 1,65 (m, 1H), 1,50 - 1,37 (m, 3H).
Isómero 4: EM (IEN) calc. para (C18H17F4N2O3) [M+H]+, 385,1, encontrado, 385,1. RMN 1H (400 MHz, CD3OD) ó 7.58 (s, 1H), 7,45 - 7,32 (m, 1H), 7,23 - 7,02 (m, 2H), 6,81 - 6,23 (m, 1H), 4,58 - 4,41 (m, 1H), 4,30 (d, J = 10,6 Hz, 1H), 2,79 (ddd, J = 5,5, 12,0, 17,3 Hz, 1H), 2,66 - 2,52 (m, 1H), 2,21 - 2,07 (m, 1H), 1,84 - 1,66 (m, 1H), 1,48 - 1,34 (m, 3H).

Los compuestos de Fórmula I (en donde, en el esquema 3, R2A, y R3A son cada uno hidrógeno y R2, R3, el anillo B, n, y cada R1 son como se describen en la fórmula I o las realizaciones alternativas descritas en el presente documento) pueden prepararse de acuerdo con el Esquema 3 mediante diversas reacciones en alcohol 3-1 (por ejemplo, alquilación, fluoración, oxidación seguido de fluoración, etc.) seguido de tratamiento con amoniaco.
EJEMPLOS 3-1A y 3-1B

3-1A: (R)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da y 3-1B: (S)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da
Etapa 1: 2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

A una mezcla de 5-(2,4-difluorofenil)-2-(hidroximetil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (Intermedio 2-4, 94 mg, 0,27 mmol) y yoduro de cobre (I) (10,3 mg, 0,05 mmol) en MeCN (10 ml) se le añadió ácido 2,2-difluoro-2-(fluorosulfonil)acético (96 mg, 0,54 mmol) y la mezcla se agitó a 45 °C durante 2,5 h en atmósfera de protección de N2. La reacción se enfrió a temperatura ambiente, se basificó a pH~8 con NaHCÜ3 acuoso saturado y agua (10 ml) y se extrajo con acetato de etilo ( 2 x 10 ml). Las fases orgánicas se secaron sobre Na2SÜ4, se filtraron y se concentraron a presión reducida para dar el producto en bruto, que se purificó por TLC prep. (gel de sílice, eluyendo con 1:1 de éter de petróleo/acetato de etilo) para dar el producto. EM (IEN) calc. para (C19H18F4NO4) [M+H]+, 400,2, encontrado, 400,2. RMN 1H (400 MHz, CD3OD) 67,61 (s, 1H), 7,37 - 7,46 (m, 1H), 7,07 - 7,20 (m, 2H), 6,30 - 6,71 (m, 1H), 4,51 - 4,61 (m, 1H), 4,39 (c, J = 7,1 Hz, 2H), 4,08 - 4,20 (m, 2H), 2,85 (ddd, J = 5,8, 11,9, 17,4 Hz, 1H), 2,57 -2,67 (m, 1H), 2,10 (td, J = 2,8, 13,8 Hz, 1H), 1,75 - 1,88 (m, 1H), 1,38 (t, J = 7,2 Hz, 3H).
Etapa 2: (S)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2,3-b1p¡r¡d¡n-7-carbox¡lato de etilo y (R)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2,3-b1p¡r¡d¡n-7-carbox¡lato de etilo

Se resolvió 2-((difluorometoxi)metil)-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo racémico (40 mg, 0,100 mmol) por SFC quiral (Columna: Chiralpak AD 250x30 mm, 5 um; Fase móvil: IPA del 30 % al 30 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 70 ml/min) para dar los dos enantiómeros.
Etapa 3: (S)-2-((difluorometoxi)metil)-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxamida y (R)-2-((difluorometoxi)metil)-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxamida

Una solución de un enantiómero de 2-((difluorometoxi)metil)-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (20 mg, 0,05 mmol) en amoniaco (10 M en MeOH) (40 ml) se agitó a 20 °C durante 16 h. La mezcla se concentró a presión reducida. El residuo se purificó por HPLC prep. (Columna: Phenomenex Gemini C18 250x21,2 mm, 5 um; Fase móvil: agua del 30 % al 60 % (que contenía NH4HCO3 10 mM)-ACN; Caudal: 25 ml/min) para dar un enantiómero del compuesto del título. EM (IEN) calc. para (C17H15F4N2O3) [M+H]+, 371,1, encontrado, 371,0. RMN 1H (400 MHz, CD3OD) 67,58 (s, 1H), 7,34 - 7,42 (m, 1H), 7,07 - 7,16 (m, 2H), 6,25 - 6,70 (m, 1H), 4,49 -4,56 (m, 1H), 4,06 - 4,17 (m, 2H), 2,82 (dd, J = 5,67, 11,93, 17,22 Hz, 1H), 2,58 (d, J = 16,04 Hz, 1H), 2,03 - 2,11 (m, 1H), 1,72 - 1,85 (m, 1H) El similar tratamiento del otro enantiómero de 2-((difluorometoxi)metil)-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo proporcionó el otro enantiómero del compuesto del título. EM (IEN) calc. para (C17H15F4N2O3) [M+H]+, 371,1, encontrado, 371,0. RMN 1H (400 MHz, CD3OD) 67,58 (s, 1H), 7,33 -7,42 (m, 1H), 7,07 - 7,17 (m, 2H), 6,27 - 6,71 (m, 1H), 4,49 - 4,56 (m, 1H), 4,06 - 4,16 (m, 2H), 2,82 (dd, J = 5,48, 11,84, 17,12 Hz, 1H), 2,58 (d, J = 16,43 Hz, 1H), 2,03 - 2,12 (m, 1H), 1,72 - 1,85 (m, 1H).
Los siguientes compuestos se prepararon de acuerdo con los procedimientos descritos para el Ejemplo 3-1A y 3-1B.

continuación

EJEMPLOS 3-5A y 3-5B

(S)-5-(2.4-d¡fluorofen¡l)-2-(metox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da y (R)-5-(2.4-difluorofen¡l)-2-(metox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxamida
Etapa 1: 5-(2.4-d¡fluorofen¡l)-2-(metox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

A una solución en agitación de 5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3.4-dih¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo (Intermedio 2-4. 150 mg. 0.43 mmol) y yodometano (67.0 mg. 0.47 mmol) en DMF seca (4 ml) se le añadió hidruro sódico (18.89 mg. 0.472 mmol) a 0 °C en atmósfera de N2. La mezcla resultante se agitó a 0 °C durante 30 min. La solución se concentró a presión reducida. el residuo se purificó por TLC prep. (gel de sílice. eluyendo con 2:1 de acetato de etilo/éter de petróleo) para proporcionar el producto. EM (IEN) calc. para (C19H20F2NO4) [M+H]+. 364.1. encontrado. 364.0.
Etapa 2: (S)-5-(2.4-d¡fluorofen¡l)-2-(metox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-blpir¡d¡n-7-carbox¡lato de etilo (R)-5-(2.4-d¡fluorofen¡l)-2-(metox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡rid¡n-7-carbox¡lato de etilo

Se separó 5-(2.4-d¡fluorofen¡l)-2-(metox¡met¡l)-3.4-dih¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo racémico (93 mg. 0.256 mmol) por SFC (Columna: Chiralpak OJ 250x30 mm. 5 um; Fase móvil: MeOH del 45 % al 45 % (que contenía DEA al 0.05 %) en CO2 ; Caudal: 60 ml/min) para proporcionar los dos enantiómeros.
Etapa 3: (S)-5-(2.4-d¡fluorofen¡l)-2-(metox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da y (R)-5-(2.4-d¡fluorofen¡h-2-ímetox¡met¡l)-3.4-d¡hidro-2H-p¡ranor2.3-b1p¡r¡d¡n-7-carboxam¡da

Una soluc¡ón de un enant¡ómero de 5-(2.4-d¡fluorofen¡l)-2-(metox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡nd¡n-7-carbox¡lato de et¡lo (41 mg. 0.11 mmol) en amon¡aco (10 M en MeoH) (15 ml) se ag¡tó a 26 °C durante 12 h. La mezcla se concentró a pres¡ón reduc¡da y el res¡duo se pur¡f¡có por TLC prep. (gel de síl¡ce. eluyendo con 2:1 de acetato de et¡lo/éter de petróleo) para proporc¡onar un enant¡ómero del compuesto del título. EM (IEN) calc. para (C17H17F2N2O3)[M+H]+. [335.11. encontrado. [335.0]. RMN 1H (400 MHz. CDCla) 6 7.71 (s. 2H). 7.26 - 7.22 (m. 1H).
6.99 - 6.89 (m. 2H). 5.50 (s. 1H). 4.46 - 4.44 (m. 1H). 3.73 - 3.62 (m. 2H). 3.44 (s. 3H). 2.74 - 2.56 (m. 2H). 2.15 - 2.02 (m. 1H). 1.85 - 1.79 (m. 1H).
El s¡m¡lar tratam¡ento del otro enant¡ómero de 5-(2.4-d¡fluorofen¡l)-2-(metox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de et¡lo proporc¡onó el otro enant¡ómero del compuesto del título. EM (IEN) calc. para (C17H17F2N2O3)[M+H]+. [335.1]. encontrado. [335.0]. RMN 1H (400 MHz. CDCla) 6 7.66 (s. 2H). 7.21 - 7.15 (m. 1H).
6.94 - 6.84 (m. 2H). 5.50 (s. 1H). 4.41 - 4.39 (m. 1H). 3.68 - 3.57 (m. 2H). 3.39 (s. 3H). 2.70 - 2.50 (m. 2H). 2.10 - 1.97 (m. 1H). 1.80 - 1.74 (m. 1H).
EJEMPLOS 3-6A y 3-6B

(S)-5-(2.4-d¡fluorofen¡l)-2-(fluoromet¡l)-3.4-d¡h¡dro-2H-p¡ranor[2.3-b1p¡r¡d¡n-7-carboxam¡da y (R)-5-(2.4-d¡fluorofen¡l)-2-(fluoromet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: 5-(2.4-d¡fluorofen¡l)-2-(fluoromet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

A una soluc¡ón de 5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carbox¡lato de et¡lo (Intermed¡o 2-4. 300 mg. 0.86 mmol) en d Cm (15 ml) a -40 °C se le añad¡ó DAs T (0.34 ml. 2.6 mmol) en DCM (3 ml). La mezcla de reacc¡ón se ag¡tó a 0 °C durante 100 m¡n en atmósfera de N2. La mezcla se ¡nact¡vó con NaHCO3 acuoso saturado (30 ml) y se extrajo con DCM (2 x 20 ml). Los extractos orgán¡cos comb¡nados se secaron con Na2SO4. se f¡ltraron y se concentraron a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (eluyendo con 20:1 a 1:3 de éter de petróleo/acetato de et¡lo) para proporc¡onar el producto. EM (IEN) calc. para (C18H17FaNO3)[M+H]+. 352.1. encontrado. 352.1
Etapa 2: (S)-5-(2.4-d¡fluorofen¡l)-2-(fluoromet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-blp¡r¡d¡n-7-carbox¡lato de et¡lo y (R)-5-(2.4-d¡fluorofen¡l)-2-(fluoromet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

Se resolvió 5-(2,4-difluorofenil)-2-(fluorometil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo racémico (990 mg, 2,82 mmol) por SFC quiral (Columna: Chiralpak Whelk-01 250x30 mm, 10 um; Fase móvil: EtOH del 50 % al 50 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 60 ml/min) para dar los dos enantiómeros.
Etapa 3: (S)-5-(2.4-d¡fluorofen¡l)-2-(fluoromet¡l)-3.4-d¡h¡dro-2H-p¡rano[2,3-b1p¡r¡d¡n-7-carboxam¡da y (R)-5-(2,4-d¡fluorofen¡l)-2-(fluoromet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da

