ES2710657T3 - Compuesto heterocíclico - Google Patents
Compuesto heterocíclico Download PDFInfo
- Publication number
- ES2710657T3 ES2710657T3 ES14800152T ES14800152T ES2710657T3 ES 2710657 T3 ES2710657 T3 ES 2710657T3 ES 14800152 T ES14800152 T ES 14800152T ES 14800152 T ES14800152 T ES 14800152T ES 2710657 T3 ES2710657 T3 ES 2710657T3
- Authority
- ES
- Spain
- Prior art keywords
- cancer
- mixture
- compound
- acid
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C*(CC(C)(C1)NC=N)CN1P Chemical compound C*(CC(C)(C1)NC=N)CN1P 0.000 description 2
- PSXLJUKMMIHADB-UHFFFAOYSA-N CN(C=NC=C1)C1=O Chemical compound CN(C=NC=C1)C1=O PSXLJUKMMIHADB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/80—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D211/84—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen directly attached to ring carbon atoms
- C07D211/86—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/20—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D239/22—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Diabetes (AREA)
- General Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
Un compuesto representado por la siguiente fórmula:**Fórmula** donde el anillo A es (1) un anillo de benceno opcionalmente sustituido con de 1 a 4 sustituyentes que son los mismos o diferentes y se seleccionan de entre: un átomo de halógeno; un grupo cicloalquilo C3-10; un grupo alcoxi C1-6; y un grupo arilo C6-14 opcionalmente sustituido con de 1 a 3 átomos de halógeno, o (2) piridina opcionalmente sustituida con de 1 a 3 sustituyentes que son los mismos o diferentes y se seleccionan de entre: un grupo cicloalquilo C3-10; un grupo alcoxi C1-6; y un grupo arilo C6-14 opcionalmente sustituido con de 1 a 3 átomos de halógeno; el anillo B es piridina o pirimidina cada una además opcionalmente sustituida con de 1 a 3 sustituyentes seleccionados de entre un átomo de halógeno y un grupo alquilo C1-6; donde cada uno de X1, X2 y X4 es un átomo de carbono y X3 es un átomo de carbono o un átomo de nitrógeno; n es 3; R1 es COOH, CONH2, un grupo tetrazolilo o un grupo dihidrooxadiazolilo o OCH2COOH y está unido a X2 del anillo B; y cada uno de R2 y R3 es un átomo de hidrógeno, o una sal del mismo.
Description
DESCRIPCION
Compuesto heterodclico
[Campo tecnico]
La presente invencion se refiere a un compuesto heterodclico que presenta una accion antagonista del receptor de la somatostatina subtipo 5 (en lo sucesivo se abreviara a veces como «SSTR5») y es util en el tratamiento, mejora o profilaxis de enfermedades o cuadros cKnicos tales como diabetes mellitus, resistencia insulmica, dislipidemia, obesidad, aterosclerosis, enfermedad cardiovascular, smdrome metabolico y neurosis.
[Antecedentes de la invencion]
La diabetes mellitus (DM) es una enfermedad que produce un aumento patologico del nivel de glucemia (concentracion de glucosa en sangre) debido a una secrecion de insulina alterada o resistencia insulmica y se sabe que supone un factor de riesgo para diversas complicaciones graves. La diabetes mellitus supuestamente se desarrolla mediante la participacion de diversos factores ambientales (falta de ejercicio, sobrealimentacion y obesidad, etc.) con base en factores geneticos. Se espera que el numero de pacientes con diabetes mellitus aumente en el futuro con el aumento de la poblacion obesa. La diabetes mellitus se clasifica en diabetes mellitus insulinodependiente (DMID) (diabetes mellitus tipo 1) y diabetes mellitus no insulinodependiente (diabetes mellitus tipo 2). La mayor parte (aproximadamente el 90 %) de pacientes con diabetes mellitus se clasifica como pacientes con diabetes mellitus tipo 2.
La diabetes mellitus tipo 1 es una enfermedad en la cual las celulas beta, que segregan insulina en los islotes pancreaticos de Langerhans, mueren debido a diversos factores geneticos y factores adquiridos. La diabetes mellitus tipo 2 es una enfermedad que es causada por cantidades insuficientes de insulina segregada como respuesta a la glucosa en las celulas beta y por una disminucion en la sensibilidad a la insulina en los tejidos perifericos (Idgado, musculo y tejido adiposo, etc.).
Como tratamiento y profilaxis de la diabetes mellitus se usan un tratamiento con dieta y un tratamiento con ejercicio, asf como medicacion.
Ejemplos de una medicacion actual habitual incluyen una medicacion que implica la administracion subcutanea de insulina, un analogo de la insulina o un analogo del GLP-1 (peptido-1 similar al glucagon) o similares y una medicacion que usa un farmaco hipoglucemico de administracion via oral. El farmaco hipoglucemico de administracion via oral incluye sulfonilureas (farmacos SU) tales como glimepirida y similares; biguanidas (farmacos BG) tales como metformina y similares; inhibidores de las alfa-glucosidasas (farmacos alfa-GI) tales como voglibosa, miglitol y similares; derivados de la tiazolidina (farmacos TZD) tales como pioglitazona y similares; inhibidores de la DPP-IV (dipeptidil peptidasa-IV) tales como Sitagliptina, Alogliptina y similares; etc.
La somatostatina se distribuye ampliamente en el sistema nervioso central incluyendo el hipotalamo y similares, los islotes pancreaticos de Langerhans y la mucosa intestinal, etc. y juega un importante papel en el control de la motilidad gastrointestinal, la secrecion de jugos digestivos y el metabolismo de la glucosa o lipfdico. Concretamente, en los organismos vivos, se sabe que la somatostatina actua de forma supresora sobre la produccion o secrecion de varias hormonas, factores de crecimiento y sustancias biologicamente activas. Las hormonas sobre las cuales la somatostatina actua de forma supresora incluyen la hormona del crecimiento (GH), hormona estimulante de la tiroides (TSH), prolactina, insulina, glucagon, gastrina, secretina, peptido YY (PYY), polipeptido inhibidor gastrico (GIP), GLP-1, GLP-2, colecistoquinina (CCK), peptido intestinal vasoactivo (VIP), oxintomodulina y similares. Ademas, la somatostatina tambien actua como paracrino en los islotes pancreaticos de Langerhans o la mucosa del tracto gastrointestinal donde las celulas delta estan en contacto con las celulas alfa y las celulas beta. La somatostatina, por tanto, tiene diversas funciones biologicas en el sistema endocrino, el sistema exocrino y el sistema nervioso, etc. El receptor de la somatostatina es un receptor acoplado a protemas G o transmembrana de siete dominios. Hasta el momento se han descubierto cinco subtipos y se denominan, respectivamente, SSTR1, SSTR2, SSTR3, SSTR4 y SSTR5 (referencia bibliografica que no es una patente 1). Entre ellas, se ha demostrado que el SSTR5 participa en la regulacion de las secreciones de insulina e incretina (referencia bibliografica que no es una patente 2).
Ademas, la referencia bibliografica de patentes 1 ha publicado que el siguiente compuesto presenta una accion antagonista del SSTR5:

donde cada Ra se selecciona independientemente del grupo constituido por un atomo de hidrogeno, un atomo de halogeno, un grupo alquilo C1-10 y un grupo alquilo C1-10 sustituido con un atomo de halogeno; R1 se selecciona del grupo constituido por un atomo de hidrogeno, un fenilo sustituido y un anillo heterodclico sustituido; R2 se selecciona del grupo constituido por un arilo sustituido y un anillo heterodclico sustituido; y n y m se seleccionan cada uno independientemente del grupo constituido por 1, 2 y 3.
Las referencias bibliograficas de patentes 2 y 3 describen que el siguiente compuesto presenta una accion inhibitoria del receptor AMPA y similares y se puede usar en el tratamiento de la enfermedad desmielinizante o enfermedad neurodegenerativa:

donde Q representa NH, O S;R1, R2, R3, R4 y R5 representan cada uno un atomo de hidrogeno, un atomo de halogeno, un grupo alquilo C1-6 o un grupo representado por la formula -X-A (donde X representa un enlace sencillo, alquileno C1-6 sustituido, -O-, -S- o similares); y A representa un grupo cicloalquilo C3-8, un grupo cicloalquenilo C3-8, un anillo heterodclico no aromatico de 5 a 14 miembros, un anillo aromatico C6-14 o un anillo heterodclico aromatico de 5 a 14 miembros, siempre que tres de R1 a R5 sean los mismos o diferentes y cada uno represente -X-A y cada uno de los dos restantes sea un atomo de hidrogeno, un atomo de halogeno o un grupo alquilo C1-6.
Las referencias bibliograficas de patentes 2 y 3 tambien describen el siguiente compuesto como un ejemplo del compuesto anteriormente mencionado en el Ejemplo 379B:

Li y col. describen el siguiente compuesto en cuanto a la conversion oxidativa de isoxazolidina en isoxazolina (referenda bibliografica que no es una patente 3):

Ademas de los mencionados anteriormente, los siguientes compuestos se conocen con los numeros de registro CAS 1381469-87-9, 1214501-97-9 y 1214473-59-2:


[Lista de referencias]
[Bibliografia de patentes]
[PTL 1] Publicacion internacional num. WO 2012/024183
[PTL 2 ] Publicacion internacional num. WO 2001/096308
[PTL 3 ] Publicacion internacional num. WO 2003/047577
[Bibliografia que no son patentes]
[NPL 1]Patel YC: «Molecular pharmacology of somatostatin receptor subtypes.» J Endocrinol Invest 20: 348 367, 1997
[NPL 2]Chisholm C y col.: «Somatostatin-28 regulates GLP-1 secretion via somatostatin receptor subtype 5 in rat intestinal cultures.» Am J Physiol Endocrinol Metab 283: E311-317, 2002
[NPL 3]Pan Li y col.: «Oxidative Conversion of Isoxazolidines to Isoxazolines.» J. Org. Chem. 63 (2): 366 369, 1998
[Resumen de la invencion]
[Problema tecnico]
Existe una demanda de desarrollo de un compuesto que presente una accion antagonista del SSTR5 y que sea util en el tratamiento, mejora o profilaxis de enfermedades o cuadros clmicos tales como diabetes mellitus, resistencia insulinica, dislipidemia, obesidad, aterosclerosis, enfermedad cardiovascular, smdrome metabolico y neurosis.
[Solucion al problema]
Los actuales inventores han descubierto por primera vez que un compuesto representado por la formula siguiente:

donde el anillo A es
(1) un anillo de benceno opcionalmente sustituido con de 1 a 4 sustituyentes que son los mismos o diferentes y se seleccionan de entre:
un atomo de halogeno; un grupo cicloalquilo C3-10; un grupo alcoxi Ci-a;
y un grupo arilo Ca-14 opcionalmente sustituido con de 1 a 3 atomos de halogeno, o (2) piridina opcionalmente sustituida con de 1 a 3 sustituyentes que son los mismos o diferentes y se seleccionan de entre:
un grupo cicloalquilo C3-10; un grupo alcoxi C1-a; y un grupo arilo Ca-14 opcionalmente sustituido con de 1 a 3 atomos de halogeno;
el anillo B es piridina o pirimidina cada una ademas opcionalmente sustituida con de 1 a 3 sustituyentes seleccionados de entre un atomo de halogeno y un grupo alquilo C1-a; donde cada uno de X1, X2 y X4 es un atomo de carbono y X3 es un atomo de carbono o un atomo de nitrogeno;
n es 3;
R1 es COOH, CONH2 , un grupo tetrazolilo o un
grupo dihidrooxadiazolilo, o OCH2COOH y esta unido a X2 del anillo B;
y cada uno de R2 y R3 es un atomo de hidrogeno,
o una sal del mismo (en lo sucesivo denominado a veces compuesto (I)) presenta una accion antagonista del SSTR5 elevada, es util en el tratamiento, mejora o profilaxis de enfermedades o cuadros clrnicos tales como diabetes mellitus, resistencia insulrnica, srndrome metabolico, dislipidemia, obesidad, aterosclerosis, enfermedad cardiovascular y neurosis y tiene una eficacia elevada. Con base en este descubrimiento, los actuales inventores han llevado a cabo estudios detallados, que dan como resultado la finalizacion de la presente invencion.
Por consiguiente, la presente invencion se refiere a
[1] un compuesto representado por la siguiente formula:

donde el anillo A es
(1) un anillo de benceno opcionalmente sustituido con de 1 a 4 sustituyentes que son los mismos o diferentes y se seleccionan de entre:
un atomo de halogeno; un grupo cicloalquilo C3-10; un grupo alcoxi C1-a;
y un grupo arilo Ca-14 opcionalmente sustituido con de 1 a 3 atomos de halogeno, o (2) piridina opcionalmente sustituida con de 1 a 3 sustituyentes que son los mismos o diferentes y se seleccionan de entre:
un grupo cicloalquilo C3-10; un grupo alcoxi C1-a; y un grupo arilo Ca-14 opcionalmente sustituido con de 1 a 3 atomos de halogeno;
el anillo B es piridina o pirimidina cada una ademas opcionalmente sustituida con de 1 a 3 sustituyentes seleccionados de entre un atomo de halogeno y un grupo alquilo C1-a, donde cada uno de X1, X2 y X4 es un atomo de carbono y X3 es un atomo de carbono o un atomo de nitrogeno;
n es 3;
R1 es COOH, CONH2 , un grupo tetrazolilo o un
grupo dihidrooxadiazolilo, o OCH2COOH y esta unido a X2 del anillo B; y
cada uno de R2 y R3 es un atomo de hidrogeno,
o una sal del mismo;
[2] el compuesto de acuerdo con el anteriormente mencionado [1] o una sal del mismo, que es acido 1-(1-((2-ciclopropil-4'-fluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridin-4-carboxflico o una sal del mismo;
[3] el compuesto de acuerdo con el anteriormente mencionado [1] o una sal del mismo, que es acido 1-(1-((a-
a
cidopropil-2,4'-difluoro-3-isopropoxibifeml-4-il)metil)pipendm-4-il)-3-etil-2-oxo-1,2-dihidropindina-4-carboxflico o una sal del mismo;
[4] el compuesto de acuerdo con el anteriormente mencionado [1] o una sal del mismo, que es acido 1-(1-((6-ddopropil-2,4'-difluoro-3-isoprapoxibifeml-4-il)metil)pipendm-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxflico o una sal del mismo;
[5] un medicamento que comprende el compuesto del anteriormente mencionado [1] o una sal del mismo;
[6] el medicamento del anteriormente mencionado [5], para uso como un antagonista del receptor 5 de la somatostatina;
[7] el medicamento del anteriormente mencionado [5], para uso en la profilaxis o tratamiento de la diabetes mellitus;
[8] el compuesto de acuerdo con el anteriormente mencionado [1] o una sal del mismo para uso en la profilaxis o tratamiento de la diabetes mellitus.
[Efectos de la invencion]
El compuesto (I) presenta una accion antagonista del SSTR5 elevada, es util en el tratamiento, mejora y profilaxis de enfermedades o cuadros clmicos tales como diabetes mellitus, resistencia insulmica, smdrome metabolico, dislipidemia, obesidad, aterosclerosis, enfermedad cardiovascular y neurosis, y tiene una eficacia elevada.
[Descripcion detallada de la invencion]
La definicion de cada sustituyente usado en la presente memoria descriptiva se describe con detalle a continuacion. A menos que se especifique lo contrario, cada sustituyente tiene la definicion siguiente.
En la presente memoria descriptiva, ejemplos del «atomo de halogeno» incluyen fluor, cloro, bromo y yodo.
En la presente memoria descriptiva, ejemplos del «grupo alquilo C1-6» incluyen metilo, etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo, tert-butilo, pentilo, isopentilo, neopentilo, 1-etilpropilo, hexilo, isohexilo, 1,1-dimetilbutilo, 2,2-dimetilbutilo, 3,3-dimetilbutilo y 2-etilbutilo.
En la presente memoria descriptiva, ejemplos del «grupo cicloalquilo C3-10» incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, ciclooctilo, biciclo[2.2.1]heptilo, biciclo[2.2.2]octilo, biciclo[3.2.1]octilo y adamantilo.
En la presente memoria descriptiva, ejemplos del «grupo arilo C6-14» incluyen fenilo, 1-naftilo, 2-naftilo, 1-antrilo, 2-antrilo y 9-antrilo.
En la presente memoria descriptiva, ejemplos del «grupo alcoxi C1-6» incluyen metoxi, etoxi, propoxi, isopropoxi, butoxi, isobutoxi, sec-butoxi, tert-butoxi, pentiloxi y hexiloxi.
La definicion de cada sfmbolo en el compuesto (I) se describe con detalle a continuacion.
El anillo A representa
(1) un anillo de benceno opcionalmente sustituido con de 1 a 4 sustituyentes seleccionados de entre:
un atomo de halogeno (preferentemente fluor, cloro); un grupo cicloalquilo C3-10 (preferentemente ciclopropilo, ciclobutilo); un grupo alcoxi C1-6 (preferentemente metoxi, etoxi, isopropoxi); y un grupo arilo C6-14 opcionalmente sustituido con de 1 a 3 atomos de halogeno (preferentemente fluor), o
(2) piridina opcionalmente sustituida con de 1 a 3 sustituyentes seleccionados de entre:
un grupo cicloalquilo C3-10 (preferentemente ciclopropilo); un grupo alcoxi C1-6 (preferentemente etoxi, isopropoxi); y un grupo arilo C6-14 (preferentemente fenilo) opcionalmente sustituido con de 1 a 3 atomos de halogeno (preferentemente fluor, cloro).
Ejemplos espedficos especialmente preferentes de anillo A incluyen un anillo de benceno opcionalmente sustituido con de 1 a 4 sustituyentes seleccionados de entre:
un atomo de halogeno (preferentemente fluor, cloro); un grupo cicloalquilo C3-10 (preferentemente ciclopropilo, ciclobutilo); un grupo alcoxi C1-6 (preferentemente metoxi, etoxi, isopropoxi); y un grupo arilo C6-14 opcionalmente sustituido con de 1 a 3 atomos de halogeno (preferentemente fluor).
El anillo B representa piridina o pirimidina cada una ademas opcionalmente sustituida con de 1 a 3 sustituyentes
seleccionados de entre un atomo de halogeno y un grupo alquilo Cm
En la presente memoria descriptiva, puesto que el anillo B es piridina o pirimidina, la formula parcial:
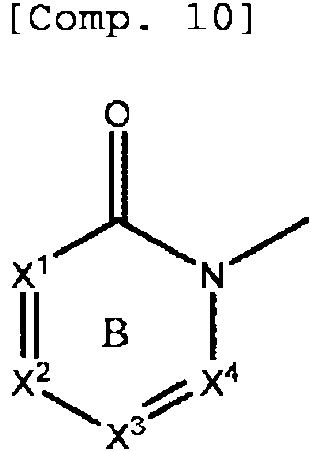
significa respectivamente

