EP0536012A1 - Procédé de débromation de dérivés dibromés du naphtol - Google Patents
Procédé de débromation de dérivés dibromés du naphtol Download PDFInfo
- Publication number
- EP0536012A1 EP0536012A1 EP92402327A EP92402327A EP0536012A1 EP 0536012 A1 EP0536012 A1 EP 0536012A1 EP 92402327 A EP92402327 A EP 92402327A EP 92402327 A EP92402327 A EP 92402327A EP 0536012 A1 EP0536012 A1 EP 0536012A1
- Authority
- EP
- European Patent Office
- Prior art keywords
- naphthol
- dibromo
- catalyst
- organic solvent
- process according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000000034 method Methods 0.000 title claims abstract description 36
- YWYAJOWYHCYKLX-UHFFFAOYSA-N 2,3-dibromonaphthalen-1-ol Chemical class C1=CC=C2C(O)=C(Br)C(Br)=CC2=C1 YWYAJOWYHCYKLX-UHFFFAOYSA-N 0.000 title 1
- 239000003054 catalyst Substances 0.000 claims abstract description 24
- 239000003960 organic solvent Substances 0.000 claims abstract description 20
- UONOETXJSWQNOL-UHFFFAOYSA-N tungsten carbide Chemical compound [W+]#[C-] UONOETXJSWQNOL-UHFFFAOYSA-N 0.000 claims abstract description 16
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 15
- 239000012429 reaction media Substances 0.000 claims abstract description 8
- 230000002378 acidificating effect Effects 0.000 claims abstract description 6
- YZCKVEUIGOORGS-UHFFFAOYSA-N Hydrogen atom Chemical compound [H] YZCKVEUIGOORGS-UHFFFAOYSA-N 0.000 claims abstract description 5
- 150000001875 compounds Chemical class 0.000 claims abstract description 5
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 19
- 125000000950 dibromo group Chemical group Br* 0.000 claims description 19
- 229910052742 iron Inorganic materials 0.000 claims description 9
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims description 8
- 229910052802 copper Inorganic materials 0.000 claims description 8
- 239000010949 copper Substances 0.000 claims description 8
- 229910052751 metal Inorganic materials 0.000 claims description 8
- 239000002184 metal Substances 0.000 claims description 8
- 239000002253 acid Substances 0.000 claims description 6
- 150000001247 metal acetylides Chemical class 0.000 claims description 5
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 5
- 239000000843 powder Substances 0.000 claims description 5
- 150000004945 aromatic hydrocarbons Chemical class 0.000 claims description 3
- 230000031709 bromination Effects 0.000 claims description 3
- 238000005893 bromination reaction Methods 0.000 claims description 3
- 150000002148 esters Chemical class 0.000 claims description 3
- 150000008282 halocarbons Chemical class 0.000 claims description 3
- 150000007522 mineralic acids Chemical class 0.000 claims description 3
- 150000007524 organic acids Chemical class 0.000 claims description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 claims description 2
- 150000002170 ethers Chemical class 0.000 claims description 2
- 235000019253 formic acid Nutrition 0.000 claims description 2
- UFHFLCQGNIYNRP-VVKOMZTBSA-N Dideuterium Chemical compound [2H][2H] UFHFLCQGNIYNRP-VVKOMZTBSA-N 0.000 claims 1
- 238000006243 chemical reaction Methods 0.000 abstract description 18
- VKESFYLPKHQOOA-UHFFFAOYSA-N 1,6-dibromonaphthalen-2-ol Chemical group C1=C(Br)C=CC2=C(Br)C(O)=CC=C21 VKESFYLPKHQOOA-UHFFFAOYSA-N 0.000 abstract description 2
- YLDFTMJPQJXGSS-UHFFFAOYSA-N 6-bromo-2-naphthol Chemical compound C1=C(Br)C=CC2=CC(O)=CC=C21 YLDFTMJPQJXGSS-UHFFFAOYSA-N 0.000 abstract description 2
- 150000002790 naphthalenes Chemical class 0.000 abstract 1
- 239000001257 hydrogen Substances 0.000 description 13
- 229910052739 hydrogen Inorganic materials 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 11
- -1 chromium carbides Chemical class 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 7
- 150000001735 carboxylic acids Chemical class 0.000 description 7
- 239000000203 mixture Substances 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 239000003708 ampul Substances 0.000 description 6
- 239000011521 glass Substances 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 5
- 239000007795 chemical reaction product Substances 0.000 description 5
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 5
- 238000007256 debromination reaction Methods 0.000 description 5
- JWAZRIHNYRIHIV-UHFFFAOYSA-N 2-naphthol Chemical compound C1=CC=CC2=CC(O)=CC=C21 JWAZRIHNYRIHIV-UHFFFAOYSA-N 0.000 description 4
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 4
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 4
- 238000010908 decantation Methods 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical class OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- ODLMAHJVESYWTB-UHFFFAOYSA-N propylbenzene Chemical compound CCCC1=CC=CC=C1 ODLMAHJVESYWTB-UHFFFAOYSA-N 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 229910052721 tungsten Inorganic materials 0.000 description 3
- 239000010937 tungsten Substances 0.000 description 3
- LEONUFNNVUYDNQ-UHFFFAOYSA-N vanadium atom Chemical compound [V] LEONUFNNVUYDNQ-UHFFFAOYSA-N 0.000 description 3
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Natural products OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N hexanedioic acid Natural products OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- YDSWCNNOKPMOTP-UHFFFAOYSA-N mellitic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(O)=O)=C(C(O)=O)C(C(O)=O)=C1C(O)=O YDSWCNNOKPMOTP-UHFFFAOYSA-N 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 229910052720 vanadium Inorganic materials 0.000 description 2
- HFVMEOPYDLEHBR-UHFFFAOYSA-N (2-fluorophenyl)-phenylmethanol Chemical compound C=1C=CC=C(F)C=1C(O)C1=CC=CC=C1 HFVMEOPYDLEHBR-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical compound C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 1
- AYFJBMBVXWNYLT-UHFFFAOYSA-N 2-bromo-6-methoxynaphthalene Chemical compound C1=C(Br)C=CC2=CC(OC)=CC=C21 AYFJBMBVXWNYLT-UHFFFAOYSA-N 0.000 description 1
- QISOBCMNUJQOJU-UHFFFAOYSA-N 4-bromo-1h-pyrazole-5-carboxylic acid Chemical compound OC(=O)C=1NN=CC=1Br QISOBCMNUJQOJU-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Natural products OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- KHLJKRBMZVNZOC-UHFFFAOYSA-N Methallenestril Chemical compound C1=C(OC)C=CC2=CC(C(CC)C(C)(C)C(O)=O)=CC=C21 KHLJKRBMZVNZOC-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 1
