ES2858404T3 - Nuevos intermedios para la preparación de clorhidrato de remifentanilo - Google Patents
Nuevos intermedios para la preparación de clorhidrato de remifentanilo Download PDFInfo
- Publication number
- ES2858404T3 ES2858404T3 ES18202878T ES18202878T ES2858404T3 ES 2858404 T3 ES2858404 T3 ES 2858404T3 ES 18202878 T ES18202878 T ES 18202878T ES 18202878 T ES18202878 T ES 18202878T ES 2858404 T3 ES2858404 T3 ES 2858404T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- remifentanil
- acid
- synthesis
- sodium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *CCC*1(CC*(CC*(*)O)CC1)C=** Chemical compound *CCC*1(CC*(CC*(*)O)CC1)C=** 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/92—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with a hetero atom directly attached to the ring nitrogen atom
- C07D211/98—Nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D211/62—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4
- C07D211/66—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4 having a hetero atom as the second substituent in position 4
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Hydrogenated Pyridines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Un compuesto de la fórmula 20 y sus sales: **(Ver fórmula)**
Description
DESCRIPCIÓN
Nuevos intermedios para la preparación de clorhidrato de remifentanilo
Se revela un nuevo intermedio para sintetizar derivados de 4-[fenil(propanoil)amino]piperidin-4-carbonitrilo sustituido en 1. Específicamente se establece un método para su uso de este intermedio en la preparación de remifentanil. El procedimiento más corto adjunto ofrece un mayor rendimiento de productos con mayor pureza en comparación con los métodos descritos en la técnica anterior.
Descripción
Campo de invención
La presente invención se refiere a un procedimiento para la síntesis de analgésicos opioides de tipo fentanilo. En particular, la presente invención describe una nueva ruta de síntesis eficaz para la preparación de clorhidrato de remifentanilo y precursores del mismo.
Estado de la técnica
El clorhidrato de remifentanilo (1, Figura 1) pertenece a la clase 4-anilidopiperidina de analgésicos opioides sintéticos. Tiene un alto grado de potencia analgésica (ED50 = 0.0044 mg/kg) y debido a su inicio rápido y duración de acción ultracorta (15 min) se convirtió en una adición clínicamente útil a la familia de analgésicos fentanilo. El remifentanil en combinación con un fármaco hipnótico se puede administrar en dosis relativamente altas debido a su rápida eliminación del plasma sanguíneo. Esto significa que no se produce acumulación con Remifentanil y su vida media sensible al contexto permanece en 4 minutos después de una perfusión de 4 horas. El remifentanil es metabolizado por esterasas plasmáticas y tisulares inespecíficas, que hidrolizan uno de los grupos éster. El ácido de Remifentanil formado tiene 1/4600 de la actividad del compuesto original. La farmacocinética de Remifentanil también ofrece una recuperación más rápida después de la cirugía.

Clorhidrato de remifentanilo
Figura 1 Estructura del clorhidrato de remifentanilo
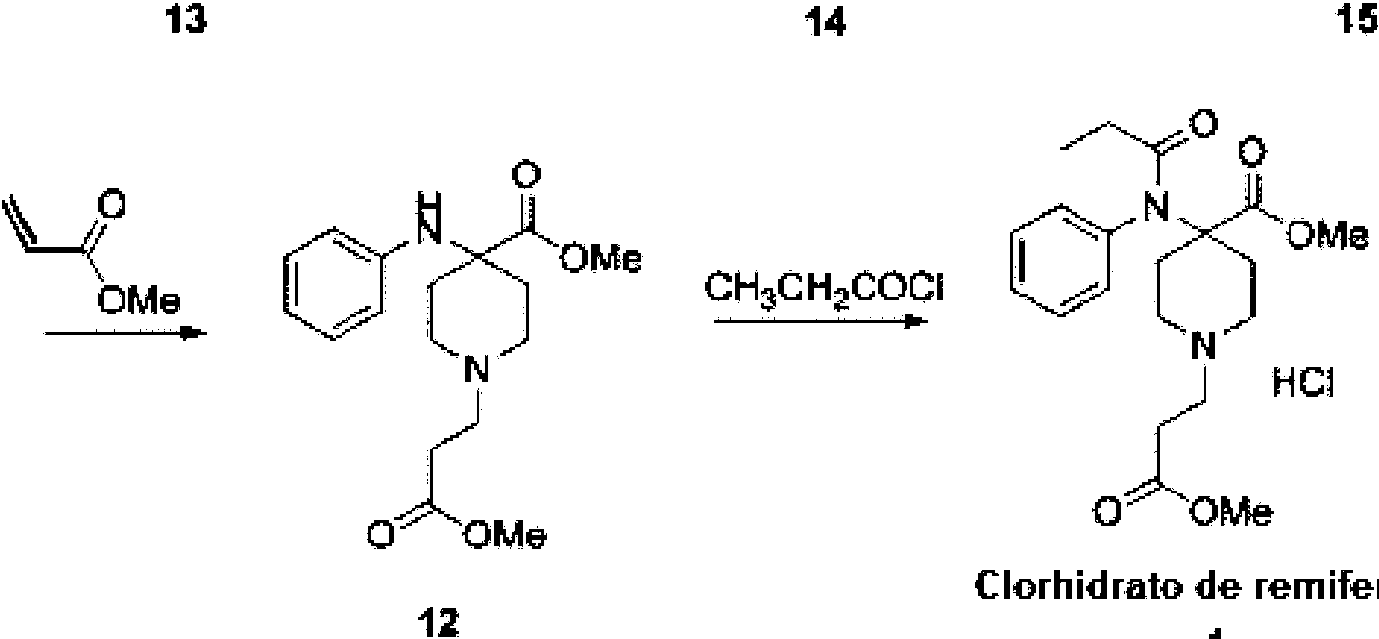
La preparación de clorhidrato de remifentanilo se describió en la Patente EP 0383579 A1 que describe las dos últimas etapas de la síntesis mostradas en el esquema 1. El intermedio 7 clave se prepara según la literatura (P.G.H. Van Daele et al. Arzneim.-Forsh. Drug. Res. 1976, 26, 1521) en 5 etapas a partir de 1-bencil-4-piperidona (2).