Una solución de un enantiómero de 5-(2,4-difluorofenil)-2-(fluorometil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (485 mg, 1,38 mmol) en amoniaco (10 M en MeOH) (50 ml) se agitó a 23 °C durante 4 h. La mezcla se concentró al vacío para formar. El sólido se lavó con CH3CN (2 ml). La mezcla se filtró y el residuo sólido se secó a presión reducida para proporcionar un enantiómero del compuesto del título. EM (IEN) calc. para (C16H14FaN2O2)[M+H]+, 323,0, encontrado 323,1. RMN 1H (400 MHz, CD3OD) ó 7,59 (s, 1H), 7,43 - 7,37 (m, 1H), 7,16 - 7,10 (m, 2H), 4,87 - 4,54 (m, 3H), 2,87 - 2,80 (m, 1H), 2,63 - 2,58 (m, 1H), 2,09 - 2,06 (m, 1H), 1,84 - 1,79 (m, 1H). El similar tratamiento del otro enantiómero de 5-(2,4-difluorofenil)-2-(fluorometil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo proporcionó el otro enantiómero del compuesto del título. EM (IEN) calc. para (C16H14FaN2O2)[M+H]+, 323,0, encontrado 323,1. RMN 1H (400 MHz, CD3OD) ó 7,59 (s, 1H), 7,43 - 7,37 (m, 1H), 7,16 - 7,10 (m, 2H), 4,87 - 4,54 (m, 3H), 2,86 - 2,81 (m, 1H), 2,63 - 2,59 (m, 1H), 2,09 - 2,04 (m, 1H), 1,82 - 1,79 (m, 1H). EJEMPLOS 3-7A y 3-7B
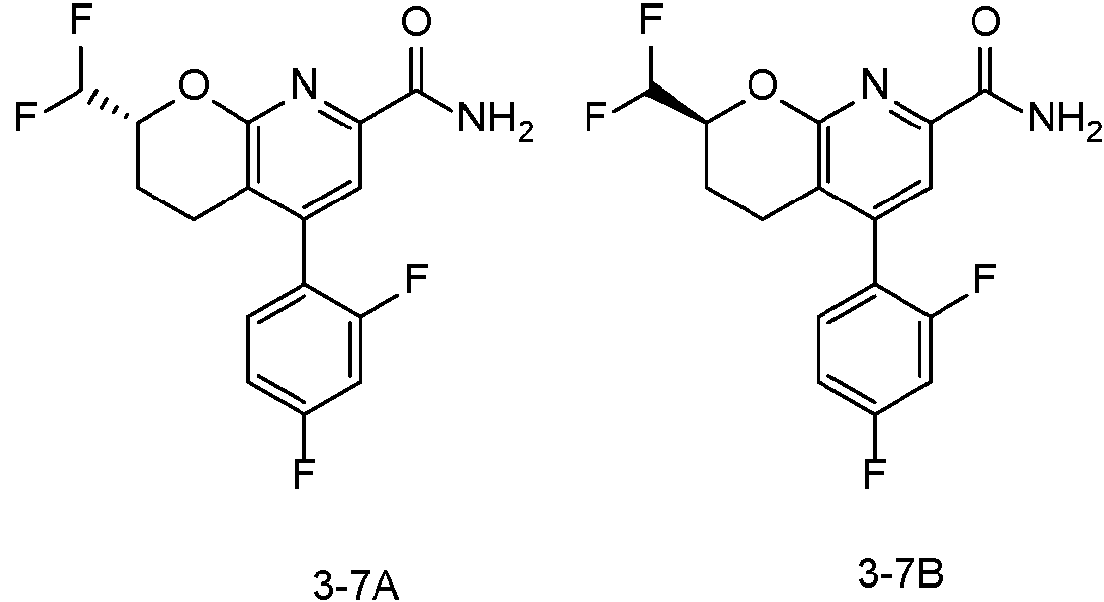
3-7A: (R)-2-(d¡fluoromet¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da y 3-7B: (S)-2-(d¡fluoromet¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: 5-(2.4-d¡fluorofen¡l)-2-form¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo (INTERMEDIO 3-1)

Una solución en agitación de 5-(2,4-difluorofenil)-2-(hidroximetil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de
etilo racémico (Intermedio 2-4, 300 mg, 0,86 mmol) en DCM (20 ml) se añadió DMP (474 mg, 1,1 mmol) a 0 °C en atmósfera de N2. La mezcla resultante se agitó a 0 °C durante 1 h. Se añadió NaHCO3 acuoso saturado (50 ml). La capa orgánica se separó, se secó sobre NaSO4 anhidro, se filtró y se concentró a presión reducida para proporcionar el producto que se usó en la siguiente etapa sin purificación adicional. EM (IEN) calc. para (C18H18F2NO5) [M+H+H2O]+, 366,1, encontrado, 366,1.
Etapa 2: 2-(d¡fluoromet¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2,3-b1p¡r¡d¡n-7-carbox¡lato de etilo

A una solución de 5-(2,4-d¡fluorofen¡l)-2-form¡l-3,4-d¡h¡dro-2H-pirano[2,3-b]p¡r¡d¡n-7-carbox¡lato de etilo (380 mg, 1,1 mmol) en DCM (20 ml) y EtOH (0,01 ml) se le añadió DAST (0,289 ml, 2,2 mmol) a 0 °C. Después de la adición, la mezcla se agitó a 24 °C durante 2 h. A la mezcla de reacción se añadió lentamente bicarbonato sódico acuoso saturado (20 ml). La capa orgánica se separó, se secó sobre sulfato sódico y se concentró a presión reducida. El residuo se purificó por TLC prep. (gel de sílice, eluyendo con 1:2 de acetato de etilo/éter de petróleo) para dar el producto. EM (IEN) calc. para (C18H16F4NO3) [M+H]+, 370,2, encontrado, 370,0.
Etapa 3: 2-(d¡fluoromet¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da

Una solución de 2-(difluorometil)-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]-piridin-7-carboxilato de etilo (81 mg, 0,22 mmol) en amoniaco (10 M en MeOH) (15 ml) se agitó a 25 °C durante 3 h. La mezcla se concentró a presión reducida para dar el producto. EM (IEN) calc. para (C16H13F4N2O2)[M+H]+, [341,1], encontrado, [341,0].
Etapa 4: (R^-td ifluorom etiD^-^^-difluorofeniD^^-dihidro^H-pirano^^-blp iridin^-carboxam ida y (S)-2-(d¡fluoromet¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da

Se separó 2-(difluorometil)-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxamida de etilo racémico (64 mg, 0,19 mmol) por SFC (Columna: Chiralpak AD 250x30 mm, 5 um; Fase móvil: MeOH del 20 % al 20 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 60 ml/min) para proporcionar los dos enantiómeros:
Un enantiómero: EM (IEN) calc. para (C16H13F4N2O2) [M+H]+, 341,1, encontrado, 341,0. RMN 1H (400 MHz, CD3OD) ó 7,63 (s, 1H), 7,43 - 7,38 (m, 1H), 7,17 - 7,11 (m, 2H), 6,22 - 5,94 (m, 1H), 4,87 - 4,54 (m, 1H), 3,30 - 2,81 (m, 1H), 2,67 - 2,63 (m, 1H), 2,19 - 2,16 (m, 1H), 1,90 - 1,84 (m, 1H).
Otro enantiómero: EM (IEN) calc. para (C16H13F4N2O2) [M+H]+, 341,1, encontrado, 341,0. RMN 1H (400 MHz, CD3OD) ó 7,63 (s, 1H), 7,43 - 7,38 (m, 1H), 7,17 - 7,11 (m, 2H), 6,22 - 5,94 (m, 1H), 4,62 - 4,54 (m, 1H), 3,30 - 2,81 (m, 1H), 2,67 - 2,63 (m, 1H), 2,19 - 2,16 (m, 1H), 1,90 - 1,84 (m, 1H).
EJEMPLOS 3-8A y 3-8B

(S)-5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoroet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da_____ y_____ (R)-5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoroet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: 5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoro-1-h¡drox¡et¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de et¡lo y 5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoro-1-((tr¡met¡ls¡l¡l)ox¡)et¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato

A una soluc¡ón de 5-(2.4-d¡fluorofen¡l)-2-form¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de et¡lo (Intermed¡o 3 1. 700 mg. 2.0 mmol). fluoruro de ces¡o (918 mg. 6.1 mmol) en THF (15 ml) se le añad¡ó tr¡met¡l(tr¡fluoromet¡l)s¡lano (860 mg. 6.1 mmol) a 0 °C en atmósfera de N2. La mezcla de reacc¡ón se ag¡tó a 0 °C durante 2 h en atmósfera de N2. La mezcla se ¡nact¡vó con H2O (15 ml) y se extrajo con EtOAc (3 x 25 ml). Las capas orgán¡cas comb¡nadas se secaron sobre Na2SO4 anh¡dro. se f¡ltraron y el f¡ltrado se concentró a pres¡ón reduc¡da. La cromatografía sobre gel de síl¡ce que eluyó con 20:1 a 2:1 de éter de petróleo:acetato de et¡lo proporc¡onó los productos. EM (IEN) calc. para (C22H25F5NO4S¡)[M+H1+. 490.5. encontrado 490.4. RMN 1H (400 MHz. CDCla) 6 7.48 (s. 1H). 6.97-7.07 (m. 1H). 6.72 6 .8 8 (m. 2H). 4.21-4.38 (m. 3H). 2.59 (s a. 1H). 2.47 (s a. 1H). 1.83-2.04 (m. 1H). 1.62-1.83 (m. 1H). 1.23 (t. J = 7.04 Hz.
3H). 0.01 (d. J = 11.35 Hz. 9H).
Etapa 2: 5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoro-1-h¡drox¡et¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

A una soluc¡ón de 5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoro-1-((tr¡met¡ls¡l¡l)ox¡)et¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de et¡lo (300 mg. 0.61 mmol) en etanol (15 ml) se le añad¡ó K2CO3 (102 mg. 0.74 mmol) a 18 °C. La mezcla se ag¡tó a 18 °C durante 40 m¡n. La mezcla se f¡ltró y el f¡ltrado se concentró al vacío y el res¡duo se pur¡f¡có por TLC prep. (gel de síl¡ce. eluyendo con 2:1 de éter de petróleo:acetato de et¡lo) para dar el producto. EM (IEN) calc. para (C19H17F5NO4 ) [M+H1+. 418.0. encontrado. 417.9. RMN 1H (400 MHz. CDCla) 6 7.68 (d. J = 10.17 Hz. 1H). 7.22 (s a.
1H). 6.86-7.06 (m. 2H). 4.53-4.66 (m. 1H). 4.43 (qu¡nt.. J = 7.24 Hz. 2H). 4.11 (c. J = 7.30 Hz. 1H). 2.81 (s a. 1H). 2.58 2.69 (m. 1H). 2.04-2.19 (m. 2H). 1.40 (c. J = 7.43 Hz. 3H).
Etapa 3: ác¡do 5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoro-1-(((met¡lt¡o)carbonot¡o¡l)ox¡)et¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxíl¡co

A una solución de 5-(2,4-difluorofenil)-2-(2,2,2-trifluoro-1-hidroxietil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (280 mg, 0,67 mmol) en THF (2 ml) se le añadió NaH (40,3 mg, 1,0 mmol) a 0 °C en atmósfera de N2. Después de agitar durante 30 min, Se añadieron disulfuro de carbono (0,10 ml, 1,68 mmol) y yodometano (0,31 ml, 5,00 mmol) y la mezcla se agitó a 0 °C en atmósfera de N2 durante 1 h. La reacción se diluyó con 15 ml de agua y 5 gotas de HCl (concentrado, 12 M), se extrajo con EtOAc (3 x 15 ml), los extractos orgánicos combinados se secaron sobre Na2SÜ4, se filtraron y se concentraron al vacío para dar el producto, que se usó en la etapa siguiente sin purificación adicional. EM (IEN) calc. para (C19H-i5F5NO4S2)[M+H]+, 480,1, encontrado, 480,2.
Etapa 4: 5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoro-1-(((met¡lt¡o)carbonot¡o¡l)ox¡)et¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxilato de etilo

