CN111529697A - 调节的免疫优势疗法 - Google Patents
调节的免疫优势疗法 Download PDFInfo
- Publication number
- CN111529697A CN111529697A CN201911266293.7A CN201911266293A CN111529697A CN 111529697 A CN111529697 A CN 111529697A CN 201911266293 A CN201911266293 A CN 201911266293A CN 111529697 A CN111529697 A CN 111529697A
- Authority
- CN
- China
- Prior art keywords
- cells
- antigen
- subdominant
- patient
- antigens
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000002560 therapeutic procedure Methods 0.000 title description 11
- 239000000427 antigen Substances 0.000 claims abstract description 295
- 108091007433 antigens Proteins 0.000 claims abstract description 294
- 102000036639 antigens Human genes 0.000 claims abstract description 294
- 210000001744 T-lymphocyte Anatomy 0.000 claims abstract description 203
- 238000012258 culturing Methods 0.000 claims abstract description 15
- 230000024932 T cell mediated immunity Effects 0.000 claims abstract description 12
- 206010028980 Neoplasm Diseases 0.000 claims description 79
- 238000000034 method Methods 0.000 claims description 66
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 58
- 210000004443 dendritic cell Anatomy 0.000 claims description 45
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 claims description 41
- 239000013612 plasmid Substances 0.000 claims description 40
- 210000000612 antigen-presenting cell Anatomy 0.000 claims description 31
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 21
- 102000004127 Cytokines Human genes 0.000 claims description 17
- 108090000695 Cytokines Proteins 0.000 claims description 17
- 102000003812 Interleukin-15 Human genes 0.000 claims description 17
- 108090000172 Interleukin-15 Proteins 0.000 claims description 17
- 102000004388 Interleukin-4 Human genes 0.000 claims description 17
- 108090000978 Interleukin-4 Proteins 0.000 claims description 17
- 230000003612 virological effect Effects 0.000 claims description 16
- 239000002609 medium Substances 0.000 claims description 14
- 102000000588 Interleukin-2 Human genes 0.000 claims description 13
- 108010002350 Interleukin-2 Proteins 0.000 claims description 13
- 108010074108 interleukin-21 Proteins 0.000 claims description 12
- 108010065805 Interleukin-12 Proteins 0.000 claims description 11
- 230000028996 humoral immune response Effects 0.000 claims description 11
- 239000013610 patient sample Substances 0.000 claims description 10
- 102000029797 Prion Human genes 0.000 claims description 6
- 108091000054 Prion Proteins 0.000 claims description 6
- 230000001580 bacterial effect Effects 0.000 claims description 6
- 239000001963 growth medium Substances 0.000 claims description 6
- 239000000523 sample Substances 0.000 claims description 6
- 108010002586 Interleukin-7 Proteins 0.000 claims description 5
- 102000000704 Interleukin-7 Human genes 0.000 claims description 5
- 150000001875 compounds Chemical class 0.000 claims description 5
- 230000003071 parasitic effect Effects 0.000 claims description 5
- 108090001005 Interleukin-6 Proteins 0.000 claims description 3
- 230000002538 fungal effect Effects 0.000 claims description 3
- 210000004027 cell Anatomy 0.000 abstract description 123
- 230000004044 response Effects 0.000 abstract description 105
- 230000028993 immune response Effects 0.000 abstract description 59
- 238000000338 in vitro Methods 0.000 abstract description 19
- 230000001225 therapeutic effect Effects 0.000 abstract description 19
- 230000008901 benefit Effects 0.000 abstract description 15
- 230000005867 T cell response Effects 0.000 abstract description 12
- 238000002054 transplantation Methods 0.000 abstract description 9
- 230000004936 stimulating effect Effects 0.000 abstract description 5
- 230000001939 inductive effect Effects 0.000 abstract description 4
- 230000010001 cellular homeostasis Effects 0.000 abstract description 3
- 230000006058 immune tolerance Effects 0.000 abstract 1
- 238000011282 treatment Methods 0.000 description 52
- 208000006454 hepatitis Diseases 0.000 description 41
- 238000003556 assay Methods 0.000 description 30
- 231100000283 hepatitis Toxicity 0.000 description 29
- 101001136981 Homo sapiens Proteasome subunit beta type-9 Proteins 0.000 description 28
- 102100035764 Proteasome subunit beta type-9 Human genes 0.000 description 28
- 201000011510 cancer Diseases 0.000 description 26
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 26
- 210000003289 regulatory T cell Anatomy 0.000 description 26
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 23
- 201000010099 disease Diseases 0.000 description 22
- 210000002540 macrophage Anatomy 0.000 description 21
- 230000030741 antigen processing and presentation Effects 0.000 description 20
- 208000015181 infectious disease Diseases 0.000 description 19
- 206010025323 Lymphomas Diseases 0.000 description 18
- 210000002950 fibroblast Anatomy 0.000 description 18
- 210000000056 organ Anatomy 0.000 description 16
- 108010031111 EBV-encoded nuclear antigen 1 Proteins 0.000 description 15
- 230000005784 autoimmunity Effects 0.000 description 15
- 230000012010 growth Effects 0.000 description 15
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 15
- 210000000987 immune system Anatomy 0.000 description 15
- 229940028885 interleukin-4 Drugs 0.000 description 15
- 208000023275 Autoimmune disease Diseases 0.000 description 14
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 14
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 14
- 210000003719 b-lymphocyte Anatomy 0.000 description 14
- 239000012894 fetal calf serum Substances 0.000 description 14
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 14
- 241001465754 Metazoa Species 0.000 description 13
- 239000011651 chromium Substances 0.000 description 12
- 239000012636 effector Substances 0.000 description 12
- 235000018102 proteins Nutrition 0.000 description 12
- 102000004169 proteins and genes Human genes 0.000 description 12
- 108090000623 proteins and genes Proteins 0.000 description 12
- 230000004083 survival effect Effects 0.000 description 12
- 241000700605 Viruses Species 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 11
- 239000008280 blood Substances 0.000 description 11
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 11
- 101710132601 Capsid protein Proteins 0.000 description 10
- 101150113776 LMP1 gene Proteins 0.000 description 10
- 241000699670 Mus sp. Species 0.000 description 10
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 10
- 238000002659 cell therapy Methods 0.000 description 10
- 230000001684 chronic effect Effects 0.000 description 10
- 210000001616 monocyte Anatomy 0.000 description 10
- 210000001519 tissue Anatomy 0.000 description 10
- 206010008909 Chronic Hepatitis Diseases 0.000 description 9
- 238000002965 ELISA Methods 0.000 description 9
- 102000003814 Interleukin-10 Human genes 0.000 description 9
- 208000002672 hepatitis B Diseases 0.000 description 9
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 9
- 238000004519 manufacturing process Methods 0.000 description 9
- 239000000203 mixture Substances 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 102000003390 tumor necrosis factor Human genes 0.000 description 9
- 102000000440 Melanoma-associated antigen Human genes 0.000 description 8
- 108050008953 Melanoma-associated antigen Proteins 0.000 description 8
- 208000037581 Persistent Infection Diseases 0.000 description 8
- 238000001574 biopsy Methods 0.000 description 8
- 230000010261 cell growth Effects 0.000 description 8
- 238000001802 infusion Methods 0.000 description 8
- 238000007918 intramuscular administration Methods 0.000 description 8
- 238000012737 microarray-based gene expression Methods 0.000 description 8
- 238000012243 multiplex automated genomic engineering Methods 0.000 description 8
- 210000002966 serum Anatomy 0.000 description 8
- 230000000638 stimulation Effects 0.000 description 8
- 208000009329 Graft vs Host Disease Diseases 0.000 description 7
- 206010061218 Inflammation Diseases 0.000 description 7
- 241000699666 Mus <mouse, genus> Species 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 230000001413 cellular effect Effects 0.000 description 7
- 208000024908 graft versus host disease Diseases 0.000 description 7
- 230000004054 inflammatory process Effects 0.000 description 7
- 239000012528 membrane Substances 0.000 description 7
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 description 6
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 description 6
