EP0303545B1 - Verfahren zur Herstellung von Phenyläthanolaminotetralinen - Google Patents
Verfahren zur Herstellung von Phenyläthanolaminotetralinen Download PDFInfo
- Publication number
- EP0303545B1 EP0303545B1 EP19880402094 EP88402094A EP0303545B1 EP 0303545 B1 EP0303545 B1 EP 0303545B1 EP 19880402094 EP19880402094 EP 19880402094 EP 88402094 A EP88402094 A EP 88402094A EP 0303545 B1 EP0303545 B1 EP 0303545B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- hydroxy
- formula
- group
- process according
- preparation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 *C(CCC1C=C2)CC1C=C2O Chemical compound *C(CCC1C=C2)CC1C=C2O 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/12—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by halogen atoms or by nitro or nitroso groups
Definitions
- the present invention relates to a process for the preparation of phenylethanolaminotetralins, more particularly by amidation of a mandelic acid with an aminotetralin and reduction, the intermediates used in this process.
- European patent 211721 describes phenylethanolaminotetralins of formula wherein X represents hydrogen, halogen, trifluoromethyl group or lower alkyl group and R represents hydrogen; a lower alkyl group unsubstituted or substituted by a cycloalkyl group containing 3 to 7 carbon atoms, a hydroxy, lower alkoxy, carboxy or lower carbalkoxy group; a cycloalkyl group containing 3 to 7 carbon atoms; or a lower alkanoyl and their pharmaceutically acceptable salts.
- the compounds of formula (A) and their pharmaceutically acceptable salts have advantageous pharmacological properties, the compounds having the OR substituent in position 7 of tetralin having shown a particularly pronounced lipolytic activity.
- the products of formula (A) can be prepared in the form of diastereoisomers or optically pure stereoisomers using one or both of the starting compounds in optically active form.
- obtaining optically active stereoisomers generally involves successive crystallizations at the level of the final product which reduce the yields and, above all, entail fairly laborious operations.
- the amide prepared by reaction of a mandelic acid with a 2-amino-7-hydroxytetraline can be O-alkylated by a lower alkyl haloacetate with very good yields and that the product thus obtained can be reduced to prepare the corresponding O-carbalcoxymethylphenylethanolaminotetralin with an overall yield higher than that of the O-alkylation described in EP 211 721.
- the present invention relates to a process for the preparation of phenylethanolaminotetralins of formula in which X is as defined above and R ° represents hydrogen or a methyl group substituted by a carboxy or lower carbalkoxy group, and their pharmaceutically acceptable salts, characterized in that a functional derivative of a mandelic acid of formula in which X is as defined above, with an aminotetralin of formula the mandelamine thus obtained of formula in which X is as defined above, is then subjected to a reduction for the transformation of its amido group into a methyleneamino group or else to a reaction, with a lower alkyl haloacetate in the presence of a basic condensing agent, said haloacetate being a bromo-, chloro- or iodoacetate; in this second hypothesis, the product obtained is subjected, in any order, to a reduction for the transformation of its amido group into a methyleneamino group and to a possible saponification of
- mandelic acid there can be used chloride, anhydride, a mixed anhydride, an active ester or the free acid suitably activated, for example, with dicyclohexylcarbodiimide or with benzotriazolyl-N- hexafluorophosphate oxytris- (dimethylamino) phosphonium (BOP).
- a mandelic acid activated with a condensing agent such as BOP is used.
- reaction of the functional derivative of mandelic acid with the 2-amino-7-hydroxytetralin of formula III above is carried out in an organic solvent such as methylene chloride, optionally in the presence of a proton acceptor, such as triethylamine.
- an equimolecular amount of mandelic acid, BOP and aminotetralin is used.
- a mandelamide of formula IV is obtained in the form of a pair of diastereoisomers (RR + RS) or (SS + SR) which, by separation, gives the pure enantiomers (RR) and (RS) or (SS) and (SR).