C crhic rato de
remite litando
Esquema 1, Síntesis de clorhidrato de remifentamlo según la Patente EP 0383579 A1
En la Patente WO 2007144391 (Kern Pharma) se describe una síntesis de 4 etapas. El material de partida 4-piperidona 8 se alquila con acrilato de metilo seguido de la introducción de la función anilina y un grupo nitrilo usando síntesis de Strecker. La acilación del nitrógeno de anilina usando cloruro de propionilo genera el precursor del clorhidrato de remifentanilo (11). En la última etapa, el grupo nitrilo se convierte en un éster metílico usando metanol y HCl, proporcionando de este modo la molécula diana.
La hidrolización de los grupos nitrilo en esta síntesis tiene bajos rendimientos debido al grupo éster que se saponifica parcialmente en las condiciones y se forman los productos secundarios. Los intermedios que contienen una función de éster metílico son propensos a la hidrólisis rápida.

Clorhiclratü de remifentamlo
Esquema 2 Preparación del clorhidrato de remifentamlo según la patente WO 2007144391
La Patente WO 2007061555 (Mallinckrodt Inc.) realizó una pequeña alteración en la síntesis Remifentani1HCI en comparación con la Patente WO 2007144391. La función nitrilo en el compuesto 10 se hidroliza primero a una amida que luego se convierte en un éster metílico. En la final se introduce la función propionilo. (Esquema 3)

Clorhidrato de remifentamlo
Esquem a 3 Síntesis de clorhidrato de remifentamlc según la patente W O 20070G1555
La síntesis de clorhidrato de remifentanilo según la Patente WO 2007087164 (Mallincrodt Inc.) comienza con 1-(carbetoxi)-4-(fenilamino)-4-piperidina carboxamida, que se hidroliza a ácido carboxílico o grupo carboxilato simultáneamente con desprotección del nitrógeno de piperidina. En la siguiente etapa se lleva a cabo la esterificación
del grupo carboxílico con metanol. La alquilación de la amina piperidina con acrilato de metilo seguida de la acilación del nitrógeno secundario con cloruro de propionilo genera el clorhidrato de remifentanilo (esquema 4).

Esquema 4 Síntesis de clorhidrato de remifentanilo según la Patente WO 2007087164
Todas las rutas de síntesis mencionadas anteriormente usaron la adición de Michael para introducir la función metil propionilo. En la Patente WO 2010053944 (Cambrex Charles City, Inc.) se trata 1-bencil-4-(N-fenilpropionamido) piperidina-4-carboxilato de metilo con ácido 4-nitrofenilsulfónico para crear la sal 17 de nosilato, que luego se hace reaccionar en condiciones básicas con hidroxipropionato de metilo activado con una función (18) nosilato. La acilación del último intermedio con cloruro de propionilo da el API final clorhidrato de remifentanilo (esquema 5).

1. Nuevos compuestos
Ahora se han encontrado dos nuevos intermedios que son útiles en la síntesis del Remifentanil. El compuesto 20 y el compuesto 21.

El uso de intermedios que contienen dos grupos nitrilo tiene muchas ventajas en comparación con la síntesis usada hasta ahora. Por ejemplo, brinda la oportunidad de excluir la adición de Michael a la cadena lateral de nitrógeno en la síntesis, eliminando de este modo el uso de reactivos alfa-beta insaturados tal como el éster de ácido acrílico en la reacción, que se sospecha que muestra propiedades citotóxicas.
El uso de intermedios que contienen dos grupos nitrilo brinda además la oportunidad de deliberar sobre los grupos éster en dos partes de la molécula dentro de una única etapa de síntesis, aumentando de este modo el rendimiento total de la síntesis total de Remifentanil.
2. Uso de un nuevo compuesto en la síntesis de Remifentanil
El compuesto 20 se puede usar de diferentes maneras para la síntesis de Remifentanil.
En una realización, el compuesto se convierte en el nuevo compuesto 21, en otra realización, el compuesto 20 se transfiere a un compuesto 12 , que ya se informó en las solicitudes de patente como se muestra anteriormente.

3. Uso del nuevo compuesto 20 en la preparación del nuevo compuesto 21
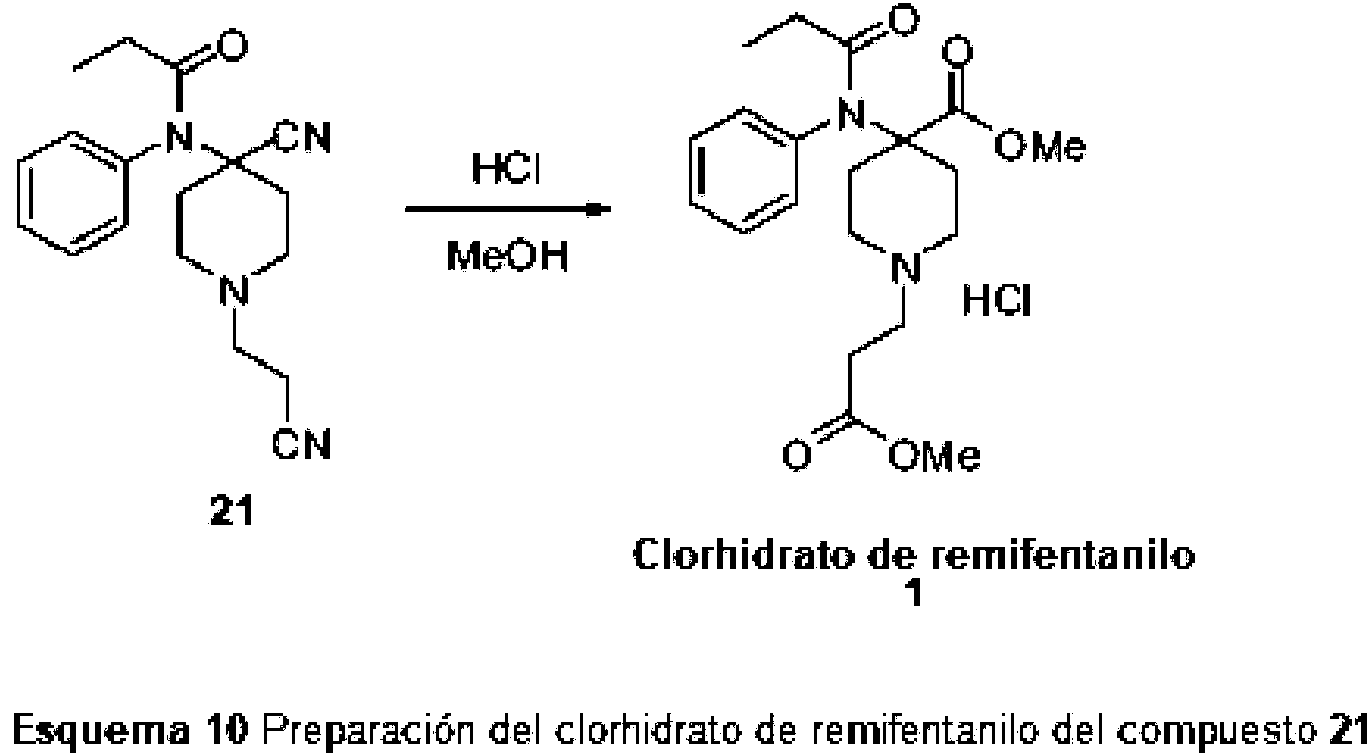
En la primera realización de la invención, el nuevo compuesto se acila mediante un agente acilante apropiado al compuesto 21.
El 1-(2-cianoetil)-4-(fenilamino)piperidina-4-carbonitrilo (20) reacciona con un agente acilante en presencia o sin presencia de una base para dar 1-(2-cianoetil)-4-[fenil(propanoil)amino]piperidina-4-carbonitrilo (21, esquema 9)

La mezcla de reacción comprende aproximadamente 1 equivalente molar a aproximadamente 10 equivalentes molares de agente acilante. En un ejemplo, la mezcla de reacción se carga con aproximadamente 1 a 3 equivalentes molares de agente acilante por 1 equivalente molar de 1-(2-cianoetil)-4-(fenilamino) piperidina-4-carbonitrilo 20.
En una realización, la reacción entre el compuesto 20 y el agente acilante se produce en presencia de un eliminador de ácido (base), en el que la mezcla de reacción comprende aproximadamente 1 equivalente molar a 3 equivalentes molares del eliminador de ácido.
La temperatura de la mezcla de reacción durante la reacción varía desde aproximadamente 0 °C a aproximadamente 80 °C. Preferiblemente, la temperatura de reacción de aproximadamente 30 °C a aproximadamente 50 °C. Se deja que la mezcla de reacción reaccione hasta varios días. En un ejemplo, el tiempo de reacción es de aproximadamente 10 horas a 30 horas.
En una realización, el agente acilante es anhídrido propiónico. En otra realización, el agente acilante es cloruro de propanoílo. Ambos reactivos muestran rendimientos comparables en la reacción.
Los ejemplos de disolventes usados en la mezcla de reacción y cristalización incluyen disolventes que son inertes a la reacción que ocurre en la etapa 3. Ejemplos de tales disolventes incluyen, pero no se limitan a, acetonitrilo, acetona, diclorometano, cloroformo, W,A/-dimetilacetam¡da. , W,W-dimetilformamida, dimetilsulfóxido, tert-butil metil éter, diisopropil éter, acetato de etilo, dicloroetano, benceno, tolueno, xileno, 1,4-dioxano, tetrahidrofurano, 2-metil tetrahidrofurano, metiletilcetona y mezclas de los mismos. En un ejemplo, la mezcla contiene diclorometano. La proporción del compuesto 20 a disolvente en peso base es de aproximadamente 1:10 a 1:50.
El eliminador de ácido puede incluir hidruros de metales, hidróxidos, carbonatos, bicarbonatos, aminas y similares.
Una vez completada la reacción, se agregan agua y base a la reacción para ajustar el pH por encima de 7. La extracción con disolvente se realiza con un disolvente orgánico. El disolvente se elimina a presión reducida para obtener el producto en bruto. El producto en bruto se puede purificar mediante cromatografía o recristalización.
En un ejemplo, el compuesto 21 se puede disolver en un disolvente orgánico al que se agrega una solución de un ácido en un disolvente para formar una sal del compuesto 21, que se puede aislar mediante procedimientos conocidos en la técnica. Los ejemplos de disolventes incluyen, pero no se limitan a acetonitrilo, acetona, diclorometano, cloroformo, W,W-dimetilformamida, dimetilsulfóxido, acetato de etilo, dicloroetano, agua, benceno, tolueno, xileno, metanol, etanol, isopropanol y mezclas de los mismos. Los ejemplos de ácidos incluyen ácido clorhídrico, ácido bromhídrico, ácido metanosulfónico, ácido 4-metilbencensulfónico, ácido sulfúrico, ácido fosfórico, ácido cítrico, ácido oxálico y similares. Luego, el compuesto 21 se convierte en remifentanil como se describe a continuación.
La siguiente tabla muestra los diferentes reactivos que se han usado para esta etapa de reacción.