- BLXXJMDCKKHMKV-UHFFFAOYSA-N Nabumetone Chemical compound C1=C(CCC(C)=O)C=CC2=CC(OC)=CC=C21 BLXXJMDCKKHMKV-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- WAIPAZQMEIHHTJ-UHFFFAOYSA-N [Cr].[Co] Chemical compound [Cr].[Co] WAIPAZQMEIHHTJ-UHFFFAOYSA-N 0.000 description 1
- MEOSMFUUJVIIKB-UHFFFAOYSA-N [W].[C] Chemical compound [W].[C] MEOSMFUUJVIIKB-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000004996 alkyl benzenes Chemical class 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000011805 ball Substances 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229910000856 hastalloy Inorganic materials 0.000 description 1
- 238000007210 heterogeneous catalysis Methods 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 229960002977 methallenestril Drugs 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 description 1
- 229960004270 nabumetone Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- 239000010955 niobium Substances 0.000 description 1
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-N o-dicarboxybenzene Natural products OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- ZFACJPAPCXRZMQ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O.OC(=O)C1=CC=CC=C1C(O)=O ZFACJPAPCXRZMQ-UHFFFAOYSA-N 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 150000003738 xylenes Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/20—Unsaturated compounds containing keto groups bound to acyclic carbon atoms
- C07C49/255—Unsaturated compounds containing keto groups bound to acyclic carbon atoms containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C37/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C39/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring
- C07C39/24—Halogenated derivatives
- C07C39/38—Halogenated derivatives with at least one hydroxy group on a condensed ring system containing two rings
Definitions
- the present invention relates to a process for debromination of dibromo derivatives of naphthol. More specifically, the invention relates to a process for monodebromation of dibromo derivatives of naphthol of formula (1): by regio-selective catalytic hydrodebromination.
- reaction products more particularly targeted by the debromination process according to the invention correspond to formula (2):
- the bromo-6 hydroxy-2 naphthalene (also called bromo-6 ⁇ naphthol) of formula (2) above is a particularly interesting and important product. It is in fact used for the synthesis of 6-bromo-2-methoxy-naphthalene (by an alkylation using dimethyl sulfate or methanol), the latter product being, for its part, widely used for obtaining naproxen or nabumetone, which are well known for their therapeutic anti-inflammatory properties, or else methallenestril, which is an estrogen (see on this subject: the Merck Index, eleventh edition, 1989, pages 1002, 1014 and 937).
- 6-bromo-2-hydroxy-naphthalene can be prepared by stoichiometric metallic reduction of 1,6-dibromo-2-hydroxy-naphthalene, according to the following reaction: where M denotes a reducing metal such as iron or tin, the above dibromo derivatives can themselves be prepared simply by direct bromination of naphthol:
- reaction yields of desired monobrominated derivatives may prove to be insufficient.
- the object of the present invention is to propose a process for the debromination of certain dibromo derivatives of naphthol which makes it possible on the one hand to obviate the abovementioned drawbacks, and on the other hand to carry out regio-selective debromination, in particular in position 1 with high efficiency.
- the reaction is carried out in the presence of iron and / or copper.
- the method according to the invention has numerous advantages and great flexibility of use. First of all, it avoids the stoichiometric consumption of reducing metals. Furthermore, unexpectedly and surprisingly, it is also highly selective, in the sense that, for the products of formula (1) above, only the bromine atom in position 1 is substituted, and this even in the event of use of a strong stoichiometric excess of hydrogen. The yields of monobrominated derivatives are high.
- the reaction can furthermore be carried out over a wide range of pressures and temperatures, and according to numerous embodiments.
- the process according to the invention has the advantage of being able to be carried out directly, without separation or prior purification, on the reaction product obtained by direct bromination of ⁇ naphthol.
- the catalysts used in the present invention are catalysts based on tungsten carbide. These catalysts can also include, in addition to tungsten carbide, another or other metal monocarbons. As other metal monocarbons, mention may in particular be made of molybdenum, vanadium, niobium, tantalum, titanium, iron and chromium carbides; these are widely described in the literature.
- the amount of these other metal carbides preferably represents from 0.01 to 50% by weight relative to the totality of the carbides present.
- tungsten carbide is already a product well known per se, it should be noted that its use as a hydrodebromination catalyst according to a process according to the invention is, for its part, completely new.
- the catalyst is either based on mass tungsten carbon or on the basis of supported tungsten carbide.
- a support it is possible in particular to use oxides, such as silica, alumina and titanium dioxide, or carbon.
- the catalyst may be in the form of a monolithic substrate (honeycomb or the like) made of tungsten carbide, or of a monolithic substrate coated with a layer of tungsten carbide, or alternatively may be presented in the form of products divided into, or coated with, tungsten carbide.
- divided form we mean pulverulent products (powders) and also the articles obtained by shaping these products (balls, pellets, pellets, granules, extruded, agglomerated, and others, of circular, oval, trilobed or multilobed section, full or hollow).
- Catalysts of the ball, tablet and other type have the advantage of being able to be separated from the reaction medium very quickly by simple decantation.
