ES2706023T3 - Administración de combinaciones de fármacos - Google Patents
Administración de combinaciones de fármacos Download PDFInfo
- Publication number
- ES2706023T3 ES2706023T3 ES08788693T ES08788693T ES2706023T3 ES 2706023 T3 ES2706023 T3 ES 2706023T3 ES 08788693 T ES08788693 T ES 08788693T ES 08788693 T ES08788693 T ES 08788693T ES 2706023 T3 ES2706023 T3 ES 2706023T3
- Authority
- ES
- Spain
- Prior art keywords
- drug
- beads
- doxorubicin
- water
- polymer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC[C@](C)(C(C1CO2)C=C(c(c(C3)c4*)nc5c4c(*)c(*)c([Rn])c5)N3C1=O)C2=O Chemical compound CC[C@](C)(C(C1CO2)C=C(c(c(C3)c4*)nc5c4c(*)c(*)c([Rn])c5)N3C1=O)C2=O 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Materials For Medical Uses (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Una composición que comprende una población de microesferas, cada una de las cuales tiene una matriz polimérica e, incorporado conjuntamente dentro de la matriz, un primer o un segundo compuesto activo farmacéuticamente, donde el polímero es un polímero no hidrosoluble e hinchable en agua que está aniónicamente cargado, donde el primer activo está catiónicamente cargado, y el segundo activo es electroestáticamente neutral y donde uno de los activos es un compuesto citotóxico, mientras que el otro activo posee actividad complementaria con el compuesto citotóxico en el tratamiento tumoral
Description
DESCRIPCIÓN
Administración de combinaciones de fármacos
La presente invención se refiere a elementos de suministro de fármacos, normalmente microesferas, formados de una matriz polimérica y de, al menos, dos fármacos, siendo estos liberables de la matriz polimérica. Los fármacos son para el tratamiento del cáncer, especialmente de tumores sólidos.
La politerapia ha sido la base para la mayoría de los desenlaces favorables en el tratamiento del cáncer, algo comprensible ya que los componentes de una combinación tienen una interacción farmacológica favorable (idéntica diana, aunque diferentes toxicidades en órganos grandes). La politerapia con múltiples fármacos consiste en un procedimiento clínicamente habitual para el tratamiento tumoral. Los tratamientos terapéuticos más habituales para el cáncer de ovario, útero, o próstata se basan en la combinación de dos fármacos antitumorales. Este tipo de terapia podría ofrecer la ventaja de aumentar la eficacia tumoricida y de reducir los efectos secundarios provocados por la alta dosis de un solo fármaco. El irinotecán está indicado en combinación con 5-FU para el tratamiento de pacientes con cáncer colorrectal avanzado y ha sido usado con éxito en combinación con otros agentes tales como epirubicina, mitomicina o capecitabina para tratar el cáncer de mama (Becerra 2004, Oncology, 18, 46-48), el cáncer de ovario (Nishino y col. 2005, Gynecol Oncol, 97, 893-897) o el cáncer gástrico (Baek y col. 2006, Br J Cancer, 25, 312-320) respectivamente.
Un cimiento más reciente para el éxito de algunos tratamientos para el cáncer implica una base mecanicista, según la cual la combinación de fármacos se usa para influir en diferentes dianas moleculares con objeto de evitar la resistencia. Una barrera para los avances más importantes en la politerapia ha sido, no obstante, la falta de comprensión al respecto de la intersección de vías de señalización clave. La sinergia podría inducirse a través del efecto de los fármacos sobre estas, además de sobre las vías paralelas. Debido a que el número de combinaciones de fármacos es ilimitado, una estrategia para determinar las combinaciones más prometedoras, además de la priorización de su evaluación, resulta crucial.
Los modelos matemáticos y las simulaciones informáticas se usan para calcular el efecto esperado de diferentes combinaciones de fármacos en un esfuerzo para entender mejor cómo podrían usarse clínicamente dichas combinaciones quimioterapéuticas. Las combinaciones favorables de fármacos sinergéticos in vivo pueden resultar en un aumento de la eficacia, una disminución de la dosis, una reducción de la toxicidad, así como en un desarrollo mínimo de resistencia. La ecuación del efecto mediana (Chou J. Biol. Chem. 1977, 252(18), 6438-6442) y la ecuación del índice de combinación (Chou-Talalay T.I.P.S. Nov. 1983450-454) consisten en un par de tratamientos cuyas curvas de isobolograma generadas representan interacciones aditivas, sinergéticas o antagonísticas provocadas por diferentes combinaciones de fármaco. El concepto subyacente y las aplicaciones de modelos de respuesta de superficie abarca el efecto conjunto de dos agentes activos que pueden verse como una superficie de respuesta tridimensional. Mientras que estos modelos para la evaluación de acción conjunto son los más integrales, también son altamente complejos y difíciles de crear. De hecho, se ha reconocido que el vínculo real entre la sinergia empírica y los mecanismos fisiológicos, moleculares y bioquímicos subyacentes resulta difícil de desentrañar.
Un sistema de alto rendimiento basado en fluorescencia, denominado ensayo de citotoxicidad basado en microscopía de imagen digital (DIMSCAN), existe para las pruebas in vitro de combinaciones de fármacos citotóxicos o citostáticos. Hay cinco principios para este tipo de pruebas: (a) el sistema de ensayo debería contar con un intervalo dinámico amplio, idealmente de 3 a 4 registros de destrucción celular; (b) el panel de línea celular debería emplear múltiples líneas celulares, lo que incluye líneas resistentes a fármacos; (c) los mecanismos principales de resistencia deberían identificarse y usarse para estructurar el panel de línea celular; (d) la exposición a los fármacos debería estar en niveles y programaciones clínicamente accesibles; y (e) debido a que la hipoxia puede antagonizar la acción del fármaco, es esencial realizar la prueba en condiciones hipóxicas. El mejor enfoque para seleccionar combinaciones de fármacos para su evaluación consiste en uno basado en dianas moleculares; los estudios deberían primero realizarse in vitro para, después, confirmarse con modelos in vivo antes de las pruebas clínicas de dicha combinación de fármacos. El antagonismo de fármacos también se usa frecuentemente para excluir las combinaciones de fármacos.
Existen conocidas interacciones sinergéticas entre moduladores novedosos de transducción de señal y agentes citotóxicos establecidos, inhibidores del ciclo celular, y los inductores de diferenciación. Mientras que el primer tipo de combinación de fármacos implica concentraciones tóxicas del agente citotóxico para producir una modesta potenciación in vitro, el segundo y tercer tipo inducen un alto grado de sinergismo a concentraciones en las que ambos agentes resultan no tóxicos (inefectivos) al tomarse individualmente. Las combinaciones de fármacos entre inhibidores de ciclo celular o estos agentes y los inductores de diferenciación también han demostrado producir una actividad sinergética significante in vitro. El número de combinaciones de moduladores de transducción de la señal
asociado con interacciones sinergéticas de muerte celular es virtualmente ilimitado. Si estas combinaciones son terapéuticamente superiores a aquellas entre agentes citotóxicos convencionales o a aquellas entre agentes novedosos y convencionales o si estas representan un nuevo paradigma para la poliquimioterapia es algo que queda por determinar.
Celator Technologies, Inc., en Canadá, ha informado de soluciones comerciales para llevar efectivas combinaciones de fármacos al ámbito clínico. Celator está trabajando en la identificación de índices cruciales en los que las combinaciones de fármacos actúan sinergéticamente para destruir las células tumorales, con el objetivo de fijar estos índices sinergéticos en un vehículo de administración basado en liposomas (CombiPlex) y diseñado para dirigirse a los tumores después de la inyección IV. Aquí, mientras que la actividad sinergética in vitro depende de índices de fármacos específicos, la actividad in vivo se basa en mantener estos índices sinergéticos. Los fármacos se incorporan de manera conjunta en los liposomas. Sus sistemas se han descrito en publicaciones de patentes, tales como WO2003028696, WO2004087115, WO2004087105, WO2004093795, WO2006055903,y en Tardi, P. G.
y col., (2007) Biochim. Biophys. Acta 1768, 678-587.
Las microesferas ofrecen una plataforma particularmente versátil para la administración de combinaciones de fármacos. Sharma y col. han investigado con microesferas para inhalación de combinaciones de fármacos en el tratamiento de la tuberculosis (Sharma R, y col., 2001, Pharm Research, 18, 1405-1410). La terapia farmacológica para la tuberculosis (TB) requiere de administración oral a largo plazo de múltiples fármacos para curar aparte de para prevenir y/o combatir la resistencia a multifármacos. Los niveles persistentes y altos de fármacos antituberculosos en sangre causados por una administración oral y prolongada de fármacos anti-TB pueden no ser necesarios ni suficientes para destruir la micobacteria que reside en macrófagos (M9). Las micropartículas biodegradables e inhalables que contienen dos o más fármacos anti-TB de primera línea, isoniazida (H), y rifampicina (R), fueron preparadas y analizadas para (i) fagocitosis por M9 de ratón (ii) administración como inhalación de polvo seco en ratas, y (iii) como diana de M9 alveolar con altas dosis de fármaco al administrarse en ratas. Los resultados demostraron que dichas microesferas con múltiples fármacos son prometedoras para una reducción tanto de dosis como de la misma frecuencia de dosis, en el alivio de la toxicidad, y con la diana en micobacterias persistentes y residentes en M9.
En otro estudio (Gupte A & Ciftci K, 2004, Int. J. Pharm., 276(1-2):93-106.), la combinación (microesferas de Paclitaxel 5-FU) se comparó con quimioterapia de fármaco único (microesferas de Paclitaxel y 5-FU) contra la línea celular de cáncer de mama metastásico (MDA-MB 435 S). Se estudiaron las características fisicoquímicas de las microesferas (es decir, eficiencia de encapsulación, distribución del tamaño de partícula, liberación in vitro, y características termales). Los resultados demostraron que la eficiencia de encapsulación del Paclitaxel fue alta (90
%) cuando el fármaco se encapsuló en micropartículas de ácido poli(láctico-co-glicólico) (PLGA) con o sin 5-fluorouracilo (5-FU). No obstante, la eficiencia de encapsulación del 5-FU resultó baja (19 %) y aumentó en un 30 % cuando el fármaco se encapsuló con Paclitaxel. El tamaño medio de partícula de las microesferas fue de 2,5 pm, siendo esféricas en forma. La liberación in vitro de tanto el 5-FU como del Paclitaxel de las microesferas resultó relativamente rápida al inicio, a lo que siguió una liberación más lenta y controlada. La actividad citotóxica de las microesferas de Paclitaxel fue mucho mayor al compararla con las microesferas que contenían 5-FU Paclitaxel o solamente 5-FU. Los resultados globales demostraron que la incorporación de Paclitaxel o 5-FU en las microesferas mejora la citotoxicidad de forma más controlada al compararla con la de los fármacos libres y que, además, debería prestarse una atención especial al combinar los fármacos que actúan en diferentes fases del ciclo celular.
También se han investigado combinaciones de fármacos en las microesferas dirigidas a restenosis (Chandy y col., 2001, Development of Poly(Lactic Acid)/Chitosan Co-Matrix Microspheres: Controlled Release of Taxol-Heparin for Preventing Restenosis, Drug delivery, 8, 78-86). La proliferación de células del músculo liso juega un importante papel en la génesis de la restenosis después de una lesión vascular o angioplastia. La liberación controlada de fármacos apropiados aislados y en combinaciones representa un enfoque a la hora de tratar las obstrucciones coronarias, la angioplastia con balón, la restenosis asociada a la trombosis, además de la calcificación. Chady y col., demostraron la posibilidad de encapsular microesferas de ácido poliáctico (PLA) cargadas con taxol dentro de esferas de heparina-quitosano para desarrollar una forma comatricial de liberación prolongada. El perfil de liberación in vitro del taxol y la heparina a partir de este sistema comatricial fue supervisado en solución salina tamponada con fosfato con un pH de 7,4 y usando un espectrofotómetro ultravioleta. La cantidad de taxol/heparina liberada fue inicialmente mucho más elevada para después continuar con un perfil de liberación lenta durante un período prolongado. La liberación repentina inicial de taxol (15,8 %) y heparina (32,7 %) de la comatriz fue modificada con recubrimientos de polietilénglicol (13,5 % y 25,4 % respectivamente, durante 24 h). A partir de estudios sobre microscopía electrónica de barrido, parece que estos fármacos salen lentamente al medio de disolución a través de microporos en la comatriz. No obstante, los microporos de la superficie se modificaron con recubrimientos de polietilénglicol (PEG) para un perfil de liberación lento y continuado. Se concluyó que esta comatriz de PLA/quitosano recubierta de PEG puede orientarse a las combinaciones de fármacos con efectos sinergéticos durante períodos prolongados para tratar la restenosis.
Se han descrito las endoprótesis cardiovasculares recubiertas con una combinación de fármacos para reducir la tasa de restenosis. La restenosis es una enfermedad hiperproliferativa. Por ejemplo, la EP-A-551162 describe una combinación de rapamicina y ácido micofenólico, un agente antiproliferativo, aunque no da detalles de cómo una endoprótesis debe impregnarse con los activos. En la EP-A-0568130, se administran rapamicina y heparina para prevenir la hiperplasia del músculo liso, p. ej., mediante la administración a partir de una endoprótesis impregnada. No se proporcionan detalles sobre los procedimientos de impregnación.
La WO-A-2003022807 describe la administración de un análogo de rapamicina desde una endoprótesis recubierta por un polímero. El fármaco se carga sumergiendo la endoprótesis recubierta de polímero en una solución del fármaco y secándolo. Además, un fármaco adicional como un agente antimiótico, antiproliferativo o antiinflamatorio puede administrarse desde la endoprótesis. Los polímeros incluyen, entre otros muchos, alcohol polivinílico, ácido hialurónico y copolímeros de metacrilato de fosforicolina (MPC), comonómeros no iónicos y monómeros de entrecruzamiento, siendo estos últimos copolímeros mencionados como ejemplo en pruebas in vitro, con un análogo de rapamicina único. En la WO2004/022124 un análogo de rapamicina y dexametasona son cargados conjuntamente en un copolímero de MPC entrecruzado con comonómero no iónico en una endoprótesis. El polímero usado en estas especificaciones tiene una hinchabilidad en agua de aproximadamente el 50 %.
El 5-Fluorouracilo (5-FU) en combinación con leucovorina (LV) supone actualmente el tratamiento estándar para el cáncer de colon. Lamprecht y col. (Eur J Pharm and Biopharm, 2005, 59, 367-371) usaron Eudragit P-4135F o Eudragit RS100 separadamente para preparar microesferas mediante un proceso de emulsificación aceite/aceite que atrapó 5-FU y LV simultáneamente. La microscopía electrónica de barrido permitió un análisis estructural, los parámetros del proceso se analizaron y se registraron los perfiles de liberación y carga del fármaco. El tamaño de partícula varió entre 123 (RS100) y 146 pm (P-4135F). En general, las tasas de encapsulación más elevadas se encontraron con RS100 (5-FU, 60,3±9,7 %; LV, 81,4±8,6 %) en comparación con P-4135F (5-FU, 48,3±2,0 %; LV, 55,4±2,7 %). Estos polímeros de Eudragit son copolímeros acrílicos que incluyen un grupo amonio cuaternario que contiene monómeros de metacrilato de alquilo. No están entrecruzados. Las micropartículas hechas de Eudragit RS100 liberaron la combinación de fármacos incorporada en 8 h. Se halló que P-4135F mantuvo la liberación de 5-FU no deseada con un pH de 6,8 inferior al 25 % en 4 h., mientras que con un pH de 7,4, se observó una liberación casi inmediata (en 15 min). Aunque la liberación fue similar con un pH de 7,4, la LV con un pH del 6,8 mostró una pérdida de fármaco inicial distintiva de aproximadamente el 60 %, así como una liberación completa en 2 h. Los análisis de MEB revelaron una presencia sustancial de cristales de LV en la superficie de la partícula, lo que provocó un efecto repentino distintivo de LV. Se concluyó que estas observaciones se relacionaban con la alta lipofilicidad de P-4135F provocando una separación entre P-4135F y LV durante el proceso de preparación.
Liu y col. (J. Pharm. Pharmacol., 55, 1063-1073, 2003) y en la WO9850018 se describe el uso de microesferas de dextrano de sulfopropilo biodegradables para una inyección intratumoral. En el procedimiento las microesferas que contienen doxorrubicina se administran juntamente con microesferas que contienen verapamilo. Después de la inyección intratumoral en ratones con microesferas cargadas de doxorrubicina, aisladas o en combinación con microesferas cargadas de verapamilo, se midió el diámetro del tumor en serie como indicación del efecto terapéutico, mientras que el peso, la apariencia, y el comportamiento de los ratones fue supervisado como una indicación de la toxicidad general. Las inyecciones intratumorales de microesferas cargadas de doxorrubicina fueron toleradas mucho mejor que la administración sistémica de concentraciones de fármaco equivalentes. Se dio un modesto retraso (hasta un 34 %) en el crecimiento tumoral en comparación con grupos que no recibieron tratamiento o bien microesferas vacías. La inyección conjunta de microesferas de verapamilo con microesferas de doxorrubicina produjo un aumento moderado en la toxicidad pero no un retraso adicional del crecimiento tumoral. Las formulaciones de microesferas de liberación controlada de agentes antineoplásicos administrados intratumoralmente se describieron como una forma eficiente de administrar altas dosis de fármaco en el tumor con poca toxicidad sistémica. Las microesferas de dextrano sulfopropiladas usadas, se habían suministrado como medios de intercambio de iones (Sephadex SP25). Estas son biodegradables. Son hinchables en agua destilada para otorgar una proporción de volumen de microesferas en el estado hinchado en relación con el estado seco de aproximadamente 12. Las microesferas tienen una capacidad de intercambio de iones de aproximadamente 2,5 meq/g con un pH de 7.
Liu y col. en (2000) J. Pharm. Sci: 8-9(6) 807-817 también han descrito formas de modificar la superficie de las microesferas sulfopropiladas con restos hidrofóbicos o con polímeros catiónicos. La modificación del polímero catiónico reduce el índice de hinchabilidad y la capacidad de intercambio de iones. La conjugación de la superficie de grupos hidrofóbicos afecta a la tasa de hinchabilidad en agua, aunque no reduce el alcance de hinchabilidad en el verapamilo en equilibrio cargado a una tasa más rápida dentro de las microesferas tratadas con polímero.
Las perlas eluyentes de doxorrubicina han demostrado ser efectivas en el tratamiento del CHC por quimioembolización (Varela y col., 2007, J Hepatology, 46, 474-81). Las microesferas para administración de
doxorrubicina en tumores sólidos mediante quimioembolización se describen en WO2004071495. Las microesferas están formadas de una matriz de alcohol polivinílico entrecruzado, cargada aniónicamente e hinchable en agua y poseen diámetros al hincharse en agua en el intervalo de 40 a 1500 pm.
Las perlas eluyentes de ibuprofeno han demostrado efectos antiinflamatorios tras la embolización en modelos animales (Borovac T, y col., 2006, J Control Release, 115, 266-74). Las microesferas para uso en el tratamiento de fibromas uterinos cargadas con ibuprofeno se describen en WO2004073688. Se describen procedimientos de carga mejorados en WO2007085615.
El ibuprofeno es un fármaco antiinflamatorio no esteroideo (AINE), que actúa por inhibición de las enzimas ciclooxigenasa 1 y 2. Muchos autores han estudiado el uso de los AINE para el tratamiento y prevención del riesgo de cáncer (Sawaoka y col. 1998, J Clin Gastroenterol, 27, 47-52; Shen y col. 1998, Cancer Research, 58, 362-366). Los fármacos AINE selectivos de la COX-2 también han demostrado una reducción en la invasión de células cancerígenas y metástasis hepáticas (Yamauchi y col. 2003, Anticancer Res, 23, 245-249; Yao y col. 2004, Br J Cancer, 90, 712-719) y, además, la inhibición de la COX-2 provocó una reducción del tamaño del tumor y un aumento en la supervivencia (Cui y col. 2005, Clin Cancer Res, 11, 8213-8221; Murata y col. 2005, Dig Dis Sci, 50, 70-75). El ibuprofeno se ha investigado en el tratamiento y prevención de un número de tumores. En WO2006013376 describimos el uso de microesferas cargadas con varios inhibidores de la COX para embolización que muestra tasas de elución y localización llevando a una expectativa de poder mostrarse algún efecto en la génesis tumoral.
En la WO2007090897, describimos procedimientos de carga de polímeros hinchables en agua, tales como microesferas, con fármacos que poseen una solubilidad muy baja, pero que son solubles en un disolvente orgánico, en el que una solución con base en disolvente del fármaco entra en contacto con la matriz polimérica sustancialmente seca, para que tenga lugar la hinchabilidad de la matriz en la solución del fármaco, seguido de precipitación del fármaco en la matriz del polímero mediante contacto con el precipitante, normalmente agua. El procedimiento es adecuado para cargar paclitaxel, dexametasona o rapamicina. La matriz polimérica se compone de un material basado en alcohol polivinílico entrecruzado, normalmente con grupos aniónicos dependientes, tales como grupos sulfonato.
En WO2006027567 describimos microesferas para la quimioembolización de tumores sólidos que comprenden un polímero hinchable en agua que está aniónicamente cargado con un pH de 7 y, electroestáticamente asociado con el polímero en forma liberable, un compuesto de camptotecina, cargado catiónicamente, tal como irinotecán. Hemos mostrado que la quimioembolización que usa estas perlas para el tratamiento de metástasis hepáticas a partir de perlas de cáncer colorrectal lleva a una reducción del volumen del tumor.
En WO 2004/071495 describimos una composición para quimioemboterapia de tumores sólidos que comprende partículas de un polímero aniónico, sintético, hinchable en agua y no hidrosoluble y, absorbido en ello, una antraciclina. Preferentemente, el polímero es un polímero basado en alcohol poli(vinílico) y el fármaco es doxorrubicina.
De acuerdo con un primer aspecto de la presente invención, se proporciona una composición que comprende una población de microesferas, cada una de las cuales tiene una matriz polimérica e, incorporado conjuntamente dentro de la matriz, un primer o un segundo compuesto activo farmacéuticamente, donde el polímero es un polímero no hidrosoluble e hinchable en agua que está aniónicamente cargado, el primer activo está catiónicamente cargado y el segundo activo es electroestáticamente neutral, y uno de los activos es un compuesto citotóxico, mientras que el otro activo posee actividad complementaria con el compuesto citotóxico en el tratamiento tumoral.
En este aspecto de la invención, el primer activo cargado catiónicamente puede ser el compuesto citotóxico. Alternativamente, el segundo activo puede ser el compuesto citotóxico, mientras que el primer activo catiónico posee actividad complementaria con el compuesto citotóxico en el tratamiento tumoral. Los activos cargados iónicamente tienden a ser hidrosolubles a un nivel relativamente alto, por ejemplo a un nivel de al menos 50 g/l, preferentemente al menos a 100 g/l. El segundo activo, el cual es electroestáticamente neutral, puede ser menos hidrosoluble, por ejemplo teniendo una baja hidrosolubilidad, menos de 10 g/l a temperatura ambiente. Hemos descubierto que para la combinación de fármacos no iónicos y catiónicos, hay poca o ninguna interferencia con la tasa de liberación o carga de cada activo con respecto al otro. Este resultado es sorprendente y permite el sencillo cálculo de dosis y proporciones adecuados de composición que se ven optimizados en eficacia. La composición contendrá partículas de un solo tipo evitando problemas de separación causando incertidumbres en la dosis.
El segundo activo posee una actividad que es complementaria con el compuesto citotóxico para el tratamiento tumoral. El compuesto puede en sí mismo ser eficaz en el tratamiento de tumores, por ejemplo es capaz de prevenir el crecimiento o reducir el tamaño de un tumor, en el caso, por ejemplo, de administrarse local o sistémicamente. El
activo puede ser, opcionalmente, un compuesto que vuelva sensible al tumor ante la actividad del primer activo. En general, la combinación de compuestos puede identificarse con una que cuenta con un índice de combinación basado en el índice de Chou-Talalay (1983, op. cit), de menos de 0,90, más preferentemente menos de 0,5, por ejemplo menos de 0,3, incluso tan bajo como 0,1 o por debajo.
Los valores para el índice de combinación son determinados, convenientemente, mediante el paquete de software Calcusyn (Biosoft), que se trata de un programa de análisis del efecto de una dosis utilizando la ecuación general para las relaciones dosis-efecto derivadas por Chou:
fa/fu = (D/Dm)m Ec. 1.
Donde D: la dosis del fármaco
Dm: la dosis mediana-efecto que significa la potencia. Se determina a partir de la intercepción de x de la gráfica mediana-efecto.
fa : la fracción afectada por la dosis
fu: la fracción no afectada, fu=1-fa
m: un exponente que significa la sigmoicidad (forma) de la curva dosis-efecto. Se determina mediante la pendiente de la gráfica mediana-efecto.
Las formas alternativas de la ecuación mediana-efecto son:
fa = 1/[1+(Dm/D)m] Ec. 2.
D =Dm[fa/<1-fa)]1/m Ec. 3.
A partir de la ecuación 2, si Dm y m son conocidas, el efecto (fa) puede determinarse para cualquier dosis (D).
A partir de la ecuación 3, si Dm y m son conocidas, la dosis (D) o (Dx) puede determinarse para cualquier efecto (fa). Así, los parámetros de Dm y m que representan la potencia y la forma, respectivamente, determinan la curva de dosis-efecto completa.
La gráfica mediana-efecto es una gráfica de x = log (D) frente a y = log (fa/fu) presentado por Chou en 1976 (Chou J. Th. Biol. 59, 253-276). Se basa en la formula logarítmica de la ecuación mediana-efecto de Chou [Ecuación 1]:
log (fa/fu) = m log (D) - m log (D„ Ec. 4
La ecuación 4 tiene la forma de una línea recta y = mx b.
Así, la pendiente produce el valor de m y la intercepción de x produce el valor del log (Dm), y así, el valor de Dm. La Dm también puede determinarse con: Dm = 10-(intercepci° n de y>/m>.
Nótese que fa/fu = fa/(1-fa) = (fu)-1-1 = [(fa)' 1-1]' 1. La bondad de ajuste para los datos en la ecuación mediana-efecto se representa mediante el coeficiente de correlación lineal r de la gráfica mediana-efecto. Normalmente, los datos experimentales a partir de los sistemas del receptor o enzima tienen una r > 0,96, a partir de un cultivo tisular una r > 0,90; y a partir de sistemas animales, una r > 0,85.
La ecuación del índice de combinación (IC) se basa en la ecuación múltiple fármaco-efecto de Chou-Talalay derivada de modelos cinéticos de enzima. Una ecuación determina únicamente el efecto aditivo más que el sinergismo o antagonismo. Sin embargo, definimos sinergismo como un efecto aditivo mayor a lo esperado, mientras que antagonismo como un efecto aditivo menor a lo esperado. Chou y Talalay en 1983 (op. cit) propusieron la designación del IC = 1 como el efecto aditivo, con lo que a partir de la ecuación múltiple fármaco-efecto de dos fármacos, obtenemos:
Una forma clásica: para fármacos mutuamente excluyentes que tienen el mismo o similar modo de acción:
CI = (D )i/(D x)i (D )2/(D x)2 Ec. 5 Una forma conservadora: para fármacos no excluyentes mutuamente que tienen modos de acción totalmente independientes:
Cl = (D)i/(Dx)i (D)2/(Dx)2 (D) i (D)2/(Dx) i (Dx)2 Ec.6
La ecuación 5 o la ecuación 6 dicta que el fármaco 1, (D)i , y el fármaco 2, (D)2, (en los numeradores) en combinación inhiben x% en el experimento presente. Así, la inhibición de x% experimentalmente observada no puede ser un número redondo sino que con la máxima frecuencia tendrá una fracción decimal. (Dx)1 y (Dx)2 (en los denominadores) de la ecuación 5 o 6 corresponden a las dosis del fármaco 1 y el fármaco 2 aislados, respectivamente, inhibiendo x%. Dx puede calcularse fácilmente a partir de la ecuación 3.
Merece la pena indicar que cada fármaco aislado debería tener una relación dosis-efecto. Ambos parámetros de potencia (Dm) y forma (m) son esenciales para determinar el sinergismo o antagonismo. Si uno de los dos fármacos no tiene efecto por sí mismo, entonces no podrá determinarse el sinergismo/antagonismo. En su lugar, podrán determinarse la potenciación (incremento, mejora) o la inhibición (supresión).
Calcusyn necesita de la relación dosis-efecto para cada fármaco y, por ello, para las combinaciones. Aunque los datos experimentales del estudio combinado se recogen preferentemente usando una proporción constante, los índices de combinación pueden, no obstante, determinarse.
El índice de combinación puede determinarse para cualquier procedimiento de prueba in vitro o in vivo. En la presente invención, la forma más conveniente de determinar el índice de combinación in vitro es usando una prueba basada en un cultivo celular, por ejemplo en una línea celular de células que representen una prueba in vitro válida para determinar la eficacia in vivo. Por ejemplo, la prueba puede determinar la citotoxicidad de los fármacos individuales y, después, de su combinación, siendo estos datos usados para calcular el índice de combinación. En la prueba, los fármacos pueden entrar en contacto con las células en la solución, o en alguna otra forma. El efecto que se medirá es la citotoxicidad, tal como una reducción en el conteo de células viables del 50 % en comparación con el control y después de un periodo dado tal como de 24 o 48 horas. No es necesario que los fármacos se incorporen dentro de la matriz polimérica, por ejemplo en forma de microesferas, con objeto de realizar la prueba. Sin embargo, se prefiere que el índice de combinación sea determinado para ser menor de 1 cuando los fármacos se administren desde la matriz polimérica, por ejemplo en forma de microesferas tal como se define en las reivindicaciones presentes. En este caso, la combinación de fármacos analizada puede implicar partículas separadas, cada una cargada con uno de los fármacos, o bien cada partícula puede estar cargada juntamente con ambos fármacos. Se describe una prueba adecuada en los Ejemplos 6 y 7 siguientes. Cuando las partículas separadas sean cargadas cada una con uno de los fármacos, deberá comprobarse que, para las partículas cargadas conjuntamente, la tasa de elución de cada fármaco no se vea afectada por la otra si se asume que el valor del IC determinado se aplicará al producto cargado conjuntamente. En este aspecto de la invención la combinación de compuestos activos farmacéuticamente es una que tiene un IC de menos de 1,0 en una prueba de citotoxicidad in vitro en una línea celular tumoral con una proporción dentro de un múltiplo de 5 (en todo caso) de la proporción en la composición. Cuando la composición sea para tratamiento de un carcinoma hepatocelular la línea celular usada para determinar el IC será preferentemente una línea celular proveniente de un carcinoma hepatocelular. Cuando la composición sea para tratar un carcinoma pancreático, la línea celular será preferentemente una línea celular proveniente de un carcinoma pancreático. El IC debería ser preferentemente el valor bajo definido cuando los compuestos activos se analicen con una proporción dentro de un múltiplo de 2 siendo, en todo caso, de la proporción en la composición.
Un agente citotóxico es uno que es tóxico directamente para las células, impidiendo su reproducción o crecimiento. Agentes citotóxicos adecuados son, por ejemplo, los compuestos de antraciclina tales como la doxorrubicina y otros compuestos descritos en el documento WO04071495, los derivados de la camptotecina, tal y como se describe en el documento WO2006027567, los taxanos, los antimetabolitos neoplásicos a base de platino tales como el 5-FU, la mercaptopurina, la capecitabina, otros antibióticos citotóxicos tales como la actinomicina D y los alcaloides de la vinca, entre los que se incluyen la vinblastina, vincristina, vindesina y vinorelbina. Ejemplos de citarabina, gemcitabina, ciclofosfamida, fludaribina, compuestos de camptotecina tales como topotecán e irinotecán, clorambucilo, busulfano, mitoxantrona, retinoides, anagrelida, etc.
El activo que proporciona actividad complementaria puede ser un compuesto citostático. Los compuestos citostáticos pueden definirse como compuestos que inhiben o suprimen la reproducción o el crecimiento celular. Los efectos citotóxicos y citostáticos de fármacos pueden determinarse por aquella persona experta en la técnica mediante el uso de pruebas adecuadas, como las basadas en líneas celulares.
Los compuestos citostáticos adecuados son rapamicina y análogos.
Compuestos adecuados cargados catiónicamente son: doxorrubicina y otros compuestos de antraciclina que poseen un grupo amino tal como se describe en el documento WO04071495 o mitoxantrona, derivados de camptotecina catiónica, tales como irinotecán, tal como se describe en el documento WO2006027567, o bien topotecán, o verapamilo.
Una clase de compuestos citotóxicos o citostáticos que se puede usar comprende la rapamicina y los análogos a la rapamicina que se dirigen a la mTOR. Tales compuestos incluyen sirolimús, temsirolimús, everolimús, biolimús, a BT-578 y AP23573. Cualquiera de los compuestos incluidos dentro del ámbito de los análogos de rapamicina descritos en el documento WO-A-2003022807, puede usarse como el análogo de rapamicina.
Cuando el activo citotóxico es rapamicina o un derivado/análogo de rapamicina, el otro activo es en una realización preferida, doxorrubicina u otro compuesto de antraciclina que posee un grupo amino tal y como se describe en el documento WO04071495.
La combinación de rapamicina y doxorrubicina como fármaco antineoplásico ha demostrado una remisión completa de un linforma de células B en un modelo de ratón a través de la inversión de la quimioresistencia en linfomas mediante la sensibilización de rapamicina, y la inducción de apoptosis mediante doxorrubicina (Wendel,H-G, y col., 2004, Nature 428, 332-337). En el tratamiento de leucemias la rapamicina ha mostrado la mejora de la apoptosis inducida por doxorrubicina (Avellino,R. y col., 2005, Blood 106(4), 1400-1406). Las combinaciones de rapamicina o un análogo con el compuesto de antraciclina, el cual tienen al menos un grupo amino, muestran un bajo índice de combinación, y resultan altamente efectivas en la presente invención.
En otra realización de este aspecto de la presente invención, el compuesto citotóxico es rapamicina o un análogo y el activo catiónico es un derivado de camptotecina que tiene un sustituyente catiónico tal como se describe en el documento WO2006027567 o en la EP-A-0321122. Preferentemente el activo es como se define en la reivindicación 6. Preferentemente A es COO. Preferentemente AR es sustituido en la posición 10.
De acuerdo con el primer aspecto de la presente invención, los dos fármacos se cargan conjuntamente en las mismas microesferas. Cuando el compuesto citotóxico es rapamicina o un análogo, el activo que tiene actividad complementaria puede, de forma alternativa, ser seleccionado de cisplatino, u otro fármaco antineoplásico a base de platino, gemcitabina, tamoxifeno o cualquier otro antiestrógeno, interferón alfa, inhibidor del receptor del factor de crecimiento endotelial, inhibidor del receptor del factor de crecimiento endotelial vascular, inhibidores de ambos VEGFR y EGFR, y Glivec. El fármaco adicional puede, de forma alternativa, ser paclitaxel o un análogo, u otro inhibidor de microtúbulos, o un inhibidor de topoisomerasa.
Las combinaciones de rapamicina con otros agentes antineoplásicos también son conocidas, y la combinación conocida descrita más adelante puede usarse en este aspecto de la invención. Cisplatino, gemcitabina, tamoxifeno, interferón alfa, el inhibidor de EGFR, el inhibidor de VEGFR/EGFR, Gleevec, todos han sido combinados con análogos de rapamicina en estudios clínicos y preclínicos, y estas combinaciones pueden usarse en el aspecto adicional de la invención.
Se han investigado las interacciones entre rapamicina y los agentes quimioterapéuticos usados como agentes de primera y segunda línea contra el cáncer de mama. Las interacciones sinergísticas, in vitro, se observaron en combinaciones con paclitaxel, carboplatino, y vinorelbina. Se observaron efectos aditivos en combinaciones con doxorrubicina y gemcitabina. La rapamicina incrementó marcadamente la apoptosis inducida por carboplatino y paclitaxel. Este efecto dependió de la secuencia y medió, al menos en parte, a través de la activación de caspasa. Además, la rapamicina incrementó la quimiosensibilidad al paclitaxel y al carboplatino en células que sobreexpresan HER2/neu, lo que sugiere un planteamiento potencial para estos tumores que actúan de forma inadecuada. La rapamicina, in vivo, combinada con paclitaxel resultó en una reducción significativa del volumen tumoral en comparación con cada agente aislado en los tumores sensibles a rapamicina. (Mondesire WH, et al., 2004, Clinical Cancer Research, 10, 7031-7042). La combinación de una rapamicina o análogo con un taxano puede usarse en el aspecto adicional de la invención.
RAD001 aumenta marcadamente la apoptosis inducida por cisplatino en la p53 de tipo silvestre, pero no en células tumorales de p53 mutante, RAD001 sensibiliza las células con respecto a cisplatino con la inhibición de la expresión de p21 inducida por la p53. Estos hallazgos apoyan la justificación molecular para la combinación de agentes perjudiciales para el ADN tales como el platino citotóxico con RAD001, mostrando que un proceso anabólico importante general puede aumentar la eficacia de un protocolo farmacológico establecido en el tratamiento de pacientes con tumores sólidos (Beuvink I y col. 2005, Cell, 120:747-759). La combinación de rapamicina o análogos con un compuesto antineoplásico de platino puede usarse en los aspectos adicionales de la invención.
CCI-779 y tamoxifeno u otro antiestrógeno pueden ser útiles si se administran de forma combinada. La vía PI3K/AKT/mTOR puede jugar un papel importante en el desarrollo de resistencia al tamoxifeno en el cáncer de mama. La activación del receptor de estrógeno puede impulsar la vía PI3K. Se ha mostrado que la interacción entre erb-1 y el ER (receptor de estrógeno) activa la vía, lo que ha sido asociado con la actividad transcripcional independiente de estrógenos y, además, las líneas celulares del cáncer de mama con AKT activado son resistentes
a los efectos inhibitorios de crecimiento del tamoxifeno. La combinación de rapamicina o un análogo con un antiestrógeno puede usarse en este aspecto adicional de la invención.
Existe un ensayo de fase III en curso que compara, en administración oral, el temsirolimús (CCI-779) más letrozol (un inhibidor de aromatasa) frente al letrozol aislado como tratamiento de primera línea entre aproximadamente 1200 mujeres postmenopáusicas con cáncer de mama avanzado. La combinación de rapamicina o un análogo con un inhibidor de aromatasa puede usarse en este aspecto adicional de la invención.
El CCI-779 y el interferón alfa han sido usados conjuntamente en el tratamiento del carcinoma de células renales (CCR). Está en marcha un extenso ensayo en fase III. La combinación de rapamicina o un análogo y un interferón alfa puede usarse en este aspecto adicional de la invención.
La rapamicina con el inhibidor EKI-785 del receptor de factor de crecimiento epidérmico (EGFR) resultó en una inhibición del crecimiento sinergística. Se obtuvieron resultados parecidos con RAD-001 y el inhibidor AEE788 de VEGFR/EGFR. La rapamicina con Glivec resultó en una inhibición del crecimiento sinergística de las líneas celulares de LMC. La combinación de rapamicina o un análogo con un receptor de factor de crecimiento o con Glivec puede usarse en este aspecto adicional de la invención.
Otras combinaciones usadas en el aspecto adicional de la invención incluyen agentes citotóxicos con inhibidores de la glicoproteína-P. Aproximadamente el 50 % de los pacientes con cáncer recibe quimioterapia, y tantos como el 75 % de estos pacientes experimentan una resistencia adquirida o intrínseca a un amplio espectro de agentes quimioterapéuticos. Este fenómeno, denominado resistencia a múltiples fármacos (RMF) es la causa más común del fracaso en la quimioterapia. Es bien sabido que la mayoría de tumores desarrollan RMF a través de la sobreexpresión de la glicoproteína (P-gp) de la bomba de expulsión del fármaco. Es conocida la inhibición de la P-gp como un medio viable de invertir esta RMF; sin embargo, los inhibidores de P-gp actuales hasta el momento han demostrado un éxito clínico limitado debido a sus limitaciones en potencia y especificidad. El verapamilo, un bloqueador de los canales de calcio, se usa habitualmente para el tratamiento de las arritmias supraventriculares, la enfermedad coronaria, y la hipertensión arterial. Es más, el verapamilo es un potente inhibidor del transporte mediado por la glicoproteína-P, y se ha demostrado que modifica la RMF en la quimioterapia contra el cáncer y en experimentos in vitro (Toffoli G, y col. 1995, Biochem Pharmacol, 50, 1245-55; Pauli-Magnus C, y col. 2000, J Pharmacol Exp Ther, 293, 376-82). La administración local de doxorrubicina y verapamilo a partir de perlas eluyentes cargadas conjuntamente puede, por ello, ofrecer beneficios sinergéticos, permitiendo altas dosis locales de agente citotóxico y la inhibición de P-gp para superar cualquier efecto de la r Mf . Así, la combinación de un antibiótico citotóxico y un inhibidor de P-gp puede usarse en el aspecto adicional de esta invención.
Otras combinaciones incluyen inhibidores de topoisomerasa, tales como tecanos con otros activos que posean un bajo índice de combinación.
Otras combinaciones tal como se describen por Celator en cualquiera de los documentos WO2003028696, WO2004087115, WO2004087105, WO2004093795, WO2006055903, y en Tardi, P. G. y col., (2007) Biochim. Biophys. Acta 1768, 678-587, cuyos contenidos se incorporan en el presente documento como referencia, pueden usarse en el aspecto adicional de la invención.
Los taxanos y antimetabolitos pueden usarse en combinación, por ejemplo, paclitaxel y 5-FU Pueden usarse 5FU y leucovorina.
Cuando los activos incluyen un derivado de camptotecina, resulta particularmente efectivo usar un derivado que posea sustituyentes catiónicos, tales como compuestos de la Fórmula general I
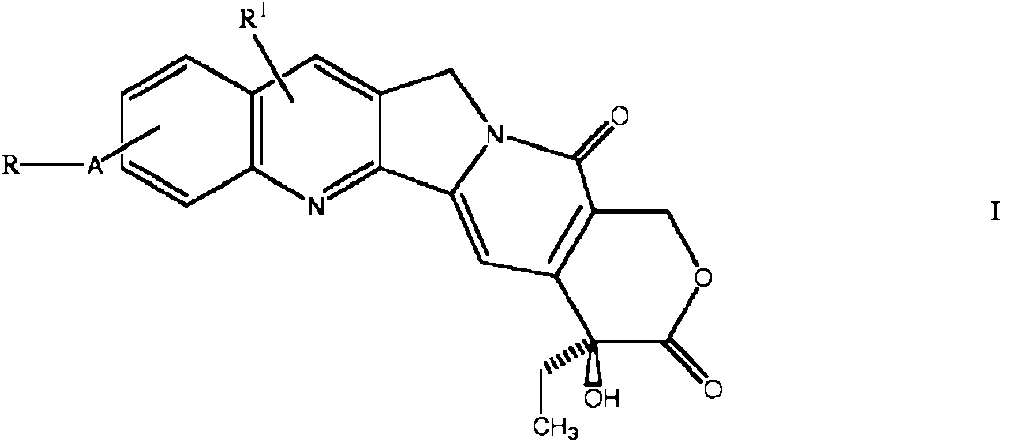
en la que R1 se selecciona de entre H, halógeno, hidroxilo y alquilo (C1-6) inferior, opcionalmente sustituido por un grupo hidroxilo, amino, alcoxi, halógeno, acilo y aciloxilo;
A es C(O)O o CH2; y
R es NR2R3 donde R2 y R3 son los mismos o diferentes y cada uno representa un átomo de hidrógeno, un grupo alquilo C1-4 sustituido o sin sustituir o un grupo hetercíclico o carbocíclico sin sustituir, o R2 y R3 junto con el átomo de nitrógeno al que están unidos forman un anillo heterocíclico sustituido que puede interrumpirse por -O-, -S- o >NR4 en el que R4 es un átomo de hidrógeno, un grupo alquilo C1-4 sustituido o sin sustituir o un grupo fenilo sustituido o sin sustituir.
y donde la agrupación -A-R está unida a un átomo de carbono situado en cualquiera de las posiciones 9, 10 u 11 en el anillo A del compuesto de camptotecina y R1 es sustituido en el anillo A o B, incluyendo las sales del mismo. En una realización los derivados de camptotecina tienen la fórmula IA

