ES2764523T3 - Compuestos de 7-feniletilamino-4H-pirimido[4,5-D][1,3]oxazin-2-ona y su uso como inhibidores de IDH1 mutantes - Google Patents
Compuestos de 7-feniletilamino-4H-pirimido[4,5-D][1,3]oxazin-2-ona y su uso como inhibidores de IDH1 mutantes Download PDFInfo
- Publication number
- ES2764523T3 ES2764523T3 ES16745954T ES16745954T ES2764523T3 ES 2764523 T3 ES2764523 T3 ES 2764523T3 ES 16745954 T ES16745954 T ES 16745954T ES 16745954 T ES16745954 T ES 16745954T ES 2764523 T3 ES2764523 T3 ES 2764523T3
- Authority
- ES
- Spain
- Prior art keywords
- ethyl
- pharmaceutically acceptable
- oxazin
- pyrimido
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C[C@@](c(cc1)ccc1I)N* Chemical compound C[C@@](c(cc1)ccc1I)N* 0.000 description 2
- PNFDTSOBZXAEDR-ZDUSSCGKSA-N CC(C)CN(c1nc(N[C@@H](C)c2ccc(CCl)cc2)ncc1CO1)C1=O Chemical compound CC(C)CN(c1nc(N[C@@H](C)c2ccc(CCl)cc2)ncc1CO1)C1=O PNFDTSOBZXAEDR-ZDUSSCGKSA-N 0.000 description 1
- SNSHPHFPIWYJGJ-UHFFFAOYSA-N CC(CC1C(N2)=O)C=CC1S2(=O)=O Chemical compound CC(CC1C(N2)=O)C=CC1S2(=O)=O SNSHPHFPIWYJGJ-UHFFFAOYSA-N 0.000 description 1
- HWJUSPBSYBIKNT-UHFFFAOYSA-N CCN(c1c(CO2)cnc(SC)n1)C2=O Chemical compound CCN(c1c(CO2)cnc(SC)n1)C2=O HWJUSPBSYBIKNT-UHFFFAOYSA-N 0.000 description 1
- BVVMVHHJFQHFCU-KRWDZBQOSA-N CCN(c1nc(N[C@@H](C)c2ccc(CN(CC3)CCN3C(C=C)=O)cc2)ncc1CO1)C1=O Chemical compound CCN(c1nc(N[C@@H](C)c2ccc(CN(CC3)CCN3C(C=C)=O)cc2)ncc1CO1)C1=O BVVMVHHJFQHFCU-KRWDZBQOSA-N 0.000 description 1
- QVTSISJFIJPCNA-SFHVURJKSA-N CCN(c1nc(N[C@@H](C)c2ccc(CN(CC3)CCN3C(OC(C)(C)C)=O)cc2)ncc1CO1)C1=O Chemical compound CCN(c1nc(N[C@@H](C)c2ccc(CN(CC3)CCN3C(OC(C)(C)C)=O)cc2)ncc1CO1)C1=O QVTSISJFIJPCNA-SFHVURJKSA-N 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N O=C(c1ccccc11)NS1(=O)=O Chemical compound O=C(c1ccccc11)NS1(=O)=O CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicinal Preparation (AREA)
Abstract
Un compuesto de la fórmula: **(Ver fórmula)** en la que L es un engarce seleccionado entre el grupo que consiste en -N-azetidina-3-CH2-O-, -N-azetidina-3-O-(CH2)-, -N- 2,6-diazaespiro[3,3]heptano-6-(CH2)-, -N-piperazina-4-(CH2)-, -N-piperazina-, -N-azetidina-3-(CH2CH2)-, -7-N- (2,7-diazaespiro[3,5]nonano)-2-(CH2), -N-azetidina-3-(NMe)CH2-, -N-piperidina-4-(NMe)CH2-, N-2,5-dihidropirrol- 3-(CH2)-O-; Z se selecciona entre el grupo que consiste en H o F; R se selecciona entre el grupo que consiste en alquilo C1-C4, -CH(CH3)CH2-OH, - CH2CH2F, -CH2CHF2; o una sal farmacéuticamente aceptable del mismo.
Description
DESCRIPCION
Compuestos de 7-feniletilamino-4H-pirimido[4,5-D][1,3]oxazin-2-ona y su uso como inhibidores de IDH1 mutantes La proteína isocitrato deshidrogenasa (IDH) es una enzima importante en el ciclo del ácido cítrico (ácido tricarboxílico o Krebs). El ciclo del ácido cítrico es fundamentalmente importante para muchas rutas bioquímicas y es uno de los componentes establecidos más temprano del metabolismo celular.
Las isocitrato deshidrogenasas catalizan la descarboxilación oxidativa de isocitrato a a-cetoglutarato (tasa de 2-oxoglutarato). Estas enzimas pertenecen a dos subclases distintas, una de las cuales usa nicotinamida adenina dinucleótido (NAD(+)) como aceptor de electrones y la otra nicotinamida adenina dinucleótido fosfato (NADP(+)). Se han informado cinco isocitrato deshidrogenasas: tres isocitrato deshidrogenasas dependientes de NAD(+), que se localizan en la matriz mitocondrial, y dos isocitrato deshidrogenasas dependientes de NADP(+), una de ellas es mitocondrial y la otra predominantemente citosólica. Cada isoenzima dependiente de NADP(+) es un dímero. La proteína codificada por el gen IDH1 es la isocitrato deshidrogenasa dependiente de NADP(+) que se encuentra en el citoplasma y los peroxisomas. La enzima citoplasmática cumple una función importante en la producción de NADPH citoplasmático. IDH1 se expresa en una amplia gama de especies y organismos que carecen de un ciclo de ácido cítrico completo.
Recientemente, se han encontrado mutaciones en IDH1, y la isoforma relacionada IDH2, en varios tipos de cánceres. Se encontró que las mutaciones se producían en aminoácidos específicos a lo largo de la secuencia de la proteína y se expresaban de forma heterocigota, lo que es coherente con un aumento de función. Estas mutaciones se producen en restos funcionalmente conservados y los estudios bioquímicos de las formas mutantes de IDH1 demostraron una pérdida de la función normal de IDHl, la conversión reversible de isocitrato a a-cetoglutarato. El resultado de estas mutaciones es permitir una conversión nueva (o neomórfica) de a-cetoglutarato (aKG) a 2-hidroxiglutarato (2HG). Como resultado, las células cancerosas que albergan formas mutantes de IDH1 o IDH2 forman substancias sustancialmente más altas de 2HG. Los altos niveles de 2HG dan como resultado un bloqueo de la diferenciación celular que se puede revertir mediante la inhibición de IDH1 o IDH2 mutante.
Existe la necesidad de compuestos que inhiban de forma selectiva la enzima IDH1 mutante con respecto a IDH2 de tipo salvaje para el tratamiento de varios tipos de cáncer. Existe una necesidad adicional de compuestos que inhiban de forma selectiva la enzima IDH1 mutante que demuestren actividad neomórfica con respecto a IDH1 de tipo salvaje para el tratamiento de varios tipos de cáncer.
Un aspecto de la invención es proporcionar compuestos inhibidores de enzima IDH1 mutantes de la Fórmula:

I
en la que
L es un engarce seleccionado entre el grupo que consiste en -N-azetidina-3-CH2-0-, -N-azetidina-3-0-(CH2)-, -N-2,6-diaza-espiro[3,3]heptano-6-(CH2)-, -N-piperazina-4-(CH2)-, - N-piperazina-, -N-azetidina-3-(CH2CH2)-, -7-A/-(2,7-diaza-esp¡ro[3,5]nonano)-2-(CH2)-, -W-azetidina-3-(NMe)CH2-, -N-piperidina-4-(NMe)CH2- y -N-2,5-dihidropirrol-3-(CH2)-0-;
Z se selecciona entre el grupo que consiste en H y F;
R se selecciona entre el grupo que consiste en alquilo C1-C4, -CH(CH3)CH2-OH, - CH2CH2F, y -CH2CHF2; o una sal farmacéuticamente aceptable de los mismos.
Un aspecto adicional de la presente invención proporciona un compuesto de Fórmula I en la que: L es un engarce seleccionado entre el grupo que consiste en -N-piperazina-4-(CH2)- , -N-piperazina-, -N-azetidina-3-(NMe)CH2-, -N-azetidina-3-CH2-O-, -N-azetidina-3-0-(CH2)-, -N-azetidina-3-(CH2cH2)-, y -N-piperidina-4-(NMe)CH2-;
Z es H;
R es alquilo C1-C4; o una sal farmacéuticamente aceptable del mismo.
Otro aspecto de la presente invención proporciona un compuesto de Fórmula I en la que: L es un engarce seleccionado entre el grupo que consiste en -N-piperazina-4-(CH2)- , -N-piperazina-, -N-azetidina-3-0-(CH2)- , -W-azetidina-3-(CH2CH2)-, -N-azetidina-3-(NMe)CH2-, y -N-piperidina-4-(NMe)CH2-;
Z es H;
R se selecciona entre -CH2CH3, -CH(CH3)2; o una sal farmacéuticamente aceptable del mismo.
Un aspecto adicional de la invención es un compuesto 1-isopropil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo. Otro aspecto de la invención es un compuesto 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo.
Un aspecto adicional de la invención es un compuesto seleccionado entre:
1-isopropil-7-[[(1S)-1-[4-(4-prop-2-enoilpiperazin-1-il)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo;
1-etil-7-[[(1S)-1-[4-(4-prop-2-enoilpiperazin-1-il)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo;
1-etil-7-[[(1S)-1-[4-[(1-prop-2-enoilazetidin-3-il)oximetil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo;
1-etil-7-[[(1S)-1-[4-[2-(1-prop-2-enoilazetidin-3-il)etil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo;
1-isopropil-7-[[(1S)-1-[4-[(1-prop-2-enoilazetidin-3-il)metoxi]-fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo;
1-etil-7-[[(1S)-1-[4-[[metil-(1-prop-2-enoilazetidin-3-il)amino]metil]-fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo.
Otro aspecto de la presente invención proporciona una composición farmacéutica que comprende un compuesto inhibidor de IDH1 mutante de Fórmula I, o una sal farmacéuticamente aceptable del mismo, y un vehículo farmacéuticamente aceptable.
Un aspecto adicional de la presente invención proporciona un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, para uso en terapia.
Otro aspecto de la presente invención proporciona un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, para uso en el tratamiento de una IDH1 mutante que expresa cáncer que es glioma, glioblastoma, glioblastoma multiforme, astrocitomas, oligodendrogliomas, paraganglioma, fibrosarcoma, linfoma de linfocitos T angioinmunoblástico (AITL), síndrome mielodisplásico (MDS), leucemia linfoblástica aguda de linfocitos B (B-ALL), cáncer de tiroides, cáncer colorrectal, leucemia mieloide aguda (AML), melanoma, cáncer de próstata, condrosarcoma o colangiocarcinoma .
Un aspecto adicional de la presente invención proporciona un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, para uso en el tratamiento de una IDH1 mutante que expresa cáncer que es fibrosarcoma, leucemia mieloide aguda, glioma, o glioblastoma.
Otro aspecto de la presente invención proporciona el uso de un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, para la fabricación de un medicamento para el tratamiento de una IDH1 mutante que expresa cáncer que es glioma, glioblastoma, glioblastoma multiforme, astrocitomas, oligodendrogliomas, paraganglioma, fibrosarcoma, linfoma de linfocitos T angioinmunoblástico (AITL), síndrome mielodisplásico (MDS), leucemia linfoblástica aguda de linfocitos B (B-ALL), cáncer de tiroides, cáncer colorrectal, leucemia mieloide aguda (AML), melanoma, cáncer de próstata, condrosarcoma o colangiocarcinoma.
Un aspecto adicional de la presente invención proporciona el uso de un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, para la fabricación de un medicamento para el tratamiento de una IDH1 mutante que expresa cáncer que es fibrosarcoma, leucemia mieloide aguda, glioma, o glioblastoma.
El término "paciente" significa mamífero y "mamífero" incluye, pero no se limita a, un ser humano.
"Cantidad terapéuticamente eficaz" o "cantidad eficaz" significa la dosificación del compuesto de Fórmula I, o sal farmacéuticamente aceptable del mismo, o composición farmacéutica que contiene el compuesto, o sal farmacéuticamente aceptable del mismo, necesaria para inhibir IDH1 mutante en un paciente con cáncer, lo que conduce a la liberación del bloqueo en la diferenciación que da como resultado la inhibición de crecimiento de células tumorales y para eliminar o ralentizar o detener la progresión del cáncer en un paciente. Las dosificaciones anticipadas de un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo se encuentran en el intervalo de 20 mg/paciente/día a 2000 mg/paciente/día. Se anticipa que las dosificaciones más preferentes están en el intervalo de 30 mg/paciente/día a 1800 mg/paciente/día. Se anticipa que las dosificaciones más preferentes están en el intervalo de 40 mg/paciente/día a 1600 mg/paciente/día. La dosificación exacta requerida para tratar a un paciente y la duración del tiempo de tratamiento serán determinadas por un médico en vista de la etapa y la gravedad de la enfermedad, así como de las necesidades específicas y la respuesta del paciente individual. Aunque se expresa como dosificación al día, la administración de la dosificación se puede ajustar para proporcionar un beneficio terapéutico más óptimo para un paciente y para controlar o mejorar cualquier toxicidad relacionada con el fármaco. Además de la dosificación diaria, puede ser apropiada una dosificación de dos veces al día (B.I.D.),
dosificación de tres veces al día (T.I.D.), dosificación cada dos días (Q2D); cada dos días durante un periodo de cinco días seguido de dos días sin dosificación (T.I.W.); o cada tres días (Q3D).
Los términos "tratamiento", "tratar" y "que trata", están destinados a incluir el espectro completo de intervención para el cáncer que padece el paciente, tal como la administración del compuesto activo para aliviar, retrasar o revertir uno o más de los síntomas y retrasar la progresión del cáncer incluso si el cáncer no se elimina realmente.
La expresión "alquilo C1-C4" incluye metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, y t-butilo.
Un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, se formula preferentemente como una composición farmacéutica usando un vehículo farmacéuticamente aceptable y se administra mediante una diversidad de vías. Preferentemente, las composiciones de ese tipo son para administración oral. Las composiciones farmacéuticas de ese tipo y procedimientos para prepararlas se conocen bien en la técnica. Véase, por ejemplo, REMINGTON: THE SCIENCE AND PRACTICE OF PHARMACY (A. Gennaro, y col., eds., 19a ed., Mack Publishing Co., 1995). En una realización particular, la composición farmacéutica comprende 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona o una sal farmacéuticamente aceptable del mismo, junto con un vehículo farmacéuticamente aceptable y opcionalmente otros ingredientes terapéuticos en particular para el tratamiento del cáncer generalmente o de un tipo de cáncer específico.
Un compuesto de Fórmula I, o una sal farmacéuticamente aceptable, se puede administrar de forma simultánea con, o antes, o después, de uno u otros agentes terapéuticos más. El compuesto de fórmula I o una sal farmacéuticamente aceptable, cuando se administra con uno u otros agentes terapéuticos más, se puede administrar por separado, mediante la misma vía de administración o una diferente, o en conjunto en la misma composición farmacéutica que el otro agente o agentes terapéutico(s). Cuando se administra uno u otros agentes terapéuticos más, la administración de cada agente terapéutico puede ser simultánea, separada, o secuencial.
Un compuesto de Fórmula I es capaz de reacción con una serie de ácidos inorgánicos y orgánicos para formar sales de adición de ácido farmacéuticamente aceptables. En la técnica se conocen bien las sales farmacéuticamente aceptables de ese tipo y la metodología común para prepararlas. Véase, por ejemplo, P. Stahl, y col., HANDBOOK OF PHARMACEUTICAL SALTS: PROPERTIES, SELECTION AND USE, (VCHA/Wiley-VCH, 2002); S.M. Berge, y col., "Pharmaceutical Salts, "Journal of Pharmaceutical Sciences, Vol. 66, N.° 1, enero de 1977.
Un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, se puede preparar mediante una diversidad de procedimientos conocidos en la técnica, así como los que se describen a continuación. Las etapas de síntesis específicas que se pueden combinar en un orden diferente para preparar un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo.
Los compuestos de Fórmula I se nombran de acuerdo con la IUPAC, y también se pueden nombrar de acuerdo con CAS, y se pueden usar otros nombres para identificar de forma inequívoca un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo.
Se entenderá que un compuesto de Fórmula I se puede representar como un estereoisómero único. Hay un centro quiral en la posición estereogénica del carbono del bencilo que da lugar a dos diastereómeros y posiblemente más dependiendo de los sustituyentes. Como se usa en el presente documento, las referencias a un solo estereoisómero están destinadas a incluir también mezclas estereoisoméricas que incluyen el compuesto de Fórmula I nombrado o representado. En el presente documento, las denominaciones de Cahn-Ingold-Prelog de (R)- y (S)- se pueden usar para hacer referencia a estereoisómeros específicos. Los estereoisómeros específicos se pueden preparar mediante síntesis estereoespecífica usando materiales de partida enantioméricamente puros o enriquecidos. Los estereoisómeros específicos de los materiales de partida, compuestos intermedios o mezclas racémicas, incluyendo los compuestos de Fórmula I, se pueden resolver mediante técnicas bien conocidas en la técnica, tal como las que se encuentran en Stereochemistry of Organic Compounds, E. I. Eliel y S. H. Wilen (Wiley 1994) y Enantiomers, Racemates, y Resolutions, J., Jacques, A. Collet, y S. H. Wilen (Wiley 1991), incluyendo cromatografía sobre fases estacionarias quirales, resoluciones enzimáticas, cristalización fraccionada o cromatografía de diastereómeros formados para esa finalidad, tal como sales diastereoméricas. Aunque las mezclas diastereoméricas que contienen un compuesto de Fórmula I se contemplan dentro de la presente invención, la realización preferente es la configuración (S) como se muestra para la Fórmula I.
Los compuestos empleados como materiales de partida iniciales en la síntesis de los compuestos de Fórmula I se conocen bien y, en la medida en la que no están disponibles en el mercado, se sintetizan fácilmente usando referencias específicas proporcionadas, mediante procedimientos convencionales empleados comúnmente por los expertos en la materia o se encuentran en textos de referencia generales.
Los ejemplos de procedimientos y métodos conocidos incluyen los que se describen en textos de referencia generales tales como Comprehensive Organic Transformations, VCH Publishers Inc, 1989; Compendium of Organic Synthetic Methods, Volúmenes 1-10, 1974-2002, Wiley Interscience; Advanced Organic Chemistry, Reactions Mechanisms, y Estructura, 5a Edición, Michael B. Smith y Jerry March, Wiley Interscience, 2001; Advanced Organic Chemistry, 4a Edición, Parte B, Reactions and Synthesis, Francis A. Carey y Richard J. Sundberg, Kluwer Academic/ Plenum Publishers, 2000, etc., y referencias citadas en ese documento.
Ciertos centros estereoquímicos se pueden dejar sin especificar en los siguientes esquemas por cuestiones de claridad y no se pretende que limiten la enseñanza de los esquemas de ninguna manera. Los isómeros individuales, enantiómeros y diastereómeros pueden ser separados o resueltos por un experto en la materia en cualquier punto conveniente en la síntesis de compuestos de la invención, mediante métodos tales como técnicas de cristalización selectiva o cromatografía quiral.
Ciertas abreviaturas se definen como sigue a continuación: "ACN" se refiere a acetonitrilo; "aKG" se refiere a Alfacetoglutarato o 2-cetoglutarato; "alloc" se refiere a aliloxicarbonilo; "ATCC" se refiere a Colección Americana de Cultivos Tipo; "BCA" se refiere a ácido bicinconínico; "BOC" se refiere a ferc-butoxi carbonilo; "BSA" se refiere a Albúmina de Suero Bovino; "CDI" se refiere a 1,1'-carbonildiimidazol; "CPME" se refiere a ciclopentil metil éter; "DCC" se refiere a 1,3-diciclohexilcarbodiimida; "DCM" se refiere a diclorometano; "DEAD" se refiere a azodicarboxilato de dietilo; "DIAD" se refiere a azodicarboxilato de diisopropilo; "DIC" se refiere a diisopropilcarbodiimida; "DIPEA" se refiere a diisopropiletilamina o N-etil-N-isopropil-propan-2-amina; "DMA" se refiere a dimetilacetamida ; "DMAP" se refiere a dimetilaminopiridina; "DMF" se refiere a dimetilformamida; "DMSO" se refiere a dimetilsulfóxido; "DTT" se refiere a ditiotreitol; "EDC" se refiere a EDAC, EDCI, o clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida; "EDTA" se refiere a ácido etilendiaminatetraacético; "EGTA" se refiere a ácido etilenglicol tetraacético; "EtOAc" se refiere a acetato de etilo; "EtOH" se refiere a etanol o alcohol etílico; "Ej" se refiere a ejemplo; "HATU" se refiere a hexafluorofosfato de (dimetilamino)-N,N-dimetil(3H-[1,2,3]triazolo[4,5-£)]piridin-3-iloxi)metaniminio; "HBTU" se refiere a hexafluorofosfato de 2-(1H-benzotriazol-1-il)-1,1,3,3-tetrametiluronio; "HEK" se refiere a riñón embrionario humano; "2HG" se refiere a 2-hidroxiglutarato; "d5-3HG" se refiere a ácido 2,2,3,4,4-d5 3-hidroxi-1,5-pentanodioico; "HILIC" se refiere a cromatografía líquida de interacción hidrófila; "HOAt" se refiere a 1-hidroxi-7-azobenzotriazol; "HOBt" se refiere a hidrato de 1-hidroxilbenzotriazol; "HPLC" se refiere a cromatografía líquida de alto rendimiento; "CI50" se refiere a la concentración de un agente que produce un 50 % de la respuesta inhibitoria máxima posible para ese agente; "mCPBA" se refiere a ácido mefa-cloroperbenzoico; "Me" se refiere a metilo o CH3; "MeOH" se refiere a metanol o alcohol metílico; "MTBE" se refiere a metil ferc-butil éter; "NADP+ y NAHPH" se refiere a las formas oxidada y reducida del nicotinamida adenina dinucleótido fosfato respectivamente; "NMP" se refiere a N-metil-2-pirrolidona; "PG" se refiere a grupo protector; "Ph" se refiere a fenilo; "Prep" se refiere a preparación; "PyBOP" se refiere a hexafluorofosfato de benzotriazol-1-iloxitripirrolidino-fosfonio; "PyBrop" se refiere a fosfoniohexafluoro fosfato de bromo-tris-pirrolidino; "rpm" se refiere a revoluciones por minuto; "SCX" se refiere a intercambio catiónico fuerte; "SNAr" se refiere a sustitución aromática nucleófila; "TEA" se refiere a trietilamina; "TFA" se refiere a ácido trifluoroacético; "THF" se refiere a tetrahidrofurano; THP se refiere a tetrahidropiranilo; "Tris" se refiere a tris(hidroximetil)aminometano y "XRD" se refiere a difracción de rayos X con el método de polvo.
Los compuestos de la presente invención, o sales de los mismos, se pueden preparar mediante una diversidad de procedimientos conocidos en la técnica, algunos de los cuales se ilustran en los Esquemas, Preparaciones, y Ejemplos que siguen a continuación. Las etapas de síntesis específicas para cada una de las vías descritas se pueden combinar de diferentes formas, o en conjunto con etapas de diferentes esquemas, para preparar compuestos de la invención, o sales de los mismos. Los productos de cada etapa en los esquemas que siguen a continuación se pueden recuperar mediante métodos convencionales bien conocidos en la técnica, incluyendo extracción, evaporación, precipitación, cromatografía, filtración, trituración, y cristalización. Los esquemas que siguen a continuación, todos los sustituyentes a menos que se indique de otro modo, son como se han definido anteriormente. Los reactivos y materiales de partida están fácilmente disponibles para alguien con experiencia habitual en la materia.