- the pulverulent type catalysts generally require, for their separation, a filtration step.
- tungsten carbides which have been synthesized according to any process known per se.
- tungsten carbides with high specific surfaces can be manufactured according to the process described in patent application PCT / FR 90/00204.
- the amount of catalyst to be used is not critical and can vary within wide limits; generally from 0.01% to 50% by weight of catalyst is used relative to the amount of dibromo derivative used.
- the reaction medium it is also possible to introduce small quantities of copper and / or iron into the reaction medium, with the aim of substantially improving the yields of the desired monobromo derivative.
- These elements can be provided in the metallic state or in the form of a salt soluble in the reaction medium.
- the amount, in moles, of iron and / or copper which is generally used is then between 0.01 and 0.1 times the amount in moles of dibromo derivative used in the reaction. It will be noted that such quantities are considerably lower than the stoichiometric quantities which it is necessary to use in accordance with the method of the abovementioned document EP-A-179,447.
- the iron and / or copper used is consumed during the reaction.
- the reaction must be carried out in a solvent medium.
- protic organic solvents of alcoholic type are not suitable for the present invention.
- the preferred organic solvents for carrying out the process according to the invention are carboxylic acids, simple or functionalized, aromatic hydrocarbons and halogenated hydrocarbons, ethers and esters.
- carboxylic acids suitable as a solvent for the present invention mention may be made of methanoic, ethanoic, propanoic, butanoic and trifluoroacetic acid.
- carboxylic acids it is understood, of course, also to cover polycarboxylic acids, simple or functionalized, which are perfectly suitable here when they are liquid under the conditions of the reaction.
- the amount of acid, in moles, which is contained in the aprotic organic solvent is generally between 0.1 and 5 times the amount in moles of dibromo derivatives involved, preferably between 0.1 and 2 times this amount.
- the amount of hydrogen to be used can vary within wide limits; it must nevertheless correspond at least to the stoichiometric quantity necessary to allow the complete substitution of half of the bromine atoms which have been brought in the form of the initial dibromo compound. There is no upper limit quantity to respect.
- hydrogen is preferably used in a molecular gas form (H 2 ).
- H 2 molecular gas form
- nascent hydrogen that is to say hydrogen formed in situ in the reaction medium by decomposition of a precursor compound such as a formate or formic acid.
- the temperatures used to conduct the reaction can vary within very wide limits.
- the reaction can be carried out either at atmospheric pressure in an open type reactor, or a trickling fixed bed, in which a continuous stream of hydrogen is bubbled, or, preferably, under autogenous pressure in a closed reactor, of the autoclave type, containing an atmosphere of hydrogen.
- the hydrogen pressure can range from 1 to 50 bars, and preferably from 5 to 20 bars.
- the reaction is preferably carried out with stirring, and this generally until complete or almost complete disappearance of the dibromo derivative of naphthol introduced as reactant.
- the monobrominated derivative obtained is separated from the reaction medium, and this by any means known per se, such as for example filtration, decantation, centrifugation, extraction or distillation.
- the catalysts and / or the solvents thus recovered, after optional purification, can then be recycled at the start of the process.
- the monobromo derivative recovered can, for its part, undergo additional purification steps, if necessary.
- TT denotes the Rate of Transformation, that is to say the ratio: quantity in mole of dibromo derivative transformed X 100 quantity in mole of dibromo derivative introduced RR denotes the Efficiency of the Reaction with respect to a reaction product given, i.e. the ratio: q u antitéen mole of a workingfo rmed X 100 mole quantity of introduced dibromo derivative RT reflects the selectivity of the reaction for a given reaction product, it is defined by the ratio RR / TT.
- the open ampoule is then introduced into a 125 ml autoclave (Hastelloy C).
- the autoclave is then purged twice with nitrogen at a pressure of 15 bar, then twice with hydrogen at 10 bar.
- Example 2 The procedure is then as in Example 1, except that the heating to 100 ° C. is only maintained for 1 hour 30 minutes.
- Example 2 The procedure is then as in Example 1, except that the heating to 100 ° C. is only maintained for 3 hours.
- HASTELLOY8 reactor 100 ml of isopropyl ether; 1.6 g hydrobromic acid; 30 g of dibromo-1,6 naphthol-2; and 4.8 g of powdered WC.
- the reactor is then purged twice with nitrogen at a pressure of 10 bar, then three times with hydrogen at 10 bar.
- the mixture is heated at 120 ° C. for 6 hours under 20 bar of hydrogen, while keeping the medium under agitation.
- the open ampoule is then introduced into a 125 ml Hastelloy HB20 autoclave.
- the autoclave is then purged twice with nitrogen at a pressure of 15 bar, then twice with 2 times 10 bar of hydrogen.
- the autoclave is then placed under 20 bar of hydrogen and this pressure is maintained throughout the duration of the reaction, this with stirring and maintaining the temperature at 120 ° C. After 4 hours of reaction, the glass bulb is removed, the carbide is decanted or filtered.