- an (R) -mandelic acid is used, preferably (R) -3-chloromandelic acid. Racemic 3-chloromandelic acid can also be used.
- the aminotetralin of formula III will preferably be used in optically active form.
- the particularly preferred aminotetralins are (R) -2-amino-7-hydroxy-1,2,3,4-tetrahydronaphthalene and (S) -2-amino-7-hydroxy-1,2,3,4-tetrahydronaphthalene .
- Racemic 2-amino-7-hydroxy-1,2,3,4-tetrahydronaphthalene can also be used.
- the mandelamide of formula IV can be directly subjected to reduction for the transformation of the amido group into a methyleneamino group or else it is treated with a lower alkyl haloacetate in an alkaline medium.
- the O-alkylation reaction is carried out according to known procedures using a chloroacetate, a bromoacetate or a lower alkyl iodoacetate, bromoacetate being preferred.
- alkaline condensing agent it is possible to use a hydroxide or a carbonate of an alkali metal, for example potassium carbonate and it can be operated in the presence of a catalyst such as potassium iodide.
- the amide IV methyl ether (lower carbalkoxy) is isolated according to conventional techniques with very good yields.
- the product thus obtained can be directly subjected to the reduction of the amido group into a methyleneamino group, or else it can be saponified to transform the lower carbalkoxy group into a carboxy group, free or salified, according to well known techniques.
- the reduction of the amido group into the methyleneamino group can be carried out on a mandelamide of formula in which X and R ° are as defined above.
- the mandelamide reduction step of formula IVa is carried out, for example, by the action of a hydride such as lithium aluminum hydride or diborane, in particular a reagent generating diborane such as the complex between borane and dimethylsulfide, hereinafter designated "borane-methylsulfide".
- a hydride such as lithium aluminum hydride or diborane
- a reagent generating diborane such as the complex between borane and dimethylsulfide, hereinafter designated "borane-methylsulfide”.
- the reaction is carried out in an organic solvent, such as tetrahydrofuran and the compound of formula I is isolated according to known techniques.
- this group can also be reduced to alcohol. It is therefore desirable, in this case, to use a reduction reagent which makes it possible to obtain, at least preferably, the selective reduction of the amide group.
- the products of formula Ia are new and have good activity on intestinal motility.
- the 2-amino-7-hydroxytetralin of formula III is prepared from the corresponding methoxytetralone of formula by reaction with benzylamine, reduction with sodium borohydride of the benzylimine thus obtained, debenzylation by catalytic hydrogenation and demethylation with hydrobromic acid at 48%.
- the two optically active forms of the aminotetralins of formula III are prepared by resolution of the racemates according to known methods, for example by salification with an optically active acid, preferably mandelic acid.
- the subject of the present invention is, according to another of its aspects, the compounds of formula IVa, in racemic form or in the form of their separate stereoisomers.
- the hydrochloride of this product has a rotary power which corresponds to that of the literature (Molecular Pharmacology, 1982, 22, 281-289).
- the hydrochloride of this product has a rotary power which corresponds to that of the literature (Molecular Pharmacology 1982, 22, 281-289).
- the purified base is dissolved in 10 ml of ethyl acetate and, by cooling, 0.35 g of SR 58339 base is obtained; mp 133-138 ° C; percentage of diastereoisomers not defined.
- the diastereoisomers (RS, SR) and (RR, SS) are not visible in chromatography and can only be separated by other techniques. Its hydrochloride, SR 58339A, is described in Example 8 of European patent 211721.
- Example 5 According to the procedure described in Example 5 and starting from 4 g of SR 58536, obtained as described in Example 1 (c), 2.4 g of pure SR 58524, crystallized from isopropanol, is obtained; m.p. 143-145 ° C
- Example 12 According to the procedure described in Example 12, starting from 4.7 g of SR 58533, obtained as described in Example 8 (c), a base is obtained in the form of a residual oil which is purified by flash chromatography eluting with a methylene chloride / methanol mixture 95/5. The purified base is dissolved in acetone, the solution is filtered and isopropanol saturated with hydrochloric acid is added. After filtration, it is crystallized twice from isopropanol. The product is cooled, filtered and washed first with isopropanol and then with acetone.
