-
HINTERGRUND
DER ERFINDUNG
-
GEBIET DER
ERFINDUNG
-
Gegenstand
der vorliegenden Erfindung sind Medikamente zur Verminderung der
Häufigkeit
der Übertragung
einer Virusinfektion, wie zum Beispiel des Human Immunodeficiency
Virus und Herpesvirus und zur Prävention
und Behandlung sexuell übertragener
Bakterieninfektionen, wie zum Beispiel Chlamydia trachomatis, durch
Verabreichung von Celluloseacetatphthalat oder Hydroxypropylmethylcellulosephthalat,
die bisher als pharmazeutische Hilfsstoffe eingesetzt wurden.
-
HINTERGRUNDINFORMATION
-
A. PHARMAZEUTISCHE HILFSSTOFFE
-
Pharmazeutische
Hilfsstoffe sind als inerte Substanzen definiert, die ein Vehikel
zur Abgabe des Medikaments bilden (Webster's Ninth New Collegiate Dictionary, Merriam-Webster
Inc. Publishers, Springfield, MA, USA, 1985, S. 432). Hilfsstoffe
wandeln folglich pharmakologische Wirkstoffe in zur Verabreichung
an Patienten geeignete pharmazeutische Dosierungsformen um. Einige
Hilfsstoffe werden auch zur Formulierung oder Herstellung von Süßwaren,
Kosmetika und Nahrungsmittelprodukten verwendet. Deshalb werden
zugelassene Hilfsstoffe, im Vergleich zu den meisten Medikamenten,
häufig
und in höherer
Dosierung verwendet. Hilfsstoffe sind auch viel billiger und lassen
sich im Vergleich zu den meisten Medikamenten leichter in sehr großem Umfang
herstellen.
-
B. SEXUELL ÜBERTRAGENE
ERKRANKUNGEN („STDs": EINE ÜBERSICHT)
-
Die
Pandemie des Human Immunodeficiency Virus (HIV) wird unterhalten
und schreitet überwiegend aufgrund
sexueller Übertragung
des Virus (Mann, J. M., Tarantola, D. J. M., Netter, T. W., „AIDS in
the World", Cambridge:
Harvard University Press, (1992)), gefördert durch eine vorherige
Infektion mit anderen STD-Pathogenen (Perine, P. L., „Sexually
Transmitted Diseases in the Tropics", Med. J. Aust., 160, (1994), 358–366), fort.
-
Die
dringende Notwendigkeit zur Verhinderung der Übertragung von STDs ist durch
die HIV/AIDS-Epidemie hervorgehoben worden, die bisher zur Infektion
von ca. 42 Millionen Menschen und zu ca. 12 Millionen Todesfällen geführt hat
(UNAIDS und WHO, Bericht über
die globale HIV/AIDS-Epidemie, Genf: Joint United Nations Programm
zu HIV/AIDS, 1. Juni 1988). Die Fakten, dass HIV-Infektionen bis
heute nicht heilbar sind, zur führenden
Todesursache unter jungen Erwachsenen geworden sind und die Lebenserwartung
in zahlreichen Ländern
vermindert haben, und die Beobachtung, dass mehrere nicht-virale
STDs eine HIV-Infektion fördern,
haben den dringenden Bedarf an neuen präventiven Ansätzen weiter
nachdrücklich
hervorgehoben.
-
Es
wurde ermittelt, dass die Behandlung von STDs (mit Ausnahme von
HIV) ein durchführbarer
und wirtschaftlich gerechtfertigter Ansatz zur Verminderung der
HIV-1-Übertragungsrate
darstellt (St. Louis, M. E., Levine, W. C., Wasserheit, J. N. et
al., „HIV
Prevention Through Early Detection and Treatment of other Sexually
Transmitted Diseases – United
States Recommendation of the Advisory Committee for HIV and STD
Prevention", Mor.
Mort. Wkly. Rep., (1998), 47 (Nr. RR-12). 1–24; Over, M., Piot, P., „Human
Immunodeficiency Virus Infection and Other Sexually Transmitted
Diseases in Developing Countries: Public Health Importance and Priorities
for Resource Allocation",
J. Infect. Dis., (1996), 174 (Suppl. 2), 162–175). Dieser vorteilhafte
Ansatz ist jedoch zur Kontrolle der Ausbreitung von STDs, einschließlich HIV-1,
nicht ausreichend.
-
In
Abwesenheit prophylaktischer Vakzine gegen STD-Pathogene und HIV
in der absehbaren Zukunft und sicheren Anti-HIV-1-Medikamenten,
die in Entwicklungsländern
preislich erschwinglich sind, müssen
andere einfache Verfahren zur Kontrolle der sexuellen Übertragung
von STD, einschließlich
HIV-1, angewendet werden. Hierzu zählen mechanische (Kondome)
und chemische Barrieremethoden und Kombinationen davon. Spermizid-Formulierungen,
von denen gezeigt wurde, dass sie STD-Pathogene in vitro inaktivieren,
wurden für
diesen Zweck in Erwägung
gezogen, ihr Nutzen bleibt jedoch basierend auf dem Outcome klinischer
Prüfungen
zur Sicherheit und Wirksamkeit zweifelhaft.
-
Die
Verwendung chemischer Barrieremethoden (topische „Mikrobizide") unter der Kontrolle
von Frauen wurde als eine Methode zur Kontrolle der sexuellen Übertragung
von HIV-1 und anderen STDs vorgeschlagen (Alexander, N. J., „Barriers
to Sexually Transmitted Diseases",
Scientific American & Medicine,
(1996), 3: 32–41).
Die schnellste Weise zur Einführung
topischer Mikrobizide in die Praxis schien die Anwendung von freiverkäuflichen
Kontrazeptiva, enthaltend das Detergens Nonoxinol-9 (N-9), zu sein.
Es wurde gezeigt, dass N-9 HIV-1 in vitro inaktiviert (Hicks, D.
R., Martin, L. S., Getchell, J. P. et al., „Inactivation of HTLV-III/LAV-infected
Cultures of Normal Human Lymphocytes by Nonoxynol-9 in vitro", Lancet, (1985),
2: 1422–1423;
Jennings, R., Clegg, A., „The
Inhibitory Effect of Spermicidal Agents on Replication of HSV-2
and HIV-1 in vitro",
J. Antimicrob. Chemother., (1993), 32: 71–82), HSV-2 (Sugarman, B.,
Mummaw, N., „Effects
of Antimicrobial Agents on Growth and Chemotaxis of Trichomonas
Vaginalis", Antimicrob.
Agents Chemother., (1988), 32: 1323–1326) und Chlamydia trachomatis
(Lyons, J. M., Ito, J. I., Jr., „Reducing the Risk of Chlamydia
Trachomatis Genital Tract Infection by Evaluating The Prophylactic
Potential of Vaginally Applied Chemicals", Clin. Infect. Dis., (1995), 21 (Suppl.
2): S174–S177).
Es wurde auch gefunden, dass N-9 cytotoxisch ist. Dieser Ansatz
scheint durch klinische Daten erschwert zu sein, die auf unerwünschte Wirkungen
einiger N-9-Formulierungen (Stafford, M. K., Ward, H., Flanagan,
A., et al., „Safety
Study of Nonoxynol-9 As A Vaginal Microbicide: Evidence of Adverse
Effects", J. Acquir.
Imm. Defic. Synd. Hum. Retrovir., (1998), 17: 327–331; Rosenstein,
I. J., Stafford, M. K., Kitchen, V. S. et al., „Effects on Normal Vaginal
Flora of Three Intravaginal Microbicidal Agents Potentially Active
Against Human Immunodeficiency Virus Type 1", J. Infect. Dis., (1998), 177: 1386–1390; Kilmarx,
P. H., Limpakarnjanarat, K., Supawitkul, S. et al., „Mucosal
Disruption Due to Use of A Widely-distributed Commercial Vaginal
Product: Potential to Facilitate HIV Transmission", AIDS, (1998), 12: 767–773) und
Wirksamkeitsmangel bei der Verminderung der Rate der heterosexuellen
HIV-1-, Gonorrhoe- und Chlamydia-Übertragung durch eine der N-9-Formulierungen
hinweisen (Roddy, R. E., Zekeng, L., Ryan, K. A. et al., „A Controlled
Trial of Nonoxynol 9 Film to Reduce Male-to-Female Transmission
of Sexually Transmitted Diseases",
N. Engl. J. Med., (1998), 339, 504–510). Dies deutet darauf hin,
dass andere mikrobizide Verbindungen als Prophylaktika gegen HIV-1
und andere STD getestet werden.
-
Unter
Berücksichtigung
der Dringlichkeit der Kontrolle der HIV-Epidemie wird die schnelle
Entwicklung anderer Mikrobizid-Formulierungen benötigt. Die
Einführung
solcher Mikrobizid-Formulierungen
in die Praxis würde
durch die Verwendung von Wirkstoffen mit einem bereits etablierten
Sicherheitsnachweis zum menschlichen Gebrauch signifikant verbessert.
-
Zur
Suche nach sicheren Mikrobizid-Formulierungen müssen Kriterien verwendet werden,
die sich von denen unterscheiden, die zum Screening auf Anti-HIV-1-Medikamente
angewendet wurden, wobei die Möglichkeit
zunimmt, dass vielversprechende Mikrobizide mit Anti-HIV-1-Aktivität bisher
zuvor während
des eingehenden Screenings auf therapeutische Anti-HIV-1-Verbindungen übersehen
worden sein könnten.
Die Kriterien zur Auswahl von Anti-HIV-Mikrobiziden, im Vergleich
zu denen für
therapeutische Anti-HIV-1-Medikamente, können wie folgt zusammengefasst
werden: (a) Unerwünschtheit
der systemischen Ausbreitung, die zur bevorzugten Erwägung von
Verbindungen mit hohem Molekulargewicht (Mw ≥ 2 kD) führt, die
am Ort der Applikation selektiv aktiv sind; (b) hoher Sicherheitsgrad
und Mangel an Nebenwirkungen (aufgrund wiederholter Verwendung durch
Gesunde im Vergleich mit der Verwendung therapeutischer Anti-HIV-1-Medikamente
durch bereits Infizierte), wobei die Sicherheit durch den Mangel
an systemischer Ausbreitung augmentiert wird; (c) Erwägung von
Verbindungen mit geringerer spezifischer antiviraler Aktivität, für die durch
höhere
Konzentrationen der Verbindungen mit etablierter Sicherheit kompensiert
werden kann und (d) gegen die früheren
Infektionsphasen gerichtete Aktivität und bevorzugt direkte Inaktivierung
des Pathogens wie durch den Begriff „Mikrobizid" impliziert ist.
-
Unter
Verbindungen, die mindestens einigen dieser Kriterien entsprechen,
befinden sich folgende: (a) sulfatierte Polysaccharide (Javan, C.
M., Gooderham, N. J., Edwards, R. J. et al., „Anti-HIV Type 1 Activity
of Sulfated Derivatives of Dextrin Against Primary Viral Isolates
of HIV Type 1 in Lymphocytes and Monocyte-Derived Macrophages", AIDS Res. Human
Retroviruses, (1997), 13, 875–880;
Stafford, M. K., Cain, D., Rosenstein, I., et al., „A Placebo-Controlled,
Double Blind Prospective Study in Healthy Female Volunteers of Dextrin Sulphate
Gel: A Novel Potential Intravaginal Virucide", J. Acquir. Immune Defic. Syndr. Hum.
Retrovirol., (1997), 14, 213–218;
Zacharopolous, V. R., Phillips, D. M., „Vaginal Formulations of Carrageenan
Protect Mice From Herpes Simplex Virus Infection", Clin. Diagn. Lab. Immunol., (1997),
4, 465–468;
Carlucci, M. J., Pujol, C. A., Ciancia, M. et al., „Antiherpetic
and Anticoagulant Properties of Carrageenans From the Red Seaweed Gigartina
Skottsbergii and Their Cyclized Derivatives: Correlation Between
Structure and Biological Activity", Int. J. Biol. Macromol., (1997), 20,
97–105)
und andere sulfonierte Polymere (die viruzide und bakterielle Aktivität dieser
Verbindungen wurde jedoch nicht etabliert, und ihre Aktivität wird ihrer
Fähigkeit
zugeschrieben, mit Targetzellen zur Inhibition des Eindringens von
Viren zu interagieren (Rusconi, S., Moonis, M., Merill, D. P. et
al., „Naphthalen
Sulfonate Polymers With CD-4-Blocking and Anti-Human Immunodeficiency
Virus Type 1 Activities",
Anticrob. Agents Chemother., (1996), 40, 234–236; McClure, M. O., Moore,
J. P., Blanc, D. F. et al., „Investigations
into the Mechanism By Which Sulfated Polysaccharides Inhibit HIV
Infection In Vitro",
AIDS Res. Hum. Retroviruses, (1992), 8, 19–26); und (b) Protegrine, die
eine Breitspektrum-Aktivität
gegen Bakterien und umhüllte
Viren aufweisen (Tamamura, H., Murakami, T., Horiuchi, S. et al., „Synthesis
of Protegrin-Related Peptides and Their Antibacterial and Anti-Human
Immunodeficiency Virus Activity",
Chem. Pharm. Bull., (Tokio), (1995), 43, 853–858; Lehrer, R. I., Ganz,
T., „Endogenous
Vertebrate Antibiotics, Defensins, Protegrins, and Other Cysteine-Rich
Antimicrobial Peptides",
Ann. N.Y., Acad. Sci., (1996), 797, 228–239; Qu, X. D., Harwig, S.
S., Shafer, W. M. et al., „Protegrin
Structure and Activity Against Neisseria Gonorrhoeae", Infect. Immun.,
(1997), 65, 636–639),
sie weisen jedoch auch eine unerwünschte Aktivität gegen
Lactobacilli auf, und ihre Applikation kann im Vergleich zu der
von sulfatierten Polymeren wirtschaftliche Nachteile aufweisen.
-
Da
erwartet würde,
dass wirksame topische „Mikrobizide" über Jahrzehnte wiederholt verwendet
würden,
sollten sie einen etablierten Sicherheitsnachweis besitzen und sollten
sich nach topischer Applikation bevorzugt nicht systemisch ausbreiten.
Sie sollten die folgenden Merkmale aufweisen: (a) preisgünstig sein,
(b) aus überall
verfügbaren
Ressourcen hergestellt werden, (c) über eine breite Spezifität verfügen, die
in der Prävention
der Übertragung
mehrerer STDs resultiert und (d) die Infektivität der entsprechenden STD-Pathogene inaktivieren.
Gemäß diesen
Anforderungen entwickelten einige dieser Erfinder vor kurzem ein
potentes Anti-HIV- und Anti-Herpesvirus-Mittel, das zur Inkorporation
in topische Gele/Cremes geeignet ist (Neurath, A. R., Jiang, S.,
Strick, N. et al., „Bovine β-Lactoglobulin
Modified by 3-Hydroxyphthalic Anhydride Blocks the CD4 Cell Receptor
for HIV", Nature
Med., (1996), 2, 230–234;
Neurath, A. R., Debnath, A. K., Strick, N. et al., „3-Hydroxyphthaloyl β-Lactoglobulin,
I, Optimization of Production and Comparison With Other Compounds
Considered for Chemoprophylaxis of Mucosally Transmitted Human Immunodeficiency
Virus Type 1", Antiviral Chem.
Chemother., (1997), 8, 131–140;
Neurath, A. R., Debnath, A. K., Strick, N. et al., „3-Hydroxyphthaloyl β-Lactoglobulin,
II, Anti-Human Immunodeficiency Virus Type 1 Activity in in vitro
Environments Relevant to Prevention of Sexual Transmission of the
Virus", Antiviral
Chem. Chemother., (1997), 8, 141–148; Neurath. A. R., Strick,
N., Li, Y-Y, „3-Hydroxyphthaloyl β-Lactoglobulin,
III. Antiviral Activity Against Herpesviruses", Antiviral Chem. Chemother., (1998),
9, 177–184;
Kokuba, H., Aurelian, L., Neurath, A. R., „3-Hydroxyphthaloyl β-Lactoglobulin,
IV, Antiviral Activity in the Mouse Model of Genital Herpesvirus
Infection", Antiviral
Chem. Chemother., (1988), 9, 353–357, durch chemische Modifikation
des bovinen Milchprodukts β-Lactoglobulin
mit 3-Hydroxyphthalsäureanhydrid.
Mögliche
Nachteile dieser antiviralen Verbindung waren die mangelnde Aktivität gegen
bakterielle STD-Pathogene.
-
C. VIRUSINFEKTIONEN
-
Human
Immunodeficiency Viren („HIV") sind als das kausale
Virus für
AIDS (Acquired Immunodeficiency Syndrom) bekannt. Die Prävalenz von
AIDS-Fällen
nimmt derzeit mit alarmierender Rate zu.
-
Zwei
verwandte Retroviren, die AIDS verursachen können, sind das Human Immunodeficiency
Virus Typ 1 (HIV-1) und Typ 2 (HIV-2). Die Genome dieser beiden
Viren sind auf der Nukleotidebene ca. 50% homolog, enthalten das
gleiche Genkomplement und scheinen die gleichen humanen Zellen über den
gleichen Mechanismus anzugreifen und abzutöten.
-
HIV-1
wurde 1983 identifiziert. Nahezu alle AIDS-Fälle in den USA lassen sich
mit einer HIV-1-Infektion in Zusammenhang bringen. HIV-2 wurde 1986
aus westafrikanischen AIDS-Patienten isoliert.
-
HIV-1
und HIV-2 sind Retroviren, bei denen das genetische Material RNA
anstelle von DNA darstellt. Die HIV-1- und HIV-2-Viren tragen eine
Polymerase (reverse Transkriptase), welche die Transkription von
viraler RNA in die doppelhelische DNA katalysiert.
-
Die
virale DNA kann als eine nicht integrierte Form in der infizierten
Zelle existieren oder kann in das Genom der Wirtszelle integriert
werden. Nach dem derzeitigen Verständnis dringt das HIV in den
T4-Lymphocyt ein, wo es seine äußere Hülle verliert,
wobei es virale RNA und reverse Transkriptase freisetzt.
-
Die
reverse Transkriptase katalysiert die Synthese eines komplementären DNA-Stranges
aus der viralen RNA-Matrize. Die DNA-Helix insertiert dann in das
Wirtsgenom, wo sie als das Provirus bekannt ist. Die integrierte
DNA kann als eine latente Infektion persistieren, die durch wenig
oder keine Virusproduktion oder den Tod von Helfer-/Inducerzellen
für eine
unbestimmte Zeitspanne gekennzeichnet ist. Wenn die virale DNA durch
den infizierten Lymphocyt transkribiert und translatiert wird, werden
neue virale RNA und Proteine zur Bildung neuer Viren produziert,
die aus der Zellmembran knospen und andere Zellen infizieren.
-
Versuchen
zur Behandlung von AIDS mit Medikamenten, die die reverse Transkriptase
inhibieren, wie zum Beispiel 3'-Azido-3'-desoxythymidin (AZT),
war nicht der erwünschte
Erfolgsgrad beschieden. Bei Verwendung von antiviralen Medikamenten
besteht überdies
ein Potenzial für
Toxizität.
Folglich besteht ein Bedarf an einem wirksamen und sicheren Mittel
zur Prävention
und Behandlung von AIDS.