- the organic phase is drawn off.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
Description
- La présente invention porte sur un procédé de débromation de dérivés dibromés du naphtol. Plus précisément, l'invention concerne un procédé de monodébromation de dérivés dibromés du naphtol de formule (1 ) :
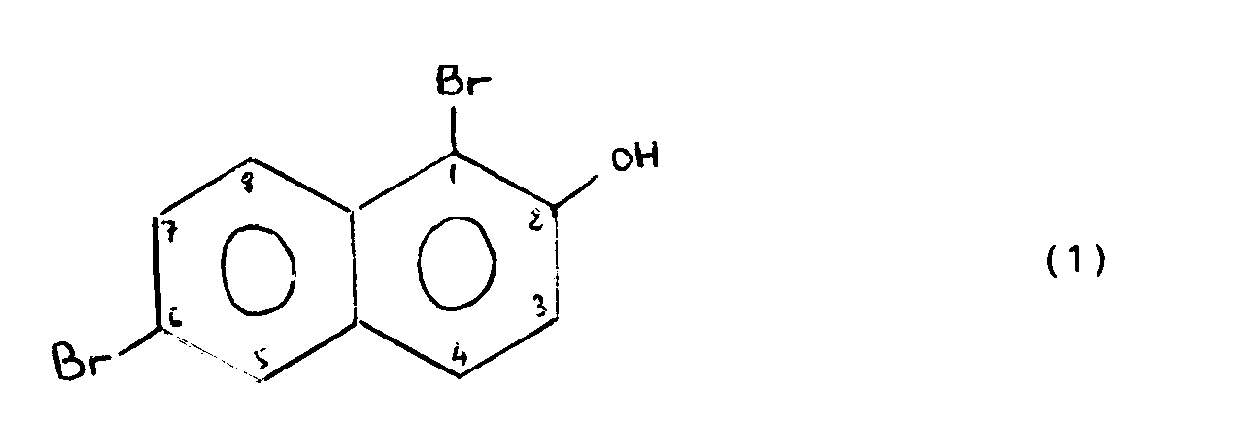
par hydrodébromation catalytique régio-sélective. - Les produits de réaction plus particulièrement visés par le procédé de débromation selon l'invention répondent à la formule (2) :

- Le bromo-6 hydroxy-2 naphtalène (aussi appelé bromo-6 β naphtol) de formule (2) ci-dessus est un produit particulièrement intéressant et important. Il est en effet utilisé pour la synthèse du bromo-6 méthoxy-2 naphtalène (par une alkylation au moyen de sulfate de diméthyle ou de méthanol), ce dernier produit étant, quant à lui, largement utilisé pour obtenir le naproxène ou la nabumétone, qui sont notoirement connus pour leurs propriétés thérapeutiques anti-inflammatoires, ou bien encore le methallenestril, qui est un oestrogène (voir à ce sujet : the Merck Index, eleventh edition, 1989, pages 1002, 1014 et 937).
- Selon l'enseignement du document EP-A-179 447, le bromo-6 hydroxy-2 naphtalène peut être préparé par réduction métallique stoëchiométrique du dibromo-1,6 hydroxy-2 naphtalène, selon la réaction suivante :

où M désigne un métal réducteur tel que fer ou étain, les dérivés dibromés ci-dessus pouvant eux-mêmes être préparés simplement par bromation directe du naphtol :
- Toutefois, la réduction des dérivés dibromés du naphtol en dérivés monobromés selon un procédé tel que décrit ci-dessus présente le désavantage, entre autres, de nécessiter une consommation importante en métal, métal qui se retrouve par ailleurs sous la forme d'un effluent perdu, difficilement valorisable, et parfois polluant, comme par exemple Fe Br2.
- En outre, les rendements de réaction en dérivés monobromés désirés, par un tel procédé, peuvent s'avérer insuffisants.
- La présente invention a pour but de proposer un procédé de débromation de certains dérivés dibromés du naphtol qui permette d'une part d'obvier aux inconvénients susmentionnés, et d'autre part de réaliser une débromation régio-sélective, en particulier en position 1, avec un rendement élevé.
- Acet effet, il est maintenant proposé un nouveau procédé de débromation de dérivés dibromés du naphtol, ledit procédé étant caractérisé par le fait que l'on fait réagir, dans un solvant organique acide et en présence d'un catalyseur à base de carbure de tungstène, (i) un dérivé dibromé du naphtol de formule :

avec (ii) de l'hydrogène moléculaire ou un composé susceptible de générer, dans le milieu de réaction, de l'hydrogène naissant. - Selon un mode préféré de réalisation du procédé selon l'invention, la réaction est conduite en présence de fer et/ou de cuivre.
- Le procédé selon l'invention présente de nombreux avantages et une grande souplesse d'utilisation. Il permet tout d'abord d'éviter la consommation stoëchiométrique de métaux réducteurs. Par ailleurs, de façon inattendue et surprenante, il est également hautement sélectif, en ce sens que, pour les produits de formule (1) ci-dessus, seul l'atome de brome en position 1 est substitué, et ceci même en cas d'utilisation d'un fort excès stoëchiométrique d'hydrogène. Les rendements en dérivés monobromés sont élevés.
- La réaction peut en outre être conduite dans une large gamme de pressions et de températures, et selon de nombreux modes de réalisation.
- Elle peut ainsi être réalisée en batch, en semicontinu, en continu, en réacteur agité, ou en lit fixe ruisselant.
- Dans tous les cas, il y a récupération et réutilisation du catalyseur, ce qui ajoute à l'économie du procédé. Compte tenu du fait que l'on travaille dans les conditions d'une catalyse hétérogène, la récupération ultérieure du catalyseur est très facile puisque pouvant être effectuée par des moyens simples, comme une filtration ou une décantation.
- Enfin, le procédé selon l'invention présente l'avantage de pouvoir être conduit directement, sans séparation ou purification préalable, sur le produit de réaction obtenu par bromation directe du β naphtol.
- Mais d'autres caractéristiques, aspects et avantages de l'invention apparaîtront encore plus clairement à la lecture de la description qui va suivre, et des exemples concrets mais non limitatifs destinés à l'illustrer.
- Les catalyseurs utilisés dans la présente invention sont des catalyseurs à base de carbure de tungstène. Ces catalyseurs peuvent également comprendre, en plus du carbure de tungstène, un autre ou d'autres monocarbures de métal. En tant qu'autres monocarbures de métal, on peut notamment citer les carbures de molybdène, de vanadium, de niobium, de tantale, de titane, de fer et de chrome ; ceux-ci sont largement décrits dans la littérature. La quantité de ces autres carbures de métal représente, de préférence, de 0,01 à 50 % en poids par rapport à la totalité des carbures présents.