- 241000700159 Rattus Species 0.000 description 6
- 210000000662 T-lymphocyte subset Anatomy 0.000 description 6
- 230000001464 adherent effect Effects 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 230000001363 autoimmune Effects 0.000 description 6
- 238000004113 cell culture Methods 0.000 description 6
- 238000012217 deletion Methods 0.000 description 6
- 230000037430 deletion Effects 0.000 description 6
- 210000002443 helper t lymphocyte Anatomy 0.000 description 6
- 238000005259 measurement Methods 0.000 description 6
- 230000007246 mechanism Effects 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 230000008685 targeting Effects 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 101710145634 Antigen 1 Proteins 0.000 description 5
- 238000011510 Elispot assay Methods 0.000 description 5
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 5
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 5
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 5
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 5
- 241000725303 Human immunodeficiency virus Species 0.000 description 5
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 description 5
- 206010027476 Metastases Diseases 0.000 description 5
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 description 5
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 description 5
- -1 but not limited to Proteins 0.000 description 5
- 230000036755 cellular response Effects 0.000 description 5
- 230000004069 differentiation Effects 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 238000011534 incubation Methods 0.000 description 5
- 238000010172 mouse model Methods 0.000 description 5
- 230000010287 polarization Effects 0.000 description 5
- 238000012545 processing Methods 0.000 description 5
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 5
- 229960002930 sirolimus Drugs 0.000 description 5
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 5
- 210000004988 splenocyte Anatomy 0.000 description 5
- 230000009885 systemic effect Effects 0.000 description 5
- 208000035473 Communicable disease Diseases 0.000 description 4
- 108010069621 Epstein-Barr virus EBV-associated membrane antigen Proteins 0.000 description 4
- 102100028972 HLA class I histocompatibility antigen, A alpha chain Human genes 0.000 description 4
- 108010075704 HLA-A Antigens Proteins 0.000 description 4
- 206010020751 Hypersensitivity Diseases 0.000 description 4
- 108060003951 Immunoglobulin Proteins 0.000 description 4
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 4
- 206010052779 Transplant rejections Diseases 0.000 description 4
- 206010046865 Vaccinia virus infection Diseases 0.000 description 4
- 208000026935 allergic disease Diseases 0.000 description 4
- 230000007815 allergy Effects 0.000 description 4
- 230000003321 amplification Effects 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- 239000011324 bead Substances 0.000 description 4
- 229960001467 bortezomib Drugs 0.000 description 4
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 4
- 238000002512 chemotherapy Methods 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 208000035475 disorder Diseases 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 230000003325 follicular Effects 0.000 description 4
- 230000001900 immune effect Effects 0.000 description 4
- 238000011493 immune profiling Methods 0.000 description 4
- 102000018358 immunoglobulin Human genes 0.000 description 4
- 238000003364 immunohistochemistry Methods 0.000 description 4
- 239000012678 infectious agent Substances 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 210000001503 joint Anatomy 0.000 description 4
- 238000003199 nucleic acid amplification method Methods 0.000 description 4
- 230000002265 prevention Effects 0.000 description 4
- 206010039073 rheumatoid arthritis Diseases 0.000 description 4
- 208000007089 vaccinia Diseases 0.000 description 4
- 102100036475 Alanine aminotransferase 1 Human genes 0.000 description 3
- 108010082126 Alanine transaminase Proteins 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- 101100455063 Caenorhabditis elegans lmp-1 gene Proteins 0.000 description 3
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 3
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 3
- 108020004414 DNA Proteins 0.000 description 3
- 101710122231 Epstein-Barr nuclear antigen 3 Proteins 0.000 description 3
- 206010015150 Erythema Diseases 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 208000005176 Hepatitis C Diseases 0.000 description 3
- 241000598436 Human T-cell lymphotropic virus Species 0.000 description 3
- 208000002454 Nasopharyngeal Carcinoma Diseases 0.000 description 3
- 239000012980 RPMI-1640 medium Substances 0.000 description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 description 3
- 108091008874 T cell receptors Proteins 0.000 description 3
- 231100000354 acute hepatitis Toxicity 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 206010003246 arthritis Diseases 0.000 description 3
- 230000007969 cellular immunity Effects 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 229910052804 chromium Inorganic materials 0.000 description 3
- 238000002591 computed tomography Methods 0.000 description 3
- 239000003636 conditioned culture medium Substances 0.000 description 3
- 230000000875 corresponding effect Effects 0.000 description 3
- 229960004397 cyclophosphamide Drugs 0.000 description 3
- 230000008030 elimination Effects 0.000 description 3
- 238000003379 elimination reaction Methods 0.000 description 3
- 239000003102 growth factor Substances 0.000 description 3
- 230000003053 immunization Effects 0.000 description 3
- 238000002649 immunization Methods 0.000 description 3
- 238000003018 immunoassay Methods 0.000 description 3
- 206010022000 influenza Diseases 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000007912 intraperitoneal administration Methods 0.000 description 3
- 210000000265 leukocyte Anatomy 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 230000035800 maturation Effects 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004962 physiological condition Effects 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 229960004641 rituximab Drugs 0.000 description 3
- 238000010186 staining Methods 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 230000008961 swelling Effects 0.000 description 3
- 238000011830 transgenic mouse model Methods 0.000 description 3
- QRXMUCSWCMTJGU-UHFFFAOYSA-L (5-bromo-4-chloro-1h-indol-3-yl) phosphate Chemical compound C1=C(Br)C(Cl)=C2C(OP([O-])(=O)[O-])=CNC2=C1 QRXMUCSWCMTJGU-UHFFFAOYSA-L 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 2
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 2
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 2
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 2
- 238000011740 C57BL/6 mouse Methods 0.000 description 2
- 241000282461 Canis lupus Species 0.000 description 2
- 208000000419 Chronic Hepatitis B Diseases 0.000 description 2
- 102000008186 Collagen Human genes 0.000 description 2
- 108010035532 Collagen Proteins 0.000 description 2
- 102000018832 Cytochromes Human genes 0.000 description 2
- 108010052832 Cytochromes Proteins 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- 206010015108 Epstein-Barr virus infection Diseases 0.000 description 2
- 241000233866 Fungi Species 0.000 description 2
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 2
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 2
- 101000883515 Homo sapiens Chitinase-3-like protein 1 Proteins 0.000 description 2
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 2
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 2
- 102000008070 Interferon-gamma Human genes 0.000 description 2
- 108010074328 Interferon-gamma Proteins 0.000 description 2
- 102000013691 Interleukin-17 Human genes 0.000 description 2
- 208000032420 Latent Infection Diseases 0.000 description 2
- 108010066345 MHC binding peptide Proteins 0.000 description 2
- 102100022430 Melanocyte protein PMEL Human genes 0.000 description 2
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 2
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 2
- 108010004729 Phycoerythrin Proteins 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 2
- VFWQQZMRKFOGLE-ZLUOBGJFSA-N Ser-Ser-Cys Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CS)C(=O)O)N)O VFWQQZMRKFOGLE-ZLUOBGJFSA-N 0.000 description 2