-
HIV-Infektionen
werden beispielsweise mittels kontaminierter intravenöser Drogennadeln
und durch sexuellen Kontakt übertragen.
Die sexuelle Übertragung
stellt die häufigste
(86%) Route der HIV-1-Infektionen unter Erwachsenen weltweit dar
(AIDS in the World, Harvard University Press, Cambridge, Mass.,
(1992)).
-
Die Übertagung
von HIV durch heterosexuellen Sex stellt für Frauen ein besonders schwerwiegendes Problem
dar. Bis zum Jahr 2000 werden schätzungsweise 90% der HIV-Infektionen
durch heterosexuellen Geschlechtsverkehr erworben.
-
Die
Verwendung von Kondomen stellt einen erheblichen Schutzgrad gegen
die Übertragung
von HIV- und Herpesvirusinfektionen während des Geschlechtsverkehrs
bereit, eine Schwierigkeit ergibt sich jedoch, wenn Kondome nicht
angewendet werden. Die Anwendung von Kondomen scheint überdies
in vielen Ländern eine
kulturell und sozial nicht akzeptierbare Praxis darzustellen.
-
Obwohl
sich Männer
durch Anwendung von Kondomen selbst vor der sexuell übertragenen
HIV- und Herpesvirusinfektion
schützen
können,
stehen sexuell aktiven Frauen keine ähnlichen Mittel zur Verfügung. Frauen
können
ihre männlichen
Sexpartner zur Verwendung eines Kondoms anhalten, sie können gegebenenfalls
jedoch keinen Erfolg damit haben. Das weibliche Kondom, das gerade
verfügbar
wird, ist teuer, und es liegen derzeit keine Hinweise vor, dass
es die sexuelle Übertragung
von HIV oder des Herpesvirus verhindert.
-
Selbst
die Unterhaltung einer monogamen sexuellen Beziehung stellt keine
Sicherheitsgarantie dar, denn wenn der männliche Partner einer Frau
infiziert wird, kann er das Virus an sie weitergeben. Und je mehr Frauen
infiziert werden, um so mehr Säuglinge
werden infiziert.
-
Derzeit
herrscht im medizinischen Bereich Frustration wegen der trostlosen
Aussichten für
ein wirksames AIDS-Vakzin in der nahen Zukunft und die schwerwiegenden
Limitationen der Medikamente, mit denen HIV wirksam und sicher bekämpft werden
kann.
-
Aufgrund
eines derzeit nicht verfügbaren
prophylaktischen Anti-HIV-Vakzins und aufgrund der Limitationen
von erzieherischen Bildungsprogrammen, wurde nach anderen Präventivverfahren
gesucht. Spermizide mit viruziden Eigenschaften wurden für diesen
Zweck in Erwägung
gezogen, ihre Applikation ist jedoch aufgrund der unerwünschten
Wirkungen kontraindiziert (Bird, K. D., „The Use of Spermicide Containing
Nonoxynol-9 in the Prevention of HIV Infection", AIDS, 5, 791–796 (1991)).
-
Die
sich derzeit im Gebrauch befindenden Anti-HIV-Medikamente oder solche,
von denen erwartet wird, dass sie in der nahen Zukunft klinisch
angewendet werden (Steele, F., „AIDS Drugs Lurch Towards
Market", Nature
Medicine, 1, 285–286
(1995)), die meist nicht auf die frühesten Phasen im Replikationszyklus
des Virus abgezielt sind, führen
zur Entstehung von Medikamenten-resistenten Mutanten und sind teuer,
was darauf hindeutet, dass ihre Applikation zur verbreiteten Verwendung
in der topischen Chemoprophylaxe unwahrscheinlich ist.
-
Zellen,
bei denen es sich um die primären
Targets für
eine sexuelle und mukosale Übertragung
von HIV, entweder in der Form von virusfreien oder virusinfizierten
Zellen, handelt, wurden nicht vollständig definiert und können divers
sein (Miller, C. J. et al., „Genital
Mucosal Transmission of Simian Immunodeficiency Virus: Animal Model
for Heterosexual Transmission of Human Immunodeficiency Virus", J. Virol., 63, 4277–4284 (1989);
Phillips, D. M. und Bourinbaiar, A. S., „Mechanism of HIV Spread from
Lymphocytes to Epithelia",
Virology, 186, 261–273
(1992); Phillips, D. M., Tan, X., Pearce-Pratt, R. und Zacharopoulos,
V. R., „An Assay
for HIV Infection of Cultured Human Cervix-derived Cells", J. Virol. Methods,
52, 1–13
(1995); Ho, J. L. et al., „Neutrophils
from Human Immunodeficiency Virus (HIV)-Seronegative Donors Induce HIV Replication from
HIV-infected Patients Mononuclear Cells and Cell Lines": An In Vitro Model
of HIV Transmission Facilitated by Chlamydia Trachomatis", J. Exp. Med., 181,
1493–1505
(1995); und Braathen, L. R. und Mork, C. in „HIV Infection of Skin Langerhans
Cells", in: Skin
Langerhans (dendritic) cells in virus infections and AIDS (Hrsg.
Becker, Y.) 131–139
(Kluwer Academic Publishers, Boston, (1991)). Solche Zellen schließen T-Lymphocyten,
Monocyten/Makrophagen und dendritische Zellen ein, was darauf hindeutet,
dass CD4-Zellrezeptoren am Vorgang der Virusübertragung beteiligt sind (Parr,
M. B. und Parr, E. L., „Langerhans
Cells and T lymphocyte Subsets in the Murine Vagina und Cervix", Biology of Reproduction,
44, 491–498
(1991); Pope, M. et al., „Conjugates
of Dendritic Cells and Memory T Lymphocytes from Skin Facilitate
Productive Infection With HIV-1",
Cell, 78, 389–398
(1994); und Wira, C. R. und Rossoll, R. M., „Antigen-presenting Cells
in the Female Reproductive Tract: Influence of Sex Hormones on Antigen
Presentation in the Vagina",
Immunology, 84, 505–508
(1995)).
-
Deshalb
wird erwartet, dass Mittel, die die HIV-CD4-Bindung blockieren,
die Virusübertragung
vermindern oder verhindern. Lösliche
rekombinante CD4 können
für diesen
Zweck nicht in Betracht gezogen werden, da zur Neutralisation der
Infektivität
der primären
HIV-Isolate hohe Konzentration erforderlich sind (Daar, E. S., Li,
X. L., Moudgil, T. und Ho, D. D., „High Concentrations of Recombinant
Soluble CD4 are Required to Neutralise Primary Human Immunodeficiency
Virus Type 1 Isolates",
Proc. Natl. Acad. Sci. USA, 87, 6574–6578 (1990), und im Fall von
SIV die Infektivität
durch CD4 gefördert
wird (Werner, A., Winskowsky, G. und Kurth, R., „Soluble CD4 Enhances Simian
Immunodeficiency Virus SIVagm, Infection", J. Virol., 64,
6252–6256
(1990)). Es wird jedoch erwartet, dass Anti-CD4-Antikörper die Übertragung von Viren unabhängig vom
Subtyp und der Variabilität
verhindern, ihre Applikation wäre
aber zu kostspielig (Daar et al., vorstehend, Watanabe, M., Boyson,
J. E., Lord, C. I. und Letvin, N. L. „Chimpanzees Immunized with
Recombinant Soluble CD4 Develop Anti-self CD4 Antibody Responses
with Anti-human Immunodeficiency Virus Activity", Proc. Natl. Acad. Sci. U.S.A, 89,
5103–5107
(1992); und Perno, C.-F., Baseler, M. W., Broder, S. und Yarchoan,
R., „Infection
of Monocytes by Human Immunodeficiency Virus Type 1 Blocked by Inhibitors
of CD4 gp120 Binding, Even in the Presence of Enhancing Antibodies", J. Exp. Med., 171,
1043–1056
(1990)).
-
Es
besteht ein Bedarf an einer sicheren und wirksamen Substanz, die
mittels eines Schaums, Gels, Schwamms oder einer anderen Form zur
Prävention,
dass HIV-1 oder HIV-2 Zellen im Körper infizieren, in die Vagina
eingeführt
werden kann. Es besteht die Hoffnung, dass eine derartige Substanz
von einer Frau ohne Kenntnis ihres Partners verwendet wird.
-
Die
Prospekte für
die nahe und möglicherweise
nicht so ferne Zukunft bei der Prävention der HIV-1-Übertragung
durch Vakzination sehen nicht gut aus. Ein kürzlich erschienener Bericht,
dass eine Vakzinierung mit inaktiviertem SIV die afrikanischen grünen Meerkatzen,
trotz einer starken Immunantwort auf SIV, nicht vor einer Infektion
mit dem homologen Virus schützte,
scheint in dieser Hinsicht nicht ermutigend zu sein (Siegel, F.,
Kurth, R. und Norley, S., (1995), „Neither Whole Inactivated
Virus Immunogen nor Passive Immunoglobulin Transfer Protects Against
SIVagm Infection in the African Green Monkey
Natural Host", J.
AIDS, 8, 217–226).
Bei der Erwägung
dieses Problems wurde der Schwerpunkt auf Versuche zum Aufbau einer
chemischen Barriere gegen eine HIV-1-Übertragung verlagert (Taylor,
(1994), „Building
a Chemical Barrier to HIV-1 Transmission", J. NIH Res., 6, 26–27).
-
Es
wurde vorgeschlagen, dass die Entwicklung von topisch applizierten
Mikrobiziden, von denen erwartet wird, dass sie die sexuelle (mukosale) Übertragung
von HIV-1 verhindern, „wirksam
gegen alle sexuell übertragenen
Erkrankungen sein müssen
und während
des Gebrauchs nicht gesehen, gerochen oder gefühlt werden sollten". Sie sollten auch
preisgünstig
und überall
verfügbar
sein, und es wurde erwartet, dass 1995 in den USA 25 Millionen USD
für ihre
Entwicklung aufgewendet werden (Taylor (1994) vorstehend). Detergenzien (Nonoxinol-9)
als ein universeller „Killer" von Pathogenen wurden
für klinische
Prüfungen
ausgewählt.
Es war jedoch nicht überraschend,
dass sich diese Verbindung für
den Wirt als schädlich
erwies.
-
Das
Targetieren der chemischen Barriere gegen die Übertragung individueller Pathogene
könnte
vielleicht die Entwicklung von Verbindungen fördern, die die Übertragung
der Human Immunodeficiency Viren verhindern. Durch die wirksame
Blockade von Rezeptoren könnte
dieses Ziel für
die Viren zum Beispiel gegebenenfalls erlangt werden. Dieses Konzept
kann durch Befunde unterstützt
werden, dass die Immunisierung von Schimpansen bzw. Rhesusaffen
mit menschlichem CD4, die im Vergleich zu CD4-Sequenzen von nicht menschlichen
Primaten mehrere Aminosäure-Punktmutationen
aufweist (Fomsgaard, A., Hirsch, V. M. und Johnson, P. R. (1992), „Cloning
and Sequences of Primate CD molecules: Diversity of the Cellular
Receptor for Simian Immunodeficiency Virus/Human Immunodeficiency
Virus", Eur. J.
Immunol., 22, 2973–2981),
entwickelte Anti-CD4-Antikörper,
die die HIV-1- und SIV-Replikation inhibierten (Watanabe, M., Levine,
C. G., Shen, L., Fisher, R. A., und Letvin, N. L. (1991), „Immunization
of Simian Immunodeficiency Virus-Infected Rhesus Monkeys with Soluble
Human CD4 Elicits an Antiviral Response", Proc. Natl. Acad. Sci. USA, 88, 4616–4620. Watanabe,
M., Chen, Z. W., Tsubota, H., Lord, C. I., Levine, C. G., und Letvin,
N. L. (1991), „Soluble
Human CD4 Elicits an Antibody Response in Rhesus Monkeys that Inhibits
Simian Immuunodeficiency Virus Replication", Proc. Natl. Acad. Sci. USA, 88, 120–124; und
Watanabe, M., Boyson, J. E., Lord, C. I. und Letvin, N. L., (1992), „Chimpanzees
Immunised with Recombinant Soluble CD4 Develop Anti-self CD4 Antibody
Responses with Anti-human Immunodefiency Virus Activity", Proc. Natl. Acad.
Sci USA, 89, 5103–5107).
-
Herpesviren
schließen
die folgenden aus Menschen isolierten Viren ein:
- (1)
Herpes-simplex-Virus 1 („HSV-1")
- (2) Herpes-simplex-Virus 2 („HSV-2")
- (3) Humanes Cytomegalievirus („HCMV")
- (4) Varicella-Zoster-Virus („VZV")
- (5) Epstein-Barr-Virus („EBV")
- (6) Humanes Herpesvirus 6 („HHV6")
- (7) Herpes-simplex-Virus 7 („HSV-7")
- (8) Herpes-simplex-Virus 8 („HSV8")
-
Herpesviren
wurden auch aus Pferden, Vieh, Schweinen (Pseudorabies-Virus („PRV") und dem porcinen
Cytomegalievirus, Hühnern
(infektiöse
Laryngotracheitis), Schimpansen, Vögeln (Marek'sche Krankheit durch Herpesvirus 1 und
2), Truthähnen
und Fischen (siehe „Herpesviridae:
A Brief Introduction",
Virology, Zweite Auflage, Hrsg. B. N. Fields, Kapitel 64, 1987 (1990))
isoliert.
-
Die
Herpes-simplex-Virusinfektion („HSV"-Infektion) stellt im Allgemeinen eine
rezidivierende Virusinfektion dar, gekennzeichnet durch das Auftreten
auf der Haut oder den Schleimhäuten
einzelner oder mehrerer Cluster mit kleinen Bläschen, die mit klarer Flüssigkeit
gefüllt
sind, auf leicht erhabenen Entzündungsbasen.
-
Das
Herpes-simplex-Virus ist ein Virus von relativ großer Größe. HSV-2
ruft häufig
Herpes labialis hervor. HSV2 kann gewöhnlich, aber nicht immer, aus
Genitalläsionen
wiedergewonnen werden. Im Allgemeinen wird HSV-2 venerisch übertragen.
-
Mindestens
20% aller Menschen in den USA wurden mit dem Herpesvirus Typ 2 (HSV-2)
infiziert, das gewöhnlich
sexuell übertragen
wird und rezidivierende Genitalulcera hervorrufen kann (Fleming,
D. T., McQuillan, G. M., Johnson, R. E. et al., „Herpes simplex virus type
2 in the United States, 1976 to 1994", N. Engl. J. Med., (1997), 337: 1105–1111; Arvin,
A. M., Prober, C. G., „Herpes
Simplex Virus Type 2 – A
Persistent Problem",
N. Engl. J. Med., (1997), 337: 1158–1159). Die Prävalenz von
HSV-2-Infektionen liegt in einigen Entwicklungsländern sogar noch höher (Nahmias,
A. J., Lee, F. K., Beckman-Nahmias, S., „Sero-epidemiological and
Sociological Patterns of Herpes-simplex Virus Infection in the World", Scand. J. Infect.
Dis., (1990), Suppl. 69: 19–36).
Obwohl die Infektion durch antivirale Medikamente behandelbar ist,
ist die wirksame Langzeitsuppression des Genitalherpes teuer (Engel,
J. P., „Long-term
Suppression of Genital Herpes",
JAMA, (1998), 280: 928–929).
Wenn man die hohe Prävalenz
von Infektionen bedenkt, ist die Wahrscheinlichkeit der weiteren Ausbreitung
des Virus durch unbehandelte Menschen und asymptomatische Träger, die
keine antivirale Therapie erhalten, extrem hoch. Andere Herpesviren,
einschließlich
das Cytomegalievirus (Krieger, J. N., Coombs, R. W., Collier, A.
C. et al., „Seminal
Shedding of Human Immunodeficiency Virus Type 1 and Human Cytomegalovirus:
Evidence For Different Immunologic Controls", J. Infect. Dis. (1995), 171: 1018–1022; van
der Meer, J. T. M., Drew, W. L., Bowden R. A. et al., „Summary
of the International Consensus Symposium on Advances in the Diagnosis,
Treatment and Prophylaxis of Cytomegalovirus Infection", Antiviral Res.,
(1996), 32: 119–140) (HCMV),
Herpesvirus 6 (Leach, C. T., Newton, E. R., McParlin, S. et al., „Human
Herpesvirus 6 Infection of the Female Genital Tract", J. Infect. Dis.,
(1994), 169: 1281–1283)
und Herpesvirus 8 (Howard, M. R., Whitby, D., Bahadur, G. et al., „Detection
of Human Herpesvirus 8 DNA in Semen from HIV-infected Individuals
But Not Healthy Semen Donors",
AIDS, (1997), 11: F15–F19),
der kausale Krankheitserreger des Kaposi-Sarkoms, werden auch sexuell übertragen.
-
Die
Zeit der initialen Herpes-simplex-Infektion ist gewöhnlich obskur,
außer
bei der ungewöhnlichen primären systemischen
Infektion, die bei Kindern vorkommt und durch generalisierte Haut-
und Schleimhautläsionen,
begleitet von schwerwiegenden Allgemeinsymptomen gekennzeichnet
ist. Lokalisierte Infektionen treten gewöhnlich in der Kindheit auf,
können
aber bis zum Erwachsenenalter verzögert sein. Es wird angenommen,
dass das Herpes-simplex-Virus in der Haut dormant bleibt und dass
herpetische Eruptionen durch Überexposition
gegenüber
Sonnenlicht, fieberhafte Erkrankungen oder physischen oder emotionalen
Stress herbeigeführt
werden; auch bestimmte Nahrungsmittel und Medikamente sind impliziert
gewesen. In vielen Fällen
kann der Trigger-Mechanismus nicht nachgewiesen werden.
-
Die
durch das Herpes-simplex-Virus verursachten Läsionen können überall auf der Haut oder auf
den Schleimhäuten
auftreten, am häufigsten
aber im Gesicht, besonders um den Mund herum oder auf den Lippen, der
Bindehaut und Kornea oder den Genitalien. Einer kurzen Prodromalperiode
mit prickelnden Beschwerden oder Jucken folgt das Auftreten kleiner,
straffer Bläschen
auf einem erythematösen
Grund. Die einzelnen Cluster können
größenmäßig von
0,5 bis 1,5 cm variieren, mehrere Gruppen können jedoch zusammenfließen. Herpes
simplex auf der Haut, die straff an den unterliegenden Strukturen
haftet (zum Beispiel der Nase, Ohren oder Finger) kann schmerzhaft
sein. Die Bläschen
können
einige Tage persistieren, dann beginnen sie auszutrocknen, wobei
sie eine dünne
gelbliche Kruste bilden. Heilung tritt im Allgemeinen innerhalb
von 10 Tagen nach dem Beginn auf. In feuchten Körperbereichen, kann die Heilung
mit einer sekundären
Entzündung
langsamer ablaufen. Die Heilung individueller Herpesläsionen findet
gewöhnlich
vollkommen statt, rezidivierende Läsionen an der gleichen Stelle
können
jedoch zu Atrophie und Narbenbildung führen.
-
Bei
mit HSV-2 infizierten Frauen können
gegebenenfalls keine Hautläsionen
auftreten, die Infektion kann vollständig auf das Innere der Vagina
beschränkt
bleiben. Die Cervix ist häufig
befallen, und es liegen zunehmend Hinweise vor, dass dies einen
Faktor bei der Entwicklung eines Cervixkarzinoms spielen könnte.