- Bien que le carbure de tungstène soit déjà un produit bien connu en soi, on notera que son utilisation comme catalyseur d'hydrodébromation selon un procédé conforme à l'invention est, quant à elle, tout à fait nouvelle.
- Le catalyseur est soit à base de carbone de tungstène massique, soit à base de carbure de tungstène supporté. Comme support, on peut notamment utiliser les oxydes, tels que la silice, l'alumine et le dioxyde de titane, ou du charbon.
- Ainsi, le catalyseur peut se présenter sous la forme d'un substrat monolithique (nid d'abeilles ou autres) en carbure de tungstène, ou d'un substrat monolithique revêtu d'une couche en carbure de tungstène, ou bien encore se présenter sous la forme de produits divisés en, ou revêtus de, carbure de tungstène. Par forme divisée, on entend des produits pulvérulents (poudres) et également les articles obtenus par mise en forme de ces produits (billes, pastilles, boulettes, granulés, extrudés, agglomérés, et autres, de section circulaire, ovale, trilobée ou multilobée, pleine ou creuse).
- Les catalyseurs de type billes, pastilles et autres, présentent l'avantage de pouvoir être séparés ultérieurement du milieu de réaction très rapidement par simple décantation. Les catalyseurs de type pulvérulent nécessitent généralement, pour leur séparation, une étape de filtration.
- Bien entendu, tous les catalyseurs précités sont choisis avec une surface spécifique convenant pour l'application considérée. Dans la pratique, on peut mettre en oeuvre un carbure de tungstène dont la surface spécifique (BET) varie de 0,1 à plusieurs centaines de m2/g, et en particulier de 1 à 300 ou 400 m2/g.
- A cet effet, on pourra utiliser soit des carbures de tungstène disponibles dans le commerce soit des carbures de tungstène que l'on aura synthétisés selon tout procédé connu en soi. Atitre d'exemples, des carbures de tungstène hautes surfaces spécifiques peuvent être fabriqués selon le procédé décrit dans la demande de brevet PCT/FR 90/00204.
- La quantité de catalyseur à utiliser n'est pas critique et peut varier dans de larges limites ; généralement on utilise de 0,01 % à 50 % en poids de catalyseur par rapport à la quantité de dérivé dibromé engagée.
- Selon l'invention, il est également possible d'introduire dans le milieu de réaction de faibles quantités de cuivre et/ou de fer, et ceci dans le but d'améliorer sensiblement les rendements en dérivé monobromé désiré. Ces éléments peuvent être apportés à l'état métallique ou sous la forme d'un sel soluble dans le milieu de réaction. La quantité, en mole, de fer et/ou de cuivre que l'on met généralement en oeuvre est alors comprise entre 0,01 et 0,1 fois la quantité en mole de dérivé dibromé engagée dans la réaction. On notera que de telles quantités sont considérablement plus faibles que les quantités stoëchiométriques qu'il est nécessaire d'utiliser conformément au procédé du document EP-A-179 447 précité. A l'inverse du catalyseur à base de carbure de tungstène, le fer et/ou cuivre engagé est consommé lors de la réaction.
- Selon l'invention, la réaction doit être conduite en milieu solvant.
- Il a été trouvé que le choix du solvant à utiliser revêtait une importance particulière, et que ce choix devait se limiter à des solvants organiques, et plus particulièrement à des solvants organiques acides.
- Selon l'invention, on doit entendre par solvants organiques acides :
- - soit des solvants organiques protiques choisis parmi les acides carboxyliques, simples ou fonctionnalisés ;
- - soit des solvants organiques aprotiques contenant au moins un acide organique ou inorganique.
- En particulier, il a été trouvé que les solvants organiques protiques de type alcoolique ne convenaient pas pour la présente invention.
- Les solvants organiques préférés pour la mise en oeuvre du procédé selon l'invention sont les acides carboxyliques, simples ou fonctionnalisés, les hydrocarbures aromatiques et les hydrocarbures halogénés, les éthers et les esters.
- A titre d'exemples non limitatifs d'acides carboxyliques convenant comme solvant pour la présente invention, on peut citer l'acide méthanoïque, éthanoïque, propanoïque, butanoïque, et trifluoroacétique. Par acides carboxyliques, on entend bien entendu couvrir également les acides polycarboxyliques, simples ou fonctionnalisés, qui conviennent ici tout à fait lorsqu'ils sont liquides dans les conditions de la réaction.
- Atitre d'exemples non limitatifs de solvants organiques aprotiques préférés convenables, on peut citer entre autres :
- - parmi les hydrocarbures aromatiques : le benzène et les alkylbenzènes (éthyl-, butyl-, propyl-benzène...), le toluène et les xylènes ;
- - parmi les hydrocarbures halogénés, en particulier fluorés et chlorés, de composés paraffiniques, cy- cloparaffiniques, et aromatiques : le dichlorométhane, le dichloro-1,2 éthane et le chlorobenzène ;
- - les éthers, tels que notamment l'éther d'isopropyle ;
- - les esters, tels que notamment les acétates et les benzates, en particulier d'alkyles, comme par exemple l'acétate d'éthyle ou le benzoate de méthyle.
- Bien entendu, il est également tout à fait possible, dans le cadre de la présente invention, d'utiliser à titre de solvant soit des mélanges d'acides carboxyliques, soit des mélanges de solvants organiques aprotiques, soit des mélanges entre des acides carboxyliques et des solvants organiques aprotiques.
- A titre d'exemples non limitatifs d'acides pouvant être contenus, seuls ou en mélanges, dans les solvants organiques aprotiques tels que susmentionnés, on peut citer :
- - parmi les acides inorganiques : l'acide phosphorique, l'acide sulfurique, les acides halogéniques, tels que l'acide chlorhydrique ou l'acide bromhydrique ;
- - parmi les acides organiques : les acides carboxyliques susmentionnés, l'acide méthanesulfonique, tri- flique, éthanesulfonique ou benzènesulfonique, oxalique, malonique, succinique, glutarique, adipique, maléique, fumarique, phtalique et méllitique.