- a base is obtained in the form of a residual oil which purified by flash chromatography, eluting with a methylene chloride / methanol mixture 95/5.
- the purified base is dissolved in acetone, the solution is filtered and isopropanol saturated with hydrochloric acid is added. After filtration, it is crystallized twice from isopropanol. The product is cooled, filtered and washed first with isopropanol and then with acetone.
- the organic phase is washed twice with 30 ml of a 2N hydrochloric acid solution and then twice with 30 ml of a saturated sodium chloride solution.
- the organic solution is dried over sodium sulfate, filtered and evaporated to dryness.
- the oil obtained is purified by flash chromatography using as solvent a mixture of ethyl acetate / cyclohexane 55/45.
- a pasty solid is obtained which is taken up with 20 ml of ethyl ether in which the product crystallizes. 10 ml of cyclohexane are added, filtered, washed with a 2/1 cyclohexane / ethyl ether mixture and dried under reduced pressure at 50 ° C.
- the aqueous phase is acidified with concentrated hydrochloric acid, extracted with ethyl acetate, the organic phase is washed with water, dried over sodium sulfate, filtered and evaporated to dryness.
- the oil thus obtained is purified by flash chromatography using as eluent a mixture of ethyl acetate / cyclohexane 70/30.
- the compound SR 58640 has a good activity on intestinal motility in the test of the isolated colon of rats (EP 255 415).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Claims (14)
- Verfahren zur Herstellung von Phenylethanolaminotetralinen der Formel




- Verfahren nach Anspruch 1, dadurch gekennzeichnet, daß man als Ausgangsprodukt eine Mandelsäure in (R)-Konfiguration verwendet.
- Verfahren nach einem der Ansprüche 1 und 2, dadurch gekennzeichnet, daß man als Ausgangsmandelsäure (R)-3-Chlormandelsäure verwendet.
- Verfahren nach Anspruch 1, dadurch gekennzeichnet, daß man als Ausgangsprodukt racemische 3-Chlormandelsäure verwendet.
- Verfahren nach einem der Ansprüche 1 bis 4, dadurch gekennzeichnet, daß man als funktionelles Derivat der Ausgangsmandelsäure die mit Benzotriazolyl-N-oxytris-(dimethylamino)-phosphonium-hexafluorphosphat aktivierte freie Säure verwendet.
- Verfahren nach einem der Ansprüche 1 bis 5, dadurch gekennzeichnet, daß man als Ausgangsaminotetralin racemisches 2-Amino-7-hydroxy-1,2,3,4-tetrahydronaphthalin verwendet.
- Verfahren nach einem der Ansprüche 1 bis 6, dadurch gekennzeichnet, daß man als Ausgangsprodukt (S)-2-Amino-7-hydroxy-1,2,3,4-tetrahydronaphthalin verwendet.
- Verfahren zur Herstellung eines Mandelsäureamids der Formel

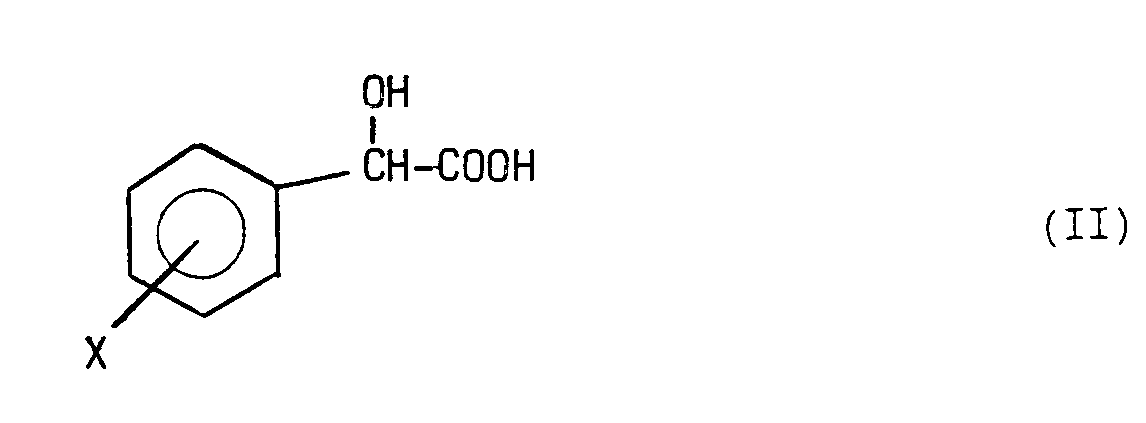

- Verfahren nach Anspruch 8 zur Herstellung des N-(7-Hydroxy-1,2,3,4-tetrahydronaphth-2-yl)-3-chlor-mandelsäureamids.