-
Kornealäsionen bestehen
häufig
aus rezidivierender herpetischer Keratitis, die sich durch ein unregelmäßiges Ulcus
dendriticum auf den superfiziellen Lagen manifestiert. Als Folgeerscheinungen
können
Narbenbildung und Sehstörungen
auftreten.
-
Bei
Kindern oder Kleinkindern können
aufgrund einer Herpesinfektion Gingivostomatitis und Vulvovaginitis
auftreten. Zu den Symptomen zählen
Reizbarkeit, Anorexia, Fieber, Entzündung und weißliche Plaques und
Ulcera im Mund. Die Primärinfektionen
können
insbesondere bei Kindern, obwohl manchmal bei älteren Kindern, eine extensive
Organbeteiligung und fatale Virämie
hervorrufen.
-
Bei
Frauen, bei denen eine HSV-2-Attacke in der Spätschwangerschaft auftritt,
kann die Infektion mit der Entwicklung einer schweren Virämie auf
den Fetus übertragen
werden. Das Herpes-simplex-Virus kann auch eine fatale Encephalitis
hervorrufen.
-
Kaposi's varicelliforme
Eruption (Ekzema herpeticum) ist eine potenziell fatale Komplikation
des atopischen Ekzems bei Kindern oder Erwachsenen. Die Exposition
von Patienten mit ausgebreiteter atopischer Dermatitis gegenüber Personen
mit aktivem Herpes simplex sollte vermieden werden.
-
Es
wurde nachgewiesen, dass kein lokales oder systemisches Chemotherapeutikum
zur Behandlung des Herpes-simplex-Virus, mit der möglichen
Ausnahme von topischem Idoxuridin (IDU) bei der herpetischen Keratitis
superficialis wirksam ist. Berichte über diese Verbindung beim kutanen
Herpes sind widersprüchlich. Zu
anderen Medikamenten, die zur Behandlung von HSV eingesetzt wurden,
gehören
Trifluorthymidin; Vidarabin (Adeninarabinosid, Ara-A), Aciclovir,
und andere Inhibitoren der viralen DNA-Synthese können bei
der herpetischen Keratitis wirksam sein. Diese Medikamente inhibieren
die Replikation des Herpes-simplex-Virus und können die klinischen Manifestationen
supprimieren. Das Herpes-simplex-Virus bleibt jedoch latent in den sensorischen
Spinalganglien, und die Rezidivrate ist bei medikamentös behandelten
und unbehandelten Patienten ähnlich.
Darüber
hinaus sind einige Medikamenten-resistente Herpesvirus-Stämme hervorgegangen.
-
Durch
das Varicella-Zoster-Virus (humanes Herpesvirus 3) verursachte Erkrankungen
schließen
Varicella (Windpocken) und Zoster (Gürtelrose) ein.
-
Das
Cytomegalievirus (humanes Herpesvirus 5) ist für die zytomegale Einschlusskrankheit
bei Kindern verantwortlich. Es gibt derzeit keine spezifische Behandlung
zur Behandlung von mit dem Cytomegalievirus infizierten Patienten.
-
Das
Epstein-Barr-Virus (humanes Herpesvirus 4) ist der kausale Krankheitserreger
der infektiösen Mononucleose
und wurde mit dem Burkitt-Lymphom und dem nasopharyngealen Karzinom
in Verbindung gebracht.
-
Zu
Herpesviren der Tiere, die ein Problem für Menschen darstellen können, gehören das
B-Virus (Herpesvirus von Altweltaffen) und das Herpesvirus der Krallenaffen
(Herpesvirus der Neuweltaffen).
-
Bei
der Suche nach preisgünstigen
antiviralen Verbindungen, die zur Verminderung der Häufigkeit
einer sexuellen Übertragung
des Human Immunodeficiency Virus Typ 1 (HIV-1) und Herpesviren (HSV)
topisch appliziert werden könnten,
entschieden sich die Anmelder, obwohl alles dagegen sprach, zum
Screening von Hilfsstoffen auf Anti-HIV-1-Aktivität und entdeckten
den erfindungsgemäßen Gegenstand,
der die Verabreichung von Celluloseacetatphthalat („CAP") oder Hydroxypropylmethylcellulosephthalat
(„HPMCP") beinhaltete.
-
D. SEXUELL ÜBERTRAGENE
ERKRANKUNGEN BAKTERIELLEN URSPRUNGS
-
Heilbare
sexuell übertragene
Erkrankungen (STDs) bakteriellen Ursprungs stellen weltweit die
häufigste
Krankheitsursache mit signifikanten gesundheitlichen, sozialen und
wirtschaftlichen Konsequenzen dar. Sie können zu langfristigen, schwerwiegenden
Komplikationen und Folgeerscheinungen führen. Die geschätzte jährliche
(1995) weltweite Inzidenz der vier wichtigsten heilbaren STDs, nämlich Syphillis,
Gonorrhoe (Neisseria gonorrhoeae), Chlamydia und Trichomoniasis,
lag bei ca. 330 Millionen (Gerbase, A. C., Rowley, J. T., Heymann,
D. H. L. et al., „Global
Prevalence and Incidence Estimates of Selected Curable STDs", Sex. Transm. Inf.,
(1998), 74, (Suppl. 1): S12–S16).
Eine andere behandelbare STD, Ulcus molle, eine ulzerative Erkrankung
der Genitalien, die von Haemophilus ducreyi verursacht wird, kommt
in Entwicklungsländern
in Afrika, Asien und Lateinamerika häufig vor, wo die Inzidenz über die
von Syphillis hinausgehen kann (Trees, D. K., Morse, S. A., „Chancroid
and Haemophilus ducreyi: An Update", Clin. Microb. Rev., (1995), 8: 357–375). Die
vorgeschlagenen Kontrollmaßnahmen
für diese
STDs schließen
Folgendes ein: Surveillance, Labordiagnose, Management des Syndroms,
Datenmonitoring. Behandlung mit antibakteriellen Mitteln, Benachrichtigung
des Partners und Entwicklung von Vakzinen (Rao, P., Mohamedali,
F. Y., Temmerman, M. et al., „Systematic
Analysis of STD Control: an Operational Model", Sex. Transm. Inf. (1998), 74 (Suppl
1): S17–S22;
Dallabetta, G. A., Gerbase, A. C., Holmes, K. K., „Problems,
Solutions, and Challenges in Syndromic Management of Sexually Transmitted
Diseases", Sex.
Transm. Inf., (1998), 74 (Suppl 1): S1–S11; Burstein, G. R., Gaydos, C.
A., Diener-West, M., „Incident
Chlamydia Trachomatis Infections Among Inner-City Adolescent Females", JAMA, (1998), 280:
521–526).
-
EP0706794 offenbart Anti-AIDS-Pharmazeutika,
umfassend einen Wirkstoff, der sich von Cellulosephthalaten unterscheidet.
Dieses Präparat
umfasst eine Beschichtung, worin das Beschichtungsmittel Hydroxypropylmethylcellulosephthalat
darstellt.
-
EP 677322 offenbart die Verwendung
von Cellulosephthalaten als Beschichtungsmaterialien.
-
US 5356634 offenbart die
Verwendung von Cellulosephthalaten als Beschichtungsmaterialien.
-
Es
wurde bisher folglich die Entwicklung eines topischen Mikrobizids
aus preisgünstigen,
weithin verfügbaren
Ressourcen mit breiten antiviralen und antibakteriellen Aktivitäten gewünscht.
-
ZUSAMMENFASSUNG
DER ERFINDUNG
-
Gegenstand
der vorliegenden Erfindung ist die Bereitstellung eines sicheren
und relativ preisgünstigen Medikamentes,
das zur Verminderung der Häufigkeit
der Übertragung
der Virusinfektionen mit dem Human Immunodeficiency Virus und Herpesvirus,
insbesondere denen, die sexuell übertragen
werden, geeignet sind.
-
Ein
weiterer Gegenstand der vorliegenden Erfindung ist die Bereitstellung
einer Zusammensetzung zur Verminderung der Häufigkeit der Übertragung
des Human Immunodeficiency Virus und Herpesvirus.
-
Ein
anderer Gegenstand der vorliegenden Erfindung ist die Bereitstellung
von Medikamenten, die zur Behandlung und Prävention von sexuell übertragenen
Bakterieninfektionen geeignet sind.
-
Die
vorstehenden Gegenstände,
zusammen mit allen Gegenständen,
Aufgaben und Vorteilen werden durch die vorliegende Erfindung erreicht.
-
Gegenstand
der vorliegenden Erfindung ist ein Medikament zur Verminderung der
Häufigkeit
einer Übertragung
und insbesondere der Prävention
der Übertragung
des Human Immunodeficiency Virus oder Herpesvirus durch Verabreichung
einer gegen das Human Immunodeficiency Virus oder gegen das Herpesvirus gerichteten
wirksamen Menge von mindestens einem Cellulosephthalat, das aus
Acetatphthalat (CAP) und Hydroxypropylmethylcellulosephthalat (HPMCP),
entweder allein oder in Kombination mit einem pharmazeutisch verträglichen
Träger
oder Verdünnungsmittel
ausgewählt
ist, an einen Menschen.
-
Gegenstand
der Erfindung ist auch eine pharmazeutische Zusammensetzung zur
Verminderung der Häufigkeit
einer Übertragung
des Human Immunodeficiency Virus oder Herpesvirus umfassend eine
gegen das Immunodeficiency Virus gerichtete wirksame Menge oder
eine gegen das Herpesvirus gerichtete wirksame Menge von mindestens
einem Cellulosephthalat, das aus Celluloseacetatphthalat und Hydroxypropylmethylcellulosephthalat
in Kombination mit einem pharmazeutisch verträglichen Träger oder einem Verdünnungsmittel
ausgewählt
ist.
-
Gegenstand
der vorliegenden Erfindung ist weiter ein Medikament zur Prävention
der Übertragung
einer sexuell übertragenen
Bakterieninfektion an einen Menschen oder Behandlung eines mit einer
sexuell übertrgenen
Bakterieninfektion infizierten Menschen, umfassend die Verabreichung
einer wirksamen antibakteriellen Menge von mindestens einem Cellulosephthalat,
das aus Acetatphthalat (CAP) und Hydroxypropylmethylcellulosephthalat
(HPMCP) entweder allein oder in Kombination mit einem pharmazeutisch
verträglichen
Träger
oder einem Verdünnungsmittel
ausgewählt
ist, an einen Menschen.
-
Gegenstand
der vorliegenden Erfindung ist auch eine pharmazeutische Zusammensetzung
zur Prävention
der Übertragung
oder zur Behandlung einer sexuell übertragenen Bakterieninfektion,
umfassend eine wirksame antibakterielle Menge aus mindestens einem
Cellulosephthalat, das aus Celluloseacetatphthalat und Hydroxypropylmethylcellulosephthalat
in Kombination mit einem pharmazeutisch verträglichen Träger oder Verdünnungsmittel
ausgewählt
ist.
-
Gegenstand
der vorliegenden Erfindung sind auch die vorstehend erwähnten Medikamente
und pharmazeutischen Zusammensetzungen, worin das Cellulosephthalat
(CAP und/oder HPMCP) in der Form einer Suspension und bevorzugt
in einer mikronisierten Form bereitgestellt wird. Eine derartige
Suspension kann weiter ein mit Wasser mischbares, weitgehend wasserfreies
Nichtlösungsmittel
für CAP
oder HPMCP, wie zum Beispiel Glycerol, einschließen.
-
KURZE BESCHREIBUNG
DER ZEICHNUNGEN
-
1 ist
eine grafische Darstellung der Inhibition (%) vs. der Celluloseacetatphthalat-Konzentration („CAP"-Konzentration) für HSV-1
und HSV-2. 1 zeigt folglich die inhibitorische
Wirkung von Celluloseacetatphthalat („CAP") auf HSV-1 und HSV-2.
-
2 ist
eine grafische Darstellung der Inhibition (%) vs. der HPMCP-Konzentration
für HSV-1
und HSV-2. Die in 2 gezeigten Ergebnisse sind
den in 1 gezeigten ähnlich.
-
3 ist
eine grafische Darstellung von HIV-1-p24-Antigen (Absorption bei
450 nm) vs. HIV-1-Verdünnung. 3 zeigt
die Disintegration von gereinigtem HIV-1 durch Behandlung mit einer „AQUATERIC"-Glycerolformulierung
(„CAP-Formulierung
I") mit oder ohne
Polyvinylpyrrolidon (PVP) und Crospovidon 5 Minuten bei 37°C, wie durch
die Freisetzung des Nukleocapsid-Antigens p24 gemessen.
-
4 ist
eine grafische Darstellung des HIV-1-p24-Antigens (Absorption bei
450 nm) vs. der Viruskonzentration. 4 zeigt
die Inaktivierung der HIV-1-Infektivität durch Behandlung mit einer „AQUATERIC"-Glycerolformulierung,
enthaltend 286 mg/ml „AQUATERIC" 5 Minuten bei 37°C, wie durch
Bildung des Nukleocapsid-Antigens p24 durch infizierte Zellen, wie
mittels ELISA gemessen, bestimmt.
-
5 ist
eine grafische Darstellung der Absorption (410 nm) vs. der Virusverdünnung. 5 zeigt
die Inaktivierung von HSV-1 und HSV-2 durch eine Suspension von „AQUATERIC" in Glycerol. Viruspräparationen wurden
1:1 mit einer Suspension aus „AQUATERIC" in Glycerol 5 Minuten
bei 37°C
gemischt.
-
6 ist
eine grafische Darstellung der Absorption (410 nm) vs. der Virusverdünnung. 6 zeigt
die Inaktivierung von HSV-1 und HSV-2 durch eine „AQUATERIC"-Glycerolformulierung
mit PVP und Crospovidon.
-
7 ist
eine grafische Darstellung, welche die Disintegration von gereinigtem
HIV-1 durch Behandlung mit einer Formulierung aus mikronisiertem
CAP in Glycerol („CAP-Formulierung
I", die hierin nachstehend definiert
ist) 5 Minuten bei 37°C
bei Anwesenheit von Samenflüssigkeit
bzw. Vollblut zeigt (nähere
Einzelheiten sind in 3 ersichtlich).
-
8 ist
eine grafische Darstellung, welche die Inaktivierung von CMV durch
die CAP-Formulierung I zeigt. Die Viruspräparationen wurden 1:1 mit der
CAP-Formulierung I 5 Minuten bei 37°C gemischt. Reihenverdünnungen
von den Viruspräparationen
wurden unter Verwendung eines auf der Quantifizierung von β-Galactosidase
basierenden Ablesesystems auf Infektivität getestet (Absorption bei
410 nm).
-
AUSFÜHRLICHE BESCHREIBUNG DER ERFINDUNG
-
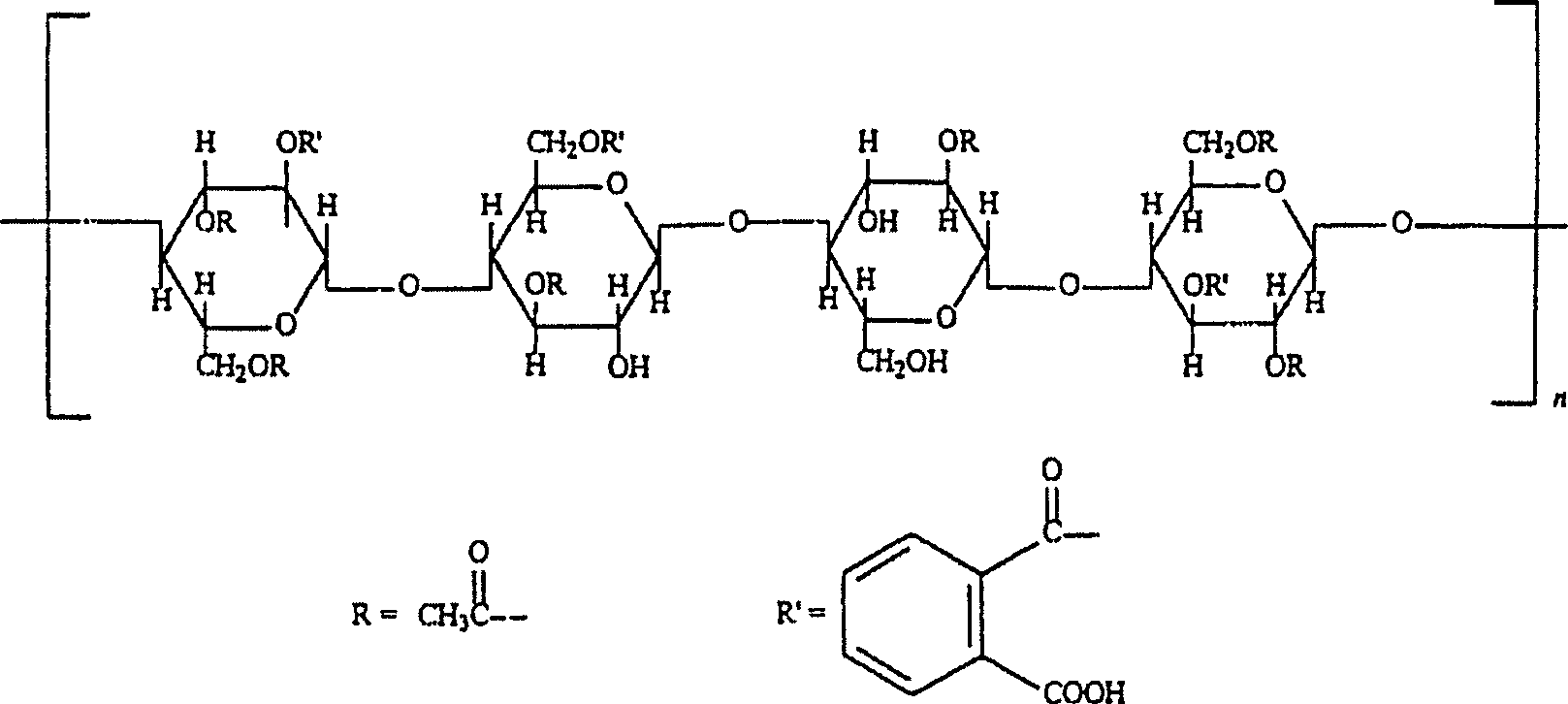
Die
vorliegende Erfindung betrifft die Verwendung von Celluloseacetatphthalat
(CAP) und/oder Hydroxypropylmethylcellulosephthalat (HPMCP) zur
Prävention
der Übertragung
von Virusinfektionen und zur Prävention
oder Behandlung von sexuell übertragenen
Bakterieninfektionen.
-
Einige
der Eigenschaften von CAP, wie im Handbook of Pharmaceutical Excipients
beschrieben, sind wie folgt zusammengefasst: FREINAMEN:
| BP: | Cellacephate |
| PhEur: | Cellulosi
acetas phthalas |
| USPNF: | Celluloseacetatphthalat |
-
SYNONYME:
-
Acetylphthalylcellulose;
CAP; Cellacefat; Celluloseacetathydrogen-1,2-benzendicarboxylat;
Celluloseacetathydrogenphthalat; Celluloseacetatmonophthalat; Celluloseacetophthalat;
Celluloseacetylphthalat.