- 108010090804 Streptavidin Proteins 0.000 description 2
- 101800001271 Surface protein Proteins 0.000 description 2
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 2
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 2
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 2
- 102000003425 Tyrosinase Human genes 0.000 description 2
- 108060008724 Tyrosinase Proteins 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 108010004469 allophycocyanin Proteins 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 230000000890 antigenic effect Effects 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 210000001124 body fluid Anatomy 0.000 description 2
- 239000010839 body fluid Substances 0.000 description 2
- 230000000973 chemotherapeutic effect Effects 0.000 description 2
- 229920001436 collagen Polymers 0.000 description 2
- 230000002596 correlated effect Effects 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 231100000321 erythema Toxicity 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 239000012737 fresh medium Substances 0.000 description 2
- 102000054350 human CHI3L1 Human genes 0.000 description 2
- 230000004727 humoral immunity Effects 0.000 description 2
- 210000002865 immune cell Anatomy 0.000 description 2
- 230000002998 immunogenetic effect Effects 0.000 description 2
- 230000016784 immunoglobulin production Effects 0.000 description 2
- 230000004957 immunoregulator effect Effects 0.000 description 2
- 238000009169 immunotherapy Methods 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 229960003130 interferon gamma Drugs 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 201000001441 melanoma Diseases 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 201000011216 nasopharynx carcinoma Diseases 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 230000002085 persistent effect Effects 0.000 description 2
- 238000007747 plating Methods 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 238000001959 radiotherapy Methods 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 210000000130 stem cell Anatomy 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 2
- 208000027930 type IV hypersensitivity disease Diseases 0.000 description 2
- 241000701161 unidentified adenovirus Species 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- 239000013603 viral vector Substances 0.000 description 2
- YTXSYWAKVMZICI-PVCZSOGJSA-N 4-(carboxymethyl)-2-[(1r)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-6-oxo-1,3,2-dioxaborinane-4-carboxylic acid Chemical compound N([C@@H](CC(C)C)B1OC(CC(O)=O)(CC(=O)O1)C(O)=O)C(=O)CNC(=O)C1=CC(Cl)=CC=C1Cl YTXSYWAKVMZICI-PVCZSOGJSA-N 0.000 description 1
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 1
- UWIQWPWWZUHBAO-ZLIFDBKOSA-N Ala-Leu-Trp Chemical compound C1=CC=C2C(C[C@H](NC(=O)[C@@H](NC(=O)[C@H](C)N)CC(C)C)C(O)=O)=CNC2=C1 UWIQWPWWZUHBAO-ZLIFDBKOSA-N 0.000 description 1
- VNFSAYFQLXPHPY-CIQUZCHMSA-N Ala-Thr-Ile Chemical compound [H]N[C@@H](C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O VNFSAYFQLXPHPY-CIQUZCHMSA-N 0.000 description 1
- YBZMTKUDWXZLIX-UWVGGRQHSA-N Arg-Leu-Gly Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)NCC(O)=O YBZMTKUDWXZLIX-UWVGGRQHSA-N 0.000 description 1
- MKJBPDLENBUHQU-CIUDSAMLSA-N Asn-Ser-Leu Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(O)=O MKJBPDLENBUHQU-CIUDSAMLSA-N 0.000 description 1
- 230000003844 B-cell-activation Effects 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 208000011691 Burkitt lymphomas Diseases 0.000 description 1
- 102100031658 C-X-C chemokine receptor type 5 Human genes 0.000 description 1
- 102100035793 CD83 antigen Human genes 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 241001227713 Chiron Species 0.000 description 1
- QCDFBFJGMNKBDO-UHFFFAOYSA-N Clioquinol Chemical compound C1=CN=C2C(O)=C(I)C=C(Cl)C2=C1 QCDFBFJGMNKBDO-UHFFFAOYSA-N 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 102000000503 Collagen Type II Human genes 0.000 description 1
- 108010041390 Collagen Type II Proteins 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 206010056370 Congestive cardiomyopathy Diseases 0.000 description 1
- 108010041986 DNA Vaccines Proteins 0.000 description 1
- 229940021995 DNA vaccine Drugs 0.000 description 1
- 201000010046 Dilated cardiomyopathy Diseases 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 208000009386 Experimental Arthritis Diseases 0.000 description 1
- DXMPMSWUZVNBSG-QEJZJMRPSA-N Gln-Asn-Trp Chemical compound C1=CC=C2C(=C1)C(=CN2)C[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CCC(=O)N)N DXMPMSWUZVNBSG-QEJZJMRPSA-N 0.000 description 1
- NNCSJUBVFBDDLC-YUMQZZPRSA-N Gly-Leu-Ser Chemical compound NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(O)=O NNCSJUBVFBDDLC-YUMQZZPRSA-N 0.000 description 1
- VPZXBVLAVMBEQI-VKHMYHEASA-N Glycyl-alanine Chemical group OC(=O)[C@H](C)NC(=O)CN VPZXBVLAVMBEQI-VKHMYHEASA-N 0.000 description 1
- 206010018691 Granuloma Diseases 0.000 description 1
- 101000691214 Haloarcula marismortui (strain ATCC 43049 / DSM 3752 / JCM 8966 / VKM B-1809) 50S ribosomal protein L44e Proteins 0.000 description 1
- 241000711549 Hepacivirus C Species 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000937544 Homo sapiens Beta-2-microglobulin Proteins 0.000 description 1
- 101000922405 Homo sapiens C-X-C chemokine receptor type 5 Proteins 0.000 description 1
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 description 1
- 101000599852 Homo sapiens Intercellular adhesion molecule 1 Proteins 0.000 description 1
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 1
- 101150106931 IFNG gene Proteins 0.000 description 1
- KIMHKBDJQQYLHU-PEFMBERDSA-N Ile-Glu-Asp Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC(=O)O)C(=O)O)N KIMHKBDJQQYLHU-PEFMBERDSA-N 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 1
- 206010023232 Joint swelling Diseases 0.000 description 1
- RCFDOSNHHZGBOY-UHFFFAOYSA-N L-isoleucyl-L-alanine Natural products CCC(C)C(N)C(=O)NC(C)C(O)=O RCFDOSNHHZGBOY-UHFFFAOYSA-N 0.000 description 1
- LZDNBBYBDGBADK-UHFFFAOYSA-N L-valyl-L-tryptophan Natural products C1=CC=C2C(CC(NC(=O)C(N)C(C)C)C(O)=O)=CNC2=C1 LZDNBBYBDGBADK-UHFFFAOYSA-N 0.000 description 1
- DAQAKHDKYAWHCG-UHFFFAOYSA-N Lactacystin Natural products CC(=O)NC(C(O)=O)CSC(=O)C1(C(O)C(C)C)NC(=O)C(C)C1O DAQAKHDKYAWHCG-UHFFFAOYSA-N 0.000 description 1
- 241000880493 Leptailurus serval Species 0.000 description 1
- KAFOIVJDVSZUMD-UHFFFAOYSA-N Leu-Gln-Gln Natural products CC(C)CC(N)C(=O)NC(CCC(N)=O)C(=O)NC(CCC(N)=O)C(O)=O KAFOIVJDVSZUMD-UHFFFAOYSA-N 0.000 description 1
- FAELBUXXFQLUAX-AJNGGQMLSA-N Leu-Leu-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CC(C)C FAELBUXXFQLUAX-AJNGGQMLSA-N 0.000 description 1
- DNDWZFHLZVYOGF-KKUMJFAQSA-N Leu-Leu-Leu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O DNDWZFHLZVYOGF-KKUMJFAQSA-N 0.000 description 1
- RXGLHDWAZQECBI-SRVKXCTJSA-N Leu-Leu-Ser Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(O)=O RXGLHDWAZQECBI-SRVKXCTJSA-N 0.000 description 1
- SBANPBVRHYIMRR-UHFFFAOYSA-N Leu-Ser-Pro Natural products CC(C)CC(N)C(=O)NC(CO)C(=O)N1CCCC1C(O)=O SBANPBVRHYIMRR-UHFFFAOYSA-N 0.000 description 1
- ZJZNLRVCZWUONM-JXUBOQSCSA-N Leu-Thr-Ala Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C)C(O)=O ZJZNLRVCZWUONM-JXUBOQSCSA-N 0.000 description 1
- 102000016200 MART-1 Antigen Human genes 0.000 description 1
- 108010010995 MART-1 Antigen Proteins 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- PTYVBBNIAQWUFV-DCAQKATOSA-N Met-Cys-Leu Chemical compound CC(C)C[C@@H](C(=O)O)NC(=O)[C@H](CS)NC(=O)[C@H](CCSC)N PTYVBBNIAQWUFV-DCAQKATOSA-N 0.000 description 1
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 1
- 241000204031 Mycoplasma Species 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- 201000002481 Myositis Diseases 0.000 description 1
- TZYWCYJVHRLUCT-VABKMULXSA-N N-benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal Chemical compound CC(C)C[C@@H](C=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)OCC1=CC=CC=C1 TZYWCYJVHRLUCT-VABKMULXSA-N 0.000 description 1
- KZNQNBZMBZJQJO-UHFFFAOYSA-N N-glycyl-L-proline Natural products NCC(=O)N1CCCC1C(O)=O KZNQNBZMBZJQJO-UHFFFAOYSA-N 0.000 description 1
- 108010066427 N-valyltryptophan Proteins 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 108010047620 Phytohemagglutinins Proteins 0.000 description 1
- 241000709664 Picornaviridae Species 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- FKYKZHOKDOPHSA-DCAQKATOSA-N Pro-Leu-Ser Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(O)=O FKYKZHOKDOPHSA-DCAQKATOSA-N 0.000 description 1