- La quantité d'acide, en mole, qui est contenue dans le solvant organique aprotique est généralement comprise entre 0,1 et 5 fois la quantité en mole de dérivés dibromés engagée, de préférence entre 0,1 et 2 fois cette quantité.
- La quantité d'hydrogène à utiliser peut varier dans de larges limites ; elle doit néanmoins correspondre au minimum à la quantité stoëchiométrique nécessaire pour permettre la substitution complète de la moitié des atomes de brome qui ont été apportés sous la forme du composé dibromé initial. Il n'y a pas de quantité limite supérieure à respecter.
- Selon l'invention, l'hydrogène est de préférence utilisé sous une forme gazeuse moléculaire (H2). On peut néanmoins également utiliser de l'hydrogène naissant, c'est à dire de l'hydrogène formé in situ dans le milieu de réaction par décomposition d'un composé précurseur tel qu'un formiate ou l'acide formique.
- Les températures mises en oeuvre pour conduire la réaction peuvent varier dans de très larges limites.
- On peut ainsi opérer de la température ambiante jusqu'à, théoriquement, la température d'ébullition du solvant utilisé, en veillant toutefois à ne pas excéder des températures où le dérivé dibromé et/ou le produit de réaction pourraient se décomposer ; dans la pratique, on travaille généralement à des températures comprises entre 20°C et 200°C, et de préférence entre 50 et 150°C.
- La réaction peut être conduite soit à pression atmosphérique dans un réacteur type ouvert, ou lit fixe ruisselant, dans lequel on fait barboter un courant continu d'hydrogène, soit, de préférence, sous pression autogène dans un réacteur fermé, du type autoclave, contenant une atmosphère d'hydrogène. Dans ce dernier cas, la pression en hydrogène peut aller de 1 à 50 bars, et de préférence de 5 à 20 bars.
- La réaction est de préférence conduite sous agitation, et ceci généralement jusqu'à disparition complète ou quasi-complète du dérivé dibromé du naphtol introduit comme réactif.
- En fin de réaction, on sépare du milieu réactionnel le dérivé monobromé obtenu, et ceci par tout moyen connu en soi, tel que par exemple filtration, décantation, centrifugation, extraction ou distillation.
- Ainsi, par exemple, on pourra procéder tout d'abord à la récupération des catalyseurs, notamment par filtration ou décantation, puis à la séparation du dérivé monobromé et de la phase solvante organique, par exemple par extraction à l'eau ou distillation.
- Les catalyseurs et/ou les solvants ainsi récupérés, après éventuellement purification, peuvent alors être recyclés en tête du procédé.
- Le dérivé monobromé récupéré peut, quant à lui, subir des étapes complémentaires de purification, si nécessaire.
- Des exemples illustrant l'invention vont maintenant être donnés.
- Dans ces exemples, TT désigne le Taux de Transformation, c'est à dire le rapport : quantité en mole de dérivé dibromé transforméeX 100 quantité en mole de dérivé dibromé introduite RR désigne le Rendement de la Réaction vis à vis d'un produit de réaction donné, c'est à dire le rapport : quantitéen mole d'un produitformé X 100 quantité en mole de dérivé dibromé introduite RT traduit la sélectivité de la réaction pour un produit de réaction donné, il est défini par le rapport RR/TT.
- Dans une ampoule en verre de 35 ml, on introduit : 1,2 g de dibromo-1,6 naphtol-2 ; 0,47 g d'une poudre de carbure de tungstène WC (granulométrie moyenne : 1 µm ; surface spécifique: 1,6 m2/g) ; et 15 ml d'acide acétique.
- On introduit ensuite l'ampoule ouverte dans un autoclave (Hastelloy C) de 125 ml. On purge alors deux fois l'autoclave par de l'azote à une pression de 15 bar, puis deux fois par de l'hydrogène à 10 bar.
- On introduit ensuite dans l'autoclave 20 bars d'hydrogène, et on chauffe pendant 4 heures à 100°C, et ceci sous agitation. En fin de réaction, on refroidit l'autoclave, on retire l'ampoule de verre, le carbure de tungstène décante et on soutire la phase organique.
- Une analyse par dosage CPG (chromatographie en phase gazeuse) avec étalon interne donne les résultats suivants :

- Dans une ampoule en verre de 35 ml, on introduit : 1,2 g de dibromo-1,6 naphtol-2 ; 40 mg de WC en poudre ; 12 mg de cuivre métallique ; et 15 ml d'acide acétique.
- On procède ensuite comme à l'exemple 1.
- L'analyse CPG donne les résultats suivants :

- Dans un SOTELEM de 300 ml, on introduit : 20 g de dibromo-1,6 naphtol-2 ; 1,29 g d'une poudre de WC ; 0,184 g de fer métallique en poudre ; et 100 ml d'acide acétique.
- On procède ensuite comme à l'exemple 1, sauf que le chauffage à 100°C n'est maintenu que pendant 1 heure 30.
- L'analyse CPG donne les résultats suivants :

- Dans un SOTELEM de 300 ml, on introduit : 100 ml de chlorobenzène ; 4 g d'acide bromhydrique ; 30 g de dibromo-1,6 naphtol-2 ; et 9,7 g de WC en poudre.
- On procède ensuite comme à l'exemple 1, sauf que le chauffage à 100°C n'est maintenu que pendant 3 heures.
- L'analyse CPG donne les résultats suivants :

- Dans un réacteur en HASTELLOY8 de 300ml, on introduit : 100ml d'oxyde d'isopropyle ; 1,6 g d'acide bromhydrique ; 30 g de dibromo-1,6 naphtol-2 ; et 4,8 g de WC en poudre. On purge alors deux fois le réacteur par de l'azote à une pression de 10 bar, puis trois fois par de l'hydrogène à 10 bar. On chauffe à 120°C pendant 6 heures sous 20 bar d'hydrogène, en maintenant le milieu sous agitation.