-
CHEMISCHE BEZEICHNUNG
UND CAS REGISTRY NUMBER:
-
- Cellulose, Acetat, 1,2-Benzendicarboxylat [9004-38-0]
- Celluloseacetatphthalat ist eine Cellulose, worin ca. die Hälfte der
Hydroxylgruppen acetyliert und ca. ein Viertel verestert sind, wobei
eine der beiden Säuregruppen
Phthalsäure
darstellt. Die andere Säuregruppe
ist frei. Siehe nachstehende Strukturformel.
-
-
Funktionskategorie:
Beschichtungsmittel.
-
APPLIKATIONEN IN DER PHARMAZEUTISCHEN
FORMULIERUNG ODER TECHNOLOGIE:
-
Celluloseacetatphthalat
wurde bisher als ein magensaftresistentes Filmüberzugsmaterial oder als ein Matrix-Bindemittel
für Tabletten
und Kapseln verwendet (Spitael, J., Kinget, R., Naessens, K., „Dissolution Rate
of Cellulose Acetate Phthalate and Brönsted Catalysis Law", Pharm. Ind., (1980),
42: 846–849;
Takenaka, H., Kawashima, Y., Lin, S-Y., „Preparation of Enteric-Coated
Microcapsules for Tabletng by Spray-Drying Technique and in vitro
Simulation of Drug Release from the Tablet in GI Tract", J. Pharm. Sci.,
(1980), 69: 1388–1392;
Stricker, H., Kulke, H., „Rate
of Disintegration and Passage of Enteric-Coated Tablets in Gastrointestinal
Tract", Pharm. Ind.,
(1981), 43: 1018–1021;
Takenaka, H., Kawashima, Y., Lin, S-Y, „Polymorphism of Spray-Dried
Microencapsulated Sulfamethoxazole with Cellulose Acetate Phthalate
and Colloidal Silica Montmorillonite, or Talc", J. Pharm. Sci., (1981), 70: 1256–1260; Maharaj,
I., Nairn, J. G., Campbell J. B., „Simple Rapid Method for the
Preparation of Enteric-Coated Microspheres", J. Pharm. Sci., (1984), 73: 39–42; Beyger, J.
W., Nairn, J. G., „Some
Factors Affecting the Microencapsulation of Pharmaceuticals with
Cellulose Acetat Phthalate",
J. Pharm. Sci., (1986), 75: 573–578;
Lin, S-Y, Kawashima, Y., „Drug
Release from Tablets Containig Cellulose Acetate Phthalate as an
Additive or Enteric-Coating Material", Pharm. Res., (1987), 4: 70–74; Thoma,
K., Hekenmüller,
H., „Effect
of Film Formers and Plasticizers on Stability of Resistance and
Disintegration Behaviour, Part 4: Pharmaceutical-Technological and
Analytical Studies of Gastric Juice Resistant Commercial Preparations", Pharmazie, (1987),
42: 837–841).
-
Derartige Überzüge sind
beständig
gegen verlängerten
Kontakt mit dem stark sauren Magensaft, werden aber weich und quellen
im leicht sauren oder neutralen intestinalen Milieu.
-
Celluloseacetatphthalat,
wenn bisher als ein Adjuvans verwendet, wurde im Allgemeinen auf
feste Dosierungsformen entweder durch Beschichtung aus organischen
oder wässrigen
Lösungsmittelsytemen
oder durch direkte Kompression appliziert. Die verwendeten Konzentrationen
betrugen 0,5 bis 9,0% des Kerngewichts. Das Zufügen von Weichmachern verbessert
die Wasserbeständigkeit
dieses Überzugsmaterials,
und derartig weich gemachte Filme sind wirksamer, als wenn Celluloseacetatphthalat als
ein Adjuvans allein verwendet wird. Celluloseacetatphthalat ist
kompatibel mit den folgenden Weichmachern: acetyliertem Monoglycerid;
Butylphthalylbutylglycolat; Dibutyltarnat; Diethylphthalat; Dimethylphthalat;
Ethylphthalylethylglycolat; Glycerin; Propylenglycol; Triacetin;
Triacetincitrat und Tripropionin. Celluloseacetatphthalat wurde
bisher auch in Kombination mit anderen Beschichtungsmitteln zur
Kontrolle der Medikamenten-Freisetzung, z. B. Ethylcellulose, verwendet.
-
BESCHREIBUNG:
-
Celluloseacetatphthalat
stellt ein hygroskopisches, weißes,
freifießendes
Pulver oder farblose Flocken dar. Es ist geschmacklos und geruchlos
oder kann einen leichten Essigsäuregeruch
aufweisen.
-
ARZNEIBUCH-SPEZIFIKATIONEN:
-
TYPISCHE EIGENSCHAFTEN:
-
Hygroskopizität: Celluloseacetatphthalat
ist hygroskopisch, und es sind Vorsichtsmaßnahmen notwendig, um eine übermäßige Absorption
von Feuchtigkeit zu vermeiden (Callahan, J. C., Cleary, G. W., Elefant,
M., Kaplan, G., Kensler, T., Nash, R. A., „Equilibrium Moisture Content
of Pharmaceutical Excipients", Drug
Dev. Ind. Pharm., (1982), 8: 355–369).
-
Schmelzpunkt:
192°C. Die
Glasübergangstemperatur
liegt bei 160–170°C (Sakellariou,
P., Rowe, R. C., White, E. F. T., „The Thermomechanical Properties
and Glass Transition Temperatures of Some Cellulose Derivatives
used in Film Coating",
Int. J. Pharmaceutics, (1985), 27: 267–277).
-
Löslichkeit:
Nahezu unlöslich
in Alkoholen, chlorierten Kohlenwasserstoffen, Kohlenwasserstoffen
und Wasser; löslich
in cyclischen Ethern, Estern, Etheralkoholen, Ketonen und gewissen
Lösungsmittel-Gemischen.
Auch löslich
in gewissen gepufferten wässrigen
Lösungen
bei höher
als pH 6. Die folgende Liste zeigt einige der Lösungsmittel und Lösungsmittel-Gemische,
in denen Celluloseacetatphthalat eine Löslichkeit von 1 in 10 Teilen
oder mehr aufweist.
Aceton
Aceton: Ethanol (1:1)
Aceton:
Methanol (1:1/1:3)
Aceton: Methylenchlorid (1:1/1:3)
Aceton:
Wasser (97:3)
Benzen: Methanol (1:1)
Diacetonalkohol
Dioxan
Ethoxyethylacetat
Ethylacetat:
Ethanol (1:1)
Ethylacetat: Propan-2-ol (1:1/1:3)
Ethylenglycolmonoacetat
Ethyllactat
Methoxyethylacetat β-Methoxyethylenalkohol
Methylacetat
Methylenchlorid:
Ethanol (3:1)
Methylethylketon
Viskosität (dynamisch):
50–90
mPas (50–90
cP) für
eine 15 gew.-%ige Lösung
in Aceton mit einem Feuchtigkeitsgehalt von 0,4%. Hierbei handelt
es sich um eine gute Beschichtungslösung mit einer honigartigen
Konsistenz, die Viskosität
wird jedoch von der Reinheit des Lösungsmittels beeinflusst.
-
STABILITÄT UND LAGERUNGSBEDINGUNGEN:
-
Cellulosacetatphthalat
hydrolysiert unter verlängerten
adversen Bedingungen, wie zum Beispiel hoher Temperatur und Feuchtigkeit,
langsam mit einer resultierenden Zunahme des Gehaltes an freier
Säure,
Viskosität
und Essigsäuregeruch.
Wenn sein Feuchtigkeitsgehalt über
ca. 6 Gew.-% liegt, tritt eine ziemlich rasche Hydrolyse auf. Celluloseacetatphthalat
ist jedoch stabil, wenn es in einem fest verschlossenen Behälter an
einem kühlen,
trockenen Ort aufbewahrt wird.
-
INKOMPATIBILITÄTEN:
-
Celluloseacetatphthalat
ist nicht kompatibel mit Eisen(II)-culfat, Eisen(III)-chlorid, Silbernitrat,
Natriumcitrat, Aluminiumsulfat, Calciumchlorid, Quecksilberchlorid,
Bariumnitrat, basischem Bleiacetat und starken Oxidationsmitteln,
wie zum Beispiel starken Alkalien und Säuren. Es ist zur Kenntnis zu
nehmen, dass eine Carbonsäuregruppe
der Phthalsäurekomponente
unverestert und für
Interaktionen frei bleibt. Demgemäß kann Inkompatibilität mit säureempfindlichen
Medikamenten auftreten (Rawlins, E. A., Hrsg., „Bentley's Textbook of Pharmaceutics", London: Baillière, Tindall
und Cox, (1977), 291).
-
HERSTELLUNGSVERFAHREN:
-
Celluloseacetatphthalat
wird durch zur Reaktion bringen des teilweisen Acetatesters von
Cellulose mit Phthalsäureanhydrid
in Gegenwart einer tertiären
organischen Base, wie zum Beispiel Pyridin, hergestellt.
-
SICHERHEIT:
-
Celluloseacetatphthalat
wird überall
in oralen Pharmazeutika verwendet und wird im Allgemeinen als nicht
toxisches Material angesehen, das frei von unerwünschten Wirkungen ist. Ergebnisse
von Langzeitfütterungsstudien
mit Celluloseacetatphthalat, an Ratten und Hunden, haben eine geringe
orale Toxizität
erkennen lassen. Die Ratten überlebten
tägliche
Fütterungen
von bis zu 30% in der Ration für
die Dauer bis zu einem Jahr, ohne eine Verminderung des Wachstums
zu zeigen. Hunde, denen täglich
16 g in der Ration für
die Dauer eines Jahres verfüttert
wurde, blieben auch normal (Hodge, H. C., „The Chronic Toxicity of Cellulose
Acetate Phthalate in Rats and Dogs", J. Pharmacol, 80, 250–255, (1944)).
-
BEHÖRDLICHER ZULASSUNGSSTATUS:
-
Eingeschlossen
in den Richtlinien der FDA zu „Arzneilich
nicht wirksamen Bestandteilen (orale Kapseln und Tabletten)". Eingeschlossen
in nicht parenteralen Medikamenten, die in Großbritannien zugelassen sind.
-
Arzneibücher: Aust,
Br, Bras, Cz, Eur, Fr, Ger, Gr, Hung, Ind, It, Jpn, Mex, Niederlande,
Nord, Port, Schweiz und USPNF.
-
Einige
der im Handbook of Pharmaceutical Excipients beschriebenen Eigenschaften
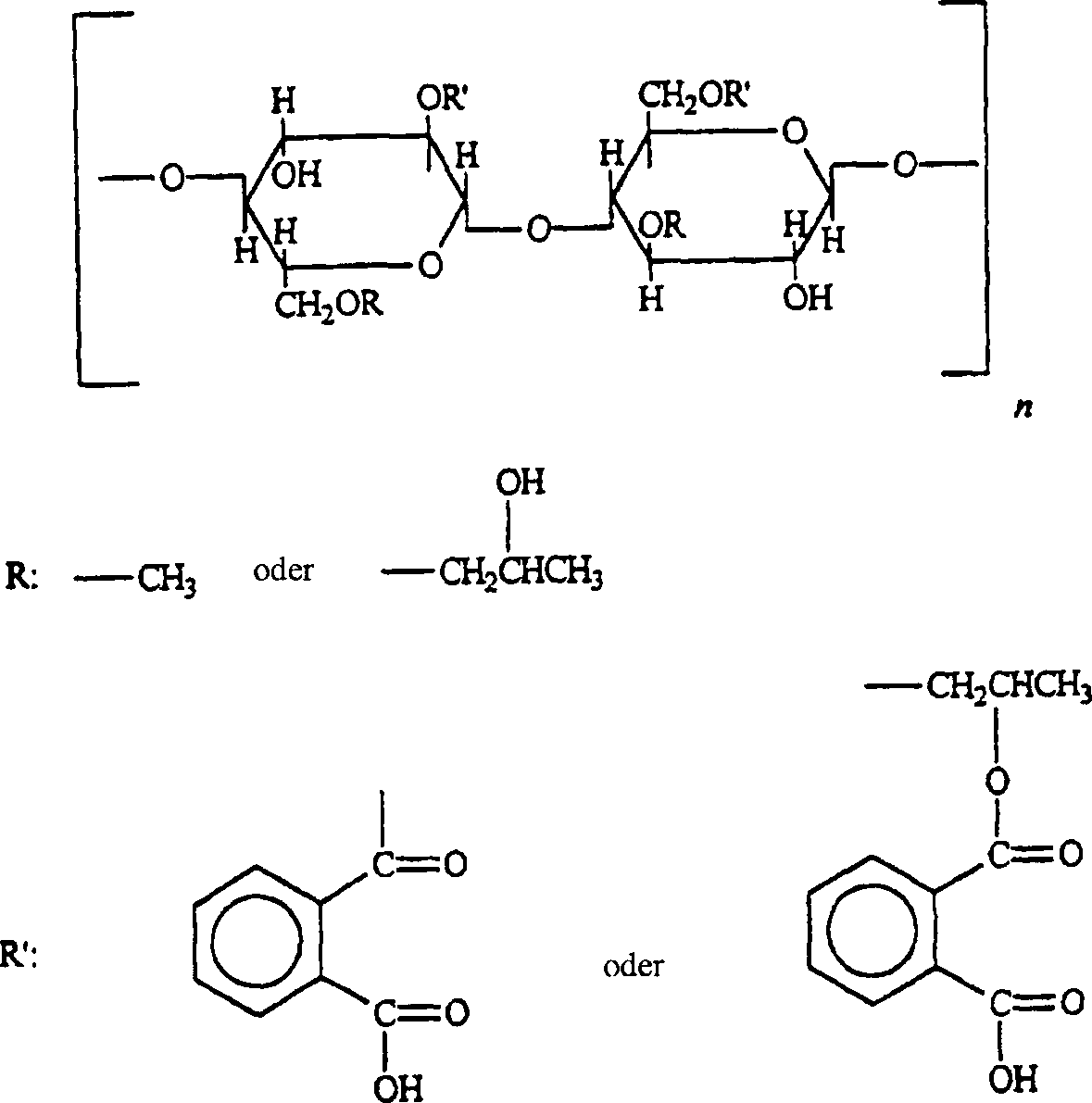
von HPMCP sind wie folgt zusammengefasst:
Freinamen: BP: Hypromellose
phthalate; PhEur: Methylhydroxypropylcellulosi phthalas und USPNF:
Hydroxypropyl methylcellulose phthalate.
Synonyme: Cellulosephthalathydroxypropylmethylether;
HPMCP; 2-Hydroxypropylmethylcellulosephthalat; Methylhydroxypropylcellulosephthalat.
Chemische
Bezeichnung und CAS Registry Number: Cellulose, Hydrogen-1,2-benzendicaboxylat,
2-Hydroxypropylmethylether [9050-31-1]
-
-
Funktionskategorie:
Beschichtungsmittel.
-
APPLIKATIONEN IN PHARMAZEUTISCHEN
FORMULIERUNGEN ODER TECHNOLOGIE
-
Hydroxypropylmethylcellulosephthalat
wurde bisher weithin in oralen pharmazeutischen Formulierungen als
ein magensaftresistentes Überzugsmaterial
für Tabletten
oder Granulate verwendet (Ehrhardt, L., Patt, L., Schindler, E., „Optimization
of Film Coating Systems",
Pharm. Ind., (1973), 35: 719–722;
Delporte, J. P., Jaminet, F., „Influence
of Formulation of Enteric-Coated Tablets on the Bioavailability
of the Drug", J.
Pharm. Belg., (1976), 31: 263–276;
Patt, L., Hartmann, V., „Solvent
Residues in Film Forming Agents",
Pharm. Ind., (1976), 38: 902–906;
Stafford, J. W., „Enteric
Film Coating Using Completely Aqueous Dissolved Hydroxypropyl Methylcellulose
Phthalate Spray Solutions",
Drug. Dev. Ind. Pharm., (1982), 8: 513–530; Thoma, K., Heckenmüller, H.,
Oschmann, R., „Resistance
and Disintegration Behaviour of Gastric Juice Resistant Drugs", Pharmazie, (1987),
42: 832–836;
Thoma, K., Heckenmüller,
H., Oschmann, R., „Impact
of Film Formers and Plasticizers on Stability of Resistance and
Disintegration Behaviour",
Pharmazie, (1987), 42: 837–841).
-
Hydroxypropylmethylcellulosephthalat
ist in Magensaft unlöslich,
quillt aber und löst
sich rasch im Dünndarm
auf. Im Allgemeinen wurden Konzentrationen von 5–10% Hydroxypropylmethylcellulosephthalat eingesetzt,
wobei das Material in entweder einem Lösungsmittelgemisch aus Dichlormethan:
Ethanol (50:50) oder Ethanol: Wasser (80:20) aufgelöst wird.
Hydroxypropylmethylcellulosephthalat kann in der Regel auf Tabletten
und Granulate ohne das Zufügen
eines Weichmachers oder anderer Filmbildner unter Verwendung etablierter
Beschichtungsverfahren appliziert werden (Rowe, R. C., „Molecular
Weight Studies on the Hydroxypropyl Methylcellulose Phthalate (HP55)", Acta. Pharm. Technol.,
(1982), 28(2): 127–130.
Das Zufügen
einer kleinen Menge Weichmacher oder Wasser kann Filmplatzprobleme
vermeiden; viele häufig
verwendete Weichmacher, wie zum Beispiel Diacetin, Triacetin, Diethyl-
und Dibutylphhalat, Castoröl,
Acetylmonoglycerid und Polyethylenglycole sind mit Hydroxypropyhnethylcellulosephthalat
kompatibel. Mit Hydroxypropylmethylcellulosephthalat beschichtete
Tabletten disintegrieren schneller als Tabletten, die mit Celluloseacetatphthalat überzogen
sind.
-
Hydroxypropylmethylcellulosephthalat
kann unter Verwendung einer Dispersion aus dem mikronisierten Hydroxypropylmethylcellulosephthalatpulver
in einer wässrigen
Dispersion aus einem geeigneten Weichmacher, wie zum Beispiel Triacetin,
Triethylcitrat oder Diethyltartrat zusammen mit einem Netzmittel
auf Tablettenoberflächen
appliziert werden (Muhammad, N. A., Boisvert, W., Harris, M. R.,
Weiss, J., „Evaluation
of Hydroxypropyl Methylcellulose Phthalate 50 as Film Forming Polymer
from Aqueous Dispersion Systems", Drug
Dev. Ind. Pharm., (1992), 18: 1787–1797).
-
Hydroxypropyhnethylcellulosephthalat
kann alleine oder in Kombination mit anderen löslichen oder unlöslichen
Bindemitteln bei der Herstellung von Granulaten mit hinhaltenden
Medikamentenfreigabeeigenschaften verwendet werden; die Freigaberate
ist pH abhängig.
Da Hydroxypropylmethylcellulosephthalat geschmacklos und in Speichel
unlöslich
ist, kann es als eine Beschichtung zur Maskierung des unangenehmen Geschmacks
einiger Tablettenformulierungen verwendet werden.
-
BESCHREIBUNG:
-
Hydroxypropylmethylcellulosephthalat
liegt als weiße
bis leicht weißliche
freifließende
Flocken oder als ein granulöses
Pulver vor. Es ist geruchlos oder hat einen leicht sauren Geruch
und hat einen kaum nachweisbaren Geschmack.