- Verfahren nach Anspruch 8 zur Herstellung des (RS,SR)-N-(7-Hydroxy-1,2,3,4-tetrahydronaphth-2-yl)-3-chlor-mandelsäureamids.
- Verfahren nach Anspruch 8 zur Herstellung des (RR,SS)-N-(7-Hydroxy-1,2,3,4-tetrahydronaphth-2-yl)-3-chlor-mandelsäureamids.
- Verfahren nach Anspruch 8 zur Herstellung des N-[(2R)-7-Hydroxy-1,2,3,4-tetrahydronaphth-2-yl]-(R)-3-chlor-mandelsäureamids.
- Verfahren nach Anspruch 8 zur Herstellung des N-[(2S)-7-Hydroxy-1,2,3,4-tetrahydronaphth-2-yl]-(R)-3-chlor-mandelsäureamids.
- Verfahren nach Anspruch 8 zur Herstellung des N-[(2S)-7-Carbethoxymethoxy-1,2,3,4-tetrahydronaphth-2-yl]-(R)-mandelsäureamids.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT88402094T ATE77364T1 (de) | 1987-08-12 | 1988-08-11 | Verfahren zur herstellung von phenylaethanolaminotetralinen. |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR8711497A FR2619378B1 (fr) | 1987-08-12 | 1987-08-12 | Procede pour la preparation de phenylethanolaminotetralines |
| FR8711497 | 1987-08-12 | ||
| FR8804219A FR2629453B1 (fr) | 1988-03-30 | 1988-03-30 | Procede pour la preparation de phenylethanolaminotetralines |
| FR8804219 | 1988-03-30 | ||
| FR8807947A FR2632636B1 (fr) | 1988-06-14 | 1988-06-14 | Procede pour la preparation de phenylethanolaminotetralines |
| FR8807947 | 1988-06-14 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0303545A2 EP0303545A2 (de) | 1989-02-15 |
| EP0303545A3 EP0303545A3 (en) | 1989-05-24 |
| EP0303545B1 true EP0303545B1 (de) | 1992-06-17 |
Family
ID=27251496
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP19880402094 Expired - Lifetime EP0303545B1 (de) | 1987-08-12 | 1988-08-11 | Verfahren zur Herstellung von Phenyläthanolaminotetralinen |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0303545B1 (de) |
| JP (1) | JP2731913B2 (de) |
| DE (1) | DE3872100T2 (de) |
| ES (1) | ES2045164T3 (de) |
| GR (1) | GR3005726T3 (de) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2648042B1 (fr) * | 1989-06-13 | 1994-06-10 | Midy Spa | Utilisation de phenylethanolaminotetralines pour la preparation de medicaments pour le traitement des affections oculaires |
| FR2653765B1 (fr) * | 1989-10-31 | 1993-08-06 | Midy Spa | Procede pour la preparation enantioselective de 2-aminotetralines. |
| FR2656607B1 (fr) * | 1989-12-29 | 1994-03-11 | Midy Spa | Phenylethanolaminomethyltetralines. |
| IE65511B1 (en) * | 1989-12-29 | 1995-11-01 | Sanofi Sa | New phenylethanolaminomethyltetralins |
| FR2669821A1 (fr) * | 1990-12-04 | 1992-06-05 | Sanofi Sa | Utilisation de phenylethanolaminotetralines pour la preparation de medicaments destines au traitement de la depression. |
| GB9107827D0 (en) * | 1991-04-12 | 1991-05-29 | Fujisawa Pharmaceutical Co | New ethanolamine derivatives,processes for the preparation thereof and pharmaceutical composition comprising the same |
| EP2098511A1 (de) | 2008-03-07 | 2009-09-09 | Solvias AG | Verfahren zur Herstellung von Verbindungen mit einer Hydronaphtalinstruktur mit einem asymmetrisch substituierten Benzolring |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2803582A1 (de) * | 1978-01-27 | 1979-08-02 | Sandoz Ag | Neue tetralinderivate, ihre herstellung und verwendung |
| IL79323A (en) * | 1985-07-10 | 1990-03-19 | Sanofi Sa | Phenylethanolaminotetralines,their preparation and pharmaceutical compositions containing them |
| DE3623941A1 (de) * | 1986-07-16 | 1988-01-28 | Bayer Ag | Substituierte amino-5,6,7,8-tetrahydronaphthyl-oxyessigsaeuren, verfahren zu deren herstellung sowie die verwendung als arzneimittel |