o

Por consiguiente, la piridina o pirimidina como anillo B incluyen respectivamente dihidropiridina (preferentemente 1,2-dihidropiridina) y dihidropirimidina (preferentemente 1,6-dihidropirimidina).
Preferentemente, ejemplos de anillo B incluyen piridina ademas opcionalmente sustituida con un grupo alquilo Cm El «atomo de halogeno» en la «piridina o pirimidina cada una ademas opcionalmente sustituida con de 1 a 3 sustituyentes seleccionados de entre un atomo de halogeno y un grupo alquilo Cm> es preferentemente bromo. El «grupo alquilo C1-6» en la «piridina o pirimidina cada una ademas opcionalmente sustituida con de 1 a 3 sustituyentes seleccionados de entre un atomo de halogeno y un grupo alquilo Cm> es preferentemente metilo, etilo o propilo. Cada uno de X1, X2 y X4 es un atomo de carbono.
X3 es un atomo de carbono o un atomo de nitrogeno y es preferentemente un atomo de carbono.
R1 es COOH, CONH2 , un grupo tetrazolilo, un grupo dihidrooxadiazolilo o OCH2COOH.
R1 es mas preferentemente COOH.
R1 esta unido a X2. Cada uno de R2 y R3 es un atomo de hidrogeno.
n es 3.
Puesto que n es 3, se forma piperidina. Preferentemente, el atomo de carbono en la posicion 4 de la piperidina esta unido al atomo de nitrogeno del anillo B como se muestra en [Comp. 13] a continuacion.
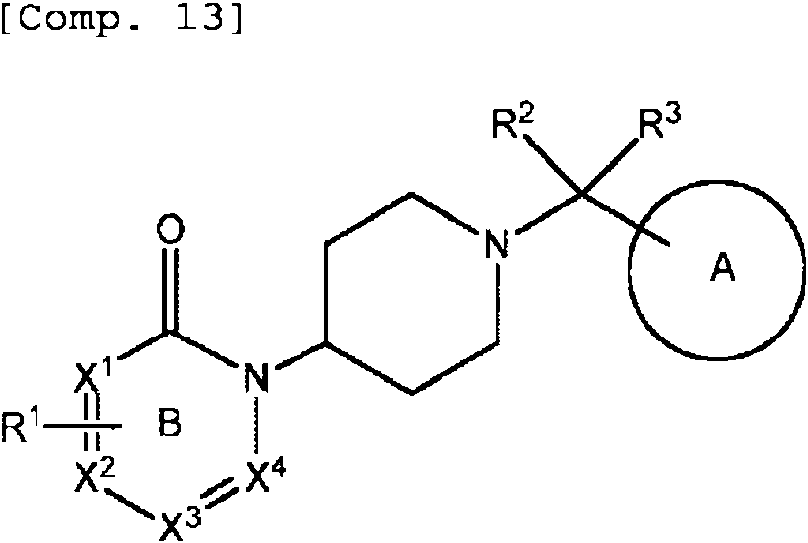
El compuesto (I) de la invencion se describe a continuacion como Compuesto A'. Ejemplos preferentes de compuesto (I) incluyen los Compuestos A, A'-1, A'-2 y A'-3 como tambien se describen a continuacion.
[Compuesto A]
Compuesto (I) donde
el anillo A es
(1) un anillo de benceno opcionalmente sustituido con de 1 a 4 sustituyentes que son los mismos o diferentes y se seleccionan de entre un atomo de halogeno (preferentemente fluor, cloro); un grupo cicloalquilo C3-10 (preferentemente ciclopropilo, ciclobutilo); un grupo alcoxi C1-6 (preferentemente metoxi, etoxi, isopropoxi); y un grupo arilo C6-14 opcionalmente sustituido con de 1 a 3 atomos de halogeno (preferentemente fluor); o
(2) piridina opcionalmente sustituida con de 1 a 3 sustituyentes que son los mismos o diferentes y se seleccionan de entre un grupo cicloalquilo C3-10 (preferentemente ciclopropilo); un grupo alcoxi C1-6 (preferentemente etoxi, isopropoxi); y un grupo arilo C6-14 opcionalmente sustituido (preferentemente fenilo opcionalmente sustituido con de 1 a 3 atomos de halogeno (preferentemente fluor));
el anillo B es piridina o pirimidina (donde cada uno de X1, X2 y X4 es un atomo de carbono y X3 es un atomo de carbono o un atomo de nitrogeno);
n es 3;
R1 es COOH, CONH2 , un grupo tetrazolilo, un grupo dihidrooxadiazolilo o OCH2COOH y esta unido a X2 del anillo B; y cada uno de R2 y R3 es un atomo de hidrogeno.
[Compuesto A']
Compuesto (I) donde
el anillo A es
(1) un anillo de benceno opcionalmente sustituido con de 1 a 4 sustituyentes que son los mismos o diferentes y se seleccionan de entre un atomo de halogeno (preferentemente fluor, cloro); un grupo cicloalquilo C3-10 (preferentemente ciclopropilo, ciclobutilo); un grupo alcoxi C1-6 (preferentemente metoxi, etoxi, isopropoxi); y un grupo arilo C6-14 opcionalmente sustituido con de 1 a 3 atomos de halogeno (preferentemente fluor); o
(2) piridina opcionalmente sustituida con de 1 a 3 sustituyentes que son los mismos o diferentes y se seleccionan de entre un grupo cicloalquilo C3-10 (preferentemente ciclopropilo); un grupo alcoxi C1-6 (preferentemente etoxi, isopropoxi); y un grupo arilo C6-14 (preferentemente fenilo) opcionalmente sustituido con de 1 a 3 atomos de halogeno (preferentemente fluor);
el anillo B es piridina o pirimidina cada una ademas opcionalmente sustituida con de 1 a 3 sustituyentes seleccionados de entre un atomo de halogeno y un grupo alquilo C1-6 (donde cada uno de X1, X2 y X4 es un atomo de carbono y X3 es un atomo de carbono o un atomo de nitrogeno);
n es 3;
R1 es COOH, CONH2 , un grupo tetrazolilo, un grupo dihidrooxadiazolilo o OCH2COOH y esta unido a X2 del anillo B; y cada uno de R2 y R3 es un atomo de hidrogeno.
[Compuesto A'-1]
Compuesto A' donde
el anillo A es un anillo de benceno opcionalmente sustituido con de 1 a 4 sustituyentes seleccionados de entre un atomo de halogeno (preferentemente fluor, cloro); un grupo cicloalquilo C3-10 (preferentemente ciclopropilo, ciclobutilo); un grupo alcoxi C1-6 (preferentemente metoxi, etoxi, isopropoxi); y un grupo arilo Ca-14 (preferentemente fenilo) opcionalmente sustituido con de 1 a 3 atomos de halogeno (preferentemente fluor).
[Compuesto A'-2]
El compuesto A'-1 donde
el anillo B es piridina ademas opcionalmente sustituida con un grupo alquilo C1-6 (preferentemente metilo o etilo), donde cada uno de X1, X2, X3 y X4 es un atomo de carbono.
[Compuesto A'-3]
El compuesto A'-1 o el compuesto A'-2 donde
R1 es COOH y esta unido a X2 del anillo B.
La sal del compuesto (I) es preferentemente una sal farmacologicamente aceptable. Ejemplos de una sal de este tipo incluyen sales con bases inorganicas, sales con base organica, sales con acido inorganico, sales con acido organico, sales con aminoacido basico o acido.
Preferentemente, ejemplos de la sal con base inorganica incluyen sales de metales alcalinos tales como sal de sodio, sal de potasio; sales de metales alcalinoterreos tales como sal de calcio, sal de magnesio; sal de aluminio; sal de amonio.
Preferentemente, ejemplos de la sal con base organica incluyen sales con trimetilamina, trietilamina, piridina, picolina, etanolamina, dietanolamina, trietanolamina, trometamina [tris(hidroximetil)metilamina], tert-butilamina, ciclohexilamina, bencilamina, diciclohexilamina, N,N-dibenciletilenodiamina.
Ejemplos preferentes de la sal con acido inorganico incluyen sales con acido clortndrico, acido bromhidrico, acido mtrico, acido sulfurico, acido fosforico.
Ejemplos preferentes de la sal con acido organico incluyen sales con acido formico, acido acetico, acido trifluoroacetico, acido ftalico, acido fumarico, acido oxalico, acido tartarico, acido maleico, acido dtrico, acido succmico, acido malico, acido metanosulfonico, acido bencenosulfonico, acido p-toluenosulfonico.
Ejemplos preferentes de la sal con un aminoacido basico incluyen sales con arginina, lisina, ornitina.
Ejemplos preferentes de la sal con un aminoacido acido incluyen sales con acido aspartico, acido glutamico.
El compuesto (I) se puede usar como un profarmaco.
Un profarmaco del compuesto (I) es un compuesto que se convierte en el compuesto (I) con una reaccion debida a un enzima, acido gastrico, etc. en el estado fisiologico del organismo vivo, es decir, un compuesto que se convierte en el compuesto (I) por oxidacion, reduccion, hidrolisis, etc. con un enzima; un compuesto que se convierte en el compuesto (I) mediante hidrolisis etc. debida a un acido gastrico, etc.
Ejemplos de un profarmaco del compuesto (I) incluyen:
un compuesto donde un grupo amino del compuesto (I) es acilado, alquilado o fosforilado (p. ej., compuesto donde un grupo amino del compuesto (I) es eicosanoilado, alanilado, pentilaminocarbonilado, (5-metil-2-oxo-1,3-dioxolen-4-il)metoxicarbonilado, tetrahidrofuranilado, pirrolidilmetilado, pivaloiloximetilatado o tertbutilado y similares);
un compuesto donde un grupo hidroxi del compuesto (I) es acilado, alquilado, fosforilado o borado (p. ej., compuesto donde un grupo hidroxi del compuesto (I) es acetilado, palmitoilado, propanoilado, pivaloilado, succinilado, fumarilado, alanilado o dimetilaminometilcarbonilado);
un compuesto donde un grupo carboxi del compuesto (I) es esterificado o amidado (p. ej., un compuesto donde un grupo carboxi del compuesto (I) es etil esterificado, fenil esterificado, carboximetil esterificado, dimetilaminometil esterificado, pivaloiloximetil esterificado, etoxicarboniloxietil esterificado, ftalidil esterificado, (5-metil-2-oxo-1,3-dioxolen-4-il)metil esterificado, ciclohexiloxicarboniletil esterificado o metilamidado); y similares. Estos compuestos se pueden producir a partir del compuesto (I) mediante un procedimiento conocido per se.
Un profarmaco del compuesto (I) tambien puede ser uno que se convierte en el compuesto (I) en un estado fisiologico, tal como se describe en el documento IYAKUHIN no KAIHATSU (Development of Pharmaceuticals), Vol.7, Design of Molecules, pp.163-198, publicado por HIROKAWA SHOTEN (1990).
En la memoria descriptiva, el profarmaco puede formar una sal. Ejemplos de una sal de este tipo incluyen aquellas ejemplificadas como la sal del compuesto (I) anteriormente mencionada.
Alternativamente, el compuesto (I) se puede marcar con un isotopo (p. ej.,3H,13C,14C,18F,35S,125I) o similar.
Ademas, el compuesto (I) puede ser un hidrato, un no hidrato, un no solvato o un solvato.
Adicionalmente, una forma con conversion en deuterio donde 1H se convierte en 2H (D) tambien esta incluida en el compuesto (I). El compuesto (I) marcado o sustituido con un isotopo se puede usar, por ejemplo, como marcador (marcador para TEP) para su uso en la tomograffa por emision de positrones (TEP) y es util en los campos del diagnostico medico y similares.
Ademas, el compuesto (I) puede ser un cocristal o sal de un cocristal farmaceuticamente aceptables. En este caso, el cocristal o sal de un cocristal significa una sustancia cristalina constituida por dos o mas sustancias concretas que son solidas a temperatura ambiente, teniendo cada una propiedades ffsicas diferentes (p. ej., estructura, punto de fusion, calor de fusion, higroscopicidad, solubilidad, estabilidad, etc.). El cocristal y la sal de un cocristal se pueden producir mediante cocristalizacion conocida per se.
El compuesto (I) (en lo sucesivo a veces abreviado simplemente como el compuesto de la presente invencion) muestra una baja toxicidad. Por tanto, el compuesto de la presente invencion se puede preparar como una composicion farmaceutica sola o mezclada con un vehnculo farmacologicamente aceptable o similar y, por tanto, usar como un agente para la profilaxis o tratamiento de diversas enfermedades anteriormente mencionadas en un mairnfero (p. ej., ser humano, raton, rata, conejo, perro, gato, animal bovino, caballo, cerdo, mono).
En este contexto, cualquiera de los diversos materiales vetnculo organicos o inorganicos que se usan habitualmente como materiales de preparacion se pueden usar como el vehnculo farmacologicamente aceptable. Estos se formulan como un excipiente, un lubricante, un agente ligante y un disgregante para preparaciones solidas o como un disolvente, un agente solubilizante, un agente de suspension, un agente isotonico, un agente amortiguador, un agente emoliente y similares para preparaciones lfquidas. Ademas, si es necesario, se pueden usar tambien aditivos de formulacion tales como un conservante, antioxidante, colorante, agente edulcorante y similares.
Ejemplos preferentes del excipiente incluyen lactosa, sacarosa, D-manitol, D-sorbitol, almidon, almidon pregelatinizado, dextrina, celulosa cristalina, hidroxipropilcelulosa poco sustituida, carboximetilcelulosa de sodio, goma arabiga, pululano, acido silfcico anhidro, silicato de aluminio sintetico y metasilicato de aluminio y magnesio.
Ejemplos preferentes del lubricante incluyen estearato de magnesio, estearato de calcio, talco y sflice coloidal. Ejemplos preferentes del agente ligante incluyen almidon pregelatinizado, sacarosa, gelatina, goma arabiga, metilcelulosa, carboximetilcelulosa, carboximetilcelulosa de sodio, celulosa cristalina, sacarosa, D-manitol, trehalosa, dextrina, pululano, hidroxipropilcelulosa, hidroxipropilmetilcelulosa y polivinilpirrolidona.
Ejemplos preferentes del disgregante incluyen lactosa, sacarosa, almidon, carboximetilcelulosa, carboximetilcelulosa de calcio, croscarmelosa de sodio, carboximetil almidon de sodio, acido silfcico anhidro ligero y hidroxipropilcelulosa poco sustituida.
Ejemplos preferentes del disolvente incluyen agua para inyeccion, suero fisiologico, solucion de Ringer, alcohol, propilenglicol, polietilenglicol, aceite de sesamo, aceite de mafz, aceite de oliva y aceite de algodon.
Ejemplos preferentes del agente solubilizante incluyen polietilenglicol, propilenglicol D-manitol, trehalosa, benzoato de bencilo, etanol, trisaminometano, colesterol, trietanolamina, carbonato de sodio, citrato de sodio, salicilato de sodio y
acetato de sodio.
Ejemplos preferentes del agente de suspension incluyen tensioactivos tales como trietanolamina de estearilo, lauril sulfato de sodio, acido laurilaminopropionico, lecitina, cloruro de benzalconio, cloruro de bencetonio, monoestearato de glicerina y similares; polfmeros hidrofilos tales como alcohol de polivinilo, polivinilpirrolidona, carboximetilcelulosa de sodio, metilcelulosa, hidroximetilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa y similares; polisorbatos y aceite de ricino polioxietileno hidrogenada.
Ejemplos preferentes del agente isotonico incluyen cloruro de sodio, glicerina, D-manitol, D-sorbitol y glucosa.
Ejemplos preferentes del agente amortiguador incluyen disoluciones amortiguadoras tales como fosfatos, acetatos, carbonatos, citratos y similares.
Ejemplos preferentes del agente emoliente incluyen alcohol de bencilo.
Ejemplos preferentes del conservante incluyen esteres de acido parahidroxibenzoico, clorobutanol, alcohol de bencilo, alcohol de fenetilo, acido deshidroacetico y acido sorbico.
Ejemplos preferentes del antioxidante incluyen sulfitos y ascorbatos.
Ejemplos preferentes del colorante incluyen tintes alimentarios de alquitran de hulla hidrosolubles (p. ej., tintes alimentarios tales como el rojo alimentario num. 2 y num. 3, amarillo alimentario num. 4 y num. 5, azul alimentario num. 1 y num. 2 y similares), tintes de laca no hidrosolubles (p. ej., sales de aluminio de los tintes de alquitran de hulla hidrosolubles anteriormente mencionados) y tintes naturales (p. ej., betacaroteno, clorofila, rojo de oxido ferrico). Ejemplos preferentes del agente edulcorante incluyen sacarina de sodio, glicirricinato de dipotasio, aspartamo y estevia.
Un medicamento que comprende el compuesto de la presente invencion se puede obtener usando el compuesto de la presente invencion solo o mezclado con un vetuculo farmacologicamente aceptable y administrar de forma segura via oral o via parenteral (p. ej., administrar via intravenosa, via intramuscular, via subcutanea, en un organo, en una cavidad nasal, via intracutanea, a traves de instilacion ocular, via intracerebral, via rectal, via vaginal, via intraperitoneal, en el interior de un tumor, en la proximidad de un tumor y similares, y administrar directamente en una lesion) a un mairnfero como una composicion farmaceutica, por ejemplo, comprimidos (incluyendo grageas, comprimidos recubiertos, comprimidos sublinguales, comprimidos bucodispersables, comprimidos gingivomaxilares y similares), pastillas, polvos, granulos, capsulas (incluyendo capsulas blandas, microcapsulas), comprimidos para chupar, jarabes, lfquidos, emulsiones, suspensiones, aerosoles, pelfculas, (p. ej., pelfculas sublinguales, pelfculas tipo parche para aplicacion en la mucosa oral), inyecciones (p. ej., inyecciones subcutaneas, inyecciones intravenosas, inyecciones intramusculares, inyecciones intraperitoneales, transfusiones, preparaciones cutaneas, pomadas, lociones, parches, supositorios (p. ej., supositorios rectales, supositorios vaginales), granulos, preparaciones nasales, preparaciones pulmonares (medicamento inhalado), gotas oculares y similares.
La composicion farmaceutica puede ser una preparacion de liberacion controlada tal como una preparacion de liberacion rapida, una preparacion de liberacion prolongada y similares (p. ej., una microcapsula de liberacion prolongada).
La composicion farmaceutica se puede producir mediante un procedimiento que se usa convencionalmente en el campo de la tecnologfa de la formulacion, por ejemplo, el procedimiento descrito en la Farmacopea Japonesa y similares.
El contenido del compuesto de la presente invencion en la composicion farmaceutica difiere dependiendo de la forma de administracion, la dosis del compuesto de la presente invencion, etc. y es, por ejemplo, de aproximadamente el 0,1 al 100 % en peso.
Durante la produccion de una preparacion via oral, el recubrimiento se puede aplicar conforme sea necesario con el fin de enmascarar el sabor, conferir propiedades entericas o durabilidad.
Ejemplos de la base de recubrimiento que se pueden usar para recubrir incluyen una base de recubrimiento con azucares, una base de recubrimiento con pelfcula acuosa, una base de recubrimiento con pelfcula enterica y una base de recubrimiento con pelfcula de liberacion prolongada.
Como base de recubrimiento con azucares, se usa la sacarosa. Ademas, se pueden usar en combinacion uno o mas
tipos seleccionados de entre talco, carbonato de calcio precipitado, gelatina, goma arabiga, pululano, cera de carnauba y similares.
Ejemplos de la base de recubrimiento con pelmula acuosa incluyen poKmeros de celulosa tales como hidroxipropil celulosa, hidroxipropilmetil celulosa, hidroxietil celulosa, metilhidroxietil celulosa etc.; poKmeros sinteticos tales como dietilaminoacetato de polivinilacetal, copolfmero E de aminoalquil metacrilato [Eudragit E (nombre comercial)], polivinilpirrolidona etc.; y polisacaridos tales como el pululano etc.
Ejemplos de la base de recubrimiento con pelmula enterica incluyen poKmeros de celulosa tales como ftalato de hidroxipropilmetil celulosa, succinato de acetato de hidroxipropilmetil celulosa, carboximetiletil celulosa, ftalato de acetato de celulosa, etc.; polfmeros acnlicos tales como copolfmero L de acido metacnlico [Eudragit L (nombre comercial)], copolfmero LD de acido metacnlico [Eudragit L-30D55 (nombre comercial)], copolfmero S de acido metacnlico [Eudragit S (nombre comercial)] etc.; y sustancias presentes en la naturaleza tales como goma laca etc. Ejemplos de la base de recubrimiento con pelmula de liberacion prolongada incluyen polfmeros de celulosa tales como etil celulosa etc.; y polfmeros acnlicos tales como copolfmero RS de aminoalquil metacrilato [Eudragit RS (nombre comercial)], suspension de copolfmero de etil acrilato-metil metacrilato [Eudragit NE (nombre comercial)] etc.
Las bases de recubrimiento anteriormente mencionadas se pueden usar despues de su mezclado con dos o mas tipos de las mismas en proporciones adecuadas. Como recubrimiento, por ejemplo, se puede usar un agente de proteccion ligera tal como oxido de titanio, oxido ferrico rojo y similares.
El compuesto de la presente invencion presenta una baja toxicidad (p. ej., toxicidad tras dosis unica, toxicidad tras tratamiento prolongado, toxicidad genetica, toxicidad para la funcion reproductora, toxicidad para la funcion pulmonar, carcinogenesis), muestra pocos efectos secundarios y se puede usar en un mairnfero como un agente para la profilaxis o tratamiento de diversas enfermedades o como un farmaco diagnostico para diversas enfermedades.
El compuesto de la presente invencion presenta una accion antagonista del SSTR5 elevada.
El compuesto de la presente invencion se puede usar como un agente para la profilaxis o tratamiento de, por ejemplo, la diabetes mellitus (p. ej., diabetes mellitus tipo 1, diabetes mellitus tipo 2, diabetes gestacional, diabetes mellitus por obesidad), obesidad (p. ej., mastocitosis maligna, obesidad exogena, obesidad por hiperinsulinismo, obesidad hiperplasica, adiposidad hipofisiaria, obesidad hipoplasica, obesidad hipotiroidea, obesidad hipotalamica, obesidad sintomatica, obesidad infantil, obesidad del tren superior, obesidad exogena, obesidad hipogonadica, mastocitosis sistemica, obesidad general, obesidad central y similares), hiperfagia, hiperlipidemia/dislipidemia (p. ej., hipertrigliceridemia, hipercolesterolemia, colesterolemia LDL elevada, colesterolemia HDL baja, hiperlipidemia posprandial), hipertension, enfermedad cardiovascular (p. ej., insuficiencia cardiaca, arritmia, cardiopatfa isquemica, valvulopatia cardiaca, arteriosclerosis), complicaciones de la diabetes [p. ej., neuropatfa, nefropatfa, retinopatfa, cardiomiopatfa diabetica, catarata, macroangiopatfa, osteopenia, coma diabetico hiperosmolar, enfermedad infecciosa (p. ej., infeccion respiratoria, infeccion del tracto urinario, infeccion gastrointestinal, infecciones de los tejidos blandos cutaneos, infeccion de los miembros inferiores), gangrena diabetica, xerostomia, hipoacusia, enfermedad cerebrovascular, alteracion de la circulacion sangumea periferica], smdrome metabolico (cuadros clmicos que presentan 3 o mas seleccionados de entre hipertriglicerid(TG)emia, colesterol(HDL-C)emia HDL baja, hipertension, obesidad abdominal y tolerancia a la glucosa alterada), sarcopenia, trastorno afectivo, disfuncion sexual, depresion, ansiedad, neurosis, arteriosclerosis, artritis de rodilla y similares.
La Sociedad Japonesa para el Diagnostico de la Diabetes publico el «Informe del comite de clasificacion y criterios diagnosticos de la diabetes mellitus» en 2010 respecto a los criterios diagnosticos de la diabetes mellitus.
De acuerdo con este informe, diabetes mellitus se refiere a un cuadro clmico que cursa con un nivel de glucemia en ayunas (concentracion de glucosa en el plasma venoso) de 126 mg/dL o superior, un valor a las 2 horas (concentracion de glucosa en el plasma venoso) de 200 mg/dL o superior con la prueba de tolerancia a la glucosa oral con 75 g (PTGO con 75 g), un nivel de glucemia eventual (concentracion de glucosa en el plasma venoso) de 200 mg/dL o superior y un valor HbA1c (valor estandar internacional) del 6,5 % o superior. Sin embargo, el HbA1c (valor estandar internacional) (%) se indica como un valor estandarizado internacional correspondiente al NGSP (Programa Nacional de Estandarizacion de la Glicohemoglobina), que es un 0,4 % superior al valor convencional de la JDS (Sociedad Japonesa de Diabetes) de HbA1c (valor JDS) (%). Ademas, un cuadro clmico que no se ajusta a la anteriormente mencionada diabetes mellitus y es un cuadro clmico que no muestra un «nivel de glucemia en ayunas (concentracion de glucosa en plasma venoso) inferior a 110 mg/dL o un valor a las 2 horas (concentracion de glucosa en el plasma venoso) inferior a 140 mg/dL en la prueba de tolerancia a la glucosa oral con 75 g (PTGO con 75 g)» (tipo normal) se denomina «tipo dudoso».
De acuerdo con el informe de la Organizacion Mundial de la Salud (OMS) del 2006, la diabetes mellitus se refiere a un cuadro clmico que cursa con un nivel de glucemia en ayunas (concentracion de glucosa en el plasma venoso) de 126 mg/dL o superior o un valor a las 2 horas (concentracion de glucosa en el plasma venoso) de 200 mg/dL o superior en la prueba de tolerancia a la glucosa oral con 75 g.
De acuerdo con los informes anteriormente mencionados, una alteracion de tolerancia a la glucosa (IGT) se refiere a un cuadro clmico que cursa con un nivel de glucemia en ayunas (concentracion de glucosa en el plasma venoso) inferior a 126 mg/dL y un valor a las 2 horas (concentracion de glucosa en el plasma venoso) de 140 mg/dL o superior e inferior a 200 mg/dL en la prueba de tolerancia a la glucosa oral con 75 g. De acuerdo con el informe de la OMS, un cuadro clmico que muestra un nivel de glucemia en ayunas (concentracion de glucosa en plasma venoso) de 110 mg/dL o superior e inferior a 126 mg/dL y un valor a las 2 horas (concentracion de glucosa en el plasma venoso) inferior a 140 mg/dL en la prueba de tolerancia a la glucosa oral con 75 g, si se ha medido, se denomina IFG (alteracion de la glucosa en ayunas).
El compuesto de la presente invencion tambien se usa como un agente para la profilaxis o tratamiento de la diabetes mellitus, diabetes mellitus de tipo dudoso, alteracion de tolerancia a la glucosa o IFG (alteracion de la glucosa en ayunas) determinadas de acuerdo con los informes anteriormente mencionados. Ademas, el compuesto de la presente invencion puede prevenir el progreso del tipo dudoso, la alteracion de tolerancia a la glucosa o la IFG (alteracion de la glucosa en ayunas) a diabetes mellitus.
El compuesto de la presente invencion tambien es util como un agente para la profilaxis o el tratamiento del smdrome metabolico. La incidencia de una enfermedad cardiovascular es significativamente elevada en pacientes con smdrome metabolico en comparacion con pacientes con una unica enfermedad relacionada con su estilo de vida. Por tanto, la profilaxis o tratamiento del smdrome metabolico es sumamente importante para prevenir enfermedades cardiovasculares.
Los criterios diagnosticos del smdrome metabolico fueron comunicados por la OMS en 1999 y por el NCEP en 2001. De acuerdo con los criterios diagnosticos de la OMS, un individuo que presenta hiperinsulinemia o tolerancia anormal a la glucosa como un requisito y dos o mas de obesidad visceral, dislipidemia (TG elevados o HDL baja) e hipertension se diagnostica con smdrome metabolico (Organizacion Mundial de la Salud: Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications. Part I: Diagnosis and Classification of Diabetes Mellitus, World Health Organization, Ginebra, 1999). De acuerdo con los criterios diagnosticos de la Grna para el tratamiento en adultos III del Programa Nacional de Educacion en Colesterol (recomendaciones sobre la cardiopatfa isquemica) de EE. UU., un individuo que presenta tres o mas de obesidad visceral, hipertrigliceridemia, colesterolemia HDL baja, hipertension y tolerancia anormal a la glucosa se diagnostica con smdrome metabolico (Programa Nacional de Educacion en Colesterol: Executive Summary of the Third Report of National Cholesterol Education Program (NCEP) Expert Panel on Detection,Evaluation, and Treatment of High Blood Cholesterol in Adults (Adults Treatment Panel III). The Journal of the American Medical Association, Vol. 285, 2486-2497, 2001).
El compuesto de la presente invencion tambien se puede usar como un agente para la profilaxis o tratamiento de, por ejemplo, osteoporosis, caquexia (p. ej., caquexia cancerosa, caquexia tuberculosa, caquexia diabetica, caquexia asociada con una hemopatfa, caquexia asociada con una endocrinopatfa, caquexia asociada con una enfermedad infecciosa o caquexia producida por un smdrome de inmunodeficiencia adquirida), esteatosis hepatica, poliquistosis ovarica, nefropatfas (p. ej., nefropatfa diabetica, glomerulonefritis, glomeruloesclerosis, smdrome nefrotico, nefroesclerosis hipertensiva, insuficiencia renal terminal), distrofia muscular, infarto de miocardio, angina de pecho, enfermedad cerebrovascular (p. ej., infarto cerebral, accidente cerebrovascular), enfermedad de Alzheimer, enfermedad de Parkinson, demencia, smdrome de resistencia insulmica, smdrome X metabolico, hiperinsulinemia, parestesia producida por hiperinsulinemia, diarrea cronica o aguda, enfermedad inflamatoria (p. ej., artritis reumatoide cronica, espondiloartritis anquilosante, enfermedad de Kashin-Bek, lumbago, gota, inflamacion posoperatoria o postraumatica, distension abdominal, neuralgia, laringofaringitis, cistitis, hepatitis (incluyendo esteatohepatitis no alcoholica), neumoma, pancreatitis, ulceras, gastritis, trastornos digestivos, lesion de la mucosa gastrica (incluyendo lesion de la mucosa gastrica producida por la aspirina)), smdrome del fondo de saco, enfermedad inflamatoria intestinal (incluyendo enfermedad inflamatoria de colon), celiaqrna (por ejemplo derivada de una enteropatfa por gluten o enfermedad celfaca), esprue tropical, esprue hipogammaglobulinemico, enteritis, enteritis regional (enfermedad de Crohn), smdrome del colon irritable asociado con diarrea, afectacion del intestino delgado (incluyendo lesiones de la mucosa del intestino delgado) y smdrome del intestino corto, esofagitis con reflujo, colitis ulcerosa, malabsorcion, insuficiencia testicular, smdrome de obesidad visceral y sarcopenia.
Ademas, el compuesto de la presente invencion tambien se puede usar como un agente para la profilaxis o tratamiento
de diversos canceres (concretamente, cancer de mama (p. ej., cancer de mama ductal invasivo, cancer de mama ductal no invasivo, cancer de mama inflamatorio, etc.), cancer prostatico (p. ej., cancer prostatico hormonodependiente, cancer prostatico no hormonodependiente, etc.), cancer pancreatico (p. ej., cancer pancreatico ductal, etc.), cancer gastrico (p. ej., adenocarcinoma papilar, adenocarcinoma mucoso, carcinoma adenoescamoso, etc.), cancer de pulmon (p. ej., carcinoma de pulmon no microdtico, carcinoma de pulmon microdtico, mesotelioma maligno, etc.), cancer de colon (p. ej., tumor estromatico gastrointestinal, etc.), cancer rectal (p. ej., tumor estromatico gastrointestinal, etc.), cancer colorrectal (p. ej., cancer colorrectal familiar, cancer colorrectal no poliposo hereditario, tumor estromatico gastrointestinal, etc.), cancer del intestino delgado (p. ej., linfoma no Hodgkin, tumor estromatico gastrointestinal, etc.), cancer esofagico, cancer duodenal, cancer de lengua, cancer farmgeo (p. ej., cancer nasofarmgeo, cancer orofarmgeo, cancer hipofarmgeo, etc.), cancer de las glandulas salivales, tumor cerebral (p. ej., astrocitoma pineal, astrocitoma pilodtico, astrocitoma difuso, astrocitoma anaplasico, etc.), neurilenoma, cancer hepatico (p. ej., cancer hepatico primario, cancer extrahepatico de las vfas biliares, etc.), cancer renal (p. ej., cancer de celulas renales, cancer de celulas de transicion de la pelvis renal y el ureter, etc.), cancer de las vfas biliares, cancer de endometrio, cancer cervical uterino, cancer ovarico (p. ej., cancer ovarico epitelial, tumor extragonadal de celulas reproductoras, tumor ovarico de celulas reproductoras, tumor ovarico de bajo potencial maligno, etc.), cancer de vejiga, cancer de uretra, cancer de piel (p. ej., melanoma (ocular) intraocular, carcinoma de celulas de Merkel, etc.), hemangioma, linfoma maligno, melanoma maligno, cancer tiroideo (p. ej., cancer tiroideo medular, etc.), cancer paratiroideo, cancer de la cavidad nasal, cancer de los senos paranasales), tumor oseo (p. ej., osteosarcoma, tumor de Ewing, sarcoma uterino, sarcoma de los tejidos blandos, etc.), angiofibroma, sarcoma de la retina, cancer de pene, tumor testicular, tumor solido pediatrico (p. ej., tumor de Wilms, tumor renal infantil, etc.), sarcoma de Kaposi, sarcoma de Kaposi producido por el sida, tumor del seno maxilar, histiocitoma fibroso, leiomiosarcoma, rabdomiosarcoma, leucemia (p. ej., leucemia mielogena aguda, leucemia linfodtica aguda, etc.), etc.).
El compuesto de la presente invencion tambien se puede usar para la prevencion secundaria o supresion de la progresion de las diversas enfermedades anteriormente mencionadas (p. ej., episodios cardiovasculares agudos tales como infarto de miocardio y similares).
La dosis del compuesto de la presente invencion se determina debidamente de acuerdo con el sujeto a quien se administra, la via de administracion, la enfermedad objetivo, los smtomas y similares. Por ejemplo, cuando el compuesto de la presente invencion se administra via oral a un paciente adulto con obesidad, una dosis unitaria habitualmente es de aproximadamente 0,01 a 100 mg/kg de peso corporal, preferentemente de aproximadamente 0,05 a 30 mg/kg de peso corporal, mas preferentemente de aproximadamente 0,5 a 10 mg/kg de peso corporal. La dosis unitaria del compuesto se administra preferentemente de una a tres veces al dfa.
El compuesto de la presente invencion se puede usar en combinacion con un farmaco (en lo sucesivo abreviado como farmacos concomitantes) tal como agentes terapeuticos para la diabetes mellitus, agentes terapeuticos para las complicaciones de la diabetes, agentes terapeuticos para la hiperlipidemia, agentes antihipertensivos, agentes antiobesidad, diureticos, agentes antitromboticos y similares con el fin de favorecer la accion del compuesto, disminuir la dosis del compuesto o similar. Respecto a esto, el momento de administracion del compuesto de la presente invencion y el del farmaco concomitante no estan limitados. Estos farmacos concomitantes pueden ser compuestos de bajo peso molecular o pueden ser macromoleculas tales como protemas, polipeptidos, anticuerpos, vacunas o similares. El compuesto de la presente invencion y el farmaco concomitante se pueden administrar simultaneamente o de forma escalonada al sujeto a quien se administra. Ademas, el compuesto de la presente invencion y el farmaco concomitante se pueden administrar como dos tipos de preparaciones que comprenden respectivamente los ingredientes activos o como una unica preparacion que comprende ambos ingredientes activos.
La dosis del farmaco concomitante se puede determinar debidamente con base en la dosis usada en situaciones sintomaticas. La proporcion de mezcla del compuesto de la presente invencion y un farmaco concomitante se puede determinar debidamente dependiendo del sujeto a quien se administra, la via de administracion, la enfermedad objetivo, los smtomas, combinaciones y similares. Cuando el sujeto de administracion es humano, por ejemplo, se pueden usar de 0,01 a 100 partes en peso de un farmaco concomitante por 1 parte en peso del compuesto de la presente invencion.