-
TYPISCHE EIGENSCHAFTEN:
-
- Schmelzpunkt: 150°C.
- Löslichkeit:
Nahezu unlöslich
in Ethanol und Wasser; wenig löslich
in Aceton und Toluen; löslich
in wässrigen Alkalien,
einem Gemisch aus gleichen Volumina Aceton und Methanol und in einem
Gemisch aus gleichen Volumina Dichlormethan und Methanol.
-
STABILITÄT UND LAGERUNGSBEDINGUNGEN:
-
Hydroxypropylmethylcellulosephthalat
ist bei Umgebungstemperatur und -feuchte chemisch und physikalisch
3–4 Jahre
und 2–3
Monate bei 40°C
und 75% relativer Feuchte stabil (Shin-Etsu Chemical Co., Ltd., Technical
Literature: Hydroxypropyl Methylcellulose Phthalate (1993). Hydroxypropylmethylcellulosephthalat ist
bei UV-Lichtexposition bis zu 3 Monate bei 25°C und 70% relativer Feuchte
stabil (Shin-Etsu Chemical Co., Ltd., Technical Literature: Hydroxypropyl
Methylcellulose Phthalate, (1993). Im Allgemeinen ist Hydroxypropylmethylcellulosephthalat
stabiler als Celluloseacetatphthalat. Bei Umgebungslagerungsbedingungen
ist Hydroxypropylmethylcellulosephthalat für einen mikrobiellen Angriff
nicht anfällig.
-
INKOMPATIBILITÄTEN:
-
Inkompatibel
mit starken Oxidationsmitteln. Ein Reißen der Filmüberzüge wurde
selten berichtet, vor allem mit Überzugstabletten,
die mikrokristalline Cellulose und Calciumcarboxymethylcellulose
enthalten. Ein Reißen
des Films trat auch auf, wenn ein Gemisch aus Aceton: Propan-2-ol
oder Dichlormethan:Propan-2-ol als Beschichtungslösungsmittel
verwendet wurde oder wenn Beschichtungen unter niedrigen Temperatur-
und Feuchtigkeitsbedingungen appliziert wurden. Das Reißen des
Films kann jedoch durch sorgfältige
Auswahl des verwendeten Beschichtungslösungsmittels unter Verwendung
eines Polymergrades mit höherem
Molekulargewicht (Rowe, R. C., „Molecular Weight Studies
on the Hydroxypropyl Methylcellulose Phthalate (HP55), Acta. Pharm.
Technol., (1982), 28(2): 127–130)
oder durch das Zufügen
eines Weichmachers, wie zum Beispiel Acetylmonoglycerid oder Triacetin,
vermieden werden. Das Zufügen
von mehr als ca. 10% Titandioxid zu einer Beschichtungslösung aus
Hydroxypropylmethylcellulosephthalat, die zur Herstellung eines
farbigen Filmüberzugs
verwendet wird, kann in Überzügen mit
verminderter Elastizität
und Magensaftbeständigkeit
resultieren (Shin-Etsu Chemical Co., Ltd., Technical Literature:
Hydroxypropyl Methylcellulose Phthalate, (1993)).
-
HERSTELLUNGSVERFAHREN:
-
Hydroxypropylmethylcelluloseacetatphthalat
wird durch die Veresterung von Hydroxypropylmethylcellulose mit
Phthalsäureanhydrid
hergestellt. Der Grad der Methoxy- und Phthalylsubstitution bestimmt
die Eigenschaften des Polymers und insbesondere des pH, bei dem
es sich in wässrigen
Medien auflöst.
-
SICHERHEIT:
-
Hydroxypropylmethylcellulosephthalat
wurde bisher weithin verwendet, primär als ein magensaftresistentes
Beschichtungsmittel in oralen pharmazeutischen Formulierungen. Chronische
und akute Tierfütterungsstudien
an mehreren verschiedenen Spezies haben keine mit Hydroxypropylmethylcellulosephthalat
in Verbindung stehende Hinweise auf oder Teratogenität oder Toxizität gezeigt
(Kitagawa, H. Kawana, H., Satoh, T., Fukuda, Y., „Acute
and Subacute Toxicities of Hydroxypropyl Methylcellulose Phthalat", Pharmacometrics, (1970),
4(6): 1017–1025;
Kitagawa, H., Satoh, T., Yokoshima, T., Nanbo, T., „Absorption,
Distribution and Excretion of Hydroxypropyl Methylcellulose Phthalat
in the Rat", Pharmacometrics,
(1971), 5(1): 1–4;
Ito, R., Toida, S., „Studies
on the Teratogenicity of a New Enteric Coating Material, Hydroxypropyl
Methylcellulose Phthalate (HPMCP) in Rats and Mice", J. Med. Soc. Toho-Univ.,
(1972), 19(5): 453–461;
Kitagawa, H., Yano, H., Fukuda, Y., „Chronic Toxicity of Hydroxypropylmethylcellulose
Phthalate in Rats",
Pharmacometrics, (1973), 7(5); 689–701; Kitagawa, H., Yokoshima,
T., Nanbo, T., Hasegawa, M., „Absorption,
Distribution, Excretion and Metabolism of 14C-Hydroxypropyl
Methylcellulose Phthalate",
Pharmacometrics, (1974), 8(8): 1123–1132. Hydroxypropylmethylcellulosephthalat
wird im Allgemeinen als ein nicht reizendes und nicht toxisches
Material angesehen.
-
LD50 (Ratte, oral): > 15 g/kg (Kitagawa et al., Pharmacometrics,
(1970), 4(6): 1017–1025).
-
Behördlicher
Zulassungsstatus: Eingeschlossen in den Richtlinien der FDA für Arzneilich
unwirksame Bestandteile (orale Kapseln und Tabletten) und eingeschlossen
unter in Großbritannien
zugelassenen nicht parenteralen Medikamenten.
-
Arzneibücher: Br,
Eur. Fr, Gr, It, Jpn, Niederlande, Port, Schweiz und USPNF.
-
Verwandte
Substanzen: Celluloseacetatphthalat; Hydroxypropylmethylcellulose.
-
Eine
besonders bevorzugte erfindungsgemäße Zusammensetzung zur topischen
Verabreichung an einen Menschen umfasst eine mikronisierte Präparation,
enthaltend CAP oder HPMCP, ein Poloxamer und destillierte acetylierte
Monoglyceride (ein Gemisch aus mikronisiertem CAP, Poloxamer und
acetylierten Monoglyceriden wird von der FMC Corporation unter dem
Warenzeichen „AQUATERIC" angeboten) in Glycerol
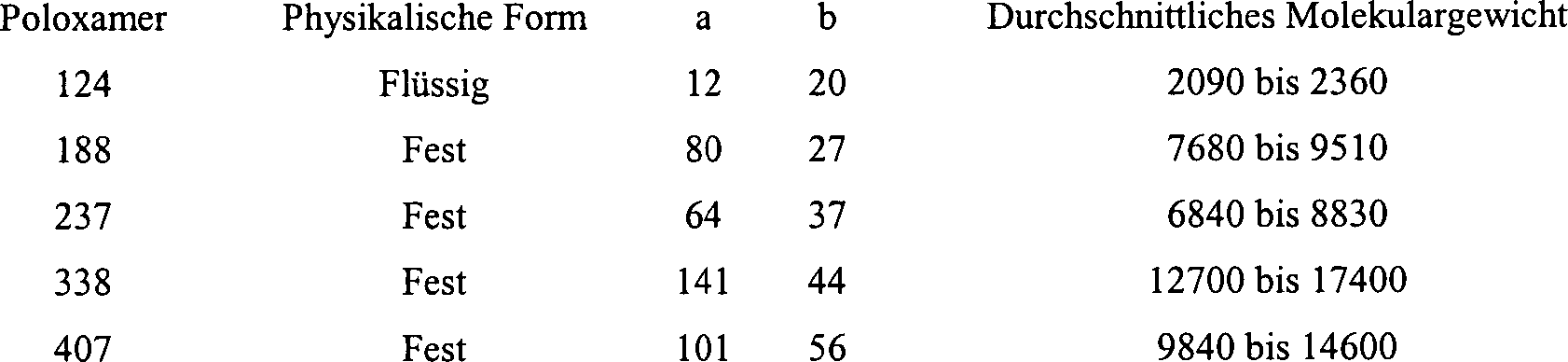
suspendiert. Ein Poloxamer stellt ein nicht ionisches Polyoxyethylen-Polyoxypropylen-Copolymer
dar. Squalan (2,6,10,15,19,23-Hexamethyltetracosan) kann anstelle
von Glycerol verwendet werden.
-
Eine
chemische Bezeichnung für
ein Poloxamer ist α-Hydro-ω-hydroxypoly(oxyethylen)-Poly(oxypropylen)-Poly(oxyethylen)-Blockcopolymer.
Die Poloxamer-Polyole stellen eine Reihe eng verwandter Blockcopolymere
aus Ethylenoxid und Propylenoxid dar, die der folgenden Formel entsprechen: HO(C2H4O)a(C3H6O)b(C2H4O)aH.
-
Im
Folgenden findet sich eine Liste von Poloxamer-Graden (USPNF XVII):
-
-
Zur
Verhinderung der Trennung der Mikrosuspension, enthaltend das CAP
oder HPMCP, das Poloxamer und die destillierten Monoglyceride, vom
Glycerol, wird das Zufügen
von Polyvinylpyrrolidon („PVP") und ein 1-Ethenyl-2-pyrrolidinon-Homopolymer
(Crospovidon) (Polyplasdone) (C6H9NO)n, Molekulargewicht > 1000000) (wasserunlösliches
synthetisches vernetztes Homopolymer von N-Vinyl-2-pyrrolidinon)
bevorzugt.
-
Der
hierin verwendete Begriff „mikronisiert" verweist auf Partikel
mit einer Partikelgröße von weniger als
35 Mikron, bevorzugt weniger als 15 Mikron, bevorzugter weniger
als 10 Mikron und am bevorzugtesten weniger als 5 Mikron.
-
In
der hierin beschriebenen Zusammensetzung, die Glycerol enthält, kann
das Glycerol mit einer Kochsalzlösung
oder Wasser ersetzt werden, so lange die Zusammensetzung bei ≤ 25°C gelagert
wird.
-
CAP
wird im Allgemeinen als ein magensaftresistentes Filmüberzugsmaterial
oder als ein Matrix-Bindemittel
für Tabletten
und Kapseln verwendet. Seine Sicherheit wurde eingehend untersucht,
und es wurde gezeigt, das es frei von unerwünschten Wirkungen ist. Vaginale
Irritationstests am Kaninchenmodell bestätigten seine Sicherheit weiter.
CAP ist eine Verbindung mit einem hohen Molekulargewicht (das Mw beträgt
ca. 60000), was darauf hindeutet, dass es sich bei topischer Applikation
nicht systemisch ausbreitet. Die Wahrscheinlichkeit, dass sich CAP über die
Applikationsstelle hinaus ausbreitet, wurde durch seine Verwendung
in mikronisierter Form weiter vermindert.
-
Die
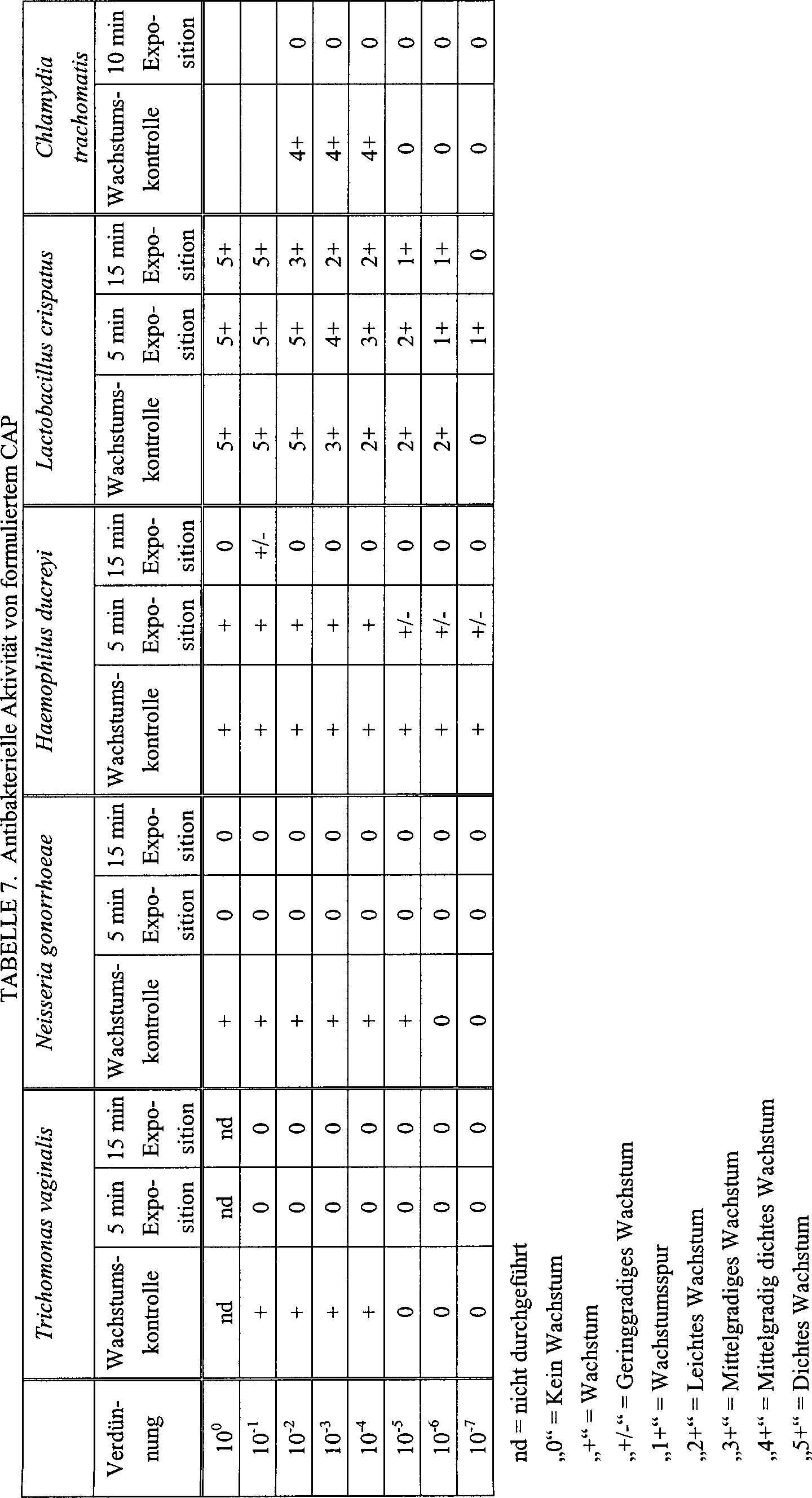
Anmelder entdeckten, dass CAP-Formulierungen gegen HIV-1, Herpesviren,
HSV-1 und HSV-2, Cytomegalievirus, Chlamydia trachomatis, Gardnerella,
Neisseria gonorrhoeae, Haemophilus ducreyi und Trichomonas vaginalis
aktiv sind.
-
Andererseits
wirkten sich die CAP-Formulierungen nicht auf die Lebensfähigkeit
von Lactobacilli aus, bei denen es sich weitgehend um essentielle
Komponenten der natürlichen
Vaginalflora handelt, die für
die Resistenz gegen einige STD-Pathogene wichtig ist (Hawes, S.
E., Hillier, S. L., Benedetti, J. et al., „Hydrogen Peroxide-Producing
Lactobacilli and Acquisition of Vaginal Infections", J. Infect. Dis.,
(1995), 172, 756–763).
-
Die
Ergebnisse ließen
erkennen, dass sich die Formulierung nicht auf die Infektivität von Papillomaviren,
einem anderen am Cervikalkarzinom beteiligten STD-Erreger auswirkt
(Franco, E. L., Villa, L. L., Ruiz, A. et al., „Transmission of Cervical
Human Papillomavirus Infection by Sexual Activity: Differences Between
Low and High Oncogenic Risk Types", J. Infect. Dis., (1995) 172, 756–763; Bosch,
F. X., Munoz, N., de Sanjose, S. et al., „Importance of Human Papillomavirus
Endemicity in the Incidence of Cervical Cancer: An Extension of the
Hypothesis on Sexual Behavior",
Cancer Epidemiol. Biomarkers & Prev.,
(1994), 3, 375–379).
-
Ohne
durch eine bestimmte Theorie der Funktionsfähigkeit gebunden sein zu wollen,
wird in Erwägung
gezogen, dass die antivirale und antibakterielle Aktivität von CAP
der hydrophoben Beschaffenheit der Phthalat-Reste am CAP-Polymer
zuschreibbar sein kann, da seine Aktivität gegen Pathogene von sexuell übertragenen
Erkrankungen wichtig zu sein scheint, weil vielen anderen getesteten
Cellulose-Derivaten sowohl die Anti-HIV-1- als auch Anti-Herpesvirus-Aktivität mangelte.
-
Eine
bevorzugte erfindungsgemäße Zusammensetzung
zur Verabreichung kann wie folgt hergestellt werden: Man löst PVP in
Glycerol auf, fügt
dann vernetztes 1-Ethenyl-2-pyrrolidinon-Homopolymer (Crospovidon)
(Crospovidon ist vernetztes Povidon) und eine Zusammensetzung, umfassend
mikronisiertes CAP und Poloxamer und acetylierte Monoglyceride zu.
Das PVP und vernetztes 1-Ethenyl-2-pyrrolidinon-Homopolymer wären in Konzentrationen
zur Stabilisierung der Suspension aus „AQUATERIC" in Glycerol ausreichend. Squalan kann
anstelle von Glycerol verwendet werden.
-
Das
erfindungsgemäße Verfahren
kann zur Prävention
der Übertragung
des Human Immunodeficiency Virus, wie zum Beispiel HIV-1 und HIV-2
und Herpesvirus bei Menschen verwendet werden. Der erfindungsgemäße Gegenstand
ist folglich wirksam bei der Prävention
der Übertragung
von HIV-1 oder HSV, wie zum Beispiel HSV-1, HSV-2, HSV-7 und HSV-8
ebenso wie vom humanen Cytomegalievirus, Varizella-Zoster-Virus, Epstein-Barr-Virus
und humanen Herpesvirus 6. Bevorzugte erfindungsgemäße Ausführungsformen
sind zur Prävention
der Übertragung
von HIV-1, HSV-1 oder HSV-2,
von denen bekannt ist, dass sie sexuell übertragen werden sowie von
HSV-8, von dem bekannt ist, dass es der kausale Erreger des Karposi-Sarkoms
ist, vorgesehen.
-
Der
erfindungsgemäße Gegenstand
betrifft auch die Prävention
der Übertragung
von oder die Behandlung einer sexuell übertragenen Bakterieninfektion,
wie zum Beispiel Syphillis, Gonorrhoe, Chlamydia, Trichomoniasis
oder eine durch Gardnerella vaginalis verursachte Infektion.
-
In
den erfindungsgemäßen Medikamenten
zur Prävention
der Übertragung
einer HIV- oder Herpesvirusinfektion in einem Menschen oder zur
Prävention
der Übertragung
oder zur Behandlung einer sexuell übertragenen Bakterieninfektion
in einem Menschen wird einem Menschen eine pharmazeutisch wirksame
antivirale Menge bzw. eine antibakterielle Menge CAP oder HPMCP
oder CAP wie auch HPMCP verabreicht. Die Zusammensetzung zur erfindungsgemäßen Verwendung
wird an eine geeignete Region des menschlichen Körpers verabreicht.