-
1988
- 1988-08-11 EP EP19880402094 patent/EP0303545B1/de not_active Expired - Lifetime
- 1988-08-11 DE DE19883872100 patent/DE3872100T2/de not_active Expired - Fee Related
- 1988-08-11 ES ES88402094T patent/ES2045164T3/es not_active Expired - Lifetime
- 1988-08-12 JP JP63202622A patent/JP2731913B2/ja not_active Expired - Lifetime
-
1992
- 1992-09-17 GR GR920402049T patent/GR3005726T3/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| DE3872100D1 (de) | 1992-07-23 |
| JP2731913B2 (ja) | 1998-03-25 |
| EP0303545A3 (en) | 1989-05-24 |
| ES2045164T3 (es) | 1994-01-16 |
| GR3005726T3 (de) | 1993-06-07 |
| JPS6466149A (en) | 1989-03-13 |
| DE3872100T2 (de) | 1993-01-28 |
| EP0303545A2 (de) | 1989-02-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0591057B1 (de) | Arylalkyl(thio)amide mit Melatonin-Rezeptor-Selektivität und Prozess zu ihrer Darstellung | |
| EP0202164B1 (de) | (Benzoyl-4-piperidino)-2-phenyl-1-alkanolderivate, ihre Herstellung und ihre Verwendung als Heilmittel | |
| EP0436435B1 (de) | Phenyläthanolaminomethyltetraline, Verfahren zu ihrer Herstellung und sie enthaltende Arzneimittel | |
| EP0303546B1 (de) | Verfahren zur O-Alkylierung von N-(Hydroxy)aralkylphenylethanolamin | |
| EP0001534B1 (de) | Pyrrolderivate, Verfahren zu ihrer Herstellung und ihre therapeutischen Verwendungen | |
| EP0515541B1 (de) | Verfahren zur herstellung von (2r,3r)-cis-beta-phenyl-glycidil-säure | |
| FR2832146A1 (fr) | Base libre de tamsulosine racemique et ses procedes de preparation | |
| EP0383686B1 (de) | Carboxyalkyl-ether von 2-Amino-7-hydroxytetralin | |
| EP0303545B1 (de) | Verfahren zur Herstellung von Phenyläthanolaminotetralinen | |
| EP0021940B1 (de) | Aminoderivate von Benzothiazol, Verfahren zu ihrer Herstellung und ihre Anwendung in der Heilkunde | |
| EP0309324A1 (de) | Verfahren zur Herstellung von N-alkylierten Aminosäuren und deren Estern, Verwendung zur Synthese von Carboxyalkyldipeptiden | |
| EP0842148B1 (de) | Benzolsulfonamidderivate, ihre herstellung und ihre therapeutische verwendung | |
| EP0634396A1 (de) | Aminosäurederivate und ihre Verwendung als Enkephalinase-Inhibitoren | |
| EP0500443B1 (de) | Phenylethanolamino- und Phenylethanolaminomethyltetraline, Verfahren zu deren Herstellung Zwischenprodukte davon und diese enthaltende pharmazeutische Zusammensetzungen | |
| EP0327455A1 (de) | Phenol-Alkyl- oder -Benzylether, Verfahren zu ihrer Herstellung und ihre therapeutische Verwendung | |
| EP0275742A1 (de) | 5-Hydroxymethyl-Derivate des 2-Oxazolidinons, deren Herstellung und deren Anwendung in der Therapie | |
| EP0998470A1 (de) | Verfahren zur herstellung von alkoxyfuranon-amin-derivaten, durch dieses verfahren erhaltene verbindungen und verwendung dieser verbindungen | |
| EP0347313B1 (de) | 2-Amino-7-hydroxytetralin-ether | |
| EP0354078B1 (de) | Derivate von Benzocyclohepten, Verfahren zu ihrer Herstellung und sie enthaltende Arzneimittel | |
| FR2632636A1 (fr) | Procede pour la preparation de phenylethanolaminotetralines | |
| US5198586A (en) | Process for the preparation of phenylethanolaminotetralins | |
| FR2629453A1 (fr) | Procede pour la preparation de phenylethanolaminotetralines | |
| FR2619378A1 (fr) | Procede pour la preparation de phenylethanolaminotetralines | |
| EP0837843B1 (de) | Verfahren zur herstellung von enantiomeren der aminoalkylaminophenylpropansäure | |