- NAIPAPCKKRCMBL-JYJNAYRXSA-N Pro-Pro-Phe Chemical compound C([C@@H](C(=O)O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H]1NCCC1)C1=CC=CC=C1 NAIPAPCKKRCMBL-JYJNAYRXSA-N 0.000 description 1
- CXGLFEOYCJFKPR-RCWTZXSCSA-N Pro-Thr-Val Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(O)=O CXGLFEOYCJFKPR-RCWTZXSCSA-N 0.000 description 1
- VDHGTOHMHHQSKG-JYJNAYRXSA-N Pro-Val-Phe Chemical compound CC(C)[C@H](NC(=O)[C@@H]1CCCN1)C(=O)N[C@@H](Cc1ccccc1)C(O)=O VDHGTOHMHHQSKG-JYJNAYRXSA-N 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 102000001183 RAG-1 Human genes 0.000 description 1
- 108060006897 RAG1 Proteins 0.000 description 1
- 239000012979 RPMI medium Substances 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- MFQMZDPAZRZAPV-NAKRPEOUSA-N Ser-Val-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CO)N MFQMZDPAZRZAPV-NAKRPEOUSA-N 0.000 description 1
- 108010008038 Synthetic Vaccines Proteins 0.000 description 1
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 210000000447 Th1 cell Anatomy 0.000 description 1
- PELIQFPESHBTMA-WLTAIBSBSA-N Thr-Tyr-Gly Chemical compound C[C@@H](O)[C@H](N)C(=O)N[C@H](C(=O)NCC(O)=O)CC1=CC=C(O)C=C1 PELIQFPESHBTMA-WLTAIBSBSA-N 0.000 description 1
- MDDYTWOFHZFABW-SZMVWBNQSA-N Trp-Gln-Leu Chemical compound C1=CC=C2C(C[C@H](N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(O)=O)=CNC2=C1 MDDYTWOFHZFABW-SZMVWBNQSA-N 0.000 description 1
- RWAYYYOZMHMEGD-XIRDDKMYSA-N Trp-Leu-Ser Chemical compound C1=CC=C2C(C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(O)=O)=CNC2=C1 RWAYYYOZMHMEGD-XIRDDKMYSA-N 0.000 description 1
- HTGJDTPQYFMKNC-VFAJRCTISA-N Trp-Thr-Leu Chemical compound C1=CC=C2C(C[C@H](N)C(=O)N[C@H](C(=O)N[C@@H](CC(C)C)C(O)=O)[C@@H](C)O)=CNC2=C1 HTGJDTPQYFMKNC-VFAJRCTISA-N 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- NKUGCYDFQKFVOJ-JYJNAYRXSA-N Tyr-Leu-Gln Chemical compound NC(=O)CC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 NKUGCYDFQKFVOJ-JYJNAYRXSA-N 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- ZLMFVXMJFIWIRE-FHWLQOOXSA-N Val-Trp-Leu Chemical compound CC(C)C[C@@H](C(=O)O)NC(=O)[C@H](CC1=CNC2=CC=CC=C21)NC(=O)[C@H](C(C)C)N ZLMFVXMJFIWIRE-FHWLQOOXSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 208000038016 acute inflammation Diseases 0.000 description 1
- 230000006022 acute inflammation Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 238000011467 adoptive cell therapy Methods 0.000 description 1
- 230000009824 affinity maturation Effects 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000000735 allogeneic effect Effects 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 230000031016 anaphase Effects 0.000 description 1
- 210000003423 ankle Anatomy 0.000 description 1
- 108010043240 arginyl-leucyl-glycine Proteins 0.000 description 1
- 230000002917 arthritic effect Effects 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 230000006472 autoimmune response Effects 0.000 description 1
- 239000003124 biologic agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 201000000053 blastoma Diseases 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- KQNZDYYTLMIZCT-KQPMLPITSA-N brefeldin A Chemical compound O[C@@H]1\C=C\C(=O)O[C@@H](C)CCC\C=C\[C@@H]2C[C@H](O)C[C@H]21 KQNZDYYTLMIZCT-KQPMLPITSA-N 0.000 description 1
- JUMGSHROWPPKFX-UHFFFAOYSA-N brefeldin-A Natural products CC1CCCC=CC2(C)CC(O)CC2(C)C(O)C=CC(=O)O1 JUMGSHROWPPKFX-UHFFFAOYSA-N 0.000 description 1
- 230000000981 bystander Effects 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- BLMPQMFVWMYDKT-NZTKNTHTSA-N carfilzomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)[C@]1(C)OC1)NC(=O)CN1CCOCC1)CC1=CC=CC=C1 BLMPQMFVWMYDKT-NZTKNTHTSA-N 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 210000000038 chest Anatomy 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 238000003759 clinical diagnosis Methods 0.000 description 1
- 229960005228 clioquinol Drugs 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 238000009096 combination chemotherapy Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 230000000139 costimulatory effect Effects 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 108010060199 cysteinylproline Proteins 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- JXSJBGJIGXNWCI-UHFFFAOYSA-N diethyl 2-[(dimethoxyphosphorothioyl)thio]succinate Chemical compound CCOC(=O)CC(SP(=S)(OC)OC)C(=O)OCC JXSJBGJIGXNWCI-UHFFFAOYSA-N 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 208000037771 disease arising from reactivation of latent virus Diseases 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 201000008184 embryoma Diseases 0.000 description 1
- 206010014599 encephalitis Diseases 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 210000003414 extremity Anatomy 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 230000030279 gene silencing Effects 0.000 description 1
- 102000054766 genetic haplotypes Human genes 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 108010013768 glutamyl-aspartyl-proline Proteins 0.000 description 1
- VPZXBVLAVMBEQI-UHFFFAOYSA-N glycyl-DL-alpha-alanine Natural products OC(=O)C(C)NC(=O)CN VPZXBVLAVMBEQI-UHFFFAOYSA-N 0.000 description 1
- 108010077515 glycylproline Proteins 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 102000047279 human B2M Human genes 0.000 description 1
- 230000008348 humoral response Effects 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 230000007124 immune defense Effects 0.000 description 1
- 230000003832 immune regulation Effects 0.000 description 1
- 238000003125 immunofluorescent labeling Methods 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 239000003547 immunosorbent Substances 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 201000006747 infectious mononucleosis Diseases 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000003960 inflammatory cascade Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 238000010212 intracellular staining Methods 0.000 description 1
- 229960003648 ixazomib Drugs 0.000 description 1
- 210000001865 kupffer cell Anatomy 0.000 description 1
- 238000009533 lab test Methods 0.000 description 1
- DAQAKHDKYAWHCG-RWTHQLGUSA-N lactacystin Chemical compound CC(=O)N[C@H](C(O)=O)CSC(=O)[C@]1([C@@H](O)C(C)C)NC(=O)[C@H](C)[C@@H]1O DAQAKHDKYAWHCG-RWTHQLGUSA-N 0.000 description 1
- 210000001821 langerhans cell Anatomy 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 238000007449 liver function test Methods 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 230000003446 memory effect Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 208000021039 metastatic melanoma Diseases 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 210000000274 microglia Anatomy 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 238000004264 monolayer culture Methods 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- JPXMTWWFLBLUCD-UHFFFAOYSA-N nitro blue tetrazolium(2+) Chemical compound COC1=CC(C=2C=C(OC)C(=CC=2)[N+]=2N(N=C(N=2)C=2C=CC=CC=2)C=2C=CC(=CC=2)[N+]([O-])=O)=CC=C1[N+]1=NC(C=2C=CC=CC=2)=NN1C1=CC=C([N+]([O-])=O)C=C1 JPXMTWWFLBLUCD-UHFFFAOYSA-N 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 201000005737 orchitis Diseases 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000007119 pathological manifestation Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 210000000680 phagosome Anatomy 0.000 description 1
- 230000001885 phytohemagglutinin Effects 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 229940034080 provenge Drugs 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000036647 reaction Effects 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 229940124551 recombinant vaccine Drugs 0.000 description 1
- 230000003252 repetitive effect Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000007390 skin biopsy Methods 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- YEENEYXBHNNNGV-XEHWZWQGSA-M sodium;3-acetamido-5-[acetyl(methyl)amino]-2,4,6-triiodobenzoate;(2r,3r,4s,5s,6r)-2-[(2r,3s,4s,5r)-3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound [Na+].CC(=O)N(C)C1=C(I)C(NC(C)=O)=C(I)C(C([O-])=O)=C1I.O[C@H]1[C@H](O)[C@@H](CO)O[C@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 YEENEYXBHNNNGV-XEHWZWQGSA-M 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 238000005728 strengthening Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 229940061353 temodar Drugs 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 230000009258 tissue cross reactivity Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 108010045269 tryptophyltryptophan Proteins 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 230000010463 virion release Effects 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 210000000707 wrist Anatomy 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
Images












Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/14—Blood; Artificial blood
- A61K35/17—Lymphocytes; B-cells; T-cells; Natural killer cells; Interferon-activated or cytokine-activated lymphocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0008—Antigens related to auto-immune diseases; Preparations to induce self-tolerance
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A61K39/245—Herpetoviridae, e.g. herpes simplex virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A61K39/29—Hepatitis virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A61K39/29—Hepatitis virus
- A61K39/292—Serum hepatitis virus, hepatitis B virus, e.g. Australia antigen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/461—Cellular immunotherapy characterised by the cell type used
- A61K39/4611—T-cells, e.g. tumor infiltrating lymphocytes [TIL], lymphokine-activated killer cells [LAK] or regulatory T cells [Treg]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/461—Cellular immunotherapy characterised by the cell type used
- A61K39/4615—Dendritic cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/462—Cellular immunotherapy characterized by the effect or the function of the cells
- A61K39/4621—Cellular immunotherapy characterized by the effect or the function of the cells immunosuppressive or immunotolerising
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/462—Cellular immunotherapy characterized by the effect or the function of the cells
- A61K39/4622—Antigen presenting cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/46433—Antigens related to auto-immune diseases; Preparations to induce self-tolerance
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464484—Cancer testis antigens, e.g. SSX, BAGE, GAGE or SAGE
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464484—Cancer testis antigens, e.g. SSX, BAGE, GAGE or SAGE
- A61K39/464486—MAGE
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464484—Cancer testis antigens, e.g. SSX, BAGE, GAGE or SAGE
- A61K39/464488—NY-ESO
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/46449—Melanoma antigens
- A61K39/464491—Melan-A/MART
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/464838—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0021—Intradermal administration, e.g. through microneedle arrays, needleless injectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0613—Cells from endocrine organs
- C12N5/0617—Thyroid and parathyroid glands
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K2035/124—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells the cells being hematopoietic, bone marrow derived or blood cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/515—Animal cells
- A61K2039/5158—Antigen-pulsed cells, e.g. T-cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55566—Emulsions, e.g. Freund's adjuvant, MF59
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/58—Medicinal preparations containing antigens or antibodies raising an immune response against a target which is not the antigen used for immunisation
- A61K2039/585—Medicinal preparations containing antigens or antibodies raising an immune response against a target which is not the antigen used for immunisation wherein the target is cancer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/31—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterized by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/38—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the dose, timing or administration schedule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/48—Blood cells, e.g. leukemia or lymphoma
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/53—Liver
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2302—Interleukin-2 (IL-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2304—Interleukin-4 (IL-4)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2306—Interleukin-6 (IL-6)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2307—Interleukin-7 (IL-7)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2312—Interleukin-12 (IL-12)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2315—Interleukin-15 (IL-15)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2321—Interleukin-21 (IL-21)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/999—Small molecules not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2502/00—Coculture with; Conditioned medium produced by
- C12N2502/11—Coculture with; Conditioned medium produced by blood or immune system cells
- C12N2502/1121—Dendritic cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2502/00—Coculture with; Conditioned medium produced by
- C12N2502/11—Coculture with; Conditioned medium produced by blood or immune system cells
- C12N2502/1157—Monocytes, macrophages
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/16011—Herpesviridae
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/16011—Herpesviridae
- C12N2710/16034—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/16011—Herpesviridae
- C12N2710/16211—Lymphocryptovirus, e.g. human herpesvirus 4, Epstein-Barr Virus
- C12N2710/16234—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2730/00—Reverse transcribing DNA viruses
- C12N2730/00011—Details
- C12N2730/10011—Hepadnaviridae
- C12N2730/10111—Orthohepadnavirus, e.g. hepatitis B virus
- C12N2730/10134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2730/00—Reverse transcribing DNA viruses
- C12N2730/00011—Details
- C12N2730/10011—Hepadnaviridae
- C12N2730/10111—Orthohepadnavirus, e.g. hepatitis B virus
- C12N2730/10171—Demonstrated in vivo effect
Abstract
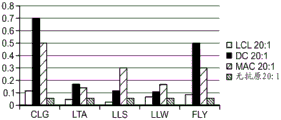
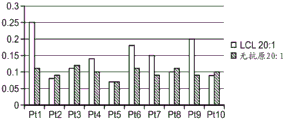
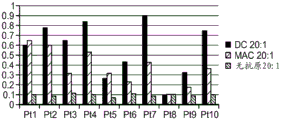
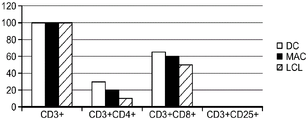
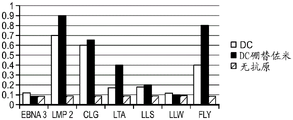
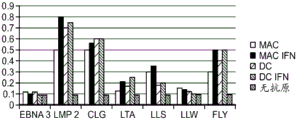
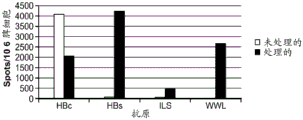
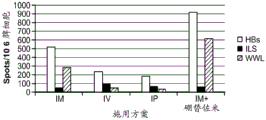
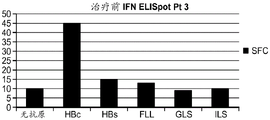
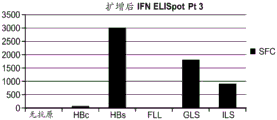
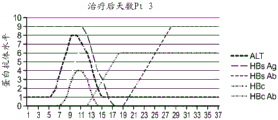
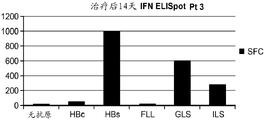
本发明涉及产生针对亚优势抗原的T细胞应答和使用细胞来治疗性改变细胞稳态和免疫应答的性质。在优选的实施方案中,细胞在患者体外产生,以避免患者的免疫环境的影响。通过在组织培养中将来自患者的T细胞针对一种或多种亚优势抗原刺激和培养,并将它们移植入患者中,如果扩增并移植了足够的细胞,则在破坏或诱导免疫耐受或另外修饰针对表达该抗原的细胞或生物的免疫应答的应答中,移植的细胞压倒了内源优势T细胞。当建立记忆细胞时,它们随后反映了该新的免疫优势等级,从而使所需的治疗效果长期持续。对亚优势抗原具有反应性的外源产生的T细胞移植重现引发并重新平衡患者的靶向细胞或生物中之前亚优势抗原的免疫应答,以产生治疗益处。
Description
本申请是申请日为2012年5月25日,标题为“调节的免疫优势疗法”的中国专利申请201280037470.4的分案申请。
本申请涉及在2011年5月26日提交的美国临时申请序号61/490,505,其内容通过引用整体并入本文。
技术领域
本发明涉及基于免疫优势等级(hierarch)的修饰通过细胞的稳态的治疗性操作,用于癌症、慢性感染、自体免疫疾病和的移植的新疗法。
背景技术
癌症的临床管理对于各种起源位置是特异性的,并且最重要的是依赖于疾病的阶段(即,肿瘤已经在何种程度上局部侵袭或通过转移传播至其他器官)。通常,手术和/或局部放疗是用于原发肿瘤的治疗选择,而化疗、单克隆抗体或细胞因子疗法或全身照射是用于转移疾病的治疗。最近,首个树突细胞疗法,Provenge,被批准用于前列腺癌,其具有延缓进展4个月的益处。诊断仍基于活组织检查的组织学分析。分子标志物有时是标准,如果它们将有助于药物(例如,赫赛汀)的选择。然而,尚未在常规临床基础上完成免疫应答概况的生成。
在初次免疫应答期间,T细胞看到抗原的环境决定了后续回忆应答的性质。初始识别事件和该初级细胞周边的微环境可以产生很多结果。如果抗原通过非专业APC呈递,在加工期间仅可释放表位的子集。如果共刺激信号,例如CD28或TNF丢失,则T细胞可能不做出反应。取决于微环境,T细胞可分化成调节性T细胞、分泌Th1细胞因子的T辅助细胞(驱动更多的具有CD8+ CTL效应物的增殖的细胞免疫应答),或分泌Th2细胞因子的T辅助细胞(驱动更多的具有B细胞的增殖和成熟以及抗体产生的体液免疫应答)。此外,免疫应答将发展,从而使对环境中的抗原的某些表位或某些抗原做出应答的T细胞生长,这是以牺牲群体中对环境中的其他表位或抗原为反应性的T细胞为代价的。因为指数型细胞生长,随着初次免疫应答的减退,这些T细胞的比例进一步增强并以记忆的形式贮存,使得在二次刺激后,个体内的免疫应答集中在可能表位的小子集上。尽管有多种机制发挥作用,作为有效的模型,生长成在群体中占优势的T细胞是对成为支配免疫应答的表位或抗原做出应答的那些。在初次免疫应答中,它们以牺牲应答亚优势表位的T细胞为代价进行生长,并且,由于记忆作用而支配后续的免疫应答。
在个体的免疫系统首次发现抗原后数天的时期中,产生出应答有限数量的优势表位的T细胞的优势群体,并且这些T细胞决定了此后对该抗原的应答的性质。尽管有多种类型的细胞参与,但与本发明相关的工作模型是,如果对优势抗原上的优势表位做出应答的T细胞生长成为反应性T细胞(例如,CD4+ :TH1、TH2、Treg、滤泡T辅助细胞、TH17、TH22、TH9;CD8+ CTL),则产生细胞或体液免疫应答。然而,如果在优势群体中的T细胞是抑制性T细胞(例如,Treg、TH17、无反应T细胞),则诱导耐受。对亚优势抗原做出应答的T细胞被对优势抗原做出应答的T细胞的克隆群体淹没。
在癌症、慢性或潜伏感染的情况下,局部抗原加工/呈递和共刺激环境影响对优势抗原的初次免疫应答,使得对肿瘤或传染物中的优势抗原的T细胞应答的平衡趋向于耐受或无效应答,而不是强的效应物应答。由于抗原加工和共刺激(CD28和细胞因子)中的差异,这会在其中树突细胞(DCs)不是普遍的抗原呈递细胞(APCs)(这不同于其中树突细胞是主要的抗原呈递细胞的身体表面)的器官中加重。还熟知,肿瘤和传染物产生的免疫抑制性环境对于强的初次免疫应答不是最佳的。或者,如果在细胞中产生的优势抗原对与其本身反应,则耐受性被破坏且继而发生自身免疫性。这样的耐受性可被存在的导致应答(对弱的亚优势抗原乃至是慢性的)的慢性或潜伏病毒破坏。病毒与自身免疫性有多重联系。在所述部位的炎症导致其他抗原的释放,同时病毒抗原为应答所述器官的T细胞提供帮助,导致自身免疫性。在初次应答中确立优势等级(hierarchy)且通过记忆强化后,患者中的免疫系统将在每次抗原存在时有效地复制相同的应答。