-
Die
Phrase „Verabreichung
an eine geeignete Region des menschlichen Körpers" schließt zum Beispiel die Applikation
des Wirkstoffs (CAP oder HPMCP oder beides) oder eine Zusammensetzung
ein, enthaltend das gleiche, das an Regionen des Körpers eines
Menschen, zum Beispiel an der Region des menschlichen Körpers verwendet
wurde, der mit einem anderen menschlichen Körper in engen Kontakt kommt,
zum Beispiel die Applikation (direkt oder indirekt) auf die männlichen
oder weiblichen Genitalien zur Prävention der Übertragung
von HIV-1-, HSV-1-, HSV-2- oder Bakterieninfektion während des
Geschlechtsverkehrs.
-
Der
Begriff „lokale
Verabreichung" schließt jedwedes
Verabreichungsverfahren ein, bei dem die Aktivität von CAP oder HPMCP oder von
beidem erfindungsgemäß verwendet
wird, ist weitgehend auf die Region des menschlichen Körpers, auf
die es appliziert wird, d. h. vaginale oder rektale (topische) Verabreichung,
beschränkt.
-
Der
erfindungsgemäße Gegenstand
ist folglich zur Bereitstellung eines Medikamentes zur Prävention der Übertragung
einer Virusinfektion, wie zum Beispiel einer HIV- oder Herpesvirusinfektion
oder zur Prävention
der Übertragung
oder zur Behandlung einer Bakterieninfektion, die durch sexuellen
Kontakt übertragen wird,
wie zum Beispiel vaginale Übertragung,
entweder während
des Geschlechtsverkehrs oder während
der Geburt (vaginale Entbindung), durch vaginale Verabreichung,
wie zum Beispiel durch Verabreichung einer Creme-, Salben-, Lotion-,
Gelee-, Lösungs-,
Emulsions- oder Schaumformulierung, enthaltend eine pharmazeutisch
wirksame Anti-HIV-1-Menge oder eine Anti-HSV-Menge oder antibakterielle
Menge von CAP (wie zum Beispiel mikronisiertes CAP) oder HPMCP (wie
zum Beispiel mikronisiertes HPMCP) oder beides, entweder allein
oder in Kombination mit einem pharmazeutisch verträglichen
Träger
oder Verdünnungsmittel,
besonders geeignet.
-
Zur
Prävention
der Übertragung
der HIV-1- oder Herpesvirusinfektion oder einer Bakterieninfektion, die
durch sexuellen Kontakt übertragen
wird, kann CAP oder HPMCP (in mikronisierter Form) oder beides auf eine
kontrazeptive Vorrichtung (zum Beispiel ein männliches oder weibliches Kondom,
ein kontrazeptives Diaphragma oder einen kontrazeptiven Schwamm,
zum Beispiel einen Polyurethanschaumschwamm) vor dem Geschlechtsverkehr
appliziert werden.
-
Als
Alternative kann CAP oder HPMCP oder beides auf einem Pessar oder
Tampon zur vaginalen Verabreichung appliziert werden. Die pharmazeutische
Formulierung zur topischen Verabreichung würde eine pharmazeutisch wirksame
Anti-HIV-Menge oder Anti-Herpesvirusmenge oder antibakterielle Menge
von CAP oder HPMCP oder beides und mindestens einen pharmazeutisch
verträglichen
topischen Träger
oder ein Verdünnungsmittel
zur Bildung einer Salbe, Creme, eines Gels, einer Lotion, Paste,
eines Gelees, Sprays oder Schaums umfassen.
-
Die
Menge (Dosierung) des Wirkstoffs (CAP oder HPMCP oder beides) in
einer topischen Formulierung zur erfindungsgemäßen Verwendung beträgt im Allgemeinen
weniger als 1000 Milligramm, bevorzugt zwischen 200 bis 800 Milligramm.
-
Die
Verabreichung des Wirkstoffs zusammen mit einem pharmazeutisch verträglichen
Verdünnungsmittel
oder Träger
ist als eine pharmazeutische Formulierung bevorzugt. Folglich ist
erfindungsgemäß auch die Verwendung
einer pharmazeutischen Formulierung oder Zusammensetzung vorgesehen,
umfassend den Wirkstoff zusammen mit einem oder mehr pharmazeutisch
verträglichen
Träger(n)
oder Verdünnungsmittel(n) und
optional anderen Prophylaktika. Der/die Träger oder das/die Verdünnungsmittel
sollten „verträglich" im Sinne von kompatibel
mit den anderen Bestandteilen der Formulierung und nicht schädlich für den Empfänger sein.
-
Pharmazeutische
Formulierungen schließen
die ein, die zur vaginalen, rektalen oder topischen Verabreichung
geeignet sind. Die Formulierungen können gegebenenfalls zweckmäßiger Weise
in diskreten Dosierungseinheiten dargereicht werden und können mittels
jedweder der im Stand der Pharmazie weithin bekannten Verfahren
hergestellt werden. Alle diese Verfahren schließen den Schritt ein, in dem
der Wirkstoff mit flüssigen
Trägern,
Gelen oder fein verteilten festen Trägern oder beidem und dann gegebenenfalls
Formen des Produkts zur gewünschten
Formulierung miteinander in Verbindung gebracht werden.
-
Zur
vaginalen Verabreichung geeignete Formulierungen können als
Pessare, Tampons, Cremes, Gele, Pasten, Gelees, Schäume oder
Sprays oder wässrige
oder ölige
Suspensionen, Lösungen
oder Emulsionen (flüssige
Formulierungen), enthaltend, außer
dem Wirkstoff, solche Träger,
wie sie im Stand der Technik geeignet sind, dargereicht werden.
Diese Formulierungen sind zum Schutz nicht nur gegen sexuelle Übertragung
einer HIV- oder HSV- oder einer sexuell übertragenen Bakterieninfektion,
sondern auch zur Prävention der
Infektion eines Säuglings
während
der Passage durch den Geburtskanal nützlich. Folglich kann die vaginale
Verabreichung vor dem Geschlechtsverkehr, während des Geschlechtsverkehrs
und unmittelbar vor der Geburt erfolgen.
-
Als
eine vaginale Formulierung kann der Wirkstoff zusammen mit einem
Spermizid verwendet werden und kann wie vorstehend besprochen mit
einem Kondom, einem Diaphragma, einem Schwamm oder anderen Kontrazeptiva
eingesetzt werden.
-
Pharmazeutische
Formulierungen und Präparate,
die zur Verabreichung geeignet sind, können zweckmäßigerweise als eine Lösung, eine
wässrige
oder ölige
Suspension oder eine Emulsion dargereicht werden. Der Wirkstoff
kann auch als ein Bolus, Electuarium oder eine Paste dargereicht
werden.
-
Flüssige Präparate zur
vaginalen Verabreichung können übliche Zusatzstoffe,
wie zum Beispiel Suspendiermittel, Emulgatoren, nicht-wässrige Vehikel
(die genießbare Öle einschließen können) oder
Konservierungsmittel, enthalten.
-
Pharmazeutische
Formulierungen, die zur rektalen Verabreichung geeignet sind, worin
der Träger
ein Feststoff ist, werden am bevorzugtesten als Suppositorien in
einer Einheitsdosis dargereicht. Geeignete Träger schließen Kakaobutter und andere
im Stand der Technik üblicherweise
verwendete Materialien ein, und die Suppositorien können zweckmäßigerweise
durch Beimischen des Wirkstoffs mit dem/den weich gemachten oder
geschmolzenen Träger(n),
gefolgt von Abkühlen
und Formen in Formwerkzeugen gebildet werden.
-
Tropfen
können
mit einer wässrigen
oder nicht wässrigen
Grundlage, umfassend ein oder mehr Dispergiermittel, Solubilisierungsmittel
und Suspendiermittel, formuliert werden. Flüssige Sprays werden zweckmäßiger Weise
aus unter Druck stehenden Packungen abgegeben.
-
Gegebenenfalls
können
die vorstehend beschriebenen Formulierungen, die zur hinhaltenden
Abgabe des Wirkstoffs angepasst sind, eingesetzt werden.
-
Die
pharmazeutischen Zusammensetzungen zur erfindungsgemäßen Verwendung
können
auch andere Wirkstoffe, wie zum Beispiel Spermizide oder antimikrobielle
Mittel, Konservierungsmittel oder andere antivirale Mittel enthalten.
-
Das
erfindungsgemäße mikronisierte
CAP und seine Suspension in Glycerol oder Squalan ergibt eine aktive
und stabile Formulierung, die antivirale Aktivität besitzt und zur topischen
Applikation zur Prävention
der sexuellen Übertragung
von HIV-1, Herpesviren und sexuell übertragenen Bakterieninfektionen
geeignet ist.
-
Der
erfindungsgemäße Gegenstand
betrifft auch pharmazeutische Zusammensetzungen, enthaltend 1 bis
25 Gew.-%, bevorzugt 5 bis 20 Gew.-% und bevorzugter 12 bis 18 Gew.-%
mikronisiertes Celluloseacetatphthalat und Glycerol und entweder
Polyvinylpyrrolidon oder Crospovidon (1-Ethenyl-2-pyrrolidinon-Homopolymer)
oder kolloidales Siliciumdioxid zur Bildung einer leicht applizierbaren
homogenen Creme.
-
Außer den
beiden vorstehend offenbarten Formulierungen kann eine halbfeste
Formulierung, enthaltend alle festen Bestandteile (d. h. "AQUATERIC", Povidon, Crospovidon
und kolloidales Siliciumdioxid) mit Glycerol und Squalan vermischt
werden, worin die Mengen aller Komponenten ausreichend sind, um
in einem halbfesten Teig oder einer kittartigen Masse zu resultieren,
der/die leicht geformt, in die gewünschten Aliquote aufgeteilt
und zum Schutz vor Umweltfaktoren (Feuchtigkeit usw.) verpackt werden
kann.
-
„AQUATERIC" kann überdies
durch eine andere Form des mikronisierten CAP ersetzt werden. Dies könnte zum
Beispiel durch Auflösen
von 100 mg CAP und 100 mg Polyvinylpyrrolidon (Povidon, PVP) pro
1 ml Dimethylsulfoxid geschehen. Nach Auflösung der festen Komponenten
wird Wasser unter effizientem, kräftigem Mischen langsam zugefügt. Dies
resultiert in der Bildung eines feinen Präzipitats von PVP enthaltendem CAP.
Das Präzipitat
wird anschließend
mit Wasser gewaschen und schließlich
gefriergetrocknet. Das feine, gefriergetrocknete Pulver kann anstelle
von "AQUATERIC" verwendet werden.
-
Das
im vorangehenden Abschnitt offenbarte Verfahren ist viel einfacher
als ein ähnliches
Verfahren, das sich Polyvinylalkohol anstelle von PVP und Aceton
anstelle von Dimethylsulfoxid zunutze macht und auch das Vorliegen
eines Mineralsalzes, wie zum Beispiel Magnesiumchlorid (USP 4.968.350
an Bindschaedler et al., "Process
for Preparing a Powder of Water-insoluble Polymer which can be Redispersed
in a Liquid Phase, the Resulting Powder and Utilization Thereof"). Die Gegenwart
von Mg++ ist auch unerwünscht, da es die Stabilität von CAP
vermindert.
-
Die
vorstehend erwähnte
Formulierung von "AQUATERIC", PVP und Crospovidon
in Glycerol ist zur topischen Applikation geeignet. Um die Formulierung
jedoch in prädeterminierten
Mengen, zusätzlich
zur Formulierung, zu applizieren (zu verabreichen), sollte eine
Messvorrichtung, z. B. ein Applikator, bereitgestellt werden.
-
Die
Inkorporation der Formulierungen, enthaltend CAP und/oder HPMCP
in Hydroxypropylmethylcellulose-Kapseln, wie zum Beispiel "VEGI CAPS" oder "VEGGIE-CAPS", hergestellt von
GS Technologies, Springville, Utah, die als vaginale Suppositorien
konfiguriert sein können,
ist vorteilhaft. Damit würden
die Kosten reduziert und mögliche
Entsorgungsprobleme vermieden. Diese Suppositorien können in
die Vagina intakt eingeführt
werden, wobei sich die Hülle
der Kapsel erweichen und nach Interaktion mit der Feuchtigkeit in
der Vagina bersten wird, wobei sie folglich die CAP- und/oder HPMCP-Formulierung
freigibt.
-
Die
vorstehend beschriebenen Hydroxypropylmethylcellulose-Kapseln können zum
Beispiel gefüllt werden,
entweder mit:
- (a) "AQUATERIC" suspendiert in Glycerol; oder
- (b) "AQUATERIC" in fester Form,
mit oder ohne zusätzliche(n)
Wirkstoffe(n), kann auch in Gelatinekapseln inkorporiert werden.
-
Die
Formulierung, enthaltend den erfindungsgemäßen Wirkstoff (CAP und/oder
HPMCP) kann in der Form einer einzelnen Kapsel vorliegen, oder die
Formulierung kann in der Form von zwei oder mehr Kapseln vorliegen,
wobei jede die gleichen oder unterschiedliche Bestandteile enthält.
-
Die
Anmelder entdeckten, dass zwei unter vielen pharmazeutischen Hilfsstoffen
eine potente Anti-HIV-1-Aktivitäts-
und Antibakterienwirkung aufweisen. Dies ist von großer Wichtigkeit,
da Hilfsstoffe keine kostspieligen Verbindungen sind. Es wird erwartet,
dass die Dosis von CAP oder HPMCP pro topischer Einzelapplikation
(ca. 300 mg) ca. 1,33 US Cents kostet. Die Applikation von CAP und/oder
HPMCP zur Verminderung der Häufigkeit
der sexuellen Übertragung
von HIV-1- und Bakterieninfektionen ist weltweit wirtschaftlich möglich, und
es wird erwartet, dass sie zur Kontrolle der weltweiten HIV-1-Epidemie
beiträgt.
-
Da
Viren mit Ausnahme von HIV-1 auch sexuell übertragen werden können, war
die Bestimmung von Interesse, ob CAP und/oder HPMCP auch die Infektion
durch solche Viren inhibieren können.
Für diese
Experimente wurden Herpesvirus Typ 1 (HSV-1) und Typ 2 (HSV-2) ausgewählt. Die
in der Figur zusammengefassten Ergebnisse deuten darauf hin, dass
CAP die Infektion von sowohl HSV-1 als auch HSV-2 inhibierte. Ähnliche
Ergebnisse wurden mit HPMCP erhalten (siehe 2).
-
Einige
der hierin dargestellten Ergebnisse wurden auf der 12. Welt-AIDS-Konferenz,
Neurath, A. R., Strick, N., Lin, K. et al., "Microbicide B195", Proceedinges of the 12th World AIDS
Conference, Genf, Schweiz, 28. Juni – 3. Juli, 1998, S. 239–242, kurz
unter dem Code "Mikrobizid
B195" zusammengefasst.
-
BEISPIELE
-
BEISPIEL 1: SCREENING
PHARMAZEUTISCHER HILFSSTOFFE AUF ANTI-HIV-AKTIVITÄT
-
Alle
Verbindungen wurden durch Messen der Inhibition der Fusion zwischen
HIV-1-infizierten und nicht infizierten Zellen zuerst auf Anti-HIV-1-Aktivität gescreent
(Jiang, S., Lin, K., Strick, N., Neurath, A. R., „Inhibition
of HIV-1 Infection by a Fusion Domain Bindung Peptide from the HIV-1
Envelope Glycoprotein gp41",
Biochem. Biophys. Res. Commun. (1993), 195, 533–538).
-
Die
Auswahl pharmazeutischer Hilfsstoffe zum Screening auf Anti-HIV-Aktivität erfolgte
aus einer Liste pharmazeutischer Hilfsstoffe, die sich aus dem Handbook
of Pharmaceutical Excipients, Hrsg. Ainley Wade und Paul J. Weller,
2. Auflage, American Pharmaceutical Association, Washington, DC,
und der Pharmaceutical Press, London (1994) herleiteten. Die ausgewählten Verbindungen
sind in der folgenden Tabelle 1A aufgelistet. Andere im Handbook
of Pharmaceutical Excipients aufgelisteten Hilfsstoffe wurden nicht
auf Anti-HIV-1-Aktivität
getestet, seit aus früheren
Studien bekannt wurde, dass sie keine derartige Aktivität aufweisen
(siehe die folgende Tabelle 2). In Wasser unlösliche Verbindungen oder Puffer
(siehe die folgende Tabelle 3), organische Verbindungen, einschließlich Ölen, Wachsen,
Lösungsmitteln
und Detergenzien, von denen bekannt ist, dass sie die Zellmembranen
und Hüllen
Lipid-enthaltender
Viren solubilisieren (siehe die folgende Tabelle 4), für Aerosol-Treibmittel
verwendete Gase (siehe die folgende Tabelle 5) und Oxidationsmittel
und Desinfektionsmittel mit antibakterieller Aktivität (siehe
die folgende Tabelle 6) wurden aus dem Screening-Verfahren ausgeschlossen.
-
Von
allen in Tabelle 1A aufgelisteten Verbindungen inhibieren überraschenderweise
nur zwei Verbindungen die Fusion zwischen HIV-1-infizierten und
nicht infizierten Zellen, was einem Verfahren zur raschen Beurteilung
der Anti-HIV-1-Aktivität
von Verbindungen entspricht. In diesem Assay wurden mit HIV-1-IIIB
infizierte H9-Zellen mit 2',7'-Bis(2-carboxyethyl)-5-(und
-6)-carboxyfluoresceinacetoxymethylester (BCECF; Molecular Probes,
Inc., Eugene, Oregon) nach den Anweisungen des Herstellers markiert.
BCECF-markierte H9/HIV-1-IIIB-Zellen (104)
wurden mit 2 × 105 nicht infizierten MT-2-Zellen gemischt.
Nach der Inkubation in einer 96-Well-Platte 1 Stunde bei 37°C wurden
die fusionierten und nicht fusionierten markierten Zellen unter einem
invertierten Fluoreszenz-Mikroskop bei einer Vergrößerung von
160 × gezählt. Es
wurden mindestens 200 BCECF-markierte Zellen gezählt, und der Anteil fusionierter
Zellen wurde bestimmt. Diese Tests wurden in An- und Abwesenheit
von zu screenenden abgestuften Verbindungsmengen durchgeführt. Alle
Experimente mit HIV-1 wurden unter den Biorisiko-Containment-Konzentrationen
nach P2 durchgeführt.
-
Die
Anti-HIV-1-Aktivität
der beiden Verbindungen, nämlich
des in Tabelle 1A aufgelisteten Celluloseacetatphthalats und Hydroxypropylmethylcellulosephthalats,
wurde anhand der folgenden zusätzlichen
Tests bestätigt
und quantifiziert: (a) Inhibition des cytopathischen Effekts (CPE)
von HIV-1 und (b) Inhibition der Produktion des HIV-1-Nukleocapsid-Antigens
(p24) (Neurath, A. R., Strick, N., Haberfield, P., Jiang, S., „Rapid Prescreening
for Antiviral Agents Against HIV-1 Based on their Inhibitory Activity
in Site-Directed Immuunoassays, II. Porphyrins Reacting with the
V3 loop of gp120",
Antivir. Chem. Chemother., (1992), 3, 55–63) (Tabelle 1B). Die beiden
Verbindungen waren für
nicht infizierte Zellen bei Konzentrationen von ≤ 2500 μg/ml nicht toxisch.