| FR2653765A1 (fr) | Procede pour la preparation enantioselective de 2-aminotetralines. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AT BE CH DE ES FR GB GR IT LI LU NL SE |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): AT BE CH DE ES FR GB GR IT LI LU NL SE |
|
| 17P | Request for examination filed |
Effective date: 19891016 |
|
| 17Q | First examination report despatched |
Effective date: 19910503 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: MIDY S.P.A. Owner name: ELF SANOFI |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE ES FR GB GR IT LI LU NL SE |
|
| REF | Corresponds to: |
Ref document number: 77364 Country of ref document: AT Date of ref document: 19920715 Kind code of ref document: T |
|
| REF | Corresponds to: |
Ref document number: 3872100 Country of ref document: DE Date of ref document: 19920723 |
|
| ITF | It: translation for a ep patent filed | ||
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) | ||
| REG | Reference to a national code |
Ref country code: GR Ref legal event code: FG4A Free format text: 3005726 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| EPTA | Lu: last paid annual fee | ||
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2045164 Country of ref document: ES Kind code of ref document: T3 |
|
| EAL | Se: european patent in force in sweden |
Ref document number: 88402094.2 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: AT Payment date: 19980723 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19980728 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 19980804 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19980811 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: ES Payment date: 19980812 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19980817 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SE Payment date: 19980818 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GR Payment date: 19980819 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: LU Payment date: 19980821 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19980831 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 19980911 Year of fee payment: 11 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19990811 Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19990811 Ref country code: AT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19990811 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19990812 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: THE PATENT HAS BEEN ANNULLED BY A DECISION OF A NATIONAL AUTHORITY Effective date: 19990830 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19990831 Ref country code: GR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19990831 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19990831 Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19990831 |
|
| BERE | Be: lapsed |
Owner name: MIDY S.P.A. Effective date: 19990831 Owner name: ELF SANOFI Effective date: 19990831 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20000301 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 19990811 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20000428 |
|
| EUG | Se: european patent has lapsed |
Ref document number: 88402094.2 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee |
Effective date: 20000301 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20000601 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20000911 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED. Effective date: 20050811 |