CN114014819A - 一种1,4,8,11-四氮杂环十四烷的制备方法 - Google Patents
一种1,4,8,11-四氮杂环十四烷的制备方法 Download PDFInfo
- Publication number
- CN114014819A CN114014819A CN202111628085.4A CN202111628085A CN114014819A CN 114014819 A CN114014819 A CN 114014819A CN 202111628085 A CN202111628085 A CN 202111628085A CN 114014819 A CN114014819 A CN 114014819A
- Authority
- CN
- China
- Prior art keywords
- tetraazacyclotetradecane
- reaction
- cooling
- oxalate
- uniformly stirring
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
Abstract
本发明提供一种1,4,8,11-四氮杂环十四烷的制备方法,包括如下步骤:S1、称取2‑甲基四氢呋喃、1,2‑双(3‑氨丙基氨基)乙烷、保护剂和水,混合,搅拌均匀;反应液旋蒸回收溶剂,用甲苯重结晶得到黄色晶体;S2、将步骤S1制得的黄色晶体、L叔丁基甲醚、1,2‑二溴乙烷混合,搅拌均匀;反应液趁热过滤,滤液冷却后加入石油醚,析晶得到浅黄色固状体产品;S3、氮气保护下,在氢氧化钾水溶液中分批加入步骤S2得到的浅黄色固体产品;反应液冷却至室温,过滤,向滤液加入二氯甲烷进行萃取,常压蒸馏回收二氯甲烷后向剩余溶液中加入石油醚,冷却析晶出1,4,8,11-四氮杂环十四烷晶体。本发明生产工艺简单,原料价格较低,总收率高,产品纯度高,经济性良好。
Description
技术领域
本发明属于精细化工领域,具体涉及一种1,4,8,11-四氮杂环十四烷的制备方法。
背景技术
大环多胺是指含有多个氨基的大环化合物,大环多胺是超分子化学中一类非常重要的主体分子,其对金属离子具有较强的配位能力,所形成的金属配合物也是一类具有独特结构和性能的化合物。由于大环多胺类的化合物及其配合物的应用十分广泛,近些年来,对它们的研究已成为新研究领域的新兴课题之一。它们不仅在过渡金属配合物方面有广泛的用途,而且在分子识别和生物医药技术方面也有着重要的应用价值。目前,其已经广泛用于化学核酸酶、生物传感器、MRI照影剂、荧光探针、DNA识别及酶模拟切割催化剂、金属分离与回收、放射免疫治疗药物、基础生物、医学等众多研究领域。
1,4,8,11-四氮杂环十四烷是大环多胺中应用最广泛的一类化合物,它也是趋化因子受体拮抗剂普乐沙福的关键原料。由于1,4,8,11-四氮杂环十四烷及其衍生物对过渡金属阳离子、重金属阳离子,铜系和铜系离子,甚至对有机或无机阴离子都表现出选择性配位性质,其准确行为依赖千其上取代基的性质,这种多样性使它们在众多领域具有广泛的应用价值。1,4,8,11-四氮杂环十四烷分子结构如下:

现有技术中陈贝贝等人在《普乐沙福的合成工艺改进》(合成化学,2015,23,774-777)中详细阐述了1,4,8,11-四氮杂环十四烷及其烷烃链上衍生物的制备方法,以1,2-双(3-氨丙基氨基)乙烷为起始原料,经对甲苯磺酰氯保护,再与相应的二对甲苯磺酸酯关环,然后在强酸性条件下脱保护,再经碱化得到产品1,4,8,11-四氮杂环十四烷或其烷烃链上衍生物。
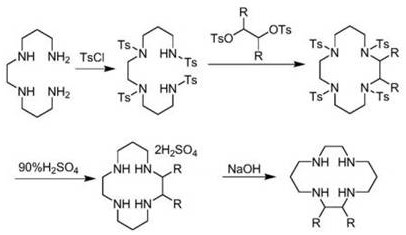
该反应过程中存在五个比较严重的问题:第一,对甲苯磺酰基保护基团位阻非常大,导致关环效率不高,文献中报道的关环收率仅为73%;第二,对甲苯磺酰基保护基团用量大,需要使用4个当量,导致整条路线的原子经济性很低;第三,对甲苯磺酰基保护的胺基需要经过碱活化才具备亲核进攻能力,导致关环步骤必须加入大量碱,导致操作工艺比较复杂,三废量也比较多;第四,脱去对甲苯磺酰基保护基团的反应条件很苛刻,需要使用高度危险的90%硫酸,于100℃进行长时间反应,该操作危险性大,对操作人员的不友好,不符合绿色生产的需求;第五,整条路线收率较低,仅为54%,经济性较差。
