ES2740802T3 - Monitorización del estadio de neoplasia linfoide usando perfiles de clonotipo - Google Patents
Monitorización del estadio de neoplasia linfoide usando perfiles de clonotipo Download PDFInfo
- Publication number
- ES2740802T3 ES2740802T3 ES16183402T ES16183402T ES2740802T3 ES 2740802 T3 ES2740802 T3 ES 2740802T3 ES 16183402 T ES16183402 T ES 16183402T ES 16183402 T ES16183402 T ES 16183402T ES 2740802 T3 ES2740802 T3 ES 2740802T3
- Authority
- ES
- Spain
- Prior art keywords
- cells
- disease
- clones
- sample
- sequence
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 223
- 238000012544 monitoring process Methods 0.000 title claims abstract description 38
- 230000009826 neoplastic cell growth Effects 0.000 title claims abstract description 6
- 238000000034 method Methods 0.000 claims abstract description 303
- 210000001744 T-lymphocyte Anatomy 0.000 claims abstract description 144
- 201000011510 cancer Diseases 0.000 claims abstract description 127
- 210000003719 b-lymphocyte Anatomy 0.000 claims abstract description 123
- 108020004511 Recombinant DNA Proteins 0.000 claims abstract description 23
- 208000019420 lymphoid neoplasm Diseases 0.000 claims abstract description 23
- 238000007481 next generation sequencing Methods 0.000 claims abstract description 7
- 210000004027 cell Anatomy 0.000 claims description 270
- 238000012163 sequencing technique Methods 0.000 claims description 185
- 108020004414 DNA Proteins 0.000 claims description 109
- 230000008707 rearrangement Effects 0.000 claims description 72
- 239000002299 complementary DNA Substances 0.000 claims description 43
- 230000000392 somatic effect Effects 0.000 claims description 29
- 238000010839 reverse transcription Methods 0.000 claims description 5
- 238000006467 substitution reaction Methods 0.000 claims description 3
- 239000000523 sample Substances 0.000 description 326
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 317
- 201000010099 disease Diseases 0.000 description 308
- 239000000427 antigen Substances 0.000 description 185
- 108091007433 antigens Proteins 0.000 description 185
- 102000036639 antigens Human genes 0.000 description 185
- 230000002596 correlated effect Effects 0.000 description 168
- 230000003321 amplification Effects 0.000 description 162
- 238000003199 nucleic acid amplification method Methods 0.000 description 162
- 238000004422 calculation algorithm Methods 0.000 description 122
- 238000012360 testing method Methods 0.000 description 116
- 238000003752 polymerase chain reaction Methods 0.000 description 108
- 150000007523 nucleic acids Chemical group 0.000 description 98
- 102000039446 nucleic acids Human genes 0.000 description 81
- 108020004707 nucleic acids Proteins 0.000 description 81
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 71
- 108091008874 T cell receptors Proteins 0.000 description 70
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 66
- 210000001519 tissue Anatomy 0.000 description 64
- 210000004698 lymphocyte Anatomy 0.000 description 61
- 239000003814 drug Substances 0.000 description 55
- 210000004369 blood Anatomy 0.000 description 54
- 239000008280 blood Substances 0.000 description 54
- 238000011282 treatment Methods 0.000 description 54
- 238000006243 chemical reaction Methods 0.000 description 51
- 108091008875 B cell receptors Proteins 0.000 description 50
- 238000001514 detection method Methods 0.000 description 50
- 230000035945 sensitivity Effects 0.000 description 50
- 108090000623 proteins and genes Proteins 0.000 description 49
- 102100023210 Necdin Human genes 0.000 description 47
- 229940079593 drug Drugs 0.000 description 46
- 208000024891 symptom Diseases 0.000 description 45
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 description 42
- 208000023275 Autoimmune disease Diseases 0.000 description 39
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 description 39
- 108091093088 Amplicon Proteins 0.000 description 36
- 230000014509 gene expression Effects 0.000 description 35
- 201000006417 multiple sclerosis Diseases 0.000 description 35
- 230000000295 complement effect Effects 0.000 description 34
- 235000018102 proteins Nutrition 0.000 description 32
- 102000004169 proteins and genes Human genes 0.000 description 32
- 210000002865 immune cell Anatomy 0.000 description 31
- 230000009266 disease activity Effects 0.000 description 30
- 239000002773 nucleotide Substances 0.000 description 30
- 125000003729 nucleotide group Chemical group 0.000 description 30
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 30
- 230000028993 immune response Effects 0.000 description 29
- 238000003753 real-time PCR Methods 0.000 description 29
- 210000000987 immune system Anatomy 0.000 description 28
- 206010039073 rheumatoid arthritis Diseases 0.000 description 28
- 239000007787 solid Substances 0.000 description 28
- 241000282414 Homo sapiens Species 0.000 description 27
- 239000003550 marker Substances 0.000 description 27
- 238000004458 analytical method Methods 0.000 description 26
- 230000005945 translocation Effects 0.000 description 26
- 208000015181 infectious disease Diseases 0.000 description 25
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 24
- 206010025323 Lymphomas Diseases 0.000 description 24
- 239000000047 product Substances 0.000 description 24
- 108091008915 immune receptors Proteins 0.000 description 23
- 102000027596 immune receptors Human genes 0.000 description 22
- 238000002483 medication Methods 0.000 description 22
- 206010020751 Hypersensitivity Diseases 0.000 description 21
- 230000035772 mutation Effects 0.000 description 21
- 230000001717 pathogenic effect Effects 0.000 description 21
- 108091028043 Nucleic acid sequence Proteins 0.000 description 19
- 238000000684 flow cytometry Methods 0.000 description 19
- 230000004054 inflammatory process Effects 0.000 description 19
- 244000052769 pathogen Species 0.000 description 19
- 230000008569 process Effects 0.000 description 19
- 108700028369 Alleles Proteins 0.000 description 18
- 238000003556 assay Methods 0.000 description 18
- 238000001574 biopsy Methods 0.000 description 18
- 230000008859 change Effects 0.000 description 18
- 239000003153 chemical reaction reagent Substances 0.000 description 18
- 230000006378 damage Effects 0.000 description 18
- 238000003745 diagnosis Methods 0.000 description 18
- 238000001114 immunoprecipitation Methods 0.000 description 18
- 210000001503 joint Anatomy 0.000 description 18
- 210000000056 organ Anatomy 0.000 description 17
- 238000002560 therapeutic procedure Methods 0.000 description 17
- 206010069754 Acquired gene mutation Diseases 0.000 description 16
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 16
- 102000004127 Cytokines Human genes 0.000 description 16
- 108090000695 Cytokines Proteins 0.000 description 16
- 206010061218 Inflammation Diseases 0.000 description 16
- 239000000203 mixture Substances 0.000 description 16
- 230000004044 response Effects 0.000 description 16
- 230000002441 reversible effect Effects 0.000 description 16
- 230000037439 somatic mutation Effects 0.000 description 16
- 210000001185 bone marrow Anatomy 0.000 description 15
- 230000000875 corresponding effect Effects 0.000 description 15
- 230000036541 health Effects 0.000 description 15
- 238000009396 hybridization Methods 0.000 description 15
- 230000001900 immune effect Effects 0.000 description 15
- 208000032839 leukemia Diseases 0.000 description 15
- 239000000463 material Substances 0.000 description 15
- 206010037844 rash Diseases 0.000 description 15
- 238000002255 vaccination Methods 0.000 description 15
- 230000001154 acute effect Effects 0.000 description 14
- 150000001413 amino acids Chemical group 0.000 description 14
- 238000002509 fluorescent in situ hybridization Methods 0.000 description 14
- 238000004519 manufacturing process Methods 0.000 description 14
- 230000006798 recombination Effects 0.000 description 14
- 238000005215 recombination Methods 0.000 description 14
- 230000002829 reductive effect Effects 0.000 description 14
- 108020004705 Codon Proteins 0.000 description 13
- 208000010201 Exanthema Diseases 0.000 description 13
- 230000008901 benefit Effects 0.000 description 13
- 239000000090 biomarker Substances 0.000 description 13
- 230000015572 biosynthetic process Effects 0.000 description 13
- 230000007423 decrease Effects 0.000 description 13
- 238000005516 engineering process Methods 0.000 description 13
- 201000005884 exanthem Diseases 0.000 description 13
- 230000001965 increasing effect Effects 0.000 description 13
- 238000005259 measurement Methods 0.000 description 13
- 210000004881 tumor cell Anatomy 0.000 description 13
- 108010074051 C-Reactive Protein Proteins 0.000 description 12
- 102100032752 C-reactive protein Human genes 0.000 description 12
- 108091034117 Oligonucleotide Proteins 0.000 description 12
- 208000026935 allergic disease Diseases 0.000 description 12
- 238000011156 evaluation Methods 0.000 description 12
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 12
- 102000018358 immunoglobulin Human genes 0.000 description 12
- 238000002493 microarray Methods 0.000 description 12
- 108090000765 processed proteins & peptides Proteins 0.000 description 12
- 229960005486 vaccine Drugs 0.000 description 12
- -1 IgH molecules Chemical class 0.000 description 11
- 108060003951 Immunoglobulin Proteins 0.000 description 11
- 208000002193 Pain Diseases 0.000 description 11
- 230000004913 activation Effects 0.000 description 11
- 239000011324 bead Substances 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 206010016256 fatigue Diseases 0.000 description 11
- 230000006870 function Effects 0.000 description 11
- 238000011068 loading method Methods 0.000 description 11
- 210000005259 peripheral blood Anatomy 0.000 description 11
- 239000011886 peripheral blood Substances 0.000 description 11
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 10
- 230000001684 chronic effect Effects 0.000 description 10
- 230000003993 interaction Effects 0.000 description 10
- 206010003246 arthritis Diseases 0.000 description 9
- 210000001124 body fluid Anatomy 0.000 description 9
- 239000010839 body fluid Substances 0.000 description 9
- 208000035475 disorder Diseases 0.000 description 9
- 230000004927 fusion Effects 0.000 description 9
- 210000000265 leukocyte Anatomy 0.000 description 9
- 238000007403 mPCR Methods 0.000 description 9
- 230000036407 pain Effects 0.000 description 9
- 230000001575 pathological effect Effects 0.000 description 9
- 230000004083 survival effect Effects 0.000 description 9
- 208000035473 Communicable disease Diseases 0.000 description 8
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 8
- 210000005006 adaptive immune system Anatomy 0.000 description 8
- 239000013566 allergen Substances 0.000 description 8
- 230000001580 bacterial effect Effects 0.000 description 8
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 8
- 230000003247 decreasing effect Effects 0.000 description 8
- 238000009826 distribution Methods 0.000 description 8
- 239000012634 fragment Substances 0.000 description 8
- 238000002955 isolation Methods 0.000 description 8
- 206010025135 lupus erythematosus Diseases 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 238000000926 separation method Methods 0.000 description 8
- 210000003491 skin Anatomy 0.000 description 8
- 210000002700 urine Anatomy 0.000 description 8
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 7
- 108700042075 T-Cell Receptor Genes Proteins 0.000 description 7
- 210000004556 brain Anatomy 0.000 description 7
- 150000001875 compounds Chemical class 0.000 description 7
- 238000011161 development Methods 0.000 description 7
- 230000018109 developmental process Effects 0.000 description 7
- 238000010790 dilution Methods 0.000 description 7
- 239000012895 dilution Substances 0.000 description 7
- 230000009610 hypersensitivity Effects 0.000 description 7
- 230000008105 immune reaction Effects 0.000 description 7
- 239000012678 infectious agent Substances 0.000 description 7
- 238000002595 magnetic resonance imaging Methods 0.000 description 7
- 239000011159 matrix material Substances 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 229960000485 methotrexate Drugs 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 6
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 6
- 102100027207 CD27 antigen Human genes 0.000 description 6
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 6
- 108010036949 Cyclosporine Proteins 0.000 description 6
- 241000701022 Cytomegalovirus Species 0.000 description 6
- 238000000018 DNA microarray Methods 0.000 description 6
- 229940123907 Disease modifying antirheumatic drug Drugs 0.000 description 6
- 238000002965 ELISA Methods 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 6
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 6
- 108091005461 Nucleic proteins Proteins 0.000 description 6
- 230000001363 autoimmune Effects 0.000 description 6
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 6
- 150000001720 carbohydrates Chemical class 0.000 description 6
- 235000014633 carbohydrates Nutrition 0.000 description 6
- 230000002759 chromosomal effect Effects 0.000 description 6
- 230000000052 comparative effect Effects 0.000 description 6
- 239000002988 disease modifying antirheumatic drug Substances 0.000 description 6
- 238000001502 gel electrophoresis Methods 0.000 description 6
- 238000012268 genome sequencing Methods 0.000 description 6
- 210000002216 heart Anatomy 0.000 description 6
- 230000003053 immunization Effects 0.000 description 6
- 238000002649 immunization Methods 0.000 description 6
- 238000003364 immunohistochemistry Methods 0.000 description 6
- 238000007901 in situ hybridization Methods 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 230000003902 lesion Effects 0.000 description 6
- 150000002632 lipids Chemical class 0.000 description 6
- 210000004185 liver Anatomy 0.000 description 6
- 210000004072 lung Anatomy 0.000 description 6
- 238000013507 mapping Methods 0.000 description 6
- 238000010208 microarray analysis Methods 0.000 description 6
- 238000004393 prognosis Methods 0.000 description 6
- 102000005962 receptors Human genes 0.000 description 6
- 108020003175 receptors Proteins 0.000 description 6
- 230000008521 reorganization Effects 0.000 description 6
- 238000004062 sedimentation Methods 0.000 description 6
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 6
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 6
- 229940124597 therapeutic agent Drugs 0.000 description 6
- 206010002198 Anaphylactic reaction Diseases 0.000 description 5
- 206010065163 Clonal evolution Diseases 0.000 description 5
- 238000001712 DNA sequencing Methods 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 208000009329 Graft vs Host Disease Diseases 0.000 description 5
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 5
- 206010052779 Transplant rejections Diseases 0.000 description 5
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 5
- 241000700605 Viruses Species 0.000 description 5
- 235000001014 amino acid Nutrition 0.000 description 5
- 230000036783 anaphylactic response Effects 0.000 description 5
- 208000003455 anaphylaxis Diseases 0.000 description 5
- 230000000890 antigenic effect Effects 0.000 description 5
- 208000006673 asthma Diseases 0.000 description 5
- 229960002170 azathioprine Drugs 0.000 description 5
- 238000004820 blood count Methods 0.000 description 5
- 239000002458 cell surface marker Substances 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 238000007796 conventional method Methods 0.000 description 5
- 230000034994 death Effects 0.000 description 5
- 231100000517 death Toxicity 0.000 description 5
- 239000000839 emulsion Substances 0.000 description 5
- 210000003743 erythrocyte Anatomy 0.000 description 5
- 239000007850 fluorescent dye Substances 0.000 description 5
- 208000024908 graft versus host disease Diseases 0.000 description 5
- 230000002757 inflammatory effect Effects 0.000 description 5
- 206010022000 influenza Diseases 0.000 description 5
- 210000003734 kidney Anatomy 0.000 description 5
- 238000007726 management method Methods 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 108020004999 messenger RNA Proteins 0.000 description 5
- 210000005036 nerve Anatomy 0.000 description 5
- 102000004196 processed proteins & peptides Human genes 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 238000011160 research Methods 0.000 description 5
- 238000012216 screening Methods 0.000 description 5
- 241000894007 species Species 0.000 description 5
- 201000005671 spondyloarthropathy Diseases 0.000 description 5
- 230000000638 stimulation Effects 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- 238000002054 transplantation Methods 0.000 description 5
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 4
- 208000003950 B-cell lymphoma Diseases 0.000 description 4
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 4
- 208000005443 Circulating Neoplastic Cells Diseases 0.000 description 4
- VVNCNSJFMMFHPL-VKHMYHEASA-N D-penicillamine Chemical compound CC(C)(S)[C@@H](N)C(O)=O VVNCNSJFMMFHPL-VKHMYHEASA-N 0.000 description 4
- 206010012735 Diarrhoea Diseases 0.000 description 4
- 208000030453 Drug-Related Side Effects and Adverse reaction Diseases 0.000 description 4
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 4
- 206010035226 Plasma cell myeloma Diseases 0.000 description 4
- 208000006045 Spondylarthropathies Diseases 0.000 description 4
- 102000004903 Troponin Human genes 0.000 description 4
- 108090001027 Troponin Proteins 0.000 description 4
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 4
- 230000002159 abnormal effect Effects 0.000 description 4
- 238000007792 addition Methods 0.000 description 4
- 230000007815 allergy Effects 0.000 description 4
- 230000003110 anti-inflammatory effect Effects 0.000 description 4
- 230000005784 autoimmunity Effects 0.000 description 4
- 229940109239 creatinine Drugs 0.000 description 4
- 208000037765 diseases and disorders Diseases 0.000 description 4
- 238000001962 electrophoresis Methods 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 201000003444 follicular lymphoma Diseases 0.000 description 4
- 230000002068 genetic effect Effects 0.000 description 4
- 210000004602 germ cell Anatomy 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 208000019622 heart disease Diseases 0.000 description 4
- XXSMGPRMXLTPCZ-UHFFFAOYSA-N hydroxychloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CCO)CC)=CC=NC2=C1 XXSMGPRMXLTPCZ-UHFFFAOYSA-N 0.000 description 4
- 229960004171 hydroxychloroquine Drugs 0.000 description 4
- 230000001506 immunosuppresive effect Effects 0.000 description 4
- 239000003018 immunosuppressive agent Substances 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 210000002414 leg Anatomy 0.000 description 4
- 230000007774 longterm Effects 0.000 description 4
- 230000003211 malignant effect Effects 0.000 description 4
- 230000008816 organ damage Effects 0.000 description 4
- 229960001639 penicillamine Drugs 0.000 description 4
- 210000004976 peripheral blood cell Anatomy 0.000 description 4
- 208000005987 polymyositis Diseases 0.000 description 4
- 230000009257 reactivity Effects 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 210000000278 spinal cord Anatomy 0.000 description 4
- 210000001179 synovial fluid Anatomy 0.000 description 4
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 3
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 3
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 3
- 208000006820 Arthralgia Diseases 0.000 description 3
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 3
- 108091012583 BCL2 Proteins 0.000 description 3
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 3
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 3
- 208000011691 Burkitt lymphomas Diseases 0.000 description 3
- 208000017667 Chronic Disease Diseases 0.000 description 3
- 208000015943 Coeliac disease Diseases 0.000 description 3
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 3
- 229930105110 Cyclosporin A Natural products 0.000 description 3
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 3
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 3
- 108010008165 Etanercept Proteins 0.000 description 3
- 208000004262 Food Hypersensitivity Diseases 0.000 description 3
- 206010016946 Food allergy Diseases 0.000 description 3
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 3
- 206010062016 Immunosuppression Diseases 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- 102000004889 Interleukin-6 Human genes 0.000 description 3
- 108090001005 Interleukin-6 Proteins 0.000 description 3
- 208000007101 Muscle Cramp Diseases 0.000 description 3
- 206010028372 Muscular weakness Diseases 0.000 description 3
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 3
- 108090000028 Neprilysin Proteins 0.000 description 3
- 102000003729 Neprilysin Human genes 0.000 description 3
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 3
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 3
- 208000033759 Prolymphocytic T-Cell Leukemia Diseases 0.000 description 3
- 208000003251 Pruritus Diseases 0.000 description 3
- 206010037660 Pyrexia Diseases 0.000 description 3
- 206010039085 Rhinitis allergic Diseases 0.000 description 3
- 206010039710 Scleroderma Diseases 0.000 description 3
- 208000021386 Sjogren Syndrome Diseases 0.000 description 3
- 108010006785 Taq Polymerase Proteins 0.000 description 3
- 206010047700 Vomiting Diseases 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 230000003044 adaptive effect Effects 0.000 description 3
- 201000010105 allergic rhinitis Diseases 0.000 description 3
- 229940035676 analgesics Drugs 0.000 description 3
- 239000000730 antalgic agent Substances 0.000 description 3
- 210000003651 basophil Anatomy 0.000 description 3
- 238000009534 blood test Methods 0.000 description 3
- 238000010322 bone marrow transplantation Methods 0.000 description 3
- 238000010804 cDNA synthesis Methods 0.000 description 3
- 239000002771 cell marker Substances 0.000 description 3
- 210000003169 central nervous system Anatomy 0.000 description 3
- 229960001265 ciclosporin Drugs 0.000 description 3
- 238000002591 computed tomography Methods 0.000 description 3
- 239000003246 corticosteroid Substances 0.000 description 3
- 229930182912 cyclosporin Natural products 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 description 3
- 230000008030 elimination Effects 0.000 description 3
- 238000003379 elimination reaction Methods 0.000 description 3
- 230000005713 exacerbation Effects 0.000 description 3
- 210000001508 eye Anatomy 0.000 description 3
- 235000020932 food allergy Nutrition 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 238000013412 genome amplification Methods 0.000 description 3
- 208000002672 hepatitis B Diseases 0.000 description 3
- 210000003630 histaminocyte Anatomy 0.000 description 3
- 238000010166 immunofluorescence Methods 0.000 description 3
- 229940072221 immunoglobulins Drugs 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 229940100601 interleukin-6 Drugs 0.000 description 3
- 210000003563 lymphoid tissue Anatomy 0.000 description 3
- 239000003147 molecular marker Substances 0.000 description 3
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 3
- 239000002751 oligonucleotide probe Substances 0.000 description 3
- 238000005457 optimization Methods 0.000 description 3
- 239000013610 patient sample Substances 0.000 description 3
- 239000011049 pearl Substances 0.000 description 3
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 3
- 238000005191 phase separation Methods 0.000 description 3
- 210000002381 plasma Anatomy 0.000 description 3
- 230000002062 proliferating effect Effects 0.000 description 3
- 239000007790 solid phase Substances 0.000 description 3
- 238000010186 staining Methods 0.000 description 3
- 238000010561 standard procedure Methods 0.000 description 3
- 150000003431 steroids Chemical class 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 239000000439 tumor marker Substances 0.000 description 3
- 238000002604 ultrasonography Methods 0.000 description 3
- 241001430294 unidentified retrovirus Species 0.000 description 3
- 230000008673 vomiting Effects 0.000 description 3
- 208000016261 weight loss Diseases 0.000 description 3
- 230000004580 weight loss Effects 0.000 description 3
- JVJFIQYAHPMBBX-UHFFFAOYSA-N 4-hydroxynonenal Chemical compound CCCCCC(O)C=CC=O JVJFIQYAHPMBBX-UHFFFAOYSA-N 0.000 description 2
- 208000024827 Alzheimer disease Diseases 0.000 description 2
- 208000008822 Ankylosis Diseases 0.000 description 2
- 206010003827 Autoimmune hepatitis Diseases 0.000 description 2
- 208000031628 Autosomal dominant spastic paraplegia type 3 Diseases 0.000 description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- 208000008035 Back Pain Diseases 0.000 description 2
- KPYSYYIEGFHWSV-UHFFFAOYSA-N Baclofen Chemical compound OC(=O)CC(CN)C1=CC=C(Cl)C=C1 KPYSYYIEGFHWSV-UHFFFAOYSA-N 0.000 description 2
- 235000017166 Bambusa arundinacea Nutrition 0.000 description 2
- 235000017491 Bambusa tulda Nutrition 0.000 description 2
- 241001330002 Bambuseae Species 0.000 description 2
- 201000004569 Blindness Diseases 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- 208000023611 Burkitt leukaemia Diseases 0.000 description 2
- 208000024172 Cardiovascular disease Diseases 0.000 description 2
- 206010050337 Cerumen impaction Diseases 0.000 description 2
- 206010008342 Cervix carcinoma Diseases 0.000 description 2
- 102000019034 Chemokines Human genes 0.000 description 2
- 108010012236 Chemokines Proteins 0.000 description 2
- 208000000094 Chronic Pain Diseases 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 208000019505 Deglutition disease Diseases 0.000 description 2
- 206010012438 Dermatitis atopic Diseases 0.000 description 2
- 206010017533 Fungal infection Diseases 0.000 description 2
- 102100024165 G1/S-specific cyclin-D1 Human genes 0.000 description 2
- 208000034826 Genetic Predisposition to Disease Diseases 0.000 description 2
- 108010072051 Glatiramer Acetate Proteins 0.000 description 2
- 208000015023 Graves' disease Diseases 0.000 description 2
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 2
- 101000980756 Homo sapiens G1/S-specific cyclin-D1 Proteins 0.000 description 2
- 102000012745 Immunoglobulin Subunits Human genes 0.000 description 2
- 108010079585 Immunoglobulin Subunits Proteins 0.000 description 2
- 102100034343 Integrase Human genes 0.000 description 2
- 108010005716 Interferon beta-1a Proteins 0.000 description 2
- 102000051628 Interleukin-1 receptor antagonist Human genes 0.000 description 2
- 108700021006 Interleukin-1 receptor antagonist Proteins 0.000 description 2
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 2
- 208000031671 Large B-Cell Diffuse Lymphoma Diseases 0.000 description 2
- 208000030289 Lymphoproliferative disease Diseases 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 208000003250 Mixed connective tissue disease Diseases 0.000 description 2
- 208000034578 Multiple myelomas Diseases 0.000 description 2
- 208000010428 Muscle Weakness Diseases 0.000 description 2
- 208000000112 Myalgia Diseases 0.000 description 2
- 206010028813 Nausea Diseases 0.000 description 2
- 206010029216 Nervousness Diseases 0.000 description 2
- 101150084044 P gene Proteins 0.000 description 2
- 238000012408 PCR amplification Methods 0.000 description 2
- 206010034277 Pemphigoid Diseases 0.000 description 2
- 208000031845 Pernicious anaemia Diseases 0.000 description 2
- 235000015334 Phyllostachys viridis Nutrition 0.000 description 2
- 206010035664 Pneumonia Diseases 0.000 description 2
- 208000035416 Prolymphocytic B-Cell Leukemia Diseases 0.000 description 2
- 206010068513 Pulmonary renal syndrome Diseases 0.000 description 2
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 2
- 208000005392 Spasm Diseases 0.000 description 2
- 230000005867 T cell response Effects 0.000 description 2
- 206010042971 T-cell lymphoma Diseases 0.000 description 2
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 2
- 208000026651 T-cell prolymphocytic leukemia Diseases 0.000 description 2
- 102000011923 Thyrotropin Human genes 0.000 description 2
- 108010061174 Thyrotropin Proteins 0.000 description 2
- 206010044565 Tremor Diseases 0.000 description 2
- 102100040247 Tumor necrosis factor Human genes 0.000 description 2
- 206010053613 Type IV hypersensitivity reaction Diseases 0.000 description 2
- 208000025865 Ulcer Diseases 0.000 description 2
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 2
- 206010047115 Vasculitis Diseases 0.000 description 2
- 208000021017 Weight Gain Diseases 0.000 description 2
- PNNCWTXUWKENPE-UHFFFAOYSA-N [N].NC(N)=O Chemical compound [N].NC(N)=O PNNCWTXUWKENPE-UHFFFAOYSA-N 0.000 description 2
- 230000001133 acceleration Effects 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 229960002964 adalimumab Drugs 0.000 description 2
- 230000004721 adaptive immunity Effects 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 230000032683 aging Effects 0.000 description 2
- 208000030961 allergic reaction Diseases 0.000 description 2
- 230000003466 anti-cipated effect Effects 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 230000003460 anti-nuclear Effects 0.000 description 2
- 210000000612 antigen-presenting cell Anatomy 0.000 description 2
- 208000010216 atopic IgE responsiveness Diseases 0.000 description 2
- 201000008937 atopic dermatitis Diseases 0.000 description 2
- 208000010668 atopic eczema Diseases 0.000 description 2
- 239000011425 bamboo Substances 0.000 description 2
- 230000004888 barrier function Effects 0.000 description 2
- 230000037429 base substitution Effects 0.000 description 2
- 108700042656 bcl-1 Genes Proteins 0.000 description 2
- 230000036772 blood pressure Effects 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 230000011712 cell development Effects 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 210000002939 cerumen Anatomy 0.000 description 2
- 201000010881 cervical cancer Diseases 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 238000009535 clinical urine test Methods 0.000 description 2
- 238000010367 cloning Methods 0.000 description 2
- 208000029742 colonic neoplasm Diseases 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 229960001334 corticosteroids Drugs 0.000 description 2
- CVSVTCORWBXHQV-UHFFFAOYSA-N creatine Chemical compound NC(=[NH2+])N(C)CC([O-])=O CVSVTCORWBXHQV-UHFFFAOYSA-N 0.000 description 2
- 230000009260 cross reactivity Effects 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 238000004163 cytometry Methods 0.000 description 2
- 230000001086 cytosolic effect Effects 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 238000004925 denaturation Methods 0.000 description 2
- 230000036425 denaturation Effects 0.000 description 2
- 230000008021 deposition Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000007847 digital PCR Methods 0.000 description 2
- 239000000428 dust Substances 0.000 description 2
- 229960000403 etanercept Drugs 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 150000002343 gold Chemical class 0.000 description 2
- 150000002344 gold compounds Chemical class 0.000 description 2
- 210000005003 heart tissue Anatomy 0.000 description 2
- 201000007474 hereditary spastic paraplegia 3A Diseases 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 230000000899 immune system response Effects 0.000 description 2
- 230000002163 immunogen Effects 0.000 description 2
- 238000013394 immunophenotyping Methods 0.000 description 2
- 229960003444 immunosuppressant agent Drugs 0.000 description 2
- 229940125721 immunosuppressive agent Drugs 0.000 description 2
- 238000009169 immunotherapy Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000000415 inactivating effect Effects 0.000 description 2
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- 229960000598 infliximab Drugs 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 208000018937 joint inflammation Diseases 0.000 description 2
- 238000011862 kidney biopsy Methods 0.000 description 2
- 229960000681 leflunomide Drugs 0.000 description 2
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- 238000007449 liver function test Methods 0.000 description 2
- 208000018769 loss of vision Diseases 0.000 description 2
- 231100000864 loss of vision Toxicity 0.000 description 2
- 210000002751 lymph Anatomy 0.000 description 2
- 210000001165 lymph node Anatomy 0.000 description 2
- 230000036210 malignancy Effects 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 239000000693 micelle Substances 0.000 description 2
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 2
- 206010063344 microscopic polyangiitis Diseases 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- DYKFCLLONBREIL-KVUCHLLUSA-N minocycline Chemical compound C([C@H]1C2)C3=C(N(C)C)C=CC(O)=C3C(=O)C1=C(O)[C@@]1(O)[C@@H]2[C@H](N(C)C)C(O)=C(C(N)=O)C1=O DYKFCLLONBREIL-KVUCHLLUSA-N 0.000 description 2
- 229960004023 minocycline Drugs 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- 210000003097 mucus Anatomy 0.000 description 2
- 201000000050 myeloid neoplasm Diseases 0.000 description 2
- 230000008693 nausea Effects 0.000 description 2
- 230000000926 neurological effect Effects 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 2
- 231100000862 numbness Toxicity 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 229940124641 pain reliever Drugs 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- 229960005489 paracetamol Drugs 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- 210000004197 pelvis Anatomy 0.000 description 2
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 2
- 210000004180 plasmocyte Anatomy 0.000 description 2
- 238000006116 polymerization reaction Methods 0.000 description 2
- 230000001124 posttranscriptional effect Effects 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 238000001959 radiotherapy Methods 0.000 description 2
- 230000004043 responsiveness Effects 0.000 description 2
- 229960004641 rituximab Drugs 0.000 description 2
- 238000005096 rolling process Methods 0.000 description 2
- 210000003131 sacroiliac joint Anatomy 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 238000010845 search algorithm Methods 0.000 description 2
- 230000001932 seasonal effect Effects 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 238000011896 sensitive detection Methods 0.000 description 2
- 238000007390 skin biopsy Methods 0.000 description 2
- 229960001940 sulfasalazine Drugs 0.000 description 2
- NCEXYHBECQHGNR-QZQOTICOSA-N sulfasalazine Chemical compound C1=C(O)C(C(=O)O)=CC(\N=N\C=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-QZQOTICOSA-N 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 201000004595 synovitis Diseases 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- 210000001541 thymus gland Anatomy 0.000 description 2
- 230000000451 tissue damage Effects 0.000 description 2
- 231100000827 tissue damage Toxicity 0.000 description 2
- XFYDIVBRZNQMJC-UHFFFAOYSA-N tizanidine Chemical compound ClC=1C=CC2=NSN=C2C=1NC1=NCCN1 XFYDIVBRZNQMJC-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 208000027930 type IV hypersensitivity disease Diseases 0.000 description 2
- 229940079023 tysabri Drugs 0.000 description 2
- 231100000397 ulcer Toxicity 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 230000004393 visual impairment Effects 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 230000004584 weight gain Effects 0.000 description 2
- 235000019786 weight gain Nutrition 0.000 description 2
- GIANIJCPTPUNBA-QMMMGPOBSA-N (2s)-3-(4-hydroxyphenyl)-2-nitramidopropanoic acid Chemical compound [O-][N+](=O)N[C@H](C(=O)O)CC1=CC=C(O)C=C1 GIANIJCPTPUNBA-QMMMGPOBSA-N 0.000 description 1
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 208000004998 Abdominal Pain Diseases 0.000 description 1
- 208000000187 Abnormal Reflex Diseases 0.000 description 1
- 206010000171 Abnormal reflexes Diseases 0.000 description 1
- 206010000830 Acute leukaemia Diseases 0.000 description 1
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 description 1
- 208000026872 Addison Disease Diseases 0.000 description 1
- 208000016683 Adult T-cell leukemia/lymphoma Diseases 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 208000035285 Allergic Seasonal Rhinitis Diseases 0.000 description 1
- 201000004384 Alopecia Diseases 0.000 description 1
- 206010001935 American trypanosomiasis Diseases 0.000 description 1
- 206010073478 Anaplastic large-cell lymphoma Diseases 0.000 description 1
- 102100040006 Annexin A1 Human genes 0.000 description 1
- 108090000663 Annexin A1 Proteins 0.000 description 1
- 208000003343 Antiphospholipid Syndrome Diseases 0.000 description 1
- 206010002921 Aortitis Diseases 0.000 description 1
- 208000036170 B-Cell Marginal Zone Lymphoma Diseases 0.000 description 1
- 208000025324 B-cell acute lymphoblastic leukemia Diseases 0.000 description 1
- 102100021631 B-cell lymphoma 6 protein Human genes 0.000 description 1
- 208000032568 B-cell prolymphocytic leukaemia Diseases 0.000 description 1
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 1
- 108700020463 BRCA1 Proteins 0.000 description 1
- 102000036365 BRCA1 Human genes 0.000 description 1
- 101150072950 BRCA1 gene Proteins 0.000 description 1
- 108700020462 BRCA2 Proteins 0.000 description 1
- 102000052609 BRCA2 Human genes 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 208000023328 Basedow disease Diseases 0.000 description 1
- 208000035462 Biphenotypic Acute Leukemia Diseases 0.000 description 1
- 206010051779 Bone marrow toxicity Diseases 0.000 description 1
- 101150008921 Brca2 gene Proteins 0.000 description 1
- 229940124292 CD20 monoclonal antibody Drugs 0.000 description 1
- 201000004085 CLL/SLL Diseases 0.000 description 1
- 101150071146 COX2 gene Proteins 0.000 description 1
- 101100114534 Caenorhabditis elegans ctc-2 gene Proteins 0.000 description 1
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 1
- 208000024699 Chagas disease Diseases 0.000 description 1
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 1
- 206010009192 Circulatory collapse Diseases 0.000 description 1
- 241000193163 Clostridioides difficile Species 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- 206010010774 Constipation Diseases 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- 241000711573 Coronaviridae Species 0.000 description 1
- 108010069514 Cyclic Peptides Proteins 0.000 description 1
- 102000001189 Cyclic Peptides Human genes 0.000 description 1
- XUIIKFGFIJCVMT-GFCCVEGCSA-N D-thyroxine Chemical compound IC1=CC(C[C@@H](N)C(O)=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-GFCCVEGCSA-N 0.000 description 1
- 102000012410 DNA Ligases Human genes 0.000 description 1
- 108010061982 DNA Ligases Proteins 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 206010011878 Deafness Diseases 0.000 description 1
- 208000006313 Delayed Hypersensitivity Diseases 0.000 description 1
- 241000725619 Dengue virus Species 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 206010012434 Dermatitis allergic Diseases 0.000 description 1
- 206010012442 Dermatitis contact Diseases 0.000 description 1
- 208000003164 Diplopia Diseases 0.000 description 1
- 208000003556 Dry Eye Syndromes Diseases 0.000 description 1
- 206010013774 Dry eye Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 102100025137 Early activation antigen CD69 Human genes 0.000 description 1
- 206010063045 Effusion Diseases 0.000 description 1
- 206010015150 Erythema Diseases 0.000 description 1
- 206010061850 Extranodal marginal zone B-cell lymphoma (MALT type) Diseases 0.000 description 1
- 206010015943 Eye inflammation Diseases 0.000 description 1
- 206010015958 Eye pain Diseases 0.000 description 1
- 206010016059 Facial pain Diseases 0.000 description 1
- 206010053172 Fatal outcomes Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102000008857 Ferritin Human genes 0.000 description 1
- 108050000784 Ferritin Proteins 0.000 description 1
- 238000008416 Ferritin Methods 0.000 description 1
- 229910052688 Gadolinium Inorganic materials 0.000 description 1
- 206010017577 Gait disturbance Diseases 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- 108010068370 Glutens Proteins 0.000 description 1
- 206010018498 Goitre Diseases 0.000 description 1
- 208000024869 Goodpasture syndrome Diseases 0.000 description 1
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 description 1
- 208000003807 Graves Disease Diseases 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 102100028976 HLA class I histocompatibility antigen, B alpha chain Human genes 0.000 description 1
- 108010058607 HLA-B Antigens Proteins 0.000 description 1
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 1
- 208000018565 Hemochromatosis Diseases 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 description 1
- 241000711549 Hepacivirus C Species 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 206010019842 Hepatomegaly Diseases 0.000 description 1
- 108091027305 Heteroduplex Proteins 0.000 description 1
- 102000029746 Histidine-tRNA Ligase Human genes 0.000 description 1
- 108700016229 Histidine-tRNA ligases Proteins 0.000 description 1
- 108010033040 Histones Proteins 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000971234 Homo sapiens B-cell lymphoma 6 protein Proteins 0.000 description 1
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 1
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 description 1
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 1
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 description 1
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 1
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 1
- 101001090860 Homo sapiens Myeloblastin Proteins 0.000 description 1
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 description 1
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 1
- 241000341655 Human papillomavirus type 16 Species 0.000 description 1
- 208000008454 Hyperhidrosis Diseases 0.000 description 1
- 208000004044 Hypesthesia Diseases 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- 206010021245 Idiopathic thrombocytopenic purpura Diseases 0.000 description 1
- 208000001718 Immediate Hypersensitivity Diseases 0.000 description 1
- 102000013463 Immunoglobulin Light Chains Human genes 0.000 description 1
- 108010065825 Immunoglobulin Light Chains Proteins 0.000 description 1
- 206010021639 Incontinence Diseases 0.000 description 1
- 206010062717 Increased upper airway secretion Diseases 0.000 description 1
- 208000027601 Inner ear disease Diseases 0.000 description 1
- 108010005714 Interferon beta-1b Proteins 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 102000000589 Interleukin-1 Human genes 0.000 description 1
- 208000005615 Interstitial Cystitis Diseases 0.000 description 1
- 206010022941 Iridocyclitis Diseases 0.000 description 1
- 206010023126 Jaundice Diseases 0.000 description 1
- 206010023198 Joint ankylosis Diseases 0.000 description 1
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 1
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 1
- 238000008214 LDL Cholesterol Methods 0.000 description 1
- 108010063045 Lactoferrin Proteins 0.000 description 1
- 102100032241 Lactotransferrin Human genes 0.000 description 1
- 208000006404 Large Granular Lymphocytic Leukemia Diseases 0.000 description 1
- 208000032004 Large-Cell Anaplastic Lymphoma Diseases 0.000 description 1
- 208000032420 Latent Infection Diseases 0.000 description 1
- 241000713666 Lentivirus Species 0.000 description 1
- 102000001109 Leukocyte L1 Antigen Complex Human genes 0.000 description 1
- 108010069316 Leukocyte L1 Antigen Complex Proteins 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 201000003791 MALT lymphoma Diseases 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- WSMYVTOQOOLQHP-UHFFFAOYSA-N Malondialdehyde Chemical compound O=CCC=O WSMYVTOQOOLQHP-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 1
- 208000035490 Megakaryoblastic Acute Leukemia Diseases 0.000 description 1
- RJQXTJLFIWVMTO-TYNCELHUSA-N Methicillin Chemical compound COC1=CC=CC(OC)=C1C(=O)N[C@@H]1C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 RJQXTJLFIWVMTO-TYNCELHUSA-N 0.000 description 1
- 108060004795 Methyltransferase Proteins 0.000 description 1
- 108700011259 MicroRNAs Proteins 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- 108020005196 Mitochondrial DNA Proteins 0.000 description 1
- 102100034256 Mucin-1 Human genes 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 208000008238 Muscle Spasticity Diseases 0.000 description 1
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 1
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 1
- 208000031888 Mycoses Diseases 0.000 description 1
- 102100034681 Myeloblastin Human genes 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 1
- 208000028389 Nerve injury Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000011644 Neurologic Gait disease Diseases 0.000 description 1
- 206010029461 Nodal marginal zone B-cell lymphomas Diseases 0.000 description 1
- 206010029803 Nosocomial infection Diseases 0.000 description 1
- 102000007999 Nuclear Proteins Human genes 0.000 description 1
- 108010089610 Nuclear Proteins Proteins 0.000 description 1
- 102100030569 Nuclear receptor corepressor 2 Human genes 0.000 description 1
- 101710153660 Nuclear receptor corepressor 2 Proteins 0.000 description 1
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 1
- 208000002366 Nut Hypersensitivity Diseases 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 206010061137 Ocular toxicity Diseases 0.000 description 1
- 208000006187 Onycholysis Diseases 0.000 description 1
- 206010031009 Oral pain Diseases 0.000 description 1
- 206010068319 Oropharyngeal pain Diseases 0.000 description 1
- 101150000187 PTGS2 gene Proteins 0.000 description 1
- 206010033661 Pancytopenia Diseases 0.000 description 1
- 208000031816 Pathologic Dilatation Diseases 0.000 description 1
- 208000037273 Pathologic Processes Diseases 0.000 description 1
- 201000011152 Pemphigus Diseases 0.000 description 1
- 241000721454 Pemphigus Species 0.000 description 1
- 208000027190 Peripheral T-cell lymphomas Diseases 0.000 description 1
- 201000007100 Pharyngitis Diseases 0.000 description 1
- 206010034960 Photophobia Diseases 0.000 description 1
- ZYFVNVRFVHJEIU-UHFFFAOYSA-N PicoGreen Chemical compound CN(C)CCCN(CCCN(C)C)C1=CC(=CC2=[N+](C3=CC=CC=C3S2)C)C2=CC=CC=C2N1C1=CC=CC=C1 ZYFVNVRFVHJEIU-UHFFFAOYSA-N 0.000 description 1
- 206010035148 Plague Diseases 0.000 description 1
- 208000007452 Plasmacytoma Diseases 0.000 description 1
- 206010035742 Pneumonitis Diseases 0.000 description 1
- 206010065857 Primary Effusion Lymphoma Diseases 0.000 description 1
- 206010036711 Primary mediastinal large B-cell lymphomas Diseases 0.000 description 1
- 208000033826 Promyelocytic Acute Leukemia Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 101710130181 Protochlorophyllide reductase A, chloroplastic Proteins 0.000 description 1
- 241000442474 Pulsatilla vulgaris Species 0.000 description 1
- 239000013614 RNA sample Substances 0.000 description 1
- 238000011529 RT qPCR Methods 0.000 description 1
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 208000035415 Reinfection Diseases 0.000 description 1
- 208000007660 Residual Neoplasm Diseases 0.000 description 1
- 241000219061 Rheum Species 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 102000002278 Ribosomal Proteins Human genes 0.000 description 1
- 108010000605 Ribosomal Proteins Proteins 0.000 description 1
- 206010039361 Sacroiliitis Diseases 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 208000019802 Sexually transmitted disease Diseases 0.000 description 1
- 208000009359 Sezary Syndrome Diseases 0.000 description 1
- 208000021388 Sezary disease Diseases 0.000 description 1
- 208000032023 Signs and Symptoms Diseases 0.000 description 1
- 206010040867 Skin hypertrophy Diseases 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- 206010041519 Spider naevus Diseases 0.000 description 1
- 208000007156 Spondylarthritis Diseases 0.000 description 1
- 241000191967 Staphylococcus aureus Species 0.000 description 1
- 102000019197 Superoxide Dismutase Human genes 0.000 description 1
- 108010012715 Superoxide dismutase Proteins 0.000 description 1
- 201000009594 Systemic Scleroderma Diseases 0.000 description 1
- 206010042953 Systemic sclerosis Diseases 0.000 description 1
- 230000020385 T cell costimulation Effects 0.000 description 1
- 208000031672 T-Cell Peripheral Lymphoma Diseases 0.000 description 1
- 201000008717 T-cell large granular lymphocyte leukemia Diseases 0.000 description 1
- 208000000389 T-cell leukemia Diseases 0.000 description 1
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 description 1
- 208000001871 Tachycardia Diseases 0.000 description 1
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 1
- 206010043521 Throat irritation Diseases 0.000 description 1
- 208000031981 Thrombocytopenic Idiopathic Purpura Diseases 0.000 description 1
- 208000034841 Thrombotic Microangiopathies Diseases 0.000 description 1
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical compound IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 description 1
- 101710183280 Topoisomerase Proteins 0.000 description 1
- 206010044245 Toxic optic neuropathy Diseases 0.000 description 1
- 241000159243 Toxicodendron radicans Species 0.000 description 1
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 1
- 241000209140 Triticum Species 0.000 description 1
- 235000021307 Triticum Nutrition 0.000 description 1
- 241000223109 Trypanosoma cruzi Species 0.000 description 1
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 206010045240 Type I hypersensitivity Diseases 0.000 description 1
- 206010046851 Uveitis Diseases 0.000 description 1
- 241000700647 Variola virus Species 0.000 description 1
- 102000013127 Vimentin Human genes 0.000 description 1
- 108010065472 Vimentin Proteins 0.000 description 1
- 208000016025 Waldenstroem macroglobulinemia Diseases 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- 241000607479 Yersinia pestis Species 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 229960004748 abacavir Drugs 0.000 description 1
- MCGSCOLBFJQGHM-SCZZXKLOSA-N abacavir Chemical compound C=12N=CN([C@H]3C=C[C@@H](CO)C3)C2=NC(N)=NC=1NC1CC1 MCGSCOLBFJQGHM-SCZZXKLOSA-N 0.000 description 1
- 229960003697 abatacept Drugs 0.000 description 1
- 206010000059 abdominal discomfort Diseases 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- FHEAIOHRHQGZPC-KIWGSFCNSA-N acetic acid;(2s)-2-amino-3-(4-hydroxyphenyl)propanoic acid;(2s)-2-aminopentanedioic acid;(2s)-2-aminopropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical compound CC(O)=O.C[C@H](N)C(O)=O.NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CCC(O)=O.OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 FHEAIOHRHQGZPC-KIWGSFCNSA-N 0.000 description 1
- 229940119059 actemra Drugs 0.000 description 1
- 208000024716 acute asthma Diseases 0.000 description 1
- 208000036677 acute biphenotypic leukemia Diseases 0.000 description 1
- 208000005652 acute fatty liver of pregnancy Diseases 0.000 description 1
- 208000013593 acute megakaryoblastic leukemia Diseases 0.000 description 1
- 208000020700 acute megakaryocytic leukemia Diseases 0.000 description 1
- 230000033289 adaptive immune response Effects 0.000 description 1
- 238000011374 additional therapy Methods 0.000 description 1
- 210000002534 adenoid Anatomy 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 201000006966 adult T-cell leukemia Diseases 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 238000011256 aggressive treatment Methods 0.000 description 1
- 238000007844 allele-specific PCR Methods 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000000961 alloantigen Effects 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 210000004381 amniotic fluid Anatomy 0.000 description 1
- 229960004238 anakinra Drugs 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 206010002449 angioimmunoblastic T-cell lymphoma Diseases 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 208000022531 anorexia Diseases 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 201000004612 anterior uveitis Diseases 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000003356 anti-rheumatic effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 230000030741 antigen processing and presentation Effects 0.000 description 1
- 239000003435 antirheumatic agent Substances 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 230000004596 appetite loss Effects 0.000 description 1
- 210000001742 aqueous humor Anatomy 0.000 description 1
- 238000000149 argon plasma sintering Methods 0.000 description 1
- 210000001188 articular cartilage Anatomy 0.000 description 1
- 230000000712 assembly Effects 0.000 description 1
- 238000000429 assembly Methods 0.000 description 1
- 238000007845 assembly PCR Methods 0.000 description 1
- 238000007846 asymmetric PCR Methods 0.000 description 1
- 201000000448 autoimmune hemolytic anemia Diseases 0.000 description 1
- 201000003710 autoimmune thrombocytopenic purpura Diseases 0.000 description 1
- 229940003504 avonex Drugs 0.000 description 1
- 229960000794 baclofen Drugs 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 208000013404 behavioral symptom Diseases 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940049706 benzodiazepine Drugs 0.000 description 1
- 229940021459 betaseron Drugs 0.000 description 1
- 230000002457 bidirectional effect Effects 0.000 description 1
- 210000000941 bile Anatomy 0.000 description 1
- 239000003124 biologic agent Substances 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 208000002352 blister Diseases 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 231100000366 bone marrow toxicity Toxicity 0.000 description 1
- 230000006931 brain damage Effects 0.000 description 1
- 231100000874 brain damage Toxicity 0.000 description 1
- 208000029028 brain injury Diseases 0.000 description 1
- 210000005252 bulbus oculi Anatomy 0.000 description 1
- 210000001217 buttock Anatomy 0.000 description 1
- 229940046731 calcineurin inhibitors Drugs 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- 229940107810 cellcept Drugs 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 210000000038 chest Anatomy 0.000 description 1
- 230000001713 cholinergic effect Effects 0.000 description 1
- 208000023819 chronic asthma Diseases 0.000 description 1
- 208000024207 chronic leukemia Diseases 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000023738 chronic lymphocytic leukemia/small lymphocytic lymphoma Diseases 0.000 description 1
- 201000006778 chronic monocytic leukemia Diseases 0.000 description 1
- 201000010902 chronic myelomonocytic leukemia Diseases 0.000 description 1
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 description 1
- 201000009151 chronic rhinitis Diseases 0.000 description 1
- 231100000749 chronicity Toxicity 0.000 description 1
- 210000004913 chyme Anatomy 0.000 description 1
- 229960002173 citrulline Drugs 0.000 description 1
- 235000013477 citrulline Nutrition 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 208000010247 contact dermatitis Diseases 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 229940038717 copaxone Drugs 0.000 description 1
- 230000000139 costimulatory effect Effects 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- 229960003624 creatine Drugs 0.000 description 1
- 239000006046 creatine Substances 0.000 description 1
- 230000001351 cycling effect Effects 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 230000002559 cytogenic effect Effects 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 206010061428 decreased appetite Diseases 0.000 description 1
- 238000012350 deep sequencing Methods 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000003831 deregulation Effects 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000007435 diagnostic evaluation Methods 0.000 description 1
- 238000012631 diagnostic technique Methods 0.000 description 1
- 230000009274 differential gene expression Effects 0.000 description 1
- 239000012470 diluted sample Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- NTGLQWGMESPVBV-UHFFFAOYSA-N diproqualone Chemical compound C1=CC=C2C(=O)N(CC(O)CO)C(C)=NC2=C1 NTGLQWGMESPVBV-UHFFFAOYSA-N 0.000 description 1
- 229950003185 diproqualone Drugs 0.000 description 1
- 238000002845 discoloration Methods 0.000 description 1
- 208000037771 disease arising from reactivation of latent virus Diseases 0.000 description 1
- 231100000676 disease causative agent Toxicity 0.000 description 1
- ZGSPNIOCEDOHGS-UHFFFAOYSA-L disodium [3-[2,3-di(octadeca-9,12-dienoyloxy)propoxy-oxidophosphoryl]oxy-2-hydroxypropyl] 2,3-di(octadeca-9,12-dienoyloxy)propyl phosphate Chemical compound [Na+].[Na+].CCCCCC=CCC=CCCCCCCCC(=O)OCC(OC(=O)CCCCCCCC=CCC=CCCCCC)COP([O-])(=O)OCC(O)COP([O-])(=O)OCC(OC(=O)CCCCCCCC=CCC=CCCCCC)COC(=O)CCCCCCCC=CCC=CCCCCC ZGSPNIOCEDOHGS-UHFFFAOYSA-L 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 101150007302 dntt gene Proteins 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 208000029444 double vision Diseases 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 210000003162 effector t lymphocyte Anatomy 0.000 description 1
- 210000001513 elbow Anatomy 0.000 description 1
- 229940073621 enbrel Drugs 0.000 description 1
- 201000002491 encephalomyelitis Diseases 0.000 description 1
- 208000037902 enteropathy Diseases 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000003628 erosive effect Effects 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- 230000000763 evoking effect Effects 0.000 description 1
- 238000007387 excisional biopsy Methods 0.000 description 1
- 210000003722 extracellular fluid Anatomy 0.000 description 1
- 230000004424 eye movement Effects 0.000 description 1
- 208000027993 eye symptom Diseases 0.000 description 1
- 210000000887 face Anatomy 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- 210000003754 fetus Anatomy 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 210000000285 follicular dendritic cell Anatomy 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000011990 functional testing Methods 0.000 description 1
- 208000024386 fungal infectious disease Diseases 0.000 description 1
- 229940084434 fungoid Drugs 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 1
- 238000002695 general anesthesia Methods 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 229960003776 glatiramer acetate Drugs 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 235000021312 gluten Nutrition 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 201000003872 goiter Diseases 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 210000002768 hair cell Anatomy 0.000 description 1
- 208000024963 hair loss Diseases 0.000 description 1
- 230000003676 hair loss Effects 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 230000010370 hearing loss Effects 0.000 description 1
- 231100000888 hearing loss Toxicity 0.000 description 1
- 208000016354 hearing loss disease Diseases 0.000 description 1
- 208000025750 heavy chain disease Diseases 0.000 description 1
- 238000005534 hematocrit Methods 0.000 description 1
- 208000007475 hemolytic anemia Diseases 0.000 description 1
- 206010066957 hepatosplenic T-cell lymphoma Diseases 0.000 description 1
- 231100000304 hepatotoxicity Toxicity 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 208000002557 hidradenitis Diseases 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 235000020256 human milk Nutrition 0.000 description 1
- 210000004251 human milk Anatomy 0.000 description 1
- 239000010800 human waste Substances 0.000 description 1
- 229940048921 humira Drugs 0.000 description 1
- 208000034783 hypoesthesia Diseases 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 230000003100 immobilizing effect Effects 0.000 description 1
- 230000006450 immune cell response Effects 0.000 description 1
- 238000011493 immune profiling Methods 0.000 description 1
- 229940127121 immunoconjugate Drugs 0.000 description 1
- 230000007233 immunological mechanism Effects 0.000 description 1
- 229940073062 imuran Drugs 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 238000007386 incisional biopsy Methods 0.000 description 1
- 206010021654 increased appetite Diseases 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 208000000509 infertility Diseases 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 231100000535 infertility Toxicity 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 230000006749 inflammatory damage Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 210000005007 innate immune system Anatomy 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 208000028774 intestinal disease Diseases 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 210000002977 intracellular fluid Anatomy 0.000 description 1
- 208000026876 intravascular large B-cell lymphoma Diseases 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 230000000366 juvenile effect Effects 0.000 description 1
- 201000002215 juvenile rheumatoid arthritis Diseases 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 229940054136 kineret Drugs 0.000 description 1
- 210000003127 knee Anatomy 0.000 description 1
- CSSYQJWUGATIHM-IKGCZBKSSA-N l-phenylalanyl-l-lysyl-l-cysteinyl-l-arginyl-l-arginyl-l-tryptophyl-l-glutaminyl-l-tryptophyl-l-arginyl-l-methionyl-l-lysyl-l-lysyl-l-leucylglycyl-l-alanyl-l-prolyl-l-seryl-l-isoleucyl-l-threonyl-l-cysteinyl-l-valyl-l-arginyl-l-arginyl-l-alanyl-l-phenylal Chemical compound C([C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CC=CC=C1 CSSYQJWUGATIHM-IKGCZBKSSA-N 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 229940078795 lactoferrin Drugs 0.000 description 1
- 235000021242 lactoferrin Nutrition 0.000 description 1
- 201000003445 large cell neuroendocrine carcinoma Diseases 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 150000002617 leukotrienes Chemical class 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 238000007834 ligase chain reaction Methods 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229940063721 lioresal Drugs 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 238000011866 long-term treatment Methods 0.000 description 1
- 208000019017 loss of appetite Diseases 0.000 description 1
- 235000021266 loss of appetite Nutrition 0.000 description 1
- 208000018883 loss of balance Diseases 0.000 description 1
- 238000005461 lubrication Methods 0.000 description 1
- 238000009593 lumbar puncture Methods 0.000 description 1
- 210000004705 lumbosacral region Anatomy 0.000 description 1
- 235000019689 luncheon sausage Nutrition 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 208000003747 lymphoid leukemia Diseases 0.000 description 1
- 208000007282 lymphomatoid papulosis Diseases 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- 206010025482 malaise Diseases 0.000 description 1
- 210000003794 male germ cell Anatomy 0.000 description 1
- 229940118019 malondialdehyde Drugs 0.000 description 1
- 210000003826 marginal zone b cell Anatomy 0.000 description 1
- 201000000638 mature B-cell neoplasm Diseases 0.000 description 1
- 208000020968 mature T-cell and NK-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000001806 memory b lymphocyte Anatomy 0.000 description 1
- 210000003071 memory t lymphocyte Anatomy 0.000 description 1
- 230000005906 menstruation Effects 0.000 description 1
- 230000003340 mental effect Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229960003085 meticillin Drugs 0.000 description 1
- 238000001531 micro-dissection Methods 0.000 description 1
- 239000002679 microRNA Substances 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 230000037230 mobility Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 230000036651 mood Effects 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 229960003816 muromonab-cd3 Drugs 0.000 description 1
- 208000016334 muscle symptom Diseases 0.000 description 1
- 230000036473 myasthenia Effects 0.000 description 1
- 229940014456 mycophenolate Drugs 0.000 description 1
- RTGDFNSFWBGLEC-SYZQJQIISA-N mycophenolate mofetil Chemical compound COC1=C(C)C=2COC(=O)C=2C(O)=C1C\C=C(/C)CCC(=O)OCCN1CCOCC1 RTGDFNSFWBGLEC-SYZQJQIISA-N 0.000 description 1
- 229960004866 mycophenolate mofetil Drugs 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- 201000005962 mycosis fungoides Diseases 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 229960005027 natalizumab Drugs 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 208000010915 neoplasm of mature B-cells Diseases 0.000 description 1
- 208000021119 neoplasm of mature T-cells or NK-cells Diseases 0.000 description 1
- 231100000417 nephrotoxicity Toxicity 0.000 description 1
- 230000008764 nerve damage Effects 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 238000007857 nested PCR Methods 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 239000002853 nucleic acid probe Substances 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- 201000010854 nut allergy Diseases 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- 238000001584 occupational therapy Methods 0.000 description 1
- 231100000327 ocular toxicity Toxicity 0.000 description 1
- 235000019645 odor Nutrition 0.000 description 1
- 229940127240 opiate Drugs 0.000 description 1
- 239000000014 opioid analgesic Substances 0.000 description 1
- 229940005483 opioid analgesics Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 229940035567 orencia Drugs 0.000 description 1
- 229940046781 other immunosuppressants in atc Drugs 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 1
- 239000005022 packaging material Substances 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 230000000242 pagocytic effect Effects 0.000 description 1
- 210000002741 palatine tonsil Anatomy 0.000 description 1
- 206010033675 panniculitis Diseases 0.000 description 1
- 239000000123 paper Substances 0.000 description 1
- 208000035824 paresthesia Diseases 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000009054 pathological process Effects 0.000 description 1
- 201000001976 pemphigus vulgaris Diseases 0.000 description 1
- 210000003516 pericardium Anatomy 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 229960002895 phenylbutazone Drugs 0.000 description 1
- VYMDGNCVAMGZFE-UHFFFAOYSA-N phenylbutazonum Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 VYMDGNCVAMGZFE-UHFFFAOYSA-N 0.000 description 1
- 210000004214 philadelphia chromosome Anatomy 0.000 description 1
- 208000026435 phlegm Diseases 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 238000000554 physical therapy Methods 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 208000010626 plasma cell neoplasm Diseases 0.000 description 1
- 210000004224 pleura Anatomy 0.000 description 1
- 210000004910 pleural fluid Anatomy 0.000 description 1
- 238000012123 point-of-care testing Methods 0.000 description 1
- 230000010287 polarization Effects 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- 210000004909 pre-ejaculatory fluid Anatomy 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 208000017426 precursor B-cell acute lymphoblastic leukemia Diseases 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000037452 priming Effects 0.000 description 1
- 208000037821 progressive disease Diseases 0.000 description 1
- 206010036807 progressive multifocal leukoencephalopathy Diseases 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 210000004915 pus Anatomy 0.000 description 1
- 238000012175 pyrosequencing Methods 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 229940038850 rebif Drugs 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 210000003289 regulatory T cell Anatomy 0.000 description 1
- 229940116176 remicade Drugs 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 206010039083 rhinitis Diseases 0.000 description 1
- 239000003161 ribonuclease inhibitor Substances 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 210000004918 root sheath Anatomy 0.000 description 1
- 238000009666 routine test Methods 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 210000003786 sclera Anatomy 0.000 description 1
- 210000002374 sebum Anatomy 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 210000000582 semen Anatomy 0.000 description 1
- 230000035807 sensation Effects 0.000 description 1
- 235000019615 sensations Nutrition 0.000 description 1
- 238000011451 sequencing strategy Methods 0.000 description 1
- 230000000405 serological effect Effects 0.000 description 1
- 238000009589 serological test Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 206010040560 shock Diseases 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 229940126586 small molecule drug Drugs 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 206010062113 splenic marginal zone lymphoma Diseases 0.000 description 1
- 238000007447 staining method Methods 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 210000004304 subcutaneous tissue Anatomy 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 230000009747 swallowing Effects 0.000 description 1
- 210000004243 sweat Anatomy 0.000 description 1
- 230000035900 sweating Effects 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 230000006794 tachycardia Effects 0.000 description 1
- 229960001967 tacrolimus Drugs 0.000 description 1
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 1
- 235000019640 taste Nutrition 0.000 description 1
- 210000001138 tear Anatomy 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- 230000035922 thirst Effects 0.000 description 1
- 229940034208 thyroxine Drugs 0.000 description 1
- XUIIKFGFIJCVMT-UHFFFAOYSA-N thyroxine-binding globulin Natural products IC1=CC(CC([NH3+])C([O-])=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-UHFFFAOYSA-N 0.000 description 1
- 229960000488 tizanidine Drugs 0.000 description 1
- 229960003989 tocilizumab Drugs 0.000 description 1
- 238000003325 tomography Methods 0.000 description 1
- 210000000515 tooth Anatomy 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 238000012549 training Methods 0.000 description 1
- 238000011277 treatment modality Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229940035722 triiodothyronine Drugs 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 239000000107 tumor biomarker Substances 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 230000009959 type I hypersensitivity Effects 0.000 description 1
- 241000712461 unidentified influenza virus Species 0.000 description 1
- 210000000689 upper leg Anatomy 0.000 description 1
- 230000002485 urinary effect Effects 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 210000005048 vimentin Anatomy 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 238000007794 visualization technique Methods 0.000 description 1
- 210000004127 vitreous body Anatomy 0.000 description 1
- 239000011800 void material Substances 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 229940000119 zanaflex Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5091—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing the pathological state of an organism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6881—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for tissue or cell typing, e.g. human leukocyte antigen [HLA] probes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6888—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5047—Cells of the immune system
- G01N33/505—Cells of the immune system involving T-cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5047—Cells of the immune system
- G01N33/5052—Cells of the immune system involving B-cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6854—Immunoglobulins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Analytical Chemistry (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Microbiology (AREA)
- Zoology (AREA)
- Physics & Mathematics (AREA)
- Wood Science & Technology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Cell Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pathology (AREA)
- Genetics & Genomics (AREA)
- General Engineering & Computer Science (AREA)
- Biophysics (AREA)
- General Physics & Mathematics (AREA)
- Medicinal Chemistry (AREA)
- Food Science & Technology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Toxicology (AREA)
- Physiology (AREA)
- Hospice & Palliative Care (AREA)
- Oncology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Peptides Or Proteins (AREA)
Abstract
Un método para monitorizar una neoplasia linfoide en un paciente con clonotipos de cáncer definidos que comprende los pasos de: (a) determinar un perfil de secuencias de ADN recombinadas en células T y/o células B de una muestra obtenida del paciente, en donde el perfil se determina mediante la secuenciación de la siguiente generación; y (b) determinar los niveles de los clonotipos de cáncer definidos en el perfil para monitorizar la neoplasia linfoide.
Description
DESCRIPCIÓN
Monitorización del estadio de neoplasia linfoide usando perfiles de clonotipo
Campo técnico
La invención se refiere en general al control de las condiciones de salud y enfermedad de un individuo mediante mediciones moleculares, y más particularmente, al control de las condiciones de salud y enfermedad de un individuo mediante la medición de perfiles de las moléculas del sistema inmune utilizando una secuenciación de ADN de alto rendimiento.
Antecedentes de la invención
El sistema inmunitario adaptativo, que comprende respuestas humorales (o mediadas por células B) y citotóxicas (o mediadas por células T), ha evolucionado para atacar características moleculares específicas en sus dianas respectivas. La aparición de una respuesta a una diana específica proporciona a un huésped una "memoria" de la misma, lo que le da la capacidad de montar una respuesta más fuerte si la misma diana apareciera en otro momento. Por lo general, cualquier proteína o polisacárido puede servir como objetivo para algún subconjunto de células de respuesta inmune adaptativa o sus productos que reconocen características moleculares específicas, o epítopos, en la diana.
Dado que la enfermedad autoinmune implica el reconocimiento por parte de algún componente del sistema inmunitario adaptativo a las autodominaciones, se han examinado aspectos del sistema inmunitario adaptativo para ayudar en el diagnóstico y pronóstico de dichas enfermedades. Usando técnicas inmunológicas estándar, el sistema inmune humoral ha sido investigado buscando autoanticuerpos circulantes. Se han identificado autoanticuerpos, como antinuclear, anti-ADNds y factor reumatoide, para varias enfermedades. Es posible que estos anticuerpos no sean patológicos en sí mismos, ni que el objetivo que reconocen en el cuerpo sea necesariamente el mismo que el analizado in vitro; sin embargo, la medición de sus niveles ayuda en el diagnóstico y en algunos casos tiene algunas implicaciones de pronóstico y tratamiento.
Otra metodología para estudiar el sistema inmune adaptativo en enfermedades autoinmunes y linfoides se basa en el análisis de la diversidad de las células inmunes adaptativas. La activación de las células inmunes adaptativas conduce a su expansión clonal. La evidencia de esta expansión clonal se obtiene generalmente mediante la amplificación del ARN o ADN de la sangre de parte de la secuencia de ácidos nucleicos que codifica la región de reconocimiento de antígeno. Por ejemplo, los cebadores de PCR para amplificar secuencias que tienen un segmento V específico de la cadena p en el receptor de células T (análogo a la cadena pesada del anticuerpo) se usan para amplificar los segmentos J o segmentos J y D conectados al segmento V específico. Cuando está presente una población celular diversa, se espera que amplifique los fragmentos con una distribución de amplicones de tamaños ligeramente diferentes, pero la expansión clonal hace que los tamaños específicos se enriquezcan y, por lo tanto, se vuelvan más intensos según se visualizan como bandas en un gel. En la técnica denominada "espectrotipificación", cada uno de los segmentos V se amplifica con los segmentos J y D para evaluar si alguno de estos amplicones muestra una expansión clonal.
Un problema de la metodología de espectrotipificación es que muchas secuencias distintas pueden tener la misma longitud y, por lo tanto, son indistinguibles. Por lo tanto, solo se puede discernir la expansión clonal dramática mediante espectrotipificación. Existe la necesidad de mejorar los métodos para diagnosticar y ayudar al pronóstico de enfermedades autoinmunes y estados de enfermedades autoinmunes, así como otras enfermedades para las cuales el sistema inmunológico juega un papel central.
Si bien la especificidad adicional en la elaboración de perfiles del sistema inmunitario sería de gran utilidad para permitir que su impacto en la salud humana se prediga mejor, se entregaría una utilidad aún mayor si se desarrollaran métodos que permitan a las células T y B específicas involucradas en los procesos de la enfermedad que se identificarán incluso si esas secuencias particulares nunca se hubieran observado. La gran diversidad del sistema inmunológico le proporciona una inmensa reserva de células potencialmente útiles, pero también presenta un desafío para el investigador que intenta utilizar este repertorio con fines predictivos. Cualquier secuencia única dirigida a un antígeno es una de una gran cantidad de personas que podrían estar involucradas y/o correlacionadas con el proceso de la enfermedad en un individuo determinado. Los métodos que identificarían cuál de las muchas células en un individuo dado están involucradas con los procesos de la enfermedad serían de gran valor para la salud humana.
El perfil de células inmunitarias también tiene utilidad en el diagnóstico y manejo de los cánceres. El tratamiento de los cánceres con frecuencia implica la evaluación de la respuesta al tratamiento, así como el seguimiento de la recurrencia de la enfermedad. Las metodologías más comunes para monitorizar la respuesta y la recurrencia del cáncer son las evaluaciones radiográficas y los biomarcadores sanguíneos. Por ejemplo, las tomografías computarizadas se usan con frecuencia para controlar la recurrencia del cáncer en múltiples enfermedades, incluido el cáncer de colon. De manera similar, los biomarcadores de proteínas, como el PSA y el CEA, son biomarcadores de la sangre que se utilizan para seguir los cánceres de próstata y colon. Los reordenamientos genómicos específicos generan otro objetivo atractivo para usaren las siguientes células cancerosas. Por ejemplo, la translocación BCR-ABL presente en la gran mayoría de los pacientes con leucemia mielógena crónica (LMC) se ha convertido en un analito
para evaluar el estado de la enfermedad. La especificidad de la translocación a las células leucémicas y su capacidad de ser analizada por la tecnología de PCR permitieron la generación de una prueba altamente específica y sensible que ahora se usa rutinariamente para monitorizar pacientes con LCM.
La generación del perfil de células inmunes (o clonotipo) se puede usar para generar marcadores para neoplasias linfoides. El cáncer en el linaje de las células linfoides es un conjunto heterogéneo de enfermedades clínicas que a menudo reflejan la etapa de desarrollo de la célula que ha sufrido la transformación en una célula cancerosa. La leucemia linfoblástica aguda (LLA) surge con mayor frecuencia en los linfocitos inmaduros. Por otro lado, el mieloma múltiple (MM) se produce en células plasmáticas que se han diferenciado para producir anticuerpos. De manera similar, los diferentes tipos de linfomas a menudo reflejan diferentes etapas de desarrollo celular. Estas enfermedades ocurren en diferentes grupos de edad, tienen diferentes pronósticos y mortalidad, y pueden tratarse con regímenes distintos.
Estas enfermedades se tratan con frecuencia con quimioterapia, radioterapia y/o trasplante de médula ósea. La recurrencia de la enfermedad luego se controla mediante diferentes métodos dependiendo de la situación clínica particular. Estos métodos incluyen la evaluación de la sangre y/o la médula ósea utilizando conteos sanguíneos estándar y morfología, citometría de flujo (FCM) utilizando marcadores de superficie celular, electroforesis de proteínas, así como técnicas moleculares como PCR y FISH. Además, los estudios radiográficos como la tomografía computarizada y la PET se utilizan con frecuencia para monitorizar la recurrencia de algunos de los cánceres linfoides. Estos métodos sufren de invasividad (médula ósea), coste y riesgo de radiación, y/o falta de sensibilidad.
Algunos marcadores moleculares específicos de una célula cancerosa detectables por PCR de una manera sensible están presentes en una fracción de las neoplasias linfoides. Por ejemplo, BCR-ABL está presente en una fracción de los pacientes con ALL y puede servir como un marcador para controlar la recaída del tumor. Desafortunadamente, para la mayoría de los pacientes no existen tales marcadores que puedan usarse para la detección sensible y específica de recaídas. La FCM se puede usar para detectar la Enfermedad Residual Mínima (ERM) que es útil para fines de pronóstico. En esta técnica que utiliza la clasificación de células activadas por flujo (FACS) de múltiples colores, una célula cancerosa puede ser identificada por la virtud de los marcadores de la superficie celular en particular que tiene. La sensibilidad de esta técnica en manos de expertos se limita a <10-4, (1 célula cancerosa en 10.000 células normales) y los marcadores presentes en un momento determinado pueden desaparecer más tarde. Por lo tanto, la FCM generalmente no es útil para detectar una recaída temprana en muestras de sangre.
La PCR proporciona una metodología sensible para la detección de secuencias específicas y se ha utilizado para detectar la reorganización particular en el receptor de células B (BCR) o receptores de células T (TCR) de la célula cancerosa (por ejemplo, véase Greenberg et al., 1995, Journal of Leukocyte Biology, 57, 856-864; US 6,416,948 B1; y Campana 2009, Hematol Oncol Clin North Am. 23(5), 1083-1098). Esta técnica aprovecha el hecho de que los receptores de células B o T en un linfocito se crean después de eventos de recombinación imperfectos que generan secuencias únicas para los diferentes linfocitos. Por ejemplo, un TCR está compuesto de cadenas TCRa y TCRp. El TCRa se crea a través de la recombinación que une una de varias regiones V diferentes a una de varias regiones J. De manera similar, el TCRp se crea a través de la recombinación que crea un segmento V, D y J en tándem. En ambos casos, la recombinación a menudo no es perfecta y algunas bases pueden eliminarse de las secuencias del segmento de la línea germinal y se pueden agregar otras bases (llamadas bases N y P). La secuencia entre los segmentos V y J se conoce como la región NDN.
Estas secuencias pueden servir como una etiqueta para estos linfocitos y su progenie. Dado que estos eventos de recombinación también ocurren en las células que finalmente se vuelven malignas, secuencias únicas de los receptores de células B y T pueden servir como marcadores para detectar las células cancerosas. La secuencia de etiquetas es específica del paciente y, de hecho, puede cambiar en el mismo paciente debido a la evolución clonal. Para definir la secuencia del receptor de células T o B de las células leucémicas para un paciente, se utiliza la muestra de leucemia diagnóstica que generalmente está muy enriquecida para el clon leucémico. Por ejemplo, el ADN del receptor de células T y/o B se amplifica a partir de una muestra de diagnóstico, y el producto se ejecuta en un gel que puede separar el ADN según el tamaño (a veces denominado " espectrotipificación"); o alternativamente se puede hacer un análisis heterodúplex. Un gran grado de sesgo de la distribución de tamaño observada indica expansión monoclonal, que luego puede confirmarse secuenciando una muestra desde el pico de separación sesgado. Sin tal secuencia subsiguiente, a menudo es difícil determinar si tal sesgo tiene orígenes monoclonales o policlonales, por ejemplo. Van Dongen et al, Publicación de Patente de los Estados Unidos 2006/0234234.
Una vez que se identifica la etiqueta de secuencia, se puede usar la PCR en tiempo real utilizando las sondas Taqman para monitorizar el nivel de esa secuencia. La región NDN usualmente no es lo suficientemente larga para abarcar los cebadores de PCR y el oligonucleótido de detección. Por lo tanto, típicamente se utilizan cebadores de PCR complementarios a las regiones V y J y una sonda Taqman que incluye algunas de las bases NDN del clon leucémico. Los cebadores proporcionan algo de especificidad, ya que amplifican solo una fracción del repertorio completo. La especificidad para el clonotipo particular es proporcionada por la hibridación de la sonda Taqman. Por lo tanto, la sensibilidad del ensayo generalmente no es tan buena como en una PCR típica (por ejemplo, BCR-ABL) donde el par de cebadores (con o sin la sonda Taqman) proporciona la especificidad. Se demostró que la sensibilidad puede ser tan alta como 10'5 para algunas secuencias, pero puede ser significativamente peor dependiendo de la especificidad de hibridación proporcionada por la sonda Taqman, cuya secuencia es complementaria a al menos parte de la región
NDN. Dada la baja sensibilidad de algunas sondas, es posible que el ensayo no funcione para ninguna de las reordenaciones en un paciente en particular. El problema de la evolución de los clones también se ha planteado anteriormente, lo que reduce aún más la posibilidad de detectar leucemia de bajo nivel. Además, esta técnica es complicada y requiere la generación de sondas Taqman específicas para cada paciente, así como una plantilla que se debe utilizar como estándares. Estos estándares específicos del paciente deben usarse cada vez que se analice la muestra del paciente. La incoherencia de la sensibilidad entre los pacientes, la naturaleza incómoda y los problemas logísticos de obtener los controles apropiados para el ensayo han limitado enormemente su uso. Por lo tanto, existe la necesidad de generar marcadores que puedan usarse para La monitorización de recaídas en pacientes con neoplasias linfoides. En algunas realizaciones, la invención descrita en el presente documento permite desarrollar un conjunto muy general, sensible y específico de marcadores para tratar pacientes con cánceres linfoides utilizando la secuenciación de células inmunitarias.
