CN111875552B - 一种胺基取代的1,2,3-三唑类化合物及其合成方法与应用 - Google Patents
一种胺基取代的1,2,3-三唑类化合物及其合成方法与应用 Download PDFInfo
- Publication number
- CN111875552B CN111875552B CN202010638309.9A CN202010638309A CN111875552B CN 111875552 B CN111875552 B CN 111875552B CN 202010638309 A CN202010638309 A CN 202010638309A CN 111875552 B CN111875552 B CN 111875552B
- Authority
- CN
- China
- Prior art keywords
- formula
- compound
- amino
- triazole
- substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/04—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles
- C07D249/06—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles with aryl radicals directly attached to ring atoms
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
- A01N43/64—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with three nitrogen atoms as the only ring hetero atoms
- A01N43/647—Triazoles; Hydrogenated triazoles
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
- A01N43/72—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with nitrogen atoms and oxygen or sulfur atoms as ring hetero atoms
- A01N43/84—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with nitrogen atoms and oxygen or sulfur atoms as ring hetero atoms six-membered rings with one nitrogen atom and either one oxygen atom or one sulfur atom in positions 1,4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Dentistry (AREA)
- Environmental Sciences (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Plant Pathology (AREA)
- Pest Control & Pesticides (AREA)
- Agronomy & Crop Science (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明涉及一种胺基取代的1,2,3‑三唑类化合物及其合成方法与应用,所述胺基取代的1,2,3‑三唑类化合物具有式I所示结构: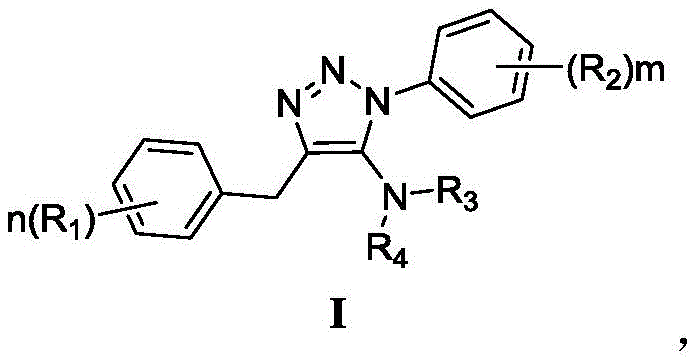 其中R1、R2各自独立地选自C1‑C3烷基、C1‑C3烷氧基、卤代C1‑C3烷基、卤素、氰基、硝基,n、m各自独立地选自0、1、2、3;R3、R4各自独立地选自C1‑C6烷基、C6‑C10芳基,或者R3与R4一起形成3至6元环,所形成的环任选包含1‑2个N、O或S原子。
其中R1、R2各自独立地选自C1‑C3烷基、C1‑C3烷氧基、卤代C1‑C3烷基、卤素、氰基、硝基,n、m各自独立地选自0、1、2、3;R3、R4各自独立地选自C1‑C6烷基、C6‑C10芳基,或者R3与R4一起形成3至6元环,所形成的环任选包含1‑2个N、O或S原子。
Description
技术领域
本发明属于活性有机化合物合成领域,具体涉及一种胺基取代的1,2,3-三唑类化合物及其合成方法与应用。
背景技术
1,2,3-三唑是药物分子中一类重要的结构基元,在多个药物分子结构中发挥了重要的作用,如他唑巴坦、卢非酰胺等。在药物分子中,这些不同的1,2,3-三唑结构,为药物的合成提供了更多的选择。在这些1,2,3-三唑生物活性分子中,胺基取代的1,2,3-三唑,是一种特殊的结构类型。这些胺基1,2,3-三唑分子中,三唑Ⅰ分子是一种具有抗肿瘤活性的分子(C.Sheng,W.Zhang,Curr.Med.Chem.2011,18,733-766);三唑II分子具有治疗炎症性肾脏疾病的潜力(S.G.Agalave,S.R.Maujan,V.S.Pore,Chem.Asian J.2011,6,2696);三唑III分子一种有效的h-NK1拮抗剂(S.G.Agalave,S.R.Maujan,V.S.Pore,Chem.Asian J.2011,6,2696),三唑IV分子具有钾离子通道调节能力(Eur.J.Med.Chem.2000,35,715)。因此,制备胺基1,2,3-三唑的方法,是药物研究和制备的重要问题,具有重要的价值。