- L'analyse CPG donne les résultats suivants :
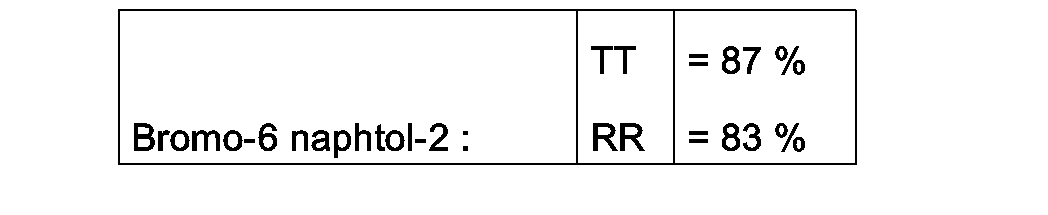
- Dans une ampoule en verre de 35 ml, on introduit: 1,5 g de dibromo-1,6 naphtol-2 ; 10 ml d'acétate d'éthyle; 0,2 g d'acide bromhydrique ; et 0,48 g de WC en poudre.
- On procède ensuite comme à l'exemple 1.
- L'analyse CPG donne Is résultats suivants :

- Dans une ampoule en verre de 35 ml, on introduit : 1,5 g de dibromo-1,6 naphtol-2 ; 10 ml de toluéne ; 0,3 g d'acide bromhydrique ; et 0,48 g de carbure mixte de tungstène et de vanadium en poudre ( le vanadium représente 0,26 % du poids total des carbures).
- On introduit ensuite l'ampoule ouverte dans un autoclave en Hastelloy HB20 de 125 ml. On purge alors deux fois l'autoclave par de l'azote à une pression de 15 bar, puis 2 fois avec 2 fois 10 bar d'hydrogène.
- On place ensuite l'autoclave sous 20 bar d'hydrogène et on maintient cette pression pendant toute la durée de la réaction, ceci sous agitation et en maintenant la température à 120°C. Après 4 heures de réaction, on retire l'ampoule de verre, le carbure est décanté ou filtré.
- On soutire la phase organique.
- L'analyse CPG donne les résultats suivants :

Claims (14)

avec (ii) de l'hydrogène moléculaire ou un composé susceptible de générer, dans le milieu de réaction, de l'hydrogène naissant.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9112268A FR2682103B1 (fr) | 1991-10-04 | 1991-10-04 | Procede de debromation de derives dibromes du naphtol. |
| FR9112268 | 1991-10-04 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0536012A1 true EP0536012A1 (fr) | 1993-04-07 |
| EP0536012B1 EP0536012B1 (fr) | 1995-01-11 |
Family
ID=9417620
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP92402327A Expired - Lifetime EP0536012B1 (fr) | 1991-10-04 | 1992-08-24 | Procédé de débromation de dérivés dibromés du naphtol |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US5256829A (fr) |
| EP (1) | EP0536012B1 (fr) |
| JP (1) | JPH0825938B2 (fr) |
| AT (1) | ATE116958T1 (fr) |
| CA (1) | CA2079731C (fr) |
| DE (1) | DE69201175T2 (fr) |
| FR (1) | FR2682103B1 (fr) |
| IL (1) | IL103317A (fr) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998050334A1 (fr) * | 1997-05-08 | 1998-11-12 | Albemarle Corporation | Production de composes methoxynaphthalenes bromes |
| WO1999042424A1 (fr) * | 1998-02-20 | 1999-08-26 | Rhodia Chimie | Procede de preparation de composes cetoniques aromatiques |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5426243A (en) * | 1994-08-17 | 1995-06-20 | Albemarle Corporation | Process for preparing 1,6-dibromo-2-naphthylene compounds |
| US5756851A (en) * | 1996-10-21 | 1998-05-26 | Albemarle Corporation | Production of nabumetone or precursors thereof |
| US6080888A (en) * | 1997-01-08 | 2000-06-27 | Albemarle Corporation | Preparation of olefinic compounds and carboxylic derivatives thereof |
| US6096920A (en) * | 1997-01-08 | 2000-08-01 | Albemarle Corporation | Preparation of carboxylic compounds and their derivatives |
| US5792886A (en) * | 1997-01-08 | 1998-08-11 | Albemarle Corporation | Production of racemic 2-(6-methoxy-2-naphthyl) propionic acid of precursors thereof |
| US6461539B1 (en) | 1999-10-18 | 2002-10-08 | Conoco Inc. | Metal carbide catalysts and process for producing synthesis gas |
| JP5357628B2 (ja) * | 2009-05-26 | 2013-12-04 | 日本特殊陶業株式会社 | セラミックヒータの製造方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2025032A (en) * | 1933-09-22 | 1935-12-24 | Du Pont | Dehalogenation of organic halides |
| EP0179447A1 (fr) * | 1984-10-23 | 1986-04-30 | Montedison S.p.A. | Procédé pour la préparation de méthoxy-2 bromo-6 naphtalène |
-
1991
- 1991-10-04 FR FR9112268A patent/FR2682103B1/fr not_active Expired - Fee Related
-
1992
- 1992-08-24 AT AT92402327T patent/ATE116958T1/de not_active IP Right Cessation
- 1992-08-24 DE DE69201175T patent/DE69201175T2/de not_active Expired - Fee Related
- 1992-08-24 EP EP92402327A patent/EP0536012B1/fr not_active Expired - Lifetime
- 1992-09-25 JP JP4279267A patent/JPH0825938B2/ja not_active Expired - Lifetime
- 1992-10-01 IL IL10331792A patent/IL103317A/en not_active IP Right Cessation
- 1992-10-02 CA CA002079731A patent/CA2079731C/fr not_active Expired - Fee Related
- 1992-10-02 US US07/955,421 patent/US5256829A/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2025032A (en) * | 1933-09-22 | 1935-12-24 | Du Pont | Dehalogenation of organic halides |
| EP0179447A1 (fr) * | 1984-10-23 | 1986-04-30 | Montedison S.p.A. | Procédé pour la préparation de méthoxy-2 bromo-6 naphtalène |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998050334A1 (fr) * | 1997-05-08 | 1998-11-12 | Albemarle Corporation | Production de composes methoxynaphthalenes bromes |
| WO1999042424A1 (fr) * | 1998-02-20 | 1999-08-26 | Rhodia Chimie | Procede de preparation de composes cetoniques aromatiques |
| FR2775284A1 (fr) * | 1998-02-20 | 1999-08-27 | Rhodia Chimie Sa | Procede de preparation de composes cetoniques aromatiques |
Also Published As
| Publication number | Publication date |
|---|---|
| DE69201175T2 (de) | 1995-05-18 |
| EP0536012B1 (fr) | 1995-01-11 |
| JPH06239784A (ja) | 1994-08-30 |
| US5256829A (en) | 1993-10-26 |
| IL103317A (en) | 1995-12-31 |
| ATE116958T1 (de) | 1995-01-15 |