Aqrn, como el agente terapeutico para la diabetes mellitus, se pueden mencionar, por ejemplo, preparaciones insulmicas (p. ej., preparaciones insulmicas de origen animal extrafdas del pancreas de bovinos o cerdos; preparaciones insulmicas de origen humano geneticamente sintetizadas usando Escherichia coli o levadura; insulina cinc; insulina protamina cinc; fragmentos o derivados de la insulina (p. ej., INS-1), preparaciones insulmicas orales), sensibilizadores a la insulina (p. ej., sensibilizadores a la insulina o una sal de la misma (preferiblemente, clorhidrato), rosiglitazona o una sal de los mismos (preferiblemente maleato), Metaglidasen, AMG-131, Balaglitazona, MBX-2044, Rivoglitazona, Aleglitazar, Chiglitazar, Lobeglitazona, PLX-204, PN-2034, GFT-505, THR-0921, los compuestos
descritos en los documented W02007/013694,W02007/018314,W02008/093639o WO2008/099794), inhibidores de la a-glucosidasa (p. ej., voglibosa, acarbosa, miglitol, emiglitato), biguanidas (p. ej., metformina, buformina o una sal de las mismas (p. ej., clorhidrato, fumarato, succinato)), secretagogos de insulina [sulfonilurea (p. ej., tolbutamida, glibenclamida, gliclazida, clorpropamida, tolazamida, acetohexamida, gliclopiramida, glimepirida, glipizida, glibuzol), repaglinida, nateglinida, mitiglinida o una sal de calcio hidratada de los mismos], inhibidores de la dipeptidil peptidasa IV (p. ej., Alogliptina o una sal de la misma (preferiblemente, benzoato), Vildagliptina, Sitagliptina, Saxagliptina, Teneligliptina, Linagliptina, Anagliptina, Melogliptina, Dutogliptina, PF-00734200, ALS2-0426, TA-6666, TS-021, KRP-104, Trelagliptina o una sal de los mismos (preferiblemente, succinato)), agonistas beta-3 (p. ej., N-5984), agonistas GPR40 (p. ej., fasiglifam, compuesto descrito en los documentos WO2004/041266,WO2004/106276,WO2005/063729,WO2005/063725,WO2005/087710,WO2005/095338,WO2007/0 13689 o WO2008/001931), agonistas de los receptores GLP-1 [p. ej., GLP-1, preparaciones con GLP-1 MR, liraglutida, exenatida, AVE-0010, BIM-51077, Aib(8,35)hGLP-1(7,37)NH2, CJC-1131, albiglutida], agonistas de la amilina (p. ej., pramlintida), inhibidores de la fosfotirosina fosfatasa (p. ej., vanadato de sodio), inhibidores de la gluconeogenesis (p. ej., inhibidores de la glucogen fosforilasa, inhibidores de la glucosa-6-fosfatasa, antagonistas del glucagon, inhibidores de la FBPasa), inhibidores del SGLT2 (cotransportador 2 de sodio-glucosa) (p. ej., dapagliflocina, AVE2268, TS-033, YM543, TA-7284, Remogliflocina, ASP1941), inhibidores del SGLT1, inhibidores de la 11-beta-hidroxiesteroide deshidrogenasa (p. ej., BVT-3498), adiponectina o agonistas de la misma, inhibidores de las IKK (p. ej., AS-2868), farmacos de mejora de la resistencia a la leptina, agonistas de los receptores de la somatostatina, activadores de las glucoquinasas (p. ej., Piragliatina, AZD1656, AZD6370, TTP-355, compuesto descrito en los documentos WO006/112549,WO007/028135,WO008/047821,WO008/050821,WO008/136428 o WO008/156757), GIP (peptido insulinotropico dependiente de la glucosa) y similares.
Como agente terapeutico para las complicaciones diabeticas, por ejemplo, se pueden mencionar inhibidores de la aldosa reductasa (p. ej., tolrestato, epalrestato, zopolrestato, fidarestato, CT-112, ranirestato (AS-3201), lidorestato), el factor neurotropico y agentes de aumento del mismo (p. ej., NGF, NT-3, BDNF, el agente de fomento de la produccion/secrecion neurotropica descrito en el documento WO01/14372 (p. ej., 4-(4-clorofenil)-2-(2-metil-1-imidazolil)-5-[3-(2-metilfenoxi)propil]oxazol), compuesto descrito en el documento WO2004/039365), farmacos que favorecen la regeneracion nerviosa (p. ej., Y-128), inhibidores de las PKC (p. ej., mesilato de ruboxistaurina), inhibidores de AGE (p. ej., ALT946, bromuro de N-fenaciltiazolio (ALT766), AlT-711, EXO-226, Piridorina, piridoxamina), agonistas de los receptores del GABA (p. ej., gabapentina, pregabalina), inhibidores de la recaptacion de la serotonina y noradrenalina (p. ej., duloxetina), inhibidores de los canales de sodio (p. ej., lacosamida), secuestrantes de oxfgeno activo (p. ej., acido tioctteo), vasodilatadores cerebrales (p. ej., tiapurida, mexiletina), agonistas de los receptores de la somatostatina (p. ej., BIM23190), inhibidores de la quinasa-1 reguladora de la senal de apoptosis (ASK-1) y similares.
Como agente terapeutico para la hiperlipidemia se pueden mencionar, por ejemplo, compuestos de estatinas (p. ej., pravastatina, simvastatina, lovastatina, atorvastatina, fluvastatina, rosuvastatina, pitavastatina, cerivastatina o una sal de las mismas (p. ej., sal de sodio, sal de calcio)), inhibidores de la smtesis del escualeno (p. ej., los compuestos descritos en el documento WO97/10224, por ejemplo, acido N-[[(3R,5S)-1-(3-acetoxi-2,2-dimetilpropil)-7-cloro-5-(2,3-dimetoxifenil)-2-oxo-1,2,3,5-tetrahidro-4,1-benzoxacepin-3-il]acetil]piperidin-4-acetico), compuestos de fibratos (p. ej., bezafibrato, clofibrato, simfibrato, clinofibrato), resinas de intercambio anionico (p. ej., colestiramina), probucol, farmacos de acido nicotmico (p. ej., nicomol, niceritrol, niaspan), icosapentato de etilo, fitosterol (p. ej., soisterol, gamma orizanol, inhibidores de la absorcion del colesterol (p. ej., zetia), inhibidores de la CETP (p. ej., dalcetrapib, anacetrapib), preparaciones de acidos grasos omega-3 (p. ej., esteres etflicos de acidos grasos omega-3 90) y similares.
Ejemplos de agentes antihipertensivos incluyen inhibidores de la enzima de conversion de angiotensina (p. ej., captoprilo, enalaprilo, delaprilo, etc.), antagonistas de la angiotensina II (p. ej., candesartan cilexetilo, candesartan, losartan, losartan potasico, eprosartan, valsartan, telmisartan, irbesartan, tasosartan, olmesartan, olmesartan medoxomilo, azilsartan, azilsartan medoxomilo), antagonistas del calcio (p. ej., manidipina, nifedipina, amlodipina, efonidipina, nicardipina, amlodipina, cilnidipina, etc.), betabloqueantes (p. ej., metopolol, atenolol, propranolol, carvedilol, pindolol), inhibidores de la renina (p. ej., aliskiren), clonidina y similares.
Ejemplos de los agentes antiobesidad incluyen inhibidores de la recaptacion de monoaminas (p. ej., fentermina, sibutramina, mazindol, fluoxetina, tesofensina), agonistas del receptor 2C de serotonina (p. ej., lorcaserina), agonistas del receptor 6 de serotonina, receptor H3 de histamina, modulador de GABA (p. ej., topiramato), antagonistas de los receptores de la MCH (p. ej., SB-568849; SNAP-7941; los compuestos descritos en los documentos WO01/82925 o WO01/87834), antagonistas del neuropeptido Y (p. ej., velneperit), antagonistas de los receptores de cannabinoides (p. ej., rimonabant, taranabant), antagonistas de la grelina, antagonistas de los receptores de grelina, inhibidores enzimaticos de la grelinaciclacion, antagonistas de los receptores de opioides (p. ej., g SK-1521498), antagonistas de
los receptores de las orexinas (p. ej., almorexant), agonistas del receptor 4 de las melanocortinas, inhibidores de la 11-beta-hidroxiesteroide deshidrogenasa (p. ej., AZD-4017), inhibidores de las lipasas pancreaticas (p. ej., orlistat, cetilistat), agonistas beta-3 (p. ej., N-5984), inhibidores de la diacilglicerol aciltransferasa 1 (DGAT1), inhibidores de la acetilCoA carboxilasa (ACC), inhibidores enzimaticos de la estearoil-CoA desaturada, inhibidores de las protemas de transferencia de trigliceridos microsomales (p. ej., R-256918), inhibidores de los cotransportadores de Na-glucosa (p. ej., JNJ-28431754, dapagliflozina, canagliflozina, remogliflozina), inhibidores del NFkappa (p. ej., HE-3286), agonistas de los PPAR (p. ej., GFT-505, DRF-11605), inhibidores de la fosfotirosina fosfatasa (p. ej., vanadato de sodio, Trodusquemin), agonistas de GPR119 (p. ej., PSN-821), activadores de la glucoquinasa (p. ej., AZD-1656), leptina, derivados de la leptina (p. ej., metreleptina), CNTF (factor neurotrofico ciliar), BDNF (factor neurotrofico derivado del cerebro), agonistas de la colecistoquinina, preparaciones de peptido-1 similar al glucagon (GLP-1), p. ej., preparaciones de GLP-1 animal extrafdas del pancreas de bovinos o cerdos; preparaciones de GLP-1 humano geneticamente sintetizadas usando Escherichia coli o levadura; fragmentos o derivados del GLP-1 (p. ej., exenatida, liraglutida)), preparaciones de amilina (p. ej., pramlintida, AC-2307), agonistas del neuropeptido Y (p. ej., PYY3-36, derivados de PYY3-36, obineptida, TM-30339, TM-30335), preparaciones de oxintomodulina: preparaciones de FGF21 (p. ej. preparaciones de FGF21 animal extrafdas del pancreas de bovinos o cerdos; preparaciones de FGF21 humano geneticamente sintetizadas usando Escherichia coli o levadura; fragmentos o derivados del FGF21), combinacion de una preparacion de liberacion prolongada de clorhidrato de naltrexona y una preparacion de liberacion prolongada de clorhidrato de bupropion, agentes anorexigenicos (p. ej., P-57) y similares.
Como el diuretico, por ejemplo, se pueden mencionar derivados de la xantina (p. ej., teobromina con salicilato de sodio, teobromina con salicilato de calcio), preparaciones de tiazida (p. ej., etiazida, ciclopentiazida, triclorometiazida, hidroclorotiazida, hidroflumetiazida, bencilhidroclorotiazida, penflutiazida, politiazida, meticlotiazida), preparaciones de antialdosterona (p. ej., espironolactona, triamtereno, canrenoato de potasio), inhibidores de la anhidrasa carbonica (p. ej., acetazolamida), agentes de clorobencenosulfonamida (p. ej., clortalidona, mefrusida, indapamida), azosemida, isosorbida, acido etacrmico, piretanida, bumetanida, furosemida y similares.
Como agente antitrombotico, por ejemplo, se pueden mencionar heparina (p. ej., heparina de sodio, heparina de calcio, enoxaparina de sodio, dalteparina de sodio), warfarina (p. ej., warfarina de potasio), farmacos antitrombina (p. ej., aragatroban, dabigatran), agentes trombolfticos (p. ej., uroquinasa, tisoquinasa, alteplasa, nateplasa, monteplasa, pamiteplasa), inhibidores de la agregacion de plaquetas (p. ej., ticlopidina clorhidrato, clopidogrel, E5555, SHC530348, cilostazol, icosapentato de etilo, beraprost sodio, sarpogrelato clorhidrato, prasugrel, ticagrelor), inhibidores del Fxa (p. ej., rivaroxaban, apixaban, edoxaban, YM150, compuesto descrito en los documentos W002/06234,W02004/048363,W02005/030740,W02005/058823 o WO2005/113504) y similares.
El momento de administracion del farmaco concomitante anteriormente mencionado no esta limitado y el compuesto de la presente invencion y el farmaco concomitante se pueden administrar simultaneamente o de forma escalonada al sujeto a quien se administra. La dosis del farmaco concomitante se puede ajustar a la dosis usada en situaciones sintomaticas y se puede determinar debidamente dependiendo del sujeto a quien se administra, la via de administracion, la enfermedad, combinaciones y similares. La forma de administracion del farmaco concomitante no esta especialmente limitada y solo es necesario que el compuesto de la presente invencion y el farmaco concomitante se combinen en el momento de su administracion. Ejemplos de tales formas de administracion incluyen las siguientes:
1) administracion de una unica preparacion obtenida mediante procesado simultaneo del compuesto de la presente invencion y el farmaco concomitante,
2) administracion simultanea de dos tipos de preparaciones del compuesto de la presente invencion y el farmaco concomitante, que se han producido de forma independiente, mediante la misma via de administracion, 3) administracion de dos tipos de preparaciones del compuesto de la presente invencion y el farmaco concomitante, que se han producido de forma independiente, mediante la misma via de administracion de forma escalonada,
4) administracion simultanea de dos tipos de preparaciones del compuesto de la presente invencion y el farmaco concomitante, que se han producido de forma independiente, mediante diferentes vfas de administracion, 5) administracion de dos tipos de preparaciones del compuesto de la presente invencion y el farmaco concomitante, que se han producido de forma independiente, mediante diferentes vfas de administracion de forma escalonada (p. ej., administracion en orden del compuesto de la presente invencion y el farmaco concomitante, o en orden inverso) y similares.
La proporcion de mezcla del compuesto de la presente invencion y un farmaco concomitante se puede determinar debidamente dependiendo del sujeto a quien se administra, la via de administracion, la enfermedad y similares.
A continuacion, se describen procedimientos para producir el compuesto de la presente invencion.
En los procedimientos de produccion que se proporcionan a continuacion, los materiales de partida o reactivos usados
en cada etapa y los compuestos obtenidos pueden, cada uno, formar una sal. Ejemplos de una sal de este tipo incluyen los mismos que la sal anteriormente mencionada del compuesto de la presente invencion y similares.
Cuando el compuesto obtenido en cada etapa es un compuesto libre, se puede convertir en una sal de interes mediante un procedimiento conocido per se. Por el contrario, cuando el compuesto obtenido en cada etapa es una sal, se puede convertir en una forma libre o en un tipo diferente de sal de interes mediante un procedimiento conocido per se. El compuesto obtenido en cada etapa se puede usar en la reaccion posterior directamente en forma de una disolucion de reaccion del mismo o despues de haber sido obtenido como un producto en bruto. Alternativamente, el compuesto obtenido en cada etapa se puede aislar y/o purificar a partir de la mezcla de reaccion mediante medios de separacion tales como concentracion, cristalizacion, recristalizacion, destilacion, extraccion con disolvente, fraccionamiento, cromatograffa y similares de acuerdo con un procedimiento convencional.
Cuando los compuestos de los materiales de partida o reactivos para cada etapa estan comercialmente disponibles, estos productos comercialmente disponibles se pueden usar directamente.
Para la reaccion en cada etapa, el tiempo de reaccion puede ser diferente dependiendo del reactivo o disolvente usado y es generalmente de 1 minuto a 48 horas, preferentemente de 10 minutos a 8 horas, a menos que se especifique lo contrario.
Para la reaccion en cada etapa, la temperatura de reaccion puede ser diferente dependiendo del reactivo o disolvente usado y es generalmente de -78C («C» representa «grados Celsius») a 300C, preferentemente -78C a 150C, a menos que se especifique lo contrario.
Para la reaccion en cada etapa, la presion puede ser diferente dependiendo del reactivo o disolvente usado y es generalmente de 1 atm a 20 atm, preferentemente de 1 atm a 3 atm, a menos que se especifique lo contrario.
Para la reaccion en cada etapa se puede usar, por ejemplo, un equipo de smtesis con microondas tales como el modelo Initiator fabricado por Biotage Japan Ltd. y similares. La temperatura de reaccion puede ser diferente dependiendo del reactivo o disolvente usado y es generalmente de 300C, preferentemente de 50c a 250C, a menos que se especifique lo contrario. El tiempo de reaccion puede ser diferente dependiendo del reactivo o disolvente usado y es generalmente de 1 minuto a 48 horas, preferentemente de 1 minuto a 8 horas, a menos que se especifique lo contrario.
Para la reaccion en cada etapa, un reactivo se usa en una cantidad de 0,5 equivalentes a 20 equivalentes, preferentemente de 0,8 equivalentes a 5 equivalentes, respecto a un sustrato, a menos que se especifique lo contrario. Cuando un reactivo se usa como un catalizador, el reactivo se usa en una cantidad de 0,001 equivalentes a 1 equivalente, preferentemente de 0,01 equivalentes a 0,2 equivalentes, respecto a un sustrato. Cuando un reactivo tambien sirve como un disolvente de la reaccion, el reactivo se usa en la cantidad del disolvente.
Para la reaccion en cada etapa, la reaccion se lleva a cabo sin un disolvente o despues de la disolucion o suspension en un disolvente adecuado, a menos que se especifique lo contrario. Ejemplos espedficos del disolvente incluyen los disolventes descritos en los Ejemplos y los siguientes:
Alcoholes: metanol, etanol, alcohol de tert-butilo, 2-metoxietanol, etc.;
Eteres: dietil eter, difenil eter, tetrahidrofurano, 1,2-dimetoxietano, etc.;
Hidrocarburos aromaticos: clorobenceno, tolueno, xileno, etc.; Hidrocarburos saturados: ciclohexano, hexano, etc.;
Amidas: N,N-dimetilformamida, N-metilpirrolidona, etc.; Hidrocarburos halogenados: diclorometano, tetracloruro de carbono, etc.;
Nitrilos: acetonitrilo, etc.;
Sulfoxidos: dimetil sulfoxido, etc.;
Bases organicas aromaticas: piridina, etc.;
Anhfdridos de acidos: anhfdrido acetico, etc.;
Acidos organicos: acido formico, acido acetico, acido trifluoroacetico, etc.;
Acidos inorganicos: acido clortndrico, acido sulfurico, etc.; Esteres: acetato de etilo, etc.;
Cetonas: acetona, metil etil cetona, etc.; y Agua.
Estos disolventes se pueden usar como una mezcla de dos o mas de los mismos en una proporcion adecuada. Cuando se usa una base para la reaccion en cada etapa, se usa, por ejemplo, cualquiera de las bases siguientes o las bases descritas en los Ejemplos.
Bases inorganicas: hidroxido de sodio, hidroxido de magnesio, etc.; Sales basicas: carbonato de sodio, carbonato de calcio, bicarbonato de sodio, etc.;
Bases organicas: trietilamina, dietilamina, piridina, 4-dimetilaminopiridina, N,N-dimetilanilina, 1,4-diazabiciclo[2.2.2]octano, 1,8-diazabiciclo[5.4.0]-7-undecano, imidazol, piperidina, etc.;
Alcoxidos metalicos: etoxido de sodio, tert-butoxido de potasio, etc.;
Hidruros de metales alcalinos: hidruro de sodio, etc.;
Amidas metalicas: amida de sodio, diisopropilamida de litio, hexametildisilazida de litio, etc.; y
Litios organicos: n-butil-litio, etc.
Cuando se usa un acido o un catalizador acido para la reaccion en cada etapa, se usa, por ejemplo, cualquiera de los acidos o catalizadores acidos siguientes o los acidos o catalizadores acidos descritos en los Ejemplos.
Acidos inorganicos: acido clorlddrico, acido sulfurico, acido mtrico, acido bromhfdrico, acido fosforico, etc.;
Acidos organicos: acido acetico, acido trifluoroacetico, acido dtrico, acido p-toluenosulfonico, acido 10 alcanforsulfonico, etc.; y
acidos de Lewis: complejo de trifluoruro de boro y dietil eter, yoduro de cinc, cloruro de aluminio anhidro, cloruro de cinc anhidro, cloruro de hierro anhidro, etc.
La reaccion en cada etapa se lleva a cabo de acuerdo con un procedimiento conocido per se, por ejemplo, el procedimiento descrito en los documentos Jikken Kagaku Koza (Encyclopedia of Experimental Chemistry in English), 5th Ed., Vol. 13-19 (editado por The Chemical Society of Japan);Shin Jikken Kagaku Koza (New Encyclopedia of Experimental Chemistry in English), Vol. 14-15 (editado por The Chemical Society of Japan);Reactions and Syntheses in the Organic Chemistry Laboratory, Revised, 2nd Ed. (L. F. Tietze, Th. Eicher, Nankodo Co., Ltd.); Revised Organic Name Reactions;The Reaction Mechanism and Essence (Hideo Togo, Kodansha Ltd.);ORGANIC SYNTHESES Collective Volume I-VII (John Wiley & Sons Inc.);Modern Organic Synthesis in the Laboratory A Collection of Standard Experimental Procedures (Jie Jack Li, OXFORD UNIVERSITY Press);Comprehensive Heterocyclic Chemistry III, Vol.
1-14 (Elsevier B.V.);Strategic Applications of Named Reactions in Organic Synthesis (traducido por Kiyoshi Tomioka, publicado por Kagaku-Dojin Publishing Company, INC);Comprehensive Organic Transformations (VCH Publishers Inc.) (1989), etc., o el procedimiento descrito en los Ejemplos, a menos que se indique lo contrario.
La reaccion de proteccion o desproteccion de un grupo funcional en cada etapa se lleva a cabo de acuerdo con un procedimiento conocido per se, por ejemplo, el procedimiento descrito en los documentos «Protective Groups in Organic Synthesis, 4th Ed.» (Theodora W. Greene, Peter G. M. Wuts), Wiley-Interscience (2007); «Protecting Groups 3rd Ed.» (P.J. Kocienski), Thieme Medical Publishers (2004), etc., o el procedimiento descrito en los Ejemplos. Ejemplos de grupos protectores para el grupo hidroxilo o el grupo hidroxilo fenolico de un alcohol o similares incluyen grupos protectores tipo eter tales como metoximetil eter, bencil eter, t-butildimetilsilil eter, tetrahidropiranil eter y similares; grupos protectores tipo esteres de acidos carboxflicos tales como ester del acido acetico y similares; grupos protectores tipo esteres de acidos sulfonicos tales como ester del acido metanosulfonico y similares; grupos protectores tipo esteres de acidos carboxflicos tales como ester del acido acetico y similares;
Ejemplos de grupos protectores para el grupo carbonilo de un aldehfdo incluyen grupos protectores tipo acetal tales como dimetil acetal y similares; grupos protectores tipo acetal dclicos tales como 1,3-dioxano dclico y similares; etc. Ejemplos de grupos protectores para el grupo carbonilo de una cetona incluyen grupos protectores tipo cetal tales como dimetil cetal y similares; grupos protectores tipo cetal dclicos tales como 1,3-dioxano dclico y similares; grupos protectores tipo oxima tales como O-metiloxima y similares; grupos protectores tipo hidrazona tales como N,N-dimetilhidrazona y similares; etc.
Ejemplos de grupos protectores para el grupo carboxilo incluyen grupos protectores tipo ester tales como metil ester y similares; grupos protectores tipo amida tales como N,N-dimetilamida y similares; etc.
Ejemplos de grupos protectores para el tiol incluyen grupos protectores tipo eter tales como benciltio eter y similares; grupos protectores tipo ester tales como ester del acido tioacetico, tiocarbonato, tiocarbamato y similares; etc.
Ejemplos de grupos protectores para el grupo amino o un anillo heterodclico aromatico tales como imidazol, pirrol, indol o similares incluyen grupos protectores tipo carbamato tales como carbamato de bencilo o similares; grupos protectores tipo amida tales como acetamida y similares; grupos protectores tipo alquilamina tales como N-trifenilmetilamina y similares; grupos protectores tipo sulfonamida tales como metanosulfonamida y similares; etc.
Un grupo protector se puede eliminar mediante un procedimiento conocido perse, por ejemplo, un procedimiento que usa acidos, bases, luz ultravioleta, hidracina, fenilhidracina, N-metilditiocarbamato de sodio, fluoruro de tetrabutilamonio, acetato de paladio o halogenuro de trialquilsililo (p. ej., yoduro de trimetilsililo, bromuro de trimetilsililo), un procedimiento de reduccion o similares.
En caso de llevar a cabo una reaccion de reduccion en cada etapa, ejemplos del agente reductor usado incluyen hidruros metalicos tales como hidruro de litio y aluminio, triacetoxi borohidruro de sodio, cianoborohidruro de sodio, hidruro de diisobutil aluminio (DIBAL-H), borohidruro de sodio, triacetoxi borohidruro de tetrametilamonio y similares; boranos tales como complejo de boranotetrahidrofurano y similares; mquel Raney; cobalto Raney; hidrogeno; acido formico; etc. Se puede usar un catalizador tal como paladio-carbon, un catalizador de Lindlar o similares en el procedimiento para reducir un triple enlace o doble enlace carbono-carbono.
En caso de llevar a cabo una reaccion de oxidacion en cada etapa, ejemplos del agente oxidante usado incluye peracidos tales como acido m-cloroperbenzoico (MCPBA), peroxido de hidrogeno, hidroperoxido de t-butilo y similares; percloratos tales como perclorato de tetrabutilamonio y similares; cloratos tales como clorato de sodio y similares; cloritos tales como clorito de sodio y similares; peryodatos tales como peryodato de sodio y similares; reactivos con yodo de elevada valencia tales como yodosilbenceno y similares; reactivos con manganeso tales como dioxido de manganeso, permanganato de potasio y similares; plomos tales como tetracetato de plomo y similares; reactivos con cromo tales como clorocromato de piridinio (PCC), dicromato de piridinio (PDC), reactivos de Jones y similares; compuestos de halogenos tales como N-bromosuccinimida (NBS) y similares; oxfgeno; ozono; complejo trioxido de azufre-piridina; tretraoxido de osmio; dioxido de selenio; 2,3-dicloro-5,6-diciano-1,4-benzoquinona (d Dq ); etc.
En caso de llevar a cabo una reaccion de ciclacion con radicales en cada etapa, ejemplos del iniciador radicalario usado incluyen compuestos azo tales como azobisisobutironitrilo (AIBN) y similares; iniciadores radicalarios hidrosolubles tales como acido 4-4'-azobis-4-cianopentanoico (ACPA) y similares; trietilboro en presencia de aire u oxfgeno; peroxido de benzoilo; etc. Ejemplos del reactivo radicalario usado incluyen tributilestannano, tristrimetilsililsilano, 1,1,2,2-tetrafenildisilano, difenilsilano, yoduro de samario y similares.
En caso de llevar a cabo una reaccion de Wittig en cada etapa, ejemplos del reactivo de Wittig usado incluyen fosforanos de alquilideno. Los fosforanos de alquilideno se pueden preparar mediante un procedimiento conocido per se, por ejemplo, la reaccion de una sal de fosfonio con una base fuerte.
En caso de llevar a cabo una reaccion de Horner-Emmons en cada etapa, ejemplos del reactivo usado incluyen esteres del acido fosfonoacetico tales como dimetilfosfonoacetato de metilo, dietilfosfonoacetato de etilo y similares; y bases tales como hidruros de metales alcalinos, litios organicos y similares.
En caso de llevar a cabo una reaccion de Friedel-Crafts en cada etapa, ejemplos del reactivo usado incluyen un acido de Lewis y un cloruro de acido o un agente alquilante (p. ej., halogenuros de alquilo, alcoholes, olefinas, etc.). Alternativamente, se puede usar un acido organico o inorganico en vez del acido de Lewis y se puede usar un anhfdrido de acido tal como anhfdrido acetico o similar en vez del cloruro de acido.
En caso de una reaccion de sustitucion nucleofila aromatica en cada etapa, se usan como reactivos un nucleofilo (p. ej., aminas, imidazol, etc.) y una base (p. ej., sales basicas, bases organicas, etc.).
En caso de llevar a cabo una reaccion de adicion nucleofila mediada por carbanion, reaccion de adicion 1,4 nucleofila mediada por carbanion (reaccion de adicion de Michael) o reaccion de sustitucion nucleofila mediada por carbanion en cada etapa, ejemplos de la base usada para generar un carbanion incluyen litios organicos, alcoxidos metalicos, bases inorganicas, bases organicas y similares.
En caso de llevar a cabo una reaccion de Grignard en cada etapa, ejemplos del reactivo de Grignard incluyen halogenuros de aril magnesio tales como bromuro de fenil magnesio y similares; y halogenuros de alquil magnesio tales como bromuro de metil magnesio y similares. El reactivo de Grignard se puede preparar mediante un procedimiento conocido per se, por ejemplo, la reaccion del halogenuro de alquilo o el halogenuro de arilo con magnesio metalico en presencia de eter o tetrahidrofurano como disolvente.
En caso de llevar a cabo una reaccion de condensacion de Knoevenagel en cada etapa, se usan como reactivos un compuesto de metileno activo rodeado de dos grupos salientes con dos electrones (p. ej. acido malonico, malonato de dietilo, malonitrilo, etc.) y una base (p. ej., bases organicas, alcoxidos metalicos, bases inorganicas).
En caso de llevar a cabo una reaccion de Vilsmeier-Haack en cada etapa, se usan como reactivos cloruro de fosforilo
y un derivado ammico (p. ej., N,N-dimetilformamida, etc.).
En caso de llevar a cabo la reaccion de azidacion de alcoholes, halogenuros de alquilo o esteres de acidos sulfonicos en cada etapa, ejemplos del agente de azidacion usado incluyen difenilfosforilazida (DPPA), azida de trimetilsililo, azida de sodio y similares.
Para la azidacion de alcoholes se usan, por ejemplo, un procedimiento que usa difenilfosforilazida y 1,8-diazabiciclo[5,4,0]undec-7-eno (DBU), un procedimiento que usa trimetilsilil azida y un acido de Lewis o similares. En caso de llevar a cabo una reaccion de aminacion reductora en cada etapa, ejemplos del agente reductor usado incluyen triacetoxi borohidruro de sodio, cianoborohidruro de sodio, hidrogeno, acido formico y similares. Cuando un sustrato es un compuesto ammico, ejemplos del compuesto carbomlico usado incluyen paraformaldelmdo, asf como aldelmdos tales como acetaldelmdo y similares y cetonas tales como ciclohexanona y similares. Cuando un sustrato es un compuesto carbomlico, ejemplos de las aminas usadas incluyen aminas primarias tales como amoniaco, metilamina y similares; aminas secundarias como dimetilamina y similares; etc.
En caso de llevar a cabo una reaccion de Mitsunobu en cada etapa, se usan como reactivos esteres de acidos azodicarboxflicos (p. ej., azodicarboxilato de dietilo (DEAD), azodicarboxilato de diisopropilo (DIAD), etc.) y trifenilfosfina.
En caso de llevar a cabo una reaccion de esterificacion, reaccion de amidacion o reaccion de formacion de urea en cada etapa, ejemplos del reactivo usado incluyen halogenuros de acilo tales como cloruro de acido, bromuro de acido y similares; anhfdridos de acido, esteres activos y acidos carboxflicos activados tales como ester de acido sulfurico y similares. Ejemplos del activador para acidos carboxflicos incluyen agentes condensadores con carbodiimida tales como clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (WSCD) y similares; agentes condensadores con triacina tales como cloruro n-hidrato de 4-(4,6-dimetoxi-1,3,5-triacin-2-il)-4-metilmorfolinio (DMT-MM) y similares; agentes condensadores con esteres del acido carbonico tales como 1,1-carbonildiimidazol (CDI) y similares; difenilfosforilazida (DPPA); sal de benzotriazol-1-iloxi-trisdimetilamino fosfonio (reactivo de BOP); yoduro de 2-cloro-1-metil-piridinio (reactivo de Mukaiyama); cloruro de tionilo; halo-formatos de alquilos inferiores tales como cloroformato de etilo y similares; hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HATU); acido sulfurico; combinaciones de los mismos; etc. En caso de usar un agente condensador con carbodiimida, se puede anadir ademas a la reaccion un aditivo tal como 1-hidroxibenzotriazol (HOBt), N-hidroxisuccinimida (HOSu), dimetilaminopiridina (DMAP) o similares.
En caso de llevar a cabo una reaccion de acoplamiento en cada etapa, ejemplos del catalizador metalico usado incluyen compuestos de paladio tales como acetato de paladio(II), tetrakis(trifenilfosfina)paladio(0), diclorobis(trifenilfosfina)paladio(II), diclorobis(trietilfosfina)paladio(II), tris(dibencilidenoacetona)dipaladio(0), cloruro de 1,1'-bis(difenilfosfino)ferroceno paladio(II) y similares; compuestos de mquel tales como tetrakis(trifenilfosfina)mquel(0) y similares; compuestos de rodio tales como cloruro de tris(trifenilfosfina)rodio(III) y similares; compuestos de cobalto; compuestos de cobre tales como oxido de cobre, yoduro de cobre(I) y similares; compuestos de platino; etc. Se puede anadir ademas una base a la reaccion. Ejemplos de una base de este tipo incluyen bases inorganicas, sales basicas y similares.
En caso de llevar a cabo una reaccion de tiocarbonilacion en cada etapa, habitualmente se usa pentasulfuro de difosforo como un agente de tiocarbonilacion. Ademas del pentasulfuro de difosforo, se puede usar un reactivo que tenga una estructura 1,3,2,4-ditiadifosfetano-2,4-disulfuro, tal como el 2,4-bis(4-metoxifenil-1,3,2,4-ditiadifosfetano-2,4-disulfuro(reactivo de Lawesson) o similares.
En caso de llevar a cabo una reaccion de Wohl-Ziegler en cada etapa, ejemplos del agente halogenante usado incluyen N-yodosuccinimida, N-bromosuccinimida (NBS), N-clorosuccinimida (NCS), bromo, cloruro de sulfurilo y similares. Se pueden aplicar a la reaccion calor, luz o un iniciador radicalario tal como peroxido de benzoilo, azobisisobutironitrilo o similares para acelerar asf la reaccion.
En caso de llevar a cabo la reaccion de halogenacion de un grupo hidroxilo en cada etapa, ejemplos del agente halogenante usado incluyen un acido halogenhfdrico y un halogenuro de acido de un acido inorganico, espedficamente acido clorhfdrico, cloruro de tionilo, oxicloruro de fosforo o similares para la cloracion y acido bromhfdrico al 48 % o similares para la bromacion. Ademas, se puede usar un procedimiento para obtener un halogenuro de alquilo a partir de un alcohol por la accion de una trifenilfosfina y tetracloruro de carbono o tetrabromuro de carbono, etc. Alternativamente, se puede usar un procedimiento para sintetizar un halogenuro de alquilo mediante dos etapas de reaccion que implica la conversion de un alcohol en un ester de acido sulfonico y la posterior reaccion con bromuro de litio, cloruro de litio o yoduro de sodio.
En caso de llevar a cabo una reaccion de Arbuzov en cada etapa, ejemplos del reactivo usado incluyen halogenuros de alquilo tales como bromoacetato de etilo y similares; y fosfitos tales como trietilfosfito, tri(isopropil)fosfito y similares. En caso de llevar a cabo una reaccion de esterificacion de sulfonas en cada etapa, ejemplos del agente sulfonante usado incluyen cloruro de metanosulfonilo, cloruro de p-toluenosulfonilo, anhndrido metanosulfonico, anhndrido ptoluenosulfonico y similares.
En caso de llevar a cabo una reaccion de hidrolisis en cada etapa, se usa un acido o una base como reactivo. Para la reaccion de hidrolisis acida de ester de t-butilo se pueden anadir acido formico, trietilsilano o similares para retener de forma reductora subproductos del cation t-butilo.
En caso de llevar a cabo una reaccion de deshidratacion en cada etapa, ejemplos del agente deshidratante usado incluyen acido sulfurico, pentaoxido de difosforo, oxicloruro de fosforo, N,N'-diciclohexilcarbodiimida, alumina, acido fosforico y similares.
El compuesto (1) se puede producir mediante un procedimiento mencionado posteriormente a partir del compuesto (2).