目前,合成胺基取代的1,2,3-三唑的方法,主要包括取含活性氢原子的腈类化合物与叠氮化合物的反应(RSC Advances,2015,5(76):62062-62066)、胺基炔烃与叠氮化合物的环加成反应(J.Org.Chem.,2015,80,2562-2572.)等。同时Zhenghu Xu等报道了方法使用羟胺酯作为亲电试剂捕获5-Cu(I)-1,2,3-三唑中间体的反应,成功地在click反应基础上,实现了5-胺基-1,2,3-三唑的合成。但是这些方法均有一定的局限性,如腈类化合物局限于苯乙腈类型化合物,产物结构也较为特殊。而胺基炔烃与羟胺酯均是一类特殊的有机分子,需要特殊的制备途径,反应步骤冗长且不易于规模化反应的实施。因此,发展便捷的原料来制备胺基取代的1,2,3-三唑,是一个药物研发中重要的问题。
本发明报道一种通过易于获取的炔丙胺类化合物与芳基叠氮的反应,在碱t-BuOK的作用下制备了式I结构的胺基取代的1,2,3-三唑。炔丙胺类化合物是一类易于获取的有机中间体,可以通过炔、胺及甲醛高效地合成。因此,本方法具有原料易得的特点,且反应不需要过渡金属的参与,操作相对简单、绿色。本发明制备的式I结构的胺基取代的1,2,3-三唑显示了较强的小麦纹枯病菌抑制活性。
发明内容
本发明提供一种式I结构的胺基取代的1,2,3-三唑类化合物或其药学上可接受的盐,其特征在于所述式I结构如下:

本发明的另一实施方案提供上述式I结构的胺基取代的1,2,3-三唑类化合物或其药学上可接受的盐,其特征在于所述R1、R2各自独立地选自甲基、乙基、甲氧基、乙氧基、三氟甲基、氟、氯、溴、碘、氰基、硝基,n、m各自独立地选自0、1、2,R3、R4各自独立地选自甲基、乙基、丙基、苯基、萘基,或者R3与R4一起形成哌啶环、四氢吡咯环、吗啉环、吡啶环、噻嗪环。
本发明的另一实施方案提供上述式I结构的胺基取代的1,2,3-三唑类化合物选自化合物1-17:

本发明的另一实施方案提供上述式I结构的胺基取代的1,2,3-三唑类化合物的合成方法,其特征在于包括如下步骤:

式II化合物与式III化合物于有机溶剂中,在碱的作用下,反应生成式I化合物。其中式II化合物、式III化合物、碱的摩尔比为1:0.6-1.0:0.7-1.0;碱选自氢氧化钾、叔丁醇钾、叔丁醇锂、叔丁醇钠,双(三甲硅基)氨基锂、氢化钠,Cs2CO3,K3PO4,K2CO3,四丁基氢氧化铵;有机溶剂选自四氢呋喃、二甲基亚砜、N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、六甲基磷酰三胺、N-甲基吡咯烷酮、二氧六环、甲苯和二甲苯中的任意一种或几种的混合溶剂;反应温度选自-10至100℃,优选0℃、10℃、室温、30℃、50℃、60℃;反应时间优选0.5-4小时。
本发明的另一实施方案提供上述式I化合物在防治小麦纹枯病中的应用。
本发明的另一实施方案提供一种药物组合物,其特征在于该药物组合物以上述式I化合物或其药学上可接受的盐作为有效成分。
具体实施方式
以下结合具体实施例对本发明作具体的介绍:
实施例1:1-苯基-4-苄基-5-N,N-二丙胺基-1,2,3-三唑的制备

室温下,在干燥的反应管中加入N,N-二丙基-3-苯基-2-丙炔胺(0.5mmol)和DMF(1mL),搅拌下加入t-BuOK(0.35mmol),反应30分钟后,加入苯基叠氮(0.35mmol),再室温反应两小时。反应结束后,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑化合物1.该产物为微红粘稠液体,产率=78%;1H NMR(400MHz,CDCl3)δ7.59(dt,J=3.7,2.1Hz,2H),7.52–7.42(m,2H),7.40(ddd,J=7.4,3.5,1.3Hz,1H),7.34–7.23(m,4H),7.22–7.14(m,1H),4.11(s,2H),3.06–2.31(m,4H),1.22(dd,J=15.2,7.5Hz,4H),0.62(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ142.4,139.3,138.9,137.0,129.0,128.8,128.6,128.4,126.3,124.8,55.3,32.0,21.3,11.2.HRMS Calcd(ESI)m/z for C21H26N4:[M+H]+335.2230,found:335.2236.
实施例2:1-间甲苯基-4-苄基-5-N,N-二丙胺基-1,2,3-三唑的制备