上述问题都严重限制了该工艺的进一步应用,也使得该产品的公斤级和百公斤级放大变得比较困难。
发明内容
本发明要解决的技术问题是提供一种1,4,8,11-四氮杂环十四烷的制备方法,解决了现有技术1,4,8,11-四氮杂环十四烷制备过程中工艺复杂、产率不高、成本高的技术问题。
为解决上述技术问题,本发明的实施例提供一种1,4,8,11-四氮杂环十四烷的制备方法,包括如下步骤:
S1、称取2-甲基四氢呋喃、1,2-双(3-氨丙基氨基)乙烷、保护剂和水,混合,搅拌均匀,保温回流反应5-8小时;反应完成后,反应液旋蒸回收溶剂,剩余物用甲苯重结晶得到黄色晶体;
S2、将步骤S1制得的黄色晶体、L叔丁基甲醚、1,2-二溴乙烷混合,搅拌均匀,保温反应4-7小时;反应完成后,反应液趁热过滤,滤液自然冷却,然后加入石油醚,冷却至0℃进行析晶,得到浅黄色固状体产品;
S3、氮气保护下,在氢氧化钾水溶液中分批加入步骤S2得到的浅黄色固体产品,控制温度低于30℃,搅拌均匀后,升温至80℃反应4-7小时,反应过程中控制pH值在9-14;反应结束后,反应液冷却至室温,过滤,向滤液加入二氯甲烷进行萃取,合并萃取液,常压蒸馏回收二氯甲烷后向剩余溶液中加入石油醚,冷却至0℃进行析晶,析出白色的1,4,8,11-四氮杂环十四烷晶体。
其中,步骤S1中,2-甲基四氢呋喃、1,2-双(3-氨丙基氨基)乙烷、草酸二乙酯和水的质量比为3-5:0.5-2:0.7-2。
其中,步骤S2中,黄色晶体、L叔丁基甲醚、1,2-二溴乙烷的质量比为0.5-1:0.8-1.2:0.8-1.2。
其中,步骤S3中,氢氧化钾水溶液的质量浓度为10%。
其中,步骤S3中,氢氧化钾和浅黄色固体产品的质量比为1:1.5-2.6。
优选的,步骤S1中,所述保护剂为是草酸二甲酯、草酸二乙酯、草酸二异丙酯、、草酸二叔丁酯、草酸二正丁酯、草酸二异丁酯、草酰氯单乙酯或草酰氯单甲酯中的一种或多种混合。
本发明的上述技术方案的有益效果如下:
本发明生产工艺简单,原料价格较低,总收率高,产品纯度高,经济性良好,能充分满足产品工业化生产的需求。
具体实施方式
为使本发明要解决的技术问题、技术方案和优点更加清楚,下面将结合具体实施例进行详细描述。
本发明提供一种1,4,8,11-四氮杂环十四烷的制备方法,包括如下步骤:
S1、称取2-甲基四氢呋喃、1,2-双(3-氨丙基氨基)乙烷、保护剂和水,混合,搅拌均匀,保温回流反应5-8小时;反应完成后,反应液旋蒸回收溶剂,剩余物用甲苯重结晶得到黄色晶体;其中,2-甲基四氢呋喃、1,2-双(3-氨丙基氨基)乙烷、草酸二乙酯和水的质量比为3-5:0.5-2:0.7-2;
步骤S1中,所述保护剂为是草酸二甲酯、草酸二乙酯、草酸二异丙酯、、草酸二叔丁酯、草酸二正丁酯、草酸二异丁酯、草酰氯单乙酯或草酰氯单甲酯中的一种或多种混合。
S2、将步骤S1制得的黄色晶体、L叔丁基甲醚、1,2-二溴乙烷混合,搅拌均匀,保温反应4-7小时;反应完成后,反应液趁热过滤,滤液自然冷却,然后加入石油醚,冷却至0℃进行析晶,得到浅黄色固状体产品;其中,黄色晶体、L叔丁基甲醚、1,2-二溴乙烷的质量比为0.5-1:0.8-1.2:0.8-1.2。
S3、氮气保护下,在质量浓度10%的氢氧化钾水溶液中分批加入步骤S2得到的浅黄色固体产品,氢氧化钾和浅黄色固体产品的质量比为1:1.5-2.6。控制温度低于30℃,搅拌均匀后,升温至80℃反应4-7小时,反应过程中控制pH值在9-14;反应结束后,反应液冷却至室温,过滤,向滤液加入二氯甲烷进行萃取,合并萃取液,常压蒸馏回收二氯甲烷后向剩余溶液中加入石油醚,冷却至0℃进行析晶,析出白色的1,4,8,11-四氮杂环十四烷晶体,收率93.7%,三步反应总收率89.1%。
本发明生产工艺简单,原料价格较低,总收率高,产品纯度高,经济性良好,能充分满足产品工业化生产的需求。
以上所述是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明所述原理的前提下,还可以作出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
Claims (6)
1.一种1,4,8,11-四氮杂环十四烷的制备方法,其特征在于,包括如下步骤:
S1、称取2-甲基四氢呋喃、1,2-双(3-氨丙基氨基)乙烷、保护剂和水,混合,搅拌均匀,保温回流反应5-8小时;反应完成后,反应液旋蒸回收溶剂,剩余物用甲苯重结晶得到黄色晶体;
S2、将步骤S1制得的黄色晶体、L叔丁基甲醚、1,2-二溴乙烷混合,搅拌均匀,保温反应4-7小时;反应完成后,反应液趁热过滤,滤液自然冷却,然后加入石油醚,冷却至0℃进行析晶,得到浅黄色固状体产品;
S3、氮气保护下,在氢氧化钾水溶液中分批加入步骤S2得到的浅黄色固体产品,控制温度低于30℃,搅拌均匀后,升温至80℃反应4-7小时,反应过程中控制pH值在9-14;反应结束后,反应液冷却至室温,过滤,向滤液加入二氯甲烷进行萃取,合并萃取液,常压蒸馏回收二氯甲烷后向剩余溶液中加入石油醚,冷却至0℃进行析晶,析出白色的1,4,8,11-四氮杂环十四烷晶体。
2.根据权利要求1所述的1,4,8,11-四氮杂环十四烷的制备方法,其特征在于,步骤S1中,2-甲基四氢呋喃、1,2-双(3-氨丙基氨基)乙烷、草酸二乙酯和水的质量比为3-5:0.5-2:0.7-2。
3.根据权利要求1所述的1,4,8,11-四氮杂环十四烷的制备方法,其特征在于,步骤S2中,黄色晶体、L叔丁基甲醚、1,2-二溴乙烷的质量比为0.5-1:0.8-1.2:0.8-1.2。
4.根据权利要求1所述的1,4,8,11-四氮杂环十四烷的制备方法,其特征在于,步骤S3中,氢氧化钾水溶液的质量浓度为10%。
5.根据权利要求1所述的1,4,8,11-四氮杂环十四烷的制备方法,其特征在于,步骤S3中,氢氧化钾和浅黄色固体产品的质量比为1:1.5-2.6。
6.根据权利要求1所述的1,4,8,11-四氮杂环十四烷的制备方法,其特征在于,步骤S1中,所述保护剂为是草酸二甲酯、草酸二乙酯、草酸二异丙酯、、草酸二叔丁酯、草酸二正丁酯、草酸二异丁酯、草酰氯单乙酯或草酰氯单甲酯中的一种或多种混合。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111628085.4A CN114014819A (zh) | 2021-12-29 | 2021-12-29 | 一种1,4,8,11-四氮杂环十四烷的制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111628085.4A CN114014819A (zh) | 2021-12-29 | 2021-12-29 | 一种1,4,8,11-四氮杂环十四烷的制备方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN114014819A true CN114014819A (zh) | 2022-02-08 |
Family
ID=80069225
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111628085.4A Pending CN114014819A (zh) | 2021-12-29 | 2021-12-29 | 一种1,4,8,11-四氮杂环十四烷的制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN114014819A (zh) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5608061A (en) * | 1995-08-02 | 1997-03-04 | Johnson Matthey Plc | Process for preparing 1,4,8,11-tetraazacyclotetradecane |
| US5811544A (en) * | 1995-08-28 | 1998-09-22 | Johnson Matthey Plc | Process for preparing 1,4,8,11-tetraazacyclotetradecane |
| CN113801123A (zh) * | 2021-10-18 | 2021-12-17 | 苏州百灵威超精细材料有限公司 | 一种1,4,8,11-四氮杂环十四烷类化合物及其中间体的制备方法 |
-
2021