-
MT-2-Zellen
(104) in 96-Well-Platten wurden mit HIV-1
(in einer Dosis, die zum Erreichen einer Infektionsmultiplizität von 0,0045
ausreicht) in 200 μl
RPMI 1640-Medium, supplementiert mit 10 Vol-% fetalem Rinderserum
(„FBS") infiziert. Nach
1 Stunde und 24 Stunden wurde die Hälfte des Kulturmediums ausgewechselt und
durch frisches Medium ersetzt. Am vierten Tag nach der Inkubation
bei 37°C
wurden 100 μl
der Kulturüberstände aus
jedem Well entnommen, und den Wells wurde ein entsprechendes Volumen
frischen Mediums zugefügt.
Die gesammelten Überstände wurden
mit einem gleichen Volumen von 5 Vol-% TRITON X-100 gemischt und
unter Verwendung eines ELISA-Kits (ELISA = Enzyme-linked Immunoassay)
von Coulter Immunology (Hialeah, FL) auf p24-Antigen untersucht.
Am sechsten Tag nach der Infektion wurde den Zellen ein Indikator
XTT-Tetrazolium-Farbstoff (1 mg/ml; 50 μl/Well; PolySciences, Inc.,
Warrington, PA) zugefügt.
Nach vier Stunden wurde intrazelluläres Formazan kolorimetrisch
bei 450 nm nach dem beschriebenen Verfahren bestimmt (Weislow, O.
S., Kiser, R., Fine, D. L. et al., „New Soluble-Formazan Assay
for HIV-1 Cytopathic Effects: Application to High-Flux Screening
of Synthetic and Natural Products for AIDS-Antiviral Activity", J. Natl. Cancer
Inst., 81, 577–586,
(1989)). Der prozentuale Anteil der Cytopathogenese wurde unter
Verwendung der folgenden Formel berechnet: 100 × [(OD450 in
der negativen Kontrolle – OD450 im Experiment)/(OD450 in
der negativen Kontrolle – OD450 in der positiven Kontrolle)]. Die negative
Kontrolle entsprach den mit Kulturmedium anstelle von HIV-1 vermischten
Zellen, während
die positive Kontrolle mit 100 CCID50 gemischte
Zellen (in der Zellkultur infektiöse Dosen) von HIV-1-IIIB, die
die MT-2-Zellen 100%ig lysierten, darstellte. Die cytopathische Wirkung
(„CPE") der Verbindungen
auf nicht infizierte Zellen wurde unter Verwendung der gleichen
Methodik gemessen.
-
Pharmazeutische
Hilfsmittel, außer
Verbindungen, die in Wasser unlöslich
sind, Detergenzien und Oxidationsmittel wurden anhand eines Assays,
wobei die Fusion zwischen infizierten und nicht infizierten Zellen gemessen
wird, auf Anti-HIV-1-Aktivität
gescreent. Nur zwei Verbindungen, Celluloseacetatphthalat (CAP)
und Hydroxypropylmethylcellulosephthalat (HPMCP) wiesen eine Inhibitionsaktivität auf. Ein
anderes polymeres Phthalat, Vinylacetatphthalat, wies keine Inhibitionsaktivität auf. Anderen
Cellulose-Derivaten, wie zum Beispiel Carboxymethylcellulose, mangelte
es auch an Aktivität.
Die beiden ausgewählten
Verbindungen, CAP und HPMCP, inhibierten auch die HIV-1-Infektion,
wie anhand der Inhibition des CPE und der Produktion des HIV-1-Nukleocapsid-Antigens
p24 gemessen (Tabelle 1B). Die beiden Verbindungen waren für nicht-infizierte Zellen
bei Konzentrationen von ≥ 2500 μg/ml nicht
toxisch. Die Anti-HIV-1-Aktivität
von CAP war besser als die von HPMCP, beide Verbindungen stellten
jedoch wünschenswerte
Ergebnisse bereit.
-
Da
Herpesviren auch häufig
sexuell übertragen
werden, war die Bestimmung von Interesse, ob CAP auch die von diesen
Viren verursachte Infektion zu inhibieren vermochte. Herpesvirus
Typ 1 (HSV-1) und Typ 2 (HSV-2) wurden ausgewählt, um diese Möglichkeit
zu testen. Die in 1 zusammengefassten Ergebnisse deuten
darauf hin, dass CAP die Infektion aufgrund von HSV-1 wie auch HSV-2
inhibierte. CAP inhibierte auf ähnliche
Weise die durch HCMV verursachte Infektion. In Bezug auf 1 wurden
Viruspräparationen
unter Verwendung eines auf der Quantifizierung von β-Galactosidase
basierenden Ablesesystems (Absorption bei 410 nm) auf Infektivität getestet.
-
TABELLE
1A. Auf Anti-HIV-1-Aktivität
getestete pharmazeutische Hilfsstoffe
-
TABELLE
1B. Auf Anti-HIV-1-Aktivität
getestete pharmazeutische Hilfsstoffe
-
TABELLE
2. Verbindungen von denen bekannt ist, dass sie keine Anti-HIV-1-Aktivität aufweisen
-
TABELLE
3. In Wasser oder Puffern unlösliche
Verbindungen
-
TABELLE
4. Zellmembranen und Hüllen
von Lipid-enthaltenden Viren solubilisierende organische Verbindungen, Öle, Wachse
und Lösungsmittel
und Detergenzien
-
-
TABELLE
5. Zum Beispiel in Aerosol-Treibgasen verwendete Gase
-
TABELLE
6. Oxidationsmittel und Desinfektionsmittel
-
BEISPIEL 2: CAP-FORMULIERUNGEN
-
Die
CAP- und HPMCP-Formulierung zur topischen vaginalen Applikation
als ein antivirales Mittel oder Viruzid zur Prävention der sexuellen Übertragung
von HIV-1 bzw. Herpesviren stellt ein schwieriges Challenge dar,
dem nur durch innovative Ansätze
begegnet werden könnte.
Sowohl CAP als auch HPMCP sind in Wasser unlöslich und können in Wasser durch Einstellen
des pH des Milieus auf ca. 6 oder darüber (Handbook of Pharmaceutical
Excipients, 2. Auflage, Hrsg. Ainley Wade und Paul J. Weller, American
Pharmaceutical Association, Washington, (1994)) oder durch die Verwendung
von geeigneten organischen Lösungsmitteln
löslich
gemacht werden. Andererseits sind vaginale Sekrete von gesunden
Frauen im gebärfähigen Alter
charakteristisch sauer (pH-Werte von 3,4 bis 6,0) (B. Voeller, D.
J. Anderson, „Heterosexual
Transmission of HIV",
JAMA, 267, 1917–1918,
(1992)). Infolge dessen würde
erwartet, dass die topische Applikation einer Formulierung, in der
entweder CAP oder HPMCP löslich
wäre (d.
h. pH = ≥ 6)
zu einem vaginalen Milieu beiträgt,
das physiologisch unerwünscht
ist. Es wurden dennoch Versuche zur Formulierung von CAP oder HPMCP
in Gelen/Cremes unternommen, die üblicherweise für vaginale
Applikationen als Feuchthaltemittel und/oder Kontrazeptiva verwendet
werden. Diese schlossen die folgenden ein: Hydroxyethylcellulosegele
(z. B. K-Y JELLY, Johnson and Johnson, Raritan, N.J.); auf Carbomer
934P basierende Gele (z. B. REPLENS, Roberts Pharmaceuticals, Inc.,
Mississauga, Ontario, Kanada; Taro-Gel, Taro Pharmaceuticals, Inc.
Bramalea, Ontario, Kanada); Hydroxypropylmethylcellulose und auf
Carbomer 934P basierende Gele (z. B. H-R Lubricating Jelly, Carter-Wallace,
Inc., New York, N.Y.); Polyglycerylmethacrylat (Gyne-Mostrin Moisturizing
Gel (Shering-Plough Healthcare Products, Inc., Mississauga, Ontario,
Kanada)), und Gele, die Carbomer 934P und Hydroxypropylmethylcellulose
allein enthalten. Alle die vorstehend erwähnten Formulierungen weisen
Wasser als ihren Hauptbestandteil auf. Wenn die Präparationen
von CAP und HPMCP in den vorstehenden Gelen 7 Tage bei 45°C beschleunigten
Stabilitätsstudien
unterworfen und anschließend
daran auf Anti-HIV-1-Aktivität
getestet wurden, wurde keine antivirale Aktivität nachgewiesen. Dies war wahrscheinlich
auf die Hydrolyse von jedem dieser Cellulosederivate zurückzuführen, die
in der Freisetzung von Essig- und Phthalsäuren resultierten und zu verminderter
Anti-HIV-1-Aktivität
führten.
-
Zur
Vermeidung dieses Problems wurde die Entscheidung getroffen, die
Cellulose-Derivate in organischen Lösungsmitteln aufzulösen (die
Experimente wurden hauptsächlich
mit CAP durchgeführt,
das im Vergleich zu HPMCP eine höhere
Anti-HIV-1-Aktivität
aufweist; siehe Tabelle 1B hierin), die einen geringen Wassergehalt
aufweisen, aber zur Kompatibilität
in vivo dennoch mit Wasser mischbar und für die vaginale Schleimhaut
nicht toxisch sind, die auf der Basis vorläufiger Studien ausgewählt wurden.
Diese Lösungsmittel schlossen
die folgenden ein: Propylenglycol, Propylencarbonat, Benzylalkohol,
Polyethylenglycol (PEG 400), Dimethylisosorbid und Ethoxydiglycol
(„TRANSCUTOL"). Die Löslichkeit
von CAP in diesen Lösungsmitteln liegt
im Bereich zwischen 5,3 und 30 Gew.-%. Zur Erhöhung der Viskosität dieser
Lösungen
war es notwendig, sie als Gele/Cremes für topische Applikatianen zu
verwenden. Den CAP-Lösungen
in den verschiedenen organischen Lösungsmitteln wurden entweder
Polyvinylpyrrolidon (PVP) und/oder andere Poloxamere (z. B. Pluronic
F68) zugefügt.
Zur Bestimmung der Eigenschaften der verschiedenen Formulierungen
nach Kontakt mit einer physiologischen Umgebung wurden sie mit Wasser
oder physiologischer Kochsalzlösung
(0,14 M NaCl) gemischt. Unter diesen Bedingungen präzipitierte
CAP an der Grenzfläche
der Formulierungen mit Wasser (Kochsalzlösung) in der Form einer großen polymeren
Masse, von der nicht erwartet würde,
dass sie antivirale Aktivität
aufweist und wäre
nicht zur topischen Applikation geeignet. Es war möglich, diesem
Problem durch Inkorporation von Verbindungen in die CAP-enthaltende
Formulierung, welche den pH nach Kontakt mit Wasser oder einer Kochsalzlösung (=
0,14 M NaCl), wie z. B. Natriumacetat oder Triethanolamin steigert,
zu begegnen. Der Einschluss der letzteren Verbindungen in die Formulierung
eliminierte oder verminderte das Problem des Auftretens großer CAP-Aggregate.
Die beschleunigten Stabilitätsstudien
(7-tägige
Inkubation bei 45°C)
mit CAP in den vorstehenden organischen pharmazeutischen Hilfsstoffen/Lösungsmitteln,
die zusätzlich die
vorstehend erwähnten
Geliermittel und Puffer enthielten, resultierten überraschend
im vollkommenen Verlust der Anti-HIV-1-Aktivität. Diese Aktivität wurde
beibehalten, wenn die Puffer ausgelassen wurden und erst kurz vor
der Initiierung des Assays auf die Anti-HIV-1-Aktivität zugefügt wurden.
Folglich ist zusammenfassend ersichtlich, dass die CAP-Formulierungen
in organischen Lösungsmitteln,
die auch einen Puffer enthalten, Formulierungen darstellen, die
für topische
Applikationen ungeeignet sind, entweder aufgrund dessen, dass der Wirkstoff,
CAP, nach Kontakt mit physiologischen Flüssigkeiten (bei Abwesenheit
in die Formulierung inkorporierter geeigneter Puffer) aus der Formulierung
in einer großen
polymeren Masse ausfallen oder in nutzlose Formulierungen umgewandelt
werden, denen aufgrund der Inaktivierung des Wirkstoffs, CAP, die
Anti-HIV-1-Aktivität
mangelt.
-
Zum
Vermeiden der vorstehenden Probleme wurde die Möglichkeit der Verwendung von
CAP in der Form einer mikronisierten Präparation in Suspension untersucht.
Dies machte die Verwendung eines Lösungsmittels erforderlich,
in dem CAP nicht löslich
ist, da anderweitig erwartet würde,
dass die erhaltenen Ergebnisse genau mit den vorstehend erwähnten gleich
wären.
Ein Lösungsmittel
mit derartigen Eigenschaften ist Wasser, in dem weder CAP noch HPMCP
löslich
sind (Handbook of Pharmaceutical Excipients, 2. Auflage, Hrsg. Ainley
Wade und Paul J. Weller, American Pharmaceutical Association, Washington
(1994)).
-
Es
wurde eine Formulierung, die Wasser und eine im Handel erhältliche
mikronisierte Form von CAP („AQUATERIC" von der FMC Corporation,
Philadelphia, PA), enthielt, die außer CAP (63 bis 70 Gew.-%)
Poloxamer und destillierte acetylierte Monoglyceride enthielt, hergestellt.
Verdickungsmittel, d. h. PVP und/oder Pluronic F68, wurden der Wasser-Suspension
aus dem „AQUATERIC" zugefügt. Wenn
dieses Gel den beschleunigten Stabilitätsstudien (7 Tage bei 45°C) unterworfen
und daran anschließend
auf Anti-HIV-1-Aktivität getestet
wurde, wurde im Wesentlichen keine antivirale Aktivität wiedergefunden.
Folglich wurde ein anderes Lösungsmittel
benötigt,
in dem das CAP („AQUATERIC") nicht löslich wäre und die
antivirale Aktivität
nicht verloren ginge. Glycerol (sehr ähnlich dem Propylenglycol,
in dem CAP bis zu ca. 30 Gew.-% löslich ist) entspricht diesen
beiden Anforderungen. Basierend auf dieser Entdeckung wurde eine
CAP-Formulierung („AQUATERIC"-Formulierung) wie
folgt hergestellt: 200 mg PVP (MG 40000, Spectrum) wurden in 1 ml
Glycerol aufgelöst.
Anschließend
daran wurden 50 mg Crospovidon (Polyplasdone INF-10, ISP Technologies)
in der Lösung suspendiert,
gefolgt vom Zufügen
von 286 mg „AQUATERIC". PVP und Crospovidon
wurden zur Verhinderung der Trennung der „AQUATERIC"-Mikrosuspension
aus Glycerol zugefügt.
Die resultierende Formulierung behielt im Verlauf der Zeit ihre
Gleichförmigkeit
und nach einem beschleunigten Stabilitätstest, der unter den vorstehend
beschriebenen Bedingungen durchgeführt wurde, auch ihre Anti-HIV-1-Aktivität bei.
-
Zusammenfassend
wird darauf hingewiesen, dass die folgenden Eigenschaften von CAP
seine Formulierung vorschrieben: (a) geringe Löslichkeit in wässrigen
Lösungen
bei pH < 6, (b)
Hydrolyse während
der Lagerung in wässrigen
Lösungen
bei Raumtemperatur und (c) Löslichkeit
in mehreren biokompatiblen organischen Lösungsmitteln, zum Beispiel
Propylencarbonat, Propylenglycol und Polyethylenglycol, aus denen
CAP nach Kontakt mit wässrigen
Lösungsmitteln
ausfällt.
Zum Vermeiden dieser Probleme wurde eine Formulierung von mikronisiertem
CAP in Glycerol (worin CAP nicht löslich ist), „CAP-Formulierung
I", hergestellt.
-
Die „CAP-Formulierung
I" ist eine Präparation
aus mikronisiertem CAP („AQUATERIC", enthaltend 66–73% CAP,
ein Polyoxyethylen-Polyoxypropylen-Blockcopolymer und destillierte
acetylierte Monoglyceride, wurde in wässrigen Medien als eine magensaftresistente
Filmüberzugsflüssigkeit
(FMC Corporation, Philadelphia, PA) verwendet); (15,9 g) wurden
mit Glycerol (70,2 g) vermischt, und Polyvinylpyrrolidon K-30 (Spectrum Quality
Products, Inc., New Brunswick, NJ) (11,1 g) und Crospovidon NF (ISP
Technologies, Inc., Wayne, NJ) (2,8 g) wurden zugefügt, um das
mikronisierte CAP in Suspension zu halten.
-
Eine
andere Formulierung, „CAP-Formulierung
II" wurde durch
Ersatz von Polyvinylpyrrolidon + Crospovidon mit kolloidalem Silciumdioxid
M-5P (Cabot Corp., Cab-O-Sil Division, Tuscola, IL), einem Hilfsstoff
mit einer erwiesenen Verwendung in vaginalen Präparaten, hergestellt. Die CAP-Formulierung
II, enthielt 23,7 g „AQUATERIC" und 7,89 g Siliciumdioxid
pro 100 g Glycerol.
-
Alle
Komponenten dieser beiden CAP-Formulierungen waren von USP-Gütegrad und
wurden zur humanmedizinischen Anwendung zugelassen.
-
BEISPIEL 3: MESSUNG DER
INHIBITORISCHEN FÄHIGKEIT
GEGEN HSV-1 UND HSV-2 UND GEGEN HCMV
-
Das
folgende Verfahren wurde zum Messen der inhibitorischen Aktivität verwendet:
500 μl Verbindungen
(bei distinkten Dosierungen) in Eagle's Minimum Essential Medium (EMEM) wurden
mit einem gleichen Volumen von angemessen verdünntem infektiösem HSV-1
oder HSV-2 gemischt. Das Gemisch wurde den ELVIS-HSV-Zellen in 24-Well-Platten
zugefügt.
Die ELVIS-Zellen wie auch die Medien wurden von Diagnostic Hybrids,
Inc, Athens, OH) bezogen.
-
ELVIS-Zellen
leiten sich von der Auswahl von G400-resistenten Kolonien nach der
Cotransfektion von Baby-Hamster-Nierenzellen mit einem Plasmid her,
das einen G418-Antibiotika-resistenten
Marker enthält
und ein Plasmid, das ein LacZ-Gen von Escherichia coli enthält, das
hinter einen induzierbaren HSV-Promotor platziert ist. Der Promotor
stammt von HSV-1UL39, das ICP6, die große Subeinheit der Ribonukleotid-Reduktase (RR1),
kodiert. Dieser Promotor weist eine Anzahl von Merkmalen auf, die
ihn zum Nachweis von HSV ideal geeignet machen. Erstens, es findet
keine konstitutive Expression aus diesem Promotor in nicht infizierten
Zellen statt. Zweitens, die Aktivierung des Promotors scheint für HSV spezifisch
zu sein. Drittens, die Expression aus diesem Promotor tritt innerhalb
von Stunden nach der Infektion auf. Viertens, dieser Promotor wird
durch das Virion-assoziierte Transaktivator-Protein VP16 stark transaktiviert.