donde P1representa un grupo protector para el grupo amino; Y1 representa un grupo sulfonato o un atomo de halogeno; y el resto de los grupos son tal como se definieron anteriormente.
El compuesto (1) se puede producir mediante una reaccion de alquilacion usando el compuesto (3), el compuesto (4) y una base. Ejemplos de la base incluyen carbonato de potasio, carbonato de sodio y similares. El compuesto (4) se puede producir mediante un procedimiento conocido per se.
De los compuestos (1), el compuesto (1-1) se puede producir mediante un procedimiento mencionado posteriormente a partir del compuesto (3).

Los compuestos (5) se pueden producir mediante un procedimiento conocido per se.
De los compuestos (1), el compuesto (1-3) se puede producir mediante un procedimiento mencionado posteriormente a partir del compuesto (1-2), donde -L1-L2CO2H es COOH o OCH2COOH y R8 es un grupo alquilo C1-6 opcionalmente sustituido.

De los compuestos (2), el compuesto (2-1) se puede producir mediante un procedimiento mencionado posteriormente a partir del compuesto (6) y el compuesto (7), donde R8 es un grupo alquilo C1-6 opcionalmente sustituido.

El compuesto (2-1) se puede producir mediante una reaccion de alquilacion usando el compuesto (6), el compuesto (7) y una base. Ejemplos de la base incluyen carbonato de potasio, carbonato de sodio, tert-butoxido de potasio y similares. El compuesto (6) y el compuesto (7) se pueden producir mediante un procedimiento conocido per se. De los compuestos (2), el compuesto (2-2) se puede producir mediante un procedimiento mencionado posteriormente a partir del compuesto (8) y el compuesto (7).

donde Y2 representa un grupo sulfonato, un atomo de hidrogeno y un grupo hidroxilo protegido y R4 es el mismo que R8, que es un grupo alquilo C1-6 opcionalmente sustituido. El compuesto (8) se puede producir mediante un procedimiento conocido per se.
El compuesto (9) se puede producir mediante una reaccion de alquilacion usando el compuesto (8), el compuesto (7) y una base. Ejemplos de la base incluyen carbonato de potasio, carbonato de sodio, tert-butoxido de potasio y
similares.
El compuesto (2-2) se puede producir mediante una reaccion de insercion de monoxido de carbono usando el compuesto (9), monoxido de carbono, una base y un catalizador de paladio. Ejemplos del catalizador de paladio incluyen acetato de paladio, un aducto en diclorometano de [1,1 '-bis(difenilfosfino)ferroceno]dicloropaladio(II), tris(dibencilidenoacetona)dipaladio y similares. Ejemplos de la base incluyen trietilamina y similares.
De los compuestos (2), el compuesto (2-3) se puede producir mediante un procedimiento mencionado posteriormente a partir del compuesto (10) y el compuesto (11).

El compuesto (12) se puede producir mediante la reaccion de condensacion del compuesto (10) y el compuesto (11). El compuesto (2-3) se puede producir mediante una reaccion de cicloadicion usando el compuesto (12), el compuesto (13) y una base. Ejemplos de la base incluyen trietilamina, N,N-diisopropiletilamina y similares. El compuesto (10) y el compuesto (11) se pueden producir cada uno mediante un procedimiento conocido per se.
De los compuestos (1), el compuesto (1-5) se puede producir mediante un procedimiento mencionado posteriormente a partir del compuesto (1-4).
[C o m p . 20 ]

El compuesto (14) se puede producir mediante una reaccion de deshidratacion usando el compuesto (1-4), un agente deshidratante y una base. Ejemplos del agente deshidratante incluyen antndrido trifluoroacetico y similares. Ejemplos de la base incluyen trietilamina y similares.
El compuesto (16) se puede producir mediante una reaccion de adicion usando el compuesto (15) y una base.
Ejemplos de la base incluyen bicarbonato de sodio y similares. Se puede usar una sal del compuesto (15).
El compuesto (1-5) se puede producir mediante la reaccion de ciclacion del compuesto (16) usando un reactivo de carbonilacion y una base. Ejemplos del reactivo de carbonilacion incluyen 1,1'-carbonilbis-1H-imidazol, difosgeno, trifosgeno, cloroformato de fenilo y similares. Ejemplos de la base incluyen 1,8-diazabiciclo[5.4.0]unde-7-ceno (DBU) y similares. El compuesto (15) se puede producir mediante un procedimiento conocido per se.
De los compuestos (9), el compuesto (9-1) se puede producir mediante un procedimiento mencionado posteriormente a partir del compuesto (17).

donde P2 representa un grupo protector para el grupo hidroxilo; M representa acido boronico (B(OR)2 o B'F3K+) o un halogenuro de cinc; R9 representa un alquilo C1-6 ; y Y3 representa un grupo sulfonato. El compuesto (18) se puede producir mediante un procedimiento conocido per se.
De los compuestos (17), el compuesto (17-1) se puede producir mediante un procedimiento mencionado posteriormente a partir del compuesto (8-1) y el compuesto (7).

donde Y4 representa un atomo de halogeno.
El compuesto (21) se puede producir mediante una reaccion de alquilacion usando el compuesto (8-1), el compuesto (7) y una base. Ejemplos de la base incluyen carbonato de potasio, carbonato de sodio, tert-butoxido de potasio y similares. El compuesto (8-1) se puede producir mediante un procedimiento conocido per se.
El compuesto (17-1) se puede producir mediante la reaccion de halogenacion del compuesto (21) con un agente halogenante. Ejemplos del agente halogenante incluyen N-yodosuccinimida (NIS), N-bromosuccinimida (NBS), N-clorosuccinimida (NCS), yodo, bromo, cloruro de sulfurilo y similares.
De los compuestos (8), el compuesto (8-2) se puede producir mediante un procedimiento mencionado posteriormente a partir del compuesto (22).