- 2021-12-29 CN CN202111628085.4A patent/CN114014819A/zh active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5608061A (en) * | 1995-08-02 | 1997-03-04 | Johnson Matthey Plc | Process for preparing 1,4,8,11-tetraazacyclotetradecane |
| US5811544A (en) * | 1995-08-28 | 1998-09-22 | Johnson Matthey Plc | Process for preparing 1,4,8,11-tetraazacyclotetradecane |
| CN113801123A (zh) * | 2021-10-18 | 2021-12-17 | 苏州百灵威超精细材料有限公司 | 一种1,4,8,11-四氮杂环十四烷类化合物及其中间体的制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108794491B (zh) | 一种枸橼酸托法替布的精制方法 | |
| CN113956312B (zh) | 一种莫匹拉韦的制备方法 | |
| CN113801123B (zh) | 一种1,4,8,11-四氮杂环十四烷类化合物及其中间体的制备方法 | |
| EP4151628A1 (en) | Preparation method for synthesizing chiral nicotine from chiral tert-butyl sulfinamide | |
| CN113416150B (zh) | 一种洛铂中间体的合成方法 | |
| CN114014819A (zh) | 一种1,4,8,11-四氮杂环十四烷的制备方法 | |
| CN114380696B (zh) | 一种盐酸特比萘芬的制备方法 | |
| CN114014864B (zh) | 一种曲拉西利化合物的制备工艺 | |
| CN110551052A (zh) | (r)-4-羟基-2-氧代-1-吡咯烷乙酸酯的制备方法 | |
| CN108164423A (zh) | 一种盐酸萘替芬的制备方法 | |
| CN111018782B (zh) | 9-氨基吖啶及其衍生物的制备方法 | |
| CN109824539B (zh) | 一种由去甲基金霉素合成替加环素的新方法 | |
| CN112047896A (zh) | 芳环基或芳杂环基四氮唑的合成方法 | |
| CN109265385B (zh) | 一种手性催化剂的合成工艺 | |
| CN102924255A (zh) | 一种液相氧化制备9-芴酮的方法 | |
| CN107602454B (zh) | 磺酰胺类化合物及其制备方法和用途 | |
| WO2019008594A1 (en) | CONTINUOUS PROCESS FOR THE PREPARATION OF 2- (1H-IMIDAZOL-4-YL) ETHANAMINE AND ITS PHARMACEUTICALLY ACCEPTABLE SALTS | |
| EP3904333A1 (en) | Method for preparing (2s,3s)-3-amino-bicyclo[2.2.2]octane-2-carboxylate | |
| CN110724098A (zh) | 一种5,7-二氯-1,2,3,4-四氢异喹啉-6-羧酸盐酸盐的合成方法 | |
| CN111995593B (zh) | 一种微通道反应器合成(s)-4-苯基-2-噁唑烷酮的方法 | |
| CN102020612A (zh) | 苄氧羰基碱基的合成方法 | |
| CN113214197B (zh) | 一种维生素c乙基醚的制备方法 | |
| CN113735716B (zh) | 一种亚精胺的制备方法 | |
| CN114891005B (zh) | 一种乌帕利斯对甲苯磺酸盐的制备工艺 | |
| CN111004141B (zh) | 一种尼达尼布中间体2-氯-n-甲基-n-(4-硝基苯基)乙酰胺合成新方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |