ES2769578T3 - Hidantoínas que modulan el procesamiento de APP mediado por BACE - Google Patents
Hidantoínas que modulan el procesamiento de APP mediado por BACE Download PDFInfo
- Publication number
- ES2769578T3 ES2769578T3 ES14751458T ES14751458T ES2769578T3 ES 2769578 T3 ES2769578 T3 ES 2769578T3 ES 14751458 T ES14751458 T ES 14751458T ES 14751458 T ES14751458 T ES 14751458T ES 2769578 T3 ES2769578 T3 ES 2769578T3
- Authority
- ES
- Spain
- Prior art keywords
- alzheimer
- disease
- administration
- compound
- subject
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C*CC[C@@](*)(C(*(*)I)=O)C(C(*)**C(C1(C)*)*=C)=C[C@@](C)(*)C1N Chemical compound C*CC[C@@](*)(C(*(*)I)=O)C(C(*)**C(C1(C)*)*=C)=C[C@@](C)(*)C1N 0.000 description 7
- NXXHDLRCKOMKQU-SREVYHEPSA-N CC/C(/C=C)=C(/C)\OC(F)F Chemical compound CC/C(/C=C)=C(/C)\OC(F)F NXXHDLRCKOMKQU-SREVYHEPSA-N 0.000 description 1
- FQJAPJHFZXDGCG-UHFFFAOYSA-N OC(C(c1ccccc1)=C1CC1)c1cc([FH+])cc(F)c1 Chemical compound OC(C(c1ccccc1)=C1CC1)c1cc([FH+])cc(F)c1 FQJAPJHFZXDGCG-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/88—Nitrogen atoms, e.g. allantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4166—1,3-Diazoles having oxo groups directly attached to the heterocyclic ring, e.g. phenytoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4168—1,3-Diazoles having a nitrogen attached in position 2, e.g. clonidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/46—8-Azabicyclo [3.2.1] octane; Derivatives thereof, e.g. atropine, cocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/70—One oxygen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/72—Two oxygen atoms, e.g. hydantoin
- C07D233/76—Two oxygen atoms, e.g. hydantoin with substituted hydrocarbon radicals attached to the third ring carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Emergency Medicine (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Hospice & Palliative Care (AREA)
- Ophthalmology & Optometry (AREA)
- Psychiatry (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Un compuesto seleccionado del grupo que consiste en **(Ver fórmula)** y **(Ver fórmula)** o una sal farmaceuticamente aceptable del mismo, un tautomero del mismo, una sal farmaceuticamente aceptable de un tautomero del mismo, un enantiomero del mismo, o una sal farmaceuticamente aceptable de un enantiomero del mismo.
Description
DESCRIPCIÓN
Hidantoínas que modulan el procesamiento de APP mediado por BACE
Referencia cruzada a solicitudes relacionadas
Esta solicitud reivindica el beneficio y la prioridad de USSN 61/763,830, presentada el 12 de febrero de 2013, que se incorpora aquí como referencia para todos los fines.
Declaración de apoyo gubernamental
[No es aplicable]
Antecedentes
El péptido beta amiloide (Ap) es un componente primario de las fibrillas y placas beta amiloides, de las que se considera que desempeñan un papel en un número creciente de patologías. Ejemplos de tales patologías incluyen, entre otros, enfermedad de Alzheimer, síndrome de Down, enfermedad de Parkinson, pérdida de memoria (incluida la pérdida de memoria asociada con la enfermedad de Alzheimer y la enfermedad de Parkinson), síntomas de déficit de atención (incluidos los síntomas de déficit de atención asociados con la enfermedad de Alzheimer, enfermedad de Parkinson y síndrome de Down), demencia (incluida la demencia presenil, demencia senil, demencia asociada con la enfermedad de Alzheimer, enfermedad de Parkinson y síndrome de Down), parálisis supranuclear progresiva, degeneración basal cortical, neurodegeneración, deterioro olfativo (incluido el deterioro olfativo asociado con Enfermedad de Alzheimer, enfermedad de Parkinson y síndrome de Down), angiopatía p-amiloide (incluida la angiopatía amiloide cerebral), hemorragia cerebral hereditaria, deterioro cognitivo leve ("MCI"), glaucoma, amiloidosis, diabetes tipo II, hemodiálisis (microglobulinas p2 y complicaciones derivadas de las mismas), enfermedades neurodegenerativas como temblores, encefalitis espongiforme bovina, enfermedad de Creutzfeld Jakob, lesión cerebral traumática y similares.
Los péptidos Ap son péptidos cortos que se producen por proteólisis de la proteína transmembrana llamada proteína precursora amiloide ("APP"). Los péptidos Ap se obtienen a partir de la escisión de APP por la actividad de la psecretasa en una posición cerca del terminal N de Ap, y por la actividad de la secretasa gamma en una posición cerca del terminal C de Ap. (La APP también se escinde por la actividad de la a-secretasa, lo que da como resultado el fragmento secretado no amiloidogénico conocido como APPa soluble). La enzima de escisión de APP del sitio beta ("BACE-1") se considera la principal proteasa de aspartilo responsable de la producción de Ap por la actividad de la p-secretasa. Se ha demostrado que la inhibición de BACE-1 inhibe la producción de Ap.
Se estima que la enfermedad de Alzheimer (EA) afecta a más de 20 millones de personas en todo el mundo y se cree que es la causa más común de demencia. A medida que la población mundial envejece, el número de personas con enfermedad de Alzheimer (EA, actualmente aproximadamente 5.4 millones en los Estados Unidos) continuará aumentando. La enfermedad de Alzheimer es una enfermedad neurodegenerativa asociada con demencia progresiva y pérdida de memoria. Dos características clave de la EA son acumulación de depósitos extracelulares que contienen péptido Ap agregado y pérdida sináptica neuronal en la EA en regiones cerebrales específicas. Aunque la patogénesis de la EA es compleja, la evidencia genética y bioquímica convincente sugiere que la sobreproducción de Ap o la falta de eliminación de este péptido es el primer evento en la cascada amiloide que conduce a la EA principalmente a través del depósito de amiloide, que se supone que está involucrado en la formación de ovillos neurofibrilares, disfunción neuronal y activación de la microglías, que caracteriza los tejidos cerebrales afectados por la EA.
La acumulación de Ap se considera el primer evento en una cascada compleja que conduce a la neurodegeneración, según se desprende de evidencia genética y bioquímica convincente. La hipótesis de la cascada amiloide (Hardy and Allsop (1991) Trends Pharmacol. Sci., 12: 383-388; Selkoe (1996) J. Biol. Chem., 271: 18295-18298; Hardy (1997) Trends Neurosci., 20: 154-159; Hardy and Selkoe (2002) Science, 297: 353-356) afirma que la sobreproducción de Ap, o la incapacidad de eliminar este péptido, conduce a EA, principalmente a través del depósito de amiloide, que se supone que está involucrado en la formación de ovillos neurofibrilares, disfunción neuronal y activación de microglías, que son características de los tejidos cerebrales afectados por EA (Busciglio et al. (1995) Neuron, 14: 879-888; Gotz et al. (1995) EMBO J., 14: 1304-1313; Lewis et al. (2001) Science, 293: 1487-1491; Hardy et al. (1985) Nat Neurosci., 1: 355-358).
Teniendo en cuenta el papel causal de Ap en la etiología de EA, se han sugerido nuevas estrategias terapéuticas que reducen los niveles de Ap o evitan la formación de las especies neurotóxicas de Ap como un método para prevenir o retrasar la progresión de la enfermedad. De hecho, el enfoque principal durante la última década ha sido inhibir la producción y agregación de Ap cerebral, aumentar el aclaramiento de Ap parenquimatoso e interferir con la muerte celular inducida por Ap.
La escisión secuencial de APP por proteasas unidas a membrana p-secretasa y Y-secretasa da como resultado la formación de Ap. Una ruta proteolítica competitiva hacia la ruta de la p-secretasa, la ruta de la a-secretasa, da como resultado la escisión de la APP dentro del dominio Ap, lo que impide la generación de Ap (Selkoe (2001) Physiol. Rev., 81: 741-766; Hussain et al. (1999) Mol. Cell. Neurosci., 14: 419-427; Sinha et al. (1999) Nature, 402: 537-540; Vassar
et al. (1999) Science, 286: 735-741). La enzima de escisión de APP del sitio p-1 (BACE1) se identificó como la actividad principal de p-secretasa que media la primera escisión de APP en la ruta p-amiloidogénica (Id.).
La BACE1 es una proteína de 501 aminoácidos que tiene homología con las proteasas aspárticas eucariotas, especialmente de la familia de las pepsinas (Yan et al. (1999) Nature, 402: 533-537). Al igual que otras proteasas aspárticas, la BACE1 se sintetiza como un zimógeno con un prodominio que es escindido por furina para liberar la proteína madura. La BACE1 es una proteína transmembrana de tipo I con un sitio activo luminal que escinde APP para liberar un ectodominio (sAPPp) en el espacio extracelular. El fragmento C-terminal restante (CTF) sufre una escisión adicional por la Y-secretasa, lo que conduce a la liberación de Ap y el dominio C-terminal intracelular de APP (AICD).
Se ha propuesto que las presenilinas son el componente enzimático principal de la Y-secretasa, cuya escisión imprecisa de APP produce un espectro de péptidos Ap que varían en longitud en unos pocos aminoácidos en el extremo C-terminal. La mayoría de Ap normalmente termina en el aminoácido 40 (Ap40), pero se ha demostrado que la variante de 42 aminoácidos (Ap42) es más susceptible a la agregación, y se ha planteado la hipótesis de que nuclea la formación de placa senil. La modulación de la Y-secretasa también puede conducir a un aumento en la variante de 38 aminoácidos (Ap38). La vía competitiva de la a-secretasa es el resultado de divisiones secuenciales por a-y ysecretasa. Se han propuesto tres metaloproteasas de la familia de desintegrina y metaloproteasa (ADAM 9, 10 y 17) como candidatas para la actividad de a-secretasa, que escinde APP en la posición 16 dentro de la secuencia Ap. Usando experimentos de sobreexpresión, se ha demostrado que ADAM-10 es la a-secretasa probable para la escisión de APP (Vassar (2002) Adv. Drug Deliv. Rev., 54: 1589-1602; Buxbaum et al. (1998) J. Biol Chem., 273: 27765-27767; Koike et al. (1999) Biochem. J., 343 (Pt 2): 371-375). Esta escisión también libera un ectodominio (sAPPa), que muestra funciones neuroprotectoras (Lammich et al. (1999) Proc. Natl. Acad. Sci. USA, 96: 3922-3927). La escisión posterior del CTF de 83 aminoácidos (C83) libera p3, que no es amiloidogénico, y AICD (Furukawa et al. (1996) J. Neurochem., 67: 1882-1896). Las funciones de estos fragmentos no están completamente aclaradas, aunque la hipótesis de AICD es mediar en la señalización intracelular.
La investigación que aclara las vías metabólicas que regulan la producción de Ap a partir de la proteína precursora amiloide (APP) indica que las secretasas que producen Ap son buenas dianas terapéuticas, ya que la inhibición de la secretasa p o y limita la producción de Ap. El hecho de que la p-secretasa inicia el procesamiento de APP y, por lo tanto, sirve como el paso limitante de la velocidad en la producción de Ap, su inhibición ha atraído esfuerzos de muchos grupos de investigación. Ejemplos de la literatura de patentes están creciendo e incluyen, por ejemplo, WO2006009653, WO2007005404, WO2007005366, WO2007038271, WO2007016012, US2005/0282826, US2007072925, WO2007149033, WO2007145568, WO2007145569, WO2007145570, WO2007145571, WO2007114771, US20070299087, WO2005/016876, WO2005/014540, WO2005/058311, WO2006/065277, WO2006/014762, WO2006/014944, WO2006/138195, WO2006/138264, WO2006/138192, WO2006/138217, WO2007/050721, WO2007/053506, WO2007/146225, WO2006/138230, WO2006/138265, WO2006/138266, WO2007/053506, WO2007/146225, WO2008/073365, WO2008/073370, WO2008/103351 US2009/041201, US2009/041202, ' WO2010/047372.
Una limitación de las estrategias inhibidoras de la proteasa es la inhibición de la escisión de todos los sustratos de una proteasa dirigida dada, como BACE o el complejo Y-secretasa. En el caso de la Y-secretasa, los sustratos que no sean APP, como Notch, generan preocupación por los posibles efectos secundarios de la inhibición de la Y-secretasa y el reciente fracaso del inhibidor de la Y-secretasa. Los problemas asociados con el uso de semagacestat, sirven para reforzar tales preocupaciones.
La BACE es una enzima clave implicada en el procesamiento de APP que conduce a la producción de Ap42 y la patología de la enfermedad de Alzheimer (EA). La BACE-1 (también llamada BACE) se ha convertido en un área de investigación popular desde su descubrimiento, y quizás ha superado a la Y-secretasa como la diana más prometedora para la investigación farmacéutica. Un problema con la Y-secretasa como objetivo es su escisión conocida de Notch, que cumple funciones importantes en el desarrollo neuronal. Los ratones inactivados con presenilina demostraron somitogénesis anormal y desarrollo esquelético axial con longitud corporal acortada, así como hemorragias cerebrales (Shen et al. (1997) Cell, 89: 629-639; Wong et al. (1997) Nature, 387: 288-292) Por el contrario, varios grupos informaron que los ratones con bloqueo de BACE1 son sanos y no muestran signos de efectos adversos (Luo et al. (2001) Nat. Neurosci., 4: 231-232; Roberds et al. (2001) Hum. Mol. Genet ., 10: 1317-1324), mientras que un grupo notó sutiles déficits neuroquímicos y cambios de comportamiento en ratones fértiles y viables (Harrison et al. (2003) Mol. Cell Neurosci., 24: 646-655). Aunque los estudios recientes han demostrado que los ratones con bloqueo de BACE1 exhiben hipomielinización de los nervios periféricos (Willem et al. (2006) Science, 314: 664-666), las consecuencias de la inhibición de BACE1 en animales adultos, donde la mielinización ya ha tenido lugar, no están claras. Recientemente se ha informado que BACE1 escinde múltiples sustratos, incluidos ST6Gal I, PSGL-1, subunidades de canales de sodio dependientes de voltaje, proteínas similares a APP (APLP), proteína relacionada con el receptor de LDL (LRP) y, más recientemente, neurregulina 1 tipo III (NRG1) (Willem et al. (2006) Science, 314: 664-666; Hu et al. (2006) Nat. Neurosci., 9: 1520-1525). Por lo tanto, las consecuencias de inhibir directamente la BACE1 aún no se entienden completamente.
El modelado molecular (Sauder et al. (2000) J. Mol. Biol., 300: 241-248) y posterior cristalografía de rayos X (Hong et al. (2000) Science, 290: 150-153; Maillard et al. (2007) J. Med. Chem., 50: 776-781) del sitio activo BAc E-1 complejado
con un inhibidor del estado de transición proporcionó información crucial sobre las interacciones BACE-1-sustrato. Estructuralmente, el sitio activo de BACE-1 es más abierto y menos hidrófobo que otras aspartil proteasas, lo que dificulta el desarrollo de candidatos eficaces de inhibidores de BACE in vivo. Si bien existe un gran esfuerzo de descubrimiento de fármacos centrado en el desarrollo de inhibidores directos de BACE, ninguno hasta ahora ha avanzado significativamente en las pruebas clínicas.
Algunos inhibidores de BACE tales como LY2811376 y CTS21166 ingresaron a las pruebas clínicas, pero no avanzaron más allá de la Fase 1 debido a razones de seguridad. El descubrimiento de otros sustratos fisiológicos de BACE plantea una gran preocupación en el desarrollo clínico de los inhibidores de BACE o los moduladores de BACE y podría ser un obstáculo significativo en el avance de estos inhibidores como terapia para la enfermedad.
El documento WO 2007/058601 A1 describe derivados de hidantoína con actividad inhibidora de BACE. De manera similar, el documento WO 2008/115552 A1 también describe derivados de hidantoína con actividad inhibidora de BACE.
Resumen
En el presente documento se describen compuestos de hidantoína que son efectivos para inhibir la actividad BACE contra APP. Sin limitarse a una teoría particular, se cree que la actividad de las hidantoínas identificadas en el presente documento parece estar asociada con la unión a BACE y/o a APP, particularmente cuando estas unidades estructurales forman un complejo BACE/APP. Por consiguiente, se cree que los compuestos descritos en el presente documento representan una nueva clase de compuestos designados en el presente documento como inhibidores de unión a APP (ABBI) y proporcionan un nuevo mecanismo para modular el procesamiento de APP. Las hidantoínas descritas en el presente documento parecen mostrar una mejor permeabilidad cerebral e inhibición funcional de BACE.
La presente invención proporciona una selección de compuestos según la reivindicación 1. Además, y únicamente con fines informativos, se proporcionan ejemplos de otros compuestos que no se consideran dentro del alcance de la invención.
Ejemplo 1: Un compuesto de acuerdo con la fórmula:

donde M es

R7 se selecciona del grupo que consiste en C=O, C=S, C-NH2 y C=NH, y el enlace representado por la línea ondulada es un enlace simple cuando R7 es C=O, C=S o C=NH, y un doble enlace cuando R7 es C-NH2; R8 y R9 se seleccionan independientemente del grupo que consiste en H, alquilo, cicloalquilo y arilo, siempre que cuando el enlace representado por la línea ondulada sea un doble enlace, entonces R9 esté ausente; R0 se selecciona del grupo que consiste en arilo, arilo sustituido, arilo disustituido, heteroarilo, heteroarilo sustituido, heteroarilo disustituido, alquilo, haloalquilo, cicloalquilo, alquenilo y alquinilo; X1 se selecciona del grupo que consiste en C-halógeno (por ejemplo, Cl o F), CH y N; A es metilo o H; R5 y R6 se seleccionan independientemente de halógeno, H, alquilo, arilo, triclorometilo y trifluorometilo; R3 y R4 están independientemente ausentes o seleccionados del grupo que consiste en alquilo, cicloalquilo, alcoxi, tioalquilo; y cuando X1 es C, entonces R0 no está monosustituido con fenilo en la posición para con -OCHF2, o una sal farmacéuticamente aceptable del mismo, un tautómero del mismo, una sal farmacéuticamente aceptable de un tautómero del mismo, un enantiómero del mismo o una sal farmacéuticamente aceptable de un enantiómero del mismo.
Ejemplo 2: El compuesto del ejemplo 1, en donde dicho compuesto es un compuesto de acuerdo con la fórmula:

Ejemplo 3 : El compuesto del ejemplo 1, en donde dicho compuesto es un compuesto de acuerdo con la Fórmula:

Ejemplo 4: El compuesto de acuerdo con uno cualquiera de los ejemplos 1-3, en donde R5 y R6 se seleccionan independientemente de halógeno, H, alquilo, triclorometilo y trifluorometilo.
Ejemplo 5: El compuesto de acuerdo con uno cualquiera de los ejemplos 1-4, en donde: X1 se selecciona del grupo que consiste en CH y N; y R5 y R6 son halógenos seleccionados independientemente.
Ejemplo 6: El compuesto de acuerdo con uno cualquiera de los ejemplos 1-5, en donde R7 es C=NH.
Ejemplo 7: El compuesto de acuerdo con uno cualquiera de los ejemplos 1-5, en donde R7 es C=O.
Ejemplo 8: El compuesto del ejemplo 6, en donde dicho compuesto es un compuesto que tiene la fórmula:

Ejemplo 9: El compuesto del ejemplo 8, en donde R5 y R6 son halógenos seleccionados independientemente. Ejemplo 10: El compuesto del ejemplo 9, en donde R5 y R6 son el mismo halógeno.
Ejemplo 11: El compuesto del ejemplo 9, en donde R5 y R6 son ambos F.
Ejemplo 12: El compuesto del ejemplo 7, en donde dicho compuesto es un compuesto que tiene la fórmula:

Ejemplo 13: El compuesto del ejemplo 7, en donde dicho compuesto es un compuesto de la fórmula:

Ejemplo 14: El compuesto del ejemplo 7, en donde dicho compuesto es un compuesto de la fórmula:

Ejemplo 15: El compuesto del ejemplo 3, en donde R7 es C=S.
Ejemplo 16: El compuesto de acuerdo con uno cualquiera de los ejemplos 1-5, en donde R7 es C-NH2 y dicho compuesto es un compuesto que tiene la fórmula:

Ejemplo 17: El compuesto del ejemplo 16, en donde dicho compuesto es un compuesto de Fórmula:

donde R1 y R2 están independientemente ausentes o seleccionados del grupo que consiste en alquilo, haloalquilo, cicloalquilo, alquenilo, alquinilo, alcoxi, tioalquilo, arilo, arilo sustituido, heteroarilo y heteroarilo sustituido; y X2, Y y Z son independientemente CH o N.
Ejemplo 18: El compuesto de acuerdo con uno cualquiera de los ejemplos 1-17, en donde R5 y R6 son halógenos diferentes.
Ejemplo 19: El compuesto de acuerdo con uno cualquiera de los ejemplos 1-17, en donde R5 y R6 son el mismo halógeno.
Ejemplo 20: El compuesto de acuerdo con uno cualquiera de los ejemplos 1-19, en donde R5 y R6 son independientemente Cl o F.
Ejemplo 21: El compuesto del ejemplo 17, en donde dicho compuesto es un compuesto que tiene la fórmula:

Ejemplo 22: El compuesto de acuerdo con uno cualquiera de los ejemplos 1-7 y 15-21, en donde X1 es CH.
Ejemplo 23: El compuesto de acuerdo con uno cualquiera de los ejemplos 1-7 y 15-22, en donde R8 es H o CH3. La presente invención proporciona un compuesto que tiene la Fórmula:

Ejemplo 25: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 26: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

La presente invención también proporciona un compuesto que tiene la fórmula:

Ejemplo 28: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 29: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 30: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 31: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 32: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 33: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 34: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 35: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

La presente invención proporciona además un compuesto que tiene la fórmula:

(FAH-17).
Ejemplo 37: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 38: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 39: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:
H-jí-j. J

Ejemplo 40: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 41: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 42: El compuesto del ejemplo 3, en donde dicho compuesto es un compuesto que tiene la Fórmula:

Ejemplo 43: Un compuesto de Fórmula:

o una sal farmacéuticamente aceptable del mismo, un tautómero del mismo, una sal farmacéuticamente aceptable de un tautómero del mismo, un enantiómero del mismo, o una sal farmacéuticamente aceptable de un enantiómero del mismo.
El compuesto de acuerdo con uno cualquiera de los compuestos FAH-1, FAH-3 o FAH-17, en donde dicho compuesto es un enantiómero S sustancialmente puro.
El compuesto de acuerdo con uno cualquiera de los compuestos FAH-1, FAH-3 o FAH-17, en donde dicho compuesto es un enantiómero R sustancialmente puro.
El compuesto de acuerdo con uno cualquiera de los compuestos FAH-1, FAH-3 o FAH-17, en donde dicho compuesto se une a APP y/o a la enzima BACE y/o a un complejo APP/BACE.
El compuesto de acuerdo con uno cualquiera de los compuestos FAH-1, FAH-3 o FAH-17, en donde dicho compuesto se une a APP e inhibe la enzima BACE.
Una formulación farmacéutica que incluye un vehículo farmacéuticamente aceptable y un compuesto de acuerdo con uno cualquiera de los compuestos FAH-1, FAH-3 o FAH-17.
Preferiblemente, dicha formulación se combina para la administración a través de una ruta seleccionada del grupo que consiste en administración oral, administración isoforética, administración transdérmica, administración parenteral, administración en aerosol, administración por inhalación, administración intravenosa y administración rectal.
Además, preferiblemente dicha formulación se compone para administración oral.
Además, Preferiblemente dicha formulación es estéril.
Preferiblemente, la formulación puede ser una formulación de dosificación unitaria.
Los compuestos y formulaciones mencionados anteriormente son adecuados para su uso en la prevención o retraso de la aparición de una afección pre-Alzheimer y/o disfunción cognitiva, y/o para mejorar uno o más síntomas de una afección pre-Alzheimer y/o disfunción cognitiva, o para prevenir o retrasar la progresión de una afección pre-Alzheimer o disfunción cognitiva a la enfermedad de Alzheimer, dicho método incluye: administrar a un sujeto que lo necesite un compuesto de acuerdo con una cualquiera de las realizaciones 1-47, o formulación de acuerdo con cualquiera de realizaciones 48-52 en una cantidad suficiente para prevenir o retrasar la aparición de una disfunción cognitiva pre-Alzheimer, y/o mejorar uno o más síntomas de una disfunción cognitiva pre-Alzheimer, y/o prevenir o retrasar la progresión de un disfunción cognitiva pre-Alzheimer para la enfermedad de Alzheimer.
En particular, dichos compuestos y formulaciones son adecuados para su uso en la prevención o retraso de la transición de una condición pre-Alzheimer cognitivamente asintomática a una disfunción cognitiva pre-Alzheimer. Los compuestos y formulaciones mencionados anteriormente son adecuados para su uso en la prevención o el retraso de la aparición de una disfunción cognitiva previa al Alzheimer.
Los compuestos y formulaciones mencionados anteriormente son adecuados para su uso en la mejora de uno o más síntomas de una disfunción cognitiva previa al Alzheimer.
Los compuestos y formulaciones mencionados anteriormente son adecuados para su uso en la prevención o retraso de la progresión de una disfunción cognitiva pre-Alzheimer a la enfermedad de Alzheimer.
Preferiblemente, dicho sujeto es un humano.
Además, preferiblemente dicho sujeto exhibe positividad de biomarcador de Ap en un sujeto humano clínicamente normal de 50 años o más.
Preferiblemente, dicho sujeto exhibe amiloidosis cerebral asintomática.
Preferiblemente, dicho sujeto exhibe amiloidosis cerebral en combinación con neurodegeneración descendente. Preferiblemente, dicho sujeto exhibe amiloidosis cerebral en combinación con neurodegeneración descendente y un deterioro cognitivo/conductual sutil.
Además, preferiblemente dicha neurodegeneración descendente está determinada por uno o más marcadores elevados de lesión neuronal seleccionados del grupo que consiste en tau y absorción de FDG.
Con fines informativos, la amiloidosis cerebral puede determinarse mediante PET o análisis de CSF y resonancia magnética estructural (IRSM).
Preferiblemente, dicho sujeto es un sujeto diagnosticado con deterioro cognitivo leve.
Además, preferiblemente dicho sujeto muestra una clasificación de demencia clínica por encima de cero y por debajo de aproximadamente 1.5.
Preferiblemente, el sujeto es humano.
Preferiblemente, el sujeto está en riesgo de desarrollar la enfermedad de Alzheimer.
Además, preferiblemente el sujeto tiene un riesgo familiar de tener la enfermedad de Alzheimer.
Aún más preferiblemente, el sujeto tiene una mutación familiar de la enfermedad de Alzheimer (EAF).
Incluso más preferiblemente, el sujeto tiene el alelo APOE £4.
Preferiblemente, la administración de dicho compuesto retrasa o evita la progresión de MCI a la enfermedad de Alzheimer.
En algunas situaciones descritas aquí, el sujeto está libre y no tiene factores de riesgo genético de la enfermedad de Parkinson o la esquizofrenia.
En algunas situaciones descritas en el presente documento, no se diagnostica que el sujeto tenga o esté en riesgo de enfermedad de Parkinson o esquizofrenia.
En algunas situaciones descritas en el presente documento, el sujeto no es diagnosticado como en riesgo de una enfermedad o trastorno neurológico distinto de la enfermedad de Alzheimer.
Preferiblemente, dicha administración produce una reducción en el CSF de los niveles de uno o más componentes seleccionados del grupo que consiste en Ap42, sAPPp, Tau total (tTau), fosfo-Tau (pTau), APPneo, Ap40 soluble, relación pTau/Ap42 y relación tTau/Ap42, y/o un aumento en el CSF de los niveles de uno o más componentes seleccionados del grupo que consiste en relación Ap42/Ap40, relación Ap42/Ap38, sAPPa, relación sAPPa/sAPPp, relación sAPPa/Ap40 y relación sAPPa/Ap42.
Preferiblemente, dicha administración produce una reducción de la carga de placa en el cerebro del sujeto.
Preferiblemente, dicha administración produce una reducción en la velocidad de formación de placa en el cerebro del sujeto.
Preferiblemente, dicha administración produce una mejora en las capacidades cognitivas del sujeto.
Preferiblemente, dicha administración produce una mejora, una estabilización o una reducción en la rata de declinación de la clasificación de demencia clínica (CDR) del sujeto.
Preferiblemente, el sujeto es un humano y dicha administración produce una mejora percibida en la calidad de vida del ser humano.
Como se indicó anteriormente, el compuesto o formulación se puede administrar a través de una ruta seleccionada del grupo que consiste en administración oral, administración isoforética, administración transdérmica, administración parenteral, administración en aerosol, administración por inhalación, administración intravenosa y administración rectal.
Como se indicó anteriormente, el compuesto o formulación se administra por vía oral.
En algunos casos, la administración es durante un período de al menos tres semanas.
En otros casos, la administración es durante un período de al menos 6 meses.
En el presente documento se describe un ejemplo de un método para mejorar uno o más síntomas de la enfermedad de Alzheimer, y/o revertir la enfermedad de Alzheimer, y/o reducir la tasa de progresión de la enfermedad de Alzheimer, incluyendo dicho método: administrar a un sujeto que así lo requiere un compuesto de acuerdo con una cualquiera de las realizaciones 1-47, o formulación de acuerdo con una cualquiera de las realizaciones 48-52 en una cantidad suficiente para mejorar uno o más síntomas de la enfermedad de Alzheimer, y/o revertir la enfermedad de Alzheimer, y/o reducir la tasa de progresión de la enfermedad de Alzheimer.
El método del ejemplo anterior, en donde dicho sujeto es un humano.
El método del ejemplo anterior, en donde dicho sujeto es un humano de al menos 50 años.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde dicho sujeto es diagnosticado con enfermedad de Alzheimer en etapa temprana.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde dicho sujeto es diagnosticado con la enfermedad de Alzheimer en la etapa media.
El método de acuerdo con cualquiera de los ejemplos anteriores, en donde dicho sujeto es diagnosticado con enfermedad de Alzheimer en etapa tardía.
El método de acuerdo con cualquiera de los ejemplos anteriores, en donde dicha administración reduce la gravedad de la enfermedad de Alzheimer.
El método de acuerdo con cualquiera de los ejemplos anteriores, en donde dicha administración mejora uno o más síntomas de la enfermedad de Alzheimer.
El método de acuerdo con cualquiera de los ejemplos anteriores, en donde dicha administración reduce la tasa de progresión de la enfermedad de Alzheimer.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde dicha administración da como resultado una reducción en el CSF de los niveles de uno o más componentes seleccionados del grupo que consiste en Ap42,
sAPPp, Tau total (tTau), fosfo-Tau (pTau), APPneo, Ap40 soluble, relación pTau/Ap42 y relación tTau/Ap42, y/o un aumento en el CSF de los niveles de uno o más componentes seleccionados del grupo que consiste en la relación Ap42/Ap40, relación Ap42/Ap38, sAPPa, relación sAPa/sAPPp, relación sAPPa/Ap40 y relación sAPPa/Ap42. es un método para prevenir o retrasar la transición de una condición pre-Alzheimer cognitivamente asintomática a una disfunción cognitiva pre-Alzheimer.
El método de acuerdo con cualquiera de los ejemplos anteriores, en donde dicha administración produce una reducción de la carga de placa en el cerebro del sujeto.
El método de acuerdo con cualquiera de los ejemplos anteriores, en donde dicha administración produce una reducción en la velocidad de formación de placa en el cerebro del sujeto.
El método según cualquiera de los ejemplos anteriores, en donde dicha administración produce una mejora en las capacidades cognitivas del sujeto.
El método según cualquiera de los ejemplos anteriores, en donde dicha administración produce una mejora, una estabilización o una reducción en la rata de declinación de la clasificación de demencia clínica (CDR) del sujeto.
El método de acuerdo con cualquiera de los ejemplos anteriores, en donde el sujeto es un humano y dicha administración produce una mejora percibida en la calidad de vida del ser humano.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde dicha administración da como resultado una amiloidosis cerebral reducida y/o neurodegeneración descendente.
El método descrito anteriormente, en donde dicha neurodegeneración descendente está determinada por uno o más marcadores de lesión neuronal seleccionados del grupo que consiste en tau, captación de FDG, disminución de sAPPalpha, aumento de sAPPbeta y Abeta.
El método descrito anteriormente, en donde dicha amiloidosis cerebral se determina mediante PET usando agentes de unión amiloide/tau, análisis de CSF y resonancia magnética estructural (sMRI).
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde dicho sujeto muestra una clasificación de demencia clínica indicativa de la enfermedad de Alzheimer.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde el sujeto tiene un riesgo familiar de tener la enfermedad de Alzheimer.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde el sujeto tiene una mutación de la enfermedad de Alzheimer familiar (EAF).
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde el sujeto tiene el alelo APOE £4.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde el sujeto está libre y no tiene factores de riesgo genético de la enfermedad de Parkinson o la esquizofrenia.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde no se diagnostica al sujeto que tiene o está en riesgo de enfermedad de Parkinson o esquizofrenia.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde el sujeto no tiene una enfermedad o trastorno neurológico distinto de la enfermedad de Alzheimer.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde no se diagnostica al sujeto que tiene o está en riesgo de una enfermedad o trastorno neurológico distinto de la enfermedad de Alzheimer.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde el compuesto se administra a través de una ruta seleccionada del grupo que consiste en administración oral, administración isoforética, administración transdérmica, administración parenteral, administración en aerosol, administración por inhalación, administración intravenosa, y administración rectal.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde el compuesto se formula para la administración a través de una ruta seleccionada del grupo que consiste en administración oral, administración isoforética, administración transdérmica, administración parenteral, administración en aerosol, administración por inhalación, intravenosa administración y administración rectal.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde el compuesto se administra por vía oral.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde la administración es durante un período de al menos tres semanas.
El método de acuerdo con cualquiera de los ejemplos anteriores, en donde la administración es durante un período de al menos 6 meses.
El método de acuerdo con uno cualquiera de los ejemplos anteriores, en donde dicho compuesto se administra en combinación con uno o más agentes seleccionados del grupo que consiste en disulfiram y/o análogos del mismo, honokiol y/o análogos del mismo, tropisetrón y/o análogos del mismo, nimetazepam y/o análogos del mismo, ésteres de tropinol y/o ésteres relacionados y/o análogos del mismo, inhibidores de la quinasa TrkA (por ejemplo, ADDN-1351) y/o análogos del mismo, agonistas del receptor D2, antagonistas del receptor adrenérgico alfal, y los inhibidores de BACE específicos de la aplicación que incluyen, pero no se limitan a galangina, un profármaco de galangina, rutina, un profármaco de rutina y otros flavonoides y profármacos flavonoides.
El método descrito anteriormente, en donde dicho compuesto se administra en combinación con tropisetrón.
Un método para ralentizar la progresión, detener o revertir la degeneración macular relacionada con la edad (DMAE) en un mamífero, dicho método incluye administrar a dicho mamífero un compuesto descrito aquí anteriormente en una cantidad suficiente para ralentizar la progresión, detener o revertir la degeneración macular relacionada con la edad en dicho mamífero.
En el presente documento se describe un método para el tratamiento de una enfermedad o trastorno asociado con la actividad BACE en un sujeto que lo necesita, en donde dicho método incluye proporcionar a dicho sujeto una cantidad terapéuticamente eficaz de un compuesto o formulación descrita anteriormente en el presente documento.
Además, dicha enfermedad o trastorno se selecciona del grupo que consiste en la enfermedad de Alzheimer; deterioro cognitivo, síndrome de Down, HCHWA-D, deterioro cognitivo, demencia senil, angiopatía amiloide cerebral y un trastorno neurodegenerativo.
Además, dicha enfermedad o trastorno se caracteriza por la producción de depósitos amiloides y/o nudos neurofibrilares.
Definiciones
A menos que se indique lo contrario, la referencia a un compuesto (por ejemplo, a una hidantoína como se describe en el presente documento) debe interpretarse en términos generales para incluir sales farmacéuticamente aceptables, profármacos, tautómeros, formas sólidas alternativas, complejos no covalentes y combinaciones de los mismos, de entidad química de la estructura representada o nombre químico.
En general, la referencia a un determinado elemento tal como hidrógeno o H debe incluir todos los isótopos de ese elemento. Por ejemplo, si un grupo R se define para incluir hidrógeno o H, también incluye deuterio y tritio. Por consiguiente, los compuestos marcados isotópicamente están dentro del alcance de esta invención.
Una sal farmacéuticamente aceptable es cualquier sal del compuesto original que sea adecuada para la administración a un animal o humano. Una sal farmacéuticamente aceptable también se refiere a cualquier sal que pueda formarse in vivo como resultado de la administración de un ácido, otra sal o un profármaco que se convierte en un ácido o sal. Una sal comprende una o más formas iónicas del compuesto, como un ácido o base conjugado, asociado con uno o más contraiones correspondientes. Las sales se pueden formar o incorporar uno o más grupos ácidos desprotonada (por ejemplo, ácidos carboxílicos), uno o más grupos básicos protonados (por ejemplo, aminas) o ambos (por ejemplo, zwitteriones).
Un profármaco es un compuesto que se convierte en un compuesto terapéuticamente activo después de la administración. Por ejemplo, la conversión puede ocurrir por hidrólisis de un grupo éster, tal como un éster alquílico C1-C6 del grupo ácido carboxílico de los presentes compuestos, o algún otro grupo biológicamente lábil. La preparación de profármacos es bien conocida en la técnica. Por ejemplo, "Prodrugs and Drug Delivery Systems", que es un capítulo en Richard B. Silverman, Organic Chemistry of Drug Design and Drug Action, 2d Ed., Elsevier Academic Press: Amsterdam, 2004, pp. 496-557, proporciona más detalles sobre el tema.
Los tautómeros son isómeros que están en equilibrio entre sí. Por ejemplo, los tautómeros pueden estar relacionados por transferencia de un protón, un átomo de hidrógeno o un ion hidruro.
A menos que se describa explícitamente la estereoquímica, se pretende que una estructura incluya todos los estereoisómeros posibles, tanto puros como en cualquier mezcla posible.
Las formas sólidas alternativas son formas sólidas diferentes a las que pueden resultar de la práctica de los procedimientos descritos en el presente documento. Por ejemplo, formas sólidas alternativas pueden ser polimorfos, diferentes tipos de formas sólidas amorfas, gafas y similares. En diversas realizaciones, se contemplan formas sólidas alternativas de cualquiera de los compuestos descritos en el presente documento.
En general, "sustituido" se refiere a un grupo orgánico como se define a continuación (por ejemplo, un grupo alquilo) en donde uno o más enlaces a un átomo de hidrógeno contenido en el mismo se reemplazan por un enlace a átomos que no son de hidrógeno o de carbono. Los grupos sustituidos también incluyen grupos en los que uno o más enlaces
a un(unos) átomo(s) de carbono o hidrógeno se reemplazan por uno o más enlaces, incluidos enlaces dobles o triples, a un heteroátomo. Por lo tanto, un grupo sustituido estará sustituido con uno o más sustituyentes, a menos que se especifique lo contrario. En algunas realizaciones, un grupo sustituido está sustituido con 1,2, 3, 4, 5 o 6 sustituyentes. Ejemplos de grupos sustituyentes incluyen: halógenos (es decir, F, Cl, Br e I); hidroxilos; grupos alcoxi, alquenoxi, alquinoxi, ariloxi, aralquiloxi, heterocicliloxi y heterociclilalcoxi; carbonilos (oxo); carboxilos; ésteres uretanos; oximas; hidroxilaminas; alcoxiaminas; aralcoxiaminas; tioles; sulfuros; sulfóxidos; sulfonas; sulfonilos; sulfonamidas; aminas; N-óxidos; hidrazinas; hidrazidas; hidrazonas; azidas; amidas ureas amidinas; guanidinas; enaminas; imidas; isocianatos; isotiocianatos; cianatos; tiocianatos; iminas grupos nitro; nitrilos (es decir, CN) y similares.
El término "alquilo" se refiere y cubre cualquiera y todos los grupos que se conocen como alquilo normal, alquilo de cadena ramificada, cicloalquilo y también cicloalquil-alquilo. Los grupos alquilo ilustrativos incluyen, pero sin limitación, metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, t-butilo, octilo y decilo. El término "cicloalquilo" se refiere a grupos hidrocarbilo saturados cíclicos, incluidos los cíclicos. Ejemplos incluyen, pero no se limitan a ciclopentilo, ciclohexilo, diciclopentilo, norbornilo, octahidronaptilo y espiro[3.4]octilo. En ciertas realizaciones, los grupos alquilo contienen 1-12 átomos de carbono (alquilo C1-12), o 1-9 átomos de carbono (alquilo C1-9), o 1-6 átomos de carbono (alquilo C1-6), o 1-5 carbono átomos (alquilo C1-5), o átomos de carbono (alquilo C1-4), o 1-3 átomos de carbono (alquilo C1-3), o 1-2 átomos de carbono (alquilo C1-2).
A modo de ejemplo, el término "grupo alquilo C W se refiere a un grupo alquilo de cadena lineal o ramificada que tiene de 1 a 6 átomos de carbono, y puede ejemplificarse por un grupo metilo, un grupo etilo, un grupo n-propilo, un grupo isopropilo, un grupo n-butilo, un grupo isobutilo, un grupo tert-butilo, un grupo sec-butilo, un grupo n-pentilo, un grupo tert-amilo, un grupo 3-metilbutilo, un grupo neopentilo , y un grupo n-hexilo.
El término "alcoxi" como se usa en el presente documento significa un grupo alquilo unido a través de un único átomo de oxígeno terminal. Un grupo "alcoxi" puede representarse como -O-alquilo donde alquilo es como se definió anteriormente. El término "ariloxi" se usa de manera similar, y puede representarse como -O-arilo, con arilo como se define a continuación. El término "hidroxi" se refiere a -OH.
De manera similar, el término "alquiltio" como se usa en el presente documento significa un grupo alquilo unido a través de un único átomo de azufre terminal. Un grupo "alquiltio" puede representarse como -S-alquilo donde alquilo es como se definió anteriormente. El término "ariltio" se usa de manera similar, y puede representarse como -S-arilo, con arilo como se define a continuación. El término "mercapto" se refiere a --SH.
Los grupos arilo son hidrocarburos aromáticos cíclicos que no contienen heteroátomos. Los grupos arilo incluyen sistemas de anillos monocíclicos, bicíclicos y policíclicos. Por lo tanto, los grupos arilo incluyen, pero no se limitan a, grupos fenilo, azulenilo, heptalenilo, bifenilenilo, indacenilo, fluorenilo, fenantrenilo, trifenilenilo, pirenilo, naftacenilo, crisenilo, bifenilo, antracenilo, indenilo, indanilo, pentalenilo y naftilo. En algunas realizaciones, los grupos arilo contienen 6-14 carbonos, y en otros de 6 a 12 o incluso 6-10 átomos de carbono en las porciones de anillo de los grupos. Aunque la expresión "grupos arilo" incluye grupos que contienen anillos fusionados, tales como sistemas de anillos aromáticos alifáticos fusionados (por ejemplo, indanilo, tetrahidronaftilo y similares), no incluye grupos arilo que tienen otros grupos, como grupos alquilo o halo, unido a uno de los miembros del anillo. Por el contrario, los grupos como el tolilo se denominan grupos arilo sustituidos. Los grupos arilo sustituidos representativos pueden estar monosustituidos o sustituidos más de una vez. Por ejemplo, los grupos arilo monosustituidos incluyen, pero no se limitan a, grupos fenilo o naftilo sustituidos con 2-, 3-, 4-, 5- o 6-, que pueden estar sustituidos con sustituyentes tales como los enumerados anteriormente.
El término "grupo heteroarilo" se refiere a un grupo heterocíclico aromático monocíclico o de anillo condensado que contiene uno o más heteroátomos seleccionados de O, S y N. Si el grupo heterocíclico aromático tiene un anillo condensado, puede incluir parcialmente grupo monocíclico hidrogenado. Ejemplos de dicho grupo heteroarilo incluyen un grupo pirazolilo, un grupo tiazolilo, un grupo isotiazolilo, un grupo tiadiazolilo, un grupo imidazolilo, un grupo furilo, un grupo tienilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo pirrolilo, un grupo imidazolilo, un grupo (1,2,3)- y (1,2,4)-triazolilo, un grupo tetrazolilo, un grupo piranilo, un grupo piridilo, un grupo pirimidinilo, un grupo pirazinilo, un grupo piridazinilo, un grupo quinolilo, un grupo isoquinolilo, un grupo benzofuranilo, un grupo isobenzofuranilo, un grupo indolilo, un grupo isoindolilo, un grupo indazolilo, un grupo benzoimidazolilo, un grupo benzotriazolilo, un grupo benzoxazolilo, un grupo benzotiazolilo, un grupo benzo[b]tiofenilo, un grupo tieno[2,3-b]tiofenilo, un grupo (1,2)- y (1,3)-benzoxatiol, un grupo cromenilo, un grupo 2-oxocromenilo, un grupo benzotiadiazolilo, un grupo quinolizinilo, un grupo ftalazinilo, un grupo naftiridinilo, un grupo quinoxalinilo, un grupo quinazolinilo, un grupo cinolinilo y un grupo carbazolilo.
Un "derivado" de un compuesto significa un compuesto modificado químicamente en donde la modificación química tiene lugar en uno o más grupos funcionales del compuesto. Sin embargo, se espera que el derivado retenga o mejore la actividad farmacológica del compuesto del que se deriva.
Como se usa en el presente documento, "administrar" se refiere a la administración local y sistémica, por ejemplo, que incluye la administración enteral, parenteral, pulmonar y tópica/transdérmica. Vías de administración para los agentes (por ejemplo, hidantoínas descritas en el presente documento, o uno o más tautómeros o estereoisómeros de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoínas, dichos estereoisómeros o dichos
tautómeros o análogos, derivados o profármacos de los mismos) que encuentran uso en los métodos descritos en el presente documento incluyen, por ejemplo, administración oral (per os (p.o.)), administración nasal o por inhalación, administración como supositorio, contacto tópico, administración transdérmica (por ejemplo, mediante un parche transdérmico), administración intratecal (IT), administración intravenosa ("iv"), administración intraperitoneal ("ip"), administración intramuscular ("im"), administración intralesional o administración subcutánea ("sc"), o la administración implantación de un dispositivo de liberación lenta, por ejemplo, una minibomba osmótica, una formulación de depósito, etc., a un sujeto. La administración puede ser por cualquier vía, incluida la parenteral y la transmucosa (por ejemplo, oral, nasal, vaginal, rectal o transdérmica). La administración parenteral incluye, por ejemplo, intravenosa, intramuscular, intraarterial, intradérmica, subcutánea, intraperitoneal, intraventricular, ionoforética e intracraneal. Otros modos de administración incluyen, entre otros, el uso de formulaciones liposómicas,, infusión intravenosa, parches transdérmicos, etc.
Los términos "administración sistémica" y "administrado sistémicamente" se refieren a un método para administrar el (los) agente(s) descrito(s) aquí o la composición a un mamífero para que el (los) agente(s) o la composición se entreguen a sitios en el cuerpo, incluyendo el sitio objetivo de la acción farmacéutica, a través del sistema circulatorio. La administración sistémica incluye, pero no se limita a, administración oral, intranasal, rectal y parenteral (por ejemplo, que no sea a través del tracto alimentario, como la administración intramuscular, intravenosa, intraarterial, transdérmica y subcutánea).
El término "coadministrar" o "administración concurrente" o "administrar conjuntamente con" cuando se usa, por ejemplo con respecto al (a los) agente(s) activo(s) aquí descrito, por ejemplo, hidantoínas descritas aquí, o un tautómero(s) o estereoisómero(s) del mismo, o sales o solvatos farmacéuticamente aceptables de dicha hidantoína(s), dicho(s) estereoisómero(s), o dicho tautómero(s), o análogos, derivados o profármacos de los mismos y un segundo agente activo (por ejemplo, un potenciador de la cognición), se refiere a la administración del (de los) agente(s) y/o el segundo agente activo de manera que ambos puedan alcanzar simultáneamente un efecto fisiológico. Los dos agentes, sin embargo, no necesitan administrarse juntos. En ciertas realizaciones, la administración de un agente puede preceder a la administración del otro. El efecto fisiológico simultáneo no necesariamente requiere la presencia de ambos agentes en la circulación al mismo tiempo. Sin embargo, en ciertas realizaciones, la administración conjunta típicamente da como resultado que ambos agentes estén presentes simultáneamente en el cuerpo (por ejemplo, en el plasma) en una fracción significativa (por ejemplo, 20% o más, preferiblemente 30% o 40% o más, más preferiblemente 50% o 60% o más, lo más preferiblemente 70% u 80% o 90% o más) de su concentración sérica máxima para cualquier dosis dada.
El término "cantidad efectiva" o "cantidad farmacéuticamente efectiva" se refiere a la cantidad y/o dosificación, y/o régimen de dosificación de uno o más agentes necesarios para lograr el resultado deseado, por ejemplo, una cantidad suficiente para mitigar en un mamífero uno o más síntomas asociados con un deterioro cognitivo leve (DCL), o una cantidad suficiente para disminuir la gravedad o retrasar la progresión de una enfermedad caracterizada por depósitos amiloides en el cerebro en un mamífero (por ejemplo, cantidades terapéuticamente efectivas), una cantidad suficiente para reducir el riesgo o retrasar la aparición, y/o reducir la gravedad final de una enfermedad caracterizada por depósitos de amiloide en el cerebro en un mamífero (por ejemplo, cantidades profilácticamente efectivas).
La expresión "hacer que se administre" se refiere a las acciones tomadas por un profesional médico (por ejemplo, un médico), o una persona que controla la atención médica de un sujeto, que controla y/o permite la administración del agente(s) en cuestión al sujeto. Hacer que se administre puede implicar el diagnóstico y/o la determinación de un régimen terapéutico o profiláctico apropiado, y/o la prescripción de agentes particulares para un sujeto. Dicha prescripción puede incluir, por ejemplo, redactar un formulario de prescripción, anotar un registro médico y similares.
Como se usa en el presente documento, los términos "tratar" y "tratamiento" se refieren a retrasar el inicio, retrasar o revertir el progreso de, reducir la gravedad o aliviar o prevenir la enfermedad o afección a la que se aplica el término, o uno o más síntomas de dicha enfermedad o afección.
El término "mitigar" se refiere a la reducción o eliminación de uno o más síntomas de esa patología o enfermedad, y/o una reducción en la tasa o retraso de aparición o gravedad de uno o más síntomas de esa patología o enfermedad, y/o la prevención de esa patología o enfermedad. En ciertas realizaciones, la reducción o eliminación de uno o más síntomas de patología o enfermedad puede incluir, pero no se limita a, reducción o eliminación de uno o más marcadores que son característicos de la patología o enfermedad (por ejemplo, de Tau total ( tTau), fosfo-Tau (pTau), APPneo, Ap40 soluble, relación pTau/Ap42 y relación tTau/Ap42, y/o un aumento en el CSF de los niveles de uno o más componentes seleccionados del grupo que consiste en la relación Ap42/Ap40, relación Ap42/Ap38, sAPPa, relación sAPPa/sAPPp, relación sAPPa/Ap40, relación sAPPa/Ap42, etc.) y/o reducción, estabilización o reversión de uno o más criterios de diagnóstico (por ejemplo, clasificación de demencia clínica (CDR)).
Como se usa en el presente documento, la expresión "que consiste esencialmente en 'se refiere a los géneros o especies de agentes farmacéuticos activos mencionados en un método o composición, y además puede incluir otros agentes que, por sí mismos, no tienen actividad sustancial para la indicación mencionada o propósito. En algunas realizaciones, la expresión "que consiste esencialmente en' excluye expresamente la inclusión de uno o más agentes adicionales que tienen actividad neurofarmacológica distinta del (de los) agente(s) recitado (por ejemplo, distinto de ASBI tales como galangina, rutina y análogos, derivados o profármacos de los mismos). En algunas realizaciones, la
expresión "que consiste esencialmente en' excluye expresamente la inclusión de uno o más agentes activos adicionales distintos del (los) agente(s) activo(s) aquí descritos (por ejemplo, distintos de ASBI tales como galangina, rutina y análogos, derivados o profármacos de los mismos) .En algunas realizaciones, la expresión "que consiste esencialmente en' excluye expresamente la inclusión de uno o más inhibidores de acetilcolinesterasa.
Los términos "sujeto", "individuo" y "paciente" se refieren indistintamente a un mamífero, preferiblemente un primate humano o no humano, pero también mamíferos domesticados (por ejemplo, caninos o felinos), mamíferos de laboratorio (por ejemplo, ratón, rata, conejo, hámster, conejillo de indias) y mamíferos agrícolas (por ejemplo, equinos, bovinos, porcinos, ovinos). En diversas realizaciones, el sujeto puede ser un humano (por ejemplo, un hombre adulto, una mujer adulta, un hombre adolescente, una mujer adolescente, un niño, una niña) bajo el cuidado de un médico u otro trabajador de la salud en un hospital, centro de atención psiquiátrica, como un paciente ambulatorio u otro contexto clínico. En ciertas realizaciones, el sujeto puede no estar bajo el cuidado o la prescripción de un médico u otro trabajador de la salud.
El término "formulación" o "formulación de fármaco" o "forma de dosificación" o "formulación farmacéutica" como se usa en el presente documento se refiere a una composición que contiene al menos un agente terapéutico o medicamento para administración a un sujeto. En ciertas realizaciones, la forma de dosificación comprende una "formulación" o "formulación de fármaco" dada y puede administrarse a un paciente en forma de pastilla, píldora, tableta, cápsula, supositorio, membrana, tira, líquido, parche, película, gel, aspersión u otra forma.
El término "membrana mucosa" se refiere generalmente a cualquiera de las membranas biológicas recubiertas de moco en el cuerpo. En ciertas realizaciones, el (los) agente(s) activo(s) descrito(s) en el presente documento pueden administrarse en el presente documento a través de cualquier membrana mucosa encontrada en el cuerpo, que incluye, pero no se limita a, mucosa bucal, perlingual, nasal, sublingual, pulmonar, rectal y vaginal. La absorción a través de las membranas mucosas de la cavidad oral y las del intestino son de interés. Por lo tanto, la absorción peroral, bucal, sublingual, gingival y palatina se contempla aquí.
El término suministro "transmucosa" de un fármaco y similares pretende abarcar todas las formas de suministro a través o a través de una membrana mucosa.
El término "bioadhesión" como se usa en el presente documento se refiere al proceso de adhesión de la(s) forma(s) de dosificación a una superficie biológica, por ejemplo, membranas mucosas.
"Suministro controlado de fármaco" se refiere a la liberación o administración de un fármaco desde una forma de dosificación dada de una manera controlada para lograr el perfil farmacocinético deseado in vivo. Un aspecto de la administración "controlada" de fármacos es la capacidad de manipular la formulación y/o la forma de dosificación para establecer la cinética deseada de liberación del fármaco.
"Suministro sostenido de fármaco" se refiere a la liberación o administración de un fármaco de una fuente (por ejemplo, una formulación de fármaco) de manera sostenida durante un período de tiempo prolongado pero específico, que puede extenderse de varios minutos a unas pocas horas, días, semanas o meses. En diversas realizaciones, el término "sostenido" se usará para referirse a la administración de niveles consistentes y/o sustancialmente constantes de fármaco durante un período de tiempo que varía de unos pocos minutos a un día, con un perfil caracterizado por la ausencia de una fase de liberación inmediata, como el obtenido de la administración IV.
El término "Tmax" como se usa en el presente documento significa el punto de tiempo de la concentración plasmática máxima observada.
El término "Cmax" como se usa en el presente documento significa la concentración plasmática máxima observada.
El término "plasma W como se usa en el presente documento significa la "vida media plasmática observada" y representa el tiempo requerido para que la concentración plasmática del fármaco alcance el 50% de su valor máximo (Cmáx). Esto facilita la determinación de la duración media de los efectos farmacológicos. Además, facilita las comparaciones directas y significativas de la duración de diferentes artículos de prueba después de la entrega a través de la misma o diferentes rutas.
El término "Relación de orientación terapéutica óptima" u "OTTR" representa el tiempo promedio que el fármaco está presente a niveles terapéuticos, definido como el tiempo dentro del cual la concentración plasmática del fármaco se mantiene por encima del 50% de la Cmax normalizada por la mitad de eliminación del fármaco-vida multiplicada por la proporción de la Cmax obtenida en la forma de dosificación de interés sobre la Cmax después de la administración IV de dosis equivalentes y se calcula mediante la fórmula:
OTTR= C IVmax/ Cmax) x (Dosis/DosisIV) (Tiempo mayor a 50% de Cmax) / (TerminalIV vida media eliminación de fármaco)
El término "substancialmente puro" significa lo suficientemente homogéneo como para parecer libre de impurezas fácilmente detectables según lo determinado por métodos estándar de análisis, tales como cromatografía en capa fina (TLC), electroforesis en gel y cromatografía líquida de alto rendimiento (HPLC), utilizadas por el expertos en la técnica
para evaluar tal pureza, o suficientemente puro de modo que una purificación adicional no altere de manera detectable las propiedades físicas o químicas del compuesto. Los expertos en la técnica conocen métodos para la purificación de los compuestos para producir compuestos sustancialmente puros químicamente. Sin embargo, un compuesto sustancialmente puro químicamente puede ser una mezcla de estereoisómeros o isómeros. En tales casos, la purificación adicional podría aumentar la actividad específica del compuesto.
El término "sustancialmente puro" cuando se usa con respecto a los enantiómeros indica que un enantiómero particular (por ejemplo, un enantiómero S o un enantiómero R) está sustancialmente libre de su estereoisómero. En diversas realizaciones, sustancialmente puro indica que un enantiómero particular es al menos 70%, o al menos 80%, o al menos 90%, o al menos 95%, o al menos 98%, o al menos 99% del compuesto purificado. Los expertos en la técnica conocen bien los métodos para producir enantiómeros sustancialmente puros. Por ejemplo, se puede obtener un estereoisómero único, por ejemplo, un enantiómero, sustancialmente libre de su estereoisómero por resolución de la mezcla racémica usando un método tal como la formación de diastereómeros usando agentes de resolución ópticamente activos (Stereochemistry of Carbon Compounds, (1962) por E.L. Eliel, McGraw Hill; Lochmuller (1975) J. Chromatogr., 113(3): 283-302). Las mezclas racémicas de compuestos quirales pueden separarse y aislarse mediante cualquier método adecuado, que incluye, pero no se limita a: (1) formación de sales iónicas, diastereoméricas con compuestos quirales y separación por cristalización fraccionada u otros métodos, (2) formación de compuestos diastereoméricos con reactivos de derivación quiral, separación de los diastereómeros y conversión en estereoisómeros puros, y (3) separación de los estereoisómeros sustancialmente puros o enriquecidos directamente en condiciones quirales. Otro enfoque para la separación de los enantiómeros es usar una columna quiral Diacel y eluir usando una fase móvil orgánica como la realizada por Chiral Technologies (www.chiraltech.com) a cambio de una tarifa por servicio.
Breve descripción de los dibujos
La figura 1 ilustra varias hidantoínas.
La figura 2 ilustra varias hidantoínas.
Figura 3 modelos de interacción propuestos de la hidantoína con la región FLAP de BACE1. El panel inferior ilustra la interacción del sustituyente 3,4 del anillo B con el FLAP, Trp76 interrumpe el enlace Trp-76 — > Tyr-71 H haciendo que Tyr-71 se voltee hacia la izquierda e interactúe con el anillo A que contiene el difluoro.
La Figura 4 ilustra la unión del inhibidor de BACE de unión de APP (ABBI) FAH-3 a eAPP575-624 medido por cribado de resonancia de plasmón superficial (SPR). La afinidad de unión de los compuestos por el ectodominio de APP se determinó usando SPR. Se ha desarrollado una técnica para medir la afinidad de los compuestos con los fragmentos del ectodominio de APP. Para los experimentos de unión del compuesto 3 se usó un sustrato TRX-eAPP575-624. El eAPP se entrecruzó con los chips CM5 Biacore (GE Healthcare). El compuesto 3 a diversas concentraciones se usó en el flujo a través del chip y la señal de resonancia de plasmón se determinó usando un Biacore T100.
La Figura 5 ilustra la inhibición de la producción de Ap por FAH-3.
La Figura 6A ilustra la selectividad de ABBI para la inhibición de la escisión de APP-BACE en comparación con la escisión de PSGL-BACE que se muestra en la Figura 6B.
Descripción detallada
En diversas realizaciones, se identifican hidantoínas que parecen inhibir el procesamiento de APP mediada por psecretasa por un mecanismo novedoso. En particular, sin limitarse a una teoría particular, se cree que estas moléculas interactúan con BACE y/o con APP y/o con un complejo BACE/APP y, por lo tanto, inhiben la escisión de BACE del sustrato de la aplicación MBP-C125, lo que resulta en la inhibición de la producción de C99 y el sustrato del péptido del sitio p (P5-P5'). Además, las diversas hidantoínas identificadas en el presente documento inhiben Ap42 en células SHSY5Y de neuroblastoma. Además, se demostró que la actividad de las hidantoínas identificadas en el presente documento parece estar asociada con la unión a BACE y/o a APP, particularmente cuando estas unidades estructurales forman un complejo BACE/APP. Por consiguiente, se cree que los compuestos descritos en el presente documento representan una nueva clase de compuestos designados en el presente documento como inhibidores de unión a APP (ABBI) y proporcionan un nuevo mecanismo para modular el procesamiento de APP. Las hidantoínas descritas en el presente documento parecen mostrar una mejor permeabilidad cerebral e inhibición funcional de BACE.
Los ABBI son específicos para la APP y/o BACE y/o el complejo APP/BACE y se cree que muestran menos efectos secundarios no deseados porque los ABBI normalmente no son activos en otros sustratos para la enzima u otros complejos enzimáticos. Con respecto a los inhibidores de la Y-secretasa, los sustratos que no sean APP, como Notch, generan preocupación por los posibles efectos secundarios de la inhibición de la Y-secretasa, y el reciente fracaso del inhibidor de la Y-secretasa, Semagacestat, sirve para reforzar tales preocupaciones. Del mismo modo, en el caso de BACE, por ejemplo, la inhibición de sustratos no APP como PSGL1 o LRP podría producir efectos secundarios adversos. Por lo tanto, un inhibidor de BACE deseable sería aquel que se uniría/interactuaría no con BACE sino con APP, o con el complejo APP/BACE que conduce a la inhibición del complejo BACE específico de APP (ABBI).
Tales ABBIs potencialmente interactuarían con el complejo APP-BACE, por ejemplo, en la membrana y evitarían su transición al complejo "activo" en los endosomas tempranos, donde a pH <5 BACE está completamente activo. Se ha demostrado que algunos anticuerpos de unión al sitio p bloquean la división de APP por BACE y también funcionan en modelos animales de EA, sin embargo, para un desarrollo farmacéutico eficaz, se prefieren las moléculas orgánicas pequeñas a las biomoléculas relativamente grandes, como los anticuerpos.
Los datos que informamos aquí sobre la identificación de los primeros ABBI demuestran que tal enfoque es factible. Sin estar vinculados a una teoría particular, los ABBI parecen inhibir la actividad de BACE al interactuar con APP, en particular cuando se encuentran en un complejo ApP/BACE, inhibiendo así la escisión de BACE de la proteína precursora amiloide (APP) pero no la escisión proteolítica de otros sustratos. Se cree que tales terapias representan una nueva clase de terapias para la enfermedad de Alzheimer (u otra enfermedad amiloidogénica).
El sitio activo de BACE1 está cubierto por aletas. Una sola aleta de 14 residuos de longitud forma una estructura de horquilla a que es perpendicular a una hendidura que alberga el sitio activo y cubre la parte central de ese sitio activo. Durante el ciclo catalítico, las aletas se abren para permitir la entrada del sustrato (APP) en la hendidura catalítica y también para liberar productos hidrolíticos. Inicialmente, las hidantoínas descritas en el presente documento se produjeron mediante la introducción de un anillo dihalo (por ejemplo, difluoro) en la amino hidantoína del Compuesto 0 (mostrado en la Figura 1) para producir el compuesto 1 (también mostrado en la Figura 1). Sin limitarse a una teoría en particular, se cree que el anillo dihalógeno introduce una interacción FLAP (por ejemplo, una interacción con los residuos FLAP Tyr-71 (a través del apilamiento pi) y Trp-76 (a través de la interacción con OCF2)) y restringe el movimiento FLAP limitar la entrada de la aplicación en el sitio activo y/o la salida de los productos de escisión (véase, por ejemplo, la Figura 3). Esto proporciona una nueva familia de inhibidores de BACE (ABCI) pequeños (PM <400) penetrantes del cerebro.
Se produjeron el Compuesto 2 y otras hidantoínas (véase, por ejemplo, los compuestos 1-5, la evaluación farmacocinética de estas hidantoínas se determinó en ensayos de absorción cerebral usando ratones NTg (véase, por ejemplo, la Tabla 1). También se determinó que el Compuesto 1 disminuyó Ap42 en los mismos animales a 5 mpk, mientras que el compuesto 3 redujo Ap a 1 mpk.
Tabla 1. Propiedades biológicas de las hidantoínas ilustrativas en comparación con BACE IV (inhibidor de p-sectretasa IV de Calbiochem (cat #565788).

Las hidantoínas contempladas en el presente documento muestran, por lo tanto, perfiles farmacocinéticos deseables y tienen la actividad deseada como lo demuestra la interacción con los complejos APP y/o BACE/APP y la disminución de Ap42.
La escisión secuencial de APP por proteasas unidas a membrana p-secretasa y Y-secretasa da como resultado la formación de Ap. La enzima de escisión de APP de sitio p-1 (BACE1) se identificó como la principal actividad de secretasa p que media la primera escisión de APP en la ruta de p-amiloidogénico. En vista de la capacidad de los compuestos ABBI descritos aquí para bloquear específicamente la actividad BACE1 en APP, se cree (y los datos presentados aquí muestran) que estos compuestos ABBI pueden reducir los niveles de Ap o prevenir la formación de las especies Ap neurotóxicas. Por consiguiente, se cree que estos compuestos previenen o retrasan la progresión de la enfermedad y/o previenen o retrasan la progresión de manifestaciones preclínicas de la vía de la enfermedad amiloidogénica.
Por consiguiente, se cree que estos agentes) (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dicha(s) hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados, o profármacos de los
mismos) pueden usarse para prevenir o retrasar la aparición de una disfunción cognitiva previa al Alzheimer, y/o para mejorar uno o más síntomas de una disfunción cognitiva previa al Alzheimer, y/o para prevenir o retrasar la progresión de una afección pre-Alzheimer o disfunción cognitiva a la enfermedad de Alzheimer, y/o para promover el procesamiento de la proteína precursora amiloide (APP) por la vía no amiloidogénica. En ciertas realizaciones, estos agentes pueden usarse en el tratamiento de la enfermedad de Alzheimer (por ejemplo, para disminuir la gravedad de la enfermedad y/o para mejorar uno o más síntomas de la enfermedad, y/o para retrasar la progresión de la enfermedad).
Métodos terapéuticos y profilácticos.
En diversas realizaciones, se proporcionan métodos terapéuticos y/o profilácticos que utilizan el (los) agente(s) activo(s), por ejemplo, hidantoínas descritas aquí, o un tautómero(s) o estereoisómero(s) de la misma, o sales o solvatos farmacéuticamente aceptables de dicho se proporcionan hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos). Típicamente, los métodos implican administrar uno o más agentes activos a un sujeto (por ejemplo, a un humano que lo necesite) en una cantidad suficiente para obtener el resultado terapéutico o profiláctico deseado.
Profilaxis
En ciertas realizaciones se utilizan agente(s) activo(s) (por ejemplo, hidantoínas descritas aquí, o un tautómero(s) o estereoisómero(s) de las mismas, o sales o solvatos farmacéuticamente aceptables de dicha hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) en diversos contextos profilácticos. Así, por ejemplo, en ciertas realizaciones, el (los) agente(s) activo(s) puede(n) usarse para prevenir o retrasar la aparición de una disfunción cognitiva pre-Alzheimer y/o para mejorar uno o más síntomas de una condición pre-Alzheimer y/o cognitiva disfunción, y/o para prevenir o retrasar la progresión de una condición previa al Alzheimer y/o disfunción cognitiva a la enfermedad de Alzheimer.
Por consiguiente, en ciertas realizaciones, los métodos profilácticos descritos en el presente documento se contemplan para sujetos identificados como "en riesgo" y/o que tienen evidencia de cambios patológicos tempranos de la enfermedad de Alzheimer (EA), pero que no cumplen los criterios clínicos para DCL o demencia . Sin limitarse a una teoría particular, se cree que incluso esta etapa "preclínica" de la enfermedad representa un continuo de individuos completamente asintomáticos con evidencia de biomarcadores que sugieren procesos fisiopatológicos EA (abreviados como AD-P, ver, por ejemplo, Sperling et al. (2011) Alzheimer's & Dementia, 1-13) en riesgo de progresión a demencia EA a individuos con biomarcadores positivos que ya están demostrando una disminución muy sutil pero que aún no cumplen con los criterios estandarizados para MCI (véase, por ejemplo, Albert et al. (2011) Alzheimer and Demencia, 1-10 (doi: 10.1016/j.jalz.2011.03.008).
Este último grupo de individuos podría clasificarse como "no normal, no MCI", pero podría designarse como "pre sintomático" o "preclínico" o "asintomático" o "premanifiesto"). En diversas realizaciones, este continuo de EA pre sintomática también puede abarcar, pero no se limita necesariamente a, (1) individuos que portan uno o más alelos de apolipoproteína E (APOE) £4 que se sabe o se cree que tienen un mayor riesgo de desarrollar EA demencia, en el momento en que son biomarcadores AD-P positivos, y (2) portadores de mutaciones autosómicas dominantes, que se encuentran en la etapa presintomática de biomarcadores positivos de su enfermedad, y que casi con certeza manifestarán síntomas clínicos y progresarán a demencia.
Se ha propuesto un modelo de biomarcadores en donde los biomarcadores de AD-P más ampliamente validados se vuelven anormales y también alcanzan un techo de manera ordenada (véase, por ejemplo, Jack et al. (2010) Lancet Neurol., 9: 119 -128.). Este modelo de biomarcador es paralelo a la secuencia fisiopatológica propuesta de (pre-AD/AD), y es relevante para el seguimiento de las etapas preclínicas (asintomáticas) de EA (véase, por ejemplo, la Figura 3 en Sperling et al. (2011) Alzheimer & Dementia, 1-13) Los biomarcadores de la amiloidosis cerebral incluyen, entre otros, reducciones en el CSF Ap42 y una mayor retención del marcador amiloide en la tomografía por emisión de positrones (PET). La tau elevada del CSF no es específica de la EA y se cree que es un biomarcador de lesión neuronal. La disminución de la captación de fluorodeoxiglucosa 18F (FDG) en PET con un patrón temporoparietal de hipometabolismo es un biomarcador de la disfunción sináptica relacionada con EA. La atrofia cerebral en la resonancia magnética estructural (MRI) en un patrón característico que involucra los lóbulos temporales mediales, las cortezas paralímbica y temporoparietal es un biomarcador de neurodegeneración relacionada con EA. Otros marcadores incluyen, entre otros, resonancia magnética volumétrica, FDG-PET o biomarcadores de plasma (véase, por ejemplo, Vemuri et al. (2009) Neurology, 73: 294-301; Yaffe et al. (2011) JAMA 305: 261 -266).
En ciertas realizaciones, los sujetos adecuados para los métodos profilácticos contemplados en el presente documento incluyen, pero no se limitan a, sujetos caracterizados por tener amiloidosis cerebral asintomática. En diversas realizaciones, estos individuos tienen evidencia de biomarcadores de acumulación de Ap con retención elevada del marcador en imágenes de amiloide PET y/o Ap42 baja en el ensayo de CSF, pero típicamente no hay evidencia detectable de alteraciones cerebrales adicionales que sugieran neurodegeneración o sintomatología cognitiva y/o conductual sutil.
Se observa que los biomarcadores de Ap de imágenes de CSF y PET disponibles actualmente proporcionan principalmente evidencia de acumulación y depósito de amiloide de formas fibrilares de amiloide. Los datos sugieren que las formas solubles u oligoméricas de Ap probablemente estén en equilibrio con las placas, que pueden servir como reservorios. En ciertas realizaciones, se contempla que hay una etapa preplaca identificable en donde solo están presentes formas solubles de Ap. En ciertas realizaciones, se contempla que las formas oligoméricas de amiloide pueden ser críticas en la cascada patológica y proporcionar marcadores útiles. Además, los cambios sinápticos tempranos pueden estar presentes antes de la evidencia de acumulación de amiloide.
En ciertas realizaciones, los sujetos adecuados para los métodos profilácticos contemplados en el presente documento incluyen, pero no se limitan a, sujetos caracterizados como amiloides positivos con evidencia de disfunción sináptica y/o neurodegeneración temprana. En diversas realizaciones, estos sujetos tienen evidencia de positividad amiloide y la presencia de uno o más marcadores de lesión neuronal relacionada con EA "descendente". Los marcadores ilustrativos, pero no limitantes de lesión neuronal incluyen, pero no se limitan a (1) tau o fosfo-tau elevados en CSF, (2) hipometabolismo en un patrón similar a EA (es decir, cingulado posterior, precuneo y/o temporoparietal cortices) en FDG-PET, y (3) adelgazamiento cortical/pérdida de materia gris en una distribución anatómica específica (es decir, cortical parietal lateral y medial, cingulado posterior y temporal lateral) y/o atrofia del hipocampo en resonancia magnética volumétrica. Otros marcadores incluyen, entre otros, medidas de fMRI de conectividad de red predeterminada. En ciertas realizaciones, la disfunción sináptica temprana, evaluada mediante técnicas de imagen funcional tales como FDG-PET y fMRI, puede ser detectable antes de la pérdida volumétrica. Sin limitarse a una teoría particular, se cree que los individuos con amiloide positivo con evidencia de neurodegeneración temprana pueden estar más abajo en la trayectoria (es decir, en etapas posteriores de EA preclínica (asintomática)).
En ciertas realizaciones, los sujetos adecuados para los métodos profilácticos contemplados en el presente documento incluyen, pero no se limitan a, sujetos caracterizados como amiloides positivos con evidencia de neurodegeneración y deterioro cognitivo sutil. Sin limitarse a una teoría particular, se cree que aquellos individuos con evidencia de biomarcadores de acumulación de amiloide, neurodegeneración temprana y evidencia de deterioro cognitivo sutil se encuentran en la última etapa de la EA preclínica (asintomática) y se están acercando a la zona límite con criterios clínicas para el deterioro cognitivo leve (DCL). Estos individuos pueden demostrar evidencia de disminución desde su propia línea base (particularmente si se toman en cuenta los indicadores de reserva cognitiva), incluso si todavía se desempeñan dentro del rango "normal" en las medidas cognitivas estándar. Sin limitarse a una teoría particular, se cree que las medidas cognitivas más sensibles, particularmente con medidas de memoria episódica desafiantes, pueden detectar un deterioro cognitivo muy sutil en individuos con amiloide positivo. En ciertas realizaciones, los criterios incluyen, pero no se limitan a, autoqueja de deterioro de la memoria u otros cambios neuroconductuales sutiles.
Como se indicó anteriormente, los sujetos/pacientes susceptibles de los métodos profilácticos descritos en el presente documento incluyen individuos en riesgo de enfermedad (por ejemplo, una patología caracterizada por la formación de placa amiloide como DCL) pero que no muestran síntomas, así como sujetos que actualmente muestran ciertos síntomas o marcadores se sabe que el riesgo de DCL y más tarde la enfermedad de Alzheimer generalmente aumenta con la edad. Por consiguiente, en sujetos asintomáticos sin otros factores de riesgo conocidos, en ciertas realizaciones, se contempla la aplicación profiláctica para sujetos mayores de 50 años, o sujetos mayores de 55 años, o sujetos mayores de 60 años, o sujetos mayores de 65 años. edad, o sujetos mayores de 70 años, o sujetos mayores de 75 años, o sujetos mayores de 80 años, en particular para prevenir o retrasar la aparición o la gravedad final del deterioro cognitivo leve (DCL), y/o disminuir o prevenir la progresión de DCL a la enfermedad de Alzheimer (EA) en etapa temprana.
En ciertas realizaciones, los métodos descritos en el presente documento son especialmente útiles para individuos que tienen un riesgo genético conocido de enfermedad de Alzheimer (u otras patologías amiloidogénicas), ya sean asintomáticos o que muestren síntomas de enfermedad. Tales individuos incluyen aquellos que tienen familiares que han experimentado DCL o EA (por ejemplo, un padre, un abuelo, un hermano) y aquellos cuyo riesgo se determina mediante el análisis de marcadores genéticos o bioquímicos. Los marcadores genéticos de riesgo para la enfermedad de Alzheimer incluyen, por ejemplo, mutaciones en el gen APP, particularmente mutaciones en la posición 717 y en las posiciones 670 y 671 denominadas mutaciones Hardy and Swedish respectivamente (véase Hardy (1997) Trends. Neurosci., 20: 154-159). Otros marcadores de riesgo incluyen mutaciones en los genes de presenilina (PS1 y PS2), antecedentes familiares de EA, que tienen la mutación familiar de la enfermedad de Alzheimer (EAF), el alelo APOE £4, hipercolesterolemia o aterosclerosis. Se revisan otros genes de susceptibilidad para el desarrollo de la enfermedad de Alzheimer, por ejemplo, en Sleegers, et al. (2010) Tendencias Genet. 26 (2): 84-93.
En algunas realizaciones, el sujeto es asintomático pero tiene factores de riesgo familiares y/o genéticos para desarrollar DCL o enfermedad de Alzheimer. En pacientes asintomáticos, el tratamiento puede comenzar a cualquier edad (por ejemplo, aproximadamente a los 20, aproximadamente 30, aproximadamente 40, aproximadamente 50 años de edad). Por lo general, sin embargo, no es necesario comenzar el tratamiento hasta que el paciente alcance al menos aproximadamente 40, o al menos aproximadamente 50, o al menos aproximadamente 55, o al menos aproximadamente 60, o al menos aproximadamente 65, o al menos aproximadamente 70 años de edad.
En algunas realizaciones, el sujeto presenta síntomas, por ejemplo, de deterioro cognitivo leve (DCL) o enfermedad de Alzheimer (EA). Las personas que actualmente padecen la enfermedad de Alzheimer pueden ser reconocidas por
la demencia característica, así como por la presencia de factores de riesgo descritos anteriormente. Además, hay varias pruebas de diagnóstico disponibles para identificar a las personas que tienen EA. Estos incluyen la medición de CSF Tau, fosfo-tau (pTau), niveles de Ap42 y fragmento de APP escindido en C-terminal (APPneo). Elevación total de Tau (tTau), fosfo-Tau (pTau), APPneo, Ap40 soluble, relación pTau/Ap42 y relación tTau/Ap42, y niveles disminuidos de Ap42, relación Ap42/Ap40, relación Ap42/Ap38, niveles de sAPpa/s relación sAPPa/sApPp, la relación sAPPa/Ap40 y la relación sAPPa/Ap42 significan la presencia de EA. En algunas realizaciones, el sujeto o paciente es diagnosticado con DCL. Los niveles aumentados de proteína de hilo neural (NTP) en orina y/o niveles aumentados de a2-macroglobulina (a2M) y/o factor de complemento H (CFH) en plasma también son biomarcadores de MCI y/o EA (véase, por ejemplo, Anoop et al. (2010) Int. J. Alzheimer's Dis.2010: 606802).
En ciertas realizaciones, los sujetos susceptibles de tratamiento pueden tener un deterioro de la memoria asociado con la edad (AAMI) o un deterioro cognitivo leve (DCL). Los métodos descritos en el presente documento son particularmente adecuados para la profilaxis y/o el tratamiento de DCL. En tales casos, los métodos pueden retrasar o prevenir la aparición de DCL, y/o reducir uno o más síntomas característicos de DCL y/o retrasar o prevenir la progresión de DCL a la enfermedad de Alzheimer en etapa temprana, media o tardía o reducir la severidad final de la enfermedad.
Deterioro cognitivo leve (DCL)
El deterioro cognitivo leve (DCL, también conocido como demencia incipiente o deterioro de la memoria aislada) es un diagnóstico que se da a las personas que tienen un deterioro cognitivo más allá de lo esperado para su edad y educación, pero que generalmente no interfiere significativamente con sus actividades diarias. (véase, por ejemplo, Petersen et al. (1999) Arch. Neurol. 56(3): 303-308). Se considera en muchos casos como una etapa límite o de transición entre el envejecimiento normal y la demencia. Aunque el DCL puede presentarse con una variedad de síntomas, cuando la pérdida de memoria es el síntoma predominante, se denomina "DCL amnésico" y con frecuencia se considera un factor de riesgo para la enfermedad de Alzheimer (véase, por ejemplo, Grundman et al. (2004) Arch. Neurol 61(1): 59-66; y en internet en en.wikipedia.org/wiki/Mild_cognitive_impairment-cite_note-Grundman-1). Cuando los individuos tienen impedimentos en dominios distintos de la memoria, a menudo se clasifica como DCL de dominio único o múltiple no amnésico y se cree que estos individuos tienen más probabilidades de convertirse a otras demencias (por ejemplo, demencia con cuerpos de Lewy). Hay evidencia que sugiere que si bien los pacientes con DCL amnésico pueden no cumplir con los criterios neuropatológicos para la enfermedad de Alzheimer, los pacientes pueden estar en una etapa de transición de la enfermedad de Alzheimer en evolución; los pacientes en esta etapa de transición hipotética demostraron amiloide difuso en la neocorteza y ovillos neurofibrilares frecuentes en el lóbulo temporal medial (véase, por ejemplo, Petersen et al. (2006) Arch. Neurol. 63(5): 665-72).
El diagnóstico de DCL típicamente implica una evaluación clínica integral que incluye observación clínica, neuroimagen, análisis de sangre y pruebas neuropsicológicas. En ciertas realizaciones, los criterios de diagnóstico para DCL incluyen, entre otros, los descritos por Albert et al. (2011) Alzheimer's & Dementia. 1-10. Como se describe en el presente documento, los criterios de diagnóstico incluyen (1) criterios clínicos básicos que podrían ser utilizados por los proveedores de atención médica sin acceso a técnicas de imagen avanzadas o análisis de líquido cefalorraquídeo, y (2) criterios de investigación que podrían utilizarse en entornos de investigación clínica, incluidos los ensayos clínicos. El segundo conjunto de criterios incorpora el uso de biomarcadores basados en imágenes y medidas de líquido cefalorraquídeo. El conjunto final de criterios para el deterioro cognitivo leve debido a EA tiene cuatro niveles de certeza, dependiendo de la presencia y la naturaleza de los hallazgos de los biomarcadores.
En ciertas realizaciones, la evaluación/diagnóstico clínico de DCL implica: (1) Preocupación que refleja un cambio en la cognición informado por el paciente o informante o clínico (es decir, evidencia histórica u observada de disminución con el tiempo); (2) Evidencia objetiva de deterioro en uno o más dominios cognitivos, típicamente incluyendo memoria (es decir, pruebas formales o de cabecera para establecer el nivel de función cognitiva en múltiples dominios); (3) Preservación de la independencia en las habilidades funcionales; (4) No demente; y en ciertas realizaciones, (5) Una etiología de DCL consistente con procesos fisiopatológicos de EA. Por lo general, se descartan causas vasculares, traumáticas y médicas de deterioro cognitivo, siempre que sea posible. En ciertas realizaciones, cuando es factible, se identifica evidencia de disminución longitudinal en la cognición. El diagnóstico se ve reforzado por una historia consistente con factores genéticos EA, donde sea relevante.
Con respecto al deterioro del dominio o dominios cognitivos, debe existir evidencia de preocupación por un cambio en la cognición, en comparación con el nivel anterior de la persona. Debe haber evidencia de un rendimiento más bajo en uno o más dominios cognitivos que sea mayor de lo que se esperaría para la edad y los antecedentes educativos del paciente. Si hay evaluaciones repetidas disponibles, entonces una disminución en el rendimiento debería ser evidente con el tiempo. Este cambio puede ocurrir en una variedad de dominios cognitivos, que incluyen memoria, función ejecutiva, atención, lenguaje y habilidades visoespaciales. Un deterioro en la memoria episódica (es decir, la capacidad de aprender y retener nueva información) se observa con mayor frecuencia en pacientes con DCL que posteriormente progresan a un diagnóstico de demencia EA.
Con respecto a la preservación de la independencia en las habilidades funcionales, se observa que las personas con DCL comúnmente tienen problemas leves para realizar tareas funcionales complejas que solían usar para realizar compras. Pueden tomar más tiempo, ser menos eficientes y cometer más errores al realizar tales actividades que en
el pasado. Sin embargo, generalmente mantienen su independencia de función en la vida diaria, con ayudas o asistencia mínimas.
Con respecto a la demencia, los cambios cognitivos deberían ser lo suficientemente leves como para que no haya evidencia de un deterioro significativo en el funcionamiento social u ocupacional. Si un individuo solo ha sido evaluado una vez, el cambio se deducirá de la historia y/o evidencia de que el rendimiento cognitivo se ve afectado más allá de lo que se hubiera esperado para ese individuo.
Las pruebas cognitivas son óptimas para evaluar objetivamente el grado de deterioro cognitivo para un individuo. Los puntajes en las pruebas cognitivas para individuos con DCL son típicamente de 1 a 1.5 desviaciones estándar por debajo de la media para su edad y sus pares de educación en datos normativos culturalmente apropiados (es decir, para el dominio o dominios deteriorados, cuando estén disponibles).
La memoria episódica (es decir, la capacidad de aprender y retener nueva información) se observa con mayor frecuencia en pacientes con DCL que posteriormente progresan a un diagnóstico de demencia EA. Hay una variedad de pruebas de memoria episódica que son útiles para identificar a aquellos pacientes con DCL que tienen una alta probabilidad de progresar a la demencia EA en unos pocos años. Estas pruebas generalmente evalúan el recuerdo inmediato y el retraso, de modo que es posible determinar la retención durante un retraso. Muchas, aunque no todas, las pruebas que han demostrado ser útiles a este respecto son pruebas de aprendizaje de listas de palabras con múltiples ensayos. Dichas pruebas revelan la tasa de aprendizaje a lo largo del tiempo, así como la cantidad máxima adquirida en el transcurso de las pruebas de aprendizaje. También son útiles para demostrar que el individuo, de hecho, está prestando atención a la tarea de recuperación inmediata, que luego puede usarse como línea base para evaluar la cantidad relativa de material retenido en la recuperación retardada. Ejemplos de tales pruebas incluyen (pero no se limitan a: la Prueba de Recordatorio Selectivo Gratuito y Guiado, la Prueba de Aprendizaje Verbal Auditivo de Rey y la Prueba de Aprendizaje Verbal California. Otras medidas de memoria episódica incluyen, pero no se limitan a: recuerdo inmediato y retrasado de un párrafo como la Memoria lógica I y II de la Escala de memoria de Wechsler revisada (u otras versiones) y el retiro inmediato y retrasado de materiales no verbales, como las subpruebas de reproducción visual de la Escala de memoria de Wechsler-Revisada I y II.
Debido a que otros dominios cognitivos pueden verse afectados entre individuos con DCL, es deseable examinar dominios además de la memoria. Estos incluyen, entre otros, funciones ejecutivas (por ejemplo, cambio de set, razonamiento, resolución de problemas, planificación), lenguaje (por ejemplo, nomenclatura, fluidez, expresión expresiva y comprensión), habilidades visoespaciales y control de la atención (por ejemplo, atención simple y dividida). Se encuentran disponibles muchas medidas neuropsicológicas clínicas para evaluar estos dominios cognitivos, que incluyen (entre otros, el Trail Making Test (función ejecutiva), el Boston Naming Test, la fluidez de letras y categorías (lenguaje), la
Como se indicó anteriormente, se pueden incorporar factores genéticos en el diagnóstico de DCL. Si se sabe que está presente una forma autosómica dominante de EA (es decir, mutación en APP, PS1, PS2), entonces el desarrollo de DCL es probablemente el precursor de la demencia EA. La gran mayoría de estos casos desarrollan EA de inicio temprano (es decir, inicio por debajo de los 65 años de edad).
Además, hay influencias genéticas en el desarrollo de la demencia EA de inicio tardío. Por ejemplo, la presencia de uno o dos alelos £4 en el gen de la apolipoproteína E (APOE) es una variante genética ampliamente aceptada como un riesgo creciente de demencia EA de inicio tardío. La evidencia sugiere que un individuo que cumple con los criterios clínicos, cognitivos y etiológicos para DCL, y que también es APOE £4 positivo, tiene más probabilidades de progresar a demencia EA en unos pocos años que un individuo sin esta característica genética. Se cree que los genes adicionales juegan un papel importante, pero más pequeño que el APOE y también confieren cambios en el riesgo de progresión a la demencia EA (véase, por ejemplo, Bertram et al. (2010) Neuron, 21: 270-281).
En ciertas realizaciones, los sujetos adecuados para los métodos profilácticos descritos en el presente documento incluyen, pero no se limitan a, sujetos identificados que tienen uno o más de los criterios clínicos centrales descritos anteriormente y/o sujetos identificados con uno o más "criterios de investigación" para DCL, por ejemplo, como se describe a continuación.
Los "criterios de investigación" para la identificación/pronóstico de DCL incluyen, pero no se limitan a, biomarcadores que aumentan la probabilidad de que el síndrome de DCL se deba a los procesos fisiopatológicos de la EA. Sin limitarse a una teoría particular, se cree que la aplicación conjunta de criterios clínicos y biomarcadores puede dar lugar a varios niveles de certeza de que el síndrome de DCL se debe a procesos fisiopatológicos de la EA. En ciertas realizaciones, se contemplan dos categorías de biomarcadores que han sido las más estudiadas y aplicadas a los resultados clínicos. Estos incluyen "Ap" (que incluye CSF Ap42 y/o imágenes de amiloide PET) y "biomarcadores de lesión neuronal" (que incluyen, entre otros, CSF tau/p-tau, hipocampo o atrofia del lóbulo temporal medial en la RM, e hipometabolismo temporoparietal/precuneus o hipoperfusión en PET o SPECT).
Sin limitarse a una teoría particular, se cree que la evidencia tanto de Ap como de lesión neuronal (ya sea un aumento en tau/p-tau o biomarcadores de imagen en un patrón topográfico característico de EA), juntos confiere la mayor probabilidad que el proceso fisiopatológico EA está presente. Por el contrario, si estos biomarcadores son negativos,
esto puede proporcionar información sobre la probabilidad de un diagnóstico alternativo. Se reconoce que los hallazgos de los biomarcadores pueden ser contradictorios y, en consecuencia, cualquier combinación de biomarcadores es indicativa (un indicador) utilizada en el contexto de un diagnóstico diferencial y no en sí misma dispositivo. Se reconoce que la gravedad variable de una anomalía puede conferir diferentes probabilidades o pronósticos, que son difíciles de cuantificar con precisión para una aplicación amplia.
Para aquellos sujetos potenciales de DCL cuyo síndrome clínico y cognitivo de DCL es consistente con EA como la etiología, la adición del análisis de biomarcadores afecta los niveles de certeza en el diagnóstico. En el ejemplo más típico en donde se ha establecido el síndrome clínico y cognitivo de DCL, incluida la evidencia de un trastorno de memoria episódica y una presunta etiología degenerativa, la causa más probable es el proceso neurodegenerativo de EA. Sin embargo, el resultado final todavía tiene grados variables de certeza. La probabilidad de progresión a demencia EA variará con la gravedad del deterioro cognitivo y la naturaleza de la evidencia que sugiere que la fisiopatología EA es la causa subyacente. Sin limitarse a una teoría en particular, se cree que los biomarcadores positivos que reflejan la lesión neuronal aumentan la probabilidad de que ocurra progresión a la demencia en unos pocos años y que los hallazgos positivos que reflejan la acumulación de Ap y la lesión neuronal juntos confieren la mayor probabilidad de que el diagnóstico sea DCL debido a EA.
Un biomarcador Ap positivo y un biomarcador positivo de lesión neuronal proporcionan una indicación de que el síndrome de DCL se debe a procesos de EA y el sujeto es muy adecuado para los métodos descritos aquí.
Un biomarcador Ap positivo en una situación en donde los biomarcadores de lesión neuronal no han sido o no pueden ser probados o un biomarcador positivo de lesión neuronal en una situación en donde los biomarcadores Ap no han sido o no pueden ser probados indican una probabilidad intermedia de que el síndrome DCL se debe a la EA. Se cree que tales sujetos son adecuados para los métodos descritos en el presente documento.
Los biomarcadores negativos tanto para Ap como para lesión neuronal sugieren que el síndrome de DCL no se debe a EA. En tales casos, los sujetos pueden no ser adecuados para los métodos descritos aquí.
Existe evidencia de que la resonancia magnética puede observar el deterioro, incluida la pérdida progresiva de materia gris en el cerebro, desde un deterioro cognitivo leve hasta la enfermedad de Alzheimer en toda regla (véase, por ejemplo, Whitwell et al. (2008) Neurology 70(7). ): 512-520). Una técnica conocida como imagen PET PiB se usa para mostrar claramente los sitios y las formas de los depósitos de beta amiloide en sujetos vivos usando un marcador C11 que se une selectivamente a dichos depósitos (véase, por ejemplo, Jack et al. (2008) Brain 131(Pt 3 ): 665-680).
En ciertas realizaciones, DCL se diagnostica típicamente cuando hay 1) Evidencia de deterioro de la memoria; 2) Preservación de las habilidades cognitivas y funcionales generales; y 3) Ausencia de demencia diagnosticada.
En ciertas realizaciones, el DCL y las etapas de la enfermedad de Alzheimer pueden identificarse/clasificarse, en parte por los puntajes de Clasificación de Demencia Clínica (CDR). El CDR es una escala de cinco puntos utilizada para caracterizar seis dominios del rendimiento cognitivo y funcional aplicables a la enfermedad de Alzheimer y las demencias relacionadas: memoria, orientación, juicio y resolución de problemas, asuntos comunitarios, hogar y pasatiempos y cuidado personal. La información para realizar cada calificación se puede obtener a través de una entrevista semiestructurada del paciente y un informante confiable o una fuente colateral (por ejemplo, un miembro de la familia).
La tabla CDR proporciona anclas descriptivas que guían al clínico en la realización de clasificaciones apropiadas basadas en datos de entrevistas y juicio clínico. Además de las calificaciones para cada dominio, se puede calcular una puntuación general de CDR mediante el uso de un algoritmo. Este puntaje es útil para caracterizar y rastrear el nivel de discapacidad/demencia de un paciente: 0 = Normal; 0.5 = Demencia muy leve; 1 = demencia leve; 2 = demencia moderada; y 3 = demencia severa. Una tabla ilustrativa de CDR se muestra en la Tabla 2.
Tabla 2. Tabla ilustrativa de clasificación de demencia clínica (CDR).


Una clasificación de CDR de ~0.5 o ~0.5 a 1.0 a menudo se considera DCL clínicamente relevante. Las calificaciones CDR más altas pueden ser indicativas de progresión a la enfermedad de Alzheimer.
En ciertas realizaciones, la administración de uno o más agentes descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o uno o más tautómeros o estereoisómeros de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) se considera efectivo cuando hay una reducción en el CSF de
los niveles de uno o más componentes seleccionados del grupo que consiste en Tau, fosfo-Tau (pTau), APPneo, Ap40 soluble, Ap42 soluble, y/o relación Ap42/Ap40, y/o cuando hay una reducción de la carga de placa en el cerebro del sujeto, y/o cuando hay una reducción en la tasa de placa formación en el cerebro del sujeto, y/o cuando hay una mejora en las habilidades cognitivas del sujeto, y/o cuando hay una mejora percibida en la calidad de vida por el sujeto, y/o cuando la tasa de aumento en la calificación de la demencia clínica se ralentiza o detiene y/o cuando la progresión de DCL a la etapa temprana de EA se ralentiza o detiene.
En algunas realizaciones, se puede determinar un diagnóstico de DCL considerando los resultados de varias pruebas clínicas. Por ejemplo, Grundman, et al. (2004) Arch Neurol 61: 59-66, informan que se puede establecer un diagnóstico de DCL con eficiencia clínica utilizando una prueba de memoria simple (recuperación de párrafo) para establecer un déficit de memoria objetivo, una medida de la cognición general (Mini-Mental State Exam (MMSE), discutido en mayor detalle a continuación) para excluir un deterioro cognitivo más amplio más allá de la memoria, y una entrevista clínica estructurada (CDR) con pacientes y cuidadores para verificar la queja de memoria del paciente y la pérdida de memoria y para garantizar que el paciente no esté demente. Los pacientes con DCL realizan, en promedio, menos de 1 desviación estándar (DE) por debajo de lo normal en medidas no cognitivas incluidas en la batería. Las pruebas de aprendizaje, atención, velocidad de percepción, fluidez de categoría y función ejecutiva pueden verse afectadas en pacientes con DCL, pero son mucho menos prominentes que el déficit de memoria.
Enfermedad de Alzheimer (EA).
En ciertas realizaciones, el (los) agente(s) (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dicha(s)hidantoína(s), dicho(s) estereoisómero(s), o dicho tautómero(s), o análogos, derivados o profármacos de los mismos) se contemplan para el tratamiento de la enfermedad de Alzheimer. En tales casos, los métodos descritos en el presente documento son útiles para prevenir o retrasar la aparición de la enfermedad de Alzheimer (EA), para reducir el gravedad de la EA cuando el sujeto ha pasado al diagnóstico clínico de EA y/o al mitigar uno o más síntomas de la enfermedad de Alzheimer.
En particular, cuando la enfermedad de Alzheimer está en una etapa temprana, los métodos pueden reducir o eliminar uno o más síntomas característicos de la EA y/o retrasar o prevenir la progresión de DCL a la enfermedad de Alzheimer en etapa temprana o tardía.
Las personas que actualmente padecen la enfermedad de Alzheimer pueden reconocerse por la demencia característica, así como por la presencia de factores de riesgo descritos anteriormente. Además, hay varias pruebas de diagnóstico disponibles para identificar a las personas que tienen EA. Las personas que actualmente padecen la enfermedad de Alzheimer pueden ser reconocidas por la demencia característica, así como por la presencia de factores de riesgo descritos anteriormente. Además, hay varias pruebas de diagnóstico disponibles para identificar a las personas que tienen EA. Estos incluyen la medición de los niveles de Tau CSF, fosfo-tau (pTau), sAPPa, sAPPp, Ap40, Ap42 y/o fragmento de APP terminalmente escindido (APPneo). Tau elevado, pTau, sAPPp y/o APPneo, y/o niveles reducidos de sAPPa, Ap40 soluble y/o Ap42 soluble, particularmente en el contexto de un diagnóstico diferencial, pueden significar la presencia de EA.
En ciertas realizaciones, los sujetos susceptibles de tratamiento pueden tener la enfermedad de Alzheimer. Las personas que padecen la enfermedad de Alzheimer también pueden ser diagnosticadas por la enfermedad de Alzheimer y los criterios de la Asociación de Trastornos Relacionados (ADRDA). Los Criterios de Alzheimer NINCDS-ADRDA fueron propuestos en 1984 por el Instituto Nacional de Trastornos Neurológicos y Comunicativos y Accidentes Cerebrovasculares y la Asociación de Enfermedades de Alzheimer y Trastornos Relacionados (ahora conocida como la Asociación de Alzheimer) y se encuentran entre los más utilizados en el diagnóstico de la enfermedad de Alzheimer (EA). McKhann et al. (1984) Neurology 34(7): 939-44. De acuerdo con estos criterios, la presencia de deterioro cognitivo y un síndrome de demencia sospechoso debe confirmarse mediante pruebas neuropsicológicas para un diagnóstico clínico de EA posible o probable. Sin embargo, la confirmación histopatológica (examen microscópico del tejido cerebral) se usa generalmente para un diagnóstico dispositivo. Los Criterios de Alzheimer NINCDS-ADRDA especifican ocho dominios cognitivos que pueden verse afectados en la EA: memoria, lenguaje, habilidades perceptivas, atención, habilidades constructivas, orientación, resolución de problemas y habilidades funcionales). Estos criterios han demostrado una buena fiabilidad y validez.
Las evaluaciones iniciales de la función del paciente pueden realizarse utilizando medidas psicométricas clásicas, tales como el Mini-Mental State Exam (Mm Se ) (Folstein et al. (1975) J. Psychiatric Research 12(3): 189-198), y Alzheimer's Disease Assessment Scale (ADAS), que es una escala integral para evaluar a los pacientes con el estado y la función de la enfermedad de Alzheimer (véase, por ejemplo, Rosen, et al. (1984) Am. J. Psychiatr., 141: 1356 1364). Estas escalas psicométricas proporcionan una medida de la progresión de la enfermedad de Alzheimer. Las escalas de vida cualitativas adecuadas también se pueden utilizar para controlar el tratamiento. La extensión de la progresión de la enfermedad se puede determinar usando un Mini-Mental State Exam (MMSE) (véase, por ejemplo, Folstein, et al. supra). Cualquier puntaje mayor o igual a 25 puntos (de 30) es efectivamente normal (intacto). Debajo de esto, los puntajes pueden indicar enfermedad de Alzheimer grave (<9 puntos), moderada (10-20 puntos) o leve (21-24 puntos).
La enfermedad de Alzheimer se puede dividir en varias etapas que incluyen: 1) deterioro cognitivo moderado (enfermedad de Alzheimer leve o en etapa temprana), 2) deterioro cognitivo moderadamente grave (enfermedad de Alzheimer moderada o en etapa intermedia), 3) deterioro cognitivo severo (Enfermedad de Alzheimer moderadamente grave o en etapa intermedia), y 4) Deterioro cognitivo muy grave (enfermedad de Alzheimer en etapa grave o tardía) como se muestra en la Tabla 3.
Tabla 3. Etapas ilustrativas de la enfermedad de Alzheimer.


En diversas realizaciones, la administración de uno o más agentes descritos en el presente documento a sujetos diagnosticados con la enfermedad de Alzheimer se considera eficaz cuando existe una reducción en los niveles de CSF de uno o más componentes seleccionados del grupo que consiste en Tau, fosfo-Tau (pTau), APPneo, Ap40 soluble, Ap42 soluble, y/o relación Ap42/Ap40, y/o cuando hay una reducción de la carga de placa en el cerebro del sujeto, y/o cuando hay una reducción en el tasa de formación de placa en el cerebro del sujeto, y/o cuando hay una mejora en las habilidades cognitivas del sujeto, y/o cuando el sujeto percibe una mejora en la calidad de vida, y/o cuando hay una reducción significativa en la calificación de demencia clínica (CDR) del sujeto, y/o cuando la tasa de aumento en la calificación de demencia clínica se ralentiza o detiene y/o cuando la progresión de EA se ralentiza o detiene (por ejemplo, cuando la transición de una etapa a otra como se enumera en la Tabla 3 se ralentiza o detiene).
En ciertas realizaciones, los sujetos susceptibles a los presentes métodos generalmente están libres de una enfermedad o trastorno neurológico distinto de la enfermedad de Alzheimer. Por ejemplo, en ciertas realizaciones, el sujeto no tiene y no está en riesgo de desarrollar una enfermedad o trastorno neurológico tal como la enfermedad de Parkinson, y/o esquizofrenia, y/o psicosis.
Agente(s) activo(s).
Los métodos descritos en el presente documento se basan, en parte, en el descubrimiento de que la administración de uno o más agentes activos (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero (s) o
estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) encuentran uso en el tratamiento y/o profilaxis de enfermedades caracterizadas por depósitos amiloides en el cerebro, por ejemplo , deterioro cognitivo leve, enfermedad de Alzheimer, degeneración macular y similares.
En ciertos ejemplos no reivindicados, el agente activo es un compuesto (por ejemplo, una hidantoína) de acuerdo con la Fórmula I:
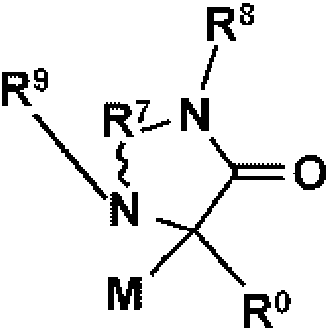
donde M es

y
R7 se selecciona del grupo que consiste en C=O, C=S, C-NH2 y C=NH, y el enlace representado por la línea ondulada es un enlace simple cuando R7 es C=O, C=S o C=NH, y un doble enlace cuando R7 es C-NH2; R8 y R9 se seleccionan independientemente del grupo que consiste en H, alquilo, cicloalquilo y arilo, siempre que cuando el enlace representado por la línea ondulada sea un doble enlace, entonces R9 esté ausente; R0 se selecciona del grupo que consiste en arilo, arilo sustituido, arilo disustituido, heteroarilo, heteroarilo sustituido, heteroarilo disustituido, alquilo, haloalquilo, cicloalquilo, alquenilo y alquinilo; X1 se selecciona del grupo que consiste en C-halógeno, CH y N; A es metilo o H; R5 y R6 se seleccionan independientemente de halógeno, H, alquilo, triclorometilo y trifluorometilo; R3 y R4 están independientemente ausentes o seleccionados del grupo que consiste en alquilo, cicloalquilo, alcoxi, tioalquilo; y cuando X1 es C, entonces R0 no está monosustituido con fenilo en la posición para con -OCHF2. También se contemplan sales farmacéuticamente aceptables de los mismos, tautómeros de los mismos, sales farmacéuticamente aceptables de un tautómero de los mismos, un enantiómero del mismo, una sal farmacéuticamente aceptable de un enantiómero del mismo, y similares.
En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la Fórmula IV:

o un compuesto de acuerdo con la fórmula V:

En ciertos ejemplos, de cualquiera de los compuestos anteriores, X1 se selecciona del grupo que consiste en C-halógeno, CH y N o del grupo que consiste en CH y N; y R5 y R6 son halógenos seleccionados independientemente. En ciertas realizaciones, de cualquiera de los compuestos anteriores, R7 es C=NH o R7 es C=O.
En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la Fórmula VI:
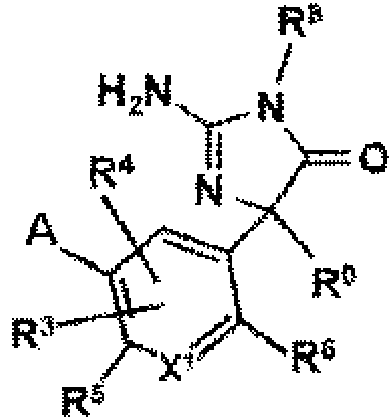
En ciertas realizaciones del compuesto de Fórmula VI, R5 y R6 son halógenos seleccionados independientemente. En ciertas realizaciones del compuesto de Fórmula VI, R5 y R6 son el mismo halógeno (por ejemplo, ambos F, ambos Cl, etc.).
En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la Fórmula V, donde dicho compuesto es un compuesto de Fórmula VII:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la Fórmula VIII:

En ciertos ejemplos, el compuesto es un compuesto de Fórmula V donde R7 es C=S.
En ciertos ejemplos, el compuesto es un compuesto donde R7 es C-NH2 y el compuesto es un compuesto de Fórmula X:

y en ciertos ejemplos de Fórmula VIII, el compuesto es un compuesto de acuerdo con la Fórmula XI:

donde R1 y R2 están independientemente ausentes o seleccionados del grupo que consiste en alquilo, haloalquilo, cicloalquilo, alquenilo, alquinilo, alcoxi, tioalquilo, arilo, arilo sustituido, heteroarilo y heteroarilo sustituido; y X2, Y y Z son independientemente CH o N. En ciertas realizaciones de cualquiera de las Fórmulas anteriores, R5 y R6 son halógenos diferentes (por ejemplo, R5=Cl y R6=F, R5=F y R6=Cl, y similares ) En ciertas realizaciones de cualquiera de las Fórmulas anteriores, R5 y R6 son el mismo halógeno (por ejemplo, ambos Cl, ambos F, etc.).
En ciertos ejemplos, el compuesto es un compuesto de Fórmula XII:
En ciertos ejemplos de cualquiera de los compuestos anteriores, X1 es CH. En ciertas realizaciones de cualquiera de los compuestos anteriores, R8 es H o CH3.
En ciertas realizaciones, el compuesto es un compuesto de acuerdo con la Fórmula XIII:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XIV:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XV:


En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XVIII:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XIX:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XX:
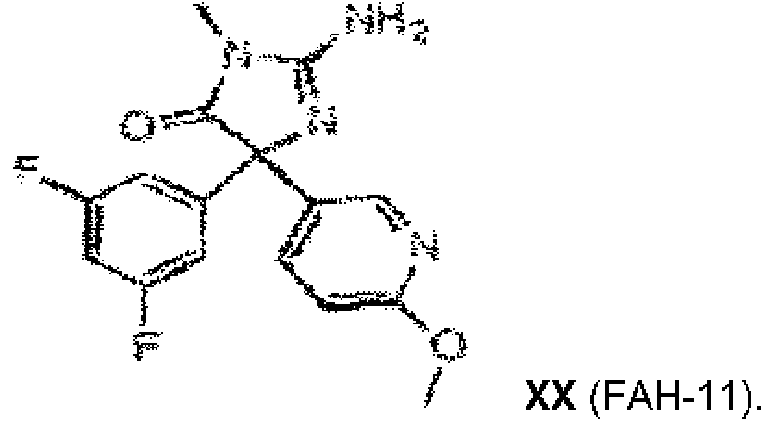
En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXI:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXII:

XXII (FAH-13). En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXIII:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXIV:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXV:

XXV (FAH-17). En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXVI:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXVII:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXVIII:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXIX:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXX:

En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la fórmula XXXI:
En ciertos ejemplos, cualquiera de las Fórmulas anteriores excluye expresamente FAH-2. En ciertos ejemplos, cualquiera de las Fórmulas anteriores excluye expresamente FAH-3. En ciertos ejemplos, cualquiera de las fórmulas anteriores excluye expresamente FAH-2 y FAH-3.
En ciertos ejemplos, el compuesto es un compuesto de acuerdo con la Fórmula XXXII:

o una sal farmacéuticamente aceptable del mismo, un tautómero del mismo, una sal farmacéuticamente aceptable de un tautómero del mismo, un enantiómero del mismo, o una sal farmacéuticamente aceptable de un enantiómero del mismo.
En ciertas realizaciones, cualquiera de los compuestos anteriores de la presente invención es un enantiómero S sustancialmente puro. En ciertas realizaciones, cualquiera de los compuestos anteriores es un enantiómero R sustancialmente puro.
Varios compuestos descritos en el presente documento incluyen los compuestos que se muestran en la Tabla 4.
Tabla 4. Ejemplos ilustrativos, pero no limitantes, de compuestos contemplados en el presente documento.




Con respecto a estos compuestos, también se contemplan sales farmacéuticamente aceptables, tautómeros, sales farmacéuticamente aceptables de un tautómero, enantiómeros de los mismos y sales farmacéuticamente aceptables de un enantiómero de los mismos. Además, se contemplan enantiómeros S sustancialmente puros o enantiómeros R sustancialmente puros de estos compuestos.
También se muestran diversas hidantoínas ilustrativas, pero no limitantes, en las Figuras 1 y 2. En ciertas realizaciones, se contemplan sales farmacéuticamente aceptables, tautómeros, sales farmacéuticamente aceptables de un tautómero, enantiómeros de los mismos y sal farmacéuticamente aceptable de un enantiómero.
Con respecto a ciertas moléculas en la Figura 1, sin limitarse a una teoría particular, se cree que el anillo B con el metilo y el OCHF2 muestra una mayor potencia con una sustitución 3,4. Se cree que este tipo de patrón de sustitución interactúa con el Trp-76 de la aleta BACE interrumpiendo la interacción del Tyr-71 de la aleta con el Trp-76 y volteando el Tyr-71 hacia la izquierda, lo que le permite interactuar con los grupos difluoro del anillo A (véase también la figura 3).
En ciertas realizaciones, el compuesto es un enantiómero "S" sustancialmente puro. En ciertas realizaciones, el compuesto es un enantiómero "R" sustancialmente puro. En ciertas realizaciones, el compuesto se une a APP y/o a la enzima BACE y/o a un complejo APP/BACE.
Los expertos en la técnica conocen métodos para preparar hidantoínas como los que se describen en el presente documento. Generalmente, en un enfoque, la hidantoína relevante (por ejemplo, una diflouoro hidantoína) se prepararía a partir de 3,4 difluoro benzaldehído transformado en diona y condensado con urea para producir la hidantoína como se describe en el Ejemplo 1.
Protocolos ilustrativos para la síntesis de FAH-1, FAH-2, FAH-3, FAH-4, FAH-5, FAH-17, sal de HCl FAH-17, FAH-22, FAH-23, FAH- 27, y FAH-28 (véase la Tabla 4) ver en los Ejemplos 1-11. La síntesis de compuestos adicionales incluidos aquí son variaciones directas de los esquemas de síntesis proporcionados aquí.
Los diversos agentes activos y esquemas de síntesis pretenden ser ilustrativos y no limitativos. Utilizando las enseñanzas proporcionadas en el presente documento, numerosas otras (por ejemplo, hidantoínas o un tautómero(s) o estereoisómero(s) de las mismas, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho(s) estereoisómero(s) o dicho(s) tautómero(s), o los análogos, derivados o profármacos de los mismos pueden ser sintetizados e identificados por un experto en la materia.
La actividad ilustrativa de ciertos inhibidores de BACE selectivos de APP descritos anteriormente se muestra en la Tabla 5.
Tabla 5. Actividad ilustrativa de ciertos inhibidores de BACE selectivos de APP descritos aquí.

Formulaciones farmacéuticas
En ciertas realizaciones, uno o más agentes activos descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) se administran a un mamífero que lo necesita, por ejemplo, a un mamífero en riesgo o que padece una patología caracterizada por el procesamiento anormal de proteínas precursoras de amiloide, un mamífero en riesgo de progresión de DCL a enfermedad de Alzheimer, y así sucesivamente. En ciertas realizaciones, el (los) agente(s) activo(s) se administran para prevenir o retrasar la aparición de una afección y/o disfunción cognitiva pre-Alzheimer, y/o para mejorar uno o más síntomas de una disfunción cognitiva pre-Alzheimer, y/o para prevenir o retrasar la progresión de una afección pre-Alzheimer o disfunción cognitiva a la enfermedad de Alzheimer, y/o promover el procesamiento de la proteína precursora amiloide (APP) por una vía no amiloidogénica.
En ciertas realizaciones, uno o más agentes activos descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dicha(s) hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) se administran a un mamífero que lo necesita, por ejemplo, a un mamífero en riesgo o que padece una patología caracterizada por el procesamiento anormal de proteínas precursoras de amiloide en condiciones aparte de la enfermedad de Alzheimer de DCL. Las condiciones ilustrativas incluyen, entre otras, síntomas de tipo EA de pacientes con síndrome de Down, glaucoma, degeneración macular (por ejemplo, Degeneración macular relacionada con la edad, DMAE), deterioro olfativo en el tratamiento de la diabetes tipo II, incluida la diabetes. asociado con la amiloidogénesis., enfermedades neurodegenerativas como tembladera, encafalopatías espongiformes bovinas (por ejemplo, EEB), lesión cerebral traumática ("TBI"), enfermedad de Creutzfeld-Jakob y similares, diabetes tipo II. Otras afecciones caracterizadas por la formación de amiloide se contempla, entre otros, la enfermedad de Huntington, carcinoma medular de tiroides, arritmias cardíacas, amiloidosis auricular aislada, aterosclerosis, artritis reumatoide, amiloide medial aórtico, prolactinomas, polineuropatía amiloide
familiar, sistema neuropático hereditario no neuropático amiloidosis, amiloidosis relacionada con diálisis, amiloidosis finlandesa, distrofia corneal del enrejado, angiopatía amiloide cerebral (p. ej., tipo islandés), amiloidosis sistémica AL, miositis esporádica por cuerpos de inclusión, demencia cerebrovascular y similares.
El (los) agente(s) activo (por ejemplo, hidantoínas descritas en el presente documento) se pueden administrar en forma "nativa" o, si se desea, en forma de sales, ésteres, amidas, profármacos, derivados y similares, siempre que la sal, el éster, la amida, el profármaco o el derivado es farmacológicamente adecuado, es decir, efectivo en el (los) presente(s) método(s). Las sales, los ésteres, las amidas, los profármacos y otros derivados de los agentes activos pueden prepararse usando procedimientos estándar conocidos por los expertos en la técnica de la química orgánica sintética y descritos, por ejemplo, en March (1992) Advanced Organic Chemistry; Reactions, Mechanisms and Structure, 4th Ed. N.Y. Wiley-Interscience, y como se describió anteriormente.
Por ejemplo, se puede preparar una sal farmacéuticamente aceptable para cualquiera de los agentes descritos aquí que tienen una funcionalidad capaz de formar una sal. Una sal farmacéuticamente aceptable es cualquier sal que retiene la actividad del compuesto original y no imparte ningún efecto nocivo o perjudicial sobre el sujeto al que se administra y en el contexto en donde se administra.
En diversas realizaciones, las sales farmacéuticamente aceptables pueden derivarse de bases orgánicas o inorgánicas. La sal puede ser un ion mono o polivalente. De particular interés son los iones inorgánicos, litio, sodio, potasio, calcio y magnesio. Las sales orgánicas pueden prepararse con aminas, particularmente sales de amonio tales como mono, di y trialquilaminas o etanol aminas. Las sales también se pueden formar con cafeína, trometamina y moléculas similares.
Los métodos para formular agentes farmacéuticamente activos como sales, ésteres, amidas, profármacos y similares son bien conocidos por los expertos en la materia. Por ejemplo, las sales se pueden preparar a partir de la base libre utilizando una metodología convencional que típicamente implica la reacción con un ácido adecuado. Generalmente, la forma básica del fármaco se disuelve en un solvente orgánico polar como metanol o etanol y el ácido se agrega al mismo. La sal resultante precipita o se puede sacar de la solución mediante la adición de un disolvente menos polar. Los ácidos adecuados para preparar sales de adición de ácido incluyen, entre otros, ácidos orgánicos, por ejemplo, ácido acético, ácido propiónico, ácido glicólico, ácido pirúvico, ácido oxálico, ácido málico, ácido malónico, ácido succínico, ácido maleico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido cinámico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido salicílico y similares, así como ácidos inorgánicos, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico y similares. Una sal de adición de ácido puede reconvertirse a la base libre mediante tratamiento con una base adecuada. Ciertas sales de adición de ácido particularmente preferidas de los agentes activos en el presente documento incluyen sales de haluro, tales como se pueden preparar usando ácidos clorhídrico o bromhídrico. A la inversa, la preparación de sales básicas de los agentes activos de esta invención se prepara de manera similar usando una base farmacéuticamente aceptable tal como hidróxido de sodio, hidróxido de potasio, hidróxido de amonio, hidróxido de calcio, trimetilamina o similares. Las sales básicas particularmente preferidas incluyen sales de metales alcalinos, por ejemplo, la sal de sodio y sales de cobre.
Para la preparación de formas salinas de fármacos básicos, el pKa del contraión es preferiblemente al menos aproximadamente 2 unidades de pH más bajo que el pKa del fármaco. De manera similar, para la preparación de formas de sal de fármacos ácidos, el pKa del contraión es preferiblemente al menos aproximadamente 2 unidades de pH más alto que el pKa del fármaco. Esto permite que el contraión lleve el pH de la solución a un nivel inferior al pHmax para alcanzar la meseta de la sal, en la cual la solubilidad de la sal prevalece sobre la solubilidad del ácido o base libre. La regla generalizada de diferencia en las unidades de pKa del grupo ionizable en el ingrediente farmacéutico activo (API) y en el ácido o la base está destinada a hacer que la transferencia de protones sea energéticamente favorable. Cuando el pKa de la API y el contraión no son significativamente diferentes, puede formarse un complejo sólido, pero puede desproporcionarse rápidamente (es decir, descomponerse en las entidades individuales del fármaco y el contraión) en un entorno acuoso.
Preferiblemente, el contraión es un contraión farmacéuticamente aceptable. Las formas de sal aniónica adecuadas incluyen, pero no se limitan a acetato, benzoato, bencilato, bitartrato, bromuro, carbonato, cloruro, citrato, edetato, edisilato, estolato, fumarato, gluceptato, gluconato, hidrobromuro, hidrocloruro, yoduro, lactato, lactobionato, malato, maleato, mandelato, mesilato, bromuro de metilo, sulfato de metilo, mucato, napsilato, nitrato, pamoato (embonato), fosfato y difosfato, salicilato y disalicilato, estearato, succinato, sulfato, tartrato, tosilato, trietiyoduro, valerato y similares, mientras que las formas de sal catiónica adecuadas incluyen, pero sin limitación, aluminio, benzatina, calcio, etilendiamina, lisina, magnesio, meglumina, potasio, procaína, sodio, trometamina, zinc y similares.
La preparación de ésteres típicamente implica la funcionalización de grupos hidroxilo y/o carboxilo que están presentes dentro de la estructura molecular del agente activo. En ciertas realizaciones, los ésteres son típicamente derivados sustituidos con acilo de grupos alcohol libres, es decir, unidades estructurales que se derivan de ácidos carboxílicos de la fórmula RCOOH donde R es alquilo, y preferiblemente es alquilo inferior. Los ésteres se pueden reconvertir en ácidos libres, si se desea, utilizando procedimientos convencionales de hidrogenólisis o hidrólisis.
Las amidas también se pueden preparar usando técnicas conocidas por los expertos en la materia o descritas en la literatura pertinente. Por ejemplo, las amidas pueden prepararse a partir de ésteres, usando reactivos de amina adecuados, o pueden prepararse a partir de un anhídrido o un cloruro de ácido por reacción con amoniaco o una alquilamina inferior.
En diversas realizaciones, los agentes activos identificados en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dicha hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados, o profármacos de los mismos) son útiles para administración parenteral, administración tópica, administración oral, administración nasal (o inhalación de otro modo), administración rectal o administración local, tal como por aerosol o transdérmicamente, para el tratamiento profiláctico y/o terapéutico de una o más de las patologías/indicaciones aquí descritas (por ejemplo, patologías caracterizadas por un exceso de formación y/o depósito de placa amiloide o procesamiento no deseado de amiloide o preamiloide).
En diversas realizaciones, los agentes activos descritos en el presente documento también se pueden combinar con un vehículo (excipiente) farmacéuticamente aceptable para formar una composición farmacológica. Los vehículos farmacéuticamente aceptables pueden contener uno o más compuestos fisiológicamente aceptables que actúan, por ejemplo, para estabilizar la composición o para aumentar o disminuir la absorción del (de los) agente(s) activo(s). Los compuestos fisiológicamente aceptables pueden incluir, por ejemplo, carbohidratos, como glucosa, sacarosa o dextranos, antioxidantes, como ácido ascórbico o glutatión, agentes quelantes, proteínas de bajo peso molecular, potenciadores de la protección y la absorción, como lípidos, composiciones que reducen la eliminación o hidrólisis de los agentes activos, o excipientes u otros estabilizadores y/o reguladores.
Otros compuestos fisiológicamente aceptables, particularmente de uso en la preparación de tabletas, cápsulas, cápsulas de gel y similares incluyen, pero no se limitan a aglutinantes, diluyentes/cargas, desintegrantes, lubricantes, agentes de suspensión y similares.
En ciertas realizaciones, para fabricar una forma de dosificación oral (por ejemplo, una tableta), un excipiente (por ejemplo, lactosa, sacarosa, almidón, manitol, etc.), un desintegrador opcional (por ejemplo, carbonato de calcio, carboximetilcelulosa de calcio, almidón de sodio glicolato, crospovidona, etc.), un aglutinante (por ejemplo, almidón alfa, goma arábiga, celulosa microcristalina, carboximetilcelulosa, polivinilpirrolidona, hidroxipropilcelulosa, ciclodextrina, etc.) y un lubricante opcional (por ejemplo, talco, estearato de magnesio, polietilenglicol 6000, etc.), por ejemplo, se añaden al componente o componentes activos (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados, o profármacos de los mismos) y la composición resultante se comprime. Cuando es necesario, el producto comprimido se reviste, por ejemplo, utilizando métodos conocidos para enmascarar el sabor o para la disolución entérica o la liberación sostenida. Los materiales de recubrimiento adecuados incluyen, pero sin limitación, etilcelulosa, hidroximetilcelulosa, POLYOX®etilenglicol, ftalato de acetato de celulosa, ftalato de hidroxipropilmetilcelulosa y Eudragit (Rohm & Haas, Alemania; copolímero metacrílico-acrílico).
Otros compuestos fisiológicamente aceptables incluyen agentes humectantes, agentes emulsionantes, agentes dispersantes o conservantes que son particularmente útiles para prevenir el crecimiento o la acción de microorganismos. Varios conservantes son bien conocidos e incluyen, por ejemplo, fenol y ácido ascórbico. Un experto en la materia apreciaría que la elección de vehículo(s) farmacéuticamente aceptable(s), que incluye un compuesto fisiológicamente aceptable, depende, por ejemplo, de la ruta de administración del (de los) agente(s) activo(s) y de las características fisicoquímicas particulares del agente activo(s)
En ciertas realizaciones, los excipientes son estériles y generalmente están libres de materia indeseable. Estas composiciones pueden esterilizarse mediante técnicas de esterilización convencionales y bien conocidas. Para varios excipientes de forma de dosificación oral, como tabletas y cápsulas, no se requiere esterilidad. El estándar USP/NF suele ser suficiente.
Las composiciones farmacéuticas pueden administrarse en una variedad de formas de dosificación unitarias dependiendo del método de administración. Las formas de dosificación unitaria adecuadas incluyen, pero no se limitan a, polvos, tabletas, píldoras, cápsulas, pastillas, supositorios, parches, aerosoles nasales, inyectables, formulaciones implantables de liberación sostenida, películas mucoadherentes, barnices tópicos, complejos lipídicos, etc.
Composiciones farmacéuticas que comprenden los agentes activos descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dicha hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) pueden fabricarse mediante procesos convencionales de mezcla, disolución, granulación, fabricación de grageas, levigación, emulsión, encapsulación, atrapamiento o liofilización. Las composiciones farmacéuticas pueden formularse de manera convencional usando uno o más vehículos, diluyentes, excipientes o auxiliares fisiológicamente aceptables que facilitan el procesamiento del (de los) agente(s) activo(s) en preparaciones que pueden usarse farmacéuticamente. La formulación adecuada depende de la ruta de administración elegida.
En ciertas realizaciones, los agentes activos descritos en el presente documento están formulados para administración oral. Para la administración oral, las formulaciones adecuadas se pueden formular fácilmente combinando el (los) agente(s) activo(s) con vehículos farmacéuticamente aceptables adecuados para administración oral bien conocidos en la técnica. Dichos vehículos permiten que el (los) agente(s) activo(s) aquí descrito se formulen como tabletas, píldoras, grageas, cápsulas, grageas, cápsulas de gel, cápsulas, líquidos, geles, jarabes, lodos, suspensiones y similares, por vía oral para un paciente a ser tratado. Para formulaciones orales sólidas como, por ejemplo, polvos, cápsulas y tabletas, los excipientes adecuados pueden incluir cargas tales como azúcares (por ejemplo, lactosa, sacarosa, manitol y sorbitol), preparaciones de celulosa (por ejemplo, almidón de maíz, almidón de trigo, almidón de arroz, almidón de patata, gelatina, goma de tragacanto, metilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa de sodio), polímeros sintéticos (por ejemplo, polivinilpirrolidona (PVP)), agentes de granulación; y agentes aglutinantes. Si se desea, se pueden agregar agentes desintegrantes, como la polivinilpirrolidona entrecruzada, agar o ácido algínico o una sal de los mismos, como el alginato de sodio. Si se desea, las formas de dosificación sólidas pueden recubrirse con azúcar o con recubrimiento entérico utilizando técnicas estándar. La preparación de partículas con recubrimiento entérico se describe, por ejemplo, en las Patentes de los Estados Unidos Nos. 4,786,505 y 4,853,230.
Para la administración por inhalación, el (los) agente(s) activo(s) se administra(n) convenientemente en forma de aerosol de paquetes presurizados o un nebulizador, con el uso de un propulsor adecuado, por ejemplo, diclorodifluorometano, triclorofluorometano, diclorotetrafluoroetano, dióxido de carbono o otro gas adecuado En el caso de un aerosol presurizado, la unidad de dosificación se puede determinar proporcionando una válvula para administrar una cantidad medida. Cápsulas y cartuchos de por ejemplo, la gelatina para usar en un inhalador o insuflador puede formularse con una mezcla en polvo del compuesto y una base en polvo adecuada tal como lactosa o almidón.
En diversas realizaciones, los agentes activos pueden formularse en composiciones rectales o vaginales tales como supositorios o enemas de retención, por ejemplo, que contienen bases de supositorios convencionales tales como manteca de cacao u otros glicéridos. Los expertos en la técnica conocen bien los métodos para formular agentes activos para el suministro rectal o vaginal (véase, por ejemplo, Allen (2007) Suppositories, Pharmaceutical Press) y generalmente implican la combinación de los agentes activos con una base adecuada (por ejemplo, hidrofílica (PEG), materiales lipofílicos como la manteca de cacao o Witepsol W45, materiales anfifílicos como Suppocire AP y glicéridos poliglicolizados, y similares). La base se selecciona y combina para un perfil de fusión/suministro deseado.
Para la administración tópica, el (los) agente(s) activo(s) descrito(s) en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de este, o sales o solvatos farmacéuticamente aceptables de dicha hidantoína(s), dicho estereoisómero( s), o dicho(s) tautómero(s), o análogos, derivados, o profármacos de los mismos) pueden formularse como soluciones, geles, ungüentos, cremas, suspensiones y similares como son bien conocidos en la técnica.
En ciertas realizaciones, los agentes activos descritos en el presente documento están formulados para administración sistémica (por ejemplo, como un inyectable) de acuerdo con métodos estándar bien conocidos por los expertos en la técnica. Las formulaciones sistémicas incluyen, entre otras, aquellas diseñadas para administración por inyección, por ejemplo, inyección subcutánea, intravenosa, intramuscular, intratecal o intraperitoneal, así como aquellas diseñadas para administración transdérmica, transmucosa oral o pulmonar. Para inyección, los agentes activos descritos en el presente documento pueden formularse en soluciones acuosas, preferiblemente en reguladores fisiológicamente compatibles tales como solución de Hanks, solución de Ringer o regulador salino fisiológico y/o en ciertas formulaciones de emulsión. La(s) solución(es) puede(n) contener agentes formuladores tales como agentes de suspensión, estabilizantes y/o dispersantes. En ciertas realizaciones, el (los) agente(s) activo(s) pueden proporcionarse en forma de polvo para constituir con un vehículo adecuado, por ejemplo, agua estéril libre de pirógenos, antes de su uso. Para la administración transmucosa, y/o para el paso de la barrera hematoencefálica, se pueden usar en la formulación penetrantes apropiados para la barrera a atravesar. Tales penetrantes son generalmente conocidos en la técnica. Las formulaciones inyectables y las formulaciones inhalables generalmente se proporcionan como una formulación estéril o sustancialmente estéril.
Además de las formulaciones descritas previamente, los agentes activos también pueden formularse como preparaciones de depósito. Dichas formulaciones de acción prolongada pueden administrarse por implantación (por ejemplo, por vía subcutánea o intramuscular) o por inyección intramuscular. Así, por ejemplo, el (los) agente(s) activo(s) puede(n) formularse con materiales poliméricos o hidrófobos adecuados (por ejemplo, como una emulsión en un aceite aceptable) o resinas de intercambio iónico, o como derivados escasamente solubles, por ejemplo, como una sal escasamente soluble .
En ciertas realizaciones, el (los) agente(s) activo(s) aquí descrito también pueden administrarse a través de la piel usando sistemas convencionales de administración transdérmica de fármacos, es decir, "parches" transdérmicos en los que el (los) agente(s) activo(s) está(n) típicamente contenido(s) dentro de una estructura laminada que sirve como un dispositivo de suministro de drogas para ser fijado a la piel. En tal estructura, la composición del fármaco está típicamente contenida en una capa, o "depósito", subyacente a una capa de respaldo superior. Se apreciará que el término "reservorio" en este contexto se refiere a una cantidad de "ingrediente(s) activo(s)" que está finalmente disponible para su administración a la superficie de la piel. Así, por ejemplo, el "depósito" puede incluir el ingrediente
o ingredientes activos en un adhesivo en una capa de respaldo del parche, o en cualquiera de una variedad de formulaciones de matriz diferentes conocidas por los expertos en la técnica. El parche puede contener un solo depósito, o puede contener múltiples depósitos.
En una realización ilustrativa, el depósito comprende una matriz polimérica de un material adhesivo de contacto farmacéuticamente aceptable que sirve para fijar el sistema a la piel durante la administración del fármaco. Ejemplos de materiales adhesivos de contacto con la piel adecuados incluyen, pero no se limitan a, polietilenos, polisiloxanos, poliisobutilenos, poliacrilatos, poliuretanos y similares. Alternativamente, el depósito que contiene el medicamento y el adhesivo de contacto con la piel están presentes como capas separadas y distintas, con el adhesivo subyacente al depósito que, en este caso, puede ser una matriz polimérica como se describió anteriormente, o puede ser un depósito de líquido o hidrogel, o puede tomar alguna otra forma. La capa de respaldo en estos laminados, que sirve como la superficie superior del dispositivo, funciona preferiblemente como un elemento estructural primario del "parche" y proporciona al dispositivo mucha de su flexibilidad. El material seleccionado para la capa de respaldo es preferiblemente sustancialmente impermeable al (a los) agente(s) activo(s) y a cualquier otro material que esté presente.
Alternativamente, se pueden emplear otros sistemas de suministro farmacéutico. Por ejemplo, los liposomas, las emulsiones y las microemulsiones/nanoemulsiones son ejemplos bien conocidos de vehículos de administración que pueden usarse para proteger y administrar compuestos farmacéuticamente activos. También se pueden emplear ciertos solventes orgánicos como el dimetilsulfóxido, aunque generalmente a costa de una mayor toxicidad.
En ciertas realizaciones, los agentes activos descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) se formulan en una nanoemulsión. Las nanoemulsiones incluyen, entre otras, nanoemulsiones de aceite en agua (O/W) y nanoemulsiones de agua en aceite (W/O). Las nanoemulsiones pueden definirse como emulsiones con diámetros medios de gota que varían de aproximadamente 20 a aproximadamente 1000 nm. Usualmente, el tamaño promedio de gota es entre aproximadamente 20 nm o 50 nm y aproximadamente 500 nm. Los términos emulsión submicrométrica (SME) y miniemulsión se usan como sinónimos.
Las nanoemulsiones ilustrativas de aceite en agua (O/W) incluyen, pero no se limitan a: micelas tensioactivas: micelas compuestas de moléculas pequeñas de tensioactivos o detergentes (por ejemplo, SDS/PBS/2-propanol); Micelas poliméricas: micelas compuestas de polímeros, copolímeros o tensioactivos de copolímero de bloque (por ejemplo, Pluronic L64/PBS/2-propanol); Micelas mixtas en las que hay más de un componente tensioactivo o en las que una de las fases líquidas (generalmente un compuesto de alcohol o ácido graso) participa en la formación de la micela (por ejemplo, ácido octanoico/PBS/EtOH); Micelas integrales: micelas combinadas en las que el (los) agente(s) activo(s) sirven como tensioactivo auxiliar, formando una parte integral de la micela; y emulsiones Pickering (fase sólida): emulsiones en las que los agentes activos están asociados con el exterior de una nanopartícula sólida (por ejemplo, nanopartículas de poliestireno/PBS/sin fase oleosa).
Las nanoemulsiones ilustrativas de agua en aceite (W/O) incluyen, pero no se limitan a: micelas tensioactivas - micelas compuestas de moléculas pequeñas de tensioactivos o detergentes (por ejemplo, dioctilsulfosuccinato/PBS/2-propanol, isopropilmidiristato/PBS/2-propanol, etc.); Micelas de polímeros: micelas compuestas de polímeros, copolímeros o tensioactivos de copolímero de bloque (por ejemplo, PLURONIC® L121/PBS/2-propanol); Micelas mezcladas: micelas en las que hay más de un componente tensioactivo o en las que una de las fases líquidas (generalmente un compuesto de alcohol o ácido graso) participa en la formación de la micela (por ejemplo, diglicérido cáprico/caprílico/PBS/EtOH); Micelas integrales: micelas mezcladas en las que el (los) agente(s) activo(s) sirven como tensioactivo auxiliar, formando una parte integral de la micela (por ejemplo, agente activo/PBS/polipropilenglicol); y emulsiones Pickering (fase sólida): emulsiones en las que los agentes activos están asociados con el exterior de una nanopartícula sólida (por ejemplo, nanopartículas de quitosano/sin fase acuosa/aceite mineral).
Como se indicó anteriormente, en ciertas realizaciones las nanoemulsiones comprenden uno o más tensioactivos o detergentes. En algunas realizaciones, el tensioactivo es un detergente no aniónico (por ejemplo, un tensioactivo de polisorbato, un éter de polioxietileno, etc.). Los tensioactivos que encuentran uso en la presente invención incluyen, pero no se limitan a tensioactivos tales como las familias de compuestos TWEEN®, TRITON® y TYLOXAPOL®.
En ciertas realizaciones, las emulsiones comprenden además uno o más compuestos que contienen halógeno catiónico, que incluyen, pero no se limitan a, cloruro de cetilpiridinio. En otras realizaciones adicionales, las composiciones comprenden además uno o más compuestos que aumentan la interacción ("potenciadores de la interacción") de la composición con microorganismos (por ejemplo, agentes quelantes como ácido etilendiaminotetraacético o ácido etilenbis (oxietilenitrilo) tetraacético en un regulador).
En algunas realizaciones, la nanoemulsión comprende además un agente emulsionante para ayudar en la formación de la emulsión. Los agentes emulsionantes incluyen compuestos que se agregan en la interfaz aceite/agua para formar una especie de membrana continua que evita el contacto directo entre dos gotas adyacentes. Ciertas realizaciones de la presente invención presentan composiciones de emulsión de aceite en agua que pueden diluirse fácilmente con agua a una concentración deseada sin perjudicar sus propiedades antipatógenas.
Además de las pequeñas gotas de aceite dispersas en una fase acuosa, ciertas emulsiones de aceite en agua también pueden contener otras estructuras lipídicas, como pequeñas vesículas lipídicas (por ejemplo., esferas lipídicas que a menudo consisten en varias bicapas lipídicas sustancialmente concéntricas separadas de entre sí por capas de fase acuosa), micelas (por ejemplo, moléculas anfifílicas en pequeños grupos de 50-200 moléculas dispuestas de manera que los grupos de la cabeza polar miren hacia afuera hacia la fase acuosa y las colas apolares estén secuestradas hacia adentro lejos de la fase acuosa), o fases lamelares (dispersiones lipídicas en las que cada partícula consiste en bicapas anfifílicas paralelas separadas por delgadas películas de agua).
Estas estructuras lipídicas se forman como resultado de fuerzas hidrófobas que conducen los residuos apolares (por ejemplo, largas cadenas de hidrocarburos) lejos del agua. Las preparaciones lipídicas anteriores pueden describirse generalmente como preparaciones lipídicas tensioactivas (SLP). Los SLP son mínimamente tóxicos para las membranas mucosas y se cree que se metabolizan dentro del intestino delgado (véase, por ejemplo, Hamouda et al., (1998) J. Infect. Disease 180: 1939).
En ciertas realizaciones, la emulsión comprende una fase oleosa discontinua distribuida en una fase acuosa, un primer componente que comprende un alcohol y/o glicerol, y un segundo componente que comprende un tensioactivo o un compuesto que contiene halógeno. La fase acuosa puede comprender cualquier tipo de fase acuosa que incluye, pero no se limita a, agua (por ejemplo, agua desionizada, agua destilada, agua corriente) y soluciones (por ejemplo, solución salina tamponada con fosfato u otros sistemas regulador). La fase oleosa puede comprender cualquier tipo de aceite, incluidos, entre otros, aceites vegetales (por ejemplo, aceite de soja, aceite de aguacate, aceite de linaza, aceite de coco, aceite de algodón, aceite de escualeno, aceite de oliva, aceite de canola, aceite de maíz, aceite de colza, aceite de cártamo y aceite de girasol), aceites animales (por ejemplo, aceite de pescado), aceite aromatizante, vitaminas insolubles en agua, aceite mineral y aceite de motor. En ciertas realizaciones, la fase oleosa comprende 30-90% en volumen de la emulsión de aceite en agua (por ejemplo, constituye 30-90% del volumen total de la emulsión final), más preferiblemente 50-80%. Las formulaciones no necesitan limitarse a tensioactivos particulares, sin embargo, en ciertas realizaciones, el tensioactivo es un tensioactivo polisorbato (por ejemplo, TWEEN 20®, TWEEN 40®, TWEEN 60® y TWEEN 80®), un fenoxipolietoxietanol (por ejemplo, TRITON® X-100, X-301, X-165, X-102 y X-200, y TYLOXAPOL®), o dodecil sulfato de sodio, y similares.
En ciertas realizaciones, está presente un componente que contiene halógeno. la naturaleza del compuesto que contiene halógeno, en algunas realizaciones, el compuesto que contiene halógeno comprende una sal de cloruro (por ejemplo, NaCl, KCl, etc.), un haluro de cetilpiridinio, un haluro de cetiltrimetilamonio, un haluro de cetildimetiletilamonio, un haluro de cetildimetilbencilamonio, haluro de cetiltributilfosfonio, haluros de dodeciltrimetilamonio, haluros de tetradeciltrimetilamonio, cloruro de cetilpiridinio, cloruro de cetiltrimetilamonio, cloruro de cetilbencildimetilamonio, bromuro de cetilpiridinio, bromuro de cetiltrimetilamonio, bromuro de cetildimetiletilamonio, bromuro de cetiltributilfosfonio, bromuro de dodeciltrimetilamonio, bromuro de tetradeciltrimetilamonio y similares.
En ciertas realizaciones, la emulsión comprende un compuesto de amonio cuaternario. Los compuestos de amonio cuaternario incluyen, pero no se limitan a, sacarinato de N-alquildimetil bencil amonio, 1,3,5-triazina-1,3,5(2H,4H,6H)-trietanol; 1-decacanaminio, N-decil-N,N-dimetil-, cloruro (o) cloruro de didecil dimetilamonio; cloruro de 2-(2-(p-(diisobuil)cresosxi)etoxi)etil dimetil bencil amonio; cloruro de 2-(2-(p-(diisobutil)fenoxi)etoxi)etil dimetil bencil amonio; cloruro de alquilo 1 o 3 bencil-1-(2-hidroxetil)-2-imidazolinio; cloruro de alquil bis(2-hidroxietil)bencil amonio; cloruro de alquil dimetil bencil amonio; cloruro de alquil dimetil 3,4-diclorobencilamonio (100% C12); cloruro de alquil dimetil 3,4-diclorobencilamonio (50% C14, 40% C12, 10% C16); cloruro de alquil dimetil 3,4-diclorobencilamonio (55% C14, 23% C12, 20% C16); cloruro de alquil dimetil bencil amonio; cloruro de alquil dimetil bencil amonio (100% C14); cloruro de alquil dimetil bencil amonio (100% C16); cloruro de alquil dimetil bencil amonio (41% C14, 28% C12); cloruro de alquil dimetil bencil amonio (47% C12, 18% C14); cloruro de alquil dimetil bencil amonio (55% C16, 20% C14); cloruro de alquil dimetil bencil amonio (58% C14, 28% C16); cloruro de alquil dimetil bencil amonio (60% C14, 25% C12); cloruro de alquil dimetil bencil amonio (61% C11, 23% C14); cloruro de alquil dimetil bencil amonio (61% C12, 23% C14); cloruro de alquil dimetil bencil amonio (65% C12, 25% C14); cloruro de alquil dimetil bencil amonio (67% C12, 24% C14); cloruro de alquil dimetil bencil amonio (67% C12, 25% C14); cloruro de alquil dimetil bencil amonio (90% C14, 5% C12); cloruro de alquil dimetil bencil amonio (93% C14, 4% C12); cloruro de alquil dimetil bencil amonio (95% C16, 5% C18); cloruro de alquil dimetil bencil amonio (y) cloruro de didecil dimetil amonio; cloruro de alquil dimetil bencil amonio (como en ácidos grasos); cloruro de alquil dimetil bencil amonio (C12-C16); cloruro de alquil dimetil bencil amonio (C12-C18); cloruro de alquil dimetil bencilo y dialquil dimetil amonio; cloruro de alquil dimetil dimetibencilamonio; bromuro de alquil dimetil etil amonio (90% C14, 5% C16, 5% C12); bromuro de alquil dimetil etil amonio (grupos mixtos de alquilo y alquenilo como en los ácidos grasos del aceite de soja); cloruro de alquil dimetil etil bencil amonio; cloruro de alquil dimetil etil bencil amonio (60% C14); cloruro de alquil dimetil isoproilbencil amonio (50% C12, 30% C14, 17% C16, 3% C18); cloruro de alquil trimetilamonio (58% C18, 40% C16, 1% C14, 1% C12); cloruro de alquil trimetilamonio (90% C18, 10% C16); cloruro de alquildimetil (etilbencil) amonio (C12-18); cloruros de di-(C8-10)-alquil dimetilamonio; cloruro de dialquil dimetilamonio; cloruro de dialquil dimetilamonio; cloruro de dialquil dimetilamonio; cloruro de dialquil metil bencil amonio; cloruro de didecil dimetil amonio; cloruro de diisodecil dimetilamonio; cloruro de dioctil dimetil amonio; cloruro de dodecil bis(2-hidroxietil)octil hidrógeno amonio; cloruro de dodecil dimetil bencil amonio; cloruro de dodecilcarbamoil metil dimetil bencil amonio; cloruro de heptadecil hidroxietilimidazolinio; hexahidro-1,3,5-tris(2-hidroxietil)-s-triazina; cloruro de miristaltonio (y) Quat RNIUM 14; polímero de cloruro de N,N-dimetil-2-hidroxipropilamonio; cloruro de n-alquil dimetil bencil amonio; cloruro de n-alquil dimetil etil bencil amonio; monohidrato de cloruro de n-tetradecil dimetil bencil amonio; cloruro de octil decil dimetil
amonio; cloruro de octil dodecil dimetil amonio; cloruro de octifenoxietoxietil dimetil bencil amonio; oxidietilenbis (cloruro de alquil dimetilamonio); compuestos de amonio cuaternario, dicoco alquildimetil, cloruro; cloruro de trimetoxisililo propil dimetil octadecil amonio; trimetoxisilil quats, cloruro de trimetil dodecilbencil amonio; cloruro de ndodecil dimetil etil bencil amonio; cloruro de n-hexadecil dimetil bencil amonio; cloruro de n-tetradecil dimetil bencil amonio; cloruro de n-tetradecil dimetil etil bencil amonio; y cloruro de n-octadecil dimetil bencil amonio.
Las formulaciones de nanoemulsión y los métodos para prepararlos son bien conocidos por los expertos en la técnica y se describen, por ejemplo, en las Patentes de los Estados Unidos Nos: 7,476,393, 7,468,402, 7,314,624, 6,998,426, 6,902,737, 6,689,371, 6,541,018, 6,464,990, 6,461,625, 6,419,946, 6,413,527, 6,375,960, 6,335,022, 6,274,150, 6,120,778, 6,039,936, 5,925,341, 5,753,241, 5,698,219, y 5,152,923 y en Fanun et al. (2009) Microemulsions: Properties and Applications (Surfactant Science), CRC Press, Boca Ratan Fl.
En ciertas realizaciones, uno o más agentes activos descritos en el presente documento pueden proporcionarse como un "concentrado", por ejemplo, en un recipiente de almacenamiento (por ejemplo, en un volumen previamente medido) listo para la dilución, o en una cápsula soluble lista para la adición a un volumen de agua, alcohol, peróxido de hidrógeno u otro diluyente.
Formulaciones de liberación prolongada (liberación sostenida)
En ciertas realizaciones, formulaciones de "liberación prolongada" de los agentes activos descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dicha hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) están contemplados. En diversas realizaciones, tales formulaciones de liberación prolongada están diseñadas para evitar los altos niveles plasmáticos máximos de las formas de dosificación oral intravenosa y de liberación inmediata convencional.
Las formulaciones ilustrativas de liberación sostenida incluyen, por ejemplo, matrices semipermeables de polímeros sólidos que contienen el agente terapéutico. Se han establecido diversos usos de materiales de liberación sostenida y son bien conocidos por los expertos en la materia. Las cápsulas de liberación sostenida pueden, dependiendo de su naturaleza química, liberar los compuestos durante algunas semanas hasta más de 100 días. Dependiendo de la naturaleza química y la estabilidad biológica del reactivo terapéutico, se pueden emplear estrategias adicionales para la estabilización.
En ciertas realizaciones, tales formulaciones de "liberación prolongada" utilizan la mucosa y pueden controlar independientemente la desintegración (o erosión) del comprimido y/o la disolución del fármaco y la liberación del comprimido a lo largo del tiempo para proporcionar un perfil de suministro más seguro. En ciertas realizaciones, las formulaciones orales de los agentes activos descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho estereoisómero( s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) proporcionan dosis repetitivas individuales que incluyen una cantidad definida del agente activo que se administra durante un período de tiempo definido.
Una formulación ilustrativa de liberación sostenida es una composición sustancialmente homogénea que comprende aproximadamente 0.01% a aproximadamente 99% p/p, o aproximadamente 0.1% a aproximadamente 95%, o aproximadamente 0.1%, o aproximadamente 1%, o aproximadamente 2%, o aproximadamente 5%, o aproximadamente 10%, o aproximadamente 15%, o aproximadamente 20% a aproximadamente 80%, o aproximadamente 90%, o aproximadamente 95%, o aproximadamente 97%, o aproximadamente 98%, o aproximadamente el 99% de los ingredientes activos (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) y uno o más mucoadhesivos (también denominados en el presente documento "bioadhesivos") que proporcionan adherencia a la mucosa objetivo del sujeto (paciente) y que pueden comprender además uno o más de los siguientes: uno o más aglutinantes que proporcionan la unión de los excipientes en una sola tableta; uno o más excipientes formadores de hidrogel; uno o más agentes de carga; uno o más lubricantes; uno o más deslizantes; uno o más solubilizantes; uno o más tensioactivos; uno o más sabores; uno o más desintegrantes; uno o más excipientes reguladores; uno o más recubrimientos; uno o más modificadores de liberación controlada; y uno o más excipientes y factores que modifican y controlan el tiempo de disolución o desintegración del fármaco y la cinética o protegen el fármaco activo frente a la degradación.
En diversas realizaciones, una forma de dosificación farmacéutica de liberación sostenida para administración transmucosa oral puede ser sólida o no sólida. En una realización ilustrativa, la forma de dosificación es un sólido que se convierte en un hidrogel después del contacto con la saliva.
Los excipientes adecuados incluyen, pero no se limitan a, sustancias añadidas a las formulaciones que se requieren para producir un producto comercial y pueden incluir, pero no se limitan a: agentes de carga, aglutinantes, tensioactivos, bioadhesivos, lubricantes, desintegrantes, estabilizadores, solubilizantes, deslizantes y aditivos o factores que afectan el tiempo de disolución o desintegración. Los excipientes adecuados no se limitan a los anteriores,
y se pueden encontrar otros portadores farmacéuticamente aceptables no tóxicos adecuados para uso en formulaciones orales en Remington's Pharmaceutical Sciences, 17a edición, 1985.
En ciertas realizaciones, las formulaciones de liberación prolongada del (de los) agente(s) activo(s) descrito aquí para la administración oral de fármacos transmucosa incluyen al menos un agente bioadhesivo (mucoadhesivo) o una mezcla de varios bioadhesivos para promover la adhesión a la mucosa oral durante la administración del fármaco. Además, los agentes bioadhesivos también pueden ser efectivos para controlar el tiempo de erosión de la forma de dosificación y/o la cinética de disolución del fármaco con el tiempo cuando la forma de dosificación se humedece. Tales sistemas de administración de fármacos mucoadhesivos son muy beneficiosos, ya que pueden prolongar el tiempo de residencia del fármaco en el sitio de absorción y aumentar la biodisponibilidad del fármaco. Los polímeros mucoadhesivos que forman hidrogeles son típicamente hidrófilos e hinchables, y contienen numerosos grupos formadores de enlaces de hidrógeno, como hidroxilo, carboxilo o amina, que favorecen la adhesión. Cuando se usan en forma seca, atraen agua de la superficie de la mucosa y se hinchan, lo que conduce a la interacción polímero/moco a través de enlaces de hidrógeno, electrostática, hidrófoba o interacción de van der Waals.
Los materiales mucoadhesivos o bioadhesivos adecuados ilustrativos incluyen, pero no se limitan a polímeros naturales, sintéticos o biológicos, lípidos, fosfolípidos y similares. Ejemplos de polímeros naturales y/o sintéticos incluyen derivados celulósicos (como metilcelulosa, carboximetilcelulosa, hidroxietilcelulosa, hidroxietilmetilcelulosa, etc.), gomas naturales (como goma guar, goma xantana, goma de algarrobilla, goma karaya, goma vee, etc.) , poliacrilatos (como CARBOPOL®, policarbofilo, etc.), alginatos, polímeros que contienen tiol, POLYOX® etilenos, polietilenglicoles (PEG) de todos los pesos moleculares (preferiblemente entre 1000 y 40.000 Da, de cualquier química, lineal o ramificada), dextranos de todos los pesos moleculares (preferiblemente entre 1000 y 40.000 Da de cualquier fuente), copolímeros de bloque, como los preparados por combinaciones de ácido láctico y glicólico (PLA, PGA, p Lg A de diversas viscosidades, pesos moleculares y relaciones de ácido láctico a glicólico) copolímeros de bloque de polietilenglicol-polipropilenglicol de cualquier número y combinación de unidades repetidas (tales como copolímeros de bloque Pl Ur On iCs®, TEKTRONIX® o GENAPOL®), combinación de los copolímeros anteriores en unidades unidas física o químicamente (por ejemplo, copolímeros PEG-PLA o PEG-PLGA). Preferiblemente, el excipiente bioadhesivo se selecciona del grupo de polietilenglicoles, POLYOX® etilenos, polímeros de ácido poliacrílico, tales como CARBOPOL® (como CARBOPOL® 71G, 934P, 971P, 974P y similares) y policarbofilos (como NOVEON® AA-1, NOVEON® CA-1, NOVEON® CA-2 y similares), celulosa y sus derivados y lo más preferiblemente es polietilenglicol, carbopol y/o un derivado celulósico o una combinación de los mismos.
En ciertas realizaciones, el excipiente mucoadhesivo/bioadhesivo está presente típicamente a 1-50% p/p, preferiblemente 1-40% p/p o lo más preferiblemente entre 5-30% p/p. Una formulación particular puede contener uno o más bioadhesivos diferentes en cualquier combinación.
En ciertas realizaciones, las formulaciones para la administración oral de fármacos transmucosa también incluyen un aglutinante o mezcla de dos o más aglutinantes que facilitan la unión de los excipientes en una única forma de dosificación. Los aglutinantes ilustrativos incluyen aglutinantes seleccionados del grupo que consiste en derivados celulósicos (como metilcelulosa, carboximetilcelulosa, hidroxietilcelulosa, hidroxietilmetilcelulosa, etc.), poliacrilatos (como CARBOPOL®, policarbofilo, etc.), POVIDONE® (todos los grados), POLYOX®® de cualquier peso o grado molecular, irradiado o no, almidón, polivinilpirrolidona (PVP), AVICEL® y similares. En ciertas realizaciones, el aglutinante está presente típicamente a 0.5-60% p/p, preferiblemente 1-30% p/p y lo más preferiblemente 1.5-15% p/p.
En ciertas realizaciones, las formulaciones también incluyen al menos un excipiente formador de hidrogel. Los excipientes formadores de hidrogel ilustrativos incluyen, pero no se limitan a los seleccionados del grupo que consiste en polietilenglicoles y otros polímeros que tienen una cadena principal de etilenglicol, ya sean homopolímeros o heteropolímeros entrecruzados, copolímeros de bloques que usan unidades de etilenglicol, como los homopolímeros de etileno POLYOX® como POLYOX®® N10/PM=100.000 POLYOX®-80/PM=200.000; POLYOX® 1105/PM=900.000; POLYOX®-301/PM=4.000.000; POLYOX®-303/PM=7.000.000, POLYOX® WSR-N-60K, todos los nombres comerciales de Union Carbide), hidroxipropilmetilcelulosa (HPMC) de todos los pesos y grados moleculares (como METOLOSE® 90SH50000, m Et OLOSE® 90SH30000, todos los cuales son nombres comerciales de Shin-Etsu Chemical Company), Poloxámeros (tales como LUTROL® F-68, LUTROL® F-127, F-105, etc., todos los nombres comerciales de Ba Sf Chemicals), g EnAPOL®, polietilenglicoles (PEG, como PEG-1500, PEG-3500, PEG-4000, PEG-6000, PEG-8000, PEG-12000, PEG-20.000, etc.), gomas naturales (goma xantana, goma de algarrobilla, etc.) y derivados de celulosa (HC, HMC, HMPC, HPC, CP, CMC), polímeros a base de ácido poliacrílico como libres o entrecruzados y combinaciones de los mismos, polímeros biodegradables como ácidos polilácticos, ácidos poliglicólicos y cualquier combinación de estos, ya sea físico mezcla o entrecruzado. En ciertas realizaciones, los componentes de hidrogel pueden estar entrecruzados. El (los) excipiente(s) formador(s) de hidrogel están típicamente presentes a 0.1-70% p/p, preferiblemente 1-50% p/p o lo más preferiblemente 1-30% p/p.
En ciertas realizaciones, las formulaciones también pueden incluir al menos un modificador de liberación controlada que es una sustancia que, tras la hidratación de la forma de dosificación, se adherirá preferiblemente a las moléculas del fármaco y, por lo tanto, reducirá la velocidad de su difusión desde la forma de dosificación oral. Dichos excipientes también pueden reducir la velocidad de absorción de agua por la formulación y, por lo tanto, permiten una disolución y liberación más prolongada del fármaco de la tableta. En general, el (los) excipiente(s) seleccionado(s) son lipófilos y
capaces de formar complejos de forma natural con los fármacos hidrófobos o lipófilos. El grado de asociación del modificador de liberación y el fármaco se puede variar alterando la relación modificador/fármaco en la formulación. Además, dicha interacción se puede potenciar adecuadamente mediante la combinación apropiada del modificador de liberación con el fármaco activo en el proceso de fabricación. Alternativamente, el modificador de liberación controlada puede ser un polímero cargado sintético o biopolímero que tenga una carga neta, ya sea positiva o negativa, y que sea capaz de unirse al activo a través de interacciones electrostáticas, modificando así su difusión a través de la tableta y/o la cinética de su permeación a través de la superficie de la mucosa. De manera similar a los otros compuestos mencionados anteriormente, dicha interacción es reversible y no implica enlaces químicos permanentes con el activo. En ciertas realizaciones, el modificador de liberación controlada puede estar presente típicamente a 0-80% p/p, preferiblemente 1-20% p/p, lo más preferiblemente 1-10% p/p.
En diversas realizaciones, las formulaciones de liberación prolongada también pueden incluir otros componentes convencionales requeridos para el desarrollo de formas de dosificación oral, que son conocidas por los expertos en la técnica. Estos componentes pueden incluir uno o más agentes de carga (como lactosa USP, Starch 1500, manitol, sorbitol, malitol u otros azúcares no reductores; celulosa microcristalina (por ejemplo, AVICEL®), deshidrato de fosfato de calcio dibásico, sacarosa y mezclas de los mismos), al menos uno o varios agentes solubilizantes (como ciclodextrinas, ajustadores de pH, sales y reguladores, tensioactivos, ácidos grasos, fosfolípidos, metales de ácidos grasos, etc.), sales metálicas y reguladores orgánicos (como acetato, citrato, tartrato, etc.) o inorgánico (fosfato, carbonato, bicarbonato, borato, sulfato, sulfito, bisulfito, metabisulfito, cloruro, etc.), sales de metales como sodio, potasio, calcio, magnesio, etc.), al menos un lubricante ( tales como ácido esteárico y cationes divalentes, tales como estearato de magnesio, estearato de calcio, etc., talco, monoestearato de glicerol y similares), uno o más deslizantes (como dióxido de silicio coloidal, dióxido de silicio precipitado, sílice pirógena (CAB-O-SIL® M-5P, marca registrada de Cabot Corporation), Stearowet y Sterotex, sílices (tales como sílices SILOID® y SILOX® - marcas registradas de Grace Davison Products, Aerosil - marca registrada de Degussa Pharma), ácidos grasos superiores, sus sales metálicas, aceites vegetales hidrogenados y similares), sabores o edulcorantes y colorantes (tales como aspartamo, manitol, lactosa, sacarosa, otros edulcorantes artificiales; óxidos férricos y escamas FD&C), aditivos para ayudar a estabilizar la sustancia farmacológica frente a productos químicos de degradación física (como antioxidantes, agentes antihidrolíticos, bloqueadores de agregación, etc. Los antioxidantes pueden incluir BHT, BHA, vitaminas, ácido cítrico, EDTA, bisulfato de sodio, metabisulfato de sodio, tiourea, metionina, tensioactivos, aminoácidos, como arginina, glicina, histidina, sales de metionina, ajustadores de pH, agentes quelantes y reguladores en forma seca o en solución), uno o más excipientes que pueden afectar la cinética de desintegración de la tableta y la liberación del fármaco de la tableta y, por lo tanto, la farmacocinética (desintegrantes como los conocidos por los expertos en la técnica y pueden seleccionarse de un grupo que consiste en almidón, tipo de carboximetilcelulosa o polivinilpirrolidona entrecruzada (como crospovidona, PVP-XL), alginatos, desintegrantes a base de celulosa (como celulosa purificada, metilcelulosa, carboximetilcelulosa sódica entrecruzada (Ac-Di-Sol) y carboximetilcelulosa), bajos en éteres hidroxipropilicos de celulosa de baja sustitución, celulosa microcristalina (como AVICEL®), resinas de intercambio iónico (como AMBRELITE® iPr 88), gomas (como agar, langosta, karaya, pectina y tragacanto), gomas guar, goma karaya, quitina y quitosano, esmecta, goma gelano, cáscara de isapghula, polacrilina de potasio (Tulsion339), desintegrantes que producen gases (como ácido cítrico y ácido tartárico junto con bicarbonato de sodio, carbonato de sodio, bicarbonato de potasio o carbonato de calcio), almidón glicolato de sodio como EXPLOTAB® y PRIMOGEL®), almidón DC y similares, al menos un polímero biodegradable de cualquier tipo útil para la liberación prolongada de fármacos. Las composiciones poliméricas ejemplares incluyen, pero no se limitan a, polianhídridos y copolímeros de ácido láctico y ácido glicólico, poli(di-lactida-co-glicólido) (PLGA), poli(ácido láctico) (PLA), poli(ácido glicólico) (PGA), poliortoésteres, proteínas y polisacáridos.
En ciertas realizaciones, el (los) agente(s) activo(s) puede(n) modificarse químicamente para modificar significativamente la farmacocinética en plasma. Esto se puede lograr, por ejemplo, mediante conjugación con poli(etilenglicol) (PEG), incluida la PEGilación específica del sitio. PEGilación, que puede mejorar el rendimiento del fármaco optimizando la farmacocinética, disminuyendo la inmunogenicidad y la frecuencia de dosificación.
Métodos para preparar una formulación de los agentes activos descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), también se proporcionan dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos para suministro GI o transmucosa oral. Un método incluye los pasos de molienda de polvo, mezcla de polvo seco y formación de tabletas mediante compresión directa. Alternativamente, se puede usar un proceso de granulación en húmedo. Tal método (como el proceso de granulación de alto cizallamiento) implica mezclar el ingrediente activo y posiblemente algunos excipientes en un mezclador. El aglutinante puede ser uno de los excipientes añadidos en estado de mezcla seca o disuelto en el fluido utilizado para la granulación. La solución o suspensión de granulación se agrega a los polvos secos en el mezclador y se mezcla hasta lograr las características deseadas. Esto generalmente produce un gránulo que tendrá características adecuadas para producir formas de dosificación con un tiempo de disolución adecuado, uniformidad de contenido y otras características físicas. Después de la etapa de granulación húmeda, el producto se seca con mayor frecuencia y/o luego se muele después del secado para obtener un porcentaje importante del producto dentro de un rango de tamaño deseado. A veces, el producto se seca después de un tamaño húmedo utilizando un dispositivo como un granulador oscilante o un molino. La granulación en seco puede procesarse para obtener un rango de tamaño aceptable mediante el primer cribado con un dispositivo de tamizado y luego moliendo las partículas de gran tamaño.
Además, la formulación puede fabricarse mediante procesos de granulación alternativos, todos conocidos por los expertos en la técnica, tales como granulación por lecho fluido por pulverización, extrusión y esferonización o granulación por rotor de lecho fluido.
Además, la forma de dosificación en tableta del (de los) agente(s) activo(s) descrito aquí puede prepararse recubriendo la tableta primaria fabricada como se describió anteriormente con recubrimientos adecuados conocidos en la técnica. Dichos recubrimientos están destinados a proteger los núcleos activos contra daños (abrasión, rotura, formación de polvo) contra las influencias a las que están expuestos los núcleos durante el transporte y el almacenamiento (humedad atmosférica, fluctuaciones de temperatura), y naturalmente estos recubrimientos de película también se pueden colorear. El efecto de sellado de las capas de película contra el vapor de agua se expresa por la permeabilidad al vapor de agua. El recubrimiento se puede realizar mediante uno de los procesos disponibles, como el recubrimiento Wurster, el recubrimiento seco, el recubrimiento de película, el recubrimiento de lecho fluido, el recubrimiento en recipiente, etc. Los materiales de recubrimiento típicos incluyen polivinilpirrolidona (PVP), copolímero de polivinilpirrolidona y acetato de vinilo (PVPVA), alcohol polivinilo (PVA), copolímero de polivinil alcohol/polietilenglicol (PVA/PEG), ftalato de acetato de celulosa, etil celulosa, goma gelano, maltodextrina, metacrilatos, metil celulosa, hidroxipropil metil celulosa (HPMC de todos los grados y pesos moleculares), carragenano, goma laca y similares.
En ciertas realizaciones, el núcleo de la tableta que comprende el (los) agente(s) activo(s) descrito aquí puede recubrirse con un material bioadhesivo y/o resistente al pH para permitir que el material, como los definidos anteriormente, mejore la bioadhesión de la tableta en la cavidad sublingual .
En ciertas realizaciones, los agentes activos descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) se formulan como complejos de inclusión. Si bien no se limita a los complejos de inclusión de ciclodextrina, se observa que la ciclodextrina es el agente más utilizado para formar complejos de inclusión farmacéuticos. Las ciclodextrinas (CD) son oligómeros cíclicos de glucosa, que típicamente contienen 6, 7 u 8 monómeros de glucosa unidos por enlaces a-1,4. Estos oligómeros se denominan comúnmente a-CD, p-CD y y-CD, respectivamente. Se conocen oligómeros superiores que contienen hasta 12 monómeros de glucosa, y se contemplan en las formulaciones descritas aquí. También se contemplan complejos de inclusión de ciclodextrina funcionalizada. Las ciclodextrinas funcionalizadas ilustrativas, pero no limitantes, incluyen, pero sin limitación, sulfonatos, sulfonatos y sulfinatos, o disulfonatos de hidroxibutenil ciclodextrina; sulfonatos, sulfonatos y sulfinatos, o disulfonatos de éteres mixtos de ciclodextrinas donde al menos uno de los sustituyentes éter es hidroxibutenil ciclodextrina. Las ciclodextrinas ilustrativas incluyen un éter de polisacárido que comprende al menos un sustituyente 2-hidroxibutenilo, en donde el al menos un sustituyente hidroxibutenilo es sulfonado y sulfinado, o disulfonado, y un éter alquilpoliglicósido que comprende al menos un sustituyente 2-hidroxibutenilo, en donde el al menos uno el sustituyente hidroxibutenilo está sulfonado y sulfinado, o disulfonado. En diversas realizaciones, se contemplan complejos de inclusión formados entre hidroxibutenil ciclodextrinas sulfonadas y uno o más de los agentes activos descritos en el presente documento. Los métodos para preparar ciclodextrinas y los complejos de inclusión de ciclodextrina se encuentran, por ejemplo, en la publicación de Patente de los Estados Unidos No: 2004/0054164 y las referencias citadas en la misma y en la publicación de Patente de los Estados Unidos No: 2011/0218173 y las referencias citadas en la misma.
Farmacocinética (PK) y atributos de formulación
Una ventaja de las formulaciones orales (GI o transmucosa) de liberación prolongada (controlada) descritas en el presente documento es que pueden mantener la concentración de fármaco en plasma dentro de una ventana terapéutica dirigida durante más tiempo que con las formulaciones de liberación inmediata, ya sea en forma de dosificación sólida o formas de dosificación a base de líquido. Los altos niveles plasmáticos máximos típicamente observados para tales formulaciones convencionales de liberación inmediata se verán reducidos por la liberación prolongada del fármaco durante 1 a 12 horas o más. Además, se evitará una disminución rápida en los niveles plasmáticos ya que el fármaco se cruzará continuamente desde la cavidad oral hacia el torrente sanguíneo durante el período de tiempo de disolución de la tableta, proporcionando así a la farmacocinética plasmática una meseta más estable. Además, las formas de dosificación descritas en el presente documento pueden mejorar la seguridad del tratamiento al minimizar los efectos secundarios potencialmente perjudiciales debido a la reducción de los picos y valles en la farmacocinética del fármaco en plasma, que comprometen la seguridad del tratamiento.
En diversas realizaciones, las formulaciones transmucosa orales del (de los) agente(s) activo(s) descrito(s) aquí diseñado para evitar los altos niveles plasmáticos máximos de formas de dosificación oral intravenosa y de liberación inmediata convencional utilizando la mucosa y controlando independientemente la desintegración (o erosión) de ambos comprimidos) y la disolución y liberación del fármaco de la tableta con el tiempo para proporcionar un perfil de administración más seguro. Las formulaciones orales descritas en el presente documento proporcionan dosis individuales repetitivas que incluyen una cantidad definida del agente activo.
Una ventaja de las formulaciones transmucosa orales bioadhesivas descritas en el presente documento es que exhiben una biodisponibilidad altamente consistente y pueden mantener la concentración de fármaco en plasma dentro de una ventana terapéutica dirigida con una variabilidad significativamente menor por una duración más larga que las formas de dosificación disponibles actualmente, ya sean formas de dosificación sólidas o formas de dosificación IV.
Además, se evita una disminución rápida de los niveles plasmáticos, ya que el fármaco cruza continuamente desde la cavidad oral o el tracto gastrointestinal hacia el torrente sanguíneo durante el tiempo de disolución de la tableta o más, proporcionando así la farmacocinética plasmática con una fase de meseta extendida como en comparación con las formas de dosificación oral de liberación inmediata convencionales. Además, las formas de dosificación descritas en el presente documento pueden mejorar la seguridad del tratamiento al minimizar los efectos secundarios potencialmente nocivos debido a la reducción relativa de los picos y valles en la farmacocinética del fármaco en plasma, lo que compromete la seguridad del tratamiento y es típico de las formas de dosificación disponibles actualmente.
En diversas realizaciones, las formulaciones bioadhesivas descritas en el presente documento pueden diseñarse para manipular y controlar el perfil farmacocinético de los agentes activos descritos en el presente documento. Como tal, las formulaciones se pueden ajustar para lograr tiempos de desintegración 'lentos' (y perfiles cinéticos de erosión) y una liberación lenta del fármaco y, por lo tanto, permiten perfiles farmacocinéticos muy prolongados que proporcionan una acción farmacológica sostenida. Aunque tales formulaciones pueden estar diseñadas para proporcionar un inicio rápido, su objetivo principal es permitir la farmacocinética y el efecto sostenido del fármaco, manteniendo los otros atributos de rendimiento de la tableta, como la bioadhesión, la reproducibilidad de la acción, la Cmax disminuida, etc.
El rendimiento y los atributos de las formulaciones transmucosa bioadhesivas de esta invención son independientes del proceso de fabricación. Se pueden usar varios procesos convencionales, bien establecidos y conocidos en la técnica para fabricar las formulaciones de la presente invención (tales como granulación en húmedo y en seco, compresión directa, etc.) sin afectar las propiedades fisicoquímicas de la forma de dosificación o el rendimiento in vivo.
Una relación matemática ilustrativa que demuestra la fase de meseta prolongada de los niveles de plasma sanguíneo medidos del (de los) agente(s) activo(s) descritos aquí, después de la administración de las formas de dosificación de la invención es el término "Relación de orientación terapéutica óptima" u "OTTR", que representa el tiempo promedio que el fármaco está presente a niveles terapéuticos, definido como el tiempo dentro del cual la concentración plasmática del fármaco se mantiene por encima del 50% de la Cmax normalizada por la vida media de eliminación del fármaco multiplicada por la proporción de la Cmax obtenida en el forma de dosificación de interés sobre la Cmax normalizada después de la administración IV de dosis equivalentes. En ciertas realizaciones, el OTTR puede calcularse mediante la fórmula:
OTTR= C IVmax/ Cmax) x (Dosis/DosisIV) (Tiempo mayor a 50% de Cmax) / (TerminalIV vida media eliminación de fármaco)
En ciertas realizaciones, el OTTR es mayor que aproximadamente 15, o mayor que aproximadamente 20, o mayor que aproximadamente 25, o mayor que aproximadamente 30, o mayor que aproximadamente 40, o mayor que aproximadamente 50.
Administración
En ciertas realizaciones, uno o más agentes activos descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho(s) estereoisómero(s) ), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) se administran a un mamífero que lo necesita, por ejemplo, a un mamífero en riesgo o que padece una patología caracterizada por el procesamiento anormal de proteínas precursoras de amiloide, un mamífero en riesgo de progresión de DCL a enfermedad de Alzheimer, y así sucesivamente. En ciertas realizaciones, el (los) agente(s) activo(s) se administran para prevenir o retrasar la aparición de una disfunción cognitiva previa al Alzheimer, y/o para mejorar uno o más síntomas de una disfunción cognitiva previa al Alzheimer, y/o para prevenir o retrasar el progresión de una afección pre-Alzheimer o disfunción cognitiva a la enfermedad de Alzheimer, y/o para promover el procesamiento de la proteína precursora amiloide (APP) por una vía no amiloidogénica. En ciertas realizaciones, se administran uno o más agentes activos para el tratamiento de la enfermedad de Alzheimer en etapa temprana, etapa media o etapa tardía, por ejemplo, para reducir la gravedad de la enfermedad y/o para mejorar uno o más síntomas de la enfermedad, y/o para retrasar la progresión de la enfermedad.
En varias realizaciones, el (los) agente(s) activo(s) descrito(s) en el presente documento (por ejemplo, hidantoína descrita en el presente documento, o un tautómero(s) o estereoisómero(s) del mismo, o sales o solvatos farmacéuticamente aceptables de dicha hidantoína(s), dicho estereoisómero( s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) pueden administrarse por cualquiera de una serie de rutas. Así, por ejemplo, pueden administrarse por vía oral, parenteral (intravenosa (IV), intramuscular (IM), depo-IM, subcutánea (SQ) y depo-SQ), sublingual, intranasal (inhalación), intratecal, transdérmica (por ejemplo, a través del parche transdérmico), tópicamente, ionoforéticamente o rectalmente. Típicamente, la forma de dosificación se selecciona para facilitar el suministro al cerebro (por ejemplo, paso a través de la barrera hematoencefálica). En este contexto, se observa que los compuestos descritos aquí se administran fácilmente al cerebro. Las formas de dosificación conocidas por los expertos en la técnica son adecuadas para la administración del compuesto.
En diversas realizaciones, los agentes activos se administran en una cantidad/régimen de dosificación suficiente para ejercer un efecto profiláctico y/o terapéuticamente útil en ausencia de efectos secundarios indeseables en el sujeto
tratado (o con la presencia de niveles aceptables y/o tipos de efectos secundarios). La cantidad específica/régimen de dosificación variará según el peso, el sexo, la edad y la salud del individuo; la formulación, la naturaleza bioquímica, la bioactividad, la biodisponibilidad y los efectos secundarios del compuesto particular.
En ciertas realizaciones, la cantidad terapéutica o profilácticamente efectiva puede determinarse empíricamente probando el (los) agente(s) en sistemas modelo in vitro e in vivo conocidos para el trastorno tratado. Se puede determinar una dosis terapéutica o profilácticamente efectiva administrando primero una dosis baja, y luego aumentando gradualmente hasta alcanzar una dosis que logre el efecto deseado con efectos secundarios mínimos o no deseados.
En ciertas realizaciones, cuando se administra por vía oral, una cantidad administrada de los agentes descritos en el presente documento es eficaz para prevenir o retrasar la aparición de una disfunción cognitiva pre-Alzheimer, y/o para mejorar uno o más síntomas de un pre disfunción cognitiva de Alzheimer y/o para prevenir o retrasar la progresión de una afección pre-Alzheimer o disfunción cognitiva a la enfermedad de Alzheimer, y/o para promover el procesamiento de la proteína precursora amiloide (APP) por una vía no amiloidogénica, y/o para tratar o prevenir la EA varía de aproximadamente 0.1 mg/día a aproximadamente 500 mg/día o aproximadamente 1.000 mg/día, o de aproximadamente 0.1 mg/día a aproximadamente 200 mg/día, por ejemplo, de aproximadamente 1 mg/día a aproximadamente 100 mg/día, por ejemplo, de aproximadamente 5 mg/día a aproximadamente 50 mg/día. En algunas realizaciones, al sujeto se le administra el compuesto a una dosis de aproximadamente 0.05 a aproximadamente 0.50 mg/kg, por ejemplo, aproximadamente 0.05 mg/kg, 0.10 mg/kg, 0.20 mg/kg, 0.33 mg/kg, 0.50 mg/kg. Se entiende que si bien un paciente puede comenzar con una dosis, esa dosis puede variar (aumentarse o disminuirse, según corresponda) con el tiempo a medida que cambia la condición del paciente. Dependiendo de las evaluaciones de resultados, se pueden usar dosis más altas. Por ejemplo, en ciertas realizaciones, se pueden administrar hasta 1000 mg/día, por ejemplo, 5 mg/día, 10 mg/día, 25 mg/día, 50 mg/día, 100 mg/día, 200 mg/día, 300 mg/día, 400 mg/día, 500 mg/día, 600 mg/día, 700 mg/día, 800 mg/día, 900 mg/día o 1000 mg/día.
En diversas realizaciones, los agentes activos descritos en el presente documento pueden administrarse por vía parenteral, por ejemplo, por IV, IM, depo-IM, SC o depo-SC. En ciertas realizaciones cuando se administra por vía parenteral, se puede administrar una cantidad terapéuticamente efectiva de aproximadamente 0.5 a aproximadamente 100 mg/día, preferiblemente de aproximadamente 5 a aproximadamente 50 mg diarios. Cuando se usa una formulación de depósito para inyección una vez al mes o una vez cada dos semanas, la dosis en ciertas realizaciones puede ser de aproximadamente 0.5 mg/día a aproximadamente 50 mg/día, o una dosis mensual de aproximadamente 15 mg a aproximadamente 1.500 mg. En parte debido al olvido de los pacientes con enfermedad de Alzheimer, se prefiere que la forma de dosificación parenteral sea una formulación depo.
En diversas realizaciones, el (los) agente(s) activo(s) descrito(s) en el presente documento pueden administrarse por vía sublingual. En algunas realizaciones, cuando se administran por vía sublingual, los compuestos y/o análogos de los mismos se pueden administrar de una a cuatro veces al día en las cantidades descritas anteriormente para la administración IM.
En diversas realizaciones, el (los) agente(s) activo(s) descrito en el presente documento pueden administrarse por vía intranasal. Cuando se administran por esta ruta, las formas de dosificación apropiadas son un aerosol nasal o polvo seco, como saben los expertos en la técnica. En ciertas realizaciones, la dosificación del compuesto y/o análogo del mismo para administración intranasal es la cantidad descrita anteriormente para administración IM.
En diversas realizaciones, el (los) agente(s) activo(s) descrito aquí puede administrarse intratecalmente. Cuando se administra por esta ruta, la forma de dosificación apropiada puede ser una forma de dosificación parenteral, como saben los expertos en la materia. En ciertas realizaciones, la dosificación del compuesto y/o análogo del mismo para administración intratecal es la cantidad descrita anteriormente para administración IM.
En ciertas realizaciones, el (los) agente(s) activo(s) descrito(s) en el presente documento pueden administrarse tópicamente. Cuando se administra por esta ruta, la forma de dosificación apropiada es una crema, pomada o parche. Cuando se administra tópicamente, la dosis es de aproximadamente 1.0 mg/día a aproximadamente 200 mg/día. Debido a que la cantidad que puede entregar un parche es limitada, se pueden usar dos o más parches. El número y el tamaño del parche no son importantes siempre que se administre una cantidad terapéuticamente efectiva de compuesto como saben los expertos en la materia. El compuesto puede administrarse rectalmente por supositorio como es conocido por los expertos en la materia. En ciertas realizaciones, cuando se administra por supositorio, la cantidad terapéuticamente efectiva es de aproximadamente 1.0 mg a aproximadamente 500 mg.
En diversas realizaciones, el (los) agente(s) activo(s) descrito en el presente documento puede administrarse mediante implantes como es conocido por los expertos en la materia. Cuando se administra el compuesto por implante, la cantidad terapéuticamente efectiva es la cantidad descrita anteriormente para la administración de depósito.
En diversas realizaciones, el (los) agente(s) activo(s) aquí descrito puede encerrarse en envases de dosis múltiples o únicas. Los agentes incluidos se pueden proporcionar en kits, por ejemplo, que incluyen partes componentes que se pueden ensamblar para su uso. Por ejemplo, un agente activo en forma liofilizada y un diluyente adecuado pueden proporcionarse como componentes separados para la combinación antes de su uso. Un kit puede incluir un agente
activo y un segundo agente terapéutico para la administración conjunta. El agente activo y el segundo agente terapéutico pueden proporcionarse como partes componentes separadas. Un kit puede incluir una pluralidad de contenedores, cada contenedor con una o más dosis unitarias de los compuestos. Los recipientes se adaptan preferiblemente para el modo de administración deseado, que incluye, pero no se limita a tabletas, cápsulas de gel, cápsulas de liberación sostenida y similares para administración oral; productos de depósito, jeringas precargadas, ampollas, viales y similares para administración parenteral; y parches, Medipads, cremas y similares para administración tópica, por ejemplo, como se describe en el presente documento.
En diversas realizaciones, las formas de dosificación se pueden administrar al sujeto 1,2, 3 o 4 veces al día. En ciertas realizaciones, se prefiere que el compuesto se administre tres o menos veces, más preferiblemente una o dos veces al día. En ciertas realizaciones, se prefiere que los agentes se administren en forma de dosificación oral.
Debería ser evidente para un experto en la materia que la dosis exacta y la frecuencia de administración dependerán de la afección particular que se está tratando, la gravedad de la afección que se está tratando, la edad, el peso, la condición física general del paciente particular, y otros medicamentos que el individuo puede estar tomando, como es bien sabido por los médicos administradores expertos en esta técnica.
Si bien las composiciones y métodos se describen en el presente documento con respecto al uso en humanos, también son adecuados para animales, por ejemplo, uso veterinario. Por lo tanto, ciertos organismos (sujetos) contemplados en el presente documento incluyen, pero no se limitan a humanos, primates no humanos, caninos, equinos, felinos, porcinos, ungulados, largomorfos y similares.
Las formulaciones y métodos de administración anteriores pretenden ser ilustrativos y no limitativos. Se apreciará que, utilizando las enseñanzas proporcionadas en el presente documento, se pueden diseñar fácilmente otras formulaciones y modos de administración adecuados.
Terapias Combinadas
En ciertas realizaciones, los agentes activos descritos en el presente documento (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de los mismos, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos de los mismos) se pueden usar en combinación con otros agentes terapéuticos o metodologías utilizadas para tratar o prevenir enfermedades caracterizadas por depósitos amiloides en el cerebro, incluyendo DCL y/o EA. Por consiguiente, en ciertas realizaciones, una composición farmacéutica que comprende al menos un agente activo descrito en el presente documento (por ejemplo, una hidantoína descrita en el presente documento, o un tautómero o estereoisómero del mismo, o sales o solvatos farmacéuticamente aceptables de dicha hidantoína, dicho estereoisómero o dicho tautómero, o un análogo, derivado o profármaco del mismo) uno junto con al menos un agente terapéutico adicional, y se contempla un vehículo o diluyente farmacéuticamente aceptable. En ciertas realizaciones, se contempla un método terapéutico o profiláctico que comprende administrar al menos un agente activo descrito en el presente documento junto con al menos un agente terapéutico adicional.
En ciertas realizaciones, los ejemplos no limitantes de agentes terapéuticos adicionales incluyen, pero sin limitación, disulfiram y/o análogos del mismo, honokiol y/o análogos del mismo, tropisetrón y/o análogos del mismo, nimetazepam y/o análogos del mismo (véase , por ejemplo, USSN 13/213,960 (Publicación de Patente de los Estados Unidos No: US-2012-0071468-A1) y PCT/US2011/048472 (Publicación de PCT No: WO 2012/024616) que se incorporan aquí como referencia para los compuestos descritos en el mismo), ésteres de tropinol y/o ésteres relacionados y/o análogos de los mismos (véase, por ejemplo, USSN 61/514,381, que se incorpora aquí como referencia para los compuestos descritos aquí), inhibidores de la quinasa TrkA (por ejemplo, ADDN-1351) y/o análogos de los mismos (véase, por ejemplo, USSN 61/525,076, que se incorpora aquí como referencia para los compuestos descritos en el mismo), agonistas del receptor D2 y antagonistas del receptor adrenérgico alfa1, e inhibidores BACE específicos de la APP (ASBI) como se describe y/o reivindica en USSN 61/728,688, presentada en noviembre 20, 2012, que se incorpora aquí como referencia para los agentes activos descritos en el presente documento, que incluyen, pero no se limitan a, galangina, un profármaco de galangina, rutina, un profármaco de rutina y otros flavonoides y profármacos flavonoides como se describe o reivindica en el mismo.
Ejemplos no limitantes de agentes terapéuticos adicionales incluyen fármacos seleccionados del grupo que consiste en: (a) fármacos útiles para el tratamiento de la enfermedad de Alzheimer y/o fármacos útiles para tratar uno o más síntomas de la enfermedad de Alzheimer, (b) fármacos útiles para inhibir la síntesis de Ap, y (c) fármacos útiles para tratar enfermedades neurodegenerativas. Ejemplos adicionales no limitantes de agentes terapéuticos adicionales para su uso en combinación con los compuestos (por ejemplo, hidantoínas) descritos en el presente documento incluyen fármacos útiles para el tratamiento, prevención, retraso del inicio, mejora de cualquier patología asociada con Ap y/o un síntoma de los mismos. Ejemplos no limitantes de patologías asociadas con Ap incluyen: enfermedad de Alzheimer, síndrome de Down, enfermedad de Parkinson, pérdida de memoria, pérdida de memoria asociada con la enfermedad de Alzheimer, pérdida de memoria asociada con la enfermedad de Parkinson, síntomas de déficit de atención, síntomas de déficit de atención asociados con la enfermedad de Alzheimer, Parkinson enfermedad y/o síndrome de Down, demencia, accidente cerebrovascular, microgliosis e inflamación cerebral, demencia presenil, demencia senil, demencia asociada con la enfermedad de Alzheimer, enfermedad de Parkinson y/o síndrome de Down, parálisis
supranuclear progresiva, degeneración basal cortical, neurodegeneración, deterioro olfativo, deterioro olfativo asociado con la enfermedad de Alzheimer, enfermedad de Parkinson y/o síndrome de Down, angiopatía p-amiloide, angiopatía amiloide cerebral, hemorragia cerebral hereditaria, deterioro cognitivo leve ("MCI"), glaucoma, amiloidosis, diabetes tipo II, hemodiálisis complicaciones (de p2 microglobulinas y complicaciones derivadas de estos en pacientes de hemodiálisis), tembladera, encefalitis espongiforme bovina, lesión cerebral traumática ("TBI") y enfermedad de Creutzfeld-Jakob, que comprende administrar a dicho paciente al menos un compuesto de hidantoína descrito aquí, o un tautómero o isómero del mismo; o sal o solvato farmacéuticamente aceptable de dicho compuesto o dicho tautómero, en una cantidad eficaz para inhibir dicha patología o patologías.
En ciertas realizaciones, tales agentes terapéuticos adicionales incluyen, pero no se limitan a, inhibidores de acetilcolinesterasa (que incluyen, sin limitación, por ejemplo, enantiómero de (-)-fenerina, tacrina, ipidacrina, galantamina, donepezil, icopezil, zanapezil, rivastigmina, huperzina A, fenerina, fisostigmina, neostigmina, piridostigmina, ambenonio, demarcarium, edrofonio, ladostigilo y ungeremina); antagonista del receptor de NMDA (que incluye sin limitaciones, por ejemplo, memantina); agonistas de los receptores muscarínicos (incluidos, entre otros, talsaclidina, AF-102B, AF-267B (NGX-267)); agonistas del receptor nicotínico (incluidos, entre otros, Ispronicline (AZD-3480)); inhibidores de beta-secretasa (incluidos, entre otros, por ejemplo, tiazolidinedionas, que incluyen rosiglitazona y pioglitazona); inhibidores de la gamma-secretasa (incluidos, entre otros, semagacestat (LY-450139), MK-0752, E-2012, BMS-708163, PF-3084014, begacestat (GSI-953) y NIC5-15); inhibidores de la agregación de Ap (incluidos, entre otros, clioquinol (PBT1), PBT2, tramiprosato (homotaurina), escilo-inositol (también conocido como escilociclohexanehexol, AZD-103 y ELND-005), inmunoterapia pasiva con fragmentos Ap (incluso sin limitaciones, por ejemplo, Bapineuzemab) y Epigalocatequina-3-galato (EGCg)); agentes antiinflamatorios tales como inhibidores de la ciclooxigenasa II; antioxidantes tales como vitamina E y ginkólidos; enfoques inmunológicos, tales como, por ejemplo, inmunización con péptido Ap o administración de anticuerpos anti-péptido Ap; estatinas y agentes neurotróficos directos o indirectos tales como Cerebrolysin™, AIT-082 (Emilieu, 2000, Arch. Neurol. 57: 454), Netrin (Luorenco (2009) Cell Death Differ., 16:655-663), miméticos de Netrin, NGF, miméticos de NGF, BDNF y otros agentes neurotróficos del futuro, agentes que promueven la neurogénesis, por ejemplo, terapia con células madre. Agentes farmacológicos adicionales útiles en el tratamiento o prevención de enfermedades caracterizadas por depósitos amiloides en el cerebro, incluidos DCL y/o EA, se describen, por ejemplo, en Mangialasche, et al. (2010) Lancet Neurol., 9: 702-716.
En ciertas realizaciones, ejemplos no limitantes adicionales de agentes terapéuticos adicionales para usar en combinación con los compuestos descritos en el presente documento incluyen: antagonistas muscarínicos (por ejemplo, agonistas m1 (tales como acetilcolina, oxotremorina, carbacol o McNa343), o antagonistas m2 de colinesterasa inhibidores (p. ej., inhibidores de acetil y/o butirilcolinesterasa como donepezil (Aricept®), galantamina (Razadyne®) y rivastigimina (Exelon®); antagonistas de los receptores de N-metil-D-aspartato (por ejemplo, NAMe NdA® (HCl de memantina); combinaciones de inhibidores de colinesterasa y antagonistas de los receptores de N-metil-D-aspartato; moduladores de gamma secretasa; inhibidores de gamma secretasa; agentes antiinflamatorios no esteroideos; agentes antiinflamatorios que pueden reducir la neuroinflamación; anticuerpos anti-amiloide (como bapineuzemab, Wyeth/Elan); vitamina E; agonistas del receptor de acetilcolina nicotínico; agonistas inversos del receptor CB1 o antagonistas del receptor CB1; antibióticos; secretagogos de la hormona del crecimiento; antagonistas de histamina H3; agonistas de AMPA; Inhibidores de PDE4; agonistas inversos de GABAa ; inhibidores de la agregación amiloide; inhibidores de la glucógeno sintasa quinasa beta; promotores de la actividad alfa secretasa; inhibidores de PDE-10; inhibidores de la tau quinasa (por ejemplo, inhibidores de GSK3beta, inhibidores de cdk5 o inhibidores de ERK); inhibidores de la agregación de Tau (por ejemplo, REMBER®; inhibidores de RAGE (por ejemplo, TTP 488 (PF-4494700)); vacuna anti-Ap; ligandos APP; agentes que regulan la insulina, agentes reductores del colesterol como los inhibidores de la HMG-CoA reductasa (por ejemplo, estatinas como Atorvastatina, Fluvastatina, Lovastatina, Mevastatina, Pitavastatina, Pravastatina, Rosuvastatina, Simvastatina) y/o inhibidores de la absorción de colesterol (como Ezetimibe), o combinaciones de inhibidores de la HMG-CoA reductasa e inhibidores de la absorción de colesterol (como, por ejemplo, VYTORIN®); fibratos (como, por ejemplo, clofibrato, clofibruro, etofibrato y clofibrato de aluminio); combinaciones de fibratos y agentes reductores del colesterol y/o inhibidores de la absorción de colesterol; agonistas de los receptores nicotínicos; niacina; combinaciones de niacina e inhibidores de la absorción de colesterol y/o agentes reductores del colesterol (por ejemplo, SIMCOR® (niacina/simvastatina, disponible de Abbott Laboratories, Inc.); agonistas de LXR; imitadores de l RP; antagonistas de los receptores H3; histón e inhibidores de la desacetilasa; inhibidores de hsp90; agonistas de 5-HT4 (por ejemplo, PRX-03140 (Epix Pharmaceuticals)); antagonistas del receptor 5-HT6; moduladores o antagonistas del receptor mGluR1; moduladores o antagonistas del receptor mGluR5; antagonistas de mGluR2/3; antagonistas del receptor de prostaglandina EP2; inhibidores de PAI-1; agentes que pueden inducir eflujo de Abeta, como gelsolina; compuesto atenuador de proteínas metálicas (por ejemplo, Pb t 2); y moduladores GPR3; y antihistamínicos como Dimebolin (por ejemplo, DIMEBON®, Pfizer).
Por consiguiente, ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente adicional terapéutico donde el agente terapéutico en la formulación y/o método es disulfiram y/o análogos de los mismos (véase, por ejemplo, USSN 13/213,960 (Publicación de Patente de los Estados nidos No: US-2012-0071468-A1), y PCT/US2011/048472 (Publicación PCT No: WO 2012/024616)).
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional en donde el agente terapéutico en la formulación y/o método es honokiol y/o análogos del mismo (véase, por ejemplo, USSN 13/213,960 (Publicación de Patente de los Estados Unidos No: US-2012-0071468-A1), y PCT/US2011/048472 (Publicación PCT No: WO 2012/024616)).
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional donde el agente terapéutico en la formulación y/o método es tropisetrón y/o análogos del mismo (véase, por ejemplo, USSN 13/213,960 (Publicación de Patente de los Estados Unidos No: US-2012-0071468-A1), y PCT/US2011/048472 ( Publicación PCT No: WO 2012/024616)).
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional donde el agente terapéutico en la formulación y/o método es tropisetrón.
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional donde el agente terapéutico en la formulación y/o método es nimetazepam y/o análogos de los mismos (véase, por ejemplo, USSN 13/213,960 (Publicación de Patente de los Estados Unidos No: US-2012-0071468-A1), y PCT/US2011/048472 (Publicación PCT No: WO 2012/024616)).
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional donde el agente terapéutico en la formulación y/o método es un éster de tropinol o un éster relacionado (véase, por ejemplo, USSN 61/514,381).
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional donde el agente terapéutico en la formulación y/o el método es un inhibidor de la quinasa TrkA (por ejemplo, ADDN-1351) y/o análogos de los mismos (véase, por ejemplo, USSN 61/525,076).
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional donde el agente terapéutico en la formulación y/o método es un agonista del receptor D2 y/o un antagonista del receptor alfa-adrenérgico.
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional donde el agente terapéutico en la formulación y/o método es un ASBI como se describe y/o reivindica en USSN 61/728,688, presentado el 20 de noviembre de 2012, que se incorpora aquí como referencia para los agentes activos descritos en el presente documento, incluidos, entre otros galangina, un profármaco de galangina, rutina, y otros flavonoides como se describe o reivindica en el mismo.
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional agente donde el agente terapéutico en la formulación y/o método es uno o más inhibidores de colinesterasa (por ejemplo, inhibidores de acetil y/o butirilcolinesterasa).
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional donde el agente terapéutico en la formulación y/o método es uno o más antagonistas muscarínicos (por ejemplo, agonistas m1 o antagonistas m2).
Ciertas realizaciones proporcionan una composición farmacéutica que comprende una cantidad eficaz de una o más hidantoínas descritas en el presente documento y un agente terapéutico adicional, y/o un método de tratamiento o profilaxis que comprende la administración de una o más hidantoínas descritas en el presente documento junto con un agente terapéutico adicional donde el agente terapéutico en la formulación y/o método es uno o más compuestos
seleccionados del grupo que consiste en inhibidores de colinesterasa (como, por ejemplo, (.+-.)-2,3-dihidro-5,6-dimetoxi-2-[[1-(fenilmetil)-4-piperidinil]metil-1]-1H-inden-1-uno hidrocloruro, es decir, clorhidrato de donepezilo, disponible como la marca ARICEPT® de clorhidrato de donepezilo), N-metil-D inhibidores del receptor de aspartato (como, por ejemplo, Namenda® (memantina HCl)); anticuerpos anti-amiloides (tales como bapineuzumab, Wyeth/Elan), inhibidores de gamma secretasa, moduladores de gamma secretasa e inhibidores de beta secretasa distintos de las hidantoínas descritas en el presente documento.
Indicaciones Adicionales
Uso de hidantoína en la degeneración macular relacionada con la edad y el glaucoma
Mientras que en diversas realizaciones, se contempla el uso de inhibidores de unión de APP-BACE (ABBI), por ejemplo, las diversas hidantoínas descritas en el presente documento, para prevenir o retrasar la aparición de una afección pre-Alzheimer y/o disfunción cognitiva, y/o mejorar uno o más síntomas de una afección pre-Alzheimer y/o disfunción cognitiva, o prevenir o retrasar la progresión de una afección pre-Alzheimer o disfunción cognitiva a la enfermedad de Alzheimer, y/o para el tratamiento de la enfermedad de Alzheimer, también se contemplan otros usos de los ABBI. En particular, en ciertas realizaciones, se contempla el uso de ABBI para el tratamiento y/o la profilaxis de la degeneración macular relacionada con la edad y/o el glaucoma.
Sin limitarse a una teoría particular, se cree que el depósito extracelular anormal de proteínas puede contribuir a la patogénesis y progresión de la degeneración macular (DMAE) relacionada con la edad, que también es el caso en la enfermedad de Alzheimer y la aterosclerosis. En ambas condiciones, los depósitos de proteínas contienen muchos constituyentes compartidos como apoE, complemento y péptidos Ap. Por ejemplo, en la DMAE humana, la deposición del péptido Ap está asociada con drusas, donde se acumula y colocaliza con componentes del complemento activados (Anderson et al. (2004) Exp. Eye. Res., 78: 243-256; Dentchev et al. ( 2003) Mol. Vis., 9: 184-190; Johnson et al. (2002) Proc Natl Acad Sci USA 99: 11830-11835.). Luibl et al. (2006) J. Clin. Invest., 116: 378-385, mostró la presencia de oligómeros amiloides potencialmente tóxicos en drusas, depósitos basales sub-RPE y RPE de ojos de donantes humanos usando un anticuerpo que reconoce específicamente la forma oligomérica de Ap. Estos oligómeros Ap no se detectaron en los ojos de donantes de control de edad coincidente sin drusas. Isas et al. (2010) Invest. Ophtalmol Vis. Sci., 51: 1304-1310, también detectó fibrillas Ap tanto solubles como maduras en drusas. Colectivamente, estos hallazgos implican a Ap en la patogénesis de la DMAE. Además, se ha detectado el péptido Ap en depósitos basales sub-RPE y lesiones neovasculares en un modelo murino de DMAE (Ding et al. (2008) Vision Res., 48: 339-345; Malek et al. (2005) Proc Natl Acad Sci USA, 102: 11900-11905). En este modelo, los ratones de reemplazo dirigidos a APOE4 humanos (ratones APOE4) alimentados con una dieta rica en grasas y rica en colesterol (HFC) (ratones APOE4-HFC) exhiben características morfológicas observadas tanto en la DMAE húmeda como en la seca. Estas características incluyen depósitos gruesos difusos de sub-RPE, depósitos focales similares a drusas que contienen lípidos y proteínas, engrosamiento de la membrana de Bruch, regiones irregulares de atrofia de RPE opuestas a áreas de degeneración de fotorreceptores y CNV (Malek et al. (2005) Proc Natl Acad Sci USA, 102: 11900-11905). Se cree que, en el modelo de ratón con DMAE APOE4-HFC, la acumulación de Ap provoca daños a nivel de RPE/coroides y se ha demostrado previamente que la administración sistémica de anticuerpos anti-Ap40 específicos puede atenuar parcialmente la disminución de la función visual exhibido en este modelo (Ding et al. (2008) Vision Res., 48: 339-345). También se ha demostrado que la inmunoterapia anti-Ap dirigida simultáneamente a Ap40 y Ap42 bloquea los cambios histopatológicos y protege completamente la función visual en ratones APOE4-HFC (Ding et al. (2011) Proc. Nat'l. Acad. Sci. USA, 108 (28): E279-E287).
Sin limitarse a una teoría particular, se cree que el procesamiento de APP a Ap en el ojo ocurre por las actividades de BACE y Y-secretasa en la retina y las capas de células epiteliales pigmentadas de la retina (RPE) y que sAPPa y Ap son secretada en el humor vítreo (véase, por ejemplo, (Prakasam et al. (2008) J. Alzh. Dis., 20: 1243-1253). El Ap se transporta adicionalmente al humor acuoso donde se mide fácilmente.
En vista de estos hallazgos, se cree que los ABBI, por ejemplo, las hidantoínas descritas en el presente documento, pueden encontrar uso en el tratamiento o la profilaxis de la degeneración macular relacionada con la edad (DMAE) y/o el glaucoma. Por consiguiente, se cree que los ABBI pueden administrarse a un sujeto para retrasar o prevenir la aparición de DMAE (y/o glaucoma), y/o para reducir uno o más síntomas de DMAE, y/o para disminuir, detener o revertir progresión de la enfermedad. En diversas realizaciones, uno o más ABBI (por ejemplo, uno o más de los agentes activos descritos en el presente documento) se administran a un sujeto (por ejemplo, un humano, un mamífero no humano) para estos fines. Como se describió anteriormente, en diversas realizaciones, el ABBI se administra a través de una ruta seleccionada del grupo que consiste en administración oral, administración isoforética, administración transdérmica, administración parenteral, administración en aerosol, administración por inhalación, administración intravenosa y administración rectal.
En ciertas realizaciones, la administración es directamente al ojo. Así, por ejemplo, en ciertas realizaciones, el (los) agente(s) pueden administrarse al ojo en forma de gotas para los ojos, mediante inyección intraocular, y similares.
Típicamente, los ABBI se administran en una cantidad efectiva para el tratamiento y/o profilaxis de DMAE o glaucoma, donde la cantidad efectiva variará según la modalidad de administración. En ciertas realizaciones, la cantidad efectiva es una cantidad suficiente para mitigar en un mamífero uno o más síntomas asociados con la degeneración macular
relacionada con la edad (DMAE). En ciertas realizaciones, la cantidad efectiva es una cantidad, una cantidad suficiente para reducir el riesgo o retrasar la aparición, y/o reducir la gravedad final de una enfermedad de DMAE (o glaucoma) caracterizada por la reducción de Ap en el humor vitreo y/o acuoso y/o los depósitos de amiloide en la retina y/o la capa de células RPE.
Sistemas de ensayo para evaluar el procesamiento de APP
Sin limitarse a una teoría particular, se cree que los agentes activos descritos en el presente documento (por ejemplo, ABBI tales como las hidantoínas descritas en el presente documento) promueven el procesamiento de ApP por la vía no amiloidogénica y/o reducen o inhiben el procesamiento de APP por la vía amiloidogénica. En la vía no amiloidogénica, la APP se escinde primero por la a-secretasa dentro de la secuencia Ap, liberando el ectodominio de APPsa ("sAPPa"). En contraste, la vía amiloidogénica se inicia cuando la p-secretasa escinde la APP en el extremo amino del Ap, liberando así el ectodominio de APPsp ("sAPPp"). El procesamiento de APP por las vías no amiloidogénicas y amiloidogénicas es conocido en la técnica y revisado, por ejemplo, por Xu (2009) J Alzheimers Dis., 16(2): 211-224, y De Strooper, et al. (2010 Nat Rev Neurol 6 (2): 99-107.
Un método para evaluar la eficacia de los agentes activos es determinar una reducción o eliminación en el nivel de procesamiento de APP por la vía amiloidogénica, por ejemplo, una reducción o eliminación en el nivel de procesamiento de APP por escisión de p-secretasa en respuesta a la administración de los agentes de interés. Los ensayos para determinar el grado de escisión de APP en el sitio de escisión de p-secretasa son bien conocidos en la técnica. Se describen ensayos ilustrativos, por ejemplo, en las Patentes de los Estados Unidos Nos. 5,744,346 y 5,942,400. Kits para determinar la presencia y los niveles en una muestra biológica de sAPPa y sAPPp, así como APPneo y Ap comercialmente disponibles, por ejemplo, de PerkinElmer.
Ensayo ABBI
La actividad del inhibidor de BACE de unión a APP (ABBI) de cualquiera de los compuestos descritos en el presente documento puede verificarse fácilmente usando, por ejemplo, ensayos descritos en el presente documento. Básicamente, en ciertas realizaciones, un par de ensayos se utilizan para identificar compuestos ABBI que inhiben la escisión BACE del sustrato de la aplicación MBP-C125, lo que resulta en la inhibición de la producción de C99 y el sustrato del péptido del sitio p (P5-P5') y también interactúa con la APP, por ejemplo, medida por análisis por resonancia de plasmón superficial (SPR).
En una realización ilustrativa, una proteína de fusión MBP-C125 APP695wt puede usarse como uno de los sustratos y el segundo sustrato puede ser el sustrato de fluorescencia P5-P5' disponible comercialmente. Cada uno de estos sustratos se incuba con BACE recombinante (R&D (cat#931-AS-050) en, por ejemplo, un formato de placa de 96 pozos. Para el sustrato MBP-C125, el producto C-99 de la escisión BACE se puede medir usando un ensayo AlphaLisa como lectura. Para el sustrato P5-5', la pérdida de fluorescencia tras la escisión de BACE puede usarse como lectura. Para el ensayo SPR, el análisis de unión de las hidantoínas a fragmentos del ectodominio de APP (eAPP) que son se realizaría recombinantemente preparado (Libeu et al. (2012) PLoS ONE 7(6): e40027). Un ABBI inhibiría la escisión BACE del MBP-C125 y/o el sustrato de fluorescencia y también se uniría al ectodominio de APP como el fragmento APP230-624.
Otros ensayos sin células
Los ensayos ilustrativos que se pueden usar para demostrar la actividad inhibitoria de los agentes activos se describen, por ejemplo, en los documentos WO 2000/017369, WO 2000/0003819 y la Patente de los Estados Unidos Nos.
5,942,400 y 5,744,346. Dichos ensayos pueden realizarse en incubaciones sin células o en incubaciones celulares usando células que expresan una alfa-secretasa y/o beta-secretasa y un sustrato de APP que tiene sitios de escisión de alfa-secretasa y beta-secretasa.
En una realización ilustrativa, los agentes de interés se ponen en contacto con un sustrato de APP que contiene sitios de escisión de alfa-secretasa y beta-secretasa de APP, por ejemplo, una APP o variante completa, un fragmento de APP, o un recombinante o sustrato de APP sintético que contiene la secuencia de aminoácidos: KM-DA o NL-DA (APP-SW), se incuba en presencia de una enzima alfa-secretasa y/o beta-secretasa, un fragmento de la misma, o un sintético o recombinante variante de polipéptido que tiene actividad alfa-secretasa o beta-secretasa y eficaz para escindir los sitios de escisión de alfa-secretasa o beta-secretasa de APP, en condiciones de incubación adecuadas para la actividad de escisión de la enzima. Los agentes que tienen la actividad deseada reducen o evitan la escisión del sustrato de APP. Los sustratos adecuados incluyen opcionalmente derivados que pueden ser proteínas de fusión o péptidos que contienen el péptido sustrato y una modificación útil para facilitar la purificación o detección del péptido o sus productos de escisión de alfa-secretasa y/o beta-secretasa. Las modificaciones útiles incluyen la inserción de un epítopo antigénico conocido para la unión de anticuerpos; la unión de un marcador o unidad estructural detectable, la unión de un sustrato de unión y similares.
Las condiciones de incubación adecuadas para un ensayo in vitro sin células incluyen, por ejemplo: aproximadamente 200 nanomolar a 10 micromolar sustrato, aproximadamente 10 a 200 enzima picomolar, y aproximadamente 0.1 nanomolar a 10 micromolar del agente(s), en acuoso solución, a un pH aproximado de 4-7, a aproximadamente 37°C, durante un período de tiempo de aproximadamente 10 minutos a 3 horas. Estas condiciones de incubación son solo
ilustrativas y pueden variarse según sea necesario para los componentes de ensayo particulares y/o el sistema de medición deseado. La optimización de las condiciones de incubación de los componentes del ensayo en particular debe tener en cuenta la enzima alfa-secretasa y/o beta-secretasa específica utilizada y su pH óptimo, cualquier enzima y/o marcador adicional que pueda usarse en el ensayo, y similares. Dicha optimización es rutinaria y no requerirá una experimentación indebida.
Otro ensayo ilustrativo utiliza un péptido de fusión que tiene proteína de unión a maltosa (MBP) fusionada a los 125 aminoácidos C-terminales de APP-SW. La porción de MBP se captura en un sustrato de ensayo mediante un anticuerpo de captura anti-MBP. La incubación de la proteína de fusión capturada en presencia de alfa-secretasa y/o beta-secretasa da como resultado la escisión del sustrato en los sitios de escisión de alfa-secretasa y/o beta-secretasa, respectivamente. Este sistema se puede usar para detectar la actividad inhibitoria de los agentes de interés. El análisis de la actividad de escisión puede ser, por ejemplo, por inmunoensayo de productos de escisión. Uno de estos inmunoensayos detecta un epítopo único expuesto en el terminal carboxi de la proteína de fusión escindida, por ejemplo, usando el anticuerpo SW192. Este ensayo se describe, por ejemplo, en la Patente de los Estados Unidos No. 5,942,400.
Ensayos celulares
Se pueden usar numerosos ensayos basados en células para evaluar la actividad del (de los) agente(s) de interés en la actividad alfa-secretasa relativa a la actividad beta-secretasa y/o el procesamiento de APP para liberar oligómeros Ap amiloidogénicos versus no amiloidogénicos. El contacto de un sustrato de APP con una enzima alfa-secretasa y/o beta-secretasa dentro de la célula y en presencia o ausencia del (de los) agente(s) puede usarse para demostrar la actividad promotora de alfa-secretasa y/o inhibidora de beta-secretasa del (de los) agente(s). Preferiblemente, el ensayo en presencia del (de los) agente(s) proporciona al menos aproximadamente 30%, lo más preferiblemente al menos aproximadamente 50% de inhibición de la actividad enzimática, en comparación con un control no inhibido.
En una realización, se usan células que expresan naturalmente alfa-secretasa y/o beta-secretasa. Alternativamente, las células se modifican para expresar una alfa-secretasa y/o beta-secretasa recombinante o enzimas variantes sintéticas, como se discutió anteriormente. El sustrato de a Pp se puede agregar al medio de cultivo y se expresa preferiblemente en las células. Células que expresan naturalmente APP, formas variantes o mutantes de APP, o células transformadas para expresar una isoforma de APP, APP mutante o variante, APP recombinante o sintética, fragmento de APP, o péptido APP sintético o proteína de fusión que contiene la alfa-secretasa y/o se pueden usar sitios de escisión de APP de beta-secretasa, siempre que se permita que la APP expresada entre en contacto con la enzima y se pueda analizar la actividad de escisión enzimática.
Las líneas celulares humanas que normalmente procesan Ap de APP proporcionan un medio útil para analizar las actividades inhibidoras del (de los) agente(s). La producción y liberación de Ap y/u otros productos de escisión en el medio de cultivo se pueden medir, por ejemplo, por inmunoensayo, como Inmunoprecipitación Western o inmunoensayo ligado a enzimas (EIA), como por ELISA.
Las células que expresan un sustrato de APP y una alfa-secretasa y/o beta-secretasa activa pueden incubarse en presencia de los agentes para demostrar la actividad enzimática relativa de la alfa-secretasa y/o beta-secretasa en comparación con un control . La actividad relativa de la alfa-secretasa con respecto a la beta-secretasa se puede medir mediante el análisis de uno o más productos de escisión del sustrato de APP. Por ejemplo, se esperaría que la inhibición de la actividad de beta-secretasa contra la aplicación de sustrato disminuya la liberación de productos de escisión de APP inducidos por beta-secretasa específicos tales como Ap (por ejemplo, Ap40 o Ap42), sAPPp y APPneo. Se esperaría que la promoción o mejora de la actividad de alfa-secretasa contra la APP de sustrato aumente la liberación de productos de escisión de APP inducidos por alfa-secretasa específicos tales como sAPPa y péptido p3.
Aunque tanto las células neurales como las no neurales procesan y liberan Ap, los niveles de actividad beta-secretasa endógena son bajos y a menudo difíciles de detectar por EIA. Por lo tanto, se prefiere el uso de tipos celulares que se sabe que tienen actividad beta-secretasa mejorada, procesamiento mejorado de APP a Ap y/o producción mejorada de Ap. Por ejemplo, la transfección de células con la forma sueca mutante de APP (APP-SW); con la forma mutante de Indiana (APP-IN); o con APP-SW-IN proporciona células que tienen una actividad beta-secretasa mejorada y producen cantidades de Ap que pueden medirse fácilmente.
En tales ensayos, por ejemplo, las células que expresan APP, alfa-secretasa y/o beta-secretasa se incuban en un medio de cultivo en condiciones adecuadas para la actividad enzimática de alfa-secretasa y/o beta-secretasa en su sitio de escisión en el sustrato de la aplicación. En la exposición de las células al (a los) agente(s), la cantidad de Ap liberada en el medio y/o la cantidad de fragmentos CTF99 de APP en los lisados celulares se reduce en comparación con el control. Los productos de escisión de APP se pueden analizar, por ejemplo, mediante reacciones inmunes con anticuerpos específicos, como se discutió anteriormente.
En ciertas realizaciones, las células preferidas para el análisis de la actividad alfa-secretasa y/o beta-secretasa incluyen células neuronales humanas primarias, células neuronales animales transgénicas primarias donde el
transgén es APP y otras células tales como las de una célula 293 estable línea que expresa APP, por ejemplo, APP-SW.
Ensayos in vivo: Modelos animales
Se pueden usar varios modelos animales para analizar la actividad de los agentes de interés en la actividad relativa de alfa-secretasa y/o beta-secretasa y/o el procesamiento de APP para liberar Ap. Por ejemplo, los animales transgénicos que expresan sustrato de APP, alfa-secretasa y/o enzima beta-secretasa se pueden usar para demostrar la actividad inhibitoria del agente o agentes. Ciertos modelos animales transgénicos se han descrito, por ejemplo, en las Patente de los Estados Unidos Nos. 5,877,399; 5.612.486; 5,387,742; 5.720.936; 5,850,003; 5.877.015 y 5.811.633, y en Ganes et al. (1995) Nature 373: 523. Se prefieren los animales que exhiben características asociadas con la fisiopatología de la EA. La administración del (de los) agente(s) a los ratones transgénicos descritos en el presente documento proporciona un método alternativo para demostrar la actividad inhibidora del (de los) agente(s). También se prefiere la administración del (de los) agente(s) en un vehículo farmacéuticamente eficaz y a través de una ruta administrativa que alcanza el tejido objetivo en una cantidad terapéutica apropiada.
La inhibición de la escisión mediada por beta-secretasa de APP en el sitio de escisión de beta-secretasa y de la liberación de Ap se puede analizar en estos animales mediante la medición de fragmentos de escisión en los fluidos corporales del animal, tales como líquido o tejidos cerebrales. Del mismo modo, la promoción o la mejora de la escisión mediada por alfa-secretasa de APP en el sitio de escisión de alfa-secretasa y la liberación de sAPPa se pueden analizar en estos animales mediante la medición de fragmentos de escisión en los fluidos corporales del animal, tales como líquido o tejidos cerebrales. En ciertas realizaciones, se prefiere el análisis de tejidos cerebrales para depósitos o placas de Ap.
Al poner en contacto un sustrato de APP con una enzima alfa-secretasa y/o beta-secretasa en presencia del (de los) agente(s) en condiciones suficientes para permitir la escisión mediada enzimáticamente de APP y/o la liberación de Ap del sustrato, deseable los agentes son efectivos para reducir la escisión de APP mediada por beta-secretasa en el sitio de escisión de beta-secretasa y/o efectivos para reducir las cantidades liberadas de Ap. Los agentes también son preferiblemente efectivos para mejorar la escisión de APP mediada por alfa-secretasa en el sitio de escisión de alfasecretasa y para aumentar las cantidades liberadas de sAPPa. Cuando tal contacto es la administración del agente o agentes a un modelo animal, por ejemplo, como se describe anteriormente, el agente o agentes son efectivos para reducir la deposición de Ap en los tejidos cerebrales del animal y para reducir el número y/o tamaño de las placas beta amiloides. Cuando dicha administración es a un sujeto humano, el (los) agente(s) es(son) eficaz(ces) para inhibir o ralentizar la progresión de la enfermedad caracterizada por mayores cantidades de Ap, ralentizar la progresión de EA en el y/o prevenir el inicio o desarrollo de EA en un paciente en riesgo de la enfermedad.
Métodos de monitorización de la eficacia clínica
En diversas realizaciones, la efectividad del tratamiento puede determinarse comparando una medida de referencia de un parámetro de enfermedad antes de la administración del (de los) agente(s) (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) del mismo, o sales o solvatos farmacéuticamente aceptables de dichas hidantoína(s), dicho(s) estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados o profármacos del mismo) se inicia en el mismo parámetro uno o más puntos de tiempo después de que el agente(s) o análogo ha sido administrado. Un parámetro ilustrativo que se puede medir es un biomarcador (por ejemplo, un péptido oligómero) del procesamiento de APP. Dichos biomarcadores incluyen, entre otros, niveles aumentados de sAPPa, p3 (Ap17-42 o Ap17-40), sAPPp, Ap40 soluble y/o Ap42 soluble en sangre, plasma, suero, orina, mucosa o líquido cefalorraquídeo ( CSF). La detección de niveles aumentados de sAPPa y/o p3, y niveles disminuidos de sAPPp y/o APPneo es un indicador de que el tratamiento es efectivo. Por el contrario, la detección de niveles reducidos de sAPPa y/o p3, y/o niveles elevados de sAPPp, APPneo, Tau o fosfo-Tau (pTau) es un indicador de que el tratamiento no es efectivo.
Otro parámetro para determinar la efectividad del tratamiento es el nivel de depósitos de placa amiloide en el cerebro. Las placas de amiloide se pueden determinar usando cualquier método conocido en la técnica, por ejemplo, como se determina por CT, PET, PIB-PET y/o MRI. Administración del (de los) agente(s) (por ejemplo, hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) del mismo, o sales o solvatos farmacéuticamente aceptables de dicha(s) hidantoína(s), dicho(s) estereoisómero(s) o dicho tautómero(s), o análogos, derivados o profármacos de los mismos) pueden dar como resultado una reducción en la tasa de formación de placa, e incluso una retracción o reducción de depósitos de placa en el cerebro. La efectividad del tratamiento también se puede determinar observando una estabilización y/o mejora de las capacidades cognitivas del sujeto. Las habilidades cognitivas se pueden evaluar utilizando cualquier método aceptado en la técnica, que incluye, por ejemplo, la clasificación de demencia clínica (CDR), el examen de estado mini mental (MMSE) o la prueba de Folstein, los criterios de evaluación enumerados en el DSM-IV (Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition) o DSM-V, y similares.
La eficacia clínica se puede controlar usando cualquier método conocido en la técnica. Los biomarcadores medibles para monitorear la eficacia incluyen, pero no se limitan a, monitorización de los niveles de sangre, plasma, suero, orina, mucosa o líquido cefalorraquídeo (CSF) de sAPPa, sAPPp, Ap42, Ap40, APPneo y p3 (por ejemplo, Ap17-42 o
Ap17-40). La detección de niveles aumentados de sAPPa y/o p3, y niveles disminuidos de sAPPp y/o APPneo son indicadores de que el régimen de tratamiento o prevención es eficaz. Por el contrario, la detección de niveles disminuidos de sAPPa y/o p3, y niveles elevados de sAPPp y/o APPneo son indicadores de que el tratamiento o el régimen de prevención no es eficaz. Otros biomarcadores incluyen Tau y fosfo-Tau (pTau). La detección de niveles disminuidos de Tau y pTau son indicadores de que el régimen de tratamiento o prevención es eficaz.
La eficacia también se puede determinar midiendo la carga de placa amiloide en el cerebro. El régimen de tratamiento o prevención se considera eficaz cuando la carga de placa amiloide en el cerebro no aumenta o se reduce. Por el contrario, el régimen de tratamiento o prevención se considera ineficaz cuando aumenta la carga de placa amiloide en el cerebro. La carga de placa amiloide se puede determinar usando cualquier método conocido en la técnica, por ejemplo, incluyendo CT, PET, PIB-PET y/o MRI.
La eficacia también se puede determinar midiendo las capacidades cognitivas del sujeto. Las habilidades cognitivas se pueden medir utilizando cualquier método conocido en la técnica. Las pruebas ilustrativas incluyen la asignación de un puntaje de Clasificación de Demencia Clínica (CDR) o la aplicación del mini examen del estado mental (MMSE) (Folstein, et al., Journal of Psychiatric Research 12 (3): 189-98). Los sujetos que mantienen el mismo puntaje o que logran un puntaje mejorado, por ejemplo, cuando aplican la CDR o MMSE, indican que el tratamiento o el régimen de prevención es eficaz. Por el contrario, los sujetos que reciben un puntaje que indica capacidades cognitivas disminuidas, por ejemplo, al aplicar la CDR o MMSE, indican que el tratamiento o el régimen de prevención no ha sido eficaz.
En ciertas realizaciones, los métodos de monitorización pueden implicar la determinación de un valor de referencia de un biomarcador o parámetro medible (por ejemplo, carga de placa amiloide o habilidades cognitivas) en un sujeto antes de administrar una dosis del (de los) agente(s) y compararlo con un valor para el mismo biomarcador o parámetro medible después del tratamiento.
En otros métodos, se determina un valor de control (por ejemplo, una desviación media y estándar) del biomarcador o parámetro medible para una población de control. En ciertas realizaciones, los individuos en la población de control no han recibido tratamiento previo y no tienen EA, MCI, ni corren el riesgo de desarrollar EA o MCl. En tales casos, si el valor del biomarcador medible o el parámetro clínico se aproxima al valor de control, entonces el tratamiento se considera eficaz. En otras realizaciones, los individuos en la población de control no han recibido tratamiento previo y han sido diagnosticados con EA o MCI. En tales casos, si el valor del biomarcador medible o el parámetro clínico se aproxima al valor de control, entonces el tratamiento se considera ineficaz.
En otros métodos, un sujeto que actualmente no está recibiendo tratamiento pero que se ha sometido a un curso previo de tratamiento es monitorizado por uno o más de los biomarcadores o parámetros clínicos para determinar si se requiere una reanudación del tratamiento. El valor medido de uno o más de los biomarcadores o parámetros clínicos en el sujeto puede compararse con un valor alcanzado previamente en el sujeto después de un curso previo de tratamiento. Alternativamente, el valor medido en el sujeto puede compararse con un valor de control (media más desviación estándar/ANOVA) determinado en la población de sujetos después de someterse a un curso de tratamiento. Alternativamente, el valor medido en el sujeto se puede comparar con un valor de control en poblaciones de profilaxis sujetos afectados que permanecen libres de síntomas de enfermedad, o poblaciones de sujetos tratados terapéuticamente que muestran una mejoría de las características de la enfermedad. En tales casos, si el valor del biomarcador medible o el parámetro clínico se aproxima al valor de control, entonces el tratamiento se considera eficaz y no necesita reanudarse. En todos estos casos, una diferencia significativa en relación con el nivel de control (por ejemplo, más de una desviación estándar) es un indicador de que el tratamiento debe reanudarse en el sujeto.
En ciertas realizaciones, la muestra de tejido para análisis es típicamente sangre, plasma, suero, orina, mucosa o líquido cefalorraquídeo del sujeto.
Kits
En diversas realizaciones, el (los) agente(s) activo (por ejemplo, hidantoínas) descritos en el presente documento pueden encerrarse en recipientes de dosis múltiples o únicas. Los agentes incluidos se pueden proporcionar en kits, por ejemplo, que incluyen partes componentes que se pueden ensamblar para su uso. Por ejemplo, un agente activo en forma liofilizada y un diluyente adecuado pueden proporcionarse como componentes separados para la combinación antes de su uso. Un kit puede incluir un agente activo y un segundo agente terapéutico para la administración conjunta. El agente activo y el segundo agente terapéutico pueden proporcionarse como partes componentes separadas. Un kit puede incluir una pluralidad de contenedores, cada contenedor con una o más dosis unitarias de los compuestos. Los recipientes se adaptan preferiblemente para el modo de administración deseado, que incluye, pero no se limita a tabletas, cápsulas de gel, cápsulas de liberación sostenida y similares para administración oral; productos de depósito, jeringas precargadas, ampollas, viales y similares para administración parenteral; y parches, medipads, cremas y similares para administración tópica, por ejemplo, como se describe en el presente documento.
En ciertas realizaciones, se proporciona un kit en donde el kit comprende uno o más compuestos de hidantoína descritos aquí, o un tautómero o estereoisómero de los mismos, o una sal o solvato farmacéuticamente aceptable de
dicho compuesto, dicho estereoisómero o dicho tautómero, preferiblemente proporcionado como una composición farmacéutica y en un recipiente o recipientes adecuados y/o con un embalaje adecuado; opcionalmente uno o más agentes activos adicionales, que si están presentes se proporcionan preferiblemente como una composición farmacéutica y en un recipiente o recipientes adecuados y/o con un embalaje adecuado; y opcionalmente instrucciones de uso, por ejemplo instrucciones escritas sobre cómo administrar el compuesto o las composiciones.
En otra realización, se proporciona un kit que comprende un único recipiente o múltiples recipientes: (a) una composición farmacéuticamente aceptable que comprende uno o más compuestos de la reivindicación 1 y/o cualquiera de los compuestos 1-10 mostrados en las Figuras 1 y 2, o un tautómero o estereoisómero del mismo, o una sal o solvato farmacéuticamente aceptable de dicho compuesto, dicho estereoisómero o dicho tautómero, opcionalmente una composición farmacéuticamente aceptable que comprende uno o más agentes terapéuticos adicionales; y opcionalmente instrucciones para usar su uso. El kit puede comprender opcionalmente el etiquetado (por ejemplo, materiales de instrucción) apropiados para el uso o usos previstos.
Como con cualquier producto farmacéutico, el (los) material(es) de embalaje y/o contenedor(es) están diseñados para proteger la estabilidad del producto durante el almacenamiento y envío. Además, los kits pueden incluir instrucciones de uso u otro material informativo que pueda aconsejar al usuario, como, por ejemplo, un médico, técnico o paciente, sobre cómo administrar adecuadamente la(s) composición(es) como tratamiento profiláctico, terapéutico o de mejora de la enfermedad de interés. En algunas realizaciones, las instrucciones pueden indicar o sugerir un régimen de dosificación que incluye, entre otros, dosis reales y procedimientos de monitorización.
En algunas realizaciones, las instrucciones pueden incluir material informativo que indica que la administración de las composiciones puede dar como resultado reacciones adversas que incluyen, pero no se limitan a, reacciones alérgicas tales como, por ejemplo, anafilaxia. El material informativo puede indicar que las reacciones alérgicas pueden presentarse solo como erupciones cutáneas pruriginosas leves o pueden ser graves e incluyen eritrodermia, vasculitis, anafilaxia, síndrome de Steven-Johnson y similares. En ciertas realizaciones, los materiales informativos pueden indicar que la anafilaxia puede ser fatal y puede ocurrir cuando se introduce cualquier proteína extraña en el cuerpo. En ciertas realizaciones, el material informativo puede indicar que estas reacciones alérgicas pueden manifestarse como urticaria o erupción cutánea y convertirse en reacciones sistémicas letales y pueden ocurrir poco después de la exposición, como, por ejemplo, dentro de los 10 minutos. El material informativo puede indicar además que una reacción alérgica puede causar que un sujeto experimente parestesia, hipotensión, edema laríngeo, cambios de estado mental, angioedema facial o faríngeo, obstrucción de las vías respiratorias, broncoespasmo, urticaria y prurito, enfermedad del suero, artritis, nefritis alérgica, glomerulonefritis, artritis temporal, eosinofilia, o una combinación de los mismos.
Mientras que los materiales de instrucción típicamente comprenden materiales escritos o impresos, no están limitados a tales. En el presente documento se contempla cualquier medio capaz de almacenar tales instrucciones y comunicarlas a un usuario final. Dichos medios incluyen, entre otros, medios de almacenamiento electrónico (por ejemplo, discos magnéticos, cintas, cartuchos, chips), medios ópticos (por ejemplo, CD ROM) y similares. Dichos medios pueden incluir direcciones de sitios de Internet que proporcionan dichos materiales de instrucción.
En algunas realizaciones, los kits pueden comprender uno o más materiales de embalaje tales como, por ejemplo, una caja, botella, tubo, vial, recipiente, pulverizador, insuflador, bolsa intravenosa (I.V.), sobre y similares; y al menos una forma de dosificación unitaria de un agente que comprende el (los) agente(s) activo(s) descrito aquí y un material de envasado. En algunas realizaciones, los kits también incluyen instrucciones para usar la composición como tratamiento profiláctico, terapéutico o de mejora para la enfermedad de interés.
En algunas realizaciones, los artículos de fabricación pueden comprender uno o más materiales de embalaje tales como, por ejemplo, una caja, botella, tubo, vial, recipiente, pulverizador, insuflador, bolsa intravenosa (I.V.), sobre y similares; y una primera composición que comprende al menos una forma de dosificación unitaria de un agente que comprende una o más hidantoínas descritas en el presente documento, o un tautómero(s) o estereoisómero(s) de la misma, o sales o solvatos farmacéuticamente aceptables de dicha hidantoína(s), dicho estereoisómero(s), o dicho(s) tautómero(s), o análogos, derivados, o profármacos del mismo dentro del material de envasado, junto con una segunda composición que comprende un segundo agente tal como, por ejemplo, un agente usado en el tratamiento y/o profilaxis de Enfermedad de Alzheimer (por ejemplo, como se describe en el presente documento), o cualquier profármaco, bacalao, metabolito, análogo, homólogo, congénere, derivados, sales y combinaciones de los mismos. En algunas realizaciones, los artículos de fabricación también pueden incluir instrucciones para usar la composición como un tratamiento profiláctico, terapéutico o de mejora para la enfermedad de interés.
Ejemplos
Ejemplo 1
Los siguientes ejemplos se ofrecen para ilustrar, pero no para limitar la invención reivindicada.
Síntesis del Compuesto 15-(3,5-difluorofenil)-5-fenilimidazolidin-2,4-diona (Hidantoína-1)
Paso 1: Síntesis de (3,5-difluorofenil)(2-fenil-1,3-ditian-2-il)metanol

Se disolvió 2-fenil-1,3-ditiano (1.791 g, 9.12 mmol) en 20 ml de THF seco y se enfrió a 0°C. Se añadió BuLi (6.84 ml, 10.94 mmol) gota a gota bajo nitrógeno y la mezcla se agitó durante 30 minutos a 0°C. Se añadió una solución de 3,5-difluorobenzaldehído (1.00 ml, 9.12 mmol) en THF (10 ml) y la mezcla se agitó durante 30 minutos, luego se calentó a temperatura ambiente durante 1 hora y se inactivó con solución saturada de cloruro de amonio. La fase orgánica se lavó con salmuera y se secó con sulfato de sodio. El disolvente se eliminó a vacío para dar metanol crudo (3,5-difluorofenil)(2-fenil-1,3-ditian-2-il) (3.15 g, 9.31 mmol, 104% de rendimiento) como un aceite amarillo espeso. El residuo se llevó al siguiente paso.
Paso 2: Síntesis de 2-(3,5-difluorofenil)-2-hidroxi-1-feniletanona

Se disolvió (3,5-difluorofenil)(2-fenil-1,3-ditian-2-il)metanol (3.15 g, 9.31 mmol) en 15 ml de acetonitrilo y 2.5 ml de agua. Se añadió lentamente bis(trifluoroacetoxi)yodobenceno (5.00 g, 11.63 mmol) en 10 ml de acetonitrilo a temperatura ambiente a la solución agitada vigorosamente. Después de 30 minutos, el análisis por TLC (25% EtOAC/hexano) indicó una reacción completa. Se añadió EtOAc (150 ml) y la mezcla se enjuagó con solución saturada de bicarbonato de sodio (50 ml) y salmuera (50 ml). Las fracciones orgánicas se secaron y el disolvente se eliminó a vacío. El producto crudo se purificó por cromatografía de columna instantánea (EtOAc/hexano al 12.5%) para dar 2-(3,5-difluorofenil)-2-hidroxi-1-feniletanona (1.10 g, 4.43 mmol, 48%) como un sólido amarillo pálido. La RMN de protones fue consistente con la estructura propuesta.
Paso 3: Síntesis de 1-(3,5-difluorofenil)-2-feniletano-1,2-diona
Se disolvió 2-(3,5-difluorofenil)-2-hidroxi-1-feniletanona (1.10 g, 4.43 mmoles) en ácido acético al 80% junto con hidrato de diacetoxi-cobre (44 mg, 0.22 mmoles) y nitrato de amonio (0.30 g, 3.75 mmol). La mezcla se calentó a reflujo durante 2.5 horas y luego se enfrió. La mezcla de reacción se vertió en acetato de etilo (50 ml) y se lavó con salmuera (2 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó. El material crudo se purificó por cromatografía en columna (5% EtOAc/hexano) para dar 1-(3,5-difluorofenil)-2-feniletano-1,2-diona (1.10 g, 4.43 mmol, cuant.) como un sólido amarillo brillante. La r Mn de protones fue consistente con la estructura propuesta.
Paso 4: Síntesis de 5-(3,5-difluorofenil)-5-fenilimidazolidin-2,4-diona

A una solución de 1-(3,5-difluorofenil)-2-feniletano-1,2-diona (0.99 g, 4.02 mmol), urea (0.435 g, 7.24 mmol) en etanol (20 ml) y agua (5 ml) se añadió NaOH sólido (0.29 g, 7.24 mmol). La mezcla de reacción se calentó a reflujo hasta que el análisis por TLC (50% EtOAc/hexano) indicó una reacción completa. La mezcla de reacción se diluyó con agua (30 ml) y se acidificó cuidadosamente con HCl 2M a pH 5. La mezcla de reacción se extrajo con acetato de etilo (100 ml) y se lavó con agua (50 ml) y salmuera (50 ml). El extracto orgánico se secó sobre sulfato de sodio, se filtró y se evaporó para dar un residuo que se trituró con mezclas de acetona y hexano para proporcionar 5-(3,5-difluorofenil)-5-fenilimidazolidin-2,4-diona (0.220 g) como un sólido que estaba altamente hidratado con agua como se juzga por espectroscopia de RMN. El sólido, después de calentar (120°C) al vacío durante la noche, proporcionó el producto deseado (0.20 g, 0.69 mmol, 17%) como un polvo blanco. 1H RMN (400 MHz, d6-DMSO) 5 ppm 11.31 (brs, 1H), 9.44 (s, 1H), 7.37 (m, 6H), 7.10 (d, J = 6.77 Hz, 2H); 13C RMN (100 MHz, d6-DMSO) 5 ppm 173.89, 163.52, 163.39, 161.06, 160.93, 155.78, 143.79, 143.70, 143.61, 139.16, 128.82, 128.44, 126.26, 110.12, 110.04, 109.93, 109.85, 104.11, 103.86, 103.60, 69.43 (nota: el acoplamiento C-F se observó en varios casos dando lugar a señales de doblete y triplete; LC (220 nm): Rt = 4.09 min, pureza LC: 95.8%, m/z (M-1): 300.3.
Ejemplo 2
Síntesis de FAH-2: 2-amino-4-(4-(difluorometoxi)fenil)-4-(3,5-difluorofenil)-1-metil-1H-imidazol-5(4H)-ona
Paso 1: Síntesis de 2-(3,5-difluorofenil)-1,3-ditiano

Se añadió gota a gota BF3.OMe2 (0.70 ml, 7.62 mmol) a una solución de 1,3-propanoditiol (0.90 ml, 8.94 mmol) y 3,5 difluorobenzaldehído (1.00 ml, 8.94 mmol) en DCM (50 ml) a 0°C. La reacción se agitó a temperatura ambiente durante 1 hora cuando la TLC (5% EtOAc/hexano) indicó una reacción completa. La mezcla de reacción se diluyó luego con DCM (50 ml), se filtró a través de Celite (y la almohadilla de Celite se lavó con DCM adicional (3 x 20 ml) y el filtrado se lavó con salmuera (50 ml), NaHCO3 saturado (3 x 50 ml), solución de KOH al 10% (50 ml), agua (50 ml) y salmuera (50 ml) y finalmente se secó sobre sulfato de sodio. El extracto orgánico se filtró y se evaporó para proporcionar 2 (3,5-difluorofenil)-1,3-ditiano (2.14 g, 9.21 mmol, 103%) como agujas cristalinas blancas. La RMN de protones fue consistente con la estructura propuesta.
Paso 2: Síntesis de (4-(difluorometoxi)fenil)(2-(3,5-difluorofenil)-1,3-ditian-2-il)metanol

Se disolvió 2-(3,5-difluorofenil)-1,3-ditiano (2.14 g, 9.21 mmol) en 20 ml de THF seco y se enfrió a 0°C. Se añadió BuLi (8.50 ml, 10.20 mmol) gota a gota bajo nitrógeno y la mezcla se agitó durante 15 minutos a 0°C. Se añadió una solución de 4-(difluorometoxi)benzaldehído (1.30 ml, 9.33 mmol) en THF (10 ml) y la mezcla se agitó durante 10 minutos, luego se calentó a temperatura ambiente durante 10 minutos y se inactivó con solución saturada de cloruro de amonio. La fase orgánica se lavó con salmuera y se secó con sulfato de sodio. El disolvente se eliminó al vacío para dar un residuo que se purificó por cromatografía instantánea en columna (EtOAc al 10%/hexano) para proporcionar (4 (difluorometoxi)fenilo)(2-(3,5-difluorofenil)-1,3-ditiano-2-il)metanol (1.90 g, 4.70 mmol, 51%) como un aceite amarillo espeso. La RMN de protones fue consistente con la estructura propuesta.
Paso 3: Síntesis de 2-(4-(difluorometoxi)fenil)-1-(3,5-difluorofenil)-2-hidroxietanona

Se disolvió (4-(difluorometoxi)fenil)(2-(3,5-difluorofenil)-1,3-ditian-2-il)metanol (1.90 g, 4.70 mmol) en 15 ml de acetonitrilo y 2.5 ml de agua. Se añadió lentamente bis(trifluoroacetoxi)yodobenceno (2.53 g, 5.87 mmol) en 10 ml de acetonitrilo a temperatura ambiente a la solución agitada vigorosamente. Después de 30 minutos, el análisis por TLC (25% EtOAC/hexano) indicó una reacción completa. Se añadió EtOAc (150 ml) y la mezcla se enjuagó con solución saturada de bicarbonato de sodio (50 ml) y salmuera (50 ml). Las fracciones orgánicas se secaron y el disolvente se eliminó a vacío. El producto crudo se purificó por cromatografía instantánea en columna (EtOAc/hexano al 12.5%) para dar 2-(4-(difluorometoxi)fenil)-1-(3,5-difluorofenil)-2-hidroxietanona (0.460 g, 1.46 mmol, 31%) como un sólido amarillo pálido. La RMN de protones fue consistente con la estructura propuesta.
Paso 4: Síntesis de 1-(4-(difluorometoxi)fenil)-2-(3,5-difluorofenil)etano-1,2-diona

Se disolvió 2-(4-(difluorometoxi)fenil)-1-(3,5-difluorofenil)-2-hidroxietanona (0.46 g, 1.46 mmol) en ácido acético al 80% junto con hidrato de diacetoxicobre (26 mg, 0.13 mmol) y nitrato de amonio (0.18 g, 2.25 mmol). La mezcla se calentó a reflujo durante 90 minutos y luego se enfrió. La mezcla de reacción se vertió en acetato de etilo (50 ml) y se lavó con salmuera (2 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó. El material crudo se hizo pasar a través de un tapón de gel de sílice, se evaporó y se hizo un azeótropo con tolueno (3 x 20 ml) para eliminar el exceso de ácido acético para dar 1-(4-(difluorometoxi)fenil)-2-(3,5-difluorofenilo)etano-1,2-diona crudo (0.456 g, 1.46 mmol, 100%) como un sólido amarillo brillante. El sólido crudo se llevó al siguiente paso.
Paso 5: Síntesis de 2-amino-4-(4-(difluorometoxi)fenil)-4-(3,5-difluorofenil)-1-metil-1H-imidazol-5(4H)-ona

A 1-(4-(difluorometoxi)fenil)-2-(3,5-difluorofenil)etano-1,2-diona (0.456 g, 1.46 mmol) en etanol (20 ml) y agua (5 ml) se añadió clorhidrato de 1-metilguanidina (0.16 g, 1.46 mmol) y carbonato de potasio (0.61 g, 4.38 mmol). La mezcla se dejó a reflujo durante 3 horas y luego se enfrió a temperatura ambiente. Los volátiles se eliminaron al vacío y el residuo se recogió en agua y se extrajo con cloroformo (50 ml). Las fracciones orgánicas se secaron con sulfato de sodio y el disolvente se eliminó al vacío. El material crudo se purificó por cromatografía en columna (EtOAc) para proporcionar 2-amino-4-(4-(difluorometoxi)fenil)-4-(3,5-difluorofenil)-1-metil-1H-imidazol-5(4H)-ona (0.172 g, 0.47 mmol) como un cristal que estaba muy contaminado con acetato de etilo según el análisis de RMN. El cristal se llevó a etanol seco (3 ml) y se colocó en capas con hexano (1 ml) para obtener una solución turbia. La solución se evaporó rotativamente para dar un aceite que se solidificó al reposar. Esto se secó durante la noche y el peso obtenido fue de 0.150 g. El sólido contenía residuos de disolvente de etanol y hexano como se juzga por análisis de RMN. Por lo tanto, los sólidos se redisolvieron en iso-propanol, se evaporaron por rotación y se secaron a alto vacío a 90°C durante la noche para proporcionar 2-amino-4-(4-(difluorometoxi)fenil)-4-(3,5-difluorofenilo)-1-metil-1H-imidazol-5(4H)-ona (0.130 g, 0.35 mmol, 24%) como un sólido blanquecino. 1H RMN (400 MHz, CDCla) 5 ppm 7.47 (d, J = 8.79 Hz, 2H), 7.10-7.00 (m, 4H), 6.70 (tt, J = 8.73, 2.32 Hz, 1H), 6.53 (t, Jh-f= 73.76 Hz, 1H), 5.45 (brs, 1H), 3.11 (s, 3H); 13C RMN (100 MHz, CDCla) 5 ppm 178.619, 164.122, 163.996, 161.650, 161.524, 155.704, 150.742, 150.714, 150.687, 145.164, 145.081, 144.991, 137.735, 128.390, 119.444, 118.328, 115.743, 113.158, 110.329, 110.255, 110.138, 110.065, 103.421, 103.169, 102.917, 77.203, 25.966(nota: el acoplamiento CF se observó en varios casos dando lugar a señales de doblete y triplete); LC (260 nm): Rt = 3,899 min, pureza LC: 96.3%, m/z (M-1): 366.
Ejemplo 3
Síntesis de FAH-3: 2-amino-4-(4-(difluorometoxi)fenil)-4-(3,5-difluorofenil)-1-metil-1H-imidazol-5(4H)-ona
Paso 1: Síntesis de 2-(3,5-difluorofenil)-1,3-ditiano

Se añadió gota a gota BF3.OMe2 (1.40 ml, 15.25 mmol) a una solución de 1,3-propanoditiol (1.81 ml, 17.87 mmol) y 3,5-difluorobenzaldehído (2.00 ml, 17.87 mmol) en DCM (50 ml) a 0°C. La reacción se agitó a temperatura ambiente durante 1 hora cuando la TLC (5% EtOAc/hexano) indicó una reacción completa. La mezcla de reacción se diluyó luego con DCM adicional (50 ml), se filtró a través de Celite, y la almohadilla de Celite se lavó con DCM adicional (3 x 20 ml). El filtrado se lavó con NaHCO3 saturado (3 x 50 ml), solución de KOH al 10% (2 x 50 ml), agua (2 x 50 ml) y salmuera (50 ml) y finalmente se secó sobre sulfato de sodio. El extracto orgánico se filtró y se evaporó para proporcionar 2-(3,5-difluorofenil)-1,3-ditiano (4.12 g, 17.73 mmol, 99%) como un sólido blanco. La RMN de protones fue consistente con la estructura propuesta.
Paso 2: Síntesis de 4-(difluorometoxi)-3-metilbenzaldehído

Intento 1:
Una solución de clorodifluoroacetato de sodio (3.23 g, 21.15 mmol) y 4-hidroxi-3-metilbenzaldehído (1.44 g, 10.58 mmol), carbonato de potasio (2.19 g, 15.87 mmol) en una mezcla de DMF (8 ml) y agua (2 ml) se calentó a 100°C durante 2 horas. La mezcla de reacción se enfrió y se concentró. HCl (1.5 ml) seguido de agua (2.1 ml). La mezcla de reacción se diluyó con agua (20 ml) y se extrajo con acetato de etilo (3 x 25 ml). El extracto orgánico se lavó con solución acuosa de LiCl al 10% (m/v) (3 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó para dar un residuo que se sometió a cromatografía instantánea (EtOAc al 15%/hexano) para dar 4-(difluorometoxi)-3-metilbenzaldehído (0.244 g, 1.31 mmol, 12%) como un aceite marrón y 4-hidroxi-3-metilbenzaldehído recuperado (1.164 g, 8.55 mmol, 81%) como un sólido marrón.
El experimento se repitió nuevamente, excepto con la ausencia de agua. En resumen, el procedimiento experimental se da a continuación:
Intento 2:
Se añadió una solución de clorodifluoroacetato de sodio (2.60 g, 17.04 mmol) y 4-hidroxi-3-metilbenzaldehído (1.16 g, 8.52 mmol) en DMF (15 ml) durante 3 horas a una solución de DMF (15 ml) que contenía carbonato de potasio (1.77 g, 12.78 mmol) a 95°C. La reacción se dejó transcurrir durante 15 minutos adicionales y luego se enfrió. La mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 50 ml). El extracto orgánico se lavó con solución acuosa de LiCl al 10% (m/v) (3 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó para dar un residuo que se sometió a cromatografía instantánea (EtOAc al 15%/hexano) para dar 4-(difluorometoxi)-3-metilbenzaldehído (021GLM-053_1(2), 1.00 g, 5.37 mmol, 63%) como un aceite amarillo. Este aceite se combinó con el del experimento anterior y se pasó a través de una columna de pipeta Pasteur eluyendo con EtOAC al 10%/hexano para dar un aceite que solidificó en reposo (1.315 g, 7.06 mmol, 67% en las dos reacciones). La RMN de protones fue consistente con la estructura propuesta.
Finalmente, repitiendo el experimento usando las condiciones en el intento 2, se aislaron 1.4 g adicionales del producto deseado.
Paso 3: Síntesis de (4-(difluorometoxi)-3-metilfenil)(2-(3,5-difluorofenil)-1,3-ditian-2-il)metanol

Se disolvió 2-(3,5-difluorofenil)-1,3-ditiano (3.12 g, 13.43 mmol) en 30 ml de THF seco y se enfrió a -10°C. Se añadió BuLi (1.6 M, 12.0 ml, 19.20 mmol) gota a gota bajo nitrógeno y la mezcla se agitó durante 15 minutos a -10°C para proporcionar una solución de color rojo sangre. Se añadió gota a gota una solución de 4-(difluorometoxi)-3-metilbenzaldehído (2.50 g, 13.43 mmol) en THF (10 ml) y la mezcla se agitó durante 15 minutos, luego se calentó a temperatura ambiente durante 10 minutos y se detuvo con solución saturada de cloruro de amonio. La fase orgánica se lavó con salmuera y se secó con sulfato de sodio. El disolvente se eliminó y el residuo se sometió a cromatografía instantánea (10% EtOAc/hexano) para dar (4-(difluorometoxi)-3-metilfenil)(2-(3,5-difluorofenil)-1,3-ditian-2-il)metanol (3.07 g, 7.22 mmol, 55%) como un aceite espeso que solidificó al reposar. La RMN fue consistente con la estructura propuesta.
Paso 4: Síntesis de 2-(4-(difluorometoxi)-3-metilfenil)-1-(3,5-difluorofenil)-2-hidroxietanona

(4-(Difluorometoxi)-3-metilfenil)(2-(3,5-difluorofenil)-1,3-ditian-2-il)metanol (3.07 g, 7.34 mmol) se disolvió en acetonitrilo (15 ml ) y agua (2.5 ml). Se añadió bis(trifluoroacetoxi)yodobenceno (3.94 g, 9.17 mmol) en acetonitrilo (10 ml) lentamente a temperatura ambiente a la solución agitada vigorosamente. Después de 20 minutos, el análisis por TLC (20% EtOAc/hexano) indicó una reacción completa. Se añadió EtOAc (150 ml) y la mezcla se enjuagó con solución saturada de bicarbonato de sodio (50 ml) y salmuera (50 ml). Las fracciones orgánicas se secaron y el disolvente se eliminó a vacío. El producto crudo se purificó dos veces por cromatografía instantánea en columna (10% EtOAc/hexano) para dar 2-(4-(difluorometoxi)-3-metilfenil)-1-(3,5-difluorofenil)-2-hidroxietanona (0.853 g, 2.60 mmol, 35%) como un aceite amarillo pálido. La RMN de protones fue consistente con la estructura propuesta.
Paso 5: Síntesis de 1-(4-(difluorometoxi)-3-metilfenil)-2-(3,5-difluorofenil)etano-1,2-diona

Se disolvió 2-(4-(difluorometoxi)-3-metilfenil)-1-(3,5-difluorofenil)-2-hidroxietanona (0.853 g, 2.60 mmol) en ácido acético al 80% junto con hidrato de diacetoxicobre (52 mg, 0.26 mmol) y nitrato de amonio (0.156 g, 1.95 mmol). La mezcla se calentó a reflujo durante 90 minutos y luego se enfrió. La mezcla de reacción se vertió en acetato de etilo (50 ml) y se lavó con salmuera (2 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó. El residuo se sometió a formación de azeótropo con tolueno para eliminar el ácido acético y el residuo (0.737 g, 2.26 mmol, 87%) se usó directamente en la siguiente etapa.
Paso 6: Síntesis de 2-amino-4-(4-(difluorometoxi)-3-metilfenil)-4-(3,5-difluorofenil)-1-metil-1H-imidazol-5(4H)-ona

A una mezcla de 1-(4-(difluorometoxi)-3-metilfenil)-2-(3,5-difluorofenil)etano-1,2-diona (737 mg, 2.26 mmol) en etanol (15 ml) y dioxano (15 ml) se añadió hidrocloruro de 1-metilguanidina (990 mg, 9.04 mmol) y se agitó a temperatura
ambiente durante 15 minutos. Se añadió carbonato de sodio (958 mg, 9.04 mmol) en agua (5 ml) y la mezcla se sumergió en un baño de aceite a 85°C y se agitó durante 3 horas. La TLC (EtOAc) indicó una reacción completa. La mezcla de reacción se enfrió a temperatura ambiente y se concentró. La purificación por cromatografía instantánea (EtOAc) proporcionó 2-amino-4-(4-(difluorometoxi)-3-metilfenil)-4-(3,5-difluorofenil)-1-metil-1H-imidazol-5(4H)-ona (0.52 g, 1.36 mmol, 60%) como un sólido amarillo. 1H RMN (400 MHz, CDCla) 5 ppm 7.34-7.24 (m, 2H), 7.08-6.97 (m, 3H), 6.74-6.66 (m, 1H), 6.47 (t, Jh-f= 74.03 Hz, 1H), 5.45 (brs, 2H), 3.11 (s, 3H), 2.25 (s, 3H); LC (260 nm): R= 3.919 min, Pureza LC: 96.1%, m/z (M+1): 382, LC (220 nm): Rt = 3.922 min, Pureza Lc : 96.8%.
Ejemplo 4
Síntesis de FAH-5: 2-amino-4-(3,5-difluorofenil)-4-(3,5-dimetilfenil)-1-metil-1H-imidazol-5(4H)-ona
Síntesis de 2-(3,5-difluorofenil)-1,3-ditiano

Se añadió gota a gota BF3.OMe2 (2.50 ml, 27.5 mmol) a una solución de 1,3-propanoditiol (3.70 ml, 36.6 mmol) y 3,5-difluorobenzaldehído (4.10 ml, 36.6 mmol) en DCM (75 ml) a 0°C. La reacción se agitó a temperatura ambiente durante 1 hora cuando la TLC (5% EtOAc/hexano) indicó una reacción completa. La mezcla de reacción se diluyó luego con DCM adicional (50 ml), se filtró a través de Celite, y la almohadilla de Celite se lavó con DCM adicional (3 x 50 ml). El filtrado se lavó con NaHCO3 saturado (3 x 100 ml), solución de KOH al 10% (2 x 100 ml), agua (100 ml) y salmuera (100 ml) y finalmente se secó sobre sulfato de sodio. El extracto orgánico se filtró a través de una almohadilla de sílice y la almohadilla de sílice se lavó con mezclas de acetato de etilo/hexano al 10% (3 x 20 ml). El extracto orgánico se evaporó para proporcionar 2-(3,5-difluorofenil)-1,3-ditiano (8.44 g, 36.3 mmol, 99%) como un sólido blanco cristalino. La RMN fue consistente con la estructura propuesta.
Síntesis de (2-(3,5-difluorofenil)-1,3-ditian-2-il)(3,5-dimetilfenil)metanol

Se disolvió 2-(3,5-difluorofenil)-1,3-ditiano (8.00 g, 34.4 mmol) en 100 ml de THF seco y se enfrió a -10°C. Se añadió BuLi (1.6 M, 34 ml, 54.4 mmol) gota a gota bajo nitrógeno y la mezcla se agitó durante 15 minutos a -10°C para proporcionar una solución marrón. Se añadió gota a gota una solución de 3,5-dimetilbenzaldehído (4.84 g, 36.1 mmol) en THF (10 ml) y la mezcla de reacción se agitó durante 15 minutos, luego se calentó a temperatura ambiente durante 30 minutos y se inactivó con solución saturada de cloruro de amonio. La fase orgánica se lavó con salmuera y se secó con sulfato de sodio. El disolvente se eliminó y el residuo se purificó por cromatografía instantánea (EtOAc al 10%/hexano) para dar (2-(3,5-difluorofenil)-1,3-ditian-2-il)(3,5-dimetilfenil)metanol (6.60 g, 18.01 mmol, 52%) como un aceite espeso que solidificó al reposar. La RMN fue consistente con la estructura propuesta.
Síntesis de 1-(3,5-difluorofenil)-2-(3,5-dimetilfenil)-2-hidroxietanona

(2-(3,5-difluorofenil)-1,3-ditian-2-il)(3,5-dimetilfenil)metanol (6.60 g, 18.01 mmol) se disolvió en una solución de acetonitrilo (75 ml) y agua (15 ml). Se añadió bis(trifluoroacetoxi)yodobenceno (9.68 g, 22.51 mmol) en varias porciones a la solución agitada vigorosamente a temperatura ambiente. Después de 60 minutos, el análisis por TLC (20% EtOAC/hexano) pareció indicar una reacción completa. Se añadió acetato de etilo (150 ml) y la mezcla se
enjuagó con solución saturada de bicarbonato de sodio (2 x 50 ml) y salmuera (50 ml). Las fracciones orgánicas se secaron sobre sulfato de sodio, se filtraron y se evaporaron. El residuo se purificó dos veces por cromatografía instantánea en columna (10% EtOAc/hexano) para dar 1-(3,5-difluorofenil)-2-(3,5-dimetilfenil)-2-hidroxietanona (2.10 g, 7.60 mmol, 42%, aproximadamente 90% de pureza por RMN) como un sólido amarillo pálido contaminado con material de partida (aproximadamente 10%); Rf (10% EtOAc/hexano): 0.20 fue idéntico tanto para el material de partida como para el producto. Sin embargo, la RMN fue consistente con la estructura propuesta del producto, que era el componente principal.
Síntesis de 1-(3,5-difluorofenil)-2-(3,5-dimetilfenil)etano-1,2-diona

1-(3,5-difluorofenil)-2-(3,5-dimetilfenil)-2-hidroxietanona (2.10 g, 7.60 mmol) se disolvió en ácido acético al 80% (10 ml) junto con hidrato de diacetoxicobre (0.15 g, 0.76 mmol) y nitrato de amonio (0.46 g, 5.70 mmol). La mezcla se calentó a reflujo durante 90 minutos y luego se enfrió. La mezcla de reacción de color verde se vertió en acetato de etilo (50 ml) y se lavó con salmuera (2 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó. El residuo se sometió a cromatografía instantánea (EtOAc al 20%/hexano) para proporcionar 1-(3,5-difluorofenil)-2-(3,5-dimetilfenil)etano-1,2-diona (1.13 g, 4.12 mmol, 54%) como un sólido amarillo. La RMN fue consistente con la estructura propuesta.
Síntesis de 2-amino-4-(3,5-difluorofenil)-4-(3,5-dimetilfenil)-1-metil-1 H-imidazol-5(4H)-ona (FAH5)

A una mezcla de 1-(3,5-difluorofenil)-2-(3,5-dimetilfenil)etano-1,2-diona (500 mg, 1,82 mmol) en etanol (15 ml) y dioxano (15 ml) se añadió clorhidrato de 1-metilguanidina (799 mg, 7.29 mmol) y se agitó a temperatura ambiente durante 15 minutos. Se añadió carbonato de sodio (773 mg, 7.29 mmol) en agua (5 ml) y la mezcla se sumergió en un baño de aceite a 85°C y se agitó durante 4 horas. La TLC (EtOAc) indicó una reacción completa. La mezcla de reacción se enfrió a temperatura ambiente y se concentró. El residuo se purificó dos veces por cromatografía en columna (EtOAc, EtOAc al 50%/hexano) y finalmente por PTLC (cloroformo) para proporcionar 2-amino-4-(3,5-difluorofenil)-4-(3,5-dimetilfenil)-1-metil-1H-imidazol-5(4H)-ona (0.20 g, 0.61 mmol, 33%) como un sólido blanco después de secar a alto vacío a 60°C durante 36 horas. 1H RMN (400 MHz, CDC3) 5 ppm 7.07 (d, J = 6.83 Hz, 2H), 7.02 (brs, 2H), 6.91 (brs, 1H), 6.69 (t, J = 8.48 Hz, 1H), 5.22 (s, 2H), 3.10 (s, 3H), 2.27 (s, 6H); 13C RMN (100 MHz, CDC3) 5 ppm 164.1, 163.9, 161.6, 161.5, 155.3, 145.4, 140.4, 138.2, 129.7, 124.5, 110.5, 110.4, 110.3, 110.2, 103.0, 77.2, 25.9, 21.41, (nótese: debido a la presencia de átomos de flúor, se observan acoplamientos J2c-f - J4c-f que dan lugar a tripletes y dobletes mal resueltos); LC (230 nm) Rt (min) = 3.97, pureza LC = 95.29%; m/z: encontrado [M+H]+ = 330.1, esperado [M+H]+ = 330.1 (C18H18F2N3O).
Ejemplo 5
Síntesis de FAH-4 (ITH002329)
Síntesis de 2-(3,5-difluorofenil)-1,3-ditiano

Se anadio BF3.OMe2 (1.40 ml, 15.25 mmol) gota a gota a una solución de 1,3-propanoditiol (1.81 ml, 17.87 mmol) y 3,5-difluorobenzaldehído (2.00 ml, 17.87 mmol) en DCM (50 ml ) a 0°C. La reacción se agitó a temperatura ambiente durante 1 hora cuando la TLC (5% EtOAc/hexano) indicó una reacción completa. La mezcla de reacción se diluyó luego con DCM adicional (50 ml), se filtró a través de Celite, y la almohadilla de Celite se lavó con DCM adicional (3 x 20 ml). El filtrado se lavó con NaHCO3 saturado (3 x 50 ml), solución de KOH al 10% (2 x 50 ml), agua (2 x 50 ml) y salmuera (50 ml) y finalmente se secó sobre sulfato de sodio. El extracto orgánico se filtró y se evaporó para proporcionar 2-(3,5-difluorofenil)-1,3-ditiano (4.12 g, 17.73 mmol, 99%) como un sólido blanco.
Síntesis de 3-(difluorometoxi)benzaldehído

Se anadió una solución de clorodifluoroacetato de sodio (12.48 g, 82 mmol) y 3-hidroxibenzaldehído (5.00 g, 40.9 mmol) en DMF (75 ml) durante 3 horas a una solución de DMF (25 ml) que contenía carbonato de potasio ( 8.49 g, 61,4 mmol) a 95°C. La reacción se dejó transcurrir durante 2 horas adicionales y luego se enfrió. La mezcla de reacción se diluyó con agua (100 ml) y se extrajo con acetato de etilo (4 x 50 ml). El extracto orgánico se lavó con una solución acuosa de LiCl al 10% (m/v) (3 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó para dar un residuo que se sometió a cromatografía instantánea (EtOAc al 15%/hexano) para dar 3-(difluorometoxi)benzaldehído (2.50 g, 14.52 mmol, 36%) como un aceite amarillo. La RMN fue consistente con la estructura propuesta.
Síntesis de (3-(difluorometoxi)fenil)(2-(3,5-difluorofenil)-1,3-ditian-2-il)metanol

El 2-(3,5-difluorofenil)-1,3-ditiano (3.37 g, 14.52 mmol) se disolvió en 30 ml de THF seco y se enfrió a -10°C. Se anadió BuLi (1.6 M, 12.0 ml, 19.20 mmol) gota a gota bajo nitrógeno y la mezcla se agitó durante 15 minutos a -10°C para proporcionar una solución de color rojo sangre. Se anadió gota a gota una solución de 3-(difluorometoxi)benzaldehído (2.50 g, 14.52 mmol) en THF (10 ml) y la mezcla se agitó durante 15 minutos, luego se calentó a temperatura ambiente durante 10 minutos y se inactivó con solución saturada de cloruro de amonio. La fase orgánica se lavó con salmuera y se secó con sulfato de sodio. El disolvente se eliminó y el residuo se sometió a cromatografía instantánea (EtOAc al 10%/hexano) para dar (3-(difluorometoxi)fenil)(2-(3,5-difluorofenil)-1,3-ditian-2-il)metanol (3.33 g, 8.23 mmol, 57%) como un aceite espeso que se solidificó al reposar. La RMN fue consistente con la estructura propuesta.
Síntesis de 2-(3-(difluorometoxi)fenil)-1-(3,5-difluorofenil)-2-hidroxietanona

(3-(Difluorometoxi)fenil)(2-(3,5-difluorofenil)-1,3-ditian-2-il)metanol (3.33 g, 8.23 mmol) se disolvió en 15 ml de acetonitrilo y 2.5 ml de agua. Se añadió lentamente bis(trifluoroacetoxi)yodobenceno (4.43 g, 10.29 mmol) en 10 ml de acetonitrilo a la solución agitada vigorosamente a temperatura ambiente. Después de 30 minutos, el análisis por TLC (20% EtOAC/hexano) indicó una reacción completa. Se añadió EtOAc (150 ml) y la mezcla se enjuagó con solución saturada de bicarbonato de sodio (50 ml) y salmuera (50 ml). Las fracciones orgánicas se secaron y el disolvente se eliminó a vacío. El producto crudo se purificó dos veces por cromatografía instantánea en columna (10% EtOAc/hexano) para dar 2-(3-(difluorometoxi)fenil)-1-(3,5-difluorofenil)-2-hidroxietanona (1.01 g, 3.21 mmol, 39%) como un aceite amarillo pálido. La RMN fue consistente con la estructura propuesta.
Síntesis de 1-(3-(difluorometoxi)fenil)-2-(3,5-difluorofenil)etano-1,2-diona

2-(3-(Difluorometoxi)fenil)-1-(3,5-difluorofenil)-2-hidroxietanona (1.00 g, 3.18 mmol) se disolvió en ácido acético al 80% (10 ml) junto con hidrato de diacetoxicobre (0.13 g, 0.64 mmol) y nitrato de amonio (0.19 g, 2.39 mmol). La mezcla se calentó a reflujo durante 90 minutos y luego se enfrió. La mezcla de reacción de color cobre se vertió en acetato de etilo (50 ml) y se lavó con salmuera (2 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó. El residuo se sometió a cromatografía instantánea (EtOAc al 20%/hexano) para proporcionar 1-(3-(difluorometoxi)fenil)-2-(3,5-difluorofenil)etano-1,2-diona (0.46 g, 1.48 mmol, 47%) como un aceite amarillo. La elución adicional de la columna proporcionó material de partida (0.40 g, 1.27 mmol, 40% de recuperación) como un aceite. El producto deseado se usó tal como se recibió.
Síntesis de 2-amino-4-(3-(difluorometoxi)fenil)-4-(3,5-difluorofenil)-1-metil-1H-imidazol-5(4H)-ona

A una mezcla de 1-(3-(difluorometoxi)fenil)-2-(3,5-difluorofenil)etano-1,2-diona (463 mg, 1.48 mmol) en etanol (15 ml) y dioxano (15 ml) se añadió hidrocloruro de 1-metilguanidina (650 mg, 5.93 mmol) y se agitó a temperatura ambiente durante 15 minutos. Se añadió carbonato de sodio (629 mg, 5.93 mmol) en agua (5 ml) y la mezcla se sumergió en un baño de aceite a 85°C y se agitó durante 3 horas. La TLC (EtOAc) indicó una reacción completa. La mezcla de reacción se enfrió a temperatura ambiente y se concentró. El residuo se purificó dos veces por PTLC (EtOAc) para proporcionar 2-amino-4-(3-(difluorometoxi)fenil)-4-(3,5-difluorofenil)-1-metiMH-imidazol-5(4H)-ona (0.22 g, 0.60 mmol, 40%) como un sólido amarillo después de secar a alto vacío a 60°C durante 48 horas. 1H RMN (400 MHz, CDCb) 5 ppm 7.38-7.29
(m, 2H), 7.24 (ancho m, 1H), 7.10-7.00 (m, 3H), 6.71 (m, 1H), 6.49 (t, Jh-f = 74.03 Hz, 1H), 5.61 (brs, 2H), 3.10 (s, 3H); 13C RMN (100 MHz, CDCla) 5 ppm 178.30, 164.13, 164.00, 161.66, 161.53, 156.03, 151.27, 151.24, 151.21, 145.00, 144.92, 144.83, 142.88, 129.90, 123.81, 118.73, 118.42, 118.17, 115.84, 113.25, 110.30, 110.22, 110.11, 110.03, 103.47, 103.22, 102.97, 74.92, 25.95 (nótese: debido a la presencia de átomos de flúor, se anotan los acoplamientos J2c-f - J4c-f que dan lugar a tripletes y dobletes); LC (220 nm): Rt = 3.85 min, pureza LC: 95,6%, m/z [M]+ = 367.9,
Ejemplo 6
FAH-17: 2-amino-4-(4-(difluorometoxi)fenil)-4-(3-fluorofenil)-1-metil-1H-imidazol-5(4H)-ona
Paso 1: Síntesis de 2-(4-(difluorometoxi)-3-metilfenil)-1,3-ditiano

Se añadió gota a gota BF3.OEt2 (4.30 ml, 34.8 mmol) a una solución de 1,3-propanoditiol (4.07 ml, 40.3 mmol) y 3-fluorobenzaldehído (5.00 g, 40.3 mmol) en DCM (201 ml) a 0°C. La reacción se agitó a temperatura ambiente durante 1 hora cuando la TLC (5% EtOAc/hexano) indicó una reacción completa. La mezcla de reacción se diluyó luego con DCM (50 ml), se filtró a través de Celite (y la almohadilla de Celite se lavó con DCM adicional (3 x 50 ml)) y el filtrado se lavó con salmuera (100 ml), NaHCO3 saturado (3 x 100 ml), solución de KOH al 10% (100 ml), agua (100 ml) y salmuera (100 ml) y finalmente se secó sobre sulfato de sodio. El extracto orgánico se filtró y se evaporó. El producto se purificó usando acetato de etilo al 5%:hexano para proporcionar 2-(3-fluorofenil)-1,3-ditiano (8.71 g, 39.0 mmol, 97%) como un aceite transparente. El aceite se usó directamente en el siguiente paso. La RMN fue consistente con la estructura propuesta.
Paso 2: Síntesis de 4-(difluorometoxi)-3-metilbenzaldehído

Se añadió una solución de clorodifluoroacetato de sodio (2.60 g, 17.04 mmol) y 4-hidroxi-3-metilbenzaldehído (1.16 g, 8.52 mmol) en DMF (15 ml) durante 3 horas a una solución de DMF (15 ml) que contenía carbonato de potasio (1.77 g, 12.78 mmol) a 95°C. La reacción se dejó transcurrir durante 15 minutos adicionales y luego se enfrió. La mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 50 ml). El extracto orgánico se lavó con solución acuosa de LiCl al 10% (m/v) (3 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó para dar un residuo que se sometió a cromatografía instantánea (EtOAc al 15%/hexano) para dar 4-(difluorometoxi)-3-metilbenzaldehído (021GLM-053_1(2), 1.00 g, 5.37 mmol, 63%) como un aceite amarillo. Este aceite se combinó con el del experimento anterior y se pasó a través de una columna de pipeta Pasteur eluyendo con EtOAC al 10%/hexano para dar un aceite que solidificó en reposo (1.315 g, 7.06 mmol, 67% en las dos reacciones). La RMN de protones fue consistente con la estructura propuesta.
Finalmente, repitiendo el experimento usando las condiciones en el intento 2, se aislaron 1.4 g adicionales del producto deseado.
Paso 3: Síntesis de (2-(4-(difluorometoxi)-3-metilfenil)-1,3-ditian-2-il)(3-fluorofenil)metanol
Se disolvió 2-(3-fluorofenil)-1,3-ditiano (4.00 g, 18.66 mmol) en THF seco (93.5 ml) y se enfrió a -10°C. Se añadió nBuLi (1.6 M, 14.00 ml, 22.40 mmol) gota a gota bajo nitrógeno y la mezcla se agitó durante 30 minutos a -10°C para proporcionar una solución roja oscura. Se añadió gota a gota una solución de 4-(difluorometoxi)-3-metilbenzaldehído (3.47 g, 18.66 mmol) en t Hf (93.5 ml) y la mezcla a -10°C y se agitó durante 15 minutos, luego se calentó a temperatura ambiente durante 1 h y se detuvo con solución saturada de cloruro de amonio (7.5 ml) seguido de dilución con EtOAc (50 ml). La fase orgánica se lavó con agua (2x20 ml), salmuera (1x20 ml) y se secó con sulfato de sodio. Después de la filtración y concentración, el producto crudo se purificó por cromatografía de columna instantánea (15% EtOAc/Hex) para dar (4-(difluorometoxi)-3-metilfenil)(2-(3-fluorofenil)-1,3-ditian-2-il)metanol (6.00 g, 14.96 mmol, 80%) como un aceite.
Paso 4: Síntesis de 2-(4-(difluorometoxi)-3-metilhenil)-1-(3-fluorohenil)-2-hidroxietanona

(4-(Difluorometoxi)-3-metilfenil)(2-(3-difluorofenil)-1,3-ditian-2-il)metanol (3.07 g, 7.34 mmol) se disolvió en acetonitrilo (15 ml) y agua (2.5 ml). Bis(trifluoroacetoxi)yodobenceno (3.94 g, 9.17 mmol) en acetonitrilo (10 ml) se añadió lentamente a temperatura ambiente a la solución agitada vigorosamente. Después de 20 minutos, el análisis por TLC (20% EtOAc/hexano) indicó una reacción completa. Se añadió EtOAc (150 ml) y la mezcla se enjuagó con solución saturada de bicarbonato de sodio (50 ml) y salmuera (50 ml). Las fracciones orgánicas se secaron y el disolvente se eliminó a vacío. El producto crudo se purificó dos veces por cromatografía de columna instantánea (10% EtOAc/hexano) para dar 2-(4-(difluorometoxi)-3-metilfenil)-1-(3-fluorofenil)-2-hidroxietanona (0.853 g, 2.60 mmol, 35%) como un aceite amarillo pálido. La RMN de protones fue consistente con la estructura propuesta.
Paso 5: Síntesis de 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-fluorofenil)etano-1,2-diona

1-(2,4-difluorofenil)-2-(4-metoxi-3-fluorofenil)etano-1,2-diona se sintetizó de acuerdo con el procedimiento representativo usando 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-fluorofenil)-2-hidroxietanona (0.500 g, 1.612 mmol) y dio 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-fluorofenil)etano-1,2-diona (0.3881 g, 78%) como un sólido amarillo. La Rm N fue consistente con la estructura propuesta.
Paso 6: Síntesis de 2-amino-4-(4-(difluorometoxi)fenil)-4-(3-fluorofenil)-1-metil-1H-imidazol-5(4H)-ona

Se añadió carbonato de potasio (0.516 g, 4.87 mmol) en agua (4.6 ml) a una mezcla de 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-fluorofenil)etano-1,2-diona (0.375 g, 1.217 mmol), clorhidrato de 1-metilguanidina (0.533 g, 4.87 mmol), dioxano (19 ml) y alcohol etílico (25 ml). La mezcla de reacción se agitó a 85°C durante 4 h. Los volátiles se eliminaron al vacío, y el residuo se tomó en cloroformo (100 ml) y se lavó con agua (2x25 ml). Los extractos orgánicos se secaron sobre MgSO4. La evaporación y purificación por cromatografía instantánea (60% EtOAc/Hex a 100% EtOAc) seguida de recristalización en CHCb/hexanos dio 2-amino-4-(4-(difluorometoxi)-3-metNfenN)-4-(3-fluorofenN)-1-metiMH-imidazol-5(4H)-ona (216 mg, 47%) como un sólido blanquecino. 1H RMN (400 MHz, CDCb) 57.37 - 7.13 (m, 5H), 6.96 (m, 2H), 6.46 (t, J = 74.1 Hz, 1H), 5.73 (s, 2H), 3.09 (s, 3H), 2.23 (s, 3H); 13C RMN (101 MHz, CDCb) 5178.87, 163.90, 161.46, 155.70, 149.21, 143.36, 138.01, 130.04, 130.02 - 129.79 (m), 125.73, 122.70 (d, J = 2.9 Hz), 118.75, 116.17, 114.45 (dd, J = 38.7, 22.0 Hz), 113.60, 75.07, 25.87, 16.35. (nótese: debido a la presencia de átomos de flúor, se
notan los acoplamientos J2c-f - J4c-f que dan lugar a tripletes y dobletes mal resueltos); LC (260 nm) Rt (min) = 3.923, pureza LC = 96%; m/z: encontrado [M+H]+ = 364.2, esperado [M+H]+ = 364.3 (C17H14F3N3O).
Ejemplo 7
(FAH-17 HCI sal)

Se disolvió 2-amino-4-(4-(difluorometoxi)-3-metilfenil)-4-(3-fluorofenil)-1-metil-1H-imidazol-5(4H)-ona (0.50 g, 1.38 mmol) en DCM anhidro (66 ml) seguido de la adición de HCl (1M en éter dietílico, 2.2 ml). La mezcla se agitó a temperatura ambiente durante 5 minutos y el disolvente se evaporó al vacío para producir 2-amino-4-(4-(difluorometoxi)-3-metilfenil)-4-(3-fluorofenil)-1-metil-1H-imidazol-5(4H)-ona clorhidrato (0.50 g, 1.20 mmol, 87%) como un sólido blanco. 1H RMN (d6-DMSO): 11.78 (brs, 1H), 9.73 (brs, 1H), 7.53-7.06 (m, 8H), 3.19 (s, 3H), 2.23 (s, 3H); 13C RMN (d6-DMSO): 176.62, 176.99, 172.57, 163.63, 161.20, 157.95, 150.07, 150.04, 140.36, 140.30, 134.40, 129.86, 126.60, 123.70, 119.52, 119.00, 116.96, 116.47, 116.26, 114.64, 114.41,70.30, 27.43, 16.40 (nótese: debido a la presencia de átomos de flúor, se notan los acoplamientos J2c-f-J4c-f que dan lugar a tripletes y dobletes mal resueltos); LC (220 nm): Rt = 3.84 min, pureza 96.5%; MS: Para C18H16F3N3O2 se espera [M+H]+ = 364.3 obtenido 364.1
Ejemplo 8
Síntesis de FAH-22
Síntesis de 2-(3-fluoro-5-metilfenil)-1,3-ditiano
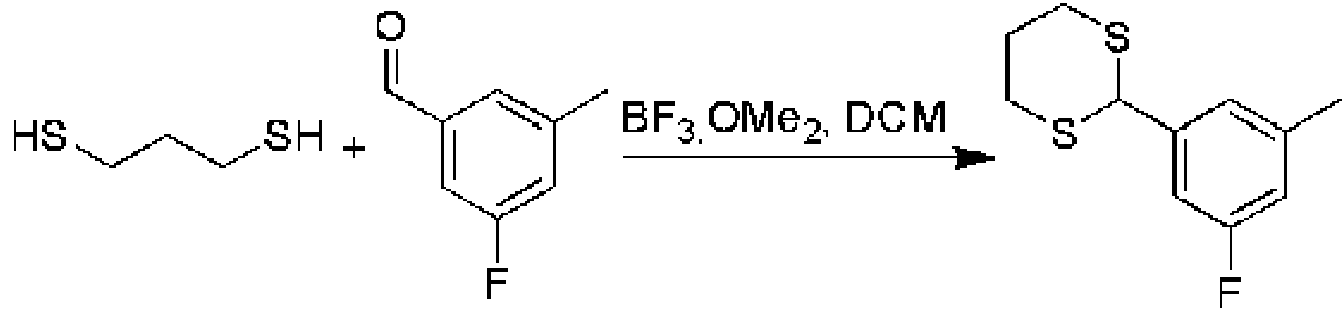
Se anadió BF3.OEt2 (2.61 ml, 21.16 mmol) gota a gota a una solución de 1,3-propanoditiol (2.48 ml, 24.47 mmol) y 3-fluoro-5-metilbenzaldehído (3.38 g, 24.47 mmol) en DCM (122 ml) a 0°C. La reacción se agitó a temperatura ambiente durante 1 hora cuando la TLC (5% EtOAc/hexano) indicó una reacción completa. La mezcla de reacción se diluyó luego con DCM (100 ml), se filtró a través de Celite (y la almohadilla de Celite se lavó con DCM adicional (3 x 100 ml)) y el filtrado se lavó con salmuera (100 ml), NaHCO3 saturado (3 x 100 ml), solución de KOH al 10% (100 ml), agua (100 ml) y salmuera (100 ml) y finalmente se secó sobre sulfato de sodio. El extracto orgánico se filtró y se evaporó para proporcionar 2-(3-fluoro-5-metilfenil)-1,3-ditiano (4.69 g, 77%) como un sólido rosa claro. El producto se usó en el siguiente paso sin purificación adicional. La RMN fue consistente con la estructura propuesta.
Síntesis de 4-(difluorometoxi)-3-metilbenzaldehído

Se añadió una solución de clorodifluoroacetato de sodio (2.60 g, 17.04 mmol) y 4-hidroxi-3-metilbenzaldehído (1.16 g, 8.52 mmol) en DMF (15 ml) durante 3 horas a una solución de DMF (15 ml) que contenía carbonato de potasio (1.77 g, 12.78 mmol) a 95°C. La reacción se dejó transcurrir durante 15 minutos adicionales y luego se enfrió. La mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 50 ml). El extracto orgánico se lavó con solución acuosa de LiCl al 10% (m/v) (3 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó para dar un residuo que se sometió a cromatografía instantánea (EtOAc al 15%/hexano) para dar 4-(difluorometoxi)-3metilbenzaldehído (021GLM-053_1(2), 1.00 g, 5.37 mmol, 63%) como un aceite amarillo. Este aceite se combinó con el del experimento anterior y se pasó a través de una columna de pipeta Pasteur eluyendo con EtOAC al 10%/hexano para dar un aceite que solidificó en reposo (1.315 g, 7.06 mmol, 67% en las dos reacciones). La RMN de protones fue consistente con la estructura propuesta. Se aislaron 1.4 g del producto deseado.
Síntesis de (4-(difluorometoxi)-3-metilfenil)(2-(3-fluoro-5-metilfenil)-1,3-ditian-2-il)metanol

Se preparó (4-(difluorometoxi)-3-metilfenil)(2-(3-fluoro-5-metilfenil)-1,3-ditian-2-il)metanol de acuerdo con el procedimiento representativo usando 2-(3-fluoro-5-metilfenil)-1,3-ditiano (0.932 g, 4.08 mmol) y 4-(difluorometoxi)-3-metilbenzaldehído (0.760 g, 4.08 mmol) lo que dio (4-(difluorometoxi)-3-metilfenilo)(2-(3-fluoro-5-metilfenil)-1,3-ditian-2-il) metanol (0.413 g, 24%) como un aceite amarillo. La RMN fue consistente con la estructura propuesta.
Síntesis de 2-(4-(difluorometoxi)-3-metilfenil)-1-(3-fluoro-5-metilfenil)-2-hidroxietanona

Se sintetizó 2-(4-(difluorometoxi)-3-metilfenil)-1-(3-fluoro-5-metilfenil)-2-hidroxietanona según el procedimiento representativo usando (4-(difluorometoxi)-3-metilfenilo)(2-(3-fluoro-5-metilfenil)-1,3-ditian-2-il)metanol (0.400 g, 0.965 mmol) y dio 2-(4-(difluorometoxi)-3-metilfenil)-1-(3-fluoro-5-metilfenil)-2-hidroxietanona (162 mg, 47%) como un sólido amarillo. La RMN fue consistente con la estructura propuesta. Nota: También se recuperó (4-(difluorometoxi)-3-metilfenil)(2-(3-fluoro-5-metilfenil)-1,3-ditian-2-il)metanona (104 mg, 22%).
Síntesis de 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-fluoro-5-metilfenil)etano-1,2-diona

Se sintetizó 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-fluoro-5-metilfenil)etano-1,2-diona de acuerdo con el procedimiento representativo usando 2-(4-(difluorometoxi)-3-metilfenil)-1-(3-fluoro-5-metilfenil)-2-hidroxietanona (0.150 g, 0.463 mmol) y dio 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-fluoro-5-metilfenil)etano-1,2-diona (0.1134 g, 74%) como un sólido amarillo. La RMN fue consistente con la estructura propuesta.
Síntesis de 2-amino-4-(4-(difluorometoxi)-3-metilfenil)-4-(3-fluoro-5-metilfenil)-1-metil-1H-imidazol-5(4H)-ona

Se añadió carbonato de potasio (0.149 g, 1.407 mmol) en agua (2.3 ml) a una mezcla de 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-fluorofenil)etano-1,2-diona, 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-fluoro-5-metilfenil)etano-1,2-diona (0.1134 g, 0.352 mmol), clorhidrato de 1-metilguanidina (0.154 g, 1.407 mmol), dioxano (5.46 ml) y alcohol etílico (7.10
ml). La mezcla de reacción se agitó a 85°C durante 1.5 h. Los volátiles se eliminaron al vacío, y el residuo se tomó en cloroformo (50 ml) y se lavó con agua (2x15 ml). Los extractos orgánicos se secaron sobre MgSO4. La evaporación y purificación cinco veces por cromatografía instantánea (metanol al 1% en acetato de etilo) dio 2-amino-4-(4-(difluorometoxi)-3-metilfenil)-4-(3-fluoro-5-metilfenil)-1-metil-1H-imidazol-5(4H)-ona (85 mg, 75%) como un sólido blanquecino. 1H RMN (400 MHz, CDCls) 57.30 (d, J = 2.0 Hz, 1H), 7.28 - 7.20 (m, 1H), 7.02 (s, 1H), 6.95 (m, 2H), 6.77 (d, J = 9.3 Hz, 1H), 6.45 (t, J = 74.1 Hz, 1H), 5.26 (s, 2H), 3.07 (s, 3H), 2.28 (s, 3H), 2.23 (s, 3H). 13C RMN (101 MHz, CDCla) 5 178.52, 163.88, 161.44, 155.75, 149.22 (t, J = 2.5 Hz), 141.88 (dd, J = 288.6, 7.8 Hz), 138.02, 130.05 (d, J = 9.4 Hz), 125.75, 123.30 (d, J = 2.5 Hz), 118.75 (d, J= 7.5 Hz), 116.21, 115.32 (d, J = 21.0 Hz), 113.63, 111.33 (d, J = 23.3 Hz), 74.72, 25.83, 21.46 (d, J= 1.8 Hz), 16.33. (nótese: debido a la presencia de átomos de flúor, se notan los acoplamientos J2c-f- J4c-f que dan lugar a tripletes y dobletes mal resueltos); LC (220 nm) Rt (min) = 4.007, pureza LC = 98%; m/z: encontrado [M+H]+ = 378.2, esperado [M+H]+ = 378.4 (C19H18F3N3O2).
Ejemplo 9
Síntesis de FAH-23: 2-amino-4-(4-(difluorometoxi)fenil)-4-(3-clorofenil)-1-metil-1H-imidazol-5(4H)-ona
Paso 1: Síntesis de 2-(3-clorofenil)-1,3-ditiano

Se añadió BF3.0Et2 (2.28 ml, 18.46 mmol) gota a gota a una solución de 1,3-propanoditiol (2.16 ml, 21.34 mmol) y 3-clorobenzaldehído (3.00 g, 21.34 mmol) en DCM (107 ml) a 0°C. La reacción se agitó a temperatura ambiente durante 1 hora cuando la TLC (5% EtOAc/hexano) indicó una reacción completa. La mezcla de reacción se diluyó luego con DCM (50 ml), se filtró a través de Celite (y la almohadilla de Celite se lavó con DCM adicional (3 x 10 ml)) y el filtrado se lavó con salmuera (100 ml), NaHC03 saturado (3 x 100 ml), solución de KOH al 10% (100 ml), agua (100 ml) y salmuera (100 ml) y finalmente se secó sobre sulfato de sodio. El extracto orgánico se filtró y se evaporó para proporcionar 2-(3-clorofenil)-1,3-ditiano (4.62 g, 92%) como un sólido incoloro. El producto se usó en el siguiente paso sin purificación adicional.
Paso 2: Síntesis de 4-(difluorometoxi)-3-metilbenzaldehído

Se añadió una solución de clorodifluoroacetato de sodio (2.60 g, 17.04 mmol) y 4-hidroxi-3-metilbenzaldehído (1.16 g, 8.52 mmol) en DMF (15 ml) durante 3 horas a una solución de DMF (15 ml) que contenía carbonato de potasio (1.77 g, 12.78 mmol) a 95°C. La reacción se dejó transcurrir durante 15 minutos adicionales y luego se enfrió. La mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 50 ml). El extracto orgánico se lavó con solución acuosa de LiCl al 10% (m/v) (3 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó para dar un residuo que se sometió a cromatografía instantánea (EtOAc al 15%/hexano) para dar 4-(difluorometoxi)-3-metilbenzaldehído (021GLM-053_1(2), 1.00 g, 5.37 mmol, 63%) como un aceite amarillo. Este aceite se combinó con el del experimento anterior y se pasó a través de una columna de pipeta Pasteur eluyendo con EtOAC al 10%/hexano para dar un aceite que solidificó en reposo (1.315 g, 7.06 mmol, 67% en las dos reacciones). La RMN de protones fue consistente con la estructura propuesta. Se aislaron 1.4 g del producto deseado.
Paso 3: Síntesis de (4-(difluorometoxi)-3-metilfenil)(2-(3-clorofenil)-1,3-ditian-2-il)metanol
Se disolvió 2-(3-dorofenil)-1,3-ditiano (0.92 g, 3.98 mmol) en THF seco (20 ml) y se enfrió a -29°C. Se añadió nBuLi (1.6 M, 2.99 ml, 4.78 mmol) gota a gota bajo nitrógeno y la mezcla se agitó durante 30 minutos a -29°C para proporcionar una solución roja oscura. Se añadió gota a gota una solución de 4-(difluorometoxi)-3-metilbenzaldehído (0.74 g, 3.98 mmol) en THF (19.8 ml) y la mezcla a -29°C y se agitó durante 15 minutos, luego se calentó a temperatura ambiente durante 1 h y se detuvo con solución saturada de cloruro de amonio (7.5 ml) seguida de dilución con EtOAc (50 ml). La fase orgánica se lavó con agua (2x20 ml), salmuera (1x20 ml) y se secó con sulfato de sodio. Después de la filtración y concentración, el producto crudo se purificó por cromatografía instantánea en columna (15% EtOAc/Hexano) para dar (2-(3-clorofenil)-1,3-ditian-2-il)(4-(difluorometoxi)-3-metilfenil)metanol (0.81 g, 1.94 mmol, 49%) como un aceite. La RMN fue consistente con la estructura propuesta.
Paso 4: Síntesis de 2-(4-(difluorometoxi)-3-metilfenil)-1-(3-clorofenil)-2-hidroxietanona

(4-(Difluorometoxi)-3-metilfenil)(2-(3-clorofenil)-1,3-ditian-2-il)metanol (3.07 g, 7.34 mmol) se disolvió en acetonitrilo (15 ml) y agua (2.5 ml). Se añadió lentamente bis(trifluoroacetoxi)yodobenceno (3.94 g, 9.17 mmol) en acetonitrilo (10 ml) a temperatura ambiente a la solución agitada vigorosamente. Después de 20 minutos, el análisis por TLC (20% EtOAc/hexano) indicó una reacción completa. Se añadió EtOAc (150 ml) y la mezcla se enjuagó con solución saturada de bicarbonato de sodio (50 ml) y salmuera (50 ml). Las fracciones orgánicas se secaron y el disolvente se eliminó a vacío. El producto crudo se purificó dos veces por cromatografía instantánea en columna (EtOAc al 10%/hexano) para dar 2-(4-(difluorometoxi)-3-metilfenil)-1-(3-clorofenil)-2-hidroxietanona (0.803 g, 2.4 mmol, 32%) como un aceite amarillo pálido. La RMN de protones fue consistente con la estructura propuesta.
Paso 5: Síntesis de 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-clorofenil)etano-1,2-diona

(2-(3-clorofenil)-1,3-ditian-2-il)(4-(difluorometoxi)-3-metilfenil)metanol (0.80 g, 1.92 mmol) se disolvió en diclorometano (24.29 ml) y tert-butanol (5.14 ml, 53.7 mmol) en atmósfera de nitrógeno. Se añadió peryodinano Dess-Martin (2.04 g, 4.80 mmol) y la reacción se agitó durante la noche a temperatura ambiente. Se añadió tiosulfato de sodio (5 ml, 1M) y las capas se separaron. La fase orgánica se lavó con hidrogenocarbonato de sodio y el disolvente se evaporó. La purificación en placa de preparación en hexano de acetato de etilo al 25% dio 1-(3-clorofenil)-2-(4- (difluorometoxi)-3-metilfenil)etano-1,2-diona (0.41 g, 1.27 mmol, 66%) como un sólido amarillo que se usó directamente en la siguiente etapa.
Paso 6: Síntesis de 2-amino-4-(4-(difluorometoxi)fenil)-4-(3-clorofenil)-1-metil-1H-imidazol-5(4H)-ona

Se añadió carbonato de potasio (0.522 g, 4.93 mmol) en agua (7.85 ml) a una mezcla de 1-(3-clorofenil)-2-(4-(difluorometoxi)-3-metilfenil)etano-1,2-diona (0.40 g, 1.23 mmol), clorhidrato de 1-metilguanidina (0.54 g, 4.93 mmol), dioxano (19.13 ml) y alcohol etílico (24.87 ml). La mezcla de reacción se agitó a 85°C durante 1.5 h. Los volátiles se eliminaron al vacío, y el residuo se tomó en cloroformo (50 ml) y se lavó con agua (2 x 15 ml). Los extractos orgánicos se secaron sobre MgSO4. Evaporación y purificación tres veces en PTLC (MeOH al 1% en EtOAc) y cromatografía en
columna (50-90% acetato de etilo: hexano) para dar 2-amino-4-(3-clorofenil)-4-(4-(difluorometoxi)-3-metilfenil)-1-metil-1H-imidazol-5(4H)-ona (0.20 g, 0.52 mmol, 43%) como un sólido blanquecino. 1H RMN (CDCb): 7.48 (s, 1H), 7.30 7.20 (m, 5H), 7.0 (s, 1H), 6.48 (t, 1H, J = 74.1 Hz), 4.50 (brs, 2H), 3.12 (s, 3H), 2.26 (s, 3H); 13C RMN (CDCla): 178.45, 155.74, 149.25, 143.22, 137.90, 134.32, 130.05, 127.12, 125.74, 125.36, 118.78, 116.17, 113.6074.73, 25.89, 16.35 (nótese: debido a la presencia de átomos de flúor, J2c-f-J4c-f acoplamientos que dan lugar a tripletes y dobletes mal resueltos); LC (220 nm): Rt = 3.95 min, pureza 96.6%; MS: Para C18H16CF2N3O2 se espera [M+H]+ = 380.8 obtenido 380.1
Ejemplo 10
Síntesis del Compuesto FAH-27: 2-amino-4-(4-(difluorometoxi)fenil)-4-(3-metilfenil)-1-metil-1H-imidazol-5(4H)-ona
Paso 1: Síntesis de 2-(3-metilfenil)-1,3-ditiano

Se añadió gota a gota BF3.OEt2 (2.67 ml, 21.60 mmol) a una solución de 1,3-propanoditiol (2.53 ml, 24.97 mmol) y 3-metilbenzaldehído (3.00 g, 24.97 mmol) en DCM (125 ml) a 0°C. La reacción se agitó a temperatura ambiente durante 1 hora cuando la TLC (5% EtOAc/hexano) indicó una reacción completa. La mezcla de reacción se diluyó luego con DCM (50 ml), se filtró a través de Celite (y la almohadilla de Celite se lavó con DCM adicional (3 x 10 ml)) y el filtrado se lavó con salmuera (100 ml), NaHCO3 saturado (3 x 100 ml), solución de KOH al 10% (100 ml), agua (100 ml) y salmuera (100 ml) y finalmente se secó sobre sulfato de sodio. El extracto orgánico se filtró y se evaporó para proporcionar 2-(m-tolil)-1,3-ditiano (4.66 g, 85%) como un sólido marrón claro. El producto se usó en el siguiente paso sin purificación adicional. La RMN de protones fue consistente con la estructura propuesta.
Paso 2: Síntesis de 4-(difluorometoxi)-3-metilbenzaldehído

Una solución de clorodifluoroacetato de sodio (2.60 g, 17.04 mmol) y 4-hidroxi-3-metilbenzaldehído (1.16 g, 8.52 mmol) en DMF (15 ml) se añadió durante 3 horas a una solución de DMF (15 ml) que contenía carbonato de potasio (1.77 g, 12.78 mmol) a 95°C. La reacción se dejó transcurrir durante 15 minutos adicionales y luego se enfrió. La mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 50 ml). El extracto orgánico se lavó con solución acuosa de LiCl al 10% (m/v) (3 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó para dar un residuo que se sometió a cromatografía instantánea (EtOAc al 15%/hexano) para dar 4-(difluorometoxi)-3-metilbenzaldehído (021GLM-053_1(2), 1.00 g, 5.37 mmol, 63%) como un aceite amarillo. Este aceite se combinó con el del experimento anterior y se pasó a través de una columna de pipeta Pasteur eluyendo con EtOAC al 10%/hexano para dar un aceite que solidificó en reposo (1.315 g, 7.06 mmol, 67% en las dos reacciones). La RMN de protones fue consistente con la estructura propuesta. Se aislaron 1.4 g del producto deseado.
Paso 3: Síntesis de (4-(difluorometoxi)-3-metilfenil)(2-(3-metilfenil)-1,3-ditian-2-il)metanol

Se disolvió 2-(m-tolil)-1,3-ditiano (2.00 g, 9.51 mmol) en THF seco (47.5 ml) y se enfrió a -29°C. Se añadió nBuLi (1.6M, 7.13 ml, 11.41 mmol) gota a gota bajo nitrógeno y la mezcla se agitó durante 30 minutos a -10°C para proporcionar una solución roja oscura. Se añadió gota a gota una solución de 4-(difluorometoxi)-3-metilbenzaldehído (1.77 g, 9.51 mmol) en THF (47.5 ml) y la mezcla a -29°C y se agitó durante 15 minutos, luego se calentó a temperatura
ambiente durante 1 h y se detuvo con solución saturada de cloruro de amonio (7.5 ml) seguido de dilución con EtOAc (50 ml). La fase orgánica se lavó con agua (2x20 ml), salmuera (1x20 ml) y se secó con sulfato de sodio. Después de la filtración y concentración, el producto crudo se purificó por cromatografía instantánea en columna (EtOAC/Hex al 15%) para dar (4-(difluorometoxi)-3-metilfenil)(2-(m-tolil)-1,3-ditian-2-il)metanol (2.73 g, 6.88 mmol, 72%) como un aceite. La RMN fue consistente con la estructura propuesta.
Paso 4: Síntesis de 2-(4-(difluorometoxi)-3-metilfenil)-1-(3-metilfenil)-2-hidroxietanona

Se disolvió (4-(Difluorometoxi)-3-metilfenil)(2-(m-tolil)-1,3-ditian-2-il)metanol (2.70 g, 6.81 mmol) en diclorometano (86 ml) y tert-butanol (18.28 ml, 191 mmol) en atmósfera de nitrógeno. Se añadió peryodinano Dess-Martin (7.22 g, 17.02 mmol) y la reacción se agitó durante la noche a temperatura ambiente. Se añadió tiosulfato de sodio (5 ml, 1M) y las capas se separaron. La fase orgánica se lavó con hidrogenocarbonato de sodio y el disolvente se evaporó. La purificación en cromatografía en columna en acetato de etilo/hexano al 5% dio 1-(4-(difluorometoxi)-3-metilfenil)-2-(mtolil)etano-1,2-diona (1.44 g, 4.75 mmol, 70%) como un sólido amarillo
Paso 5: Síntesis de 1-(4-(difluorometoxi)-3-metilfenil-2-(3-metilfenil)etano-1,2-diona

(4-(Difluorometoxi)-3-metilfenil)(2-(m-tolil)-1,3-ditian-2-il)metanol (2.70 g, 6.81 mmol) se disolvió en diclorometano (86 ml) y tert-butanol (18.28 ml, 191 mmol) en atmósfera de nitrógeno. Se añadió peryodinano Dess-Martin (7.22 g, 17.02 mmol) y la reacción se agitó durante la noche a temperatura ambiente. Se añadió tiosulfato de sodio (5 ml, 1M) y las capas se separaron. La fase orgánica se lavó con hidrogenocarbonato de sodio y el disolvente se evaporó. La purificación en cromatografía en columna en acetato de etilo/hexano al 5% dio 1-(4-(difluorometoxi)-3-metilfenil)-2-(mtolil)etano-1,2-diona (1.44 g, 4.75 mmol, 70%) como un sólido amarillo y se utilizó directamente en la siguiente etapa.
Paso 6: Síntesis de 2-amino-4-(4-(difluorometoxi)fenil)-4-(3-metilfenil)-1-metil-1H-imidazol-5(4H)-ona
clorhidrato de 1-metilguanidma
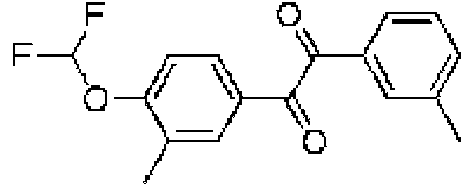

Se añadió carbonato de potasio (1.81 g, 17.09 mmol) en agua (27.23 ml) a una mezcla de 1-(4-(difluorometoxi)-3-metilfenil)-2-(m-tolil)etano-1,2-diona (1.30 g, 4.27 mmol), clorhidrato de 1-metilguanidina (1.87 g, 17.09 mmol), dioxano (66.3 ml) y alcohol etílico (86 ml). La mezcla de reacción se agitó a 85°C durante 1.5 h. Los volátiles se eliminaron al vacío, y el residuo se tomó en cloroformo (50 ml) y se lavó con agua (2 x 15 ml). Los extractos orgánicos se secaron sobre MgSO4. Evaporación y purificación tres veces por PTLC (MeOH al 1% en EtOAc) y una vez por cromatografía en columna (50-90% acetato de etilo: hexano) para dar 2-amino-4-(4-(difluorometoxi)-3-metilfenil)-1-metil-4-(m-tolil)-1H-imidazol-5(4H)-ona (0.39 g, 1.07 mmol, 25%) como un sólido blanquecino. 1H r Mn (CDCb): 7.34 (s, 1H), 7.24 7.10 (m, 5H), 6.95 (d, 1H, J = 8 Hz), 6.47 (t, 1H, J = 74.1 Hz), 6.05 (brs, 2H), 3.04 (s, 3H), 2.30 (s, 3H), 2.23 (s, 3H); 13C RMN (CDCla): 178.55, 155.97, 149.14, 149.12, 149.09, 141.16, 138.14, 130.20, 128.35, 127.56, 125.94, 124.16, 118.83, 118.61, 116.25, 113.67, 74.74, 25.70, 21.52, 16.32 (nótese: debido a la presencia de átomos de flúor, se observan acoplamientos J2c-f- J4c-f que dan lugar a tripletes y dobletes mal resueltos); LC (220 nm): Rt = 3.85 min, pureza 97.3%; MS: Para C19H19F2N3O2 se espera [M+H]+ = 360.4 obtenido 360.2
Ejemplo 11
Síntesis de FAH-28: 2-amino-4-(4-(difluorometoxi)fenil)-4-(3-(trifluorometil)fenil)-1-metil-1H-imidazol-5(4H)-ona
Paso 1: Síntesis de 2-(3-trifluorometil)fenil)-1,3-ditiano

Se añadió BF3.OEt2 (1.84 ml, 14.90 mmol) gota a gota a una solución de 1,3-propanoditiol (1.74 ml, 17.23 mmol) y 3-(trifluorometil)benzaldehído (3.00 g, 17.23 mmol) en DCM (86 ml) a 0°C. La reacción se agitó a temperatura ambiente durante 1 hora cuando la TLC (5% EtOAc/hexano) indicó una reacción completa. La mezcla de reacción se diluyó luego con DCM (50 ml), se filtró a través de Celite (y la almohadilla de Celite se lavó con DCM adicional (3 x 10 ml)) y el filtrado se lavó con salmuera (100 ml), NaHCO3 saturado (3 x 100 ml), solución de KOH al 10% (100 ml), agua (100 ml) y salmuera (100 ml) y finalmente se secó sobre sulfato de sodio. El extracto orgánico se filtró y se evaporó para proporcionar 2-(3-(trifluorometil)fenil)-1,3-ditiano (4.62 g, 17.30 mmol, 100%) como un sólido incoloro. El producto se usó en el siguiente paso sin purificación adicional.
Paso 2: Síntesis de 4-(difluorometoxi)-3-metilbenzaldehído

Se añadió una solución de clorodifluoroacetato de sodio (2.60 g, 17.04 mmol) y 4-hidroxi-3-metilbenzaldehído (1.16 g, 8.52 mmol) en DMF (15 ml) durante 3 horas a una solución de DMF (15 ml) que contenía carbonato de potasio (1.77 g, 12.78 mmol) a 95°C. La reacción se dejó transcurrir durante 15 minutos adicionales y luego se enfrió. La mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 50 ml). El extracto orgánico se lavó con solución acuosa de LiCl al 10% (m/v) (3 x 25 ml), se secó sobre sulfato de sodio, se filtró y se evaporó para dar un residuo que se sometió a cromatografía instantánea (EtOAc al 15%/hexano) para dar 4-(difluorometoxi)-3-metilbenzaldehído (021GLM-053_1(2), 1.00 g, 5.37 mmol, 63%) como un aceite amarillo. Este aceite se combinó con el del experimento anterior y se pasó a través de una columna de pipeta Pasteur eluyendo con EtOAC al 10%/hexano para dar un aceite que solidificó en reposo (1.315 g, 7.06 mmol, 67% en las dos reacciones). La RMN de protones fue consistente con la estructura propuesta. Se aislaron 1.4 g del producto deseado.
Paso 3: Síntesis de (4-(difluorometoxi)-3-metilfenil)(2-(3-(trifluorometil)fenil)-1,3-ditian-2-il)metanol

Se disolvió 2-(3-(trifluorometil)fenil)-1,3-ditiano (2.00 g, 7.57 mmol) (021STM-080) en THF seco (38 ml) y se enfrió a -29°C. Se añadió nBuLi (1.6 M, 5.67 ml, 9.08 mmol) gota a gota bajo nitrógeno y la mezcla se agitó durante 30 minutos a -29°C para proporcionar una solución roja oscura. Se añadió gota a gota una solución de 4-(difluorometoxi)-3-metilbenzaldehído (1.41 g, 7.57 mmol) en THF (38 ml) y la mezcla a -29°C y se agitó durante 15 minutos, luego se calentó a temperatura ambiente durante 1 h y se detuvo con solución saturada de cloruro de amonio (7.5 ml) seguido de dilución con EtOAc (50 ml). La fase orgánica se lavó con agua (2x20 ml), salmuera (1x20 ml) y se secó con sulfato de sodio. Después de la filtración y concentración, el producto crudo se purificó por cromatografía instantánea en columna (5-15% EtOAc/Hex) para dar (4-(difluorometoxi)-3-metilfenil)(2-(3-(trifluorometil)fenil)-1,3-ditian-2-il)metanol (2.29 g, 4.55 mmol, 60%) como un aceite dorado.
Paso 4: Síntesis de 2-(4-(difluorometoxi)-3-metilfenil)-1-(3-(trifluorometil)fenil)-2-hidroxietanona

Se disolvió (4-(difluorometoxi)-3-metilfenil)(2-(3-trifluorometilfenil)-1,3-ditian-2-il)metanol (3.07 g, 7.34 mmol) en acetonitrilo (15 ml) y agua (2.5 ml). Bis(trifiuoroacetoxi)yodobenceno (3.94 g, 9.17 mmol) en acetonitrilo (10 ml) se añadió lentamente a temperatura ambiente a la solución agitada vigorosamente. Después de 20 minutos, el análisis por TLC (20% EtOAc/hexano) indicó una reacción completa. Se añadió EtOAc (150 ml) y la mezcla se enjuagó con solución saturada de bicarbonato de sodio (50 ml) y salmuera (50 ml). Las fracciones orgánicas se secaron y el disolvente se eliminó a vacío. El producto crudo se purificó dos veces por cromatografía instantánea en columna (EtOAc al 10%/hexano) para dar 2-(4-(difluorometoxi)-3-trifluorometilfenil)-1-(3-metilfenil)-2-hidroxietanona (0.803 g, 2.4 mmol , 32%) como un aceite amarillo pálido. La RMN de protones fue consistente con la estructura propuesta.
Paso 5: Síntesis de 1-(4-(difluorometoxi)-3-(trifluorometil)fenil)-2-(3-(trifluorometil)fenil)etano-1,2-diona

Se disolvió (4-(difluorometoxi)-3-metilfenil)(2-(3-(trifluorometil)fenil)-1,3-ditian-2-il)metanol (2.20 g, 4.88 mmol) en diclorometano (61.8 ml) y tert-butanol (13.08 ml, 137 mmol) en atmósfera de nitrógeno. Se añadió peryodinano Dess-Martin (5.18 g, 12.21 mmol) y la reacción se agitó durante la noche a temperatura ambiente. Se añadió tiosulfato de sodio (5 ml, 1M) y las capas se separaron. La fase orgánica se lavó con hidrogenocarbonato de sodio y el disolvente se evaporó. La purificación en cromatografía en columna en acetato de etilo al 5%/hexano dio 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-(trifluorometil)fenil)etano-1,2-diona (1.20 g, 3.35 mmol, 69%) como un sólido amarillo se utilizó directamente en la siguiente etapa.
Paso 6: Síntesis de 2-amino-4-(4-(difluorometoxi)fenil)-4-(3-(trifluorometil)fenil)-1-metil-1H-imidazol-5(4H)-ona

Se añadió carbonato de potasio (1.36 g, 12.84 mmol) en agua (20.46 ml) a una mezcla de 1-(4-(difluorometoxi)-3-metilfenil)-2-(3-(trifluorometil)fenil)etano-1,2-diona (1.15 g, 3.21 mmol), clorhidrato de 1-metilguanidina (1.41 g, 12.84 mmol), dioxano (49.8 ml) y alcohol etílico (64.8 ml). La mezcla de reacción se agitó a 85°C durante 1.5 h. Los volátiles se eliminaron al vacío, y el residuo se tomó en cloroformo (50 ml) y se lavó con agua (2 x 15 ml). Los extractos orgánicos se secaron sobre MgSO4. Evaporación y purificación tres veces por PTLC (MeOH al 1% en EtOAc) y una vez por cromatografía en columna (50-90% acetato de etilo: hexano) para dar 2-amino-4-(3,5-difluorofenil)-4-(6-metoxipiridin-3-il)-1-metil-1H-imidazol-5(4H)-ona (0.18g, 0.44 mmol, 14%) como un sólido blanquecino. 1H r Mn (CDCla): 7.80 (s, 1H), 7.70 (d, 1H, J = 8 Hz), 7.55 (d, 1H, J = 8 Hz), 7.45-7.41 (m, 1H), 7.32 (s, 1H), 7.28-7.24 (m, 1H), 7.0 (d, 1H, J = 8 Hz), 6.48 (t, 1H, J = 74.1 Hz), 5.64 (brs, 2H), 3.10 (s, 3H), 2.26 (s, 3H); 13C RMN (CDCla): 178.78, 156.18, 156.09, 156.07, 149.23, 142.39, 138.11, 130.71, 130.13, 128.99, 125.72, 122.72, 118.73, 116.15, 113.57, 75.05, 25.84, 16.33 (nótese: debido a la presencia de átomos de flúor, se observan acoplamientos J2c-f- J4c-f que dan lugar a tripletes y dobletes mal resueltos); LC (220 nm): Rt = 4.04 min, pureza 96.6%; MS: Para C19H16F5N3O2 se espera [M+H]+ = 414.3 obtenido 414.1
Ejemplo 12
Análisis SPR
Las superficies de las dos celdas de flujo FC1 y FC2 de un chip de dextrano carboximetilado (CM-5) se lavaron secuencialmente con NaOH 50 mM, HCl 1 mM, H3PO4 al 0.05% y fosfato de sodio 20 mM pH 7.4, cloruro de sodio 125 mM en paralelo utilizando un caudal de 30 |jl/min durante 1 minuto utilizando un Biacore T-100 (GE Healthcare). La proteína de fusión se inmovilizó mediante acoplamiento de amina usando fosfato 20 mM, cloruro de sodio 125 mM,
pH 7.4 en FC2. Esta proteína de fusión TRX-eAPP575-624 contiene una fusión de tiorredoxina (TRX) y los residuos 575 624 del ectodominio APP (20-kDa). La proteína de fusión es producida como se describe en Libeu et al (JAD 2011). La proteína se concentró a 2 mg/ml en fosfato 20 mM, pH 6.5, cloruro de sodio 125 mM y luego se disolvió a una concentración de 50 |jg por ml en acetato de sodio 20 mM, pH 5.0. FC1 sirvió como celda de referencia después de una inmovilización simulada con regulador solo. Para todas las células, la velocidad de flujo fue de 10 j l por minuto. El chip se bloqueó con etanolamina 1M (pH 8.5). Los valores finales de RU se determinaron para los inhibidores de BACE que se unían a TRX-eAPP575-624 haciendo fluir concentraciones variables del inhibidor en DMSO a 50 jM . Los compuestos se diluyeron de soluciones 10 mM en DMSO a 50 jM en DMSO al 1%, fosfato de sodio 20 mM, pH 7.4, cloruro de sodio 125 mM, Tween al 0.05% y luego se diluyeron en serie en 1.5 durante 10 pasos. Se registraron trazas de unión para cada dilución con una fase de unión de 60 segundos y una fase de disociación de 240 segundos. Cada ciclo se realizó a 20°C con un caudal constante de 20 jl/min. Se aplicaron 240 segundos adicionales de flujo de regulador a 60 j l por minuto a través de las células como fase de regeneración para facilitar la disociación completa del compuesto de la proteína. Los sensogramas se obtuvieron restando las señales de referencia y de regulador utilizando el software de evaluación Biacore T100. Las curvas de unión se modelaron con el PRISM (Graphpad Inc).
Ejemplo 13
Métodos experimentales para medir Ap42 en células SH-SY5Y
Ensayo de prueba in vitro de Abeta:
Se sembraron células de neuroblastoma SH-SY5Y a 50.000 células/pozo en una placa de 96 pozos durante 24 h. Luego se cambió su medio por medio nuevo suplementado con la concentración deseada del compuesto de hidantoína (por ejemplo, el compuesto 3). Después de 24 h, se añadieron 20 j l del medio a 2 j l del inhibidor de proteasa completo con EDTA 1 jM y se mantuvo a 4°C hasta el análisis usando el ensayo ELISA a continuación.
Ensayos de ELISA:
También se usaron kits ELISA de Invitrogen para cuantificar Ap1-42 (KHB3544) por duplicado. Para el ELISA ultrasensible Ap 1-42, las muestras se diluyeron 1:2 (50 j l CSF más 50 j l de regulador diluyente estándar proporcionado por el kit). Los ensayos se realizaron de acuerdo con las instrucciones del fabricante. En resumen, los estándares y las muestras se agregaron a una placa recubierta previamente con un anticuerpo de captura monoclonal específico para el extremo amino terminal de Hu Ap. Las muestras se coincubaron con un anticuerpo específico de detección de conejo (Ab) para el extremo carboxilo terminal de la especie AI3 se ensayó durante 3 horas a temperatura ambiente durante la noche a 4° (Ap 1-42) con balanceo suave. Después del lavado, de conejo unidos Ab se detectó mediante un marcado con peroxidasa de rábano picante anticonejo Ab secundario. Después de lavar nuevamente, se detectó colorimétricamente el Ab de HRP-anti conejo unido (Spectramax 190, Molecular Devices) mediante la adición de una solución de sustrato. 1 mM de 4-(2-aminoetil) bencenosulfonilo fluoruro hidrocloruro de inhibidor (AEBSF) proteasa (101500, Calbiochem) se añadió a los estándares y muestras.
Ejemplo 14
Prueba de captación cerebral (PK)
En general, la exposición CNS de las hidantoínas se realizaron como sigue: Los estudios consistió en colección de plasma heparinizado y el cerebro después del tratamiento con las hidantoínas, siguiente (sc) la administración subcutánea de las moléculas en 10 mg/kg. Los niveles plasmáticos y cerebrales de los compuestos se determinaron mediante la metodología cuantitativa LC/MS/MS, realizada en Integrated Analytical Solutions (en Internet en ianalytical.net). Las muestras de plasma se precipitaron con acetonitrilo: metanol (1:1) cóctel que contiene un patrón interno. Las muestras de cerebro se homogeneizaron directamente en acetato de etilo o extraen de 5M homogeneizados de guanidina utilizando el método de líquido-líquido. El sobrenadante resultante se evaporaron a sequedad y se sometió a la LC/MS/MS análisis. Para cada compuesto se utilizaron 3 ratones para el análisis. Luego se calcularon las relaciones cerebro-plasma y los niveles cerebrales.
Ejemplo 15
Selectividad de ABBI: inhibición de APP-BACE versus escisión de PSLG1-BACE o NRG1-BACE
Ensayo P5-P5':
Para determinar el sustrato APP-BACE1 IC50 Sigma BACE1 (7-metoxicumarina-4-acetil-[Asn670, Lue671]-amiloide b/A4 Precursor Protein 770 Fragment 667-676-(2,4 dinitrofenil) Lys-Arg-Arg se usó sal de trifluoroacetato de amida), se siguió el protocolo del fabricante. Brevemente, se incubaron 0.01 unidades de BACE1 durante 1 hora a temperatura ambiente con un inhibidor de BACE, luego se añadió el sustrato a cada pozo y se leyó la fluorescencia inmediatamente y cada 30 minutos durante 2 horas. La actividad se determinó dividiendo la fluorescencia en un [inhibidor de BACE] específico por la fluorescencia en [inhibidor de BACE]=0 jM , el % de actividad se representó frente al log [inhibidor de BACE] para determinar la APP-bAc E1 usando GraphPad Prism 5 (Figura 6A)
Ensayos PSGL1 y NRG1:
En resumen, para determinar las células PSGL-1-BACE1 IC50 IC50 HEK 293 se sembraron en placas de 24 pozos y se cotransfectaron transitoriamente con constructos PSGL1/lacZ o NRG1/1acZ usando Lipofectamine 2000; se siguió el protocolo del fabricante. Dos horas después de agregar el complejo ADN-lípido a las células, se agregó un inhibidor de BACE a cada pozo, luego las células se incubaron durante la noche a 37°C y 5% de CO2. El medio cultivado se recogió para determinar NRG1 o PSGL1, y las células se lisaron para medir los niveles de lacZ. El protocolo estándar del kit Sigma SEAP se realizó en el medio cultivado para detectar niveles de PSGL1 o NRG1. Se siguieron las instrucciones del kit Promega para determinar la concentración de lacZ. La relación de PSGL1/lacz vs [inhibidor de BACE] o se trazaron para determinar el BACE1-IC50 en cada uno de los sustratos usando GraphPad Prism 5 (Figura 6B). El ABBI FAH17 muestra una selectividad >200 veces para la aplicación sobre PSGL1. Pruebas similares muestran que FAH17 es >10 veces selectivo para APP sobre NRG1.
Se entiende que los ejemplos y realizaciones descritos en el presente documento son solo para fines ilustrativos.
Claims (14)
1. Un compuesto seleccionado del grupo que consiste en

o una sal farmacéuticamente aceptable del mismo, un tautómero del mismo, una sal farmacéuticamente aceptable de un tautómero del mismo, un enantiómero del mismo, o una sal farmacéuticamente aceptable de un enantiómero del mismo.
2. El compuesto de la reivindicación 1, en donde dicho compuesto es un compuesto de acuerdo con la fórmula:

o una sal farmacéuticamente aceptable del mismo, un tautómero del mismo, una sal farmacéuticamente aceptable de un tautómero del mismo, un enantiómero del mismo, o una sal farmacéuticamente aceptable de un enantiómero del mismo.
3. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1-2, en donde dicho compuesto es un enantiómero R sustancialmente puro o un enantiómero S sustancialmente puro.
4. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1-3, en donde
dicho compuesto se une a APP y/o a la enzima BACE y/o a un complejo APP/BACE; o
dicho compuesto se une a la APP e inhibe la enzima BACE.
5. Una formulación farmacéutica que comprende un vehículo farmacéuticamente aceptable y un compuesto de acuerdo con una cualquiera de las reivindicaciones 1-4.
6. La formulación de la reivindicación 5, en donde dicha formulación se combina para la administración a través de una ruta seleccionada del grupo que consiste en administración oral, administración isoforética, administración transdérmica, administración parenteral, administración en aerosol, administración por inhalación, administración intravenosa y administración rectal.
7. La formulación de la reivindicación 5, en donde dicha formulación es compuesta para administración oral.
8. La formulación de la reivindicación 5, en donde dicha formulación es estéril.
9. La formulación de acuerdo con una cualquiera de las reivindicaciones 5-8, en donde dicha formulación es una formulación de dosificación unitaria.
10. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1-4, o la formulación de acuerdo con una cualquiera de las reivindicaciones 5-9 para el uso en terapia.
11. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1-4, o la formulación de acuerdo con una cualquiera de las reivindicaciones 5-10 para el uso en la prevención o retardo de la aparición de una afección pre-Alzheimer y/o disfunción cognitiva, y/o mejorar uno o más síntomas de una afección pre-Alzheimer y/o disfunción cognitiva, o prevenir o retrasar la progresión de una afección pre-Alzheimer o disfunción cognitiva a la enfermedad de Alzheimer, o para el uso en un método de tratamiento de la enfermedad de Alzheimer, o para el uso en un método de tratamiento de una enfermedad o trastorno asociado con la actividad de BACE, o para el uso en desacelerar la progresión, detener o revertir la degeneración macular relacionada con la edad (DMAE) en un mamífero; en donde dicho uso comprende administrar a un sujeto que lo necesita una cantidad efectiva de dicho compuesto o formulación.
12. El compuesto o formulación para su uso de la reivindicación 11, en donde dicho compuesto o formulación es para uso terapéutico para prevenir o retrasar la aparición de una afección pre-Alzheimer y/o disfunción cognitiva, y/o mejorar uno o más síntomas de una afección pre-Alzheimer y/o disfunción cognitiva, o prevenir o retrasar la progresión de una afección pre-Alzheimer o disfunción cognitiva a la enfermedad de Alzheimer en donde:
dicho sujeto exhibe positividad de biomarcador de Ap en un sujeto humano clínicamente normal de 50 años o más; y/o
dicho sujeto presenta amiloidosis cerebral asintomática, opcionalmente en combinación con neurodegeneración descendente y/o deterioro cognitivo/conductual sutil; y/o
dicho sujeto es un sujeto diagnosticado con deterioro cognitivo leve, opcionalmente con una calificación de demencia clínica superior a cero e inferior a aproximadamente 1.5; o
el sujeto está en riesgo de desarrollar la enfermedad de Alzheimer, preferiblemente el sujeto tiene un riesgo familiar de tener la enfermedad de Alzheimer, más preferiblemente el sujeto tiene una mutación de la enfermedad de Alzheimer familiar (EAF), aún más preferiblemente el sujeto tiene el alelo APOE £4; y/o
dicha administración produce una reducción en los niveles de CSF de uno o más componentes seleccionados del grupo que consiste en Ap42, sAPPp, Tau total (tTau), fosfo-Tau (pTau), APPneo, Ap40 soluble, relación pTau/Ap42 y relación tT Au/Ap42, y/o un aumento en el CSF de los niveles de uno o más componentes seleccionados del grupo que consiste en relación Ap42/Ap40, relación Ap42/Ap38, sAPPa, relación sAPPa/sAPPp, relación sAPPa/Ap40 y relación sAPPa/Ap42; y/o
dicha administración produce una reducción de la carga de placa en el cerebro del sujeto; y/o
dicha administración produce una reducción en la tasa de formación de placa en el cerebro del sujeto; y/o dicha administración produce una mejora en las habilidades cognitivas del sujeto; y/o
dicha administración produce una mejora, una estabilización o una reducción en la rata de declinación de la calificación de demencia clínica (CDR) del sujeto; y/o
dicha administración produce una mejora percibida en la calidad de vida del ser humano.
13. El compuesto o formulación para el uso de la reivindicación 11, en donde dicho compuesto o formulación es para mejorar uno o más síntomas de la enfermedad de Alzheimer, y/o revertir la enfermedad de Alzheimer, y/o reducir la tasa de progresión de la enfermedad de Alzheimer, en donde:
dicho sujeto es un humano de 50 años o más; y/o
dicho sujeto es diagnosticado con enfermedad de Alzheimer en etapa temprana; o
dicho sujeto es diagnosticado con la enfermedad de Alzheimer en etapa intermedia; o
dicho sujeto es diagnosticado con enfermedad de Alzheimer en etapa tardía; y/o
dicha administración reduce la gravedad de la enfermedad de Alzheimer; o
dicha administración mejora uno o más síntomas de la enfermedad de Alzheimer; o
dicha administración reduce la tasa de progresión de la enfermedad de Alzheimer; y/o
dicha administración produce una reducción en los niveles de CSF de uno o más componentes seleccionados del grupo que consiste en Ap42, sAPPp, Tau total (tTau), fosfo-Tau (pTau), APPneo, Ap40 soluble, relación pTau/Ap42 y
relación tTau/Ap42, y/o un aumento en el CSF de los niveles de uno o más componentes seleccionados del grupo que consiste en relación Ap42/Ap40, relación Ap42/Ap38, sAPPa, relación sAPPa/sAPPp, relación sAPPa/Ap40 y relación sAPPa/Ap42; y/o
dicha administración produce una reducción de la carga de placa en el cerebro del sujeto; y/o
dicha administración produce una reducción en la tasa de formación de placa en el cerebro del sujeto; y/o
dicha administración produce una mejora en las habilidades cognitivas del sujeto; y/o
dicha administración produce una mejora, una estabilización de, o una reducción en la rata de declinación de la calificación de demencia clínica (CDR) del sujeto; y/o
dicha administración produce una mejora percibida en la calidad de vida por parte del sujeto; y/o
dichos resultados de administración reducen la amiloidosis cerebral y/o la neurodegeneración descendente; y/o dicho sujeto muestra una calificación de demencia clínica indicativa de la enfermedad de Alzheimer; y/o
dicho sujeto tiene un riesgo familiar de tener la enfermedad de Alzheimer, preferiblemente el sujeto tiene una mutación de la enfermedad de Alzheimer familiar (EAF), más preferiblemente el sujeto tiene el alelo APOE £4.
14. El compuesto o formulación para el uso de la reivindicación 11, en donde dicho compuesto o formulación es para el tratamiento de una enfermedad o trastorno asociado con la actividad de BACE en un sujeto que lo necesita, en donde dicha enfermedad o trastorno se selecciona del grupo que consiste en Enfermedad de Alzheimer, deterioro cognitivo, síndrome de Down, HCHWA-D, declinación cognitiva, demencia senil, angiopatía amiloide cerebral y un trastorno neurodegenerativo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361763830P | 2013-02-12 | 2013-02-12 | |
| PCT/US2014/016100 WO2014127042A1 (en) | 2013-02-12 | 2014-02-12 | Hydantoins that modulate bace-mediated app processing |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2769578T3 true ES2769578T3 (es) | 2020-06-26 |
Family
ID=51354522
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14751458T Active ES2769578T3 (es) | 2013-02-12 | 2014-02-12 | Hidantoínas que modulan el procesamiento de APP mediado por BACE |
| ES19199279T Active ES2975235T3 (es) | 2013-02-12 | 2014-02-12 | Hidantoínas que modulan el procesamiento de APP mediado por BACE |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19199279T Active ES2975235T3 (es) | 2013-02-12 | 2014-02-12 | Hidantoínas que modulan el procesamiento de APP mediado por BACE |
Country Status (11)
| Country | Link |
|---|---|
| US (6) | US20140371283A1 (es) |
| EP (2) | EP3653609B1 (es) |
| JP (2) | JP6471100B2 (es) |
| KR (2) | KR102220259B1 (es) |
| CN (1) | CN104995176B (es) |
| AU (1) | AU2014216390B2 (es) |
| CA (1) | CA2899938C (es) |
| DK (1) | DK2956443T3 (es) |
| ES (2) | ES2769578T3 (es) |
| IL (1) | IL240555B (es) |
| WO (1) | WO2014127042A1 (es) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2899938C (en) | 2013-02-12 | 2021-10-19 | Buck Institute For Research On Aging | Hydantoins that modulate bace-mediated app processing |
| CA2976258C (en) | 2015-02-18 | 2024-02-20 | Buck Institute For Research On Aging | Triazolopyridines and triazolopyrimidines that lower stress-induced p-tau |
| EP3341362B1 (en) | 2015-08-27 | 2023-07-05 | Nantneuro, LLC | Compositions for app-selective bace inhibition and uses therfor |
| WO2017096049A1 (en) | 2015-12-03 | 2017-06-08 | The University Of North Carolina At Pembroke | Materials for cathepsin b enhancement and methods of use |
| DK3503885T3 (da) * | 2016-08-19 | 2022-06-20 | Foresee Pharmaceuticals Co Ltd | Farmaceutisk præparat og fremgangsmåder til anvendelse heraf |
| US10532102B2 (en) | 2016-08-19 | 2020-01-14 | Foresee Pharmaceuticals Co., Ltd. | Pharmaceutical composition and methods of uses |
| CN107468690B (zh) | 2017-08-11 | 2020-01-31 | 北京卓凯生物技术有限公司 | 4-氧-烷基化特特拉姆酸类化合物及其制备方法与应用 |
| CN107353239B (zh) | 2017-08-11 | 2019-06-18 | 北京卓凯生物技术有限公司 | 4-氧-烷基化特特拉姆酸类化合物及其制备方法 |
| US20210101879A1 (en) * | 2018-03-01 | 2021-04-08 | The Regents Of The University Of California | AMYLOID PROTEIN-SELECTIVE BACE INHIBITORS (ASBIs) FOR ALZHEIMER?S DISEASE |
| CN110652516A (zh) * | 2018-06-29 | 2020-01-07 | 上海绿谷制药有限公司 | 甘露糖醛二酸的组合物在治疗帕金森氏症中的应用 |
| CN121714327A (zh) * | 2018-12-04 | 2026-03-24 | 脑部保护私人有限公司 | 包括用于治疗病症的可植入阻尼装置和治疗剂的组合疗法及相关系统和使用方法 |
| CN113727658A (zh) | 2018-12-04 | 2021-11-30 | 脑部保护私人有限公司 | 包括用于治疗病症的可植入阻尼装置和治疗剂的组合疗法及相关系统和使用方法 |
| US11926599B1 (en) | 2023-10-23 | 2024-03-12 | King Faisal University | 1-(2-chlorobenzylideneamino)-5,5-diphenylimidazolidine-2,4-dione as an antimicrobial compound |
Family Cites Families (103)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4995971A (es) * | 1973-01-27 | 1974-09-11 | ||
| DE2448869A1 (de) | 1973-10-19 | 1975-09-04 | Mcneilab Inc | 4-oxo-2-imidazolidinylidenharnstoffe, verfahren zu ihrer herstellung und arzneimittel |
| US4025517A (en) * | 1975-06-23 | 1977-05-24 | Mcneil Laboratories, Incorporated | 4-Oxo-2-hexahydropyrimidinylidene ureas |
| JPS554305A (en) * | 1978-06-13 | 1980-01-12 | Nippon Zoki Pharmaceut Co Ltd | Remedy for disease caused by stress and its preparation |
| US4864028A (en) | 1983-09-14 | 1989-09-05 | Alcon Laboratories, Inc. | Spiro-tricyclicaromatic succinimide derivatives |
| US5070100A (en) | 1983-09-14 | 1991-12-03 | Alcon Laboratories, Inc. | Spiro-tricyclicaromatic succinimide derivatives |
| GB2189698A (en) | 1986-04-30 | 1987-11-04 | Haessle Ab | Coated omeprazole tablets |
| GB2189699A (en) | 1986-04-30 | 1987-11-04 | Haessle Ab | Coated acid-labile medicaments |
| CH677886A5 (es) | 1989-06-26 | 1991-07-15 | Hans Georg Prof Dr Weder | |
| ES2217250T3 (es) | 1990-06-15 | 2004-11-01 | Scios Inc. | Mamifero transgenico, no humano que muestra la patologia de formacion amiloides de la enfermedad de alzheimer. |
| ES2236682T5 (es) | 1991-01-21 | 2011-03-31 | Elan Pharmaceuticals, Inc. | Ensayo y modelo para la enfermedad de alzheimer. |
| AU671093B2 (en) | 1992-01-07 | 1996-08-15 | Elan Pharmaceuticals, Inc. | Transgenic animal models for alzheimer's disease |
| US5604102A (en) | 1992-04-15 | 1997-02-18 | Athena Neurosciences, Inc. | Methods of screening for β-amyloid peptide production inhibitors |
| AU702293B2 (en) | 1993-10-27 | 1999-02-18 | Athena Neurosciences, Inc. | Transgenic animals harboring APP allele having Swedish mutation |
| US5877399A (en) | 1994-01-27 | 1999-03-02 | Johns Hopkins University | Transgenic mice expressing APP-Swedish mutation develop progressive neurologic disease |
| ES2094688B1 (es) | 1994-08-08 | 1997-08-01 | Cusi Lab | Manoemulsion del tipo de aceite en agua, util como vehiculo oftalmico y procedimiento para su preparacion. |
| FR2730932B1 (fr) | 1995-02-27 | 1997-04-04 | Oreal | Nanoemulsion transparente a base de lipides amphiphiles non-ioniques fluides et utilisation en cosmetique ou en dermopharmacie |
| JPH11507538A (ja) | 1995-06-07 | 1999-07-06 | アテナ ニューロサイエンシズ インコーポレイティド | β−セクレターゼ、β−セクレターゼに対する抗体、及びβ−セクレターゼ阻害を検出するためのアッセイ |
| US5744346A (en) | 1995-06-07 | 1998-04-28 | Athena Neurosciences, Inc. | β-secretase |
| FR2742676B1 (fr) | 1995-12-21 | 1998-02-06 | Oreal | Nanoemulsion transparente a base de tensioactifs silicones et utilisation en cosmetique ou en dermopharmacie |
| FR2755854B1 (fr) | 1996-11-15 | 1998-12-24 | Oreal | Nanoemulsion a base de lipides amphiphiles non-ioniques et cationiques et utilisations |
| FR2760970B1 (fr) | 1997-03-18 | 2000-03-10 | Oreal | Nanoemulsions a base de lipides amphiphiles non-ioniques et de silicones aminees et utilisations |
| WO2000003819A1 (en) | 1998-07-17 | 2000-01-27 | The Penn State Research Foundation | Full form roll finishing technique |
| PL205294B1 (pl) | 1998-09-24 | 2010-03-31 | Upjohn Co | Izolowany lub oczyszczony polipeptyd, oczyszczony lub izolowany polinukleotyd, wektor, wektor ekspresyjny, komórka gospodarza, sposób wytwarzania polipeptydu, sposoby identyfikowania czynnika obniżającego, modulującego lub hamującego aktywność, zastosowanie takiego czynnika, izolowany kwas nukleinowy, obejmujący go wektor i ich zastosowanie, sposoby zmniejszania komórkowego wytwarzania beta amyloidu i zastosowanie antysensownego reagenta |
| FR2787027B1 (fr) | 1998-12-14 | 2001-01-12 | Oreal | Nanoemulsion a base d'esters gras de sucre ou d'ethers gras de sucre et ses utilisations dans les domaines cosmetique, dermatologique et/ou ophtalmologique |
| FR2787026B1 (fr) | 1998-12-14 | 2001-01-12 | Oreal | Nanoemulsion a base d'esters mixtes d'acide gras ou d'alcool gras, d'acide carboxylique et de glyceryle, et ses utilisations dans les domaines cosmetique, dermatologique et/ou ophtalmologique |
| FR2787325B1 (fr) | 1998-12-17 | 2001-01-26 | Oreal | Nanoemulsion a base d'esters gras de sorbitan oxyethylenes ou non oxyethylenes, et ses utilisations dans les domaines cosmetique, dermatologique et/ou ophtalmologique |
| FR2787326B1 (fr) | 1998-12-17 | 2001-01-26 | Oreal | Nanoemulsion a base d'esters gras de glycerol, et ses utilisations dans les domaines cosmetique, dermatologique et/ou ophtalmologique |
| FR2787728B1 (fr) | 1998-12-23 | 2001-01-26 | Oreal | Nanoemulsion a base d'esters gras d'acide phosphorique, et ses utilisations dans les domaines cosmetique, dermatologique, pharmaceutique et/ou ophtalmologique |
| FR2787703B1 (fr) | 1998-12-29 | 2001-01-26 | Oreal | Nanoemulsion a base d'ethers gras ethoxyles ou d'esters gras ethoxyles, et ses utilisations dans les domaines cosmetique, dermatologique et/ou ophtalmologique |
| FR2788007B1 (fr) | 1999-01-05 | 2001-02-09 | Oreal | Nanoemulsion a base de copolymeres blocs d'oxyde d'ethylene et d'oxyde de propylene, et ses utilisations dans les domaines cosmetique, dermatologique et/ou ophtalmologique |
| FR2788449B1 (fr) | 1999-01-14 | 2001-02-16 | Oreal | Nanoemulsion a base de citrates d'alkylether, et ses utilisations dans les domaines cosmetique, dermatologique, pharmaceutique et/ou ophtalmologique |
| FR2789076B1 (fr) | 1999-02-02 | 2001-03-02 | Synthelabo | Derives de alpha-azacyclomethyl quinoleine, leur preparation et leur application en therapeutique |
| US7161962B1 (en) | 1999-05-27 | 2007-01-09 | Nuera Communications, Inc. | Method and apparatus for coding modem signals for transmission over voice networks |
| FR2811564B1 (fr) | 2000-07-13 | 2002-12-27 | Oreal | Nanoemulsion contenant des polymeres non ioniques, et ses utilisations notamment dans les domaines cosmetique, dermatologique, pharmaceutique et/ou ophtalmologique |
| AU2002241823A1 (en) | 2001-01-11 | 2002-07-24 | Eastman Chemical Company | Cyclodextrin sulfonates, guest inclusion complexes, methods of making the same and related materials |
| FR2819427B1 (fr) | 2001-01-18 | 2003-04-11 | Oreal | Nanoemulsion translucide, son procede de fabrication et ses utilisations dans les domaines cosmetique, dermatologique et/ou ophtalmologique |
| JP2006512401A (ja) | 2001-06-05 | 2006-04-13 | ザ リージェンツ オブ ザ ユニバーシティ オブ ミシガン | ナノエマルジョンワクチン |
| US7476393B2 (en) | 2002-11-29 | 2009-01-13 | L'oreal | Process for the preparation of a cationic nanoemulsion, and cosmetic composition |
| AR045219A1 (es) | 2003-08-08 | 2005-10-19 | Pharmacopeia Inc | Aminas ciclicas inhibidoras de base - 1 que poseen un sustiuyente heterociclico |
| WO2005016876A2 (en) | 2003-08-08 | 2005-02-24 | Schering Corporation | Cyclic amine bace-1 inhibitors having a benzamide substituent |
| US7592348B2 (en) | 2003-12-15 | 2009-09-22 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| EP2335701B1 (en) | 2003-12-15 | 2012-07-11 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7763609B2 (en) | 2003-12-15 | 2010-07-27 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| US7468402B2 (en) | 2004-03-17 | 2008-12-23 | Baker Hughes Incorporated | Polymeric nanoemulsion as drag reducer for multiphase flow |
| JP2008502880A (ja) | 2004-04-02 | 2008-01-31 | メルク エンド カムパニー インコーポレーテッド | アミロイド前駆体タンパク質又はβアミロイド断片に結合する物質を検出する方法及び結合化合物 |
| US7618978B2 (en) | 2004-04-22 | 2009-11-17 | Eli Lilly And Company | Amides as BACE inhibitors |
| IL162288A0 (en) | 2004-06-01 | 2005-11-20 | Future Products Man S A | Compositions and methods for treating neurodegenerative disorders |
| RU2006144075A (ru) | 2004-06-16 | 2008-07-27 | Вайет (Us) | ДИФЕНИЛИМИДАЗОПИРИМИДИН И -ИМИДАЗОЛАМИНЫ КАК ИНГИБИТОРЫ β-СЕКРЕТАЗЫ |
| BRPI0512213A (pt) * | 2004-06-16 | 2008-02-19 | Wyeth Corp | método para tratamento, prevenção ou melhora de uma doença ou distúrbio; composição farmacêutica; processo para a preparação de um composto; e uso de um composto |
| CA2574218A1 (en) | 2004-07-22 | 2006-02-09 | Schering Corporation | Substituted amide beta secretase inhibitors |
| CA2575340A1 (en) | 2004-07-28 | 2006-02-09 | Schering Corporation | Macrocyclic beta-secretase inhibitors |
| SV2006002232A (es) | 2004-09-21 | 2006-05-25 | Lilly Co Eli | Inhibidores bace ref. x-16940 |
| GB0500683D0 (en) | 2005-01-13 | 2005-02-23 | Novartis Ag | Organic compounds |
| KR20070102514A (ko) | 2005-01-13 | 2007-10-18 | 노파르티스 아게 | Bace 억제제로서 유용한 마크로시클릭 화합물 |
| EP1896406A2 (en) | 2005-06-14 | 2008-03-12 | Shering Corporation | The preparation and use of compounds as protease inhibitors |
| WO2006138265A2 (en) | 2005-06-14 | 2006-12-28 | Schering Corporation | Heterocyclic aspartyl protease inhibitors, preparation and use thereof |
| ATE489370T1 (de) | 2005-06-14 | 2010-12-15 | Schering Corp | Herstellung und verwendung von verbindungen als aspartylproteasehemmer |
| PE20070135A1 (es) | 2005-06-14 | 2007-03-09 | Schering Corp | Compuestos heterociclicos como inhibidores de aspartil proteasas |
| AU2006259675A1 (en) | 2005-06-14 | 2006-12-28 | Schering Corporation | Aspartyl protease inhibitors |
| KR20080025079A (ko) | 2005-06-14 | 2008-03-19 | 쉐링 코포레이션 | 아스파르틸 프로테아제 억제제 |
| AR053902A1 (es) | 2005-06-14 | 2007-05-23 | Schering Corp | Inhibidores de aspartil proteasa heterociclicos macrociclicos |
| AU2006266167A1 (en) | 2005-06-30 | 2007-01-11 | Wyeth | Amino-5-(6-membered)heteroarylimidazolone compounds and the use thereof for beta-secretase modulation |
| TW200738683A (en) | 2005-06-30 | 2007-10-16 | Wyeth Corp | Amino-5-(5-membered)heteroarylimidazolone compounds and the use thereof for β-secretase modulation |
| TW200730523A (en) | 2005-07-29 | 2007-08-16 | Wyeth Corp | Cycloalkyl amino-hydantoin compounds and use thereof for β-secretase modulation |
| JP2009509957A (ja) * | 2005-09-26 | 2009-03-12 | ワイス | β−セクレターゼ阻害剤としてのアミノ−5−[4−(ジフルオロメトキシ)フェニル]−5−フェニルイミダゾロン化合物 |
| ATE478070T1 (de) | 2005-10-27 | 2010-09-15 | Schering Corp | Heterozyklische aspartyl-proteasehemmer |
| JP2009513670A (ja) | 2005-10-31 | 2009-04-02 | シェーリング コーポレイション | アスパルチルプロテアーゼインヒビター |
| WO2007058601A1 (en) * | 2005-11-21 | 2007-05-24 | Astrazeneca Ab | Novel 2-amino-imidazole-4-one compounds and their use in the manufacture of a medicament to be used in the treatment of cognitive impairment, alzheimer’s disease, neurodegeneration and dementia |
| GB0602951D0 (en) | 2006-02-14 | 2006-03-29 | Novartis Ag | Organic Compounds |
| CN101460480A (zh) | 2006-04-05 | 2009-06-17 | 阿斯利康(瑞典)有限公司 | 2-氨基嘧啶-4-酮类化合物及其用于治疗或预防Aβ相关病理的用途 |
| GB0611064D0 (en) | 2006-06-05 | 2006-07-12 | Novartis Ag | Organic compounds |
| PE20080155A1 (es) | 2006-06-12 | 2008-03-10 | Schering Corp | Compuestos heterociclicos como inhibidores de aspartil-proteasa |
| TW200815449A (en) | 2006-06-14 | 2008-04-01 | Astrazeneca Ab | Novel compounds II |
| TW200808796A (en) | 2006-06-14 | 2008-02-16 | Astrazeneca Ab | New compounds III |
| TW200815447A (en) | 2006-06-14 | 2008-04-01 | Astrazeneca Ab | Novel compounds IV |
| TW200815443A (en) | 2006-06-14 | 2008-04-01 | Astrazeneca Ab | Novel compounds I |
| TW200815349A (en) | 2006-06-22 | 2008-04-01 | Astrazeneca Ab | New compounds |
| JP2009544599A (ja) | 2006-07-20 | 2009-12-17 | ノバルティス アクチエンゲゼルシャフト | Bace阻害剤として有用な大環状化合物 |
| BRPI0715437A2 (pt) | 2006-07-20 | 2013-04-16 | Novartis Ag | lactamas macrocÍclicas |
| JP2010522691A (ja) | 2006-11-23 | 2010-07-08 | ノバルティス アーゲー | 2−ヒドロキシ−1,3−ジアミノプロパン誘導体 |
| JP2010512389A (ja) | 2006-12-12 | 2010-04-22 | シェーリング コーポレイション | アスパルチルプロテアーゼ阻害剤 |
| US20100016341A1 (en) | 2006-12-12 | 2010-01-21 | Zhaoning Zhu | Aspartyl protease inhibitors containing a tricyclic ring system |
| TW200831091A (en) | 2006-12-20 | 2008-08-01 | Astrazeneca Ab | New compounds |
| JP2010521457A (ja) | 2007-03-12 | 2010-06-24 | メルク・シャープ・エンド・ドーム・コーポレイション | 単環式アニリドスピロラクタムcgrp受容体アンタゴニスト |
| CL2008000784A1 (es) | 2007-03-20 | 2008-05-30 | Wyeth Corp | Compuestos amino-5-[-4-(diflourometoxi) fenil sustituido]-5-fenilmidazolona, inhibidores de b-secretasa; composicion farmaceutica que comprende a dichos compuestos; y su uso para tratar alzheimer, deterioro cognitivo, sindrome de down, disminucion co |
| TW200902499A (en) | 2007-05-15 | 2009-01-16 | Astrazeneca Ab | New compounds |
| US20090041201A1 (en) | 2007-08-06 | 2009-02-12 | Carestream Health, Inc. | Alignment apparatus for imaging system |
| WO2009024615A1 (en) | 2007-08-23 | 2009-02-26 | Novartis Ag | Aminobenzyl-substituted cyclic sulfones useful as bace inhibitors |
| US20090062361A1 (en) | 2007-08-30 | 2009-03-05 | Allergan, Inc. | Therapeutic hydantoins |
| TWI431004B (zh) | 2008-05-02 | 2014-03-21 | Lilly Co Eli | Bace抑制劑 |
| KR20110050616A (ko) | 2008-07-10 | 2011-05-16 | 노파르티스 아게 | Bace 억제제로서 유용한 아미노벤질 치환을 갖는 시클릭 술폰 |
| CN102186841A (zh) | 2008-10-22 | 2011-09-14 | 盐野义制药株式会社 | 具有bace1抑制活性的2-氨基嘧啶-4-酮及2-氨基吡啶衍生物 |
| BRPI0823254A2 (pt) | 2008-11-11 | 2015-06-23 | Inst Materia Medica Cams | Complexos de inclusão de pinocembrina com ciclodextrina ou seus derivados |
| AR077277A1 (es) | 2009-07-09 | 2011-08-17 | Lilly Co Eli | Compuestos de biciclo (1,3)tiazin-2-amina formulacion farmaceutica que lo comprende y su uso para la manufactura de un medicamento util para el tratamiento de la enfermedad de alzheimer |
| UA108363C2 (uk) | 2009-10-08 | 2015-04-27 | Похідні імінотіадіазиндіоксиду як інгібітори bace, композиція на їх основі і їх застосування | |
| WO2011044187A1 (en) | 2009-10-08 | 2011-04-14 | Schering Corporation | Iminothiadiazine dioxide compounds as bace inhibitors, compositions, and their use |
| ES2705027T3 (es) | 2010-08-19 | 2019-03-21 | Buck Institute For Age Res | Métodos de tratamiento del deterioro cognitivo leve (DCL) y trastornos relacionados |
| US8415483B2 (en) | 2010-12-22 | 2013-04-09 | Astrazeneca Ab | Compounds and their use as BACE inhibitors |
| US20120165346A1 (en) | 2010-12-22 | 2012-06-28 | Astrazeneca Ab | Compounds and their use as BACE inhibitors |
| US8877744B2 (en) | 2011-04-04 | 2014-11-04 | Hoffmann-La Roche Inc. | 1,4-Oxazepines as BACE1 and/or BACE2 inhibitors |
| CA2899938C (en) | 2013-02-12 | 2021-10-19 | Buck Institute For Research On Aging | Hydantoins that modulate bace-mediated app processing |
| EP3341362B1 (en) * | 2015-08-27 | 2023-07-05 | Nantneuro, LLC | Compositions for app-selective bace inhibition and uses therfor |
-
2014
- 2014-02-12 CA CA2899938A patent/CA2899938C/en active Active
- 2014-02-12 AU AU2014216390A patent/AU2014216390B2/en active Active
- 2014-02-12 EP EP19199279.1A patent/EP3653609B1/en active Active
- 2014-02-12 CN CN201480007825.4A patent/CN104995176B/zh active Active
- 2014-02-12 US US14/179,310 patent/US20140371283A1/en not_active Abandoned
- 2014-02-12 WO PCT/US2014/016100 patent/WO2014127042A1/en not_active Ceased
- 2014-02-12 KR KR1020197035067A patent/KR102220259B1/ko active Active
- 2014-02-12 ES ES14751458T patent/ES2769578T3/es active Active
- 2014-02-12 ES ES19199279T patent/ES2975235T3/es active Active
- 2014-02-12 DK DK14751458.2T patent/DK2956443T3/da active
- 2014-02-12 KR KR1020157021979A patent/KR102051485B1/ko active Active
- 2014-02-12 EP EP14751458.2A patent/EP2956443B1/en active Active
- 2014-02-12 JP JP2015557226A patent/JP6471100B2/ja active Active
- 2014-02-12 US US14/764,107 patent/US10202355B2/en active Active
-
2015
- 2015-08-12 IL IL240555A patent/IL240555B/en active IP Right Grant
- 2015-10-30 US US14/928,775 patent/US9926280B2/en active Active
-
2017
- 2017-12-06 US US15/833,862 patent/US10766867B2/en active Active
-
2019
- 2019-01-17 JP JP2019005905A patent/JP6773820B2/ja active Active
- 2019-02-08 US US16/271,681 patent/US20200017449A1/en not_active Abandoned
-
2020
- 2020-01-10 US US16/740,110 patent/US11091444B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2769578T3 (es) | Hidantoínas que modulan el procesamiento de APP mediado por BACE | |
| US10835546B2 (en) | App specific BACE inhibitors (ASBIs) and uses thereof | |
| JP6830895B2 (ja) | ストレス誘発性p−tauを低下させるトリアゾロピリジン及びトリアゾロピリミジン | |
| EP3183229A1 (en) | Apoe4-targeted theraputics that increase sirt1 |