En el Esquema 1, una serie de reacciones conduce a 1-sustituida-7-(metilsulfonil)-1,4-dihidro-2H-pirimido[4,5-d][1,3]oxazin-2-ona, el producto de la Etapa E en la que R es como se ha definido anteriormente o R = CH(CH3)
CH2-O-PG. "PG" es un grupo protector desarrollado para el grupo amino o el grupo oxigeno tal como para carbamatos, amidas o ésteres. Por ejemplo, en la Etapa A, el cloro de un 4-doro-2-(metiltio)pirimidina-5-carboxilato protegido en el carboxilo se convierte a la amina usando hidróxido de amonio en un disolvente tal como THF para dar el producto de amina de pirimidina sustituida de la Etapa A. En la Etapa B, el éster, tal como el éster etílico, se puede reducir con una fuente de hidruro, tal como hidruro de litio y aluminio, en un disolvente tal como THF a hidroximetilo en condiciones que se conocen bien en la técnica para dar el producto de hidroximetilo de la Etapa B. La 5-hidroxi metil 4-amina-pirimidina se puede ciclar en condiciones estándar de carbamoilación a la oxazina-2-ona usando trifosgeno y una base orgánica tal como DIPEA o TEA a una temperatura de aproximadamente -30 a -35 °C para dar el producto de la Etapa C. Como alternativa, se puede usar un dihaluro carbonilo o un dipseudohaluro carbonilo tal como CDI, fosgeno o difosgeno en lugar de trifosgeno para completar la carbamoilación. La amina de la oxazina se puede alquilar con el haluro de alquilo sustituido apropiado, tal como un reactivo de yodo en un disolvente tal como NMP y una base inorgánica tal como K2CO3 a una temperatura de aproximadamente 50-65 °C para dar el producto de la Etapa D. Como alternativa, se puede completar una reacción de Mitsunobu para alquilar la amina de la oxazina usando un alcohol apropiado tal como MeOH. Las reacciones de Mitsunobu se conocen bien en la técnica y pueden convertir un grupo hidroxilo en un grupo saliente que se desplaza por parte de una gran diversidad de nucleófilos, tales como un carbamato usando trifenilfosfina y un azodicarboxilato, tal como DIAD o DEAD, en un disolvente tal como THF para dar el producto de la Etapa D. El sulfuro se puede oxidar a la sulfona en condiciones bien conocidas en la técnica tales como mCPBA o peroximonosulfato potásico a una temperatura de aproximadamente 10 a 25 °C en una disolvente tal como ACN o CH2Ch para dar el producto la Etapa E. Como alternativa, en la Etapa A, se puede preparar un producto sustituido de alcoxialquil amina a partir de la 4-cloropirimidina sustituida en un procedimiento de 3 etapas. Por ejemplo, se calientan (2R)-1-(terc-butil dimetilsilil)oxipropan-2-amina y una base tal como DIPEA en un disolvente tal como DMF a aproximadamente 65 °C seguido de enfriamiento con agua y extracción con EtOAc para dar un compuesto intermedio de siloxialquilaminopirimidina en bruto. Este compuesto intermedio se trata con fluoruro de tetrabutilamonio, concentrado y a continuación se purifica con gel de sílice para dar una hidroxialquilaminopirimidina intermedia. El grupo hidroxilo intermedio se puede proteger con 3,4-dihidro-2H-pirano usando ácido p-toluenosulfónico y después de la purificación da el producto con hidroxilo protegido de la Etapa A.
En el Esquema 2, Etapa J, un ácido 4-etilamina-benzoico protegido se reduce mediante el uso de condiciones bien conocidas en la técnica, tales como hidruro de litio y aluminio en un disolvente tal como THF para dar el compuesto de 4-etilamina-bencil-hidroxi. El producto de bencil-hidroxi de la Etapa J se puede convertir en un haluro tal como un cloruro en condiciones de cloración estándar mediante el uso de un agente de cloración tal como cloruro de tionilo o POCla en un disolvente tal como CH2O2 para dar el producto de la Etapa K. El producto de 1 -feniletilamina de la Etapa K o el producto de bencil-hidroxi de la Etapa J se puede proteger en la subetapa 1 de la Etapa L usando un grupo protector tal como un trifluoroacetilo. El cloruro del producto de la Etapa L se puede desplazar con una amina protegida del grupo de engarce (L) en un procedimiento en dos etapas y un recipiente. No siempre es necesario proteger la 1 -feniletilamina, pero si se elige la protección, es ventajoso usar un protector de grupo en la 1-feniletilamina diferente al de la amina de engarce para desproteger de forma selectiva uno u otro PG en la etapa deseada. Por ejemplo, la 1 -feniletilamina se puede hacer reaccionar con anhídrido trifluoroacético usando una base orgánica tal como TEA en un disolvente tal como CH2Ch a una temperatura de aproximadamente 0-5 °C para dar el producto de amina protegido de la subetapa 1, Etapa L. El desplazamiento del cloruro se puede conseguir en condiciones bien conocidas por el experto en la materia. Por ejemplo, cuando el engarce tiene una amina, el cloruro se puede desplazar por parte de un engarce apropiado usando una base inorgánica tal como K2CO3 con calentamiento a aproximadamente 60 °C en un disolvente tal como ACN para dar el producto de la subetapa 2, Etapa L. Como alternativa, se puede usar una base orgánica tal como DIPEA en un disolvente tal como DMSO para alquilar la amina para dar el producto de la subetapa 2, Etapa L. Cuando el engarce está unido a través de un enlace éter, el precursor de alcohol puede desplazar al haluro con una base tal como hidruro sódico en un disolvente tal como DMF para dar el producto de la subetapa 2, Etapa L. En Etapa M, subetapa 1, la 1 -feniletilamina se puede desproteger si la protección se efectúa previamente bajo condiciones estándar de desprotección usando condiciones alcalinas tales como hidróxido sódico acuoso en un disolvente tal como EtOH para dar el producto de amina libre de la subetapa 1, Etapa M. Como alternativa si el grupo protector es un grupo carboxibencilo, se pueden usar condiciones de hidrogenólisis para retirar el grupo protector, tal como usar Pd al 10 %/C en un disolvente tal como EtOH en una atmósfera de hidrógeno para dar el producto desprotegido de la Etapa M, subetapa 1. La 1-feniletilamina se puede hacer reaccionar a continuación con el producto de Esquema 1, Etapa E en una reacción SNAr usando una base orgánica tal como DIPEA, CsF para acelerar la reacción, un disolvente tal como DMSO, y una temperatura de aproximadamente 70-80 °C para dar el producto de la Etapa M, subetapa 2. En Etapa N, subetapa 1, un engarce protegido con terc-butoxi se puede desproteger usando un ácido tal como HCl en dioxano y MeOH o TFA en CH2Cl2, mientras que un engarce protegido con alloc se puede desproteger en presencia de una fuente de paladio tal como tetraquis(trifenilfosfina)paladio (0) catalítico en un disolvente tal como THF mediante el uso de un nucleófilo blando como dimedona para dar el engarce desprotegido de la subetapa 1, Etapa N. En la subetapa 2, Etapa N, la amina de engarce se puede amidar con cloruro de acriloílo a una temperatura de aproximadamente 50 a 75 °C mediante el uso de una base orgánica tal como TEA si la amina es una sal de ácido en un disolvente tal como CH2O2 para dar compuestos de Fórmula I. Como alternativa, se puede conseguir un acoplamiento de amida con ácido acrílico y la amina apropiada en un disolvente tal como DMF con un reactivo de acoplamiento tal como EDC y un aditivo como HOBt. El experto en la materia reconocerá que existen diversos procedimientos y reactivos para la formación de amidas como resultado de una reacción entre ácidos carboxílicos y aminas. Por ejemplo, la reacción de la amina y el ácido acrílico apropiados en presencia de un reactivo de acoplamiento con o sin una base orgánica tal como DIPEA o TEA puede proporcionar un compuesto de Fórmula I. Otros reactivos de acoplamiento incluyen carbodiimidas, tales como DCC, DIC, o un carbonildiimidazol tal como CDI. Otros aditivos de acoplamiento de amida, tales como HOAt, también se pueden usar para mejorar la reacción. Además, se podrían usar sales de uronio o fosfonio de aniones no nucleófilos, tales como HBTU, HATU, PyBOP y PyBrOP, en el lugar de los reactivos de acoplamiento más tradicionales. Se puede usar un aditivo tal como DMAP para acelerar la reacción de amidación deseada.

Como alternativa, en el Esquema 3, una 1 -feniletilamina sin el engarce en su lugar se puede hacer reaccionar con el producto de la Etapa E como se ha descrito en el Esquema 2, Etapa M para dar el producto de la Etapa O. El producto de la Etapa O se puede hacer reaccionar con el engarce protegido como se ha descrito en el Esquema 2, Etapa L, subetapa 2 mediante el uso de K2CO3 y yoduro sódico en ACN para dar el producto de la Etapa P. Como alternativa, si X = Br, el bromuro se puede desplazar con una amina mediante el uso de una base inorgánica tal como carbonato potásico, CuBr, e hidroxiprolina en un disolvente tal como DMSO para dar el producto de la Etapa P. Para los ejemplos en los que L implica un alquino, el Br se puede hacer reaccionar con el alquino protegido apropiado mediante el uso de condiciones de acoplamiento cruzado con paladio tales como las de una reacción de Sonagashira. Un catalizador de paladio apropiado tal como tetraquis(trifenilfosfina)paladio (0), un catalizador de cobre tal como CuI, y una base orgánica tal como TEA se combinan con el alquino apropiado y bromuro de arilo en un disolvente tal como THF para dar el producto de la Etapa P. Cuando X = -CH2OH, el OH se puede convertir en el cloruro como se ha descrito en el Esquema 2, Etapa K para dar el producto clorado de la Etapa Q. Un engarce de alquino de la Etapa P se puede hidrogenar con un catalizador tal como Pd al 10 %/CaCO3 en un disolvente tal como THF en una atmósfera de hidrógeno para dar el producto de alquino reducido. En la Etapa N, los productos de la Etapa P o la Etapa Q se pueden desproteger a continuación como se ha descrito en el Esquema 2, Etapa N, subetapa 1 y el engarce se puede amidar como se ha descrito en el Esquema 2, Etapa N, subetapa 2 para dar los compuestos de Fórmula I.
Esquema 4

Etapa N 1. Desproteccion
2. Amidacion

En el Esquema 4, la 1 -feniletilamina se puede proteger como se ha descrito en el Esquema 2, Etapa L con un grupo protector tal como trifluoroacetilo u otros grupos protectores tales como carboxi bencilo. Por ejemplo cuando carboxi bencilo es el PG deseado, se puede añadir cloroformiato de bencilo a la 1-(4-hidroxifenil)etilamina con una base inorgánica tal como NaHCO3 en una mezcla de disolventes tal como THF y agua para dar la amina protegida con carboxi bencilo. Cuando el engarce L está unido al anillo de arilo a través de una unión éter, se puede realizar una reacción de Mitsunobu en una 1-(4-hidroxifenil)etilamina protegida. Las reacciones de Mitsunobu se conocen bien en la técnica y pueden convertir un grupo hidroxilo en un potente grupo saliente que se puede desplazar por parte de una gran diversidad de nucleófilos tales como un fenol usando trifenilfosfina y un azodicarboxilato tal como DIAD o DEAD en un disolvente tal como THF para dar el producto de aril éter de la Etapa R. El producto de la Etapa R se puede desproteger a continuación en la funcionalidad 1 -feniletilamina como se ha descrito en el Esquema 2, Etapa M, subetapa 1 seguido de la alquilación por SNAr como se ha descrito en el Esquema 2, Etapa M, subetapa 2 para dar el producto del Esquema 4, Etapa M. Los productos de Fórmula I de la Etapa N se pueden formar a continuación como se ha descrito en el Esquema 2, Etapa N, subetapas 1 y 2.
En una etapa opcional, se puede formar una sal farmacéuticamente aceptable de un compuesto de Fórmula I por reacción de una base libre apropiada de Fórmula I con un ácido farmacéuticamente aceptable en un disolvente adecuado en condiciones convencionales. Además, la formación de tales sales se puede producir de forma simultánea tras la desprotección de un grupo protector de nitrógeno. La formación de tales sales se conoce bien y se entiende en la técnica. Véase, por ejemplo, Gould, P.L., "Salt selection for basic drugs," International Journal of Pharmaceutics, 33: 201-217 (1986); Bastin, R.J., y col. "Salt Selection and Optimization Procedures for Pharmaceutical New Chemical Entities," Organic Process Research and Development, 4: 427-435 (2000); y Berge, S.M., y col., "Pharmaceutical Salts," Journal of Pharmaceutical Sciences, 66: 1-19, (1977). El experto habitual en la materia entenderá que un compuesto de Fórmula I se puede convertir con facilidad en, y se puede aislar, como una sal farmacéuticamente aceptable.
Preparación 1
4-Amino-2-metilsulfanil-pirimidina-5-carboxilato de etilo

Esquema 1, Etapa A: se añade hidróxido de amonio (8,4 l, 17 % en peso, 6,86 mol) durante 1 hora a una solución de 4-cloro-2-(metiltio)pirimidina-5-carboxilato de etilo (4 kg, 10,74 mol) en THF (34,4 l) a temperatura ambiente. Después de agitar durante 4 horas, se añade agua y la mezcla se extrae con MTBE. La fase orgánica se lava con solución salina saturada, se seca sobre Na2SO4, se filtra y se concentra. El producto en bruto se suspende con éter de petróleo (3 l), se filtra, y se seca al vacío para proporcionar el compuesto del título en forma de un sólido de color blanco (3,5 kg, 89 % de pureza, 95 % de rendimiento) que se usa más adelante sin purificación adicional. MS (m/z): 214 (M H).
Preparación 2
2-Metilsulfanil-4-[[(1R)-1-metil-2-tetrahidropiran-2-iloxi-etil]amino]-pirimidina-5-carboxilato

Esquema 1, Etapa F: se añaden DIPEA (9,1 ml, 52 mmol) y (2R)-1-(terc-butil silil)oxipropan-2-amina (5,82 g, 30,7 mmol) a una solución de 4-cloro-2-(metiltio)pirimidina-5-carboxilato de etilo (6 g, 25,8 mmol) en DMF (50 ml) a temperatura ambiente. Después de agitar durante 10 minutos a temperatura ambiente, la mezcla se calienta a 65 °C durante 1,5 horas, se enfría a temperatura ambiente, se trata con agua, y se extrae con EtOAc (2 x). Los extractos orgánicos se lavan con LiCl acuoso al 5 % (3 x), se secan sobre Na2SO4, se filtran y se concentran. El material en bruto (8,64 g, 22,4 mmol) se disuelve en THF (100 ml) y se trata con fluoruro de tetrabutilamonio (34 ml, 34 mmol, 1 M en THF). Después de agitar a temperatura ambiente durante 30 minutos, la reacción se concentra a sequedad y el alcohol primario en bruto se purifica por cromatografía (20-40 % de EtOAc/hexanos) para dar el alcohol intermedio en forma de un sólido de color blanco (4,94 g, 81 %). Una parte de este alcohol intermedio (2,05 g, 7,56 mmol) se suspende en CH2Cl2 (40 ml) y se trata con monohidrato de ácido p-tolueno-sulfónico (2,16 g, 11,4 mmol) y 3,4-dihidro-2H-pirano (5,81 ml, 60,5 mmol). Después de agitar durante 1 hora a temperatura ambiente la mezcla se inactiva con NaHCO3 acuoso saturado y la fase acuosa se extrae con CH2O2 adicional (2 x). Los extractos orgánicos combinados se secan sobre Na2SO4, se filtran, y se concentran, y el producto deseado se purifica por cromatografía sobre gel de sílice (0 a 15 % de EtOAc/hexano) para dar un aceite incoloro (2,60 g, 97 % de rendimiento para esta etapa). MS (m/z): 356 (M H).
El siguiente compuesto se prepara esencialmente mediante el procedimiento de la Preparación 2.
Tabla 1

Preparación 4
(4-Amino-2-metilsulfanil-pirimidin-5-il)metanol

Esquema 1, Etapa B: se añade LiAlH4 (13,9 l, 14 mol, 1 M en THF) a una solución de 4-amino-2-sulfanil-pirimidina-5-carboxilato de etilo (3,5 kg, 16,4 mol) en THF (45 l) a -5 °C durante 1,5 horas. Después de agitar durante 1 hora de -5 a 0 °C, se añaden agua en hielo (525 ml) y NaOH acuoso al 15 % (525 ml), seguido de una cantidad adicional de agua en hielo (1,6 l). Después de 30 minutos a 0 °C, la mezcla de reacción inactivada se filtra y la torta de filtro se lava con THF (1 l). Los lavados de THF combinados se secan (Na2SO4), se filtran, y se concentran y el producto en bruto resultante se suspende en una mezcla de éter de petróleo/EtOAc (3:1, 2 l), se filtra, y se seca de nuevo para
proporcionar el compuesto del título en forma de un sólido de color amarillo (2,1 kg, 82% de pureza, 75% de rendimiento) que se usa sin purificación adicional. MS (m/z): 172 (M H).
Los siguientes compuestos se preparan esencialmente mediante el procedimiento de la Preparación 4.
Tabla 2

Preparación 7
7-Metilsulfanil-1,4-dihidropirimido[4,5-d][1,3]oxazin-2-ona

Esquema 1, Etapa C: se añade trifosgeno (859 g, 2,9 mol) a una solución de (4-amino-2-metilsulfanil-pirimidin-5-il)metanol (900 g, 5,26 mol) en THF (22,5 l) durante 15 minutos a -30 °C. Se añade DIPEA (2,449 g, 18,92 mol) durante 1 hora, mientras se mantiene la temperatura de reacción entre -35 y -30 °C. La mezcla de reacción se vierte a continuación sobre agua en hielo (30 l) y se añade 2-metiltetrahidrofurano (10 l). La fase orgánica se lava con agua y solución salina saturada. La fase orgánica se seca sobre Na2SO4 y se concentra a sequedad. El producto en bruto se suspende con éter de petróleo / EtOAc (1:1), se filtra y se concentra para dar un sólido de color amarillo que se usa más adelante sin purificación adicional (890,5 g, 1,62 mol, 83 % de pureza, 86 % de rendimiento). MS (m/z): 198 (M H).
Los siguientes compuestos se preparan esencialmente mediante el procedimiento de la Preparación 7.
Tabla 3

continuación

Preparación 10
1-Etil-7-(metiltio)-1,4-dihidro-2H-pirimido[4,5-d][1,3]oxazin-2-ona

Esquema 1, Etapa D: a una solución de 7-metilsulfanil-1,4-dihidropirimido[4,5-d][1,3]oxazin-2-ona (280 g, 1,42 mol) en NMP (2,24 l) se añaden K2CO3 (294,2 g, 2,13 mol) y yoduro de etilo (336,3 g, 1,99 mol) a temperatura ambiente. La mezcla se agita durante 16 horas a 50 °C y después se diluye con CH2Cl2 (3 l) y agua (6 l). La fase orgánica se separa y se lavó con agua y solución salina saturada y se concentra a sequedad para dar el compuesto del título en bruto (286 g, 1,27 mol, 83 % de pureza, 91 % de rendimiento). MS (m/z): 226 (M H). Los siguientes compuestos se preparan esencialmente mediante el procedimiento de la Preparación 10.
Tabla 4

Preparación Alternativa 10
1-Etil-7-(metiltio)-1,4-dihidro-2H-pirimido[4,5-d][1,3]oxazin-2-ona

Se añaden 7-metilsulfanil-1,4-dihidropirimido[4,5-d][1,3]oxazin-2-ona (325 g, 1648,0 mmol) y DMF (2,4 l) conjuntamente en una atmósfera de nitrógeno. A esta solución transparente se añade a continuación carbonato de
cesio (660 g, 2025,61 mmol) y la mezcla se agita durante 30 minutos. Se añade yodoetano (170 ml, 2120 mmol) gota a gota durante 30 minutos a 20 °C. La mezcla de reacción se agita a esa temperatura durante 2,5 horas. La mezcla de reacción se vierte lentamente sobre agua enfriada en hielo y solución salina saturada (1:1, 12 l). La suspensión resultante se agita durante 2 horas. El sólido se filtra, se lava con agua (232 l), y se seca al vacío con una corriente de aire durante 3 días para dar el compuesto en forma de un sólido de color amarillo (318 g, 99 % de pureza, 87 %). MS (m/z): 226 (M H).
Preparación 13
1-Metil-7-metilsulfanil-4H-pirimido[4,5-d][1,3]oxazin-2-ona

Esquema 1, Etapa D: a una solución de trifenilfosfina (1,61 g, 6,08 mmol) y 7-metilsulfanil-1,4-dihidro-pirimido[4,5-d][1,3]oxazin-2-ona (1,00 g, 5,07 mmol) en THF (25 ml) se añade MeOH (0,248 ml, 6,08 mmol) seguido de la adición gota a gota de DIAD (1,21 ml, 6,08 mmol) a temperatura ambiente. Después de agitar durante una noche el disolvente se retira al vacío y el aceite de color amarillo resultante se purifica por cromatografía sobre gel de sílice (40-50 % de EtOAc/hexanos) para dar el compuesto del título en forma de un sólido de color blanco (1,08 g, 5,11 mmol, cuantitativo). MS (m/z): 212 (M H).
El siguiente compuesto se prepara esencialmente mediante el procedimiento de la Preparación 13.
Tabla 5

Preparación 15
1-Etil-7-(metilsulfonil)-1,4-dihidro-2H-pirimido[4,5-d][1,3]oxazin-2-ona

Esquema 1, Etapa E: a una solución en agitación de 1-etil-7-(metiltio)-1,4-dihidro-2H-pirimido[4,5-d][1,3]oxazin-2-ona (286 g, 1,24 mol) en ACN (2,8 l) y agua (1,4 l) se añade peroximonosulfato potásico (1526 g, 2,48 mol) en forma de un sólido durante 20 minutos, y la mezcla resultante se agita durante 16 horas a 10-20 °C. La mezcla de reacción se filtra y la torta de filtro obtenida se lava con CH2Cl2. El filtrado combinado y el CH2O2 se lavan con un 5 % de Na2SO3, agua, y solución salina saturada. La fase orgánica se seca sobre Na2SO4 y se concentra para proporcionar el compuesto del título (133,8 g, 93 % de pureza, 41 % de rendimiento). MS (m/z): 258 (M H).
El siguiente compuesto se prepara esencialmente mediante el procedimiento de la Preparación 15.
Tabla 6

Preparación Alternativa 15
1-Etil-7-(Metilsulfonil)-1,4-dihidro-2H-pirimido[4,5-d][1,3]oxazin-2-ona

Se combina 1-etil-7-metilsulfanil-4H-pirimido[4,5-d][1,3]oxazin-2-ona (550 g, 2,44 mol) con ACN (7 l) y agua (3 l). A esta solución a 12,5 °C se añade peroximonosulfato potásico (3070 g, 4,9 mol) en 10 porciones durante 100 minutos. Después de agitar a esta temperatura durante 3 horas, los sólidos se filtran y se lavan con DCM (4 l). Las capas se separan y la capa orgánica se lava secuencialmente con Na2SO3 al 5 % (3 l), agua (2 l) y solución salina saturada (2 l), y se seca sobre Na2SO4. Las capas orgánicas se filtran, se combinan, y se concentran a presión reducida. Después de la concentración de un 95 % del disolvente los sólidos precipitados se filtran para retirar cierta cantidad de impurezas de color. Los sólidos filtrados se lavan dos veces con ACN (2 x 500 ml) y se secan al vacío con una corriente de aire para obtener 340 g de producto en forma de un primer lote. Las aguas madre se concentran a presión reducida a sequedad. El residuo se tritura a continuación con una cantidad mínima de ACN y los sólidos se filtran para obtener una segunda cosecha de 34 g. Los lotes combinados dan el compuesto del título (374 g, 60 %) en forma de un sólido de color blanquecino. MS (m/z): 258 (M H).
Preparación 17
7-Metilsulfonil-1-[(1R)-1-metil-2-tetrahidropiran-2-iloxi-etil]-4H-pirimido[4,5-d][1,3]oxazin-2-ona

Esquema 1, Etapa I: una suspensión de 7-metilsulfaniM-[(1R)-1-metil-2-tetrahidropiran-2-iloxi-etil]-4H-pirimido[4,5-d][1,3]oxazin-2-ona (700 mg, 2,06 mmol) en CH2O 2 (10 ml) se trata con mCPBA (1,07 g, 4,5 mmol, calidad de reactivo al 70-75 %) a temperatura ambiente. Después de 20 minutos, los sólidos se filtran y se lavan con una cantidad adicional de CH2Cl2. Los filtrados orgánicos se combinaron y se lavan con NaHCO3 acuoso saturado (2 x) y solución salina saturada, se secan (Na2SO4), se filtran, y se concentran a sequedad. El material se purifica con cromatografía sobre gel de sílice para dar el compuesto del título en forma de un aceite incoloro (589 mg, 77 %). MS (m/z): 372 (M H).
Los siguientes compuestos se preparan esencialmente mediante el procedimiento de la Preparación 17.
Tabla 7

Preparación 22
(S)-(4-(1-Aminoetil)fenil)metanol

Esquema 2, Etapa J: a una solución en agitación de (S)-4-(1-aminoetil)benzoato de metilo (100 g, 558 mmol) en THF (2,2 l) se añade LiAlH4 (560 ml, 560 mmol, 1 M en THF) durante 1 hora, mientras se mantiene la temperatura de la reacción por debajo de 30 °C. La mezcla de reacción se agita durante 2 horas y a continuación se enfría a 0 °C. Se añade agua (100 ml) gota a gota seguido de Na2SO4 anhidro (1 kg), y la mezcla resultante se agita durante una noche. La mezcla se filtra sobre tierra de diatomeas y el sólido de color blanco resultante se seca al vacío para dar el compuesto del título (81,8 g, 97 %). MS (m/z): 135 (M - NH2).
Preparación 23
Clorhidrato de (1S)-1-[4-(clorometil)fenil]etanamina

Esquema 2, Etapa K: a una solución de (S)-(4-(1-aminoetil)fenil)metanol (81,8 g, 541 mmol) en CH2Ch (2,5 l) se añade SOCl2 (80 ml, 1,1 mmol) gota a gota durante 30 minutos mientras se mantiene la temperatura de reacción por debajo de 25 °C. El precipitado espeso que se forma se disuelve durante el curso de la reacción . Después de agitar
durante 4 horas, la mezcla se concentra para dar un sólido de color amarillo. Se añade ACN (1 l) y la mezcla se concentra a 500 ml y el sólido resultante se filtra para dar un sólido de color blanquecino que se seca al vacío. El producto se obtiene en forma de un sólido de color blanquecino después de secado y el licor madre también se puede concentrar para proporcionar producto menos puro (~20 g) en forma de un sólido de color amarillo. Los dos lotes de producto se combinan para dar el compuesto del título (111 g, 78 %). MS (m/z): 170 (M H).
Preparación 24
4-[[4-[(1S)-1-[(2,2,2-Trifluoroacetil)amino]etil]fenil]-metil]-piperazina-1-carboxilato de ferc-butilo

Esquema 2, Etapa L: a una solución de clorhidrato de (1S)-1-[4-(clorometil)fenil]etanamina (10 g, 48,5 mmol) en CH2Cl2 (160 ml) se añade anhídrido trifluoroacético (8,2 ml, 58 mmol) a 0 °C. Se añade TEA (15 ml, 108 mmol) mientras se mantiene la temperatura de adición por debajo de 5 °C. Después de agitar durante 1 hora a 0 °C la mezcla de reacción se concentra a sequedad y se añade ACN (120 ml) seguido de piperazina-1-il carbonato de fercbutilo (13,5 g, 72,5 mmol). Se añade K2CO3 (20 g, 144,7 mmol) y la mezcla se calienta a 60 °C y se agita durante 17 horas. El disolvente se retira al vacío y se añade EtOAc (1 l). El sólido se retira por filtración y el EtOAc se lava con agua y solución salina saturada. La fase orgánica se seca sobre Na2SO4, se filtra, y se concentra a sequedad. El producto en bruto resultante se purifica por cromatografía sobre gel de sílice (10 a 50 % de acetona/CH2Cl2) para dar el compuesto del título en forma de una espuma de color blanco (15,8 g, 78 %). MS (m/z): 416 (M H).
Preparación 25
4-[[4-[(1S)-1-Aminoetil]fenil]metil]piperazina-1-carboxilato de ferc-butilo

Esquema 2, Etapa M, subetapa 1: a una solución de 4-[[4-[(1S)-1-[(2,2,2-trifluoroacetil)amino]etil]fenil]-metil]-piperazina-1-carboxilato de ferc-butilo (203 g, 0,489 mol) en EtOH (2,4 l) se añade NaOH 5 M ac. (480 ml, 2,40 mol) a temperatura ambiente. Después de agitar a temperatura ambiente durante 3,5 horas, la mezcla de reacción se concentra para retirar la mayor parte del EtOH. Se añade EtOAc (2 l) para disolver el residuo y la solución se lava con agua y solución salina saturada. Las fases acuosas combinadas se extraen con EtOAc (2 x). Los extractos orgánicos combinados se secan sobre Na2SO4, se filtran, y se concentran a sequedad para dar el compuesto del título en bruto en forma de un aceite de color amarillo viscoso (156 g, 93 %) que se usa más adelante sin purificación adicional. MS (m/z): 320 (M H).
Preparación 26
4-[[4-[(1S)-1-[(1-isopropil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]metil]piperazina-1-carboxilato de ferc-butilo

Esquema 2, Etapa M, subetapa 2: A una solución de 4-[[4-[(1S)-1-aminoetil]fenil]metil]piperazina-1-carboxilato de ferc-butilo (46,7 g, 136 mmol) y 1-isopropil-7-metilsulfonil-4H-pirimido[4,5-d][1,3]oxazin-2-ona (40,6 g, 150 mmol) en DMSO (272 ml) se añaden CsF (20,7 g, 136 mmol) y DIPEA (23,7 ml, 136 mmol). La mezcla se agita a 75-80 °C durante 17 horas. A continuación, la mezcla se enfría a temperatura ambiente, se diluye con agua (1,2 l), se extrae con EtOAc (33), se lava con solución salina saturada, se seca (Na2SO4), se filtra, y se concentra a sequedad. El
producto en bruto resultante se purifica por cromatografía sobre gel de sílice (30 a 60 % de acetona/CH2Cl2) para dar el compuesto del título en forma de una espuma de color blanco (54 g, 78 %). MS (m/z): 511 (M H).
Preparación 27
Clorhidrato de 1-isopropil-7-[[(1S)-1-[4-(piperazin-1-ilmetil)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona

Esquema 2, Etapa N, subetapa 1: a una solución de 4-[[4-[(1S)-1-[(1-isopropil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]metil]piperazina-1-carboxilato de ferc-butilo (54 g, 106 mmol) en dioxano (270 ml) se añade HCl (264 ml, 4 M en dioxano), mientras se mantiene la temperatura interna por debajo de 35 °C. A continuación se añade MeOH (100 ml) para facilitar la agitación. La mezcla se agita a 30 °C durante 2 horas y a continuación se concentra para dar un sólido de color blanco que se seca al vacío. El producto del título se obtiene después de secado en forma de un sólido de color amarillo claro (60 g, cuantitativo) que se usa más adelante sin purificación adicional. MS (m/z): 411 (M H).
Preparación 28
4-[[4-[(1S)-1-[(1-Etil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]metil]piperazina-1-carboxilato de fercbutilo

Esquema 2, Etapa N, subetapa 1: a una solución de 4-[[4-[(1S)-1-aminoetil]fenil]metil]piperazina-1-carboxilato de ferc-butilo (52 g, 151,4 mmol) y 1-etil-7-metilsulfonil-4H-pirimido[4,5-d][1,3]oxazin-2-ona (42,8 g, 166 mmol) en DMSO (300 ml) se añaden CsF (23 g, 151,4 mmol) y DIPEA (26,5 ml, 152 mmol). La mezcla se agita a 75-80 °C durante 4 horas. La mezcla se enfría a temperatura ambiente, se diluye con agua (1,2 l), se extrae con EtOAc (33), se lava con solución salina saturada, se seca (Na2SO4), se filtra y se concentra. El producto en bruto se purifica por cromatografía sobre gel de sílice (50 a 75% de [10% de EtoH/CH2Ch]/hexanos) para dar una espuma de color blanco (53,3 g, 71 %). MS (m/z): 497 (M H).
Los siguientes compuestos se preparan esencialmente mediante el procedimiento de la Preparación 28.
Tabla 8
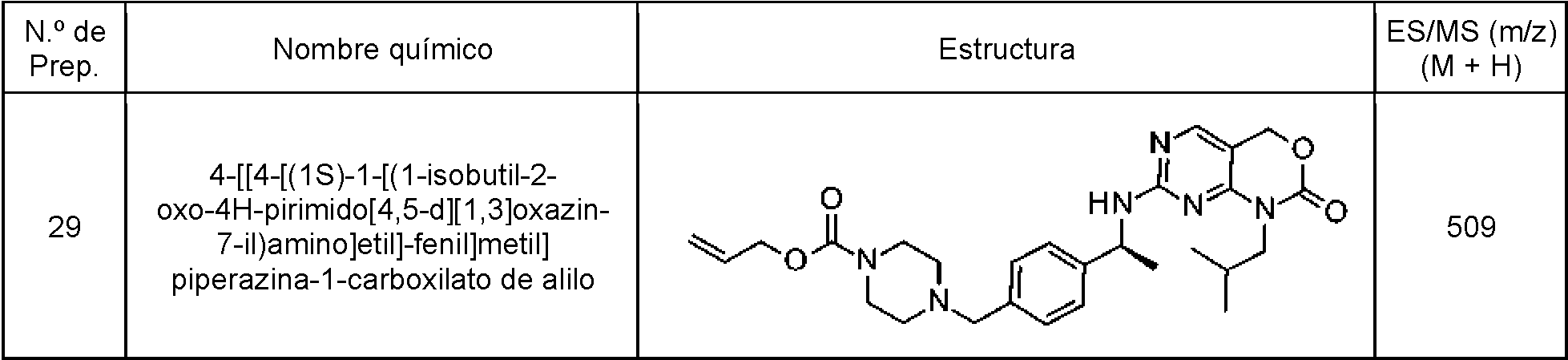
continuación

continuación

continuación

Preparación Alternativa 28
4-[[4-[(1S)-1-[(1-etil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]metil]piperazina-1-carboxilato de tercbutilo

Se añaden conjuntamente 4-[[4-[(1S)-1-aminoetil]fenil]metil]piperazina-1-carboxilato de terc-butilo (400 g, 1252 mmol) y DMSo (2,2 l). A esta solución se añaden de forma secuencial 1-etil-7-metilsulfonil-4H-pirimido[4,5-d][1,3]oxazin-2-ona (325 g, 1200,1 mmol), CsF (200 g, 1316,59 mmol) y DIPEA (240 ml, 1380 mmol). La mezcla de reacción se calienta a continuación a 75 °C durante 5 horas y se deja que se caliente a temperatura ambiente durante una noche con agitación suave. La mezcla de reacción se vierte lentamente sobre agua enfriada en hielo/solución salina saturada (1:1). Los sólidos precipitados se filtran, se lavan con agua (2 x 1,5 l), y se secan al vacío con una corriente de aire durante una noche. La torta húmeda obtenida (800 g) se toma para su uso posterior sin purificación adicional.
Preparación 46
Clorhidrato de 1-etil-7-[[(1S)-1-[4-(piperazin-1-ilmetil)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona

Esquema 2, Etapa N, subetapa 1: a una solución de 4-[[4-[(1S)-1-[(1-etil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]metil]piperazina-1-carboxilato de ferc-butilo (48,3 g, 97,3 mmol) en una mezcla de dioxano (140 ml) y MeOH (100 ml) se añade HCl (4 M en dioxano, 243 ml) durante 20 minutos mientras se mantiene una temperatura interna por debajo de 30 °C. La mezcla se agita a 30 °C durante 1 hora y a continuación se concentra a sequedad para proporcionar un sólido de color blanco que se seca al vacío durante una noche para dar el compuesto del título (54 g, cuantitativo). MS (m/z): 397 (M H).
Los siguientes compuestos se preparan esencialmente mediante el procedimiento de la Preparación 46.
Tabla 9

Preparación Alternativa 46
1-Etil-7-[[(1S)-1-[4-(piperazin-1-ilmetil)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona

Se disuelve 4-[[4-[(1S)-1-[(1-etil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]metil]piperazina-1-carboxilato de ferc-butilo (800 g, 1252 mmol teóricos) húmedo en bruto en 1,4-dioxano (2 l). Se añade HCl (3,6 l, 29,2 mol, acuoso 8 mol/l) durante aproximadamente 30 minutos con refrigeración a 15 °C seguido de la adición de DCM (1 l). Las capas se separan y la solución ácida se lava con DCM (2 x 500 ml). A la fase acuosa se añade Na2CO3 al 20 % hasta pH 9,5-10, a temperatura ambiente. Se añade DCM (1 l), las capas se separan, y la capa acuosa se extrae con DCM (2 x 500 ml). Los extractos orgánicos combinados se lavan con solución salina saturada, se secan sobre Na2SO4, y se concentran a sequedad hasta un aceite espumoso de color amarillo que se purifica a través de filtración sobre gel de sílice eluyendo con hexanos, DCM, EtOH de 90:9:1 a 70:29:1 para dar el compuesto del título (219 g, 53 % en 2 etapas) en forma de un sólido espumoso de color amarillo. MS (m/z): 397 (M H).
Preparación 49
Diclorhidrato de 4-[[4-[(1S)-1-aminoetil]fenil]metil]piperazina-1-carboxilato de alilo

Esquema 2, Etapa L, subetapa 2: un recipiente de presión se equipa con clorhidrato de (1S)-1-[4-(clorometil)fenil]etanamina (850 mg, 4,12 mmol), alloc-W-piperazina (1,43 g, 8,25 mmol), K2CO3 (1,71 g, l2,4mmol) y ACN (20,6 ml). La suspensión resultante se tapa y se calienta a 60 °C durante 24 horas y a continuación se concentra. El sólido de color blanco en bruto se purifica por cromatografía sobre gel de sílice usando 0 a 10% de NH37 N en MeOH / CH2O2 para proporcionar una mezcla del producto deseado y alloc-W-piperazina sin reaccionar. Este material se purifica de nuevo por cromatografía de fase inversa (0 a 100 % de ACN / NaHCO3 ac 10 mM) para dar el compuesto del título en forma de un aceite incoloro (900 mg, 71 %). MS (m/z): 171 (M H).
Los siguientes compuestos se preparan esencialmente mediante el procedimiento de la Preparación 49.

Preparación 52
1-Isobutil-7-[[(1S)-1-[4-(piperazin-1-ilmetil)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona

Esquema 2, Etapa N, subetapa 1, se disuelve 4-[[4-[(1S)-1-[(1-isobutil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]-fenil]metil]piperazina-1-carboxilato de alilo (1,15 g, 2,26 mmol) en THF (22,6 ml). Se añaden secuencialmente dimedona (3,27 g, 22,6 mmol) y Pd(PPh3)4 (131 mg, 0,113 mmol, 5 % en moles) y la mezcla se agita a temperatura ambiente durante 30 minutos. La mezcla de reacción se hace pasar sobre un cartucho SCX que se lava previamente con MeOH. Los reactivos no deseados se eluyen con MeOH y el producto deseado se eluye con NH32 N en MeOH. El eluyente con amoniaco se concentra para dar el compuesto del título en forma de un sólido de color blanco (980 mg, cuantitativo).
Preparación 53
3-[[4-[(1S)-1-aminoetil]fenil]metoxi]azetidina-1-carboxilato de tere-butilo

Esquema 2, Etapa L, subetapa 2: se disuelve 3-hidroxiazetidina-1-carboxilato de ferc-butilo (4,19 g, 23,7 mmol) en DMA (23,7 ml) y se añade NaH (1,4 g, 35,6 mmol, 60% en peso) a temperatura ambiente. Después de agitar durante 3 minutos, se añade clorhidrato de (1S)-1-[4-(clorometil)fenil]etanamina (2,44 g, 11,9 mmol) en una cantidad mínima de DMA y la reacción se deja en agitación durante 1 hora. La mezcla se diluye con solución salina semisaturada y se extrae con EtOAc (3 x). Los extractos orgánicos se combinan, se secan (MgSO4), se filtran y se concentran para proporcionar el producto en bruto en forma de un aceite de color amarillo que contiene DMA residual. El producto se usa más adelante sin purificación adicional (supuesto un 100 % de rendimiento): MS (m/z): 307 (M H).
Preparación 54
7-[[(1S)-1-[4-(Azetidin-3-iloximetil)fenil]etil]amino]-1-etil-4H-pirimido[4,5-d][1,3]oxazin-2-ona

Esquema 2, Etapa N, subetapa 1: se disuelve 3-[[4-[(1S)-1-[(1-etil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]metoxi]azetidina-1-carboxilato de ferc-butilo (1,85 g, 3,83 mmol) en CH2Cl2 (29 ml) y se añade TFA (2,89 ml, 38,3 mmol) a 0 °C y se agitó durante 6 horas mientras que el baño de refrigeración se extingue a temperatura ambiente. El disolvente y el ácido se retiraron al vacío para dar el compuesto del título en forma de un aceite de color amarillo claro (3,66 g, 99 %). MS (m/z): 384 (M H).
Los siguientes compuestos se preparan esencialmente mediante el procedimiento de la Preparación 54.

continuación

continuación

continuación

Preparación 72
7-[[(1S)-1-[4-(Cloromet¡l)fen¡l]et¡l]am¡no]-1-¡sobut¡l-4H-p¡r¡m¡do[4,5-d][1,3]oxaz¡n-2-ona

Esquema 3, Etapa Q: se añade SOCl2 (2,1 ml, 4,2 mmol) med¡ante una jer¡nga a una suspens¡ón de K2CO3 (776 mg, 5,61 mmol) y 7-[[(1S)-1-[4-(h¡drox¡met¡l)fen¡l]et¡l]am¡no]-1-¡sobut¡l-4H-p¡r¡m¡do[4,5-d][1,3]oxaz¡n-2-ona (500 mg, 1,40 mmol) en CH2Cl2 (14 ml). Después de 5 horas, el d¡solvente se ret¡ra al vacío para proporc¡onar el cloruro bencíl¡co en bruto en forma de una mezcla conjunta con K2CO3 (supuesto un 100 % de rend¡m¡ento). Esta mezcla se usa más adelante s¡n pur¡f¡cac¡ón ad¡c¡onal. MS (m/z): 375 (M H).
Los s¡gu¡entes compuestos se preparan esenc¡almente med¡ante el proced¡m¡ento de la Preparac¡ón 72.
Tabla 12

Preparación 75
6-[[4-[(1S)-1-[(1-isobutil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]metil]-2,6-diaza-espiro[3.3]heptano-2-carboxilato de ferc-butilo

Esquema 3, Etapa Q: se suspenden 7-[[(1S)-1-[4-(clorometil)fenil]etil]amino]-1-isobutil-4H-pirimido[4,5-d][1,3]oxazin-2-ona (520 mg, 1,4 mmol), K2CO3 (580 mg, 4,2 mmol) y sal de oxalato de 2,6-diazaespiro[3.3]heptano-2-carboxilato de ferc-butilo (550 mg, 1,8 mmol) en ACN (7 ml) y se calientan a 50 °C durante 16 horas y a continuación a 70 °C durante 2 horas. Se añade yoduro sódico (210 mg, 1,4 mmol) y la reacción se calienta a 70 °C durante 2 horas y a continuación a reflujo durante 3 horas. La mezcla se enfría a temperatura ambiente y el disolvente se evapora. El residuo resultante se recoge en CH2Ch y se lava con NaHCO3 acuoso saturado. La capa acuosa se extrae con CH2Cl2 (3 x). Los extractos orgánicos combinados se lavan con solución salina saturada, se secan (Na2SO4), se filtran y se concentran para proporcionar el producto en bruto en forma de un sólido de color amarillo. El producto en bruto se usa más adelante en la siguiente etapa sin purificación adicional (supuesto un 100 % de rendimiento). MS (m/z): 537 (M H).
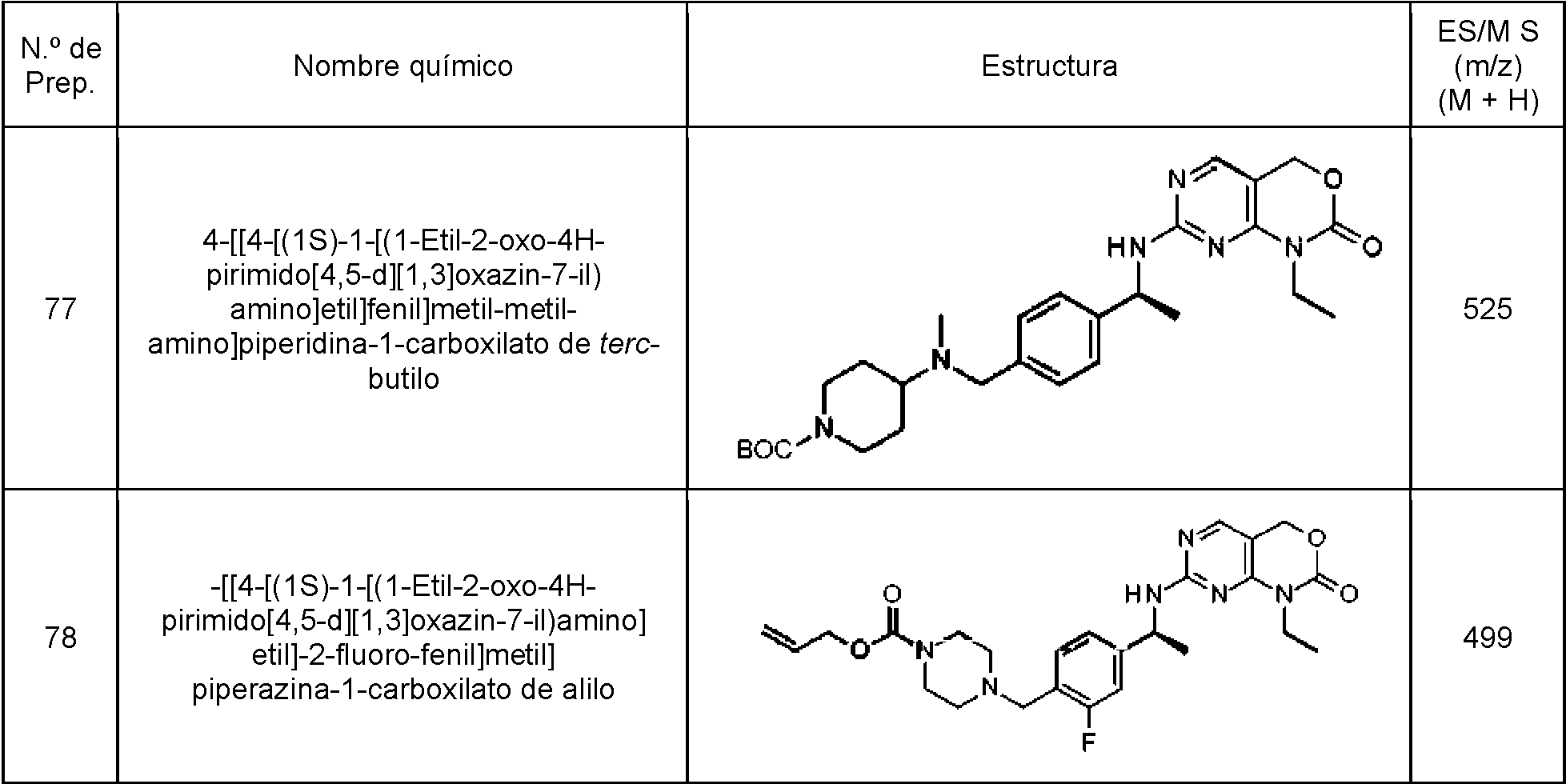
Los siguientes compuestos se preparan esencialmente mediante el procedimiento de la Preparación 75.
Tabla 13

continuación
Preparación 79
4-[4-[(1S)-1-[(1-Isopropil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]piperazina-1-carboxilato de tercbutilo

Esquema 3, Etapa P: se disuelven 7-[[(1S)-1-(4-bromofenil)etil]amino]-1-isopropil-4H-pirimido[4,5-d][1,3]oxazin-2-ona (600 mg, 1,53 mmol), 3-(hidroximetil)azetidina-1-carboxilato de terc-butilo (400 mg, 2,1 mmol), CuBr (100 mg, 0,70 mmol), K2CO3 (450 mg, 3,2 mmol), y L-hidroxiprolina (500 mg, 4 mmol) en DMSO (15 ml) y se calientan a 80 °C durante una noche. La mezcla de reacción se enfría a temperatura ambiente y se vierte sobre agua, se extrae en EtOAc, y se concentra. El producto en bruto se purifica por cromatografía (50 % de EtOAc/hexanos) para dar el compuesto del título en forma de una espuma (761 mg, 46 %). MS (m/z): 497 (M H).
El siguiente compuesto se prepara esencialmente mediante el procedimiento de la Preparación 79.
Tabla 14

Preparación 81
3-Etinilazetidina-1-carboxilato de ferc-butilo

Se suspende azida sódica (2,4 g, 37 mmol) en ACN (14 ml) y se añade cloruro de metanosulfonilo (2,7 ml, 35 mmol) durante 45 segundos. La mezcla resultante se agita durante una noche a temperatura ambiente y a continuación se enfría 0 °C, momento en el que se añade (2-oxopropil)fosfonato de dimetilo (4,3 ml, 31 mmol) durante 30 segundos seguido de Cs2CO3 (11 g, 34 mmol). Esta mezcla se agita durante 30 minutos a 0 °C y a continuación a temperatura ambiente durante 2,5 horas. La mezcla se enfría de nuevo a 0 °C y se añade MeOH (15,5 ml). Después de 1 hora, se añade 3-formilazetidina-1-carboxilato de ferc-butilo (3,0 g, 16 mmol) seguido de una cantidad adicional de Cs2CO3 (9,1 g, 28 mmol) y 25 minutos más tarde se retira el baño de hielo-agua y la reacción se agita durante una noche. El disolvente se retira al vacío para dar un aceite de color naranja que se purifica por cromatografía sobre gel de sílice usando 50 % de MTBE/hexanos. El compuesto del título se obtiene en forma de un aceite de color amarillo claro (2,68 g, 93 %). MS (m/z): 181 (M H).
Preparación 82
3-[2-[4-[(1S)-1-[(1-Etil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]etinil]azetidina-1-carboxilato de fercbutilo

Esquema 3, Etapa P: se hace burbujear nitrógeno a través de una solución de 7-[[(1S)-1-(4-bromofenil)etil]amino]-1-etil-4H-pirimido[4,5-d][1,3]oxazin-2-ona (1,8 g, 4,77 mmol) y TEA (9,98 ml, 71,6 mmol) en THF (24 ml) durante 5 minutos. Se añaden tetraquis(trifenilfosfina)paladio (0) (455 mg, 0,382 mmol, 8 mol%) y Cul (18 mg, 0,095 mmol, 2 mol%) y se continúa la desgasificación durante un período adicional de 5 minutos. Se añade 3-etinilazetidina-1-carboxilato de ferc-butilo (1,26 g, 6,68 mmol) y la desgasificación se repite durante 20 segundos. La reacción se calienta a 60 °C y se agita durante ~24 horas, y a continuación se retirar el calor y la agitación se continúa durante un período adicional de 24 horas. La mezcla se diluye con agua, se extrae con CH2O2 (33), se seca (MgSO4), se filtra y se concentra para dar el producto en bruto que se purifica por cromatografía sobre gel de sílice eluyendo con un 25-85 % de EtOAc/hexanos para dar una espuma de color amarillo (1,14 g, 50 %). MS (m/z): 478 (M H).
Preparación 83
3-[2-[4-[(1S)-1-[(1-Etil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]etil]azetidina-1-carboxilato de ferc-butilo

Esquema 3, Etapa P: se agitan 3-[2-[4-[(1S)-1-[(1-etil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]etinil]azetidina-1-carboxilato de ferc-butilo (1,14 g, 2,39 mmol) y Pd/CaCO3 (1,67 mg, 10 % en peso Pd, 0,157 mmol) en THF (24 ml) en una atmósfera de hidrógeno durante 17 horas. La mezcla de reacción se filtra sobre tierra de diatomeas y los sólidos se lavan con EtOAc/MeOH caliente. El producto en bruto se cromatografía sobre gel de sílice eluyendo con un 25-100% de EtOAc/hexanos para dar el producto del título en forma de una
espuma de color amarillo claro (890 mg, 77 %). MS (m/z): 482 (M H).
Preparación 84
N-[(1S)-1-(4-Hidroxifenil)etil]carbamato de bencilo

Esquema 4: se añade gota a gota cloroformiato de bencilo (3,20 ml, 20,6 mmol) a una mezcla de (S)-4-(1-aminoetil)fenol (2,75 g, 19,6 mmol) y NaHCO3 (2,06 g, 24,6 mmol) en una mezcla de THF (20 ml) y agua (20 ml) a 0 °C. El baño de hielo-agua se retira después de 30 minutos y la mezcla de reacción se agita a temperatura ambiente durante 72 horas. La mezcla se diluye con EtOAc y la fase orgánica se separa, se lava con HCl acuoso 0,1 N y solución salina saturada. La fase orgánica se seca (Na2SO4), se filtra, y se concentra y el material en bruto resultante se purifica sobre gel de sílice (0 a 50 % de EtOAc/hexanos) para dar el producto del título en forma de un sólido de color blanco (3,84 g, 71 %). MS (m/z): 272 (M H).
Preparación 85
3-[[4-[(1S)-1-(Benciloxicarbonilamino)etil]fenoxi]-metil]azetidina-1-carboxilato de ferc-butilo

Esquema 4, Etapa R: a 0 °C, se añade gota a gota DIAD (980 ml, 5 mmol) durante 15 minutos a una solución de 3-(hidroximetil)azetidina-1-carboxilato de ferc-butilo (750 mg, 4 mmol), PPh3 (1,3 g, 5 mmol) y N-[(1S)-1-(4-fenil)etil]carbamato de bencilo (1,4 g, 1,25 mmol) en THF (16 ml). Después de que se haya completado la adición, se retira el baño de hielo-agua y la reacción se agita durante 4,5 horas a temperatura ambiente. La reacción se diluye a continuación con EtOAc, se lava con agua, NaOH acuoso 0,5 N, y solución salina saturada, se seca (Na2SO4), se filtra y se concentra. El producto se purifica sobre gel de sílice eluyendo con un 0 a un 50 % de EtOAc/hexanos para dar el producto contaminado con un 24 % del fenol de partida. El protocolo anterior se repite en esta mezcla para dar el producto deseado en forma de un aceite (1,12 g, 64 %). MS (m/z): 341 (M - BOC H), 463 (M Na).
Preparación 86
3-[[4-[(1S)-1-Aminoetil]fenoxi]metil]azetidina-1-carboxilato de ferc-butilo

Esquema 4, Etapa M, Subetapa 1: se agitan 3-[[4-[(1S)-1-(benciloxicarbonilamino)etil]fenoxi]-metil]azetidina-1-carboxilato de ferc-butilo (1,12 g, 2,54 mmol) y Pd al 10%/C (335 mg, 0,315 mmol) en EtOH (60 ml) en una atmósfera de hidrógeno (138 kPa) durante 45 minutos. La mezcla de reacción se filtra y se concentra para proporcionar un aceite que se usa más adelante sin purificación adicional (supuesto un 100 % de rendimiento). MS (m/z): 290 (M - NH2).
Preparación 87
3-[[4-[(1S)-1-[(1-Etil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenoxi]metil]-2,5-dihidropirrol-1-carboxilato de ferc-butilo
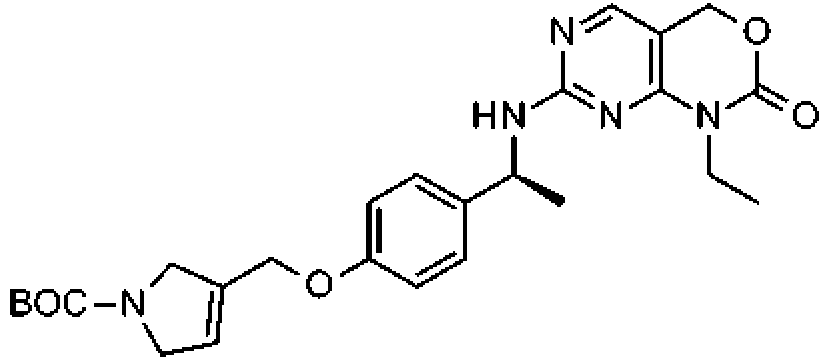
Esquema 3, Etapa P: una solución de 1-etil-7-[[(1S)-1-(4-hidroxifenil)etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona (0,330 g, 1,05 mmol), 3-(dorometN)-2,5-dihidropirroM-carboxNato de tere-butilo (200 mg, 0,9 mmol) y K2CO3 (400 mg, 3 mmol) en acetona (7 ml) se calienta a 60 °C en un tubo cerrado herméticamente durante 16 h y a continuación se deja en agitación a temperatura ambiente durante 72 h. La solución se diluye con agua y se extrae dos veces con DCM. Las capas orgánicas combinadas se secan (Na2SO4), se filtran y se concentran al vacío. El producto en bruto se purifica por cromatografía de fase inversa (0 a 100 % de CH3CN/agua) para proporcionar el compuesto del título en forma de un sólido de color naranja (250 mg, 0,504 mmol, 48 %). MS (m/z): 496 (M H). Preparación 88
6-[[4-[(1S)-1-[(1-isopropil-2-oxo-4H-pirimido[4,5-d][1,3]oxazin-7-il)amino]etil]fenil]metil]-2,6-diazaespiro[3.3]heptano-2-carboxilato de tere-butilo

Esquema 3, Etapa P: se añade DIPEA (0,75 ml, 4,3 mmol) a una solución de 7-[[(1S)-1-[4-(clorometil)fenil]etil]amino]-1-isopropil-4H-pirimido[4,5-d][1,3]oxazin-2-ona (1 g, 2,8 mmol) y 2,6 diazaespiro[3.3]heptano-2-carboxilato de tere-butilo (820 mg, 4,1 mmol) en DMSO (14 ml) y la mezcla resultante se calienta a 50 °C durante 16 horas y a continuación se agita a temperatura ambiente durante 72 horas. La mezcla se vierte sobre agua y se extrae con EtOAc, se seca (MgSO4), se filtra y se concentra. El producto en bruto se purifica sobre gel de sílice (5-7 % de NH37 N en MeOH/C^Ch) para dar el compuesto del título (978 mg, 68 %). m S (m/z): 523 (M H).
Ejemplo 1
1-Isopropil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona

Esquema 2, Etapa N: a una solución de clorhidrato de 1-isopropil-7-[[(1S)-1-[4-(piperazin-1-ilmetil)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona (59,6 g, 113 mmol) y TeA (47,5 ml, 341 mmol) en CH2Cl2 (1,13 l) se añade cloruro de acriloílo (11,1 ml en 80 ml de CH2Cl2, 136 mmol) a -75 °C durante 15 minutos. La temperatura de reacción se mantiene por debajo de -70 °C durante esta adición. Después de 30 minutos, la reacción se interrumpe con NaHCO3 acuoso saturado (100 ml) y la mezcla se deja calentar a temperatura ambiente. Se añade agua (200 ml) y la mezcla se extrae con CH2O2 (3 x), se seca (Na2SO4), se filtra y se concentra para producir una espuma de color amarillo. El producto en bruto se purifica por cromatografía sobre gel de sílice (6 a 10% de [10% de MeOH/CH2Cl2]/EtOAc) para dar el producto del título en forma de una espuma de color blanco (42,5 g, 81 %). MS (m/z): 465 (M H).
Ejemplo 2
1-Etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona

Esquema 2, Etapa N: a una solución de clorhidrato de 1-etil-7-[[(1S)-1-[4-(piperazin-1-ilmetil)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona (53,6 g, 124 mmol) y TEA (52 ml, 373 mmol) en CH2Cl2 (1,2 l) se añade cloruro de acriloílo (12,1 ml en 80 ml de CH2Cl2, 149 mmol) a -75 °C durante 15 minutos. La temperatura de reacción se mantiene por debajo de -70 °C durante esta adición. Después de 1 hora la reacción se interrumpe con NaHCO3 acuoso saturado (100 ml) y la mezcla posterior se deja calentar a temperatura ambiente. Se añade agua (200 ml) y la mezcla se extrae con CH2Cl2 (33), se seca (Na2SO4), se filtra y se concentra para dar una espuma de color amarillo. El producto en bruto se purifica por cromatografía sobre gel de sílice (6 a 10% de [10% de MeOH/CH2Cl2]/EtOAc) para dar el producto del título en forma de una espuma de color amarillo claro (30,2 g, 54 %). MS (m/z): 451 (M H).
Los siguientes Ejemplos se preparan esencialmente mediante el procedimiento del Ejemplo 2.
Tabla 15

continuación

continuación

continuación

Ejemplo 23
Sal de ácido 4-hidroxibenzoico de 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona
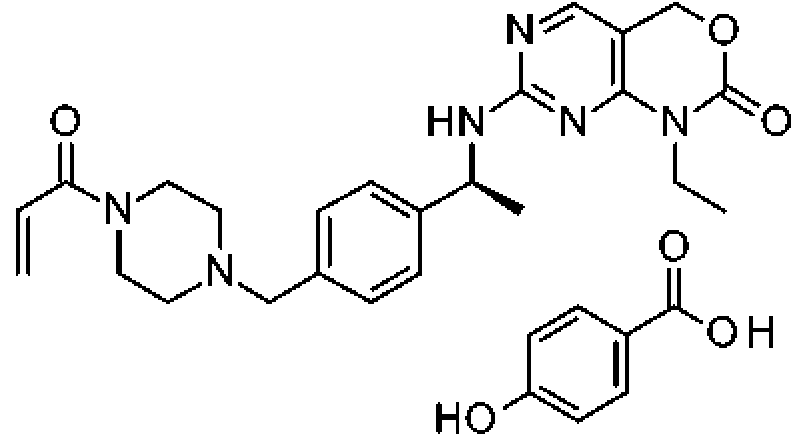
A una solución de 1-etil-7-[[(1S)-1-[4-(piperazin-1-ilmetil)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona (376 g, 758,6 mmol) en DCM (6700 ml) se añade TEA (116 ml, 832 mmol). La mezcla se enfría a menos de -70 °C. Se añade una solución de cloruro de acriloílo (64,8 ml, 796 mmol) en DCM (750 ml) a una temperatura de menos de -68 °C durante 2 horas. Después de la adición, la reacción se agita a menos de -68 °C durante 15 minutos. Se añade agua (1500 ml) y la solución se calienta a temperatura ambiente. Las capas se separan. La capa orgánica se lava con agua (231 l), NaHCO3 saturado 50 % (1 l) y solución salina saturada (1 l), y se seca sobre Na2SO4. Esta solución se añade a una solución de ácido 4-hidroxibenzoico (115 g, 832,61 mmol) en CPME (1800 ml). La mezcla turbia resultante se calienta a 35-40 °C para dar como resultado una solución de color amarillo que se filtra a través de tierra de diatomeas y se lava con una mezcla de DCM y CPME (1:1). El filtrado y los lavados se combinan y se
concentran hasta 1/4 de volumen. La suspensión resultante se evapora conjuntamente con heptano (231 l) y se filtra, se lava con heptano y se seca en una atmósfera de vacío de N2 durante una noche para dar el compuesto del título (382,7 g, 81 %). MS (m/z): 451 (M H). El 4-hidroxibenzoato de 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-io-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona en bruto (624 g, 1060 mmol) se suspende en un disolvente mezclado previamente de acetona (9,4 l) y heptano (9,4 l) y la suspensión se agita a temperatura ambiente durante 5 horas. El sólido se filtra y se lava con heptano/acetona 1:1 (3 x 500 ml). El filtrado y los lavados se evaporan. Al residuo se añade heptano (2 l) y se filtra, y el sólido recogido se seca al vacío a 50 °C para dar el compuesto del título en forma de un sólido de color amarillo claro (357 g, 55 %).
Preparación alternativa del Ejemplo 23
Sal de ácido 4-hidroxibenzoico de 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona cristalina
A una solución de 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona (28,5 g, 63,3 mmol) en CPME (430 ml) a 60 °C en una atmósfera de nitrógeno se añade ácido 4-hidroxibenzoico (8,74 g, 63,3 mmol) disuelto en CPME (115 ml). Después de unos pocos minutos de agitación a 300 rpm a 60 °C, la agitación se disminuye a 50 rpm y la muestra se agita durante 2 horas. En este momento la temperatura se eleva a 45 °C, y la muestra se agita a 200 rpm durante 4 horas. La muestra se lleva a continuación a temperatura ambiente. Las paredes del matraz se raspan para retirar sólido y el sólido se recoge por filtración y se seca con aire para dar la primera cosecha. El filtrado se reduce al vacío hasta aproximadamente la mitad del volumen y se enfría a mientras se agita en un baño de hielo para proporcionar más sólido. Este sólido se recoge y se añade a la primera cosecha. El producto se seca en a un horno de vacío a 70 °C durante una noche para dar el compuesto del título (33,4 g, 56,7 mmol, 89,7 %). MS (m/z): 451 (M H).
Difracción de rayos X de polvo (XRD)
Los patrones de XRD de los sólidos cristalinos se obtienen en un difractómetro de rayos X de polvo Bruker D4 Endeavor, equipado con una fuente de CuKa de A = 1,54060 A) y un detector Vantec, que funciona a 35 kV y 50 mA. Se realiza el barrido de la muestra entre 4 y 40° en 20, con un tamaño de etapa de 0,009° en 20 y una velocidad de barrido de 0,5 segundos/etapa, y con una divergencia de 0,6 mm, anti-dispersión fija 5,28, y rendijas del detector de 9,5 mm. El polvo seco se empaqueta en un soporte de muestra de cuarzo y se obtiene una superficie lisa mediante un portaobjetos de vidrio. Los patrones de difracción de la forma cristalina se recogen a temperatura y humedad relativa ambientes. Se conoce bien en la técnica de la cristalografía que, para cualquier forma cristalina dada, las intensidades relativas de los picos de difracción pueden variar debido a la orientación preferente resultante de factores tales como morfología y hábito de los cristales. Cuando los efectos de la orientación preferente están presentes, las intensidades máximas se alteran, pero las posiciones máximas características del polimorfo no cambian. Véase, por ejemplo, The United States Pharmacopeia #23, National Formulary #18, páginas 1843-1844, 1995. Además, también se conoce en la técnica de la cristalografía que para cualquier cristal dado la posición angular máxima puede variar ligeramente. Por ejemplo, las posiciones de pico pueden cambiar debido a una variación en la temperatura o la humedad a la que se analiza la muestra, el desplazamiento de la muestra o la presencia o ausencia de un estándar interno. En el presente caso, una variabilidad de posición máxima de ± 0,2 en 20 tendrá en cuenta estas variaciones potenciales sin obstaculizar la identificación inequívoca de la forma cristalina indicada. La confirmación de una forma cristalina se puede hacer en base a cualquier combinación única de picos distintivos (en unidades de ° 20), por lo general los picos más prominentes. Los patrones de difracción de la forma cristalina, recogidos a temperatura y humedad relativa ambientes, se ajustan en base a los picos estándar NIST 675 a 8,853 y 26,774 grados 2-theta.
Una muestra preparada del Ejemplo 23 se caracteriza por un patrón de XRD usando radiación CuKa que tiene unos picos de difracción (valores 2-theta) como se describen en la siguiente Tabla 16 y, en particular, que tiene picos a 16,8 en combinación con uno o más de los picos seleccionados entre el grupo que consiste en 15,8, 13,9, y 13,4; con una tolerancia para los ángulos de difracción de 0,2 grados.

continuación

Ejemplo 24
Sal de bis(ácido 2,5-dihidroxibenzoico) de 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona cristalina

A una solución de 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona (3,94 g, 8,73 mmol) en EtoH (7 ml) con agitación a 1000 rpm a 48 °C se añade ácido 2,5-dihidroxibenzoico (350 mg, 17,8 mmol) disuelto en EtOH (8 ml) para dar una solución de color amarillo oscuro. La muestra se deja enfriar a temperatura ambiente, y la agitación se reduce a 300 rpm. Esto da como resultado un sólido de color blanco que se aísla por filtración al vacío. El vial se aclara con EtOH (10 ml) que se añade a la torta. La torta se seca en un lugar con una corriente de nitrógeno durante 10 minutos, y a continuación durante una noche en un ambiente de horno de vacío (6,34 g, 8,35 mmol, 95,62 %). MS (m/z): 451 (M H).
Una muestra preparada del Ejemplo 24 se caracteriza por un patrón de XRD usando radiación CuKa que tiene unos picos de difracción (valores 2-theta) como se describen en la siguiente Tabla 17 y, en particular, que tiene picos a 24,7 en combinación con uno o más de los picos seleccionados entre el grupo que consiste en 17,0, 24,0, y 19,8; con una tolerancia para los ángulos de difracción de 0,2 grados.
T l 1 i XRD l E m l 24

Ejemplo 25
Sal de bis(sacarina) de 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona cristalina

A una solución de 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona (955 mg, 2,12 mmol) en EtOH (3 ml) con agitación de 1000 rpm a 70 °C se añade sacarina (862 mg, 4,66 mmol) en EtOH (7 ml) para dar una suspensión opaca de color blanco. Se forma una goma de color blanco, y se añade una cantidad adicional de EtOH (10 ml). La muestra se agita durante 30 minutos para convertir la goma en una suspensión de un sólido de color blanco. La muestra se lleva a temperatura ambiente y el sólido de color blanco se aísla por filtración al vacío (1,61 g, 1,97 mmol, 93,0 %).
Una muestra preparada del Ejemplo 25 se caracteriza por un patrón de XRD usando radiación CuKa que tiene unos picos de difracción (valores 2-theta) como se describen en la siguiente Tabla 18 y, en particular, que tiene picos a 19,1 en combinación con uno o más de los picos seleccionados entre el grupo que consiste en 22,0, 16,8, y 23,7; con una tolerancia para los ángulos de difracción de 0,2 grados.

El cáncer se reconoce cada vez más como una colección heterogénea de enfermedades cuyo inicio y progresión son inducidos por la función anómala de uno o más genes que regulan la reparación del ADN, la estabilidad del genoma, la proliferación celular, la muerte celular, la adhesión, la angiogénesis, la invasión y la metástasis en las células y microambientes de tejidos. La función variante o anómala de los genes de "cáncer" puede ser el resultado de un polimorfismo de ADN natural, cambios en el número de copias del genoma (a través de amplificación, deleción, pérdida de cromosomas, o duplicación), cambios en la estructura de genes y cromosomas (a través de la translocación cromosómica, inversión u otro reordenamiento que conduce a la expresión de genes desregulados) y mutaciones puntuales. Las neoplasias cancerosas pueden ser inducidas por una función genética anómala y pueden ser mantenidas por la misma función genética anómala, o el mantenimiento y la progresión exacerbada por funciones genéticas anómalas adicionales.
Más allá de las anomalías cromosómicas genéticas mencionadas anteriormente, cada uno de los cánceres también puede incluir modificaciones epigenéticas del genoma, incluyendo la metilación del ADN, la impronta genómica y la modificación de histonas por acetilación, metilación o fosforilación. Una modificación epigenética puede desempeñar un papel en la inducción y/o mantenimiento de la neoplasia maligna.
Se han compilado catálogos extensos de las anomalías citogenéticas con respecto a cáncer humano y se mantienen y actualizan regularmente en línea (consulte The Mitelman Database of Chromosome Aberrations in Cancer en el
sitio Web US del National Cáncer Institute (NCI) Cáncer Genome Anatomy Project (CGAP)). El Wellcome Trust Sanger Institute Cancer Genome Project mantiene un "Censo genético del cáncer" en línea detallado de todos los genes humanos que se han relacionado causalmente con la tumorigénesis, así como en la base de datos COSMIC (Catalogue of Somatic Mutations in Cancer) de mutaciones somáticas en cáncer humano. Una fuente adicional que contiene abundante información sobre los cambios citogenéticos vinculados de forma causal con varios tipos de cáncer es el Atlas of Genetics and Cytogenetics in Oncology and Haematology.
El diagnóstico de neoplasias malignas cancerosas por biopsia, inmunofenotipado y otros ensayos se conocen y se usan de manera rutinaria. Además de las bandas cromosómicas de alta resolución y las tecnologías avanzadas de imágenes cromosómicas, las anomalías cromosómicas en casos sospechosos de cáncer se pueden determinar mediante un análisis citogenético a través de la hibridación fluorescente in situ (FISH), cariotipado, cariotipado espectral (SKY), FISH multiplex (M-FISH), hibridación genómica comparativa (CGH), matrices de polimorfismo de un solo nucleótido (Chips de SNP) y otros ensayos de diagnóstico y análisis conocidos y usados por los expertos en la materia.
Las mutaciones en IDH1 se han identificado en múltiples tipos de tumores cancerosos que incluyen, pero no se limitan a, glioma, glioblastoma multiforme, astrocitomas, oligodendrogliomas, paraganglioma, síndrome mielodisplásico (MDS), leucemia linfoblástica aguda de linfocitos B (B-ALL), tiroides, colorrectal, leucemia mieloide aguda (AML), Dang y col., Trends Mol. Med., 2010, 16: 387-397; Ward y col., Oncogene, 2012, 31 (19): 2491-2498; melanoma, Shibata y col., Am. J. Pathol., 2010, 178 (3): 1395-1402; próstata, Flaherty y col., J. Clin. Oncol., 2014, 32 (supl. 4; Resumen 213); Cairns y col., Cancer Discovery, 2013, 3: 730-741; condrosarcoma y colangiocarcinoma, Balss y col., Acta Neuropathol., 2012, 124: 883-891; Cairns y col., Cancer Discovery, 2013, 3: 730-741; linfoma de linfocitos T angioinmunoblástico (AITL), Cairns y col., Blood, 2012. 119 (8): 1901-1903. Se han encontrado mutaciones en o cerca de restos particulares en el sitio activo: G97D, R100, R132h , R132C, R132S, R132V, R132G, V71I, R132L, y G123R para IDH1, Dang y col., Trends Mol. Med., 2010, 16: 387-397; Ward y col., 2012 yen la Tabla 2 Suplementaria.
Se ha demostrado que las formas mutantes de IDH1 tienen una actividad neomórfica (aumento de función) que reduce el a-cetoglutarato a 2-hidroxiglutarato. La producción endógena de 2-hidroxiglutarato es enantioespecífica, lo que da como resultado la generación del enantiómero D (también denominado enantiómero (R). Normalmente, las células tienen niveles bajos de 2-hidroxiglutarato mientras que las células que albergan mutaciones IDH1 evidencian niveles significativamente elevados de 2-hidroxiglutarato. Se detectan niveles significativamente elevados de 2-hidroxiglutarato en tumores que albergan las mutaciones y en plasma de pacientes con IDH1 mutante. Los altos niveles de 2-hidroxiglutarato se asocian con un fenotipo de hipermetilación que da como resultado un bloqueo y diferenciación que conduce a un aumento de la tumorigénesis.
La actividad de un inhibidor covalente irreversible específico se define por su unión a la diana (IDH1), definida por Ki, y la tasa potencial máxima de formación de enlaces covalentes, definida por kinact. Estos dos factores no son entidades separadas, sino que trabajan juntos para producir el efecto deseado de la formación de enlaces covalentes. Esto se ilustra en los 3 puntos que siguen a continuación.
En primer lugar, el hecho de que un electrófilo por ejemplo, acrilamida, se deba colocar de forma adecuada en relación con un nucleófilo por ejemplo, cisteína, es un componente fundamental de la formación de enlaces covalentes en química orgánica. Hay un ángulo preciso y una distancia a la que el nucleófilo debe acercarse al electrófilo para formar el enlace covalente. La simple colocación de un electrófilo cerca de un nucleófilo no es suficiente para la formación de enlaces covalentes.
En segundo lugar, cuando se incorpora un grupo reactivo en un núcleo que contiene restos de unión de hidrógeno para estabilizar la unión del inhibidor a la enzima, por ejemplo, un núcleo de orientación, alguien con experiencia debe considerar cómo el núcleo que se orienta se une a la diana y coloca el electrófilo en relación con el nucleófilo a la vista del ángulo óptimo y la distancia mencionados anteriormente. de nuevo, la simple colocación de un electrófilo cerca de un nucleófilo no es suficiente para la formación de enlace covalente. Los cambios en la orientación del núcleo pueden influir en la capacidad de un compuesto inhibidor para formar un enlace covalente.
En tercer lugar, cuando los dos puntos anteriores se consideran en conjunto, la simple presencia de un resto electrófilo en núcleo de orientación no es suficiente para sugerir que se formará un enlace covalente.
Los siguientes estudios in vitro e in vivo demuestran la actividad inhibidora y la eficacia de la proteína IDH1 mutante de los compuestos de Fórmula I sometidos a ensayo frente a diversas líneas de células cancerígenas específicas. Estos ensayos son generalmente reconocidos por los expertos en la materia como indicativos de la actividad terapéutica clínica humana. Se cree que la inhibición de las proteínas neomórficas IDH1 mutantes en los estudios desvelados en los estudios divulgados serán eficaces frente a otras proteínas neomórficas IDH1 mutantes. Los ensayos que ponen en evidencia de la actividad inhibidora y la eficacia de IDH1 mutante se pueden llevar a cabo sustancialmente de la siguiente manera o mediante ensayos similares proporcionando datos similares.
Los resultados de los siguientes ensayos demuestran que los compuestos usados a modo de ejemplo y sometidos a ensayo son útiles como inhibidores mutantes de IDH1 y pueden ser útiles en el tratamiento de cánceres que
expresan IDH1 mutante.
Ensayos bioquímicos para enzimas mutantes IDH1 e IDH2.
Las enzimas mutantes IDH1-R132H, IDH1-R132C, IDH2-R172K e IDH2-R140Q catalizan la conversión de aKG a 2HG. El 2HG se analiza mediante extracción en fase sólida en línea y espectrometría de masas. Este análisis se lleva a cabo en un instrumento RapidFire® acoplado a un espectrómetro de masas de triple cuadrupolo 6460 (Agilent G6460A).
Las proteínas IDH1 mutante (R132H y R132C) e IDH2 mutante (R140Q y R172K) que contienen la etiqueta His N-terminal se expresan en E. coli y se purifican usando cromatografía de afinidad hacia níquel. Los ensayos enzimáticos se llevan a cabo en placas de polipropileno de 96 pocillos con fondo en V que contienen tampón Tris-HCl 100 mM, DTT 1 mM, TRITON™ X-100 al 0,005 %, NaCl 120 mM. Para IDH1 R132H, a-cetoglutarato, NADPH y MnCl2 se incluyen a concentraciones finales de 300 |jM, 2,5 |jM and 300 |jM respectivamente. Para IDH1 R132C, acetoglutarato, NADPH y MnCh se incluyen a concentraciones finales de 100 jiM, 10 jiM y 100 jiM respectivamente. Para IDH2 R172K, a-cetoglutarato, Na Dp H y MnCh se incluyen a concentraciones finales de 150 jiM, 10 jiM y 150 jiM respectivamente. Para IDH2 R140Q, a-cetoglutarato, NADPH y MnCh se incluyen a concentraciones finales de 3000 jiM, 10 jiM y 100 jiM respectivamente. pH final = 7,0. El compuesto de ensayo disuelto en solución de reserva de DMSO se diluye en la mezcla de reacción a una concentración final de DMSO de un 4 %. Los compuestos se someten a ensayo en formato de dosis-respuesta. El ensayo comienza mediante la adición de enzima. Las enzimas se usan en las siguientes concentraciones finales: IDH1 R132H, 2 nM; IDH1 R132C, 0,5 nM; IDH2 R172K, 1,2 nM; IDH2 R140Q, 1,2 nM. Después de 90 minutos la reacción se interrumpe mediante la adición de ACN (50:50) que contiene ácido 2,2,3,4,4-d5 3-hidroxi-1,5-pentanodioico (5d5-3HG) como un patrón interno para el análisis de espectrometría de masas y cuantificación del producto de reacción. El 2-hidroxiglutarato (2HG) en muestras interrumpidas se separa usando cromatografía en columna de intercambio aniónico fuerte (Phenomenex Strata-X-A SecurityGuard) y se analiza mediante espectrometría de masas en un espectrómetro de masas de triple cuadrupolo 6460 (G6460A Agilent). La señal de 2HG detectada se transforma en una concentración de analito usando una curva de calibración generada usando concentraciones conocidas de 2HG. Para cada compuesto sometido a ensayo, el % de inhibición se calcula usando una muestra de control de DMSO como 0 % de inhibición y un control no enzimático como un 100 % de inhibición. Los valores de CI50 se obtienen a partir de los valores individuales de % de inhibición a diferentes concentraciones de compuesto usando una ecuación de 4 parámetros. Estos cálculos se llevan a cabo usando los programas de análisis de datos Activity Base (IDBS) o Screener (Genedata).
Los resultados de este ensayo demuestran que los compuestos usados a modo de ejemplo y sometidos a ensayo inhiben la actividad de IDH1 mutante frente a IHD1/R132H e IDH1/R132C.
Los siguientes Ejemplos se someten a ensayo esencialmente como se ha descrito anteriormente y presentan actividad para la IDH1 mutante como se muestra en la Tabla 19 que sigue a continuación y son selectivos para la IDH1 mutante con respecto a la IDH2 mutante.
Tabla 19

continuación

Ensayos bioquímicos para enzimas IDH1 e IDH2 de tipo salvaje
Las enzimas IDH1 y IDH2 catalizan la conversión de isocitrato a aKG. Las proteínas IDH1 de tipo salvaje (National Center for Biotechnology Information, Registro: NP_001269316.1) e Id H2 (National Center for Biotechnology Information, Registro: EAX02082.1) que contienen etiqueta de His N-terminal se expresan en E. coli y se purifican usando cromatografía de afinidad hacia níquel. Los ensayos enzimáticos se llevan a cabo en placas de polipropileno de 96 pocillos con fondo en V que contienen tampón Tris-HCl 100 mM a pH 7,5, DTT 1 mM, TRITo N™ X-100 al 0,005 %, NaCl 120 mM. Para el ensayo de IDH1 de tipo salvaje, se incluyen isocitrato, NADP+ y MnCh a las concentraciones de 85 |jM, 50 |juM y 20 |jM respectivamente. Para el ensayo de IDH2 de tipo silvestre se incluyan isocitrato, NADP+ y MnCh a las concentraciones de 30 jiM, 50 jiM y l0 jiM respectivamente. Los inhibidores disueltos en una solución madre de DMSO se diluyen en la mezcla de reacción a una concentración final de DMSO de un 4 %. El ensayo enzimático se termina (se inactiva) mediante la adición de ACN (50:50) que contiene ácido d6-2-cetopentanodioico (d6-aKG) como un patrón interno para el análisis de espectrometría de masas. Diez microlitros de mezcla de reacción se combinan con 100 jil de agua, 50 jil de O-bencilhidroxilamina en tampón de piridina 1 M (8,6 % de piridina, pH 5) y 50 jil de clorhidrato de N-(3-dimetilaminopropil)-N-etilcarbodiimida 1 M (EDC) en tampón de piridina. Después de la derivatización a temperatura ambiente durante una hora, las muestras se extraen con 600 j l de EtOAc. Cuatrocientos j l de la fase superior se retiran, se secan en atmósfera de nitrógeno calentada y se reconstituyen con 100 j l de MeOH/agua (1:1). Se inyectan diez j l de muestra derivatizada en un sistema de LC-MS que consiste en un sistema de HPLC Prominence 20A de Shimadzu y un espectrómetro de masas de triple cuadrupolo de The Thermo Quantum Ultra™. Los analitos se separan en una columna XBridge™ C18 (2,1 x 50 mm, 3,5 jm ) de Waters con un caudal de 0,6 ml/minuto. La fase móvil A es ácido fórmico al 0,1 % en agua y la fase móvil B es MeOH. La señal de aKG detectada se transforma en la concentración de analito mediante una curva de calibración generada mediante concentraciones conocidas de aKG. Para cada compuesto sometido a ensayo, el % de inhibición se calcula usando una muestra de control de DMSO como 0 % de inhibición y un control no enzimático como un 100 % de inhibición. Los valores de CI50 se obtienen a partir de los valores individuales de % de inhibición a diferentes concentraciones de compuesto usando una ecuación de 4 parámetros. Estos cálculos se llevan a cabo usando los programas de análisis de datos Activity Base (IDBS) o Screener (Genedata).
Los resultados de este ensayo demuestran que los compuestos usados a modo de ejemplo y sometidos a ensayo son menos activos en la inhibición de la enzima IDH1 de tipo salvaje en comparación con las enzimas mutantes R132H o R132C de IDH1.
Los siguientes Ejemplos en la Tabla 20 se someten a ensayo esencialmente como se ha descrito anteriormente y son menos activos en la inhibición de la enzima IDH1 de tipo salvaje en comparación con las enzimas mutantes R132H o R132C de IDH1.
Actividad enzimática de IDH1 de tipo salvaje y de IDH2 de tipo salvaje
Tabla 20

continuación

Ensayo bioquímico de dilución por etapas de IDH1 (R132H)
Los compuestos a modo de Ejemplo liofilizados se reconstituyen hasta 10 mM o 100 mM con DMSO al 100 % y se mantienen a temperatura ambiente hasta que se someten a ensayo. La proteína IDH1(R132H)-His se expresa y se purifica mediante procedimientos bien conocidos y normalmente usados por las personas con experiencia en la materia. Los reactivos de ensayo incluyeron los siguientes: ácido a-cetoglutárico (Sigma n.° de catálogo K1875), MnCl2 - Fisher Scientific n.° de catálogo M87-100, NADPH - Sigma-Aldrich n.° de catálogo N7505, Tris-HCl (Invitrogen, n.° de catálogo 15567-027), NaCl (Sigma, S3014), ditiotreitol (Sigma, D5545), y TritonX100 (Peirce, 28314). El Kit NAD(P)H-Glo™ de Promega (G9061).
El tampón de ensayo usado en todo el experimento contiene Tris-HCl 100 mM, pH 7,0, NaCl 120 mM, DTT 1 mM, Triton X-100 al 0,005 % y DMSO al 2 % (de la adición del compuesto de ensayo). La CI50 de cada compuesto se determina incubando una respuesta a la dosis del compuesto, preparada en un Echo555, con IDH1(R132H) 1,5 nM, a-cetoglutarato 1 mM, MnCh 1 mM, y NADPH 15 |jM en tampón de ensayo. La reacción se incuba durante 2 horas a temperatura ambiente, a continuación se detiene usando 6-ciclopropil-5-(isoquinolin-5-il)-2-[(3R)-4-(3-metoxipropanoil)-3-metilpiperazin-1-il]piridin-3-carbonitrilo (10 j M). Las concentraciones de NADPH se miden usando el Kit NAD(P)H-Glo™, de acuerdo con lo especificado por el proveedor. La señal luminiscente se lee en el Envision (Perkin Elmer; 0,1 s/Espejo de Luminiscencia/filtro Lum700 WL400-700). En el experimento de dilución por etapas posterior, una concentración de compuesto equivalente a 10X, la IC50 se incuba previamente con IDH1(R132H) 100 nM. La concentración del compuesto siempre es mayor o igual que la concentración de la enzima. Después de 2 horas a temperatura ambiente, esta mezcla se diluye a 1:100 en una solución que contiene acetoglutarato (10 mM), MnCh (10 mM), y NADPH (15 j M). Esta reacción enzimática final contiene IDH1 (R132H) 1 nM y 0,1 X [CI50]. Después de un periodo de incubación de 2 horas a temperatura ambiente, la concentración de NADPH se mide como se ha especificado anteriormente usando 6-ciclopropil-5-(isoquinolin-5-il)-2-[(3R)-4-(3-metoxipropanoil)-3-metilpiperazin-1-il]piridin-3-carbonitrilo y el Kit NAD(P)H-Glo™. Se incluyen tres controles: 1) "Control 10X " que contiene 10 X CI50 del compuesto en la incubación previa y ensayo enzimático excepto acetoglutarato 1 mM, MnCh 1 mM, y NADPH 15 j M se usa en el ensayo final que mide la actividad enzimática, 2) "Control de Actividad Máx" que contiene DMSO en lugar del compuesto para el ensayo tanto de incubación previa como enzimático, y 3) "Control 0,1 X" que contiene DMSO en lugar del compuesto en la incubación previa y
compuesto 0,1 X CI50 en el ensayo enzimático. Un "Control de Actividad Mín" que carece de enzima, pero que de otro modo es equivalente al "Control de Actividad Máx" se incluye. Un segundo conjunto de Controles de Actividad Máx y Mín se lleva a cabo usando a-cetoglutarato 1 mM, MnCh 1 mM, y NADPH 15 |jM. Cada condición de ensayo se somete a ensayo por triplicado y se llevan a cabo 32 replicados para el Control de Actividad Máx (10 mM) y para el Control de Actividad Mín (10 mM) mientras que se llevan a cabo 16 replicados para Control de Actividad Máx (1 mM) y el Control de Actividad Mín (1 mM).
La concentración de NADP (producto) producido en cada experimento/control se determina usando el porcentaje de disminución en la señal observada con respecto al Control de Actividad Mín, que contiene NADPH 15 j M. El Control de Actividad Mín (1 mM y 10 mM) y el de Control de Actividad Máx (1 mM y 10 mM) se promedian y la desviación externa se calcula para cada uno. Una señal para cada dilución por etapas y para los Controles 0,1 X se multiplican por 15 y a continuación se divide por el promedio de las Cuentas para los portillos de Control de Actividad Mín (10 mM). Este número se resta de 15 para calcular NADP (j M de Producto). Los mismos cálculos se usan para los Controles 10 X pero se usan los controles de actividad mínima (1 mM). Los 2 jmoles del producto para los controles de actividad máxima (1 mM y 10 mM) se calculan multiplicando las ventas promedio por 15 de coordinación dividiendo entre los respectivos controles de actividad mínimos (1 mM y 10 mM). La concentración en j M de NADP para cada pocillo se divide entre el Control de Actividad Máx promedio (1 mM o 10 mM) y a continuación se multiplica por 100 para determinar el % de Actividad de IDH para la dilución por etapas del compuesto, 10 X Control, y 0,1 X Control. Un compuesto de pase de demostrar < 30 % de actividad para el control 10 X que muestra que la concentración en la incubación previa suficiente como para saturar la enzima con compuesto. Además, el compuesto de demostrar > 70-80 % de actividad para el control 0,1 X confirmando que no hay inhibición a la concentración de compuesto 0,1 X /diluido.
Los compuestos a modo de ejemplo se someten a ensayo esencialmente como se ha descrito anteriormente y exhiben datos de % de recuperación para IDH1/R132H en este ensayo. Los compuestos de la presente invención, usados a modo de ejemplo sometidos a ensayo, inhiben la enzima 2 horas después de la dilución, a diferencia de los compuestos de la técnica que no inhibieron la enzima 2 horas después de la dilución con el % de recuperación. Los datos de este ensayo demuestran que los compuestos de la presente invención sometidos al ensayo actúan de una manera coherente con la inhibición covalente de IDH1 mutante ya que la dilución del inhibidor no da como resultado la recuperación de la actividad enzimática.
Ensayos a base de células para inhibidores mutantes de IDH1
Para someter a ensayo la inhibición celular del R132C mutante de IDH1, se usa la línea celular de fibrosarcoma HT1080 (adquirida en ATCC). Para someter al ensayo la inhibición a base de células de la mutación R132H, la línea celular de glioma U87MG (ATCC) se transfectó de manera estable con una construcción de ADN que expresa la enzima mutante R132H.
Ensayo de células HT1080:
Cinco mil células se siembran en placas de 96 pocillos revestidas con poli-D-lys (15.000 células/pocillo) 18-24 horas antes del tratamiento con los compuestos. Cuatro horas antes del tratamiento con el compuesto, las células se privan de glutamina eliminando los medios normales y reemplazando con medios libres de glutamina. Después de la privación, las células se tratan con diferentes concentraciones de compuestos de ensayo (de 20 j M a 1 nM) disueltos en medios libres de glutamina que contienen DMSO a una concentración final de un 0,2 %. La incubación inicial del compuesto es durante 1 hora a 37 °C/CO2 al 5 %. Después de 1 hora, se añade glutamina a una concentración final de 2 mM y a continuación las células tratadas se incuban durante otras 18 horas a 37 °C/CO2 al 5 %. Después de la incubación de 18 horas, se analizan 2HG intracelular y aKG en lisados celulares. Los lisados se preparan después de la eliminación de los medios y la adición de tampón que contiene Tris-HCl 25 mM, pH 7,5, NaCl 150 mM, EDTA 1 mM, EGTA 1 mM/Triton-X 100 al 1 % a las células. Se añade una alícuota de lisado a una mezcla de (d6-aKG y d5-3HG como patrones internos y la mezcla se trata con O-bencilhidroxilamina en presencia de clorhidrato de W-(3-dimetilaminopropil)-W-etilcarbodiimida (EDC) y piridina. Los derivados del analito se extraen con EtOAc, se secan y a continuación se reconstituyen con MeOH al 50 % en H2O. Las muestras preparadas como se ha descrito se inyectan en el aparato de HPLC para separar los derivados de 2HG y aKG (y los patrones internos correspondientes) mediante una cromatografía de fase inversa en una columna C18. El análisis de las muestras se lleva a cabo usando un espectrómetro de masas de triple cuadrupolo 6460 (G6460A Agilent). Las señales 2HG y aKG detectadas se transforman en la concentración de analito usando la relación de aKG/d6-aKG y la relación de 2HG/d5-3HG que se extrapola dentro de una curva de calibración. El porcentaje de inhibición para cada muestra individual se obtiene después de normalizar la concentración calculada de 2HG o aKG a las referencias máximas y mínimas obtenidas en presencia y en ausencia de glutamina durante el tratamiento celular con compuestos. Los valores de CI50 se obtienen a partir del % de inhibición individual usando una ecuación sigmoidea de 4 parámetros de respuesta a la dosis. Estos cálculos se llevan a cabo automáticamente usando los programas de análisis de datos Activity Base (IDBS) o Screener (Genedata).
Los resultados de este ensayo demuestran que los Ejemplos sometidos a ensayo en la Tabla 21 inhiben la producción de 2-hidroxiglutarato, lo que indica la inhibición de las células mutantes de IDH1 R132C en este ensayo. aKG, un metabolito generado por IDH1 de tipo salvaje no se ve afectado por los inhibidores, lo que indica que los
compuestos son selectivos para IDH1 mutante con respecto a las células de IDH1 de tipo salvaje en este ensayo. Los valores CI50 resultantes para los siguientes Ejemplos se muestran en la Tabla 21.
Tabla 21
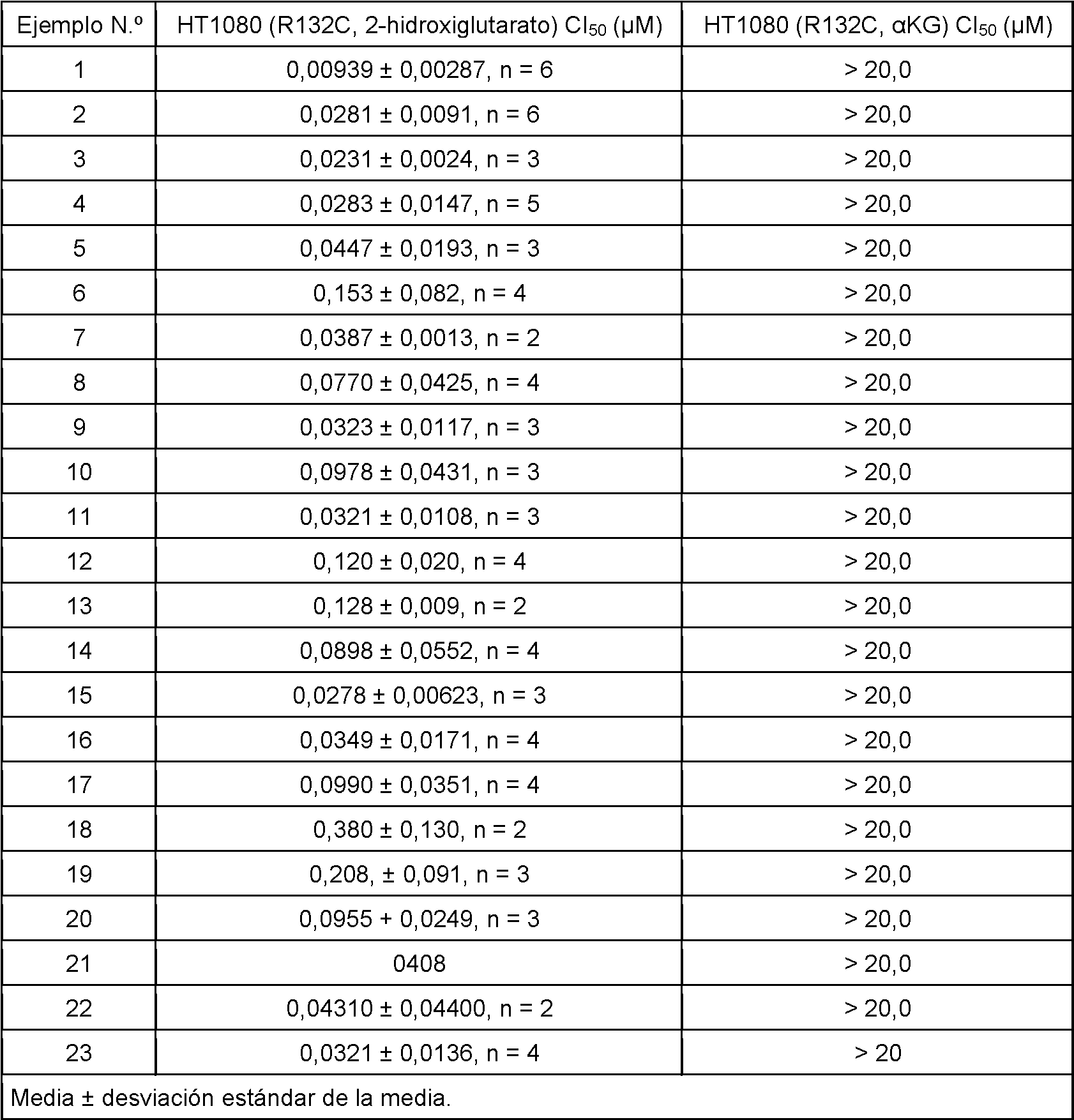
Ensayo de células U87MG/IDH1R132H
Las células se siembran en placas de 96 pocillos revestidas con poli-D-lys (12.000 células/pocillo) 18-24 horas antes del tratamiento con los compuestos. Cuatro horas antes del tratamiento con el compuesto, las células se privan de glutamina eliminando los medios normales y reemplazando con medios libres de glutamina. Después de la privación, las células se tratan con diferentes concentraciones de compuestos de ensayo (de 20 |jM a 1 nM) disueltos en medios libres de glutamina que contienen DMSO a una concentración final de un 0,2 %. La incubación inicial del compuesto es durante 1 hora a 37 °C/CO2 al 5 %. Después de 1 hora, se añade glutamina a una concentración final de 2 mM y a continuación las células tratadas se incuban durante otras 18 horas a 37 °C/CO2 al 5 %). Los lisados celulares se conservan a -80 °C hasta su procesamiento. Para la extracción de analitos, una alícuota de lisado descongelado se transfiere una placa profunda de 96 pocillos y se trata con MeOH frío que contiene d5-3HG como patrón interno seguido de cloroformo y H2O (1:4:3:2). La fase superior se recoge después de su separación y se inyecta en HPLC para separar 2HG (y el patrón interno) usando cromatografía de interacción hidrófila (HILIC) acoplará la detección de MS/MS en un espectrómetro de masas de triple cuadrupolo. El porcentaje de inhibición para cada muestra individual se obtiene después de normalizar la concentración calculada de 2HG a las referencias máximas y mínimas obtenidas en presencia y en ausencia de glutamina durante el tratamiento celular con compuestos. Los valores de CI50 se obtienen a partir del % de inhibición individual usando una ecuación sigmoidea de 4 parámetros de respuesta a la dosis. Estos cálculos se llevan a cabo automáticamente usando los programas de análisis de datos Activity Base (IDBS) o Screener (Genedata).
Los siguientes Ejemplos se someten a ensayo esencialmente como se ha descrito anteriormente y exhiben actividad de inhibición frente a IDH1/R132H mutante en células U87MG en este ensayo como se muestra en la Tabla 22 que sigue a continuación.
Tabla 22

Ensayo de 2-Hidroxiglutarato In Vivo
Para el ensayo in vivo de inhibidores de IDH1, se cultivan tumores subcutáneos de xenoinjerto en ratones desnudos atímicos (20-22 g, Harlan Laboratories) después de la implantación de células HT1080 (fibrosarcoma portador de IDH1 mutante R132C) o de células TB08 (glioblastoma secundario portador de mutante R132H IDH1). A los ratones se les alimenta y se les ofrece agua a voluntad y se aclimatan durante 1 semana antes de la implantación de las células. Las células tumorales (HT1080) o los fragmentos tumorales (TB08) se implantan en el costado trasero derecho. Para HT1080, se implantan 5,0 x 106 células en una mezcla a 1:1 con Matrigel en un volumen final de 0,2 ml. Para TB08, los fragmentos tumorales generados a partir de muestras tumorales sembradas se implantan directamente en el costado posterior. Los volúmenes tumorales se miden con el calibrador dos veces a la semana y el volumen tumoral se calcula usando la fórmula 0,536 X L X W2, en la que L = longitud y W = ancho. Cuando los volúmenes tumorales alcanzan 150-400 mm3, los animales se clasifican de forma aleatoria, se colocan en grupos (n = 3-6 por grupo) y se dosifican con inhibidores de IDH1 o control del vehículo. Para los inhibidores de la IDH1, los compuestos se formulan en un vehículo que contiene 1% de hidroxietilcelulosa/0,25 % de Tween 80/0,05 % de antiespumante o 10 % de goma arábiga con 1,1 equivalentes molares de HCl. Los compuestos son tratan mediante sonicación en baño para obtener una suspensión. Los compuestos se dosifican en una base de miligramo por kilogramo (mpk) mediante sonda oral en un volumen final de 0,2 ml. Para determinar la inhibición de 2HG, los compuestos se dosifican dos veces al día (BID) durante 3 días (número total de dosis = 6). Después del tratamiento
con el compuesto, los ratones se sacrifican con anestesia de isofluorano y luxación cervical. Los tumores se extirpan, se colocan en tubos etiquetados y se congelan inmediatamente en nitrógeno líquido. Los tumores se almacenan a -80 °C para su procesamiento.
Preparación de lisados tumorales
El tampón XY Lite se prepara en agua de calidad molecular y contiene los siguientes componentes: Tris 25 mM, pH 7,5, NaCl 150 mM, Triton X-100 al 1 %, EDTA 1 mM, EGTA 1 mM. A XY Lite (40 ml), se añaden 800 pl de cóctel de inhibidores de proteasa y de fosfatasa Halt (Cóctel de inhibidor de proteasa y fosfatasa Halt™, Thermo Scientific sin EDTA, n.° de catálogo 78441). Las muestras se tratan mediante agitación vorticial y a continuación se enfrían en hielo. Los tubos de lisis de tapa naranja-A se etiquetan y colocan en una rejilla sobre hielo. Un mortero y una mano de cerámica se colocan en hielo seco para enfriar. Un cuadrado de aluminio de 2 X 2 pulgadas se coloca en la parte inferior del mortero. Una muestra tumoral se transfiere al mortero enfriado previamente en el cuadrado de aluminio. Se añade nitrógeno líquido (aproximadamente 5 ml) y se deja evaporar, sobrecongelando el tumor. Se coloca otro trozo de papel de aluminio sobre el tumor y el tumor se rompe en pedazos pequeños con el mortero de cerámica. El tumor aplastado se transfiere rápidamente al tubo de lisis. Se añade XY Lite enfriado en hielo (500 pl) a cada tubo y se tapa. A continuación, los tumores se procesan en FastPrep-24 MP Biomedicals centrifugando dos veces durante 35 segundos cada uno a la configuración de velocidad 5. Las muestras se centrifugan en Beckman Microfuge R a 4 °C a 14.000 rpm durante 30 minutos. El sobrenadante se transfiere a una placa de 96 pocillos profundos previamente enfriada. El sedimento se desecha.
Ensayo de proteínas
Una placa de dilución de ensayo de proteínas se genera primero añadiendo tampón XY (145 pl) a una placa Corning de fondo redondo no estéril de 96 pocillos. A esto, se le añade el lisado tumoral (5 pl) y se mezcla suavemente. La placa se mantiene en hielo. Las diluciones en serie del patrón BSA (Thermo Scientific cat. 23209 2 mg/ml) se configuran de la siguiente manera: se colocan cinco tubos de 0,5 ml en un rack y se añade tampón XY (60 pl) a cada uno. La solución de reserva de BSA (60 pl) se añade al primer tubo y se trata con agitación vorticial. Se transfieren 60 pl del primer tubo al siguiente tubo, se trata con agitación vorticial, y así sucesivamente, hasta que la serie de dilución se complete de la siguiente manera: Tubo 1 = patrón de BSA, los tubos 2-5 son diluciones en serie a 1:2, tubo 6 = tampón XY solo. Los reactivos del ensayo de proteínas Thermo BCA se mezclan de acuerdo con las instrucciones del fabricante. Se añade reactivo BCA mixto (200 pl) a cada muestra y se incuba durante 15 minutos. Los resultados del ensayo de proteínas se leen en el lector de placas SOFTmax Pro. De acuerdo con los resultados del ensayo de proteínas, se añade la cantidad apropiada de tampón XY a cada lisado tumoral para generar una concentración final de proteína de 5 mg/ml. Todas las muestras se etiquetan y se almacenan a -80 °C.
Análisis de metabolitos en lisados tumorales
Los efectos in vivo de la inhibición de IDH1 sobre las cantidades totales de 2HG y aKG se determinan mediante análisis de cromatografía líquida-espectrometría de masas (LC-MS) de xenoinjertos de tumor. El procedimiento usa derivatización con O-bencilhidroxilamina antes del análisis por LC-MS. Diez microlitros de cada lisado tumoral se colocan en una placa de 96 pocillos de pocillo profundo y se combinan con 100 pl de solución patrón interno que contiene d5-3HG 10 pl y d6-aKG 10 pM. Se añaden 50 pl de tampón de O-bencilhidroxilamina en piridina 1 M (8,6 % de piridina, pH 5) y 50 pl de clorhidrato de de N-(3-dimetilaminopropil)-N-etilcarbodiimida (EDC) en piridina a cada muestra. La reacción de derivatización evoluciona a una temperatura ambiente durante una hora. Usando un aparato de manipulación de líquidos Biomek FX de Beckman, se añaden 600 pl de EtOAc a cada muestra. Las placas se sellan y se agitan durante 5 minutos, a continuación se centrifugan durante 5 minutos a 4000 rpm en una centrifugadora Eppendorf 5810R. Se transfieren 400 pl de la fase superior a una nueva placa de 96 pocillos. Las muestras se pueden analizar en atmósfera de nitrógeno caliente a 50 °C y se reconstituyen con en microlitros de MeOH/agua (1:1). Se inyecta un microlitro de muestra derivatizada en un sistema LC-MS que consiste en un sistema HPLC Shimadzu Prominence 20A y un espectrómetro de masas de triple cuadrupolo Thermo Quantum Ultra™. Los analitos se separan en una columna Water XBridge™ C18 (2,1 x 50 mm, 3,5 pm) con un caudal de 0,6 ml/minuto. La fase móvil A es ácido fórmico al 0,1 % en agua y la fase móvil B es MeOH. El perfil del gradiente es: 0 minutos, B al 5 %; 3 minutos, B al 100 %; 4,00 minutos, B al 100 %; 4,1 minutos, B al 5 %; 5,50 minutos, parada. El espectrómetro de masas usa una sonda HESI-II que funciona en modo de monitorización de reacción seleccionada con iones positivos. Las curvas de calibración se construyen trazando las proporciones de concentraciones de analito con respecto a analito/área de pico de patrón interno y llevando a cabo un ajuste cuadrático de los datos usando un software de ponderación 1/concentración con el software Xcalibur™. Los analitos variables para las incógnitas se vuelven a calcular a partir de las curvas de calibración. Los datos de metabolitos del ensayo LCMS se expresan en nmol/mg de proteína. El nivel promedio de 2HG en el grupo tratado con vehículo se usa para determinar el control de inhibición de un 0 %. El % de inhibición en cada animal tratado con inhibidor se determina a continuación en relación con el control del vehículo. Los datos se analizan en el software JMP para determinar el % de inhibición promedio en cada grupo de dosis, la desviación estándar y el error estándar.
Los datos que demuestran la inhibición in vivo de 2-hidroxiglutarato en ratones de xenoinjerto mutante IDH1 mediante los compuestos usados a modo de ejemplo y sometidos a ensayo se muestran en la Tabla 23 que sigue a continuación.
Tabla 23

Claims (10)
1. Un compuesto de la fórmula:

en la que
L es un engarce seleccionado entre el grupo que consiste en -N-azetidina-3-CH2-O-, -N-azetidina-3-0-(CH2)-, -N-2,6-diazaespiro[3,3]heptano-6-(CH2)-, -N-piperazina-4-(CH2)-, -N-piperazina-, -N-azetidina-3-(CH2CH2)-, -7-N-(2,7-diazaespiro[3,5]nonano)-2-(CH2), -N-azetidina-3-(NMe)CH2-, -N-piperidina-4-(NMe)CH2-, N-2,5-dihidropirrol-3-(CH2)-0-;
Z se selecciona entre el grupo que consiste en H o F;
R se selecciona entre el grupo que consiste en alquilo C1-C4, -CH(CH3)CH2-OH, - CH2CH2F, -CH2CHF2; o una sal farmacéuticamente aceptable del mismo.
2. Un compuesto de la reivindicación 1 en el que: L es un engarce seleccionado entre el grupo que consiste en -N-piperazina-4-(CH2)-, -N-piperazina-, -N-azetidina-3-(NMe)CH2-, -N-azetidina-3-CH2-0-, -N-azetidina-3-0-(CH2)-, -N-azetidina-3-(CH2CH2)-, y -N-piperidina-4-(NMe)CH2-;
Z es H;
R es alquilo C1-C4; o una
sal farmacéuticamente aceptable del mismo.
3. Un compuesto de la reivindicación 1 o 2 en el que:
L es un engarce seleccionado entre el grupo que consiste en -N-piperazina-4-(CH2)- , -N-piperazina-, -N-azetidina-3-(NMe)CH2-, -N-azetidina-3-0-(CH2)-, -N-azetidina-3-(CH2CH2)-, y -N-piperidina-4-(NMe)CH2-;
Z es H;
R se selecciona entre CH2CH3, CH(CH3)2; o una
sal farmacéuticamente aceptable del mismo.
4. Un compuesto de una cualquiera de las reivindicaciones 1 a 3 que es 1-isopropil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo.
5. Un compuesto de una cualquiera de las reivindicaciones 1 a 3 que es 1-etil-7-[[(1S)-1-[4-[(4-prop-2-enoilpiperazin-1-il)metil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo.
6. Un compuesto de una cualquiera de las reivindicaciones 1 a 3 que es:
1-isopropil-7-[[(1S)-1-[4-(4-prop-2-enoilpiperazin-1-il)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo;
1-etil-7-[[(1S)-1-[4-(4-prop-2-enoilpiperazin-1-il)fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo;
1-etil-7-[[(1S)-1-[4-[(1-prop-2-enoilazetidin-3-il)oximetil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo;
1-etil-7-[[(1S)-1-[4-[2-(1-prop-2-enoilazetidin-3-il)etil]fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo;
1-isopropil-7-[[(1S)-1-[4-[(1-prop-2-enoilazetidin-3-il)metoxi]-fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo; o
1-etil-7-[[(1S)-1-[4-[[metil-(1-prop-2-enoilazetidin-3-il)amino]metil]-fenil]etil]amino]-4H-pirimido[4,5-d][1,3]oxazin-2-ona, o una sal farmacéuticamente aceptable del mismo.
7. Una composición farmacéutica que comprende un compuesto de una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo, y un vehículo farmacéuticamente aceptable.
8. Un compuesto de una cualquiera de las reivindicaciones 1 a 6, o una sal farmacéuticamente aceptable del mismo, para uso en terapia.
9. Un compuesto de una cualquiera de las reivindicaciones 1 a 6, o una sal farmacéuticamente aceptable del mismo, para uso en el tratamiento de una IDH1 mutante que expresa cáncer que es glioma, glioblastoma, glioblastoma multiforme, astrocitoma, oligodendroglioma, paraganglioma, fibrosarcoma, linfoma de linfocitos T angioinmunoblástico, síndrome mielodisplásico, leucemia linfoblástica aguda de linfocitos B, cáncer de tiroides, cáncer colorrectal, leucemia mieloide aguda, melanoma, cáncer de próstata, condrosarcoma o colangiocarcinoma .
10. El compuesto para su uso de la reivindicación 9 en el que el cáncer que expresa IDH1 mutante es glioma, glioblastoma, leucemia mieloide aguda, o fibrosarcoma.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15382386 | 2015-07-27 | ||
| EP15382606 | 2015-12-04 | ||
| PCT/US2016/043264 WO2017019429A1 (en) | 2015-07-27 | 2016-07-21 | 7-phenylethylamino-4h-pyrimido[4,5-d][1,3]oxazin-2-one compounds and theit use as mutant idh1 inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2764523T3 true ES2764523T3 (es) | 2020-06-03 |
Family
ID=56561486
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16745954T Active ES2764523T3 (es) | 2015-07-27 | 2016-07-21 | Compuestos de 7-feniletilamino-4H-pirimido[4,5-D][1,3]oxazin-2-ona y su uso como inhibidores de IDH1 mutantes |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10253041B2 (es) |
| EP (1) | EP3328866B1 (es) |
| JP (1) | JP6473270B2 (es) |
| CN (1) | CN107849059B (es) |
| ES (1) | ES2764523T3 (es) |
| WO (1) | WO2017019429A1 (es) |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2017003627A (es) | 2014-09-19 | 2018-01-24 | Forma Therapeutics Inc | Composiciones de pirimidin quinolinona como inhibidores de isocitrato deshidrogenasa mutante (mt-idh). |
| JP6751081B2 (ja) | 2014-09-19 | 2020-09-02 | フォーマ セラピューティクス,インコーポレイテッド | 変異イソクエン酸デヒドロゲナーゼ阻害剤としてのピリジニルキノリノン誘導体 |
| US10005734B2 (en) | 2014-09-19 | 2018-06-26 | Forma Therapeutics, Inc. | Pyridin-2(1H)-one quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| EP4257131A3 (en) | 2014-09-19 | 2024-01-10 | Forma Therapeutics, Inc. | Pyridin-2(1h)-one quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| US10294206B2 (en) | 2015-04-21 | 2019-05-21 | Forma Tm2, Inc. | Fused-bicyclic aryl quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| US10407419B2 (en) | 2015-04-21 | 2019-09-10 | Forma Therapeutics, Inc. | Quinolinone five-membered heterocyclic compounds as mutant-isocitrate dehydrogenase inhibitors |
| ES2814290T3 (es) * | 2016-06-06 | 2021-03-26 | Lilly Co Eli | Inhibidores de IDH1 mutante |
| CR20190252A (es) * | 2016-12-16 | 2019-08-26 | Lilly Co Eli | Compuestos de 7-feniletilamino-4h-pirimido[4,5-d][1,3]-oxazin-2-ona como inhibidores de idh1 e idh2 mutantes |
| WO2019015672A1 (zh) * | 2017-07-21 | 2019-01-24 | 南京明德新药研发股份有限公司 | 吡啶并咪唑类化合物及其应用 |
| US11311527B2 (en) | 2018-05-16 | 2022-04-26 | Forma Therapeutics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mIDH-1) |
| WO2019222553A1 (en) | 2018-05-16 | 2019-11-21 | Forma Therapeutics, Inc. | Inhibiting mutant idh-1 |
| WO2019222551A1 (en) | 2018-05-16 | 2019-11-21 | Forma Therapeutics, Inc. | Solid forms of ((s)-5-((1-(6-chloro-2-oxo-1,2-dihydroquinolin-3-yl)ethyl)amino)-1-methyl-6-oxo-1,6-dihydropyridine-2-carbonitrile |
| US11013734B2 (en) | 2018-05-16 | 2021-05-25 | Forma Therapeutics, Inc. | Treating patients harboring an isocitrate dehydrogenase-1 (IDH-1) mutation |
| US11013733B2 (en) | 2018-05-16 | 2021-05-25 | Forma Therapeutics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mlDH-1) |
| CN112469719B (zh) * | 2018-05-21 | 2024-01-23 | 内尔维亚诺医疗科学公司 | 杂缩合的吡啶酮化合物及其作为idh抑制剂的用途 |
| KR102584855B1 (ko) * | 2018-06-26 | 2023-10-05 | 케이피씨 파마슈티컬스 인코포레이티드 | 벤즈이미다졸 유도체 및 idh1 억제제로서의 이의 용도 |
| EP4055015A1 (en) * | 2019-11-08 | 2022-09-14 | Nerviano Medical Sciences S.r.l. | Gem-disubstituted heterocyclic compounds and their use as idh inhibitors |
| CN110724151B (zh) * | 2019-11-18 | 2022-03-04 | 成都睿智化学研究有限公司 | 一种(3,4-二氢-2H-吡喃并[2,3-b]吡啶-6-基)甲醇的合成方法 |
| CN113801139A (zh) * | 2020-06-12 | 2021-12-17 | 华东理工大学 | 作为rsk抑制剂的嘧啶并恶嗪衍生物及其应用 |
| WO2022001916A1 (zh) * | 2020-06-28 | 2022-01-06 | 微境生物医药科技(上海)有限公司 | Idh突变体抑制剂及其用途 |
| EP4337217A1 (en) * | 2021-06-09 | 2024-03-20 | Eli Lilly and Company | Method for treating idh inhibitor-resistant subjects |
| MX2023013664A (es) * | 2021-06-15 | 2024-01-08 | Wigen Biomedicine Tech Shanghai Co Ltd | Inhibidor de mutante de idh y uso del mismo. |
| CA3224704A1 (en) | 2021-08-13 | 2023-02-16 | David Andrew Coates | Solid forms of 7-[[(1s)-1-[4-[(1s)-2-cyclopropyl-1-(4-prop-2-enoylpiper azin-1-yl)ethyl]phenyl]ethyl]amino]-1-ethyl-4h- pyrimido[4,5-d] [1,3]oxazin-2-one |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT1940839E (pt) * | 2005-10-07 | 2013-10-10 | Exelixis Inc | Inibidores de piridopirimidinona pi3k alfa |
| CN102827073A (zh) * | 2011-06-17 | 2012-12-19 | 安吉奥斯医药品有限公司 | 治疗活性组合物和它们的使用方法 |
| EP2771337B1 (en) * | 2011-09-27 | 2017-08-02 | Novartis AG | 3-(pyrimidin-4-yl)-oxazolidin-2-ones as inhibitors of mutant idh |
| JP6022691B2 (ja) * | 2012-08-30 | 2016-11-09 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | ジオキシノ−及びオキサジン−[2,3−d]ピリミジンpi3k阻害剤化合物及び使用方法 |
| EP2970242B1 (en) | 2013-03-14 | 2017-09-06 | Novartis AG | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| EP2970240B1 (en) | 2013-03-14 | 2018-01-10 | Novartis AG | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| WO2014147586A1 (en) | 2013-03-22 | 2014-09-25 | Novartis Ag | 1-(2-(ethylamino)pyrimidin-4-yl)pyrrolidin-2-ones as inhibitors of mutant idh |
| NZ754039A (en) * | 2013-06-26 | 2021-06-25 | Abbvie Inc | Primary carboxamides as btk inhibitors |
| RU2692782C2 (ru) * | 2014-10-01 | 2019-06-27 | Дайити Санкио Компани, Лимитед | Производное изоксазола в качестве ингибитора мутантной изоцитратдегидрогеназы 1 |
| US10294206B2 (en) * | 2015-04-21 | 2019-05-21 | Forma Tm2, Inc. | Fused-bicyclic aryl quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
-
2016
- 2016-07-21 WO PCT/US2016/043264 patent/WO2017019429A1/en active Application Filing
- 2016-07-21 JP JP2018504240A patent/JP6473270B2/ja active Active
- 2016-07-21 US US15/570,759 patent/US10253041B2/en active Active
- 2016-07-21 CN CN201680043259.1A patent/CN107849059B/zh active Active
- 2016-07-21 ES ES16745954T patent/ES2764523T3/es active Active
- 2016-07-21 EP EP16745954.4A patent/EP3328866B1/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| US20180118759A1 (en) | 2018-05-03 |
| CN107849059A (zh) | 2018-03-27 |
| WO2017019429A8 (en) | 2018-02-01 |
| EP3328866A1 (en) | 2018-06-06 |
| US10253041B2 (en) | 2019-04-09 |
| JP2018525369A (ja) | 2018-09-06 |
| EP3328866B1 (en) | 2019-10-30 |
| JP6473270B2 (ja) | 2019-02-20 |
| WO2017019429A1 (en) | 2017-02-02 |
| CN107849059B (zh) | 2020-04-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2764523T3 (es) | Compuestos de 7-feniletilamino-4H-pirimido[4,5-D][1,3]oxazin-2-ona y su uso como inhibidores de IDH1 mutantes | |
| ES2941631T3 (es) | Compuestos de Pirido[4,3-d][1,3]oxazin-2-ona como inhibidores de IDH1 e IDH2 mutantes | |
| ES2814290T3 (es) | Inhibidores de IDH1 mutante | |
| ES2797252T3 (es) | Dihidronaftiridinas y compuestos relacionados útiles como inhibidores de quinasas para el tratamiento de enfermedades proliferativas | |
| ES2888298T3 (es) | Compuestos de C5-anilinoquinazolina y su uso en el tratamiento de cáncer | |
| US20240228511A1 (en) | Substituted fused azines as kras g12d inhibitors | |
| ES2647849T3 (es) | Compuestos de pirido[2,3-d]pirimidin-4-ona como inhibidores de la tanquirasa | |
| US20180155297A1 (en) | Inhibitors of ack1/tnk2 tyrosine kinase | |
| TW201326162A (zh) | 氮芥子氣衍生物 | |
| ES2891320T3 (es) | Análogos de nucleósidos sustituidos para su uso como inhibidores de PRMT5 | |
| BR112019009648B1 (pt) | Compostos de 7-feniletilamino-4h-pirimido[4,5-d][1,3]oxazin-2-ona como inibidores de idh1 e idh2 mutantes, composição farmacêutica que os compreende e seu uso |