A una solución de ácido 5-(2,4-difluorofenil)-2-(2,2,2-trifluoro-1-(((metiltio)carbonotioil)oxi)etil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxílico (430 mg, 0,90 mmol) en EtOH seco (10 ml) se le añadió dicloruro sulfuroso (320 mg, 2,69 mmol) en atmósfera de N2. La mezcla se agitó a 85 °C en atmósfera de N2 durante 2,5 h. La mezcla se inactivó con H2O (15 ml), después la mezcla se ajustó a pH = 8 con NaHCO3 sólido. La mezcla se extrajo con EtOAc (3 x 20 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. La cromatografía sobre gel de sílice que eluyó con 10:1 a 5:1 de éter de petróleo:acetato de etilo proporcionó el producto. EM (IEN) calc. para (C21H-i9F5NO4S2)[M+H]+, 508,4, encontrado, 508,3. RMN 1H (400 MHz, CDCla) 67,65 (d, J = 2,74 Hz, 1H), 7,17-7,23 (m, 1H), 6,87-7,04 (m, 2H), 4,68-4,82 (m, 1H), 4,42 (dc, J = 2,35, 7,04 Hz, 2H), 2,70 2,89 (m, 1H), 2,66 (s a, 1H), 2,60 (s, 3H), 2,07-2,25 (m, 1H), 1,80-2,00 (m, 1H), 1,39 (dt, J = 1,96, 7,04 Hz, 3H).
Etapa 5: 5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoroet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡rid¡n-7-carbox¡lato de etilo

A una solución de 5-(2,4-difluorofenil)-2-(2,2,2-trifluoro-1-(((metiltio)carbonotioil)oxi)etil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (230 mg, 0,45 mmol) y tributilestannano (1,18 g, 4,05 mmol) en tolueno (10 ml) se le añadió azobisisobutironitrilo (20 mg, 0,12 mmol) en atmósfera de N2. La mezcla de reacción se calentó a 100 °C y se agitó en atmósfera de N2 durante 1 h. La reacción se diluyó con 20 ml de agua, se extrajo con EtOAc (20 ml x 3), las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para dar el producto en bruto. El residuo se purificó por cromatografía sobre gel de sílice (eluyendo con 10:1 a 5:1 de éter de petróleo:acetato de etilo) para dar el producto. EM (IEN) calc. para (C19H17F5n O3)[M+H]+, 402,1, encontrado, 402,1. RMN 1H (400 MHz, CDCla) 67,65 (s, 1H), 7,18-7,24 (m, 1H), 6,87-7,04 (m, 2H), 4,58 (td, J = 5,23; 10,27 Hz, 1H), 4,42 (c, J = 7,04 Hz, 2H), 2,71-2,94 (m, 2H), 2,42-2,65 (m, 2H), 2,12-2,24 (m, 1H), 1,69-1,86 (m, 1H), 1,39 (t, J = 7,04 Hz, 3H).
Etapa 6: (S)-5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoroet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-blp¡rid¡n-7-carbox¡lato de etilo y R)-5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoroet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

Se resolvió 5-(2,4-difluorofenil)-2-(2,2,2-trifluoroetil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo racémico (120 mg, 0,3 mmol) a través de SFC quiral (Columna: Chiralpak IC 250x30 mm, 10 um; Fase móvil: IPA del 30 % al 30 % (que contenía DEA al 0,05 % en CO2 ; Caudal: 6 ml/min) para dar los dos enantiómeros que se usaron en la siguiente etapa sin purificación adicional.
Etapa 7: (S)-5-(2.4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoroet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da y (R)-5-(2,4-d¡fluorofen¡l)-2-(2.2.2-tr¡fluoroet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da

Una solución de un enantiómero de 5-(2,4-difluorofenil)-2-(2,2,2-trifluoroetil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (45 mg, 0,11 mmol) en amoniaco (10 M en MeOH) (20 ml) se agitó a 10 °C durante 20 h. La mezcla se concentró y se purificó por HPLC prep. (Columna: Phenomenex Synergi C18 150x30 mm, 4 um; Fase móvil: agua del 43 % al 63 % (que contenía HCl al 0,05 %)-ACN; Caudal: 25 ml/min) para dar un enantiómero del compuesto del título. EM (IEN) calc. para (C17H14FsN2O2)[M+H]+, 373,1, encontrado, 372,9. RMN 1H (400 MHz, CD3OD) ó 7,59 (s a, 1H), 7,38 (d, J = 6,26 Hz, 1H), 7,01-7,18 (m, 2H), 4,65 (s a, 1H), 2,49-2,89 (m, 4H), 2,05-2,24 (m, 1H), 1,79 (s a, 1H). El similar tratamiento del otro enantiómero de 5-(2,4-difluorofenil)-2-(2,2,2-trifluoroetil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo proporcionó el otro enantiómero del compuesto del título. EM (IEN) calc. para (C17H14F5N2O2)[M+H]+, 373,1, encontrado 372,9. RMN 1H (400 MHz, CD3OD) ó 7,60 (s, 1H), 7,35-7,45 (m, 1H), 7,08 7,18 (m, 2H), 4,67 (s a, 1H), 2,54-2,90 (m, 4H), 2,09-2,20 (m, 1H), 1,73-1,88 (m, 1H) EJEMPLO 3-9A, 3-9B, 3-9C y 3-9D

(2S.3R)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da.
(2S.3S)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da.
(2R.3R)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da_____ y
(2R.3S)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: etil 2S.3R)-. (2S.3S)-. (2R.3R) o (2R.3S)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato

A una soluc¡ón de un ¡sómero de 5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de et¡lo (Intermed¡o 2-5A. 2-5B. 2-5C o 2-5D) 140 mg. 0.39 mmol) en aceton¡tr¡lo (8 ml) se le añad¡ó yoduro de cobre (I) (14.7 mg. 0.077 mmol) y la mezcla se calentó a 45 °C en atmósfera de N2. Se añad¡ó gota a gota ác¡do 2.2-d¡fluoro-2-(fluorosulfon¡l)acét¡co (0.2 ml. 1.9 mmol). la mezcla se ag¡tó a 45 °C durante 2 h. La mezcla se enfr¡ó en un baño de h¡elo y se añad¡ó NaHCÜ3 acuoso saturado (15 ml). La capa acuosa se extrajo con EtOAc (25 ml x 3). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (sat. 20 ml). se secaron sobre Na2SO4. se f¡ltraron y se concentraron a pres¡ón reduc¡da para dar el producto en bruto. El res¡duo se pur¡f¡có por TLC prep. (gel de síl¡ce. eluyendo con 2:1 de éter de petróleo:acetato de et¡lo) para dar un ¡sómero del producto. EM (IEN) calc. para (C20H20F4 NO4) [M+H1+. 414.1. encontrado. 414.3. RMN 1H (400 MHz. CDCla) 67.59 (d. J = 6.26 Hz. 1H). 7.13-7.19 (m.
1H). 6.85-7.01 (m. 2H). 5.99-6.48 (m. 1H). 4.37 (c. J = 7.04 Hz. 2H). 3.93-4.26 (m. 3H). 2.01-2.61 (m. 3H). 1.24-1.41 (m. 3H). 0.77-1.06 (m. 3H).
El s¡m¡lar tratam¡ento de los otros ¡sómeros de 5-(2.4-d¡fluorofen¡l)-2-(h¡drox¡met¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carbox¡lato de et¡lo proporc¡onó los otros ¡sómeros del producto.
Etapa 2: (2S.3R)-. (2S.3S)-. (2R.3R) o (2R.3S)-2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da

Una soluc¡ón de un ¡sómero de 2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de et¡lo (13 mg. 0.03 mmol) en amon¡aco (10 M en MeOH) (10 ml) se ag¡tó a temperatura amb¡ente durante 10 h. La mezcla se concentró y el res¡duo se pur¡f¡có por HPLC prep. (Columna: Phenomenex Synerg¡ C18 150x30 mm. 4 um; Fase móv¡l: agua del 40 % al 60 % (que contenía TFA al 0.1 %)-ACN; Caudal: 25 ml/m¡n) para dar un ¡sómero del compuesto del título. EM (IEN) calc. para (C18H17F4 N2O3XM+H1+. 385.1. encontrado. 385.0. RMN 1H (400 MHz. CD3OD) 67.59 (s. 1H). 7.28-7.45 (m. 1H). 6.99-7.21 (m. 2H). 6.23-6.71 (m. 1H). 4.58 (s a. 1H). 4.01-4.19 (m. 2H). 2.88-3.03 (m. 1H). 2.23-2.44 (m. 2H). 0.90 (d. J = 6.26 Hz. 3H).
El s¡m¡lar tratam¡ento de los otros ¡sómeros de 2-((d¡fluorometox¡)met¡l)-5-(2.4-d¡fluorofen¡l)-3-met¡l-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de et¡lo proporc¡onó los otros ¡sómeros del compuesto del título.
Isómero 2: EM (IEN) calc. para (C18H17F4N2O3 XM+H1+. 385.1. encontrado. 385.0. RMN 1H (400 MHz. CD3OD) 6 7.59 (s a. 1H). 7.29-7.44 (m. 1H). 7.04-7.19 (m. 2H). 6.24-6.73 (m. 1H). 4.59 (s a. 1H). 4.10 (s a. 2H). 2.97 (s a.
1H). 2.32 (s a. 2H). 0.90 (d. J = 5.87 Hz. 3H).
Isómero 3: EM (IEN) calc. para (C18H17F4N2O3 XM+H1+. 385.1. encontrado. 385.0. RMN 1H (400 MHz. CD3OD) 6 7.57 (s a. 1H). 7.38 (d. J = 5.09 Hz. 1H). 7.12 (d. J = 7.43 Hz. 2H). 6.22-6.71 (m. 1H). 4.07-4.32 (m. 3H). 2.40-2.69 (m. 2H). 2.06 (s a. 1H). 1.04 (d. J = 4.30 Hz. 3H).
Isómero 4: EM (IEN) calc. para (C18H17F4N2O3 XM+H1+. 385.1. encontrado. 385.0. RMN 1H (400 MHz. CD3OD) 6 7.58 (s. 1H). 7.29-7.46 (m. 1H). 7.04-7.20 (m. 2H). 6.25-6.68 (m. 1H). 4.10-4.31 (m. 3H). 2.41-2.69 (m. 2H). 2.07 (s a. 1H). 1.05 (d. J = 6.65 Hz. 3H).
Esquem a 4
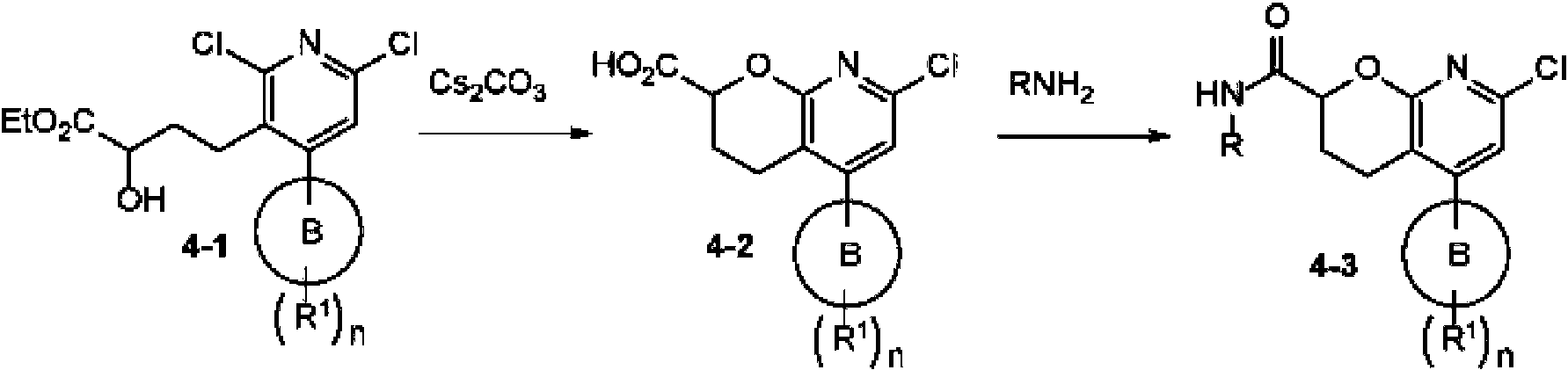
alquilacion
reducción