donde R10 representa alquilo Ci-6.
El compuesto (24) se puede producir mediante una reaccion de transferencia-adicion usando el compuesto (22), una base y el compuesto (23). Ejemplos de la base incluyen diisopropilamida de litio, hexametildisilazida de litio, n-butillitio y similares. El compuesto (22) y el (23) se pueden producir cada uno mediante un procedimiento conocido per se. El compuesto (8-2) se puede producir mediante una reaccion de hidroxilacion usando el compuesto (24) y un acido. Ejemplos del acido incluyen acido clortffdrico, acido sulfurico y similares.
La presente invencion se explica con detalle a continuacion en referencia a los siguiente Ejemplos, Ejemplos de ensayos y Ejemplos de formulacion, que no se deben interpretar como limitativos. Ademas, la presente invencion se puede modificar sin apartarse del alcance de la invencion.
El termino «temperatura ambiente» en los siguientes ejemplos indica el intervalo, generalmente, de aproximadamente 10C a aproximadamente 35C. Una proporcion usada para un disolvente mezclado indica una proporcion en volumen, a menos que se especifique lo contrario. % indica % en peso, a menos que se especifique lo contrario.
El termino «NH» en cromatograffa en columna con gel de sflice indica que se uso un gel de sffice con enlaces aminopropilsilano. El termino «diol» en cromatograffa en columna con gel de sffice indica que se uso un gel de sffice con enlaces 3-(2,3-dihidroxipropoxi)propilsilano. El termino «C18» en HPLC (cromatograffa de ffquidos de alta resolucion) indica que se uso un gel de sffice con enlaces octadecilo. Una proporcion usada para los disolventes de elucion indica una proporcion en volumen, a menos que se especifique lo contrario.
Las abreviaturas descritas a continuacion se usan en los Ejemplos siguientes.
THF: tetrahidrofurano
DMF: dimetilformamida
DMA: dimetilacetamida
DME: 1,2-dimetoxietano
DMSO: dimetil sulfoxido
El espectro RMN 1H (espectro de resonancia magnetica nuclear de protones) se obtuvo mediante RMN con transformada de Fourier. Se uso un equipo ACD/SpecManager (nombre comercial) o similar. No se ha hecho mencion de los picos muy anchos de los protones de un grupo carboxilo, un grupo hidroxilo, un grupo amino y similares. Otras abreviaciones usadas en el presente documento tienen los significados que se describen a continuacion. s: singulete
d: doblete
t: triplete
q: cuatriplete
quin: quintuplete
m: multiplete
br: ancho
J: constante de acoplamiento
Hz: hercio
CDCb: cloroformo deuterado
DMSO-d6: d6-dimetil sulfoxido
CD3OD: metanol deuterado
1H-NMR: resonancia magnetica nuclear de protones
TFA: acido trifluoroacetico
El EM (espectro de masas) se midio usando CL/EM (espectrometro de masas con cromatograffa de ffquidos). Se uso ESI (ionizacion por electrospray) o APCI (ionizacion qmmica a presion atmosferica) como tecnica de ionizacion. No se usaron ni un modo positivo (ESI+) ni un modo negativo (ESI-) como modo de ionizacion y no se describio ningun dato. Los datos se indicaron como el valor medido real (hallado). En general, se observaron picos ionicos moleculares. En caso de un compuesto que tenga un grupo tert-butoxicarbonilo (-Boc), se puede observar un pico de iones fragmentados derivado de la eliminacion del grupo tert-butoxicarbonilo o el grupo tert-butilo (-tBu). En caso de un compuesto que tenga un grupo hidroxilo (-OH), se puede observar un pico de iones fragmentados derivado de la eliminacion de H2O. En caso de una sal, generalmente se observa un pico de ion molecular o un pico de ion fragmentado de una forma libre.
Ejemplo 1
Acido 1-(1-(4-CiclobutM-5-ciclopropM-2-etoxibencM)piperidm-4-M)-2-oxo-1,2-dihidropiridma-4-carboxiMco A) 2-(Benciloxi)-4-ciclobutilbenzoato de metilo
Se anadieron yodo (40 mg) y 1,2-dibromoetano (0,5 mL) a una mezcla de magnesio (11,8 g) y eter diefflico anhidro (250 mL), a continuacion, se lentamente b arñomadoióciclobutano (44,0 g) a la misma y la mezcla se agito a 40C durante 2 horas. Se una di asñoaludcióion en THF (450 mL) de cloruro de cinc (54 g) preparada a 160C a elevado vado a la mezcla de reaccion a 0C y la mezcla se agito a la misma temperatura que antes durante 1 hora. Se desgasifico una mezcla de 2-(benciloxi)-4-yodobenzoato de metilo (20 g), tris(dibencilidenoacetona)dipaladio(0) (4,97 g), 4,5-bis(difenilfosfino)-9,9-dimetilxanteno (3,14 g) y THF (500 mL) con una corriente de argon. El reactivo de cinc (350 mL) preparado anteriormente se a la misma y la mezcla añ saed aiógito durante la noche a 60C. La mezcla de reaccion se filtro sobre celita. El disolvente se concentro a presion reducida. Se anadio agua al residuo obtenido, seguido de extraccion con acetato de etilo tres veces. Las fases organicas combinadas se lavaron con agua y se secaron sobre sulfato de sodio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (9,5 g).
EM (ESI+): [M+H]+ 297,3.
B) 2-(benciloxi)-5-bromo-4-ciclobutilbenzoato de metilo
Se añadió carbonato de sodio (7,51 g) a una disolucion en diclorometano (150 mL) de 2-(benciloxi)-4-ciclobutilbenzoato de metilo (14 g), a continuacion, se lentamente a la misma una disolucion a eñnad dióiclorometano (25 mL) de bromo (3,17 mL) a 10C y la mezcla se agito a la misma temperatura que antes durante 1 hora. Se anadio una disolucion acuosa saturada de bicarbonato de sodio a la mezcla de reaccion, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se lavo con una disolucion acuosa saturada de bisulfito de sodio y salino saturado en este orden y se seco sobre sulfato de sodio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (12,2 g).
EM (ESI+): [M+H]+ 375,1.
C) 2-(benciloxi)-4-ciclobutil-5-ciclopropilbenzoato de metilo
Se anadieron acido ciclopropilboronico (6,18 g) y carbonato de sodio (15,3 g) a una disolucion mezclada de 2-(benciloxi)-5-bromo-4-ciclobutilbenzoato de metilo (13,5 g) en tolueno (240 mL) y agua (60 mL) y la mezcla se desgasifico durante 20 minutos con una corriente de argon. A continuacion, se anadieron tris(dibencilidenoacetona)dipaladio(0) (6,25 g) y diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (2,80 g) a la misma y la mezcla se desgasifico adicionalmente durante 5 minutos. La mezcla de reaccion se agito a 100C durante 16 horas. La mezcla de reaccion se filtro sobre celita y la celita se lavo con acetato de etilo. Las fases organicas combinadas se lavaron con agua y salino saturado en este orden y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (8,5 g).
EM (ESI+): [M+H]+ 337,3.
D) 4-ciclobutil-5-ciclopropil-2-hidroxibenzoato de metilo
Se desgasifico una disolucion en metanol (100 mL) de 2-(benciloxi)-4-ciclobutil-5-ciclopropilbenzoato de metilo (8,5 g) durante 20 minutos con una corriente de nitrogeno. A continuacion, se anadio paladio sobre carbon al 10 % (que contema el 55 % de agua, 1,1 g) a la misma y la mezcla se agito a temperatura ambiente durante 3 horas en atmosfera de hidrogeno. La mezcla de reaccion se filtro sobre celita y el disolvente se destilo a presion reducida para obtener el compuesto del fftulo (6,0 g).
EM (ESI+): [M+H]+ 247,2.
E) 4-ciclobutil-5-ciclopropil-2-etoxibenzoato de metilo
Se anadieron carbonato de cesio (3,44 g) y yodoetano (0,55 mL) a una disolucion en DMF (25 mL) de 4-ciclobutil-5-ciclopropil-2-hidroxibenzoato de metilo (1,3 g) con enfriamiento en hielo y la mezcla se agito durante la noche a temperatura ambiente. La mezcla de reaccion se diluyo con agua, seguido de extraccion con acetato de etilo dos veces. Las fases organicas combinadas se lavaron con agua y salino saturado en este orden y se secaron sobre
sulfato de sodio anhidro y, a continuacion, el disolvente se destilo a presion reducida para obtener el compuesto del t^tulo (1,2 g).
EM (ESI+): [M+H]+ 275,5.
F) 4-Cidobutil-5-cidopropM-2-etoxibenzaldehido
Se añadió hidruro de litio y aluminio (1 M disolucion en THF, 12,0 mL) a 0C a una disolucion en THF (50 mL) de 4-ciclobutil-5-ciclopropil-2-etoxibenzoato de metilo (3,0 g). La mezcla de reaccion se agito a temperature ambiente durante 2 horas. Se una dis aoñluacdioión acuosa saturada de sulfato de sodio a la mezcla de reaccion a 0C, seguido de extraccion con acetato de etilo. Las fases organicas combinadas se lavaron con agua y salino saturado en este orden y se secaron sobre sulfato de sodio anhidro y, a continuacion, el disolvente se destilo a presion reducida.
Se añadió dioxido de manganeso (8,4 g) en pequenas cantidades a temperature ambiente a una disolucion en acetona (40 mL) del residuo obtenido y la mezcla se agito durante la noche a temperatura ambiente. La mezcla de reaccion se filtro sobre celita y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (1,8 g).
EM (ESI+): [M+H]+ 245,5.
G) 4-(4-Bromo-2-oxopiridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo
Se añadió tert-butoxido de potasio (7,09 g) a temperatura ambiente a una mezcla de 4-bromopiridin-2(1H)-ona (10 g) en DME (287 mL). La mezcla de reaccion se agito a temperatura ambiente durante 10 minutos. A continuacion, se anadieron a la misma carbonato de potasio (15,9 g) y 4-(((4-metilfenil)sulfonil)oxi)piperidina-1-carboxilato de tert-butilo (37,8 g) y la mezcla se agito a 85C durante 1 dfa, a 95C durante 1 dfa y a 100C durante 1 dfa en atmosfera de nitrogeno. Se salin aoña sdaitóurado a la mezcla de reaccion, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (14,7 g).
EM (ESI+): [M+H-tBu]+ 301,1.
H) 1-(1-(Tert-butoxicarbonil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se agito una mezcla de 4-(4-bromo-2-oxopiridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo (14,7 g), trietilamina (11,5 mL), metanol (50 mL), dicloruro de 1,1'-bis(difenilfosfino)ferroceno-paladio(II) (3,01 g) y DMF (137 mL) a 80C durante 4 horas en atmosfera de monoxido de carbono de 0,3 MPa. Se anadio agua a la mezcla de reaccion, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se lavo con salino saturado dos veces y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) y cromatograffa en columna con gel de sflice (NH, hexano/acetato de etilo) para obtener el compuesto del tftulo (8,32 g).
EM (ESI+): [M+H-tBu]+ 281,2.
I) 1-(1-(4-Ciclobutil-5-ciclopropil-2-etoxibencil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se agito una mezcla de 1-(1-(tert-butoxicarbonil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (500 mg) y acido formico (2 mL) a 70C durante 1 hora y, a continuacion, el disolvente se destilo a presion reducida. Se anadieron 4-Ciclobutil-5-ciclopropil-2-etoxibenzaldehffdo (436 mg), THF (3 mL) y DMA (1 mL) al residuo obtenido y la mezcla se agito a temperatura ambiente durante 10 minutos. A continuacion, se anadio triacetoxiborohidruro de sodio (945 mg) a la misma y la mezcla se agito durante la noche a temperatura ambiente.Se anadio una disolucion acuosa saturada de bicarbonato de sodio a la mezcla de reaccion, seguido de extraccion con acetato de etilo dos veces. Las fases organicas combinadas se lavaron con salino saturado y se secaron sobre sulfato de magnesio anhidro y el disolvente se destilo a presion reducida.El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (567 mg).
EM (ESI+): [M+H]+ 465,4.
J) Acido 1-(1-(4-ciclobutil-5-ciclopropil-2-etoxibencil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxflico Se agito una mezcla de 1-(1-(4-ciclobutil-5-ciclopropil-2-etoxibencil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (567 mg), una disolucion acuosa de hidroxido de sodio 1 M (3 mL) y etanol (3 mL) a 70c durante 2 horas. Se ajusto el pH de la mezcla de reaccion hasta 4 usando acido clorhftdrico 1 M y se agito a temperatura ambiente durante 30 minutos. El solido resultante se recogio mediante filtracion y recristalizacion (DMSO/etanol) para obtener el compuesto del tftulo (358 mg).
RMN 1H (300 MHz, DMSO-da) delta 0,45-0,55 (2H, m), 0,80-0,91 (2H, m), 1,33 (3H, t, J = 6,9 Hz), 1,66-2,39 (13H, m), 2,97 (2H, d, J = 11,5 Hz), 3,51 (2H, s), 3,76-3,95 (1H, m), 4,05 (2H, q, J = 7,0 Hz), 4,53-4,74 (1H, m), 6,55 (1H, dd, J = 7,1, 1,8 Hz), 6,81 (2H, d, J = 2,3 Hz), 6,90 (1H, s), 7,84 (1H, d, J = 7,3 Hz).
Ejemplo 2
Acido 1-(1-((5-CidopropN-6-(4-fluorofenM)-2-isopropoxipiridm-3-N)metM)piperidm-4-N)-2-oxo-1,2-dihidropiridm-4-carboxilico
A) 6-(4-Fluorofenil)-2-hidroxinicotinonitrilo
Se anadieron en este orden 2-cianoacetamida (4,31 g) y 3-(dimetilamino)-1-(4-fluorofenil)prop-2-en-1-ona (9,00 g) a una mezcla de hidruro de sodio (60% aceite, 4,10 g) y DMF (90 mL) y la mezcla resultante se agito a 105C durante 2 horas. El disolvente se destilo. Se agua al residuo obten aidñoad yi,ó a continuacion, la mezcla se acidifico mediante la adicion de acido acetico y se agito a 70C durante 15 minutos. Se anadio metanol a la mezcla de reaccion para lograr una suspension y el solido depositado se lavo con acetato de etilo para obtener el compuesto del tftulo (9,98 g). RMN 1H (300 MHz, DMSO-d6) delta 6,78 (1H, d, J = 7,2 Hz), 7,38 (2H, t, J = 8,9 Hz), 7,89 (2H, dd, J = 8,8, 5,4 Hz), 8,19 (1H, d, J = 7,6 Hz).
B) 5-Bromo-6-(4-fluorofenil)-2-hidroxinicotinonitrilo
Se añadió N-bromosuccinimida (3,66 g) a una mezcla de 6-(4-fluorofenil)-2-hidroxinicotinonitrilo (4,00 g), THF (30 mL) y metanol (30 mL) y la mezcla resultante se agito a temperatura ambiente durante 10 minutos. El disolvente se disolvio y el residuo obtenido se suspendio en un disolvente mezclado de agua, acetato de etilo y hexano. A continuacion, el solido obtenido se lavo con hexano para obtener el compuesto del tftulo (5,18 g).
RMN 1H (300 MHz, DMSO-d6) delta 7,28-7,44 (2H, m), 7,63 (2H, dd, J = 8,6, 5,5 Hz), 8,54 (1H, s), 13,11 (1H, brs).
C) 5-Bromo-6-(4-fluorofen il)-2-isopropoxin icotinonitrilo
Se añadió 2-bromopropano (3,32 mL) a una mezcla de 5-bromo-6-(4-fluorofenil)-2-hidroxinicotinonitrilo (5,18 g), carbonato de potasio (4,89 g) y DMF (30 mL) y la mezcla resultante se agito a 80C durante 30 minutos. Se anadio agua a la mezcla de reaccion, seguido de extraccion con acetato de etilo. La fase organica se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (5,92 g).
RMN 1H (300 MHz, CDCla) delta 1,41 (6H, d, J = 6,2 Hz), 5,27-5,56 (1H, m), 7,16 (2H, t, J = 8,7 Hz), 7,66-7,82 (2H, m), 8,08 (1H, s).
D) 5-Bromo-6-(4-fluorofenil)-2-isopropoxinicotinato de metilo
Se añadió una disolucion acuosa 8 M de hidroxido de potasio (22,1 mL) a una mezcla de 5-bromo-6-(4-fluorofenil)-2-isopropoxinicotinonitrilo (5,92 g) y etanol (50 mL) y la mezcla de reaccion resultante se agito durante la noche a 100C. La mezcla de reaccion se neutralizo con acido clorhffdrico 6 M a 0C, seguido de extraccion con acetato de etilo. La fase organica se seco sobre sulfato de magnesio anhidro y, a continuacion, se destilo el disolvente a presion reducida. Se anadieron carbonato de potasio (4,88 g) y yodometano (1,66 mL) a una mezcla del residuo obtenido y DMF (30 mL) y la mezcla de reaccion se agito a 60C durante 30 minutos. Se agua a la mezcla de reaccion, segu aiñdaodi dóe extraccion con acetato de etilo. La fase organica se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (3,65 g). RMN 1H (300 MHz, CDCla) delta 1,40 (6H, d, J = 6,1 Hz), 3,91 (3H, s), 5,35-5,51 (1H, m), 7,14 (2H, t, J = 8,7 Hz), 7,78 (2H, dd, J = 8,9, 5,4 Hz), 8,38 (1H, s).
E) 5-ciclopropil-6-(4-fluorofenil)-2-isopropoxinicotinato de metilo
Se añadió tris(dibencilidenoacetona)dipaladio(0) (635 mg) a una mezcla de 5-bromo-6-(4-fluorofenil)-2-isopropoxinicotinato de metilo (3,65 g), acido ciclopropilboronico (2,55 g), diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (610 mg), una disolucion acuosa 2 M de carbonato de sodio (14,9 mL) y tolueno (25 mL) y la mezcla resultante se agito a 100C durante 2 horas en atmosfera de argon. La mezcla de reaccion se dejo enfriar hasta temperatura ambiente y se vertio en agua, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo
obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (3,18 g).
RMN 1H (300 MHz, CDCb) delta 0,60-0,68 (2H, m), 0,79-0,99 (2H, m), 1,39 (6H, d, J = 6,1 Hz), 1,87-2,00 (1H, m), 3,89 (3H, s), 5,37-5,53 (1H, m), 7,14 (2H, t, J = 8,7 Hz), 7,71-7,80 (2H, m), 7,82 (1H, s).
F) 5-Ciclopropil-6-(4-fluorofeml)-2-isopropoximcotmaldehido
Se añadió una disolucion en THF (20 mL) de 5-ciclopropil-6-(4-fluorofenil)-2-isopropoxinicotinato de metilo (3,17 g) a una suspension en THF (20 mL) de hidruro de litio y aluminio (365 mg) con enfriamiento en hielo en atmosfera de nitrogeno. Despues de agitacion a la misma temperatura que antes durante 30 minutos, se anadieron a la misma agua (0,35 mL) y una disolucion acuosa al 15 % de hidroxido de sodio (0,35 mL) y la mezcla se agito durante 5 minutos. A continuacion, se adicio anñaalmdióente agua (1,05 mL) a la misma. La mezcla de reaccion se agito durante 30 minutos y, a continuacion, se filtro. El filtrado se concentro a presion reducida. Se anadio dioxido de manganeso (8,36 g) a una disolucion en tolueno (30 mL) del residuo obtenido y la mezcla se agito a 60C durante 1 hora en atmosfera de nitrogeno. La mezcla de reaccion se filtro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (2,45 g).RMN 1H (300 MHz, CDCla) delta 0,58-0,72 (2H, m), 0,87-0,99 (2H, m), 1,40 (6H, d, J = 6,2 Hz), 1,81-2,01 (1H, m), 5,35-5,66 (1H, m), 7,15 (2H, t, J = 8,7 Hz), 7,69-7,84 (3H, m), 10,36 (1H, s).
G) 1-(1-((5-ciclopropil-6-(4-fluorofenil)-2-isopropoxipiridin-3-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridin-4-carboxilato de metilo
La mezcla de 1-(1-(tert-butoxicarbonil)piperidin-4-il)-2- oxo-1,2-dihidropiridin-4-carboxilato de metilo (150 mg) y acido formico se agito a 70C durante 1 hora. A continuacion, el disolvente se destilo a presion reducida. Se anadieron 5 Ciclopropil-6-(4-fluorofenil)-2-isopropoxinicotinaldel'ffdo (174 mg) y THF (2 mL) al residuo obtenido y la mezcla se agito a temperatura ambiente durante 10 minutos. A continuacion, se triacetoxiborohidruro de sodio (378 m agñ)ad aió la misma y la mezcla se agito durante la noche a temperatura ambiente. Se anadio una disolucion acuosa saturada de bicarbonato de sodio a la mezcla de reaccion, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (189 mg).
EM (ESI+): [M+H]+ 520,4.
H) Acido 1-(1-((5-CiclopropM-6-(4-fluorofeml)-2-isopropoxipiridm-3-il)metil)piperidm-4-il)-2-oxo-1,2-dihidropiridina-4-carboxnico
Se agito una mezcla de 1-(1-((5-ciclopropil-6-(4-fluorofenil)-2-isopropoxipiridin-3-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (189 mg), una disolucion acuosa de hidroxido de sodio 1 M (1,5 mL) y etanol (3 mL) a 70C durante 1 hora. La mezcla de reaccion obtenida se neutralizo con acido clorhfdrico 1 M y se agito durante la noche a temperatura ambiente. El solido resultante se recogio mediante filtracion y recristalizacion (etanol/acetato de etilo) para obtener el compuesto del fftulo (91 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,57 (2H, d, J = 5,7 Hz), 0,87 (2H, d, J = 6,7 Hz), 1,30 (6H, d, J = 6,1 Hz), 1,68 1,78 (2H, m), 1,82-1,98 (3H, m), 2,24 (2H, d, J = 17,3 Hz), 2,99 (2H, d, J = 9,0 Hz), 3,51 (2H, s), 4,64 (1H, d, J = 11,4 Hz), 5,19-5,32 (1H, m), 6,57 (1H, d, J = 6,9 Hz), 6,83 (1H, d, J = 1,5 Hz), 7,29 (2H, t, J = 8,9 Hz), 7,37 (1H, s), 7,74 (2H, dd, J = 8,7, 5,7 Hz), 7,90 (1H, d, J = 6,9 Hz).
Ejemplo 3
Acido 1-(1-((2-ciclopropil-4'-fluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridin-4-carboxftico
A) 4-yodo-2-isopropoxibenzoato de metilo
Se añadió 2-bromopropano (6,49 mL) a una suspension en DMF (70 mL) de 2-hidroxi-4-yodobenzoato de metilo (12,8 g) y carbonato de potasio (12,7 g) y la mezcla se agito a 60C durante 2 horas. La mezcla de reaccion se dejo enfriar hasta temperatura ambiente y, a continuacion, se vertio agua a la misma, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (NH, hexano/acetato de etilo) para obtener el compuesto del fftulo (14,0 g).
RMN 1H (300 MHz, CDCb) delta 1,37 (6H, d, J = 6,0 Hz), 3,86 (3H, s), 4,41-4,67 (1H, m), 7,29-7,35 (2H, m), 7,46 (1H,
d, J = 8,6 Hz).
B) 4'-Fluoro-3-isopropoxibifenil-4-carboxilato de metilo
Se agito una mezcla de 4-yodo-2-isopropoxibenzoato de metilo (7,50 g), acido (4-fluorofenil)boronico (6,56 g), dicidohexil(2',6'-dimetoxibifenil-2-il)fosfina (1,44 g), una disolucion acuosa de carbonato de sodio 2 M (35,1 mL), tris(dibencilidenoacetona)dipaladio(0) (1,50 g) y tolueno (50 mL) a 100C durante 2 horas en atmosfera de argon. Se dejo enfriar la mezcla de reaccion hasta temperatura ambiente. A continuacion, la fase organica se separo y se hizo pasar a traves de una columna de gel de sflice (NH) corta y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (6,61 g).
RMN 1H (300 MHz, CDCb) delta 1,41 (6H, d, J = 6,0 Hz), 3,90 (3H, s), 4,56-4,78 (1H, m), 7,07-7,19 (4H, m), 7,49-7,59 (2H, m), 7,80-7,90 (1H, m).
C) 2-bromo-4'-fluoro-5-isopropoxibifenil-4-carboxilato de metilo
Se añadió acido dibromoisocianurico (4,60 g) a una mezcla de 4'-fluoro-3-isopropoxibifenil-4-carboxilato de metilo (6,61 g) y DMF (60 mL) y la mezcla resultante se agito a temperatura ambiente durante 5 horas. Se anadio agua a la mezcla de reaccion, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (7,53 g).
RMN 1H (300 MHz, CDCla) delta 1,37 (6H, d, J = 6,0 Hz), 3,90 (3H, s), 4,45-4,69 (1H, m), 6,91 (1H, s), 7,06-7,18 (2H, m), 7,32-7,44 (2H, m), 8,05 (1H, s).
D) (2-Ciclopropil-4'-fluoro-5-isopropoxibifenil-4-il)metanol
Se agito durante la noche una mezcla de 2-bromo-4'-fluoro-5-isopropoxibifenil-4-carboxilato de metilo (7,53 g), acido ciclopropilboronico (4,40 g), diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (1,26 g), una disolucion acuosa de carbonato de sodio 2 M (30,8 mL), tris(dibencilidenoacetona)dipaladio(0) (1,31 g) y tolueno (150 mL) a 100C en atmosfera de argon. Se dejo enfriar la mezcla de reaccion hasta temperatura ambiente. A continuacion, la fase organica se separo, se lavo con salino saturado y se hizo pasar a traves de una columna de gel de sflice (NH) corta y el disolvente se destilo a presion reducida. Se una disolucion e anñ TaHdiFó (50 mL) del residuo obtenido a una suspension en THF (50 mL) de hidruro de litio y aluminio (2,00 g) con enfriamiento en hielo en atmosfera de nitrogeno. Despues de agitacion a la misma temperatura que antes durante 30 minutos, se anadieron a la misma agua (2 mL) y una disolucion acuosa al 15 % de hidroxido de sodio (2 mL) y la mezcla se agito durante 5 minutos. Se anadio ademas agua (6 mL) a la mezcla de reaccion y la mezcla se agito durante 30 minutos y, a continuacion, se filtro. El filtrado se concentro a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (5,75 g).
RMN 1H (300 MHz, CDCb) delta 0,55-0,64 (2H, m), 0,68-0,83 (2H, m), 1,31-1,40 (6H, m), 1,67-1,89 (1H, m), 2,45 (1H, t, J = 6,6 Hz), 4,51-4,64 (1H, m), 4,66 (2H, d, J = 6,6 Hz), 6,73 (1H, s), 6,86 (1H, s), 7,05-7,15 (2H, m), 7,34-7,45 (2H, m).
E) 2-CiclopropM-4'-ffuoro-5-isopropoxibifeml-4-carbaldehido
Se añadió dioxido de manganeso (16,6 g) a una disolucion en tolueno (80 mL) de (2-ciclopropil-4'-fluoro-5-isopropoxibifenil-4-il)metanol (5,75 g) y la mezcla se agito a 60C durante 1 hora en atmosfera de nitrogeno. La mezcla de reaccion se filtro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (4,54 g).
RMN 1H (300 MHz, CDCb) delta 0,61-0,72 (2H, m), 0,75-0,85 (2H, m), 1,39 (6H, d, J = 6,0 Hz), 1,71 (1H, tt, J = 8,4, 5,4 Hz), 4,54-4,76 (1H, m), 6,83 (1H, s), 7,07-7,20 (2H, m), 7,35-7,50 (3H, m), 10,46 (1H, s).
F) 1-(1-((2-Ciclopropil-4'-fluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridin-4-carboxilato de metilo
Se añadió una mezcla de 1-(1-(tert-butoxicarbonil)piperidin-4-il)-2-oxo-1,2-dihidropiridin-4-carboxilato de metilo (170 mg) y acido formico (2 mL) y la mezcla se agito a 70C durante 1 hora. A continuacion, el disolvente se destilo. Se anadieron 2-Ciclopropil-4'-fluoro-5-isopropoxibifenil-4-carbaldel'ffdo (181 mg) y THF (2 mL) al residuo obtenido y la mezcla se agito a temperatura ambiente durante 10 minutos.
A continuacion, se triaceto axñiabdoiróohidruro de sodio (428 mg) a la misma y la mezcla se agito durante la noche a temperatura ambiente. Se una disolu acñioandió acuosa saturada de bicarbonato de sodio a la disolucion de reaccion obtenida, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (103 mg).
EM (ESI+): [M+H]+ 519,4.
G) Acido 1-(1-((2-Cidopropil-4'-fluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilico
Se agito una disolucion mezclada de 1-(1-((2-ciclopropil-4'-fluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (103 mg), una disolucion acuosa de hidroxido de sodio 1 M (1 mL) y etanol (2 mL) a 70C durante 1 hora. La disolucion de reaccion obtenida se neutralizo con acido clorhffdrico 1 M y, en este estado, se agito a temperatura ambiente durante 3 horas. El solido resultante se recogio mediante filtracion y recristalizacion (acetato de etilo) para obtener el compuesto del tftulo (70,3 mg).
RMN 1H (300 MHz, DMSO-da) delta 0,49-0,59 (2H, m), 0,71-0,84 (2H, m), 1,26 (6H, d, J = 5,9 Hz), 1,67-1,79 (3H, m), 1,89 (2H, d, J = 10,9 Hz), 2,22 (2H, t, J = 12,6Hz), 3,02 (2H, d, J = 11,0 Hz), 3,55 (2H, s), 4,54-4,73 (2H, m), 6,57 (1H, dd, J = 7,1, 1,8 Hz), 6,78 (1H, s), 6,82 (1H, d, J = 1,7 Hz), 6,98 (1H, s), 7,27 (2H, t, J = 8,9 Hz), 7,49 (2H, dd, J = 8,7, 5,7 Hz), 7,87 (1H, d, J = 7,3 Hz).
Ejemplo 4
Acido 1-(1-(4,5-Diciclopropil-2-isopropoxibencil)piperidm-4-M)-2-oxo-1,2-dihidropiridma-4-carboxnico
A) 4-yodo-2-isopropoxibenzoato de metilo
Se añadió 2-yodopropano (6,42 g) a temperatura ambiente a una mezcla de 2-hidroxi-4-yodobenzoato de metilo (7,00 g), carbonato de potasio (6,96 g) y DMF (100 mL) y la mezcla resultante se agito a 70C durante 2 dfas en atmosfera de nitrogeno. Se agua a la mez acñlaad dióe reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (7,92 g).
RMN 1H (300 MHz, CDCla) delta 1,37 (6H, d, J = 6,0 Hz), 3,86 (3H, s), 4,49-4,63 (1H, m), 7,28-7,33 (2H, m), 7,46 (1H, d, J = 8,5 Hz).
B) 4-ciclopropil-2-isopropoxibenzoato de metilo
Se anadieron acido ciclopropilboronico (3,19 g), una disolucion acuosa de carbonato de sodio 2 M (37 mL), tris(dibencilidenoacetona)dipaladio(0) (1,59 g) y diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (1,52 g) a temperatura ambiente a una disolucion en tolueno (100 mL) de 4-yodo-2-isopropoxibenzoato de metilo (7,92 g) y la mezcla se agito a 100C durante 15 horas en atmosfera de argon. Se agua a la mezcla de reaccion añ aa tdeimóperatura ambiente. La mezcla se filtro sobre celita y, a continuacion, el filtrado se sometio a extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (5,43 g).
RMN 1H (300 MHz, CDCla) delta 0,70-0,76 (2H, m), 0,98-1,05 (2H, m), 1,36 (6H, d, J = 6,1 Hz), 1,82-1,93 (1H, m), 3,85 (3H, s), 4,49-4,63 (1H, m), 6,62 (1H, dd, J =8,1, 1,6 Hz), 6,69 (1H, d, J = 1,5 Hz), 7,69 (1H, d, J = 8,1 Hz).
C) 5-bromo-4-ciclopropil-2-isopropoxibenzoato de metilo
Se añadió acido dibromoisocianurico (3,99 g) a temperatura ambiente a una disolucion en DMF (80 mL) de 4-ciclopropil-2-isopropoxibenzoato de metilo (5,43 g) y la mezcla se agito a 90C durante 1 hora en atmosfera de nitrogeno. Se una a dñiasdoilóucion acuosa de tiosulfato de sodio a la mezcla de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (6,89 g).RMN 1H (300 MHz, CDCb) delta 0,65-0,72 (2H, m), 1,04-1,11 (2H, m), 1,34 (6H, d, J = 6,1 Hz), 2,12-2,23 (1H, m), 3,86 (3H, s), 4,42-4,56 (1H, m), 6,50 (1H, s), 7,96 (1H, s).
D) 4,5-Diciclopropil-2-isopropoxibenzoato de metilo
Se anadieron acido ciclopropilboronico (2,83 g), una disolucion acuosa de carbonato de sodio 2 M (33 mL), tris(dibencilidenoacetona)dipaladio(0) (1,41 g) y diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (1,36 g) a temperature ambiente a una disolucion en tolueno (100 mL) de 5-bromo-4-ciclopropil-2-isopropoxibenzoato de metilo (6,89 g) y la mezcla se agito a 100C durante 15 horas en atmosfera de argon. Se agua a la mezcla de reaccion a añadió temperatura ambiente. La mezcla se filtro sobre celita y, a continuacion, el filtrado se sometio a extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (5,90 g).
RMN 1H (300 MHz, CDCla) delta 0,62-0,72 (4H, m), 0,88-0,96 (2H, m), 0,98-1,06 (2H, m), 1,33 (6H, d, J = 6,1 Hz), 1,99-2,11 (1H, m), 2,21-2,32 (1H, m), 3,85 (3H, s), 4,40-4,53 (1H, m), 6,51 (1H, s), 7,43 (1H, d, J = 0,4 Hz).
E) (4,5-Diciclopropil-2-isopropoxifenil)metanol
Se añadió una disolucion en THF (15 mL) de 4,5-diciclopropil-2-isopropoxibenzoato de metilo (5,90 g) a 0C a una mezcla de hidruro de litio y aluminio (1,71 g) y THF (85 mL) y la mezcla resultante se agito a temperatura ambiente durante 30 minutos en atmosfera de nitrogeno. Se anadieron, en este orden, agua (1,8 mL), una disolucion acuosa de hidroxido de sodio 1 M (1,8 mL) y agua (5,4 mL) a la mezcla de reaccion a 0C. La mezcla se filtro sobre celita y, a continuacion, el filtrado se concentro a presion reducida. El residuo obtenido se purifico mediante cromatograffa con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (5,22 g).RMN 1H (300 MHz, CDCb) delta 0,59-0,68 (4H, m), 0,86-1,00 (4H, m), 1,33 (6H, d, J = 6,0 Hz), 2,02-2,13 (1H, m), 2,15-2,27 (1H, m), 2,41 (1H, t, J = 6,5 Hz), 4,48-4,64 (3H, m), 6,49 (1H, s), 6,86 (1H, s).
F) 4,5-Diciclopropil-2-isopropoxibenzaldehido
Se añadió a temperatura ambiente dioxido de manganeso (14,7 g) a una disolucion en tolueno (80 mL) de (4,5 diciclopropil-2-isopropoxifenil)metanol (5,22 g) y la mezcla se agito a 80C durante 1 hora en atmosfera de nitrogeno. La mezcla de reaccion se filtro sobre celita y, a continuacion, se concentro el filtrado. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (4,88 g).
RMN 1H (300 MHz, CDCla) delta 0,64-0,76 (4H, m), 0,88-0,96 (2H, m), 1,03-1,11 (2H, m), 1,36 (6H, d, J = 6,0 Hz), 1,98-2,09 (1H, m), 2,25-2,37 (1H, m), 4,53-4,66 (1H, m), 6,49 (1H, s), 7,47 (1H, d, J = 0,6 Hz), 10,37 (1H, s).
G) 1-(1-(4,5-diciclopropil-2-isopropoxibencil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo Se agito una mezcla de 1-(1-(tert-butoxicarbonil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (536 mg) en acido formico (8 mL) a 70C durante 30 minutos en atmosfera de nitrogeno y, a continuacion, el disolvente se destilo a presion reducida. Se triacetoxiborohidrur aoña ddeió sodio (676 mg) a una mezcla del residuo obtenido y 4,5-diciclopropil-2-isopropoxibenzaldel‘ndo (476 mg) en THF (10 mL) y la mezcla se agito a temperatura ambiente durante 15 horas en atmosfera de nitrogeno. Se una disolucion acuosa satu arñaaddaió de bicarbonato de sodio a la mezcla de reaccion, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (429 mg).
EM (ESI+): [M+H]+ 465,3.
H) Acido 1-(1-(4,5-DiciclopropM-2-isopropoxibencil)piperidm-4-il)-2-oxo-1,2-dihidropiridma-4-carboxnico
Se añadió una disolucion acuosa de hidroxido de sodio 2 M (3 mL) a una disolucion en etanol (8 mL) de 1-(1-(4,5-diciclopropil-2-isopropoxibencil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (426 mg) y la mezcla se agito a 90C durante 2 horas en atmosfera de nitrogeno. El disolvente de la mezcla de reaccion se destilo a presion reducida. El residuo obtenido se disolvio en agua y la disolucion se neutralizo con acido clorlffdrico 2 M. El solido resultante se recogio mediante filtracion y se disolvio en acetato de etilo y THF. El disolvente de la disolucion obtenida se destilo a presion reducida. El residuo obtenido se cristalizo (acetato de etilo/hexano) y se recristalizo (etanol/hexano) para obtener el compuesto del fftulo (286 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,53-0,59 (2H, m), 0,62-0,69 (2H, m), 0,85-0,98 (4H, m), 1,23 (6H, d, J = 5,9 Hz), 1,67-1,79 (2H, m), 1,82-1,98 (2H, m), 2,01-2,12 (1H, m), 2,14-2,34 (3H, m), 3,00 (2H, d, J = 11,4 Hz), 3,53 (2H, s),
4,48-4,75 (2H, m), 6,47 (1H, s), 6,57 (1H, dd, J = 7,2, 1,8 Hz), 6,81 (1H, d, J = 1,7 Hz), 6,94 (1H, s), 7,82 (1H, d, J = 7,2 Hz).
Ejemplo 5
Acido 1-(1-((2-Cloro-6-cidopropM-2\4'-difluoro-3-isopropoxibifeml-4-M)metM)piperidm-4-M)-2-oxo-1,2-dihidropiridin a-4-carboxilico
A) 3-(Benziloxi)-2',4'-difluorobifenil-4-carboxilato de metilo
Se agito una mezcla de 2-hidroxi-4-yodobenzoato de metilo (22 g), acido 2,4-difluorofenilboronico (25 g), una disolucion acuosa de carbonato de sodio 2 M (119 mL), tris(dibencilidenoacetona)dipaladio(0) (5,07 g) y dicidohexil(2',6'-dimetoxibifenil-2-il)fosfina (4,87 g) en tolueno (150 mL) a 100C durante 2 horas. La mezcla de reaccion se vertio en agua y la mezcla se filtro sobre celita. A continuacion, el filtrado se sometio a extraccion con acetato de etilo. La fase organica obtenida se lavo con salino saturado y se hizo pasar a traves de gel de s l^ice basico y el disolvente se destilo a presion reducida. Se a temperatura ambiente brom auñroad dieó bencilo (10,4 mL) a una mezcla del residuo obtenido, carbonato de potasio (21,9 g) y DMF (100 mL) y la mezcla se agito a 70C durante 1 hora en atmosfera de nitrogeno. Se agua a la m aeñzacldaió de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (30,5 g).
RMN 1H (300 MHz, CDCla) delta 3,92 (3H, s), 5,22 (2H, s), 6,85-7,02 (2H, m), 7,09-7,19 (2H, m), 7,28-7,45 (3H, m), 7,47-7,56 (2H, m), 7,62 (1H, dd, J = 6,8, 2,9 Hz), 7,90 (1H, d, J = 8,0 Hz).
B) 5-(benciloxi)-2-bromo-2',4'-difluorobifenil-4-carboxilato de metilo
Se añadió a temperatura ambiente acido dibromoisocianurico (15,9 g) a una disolucion en DMF (150 mL) de 3-(benciloxi)-2',4'-difluorobifenil-4-carboxilato de metilo (28,0 g) y la mezcla se agito durante la noche a temperatura ambiente. Se ag auñaad aió la mezcla de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (34,0 g). RMN 1H (300 MHz, CDCla) delta 3,92 (3H, s), 5,15 (2H, s), 6,87-7,01 (3H, m), 7,10-7,55 (6H, m), 8,11 (1H, s).
C) 5-(benziloxi)-2-ciclopropil-2',4'-difluorobifenil-4-carboxilato de metilo
Se agito una mezcla de 5-(benciloxi)-2-bromo-2',4'-difluorobifenil-4-carboxilato de metilo (34,3 g), acido ciclopropilboronico (17,0 g), una disolucion acuosa de carbonato de sodio 2 M (119 mL), tris(dibencilidenoacetona)dipaladio(0) (5,07 g) y diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (4,87 g) en tolueno (150 mL) durante la noche a 100C. La mezcla de reaccion se vertio en agua, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con salino saturado y se hizo pasar a traves de gel de sflice basico y el disolvente se destilo a presion reducida.
El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (20,3 g).
RMN 1H (300 MHz, CDCb) delta 0,54-0,63 (2H, m), 0,70-0,80 (2H, m), 1,59-1,72 (1H, m), 3,87-3,93 (3H, m), 5,14 (2H, s), 6,83-7,01 (3H, m), 7,18-7,41 (4H, m), 7,44-7,51 (3H, m).