正在进行的针对优势表位的免疫应答能够减少对亚优势表位的应答(Wolpert EZ1998, Kedl RM 2003)。优势/亚优势等级在一定程度上是可改变的。例如,消除或沉默针对优势表位的T细胞应答,可以导致出现此前无法检测的针对亚优势表位的应答(VanderMost RG等人,1997, Andreansky SS等人,2005)。类似地,删除表位中的优势序列没有消除对所述抗原的应答,而是导致宿主对此前的亚优势表位的更强应答(Allan JE andDoherty PC 1985, Mylin LM等人,2000)。
发明内容
本发明涉及基于免疫优势等级的修饰通过细胞的稳态的治疗性操作,用于癌症、慢性感染、自体免疫疾病和移植的新疗法。
本发明公开了用于重新平衡对抗原的免疫应答的新方法,以特别是在癌症、慢性和潜伏感染、自身免疫性和移植中提供显著的治疗益处。通过对受控微环境中的亚优势表位和亚优势抗原产生免疫应答,本发明从根本上将对疾病的免疫应答的性质改变为提供治疗益处的那种。它能够在对抗原已经发生的在先免疫应答之前或之后,或者甚至在免疫应答正在进行的情况下,改变免疫应答的平衡。
本发明的特征在于一种方法,其包括鉴定在患者样品中的优势抗原或表位和亚优势抗原或表位,培养能够识别所述亚优势抗原或表位的T细胞,和使用有效数量的T细胞治疗患者以改变所述患者的免疫优势等级。
本发明的特征还在于改变患者的免疫优势等级的方法,其包括鉴定患者样品中的至少一种亚优势抗原或表位,培养能够识别所述亚优势抗原或表位的T细胞,和使用有效数量的那些T细胞治疗所述患者以提供治疗益处。
在一些方面,本发明进一步包括在不存在优势抗原或表位的情况下培养T细胞。在其他方面,本发明进一步包括在不存在或存在富集抑制性T细胞或应答性T细胞的试剂的情况下培养T细胞。此类试剂抗原可以,但不限于,生长因子、激素或其他免疫细胞。
在一些方面,本发明进一步包括通过皮内施用来施用有效量的T细胞。
在其他方面,本发明进一步包括在使用培养的T细胞治疗患者之前,使用调节剂预先治疗所述患者以减少内源T细胞的数量。所述调节剂可以是,但不限于化疗剂。
在一些方面,所述T细胞从患者先体外后体内(ex vivo)提供。
所述亚优势抗原或亚优势表位是,例如,针对该抗原或表位的细胞或体液免疫应答不可检测或仅低水平可测的抗原或表位。或者,所述亚优势抗原或亚优势表位是与优势抗原或优势表位相比,激发较弱的耐受或免疫应答的抗原或表位。所述亚优势抗原是,例如,病毒抗原、真菌抗原、细菌抗原、寄生虫抗原、朊病毒抗原、肿瘤抗原或与自身免疫性、变态反应、炎症、器官移植排斥或移植物抗宿主病相关的抗原。所述病毒抗原是,例如,慢性或潜伏病毒抗原。所述病毒抗原来自EBV、HPV、HSV、VZV、乙型肝炎、丙型肝炎、HIV、HTLV、CMV、RSV或流感。所述肿瘤抗原是,例如,肿瘤相关抗原、肿瘤特异性抗原,或与癌症干细胞或转移相关的抗原。
本发明的特征还在于一种方法,其用于鉴定患者样品中的优势抗原或表位和/或亚优势抗原或表位,培养能够识别所述亚优势抗原或表位的T细胞,其中所述T细胞是抑制性T细胞,和使用有效量的所述T细胞治疗患者以改变所述患者的免疫优势等级,由此在所述患者中诱导耐受性以治疗或预防自体免疫疾病、变态反应、炎症、器官移植排斥或移植物抗宿主病。
本发明的特征还在于一种方法,其用于鉴定患者样品中的优势抗原或表位和/或亚优势抗原或表位,培养能够识别所述亚优势抗原或表位的T细胞,其中所述T细胞是应答性T细胞,和使用有效量的所述T细胞治疗患者以改变所述患者的免疫优势等级,从而在所述患者中诱导细胞毒性免疫反应以治疗或预防感染或癌症。所述感染是,例如,细菌性、病毒性、寄生虫或朊病毒感染。
在任意的本发明的方法中,疾病、感染、癌症或医疗状况的治疗或预防包括缓解或改善疾病、感染、癌症或医疗状态的至少一种症状。治疗益处包括疾病、感染、癌症或医疗状态的至少一种症状的任何缓解、改善、改进、预防或治疗。
在一些方面,所述患者样品是血液样品。
本发明的特征还在于一种方法,其进一步包括通过测定耐受性或对所述亚优势抗原或表位响应的体液或细胞免疫应答而重新确定患者的概况,从而确定所述疗法是否成功地使免疫应答重新平衡。
除非另有定义,否则本文使用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常理解相同的含义。下文描述了适合的方法和材料,尽管与本文所述那些类似或等效的方法和材料可以用于本发明的实施。本文提到的所有出版物、专利申请、专利和其他参考文献均明确地通过引用整体并入。在冲突的情况下,以本说明书,包括定义为准。此外,本文所述的材料、方法和实施例仅为说明性并且不意图限定。
本发明的其他特征和优点将体现于并包含于下文详细的说明书和权利要求书。
附图说明
图1A、1B和1C显示了51Cr释放测定的结果。
图2显示了51Cr释放测定的结果。
图3A和3B显示了51Cr释放测定的结果。图3C显示了活APCs的百分比。
图4显示了通过ELISPOT(酶联免疫斑点法)测量的产生IFNγ的细胞。
图5、6A、6B、7和8显示了51Cr释放测定的结果。
图9显示了慢性乙型肝炎的小鼠模型。
图10显示了小鼠模型的治疗。
图11显示了对HBs和HBc的T细胞应答。
图12和13显示了通过施用方法的应答。
图14显示了抗原的等级。
图15显示了ICS的应答。
图16显示了抗原的等级。
图17显示了在急性发作(flair),随后清除肝炎后的免疫应答。
图18显示了在清除之后的抗原的等级。
图19显示了T细胞完全溶解了患者的肝细胞癌(治疗前—左图;治疗后8周—右图)。
图20显示了存在于患者肿瘤中的抗原。
图21显示了对NY-ESO-1抗原的细胞应答。
图22显示了在根据本发明的治疗后存在的抗原。
图23显示了免疫优势等级的重新平衡。
图24是在根据本发明的T细胞治疗之前和之后的CT扫描。
图25显示了治疗后的存活,无进展。
图26显示了通过本发明的治疗与利妥昔单抗(Rituxan)+ CHOP的比较。
图27显示了施用给动物的T细胞的表征。
图28显示了对于试验小鼠的临床疾病得分。
图29显示了在试验小鼠中的关节炎发生率。
图30A、30B和30C显示了正常大鼠、接种人蛋白聚糖的大鼠和用T细胞治疗的大鼠的组织病理学。
图31是用于本发明的治疗的生物反应器的示意图。
具体实施方案
A. 定义
术语“抗体”是以最广的含义使用,并且具体是涵盖人、非人(例如小鼠)及人源化单克隆抗体(包括全长单克隆抗体)、多克隆抗体、多特异性抗体(例如双特异性抗体)和抗体片段,只要它们显示期望的生物活性。
术语“抗原”是指能够刺激抗体产生或动物中的T细胞应答的化合物、组合物或物质,包括注射或吸收到动物中的组合物。抗原与特异性体液或细胞免疫的产物(包括由异源免疫原诱导的那些)反应。术语“抗原”包括所有相关的抗原性表位。
“抗原呈递细胞”或“APCs”是用于向T细胞呈递抗原的免疫系统的细胞。APC包括树突细胞、单核细胞、巨噬细胞、边缘区Kupffer细胞、小胶质神经细胞、朗氏细胞、T细胞和B细胞(参见,例如Rodriguez-Pinto and Moreno (2005) Eur. J. Immunol. 35:1097-1105)。
“自体免疫”、“自体免疫疾病”、“自体免疫状况”或“自体免疫失调”是指与改变的免疫稳态相关的一组持续性的器官特异性或系统性临床症状或征候,所述改变的免疫稳态通过表达的自体免疫组库(autoimmune repertoires)的定性和/或定量缺陷来表现。自体免疫疾病的病理学的表现是由自体免疫应答诱导的结构或功能性破坏的结果。自体免疫疾病的特征在于体液(例如,抗体介导的)、细胞(例如,细胞毒性T淋巴细胞介导的),或对自体抗原上的表位的两种类型的免疫应答的组合。受影响的个体的免疫系统活化指向呈递这些特异性自体抗原的细胞和组织的炎性级联。受攻击的抗原、组织、细胞类型或器官的破坏引起疾病的症状。
术语“癌症”是指通过不受控的细胞生长表征的疾病或病变。癌症的实例包括,但不限于癌、淋巴瘤、母细胞瘤和肉瘤。具体癌症的实例包括,但不限于,肺癌、结肠癌、乳腺癌、睾丸癌、胃癌、胰腺癌、卵巢癌、肝癌、膀胱癌、结直肠癌和前列腺癌。对于本领域技术人员已知的其他癌症也在考虑之内。
“优势抗原”或“优势表位”是指激发强的耐受性或免疫应答的抗原或表位,其可表征为对该抗原或表位特异性的T细胞的存在量大于应答性T细胞的总数的70%。
术语“表位”是指参与具体免疫球蛋白的识别的一组氨基酸残基,或者在T细胞的背景下,是指对于T细胞受体蛋白和/或主要组织相容性复合体(MHC)受体的识别所必需的那些残基。在体外或体内的免疫系统的背景中,表位是共同形成由免疫球蛋白、T细胞受体或HLA分子识别的位点的分子的集合的特征,例如初级、二级和三级肽结构和电荷。
“肝炎”是指由肝的炎症定义的医学状况。
“人白细胞抗原”或“HLA”是人I型或II型主要组织相容性复合体(MHC)蛋白(参见,例如Stites等人,IMMUNOLOGY, 8TH ED., Lange Publishing, Los Altos, Calif.(1994))。
“免疫应答”是指免疫系统的细胞,例如B细胞、T细胞或单核细胞,对刺激物的应答。在一个实施方案中,所述应答对于具体抗原是特异性的(“抗原特异性应答”)。在一个实施方案中,免疫应答是T细胞应答,例如CD4+应答或CD8+应答。在另一个实施方案中,所述应答是B细胞应答,并且导致特异性抗体的产生。
免疫优势是以下观察结果:尽管在抗原中有大量的可能的表位(抗原片段),但免疫系统将其应答集中于有限数量的表位,并且能够安排为可重现的等级 (Sercarz等人,1993)。对于人工抗原、人病毒,包括流感和牛痘,以及细胞内细菌的免疫应答均呈免疫优势((Chen WS 1994, Belze GT等人,2000, Chen W 2000, Tscharke DC 2005)。免疫优势的最后结果由多个步骤决定,包括MHC结合亲和力、细胞加工以产生适当的MHC结合肽的效率、识别MHC结合肽与MHC之间的复合物的TCRs的可得性、随后是细胞免疫调节机制(YewdellJW 2006, Sette A等人,2009)。
“淋巴细胞”是指参与机体的免疫防御的一类白细胞。有两种主要类型的淋巴细胞:B细胞和T细胞。
“主要组织相容性复合体”或“MHC”是旨在包含在不同物种中描述的组织相容性抗原系统的通用名称,包括人白细胞抗原(“HLA”)。
“亚优势抗原”或“亚优势表位”是指与优势抗原或优势表位相比,激发较弱的耐受或免疫应答的抗原或表位。
术语“治疗”是指响应患者表现的或有待于在患者中预防的疾病、病变或生理状况而进行的临床干预。治疗的目的包括缓解和/或预防症状,以及减缓、阻止或逆转疾病、病变或状况的进展。“治疗”是指治疗性治疗和预防性或防御性措施二者。需要治疗的那些包括已经受到疾病或病变或不良的生理状况影响的那些,以及其中,有待于预防所述疾病或病变或不良生理状况的那些。
“肿瘤”是指所有的瘤细胞生长和增殖,无论是恶性的还是良性的,以及所有的癌症前期和癌细胞和组织。
“_-聚体”是指在目标抗原中发生的_个氨基酸的线性序列。
B. 识别/区分亚优势表位与优势表位的测定
首先,对于患者的肿瘤或感染,测定一组肿瘤相关性、病毒性或其他抗原的存在。这通常通过对肿瘤活组织切片上的免疫组织化学完成,或在恶性血液病中,通过FACS完成。对于患者的肿瘤或感染,测定一组肿瘤相关性、病毒性或其他抗原的存在。这通常通过对肿瘤活组织切片上的免疫组织化学完成,或在恶性血液病中,通过FACS完成。抽取患者的血液,并测试存在的对抗原的体液和细胞免疫应答二者。主动选择对该抗原的免疫应答不可检测或在低水平检测的抗原,以在体外生长T细胞。在这些T细胞生长后,测试它们对重新输注的抗原的应答,并且可测定患者的血液对所述抗原的应答。通过这种方式,可有效地重新平衡患者的免疫应答以提供治疗益处。在优选的实施方案中,体液免疫性的测试可包括但不限于ELISA测定。在优选的实施方案中,细胞免疫性的测定可包括但不限于对于细胞因子的细胞内细胞因子染色(ICS),包括但不限于干扰素γ (IFNγ)和肿瘤坏死因子α (TNFα)。应答性T细胞的亚群(例如,CD8、CD4、Treg)也可在此测定中进行测定。或者,细胞免疫性的测定可包括但不限于用于IFNγ或TNFα的ELISPOT测定。在可选的实施方案中,ELISPOT或ICS可测定IL-4、IL-12 (TH2和TH1)、IL-10 (Treg)或IL-21(滤泡T辅助细胞亚群)。在又一个实施方案中,细胞免疫分析测定可以是用于这些或其他细胞因子的细胞内染色(ICS)。所测试的抗原可以是全抗原、表位删除的抗原,或优势或亚优势表位。在表位的情况下,可使用生物信息学软件预测可与患者的MHC结合的表位,并且,随后测定这些表位。在一个实施方案中,这种软件是Net MHCpan或在测定部分描述的共有表位免疫信息学软件。在表位的情况下,四聚体结合可用作量化CTL的可选测定,前提是HLA类型是已知的。包括肽的四聚体与细胞组合且将细胞染色,并且使用FACS测定识别每种四聚体的细胞%。如果患者具有已知的HLA类型,例如HLA A2,这是有用的。然而,优选的方法是用于体液应答的ELISA和用于细胞应答的ICS或ELISPOT。在优选的实施方案中,从外周血产生CTL。或者,从肿瘤浸润淋巴细胞(TIL)或从注射部位周围的DTH产生CTL。
可以通过在包含10% FCS (CSL)的RPMI培养基中切碎的4 mm从皮肤钻取的活组织,制备DTH浸润淋巴细胞。使用1 μg/ml植物凝集素(Sigma)刺激单细胞悬液,并与经过辐照的自体PBMC以及10 IU/ml IL-2 (Cetus)和10 ng/ ml IL-7 (Peprotech, Rocky Hill,NJ)共培养。每2-3天补充培养基。
对于体液免疫应答概况分析,对来自患者的血清进行从1/100至1/100000的1:4连续稀释,并且用于使用纯化重组肿瘤抗原(通常在大肠杆菌中制备)的标准ELISA中。可测试2中至10000+种抗原。将1微克的每种纯化蛋白吸附到微孔板(Nunc),在4°C过夜。使用PBS清洗平板并使用2% FCS/PBS封闭。将患者血清在2% FCS/PBS中稀释并添加2小时。清洗平板并添加山羊抗人IgG-AP (Southern Biotechnology Assoc)。清洗平板,并与Attophose底物(JBL Bioscientific)温育25分钟,并立即读取(CytoFluor 2350, Millipore)。读取的是UV吸光度。
有两种用于细胞应答概况分析的方法。第一种方法包括用于IFNγ的酶联免疫吸附斑点(“ELISPOT”)测定。在4°C,使用5-15 μg/ml的抗IFNγ单克隆抗体1-DIK (MABTECH,Stockholm, Sweden)包被聚偏氟乙烯后背的96孔板(Millipore, Bedford, MA)过夜。清洗孔,并使用5%人AB血清(Valeant Pharm)封闭。每孔添加5×106个PBMCs (或测试在体外扩增后进行时,添加5x 105个CTL)和来自每种抗原的肽混合物各2 μM。在37°C,5% CO2下温育过夜(18小时)。弃去细胞,并使用包含0.05% Tween 20的PBS清洗孔。将1 μg/ml生物素化的抗IFNγ单克隆抗体7-B6-1 (MABTECH)在室温下温育2-4小时,随后再与链霉亲和素缀合的碱性磷酸酶(MABTECH或Sigma Aldrich)温育2小时。随后,与来自alk-phos底物试剂盒(Bio-Rad Richmond, CA)的5-溴-4-氯-3-吲哚磷酸酯和硝基蓝四唑反应30分钟。使用解剖显微镜(SZ CTV Olympus microscope)进行斑点计数。也可以在AIDELISPOT计数器(Autoimmun Diagnostika, Strassberg, 德国)上进行斑点计数。每个斑点是一个细胞,报告为斑点形成细胞(SFC)/105个PBMC。可使用10 μg/ml PHA作为阳性对照;没有肽的单独细胞可充当阴性对照。
第二种方法包括用于IFNγ和TNFα的细胞内细胞因子染色。对于每种所研究的表位或抗原,将5×106个PBMCs (或测试在体外扩增后进行时,5x 105个CTL)与肽一起(终浓度10-5至10-9 M)平铺到96孔板的100 μl PBS 1% FCS中。在IL-2 (150 U/Ml)、50 μM β巯基乙醇和布雷菲德菌素A (1 μg/ ml) 或Golgi Plug (BD Biosciences, San Diego, CA)(后两种组分用于增加IFNγ或TNFα在应答性细胞中的累积)的存在下温育6小时后,将细胞沉淀,在200 ml PBS 1% FCS中清洗,并且随后在4°C使用对于表面抗原的染料(CD4异硫氰酸荧光素和CD8别藻蓝蛋白0.25 μg/ml (Pharmingen, Becton Dickinson))标记30分钟(在冰上30分钟)。清洗后,使用Cytofix/Cytoperm将细胞在冰上透化20分钟,并且随后使用藻红蛋白缀合的抗IFNγ (0.4 μg/ml)或抗TNFα (0.8 μg/ml)抗体(Pharmingen, BectonDickinson)染色。随后,清洗细胞,固定并悬浮在PBS 1% FCS中,并且在FACScan流式细胞仪上测试,并使用Cell Quest软件分析。或者,可使用FACS Canto (Becton Dickinson)。可测试其他细胞因子,包括但不限于IL-12和IL-4,以测量TH1或TH2亚群。给出T细胞的较宽测定的细胞因子组可以测量IL-12、IFN g、IL-4、IL-10和IL-17。滤泡T辅助细胞可作为CD4+,CXCR5+, ICOS+细胞测量。B细胞可作为 CD19+和B220+细胞测量。T细胞中的IL-21应当与B细胞活化和抗体的亲和力成熟相关,因此,这也可以用于本研究。作为分析IFNγ、IL-4 (BDBiosciences) IL-12、IL-10、IL-17 (R&D Systems)和IL-21抗体(R&D systems)概况的备选方案,也可使用ELISPOT。ICS实际上分析出对不同抗原或表位做出应答的CD8或CD4 T细胞的%概况。也可分析其他细胞亚群,包括Treg。可以使用细胞色素标记的CD25单克隆抗体作为大多数Treg的表面标记。或者,使用来自BD Biosciences的细胞色素标记的人FoxP3单克隆抗体克隆259D/C7,对预透化后的细胞进行染色以测量Treg细胞%及其状态。还可测定IL-10。
NetMHCpan是用于与HLA-A和-B结合的肽的定量预测的生物信息学方法(NielsenM 2007)。还开发出共有表位预测方法(Mouaftsi M 2006)。这些方法可用于分选对于抗原的所有的潜在的MHC I表型,并排序前1%的肽,并由此预测表位。随后,可将这些预测的表位合成为9-10聚体的肽并测试(例如,对于患者的PBMC,或在转基因小鼠中对于目标的HLA类型)。
具有特异性MHC (例如HLA A A2)的四聚体与8聚体的肽表位一起合成(在I型MHC的情况下),且在II型MHC的情况下,与15聚体肽表位一起合成。在室温下使用1/200稀释的四聚体将培养的细胞染色20分钟;随后,添加抗CD8抗体并进一步染色30分钟。随后,清洗细胞并在FACS Calibur (BD Biosciences)。
治疗后,通过测定耐受性和对亚优势抗原或表位的体液或细胞免疫应答而重新分析所述患者概况,以确定治疗是否成功地使免疫应答重新平衡。
C.重新平衡免疫应答
在本发明的一个实施方案中,可通过对亚优势抗原或亚优势表位先体外后体内培养来自患者的T细胞,随后将这些T细胞输注或施用到所述患者中,而重新平衡免疫应答。针对亚优势抗原或表位的T细胞可在组织培养物中先体外后体内生长(脱离患者的免疫调节环境)。在培养足够的细胞以淹没此前的优势细胞后,将细胞重新输注到患者体内以改变细胞平衡并治疗性地转变优势等级。在进一步的优选实施方案中,作为治疗从头引入的该数量的T细胞的这个数量大于应答所述抗原、传染物、肿瘤或器官的T细胞的5%。可通过使用减少内源T细胞数量的调节剂对所述患者进行预治疗(即,化疗)而进一步改变这个比例以有利于输注的细胞。