Pd(dppf)Cl2
-:O H 
después, desproteccion
(si R' = H deseado) 
Los compuestos de Fórmula I (en donde, en el esquema 4, R es -haloalquilo (C1-4), R' es H o metilo, el anillo B, n, y cada R1 son como se describen en la Fórmula I o las realizaciones alternativas descritas en el presente documento) pueden prepararse de acuerdo con el Esquema 4 a través de la ciclación de 4-1 para generar el ácido 4-2. Esto fue seguido de la formación de amida y reducción a la amina. La alquilación posterior (o protección para aminas secundarias) seguido de carbonilación y tratamiento con amoniaco proporcionó las aminas terciarias (una desprotección final proporcionó aminas secundarias).
EJEMPLOS 4-1A y 4-1B
(S)-5-(2.4-d¡fluorofen¡l)-2-((met¡l(2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da y (R)-5-(2.4-d¡fluorofen¡l)-2-((met¡l(2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da Etapa 1: 7-cloro-5-(2.4-d¡fluorofen¡l)-N-(2.2.2-tr¡fluoroet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-carboxam¡da

A una soluc¡ón de ác¡do 7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-carboxíl¡co (Intermed¡o 3-2.
0.8 g. 2.5 mmol) y HATU (1.2 g. 3.7 mmol) en DMF (10 ml). La mezcla se ag¡tó a 25 °C durante 10 m¡n en atmósfera de N2. Después. se añad¡ó DIPEA (0.68 g. 4.9 mmol) segu¡do de la ad¡c¡ón lenta de 2.2.2-tr¡fluoroetanam¡na (0.37 g.
3.7 mmol). La mezcla se ag¡tó a 25 °C durante 2 h. La mezcla se vert¡ó en H2O (100 ml) y se extrajo con EtOAc (30 ml x 3). Las capas orgán¡cas comb¡nadas se pur¡f¡caron por cromatografía sobre gel de síl¡ce [columna ISCO®:4 g SepaFlash eluyendo con acetato de et¡lo del 0 % al 20 %/éter de petróleo] para dar el producto. EM (IEN) calc. para (C17H13ClF5N2O2)[M+H]+.407.0 encontrado. 407.0
Etapa 2: N-((7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-¡l)met¡l)-2.2.2-tr¡fluoroetanam¡na

A una soluc¡ón de 7-cloro-5-(2.4-d¡fluorofen¡l)-N-(2.2.2-tr¡fluoroet¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-carboxam¡da (0.45 g. 1.1 mmol) y BH3 S(CH3)2 (44 mmol) en Th F (10 ml). La mezcla se ag¡tó a 0 °C durante 10 m¡n. después a 45 °C durante otras 2 h. La mezcla se ¡nact¡vó con CH3OH (40 ml) y se concentró a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce [columna ISCO®:4 g SepaFlash eluyendo con acetato de et¡lo del 0 % al 20 %/éter de petróleo1 para dar el producto. Em (IEN) calc. para (C17H15C F 5 N2OXM+H1+. 392.9 encontrado. 392.9 Etapa 3: N-((7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-¡l)met¡l)-2.2.2-tr¡fluoro-N-met¡letanam¡na

Una soluc¡ón de N-((7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-¡l)met¡l)-2.2.2-tr¡fluoroetanam¡na (0.1 g. 0.26 mmol) y CH2O (0.016 g. 0.51 mmol) en DCE (10 ml) se ag¡tó a 25 °C. Se añad¡ó NaBH(OAc)3 (0.11 g.
0.51 mmol) a la soluc¡ón segu¡do de una gota de AcOH. La mezcla se ag¡tó a 25 °C durante 8 h. La mezcla se vert¡ó en H2O (20 ml). el pH se ajustó a ~9 con la ad¡c¡ón de NaHCO3 y la mezcla se extrajo con EtOAc (20 ml x 3). Los extractos orgán¡cos comb¡nados se secaron sobre Na2SO4 anh¡dro. se f¡ltraron y se concentraron. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce [columna ISCO®:4 g SepaFlash eluyendo con acetato de et¡lo del 0 % al 20 %/éter de petróleo1 para dar el producto. EM (IEN) calc. para (C16H19ClF5N2O)[M+H1+. 407.0 encontrado. 407.0. Etapa 4: 5-(2.4-d¡fluorofen¡l)-2-((met¡l(2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

A una solución en agitación de N-((7-cloro-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-2-il)metil)-2,2,2-trifluoro-N-metiletanamina (0,13 g, 0,32 mmol) en EtOH (20 ml) se le añadieron Pd(dppf)Cl2 (0,023 g, 0,032 mmol) y KOAc (0,064 g, 0,64 mmol). La mezcla se calentó a 60 °C y se agitó durante 18 h en 0,34 MPa (50 psi) de CO. La mezcla se concentró a presión reducida y la solución resultante se diluyó con agua (20 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron. El residuo se purificó por TLC prep. (gel de sílice, eluyendo con 4:1 de éter de petróleo/acetato de etilo) para proporcionar el producto. EM (IEN) calc. para (C21H22F5 N2O3) [M+H]+, 445,1, encontrado, 445,0. RMN 1H (400 Mh z , CDCla) ó 7,56 (s, 1H), 7,24 - 7,11 (m, 1H), 6,99 - 6,79 (m, 2H), 4,36 (c, J = 7,0 Hz, 2H), 4,28 (td, J = 4,9; 9,8 Hz, 1H), 3,10 (c, J = 9,0 Hz, 2H), 3,04 - 2,91 (m, 1H), 2,78 (dd, J = 6,7; 13,3 Hz, 1H), 2,49 (s, 3H), 2,16 - 2,02 (m, 1H), 1,74 -1,56 (m, 1H), 1,33 (t, J = 7,2 Hz, 3H)
Etapa 5: (S)-5-(2.4-d¡fluorofen¡l)-2-((met¡l(2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxilato de etilo y (R)-5-(2,4-difluorofenil)-2-((metil(2,2,2-trifluoroetil)amino)metil)-3,4-dihidro-2H-pirano[2,3-blpiridin-7-carboxilato de etilo
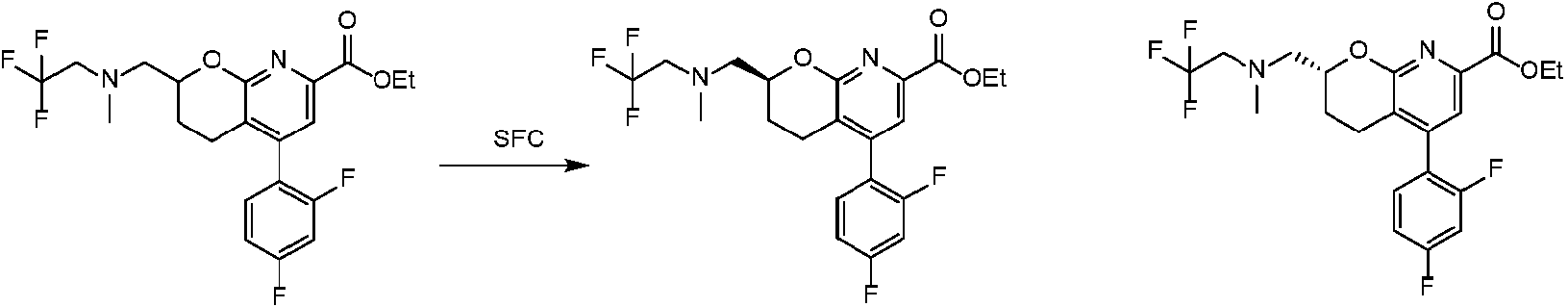
Se resolvió -5-(2,4-difluorofenil)-2-((metil(2,2,2-trifluoroetil)amino)metil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo racémico (100 mg, 0,28 mmol) por SFC quiral (Columna: Chiralpak AD 250x30 mm, 5 um; Fase móvil: IPA del 20 % al 20 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 60 ml/min) para proporcionar los dos enantiómeros. EM (IEN) calc. para (C21H22F5N2O3) [M+H]+, 445,1, encontrado, 445,0. EM (IEN) calc. para (C21H22F5 N2O3) [M+H]+, 445,1, encontrado, 445,0.
Etapa 6:
(S)-5-(2.4-d¡fluorofen¡l)-2-((met¡l(2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da y (R)-5-(2.4-d¡fluorofen¡l)-2-((met¡l(2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da

Una solución de un enantiómero de 5-(2,4-difluorofenil)-2-((metil(2,2,2-trifluoroetil)amino)metil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (40 mg, 0,09 mmol) en amoniaco (10 M en MeOH) (40 ml) se agitó a 20 °C durante 16 h. La mezcla se concentró a presión reducida. El residuo se purificó por TLC prep. para proporcionar un enantiómero del compuesto del título. EM (IEN) calc. para (C19H19F5N3O2) [M+H]+, 416,1, encontrado, 416,3.
El similar tratamiento del otro enantiómero de 5-(2,4-difluorofenil)-2-((metil(2,2,2-trifluoroetil)amino)metil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo proporcionó el otro enantiómero del compuesto del título. EM (IEN) calc. para (C19H19F5N3O2) [M+H]+, 416,1, encontrado, 416,3.
EJEMPLOS 4-2A y 4-2B

(S)-5-(2.4-d¡fluorofen¡l)-2-(((2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da y (R)-5-(2.4-d¡fluorofen¡l)-2-(((2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carboxam¡da
Etapa 1: ((7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-¡l)met¡l)(2.2.2-tr¡fluoroet¡l)carbamato de terc-but¡lo

A una soluc¡ón en ag¡tac¡ón de N-((7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-¡l)met¡l)-2.2.2-tr¡fluoroetanam¡na (150 mg. 0.38 mmol) en DCM (10 ml) se le añad¡eron d¡carbonato de d¡-ferc-but¡lo (194 mg.
3.3 mmol). Et3N (820 mg. 3.8 mmol) y N.N-d¡met¡lp¡r¡d¡n-4-am¡na (4.7 mg. 0.04 mmol). Después. la mezcla se ag¡tó a 25 °C durante 16 h. La soluc¡ón se concentró a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por cromatografía sobre gel de síl¡ce (eluyendo con 1:0 a 20:1 de éter de petróleo/acetato de et¡lo) para proporc¡onar el producto. EM (IEN) calc. para (C22H23C F 5N2O3) [M+H1+. 493.12. encontrado. 493.
Etapa 2: 2-(((ferc-butox¡carbon¡l)(2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-7-carbox¡lato de etilo

A una soluc¡ón en ag¡tac¡ón de N-((7-cloro-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-¡l)met¡l)-2.2.2-tr¡fluoroetanam¡na (71 mg. 0.14 mmol) en EtOH (15 ml) se le añad¡eron Pd(dppf)Cl2 (10.6 mg. 0.014 mmol) y KOAc (28 mg. 0.29 mmol). Después. la soluc¡ón se calentó a 60 °C y se ag¡tó durante 18 h a 0.34 MPa (50 ps¡) de CO. La soluc¡ón se concentró a pres¡ón reduc¡da. La soluc¡ón resultante se d¡luyó con agua (20 ml) y se extrajo con EtOAc (20 ml x 3). Las capas orgán¡cas comb¡nadas se secaron sobre Na2SO4. se f¡ltraron y se concentraron a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por TLC prep. (gel de síl¡ce. eluyendo con 3:1 de éter de petróleo:acetato de et¡lo) para proporc¡onar el producto. EM (IEN) calc. para (C25H28F5N2O5) [M+H1+. 531.18. encontrado. 531.3.
Etapa 3: ((7-carbamo¡l-5-(2.4-d¡fluorofen¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b1p¡r¡d¡n-2-¡l)met¡l)(2.2.2-tr¡fluoroet¡l)carbamato de terc-but¡lo
2-(((Terc-butoxicarbonil)(2,2,2-trifluoroetil)amino)metil)-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-7-carboxilato de etilo (53 mg, 0,1 mmol) en NH3/MeOH (10 M, 15 ml) se agitó a 25 °C durante 16 h. La mezcla se concentró a presión reducida para dar el producto que se usó en etapas posteriores sin purificación adicional. EM (IEN) calc. para (C23H25F5 N3O4) [M+H]+, 502,17, encontrado, 502,2.
Etapa_________ 4 _________ (S)-((7-carbamoil-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-2-il)metil)(2,2,2-trifluoroetil)carbamato de ferc-butilo y (R)-((7-carbamoil-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-2-¡l)met¡l)(2.2.2-tr¡fluoroet¡l)carbamato de ferc-butilo

Se separó ((7-carbamoil-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-2-il)metil)(2,2,2-trifluoroetil)carbamato de ferc-butilo (40 mg, 0,08 mmol) por SFC (Columna: Chiralpak AS 250x30 mm, 5 um; Fase móvil: MeOH del 10 % al 10 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 60 ml/min) para proporcionar los dos enantiómeros.
Etapa 4: (S)-5-(2.4-d¡fluorofen¡l)-2-(((2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-3.4-d¡h¡dro-2H-p¡rano[2.3-b]p¡r¡d¡n-7-carboxam¡da y (R)-5-(2.4-d¡fluorofen¡l)-2-(((2.2.2-tr¡fluoroet¡l)am¡no)met¡l)-3.4-d¡h¡dro-2H-p¡ranor2,3-b1p¡r¡d¡n-7-carboxam¡da

Un enantiómero de ((7-carbamoil-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-2-il)metil)(2,2,2-trifluoroetil)carbamato de ferc-butilo (20 mg, 0,04 mmol) se agitó en DCM (3 ml) y TFA (0,5 ml) a 25 °C durante 1 h. La mezcla de reacción se concentró a presión reducida para proporcionar un enantiómero del compuesto del título. EM (IEN) calc. para (C18H17F5 N3O2) [M+H]+, 402,1, encontrado, 402,1.
El similar tratamiento del otro enantiómero de ((7-carbamoil-5-(2,4-difluorofenil)-3,4-dihidro-2H-pirano[2,3-b]piridin-2-il)metil)(2,2,2-trifluoroetil)carbamato de ferc-butilo proporcionó el otro enantiómero del compuesto del título. Em (IEN) calc. para (C18H17F5N3O2) [M+H]+, 402,1, encontrado 402,1.
EJEMPLOS 5A y 5B

(5S.8R)-4-(2.4-d¡fluorofen¡l)-5.6.7.8-tetrah¡dro-5,8-epox¡qu¡nol¡na-2-carboxam¡da y (5R,8S)-4-(2,4-difluorofenil)-5.6.7.8-tetrah¡dro-5,8-epox¡qu¡nol¡na-2-carboxam¡da
Etapa 1: 3-(trimetilsilil)piridin-2-ol

A una solución de diisopropilamina (31,0 ml, 218 mmol) en THF (200 ml) se le añadió n-butillitio (95 ml, 238 mmol) a -78 °C. Después de agitar durante 1 h, una solución de piridin-2-ol (9 g, 95 mmol) en THF (100 ml) se añadió a -78 °C. La mezcla se calentó gradualmente a 0 °C durante 1 h. Se añadió clorotrimetilsilano (13,20 ml, 104 mmol) a la mezcla a 0 °C. La mezcla resultante se calentó gradualmente a 25 °C durante 14 h. La reacción se interrumpió con H2O (300 ml) y se extrajo con EtOAc (3 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 40 g SepaFlash®, eluyendo con acetato de etilo al 0-30 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (CaH^NOSi) [M+H]+, 168,1, encontrado, 168,0. RMN 1H (400 MHz, CDCla) ó 12,07 (s a, 1H), 7,54 (dd, J = 1,8; 6,5 Hz, 1H), 7,33 (dd, J = 2,0; 6,3 Hz, 1H), 6,23 (t, J = 6,5 Hz, 1H), 0,28 (s, 9H).
Etapa 2: trifluorometanosulfonato de 3-(trimetilsilil)piridin-2-ilo

A una solución de 3-(trimetilsilil)piridin-2-ol (13 g, 78 mmol) en piridina (100 ml) se le añadió trifluorometanosulfónico anhídrido (16 ml, 95 mmol) a 0 °C en atmósfera de N2 durante 20 min. La mezcla se calentó gradualmente a 25 °C y se agitó durante otras 14 h. La mezcla de reacción se concentró a presión reducida y el residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 120 g SepaFlash®, eluyendo con acetato de etilo al 0-10 %/éter de petróleo) para dar el producto. EM (IEN) calc. para (C9H13FaNO3SSi) [M+H]+, 300,0, encontrado, 300,0. RMN 1H (400 MHz, CDCla) ó 8,33 (dd, J = 2,0; 4,7 Hz, 1H), 7,92 (dd, J = 1,6; 7,0 Hz, 1H), 7,31 (dd, J = 5,1,7,0 Hz, 1H), 0,38 (s, 9H).
Etapa 3: 5.8-d¡h¡dro-5,8-epox¡qu¡nol¡na

A una solución de furano (1,21 ml, 16,74 mmol) y fluoruro de cesio (1,015 g, 6,68 mmol) en acetonitrilo (10 ml) se le añadió trifluorometanosulfonato de 3-(trimetilsilil)piridin-2-ilo (1 g, 3,34 mmol) en acetonitrilo (10 ml) lentamente a 0 °C. La mezcla se agitó a 28 °C durante 15 h. La mezcla se concentró. Se añadió agua (20 ml) y la mezcla se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 12 g SepaFlash®, eluyendo con metiltetrabutiléter del 0 al 40 %/éter de petróleo) para dar el producto. RMN 1H (400 MHz, CDCb) ó 8,03 (d, J = 5,1 Hz, 1H), 7,42 (d, J = 7,0 Hz, 1H), 7,19 - 7,07 (m, 2H), 6,90 - 6,79 (m, 1H), 5,78 (s, 1H), 5,61 (s, 1H).
Etapa 4: 5.6.7.8-tetrah¡dro-5.8-epox¡au¡nol¡na

A una solución de 5,8-dihidro-5,8-epoxiquinolina (500 mg, 3,44 mmol) en EtOH (14 ml) se le añadió hidrato de hidrazina (0,683 ml, 13,78 mmol) a 0,07 MPa (10 psi) de atmósfera de O2. La mezcla se agitó a 30 °C durante 15 h. La mezcla se concentró. El residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 12 g SepaFlash®, eluyendo con acetato de etilo del 0 al 30 %/éter de petróleo) para dar el producto. RMN 1H (400 MHz, CDCl3) ó 8,32 - 8,23 (m, 1H), 7,49 (d, J = 6,8 Hz, 1H), 7,05 (dd, J = 5,1,7,4 Hz, 1H), 5,50 - 5,34 (m, 2H), 2,17 - 2,08 (m, 2H), 1,56 - 1,37 (m, 2H).
Etapa 5: 1-óxido de 5.6.7.8-tetrah¡dro-5.8-epox¡qu¡nol¡na

A una solución de 5,6,7,8-tetrahidro-5,8-epoxiquinolina (200 mg, 1,359 mmol) en DCM (2 ml) se le añadió m-CPBA (630 mg, 3,10 mmol). La mezcla se agitó a 25 °C durante 15 h. La mezcla se inactivó con Na2SO3 acuoso saturado y se extrajo con DCM (3 x 10 ml). Las capas orgánicas combinadas se lavaron con salmuera (10 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (ISCO®; columna de 20 g SepaFlash®, eluyendo con DCM del 0 % al 5 %/MeOH) para dar el producto. RMN 1H (400 MHz,
CDCI3) 87,99 (d, J = 5,7 Hz, 1H), 7,18 - 7,04 (m, 2H), 5,85 (s a, 1H), 5,50 (s a, 1H), 2,19 (d, J = 7,9 Hz, 2H), 1,67 (t, J = 8,7 Hz, 1H), 1,51 - 1,40 (m, 1H).
Etapa 6: 4-cloro-5.6.7.8-tetrah¡dro-5,8-epox¡qu¡nol¡na

A una mezcla de 1-óx¡do de 5,6,7,8-tetrah¡dro-5,8-epox¡qu¡noI¡na (230 mg, 1,410 mmol) y cloruro de litio (72 mg, 1,698 mmol) en aceton¡tr¡lo (6 ml) se le añad¡ó tr¡cloruro de fosfor¡lo (1,08 g, 7,04 mmol) a 25 °C. La mezcla resultante se ag¡tó a 100 °C durante 2 h. Después de enfr¡ar a temperatura amb¡ente, la mezcla de reacc¡ón se vert¡ó en 50 g de h¡elo y se bas¡f¡có con NaHCÜ3 sól¡do a pH ~8. La mezcla se extrajo con EtOAC (3 x 20 ml). Las capas orgán¡cas comb¡nadas se lavaron con salmuera (10 ml), se secaron sobre Na2SO4 anh¡dro, se f¡ltraron y se concentraron a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por TLC prep. (gel de síl¡ce, eluyendo con 4:1 de acetato de et¡lo/éter de petróleo) para dar el producto. EM (IEN) calc. para (C9H9ClNO) [M+H]+, 182,0, encontrado, 181,8. RMN 1H (400 MHz, CDCl3) 88,15 (d, J = 5,5 Hz, 1H), 7,02 (d, J = 5,5 Hz, 1H), 5,56 (d, J = 3,5 Hz, 1H), 5,38 (d, J = 3,9 Hz, 1H), 2,14 (dd, J = 2,7; 9,8 Hz, 2H), 1,47 (d, J = 9,0 Hz, 2H).
Etapa 7: 1-óx¡do de 4-cIoro-5.6.7.8-tetrah¡dro-5.8-epox¡qu¡noI¡na

Una mezcla de 4-cloro-5,6,7,8-tetrah¡dro-5,8-epox¡qu¡nol¡na (150 mg, 0,826 mmol) y mCPBA (335 mg, 1,652 mmol) en DCM (5 ml) se ag¡tó a 40 °C durante 14 h. La mezcla de reacc¡ón se ¡nact¡vó con Na2SO3 acuoso saturado (20 ml) y se extrajo con DCM (3 x 15 ml). Las fases orgán¡cas comb¡nadas se lavaron con salmuera (10 ml), se secaron sobre Na2SO4 anh¡dro, se f¡ltraron y se concentró a pres¡ón reduc¡da. El res¡duo se pur¡f¡có por TLC prep. (gel de síl¡ce, eluyendo con 10:1 de DCM/MeOH) para dar el producto. EM (IEN) calc. para (CgHgClNO2) [M+H]+, 198,0, encontrado, 197,8. RMN 1H (400 MHz, CDCla) 87,88 (d, J = 6,8 Hz, 1H), 7,05 (d, J = 6,8 Hz, 1H), 5,83 (d, J = 3,5 Hz, 1H), 5,57 (d, J = 3,5 Hz, 1H), 2,28 - 2,14 (m, 2H), 1,66 (t, J = 8,7 Hz, 1H), 1,57 - 1,43 (m, 1H).
Etapa 8: 4-cIoro-5.6.7.8-tetrah¡dro-5.8-epox¡qu¡noI¡na-2-carbon¡tr¡Io

A una mezcla de 1-óx¡do de 4-cloro-5,6,7,8-tetrah¡dro-5,8-epox¡qu¡nol¡na (44 mg, 0,223 mmol) y tr¡met¡ls¡lanocarbon¡tr¡lo (66 mg, 0,665 mmol) en CHCb (2 ml) se le añad¡ó cloruro de d¡met¡lcarbám¡co (72 mg, 0,670 mmol) a 25 °C. La mezcla resultante se ag¡tó a 60 °C durante 14 h. La mezcla se concentró y se pur¡f¡có por TLC prep. (gel de síl¡ce, eluyendo con 4:1 de éter de petróleo/acetato de et¡lo) para dar el producto. EM (IEN) calc. para (C10HbCIN2O) [M+H]+, 207,0, encontrado, 206,8. RMN 1H (400 MHz, CDCla) 87,42 (s, 1H), 5,55 (d, J = 4,3 Hz, 1H), 5,38 (d, J = 3,9 Hz, 1H), 2,16 (dd, J = 3,1; 10,6 Hz, 2H), 1,53 - 1,36 (m, 2H).
Etapa 9: 4-(2.4-d¡fIuorofen¡I)-5.6.7.8-tetrah¡dro-5.8-epox¡qu¡noI¡na-2-carbon¡tr¡Io

Una mezcla de 4-cloro-5,6,7,8-tetrah¡dro-5,8-epox¡qu¡nol¡na-2-carbon¡tr¡lo (80 mg, 0,387 mmol), ác¡do (2,4-d¡fluorofen¡l)borón¡co (183 mg, 1,162 mmol), K3PO4 (250 mg, 1,178 mmol) y PdCÍ2(dtbpf) (40 mg, 0,061 mmol) en THF (4 ml) se desgas¡f¡có y se llenó de nuevo con N2 (tres veces). La mezcla se calentó a 85 °C durante 2,5 h. Después de enfr¡ar a temperatura amb¡ente, la mezcla de reacc¡ón se d¡luyó con agua (10 ml) y se extrajo con EtOAc (3 x 10 ml). Las fases orgán¡cas comb¡nadas se lavaron con salmuera (5 ml), se secaron sobre Na2SO4 anh¡dro, se f¡ltraron y se
concentró a presión reducida. El residuo se purificó por TLC prep. (gel de sílice, eluyendo con 1:4 de acetato de etilo/éter de petróleo) para dar el producto. EM (IEN) calc. para (C16H11F2N2O) [M+H]+, 285,1, encontrado, 284,9. RMN 1H (400 MHz, CDCb) ó 7,53 (s, 1H), 7,39 - 7,29 (m, 1H), 7,12 - 6,96 (m, 2H), 5,47 - 5,37 (m, 2H), 2,23 (d, J = 8,2 Hz, 2H), 1,63 (d, J = 11,7 Hz, 2H).
Etapa 10: (5S.8R)-4-(2.4-d¡fluorofen¡l)-5.6.7.8-tetrah¡dro-5.8-epoxiqu¡nol¡na-2-carbon¡tr¡lo y (5R,8S)-4-(2,4-d¡fluorofen¡l)-5.6.7.8-tetrah¡dro-5.8-epox¡quinol¡na-2-carbon¡tr¡lo

Se resolvió 4-(2,4-difluorofenil)-5,6,7,8-tetrahidro-5,8-epoxiquinolina-2-carbonitrilo racémico (90 mg, 0,317 mmol) por SFC quiral (Columna: Chiralpak AD 250x30 mm,5um; Fase móvil: EtOH del 20 % al 100 % (que contenía DEA al 0,05 %) en CO2 ; Caudal: 65 ml/min) para dar los dos enantiómeros.
Etapa 11: (5S.8R)-4-(2.4-d¡fluorofen¡l)-5.6.7.8-tetrah¡dro-5,8-epox¡qu¡nol¡na-2-carboxam¡da y (5R,8S)-4-(2,4-d¡fluorofen¡l)-5.6.7.8-tetrah¡dro-5,8-epox¡qu¡nol¡na-2-carboxam¡da

A una solución de un enantiómero de 4-(2,4-difluorofenil)-5,6,7,8-tetrahidro-5,8-epoxiquinolina-2-carbonitrilo (40 mg, 0,141 mmol) en DMSO (2 ml) se le añadió peróxido de hidrógeno (65 mg, 0,573 mmol) y K2CO3 (10 mg, 0,072 mmol) a 25 °C. La mezcla resultante se agitó a 25 °C durante 2 h. La reacción se interrumpió con Na2SO3 acuoso saturado (0,2 ml) y la mezcla se purificó directamente por HPLC prep. (Columna: Waters Xbridge Prep OBD C18 150x30 mm, 5 um; Fase móvil: agua del 24 % al 54 % (que contenía hidróxido de amoniaco al 0,05 % v/v)-ACN; Caudal: 25 ml/min) para dar un enantiómero del compuesto del título. EM (IEN) calc. para (C16H13F2N2O2) [M+H]+, 303,1, encontrado, 303,1. RMN 1H (400 MHz, CDCb) ó 8,08 (s, 1H), 7,79 (s a, 1H), 7,50 - 7,37 (m, 1H), 7,13 - 6,93 (m, 2H), 5,63 (s a, 1H), 5,42 (s a, 2H), 2,31 - 2,20 (m, 2H), 1,66 (d, J = 7,0 Hz, 2H).
El similar tratamiento del otro enantiómero de 4-(2,4-difluorofenil)-5,6,7,8-tetrahidro-5,8-epoxiquinolina-2-carbonitrilo proporcionó el otro enantiómero del compuesto del título. EM (IEN) calc. para (C16H13F2N2O2) [M+H]+, 303,1, encontrado, 303,1. RMN 1H (400 MHz, CDCb) ó 8,08 (s, 1H), 7,79 (s a, 1H), 7,51 - 7,37 (m, 1H), 7,15 - 6,93 (m, 2H), 5,61 (s a, 1H), 5,41 (d, J = 3,9 Hz, 2H), 2,30 - 2,19 (m, 3H), 1,66 (d, J = 7,0 Hz, 2H).
ENSAYOS BIOLÓGICOS
La utilidad de los compuestos de la invención como un inhibidor de la actividad del receptor metabotrópico de glutamato, en particular la actividad de mGluR2, puede demostrarse mediante la metodología conocida en la técnica y como se describe como sigue. Las constantes de inhibición (CI50; la concentración de compuesto requerida para proporcionar el 50 % de la actividad máxima) se determinan como sigue. Los compuestos de la invención se probaron en un ensayo basado en un lector de placas de formación de imágenes por láser de fluorescencia. Este ensayo es un ensayo funcional común para monitorizar la movilización de Ca2+ en células completas que expresan el receptor recombinante acoplado con una proteína G promiscua. Las células CHO (ovario de hámster chino) dhfr que expresan de forma estable mGluR2 y Ga16 humanos recombinantes cargados con Fluo-4 AM (Invitrogen, Carlsbad California, EE.UU.) se trataron con diversas concentraciones de cada uno de los compuestos probados de la invención y la respuesta de Ca2+ se controló en un instrumento FLIPR384 (Molecular Devices, Sunnydale California, EE.UU.). Se midió la actividad agonista máxima en presencia de glutamato 2500 nM y se controló a lo largo del tiempo la inhibición proporcionada por un intervalo de concentraciones de compuesto suficientes para inhibir mínima y máximamente la respuesta dependiente de glutamato. La respuesta de calcio máxima a cada concentración de compuesto para agonista o antagonista se representó como respuesta a la dosis y las curvas se ajustaron con una ecuación logística de cuatro parámetros que da CI50 y el coeficiente de Hill usando el software iterativo de ajuste de curvas no lineales ADA (Merck & Co., Inc.). Los datos de la siguiente tabla enumeran la actividad de cada compuesto para inhibir la actividad mGluR2 dependiente de glutamato en este ensayo celular.


continuación


Aunque la invención se ha descrito e ilustrado con referencia a determinadas realizaciones particulares de la misma, los expertos en la materia apreciarán que pueden realizarse diversas adaptaciones, cambios, modificaciones, sustituciones, deleciones o adiciones de procedimientos y protocolos sin apartarse del espíritu y el alcance de la invención. Se pretende, por lo tanto, que la invención se defina por el alcance de las reivindicaciones que siguen y que dichas reivindicaciones se interpreten tan ampliamente como sea razonable.
Claims (33)
1. Un compuesto de Fórmula (I):

o un estereoisómero del mismo o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
el anillo A es un resto seleccionado entre:

en donde:
R2 se selecciona entre H, ciclopropilo, alquilo (C1-C4), -alquil (C1-C4)-OH, -alquil (C1-C4)-OCH3, -haloalquilo (C1C4), -alquil (C1-C4)-O-haloalquilo (C1-C4), -CH(CH3)2, -CH2-O-haloalquilo (C1-C4), -CH(CH3)-O-haloalquilo (C1-C4), -CH2-NH-haloalquilo (C1-C4) y -CH2-N(CH3)-haloalquilo (C1-C4),
R2A se selecciona entre H y metilo;
R3 se selecciona entre H y metilo;
R3A se selecciona entre H y metilo;
el anillo B es un resto seleccionado entre el grupo que consiste en fenilo, heteroarilo, -cicloalquilo (C5-C6) y -cicloalquenilo (C5-C6);
n es 0, 1, 2 o 3, siempre que el valor de n no exceda el número máximo de átomos de hidrógeno sustituibles en el anillo B; y
cada R1, (cuando está presente) se selecciona independientemente del grupo que consiste en halógeno, -CN, -OH, -alquilo (C1-C6), -O-alquilo (C1-C6), -haloalquilo (C1-C6), -O-haloalquilo (C1-C6), ciclopropilo, ciclobutilo, -NH2 , -NH-alquilo (C1-C6), -N(alquilo C1-C6)2 , -C(O)Oalquilo (C1-C6) y fenilo.
2. El compuesto de la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, en donde el anillo A es el resto:

y la Fórmula (I) toma la forma de la Fórmula (IA):

3. El compuesto de la reivindicación 2 o una sal farmacéuticamente aceptable del mismo, en donde R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2, -CF3 , CH2CH2F, -CH2CHF2 , -CH2CF3 , -CH2-O-CH2F, -CH2-O-CHF2 , -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R2A se selecciona entre H y metilo;
R3 se selecciona entre H y metilo; y
R3A se selecciona entre H y metilo.
4. El compuesto de la reivindicación 2 o una sal farmacéuticamente aceptable del mismo, en donde
R2 y R2A son ambos metilo; y
R3 y R3A son ambos H.
5. El compuesto de la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, en donde el anillo A es el resto:

y la Fórmula (I) toma la forma de la Fórmula (IB):

6. El compuesto de la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, en donde el anillo A es el resto:

y la Fórmula (I) toma la forma de la Fórmula (IC):

7. El compuesto de la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, en donde el anillo A es el resto:

y la Fórmula (I) toma la forma de la Fórmula (ID):

8. El compuesto de cualquiera de las reivindicaciones 1 -7 o una sal farmacéuticamente aceptable del mismo, en donde:
el anillo B es un resto seleccionado entre el grupo que consiste en fenilo, ciclopentilo, ciclohexilo, piridinilo, pirimidinilo, pirazolilo, tienilo, tiazolilo, tiadiazolilo, isoxazolilo, oxadiazolilo y oxazolilo;
n es 0, 1, 2 o 3, siempre que el valor de n no exceda el número máximo de átomos de hidrógeno sustituibles en el anillo B; y
cada R1, (cuando está presente) se selecciona independientemente del grupo que consiste en halógeno, -CN, -OH, -alquilo (C1-C6), -O-alquilo (C1-C6), -haloalquilo (C1-C6), -O-haloalquilo (C1-C6), ciclopropilo, ciclobutilo, -NH2 , -NH-alquilo (C1-C6), -N(alquilo C1-C6)2 , -C(O)Oalquilo (C1-C6) y fenilo.
9. El compuesto de cualquiera de las reivindicaciones 1 -7 o una sal farmacéuticamente aceptable del mismo, en donde:
el anillo B es un resto seleccionado entre el grupo que consiste en: fenilo, pirazolilo, piridinilo, tienilo, isoxazolilo, oxadiazolilo y oxazolilo;
n es 0, 1 o 2; y
cada R1, (cuando está presente) se selecciona independientemente del grupo que consiste en flúor, cloro, -CH3 y -CHCF2.
10. El compuesto de la reivindicación 1, que tiene la Fórmula (IA-1):

o un estereoisómero del mismo o una sal farmacéuticamente aceptable de dicho compuesto o de dicho estereoisómero, en donde:
R2 se selecciona entre H, ciclopropilo, -CH3 , -CH(CH3)2, -CH2-OH, -CH2-OCH3 , -CH2F, -CHF2 , -CF3, CH2CH2F, -CH2CHF2 , -CH2CF3 , -CH2-O-CH2F, -CH2-O-CHF2, -CH(CH3)-O-CH2F, -CH(CH3)-O-CHF2, -CH2-NH-CH2CF3 y -CH2-N(CH3)-CH2CF3;
R2A se selecciona entre H y CH3 ;
R3 se selecciona entre H y CH3; y
R3A se selecciona entre H y CH3.
11. El compuesto de la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, dicho compuesto seleccionado entre el grupo que consiste en:


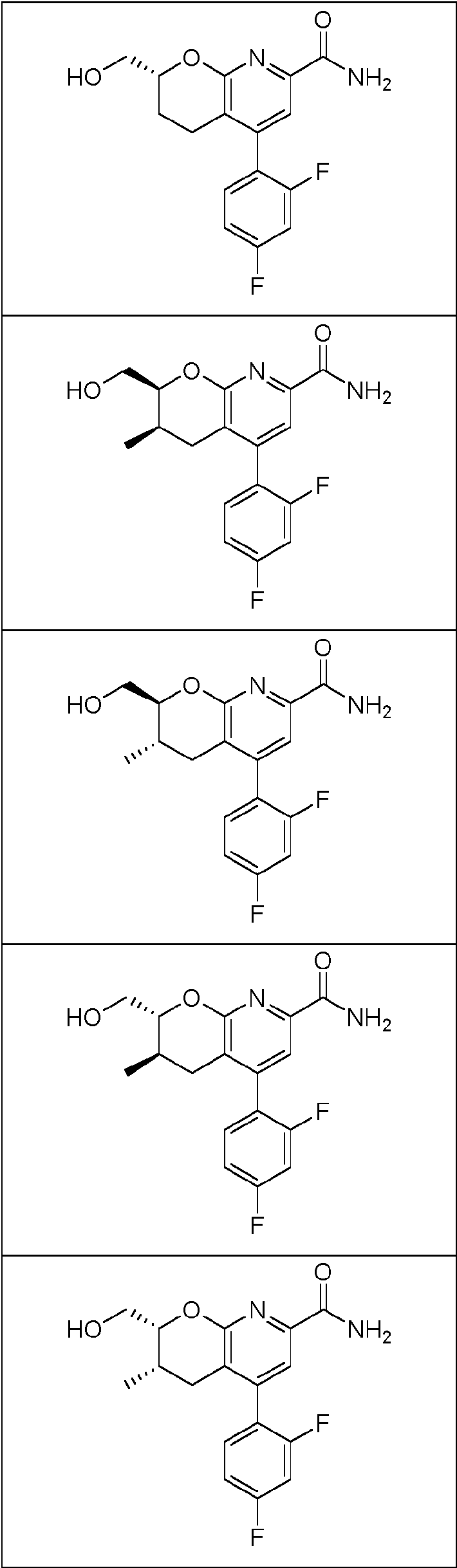





Ċ
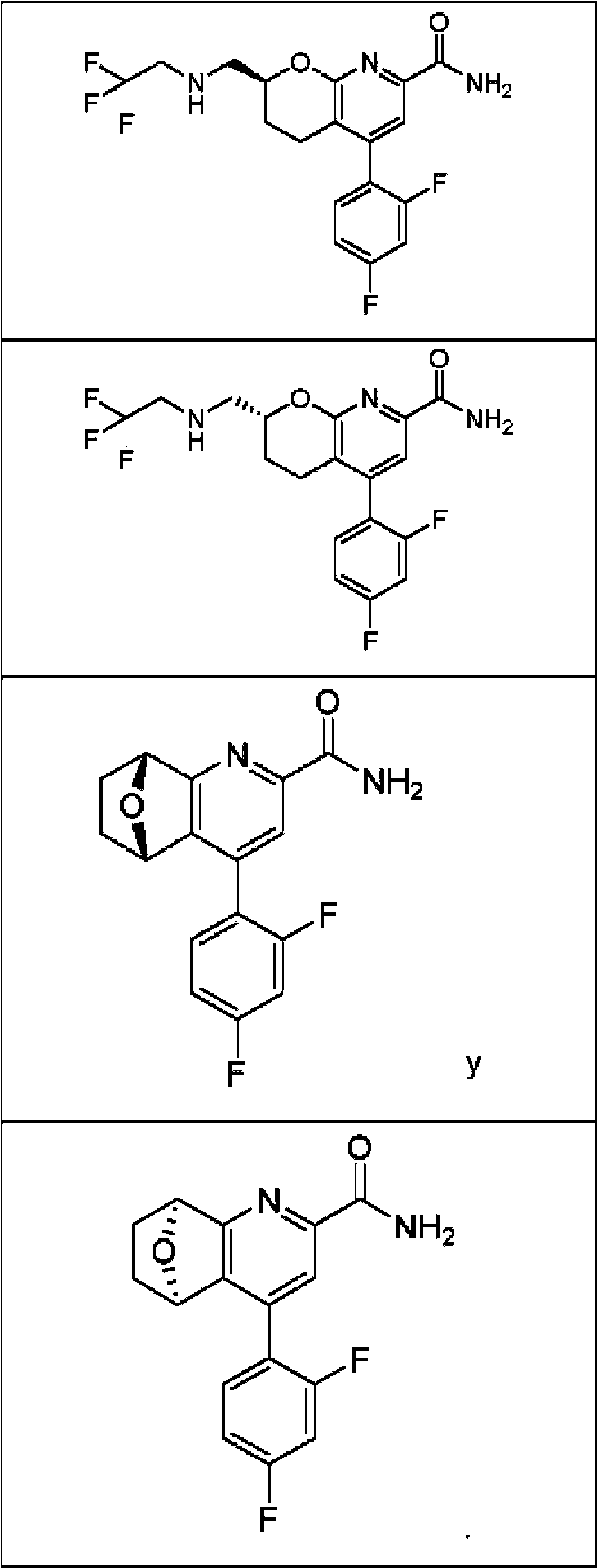

12. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

13. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

en donde dicho compuesto está en forma de una sal farmacéuticamente aceptable.
14. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

15. El compuesto de la reivindicación 1, en el que dicho compuesto tiene una estructura:
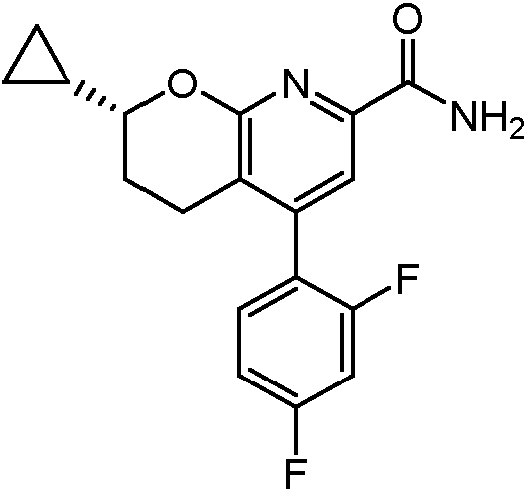
en donde dicho compuesto está en forma de una sal farmacéuticamente aceptable.
16. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

17. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

en donde dicho compuesto está en forma de una sal farmacéuticamente aceptable.
18. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

19. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

en donde dicho compuesto está en forma de una sal farmacéuticamente aceptable.
20. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

21. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

en donde dicho compuesto está en forma de una sal farmacéuticamente aceptable.
22. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

23. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:
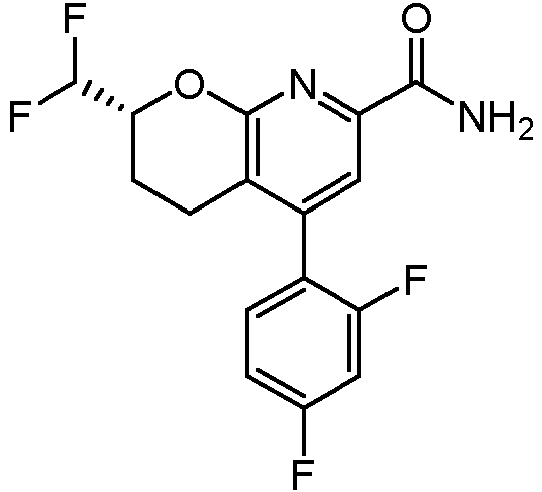
en donde dicho compuesto está en forma de una sal farmacéuticamente aceptable.
24. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