D) 2-Ciclopropil-2',4'-difluoro-5-hidroxibifenil-4-carboxilato de metilo
Se agito una mezcla de 5-(benciloxi)-2-ciclopropil-2',4'-difluorobifenil-4-carboxilato de metilo (20,3 g), paladio sobre carbon al 10 % (que contiene el 55 % de agua, 10,0 g) y THF (100 mL) a temperatura ambiente durante 1 hora en atmosfera de hidrogeno a la presion de un globo. El catalizador se filtro y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (15,0 g).
EM (ESI+): [M+H]+ 305,2.
E) 2-cloro-6-ciclopropil-2',4'-difluoro-3-isopropoxibifenil-4-carboxilato de metilo
Se añadi Nó-clorosuccinimida (1,58 g) en pequenas cantidades a temperatura ambiente a una disolucion en DMF
(30 mL) de 2-ciclopropil-2',4'-difluoro-5-hidroxibifenil-4-carboxilato de metilo (3,0 g) y, a continuacion, la mezcla se agito a temperatura ambiente durante 2 horas. Se agua a la mezcla de reac acñioandió a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con salino saturado dos veces y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. Se anadio 2 yodopropano (2,95 mL) a una mezcla del residuo obtenido, carbonato de potasio (4,09 g) y DMF (40 mL) y la mezcla se agito a 80C durante 1 hora. Se agua a la mezcla añ dead rieóaccion, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con salino saturado dos veces y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (3,10 g).
RMN 1H (300 MHz, CDCb) delta 0,54-0,68 (2H, m), 0,70-0,78 (2H, m), 1,32 (6H, d, J = 6,1 Hz), 1,43-1,54 (1H, m), 3,89-3,96 (3H, m), 4,26-4,46 (1H, m), 6,86-7,03 (2H, m), 7,13-7,23 (1H, m), 7,28 (1H, s).
F) (2-cloro-6-ciclopropil-2',4'-difluoro-3-isopropoxibifenil-4-il)metanol
Se añadi hóidruro de diisobutilaluminio (disolucion 1,5 M en tolueno, 16,3 mL) a 0C a una disolucion en THF (30 mL) de 2-cloro-6-ciclopropil-2',4'-difluoro-3-isopropoxibifenil-4-carboxilato de metilo (3,10 g) y, a continuacion, la mezcla se agito a 0C durante 20 minutos en atmosfera de nitrogeno. Se a la mezcla de reaccion sulfato añ daedió sodio decahidratado y la mezcla se agito durante 1 hora. Despues de filtracion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (2,18 g).
RMN 1H (300 MHz, CDCla) delta 0,50-0,76 (4H, m), 1,35 (6H, dd, J = 6,1, 4,6 Hz), 1,42-1,55 (1H, m), 2,16-2,34 (1H, m), 4,57 (1H, dt, J = 12,3, 6,2 Hz), 4,65-4,84 (2H, m), 6,82-7,02 (3H, m), 7,19 (1H, td, J = 8,3, 6,5 Hz).
G) 2-Cloro-6-cidopropM-2\4'-diffuoro-3-isopropoxibifenM-4-carbaldehido
Se añadió a temperatura ambiente dioxido de manganeso (5,37 g) a una disolucion en tolueno (30 mL) de (2-cloro-6-ciclopropil-2',4'-difluoro-3-isopropoxibifenil-4-il)metanol (2,18 g) y la mezcla se agito a 60C durante 1 hora en atmosfera de nitrogeno. El solido se filtro y, a continuacion, el filtrado se concentro a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (1,67 g).
RMN 1H (300 MHz, CDCla) delta 0,61-0,84 (4H, m), 1,39 (6H, dd, J = 6,1, 3,1 Hz), 1,43-1,55 (1H, m), 4,39-4,65 (1H, m), 6,86-7,06 (2H, m), 7,20 (1H, td, J = 8,3, 6,4 Hz), 7,40 (1H, s), 10,39 (1H, s).
H) 1-(1-((2-doro-6-cidopropil-2',4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se agito una mezcla de 1-(1-(tert-butoxicarbonil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (400 mg) y acido formico (6 mL) a 70C durante 1 hora en atmosfera de nitrogeno y, a continuacion, el disolvente se destilo a presion reducida. Se triacetoxiborohid aruñraodi dóe sodio (504 mg) a una mezcla del residuo obtenido y 2 cloro-6-ciclopropil-2',4'-difluoro-3-isopropoxibifenil-4-carbaldel'ffdo (459 mg) en THF (10 mL) y DMA (2 mL) y la mezcla se agito a temperatura ambiente durante 16 horas en atmosfera de nitrogeno. Se anadio una disolucion acuosa saturada de bicarbonato de sodio a la mezcla de reaccion, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (601 mg). EM (ESI+): [M+H]+ 571,9.
I) Acido 1-(1-((2-Cloro-6-cidopropil-2',4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridin a-4-carboxilico
Se añadió una disolucion acuosa de hidroxido de sodio 2 M (2 mL) a una disolucion en etanol (8 mL) de 1-(1-((2-cloro-6-ciclopropil-2',4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (590 mg) y la mezcla se agito a 80c durante 4 horas en atmosfera de nitrogeno. La mezcla de reaccion se concentro hasta sequedad a presion reducida. El residuo obtenido se disolvio en agua y la disolucion se neutralizo con acido clorhfdrico 2 M. El solido resultante se recogio mediante filtracion y recristalizacion (DMSO/etanol) para obtener el compuesto del fftulo (157 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,48-0,80 (4H, m), 1,27 (6H, d, J = 6,1 Hz), 1,37-1,52 (1H, m), 1,69-1,98 (4H, m), 2,05-2,35 (2H, m), 2,89-3,04 (2H, m), 3,60 (2H, s), 4,37-4,51 (1H, m), 4,56-4,77 (1H, m), 6,57 (1H, dd, J = 7,1, 1,9 Hz), 6,84 (1H, d, J = 1,8 Hz), 7,07 (1H, s), 7,13-7,30 (1H, m), 7,32-7,48 (2H, m), 7,89 (1H, d, J = 7,2 Hz).
Ejemplo 6
Acido 1-(1-((2-Ciclopropil-2',4'-difluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridin a-4-carboxilico
A) 2',4'-difluoro-3-isopropoxibifenil-4-carboxilato de metilo
Se agito una mezcla de 4-yodo-2-isopropoxibenzoato de metilo (4,10 g), acido (2,4-difluorofenil)boronico (4,04 g), diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (0,789 g), una disolucion acuosa de carbonato de sodio 2 M (19,2 mL), tris(dibencilidenoacetona)dipaladio(0) (0,821 g) y tolueno (50 mL) a 100C durante 2 horas. La mezcla de reaccion se vertio en agua a temperature ambiente y, a continuacion, la mezcla se hizo pasar a traves de celita, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con salino saturado y se hizo pasar a traves de una columna de gel de sflice (NH) corta y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (3,90 g).
RMN 1H (300 MHz, CDCb) delta 1,40 (6H, d, J = 6,0 Hz), 3,90 (3H, s), 4,52-4,70 (1H, m), 6,83-7,01 (2H, m), 7,04-7,15 (2H, m), 7,33-7,49 (1H, m, J = 6,4 Hz), 7,83 (1H, d, J = 8,0 Hz).
B) 2-Bromo-2',4'-difluoro-5-isopropoxibifenil-4-carboxilato de metilo
Se añadió acido dibromoisocianurico (2,19 g) a una mezcla de 2',4'-difluoro-3-isopropoxibifenil-4-carboxilato de metilo (3,90 g) y DMF (40 mL) y la mezcla se agito a temperatura ambiente durante 3 horas. Se anadio agua a la mezcla de reaccion, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (4,90 g).
RMN 1H (300 MHz, CDCla) delta 1,37 (6H, d, J = 6,0 Hz), 3,90 (3H, s), 4,41-4,63 (1H, m), 6,85-7,02 (3H, m), 7,17-7,33 (1H, m), 8,04 (1H, s) .
C) (2-Ciclopropil-2',4'-difluoro-5-isopropoxibifenil-4-il)metanol
Se agito durante la noche una mezcla de 2-bromo-2',4'-difluoro-5-isopropoxibifenil-4-carboxilato de metilo (4,90 g), acido ciclopropilboronico (3,28 g), diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (0,783 g), una disolucion acuosa de carbonato de sodio 2 M (19,1 mL), tris(dibencilidenoacetona)dipaladio(0) (0,815 g) y tolueno (50 mL) a 100C. La fase organica se separo de la mezcla de reaccion, se lavo con salino saturado y se hizo pasar a traves de una columna de gel de sflice (NH) corta y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo). Se anadio una disolucion en THF (50 mL) de este producto purificado a una suspension en THF (50 mL) de hidruro de litio y aluminio (0,474 g) con enfriamiento en hielo. Despues de agitacion a la misma temperatura que antes durante 30 minutos, se anadieron a la misma agua (0,5 mL) y una disolucion acuosa al 15 % de hidroxido de sodio (0,5 mL) y la mezcla se agito durante 5 minutos. Se anadio ademas agua (1,5 mL) a la mezcla de reaccion y la mezcla se agito durante 30 minutos y, a continuacion, se filtro. El filtrado se concentro a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (3,97 g).
RMN 1H (300 MHz, CDCb) delta 0,48-0,60 (2H, m), 0,66-0,79 (2H, m), 1,35 (6H, d, J = 6,0 Hz), 1,59-1,72 (1H, m), 2,45 (1H, t, J = 6,5 Hz), 4,47-4,63 (1H, m), 4,67 (2H, d, J = 6,4 Hz), 6,71 (1H, s), 6,86-6,99 (3H, m), 7,19-7,36 (1H, m).
D) 2-CiclopropM-2\4'-diffuoro-5-isopropoxibifeml-4-carbaldehido
Se añadió dioxido de manganeso (10,8 g) a una disolucion en tolueno (30 mL) de (2-ciclopropil-2',4'-difluoro-5-isopropoxibifenil-4-il)metanol (3,97 g) y la mezcla de reaccion se agito a 60C durante 1 hora en atmosfera de nitrogeno. La mezcla de reaccion se filtro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (3,53 g).
RMN 1H (300 MHz, CDCb) delta 0,56-0,66 (2H, m), 0,70-0,80 (2H, m), 1,39 (6H, d, J = 6,1 Hz), 1,58-1,70 (1H, m), 4,52-4,74 (1H, m), 6,82 (1H, s), 6,87-7,04 (2H, m), 7,28-7,35 (1H, m), 7,48 (1H, s), 10,47 (1H, s).
E) 4-(4-(benciloxi)-2-oxopiridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo
Se añadió a temperatura ambiente tert-butoxido de potasio (5,05 g) a una disolucion en DME (70 mL) de 4 (benciloxi)piridin-2(1H)-ona (7,55 g). La mezcla de reaccion se agito a temperatura ambiente durante 10 minutos. A
continuacion, se anadieron a la misma carbonato de potasio (10,4 g) y 4-(((4-metilfenil)sulfonil)oxi)piperidina-1-carboxilato de tert-butilo (20,0 g) y la mezcla se agito a 100c durante 1 dfa en atmosfera de nitrogeno. A continuacion, se añadió DME (30 mL) a la misma y la mezcla se agito adicionalmente durante el fin de semana a 100C. Se anadio salino saturado a la mezcla de reaccion, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (4,40 g).
EM (ESI+): [M+H]+ 385,3.
F) 4-(4-(benciloxi)-3-yodo-2-oxopiridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo
Se añadió a temperatura ambiente N-yodosuccinimida (2,85 g) a una disolucion en acido acetico (57,6 mL) de 4-(4-(benciloxi)-2-oxopiridin-1(2H)-il)piperidina-1-carboxilato (4,43 g) de tert-butilo y la mezcla se agito a temperatura ambiente durante 2 horas. La mezcla de reaccion se alcalinizo mediante la adicion de una disolucion acuosa saturada de bicarbonato de sodio, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (4,83 g).
EM (ESI+): [M+H]+ 511,1.
G) 4-(4-(benciloxi)-2-oxo-3-vinilpiridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo
Se agito una mezcla de 4-(4-(benciloxi)-3-yodo-2-oxopiridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo (2,4 g), vinil trifluoroborato de potasio (0,945 g), trietilamina (1,3 mL), un aducto en diclorometano de dicloruro de 1,1' bis(difenilfosfino)ferroceno-paladio(Il) (0,269 g) y etanol (47 mL) durante la noche a 90C en atmosfera de nitrogeno. Despues de enfriamiento hasta temperatura ambiente, el disolvente se destilo a presion reducida. Se anadieron agua y acetato de etilo al residuo obtenido y la fase organica se separo. La fase organica obtenida se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (NH, hexano/acetato de etilo) para obtener el compuesto del tftulo (1,48 g).
EM (ESI+): [M+H]+ 411,2.
H) 4-(3-etil-4-hidroxi-2-oxopiridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo
Se añadi póaladio sobre carbon al 10% (que contiene el 55 % de agua, 0,767 g) a una disolucion en etanol (30 mL) de 4-(4-(benciloxi)-2-oxo-3-vinilpiridin-1 (2H)-il)piperidina-1-carboxilato de tert-butilo (1,48 g) y la mezcla se agito a temperatura ambiente durante 3 horas en atmosfera de hidrogeno. La mezcla de reaccion se filtro sobre celita y, a continuacion, el disolvente del filtrado se destilo a presion reducida. El residuo obtenido se hizo pasar a traves de gel de sflice y, a continuacion, el disolvente se destilo a presion reducida para obtener el compuesto del tftulo (1,12 g). EM (ESI+): [M+H-tBu]+ 267,1.
I) 4-(3-etil-2-oxo-4-(((trifluorometil)sulfonil)oxi)piridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo
Se añadió anhfdrido trifluorometanosulfonico (1,76 mL) a 0C a una mezcla de 4-(3-etil-4-hidroxi-2-oxopiridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo (1,12 g) en piridina (17,4 mL) y la mezcla se agito a 0C durante 30 minutos en atmosfera de nitrogeno. Se agua a la m aisñmadaió y, a continuacion, el disolvente se destilo a presion reducida. Se anadio agua de nuevo al residuo, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con acido clorhfdrico 1 M y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (1,14 g). EM (ESI+): [M+H]+ 399,1.
J) 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se agito una mezcla de 4-(3-etil-2-oxo-4-(((trifluorometil)sulfonil)oxi)piridin-1(2H)-il)piperidina-1-carboxilato de tertbutilo (1,14 g), trietilamina (0,699 mL), metanol (3,05 mL), dicloruro de 1,1'-bis(difenilfosfino)ferroceno-paladio(II) (0,184 g) y DMF (12,5 mL) a 90C durante 4 horas en atmosfera de monoxido de carbono a 0,5 MPa. Se anadio agua a la mezcla de reaccion, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se lavo con salino saturado dos veces y se seco sobre sulfato de magnesio anhidro y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (NH, hexano/acetato de etilo) para obtener el compuesto del tftulo (0,798 g). EM (ESI+): [M+H-tBu]+ 309,1.
K) 1-(1-((2-ciclopropil-2',4'-difluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se agito una mezcla de 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (150 mg) y acido formico (3 mL) a 70C durante 30 minutes y, a continuacion, el disolvente se destilo a presion reducida. Se anadieron 2-ciclopropil-2',4'-difluoro-5-isopropoxibifenil-4-carbaldel'ndo (156 mg) y DMA (2 mL) al residuo obtenido y la mezcla se agito a temperature ambiente durante 10 minutos. A continuacion, se anadio triacetoxiborohidruro de sodio (174 mg) a la misma y la mezcla se agito durante la noche a temperatura ambiente. Se anadio una disolucion acuosa saturada de bicarbonato de sodio a la mezcla de reaccion, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (208 mg).
EM (ESI+): [M+H]+ 565,3.
L) Acido 1-(1-((2-Ciclopropil-2',4'-difluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridin a-4-carboxilico
Se agito una mezcla de 1-(1-((2-ciclopropil-2',4'-difluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (208 mg), una disolucion acuosa de hidroxido de sodio 1 M (3 mL) y etanol (6 mL) a 70C durante 1 hora. Se ajusto el pH de la mezcla de reaccion hasta 3 usando acido clortedrico 1 M, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se seco sobre sulfato de sodio anhidro y el disolvente se destilo a presion reducida. El residuo obtenido se recristalizo (2-propanol/hexano) para obtener el compuesto del tftulo (156 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,65 (2H, d, J = 3,6 Hz), 0,70-0,79 (2H, m), 1,05 (3H, t, J = 7,3 Hz), 1,30 (6H, d, J = 5,9 Hz), 1,50-1,62 (1H, m), 1,92-2,07 (2H, m), 2,16-2,37 (2H, m), 2,67 (2H, q, J = 7,2 Hz), 3,20-3,40 (2H, m), 3,45 (2H, brs), 4,21 (2H, brs), 4,63-4,77 (1H, m), 5,02 (1H, brs), 6,43 (1H, d, J = 7,3 Hz),6,92 (1H, s), 7,20 (1H, td, J = 8,3, 2,4 Hz), 7,27 (1H, s), 7,32-7,52 (3H, m).
Ejemplo 7
Acido 1-(1-((6-Ciclopropil-2,2',4'-trifluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridin a-4-carboxftico
A) 2,2',3,4'-Tetrafluorobifenil-4-carbaldehido
Se anadieron acido (2,4-difluorofenil)boronico (5,39 g), una disolucion acuosa de carbonato de sodio 2 M (34,1 mL), tris(dibencilidenoacetona)dipaladio(0) (1,46 g) y diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (1,40 g) a temperatura ambiente a una disolucion en tolueno (150 mL) de 4-bromo-2,3-difluorobenzaldel‘ndo (5,03 g) y la mezcla se agito a 100C durante 16 horas en atmosfera de nitrogeno. Se agua a la mezcla de reaccion a t aeñmapdeióratura ambiente y la mezcla se filtro sobre celita. A continuacion, el filtrado se sometio a extraccion con acetato de etilo. La fase organica se separo, se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (5,49 g).
RMN 1H (300 MHz, CDCla) delta 6,93-7,06 (2H, m), 7,23-7,30 (1H, m), 7,35-7,44 (1H, m), 7,70 (1H, ddd, J = 8,1, 6,2, 1,8 Hz), 10,39 (1H, d, J = 0,6 Hz).
B) 2,2',4'-Trifluoro-3-metoxibifeml-4-carbaldehido
Se añadi aó temperatura ambiente metoxido de sodio (disolucion en metanol al 28%, 5,64 g) a una disolucion en metanol (120 mL) de 2,2',3,4'-tetrafluorobifenil-4-carbaldel'ndo (4,95 g) y la mezcla se calento a reflujo durante 16 horas en atmosfera de nitrogeno. A continuacion, el disolvente se destilo a presion reducida. El residuo se diluyo con acetato de etilo y agua, seguido de extraccion con acetato de etilo. La fase organica se separo, se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida para obtener el compuesto del tftulo (5,14 g).
RMN 1H (300 MHz, CDCla) delta 4,13 (3H, d, J = 2,6 Hz), 6,91-7,05 (2H, m), 7,09-7,16 (1H, m), 7,32-7,43 (1H, m), 7,67 (1H, dd, J = 8,1, 1,4 Hz), 10,43 (1H, d, J = 0,8 Hz).
C) 2,2',4'-Trifluoro-3-hidroxibifenil-4-carbaldehido
Se añadió a temperature ambiente acido bromhffdrico al 48 % (22,0 mL) a una disolucion en acido acetico (120 mL) de 2,2',4'-trifluoro-3-metoxibifenil-4-carbaldel'ffdo (5,14 g) y la mezcla se agito a 120C durante 16 horas en atmosfera de nitrogeno. A continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se neutralizo con una disolucion acuosa de hidroxido de sodio 1 M a temperature ambiente, seguido de extraccion con acetato de etilo. La fase organica se separo, a continuacion, se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se hizo pasar a traves de una columna corta de gel de sflice y, a continuacion, el disolvente se destilo a presion reducida para obtener el compuesto del tftulo (4,60 g).
RMN 1H (300 MHz, CDCb) delta 6,91-7,05 (3H, m), 7,34-7,46 (2H, m), 9,96 (1H, d, J = 1,8 Hz), 11,07 (1H, s).
D) 6-Bromo-2,2',4'-trifluoro-3-hidroxibifenM-4-carbaldeMdo
Se añadió a temperatura ambiente acido dibromoisocianurico (2,25 g) a una disolucion en DMF (90 mL) de 2,2',4'-trifluoro-3-hidroxibifenil-4-carbaldel'ffdo (3,29 g) y la mezcla se agito a la misma temperatura que antes durante 3 horas en atmosfera de nitrogeno. Se una disolucion a acñuaodsióa saturada de tiosulfato de sodio a la mezcla de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica se separo, a continuacion, se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) y cromatograffa en columna con gel de sflice (diol, hexano/acetato de etilo) para obtener el compuesto del tftulo (3,15 g).
RMN 1H (300 MHz, CDCla) delta 6,93-7,06 (2H, m), 7,22-7,32 (1H, m), 7,71 (1H, d, J = 1,9 Hz), 9,92 (1H, d, J = 1,9 Hz), 10,90 (1H, brs).
E) 6-Bromo-3-isopropoxi-2,2\4'-trifluorobifenM-4-carbaldehido
Se añadió a temperatura ambiente 2-yodopropano (461 mg) a una mezcla de 6-bromo-2,2',4'-trifluoro-3-hidroxibifenil-4-carbaldelffdo (598 mg), carbonato de potasio (499 mg) y DMF (10 mL) y la mezcla se agito a 60C durante 3 horas en atmosfera de nitrogeno. Se agua a la me azcñlaadi dóe reaccion, seguido de extraccion con acetato de etilo. La fase organica se separo, a continuacion, se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (637 mg).
RMN 1H (300 MHz, CDCla) delta 1,39 (6H, dd, J = 6,0, 3,1 Hz), 4,57-4,71 (1H, m), 6,91-7,07 (2H, m), 7,23-7,33 (1H, m), 7,94 (1H, d, J = 1,8 Hz), 10,38 (1H, s).
F) 6-CidopropM-3-isopropoxi-2,2\4'-trifluorobifeml-4-carbaldehido
Se anadieron acido ciclopropilboronico (424 mg), una disolucion acuosa de carbonato de sodio 2 M (3,29 mL), tris(dibencilidenoacetona)dipaladio(0) (151 mg) y diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (135 mg) a temperatura ambiente a una disolucion en tolueno (15 mL) de 6-bromo-3-isopropoxi-2,2',4'-trifluorobifenil-4-carbaldel'ffdo (615 mg) y la mezcla se agito a 100C durante 16 horas en atmosfera de argon. Se anadio agua a la mezcla de reaccion y, a continuacion, la mezcla se filtro sobre celita. El filtrado se sometio a extraccion con acetato de etilo. La fase organica se separo, a continuacion, se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (529 mg). RMN 1H (300 MHz, DMSO-d6) delta 0,59-0,72 (2H, m), 0,74-0,87 (2H, m), 1,31 (6H, dd, J = 6,0, 2,6 Hz), 1,50-1,62 (1H, m), 4,42-4,54 (1H, m), 7,19 (1H, d, J = 1,1 Hz), 7,27 (1H, tdd, J = 8,5, 2,5, 0,9 Hz), 7,41-7,50 (1H, m), 7,56 (1H, td, J = 8,5, 6,5 Hz), 10,29 (1H, s).
G) 1-(1-((6-ciclopropil-2,2',4'-trifluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se añadió acido formico (3 mL) a 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (225 mg) y la mezcla se agito a 70C durante 30 minutos. A continuacion, el disolvente se destilo. Se anadieron 6-Ciclopropil-2,2',4'-trifluoro-3-isopropoxibifenil-4-carbaldel'ffdo (206 mg), THF (3 mL) y DMA (1 mL) al residuo obtenido y la mezcla se agito a temperatura ambiente durante 10 minutos.
A continuacion, se triaceto axñiabdoiróohidruro de sodio (261 mg) a la misma y la mezcla se agito durante la noche a temperatura ambiente. Se una disol auñcaiodnió acuosa saturada de bicarbonato de sodio a la mezcla de reaccion obtenida, seguido de extraccion con acetato de etilo dos veces. La fase organica obtenida se lavo con salino saturado y, a continuacion, se seco sobre sulfato de magnesio anhidro y el disolvente se destilo a presion reducida. El residuo
obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (161 mg). EM (ESI+): [M+H]+ 583,3.
H) Acido 1-(1-((6-Ciclopropil-2,2',4'-trifluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxiMco
Se agito una mezcla de 1-(1-((6-ciclopropil-2,2',4'-trifluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo ( l6 l mg), una disolucion acuosa de hidroxido de sodio 1 M (1 mL) y etanol (2 mL) a 70C durante 1 hora. Se ajusto el pH de la mezcla de reaccion hasta 3 usando acido clortftdrico 1 M, seguido de extraccion con acetato de etilo dos veces. La fase organica se separo y se seco sobre sulfato de sodio anhidro y se destilo el disolvente a presion reducida. El residuo obtenido se cristalizo (2-propanol/hexano) para obtener el compuesto del tftulo (123 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,64-0,85 (4H, m), 1,01-1,10 (3H, m), 1,29 (6H, d, J = 6,0 Hz), 1,46-1,60 (1H, m), 1,84-2,01 (2H, m), 2,04-2,34 (2H, m), 2,67 (2H, q, J = 7,1 Hz), 2,70-3,45 (4H, m), 3,90-4,29 (2H, m), 4,33-4,55 (1H, m), 4,95 (1H, brs), 6,41 (1H, d, J = 7,2 Hz), 7,02-7,30 (2H, m), 7,42 (1H, td, J = 9,7, 2,5Hz), 7,47-7,57 (2H, m), 12,76 13,80 (1H, m) .
Ejemplo 43
Acido 1-(1-((6-Ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridin a-4-carboxilico
A) 1-Bromo-2-fluoro-3-(metoximetoxi)benceno
Se añad cióloro(metoxi)metano (11,8 mL) a 0C a una disolucion en THF (150 mL) de 3-bromo-2-fluorofenol (15,0 g) y N,N-diisopropiletilamina (41,1 mL). La mezcla de reaccion se agito durante la noche a temperatura ambiente en atmosfera de nitrogeno y, a continuacion, se filtro sobre celita. El filtrado obtenido se lavo con acido clortftdrico 1 M y una disolucion acuosa saturada de bicarbonato de sodio en este orden y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se hizo pasar a traves de una columna corta de gel de sflice (NH, hexano/acetato de etilo) y, a continuacion, el disolvente se destilo a presion reducida para obtener el compuesto del tftulo (17,9 g). RMN 1H (300 MHz, DMSO-d6) delta 3,41 (3H, s), 5,27 (2H, s), 7,06-7,14 (1H, m), 7,24-7,34 (2H, m).
B) 2,4'-Difluoro-3-(metoximetoxi)bifenilo
Se agito una mezcla de 1-bromo-2-fluoro-3-(metoximetoxi)benceno (17,9 g), acido (4-fluorofenil)boronico (16,0 g), tris(dibencilidenoacetona)dipaladio(0) (3,49 g), una disolucion acuosa de carbonato de sodio 2 M (114 mL), diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (3,13 g) y tolueno (180 mL) a 100C durante 2 horas en atmosfera de nitrogeno. La mezcla de reaccion se enfrio hasta temperatura ambiente y, a continuacion, se hizo pasar por celita. La fase organica se separo y el disolvente se destilo a presion reducida. El residuo obtenido se hizo pasar a traves de una columna corta de gel de sflice (NH) y, a continuacion, el disolvente se destilo a presion reducida para obtener el compuesto del tftulo (19,0 g).
RMN 1H (300 MHz, DMSO-d6) delta 3,44 (3H, s), 5,28 (2H, s), 7,08-7,36 (5H, m), 7,54-7,63 (2H, m).
C) 2,4'-Difluoro-3-(metoximetoxi)bifenil-4-carbaldehido
Se añadió n-butil-litio (disolucion en hexano 1,6 M, 52,2 mL) a -78C a una disolucion anhidra en THF (200 mL) de 2,4'-difluoro-3-(metoximetoxi)bifenilo (19,0 g) en atmosfera de argon y, a continuacion, la mezcla se agito a -78C durante 1 hora. Se a DñMadFió (12,9 mL) a la misma y, a continuacion, la mezcla se agito durante 3 horas en atmosfera de argon mientras se calentaba desde -78C a 0C. Se una disolucion acuosa sat auñraaddaió de cloruro de amonio a la mezcla de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica se separo, a continuacion, se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se solidifico con hexano para obtener el compuesto del tftulo (16,4 g). RMN 1H (300 MHz, DMSO-d6) delta 3,54 (3H, s), 5,31 (2H, d, J = 0,8 Hz), 7,32-7,49 (3H, m), 7,62-7,73 (3H, m), 10,31 (1H, d, J = 0,8 Hz).
D) 2,4'-Difluoro-3-hidroxibifenil-4-carbaldehido
Se añadió acido clortftdrico 6 M (49,1 mL) a una disolucion en etanol (160 mL) de 2,4'-difluoro-3-(metoximetoxi)bifenil-4-carbaldehftdo (16,4 g). La mezcla de reaccion se agito a 50C durante 30 minutos y ademas a temperatura ambiente durante 2 horas. El solido resultante se recogio mediante filtracion y, a continuacion, se lavo con etanol-agua y agua
en este orden para obtener el compuesto del tftulo (12,7 g).
RMN 1H (300 MHz, DMSO-da) delta 7,10 (1H, dd, J = 7,8, 6,8 Hz), 7,31-7,40 (2H, m), 7,56 (1H, dd, J = 8,2, 1,4 Hz), 7,62-7,70 (2H, m), 10,29 (1H, s), 11,02 (1H, s).
E) 6-Bromo-2,4'-difluoro-3-hidroxibifenM-4-carbaldehido
Se añadió acido dibromoisocianurico (8,87 g) a temperature ambiente a una disolucion en DMF (75 mL) de 2,4'-difluoro-3-hidroxibifenil-4-carbaldeh^do (12,7 g) y la mezcla se agito a temperature ambiente durante 30 minutos. Se anadio agua (50 mL) gota a gota a la mezcla de reaccion y la mezcla se agito a temperatura ambiente durante 1 hora. El solido resultante se recogio mediante filtracion, a continuacion, se lavo con DMF-agua y se seco para obtener el compuesto del tftulo (17,6 g).
RMN 1H (300 MHz, DMSO-d6) delta 7,27-7,50 (4H, m), 7,77 (1H, d, J = 1,8 Hz), 10,27 (1H, s), 11,15 (1H, s).
F) 6-Bromo-2,4'-difluoro-3-isopropoxibifenil-4-carbaldehido
Se agito una mezcla de 6-bromo-2,4'-difluoro-3-hidroxibifenil-4-carbaldel'ndo (3,50 g), 2-yodopropano (5,70 g), carbonato de potasio (3,09 g) y DMF (15 mL) a 50C durante 1 hora. La mezcla de reaccion se vertio en agua, seguido de extraccion con acetato de etilo dos veces. La fase organica se separo, a continuacion, se lavo con salino saturado y se seco sobre sulfato de magnesio anhidro y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (2,94 g).
RMN 1H (300 MHz, DMSO-d6) delta 1,34 (6H, dd, J = 6,1, 0,7 Hz), 4,50-4,64 (1H, m), 7,32-7,42 (2H, m), 7,43-7,51 (2H, m), 7,83 (1H, d, J = 1,8 Hz), 10,26 (1H, s).
G) 6-CidopropM-2,4'-difluoro-3-isopropoxibifenM-4-carbaldehido
Se agito una mezcla de 6-bromo-2,4'-difluoro-3-isopropoxibifenil-4-carbaldel'ndo (2,94 g), acido ciclopropilboronico (1,42 g), tris(dibencilidenoacetona)dipaladio(0) (0,531 g), una disolucion acuosa de carbonato de sodio 2 M (12,4 mL), diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (0,510 g) y tolueno (30 mL) durante la noche a 100C en atmosfera de nitrogeno. La mezcla de reaccion se diluyo con acetato de etilo y agua y, a continuacion, se filtro sobre celita. La fase organica se separo del filtrado y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (2,38 g).
RMN 1H (300 MHz, DMSO-d6) delta 0,62-0,70 (2H, m), 0,75-0,84 (2H, m), 1,31 (6H, dd, J = 6,1, 0,7 Hz), 1,52-1,65 (1H, m), 4,43-4,53 (1H, m), 7,15 (1H, d, J = 1,2 Hz), 7,31-7,40 (2H, m), 7,43-7,51 (2H, m), 10,28 (1H, s).
H) 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se añadi aócido formico (2 mL) a 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (300 mg) y la mezcla se agito a 70C durante 1 hora. A continuacion, el disolvente se destilo a presion reducida. Se t ariñaacedtióoxiborohidruro de sodio (262 mg) a una mezcla del residuo obtenido, 6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-carbaldel'ffdo (260 mg) y THF (5,0 mL) y la mezcla se agito durante la noche a temperatura ambiente. Se una disolu acñiaodnió acuosa saturada de bicarbonato de sodio a la mezcla de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica se separo y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (350 mg). EM (ESI+): [M+H]+ 565,4.
I) Acido 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxnico
Se añadió a temperatura ambiente una disolucion acuosa de hidroxido de sodio 2 M (1,54 mL) a una disolucion en metanol (3 mL) de 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (348 mg) y la mezcla se agito a 50C durante 1 hora. La mezcla de reaccion se enfrio hasta temperatura ambiente y se neutralizo con acido clorlffdrico 2 M y el disolvente se destilo a presion reducida. El residuo obtenido se cristalizo a partir del agua y el solido obtenido se recristalizo (etanol/eter de diisopropilo) para obtener el compuesto del tftulo (240 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,56-0,64 (2H, m), 0,72-0,81 (2H, m), 0,98-1,08 (3H, m), 1,26 (6H, d, J = 6,0 Hz), 1,51-1,64 (1H, m), 1,68-1,92 (4H, m), 2,17 (2H, t, J = 10,5 Hz), 2,65 (2H, q, J = 7,3 Hz), 2,98 (2H, d, J = 11,1 Hz), 3,56 (2H, s), 4,29-4,41 (1H, m), 4,63-4,78 (1H, m), 6,33 (1H, d, J = 7,3 Hz), 6,84 (1H, s), 7,24-7,36 (2H, m), 7,37-7,46 (2H,
m), 7,67 (1H, d, J = 7,4 Hz).
Ejemplo 44
Acido 1-(1-((6-ciclopropil-3-etoxi-2,4'-difluorobifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilico
A) 6-Ciclopropil-3-etoxi-2,4'-difluorobifenil-4-carbaldehido
Se agito una mezcla de 6-bromo-2,4'-difluoro-3-hidroxibifenil-4-carbaldelffdo (2,60 g), acido ciclopropilboronico (1,43 g), tris(dibencilidenoacetona)dipaladio(0) (0,760 g), una disolucion acuosa de carbonato de sodio 2 M (12,5 mL), diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (0,682 g) y tolueno (70 mL) durante la noche a 100C en atmosfera de nitrogeno. La mezcla de reaccion se diluyo con acetato de etilo y agua y, a continuacion, se filtro sobre celita. La fase organica se separo del filtrado y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (1,60 g).
EM (ESI-): [M-H]- 273,0.
B) 6-Ciclopropil-3-etoxi-2,4'-difluorobifenil-4-carbaldehido
Se agito una mezcla de 6-ciclopropil-3-etoxi-2,4'-difluorobifenil-4-carbaldel'Hdo (530 mg), yodoetano (603 mg), carbonato de potasio (534 mg) y acetona (10 mL) a 50C durante 2 horas y, a continuacion, se filtro sobre celita. El filtrado se concentro a presion reducida y, a continuacion, el residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (400 mg).
EM (ESI+): [M+H]+ 303,1.
C) 1-(1-((6-Ciclopropil-3-etoxi-2,4'-difluorobifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se añadióacido formico (2 mL) a 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (300 mg) y la mezcla se agito a 70C durante 1 hora. A continuacion, el disolvente se destilo a presion reducida. Se tr aiañcaedtoióxiborohidruro de sodio (262 mg) a una mezcla del residuo obtenido, 6-ciclopropil-3-etoxi-2,4 '-difluorobifenil-4-carbaldelffdo (249 mg) y THF (5,0 mL) y la mezcla se agito durante la noche a temperatura ambiente. Se u anñaad diiósolucion acuosa saturada de bicarbonato de sodio a la mezcla de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica se separo y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (387 mg). EM (ESI+): [M+H]+ 551,7.
D) Acido 1-(1-((6-Cidopropil-3-etoxi-2,4'-difluorobifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxHico
Se añadió una disolucion acuosa de hidroxido de sodio 2 M (1,73 mL) a una disolucion en metanol (3 mL) de 1-(1-((6-ciclopropil-3-etoxi-2,4'-difluorobifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (382 mg) y la mezcla se agito a 50C durante 1 hora. La mezcla de reaccion se enfrio hasta temperatura ambiente y se neutralizo con acido clorhfdrico 2 M y el disolvente se destilo a presion reducida. El residuo obtenido se cristalizo a partir del agua y el solido obtenido se recristalizo (etanol/eter de diisopropilo/hexano) para obtener el compuesto del fftulo (211 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,56-0,65 (2H, m), 0,72-0,80 (2H, m), 0,99-1,08 (3H, m), 1,32 (3H, t, J = 7,0 Hz), 1,52-1,64 (1H, m), 1,69-1,90 (4H, m), 2,19 (2H, t, J = 10,5 Hz), 2,65 (2H, q, J = 7,2 Hz), 2,99 (2H, d, J = 11,3 Hz), 3,55 (2H, s), 4,04 (2H, q, J = 7,0 Hz), 4,64-4,78 (1H, m), 6,33 (1H, d, J = 7,3 Hz), 6,81 (1H, s), 7,26-7,35 (2H, m), 7,38-7,46 (2H, m), 7,66 (1H, d, J = 7,3 Hz).
Ejemplo 45
Acido 1-(1-((6-ciclopropil-3-etoxi-2,2',4'-trifluorobifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilico
A) 6-Bromo-3-etoxi-2,2',4'-trifluorobifenil-4-carbaldehido
Se añadió a temperatura ambiente yodoetano (0,77 g) a una mezcla de 6-bromo-2,2',4'-trifluoro-3-hidroxibifenil-4carbaldehffdo (1,09 g), carbonato de potasio (0,91 g) y DMF (20 mL) y la mezcla se agito a 60C durante 2 horas en atmosfera de nitrogeno. Se agua a la m aeñzacldaió de reaccion a temperature ambiente, seguido de extraccion con acetato de etilo. La fase organica se separo, se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (1,07 g).
RMN 1H (300 MHz, CDCla) delta 1,45 (3H, td, J = 7,0, 0,8 Hz), 4,29-4,39 (2H, m), 6,92-7,06 (2H, m), 7,23-7,32 (1H, m), 7,93 (1H, d, J = 1,8 Hz), 10,39 (1H, s).
B) 6-CiclopropM-3-etoxi-2,2\4'-triffuorobifenM-4-carbaldehido
Se anadieron a temperatura ambiente acido ciclopropilboronico (0,77 g), una disolucion acuosa de carbonato de sodio 2 M (5,94 mL), tris(dibencilidenoacetona)dipaladio(0) (0,272 g) y diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (0,244 g) a una disolucion en tolueno (20 mL) de 6-bromo-3-etoxi-2,2',4'-trifluorobifenil-4-carbaldel'ffdo (1,07 g) y la mezcla se agito a 100C durante 15 horas en atmosfera de argon. Se agua a la mezcla de reaccion a tempe arñaatudrióa ambiente y, a continuacion, la mezcla se filtro sobre celita. El filtrado se sometio a extraccion con acetato de etilo. La fase organica se separo, a continuacion, se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (904 mg).RMN 1H (300 MHz, CDCla) delta 0,59-0,67 (1H, m), 0,67-0,76 (1H, m), 0,77-0,83 (2H, m), 1,43 (3H, td, J = 7,1, 0,7 Hz), 1,50-1,63 (1H, m), 4,28 (2H, qt, J = 7,0, 1,3 Hz), 6,92-7,05 (2H, m), 7,24-7,36 (2H, m), 10,41 (1H, s). C) 1-(1-((6-Ciclopropil-3-etoxi-2,2',4'-trifluorobifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxiilato de metilo