Sería ventajoso para muchos campos, incluidos particularmente los campos de cáncer autoinmune y linfoide, si hubiera ensayos disponibles para evaluar perfiles de clonotipo de individuos que fueran más sensibles y completos que las técnicas actuales y que fueran aplicables en general sin la necesidad de fabricar reactivos individualizados.
Resumen de la invención
La invención proporciona un método para monitorizar una neoplasia linfoide en un paciente con clonotipos de cáncer definidos que comprende las etapas de: (a) determinar un perfil de secuencias de ADN recombinadas en células T y/o células B de una muestra obtenida del paciente, en donde el perfil se determina usando secuenciación de próxima generación; y (b) determinar los niveles de los clonotipos de cáncer definidos en el perfil para monitorizar la neoplasia linfoide.
En el presente documento se describe un método para controlar una enfermedad que comprende las etapas de (a) identificar uno o más clonotipos específicos de pacientes correlacionados con una enfermedad mediante la determinación de un perfil de clonotipo a partir de una muestra de linfocitos en un tejido relacionado con la enfermedad, comprendiendo la muestra un repertorio de clonotipos del tejido relacionado con la enfermedad; (b) determinar un perfil de clonotipo a partir de una muestra de células de sangre periférica para identificar una presencia, ausencia y/o nivel de uno o más clonotipos específicos del paciente correlacionados con la enfermedad, tal muestra de sangre periférica que comprende un repertorio de clonotipos; y (c) repetir el paso (b) para controlar la enfermedad o afección en el paciente. En un método, la etapa de identificación incluye la determinación de un perfil de clonotipo a partir de una muestra de linfocitos en un tejido no relacionado con la enfermedad en el mismo paciente y la comparación de dicho perfil de clonotipo con el de dicho tejido relacionado con la enfermedad para identificar dicho uno o más clonotipos específicos del paciente. Las enfermedades que pueden controlarse incluyen, entre otras, trastornos proliferativos linfoides, tumores sólidos, enfermedades infecciosas y enfermedades autoinmunes. El tamaño del repertorio puede variar ampliamente dependiendo de aplicaciones particulares; pero en un método, un repertorio incluye cada clonotipo en una muestra de un individuo presente con una frecuencia de .01 por ciento o más con una probabilidad de noventa y nueve por ciento. En otros métodos, un repertorio incluye cada clonotipo en una muestra de un individuo presente con una frecuencia de 0.001 por ciento o más con una probabilidad de noventa y nueve por ciento.
También se describe en el presente documento un método para monitorizar una enfermedad que comprende las etapas de un método para monitorizar una enfermedad en un paciente, el método comprende las etapas de: (a) determinar un perfil de clonotipo a partir de una muestra de linfocitos de un individuo afectado por un enfermedad y un perfil de clonotipo del mismo individuo de una muestra de linfocitos enriquecidos con base en marcadores de superficie celular asociados con la enfermedad para identificar uno o más clonotipos específicos del paciente correlacionados con la enfermedad, en donde cada una de las muestras comprende un repertorio de clonotipos de las poblaciones de linfocitos enriquecidos y no enriquecidos; (b) determinar un nivel de cada uno de los uno o más clonotipos específicos del paciente en un perfil de clonotipo a partir de una muestra de células de sangre periférica, teniendo dicha muestra un volumen definido y que comprende un repertorio de clonotipos de los mismos; y (c) repetir el paso (b) para controlar la enfermedad o afección en el paciente.
En otros casos de los métodos anteriores, los pasos respectivos para determinar un repertorio a partir de una muestra de células de sangre periférica comprenden además incluir como uno o más clonotipos específicos del paciente cualquier clonotipo no registrado previamente que sea un clonotipo filogénico de uno o más clonotipos específicos del paciente. Cuando la enfermedad es un trastorno proliferativo linfoide, el uno o más clonotipos específicos para el paciente de tales métodos pueden incluir además mutaciones adicionales relacionadas con el cáncer y reordenamientos genéticos, como los reemplazos de la región V (descritos más detalladamente a continuación) fácilmente identificados por los métodos descritos en este documento.
También se describe en el presente documento un método para medir simultáneamente números de linfocitos y niveles de expresión de clonotipos en una muestra que comprende los pasos de: (i) obtener de un individuo una muestra que comprende células T y/o células B; (ii) secuenciación de moléculas individuales aisladas espacialmente derivadas del ADN genómico de dichas células, comprendiendo tales moléculas individuales aisladas espacialmente una serie de clonotipos correspondientes a una serie de linfocitos en la muestra; (iii) secuenciación de moléculas individuales aisladas espacialmente derivadas de ARN de dichas células, comprendiendo tales moléculas individuales aisladas
espacialmente números de clonotipos correspondientes a los niveles de expresión de los mismos en los linfocitos de la muestra; y (iv) determinar los niveles de expresión del clonotipo en los linfocitos de la muestra comparando para cada clonotipo el número determinado a partir de moléculas individuales aisladas derivadas del ADN genómico de dichas células y el número determinado a partir de moléculas individuales aisladas derivadas del ARN de dichas células.
La invención supera varias deficiencias en la técnica anterior al proporcionar, entre otras ventajas, métodos basados en secuencias para medir clonotipos de sensibilidad mucho mayores correlacionadas con enfermedades o afecciones de salud. La invención proporciona además dichos ensayos en un formato general aplicable a cualquier paciente sin la necesidad de fabricar reactivos individualizados o específicos para el paciente. Tales avances tienen aplicaciones particularmente útiles en las áreas de autoinmunidad y cánceres linfoides. En esta última área, la invención proporciona además métodos de ensayo y monitorización que son capaces de detectar y rastrear no solo niveles muy bajos de clonotipos correlacionados con la enfermedad, sino también aquellos clonotipos que han sufrido modificaciones que escaparían a la detección por metodologías anteriores. Esta última característica es de gran valor, por ejemplo, en La monitorización de la enfermedad residual mínima en los cánceres linfoides.
Breve descripción de los dibujos
Las características novedosas de la invención se exponen con particularidad en las reivindicaciones adjuntas. Una mejor comprensión de las características y ventajas de la presente invención se obtiene por referencia a la siguiente descripción detallada que establece realizaciones ilustrativas, en las que se utilizan los principios de la invención, y los dibujos adjuntos de los cuales:
La figura 1A es un diagrama de flujo de un ejemplo de un método para determinar perfiles de clonotipo.
La figura 1B ilustra la distribución relativa de las mutaciones somáticas que se producen en las inmunoglobulinas producidas por las células B.
Las figuras 2A-2B muestran un esquema de PCR de dos etapas para amplificar los genes TCRp.
La figura 3A ilustra un producto de PCR que se amplificó utilizando el esquema de las figuras 2A-2B, que se someterá a una PCR secundaria para agregar la amplificación de puente y los sitios de unión del cebador de secuenciación para la secuenciación basada en Solexa.
La figura 3B ilustra detalles de un ejemplo de determinación de una secuencia de nucleótidos del producto de PCR de la figura 3A.
La figura 3C ilustra detalles de otro ejemplo de determinación de una secuencia de nucleótidos del producto de PCR de la figura 3A.
La figura 4A ilustra un esquema de PCR para generar tres plantillas de secuenciación a partir de una cadena de IgH en una sola reacción. Las figuras
4B-4C ilustra un esquema de PCR para generar tres plantillas de secuenciación a partir de una cadena de IgH en tres reacciones separadas, después de lo cual los amplicones resultantes se combinan para una PCR secundaria para agregar los sitios de unión del cebador P5 y P7.
La figura 4D ilustra las ubicaciones de las lecturas de secuencia generadas para una cadena de IgH. La figura 4E ilustra el uso de la estructura de codones de las regiones V y J para mejorar las llamadas de base en la región NDN.
La figura 5 muestra datos que evidencian la reproducibilidad de las PCR multiplex de la invención.
La figura 6 muestra datos que demuestran que las PCR multiplex de la invención introducen un sesgo de amplificación mínimo.
La figura 7A muestra el log10 de la frecuencia de cada clonotipo en las dos muestras duplicadas utilizando Accuprime y ADNc correspondiente a 500 ng de ARN como plantilla de entrada.
La figura 7B representa el log10 de la frecuencia de cada clonotipo utilizando un ADNc correspondiente a 500 ng de ARN como plantilla de entrada y Accuprime (eje X) o Taq de alta fidelidad (eje Y).
La figura 7C muestra el log10 de la frecuencia de cada clonotipo utilizando un ADNc correspondiente a 50 ng de ARN como plantilla de entrada y Accuprime (eje X) o Taq de alta fidelidad (eje Y).
La figura 8 el número de moléculas TCRp de muestras. 8A y 8B muestran datos de la amplificación de IgH a partir de ADN genómico.
La figura 9 muestra datos que indican que las amplificaciones multiplex de acuerdo con la invención tienen un sesgo de amplificación mínimo.
La figura 10 muestra datos que comparan clonotipos de dos individuos.
Descripción detallada de la invención
Un aspecto de esta invención utiliza tecnologías de secuenciación de próxima generación para evaluar los niveles de reordenamientos de TCR o BCR en una población de linfocitos. Estas tecnologías de secuenciación pueden obtener 1 millón o más de lecturas de una muestra a un coste razonable. Un clonotipo presente a una frecuencia de 1/1.000.000 o inferior aún puede ser detectado de una manera específica usando estas tecnologías. La amplificación multiplex para amplificar todos los diferentes tipos de secuencias de una parte específica del gen o transcripción se puede lograr a partir de una muestra de sangre o ADN de médula ósea. Por ejemplo, para amplificar secuencias de IgH, se pueden usar varios cebadores complementarios de todos los segmentos V y alelos conocidos, junto con varios cebadores complementarios de todos los segmentos J y alelos. La figura 1A ilustra los pasos de un método de este tipo para una realización que emplea una clase de secuenciadores de ADN (por ejemplo, secuenciación Solexa por síntesis, como se describe a continuación) para perfilar los clonotipos de una muestra. Se obtiene una muestra que contiene células B o células T (100), después de lo cual el ADN o el ARN se extrae y amplifica (102) en una reacción que amplifica de forma preferente los clonotipos y une secuencias terminales para la posterior amplificación y secuenciación. Las moléculas individuales de los clonotipos amplificados se distribuyen al azar sobre una superficie sólida (104), como una superficie de vidrio, que se ha configurado para permitir una segunda amplificación in situ para producir poblaciones clonales (o polonias) de cada molécula individual (106). Las moléculas de cada polonia se secuencian (108), por ejemplo, utilizando una técnica de secuenciación por síntesis, después de lo cual los tipos y abundancias de las secuencias se tabulan para formar un perfil de clonotipo (110), o equivalentemente un perfil de repertorio. El método se puede realizar con un pequeño sesgo de amplificación entre las diferentes secuencias. El ARN de los genes TCRp e IgH se puede amplificar con solo pequeñas diferencias en las eficiencias de los diferentes cebadores V, lo que valida la posibilidad de hacer lo mismo con el ADN. Este esquema puede mejorar los problemas de la lectura en tiempo real para la detección de reordenamientos de TCR y/o BCR de bajo nivel.
La sensibilidad se determina mediante estadística de recuentos (es decir, la sensibilidad aumenta al aumentar el tamaño de las muestras de las células y la secuenciación) y la amplificación equivalente (es decir, los clonotipos que tienen secuencias variadas pueden amplificarse sin un sesgo significativo en una reacción de amplificación múltiple, como una PCR, como se ilustra más adelante). Dado que la sensibilidad está limitada en última instancia por las estadísticas de conteo, para obtener una mayor sensibilidad, simplemente se pueden obtener más células (es decir, muestras más grandes) y más lecturas de secuenciación. Con suficientes lecturas de secuenciación, la sensibilidad está limitada por el número de linfocitos en la muestra. En contraste, la sensibilidad para el ensayo de PCR en tiempo real está limitada por el fondo. Además, los clones específicos de un paciente se pueden determinar mediante la secuenciación de una muestra diagnóstica de leucemia o linfoma. Una vez que se determina el clonotipo, su nivel se puede determinar en muestras en puntos de tiempo posteriores. En algunas realizaciones preferidas, no es necesario que las sondas o cebadores específicos del paciente o la utilización de plantillas específicas del paciente se ejecuten como estándares. En su lugar, a los clones específicos del paciente les sigue el almacenamiento de los datos relativos a las secuencias relevantes para cada paciente, y el mismo ensayo funciona para todos los pacientes.
En general, se describen en el presente documento métodos para aplicar técnicas de secuenciación de ácidos nucleicos a la tarea de controlar el repertorio de células de inmunidad adaptativa para perfilar el sistema inmune. Los perfiles del sistema inmunitario generado pueden usarse para el diagnóstico de enfermedades y trastornos, y para el diagnóstico de estados de enfermedades y trastornos. Los métodos de perfil inmunológico de la invención proporcionada se pueden usar para controlar enfermedades y trastornos y evaluar el tratamiento de enfermedades y trastornos. Las enfermedades y trastornos en los que se pueden aplicar los métodos de la invención para incluir la enfermedad autoinmune, incluido lupus eritematoso sistémico (SLE), esclerosis múltiple (EM), artritis reumatoide (AR) y espondilitis anquilosante (EA). Los métodos descritos en este documento pueden aplicarse al diagnóstico, monitorización y tratamiento del rechazo de trasplantes y el envejecimiento inmunitario. Además, los métodos de perfiles inmunitarios de la invención proporcionada se pueden usar para diagnosticar, controlar y tratar otras enfermedades relacionadas con el sistema inmunitario, incluido cáncer y enfermedades infecciosas.
La secuenciación de moléculas amplificadas individuales puede distinguir diferentes secuencias y, por lo tanto, tiene la sensibilidad para detectar cambios cuantitativos en la expansión clonal. En general, en un caso, se proporciona un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B. El método puede comprender etapas que incluyen el aislamiento de muestras de un sujeto, una o más rondas de amplificación de ácidos nucleicos, el aislamiento espacial de ácidos nucleicos individuales y la secuenciación de ácidos nucleicos. Los ácidos nucleicos pueden ser ADN o ARN. Las secuencias de ADN recombinadas en células T y/o células B pueden denominarse clonotipos.
En una instancia, se proporciona un método para determinar uno o más clonotipos correlacionados en un sujeto o individuo. En otro caso, se proporciona un método para desarrollar un algoritmo que puede predecir uno o más clonotipos correlacionados en cualquier muestra de un sujeto con una enfermedad. En otro caso, se proporciona un método para descubrir uno o más clonotipos correlacionados para un individuo que usa un algoritmo que puede predecir uno o más clonotipos correlacionados en cualquier muestra de un sujeto. En otro caso, se proporciona un método para generar un algoritmo que calcula una puntuación de actividad de la enfermedad. En otro caso, se proporciona un método para controlar el estado de enfermedad de un individuo.
El perfil de repertorio de células T y B puede ser valioso para enfermedades con aspectos inflamatorios. Esta inflamación a menudo se debe a una reacción autoinmune y/o de hipersensibilidad. Estas enfermedades incluyen enfermedades cardiovasculares, enfermedad de Alzheimer y preclamsia. La inflamación también se ha asociado con estados metabólicos anormales, como la obesidad y la diabetes. Existen otras enfermedades relacionadas con la inflamación. En un aspecto de la invención, un segmento de ácido nucleico de células B recombinadas se amplifica mediante una PCR con una pluralidad de cebadores directos o una pluralidad de cebadores inversos para generar un conjunto anidado de plantillas (consulte las figuras 4A y 4B y sus descripciones a continuación). Las plantillas de un conjunto de este tipo pueden amplificarse aún más en una superficie para formar amplicones separados (por ejemplo, mediante PCR de puente utilizando un instrumento cBot, Illumina, San Diego, CA). Las plantillas del mismo conjunto anidado pueden asociarse entre sí mediante lecturas de secuencia generadas en sus extremos comunes. Los conjuntos de plantillas anidadas permiten utilizar una secuenciación química con tasas de error relativamente altas para analizar secuencias más largas de lo que sería posible, mientras que al mismo tiempo se mantienen puntuaciones de alta calidad promedio en toda la longitud de la secuencia. Los conjuntos anidados también aseguran que al menos una secuencia leída se obtiene de una región V, incluso si se ha sometido a hipermutación somática. En una realización, las químicas de secuenciación se pueden usar para analizar ácidos nucleicos altamente variables, como las moléculas de IgH, que tienen tasas de error no mejores que las siguientes: el 0.2 por ciento de las lecturas de secuencia contienen al menos un error en las posiciones 1-50; 0.2-1.0 por ciento de las lecturas de secuencia contienen al menos un error en las posiciones 51-75; 0.5-1.5 por ciento de las lecturas de secuencia contienen al menos un error en las posiciones 76-100; y el 1-5 por ciento de las lecturas de secuencia contienen al menos un error en las posiciones 101-125. En otra realización, los sitios de unión del cebador de secuenciación se colocan de manera que cuando se extienden producen una serie de lecturas de secuencia donde cada lectura de secuencia, excepto la última, se superponen con su sitio de unión del cebador y/o secuencia inmediatamente adyacentes corriente abajo, proporcionando así una cobertura de secuencia continua con puntuaciones de mayor calidad que serían posibles si se utilizara una sola plantilla larga para generar una sola lectura de secuencia larga.
I. Divulgación adicional
Se describe un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B que comprende: obtener una muestra de un sujeto que comprende células T y/o células B, aislar espacialmente moléculas individuales de ADN genómico de dichas células; secuenciar dichas moléculas individuales aisladas espacialmente de ADN genómico, y determinar los niveles de diferentes secuencias de dicha muestra para generar dicho perfil de secuencias de ADN recombinadas. Se proporciona un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B que comprende: obtener una muestra de un sujeto que comprende células T y/o células B, aislar espacialmente moléculas individuales de ADN genómico de dicho células, amplificando dichas moléculas individuales de ADN genómico, secuenciar dicho ADN amplificado, y determinando los niveles de diferentes secuencias de dicha muestra para generar dicho perfil de secuencias de ADN recombinadas. Se proporciona un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B que comprende: obtener una muestra de un sujeto que comprende células T y/o células B, amplificar ADN genómico de dichas células, aislar espacialmente moléculas individuales de dicho ADN amplificado, secuenciando dichas moléculas individuales espacialmente aisladas de ADN amplificado; y determinar los niveles de diferentes secuencias de dicha muestra para generar dicho perfil de secuencias de ADN recombinadas. Se proporciona un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B que comprende: obtener una muestra de un sujeto que comprende células T y/o células B, amplificar ADN genómico de dichas células, aislar espacialmente moléculas individuales de dicho ADN amplificado, que vuelven a amplificar dichas moléculas de ADN amplificado, secuenciar dichas moléculas de ADN amplificadas, y determinan los niveles de diferentes secuencias de dicha muestra para generar dicho perfil de secuencias de a Dn recombinadas. Se proporciona un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B que comprende: obtener una muestra de un sujeto que comprende células T y/o células B, amplificar ADN genómico de dichas células, aislar espacialmente moléculas individuales de dicho ADN amplificado, que vuelven a amplificar dichas moléculas de ADN amplificado, secuenciar dichas moléculas de ADN amplificadas, y determinan los niveles de diferentes secuencias de dicha muestra para generar dicho perfil de secuencias de a Dn recombinadas. Se proporciona un método para determinar un perfil de secuencias de ADN recombinado en células T y/o células B que comprende: obtener una muestra de un sujeto que comprende células T y/o células B, transcribir de forma inversa el ARN de dichas células para formar el ADNc, que aísla espacialmente moléculas individuales de dicho ADNc, opcionalmente vuelve a amplificar dichas moléculas individuales espacialmente aisladas de ADNc, secuenciar dicho ADNc y/o ADNc reamodificado; y determinar los niveles de diferentes secuencias de dicha muestra para generar dicho perfil de secuencias de ADN recombinadas. Se proporciona un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B que comprende: obtener una muestra de un sujeto que comprende células T y/o células B; aislar espacialmente células individuales en dicha muestra, secuenciar moléculas individuales de nucleico de dichas células; y determinar los niveles de diferentes secuencias de dicha muestra para generar dicho perfil de secuencias de ADN recombinadas. En un caso, dicha amplificación y/o reamplificación comprende PCR, PCR multiplex, TMA, NASBAo LAMP. En otro caso, dicho aislamiento espacial comprende separar dicho ADN o ADNc en dos dimensiones sobre un soporte sólido, separar dicho ADN o ADNc en tres dimensiones en una solución con micelas, o separar moléculas utilizando cámaras de microrreacción. En otro caso, dicha amplificación y/o reamplificación es mediante el crecimiento de bacterias que albergan ADN o ADNc subclonado, la amplificación de ADN o ADNc en un portaobjetos, o la amplificación de ADN o ADNc en una perla. En otro caso, dicha secuencia comprende la secuencia didesoxi. En otro caso, dicha secuencia
comprende la secuenciación por síntesis utilizando nucleótidos marcados terminados de manera reversible. En otro caso, dicha secuencia comprende la detección de la liberación de pirofosfato en la incorporación de nucleótidos. En otro caso, dicha secuencia comprende la hibridación específica de alelo con una biblioteca de sondas oligonucleotídicas marcadas. En otro caso, dicha secuenciación comprende la secuenciación por síntesis utilizando una hibridación específica de alelo con una biblioteca de sondas oligonucleotídicas marcadas, seguida de la ligación de dichas sondas. En otro caso, dicha secuencia comprende la supervisión en tiempo real de la incorporación de nucleótidos marcados durante una etapa de polimerización. En otro caso, dichas secuencias de ADN recombinadas comprenden genes de receptores de células T y/o genes de inmunoglobulina. En otro caso, dicha secuenciación comprende la secuenciación de un subconjunto de las secuencias clonales completas de genes de inmunoglobulina y/o receptores de células T. En otro caso, dicho subconjunto de la secuencia clonal completa comprende la unión V-D, la unión D-J de un gen receptor de inmunoglobulina o de células T, la región variable completa de un gen receptor de inmunoglobulina o célula T, la región de reconocimiento de antígeno o la complementariedad determinando la región 3 (CDR3). En otro caso, dichos genes receptores de células T comprenden genes p del receptor de células T. En otro caso, dichos genes de inmunoglobulina comprenden genes pesados de inmunoglobulina. En otro caso, dicha amplificación o reamplificación comprende una pluralidad de cebadores complementarios a los segmentos V y un cebador complementario a un segmento C. En otro caso, dicha amplificación o reamplificación comprende una pluralidad de cebadores complementarios a los segmentos V y una pluralidad de cebadores complementarios a los segmentos C. En otro caso, dicha pluralidad de cebadores complementarios a los segmentos V comprende al menos tres cebadores diferentes para cada segmento V y la pluralidad de cebadores complementarios a los segmentos C comprende al menos 1, al menos 2, al menos 3, al menos 4, al menos 5, o al menos 6 cebadores. En otro caso, dichas células T o B son subconjuntos de las células T y B totales. En otro caso, dicho subconjunto de células T son células CD4+, CD8+ o células altas en CD27. En otro caso, dicha muestra comprende al menos 100.000, al menos 500.000, al menos 750.000, de al menos 1.000.000 de células T. En otro caso, dicha secuencia comprende al menos 1000 lecturas por corrida, al menos 10.000 lecturas por corrida, al menos 100.000 lecturas por corrida, o al menos 1.000.000 lecturas por corrida. En otro caso, dicha secuencia comprende la generación de aproximadamente 30 pb, aproximadamente 40 pb, aproximadamente 50 pb, aproximadamente 60 pb, aproximadamente 70 pb, aproximadamente 80 pb, aproximadamente 90 pb, aproximadamente 100 pb, aproximadamente 110 o aproximadamente 120 pb por lectura. En otro caso, dicha muestra se toma cuando el sujeto se encuentra en un estado de exacerbación de una enfermedad autoinmune. En otro caso, dicha muestra se toma de un sujeto que tiene o se sospecha que tiene lupus eritematoso sistémico. Se proporciona un método para determinar uno o más clonotipos correlacionados en un sujeto que comprende: generar uno o más perfiles de clonotipo por secuenciación de ácidos nucleicos, moléculas individuales aisladas espacialmente de al menos una muestra del sujeto, en donde la al menos una muestra está relacionada con un primer estado de la enfermedad y determinación de uno o más clonotipos correlacionados en el sujeto en función de uno o más perfiles de clonotipo. En un caso, dicha al menos una muestra proviene de un tejido afectado por la enfermedad. En otro caso, dicha determinación de uno o más clonotipos correlacionados comprende comparar perfiles de clonotipo de al menos dos muestras. En otro caso, el primer estado de la enfermedad es un estado máximo de la enfermedad. En otro caso, dicho uno o más clonotipos correlacionados están presentes en el estado máximo de la enfermedad. En otro caso, dicho uno o más clonotipos correlacionados están ausentes en el estado máximo de la enfermedad. En otro caso, dicho uno o más clonotipos correlacionados son altos en el estado máximo de la enfermedad. En otro caso, dicho uno o más clonotipos correlacionados son bajos en el estado máximo de la enfermedad. En otro caso, dicha muestra comprende células T y/o células B. En otro caso, dichas células T y/o células B comprenden un subconjunto de células T y/o células B. En otro caso, dicho subconjunto de células T y/o células B se enriquece mediante la interacción con un marcador. En otro caso, dicho marcador es un marcador de superficie celular en el subconjunto de células T y/o células B. En otro caso, dicho subconjunto de células T y/o células B interactúan con un antígeno específicamente presente en la enfermedad. En otro caso, la enfermedad es lupus eritematoso sistémico o esclerosis múltiple. Se describe un método para desarrollar un algoritmo que puede predecir uno o más clonotipos correlacionados en cualquier muestra de un sujeto con una enfermedad que comprende: a) generar una pluralidad de perfiles de clonotipos a partir de un conjunto de muestras, en donde las muestras son relevantes para la enfermedad, b) identificar uno o más clonotipos correlacionados a partir del conjunto de muestras, c) usar parámetros de secuencia y/o datos funcionales de uno o más clonotipos correlacionados identificados en b) para desarrollar el algoritmo que puede predecir los clonotipos correlacionados en cualquier muestra de un sujeto con la enfermedad. En una instancia, el conjunto de muestras se toma de uno o más tejidos afectados por la enfermedad. En otro caso, dicha identificación de uno o más clonotipos correlacionados comprende comparar perfiles de clonotipos de al menos dos muestras. En otro caso, dichos datos funcionales incluyen la capacidad de unión de marcadores en células T y/o la superficie de las células B o la interacción con el antígeno por una célula T o una célula B. En otro caso, dichos parámetros de secuencia comprenden secuencia de ácidos nucleicos y secuencia de aminoácidos predicha. En otro caso, las muestras son de uno o más individuos en un estadio pico de la enfermedad. En otro caso, dicho uno o más clonotipos correlacionados están presentes en el estado máximo de la enfermedad. En otro caso, dicho uno o más clonotipos correlacionados se encuentran en un nivel alto en el estado máximo de la enfermedad. En otro caso, dicho uno o más clonotipos correlacionados se encuentran en un nivel bajo en el estado máximo de la enfermedad. En otro caso, el uno o más clonotipos correlacionados están ausentes en el estado máximo de la enfermedad. En otro caso, la enfermedad es lupus eritematoso sistémico o esclerosis múltiple. En otro caso, se proporciona un método para descubrir uno o más clonotipos correlacionados para un individuo, que comprende ingresar un perfil de clonotipo de una muestra del individuo en un algoritmo, y usar el algoritmo para determinar uno o más clonotipos correlacionados para el individuo. En una instancia, el algoritmo es un algoritmo que puede predecir uno o más clonotipos correlacionados en cualquier muestra de un sujeto con una enfermedad que comprende, dicho
algoritmo se desarrolla al: a) generar una pluralidad de perfiles de clonotipo a partir de un conjunto de muestras, en donde las muestras son relevantes para la enfermedad, b) identificar uno o más clonotipos correlacionados del conjunto de muestras, c) usar parámetros de secuencia y/o datos funcionales de uno o más clonotipos correlacionados identificados en b) para desarrollar un algoritmo que pueda predecir correlacionando clonotipos en cualquier muestra de un sujeto con la enfermedad. En una instancia, dicha muestra se toma en un estado máximo de enfermedad. En otro caso, la muestra se toma de tejido afectado por enfermedad. Se describe un método para generar un algoritmo que calcula un puntaje de actividad de la enfermedad que comprende: desarrollar un algoritmo que usa un conjunto de factores para combinar los niveles de los clonotipos correlacionados en un puntaje de actividad de la enfermedad, comparar el puntaje de actividad de la enfermedad con los datos clínicos relacionados con el estado de la enfermedad. y optimizar los factores para maximizar la correlación entre los datos clínicos y el puntaje de actividad de la enfermedad. En un caso, se proporciona un método para monitorizar el estado de la enfermedad de un individuo que comprende: a) determinar un perfil de clonotipo a partir de una muestra del individuo, b) ingresar la información del perfil de clonotipo de a) en un algoritmo que calcula la puntuación de actividad de la enfermedad, donde se genera un algoritmo al desarrollar un algoritmo que usa un conjunto de factores para combinar los niveles de los clonotipos correlacionados en un puntaje de actividad de la enfermedad, comparando el puntaje de actividad de la enfermedad con los datos clínicos relacionados con el estado de la enfermedad y optimizando los factores para maximizar la correlación entre los datos clínicos y el puntaje de actividad de la enfermedad, y c) el uso del algoritmo que calcula el puntaje de actividad de la enfermedad para generar un puntaje predictivo del estado de la enfermedad del individuo. En otro caso, el método para monitorizar el estado de la enfermedad de un individuo comprende además determinar uno o más clonotipos correlacionados en el individuo, e ingresar la información del uno o más clonotipos correlacionados en el algoritmo. En otro caso, dicha determinación de uno o más clonotipos correlacionados en el individuo comprende a) generar uno o más perfiles de clonotipo mediante la secuenciación de ácidos nucleicos moléculas individuales aisladas espacialmente de al menos una muestra del sujeto, en donde la al menos una muestra está relacionada con un primer estado de la enfermedad, y b) determinar uno o más clonotipos correlacionados en el sujeto basándose en uno o más perfiles de clonotipo. En otro caso, dicha determinación de uno o más clonotipos correlacionados en el individuo comprende a) ingresar un perfil de clonotipo de una muestra del individuo en un algoritmo que puede predecir uno o más clonotipos correlacionados, en donde dicho algoritmo puede predecir uno o más clonotipos correlacionados es desarrollado por i) generación de una pluralidad de perfiles de clonotipo a partir de un conjunto de muestras, en donde las muestras son relevantes para la enfermedad, ii) identificación de uno o más clonotipos correlacionados del conjunto de muestras, iii) uso de parámetros de secuencia y/o datos funcionales de uno o más clonotipos correlacionados identificados en ii) para desarrollar el algoritmo que puede predecir los clonotipos correlacionados en cualquier muestra de un sujeto con la enfermedad, y c) usar el algoritmo que puede predecir uno o más clonotipos correlacionados para determinar uno o más clonotipos correlativos para el individuo. En otro caso, la enfermedad es lupus eritematoso sistémico o esclerosis múltiple. Se describe un método para determinar uno o más clonotipos de células T o B correlacionados que comprenden: a) dividir una muestra de células de un sujeto en al menos dos muestras, b) generar uno o más perfiles de clonotipo por secuenciación de ácido nucleico moléculas aisladas individualmente a partir de una de las muestras de células del sujeto, c) enriquecimiento de otra muestra de células de este sujeto basándose en al menos un parámetro molecular de las células, d) generación de uno o más perfiles de clonotipo mediante secuenciación de ácidos nucleicos moléculas aisladas individualmente de la muestra enriquecida del sujeto, y e) identificación de al menos un clonotipo basado en clonotipos cuya abundancia dentro de la muestra ha sido alterada entre la muestra enriquecida y la muestra no enriquecida. En otro caso, el parámetro molecular es un marcador de superficie celular. En otro caso, el enriquecimiento se realiza capturando células utilizando un marcador de afinidad inmovilizado en fase sólida. En otro caso, la superficie sólida es un conjunto de cuentas. En otro caso, la superficie sólida es una columna. En otro caso, el marcador se marca usando un resto fluorescente. En otro caso, el enriquecimiento se realiza mediante citometría de flujo utilizando la etiqueta fluorescente. En otro caso, las células son linfocitos B y el enriquecimiento se realiza utilizando antígenos que se unen al receptor de células B. En otro caso, el enriquecimiento se realiza mediante la captura de una superficie sólida sobre la cual se inmovilizan los antígenos. En otro caso, el antígeno se utiliza para marcar los linfocitos B y el enriquecimiento se realiza mediante citometría de flujo utilizando esta etiqueta. En otro caso, las células son linfocitos T y el enriquecimiento se realiza mediante un método que permite que las células T que reaccionan a un antígeno específico se marquen y se enriquezcan mediante citometría de flujo. En otro caso, las células T se marcan utilizando el método de tinción de citoquinas intracelulares. En otro caso, las células T se marcan utilizando el método de captura de citoquinas. En otro caso, el parámetro molecular del receptor de células B que es capaz de unirse a al menos un antígeno que es específico a un patógeno. En otro caso, el parámetro molecular es el receptor de células T que es capaz de unirse al menos al antígeno que es específico de un patógeno. En otro caso, la muestra se toma de un paciente que ha estado expuesto a un patógeno en un primer momento. Se describe un método para determinar un conjunto de clonotipos en un individuo que se correlaciona con una reacción inmune a un patógeno, que comprende: a) dividir una muestra de células de un sujeto en al menos dos muestras b) generar uno o más perfiles de clonotipo por ácido nucleico secuenciación individualmente de moléculas aisladas espacialmente de una muestra de células del sujeto, c) enriquecimiento de otra muestra de células de este sujeto basada en la capacidad de las células para unirse a al menos un antígeno del patógeno d) generación de uno o más perfiles de clonotipos por ácido nucleico secuenciar individualmente las moléculas aisladas espacialmente de la muestra enriquecida del sujeto y, e) identificar al menos un clonotipo correlacionado basado en clonotipos cuya abundancia dentro de la muestra ha sido alterada entre la muestra enriquecida y la muestra no enriquecida. Se describe un método para determinar un conjunto de clonotipos en un individuo que se correlaciona con una reacción inmune a un tumor que comprende: a) dividir una muestra de células de un sujeto en al menos dos muestras b) generar uno o más perfiles de clonotipo por ácido nucleico
secuenciación individual de moléculas aisladas espacialmente de una muestra de células del sujeto c) enriquecimiento de otra muestra de células de este sujeto en función de la capacidad de las células para unirse al menos un autoantígeno presente en el tumor d) generación de uno o más perfiles de clonotipos por ácido nucleico secuenciar moléculas aisladas individualmente espacialmente de la muestra enriquecida del sujeto e) identificación de al menos un clonotipo correlacionado basado en clonotipos cuya abundancia dentro de la muestra ha sido alterada entre la muestra enriquecida y la muestra no enriquecida. En otro caso, los niveles de clonotipos correlacionados se utilizan para evaluar el riesgo de que el individuo tenga un tumor. En otro caso, se sabe que los antígenos están presentes en un tumor que ya ha ocurrido en ese individuo y los clonotipos correlacionados se utilizan para evaluar el riesgo de recurrencia del tumor. En otro caso, se sabe que los antígenos están presentes en tumores en otros individuos y los clonotipos se utilizan para evaluar el riesgo de cáncer en un paciente que no ha detectado un tumor previamente. Se describe un método para determinar un conjunto de clonotipos en un individuo que se correlaciona con una reacción inmune a un agente terapéutico que comprende: a) dividir una muestra de células de un sujeto en al menos dos muestras b) generar uno o más perfiles de clonotipos por núcleos de secuenciación de moléculas ácidas aisladas espacialmente individualmente de una muestra de células del sujeto c) enriquecimiento de otra muestra de células de este sujeto en función de la capacidad de las células para unirse al menos a un antígeno contenido en el agente terapéutico d) generación de uno o más perfiles de clonotipo mediante secuenciación de ácidos nucleicos moléculas aisladas espacialmente individualmente de la muestra enriquecida del sujeto y e) identificación de al menos un clonotipo correlacionado basado en clonotipos cuya abundancia dentro de la muestra ha sido alterada entre la muestra enriquecida y la muestra no enriquecida. En otro caso, los niveles de clonotipos correlacionados se utilizan para evaluar el riesgo de que el individuo muestre hipersensibilidad a un agente terapéutico. Se describe un método para determinar un conjunto de clonotipos en un individuo que se correlaciona con una reacción inmune a un agente terapéutico que comprende: a) dividir una muestra de células de un sujeto en al menos dos muestras b) generar uno o más perfiles de clonotipos por núcleos secuenciación ácida moléculas aisladas espacialmente individualmente de una muestra de células del sujeto c) enriquecimiento de otra muestra de células de este sujeto en función de la capacidad de las células para unirse al menos a un antígeno contenido en el agente terapéutico d) generación de uno o más perfiles de clonotipo mediante secuenciación de ácidos nucleicos moléculas aisladas espacialmente individualmente de la muestra enriquecida del sujeto y e) identificación de al menos un clonotipo correlacionado basado en clonotipos cuya abundancia dentro de la muestra ha sido alterada entre la muestra enriquecida y la muestra no enriquecida. En otro caso, los niveles de clonotipos correlacionados se utilizan para evaluar el riesgo de que el individuo muestre hipersensibilidad a un agente terapéutico. Se describe un método para determinar un conjunto de clonotipos en un individuo que se correlaciona con una reacción inmune a una placa arterial que comprende: a) dividir una muestra de células de un sujeto en al menos dos muestras b) generar uno o más perfiles de clonotipos por nucleico secuenciación ácida moléculas aisladas espacialmente individualmente de una muestra de células del sujeto c) enriquecimiento de otra muestra de células de este sujeto en función de la capacidad de las células para unirse al menos a un antígeno presente en la placa arterial d) generación de uno o más perfiles de clonotipo por nucleico secuenciación ácida de moléculas individualmente aisladas espacialmente de la muestra enriquecida del sujeto y e) identificación de al menos un clonotipo correlacionado basado en clonotipos cuya abundancia dentro de la muestra ha sido alterada entre la muestra enriquecida y la muestra no enriquecida. En otro caso, los niveles de clonotipos correlacionados se utilizan para evaluar el riesgo de que el individuo tenga una enfermedad cardiovascular. En otro caso, los niveles de clonotipos correlacionados se utilizan para evaluar el riesgo de que la placa arterial sea inestable.
Se describe un método para determinar un identificador de secuencia encontrado en células involucradas en una neoplasia linfoide que comprende: a) obtener una muestra de células del individuo afectado en donde se sabe que existen células cancerosas b) generar uno o más perfiles de clonotipo relacionados con at al menos un reordenamiento genómico de células inmunes mediante la secuenciación de ácidos nucleicos moléculas aisladas espacialmente individualmente de las células en la muestra c) que identifican el identificador de secuencia como la secuencia del clonotipo asociado con el tumor. En otro caso, la muestra procede de la médula ósea del paciente. En otro caso, la muestra es de la sangre del paciente. En otro caso, la muestra proviene de una biopsia de un tumor linfoide sólido. En otro caso, la reorganización genómica de las células inmunitarias es un reordenamiento VDJ de IgH en una célula B.
En otro caso, el reordenamiento genómico de las células inmunitarias es un reordenamiento DJ de IgH en una célula
B. En otro caso, el reordenamiento genómico de las células inmunitarias es un reordenamiento VJ de IgK en una célula
B. En otro caso, el reordenamiento genómico de las células inmunitarias es un reordenamiento VJ de IgL en una célula
B. En otro caso, el reordenamiento genómico de las células inmunitarias es un reordenamiento VDJ de TCR p en una célula T. En otro caso, el reordenamiento genómico de células inmunes es un reordenamiento de DJ de TCR p en una célula T. En otro caso, el reordenamiento genómico de células inmunes es un reordenamiento VJ de TCR a en una célula T. En otro caso, el reordenamiento genómico de células inmunes es un reordenamiento VJ de TCR A en una célula T. En otro caso, la reorganización genómica de las células inmunitarias es un reordenamiento VDJ de TCR 8 en una célula T. En otro caso, el reordenamiento genómico de células inmunes es un reordenamiento VD de TCR 8 en una célula T. En otro caso, el reordenamiento genómico de las células inmunitarias es una translocación de un segmento J de IgH a otra región del genoma. En otro caso, el reordenamiento genómico de las células inmunitarias es una translocación de cualquier segmento J a otra región del genoma. En otro caso, la identificación del clonotipo asociado a tumor se realiza por frecuencia de clonotipo. En otro caso, la identificación del clonotipo asociado a tumor se realiza por frecuencia de clonotipo. En otro caso, la identificación del clonotipo asociado al tumor se realiza mediante la detección del reordenamiento de linaje cruzado. En otro caso, la identificación del clonotipo asociado a tumor se realiza mediante la identificación de reordenamientos no funcionales. En otro caso, la identificación del clonotipo asociado a tumor se realiza asociando clonotipos celulares con al menos un marcador molecular asociado con el tumor. Se describe un método para determinar los niveles de células tumorales linfoides circulantes dentro de un
individuo cuyo tumor se ha asociado con un identificador único de secuencia en un primer momento que involucra: a) obtener una muestra de células del paciente b) generar una o más perfiles clonotipos relacionados con al menos un reordenamiento genómico de células inmunitarias mediante la secuenciación de ácidos nucleicos moléculas aisladas espacialmente individualmente de las células en la muestra c) determinar los niveles de células tumorales a partir del nivel de los clonotipos asociados con el identificador de secuencia. Se describe un método para determinar los niveles de células tumorales linfoides circulantes dentro de un individuo cuyo tumor se ha asociado con un identificador único de secuencia en un primer momento que involucra: a) obtener una muestra de células del paciente b) enriquecer las células con base en al menos un marcador molecular c) generación de uno o más perfiles de clonotipo relacionados con al menos un reordenamiento genómico de células inmunes mediante secuenciación de ácidos nucleicos moléculas aisladas espacialmente individualmente de las células en la muestra d) determinando los niveles de células tumorales a partir del nivel de Los clonotipos asociados con el identificador de secuencia. En otro caso, la muestra es una muestra de sangre. En otro caso, la muestra es una muestra de médula ósea. En otro caso, la muestra es una muestra de linfa. En otro caso, la muestra es una muestra de tejido. En otro caso, las células se marcan de forma fluorescente y se enriquecen utilizando citometría de flujo. En otro caso, las células se enriquecen mediante la unión a un soporte sólido. En otra instancia, el clonotipo se define como el clonotipo que contiene el identificador de secuencia único. En otro caso, se determina que los clonotipos son aquellos clonotipos que probablemente hayan resultado de mutaciones y reordenamientos en el identificador de secuencia. En otro caso, los niveles de células tumorales circulantes y/o el cambio en los niveles de las células tumorales circulantes se utilizan en un algoritmo para producir una puntuación que se correlaciona con el riesgo de tener una recurrencia clínica tumoral. En otro caso, los niveles de células tumorales circulantes y/o el cambio en los niveles de las células tumorales circulantes se utilizan para tomar una decisión de tratamiento.
II. Métodos para determinar los perfiles de clonotipo
Los métodos descritos en el presente documento se pueden usar para generar perfiles de secuencias de ADN recombinadas, o clonotipos, en una muestra de un sujeto. En un caso, se describe un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B, incluida la obtención de una muestra de un sujeto que comprende células T y/o células B, aislando moléculas individuales de ADN genómico a partir de dichas células, secuenciando las moléculas individuales aisladas de ADN genómico y determinando los niveles de diferentes secuencias de la muestra para generar dicho perfil de secuencias de ADN recombinadas.
En otro caso, se describe un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B, incluida la obtención de una muestra de un sujeto que comprende células T y/o células B, aislando moléculas individuales de ADN genómico a partir de las células, amplificando las moléculas individuales del ADN genómico, secuenciando el ADN amplificado y determinando los niveles de diferentes secuencias de la muestra para generar dicho perfil de secuencias de ADN recombinadas.
En otro caso, se describe un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B, incluida la obtención de una muestra de un sujeto que comprende células T y/o células B, amplificando el ADN genómico a partir de las células, aislando moléculas individuales del ADN amplificado, secuenciando las moléculas individuales aisladas del ADN amplificado y determinando los niveles de diferentes secuencias de la muestra para generar el perfil de secuencias de ADN recombinadas.
En otro caso, se describe un método para determinar un perfil de secuencias de ADN recombinadas en células T y/o células B, incluida la obtención de una muestra de un sujeto que incluye células T y/o células B, amplificando el a Dn genómico de las células, aislando moléculas individuales del ADN amplificado, volviendo a amplificar las moléculas de ADN amplificado, secuenciando las moléculas de ADN redamplificadas y determinando los niveles de diferentes secuencias de la muestra para generar el perfil de secuencias de ADN recombinadas.
En otro caso, se describe un método para determinar un perfil de secuencias de ADN recombinado en células T y/o células B, que incluye obtener una muestra de un sujeto que comprende células T y/o células B, aislando el ARN a partir de dicha muestra, transcripción reversa del ARN de dichas células para formar ADNc, aislando moléculas individuales de dicho ADNc, opcionalmente volver a amplificar dicho ADNc, secuenciar dichas moléculas individuales aisladas de dicho ADNc o a Dn reamplificado, y determinar los niveles de diferentes secuencias a partir de dicha muestra para generar dicho perfil de secuencias de ADN recombinadas.
En otro caso, se describe un método para determinar un perfil de secuencias de ADN recombinado en células T y/o células B, que incluye obtener una muestra de un sujeto que incluye células T y/o células B, aislando individuos moléculas de ARN de dicha muestra, secuenciando las moléculas individuales de ARN y determinando los niveles de diferentes secuencias de dicha muestra para generar el perfil de secuencias de ADN recombinadas.
Temas y muestras
Los métodos de la invención proporcionada pueden usar muestras de sujetos o individuos (por ejemplo, pacientes). El sujeto puede ser un paciente, por ejemplo, un paciente con una enfermedad autoinmune. El sujeto puede ser un paciente con una enfermedad infecciosa o cáncer, como una leucemia o un linfoma. El sujeto puede ser un mamífero, por ejemplo, un humano. El sujeto puede ser masculino o femenino. El sujeto puede ser un bebé, un niño o un adulto.
En algunas realizaciones el sujeto ya no está vivo. En algunas realizaciones el sujeto está vivo. El sujeto puede ser un individuo que fue expuesto a un arma biológica.
El sujeto también podría ser un animal no humano. El animal no humano podría ser una mascota doméstica o un animal de granja. El animal no humano podría ser un perro, un gato, una vaca, un caballo, una cabra o un cerdo. El animal no humano podría ser un animal clonado. El animal no humano podría estar involucrado en la producción de productos farmacéuticos.
Las muestras utilizadas en los métodos de la invención proporcionada pueden incluir, por ejemplo, un fluido corporal de un sujeto, incluido un fluido amniótico que rodea un feto, humor acuoso, bilis, sangre y plasma sanguíneo, cerumen (cera de oído), fluido de Cowper o líquido preeyaculatorio, quilo, quimo, eyaculación femenina, líquido intersticial, linfa, menstruación, leche materna, moco (incluyendo moco y flemas), líquido pleural, pus, saliva, sebo (aceite de la piel), semen, suero, sudor, lágrimas, orina, lubricación vaginal, vómito, agua, heces, fluidos corporales internos, incluido el líquido cefalorraquídeo que rodea el cerebro y la médula espinal, el líquido sinovial que rodea las articulaciones óseas, el líquido intracelular es el líquido dentro de las células y el humor vítreo, los fluidos del globo ocular. En una realización, la muestra es una muestra de sangre. La muestra de sangre puede ser de aproximadamente 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5 o 5.0 mL. La muestra puede ser líquido cefalorraquídeo (LCR) cuando el sujeto tiene esclerosis múltiple, líquido sinovial cuando el sujeto tiene artritis reumatoide y biopsia de la piel (u otro órgano) cuando el sujeto tiene lupus sistémico. En una realización, el clonotipo puede identificarse a partir del fluido/tejido corporal disponible que sea más probable que refleje la patología, seguido de un seguimiento posterior de los niveles de los clonotipos que forman un fluido corporal diferente, por ejemplo, la sangre. Las muestras también pueden incluir disolventes en los que se ha disuelto material biológico. Las muestras se pueden analizar en un momento en que una enfermedad está inactiva. Las muestras se pueden analizar en un momento en que una enfermedad está activa. Las muestras se pueden obtener en un momento en que una enfermedad está inactiva. Las muestras se pueden obtener en un momento en que una enfermedad está activa. La muestra puede ser obtenida por un proveedor de atención médica, por ejemplo, un médico, un asistente médico, una enfermera, un veterinario, un dermatólogo, un reumatólogo, un dentista, un paramédico o un cirujano. La muestra puede ser obtenida por un técnico de investigación. La muestra puede ser proporcionada por el sujeto. La muestra se puede proporcionar de forma anónima. La muestra se puede proporcionar a través del correo. La muestra puede ser proporcionada por una agencia de aplicación de la ley o por un investigador. Se puede obtener más de una muestra de un sujeto.
La muestra puede ser una biopsia, por ejemplo, una biopsia de piel. La biopsia puede ser de, por ejemplo, cerebro, hígado, pulmón, corazón, colon, riñón o médula ósea. Cualquier técnica de biopsia utilizada por los expertos en la técnica puede usarse para aislar una muestra de un sujeto. Por ejemplo, una biopsia puede ser una biopsia abierta, en la cual se usa anestesia general. La biopsia puede ser una biopsia cerrada, en la cual se realiza un corte más pequeño que en una biopsia abierta. La biopsia puede ser una biopsia de núcleo o incisional, en la que se extrae una parte del tejido. La biopsia puede ser una biopsia por escisión, en la que se realizan intentos para extirpar una lesión completa. La biopsia puede ser una biopsia por aspiración con aguja fina, en la cual se extrae una muestra de tejido o líquido con una aguja.
La muestra se puede obtener a partir de material corporal que es dejado por un sujeto. Dicho material desechado puede incluir residuos humanos. El material desechado también podría incluir células de la piel, sangre, dientes o cabello.
La muestra puede incluir células inmunes, por ejemplo, las células inmunes pueden incluir células T y/o células B. Las células T (linfocitos T) incluyen, por ejemplo, células que expresan receptores de células T. Las células T incluyen células T auxiliares (células T efectoras o células Th), células T citotóxicas (CTL), células T de memoria y células T reguladoras. La muestra puede incluir una sola celda en algunas aplicaciones (por ejemplo, una prueba de calibración para definir células T relevantes) o, más generalmente, al menos 1.000, al menos 10.000, al menos 100.000, al menos 250.000, al menos 500.000, al menos 750.000 o al menos 1.000.000 de células T.
Las células B incluyen, por ejemplo, células B plasmáticas, células B de memoria, células B1, células B2, células B de la zona marginal y células B foliculares. Las células B pueden expresar inmunoglobulinas (anticuerpos, receptor de células B). La muestra puede incluir una sola celda en algunas aplicaciones (por ejemplo, una prueba de calibración para definir células B relevantes) o, más generalmente, al menos 1.000, al menos 10.000, al menos 100.000, al menos 250.000, al menos 500.000, al menos 750.000 o al menos 1.000.000 células B.
La muestra puede incluir ácido nucleico, por ejemplo, ADN (por ejemplo, ADN genómico o ADN mitocondrial) o ARN (por ejemplo, ARN mensajero o microARN). El ácido nucleico puede ser ADN o ARN libre de células, por ejemplo, extraído del sistema circulatorio, Vlassov et al., Curr. Mol. Med., 10: 142-165 (2010); Swarup et al, FEBS Lett., 581: 795-799 (2007). En los métodos de la invención proporcionada, la cantidad de ARN o ADN de un sujeto que se puede analizar incluye, por ejemplo, tan baja como una sola célula en algunas aplicaciones (por ejemplo, una prueba de calibración) y hasta 10 millones de células o más, lo que se traduce en un rango de ADN de 6pg-60ug y ARN de aproximadamente 1pg-10ug.
Como se explica más detalladamente a continuación (Definiciones), una muestra de linfocitos es lo suficientemente grande como para que sustancialmente cada célula T o célula B con un clonotipo distinto se represente allí, formando
así un repertorio (como se usa el término en este documento). En una instancia, se toma una muestra que contiene una probabilidad de noventa y nueve por ciento de cada clonotipo de una población presente con una frecuencia de 0.001 por ciento o mayor. En otro caso, se toma una muestra que contiene una probabilidad de noventa y nueve por ciento de cada clonotipo de una población presente con una frecuencia de 0.0001 por ciento o mayor. En una realización, una muestra de células B o células T incluye al menos medio millón de células, y en otra realización dicha muestra incluye al menos un millón de células.
Cuando una fuente de material de la que se toma una muestra es escasa, como, por ejemplo, muestras de estudios clínicos o similares, el ADN del material puede amplificarse mediante una técnica sin polarización, como la amplificación del genoma completo (WGA), desplazamiento múltiple amplificación (MDA); o técnica similar, por ejemplo. Hawkins et al., Curr. Opin. Biotech., 13: 65-67 (2002); Dean et al, Genome Research, 11: 1095-1099 (2001); Wang et al, Nucleic Acids Research, 32: e76 (2004); Hosono et al, Genome Research, 13: 954-964 (2003); y similares.
Las muestras de sangre son de particular interés, especialmente en el control de neoplasias linfoides, como linfomas, leucemias o similares, y pueden obtenerse utilizando técnicas convencionales, por ejemplo, Innis et al, editors, PCR Protocols (Academic Press, 1990); o similar. Por ejemplo, los glóbulos blancos se pueden separar de las muestras de sangre utilizando técnicas convencionales, por ejemplo, Kit RosetteSep (Stem Cell Technologies, Vancouver, Canadá). Las muestras de sangre pueden variar en volumen de 100 pL a 10 mL; en un aspecto, los volúmenes de muestra de sangre están en el rango de 200 100 pL a 2 mL. El ADN y/o el ARN se pueden extraer de dicha muestra de sangre usando técnicas convencionales para usar en los métodos de la invención, por ejemplo, DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA). Opcionalmente, subconjuntos de glóbulos blancos, por ejemplo, los linfocitos, se pueden aislar aún más utilizando técnicas convencionales, por ejemplo, clasificación celular activada por fluorescencia (FACS) (Becton Dickinson, San Jose, CA), clasificación celular activada magnéticamente (MACS) (Miltenyi Biotec, Auburn, CA), o similares.
En otras realizaciones, los ácidos nucleicos se analizan a partir de una muestra de un subconjunto de células. Se puede emplear un método para separar células, por ejemplo, usando un marcador de superficie celular. Por ejemplo, las células se pueden aislar por citometría de flujo de clasificación celular, clasificación de flujo, clasificación de células activadas por fluorescencia (FACS), separación basada en perlas como la clasificación de células magnéticas (MACS; por ejemplo, utilizando partículas magnéticas recubiertas de anticuerpos), separación basada en el tamaño (por ejemplo, un tamiz, una serie de obstáculos o un filtro), clasificación en un dispositivo para microfluidos, separación basada en anticuerpos, sedimentación, adsorción por afinidad, extracción por afinidad o centrifugación en gradiente de densidad. Las células se pueden purificar mediante microdisección por captura láser. La clasificación puede basarse en el tamaño de la célula, la morfología o los marcadores intracelulares o extracelulares. Los métodos para aislar o clasificar células tumorales se describen, por ejemplo, en Nagrath S. et al. (2007) Nature 450: 1235-1239; las Patentes de los Estados Unidos números 6008002, 7232653 y 7332288; Publicación PCT No. WO2008157220A1; y las Solicitudes de Patente de los Estados Unidos Nos. US20080138805A1 y US20090186065; y Rosenberg R. et al. (2002) Citometría 49: 150-158.
El subconjunto de células puede ser un subconjunto de células T y/o células B. El subconjunto de células T puede ser células altas CD4+, CD8+ o CD27. Cócteles de anticuerpos para marcar y/o separar una gran variedad de subconjuntos de células T y células B están disponibles comercialmente de proveedores como Quest Diagnostic (San Juan Capistrano, CA); Dako (Dinamarca); y similares. Los siguientes son ejemplos de kits disponibles para subconjuntos relacionados con la enfermedad (donde se enumeran la especificidad antigénica de los anticuerpos: leucemia/linfoma linfoblástico precursor B (CD19, CD79a (citoplasmático), CD20, CD10, TDt, HLADR, CD34, IgM (citoplasmático); linfoma difuso de células B grandes (CD20, CD19, CD22, CD79a, CD30); linfoma folicular (CD20, CD10, CD10, BCL2, BCL6); leucemia de células del manto (CD19, CD20, CD5, CD23-, BCL1); y similares.
La clasificación de células activadas por fluorescencia (FACS) utiliza la dispersión de la luz y las características fluorescentes para clasificar las células. Se puede impartir una propiedad fluorescente en una célula usando, por ejemplo, sondas de ácido nucleico o anticuerpos conjugados con un tinte fluorescente. Una suspensión celular puede formar una corriente de líquido que fluye. El flujo de células forma gotas que contienen aproximadamente una célula por gota. Antes de que la corriente forme gotas, se mide una característica fluorescente de cada célula. Una carga se coloca en un anillo de carga eléctrica antes de la medición de la intensidad de la fluorescencia y la carga opuesta se transporta en la gota cuando se rompe de la corriente. Las gotas cargadas pasan a través de dos placas de desviación de alto voltaje que desvían las gotas a diferentes contenedores según su carga. La carga se puede aplicar directamente al flujo y la interrupción de la caída retiene el cargo del mismo signo que el flujo. El flujo se vuelve entonces a neutral después de que la caída se interrumpa.
La inmunofluorescencia directa o indirecta se puede utilizar en FACS. En la inmunofluorescencia directa, un anticuerpo se conjuga directamente con un tinte fluorescente. En la inmunofluorescencia indirecta, el anticuerpo primario no está marcado, y un anticuerpo secundario se conjuga con un tinte fluorescente.
Como las recombinaciones de identificación están presentes en el ADN de cada célula de inmunidad adaptativa, así como en sus transcritos de ARN asociados, se puede secuenciar ARN o ADN en los métodos de la invención proporcionada. Una secuencia recombinada de una célula T o célula B que codifica un receptor de célula T o una molécula de inmunoglobulina, o una porción del mismo, se denomina clonotipo. El ADN o el ARN pueden corresponder
a secuencias de genes del receptor de células T (TCR) o genes de inmunoglobulina (Ig) que codifican anticuerpos. Por ejemplo, el ADN y el ARN pueden corresponder a secuencias que codifican las cadenas a, p, y o 8 de un TCR. En la mayoría de las células T, el TCR es un heterodímero que consiste en una cadena a y una cadena p. La cadena TCRa se genera por recombinación VJ, y el receptor de la cadena p se genera por recombinación V(D)J. Para la cadena TCRp, en humanos hay 48 segmentos V, 2 segmentos D y 13 segmentos J. Se pueden eliminar varias bases y agregar otras (llamadas nucleótidos N y P) en cada una de las dos uniones. En una minoría de células T, los TCR consisten en cadenas 8 delta y y. La cadena TCR y se genera por recombinación VJ, y la cadena TCR se genera por recombinación V(D)J (Kenneth Murphy, Paul Travers and Mark Walport, 7a edición de Janeway Immunology, Garland Science, 2007).
El ADN y el ARN analizados en los métodos de la invención pueden corresponder a secuencias que codifican inmunoglobulinas de cadena pesada (IgH) con regiones constantes (a, 8, £, y o |j) o inmunoglobulinas de cadena ligera (IgK o IgL) con regiones constantes A o k. Cada anticuerpo tiene dos cadenas ligeras idénticas y dos cadenas pesadas idénticas. Cada cadena está compuesta por una constante (C) y una región variable. Para la cadena pesada, la región variable se compone de una variable (V), diversidad (D) y segmentos de unión (J). Varias secuencias distintas que codifican para cada tipo de estos segmentos están presentes en el genoma. Un evento específico de recombinación VDJ ocurre durante el desarrollo de una célula B, marcando esa célula para generar una cadena pesada específica. La diversidad en la cadena ligera se genera de manera similar, excepto que no hay una región D, por lo que solo hay una recombinación VJ. La mutación somática a menudo ocurre cerca del sitio de la recombinación, causando la adición o eliminación de varios nucleótidos, lo que aumenta aún más la diversidad de las cadenas pesadas y ligeras generadas por las células B. La posible diversidad de los anticuerpos generados por una célula B es entonces el producto de las diferentes cadenas pesadas y ligeras. Las regiones variables de las cadenas pesada y ligera contribuyen a formar la región o sitio de reconocimiento (o unión) del antígeno. A esta diversidad se agrega un proceso de hipermutación somática que puede ocurrir después de que una respuesta específica se monta contra algún epítopo.
Como se mencionó anteriormente, de acuerdo con la invención, los cebadores pueden seleccionarse para generar amplicones de subconjuntos de ácidos nucleicos recombinados extraídos de linfocitos. Dichos subconjuntos pueden denominarse en el presente documento como "regiones somáticamente reorganizadas". Las regiones reorganizadas somáticamente pueden comprender ácidos nucleicos procedentes de linfocitos en desarrollo o completamente desarrollados, en los que los linfocitos en desarrollo son células en las que no se ha completado la reorganización de genes inmunes para formar moléculas que tienen regiones V(D)J completas. Las regiones sombreadas, reorganizadas somáticamente, a modo de ejemplo incluyen moléculas de IgH incompletas (tales como moléculas que contienen solo regiones D-J), moléculas de TCR8 incompletas (tales como moléculas que contienen solo regiones D-J) e IgK inactiva (por ejemplo, que comprenden regiones Kde-V). También se encuentran reordenamientos incompletos en células inmunes completamente desarrolladas asociadas con el segundo cromosoma en una célula en la que el primer cromosoma formó un reordenamiento productivo.
Control de cantidades de muestra y estimación de números de celda
El muestreo adecuado de las células es un aspecto importante de la interpretación de los datos del repertorio, como se describe más adelante en las definiciones de "clonotipo" y "repertorio". Por ejemplo, comenzar con 1.000 células crea una frecuencia mínima a la que el ensayo es sensible, independientemente de cuántas lecturas de secuencia se obtengan. Por lo tanto, en este documento se describen métodos para cuantificar el número de moléculas de receptores inmunes de entrada. Esto se ha implementado para secuencias TCRp e IgH. En cualquier caso, se utiliza un conjunto de cebadores que son capaces de amplificar todas las secuencias diferentes. Para obtener un número absoluto de copias, se realiza una PCR en tiempo real con el multiplex de cebadores junto con un estándar con un número conocido de copias de receptores inmunitarios. En la figura 9 se muestra un ejemplo de datos de PCR en tiempo real asociados con el ejemplo de vacunación de ratón. Esta medición de PCR en tiempo real se puede realizar a partir de la reacción de amplificación que posteriormente se secuenciará o se puede realizar en una alícuota separada de la misma muestra. En el caso del ADN, el número absoluto de moléculas de receptor inmune reorganizadas se puede convertir fácilmente en número de células (dentro de 2 veces, ya que algunas células tendrán 2 copias reorganizadas del receptor inmunitario específico evaluado y otras tendrán una). En el caso de ADNc, el número total medido de moléculas reorganizadas en la muestra en tiempo real se puede extrapolar para definir el número total de estas moléculas utilizadas en otra reacción de amplificación de la misma muestra. Además, este método se puede combinar con un método para determinar la cantidad total de ARN para definir el número de moléculas de receptor inmunes reorganizadas en una cantidad unitaria (por ejemplo, 1 jg ) de ARN, asumiendo una eficiencia específica de la síntesis de ADNc. Si se mide la cantidad total de ADNc, no es necesario considerar la eficiencia de la síntesis del ADNc. Si también se conoce el número de células, se pueden calcular las copias del receptor inmune reorganizadas por célula. Si no se conoce el número de células, uno puede estimarlo a partir del ARN total, ya que las células de un tipo específico generalmente generan una cantidad comparable de ARN. Por lo tanto, a partir de las copias de moléculas de receptores inmunes reorganizadas por 1 jg , se puede estimar el número de estas moléculas por célula.
Una desventaja de realizar una PCR en tiempo real separada de la reacción que se procesaría para la secuenciación es que podría haber efectos inhibitorios que son diferentes en la PCR en tiempo real de la otra reacción, ya que se pueden utilizar diferentes enzimas, ADN de entrada y otras condiciones. El procesamiento de los productos de la PCR en tiempo real para la secuenciación mejoraría este problema. Sin embargo, un número bajo de copias usando la PCR
en tiempo real puede deberse a un número bajo de copias o a efectos inhibitorios u otras condiciones subóptimas en la reacción.
Otra metodología que puede utilizarse es agregar una cantidad conocida de moléculas reorganizadas de receptor inmune únicas con una secuencia conocida, es decir, cantidades conocidas de uno o más estándares internos, al ADNc o ADN genómico de una muestra de cantidad desconocida. Al contar el número relativo de moléculas que se obtienen para la secuencia agregada conocida en comparación con el resto de las secuencias de la misma muestra, se puede estimar el número de moléculas de receptores inmunes reorganizadas en la muestra de ADNc inicial. (Estas técnicas para el recuento molecular son bien conocidas, por ejemplo, Brenner et al, Patente de los Estados Unidos 7,537,897. Los datos de la secuenciación de la secuencia única agregada se pueden usar para distinguir las diferentes posibilidades si también se está utilizando una calibración de PCR en tiempo real. Un número de copias bajo de receptor inmune reorganizado en el ADN (o ADNc) crearía una alta proporción entre el número de moléculas para la secuencia enriquecida en comparación con el resto de las secuencias de la muestra. Por otro lado, si el bajo número de copias medido por PCR en tiempo real se debe a una ineficiencia en la reacción, la proporción no sería alta.
En el presente documento se describen métodos para medir la expresión de un clonotipo a nivel celular. Es decir, como se señaló anteriormente, los clonotipos se pueden usar para contar linfocitos; por lo tanto, midiendo los clonotipos derivados de ADN genómico y los mismos clonotipos derivados de ARN, se puede determinar la expresión de clonotipos basada en células. Un método para medir simultáneamente los números de linfocitos y los niveles de expresión del clonotipo en una muestra puede comprender los pasos de: (a) obtener de un individuo una muestra que comprende células T y/o células B; (b) secuenciar moléculas individuales aisladas espacialmente derivadas del ADN genómico de dichas células, comprendiendo tales moléculas individuales aisladas espacialmente una serie de clonotipos correspondientes a una serie de linfocitos en la muestra; (c) secuenciar moléculas individuales aisladas espacialmente derivadas de ARN de dichas células, comprendiendo tales moléculas individuales aisladas espacialmente números de clonotipos correspondientes a sus niveles de expresión en los linfocitos de la muestra; y (d) determinar los niveles de expresión del clonotipo en los linfocitos de la muestra comparando para cada clonotipo el número determinado a partir de moléculas individuales aisladas derivadas de ADN genómico de dichas células y el número determinado a partir de moléculas individuales aisladas derivadas de ARN de dichas células. El ADN genómico y el ARN se extraen fácilmente de la misma muestra utilizando kits disponibles en el mercado, como el Mini Kit AllPrep ADN/ARN (Qiagen GmbH, Alemania). Como se mencionó anteriormente, en un caso, la etapa de determinación incluye la determinación de dicho número de linfocitos en dicha muestra agregando una cantidad conocida de un patrón interno a dicho ADN genómico. En otro caso, donde, por ejemplo, la muestra es sangre periférica, la muestra tiene un volumen definido que permite determinar una concentración de dichos linfocitos en dicha muestra. Típicamente, dicho volumen definido está en el rango de 1 mL a 50 mL, y más generalmente, en el rango de 1 mL a 10 mL. En otro caso, los números del mismo clonotipo derivado del ADN genómico y el ARN se comparan simplemente dividiendo el número de clonotipos determinados a partir de las moléculas individuales aisladas derivadas del ARN por el número de clonotipos determinados a partir de las moléculas individuales aisladas derivadas de dicho ADN genómico. Dichos dos conjuntos de clonotipos se distinguen fácilmente en la misma secuenciación mediante el uso de etiquetas, particularmente etiquetas de oligonucleótidos que se unen durante el proceso de preparación de la muestra. Para la secuenciación basada en Solexa, tales etiquetas pueden incorporarse con las etiquetas utilizadas para identificar diferentes muestras (por ejemplo) agregando un solo nucleótido a la etiqueta para indicar ADN o ARN, o simplemente usando una etiqueta adicional para que cada muestra de paciente se marque con dos etiquetas, una para la fracción de ADN genómico y otra para la fracción de ARN. Por lo tanto, dicha etapa de secuenciación de dichas moléculas individuales aisladas espacialmente derivadas de dicho ARN puede incluir marcar cada una de dichas moléculas individuales aisladas espacialmente con una primera etiqueta que indique su origen de ARN y dicha etapa de secuenciación de dichas moléculas individuales aisladas espacialmente derivadas de dicho ADN genómico pueden incluir etiquetar cada una de dichas moléculas individuales aisladas espacialmente con una segunda etiqueta que indica su origen de ADN genómico de modo que la primera etiqueta se distinga de la segunda etiqueta. En una instancia, tales etiquetas son etiquetas de oligonucleótidos distintas que se identifican mediante secuenciación.
Del mismo modo, los métodos descritos en el presente documento se pueden usar para proporcionar simultáneamente (es decir, con base en las mediciones en una sola muestra) el número de linfocitos y la clonalidad. Dicho método puede implementarse con los siguientes pasos: (a) obtener de una persona una muestra que comprende células T y/o células B; (b) secuenciar moléculas individuales aisladas espacialmente derivadas del ácido nucleico de dichas células, comprendiendo tales moléculas individuales aisladas espacialmente una serie de clonotipos correspondientes a una serie de linfocitos en la muestra; (c) determinar el número de linfocitos a partir del número de moléculas individuales aisladas espacialmente; (d) determinar las abundancias de diferentes secuencias de las moléculas individuales aisladas espacialmente para generar un perfil de clonotipo y una medida de clonalidad basada en ellas. El ácido nucleico de los linfocitos puede ser ADN genómico y/o ARN; sin embargo, preferiblemente el ácido nucleico es ADN genómico. De manera similar a lo anterior, en un caso, la etapa de determinar dicho número incluye además determinar dicho número de linfocitos en dicha muestra agregando una cantidad conocida de un estándar interno a dicho ADN genómico. Y de manera similar, cuando la muestra es una muestra de sangre periférica, tiene un volumen definido, de modo que se determina la concentración de dichos linfocitos en dicha muestra. En algunos casos de lo anterior, solo se emplean células B y en otras realizaciones solo se emplean células T.
Amplificación de poblaciones de ácidos nucleicos
Como se indica a continuación, los amplicones de las poblaciones diana de ácidos nucleicos pueden generarse mediante una variedad de técnicas de amplificación. En un aspecto de la invención, la PCR múltiple se usa para amplificar miembros de una mezcla de ácidos nucleicos, particularmente mezclas que comprenden moléculas inmunes recombinadas tales como receptores de células T, receptores de células B, o porciones de las mismas. La guía para llevar a cabo PCRs multiplex de tales moléculas inmunes se encuentra en las siguientes referencias: Morley, Patente de los Estados Unidos 5,296,351; Gorski, Patente de los Estados Unidos 5,837,447; Dau, Patente de los Estados Unidos 6,087,096; Von Dongen et al, Publicación de Patente de los Estados Unidos 2006/0234234; Publicación de Patente Europea EP 1544308B1; y similares. Las referencias anteriores describen la técnica denominada "espectrotipificación", donde una población de moléculas inmunes se amplifica por PCR multiplex, después de lo cual las secuencias del amplicón resultante se separan físicamente, por ejemplo, por electroforesis, para determinar si existe una clase de tamaño predominante. Tal clase indicaría una población clonal predominante de linfocitos que, a su vez, sería indicativa de un estado de enfermedad. En la espectrotipificación, es importante seleccionar cebadores que muestren poca o ninguna reactividad cruzada (es decir, que no se acoplen a los sitios de unión de otros cebadores); de lo contrario, puede haber una representación falsa de clases de tamaño en el amplicón. En la presente invención, siempre que los ácidos nucleicos de una población se amplifiquen uniformemente, la reactividad cruzada de los cebadores es admisible porque las secuencias de los ácidos nucleicos amplificados se analizan en la presente invención, no simplemente sus tamaños. Como se describe más detalladamente a continuación, en un aspecto, la etapa de aislar espacialmente las moléculas individuales de ácido nucleico se logra al realizar una amplificación múltiple primaria de una región preseleccionada somáticamente reorganizada o una parte de la misma (es decir, secuencias diana) utilizando cebadores directos e inversos que tienen colas no complementarias a las secuencias diana para producir un primer amplicón cuyas secuencias miembros tienen secuencias comunes en cada extremo que permiten una manipulación adicional. Por ejemplo, dichos extremos comunes pueden incluir sitios de unión del cebador para la amplificación continuada utilizando solo un cebador directo único y un cebador inverso único en lugar de los múltiplos de cada uno, o para la amplificación en puente de moléculas individuales en una superficie sólida, o similar. Dichos extremos comunes pueden agregarse en una sola amplificación como se describe anteriormente, o pueden agregarse en un procedimiento de dos pasos para evitar dificultades asociadas con la fabricación y el control de calidad de las mezclas de cebadores largos (por ejemplo, 50-70 bases o más). En un proceso de dos pasos de este tipo (descrito más detalladamente a continuación e ilustrado en las figuras 4A-4B), la amplificación primaria se lleva a cabo como se describió anteriormente, excepto que las colas del cebador tienen una longitud limitada para proporcionar solo sitios de unión del cebador directo e inverso En los extremos de las secuencias del primer amplicón. Luego se lleva a cabo una amplificación secundaria utilizando cebadores de amplificación secundarios específicos para estos sitios de unión del cebador para agregar secuencias adicionales a los extremos de un segundo amplicón. Los cebadores de amplificación secundarios tienen colas no complementarias a las secuencias diana, que forman los extremos del segundo amplicón y que se pueden usar en relación con la secuenciación de los clonotipos del segundo amplicón. En una realización, tales secuencias agregadas pueden incluir sitios de unión de cebadores para generar lecturas de secuencias y sitios de unión de cebadores para llevar a cabo una PCR de puente sobre una superficie sólida para generar poblaciones clonales de moléculas individuales aisladas espacialmente, por ejemplo, cuando se usa una secuenciación basada en Solexa. En esta última metodología una muestra de secuencias del segundo amplicón se dispone sobre una superficie sólida que tiene oligonucleótidos complementarios que se pueden unir a las secuencias de la muestra, después de lo cual se implementan ciclos de extensión del cebador, desnaturalización y fusionamiento hasta que las poblaciones clonales de plantillas están formadas. Preferiblemente, el tamaño de la muestra se selecciona de manera que (i) incluya una representación efectiva de clonotipos en la muestra original, y (ii) la densidad de las poblaciones clonales en la superficie sólida se encuentre en un rango que permita una determinación de secuencia inequívoca de clonotipos.
Además de garantizar que la muestra contenga suficientes células para ser representativa de la muestra original, es importante que los amplicones generados por la reacción de PCR multiplex sean representativos de las células en la reacción. Para lograr esto, se deben seleccionar las condiciones del cebador de modo que se produzca la amplificación de cada célula en la reacción.
Las secuencias de TCR o BCR o porciones de las mismas pueden amplificarse a partir de ácido nucleico en una reacción multiplexada utilizando al menos un cebador que se acopla a la región C y uno o más cebadores que pueden acoplarse a uno o más segmentos V (como se ilustra en las figuras 2A-2B y las figuras 4A-4B y se explican más detalladamente a continuación). El número de cebadores que se reasocian a segmentos V en una reacción multiplexada puede ser, por ejemplo, al menos 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 u 80. El número de cebadores que se aparean a segmentos V en una reacción multiplex puede ser, por ejemplo, 10-60, 20-50, 30-50, 40 50, 20-40, 30-40, o 35-40. Los cebadores pueden recocer a diferentes segmentos en V. Para los genes IgH, debido a la posibilidad de mutaciones somáticas en los segmentos V, se pueden usar múltiples cebadores que se fusionen a cada segmento V; por ejemplo, 1, 2, 3, 4 o 5 cebadores por segmento V. El número de cebadores que se reasocian a segmentos C en una reacción multiplexada puede incluir, por ejemplo, al menos 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, o 15. El número de cebadores que se fusionan a los segmentos C en una reacción multiplexada puede ser 1-10, 2-9, 3-8, 4-7, 3-8 o 3-6. La amplificación de los genes TCR o inmunoglobulina puede ocurrir como se describe en el Ejemplo 3 y/o Ejemplo 4.