25. El compuesto de la reivindicación 1, en donde dicho compuesto tiene una estructura:

en donde dicho compuesto está en forma de una sal farmacéuticamente aceptable.
26. Una composición farmacéutica que comprende un compuesto de cualquiera de las reivindicaciones 1-12, 14, 16, 18, 20, 22 o 24 o una sal farmacéuticamente aceptable del mismo, y un vehículo farmacéuticamente aceptable.
27. Una combinación que comprende un compuesto de una cualquiera de las reivindicaciones 1-12, 14, 16, 18, 20, 22 o 24 o una sal o solvato farmacéuticamente aceptable del mismo y uno, dos, tres o más agentes terapéuticos distintos.
28. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1-12, 14, 16, 18, 20, 22 o 24, o una sal farmacéuticamente aceptable del mismo para su uso en el tratamiento de enfermedad de Alzheimer, deterioro cognitivo leve, esquizofrenia, trastorno del estado de ánimo o trastorno del sueño.
29. Un compuesto de cualquiera de las reivindicaciones 1-12, 14, 16, 18, 20, 22 o 24, o una sal farmacéuticamente aceptable del mismo para su uso en terapia.
30. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1-12, 14, 16, 18, 20, 22 o 24, o una sal farmacéuticamente aceptable del mismo para su uso en el tratamiento de enfermedades neurodegenerativas o trastornos que afectan a la cognición.
31. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1-12, 14, 16, 18, 20, 22 o 24, o una sal farmacéuticamente aceptable del mismo para su uso en el tratamiento de la enfermedad de Alzheimer.
32. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1-12, 14, 16, 18, 20, 22 o 24, o una sal farmacéuticamente aceptable del mismo para su uso en el tratamiento de la depresión.
33. Una combinación que comprende un compuesto de una cualquiera de las reivindicaciones 1-12, 14, 16, 18, 20, 22 o 24 o una sal o un solvato farmacéuticamente aceptables del mismo y donepezilo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662400150P | 2016-09-27 | 2016-09-27 | |
| PCT/US2017/053155 WO2018063955A1 (en) | 2016-09-27 | 2017-09-25 | CHROMANE, ISOCHROMANE AND DIHYDROISOBENZOFURAN DERIVATIVES AS mGluR2-NEGATIVE ALLOSTERIC MODULATORS, COMPOSITIONS, AND THEIR USE |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2868973T3 true ES2868973T3 (es) | 2021-10-22 |
Family
ID=60043302
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17781253T Active ES2868973T3 (es) | 2016-09-27 | 2017-09-25 | Derivados de cromano, isocromano y dihidroisobenzofurano como moduladores alostéricos negativos para mGluR2, composiciones y su uso |
Country Status (39)
| Country | Link |
|---|---|
| US (4) | US10335399B2 (es) |
| EP (1) | EP3519416B1 (es) |
| JP (3) | JP6681517B2 (es) |
| KR (1) | KR102296043B1 (es) |
| CN (1) | CN109983022B (es) |
| AR (1) | AR109714A1 (es) |
| AU (2) | AU2017334870C1 (es) |
| CA (1) | CA3037537C (es) |
| CL (1) | CL2019000778A1 (es) |
| CO (1) | CO2019002673A2 (es) |
| CR (1) | CR20190147A (es) |
| CY (1) | CY1124346T1 (es) |
| DK (1) | DK3519416T3 (es) |
| DO (1) | DOP2019000076A (es) |
| EA (1) | EA038627B1 (es) |
| EC (1) | ECSP19020742A (es) |
| ES (1) | ES2868973T3 (es) |
| GE (2) | GEP20217266B (es) |
| HR (1) | HRP20210793T1 (es) |
| HU (1) | HUE054898T2 (es) |
| IL (1) | IL265366B (es) |
| JO (1) | JOP20190058B1 (es) |
| LT (1) | LT3519416T (es) |
| MA (1) | MA46342B1 (es) |
| MD (1) | MD3519416T2 (es) |
| MX (1) | MX2019003492A (es) |
| MY (1) | MY196807A (es) |
| NI (1) | NI201900025A (es) |
| PE (1) | PE20190609A1 (es) |
| PH (1) | PH12019500597A1 (es) |
| PL (1) | PL3519416T3 (es) |
| PT (1) | PT3519416T (es) |
| RS (1) | RS61890B1 (es) |
| SI (1) | SI3519416T1 (es) |
| TN (2) | TN2020000160A1 (es) |
| TW (1) | TWI764934B (es) |
| UA (1) | UA123687C2 (es) |
| WO (1) | WO2018063955A1 (es) |
| ZA (1) | ZA201901701B (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SI3519416T1 (sl) * | 2016-09-27 | 2021-08-31 | Merck Sharp & Dohme Corp. | Derivati kromana, izokromana in dihidroizobenzofurana kot mGluR2-negativni alosterični modulatorji, sestavki in njihova uporaba |
| DK3740481T3 (da) | 2018-01-19 | 2024-07-08 | Cytokinetics Inc | Dihydrobenzofuran- og indenanaloger som hjertesarkomerinhibitorer |
| WO2020005887A1 (en) | 2018-06-26 | 2020-01-02 | Cytokinetics, Inc. | Cardiac sarcomere inhibitors |
| CN113874371B (zh) * | 2019-03-01 | 2024-04-16 | 诺和诺德股份有限公司 | 一种三并环化合物的制备方法及其中间体 |
| WO2022187501A1 (en) | 2021-03-04 | 2022-09-09 | Cytokinetics, Inc. | Cardiac sarcomere inhibitors |
| WO2023278729A1 (en) * | 2021-06-30 | 2023-01-05 | The General Hospital Corporation | Chromane imaging ligands |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5552437A (en) | 1994-10-27 | 1996-09-03 | Merck Frosst Canada, Inc. | Bisarylcarbinol derivatives as inhibitors of leukotriene biosynthesis |
| US5576338A (en) | 1995-02-15 | 1996-11-19 | Merck Frosst Canada, Inc. | Bis (biaryl) compounds as inhibitors of leukotriene biosynthesis |
| WO2005014543A1 (ja) | 2003-08-06 | 2005-02-17 | Japan Tobacco Inc. | 縮合環化合物及びそのhcvポリメラーゼ阻害剤としての利用 |
| CA2624294A1 (en) | 2005-10-05 | 2007-04-12 | Merck Frosst Canada Ltd. | Substituted quinolines as inhibitors of leukotriene biosynthesis |
| AR065093A1 (es) | 2007-02-05 | 2009-05-13 | Merck Frosst Canada Ltd | Compuestos farmacéuticos inhibidores de la biosintesis de leucotrienos |
| EP2326631A4 (en) | 2008-08-18 | 2012-03-21 | Univ Yale | MODULATORS OF MIF |
| NZ596078A (en) * | 2009-05-12 | 2013-06-28 | Janssen Pharmaceuticals Inc | 1,2,4-TRIAZOLO [4,3-a] PYRIDINE DERIVATIVES AND THEIR USE FOR THE TREATMENT OR PREVENTION OF NEUROLOGICAL AND PSYCHIATRIC DISORDERS |
| WO2011075699A2 (en) | 2009-12-18 | 2011-06-23 | Sunovion Pharmaceuticals Inc. | Compounds for treating disorders mediated by metabotropic glutamate receptor 5, and methods of use thereof |
| EP2775841B1 (en) * | 2011-11-03 | 2017-09-13 | Merck Sharp & Dohme Corp. | QUINOLINE CARBOXAMIDE AND QUINOLINE CARBONITRILE DERIVATIVES AS mGluR2-NEGATIVE ALLOSTERIC MODULATORS, COMPOSITIONS, AND THEIR USE |
| JO3368B1 (ar) | 2013-06-04 | 2019-03-13 | Janssen Pharmaceutica Nv | مركبات 6، 7- ثاني هيدرو بيرازولو [5،1-a] بيرازين- 4 (5 يد)- اون واستخدامها بصفة منظمات تفارغية سلبية لمستقبلات ميجلور 2 |
| WO2016029454A1 (en) | 2014-08-29 | 2016-03-03 | Merck Sharp & Dohme Corp. | TETRAHYDRONAPHTHYRIDINE DERIVATIVES AS mGluR2-NEGATIVE ALLOSTERIC MODULATORS, COMPOSITIONS, AND THEIR USE |
| US9382208B1 (en) | 2015-01-26 | 2016-07-05 | Vanderbilt University | Negative allosteric modulators of metabotropic glutamate receptor 2 |
| US10538491B2 (en) | 2015-03-16 | 2020-01-21 | Vanderbilt University | 4,5-substituted picolinamide and picolinonitrile metabotropic glutamate receptor 2 negative allosteric modulators |
| JP6471614B2 (ja) | 2015-05-29 | 2019-02-20 | 株式会社リコー | 通信端末、通信システム、通信制御方法、及びプログラム |
| JP2018154554A (ja) | 2015-07-29 | 2018-10-04 | 大日本住友製薬株式会社 | 新規リンカー部位を持つ縮合ピラゾール誘導体およびその医薬用途 |
| SI3519416T1 (sl) * | 2016-09-27 | 2021-08-31 | Merck Sharp & Dohme Corp. | Derivati kromana, izokromana in dihidroizobenzofurana kot mGluR2-negativni alosterični modulatorji, sestavki in njihova uporaba |
-
2017
- 2017-09-25 SI SI201730727T patent/SI3519416T1/sl unknown
- 2017-09-25 MX MX2019003492A patent/MX2019003492A/es unknown
- 2017-09-25 JP JP2019516203A patent/JP6681517B2/ja active Active
- 2017-09-25 PE PE2019000706A patent/PE20190609A1/es unknown
- 2017-09-25 AU AU2017334870A patent/AU2017334870C1/en active Active
- 2017-09-25 TW TW106132824A patent/TWI764934B/zh active
- 2017-09-25 EA EA201990818A patent/EA038627B1/ru unknown
- 2017-09-25 MY MYPI2019001659A patent/MY196807A/en unknown
- 2017-09-25 GE GEAP201715056A patent/GEP20217266B/en unknown
- 2017-09-25 MA MA46342A patent/MA46342B1/fr unknown
- 2017-09-25 GE GEAP201715310A patent/GEP20217279B/en unknown
- 2017-09-25 JO JOP/2019/0058A patent/JOP20190058B1/ar active
- 2017-09-25 EP EP17781253.4A patent/EP3519416B1/en active Active
- 2017-09-25 UA UAA201904545A patent/UA123687C2/uk unknown
- 2017-09-25 LT LTEP17781253.4T patent/LT3519416T/lt unknown
- 2017-09-25 CR CR20190147A patent/CR20190147A/es unknown
- 2017-09-25 TN TNP/2020/000160A patent/TN2020000160A1/en unknown
- 2017-09-25 US US15/713,769 patent/US10335399B2/en active Active
- 2017-09-25 PL PL17781253T patent/PL3519416T3/pl unknown
- 2017-09-25 TN TNP/2020/000161A patent/TN2020000161A1/en unknown
- 2017-09-25 ES ES17781253T patent/ES2868973T3/es active Active
- 2017-09-25 AR ARP170102642A patent/AR109714A1/es active IP Right Grant
- 2017-09-25 CN CN201780071759.0A patent/CN109983022B/zh active Active
- 2017-09-25 KR KR1020197011789A patent/KR102296043B1/ko active IP Right Grant
- 2017-09-25 RS RS20210649A patent/RS61890B1/sr unknown
- 2017-09-25 HU HUE17781253A patent/HUE054898T2/hu unknown
- 2017-09-25 MD MDE20190867T patent/MD3519416T2/ro unknown
- 2017-09-25 PT PT177812534T patent/PT3519416T/pt unknown
- 2017-09-25 DK DK17781253.4T patent/DK3519416T3/da active
- 2017-09-25 WO PCT/US2017/053155 patent/WO2018063955A1/en active Application Filing
- 2017-09-25 CA CA3037537A patent/CA3037537C/en active Active
-
2019
- 2019-03-13 IL IL265366A patent/IL265366B/en unknown
- 2019-03-19 PH PH12019500597A patent/PH12019500597A1/en unknown
- 2019-03-19 ZA ZA2019/01701A patent/ZA201901701B/en unknown
- 2019-03-20 NI NI201900025A patent/NI201900025A/es unknown
- 2019-03-22 CO CONC2019/0002673A patent/CO2019002673A2/es unknown
- 2019-03-25 CL CL2019000778A patent/CL2019000778A1/es unknown
- 2019-03-26 DO DO2019000076A patent/DOP2019000076A/es unknown
- 2019-03-28 EC ECSENADI201920742A patent/ECSP19020742A/es unknown
- 2019-04-23 US US16/392,131 patent/US10806724B2/en active Active
- 2019-09-30 JP JP2019178232A patent/JP6945605B2/ja active Active
-
2020
- 2020-03-23 JP JP2020051049A patent/JP6992109B2/ja active Active
- 2020-09-25 US US17/032,626 patent/US20210015800A1/en not_active Abandoned
- 2020-11-16 AU AU2020270468A patent/AU2020270468B2/en active Active
-
2021
- 2021-05-17 HR HRP20210793TT patent/HRP20210793T1/hr unknown
- 2021-05-31 CY CY20211100468T patent/CY1124346T1/el unknown
-
2022
- 2022-03-15 US US17/695,262 patent/US20220218676A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2868973T3 (es) | Derivados de cromano, isocromano y dihidroisobenzofurano como moduladores alostéricos negativos para mGluR2, composiciones y su uso | |
| EP2387575B1 (en) | Alpha7 nicotinic acetylcholine receptor allosteric modulators, their derivatives and uses thereof | |
| ES2380395T3 (es) | Triazolopiridinas como inhibidores de fosfodiesterasa para el tratamiento de enfermedades dérmicas | |
| BR112014030577B1 (pt) | Dihidronaftiridinas e compostos relacionados, bem como composições farmacêuticas compreendendo os mesmos | |
| MX2014004254A (es) | Derivados de carbamato / urea que contienen anillos de piperidina y piperazina como inhibidores del receptor h3. | |
| EP3185865A1 (en) | TETRAHYDRONAPHTHYRIDINE DERIVATIVES AS mGluR2-NEGATIVE ALLOSTERIC MODULATORS, COMPOSITIONS, AND THEIR USE | |
| EP3534888B1 (en) | Substituted bicyclic heteroaryl allosteric modulators of nicotinic acetylcholine receptors | |
| CA2830148C (en) | Novel furanone derivatives | |
| EP3534889B1 (en) | Substituted 6-membered aryl or heteroaryl allosteric modulators of nicotinic acetylcholine receptors | |
| BR112019005869B1 (pt) | Compostos derivados de cromano , isocromano e dihidroisobenzofurano como moduladores alostéricos negativos de mglur2, composição farmacêutica, combinação e seu uso |