Se agito una mezcla de 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (300 mg) y acido formico (6 mL) a 70C durante 30 minutos en atmosfera de nitrogeno y, a continuacion, el disolvente se destilo a presion reducida. Se triacetoxiborohidru aroña ddeió sodio (349 mg) a una mezcla del residuo obtenido, 6-ciclopropil-3-etoxi-2,2',4'-trifluorobifenil-4-carbaldelffdo (296 mg) y THF (8 mL) y la mezcla se agito a temperatura ambiente durante 16 horas en atmosfera de nitrogeno. Se una disolucion acuosa saturada de añ baicdaiórbonato de sodio a la mezcla de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica se separo, a continuacion, se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (282 mg).
EM (ESI+): [M+H]+ 569,3.
D) Acido 1-(1-((6-ciclopropN-3-etoxi-2,2',4'-trifluorobifenM-4-M)metM)piperidm-4-N)-3-etM-2-oxo-1,2-dihidropiridin a-4-carboxilico
Se añadi uóna disolucion acuosa de hidroxido de sodio 2 M (1,5 mL) a una disolucion en etanol (6 mL) de 1-(1-((6-ciclopropil-3-etoxi-2,2',4'-trifluorobifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (264 mg), y la mezcla se agito a 80C durante 3 horas en atmosfera de nitrogeno. A continuacion, el disolvente se destilo a presion reducida. Se agua al residuo y añ laad mióezcla se neutralizo con acido clorhfdrico 2 M, seguido de una extraccion con una disolucion mezclada de acetato de etilo y THF. La fase organica se separo, a continuacion, se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y el disolvente se destilo a presion reducida. El residuo obtenido se cristalizo (hexano/acetato de etilo) y ademas se recristalizo (hexano/etanol) para obtener el compuesto del fftulo (173 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,55-0,63 (2H, m), 0,72-0,80 (2H, m), 1,04 (3H, t, J = 7,3 Hz), 1,32 (3H, t, J = 7,0 Hz), 1,47-1,59 (1H, m), 1,68-1,93 (4H, m), 2,14-2,28 (2H, m), 2,66 (2H, q, J = 7,2 Hz), 2,99 (2H, d, J = 11,5 Hz), 3,52-3,62 (2H, m), 4,04 (2H, q, J = 7,0 Hz), 4,64-4,79 (1H, m), 6,33 (1H, d, J = 7,3 Hz), 6,86 (1H, s), 7,17-7,26 (1H, m), 7,40 (1H, td, J = 9,7, 2,5 Hz), 7,50 (1H, td, J = 8,5, 6,7 Hz), 7,66 (1H, d, J = 7,4 Hz).
Ejemplo 47
Acido 1-(1-(4,5-diciclopropil-3-fluoro-2-isopropoxibencil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilico
A) 4-Ciclopropil-2,3-difluorobenzaldehido
Se anadieron a temperatura ambiente acido ciclopropilboronico (1,77 g), una disolucion acuosa de carbonato de sodio
2 M (21 mL), tris(dibencilidenoacetona)dipaladio(0) (0,882 g) y diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (0,847 g) a una disolucion en tolueno (70 mL) de 4-bromo-2,3-difluorobenzaldel‘ffdo (3,04 g) y la mezcla se agito a 100C durante 15 horas en atmosfera de argon. Se agua a la mezcla a dñea rdeiaóccion a temperatura ambiente y la mezcla se filtro sobre celita. A continuacion, el filtrado se sometio a extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (2,18 g).
RMN 1H (300 MHz, CDCb) delta 0,82-0,90 (2H, m), 1,11-1,20 (2H, m), 2,14-2,25 (1H, m), 6,70 (1H, ddd, J = 8,2, 6,3, 1,7 Hz), 7,51 (1H, ddd, J = 8,3, 6,3, 1,8 Hz), 10,27 (1H, d, J = 0,6 Hz).
B) 4-Ciclopropil-3-fluoro-2-metoxibenzaldehido
Se añadió a temperatura ambiente metoxido de sodio (disolucion en metanol al 28 %, 3,43 g) a una disolucion en metanol (100 mL) de 4-ciclopropil-2,3-difluorobenzaldel'ffdo (2,16 g) y la mezcla se agito a 70C durante 15 horas en atmosfera de nitrogeno. Se agua a la mez acñlaad dieó reaccion y se destilo el metanol a presion reducida. El residuo se sometio a extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (2,11 g).
RMN 1H (400 MHz, CDCla) delta 0,79-0,84 (2H, m), 1,07-1,14 (2H, m), 2,13-2,22 (1H, m), 4,08 (3H, d, J = 2,4 Hz), 6,59 (1H, dd, J = 8,2, 6,4 Hz), 7,50 (1H, dd, J = 8,3, 1,4 Hz), 10,31 (1H, d, J = 0,6 Hz).
C) 4-CidopropM-3-fluoro-2-hidroxibenzaldehido
Se añadi aó -78C tribromuro de boro (disolucion en diclorometano 1 M, 18,5 mL) a una disolucion en diclorometano (70 mL) de 4-ciclopropil-3-fluoro-2-metoxibenzaldel'ffdo (1,79 g) y la mezcla se agito a la misma temperatura que antes durante 2 horas en atmosfera de nitrogeno y, a continuacion, se agito a 0C durante 1 hora. Se anadio una disolucion acuosa saturada de bicarbonato de sodio a la mezcla de reaccion a 0C y se destilo el diclorometano a presion reducida. El residuo obtenido se sometio a extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (1,50 g).
RMN 1H (300 MHz, CDCb) delta 0,81-0,89 (2H, m), 1,09-1,18 (2H, m), 2,17-2,28 (1H, m), 6,44 (1H, dd, J = 8,2, 5,9 Hz), 7,23 (1H, dd, J = 8,2, 1,3 Hz), 9,82 (1H, d, J = 1,9 Hz), 11,02 (1H, s).
D) 5-Bromo-4-ciclopropil-3-fluoro-2-hidroxibenzaldehido
Se añadió a temperatura ambiente acido dibromoisocianurico (1,43 g) a una disolucion en DMF (50 mL) de 4-ciclopropil-3-fluoro-2-hidroxibenzaldel'ffdo (1,50 g) y la mezcla se agito a la misma temperatura que antes durante 3 horas en atmosfera de nitrogeno. Se una disolucion acu aoñsaad sióaturada de tiosulfato de sodio a la mezcla de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (1,73 g).
RMN 1H (400 MHz, CDCb) delta 1,04-1,18 (4H, m), 1,89-1,98 (1H, m), 7,55 (1H, d, J = 1,9 Hz), 9,80 (1H, d, J = 1,9 Hz), 10,81 (1H, s).
E) 4,5-DicidopropM-3-ffuoro-2-hidroxibenzaldehido
Se anadieron a temperatura ambiente acido ciclopropilboronico (1,72 g), una disolucion acuosa de carbonato de sodio 2 M (13 mL), tris(dibencilidenoacetona)dipaladio(0) (0,611 g) y diciclohexil(2',6'-dimetoxibifenil-2-il)fosfina (0,548 g) a una disolucion en tolueno (50 mL) de 5-bromo-4-ciclopropil-3-fluoro-2-hidroxibenzaldel'ffdo (1,73 g) y la mezcla se agito a 100C durante 15 horas en atmosfera de argon. Se agua a la mezcla de reaccion a tem apñearadtiuóra ambiente y la mezcla se filtro sobre celita. A continuacion, el filtrado se sometio a extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tftulo (1,31 g).RMN 1H (300 MHz, CDCb) delta 0,62-0,69 (2H, m), 0,94-1,02 (2H, m), 1,04-1,12 (4H, m), 1,97-2,17 (2H, m), 6,97 (1H, dd, J = 1,4, 0,8 Hz), 9,79 (1H, d, J = 1,9 Hz), 10,78 (1H, s).
F) 4,5-DiciclopropM-3-fluoro-2-isopropoxibenzaldehido
Se añadió a temperature ambiente 2-yodopropano (862 mg) a una mezcla de 4,5-diciclopropil-3-fluoro-2-hidroxibenzaldehndo (744 mg), carbonato de potasio (934 mg) y DMF (20 mL), y la mezcla se agito a 60C durante 2 horas en atmosfera de nitrogeno. Se agua a la mezcla de a rñeaadcicóion, seguido de una extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida.
El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del tttulo (865 mg).
RMN 1H (300 MHz, CDCla) delta 0,67-0,75 (2H, m), 0,90-1,02 (4H, m), 1,03-1,12 (2H, m), 1,34 (6H, dd, J = 6,1,0,8 Hz), 1,88-2,00 (1H, m), 2,11-2,23 (1H, m), 4,41-4,56 (1H, m), 7,19 (1H, d, J = 1,0 Hz), 10,30 (1H, s).
G) 1-(1-(4,5-Diciclopropil-3-fluoro-2-isopropoxibencil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se agito una mezcla de 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (360 mg) en acido formico (8 mL) a 70C durante 30 minutos en atmosfera de nitrogeno y, a continuacion, el disolvente se destilo a presion reducida. Se a temperatura ambie anñtaed tiróiacetoxiborohidruro de sodio (418 mg) a una mezcla del residuo obtenido y 4,5-diciclopropil-3-fluoro-2-isopropoxibenzaldel'ndo (287 mg) en THF (10 mL) y la mezcla se agito a la misma temperatura que antes durante 15 horas en atmosfera de nitrogeno. Se anadio una disolucion acuosa saturada de bicarbonato de sodio a la mezcla de reaccion, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (381 mg).
EM (ESI+): [M+H]+ 511,3.
H) Acido 1-(1-(4,5-Diciclopropil-3-fluoro-2-isopropoxibencil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilico
Se añadi uóna disolucion acuosa de hidroxido de sodio 2 M (2 mL) a una disolucion en etanol (8 mL) de 1-(1-(4,5-diciclopropil-3-fluoro-2-isopropoxibencil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (358 mg), y la mezcla se agito a 80C durante 3 horas en atmosfera de nitrogeno. El disolvente de la mezcla de reaccion se destilo a presion reducida. El residuo obtenido se disolvio en agua y la disolucion se neutralizo con acido clorhffdrico 2 M, seguido de una extraccion con una disolucion mezclada de acetato de etilo y THF. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se cristalizo (acetato de etilo/hexano) y ademas se recristalizo (acetato de etilo/hexano) para obtener el compuesto del tftulo (258 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,58-0,65 (2H, m), 0,67-0,75 (2H, m), 0,91-1,06 (7H, m), 1,23 (6H, d, J = 5,9 Hz), 1,63-1,89 (5H, m), 2,03-2,27 (3H, m), 2,63 (2H, q, J = 7,2 Hz), 2,91 (2H, d, J = 11,6 Hz), 3,46 (2H, s), 4,20-4,35 (1H, m), 4,60-4,75 (1H, m), 6,27 (1H, d, J = 7,2 Hz), 6,73 (1H, s), 7,59 (1H, d, J = 7,3 Hz).
Ejemplo 49
Acido 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxnico
A) 4-Bromo-3-fluoro-2-metoxibenzaldehido
Se añadió metoxido de sodio (disolucion en metanol al 28 %, 69,1 g) a una disolucion de 4-bromo-2,3-difluorobenzaldehffdo (52,8 g) en metanol (600 mL) a temperatura ambiente. La mezcla se calento a reflujo en atmosfera de nitrogeno durante 2 horas y se concentro al vado hasta aproximadamente 1/4 de su volumen. La mezcla se diluyo con acetato de etilo y agua. La mezcla se extrajo con acetato de etilo. La fase organica se separo, se lavo con agua y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado para proporcionar el compuesto del tftulo (52,3 g).
RMN 1H (300 MHz, CDCla) delta 4,12 (3H, d, J = 2,9 Hz), 7,34 (1H, dd, J = 8,5, 5,7 Hz), 7,50 (1H, dd, J = 8,5, 1,6 Hz), 10,34 (1H, s).
B) 4-Bromo-3-fluoro-2-hidroxibenzaldehido
Se añadi aócido bromhffdrico al 48 % (254 mL) a una disolucion de 4-bromo-3-fluoro-2-metoxibenzaldel'ndo (52,3 g)
en acido acetico (350 mL) a temperature ambiente. La mezcla se agito a 120C en atmosfera de nitrogeno durante 16 horas y se concentro al vado. El residuo se diluyo con acetato de etilo y agua. La mezcla se extrajo con acetato de etilo. La fase organica se separo, se lavo con agua y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado. El residuo se trituro con eter de diisopropilo y se recogio mediante filtracion para proporcionar el compuesto del tftulo (34,6 g).
RMN 1H (300 MHz, DMSO-d6) delta 7,26 (1H, dd, J = 8,5, 5,9 Hz), 7,42 (1H, dd, J = 8,5, 1,4 Hz), 10,25 (1H, s), 11,36 (1H, brs).
C) 2,4'-Difluoro-3-hidroxibifernl-4-carbaldehido
Se anadieron acido (4-fluorofenil)boronico (33,2 g), una disolucion acuosa de carbonato de sodio 2M (237 mL), acetato de paladio (II) (2,49 g) y diciclohexilfosfino-2',6'-dimetoxi-1, 1 '-bifenil (9,74 g) a una disolucion de 4-bromo-3-fluoro-2-hidroxibenzaldelddo (34,6 g) en DME (350 mL) a temperature ambiente. La mezcla se agito a 100C en atmosfera de argon durante 16 horas. La mezcla se enfrio hasta temperatura ambiente. Se anadio agua (700 mL) a la mezcla de reaccion. La mezcla se concentro al vado para eliminar el DME. El precipitado se recogio mediante filtracion y se lavo con agua. El filtrado acuoso se dejo aparte para purificacion adicional. A continuacion, el solido se lavo con acetato de etilo. El solido se a una me azñcaladi dóe acetato de etilo y acido clorlddrico 1 M. La mezcla se agito a temperatura ambiente durante 1 hora y se filtro sobre celita. El filtrado se extrajo con acetato de etilo. La fase organica se separo, se lavo con agua y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado para proporcionar el compuesto del tftulo (20,6 g).
El filtrado acuoso se neutralizo con acido clorlddrico 1 M y se extrajo con acetato de etilo. La fase organica se separo, se lavo con agua y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado. El residuo se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para proporcionar el compuesto del tftulo (8,96 g).
RMN 1H (300 MHz, DMSO-d6) delta 7,05-7,15 (1H, m), 7,30-7,42 (2H, m), 7,56 (1H, dd, J = 8,2, 1,3 Hz), 7,61-7,72 (2H, m), 10,29 (1H, s), 11,02 (1H, brs).
D) 6-Bromo-2,4'-difluoro-3-hidroxibifenil-4-carbaldehido
Se añad aiócido dibromoisocianurico (24,8 g) a una disolucion de 2,4'-difluoro-3-hidroxibifenil-4-carbaldel'ndo (33,7 g) en DMF (400 mL) a temperatura ambiente. La mezcla se agito a la misma temperatura en atmosfera de nitrogeno durante 3 horas. La mezcla se neutralizo con una disolucion acuosa saturada de tiosulfato de sodio a temperatura ambiente y se extrajo con acetato de etilo. La fase organica se separo, se lavo con agua y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado. El residuo se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para proporcionar el compuesto del tftulo (33,1 g).
RMN 1H (300 MHz, DMSO-d6) delta 7,32-7,48 (4H, m), 7,77 (1H, d, J = 1,7 Hz), 10,27 (1H, s), 11,28 (1H, brs).
E) 6-Bromo-2,4'-difluoro-3-isopropoxibifenil-4-carbaldehido
Se añadi 2ó-yodopropano (21,7 g) a una mezcla de 6-bromo-2,4'-difluoro-3-hidroxibifenil-4-carbaldel‘ftdo (26,7 g) y carbonato de potasio (23,6 g) en DMF (250 mL) a temperatura ambiente. La mezcla se agito a 60C en atmosfera de nitrogeno durante 3 horas. La mezcla se enfrio con agua a temperatura ambiente y se extrajo con acetato de etilo. La fase organica se separo, se lavo con agua y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado. El residuo se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para proporcionar el compuesto del tftulo (28,5 g).
RMN 1H (300 MHz, DMSO-d6) delta 1,34 (6H, d, J = 6,0 Hz), 4,50-4,65 (1H, m), 7,32-7,42 (2H, m), 7,43-7,53 (2H, m), 7,83 (1H, d, J = 1,7 Hz), 10,26 (1H, s).
F) 6-CidopropM-2,4'-difluoro-3-isopropoxibifenM-4-carbaldehido
Se anadieron acido ciclopropilboronico (13,8 g), diciclohexil(2',6'-dimetoxi-[1,1'-bifenil]-2-il)fosfina (6,58 g), tris(dibencilidenoacetona)dipaladio (0) (7,34 g) y una disolucion acuosa de carbonato de sodio 2M (120 mL) a una disolucion de 6-bromo-2,4'-difluoro-3-isopropoxibifenil-4-carbaldel‘ndo (28,5 g) en tolueno (250 mL) a temperatura ambiente. La mezcla se agito a 100C en atmosfera de argon durante 16 horas. La mezcla se filtro sobre celita. El filtrado se extrajo con acetato de etilo. La fase organica se separo, se lavo con agua y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado. El residuo se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para proporcionar el compuesto del tftulo (24,8 g).
RMN 1H (300 MHz, DMSO-d6) delta 0,61-0,70 (2H, m), 0,75-0,85 (2H, m), 1,31 (6H, d, J = 6,1 Hz), 1,59 (1H, tt, J = 8,4, 5,3 Hz), 4,41-4,56 (1H, m), 7,15 (1H, d, J = 1,0 Hz), 7,30-7,41 (2H, m), 7,43-7,52 (2H, m), 10,28 (1H, s).
G) 4-(((4-metilfenil)sulfonil)oxi)piperidina-1-carboxilato de tert-butilo
Se añadió cloruro de 4-metilbencenosulfonilo (52,1 g) a una disolucion de 4-hidroxipiperidina-1-carboxilato de tertbutilo (50,0 g) en piridina (248 mL) a temperatura ambiente. Se agito la mezcla a temperatura ambiente durante la noche. Despues de la evaporacion del disolvente, se anadieron al residuo agua y acetato de etilo. La fase organica se separo, se lavo con acido clortftdrico 1 M y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado. El residuo se trituro en eter de diisopropilo a 0C («C» representa «grados Celsius») para proporcionar el compuesto del tftulo (76,1 g).
RMN 1H (300 MHz, CDCb) delta 1,43 (9H, s), 1,57-1,88 (4H, m), 2,45 (3H, s), 3,14-3,32 (2H, m), 3,49-3,68 (2H, m), 4,61-4,73 (1H, m), 7,34 (2H, d, J = 8,0 Hz), 7,80 (2H, d, J = 8,3 Hz) .
H) 2-Fluoro-4-yodo-3-metilpiridina
Se añadió n-butil litio (disolucion en hexano 1,6 M, 53,8 mL) a una disolucion de diisopropilamina (8,71 g) en THF (200 mL) a -10C. Despues de ser agitada a la misma temperatura en atmosfera de nitrogeno durante 1 hora, se anadio una disolucion de 2-fluoro-3-yodopiridina (18,3 g) en THF (70 mL) a la mezcla de reaccion a -78C. La mezcla se agito a la misma temperatura en atmosfera de nitrogeno durante 1 hora. Se anadio una disolucion de yodometano (12,8 g) en THF (30 mL) a la mezcla de reaccion a -78C. La mezcla se agito a la misma temperatura en atmosfera de nitrogeno durante 2 horas. La mezcla se neutralizo con una disolucion acuosa saturada de cloruro de amonio a 0C y se extrajo con acetato de etilo. La fase organica se separo, se lavo con agua y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado. El residuo se hizo pasar a traves de sflice NH y se concentro al vado. El residuo se purifico mediante cromatografta en columna con gel de sflice (hexano/acetato de etilo) para proporcionar el compuesto del tftulo (17,2 g).
RMN 1H (300 MHz, CDCla) delta 2,39 (3H, d, J = 1,5 Hz), 7,57-7,63 (1H, m), 7,65-7,70 (1H, m).
I) 4-Yodo-3-metilpiridin-2(1 H)-ona
Se añadió acido clortftdrico 12 M (13 mL) a una disolucion de 2-fluoro-4-yodo-3-metilpiridina (12,2 g) en DME (15 mL) a temperatura ambiente. La mezcla se agito a 100C durante 1,5 horas con una trampa alcalina. Se anadio agua (150 mL) a la mezcla de reaccion a temperatura ambiente. Se agito la mezcla a la misma temperatura durante 2 horas. El precipitado se recogio mediante filtracion y se lavo con agua. Se agito una suspension del solido en etanol (20 mL) a 80C durante 30 minutos. Se eter de diis aoñparodpióilo (150 mL) a la mezcla. Se agito la mezcla a temperatura ambiente durante 30 minutos. El precipitado se recogio mediante filtracion y se lavo con eter de diisopropilo para proporcionar el compuesto del tftulo (9,82 g).
RMN 1H (300 MHz, DMSO-d6) delta 2,14 (3H, s), 6,55 (1H, d, J = 6,9 Hz), 6,96 (1H, dd, J = 6,9, 0,7 Hz), 11,68 (1H, brs).
J) 4-(4-yodo-3-metil-2-oxopiridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo
Se añadió 4-(((4-metilfenil)sulfonil)oxi)piperidina-1-carboxilato de tert-butilo (21,7 g) a una mezcla de 4-yodo-3-metilpiridin-2(1H)-ona (8,98 g) y carbonato de potasio (21,1 g) en 1-metoxi-2-(2-metoxietoxi)etano (160 mL) a temperatura ambiente. La mezcla se agito a 100C en atmosfera de nitrogeno durante 22 horas. La mezcla se enfrio con agua a temperatura ambiente y se extrajo con acetato de etilo. La fase organica se separo, se lavo con agua y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado. Se anadio tolueno al residuo. La mezcla se concentro al vado. El residuo se purifico mediante cromatografta en columna con gel de sflice (hexano/acetato de etilo) para proporcionar el compuesto del tftulo (12,9 g).
RMN 1H (300 MHz, CDCb) delta 1,47 (9H, s), 1,57-1,72 (2H, m), 1,88 (2H, d, J = 13,4 Hz), 2,35 (3H, s), 2,87 (2H, t, J = 12,7 Hz), 4,29 (2H, d, J = 11,1 Hz), 4,99 (1H, tt, J = 12,3, 3,8 Hz), 6,63 (1H, d, J = 7,4 Hz), 6,81 (1H, d, J = 7,4 Hz).
K) 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se añadió [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) (416 mg), trietilamina (1,59 mL) y metanol (6,91 mL) a una disolucion de 4-(4-yodo-3-metil-2-oxopiridin-1(2H)-il)piperidina-1-carboxilato de tert-butilo (2,38 g) en DMF (15 mL) a temperatura ambiente. La mezcla se agito a 90C a 0,5 MPa de atmosfera de monoxido de carbono durante 7 horas. La mezcla se enfrio con agua a temperatura ambiente y se filtro sobre celita. El filtrado se extrajo con acetato de etilo. La fase organica se separo, se lavo con agua y agua salada, se seco sobre sulfato de magnesio anhidro y se concentro al vado. El residuo se hizo pasar a traves de gel de sflice (NH, hexano/acetato de etilo) y se concentro al vado. El residuo se trituro con eter de diisopropilo/hexano y se recogio mediante filtracion para proporcionar el compuesto del tftulo (1,77 g).
RMN 1H (300 MHz, CDCb) delta 1,48 (9H, s), 1,60-1,76 (2H, m), 1,89 (2H, d, J = 12,8 Hz), 2,37 (3H, s), 2,81-2,99 (2H,
m), 3,90 (3H, s), 4,28 (2H, brs), 5,00-5,16 (1H, m), 6,48 (1H, d, J = 7,4 Hz), 7,18 (1H, d, J = 7,4 Hz).
L) 3-metil-2-oxo-1-(piperidin-4-il)-1,2-dihidropiridina-4-carboxilato de metilo clorhidrato
A cloruro de hidrogeno 2M (disolucion en 2-propanol 2,85 L) se 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-m aeñtial-dió 2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (200 g) a temperature ambiente. Se agito la mezcla a temperature ambiente durante 18 horas. Se eter de diisopr aoñpaildoió (11,5 L) a la mezcla de reaccion y el precipitado se recogio mediante filtracion, se lavo con eter de diisopropilo (2,86 L) para proporcionar el compuesto del tftulo (163,2 g).
RMN 1H (300 MHz, DMSO-d6) delta 1,93 (2H, d, J = 11,3 Hz), 2,06-2,25 (5H, m), 3,02-3,18 (2H, m), 3,40 (2H, d, J = 12,8 Hz), 3,84 (3H, s), 4,98 (1H, tt, J = 12,2, 3,9 Hz), 6,48 (1H, d, J = 7,6 Hz), 7,50 (1H, d, J = 7,2 Hz), 9,08 (2H, brs).
M) 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo clorhidrato
Se añad tiróietilamina (35,3 g) a una suspension de 3-metil-2-oxo-1-(piperidin-4-il)-1,2-dihidropiridina-4-carboxilato de metilo clorhidrato (100 g) en THF (2,00 L) a temperatura ambiente. Despues de ser agitada a la misma temperatura durante 30 minutos, se 6-ciclop arñoapdili-ó2,4'-difluoro-3-isopropoxibifenil-4-carbaldel'ndo (121 g) a la mezcla de reaccion. Se agito la mezcla a temperatura ambiente durante 30 minutos. Se anadieron a la mezcla de reaccion triacetoxihidroborato de sodio (111 g) y acido acetico (20,9 g) Se agito la mezcla a temperatura ambiente durante 16 horas. Se anadieron una disolucion acuosa saturada de hidrogeno carbonato de sodio (1,30 L) y agua (1,30 L) a la mezcla a temperatura ambiente. La mezcla se extrajo con acetato de etilo (10 L). La fase organica se separo, se lavo con agua salada (4,00 L), se seco sobre sulfato de sodio anhidro y se concentro al vado. El residuo se disolvio en acetato de etilo (1,00 L). Se cloruro de hidro agñeandoió 4 M (disolucion en acetato de etilo, 87 ml) a la mezcla a temperatura ambiente. A continuacion, se anadieron germenes de cristalizacion a la mezcla. La mezcla se diluyo con eter de diisopropilo (2,00 L) y se agito a temperatura ambiente durante 1 hora. El precipitado se recogio mediante filtracion y se lavo con eter de diisopropilo (1,50 L) para proporcionar el compuesto del tftulo (183,7 g).
RMN 1H (300 MHz, DMSO-d6) delta 0,74-0,89 (4H, m), 1,30 (6H, d, J = 6,0 Hz), 1,48-1,64 (1H, m), 1,93-2,06 (2H, m), 2,18 (3H, s), 2,29-2,47 (2H, m), 3,24-3,40 (2H, m), 3,41-3,53 (2H, m), 3,84 (3H, s), 4,30 (2H, d, J = 4,9 Hz), 4,37-4,50 (1H, m), 4,99-5,17 (1H, m), 6,50 (1H, d, J = 7,2 Hz), 7,25-7,39 (3H, m), 7,39-7,51 (3H, m), 10,90 (1H, brs).
N) Acido 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxobifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridma-4-carboxiMco
Se añadió una disolucion acuosa de hidroxido de sodio 2 M (690 mL) a una disolucion de 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo clorhidrato (270 g) en metanol (1,35 L) a temperatura ambiente. La mezcla se agito a 50C durante 2 horas y se enfrio hasta temperatura ambiente. Se agua (2,70 añ La)d aió la mezcla de reaccion. La mezcla se concentro al vado para eliminar el metanol y se neutralizo con acido clorhfdrico 2 M (460 mL) a temperatura ambiente. Se anadieron germenes de cristalizacion a la mezcla. Se agito la mezcla a temperatura ambiente durante 17 horas. El precipitado se recogio mediante filtracion, se lavo con agua (7,00 L) y se seco al vado a 65C durante 24 horas para proporcionar el compuesto del tftulo (245,3 g).
RMN 1H (300 MHz, DMSO-d6) delta 0,55-0,65 (2H, m), 0,71-0,84 (2H, m), 1,26 (6H, d, J = 5,7 Hz), 1,51-1,65 (1H, m), 1,68-1,96 (4H, m), 2,10-2,26 (5H, m), 2,98 (2H, d, J = 11,3 Hz), 3,56 (2H, s), 4,26-4,43 (1H, m), 4,61-4,79 (1H, m), 6.36 (1H, d, J = 7,6 Hz), 6,84 (1H, s), 7,25-7,36 (2H, m), 7,38-7,49 (2H, m), 7,67 (1H, d, J = 7,2 Hz).
O) Acido 1-(1-((6-Ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridin a-4-carboxilico
Se disolvio acido 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxflico (350 g) en una disolucion acuosa en etanol al 90 % (700 mL) a 70C. La mezcla se filtro a traves de un papel de filtro con acetato de etilo (1,00 L). Se acetato de etilo (4,25 L) al filtrado a añ 6a5dCió (temperatura interior). Se anadieron germenes de cristalizacion a la mezcla. La mezcla se enfrio lentamente hasta 45C (temperatura interior) y se agito a la misma temperatura durante 18 horas. La mezcla se enfrio hasta 30C. El precipitado se recogio mediante filtracion, se lavo con acetato de etilo (1,50 L) y se seco al vado a 70C para proporcionar el compuesto del tftulo (306 g).
RMN 1H (300 MHz, DMSO-d6) delta 0,55-0,64 (2H, m), 0,72-0,82 (2H, m), 1,26 (6H, d, J = 6,0 Hz), 1,51-1,64 (1H, m), 1,67-1,94 (4H, m), 2,11-2,26 (5H, m), 2,99 (2H, d, J = 11,3 Hz), 3,57 (2H, s), 4,27-4,43 (1H, m), 4,62-4,78 (1H, m), 6.37 (1H, d, J = 7,2 Hz), 6,84 (1H, s), 7,25-7,36 (2H, m), 7,37-7,48 (2H, m), 7,67 (1H, d, J = 7,2 Hz).
pf 144,8-145,7C
Ejemplo 50
Acido 1-(1-((6-Ciclopropil-3-etoxi-2,4'-difluorobifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxilico
A) 1-(1-((6-Ciclopropil-3-etoxi-2,4'-difluorobifenil-4-il)meti)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se agito una mezcla de 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (306 mg) y acido formico (8 mL) a 70C durante 30 minutos en atmosfera de nitrogeno y, a continuacion, el disolvente se destilo a presion reducida. Se a temperatura ambie anñtaed tiróiacetoxiborohidruro de sodio (370 mg) a una mezcla del residuo obtenido y 6-ciclopropil-3-etoxi-2,4'-difluorobifenil-4-carbaldel'ffdo (290 mg) y THF (8 mL) y la mezcla se agito a la misma temperatura que antes durante 15 horas en atmosfera de nitrogeno. Se anadio una disolucion acuosa saturada de bicarbonato de sodio a la mezcla de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. La fase organica obtenida se lavo con agua y salino saturado en este orden y se seco sobre sulfato de magnesio anhidro y, a continuacion, el disolvente se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (301 mg).
RMN 1H (300 MHz, CDCla) delta 0,58-0,65 (2H, m), 0,74-0,82 (2H, m), 1,39 (3H, t, J = 7,0 Hz), 1,57-1,67 (1H, m), 1,72 1,94 (4H, m), 2,28 (2H, td, J = 11,6, 2,5 Hz), 2,37 (3H, s), 3,05 (2H, d, J = 11,8 Hz), 3,58 (2H, s), 3,90 (3H, s), 4,04 4,13 (2H, m), 4,87-5,01 (1H, m), 6,47 (1H, d, J = 7,4 Hz), 6,71 (1H, d, J = 1,3 Hz), 7,09-7,18 (2H, m), 7,27-7,37 (3H, m) .
B) Acido 1-(1-((6-Ciclopropil-3-etoxi-2,4'-difluorobifeml-4-il)metil)piperidm-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxnico
Se añadió una disolucion acuosa de hidroxido de sodio 2 M (2 mL) a temperatura ambiente a una disolucion en etanol (8 mL) de 1-(1-((6-ciclopropil-3-etoxi-2,4'-difluorobifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (283 mg) y la mezcla se agito a 80C durante 3 horas en atmosfera de nitrogeno. A continuacion, el disolvente se destilo a presion reducida. Se agua al residuo obtenido y la m añeazcdliaó se neutralizo con acido clorlffdrico 2 M. El cristal depositado se recogio mediante filtracion y se disolvio en etanol. La disolucion obtenida se filtro y, a continuacion, el filtrado se concentro a presion reducida. El residuo obtenido se cristalizo (hexano/acetato de etilo) y ademas se recristalizo (hexano/acetato de etilo) para obtener el compuesto del fftulo (151 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,57-0,65 (2H, m), 0,72-0,80 (2H, m), 1,33 (3H, t, J = 7,0 Hz), 1,51-1,63 (1H, m), 1,71-1,98 (4H, m), 2,17 (3H, s), 2,22-2,36 (2H, m), 3,03 (2H, d, J = 11,4 Hz), 3,62 (2H, s), 4,04 (2H, q, J = 7,0 Hz), 4,65-4,80 (1H, m), 6,38 (1H, d, J = 7,3 Hz), 6,83 (1H, s), 7,26-7,36 (2H, m), 7,38-7,47 (2H, m), 7,65 (1H, d, J = 7,4 Hz).
Ejemplo 51
Acido 1-(1-((2-ciclopropil-4'-fluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilico
A) 1-(1-((2-ciclopropil-4'-fluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo
Se agito una mezcla de 1-(1-(tert-butoxicarbonil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxilato de metilo (300 mg) y acido formico (2 mL) a 70C durante 1 hora en atmosfera de nitrogeno y, a continuacion, el disolvente se destilo a presion reducida. Se triacetoxiborohid arñuarodió de sodio (262 mg) a una mezcla del residuo obtenido, 2-ciclopropil-4'-fluoro-5-isopropoxibifenil-4-carbaldel'ffdo (246 mg) y THF (5 mL) y la mezcla se agito durante la noche a temperatura ambiente. Se una disoluc aioñnad aicóuosa saturada de bicarbonato de sodio a la mezcla de reaccion a temperatura ambiente, seguido de extraccion con acetato de etilo. El disolvente de la fase organica se destilo a presion reducida. El residuo obtenido se purifico mediante cromatograffa en columna con gel de sflice (hexano/acetato de etilo) para obtener el compuesto del fftulo (390 mg).
EM (ESI+): [M+H]+ 547,5.
B) Acido 1-(1-((2-Ciclopropil-4'-fluoro-5-isopropoxibifeml-4-il)metil)piperidm-4-il)-3-etil-2-oxo-1,2-dihidropiridin a-4-carboxilico
Se añadió a temperatura ambiente una disolucion acuosa de hidroxido de sodio 2 M (1 mL) a una disolucion en metanol (4 mL) de 1-(1-((2-ciclopropil-4'-fluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4
carboxilato de metilo (390 mg) y la mezcla se agito a 60C durante 1 hora en atmosfera de nitrogeno y, a continuacion, se neutralizo con acido clorhftdrico 1 M. La mezcla de reaccion se agito a temperatura ambiente durante 30 minutos y, a continuacion, el cristal depositado se recogio mediante filtracion y se recristalizo (eter de diisopropilo/etanol) para obtener el compuesto del tftulo (240 mg).
RMN 1H (400 MHz, DMSO-d6) delta 0,51-0,58 (2H, m), 0,72-0,79 (2H, m), 1,03 (3H, t, J = 7,3 Hz), 1,26 (6H, d, J = 6,0 Hz), 1,69-1,79 (3H, m), 1,80-1,92 (2H, m), 2,20 (2H, t, J = 11,3 Hz), 2,65 (2H, q, J = 7,2 Hz), 3,01 (2H, d, J = 11,2 Hz), 3,54 (2H, s), 4,55-4,64 (1H, m), 4,64-4,75 (1H, m), 6,32 (1H, d, J = 7,2 Hz), 6,77 (1H, s), 6,98 (1H, s), 7,26 (2H, t, J = 8,9 Hz), 7,44-7,53 (2H, m), 7,65 (1H, d, J = 7,3 Hz).
Ejemplo 60
Acido 1-(1-((6-Ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridma-4-carboxilico clorhidrato
A) Acido 1-(1-((6-Ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridma-4-carboxilico clorhidrato
Se disolvio acido 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carbox^lico (15,0 g) en isopropanol (150 mL) a 60C. La mezcla se enfrio hasta temperatura ambiente. Se añad cióloruro de hidrogeno 2 M (disolucion en isopropanol, 21,0 mL) a la mezcla a temperatura ambiente. Despues de ser agitada a temperatura ambiente durante 30 minutos, se anadieron eter de diisopropilo (300 mL) y germenes de cristalizacion a la mezcla. La mezcla se agito durante 1 hora en atmosfera de nitrogeno. El precipitado se recogio mediante filtracion, se lavo con eter de diisopropilo (150 mL) y heptano (100 mL) y, a continuacion, se seco a 60C para proporcionar el compuesto del tftulo (14,3 g).
RMN 1H (300 MHz, DMSO-d6) delta 0,74-0,88 (4H, m), 1,30 (6H, d, J = 6,0 Hz), 1,49-1,62 (1H, m), 1,98 (2H, d, J = 12,1 Hz), 2,19 (3H, s), 2,37 (2H, d, J = 11,3 Hz), 3,21-3,52 (4H, m), 4,30 (2H, brs), 4,43 (1H, dt, J = 12,0, 5,9 Hz), 5,07 (1H, brs), 6,48 (1H, d, J = 7,6 Hz), 7,26 (1H, s), 7,30-7,38 (2H, m), 7,39-7,48 (3H, m), 10,86 (1H, brs), 13,57 (1H, brs).
B) Acido 1-(1-((6-Ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxnico clorhidrato
Se disolvio acido 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridin a-4-carbox^lico clorhidrato (20,0 g) en isopropanol/2-butanona (190 mL/190 mL) a 80C. La mezcla se filtro a traves de un papel de filtro con isopropanol/2-butanona (10 mL/10 mL). Se anadieron heptano (250 mL) y germenes de cristalizacion a la mezcla a 60C. A continuacion, se heptano (350 mL) a la mezcla a de 55 a aña 6d0iCó. Se agito la mezcla a la misma temperatura durante 1,5 horas y se enfrio hasta temperatura ambiente durante 2 horas. La mezcla se agito a 12C durante 1 hora. El precipitado se recogio mediante filtracion y se seco al vacfo a 60C durante 8 horas para proporcionar el compuesto del tftulo (18,2 g).
RMN 1H (300 MHz, DMSO-d6) delta 0,73-0,91 (4H, m), 1,30 (6H, d, J = 6,0 Hz), 1,48-1,62 (1H, m), 1,97 (2H, d, J = 11,7 Hz), 2,19 (3H, s), 2,33-2,48 (2H, m), 3,22-3,53 (4H, m), 4,30 (2H, brs), 4,42 (1H, dt, J = 12,1,6,0 Hz), 5,10 (1H, t, J = 12,1 Hz), 6,48 (1H, d, J = 7,2 Hz), 7,27-7,39 (3H, m), 7,39-7,49 (3H, m), 11,18 (1H, brs), 13,56 (1H, brs). pf 242C
Ejemplo 61
Acido 1-(1-((6-Ciclopropil-2,4'-difluoro-3-isopropoxobifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxilico 1/2 hidrosulfato
Se añadió acido sulfurico 0,5 M (2 mL) a una disolucion de acido 1-(1-((6-Ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxflico (515 mg) en etanol (5 mL) a temperatura ambiente. La mezcla se agito a la misma temperatura en atmosfera de nitrogeno durante 1 hora. Se anadio eter de diisopropilo (15 mL) a la mezcla de reaccion. La mezcla se agito a temperatura ambiente en atmosfera de nitrogeno durante 1 hora. El precipitado se recogio mediante filtracion. El solido se recristalizo a partir de etanol-heptano para proporcionar el compuesto del tftulo (422 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,59-0,71 (2H, m), 0,75-0,86 (2H, m), 1,29 (6H, d, J = 6,0 Hz), 1,51-1,65 (1H, m), 1,80-2,13 (4H, m), 2,18 (3H, s), 3,31 (4H, brs), 3,94 (2H, brs), 4,35-4,50 (1H, m), 4,83 (1H, brs), 6,42 (1H, d, J = 7,3 Hz), 6,93 (1H, s), 7,26-7,37 (2H, m), 7,38-7,48 (2H, m), 7,52-7,64 (1H, m).
pf 233C
Ejemplo 62
Acido 1-(1-((6-Ciclopropil-2,4'-difluoro-3-isopropoxobifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxilico, maleato
Se añadió acido maleico (111 mg) a una disolucion de acido 1-(1 -((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carbox^lico (513 mg) en etanol (5 mL) a temperatura ambiente. La mezcla se agito a la misma temperatura en atmosfera de nitrogeno durante 1 hora y se concentro al vado. El residuo se cristalizo a partir de acetato de etilo-hexano para proporcionar un solido incoloro. El solido se recristalizo a partir de etanol-heptano para proporcionar el compuesto del titulo (573 mg).
RMN 1H (300 MHz, DMSO-d6) delta 0,63-0,72 (2H, m), 0,77-0,86 (2H, m), 1,30 (6H, d, J = 6,0 Hz), 1,53-1,67 (1H, m), 1,87-2,16 (4H, m), 2,19 (3H, s), 3,31 (4H, brs), 4,11 (2H, brs), 4,44 (1H, brs), 4,89 (1H, brs), 6,07 (2H, s), 6,45 (1H, d, J = 7,3 Hz), 6,98 (1H, s), 7,28-7,38 (2H, m), 7,39-7,54 (3H, m).
pf 193C
Los compuestos de los Ejemplos 8 a 42, 46, 48 y 52 a 59 que se muestran en la tabla posterior se llevaron a cabo de acuerdo con los procedimientos mostrados anteriormente en los Ejemplos o procedimientos equivalentes a los mismos.
Los compuestos de los Ejemplos llevados a cabo de acuerdo con los procedimientos de produccion anteriormente mencionados y los procedimientos mostrados en los Ejemplos o procedimientos equivalentes a los mismos se muestran en la Tabla 1 posterior. La tabla 1 incluye los nombres de los compuestos, formulas estructurales y datos de los valores medidos reales de EM de los compuestos de los Ejemplos. Los Ejemplos 14, 26, 34, 37 y 38 se proporcionan con fines comparativos. Los valores medidos reales de e M se indican mediante los valores hallados en modo positivo (ESI+) o en modo negativo (ESI-).
[Tabla 1-1]