La región por amplificar puede incluir la secuencia clonal completa o un subconjunto de la secuencia clonal, incluida la unión V-D, la unión D-J de un gen receptor de inmunoglobulina o de células T, la región variable completa de un gen receptor de inmunoglobulina o célula T, la región de reconocimiento de antígeno, o una CDR, por ejemplo, la región 3 determinante de complementariedad (CDR3).
La secuencia de TCR o inmunoglobulina puede amplificarse usando una etapa de amplificación primaria y una secundaria. Cada uno de los diferentes pasos de amplificación puede comprender diferentes cebadores. Los diferentes cebadores pueden introducir secuencias no presentes originalmente en la secuencia del gen inmune. Por ejemplo, el procedimiento de amplificación puede agregar nuevos sitios de unión de cebadores a los extremos de las secuencias diana para convertir una amplificación multiplex en una amplificación de un solo complejo o el procedimiento de amplificación puede agregar una o más etiquetas al extremo 5' y/o 3' de la amplificación TCR o secuencia de inmunoglobulina (como se ilustra en las figuras 3A-3C). La etiqueta puede ser una secuencia que facilita la secuenciación posterior del ADN amplificado. La etiqueta puede ser una secuencia que facilita la unión de la secuencia amplificada a un soporte sólido.
Otros métodos de amplificación pueden no emplear ningún cebador en la región V. En su lugar, se puede usar un cebador específico del segmento C y un cebador genérico se puede colocar en el otro lado (5'). El cebador genérico se puede agregar a la síntesis de ADNc a través de diferentes métodos, incluidos los métodos bien descritos de cambio de cadena. De manera similar, el cebador genérico se puede adjuntar después de la creación de ADNc a través de diferentes métodos, incluida la ligación.
Otros medios para amplificar el ácido nucleico que pueden usarse en los métodos de la invención incluyen, por ejemplo, PCR de transcripción reversa, PCR en tiempo real, PCR cuantitativa en tiempo real, PCR digital (dPCR), PCR de emulsión digital (dePCR), PCR clonal, polimorfismo de longitud de fragmento amplificado (AFLP PCR), PCR específica de alelo, PCR de ensamblaje, PCR asimétrica (en la que se utiliza un gran exceso de cebadores para una cadena seleccionada), PCR de colonias, amplificación dependiente de la helicasa (HDA), PCR de inicio rápido, PCR inversa (IPCR), PCR in situ, PCR larga (extensión del ADN superior a aproximadamente 5 kilobases), PCR múltiple, PCR anidada (usa más de un par de cebadores), PCR de una sola célula, PCR de toma de contacto, PCR isotérmica mediada por bucle (LAMP) y amplificación basada en la secuencia de ácidos nucleicos (NASBA). Otros esquemas de amplificación incluyen: reacción en cadena de la ligasa, amplificación de ADN ramificada, amplificación de círculo rodante, amplificación de círculo a círculo, amplificación SPIA, amplificación de objetivo por captura y ligación (TACL), y amplificación RACE.
La información en el ARN de una muestra se puede convertir en ADNc utilizando la transcripción reversa. Se pueden usar cebadores PolyA, cebadores aleatorios y/o cebadores específicos de genes en reacciones de transcripción reversa de acuerdo con los protocolos convencionales.
Después de la amplificación del ADN del genoma (o la amplificación del ácido nucleico en forma de ADNc por el ARN de transcripción reversa), las moléculas individuales de ácido nucleico se pueden aislar, opcionalmente volver a amplificar y luego secuenciar individualmente. Protocolos de amplificación ejemplares pueden encontrarse en van Dongen et al, Leukemia, 17: 2257-2317 (2003) o van Dongen et al, Publicación de Patente de los Estados Unidos 2006/0234234. En resumen, un protocolo ejemplar es el siguiente: Regulador de reacción: ABI Buffer II o ABI Gold Buffer (Life Technologies, San Diego, CA); volumen de reacción final de 50 pL; 100 ng de muestra de ADN; 10 pmol de cada cebador (sujeto a los ajustes de amplificación de balance como se describe a continuación); dNTPs a una concentración final de 200 pM; MgCh a una concentración final de 1.5 mM (sujeto a optimización dependiendo de las secuencias diana y la polimerasa); Taq polimerasa (1-2 U/tubo); condiciones de ciclización: preactivación 7 min a 95°C; fusionamiento a 60°C; tiempos de ciclización: desnaturalización 30s; 30s de fusionamiento; extensión 30s.
Las polimerasas que pueden usarse para la amplificación en los métodos de la invención están disponibles comercialmente e incluyen, por ejemplo, polimerasa Taq, polimerasa AccuPrime o Pfu. La elección de la polimerasa por usar puede basarse en la preferencia entre la fidelidad o la eficiencia.
Los métodos para el aislamiento de ácidos nucleicos de un grupo incluyen la separación espacial de las moléculas en dos dimensiones sobre un sustrato sólido (por ejemplo, un portaobjetos de vidrio), la separación espacial de las moléculas en tres dimensiones en una solución dentro de las micelas (como puede ser logrado usando emulsiones de aceite con o sin inmovilizar las moléculas en una superficie sólida como las perlas), o usando cámaras de microrreacción en, por ejemplo, chips para microfludos o nanofluidos. Se puede usar una dilución para asegurar que, en promedio, una sola molécula esté presente en un volumen, región espacial, perla o cámara de reacción dados. La guía para tales métodos de aislamiento de moléculas de ácido nucleico individuales se encuentra en las siguientes referencias: Sambrook, Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Laboratory Press, 2001s); Shendure et al, Science, 309: 1728-1732 (incluido material suplementario) (2005); Patente de los Estados Unidos 6,300,070; Bentley et al, Nature, 456: 53-59 (incluido material suplementario) (2008); Patente de los Estados Unidos 7,323,305; Matsubara et al, Biosensors & Bioelectronics, 20: 1482-1490 (2005): Patente de los Estados Unidos 6,753,147; y similares.
La PCR en tiempo real, la tinción con Picogreen, la electroforesis de nanofluidos (por ejemplo, LabChip) o las mediciones de absorción UV se pueden usar en un paso inicial para evaluar la cantidad funcional de material amplificable.
Los métodos para la reamplificación de ácidos nucleicos incluyen el crecimiento bacteriano de colonias aisladas transformadas con ácido nucleico, la amplificación en un portaobjetos (por ejemplo, colonias de PCR (polonias)), y la amplificación en una perla (por ejemplo, en una PCR de emulsión). Se puede usar el mismo método para amplificar y volver a amplificar el ácido nucleico o se puede usar un método diferente para amplificar y reamplificar el ácido nucleico.
En ciertas realizaciones, las etapas de subclonación incluyen una etapa en la que un cebador común se une al ADN o ARN a través de una etapa de amplificación o ligación. Este cebador se usa luego para amplificar los clones y como una secuencia de reconocimiento para la hibridación de un cebador para la secuenciación (por ejemplo, como se ilustra en las figuras 2A-2B y 4A-4B, y se explica más detalladamente a continuación).
En un aspecto, las amplificaciones multiplex se llevan a cabo de modo que las cantidades relativas de secuencias en una población de partida son sustancialmente las mismas que las de la población amplificada, o amplicón. Es decir, las amplificaciones multiplex se llevan a cabo con un mínimo sesgo de amplificación entre las secuencias miembros de una población de muestra. En una realización, tales cantidades relativas son sustancialmente iguales si cada cantidad relativa en un amplicón está dentro de cinco veces su valor en la muestra inicial. En otra realización, tales cantidades relativas son sustancialmente iguales si cada cantidad relativa en un amplicón está dentro de dos veces su valor en la muestra inicial. Como se explica más detalladamente a continuación, el sesgo de amplificación en la PCR se puede detectar y corregir usando técnicas convencionales, de modo que se pueda seleccionar un conjunto de cebadores de PCR para un repertorio predeterminado que proporcione una amplificación no sesgada de cualquier muestra.
Con respecto a muchos repertorios basados en secuencias TCR o BCR, una amplificación multiplex utiliza opcionalmente todos los segmentos V. La reacción está optimizada para intentar obtener una amplificación que mantenga la abundancia relativa de las secuencias amplificadas por diferentes cebadores de segmento V. Algunos de los cebadores están relacionados y, por lo tanto, muchos de los cebadores pueden "hablar de manera cruzada", amplificando las plantillas que no se combinan perfectamente con él. Las condiciones se optimizan para que cada plantilla pueda amplificarse de una manera similar, independientemente de qué cebador lo amplificó. En otras palabras, si hay dos plantillas, luego de 1.000 veces la amplificación, ambas plantillas se pueden amplificar aproximadamente 1.000 veces, y no importa que para una de las plantillas la mitad de los productos amplificados tenga un cebador diferente debido a la interferencia. En el análisis posterior de los datos de secuenciación, la secuencia del cebador se elimina del análisis y, por lo tanto, no importa qué cebador se use en la amplificación siempre que las plantillas se amplifiquen por igual.
Como la cantidad de cada plantilla no se conoce en una población de ADNc generada a partir de ARNm, se puede generar un conjunto de estándares utilizando PCRs de dúplex simples de una población de ADNc de clonotipos. Esto se llevó a cabo para un repertorio de clonotipos TCRp. El producto en cada una de 34 PCR (utilizando los cebadores de reacciones separadas del ejemplo 3) comprendía una pluralidad de secuencias con un cebador en V. Los diferentes productos se cuantificaron cuidadosamente para crear un conjunto de estándares en la misma concentración. Se utilizó un conjunto de los 34 cebadores y se realizaron 34 PCR en tiempo real utilizando el conjunto de cebadores y cada una de las secuencias estándar como plantilla. Idealmente, sin sesgos, todos los 34 estándares mostrarán la misma eficiencia de amplificación mediante PCR en tiempo real. Eso sugiere que cada secuencia se amplifica por igual, aunque la presencia de interferencias hace que no esté claro qué cebadores están llevando a cabo la amplificación. Esta optimización es coherente con el objetivo de tener una amplificación igual independientemente de los cebadores reales que se incorporan en el producto de amplificación. El aumento de la concentración total del conjunto de cebadores redujo significativamente el rango dinámico como se esperaba al aumentar la eficiencia de la amplificación. Además, para las plantillas que parecían amplificarse más eficientemente que el promedio, se redujo la concentración de su imprimador perfectamente combinado en el conjunto. A la inversa, para las plantillas que se amplificaron ineficientemente, se incrementó la concentración de su cebador perfectamente emparejado. Esta optimización demostró que todas las plantillas se amplifican dentro de dos veces la amplificación promedio.
El sesgo de la amplificación también se puede evitar mediante la realización de una amplificación de dos etapas (como se ilustra en las figuras 2A-2B) en donde se implementa un pequeño número de ciclos de amplificación en una primera etapa, o primaria, utilizando cebadores que tienen colas no complementarias con la secuencia objetivo. Las colas incluyen los sitios de unión del cebador que se agregan a los extremos de las secuencias del amplicón primario, de modo que dichos sitios se usan en una la segunda etapa de amplificación utilizando solo un cebador directo único y un cebador inverso único, eliminando así la causa principal del sesgo de amplificación. Preferiblemente, la PCR primaria tendrá un número suficientemente pequeño de ciclos (por ejemplo, 5-10) para minimizar la amplificación diferencial de los diferentes cebadores. La amplificación secundaria se realiza con un par de cebadores y, por lo tanto, el problema de la amplificación diferencial es mínimo. El uno por ciento de la PCR primaria se lleva directamente a la PCR secundaria. Treinta y cinco ciclos (equivalentes a -28 ciclos sin la etapa de dilución de 100 veces) utilizados entre las dos amplificaciones fueron suficientes para mostrar una amplificación robusta independientemente de si la ruptura de los ciclos fue: un ciclo primario y 34 secundarios o 25 primarios y 10 secundarios. Aunque idealmente hacer solo 1
ciclo en la PCR primaria puede disminuir el sesgo de amplificación, existen otras consideraciones. Un aspecto de esto es la representación. Estajuega un papel cuando la cantidad de entrada inicial no está en exceso al número de lecturas obtenidas en última instancia. Por ejemplo, si se obtienen 1.000.000 lecturas y comenzando con 1.000.000 de moléculas de entrada, entonces tomar solo la representación de 100.000 moléculas a la amplificación secundaria degradaría la precisión de estimar la abundancia relativa de las diferentes especies en la muestra original. La dilución de 100 veces entre los 2 pasos significa que la representación se reduce a menos que la amplificación primaria de PCR genere significativamente más de 100 moléculas. Esto indica que se puede usar un mínimo de 8 ciclos (256 veces), pero más cómodamente 10 ciclos (aproximadamente 1.000 veces). La alternativa a eso es llevar más del 1% de la PCR primaria a la secundaria, pero debido a la alta concentración de cebador utilizado en la PCR primaria, se puede usar un gran factor de dilución para garantizar que estos cebadores no interfieran en la amplificación y empeorar el sesgo de amplificación entre secuencias. Otra alternativa es agregar un paso de purificación o enzimático para eliminar los cebadores de la PCR primaria para permitir una dilución menor. En este ejemplo, la PCR primaria fue de 10 ciclos y la segunda de 25 ciclos.
La reproducibilidad de una PCR multiplex se puede evaluar de la siguiente manera, como se ejemplifica con el conjunto de prueba de cebadores del Ejemplo 2. Se realizan dos reacciones de PCR primaria utilizando la serie de prueba de cebadores, por ejemplo. los cebadores TCRp agrupados y el cebador C (del Ejemplo 2) y una muestra de ADNc como plantilla. La abundancia relativa en cada plantilla amplificada se evalúa mediante PCR en tiempo real. Utilizando cada uno de los dos productos amplificados como plantilla, se realizaron treinta y cuatro reacciones de PCR en tiempo real diferentes utilizando el cebador C y uno de los cebadores V en cada reacción. Los datos mostrados en la figura 5 demuestran que la abundancia relativa determinada por PCR en tiempo real fue altamente reproducible utilizando todos los cebadores V para dos muestras, lo que indica que la amplificación multiplexada es altamente reproducible. El número de ciclo (valor Ct) para cada una de las amplificaciones de PCR en tiempo real que utilizan el producto de amplificación multiplexado como plantilla se muestra en el eje X y el segundo producto de amplificación multiplexado como plantilla se muestra en el eje Y.
La cantidad de sesgo de amplificación de un conjunto de cebadores puede evaluarse utilizando el siguiente procedimiento, que se ejemplifica utilizando el conjunto de cebadores del Ejemplo 2. El conjunto de cebadores de prueba (como anteriormente) se usa para amplificar un ADNc como plantilla (por ejemplo, obtenido de ARNm extraído de linfocitos). La cantidad de plantilla amplificada por cada uno de los 34 cebadores diferentes (junto con el cebador del segmento C) se determina usando PCR en tiempo real y esa cantidad se compara con la cantidad amplificada utilizando los mismos cebadores con el ADNc. Dado que puede haber interferencias, incluso si la abundancia relativa entre las secuencias internas en el producto amplificado y el ADNc fuera la misma, solo se pueden detectar diferencias significativas en la amplificación utilizando esta lectura. Esta posibilidad se puede probar sintetizando la colección de cebadores para amplificar segmentos internos de varias secuencias de ADNc de inicio. Por ejemplo, se diseñaron 12 oligos que, cuando se usan con cebador de segmento C, pueden amplificar secuencias internas a los cebadores de segmento V anteriores. Si hay un sesgo de amplificación mínimo, entonces la concentración de estas secuencias internas debería cambiar poco entre el ADNc de inicio y los productos amplificados. Los datos de este ejemplo se muestran en la figura 6. Allí, se usó una muestra de ADNc como plantilla para una amplificación multiplexada utilizando los cebadores TCRp agrupados y el cebador C (del Ejemplo 2). El cebador C y los cebadores internos posteriores se utilizaron para la amplificación inicial del material de plantilla de la amplificación multiplex. De manera similar, se usó PCR en tiempo real para evaluar la abundancia relativa de estas mismas secuencias en el ADNc. Si la amplificación multiplexada tuviera algún sesgo significativo, la abundancia relativa en el material amplificado sería muy diferente de la del ADNc. Como puede verse en la figura 6, se observó una alta correlación que demuestra un sesgo de amplificación mínimo en la amplificación multiplexada. El número de ciclo (valor Ct) para cada una de las amplificaciones de PCR en tiempo real utilizando cebadores internos, y el ADNc y el producto de amplificación multiplexado como plantilla se muestran en los ejes X e Y, respectivamente.
La amplificación inicial se puede realizar a partir de ADN o ARN (por ejemplo, después de la conversión a ADNc).
Secuenciación de poblaciones de ácidos nucleicos
Las técnicas de secuenciación de ADN para su uso en los métodos de la invención incluyen la secuenciación por síntesis utilizando nucleótidos marcados terminados de manera reversible, pirosecuenciación, secuenciación 454, hibridación específica de alelo a una biblioteca de sondas de oligonucleótidos marcadas, secuenciación por síntesis utilizando hibridación específica de alelo a una biblioteca de clones marcados a esto le sigue la ligación, monitorización en tiempo real de la incorporación de nucleótidos marcados durante una etapa de polimerización, la secuenciación polónica y la secuenciación SOLiD. La secuenciación de las moléculas separadas se ha demostrado más recientemente mediante reacciones de extensión simple o secuencial utilizando polimerasas o ligasas, así como mediante hibridaciones diferenciales únicas o secuenciales con bibliotecas de sondas. Estas reacciones se han realizado en muchas secuencias clonales en paralelo, incluidas demostraciones en aplicaciones comerciales actuales de más de 100 millones de secuencias en paralelo. Estas metodologías de secuenciación pueden utilizarse para estudiar el repertorio de receptores de células T (TCR) y/o receptores de células B (BCR). En un aspecto de la invención, se emplean métodos de secuenciación de alto rendimiento que comprenden una etapa de aislamiento espacial de moléculas individuales en una superficie sólida donde se secuencian en paralelo. Dichas superficies sólidas pueden incluir superficies no porosas (como en la secuenciación Solexa, por ejemplo, Bentley et al, Nature, 456: 53-59 (2008) o la secuenciación genómica completa, por ejemplo, Drmanac et al, Science, 327: 78-81 (2010)),
matrices de pozos, que pueden incluir plantillas unidas a perlas o partículas (como con 454, por ejemplo, Margulies et al, Nature, 437: 376-380 (2005) o secuenciación por torrente de iones, Publicación de Patente de los Estados Unidos 2010/0137143 o 2010/0304982), membranas micromaquinadas (como con la secuenciación SMRT, por ejemplo, Eid et al, Science, 323: 133-138 (2009)), o matrices de perlas (como con la secuenciación SOLiD o la secuencia de polonia, por ejemplo, Kim et al, Science, 316: 1481-1414 (2007)). En otro aspecto, tales métodos comprenden amplificar las moléculas aisladas antes o después de que se aíslen espacialmente en una superficie sólida. La amplificación previa puede comprender una amplificación basada en emulsión, como la PCR en emulsión o la amplificación de círculo rodante. De particular interés es la secuenciación basada en Solexa donde las moléculas individuales de la plantilla se aíslan espacialmente en una superficie sólida, después de lo cual se amplifican en paralelo mediante PCR puente para formar poblaciones clonales separadas, o agrupaciones, y luego se secuencian, como se describe en Bentley et al (citado anteriormente) y en las instrucciones del fabricante (por ejemplo, el kit de preparación de muestras TruSeq™ y la hoja de datos, Illumina, Inc., San Diego, CA, 2010); y más adelante en las siguientes referencias: Patentes de los Estados Unidos 6,090,592; 6,300,070; 7,115,400; y EP0972081B1. En una realización, las moléculas individuales dispuestas y amplificadas sobre una superficie sólida forman agrupaciones en una densidad de al menos 105 agrupaciones por cm2; o en una densidad de al menos 5x105 por cm2; o en una densidad de al menos 106 agrupamientos por cm2. En una realización, se emplean químicas de secuenciación que tienen tasas de error relativamente altas. En tales realizaciones, las puntuaciones de calidad promedio producidas portales químicas son funciones que disminuyen de manera monotónica las longitudes de lectura de secuencia. En una realización, tal disminución corresponde al 0.5 por ciento de las lecturas de secuencia tienen al menos un error en las posiciones 1 75; 1 por ciento de las lecturas de secuencia tienen al menos un error en las posiciones 76-100; y el 2 por ciento de las lecturas de secuencia tienen al menos un error en las posiciones 101-125.
En un aspecto, para cada muestra de un individuo, la técnica de secuenciación utilizada en los métodos de la invención genera secuencias de al menos 1000 clonotipos por ejecución; en otro aspecto, tal técnica genera secuencias de al menos 10.000 clonotipos por ejecución; en otro aspecto, tal técnica genera secuencias de al menos 100.000 clonotipos por ejecución; en otro aspecto, tal técnica genera secuencias de al menos 500.000 clonotipos por ejecución; y en otro aspecto, tal técnica genera secuencias de al menos 1.000.000 de clonotipos por ejecución. En aún otro aspecto, tal técnica genera secuencias de entre 100.000 y 1.000.000 de clonotipos por corrida por muestra individual.
La técnica de secuenciación utilizada en los métodos de la invención proporcionada puede generar aproximadamente 30 pb, aproximadamente 40 pb, aproximadamente 50 pb, aproximadamente 60 pb, aproximadamente 70 pb, aproximadamente 80 pb, aproximadamente 90 pb, aproximadamente 100 pb, aproximadamente 110 pb, aproximadamente 120 bp por lectura, aproximadamente 150 bp, aproximadamente 200 bp, aproximadamente 250 bp, aproximadamente 300 bp, aproximadamente 350 bp, aproximadamente 400 bp, aproximadamente 450 bp, aproximadamente 500 bp, aproximadamente 550 bp, o aproximadamente 600 bp por lectura.
La técnica de secuenciación utilizada en los métodos de la invención proporcionada puede generar al menos 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 150, 200, 250, 300, 350, 400, 450, 500, 550, o 600 pb por lectura. Se puede obtener un perfil de clonotipo basado en la secuencia de un individuo usando los siguientes pasos: (a) obtener una muestra de ácido nucleico de células T y/o células B del individuo; (b) aislar espacialmente moléculas individuales derivadas de dicha muestra de ácido nucleico, comprendiendo las moléculas individuales conjuntos de plantillas anidadas, cada una generada a partir de un ácido nucleico en la muestra y cada una de ellas conteniendo una región reorganizada somáticamente o una porción de la misma, siendo cada serie anidada capaz de producir una pluralidad de lecturas de secuencias extendiéndose cada una en la misma dirección y cada una partiendo de una posición diferente en el ácido nucleico desde el cual se generó el conjunto anidado; (c) secuenciar dichas moléculas individuales espacialmente aisladas; y (d) determinar las abundancias de diferentes secuencias de las moléculas de ácido nucleico de la muestra de ácido nucleico para generar el perfil de clonotipo. En una realización, la etapa de secuenciación incluye producir una pluralidad de lecturas de secuencia para cada uno de los conjuntos anidados. En otra realización, cada una de las regiones reorganizadas somáticamente comprende una región V y una región J, y cada una de la pluralidad de lecturas de secuencias comienza desde una posición diferente en la región V y se extiende en la dirección de su región J asociada. En otra realización, la etapa de secuenciación comprende la secuenciación bidireccional de cada una de las moléculas individuales aisladas espacialmente para producir al menos una lectura de secuencia de avance y al menos una lectura de secuencia reversa. Además de la última realización, al menos una de las lecturas de secuencia de avance y al menos una de las lecturas de secuencia reversa tienen una región de solapamiento tal que las bases de dicha región de superposición están determinadas por una relación complementaria inversa entre dichas lecturas de secuencia. En otra realización más, cada una de las regiones reorganizadas somáticamente comprende una región V y una región J y la etapa de secuenciación incluye además determinar una secuencia de cada una de las moléculas de ácido nucleico individuales a partir de una o más de sus lecturas de secuencia de avance y al menos una lectura de secuencia reversa comenzando desde una posición en una región J y extendiéndose en la dirección de su región V asociada. En otra realización, las moléculas individuales comprenden ácidos nucleicos seleccionados del grupo que consiste en moléculas de IgH completas, moléculas de IgH incompletas, IgK completas, moléculas inactivas de IgK, moléculas TCRp, moléculas TCRy, moléculas TCR5 completas y moléculas TCR5 incompletas. En otra realización, la etapa de secuenciación comprende generar las lecturas de secuencia que tienen puntuaciones de calidad que disminuyen de manera monótona. Además de la última realización, las puntuaciones de calidad monotónicamente decrecientes son tales que las lecturas de secuencia tienen tasas de error no mejores que las siguientes: 0.2 por ciento de las lecturas de secuencia contienen al menos un error en las posiciones de base 1 a 50, 0.2 a 1.0 por ciento de las
lecturas de secuencia contienen al menos un error en las posiciones 51-75, 0.5 a 1.5 por ciento de las lecturas de secuencia contienen al menos un error en las posiciones 76-100.
Como se indica a continuación en la definición de repertorio, pueden secuenciarse diferentes regiones predeterminadas de inmunoglobulina o genes de receptores de células T. En algunas realizaciones, la secuencia completa de las regiones variables se puede secuenciar para identificar y cuantificar un clonotipo.
Se puede secuenciar un subconjunto único de las secuencias clonales completas. En algunas realizaciones, los nucleótidos que comprenden las uniones VD y DJ se secuencian para identificar y cuantificar de forma única un clonotipo. En otras realizaciones, el fragmento que puede secuenciarse es la región variable completa. En otra realización más, se secuencia la región de reconocimiento de antígeno o la región 3 determinante de complementariedad (CDR3). Un fragmento que contiene la CDR3 completa o la región variable completa se puede amplificar para permitir la secuenciación de la CDR3 que comprende partes de los segmentos V, D y J.
En una realización, solo la CDR3 está amplificada y secuenciada. La amplificación y secuenciación de la CDR3 se puede lograr utilizando cebadores específicos para una o más secuencias del segmento V (así como uno o más cebadores en el otro lado del amplicón en el segmento C). Los cebadores para cada uno de los segmentos V se pueden utilizar en una o más reacciones de amplificación que conducen a la amplificación del repertorio completo de secuencias. Este repertorio de secuencias se puede mezclar y someter a separación, con o sin amplificación, y secuenciar usando cualquiera de las técnicas de secuenciación descritas. Cuando la amplificación con los distintos cebadores V se realiza en tubos separados, el número de moléculas que llevan los diferentes segmentos V se puede "normalizar" debido a la saturación de la PCR. Por ejemplo, si un segmento V en particular tenía una o varias expansiones clonales que llevan a su representación más que a otros segmentos, esta información puede borrarse o disminuirse, ya que la reacción de la PCR para cada segmento puede ser saturada o cercana a ella. La PCR en tiempo real se puede usar para cuantificar qué cantidad de cada segmento V está presente. Se puede secuenciar la CDR3 completa, o se puede secuenciar un subconjunto de la secuencia CDR3.
En una realización, solo se analiza un subconjunto de clonotipos. Esto se puede lograr amplificando con un cebador específico para el subconjunto de clonotipos, por ejemplo, un cebador que sea específico para el segmento V. Los clonotipos únicos se pueden identificar mediante la secuenciación con lecturas contiguas largas que proporcionan conectividad completa. En algunas realizaciones, cuando están presentes varias secuencias de interés, una corta longitud de lectura en solo una de las uniones puede generar etiquetas degeneradas que no son exclusivas de un clonotipo específico pero que se comparten entre múltiples clonotipos. Por ejemplo, la secuenciación en la unión V/J puede agrupar todas las secuencias con el mismo V/J independientemente del segmento D como un clonotipo. La información sobre la conectividad total de todos los segmentos permite distinguir las secuencias que pueden compartir los mismos segmentos V y J, pero que están conectadas a diferentes segmentos D, por ejemplo.
Determinación de clonotipos a partir de datos de secuencia
En un aspecto de la invención, las secuencias de clonotipos (incluidas, entre otras, las derivadas de IgH, TCRa, TCRp, TCRy, TCR8 y/o IgLK (IgK)) se pueden determinar combinando información de una o más lecturas de secuencias, por ejemplo, a lo largo de las regiones V(D)J de las cadenas seleccionadas. En otro aspecto, las secuencias de clonotipos se determinan combinando información de una pluralidad de lecturas de secuencia. (Como se usa en el presente documento, una "lectura de secuencia" es una secuencia de datos generada por una técnica de secuenciación a partir de la cual se determina una secuencia de nucleótidos. Típicamente, las lecturas de secuencia se realizan extendiendo un cebador a lo largo de un ácido nucleico plantilla, por ejemplo, con un ADN polimerasa o un ADN ligasa. Los datos se generan al registrar señales, como señales ópticas, químicas (por ejemplo, cambio de pH) o eléctricas, asociadas con dicha extensión.) Tales pluralidades de lecturas de secuencia pueden incluir una o más lecturas de secuencia a lo largo de una cadena de sentido (es decir, lecturas de secuencia "de avance") y una o más lecturas de secuencia a lo largo de su hebra complementaria (es decir, lecturas de secuencia "reversa"). Cuando se generan múltiples lecturas de secuencia a lo largo de la misma cadena, primero se generan plantillas separadas amplificando moléculas de muestra con cebadores seleccionados para las diferentes posiciones de las lecturas de secuencia. Este concepto se ilustra en la figura 4A donde se emplean cebadores (404, 406 y 408) para generar amplicones (410, 412 y 414, respectivamente) en una sola reacción. Dichas amplificaciones se pueden llevar a cabo en la misma reacción o en reacciones separadas. En un aspecto, cuando se emplea la PCR, se usan reacciones de amplificación separadas para generar las plantillas separadas que, a su vez, se combinan y se usan para generar múltiples lecturas de secuencia a lo largo de la misma cadena. Esta última metodología es preferible para evitar la necesidad de equilibrar las concentraciones de cebadores (y/u otros parámetros de reacción) para asegurar una amplificación igual de las múltiples plantillas (algunas veces referidas aquí como "amplificación balanceada" o "amplificación no sesgada"). La generación de plantillas en reacciones separadas se ilustra en las figuras 4B-4C. Allí, una muestra que contiene IgH (400) se divide en tres porciones (472, 474 y 476) que se agregan a PCRs separadas utilizando los cebadores de la región J (401) y los cebadores de la región V (404, 406 y 408, respectivamente) para producir amplicones (420, 422 y 424, respectivamente). Los últimos amplicones se combinan (478) en la PCR secundaria (480) utilizando los cebadores P5 y P7 para preparar las plantillas (482) para la PCR de puente y la secuenciación en un secuenciador Illumina GA o un instrumento similar.
Las lecturas de secuencia pueden tener una gran variedad de longitudes, dependiendo en parte de la técnica de secuenciación que se esté empleando. Por ejemplo, para algunas técnicas, pueden surgir varias compensaciones en su implementación, por ejemplo, (i) el número y la longitud de las lecturas de secuencia por plantilla y (ii) el coste y la duración de una operación de secuenciación. En una realización, las lecturas de secuencia están en el intervalo de 20 a 400 nucleótidos; en otra realización, las lecturas de secuencia están en un intervalo de 30 a 200 nucleótidos; en otra realización más, las lecturas de secuencia están en el intervalo de 30 a 120 nucleótidos. En una realización, se generan de 1 a 4 lecturas de secuencia para determinar la secuencia de cada clonotipo; en otra realización, se generan de 2 a 4 lecturas de secuencia para determinar la secuencia de cada clonotipo; y en otra realización, se generan de 2 a 3 lecturas de secuencia para determinar la secuencia de cada clonotipo. En las realizaciones anteriores, los números dados son exclusivos de las lecturas de secuencia utilizadas para identificar muestras de diferentes individuos. Las longitudes de las diversas lecturas de secuencia utilizadas en las realizaciones descritas a continuación también pueden variar en función de la información que la lectura pretende captar; por ejemplo, la ubicación de inicio y la longitud de una secuencia leída pueden diseñarse para proporcionar la longitud de una región NDN así como su secuencia de nucleótidos; por lo tanto, se seleccionan las lecturas de secuencia que abarcan toda la región NDN. En otros aspectos, una o más lecturas de secuencia abarcan las regiones D y/o NDN.
Las secuencias de los clonotipos se pueden determinar en parte al alinear las lecturas de secuencia con una o más secuencias de referencia de la región V y una o más secuencias de referencia de la región J, y en parte mediante la determinación de la base sin alineación con las secuencias de referencia, como en la región n Dn altamente variable. Se puede aplicar una variedad de algoritmos de alineación a las lecturas de secuencia y secuencias de referencia. Por ejemplo, la guía para seleccionar métodos de alineación está disponible en Batzoglou, Briefings in Bioinformatics, 6: 6-22 (2005). En un aspecto, cada vez que las lecturas V o las lecturas C (descritas con mayor detalle más adelante) se alinean con las secuencias de referencia de las regiones V y J, se emplea un algoritmo de búsqueda de árbol, por ejemplo. Cormen et al, Introduction to Algorithms, tercera edición (The MIT Press, 2009). Las estructuras de codón de las secuencias de referencia V y J pueden usarse en un proceso de alineación para eliminar los errores de secuenciación y/o para determinar un nivel de confianza en la alineación resultante, como se describe más detalladamente a continuación. En otro aspecto, un final de al menos una lectura directa y un final de al menos una lectura inversa se superpone en una región solapada (por ejemplo, 308 en la figura 3B), de modo que las bases de las lecturas están en una relación complementaria reversa de una con la otra. Así, por ejemplo, si una lectura directa en la región de superposición es "5'-acgttgc", entonces una lectura inversa en una relación complementaria inversa es "5'-gcaacgt" dentro de la misma región de superposición. En un aspecto, las bases dentro de dicha región de superposición se determinan, al menos en parte, a partir de dicha relación complementaria inversa. Es decir, la probabilidad de una llamada de base (o un puntaje de calidad relacionado) en una posible región de superposición aumenta si conserva, o es consistente con, una relación complementaria inversa entre las dos lecturas de secuencia. En un aspecto, los clonotipos de las cadenas TCR p e IgH (ilustradas en la figura 3B) se determinan por al menos una secuencia leída que comienza en su región J y se extiende en la dirección de su región V asociada (referida aquí como una "lectura C" "(304)) y al menos una secuencia leída que comienza en su región V y se extiende en la dirección de su región J asociada (referida aquí como una" lectura V "(306)). La región de solapamiento (308) puede o no abarcar la región NDN (315) como se muestra en la figura 3B. La región de solapamiento (308) puede estar completamente en la región J, completamente en la región NDN, completamente en la región V, o puede abarcar un límite de región NDN-región o un límite de región V-NDN, o ambos límites (como se ilustra en la figura 3B). Normalmente, tales lecturas de secuencia se generan al extender los cebadores de secuenciación, por ejemplo, (302) y (310) en la figura 3B, con una polimerasa en una reacción de secuenciación por síntesis, por ejemplo, Metzger, Nature Reviews Genetics, 11: 31-46 (2010); Fuller et al, Nature Biotechnology, 27: 1013-1023 (2009). Los sitios de unión para los cebadores (302) y (310) están predeterminados, de modo que pueden proporcionar un punto de inicio o punto de anclaje para la alineación inicial y el análisis de las lecturas de secuencia. En una realización, una lectura de C se posiciona de manera que abarca la región D y/o NDN de la cadena TCR p o IgH e incluye una parte de la región V adyacente, por ejemplo, como se ilustra en las figuras 3B y 3C. En un aspecto, la superposición de la lectura V y la lectura C en la región V se utiliza para alinear las lecturas entre sí. En otras realizaciones, tal alineación de lecturas de secuencia no es necesaria, por ejemplo, con cadenas TCRp, de modo que una lectura V puede ser solo lo suficientemente larga para identificar la región V particular de un clonotipo. Este último aspecto se ilustra en la figura 3C. La lectura de secuencia (330) se usa para identificar una región V, con o sin superposición de otra lectura de secuencia, y otra lectura de secuencia (332) atraviesa la región NDN y se usa para determinar la secuencia de la misma. La porción (334) de lectura de secuencia (332) que se extiende a la región V se utiliza para asociar la información de secuencia de lectura de secuencia (332) con la de lectura de secuencia (330) para determinar un clonotipo. Para algunos métodos de secuenciación, como las metodologías base por base como el método de secuenciación Solexa, el tiempo de ejecución de la secuenciación y los costes de los reactivos se reducen al minimizar el número de ciclos de secuenciación en un análisis. Opcionalmente, como se ilustra en la figura 3B, el amplicón (300) se produce con la etiqueta de muestra (312) para distinguir entre los clonotipos que se originan de diferentes muestras biológicas, por ejemplo, diferentes pacientes. La etiqueta de muestra (312) se puede identificar al recocer un cebador con la región de unión del cebador (316) y extenderlo (314) para producir una secuencia leída a través de la etiqueta (312), a partir de la cual se decodifica la etiqueta (312) de muestra.
La cadena de IgH es más difícil de analizar que la cadena TCRp debido a al menos dos factores: i) la presencia de mutaciones somáticas dificulta el mapeo o la alineación, y ii) la región NDN es más grande, por lo que a menudo no es posible mapear una porción del segmento V a la lectura de C. En un aspecto de la invención, este problema se
resuelve utilizando una pluralidad de conjuntos de cebadores para generar lecturas V, que se ubican en diferentes ubicaciones a lo largo de la región V, preferiblemente de modo que los sitios de unión del cebador no estén superpuestos y separados, y con al menos un sitio de unión a cebador adyacente a la región NDN, por ejemplo, en una realización de 5 a 50 bases de la unión V-NDN, o en otra realización de 10 a 50 bases de la unión V-NDn . La redundancia de una pluralidad de conjuntos de cebadores minimiza el riesgo de no detectar un clonotipo debido al fallo de uno o dos cebadores que tienen sitios de unión afectados por mutaciones somáticas. Además, la presencia de al menos un sitio de unión del cebador adyacente a la región n Dn hace que sea más probable que una lectura V se superponga con la lectura C y, por lo tanto, extienda la longitud de la lectura C. Esto permite la generación de una secuencia continua que abarca todos los tamaños de regiones NDN y que también puede asignar sustancialmente las regiones V y J completas en ambos lados de la región NDN. Las realizaciones para llevar a cabo tal esquema se ilustran en las figuras 4A y 4D. En la figura 4A, una muestra que comprende cadenas de IgH (400) se secuencian generando una serie de amplicones para cada cadena al amplificar las cadenas con un solo conjunto de cebadores de la región J (401) y una pluralidad (se muestran tres) de conjuntos de la región V (402) cebadores (404, 406, 408) para producir una pluralidad de amplicones anidados (por ejemplo, 410, 412, 416) que comprenden la misma región NDN y que tienen diferentes longitudes que abarcan sucesivamente porciones más grandes (411, 413, 415) de V región (402). Los miembros de un conjunto anidado se pueden agrupar después de la secuenciación señalando la identificación (o identidad sustancial) de sus respectivas regiones NDN, J y/o C, lo que permite la reconstrucción de un segmento V(D)J más largo de lo que sería de otro modo para una plataforma de secuenciación con longitud de lectura y/o calidad de secuencia limitadas. En una realización, la pluralidad de conjuntos de cebadores puede ser un número en el intervalo de 2 a 5. En otra realización, la pluralidad es 2-3; y aún otra realización más, la pluralidad es 3. Las concentraciones y posiciones de los cebadores en una pluralidad pueden variar ampliamente. Las concentraciones de los cebadores de la región V pueden o no ser las mismas. En una realización, el cebador más cercano a la región NDN tiene una mayor concentración que los otros cebadores de la pluralidad, por ejemplo. para asegurar que los amplicones que contienen la región n Dn están representados en el amplicón resultante. Se pueden usar uno o más cebadores (por ejemplo, 435 y 437 en la figura 4B) adyacentes a la región NDN (444) para generar una o más lecturas de secuencia (por ejemplo, 434 y 436) que se superponen con la lectura de secuencia (442) generada por la región J cebador (432), lo que mejora la calidad de las llamadas de base en la región de solapamiento (440). Las lecturas de secuencia de la pluralidad de cebadores pueden o no solaparse con el sitio de unión del cebador posterior adyacente y/o la lectura de secuencia posterior adyacente. En una realización, las lecturas de secuencia proximales a la región NDN (por ejemplo, 436 y 438) se pueden usar para identificar la región V particular asociada con el clonotipo. Dicha pluralidad de cebadores reduce la probabilidad de una amplificación incompleta o fallida en caso de que uno de los sitios de unión del cebador esté hipermutado durante el desarrollo de inmunoglobulina. También aumenta la probabilidad de que la diversidad introducida por la hipermutación de la región V se capture en una secuencia de clonotipo. Se puede realizar una PCR secundaria para preparar los amplicones anidados para la secuenciación, por ejemplo, amplificando con los cebadores P5 (401) y P7 (404, 406, 408) como se ilustra para producir amplicones (420, 422 y 424), que pueden distribuirse como moléculas individuales en una superficie sólida, donde se amplifican aún más por PCR de puente, o técnica similar.
La llamada a la base en las regiones NDN (particularmente de las cadenas IgH) se puede mejorar utilizando la estructura de codón de las regiones J y V flanqueantes, como se ilustra en la figura 4C. (Tal como se usa en el presente documento, "estructura de codón" significa los codones del marco de lectura natural de segmentos de transcripciones de TCR o BCR o genes fuera de las regiones NDN, por ejemplo, la región V, la región J, o similares.) Hay un amplicón (450), que es una vista ampliada del amplicón de la figura 4B, se muestra junto con las posiciones relativas de lectura C (442) y lectura adyacente V (434) anterior y las estructuras de codón (452 y 454) de la región V (430) y la región J (446), respectivamente, a continuación. De acuerdo con este aspecto de la invención, después de que las estructuras de los codones (452 y 454) se identifican mediante alineación convencional con las secuencias de referencia V y J, las bases en la región NDN (456) se denominan (o identifican) una base a la vez que se mueven desde la región J (446) hacia la región V (430) y en la dirección opuesta desde la región V (430) hacia la región J (446) usando lecturas de secuencia (434) y (442). En condiciones biológicas normales, solo las secuencias TCR o IgH recombinadas que tienen codones de marco de la región V a través de la región NDN y a la región J se expresan como proteínas. Es decir, de las variantes generadas somáticamente, las únicas expresadas son aquellas cuyos fotogramas de codones de región J y V están en marco unos con otros y permanecen en marco a través de la región NDN. (Aquí, los cuadros correctos de las regiones V y J se determinan a partir de secuencias de referencia). Si se identifica una secuencia fuera de marco basada en una o más llamadas de base de baja calidad, el clonotipo correspondiente se marca para una reevaluación o como una posible anomalía relacionada con la enfermedad. Si la secuencia identificada es en marco y se basa en llamadas de base de alta calidad, entonces hay una mayor confianza de que el clonotipo correspondiente se ha llamado correctamente. Por consiguiente, se describe en este documento un método para determinar los clonotipos basados en V(D)J a partir de lecturas de secuencia bidireccionales que comprenden las etapas de: (a) generar al menos una lectura de secuencia de la región J que comienza en una región J y se extiende hasta una región NDN y al menos una lectura de la secuencia de la región V que comienza en las regiones V y se extiende hacia la región NDN, de modo que la lectura de la secuencia de la región J y la lectura de la secuencia de la región V se solapan en una región de solapamiento, y la región J y la región V tienen cada una estructura de codón; (b) determinar si la estructura de codones de la región J extendida en la región NDN está en marco con la estructura de codones de la región V extendida hacia la región NDN. En otro caso, el paso de generar incluye generar al menos una lectura de secuencia de la región V que comienza en la región V y se extiende a través de la región NDN a la
región J, de modo que la lectura de la secuencia de la región J y la lectura de la secuencia de la región V se superponen en una región de superposición.
Análisis de lecturas de secuencia. Lectura de secuencias coalescentes en clonotipos. La construcción de clonotipos a partir de datos de lectura de secuencia depende en parte del método de secuenciación utilizado para generar dichos datos, ya que los diferentes métodos tienen diferentes longitudes de lectura esperadas y calidad de datos. En una metodología, se emplea un secuenciador Solexa para generar datos de lectura de secuencia para su análisis. En una realización, se obtiene una muestra que proporciona al menos 0.5-1.0x106 linfocitos para producir al menos 1 millón de moléculas de plantilla, que después de la amplificación opcional pueden producir un millón o más de poblaciones clonales correspondientes de moléculas de plantilla (o agrupaciones). Para la mayoría de las metodologías de secuenciación de alto rendimiento, incluida la metodología de Solexa, tal muestreo excesivo a nivel de grupo es deseable para que cada secuencia de plantilla se determine con un alto grado de redundancia para aumentar la precisión de la determinación de la secuencia. Para implementaciones basadas en Solexa, preferiblemente la secuencia de cada plantilla independiente se determina 10 veces o más. Para otras metodologías de secuenciación con diferentes longitudes de lectura y calidad de datos esperadas, se pueden usar diferentes niveles de redundancia para una precisión comparable de la determinación de la secuencia. Los expertos en la técnica reconocen que los parámetros anteriores, por ejemplo, el tamaño de la muestra, la redundancia y similares son opciones de diseño relacionadas con aplicaciones particulares.
La reducción de un conjunto de lecturas para una muestra dada en sus clonotipos distintos y el registro de la cantidad de lecturas para cada clonotipo sería un problema computacional trivial si la tecnología de secuenciación estuviera libre de errores. Sin embargo, en presencia de errores de secuenciación, cada clonotipo está rodeado por una "nube" de lecturas con un número variable de errores con respecto a la secuencia del clonotipo real. Cuanto mayor sea el número de errores, menor será la densidad si la nube circundante, es decir, la nube cae en densidad a medida que se aleja del clonotipo en el espacio de secuencia. Hay una variedad de algoritmos disponibles para convertir las lecturas de secuencia en clonotipos. En un aspecto, la fusión de las lecturas de secuencias depende de tres factores: el número de secuencias obtenidas para cada uno de los dos clonotipos de interés; el número de bases en que difieren; y la calidad de la secuenciación en las posiciones en las que son discordantes. Se evalúa un índice de probabilidad que se basa en las tasas de error esperadas y la distribución binomial de errores. Por ejemplo, dos clonotipos, uno con 150 lecturas y el otro con 2 lecturas con una diferencia entre ellos en un área de mala calidad de secuenciación probablemente se fusionarán ya que es probable que se generen por error de secuenciación. Por otro lado, dos clonotipos, uno con 100 lecturas y el otro con 50 lecturas con dos diferencias entre ellos no se combinan, ya que se considera que es poco probable que se generen por error de secuenciación. En una realización de los métodos de la invención, el algoritmo descrito a continuación se puede usar para determinar clonotipos a partir de lecturas de secuencia.
Esta nube de lecturas que rodean a cada clonotipo se puede modelar utilizando la distribución binomial y un modelo simple para la probabilidad de un solo error de base. Este último modelo de error se puede inferir del mapeo de los segmentos V y J o del propio algoritmo de búsqueda de clonotipos, a través de la autoconsistencia y la convergencia. Se construye un modelo para la probabilidad de que una secuencia de 'nube' Y dada con recuentos de lectura C2 y E (con respecto a la secuencia X) sea parte de una secuencia de clonotipo X verdadera con recuento de lecturas perfectas C1 bajo el modelo nulo de que X es el único clonotipo verdadero en esta región del espacio de secuencia. Se decide si unir o no la secuencia Y en el clonotipo X según los parámetros C1, C2 y E. Para cualquier C1 y E dados, se calcula previamente un valor máximo C2 para decidir fusionar la secuencia Y. Los valores máximos para C2 se eligen de modo que la probabilidad de no fusionar Y bajo la hipótesis nula de que Y es parte del clonotipo X es menor que algún valor P después de integrarse en todas las secuencias posibles Y con error E en la vecindad de la secuencia X. El valor P controla el comportamiento del algoritmo y hace que la fusión sea más o menos permisiva.
Si una secuencia Y no se fusiona en el clonotipo X porque su recuento de lectura está por encima del umbral C2 para fusionarse en el clonotipo X, entonces se convierte en un candidato para sembrar clonotipos separados. El algoritmo también se asegura de que cualquier otra secuencia Y2, Y3, etc. que estén 'más cerca' de esta secuencia Y (que se consideró independiente de X) no se agreguen en X. Este concepto de 'proximidad' incluye ambos conteos de errores con respecto a Y y X y el recuento de lectura absoluta de X e Y, es decir, se modela de la misma manera que el modelo anterior para la nube de secuencias de error alrededor del clonotipo X. De esta manera, las secuencias de "nube" se pueden atribuir adecuadamente a su clonotipo correcto si están "cerca" de más de un clonotipo.
El algoritmo avanza de manera descendente comenzando con la secuencia X con el recuento de lecturas más alto. Esta secuencia siembra el primer clonotipo. Las secuencias vecinas se unen en este clonotipo si sus recuentos están por debajo de los umbrales precalculados (ver arriba), o se dejan solas si están por encima del umbral o están "más cerca" de otra secuencia que no se fusionó. Después de buscar todas las secuencias vecinas dentro de un recuento máximo de errores, el proceso de coalescer las lecturas en el clonotipo X se termina. Sus lecturas y todas las lecturas que se han unido en ella se contabilizan y eliminan de la lista de lecturas disponibles para realizar otros clonotipos. La siguiente secuencia se mueve con el recuento de lecturas más alto. Las lecturas vecinas se unen en este clonotipo como se indicó anteriormente y este proceso continúa hasta que no haya más secuencias con recuentos de lecturas por encima de un umbral dado, por ejemplo, hasta que todas las secuencias con más de 1 conteo se hayan utilizado como semillas para los clonotipos.
En otra realización del algoritmo anterior, se puede agregar una prueba adicional para determinar si unir una secuencia candidata Y en un clonotipo X existente, que Nótese la puntuación de calidad de las lecturas de secuencias relevantes. El (los) puntaje(s) de calidad promedio(s) son determinados para la secuencia(es) Y (promediado en todas las lecturas con la secuencia Y) cuando las secuencias Y y X difieren. Si el puntaje promedio está por encima de un valor predeterminado, entonces es más probable que la diferencia indique un clonotipo realmente diferente que no debe fusionarse y si el puntaje promedio está por debajo de ese valor predeterminado, entonces es más probable que la secuencia Y esté causada por errores de secuenciación y por lo tanto debe unirse en X.
Árbol de secuencias. El algoritmo anterior de lecturas coalescentes en clonotipos depende de tener una forma eficiente de encontrar todas las secuencias con menos de E errores de alguna secuencia de entrada X. Este problema se resuelve utilizando un árbol de secuencias. La implementación de este árbol tiene algunas características inusuales en las que los nodos del árbol no están restringidos a ser letras individuales de ADN. Los nodos pueden tener secuencias arbitrariamente largas. Esto permite un uso más eficiente de la memoria del ordenador.
Todas las lecturas de una muestra dada se colocan en el árbol de secuencia. Cada nodo de la hoja contiene punteros a sus lecturas asociadas. Corresponde a una secuencia única dada al retroceder en el árbol desde la hoja hasta el nodo raíz. La primera secuencia se coloca en un árbol simple con un nodo raíz y un nodo de hoja que contiene la secuencia completa de la lectura. Las secuencias se agregan una por una. Para cada secuencia agregada, se forma una nueva rama en el último punto de la secuencia común entre la lectura y el árbol existente o agrega la lectura a un nodo de hoja existente si el árbol ya contiene la secuencia.
Una vez colocadas todas las lecturas en el árbol, es fácil utilizar el árbol para los siguientes propósitos: 1. Recuento de lecturas más alto: la clasificación de nodos de hojas por cuenta de lecturas nos permite encontrar el nodo de hojas (es decir, la secuencia) con la mayoría de las lecturas. 2. Búsqueda de hojas vecinas: para cualquier secuencia, se pueden buscar todas las rutas a través del árbol que tienen menos de X errores con respecto a esta secuencia. Se inicia una ruta en la raíz y se ramifica esta ruta en rutas separadas que continúan a lo largo del árbol. Se anota el recuento actual de errores de cada ruta a lo largo del árbol. Cuando el recuento de errores supera los errores máximos permitidos, se termina la ruta dada. De esta manera, se podan partes grandes del árbol lo antes posible. Esta es una forma eficiente de encontrar todas las rutas (es decir, todas las hojas) dentro de X errores de cualquier secuencia dada.
Hipermutaciones somáticas. En una realización, los clonotipos basados en IgH que se han sometido a hipermutación somática se determinan como sigue. Una mutación somática se define como una base secuenciada que es diferente de la base correspondiente de una secuencia de referencia (del segmento relevante, generalmente V, J o C) y que está presente en un número estadísticamente significativo de lecturas. En una realización, las lecturas en C se pueden usar para encontrar mutaciones somáticas con respecto al segmento J mapeado y, de la misma manera, lecturas en V para el segmento en V. Solo se utilizan partes de las lecturas C y V que se asignaron directamente a los segmentos J o V o que estaban dentro de la extensión del clonotipo hasta el límite de NDN. De esta manera, se evita la región NDN y no se usa la misma 'información de secuencia' para el hallazgo de mutación que se usó previamente para la determinación del clonotipo (para evitar clasificar erróneamente como nucleótidos mutaciones que en realidad son solo regiones NDN recombinadas diferentes). Para cada tipo de segmento, el segmento mapeado (alelo principal) se usa como un andamio y se consideran todas las lecturas que se han mapeado a este alelo durante la fase de mapeo de lectura. Cada posición de las secuencias de referencia donde al menos una lectura se ha mapeado se analiza para detectar mutaciones somáticas. En una realización, los criterios para aceptar una base no de referencia como una mutación válida incluyen lo siguiente: 1) al menos N lee con la base de mutación dada, 2) al menos una fracción dada N/M lee (donde M es el total número de lecturas mapeadas en esta posición base) y 3) un recorte estadístico basado en la distribución binomial, el puntaje Q promedio de las lecturas N en la base de mutación, así como el número (M-N) de lecturas con una base sin mutación. Preferiblemente, los parámetros anteriores se seleccionan de modo que la tasa de descubrimiento falso de mutaciones por clonotipo sea menor que 1 en 1000, y más preferiblemente, menor que 1 en 10000.
Clonotipos filogénicos (Clanes). En algunas enfermedades, como los cánceres, que incluyen trastornos proliferativos linfoides, un solo progenitor de linfocitos puede dar lugar a muchos progenitores de linfocitos relacionados, cada uno de los cuales posee y/o expresa un TCR o BCR ligeramente diferente, y por lo tanto un clonotipo diferente, debido a hipermutación somática o con mutaciones somáticas relacionadas con la enfermedad, como sustituciones de bases, reordenamientos aberrantes o similares. Las células que producen dichos clonotipos se denominan aquí clones filogénicos, y un conjunto de dichos clones relacionados se denominan aquí como un "clan". Del mismo modo, los clonotipos de clones filogénicos se denominan clonotipos filogénicos y un conjunto de clonotipos filogénicos puede denominarse un clan de clonotipos. En un aspecto, los métodos de la invención comprenden monitorizar la frecuencia de un clan de clonotipos (es decir, la suma de las frecuencias de los clonotipos filogénicos constituyentes del clan), en lugar de la frecuencia de un clonotipo individual. (La expresión "uno o más clonotipos específicos del paciente" abarca el concepto de clanes). Los clonotipos filogénicos pueden identificarse por una o más medidas de relación con un clonotipo primario. En una realización, los clonotipos filogénicos se pueden agrupar en el mismo clan por porcentaje de homología, como se describe más detalladamente a continuación. En otra realización, los clonotipos filogénicos se identifican mediante el uso común de regiones V, regiones J y/o regiones NDN. Por ejemplo, un clan puede definirse por clonotipos que tienen regiones J y ND comunes pero regiones V diferentes (a veces referidas como "reemplazo VH"); o puede definirse por clonotipos que tienen las mismas regiones V y J (mutadas idénticamente por sustituciones
de bases de sus respectivas secuencias de referencia) pero con diferentes regiones NDN; o puede definirse por un clonotipo que ha sufrido una o más inserciones y/o eliminaciones de 1-10 bases, o de 1-5 bases, o de 1-3 bases, para generar miembros del clan. En otra realización, los clonotipos se asignan al mismo clan si satisfacen los siguientes criterios: i) se asignan a los mismos segmentos de referencia V y J, con los mapeos que se producen en las mismas posiciones relativas en la secuencia del clonotipo, y ii) sus regiones NDN son sustancialmente idénticas. "Sustancial" en referencia a la pertenencia al clan significa que se permiten algunas pequeñas diferencias en la región NDN porque pueden haberse producido mutaciones somáticas en esta región. Preferiblemente, en una realización, para evitar llamar falsamente una mutación en la región NDN, si una sustitución de base se acepta como una mutación relacionada con el cáncer depende directamente del tamaño de la región NDN del clan. Por ejemplo, un método puede aceptar un clonotipo como miembro de un clan si tiene una diferencia de una base de las secuencias de NDN de clan como una mutación relacionada con el cáncer si la longitud de las secuencias de NDN de clan es m nucleótidos o más, por ejemplo, 9 nucleótidos o más, de lo contrario no se acepta, o si tiene una diferencia de dos bases de las secuencias de NDN de clan como mutaciones relacionadas con el cáncer si la longitud de la secuencia de NDN de clan es de n nucleótidos o más, por ejemplo, 20 nucleótidos o más, de lo contrario no se acepta. En otra realización, los miembros de un clan se determinan utilizando los siguientes criterios: (a) V lee mapas a la misma región V, (b) C lee mapas a la misma región J, (c) región NDN sustancialmente idéntica (como se describió anteriormente), y (d) la posición de la región NDN entre el límite V-NDN y el límite J-NDN es la misma (o de manera equivalente, el número de adiciones de base descendentes a D y el número de las adiciones de base aguas arriba a D son las mismas). Tal como se usa en el presente documento, el término "lectura C" puede referirse a una lectura generada a partir de un cebador de secuenciación que se aparea en una región C (en el caso de usar una muestra de ARN) o en una región J (en el caso de usar un ADN muestra). Como se explica en otro lugar, esto se debe a que una región C se une con una región J en un proceso de empalme postranscripcional.
Los clonotipos filogénicos de una sola muestra pueden agruparse en clanes y los clanes de muestras sucesivas adquiridas en diferentes momentos pueden compararse entre sí. En particular, en un aspecto de la invención, los clanes que contienen clonotipos correlacionados con una neoplasia linfoide se identifican entre los clonotipos determinados a partir de cada muestra en cada momento. El conjunto (o clan) de correlacionar clonotipos de cada punto de tiempo se compara con el de la muestra inmediatamente anterior para determinar el estado de la enfermedad, por ejemplo, determinando en clanes sucesivos si la frecuencia de un clonotipo particular aumenta o disminuye, si aparece un nuevo clonotipo correlativo que se sabe a partir de estudios de población o bases de datos que se correlaciona, o similar. Un estado determinado podría ser la remisión continua, la recaída incipiente, la evidencia de una mayor evolución clonal o similares.
Uso de isotipos. Los perfiles de clonotipos pueden incluir información de uso de isotipos. Cada vez que se determinan los clonotipos basados en IgH o TCRp a partir del ARN, el empalme postranscripcional une las regiones C a las regiones J, como se ilustra en la figura 3B. Los cebadores de secuenciación se pueden usar para generar lecturas de C (por ejemplo, 304) se fusionan a un sitio de unión del cebador predeterminado (302) en la región C (307) en la unión con la región J (309). Si el sitio de unión del cebador (302) se selecciona de modo que la lectura de C (304) incluya una porción (305) de la región C (307), entonces se puede determinar la identidad de la región C (307) que, a su vez, permite determinar el isotipo del BCR sintetizado. En una realización, el sitio de unión del cebador (302) se selecciona de modo que la lectura de C (304) incluya al menos seis nucleótidos de la región C (307); en otra realización, el sitio de unión del cebador (302) se selecciona de modo que la lectura de C (304) incluya al menos 8 nucleótidos de la región C (307). Cada clonotipo determinado de acuerdo con esta realización incluye información de secuencia de la parte (305) de su región C correspondiente y a partir de dicha información de secuencia se determina su isotipo correspondiente. Los clonotipos correlacionados pueden tener un primer isotipo en el momento en que se determinan inicialmente, pero pueden cambiar a otro tipo de isotipo durante el tiempo que están siendo monitoreados. Esta realización es capaz de detectar tales interruptores al observar los clonotipos no registrados previamente que tienen secuencias idénticas a los clonotipos correlacionados, excepto por la secuencia de la porción (305) que corresponde a un isotipo diferente.
Se espera que el error de la PCR se concentre en algunas bases que mutaron en los primeros ciclos de la PCR. Se espera que el error de secuenciación se distribuya en muchas bases, aunque sea totalmente aleatorio, ya que es probable que el error tenga algunos sesgos sistemáticos. Se supone que algunas bases tendrán un error de secuenciación a una tasa más alta, digamos 5% (5 veces el promedio). Dados estos supuestos, el error de secuenciación se convierte en el tipo de error dominante. Distinguir los errores de PCR de la aparición de clonotipos altamente relacionados desempeñará un papel en el análisis. Dada la importancia biológica para determinar que hay dos o más clonotipos altamente relacionados, se toma una metodología conservadora para hacer tales llamadas. Se considera la detección de una cantidad suficiente de los clonotipos menores para asegurarnos con alta confianza (por ejemplo, 99.9%) de que hay más de un clonotipo. Por ejemplo, de los clonotipos que están presentes en 100 copias/1.000.000, la variante menor se detecta 14 o más veces para que se designe como un clonotipo independiente. De manera similar, para los clonotipos presentes en 1.000 copias/1.000.000, la variante menor puede ser detectada 74 o más veces para ser designada como un clonotipo independiente. Este algoritmo puede mejorarse utilizando el puntaje de calidad base que se obtiene con cada base secuenciada. Si la relación entre el puntaje de calidad y la tasa de error se valida más arriba, entonces, en lugar de emplear la tasa de error conservadora del 5% para todas las bases, se puede usar el puntaje de calidad para decidir la cantidad de lecturas que deben estar presentes para llamar a un clonotipo independiente. La puntuación de calidad media de la base específica en todas las lecturas se puede
utilizar, o más rigurosamente, la probabilidad de ser un error se puede calcular dada la puntuación de calidad de la base específica en cada lectura, y luego las probabilidades se pueden combinar (asumiendo que independencia) para estimar el número probable de errores de secuencia para esa base. Como resultado, hay diferentes umbrales para rechazar la hipótesis del error de secuencia para diferentes bases con diferentes puntuaciones de calidad. Por ejemplo, para un clonotipo presente a 1.000 copias por 1.000.000, la variante menor se designa como independiente cuando se detecta 22 y 74 veces si la probabilidad de error fue 0.01 y 0.05, respectivamente.
III. Correlación entre clonotipos y algoritmos médicos
En el presente documento se describen métodos para identificar clonotipos cuya presencia, ausencia y/o nivel se correlacionan con un estado de enfermedad y para usar dicha información para tomar decisiones de diagnóstico o pronóstico. En un método, la información de los perfiles de clonotipo, que se puede acoplar con otra información médica, como los niveles de expresión de los genes sin TCR o sin BCR, la condición fisiológica o similares, se presenta a los pacientes o profesionales de la salud en el contexto de un algoritmo; es decir, un conjunto de uno o más pasos en los que se evalúan los resultados de las pruebas y/o exámenes y (i) se determina un curso de acción o se toma una decisión sobre la salud o el estado de la enfermedad o (ii) una serie de las decisiones se toman de acuerdo con un diagrama de flujo, o una estructura similar de toma de decisiones, que conduce a un curso de acción, o una decisión sobre la salud o el estado de la enfermedad. Los algoritmos descritos en este documento pueden variar ampliamente en formato. Por ejemplo, un algoritmo puede simplemente sugerir que un paciente debe ser tratado con un medicamento, si un determinado clonotipo, o un subconjunto de clonotipos, excede una proporción predeterminada en un perfil de clonotipo, o aumenta en proporción a más de una tasa predeterminada entre mediciones de monitorización. Aún más simple, un algoritmo puede simplemente indicar que existe una correlación positiva entre el estado de una enfermedad y un nivel de uno o más clonotipos y/o una función de TCR o BCR codificados por uno o más clonotipos. Algoritmos más complejos pueden incluir información fisiológica del paciente además de información de uno o más perfiles de clonotipo. Por ejemplo, en trastornos complejos, como algunos trastornos autoinmunes, la información del perfil de clonotipo se puede combinar en un algoritmo con otros datos del paciente, como el curso previo del tratamiento, la presencia, la ausencia o la intensidad de los síntomas, por ejemplo, erupción cutánea, inflamación articular, expresión de genes particulares, o similares. En una instancia, un algoritmo para usar con La monitorización de trastornos linfoides proporciona un valor fraccional predeterminado por encima del cual la proporción de un clonotipo (y/o clonotipos relacionados evolutivamente) en un perfil de clonotipo de una muestra (como una muestra de sangre) indica una recaída de enfermedad o resistencia a un tratamiento. Dichos algoritmos pueden consistir en o incluir medidas convencionales de clonalidad TCR o BCR. En otro caso, un algoritmo para usar con La monitorización de trastornos autoinmunes proporciona uno o más valores fraccionados predeterminados por encima de los cuales una proporción de clonotipos en un perfil de clonotipo que codifica TCR o BCR específicos para uno o más antígenos predeterminados, respectivamente, indica el inicio de una erupción autoinmune.
A. Correlación versus clonotipos no correlacionados
Los métodos descritos aquí proporcionan medios para distinguir a) correlacionar los clonotipos (que pueden ser aquellos clonotipos cuyo nivel se correlaciona con la enfermedad) de b) los clonotipos no correlacionados (que pueden ser aquellos clonotipos cuyos niveles no se correlacionan con la enfermedad). En un caso, un clonotipo correlativo puede mostrar una correlación positiva o negativa con la enfermedad. En otro caso, un clonotipo presente en un estado máximo de una enfermedad, pero no presente en un estado no máximo de una enfermedad puede ser un clonotipo correlativo (correlación positiva con la enfermedad). En otro caso, un clonotipo que es más abundante (es decir, está presente en un nivel más alto de moléculas) en un estado máximo (o estadio) de una enfermedad que en un estado no máximo de la enfermedad puede ser un clonotipo correlacionado (correlación positiva con la enfermedad). En otro caso, un clonotipo ausente en un estado máximo de una enfermedad, pero presente durante un estado no máximo de la enfermedad puede ser un clonotipo correlacionado (correlación negativa con la enfermedad). En otro caso, un clonotipo que es menos abundante en un estado máximo de una enfermedad que en un estado no máximo de una enfermedad puede ser un clonotipo correlacionado (correlación negativa con la enfermedad). En otro caso, un clonotipo correlacionado para un individuo está determinado por un algoritmo.
B. Descubrimiento de clonotipos correlacionados y no correlacionados mediante una prueba de calibración sin un estudio de población.
Los clonotipos correlacionados pueden identificarse observando los clonotipos presentes en algunas muestras que tienen relevancia para un estado de enfermedad. Esta muestra podría ser sangre de una muestra en un estado máximo de la enfermedad (por ejemplo, una muestra de sangre de un paciente con EM o lupus durante una erupción aguda), o podría ser de un tejido afectado por la enfermedad o relacionado con la enfermedad, es decir, enriquecido en las células T y B involucradas en la enfermedad para ese individuo, como una inflamación o un tumor. Ejemplos de estos tejidos podrían ser las biopsias renales de pacientes con lupus con inflamación renal, líquido cefalorraquídeo (LCR) en pacientes con EM durante una erupción, líquido sinovial para pacientes con artritis reumatoide o muestras de tumores de pacientes con cáncer. En todos estos ejemplos, es probable que los tejidos contengan células T y B relevantes que estén relacionadas con la enfermedad (aunque no necesariamente los agentes causales). Es notable que, si este método se utiliza para identificar los clonotipos que son relevantes para la enfermedad, solo serán relevantes para el individuo en cuya muestra se detectaron. Como resultado, se necesita una prueba de calibración específica para usar este método para identificar clonotipos correlacionados en cualquier individuo con una
enfermedad. Es decir, los clonotipos correlacionados pueden descubrirse o determinarse generando un perfil de clonotipo a partir de una muestra tomada de un tejido directamente afectado por una enfermedad (o referido en este documento como "tejido relacionado con la enfermedad"). Dicha determinación puede incluir además generar un perfil de clonotipo a partir de una muestra tomada de un tejido no afectado por, o relevante para, una enfermedad (a veces referido aquí como un "tejido no relacionado con la enfermedad"), luego comparar el clon anterior y el último. perfiles para identificar clonotipos correlacionados como aquellos que se encuentran en un nivel alto, bajo o funcionalmente distintos, por ejemplo, codifique TCR o BCR específicos para un antígeno particular. Dicha determinación se puede hacer identificando los clonotipos presentes en un perfil de clonotipo de un tejido afectado, o relacionado con la enfermedad, a una frecuencia más alta que los mismos clonotipos en un perfil de clonotipo de tejido no afectado o no relacionado con la enfermedad.
Se describe un método para determinar uno o más clonotipos correlacionados en un sujeto. El método puede incluir pasos para a) generar uno o más perfiles de clonotipo por secuenciación de ácidos nucleicos, moléculas individuales aisladas espacialmente de al menos una muestra del sujeto, en donde la al menos una muestra está relacionada con un primer estado de la enfermedad, y b) determinar uno o más clonotipos correlacionados en el sujeto en función de uno o más perfiles de clonotipo.
En una instancia, al menos una muestra proviene de un tejido afectado por la enfermedad. En otro caso, dicha determinación de uno o más clonotipos correlacionados comprende comparar perfiles de clonotipo de al menos dos muestras. En otro caso, el primer estado de la enfermedad es un estado máximo de la enfermedad. En otro caso, uno o más clonotipos correlacionados están presentes en el estado máximo de la enfermedad. En otro caso, el uno o más clonotipos correlacionados están ausentes en el estado máximo de la enfermedad. En otro caso, uno o más clonotipos correlacionados son altos en el estado máximo de la enfermedad. En otro caso, uno o más clonotipos correlacionados son bajos en el estado máximo de la enfermedad. En otro caso, la muestra comprende células T y/o células B. En otro caso, las células T y/o las células B comprenden un subconjunto de células T y/o células B. En otro caso, el subconjunto de células T y/o células B se enriquece mediante la interacción con un marcador. En otro caso, el marcador es un marcador de superficie celular en el subconjunto de células T y/o células B. En otro caso, el subconjunto de células T y/o células B interactúa con un antígeno específicamente presente en la enfermedad. Por ejemplo, en el caso de trastornos linfoproliferativos, como los linfomas, se puede obtener una muestra de calibración de tejidos linfoides, de lesiones causadas por el trastorno, por ejemplo, lesiones metastásicas o de tejidos afectados indirectamente por el trastorno por enriquecimiento como se sugirió anteriormente. Para las neoplasias linfoides, existe una amplia guía disponible y kits disponibles comercialmente para la inmunofenotipificación y el enriquecimiento de linfocitos relacionados con enfermedades, por ejemplo, "U.S.-Canadian consensus recommendations on the immunophenotypic analysis of haematologic neoplasia by flow cytometry", Cytometry, 30: 214-263 (1997); Paneles de anticuerpos MultiMix™ para inmunofenotipificación de leucemia y linfoma por citometría de flujo (Dako, Dinamarca); y similares. Los tejidos linfoides incluyen ganglios linfáticos, bazo, amígdalas, adenoides, timo y similares.
En un caso, la enfermedad es una enfermedad autoinmune. En otro caso, la enfermedad autoinmune es el lupus eritematoso sistémico, la esclerosis múltiple, la artritis reumatoide o la espondilitis anquilosante.
En algunos casos, los clonotipos correlacionados se identifican observando los clonotipos presentes en alguna muestra que tiene relevancia para un estado que no sea un estado de enfermedad. Estos estados podrían incluir la exposición a antígenos que no causan enfermedades, como reacciones alérgicas subsintomáticas a pólenes locales. Esto podría usarse para identificar si una persona había regresado recientemente a una geografía que contenía el antígeno. Los estados podrían incluir la exposición a un antígeno relacionado con un proceso industrial o la fabricación o producción de agentes de bioterrorismo.
C. Descubrimiento de clonotipos correlacionados y no correlacionados utilizando un estudio de población.
Se describe un método para identificar clonotipos correlacionados utilizando un estudio de población. La utilidad del estudio poblacional es que permite que la información específica sobre correlacionar los clonotipos que se han determinado en individuos con resultados de estado de enfermedad conocidos se generalice para permitir que dichos clonotipos correlacionados se identifiquen en todos los sujetos futuros sin la necesidad de una prueba de calibración. El conocimiento de un conjunto específico de clonotipos correlacionados se puede usar para extraer reglas sobre los atributos probables (parámetros) de los clonotipos que se correlacionarán en sujetos futuros. Dicho método se implementa con los siguientes pasos: (a) generar perfiles de clonotipo para cada uno de un conjunto de muestras de tejidos afectados por, o relevantes a, una enfermedad; (b) determinar clonotipos que se encuentran en un nivel alto o bajo en relación con los mismos clonotipos en muestras de tejidos no afectados o que son funcionalmente distintos de los clonotipos en muestras de tejidos no afectados. Como se usa en este documento, en un aspecto, "funcionalmente distinto" en referencia a los clonotipos significa que los TCR o BCR codificados por uno son específicos para un antígeno, proteína o complejo diferente que el otro. Opcionalmente, el método anterior puede incluir además un paso para desarrollar un algoritmo para predecir los clonotipos correlacionados en cualquier muestra a partir de la información de secuencia de los clonotipos determinados en los pasos (a) y/o (b) anteriores o de los datos funcionales, es decir, una determinación de que los clonotipos recién medidos codifican TCR o BCR específicos para un antígeno, proteína o complejo específico para la enfermedad en observación.
En relación con lo anterior, uno o más clonotipos específicos del paciente pueden identificarse haciendo coincidir los clonotipos determinados en una o más mediciones iniciales ("clonotipos determinados") con clonotipos que se sabe que están relacionados con dicha enfermedad, que pueden estar disponibles a través de un estudio de población, base de datos, o similares. En un caso, la coincidencia de tales clonotipos comprende encontrar una identidad entre una secuencia de aminoácidos codificada por el clonotipo determinado y la de una secuencia de aminoácidos codificada por un clonotipo que se sabe que se correlaciona con la enfermedad, o una variante sustancialmente idéntica, el último clonotipo. Como se usa en el presente documento, "variante sustancialmente idéntica", en un caso, significa que las secuencias que se comparan o combinan son al menos 80 por ciento idénticas, o al menos 90 por ciento idénticas, ya sea una secuencia de ácidos nucleicos o una secuencia de aminoácidos. En otro caso, una variante sustancialmente idéntica significa que se diferencian en 5 o menos adiciones, eliminaciones y/o sustituciones de aminoácidos o bases. En otro caso, la coincidencia de tales clonotipos comprende encontrar identidad entre el clonotipo determinado y una secuencia de ácidos nucleicos de un clonotipo conocido por estar correlacionado con la enfermedad, o una variante sustancialmente idéntica del último clonotipo. En otro caso más, la coincidencia de tales clonotipos comprende encontrar identidad entre el clonotipo determinado y una secuencia de ácidos nucleicos de un clonotipo que se sabe que está relacionado con la enfermedad, o una variante sustancialmente idéntica del último clonotipo.
En el presente documento se describen métodos que incluyen la identificación de clonotipos correlacionados y no correlacionados mediante la secuenciación del repertorio de células inmunitarias en un estudio de muestras de pacientes con enfermedad(s) y controles opcionalmente sanos en diferentes momentos y, en el caso de los pacientes con una enfermedad, en diferentes (y conocidos) estados del curso de la enfermedad caracterizados por datos clínicos. La enfermedad puede ser, por ejemplo, una enfermedad autoinmune. Los clonotipos cuyo nivel se correlaciona con las medidas de la enfermedad en estos diferentes estados se pueden usar para desarrollar un algoritmo que predice la identidad de un conjunto mayor de secuencias que se correlacionarán con la enfermedad, a diferencia de aquellos que no se correlacionarán con la enfermedad en todos los individuos. A diferencia del caso de la prueba de calibración, las secuencias correlacionadas no deben haber estado presentes en el estudio de descubrimiento, pero se pueden predecir en función de estas secuencias. Por ejemplo, una secuencia de correlación puede ser una secuencia de ADN del gen TCR que codifica la misma secuencia de aminoácidos que la secuencia de ADN de un clonotipo identificado en el estudio de descubrimiento. Además, el algoritmo que puede predecir uno o más clonotipos correlacionados se puede usar para identificar clonotipos en una muestra de cualquier individuo y no es en modo alguno exclusivo de un individuo dado, lo que permite predecir los clonotipos correlacionados en una muestra novedosa sin conocimiento previo de los clonotipos presentes en ese individuo.
Se describe un método para desarrollar un algoritmo que predice uno o más clonotipos correlacionados en cualquier muestra de un sujeto con una enfermedad que comprende: a) generar una pluralidad de perfiles de clonotipo a partir de un conjunto de muestras, en donde las muestras son relevantes para la enfermedad, b) identificar uno o más clonotipos correlacionados del conjunto de muestras, c) usar parámetros de secuencia y/o datos funcionales de uno o más clonotipos correlacionados identificados en b) para desarrollar un algoritmo que pueda predecir clonotipos correlacionados en cualquier muestra de un sujeto con la enfermedad.
En un caso, el conjunto de muestras se toma de uno o más tejidos afectados por la enfermedad.