donde R1 es H, alquilo (C1-6) inferior, opcionalmente sustituido por un grupo hidroxilo, amino, alcoxi, halógeno, acilo y aciloxilo, por halógeno; y
R es NR2R3 donde R2 y R3 son los mismos o diferentes y cada uno representa un átomo de hidrógeno, un grupo alquilo C1-4 sustituido o sin sustituir o un grupo hetercíclico o carbocíclico sin sustituir, o R2 y R3 junto con el átomo de nitrógeno al que están unidos forman un anillo heterocíclico sustituido que puede interrumpirse por -O-, -S- o >NR4 en el que R4 es un átomo de hidrógeno, un grupo alquilo C1-4 sustituido o sin sustituir o un grupo fenilo sustituido o sin sustituir.
y donde la agrupación -O-CO-R está unida a un átomo de carbono situado en cualquiera de las posiciones 9, 10 u 11 en el anillo A del compuesto de camptotecina, incluyendo las sales del mismo.
Se prefiere que la agrupación -O-CO-R esté unida en la posición 10.
R1 es preferentemente alquilo C1-4, con la máxima preferencia etilo, y m es preferentemente 1.
Un átomo de halógeno R es, por ejemplo, F, CI, Br o I, preferentemente F o CI. De R1 a R4 puede ser metilo, etilo, propilo, isopropilo, n-butilo, isobutilo y t-butilo, preferentemente metilo.
Los sustituyentes en R y R1 son preferentemente seleccionados de entre átomos de halógeno, hidroxi alcoxi C1-4, fenoxi, COOR6 , SO3R6 y PO3(R6)2, arilo,