Bereits so früh
wie sechs Stunden nach der Infektion können HSV-infizierte Zellen
mittels histochemischer Färbung
auf β-Galactosidase-Aktivität nachgewiesen
werden (Stabell E. C. und Olivo P. D., „Isolation of a Cell Line
for Rapid and Sensitive Histochemical Assay for the Detection of
Herpes Simplex Virus",
J. Virological Methods, 38, 195–204,
(1992)).
-
Vierundzwanzig
Stunden nach der HSV-Infektion wurden die ELVIS-Zellen bei An- und
Abwesenheit von abgestuften Quantitäten der Testverbindungen mit
TRITON X-100 lysiert, und die β-Galactosidase
in den Zelllysaten wurde anhand eines ELISA-Kits, das von Five Prime → Three Prime,
Inc. (Boulder, CO) bezogen wurde, bestimmt. Dieses ELISA-Kit ist
zum Nachweis und zur Quantifizierung von Pikogramm-Mengen von β-Galactosidase-Protein
von Escherichia coli, das in transformierten Bakterien oder eukaryotischen
Zellen und Geweben exprimiert wird, in der Lage. Das Verfahren basiert
mehr auf dem Nachweis des β-Galactosidase-Proteins
als auf der enzymatischen Aktivität. Die β-Galactosidase aus E. coli stellt
ein tetrameres Enzym dar, das sich aus vier identischen Subeinheiten
zusammensetzt. Die individuellen Subeinheiten weisen keine Enzymaktivität auf und
sind deshalb mithilfe von Standard-Enzymaktivitätsassays nicht nachweisbar.
Das Five Prime → Three
Prime ELISA-Kit auf β-Galactosidase
begegnet dieser Limitation durch den Nachweis des eigentlichen Proteins,
das exprimiert wird.
-
Die überraschende
Schlussfolgerung der beschriebenen Experimente bestand darin, dass
Cellulaseacetatphthalat und Hydroxypropylmethylcellulosephthalat
unter allen den getesteten Hilfsstoffen einzigartig waren, insoweit,
dass sie eine potente antivirale Aktivität sowohl gegen HIV-1 als auch
HSV-1 und HSV-2 und gegen andere Viren aufweisen, die zur Herpesvirus-Gruppe
gehören.
-
Als
Alternative wurde die antivirale Aktivität gegen HSV-1 unter Verwendung
von HSV vgCL5, einem rekombinanten Virus gemessen, in dem sich die
Expression von β-Galactosidase
(„β-gal") unter der Kontrolle der
Regulationsregion des späten
Gens gC von HSV-1 befindet (Weir, J. P., Steffy, K. R., Sethna, M., „An Insertion
Vector for the Analysis of Gene Expression During Herpes Simplex
Virus Infection",
Gene 91990), 89, 271–274).
-
In
diesem Test wurden 50 μl
MEM-Gewebekulturmedium, enthaltend 5% FBS und abgestufte Konzentrationen
von CAP mit 100 μl
Vero-Zellen im gleichen FBS-enthaltenden Medium (106 Zellen/ml)
30 Minuten bei 25°C
gemischt. Daran anschließend
wurden 50 μl
HSV vgCL5 bei einer Verdünnung,
die ausreicht, um ca. 50% der Zellen zu infizieren, wie anhand der
Färbung
in situ auf β-Galactosidase
bestimmt, zugefügt,
und das Gemisch wurde in Wells von 96-Well-Platten gegeben. Nach
der 24-stündigen
Inkubation bei 37°C
wurden die Zellen entweder sofort lysiert oder 1 bis 5 Tage zur
Lagerung eingefroren und im Anschluss daran durch Zufügen zu den
Zellen und Medium, 50 μl
von 2,5 Vol-% TRITON X-100, enthaltend Protease-Inhibitoren (PMSF, Leupeptin
und Pepstatin, alle bei 10 μg/ml)
lysiert. Die β-Galactosidase
in den lysierten Präparationen
wurde mithilfe des ELISA-Kits von Five Prime → Three Prime nachgewiesen.
-
Die
inhibitorische Aktivität
gegen das humane Cytomegalievirus (HCMV) wurde unter Verwendung
des HCMV-Stammes RC-256 (ATCC VR-2536), eine Rekombinante von HCMV
Towne, enthaltend das LacZ-Gen von E. coli, gemessen (Spaete, R.
R., Mocarski, E. S., „Regulation
of Cytomegalovirus Gene Expression: α and β Promotors Are Trans Activated
by Viral Functions in Permissive Human Fibroblasts", J. Virol., (1985), 56,
135–143).
-
Die
Inhibitionswirkung auf CMV bei der Replikation des β-Galactosidase
exprimierenden HCMV RC-256 wurde unter Bedingungen ähnlich denen
gemessen, die für
HSV vgCL5 beschrieben wurden, außer dass die Zell-Virus-Gemische
vor dem Messen der β-Galactosidase-Konzentrationen
48 Stunden bei 37°C
gehalten und humane Vorhaut-Fibroblastenzellen (HFF-Zellen) verwendet
wurden.
-
BEISPIEL 4: VIRUZIDE AKTIVITÄT DER CAP-FORMULIERUNGEN
-
Sowohl
die Anti-HIV-1-Aktivität
als auch die Anti-HSV-1- und Anti-HSV-2-Aktivitäten der CAP-Formulierung, vor
und nach dem Stabilitätstest,
entsprachen dem CAP-Gehalt in der Präparation und den in Tabelle 1B
und 1 und 2 gezeigten Ergebnissen für HIV-1
bzw. Herpesviren.
-
Wenn
eine HIV-1-Präparation
mit entweder einem gleichen Volumen einer Suspension aus „AQUATERIC" in Glycerol oder
mit einem gleichen Volumen der vorstehend erwähnten Formulierung 5 Minuten
bei 37°C 1:1
gemischt wurde, trat ein vollkommener Verlust der HIV-1-Infektivität auf (4).
Die Inaktivierung der HIV-1-Infektivität kann der vollständigen Zerstörung der
HIV-1-Virionen zugeschrieben werden, wie durch die quantitative
Freisetzung des internen Nukleocapsid-Antigens p24 nachgewiesen
wurde (3). Die Infektivität sowohl von HSV-1 als auch
HSV-2 wurde anhand von Suspensionen aus „AQUATERIC" in Glycerol oder der „AQUATERIC"-Glycerol-Formulierung
mit PVP und Crospovidon (5 und 6) ähnlich zerstört.
-
In
Bezug auf 3 wurden Reihenverdünnungen
von unbehandelten und behandelten HIV-1 mittels ELISA auf p24 getestet.
Als positive Kontrolle wurden auch mit dem Detergens NP40 behandelte
HIV-1 getestet. Die mit der „AQUATERIC"-Glycerol-Formulierung,
enthaltend PVP und Crospovidon und NP40, erhaltenen Ergebnisse waren
identisch und wiesen auf eine ca. 100fache Zunahme des freigesetzten
p24-Antigens im Vergleich zu den Hintergrundkonzentrationen auf,
die dem unbehandelten Virus entsprachen. Die Infektivität von HIV-1
wurde auch mithilfe der Behandlung mit der „AQUATERIC"-Glycerolformulierung, enthaltend PVP und
Crospovidon, eliminiert.
-
Bezüglich 5 wurden
Reihenverdünnungen
der Viruspräparationen
vor und nach der Behandlung mit „AQUATERIC" unter Verwendung von zwei verschiedenen
Ablesesystemen basierend auf der Quantifizierung von β-Galactosidase
(Absorption bei 410 nm) auf Infektivität getestet.
-
Unter
Bezugnahme auf 6 wurden die Viruspräparationen
mit einer „AQUATERIC"-Glycerolformulierung
mit PVC und Crospovidon 5 Minuten bei 37°C 1:1 gemischt. Reihenverdünnungen
der Viruspräparationen
wurden unter Verwendung eines Ablesesystems basierend auf der Quantifizierung
von β-Galactosidase (Absorption
bei 410 nm) auf Infektivität
getestet.
-
CAP
als ein Essigsäureester
von Cellulose kann in wässrigem
Milieu der Hydrolyse unterliegen. Deshalb wurden CAP-Formulierungen
in organischen Lösungsmitteln
untersucht. Die besten Ergebnisse wurden mittels Dispersion mikronisierter
CAP, enthaltend außer
CAP ein Polyoxyethylen-Polyoxypropylen-Blockcopolymer und destillierte
acetylierte Monoglyceride („AQUATERIC") in Glycerol, dem
Povidon und Crospovidon zur Stabilisierung der Suspension (CAP-Formulierung
I) zugefügt
wurde, erhalten. Eine Alternative, CAP-Formulierung II, enthaltend
kolloidales Siliciumdioxid, anstelle von Povidon und Crospovidon,
wurde auch hergestellt. Mit dem Mischen dieser Formulierungen mit
einem gleichen Volumen einer Suspension von infektiösem HIV-1
war es möglich
zu zeigen, dass diese Behandlung zur Disintegration der HIV-1-Partikel
führte,
was durch die Freisetzung des Nukleocapsid-Antigens p24 messbar
war (3). Die CAP-Formulierung war bei der Freisetzung
des Nukleocapsid-Antigens aus den Viruspartikeln genauso wirksam
wie das Detergens NP40. Ähnliche
Ergebnisse wurden erhalten, wenn den HIV-CAP-Formulierungsgemischen
Samenflüssigkeit
oder Vollblut zugefügt
wurde (7).
-
Die
CAP-Formulierungen inaktivierten auch die Infektivität von HSV-1
und HSV-2 (6). Ähnliche Ergebnisse wurden unter
Verwendung von HCMV (8) erhalten. Das Zufügen von
Samenflüssigkeit
oder Vollblut zur CAP-Formulierung griff nicht störend in
ihre virusinaktivierenden Eigenschaften unter den verwendeten Bedingungen
ein, was darauf hindeutete, dass das mikrobizide Potenzial von CAP
unter analogen Bedingungen in vivo aufrechterhalten bleibt.
-
BEISPIEL 5: INAKTIVIERUNG
MEHRERER SEXUELL ÜBERTRAGENER
PATHOGENE DURCH CAP-FORMULIERUNGEN
-
Zur
Untersuchung der Wirkung des formulierten CAP auf HIV-1, HSV-1,
HSV-2 und HCMV wurden die entsprechenden Virus-Suspensionen mit
einem gleichen Volumen der CAP-Formulierungen I und II (jeweils bei
37°C vorgewärmt) vermischt.
Nach der 5-minütigen
Inkubation bei 37°C
wurden die Suspensionen durch ein 0,45-μm-Filter filtriert und anschließend zur
Entfernung von suspendiertem CAP zentrifugiert. Die Überstandsflüssigkeiten
wurden auf Virusinfektivität
getestet. Diese Trennung konnte unter Verwendung der CAP-Formulierung
II nicht erreicht werden, und die entsprechenden Virus-CAP-Formulierungsgemische
wurden direkt auf Virus-Infektivität getestet. In Kontrollexperimenten
wurden Formulierungen, denen das CAP („AQUATERIC") mangelte, gemischt mit den entsprechenden
Viruspräparationen
bzw. PBS (0,14 NaCl, 0,01 M Phosphat pH 7,2), verwendet. Reihenverdünnungen
(2fach) der entsprechenden Viruspräparationen wurden unter Verwendung
von β-Galactosidase-Ablesesystemen
auf Herpesviren, wie vorstehend beschrieben, auf HIV-1- und Herpesvirus-Infektivität getestet.
In einigen Experimenten wurden Samenflüssigkeit (0,6 ml; bezogen von
den New England Immunology Associates, Cambridge, MA) oder heparinisiertes
humanes Vollblut (0,11 ml) den CAP-Formulierungen (1 ml) zugefügt, gefolgt
vom Zufügen
des Virus (1 ml), um ihre Wirkung auf die Virusinaktivierung zu
bewerten.
-
Zur
Bestimmung, ob sich die CAP-Formulierungen gegebenenfalls auf die
Integrität
von HIV-1 auswirken, wurde eine Präparation aus gereinigtem Virus
(Stamm IIIB; 5 μl;
1,52 × 1010 Viruspartikel/ml; Advanced Biotechnologies,
Columbia, MD) mit 45 μl
0,14 M NaCl und 50 μl
der entsprechenden CAP-Formulierung, jeweils auf 37°C vorgewärmt, gemischt.
Nach 5 Minuten bei 37°C
wurden 400 μl
0,14 M NaCl zugefügt,
und das Gemisch wurde durch ein 0,45-μm-Filter mit 0,14 M NaCl, 0,01
M Tris (pH 7,2), 0,02 NaN3 (TS; Testlösung) vorgewaschen.
Reihenverdünnungen
(5fach) vom Filtrat wurden mittels ELISA, wie vorstehend beschrieben, auf
das p24-Antigen getestet. In Kontrollexperimenten wurden entweder
unbehandelte HIV-1-IIIB oder mit dem Detergens NP40 (Endkonzentration
5 μg/ml,
5 Minuten bei 37°)
behandelte Viren gleichmäßig verdünnt und auf
das p24-Antigen getestet. Ähnliche
Experimente wurden mit den CAP-Formulierungen in Gegenwart von Samenflüssigkeit
oder Vollblut (Anteile sind vorstehend angegeben) durchgeführt. Wenn
den CAP-Formulierungen Vollblut zugefügt wurde, wurden die verdünnten Präparationen
vor der Filtration durch 0,45-μm-Filter bei
2000 × g
5 Minuten zentrifugiert.
-
BEISPIEL 6: INAKTIVIERUNG
BAKTERIELLER PATHOGENE
-
Zum
Testen der Wirkung der CAP-Formulierung I (einer Präparation
aus mikronisiertem CAP („AQUATERIC", enthaltend 66–73% CAP,
ein Polyoxyethylen-Polyoxypropylen-Blockcopolymer und destillierten
acetylierten Monoglyceriden, verwendet in wässrigen Medien als eine magensaftresistente
Filmüberzugsflüssigkeit
(FMC Corporation, Philadelphia, PA)), wurden (15,9 g) mit Glycerol
(70,2 g) gemischt und Polyvinylpyrrolidon K-30 (Spectrum Quality
Products, Inc., New Brunswick, NJ) (11,1 g) und Crospovidon NF (ISP
Technologies, Inc., Wayne, NJ) (2,8 g) wurden zugefügt, um das
mikronisierte CAP in Suspension zu halten), auf verschiedene bakterielle
Pathogene von sexuell übertragenen
(STD) Erkrankungen bzw. Lactobacilli, wurden gleiche Volumina der
CAP-Formulierung und einer Bakteriensuspension (108 bis
109 Zellen/ml in 0,14 M NaCl) gemischt und
entweder 5 oder 15 Minuten bei 37°C
inkubiert. Im Anschluss daran wurden 50 μl der 0,43 M Na3PO4·12H2O pro 1 ml der Suspension zugefügt. Nach
5 Minuten bei Raumtemperatur und intermittierendem Mischen wurden
die neutralisierten Suspensionen in PBS oder geeigneten Nährbouillonmedien
um in einer Reihenverdünnung
das 10fache verdünnt.
Ein Volumen zu 20 μl
von jeder Verdünnung
wurde pro Well einer Mikrotiterplatte, enthaltend Wachstumsmedien,
oder auf eine entsprechende Agarplatte inokuliert und unter geeigneten
Bedingungen zur Überwachung
des Bakterienwachstums inkubiert. Eingeschlossen in jedem Experiment
war eine positive Antibiotika-Kontrolle, um die Inhibition des Bakterienwachstums
nachzuweisen und eine Kontrolle ohne die Formulierung oder das Antibiotikum,
um das Wachstum der Bakterien zu überwachen.
-
Die
verwendeten Bakterienstämme,
die entsprechenden Wachstumsmedien und die Kontrollantibiotika waren
wie folgt: Lactobacillus crispatus, ATCC 33820 (Lactobacillus-Nährbouillon;
Tetracyclin 0,5 bis 64 μg/ml);
Neisseria gonorrhoeae, ATCC 49226 (GC-Medien [Becton Dickinson Microbiology
Systems, Sparks, MD), supplementiert mit 500 μl einer 2%igen Hämoglobin-Stammlösung und
10 ml "ISOVITALEX" (Becton Dickinson
Microbiology Systems, Sparks, MD) pro Liter Medien; Ceftriaxon zu
0,005 bis 0,128 μg/ml);
Haemophilus ducreyi, ATCC 33940 (revidiertes Ducreyi-Medium, Kulturmedium
1724 der American Type Culture Collection (ATCC), supplementiert
mit 10% FBS (Life Technologies, Gaithersburg, MD) und 1% "ISOVITALEX" (Becton Dickinson
Microbiology Systems, Sparks, MD); Tetracyclin 0,125 bis 64 μg/ml; Trichomonas
vaginalis, ATCC 30092 (modifizierte Fuji-Medien (Ohkawa M., Yamaguchi,
K., Tokunaga, S. et al., "The
Incidence of Trichomonas Vaginalis in Chronic Prostatitis Patients
Determined by Culture Using a Newly Modified Liquid Medium", J. Infect. Dis.,
(1992), 166, 1205–1206);
Metronidazol 1 bis 128 μg/ml).
-
Die
von der Environmental Protection Agency etablierten Richtlinien
(Efficacy Data Requirements: Virucides DISITSS-7, 12. Nov. 1981,
Modifying ADAC Methods 4.007–4.014,
Office of Pesticide Research, Environmental Protection Agency, Washington,
D. C.) wurden zur Untersuchung der Wirkung der CAP-Formulierung
I auf Chlamydia trachomatis (Stamm LGV Typ III; ATCC VR-903) befolgt.
Nach der Inkubation mit der Formulierung und Neutralisation, wurde
die Präparation
in DMEM-Wachstumsmedien, enthaltend 3,6 mM L-Glutamin, 45 μg/ml Gentamicin
und 8,9% FBS verdünnt,
und es wurden Reihenverdünnungen
(10fach) hergestellt. Die Verdünnungen
wurden den McCoy-Zellmonolayers (ATCC CRL 1996) zugefügt, und
die Infektion wurde mittels mikroskopischer Beobachtung von CPE
oder durch Färbung
von Einschluss- oder Elementarkörperchen
mittels Iodfärbung
nach 7-tägiger
Inkubation bei 37°C
mit CO2 bestimmt.
-
Folglich
wurde die vorstehend beschriebene CAP-Formulierung I auf ihre Inaktivierungswirkung
auf die folgenden sexuell übertragenen
Pathogene getestet: Chlamydia trachomatis, Trichomonas vaginalis,
Neisseria gonorrhoeae, Haemophilus ducreyi und auf Lactobacillus
crispatus. Die in der folgenden Tabelle 7 gezeigten Ergebnisse weisen
darauf hin, dass alle Bakterien, außer Lactobacillus crispatus,
die Kapazität
zur Replikation nach Exposition gegenüber der CAP-Formulierung verloren.
Die Aktivität
der CAP-Formulierung gegen Treponema palladium wurde nicht getestet,
weil geeignete In-vitro-Assays nicht zur Verfügung standen. Die CAP-Formulierung
war aktiv gegen vier wichtige nicht-virale STD-Pathogene, wirkte
sich aber nicht auf Lactobacilli, eine wesentliche Komponente der
normalen vaginalen Flora, aus.
-