En otro caso, la identificación de uno o más clonotipos correlacionados comprende comparar perfiles de clonotipo de al menos dos muestras. En otro caso, los datos funcionales incluyen la capacidad de unión de marcadores en células T y/o células B o la interacción con el antígeno por una célula T o una célula B. En otro caso, dichos parámetros de secuencia comprenden secuencia de ácidos nucleicos y secuencia de aminoácidos predicha. En otro caso, las muestras son de uno o más individuos en un estadio pico de la enfermedad. En otro caso, dicho uno o más clonotipos correlacionados están presentes en el estado máximo de la enfermedad. En otro caso, dicho uno o más clonotipos correlacionados se encuentran en un nivel alto en el estado máximo de la enfermedad. En otro caso, uno o más clonotipos correlacionados se encuentran en un nivel bajo en el estado máximo de la enfermedad. En otro caso, uno o más clonotipos correlacionados están ausentes en el estado máximo de la enfermedad.
En un caso, la enfermedad es una enfermedad autoinmune. En otro caso, la enfermedad autoinmune es el lupus eritematoso sistémico, la esclerosis múltiple, la artritis reumatoide o la espondilitis anquilosante.
Se describe un método para descubrir uno o más clonotipos correlacionados para un individuo, que comprende a) ingresar un perfil de clonotipo de una muestra del individuo en un algoritmo, y b) usar el algoritmo para determinar uno o más clonotipos correlacionados para el individuo. El algoritmo puede ser un algoritmo desarrollado por: a) generación de una pluralidad de perfiles de clonotipo a partir de un conjunto de muestras, en donde las muestras son relevantes para la enfermedad, b) identificación de uno o más clonotipos correlacionados del conjunto de muestras, y c) uso de parámetros de secuencia y/o datos funcionales de uno o más clonotipos correlacionados identificados en b) para desarrollar el algoritmo que puede predecir los clonotipos correlacionados en cualquier muestra de un sujeto con la enfermedad.
En algunos casos, los clonotipos correlacionados se identifican como clonotipos presentes en poblaciones que han sido expuestas a un antígeno que tiene relevancia para un estado distinto del estado de enfermedad. Este estado podría incluir la exposición a antígenos que no causan enfermedades, como reacciones alérgicas subsintomáticas a
pólenes locales. Esto podría usarse para identificar si una persona había viajado recientemente a una geografía que contenía el antígeno. Los estados podrían incluir la exposición a un antígeno relacionado con un proceso industrial o la fabricación o producción de agentes de bioterrorismo.
D. Descubrir clonotipos correlacionados y no correlacionados mediante una prueba de calibración combinada con un estudio de población.
Los clonotipos correlacionados pueden identificarse mediante el uso de una prueba de calibración combinada con un estudio de población. El estudio de población no da como resultado un algoritmo que permita predecir clonotipos en cualquier muestra, sino que permite que se desarrolle un algoritmo para predecir clonotipos correlacionados en cualquier muestra de un sujeto para el que se haya generado un perfil de clonotipo de calibración particular. Un ejemplo de esto podría ser el desarrollo de un algoritmo que predeciría la correlación de los clonotipos en un paciente con lupus basándose en el perfil del clonotipo medido a partir de una muestra de sangre en cualquier estadio de la enfermedad luego de haberse realizado un análisis de sangre durante un estado de erupción clínica que se utilizó para calibrar el algoritmo. Por lo tanto, los clonotipos correlacionados se pueden identificar en los pasos: (a) generar perfiles de clonotipos a partir de un conjunto de muestras de tejidos relevantes o afectados por una enfermedad para identificar un conjunto de clonotipos asociados con la enfermedad ya sea por nivel y/o por función y para identificar una relación entre dicho nivel y/o función y el estado de la enfermedad; (b) medir un perfil de clonotipo de una muestra de un tejido de un primer estado de la enfermedad; (c) determinar un clonotipo correlacionado a partir de la relación del paso (a). Los clonotipos correlacionados pueden identificarse en los pasos: (a) generar perfiles de clonotipo a partir de un conjunto de muestras de tejidos relevantes o afectados por una enfermedad para identificar un conjunto de clonotipos asociados con la enfermedad por nivel y/o por función e identificar un relación entre tal nivel y/o función y estado de la enfermedad; (b) medir un perfil de clonotipo de calibración en un nuevo sujeto en una estadio de enfermedad relevante en un estadio pico o de tejido afectado por enfermedad o en un estado funcionalmente caracterizado; (c) determinar un clonotipo correlacionado a partir de la relación del paso (a).
En el presente documento se describen métodos para identificar clonotipos correlacionados y no correlacionados mediante la secuenciación del repertorio de células inmunitarias en un estudio de muestras de pacientes con enfermedades y controles opcionalmente sanos en diferentes momentos y, en el caso de los pacientes con una enfermedad, en diferentes (y conocidos) estados del curso de la enfermedad caracterizado por datos clínicos. Los clonotipos que se encuentran a una frecuencia (o nivel) diferente en el primer estado que en el segundo estado se utilizan para desarrollar un algoritmo que predice cuál de las secuencias encontradas en los repertorios de cada individuo en el primer estado de enfermedad se correlacionará con la enfermedad en el estado posterior en cada individuo, a diferencia de aquellos que no se correlacionarán con la enfermedad en ese individuo. A diferencia del caso de la prueba de calibración sola, las secuencias correlacionadas pueden ser un subconjunto de todas las secuencias encontradas diferentes entre los estados de enfermedad. También es posible que los clonotipos correlacionados no se encuentren en la muestra de calibración, pero se pronostica en función del algoritmo que se correlaciona si aparecen en una muestra futura. Como ejemplo, se puede predecir que un clonotipo que codifica la misma secuencia de aminoácidos que un clonotipo encontrado en una muestra de calibración es un clonotipo correlativo basado en el algoritmo que resulta del estudio de población. A diferencia de las realizaciones anteriores, el algoritmo se desarrolla para predecir los clonotipos correlacionados con base en un perfil de clonotipo de calibración que es un perfil de clonotipo generado en el individuo para el cual se deben predecir los clonotipos correlacionados en un estado específico de la enfermedad. En este caso, el algoritmo no se puede usar para generar clonotipos correlacionados en un individuo en particular hasta que se haya medido un perfil de clonotipo de calibración específico. Después de que este perfil de calibración se haya medido en un sujeto en particular, todos los clonotipos correlacionados subsiguientes pueden predecirse basándose en la medición de los perfiles de clonotipo en ese individuo.
Se describe un método para descubrir uno o más clonotipos correlacionados para un individuo, que comprende a) ingresar un perfil de clonotipo de una muestra del individuo en un algoritmo, y b) usar el algoritmo para determinar uno o más clonotipos correlacionados para el individuo. El algoritmo puede ser un algoritmo desarrollado por: a) generación de una pluralidad de perfiles de clonotipo a partir de un conjunto de muestras, en donde las muestras son relevantes para la enfermedad, b) identificación de uno o más clonotipos correlacionados del conjunto de muestras, y c) uso de parámetros de secuencia y/o datos funcionales de uno o más clonotipos correlacionados identificados en b) para desarrollar un algoritmo que pueda predecir clonotipos correlacionados en cualquier muestra de un sujeto con la enfermedad. En una instancia, la muestra se toma en un estado máximo de enfermedad. En otro caso, la muestra se toma de tejido afectado por enfermedad.
En algunos casos, se realizan clonotipos correlacionados y no correlacionados utilizando una prueba de calibración combinada con un estudio de población para clonotipos presentes en poblaciones que han sido expuestas a un antígeno que tiene relevancia para un estado distinto del estado de enfermedad. Este estado podría incluir la exposición a antígenos que no causan enfermedades, como reacciones alérgicas subsintomáticas a pólenes locales. Esto podría usarse para identificar si una persona había viajado recientemente a una geografía que contenía el antígeno. Los estados podrían incluir la exposición a un antígeno relacionado con un proceso industrial o la fabricación o producción de agentes de bioterrorismo.
E1. Parámetros relacionados con la secuencia que pueden utilizarse para predecir clonotipos correlacionados
Para llevar a cabo un estudio de población, se puede usar un conjunto de entrenamiento para comprender las características de correlación de clonotipos mediante la prueba de varios parámetros que pueden distinguir aquellos correlacionados de clonotipos de aquellos que no lo hacen. Estos parámetros incluyen la secuencia o los segmentos específicos V, D y J utilizados. En un caso, se muestra que es más probable que los segmentos V específicos se correlacionen con algunas enfermedades, como ocurre si los clonotipos de una enfermedad específica reconocen los epítopos relacionados y, por lo tanto, pueden tener una similitud de secuencia. Otros parámetros incluidos en otros casos incluyen la extensión de la hipermutación somática identificada y el nivel de un clonotipo en el pico de un episodio y su nivel cuando la enfermedad está relativamente inactiva. Otros parámetros que pueden predecir los clonotipos correlacionados incluyen sin limitación: 1) motivos de secuencia que incluyen la región V o J, una combinación VJ, secuencias cortas en la región DJ; 2) Longitud de secuencia del clonotipo; 3) Nivel del clonotipo, incluido el nivel absoluto (número de clones por millón de moléculas) o el nivel de rango; 4) Similitud de secuencia de aminoácidos y ácidos nucleicos con otros clonotipos: la frecuencia de otros clonotipos altamente relacionados, incluidos aquellos con cambios silenciosos (diferencias de nucleótidos que codifican para los mismos aminoácidos) o aquellos con cambios de aminoácidos conservativos; 5) Para los BCR, el nivel de mutaciones somáticas en el clonotipo y/o el número de clonotipos distintos que difieren por mutaciones somáticas de algunos clonotipos de línea germinal; 6) clonotipos cuyas proteínas asociadas tienen estructuras tridimensionales similares.
E2. Bases de datos de clonotipos que codifican anticuerpos específicos para un antígeno
Estos clonotipos correlacionados pueden codificar inmunoglobulinas o TCR que son específicos para uno o más epítopos de uno o más antígenos. Por lo tanto, la correlación de los clonotipos se puede determinar comparando los clonotipos medidos con las entradas de una base de datos que comprende sustancialmente todos los clonotipos posibles con uno o más antígenos seleccionados (es decir, una "base de datos de clonotipos específica de antígeno"). Dichas bases de datos pueden construirse mediante la secuenciación de regiones seleccionadas de secuencias codificantes de anticuerpos de linfocitos que producen anticuerpos con especificidad por los antígenos o epítopos de interés, o dichas bases de datos pueden poblarse realizando experimentos de unión con fagos que expresan y muestran anticuerpos o fragmentos de los mismos en sus superficies. El último proceso se lleva a cabo fácilmente como se describe en Niro et al, Nucleic Acids Research, 38(9): e110 (2010). En resumen, el método puede comprender los siguientes pasos: (a) un antígeno de interés, por ejemplo, la proteína central del VHC está unida a un soporte sólido, (b) una biblioteca de anticuerpos codificada por fagos se expone al antígeno en condiciones de unión a anticuerpos, de modo que una fracción de los anticuerpos codificados en fagos se une al antígeno unido y otra fracción permanece libre, y (c) recopilar y secuenciar los anticuerpos codificados en fagos que se unen para crear entradas de una base de datos de clonotipos correlacionados. Los anticuerpos codificados en fagos unidos se secuencian convenientemente usando una técnica de secuenciación de ADN de alto rendimiento como se describe anteriormente. En un caso, los clonotipos del método codifican compuestos de unión de fragmentos de cadena variable (scFv). Se pueden usar condiciones de unión a anticuerpos de diferentes condiciones. Las secuencias de ácido nucleico determinadas a partir del fago unido pueden tabularse e introducirse en la base de datos de clonotipo específica de antígeno apropiada.
F. Datos funcionales para refinar la determinación de clonotipos correlacionados.
Los datos funcionales pueden usarse para ayudar a identificar clonotipos correlacionados. Por ejemplo, las células T y/o las células B que contienen ciertos marcadores que están enriquecidos en células que contienen clonotipos correlacionados se pueden capturar a través de métodos estándar como FACS o MACS. En otro caso, el marcador es un marcador de superficie celular. En otro caso, la reactividad de las células T y/o de las células B a un antígeno relevante para la patología o al tejido afectado sería una buena evidencia de la relevancia patológica de un clonotipo.
En otro caso, la secuencia de los clonotipos candidatos se puede sintetizar y poner en el contexto del TCR completo o BCR y evaluar la reactividad relevante. Alternativamente, los fragmentos amplificados de las diferentes secuencias pueden usarse como entrada para las técnicas de presentación de fagos, ribosomas o ARN. Estas técnicas pueden seleccionar las secuencias con la reactividad relevante. La comparación de los resultados de la secuenciación de aquellos antes y después de la selección puede identificar los clones que tienen la reactividad y, por lo tanto, es probable que sean patológicos. En otro caso, las técnicas de visualización específicas (por ejemplo, la visualización de fagos, ribosomas o ARN) se pueden utilizar en un formato de matriz. Las moléculas individuales (o las amplificaciones de estas moléculas individuales) que llevan secuencias individuales de TCR o BCR (por ejemplo, secuencias de CDR3) pueden agruparse como fagos, ribosomas o ARN. Luego se pueden estudiar los antígenos específicos para identificar la secuencia(s) que codifican los péptidos que los unen. Los péptidos que se unen a los antígenos relevantes para la enfermedad probablemente sean patológicos.
G. Generación de un algoritmo de carga inmune
Se puede usar un algoritmo para calcular una Carga Inmune, un valor o puntaje dado por una función de los niveles de clonotipos correlacionados y no correlacionados. La Carga Inmune se puede utilizar para tomar una decisión clínica. Usando datos de un experimento (por ejemplo, un experimento que comprende muestras de sujetos en un primer estado de una enfermedad y muestras de sujetos en un segundo estado de la enfermedad), se puede desarrollar un algoritmo que combine la información sobre los niveles de la correlación y clonotipos no correlacionados en una sola puntuación (Carga inmune). Los parámetros de este algoritmo se pueden ajustar para maximizar la correlación entre
la carga inmune y los datos clínicos. Por ejemplo, los datos clínicos pueden ser una medida clínica de la gravedad de la enfermedad (por ejemplo, la extensión de las lesiones en una resonancia magnética para un paciente con esclerosis múltiple). Por lo tanto, en una instancia, una carga inmune se puede calcular mediante los pasos: (a) desarrollo de un algoritmo que usa un conjunto de factores para combinar los niveles de los clonotipos correlacionados en una única puntuación de actividad de la enfermedad; (b) comparar la puntuación generada en el paso (a) con los datos clínicos sobre el estado de la enfermedad; y (c) optimizar los factores para maximizar la correlación entre los datos clínicos y la puntuación de actividad de la enfermedad.
Los clonotipos correlacionados utilizados en la generación de un algoritmo de carga inmune pueden generarse usando una prueba de calibración, un estudio de población o una prueba de calibración y un estudio de población como se describió anteriormente.
Algunos de los factores que pueden considerarse al combinar los clonotipos correlacionados son el número de clonotipos correlacionados, su nivel, su tasa de cambio (velocidad) y la tasa de cambio en la velocidad (aceleración). Otros factores que deben evaluarse incluyen el nivel de los clonotipos en el pico del episodio y en el estado de enfermedad inactiva.
En un caso, la carga inmune generada se relaciona con una enfermedad autoinmune. Una carga de este tipo se puede denominar AutoImm Load.
Se describe un método para generar un algoritmo que calcula un puntaje de actividad de la enfermedad, que comprende: a) desarrollar un algoritmo que usa un conjunto de factores para combinar los niveles de los clonotipos correlacionados en un puntaje de actividad de la enfermedad, b) comparar el puntaje de actividad de la enfermedad con los datos clínicos con respecto al estado de la enfermedad, y c) optimizando los factores para maximizar la correlación entre los datos clínicos y la puntuación de actividad de la enfermedad.
H. Monitorización de la enfermedad utilizando el algoritmo de carga.
I. Monitorización de la enfermedad sin una prueba de calibración.
Los clonotipos y el algoritmo de carga inmune se pueden determinar mediante un estudio de población. La carga inmune se puede usar directamente sin tener que calibrar primero al paciente individual. Esta prueba se puede hacer cuando el paciente está en cualquier estado de enfermedad. Esta prueba se puede utilizar para generar clonotipos específicos correlacionados y no correlacionados en función del algoritmo desarrollado anteriormente. La carga inmune se puede calcular utilizando el segundo algoritmo generado en un estudio de población. Esta puntuación puede ser utilizada clínicamente. Se puede llevar a cabo una prueba de monitorización sin utilizar una prueba de calibración mediante los siguientes pasos: (a) medición de los clonotipos de un paciente en el momento en que el paciente debe ser monitoreado; y (b) usar los clonotipos correlacionados pronosticados por la prueba del algoritmo de descubrimiento y los datos de la prueba de monitorización para generar una puntuación que refleje el estado de la enfermedad del paciente usando el algoritmo de monitorización.
Se describe un método para controlar el estado de enfermedad de un individuo que comprende: a) determinar un perfil de clonotipo a partir de una muestra de un sujeto, b) ingresar la información del perfil de clonotipo de a) en un algoritmo, y c) usar el algoritmo para generar un puntaje predictivo del estado patológico del individuo. El algoritmo puede ser un algoritmo generado por a) el desarrollo de un algoritmo que usa un conjunto de factores para combinar los niveles de los clonotipos correlacionados en un puntaje de actividad de la enfermedad, b) comparar el puntaje de actividad de la enfermedad con los datos clínicos con respecto al estado de la enfermedad, y c) optimizar los factores para maximizar la correlación entre los datos clínicos y la puntuación de actividad de la enfermedad.
2. Monitorización de la enfermedad mediante una prueba de calibración.
Los clonotipos correlacionados y el algoritmo de carga inmune pueden determinarse utilizando una prueba de calibración o una prueba de calibración y un estudio de población. La carga inmune se puede utilizar en la clínica realizando primero una prueba de calibración. Esta prueba se puede realizar cuando el paciente se encuentra en un estado similar al primer estado utilizado en el estudio que generó los clonotipos correlacionados y no correlacionados que se utilizan en el algoritmo de carga inmune. Por ejemplo, este estado puede ser un estado exacerbado de una enfermedad autoinmune si es así como se derivó el algoritmo de carga inmune. Esta prueba de calibración se puede usar para generar los clonotipos correlacionados específicos y no correlacionados que se utilizarán en las pruebas de monitorización de enfermedades posteriores. En un momento posterior en el tratamiento de este paciente, se realiza otra prueba en el paciente y se puede calcular la Carga Inmune utilizando el algoritmo generado en el estudio de descubrimiento y la lista de niveles de clonotipo generados en la prueba de calibración específica de este paciente. Este puntaje de Carga inmunológica se puede usar clínicamente. En una instancia, una prueba de monitorización que utiliza una prueba de calibración comprende los siguientes pasos: (a) probar a un paciente en estado de enfermedad 1 para determinar un perfil de clonotipo; (b) medir los clonotipos de un paciente en un momento posterior (el momento en donde el paciente debe ser monitoreado); (c) usar el algoritmo de monitorización para generar un puntaje de enfermedad que refleje un estado de enfermedad del perfil de clonotipo del estado de enfermedad 1 a partir de la prueba de calibración e información de una prueba de tiempo posterior.
Se describe un método para monitorizar el estado de enfermedad de un individuo que comprende: a) determinar un perfil de clonotipo a partir de una muestra de un sujeto, b) ingresar la información del perfil de clonotipo de a) en un algoritmo, y c) usar el algoritmo para generar una puntuación predictiva del estado patológico del individuo. El algoritmo puede ser un algoritmo generado a) que desarrolla un algoritmo que usa un conjunto de factores para combinar los niveles de los clonotipos correlacionados en un puntaje de actividad de la enfermedad, b) que compara el puntaje de actividad de la enfermedad con los datos clínicos relacionados con el estado de la enfermedad, y c) que optimiza los factores para maximizar la correlación entre los datos clínicos y el puntaje de actividad de la enfermedad. En otro caso, el método puede comprender además determinar uno o más clonotipos correlacionados en el individuo mediante cualquiera de los métodos descritos en el presente documento, e ingresar información del uno o más clonotipos correlacionados en el algoritmo.
En un caso, la enfermedad es una enfermedad autoinmune. En otro caso, la enfermedad autoinmune es el lupus eritematoso sistémico, la esclerosis múltiple, la artritis reumatoide o la espondilitis anquilosante.
3. Otros factores relacionados con el uso de la Carga Inmune.
La misma carga inmune puede significar cosas diferentes para diferentes pacientes. Por un lado, debe considerarse el cuadro clínico completo de un paciente. Desde una perspectiva de prueba, se puede considerar la velocidad (tasa de cambio de la carga inmune a lo largo del tiempo) y la aceleración (tasa de cambio de la velocidad a lo largo del tiempo) además del nivel de la carga inmune al tomar decisiones clínicas. Por ejemplo, si la puntuación de AutoImm Load está aumentando (alta velocidad) puede ser predictiva de una erupción incipiente en una enfermedad autoinmune.
Las pruebas adicionales que se pueden integrar en la puntuación de carga, por ejemplo, una puntuación de carga automática, incluyen, por ejemplo, la tasa de sedimentación eritrocítica (ESR), los niveles de proteína C reactiva (CRP), el Anti-ADNds, otros títulos de autoanticuerpo, niveles de complemento, niveles de proteína en la orina, proporción de creatina/proteína en la orina, niveles de creatinina, niveles de nitrógeno ureico en la sangre (BUN), niveles de plaquetas, recuentos de glóbulos blancos, hematorcrito (Hct), Hb, resultados del análisis de orina. Otras pruebas relacionadas con el SLE que se pueden integrar incluyen, por ejemplo, el nivel de CD27, el nivel de células CD27++, los genes que responden a INF (Baechler, EC et al. (2003) Proc. Natl. Acad. Sci. 100: 2610-2615), y la puntuación de quimioquinas (Bauer JW et al. (2009) Arthritis Rheum. 60: 3098-3107). Otras pruebas no relacionadas con el lupus incluyen, por ejemplo, prueba de la hormona estimulante de la tiroides (TSH), prueba de triyodotironina (T3), prueba de tiroxina (T4), pruebas de función hepática (LFT), otros autoanticuerpos, prueba de calprotectina, prueba de lactoferrina y análisis de fluidos sinoviales. Las pruebas adicionales pueden incluir pruebas de imágenes, que incluyen, por ejemplo, resonancia magnética, tomografía computarizada, rayos X y ultrasonido.
I. El uso de tecnologías de secuenciación en combinación con enriquecimientos celulares parciales como parte de un paso de calibración para encontrar clonotipos correlacionados.
Existen varias tecnologías que se pueden usar para separar células en sangre o tejido con base en marcadores celulares. Estos incluyen separaciones en fase sólida, tales como perlas o columnas en las que se inmovilizan reactivos de afinidad específicos, como los anticuerpos. La separación de la fase líquida se puede lograr utilizando técnicas como la citometría de flujo en la que se utilizan reactivos marcados que se unen específicamente a marcadores celulares seleccionados para dirigir un dispositivo de flujo cerrado en donde se pueden usar marcadores fluorescentes específicos para separar las células etiquetadas de ese modo. La leucoféresis es otra técnica de separación en fase líquida que se puede usar para enriquecer las poblaciones de leucocitos de la sangre, por ejemplo, Shelat, Am. J. Med. 123: 777-784 (2010); Patente de los Estados Unidos 5,846,928; o similar, después de lo cual se puede llevar a cabo un enriquecimiento adicional por medio de marcadores de superficie celular.
A veces será ventajoso perfilar un subconjunto de células T y/o B al calibrar un algoritmo para predecir clonotipos correlacionados y/o medir los niveles de estos clonotipos para medir la carga de una enfermedad. Esto se puede hacer usando los métodos descritos anteriormente para una variedad de marcadores de superficie o internos. Un desafío de usar tales métodos radica en el hecho de que la selectividad para un marcador dado nunca es perfecta para un marcador dado. Como resultado, es poco probable que el enriquecimiento conduzca a una población pura de las células seleccionadas. Otro método para lograr el enriquecimiento de subconjuntos de células sin la separación de células es hacer crecer selectivamente el subconjunto de células de interés. Por ejemplo, las células T pueden activarse con antígeno in vitro, y se puede permitir que las células activadas se dividan y aumenten en número, lo que lleva a su enriquecimiento.
Los perfiles de clonaciones de las poblaciones de células T y/o B pueden generarse mediante la secuenciación de moléculas individuales aisladas espacialmente, tanto antes como después del enriquecimiento para al menos un marcador celular. Los dos perfiles se comparan para determinar qué clonotipos tienen frecuencias significativamente alteradas entre las poblaciones de células enriquecidas y no enriquecidas para identificar los clonotipos que están asociados con las células que llevan los marcadores seleccionados en el enriquecimiento. La ventaja de utilizar el método de secuenciación para lograr esta identificación es que los clonotipos pueden identificarse incluso si el enriquecimiento es relativamente pobre, ya que las frecuencias de clonotipo pueden medirse muy bien con suficiente profundidad de secuencia. Esto, a su vez, permitirá una variedad de metodologías de enriquecimiento que se utilizarán
de manera rutinaria y económica, ya que no serán necesarios los costosos enriquecimientos múltiples diseñados para alcanzar la pureza.
En un caso, tal secuenciación de receptores de células T y B antes y después del enriquecimiento celular podría usarse como parte de un algoritmo de calibración para determinar qué clonotipos están correlacionados. En este caso, las células se secuencian antes y después del enriquecimiento para los marcadores que son relevantes para la enfermedad en un primer estado de enfermedad. Ejemplos de este primer estado de enfermedad podrían ser: muestras de sangre en un estado máximo de la enfermedad, muestras de tejido afectado, muestras de tejido afectado en un estado máximo de la enfermedad, etc. Los clonotipos de la fracción celular que contiene y no contienen el marcador celular son así obtenidos y se pueden utilizar como entradas en un algoritmo que luego se utiliza para determinar los clonotipos correlacionados en ese individuo.
En otro caso, el enriquecimiento no solo se realiza en una muestra recolectada durante un primer estado de enfermedad, sino en muestras posteriores del mismo individuo. De esta manera, los clonotipos correlacionados se determinan dentro de las subfracciones de las células en cualquier momento.
En otro caso, los marcadores celulares se usan en conjugación con secuenciación para evaluar no solo la frecuencia de clonotipos relevantes, sino también su estado funcional. En este caso, los marcadores celulares proporcionan información más allá de la identidad de los clonotipos correlacionados, ya que refinan la predicción del estado de la enfermedad obtenida por la frecuencia de los clonotipos correlacionados. Al secuenciar antes y después del enriquecimiento con marcadores específicos, se determina la frecuencia de un clonotipo específico. Además, se determina la frecuencia y la fracción de las células que contienen este clonotipo específico junto con otro marcador de células. Considere, dos pacientes con la misma frecuencia de clonotipos relevantes para algún estado clínico, pero con una frecuencia diferente de marcadores celulares particulares (por ejemplo, marcadores de activación) en las células que contienen estos clonotipos. Estos pacientes pueden tener diferentes actividades de la enfermedad a pesar de tener la misma frecuencia de los clonotipos relevantes.
Los marcadores celulares pueden ser marcadores de activación celular. En general, los marcadores pueden determinarse midiendo la expresión de genes utilizando una población de células T y/o B que se sabe que se correlacionan con la enfermedad. Estos marcadores podrían ser marcadores de la superficie celular o células expresadas dentro de la célula.
En un caso de esta invención, se enriquecen las células que muestran tener afinidad por un antígeno que se sabe que es relevante para una enfermedad particular. Hay varios métodos para hacer esto.
En otro caso, las células de interés son células B que interactúan con un antígeno específico. En este caso, existirán células B que tienen secuencias de receptores de células B que se unen a este antígeno específico. Estos receptores de células B pueden utilizarse, así como marcadores de superficie celular que pueden enriquecerse utilizando reactivos específicos de antígeno. En un caso, las perlas o columnas en las que se inmoviliza el antígeno pueden usarse para enriquecer las células que expresan receptores de células B específicos para este antígeno. En otro caso, el antígeno se vuelve multimérico, por ejemplo, tetramérico, para aumentar la afinidad de las células que expresan los receptores de células B apropiados. En otro caso, estas células pueden marcarse utilizando un reactivo de antígeno marcado de forma fluorescente. Estas células se pueden enriquecer luego utilizando un método de citometría de flujo que se clasifica según la etiqueta fluorescente. Este proceso se puede realizar en combinación con otros marcadores de células B en la metodología de citometría de flujo. En otra realización, el reactivo de antígeno fluorescente se hace multiméri
Se puede definir la intensidad de la interacción de los diferentes clonotipos con el antígeno específico. El grado de enriquecimiento de un clonotipo por la interacción del antígeno proporciona una medida de la fuerza de la interacción. Como resultado, en lugar del nivel tradicional de "título" de un anticuerpo, se puede obtener información más detallada. Específicamente, se puede determinar la frecuencia de diferentes clonotipos con diferente avidez. En un caso, el antígeno utilizado en el enriquecimiento es una sola especie molecular. En otro caso, el antígeno es una mezcla compleja de antígenos que son relevantes para una enfermedad. El antígeno puede ser un tipo de célula o una mezcla de tipos de células.
En otro caso, las células de interés son células T que interactúan con un antígeno específico en el contexto de una molécula de MHC. En un caso, el péptido complejado con las moléculas MHC se utiliza para capturar las células relevantes. Los tetrámeros del complejo MHC-péptido se han utilizado previamente con éxito para este propósito. En otro caso, la sangre o el tejido relevante que contiene células capaces de presentar antígenos y las células T se incuba con el antígeno para permitir que los péptidos se presenten a las células T. Las células que se activan mediante la unión a estos antígenos se pueden enriquecer con alguna característica de activación. Potencialmente, cualquier característica de activación puede ser utilizada como proliferación celular, migración de leucocitos, citotoxicidad y/o expresión de marcadores de activación. Por ejemplo, las células activadas proliferan y se les puede permitir que se dividan y se enriquezcan. De manera similar, las celdas activadas pueden expresar algunos marcadores que pueden usarse para capturarlos. Estos marcadores pueden ser marcadores de superficie o algún marcador interno como citoquinas, como INFy, IL-2 o IL-17. Las células que expresan marcadores de superficie se pueden capturar fácilmente usando diferentes técnicas, como FACS o perlas recubiertas con anticuerpos contra el marcador de superficie.
También se han desarrollado técnicas para capturar células que expresan marcadores intracelulares, particularmente citoquinas. Una técnica se llama tinción intracelular de citoquinas. En este método, las citoquinas específicas del proceso inmunitario en cuestión se encuentran atrapadas dentro de las células T que posteriormente se permeabilizan permitiendo que estas citoquinas específicas se marquen utilizando anticuerpos fluorescentes. Estas células marcadas se pueden enriquecer luego utilizando un método de citometría de flujo. Otro método, la captura de citoquinas, utiliza anticuerpos híbridos que tienen especificidad dual. Una de las especificidades es para algún marcador genérico en todas las células T (como la molécula MHC) y la otra es para la citoquina de interés, por ejemplo, INFy, IL-2 u 11-17. La especificidad genérica se une al anticuerpo en la superficie de todas las células T, y las citoquinas liberadas de las células T luego son capturadas por los anticuerpos unidos a las mismas células. Los anticuerpos fluorescentes contra la citoquina relevante se pueden usar para permitir la captura de las células relevantes utilizando FACS.
Se puede definir la intensidad de la interacción de los diferentes clonotipos con el antígeno específico. El grado de enriquecimiento de un clonotipo por la interacción del antígeno proporciona una medida de la fuerza de la interacción. Por lo tanto, se puede determinar la frecuencia de diferentes clonotipos con diferente avidez.
En un caso, el antígeno utilizado en el enriquecimiento es una sola especie molecular. En otro caso, el antígeno es una mezcla compleja de antígenos que son relevantes para una enfermedad. La mezcla compleja de antígenos puede ser un tipo de célula o una mezcla de tipos de células.
J. Enriquecimiento de antígenos para la detección de recurrencia de infección latente
A menudo es útil en enfermedades infecciosas no solo medir la presencia o ausencia de un patógeno, sino también medir y monitorizar la respuesta inmune a este patógeno. Como resultado, la medición de anticuerpos generados por el sistema inmunitario contra antígenos patógenos específicos es una metodología en la práctica clínica habitual. Sin embargo, tales respuestas inmunes al antígeno patógeno específico medido por anticuerpos no proporcionan una visión completa de la respuesta inmune al antígeno. Los anticuerpos medidos pueden ser el producto de muchos clones de células B diferentes, cada uno de los cuales expresa un anticuerpo ligeramente diferente, cada uno de los cuales puede llevar información ligeramente diferente sobre el estado de la enfermedad. Además, las respuestas de las células T a estos antígenos no se están midiendo en absoluto.
La respuesta inmune de un paciente a una infección patógena podría perfilarse de manera muy completa usando los métodos descritos en el presente documento. En un caso, la respuesta de las células B puede medirse de manera integral en un individuo infectado con un patógeno utilizando un enriquecimiento de células B en un punto del curso de la enfermedad para determinar los clonotipos de células B que son relevantes para la respuesta del anticuerpo a esa infección. Para lograr esto, las células B que están involucradas en una respuesta inmune a un patógeno se identificarían realizando un enriquecimiento utilizando antígenos presentes en el patógeno en cuestión. Estos antígenos pueden ser una sola especie de antígeno, un conjunto de distintas especies de antígenos o una mezcla compleja de antígenos del patógeno, incluidas las células completas del patógeno. Dichos antígenos se inmovilizan luego sobre una superficie sólida o se marcan con fluorescencia y el enriquecimiento se lleva a cabo utilizando un protocolo de unión basado en perlas, un protocolo de unión basado en columna o un método de citometría de flujo en el caso en donde los antígenos están marcados con fluorescencia. Las células del paciente se perfilan antes y después del enriquecimiento separando las moléculas de ADN o ARN individuales del receptor de células B en dos dimensiones y secuenciando las moléculas individuales para formar un perfil de clonotipo BCR. Las secuencias de clonotipos que muestran un cambio de frecuencia significativo entre los dos perfiles de clonotipos son candidatas para ser clonotipos responsables de una respuesta inmune al antígeno(s). Opcionalmente, se pueden desarrollar algoritmos adicionales para refinar la predicción de qué clonotipos probablemente sean relevantes para esta respuesta inmune específica. Estos algoritmos pueden usar parámetros de secuencia tales como frecuencia, longitud de secuencia, similitud de secuencia de aminoácidos, similitud con otras secuencias de clonotipo similares, incluidas las creadas por hipermutaciones somáticas, etc.
En un caso preferido, la captura de antígeno se realiza en un punto de calibración en el tiempo para identificar las células B relevantes y no se realiza y los puntos de tiempo de perfilado posteriores en los que todas las células B se perfilan sin enriquecimiento.
En otro caso, se mide la respuesta de las células T. Para lograr esto, las células T que están involucradas en una respuesta inmune a un patógeno se identificarían realizando un enriquecimiento utilizando antígenos presentes en el patógeno en cuestión. Estos antígenos pueden ser una sola especie de antígeno, un conjunto de distintas especies de antígenos o una mezcla compleja de antígenos del patógeno, incluidas las células completas del patógeno. En un caso, se usan tetrámeros del complejo MHC-antígeno para marcar de manera fluorescente las células T. En otro caso, tales antígenos se agregan a la sangre de un paciente al menos un primer momento y se incuban para permitir que los péptidos antigénicos sean presentados por las células presentadoras de antígenos de este individuo. En ambos casos, en moléculas de ADN o ADN aisladas espacialmente de muestras de sangre se perfilan antes y después del enriquecimiento de estas células T utilizando tetrámeros del complejo MHC-antígeno, el método de tinción de citoquinas internas o la captura de citoquinas y la clasificación FACS. Las secuencias de clonotipos de células T que muestran un cambio de frecuencia significativo entre los dos perfiles de clonotipos son candidatas para ser clonotipos responsables de una respuesta inmune al antígeno(s). Opcionalmente, se pueden desarrollar algoritmos adicionales para refinar la predicción de qué clonotipos probablemente sean relevantes para esta respuesta inmune específica.
Estos algoritmos pueden usar parámetros de secuencia tales como frecuencia, longitud de secuencia, similitud de secuencia de aminoácidos, similitud con otras secuencias de clonotipo similares, etc.
En un caso preferido, la captura de antígeno se realiza en un punto de calibración en el tiempo para identificar las células B relevantes y no se realiza y los puntos de tiempo de perfilado posteriores en los que todas las células B se perfilan sin enriquecimiento.
IV. Determinación de estados de enfermedad
Debido a que el sistema inmunológico es tan fundamental para la salud humana, la capacidad de medir las respuestas inmunitarias tiene amplias aplicaciones en la medicina. El sistema inmunológico se puede utilizar para comprender el estado de la enfermedad subyacente cuando está mediado por el sistema inmunitario. Esto permite un conjunto muy poderoso de aplicaciones de diagnóstico y pronóstico que utilizan los perfiles inmunes para informar los riesgos de una amplia variedad de resultados clínicos y permiten a los médicos intervenir de manera más efectiva.
A. Utilidad del perfil inmunológico en el tratamiento de enfermedades autoinmunes
Los métodos descritos en el presente documento pueden usarse para diagnosticar y tratar una enfermedad autoinmune en un sujeto. La enfermedad autoinmune implica que las células inmunitarias adaptativas escapen al proceso habitual que confiere autoinmunidad y ataca a algunos objetivos en el tejido corporal. Las enfermedades autoinmunes incluyen, por ejemplo, encefalomielitis aguda diseminada, enfermedad de Addison, espondilitis anquilosante, síndrome de anticuerpos antifosfolípidos, anemia hemolítica autoinmune, hepatitis autoinmune, enfermedad autoinmune del oído interno, enfermedad de Behget, penfigoide biliar, enfermedad celíaca, enfermedad de Chagas, enfermedad pulmonar obstructiva crónica, dermatomiositis, diabetes mellitus tipo 1, síndrome de Goodpasture, enfermedad de Graves, síndrome de Guillain-Barré, tiroiditis de Hashimoto, hidradenitis supurativa, púrpura trombocitopénica idiopática, cistitis intersticial, esclerosis múltiple, miastenia grave, neuromiotonía, pénfigo vulgaris, anemia perniciosa, polimiositis, cirrosis biliar primaria, artritis reumatoide, esclerodermia, lupus eritematoso sistémico, síndrome de Sjogren y síndromes de vasculitis. Las estadios de estas enfermedades autoinmunes se pueden diagnosticar utilizando los métodos descritos en este documento. Se pueden sugerir tratamientos a un sujeto según la estadio de la enfermedad autoinmune.
La información clínica con respecto a un sujeto con una enfermedad autoinmune, o que se sospecha que tiene una enfermedad autoinmune, se puede usar para determinar el estado de la enfermedad (o la Autolmm Load). La información clínica se puede utilizar para identificar patrones de un perfil de clonotipo que se correlacionan con un estado de enfermedad. La información clínica puede incluir, por ejemplo, altura, peso, color de ojos, edad, sexo, grupo étnico, presión arterial, niveles de colesterol LDL, niveles de colestero1HDL, historial médico familiar e información de marcadores moleculares.
La información clínica puede incluir síntomas de una o más enfermedades autoinmunes. Para la hepatitis autoinmune, los síntomas pueden incluir fatiga, hepatomegalia, ictericia, prurito, erupción cutánea, artralgia, malestar abdominal, angiomas de araña, náuseas, vómitos, anorexia, orina oscura, heces pálidas o grises. Los síntomas pueden incluir erupción cutánea (manchas, descoloración azulada-púrpura en la cara, cuello, hombros, parte superior del pecho, codos, rodillas, nudillos y espalda) acompañando o precediendo debilidad muscular, disfagia, mialgia, fatiga, pérdida de peso y la fiebre baja. Para la enfermedad de Graves, los síntomas pueden incluir pérdida de peso debido al aumento del gasto de energía, aumento del apetito, ritmo cardíaco y presión arterial, y temblores, nerviosismo y sudoración. Para la tiroiditis de Hashimoto, los síntomas pueden incluir retraso mental y físico, mayor sensibilidad al frío, aumento de peso, engrosamiento de la piel, bocio. Para la enfermedad mixta del tejido conectivo (MCTD), los síntomas pueden incluir características de lupus eritematoso sistémico (SLE), escleroderma y polimiositis. Para el penfigoide, los síntomas ampollares (SA) pueden incluir ronchas de prurito leve a ampollas graves e infecciones, ampollas orales o esofágicas. Para el pénfigo, los síntomas pueden incluir ampollas en la piel y las membranas mucosas. Para la anemia perniciosa, los síntomas pueden incluir dificultad para respirar, fatiga, palidez, taquicardia, inapetencia, diarrea, hormigueo y entumecimiento de las manos y los pies, dolor de boca y marcha inestable. Para la polimiositis (PM), los síntomas pueden incluir debilidad muscular, disfagia y mialgia. Para la cirrosis biliar primaria (PBC), los síntomas pueden incluir fatiga y prurito. Para la esclerodermia (esclerosis sistémica), los síntomas pueden incluir inflamación e hinchazón de los dedos o las manos, engrosamiento de la piel, úlceras en la piel de los dedos, rigidez en las articulaciones de las manos, dolor, irritación de garganta y diarrea. Para el síndrome de Sjogren, los síntomas pueden incluir sequedad de los ojos y la boca, glándulas del cuello inflamadas, dificultad para tragar o hablar, sabores u olores inusuales, sed y úlceras en la lengua. Para el lupus eritematoso sistémico (SLE), los síntomas pueden incluir fiebre, pérdida de peso, pérdida de cabello, llagas en la boca y la nariz, malestar, convulsiones y síntomas de enfermedades mentales, inflamación articular similar a la AR, erupción de la mariposa en la nariz y las mejillas, sensibilidad extrema al frío en manos y pies. Para los síndromes de vasculitis, por ejemplo, granulomatosis de Wegener, glomerulonefritis idiopática creciente (ICGN), poliarteritis microscópica (MPA), síndrome renal pulmonar (PRS), los síntomas pueden incluir fatiga, debilidad, fiebre, artralgia, dolor abdominal, problemas renales y problemas neurológicos. La información clínica puede ser de uno o más sujetos en uno o más puntos de tiempo.
La información clínica puede incluir información sobre las respuestas de un sujeto con una enfermedad autoinmune a uno o más tratamientos que el sujeto ha recibido.
La utilidad clínica de AutoImm Load se discute a continuación para enfermedades autoinmunes específicas. Otra realización de esta invención contempla la combinación de las pruebas de perfil inmunológico con otros marcadores que ya están en uso para la detección de la actividad de la enfermedad en estas enfermedades para permitir pruebas con mayor sensibilidad y especificidad. Se pueden usar otros identificadores o marcadores moleculares para calcular la AutoImm Load o para determinar el estado de la enfermedad. Los identificadores moleculares pueden incluir ácidos nucleicos, proteínas, carbohidratos y lípidos, y perfiles de expresión de ácidos nucleicos o proteínas. Los identificadores moleculares pueden ser de origen humano o no humano (por ejemplo, bacteriano). Los identificadores o marcadores pueden determinarse mediante técnicas que incluyen, por ejemplo, hibridación genómica comparativa (CGH), análisis de micromatrices cromosómicas (CMA), perfiles de expresión, micromatrices de ADN, micromatrices de oligonucleótidos de alta densidad, matriz de expresión de ARN de genoma completo, micromatriz de péptidos, ensayo inmunoenzimático (ELISA), secuenciación del genoma, número de copias (CNV), análisis de polimorfismo de nucleótidos pequeños (SNP), inmunohistoquímica, hibridación in situ, hibridación in situ fluorescente (FISH), PCR, Inmunoprecipitación Western, Inmunoprecipitación Southern, SDS-PAGE, electroforesis en gel y inmunoprecipitación Northern.
Para el lupus eritematoso sistémico, los marcadores pueden incluir niveles de tasa de sedimentación eritrocítica (ESR), niveles de proteína C reactiva (CRP), Anti-ADNds, otros títulos de autoanticuerpos, niveles de complemento, niveles de proteína en la orina, proporción de proteína en orina/creatinina, niveles de creatinina, niveles de nitrógeno ureico en sangre (BUN), niveles de plaquetas, recuentos de glóbulos blancos (WBC), hematocrito (Hct), Hb y resultados de análisis de orina. Otras pruebas que están relacionadas, por ejemplo, con el SLE que se pueden integrar, incluyen, por ejemplo, el nivel de CD27, el nivel de células CD27++, los genes que responden a INF y la puntuación de quimioquinas.
1. Lupus eritematoso sistémico (SLE)
Los métodos descritos en el presente documento se pueden usar para determinar los estados o estadios del lupus eritematoso sistémico (SLE o lupus). El SLE es una afección autoinmune grave que a menudo afecta a adultos jóvenes (en su mayoría mujeres). Se caracteriza por procesos inflamatorios que pueden afectar a muchos órganos, como la piel, las articulaciones, los riñones, los pulmones, el corazón y el sistema nervioso central, lo que conduce a discapacidades frecuentes y, en ocasiones, a la muerte. La enfermedad sigue un curso muy impredecible marcado por períodos de estallido seguidos por períodos de remisión inactivos.
Sin embargo, los pacientes diagnosticados con SLE son atendidos regularmente por un reumatólogo y tratados con una variedad de medicamentos severos. Estos medicamentos incluyen esteroides como prednisona y otros inmunosupresores como Cellcept (micofenolato mofetil). Si bien estos medicamentos pueden reducir el daño a los órganos, contienen efectos secundarios significativos, incluido el riesgo de infección e infertilidad. La falta de fiabilidad para algunos de los síntomas (por ejemplo, dolor y fatiga) y el curso impredecible de la enfermedad dificultan la adaptación de las dosis de los medicamentos, lo que resulta en un tratamiento excesivo de algunos pacientes y en el tratamiento insuficiente de otros. Como resultado, el tratamiento del SLE plantea importantes desafíos terapéuticos para el clínico.
Hay una serie de métodos estándar que un médico puede usar para evaluar la actividad del SLE. El estado de la enfermedad puede medirse observando los síntomas clínicos de la enfermedad. Estos métodos incluyen la evaluación de los signos (por ejemplo, erupción cutánea) y los síntomas (por ejemplo, dolor y fatiga de las articulaciones), así como los resultados de laboratorio (por ejemplo, la proporción de proteína/creatinina en la orina, el anticuerpo anti-ADNds y los recuentos sanguíneos). Sin embargo, estos marcadores clínicos pueden ser indicadores de retraso del estado de la enfermedad y, como tales, los pacientes pueden responder solo después de semanas o meses de tratamiento. Además, en algunos casos los síntomas pueden ser difíciles de evaluar con precisión (por ejemplo, dolor y fatiga). Otros marcadores de inflamación, por ejemplo, el anticuerpo anti-ADNds, el nivel de complemento (por ejemplo, C3), la proteína C reactiva (CRP) y la tasa de sedimentación de eritrocitos (ESR) generalmente carecen de especificidad y/o sensibilidad. Los métodos invasivos, como la biopsia de riñón, no son prácticos para el uso de rutina. Como resultado, los médicos realizan pruebas bastante frecuentes de sus pacientes sin una medida perfecta del estado de la enfermedad. Los síntomas clínicos y la evaluación de laboratorio se integran en medidas como el Índice de Actividad de la Enfermedad de Lupus Eritematoso Sistémico (SLEDAI) y la Evaluación Global del Médico (PGA). Estas medidas no se realizan de forma rutinaria en la práctica clínica y, a menudo, se quedan cortas en varias situaciones clínicas.
Se pueden identificar los clonotipos que representan diferentes perfiles inmunes para diferentes estados de enfermedad. Luego se realiza un seguimiento del estado de la enfermedad comparando el perfil inmunitario identificado con el perfil inmunitario actual del paciente. La enfermedad puede ser lupus. Los estados de enfermedad pueden ser períodos de exacerbación y períodos de no exacerbación. Este perfil inmunológico se puede usar como un indicador temprano de un estado de destello. Esto puede impulsar decisiones de tratamiento.
[0171 En la sección de ejemplos se analizan con mayor detalle ejemplos específicos de la utilidad de AutoImm Load para realizar intervenciones terapéuticas en SLE, junto con estudios de habilitación específicos que determinan la AutoImm Load.
Los clonotipos correlacionados con SLE pueden relacionarse con anticuerpos específicos para antígenos propios. Por consiguiente, un método para determinar la probabilidad de que un individuo tenga lupus eritematoso sistémico comprende los siguientes pasos: (a) determinar un perfil de clonotipos a partir de una muestra de células B del individuo, comprendiendo la muestra un repertorio de sus clonotipos; y (b) comparar los clonotipos del perfil con los clonotipos de una base de datos de clonotipos específicos de antígeno para determinar un nivel de coincidencia de clonotipos, determinando así una probabilidad de lupus eritematoso sistémico, la base de datos de clonotipos específica de antígeno que incluye sustancialmente todos los clonotipos de cadenas de inmunoglobulinas humanas específicas para el uno o más antígenos seleccionados del grupo que consiste en ADN bicatenario, malondialdehído, 4-hidroxinonenal, superóxido dismutasa, nitrotirosina, cardiolipina, proteína ribosomal P, fosfolípido, proteína central de la pequeña proteína nuclear ribonuclear (antígeno de Smith), histona, proteína ribonuclear U1 pequeña del núcleo, topoisomerasa tipo I, proteínas centroméricas, proteína ribonuclear SS-A, proteína ribonuclear s S-B y histidina-ARNt ligasa.
2. Esclerosis múltiple (EM)
Los métodos descritos en el presente documento también se pueden usar para determinar estados o estadios de esclerosis múltiple (EM). La EM es una enfermedad autoinmune que afecta el cerebro y la médula espinal (sistema nervioso central). Los síntomas varían, porque la ubicación y la gravedad de cada ataque pueden ser diferentes. Los episodios pueden durar días, semanas o meses. Estos episodios se alternan con períodos de síntomas reducidos o sin síntomas (remisiones). Es común que la enfermedad regrese (recaída). Sin embargo, la enfermedad puede continuar empeorando sin períodos de remisión.
Debido a que los nervios en cualquier parte del cerebro o la médula espinal pueden dañarse, los pacientes con esclerosis múltiple pueden tener síntomas en muchas partes del cuerpo. Los síntomas musculares incluyen, por ejemplo, pérdida de equilibrio, entumecimiento o sensación anormal en cualquier área, dolor debido a espasmos musculares, dolor en los brazos o piernas, problemas para mover los brazos o piernas, problemas para caminar, problemas de coordinación y movimientos pequeños, arrastrando los pies o habla difícil de entender, temblor en uno o más brazos o piernas, espasmo incontrolable de los grupos musculares (espasticidad muscular) y debilidad en uno o más brazos o piernas.
Los síntomas oculares incluyen, por ejemplo, visión doble, malestar ocular, movimientos oculares rápidos e incontrolables y pérdida de la visión (generalmente afecta un ojo a la vez).
Otros síntomas cerebrales y nerviosos incluyen, por ejemplo, disminución de la capacidad de atención, disminución del juicio, disminución de la memoria, depresión o sentimientos de tristeza, mareos y problemas de equilibrio, dolor facial, pérdida de la audición y fatiga.
Los síntomas del intestino y la vejiga incluyen, por ejemplo, estreñimiento, dificultad para comenzar a orinar, necesidad frecuente de orinar, escape de heces, fuerte deseo de orinar y escape de orina (incontinencia).
No hay cura conocida para la esclerosis múltiple en este momento. Sin embargo, existen terapias que pueden retardar la enfermedad. El objetivo del tratamiento es controlar los síntomas y ayudar al paciente a mantener una calidad de vida normal.
Los medicamentos utilizados para disminuir la progresión de la esclerosis múltiple pueden incluir, por ejemplo, moduladores inmunitarios para ayudar a controlar el sistema inmunológico, incluidos los interferones (Avonex, Betaseron o Rebif), los anticuerpos monoclonales (Tysabri), el acetato de glatiramer (Copaxone), la mitoxantrona (Novantrone), metotrexato, azatioprina (Imuran), ciclofosfamida (Cytoxan) y natalizumab (Tysabri). Los esteroides se pueden usar para disminuir la gravedad de los ataques.
Los medicamentos para controlar los síntomas pueden incluir, por ejemplo, medicamentos para reducir espasmos musculares como Lioresal (Baclofen), tizanidina (Zanaflex) o una benzodiazepina, medicamentos colinérgicos para reducir los problemas urinarios, antidepresivos para el estado de ánimo o síntomas de comportamiento y amantadina para la fatiga.
La EM afecta a las mujeres más que a los hombres. El trastorno generalmente comienza entre los 20 y los 40 años, pero se puede observar a cualquier edad. La EM es una enfermedad progresiva, lo que significa que el daño a los nervios (neurodegeneración) empeora con el tiempo. La rapidez con que la EM empeora varía de persona a persona. La inflamación ocurre cuando las células inmunes del cuerpo atacan el sistema nervioso. Los episodios repetidos de inflamación pueden ocurrir a lo largo de cualquier área del cerebro y la médula espinal. Las personas con antecedentes familiares de EM y las que viven en un área geográfica con una mayor tasa de incidencia de EM tienen un mayor riesgo de la enfermedad.
Los síntomas de la EM pueden imitar a los de muchos otros trastornos del sistema nervioso. La enfermedad se diagnostica descartando otras afecciones. Las personas que tienen una forma de EM llamada recaída-remisión pueden tener un historial de al menos dos ataques, separados por un período de síntomas reducidos o sin síntomas. El proveedor de atención médica puede sospechar EM si hay una disminución en la función de dos partes diferentes
del sistema nervioso central (como los reflejos anormales) en dos momentos diferentes. Un examen neurológico puede mostrar una función nerviosa reducida en un área del cuerpo o diseminarla en muchas partes del cuerpo.
Las pruebas para diagnosticar esclerosis múltiple incluyen, por ejemplo, pruebas de líquido cefalorraquídeo, incluidas bandas oligoclonales de LCR, resonancia magnética de la cabeza, punción lumbar (punción espinal), estudio de la función nerviosa (prueba de potencial evocado) y resonancia magnética de la columna vertebral.
Al igual que otras enfermedades autoinmunes, la EM sigue un curso impredecible con erupciones agudas y períodos de remisión. Hay un número creciente de terapias, cada una con efectos secundarios que van desde graves (aumento de peso y depresión) hasta mortales (pancitopenia e infecciones por PML), efectividad variable en diferentes pacientes y altos costes. Al mismo tiempo, la falta de pruebas de rutina altamente precisas y específicas de la actividad de la enfermedad de la EM hace que el desafío de administrar la terapia de manera efectiva sea complicado. Los episodios clínicos se pueden separar por largos períodos de tiempo (hasta años en la enfermedad en estadio temprano) incluso sin tratamiento. Además, los medicamentos disponibles reducen la probabilidad de recaída, pero no los previenen por completo. Por lo tanto, la actividad de la enfermedad es difícil de evaluar y, por lo tanto, existe una medida inadecuada a corto plazo de la actividad de la enfermedad que podría usarse para medir si una terapia específica está mostrando eficacia en un paciente dado al medir la reducción en el número o la gravedad de las recaídas. La única otra prueba disponible para monitorizar la actividad de la enfermedad es la RM cerebral para rastrear el estado de las lesiones como se revela con la ayuda de agentes que aumentan el contraste, como el gadolinio. Sin embargo, estas imágenes ofrecen solo una visión integrada del daño cerebral y carecen de especificidad y resolución de tiempo. Intentar usar imágenes de resonancia magnética para seguir el curso de la enfermedad en escalas de tiempo menores a un año no es práctico dados los costes, la falta de especificidad y los peligros de la exposición excesiva al contraste. Como resultado, los pacientes a menudo son tratados con grandes gastos durante períodos prolongados de tiempo sin una retroalimentación efectiva que le permita al médico modificar la dosis y/o cambiar las terapias adicionales.
Se pueden identificar los clonotipos que representan diferentes perfiles inmunes para diferentes estados de enfermedad. Luego se realiza un seguimiento del estado de la enfermedad comparando el perfil inmunitario identificado con el perfil inmunitario actual de los pacientes. La enfermedad puede ser EM. Los estados de enfermedad pueden ser períodos de remisión y períodos activos. Este perfil inmunológico se puede usar como un indicador temprano de un período de remisión o de no remisión. Esto puede impulsar decisiones de tratamiento.
3. Artritis reumatoide (AR)
Los métodos pueden utilizarse para medir el estado de la enfermedad en pacientes con artritis reumatoide. La artritis reumatoide (Ar ) es un trastorno inflamatorio crónico sistémico que puede afectar a muchos tejidos y órganos, pero principalmente ataca las articulaciones, produciendo una sinovitis inflamatoria que a menudo progresa hacia la destrucción del cartílago articular y la anquilosis de las articulaciones. La artritis reumatoide también puede producir inflamación difusa en los pulmones, pericardio, pleura y esclerótica, y también lesiones nodulares, más comunes en el tejido subcutáneo debajo de la piel. Aunque la causa de la artritis reumatoide es desconocida, la autoinmunidad desempeña un papel fundamental en su cronicidad y progresión.
Alrededor del 1% de la población mundial está afectada por artritis reumatoide, las mujeres tres veces más a menudo que los hombres. El inicio es más frecuente entre las edades de 40 y 50, pero las personas de cualquier edad pueden verse afectadas. Puede ser una condición incapacitante y dolorosa, que puede llevar a una pérdida sustancial de funcionamiento y movilidad. La AR se diagnostica principalmente en los síntomas y signos, pero también se puede diagnosticar con análisis de sangre (especialmente una prueba llamada factor reumatoide) y radiografías. El diagnóstico y el tratamiento a largo plazo suelen ser realizados por un reumatólogo, un experto en enfermedades de las articulaciones y los tejidos conectivos.
Varios tratamientos están disponibles. El tratamiento no farmacológico incluye fisioterapia, ortesis y terapia ocupacional. Se pueden usar analgesia (analgésicos) y medicamentos antiinflamatorios, incluidos los esteroides, para suprimir los síntomas, mientras que los medicamentos antirreumáticos modificadores de la enfermedad (DMARD) se pueden usar para inhibir o detener el proceso inmunitario subyacente y prevenir el daño a largo plazo. En los últimos tiempos, el nuevo grupo de productos biológicos ha aumentado las opciones de tratamiento.
Cuando se sospecha clínicamente de AR, se pueden realizar estudios inmunológicos, como probar la presencia del factor reumatoide (RF, un anticuerpo específico). Un RF negativo no descarta la RA; más bien, la artritis se llama seronegativa. Este es el caso en aproximadamente el 15% de los pacientes. Durante el primer año de enfermedad, es más probable que el factor reumatoide sea negativo y que algunas personas se conviertan en estado seropositivo con el tiempo. La RF también se observa en otras enfermedades, por ejemplo, el síndrome de Sjogren, y en aproximadamente el 10% de la población sana, por lo tanto, la prueba no es muy específica.
Debido a esta baja especificidad, se han desarrollado nuevas pruebas serológicas, que analizan la presencia de los llamados anticuerpos de proteínas anticitrulinados (ACPA). Al igual que la RF, estas pruebas son positivas solo en una proporción (67%) de todos los casos de AR, pero rara vez son positivas si la RA no está presente, lo que le da una especificidad de alrededor del 95%. Al igual que con la RF, existe evidencia de que las ACPa están presentes en muchos casos incluso antes de la aparición de la enfermedad clínica.
Las pruebas más comunes para ACPA son la prueba anti-CCP (péptido cíclico citrulinado) y la prueba anti-MCV (anticuerpos contra la vimentina citrulina mutada). Recientemente, se ha desarrollado una prueba serológica de punto de atención (POCT) para la detección temprana de la AR. Este ensayo combina la detección de factor reumatoide y anti-MCV para el diagnóstico de artritis reumatoide y muestra una sensibilidad del 72% y una especificidad del 99.7%.
Además, se pueden realizar varios otros análisis de sangre para detectar otras causas de artritis, como el lupus eritematoso. La tasa de sedimentación eritrocítica (ESR), la proteína C reactiva, el hemograma completo, la función renal, las enzimas hepáticas y otras pruebas inmunológicas (por ejemplo, anticuerpo antinuclear/ANA) se realizan en esta etapa. Los niveles elevados de ferritina pueden revelar hemocromatosis, una RA mímica, o ser un signo de la enfermedad de Still, una variante seronegativa, generalmente juvenil, de reumatoide.
El término fármaco antirreumático modificador de la enfermedad (DMARD, por sus siglas en inglés) originalmente significaba un fármaco que afecta a medidas biológicas como la ESR y los niveles de hemoglobina y autoanticuerpos, pero ahora se usa generalmente para significar un fármaco que reduce la tasa de daño al hueso y al cartílago. Se ha descubierto que los DMARD producen remisiones sintomáticas duraderas y retrasan o detienen la progresión. Esto es significativo, ya que tal daño suele ser irreversible. Los antiinflamatorios y los analgésicos mejoran el dolor y la rigidez, pero no evitan el daño articular ni ralentizan la progresión de la enfermedad.
Hay un reconocimiento creciente entre los reumatólogos de que el daño permanente en las articulaciones se produce en un estadio muy temprano de la enfermedad. En el pasado, era común comenzar la terapia con solo un medicamento antiinflamatorio y evaluar la progresión clínicamente y el uso de rayos X. Si hubiera evidencia de que el daño articular comenzaba a ocurrir, entonces se prescribiría un DMARD más potente. El ultrasonido y la RMN son métodos más sensibles para obtener imágenes de las articulaciones y han demostrado que el daño articular se produce mucho antes y en más pacientes de lo que se pensaba. Las personas con rayos X normales a menudo tendrán erosiones detectables por ultrasonido que los rayos X no pudieron demostrar. El objetivo ahora es tratar antes de que ocurra el daño.
Puede haber otras razones por las que el inicio temprano de los DMARD es beneficioso para prevenir daños estructurales en las articulaciones. Desde los estadios más tempranos de la enfermedad, las articulaciones están infiltradas por células del sistema inmunológico que se comunican entre sí de maneras que pueden involucrar una variedad de ciclos de retroalimentación positiva (se ha observado que una sola inyección de corticosteroides puede abortar la sinovitis en una articulación particular durante largos períodos). La interrupción de este proceso lo antes posible con un DMARD efectivo (como el metotrexato) parece mejorar el resultado de la AR durante años después. Retrasar la terapia por unos pocos meses después de la aparición de los síntomas puede dar lugar a peores resultados a largo plazo. Por lo tanto, existe un interés considerable en establecer la terapia más efectiva con la artritis temprana, cuando el paciente responde mejor a la terapia y tiene más que ganar.
Los fármacos tradicionales de pequeña masa molecular utilizados para tratar la artritis incluyen, por ejemplo, DMARD sintetizados químicamente: azatioprina, ciclosporina (ciclosporina A), D-penicilamina, sales de oro, hidroxicloroquina, leflunomida, metotrexato (MTX), minociclina y sulfasalazina SSZ). Los fármacos citotóxicos incluyen ciclofosfamida.
Los eventos adversos más comunes se relacionan con la toxicidad del hígado y la médula ósea (MTX, SSZ, leflunomida, azatioprina, compuestos de oro, D-penicilamina), toxicidad renal (ciclosporina A, sales de oro parenteral, D-penicilamina), neumonitis (MTX), reacciones alérgicas de la piel (compuestos de oro, SSZ), autoinmunidad (D-penicilamina, SSZ, minociclina) e infecciones (azatioprina, ciclosporina A). La hidroxicloroquina puede causartoxicidad ocular, aunque esto es poco frecuente, y debido a que la hidroxicloroquina no afecta la médula ósea ni el hígado, a menudo se considera el FAME con la menor toxicidad. Desafortunadamente, la hidroxicloroquina no es muy potente y, por lo general, es insuficiente para controlar los síntomas por sí sola.
Los agentes biológicos (biológicos) se pueden producir mediante ingeniería genética e incluyen, por ejemplo, los bloqueadores del factor de necrosis tumoral alfa (TNFa) - etanercept (Enbrel), infliximab (Remicade), adalimumab (Humira), interleucina 1 (IL-1) - anakinra (Kineret), anticuerpos monoclonales contra las células B - rituximab (Rituxan), bloqueador de la coestimulación de células T - abatacept (Orencia), interleuquina 6 (IL-6) bloqueadores - tocilizumab (un anticuerpo anti-receptor de IL-6) (RoActemra, Actemra).
Los agentes antiinflamatorios incluyen, por ejemplo, glucocorticoides, fármacos antiinflamatorios no esteroideos (AINE, la mayoría también actúan como analgésicos). Los analgésicos incluyen, por ejemplo, paracetamol (acetaminofén en Estados Unidos y Canadá), opiáceos, diproqualona y lidocaína tópica.
El desafío de tratar la AR radica en el hecho de que la enfermedad es una enfermedad crónica a largo plazo que puede resultar en una discapacidad difícil para la cual existe una gran variedad de tratamientos, cada uno de los cuales tiene inconvenientes importantes. Muchos de los DMARD someten a los pacientes a efectos secundarios significativos, incluido un mayor riesgo de infecciones graves, cáncer o incluso enfermedades autoinmunes. Además, los medicamentos derivados biológicamente son muy caros, y el paciente puede ser sometido a inyecciones frecuentes.
Un médico que inicia la terapia para un paciente se enfrenta a muchas opciones posibles. Sería deseable obtener una retroalimentación rápida una vez que el paciente comience la terapia para comprender si el paciente está respondiendo a la terapia que se elige antes de que se presente la manifestación clínica. Las imágenes no son sensibles y son caras
y muchos marcadores de sangre, como la PCR, carecen de sensibilidad suficiente. Una prueba que le permitiría al médico determinar rápidamente el estado de la enfermedad le permitiría ajustar la terapia rápidamente a una terapia más efectiva, evitando que el paciente sufra daños adicionales en las articulaciones y utilizando las terapias disponibles.
Un paciente que no ha experimentado ninguna erupción aguda desde el inicio del tratamiento puede, de hecho, experimentar un daño inflamatorio continuo en las articulaciones que no se ha manifestado clínicamente. Una prueba que le permitiría al médico diferenciar este estado del fondo permitiría ajustar la terapia para intentar acercar al paciente a un estado en donde no se experimenta daño articular continuo.
Los ejemplos específicos de cómo se puede usar AutoImm Load en el manejo de pacientes con AR se describen con más detalle en la sección de ejemplos de este documento.
Se pueden identificar los clonotipos que representan diferentes perfiles inmunes para diferentes estados de enfermedad. Luego se realiza un seguimiento del estado de la enfermedad comparando el perfil inmunitario identificado con el perfil inmunitario actual de los pacientes. La enfermedad es la AR. Los estados de enfermedad pueden ser, pero no están limitados a períodos de alta inflamación y una línea de base. Estos perfiles inmunes se utilizan para impulsar las decisiones de tratamiento.
4. Espondilitis anquilosante
Los métodos pueden usarse para detectar la actividad de la enfermedad para la espondilitis anquilosante. La espondilitis anquilosante (AS, del griego ankylos, doblado; spondylos, vértebras), anteriormente conocida como enfermedad de Bechterew, síndrome de Bechterew y enfermedad de Marie Strümpell, una forma de espondiloartritis, es una artritis inflamatoria crónica y una enfermedad autoinmune. Afecta principalmente a las articulaciones de la columna vertebral y al sacroilium en la pelvis, lo que provoca la fusión final de la columna vertebral. Es un miembro del grupo de espondiloartropatías con una fuerte predisposición genética. La fusión completa da como resultado una rigidez completa de la columna vertebral, una condición conocida como columna vertebral de bambú.
El paciente típico es un hombre joven, de 18 a 30 años de edad, cuando aparecen los síntomas de la enfermedad por primera vez, con dolor crónico y rigidez en la parte inferior de la columna vertebral o, a veces, toda la columna vertebral, a menudo con dolor referido a una u otra nalga o la parte posterior del muslo de la articulación sacroilíaca. Los hombres se ven más afectados que las mujeres en una proporción de aproximadamente 3:1, y la enfermedad generalmente toma un curso más doloroso en los hombres que en las mujeres. En el 40% de los casos, la espondilitis anquilosante se asocia con una inflamación ocular (iridociclitis y uveítis) que causa enrojecimiento, dolor ocular, pérdida de visión, flotadores y fotofobia. Otro síntoma común es la fatiga generalizada y, a veces, las náuseas. Con menos frecuencia, pueden aparecer aortitis, fibrosis apical de pulmón y ectasia de las vainas de la raíz del nervio sacro. Al igual que con todas las espondiloartropatías seronegativas, puede ocurrir el levantamiento de las uñas (onicolisis).
No hay una prueba directa para diagnosticar EA. El examen clínico y los estudios de rayos X de la columna vertebral, que muestran cambios espinales característicos y sacroilitis, son las principales herramientas de diagnóstico. Un inconveniente del diagnóstico de rayos X es que los signos y síntomas de la EA generalmente se han establecido hasta 8-10 años antes de que se produzcan cambios evidentes en los rayos X en una radiografía de película simple, lo que significa un retraso de hasta 10 años antes de poder introducir terapias adecuadas. Las opciones para un diagnóstico más temprano son la tomografía y la resonancia magnética de las articulaciones sacroilíacas, pero la confiabilidad de estas pruebas aún no está clara. La prueba de Schober es una medida clínica útil de la flexión de la columna lumbar realizada durante el examen.
Durante los períodos inflamatorios agudos, los pacientes con EA a veces mostrarán un aumento en la concentración sanguínea de proteína C reactiva (PCR) y un aumento en la tasa de sedimentación de eritrocitos (ESR), pero hay muchos con EA cuyas tasas de PCR y ESR no aumentan, por lo tanto, los resultados normales de CRP y ESR no siempre se corresponden con la cantidad de inflamación que realmente tiene una persona. Aveces, las personas con EA tienen resultados de nivel normal, sin embargo, están experimentando una cantidad significativa de inflamación en sus cuerpos.
La espondilitis anquilosante (AS, del griego ankylos, doblado; spondylos, vértebras), conocida anteriormente como enfermedad de Bechterew, síndrome de Bechterew y enfermedad de Marie Strümpell, una forma de espondiloartritis, es una enfermedad crónica, inflamatoria y autoinmunitaria. Afecta principalmente a las articulaciones de la columna vertebral y al sacroilium en la pelvis, lo que provoca la fusión final de la columna vertebral.
Es un miembro del grupo de espondiloartropatías con una fuerte predisposición genética. La fusión completa da como resultado una rigidez completa de la columna vertebral, una condición conocida como columna vertebral de bambú.
Hay tres tipos principales de medicamentos que se usan para tratar la espondilitis anquilosante.: 1) Medicamentos antiinflamatorios, que incluyen AINE como el ibuprofeno, la fenilbutazona, la indometacina, el naproxeno y los inhibidores de la COX-2, que reducen la inflamación y el dolor. La evidencia clínica también ha demostrado que los analgésicos opioides son muy efectivos para aliviar el tipo de dolor crónico que experimentan comúnmente quienes sufren de EA, especialmente en formulaciones de liberación prolongada. 2) DMARD, como la ciclosporina, el
metotrexato, la sulfasalazina y los corticosteroides, utilizados para reducir la respuesta del sistema inmunitario a través de la inmunosupresión; 3) Los bloqueadores del TNFa (antagonistas), como etanercept, infliximab y adalimumab (también conocidos como biologías), están indicados para el tratamiento y son inmunosupresores efectivos como en otras enfermedades autoinmunes;
Se ha demostrado que los bloqueadores del TNFa son el tratamiento más prometedor, lo que retrasa el progreso de la EA en la mayoría de los casos clínicos, lo que ayuda a muchos pacientes a recibir una reducción significativa, aunque no la eliminación, de su inflamación y dolor. También se ha demostrado que son altamente efectivos para tratar no solo la artritis de las articulaciones, sino también la artritis espinal asociada con la EA. Un inconveniente, además del coste a menudo alto, es el hecho de que estos medicamentos aumentan el riesgo de infecciones. Por esta razón, el protocolo para cualquiera de los bloqueadores de TNF-a incluye una prueba de tuberculosis (como Mantoux o Heaf) antes de comenzar el tratamiento. En caso de infecciones recurrentes, incluso dolor de garganta recurrente, la terapia puede suspenderse debido a la inmunosupresión involucrada. Se recomienda a los pacientes que toman los medicamentos contra el TNF que limiten su exposición a otras personas que tienen o pueden estar portando un virus (como un resfriado o influenza) o que pueden tener una infección bacteriana o por hongos.
Los efectos de EA producen síntomas que son muy comunes en las poblaciones sanas. Por ejemplo, un paciente que se presenta quejándose de dolor severo de espalda no necesita estar experimentando una erupción de EA, sino que simplemente puede tener un dolor de espalda de rutina. El médico se ve obligado a tomar una decisión sobre si tratar estos síntomas con medicamentos caros con efectos secundarios potencialmente graves sin una visión muy precisa del estado de la enfermedad. CRP y ESR no proporcionan una visión muy precisa del estado de la enfermedad. Al mismo tiempo, el curso de la enfermedad no tratada puede resultar en un daño espinal debilitante a largo plazo. Este estado de cosas conduce a un desafío clínico difícil y se utiliza un tratamiento excesivo significativo. La disponibilidad de una medida objetiva que refleje la actividad de la enfermedad puede ser de gran ayuda en el manejo de pacientes con EA.
Se pueden identificar los clonotipos que representan diferentes perfiles inmunes para diferentes estados de enfermedad. Luego se realiza un seguimiento del estado de la enfermedad comparando el perfil inmunitario identificado con el perfil inmunitario actual de los pacientes. La enfermedad es EA. Los estados de enfermedad pueden ser, pero no están limitados a períodos de alta inflamación y una línea de base. Estos perfiles inmunes se utilizan para impulsar las decisiones de tratamiento.
B. Utilidad del perfil inmunológico en la detección del cáncer
Estos métodos pueden usarse para medir el riesgo de cáncer. El cáncer se ha convertido en la principal causa de muerte en el mundo industrializado. Por lo tanto, los métodos de tratamiento del cáncer están en gran necesidad. Se están intentando muchas metodologías para el tratamiento del cáncer, incluido el desarrollo de nuevos fármacos de molécula pequeña, así como anticuerpos dirigidos contra el tumor.
Un conjunto de métodos que se ha propuesto es la inmunoterapia. La monitorización del tumor es una de las funciones de las células del sistema inmunológico. Existen varias categorías de antígenos tumorales que son reconocidos por el sistema inmunológico. La primera categoría está formada por antígenos que son nuevos generados por la mutación somática (mutación puntual o una translocación) en el tumor. Otra categoría consiste en antígenos de proteínas que solo se expresan en células germinales masculinas que no expresan moléculas MHC. La desregulación de la expresión génica en muchos tumores puede permitir que algunos de estos antígenos se expresen. Una tercera categoría incluye antígenos de proteínas expresadas solo en tejidos particulares. La cuarta categoría comprende antígenos que están sobreexpresados significativamente en el tejido tumoral. Finalmente, la quinta categoría incluye antígenos que resultan de una modificación postraducción anormal.
Una de las propiedades de los tumores es su capacidad para escapar de la eliminación efectiva por parte del sistema inmunológico. Se piensa que las nuevas mutaciones adquiridas en el tumor le permiten pasar de la fase de equilibrio (donde el tumor no se elimina completamente, pero su crecimiento se mantiene bajo control) a la fase de escape donde el tumor crece sin un control efectivo del sistema inmunitario. Hay muchos mecanismos que los tumores emplean para escapar del sistema inmunológico. Estos mecanismos incluyen la falta de péptidos antigénicos específicos, o las moléculas coestimuladoras que pueden activar las células T. Otros mecanismos incluyen la secreción tumoral de factor que inhibe las células T y la creación de un sitio privilegiado inducido por el tumor mediante la creación de una barrera física que separa el tumor de los linfocitos. La inducción del sistema inmunitario para combatir mejor el tumor como una estrategia para tratar el cáncer se está estudiando y probando de múltiples maneras. Una metodología es la terapia adoptiva de células T. Esta metodología se centra en identificar las células T que se dirigen a los antígenos tumorales mediante el aislamiento de células que se infiltran en el tumor y/o reaccionan a un antígeno tumoral específico. Estas células T pueden crecer in vitro en condiciones que mejoren su efectividad, como el uso de IL-2 y/o células presentadoras de antígenos. Las células expandidas se infunden de nuevo a la sangre del paciente. Otra metodología es el uso de retrovirus que contienen TCR específico de tumor. Estos retrovirus pueden infundirse en el paciente en células especiales que más tarde secretan el retrovirus, lo que le permite infectar las células T que luego comienzan a expresar el TCR específico del tumor. Finalmente, una metodología común es el uso de la vacunación. La premisa de esta metodología terapéutica es que la inmunización del paciente con uno o más de los antígenos tumorales estimulará la capacidad del sistema inmunitario para combatir el tumor. La inmunización a
menudo se realiza con el uso de un adyuvante como Bacille Calmette-Guerin (BCG). Esta metodología ha tenido éxito en la prevención del cáncer inducido por virus, como lo demuestra la capacidad para prevenir los cánceres de cuello uterino inducidos por el VPH-16 y el VPH-18. Sin embargo, esto ha tenido menos éxito en el tratamiento de otros tumores.