本发明包括优化针对组织培养中的亚优势抗原的T细胞的生长的方法。在一个实施方案中,所述细胞在不存在优势抗原的情况下生长。这通过选择未曾暴露于优势抗原的专业抗原呈递细胞和将抗原修饰以消除优势表位或限制抗原加工能力的其他组分而完成。
由于治疗方法需要富集对于亚优势抗原/表位且理想地对亚优势抗原/表位完全反应性的T细胞,所以用于此类T细胞的生长的有效方法是重要的。针对优势表位和抗原的其他T细胞的生长增加了在培养物中产生足够的特异性细胞以改变此比例所需的时间。此外,生长出的对于优势表位的细胞妨碍在重新输注后正确比例的实现。因此,已开发出特异性地限制优势抗原或表位的引入的T细胞培养方法。例如,尽管该方法已广泛应用于所有肿瘤,但它对于EBV恶性肿瘤具有明显的优势,因为它不使用EBV转化的B细胞(其表达EBV优势抗原/表位)。在施用其中显著%的CTL是应答亚优势抗原/表位的细胞时,其也更为可靠。
在体外,从来自患者的外周血单核细胞(PBMCs)产生单核细胞衍生的树突细胞。在优选的实施方案中,将PBMC在组织培养烧瓶中铺板2小时以允许单核细胞粘附。在可选的实施方案中,可使用CD14+磁珠从PBMC分离树突细胞(Miltenyi Biotec, Auburn, CA)。此时,除去未粘附的细胞,并在-80°C冷冻以供后续作为T细胞的来源使用。使用白介素-4 (IL-4)和粒细胞-巨噬细胞集落刺激因子(GM-CSF)处理粘附的单核细胞,导致在约1周内分化成不成熟的树突细胞。随后使用肿瘤坏死因子(TNF)或巨噬细胞条件培养基处理2天,使iDC进一步分化成为成熟的树突细胞。随后,使用包含亚优势抗原的肽或质粒对这些细胞进行脉冲处理2小时,并且,随后将 PBMC解冻并添加到脉冲处理的树突细胞。数小时后,将细胞汇集并重新悬浮在包含IL-2或IL-15 (优选IL-15)的培养基中,以在体外产生已识别所述抗原的T细胞的扩增。对于某些方案,添加IL-7和IL-15以提高T细胞存活。对于其他方案,调整培养条件以优化T细胞的某些亚群的生长。例如,可添加IL-12以极化成TH1细胞。或者,可添加IL-4极化成TH2。在某些方案中,可添加 IL-6以阻止Treg的生长。在可用于自体免疫或器官移植应用中的又一个变化中,可添加低水平的IL-2+雷帕霉素以加速Treg的生长。更详细的方案列出在多个实施例中,并且实施例3、图3中描述了使用将细胞极化成某些T细胞亚群的方案产生的细胞的体内比较。如果T细胞生长在组织培养烧瓶中,则必须在第14日和21日更换培养基。然而,在优选实施方案中,可使用生物反应器以减少这种需求,例如,气体通透性生物反应器,例如Grex (Wilson Wolf)或Hyperstack (Corning)。通常,与使用传统方法的12-24周相比,可在2-6周内产生向患者施用的足够的细胞。
在本发明的另一个实施方案中,本发明人已开发了用于过继转移针对其优势表位已经被删除的抗原的T细胞,并且证实产生更多的针对亚优势表位的T细胞。可使用此类蛋白或相应的DNA疫苗以产生对亚优势表位具有广泛免疫应答的T细胞。这种方法应广泛地适用于多种类型的疾病以实现对亚优势表位的免疫反应的平衡,包括但不限于EBV、癌症、HIV或肝炎。在另一个实施方案中,使用抗原或编码它的质粒/重组疫苗对患者进行免疫接种以诱导对亚优势抗原的新的广泛免疫反应。在另一个实施方案中,可以在使用亚优势抗原的免疫接种后施用亚优势反应性T细胞以强化所述应答。在又一个实施方案中,可治疗性或预防性地使用所述方法,其中可以确定患者的免疫概况以测量患病的风险,并且随后可以使用本文公开的任何方法以适当的亚优势抗原对所述患者进行初免(prime)。
在可选的实施方案中,生长的T细胞诱导耐受性以预防或治疗自体免疫疾病、变态反应、炎症、器官移植排斥或移植物抗宿主病。取决于期望的T细胞的类型,可调整培养条件以优先生长或富集相关亚群,包括但不限于CD8、CD4、TH1、TH2或Treg。例如,T细胞可在存在或不存在某些生长因子、细胞因子、药物、小分子或其他免疫细胞的情况下生长。在优选的实施方案中,在存在经刺激的专业抗原呈递细胞(例如,单核细胞衍生的树突细胞、巨噬细胞或EBV永生化B细胞)的情况下,从组织培养物中的外周血单核细胞(PBMC)产生亚优势抗原反应性T细胞。
在另一个实施方案中,使用各种技术以调整抗原加工,从而有利于亚优势表位。在本发明的一个实施方案中,这通过修饰抗原以消除优势表位、抑制抗原加工的区域,或限制同时呈递至抗原呈递细胞的优势或亚优势表位的数量而实现。这些修饰提高了识别的亚优势表位的应答和多样性(实施例1,图5)。在可选的实施方案中,可使用病毒载体,例如腺病毒或牛痘苗,将修饰的LMP1、LMP2和EBNA-1序列递送至APC。在其他抗原(LMP1和EBNA-1)的情况下,消除导致不良抗原加工的蛋白区域极大地增强了对这些抗原上的亚优势表位的免疫应答(实施例1,图6)。可使用肽、蛋白、质粒或病毒载体,例如腺病毒或牛痘苗,将修饰的LMP1、LMP2和EBNA-1序列递送至APC。
在可选的实施方案中,可在CTL产生期间向APC和抗原添加蛋白酶体拮抗剂以增加识别的亚优势表位的数量并且增强对亚优势抗原的应答(实施例2,图7)。有具有不同机制的很多可得的蛋白酶体拮抗剂(例如,硼替佐米、氯碘羟喹、乳胞素、环氧霉素(epoxomycin)、MG-132、MLN9708、卡非佐米 (carfilzomib, PR-171)。
在可选的实施方案中,可施用与抗体在复合物中的抗原,所述抗体对于针对亚优势决定簇的应答具有不同的同种型。与抗原一起注射的抗体结合期望的T细胞表位侧翼的决定簇以靶向专业抗原呈递细胞,并引导对侧翼表位的抗原加工。
在本发明的另一个实施方案中,使用包含亚优势表位或抗原的质粒产生所施用T细胞,或者将包含亚优势表位或抗原的质粒直接通过各种施用途径与IFNγ、IL-21或其他细胞因子组合而直接施用,或在经脉冲处理的树突细胞上直接施用,以诱导对亚优势抗原的应答。抗原在T细胞刺激之前诱导IFNγ或其他细胞因子,以增强对亚优势表位的T细胞反应性,并改变免疫优势等级。
在另一个实施方案中,调整施用途径以改变免疫优势等级。牛痘苗应答的施用途径决定了优势决定簇的程度。已发现,在腹膜内施用时,优势决定簇仅占应答的四分之一,相比之下,在皮内施用的情况下,优势决定簇占应答的一半(Tscharke DC等人,2006,Tscharke DC等人,2005)。如实施例3、图11中所示,与IP或IV途径相比,通过IM途径施用抗原产生了对亚优势表位的更强的应答和更广的应答。因此,调整施用途径是本发明人主张的用于调节免疫优势等级的另一个体内机制。在优选的实施方案中,通过皮内施用递送培养的T细胞。通过靶向不同的APC (例如,巨噬细胞、树突细胞),施用途径改变了优势等级(实施例3,图12)。
在优选的实施方案中,所述抗原是病毒抗原,特别是其上带有亚优势表位的潜伏病毒抗原或慢性病毒抗原。例如,所述病毒抗原来自选自包含以下的组的病毒:EBV、HSV、VZV、乙型肝炎和丙型肝炎、HIV和HTLV。病毒抗原是,例如EBV LMP1、LMP2、EBNA-1、HPV E6或HPV E7。例如,所述病毒抗原与EBV、HSV、VZV、乙型肝炎和丙型肝炎、HIV,和HTLV、CMV、RSV或流感相关。在另一个实施方案中,所述抗原是在其他慢性和潜伏传染物(例如与细菌、真菌、寄生虫或朊病毒相关的试剂)上的抗原。在又一个实施方案中,所述抗原是肿瘤抗原,包括但不限于:肿瘤相关抗原、肿瘤特异性抗原、与癌干细胞或转移相关的抗原。在其他实施方案中,所述抗原与自体免疫性、变态反应、炎症或器官抑制排斥或移植物抗宿主病相关。
在一个实施方案中,通过鉴定患者样品中的优势抗原或表位和亚优势抗原或表位,培养能够识别所述亚优势抗原或表位的T细胞,并使用有效量的所述T细胞治疗患者,而改变所述患者的免疫优势等级。
在一个实施方案中,通过鉴定患者样品中的至少一种亚优势抗原或表位,培养能够识别所述亚优势抗原或表位的T细胞,和使用有效数量的那些T细胞治疗所述患者来改变患者的免疫优势等级,从而提供治疗益处。
在另一个实施方案中,所述T细胞是反应性T细胞,并且使用有效量的所述T细胞治疗患者以改变所述患者的免疫优势等级,由此诱导所述患者中的细胞毒性免疫应答以用于治疗或预防感染或癌症。所述感染是,例如,细菌性、病毒性、寄生虫或朊病毒感染。
在任意的本发明的方法中,疾病、感染、癌症或医疗状况的治疗或预防包括缓解或改善所述疾病、感染、癌症或医疗状态的至少一种症状。
D. 治疗方法
1. 癌症
优势和亚优势抗原的免疫概况分析的临床使用的工作流程
步骤1:肿瘤活检(免疫组织化学)或血液(IHC、FACS或ELISA)抗原1、2、3、4、5、6、7、8、10
结果:抗原1、2和6在肿瘤上,组中的其他则不在肿瘤上
步骤2:免疫应答概况分析
体液概况分析
对血清的ELISA
细胞概况分析
对由每种抗原刺激的PBMC进行ELISPOT或ICS (至少对于IFNg,还有IL-10、IL-4和IL-12、IL-21以测定T细胞亚群)
结果:抗原1:强应答(优势);抗原2和6:无/中度应答(亚优势)
步骤3:在体外培养对亚优势抗原的T细胞(CD8和CD4)
结果:T细胞对抗原2和6应答
步骤4 (一旦治疗充分确定,则为任选的):使用细胞免疫概况分析(ELISPOT或ICS)确定 >5%的生长的T细胞对亚优势抗原应答
结果:25%的T细胞对抗原2和6应答
在继续进行步骤5之前,提供至少5%的对亚优势抗原应答的T细胞
步骤5:在进行或不进行预先调节(例如,环磷酰胺)的情况下,将细胞IV输注到患者中
步骤6 (一旦治疗充分确定,则成为任选的):在输注后2-3周从血液分离PBMC并分析免疫应答概况
结果:细胞概况分析:抗原1:无/中度应答;抗原2&:对至少一个抗原的强应答(优势)
步骤7:评估临床应答
RECIST (CR, PR);存活或无进展存活
结果:改进的应答率和存活
在实施例4中,使用这个系统性方法治疗黑色素瘤。在实施例5中,将该方法应用于用多质粒LMP2治疗淋巴瘤。实际上,T细胞治疗改变了淋巴瘤的天然过程,从复发-缓解的过程改变为持久缓解的过程。这种抗原以及删除的LMP1和EBNA-1也可以用于治疗包括EBV抗原的其他肿瘤,包括鼻咽癌、伯基特淋巴瘤、CLL、霍奇金病和除此之外的一些胃癌等。在实施例3中,使用类似的方法治疗肝细胞癌。这些实施例通过引用并入本发明,并且证实了免疫应答向亚优势抗原的重新平衡可广泛地应用于所有的癌症形式。
2.一般使用肿瘤上的大量亚优势抗原以产生泛(pan)肿瘤类型的治疗
在另一个实施方案中,本发明人提出,如果能够鉴定对于具体类型的肿瘤的足够的亚优势抗原,则可以使用对于多种亚优势抗原的T细胞治疗该类型的肿瘤,而无需对患者进行测试。类似地,靶向相同肿瘤上的多种抗原将降低抗性,例如组合化疗。例如,开发靶向亚优势抗原EBV LMP2 (其在40%的淋巴瘤上)和存活(其在50%的淋巴瘤上)的单一T细胞系的能力将允许使用对这两种抗原特异性的T细胞系靶向约80%的淋巴瘤。实施例5中证实了泛淋巴瘤产品的临床测试。经过3年,使用泛淋巴瘤产品治疗的患者的无进展存活,与治疗前测试抗原的产品相当。这可以是因为,与组合化疗类似,CTL对在相同肿瘤上的多种抗原的应答可减少逃脱的可能性。使用单一T细胞产物治疗所有淋巴瘤的能力(不是必须仅治疗对单个抗原阳性的亚群)是新的产品构思。在本发明的另一个实施方案中,本发明人还主张用于实际上任何癌症的泛癌症产品。
3. 慢性感染
肝炎和肝细胞癌
在慢性肝炎患者中,不产生HBs抗体而产生HBc抗体 (Ganem D等人,2004)。在急性肝炎患者中,产生对两种抗原的抗体。>90%的新生儿和30%的1-5岁的儿童发展出慢性形式,而>90%的成人此时剧烈地清除所述病毒。95%的肝细胞癌与乙肝病毒的慢性感染相关。HBsAg通常在HCC的细胞表面。基于这些观察结果,本发明人选择研究HBs在肝炎和肝细胞癌患者中是否是亚优势的,以及T细胞的生长是否能够产生针对HBs抗原的CTL,其能够重新平衡对于亚优势抗原的免疫应答,并且可以是治疗性的。
基于这个发现,本发明人施用针对HBs Ag培养的CTL,以治疗肝炎动物模型中的慢性肝炎,并且最终治疗与HBV相关的HCC患者中的慢性肝炎。在动物模型中,施用并测试了HBVtgRAG细胞。在接受对照细胞的动物中,慢性肝炎发展。此外,当动物发展肝炎并且接受针对亚优势抗原的CTL时,动物发展出急性肝炎但清除了肝炎病毒(实施例3、图9-11)。
基于所述振奋人心的动物数据,使用T细胞重新平衡疗法治疗肝炎患者。
优势和亚优势抗原的免疫概况分析的临床使用的工作流程
步骤1:肝炎表面抗原和肝炎核心抗原的免疫应答概况分析
体液分析 细胞分析
对血清的ELISA 对由抗原刺激的PBMC进行ELISPOT 或ICS (至少对于IFNγ,还有IL-10、IL-4和IL-12、IL-21以测定T细胞亚群)
结果:Hbc强应答(优势); Hbs 无/中度应答(亚优势)
步骤2:在体外培养针对亚优势抗原的T细胞(CD8和CD4)
结果:T细胞对HBs抗原应答
步骤3 (一旦治疗充分确定,则为任选的):使用细胞免疫概况分析(ELISPOT或ICS)确定 >5%的生长的T细胞对亚优势抗原应答
结果:25%的T细胞对HBs抗原应答
在继续进行步骤4之前,提供至少5%的对亚优势抗原应答的T细胞
步骤4:在进行或不进行预先调节(例如,环磷酰胺)的情况下,将细胞IV输注到患者中
步骤5 (一旦治疗充分确定,则成为任选的):在输注后2-3周从血液分离PBMC并分析免疫应答概况
结果:细胞分析:HBc 无/中度应答;HBs 强应答(优势)
步骤6:评估临床应答
结果:感染的清除
对具有HBV和HCC的5名患者进行免疫概况分析。如所预期的,ELISA证实对高滴度的针对乙型肝炎核心抗原(HBc Ag)的抗体,但没有针对乙型肝炎表面抗原(HBs Ag)的抗体(实施例3,图17)。然而,在将PBMC诱导成树突细胞,使用乙型肝炎核心抗原(HBc Ag)和乙型肝炎表面抗原(HBs Ag)进行脉冲处理,并用于从来自相同患者的PBMC生长CTL时,观察到预想不到的结果。通过产生IFN的T细胞在ELISPOT上的频率,观察到等级如下:无抗原(10 sfc)<HBsAg(15 sfc)<HBcAg(45 sfc) (实施例3,图14)。这表明,相对于肝炎核心抗原,乙型肝炎表面抗原的确是亚优势的。此外,通过ELISPOT (实施例3、图14)且通过ICS (实施例3、图15),显示HBs的一个表位(FLL)是优势的。将包含这个表位的肽从用于培养来自患者细胞的肽混合物中排除。
在HCC患者中,接受针对亚优势HBs (实施例3,图16)的T细胞的3/5个患者清除了HBs抗原,并且证实在肝功能测试中丙氨酸转氨酶 (ALT)瞬时提高之后,对HCC的CR (实施例3,图17和19)。当测试在施用后2周来自这些患者的PBMC时,优势应答针对 HBs而不是针对HBc,并且针对此前HBs的亚优势表位(实施例3,图18)表明患者已经重新平衡了他们的免疫优势等级。作为治疗和免疫优势等级转化的结果,提供了治疗益处(实施例3,图17和19)。
因此,针对亚优势抗原的T细胞过继免疫治疗可以用于根除慢性病毒并治疗其上存在慢性病毒的癌症。此外,指导针对亚优势抗原和表位的T细胞的产生的免疫分析为多种疾病开拓了新的治疗道路。
实际上,可以治疗任何病毒、细菌、真菌、朊病毒、寄生虫或其他传染病,包括但不限于丙型肝炎病毒和HIV病毒。使用重新平衡,还可以治疗其他慢性传染病。例如,结核分枝杆菌是很多患者中的慢性感染,但只有在免疫系统受到压制时恢复活动。在其潜伏状态中,其包含在肉芽肿中,通常在肺中。最近,分枝杆菌吞噬体的蛋白质组研究表明,MTB抑制树突细胞中抗原呈递的程度大于巨噬细胞(Li等人,2011)。为此,本发明人提出重新平衡免疫系统以应答亚优势表位是用于实际上任何癌症或传染病的广泛治疗方法。
4. 自体免疫性
流行病学研究证实,患者发生多发性硬化的风险与EBV抗体滴度相关。通过在其感染EBV之前跟踪几十万个体,并且在初始感染后对其数年随访,Ascherio等人得以研究发生MS的305名患者。他们的风险在EBV感染后剧烈提高(Ascherio A等人,2010)。在与同样是EBV携带者的健康个体相比时,相对于其他EBV抗原,MS患者中针对EBNA-1 400-641应答的记忆CTL增加(Lunemann JD等人,2006)。看起来,EBNA-1特异性Th1细胞能够通过自体抗原的交叉识别或旁观者机制维持自体免疫性。此外,转基因小鼠研究表明,表达LMP2a的B细胞绕过正常耐受性检查点,并强化自体免疫疾病的发展(Swanson-Mungerson M 2007)。因此,尽管本发明人已经研究了对EBV亚优势表位反应性的T细胞作为对EBV和EBV相关癌症的治疗,但对EBV亚优势表位反应性的T细胞也可用于恢复自体免疫性中的平衡。通过培养并引入针对EBV潜伏抗原(EBNA-1、LMP1和LMP2)应答的T细胞,本发明人提出,可以重新平衡免疫应答,在具有EBV相关性的自体免疫疾病中重新引入耐受性。对于与其他病毒相关的其他自体免疫疾病也是如此。例如,小核糖核酸病毒,例如库克萨基病毒B3导致心肌炎/扩张型心肌病、I型糖尿病、脑炎,肌炎,睾丸炎,肝炎。
在又一个实施方案中,可以产生针对与器官相关的抗原的T细胞,在所述器官中发生了器官特异性自身免疫。由于免疫优势现象涉及所有在任一时刻加工的抗原的表位,因此知晓在那一时刻驱动自身免疫的准确的自身抗原并不是完全必需的需求。例如,产生针对胶原的反应性亚优势表位的T细胞并将其引入至类风湿关节炎患者的发炎关节中,正在进行的免疫应答将重新平衡并且将恢复耐受。相反且意料不到的是认为引入有活性的T细胞至炎性部位将是有利的,但根据我们的操作模型,在该部位上免疫应答的重新平衡将对异常的免疫应答恢复适合的控制。自身免疫疾病的治疗是本发明的另一个实施方案。
5.Treg体外扩增
免疫调节原则之一是Tregs与T细胞的其他亚群的平衡。本发明的另一个实施方案是使先体外后体内T细胞生长向Treg细胞极化。在这种情况下,代替靶向亚优势表位,产生对优势表位的Treg作为另一种重新平衡免疫应答的方式。在一个实施方案中,将这样产生的Treg T细胞用作单独的治疗产品。在另一个实施方案中,将Treg亚群与来自对亚优势表位或亚优势抗原生长的其他亚群的T细胞组合使用。尽管已经使用FACS分选随后用抗CD23和抗CD28包被的珠扩增将Tregs以非抗原特异性方式扩增(Putnam等 2009),但到目前为止还未有人将Tregs以抗原特异性方式扩增用于过继免疫治疗。本文描述了用于建立特异性对于不同抗原,尤其是在自身免疫疾病、移植物抗宿主病或移植排斥中观察到的免疫应答中优势的那些抗原的Tregs的方法。