室温下,在干燥的反应管中加入N,N-二丙基-3-苯基-2-丙炔胺(0.5mmol),间甲基苯基叠氮(0.5mmol)和N,N-二甲基乙酰胺(2mL),搅拌下加入t-BuOLi(0.5mmol),50℃下反应1小时。反应结束后,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑.该产物为微红粘稠液体橙色油状液体,产率=79%;1H NMR(400MHz,CDCl3)δ7.39(dd,J=7.0,6.3Hz,2H),7.34(d,J=7.5Hz,1H),7.32–7.27(m,3H),7.27–7.24(m,1H),7.22(d,J=7.5Hz,1H),7.20–7.15(m,1H),4.10(s,2H),2.76–2.66(m,4H),2.39(s,3H),1.23(ddd,J=9.6,7.5,5.8Hz,4H),0.64(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ142.4,139.4,139.0,138.8,136.9,129.5,128.7,128.6,128.4,126.3,125.5,121.8,55.2,32.0,21.3,11.2.HRMS Calcd(ESI)m/zfor C22H28N4:[M+H]+349.2387,found:349.2384.
实施例3:1-(4-甲氧基苯基)-4-苄基-5-N,N-二丙胺基-1,2,3-三唑的制备:

0℃下,在干燥的反应管中加入N,N-二丙基-3-苯基-2-丙炔胺(0.5mmol)和四氢呋喃(2mL),搅拌下加入t-BuONa(0.5mmol),反应30分钟后,加入4-甲氧基苯基叠氮(0.5mmol),再10℃下反应两小时。反应结束后,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑.该产物为红色固体,产率=60%,熔点:43.5-46℃.1H NMR(400MHz,CDCl3)δ7.50–7.44(m,2H),7.31–7.24(m,4H),7.19(ddd,J=11.1,5.5,3.0Hz,1H),7.00–6.93(m,2H),4.09(s,2H),3.85(s,3H),2.75–2.63(m,4H),1.22(ddd,J=9.8,7.5,5.8Hz,4H),0.64(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ159.8,142.4,139.4,138.8,130.1,128.6,128.4,126.3,126.3,114.1,55.5,55.3,32.0,21.3,11.2.HRMS Calcd(ESI)m/z for C22H28N4O:[M+H]+365.2336,found:365.2328.
实施例4:(4-乙氧基苯基)-4-苄基-5-N,N-二丙胺基-1,2,3-三唑:

室温下,在干燥的反应管中加入N,N-二丙基-3-苯基-2-丙炔胺(0.5mmol)和六甲基磷酰三胺(2mL),搅拌下加入t-BuOK(0.4mmol),反应30分钟后,加入4-乙氧基苯基叠氮(0.35mmol),再30℃下反应两小时。反应结束后,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑.红色油状液体,产率=79%;1H NMR(400MHz,CDCl3)δ7.48–7.43(m,2H),7.31–7.24(m,4H),7.18(td,J=6.0,2.3Hz,1H),6.98–6.92(m,2H),4.09(d,J=2.5Hz,2H),4.07–4.03(m,2H),2.74–2.64(m,4H),1.42(t,J=7.0Hz,3H),1.23(dt,J=9.6,6.3Hz,4H),0.63(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ159.2,142.4,139.4,138.7,129.9,128.6,128.4,126.3,126.3,114.6,63.8,55.3,32.0,21.3,14.7,11.2.HRMS Calcd(ESI)m/z forC23H30N4O:[M+H]+379.2492,found:379.2488.
实施例5:1-(4-溴苯基)-4-苄基-5-N,N-二丙胺基-1,2,3-三唑:

室温下,在干燥的反应管中加入N,N-二丙基-3-苯基-2-丙炔胺(0.5mmol)和二甲基亚砜(2mL),搅拌下加入KOH粉末(0.4mmol),反应30分钟后,加入4-溴苯基叠氮(0.5mmol),再室温下反应两小时。反应结束后,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑.橙色固体,产率=54%,熔点:39-41.5℃;1H NMR(400MHz,CDCl3)δ7.62–7.58(m,2H),7.57–7.53(m,2H),7.31–7.24(m,4H),7.23–7.16(m,1H),4.10(s,2H),2.78–2.68(m,4H),1.27–1.19(m,4H),0.64(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ142.3,139.2,139.1,136.0,132.2,128.6,128.4,126.4,125.9,122.5,55.3,32.0,21.2,11.3.HRMS Calcd(ESI)m/z for C21H25BrN4:[M+H]+413.1335,found:413.1329.
实施例6:1-(邻氯苯基)-4-苄基-5-N,N-二丙胺基-1,2,3-三唑:

室温下,在干燥的反应管中加入N,N-二丙基-3-苯基-2-丙炔胺(0.5mmol),加入2-氯苯基叠氮(0.5mmol)和二氧六环(2mL),搅拌下加入Cs2CO3粉末(0.4mmol),50℃下反应2小时,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑.浅黄色油状液体,产率=65%;1H NMR(400MHz,CDCl3)δ7.56–7.53(m,1H),7.46–7.38(m,2H),7.37(d,J=1.8Hz,1H),7.28(t,J=4.8Hz,4H),7.23–7.17(m,1H),4.12(s,2H),2.73–2.60(m,4H),1.24–1.17(m,4H),0.61(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ144.0,139.4,137.6,134.7,132.5,131.1,130.4,129.7,128.6,128.4,127.3,126.3,54.9,31.9,21.5,11.1.HRMS Calcd(ESI)m/z forC21H25ClN4:[M+H]+369.1841,found:369.1834.
实施例7:1-(4-氰基苯基)-4-苄基-5-N,N-二丙胺基-1,2,3-三唑:

室温下,在干燥的反应管中加入N,N-二丙基-3-苯基-2-丙炔胺(0.5mmol)和六甲基磷酰三胺(2mL),搅拌下加入t-BuOK(0.4mmol),20分钟后加入4-氰基苯基叠氮,在室温下反应30分钟,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑.无色固体,产率=50%,熔点:64-67.5℃;1H NMR(400MHz,CDCl3)δ7.98–7.91(m,2H),7.82–7.73(m,2H),7.33–7.25(m,4H),7.24–7.16(m,1H),4.12(s,2H),2.85–2.69(m,4H),1.29–1.22(m,4H),0.66(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ142.4,140.4,139.8,138.7,133.1,128.6,128.5,126.5,124.1,118.0,112.1,55.5,32.0,21.1,11.3.HRMS Calcd(ESI)m/z for C22H25N5:[M+H]+360.2183,found:360.2179.
实施例8:1-(4-硝基苯基)-4-苄基-5-N,N-二丙胺基-1,2,3-三唑:

室温下,在干燥的反应管中加入N,N-二丙基-3-苯基-2-丙炔胺(0.5mmol),加入4-硝基苯基叠氮(0.5mmol)和二甲基亚砜(2mL),搅拌下加入NaH(0.4mmol),50℃下反应1小时,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑.红色固体,产率=28%,熔点:75.5-78.5℃;1H NMR(400MHz,CDCl3)δ8.35(d,J=9.0Hz,2H),8.04(d,J=9.0Hz,2H),7.31–7.26(m,4H),7.21(ddd,J=14.8,8.8,5.9Hz,1H),4.12(s,2H),2.85–2.68(m,4H),1.27(dd,J=15.6,7.4Hz,4H),0.67(t,J=7.3Hz,6H).13C NMR(100MHz,CDCl3)δ147.0,142.5,141.9,139.9,138.7,128.6,128.5,126.6,124.6,124.0,55.5,32.0,21.1,11.3.HRMS Calcd(ESI)m/z for C21H25N5O2:[M+H]+380.2081,found:380.2086.
实施例9:1-(3-三氟甲基苯基)-4-苄基-5-N,N-二丙胺基-1,2,3-三唑:

室温下,在干燥的反应管中加入N,N-二丙基-3-苯基-2-丙炔胺(0.5mmol),和甲苯(2mL),搅拌下加入K3PO4(1.0mmol),加入3-三氟甲基苯基叠氮(0.5mmol),50℃下反应2小时,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑。黄色油状液体,产率=39%;1H NMR(400MHz,CDCl3)δ8.03(s,1H),7.92(d,J=8.0Hz,1H),7.68(d,J=7.9Hz,1H),7.62(t,J=7.9Hz,1H),7.28(t,J=6.6Hz,4H),7.24–7.17(m,1H),4.13(s,2H),2.82–2.64(m,4H),1.29–1.21(m,4H),0.65(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ142.6,139.4,139.0,137.4,132.1,131.7,131.4,131.1,129.8,129.7,128.6,128.5,127.6,127.4,126.5,125.3,125.2,125.2,125.1,121.3,121.2,121.2,121.1,119.5,55.3,32.0,21.2,11.2.HRMS Calcd(ESI)m/z for C22H25F3N4:[M+H]+403.2104,found:403.2097.
实施例10:1-苯基-4-(4-甲基苄基)-5-N,N-二丙胺基-1,2,3-三唑:

室温下,在干燥的反应管中加入N,N-二丙基-3-(4-甲基苯基)-2-丙炔胺(0.5mmol),和二甲苯(2mL),搅拌下加入K3PO4(1.0mmol),加入苯基叠氮(0.5mmol),50℃下反应2小时,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑。黄色油状液体,产率=70%;1HNMR(400MHz,CDCl3)δ7.63–7.56(m,2H),7.50–7.37(m,3H),7.18(d,J=8.0Hz,2H),7.09(d,J=7.9Hz,2H),4.06(s,2H),2.75–2.69(m,4H),2.30(s,3H),1.24(ddd,J=9.8,7.5,5.7Hz,4H),0.64(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ142.3,139.2,137.0,136.2,135.8,129.1,129.0,128.7,128.5,124.8,55.3,31.6,21.3,21.0,11.2.HRMS Calcd(ESI)m/z forC22H28N4:[M+H]+349.2387,found:349.2383.
实施例11:1-苯基-4-(4-甲氧基苄基)-5-N,N-二丙胺基-1,2,3-三唑:

室温下,在干燥的反应管中加入N,N-二丙基-3-(4-甲氧苯基)-2-丙炔胺(0.5mmol),苯基叠氮(0.5mmol)和N,N-二甲基乙酰胺(2mL),搅拌下加入t-BuOK(0.5mmol),室温下反应1小时。反应结束后,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑.黄色固体,产率=80%,熔点:50-53.5℃;1H NMR(400MHz,CDCl3)δ7.64–7.58(m,2H),7.51–7.41(m,3H),7.23(d,J=8.5Hz,2H),6.85(d,J=8.6Hz,2H),4.06(s,2H),3.79(s,3H),2.80–2.67(m,4H),1.26(dd,J=15.2,7.5Hz,4H),0.66(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ158.1,142.2,139.2,136.9,131.3,129.5,128.9,128.6,124.7,113.8,55.2,31.0,21.2,11.2.HRMS Calcd(ESI)m/z for C22H28N4O:[M+H]+365.2336,found:365.2331.
实施例12:1-苯基-4-(4-氯苄基)-5-N,N-二丙胺基-1,2,3-三唑:

室温下,在干燥的反应管中加入N,N-二丙基-3-苯基-2-丙炔胺(0.5mmol)和DMF(1mL),搅拌下加入t-BuOK(0.35mmol),反应30分钟后,加入苯基叠氮(0.35mmol),再室温反应两小时。反应结束后,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑。橙色油状液体,产率=59%;1H NMR(400MHz,CDCl3)δ7.61–7.56(m,2H),7.51–7.43(m,3H),7.29–7.23(m,4H),4.06(s,2H),2.77–2.67(m,4H),1.25–1.19(m,4H),0.66(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ142.4,138.2,137.8,136.9,132.2,130.0,129.1,128.9,128.5,124.8,55.3,29.7,21.3,11.2.HRMS Calcd(ESI)m/z for C21H25ClN4:[M+H]+369.1841,found:369.1837.
实施例13:1-苯基-4-(4-氟苄基)-5-N,N-二丙胺基-1,2,3-三唑:

室温下,在干燥的反应管中加入N,N-二丙基-3-(4-氟苯基)-2-丙炔胺(0.5mmol),和二甲苯(2mL),搅拌下加入t-BuOK(0.5mmol),加入苯基叠氮(0.5mmol),50℃下反应1小时,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑。橙红色油状液体,产率=63%;1H NMR(400MHz,CDCl3)δ7.62–7.56(m,2H),7.48(ddd,J=7.6,5.0,1.6Hz,2H),7.45–7.39(m,1H),7.29–7.22(m,2H),6.98(ddd,J=10.8,5.9,2.5Hz,2H),4.07(s,2H),2.79–2.63(m,4H),1.27–1.21(m,4H),0.65(t,J=7.4Hz,6H).13C NMR(100MHz,CDCl3)δ162.8,160.4,142.4,138.7,136.9,135.0,134.9,130.1,130.0,129.0,128.8,124.8,115.3,115.0,55.3,31.1,21.3,11.2.HRMS Calcd(ESI)m/z for C21H25FN4:[M+H]+353.2136,found:353.2122.
实施例14:1-苯基-4-苄基-5-(N-甲基苯胺基)-1,2,3-三唑:

室温下,在干燥的反应管中加入N-甲基-N-苯基-3-苯基-2-丙炔胺(0.5mmol),和N,N-二甲基甲酰胺(2mL),搅拌下加入t-BuOK(0.4mmol),加入苯基叠氮(0.5mmol),室温下反应1小时,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑。
橙红色油状液体,产率=53%;1H NMR(400MHz,CDCl3)δ7.52–7.46(m,2H),7.43–7.35(m,3H),7.26–7.13(m,5H),7.08–7.00(m,2H),6.88(t,J=7.3Hz,1H),6.55(d,J=7.9Hz,2H),3.86(s,2H),2.62(s,3H).13C NMR(100MHz,CDCl3)δ146.2,141.8,138.2,137.9,135.9,129.4,129.4,129.0,128.8,128.3,126.3,122.9,119.4,113.1,37.3,31.5.HRMSCalcd(ESI)m/z for C22H20N4:[M+H]+341.1761,found:341.1753.
实施例15:1-苯基-4-苄基-5-吗啉基-1,2,3-三唑:

室温下,在干燥的反应管中加入4-(3-苯基-2-丙炔基)吗啡啉(0.5mmol),和N,N-二甲基甲酰胺(2mL),搅拌下加入t-BuOK(0.4mmol),加入苯基叠氮(0.5mmol),室温下反应1小时,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑。浅黄色油状液体,产率=77%;1H NMR(400MHz,CDCl3)δ7.69–7.64(m,2H),7.50(tt,J=8.7,2.0Hz,2H),7.46–7.41(m,1H),7.31–7.25(m,4H),7.23–7.17(m,1H),4.18(s,2H),3.60–3.52(m,4H),2.83(dd,J=5.5,3.8Hz,4H).13C NMR(100MHz,CDCl3)δ142.1,139.7,137.6,136.6,129.2,129.0,128.6,128.4,126.5,124.3,66.9,50.5,31.9.HRMS Calcd(ESI)m/z for C19H20N4O:[M+H]+321.1710,found:321.1705.
实施例16:1-苯基-4-苄基-5-哌啶基-1,2,3-三唑:

室温下,在干燥的反应管中加入1-(3-苯基-2-丙炔基)哌啶(0.5mmol),和N,N-二甲基甲酰胺(2mL),搅拌下加入t-BuOK(0.4mmol),加入苯基叠氮(0.5mmol),室温下反应1小时,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑。无色针状晶体,产率=80%,熔点:83.5-86℃;1H NMR(400MHz,CDCl3)δ7.72–7.67(m,2H),7.50–7.44(m,2H),7.43–7.37(m,1H),7.32–7.25(m,4H),7.23–7.16(m,1H),4.17(s,2H),2.84–2.72(m,4H),1.50–1.35(m,6H).13C NMR(100MHz,CDCl3)δ143.5,140.1,137.1,136.9,129.0,128.6,128.5,128.5,126.3,124.0,51.6,32.0,26.0,23.6.HRMS Calcd(ESI)m/z for C20H22N4:[M+H]+319.1917,found:319.1914.
实施例17:1-苯基-4-苄基-5-吡咯烷基-1,2,3-三唑:

室温下,在干燥的反应管中加入1-(3-苯基-2-丙炔基)四氢吡咯(0.5mmol),和N,N-二甲基甲酰胺(2mL),搅拌下加入t-BuOK(0.4mmol),加入苯基叠氮(0.5mmol),室温下反应1小时,反应液以乙酸乙酯(20mL)萃取。有机相用水洗(3×40mL),饱和食盐水洗(1×30mL),用无水硫酸钠干燥,过滤后,将所得滤液在真空下蒸发浓缩,残余物再通过硅胶柱色谱法分离,得到相应的1,4,5-三取代-5-胺基-1,2,3-三唑。
橙红色油状液体,产率=72%;1H NMR(400MHz,CDCl3)δ7.64–7.57(m,2H),7.51–7.44(m,2H),7.41(ddd,J=7.4,3.5,1.2Hz,1H),7.29–7.22(m,4H),7.20(d,J=7.1Hz,1H),4.19(s,2H),2.97(dd,J=7.6,5.4Hz,4H),1.71–1.61(m,4H).13C NMR(100MHz,CDCl3)δ141.6,140.3,137.3,134.6,129.1,128.6,128.5,128.3,126.2,124.3,50.6,32.3,25.44.HRMS Calcd(ESI)m/z for C19H20N4:[M+H]+305.1761,found:305.1762.
实施例18
采用菌丝生长法(NY/T 1156.2-2006)测试化合物1-17在浓度100μg/mL时对小麦纹枯病菌离体抑制活性。按照如下公式(1)、(2)计算抑制率。
公式(1):D=D1-D2,其中D表示菌落增长直径,D1表示菌落直径,D2表示菌饼直径;
公式(2):I=(D0-Dt)/D0×100%,其中I表示菌丝生长抑制率,D0表示空白对照菌落增长直径,Dt表示化合物1-17处理菌落增长直径。
结果表明,化合物1-17在100μg/mL浓度下,对小麦纹枯病菌离体抑制活性在82.3%-94.7%之间。其中化合物9活性最高,化合物15活性最低。
Claims (10)
1.一种式I结构的胺基取代的1,2,3-三唑类化合物或其药学上可接受的盐,其特征在于所述式I结构如下:

2.权利要求1所述的式I结构的胺基取代的1,2,3-三唑类化合物或其药学上可接受的盐,其特征在于所述R1选自甲基、乙基、甲氧基、乙氧基、氟、氯、溴、碘,R2选自甲基、乙基、甲氧基、乙氧基、三氟甲基、氟、氯、溴、碘、氰基、硝基。
3.权利要求1-2任一项所述的式I结构的胺基取代的1,2,3-三唑类化合物,其特征在于选自化合物1-17:

4.权利要求1所述的式I结构的胺基取代的1,2,3-三唑类化合物的合成方法,其特征在于包括如下步骤:

式II化合物与式III化合物于有机溶剂中,在碱的作用下,反应生成式I化合物;所述碱选自氢氧化钾、叔丁醇钾、叔丁醇锂、叔丁醇钠、 双(三甲硅基)氨基锂、氢化钠、 Cs2CO3、K3PO4、 K2CO3、 四丁基氢氧化铵。
5.权利要求4所述的合成方法,其特征在于式II化合物、式III化合物、碱的摩尔比为1:0.6-1.0:0.7-1.0。
6.权利要求4所述的合成方法,其特征在于有机溶剂选自四氢呋喃、二甲基亚砜、N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、六甲基磷酰三胺、N-甲基吡咯烷酮、二氧六环、甲苯和二甲苯中的任意一种或几种的混合溶剂。
7.权利要求4所述的合成方法,其特征在于反应温度选自-10至100℃,反应时间为0.5-4小时。
8.权利要求7所述的合成方法,其特征在于反应温度选自0℃、10℃、室温、30℃、50℃、60℃。
9.权利要求1-3任一项所述的式I化合物或其药学上可接受的盐在防治小麦纹枯病中的应用。
10.一种药物组合物,其特征在于该药物组合物以权利要求1-3任一项所述的式I化合物或其药学上可接受的盐作为有效成分。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010638309.9A CN111875552B (zh) | 2020-07-03 | 2020-07-03 | 一种胺基取代的1,2,3-三唑类化合物及其合成方法与应用 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010638309.9A CN111875552B (zh) | 2020-07-03 | 2020-07-03 | 一种胺基取代的1,2,3-三唑类化合物及其合成方法与应用 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN111875552A CN111875552A (zh) | 2020-11-03 |
| CN111875552B true CN111875552B (zh) | 2022-06-21 |
Family
ID=73150922
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010638309.9A Expired - Fee Related CN111875552B (zh) | 2020-07-03 | 2020-07-03 | 一种胺基取代的1,2,3-三唑类化合物及其合成方法与应用 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN111875552B (zh) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5064844A (en) * | 1988-07-08 | 1991-11-12 | Schering Agrochemicals Limited | 1,2,3-triazole insecticides |
| CA2494263A1 (en) * | 2002-08-22 | 2004-03-04 | Syngenta Participations Ag | Microbiocidal (e.g. fungicidal) 1,2,3-triazole derivatives |
| CN106146418A (zh) * | 2015-04-19 | 2016-11-23 | 海南师范大学 | 一种nh-1,2,3-三唑化合物的合成方法 |
| CN108794412A (zh) * | 2018-09-10 | 2018-11-13 | 海南师范大学 | 一种4,5-二芳基-2h-1,2,3-三唑化合物的制备方法 |
-
2020
- 2020-07-03 CN CN202010638309.9A patent/CN111875552B/zh not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5064844A (en) * | 1988-07-08 | 1991-11-12 | Schering Agrochemicals Limited | 1,2,3-triazole insecticides |
| CA2494263A1 (en) * | 2002-08-22 | 2004-03-04 | Syngenta Participations Ag | Microbiocidal (e.g. fungicidal) 1,2,3-triazole derivatives |
| CN106146418A (zh) * | 2015-04-19 | 2016-11-23 | 海南师范大学 | 一种nh-1,2,3-三唑化合物的合成方法 |
| CN108794412A (zh) * | 2018-09-10 | 2018-11-13 | 海南师范大学 | 一种4,5-二芳基-2h-1,2,3-三唑化合物的制备方法 |
Non-Patent Citations (2)
| Title |
|---|
| The Use of Propargylamines to Synthesize Amino-1,2,3-triazoles via Cycloaddition of Azides with Allenamines;Shanguang Qiu,等;《Synthesis》;20220201;第2175-2184页 * |
| 无过渡金属参与的1,2,3-三唑合成进展;陈昱学,等;《有机化学》;20160503;第1779-1789页 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN111875552A (zh) | 2020-11-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101490014B (zh) | 苯并咪唑和氮杂苯并咪唑的区域选择性钯催化合成 | |
| JP7446316B2 (ja) | 置換ピロリジンアミドiii | |
| RU2734760C2 (ru) | Противоинфекционные соединения | |
| CN106132947B (zh) | 化合物的制备方法 | |
| AU2009200478A1 (en) | Metabotropic glutamate receptor-5 modulators | |
| JP6442005B2 (ja) | ベンゾオキサゼピン化合物の作製方法 | |
| NZ582796A (en) | Pyrazol-indazol-pyridin derivatives as P38 kinase inhibitors | |
| WO2015145371A1 (en) | Ror-gamma modulators and uses thereof | |
| TWI754172B (zh) | 作為ccr6抑制劑之n取代二氧代環丁烯基胺基-3-羥基-吡啶醯胺 | |
| EA034196B1 (ru) | Производные иминотетрагидропиримидинона в качестве ингибиторов плазмепсина v | |
| KR102696304B1 (ko) | Tnf의 억제제로서 유용한 헤테로시클릭 화합물 | |
| UA100867C2 (uk) | Похідна (аза)індолу, заміщена в положенні 5, фармацевтична композиція, що містить її, проміжні сполуки та спосіб їх одержання | |
| WO2013092467A1 (en) | 7-azaindole inhibitors of crac | |
| CN115677696B (zh) | 一种苯并萘啶衍生物的制备方法 | |
| JP2025156455A (ja) | チアゾロ[5,4-b]ピリジンMALT-1阻害剤 | |
| WO2025138874A1 (zh) | 可见光驱动合成2-芳基-2h-苯并三唑化合物的方法及其应用 | |
| CN111875552B (zh) | 一种胺基取代的1,2,3-三唑类化合物及其合成方法与应用 | |
| EP3087074A1 (en) | Benzene sulfonamides as ccr9 inhibitors | |
| EP1276742B1 (en) | Process for the preparation of pyrazolopyridine derivatives | |
| WO2023280970A1 (en) | Inhibitors of alpha-hemolysin of staphylococcus aureus | |
| JP7094942B2 (ja) | ベンズイミダゾール誘導体の製造方法 | |
| AU2016344118A1 (en) | Heteroaryl substituted benzoic acids as rorgammat inhibitors and uses thereof | |
| JPH061776A (ja) | 置換ピラジンカルボニトリルの製造方法 | |
| CN110003080B (zh) | 一种含硒化合物及其制备方法与用途 | |
| JP2026511081A (ja) | Atmキナーゼ阻害剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20220621 |
|
| CF01 | Termination of patent right due to non-payment of annual fee |