Gran parte de la mejora en la mortalidad debida al cáncer se debe a la disponibilidad de mejores métodos de detección temprana que llevan, por ejemplo, a tasas reducidas de mortalidad en cáncer de mama y cáncer de cuello uterino. La mutabilidad de los tumores hace que su tratamiento temprano sea mucho más efectivo que cuando se detectan tarde. Tradicionalmente, la búsqueda de biomarcadores de detección de cáncer usualmente involucraba la búsqueda de marcadores que estén altamente expresados en el cáncer y estén en un nivel bajo o ausente en el tejido normal. Esto ha llevado a la identificación de varios marcadores tumorales, como el PSA. Un problema con la detección temprana del cáncer es que el mayor valor para la detección del cáncer ocurre cuando la detección del biomarcador es más difícil, es decir, el tumor es muy pequeño. Por lo tanto, para tener un biomarcador de detección de cáncer efectivo que pueda distinguir a los pacientes con tumores pequeños de los que no lo tienen, es necesario que haya una diferencia tremenda en la expresión entre el tumor y el tejido normal debido a la gran diferencia de tamaño entre el tumor y el tejido normal. Además, el marcador debe "derramarse" eficientemente en la sangre u otro fluido corporal para permitir la detección utilizando una técnica no invasiva.
Un nuevo mecanismo para la detección del cáncer utilizando la respuesta de las células inmunitarias se describe aquí. En este sentido, la detección del cáncer no se logra mediante la detección de un marcador producido por el propio tumor, sino por la respuesta del sistema inmunitario al tumor. Específicamente, el perfil de TCR y/o BCR puede proporcionar una idea de si el cuerpo está respondiendo a un tumor o no. Esto puede mejorar algunos de los problemas con los biomarcadores actuales. Primero, la respuesta inmune es una señal de amplificación que puede ser más fácil de detectar. Los segundos linfocitos pasan a través de la sangre regularmente y, por lo tanto, el biomarcador relevante puede presentarse fácilmente y ser detectable en la sangre periférica que el biomarcador tumoral tradicional. Finalmente, el problema del "biomarcador" de fondo generado por el tejido normal se reduce considerablemente. La gran diversidad de células T y/o B proporciona una forma de detectar biomarcadores relevantes con alta sensibilidad y especificidad, en particular con la reciente disponibilidad de métodos de alto rendimiento para la secuenciación de ADN. La metodología de utilizar la respuesta del sistema inmunitario al cáncer para detectarla aprovecha los fundamentos establecidos en este campo con la promesa de la inmunoterapia. Sin embargo, el riesgo para las dos aplicaciones es probablemente bastante diferente. Para utilizar la respuesta inmune al cáncer para su detección, no es necesario que el clonotipo específico sea eficaz en el tratamiento del tumor, sino que esté asociado con la respuesta inmune al tumor.
Las pruebas de perfil inmunológico se pueden combinar con otros marcadores que ya están en uso para la detección de cáncer para permitir pruebas con mayor sensibilidad y especificidad. Se pueden usar otros identificadores o marcadores moleculares para calcular el algoritmo de carga o para determinar el estado de la enfermedad. Los identificadores moleculares pueden incluir ácidos nucleicos, proteínas, carbohidratos y lípidos, y perfiles de expresión de ácidos nucleicos o proteínas. Los identificadores moleculares pueden ser de origen humano o no humano (por ejemplo, bacteriano). Los identificadores o marcadores pueden determinarse mediante técnicas que incluyen, por ejemplo, hibridación genómica comparativa (CGH), análisis de micromatrices cromosómicas (CMA), perfiles de expresión, micromatrices de ADN, micromatrices de oligonucleótidos de alta densidad, matriz de expresión de ARN de genoma completo, micromatriz de péptidos, ensayo inmunoenzimático (ELISA), secuenciación del genoma, número de copias (CNV), análisis de polimorfismo de nucleótidos pequeños (SNP), inmunohistoquímica, hibridación in situ, hibridación in situ fluorescente (FISH), PCR, Inmunoprecipitación Western, Inmunoprecipitación Southern, SDS-PAGE, electroforesis en gel y inmunoprecipitación Northern.
C. Utilidad del perfil inmunológico en la medicina de trasplante.
Estos métodos se pueden utilizar para detectar el rechazo inmune de los órganos trasplantados. El trasplante de órganos se ha convertido en una parte integral de la medicina con más de 25.000 trasplantes de órganos sólidos (riñón, hígado, corazón, páncreas y pulmón) y más de 15.000 trasplantes de médula ósea que se realizan en los Estados Unidos por año. Estos son procedimientos generalmente complicados que se realizan en los centros de atención terciaria. Para minimizar el riesgo de rechazo del trasplante, los pacientes a menudo reciben inmunosupresión durante largos períodos de tiempo, lo que los somete al riesgo de cáncer e infecciones. Además, muchos trasplantes se rechazan de forma aguda o años después del trasplante. A pesar de estos problemas, el trasplante de órganos sigue siendo una modalidad de tratamiento esencial, ya que los pacientes con insuficiencia orgánica tienen pocas alternativas.
El rechazo al trasplante de órganos sólidos se produce principalmente debido a la respuesta del sistema inmunitario adaptativo al órgano trasplantado. Esto se debe a la presencia de aloantígenos en el injerto que son reconocidos por el sistema inmunitario del huésped. El rechazo puede ocurrir en tres fases diferentes. La primera es la fase hiperaguda a los pocos minutos del trasplante donde los anticuerpos preformados montan una respuesta al injerto. El segundo es el rechazo agudo que se produce en las primeras semanas o meses después del trasplante. El último es el rechazo crónico que puede ocurrir años después del trasplante. Teniendo en cuenta estos riesgos, se han tomado medidas para minimizar las diferencias inmunogénicas entre el donante y el receptor. Por ejemplo, el riesgo de la reacción hiperaguda se reduce considerablemente cuando el donante y el receptor se comparan para sus subtipos ABO y se
analizan para determinar la compatibilidad cruzada (determinar si el receptor tiene anticuerpos que reaccionan con los leucocitos del donante). De manera similar, se realiza una concordancia cuidadosa para la HistoCompatability principal (MHC) para reducir el rechazo agudo. Sin embargo, dado que las moléculas de MHC son muy polimórficas, es muy difícil encontrar una coincidencia perfecta. Los gemelos monocigóticos tienen una combinación perfecta de MHC. De manera similar, se espera que % hermanos tengan una coincidencia perfecta de MHC. Los individuos no relacionados que tienen los mismos alelos detectados según la prueba clínica a menudo tienen diferencias debido a otros sitios polimórficos que no se prueban en la práctica clínica habitual. Sin embargo, incluso con la compatibilidad perfecta de MHC con los hermanos, todavía existe un riesgo significativo de rechazo debido a la existencia de antígenos de histocompatibilidad menores, y, de hecho, el rechazo agudo es muy común en más de la mitad de los injertos.
Uno podría imaginar que las pruebas más agresivas del locus MHC, así como la identificación y el emparejamiento de los antígenos de histocompatibilidad menores, mejorarán significativamente el rechazo del injerto y posiblemente las tasas de supervivencia. Si bien eso podría ser cierto, el número limitado de órganos disponibles de donantes disponibles hace que esta tarea sea poco práctica, ya que las pruebas más agresivas pueden retrasar significativamente la identificación de un injerto apropiado para cada paciente. Por lo tanto, gran parte del progreso que se ha producido en el campo del trasplante fue en el uso de agentes inmunosupresores para prevenir y tratar el rechazo. Actualmente, se utilizan muchos medicamentos para este fin, incluidos: azatioprina, corticosteroides, ciclosporina, tacrolimus, ácido micofenolato, sirolimus, muromonab-CD3, anticuerpo monoclonal anti-CD25, anticuerpo monoclonal anti-CD20 e inhibidores de la calcineurina.
El trasplante de médula ósea se usa más frecuentemente para el tratamiento de la leucemia y el linfoma. Normalmente, el receptor se somete a un régimen agresivo de radiación y/o quimioterapia para disminuir la carga del tumor antes del trasplante. Las células T maduras del donante pueden atacar algunos de los tejidos del huésped en el rechazo inverso que se denomina enfermedad de injerto contra huésped (GVHD). Esto se manifiesta a menudo por erupción cutánea, diarrea y enfermedad hepática. La combinación cuidadosa de MHC puede mejorar, pero no eliminar este problema. Una solución es el agotamiento de la médula ósea del donante in vitro de células T maduras que son en última instancia responsables de la GVHD. Un problema con esto es que el mismo fenómeno que causa la GVHD puede ser responsable de algunos de los efectos terapéuticos del trasplante de médula ósea a través del efecto de injerto contra leucemia cuando las células T del donante atacan las células cancerosas restantes. Además, el agotamiento de las células T del donante puede exponer al paciente al riesgo de ser inmunodificente. Por lo tanto, el riesgo y los beneficios deben ser equilibrados al considerar. Por lo tanto, los pacientes a menudo se tratan con inmunosupresores para prevenir y tratar la GVHD.
El tratamiento actual de la médula ósea, pero aún más para el trasplante de órganos sólidos, depende en gran medida del tratamiento con agentes inmunosupresores fuertes. Sin embargo, dado que estos medicamentos tienen riesgos significativos, se utilizan de una manera para equilibrar el riesgo y el beneficio. Sin embargo, dado que el riesgo para un paciente específico en un momento determinado no se comprende bien, los pacientes se tratan con la dosis en la que el riesgo y los beneficios se equilibran para el paciente promedio. Las pruebas que pueden predecir futuros eventos de rechazo pueden ser muy útiles para adaptar el tratamiento a los pacientes en el momento adecuado en que lo necesiten. Esto puede resultaren una reducción de las dosis inmunosupresoras o en algunos de los pacientes, al mismo tiempo que mejora la tasa de rechazo y, con suerte, la supervivencia del injerto.
Las pruebas de perfil inmunológico se pueden combinar con otros marcadores que ya están en uso para la detección del rechazo de trasplantes para permitir pruebas con mayor sensibilidad y especificidad. Se pueden usar otros identificadores o marcadores moleculares para calcular el algoritmo de carga o para determinar el estado de la enfermedad. Los identificadores moleculares pueden incluir ácidos nucleicos, proteínas, carbohidratos y lípidos, y perfiles de expresión de ácidos nucleicos o proteínas. Los identificadores moleculares pueden ser de origen humano o no humano (por ejemplo, bacteriano). Los identificadores o marcadores pueden determinarse mediante técnicas que incluyen, por ejemplo, hibridación genómica comparativa (CGH), análisis de micromatrices cromosómicas (CMA), perfiles de expresión, micromatrices de ADN, micromatrices de oligonucleótidos de alta densidad, matriz de expresión de ARN de genoma completo, micromatriz de péptidos, ensayo inmunoenzimático (ELISA), secuenciación del genoma, análisis del número de copias (CNV), polimorfismo de nucleótidos pequeños (SNP) análisis, inmunohistoquímica, hibridación in situ, hibridación in situ fluorescente (FISH), PCR, Inmunoprecipitación Western, inmunoprecipitación Southern, SDS-PAGE, gel de electroforesis e inmunoprecipitación Northern.
D. Utilidad del perfil inmunológico en el tratamiento de infecciones.
Estos métodos son útiles para guiar el tratamiento de infecciones, particularmente cuando estas infecciones pueden existir en estados activos y latentes. La llegada de los antibióticos para el tratamiento de enfermedades infecciosas en el último siglo ha tenido un gran impacto en la esperanza de vida. Durante la última década, las técnicas de diagnóstico molecular han adquirido un papel cada vez más importante en el diagnóstico y tratamiento de las enfermedades infecciosas. La excelente sensibilidad y especificidad que proporciona la amplificación de ácidos nucleicos ha permitido la aplicación de estas técnicas a un número creciente de aplicaciones. Muchas de las aplicaciones se utilizan para la evaluación diagnóstica de la presencia o ausencia de agentes infecciosos. Por ejemplo, la prueba de enfermedades de transmisión sexual a menudo se realiza mediante una prueba molecular que emplea una técnica de amplificación de ácido nucleico. Otro conjunto de aplicaciones implica la evaluación de la "carga" de la infección en un paciente con un agente infeccioso ya diagnosticado. Un ejemplo de ello es la evaluación de la carga viral del VIH en pacientes con
diagnóstico de SIDA. Esta prueba ayuda al médico a determinar si el estado de la enfermedad del paciente y, por tanto, puede proporcionar orientación sobre la eficacia del régimen de tratamiento que se está utilizando.
Aveces es útil no solo considerar el nivel del agente infeccioso sino también la respuesta inmune al agente infeccioso. Un ejemplo en donde la respuesta inmunitaria a la infección se usa habitualmente en la práctica clínica es en la hepatitis B. Un aspecto de las pruebas de hepatitis B se basa en la detección del agente infeccioso mediante la detección de antígenos de hepatitis B mediante un ensayo de amplificación de ácido nucleico. Además, es habitual en la práctica clínica habitual detectar la presencia de diferentes anticuerpos dirigidos contra el virus de la hepatitis B. La presencia de IgM anti-HBc por lo general ocurre en un entorno de infección aguda, la aparición de IgG anti-HBc indica que la infección es crónica. De manera similar, la aparición de anticuerpos anti-HBs indica la eliminación de la infección.
El valor de la evaluación de la respuesta inmune a una infección se puede aprovechar junto con la sensibilidad y especificidad de las pruebas moleculares. Esto puede ser particularmente útil para enfermedades infecciosas que son crónicas donde el agente infeccioso permanece latente en el cuerpo. El perfil del TCR y/o BCR se puede usar para evaluar la respuesta inmune a una infección. La secuenciación se puede utilizar para obtener un perfil del TCR y/o BCR que permite la detección de clonotipos particulares con alta sensibilidad y especificidad. Para determinar los clonotipos específicos que se correlacionan con la enfermedad, se conciben varias metodologías.
Las pruebas de perfil inmunológico se pueden combinar con otros marcadores que ya están en uso para la detección de agentes infecciosos para permitir pruebas con mayor sensibilidad y especificidad. Se pueden usar otros identificadores o marcadores moleculares para calcular el algoritmo de carga o para determinar el estado de la enfermedad. Los identificadores moleculares pueden incluir ácidos nucleicos, proteínas, carbohidratos y lípidos, y perfiles de expresión de ácidos nucleicos o proteínas. Los identificadores moleculares pueden ser de origen humano o no humano (por ejemplo, bacteriano). Los identificadores o marcadores pueden determinarse mediante técnicas que incluyen, por ejemplo, hibridación genómica comparativa (CGH), análisis de micromatrices cromosómicas (CMA), perfiles de expresión, micromatrices de ADN, micromatrices de oligonucleótidos de alta densidad, matriz de expresión de ARN de genoma completo, micromatriz de péptidos, ensayo inmunoenzimático (ELISA), secuenciación del genoma, número de copias (CNV), análisis de polimorfismo de nucleótidos pequeños (SNP), inmunohistoquímica, hibridación in situ, hibridación in situ fluorescente (FISH), PCR, Inmunoprecipitación Western, Inmunoprecipitación Southern, SDS-PAGE, electroforesis en gel y inmunoprecipitación Northern.
E. Utilidad del perfil inmunológico en el tratamiento de pacientes ancianos.
Estos métodos tienen utilidad en La monitorización del estado del sistema inmunológico en personas de edad avanzada. Las personas mayores sufren una disminución en el sistema inmunológico llamada inmunosenescencia que afecta su capacidad para responder a las infecciones y aumentar las respuestas efectivas a las vacunas (Weinberger et al., 2008). Esto se desprende de las altas tasas de mortalidad por neumonía en ancianos (Office for National Statistics, 2005) y su susceptibilidad a las infecciones adquiridas en el hospital, como Clostridium difficile y Staphylococcus aureus resistente a la meticilina (Health Protection Agency, 2008). Además, se cree que la disminución de la capacidad del sistema inmunitario explica el aumento de la tasa de cáncer en los ancianos. Además, la inmunosenescencia puede contribuir a otras enfermedades importantes de los ancianos con un componente importante de los procesos inflamatorios, como el Alzheimer y la enfermedad cardíaca. La capacidad de predecir qué individuos tienen mayor riesgo de estos resultados mortales sería útil para los médicos de geriatría, ya que toman decisiones clínicas sobre la vacunación, el tratamiento agresivo de infecciones y la hospitalización.
Muchos aspectos del sistema inmunitario innato y adaptativo están alterados en la inmunosenescencia. Las células T pierden capacidad de respuesta, los macrófagos tienen una capacidad de presentación de antígeno disminuida y la secreción de citoquinas alterada, las células asesinas naturales tienen una toxicidad reducida, las células dendríticas foliculares no pueden presentar el antígeno de manera eficiente y los neutrófilos pierden la capacidad fagocítica. Hay un grupo más pequeño de células T y B ingenuas y un aumento en el grupo de memoria y efector que conduce a una diversidad reducida de repertorios de células T y B que conducen a la reducción de la capacidad del sistema inmune adaptativo para responder a nuevos antígenos. En particular, los repertorios de células T que están asociados con el citomegalovirus (CMV) aumentan considerablemente y hasta el 45% del repertorio de células T puede estar dedicado a él. Se ha observado que estas expansiones son menos pronunciadas en los centenarios.
Los estudios han sugerido que los marcadores inmunitarios pueden predecir la supervivencia en los ancianos. Se ha demostrado que el grado de diversidad del repertorio de células B predice la supervivencia en los ancianos al menos en una población. Aunque se demostró que estas diferencias globales en la diversidad de TCR y BCR predecían los resultados clínicos, estos marcadores carecen de especificidad. Un análisis más profundo de los datos del repertorio puede proporcionar una precisión de predicción significativamente mayor. Por ejemplo, las expansiones que responden a CMV pueden tener un significado diferente a otras expansiones.
El ARN de las células T y B que se encuentra en la sangre periférica se puede obtener de una cohorte longitudinal de pacientes envejecidos cuyas historias clínicas se siguen durante varios años. Los genes TCRa y TCRp y los genes IgH, IgK e IgL pueden amplificarse en cada una de estas cohortes en varios puntos temporales en sus historias clínicas. Los perfiles de los pacientes con supervivencia larga se comparan con los pacientes con supervivencia corta. Primero,
se puede obtener una medida global de la diversidad. Esto incluirá no solo el número de clonotipos diferentes identificados sino también su diversidad. Por ejemplo, ¿el uso del segmento V, D, J es el mismo en los dos grupos o es un grupo más restringido en su uso? Por ejemplo, dos muestras pueden tener el mismo número de clonotipos independientes pero los clonotipos para una de las dos muestras no cubren muchos de los segmentos V. Es lógico esperar que esta muestra sea menos versátil para responder a un nuevo antígeno en comparación con la otra muestra cuyos clonotipos se distribuyen entre todos los segmentos V.
Además de la diversidad global, se determina si los clonotipos expandidos en pacientes que tuvieron una larga supervivencia se pueden distinguir sobre la base de algún parámetro de secuencia en comparación con los clonotipos en pacientes que tuvieron una supervivencia corta. Esta metodología puede complementarse observando los clonotipos que responden a antígenos específicos. Por ejemplo, dada la evidencia disponible, la identificación de los clonotipos sensibles a CMV puede tener poder predictivo. La captura de los clonotipos de células T que son reactivos al CMV en un estudio de descubrimiento se puede hacer desde un grupo de pacientes ancianos y sanos. Se pueden estudiar las secuencias de estos clonotipos para identificar los parámetros que los distinguen de otros clonotipos. Usando este algoritmo predictivo de clonotipos de CMV con la cohorte longitudinal descrita anteriormente, se puede evaluar si agregar esta información puede aumentar la capacidad de predecir el paciente que sobrevive durante largo tiempo y el que no.
Las pruebas de perfil inmunológico pueden combinarse con otros marcadores que ya están en uso para la detección de la salud en la población envejecida para permitir pruebas con mayor sensibilidad y especificidad. Se pueden usar otros identificadores o marcadores moleculares para calcular el algoritmo de carga o para determinar el estado de la enfermedad. Los identificadores moleculares pueden incluir ácidos nucleicos, proteínas, carbohidratos y lípidos, y perfiles de expresión de ácidos nucleicos o proteínas. Los identificadores moleculares pueden ser de origen humano o no humano (por ejemplo, bacteriano). Los identificadores o marcadores pueden determinarse mediante técnicas que incluyen, por ejemplo, hibridación genómica comparativa (CGH), análisis de micromatrices cromosómicas (CMA), perfiles de expresión, micromatrices de ADN, micromatrices de oligonucleótidos de alta densidad, matriz de expresión de ARN de genoma completo, micromatriz de péptidos, ensayo inmunoenzimático (ELISA), secuenciación del genoma, número de copias (CNV), análisis de polimorfismo de nucleótidos pequeños (SNP), inmunohistoquímica, hibridación in situ, hibridación in situ fluorescente (FISH), PCR, Inmunoprecipitación Western, Inmunoprecipitación Southern, SDS-PAGE, electroforesis en gel y inmunoprecipitación Northern.
F. Utilidad del perfil inmunológico en la administración de vacunas
Estos métodos tienen utilidad en la administración de vacunas. El uso de la vacunación ha conducido a una gran reducción en la tasa de infecciones de múltiples organismos. Una enfermedad infecciosa que continúa teniendo un impacto significativo en la salud con más de 30.000 muertes al año en los Estados Unidos es la influenza. La vacunación contra la influenza debe realizarse anualmente, ya que la cepa muta rápidamente. La mayoría de las secuelas severas de la enfermedad ocurren en los ancianos. Desafortunadamente, los ancianos a menudo experimentan una inmunosenescencia que los hace inadecuadamente sensibles a la vacunación.
Para distinguir a los pacientes que responden a la vacunación de los que no lo son, se debe realizar un estudio de descubrimiento. En esta población, se dispone de muestras de sangre antes y después de la vacunación (en uno o más de ellos) para una cohorte de pacientes vacunados contra la influenza con resultado conocido de la influenza (es decir, si luego se los protegió de la infección o no). La secuencia de TCR y/o BCR se puede obtener de estas muestras. Se determinan los clonotipos que se enriquecen después de la vacunación en cada paciente. Los clonotipos enriquecidos en pacientes que respondieron a la vacunación se comparan luego con un conjunto de clonotipos de control (por ejemplo, el resto de los clonotipos en el mismo conjunto de pacientes) para distinguir los clonotipos correlacionados de otros clonotipos. El algoritmo para predecir estos clonotipos se usa luego para predecir la correlación de los clonotipos entre los pacientes que no respondieron a la vacunación. Los pacientes que no respondieron pueden generar el mismo tipo de clonotipos que los que respondieron, pero en niveles más bajos. Alternativamente, podría ser que los no respondedores generen una clase distinta de clonotipos. El número de clonotipos correlacionados identificados en el no respondedor puede distinguir estas dos posibilidades.
La capacidad de respuesta de un individuo a una vacuna se puede monitorizar obteniendo primero una muestra de linfocitos del individuo después de la vacunación a partir de la cual se aíslan los linfocitos reactivos a la vacuna. Dicho aislamiento se lleva a cabo fácilmente para las células B utilizando una purificación de afinidad convencional con material antigénico de la vacuna o en relación con ella, como restos de captura unidos a un soporte sólido tal como perlas magnéticas. El aislamiento de las células T también se puede llevar a cabo con medios convencionales, por ejemplo, Patentes de los Estados Unidos 7,776,562; 7,125,964; 5,635,363; o similar.
Se genera un perfil de clonotipo a partir de la muestra aislada de linfocitos para obtener un conjunto de clonotipos correlacionados. En puntos de tiempo subsiguientes, se obtienen muestras de sangre periférica de los perfiles individuales y se generan los clonotipos. La tasa de cambio de la frecuencia de correlación de los clonotipos en las muestras posteriores se controla para determinar la capacidad de respuesta del individuo a la vacunación. Dicho método para monitorizar la capacidad de respuesta a una vacuna se puede implementar con los siguientes pasos: (a) enriquecer después de una vacuna una muestra de linfocitos de la sangre periférica de un individuo para obtener una muestra de linfocitos sensibles a la vacuna; (b) determinar un perfil de clonotipo a partir de la muestra de linfocitos
sensibles a la vacuna para identificar uno o más clonotipos específicos de pacientes correlacionados con la respuesta de la vacuna; y (c) determinar un nivel de cada uno de los uno o más clonotipos específicos del paciente en un perfil de clonotipo a partir de una muestra de células de sangre periférica obtenidas en uno o más momentos posteriores para controlar la respuesta del individuo a la vacunación. La capacidad de respuesta puede determinarse por el aumento en la cantidad o la frecuencia de uno o más clonotipos específicos del paciente en los perfiles de clonotipo medidos posteriormente.
Con los clonotipos correlacionados identificados, se construye un algoritmo para generar una puntuación para predecir la probabilidad de inmunización. Los datos de los perfiles de los que responden a la vacuna y aquellos que no responden se utilizan para generar este algoritmo. Este algoritmo se puede usar para predecir la probabilidad de inmunización en el siguiente paciente utilizando los clonotipos correlacionados predichos de una muestra obtenida después de la inmunización. La predicción se realiza mediante la aplicación de otro algoritmo que también se ha generado en el estudio de descubrimiento. Opcionalmente, se puede ayudar (o sustituir) con datos de la calibración previa para limitar la búsqueda de correlacionar los clonotipos con aquellos que se enriquecieron después de la inmunización.
Las pruebas de perfil inmunológico se pueden combinar con otros marcadores que ya están en uso para la detección de la respuesta a la vacunación para permitir pruebas con mayor sensibilidad y especificidad. Se pueden usar otros identificadores o marcadores moleculares para calcular el algoritmo de carga o para determinar el estado de la enfermedad. Los identificadores moleculares pueden incluir ácidos nucleicos, proteínas, carbohidratos y lípidos, y perfiles de expresión de ácidos nucleicos o proteínas. Los identificadores moleculares pueden ser de origen humano o no humano (por ejemplo, bacteriano). Los identificadores o marcadores pueden determinarse mediante técnicas que incluyen, por ejemplo, hibridación genómica comparativa (CGH), análisis de micromatrices cromosómicas (CMA), perfiles de expresión, micromatrices de ADN, micromatrices de oligonucleótidos de alta densidad, matriz de expresión de ARN de genoma completo, micromatriz de péptidos, ensayo inmunoenzimático (ELISA), secuenciación del genoma, número de copias (CNV), análisis de polimorfismo de nucleótidos pequeños (SNP), inmunohistoquímica, hibridación in situ, hibridación in situ fluorescente (FISH), PCR, Inmunoprecipitación Western, Inmunoprecipitación Southern, SDS-PAGE, electroforesis en gel y inmunoprecipitación Northern.
G. Utilidad del perfil inmunológico en el control de la hipersensibilidad inmunitaria (alergia)
El sistema inmunitario adaptativo ha evolucionado para responder a los antígenos que están asociados con patógenos. Como en el caso de las enfermedades autoinmunes, el sistema inmunológico a veces puede tener el objetivo equivocado. Mientras que, en las enfermedades autoinmunes, el sistema inmunitario se dirige al antígeno propio, en las reacciones de hipersensibilidad, responde a estímulos inofensivos como los medicamentos, el polvo y los alimentos. La hipersensibilidad es muy común ya que hasta el 50% de la población de los Estados Unidos es alérgica a un estímulo ambiental y es causada por mecanismos. La hipersensibilidad se divide en 4 tipos. La hipersensibilidad tipo I es la hipersensibilidad de tipo inmediato y está mediada por IgE. El tipo II a menudo se debe a la unión del anticuerpo IgG al antígeno asociado a la superficie celular. Por ejemplo, un fármaco inofensivo que se une a la superficie de la célula puede convertir a la célula en un objetivo para la IgG antifármacos en pacientes que tienen estos anticuerpos. El tipo III es causado por la deposición de complejos antígeno-anticuerpo en los tejidos. Esto ocurre, por ejemplo, cuando la cantidad de antígeno es grande, lo que da lugar a pequeños complejos inmunes que no pueden eliminarse de manera eficiente y que, en cambio, se depositan en las paredes de los vasos sanguíneos. La sensibilidad de tipo IV es un tipo de hipersensibilidad retardada mediada por las células T. Los tipos I y IV tienen el mayor impacto en la salud humana.
En la reacción de hipersensibilidad de Tipo I, el paciente se sensibiliza a un antígeno inofensivo (alérgeno) produciendo un anticuerpo IgE contra él. La exposición posterior al alérgeno induce la activación de células de unión a IgE, como los mastocitos y los basófilos. Una vez activadas, estas células causan la reacción alérgica a través de la inducción de un proceso inflamatorio al secretar sustancias químicas almacenadas y sintetizar citoquinas, leucotrienos y prostaglandinas. La dosis y la vía de entrada del alérgeno determinan la magnitud de la reacción alérgica que puede ir desde los síntomas de la rinitis alérgica hasta el colapso circulatorio que pone en peligro la vida en la anafilaxia. A menudo, la reacción aguda de Tipo I es seguida por otra fase tardía que desempeña un papel en muchos de los procesos patológicos resultantes. La fase tardía del reclutamiento de células T auxiliares y otras células inflamatorias es esencialmente una reacción de hipersensibilidad de tipo IV. Algunas reacciones alérgicas de tipo I incluyen rinoconjuntivitis estacional (fiebre del heno), alergia alimentaria, anafilaxia inducida por fármacos, dermatitis atópica (eczema) y asma. Estas son condiciones muy comunes con una prevalencia creciente que causa costes significativos, así como morbilidad y mortalidad. Por ejemplo, el asma es una enfermedad crónica que inflige al 7% de la población de los Estados Unidos y causa aproximadamente 4.000 muertes por año. Algunas de estas enfermedades tienen algunos aspectos relacionados. Por ejemplo, los pacientes con dermatitis atópica tienen un riesgo significativamente mayor detener asma. La alergia a los alimentos puede causar vómitos y diarrea, pero también puede causar anafilaxis en un número significativo de pacientes: 30.000 casos que producen ~200 muertes por año en los Estados Unidos algunos de los mismos alérgenos que activan los mastocitos submucosos en la nariz que causan síntomas de rinitis alérgica también pueden activar los mastocitos en las vías respiratorias inferiores que causan la constricción bronquial, un síntoma típico del asma. Algunas reacciones de hipersensibilidad de tipo IV son dermatitis de contacto (por ejemplo, hiedra venenosa), rinitis crónica, asma crónica y enfermedad celíaca. La enfermedad celíaca es una enfermedad crónica causada por una alergia alimentaria no mediada por IgE. Es una enfermedad del intestino delgado causada
por la respuesta alérgica contra el gluten, un componente presente en el trigo y otros alimentos. Más del 95% de los pacientes celíacos tienen un alelo MHC específico de clase II, el HLA-DQ2.
El tratamiento de las reacciones de hipersensibilidad difiere, pero a menudo tenían dos aspectos: el tratamiento agudo y el tratamiento o la prevención crónicos. Algunas de estas afecciones pueden ser potencialmente mortales (anafilaxia y asma aguda) e implican atención médica inmediata. El manejo crónico en general implica tratar de evitar el alérgeno específico. Esto puede ser efectivo cuando el alérgeno se puede identificar claramente (por ejemplo, alergia a las nueces), pero puede ser difícil cuando el alérgeno está presente en el medio ambiente, como el polen o el polvo. Por lo tanto, el tratamiento crónico con medicamentos a menudo se utiliza para algunas de estas enfermedades (por ejemplo, asma y rinitis alérgica). El nivel de efectividad del manejo del tratamiento se prueba en última instancia cuando el paciente se vuelve a exponer al alérgeno(s). Por lo tanto, algunos pacientes pueden estar sujetos a un tratamiento excesivo o insuficiente. Lo ideal sería que estuviera disponible una prueba que evalúe la actividad de la enfermedad y el grado en que el paciente es propenso a desarrollar una respuesta de hipersensibilidad. Tal prueba permitiría adaptar el tratamiento a las necesidades individuales del paciente.
H. Detección de neoplasias linfoides.
Un aspecto de esta invención utilizará tecnologías de secuenciación de próxima generación para evaluar el nivel de reordenamientos específicos de TCR o BCR en cánceres de los linfocitos. Estas tecnologías de secuenciación pueden obtener lecturas de secuencia de 1 millón o más de moléculas de TCR o BCR aisladas individualmente a un coste razonable. Una secuencia presente en 1/1.000,000 o inferior aún se puede detectar de una manera específica utilizando estas tecnologías, lo que permite que una célula cancerosa asociada con un reordenamiento de TCR o BCR particular sea detectada a ese nivel. La amplificación multiplex para amplificar todos los diferentes tipos de secuencias para un gen específico puede realizarse a partir de la sangre o el ADN de la médula ósea. Por ejemplo, para amplificar secuencias de IgH, se pueden usar varios cebadores complementarios de todos los segmentos V y alelos conocidos, junto con varios cebadores complementarios de todos los segmentos J y alelos. Es importante que ocurra poco sesgo de amplificación entre las diferentes secuencias. Se ha demostrado que se puede amplificar a partir del ARN los genes TCRp e IgH con solo pequeñas diferencias en las eficiencias de los diferentes cebadores V, y así validar la posibilidad de hacer lo mismo con el ADN que permitirá la evaluación de las células cancerosas incluso cuando el TCR o BCR no se está expresando.
La sensibilidad de esta invención está determinada por las estadísticas de recuento de las moléculas individuales aisladas espacialmente teniendo en cuenta cualquier sesgo en la amplificación de diferentes clonotipos. Por lo tanto, se anticipa que esta metodología ofrecerá más sensibilidad y estará menos sujeto a diferencias de sensibilidad para diferentes secuencias de TCR o BCR de células tumorales en comparación con los ensayos de PCR en tiempo real desarrollados para el mismo propósito. Además, para obtener una mayor sensibilidad, simplemente se pueden obtener más lecturas de secuenciación. Dado que los costes de secuenciación continúan bajando, anticipamos que la sensibilidad a un coste determinado continuará mejorando. Con suficientes lecturas de secuenciación, la sensibilidad se ve limitada por el número de linfocitos en la muestra. En contraste, la sensibilidad para el ensayo de PCR en tiempo real está limitada por los antecedentes causados por amplificaciones no específicas e hibridaciones de cualquier sonda que pueden ser sustanciales.
Con el fin de utilizar esta invención para controlar los cánceres linfoides, se pueden determinar los clones específicos de un paciente mediante la secuenciación de una muestra diagnóstica de leucemia o linfoma; es decir, los clonotipos específicos del paciente se determinan generando un perfil de clonotipo a partir de una muestra de un tejido relacionado con la enfermedad, donde los linfocitos sensibles a la enfermedad se encuentran en un estado enriquecido. Una vez que se determina el perfil de clonotipo, los niveles de los clonotipos relacionados con la enfermedad se pueden determinar por comparación con un perfil de clonotipo de una muestra de un tejido que no está asociado con la enfermedad. Posteriormente, los niveles de los clonotipos relacionados con la enfermedad se determinan en los perfiles de los clonotipos a partir de muestras de ese paciente en puntos de tiempo posteriores en el curso de la enfermedad. Preferiblemente, tales muestras posteriores se toman de tejidos a los que se accede convenientemente, tales como sangre periférica. Se pueden usar las células en la muestra de sangre y, alternativamente, se puede usar ADN o ARN del plasma libre de células. No hay ningún requisito para que las sondas específicas de un paciente o la utilización de plantillas específicas de un paciente se ejecuten como estándares, como se exige en la tecnología actual. En esta realización de la invención, los clonotipos específicos de pacientes que se identifican pueden seguirse obteniendo repertorios de secuencia completos y midiendo de manera informativa los clonotipos correlacionados relevantes basándose en la coincidencia de las secuencias obtenidas con los datos almacenados con respecto a las secuencias relevantes para cada paciente. Los tejidos relacionados con enfermedades para los linfomas pueden incluir tejidos linfoides, médula ósea, sangre periférica y similares.
Identificación de clonotipos de cáncer. Para utilizar los métodos de secuenciación para controlar el cáncer, es fundamental definir los clonotipos de cáncer en cada individuo. Para pruebas secundarias (aplicación de recurrencia y pronóstico para pacientes diagnosticados con neoplasias linfoides), la identificación del clonotipo de cáncer a menudo puede ser bastante sencilla. Por ejemplo, las muestras de sangre o de médula ósea de pacientes con leucemia en el momento del diagnóstico exhiben el clonotipo de cáncer como el clonotipo más frecuente en la muestra. En otros casos de pruebas secundarias (por ejemplo, biopsia de algunas muestras de linfoma), el clon del cáncer puede no estar presente en niveles muy altos. Otros clonotipos que son reactivos a una variedad de antígenos,
incluidos los que están atacando el cáncer, pueden tener tumores, y se encuentran con mayor frecuencia. Si el nivel del clonotipo no es suficiente por sí mismo para determinar el clonotipo del cáncer, se pueden utilizar otros criterios. Se pueden usar varios métodos descritos a continuación para identificar los clonotipos de cáncer.
Reorganización del linaje cruzado. Algunos tipos de reordenamientos poco comunes son comunes en algunos tipos de cáncer y, por lo tanto, se pueden usar para asociarlos con un tumor. Por ejemplo, los reordenamientos de linaje cruzado, como el receptor de células T (a, p, y, y/o5) en las células B o el receptor de células B (IgH, IgK, y/o IgL) en las células T son comunes, especialmente en a Ll . La presencia de reordenamientos de linajes cruzados es probable que apoye un origen maligno del clonotipo.
Secuenciación de reordenamiento de linaje cruzado usando secuenciación de tipos celulares específicos. Alternativamente, las células de un tipo pueden aislarse mediante métodos estándar (por ejemplo, perlas magnéticas y FACS) que utilizan la presencia de ciertos antígenos en la superficie de las células. Se puede hacer una secuenciación para evaluar la presencia de un reordenamiento de linaje cruzado. Por ejemplo, se pueden aislar células B y se puede realizar la secuenciación de TRCp. La presencia de una secuencia de TCRp específica enriquecida sería consistente con el cáncer. La secuenciación puede realizarse antes y después del enriquecimiento, lo que permite la determinación del nivel sin enriquecimiento y el grado de enriquecimiento.
Receptor inmune inactivo. Otra característica que puede ser útil para distinguir las células malignas de otras es que las células no malignas necesitan tener un receptor de células inmunitarias activo. Los linfocitos proliferan en reacción a los antígenos y pueden alcanzar niveles altos. Por lo tanto, alcanzar niveles altos para un clonotipo normal (no canceroso) depende de un receptor inmune activo. La identificación en una biopsia de una secuencia no funcional de alto nivel no es suficiente para identificar el cáncer, ya que se pueden encontrar reordenamientos no funcionales en la misma célula que en los funcionales debido a reordenamientos no funcionales del segundo alelo dentro de una célula que contiene un reordenamiento funcional. El uso de ARN puede permitir la desambiguación de este punto, ya que las células tumorales pueden continuar expresando reordenamientos no funcionales, mientras que es poco probable que esto ocurra en células normales, pero en general son útiles métodos más definitivos. Hay características adicionales que pueden distinguir las secuencias no funcionales en el cáncer. Por ejemplo, algunos de los cánceres inmaduros, por ejemplo, ALL, a menudo tienen solo la IgH reorganizada pero no IgK o IgL. No es probable que este patrón logre alta frecuencia en ausencia de cáncer. Estos reordenamientos no funcionales pueden evaluarse utilizando las siguientes técnicas.
Enlace estadístico en una serie de muestras diluidas. Una alternativa al enlace físico es buscar el enlace de mapeo como se describe anteriormente. En este caso, solo se está probando un gen (por ejemplo, IgH) y la pregunta es cuáles son las secuencias no funcionales vinculadas, es decir, cuál es la secuencia del otro alelo de IgH en la misma célula. Los alelos no funcionales de alta frecuencia vinculados a un segundo alelo no funcional son consistentes con el logro de alta frecuencia debido al cáncer.
Secuenciación de un tipo de célula específico. Este patrón también puede identificarse capturando células que llevan un marcador y evaluando la secuencia del otro. Por ejemplo, uno puede usar FCM para capturar células que son IgK e IgL negativas. La secuenciación de IgH antes y después del enriquecimiento de FCM puede identificar clones enriquecidos en esta población. No se espera que las células que son IgK e IgL negativas alcancen una alta frecuencia y su presencia es consistente con un cáncer inmaduro, como ALL.
Inactivación de las hipermutaciones somáticas. Se puede encontrar un patrón alternativo en los linfomas de células B cuyos clonotipos se han sometido a hipermutaciones somáticas. Algunos de estos clones pueden tener una variabilidad interclonal, en la cual las células cancerosas comprenden varios clones con diferentes mutaciones dentro de ellos. Algunos de los clones resultantes pueden tener mutaciones inactivadoras. Para una hipermutación somática normal dirigida por un antígeno, es improbable que un clon con una hipermutación somática inactivadora sea seleccionado y expandido. La presencia de tales clones es consistente con el cáncer.
Secuenciación de fracciones de células portadoras de marcadores de cáncer. En los casos en que se conocen marcadores de cáncer, se puede realizar una FCM para enriquecer las células cancerosas con este marcador. La secuenciación del repertorio de receptores inmunes se puede realizar antes y después del enriquecimiento. Los clonotipos que se enriquecen son probablemente asociados con el clonotipo de cáncer. Por ejemplo, las células de linfoma pueden enriquecerse utilizando FACS para aislar células que llevan los marcadores particulares (más convenientemente marcadores de superficie) relevantes para el tumor. La secuenciación de BCR antes y después del enriquecimiento identificaría fácilmente el clonotipo enriquecido y, por extensión, el clonotipo de cáncer.
Alternativamente, la asociación de un marcador puede evaluarse a nivel de ADN o ARN. Esto se puede lograr por varios medios, incluyendo la PCR vinculada o la asociación estadística con células diluidas en serie como se describe anteriormente. La cuantificación del enlace de estos marcadores permitirá un mejor rendimiento del ensayo, ya que muchos marcadores están sobreexpresados en las células cancerosas, pero aún están presentes en algún nivel en las células normales. Para tener en cuenta esto, la PCR vinculada se puede hacer utilizando tres genes: el receptor inmune, el marcador del cáncer y el gen de control. El gen del receptor inmunitario puede vincularse con cualquiera de los otros dos genes y la fracción de moléculas enlazadas que es el resultado de un enlace entre la reorganización del receptor y el marcador de cáncer puede indicar el nivel de expresión de este marcador de cáncer.
Detección de translocaciones. Además de servir como marcador de células que se han vuelto cancerosas, la IgH es a menudo uno de los dos socios patológicos de translocación en las neoplasias linfoides. Un ejemplo es la t(11:14) que coloca el segmento J de IgH cerca del gen de la ciclina D 1 (CCND1), lo que resulta en su sobreexpresión. Este reordenamiento que se conoce como BCL1-IgH ocurre en hasta un 60-70% del linfoma de células del manto, así como en otras neoplasias linfoides (por ejemplo, 20% de mieloma múltiple). Otro ejemplo es t(14:18) que coloca el segmento J de IgH cerca de BCL2 dando como resultado su sobreexpresión. Esta reorganización se produce en hasta el 90% de los linfomas foliculares y en el 20% de los linfomas de células B grandes. Estos reordenamientos se identifican típicamente por citogenética, transferencia de Southern o FISH. La PCR tiene el potencial de identificar un reordenamiento con una sensibilidad y especificidad muy altas, como lo demuestra BCR-ABL para la detección del cromosoma Filadelfia. Se han utilizado diferentes técnicas de PCR para evaluar las translocaciones relacionadas con el linfoma, y la PCR en tiempo real recientemente introducida (por ejemplo, para BCL2-IgH) es probablemente la más avanzada. Hay algunas características de BCL1-IgH y BCL2-IgH que hacen que su detección sea menos sensible y específica que la de BCR-ABL. Primero, a diferencia de BCR-ABL, BCL1-IgH y BCL2-IgH no generan una proteína de fusión, y no hay un evento de empalme que genere una estructura molecular predecible. En cambio, los puntos de interrupción pueden abarcar una gran región. Existen puntos de interrupción comunes que permiten la detección de hasta el 88% de BCL2-IgH mediante una combinación de cebadores y ~40% de BCL1-IgH. Esto hace que se pierdan algunos pacientes que tienen la translocación. En segundo lugar, estos reordenamientos pueden estar presentes en individuos normales que nunca contraerían cáncer. Por ejemplo, la translocación de BLC2-IgH se ha encontrado en el nivel de ~10'5 en una gran fracción de los individuos normales con más de ~4% portando BCL2-IgH a una frecuencia de >1/25K. La frecuencia de BCL2-IgH aumenta con la edad. También se plantea la hipótesis de que diferentes personas pueden tener distintos niveles de translocación "de fondo". Presumiblemente, la presencia de esta translocación en una muestra normal se debe al hecho de que la tumorogénesis es un proceso de varios pasos y que la BCL2-IgH no es suficiente para que surjan tumores. La presencia de este fondo de bajo nivel pone un límite a la sensibilidad de detección.
Se puede secuenciar la amplificación con un conjunto de los cebadores J complementarios a todos los segmentos J y los cebadores complementarios a las regiones aguas arriba de los puntos de interrupción de translocación BCL1 o BCL2. Esto puede generar un método para la detección sensible de estas translocaciones y las células cancerosas en las que aparecen. Primero, la secuenciación profunda de moléculas individuales aisladas (por ejemplo, 100K o 1 millón de lecturas) puede permitir la detección de las secuencias apropiadas de un pequeño número de células En un fondo de amplificaciones de otros loci. Además, el problema de las translocaciones de fondo en individuos normales puede mejorar el problema que sufre la PCR en tiempo real. Hay evidencia de que, al menos en algunos casos, las translocaciones de fondo no son clonales, sino que aparecen repetidamente en el mismo paciente. Mediante la secuenciación, se pueden distinguir los diferentes eventos de translocación para obtener la frecuencia de los eventos de translocación independientes. Dado que el punto de interrupción de las diferentes translocaciones es probable que sean distintos, los eventos de translocación se pueden distinguir entre sí. De forma alternativa o adicional, se puede realizar una PCR de enlace que utiliza la translocación con un gen receptor de células B o T para proporcionar un código de barras único para la translocación. La vinculación también se puede hacer estadísticamente usando un conjunto de muestras de dilución como se describe anteriormente.
La monitorización en serie del nivel de la translocación para detectar puntos en los que aumentan en frecuencia puede ser útil en la detección temprana del cáncer, así como para la detección de recurrencia. En este último caso, el punto de ruptura específico relevante para el paciente puede identificarse a partir de la biopsia de diagnóstico. Este concepto de distinguir las translocaciones por sus puntos de ruptura a través de la secuenciación y, por lo tanto, distinguir el fondo del cáncer puede extenderse a otras translocaciones que involucren IgH (por ejemplo, t(8:14)) o todas las otras translocaciones en neoplasias linfoides u otros cánceres.
Cambiando los niveles de células cancerosas y la probabilidad de cáncer. La mera presencia de secuencias que apuntan a la existencia de células tumorales remanentes puede no predecir por sí sola una recaída clínica. Por ejemplo, se puede lograr un estado estable de nivel tumoral mediante las fuerzas de equilibrio de la proliferación de células tumorales y la respuesta inmune al tumor. Se anticipa que además del nivel absoluto de un clonotipo, su tasa de cambio puede ser informativa para predecir la probabilidad de recaída. Por ejemplo, considere dos pacientes, cada uno con el nivel X de sus respectivos clonotipos de cáncer específicos del paciente. Si el nivel en pruebas previas para uno de los pacientes ha sido consistentemente X y el nivel para el otro paciente en pruebas previas ha sido considerablemente más bajo que la probabilidad de que el segundo paciente desarrolle una recaída podría ser más alto que para el primer paciente.
De manera similar, se pueden usar datos adicionales relacionados con el estado de la célula que contiene el clonotipo relacionado con el cáncer para predecir la probabilidad de recurrencia. Por ejemplo, la presencia de ciertos marcadores (superficiales o no superficiales) puede ser una indicación del estado funcional de la célula y, por lo tanto, de la probabilidad de recurrencia. La secuenciación antes y después de la captura de células con los marcadores relevantes puede determinar la fracción de células con el clonotipo de cáncer que lleva los marcadores relevantes. De forma similar, algunos marcadores relevantes para la probabilidad de recurrencia (por ejemplo, la expresión de algún gen relacionado con el crecimiento celular) se pueden evaluar a nivel de ARN. Esto se puede hacer mediante varios métodos que incluyen la vinculación de la PCR o estadísticamente por dilución celular como se describe anteriormente. Finalmente, es posible que el nivel de ARN específico del receptor inmune en la célula tumoral pueda tener una consecuencia funcional y una asociación con la probabilidad de recurrencia. Este nivel puede evaluarse realizando
una PCR de enlace entre un gen de control 1 que puede vincularse con el reordenamiento del receptor inmune o el gen de control 2. La fracción relativa de los dos productos puede ser indicativa de la cantidad relativa de ARN en la célula. Otro método consiste en comparar el nivel de ARN con el nivel de ADN de la reorganización del receptor inmune. La frecuencia del clonotipo específico del cáncer en el ADN identifica el nivel relativo del clonotipo específico del cáncer. La frecuencia del mismo clonotipo se puede evaluar a partir del ARN, y se puede seguir la frecuencia relativa en el ARN y en el ADN. Un cambio en esta frecuencia relativa puede ser indicativo de un cambio en la probabilidad de recurrencia.
Reacción inmune al cáncer linfoide. Además de monitorizar el clonotipo de cáncer y su posible progenie, también se puede evaluar la respuesta inmune al tumor. Se puede identificar clonotipos que probablemente estén montando una respuesta contra el tumor. Por ejemplo, los clonotipos de células B o T que están enriquecidos en la biopsia de la biopsia diagnóstica de los ganglios linfáticos pueden ser el resultado de una respuesta inmune al tumor. Además, se pueden realizar pruebas funcionales para identificar las células T que interactúan con algún antígeno tumoral. Estos pueden ser antígenos específicos o pueden ser las propias células tumorales. Por ejemplo, la secuenciación del TCR antes y después de la estimulación con antígeno o células tumorales puede identificar las células T relevantes en virtud de su enriquecimiento después de la estimulación. El nivel de estas células T en las muestras de sangre posteriores del paciente puede ser útil para predecir la recaída. Por ejemplo, considere el caso mencionado anteriormente en donde se detectó de manera estable un nivel X específico del clonotipo de cáncer. Este es probablemente el resultado de equilibrar el crecimiento del tumor con la respuesta inmune al tumor. Si en algún momento se reduce la respuesta inmune al tumor, es posible anticipar que el tumor recaerá. La respuesta inmune al tumor se puede cuantificar a través de la determinación del nivel de los clonotipos de células T que se ha determinado que son capaces de atacar el tumor.
Integración de la secuenciación con otros marcadores celulares. La detección del clonotipo específico del cáncer se puede realizar a través de la secuenciación de los reordenamientos de los receptores inmunitarios como se describió anteriormente. La presencia de marcadores (superficiales o no superficiales) que son relevantes para las células cancerosas se puede usar para capturar subpoblaciones de células que se secuencian posteriormente. La combinación de usar la captura y secuenciación específica del marcador antes y después de la captura puede proporcionar información adicional. Primero, esto puede usarse para identificar los clonotipos que están enriquecidos y, por lo tanto, es probable que sean los clonotipos específicos del cáncer como se describió anteriormente. Además, las células de secuenciación que tienen marcadores específicos del cáncer pueden llevar a sensibilidades más altas. Con un marcador perfecto, solo se necesitan hacer pocas lecturas para detectar clonotipos específicos de cáncer en millones de otras células. La mayoría de los marcadores no funcionan perfectamente y se genera un fondo significativo (es decir, células no cancerosas) por su captura. Sin embargo, el enriquecimiento de las células cancerosas por estos marcadores puede llevar a una sensibilidad equivalente con menos lecturas de secuenciación o a una mejor sensibilidad que hacer la misma cantidad de lecturas de secuenciación sin enriquecimiento. Por ejemplo, con 1 millón de lecturas de secuencia, se pueden evaluar aproximadamente 1 millón de células que se capturan con un marcador específico para el cáncer. Esto corresponde a más células que estaban presentes antes de la captura y, por lo tanto, mejor sensibilidad. Finalmente, el uso de marcadores puede proporcionar aspectos funcionales relacionados con la biología y el pronóstico del tumor. Puede haber algún nivel de células tumorales en la sangre de diferentes muestras, pero el marcadorfuncional en las células puede distinguirlas muestras que indican una alta probabilidad de recurrencia de aquellas que predicen una baja probabilidad de recurrencia. Por ejemplo, las muestras pueden secuenciarse antes y después de la captura con un marcador relevante y se puede evaluar el porcentaje de las secuencias de clonotipo con el marcador específico. Se puede predecir que dos muestras con el mismo nivel total de clonotipos específicos de cáncer, pero diferentes fracciones de las células que llevan el marcador relevante, tienen diferentes probabilidades de recurrencia.
Evolución de los clones y detección de clones filogénicos (clanes) y otras mutaciones relacionadas con el cáncer
Como se mencionó anteriormente, los métodos de la invención monitorean un nivel de un clan de clonotipos en lugar de un clonotipo individual. Esto se debe a los fenómenos de la evolución clonal, por, ejemplo Campbell et al., Proc. Natl Acad Sci., 105: 13081-13086 (2008); Gerlinger et al., Br. J. Cancer, 103: 1139-1143 (2010). La secuencia de un clon que está presente en la muestra de diagnóstico puede no ser exactamente igual a la de una muestra posterior, como la que se toma en una recaída de la enfermedad. Por lo tanto, si uno sigue la secuencia de clonotipo exacta que coincide con la secuencia de muestra de diagnóstico, la detección de una recaída podría fallar. Dichos clones evolucionados se detectan e identifican fácilmente mediante secuenciación. Por ejemplo, muchos de los clones evolucionados emergen por reemplazo de la región V (llamado reemplazo VH). Estos tipos de clones evolucionados se pierden con las técnicas de p Cr en tiempo real, ya que los cebadores se dirigen al segmento V incorrecto. Sin embargo, dado que la unión D-J permanece intacta en el clon evolucionado, puede detectarse e identificarse en esta invención utilizando la secuenciación de moléculas individuales aisladas espacialmente. Además, la presencia de estos clonotipos relacionados a una frecuencia apreciable en la muestra de diagnóstico aumenta la probabilidad de la relevancia del clonotipo. De manera similar, el desarrollo de hipermutaciones somáticas en la secuencia del receptor inmune puede interferir con la detección de la sonda de PCR en tiempo real, pero los algoritmos apropiados aplicados a la lectura de secuenciación (como se describió anteriormente) aún pueden reconocer un clonotipo como un clonotipo en evolución. Por ejemplo, se pueden reconocer las hipermutaciones somáticas en los segmentos V o J. Esto se hace mediante el mapeo de los clonotipos a las secuencias V y J de la línea germinal más cercanas. Las diferencias de las secuencias de la línea germinal se pueden atribuir a hipermutaciones somáticas. Por lo tanto, los clonotipos que
evolucionan a través de hipermutaciones somáticas en los segmentos V o J pueden detectarse e identificarse fácilmente. Pueden predecirse hipermutaciones somáticas en la región NDN. Cuando el segmento D restante es lo suficientemente largo como para ser reconocido y mapeado, cualquier mutación somática en él puede ser fácilmente reconocida. Las hipermutaciones somáticas en las bases N+P (o en el segmento D que no es asignable) no pueden reconocerse con certeza ya que estas secuencias pueden modificarse en células recién recombinadas que pueden no ser progenie del clonotipo canceroso. Sin embargo, los algoritmos se construyen fácilmente para identificar cambios en la base que tienen una alta probabilidad de ser debidos a una mutación somática. Por ejemplo, un clonotipo con los mismos segmentos V y J y una diferencia de 1 base en la región NDN del clon(es) original(es) tiene una alta probabilidad de ser el resultado de una recombinación somática. Esta probabilidad puede aumentarse si hay otras hipermutaciones somáticas en los segmentos V y J porque esto identifica este clonotipo específico como uno que ha sido objeto de hipermutación somática. Por lo tanto, la probabilidad de que un clonotipo sea el resultado de una hipermutación somática de un clonotipo original se puede calcular utilizando varios parámetros: el número de diferencias en la región NDN, la longitud de la región n Dn , así como la presencia de otras hipermutaciones somáticas en la V y/o segmentos J.
Los datos de evolución clonal pueden ser informativos. Por ejemplo, si el clon principal es un clon evolucionado (uno que estuvo ausente previamente y, por lo tanto, no se registró previamente), esto es una indicación de que el tumor ha adquirido nuevos cambios genéticos con posibles ventajas selectivas. Esto no quiere decir que los cambios específicos en el receptor de las células inmunitarias sean la causa de la ventaja selectiva, sino que pueden representar un marcador para ello. Los tumores cuyos clonotipos han evolucionado pueden asociarse potencialmente con un pronóstico diferencial. En un aspecto de la invención, un clonotipo o clonotipos que se utilizan como biomarcadores específicos de un paciente de una enfermedad, como una neoplasia linfoide, por ejemplo, una leucemia, incluye clonotipos no registrados previamente que son mutantes somáticos del clonotipo o clonotipos que se están monitoreando. En otro aspecto, siempre que cualquier clonotipo no registrado previamente sea al menos el noventa por ciento homólogo a un clonotipo existente o grupo de clonotipos que sirven como biomarcadores específicos del paciente, dicho clonotipo homólogo se incluye con o en el grupo de clonotipos que se está monitoreando en el futuro. Es decir, si se identifican uno o más clonotipos específicos de pacientes en una neoplasia linfoide y se usan para monitorizar periódicamente la enfermedad (por ejemplo, al realizar mediciones en muestras de sangre adquiridas de manera menos invasiva) y si en el curso de una de esas mediciones se produce una nueva (previamente no registrado) se detecta un clonotipo que es una mutación somática de un clonotipo del conjunto actual, luego se agrega al conjunto de clonotipos específicos del paciente que se monitorean para mediciones posteriores. En una realización, si dicho clonotipo no registrado previamente es al menos el noventa por ciento homólogo con un miembro del conjunto actual, entonces se agrega al conjunto específico de biomarcadores de clonotipo para la siguiente prueba realizada en el paciente; es decir, dicho clonotipo no registrado previamente se incluye en el clan del miembro del conjunto actual de clonotipos del cual se derivó (basado en el análisis anterior de los datos del clonotipo). En otra realización, tal inclusión se lleva a cabo si el clonotipo no registrado previamente es al menos un noventa y cinco por ciento homólogo con un miembro del conjunto actual. En otra realización, tal inclusión se lleva a cabo si el clonotipo no registrado previamente es al menos el noventa y ocho por ciento homólogo con un miembro del conjunto actual.
También es posible que una célula evolucione a través de un proceso que reemplaza la región NDN pero preserva los segmentos V y V junto con sus mutaciones acumuladas. Dichas células pueden identificarse como clonotipos de cáncer no grabados previamente mediante la identificación del segmento V y J común, siempre y cuando contengan un número suficiente de mutaciones para que la probabilidad de que estas mutaciones sean derivadas independientemente sea pequeña. Una restricción adicional puede ser que la región NDN sea de tamaño similar al clon previamente secuenciado.
Evaluación de un número suficiente de células.
La sensibilidad del ensayo está limitada por el número de células que generan la plantilla de ácido nucleico que se usa en la reacción de amplificación. Por lo general, ~6pg de ADN está presente en cada célula. Por lo tanto, para tener una sensibilidad de 1/1000.000, se necesitan ~6 |jg de ADN. Sin embargo, en la sangre periférica solo una fracción de las células son células B y, por lo tanto, ~6 jg de ADN de la sangre periférica pueden tener solo -100.000 células B. Para obtener una mayor sensibilidad se pueden usar mayores cantidades de ADN. Un problema puede ser que a medida que se usa más ADN, el efecto de los inhibidores purificados con el ADN puede ser más profundo y se puede observar una variación de muestra a muestra. Obtener una población más pura de células puede mejorar este problema. La generación de células mononucleares de sangre periférica (PBMC) se realiza con frecuencia en entornos clínicos. - 6 jg de ADN de PBMC puede tener -250.000-300.000 células B. La captura de células B específicamente también se puede hacer para obtener más células B por jg de ADN utilizado.
Se pueden seguir más de un reordenamiento de los receptores inmunes para maximizar la sensibilidad y mejorar el problema de la evolución clonal. Por lo tanto, si se siguen 3 reordenamientos, la división de las celdas disponibles disminuirá la sensibilidad en el análisis de cada reordenamiento. Por lo tanto, la amplificación del ADN (o ARN) de manera que los 3 loci de los reordenamientos se amplifiquen antes de que la división mejore este problema. Las metodologías de amplificación del genoma completo se han empleado anteriormente y se pueden usar aquí para lograr la amplificación de los tres loci antes de la división. Alternativamente, la amplificación para los loci específicos en una reacción se puede emplear para lograr la misma tarea. En este caso, una división posterior para amplificar
cada reorganización por separado es opcional. La amplificación del genoma completo antes de la amplificación del reordenamiento del receptor inmune específico también puede ser útil cuando se emplea para evaluar solo un reordenamiento del receptor inmune. Por ejemplo, la evaluación de la IgH a menudo se complica con hipermutaciones somáticas, lo que hace que el uso de múltiples conjuntos de cebadores a menudo sea deseable. En este caso, la amplificación del genoma completo antes de la división del ácido nucleico de entrada entre diferentes reacciones con diferentes conjuntos de cebadores no conduce necesariamente a una sensibilidad mejorada para detectar el clonotipo específico del cáncer. En este caso, las diferentes reacciones (por ejemplo, 3) evalúan el repertorio completo del ácido nucleico de entrada disponible y, por lo tanto, no habría ninguna ventaja de la amplificación del genoma completo (aparte de preservar el ADN para otra interrogación). Sin embargo, esto no es cierto cuando se produce hipermutación somática en la secuencia complementaria a un cebador. Por ejemplo, si solo hay una molécula de ADN que representa el clonotipo específico del cáncer, iría a una de las tres reacciones. Una hipermutación somática impide que este clonotipo específico se amplifique. Por otro lado, la amplificación del genoma completo protegería contra eso, ya que la molécula única inicial de secuencia específica del cáncer se amplifica y, por lo tanto, estaría presente en los 3 tubos. Entonces, aunque la frecuencia del clonotipo específico del cáncer no aumenta en la plantilla de entrada para la amplificación del locus, el hecho de que esté presente en los 3 tubos es una ventaja. En lugar de la amplificación del genoma completo, se puede realizar una amplificación específica utilizando metodologías como la PCR de largo alcance o el uso de cebadores de los tres conjuntos de cebadores o la amplificación preliminar.
Detección de neoplasias linfoides. Los métodos anteriores se aplican al seguimiento de los pacientes después del diagnóstico inicial; sin embargo, esta invención se aplica también al cribado del cáncer. La detección del cáncer primario ha sido una fuerza importante para reducir la mortalidad. La detección temprana de neoplasias linfoides puede conducir a una gran mejora en las tasas de supervivencia de estos cánceres. Se ha demostrado, al menos en la leucemia linfocítica aguda y crónica, que los clonotipos específicos de cáncer pueden detectarse años antes del diagnóstico definitivo. Es posible que los clonotipos de linfoma también se puedan detectar antes de lo que es posible utilizando las metodologías de diagnóstico actuales. La detección de los clonotipos de cáncer con fines de detección (es decir, antes de que se produzca un tumor primario) se puede hacer usando esta invención mediante la secuenciación de los receptores de células inmunitarias como se describe anteriormente. Es probable que el clonotipo de cáncer en cada paciente sea único, y está claro que no se sabe a priori la secuencia que se debe examinar en cada paciente. Sin embargo, muchos de los métodos mencionados anteriormente se pueden usar en la sangre de un paciente que aún no ha sido diagnosticado con cáncer para identificar los clonotipos que probablemente estén asociados con el cáncer y sus niveles y los cambios en estos niveles se pueden usar para evaluar el riesgo de que un paciente desarrolle cáncer clínico.
Tipos de neoplasias linfoides. Los métodos de la invención proporcionada se pueden usar para controlar neoplasias linfoides, por ejemplo, linfoma o leucemia. Las neoplasias de células B maduras pueden incluir, por ejemplo, leucemia linfocítica crónica/linfoma linfocítico pequeño, leucemia prolinfocítica de células B, linfoma linfoplasmocítico (como la macroglobulinemia de Waldenstrom), linfoma esplénico de la zona marginal, neoplasias de células plasmáticas (mieloma de células plasmáticas, plasmacitoma, enfermedades de deposición de inmunoglobulinas monoclonales y enfermedades de cadena pesada), linfoma extranodal de células de la zona B marginal (linfoma MALT), linfoma nodular de la zona marginal de células B (NMZL), linfoma folicular, linfoma de células del manto, linfoma difuso de células grandes B, linfoma mediastínico (tímico) de células B grandes, linfoma intravascular de células grandes B, linfoma de derrame primario y linfoma/leucemia de Burkitt.
Las neoplasias de células T maduras pueden incluir, por ejemplo, leucemia prolinfocítica de células T, leucemia linfocítica granular de células T grandes, leucemia/linfoma de células T adultas, linfoma de células T extranodal (tipo nasal), linfoma de células T de tipo enteropatía, linfoma de células T hepatosplénica, micosis fungoides/síndrome de Sezary, trastornos linfoproliferativos primarios cutáneos de células T c D30 positivas, linfoma anaplásico cutáneo de células grandes, papulosis linfomatoide, linfoma de células T angioinmunoblásticas, linfoma periférico de células T (no especificado) o linfoma anaplásico de células grandes.
Los métodos de la invención proporcionada se pueden usar para controlar la leucemia aguda o la leucemia crónica. La leucemia puede ser leucemia linfoblástica aguda (LLA) (por ejemplo, leucemia linfoblástica aguda precursora B, leucemia linfoblástica aguda precursora T, leucemia de Burkitt y leucemia bifenotípica aguda); leucemia linfocítica crónica (CLL) (por ejemplo, leucemia prolinfocítica de células B); leucemia mielógena aguda (LMA) (por ejemplo, leucemia promielocítica aguda, leucemia mieloblástica aguda y leucemia megacarioblástica aguda); leucemia mielógena crónica (CML) (por ejemplo, leucemia monocítica crónica); leucemia de células pilosas; leucemia prolinfocítica de células T (T-PLL); o leucemia linfocítica granular de gran tamaño.
I. Cribado del cáncer
Otro ejemplo de una situación en la que se pueden usar perfiles de células T y/o B específicas de antígeno para informar situaciones clínicas es el uso de autoantígenos de cáncer específicos en el cribado del cáncer. Las células cancerosas a menudo producen moléculas anormales que son antigénicas y provocan una respuesta inmune. Dichas moléculas se pueden detectar directamente en la sangre o en los fluidos corporales para evaluar la probabilidad de que un paciente desarrolle cáncer. La sensibilidad de estos métodos ha demostrado ser un factor limitante, sin embargo, ya que estos antígenos se encuentran en concentraciones muy pequeñas en la sangre u otros fluidos corporales cuando los cánceres se encuentran en un estadio temprano, cuando es más probable que sean tratables
y asintomáticos. Sin embargo, estas moléculas antigénicas pueden provocar una respuesta inmune que, aunque puede ser insuficiente para controlar el crecimiento del tumor, puede tener la fuerza suficiente para poder medirse como una forma de detectar cáncer temprano. Se han detectado anticuerpos contra antígenos específicos para, por ejemplo, células de cáncer de pulmón y de mama, y se pueden usar como un medio para detectar estos cánceres utilizando los propios antígenos para capturar y detectar los anticuerpos (M. Nesterova et al., Biochimica et Biophysica Acta 2006: 1762: 398-403). Como se describió anteriormente, estos ensayos carecen de la capacidad de diferenciar entre diferentes clones de anticuerpos y omiten cualquier posible respuesta inmune de las células T a los antígenos tumorales. Los métodos descritos anteriormente se pueden usar para enriquecer tanto las células T como las células B que expresan TCR o BCR que se unen a antígenos que se sabe que existen en las células cancerosas. Usando los métodos descritos en el presente documento, se pueden generar frecuencias de clonotipo antes y después del enriquecimiento de T/BCR específico del antígeno para identificar células T o B que probablemente formen parte de una respuesta inmune a estos antígenos en un individuo en particular.
Hay varias formas en que este método podría implementarse clínicamente. En primer lugar, la muestra de sangre de un individuo cuyo riesgo de desarrollar un tumor peligroso se evalúa y se analiza en busca de perfiles completos de clonotipo de células T y B antes y después del enriquecimiento de las células que se unen a un conjunto específico de antígenos del cáncer. Estos antígenos podrían ser una sola especie de antígeno, un conjunto de antígenos o una mezcla compleja de antígenos o podrían ser la mezcla completa de material de un solo tumor de una mezcla de tumores. Estos antígenos podrían incluir p53, c-myc, NY-ESO-1, BRCA1, BRCA2, HER2, MUC1, CAGE, Sox2, GBU4-5, Annexin1, cox-2. Los clones que probablemente estén asociados con una respuesta inmune a estos antígenos podrían identificarse por un cambio significativo en la frecuencia antes y después del enriquecimiento, además de otros parámetros que incluyen parámetros de secuencia de aminoácidos y ácidos nucleicos, longitud, uso del segmento, etc. el riesgo de desarrollar cáncer podría determinarse en función de un solo punto de tiempo de medición de este tipo o al menos desde un segundo punto de tiempo en donde se usan los cambios en la frecuencia de estos clonotipos para calcular el riesgo de cáncer. El enriquecimiento podría realizarse en un primer momento para identificar los clonotipos correlacionados específicos del antígeno y estos clonotipos correlacionados específicos del antígeno cuantificados en los puntos temporales posteriores en los que no se lleva a cabo el enriquecimiento para calcular una puntuación de riesgo.
Un estudio de población también podría usarse para generar un algoritmo para predecir clonotipos correlacionados a partir del enriquecimiento específico de antígeno. En este estudio de población, se obtiene más de un individuo con riesgos conocidos de cáncer y se identifican los clonotipos específicos de antígeno mediante las técnicas de esta invención. Esta población se utiliza para generar un algoritmo que se puede usar para predecir clonotipos específicos de antígeno que son relevantes para la enfermedad en un nuevo individuo cuyo riesgo de cáncer es desconocido.
J. Reacciones adversas a los medicamentos.
Los beneficios del tratamiento farmacológico a menudo se equilibran con sus efectos adversos. La mayoría de las reacciones adversas a los medicamentos (ADR) son bastante predecibles y dependen de la dosis. Otras ADR son idiosincrásicos, y muchos de estos son causados por mecanismos inmunológicos. La predisposición a varios de estos ADR está asociada a genotipos específicos de HLA. Lo ideal es que los pacientes sepan antes de recibir el medicamento que están predispuestos a tener un ADR. Recientemente, la FDA agregó en la etiqueta de la medicación contra el VIH, Abacavir, una recomendación para probar el alelo HLA-B*5701 antes de iniciar el tratamiento ya que los pacientes con este genotipo están predispuestos a la reacción de hipersensibilidad al medicamento. En los casos en los que no es posible decirle a un paciente que están predispuestos a tener una ADR, es conveniente detectar evidencia de la ADR antes de que aparezca cualquier síntoma mediante análisis de sangre. Hay varios métodos para diagnosticar ADR relacionados con el sistema inmunológico. Existen varios métodos in vivo (pruebas cutáneas, pruebas intradérmicas, pruebas de parches y pruebas de provocación de fármacos) que reproducen la alergia al fármaco al exponer al paciente al fármaco. Los métodos in vitro incluyen evaluar la activación de basófilos, la IgE específica del fármaco y la prueba de estimulación de linfocitos específicos del fármaco. Se utilizan diferentes versiones de las pruebas de activación de linfocitos específicos del fármaco para evaluar diferentes propiedades de la activación de linfocitos. Estos incluyen estimulación de linfocitos, migración de linfocitos, toxicidad de linfocitos y pruebas de transformación de linfocitos. Algunas variantes de las pruebas incluyen la evaluación de marcadores de activación como CD69 o el nivel de citoquina liberado. Generalmente, todos estos métodos se utilizan para diagnosticar a pacientes que ya tenían una alergia y no para predecir la reacción de hipersensibilidad. Problemas adicionales plagan las diferentes técnicas. Por ejemplo, algunas de las pruebas in vivo tienen algún riesgo de respuesta alérgica grave en el paciente. La activación de basófilos carece de la especificidad para el antígeno relevante y la IgE específica del fármaco es relevante solo para aquellos tipos de alergia que involucran a la IgE (por ejemplo, anemia hemolítica y anafilaxia). Por lo tanto, es necesario un método que sea capaz de predecir la rA l antes de la administración del fármaco o antes de que aparezcan los síntomas. La evaluación del repertorio de células T y/o B puede generartal prueba. Algunos de los métodos in vitro se pueden usar para identificarlos clonotipos que interactúan con el medicamento. Por ejemplo, la prueba de estimulación de linfocitos se puede realizar con el medicamento específico para identificar los clonotipos que interactúan con el medicamento.
K. Detección de dañotisular
Se ha demostrado que el uso de marcadores moleculares en sangre y fluidos corporales y/o tejidos puede proporcionar información crítica sobre órganos potencialmente dañados que pueden indicar el diagnóstico de enfermedades e intervenciones terapéuticas. Un ejemplo de dicho marcador es la detección de la proteína troponina en la sangre como parte del diagnóstico de enfermedad cardíaca. La troponina es una molécula que es altamente específica para los tejidos del corazón que está contenida en gran parte dentro de las células en el tejido del corazón y se encuentra en niveles muy bajos en la circulación para individuos con corazones sanos. Sin embargo, cuando se produce una enfermedad cardíaca, la muerte celular y la apoptosis provocan el derrame de esta y otras moléculas en el torrente sanguíneo, donde la detección sensible mediante ensayos ELISA puede revelar niveles elevados de troponina que están claramente asociados con la enfermedad cardíaca.
Este paradigma puede extenderse fácilmente a otros tejidos que podrían sufrir daños similares, pero tales técnicas están limitadas por la capacidad de los investigadores para identificar marcadores que son ambos suficientemente específicos para un tejido dado para proporcionar información relevante para el diagnóstico y suficientemente abundante en las fases iniciales del daño tisular para proporcionar información en un momento clínicamente útil en el curso de una enfermedad que podría causar daño a los órganos.
Se ha demostrado que, si bien los marcadores de superficie presentes en las células humanas no producen reacciones inmunitarias, las moléculas contenidas internamente dentro de las células pueden ser inmunógenas cuando se liberan en el torrente sanguíneo. Al igual que en el caso de la detección de autoantígenos cancerosos, estos autoantígenos relacionados con el daño a los órganos podrían detectarse de manera más sensible indirectamente a través de la detección de células inmunes que son reactivas a estos antígenos de lo que sería posible mediante la detección directa de antígenos del tipo que se realiza en el caso de troponina.
Esto se puede usar para proporcionar una visión diagnóstica de los niveles de daño de órgano en un individuo. Las células T y/o B que son reactivas a antígenos que son específicas de las moléculas encontradas dentro de un tipo particular de tejido humano pueden enriquecerse utilizando los métodos descritos anteriormente. Los cambios en la frecuencia en los clonotipos de células T y/o B antes y después del enriquecimiento pueden usarse para determinar qué clonotipos probablemente reaccionarán a estos antígenos. Este método se puede combinar con algoritmos de secuencia que utilizan parámetros de secuencia para determinar cuáles de estos clonotipos enriquecidos tienen más probabilidades de reaccionar a estos antígenos. Los estudios de población que involucran a más de un individuo con daño en los órganos conocidos cuyos antotipos correlacionan clonotipos asociados se han identificado empíricamente también se pueden usar para informar un algoritmo que se puede usar para refinar estas predicciones mediante la identificación de características de secuencia que a menudo se asocian con estos clonotipos correlacionados en los siguientes individuos.
La correlación del clonotipo específico del antígeno se puede hacer al menos en un primer momento para identificar y predecir los clonotipos correlacionados. La sangre o los fluidos corporales muestreados en los puntos de tiempo subsiguientes se pueden perfilar con o sin enriquecimiento específico de antígeno para medir los niveles de estos clonotipos correlacionados que se pueden usar para generar una puntuación de daño en el órgano que correlaciona el grado de daño a un órgano específico en ese individuo En ese tiempo. Los niveles de estos clonotipos se pueden usar para establecer esta puntuación, al igual que los cambios en estos niveles a lo largo del tiempo.
Los antígenos para tejidos específicos se pueden usar en esta realización. Los tejidos pueden ser: corazón, pulmón, hígado, intestino, páncreas, esófago, estómago, riñón, nervios, testículos, ovario, próstata, timo, placenta, útero, etc. Los antígenos para cada uno de estos tejidos podrían ser un conjunto selecto de productos genéticos que se sabe que se expresan específicamente en estos tejidos. Estos productos genéticos expresados específicamente podrían ser el resultado de observar la expresión génica diferencial entre estos órganos y otros órganos. Los antígenos pueden ser un antígeno único, un conjunto de antígenos o mezclas complejas de materiales hasta e incluyendo material de células completas de los tejidos en cuestión.
L. Identificación de la exposición a antígenos locales
Los métodos descritos pueden usarse para generar una base de datos de perfiles inmunes relacionados con una ubicación geográfica particular basada en antígenos locales. Estos antígenos podrían ser, pero no se limitan a, pólenes locales. Estos antígenos podrían tener un componente estacional. Una vez que los perfiles inmunitarios se generan ubicaciones geográficas, un sujeto tendrá su perfil inmunitario actual en comparación con la base de datos. Dicha comparación se utiliza para determinar si un sujeto ha estado expuesto recientemente a los antígenos locales. En una instancia, este método se usa para probar si un sujeto estaba en una ubicación sospechosa. En otro caso, este método se utiliza para identificar lugares donde es probable que el sujeto haya estado sin una sospecha preconcebida de dónde ha estado el sospechoso. En consecuencia, un método para determinar la exposición de un individuo a uno o más antígenos puede comprender los pasos de (a) determinar un perfil de clonotipos a partir de una muestra de células B y/o células T del individuo, tal muestra que comprende un repertorio de clonotipos en esto; y (b) comparar clonotipos del perfil con clonotipos de una base de datos de clonotipos específicos de antígeno para determinar un nivel de coincidencia de clonotipos, determinando así un nivel de exposición al antígeno, la base de datos de clonotipos específicos de antígeno que incluye sustancialmente todos los clonotipos de TCR humano y/o cadenas de inmunoglobulina específicas para uno o más antígenos. En un caso, el uno o más antígenos consisten en antígenos de un patógeno. En otro caso, el patógeno es un virus. En otro caso, dicho virus es un virus de la influenza, un virus
de la viruela, un virus de la hepatitis C, un coronavirus, un virus del dengue o un lentivirus. En un caso, dicha base de datos de clonotipos específicos de antígeno consiste en clonotipos que consisten en sustancialmente todas las regiones CDR3 de las cadenas TCRp e IgH humanas.
M. Identificación de la exposición a antígenos relacionados con el bioterrorismo.
Los métodos descritos se pueden usar para generar una base de datos de perfiles inmunes relacionados con antígenos que probablemente estén relacionados con la producción de compuestos relacionados con el bioterrorismo. Estos antígenos podrían ser, pero no están limitados a, vectores virales capaces de ser armados. Una vez que se generen los perfiles inmunitarios, el sujeto tendrá su perfil inmunitario actual en comparación con la base de datos. Dicha comparación se usa para determinar si un sujeto ha estado expuesto recientemente a los compuestos relacionados con el bioterrorismo. En un caso, este método se usa para probar si un sujeto estuvo expuesto a un compuesto sospechoso en particular. En otro caso, este método se utiliza para identificar si es probable que el sujeto haya estado expuesto a una lista de compuestos potenciales sin una sospecha preconcebida de qué compuestos se esperan. En una instancia, el perfil inmune que se va a probar se genera después de que se haya encontrado un agente biológico. Por ejemplo, las autoridades identifican a una persona sospechosa de un ataque de bioterrorismo. Se obtiene una muestra del sospechoso y se obtiene un perfil inmunitario utilizando los métodos descritos anteriormente. Este perfil se compara estadísticamente con una base de datos que contiene muchos perfiles de muestra. Los perfiles de muestra incluyen perfiles inmunitarios que representan ciertos antígenos relacionados con bioterrorismo. Los perfiles de muestra incluyen perfiles inmunes que representan ciertos antígenos o combinaciones de antígenos que existen solo en ubicaciones geográficas particulares en determinadas épocas del año. La comparación del perfil inmunológico del sospechoso con esta base de datos proporciona evidencia de que el sospechoso se encontraba en una ubicación geográfica particular durante un período de tiempo particular y que el sospechoso estuvo expuesto a ciertos antígenos relacionados con el bioterrorismo. Esta evidencia se utiliza para dirigir la investigación y durante el procesamiento.
Kits
En la comercialización de los métodos descritos en el presente documento, los kits para la amplificación de regiones específicas reorganizadas somáticamente o porciones de las mismas son particularmente útiles. Dichos kits pueden ser para llevar a cabo una o dos PCR en etapas (como se describió anteriormente) para amplificar una región predeterminada reorganizada somáticamente o una porción de la misma con el fin de preparar una muestra de clonotipos para el análisis de secuencias. Un kit típicamente comprende uno o más reactivos, tales como, sin limitación, cebadores de ácido nucleico, envasados en un recipiente, como, por ejemplo, un vial, tubo o botella, en un paquete adecuado para su distribución comercial, tal como, sin limitación, una caja, una bolsa sellada, un blíster o una caja de cartón.
Normalmente, el paquete contiene una etiqueta o un prospecto de empaque que indica que los reactivos empaquetados se pueden usar en un método para generar un perfil de clonotipo a partir de una muestra de tejido de un paciente. Tal como se usa en el presente documento, "materiales de empaque" incluye cualquier artículo usado en el empaque para la distribución de reactivos en un kit, incluidos, entre otros, recipientes, viales, tubos, botellas, bolsas, envases de blíster, marcas, etiquetas, hojas de instrucciones e insertos para paquetes. Un ejemplo de dicho kit incluye los reactivos necesarios para la amplificación en un tubo de secuencias TCRp de células T extraídas de ADN o a Rn de un paciente o linfocitos de sangre periférica de un paciente o médula ósea de un paciente, como se describe anteriormente. Otro ejemplo de tal kit incluye los reactivos necesarios para la amplificación en una pluralidad de secuencias IgH de tubos de células B extraídas de ADN o ARN de un paciente o linfocitos de sangre periférica de un paciente o médula ósea de un paciente, como se describe anteriormente. En el último ejemplo, los reactivos necesarios incluyen una pluralidad de conjuntos de cebadores para generar conjuntos anidados de plantillas, como se describió anteriormente. Normalmente, dicha pluralidad es 2 o 3 o 4. Para el último ejemplo, en un caso, se proporcionan tres conjuntos de cebadores; y más específicamente, se proporcionan los siguientes tres conjuntos de cebadores: el conjunto 1 comprende los cebadores directos de la Tabla 5 y los cebadores inversos de la Tabla 8; el conjunto 2 comprende cebadores directos de la Tabla 6 y cebadores inversos de la Tabla 8; el conjunto 3 comprende los cebadores directos de la Tabla 7 y los cebadores inversos de la Tabla 8. En otro ejemplo, el kit incluiría los reactivos descritos anteriormente, incluidos uno o más conjuntos de cebadores de PCR y una polimerasa de ADN termoestable, como la polimerasa Taq, y sí las secuencias se amplifican a partir de ARN, una transcriptasa inversa. Los cebadores pueden estar presentes en cantidades que produzcan una amplificación equilibrada de secuencias de clonotipos individuales en una muestra de paciente, como se describió anteriormente. Se pueden proporcionar cantidades de cebadores para garantizar una amplificación equilibrada de los clonotipos. Este equilibrio de PCR multiplex es bien conocido por los expertos en la técnica e incluye, entre otros, ajustar las concentraciones de cebadores en la reacción y/o seleccionar las posiciones y longitudes de los cebadores en una región de interés para aumentar o disminuir la velocidad de fusionamiento de cebadores individuales. En un caso, las cantidades de cebadores se seleccionan de modo que en la PCR sus concentraciones sean tales que la velocidad a la que cada cebador se reasiente con su sitio de unión del cebador sea sustancialmente idéntica. En otro caso, las cantidades de cebadores se seleccionan de modo que cada secuencia en una muestra se amplifique a una cantidad que esté dentro de 2 veces la cantidad promedio de amplificación de una muestra aleatoria de clonotipos. En otro caso más, dicha muestra aleatoria contiene al menos 100 clonotipos.
Las polimerasas y transcriptasas de ADN termoestables están disponibles comercialmente de una variedad de fabricantes. Los materiales adicionales en el kit pueden incluir: tubos de reacción o viales adecuados, una composición de barrera, típicamente una perla de cera, que incluye opcionalmente magnesio; mezclas de reacción (a menudo concentradas, por ejemplo 2X, 5X, 10X o 20X) para las etapas de PCR, incluidos los reguladores y reactivos necesarios, como los dNTP; agua libre de nucleasa o ARNasa; inhibidor de la ARNasa; ácido(s) nucleico(s) de control (es decir, estándares internos) y/o cualquier regulador, compuesto, cofactor, constituyentes iónicos, proteínas y enzimas, polímeros y similares adicionales que puedan usarse en reacciones de PCR multiplex.
Los componentes de un kit se empaquetan de cualquier manera que sea comercialmente practicable. Por ejemplo, los cebadores de PCR y/o la transcriptasa inversa se pueden empaquetar individualmente para facilitar la flexibilidad en la configuración del ensayo, o en conjunto para aumentar la facilidad de uso y reducir la contaminación. De manera similar, los reguladores, sales y cofactores se pueden empaquetar por separado o juntos. Los kits también pueden incluir reactivos y componentes mecánicos adecuados para la extracción manual o automatizada de ácido nucleico de una muestra de tejido. Estos reactivos son conocidos por los expertos en la técnica y típicamente son una cuestión de elección de diseño. Por ejemplo, en una instancia de un proceso automatizado, el tejido se rompe por ultrasonidos en una solución de lisis adecuada provista en el kit.
Ejemplo 1
Determinación del estado de una enfermedad autoinmune.
Se toma una muestra de líquido cefalorraquídeo (LCR) y sangre de un paciente con un pico de episodio de esclerosis múltiple. Las células CD4+ se aíslan del CSF y de la sangre, y la CDR3 del gen del receptor p de las células T se amplifica por PCR. Los fragmentos amplificados se amplifican adicionalmente para agregar sitios de unión del cebador de amplificación puente y sitios de unión del cebador de secuenciación para la secuenciación de Solexa. Las regiones variables del gen del receptor p de células T se secuencian para identificar los clonotipos. La información de secuencia se utiliza para generar un perfil de clonotipo para el paciente.
Se toma otra muestra de sangre cuando el paciente se encuentra en un estado relativamente inactivo de esclerosis múltiple. Se repite el mismo procedimiento anterior para generar un perfil de clonotipo. Los clonotipos patológicos se identifican como aquellos que son altos en el episodio máximo y disminuyeron significativamente en el estado inactivo. Otra muestra de sangre se toma del paciente en un estado posterior. En este momento, solo una fracción de las regiones CDR3 del gen p del receptor p de células T se amplifican y luego se secuencian. Este subconjunto contiene los clonotipos patológicos. El nivel de los diversos clonotipos se determina para evaluar el estado de la enfermedad del paciente.
Ejemplo 2
Análisis de repertorio de TCRp: Estrategia de amplificación y secuenciación
En este ejemplo, se analizan las cadenas de TCRp. El análisis incluye la amplificación, la secuenciación y el análisis de las secuencias de TCRp. Un cebador AGCGa Cc TCGGGTGGGAACA (SEQ ID NO: 1) es complementario a una secuencia común en Cp1 y Cp2, y hay cebadores de 34 V (Tabla 1) capaces de amplificar todos los segmentos de 48 V. Cp1 o Cp2 difieren entre sí en la posición 10 y 14 de la unión J/C. El cebador para Cp1 y Cp2 termina en la posición 16 pb y no tiene preferencia por Cp1 o Cp2.
Los cebadores de 34 V se modifican a partir de un conjunto original de cebadores descrito en Van Dongen et al, Publicación de Patente de Estados Unidos 2006/0234234.
Tabla 1.



El analizador de genoma Illumina se utiliza para secuenciar el amplicón producido por los cebadores anteriores. Se realiza una amplificación de dos etapas en las transcripciones de ARN mensajero (200), como se ilustra en las figuras 2A-2B, la primera etapa utiliza los cebadores anteriores y una Segunda etapa para agregar cebadores comunes para la amplificación y secuenciación de puentes. Como se muestra en la figura 2A, se realiza una PCR primaria utilizando en un lado un cebador de 20 pb (202) cuyo extremo 3' está a 16 bases de la unión J/C (204) y que es perfectamente complementario a Cp1 (203) y los dos alelos de Cp2. En la región V (206) de los transcritos de Ar N (200), se proporciona un conjunto de cebadores (212) que contiene secuencias de cebadores complementarias a las diferentes secuencias de la región V (34 en un caso). Los cebadores del conjunto (212) también contienen una cola no complementaria (214) que produce el amplicón (216) que tiene un sitio de unión del cebador (218) específico para los cebadores P7 (220). Después de una PCR multiplex convencional, se forma un amplicón (216) que contiene la porción altamente diversa de la región J(D)V (206, 208 y 210) de los transcritos de ARNm y los sitios de unión del cebador comunes (203 y 218) para una amplificación secundaria para agregar una etiqueta de muestra (221) y cebadores (220 y 222) para la formación de conglomerados mediante p Cr de puente. En la PCR secundaria, en el mismo lado de la plantilla, se usa un cebador (222 en la figura 2B y denominado en este documento "C10-17-P5") que tiene en su
extremo 3' la secuencia de las 10 bases más cercanas a la unión J/C, seguido de 17 pb con la secuencia de posiciones 15-31 desde la unión J/C, seguido de la secuencia P5 (224), que desempeña un papel en la formación de agrupaciones mediante PCR de puente en la secuenciación de Solexa. (Cuando el cebador C10-17-P5 (222) se acopla a la plantilla generada a partir de la primera PCR, se crea un bucle de 4 pb (posición 11-14) en la plantilla, ya que el cebador se hibrida a la secuencia de las 10 bases más cercanas a la unión J/C y las bases en las posiciones 15-31 desde la unión J/C. El bucle de las posiciones 11-14 elimina la amplificación diferencial de las plantillas que llevan Cp1 o Cp2. La secuenciación se realiza luego con un cebador complementario a la secuencia de 10 bases más cercanas a la unión J/C y bases en las posiciones 15-31 desde la unión J/C (este cebador se llama C'). El cebador C10-17-P5 se puede purificar por HPLC para garantizar que todo el material amplificado tiene extremos intactos que pueden ser utilizados eficientemente en la formación de agrupaciones).
En la figura 2A, la longitud del saliente en los cebadores V (212) es preferiblemente de 14 pb. La PCR primaria se ayuda con un voladizo más corto (214). Alternativamente, por el bien de la PCR secundaria, el saliente en el cebador V se utiliza en la PCR primaria el mayor tiempo posible porque la PCR secundaria se está cebando desde esta secuencia. Se investigó un tamaño mínimo de voladizo (214) que soporta una PCR secundaria eficiente. Se realizaron dos series de cebadores V (para dos segmentos V diferentes) con tamaños de voladizo de 10 a 30 con pasos de 2 pb. Usando las secuencias sintéticas apropiadas, se realizó la primera PCR con cada uno de los cebadores de la serie y se realizó una electroforesis en gel para mostrar que todos los amplificados. Con el fin de medir la eficiencia de la segunda amplificación por PCR, se realizó la PCR en tiempo real SYBR verde utilizando como plantilla los productos de la PCR de las diferentes primeras reacciones de la PCR y como cebadores Read2-tag1-P7 y Read2-tag2-P7. Surgió una imagen consistente utilizando las 4 series de datos en tiempo real (2 PCR primarias con dos segmentos V diferentes y dos PCR secundarias con cebadores diferentes que contienen dos etiquetas diferentes). Hubo una mejora en la eficiencia entre los tamaños de voladizo de 10 y 14 pb. Sin embargo, hubo poca o ninguna mejora en la eficiencia con un saliente de más de 14 pb. La eficiencia se mantuvo alta a medida que el voladizo llegó a sertan pequeño como 14 pb debido a la alta concentración de cebadores, lo que permite que los 14 pb sean una plantilla de cebado suficiente a una temperatura mucho mayor que su temperatura de fusión. Al mismo tiempo, la especificidad se mantuvo porque la plantilla no era todo el ADNc, sino un producto de PCR de baja complejidad en donde todas las moléculas tenían un saliente de 14 pb.
Como se ilustra en la figura 2A, la PCR primaria utiliza 34 cebadores V diferentes (212) que se aparean a la región V (206) de las plantillas de ARN (200) y contienen un saliente común de 14 pb en la cola 5'. El 14 pb es la secuencia parcial de uno de los cebadores de secuenciación de Illumina (denominado cebador de lectura 2). El cebador de amplificación secundario (220) en el mismo lado incluye la secuencia P7, una etiqueta (221) y la secuencia del cebador de Lectura 2 (223) (este cebador se llama Read2_tagX_P7). La secuencia P7 se utiliza para la formación de agrupaciones. El cebador de lectura 2 y su complemento se utilizan para secuenciar el segmento V y la etiqueta, respectivamente. Se crea un conjunto de 96 de estos cebadores con etiquetas numeradas del 1 al 96 (ver más abajo). Estos cebadores se purifican por HPLC para garantizar que todo el material amplificado tenga extremos intactos que se pueden utilizar de manera eficiente en la formación de grupos.
Como se mencionó anteriormente, el cebador de la segunda etapa, C-10-17-P5 (222, figura 2B) ha interrumpido la homología con la plantilla generada en la PCR de la primera etapa. La eficacia de la amplificación utilizando este cebador ha sido validada. Un cebador alternativo al C-10-17-P5, denominado CsegP5, tiene una homología perfecta con el cebador de la primera etapa C y una cola 5' que lleva P5. La eficiencia del uso de C-10-17-P5 y CsegP5 en la amplificación de las plantillas de PCR de la primera etapa se comparó mediante la realización de la PCR en tiempo real. En varias réplicas, se encontró que la PCR que usa el cebador C-10-17-P5 tuvo poca o ninguna diferencia en la eficiencia en comparación con la PCR que usa el cebador CsegP5.
El amplicón (300) resultante de la amplificación de 2 etapas ilustrada en las figuras 2A-2B tiene la estructura utilizada típicamente con el secuenciador Illumina como se muestra en la figura 3A. Dos cebadores que se aparean a la parte más externa de la molécula, los cebadores Illumina P5 (AATGATACGGCGACCACCGAG) (SEQ ID NO: 36) y P7 (CAAGCAGAAGACGGCATACGAGAT) (SEQ ID NO: 37) se utilizan para la amplificación de la fase sólida de la molécula (formación de grupos). Se hacen tres lecturas de secuencia por molécula. La primera lectura de 100 pb se realiza con el cebador C', que tiene una temperatura de fusión adecuada para el proceso de secuenciación de Illumina. La segunda lectura solo tiene 6 pb de longitud y tiene el único propósito de identificar la etiqueta de muestra. Se genera utilizando el cebador de la etiqueta Illumina (AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC) (SEQ ID NO: 38). La lectura final es el cebador de Lectura 2, un cebador de Illumina con la secuencia GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT (SEQ ID NO: 39). Usando este cebador, se genera una lectura de 100 pb en el segmento V a partir de la primera secuencia del cebador V de PCR.
Se diseñó un conjunto de etiquetas de secuencia de 6 pb para distinguir diferentes muestras ejecutadas en el mismo carril de secuenciación, donde cada etiqueta es diferente de todas las demás etiquetas en el conjunto por al menos 2 diferencias. Las 2 diferencias previenen la asignación errónea de una lectura a la muestra incorrecta si hay un error de secuencia. La alineación realizada para comparar las brechas permitidas por las etiquetas y, por lo tanto, un error de eliminación o inserción por secuenciación tampoco asignará la lectura a la muestra incorrecta. Las características adicionales al seleccionar las etiquetas fueron limitar las ejecuciones de base única (4 A o T y 3 G o C), así como no tener similitud con los cebadores de Illumina. En total se generaron 143 etiquetas, se utilizan 96 de ellas.
Secuenciación de TCRp. Se utilizaron seis amplificaciones multiplexadas con los oligos combinados y una muestra de ADNc como plantilla. Tres de cada una de las amplificaciones se realizaron con Accuprime y otras tres con Taq de alta fidelidad. Dos amplificaciones con cada enzima utilizaron ADNc que corresponden a 500 ng de ARN inicial, y una amplificación con cada enzima usó 10 veces menos ADNc. Para cada una de las seis reacciones, se realizó una PCR primaria y secundaria y el material amplificado se secuenció utilizando la plataforma Illumina y el esquema descrito anteriormente. Se obtuvieron secuencias de 100 pb de cada lado. El análisis primario de los datos se realizó utilizando los mismos conceptos que se describen a continuación.
Para evaluar la reproducibilidad del ensayo, se determinó si los niveles de clonotipo son consistentes en los experimentos duplicados. Como se muestra en las figuras 5A-5C, se obtiene una alta correlación cuando se utiliza la misma enzima y la cantidad de ADNc de entrada inicial (cada una de las 2 comparaciones tuvo r2=0.944). Cuando se usaron diferentes enzimas, la correlación empeora (correlación media para las 4 combinaciones posibles r2=0.931), y solo se reduce moderadamente (r2=0.924) cuando las 2 enzimas se usaron para amplificar un ADNc de entrada más pequeño (correspondiente a solo 50 ng ARN).
En las figuras 5A-5C, se identificaron secuencias idénticas en cada muestra. Luego, para lidiar con los errores de secuenciación, algunos clonotipos se fusionaron para formar clonotipos más grandes utilizando las metodologías generales descritas en la sección de análisis primario de secuencia. Los recuentos de los clonotipos se calcularon en cada muestra. Una fracción de los clonotipos (que no se muestra en la figura) estaban presentes en una muestra, pero no en otra, probablemente debido al algoritmo que los unió con otro clonotipo en una muestra, pero no en la otra. La frecuencia de los clonotipos en una muestra se calcula luego como su número de recuentos dividido por el número total de lecturas obtenidas para esa muestra. Por ejemplo, si se observan 1.000 conteos para un clonotipo en una muestra con 1.000.000 lecturas, su frecuencia se calcula como 0.1%. La figura 7A muestra el log-10 de la frecuencia de cada clonotipo en las dos muestras duplicadas utilizando Accuprime y ADNc correspondiente a 500 ng de ARN como plantilla de entrada. La correlación (r2) entre estos duplicados es 0.944. La figura 7B representa el log-10 de la frecuencia de cada clonotipo utilizando un ADNc correspondiente a 500 ng de ARN como plantilla de entrada y Accuprime (eje X) o Taq de alta fidelidad (eje Y). Hay 4 comparaciones con esta combinación con una correlación mediana r2=0.931. El que se muestra en la figura tiene r2=0.929. La figura 7C muestra el log-10 de la frecuencia de cada clonotipo utilizando un ADNc correspondiente a 50 ng de ARN como plantilla de entrada y Accuprime (eje X) o Taq de alta fidelidad (eje Y). La correlación observada r2=0.924.
Ejemplo 3
Análisis del repertorio de IgH: Estrategia de amplificación y secuenciación
En este ejemplo, se usan tres cebadores para amplificar las regiones V de las moléculas de IgH. Preferiblemente, los cebadores están en regiones que evitan las CDR, que tienen la frecuencia más alta de mutaciones somáticas. Se realizan tres reacciones de amplificación diferentes. En cada reacción, cada uno de los segmentos V se amplifica por uno de los tres cebadores y todos usarán los mismos cebadores del segmento C. Los cebadores en cada una de las reacciones separadas son aproximadamente la misma distancia de la articulación V-D y distancias diferentes con respecto a los cebadores en diferentes reacciones, de modo que los cebadores de lastres reacciones están separados a lo largo del segmento V. Suponiendo que la última posición del segmento V es 0, entonces el primer conjunto de cebadores (cuadro A) tiene el extremo 3' en aproximadamente -255, el segundo conjunto (cuadro B) tiene el extremo 3' en aproximadamente -160, y el tercer conjunto (cuadro C) tiene el extremo 3' a aproximadamente -30. Dada la homología entre varios segmentos V, para amplificar todos los segmentos 48V y los muchos alelos conocidos (según lo define el sistema internacional de información ImMunoGeneTics <<http://imgt.cines.fr/>>) 23, 33 y 32 cebadores en los cuadros A, B y C respectivamente, es necesario. La lista de cebadores se muestra en las Tablas 2, 3 y 4.
Tabla 2


Tabla 3 Cebadores de marco B


Tabla 4 Cebadores de marco C


En el lado del segmento C, dos secuencias con una diferencia de base entre ellas (GCCAGGGGGAAGACCGATGG (SEQ ID NO: 128) y GCCAGGGGGAAGACGGATGG) (SEQ ID NO: 129) cubren los cuatro segmentos y los múltiples alelos conocidos de IgG. Se utiliza un esquema similar a las dos etapas de la PCR para los genes TCRp.
En el lado V, se utiliza el mismo saliente de 5' 14 pb en cada uno de los cebadores en V. En la PCR secundaria, se emplea el mismo cebador Read2-tagX-P7 en el lado V. En el lado C se usa una estrategia similar a la utilizada con la amplificación de TCRp para evitar variantes entre los diferentes segmentos de IgG y sus alelos conocidos. La secuencia del cebador (AATGATACGGCGACCACCGAGATCTGGGAAGACGATGGGCCCTTGGTGGA) (SEQ ID NO: 130) comprende la secuencia del segmento C de las posiciones 3-19 y 21-28 y omite la posición 20 que tiene una base diferente en al menos uno de los diferentes alelos IgG y La secuencia para P5 que se puede usar para la formación de los grupos como se muestra en la figura 4A.
Se llevó a cabo una PCR multiplexada utilizando tres grupos de cebadores correspondientes a las tres tramas utilizando ADNc como plantilla. Después de las PCR primarias y secundarias, los productos se ejecutaron en un gel de agarosa. Se obtuvieron bandas individuales con los tamaños relativos apropiados de los tres grupos.
En un caso, tres reacciones diferentes de una sola muestra se mezclan en una proporción equimolary se someten a secuenciación. La secuenciación se realiza desde ambas direcciones utilizando los dos cebadores Illumina, como se describe anteriormente. Se secuencian 100 pb de cada lado. Las secuencias máximas de la línea germinal que abarcan los segmentos D+J son -30 pb más largas para BCR que para TCR. Por lo tanto, si el resultado neto de la eliminación y adición de nucleótidos en las uniones (nucleótidos N y P) genera una distribución similar para IgH y TCRp, en promedio 90 pb y un máximo de 120 pb de secuencia después del segmento C es suficiente para alcanzar el 3' de el segmento V. Por lo tanto, en la mayoría de los casos, la secuencia del cebador C es suficiente para alcanzar el segmento V. La secuenciación de uno de los adaptadores Illumina identifica el segmento V utilizado, así como las hipermutaciones somáticas en los segmentos V. Se secuencian diferentes piezas de los segmentos V dependiendo de cuál de las tres reacciones de amplificación se originó la secuencia. La secuencia completa del BCR puede alinearse a partir de diferentes lecturas que se originaron a partir de diferentes reacciones de amplificación. La reacción de secuenciación desde un extremo que muestra la secuencia completa de CDR3 facilita enormemente la alineación precisa de diferentes lecturas.
Ejemplo 4
Análisis de repertorio de TCR e IgH en muestras de pacientes con SLE
Primero se probará si hay clonotipos que se correlacionan con la actividad de la enfermedad en pacientes. En segundo lugar, se define un conjunto de características de secuencia y/o marcadores de superficie celular que distinguen los clonotipos que se correlacionan con la enfermedad de aquellos que no lo hacen. En tercer lugar, se mide el grado en
que el análisis del clonotipo proporciona información clínicamente útil, como la correlación con el resultado a corto plazo (por ejemplo, 3 meses).
1. Presencia de clonotipos correlacionados con enfermedad
Hay dos tareas principales: identificar clonotipos correlacionados y medir la actividad de la enfermedad desde su nivel. Estas tareas se pueden realizar en un entorno clínico en dos pasos para cada paciente:
1) Se puede realizar una prueba de calibración para determinar la identidad de los clonotipos correlacionados para el paciente específico. Esto puede hacerse secuenciando el ARN de IgH y TCRp (o la secuencia TCRa-TCRp enlazada de una sola célula) para cada paciente en el momento de un pico de un episodio, momento en el cual el nivel de clonotipo correlacionado puede alcanzar sus niveles más altos.
2) Se puede realizar una prueba de monitorización para determinar el nivel de los clonotipos correlacionados en un punto de tiempo posterior a la prueba de calibración. Esto puede hacerse secuenciando el ARN de IgH y TCRp y determinando el nivel de los clonotipos correlacionados específicos que se identificaron en la muestra de calibración del mismo paciente. El nivel de los clonotipos correlacionados se utiliza para calcular la actividad de la enfermedad en estos puntos.
La amplificación, la secuenciación y el desarrollo del análisis primario como se describió anteriormente se utilizan para evaluar las muestras de pacientes. Específicamente, se evalúa un conjunto de pacientes con lupus eritematoso sistémico (SLE) que tienen un período de seguimiento de un año y muestras de sangre en serie durante este período. Estos pacientes fueron atendidos por la Dra. Michele Petri en la Escuela de Medicina Johns Hopkins cada tres meses durante un año, y las medidas clínicas de la actividad de la enfermedad incluyen el Índice de Actividad de la Enfermedad del Lupus Eritematoso Sistémico (SLEDAI), la Evaluación Global del Médico (PGA) así como múltiples pruebas de laboratorio que incluyen C3 (Complemento 3) y niveles de ADN anti-d están disponibles para todas las visitas de todos los pacientes. Los medicamentos que se administran a los pacientes incluyen prednisona, plaquenil, AINE, AINE, dosis de ácido acetilsalicílico (ASA), plavix, diuréticos, inhibidores de la ECAo bloqueadores del receptor de angiotensina (BRA), bloqueador de canales de Ca, Triam y solumedrol. Se estudian pacientes que tuvieron al menos una vez durante el seguimiento, un cambio significativo en la actividad de la enfermedad según lo definido por un cambio de 3 puntos en la SLEDAI o un cambio de 1 punto en la PGA. En general, hay 181 pacientes (con un total de 815 muestras de sangre) que cumplen con estos criterios. El ARN de todas estas muestras de sangre se somete a PCR multiplex utilizando los cebadores descritos anteriormente para amplificar las secuencias que abarcan CDR3 en IgH y TCRp. Todos los materiales amplificados se secuencian (a un millón de lecturas) y se determina la abundancia de diferentes clonotipos.
Usando los datos clínicos, secuenciación, se identifican las características que distinguen los clonotipos cuyo nivel se correlaciona con la actividad de la enfermedad de aquellos que no lo hacen. En segundo lugar, se desarrolla un algoritmo para determinar la actividad de la enfermedad utilizando el perfil de IgH y TCRp en la sangre.
2. Identificación de características de clonotipos correlacionados.
Se anticipa que los clonotipos que son relevantes para la enfermedad aumentan en el momento de una alta actividad de la enfermedad. Sin embargo, no todos los clonotipos enriquecidos en un punto de alta actividad de la enfermedad se correlacionan necesariamente con la enfermedad. Por ejemplo, en un paciente en particular puede haber 10 clonotipos enriquecidos en el punto de alta actividad de la enfermedad, pero solo 5 se correlacionan con la enfermedad. Para identificar estos clonotipos relevantes, se estudia un subconjunto de clonotipos que se correlacionan claramente con la enfermedad y otro conjunto que claramente no se correlaciona con la enfermedad. Se investigan las características que distinguen esas dos clases de clonotipos.
Todos los pacientes tendrán al menos un cambio significativo en la actividad de la enfermedad durante el año de seguimiento en este diseño experimental. Se analizan los clonotipos de IgH y TCR obtenidos en el pico de actividad de la enfermedad en cada paciente. Se seleccionan conjuntos de clonotipos correlacionados y no correlacionados entre aquellos con los clonotipos de nivel más alto. Por lo tanto, el primer paso es definir los clonotipos que se encuentran en un nivel alto. Los criterios específicos para elegir los clonotipos que entrarán en el análisis incluirán una combinación de rango de frecuencia del clonotipo y el nivel de clonotipo (número de lecturas de clonotipo por millón), así como evidencia de que el clonotipo no pertenece a la distribución de clonotipos de baja frecuencia.
Este conjunto de clonotipos de cada muestra de paciente, denominados clonotipos de alta prevalencia (HPC) se analiza más a fondo. Se evalúa la correlación del nivel de cada uno de estos clonotipos con medidas clínicas. Se calcula la correlación de la puntuación SLEDAI con el nivel de clonotipo. Para cada paciente hay 4-5 puntos de estudio que pueden usarse para evaluar la correlación de SLEDAI con el nivel de cada HPC. Se investiga la distribución de estas correlaciones obtenidas. Se anticipa que la mayoría de las HPC tendrán una baja correlación con SLEDAI. Se investiga si en el extremo de alta correlación hay un exceso de lo que se espera que se genere de forma aleatoria. Por ejemplo, con 4 y 5 puntos de datos, se espera que -2.5% y ~0.6% de los niveles de correlación (r2) sean >0.9 por casualidad. Una mayor proporción de HPC con r2>0.9 indica la presencia de un clonotipo que se correlaciona con la enfermedad. Además de comparar el número de clonotipos correlacionados con la expectativa aleatoria, se realiza un análisis de permutación donde se calcula la correlación de las puntuaciones SLEDAI de un paciente y el nivel de HPC
individuales de otro. La distribución de las correlaciones generadas a partir de esta permutación se puede utilizar como la correlación de "fondo". (Para asegurar su validez, se confirma que hay poca correlación entre SLEDAI entre diferentes pacientes). El exceso de correlación en el extremo de alta correlación, por ejemplo, r2>0.9 indicará la presencia de clonotipos que se correlacionan con la enfermedad. Se seleccionan los clonotipos de correlación más altos como el conjunto de clonotipos de correlación. Debido a que se conoce el número de HPC que tiene una correlación por casualidad mayor que un umbral establecido (a partir del cálculo mediante suposición aleatoria o mediante el análisis de permutación descrito anteriormente), el umbral para definir el clonotipo correlativo se puede establecer de tal manera que tenga un 10% de falsa tasa de descubrimiento, es decir, el 10% del conjunto de clonotipos correlacionados se correlaciona por casualidad. Se selecciona un conjunto de HPC que tienen muy poca correlación con la puntuación SLEDAI. Estos servirán como el conjunto de clonotipos no correlacionados. Estos 2 conjuntos de clonotipos pueden analizarse más a fondo para identificar las características que pueden distinguirlos. Estas características se pueden buscar en nuevas muestras para identificar los clonotipos que probablemente estén relacionados con la actividad de la enfermedad en estas muestras. Los niveles en sangre de estos clonotipos se pueden seguir para determinar la actividad de la enfermedad.
Una complicación surge de la premisa de que el nivel de clonotipo puede cambiar antes de que lo haga la actividad de la enfermedad. Por lo tanto, es posible que al intentar estudiar solo HPC que se correlacionan altamente con SLEDAI, se puedan eliminar los clonotipos clínicamente útiles que cambian antes que SLEDAI. Se selecciona otro conjunto de clonotipos que se correlacionan con una puntuación de SLEDAI modificada (MSLEDAI). MSLEDAI es el mismo que SLEDAI en todos los puntos de estudio, excepto aquellos justo antes de un cambio significativo. Para esos puntos de datos, la puntuación de MSLEDAI es el promedio entre la puntuación de SLEDAI en ese momento y el siguiente punto de estudio. Los clonotipos que cambian antes de SLEDAI probablemente muestren una mejor correlación con MSLEDAI que con SLEDAI. Es informativo calcular el número excesivo de HPC que tienen una alta correlación con MSLEDAI de lo esperado por las expectativas generadas al azar o por permutación.
Se identificarán las características que distinguen los clonotipos correlacionados de aquellos que no se correlacionan. El análisis se realiza de manera exacta para aquellos clonotipos que se correlacionan con SLEDAI o MSLEDAI. En cualquier caso, el objetivo sería que este conjunto de características recapitule correctamente esta clasificación permitiendo la identificación de clonotipos correlacionados en el siguiente conjunto de muestras. Se espera que cada paciente tenga un conjunto único de clonotipos correlacionados, pero el estudio de entrenamiento está diseñado para generar las reglas que predicen los clonotipos correlacionados a partir de una muestra de calibración (con alta actividad de la enfermedad). Se pueden probar dos tipos generales de parámetros: aquellos que se obtienen a partir de los datos de la secuencia en sí, y aquellos que pueden usar experimentación adicional. La experimentación adicional puede incluir la evaluación de diferentes células con diferente superficie celular u otros marcadores. Aquí hay algunos tipos de parámetros que se investigan: 1) Motivo de la secuencia: el motivo puede ser una región V o J específica, una combinación de VJ o secuencias cortas en la región DJ asociada con un clonotipo que se correlaciona. 2) Tamaño del clonotipo. 3) Nivel: Nivel absoluto (número de lecturas por millón) o nivel de rango. 4) Similitud con otros clonotipos: la presencia de otros clonotipos altamente relacionados, como aquellos con cambios silenciosos (diferencias de nucleótidos que codifican para los mismos aminoácidos) o aquellos con cambios de aminoácidos conservativos. 5) Para los BCR, el nivel de mutaciones somáticas en el clonotipo y/o el número de clonotipos distintos que difieren en las mutaciones somáticas de algunos clonotipos de línea germinal.
Cada uno de estos parámetros se estudia individualmente para su asociación con clonotipos correlacionados. Se establece un umbral de 0.05 (no corregido para pruebas múltiples) para eliminar los factores que probablemente no contribuyan a la predicción de los clonotipos correlacionados. Dados los múltiples parámetros, se realizan muchas pruebas para generar múltiples resultados positivos por casualidad. Sin embargo, el objetivo principal de este paso es filtrar los parámetros a un conjunto más pequeño. El conjunto de parámetros positivos se utilizará para crear un algoritmo para clasificar los dos conjuntos de clonotipos. Se emplea un algoritmo de aprendizaje automático que utiliza los diferentes parámetros para clasificar los dos conjuntos de clonotipos. Para minimizar el riesgo de sobreajuste, se utiliza la técnica de validación cruzada. Usando este algoritmo, cada clonotipo obtendrá una puntuación que corresponde a la probabilidad de que sea un clonotipo correlativo. Luego se colocará un umbral para clasificar los clonotipos por encima de él como correlatos y aquellos por debajo como no correlacionados. La precisión de la clasificación puede estimarse mediante la técnica de validación cruzada; por ejemplo, los clonotipos se colocan en grupos iguales y el algoritmo utiliza todos los clonotipos, excepto un grupo. Los clonotipos en el último grupo (grupo de prueba) se clasifican luego utilizando el algoritmo que se obtuvo utilizando el resto de los clonotipos. Esto se repite tantas veces como el número de grupos, y en cada iteración todos los grupos, excepto uno, se utilizan para la capacitación y un grupo se clasifica. La precisión del algoritmo se puede estimar a partir de la precisión promedio de las diferentes clasificaciones en las diferentes iteraciones. Es de destacar que en todas estas iteraciones el algoritmo exacto sería ligeramente diferente. La precisión de la clasificación es entonces una estimación, ya que no se encuentra en el algoritmo final sino en un conjunto de algoritmos relacionados generados con datos de entrenamiento de todos los clonotipos, excepto uno.
En última instancia, se generan dos algoritmos entrenados en dos conjuntos de clonotipos correlacionados diferentes: uno correlaciona con SLEDAI y el otro correlaciona con MSLEDAI. Incluso si los clonotipos en el conjunto de entrenamiento son diferentes, el algoritmo resultante puede o no ser muy diferente, dependiendo de si estos clonotipos provienen de dos poblaciones distintas. Se comparan los algoritmos. Además, estos algoritmos se utilizan para identificar clonotipos correlacionados que no estaban inicialmente en el conjunto de entrenamiento. Los clonotipos
identificados en los dos algoritmos se comparan, y si los clonotipos iniciales en los dos conjuntos de entrenamiento eran de la misma población, es probable que los clonotipos identificados sean muy similares. A menos que los resultados del algoritmo fueran bastante similares, ambos algoritmos se llevan a cabo para identificar clonotipos correlacionados con el fin de medir la actividad de la enfermedad de lupus.
Otras metodologías experimentales pueden aumentar el poder de la secuenciación en la identificación de clonotipos que se correlacionan con enfermedades. Los clonotipos correlacionados pueden enriquecerse en células con alguna superficie u otros marcadores. Por ejemplo, las células B con altos niveles de CD27 se conocen en pacientes con lupus activo y, por lo tanto, podría ser que los clones de correlación puedan enriquecerse en la población de células CD27. Si se confirma que esto es cierto, la predicción de los clonotipos correlacionados se puede mejorar haciendo un enriquecimiento para las células con altos niveles de CD27. Específicamente, se puede realizar una reacción de secuenciación en las secuencias de IgH de todas las células B en la muestra de sangre, así como de las células B con CD27 alto. Se espera que los clonotipos correlacionados estén presentes con mayor frecuencia en la población con CD27 alta que en toda la muestra de sangre.
3. Usar los perfiles de IgH y TCRp para determinar la actividad de la enfermedad lupus
La sección anterior describió el análisis basado en el clonotipo para identificar las características de los clonotipos correlacionados. Además, para ese análisis, solo una fracción de todas las HPC se usaron para designar claramente los clonotipos como correlacionados o no correlacionados. Esta sección describe el análisis que se encuentra al nivel del paciente con el objetivo de calcular una medida de la actividad de la enfermedad, que se llamará puntuación Autolmm (AI). El algoritmo desarrollado según la sección anterior se aplica para identificar clonotipos correlacionados entre todas las HPC. Se determina el nivel de estas HPC correlacionadas. El nivel de los clonotipos correlacionados se puede normalizar al número total de clonotipos de TCR, así como a las HPC que se predice que no se correlacionarán con la enfermedad. El nivel de estos clonotipos correlacionados en diferentes puntos de tiempo se utiliza para calcular la puntuación de AI en estos diferentes puntos.
En pacientes con más de un clonotipo correlacionado, se combina la información con respecto al nivel de estos clonotipos diferentes. Además, se integran los datos de los clonotipos de IgH y TCRp. Se intentan diferentes algoritmos para hacer la combinación. Por ejemplo, se estudia el promedio, la mediana, la suma y el nivel de clonotipo de correlación más alto. El nivel de clonotipo puede ser su simple conteo de lectura lineal, el logaritmo de esa o alguna otra conversión. Potencialmente, puede ser la diferencia entre clonotipos correlacionados y no correlacionados. Además, se pueden utilizar métodos para el promedio ponderado. La ponderación puede basarse en la probabilidad de que un clonotipo esté correlacionado.
Para evaluar cuál de los modelos es óptimo, todos los modelos se evalúan para identificar el que genera la mayor correlación entre la puntuación de AI y la puntuación de SLEDAI. Para este análisis, la correlación de las puntuaciones de SLEDAI y AI se realiza en todos los datos obtenidos de todos los puntos de estudio de todos los pacientes. Para estimar y mejorar el grado de sobrealimentación, se utiliza la técnica de validación cruzada. El nivel de correlación medido refleja la relación "transversal" entre las puntuaciones de AI y SLEDAI. Además de SLEDAI, se estudia la correlación con otras medidas clínicas como los niveles de C3 y anticuerpos anti- ADNds, así como la proteína en orina/creatinina sérica para pacientes con manifestación renal y recuentos sanguíneos en pacientes con afectación hematológica. La correlación puede deberse a la clasificación de los pacientes en actividad alta y baja de la enfermedad, y no es necesariamente un reflejo de la AI que se correlaciona con la puntuación SLEDAI dentro de un paciente. Para demostrarlo, se realiza una evaluación "longitudinal".
4. Análisis longitudinal.
En el análisis longitudinal, se evalúan dos preguntas generales: ¿la puntuación de AI en un punto del estudio predice la actividad de la enfermedad en el mismo punto y el puntaje de AI en un punto de estudio predice la actividad de la enfermedad en un punto posterior, por ejemplo, el siguiente punto de estudio 3 meses después?.
La relación entre las puntuaciones de AI y SLEDAI en el mismo punto de estudio se evalúa de dos maneras. Primero se calcula la correlación de la IA y la SLEDAI en cada paciente, y luego se calcula el nivel de correlación medio y medio del paciente. Si la correlación observada en el análisis de corte transversal anterior se debe a la clasificación de pacientes con actividad de enfermedad alta y baja y no cambia la actividad de la enfermedad en pacientes individuales, entonces la correlación longitudinal en pacientes individuales probablemente sea baja. Un nivel alto de correlación mediana del paciente sugiere que la IA refleja la puntuación SLEDAI a nivel de paciente individual. Además de la correlación de las puntuaciones de AI y SLEDAI, se evalúa la correlación de la IA con otras medidas relevantes como C3 y anticuerpos anti-ADN de ADN, así como la proteína en orina/creatinina sérica para pacientes con manifestación renal y recuentos sanguíneos en pacientes con compromiso hematológico.
Otra forma de demostrar la capacidad de la puntuación de AI para medir los cambios en la actividad de la enfermedad en pacientes individuales es determinando su precisión para distinguir los estados de alta actividad de baja enfermedad en los mismos pacientes. Para cada uno de los 181 pacientes, los dos puntos de estudio cuando se selecciona SLEDAI donde se encuentra en el nivel más alto (que se llamará HDAP para el punto de actividad de la enfermedad alta) y en los niveles más bajos (que se llamará LDAP para el punto de actividad de la enfermedad de
baja). Se compara la distribución de la AI de todos los HDAP con la de la AI de todos los LDAP y se calcula el valor p de que son diferentes. Además, se evalúa la frecuencia con la que la IA en HDAP es mayor que la LDAP en cada paciente. Si la IA no cambia con la actividad de la enfermedad en un paciente individual, se espera que la IA en HDAP sea mayor que en LDAP solo el 50% de las veces. Se realiza otro análisis donde se determina la fracción de veces que la IA en HDAP es mayor que en LDAP por una diferencia significativa (es decir, por encima de la variación probable de AI). Para medir la fluctuación de la IA, se utilizan todos los puntos de estudio de todos los pacientes, y se puede calcular la desviación estándar (y la desviación estándar relativa) de la IA en los diferentes intervalos de los valores de SLEDAI. Esto generará una desviación estándar relativa en todos los pacientes (AI-RSDall) y este valor puede o no depender de SLEDAI (es decir, el AI-RSDall puede ser diferente en diferentes valores de SLEDAI). Se puede calcular la proporción de pacientes en los que la IA en HDAP es más alta que la AI en LDAP por un número específico (por ejemplo, 2) de AI-RSDall. Puede haber cierto sesgo sistemático en donde la IA calculada en algunos pacientes es consistentemente más alta (o más baja) que lo que se espera del puntaje SLEDA. Por lo tanto, AI-RSDall es una combinación de la fluctuación intrínseca de la IA dentro de un paciente, así como la diferencia sistemática de la IA en pacientes con SLEDAI similar. La fluctuación intrínseca de la IA se puede calcular dentro de un paciente calculando la desviación estándar (y la desviación estándar relativa) de las puntuaciones de la AI entre los puntos del estudio con valores SLEDAI similares (<2 puntos de diferencia) dentro de un paciente. La mediana entre todos los pacientes de la desviación estándar relativa se puede calcular (AI-RSDpt-med). La proporción de pacientes en los que la IA en HDAP es mayor que la IA en LDAP por un número específico (por ejemplo, 2) de AI-RSDpt-med puede evaluarse.
Después de la demostración de que la IA efectivamente fluctúa con SLEDAI en pacientes individuales, se evalúa si IA puede predecir SLEDAI en el siguiente punto de estudio, 3 meses después. Para evaluar, se puede cuantificar el nivel de correlación entre la puntuación de iA i en el momento 0 y la puntuación de SLEDAI en el momento 3 meses. La correlación se puede calcular a nivel del paciente y luego se puede obtener la correlación mediana del paciente. Otra forma de demostrar la capacidad de la IA para predecir la actividad de la enfermedad en el futuro cercano es evaluar la sensibilidad y especificidad de la IA para predecir la actividad de la enfermedad 3 meses en el futuro. Clínicamente, los pacientes que se encuentran bien en su manejo actual pueden distinguirse de los que no lo hacen. El estado de un paciente en un momento determinado se clasifica en una de dos clases: Control deficiente (PC) e incluye pacientes que en 3 meses tendrán una actividad de enfermedad alta (SLEDAI> 6 puntos) y/o una exacerbación (aumento de SLEDAI en 3 puntos) y Good Control (GC) e incluyen pacientes que en 3 meses tendrán una actividad de la enfermedad baja o moderada (SLEDAI <6) y/o una reducción significativa de la actividad de la enfermedad (SLEDAI disminuye en 3 puntos). Luego se puede evaluar la sensibilidad de la clasificación y obtener la especificidad utilizando diferentes umbrales de IA. Se puede generar una curva ROC que describa el desempeño de la lA en la predicción del estado del paciente (PC o GC) 3 meses antes de tiempo. El rendimiento obtenido por esta prueba se compara con el de las medidas clínicas estándar que incluyen SLEDAI, niveles de Anti-ADNds y C3.
También se llevará a cabo un análisis para evaluar la capacidad de la IA para predecir cambios en las puntuaciones de SLEDAI 3 meses después. Usando datos de todos los puntos de estudio de todos los pacientes, la relación entre las puntuaciones de IA y SLEDAI se puede trazar para identificar el nivel de correlación "transversal" como se discutió anteriormente. Esto determina la relación entre SLEDAI y IA en el mismo punto de estudio. Esta relación se ajusta a una ecuación que permite la predicción de la puntuación SLEDAI dada una puntuación IA (o viceversa). Si la IA predice las erupciones, los cambios en SLEDAI en algún punto del estudio 1 van precedidos por los cambios en la IA en el punto 0. Por lo tanto, si se produce una erupción entre los puntos 0 y 1, la puntuación de la IA en el punto 0 (que se llamará Almeas) es mayor que lo que se espera (que se llamará Alexp) dado el SLEDAI en el punto de estudio 0. Por otro lado, sin cambios en la actividad de la enfermedad entre el punto de estudio 0 y el punto de estudio 1, la puntuación de IA en el punto 0 es muy similar a la esperada dada la SLEDAI en el punto de estudio 0. El cambio relativo de IA (Rel-AI-diff) se puede calcular dividiendo la diferencia de Almeas y Alexp por Almeas. La sensibilidad y la especificidad de la IA para predecir un cambio significativo en SLEDAI 3 meses más tarde se pueden evaluar utilizando diferentes umbrales de Rel-AI-diff. Los umbrales pueden ser bidireccionales, por lo que si el Rel-AI-diff en un punto de estudio específico es más alto que un umbral específico, se prevé una erupción y, de manera similar, si es más bajo que el negativo del umbral específico, se espera una reducción significativa de SLEDAI. Por otro lado, cuando el Rel-Al-diff en un punto de estudio se encuentra entre el umbral y su negativo, no se esperan cambios significativos en la actividad de la enfermedad. Se puede generar una curva ROC que muestre el comercio de sensibilidad y falsos positivos utilizando muchos umbrales diferentes de Rel-Al-diff. Se pueden generar curvas ROC similares utilizando medidas clínicas estándar que incluyen SLEDAI, niveles de Anti-ADNds y C3.
Si la fluctuación de la IA varía en diferentes valores de SLEDAI, el análisis anterior se refina. Una sección anterior describió el cálculo de AI-RSDall y AI-RSDpt-med y mencionó evaluar si cambian a diferentes valores SLEDAI. Si lo hacen, entonces el análisis de ROC se puede hacer como se describe anteriormente, pero en lugar de usar umbrales diferentes de Rel-Al-diff, se usan umbrales diferentes de AI-RSDall y AI-RSDpt-med. Se compara el rendimiento obtenido por la prueba con el de las medidas clínicas estándar que incluyen SLEDAI, niveles de Anti-ADNds y C3.
En el análisis anterior, se intentan predecir el SLEDAI en el punto 1 a partir de la puntuación de IA en el punto 0. Es probable que además del nivel absoluto en el punto 0, el cambio de IA del punto -1 al 0 sea informativo en predicción de SLEDAI en el punto 1. Por ejemplo, considere a un paciente que tiene en el punto de estudio -1 una puntuación de IA de X-1, y en el punto 0 la puntuación de IA se incrementa aun nuevo valor X0 que es sensiblemente más alto que X-1. Este paciente puede tener una probabilidad más alta de erupción en el punto 1 que un paciente cuya IA se ha mantenido estable en X0 en los puntos de estudio -1 y 0. Este concepto de cambio o velocidad de IA se incorpora para
generar un puntaje de IA modificado (MAI). Para generar un MAI en el punto 0, se necesita la puntuación de IA en el punto -1 y en el punto 0, y, por lo tanto, un punto de datos por paciente no tendrá un MAI asociado. La fórmula específica para incorporar la velocidad en el cálculo de IA para obtener MAI está optimizada. Esta optimización puede realizarse mediante la maximización de la correlación de MAI y SLEDAI tres meses después. El diseño de validación cruzada se utiliza para evaluar y controlar el grado de sobrealimentación. La correlación se puede hacer para los puntos de datos de todas las muestras, pero también se puede hacer a nivel de paciente y se puede evaluar la correlación media entre todos los pacientes. Esta última metodología mejora el problema de algunos pacientes que tienen un sesgo sistemático de puntaje AI demasiado bajo o demasiado alto. Usando MAI, se puede realizar el mismo tipo de análisis de ROC que se mencionó para la IA para evaluar su capacidad para predecir SLEDAI 3 meses después. Primero, de manera análoga a lo que se describe para la IA, se puede hacer un análisis para mostrar la capacidad de MAI en el punto 0 para distinguir los estados de PC y GC en el punto 1. Además, un análisis similar al descrito para AI para evaluar la capacidad de El MAI en el punto 0 se puede realizar para predecir un cambio significativo en la actividad de la enfermedad (3 puntos en SLEDAI) entre los puntos 0 y 1. Para este último análisis, se pueden utilizar diferentes umbrales de Rel-AI-diff, AI-RSDall o AI-RSDpt-med. usado. El rendimiento de MAI se compara con el de AI para determinar si la adición del factor de velocidad es útil.
Una complicación del estudio descrito es que los cambios de tratamiento se realizan para diferentes pacientes durante el período de seguimiento del estudio. Es probable que esto complique la predicción de la actividad de la enfermedad. Por ejemplo, considere dos pacientes con la misma puntuación de AI en el punto 0 y uno de esos pacientes tuvo una reducción de la medicación al mismo tiempo. Es probable que la probabilidad de que este paciente tenga un aumento en la actividad de la enfermedad en el punto 1 sea mayor que para el paciente que no cambió los medicamentos en el punto 0. Esto puede llevar a una subestimación del rendimiento de la IA. Una forma de aliviar eso es eliminar todos los puntos con cambios significativos en la medicación del estudio. Otra es modificar la puntuación de la IA para incluir si un paciente tiene un cambio de medicación y crear una IA modificada con la medicación. Entonces, en el ejemplo anterior con los dos pacientes, el que tenga el cambio de medicación tendrá una IA más alta modificada por la medicación.
5. Integración con otros marcadores predictivos.
La capacidad predictiva del marcador de actividad de la enfermedad se puede maximizar. Por lo tanto, se prueba la capacidad predictiva de la información del repertorio de TCR/BCR integrada con otros marcadores. Estos marcadores incluyen marcadores estándar utilizados en la clínica, como los niveles de Anti-ADNds y C3. También incluirá otros marcadores que se publiquen. Por ejemplo, ya se ha demostrado que un panel de quimioquinas tiene cierta capacidad predictiva utilizando el mismo conjunto de pacientes que se usa. Se evaluará si este panel aumentará la capacidad predictiva del repertorio de TCR y BCR. El primer paso es integrar el puntaje AI con la medida adicional para generar un puntaje AI expandido (EAI). Se pueden evaluar diferentes formas de hacer la integración, y esto se puede optimizar mediante la maximización de la correlación de EAI y SLEDAI tres meses después. El diseño de validación cruzada se utiliza para evaluar y controlar el grado de sobrealimentación. Usando EAI, la capacidad para predecir la actividad de la enfermedad 3 meses después se evalúa por su capacidad para distinguir GC de PC y para predecir cambios en la actividad de la enfermedad. El desempeño en la medición de la actividad de la enfermedad y el cambio en la actividad de la enfermedad se puede describir a través del análisis ROC como se describió anteriormente.
6. Validación
El número de variables que se están probando es alto en comparación con el número de muestras. Esto puede prestarse a sobreajuste, con resultados inicialmente prometedores que no pueden validarse en estudios posteriores. Se utiliza una metodología de validación cruzada en la capacitación para obtener una medida del alcance del exceso de equipamiento. Sin embargo, una validación en un conjunto independiente de muestras está involucrada en un trabajo posterior. Esto no es parte de esta propuesta, pero este marcador puede ser clínicamente aplicable. Utilizando los datos obtenidos anteriormente, se puede determinar si AI, MAI o EAI deben validarse y la forma específica de calcular la medida de interés. Se toma un algoritmo específico para la validación. Además, se especifican uno o más puntos finales específicos. La sensibilidad y la especificidad de la IA se pueden evaluar en la capacidad de distinguir GC de PC 3 meses después para evaluar la capacidad de la IA para predecir la actividad de la enfermedad. En otro ejemplo, se puede evaluar la sensibilidad y especificidad de la IA para predecir un cambio significativo en la actividad de la enfermedad en 3 meses utilizando un umbral específico de Rel-AI-diff.
Ejemplo 5
Medición de la respuesta de un paciente con SLE a la farmacoterapia
Los métodos descritos en el presente documento se usan para medir la respuesta de un paciente con SLE a la terapia con medicamentos. La determinación de si un paciente con SLE que recibe un medicamento costoso con efectos secundarios graves responde al medicamento desempeña un papel en la atención del paciente y también para hacer que la administración de dicha atención sea rentable. Muchos indicadores clínicos de actividad de la enfermedad responden al tratamiento de forma imprecisa y después de un período de hasta varios meses. Durante este tiempo, la enfermedad puede progresar y los efectos secundarios pueden agregar complicaciones a la terapia. Una comprensión rápida de la respuesta al fármaco permitiría a los pacientes cambiar a terapias más efectivas más rápidamente.
En este ejemplo, una mujer afroamericana de 35 años con un diagnóstico previo de lupus se presenta a su reumatólogo habitual. El estado de la enfermedad del paciente se evalúa trimestralmente a través de una evaluación clínica completa además de las pruebas de laboratorio que incluyen la medición de C3, los niveles de anticuerpos Anti-ADNds, los recuentos sanguíneos y el análisis de orina. Durante una visita, el paciente se queja de lesiones en la piel y fatiga, y el análisis de orina muestra evidencia de proteinuria y/o cilindros celulares. El reumatólogo refiere al paciente a un nefrólogo para una biopsia de riñón para evaluar el estado inflamatorio del riñón y ordena creatinina sérica y una proporción de creatinina en orina de 24 horas para evaluar el grado de deterioro de la función renal. Una biopsia de riñón muestra evidencia de nefritis lúpica difusa, mientras que la prueba de la proteína de la orina a la creatinina revela evidencia de síndrome nefrótico (proporción de proteína de la orina a creatinina de 3.6). Sobre la base de esta información, se proporciona un diagnóstico de nefritis lúpica aguda y el paciente comienza un tratamiento farmacológico. Hay varios fármacos posibles que pueden ser elegidas en este punto. Los inmunomoduladores como el micofenolato mofetilo (Cellcept) se usan a menudo, aunque a veces en los casos graves se prescriben medicamentos como el metotrexato, la azatiopurina (Imuran), la ciclofosfamida (cytoxan). Rituximab (Rituxan) también se usa en ocasiones como segunda o tercera opción. Uno de estos medicamentos se usa a menudo en combinación con un esteroide sistémico como la prednisona o la metilprednisolona para suprimir los síntomas agudos. Aquí, el micofenolato de mofetilo se prescribe a 150 mg por día junto con 60 mg de prednisona. Dados los muchos efectos secundarios de los esteroides, incluido el riesgo de osteoporosis, hiperglucemia, aumento de peso y otros síntomas de Cushingoide a largo plazo, la dosis de prednisona del paciente se reduce en más de ~6 semanas si el cuadro clínico lo permite.
La primera pregunta que se determina es si el paciente está respondiendo a la terapia, y como resultado, la dosis de esteroides puede reducirse adecuadamente. Por lo tanto, durante este período, se realiza un seguimiento de la creatinina sérica del paciente, así como de la proteína de la orina y la creatinina, para garantizar que el paciente responde a los medicamentos. Se puede hacer una biopsia renal frecuente para detectar si el daño inflamatorio se está revirtiendo; sin embargo, el uso rutinario de la biopsia de riñón conlleva un riesgo demasiado grande y es demasiado invasivo para ser práctico. Los marcadores basados en sangre actuales que se utilizan para evaluar el estado inflamatorio son de uso limitado para tomar esta decisión, ya que no están lo suficientemente bien correlacionados con la enfermedad subyacente como para confiar en el aumento de los efectos secundarios que acompañan a las dosis altas de esteroides. Los marcadores de la función del suero y la orina pueden tener algún retraso en la detección de mejoría en el estado inflamatorio y, por lo tanto, los esteroides pueden disminuirse antes de que estos marcadores muestren un cambio definitivo y, por lo tanto, extiendan el período de erupción renal. Una disminución más lenta, informada por marcadores más sensibles, en estos casos podría haber acortado el período de exacerbación evitando daños adicionales en el tejido renal. Después de la reducción de esteroides a una dosis de mantenimiento de aproximadamente 10 mg, el paciente puede mostrar niveles persistentemente elevados de proteína en la orina y la proporción alta de proteína en orina de creatinina de 2, y el médico ahora debe decidir si cambiará de Cellcept a otro medicamento. Argumentar a favor de esto es la evidencia continua de pérdida de la función renal, pero sin una medida precisa del estado renal inflamatorio, puede ser difícil saber si la enfermedad en sí está en remisión, sin embargo, ha hecho algún nivel de daño renal irreversible que resulta en estos niveles persistentes de proteinuria. Aquí nuevamente, los marcadores basados en sangre existentes son imperfectamente informativos y otras biopsias de riñón no son prácticas. Esta decisión se vería enormemente ayudada por una medida precisa del estado de la enfermedad basada en sangre.
Autolmm Load sería muy útil en esta situación para evaluar la respuesta al tratamiento midiendo la actividad de la enfermedad ya sea solo o en combinación con otros marcadores de la actividad de la enfermedad. Se desarrolló un algoritmo para Autolmm Load utilizando el estudio descrito anteriormente. Los clonotipos correlacionados que se utilizan para calcular AutoImm se miden mediante una prueba de calibración. Esta prueba de calibración se realiza utilizando sangre de un paciente en el momento de mayor actividad de la enfermedad, por ejemplo, al inicio de la terapia. La prueba de calibración se realiza con sangre o alternativamente con el tejido afectado (por ejemplo, biopsia renal o biopsia de piel). En un momento posterior en donde se evaluará la respuesta al tratamiento, se tomará una muestra de sangre y se usará junto con la prueba de calibración para medir AutoImm. Esto se utiliza para tomar una decisión de tratamiento. Si los clonotipos correlacionados se derivan de un estudio de poblaciones, no es necesario realizar la prueba de calibración y una prueba de sangre en el momento en que se evalúa la respuesta al tratamiento es suficiente para medir la AutoImm Load para informar la decisión del tratamiento.
Ejemplo 6
Determinación del tiempo apropiado para reducir o interrumpir la terapia para un paciente con SLE
Los métodos descritos en el presente documento pueden usarse para determinar el tiempo apropiado para reducir o interrumpir la terapia para un paciente con SLE. Además del lapso de tiempo que pueden exhibir las medidas clínicas de la actividad de la enfermedad, una dificultad adicional reside en la falta de sensibilidad de estas medidas. Sin embargo, la enfermedad subclínica puede dar como resultado una reaparición de la enfermedad si la terapia se reduce demasiado pronto. Como resultado de esto, los cursos de terapia inmunosupresora se administran normalmente durante un período de tiempo que es mucho más prolongado que el necesario para que el paciente promedio se asegure de que el riesgo de reerupción sea bajo para el paciente promedio, pero aún puede ser lo suficientemente largo para el final de la cola de la distribución. Por lo tanto, en la mayoría de los pacientes se está produciendo un tratamiento excesivo significativo, causando efectos secundarios y costes, mientras que el tratamiento insuficiente de
algunos pacientes causa reerupciones potencialmente prevenibles. Un método que pudiera medir la actividad subclínica que predijera el riesgo de reerupción permitiría que la terapia se redujera en función de dichas medidas en lugar de depender del tratamiento excesivo por diseño.
En este ejemplo, el paciente del Ejemplo 7 está en tratamiento con prednisona y micofenolato mofetilo durante un período de 6 meses y la proporción de proteína en orina y creatinina en la orina vuelve a un nivel de 0.5. Este nivel se mantiene por encima del nivel de referencia esperado en individuos sanos, pero no está claro que este nivel no se deba a algún daño renal que no sea reversible. Otras medidas clínicas de inflamación son normales y el paciente no informa ningún otro síntoma. Al mismo tiempo, el paciente está experimentando niveles moderados de náuseas y aumento de peso como posibles efectos secundarios de los medicamentos que, además, tienen efectos secundarios graves a largo plazo. El médico se enfrenta a una decisión difícil: equilibrar el miedo a reducir el Cellcept y/o el esteroide demasiado rápido, lo que podría resultar en una inflamación renal renovada y posiblemente en un daño renal irreversible a largo plazo y en las reacciones adversas que pueden ocurrir debido a los medicamentos. Aquí, nuevamente, una evaluación inequívoca del estado de la enfermedad sin tener que realizar una biopsia renal desempeñaría un papel en la toma de esta decisión. Se recomienda intentar reducir los esteroides a través de ensayos repetidos de esteroides que conducen a la recurrencia del mismo dilema clínico. De hecho, esta pregunta surge cada vez que el paciente está en remisión y el paciente toma esteroides o inmunomoduladores.
AutoImm Load sería muy útil en esta situación para evaluar si disminuir o no la terapia mediante la medición de la actividad de la enfermedad ya sea solo o en combinación con otros marcadores de la actividad de la enfermedad. Se desarrolló un algoritmo para AutoImm Load utilizando el estudio descrito anteriormente. Los clonotipos correlacionados que se utilizan para calcular la AutoImm Load se miden mediante una prueba de calibración. Esta prueba de calibración se realiza utilizando sangre de un paciente en el momento de mayor actividad de la enfermedad, por ejemplo, al inicio de la terapia. La prueba de calibración podría realizarse con sangre o alternativamente con el tejido afectado (por ejemplo, biopsia de riñón o biopsia de piel). En un momento posterior en donde se evaluará el nivel de actividad de la enfermedad, se puede tomar una muestra de sangre y utilizarla junto con la prueba de calibración para medir la AutoImm Load. Esto se utiliza para tomar una decisión de tratamiento y para evaluar si el paciente tiene alguna actividad detectable de la enfermedad. Si los clonotipos correlacionados se derivan de un estudio de poblaciones, no es necesario realizar la prueba de calibración y una prueba de sangre en el momento en que se evalúa la respuesta al tratamiento es suficiente para medir la AutoImm Load para informar la decisión del tratamiento.
Ejemplo 7
Predicción de erupciones en un paciente con SLE
Un desafío en el tratamiento de pacientes con SLE es el hecho de que aparecen erupciones sin advertencia, lo que frena los esfuerzos de los médicos para tratar la enfermedad de manera preventiva. La espera de que ocurran erupciones antes de comenzar el tratamiento somete a los pacientes a síntomas clínicos potencialmente destructivos, puede implicar una hospitalización costosa e inconveniente, y puede causar daños a los órganos a largo plazo, a la vez que también requiere intervenciones terapéuticas agresivas que estén llenas de efectos secundarios. Un paradigma mucho más deseable sería un paradigma terapéutico en donde las erupciones se detecten en una fase subclínica, momento en donde se podría administrar la terapia de forma proactiva, lo que ahorraría un sufrimiento significativo para el paciente, lo que resultaría en hospitalizaciones menos costosas y, en última instancia, permitiría un mejor pronóstico a largo plazo para los pacientes.
El paciente del Ejemplo 7 se está recuperando de la exacerbación aguda descrita anteriormente, y el paciente está disminuido de todas las terapias, excepto Plaquinil y una dosis baja de 5 mg de Prednisona. Sin embargo, este paciente sigue teniendo un alto riesgo de sufrir otro episodio inflamatorio. Como resultado, este paciente permanecerá bajo la atención de un reumatólogo que continuará siguiendo los síntomas clínicos y las pruebas de laboratorio del paciente. Desafortunadamente, estos síntomas y pruebas no proporcionan una advertencia temprana para una erupción inminente hasta que los pacientes realmente han mostrado síntomas clínicos de erupción y la secuencia se repite. Se podría incluir un marcador altamente específico de aumento de la actividad subclínica en la evaluación clínica de rutina del paciente para detectar signos inequívocos de una erupción que pueda alcanzar un estadio clínicamente detectable en los siguientes 1-3 meses. Las terapias iniciales más tempranas podrían hacer que la exacerbación sea menos grave y permitir que el tratamiento se realice con menos daño a los órganos a largo plazo o menos esteroides que lo que actualmente es el caso.
Autolmm Load sería muy útil en esta situación para evaluar la probabilidad de una erupción incipiente al medir la actividad de la enfermedad sola o en combinación con otros marcadores de actividad de la enfermedad. Esta puntuación, ya sea por sí misma o la tasa de aumento (velocidad) o la aceleración de esta puntuación, se puede utilizar para evaluar la probabilidad de progresión a una erupción. Se podría desarrollar un algoritmo para Autolmm Load utilizando el estudio descrito anteriormente. Los clonotipos correlacionados que se utilizan para calcular AutoImm podrían medirse utilizando una prueba de calibración. Esta prueba de calibración podría realizarse utilizando sangre de un paciente en el momento de mayor actividad de la enfermedad, por ejemplo, al inicio de la terapia. La prueba de calibración podría realizarse con sangre o alternativamente con el tejido afectado (por ejemplo, biopsia de riñón o biopsia de piel). En un momento posterior en donde se evaluará la respuesta al tratamiento, se puede tomar una muestra de sangre y usarla junto con la prueba de calibración para medir AutoImm. Esto se puede utilizar para tomar
una decisión de tratamiento. Si los clonotipos correlacionados se derivan de un estudio de poblaciones, no es necesario realizar la prueba de calibración y una prueba de sangre en el momento en que se debe evaluar el riesgo de erupción es suficiente para medir la Autolmm Load para informar la decisión de tratamiento.
Ejemplo 8
Medida objetiva para evaluar los síntomas subjetivos de pacientes con SLE
El SLE afecta a muchos órganos y produce muchos síntomas potenciales, incluidos algunos que son muy comunes en las poblaciones sanas. Por ejemplo, si un paciente con SLE se queja de dolor de cabeza, el dolor de cabeza puede ser un signo de lupus del SNC o puede deberse al dolor de cabeza común. De manera similar, si los pacientes con SLE se quejan de empeoramiento de la fatiga durante un período de tiempo, el empeoramiento de la fatiga puede deberse al deterioro de su enfermedad o puede deberse a la depresión u otras causas. La disponibilidad de una medida objetiva que refleje la actividad de la enfermedad puede ser de gran ayuda en el manejo de los pacientes con SLE.
El paciente en el Ejemplo 7 presenta al reumatólogo quejas principales de dolor de cabeza, fatiga y dificultad con la concentración. El dolor de cabeza del paciente es recurrente y solo mejora transitoriamente con el tratamiento con Motrin. De lo contrario, el SLE del paciente está bien controlado. Los factores estresantes psicosociales relevantes en la vida de la paciente incluyen que ella está pasando por un divorcio. Los médicos se encuentran en un dilema cuando se enfrentan a pacientes con SLE con síntomas que no son específicos del SLE y son comunes en la población general. ¿El paciente sufre de lupus del SNC? ¿O podría sufrir otras causas comunes de sus síntomas, como la depresión? Actualmente, las pruebas de laboratorio actuales carecen de la sensibilidad y la especificidad en que se puede confiar para distinguir estas posibilidades. Una prueba confiable para medir la actividad de la enfermedad del SLE se puede utilizar rutinariamente para ayudar a distinguir las dos posibilidades.
Autolmm Load sería muy útil en esta situación para evaluar objetivamente la actividad de la enfermedad, ya sea solo o en combinación con otros marcadores de la actividad de la enfermedad. Se desarrolló un algoritmo para Autolmm Load utilizando el estudio descrito anteriormente. Los clonotipos correlacionados que se utilizan para calcular AutoImm se medirán mediante una prueba de calibración. Esta prueba de calibración se realiza utilizando sangre de un paciente en el momento de mayor actividad de la enfermedad, por ejemplo, al inicio de la terapia. La prueba de calibración se realiza con sangre o alternativamente con el tejido afectado (por ejemplo, biopsia renal o biopsia de piel). En un momento posterior en donde se evaluará la actividad objetiva de la enfermedad, se puede tomar una muestra de sangre y utilizarla junto con la prueba de calibración para medir AutoImm. Esto se utiliza para tomar una decisión de tratamiento. Si los clonotipos correlacionados se derivan de un estudio de poblaciones, no es necesario realizar la prueba de calibración y una prueba de sangre en el momento en que se evalúa la actividad objetiva de la enfermedad es suficiente para medir la Autolmm Load para informar la decisión de tratamiento.
Ejemplo 9
Medición de la respuesta al tratamiento farmacológico de un paciente con EM
Como se indicó anteriormente, uno de los principales desafíos en la terapia de la EM es medir qué tan bien y si un paciente está respondiendo a una terapia farmacológica. Durante la enfermedad progresiva y en estadio tardío, existen evaluaciones clínicas, como la puntuación del estado de discapacidad expandida (EDSS), que mide el grado de discapacidad física que ha resultado de la enfermedad. Sin embargo, estas evaluaciones no son útiles en el estadio temprano ni en la enfermedad recurrente/remitente. Los parámetros clínicos alrededor de las recaídas se pueden usar para evaluar la progresión de la enfermedad, pero estos son indicadores precisos y retardados, ya que los pacientes pueden pasar varios años entre las recaídas, durante los cuales se pueden obtener pocas evidencias de las evaluaciones clínicas. Por último, se pueden usar imágenes cerebrales como la RMN con gadolinio para examinar lesiones cerebrales. Los pacientes con EM normalmente reciben una RMN de este tipo anualmente. Sin embargo, tales imágenes carecen de especificidad. Además, como medida del daño cerebral integrado, no son buenas medidas de la actividad actual de la enfermedad, sino que reflejan la historia de la enfermedad y su impacto en el cerebro.
Si bien es cierto que el paradigma actual del tratamiento clínico para la EM es que los pacientes diagnosticados con enfermedad remitente recurrente deben recibir un tratamiento continuo para retrasar la aparición de la enfermedad progresiva, el repertorio cada vez mayor de medicamentos aprobados para tratar la EM hace que no haya retroalimentación biológica. Cada vez más problemático. La lista que se muestra arriba de los medicamentos aprobados para tratar la EM continúa haciéndose más larga a medida que la inversión sustancial en terapias para la EM comienza a dar frutos. Cada uno de estos medicamentos tiene efectos secundarios graves y su administración es muy costosa, con costes de $ 30.000 a $ 100.000 por año de tratamiento. Los pacientes que no están bien manejados harán una transición más rápida a una enfermedad progresiva que es debilitante y causa costosas intervenciones de atención médica, incluidas hospitalizaciones y atención a largo plazo. Por lo tanto, se puede permitir que el paciente reciba una terapia óptima al principio del tratamiento.
Ejemplo de utilidad clínica
Perfil del paciente: Una mujer de 30 años de edad acude al hospital con discapacidad visual monocular con dolor. Se le administra una evaluación neurológica y una punción lumbar para obtener líquido cefalorraquídeo que se usa para evaluar si hay células T clonales presentes. También es referida a una resonancia magnética cerebral. Sobre la base de estas pruebas, se realiza un diagnóstico de EM. Se le receta Betaseron 250 mcg por inyección para que se administre por vía subcutánea cada dos días. En una visita de seguimiento seis meses después, el paciente se queja de depresión y aumento de peso. No se han reportado más eventos neurológicos al médico. El médico se enfrenta ahora a un dilema clínico. ¿Debe el médico mantener la terapia a medida que se administra? ¿Debería usarse una nueva terapia? ¿Debería el médico ordenar una MRI incurriendo en costes y sometiendo al paciente a una exposición de contraste adicional? ¿El médico debe esperar hasta que la próxima RM programada muestre nuevas lesiones? ¿Debería esperar el médico para véase si reaparecen las erupciones? Todas estas decisiones se beneficiarían de una medida inequívoca de si la enfermedad está activa o no.
Autolmm Load sería muy útil en esta situación para evaluar la respuesta al tratamiento midiendo la actividad de la enfermedad ya sea solo o en combinación con otros marcadores de la actividad de la enfermedad. Se desarrolló un algoritmo para Autolmm Load utilizando los estudios descritos en este documento. Los clonotipos correlacionados que se utilizan para calcular AutoImm se miden mediante una prueba de calibración. Esta prueba de calibración se realiza utilizando sangre de un paciente en el momento de mayor actividad de la enfermedad, por ejemplo, al inicio de la terapia. La prueba de calibración podría realizarse con sangre o alternativamente con el tejido afectado (por ejemplo, CSF). En un momento posterior en donde se evaluará la respuesta al tratamiento, se puede tomar una muestra de sangre y usarla junto con la prueba de calibración para medir AutoImm. Esto se puede utilizar para tomar una decisión de tratamiento. Si los clonotipos correlacionados se derivan de un estudio de población, no es necesario realizar la prueba de calibración y una prueba de sangre en el momento en que se evalúa la respuesta al tratamiento es suficiente para medir la Autolmm Load para informar la decisión del tratamiento.
Ejemplo 10
Predicción de las erupciones de MS
Como en todas las enfermedades autoinmunes, la mejora de las es un objetivo principal de la terapia. Las erupciones no solo son debilitantes para el paciente y costosas de tratar, sino que se cree cada vez más que cada erupción contribuye a la progresión de la enfermedad a largo plazo y no reversible. Se pueden usar varias terapias para controlar las erupciones incipientes como la metilprednisolona IV o la prednisona oral. Tales medicamentos tienen efectos secundarios significativos y, como tales, no se recetan sin evidencia de una erupción activa. Una medida del aumento de la actividad subclínica que se correlacionó con las erupciones clínicos subsiguientes se podría usar para informar a este tipo de tratamiento proactivo contra erupciones que podría dar lugar a erupciones más cortas y menos dañinas. Además, existen terapias que demuestran una alta eficacia clínica para la reducción de erupciones que conllevan riesgos de efectos secundarios muy significativos y letales. Uno de esos fármacos es Tysabri, un fármaco que se ha demostrado que mejora los resultados clínicos y aumenta el riesgo de infecciones cerebrales mortales como la LMP. Estos riesgos han reducido el valor de dichos medicamentos para la terapia de última línea cuando otros medicamentos ya no están controlando la progresión y han limitado el valor de estos medicamentos como tratamientos crónicos. Una prueba que podría predecir cuándo el estado de exacerbación es incipiente podría aumentar la utilidad de dichos medicamentos, ya que podrían usarse de manera similar a los esteroides para controlar los períodos agudos de exacerbación y minimizar los riesgos de efectos secundarios letales.
Ejemplo de utilidad clínica
El paciente del Ejemplo 11 está con Betaseron durante 3 años y reporta una erupción clínica que dura una semana. La MRI del paciente al final del año muestra lesiones nuevas significativas (múltiples lesiones discretas de tamaño variable ovoidales dirigidas perpendicularmente a T2W y FLAIR hiperintensas (placas), que aparecen isohipointensas en imágenes T1W e hiperintensas en imágenes T2W que incluyen regiones bilaterales periventriculares y subcorales de materia blanca, incluida la interfaz calloso-septal). El médico está preocupado de que el paciente tenga un alto riesgo de erupciones durante los próximos 12 meses. Se presenta un dilema clínico. ¿El médico espera que aparezcan más síntomas clínicos para intervenir con terapia adicional? ¿Debería el médico cambiar de terapia? Si es así, ¿debería usarse otra clase de inyectables como la copaxona o debería usarse una nueva clase de terapia como Tysabri? ¿Deberían recetarse esteroides? Una prueba que podría monitorizar la actividad subclínica de la enfermedad y mostrar cuándo está aumentando la enfermedad y cuándo es probable que se produzca una erupción podría usarse para ayudar a tomar estas decisiones clínicas.
AutoImm Load sería muy útil en esta situación para evaluar el riesgo de erupción al medir la actividad de la enfermedad ya sea solo o en combinación con otros marcadores de actividad de la enfermedad. Se podría desarrollar un algoritmo para AutoImm Load utilizando los estudios descritos aquí. Los clonotipos correlacionados que se utilizan para calcular la AutoImm Load podrían medirse utilizando una prueba de calibración. Esta prueba de calibración podría realizarse utilizando sangre de un paciente en el momento de mayor actividad de la enfermedad, por ejemplo, al inicio de la terapia. La prueba de calibración podría realizarse con sangre o alternativamente con el tejido afectado (por ejemplo, CSF). En un momento posterior en donde se evaluará el riesgo de erupción, se puede tomar una muestra de sangre y usarla junto con la prueba de calibración para medir la AutoImm Load. Esto se puede utilizar para tomar una decisión de tratamiento. Si los clonotipos correlacionados se derivan de un estudio de población, no es necesario realizar la
prueba de calibración y una prueba de sangre en el momento en que se debe evaluar el riesgo de erupción es suficiente para medir la AutoImm Load para informar la decisión de tratamiento.
Ejemplo 11
Monitorización de la terapia de cumplimiento de la EM.
Debido a la relativa frecuencia de los síntomas clínicos en los primeros estadios de la enfermedad, las interacciones entre un paciente y su médico no son muy frecuentes. Al mismo tiempo, las terapias que se prescriben son costosas e inconvenientes para el paciente, e incluyen autoinyecciones que pueden causar reacciones dolorosas y efectos secundarios. Como resultado, existe un grado significativo de incumplimiento de los regímenes terapéuticos que son difíciles de controlar para un médico ya que las interacciones entre el paciente y el médico no son rutinarias. Una prueba que podría medir el estado de la enfermedad subclínica permitiría al médico y al paciente véase de forma rutinaria qué tan bien controlada está la enfermedad subyacente. Tales métodos han demostrado ser muy efectivos en los pacientes con VIH para motivarlos a seguir la terapia de manera efectiva. Una prueba de sangre que se realizó trimestralmente permitiría al médico véase al paciente y medir el estado de la enfermedad.
AutoImm Load sería muy útil en esta situación para evaluar el cumplimiento de la terapia midiendo la actividad de la enfermedad ya sea solo o en combinación con otros marcadores de actividad de la enfermedad. Se desarrolló un algoritmo para AutoImm Load utilizando los estudios descritos aquí. Los clonotipos correlacionados que se utilizan para calcular la AutoImm Load se miden mediante una prueba de calibración. Esta prueba de calibración se realiza utilizando sangre de un paciente en el momento de mayor actividad de la enfermedad, por ejemplo, al inicio de la terapia. La prueba de calibración podría realizarse con sangre o alternativamente con el tejido afectado (por ejemplo, CSF). En un momento posterior en donde se evaluará el cumplimiento de la terapia, se tomará una muestra de sangre y se usará junto con la prueba de calibración para medir AutoImm. Esto se utiliza para tomar una decisión de tratamiento y para guiar mejor al paciente hacia un mejor cumplimiento. Si los clonotipos correlacionados se derivan de un estudio de población, no es necesario realizar la prueba de calibración y una prueba de sangre en el momento en que se debe evaluar el cumplimiento de la terapia es suficiente para medir la Autolmm Load para informar la decisión del tratamiento.
Ejemplo 12
Amplificación de las secuencias de TCRp e IgH de ratón
Se desarrolló un esquema de amplificación y secuenciación para TCRp e IgH de ratón que es similar al desarrollado para humanos. Se aplican métodos similares para minimizar las diferencias en la eficiencia de amplificación de diferentes secuencias y técnicas de validación similares utilizando picos y la técnica 5' RACE descrita anteriormente. La cantidad de entrada mínima de ADNc se determina en una metodología similar a la descrita para muestras humanas. Una diferencia en el esquema de amplificación entre el ratón y los humanos es que los dos segmentos C para TCRp en el ratón no tienen ningún polimorfismo en los 50 pb más cercanos a la unión J/C. Por lo tanto, en el esquema, el cebador para la amplificación de la primera etapa se coloca en las posiciones 25-50 y para la segunda etapa de amplificación el cebador se coloca en las posiciones 1-25, y el cebador tendrá una cola 5' para el último cebador que contiene la secuencia P5. Las diferentes secuencias mejorarán la especificidad y son similares a la estrategia utilizada en los humanos, excepto que no hay necesidad de "intercalar" ninguna base para polimorfismos.
Ejemplo 13
Análisis primario de datos de secuencia de ratón
El marco de análisis que se usa para el análisis de datos de ratón es similar al descrito anteriormente para los datos humanos. Una diferencia es que las muestras de ratón se secuencian a menos profundidad que las muestras humanas. Se anticipa que las muestras de sangre del ratón son de 100 pl. En 100 pl de sangre hay linfocitos -100K y, por lo tanto, la secuenciación a una profundidad mucho mayor que 100K no mejora significativamente la precisión. Por lo tanto, solo se obtienen 100K lecturas para cada muestra de ratón. A pesar de que el número de lecturas es más pequeño para los ratones que para los humanos, se toma una muestra de una fracción más grande del total de ratones y de los linfocitos sanguíneos. Se espera que el número total de linfocitos de ratón sea más de 3 órdenes de magnitud menor que el de los humanos. De manera similar, 100 pl de sangre proporcionarán un mejor muestreo (-10%) de los linfocitos en la sangre del ratón en el momento en comparación con el muestreo obtenido con 10 ml de sangre humana (0.2%).
Ejemplo 14
Análisis de repertorio de IgH y TCR en modelo SLE de ratón
Se usa un modelo de ratón de SLE para estudiar la relación entre el repertorio de TCR/BCR y la actividad de la enfermedad. El modelo de ratón es el B6 con los loci slel y sle3 de NZM2410. Estos ratones B6.slel.sle3 (BSS) desarrollan nefritis de tipo SLE de manera espontánea. Se estudian tres tipos de cohortes. Para todos los puntos de estudio, se obtiene BUN en sangre, creatinina y autoanticuerpos antinucleares, proteína en la orina y nivel de
creatinina. Se determina si una puntuación generada a partir del repertorio de TCR/BCR en sangre se correlaciona bien con estos índices medidos de enfermedad renal. La primera cohorte es similar a la cohorte humana descrita donde se recogen muestras de sangre longitudinales junto con la evaluación de la función renal. Específicamente, se siguen 7 ratones BSS mensualmente hasta el mes 8. Al final, estos ratones se sacrifican y, además de la sangre, se analizan el bazo y el tejido renal. Como control, se evalúan 5 ratones B6 de manera similar. La segunda cohorte es una sección transversal donde se sacrifican diferentes cohortes de animales en momentos específicos y se analizan muestras de bazo, riñón y sangre en ese momento. Específicamente, se sacrifican 5 ratones BSS cada mes y se analizan la sangre, el bazo y el riñón. Como control, dos ratones de control B6 se evalúan de la misma manera. Finalmente, una tercera cohorte se trata con esteroides después de la aparición de la enfermedad y la evaluación de la nefritis y luego se obtienen muestras de sangre de forma regular. Específicamente, a los 4 meses de edad, 20 ratones que tienen la enfermedad se tratan con esteroides y luego cada dos semanas durante los próximos 4 meses se extrae sangre para el análisis del repertorio de TCR/BCR y la evaluación de la función renal. Como control, 5 ratones BSS se tratan con placebo y se siguen de manera similar. El análisis del repertorio de TCR y BCR se realiza desde todos los puntos de estudio (es decir, diferentes puntos de tiempo y diferentes tejidos para el mismo punto de tiempo). El análisis incluirá PCR de 2 etapas, procesamiento de secuenciación y análisis de datos primarios como se describe anteriormente.
Ejemplo 15
Identificación y dinámica de clonotipos que se correlacionan con el SLE del ratón
En primer lugar, se identifica un conjunto de clonotipos que se correlacionan con la función renal. Como medida de la función renal, se puede usar la proporción de proteína/creatinina en la orina, creatinina sérica o niveles de BUN. En la primera y tercera cohortes, se puede evaluar la correlación del nivel sanguíneo de cada clonotipo de HPC con cada una de las tres medidas. De manera similar a loque se describe en humanos, se puede evaluar si hay un gran aumento en el número de clonotipos con una alta correlación a 1, 2, o si las 3 medidas de la función renal se superan a las expectativas aleatorias (o pruebas de permutación). Dada esa expectativa aleatoria, el umbral de correlación se elige donde se espera que solo el 10% de los clonotipos con un nivel de correlación por encima de ese umbral tenga el nivel de correlación observado por casualidad (10% de descubrimiento falso). Estos clonotipos se centran en, y este conjunto se define como "clonotipos correlacionados".
Además de este método estadístico para identificar clonotipos correlacionados, los clonotipos podrían identificarse como relevantes para la enfermedad mediante un método "funcional" de enriquecimiento de clonotipos específicos en tejido renal. Por el método funcional, se puede identificar un conjunto de clonotipos en la cohorte 2 que puede ser relevante para la enfermedad, y estos se llaman clonotipos correlacionados funcionalmente identificados. Se puede evaluar el grado de superposición entre la definición "estadística" y la definición "funcional" de correlacionar clonotipos. Las cohortes 1 y 3 tienen muestras de riñón recolectadas en el último punto de tiempo. Se puede evaluar si los clonotipos enriquecidos en estas muestras de riñón están presentes en la sangre y se encuentran entre los clonotipos con mayor correlación con la función renal.
Se puede evaluar la dinámica de los clonotipos correlacionados (identificados estadísticamente y funcionalmente). Por ejemplo, utilizando datos de la cohorte 2, el curso temporal del aumento y la caída (si corresponde) de sus niveles se evalúa en los tres compartimentos: riñón, sangre y bazo.
En los clonotipos correlacionados estadísticamente identificados, un subconjunto de los clonotipos correlacionados se identificaría en virtud de su correlación con la función renal. Los clonotipos correlacionados pueden identificarse sin conocer los datos de la función renal. En otras palabras, se pueden comprender las características que distinguen los clonotipos correlacionados de aquellos que son irrelevantes para la enfermedad. Para hacer eso, un conjunto de clonotipos con baja correlación con la función renal se identifica como clonotipos de control no correlacionados.
Características de los clonotipos que se correlacionan con la enfermedad. Después de identificar los dos conjuntos de clonotipos, correlacionando y no correlacionando, se buscan las características que distinguen estos dos conjuntos. Se realizan análisis separados y combinados utilizando los clonotipos correlacionados identificados estadística y funcionalmente. Se evalúa el mismo tipo de características estudiadas en humanos, por ejemplo, el nivel del clonotipo, la presencia de motivos de secuencia particulares y la secuencia de otros clonotipos relacionados. Como se describe para el estudio en humanos, existe un riesgo significativo de sobrealimentación y, por lo tanto, es necesario emplear una técnica de validación cruzada o conjuntos de entrenamiento y pruebas separados.
Una utilidad para el experimento con ratones es la disponibilidad de células que permiten evaluar si los clonotipos correlacionados están enriquecidos en un subtipo específico de células. Se estudia si los clonotipos correlacionados están enriquecidos en algunos subtipos de células; se puede realizar una secuenciación desde el conjunto completo de linfocitos y desde el subtipo específico donde se enriquecen los clonotipos correlacionados, y este criterio de enriquecimiento se puede usar como una característica adicional para distinguir los clonotipos correlacionados de otros clonotipos irrelevantes para la enfermedad. Con el fin de saber qué subtipos celulares se enriquecen, se toman dos metodologías: hipótesis impulsadas y sin hipótesis. El primero es probar una docena de marcadores de superficie candidatos en células T o B en un conjunto de muestras. Por ejemplo, un candidato es CD69 en las células T para seleccionar las células T activadas. Para las células B, los estudios han demostrado el aumento de células CD27high
en el SLE activo, y por lo tanto es un buen candidato para un marcador de células que pueden tener un enriquecimiento de los clonotipos correlacionados. En cada uno de estos experimentos, los subtipos celulares específicos se purifican a través de FACS. Luego se realiza una reacción de secuenciación para el ADNc del complemento completo de los linfocitos, así como para el ADNc de los linfocitos que se purificaron mediante FACS a partir de una colección de diferentes muestras. Se evalúa si los dos conjuntos de clonotipos correlacionados y no correlacionados están presentes en diferentes proporciones en el complemento completo de linfocitos en comparación con el subconjunto purificado por FACS. Los marcadores que tienen una gran diferencia pueden ser útiles para identificar clonotipos correlacionados. El enriquecimiento de clonotipos en subtipos de células con estos marcadores se utiliza además de los parámetros de secuencia para detectar clonotipos correlacionados.
En la metodología libre de hipótesis, se buscan marcadores que se expresan diferencialmente en células con un clonotipo correlacionado de otras células. Se seleccionan algunos casos en los que un clonotipo de TCR específico se correlaciona claramente con la enfermedad, y los casos en los que ese clonotipo está muy enriquecido, representa la mayoría de los clonotipos con el mismo segmento V. El FACS se realiza utilizando anticuerpos contra el segmento V específico (los anticuerpos contra todos los segmentos V están disponibles comercialmente) para seleccionar una población que esté altamente enriquecida para las células que llevan el clonotipo correlativo. El a Rn se puede preparar a partir de estas células y la expresión de todos los genes se puede estudiar realizando un experimento en serie. Como control, se puede usar ARN total de linfocitos y/o ARN de células purificadas con FACS que llevan otro segmento V irrelevante. Se pueden buscar los marcadores que distinguen al máximo la muestra obtenida del segmento V purificado por FACS con el clonotipo correlativo de los controles. Se pueden encontrar marcadores, incluidos los marcadores de superficie (ya que es mucho más fácil hacer FACS con proteínas de superficie) que distinguen a las dos poblaciones. Si se observa un marcador de ARN consistente de muestras de varios ratones, se valida al nivel de proteína. Usando las mismas muestras, los anticuerpos contra la proteína marcadora se usan en un ensayo FACS para purificar las células que llevan la proteína marcadora. Se puede probar más de un marcador para aumentar la posibilidad de validar uno de ellos. El TCR y/o BCR de las células purificadas se secuencian. Si los resultados del ARN se mantienen en el nivel de la proteína, los clonotipos correlacionados deben enriquecerse en el subconjunto purificado de células. Después de validar que los resultados de ARN aún se mantienen en el nivel de proteína, los resultados se validan en otras muestras. Las muestras que no se sometieron al análisis de matriz se someten a análisis FACS utilizando el anticuerpo para la(s) proteína(s) marcadora(s). El TCR y/o BCR de las células purificadas se secuencian. Se evalúa si los clonotipos correlacionados están enriquecidos en las células purificadas mediante el uso de anticuerpos para el marcador(s) específico(s). Esto validará la utilidad del marcador(s) en la identificación de clonotipos correlacionados.
Ejemplo 16
Uso del repertorio de IgH y TCRp para medir la actividad de la enfermedad
El algoritmo para correlacionar clonotipos desde arriba puede aplicarse para identificar en todas las muestras de cohortes 1 y 3 correlacionar clonotipos en virtud de su secuencia y/o marcadores. Usando el nivel de los clonotipos correlacionados en cada paciente, se puede generar una puntuación de IA que se correlaciona con una medida de la función renal. Como se describió anteriormente, existe un riesgo de sobreajuste y es necesario emplear la técnica de validación cruzada y/o el conjunto de entrenamiento y prueba separado. La correlación de la IA y las medidas de la función renal se pueden evaluar de manera transversal (todos los puntos de estudio de todos los ratones). La cuestión de si los cambios en la puntuación de la IA en un ratón individual también se pueden evaluar cuando cambia la función renal. Esto se puede evaluar comparando la IA de la función renal alta y baja en el mismo animal de una manera similar a la que se describe en los seres humanos.
Ejemplo 17
Seguimiento de la recidiva metastásica en pacientes con cáncer de colon
Muchos cánceres que se detectan en un estadio tratable aún conllevan un riesgo continuo para el paciente de recurrencia de tumor metastásico. Tales recurrencias a menudo se detectan tarde y en estadios no tratables pueden ser fatales para los pacientes. Un ejemplo de tal situación es el del cáncer de colon recurrente. A pesar de los programas de detección de cáncer de colon cada vez más agresivos, el cáncer de colon representa una de las neoplasias malignas más comunes en los Estados Unidos. Aproximadamente 150.000 pacientes por año son diagnosticados con cáncer de colon en estadios serios pero tratables (Estadio II y Estadio III). Estos pacientes se tratan con resección tumoral seguida de un tratamiento de quimioterapia. Si bien estos tratamientos son generalmente efectivos, sin embargo, existe una posibilidad significativa de que estos pacientes tengan recurrencias metastásicas del tumor primario en los años posteriores al tratamiento. El 50% de los pacientes en estadio III, por ejemplo, tendrá una recurrencia dentro de los 5 años de la cirugía. Estas recurrencias pueden ser aisladas (por ejemplo, en el colon o el hígado) o multifocales. En cualquier caso, pero especialmente si están aislados, detectarlos en un estadio temprano puede jugar un papel en maximizar las posibilidades de éxito de la terapia (cirugía y/o quimioterapia).
Actualmente hay dos pruebas utilizadas en la monitorización posterior al tratamiento. La tomografía computarizada del abdomen y el tórax se usa para identificar los tumores visibles en estas imágenes. Normalmente, estas exploraciones se realizan a intervalos de 6 a 12 meses durante los primeros 5 años posteriores a la terapia. Si bien estas
exploraciones pueden revelar tumores malignos en estadio temprano, allí se discute la efectividad clínica. Las desventajas de estas exploraciones incluyen el hecho de que someten a los pacientes a cantidades significativas de radiación que pueden causar tumores adicionales y un gasto significativo. Se ha demostrado que otra prueba basada en sangre tiene algún valor: la prueba CEA. Esta prueba de anticuerpos mide el nivel de una proteína en el suero que es específica de algunos tumores de colon. El inconveniente de las pruebas de CEA es su falta de sensibilidad (<60% de los pacientes con tomografías computarizadas positivas tienen una prueba de CEA positiva).
Los linfocitos obtenidos del tumor primario resecado se usan para desarrollar un perfil inmune que se puede usar para agregar sensibilidad a una prueba basada en sangre para los TCR de recurrencia temprana del cáncer (y/o BCR) de los linfocitos encontrados en el tumor resecado que se pueden amplificar y secuenciar. Los clonotipos que están enriquecidos en la muestra del tumor son probablemente relevantes para la respuesta inmune al tumor. Se pueden utilizar extracciones de sangre posteriores del paciente para evaluar el nivel de estos clonotipos. Un aumento en el nivel de estos clonotipos puede indicar una respuesta inmune a la recurrencia de un tumor. En este caso, la detección de la respuesta inmune puede ser más sensible que la detección del marcador tumoral en sí.
Estudio de descubrimiento para la detección de recurrencia de cáncer usando una prueba de calibración. Se realiza un estudio de descubrimiento para determinar la probabilidad de detección de recurrencia dado el perfil de TCR en sangre (y/o BCR). Para este estudio se utilizan muestras tomadas de muestras de tumores resecados, así como muestras de sangre de seguimiento de pacientes con resultados conocidos. Se secuencia el TCR (y/o BCR) de todas estas muestras. Los candidatos para los clonotipos correlacionados son aquellos que están presentes en los datos de TCR (y/o BCR) de las muestras de tumores. Dados los resultados conocidos en este estudio de capacitación, utilizando las técnicas de validación cruzada estándar, un modelo que genera una puntuación (riesgo de recurrencia) dado el nivel de los diferentes clonotipos se concibe. Por lo tanto, esta puntuación de recurrencia se calcula en un nuevo paciente midiendo los clonotipos en el tumor resecado (punto de calibración) y los datos de los clonotipos encontrados en la sangre del mismo paciente en un momento posterior durante la monitorización de la recurrencia. El uso de los datos del tumor permite una gran reducción en el número de clonotipos presentes en la sangre que se consideran en este análisis.
Estudio de descubrimiento para la detección de recurrencia de cáncer mediante una prueba de calibración y un estudio de población. Es probable que no todos los clonotipos enriquecidos en la muestra del tumor sean relevantes para la respuesta inmune al tumor. Puede haber algunos linfocitos que se expandieron localmente debido a una condición inflamatoria favorable. En otro caso, el estudio de descubrimiento se realiza utilizando las mismas muestras, pero el estudio se utiliza para identificar los parámetros que distinguen los clonotipos "correlacionados" de los "no correlacionados". Estos parámetros pueden incluir 1) Motivo de secuencia: El motivo puede ser una región V o J específica, una combinación de VJ o secuencias cortas en la región DJ que está asociada con un clonotipo que se correlaciona; 2) Tamaño del clonotipo; 3) Nivel: nivel absoluto (número de lecturas por millón) o nivel de rango; 4) Similitud con otros clonotipos: la presencia de otros clonotipos altamente relacionados, como aquellos con cambios silenciosos (diferencias de nucleótidos que codifican para los mismos aminoácidos) o aquellos con cambios de aminoácidos conservativos; 5) Para los BCR, el nivel de mutaciones somáticas en el clonotipo y/o el número de clonotipos distintos que difieren en las mutaciones somáticas de un clonotipo de línea germinal. 6) Presencia en una célula portadora de un marcador específico. Este estudio da como resultado un algoritmo que puede predecir qué clonotipos probablemente se correlacionarán con la recurrencia del cáncer en la sangre dado un conjunto específico de clonotipos presentes en una muestra tumoral determinada. Estos clonotipos se utilizan para desarrollar una puntuación de riesgo de recurrencia de la misma manera que se describió anteriormente.
Estudio de descubrimiento para la detección de recurrencia de cáncer mediante un estudio de población. En otro caso, los clonotipos medidos en el tumor resecado se utilizan para generar un modelo que predice la correlación de los clonotipos en muestras aún no vistas. Este modelo también se puede usar para generar una puntuación de riesgo de recurrencia de manera análoga a la descrita anteriormente. En este modelo no habría necesidad de medir los clonotipos en el tejido canceroso resecado en un nuevo paciente sometido a monitorización de recurrencia, sino que el riesgo de recurrencia podría evaluarse simplemente midiendo los clonotipos en una muestra de sangre determinada.
Estudio de descubrimiento para la detección de cáncer de colon primario mediante un estudio de población. Como extensión, la detección de cánceres primarios se logra utilizando la misma metodología. Con los cánceres primarios, no se ha resecado un tumor que se pueda usar para enriquecer los clonotipos relevantes. Sin embargo, incluso en presencia de datos de resección tumoral, la secuencia adicional y otros parámetros deben utilizarse para identificar los clonotipos relevantes y generar una puntuación para la probabilidad de detección de cáncer. Por lo tanto, por extensión, si el algoritmo es lo suficientemente predictivo, se puede detectar el cáncer de la sangre (u otro fluido corporal) sin los datos del tumor resecado. En este caso, debe estar disponible un estudio de descubrimiento con muestras de sangre de pacientes que preceden su diagnóstico de cáncer primario.
De manera análoga a la descrita anteriormente, los parámetros (secuencia y otros) se pueden identificar para predecir los clonotipos que se correlacionan con la respuesta del sistema inmune al tumor. Luego se puede usar un modelo para generar una puntuación de riesgo de cáncer que predice el riesgo de progresión al cáncer de colon. Este algoritmo se puede aplicar a la muestra de sangre de un nuevo paciente para medir el riesgo de cáncer de colon primario.
E je m p lo 18
Monitorización del rechazo en pacientes con trasplante cardíaco
Los trasplantes de corazón son un procedimiento relativamente poco frecuente ya que el suministro de órganos es muy limitado. Se realizan 3.500 trasplantes de corazón cada año en todo el mundo. Cada procedimiento es muy costoso y los órganos que se utilizan no tienen precio. Como resultado, los pacientes que reciben estos órganos reciben un tratamiento extremadamente proactivo. Para medir el estado de la reacción inmune al órgano donado en un momento en que las intervenciones con inmunosupresores pueden ser efectivas, los pacientes reciben biopsias periódicas del corazón para medir la inflamación del órgano. Sobre la base de estas pruebas, se pueden administrar cursos agresivos de inmunosupresores. Estos procedimientos tienen varias limitaciones. Como procedimientos quirúrgicos invasivos tienen riesgos para el paciente. Además, son caros y solo se pueden hacer en intervalos poco frecuentes. Se ha demostrado que las pruebas basadas en sangre basadas en el perfil de la expresión de un panel de 11 genes de prueba (Allomap) son bastante sensibles para detectar el rechazo de órganos, pero carecen de la sensibilidad suficiente como para reemplazar una biopsia y, en cambio, se utilizan para decidir cuándo hacer una biopsia En una instancia, los perfiles t Cr (y/o BCR) se utilizan para evaluar el estado de "rechazo" y generar una puntuación de riesgo de rechazo que predice la probabilidad de rechazo en un período de tiempo específico. Se concibe que se puede realizar un estudio de descubrimiento para determinar la probabilidad de rechazo dado el perfil de TCR en sangre (y/o BCR). Esto se puede usar en la clínica para informar las terapias inmunosupresoras que se están utilizando.
Descubrimiento de correlación de clonotipos utilizando un estudio de población. En este caso, se utiliza una población de pacientes postrasplante con muestras de sangre con resultado clínico conocido. Se secuencia la TCR (y/o BCR) de todas estas muestras y se utiliza la correlación de los clonotipos individuales con el resultado de rechazo para distinguir la correlación de los clonotipos no correlacionados. Posteriormente, se derivan parámetros que distinguen esas dos clases de clonotipos. Estos parámetros pueden incluir 1) Motivo de secuencia: el motivo puede ser una región V o J específica, una combinación de VJ o secuencias cortas en la región DJ que está asociada a la correlación de un clonotipo; 2) Tamaño del clonotipo; 3) Nivel: nivel absoluto (número de lecturas por millón) o nivel de rango; 4) Similitud con otros clonotipos: la presencia de otros clonotipos altamente relacionados, como aquellos con cambios silenciosos (diferencias de nucleótidos que codifican para los mismos aminoácidos) o aquellos con cambios de aminoácidos conservativos; 5) Para los BCR, el nivel de mutaciones somáticas en el clonotipo y/o el número de clonotipos distintos que difieren en las mutaciones somáticas de un clonotipo de línea germinal. 6) Presencia en una célula portadora de un marcador específico. Un método alternativo o complementario para definir el clonotipo correlativo y no correlativo vendría si las muestras del estudio tienen muestras de biopsia del injerto, particularmente si estaba en rechazo activo. Se espera que en ese momento haya un gran enriquecimiento de los clonotipos correlacionados. Los parámetros para distinguir estos de los otros clonotipos se identifican como se discutió anteriormente.
Los datos del perfil de las muestras de sangre se utilizan para predecir la probabilidad de rechazo. Dados los resultados conocidos en este estudio de capacitación, se puede diseñar un modelo que utilice las técnicas de validación cruzada estándar que genera una puntuación de riesgo de rechazo dado el nivel de los diferentes clonotipos. Dado el perfil en una nueva muestra de sangre de TCR (y/o BCR) en un punto específico, se puede generar un puntaje de riesgo de rechazo relacionado con la probabilidad de rechazo.
Descubrimiento de correlación de clonotipos utilizando una prueba de calibración. En otro caso, se implementa un método para identificar clonotipos correlacionados utilizando una prueba de calibración para cada paciente. Este método consiste en tomar una primera muestra de biopsia después del trasplante. La presencia de material de biopsia del injerto posterior al trasplante ofrece la posibilidad de analizar los TCR de la muestra de biopsia para identificar los clonotipos correlacionados según la definición de los que prevalecen en esta muestra. Este conjunto de clonotipos se sigue en la sangre y se genera una puntuación para la probabilidad de rechazo. El algoritmo para generar la puntuación de riesgo de rechazo se deriva a través de un estudio de descubrimiento que es similar al descrito anteriormente que utiliza los datos clínicos disponibles y los niveles de los clonotipos correlacionados para generar una puntuación de riesgo de rechazo que se aproxima a la probabilidad de rechazo.
En este caso, se realiza una prueba de calibración específica utilizando material de una primera biopsia posterior al trasplante, pero se pueden reemplazar otras biopsias mediante el uso de muestras de sangre cuyos clonotipos podrían usarse junto con esta prueba de calibración para medir la puntuación de riesgo de rechazo.
Además de la biopsia de injerto, las muestras de sangre antes del trasplante sirven como otro punto de calibración. Es poco probable que los clonotipos que prevalecen en esta muestra estén relacionados con el rechazo que representa más bien la historia de antígenos anteriores que el paciente ha visto. Por lo tanto, al considerar las muestras de sangre después del trasplante, se pueden restar los clonotipos que estaban presentes antes del trasplante para determinar los clonotipos correlacionados. Estos clonotipos se utilizan para generar un modelo de riesgo de rechazo.
En este caso, se podrían usar dos pruebas de calibración: una antes del trasplante y otra de una biopsia después del trasplante. Estas calibraciones podrían utilizarsejunto con los clonotipos derivados de un análisis de sangre para medir el riesgo de rechazo.
Descubrimiento de correlación de clonotipos utilizando una prueba de calibración y un estudio de población. En otro caso, la identificación de los clonotipos correlacionados se puede lograr a través de una combinación de las
metodologías anteriores. Específicamente, esto se puede lograr utilizando el estudio de población para generar un algoritmo para predecir clonotipos correlacionados. Además, se puede lograr a través de los datos de calibración del mismo paciente mediante biopsia de injerto y/o muestras de sangre antes del trasplante. Una instancia más preferida empleará ambas metodologías: algoritmo construido por la población y calibración individual para identificar con mayor precisión los clonotipos correlacionados. Luego se genera una puntuación de riesgo de rechazo utilizando el nivel de estos clonotipos para predecir la probabilidad de rechazo mediante el uso del estudio de población como un conjunto de capacitación.
En este caso, se pueden usar dos pruebas de calibración: una antes del trasplante y una de una biopsia después del trasplante. Estas calibraciones podrían utilizarsejunto con los clonotipos derivados de un análisis de sangre para medir el riesgo de rechazo.
La predicción de GVHD se puede hacer de una manera muy similar con el mismo concepto del estudio de población para generar un algoritmo para predecir clonotipos correlacionados. También se puede generar la calibración "negativa" a partir de la muestra de donante previa al trasplante. Es probable que una metodología que utilice tanto el algoritmo como la calibración sea más predictivo de los clonotipos correlacionados. Se puede generar un algoritmo para calcular una puntuación de la probabilidad de GVHD dado el nivel de los clonotipos correlacionados utilizando un estudio de población de la manera descrita anteriormente. Este algoritmo se puede usar para predecir la probabilidad de GVHD en el siguiente grupo de pacientes.
Ejemplo 19
Monitorización de la infección por PML en pacientes con EM tratados con natalizumab
El perfil de TCR y/o BCR se puede usar para detectar leucoencefalopatía multifocal progresiva (LMP) subclínica en pacientes con e M. La PML es una enfermedad grave y, a menudo, mortal que causa enfermedad desmielinizante a menudo rápidamente progresiva al matar oligodendrocitos que sintetizan la mielina. Es causada por el virus JC que está presente en una fase latente en la mayoría de la población. En una fracción de la población inmunosuprimida (por ejemplo, el SIDA), el virus se reactiva, lo que lleva al desarrollo de esta grave enfermedad. Además, algunos pacientes que están siendo inmunodeprimidos a través del uso de medicamentos como los pacientes después de un trasplante también pueden desarrollar LMP. Algunos medicamentos específicos se han relacionado con el riesgo de LMP en poblaciones específicas de pacientes. Por ejemplo, natalizumab (Tysabri) se asoció con el desarrollo de más de 10 casos de LMP entre pacientes con esclerosis múltiple (EM) que llevó a su retiro del mercado por un período de tiempo. Natalizumab es bien aceptado como más efectivo que los otros medicamentos aprobados por la FDA para la esclerosis múltiple, pero su uso se ha visto limitado por el temor al desarrollo de la LMP. Una vez que se sospecha la presencia de LMP, se puede realizar una plasmaféresis para reducir la concentración del fármaco en el paciente. La superposición entre los síntomas de EM y LMP a veces puede retrasar la detección de LMP. Se necesita urgentemente la detección temprana de LMP subclínica.
Estos clonotipos se pueden discernir a partir de muestras de sangre de una población donde algunos pacientes desarrollaron LMP. Esta población se puede utilizar para identificar los clonotipos que se correlacionan con el desarrollo posterior de la LMP. Con la disponibilidad de estos clonotipos, se puede generar un algoritmo para identificar los parámetros que los distinguen de otros clonotipos.
Descubrimiento de correlación de clonotipos utilizando un estudio de población. En este caso, se genera un algoritmo para predecir los clonotipos que son relevantes para la aparición de LMP. El algoritmo se puede entrenar en un conjunto de clonotipos que se consideran correlacionados con la enfermedad. Se pueden usar muestras de sangre (u otro fluido corporal) en un estudio de descubrimiento de una población de pacientes con una infección latente con el virus JC, algunos de los cuales desarrollan LMP. Se puede secuenciar el TCR (y/o BCR) de todas estas muestras y se puede usar la correlación de clonotipos individuales con el resultado de reactivación del agente infeccioso para distinguir la correlación de los clonotipos no correlacionados. Se pueden identificar los parámetros que distinguen esas dos clases de clonotipos. Estos parámetros pueden incluir 1) Motivo de secuencia: el motivo puede ser una región V o J específica, una combinación de VJ o secuencias cortas en la región DJ que está asociada a la correlación de un clonotipo; 2) Tamaño del clonotipo; 3) Nivel: nivel absoluto (número de lecturas por millón) o nivel de rango; 4) Similitud con otros clonotipos: la presencia de otros clonotipos altamente relacionados, como aquellos con cambios silenciosos (diferencias de nucleótidos que codifican para los mismos aminoácidos) o aquellos con cambios de aminoácidos conservativos; 5) Para los BCR, el nivel de mutaciones somáticas en el clonotipo y/o el número de clonotipos distintos que difieren en las mutaciones somáticas de un clonotipo de línea germinal. 6) Presencia en una célula portadora de un marcador específico. Un método alternativo o complementario para definir el clonotipo correlativo y no correlativo provendría de un conjunto de pacientes que están desarrollando una respuesta inmune al mismo agente infeccioso. Los clonotipos enriquecidos (particularmente aquellos que se encuentran en un nivel significativamente más alto que antes de la respuesta inmune) en estos pacientes pueden considerarse correlatos y se pueden identificar los parámetros que los distinguen de otros clonotipos.
De manera similar, los clonotipos correlacionados pueden identificarse a partir de muestras de pacientes con LMP activa o de estudios in vitro para identificar los clonotipos que responden al antígeno del virus JC. Los clonotipos que responden pueden originarse en uno o varios sujetos que pueden estar sanos o infectados con el agente infeccioso.
Estos clonotipos se pueden considerar correlacionados y se pueden identificar los parámetros que los distinguen de otros clonotipos.
Los datos de perfil de las muestras en el estudio de descubrimiento se usan para predecir la probabilidad de reactivación. Dados los resultados conocidos en este estudio de capacitación, se puede diseñar utilizando las técnicas de validación cruzada estándar, un modelo que genera una puntuación de riesgo de LMP dado el nivel de los diferentes clonotipos. Por lo tanto, dado el perfil en una muestra de sangre de TCR (y/o BCR) en un punto específico, se puede generar una puntuación relativa a la probabilidad de reactivación. Este algoritmo ahora se puede usar con datos de un nuevo paciente para predecir los clonotipos correlacionados del paciente, así como para generar una puntuación de riesgo de LMP para la probabilidad de reactivación.
De manera muy similar, se pueden estudiar otros resultados relacionados con la infección. Por ejemplo, además de la reactivación de una infección latente, se puede evaluar la eliminación de la infección. Además, dado el repertorio de TCR y/o BCR, uno puede evaluar la probabilidad de tener inmunidad contra un agente infeccioso específico.
Ejemplo 20
Monitorización de reactivación de infecciones latentes
El perfil de TCR y BCR se puede usar para controlar infecciones que tienen periodos de infección aguda seguidos de latencia y reactivación. Los ejemplos de tales enfermedades incluyen la hepatitis B y C, así como los virus del herpes. Predecir las infecciones en un estadio temprano sería deseable.
Descubrimiento de clonotipos correlacionados usando una prueba de calibración. Se puede implementar un método para identificar clonotipos correlacionados utilizando una prueba de calibración para cada paciente. La presencia de una muestra biológica del mismo paciente en un momento anterior en donde el paciente tenía una respuesta inmune al agente infeccioso puede servir para identificar los clonotipos correlacionados. Este conjunto de clonotipos se puede seguir en la sangre y se genera una puntuación de Riesgo de Reactivación para la probabilidad de reactivación. El algoritmo para generar la puntuación se deriva a través de un estudio de descubrimiento que es similar al descrito anteriormente que utiliza los datos clínicos disponibles y los conteos de los clonotipos correlacionados para generar una puntuación de Riesgo de Reactivación que se aproxima a la probabilidad de reactivación. Para utilizar este puntaje, una muestra tomada de un nuevo paciente en la práctica clínica durante un período de infección aguda. Estos datos se utilizarían junto con una muestra posterior tomada durante el período latente para medir el riesgo de reactivación con fines clínicos.
Descubrimiento de correlación de clonotipos utilizando una prueba de calibración y un estudio de población. La identificación de los clonotipos correlacionados se puede lograr a través de una combinación de las metodologías anteriores. Específicamente, esto se puede lograr utilizando el estudio de población para generar un algoritmo para predecir clonotipos correlacionados. Los clonotipos correlacionados pueden obtenerse a partir de un estudio de población de pacientes con resultado conocido de la infección y/o un conjunto de pacientes con respuesta inmune activa al agente infeccioso, y/o de experimentos in vitro para identificar clonotipos reactivos con el agente infeccioso. Además, se puede lograr a través de los datos de calibración del mismo paciente utilizando puntos de datos más antiguos en el momento de una respuesta inmune activa contra el agente infeccioso relevante. Preferiblemente, se emplearán ambas metodologías: algoritmo construido por la población y calibración individual para identificar con mayor precisión los clonotipos correlacionados. Luego se genera una puntuación de riesgo de reactivación utilizando el nivel de estos clonotipos para predecir la probabilidad de reactivación mediante el uso del estudio de población como un conjunto de entrenamiento. Para utilizar esta puntuación, se perfila una muestra tomada de un paciente nuevo en la clínica durante un período de infección aguda. Estos datos se utilizarían junto con una muestra posterior tomada durante el período latente para medir el riesgo de reactivación con fines clínicos. Se puede emplear una estructura similar para estudiar la eliminación de agentes infecciosos y/o la inmunidad a ella.
Ejemplo 21
Monitorización de la respuesta alérgica durante la inmunoterapia
La rinitis alérgica es una afección común que afecta a ~11% de la población de los Estados Unidos. Es típicamente una alergia al polen o al polvo. Eliminar la exposición es difícil e implica un esfuerzo de monitorización. Los tratamientos más comunes utilizados en la rinitis crónica son los descongestivos, los antihistamínicos y los esteroides nasales. En casos severos se realiza inmunoterapia. El objetivo de la inmunoterapia es desensibilizar al paciente. Primero, se hace un desafío con muchos alérgenos potenciales para identificar el alérgeno específico al que el paciente está reaccionando. Luego, al paciente se le inyecta una cantidad cada vez mayor de alergeno durante un período de meses a años hasta que se alcanza una dosis de mantenimiento, y luego el tratamiento continúa durante varios años. Por lo general, el paciente puede sentir una mejoría en los síntomas dentro de los 3 a 6 meses, pero también puede llegar hasta los 12 a 18 meses, pero una gran parte de los pacientes no se benefician del tratamiento o tienen recaídas. Una razón para el lento aumento de la dosis es el riesgo de anafilaxis si el paciente recibe una dosis alta de alérgeno antes de que él/ella esté lo suficientemente sensibilizado.
Los perfiles TCR (y/o BCR) se pueden usar para evaluar el estado de la enfermedad en la rinitis alérgica y generar un puntaje de alergia que predice cuán propenso al paciente a presentar una respuesta alérgica en caso de que esté expuesto al alérgeno relevante. Se concibe que se puede realizar un estudio de descubrimiento para determinar la probabilidad de respuesta alérgica dado el perfil de t Cr en sangre (y/o BCR). Esto puede ser utilizado en la adaptación del tratamiento de inmunoterapia. La posible decisión clínica puede ser interrumpir el tratamiento si se considera ineficaz, continuar con el régimen de inyección o acelerar el tratamiento para alcanzar la dosis de mantenimiento más rápido.
Descubrimiento de correlación de clonotipos utilizando un estudio de población. En este caso, se puede utilizar una población de pacientes con rinitis alérgica en inmunoterapia con muestras de sangre con resultado clínico conocido. Se puede secuenciar el TCR (y/o BCR) de todas estas muestras y se puede usar la correlación de clonotipos individuales con el resultado de alergia para distinguir la correlación de los clonotipos no correlacionados. Posteriormente, se pueden derivar parámetros que distinguen esas dos clases de clonotipos. Estos parámetros pueden incluir 1) Motivo de secuencia: el motivo puede ser una región V o J específica, una combinación de VJ o secuencias cortas en la región DJ que está asociada a la correlación de un clonotipo; 2) Tamaño del clonotipo; 3) Nivel: nivel absoluto (número de lecturas por millón) o nivel de rango; 4) Similitud con otros clonotipos: la presencia de otros clonotipos altamente relacionados, como aquellos con cambios silenciosos (diferencias de nucleótidos que codifican para los mismos aminoácidos) o aquellos con cambios de aminoácidos conservativos; 5) Para los BCR, el nivel de mutaciones somáticas en el clonotipo y/o el número de clonotipos distintos que difieren en las mutaciones somáticas de un clonotipo de línea germinal. 6) Presencia en una célula portadora de un marcador específico. Un método alternativo o complementario para definir el clonotipo correlativo y no correlativo utilizaría una biopsia de material de prueba de alergia positiva de pacientes positivos para un alérgeno específico. En el sitio de inyección del alérgeno se espera que haya un gran enriquecimiento de los clonotipos correlacionados. Los parámetros para distinguirlos de los otros clonotipos se pueden identificar como se discutió anteriormente.
Los datos del perfil de las muestras de sangre se utilizan para predecir el estado de alergia. Dados los resultados conocidos en este estudio de entrenamiento, se puede diseñar un modelo que utilice las técnicas de validación cruzada estándar que generan un puntaje de alergia dado el nivel de los diferentes clonotipos. Dado el perfil en una nueva muestra de sangre de TCR (y/o BCR) en un punto específico, se puede generar un puntaje de alergia para estimar el grado en que este paciente es propenso a presentar una respuesta alérgica.
Descubrimiento de clonotipos correlacionados usando una prueba de calibración. Se puede implementar un método para identificar clonotipos correlacionados utilizando una prueba de calibración para cada paciente. Este método consiste en tomar una muestra de biopsia de un sitio con una respuesta alérgica positiva que se debe tomar del paciente. Esto puede ser de la prueba de alergia inicial que se realizó para determinar el alérgeno específico al que el paciente responde o muestra en el sitio de cualquier otra inyección de tratamiento. Esto se puede hacer más de una vez para asegurar que se sigan los clonotipos adecuados en caso de que exista cierta propagación de epítopos. El TCR y/o BCR de estas muestras de biopsia se pueden usar para identificar los clonotipos correlacionados según lo definido por los que prevalecen en esta muestra. Este conjunto de clonotipos se puede seguir en la sangre y se genera una puntuación para la probabilidad de respuesta alérgica. El algoritmo para generar el puntaje de alergia se deriva a través de un estudio de descubrimiento que es similar al descrito anteriormente que utiliza los datos clínicos disponibles y los niveles de los clonotipos correlacionados para generar un puntaje de alergia que estima el estado de alergia.
Descubrimiento de correlación de clonotipos utilizando una prueba de calibración y un estudio de población. La identificación de los clonotipos correlacionados se puede lograr a través de una combinación de las metodologías anteriores. Específicamente, esto se puede lograr utilizando el estudio de población para generar un algoritmo para predecir clonotipos correlacionados. Además, se puede lograr a través de los datos de calibración del mismo paciente utilizando una biopsia de un sitio con una respuesta alérgica positiva. Preferiblemente, se emplearán ambas metodologías: algoritmo construido por la población y calibración individual para identificar con mayor precisión los clonotipos correlacionados. Luego se genera un puntaje de alergia utilizando el nivel de estos clonotipos para predecir el estado de la alergia a través del uso del estudio de población como un conjunto de entrenamiento.
Ejemplo 22
Amplificación de secuencias de IgH a partir de ADN genómico
En este ejemplo, se describe la amplificación de secuencias de IgH a partir de ADN genómico. Dicha amplificación es ventajosa porque (1) el nivel de un clonotipo en el ADN genómico puede convertirse fácilmente en un número de células, y (2) en algunas neoplasias linfoides, el ARN no puede expresarse para el reordenamiento del receptor inmune relevante.
La amplificación del reordenamiento del receptor inmune es importante para la detección de neoplasias linfoides. Las neoplasias de células B son más comunes que los tumores de células T e IgH es el receptor inmunitario reordenado más común en las neoplasias de células B. Debido a la hipermutación somática, la confiabilidad de la amplificación de IgH a partir del ADN genómico puede aumentar al amplificar con múltiples cebadores para cada segmento V, aunque existe un riesgo de amplificación diferencial. En la amplificación del ADN genómico, se utilizaron los mismos cebadores
V que se usaron en la amplificación del ADNc. Cada segmento V se amplifica mediante 3 cebadores (en 3 regiones distintas del segmento V: A, B y C) en tres reacciones diferentes (Tablas 5-7, respectivamente) (ver figura 4A).
Tabla 5

Tabla 6


Tabla 7

La amplificación de IgH a partir del ADN genómico tiene varias diferencias con respecto a su amplificación del ADNc. El segmento C se une a la región VDJ a través de empalme y, por lo tanto, las secuencias del segmento C se pueden usar para la amplificación de ADNc pero no el ADN genómico. El uso del segmento C permite el uso de dos cebadores distintos en las amplificaciones 1a y 2a, lo que aumenta la especificidad. Para la amplificación del ADN genómico, se optó por utilizar cebadores que sean complementarios a las secuencias J (Tabla 8).
Tabla 8


Estos cebadores abarcan los límites exón-intrón, y los cuatro cebadores utilizados amplifican los diferentes segmentos J y alelos descritos en la base de datos IMGT. Los cebadores de la segunda etapa no tienen secuencias complementarias a las secuencias genómicas.
El uso de los cebadores J sobre los cebadores de región constante complementarios a la región constante de IgG permite la evaluación de las otras clases (IgM, IgD, IgA e IgE).
En el caso de ADNc, existe la opción de utilizar el cebador J o los cebadores de región constante. Se pueden usar varios cebadores de región constante para amplificar todas las clases y secuenciar algunas de las regiones constantes antes de ingresar en la secuencia J para vincular la información sobre el clonotipo y su clase específica. Las lecturas de secuenciación para muchas de las tecnologías de secuenciación son cortas y serían difíciles de lograr. Una de las plataformas actuales en el mercado (454 Roche) tiene una lectura más larga, pero tiene un rendimiento más bajo que otras plataformas. A medida que estas tecnologías se desarrollan más, esta opción se hace posible. Con las lecturas cortas actuales (<100 pb), nuestro trabajo en el ensayo de ADN genómico sugiere que para la amplificación a partir de ADNc se puede hacer utilizando tanto la metodología de cebado J como el de C. Se puede implementar la amplificación utilizando cebadores J a partir de ADNc. Sin embargo, dado que el segmento exónico de estos cebadores puede ser demasiado corto para la amplificación específica del ADNc, potencialmente la primera etapa de la PCR se puede hacer usando un conjunto de cebadores de región constante que abarque todas las diferentes clases (y los cebadores del segmento V en el otro lado como se ha demostrado). Luego, la segunda etapa de la PCR se puede hacer con los cebadores J, que son lo suficientemente largos para tener una alta especificidad para la segunda etapa de la PCR que utiliza una plantilla de baja complejidad. Los productos son luego secuenciados. Como se indicó anteriormente, la desventaja en comparación con el esquema demostrado para la IgG es que las mutaciones somáticas en la secuencia J pueden inhibir la amplificación. La ventaja es que todas las diferentes clases se evalúan, aunque la información sobre la clase de cada clonotipo no está completamente determinada. Potencialmente, se puede hacer una amplificación específica de clase IgG, IgM, IgD, IgA o IgE y comparar con la imagen general obtenida al usar todos los cebadores seguidos de J primer. Por ejemplo, se puede comparar el perfil de clonotipo obtenido a partir de la amplificación de IgG con el de todos los cebadores seguidos por el cebador J. La diferencia probablemente se deba a la mutación somática en el cebador J (que puede identificarse fácilmente en la reacción usando el cebador IgG) y los clonotipos de las otras clases, que luego pueden cuantificarse.
El uso de cebadores J en el ADNc también permite la comparación directa entre los resultados del ADNc y del ADN genómico. Esto proporcionaría información de nivel de expresión a nivel de clonotipo y, de hecho, puede tener relevancia funcional. La comparación del perfil de clonotipo de ADNc y ADN genómico de la misma sangre u otra muestra biológica identifica los clonotipos que tienen diferentes frecuencias que indican una expresión por célula inusualmente alta o baja. Esta información funcional puede utilizarse para predecir si es probable que un clonotipo se esté correlacionando con la enfermedad o no. Además, el nivel de expresión por célula de un clonotipo que se correlaciona con una enfermedad puede usarse para determinar la actividad de la enfermedad o la probabilidad de un resultado de la enfermedad. Por ejemplo, obtener el mismo nivel en el ensayo de ADNc para un clonotipo correlacionado en dos individuos puede indicar que los pacientes tienen diferentes actividades de enfermedad si el nivel de expresión por célula de los clonotipos (determinado por comparación con el perfil del clonotipo del ADN genómico) es diferente.
La segunda etapa de la PCR consiste en unir las secuencias necesarias para la amplificación. Los cebadores utilizados en la Segunda etapa se enumeran en la Tabla 9.
Tabla 9

La amplificación es posible usando los cebadores anteriores y otras secuencias que son sustancialmente similares. Las figuras 8A-8B muestran ejemplos de tales amplificaciones, que tuvieron éxito al menos en el rango del ADN genómico 50-2.000 |jg en 20 j l de ADN genómico de entrada.
El ensayo debe adaptarse a un amplio rango dinámico de ADN. Es posible que las muestras de biopsia no tengan una gran cantidad de material, pero dado que es probable que el tumor esté muy enriquecido, no hay necesidad de una gran cantidad de material de partida. Por otro lado, un millón de células tendrán ~6 jg de ADN genómico. Las PBMC que contienen 1 millón de células B probablemente tengan ~20 jg de ADN genómico. Para poder evaluar 1 millón de células B, se utilizan ~6.6 jg de ADN genómico en cada una de las 3 reacciones de PCR. Es de destacar que, si hay una mutación somática en la secuencia complementaria a uno de los cebadores, en este ejemplo solo se interrogan células -660K B. Es útil si el ensayo funciona en el rango de 50 a 10.000 ng. Se ha demostrado que el ensayo funciona en el rango de 50-2.000 ng de a Dn en 20 jl. Al ampliar la reacción a 100 jl, se pueden usar l0 jg de ADN.
Ejemplo 23
Monitorización de la leucemia linfoblástica aguda (LLA)
La enfermedad residual mínima (ERM) es un factor pronóstico importante para la estratificación de la LLA infantil. Por lo general, la ERM se analiza en la médula ósea en las pocas semanas posteriores a la terapia de inducción. La detección más sensible de las células leucémicas puede permitir la monitorización de la recurrencia del cáncer en la sangre.
El perfil del clonotipo para evaluar el nivel del clonotipo tumoral en la sangre se utiliza para detectar las células leucémicas con sensibilidad.
La calibración se identifica a través de la interrogación de una muestra con alta carga leucémica. Las células leucémicas suelen estar presentes con alta frecuencia en muestras de diagnóstico (sangre o médula ósea). La muestra de diagnóstico a menudo se secuencia para varias reordenaciones.
Si el tumor es una célula B, se puede evaluar la IgH completamente reorganizada, la IgH parcial reorganizada con D-J, IgK, incluidas las reorganizaciones de Kde.
Los reordenamientos de linajes cruzados ocurren frecuentemente, y el más frecuente es el TCRp parcialmente reorganizado (V-D) o (D-D). Para las células T, se produce un reordenamiento frecuente de TCRa y TCRp, y a una frecuencia más baja de TCRa. La secuenciación del repertorio de clonotipos para estos reordenamientos diferentes identifica el tumor particular, el reordenamiento presente en el tumor. El nivel en sangre de la secuencia específica puede entonces ser monitoreado.
La prueba de monitorización puede involucrar solo los tipos de reordenamientos relevantes. Por ejemplo, si los reordenamientos del tumor identificados en la muestra diagnóstica son IgH e IgK, entonces la IgH y la IgK se amplificarían y secuenciarían en muestras de sangre posteriores. El ADN de hasta aproximadamente 1 millón de células B de estas muestras puede usarse para amplificar IgH e IgK y se pueden obtener hasta aproximadamente 1 millón o más de lecturas de secuenciación, lo que da una sensibilidad de ensayo de 1 en un millón de células B es ~ a una sensibilidad de 1 en 10 millones de glóbulos blancos. Con esta gran sensibilidad, es probable que las células leucémicas se detecten significativamente antes de una recaída franca.
La evolución clonal ha sido descrita en ALL. Esto puede ocurrir a través de la sustitución de V u otros mecanismos. Para detectar la evolución, identificaremos los clonotipos relacionados con los presentes en la muestra de diagnóstico. Por ejemplo, se identificarán los clonotipos con la misma unión D-J, pero con una V diferente. La presencia de estos clonotipos relacionados a una frecuencia apreciable en la muestra de diagnóstico aumenta la probabilidad de la relevancia del clonotipo. Después de más de un reordenamiento (para este ejemplo, el tumor IgH e IgK) también puede mejorar este problema.
La mera presencia de células leucémicas puede no ser necesariamente suficiente para predecir la recaída. Se realizó un estudio de descubrimiento con muestras de sangre longitudinales para pacientes con resultados conocidos. Se evaluará en estas muestras si la mera presencia de un clonotipo leucémico es suficiente para predecir una recaída algún tiempo después. Además, evaluaremos el cambio en la frecuencia de estos clonotipos como un predictor de recaída. Además de la frecuencia de clonotipo, los marcadores en las células leucémicas pueden ser indicativos de recaída. La secuenciación se puede realizar antes y después del enriquecimiento de las células con el marcador correspondiente. Por lo tanto, se determina la frecuencia total del clonotipo. Además, se determina la fracción de estas células con el marcador relevante, lo que permite una estimación más precisa del riesgo de recaída.
De acuerdo con lo anterior, en un aspecto los métodos de la invención proporcionan ensayos para MRD basados en mediciones de perfil de clonotipo. Dichos ensayos incluyen el uso de perfiles de clonotipos para (i) monitorizar la presencia y abundancia de clonotipos específicos del paciente asociados con la enfermedad, incluidos los clones evolucionados, (ii) proporcionar recuentos de linfocitos de los cuales se derivan los clonotipos y (iii) proporcionar una medida de clonalidad (es decir, una medida de qué tan "sesgado" es un perfil para uno o unos pocos clonotipos). En un aspecto, se proporciona un ensayo que da valores para (i) y (ii) simultáneamente; y en otro aspecto, se proporciona un ensayo que proporciona las tres cantidades simultáneamente de una única medición del perfil de clonotipo de un paciente.
Ejemplo 24
Monitorización del rechazo de trasplante de un órgano sólido
El rechazo de un trasplante de órgano sólido puede ocurrir a través de dos vías distintas: presentación directa e indirecta. La vía directa utiliza las células presentadoras de antígenos donantes que se transfieren con el trasplante. Los receptores de células T están reconociendo en este caso al donante HLA. La vía indirecta por otro lado, ocurre algún tiempo después. En este caso, los péptidos donantes se presentan por el receptor HLA a las células T.
Las muestras de biopsia del órgano trasplantado se pueden usar para identificar secuencias relevantes para la calibración de los receptores de células T (o B) relevantes en el rechazo de trasplantes. Los clonotipos enriquecidos en una biopsia del órgano trasplantado se pueden comparar con la sangre en el momento del rechazo para identificar los clonotipos que son relevantes para el rechazo. El nivel de estos clonotipos en la sangre se monitoriza para predecir el estado del rechazo.
También se realiza calibración específica de antígeno. Para identificar los clonotipos relevantes para el rechazo por vía directa, los linfocitos del donante se irradian y se mezclan con PBMC receptoras. Las PBMC receptoras capaces de reconocer los linfocitos del donante se activan. El aislamiento de estas células activadas se realiza mediante cualquiera de varias técnicas. Por ejemplo, estas células se aíslan en virtud de la liberación de citoquinas mediante tinción intracelular de citoquinas o técnicas de captura de citoquinas. En su lugar (o, además) para el aislamiento, se permite que las células se repliquen in vitro. La comparación de secuencias de las células aisladas (y/o replicadas) con las secuencias de preactivación identifica los clonotipos que interactúan con las PBMC donantes. También se realizan los mismos procedimientos de activación de células T sin la adición de antígeno seguido de secuenciación para restar el fondo potencial de la activación independiente del antígeno. El nivel en sangre de los clonotipos relevantes para el rechazo del trasplante se controla luego para evaluar la actividad de rechazo de la vía directa. Luego se utiliza una medida de la diversidad general para monitorizar el nivel de rechazo por la vía directa.
Para identificar los clonotipos relevantes para el rechazo por la vía indirecta, los antígenos del donante deben presentarse en el contexto del HLA receptor. Dado que el HLA del donante es a menudo un antígeno importante, el HLA del donante se incuba con células presentadoras de antígenos que pueden presentar péptidos de1 HLA del donante en el contexto del HLA del receptor a las células T del receptor. De una manera similar a lo que se describe anteriormente, estas células se aíslan y replican para identificar los clonotipos que interactúan con e1HLA del donante en la ruta indirecta. Alternativamente, se realizan los mismos procedimientos de activación de células T sin la adición de antígeno seguido de secuenciación para restar el fondo potencial de la activación independiente del antígeno. Alternativamente, se utilizan células donantes y no solo HLA como fuente de antígeno. Las células donadoras se preparan de una manera que facilita que las células presentadoras de antígenos receptores presentan el antígeno donante en el contexto del receptor HLA. Esto se realiza mediante varios métodos alternativos, incluida la lisis con varios ciclos de congelación y descongelación o mediante sonicación antes de la adición a las células presentadoras de antígenos. Los clonotipos que son activados por estos antígenos en el contexto del receptor HLA, se identifican mediante la secuenciación de las células aisladas y/o replicadas como se describió anteriormente. Los mismos procedimientos de activación de células T sin la adición de antígeno seguido de secuenciación para restar el fondo potencial de la activación independiente del antígeno, se realiza alternativamente. Una vez que se identifican estos clonotipos, se monitoriza su nivel de sangre para evaluar la actividad de rechazo de la vía indirecta.
Ejemplo 25
Recurrencia del cáncer
La recurrencia del cáncer se detecta mediante la detección de la respuesta inmune al tumor. El nivel de clonotipos de células T y B que son relevantes para el tumor se usa para detectar la recurrencia del cáncer. El aumento en el nivel sanguíneo de las células T y B relevantes (o la frecuencia de los clonotipos relevantes de ADNc obtenido en la sangre) se detecta e indica el reconocimiento por parte del sistema inmunitario de la recurrencia del tumor.
La reducción de estos niveles también se detecta e indica el éxito del tumor para evadir el sistema inmunológico y, por lo tanto, el desarrollo de recurrencia del cáncer.
Los marcadores celulares cambian en las células que contienen clonotipos relevantes que se detectan e indican la recurrencia del tumor en ausencia de un cambio en la frecuencia de los clonotipos relevantes. Estos últimos casos pueden ser un reflejo del efecto tumoral en las células inmunes para hacerlas ineficaces o anérgicas.
Para determinar los clonotipos relevantes en un individuo, se utilizan muestras del tumor original y algoritmos desarrollados en estudios de población. Alternativamente, el antígeno específico del tumor se usa para definir los clonotipos que interactúan con las células tumorales. Por ejemplo, las células B o T que interactúan con algunos antígenos específicos del tumor se capturan y secuencian antes y después de este enriquecimiento para determinar los clonotipos específicos que interactúan con los antígenos específicos.
La experimentación in vitro se realiza utilizando técnicas como las analizadas anteriormente (por ejemplo, la unión a tetrámeros, la tinción intracelular de citoquinas o la captura de citoquinas) para determinar en una muestra de un paciente los clonotipos específicos que interactúan con los antígenos específicos del tumor. Una vez que se definen estos clonotipos, su nivel se controla en otras muestras de sangre. El cambio en el nivel de estos clonotipos indica una recurrencia del tumor.
Los clonotipos relevantes para el cáncer en un individuo se identifican mediante un ensayo in vitro para determinar los clonotipos que interactúan con el tumor. Las células tumorales se lisan utilizando ciclos repetidos de congelación y descongelación o sonicación. Esta preparación se agrega a células presentadoras de antígeno autólogas (o a PBMC autólogas que contienen células presentadoras de antígeno y células T). La mezcla se agrega a las células T autólogas y los clonotipos que se activan por antígeno se identifican por secuenciación de células T aisladas y/o replicadas y se comparan con la secuencia del material no enriquecido como se describe anteriormente. El mismo procedimiento de activación de células T se realiza sin la adición de antígeno seguido de secuenciación para, opcionalmente, restar el fondo potencial de la activación independiente del antígeno. Una vez que se determinan los clonotipos relevantes, se controla su nivel en la sangre para evaluar la probabilidad de recurrencia.
Cuando el cáncer es de células que son capaces de presentar antígeno, las células tumorales pueden opcionalmente no ser lisadas ya que pueden servir como células presentadoras de antígeno. El linfoma, un tumor de células B que puede presentar antígenos en algunos casos, sirve como célula presentadora de antígenos. Las células tumorales se activan opcionalmente in vitro para mejorar su capacidad de presentación de antígenos. Estos tumores se mezclan con células T autólogas (o PBMC). La secuenciación de los clonotipos de células T antes y después del enriquecimiento identifica los clonotipos relevantes para el cáncer. El nivel de estos clonotipos se controla en la sangre para determinar el riesgo de recurrencia.
El nivel del clonotipo en un tipo específico de células, por ejemplo, aquellas con marcadores de superficie específicos se monitorea para detectar células cancerosas que pueden evadir el sistema inmunológico. Entonces, para dos pacientes con el mismo nivel del clonotipo relevante, luego, dependiendo de los marcadores contenidos en la célula que contiene los clonotipos, un paciente puede tener una mayor probabilidad de recurrencia en comparación con otro. Para obtener la información, la secuenciación puede realizarse antes y después del enriquecimiento de las células
con un marcador en particular. Por lo tanto, se puede medir el total, así como la fracción y el número de células de clonotipo con el marcador en particular.
Ejemplo 26
Seguimiento de la infección por hepatitis C
La infección aguda de la hepatitis C suele ir acompañada de una respuesta inmune que es capaz de eliminar la infección en ~15% de los casos. Se ha demostrado que la capacidad para eliminar la infección está asociada con ciertos genotipos de HLA. En la mayoría de los casos, el virus no se elimina y se produce una infección crónica. Durante esta infección crónica, el virus es capaz de evadir la respuesta inmune que es probablemente responsable de gran parte del daño hepático resultante. El tratamiento más efectivo para la enfermedad es el interferón. Este tratamiento mata el virus al menos en parte a través de la activación de la respuesta inmune. Por lo tanto, La monitorización de la respuesta inmune puede ser útil en diferentes estados durante el curso de la enfermedad. Durante la fase aguda, evaluar la extensión de la reacción inmune puede ser útil para predecir quién tiene la probabilidad de eliminar el virus. Durante la fase crónica, la medición del nivel de la respuesta inmune puede proporcionar una indicación del grado de inflamación del hígado. Finalmente, la evaluación de la reacción inmune durante el tratamiento con interferón puede proporcionar una indicación temprana de si el tratamiento es efectivo. La evaluación de la respuesta inmune se puede realizar mediante la medición del repertorio de células T y B mediante la secuenciación como se describe anteriormente.
La identificación de los clonotipos relevantes para la hepatitis C en cada individuo se realiza mediante varios métodos. Los antígenos de la hepatitis C se utilizan como péptidos individuales, una mezcla de péptidos, proteínas o el virus completo. Las células T y/o las células B que interactúan con el antígeno se identifican mediante la evidencia del enriquecimiento de los clonotipos en células activadas por antígenos en comparación con su nivel en el resto de las células. Además, la biopsia hepática se realiza opcionalmente para estos pacientes durante el curso del tratamiento. Eso proporciona medios adicionales o alternativos para identificar los clonotipos relevantes para la hepatitis C. Es probable que los clonotipos significativamente más enriquecidos en el hígado en comparación con la sangre sean relevantes al menos en relación con el proceso inflamatorio en el hígado. Por lo tanto, sus niveles en la sangre en puntos posteriores se monitorean para evaluar la actividad inflamatoria del hígado. Finalmente, los estudios de descubrimiento en una población de pacientes pueden indicar un conjunto de secuencias o motivos que son relevantes para la hepatitis C. En este estudio de población, los clonotipos relevantes se identifican en virtud de su correlación con la enfermedad o el enriquecimiento en la biopsia hepática y se descubren los algoritmos para distinguir estos clonotipos de otros. Algunos de los criterios incluyen la frecuencia del clonotipo, el rango, la similitud de secuencia de múltiples clonotipos o el motivo de la secuencia, así como la presencia de algún marcador celular.
La tipificación HLA se utiliza como un método de estratificación. Los motivos específicos son predictivos solo en el contexto de tipos específicos de HLA.
Las células infectadas con virus a veces pueden evadir el sistema inmunológico. Por lo tanto, se realiza La monitorización del nivel del clonotipo en un tipo específico de células, por ejemplo, aquellas con marcadores de superficie específicos. Entonces, para dos pacientes con el mismo nivel del clonotipo relevante, luego, dependiendo de los marcadores contenidos en la célula que contiene los clonotipos, un paciente está montando una respuesta más rigurosa que el otro. Por ejemplo, el efecto del tratamiento con interferón puede ser tanto en el cambio cualitativo como en el cuantitativo en las células del clonotipo. Por lo tanto, es importante obtener el nivel del clonotipo y definir si tienen marcadores de células específicos. Para obtener la información, la secuenciación se realiza antes y después del enriquecimiento de las células con un marcador en particular. Por lo tanto, se puede medir el total, así como la fracción y el número de células de clonotipo con el marcador en particular.
Ejemplo 27
Hipersensibilidad a los fármacos
La identificación de clonotipos que son relevantes para una hipersensibilidad a un fármaco específico se realiza mediante estudios de población. En estos estudios, se identifican los clonotipos que se correlacionan con el ADR y las características que los distinguen de los otros clonotipos se identifican mediante diferentes criterios como la frecuencia, el rango, el cambio relativo antes y después del tratamiento, la similitud de secuencia de múltiples clonotipos, el motivo de la secuencia y la presencia de marcador celular. El motivo de secuencia puede ser dependiente de HLA cuando se determina que diferentes motivos son relevantes para diferentes secuencias de HLA correspondientes.
Otro método para la identificación de clonotipos relevantes para la hipersensibilidad a fármacos es por interacción con el antígeno. Los medicamentos y/o su(s) metabolito(s) se utilizan para capturar las células B que interactúan con él. De manera similar, el fármaco o sus metabolitos se incuban con células presentadoras de antígeno autólogas de manera óptima antes o al mismo tiempo de la adición de células T. Las células T activadas se aíslan o replican utilizando algunos de los métodos descritos anteriormente para obtener células enriquecidas con antígeno. Estas células enriquecidas con antígeno luego se secuencian y los clonotipos enriquecidos en estas células en comparación con las células no enriquecidas se identifican como relevantes para la interacción del fármaco.
Los mismos procedimientos de activación de células T sin la adición de antígeno también se realizan seguidos de una secuenciación para restar el fondo potencial de la activación independiente del antígeno farmacológico.
Esta prueba de calibración se realiza antes o después de tomar el medicamento. Las muestras de sangre antes y después de tomar el fármaco y el aumento in vivo en el nivel del clonotipo se utilizan como criterios adicionales para definir los clonotipos relevantes. Una vez que se identifican estos clonotipos, se monitorean para predecir la probabilidad de una hipersensibilidad al fármaco. La activación de los linfocitos con fármacos se utiliza simplemente para definir los clonotipos relevantes y el nivel en sangre de estos clonotipos se puede monitorizar en muestras posteriores mediante la secuenciación que genera un método sensible y específico para predecir las ADR. Los fármacos que se utilizan son moléculas pequeñas o biológicas, como los anticuerpos. De manera similar, se usa un metabolito farmacológico o una combinación de metabolitos para identificar las células T que interactúan con él como se describió anteriormente. El metabolito se genera por síntesis química o se purifica a partir de una muestra biológica. Por ejemplo, el fármaco se introduce en un organismo y los metabolitos del fármaco se purifican para su uso en el ensayo. Los metabolitos también se obtienen procesando el fármaco por células in vitro.
El nivel del clonotipo en un tipo específico de células, por ejemplo, aquellas con marcadores de superficie específicos, se monitoriza. El aumento en el nivel de clonotipo en células que llevan marcadores de activación después de la introducción del fármaco es más indicativo de hipersensibilidad al fármaco que si el marcador de activación no es presente. Para obtener la información, la secuenciación se realiza antes y después del enriquecimiento de las células con un marcador en particular. Por lo tanto, se mide el total, así como la fracción y el número de células de clonotipo con el marcador en particular.
Se predice una ADR sin tomar el fármaco. Se detecta un alto nivel de clonotipos que interactúan con el fármaco o la presencia de un motivo de secuencia que probablemente genere una fuerte respuesta al fármaco para predecir la ADR sin administrar el medicamento al paciente.
Se identifican características de respuesta inmunitaria similares de fármacos con ADR idiosincrásicas relacionadas con el sistema inmunitario. Estos medicamentos pueden tener clonotipos de alta frecuencia que interactúan con los medicamentos o sus metabolitos. Los medicamentos que probablemente tengan ADR relacionados con el sistema inmunológico se identifican después de la administración en solo un pequeño número de pacientes.
La respuesta subclínica de un fármaco causante de ADR se detecta mediante la determinación de un aumento en los clonotipos específicos del fármaco (y/o su metabolito) después de la administración del medicamento.
Los fármacos que probablemente tengan una ADR relacionada con el sistema inmunitario se identifican mediante el seguimiento del aumento en los clonotipos correspondientes a las ARD después de la administración en un pequeño número de pacientes.
Ejemplo 28
Métodos para la estratificación del riesgo en la enfermedad vascular carotidea
La inflamación involucrada en la formación y la estabilidad de las placas se detecta en un paciente. La respuesta inmune específica a la inflamación vascular se usa para indicar el riesgo de desestabilización de la placa. Los antígenos específicos relevantes en la reacción inmune en el ICA (incluidas las LDL modificadas u oxidadas y la proteína de choque térmico) se utilizan para identificar los clonotipos específicos relevantes para la reacción inmune en ICA. Usando procedimientos similares a los descritos anteriormente, se identifican los clonotipos de células T o B que interactúan con los antígenos específicos. Los niveles de los clonotipos identificados se monitorean para evaluar el riesgo de desestabilización de la placa ICA.
Los clonotipos que son relevantes para la desestabilización de la placa ICA también se identifican utilizando un algoritmo que se genera a partir de un estudio de población que identifica características que distinguen los clonotipos relevantes de otros. Estos clonotipos relevantes se identifican en el estudio de población en virtud de su correlación con la desestabilización de la placa o por su enriquecimiento significativo en la placa ICA (por ejemplo, obtenida de la endarterectomía carotidea) en comparación con la sangre. Los motivos de secuencia pueden ser específicos para distintos genotipos de HLA correspondientes. Los algoritmos desarrollados se utilizan para predecir los clonotipos relevantes de desestabilización de la placa ICA en otros pacientes.
Ejemplo 29
Análisis de repertorio de TCR en ratones EAE
Se trataron 10 ratones de la cepa SJL con el péptido 139-151 junto con adyuvante completo de Freund (CFA) usando un protocolo disponible comercialmente, por ejemplo, Laboratorios Hooke (Lawrence, MA). Ocho de estos ratones desarrollaron encefalitis autoinmune experimental (EAE), un modelo de esclerosis múltiple en ratones, y los otros dos no lo hicieron. Además, 2 ratones de la misma cepa fueron tratados solo con CFA. Para cada ratón, se obtuvieron puntuaciones diarias de enfermedad durante 61 días después de la inyección. El rango de puntuación fue de 0 a 5. Se obtuvieron muestras de sangre antes de la inyección y días específicos después. En general, se obtuvieron 11
muestras de sangre de cada ratón y los ratones se sacrificaron en el día 62 o 63 y se obtuvieron bazo, ganglios linfáticos y médula espinal. La sangre y el tejido se almacenaron inmediatamente en sangre animal, protegiendo el reactivo de ARN y ARN posterior, respectivamente. El ARN se extrajo de las muestras de sangre utilizando el kit de extracción de sangre Qiagen animal protect y las muestras de tejido se homogeneizaron mecánicamente, y el ARN se preparó utilizando el minikit ARN Qiagen Plus. El ADNc se produjo a partir de cada muestra usando el kit de síntesis de ADNc de Vilo (Life technologies). El repertorio de TCRp de cada una de las muestras se amplificó utilizando los cebadores de la Tabla 10.
Tabla 10

Se realizó una segunda PCR en cada una de las muestras utilizando cebadores en la Tabla 7. Cada muestra se amplificó con un par de cebadores que amplificaron el conjunto completo de amplicones de PCR de la primera etapa, pero que también incluían una etiqueta de secuencia individual que luego podría permitir que muestras individuales Identificarse en una mezcla de datos de secuencia. Se realiza una tercera PCR utilizando un par de cebadores para todas las muestras (Tabla 7). El propósito de la tercera PCR era asegurar la integridad de las secuencias terminales. La estructura del producto de la PCR se muestra en las figuras 2A-2B y en la figura 3A. Estas reacciones de PCR se agruparon en conjuntos de 64, de modo que cada muestra con el conjunto se indexó por una de las etiquetas únicas incorporadas en la segunda etapa de la PCR. Las moléculas de estos grupos se desnaturalizaron y se separaron en dos dimensiones en una superficie sólida mediante hibridación con una célula de flujo que contenía oligonucleótidos que se hibridan con las secuencias terminales de los productos amplificados. Las moléculas hibridadas se amplificaron luego mediante una amplificación de puente para formar agrupamientos en la superficie bidimensional, de modo que cada agrupación contenía aproximadamente 1.000 moléculas, cada una de las cuales era el resultado de una amplificación de una sola molécula de la agrupación de PCR. Luego se usó un método para escindir y liberar una de las dos cadenas de cada una de estas moléculas, dejando una única plantilla de cadena. Luego se hibridó un cebador
de secuenciación con los grupos. Luego se llevaron a cabo rondas iterativas de secuenciación que involucraban: la introducción de 4 nucleótidos terminados químicamente marcados con fluorescencia de cada base de ácido nucleico y una polimerasa y reguladores, de modo que se produciría una incorporación de un solo nucleótido para la posición activa del producto de extensión que era complementaria a cada nucleótido; una etapa de lavado, una exploración de fluorescencia de la superficie para medir qué agrupaciones incorporaron qué colorante fluorescente, la introducción de un químico de escisión que liberó las moléculas de terminación de las bases incorporadas junto con la etiqueta fluorescente para permitir los ciclos subsiguientes; una etapa de lavado. Estos pasos se repitieron -100 veces para revelar la secuencia de cada grupo (lectura 1). La cadena sintetizada se eliminó luego por desnaturalización. Luego se introdujo un segundo cebador y se reiteró el proceso de secuenciación para leer una etiqueta de 6 bases. Esta cadena sintetizada se eliminó luego por desnaturalización. A continuación, se dejó que la cadena de plantilla original se hibridara de nuevo con los cebadores de superficie inmovilizados que se extendieron para volver a formar los grupos de doble cadena. En este punto, la hebra original se escindió dando como resultado agrupaciones formadas a partir de los complementos de las agrupaciones monocatenarias originales. Se introdujo un tercer cebador de secuenciación que se hibridó con esta hebra y la secuenciación se iteró en ~60 ciclos para obtener secuencias de la hebra inversa de los amplicones (lectura 2). Las secuencias resultantes contribuyeron -100.000 a 200.000 lecturas por muestra después de haber sido ordenadas con base en las secuencias de etiquetas.
Las secuencias obtenidas se mapearon primero en regiones específicas de los segmentos V y J. Específicamente, los primeros 27 pb de la lectura 1 se asignaron a los últimos 27 pb de los diferentes segmentos J del ratón. De forma similar, las secuencias iniciales de la lectura 2 se asignaron a las secuencias de los cebadores utilizados. Cuando una secuencia se asigna a uno de los cebadores, la secuencia de la lectura 2 que sigue al cebador se asigna a los diferentes segmentos V del ratón. Dado que se espera que la lectura 1 alcance el segmento V, también intentamos mapearlo para determinar el marco y la secuencia de aminoácidos. Las posiciones 81-95 de la lectura 1 se asignaron a la mayoría de 3' 60 pb del segmento V específico al que se asigna la lectura 2. Si una secuencia no tiene una identidad sustancial con ninguna de las secuencias J y V, se eliminaría. Además, los mapas de J y V se extienden en lectura 1. Ya que los mapas específicos de J y V se mapean para cada grupo, se puede evaluar si las bases fuera de la secuencia originalmente mapeada son consistentes con la secuencia de V y J. mapeadas. Estas secuencias dejarán de ser consistentes cuando se alcancen las bases que resultan de la replicación sin plantilla (N bases) o el segmento D. Las diferencias entre las secuencias de lectura y la secuencia V o J en las regiones originalmente mapeadas o extendidas se consideran debido a un error y se "corrigen".
Las lecturas que tienen secuencias idénticas para las posiciones 28-80 se consideran un clonotipo. Los clonotipos que tienen una secuencia muy similar se evalúan para determinar si es probable que sean clonotipos independientes o uno clonotipos que se separaron debido a un error de PCR y/o de secuenciación. Se ha ideado un algoritmo que incorpora la frecuencia de los dos clonotipos en consideración, el número de diferencias entre ellos y la probabilidad de error en esa posición para determinar si los dos clonotipos deben fusionarse en un clonotipo o no. Cuando uno de los dos clonotipos tiene una frecuencia muy alta y el otro es raro y solo hay una diferencia de base hacia el final de la lectura (que generalmente tiene más errores), entonces los dos clonotipos son probablemente el resultado de un error y son luego se unió. Por otro lado, la presencia de dos clonotipos con una frecuencia similar y con tres diferencias entre ellos indica que estos dos clonotipos son genuinamente independientes y no están unidos. Luego se calcula la frecuencia de cada clonotipo de TCRa
Un clonotipo público estaba presente en los 12 ratones. Este clonotipo tenía la firma de un clonotipo reactivo a CFA. Fue indetectable en cualquiera de los ratones antes de la inyección o en el día 5, pero aumenta dramáticamente a alta frecuencia en puntos de tiempo posteriores. Múltiples secuencias de nucleótidos a través de ratones y dentro de un solo ratón codificado para la misma secuencia de aminoácidos de este clonotipo. En general, y luego de eliminar los clonotipos observados en menos de 3/10 puntos de tiempo para un ratón (el punto de tiempo número 8 se eliminó de todos los ratones, ya que había algunas características inusuales en algunos de los ratones), hay 23 observaciones de clonotipos con esta secuencia de aminoácidos (19 de las cuales se presentan a una frecuencia media >10-4) con 10 secuencias de nucleótidos distintas en los 12 ratones (8 secuencias distintas para los 19 clonotipos de alta frecuencia) (ver tabla 10). Además, hay clonotipos relacionados adicionales (con solo una diferencia de aminoácidos) que también tienen el mismo patrón de ser bajos antes de la inyección y altos después de 10 días de la inyección. Esto proporciona una evidencia abrumadora de un clonotipo que responde al CFA.
Tabla 11




El clonotipo público descrito en el ejemplo anterior se validó observando repertorios de TCR de ratones adicionales. Específicamente, se evaluó la presencia del clonotipo en 11 ratones tratados con PLP y 3 tratados simulados. El clonotipo estuvo presente en 12/14 ratones. Además, el clonotipo relacionado mencionado en el ejemplo anterior (con una diferencia de aminoácidos) también se vio repetidamente. Hubo 15 observaciones (diferentes secuencias de nucleótidos en el mismo animal o en diferentes animales) para el clon con una diferencia de aminoácidos. El clonotipo no se vio en ninguno de los animales antes de la vacunación. El patrón de frecuencia también fue bastante similar a lo que se vio en el primer grupo de animales. Esto valida que el clonotipo público identificado es de hecho común a la mayoría de los ratones que fueron tratados con el adyuvante de Freund.
El uso del tejido afectado para ayudar a determinar los clonotipos relevantes para la enfermedad. Se encontraron clonotipos correlacionados con la actividad de la enfermedad en el modelo de eAe anterior. Sin embargo, no es obvio si los clonotipos eran específicos del aspecto adyuvante de la vacunación o del péptido PLP. La presencia o ausencia de los clonotipos en las muestras tratadas de forma simulada fue un método para distinguir las dos posibilidades. Como se reveló anteriormente, el tejido afectado se puede usar para determinar los clonotipos relevantes para la enfermedad. En este caso, el tejido de la médula espinal estaba disponible. Los clonotipos se filtraron de los datos que tenían una frecuencia al menos 3 veces mayor en la médula espinal que las otras tres muestras obtenidas al mismo tiempo (bazo, ganglios linfáticos y sangre). Luego se examinaron los clonotipos que se correlacionaron con la puntuación de la enfermedad. Se identificó un clonotipo que apareció más de una vez. La secuencia del clonotipo fue LYCTCSALGGSSYEQYF (secuencia A) (SEQ ID NO: 194). Esta secuencia se buscó e identificó en los 8 ratones con la enfermedad, dos ratones que fueron tratados con PLP pero no desarrollaron la enfermedad y no se detectó en los dos ratones tratados de forma simulada. El patrón de frecuencia del clonotipo en las muestras de sangre en serie difirió entre los ratones. Sin embargo, en todos los ratones el clonotipo estaba ausente antes de la vacunación. Además, el patrón de mayor frecuencia en la médula espinal que el otro tejido en el mismo momento se demostró en todos excepto en un ratón.
Dado el riesgo de sobrealimentación, se examinó un conjunto independiente de ratones para validar estos hallazgos. Se examinó la presencia de la secuencia A en ratones adicionales. Específicamente, la presencia de la secuencia A se probó en las siguientes circunstancias: en ocho ratones tratados con PLP adicionales que desarrollaron EAE, tres ratones tratados con PLP que no desarrollaron EAE y tres ratones tratados de forma simulada. Entre los 11 ratones tratados con PLP, el clonotipo estaba presente en ocho ratones. Dos de los tres que no tenían el clonotipo no desarrollaron enfermedad. El clonotipo no se encontró en los tres ratones tratados de forma simulada ni en ninguno de los ratones tratados con PLP en el punto antes de la vacunación. Además, se demostró en todos los animales el patrón de mayor frecuencia en la médula espinal que en los otros tejidos del mismo momento. En todas las muestras (el descubrimiento inicial y la validación posterior) entre 21 animales tratados con PLP, 18 tienen los clones, pero ninguno antes de la vacunación. Además, ninguno de los cinco ratones tratados simuladamente tiene la secuencia A. Esto proporciona una evidencia sólida para un clonotipo de EAE público que es específico de la enfermedad.
Los clonotipos adicionales que se correlacionan con la enfermedad y están enriquecidos en la médula espinal parecen tener secuencias muy similares entre sí. Estos clonotipos semiprivados son clonotipos adicionales específicos de la enfermedad. Estos pueden identificarse de manera más completa a través de la utilización de un algoritmo de búsqueda de motivos para determinar secuencias compartidas entre clonotipos enriquecidos en la médula espinal y correlacionados con la enfermedad.
Ejemplo 30
Hipermutaciones somáticas identificadas en perfiles de clonotipo de IgH
Se realizaron tres conjuntos de amplificaciones utilizando los cebadores del segmento V de IgH enumerados en la tabla 5 y los cebadores complementarios a la secuencia constante de IgG que también se describen anteriormente. Se usaron ADNc de 7 muestras normales, 7 muestras de pacientes con esclerosis múltiple y 4 muestras con SLE como plantillas para la amplificación. Después de la segunda etapa de la PCR que introdujo una etiqueta única para cada muestra, los productos se aislaron espacialmente y se sometieron a secuenciación.
Las secuencias se mapearon entonces en segmentos V y J individuales y se ensamblaron en clonotipos utilizando las metodologías descritas anteriormente. Se buscaron pruebas de frecuentes clonotipos altamente relacionados que pueden ser el resultado de la hipermutación somática en estas muestras. Los datos en la tabla 11 muestran un ejemplo sorprendente en uno de los pacientes con esclerosis múltiple. En este ejemplo, se identificaron 12 clonotipos distintos. Estos clonotipos de 12 nucleótidos codifican 3 secuencias de aminoácidos altamente relacionadas. Dos de las secuencias de aminoácidos tienen una frecuencia muy alta (> 1%) y difieren en un aminoácido conservado (Lisina frente a Arginina).
Tabla 12



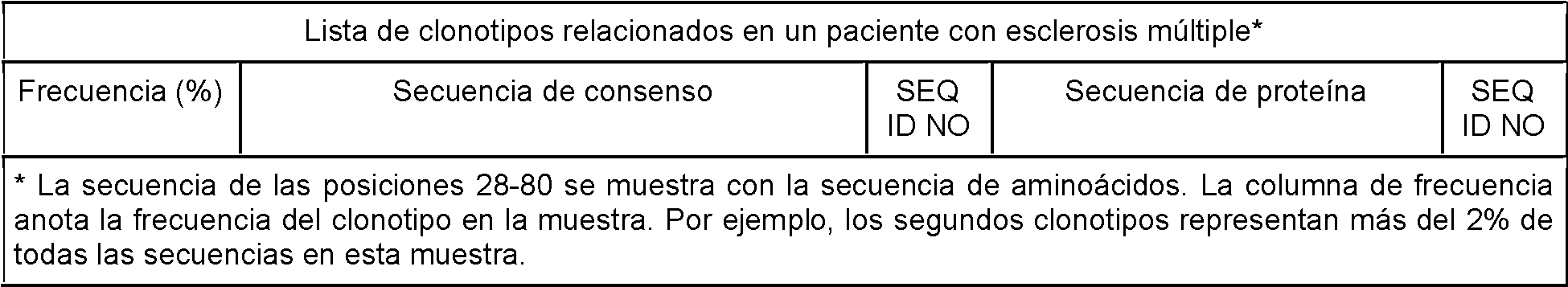
Ejemplo 31
Uso forense de perfiles inmunitarios
Los perfiles de clonotipo para receptores de células T y/o B pueden emplearse para la identificación humana y animal. La tremenda diversidad de estos perfiles de clonotipos proporciona una firma única del individuo. Esto se ejemplifica mediante los perfiles de la figura 10. En este ejemplo, las secuencias de TCRp se amplificaron mediante p Cr de transcriptasa inversa a partir de ARNm extraído de la sangre de dos individuos diferentes. Los cebadores complementarios a las regiones V se enumeran en la Tabla 13. Los cebadores de la segunda etapa de amplificación son los mismos que los del Ejemplo 2. Los productos se secuenciaron y la frecuencia de cada clonotipo se determinó la frecuencia. Como puede verse en la figura 10, la gran mayoría de los clonotipos de los individuos son diferentes, los clonotipos de un individuo (muestra 122) colocados casi exclusivamente a lo largo del eje X y los clonotipos del otro individuo (muestra 140) colocados casi exclusivamente a lo largo del eje Y. Solo unos 25 clonotipos parecen estar compartidos, como lo indican los puntos de datos fuera del eje. Esta disparidad de uso del clonotipo entre individuos ha sido corroborada por Warren et al, Genome Research, Epub (24 de febrero de 2011). En un aspecto de la invención, esta disparidad en el uso del clonotipo proporciona un método para distinguir individuos que comprende los pasos: (a) obtener un perfil de clonotipo de una primera muestra, (b) obtener un perfil de clonotipo de una segunda muestra, y (c) determinar si la primera muestra y la segunda son del mismo individuo midiendo el grado en donde se superpone el uso de clonotipos.
Tabla 13
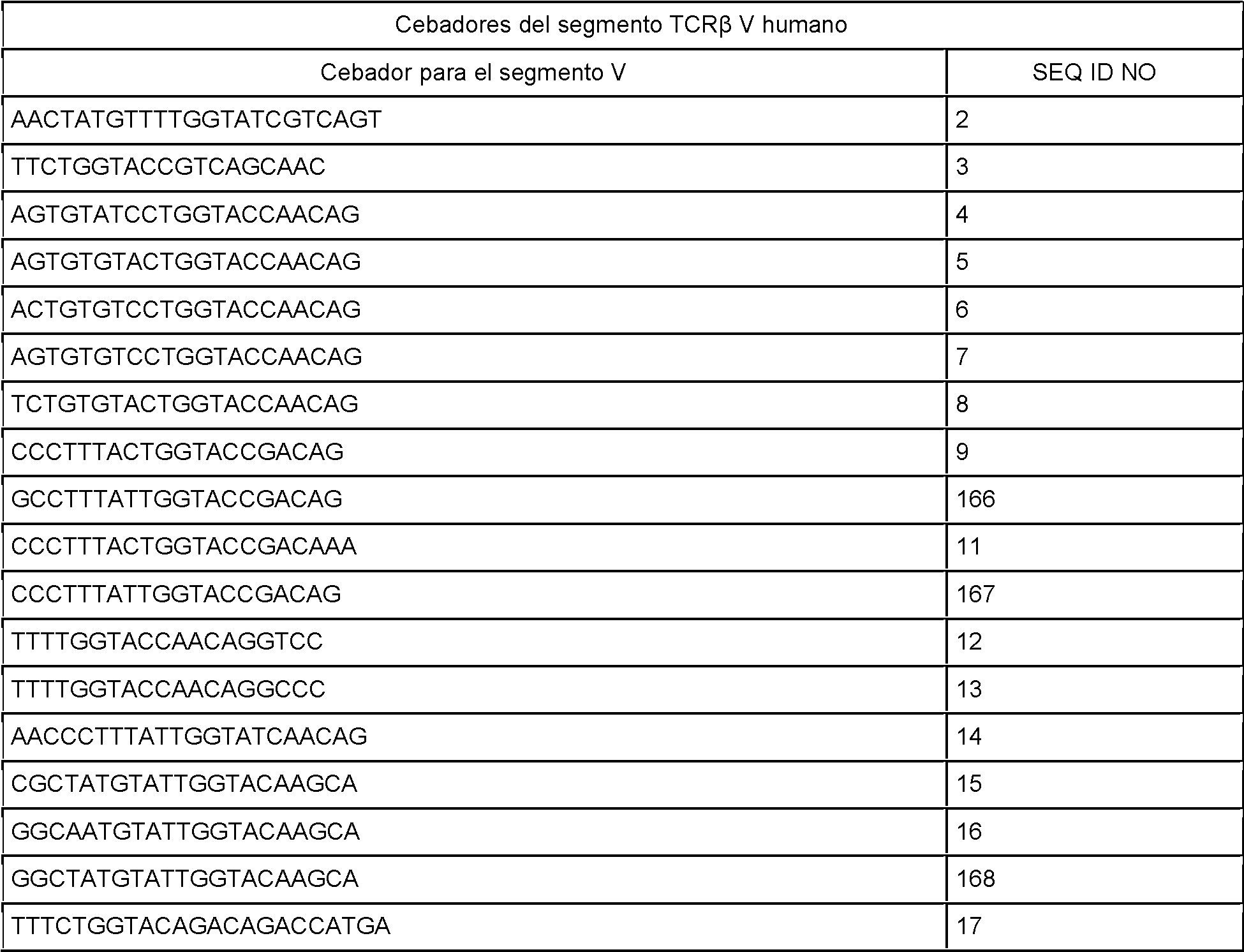

De los 342 clonotipos en sangre de la primera muestra (muestra 122) a una frecuencia >10-4, solo se detecta uno en la segunda persona (muestra 140). Recíprocamente, de los 505 clonotipos presentes a una frecuencia >10-4 en la muestra 144, 3 se detectan en la muestra 122. Como control para la fluctuación aleatoria de la medición, de los mismos 505 clonotipos, 504 clonotipos estaban presentes en una muestra de amplificación replicada. Esto demuestra la posibilidad de que un perfil de clonotipo completo sea un identificador potencialmente extremadamente específico. Por supuesto, la naturaleza de estos perfiles es que no son estables en el tiempo, ya que las nuevas reacciones inmunitarias agregarán nuevos clonotipos al espectro. Sin embargo, estos procesos no alteran estos perfiles muy rápidamente. Se puede ver que, si bien la frecuencia precisa de los clonotipos individuales cambia con el tiempo, el conjunto de clonotipos presentes en una frecuencia mensurable es probablemente mucho mayor que lo que se encontraría en un segundo individuo. Se pueden desarrollar algoritmos para definir la fracción y el número de clonotipos idénticos que serían necesarios para determinar que dos especímenes son del mismo individuo. Además, dado que esta diversidad se encuentra en un gen con una gran cantidad de función activa en contraste con la diversidad de microsatélites, es posible extraer información de identificación potencialmente relevante sin necesidad de tener una muestra compatible de una muestra de donante identificada. La información sobre la salud de este individuo, su historial de vacunación, etc. se puede medir a partir de la información del clonotipo.
Definiciones
A menos que se especifique lo contrario en el presente documento, los términos y símbolos de la química de los ácidos nucleicos, la bioquímica, la genética y la biología molecular utilizados en este documento siguen los de los tratados y textos estándar en este campo, por ejemplo. Kornberg and Baker, ADN Replication, segunda edición (W.H. Freeman, Nueva York, 1992); Lehninger, Biochemistry, segunda edición (Worth Publishers, Nueva York, 1975); Strachan and Read, Human Molecular Genetics, segunda edición (Wiley-Liss, Nueva York, 1999); Abbas et al, Cellular and Molecular Immunology, 6a edición (Saunders, 2007).
"Amplicón" significa el producto de una reacción de amplificación de polinucleótidos; es decir, una población clonal de polinucleótidos, que pueden ser monocatenarias o bicatenarias, que se replican a partir de una o más secuencias de partida. La una o más secuencias de inicio pueden ser una o más copias de la misma secuencia, o pueden ser una mezcla de diferentes secuencias. Preferiblemente, los amplicones se forman mediante la amplificación de una única secuencia de inicio. Los amplicones pueden producirse mediante una variedad de reacciones de amplificación cuyos productos comprenden réplicas de uno o más ácidos nucleicos de partida o diana. En un aspecto de los métodos de la invención, las reacciones de amplificación que producen amplicones son "impulsadas por la plantilla", ya que el
emparejamiento de bases de los reactivos, ya sea nucleótidos u oligonucleótidos, tiene complementos en un polinucleótido plantilla que se requieren para la creación de productos de reacción. En un aspecto de los métodos de la invención, las reacciones dirigidas por la plantilla son extensiones de cebador con una polimerasa de ácido nucleico u ligaciones de oligonucleótidos con una ligasa de ácido nucleico. Tales reacciones incluyen, pero no se limitan a, reacciones en cadena de polimerasa (PCR), reacciones de polimerasa lineal, amplificación basada en la secuencia de ácidos nucleicos (NASBA), amplificaciones de círculo rodante y similares, descritas en las siguientes referencias: Mullis et al, Patentes de los Estados Unidos 4,683,195; 4,965,188; 4,683,202; 4,800,159 (PCR); Gelfand et al, Patente de los Estados Unidos 5,210,015 (PCR en tiempo real con sondas "taqman"); Wittwer et al, Patente de los Estados Unidos 6,174,670; Kacian et al, Patente de los Estados Unidos 5,399,491 ("n As BA"); Lizardi, Patente de los Estados Unidos 5,854,033; Aono et al, Patente Japonesa pública JP 4-262799 (amplificación de círculo rodante); y similares. En un aspecto de los métodos de la invención, los amplicones se producen mediante PCR. Una reacción de amplificación puede ser una amplificación "en tiempo real" si se dispone de una química de detección que permita medir un producto de reacción a medida que avanza la reacción de amplificación, por ejemplo "PCR en tiempo real" que se describe a continuación, o "NASBA en tiempo real", como se describe en Leone et al, Nucleic Acids Research, 26: 2150-2155 (1998), y referencias similares. Como se usa en el presente documento, el término "amplificación" significa realizar una reacción de amplificación. Una "mezcla de reacción" significa una solución que contiene todos los reactivos necesarios para realizar una reacción, que puede incluir, entre otros, agentes de regulación para mantener el pH en un nivel seleccionado durante una reacción, sales, cofactores, eliminadores y similares.
"Clonalidad" como se usa en este documento significa una medida del grado en que la distribución de abundancias de clonotipos entre clonotipos de un repertorio está sesgada a uno o unos pocos clonotipos. A grandes rasgos, la clonalidad es una medida inversa de la diversidad de clonotipos. Muchas medidas o estadísticas están disponibles en la ecología que describe las relaciones de abundancia de especies que se pueden usar para las medidas de clonalidad de acuerdo con la invención, por ejemplo. Capítulos 17 y 18, en Pielou, An Introduction to Mathematical Ecology, (Wiley-Interscience, 1969). En un aspecto, una medida de clonalidad utilizada con los métodos de la invención es una función de un perfil de clonotipo (es decir, el número de clonotipos distintos detectados y sus abundancias), de modo que después de medir un perfil de clonotipo, la clonalidad se puede calcular a partir de él para dar un solo número. Una medida de clonalidad es la medida de Simpson, que es simplemente la probabilidad de que dos clonotipos dibujados al azar sean iguales. Otras medidas de clonalidad incluyen medidas basadas en información y el índice de diversidad de McIntosh, revelado en Pielou (citado anteriormente).
"Clonotipo" significa una secuencia de nucleótidos recombinada de una célula T o célula B que codifica un receptor de célula T (TCR) o receptor de célula B (BCR), o una porción del mismo. En un aspecto de los métodos de la invención, una colección de todos los clonotipos distintos de una población de linfocitos de un individuo es un repertorio de dicha población, por ejemplo, Arstila et al, Science, 286: 958-961 (1999); Yassai et al, Immunogenetics, 61: 493 502 (2009); Kedzierska et al., Mol. Immunol., 45(3): 607-618 (2008); y similares. Como se usa en este documento, "perfil de clonotipo" o "perfil de repertorio", es una tabla de clonotipos de una muestra de células T y/o células B (como una muestra de sangre periférica que contiene dichas células) que incluye sustancialmente todos los clonotipos del repertorio y sus abundancias relativas. "Perfil de clonotipo", "perfil de repertorio" y "repertorio" se usan aquí de manera intercambiable. (Es decir, el término "repertorio", como se explica más detalladamente a continuación, significa un repertorio medido a partir de una muestra de linfocitos). En un aspecto de los métodos de la invención, los clonotipos comprenden porciones de una cadena pesada de inmunoglobulina (IgH) o una cadena p de TCR. En otros aspectos de los métodos de la invención, los clonotipos pueden basarse en otras moléculas recombinadas, tales como cadenas ligeras de inmunoglobulina o cadenas TCRa, o porciones de las mismas.
Las "regiones determinantes de la complementariedad" (CDR) significan regiones de una inmunoglobulina (es decir, un anticuerpo) o un receptor de células T donde la molécula complementa la conformación de un antígeno, determinando así la especificidad de la molécula y el contacto con un antígeno específico. Los receptores de células T y las inmunoglobulinas tienen cada uno tres CDR: CDR1 y CDR2 se encuentran en el dominio variable (V), y CDR3 incluye algunos de V, todos de diversos (D) (solo cadenas pesadas) y conjuntos (J), y algunos de los dominios constantes (C).
"Estándar interno" significa una secuencia de ácidos nucleicos que se amplifica en la misma reacción de amplificación que uno o más polinucleótidos objetivo para permitir la cuantificación absoluta o relativa de los polinucleótidos objetivo en una muestra. Un estándar interno puede ser endógeno o exógeno. Es decir, un estándar interno puede ocurrir naturalmente en la muestra, o puede agregarse a la muestra antes de la amplificación. Se pueden agregar múltiples secuencias estándar internas exógenas a una mezcla de reacción en una serie de concentraciones predeterminadas para proporcionar una calibración con la cual se puede comparar un amplicón objetivo para determinar la cantidad de su polinucleótido objetivo correspondiente en una muestra. La selección del número, las secuencias, las longitudes y otras características de los estándares internos exógenos es una elección de diseño de rutina para un experto en la técnica. Preferiblemente, los estándares internos endógenos, también denominados en el presente documento como "secuencias de referencia", son secuencias naturales de una muestra que corresponden a genes regulados mínimamente que muestran un nivel de transcripción constante e independiente del ciclo celular, por ejemplo, Selvey et al, Mol. Cell Probes, 15: 307-311 (2001). Las secuencias de referencia ejemplares incluyen, pero no se limitan a, secuencias de los siguientes genes: GAPDH, p2-microglobulina, 18S ribosomal ARN y p-actina (aunque véase Selvey et al, citado anteriormente).
"Kit" se refiere a cualquier sistema de entrega para entregar materiales o reactivos para llevar a cabo un método de la invención. En el contexto de los ensayos de reacción, dichos sistemas de entrega incluyen sistemas que permiten el almacenamiento, transporte o entrega de reactivos de reacción (por ejemplo, cebadores, enzimas, etc. en los contenedores apropiados) y/o materiales de apoyo (por ejemplo, reguladores, instrucciones escritas para realizar el ensayo, etc.) de un lugar a otro. Por ejemplo, los kits incluyen uno o más recintos (por ejemplo, cajas) que contienen los reactivos de reacción relevantes y/o materiales de soporte. Dichos contenidos se pueden entregar al destinatario deseado juntos o por separado. Por ejemplo, un primer recipiente puede contener una enzima para usar en un ensayo, mientras que un segundo recipiente contiene cebadores.
"Neoplasia linfoide" significa una proliferación anormal de linfocitos que pueden ser malignos o no malignos. Un cáncer linfoide es una neoplasia linfoide maligna. Las neoplasias linfoides son el resultado de, o están asociadas a, trastornos linfoproliferativos, que incluyen, entre otros, linfoma folicular, leucemia linfocítica crónica (LLC), leucemia linfocítica aguda (LLA), leucemia de células pilosas, linfomas, mieloma múltiple, trastorno linfoproliferativo postrasplante,, linfoma de células del manto (MCL), linfoma difuso de células B grandes (DLBCL), linfoma de células T, o similares, por ejemplo Jaffe et al, Blood, 112: 4384-4399 (2008); Swerdlow et al., Clasificación de la OMS de tumores de tejidos hematopoyéticos y linfoides (p. 4) (IARC Press, 2008).
"Enfermedad residual mínima" significa células cancerosas restantes después del tratamiento. El término se usa con más frecuencia en relación con el tratamiento de linfomas y leucemias.
"Porcentaje homólogo", "porcentaje idéntico", o términos similares utilizados en referencia a la comparación de una secuencia de referencia y otra secuencia ("secuencia de comparación") significa que en una alineación óptima entre las dos secuencias, la secuencia de comparación es idéntica a la secuencia de referencia en varias posiciones de subunidades equivalentes al porcentaje indicado, siendo las subunidades nucleótidos para las comparaciones de polinucleótidos o aminoácidos para las comparaciones de polipéptidos. Como se usa en este documento, una "alineación óptima" de secuencias que se comparan es una que maximiza las coincidencias entre subunidades y minimiza el número de huecos empleados en la construcción de una alineación. El porcentaje de identidades se puede determinar con implementaciones de algoritmos disponibles comercialmente, como la descrita por Needleman and Wunsch, J. Mol. Biol., 48: 443-453 (1970) (programa "GAP" de Wisconsin Sequence Analysis Package, Genetics Computer Group, Madison, WI), o similares. Otros paquetes de software en la técnica para construir alineaciones y calcular el porcentaje de identidad u otras medidas de similitud incluyen el programa "BestFit", basado en el algoritmo de Smith y Waterman, Advances in Applied Mathematics, 2: 482-489 (1981) (Wisconsin Sequence Analysis Package, Genetics Computer Group, Madison, WI). En otras palabras, por ejemplo, para obtener un polinucleótido que tenga una secuencia de nucleótidos al menos el 95 por ciento idéntica a una secuencia de nucleótidos de referencia, hasta el cinco por ciento de los nucleótidos en la secuencia de referencia puede ser eliminado o sustituido con otro nucleótido, o un número de nucleótidos hasta el cinco por ciento del número total de nucleótidos en la secuencia de referencia pueden insertarse en la secuencia de referencia.
"Reacción en cadena de la polimerasa", o "PCR", significa una reacción para la amplificación in vitro de secuencias de ADN específicas mediante la extensión del cebador simultánea de cadenas complementarias de ADN. En otras palabras, la PCR es una reacción para realizar múltiples copias o réplicas de un ácido nucleico objetivo flanqueado por sitios de unión del cebador, dicha reacción comprende una o más repeticiones de los siguientes pasos: (i) desnaturalizar el ácido nucleico objetivo, (ii) fusionamiento cebadores a los sitios de unión del cebador, y (iii) extensión de los cebadores por una polimerasa de ácido nucleico en presencia de nucleósidos trifosfatos. Normalmente, la reacción se realiza en ciclos a diferentes temperaturas optimizadas para cada paso en un instrumento de termociclador. Las temperaturas particulares, las duraciones en cada paso y las tasas de cambio entre los pasos dependen de muchos factores bien conocidos por los expertos en la técnica, por ejemplo, ejemplificado por las referencias: McPherson et al, editors, PCR: A Practical Approach and PCR2: A Practical Approach (IRL Press, Oxford, 1991 y 1995, respectivamente). Por ejemplo, en una PCR convencional que utiliza ADN polimerasa Taq, un ácido nucleico diana bicatenario se puede desnaturalizar a una temperatura >90° C, los cebadores se fusionan a una temperatura en el rango de 50-75°C y los cebadores se extienden a una temperatura en el rango 72-78°C. El término "PCR" abarca formas derivadas de la reacción, que incluyen, pero no se limitan a, RT-PCR, PCR en tiempo real, PCR anidada, PCR cuantitativa, PCR multiplexada y similares. Los volúmenes de reacción varían desde unos pocos cientos de nanolitros, por ejemplo 200 nL, hasta unos pocos cientos de pL, por ejemplo 200 pL. "PCR de transcripción reversa", o "RT-PCR", significa una PCR que está precedida por una reacción de transcripción reversa que convierte un ARN objetivo en un ADN monocatenario complementario, que luego se amplifica, por ejemplo, Tecott et al, Patente de los Estados Unidos 5,168,038. "PCR en tiempo real" significa una PCR para la cual se monitoriza la cantidad de producto de reacción, es decir, un amplicón, a medida que avanza la reacción. Hay muchas formas de PCR en tiempo real que difieren principalmente en los productos químicos de detección utilizados para monitorizar el producto de reacción, por ejemplo, Gelfand et al, Patente de los EE.UU. 5,210,015 ("taqman"); Wittwer et al., Patentes de los Estados Unidos 6,174,670 y 6,569,627 (colorantes intercalantes); Tyagi et al, Patente de los Estados Unidos 5,925,517 (balizas moleculares). Las químicas de detección para la PCR en tiempo real se revisan en Mackay et al, Nucleic Acids Research, 30: 1292-1305 (2002). "PCR anidada" significa una PCR de dos etapas en donde el amplicón de una primera PCR se convierte en la muestra para una segunda PCR utilizando un nuevo conjunto de cebadores, al menos uno de los cuales se une a una ubicación interior del primer amplicón. Como se usa en este documento, "cebadores iniciales" en referencia a una reacción de amplificación anidada significa los cebadores utilizados para generar un primer amplicón, y "cebadores secundarios" significa el uno o más cebadores utilizados para generar un segundo
amplicón, o anidado. "PCR multiplexada" significa una PCR en la que múltiples secuencias diana (o una única secuencia diana y una o más secuencias de referencia) se llevan a cabo simultáneamente en la misma mezcla de reacción, por ejemplo, Bernard et al, Anal. Biochem., 273: 221-228 (1999) (PCR en tiempo real de dos colores). Normalmente, se emplean distintos conjuntos de cebadores para cada secuencia que se amplifica. Típicamente, el número de secuencias diana en una PCR múltiple está en el rango de 2 a 50, o de 2 a 40, o de 2 a 30. "PCR cuantitativa" significa una PCR diseñada para medir la abundancia de uno o más secuencias diana en una muestra o espécimen. La PCR cuantitativa incluye tanto la cuantificación absoluta como la cuantificación relativa de dichas secuencias diana. Las mediciones cuantitativas se realizan utilizando una o más secuencias de referencia o estándares internos que pueden analizarse por separado o junto con una secuencia objetivo. La secuencia de referencia puede ser endógena o exógena a una muestra o espécimen, y en este último caso, puede comprender una o más plantillas competidoras. Las secuencias de referencia endógenas típicas incluyen segmentos de transcripciones de los siguientes genes: p-actina, GAPDH, p2-microglobulina, ARN ribosomal y similares. Las técnicas para la PCR cuantitativa son bien conocidas por los expertos en la técnica, como se ejemplifica en las siguientes referencias: Freeman et al, Biotechniques, 26: 112-126 (1999); Becker-Andre et al., Nucleic Acids Research, 17: 9437-9447 (1989); Zimmerman et al, Biotechniques, 21: 268-279 (1996); Diviacco et al, Gene, 122: 3013-3020 (1992); Becker-Andre et al., Nucleic Acids Research, 17: 9437-9446 (1989); y similares.
"Cebador" significa un oligonucleótido, ya sea natural o sintético, capaz de, al formar un dúplex con una plantilla de polinucleótido, actuar como un punto de inicio de la síntesis de ácido nucleico y extenderse desde su extremo 3' a lo largo de la plantilla para que se forme el dúplex. La extensión de un cebador generalmente se lleva a cabo con una polimerasa de ácido nucleico, como una polimerasa de ADN o ARN. La secuencia de nucleótidos agregada en el proceso de extensión está determinada por la secuencia del polinucleótido de plantilla. Normalmente los cebadores se extienden por un ADN polimerasa. Los cebadores usualmente tienen una longitud en el rango de 14 a 40 nucleótidos, o en el rango de 18 a 36 nucleótidos. Los cebadores se emplean en una variedad de reacciones de amplificación nucleica, por ejemplo, reacciones de amplificación lineal usando un cebador único, o reacciones en cadena de la polimerasa, empleando dos o más cebadores. La guía para seleccionar las longitudes y secuencias de los cebadores para aplicaciones particulares es bien conocida por los expertos en la técnica, como lo demuestran las siguientes referencias: Dieffenbach, editor, PCR Primer: A Laboratory Manual, 2a edición (Cold Spring Harbor Press, Nueva York, 2003).
"Nivel de calidad" significa una medida de la probabilidad de que una asignación de base en una ubicación de secuencia particular sea correcta. Los expertos en la materia conocen a menudo una variedad de métodos para calcular puntuaciones de calidad para circunstancias particulares, como, por ejemplo, para bases llamadas como resultado de diferentes secuencias químicas, sistemas de detección, algoritmos de llamada de bases, etc. En general, los valores de la puntuación de calidad están relacionados monótonamente con las probabilidades de una llamada de base correcta. Por ejemplo, un puntaje de calidad, o Q, de 10 puede significar que hay una probabilidad del 90 por ciento de que una base se llame correctamente, una Q de 20 puede significar que hay una probabilidad del 99 por ciento de que una base se llame correctamente, y así sucesivamente. Para algunas plataformas de secuenciación, en particular las que usan químicos de secuenciación por síntesis, las puntuaciones de calidad promedio disminuyen en función de la longitud de la lectura de secuencia, de modo que las puntuaciones de calidad al comienzo de una lectura de secuencia son más altas que las que se encuentran al final de una lectura de secuencia, dichas disminuciones se deben a fenómenos tales como extensiones incompletas, extensiones de arrastre, pérdida de plantilla, pérdida de polimerasa, fallas en el límite, fallas de desprotección y similares.
"Repertorio", o "repertorio inmune", significa un conjunto de distintas secuencias de nucleótidos recombinados que codifican receptores de células T (TCR) o receptores de células B (BCR), o fragmentos de los mismos, respectivamente, en una población de linfocitos de un individuo, en donde las secuencias de nucleótidos del conjunto tienen una correspondencia uno a uno con linfocitos distintos o sus subpoblaciones clonales para prácticamente todos los linfocitos de la población. Una población de linfocitos a partir de la cual se determina un repertorio puede tomarse de uno o más muestras de tejidos, como una o más muestras de sangre. Una secuencia de nucleótidos miembro de un repertorio se denomina aquí como un "clonotipo". En un aspecto de los métodos de la invención, los clonotipos de un repertorio comprenden cualquier segmento de ácido nucleico común a una población de células T o células B que ha sufrido una recombinación somática durante el desarrollo de TCR o BCR, incluidos los normales o aberrantes (por ejemplo, asociados con cánceres) moléculas precursoras de los mismos, que incluyen, entre otros, cualquiera de los siguientes: una cadena pesada de inmunoglobulina (IgH) o subconjuntos de la misma (por ejemplo, una región variable de IgH, una región CDR3 o similar), moléculas de IgH incompletas, una inmunoglobulina cadena ligera o subconjuntos de la misma (por ejemplo, una región variable, región CDR o similar), cadena a del receptor de células T o subconjuntos de la misma, cadena p del receptor de células T o subconjuntos de la misma (por ejemplo, región variable, CDR3, región V(D)J, o similar, una CDR (que incluye CDR1, CDR2 o CDR3, ya sea de TCR o BCR, o combinaciones de dichas CDR), regiones V(D)J de TCR o BCR, regiones hipermutadas de regiones variables de IgH, o similares. En un aspecto de los métodos de la invención, los segmentos de ácido nucleico que definen los clonotipos de un repertorio se seleccionan de modo que su diversidad (es decir, el número de secuencias de ácido nucleico distintas en el conjunto) sea lo suficientemente grande como para que sustancialmente cada célula T o célula B o su clon en un individuo lleva una secuencia de ácidos nucleicos única de dicho repertorio. Es decir, de acuerdo con la invención, un profesional puede seleccionar para definir clonotipos un segmento o región particular de ácidos nucleicos recombinados que codifican TCR o BCR que no reflejan la diversidad completa de una población de células T o células
B; sin embargo, preferiblemente, los clonotipos se definen de manera que reflejen la diversidad de la población de células T y/o células B de las que se derivan. Es decir, preferiblemente cada clon diferente de una muestra tiene un clonotipo diferente. (Por supuesto, en algunas aplicaciones, habrá múltiples copias de uno o más clonotipos particulares dentro de un perfil, como en el caso de muestras de pacientes con leucemia o linfoma). En otros aspectos de los métodos de la invención, la población de linfocitos que corresponde a un repertorio puede ser células B en circulación, o pueden ser células T en circulación, o pueden ser subpoblaciones de cualquiera de las poblaciones anteriores, incluidas, entre otras, células T CD4+, o células T CD8+, u otras subpoblaciones definidas por marcadores de superficie celular, o similares. Dichas subpoblaciones se pueden adquirir tomando muestras de tejidos particulares, por ejemplo, médula ósea, o ganglios linfáticos, o similares, o clasificando o enriqueciendo células de una muestra (como sangre periférica) en función de uno o más marcadores de la superficie celular, tamaño, morfología o similares. En otros aspectos de los métodos de la invención, la población de linfocitos que corresponde a un repertorio puede derivarse de tejidos de enfermedades, tales como un tejido tumoral, un tejido infectado o similares. En una realización, un repertorio que comprende cadenas p de TCR humanas o fragmentos de las mismas comprende varias secuencias de nucleótidos distintas en el intervalo de 0.1 x 106 a 1.8 x 106, o en el intervalo de 0.5 x 106 a 1.5 x 106, o en el intervalo de 0.8 x 106 a 1.2 x 106 En otra realización, un repertorio que comprende cadenas de IgH humanas o fragmentos de las mismas comprende varias secuencias de nucleótidos distintas en el intervalo de 0.1 x 106 a 1.8 x 106, o en el intervalo de 0.5 x 106 a 1.5 x 106, o en el rango de 0.8 x 106 a 1.2 x 106. En una realización particular, un repertorio comprende un conjunto de secuencias de nucleótidos que codifican sustancialmente todos los segmentos de la región V(D)J de una cadena IgH. "Sustancialmente todos", como se usa en este documento, puede significar que cada segmento que tiene una abundancia relativa de 0.001 por ciento o más; o alternativamente, "sustancialmente todo" como se usa en este documento significa cada segmento que tiene una abundancia relativa de 0.0001 por ciento o más. En otra realización particular, un repertorio comprende un conjunto de secuencias de nucleótidos que codifica sustancialmente todos los segmentos de la región V(D)J de una cadena p de TCR. En otra realización, un repertorio comprende un conjunto de secuencias de nucleótidos que tienen longitudes en el intervalo de 25-200 nucleótidos y que incluyen segmentos de las regiones V, D y J de una cadena p de TCR. En otra realización, un repertorio comprende un conjunto de secuencias de nucleótidos que tienen longitudes en el intervalo de 25-200 nucleótidos y que incluyen segmentos de las regiones V, D y J de una cadena de IgH. En otra realización, un repertorio comprende varias secuencias de nucleótidos distintas que es sustancialmente equivalente a la cantidad de linfocitos que expresan una cadena de IgH distinta. En otra realización, un repertorio comprende varias secuencias de nucleótidos distintas que es sustancialmente equivalente a la cantidad de linfocitos que expresan una cadena p de TCR distinta. En otra realización más, "sustancialmente equivalente" significa que con un noventa y nueve por ciento de probabilidad, un repertorio de secuencias de nucleótidos incluirá una secuencia de nucleótidos que codifique un IgH o TCR p o una porción del mismo transportada o expresada por cada linfocito de una población de un individuo en una frecuencia de 0.001 por ciento o mayor. En otra realización más, "sustancialmente equivalente" significa que, con un noventa y nueve por ciento de probabilidad, un repertorio de secuencias de nucleótidos incluirá una secuencia de nucleótidos que codifique una IgH o TCR p o una porción del mismo transportada o expresada por cada linfocito presente a una frecuencia de 0.0001 por ciento o mayor. Los conjuntos de clonotipos descritos en las dos oraciones anteriores a veces se denominan en el presente documento como representativos del "repertorio completo" de las secuencias de IgH y/o TCRp. Como se mencionó anteriormente, al medir o generar un perfil de clonotipo (o perfil de repertorio), se obtiene una muestra suficientemente grande de linfocitos para que dicho perfil proporcione una representación razonablemente precisa de un repertorio para una aplicación particular. En un aspecto de los métodos de la invención, se emplean muestras que comprenden de 105 a 107 linfocitos, especialmente cuando se obtienen de muestras de sangre periférica de 1 a 10 mL.
"Etiqueta de secuencia" (o "etiqueta") significa un oligonucleótido que está unido a un polinucleótido o plantilla y se utiliza para identificar y/o rastrear el polinucleótido o plantilla en una reacción. Una etiqueta de oligonucleótido puede estar unida al extremo 3'- o 5'- de un polinucleótido o plantilla o puede insertarse en el interior de dicha plantilla de polinucleótido para formar un conjugado lineal, en algún momento denominado aquí "polinucleótido marcado" o "plantilla etiquetada", o "conjugado de polinucleótido etiqueta", o similar. Las etiquetas de oligonucleótidos pueden variar ampliamente en tamaño y composiciones; las siguientes referencias proporcionan una guía para seleccionar conjuntos de etiquetas de oligonucleótidos apropiadas para realizaciones particulares: Brenner, Patente de los Estados Unidos 5,635,400; Brenner et al., Proc. Natl Acad Sci., 97: 1665-1670 (2000); Church et al, Publicación de Patente Europea 0 303 459; Shoemaker et al, Nature Genetics, 14: 450-456 (1996); Morris et al, publicación de Patente Europea 0799897A1; Wallace, Patente de los Estados Unidos 5,981,179; y similares. Las longitudes y composiciones de las etiquetas de oligonucleótidos pueden variar ampliamente, y la selección de longitudes y/o composiciones particulares depende de varios factores que incluyen, sin limitación, cómo se usan las etiquetas para generar una lectura, por ejemplo, mediante una reacción de hibridación o mediante una reacción enzimática, como la secuenciación; si están etiquetados, por ejemplo, con un colorante fluorescente o similar; el número de etiquetas de oligonucleótidos distinguibles requeridas para identificar inequívocamente un conjunto de polinucleótidos, y similares, y qué tan diferentes deben ser las etiquetas de un conjunto para garantizar una identificación confiable, por ejemplo, libertad de hibridación cruzada o identificación errónea de errores de secuenciación. Las etiquetas de oligonucleótidos pueden tener una longitud dentro de un rango de 2 a 36 nucleótidos, o de 4 a 30 nucleótidos, o de 8 a 20 nucleótidos, o de 6 a 10 nucleótidos, respectivamente. Se pueden usar conjuntos de etiquetas en las que cada etiqueta de oligonucleótido de un conjunto tiene una secuencia de nucleótidos única que difiere de la de todas las demás etiquetas del mismo conjunto en al menos dos bases; alternativamente, se usan conjuntos de etiquetas en las que la secuencia de cada etiqueta de un conjunto difiere de la de todas las demás etiquetas del mismo conjunto en al menos tres bases.
Claims (11)
1. Un método para monitorizar una neoplasia linfoide en un paciente con clonotipos de cáncer definidos que comprende los pasos de:
(a) determinar un perfil de secuencias de ADN recombinadas en células T y/o células B de una muestra obtenida del paciente, en donde el perfil se determina mediante la secuenciación de la siguiente generación; y
(b) determinar los niveles de los clonotipos de cáncer definidos en el perfil para monitorizar la neoplasia linfoide.
2. El método de la reivindicación 1, que comprende además la etapa de determinar los niveles de clones evolucionados a partir de los clonotipos de cáncer definidos.
3. El método de la reivindicación 2, en donde los clones evolucionados a partir de los clonotipos de cáncer definidos incluyen clones evolucionados a partir de los clonotipos de cáncer definidos por sustitución de VH.
4. El método de la reivindicación 3, en donde el reemplazo de VH se identifica detectando la unión D-J.
5. El método de la reivindicación 2, en donde los clones evolucionados a partir de los clonotipos de cáncer definidos incluyen clones evolucinados a partir de los clonotipos de cáncer definidos por hipermutación somática.
6. El método de la reivindicación 5, en donde los clones evolucionados a partir de los clonotipos de cáncer definidos mediante hipermutación somática se calculan basándose en:
a) el número de diferencias en la región NDN,
b) la longitud de la región NDN, y
c) la presencia de otras hipermutaciones somáticas en los segmentos V y/o J.
7. El método de una cualquiera de las reivindicaciones 1 a 6, en donde la etapa (a) comprende la secuenciación del ADN genómico de las células utilizando secuenciación de próxima generación.
8. El método de una cualquiera de las reivindicaciones 1 a 6, en donde la etapa (a) comprende amplificar el ADN genómico de las células y secuenciar el ADN amplificado utilizando secuenciación de próxima generación.
9. El método de una cualquiera de las reivindicaciones 1 a 6, en donde la etapa (a) comprende la transcripción reversa del ARN de las células para formar un ADNc y la secuenciación del ADNc utilizando secuenciación de próxima generación.
10. El método de una cualquiera de las reivindicaciones 1 a 9, en donde dichas secuencias recombinadas comprenden un reordenamiento genómico seleccionado del grupo que consiste en un reordenamiento VDJ de IgH, un reordenamiento DJ de IgH, un reordenamiento VJ de IgK, un reordenamiento VJ de IgL, un reordenamiento VDJ de TCR p, un reordenamiento DJ de TCR p, un reordenamiento VJ de TCR a, un reordenamiento VJ de TCR A, un reordenamiento VDJ de TCR 8, y un reordenamiento VD de TCR 8.
11. El método de una cualquiera de las reivindicaciones 1 a 10, en donde el método es para detectar la recurrencia de una neoplasia linfoide en el paciente.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US33217510P | 2010-05-06 | 2010-05-06 | |
| US45574310P | 2010-10-25 | 2010-10-25 | |
| US201161446822P | 2011-02-25 | 2011-02-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2740802T3 true ES2740802T3 (es) | 2020-02-06 |
Family
ID=44903937
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16183402T Active ES2740802T3 (es) | 2010-05-06 | 2011-05-04 | Monitorización del estadio de neoplasia linfoide usando perfiles de clonotipo |
| ES11777704.5T Active ES2593614T3 (es) | 2010-05-06 | 2011-05-04 | Monitorización de estados de salud y enfermedad usando perfiles de clonotipos |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES11777704.5T Active ES2593614T3 (es) | 2010-05-06 | 2011-05-04 | Monitorización de estados de salud y enfermedad usando perfiles de clonotipos |
Country Status (9)
| Country | Link |
|---|---|
| EP (4) | EP3456847B1 (es) |
| JP (4) | JP6085249B2 (es) |
| CN (2) | CN104673899A (es) |
| AU (1) | AU2011249041B2 (es) |
| CA (1) | CA2798431C (es) |
| DK (2) | DK2567226T3 (es) |
| ES (2) | ES2740802T3 (es) |
| SG (2) | SG185128A1 (es) |
| WO (2) | WO2011139371A1 (es) |
Families Citing this family (77)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9528160B2 (en) | 2008-11-07 | 2016-12-27 | Adaptive Biotechnolgies Corp. | Rare clonotypes and uses thereof |
| US9365901B2 (en) | 2008-11-07 | 2016-06-14 | Adaptive Biotechnologies Corp. | Monitoring immunoglobulin heavy chain evolution in B-cell acute lymphoblastic leukemia |
| WO2010053587A2 (en) | 2008-11-07 | 2010-05-14 | Mlc Dx Incorporated | Methods of monitoring conditions by sequence analysis |
| US8748103B2 (en) | 2008-11-07 | 2014-06-10 | Sequenta, Inc. | Monitoring health and disease status using clonotype profiles |
| US8691510B2 (en) | 2008-11-07 | 2014-04-08 | Sequenta, Inc. | Sequence analysis of complex amplicons |
| US9394567B2 (en) | 2008-11-07 | 2016-07-19 | Adaptive Biotechnologies Corporation | Detection and quantification of sample contamination in immune repertoire analysis |
| US9506119B2 (en) | 2008-11-07 | 2016-11-29 | Adaptive Biotechnologies Corp. | Method of sequence determination using sequence tags |
| US8628927B2 (en) | 2008-11-07 | 2014-01-14 | Sequenta, Inc. | Monitoring health and disease status using clonotype profiles |
| PL2387627T3 (pl) | 2009-01-15 | 2016-10-31 | Profilowanie odporności adaptacyjnej i wytwarzanie przeciwciał monoklonalnych | |
| KR20120044941A (ko) | 2009-06-25 | 2012-05-08 | 프레드 헛친슨 켄서 리서치 센터 | 적응 면역의 측정방법 |
| US9043160B1 (en) | 2009-11-09 | 2015-05-26 | Sequenta, Inc. | Method of determining clonotypes and clonotype profiles |
| DK2652155T3 (en) | 2010-12-16 | 2017-02-13 | Gigagen Inc | Methods for Massive Parallel Analysis of Nucleic Acids in Single Cells |
| KR102759126B1 (ko) | 2010-12-30 | 2025-01-24 | 파운데이션 메디신 인코포레이티드 | 종양 샘플의 다유전자 분석의 최적화 |
| WO2012097374A1 (en) * | 2011-01-14 | 2012-07-19 | Cb Biotechnologies, Inc. | Immunodiversity assessment method and its use |
| US10385475B2 (en) | 2011-09-12 | 2019-08-20 | Adaptive Biotechnologies Corp. | Random array sequencing of low-complexity libraries |
| CA2853088C (en) | 2011-10-21 | 2018-03-13 | Adaptive Biotechnologies Corporation | Quantification of adaptive immune cell genomes in a complex mixture of cells |
| EP3904536B1 (en) * | 2011-12-09 | 2025-10-29 | Adaptive Biotechnologies Corporation | Diagnosis of lymphoid malignancies and minimal residual disease detection |
| US9499865B2 (en) | 2011-12-13 | 2016-11-22 | Adaptive Biotechnologies Corp. | Detection and measurement of tissue-infiltrating lymphocytes |
| JP2015504661A (ja) * | 2011-12-20 | 2015-02-16 | シーケンタ インコーポレイテッド | 免疫レパートリー解析による、濾胞性リンパ腫からびまん性大細胞型b細胞リンパ腫への形質転換のモニタリング |
| US10077478B2 (en) | 2012-03-05 | 2018-09-18 | Adaptive Biotechnologies Corp. | Determining paired immune receptor chains from frequency matched subunits |
| JP2015512655A (ja) * | 2012-04-13 | 2015-04-30 | シーケンタ インコーポレイテッド | 免疫レパートリ分析におけるサンプル混入の検出および定量化 |
| KR101621643B1 (ko) | 2012-05-08 | 2016-05-16 | 어댑티브 바이오테크놀로지스 코포레이션 | 다중 pcr 반응에서 증폭 바이어스를 측정 및 교정하기 위한 조성물 및 방법 |
| WO2013181428A2 (en) * | 2012-05-31 | 2013-12-05 | Sequenta, Inc. | Predicting relapse of chronic lymphocytic leukemia patients treated by allogeneic stem cell transplantation |
| EP2861761A1 (en) * | 2012-06-15 | 2015-04-22 | Adaptive Biotechnologies Corporation | Uniquely tagged rearranged adaptive immune receptor genes in a complex gene set |
| WO2014026031A1 (en) * | 2012-08-10 | 2014-02-13 | Sequenta, Inc. | High sensitivity mutation detection using sequence tags |
| EP3640343B1 (en) | 2012-10-01 | 2026-01-07 | Adaptive Biotechnologies Corporation | Immunocompetence assessment by adaptive immune receptor diversity and clonality characterization |
| US20150247198A1 (en) * | 2012-10-19 | 2015-09-03 | Sequenta, Inc. | Monitoring clonotypes of plasma cell proliferative disorders in peripheral blood |
| WO2015160439A2 (en) | 2014-04-17 | 2015-10-22 | Adaptive Biotechnologies Corporation | Quantification of adaptive immune cell genomes in a complex mixture of cells |
| US20150299800A1 (en) * | 2012-10-22 | 2015-10-22 | Sequenta, Inc. | Monitoring mantle cell lymphoma clonotypes in peripheral blood after immunotransplant |
| CA2900776C (en) * | 2013-02-11 | 2023-08-15 | Cb Biotechnologies, Inc. | Method for evaluating an immunorepertoire |
| EP2959020B1 (en) * | 2013-02-22 | 2019-09-18 | Adaptive Biotechnologies Corporation | Method to select rare clonotypes |
| CA2906218A1 (en) * | 2013-03-15 | 2014-09-18 | Adaptive Biotechnologies Corporation | Uniquely tagged rearranged adaptive immune receptor genes in a complex gene set |
| US9816088B2 (en) | 2013-03-15 | 2017-11-14 | Abvitro Llc | Single cell bar-coding for antibody discovery |
| US9708657B2 (en) | 2013-07-01 | 2017-07-18 | Adaptive Biotechnologies Corp. | Method for generating clonotype profiles using sequence tags |
| CN106103711A (zh) * | 2013-11-21 | 2016-11-09 | 组库创世纪株式会社 | T细胞受体和b细胞受体库分析系统及其在治疗和诊断中的应用 |
| ES2741740T3 (es) | 2014-03-05 | 2020-02-12 | Adaptive Biotechnologies Corp | Métodos que usan moléculas sintéticas que contienen segmentos de nucleótidos aleatorios |
| US10066265B2 (en) | 2014-04-01 | 2018-09-04 | Adaptive Biotechnologies Corp. | Determining antigen-specific t-cells |
| US10202640B2 (en) | 2014-05-07 | 2019-02-12 | The Board Of Trustees Of The Leland Stanford Junior University | Single cell analysis of T cells using high-throughput multiplex amplification and deep sequencing |
| CN107002076B (zh) | 2014-09-15 | 2021-11-23 | 阿布维特罗有限责任公司 | 高通量核苷酸文库测序 |
| EP3715455A1 (en) | 2014-10-29 | 2020-09-30 | Adaptive Biotechnologies Corp. | Highly-multiplexed simultaneous detection of nucleic acids encoding paired adaptive immune receptor heterodimers from many samples |
| US10246701B2 (en) | 2014-11-14 | 2019-04-02 | Adaptive Biotechnologies Corp. | Multiplexed digital quantitation of rearranged lymphoid receptors in a complex mixture |
| WO2016086029A1 (en) | 2014-11-25 | 2016-06-02 | Adaptive Biotechnologies Corporation | Characterization of adaptive immune response to vaccination or infection using immune repertoire sequencing |
| EP3227464B1 (en) | 2014-12-05 | 2022-04-20 | Foundation Medicine, Inc. | Multigene analysis of tumor samples |
| ES3002665T3 (en) | 2015-02-24 | 2025-03-07 | Adaptive Biotechnologies Corp | Methods for diagnosing infectious disease and determining hla status using immune repertoire sequencing |
| AU2016242967B2 (en) | 2015-04-01 | 2021-07-01 | Adaptive Biotechnologies Corp. | Method of identifying human compatible T cell receptors specific for an antigenic target |
| US9422547B1 (en) | 2015-06-09 | 2016-08-23 | Gigagen, Inc. | Recombinant fusion proteins and libraries from immune cell repertoires |
| CN106557536B (zh) * | 2015-09-30 | 2021-12-21 | 松下知识产权经营株式会社 | 控制方法 |
| US10130658B2 (en) * | 2015-12-18 | 2018-11-20 | Provectus Pharmatech, Inc. | Method of ex vivo enhancement of immune cell activity for cancer immunotherapy with a small molecule ablative compound |
| US10428325B1 (en) | 2016-09-21 | 2019-10-01 | Adaptive Biotechnologies Corporation | Identification of antigen-specific B cell receptors |
| CN107955831A (zh) * | 2016-10-13 | 2018-04-24 | 深圳华大基因研究院 | 用于淋巴细胞定量检测的标记物和淋巴细胞定量检测方法 |
| GB201618485D0 (en) * | 2016-11-02 | 2016-12-14 | Ucl Business Plc | Method of detecting tumour recurrence |
| IL317050A (en) * | 2016-12-09 | 2025-01-01 | Regeneron Pharma | Systems and methods for sequencing T cell receptors and their uses |
| RU2674994C2 (ru) * | 2016-12-29 | 2018-12-14 | Федеральное государственное бюджетное учреждение науки институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова Российской академии наук (ИБХ РАН) | Способ определения маркеров наличия опухолевых т-лимфобластов |
| EP4212631A1 (en) | 2017-09-01 | 2023-07-19 | Life Technologies Corporation | Compositions and methods for immune repertoire sequencing |
| TWI814745B (zh) | 2017-10-10 | 2023-09-11 | 國立大學法人廣島大學 | 使用效應T細胞(Teff)抗原受體之抗原特異性控制性T細胞(Treg)之製出技術 |
| EP3702453A4 (en) * | 2017-10-10 | 2021-07-21 | Hiroshima University | FULL REPLACEMENT PROCEDURE FOR T-CELL RECEPTOR USING PLATINUM |
| US11254980B1 (en) | 2017-11-29 | 2022-02-22 | Adaptive Biotechnologies Corporation | Methods of profiling targeted polynucleotides while mitigating sequencing depth requirements |
| WO2019196088A1 (en) * | 2018-04-13 | 2019-10-17 | Syz Cell Therapy Co. | Methods of obtaining tumor-specific t cell receptors |
| CA3102460A1 (en) | 2018-06-11 | 2019-12-19 | Foundation Medicine, Inc. | Compositions and methods for evaluating genomic alterations |
| US11603565B2 (en) * | 2018-06-15 | 2023-03-14 | Roche Sequencing Solutions, Inc. | System for identification of antigens recognized by T cell receptors expressed on tumor infiltrating lymphocytes |
| US12473663B2 (en) | 2018-07-18 | 2025-11-18 | Life Technologies Corporation | Compositions and methods for immune repertoiresequencing |
| CN109002689B (zh) * | 2018-07-23 | 2020-10-09 | 西安交通大学医学院第一附属医院 | T细胞数据处理方法及装置 |
| WO2020102721A1 (en) * | 2018-11-16 | 2020-05-22 | Rapa Therapeutics, Llc | Immune monitoring of neuro-inflammatory amyotrophic lateral sclerosis (als) |
| US20220073963A1 (en) * | 2018-12-20 | 2022-03-10 | The Johns Hopkins University | Compositions and methods for detecting and treating type 1 diabetes and other autoimmune diseases |
| WO2020191365A1 (en) | 2019-03-21 | 2020-09-24 | Gigamune, Inc. | Engineered cells expressing anti-viral t cell receptors and methods of use thereof |
| CN110988364A (zh) * | 2019-12-31 | 2020-04-10 | 天津美瑞特医疗科技有限公司 | 一种应用流式细胞术检测移植后gvhd相关细胞因子的方法及检测试剂盒 |
| WO2021151114A1 (en) * | 2020-01-22 | 2021-07-29 | Life Technologies Corporation | Immune repertoire biomarkers in autoimmune disease and immunodeficiency disorders |
| CN111808195A (zh) * | 2020-06-30 | 2020-10-23 | 中国科学院心理研究所 | 抗n-甲基-d-天冬氨酸受体脑炎的b细胞抗体基因获取方法及其免疫组库研究 |
| CN111524548B (zh) * | 2020-07-03 | 2020-10-23 | 至本医疗科技(上海)有限公司 | 用于检测igh重排的方法、计算设备和计算机存储介质 |
| BR112023019195A2 (pt) | 2021-03-24 | 2023-10-17 | Genentech Inc | Edição eficiente do gene tcr em linfócitos t |
| CN113637741B (zh) * | 2021-09-29 | 2023-12-01 | 成都二十三魔方生物科技有限公司 | 一种早发性白发遗传风险基因检测试剂盒、以及一种早发性白发遗传风险评估系统和方法 |
| WO2024257764A1 (ja) * | 2023-06-12 | 2024-12-19 | 国立大学法人神戸大学 | 免疫反応の評価方法 |
| WO2025122939A1 (en) * | 2023-12-08 | 2025-06-12 | Dana-Farber Cancer Institute, Inc. | Detection and classification of b-cell malignancies |
| CN117867121A (zh) * | 2024-01-12 | 2024-04-12 | 广州华银医学检验中心有限公司 | 一种检测肿瘤免疫组库微小残留的引物组合和方法 |
| WO2025243303A1 (en) * | 2024-05-22 | 2025-11-27 | Technion Research & Development Foundation Limited | Improved methods of determining immune age |
| CN120432008B (zh) * | 2025-05-07 | 2026-02-06 | 博德生物技术(德州)有限公司 | CellRanger单细胞RNA测序数据分析方法 |
| CN120290752B (zh) * | 2025-06-13 | 2025-09-16 | 北京大学成都前沿交叉生物技术研究院 | 捕获探针、包含其的试剂盒及利用其在空间分辨率下检测t细胞和b细胞免疫组库中的用途 |
Family Cites Families (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4965188A (en) | 1986-08-22 | 1990-10-23 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences using a thermostable enzyme |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4800159A (en) | 1986-02-07 | 1989-01-24 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences |
| US4942124A (en) | 1987-08-11 | 1990-07-17 | President And Fellows Of Harvard College | Multiplex sequencing |
| US5168038A (en) | 1988-06-17 | 1992-12-01 | The Board Of Trustees Of The Leland Stanford Junior University | In situ transcription in cells and tissues |
| JP2781438B2 (ja) | 1988-10-20 | 1998-07-30 | モーリィ,アリグザンダー,アラン | 白血病及びリンパ腫におけるモノクローナリテイーの診断法 |
| CA2020958C (en) | 1989-07-11 | 2005-01-11 | Daniel L. Kacian | Nucleic acid sequence amplification methods |
| US5210015A (en) | 1990-08-06 | 1993-05-11 | Hoffman-La Roche Inc. | Homogeneous assay system using the nuclease activity of a nucleic acid polymerase |
| JP3080178B2 (ja) | 1991-02-18 | 2000-08-21 | 東洋紡績株式会社 | 核酸配列の増幅方法およびそのための試薬キット |
| US5981179A (en) | 1991-11-14 | 1999-11-09 | Digene Diagnostics, Inc. | Continuous amplification reaction |
| US5837447A (en) | 1992-04-15 | 1998-11-17 | Blood Center Research Foundation, Inc., The | Monitoring an immune response by analysis of amplified immunoglobulin or T-cell-receptor nucleic acid |
| US5925517A (en) | 1993-11-12 | 1999-07-20 | The Public Health Research Institute Of The City Of New York, Inc. | Detectably labeled dual conformation oligonucleotide probes, assays and kits |
| US6090592A (en) | 1994-08-03 | 2000-07-18 | Mosaic Technologies, Inc. | Method for performing amplification of nucleic acid on supports |
| US5604097A (en) | 1994-10-13 | 1997-02-18 | Spectragen, Inc. | Methods for sorting polynucleotides using oligonucleotide tags |
| US5635363A (en) | 1995-02-28 | 1997-06-03 | The Board Of Trustees Of The Leland Stanford Junior University | Compositions and methods for the detection, quantitation and purification of antigen-specific T cells |
| US6087096A (en) | 1995-11-13 | 2000-07-11 | Dau; Peter C. | Method of intrafamily fragment analysis of the T cell receptor α and β chain CDR3 regions |
| US5854033A (en) | 1995-11-21 | 1998-12-29 | Yale University | Rolling circle replication reporter systems |
| US20020076725A1 (en) * | 1996-03-13 | 2002-06-20 | Tomoko Toyosaki-Maeda | Human t cell clone specific for rheumatoid arthritis |
| US6458530B1 (en) | 1996-04-04 | 2002-10-01 | Affymetrix Inc. | Selecting tag nucleic acids |
| WO1997046706A1 (en) * | 1996-06-03 | 1997-12-11 | University Of Alberta | Methods for detection of rearranged dna |
| EP0912766B2 (en) | 1996-06-04 | 2011-12-14 | University of Utah Research Foundation | Monitoring hybridization during pcr |
| US5846928A (en) | 1996-08-01 | 1998-12-08 | Pasken Products Co., Ltd. | Method for treating cancer patients |
| ATE347103T1 (de) | 1996-09-06 | 2006-12-15 | Ortho Mcneil Pharm Inc | Reinigung von antigenspezifischen t-zellen |
| GB9626815D0 (en) * | 1996-12-23 | 1997-02-12 | Cemu Bioteknik Ab | Method of sequencing DNA |
| EP0972081B1 (en) * | 1997-04-01 | 2007-06-13 | Solexa Ltd. | Method of nucleic acid amplification |
| US6008002A (en) | 1997-09-29 | 1999-12-28 | Bodey; Bela | Immunomagnetic detection and isolation of cancer cells |
| JP2002503814A (ja) | 1998-02-12 | 2002-02-05 | イムニベスト・コーポレイション | 循環ガン細胞の迅速かつ効率的な単離のための方法および試薬 |
| DE19833738A1 (de) | 1998-07-27 | 2000-02-03 | Michael Giesing | Verfahren zur Isolierung von Krebszellen aus zellhaltigen Körperflüssigkeiten sowie Sets zur Durchführung dieses Verfahrens |
| AR021833A1 (es) | 1998-09-30 | 2002-08-07 | Applied Research Systems | Metodos de amplificacion y secuenciacion de acido nucleico |
| US6300070B1 (en) | 1999-06-04 | 2001-10-09 | Mosaic Technologies, Inc. | Solid phase methods for amplifying multiple nucleic acids |
| US6440706B1 (en) | 1999-08-02 | 2002-08-27 | Johns Hopkins University | Digital amplification |
| EP1227321A1 (en) | 2000-12-28 | 2002-07-31 | Institut für Bioanalytik GmbH | Reversible MHC multimer staining for functional purification of antigen-specific T cells |
| GB0128153D0 (en) | 2001-11-23 | 2002-01-16 | Bayer Ag | Profiling of the immune gene repertoire |
| ATE483822T1 (de) * | 2002-10-11 | 2010-10-15 | Univ Erasmus | Primer für nukleinsäureamplifikation in pcr- basierten klonalitätsstudien |
| CN101128601B (zh) | 2003-01-29 | 2011-06-08 | 454生命科学公司 | 核酸扩增和测序的方法 |
| AU2003902299A0 (en) | 2003-05-13 | 2003-05-29 | Flinders Medical Centre | A method of analysing a marker nucleic acid molecule |
| ATE421998T1 (de) | 2003-12-15 | 2009-02-15 | Pasteur Institut | Ermittlung des repertoires von b-lymphozyten populationen |
| US8298756B2 (en) | 2004-08-11 | 2012-10-30 | Albert Einstein College Of Medicine Of Yeshiva University | Isolation, gene expression, and chemotherapeutic resistance of motile cancer cells |
| US8426146B2 (en) * | 2005-06-03 | 2013-04-23 | University of Pittsburgh—of the Commonwealth System of Higher Education | Multiparameteric method for assessing immune system status |
| US7993821B2 (en) | 2005-08-11 | 2011-08-09 | University Of Washington | Methods and apparatus for the isolation and enrichment of circulating tumor cells |
| US7375211B2 (en) * | 2005-11-18 | 2008-05-20 | Kou Zhong C | Method for detection and quantification of T-cell receptor Vβ repertoire |
| WO2007087312A2 (en) | 2006-01-23 | 2007-08-02 | Population Genetics Technologies Ltd. | Molecular counting |
| CN101225441B (zh) * | 2007-12-05 | 2010-12-01 | 浙江大学 | 一种检测克隆特异性t淋巴细胞tcr bv cdr3基因组成的方法 |
| US11051733B2 (en) | 2008-01-18 | 2021-07-06 | Wake Forest University Health Sciences | Isolating and purifying cells for therapy |
| EP2088205A1 (en) * | 2008-02-11 | 2009-08-12 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | PSMB10: A diagnosis marker and therapeutic target of chronic rejection. |
| US20090226975A1 (en) * | 2008-03-10 | 2009-09-10 | Illumina, Inc. | Constant cluster seeding |
| US20100137143A1 (en) | 2008-10-22 | 2010-06-03 | Ion Torrent Systems Incorporated | Methods and apparatus for measuring analytes |
| WO2010053587A2 (en) | 2008-11-07 | 2010-05-14 | Mlc Dx Incorporated | Methods of monitoring conditions by sequence analysis |
| US8574835B2 (en) | 2009-05-29 | 2013-11-05 | Life Technologies Corporation | Scaffolded nucleic acid polymer particles and methods of making and using |
| WO2011106738A2 (en) * | 2010-02-25 | 2011-09-01 | Fred Hutchinson Cancer Research Center | Use of tcr clonotypes as biomarkers for disease |
-
2011
- 2011-05-04 ES ES16183402T patent/ES2740802T3/es active Active
- 2011-05-04 EP EP18201137.9A patent/EP3456847B1/en active Active
- 2011-05-04 AU AU2011249041A patent/AU2011249041B2/en active Active
- 2011-05-04 CN CN201510054401.XA patent/CN104673899A/zh active Pending
- 2011-05-04 JP JP2013509047A patent/JP6085249B2/ja active Active
- 2011-05-04 CA CA2798431A patent/CA2798431C/en active Active
- 2011-05-04 JP JP2013509046A patent/JP6158080B2/ja active Active
- 2011-05-04 EP EP11777704.5A patent/EP2567226B1/en active Active
- 2011-05-04 ES ES11777704.5T patent/ES2593614T3/es active Active
- 2011-05-04 WO PCT/US2011/000791 patent/WO2011139371A1/en not_active Ceased
- 2011-05-04 SG SG2012081527A patent/SG185128A1/en unknown
- 2011-05-04 EP EP23155863.6A patent/EP4219759A3/en active Pending
- 2011-05-04 EP EP16183402.3A patent/EP3144673B1/en not_active Revoked
- 2011-05-04 CN CN201180033142.2A patent/CN103097888B/zh active Active
- 2011-05-04 WO PCT/US2011/000792 patent/WO2011139372A1/en not_active Ceased
- 2011-05-04 DK DK11777704.5T patent/DK2567226T3/en active
- 2011-05-04 DK DK16183402.3T patent/DK3144673T3/da active
- 2011-05-04 SG SG10201503534RA patent/SG10201503534RA/en unknown
-
2017
- 2017-06-07 JP JP2017112502A patent/JP6246971B2/ja active Active
- 2017-11-15 JP JP2017219812A patent/JP6533272B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| DK3144673T3 (da) | 2019-09-09 |
| WO2011139372A1 (en) | 2011-11-10 |
| WO2011139371A1 (en) | 2011-11-10 |
| EP3456847A1 (en) | 2019-03-20 |
| EP3144673B1 (en) | 2019-07-10 |
| DK2567226T3 (en) | 2016-10-10 |
| JP6533272B2 (ja) | 2019-06-19 |
| EP2567226A4 (en) | 2013-08-28 |
| EP2567226A1 (en) | 2013-03-13 |
| EP2567226B1 (en) | 2016-08-10 |
| AU2011249041A1 (en) | 2012-11-22 |
| JP6246971B2 (ja) | 2017-12-13 |
| JP2013524849A (ja) | 2013-06-20 |
| CA2798431C (en) | 2018-10-23 |
| JP2017205112A (ja) | 2017-11-24 |
| SG185128A1 (en) | 2012-12-28 |
| JP6158080B2 (ja) | 2017-07-05 |
| EP3456847B1 (en) | 2024-12-25 |
| CN103097888A (zh) | 2013-05-08 |
| ES2593614T3 (es) | 2016-12-12 |
| JP2013524848A (ja) | 2013-06-20 |
| EP4219759A2 (en) | 2023-08-02 |
| SG10201503534RA (en) | 2015-06-29 |
| EP3144673A1 (en) | 2017-03-22 |
| JP2018068300A (ja) | 2018-05-10 |
| AU2011249041B2 (en) | 2014-10-30 |
| JP6085249B2 (ja) | 2017-02-22 |
| EP3456847C0 (en) | 2024-12-25 |
| CA2798431A1 (en) | 2011-11-10 |
| EP4219759A3 (en) | 2023-09-06 |
| CN103097888B (zh) | 2016-03-09 |
| CN104673899A (zh) | 2015-06-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2740802T3 (es) | Monitorización del estadio de neoplasia linfoide usando perfiles de clonotipo | |
| US20250129420A1 (en) | Monitoring Health and Disease Status Using Clonotype Profiles | |
| US11021757B2 (en) | Monitoring health and disease status using clonotype profiles | |
| US10519511B2 (en) | Monitoring health and disease status using clonotype profiles | |
| US8748103B2 (en) | Monitoring health and disease status using clonotype profiles | |
| AU2015200383A1 (en) | Monitoring health and disease status using clonotype profiles |