4. Uso del compuesto 20 en la preparación del compuesto 12
En otra realización de la invención, el nuevo compuesto 20 se convierte en una etapa en el conocido compuesto 12:

La síntesis del compuesto 12 es una reacción de un solo recipiente que tiene lugar en una única mezcla de reacción en la que no se aísla ningún producto intermedio amida. La reacción se puede realizar como se describe, por ejemplo, en el documento WO2007061555. En una primera reacción, el compuesto 20 se hidroliza con un ácido y agua para formar un producto intermedio amida in situ. La mezcla de reacción puede comprender opcionalmente un disolvente.
En una realización, la mezcla de reacción comprende aproximadamente 3 equivalentes molares a aproximadamente 10 equivalentes molares del ácido por 1 equivalente molar del compuesto 20. En otra realización, la mezcla de reacción comprende aproximadamente 3 equivalentes molares a aproximadamente 5 equivalentes molares del ácido a 1 equivalente molar del compuesto 20
En una realización, la temperatura de la mezcla de reacción es desde aproximadamente -10 °C a aproximadamente 40 °C. En otro ejemplo más, la temperatura de la mezcla de reacción es desde aproximadamente 15 °C a aproximadamente 35 °C. En otro ejemplo más, la temperatura de la mezcla de reacción es desde aproximadamente 10 °C a aproximadamente 30 °C.
Se deja reaccionar la mezcla de reacción hasta un par de días. En un ejemplo, la reacción se lleva a cabo hasta aproximadamente 24 horas. En otro ejemplo, el tiempo de reacción es desde aproximadamente 2 horas a 8 horas.
La fuente de ácido se puede seleccionar de ácidos orgánicos o inorgánicos para ajustar el pH de la mezcla de reacción por debajo de aproximadamente 7. En una realización, el ácido se selecciona de ácido acético, ácido clorhídrico, ácido sulfúrico, ácido metansulfónico, ácido fosfórico, ácido oxálico, y similares. En un ejemplo, la concentración de ácido está entre 10 % y aproximadamente 99 %, preferiblemente entre 70 % y aproximadamente 99 %, y el resto comprende agua. En otro ejemplo más, el ácido se selecciona de ácido sulfúrico o ácido metansulfónico.
En una realización, la mezcla de reacción contiene un disolvente seleccionado entre los disolventes orgánicos descritos anteriormente para el esquema 2. En un ejemplo, el disolvente comprende entre aproximadamente 10 % y aproximadamente 99 % del ácido.
Si la reacción tiene lugar en condiciones anhidras, se usa una cantidad en exceso de alcohol como disolvente en la mezcla de reacción. En una realización, el alcohol es un alcohol alifático que tiene de 1 a 3 carbonos.
En una segunda etapa, se agrega un alcohol a la mezcla de reacción, en la que. El producto intermedio amida se esterifica para formar el compuesto 12.
En otra realización, el compuesto de nitrilo se puede agregara una mezcla de alcohol y ácido en una etapa para formar el correspondiente éster 12 mediante una sal Pinner.
En una realización, se agregan aproximadamente 10 partes a aproximadamente 50 partes de alcohol a la mezcla de reacción. En un ejemplo, se agregan aproximadamente 10 partes a aproximadamente 20 partes de alcohol a la mezcla de reacción.
En una realización, la temperatura de la mezcla de reacción es desde aproximadamente -10 °C a aproximadamente 75 °C. En otro ejemplo, la temperatura de la mezcla de reacción es desde aproximadamente 40 °C a aproximadamente 65 °C. Se deja reaccionar la mezcla de reacción durante aproximadamente 24 horas a aproximadamente 150 horas. En otro ejemplo, el tiempo de reacción es desde aproximadamente 60 horas a aproximadamente 100 horas.
El compuesto 12 se puede aislar usando procedimientos de aislamiento conocidos en la técnica tales como los descritos para los esquemas anteriores.
5. Uso del nuevo compuesto 21 en la preparación de Remifentanil
En la etapa final, 1-(2-cianoetil)-4-[fenil(propanoil)amino]piperidina-4-carbonitrilo (21) reacciona con metanol en condiciones ácidas para dar el clorhidrato 1-(3-metoxi-3-oxopropil)-4-[fenil (propanoil)amino] piperidina-4-carboxilato de metilo (clorhidrato de remifentanilo, 1 )
En una realización, la mezcla de reacción comprende de aproximadamente 5 equivalentes molares a aproximadamente 20 equivalentes molares de ácido clorhídrico por 1 equivalente molar de 1-(2-cianoetil)-4-[fenil(propanoil)amino] piperidina-4-carbonitrilo. 21
En una realización, se usa metanol como disolventes y acetona, acetonitrilo, metanol, etanol, isopropanol, metiletilcetona se usan como disolventes para la cristalización. La proporción del compuesto 21 a disolvente en peso base es de 1:2 a 1 :10.
La temperatura de la mezcla de reacción durante la reacción varía desde aproximadamente 0 °C a aproximadamente 50 °C. Se deja reaccionar la mezcla de reacción durante varios días. En un ejemplo, el tiempo de reacción es desde aproximadamente 10 horas a 30 horas.
El compuesto 1 se puede aislar usando procedimientos de aislamiento conocidos en la técnica.
La siguiente tabla muestra los diferentes reactivos que se han usado para esta etapa de reacción.

6. Preparación del nuevo compuesto 20
El compuesto 20 se puede sintetizar en una etapa a partir del 3-(4-oxopiperidin-1-il) propanonitrilo 19 comercialmente disponible con fenilamina en presencia de un compuesto de cianuro y un ácido.
El siguiente esquema 8 ilustra el procedimiento en el que el 3-(4-oxo-piperidina-1-il) propanonitrilo reacciona con anilina y un reactivo que contiene cianuro en presencia de un ácido para dar el 1-(2-cianoetil)-4-(fenilamino) piperidina-4-carbonitrilo 20

Esquem a 8 Preparación del producto intermedio 20
En una realización, la mezcla de reacción comprende de aproximadamente 1 equivalente molar a aproximadamente 3 equivalentes molares de anilina, aproximadamente 1 equivalente molar a aproximadamente 3 equivalentes molares del compuesto de cianuro y aproximadamente 2 equivalentes molares a 5 equivalentes molares de ácido a 1 equivalente molar de 19. Preferiblemente, la mezcla de reacción se carga con aproximadamente 1 a 1.5 equivalentes de anilina, aproximadamente 1 a 1.5 equivalentes de compuesto de cianuro y aproximadamente 3-4 equivalentes de ácido por 1 equivalente de 3-(4-oxo-piperidina-1-il) propanonitrilo. La proporción del compuesto 19 al disolvente en peso base es de 1:5 a 1:20.
La temperatura de la mezcla de reacción durante la reacción varía desde aproximadamente 0 °C a aproximadamente 80 °C Preferiblemente, la temperatura de reacción varía desde aproximadamente 20 °C a aproximadamente 60 °C. Se deja reaccionar la mezcla de reacción hasta varios días. En un ejemplo, el tiempo de reacción es desde aproximadamente 2 horas a 5 horas.
Los ejemplos no limitantes de compuestos de cianuro son cianuro de sodio, cianuro de potasio, cianuro de trimetilsililo, cianuro de hidrógeno y similares.
Por razones económicas, se prefiere el cianuro de sodio.
El ácido puede incluir cualquier ácido orgánico e inorgánico para ajustar el pH por debajo de 7. Los ejemplos no limitantes de ácidos incluyen ácido acético, ácido clorhídrico, ácido sulfúrico, ácido fosfórico, ácido oxálico y similares. En una realización, se usa ácido acético para ajustar el pH por debajo de 7.
Los ejemplos de disolventes usados en la mezcla de reacción incluyen, pero no se limitan a, acetonitrilo, diclorometano, cloroformo, W,W-dimetilformamida, dimetilsulfóxido, acetato de etilo, dicloroetano, agua, benceno, tolueno, xileno, metanol, etanol, isopropanol y mezclas de los mismos.
El compuesto 20 se puede aislar mediante extracción con disolvente o precipitación de la mezcla de reacción. El compuesto 20 se puede purificar adicionalmente usando un procedimiento de purificación conocido en la técnica. En un ejemplo, el compuesto 20 se puede disolver en un disolvente orgánico al que se agrega una solución de un ácido en un disolvente para formar una sal del compuesto 20, que se puede aislar mediante procedimientos conocidos en la técnica. Los ejemplos de disolventes incluyen, pero no se limitan a acetonitrilo, acetona, diclorometano, cloroformo, W,W-dimetilformamida, dimetilsulfóxido, acetato de etilo, dicloroetano, agua, benceno, tolueno, xileno, metanol, etanol, isopropanol y mezclas de los mismos. Los ejemplos de ácidos incluyen ácido clorhídrico, ácido bromhídrico, ácido metanosulfónico, ácido 4-metilbencensulfónico, ácido sulfúrico, ácido fosfórico, ácido cítrico, ácido oxálico y similares.
La siguiente tabla muestra los diferentes reactivos que se han usado para esta etapa de reacción.


7. Preparación del compuesto 19
El esquema 7 siguiente ilustra la reacción en la que el clorhidrato de 4-piperidona monohidrato reacciona con el derivado 18 de propionitrilo para dar 3-(4-oxo-piperidin-1-il) propanonitrilo.