NR8R9 y CONR8R9, QAOR5 , QANR8R9 y QAQR5 en el que R5 es arilo o alquilo C1-4; R6 es hidrógeno, alcoxi C1-4 o alquilo C1-4 halógeno; R7 es hidrógeno, halógeno o alquilo C1-4; R8 y R9 son los mismos o diferentes y cada uno es H, o alquilo C1-4 o R8 y R9 representan ambos alcanediilo C3-6;
Q es OCO, o -Co O- y A es alcanediilo C2-4.
Preferentemente R es NR3 R3 donde R2 y R3 junto con el átomo de hidrógeno forman un anillo de 5 o 6 miembros, preferentemente un anillo saturado, con sustituyentes opcionales.. Un sustituyente es preferentemente -NR8R9. En tal sustituyente R8 y R9 preferentemente juntos son alcanediilo C4-5. Tales grupos son básicos y tienden a estar catiónicamente cargados con un pH de 7. Con la máxima preferencia R es

Otra familia de compuestos adecuados tiene la Fórmula general II

en la que R20 y R23 son cada uno hidroxi o hidrógeno o juntos son CH2OCH2 ;
uno de R21 y R22 es H y el otro es CH2NR24R25 donde R23 y R24 son los mismos o diferentes y cada uno representa un átomo de hidrógeno, un grupo alquilo C1-4 sustituido o sin sustituir o un grupo hetercíclico o carbocíclico sin sustituir, o R23 y R24 junto con el átomo de nitrógeno al que están unidos forman un anillo heterocíclico sustituido que puede interrumpirse por -O-, -S- o >NR4 en el que R4 es un átomo de hidrógeno, un grupo alquilo C1-4 sustituido o sin sustituir o un grupo fenilo sustituido o sin sustituir; incluyendo sales y derivados cuaternarios de los mismos. Un ejemplo de un compuesto adecuado de esta reivindicación es topotecán, en el que R20 es hidroxilo, R22 y R23 son hidrógeno, R21 es CH2NR24R25y R24 y R25 son ambos metilo.
La proporción del primer activo con respecto al segundo activo está preferentemente en el intervalo de 100:1 a 1:100, preferentemente en el intervalo de 5:1 a 1:5. Las proporciones adecuadas pueden determinarse por aquella persona experta en la materia, por ejemplo mediante la gestión de información a partir de determinaciones del índice de combinación. Generalmente, las proporciones deberían seleccionarse a fin de optimizar el efecto sinergístico. Preferentemente la proporción de equivalentes de anión en el polímero con respecto al catión en el primer activo debería estar en el intervalo de 2:1 a 1:1, preferentemente aproximadamente 1:1. Con la selección de estas proporciones puede lograrse una carga óptima, mientras se minimiza el primer efecto tras la administración inicial, y se proporciona una liberación ampliada y optimizada.
El segundo activo usado en la invención puede ser, de forma alternativa, un inhibidor de la COX. Los inhibidores de la COX, por ejemplo, los AINE, podrían actuar como agentes antiinflamatorios y analgésicos, dirigiéndose a la inflamación y el dolor asociados al procedimiento de quimioembolización. Además, hasta el 50 % de las células de un tumor pueden ser células inflamatorias. Por otra parte, ha habido numerosos estudios que vinculan la inflamación con el avance del tumor y la angiogénesis. En consecuencia, una combinación de un agente citotóxico tal como doxorrubicina, con un inhibidor de la COX puede tener efectos altamente deseables. Como se indicó anteriormente, el ibuprofeno ha sido incluso propuesto por tener efectos positivos en la reducción de la invasión de células cancerígenas y en las metástasis hepáticas.
El polímero es un material no hidrosoluble. Aunque puede ser biodegradable, a fin de que el fármaco pueda ser liberado sustancialmente por erosión de la matriz polimérica para así liberar el fármaco desde la superficie, preferentemente el polímero es bioestable sustancialmente (es decir, no biodegradable).
El polímero es hinchable en agua. El polímero hinchable en agua útil en la invención preferentemente posee un equilibrio en el contenido de agua, cuando se hincha en agua a 37° C, cuantificado mediante un análisis gravimétrico, en el intervalo de entre 40 a 99 % en peso, preferentemente de 75 a 95 %.
Generalmente, el polímero está entrecruzado covalentemente, aunque puede ser apropiado para el polímero que esté entrecruzado iónicamente, al menos en parte, o incluso entrecruzado hidrofóbicamente. En algunas realizaciones puede ser adecuado usar polímeros que deriven de fuentes naturales, tales como albumina, alginato, gelatina, almidón, quitosano o colágeno, todos ellos han sido usados como agentes embólicos. En otras realizaciones el polímero carece sustancialmente de derivados o polímeros en su forma natural. Están preferentemente formados por polimerización de monómeros etilénicamente insaturados en presencia de
monómeros de entrecruzamiento difuncionales o de funcionalidad más alta. Los monómeros etilénicamente insaturados pueden incluir un monómero (incluyendo uno zwitteriónico) iónico.
Se pueden usar copolímeros de metacrilato de hidroxietilo, ácido acrílico y monómero de entrecruzamiento, tales como el dimetacrilato de etilenglicol o la metilenbisacrilamida, tal y como se usa para las lentes de contacto a base de etafilcon A. También pueden usarse copolímeros de N-acriloil-2-amino-2-hidroximetil-propano-1,3-diol y N,N-bisacrilamida.
Otros polímeros sintéticos adecuados son los polímeros estirénicos entrecruzados, p. ej., con sustituyentes iónicos, del tipo usado como medio de separación o bien como medio de intercambio de iones.
Otro tipo de polímero sintético que se puede usar para formar la matriz hinchable en agua y no hidrosoluble es el alcohol polivinílico entrecruzado usando agentes de entrecruzamiento de tipo aldehído tales como el glutaraldehído. Para tales productos, el alcohol polivinílico (PVA) puede volverse iónico, mediante aporte de grupos iónicos colgantes mediante reacción de un grupo funcional iónico que contenga un compuesto con los grupos hidroxilo. Ejemplos de grupos funcionales adecuados para reacción con los grupos hidroxilo son los agentes acilantes, tales como ácidos carboxílicos o derivados de los mismos, u otros grupos ácidos que puedan formar ésteres.
La invención resulta de especial valor cuando la matriz polimérica está formada a partir de un macrómero de alcohol polivinílico, que tiene más de un grupo etilénicamente insaturado colgante por molécula, mediante polimerización radical de los grupos etilénicos. Preferentemente el macrómero de PVA está copolimerizado con monómeros etilénicamente insaturados, por ejemplo, incluyendo un monómero no iónico y/o iónico que incluye un monómero aniónico.
El macrómero de PVA se puede formar, por ejemplo, aportando un polímero de PVA, de un peso molecular adecuado, tal como en el rango de 1000 a 500000 D, preferentemente de 10000 a 100000 D, con grupos acrílicos o vinílicos colgantes. Los grupos acrílicos colgantes se pueden aportar, por ejemplo, mediante reacción del ácido acrílico o metacrílico con pVa para formar enlaces éster a través de alguno de los grupos hidroxilo. Otros procedimientos de fijación de grupos vinílicos capaces de polimerizar para dar un alcohol polivinílico se describen en, por ejemplo, los documentos US 4.978.713 y, preferentemente, en US 5.508.317 y 5.583.163. Por tanto, el macrómero preferido comprende un esqueleto principal de alcohol polivinílico al que se conecta, a través de un enlace acetal cíclico, un resto (alqu)acrilaminoalquilo. El Ejemplo 1 describe la síntesis de un ejemplo de tal macrómero conocido con el nombre aprobado de nelfilcón B. Preferentemente, los macrómeros de PVA tienen aproximadamente de 2 a 20 grupos etilénicos colgantes por molécula, por ejemplo de 5 a 10.
Cuando los macrómeros de PVA se copolimerizan con monómeros etilénicamente insaturados, incluyendo un monómero iónico, el monómero iónico tiene preferentemente la Fórmula general II
Y1BQ1 II
en que Y1 se selecciona de entre

CH2=C(R10)-CH2-O-, CH2=C(R10)-CH2 OC(O)-, CH2=C(R10)OC(O)-, CH2=C(R10)-O-, CH2=C(R10)CH2OC(O)N(R11)-, R12OOCCR10=CR10C(O)-O-, R10CH=CHC(O)O-, R10CH=C(COOR12)CH2-C(O)-O-,

donde:
R10 es hidrógeno o un grupo alquilo C1-C4;
R11 es hidrógeno o un grupo alquilo C1-C4;
R12 es hidrógeno o un grupo alquilo C1-4 o BQ1 donde B y Q1 se definen a continuación;
A1 es -O- o -NR11-;
K1 es un grupo -(CH2)rOC(O)-, -(CH2)rC(O)O-, - (CH2)rOC(O)O-, - (CH2)rNR13-, -(CH2)rNR13C(O)-, -(CH2)rC(O)NR13-, -(CH2)rNR13C(O)O-, - (CH2)rOC(O)NR13-, -(CH2)rNR13C(O)NR13- (en el que los grupos R13 son iguales o diferentes), -(CH2)rO-, -(CH2)rSO3-, u, opcionalmente en combinación con B, un enlace de valencia y r es de 1 a 12 y R13 es hidrógeno o un grupo alquilo C1-C4;
B es una cadena alcanodiilo, oxaalquileno, alcanodiiloxaalcanodiilo o alcanodiiloligo(oxaalcanodiilo) lineal oramificada que contiene opcionalmente uno o más átomos de flúor hasta e incluyendo cadenas perfluoradas o, si Q1 o Y1 contiene un átomo de carbono terminal unido a B, un enlace de valencia; y
Q1 es un grupo aniónico.
Un grupo aniónico Q1 puede ser, por ejemplo, un grupo carboxilato, carbonato, sulfonato, sulfato, nitrato, fosfonato ofosfato. El monómero puede polimerizarse como ácido libre o en forma de sal. Preferentemente, el pKa del ácidoconjugado es menor de 5.
Un grupo catiónico Q1 adecuado preferentemente un grupo N+R143 , P+R153 o S+R152 en que los grupos R14 son iguales o diferentes y son cada uno hidrógeno, alquilo C1-4 o arilo (preferentemente fenilo) o dos de los grupos R14 junto con el heteroátomo al que están enlazados forman un anillo heterocíclico saturado o insaturado que contiene de 5 a 7 átomos y los grupos R15 son cada uno OR14 o R14. Preferentemente, el grupo catiónico es permanentemente catiónico, es decir cada R14 es distinto de hidrógeno. Preferentemente, un grupo catiónico Q es N+R143 , en que cada R14 es alquilo C1-4, preferentemente metilo.
Un grupo zwitteriónico Q1 puede tener una carga global, por ejemplo, por tener un centro divalente de carga aniónica y un centro monovalente de carga catiónica o viceversa, o por tener dos centros de carga catiónica y un centro de carga aniónica o viceversa. Sin embargo, preferentemente el ion dipolar no tiene carga global y con la máxima preferencia tiene un centro de carga catiónica monovalente y un centro de carga aniónica monovalente.
Se describen ejemplos de grupos zwitteriónicos que pueden usarse como Q1 en la presente invención en el documento WO-A-0029481.
Cuando el monómero etilénicamente insaturado incluye un monómero zwitteriónico, por ejemplo, este puede aumentar la hidrofilicidad, lubricidad, biocompatibilidad y/o hemocompatibilidad de las partículas. Se describen monómeros zwitteriónicos en las publicaciones previas Wo -A-9207885, WO-A-9416748, WO-A-9416749 y WO-A-9520407. Preferentemente, es un monómero zwitteriónico la sal interna de etilfosfato de 2-metacriloiloxi-2'-trimetilamonio (MPC).
En el monómero de Fórmula general I, preferentemente Y1 es un grupo CH2=CR10COA- en que R10 es H o metilo,preferentemente metilo, y en que A1 es preferentemente NH. B es preferentemente un grupo alcanodiilo de 1 a 12, preferentemente 2 a 6 átomos de carbono. Tales monómeros son monómeros acrílicos.
En el monómero etilénicamente insaturado puede haber incluido un monómero diluyente, por ejemplo, un monómero no iónico. Tal monómero puede ser útil para controlar el pKa de los grupos ácidos, para controlar la hidrofilicidad o hidrofobicidad del producto, para proporcionar regiones hidrofóbicas en el polímero o simplemente para actuar como diluyente inerte. Ejemplos de monómeros diluyentes no iónicos son, por ejemplo, los (alqu)acrilatos y las (alqu)acrilamidas, especialmente compuestos tales que tengan grupos alquilo con 1 a 12 átomos de carbono, los alquil(alqu)acrilatos y -(alqu)acrilamidas hidroxi y dihidroxi sustituidos, las lactamas vinílicas, el estireno y otros monómeros aromáticos.
En la matriz polimérica, el nivel aniónico está preferentemente en el intervalo de 0,1 a 10 meq g-1, preferentemente almenos 1,0 meq g-1. Los aniones preferidos derivan de ácidos fuertes, tales como sulfatos, sulfonatos, fosfatos y fosfonatos.
Cuando el macrómero de PVA copolimeriza con otros monómeros etilénicamente insaturados, la proporción en peso de macrómero de PVA con respecto a otro monómero se encuentra preferentemente en el intervalo de 50:1 a 1:5, más preferentemente en el intervalo de 20:1 a 1:2. En el monómero etilénicamente insaturado, el monómero aniónico está presente preferentemente en una cantidad en el intervalo del 10 al 100 % molar, preferentemente al menos el 25 % molar.
El polímero entrecruzado se puede formar como tal en forma de microesfera, por ejemplo mediante polimerización
de gotas de monómero en una fase dispersa de un vehículo inmiscible continuo. Se conocen ejemplos de polimerizaciones de agua en aceite para producir partículas que tengan el tamaño deseado cuando se hinchen. Por ejemplo, el documento US 4.224.427 describe procedimientos de formación de perlas esféricas uniformes (microesferas) de hasta 5 mm de diámetro mediante dispersión de monómeros hidrosolubles en una fase solvente continua, en presencia de agentes de suspensión. Pueden estar presentes estabilizantes y tensioactivos para permitir controlar el tamaño de las partículas de la fase dispersa. Después de la polimerización, las microesferas entrecruzadas se recuperan mediante medios conocidos, se lavan y, opcionalmente, se esterilizan. Preferentemente, las partículas, por ejemplo, microesferas, se hinchan en un líquido acuoso y se clasifican en función de su tamaño. Los procedimientos para cargar los dos activos en una microesfera se seleccionan de acuerdo con la naturaleza de los activos y los niveles de carga necesarios. Cuando son cargados simultáneamente, esto resulta adecuado a partir de una solución que contenga ambos activos en combinación uno con el otro. Los procedimientos de carga secuenciales pueden incluir poner en contacto uno de los activos en una solución acuosa con polímero a un nivel o durante un tiempo tal que el intercambio de iones dentro de la matriz polimérica tenga lugar hasta tal punto que una proporción de grupos aniónicos del polímero permanezca independiente del activo catiónico. En una segunda etapa, opcionalmente después de la eliminación del primer activo no absorbido, una solución del segundo activo, normalmente en agua, se pone en contacto con las microesferas parcialmente cargadas durante un período de tiempo que permita que el intercambio de iones tenga lugar y que el segundo fármaco pueda cargarse dentro del polímero.
Cuando uno de los activos es un compuesto que tiene baja solubilidad en agua, se usa una técnica diferente. Por ejemplo, puede ser posible cargar los activos secuencialmente, por ejemplo poniendo en contacto las microesferas con las soluciones de cada fármaco en disolventes adecuados, lo que permite que las microesferas se hinchen y absorban el activo dentro de la matriz polimérica. En una realización, un activo con baja solubilidad en agua es cargado dentro del polímero primero. En otra realización, el activo cargado catiónicamente es cargado primero. Puede ser posible seleccionar un disolvente o un sistema de disolventes en el que tanto un activo cargado catiónicamente como un activo neutral, que tiene baja hidrosolubilidad, sean solubles, siendo el disolvente uno que actúe para hinchar el polímero. Por ejemplo, cuando los activos sean ambos solubles en alcohol seleccionado, el alcohol puede usarse para disolver los activos juntos, y para hinchar la matriz polimérica que es hinchable en el alcohol seleccionado.
Preferentemente el procedimiento de carga incluye las etapas:
poner en contacto las microesferas que comprenden una matriz de un polímero entrecruzado cargado aniónicamente, hinchable en agua y no hidrosoluble en una forma sustancialmente seca con una solución de un fármaco que tiene una solubilidad en agua menor de 10 g/l a temperatura ambiente en un primer disolvente orgánico, donde una solución de fármaco en disolvente se impregna dentro de las microesferas;
separar la solución de fármaco que no ha impregnado las microesferas;
poner en contacto las microesferas impregnadas con el líquido acuoso en el cual el fármaco es precipitado en el núcleo de las microesferas;
poner en contacto las microesferas que contienen el fármaco precipitado con una solución acuosa de un fármaco iónico que tiene una carga opuesta a la del polímero para permitir la carga del intercambio de iones; y recuperar las microesferas cargadas duales, donde se han seleccionado los fármacos como se describió anteriormente.
En este procedimiento, hemos hallado que podemos alcanzar niveles elevados de carga para tanto el fármaco no hidrosoluble como para el fármaco catiónico. El nivel de carga del fármaco catiónico es tan elevado como el de las microesferas en neto. Cuando el procedimiento de carga se lleva a cabo con el fármaco iónico siendo cargado primero y el fármaco no hidrosoluble siendo cargado segundo, los niveles de carga posibles que se alcanzan para el segundo, el fármaco neutral, resultan inferiores a los de las microesferas en neto. El efecto se muestra adicionalmente en el Ejemplo 4 más adelante.
Creemos que es la primera vez que un polímero que administra fármaco cargado iónicamente e hinchable en agua ha sido cargado con una combinación de fármaco no hidrosoluble y un fármaco que tiene una carga opuesta a la del polímero. De acuerdo con un aspecto adicional (referido en lo sucesivo como el segundo aspecto) de la invención, una matriz de administración de fármaco que comprende un polímero cargado iónicamente, hinchable en agua y no hidrosoluble que es hinchable en agua a temperatura ambiente hasta un contenido de agua en equilibrio con el intervalo del 40 al 99 % por peso en base al polímero más agua puesto en contacto con una solución de fármaco que tiene una solubilidad en agua menor de 10 g/l a temperatura ambiente en un disolvente orgánico que es capaz de hinchar el polímero, en el cual la solución se impregna dentro de la matriz;
se separa cualquier solución de fármaco que no ha impregnado la matriz polimérica;
la matriz impregnada se pone en contacto con el agua en la cual el fármaco es precipitado en la matriz;
la matriz que tiene el fármaco precipitado no hidrosoluble se pone en contacto con una solución acuosa del fármaco cargado iónicamente en el que la matriz se hincha y tiene lugar el intercambio de iones con los contraiones asociados con el polímero y el fármaco es unido electroestáticamente en la matriz polimérica.
Opcionalmente la matriz cargada con el fármaco de baja solubilidad es subsecuentemente enjuagada y/o secada. Las etapas subsecuentes hasta la puesta en contacto con la solución acuosa del fármaco cargado iónicamente pueden ser descrita en el documento WO2007090897 y los componentes y condiciones preferidas y descritas en esa especificación pueden usarse en las primeras etapas en el procedimiento de esta realización, que incluye los fármacos. La matriz puede tener la forma de una microesfera de acuerdo con el primer aspecto de la invención. Alternativamente, puede ser un dispositivo de administración de fármaco tal como un implante en la forma de una aguja, placa, disco, hilo o película de polímero. Alternativamente, se trata de un recubrimiento sobre un dispositivo que se implanta tal como una endoprótesis, catéter, aguja, tubo, injerto, válvula, alambre, etc.
El polímero puede ser un polímero aniónico como el usado en el primer aspecto de la invención descrito anteriormente. Alternativamente, puede ser un polímero catiónico, por ejemplo que tiene también grupos colgantes zwitteriónicos, tal como se describe en el documento WO0152915. Alternativamente, puede ser otro polímero de hidrogel iónico usado en el dispositivo de administración de fármaco tal como un material a base de sulfato de dextrano tal como se describe en Liu y col., (2003) op. cit.
Preferentemente es sintético, aunque puede ser, de forma alternativa, un polímero en forma natural, basado en un proteína o polisacárido, tal como colágeno, ácido hialurónico, sulfato de dextrano, alginato, una albúmina o polímero celulósico.
En el procedimiento del segundo aspecto de la invención, la matriz cargada con el primer fármaco puede secarse antes en entrar en contacto con la solución acuosa del segundo fármaco. Después del intercambio de iones con el segundo fármaco, puede eliminarse el exceso de solución del fármaco antes del enjuagado. Este aspecto de la invención posee un valor particular cuando el fármaco no hidrosoluble es rapamicina o un análogo como se describió anteriormente, o un taxano tal como paclitaxel, o dexametasona.
En una realización alternativa de procedimiento de carga para producir el producto del primer aspecto de la invención, el activo, que no está cargado, puede ser un material que es cristalino a temperatura ambiente, pero para el que sería deseable tratar de no estar en forma cristalina. Tales fármacos incluyen, por ejemplo, ibuprofeno. Para estos fármacos, la cristalización del fármaco puede inhibirse mediante la incorporación de un inhibidor de cristalización, tal como un aceite, por ejemplo un aceite mineral o un aceite vegetal, opcionalmente un aceite que actúe como agente de contraste, por ejemplo lipiodol u otro aceite yodado. La adecuación al procedimiento implica poner en contacto microesferas que comprenden una matriz de un polímero entrecruzado iónicamente cargado, hinchable en agua e hidrosoluble con una solución del fármaco cristalizable en un disolvente orgánico capaz de penetrar en las microesferas, la solución contiene además un modificador cristalino que inhibe la cristalización del fármaco, y, en una etapa subsecuente, poner en contacto las microesferas cargadas con el fármaco cristalizable con una solución acuosa de un fármaco iónico con una carga opuesta a la de la matriz polimérica en una cantidad y durante un tiempo adecuados para cargar el fármaco mediante intercambio de iones. Preferentemente en el procedimiento, después de la carga del fármaco cristalizable, las perlas se lavan con agua o una solución acuosa a fin de eliminar el fármaco cristalizable no absorbido.
En la invención de los aspectos primero o segundo de la invención, las microesferas pueden ser adecuadas para administración en un procedimiento de embolización. Las microesferas, por consiguiente, son de un tamaño que las convierte en adecuadas para uso en embolización, dependiendo del ser vivo tratado, y la localización del tumor, y, de esta forma, del tamaño de los vasos sanguíneos que irriguen. Por ejemplo, las microesferas embólicas preferentemente poseen un tamaño al hincharse en agua deionizada a temperatura ambiente (en neto) de 40 pm hasta 1500 pm, preferentemente en el intervalo de 100 pm hasta 1200 pm. Alternativamente las microesferas pueden ser adecuadas para la administración como un depósito de fármaco, por ejemplo que puede liberar fármaco durante un largo período en el tejido circundante, y opcionalmente en la circulación. En otra realización, las microesferas son adecuadas para inyección intratumoral. Las microesferas con tamaños en el intervalo mencionado anteriormente para embolización pueden también ser adecuadas para administración intratumoral o para administración en los márgenes de resección después de la resección del tumor.
Los procedimientos para cargar activos hidrosolubles, y catiónicamente cargados, en los polímeros de matriz aniónica como se describieron anteriormente para uso con microesferas, también pueden ser aplicables para cargar
tales tipos de fármaco en otros elementos de administración de fármaco con base en hidrogel. En el segundo aspecto de la invención los dos fármacos son preferentemente seleccionados para ser efectivos terapéuticamente en el tratamiento tumoral, y preferentemente tienen un índice de combinación como se ha definido anteriormente en el presente documento de menos de 0,9, preferentemente menos de 0,7, por ejemplo menos de 0,5, preferentemente menos de 0,3, y con la máxima preferencia menos de 0,1.
El fármaco iónico en el segundo aspecto de la invención puede ser una antraciclina catiónica como se describió en el documento WO04071495, o un derivado de camptotecina tal como se describió en el documento WO2006027567. Alternativamente el fármaco puede ser aniónico tal como se describió en el documento WO0152915.
La presente invención se ilustra en los ejemplos adjuntos. Los ejemplos se refieren a varias figuras que se relacionan brevemente con los resultados posteriores:
La Figura 1 muestra el perfil de carga de perlas cargadas con doxorrubicina (Muestra 1) o irinotecán (Muestra 2) con un nivel de 25 mg.ml-1 de perlas tras carga con el segundo fármaco a un nivel de saturación, n=1, tal como se describe en el Ejemplo 2.
La Figura 2 muestra el perfil de carga de irinotecán y doxorrubicina combinado en las perlas. Perfil de carga de perlas cargadas con 25 mg de irinotecán durante 1 hora y después con 25 mg de doxorrubicina (Muestra 3), o con 25 mg de doxorrubicina durante una 1 h y después con 25 mg de irinotecán (Muestra 4) y perfil de carga de perlas cargadas con una mezcla de 25 mg de doxorrubicina y 25 mg de irinotecán (Muestra 5), n=1, como se describe en el Ejemplo 2;
La Figura 3 muestra el tamaño de las muestras cargadas con una combinación de doxorrubicina e irinotecán mediante procedimientos diferentes, n=1, como se describe en el Ejemplo 2;
La Figura 4 muestra el perfil de elución de doxorrubicina e irinotecán eluidos en PBS (solución salina tamponada con fosfato) a temperatura ambiente (RT) a partir de las mismas perlas. La Muestra 1 se compone de perlas cargadas con doxorrubicina en un nivel de 25 mg.ml-1 de perlas después de cargarlas con irinotecán a un nivel de saturación, la Muestra 2 se compone de perlas cargadas con irinotecán a un nivel de 25 mg ml-1 de perlas después de cargarlas con doxorrubicina a un nivel de saturación. La Muestra 3 se compone de perlas cargadas con 25 mg de irinotecán durante 1 hora y después con 25 mg de doxorrubicina, la Muestra 4 se compone de perlas cargadas con 25 mg de doxorrubicina durante 1 hora y después con 25 mg de irinotecán y la Muestra 5 se compone de perlas cargadas con una mezcla de 25 mg de doxorrubicina y 25 mg de irinotecán, n=1, como se describe en el Ejemplo 2.
La Figura 5 muestra el perfil de liberación de microesferas cargadas con doxorrubicina e ibuprofeno en 200 ml de PBS a temperatura ambiente (RT) sobre una mezcladora de rodillos durante 24 h, n=1, como se describe en el Ejemplo 3.
La Figura 6 es un diagrama de flujo que muestra dos procedimientos de carga de rapamicina y doxorrubicina en perlas, como se describe en el Ejemplo 4.
La Figura 7 muestra la distribución del tamaño de perlas cargadas de fármaco y no cargadas, como se describe en el Ejemplo 4.
Las Figura 8A-D muestran una elución de doxorrubicina y rapamicina a partir de perlas cargadas hasta 200 ml de PBS, a 25° C. (A) Elución de fármaco de las perlas realizada según el Procedimiento 1. (B) Datos de elución de (A) presentados en porcentaje. (C) Elución de fármaco de perlas realizada mediante el Procedimiento 2. (D) Datos de elución de (C) presentados en porcentaje. En (A) y (C), la cantidad de fármaco eluido fue corregida hasta por volumen de perlas sin fármaco, como se describe en el Ejemplo 4.
La Figura 9 muestra el procedimiento de preparación de perla de ibuprofeno-irinotecán, como se describe en el Ejemplo 5.
La Figura 10 muestra la distribución del tamaño de perlas cargadas con ibuprofeno e irinotecán y con ibuprofeno, como se describe en el Ejemplo 5.
La Figura 11 muestra la elución de irinotecán de perlas con la combinación de ibuprofeno e irinotecán. Condición de la elución: 0,24 ml de perlas (cargadas), 200 ml de PBS, a 25° C, como se describe en el Ejemplo 5.
La Figura 12 muestra la elución de ibuprofeno de perlas con la combinación de ibuprofeno e irinotecán. Condición de la elución: 0,24 ml de perlas (cargadas), 200 ml de PBS, a 25° C, como se describe en el Ejemplo 5.
La Figura 13 muestra la elución de doxorrubicina de Perlas frente al Tiempo de elución. (Barras de error: 1 de DE, n=6), como se describe en el Ejemplo 6.
La Figura 14 muestra el porcentaje de células HepG2 viables en comparación con células no tratadas con respecto al tiempo de exposición y al número de perlas a partir del ensayo con LDH. (Barras de error: 1 de DE, n=6), como se describe en el Ejemplo 6.
La Figura 15 muestra el porcentaje de células HepG2 viables para rapamicina, doxorrubicina y la combinación (Barras de error: 1 de DE, n=5), resultados a partir de un ensayo LDH, como se describe en el Ejemplo 7.
La Figura 16 muestra el porcentaje de células HepG2 viables para rapamicina, doxorrubicina y la combinación -ampliación a escala (Barras de error: 1 de DE, n=5), resultados a partir de un ensayo LDH, como se describe en el Ejemplo 7.
La Figura 17 muestra el porcentaje de células HepG2 viables para rapamicina, doxorrubicina y la combinación
(Barras de error: 1 de EEM, n=3), resultados a partir de un ensayo MTS, como se describe en el Ejemplo 7.
La Figura 18 muestra el porcentaje de células HepG2 viables para rapamicina, doxorrubicina y la combinación -ampliación a escala (Barras de error: 1 de EEM, n=3), resultados a partir de un ensayo MTS, como se describe en el Ejemplo 7.
La Figura 19 muestra el % de viabilidad (escala logarítmica) de células HepG2 después de una exposición de 72 horas a combinaciones de microesferas cargadas de fármaco (ensayo MTS, n=6, ±DE), como se describe en el Ejemplo 8.
Las Figuras 20 a 22 muestran el porcentaje de células PSN1 después de una exposición de 24, 48 y 72 horas (respectivamente) a perlas eluyentes con carga de fármaco única o dual (n=7 /-DE) como se describe en el Ejemplo 9.
Las Figuras 23 a 25 muestran el porcentaje de células HepG2 después de una exposición de 24, 48 y 72 horas (respectivamente) a perlas eluyentes con carga de fármaco única o dual (n=4 /-DE) como se describe en el Ejemplo 9.
Las Figuras 26 y 28 muestran el porcentaje de células HepG2 después de una exposición de 24, 48 y 72 horas (respectivamente) a perlas eluyentes con carga de fármaco única o dual (n=4 /-DE) como se describe en el Ejemplo 10.
Ejemplo 1: Preparación de perlas eluyentes de fármaco
Las microesferas son sintetizadas como se describe en el Ejemplo de referencia del documento WO2006027567 para producir formulaciones de AMPS alto y de AMPS bajo.
Ejemplo 2: Carga y elución de combinaciones de doxorrubicina e irinotecán
Se cargaron cinco muestras de perlas con doxorrubicina e irinotecán con AMPS alto de 500 a 700 pm del Ejemplo 1 después de diferentes enfoques:
Muestra 1: Se cargaron 3,5 ml de solución de irinotecán (Campto, Pfizer, 20 mg.mL-1= 70 mg) dentro de 1 ml de perlas durante 48 horas a fin de saturar todos los grupos sulfonato. Después, se eliminó la solución reducida y las perlas cargadas se lavaron con un volumen conocido de agua deionizada para quitar el fármaco no unido. La cantidad de irinotecán cargado en las perlas se calculó mediante reducción, como una diferencia entre las cantidades iniciales y finales en la solución reducida y se cuantificaron todos los lavados mediante UV con una longitud de onda de 369 nm. Posteriormente se añadió 1 ml de doxorrubicina (de Dabur Oncology-25 mg.ml-1) a las mismas perlas y se determinó el perfil de carga.
Muestra 2: De forma semejante a la Muestra 1, aunque se añadieron 2,8 ml de solución de doxorrubicina (25 mg.ml-1 = 70 mg) a las perlas en lugar de solución de irinotecán en la primera etapa, para saturar todos los grupos sulfonato. Se cuantificaron todas las soluciones mediante UV con una longitud de onda de 483 nm, además de la cantidad de fármaco cargado en las perlas que se determinó por reducción. Después de cargar las perlas y lavarlas con agua deionizada, en la segunda etapa, se añadió 1,25 ml de solución de irinotecán (20 mg.ml-1 = 25 mg) a las mismas perlas y se determinó el perfil de carga.
Muestra 3: Se cargó 1,25 ml de solución de irinotecán (20 mg.ml-1 = 25 mg) en 1 ml de perlas y después de 1 hora, se añadió 1 ml de doxorrubicina (25 mg.ml-1) a las mismas perlas. No se realizaron lavados intermedios ni en esta ni en las siguientes muestras.
Muestra 4: Se cargó 1 ml de doxorrubicina (25 mg.ml-1) en 1 ml de perlas y después de 1 hora, se añadió 1,25 ml de solución de irinotecán (20 mg.ml-1= 25 mg) a las mismas perlas.
Muestra 5: Se mezcló 1,25 ml de solución de irinotecán (20 mg.ml-1 = 25 mg) con 1 ml de doxorrubicina (25 mg.ml-1) y después se añadió a 1 ml de perlas.
Para determinar los perfiles de carga de todas las muestras, se retiró una alícuota de 50 pl de cada vial a los 5, 15, 30 y 45 minutos después de añadir cada fármaco, se diluyó como convino y se leyó mediante UV a 483 y 369 nm. La cantidad cargada en cada momento temporal fue determinada mediante reducción usando las curvas estándar correspondientes.
Después de cargarlas con ambos fármacos, se dimensionaron todas las muestras en sus soluciones de carga usando la videocámara Colorview III para después tomar fotografías. Para eso, se cuantificaron al menos 100 perlas. Se normalizaron estos datos y se generaron los correspondientes histogramas.
Después de ser cuantificadas, se eluyeron todas las muestras. Aquí, las soluciones de carga restantes fueron eliminadas mediante el uso de una pipeta Pasteur para después transferir las perlas cargadas a matraces color café que contenían 200 ml de PBS (Inverclyde Biologicals). A los 5, 15, 30, 45, 60 y 90 minutos, y a las 2, 4 y 24 horas, se retiró una alícuota del medio de elución (4 ml) sustituyéndola por 4 ml de PBS fresco para mantener el volumen. Todas las muestras se leyeron con UV a 483 y 369 nm de longitudes de onda, usando las correspondientes curvas
estándar para calcular la cantidad eluida en cada momento temporal.
Cálculos:
Para calcular la carga y elución de ambos fármacos a partir de las mismas perlas, se presentaron los siguientes supuestos:
Absorbencia a 483 nm = Absorbencia de doxorrubicina a 483 nm.
Se observó en la lectura de barrido por UV de la solución de irinotecán que este fármaco no absorbe a esa longitud de onda.
Absorbencia de 369 nm = Absorbencia de doxorrubicina a 369 nm Absorbencia de irinotecán a 369 nm.
La lectura de doxorrubicina mostró cierta absorbencia a 369 nm de longitud de onda y aquí se tuvo en cuenta que no existe interacción entre la absorbencia de doxorrubicina e irinotecán al ser cuantificadas al mismo tiempo. La lectura de la mezcla no muestra ninguna interacción notable entre los espectros de absorbencia de ambos fármacos.
Estos dos supuestos se aplicaron a todos los cálculos. Todas las muestras fueron leídas tanto a 483 como a 369 nm de longitud de onda. Mediante la absorbencia y la curva estándar de doxorrubicina a 483 nm, se calculó la concentración de doxorrubicina en cada muestra y usando estas concentraciones y la curva estándar de doxorrubicina a 369 nm, se calculó la absorbencia de doxorrubicina a esta longitud de onda. A continuación, sustrayendo este valor de la absorbencia total a 369 nm, se calculó la absorbencia a 369 nm y, de este modo, la concentración de irinotecán.
Carga de irinotecán y doxorrubicina en las mismas perlas
El objetivo de estos experimentos fue valorar y comparar el comportamiento de carga y elución de diferentes combinaciones de doxorrubicina e irinotecán a fin de determinar sus mecanismos de interacción con las perlas, además de para evaluar posibles interacciones entre fármacos. Si ambos fármacos pudieran incorporarse con éxito en las mismas perlas, sería sencillo conseguir la administración local de ambos fármacos antitumorales simultáneamente, de ser así necesario.
La doxorrubicina y el irinotecán se cargaron siguiendo los diferentes enfoques detallados anteriormente, presentándose sus resultados como sigue:
Como se vio en la Figura 1, si se carga irinotecán primero a su máxima carga de unión y después se añade la doxorrubicina al sistema (Muestra 1), se desplaza parte (~ 17%) del irinotecán, aunque la mayor parte del fármaco es retenida en las perlas. Si se carga doxorrubicina primero a su máxima capacidad de unión y después se añade irinotecán al sistema (Muestra 2), un porcentaje más pequeño de doxorrubicina (< 8%) se ve desplazado por el irinotecán. Estos datos se corresponden con el hecho de que los valores Kb para doxorrubicina son ligeramente más elevados que aquellos calculados para irinotecán (González y col. 2006) siendo las liberaciones de doxorrubicina mucho más lentas, y hasta un punto más lentas que las de irinotecán teniendo el mismo volumen de solución. En ambos casos, no obstante, la cantidad de fármaco desplazado es poca, lo que indica que ambas interacciones son estables después de la adición del segundo fármaco. Esto podría explicarse como consecuencia de los pequeños volúmenes de solución usados, saturados con facilidad, durante el procedimiento de carga y por la voluminosa molécula de fármaco inicialmente unido que impide estéricamente el acercamiento del segundo fármaco a los lugares de unión, un fenómeno que no experimentarían pequeños iones, como el caso de Na+ o K+ . Estos dos factores contribuirán al pequeño porcentaje de fármaco desplazado por el segundo fármaco añadido.
Las Muestras 3 y 4 en la Figura 2 muestran que si las perlas son primero cargadas con 25 mg de un fármaco y después se añade 25 mg de otro fármaco, se desplaza una muy pequeña cantidad (< 5%) del fármaco unido al añadir el segundo fármaco, independientemente del orden. Estos resultados son de esperar, ya que la capacidad máxima de carga de las perlas es ~ 40 mg y 52 mg para la doxorrubicina e irinotecán respectivamente y, además, los lugares de unión de las perlas no están saturados cuando se añade el segundo fármaco (inicialmente, se añaden 25 mg de fármaco a las perlas para después añadir otros 25 mg del otro fármaco).
La Muestra 5 en la Figura 2 muestra que si ambos fármacos se cargan al mismo tiempo, la tasa de carga y la cantidad total cargada resultan ligeramente más elevadas para doxorrubicina que para irinotecán (después de 2 horas, se carga el 94 % del total de doxorrubicina añadida (25 mg), siendo el 91 % del total de irinotecán añadido (25 mg)). Esta pequeña diferencia podría explicarse como resultado de sus valores Kb, ya que el valor Kb de doxorrubicina (5,58*108-1.02*109) es ligeramente superior que para irinotecán (8,73*107-3,62*108).
Volumen de irinotecán y doxorrubicina cargados en las mismas perlas
No se detectaron diferencias significativas entre las perlas cargadas con ambos fármacos desde los diferentes planteamientos (Figura 3).
Elución de irinotecán y doxorrubicina cargados en las mismas perlas
La Figura 4 muestra que todas las muestras siguen tendencias parecidas de elución, y que la doxorrubicina se libera muy lentamente (< 25 % en 24 horas) en comparación al irinotecán (> 99 % mg en < 4 horas) cuando son eluidos en
200 ml de PBS.
En las Muestras 1 y 2, cuando un fármaco fue cargado hasta el nivel de saturación antes de añadir el otro fármaco, el segundo fármaco a cargar se liberó más rápido que en la Muestra 3 y 4, donde ambos fármacos fueron cargados en las perlas al mismo tiempo por debajo del nivel de saturación. Esto podría explicarse por la naturaleza de unión parcial del segundo fármaco a cargar, debido a que todos los grupos sulfonato son saturados (Muestra 1 y 2). Una pequeña cantidad del fármaco cargado segundo estaría unida a las perlas debido al desplazamiento del primero, en su mayor parte fármaco unido. La mayor parte del fármaco cargado sería libre (no unida) dentro de las perlas y se eluiría más rápidamente que el fármaco unido.
En conclusión, cuando se cargan doxorrubicina e irinotecán al mismo tiempo, o consecutivamente, en las perlas, no se observó ningún cambio en el comportamiento de liberación del fármaco acompañante. La presencia de un fármaco adicional no interfiere en la carga ni modifica el comportamiento de liberación del fármaco acompañante.
Cada fármaco se eluye como se espera a partir de los perfiles de elución del fármaco individual, generados para cada fármaco aislado.
Los perfiles de UV de las soluciones eluidas se llevaron a cabo y compararon con una mezcla no cargada de doxorrubicina e irinotecán. El aspecto de los picos, así como los valores máximos, no variaron; lo que confirma que no se da interacción o modificación aparente de ninguno de los fármacos dentro de las perlas.
Estos experimentos demuestran que es posible cargar y eluir ambos fármacos al mismo tiempo a partir de las mismas perlas. Se espera una liberación sostenida, de ambos fármacos, a partir de este producto. Se espera que la combinación de estos dos fármacos antitumorales alcance un efecto terapéutico más marcado que el tratamiento con una fármaco individual de dosis elevada y que, además, disminuya los efectos secundarios de ambos fármacos.
Este producto potencial será evaluado in vivo para determinar su efecto como sinergético, aditivo o antagonista. Este efecto dependerá tanto de las proporciones moleculares de agentes de quimioterapia individuales, como de los programas de administración (Aschele y col. 1998). Los estudios in vivo se llevarán a cabo para evaluar las dosis diferentes de los agentes con el objetivo de determinar la mejor proporción. Estos experimentos revelaron que ambos fármacos podrían ser cargados y eluidos con éxito dentro de las mismas perlas sin alterar los perfiles de liberación de los fármacos de forma individual, sugiriendo la posibilidad de cargar más de un fármaco catiónico en las perlas.
Ejemplo 3: Combinación de ibuprofeno y doxorrubicina en las mismas perlas
Las soluciones de ibuprofeno y doxorrubicina se cargaron simultáneamente dentro de perlas con AMPS bajo. Para lograr esto, se cargó una muestra de 1 ml de perlas de entre 500 y 700 pm con ibuprofeno (de acuerdo con los ejemplos en el documento WO2004073688). Se eliminó el exceso de solución envasada mediante una pipeta Pasteur y se añadieron 2 ml de solución de doxorrubicina (5 mg.mf1). Después de 24 h, se retiró una alícuota de 50 pl de la solución reducida y se cuantificó mediante UV a 263 nm y a 483 nm. La cantidad de doxorrubicina cargada en las perlas fue calculada por reducción, como la diferencia entre las cantidades iniciales y finales en la solución reducida (los cálculos se explican más adelante).
Después de cargar ambos fármacos, se retiró la solución de carga reducida y las perlas cargadas se transfirieron a matraces de color café que contenían 200 ml de PBS. A los 5, 45 y 90 minutos, y a las 3, 5 y 24 horas, se retiraron 2 ml del medio de elución sustituyéndolo por 2 ml de PBS fresco para mantener el volumen. Todas las muestras se leyeron mediante UV a 263 y 483 nm y la cantidad de cada fármaco eluida en cada momento temporal se calculó usando las curvas estándar correspondientes.
Cálculos:
Para calcular la carga y elución de ambos fármacos a partir de las mismas perlas, se presentaron los siguientes supuestos:
Absorbencia a 483 nm = Absorbencia de doxorrubicina a 483 nm.
Los barridos por UV de solución de ibuprofeno y doxorrubicina mostraron que únicamente la doxorrubicina absorbe a esta longitud de onda.
Absorbencia de 263 nm = Absorbencia de doxorrubicina a 263 nm Absorbencia de ibuprofeno a 263 nm.
El barrido por UV de doxorrubicina mostró cierta absorbencia con una longitud de onda de 263 nm. El supuesto resultante indica que no existe interacción entre la absorbencia de doxorrubicina e ibuprofeno al cuantificarlos en el mismo momento.
Estos dos supuestos se aplicaron a todos los cálculos. Todas las muestras fueron leídas a ambas longitudes de onda (263 nm y 483 nm). Mediante la absorbencia a 483 nm, se calculó la concentración de doxorrubicina en cada muestra y, después, usando estas concentraciones y la curva estándar de doxorrubicina a 263 nm, se calculó la absorbencia de doxorrubicina a 263 nm. Mediante la sustracción de este valor a partir de la absorbencia total a 263 nm, se calculó la concentración de ibuprofeno usando la curva estándar de ibuprofeno.
Carga y liberación de las combinaciones de doxorrubicina e ibuprofeno en perlas
En el primer conjunto de experimentos, se combinaron doxorrubicina e ibuprofeno dentro de las mismas perlas y se evaluó su comportamiento de liberación.
Inicialmente, las perlas liofilizadas, como se describen en el documento WO2004073688, fueron cargadas con solución de etanol / ibuprofeno mediante evaporación del disolvente y en el autoclave durante 15 minutos a 121° C. Las perlas rehidratadas y cargadas de ibuprofeno se cargaron, después, con doxorrubicina mediante inmersión en solución con doxorrubicina (5 mg.ml-1) durante 24 horas. En el experimento, se cargó una cantidad máxima de 8,3 mg de doxorrubicina en las perlas, calculada aquella por reducción de la concentración de la solución de carga. Las perlas con AMPS bajo tienen una capacidad máxima para doxorrubicina de -14 mg.ml-1 al ser cargadas con este fármaco aislado. Esta cantidad máxima está limitada por el número de sitios de unión disponibles en las perlas. Si se añadiera una cantidad de fármaco mayor a la muestra, aquella no se cargaría ya que todos los grupos sulfonato dentro de las perlas se habrían saturado.
La liberación de ambos fármacos a partir de las mismas perlas se supervisó mediante espectrofotometría de UV al mismo tiempo; el perfil de elución para ambos fármacos liberados se presenta en la Figura 5. Ambos fármacos generaron perfiles de liberación similares en relación con los exhibidos para perlas cargadas con sólo uno de los fármacos y, además, no se observó interacción entre ambos fármacos. Este experimento mostró que ambos fármacos pueden cargarse y liberarse dentro de la misma perla, lo que ofrece una plataforma para la liberación sostenida de ambos fármacos simultáneamente. Esto podría ser beneficioso con la forma de un dispositivo de administración de fármaco, en suma a los efectos terapéuticos de ambos fármacos con la administración de un producto. Este tratamiento podría reducir los requisitos de los fármacos analgésicos posteriores a los procedimientos, y la dosis del fármaco más tóxico, si se demuestran los resultados antitumorales sinergéticos por la combinación de ambos fármacos.
Ejemplo 4: Combinación de doxorrubicina y rapamicina en las mismas perlas
Procedimientos de carga: Tal como se muestra en la Figura 6, se probaron dos procedimientos de carga).
Procedimiento 1:
Se cargó 1 ml, de suspensión de perla de AMPS alto no cargado, con solución de doxorrubicina (5 ml de 10,07 mg/ml, carga objetivo de 50 mg/ml por perla) durante una noche. Las perlas cargadas de doxorrubicina se lavaron cinco veces con 1 ml de DMSO para eliminar el agua superflua, y la solución de 1 ml de DMSO de rapamicina a 60 mg/ml se mezcló con las perlas de doxorrubicina durante aproximadamente 30 minutos. Posteriormente, después de retirar la solución de DMSO de rapamicina superflua, se lavaron las perlas con 5 ml de solución salina hasta no detectarse partículas blancas de fármaco.
Procedimiento 2:
Una suspensión de perla de 1 ml se lavó cinco veces con 1 ml de DMSO y se mezcló con 1 ml de solución de DMSO de rapamicina a 60 mg/ml durante 30 minutos. Después, las perlas cargadas se lavaron con 5 ml de solución salina hasta no detectarse partículas blancas de fármaco. 0,5 ml de suspensión de perla cargada con rapamicina se mezcló en una mezcladora de rodillos con 2,5 ml de solución de doxorrubicina a 10,07 mg/ml durante una noche, la carga de doxorrubicina objetivo es de 50 mg/ml.
Prueba de carga de doxorrubicina: La carga de doxorrubicina se cuantificó mediante el procedimiento de reducción tanto en agua como en solución de DMSO. Se extrajo la solución de carga y se diluyó 10 veces para cuantificarse mediante UV a 483 nm a fin de determinar la concentración del residuo de doxorrubicina en la solución.
Prueba de carga de rapamicina: Para las perlas sólo con rapamicina, se extrajeron 0,5 ml de perlas cargadas de rapamicina (a partir de la primera etapa, Procedimiento 1) mediante 1 ml de DMSO, cinco veces, acumulándose la solución en un matraz volumétrico de 10 ml. La concentración de rapamicina se determinó por UV a 279 nm.
Para las perlas con la combinación de doxorrubicina y rapamicina, 0,26 ml de perlas cargadas de fármaco (a partir de la segunda etapa, Procedimiento 2) se extrajeron con 1 ml de DMSO 4 veces, recogiéndose la extracción en un matraz volumétrico de 5 ml a fin de añadir DMSO para superar la solución hasta la marca. La concentración de rapamicina se determinó por sustracción de la absorbencia de doxorrubicina a 279 nm a partir de la absorbencia total a 279 nm, en la que la absorbencia de doxorrubicina a 279 nm se calculó a partir de aquella a 483 nm de acuerdo con el barrido de las soluciones de concentración conocida.
Determinación de pérdida de doxorrubicina: La pérdida de doxorrubicina durante el lavado con DMSO de las perlas cargadas antes de la carga adicional de rapamicina (Procedimiento 2) fue determinada mediante la cuantificación de la solución de lavado de DMSO acumulada en un matraz volumétrico a 483 nm.
Elución de perlas cargadas de fármaco: La elución de perlas cargadas de fármaco se llevó a cabo mezclando las perlas con 200 ml de PBS (Inverclyde, Bellshill, RU) y con un rodillo en una mezcladora de rodillo. En un momento temporal determinado, se extrajo la solución (100 ml o 150 ml) para la determinación de la concentración mediante UV y se añadió el mismo volumen de PBS fresco para mantenerlo en condiciones de sedimentación. La determinación de la concentración de rapamicina siguió el mismo principio que el utilizado en la sección de Prueba de carga de rapamicina.
Dimensionamiento y análisis de imagen Las perlas cargadas se colocaron bajo un microscopio Olympus, se contabilizaron aproximadamente 200 perlas para el estudio de distribución por tamaño mediante software (AnalySIS 5.0 Soft Image System GmbH), y se realizaron fotos con diferente fondo de contraste/brillo para un estudio morfológico.
Curva estándar de doxorrubicina y rapamicina La absorbencia máxima de rapamicina es con 279 nm de longitud de onda, superponiéndose con la absorbencia de doxorrubicina. A 483 nm, en la longitud de onda de absorbencia máxima de doxorrubicina, la rapamicina carece de absorción. Por ello, la concentración de rapamicina en un sistema bicomponente (doxorrubicina y rapamicina) podría determinarse con la primera medición de las soluciones estándar de doxorrubicina a ambas longitudes de onda, y, después, con la sustracción de la absorbencia de doxorrubicina a partir de la absorbencia de la mezcla. Las curvas estándar vienen de doxorrubicina a 279 y 483 nm en agua y en DMSO.
La concentración de rapamicina después de la sustracción de la absorbencia de doxorrubicina puede determinarse mediante la relación dada subyacente a 279 nm:
Absorbencia = 56,18 x Concentración (mg/ml) [en DMSO]
Absorbencia = 44,64 x Concentración (mg/ml) [en agua]
Comparación de la carga de fármaco en los dos procedimientos de carga
En la primera etapa del Procedimiento 1, la rapamicina se cargó a 28,48 mg/ml, la eficiencia de carga fue de aproximadamente el 47,5 % (Tabla 1). En comparación con la carga de rapamicina en la segunda etapa del Procedimiento 2, se cargaron 37,22 mg/ml de rapamicina, con una eficiencia de carga del 62,0 %. La significativamente alta eficiencia de carga fue consistente con la observación de que las perlas cargadas de doxorrubicina se hinchaban más, siendo aspirada hacia el interior de las perlas la mayor parte de la solución de carga de rapamicina. Desde un punto de vista mecanicista, las perlas cargadas de doxorrubicina tenían un tamaño menor, y tendían a hincharse más en disolventes polares como el agua y el DMSO debido al efecto de sal reducida que se observó en la solución salina o en la solución de carga de doxorrubicina.
Para las perlas cargadas con rapamicina (28,48 mg/ml), se halló que la capacidad de carga de doxorrubicina era 46,02 mg/ml, similar a la de las perlas sin rapamicina. Durante la carga de doxorrubicina, no se observó ninguna filtración de rapamicina. Este experimento sugiere que la precarga de rapamicina no afecta a la carga posterior de
doxorrubicina, lo que significa que los dos fármacos ocupan diferentes lugares dentro de las perlas. Las moléculas de doxorrubicina tienden a unirse al sitio de los grupos sulfonato por interacción iónica; sin embargo, la rapamicina se queda dentro del espacio intersticial dentro del hidrogel mediante la expulsión de agua fuera y formando pequeños cristales del fármaco.
T l 1. r r mi in x rr i in m i n r imi n

En el Procedimiento 2, el lavado de las perlas cargadas de doxorrubicina con DMSO antes de la carga de rapamicina provocó una pérdida de doxorrubicina de aproximadamente 6,71 mg/ml, aunque este procedimiento tuvo una carga de rapamicina más elevada. El índice final de doxorrubicina respecto a rapamicina fue de 1,62 para las perlas a partir del Procedimiento 1, y de 1,05 para las perlas a partir del Procedimiento 2.
Desde un punto de vista operacional, el Procedimiento 1 resulta un procedimiento relativamente más limpio y produce únicamente solución de desecho de rapamicina. El Procedimiento 2 produce solución de desecho tanto de doxorrubicina-DMSO como de rapamicina. La carga de doxorrubicina en el Procedimiento 1 es casi completa, siendo también este Procedimiento 1 relativamente más fácil de procesar. A pesar de la alta carga de rapamicina en el Procedimiento 2, se recomienda el Procedimiento 1.
Distribución de tamaño y análisis de imagen de las perlas cargadas de fármaco
Se hacen fotografías después de la carga de rapamicina y rapamicina-doxorrubicina según el Procedimiento 1. Cabe suponer que estas perlas conservan bien su forma esférica y la suavidad superficial después de la carga de fármaco. Se observaron algunas pequeñas partículas de rapamicina, residuos resultantes de la etapa primera. Las perlas cargadas de doxorrubicina experimentaron una leve pérdida de su forma esférica. Después de la carga adicional con rapamicina, la forma se recuperó bien y sus superficies se mantenían suaves. Si se compara con las perlas finales a partir del Procedimiento 1, las perlas del Procedimiento 2 poseen un color rojo superficial, que puede deberse a la pérdida de doxorrubicina después del lavado con DMSO, o bien debido a una cobertura posterior de rapamicina cargada.
La caracterización en tamaño por DC Bead y las perlas cargadas de fármaco se indican en la Tabla 2, mientras que la distribución del tamaño se muestra en la Figura 7. Según la Tabla 2, las perlas cargadas de rapamicina en el Procedimiento 1 poseen unos tamaños ligeramente mayores en comparación con las perlas no cargadas. Después de la carga con doxorrubicina, el tamaño medio disminuyó aproximadamente 100 pm. En el Procedimiento 2, las perlas cargadas de doxorrubicina muestran el tamaño medio más pequeño. Después de la carga con rapamicina, el tamaño se incrementó a aproximadamente 479 pm, cerca del tamaño final de perla según el Procedimiento 1. Mientras tanto, los datos de desviación estándar en la Tabla 2 indican que el intervalo en la distribución del tamaño casi no tiene cambios durante la carga de fármaco. Por consiguiente, los dos procedimientos originan perlas finales con casi el mismo intervalo de tamaño (Figura 7).
Tabla 2. Caracterización del tamaño ara las erlas car adas^ de fármaco las no car adas


Elución de fármaco a partir de perlas cargadas
La Figura 8 muestra los datos de elución para doxorrubicina y rapamicina a partir de perlas cargadas dentro de PBS. En este experimento, el medio de elución fue renovado después de un tiempo determinado para imitar las condiciones de sedimentación. A partir de la Figura 8 (B) y (D), los perfiles de elución de fármaco de las perlas realizados por los diferentes procedimientos son muy similares: la liberación de rapamicina fue de aproximadamente el 33 % después de una elución de 30 horas, mientras que la liberación de doxorrubicina fue de entre el 41 y el 46 %.
Comparando la Figura 8 (A) y (C), la doxorrubicina a partir de las perlas realizadas por el Procedimiento 1 se eluyó ligeramente más rápido que las perlas realizadas por el Procedimiento 2. Para la elución de rapamicina, se observó el fenómeno inverso. Esto puede deberse a que el fármaco cargado más tarde cubrió o bloqueó la vía del primer fármaco, que también fue usada para la primera elución del fármaco.
Ejemplo 5: Combinación de irinotecán e ibuprofeno en la misma perla
Carga de fármaco: El procedimiento de carga de fármaco se muestra en la Figura 9, en la cual las perlas con AMPS no cargadas fueron lavadas con DMSO para eliminar el agua atrapada dentro del hidrogel. Una mezcla de ibuprofeno solubilizado en el DMSO y aceite de ricino como inhibidor de cristalización fueron posteriormente mezclados con la suspensión de perla, manteniéndolo durante aproximadamente 10 minutos. Después de la extracción de la solución de carga, la solución salina se usó para lavar las perlas cargadas hasta que no se observaron partículas de fármaco ni gotas grandes de aceite. La carga de irinotecán se logró mediante la simple mezcla de las perlas cargadas de ibuprofeno con la solución de irinotecán (10 mg/ml) con la mezcladora de rodillo durante una noche. Las perlas cargadas finales se mantuvieron en la solución de agua como producto.
La matriz de la muestra de perlas de ibuprofeno e irinotecán se indica en la Tabla 3.
Tabla 3. Matriz de la muestra de car a de^ ibu rofeno e irinotecán

Determinación del contenido de fármaco en las perlas
1) . Ibuprofeno. El contenido del ibuprofeno cargado se determinó mediante un procedimiento de HPLC La suspensión de las perlas cargadas de fármaco con el volumen conocido se extrajo con DMSO (1 ml) de cinco a seis veces. La solución de DMSO recogida se llenó hasta la marca en un matraz volumétrico de 10 ml, y se midió la concentración de ibuprofeno con una HPLC.
2) . Irinotecán El contenido de irinotecán en las perlas cargadas se midió mediante un procedimiento de reducción. Entre 1 y 1,1 ml de perlas cargadas de ibuprofeno se mezclaron con rodillo con 4 ml de solución de irinotecán a 10 mg/ml para dirigirlo a la carga con perlas a 40 mg/ml. La solución de carga se diluyó con agua y se cuantificó mediante UV a 369 nm. En comparación con la curva estándar, puede calcularse la concentración de irinotecán restante en la solución de carga.
Elución de perlas cargadas de fármaco:
La elución de perlas cargadas de ibuprofeno e irinotecán se llevó a cabo mezclando perlas de 0,24 ml con 200 ml de PBS (Inverclyde, Bellshill, RU) y mediante un rodillo en una mezcladora de rodillo. En un momento temporal determinado, se extrajo la solución (5 ml) para la determinación de la concentración mediante UV y se añadió el mismo volumen de PBS fresco para mantenerlo en condiciones de sedimentación.
Dimensionamiento y análisis de imagen
Las perlas cargadas se colocaron bajo un microscopio Olympus, se contabilizaron aproximadamente 200 perlas para el estudio de distribución por tamaño mediante software (AnalySIS 5.0 Soft Image System GmbH), y se realizaron fotos con diferente fondo de contraste/brillo para un estudio morfológico.
Determinación de concentración de la combinación de ibuprofeno e irinotecán
Para determinar la concentración de cada componente en una solución de fármaco combinada, se midieron las curvas estándar para la absorción de concentraciones predeterminadas de ibuprofeno e irinotecán a 263, 272 y 369 nm en agua. El principio es determinar la concentración de irinotecán primero a partir de la absorbencia a 369 nm, cuando el ibuprofeno carece de absorción. Después, la concentración de ibuprofeno podría calcularse mediante la sustracción de la absorbencia de irinotecán a 272 nm a partir del espectro de la mezcla de fármaco. Con el uso de 272 nm en lugar de los 263 nm usados habitualmente se reduce la posible interferencia de residuos de DMSO en la carga de fármaco. Sin embargo, se halló que la absorbencia de ibuprofeno es mucho menor en comparación con la de irinotecán debido a una solubilidad baja en agua del ibuprofeno, estando el pico de ibuprofeno cubierto completamente por irinotecán. Por consiguiente, la forma de usar UV para determinar la concentración de ibuprofeno en la mezcla de ibuprofeno e irinotecán no resulta precisa. En este estudio, la concentración de irinotecán se cuantificó mediante UV, mientras que la concentración de ibuprofeno se hizo mediante HPLC.
Carga de ibuprofeno e irinotecán en perlas
Todas las muestras cargadas con ibuprofeno en la etapa primera retuvieron su forma esférica y no parecieron formarse cristales de ibuprofeno. Después de la carga de irinotecán, el Ejemplo 5-1 con alta carga de ibuprofeno tuvo gran cantidad de cristales de ibuprofeno que aparecieron en la superficie de las perlas. En comparación, en el Ejemplo 5-4, con la misma cantidad de ibuprofeno, no se observaron cristales, lo que es debido al efecto del alto contenido en inhibidor de cristalización de aceite de ricino que existía dentro de las perlas. Por consiguiente, el aceite de ricino actúa como un excipiente para impedir la formación de cristales de ibuprofeno con la inhibición en la formación de los mismos.
La Tabla 4 indica la carga de fármaco en cada muestra. La carga de irinotecán en todas las muestras es cercana al 100 % de la capacidad de carga máxima para irinotecán en las perlas no cargadas, estando cerca de 50 a 60 mg/ml, habiendo por debajo de ella una eficiencia de carga superior al 98 %.
T l 4. r f rm n rl n r

La carga de ibuprofeno es relativamente baja, y la eficiencia de carga estimada se encuentra entre el 29 % y el 58 %. Un objetivo de carga más bajo para ibuprofeno otorga relativamente una eficiencia de carga más elevada. La razón de la carga de ibuprofeno relativamente baja se debe que el tamaño de la perla encoge cuando las perlas entran en contacto con una solución rica en aceite de ricino. De este modo, la carga de fármaco disminuyó con un menor volumen de las perlas
Distribución del tamaño de las perlas cargadas de fármaco
En la Figura 10, se muestra la distribución del tamaño de las perlas cargadas de fármaco. Los parámetros
correspondientes se indican en la Tabla 5. Las perlas cargadas de ibuprofeno casi conservaron el mismo tamaño siendo de DC Bead y sin la carga de fármaco, en el intervalo de 500 a 700 pm. Sin embargo, el tamaño disminuyó marcadamente con la carga de irinotecán debido a un aumento en la hidrofobicidad de las perlas cargadas de fármaco, la disminución del tamaño medio es de aproximadamente 140 a 150 pm. Es de destacar que las perlas con una variación de la carga de ibuprofeno tienen casi el mismo tamaño medio y la misma distribución de tamaño.

Morfología de las perlas cargadas de fármaco
Una micrografía de las perlas cargadas de ibuprofeno del Ejemplo 5.1 demuestra que la estructura de las perlas cargadas consiste en un área central grande con ibuprofeno y aceite de ricino, junto con una capa en la superficie de la perla libre de fármaco y aceite. El aceite de ricino atrapado dentro de las perlas hace que en la fase hidrofóbica se retenga ibuprofeno y se impida la formación de cristales de ibuprofeno. El resto de Ejemplos 5-2, 5.3 y 5.4 con ibuprofeno en la etapa primera de carga cuentan con la misma estructura mostrada aquí. Además, se observaron algunas pequeñas gotas de aceite entre las perlas, que resultaron ser los residuos de la mezcla de aceite de ricino e ibuprofeno en la solución.
Cuando se cargó irinotecán mediante mezcla con rodillo durante una noche, se observaron cristales de ibuprofeno sobre la superficie de las perlas en el Ejemplo 5.1, además de darse una agregación de perlas ocurrida como resultado de la formación de cristales. No se halló cristalización en otras muestras. Este fenómeno es debido al contenido más elevado de ibuprofeno en las perlas, y sin suficiente aceite de ricino para impedir la formación de cristales, cuando se cargó el irinotecán. Aunque el irinotecán interactúa con el grupo AMPS en las perlas, es todavía hidrofóbico y puede tender a disolverse parcialmente en el aceite de ricino, lo que podría reducir la solubilidad del ibuprofeno. En el producto del Ejemplo 5.4, con un nivel más elevado de aceite de ricino y una alta carga de ibuprofeno, después de la carga de irinotecán, no se observaron cristales de ibuprofeno.
Elución de perlas cargadas de fármaco:
Las Figuras 11 y 12 muestran el perfil de elución de irinotecán e ibuprofeno a partir de las perlas dentro de PBS a 25° C. En la Figura 11, la tasa de elución de irinotecán con diferente contenido de aceite de ricino y la carga de ibuprofeno son casi las mismas. La tasa de elución de irinotecán es también comparable con la de irinotecán aislado en la que casi el 100 % del fármaco fue eluido después de 5 horas. Esto indica que el aceite de ricino y la carga de ibuprofeno no tienen efecto en la elución de irinotecán, y que no existe interacción entre ibuprofeno e irinotecán. En la Figura 12, la elución de ibuprofeno se completó después de 1 o 2 horas en PBS. En comparación con la elución de ibuprofeno a partir de perlas cargadas de un único fármaco, la elución, una vez más, resulta similar ya que no se da interacción fármaco a fármaco.
Ejemplo 6: Evaluación de citotoxicidad in vitro de perlas cargadas de doxorrubicina usando células con CHC (referencia).
Las perlas de doxorrubicina (500-700 pm) se prepararon de acuerdo con los procedimientos expuestos en el Ejemplo 2 del documento WO04071495. Se monitorizó la elución de perlas cargadas de doxorrubicina en medios de
cultivo celular con una línea celular (HepG2) de carcinoma (de hígado) hepatocelular en humanos. Se utilizó un ensayo Promega con lactato deshidrogenasa (LDH) para evaluar los efectos de citotoxicidad sobre la línea celular HepG2 en los momentos temporales definidos por 24, 48 y 72 horas.
Preparación del ensayo
En condiciones asépticas y en tres placas estériles de 96 pocillos con fondo plano (Cellstar®, 655180) fueron sedimentadas 20.000 células de HepG2 en un medio de 200 pl dejándose en una incubadora (37 °C, 5% de CO2 ) durante 20 horas. Después de la incubación, el medio se aspiró cuidadosamente de todos los pocillos, para luego añadirse 200 pl de un medio de PRF a los pocillos. Se contabilizaron, una, tres y diez perlas precargadas de doxorrubicina (37,5 mg por cada ml de perlas) en condiciones asépticas dentro de 200 pl de medio de cultivo celular libre de rojo de fenol en una placa de 96 pocillos.
Ensayo de elución de doxorrubicina
La placa se incubó a 37oC, con un 5% de CO2. En los momentos temporales de 24, 48 y 72 horas, 100 pl de cada pocillo se sometieron a barrido a 490 nm (Biotek, ELX800). La elución se calculó mediante la comparación de la absorbencia con la curva de calibración estándar.
En periodos de tiempo mayores y con mayores números de perla, se liberó más doxorrubicina en el medio de cultivo celular (Figura 13). El perfil de elución es diferente para diferentes números de perla; la meseta para 10 perlas en un periodo temporal posterior puede deberse a la saturación del medio de cultivo celular con doxorrubicina.
Ensayo de viabilidad celular
Este ensayo mide la cantidad del LDH liberado a partir de las células restantes viables después de que se han lavado los residuos celulares muertos. El LDH es liberado después de que las membranas de células viables se han visto alteradas mediante la congelación de las células.
En cada momento temporal, se aspiró cuidadosamente el medio y las perlas de la placa original y se añadieron 200 pl de PBS estéril, retirándose tres veces para lavar las células de cualquier cantidad residual de doxorrubicina y desecho celular sobrante. Antes de someter la placa a -80oC durante una hora, se añadieron 200 pl de PRF; esto alteró intencionalmente todas las membranas de células, liberando todas las enzimas de lactato deshidrogenasa a partir de las células vivas restantes. Después de su retirada del congelador, la placa se colocó inmediatamente en una incubadora bajo condiciones estándar (37oC, 5% CO2) durante treinta minutos. Se eliminaron 50 pl del sobrenadante de todos los pocillos dentro de sus respectivos pocillos nuevos, seguido de 50 pl de la mezcla (Promega) del sustrato del ensayo de LDH. La placa original con células se conservó para análisis posteriores. La placa nueva se dejó a temperatura ambiente y sin iluminación. Después de 30 minutos, se añadieron 50 pl de solución quelante (ácido acético (1M)) para detener la reacción. Se leyó entonces la placa usando un lector de placas (Biotek) a 490 nm, en caso de que la lectura resultara demasiado elevada (>3), se diluyó la solución usando 75 pl de sobrenadante y 75 pl de medio de PRF. El porcentaje de células alteradas fue calculado.
El número de células viables disminuye a medida que aumenta la exposición y el número de perlas de DC precargadas de doxorrubicina (Figura 14).
Ejemplo 7: Citotoxicidad de las combinaciones de perlas de doxorrubicina y perlas de rapamicina e in vitro Se usó un ensayo de LDH para evaluar los efectos de citotoxicidad en la línea celular HepG2 en los momentos temporales de 24, 48 y 72 horas con diferentes combinaciones de perlas de rapamicina/doxorrubicina.
Las perlas de rapamicina (500-700 pm) se cargaron y analizaron tal como se describe en PCT/EP2007/50690. La carga resultante fue 21,4 mg/ml.
Las perlas de doxorrubicina (500-700pm) se cargaron con 37,5 mg por ml de perlas tal como se describe en el documento WO04071495.
Preparación del ensayo
En condiciones asépticas y en tres placas estériles de 96 pocillos con fondo plano (Cellstar®, 655180) fueron sedimentadas 20.000 células de HepG2 en un medio de 200 pl dejándose en una incubadora (37 °C, 5% de CO2 ) durante 20 horas.
Después de la incubación, el medio se aspiró cuidadosamente de todos los pocillos, para luego añadirse 200pl de un
medio de PRF a los pocilios.
Las perlas precargadas de doxorrubicina y las perlas precargadas de rapamicina se contabilizaron en condiciones asépticas dentro de 200 pl de medio de cultivo celular libre de rojo de fenol en una placa de 96 pocillos. El número de cada tipo de perlas colocado en cada pocillo puede verse en la Tabla 5 (n=6). Se realizó un control de cada tipo de fármaco junto con el ensayo (de 1 a 5 perlas de un tipo de fármaco únicamente).

Después de una exposición de 72 horas a las células, como se describe en el Ejemplo 6, se realizó un ensayo de LDH sobre los controles y las muestras en la Tabla 6.
También se realizó un ensayo MTS en los pocillos. A las 72 horas, se retiró cuidadosamente el medio de todos los pocillos y se lavó cada pocillo tres veces con 200 pl de PBS estéril. Después de añadir 100 pl de medio de PRF a cada pocillo, se añadieron también 20 pl de la solución MTS preparada (Promega). Tras una incubación de dos horas (37° C, 5% CO2) se realizó una lectura de la placa a 490 nm usando el lector de placas y como en el ensayo de LDH, si la lectura resultó demasiado elevada entonces se diluyeron las muestras (75 pl de medio de PRF: muestras de 75 pl).
Sinergia de rapamicina y doxorrubicina
El experimento de viabilidad celular con las perlas de rapamicina y las perlas de doxorrubicina, en diferentes proporciones, permite que se establezca la sinergia de cualquier fármaco. Una sola perla de rapamicina y una sola perla de doxorrubicina juntas en un pocillo dejaron menos células viables que dos perlas de un tipo de fármaco. Esta tendencia se detecta a través de las combinaciones de perlas analizadas (Figura 15 y 16). Se observa una tendencia parecida con el ensayo MTS (Figuras 17 y 18).
Gracias a Calcusyn versión 2.1 (Biosoft), un valor del índice de combinación puede establecerse para cada combinación usando los datos del ensayo de LDH (Tabla 7). El valor del índice de combinación se refiere a una escala (Tabla 7). Usando estas descripciones, se observa un fuerte sinergismo según estas condiciones.
Tabla 6: Descripción y valores del índice de combinación (IC). (Fa = fracción afectada por la dosis, 1= 100%
=

Tabla 7: Descri ción valores del índice de combinación

Como puede verse en todas las combinaciones de doxorrubicina-rapamicina produjeron un IC que indicaba un sinergismo muy fuerte entre los dos fármacos. Ambos ensayos muestran sinergia entre ambos fármacos. Los datos no muestran una proporción preferida, aunque aparece sinergia para todas las combinaciones.
Ejemplo 8: Citotoxicidad in vitro de perlas cargadas de doxorrubicina y rapamicina, individuales y en combinación.
El Ejemplo 7 se repitió sustancialmente como se describió, con un número ampliado de perlas de uno de los fármacos. Los resultados del ensayo MTS, es decir, indicando el % de viabilidad celular (escala logarítmica) de las células HepG2 después de una exposición de 72 horas, concluyeron como la media de 6 muestras y se muestran en la Figura 19.
Resultados
Cuando fue eluida individualmente, la doxorrubicina resultó más citotóxica para las células que la rapamicina (Figura 1). Sin embargo, al usarse en combinación, se observó una reducción mayor en la viabilidad celular. Los índices de combinación para todas las combinaciones de perlas son <0,1; esto indica una fuerte sinergia positiva entre los dos fármacos eluidos.
Ejemplo 9: Sinergia de rapamicina y doxorrubicina en líneas celulares de carcinoma pancreático humano (PSN1) y de carcinoma hepatocelular humano (HepG2)
in-vitro
Se usó un ensayo MTS para evaluar los efectos citotóxicos y sinergísticos de las perlas con carga dual en las células HepG2 y PSN1. Las perlas cargadas de rapamicina y las perlas cargadas de doxorrubicina fueron las mismas que las descritas en el Ejemplo 7. Las perlas cargadas con carga dual de doxorrubicina y rapamicina se cargaron y analizaron, como se describe en el Ejemplo 4, Procedimiento 2, usando una solución de 1 ml de rapamicina a 60 mg/ml y una solución de 1 ml de doxorrubicina a 37,5 mg/ml. La carga resultante en las perlas con carga dual fue de rapamicina a 21,4 mg/ml y de doxorrubicina a 35,5 mg/ml.
Preparación del ensayo
El ensayo se preparó como se describe en el Ejemplo 7 con la utilización de células HepG2 y placas adicionales de células PSN1 para su análisis.
Viabilidad celular
Las perlas precargadas de doxorrubicina, las perlas precargadas de rapamicina y las perlas con precarga dual de rapamicina y doxorrubicina se contabilizaron (1, 2 o 3) y se añadieron a las 20.000 células preparadas tipo PSN1 o HepG2 en el medio de cultivo celular, para después colocarse en una incubadora (37oC, 5% de CO2 ).
Se realizó un ensayo MTS (descrito en el Ejemplo 7) después de una exposición de 24, 48 y 72 horas.
La combinación de perlas con carga dual mostró una muerte celular mayor que en las células con carga única en todos los momentos temporales tanto para las células PSN1 (Figuras 20-22) como para las células HspG2 (Figuras 23-25).
Ejemplo 10: Perla con carga dual comparada con las dos perlas con carga única
Se utilizó un ensayo MTS para demostrar que no había diferencia entre los efectos citotóxicos de una perla con carga dual de rapamicina y doxorrubicina en comparación con dos perlas con carga única (una cargada de doxorrubicina y la otra cargada de rapamicina).
Las perlas cargadas de fármaco usadas fueron los mismos lotes que los descritos en el Ejemplo 9.
El ensayo se configuró como se describe en el Ejemplo 7 con el uso de células HepG2. Se añadieron de una a cuatro perlas con carga dual a las 20.000 células HepG2 preparadas en 200 pl de medio y se añadió la dosis equivalente, usando perlas con carga única, al otro pocillo en el mismo volumen de medio. Las placas se colocaron en una incubadora (a 37oC, 5% de CO2).
Se realizó un ensayo MTS (descrito en el Ejemplo 7) después de una exposición de 24, 48 y 72 horas.
El efecto citotóxico de las perlas eluyentes de fármaco resultó similar sin tener en cuenta si los fármacos eran cargados en una perla o en dos perlas separadas (Figuras 26-28) y se añadieron a las 20.000 células preparadas tipo PSN1 o HepG2 en el medio de cultivo celular, para después colocarse en una incubadora (37° C, 5% de CO2). Se realizó un ensayo MTS (descrito en el Ejemplo 7) después de una exposición de 24, 48 y 72 horas.
La combinación de perlas con carga dual mostró una muerte celular mayor que en las células con carga única en
todos los momentos temporales tanto para las células PSN1 (Figuras 20-22) como para las células HspG2 (Figuras 23-25).
Ejemplo 10: Perla con carga dual comparada con las dos perlas con carga única
Se utilizó un ensayo MTS para demostrar que no había diferencia entre los efectos citotóxicos de una perla con carga dual de rapamicina y doxorrubicina en comparación con dos perlas con carga única (una cargada de doxorrubicina y la otra cargada de rapamicina).
Las perlas cargadas de fármaco usadas fueron los mismos lotes que los descritos en el Ejemplo 9.
El ensayo se configuró como se describe en el Ejemplo 7 con el uso de células HepG2. Se añadieron de una a cuatro perlas con carga dual a las 20.000 células HepG2 preparadas en 200 pl de medio y se añadió la dosis equivalente, usando perlas con carga única, al otro pocillo en el mismo volumen de medio. Las placas se colocaron en una incubadora (a 37° C, 5% de CO2).
Se realizó un ensayo MTS (descrito en el Ejemplo 7) después de una exposición de 24, 48 y 72 horas.
El efecto citotóxico de la perlas eluyentes de fármaco resultó similar sin tener en cuenta si los fármacos eran cargados en una perla o en dos perlas separadas (Figuras 26-28).
Claims (23)
1. Una composición que comprende una población de microesferas, cada una de las cuales tiene una matriz polimérica e, incorporado conjuntamente dentro de la matriz, un primer o un segundo compuesto activo farmacéuticamente, donde el polímero es un polímero no hidrosoluble e hinchable en agua que está aniónicamente cargado,
donde el primer activo está catiónicamente cargado, y el segundo activo es electroestáticamente neutral y donde uno de los activos es un compuesto citotóxico, mientras que el otro activo posee actividad complementaria con el compuesto citotóxico en el tratamiento tumoral.
2. Una composición de acuerdo con la reivindicación 1, donde el primer compuesto catiónicamente cargado es un agente citotóxico.
3. Una composición de acuerdo con la reivindicación 1, en la que el segundo activo es hidrosoluble hasta un grado menor de 10 g/l a temperatura ambiente.
4. Una composición de acuerdo con cualquier reivindicación anterior, en la que el agente citotóxico es mitoxantrona o un compuesto de antraciclina que tiene un grupo amino, de la Fórmula general IV:

5. Una composición de acuerdo con la reivindicación 4, en la que la antraciclina es doxorrubicina.
6. Una composición de acuerdo con cualquier reivindicación anterior, en la que el activo catiónico es un compuesto de la Fórmula general I

en la que R1 se selecciona de entre H, halógeno, hidroxilo y alquilo (C1-6) inferior, opcionalmente sustituido por un grupo hidroxilo, amino, alcoxi, halógeno, acilo y aciloxilo;
A es C(O)O o CH2; y
R es NR2R3 donde R2 y R3 son los mismos o diferentes y cada uno representa un átomo de hidrógeno, un grupo alquilo C1-4 sustituido o sin sustituir o un grupo hetercíclico o carbocíclico sin sustituir, o R2 y R3 junto con el átomo de nitrógeno al que están unidos forman un anillo heterocíclico sustituido que puede interrumpirse por -O-, -S- o >NR4 en el que R4 es un átomo de hidrógeno, un grupo alquilo C1-4 sustituido o sin sustituir o un grupo fenilo
sustituido o sin sustituir; y donde la agrupación -A-R está unida a un átomo de carbono situado en cualquiera de las posiciones 9, 10 u 11 en el anillo A del compuesto de camptotecina y R1 es sustituido en el anillo A o B, incluyendo las sales del mismo.
7. Una composición de acuerdo con la reivindicación 6, en la que el activo catiónico es doxorrubicina.
8. Un compuesto de acuerdo con la reivindicación 3, en la que el compuesto de baja hidrosolubilidad es rapamicina o una análogo de rapamicina, preferentemente rapamicina.
9. Una composición de acuerdo con cualquier reivindicación anterior, en la que el o cada polímero es no biodegradable.
10. Una composición de acuerdo con cualquier reivindicación anterior, en la que el o cada polímero es hinchable en agua hasta un contenido de agua en equilibrio cuando se hincha en agua a 37° C en el intervalo del 40 al 99 %, preferentemente del 75 al 95 %.
11. Una composición de acuerdo con cualquier reivindicación anterior, en la que el o cada polímero es totalmente sintético.
12. Una composición de acuerdo con la reivindicación 11, en la que el o cada polímero es alcohol polivinílico entrecruzado.
13. Una composición de acuerdo con la reivindicación 12, donde la matriz polimérica está formada a partir de un macrómero de alcohol polivinílico, que tiene más de un grupo etilénicamente insaturado colgante por molécula, mediante polimerización radical de los grupos etilénicos.
14. Una composición de acuerdo con la reivindicación 13, donde el macrómero está copolimerizado con monómeros etilénicamente insaturados, opcionalmente incluyendo un monómero no iónico y/o iónico que incluye un monómero aniónico.
15. Una composición de acuerdo con la reivindicación 14, en la que los monómeros etilénicamente insaturados incluyen un monómero iónico que tiene la Fórmula III general
Y1BQ1 III
en que Y1 se selecciona de entre

CH2=C(R10)-CH2-O-, CH2=C(R10)-CH2 OC(O)-, CH2=C(R10)OC(O)-, CH2=C(R10)-O-, CH2=C(R10)CH2OC(O)N(R11)-, R12OOCCR10=CR10C(O)-O-, R10CH=CHC(O)O-, R10CH=C(COOR12)CH2-C(O)-O-,

donde:
R10 es hidrógeno o un grupo alquilo C1-C4;
R11 es hidrógeno o un grupo alquilo C1-C4;
R12 es hidrógeno o un grupo alquilo C1-4 o BQ1 donde B y Q1 se definen a continuación;
A1 es -O- o -NR11-;
K1 es un grupo -(CH2)rOC(O)-, -(CH2)rC(O)O-, - (CH2)rOC(O)O-, - (CH2)rNR13-, -(CH2)rNR13C(O)-, -(CH2)rC(O)NR13-, -(CH2)rNR13C(O)O-, - (CH2)rOC(O)NR13-, -(CH2)rNR13C(O)NR13- (en el que los grupos R13 son iguales o diferentes), (CH2)rO-, -(CH2)rS03-, u, opcionalmente en combinación con B, un enlace de valencia y r es de 1 a 12 y R13 es hidrógeno o un grupo alquilo C1-C4 ;
B es una cadena alcanodiilo, oxaalquileno, alcanodiiloxaalcanodiilo o alcanodiiloligo(oxaalcanodiilo) lineal oramificada que contiene opcionalmente uno o más átomos de flúor hasta e incluyendo cadenas perfluoradas o, si Q1 o Y1 contiene un átomo de carbono terminal unido a B, un enlace de valencia; y
Q1 es un grupo aniónico.
16. Una composición de acuerdo con la reivindicación 15, en la que Q1 es un grupo aniónico, preferentemente un grupo carboxilato, carbonato, sulfonato, sulfato, nitrato, fosfonato o fosfato.
17. Un procedimiento para cargar dos activos juntamente dentro de microesferas que implica las etapas de:
Poner en contacto las microesferas que comprenden una matriz de un polímero entrecruzado cargado aniónicamente, hinchable en agua y no hidrosoluble en una forma sustancialmente seca con una solución de un fármaco que tiene una solubilidad en agua menor de 10 g/l a temperatura ambiente en un primer disolvente orgánico, donde una solución de fármaco en disolvente se impregna dentro de las microesferas;
separar la solución de fármaco que no ha impregnado las microesferas;
poner en contacto las microesferas impregnadas con el líquido acuoso en el cual el fármaco es precipitado en el núcleo de las microesferas;
poner en contacto las microesferas que contienen el fármaco precipitado con una solución acuosa de un fármaco iónico que tiene una carga opuesta a la del polímero para permitir la carga del intercambio de iones; y recuperar las microesferas con carga dual, donde los fármacos son seleccionados como en cualquiera de las reivindicaciones 1 a 10.
18. Un procedimiento para cargar dos activos farmacéuticos conjuntamente dentro de una matriz de administración de fármaco que comprende las etapas de:
una matriz de administración de fármaco que comprende un polímero cargado iónicamente, hinchable en agua y no hidrosoluble que es hinchable en agua a temperatura ambiente hasta un contenido de agua en equilibrio con el intervalo del 40 al 99 % por peso en base al polímero más agua que es puesto en contacto con una solución de fármaco que tiene una solubilidad en agua menor de 10 g/l a temperatura ambiente en un disolvente orgánico que es capaz de hinchar el polímero, en el cual la solución se impregna dentro de la matriz;
se separa cualquier solución de fármaco que no ha impregnado la matriz polimérica;
la matriz impregnada se pone en contacto con el agua en la cual el fármaco es precipitado en la matriz;
la matriz que tiene el fármaco precipitado que tiene baja solubilidad en agua se pone en contacto con una solución acuosa de un fármaco cargado iónicamente en el que la matriz se hincha y tiene lugar el intercambio de iones con los contraiones asociados con el polímero y el fármaco cargado es unido electroestáticamente en la matriz polimérica.
19. Un procedimiento de acuerdo con la reivindicación 17 o 18, en la que el activo que tiene una hidrosolubilidad de menos de 10 g/l es rapamicina o un análogo.
20. Un procedimiento de acuerdo con la reivindicación 18, en la que el activo cargado iónicamente es un compuesto de mitoxantrona o antraciclina que tiene un grupo amino, preferentemente doxorrubicina.
21. Una composición de acuerdo con cualquiera de las reivindicaciones 1 a 16 para uso en un procedimiento para tratar un tumor sólido mediante administración a un animal que padece de un tumor de tal forma que el primer y segundo activo son cada uno administrados en el tumor diana.
22. Una composición para uso en un procedimiento de acuerdo con la reivindicación 21, para inyección intratumoral, o para inyección en el margen de resección tras la eliminación del tumor mediante cirugía o donde la matriz tiene la forma de microesferas, que tienen diámetros en el intervalo de 100 a 1500 um al hincharse en agua destilada a 37 °C para la embolización de un vaso sanguíneo que irriga el tumor.
23. Una composición para uso en un procedimiento de acuerdo con la reivindicación 22, en la que el tumor es un tumor hepático.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0716003A GB0716003D0 (en) | 2007-08-16 | 2007-08-16 | Delivery of drug combinations |
| EP07121158 | 2007-11-20 | ||
| PCT/GB2008/050722 WO2009022190A1 (en) | 2007-08-16 | 2008-08-18 | Delivery of drug combinations |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2706023T3 true ES2706023T3 (es) | 2019-03-27 |
Family
ID=39941549
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES08788693T Active ES2706023T3 (es) | 2007-08-16 | 2008-08-18 | Administración de combinaciones de fármacos |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20110229572A1 (es) |
| EP (1) | EP2190409B9 (es) |
| JP (3) | JP2010536738A (es) |
| ES (1) | ES2706023T3 (es) |
| TR (1) | TR201820076T4 (es) |
| WO (1) | WO2009022190A1 (es) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1985286A1 (en) * | 2007-04-24 | 2008-10-29 | Biocompatibles UK Limited | Microspheres for treatment of brain tumours |
| JP6138140B2 (ja) * | 2011-10-25 | 2017-05-31 | ネクター セラピューティクス | 癌患者の治療 |
| RU2697480C2 (ru) * | 2014-03-06 | 2019-08-14 | Юниверсити Оф Саутерн Калифорния | Применение режима краткосрочного голодания в сочетании с ингибиторами киназ для усовершенствования традиционной химио-лекарственной эффективности и пригодности и обращения вспять побочных эффектов от киназ в нормальных клетках и тканях |
| WO2019164397A1 (en) * | 2018-02-23 | 2019-08-29 | Van Rijn Beheer B.V. | Porous embolization microspheres comprising drugs |
| WO2020077170A1 (en) | 2018-10-12 | 2020-04-16 | University Of Pittsburgh- Of The Commonwealth System Of Higher Education | Small polymeric carriers for delivery of agents |
| CN110327300B (zh) * | 2019-07-23 | 2020-08-07 | 赵修文 | 一种负载药物的聚乙烯醇微球 |
| WO2022010850A1 (en) * | 2020-07-06 | 2022-01-13 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | Polymeric carriers for delivery of therapeutic agents |
| WO2022047544A1 (en) * | 2020-09-04 | 2022-03-10 | IP Cornerstone Pty Ltd | Minimally invasive treatment of osteoarthritis and other conditions |
| EP4337219A1 (en) * | 2021-05-12 | 2024-03-20 | Neupharma, Inc. | Certain chemical entities, compositions, and methods |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2762140B1 (en) * | 2001-02-19 | 2017-03-22 | Novartis AG | Treatment of solid brain tumours with a rapamycin derivative |
| JP4778679B2 (ja) * | 2001-10-03 | 2011-09-21 | セレーター ファーマシューティカルズ インコーポレイテッド | 併用薬剤を送達するための組成物 |
| US7850990B2 (en) * | 2001-10-03 | 2010-12-14 | Celator Pharmaceuticals, Inc. | Compositions for delivery of drug combinations |
| WO2003074026A1 (en) * | 2002-03-07 | 2003-09-12 | Biocompatibles Uk Limited | Drug carriers comprising amphiphilic block copolymers |
| US20040161466A1 (en) * | 2003-02-14 | 2004-08-19 | Biocompatibles Uk Limited | Chemoembolisation |
| KR101157260B1 (ko) * | 2003-02-12 | 2012-06-15 | 바이오컴페터블즈 유케이 리미티드 | 고형 종양의 화학색전술용 조성물 |
| JP2006522026A (ja) * | 2003-04-02 | 2006-09-28 | セレーター ファーマスーティカルズ、インク. | カンプトセシンとフルオロピリミジンとを組み合わせた組成物 |
| EP3085361A1 (en) * | 2004-09-07 | 2016-10-26 | Biocompatibles UK Limited | Compositions comprising camptothecins in microspheres |
| US8663673B2 (en) * | 2005-07-29 | 2014-03-04 | Surmodics, Inc. | Devices, articles, coatings, and methods for controlled active agent release or hemocompatibility |
-
2008
- 2008-08-18 EP EP08788693.3A patent/EP2190409B9/en not_active Not-in-force
- 2008-08-18 ES ES08788693T patent/ES2706023T3/es active Active
- 2008-08-18 US US12/672,031 patent/US20110229572A1/en active Pending
- 2008-08-18 JP JP2010520637A patent/JP2010536738A/ja active Pending
- 2008-08-18 WO PCT/GB2008/050722 patent/WO2009022190A1/en active Application Filing
- 2008-08-18 TR TR2018/20076T patent/TR201820076T4/tr unknown
-
2014
- 2014-01-30 JP JP2014016185A patent/JP2014114307A/ja active Pending
-
2016
- 2016-06-08 JP JP2016114725A patent/JP2016216465A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009022190A1 (en) | 2009-02-19 |
| JP2016216465A (ja) | 2016-12-22 |
| EP2190409B1 (en) | 2018-10-24 |
| EP2190409B9 (en) | 2019-03-06 |
| US20110229572A1 (en) | 2011-09-22 |
| EP2190409A1 (en) | 2010-06-02 |
| TR201820076T4 (tr) | 2019-01-21 |
| JP2010536738A (ja) | 2010-12-02 |
| JP2014114307A (ja) | 2014-06-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2706023T3 (es) | Administración de combinaciones de fármacos | |
| Wolinsky et al. | Local drug delivery strategies for cancer treatment: gels, nanoparticles, polymeric films, rods, and wafers | |
| JP5792691B2 (ja) | 塞栓剤からの薬物送達 | |
| JP5559035B2 (ja) | 脳腫瘍の治療のための微小球 | |
| ES2672625T3 (es) | Carga de fármacos hidrófobos en sistemas de suministro poliméricos hidrófilos | |
| JP5960751B2 (ja) | 膵がんの治療 | |
| ES2542242T3 (es) | Composición de quimioembolización que comprende agentes antiangiogénicos | |
| Doddapaneni et al. | A three-drug nanoscale drug delivery system designed for preferential lymphatic uptake for the treatment of metastatic melanoma | |
| Hao et al. | 3D printing‐based drug-loaded implanted prosthesis to prevent breast cancer recurrence post‐conserving surgery | |
| Lin et al. | Development of a 5-fluorouracil-loaded PLGA microsphere delivery system by a solid-in-oil-in-hydrophilic oil (S/O/hO) novel method for the treatment of tumors | |
| Kuche et al. | Synergistic anticancer therapy via ferroptosis using modified bovine serum albumin nanoparticles loaded with sorafenib and simvastatin | |
| Pande | Formulation and Characterization of Dual Drug-Loaded Liposomes for Treatment of Pancreatic Cancer | |
| Altintas et al. | Targeted delivery of kinase inhibitors: a nanomedicine approach for improved selectivity in cancer | |
| Lewis | Drug eluting beads in the treatment of liver cancer | |
| CZ307237B6 (cs) | Supramolekulární komplex oxycelulózové matrice s taxolovým derivátem s postupným uvolňováním taxolového derivátu a jeho použití | |
| JP2018505200A (ja) | 腫瘍を治療するためのアクリフラビンの局所送達形態 |