| CA2079731C (fr) | 1997-01-07 |
| JPH0825938B2 (ja) | 1996-03-13 |
| FR2682103B1 (fr) | 1994-03-11 |
| DE69201175D1 (de) | 1995-02-23 |
| CA2079731A1 (fr) | 1993-04-05 |
| IL103317A0 (en) | 1993-03-15 |
| FR2682103A1 (fr) | 1993-04-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1890990B1 (fr) | Procede de fabrication d'acides carboxyliques | |
| EP0452182B1 (fr) | Procédé d'obtention d'au moins une oléfine tertiaire par décomposition de l'éther correspondant | |
| EP0536012B1 (fr) | Procédé de débromation de dérivés dibromés du naphtol | |
| EP0044260A2 (fr) | Procédé de préparation de polyphénols comportant éventuellement un groupement aldéhyde | |
| CA2054578C (fr) | Procede de debromation de derives dibromes du naphtalene | |
| FR2846651A1 (fr) | Procede de fabrication d'acides carboxyliques | |
| CA2758475A1 (fr) | Procede de preparation d'un terpenylcyclohexanol | |
| CH668064A5 (fr) | Procede de preparation de derives bromes du diphenylether. | |
| WO1996037454A1 (fr) | Procede de preparation de 3-carboxy-4-hydroxybenzaldehydes et derives | |
| FR2525587A1 (fr) | Preparation de resorcinol et de resorcinols substitues par deshydrogenation en phase liquide, de 1,3-diones cycliques derivees de la cyclisation en phase vapeur d'esters d'acides delta-cetocarboxyliques | |
| EP1390338A1 (fr) | Procede d'oxydation d'hydrocarbures | |
| WO2001066502A1 (fr) | Procede d'oxydation d'hydrocarbures en acides | |
| FR2754533A1 (fr) | Procede de preparation selective d'un acide 2-hydroxybenzoique et d'un 4-hydroxybenzaldehyde et derives | |
| EP1440050A2 (fr) | Procede de decomposition catalytique des hydroperoxydes organiques | |
| CH643226A5 (fr) | Procede de fabrication de composes carbonyles par oxydation de composes olefiniques. | |
| FR2828194A1 (fr) | Procede d'oxydation d'hydrocarbures en acides | |
| FR2740708A1 (fr) | Procede de preparation d'un catalyseur bi-metallique ruthenium/etain et son utilisation dans un procede de preparation d'aldehydes et de leurs derives | |
| EP0055198B1 (fr) | Procédé de préparation de phénols métachlorés | |
| CH666677A5 (fr) | Procede de valorisation sous forme de produits perbromes de produits sous-bromes obtenus en solutions. | |
| WO2012120017A1 (fr) | Préparation d'éthers de (poly)glycérol | |
| WO1996038400A1 (fr) | Procede de preparation d'un compose aromatique meta-dihydroxyle | |
| WO1982002549A1 (fr) | Procede de preparation d'hydroxybenzaldehydes | |
| WO2001066506A1 (fr) | Procede d'oxydation d'hydrocarbures en acides | |
| CH666680A5 (fr) | Procede de preparation de decabromodiphenylether. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 19920827 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LI LU NL PT SE |
|
| 17Q | First examination report despatched |
Effective date: 19940318 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE PATENT HAS BEEN GRANTED |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LI LU NL PT SE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19950111 Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 19950111 Ref country code: ES Free format text: THE PATENT HAS BEEN ANNULLED BY A DECISION OF A NATIONAL AUTHORITY Effective date: 19950111 Ref country code: DK Effective date: 19950111 Ref country code: AT Effective date: 19950111 |
|
| REF | Corresponds to: |
Ref document number: 116958 Country of ref document: AT Date of ref document: 19950115 Kind code of ref document: T |
|
| ITF | It: translation for a ep patent filed | ||
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D Free format text: 62509 |
|
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) |
Effective date: 19950123 |
|
| REF | Corresponds to: |
Ref document number: 69201175 Country of ref document: DE Date of ref document: 19950223 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Effective date: 19950411 Ref country code: PT Effective date: 19950411 |
|
| NLV1 | Nl: lapsed or annulled due to failure to fulfill the requirements of art. 29p and 29m of the patents act | ||
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FD4D Ref document number: 62509 Country of ref document: IE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19950831 Ref country code: LI Effective date: 19950831 Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19950831 Ref country code: CH Effective date: 19950831 Ref country code: BE Effective date: 19950831 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| 26N | No opposition filed | ||
| BERE | Be: lapsed |
Owner name: POTASSE ET PRODUITS CHIMIQUES Effective date: 19950831 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: IF02 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20040818 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20040819 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20040930 Year of fee payment: 13 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED. Effective date: 20050824 Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20050824 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20060301 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20050824 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20060428 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST Effective date: 20060428 |