En una realización, el compuesto 8 se mezcla en una mezcla de reacción con el derivado 18 de propionitrilo con un grupo saliente en la posición 3 en presencia de un disolvente y una base para formar el intermedio 19. La mezcla de reacción comprende de aproximadamente 1 equivalente molar a aproximadamente 6 equivalentes molares de 18 y aproximadamente 1 equivalente molar por aproximadamente 6 equivalentes molares de base por 1 equivalente molar de 8. Preferiblemente, la mezcla de reacción se carga con aproximadamente 2 a 3 equivalentes de 18 y aproximadamente 2 a 4 equivalentes de base a 1 equivalente de clorhidrato de 4-piperidona monohidrato. La proporción del compuesto 8 a disolvente en peso base es de 1: 3 a 1:20.
La temperatura de la mezcla de reacción durante la reacción varía desde aproximadamente 0 °C a aproximadamente 80 °C. Se deja reaccionar la mezcla de reacción desde aproximadamente 6 h a aproximadamente 48 h. En un ejemplo, el tiempo de reacción es desde aproximadamente 18 horas a 24 horas.
Los ejemplos del derivado 18 de propionitrilo incluyen 3-cloropropionitrilo, 3-bromopropionitrilo, 3-yodopropionitrilo, 2-cianoetil metanosulfonato, 2-cianoetil 4-metilbencenosulfonato.
Los ejemplos de la base incluyen hidróxido de sodio, hidróxido de potasio, carbonato de sodio, carbonato de potasio, alcóxidos metálicos, amidas metálicas, hidruros metálicos y aminas.
Los ejemplos de disolventes usados en la mezcla de reacción incluyen, pero no se limitan a, acetonitrilo, acetona, metiletilcetona, diclorometano, cloroformo, W,W-dimetilacetamida, W,Ñ-dimetilformamida, dimetilsulfóxido, acetato de etilo, dicloroetano, benceno, tolueno, xileno, tetrahidrofurano, 2-metil tetrahidrofurano y mezclas de los mismos.
En una realización, el compuesto 19 se puede aislar mediante un procedimiento de extracción y aislamiento con disolvente conocido en la técnica. Tal aislamiento puede incluir la evaporación del disolvente para recuperar el producto aceitoso en bruto.
En algunos ejemplos, el compuesto 19 se puede purificar adicionalmente mediante destilación o cromatografía. En un ejemplo, el compuesto 19 se puede disolver en un disolvente orgánico al que se agrega una solución de un ácido en un disolvente para formar una sal del compuesto 19, que se puede aislar mediante procedimientos conocidos en la técnica. Los ejemplos de disolventes incluyen, pero no se limitan a acetonitrilo, acetona, diclorometano, cloroformo,
^W-dimetilformamida, dimetilsulfóxido, acetato de etilo, dicloroetano, agua, benceno, tolueno, xileno, metanol, etanol, isopropanol y mezclas de los mismos. Los ejemplos de ácidos incluyen ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido fosfórico, ácido cítrico, ácido oxálico y similares.
La siguiente tabla muestra los diferentes reactivos que se han usado para esta etapa de reacción.

8. Uso de los nuevos compuestos en la síntesis de Remifentanil
Siguiendo lo mencionado anteriormente, se describe en la presente invención un nuevo método para la preparación de clorhidrato de remifentanilo. Hemos encontrado que ambos grupos éster metílico presentes en el clorhidrato de remifentanilo se pueden obtener a partir de funciones nitrilo en la etapa final. La síntesis comienza a partir de 4 piperidona.H2O.HCl y el API objetivo se obtiene después de 4 etapas de reacción (esquema 6).

Los siguientes ejemplos se proporcionan para ilustrar más completamente la presente invención.
Ejemplo 1
Síntesis de 3-(4-oxop¡per¡d¡n-1-¡l)propanon¡tr¡lo
Se suspendieron 5.10 mL (2 eq.) de 3-cloropropionitrilo, 5.0 g de clorhidrato de 4-piperidona monohidrato y 13.5 g (3 eq.) de K2CO3 en 40 mL de metiletilcetona. La mezcla se calentó a 80 °C, durante 18 h. Los sólidos se separaron por filtración y el filtrado se concentró al vacío. Luego se disolvió en 30 mL de agua destilada y se extrajo con acetato de etilo. La fase orgánica se secó sobre Na2SO4 y se concentró al vacío: se obtuvieron 4.21 g de 3-(4-oxopiperidin-1-il) propanonitrilo como un líquido amarillo.
Ejemplo 2
Síntesis de 1-(2-cianoetil)-4-(fenilamino) piperidina-4-carbonitrilo
Se agregaron gota a gota 1.69 g (1.05 eq.) de NaCN disuelto en 4.5 mL de agua destilada (RT, ~ 30 min.) a una solución agitada de 5 g de 3-(4-oxopiperidin-1-il) propanonitrilo, 4.8 mL (1.6 eq.) de anilina y 4.9 mL (2.6 eq.) de ácido acético en 25 mL de metanol. La solución se calentó a 60 °C, durante aprox. 20 h. y luego se enfrió a 0 °C. Se agregó gota a gota una solución de NaOH (33 % p/v) mientras se formaba un precipitado de color blanco (aproximadamente 7 mL). Se agregó agua destilada (10 mL) y la suspensión se agitó a 0 °C, durante 6 h. El precipitado se separó por filtración, se lavó con una mezcla de H2O:MeOH (1:1) y se dejó secar. Rendimiento: 6.5 g (78 %) de polvo de color blanco crema.
Ejemplo 3
Síntesis de A/-(4-ciano-1-(2-cianoetil) piperidin-4-il)-W-fenilpropionamida
Se agregaron gota a gota 12.4 mL (3 eq.) de cloruro de propionilo a una solución enfriada y agitada de 12 g de 1-(2-cianoetil)-4-(fenilamino) piperidina-4-carbonitrilo en 120 ml de diclorometano. En poco tiempo se formó un precipitado de color blanco. La mezcla se mantuvo a reflujo durante la noche durante aprox. 20 h. y luego se dejó alcanzar RT. Se agregaron gota a gota 6,5 mL (1 eq.) de trietilamina.
La mezcla se volvió transparente y gradualmente opaca nuevamente. Después de agitar a RT, durante la noche (20 h), se agregaron 120 mL de agua destilada. La fase orgánica separada se lavó con 120 mL de solución sat. Solución de Na2CO3, 120 ml de salmuera, secada sobre Na2SO4 y concentrada al vacío: se obtuvieron 8.2 g de materia en bruto de color marrón claro. El producto en bruto se cristalizó en 40 mL de isopropanol. El precipitado se filtró, se lavó con isopropanol frío y se dejó secar. Rendimiento: 7.25 g (49 %) de polvo de color blanco.
Síntesis de Remifentanil HCI
Ejemplo 4
Se agregaron 30 mL (15 eq.) de solución de HCl al 35 % en metanol a 5 g de N-(4-ciano-1-(2-cianoetil) piperidin-4-il)-N-fenilpropionamida y la mezcla se agitó a temperatura ambiente. Después de 20 h., se agregaron 5 mL de metanol y la suspensión se agitó durante 5 h más a temperatura ambiente. Luego se filtró, el precipitado se lavó con 5 mL de isopropanol frío y se dejó secar. El producto en bruto contiene un residuo inorgánico (NH4Cl) que se puede filtrar durante la cristalización. Se suspendieron 5.3 g del producto en bruto en 75 mL de isopropanol (13-15 mL/g) y se sometieron a reflujo durante 15 min. Mientras estaba caliente, el sólido inorgánico se filtró y se lavó con 20 mL de isopropanol caliente. El filtrado se calentó a reflujo de nuevo durante 5 min, se dejó que alcanzara RT y se enfrió a 5 °C, durante 2 h. Se filtró el cristal, se lavó con isopropanol y se dejó secar. Rendimiento: 2.18 g de cristales en polvo de color blanco.
Ejemplo 5
Se agregaron 30 mL (15 eq.) de una solución de HCl al 35 % en metanol a 5 g de N-(4-ciano-1-(2-cianoetil) piperidin-4-il)-N-fenilpropionamida y la mezcla se agitó a temperatura ambiente. Después de 20 h., se agregaron 5 ml de metanol y la suspensión se agitó durante 5 h más a temperatura ambiente. Luego se filtró, el precipitado se lavó con 5 mL de isopropanol frío. El producto en bruto se agregó lentamente a 80 mL de solución sat. de Na2CO3. El producto se extrajo con 2x40 mL de acetato de etilo. Las fases orgánicas se combinaron, se secaron sobre Na2SO4 y se concentraron a presión reducida. La base de remifentanil obtenida se disolvió en 15 mL de metanol, se enfrió a 5 °C y se agregó lentamente HCl gaseoso (1.5 eq.). La sal de clorhidrato precipitada se separó por filtración. Rendimiento: 4.5 g de cristales en polvo de color blanco.
Claims (15)
1. Un compuesto de la fórmula 20 y sus sales:

2. Uso del compuesto 20 para la síntesis de Remifentanil (1).
3. Uso del compuesto 20 según la reivindicación 2 en el que el compuesto 20 se convierte primero en el compuesto 2 1.

4. Uso del compuesto 20 según la reivindicación 3, donde el agente acilante es anhídrido de propión o cloruro de propionilo.
5. Uso del compuesto 20 según la reivindicación 4, en el que se usa un eliminador de ácido seleccionado de trietilamina, morfolina, piperidina, piridina u otra base orgánica.
6. Uso del compuesto 20 según la reivindicación 3 en el que el compuesto 21 se convierte en Remifentanil (1) 7. Uso del compuesto 20 para la síntesis de Remifentanil según la reivindicación 2 en el que el compuesto 20 se convierte primero en el compuesto 12.

8. Uso del compuesto 20 según la reivindicación 7 en el que el compuesto 12 se convierte en Remifentanil (1) 9. Método para la preparación de un compuesto según la reivindicación 1 en el que el compuesto se prepara mediante reacción del compuesto 19 con fenilamina en presencia de un compuesto de cianuro.

10. Método según la reivindicación 9 en el que el compuesto de cianuro es cianuro de sodio.
11. Método para la preparación del compuesto 19 en el que el compuesto se prepara mediante la alquilación del clorhidrato de 4-piperidona monohidrato con un agente alquilante apropiado en presencia de un catalizador básico.
12. Método según la reivindicación 11 en el que el agente alquilante apropiado es 3-cloropropionitrilo, 3-bromopropionitrilo, 3-yodopropionitrilo, 2-cianoetil metanosulfonato, 2-cianoetil 4-metilbencenosulfonato,
13. Método según la reivindicación 12 en el que un catalizador básico es carbonato de sodio o potasio, hidrogenocarbonato de sodio, hidróxido de sodio o potasio, amida de sodio, tert-butóxido de potasio, etóxido de sodio,
14. Método según la reivindicación 13 en el que el disolvente usado es acetona, metiletilcetona, isopropanol, acetonitrilo, W,A/-dimet¡lacetamida, W,A/-dimetilformamida, tetrahidrofurano, 2-metil tetrahidrofurano,
15. Uso de un compuesto según la reivindicación 1 para la síntesis de clorhidrato de remifentanilo según el siguiente esquema:

Clorhidrato de remifentanilo
1
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18202878.7A EP3643704B1 (en) | 2018-10-26 | 2018-10-26 | New intermediates for the preparation of remifentanil hydrochloride |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2858404T3 true ES2858404T3 (es) | 2021-09-30 |
Family
ID=64051418
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18202878T Active ES2858404T3 (es) | 2018-10-26 | 2018-10-26 | Nuevos intermedios para la preparación de clorhidrato de remifentanilo |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US10844014B2 (es) |
| EP (1) | EP3643704B1 (es) |
| DK (1) | DK3643704T3 (es) |
| ES (1) | ES2858404T3 (es) |
| HR (1) | HRP20210376T1 (es) |
| PL (1) | PL3643704T3 (es) |
| PT (1) | PT3643704T (es) |
| RS (1) | RS61517B1 (es) |
| SI (1) | SI3643704T1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB202010168D0 (en) * | 2020-07-02 | 2020-08-19 | Johnson Matthey Plc | Process for preparing remifentanil hydrochloride |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5019583A (en) | 1989-02-15 | 1991-05-28 | Glaxo Inc. | N-phenyl-N-(4-piperidinyl)amides useful as analgesics |
| WO2007061555A1 (en) | 2005-11-17 | 2007-05-31 | Mallinckrodt Inc. | Process for synthesizing remifentanil |
| AU2007208438A1 (en) | 2006-01-24 | 2007-08-02 | Mallinckrodt Inc. | Process for synthesizing remifentanil |
| EP1867635A1 (en) | 2006-06-15 | 2007-12-19 | Kern Pharma, S.L. | Process for preparing remifentanil, intermediates thereof, use of said intermediates and processes for their preparation |
| US8299258B2 (en) | 2008-11-04 | 2012-10-30 | Cambrex Charles City | Method of making piperidine derivatives |
-
2018
- 2018-10-26 SI SI201830231T patent/SI3643704T1/sl unknown
- 2018-10-26 ES ES18202878T patent/ES2858404T3/es active Active
- 2018-10-26 PT PT182028787T patent/PT3643704T/pt unknown
- 2018-10-26 EP EP18202878.7A patent/EP3643704B1/en active Active
- 2018-10-26 RS RS20210271A patent/RS61517B1/sr unknown
- 2018-10-26 PL PL18202878T patent/PL3643704T3/pl unknown
- 2018-10-26 DK DK18202878.7T patent/DK3643704T3/da active
-
2019
- 2019-10-25 US US16/664,464 patent/US10844014B2/en active Active
-
2021
- 2021-03-03 HR HRP20210376TT patent/HRP20210376T1/hr unknown
Also Published As
| Publication number | Publication date |
|---|---|
| PL3643704T3 (pl) | 2021-06-14 |
| SI3643704T1 (sl) | 2021-04-30 |
| US10844014B2 (en) | 2020-11-24 |
| US20200131127A1 (en) | 2020-04-30 |
| RS61517B1 (sr) | 2021-03-31 |
| EP3643704B1 (en) | 2020-12-16 |
| DK3643704T3 (da) | 2021-03-15 |
| PT3643704T (pt) | 2021-03-08 |
| HRP20210376T1 (hr) | 2021-04-16 |
| EP3643704A1 (en) | 2020-04-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7531673B2 (en) | Preparation of amino acid amides | |
| JP2895145B2 (ja) | 鎮痛剤として用いられるn―フエニル―n―(4―ピペリジニル)アミド | |
| KR20080108291A (ko) | P 물질 길항제로서 유용한 아실아미노알케닐렌 아미드의 합성 | |
| US20190071441A1 (en) | Pyrazole derivative manufacturing method | |
| RU2039043C1 (ru) | Производное n-замещенного 1-гексил-4-фенил-4-пиперидинкарбоксамида или его фармацевтически приемлемая соль и способ его получения | |
| ES2343212T3 (es) | Procedimiento para preparar remifentanilo, intermedios del mismo, uso de dichos intermedios y procedimientos para su preparacion. | |
| ES2858404T3 (es) | Nuevos intermedios para la preparación de clorhidrato de remifentanilo | |
| US20170267633A1 (en) | Novel crystalline arylalkylamine compound and process for producing the same | |
| CA2333689A1 (en) | New neurokinin antagonists, processes for preparing them and pharmaceutical compositions containing said compounds | |
| JP5663743B2 (ja) | モチリン受容体作動活性を有するオキシインドール誘導体 | |
| US20080319196A1 (en) | Process for Synthesizing Remifentanil | |
| US8334389B2 (en) | Process for the preparation of pleuromutilins | |
| WO2020053795A2 (en) | Process for the preparation of acalabrutinib and its intermediates | |
| ES2375384T3 (es) | Asimadolina para el tratamiento del síndrome de colon irritable (irritable bowel syndroes). | |
| JP2012519153A5 (es) | ||
| WO2012117410A1 (en) | A process for the preparation of n-[2-[(acetylthio) methyl]-1-oxo-3-phenylpropyl] glycine phenyl methyl ester and intermediates thereof | |
| TWI386395B (zh) | 哌啶衍生物的立體有擇合成 | |
| US8664443B2 (en) | Process for the preparation of (1S, 3S, 5S)-2-[2(S)-2-amino-2-(3-hydroxy-1-adamantan-1-yl) acetyl]-2-azabicyclo [3.1.0] hexane-3-carbonitrile | |
| AU2004295093A1 (en) | Delta-amino-gamma-hydroxy-omega-aryl-alkanoic acid amides and use as renin inhibitors | |
| JPWO2012026529A1 (ja) | イソキノリン誘導体又はその塩の新規製造方法 | |
| AU2004266247A1 (en) | 1-carbamoylcycloalkylcarboxylic acid compounds, processes for making and uses thereof | |
| ZA200903939B (en) | Azabicyclic compounds, a process for their preparation and pharmaceutical compositions containing them | |
| JP2008019168A (ja) | 2−シアノ−4−フルオロピロリジン誘導体の製造法 | |
| JP2014114221A (ja) | ベンズイミダゾール誘導体の製造方法及びその中間体 | |
| US9908858B2 (en) | Method for the synthesis of a hydrazine that can be used in the treatment of the papilloma virus |