0 [Tabla 1-2]

[Tabla 1-3]

[Tabla 1-4]


[Tabla 1-5]
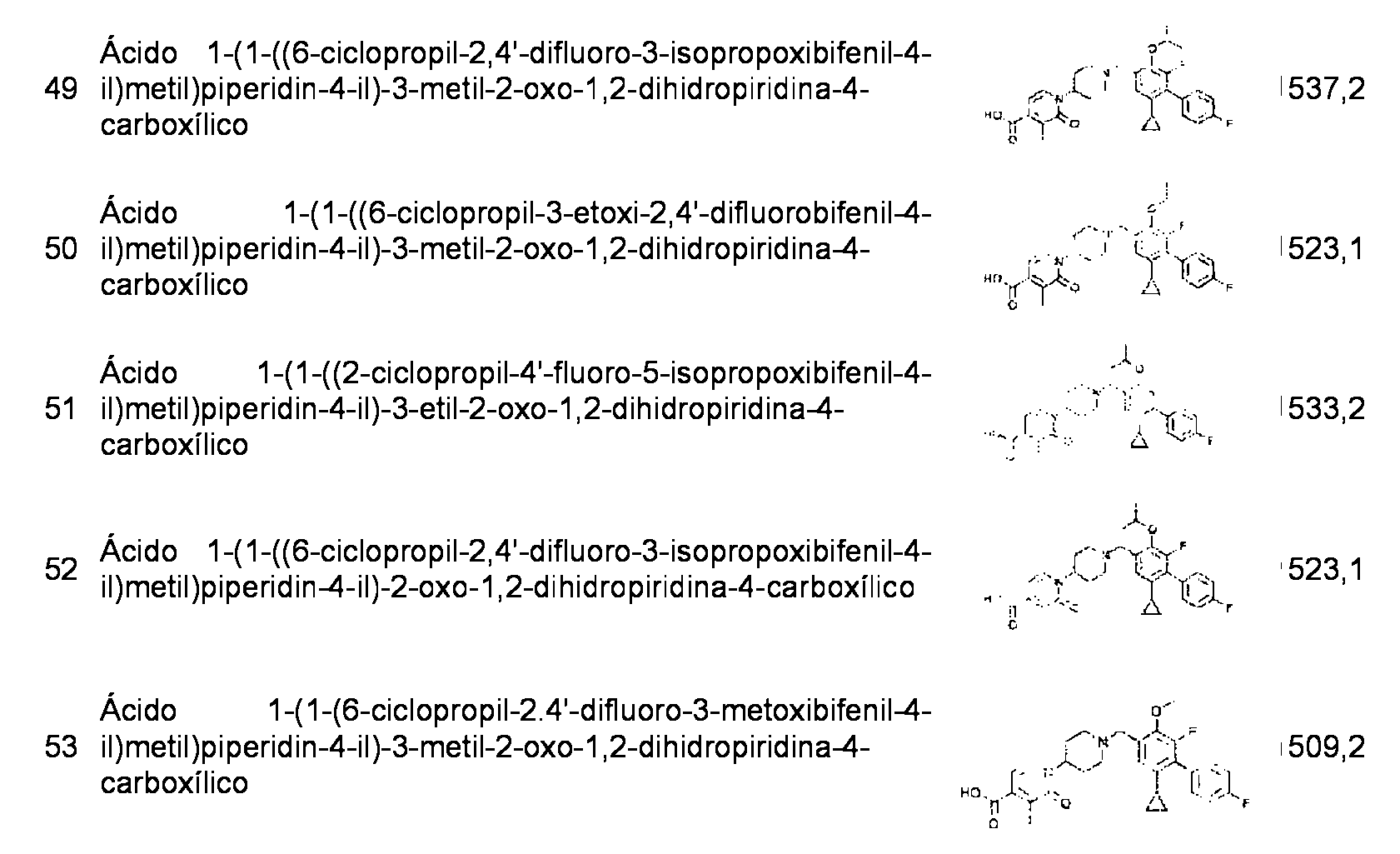

[Tabla 1-6]

Ejemplo de ensayo 1
Evaluacion de la actividad antagonista del SSTR5 humano usando la concentracion de cAMP intracelular como indice
La concentracion de cAMP intracelular se midio usando un kit HTRF cAMP dynamic 2 (Cisbio Bioassays). Se anadio cada compuesto de ensayo diluido con una disolucion amortiguadora (HBSS (Invitrogen Corp.) que contema HEPES 5 mM (pH 7,5) (Invitrogen Corp.), BSA sin acidos grasos al 0,1 % (Sigma-Aldrich Corp.) y IBMx 500 u(«u» representa «micro»)M (Wako Pure Chemical Industries, Ltd.)) con una concentracion de 2 uL/pocillo a una placa blanca de 384 pocillos (Greiner Bio-One Co., Ltd.) para proporcionar la concentracion final de 1 uM. Se descongelo una reserva congelada de celulas CHO (dhfr-) que expresan de forma estable el gen SSTR5 humano (identificador num. NM_001053) en un bano con termostato a 37C y se suspendieron en un medio de cultivo (MEM alfa (Wako Pure Chemical Industries, Ltd.), suero dializado al 10 % (Gemini BioProducts) y 50 ug/mL de gentamicina (Invitrogen Corp.). Despues de centrifugacion de la suspension de celulas, las celulas se resuspendieron en una disolucion amortiguadora
de ensayo y se anadieron con una concentracion de 2 uL/pociMo (aproximadamente 4.000 celulas/pocillo) a la placa. El compuesto y las celulas se incubaron durante 15 minutos y, a continuacion, se anadieron a los mismos una disolucion amortiguadora de ensayo que contema somatostatina 28 (Toray Research Center) 0,1 nM (concentracion final) y forskolina (Wako Pure Chemical Industries, Ltd.) 0,3 uM (concentracion final) con una concentracion de 2 uL/pocillo, seguido de una incubacion a temperatura ambiente durante 30 minutos. Se anadieron a la misma cAMP-d2 y anti-cAMP-criptato con una concentracion de 3 uL/pocillo. La placa se dejo reposar a temperatura ambiente durante 60 minutos. A continuacion, se midio la intensidad por transferencia de energfa por resonancia de fluorescencia (FRET) usando un equipo Multi-label reader Envision (PerkinElmer). La intensidad FRET de los pocillos suplementados con el compuesto de ensayo se convirtio a concentracion de cAMP usando una curva de calibracion preparada a partir de la intensidad FRET de un grupo de pocillos que conteman una disolucion amortiguadora suplementada con una concentracion arbitraria de cAMp . La actividad inhibitoria de cada compuesto se calculo de acuerdo con la siguiente expresion:
Act i v i da d i nhi bi dor a (%) = (C - B) / (A - B) xl OO A: concentracion de cAMP calculada a partir de los pocillos suplementados con forskolina 0,3 uM
B: concentracion de cAMP calculada a partir de los pocillos suplementados con forskolina 0,3 uM y somatostatina 280,1 nM
C: concentracion de cAMP calculada a partir de los pocillos suplementados con forskolina 0,3 uM, somatostatina 28 0,1 nM y compuesto de ensayo 1 uM
La Tabla 2 muestra la tasa de inhibicion (%) frente a SSTR5 para una concentracion de 1 uM del compuesto de ensayo.
[Tabla 2]

Como es evidente a partir de la Tabla 2, el compuesto de la presente invencion exhibio una accion antagonista del SSTR5 elevada.
Ejemplo de ensayo 2
Efecto antidiabetico en ratones
En este estudio se usaron ratones hembra KK-Ay/Ta (Clea Japan Inc.), un modelo de diabetes tipo 2. A la edad de 7 semanas, se extrajeron muestras de sangre de la vena de la cola a las 8:00 am y los animales se dividieron en grupos separados (n=8) con base en la glucohemoglobina (GHb), glucosa plasmatica, insulina, niveles de trigliceridos y peso
corporal. El vehnculo (0,5 % (p/v) metilcelulosa) o los compuestos (suspendidos en el vehnculo) se administraron v^ a oral una vez al dfa durante 2 semanas. Despues de 2 semanas de tratamiento, se determino la GHb mediante un equipo de analisis automatico HLC-723G8 (TOSOH, Japon).
[Tabla 3]

Ejemplo de formulacion 1 (produccion de capsula)

Los ingredientes 1), 2), 3) y 4) se mezclan y se introducen en una capsula de gelatina.
Ejemplo de formulacion 2 (produccion de comprimido)

Las cantidades totales de ingredientes 1), 2) y 3) y 30 g del ingrediente 4) se amasan con agua y se granulan despues de secado al vado. Los polvos granulados se mezclan con 14 g del ingrediente 4) y con 1 g del ingrediente 5). La mezcla se comprime usando un equipo de compresion. De esta forma, se obtienen 1000 comprimidos que contienen cada uno 30 mg del compuesto del Ejemplo 1.
[Aplicabilidad industrial]
El compuesto de la presente invencion presenta una accion antagonista del receptor de la somatostatina subtipo 5 y es util en la profilaxis o tratamiento de la diabetes mellitus y la obesidad.
Claims (8)
1. Un compuesto representado por la siguiente formula:

donde el anillo A es
(1) un anillo de benceno opcionalmente sustituido con de 1 a 4 sustituyentes que son los mismos o diferentes y se seleccionan de entre:
un atomo de halogeno; un grupo cicloalquilo C3-10; un grupo alcoxi Ci-a; y un grupo arilo Ca-14 opcionalmente sustituido con de 1 a 3 atomos de halogeno, o
(2) piridina opcionalmente sustituida con de 1 a 3 sustituyentes que son los mismos o diferentes y se seleccionan de entre:
un grupo cicloalquilo C3-10; un grupo alcoxi C1-a; y un grupo arilo Ca-14 opcionalmente sustituido con de 1 a 3 atomos de halogeno;
el anillo B es piridina o pirimidina cada una ademas opcionalmente sustituida con de 1 a 3 sustituyentes seleccionados de entre un atomo de halogeno y un grupo alquilo C1-a;
donde cada uno de X1, X2 y X4 es un atomo de carbono y X3 es un atomo de carbono o un atomo de nitrogeno; n es 3;
R1 es COOH, CONH2 , un grupo tetrazolilo o un grupo dihidrooxadiazolilo o OCH2COOH y esta unido a X2 del anillo B;
y cada uno de R2 y R3 es un atomo de hidrogeno,
o una sal del mismo.
2. El compuesto de acuerdo con la reivindicacion 1 o una sal del mismo, que es acido 1-(1-((2-ciclopropil-4'-fluoro-5-isopropoxibifenil-4-il)metil)piperidin-4-il)-2-oxo-1,2-dihidropiridin-4-carboxflico o una sal del mismo.
3. El compuesto de acuerdo con la reivindicacion 1 o una sal del mismo, que es acido 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-etil-2-oxo-1,2-dihidropiridina-4-carboxflico o una sal del mismo.
4. El compuesto de acuerdo con la reivindicacion 1 o una sal del mismo, que es acido 1-(1-((6-ciclopropil-2,4'-difluoro-3-isopropoxibifenil-4-il)metil)piperidin-4-il)-3-metil-2-oxo-1,2-dihidropiridina-4-carboxflico o una sal del mismo.
5. Un medicamento que comprende el compuesto de la reivindicacion 1 o una sal del mismo.
6. El medicamento de la reivindicacion 5, para uso en el tratamiento de la diabetes mellitus (p. ej., diabetes mellitus tipo 1, diabetes mellitus tipo 2, diabetes gestacional, diabetes mellitus por obesidad), obesidad (p. ej., mastocitosis maligna, obesidad exogena, obesidad por hiperinsulinismo, obesidad hiperplasica, adiposidad hipofisiaria, obesidad hipoplasica, obesidad hipotiroidea, obesidad hipotalamica, obesidad sintomatica, obesidad infantil, obesidad del tren superior, obesidad exogena, obesidad hipogonadica, mastocitosis sistemica, obesidad general, obesidad central y similares), hiperfagia, hiperlipidemia/dislipidemia (p. ej., hipertrigliceridemia, hipercolesterolemia, colesterolemia LDL elevada, colesterolemia HDL baja, hiperlipidemia posprandial), hipertension, enfermedad cardiovascular (p. ej., insuficiencia cardiaca, arritmia, cardiopatfa isquemica, valvulopatfa cardiaca, arteriosclerosis), complicaciones de la diabetes [p. ej., neuropatfa, nefropatfa, retinopatfa, cardiomiopatfa diabetica, catarata, macroangiopatfa, osteopenia, coma diabetico hiperosmolar, enfermedad infecciosa (p. ej., infeccion
respiratoria, infeccion del tracto urinario, infeccion gastrointestinal, infecciones de los tejidos blandos cutaneos, infeccion de los miembros inferiores), gangrena diabetica, xerostomia, hipoacusia, enfermedad cerebrovascular, alteracion de la circulacion sangumea periferica], smdrome metabolico (cuadros clmicos que presentan 3 o mas seleccionados de entre hipertriglicerid(TG)emia, colesterol(HDL-C)emia HDL baja, hipertension, obesidad abdominal y tolerancia a la glucosa alterada), sarcopenia, trastorno afectivo, disfuncion sexual, depresion, ansiedad, neurosis, arteriosclerosis, artritis de rodilla, osteoporosis, caquexia (p. ej., caquexia cancerosa, caquexia tuberculosa, caquexia diabetica, caquexia asociada con una hemopatia, caquexia asociada con una endocrinopatfa, caquexia asociada con una enfermedad infecciosa o caquexia producida por un smdrome de inmunodeficiencia adquirida), esteatosis hepatica, poliquistosis ovarica, nefropatfas (p. ej., nefropatfa diabetica, glomerulonefritis, glomeruloesclerosis, smdrome nefrotico, nefroesclerosis hipertensiva, insuficiencia renal terminal), distrofia muscular, infarto de miocardio, angina de pecho, enfermedad cerebrovascular (p. ej., infarto cerebral, accidente cerebrovascular), enfermedad de Alzheimer, enfermedad de Parkinson, demencia, smdrome de resistencia insulmica, smdrome X metabolico, hiperinsulinemia, parestesia producida por hiperinsulinemia, diarrea cronica o aguda, enfermedad inflamatoria (p. ej., artritis reumatoide cronica, espondiloartritis anquilosante, enfermedad de Kashin-Bek, lumbago, gota, inflamacion posoperatoria o postraumatica, distension abdominal, neuralgia, laringofaringitis, cistitis, hepatitis (incluyendo esteatohepatitis no alcoholica), neumorna, pancreatitis, ulceras, gastritis, trastornos digestivos, lesion de la mucosa gastrica (incluyendo lesion de la mucosa gastrica producida por la aspirina)), smdrome del fondo de saco, enfermedad inflamatoria intestinal (incluyendo enfermedad inflamatoria de colon), celiaqrna (por ejemplo derivada de una enteropatfa por gluten o enfermedad celfaca), esprue tropical, esprue hipogammaglobulinemico, enteritis, enteritis regional (enfermedad de Crohn), smdrome del colon irritable asociado con diarrea, afectacion del intestino delgado (incluyendo lesiones de la mucosa del intestino delgado) y smdrome del intestino corto, esofagitis con reflujo, colitis ulcerosa, malabsorcion, insuficiencia testicular, smdrome de obesidad visceral, sarcopenia, y cancer como cancer de mama (p. ej., cancer de mama ductal invasivo, cancer de mama ductal no invasivo, cancer de mama inflamatorio, etc.), cancer prostatico (p. ej., cancer prostatico hormonodependiente, cancer prostatico no hormonodependiente, etc.), cancer pancreatico (p. ej., cancer pancreatico ductal, etc.), cancer gastrico (p. ej., adenocarcinoma papilar, adenocarcinoma mucoso, carcinoma adenoescamoso, etc.), cancer de pulmon (p. ej., carcinoma de pulmon no microcftico, carcinoma de pulmon microdtico, mesotelioma maligno, etc.), cancer de colon (p. ej., tumor estromatico gastrointestinal, etc.), cancer rectal (p. ej., tumor estromatico gastrointestinal, etc.), cancer colorrectal (p. ej., cancer colorrectal familiar, cancer colorrectal no poliposo hereditario, tumor estromatico gastrointestinal, etc.), cancer del intestino delgado (p. ej., linfoma no Hodgkin, tumor estromatico gastrointestinal, etc.), cancer esofagico, cancer duodenal, cancer de lengua, cancer farmgeo (p. ej., cancer nasofarmgeo, cancer orofarmgeo, cancer hipofarmgeo, etc.), cancer de las glandulas salivales, tumor cerebral (p. ej., astrocitoma pineal, astrocitoma pilodtico, astrocitoma difuso, astrocitoma anaplasico, etc.), neurilenoma, cancer hepatico (p. ej., cancer hepatico primario, cancer extrahepatico de las vfas biliares, etc.), cancer renal (p. ej., cancer de celulas renales, cancer de celulas de transicion de la pelvis renal y el ureter, etc.), cancer de las vfas biliares, cancer de endometrio, cancer cervical uterino, cancer ovarico (p. ej., cancer ovarico epitelial, tumor extragonadal de celulas reproductoras, tumor ovarico de celulas reproductoras, tumor ovarico de bajo potencial maligno, etc.), cancer de vejiga, cancer de uretra, cancer de piel (p. ej., melanoma (ocular) intraocular, carcinoma de celulas de Merkel, etc.), hemangioma, linfoma maligno, melanoma maligno, cancer tiroideo (p. ej., cancer tiroideo medular, etc.), cancer paratiroideo, cancer de la cavidad nasal, cancer de los senos paranasales), tumor oseo (p. ej., osteosarcoma, tumor de Ewing, sarcoma uterino, sarcoma de los tejidos blandos, etc.), angiofibroma, sarcoma de la retina, cancer de pene, tumor testicular, tumor solido pediatrico (p. ej., tumor de Wilms, tumor renal infantil, etc.), sarcoma de Kaposi, sarcoma de Kaposi producido por el sida, tumor del seno maxilar, histiocitoma fibroso, leiomiosarcoma, rabdomiosarcoma, y leucemia (p. ej., leucemia mielogena aguda, leucemia linfodtica aguda).
7. El medicamento para uso de la reivindicacion 6 donde el uso es en la profilaxis o tratamiento de la diabetes mellitus.
8. El compuesto de acuerdo con la reivindicacion 1 o una sal del mismo para uso en la profilaxis o tratamiento de la diabetes mellitus.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013224093 | 2013-10-29 | ||
| PCT/JP2014/005438 WO2015064083A1 (en) | 2013-10-29 | 2014-10-28 | Heterocyclic compound |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2710657T3 true ES2710657T3 (es) | 2019-04-26 |
Family
ID=51932560
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14800152T Active ES2710657T3 (es) | 2013-10-29 | 2014-10-28 | Compuesto heterocíclico |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US9120777B2 (es) |
| EP (1) | EP3063139B1 (es) |
| JP (1) | JP6450755B2 (es) |
| AR (2) | AR098215A1 (es) |
| CY (1) | CY1121279T1 (es) |
| DK (1) | DK3063139T3 (es) |
| ES (1) | ES2710657T3 (es) |
| HR (1) | HRP20190133T1 (es) |
| HU (1) | HUE041792T2 (es) |
| LT (1) | LT3063139T (es) |
| PL (1) | PL3063139T3 (es) |
| PT (1) | PT3063139T (es) |
| RS (1) | RS58321B1 (es) |
| SI (1) | SI3063139T1 (es) |
| TR (1) | TR201901986T4 (es) |
| TW (1) | TWI668214B (es) |
| UY (1) | UY35801A (es) |
| WO (1) | WO2015064083A1 (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2816032A4 (en) * | 2012-02-13 | 2015-09-30 | Takeda Pharmaceutical | AROMATIC CORE COMPOUND |
| WO2014142363A1 (en) * | 2013-03-14 | 2014-09-18 | Takeda Pharmaceutical Company Limited | Spiro azetidine isoxazole derivatives and their use as sstr5 antagonists |
| JO3442B1 (ar) | 2013-10-07 | 2019-10-20 | Takeda Pharmaceuticals Co | مضادات ذات نوع فرعي من مستقبل سوماتوستاتين 5 (sstr5) |
| JP2023507508A (ja) * | 2019-12-19 | 2023-02-22 | ユニヴェルシテ・ドゥ・ストラスブール | シグマ-1受容体リガンド及びその使用 |
| CN116354961A (zh) * | 2021-12-27 | 2023-06-30 | 中国科学院上海药物研究所 | 生长抑素受体5拮抗剂及其药物组合物及用途 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5356906A (en) | 1989-10-27 | 1994-10-18 | The Du Pont Merck Pharmaceutical Company | (N-phthalimidoalkyl) piperidines useful as treatments for psychosis |
| NZ522773A (en) | 2000-06-12 | 2005-06-24 | Eisai Co Ltd | 1,2-dihydropyridine compounds, manufacturing method thereof and use thereof |
| GB0129260D0 (en) | 2001-12-06 | 2002-01-23 | Eisai London Res Lab Ltd | Pharmaceutical compositions and their uses |
| BRPI0408332A (pt) | 2003-03-14 | 2006-03-21 | Ono Pharmaceutical Co | derivados heterocìclicos contendo nitrogênio e medicamentos contendo os mesmos como ingrediente ativo |
| AU2004289690A1 (en) | 2003-11-10 | 2005-05-26 | Schering Aktiengesellschaft | Benzylether amine compounds useful as CCR-5 antagonists |
| CN101506165B (zh) * | 2006-08-15 | 2012-07-04 | 霍夫曼-拉罗奇有限公司 | 苯基、吡啶和喹啉衍生物 |
| US7862825B2 (en) | 2007-02-21 | 2011-01-04 | Mladen Vranic | Method of controlling tight blood glucose by somatostatin receptor antagonists |
| US7799806B2 (en) | 2007-04-04 | 2010-09-21 | Hoffmann-La Roche Inc. | Substituted n-benzyl piperidines as somatostatin receptor modulators |
| US20080306116A1 (en) | 2007-06-08 | 2008-12-11 | Christ Andreas D | Aryloxazole, aryloxadiazole and benzimidazole derivatives |
| TW200918062A (en) | 2007-09-12 | 2009-05-01 | Wyeth Corp | Azacyclylisoquinolinone and-isoindolinone derivatives as histamine-3 antagonists |
| CA2759651A1 (en) | 2009-05-07 | 2010-11-11 | Merck Sharp & Dohme Corp. | Substituted spirocyclic amines useful as antidiabetic compounds |
| WO2012024183A1 (en) | 2010-08-18 | 2012-02-23 | Merck Sharp & Dohme Corp. | Spiroxazolidinone compounds |
| EP2643311A1 (en) | 2010-11-26 | 2013-10-02 | Lupin Limited | Bicyclic gpr119 modulators |
| WO2014142363A1 (en) | 2013-03-14 | 2014-09-18 | Takeda Pharmaceutical Company Limited | Spiro azetidine isoxazole derivatives and their use as sstr5 antagonists |
-
2014
- 2014-10-28 AR ARP140104043A patent/AR098215A1/es active IP Right Grant
- 2014-10-28 TR TR2019/01986T patent/TR201901986T4/tr unknown
- 2014-10-28 UY UY0001035801A patent/UY35801A/es not_active Application Discontinuation
- 2014-10-28 TW TW103137143A patent/TWI668214B/zh active
- 2014-10-28 SI SI201431055T patent/SI3063139T1/sl unknown
- 2014-10-28 RS RS20190190A patent/RS58321B1/sr unknown
- 2014-10-28 DK DK14800152.2T patent/DK3063139T3/en active
- 2014-10-28 EP EP14800152.2A patent/EP3063139B1/en active Active
- 2014-10-28 PL PL14800152T patent/PL3063139T3/pl unknown
- 2014-10-28 WO PCT/JP2014/005438 patent/WO2015064083A1/en active Application Filing
- 2014-10-28 PT PT14800152T patent/PT3063139T/pt unknown
- 2014-10-28 US US14/525,593 patent/US9120777B2/en active Active
- 2014-10-28 HU HUE14800152A patent/HUE041792T2/hu unknown
- 2014-10-28 JP JP2016526960A patent/JP6450755B2/ja active Active
- 2014-10-28 LT LTEP14800152.2T patent/LT3063139T/lt unknown
- 2014-10-28 ES ES14800152T patent/ES2710657T3/es active Active
-
2019
- 2019-01-21 HR HRP20190133TT patent/HRP20190133T1/hr unknown
- 2019-02-14 CY CY20191100202T patent/CY1121279T1/el unknown
-
2022
- 2022-12-29 AR ARP220103645A patent/AR128158A2/es unknown
Also Published As
| Publication number | Publication date |
|---|---|
| TW201542539A (zh) | 2015-11-16 |
| TR201901986T4 (tr) | 2019-03-21 |
| WO2015064083A1 (en) | 2015-05-07 |
| RS58321B1 (sr) | 2019-03-29 |
| EP3063139B1 (en) | 2018-11-21 |
| PT3063139T (pt) | 2019-02-22 |
| AR128158A2 (es) | 2024-03-27 |
| JP6450755B2 (ja) | 2019-01-09 |
| AR098215A1 (es) | 2016-05-18 |
| SI3063139T1 (sl) | 2019-03-29 |
| LT3063139T (lt) | 2019-02-25 |
| UY35801A (es) | 2015-06-30 |
| DK3063139T3 (en) | 2019-03-11 |
| CY1121279T1 (el) | 2020-05-29 |
| HUE041792T2 (hu) | 2019-05-28 |
| JP2016535040A (ja) | 2016-11-10 |
| TWI668214B (zh) | 2019-08-11 |
| US20150119412A1 (en) | 2015-04-30 |
| HRP20190133T1 (hr) | 2019-03-22 |
| EP3063139A1 (en) | 2016-09-07 |
| PL3063139T3 (pl) | 2019-05-31 |
| US9120777B2 (en) | 2015-09-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6529649B2 (ja) | 縮合複素環化合物 | |
| ES2687102T3 (es) | Antagonistas del receptor de somatostatina subtipo 5 (SSTR5) | |
| ES2710657T3 (es) | Compuesto heterocíclico | |
| IL269347B2 (en) | ip6k inhibitors | |
| US10017487B2 (en) | Fused heterocyclic compound | |
| ES2629729T3 (es) | Derivados de espiro azetidina asoxazol y uso de los mismos como antagonistas de sstr5 | |
| WO2016121782A1 (ja) | スルホンアミド化合物 |