通过FACS分选(BD FACS Aria II高速细胞分选仪)使用无菌技术在GMP清洁房间内用CD4-PerCP( SK3)、CD127-PE (hIL-7R-M21)、CD25 APC (2A3)、CD45RA-PE.Cy7 (L48)和CD45RO-PE.Cy5 (UCHLI)从自身免疫或移植患者的PBMC中分离Tregs。分选CD4+CD127 lo/-CD25+和CD4+CD127 lo/- T细胞并收集于3ml含有10%人热失活混合AB血清(ValleyBiomedical, Winchester, VA)的X-Vivo 15培养基 (Lonza, Walkersville, MD)中。(或者,可以使用包被有相同抗体的磁珠分离Tregs(Miltenyi Biotec, Auburn, CA))。将这些细胞以2.5×105 Tregs/孔铺于24孔板(Costar, Cambridge, MA)中,每孔含有从PBMC制备的树突细胞(如上文对T细胞刺激所述),所述PBMC已经用以Treg:APC比为1:5的优势抗原预先脉冲。18小时孵育后,将细胞在第1-7天在培养中接受雷帕霉素 (100ng/ml; Wyeth,Madison, NJ)。在第2天,将培养体积加倍并加入300单位/ml IL-2(Chiron, Emeryville,CA)。将细胞重悬于新鲜培养基中并在第2、5、7、9和12天加入IL-2。在第9天用肽脉冲的树突细胞再刺激细胞。或者,抗CD23/抗CD28包被的微珠(Invitrogen, Carlsbad, CA)可以用于该第二次刺激。此外,气体渗透生物反应器(诸如Grex)可以用于进行该细胞生长,并在封闭系统内有更少操作,并且改善生长的动力学。在另一种变化中,将IL-10与包被的珠在刺激时一同加入以驱动Treg向Treg1(其分泌高水平的IL-10并调节TH1和Th2应答)的进一步分化。
自身免疫中优势和亚优势抗原的免疫概况分析的临床使用的工作流程
步骤1:免疫应答概况分析
体液概况 细胞概况
ELISA或血清 在用每一种抗原刺激的PBMCs上的ELISPOT或ICS (至少对于IFNγ,还有IL-10、IL-4I和L-12、IL-21以测定T细胞亚群)
结果:抗原1强应答(优势); 抗原2无/中度应答(亚优势)
步骤2:体外生长针对亚优势抗原的T细胞(CD8和CD4);体外生长针对优势抗原的Treg
结果:T细胞对亚优势抗原应答;Treg对优势抗原应答
步骤3(一旦良好建立了治疗,则任选):使用细胞免疫概况分析(ELISPOT或ICS)确定>5% T细胞对亚优势抗原应答生长,并且Treg对优势抗原生长
结果:25%的T细胞对抗原应答
提供对亚优势抗原的至少5%的T细胞应答和/或对优势抗原的至少5%的Treg细胞应答,进行步骤4
步骤4:IV将细胞输注入具有或不具有在前调节(例如环磷酰胺)的患者中
步骤5(一旦良好建立了治疗,则可以成为任选):从输注后2-3周的血液中分离PBMC并进行免疫应答概况分析
结果:细胞概况抗原1无/中度效应物应答;抗原2和强Treg应答(优势)
步骤6:评估临床应答
在自身免疫中(MS-发作下降,类风湿关节炎-关节肿胀减少,哮喘-发作数目下降,早期I型糖尿病,胰维持),在移植中-器官排斥率和移植物抗宿主病下降
结果:改善的临床结果
6. 在自身免疫或移植中施用针对优势抗原的Tregs
在又一个实施方案中,针对优势抗原的Treg能够与TH1、TH2或CTL亚群的T细胞(其自身对亚优势抗原应答)组合。此类组合将更加充分转换应答向亚优势表位的平衡。本发明人已经证明关节炎中两种类型的T细胞之间的协同作用(实施例6)。
7. 移植
在可选实施方案中,使用上述Treg培养条件从健康供体的PBMC用来自其他健康供体的树突细胞(或受照射的PBMC)培养Treg。以这种方式,建立了每一种MHC的同种异体反应性Treg系并储存。如上我们所讨论,用针对20-50种MHC单元型产生的Treg系能够覆盖80%的MHC。这些系的每一种能够以单次剂量的等分试样冷冻于–80摄氏度。当进行器官或BMT时,还将5×107细胞/m2的对错配的MHC有反应性的Treg移植入患者中。以这种方式,减缓了同种异体排斥或移植物抗宿主病。
8. 自动免疫概况分析测定和封闭系统细胞培养设备
气体渗透膜设备是培养技术的优选实施方案。由于膜是气体可渗透的,因此培养规模由培养细胞所需的膜的表面积和培养基的体积所决定。这些气体渗透设备的实例包括Hyperstack (Corning)或Grex (Wilson Wolf)。这类生物反应器的有用特色之一是细胞培养过程是线性比例可变的。作为我们的方法标准化的部分,我们已经设计了在生产套件中使用的可滑入标准CO2培养箱内的生物反应器版本。在独立的实施方案中,我们已经设计了适合于标准组套的用于在自动生产设施中温暖房间的生物反应器。旨在自动化的该生物反应器制成两种大小:一种用于对单个患者的自体细胞生长,而第二种更大的版本用于同种异体T细胞系的商品化生产。在改进的方法中,将细胞培养设备为了自动化而修饰成矩形形状,以滑入标准CO2培养箱中的槽内。这是明显的优点,因为气体渗透膜在瓶的底部并如果它位于架上则会被堵塞。通过使瓶成为架,存在对膜的更好的气流。关键属性是瓶侧面的凸缘,其允许它们托住生物反应器的重量。在一个实施方案中,生物反应器放置入不锈钢框内以形成插入架支撑物中的板。在另一个实施方案中,将它们塑成塑料部分。在实施方案之一中,它们具有完整架的底座。在另一个实施方案中,它们包含架的½、¼或1/3或1/5。在优选的实施方案中,培养箱由New Brunswick、Forma、ThermoElectron、Nuaire、ESCO制造。在优选的实施方案中,培养箱可以是空气套或水套的或者是其他设计。在又一个实施方案中,它们适合于金属框的架内或在温暖房间的平面上。在另一个优选的实施方案中,该平面可移动并由机器处理。在又一个实施方案中,生物反应器适用于多种商品化处理设备,包括但不限于在收集细胞前使细胞回到悬浮状态的振荡器。
每一生物反应器是封闭系统,其具有接触接口以引入培养基、组分和细胞,并且从该接口收获细胞用于冷冻和质量控制。在优选的实施方案中,这些接口是使流体和细胞进出生物反应器的管,所述接口的盖具有达到生物反应器的底部的一体的固体管。在可选实施方案中,接口是无菌的橡胶片,针或其他探针可以经其插入生物反应器中以手动或使用自动化机器来注射或移除流体细胞或其他试剂。将该设备用条形码编号以对其追踪,从而每一患者和每一细胞系将具有其自身指定的生物反应器。当该生物反应器使用后即可将其丢弃。还将生物反应器大小设计为适合自动化细胞培养中的标准机器人自动化设备,包括但不限于组套、运输器和摇动搅拌器。图31是此类生物反应器的实例。
如同生物反应器,还将商品化多重免疫概况分析测定设计为能够测量抗体的效价和针对抗原组的T细胞应答。为了此目的,使用ELISPOT测定,这是由于可以应用标准96孔板形式。在优选的实施方案中,使用AIDELISPOT读数器(Autoimmun Diagnostika,Strassberg, Germany)来计数点。或者,使用96孔板来输入细胞至FACS进行ICS测定。在任何情况下,每一患者具有它们自身指定的96孔板,其经条形码编号并且所有板均是一次性的。这些商品化测定和制造方法是本发明的实施方案。
实施例
1. 实施例1:EBV潜在感染、淋巴瘤和鼻咽癌
如同由血液中的抗体所测量的,全世界人口的90%已经暴露于EBV(单核细胞增多症中的致病性病毒)。EBV在B细胞中变为潜伏的并且关闭了大部分它的蛋白,但表达非常低水平的潜伏抗原LMP1、LMP2及有时表达EBNA-1。这些蛋白是弱免疫原性的,但对于维持病毒甚至在它的潜伏状态中是必需的。由于它们主要来源于B细胞,因此40%的淋巴瘤对EBV潜伏抗原测试为阳性。因此,这些抗原可以用作在过继细胞治疗中产生CTL应答的靶标。此外,鼻咽癌也如同其他肿瘤(例如,-10%的胃癌)一样表达EBV潜伏抗原。尽管CTL已经用于治疗EBV淋巴瘤,但目前使用转染有EBV作为重复刺激的B细胞的产生方法是耗时(3-6个月)且繁琐的。此外,以这种方式产生的针对LMP2的CTL产生T细胞批次的仅一半在产生后对LMP2具有可检测的应答。本发明人相信对此的原因是由于来自EBV蛋白的优势表位存在于EBV转化的产生CTL的LCL细胞中,其在50%的时间过度生长针对LMP2的细胞。在患有巨大肿瘤的患者中,52%用通过传统方法产生的CTL治疗的患者具有完全应答。尽管现有技术认为所有CTL无论存在或缺少对LMP2的应答均是等效的,但本发明人认为这可以是可变的临床应答的一种原因。此外,本发明的操作模型预测这是真正的原因。因此,增加针对LMP2的CTL产生效率可以在临床上是重要的。
尽管CTL制备的其他方法也包含在本发明内,但使用以下方法来产生以下实验数据。从患者收集40 ml-100 ml的外周血在Vacutainer管中。将外周血单核细胞(PBMC)通过在Lymphoprep (Nycomed, Oslo, Norway)上离心分离,在包含2mM L谷氨酰胺、100 IU/ml青霉素、100 μg 链霉素/ml及10%胎牛血清(FCS) (5×106细胞/ml)的RPMI 1640 (Gibco,Grand Island, NY)中重悬,并以107细胞/孔接种于6孔板(Costar Corp, Cambridge, MA)中。在37摄氏度下2小时后,将未粘附的细胞去除并重悬于置于试管内含有10%聚乙二醇(PEG)的FCS中,在干冰上冷冻并储存于-80冰箱中。将仍在6孔板中的粘附细胞在添加有每ml 50 ng的GM-CSF和1000 U的IL-4的RPMI + 10% FCS中培养。在第2天和第4天用包含上述相同生长因子的新鲜培养基更换一半培养基。在第6天,用所述培养基将培养基完全更换,并加入25%体积的巨噬细胞条件培养基以刺激成熟。巨噬细胞条件培养基由粘附于免疫球蛋白包被的板(由将PBS中的免疫球蛋白铺板并在4摄氏度下孵育过夜制备)上的PBMCs在RPMI 10% FCS中37摄氏度下24小时,收集上清液,经0.2 mm孔径膜(Acrodisc, GelmanSciences)过滤来制备,并且使用前可以在-20摄氏度下储存至多8周。未粘附的细胞在2天后收集并用作树突细胞的来源。进行用单克隆抗体对表面标志物包括CD54、CD80、CD83和CD86的免疫荧光染色以确保树突细胞质量(>50%的细胞+)。
将DC刺激细胞在37摄氏度下预先暴露于在添加有3 μg/ml的人β2微球蛋白的无血清RPMI 1640中的浓度为μg/ml的蛋白(肽为50)中2小时。随后将它们洗涤并以105细胞/ 2ml孔接种于添加有IL-7 5 ng/ml的RPMI 10% FCS中。将2 ×106 PBMC加入每一孔中作为应答细胞,其与刺激细胞比例为20:1。在第14天和第21天将培养物用在添加有20 U/ml的IL-2的RPMI 10% FCS中的负荷有自体肽的树突细胞再刺激(如需要,则分至额外的孔中)。
用于治疗患者的CTL释放测试包括生存力>70%、7天后对细菌和真菌的阴性培养、低于5 EU/ml的内毒素检测、支原体的阴性结果、在51Cr释放测定中以20:1比例的受体淋巴母细胞的杀死低于20%、低于2%的CD19+ B细胞、低于2%的CD14+单核细胞和HLA特性。
将多克隆T细胞群收集并用作5小时铬释放测定的效应物。对于铬释放测定,从CTL供体的皮肤活组织检查中建立成纤维细胞的单层培养,并且暴露于重组牛痘病毒(2x 106细胞/9 cm培养皿)。转染后18小时收集细胞并用51CrO4标记1小时,洗涤三次,并用作5小时铬释放测定中的靶标。在γ计数器上计数前,将测定的上清液收集入1%甲醛中。
a实验1:当树突细胞或活化的巨噬细胞代替LCL用作抗原呈递细胞时,对亚优势表位应答的T细胞的相对频率增加。
使用3种不同的抗原呈递细胞作为刺激细胞从10位患者中制备T细胞系:EBV转化的淋巴母细胞细胞系(LCL)呈递和扩增;树突细胞(DC)呈递及细胞因子扩增;IFNγ 巨噬细胞(MAC)呈递及细胞因子扩增。所述3种中的每一种在抗原呈递过程中用3种质粒的混合物刺激,所述3种质粒表达EBNA-1 (具有aa 90-325缺失的EBNA-1)、LMP1 (具有aa 1-43和aa260-315缺失的LMP1)和LMP2 (LMP2A在2种质粒中,一种表达aa 1-399并且第二种质粒表达aa 400-497)的亚优势表位。将使用本发明的方案这样产生的T细胞系随后在51Cr释放测定中研究。
图1A和1B:分别以20:1和10:1的两种不同的效应物:靶标(E/T)(CTL系:HLA匹配成纤维细胞)比例的51Cr释放,HLA匹配的成纤维细胞用来自所示抗原的肽混合物预先脉冲,使用不同的指示的APC从代表性患者中扩增CTL以培养CTL系。
结论:树突细胞(DC)和巨噬细胞(MAC)比EBV转化的B细胞(LCL)更选择性地刺激针对亚优势抗原的CTL的生长。
图1C:以三种不同的效应物:靶标(E/T)比(CTL: HLA匹配的成纤维细胞)的51Cr释放,HLA匹配的成纤维细胞用来自血液采集后直接得自相同患者的PBMC(培养前)的指示的抗原CTL的肽混合物预先脉冲。
结论:患者对于EBV优势抗原具有某些应答,但相比于T细胞培养后具有明显更高的E/T比。
图2:以20:1的效应物:靶标(E/T)比(CTL系:HLA匹配成纤维细胞)的51Cr释放,HLA匹配的成纤维细胞用代表来自LMP2的特定HLA A2限定亚优势表位的肽预先脉冲,并且CTLs使用上述三种不同方法从相同患者中制成。
结论:树突细胞和巨噬细胞均比LCL导致针对更大数目的亚优势表位的应答和更高水平的CTL活性,并且对不同表位的应答强度在树突细胞和巨噬细胞之间不同。
图3A和3B:以20:1的效应物:靶标(E/T)比(CTL系:HLA匹配成纤维细胞)的51Cr释放,HLA匹配的成纤维细胞用来自LMP2的肽预先脉冲:LCL刺激在图3A中,并且DC/MAC刺激在图3B中。
结论:相比于LCL其仅导致50%的来自患者的CTL系具有可检测的LMP2应答,树突细胞和巨噬细胞导致90%的患者中的CTL细胞具有可检测的LMP2应答。因此,DC/MAC处理更有效且更可重复地产生针对亚优势抗原和表位的T细胞。
图3C:使用巨噬细胞、树突细胞或LCL细胞作为抗原呈递细胞培养的CTL系中为CD4+、CD8+和CD25+ 的存活CD3+细胞%。将系用针对CD3、CD4、CD8和CD25的抗体染色并通过流式细胞术分析。
结论:尽管所有方法建立CD8+细胞而非Tregs,但使用树突细胞和巨噬细胞作为APC似乎相对于用LCLs产生的增加了CD4+细胞的%。
ELISpot测定:
进行ELISPOT测定以确定针对特定抗原产生的T细胞数目。用15 μg/ml的抗IFNγ 单克隆抗体1-DIK (MABTECH, Stockholm, Sweden)包被ELISPOT γ IFN 96孔聚亚乙烯二氟衬板(Millipore, Bedford, MA)。将5×106 PBMCs及每种蛋白各2 μM的肽混合物加入每孔中,并在37摄氏度5% CO2 下孵育过夜。丢弃细胞,并将1 μg/ml生物素化的IFNγ 单克隆抗体7-B6-1 (MABTECH)在室温下孵育2-4小时,随后加入链霉抗生物素缀合的碱性磷酸酶(MABTECH)再孵育2小时。用来自alk-phos底物试剂盒(Bio-Rad Richmond, CA)的5-溴-4-氯-3-吲哚磷酸酯和硝基蓝四唑反应30分钟后,根据样品的数目,使用解剖显微镜或AIDELISPOT读取器(Autoimmun Diagnostika, Strassberg, Germany)对点计数。每一个点为报道为斑点形成细胞(SFC)/ 105 PBMC的细胞。在这些测定中,阳性对照为细胞+ 10 μg/ml PHA,并且阴性对照为单独的不含肽的细胞
图4:由ELISPOT测量的体外培养的IFN产生细胞(SFC) /105 T细胞。
结论:树突细胞和巨噬细胞相比于LCL更选择性地扩增针对亚优势抗原的T细胞。
优化抗原以建立针对亚优势表位的应答。
选择LMP2用于优化,这是由于它是相对于EBNA-3的亚优势抗原,并且如使用共同软件和PBMC测试所鉴定的,所识别的EBV亚优势表位的大部分似乎均在该蛋白质上。为了增加识别的亚优势表位数目,将LMP2分开至两个或更多个质粒上。以下质粒构建于p穿梭或pUC19质粒中,并在CMV启动子和人工ATG和多聚A的控制下:
质粒1 LMP2B (aa 1-497)
质粒2 LMP2A 第一外显子(aa 1-119)
质粒3 LMP2A 第二外显子 (aa 120-497)
质粒4 LMP2A (aa 120-399)
质粒5 LMP2A (aa 400-497)
质粒6 LMP2A (aa 120-440)
质粒7 LMP2A (aa440-497)
质粒8 LMP2A (aa 1-399)
质粒9 LMP2A (aa400-497)
使用标准程序在SCS110细菌菌株(Stratagene, La Jolla, CA)中产生所有质粒,并且用Endo free Plasmid Maxi试剂盒(Qiagen, Hilden, Germany)纯化。在成熟后使用AmaxaDC Nucleofection Kit (Amaxa, Koeln, Germany)用2-20 μg质粒DNA/106细胞转染抗原呈递细胞24小时。
在LMP2上的T细胞免疫亚优势表位包括但不限于:
LLW 329-337 LLWTLVVLL HLA A 2.01
CLG 426-434 CLGGLLTMV HLA A 2.01
IED 200-208 IEDPPFNSL HLA B 40.01
SSC 340-350 SSCSSCPLSKI HLA A11.01
TYG 419-427 TYGPVFMCL HLA A24.02
LLS 447-455 LLSAWILTA HLA A2
LTA 453 461 LTAGFLIFL HLA A2
FLY 356-364 FLYALALL HLA A2
合成这些表位的肽并用于在51C释放测定中测试CTL对从PMBC供体的皮肤分离的表达HLA-A2的成纤维细胞的应答。
图5:以20:1的效应物:靶标(E/T)比(CTL系:HLA匹配成纤维细胞)的51Cr释放,HLA匹配的成纤维细胞用来自LMP2的肽预先脉冲:CTL系用分至1、2、3或4个质粒上的LMP2建立(1P: 497 aa LMP2B; 2P: 第1质粒上的LMP2A 第一外显子(119 aa), LMP2A 第二外显子(378 aa); 3P: 在第1质粒上的LMP2A 第一外显子(119 aa), 在第2质粒上的LMP2A (120-399 aa)和在第3质粒上的LMP2A (400-497 aa); 4P: 在第1质粒上的LMP2A第一外显子(119 aa), 在第2质粒上的LMP2A (120-399 aa),在第3质粒上的LMP2A (400-440 aa),在第4质粒上的LMP2A (440-497 aa); 2P 2: 质粒1 (aa 120-440), 质粒2 (aa 440-497))。
结论:将LMP2分至至少两个质粒上(一个含有aa 400-497,另一个含有399之前的残基)导致针对更大数目的亚优势表位的更强应答。
从EBNA-1序列中删除对应于Gly Ala重复结构域的EBNA-1氨基酸90-325,并插入p穿梭质粒中在CMV启动子的控制下。选择该序列删除是由于证明它抑制肽加工。将以下HLA-A2限制肽用于评估应答:VLK 574-582 HLA A2。
此外,制备删除aa 1-43(以避免它的聚集/保护影响蛋白酶体加工)和删除260-315(5个拷贝的11个氨基酸串联重复)的LMP1序列。将这些序列构建在p穿梭质粒中在CMV启动子控制下,并与野生型LMP1比较。为了评估应答的广度,制备并测试以下LMP1的HLA-A2限制表位:
YLL 125-133 YLLEMLWRL HLA A2
YLQ 159-167 YLQQNWWTL HLA A2
TLL 166-174 TLLVDLLWLL HLA A2
LLV 167-175 LLVDLLWLL HLA A2
LLL 92-100 LLLIALWNL HLA A2
RLG 132-140 RLGATIWQL HLA A2
图6A和6B:以20:1的效应物:靶标(E/T)比(CTL系:HLA匹配的成纤维细胞)的51Cr释放,HLA匹配的成纤维细胞用来自EBNA-1或LMP1的肽预先脉冲:比较用EBNA-1野生型与EBNA-1删除所建立的CTL系,以及比较用LMP1野生型与LMP1删除所建立的CTL系。
结论:删除蛋白的避免抗原加工的某些区域导致针对更大数目的亚优势表位的更强应答。
b.实施例2
在以下实验中,将T细胞如上文实施例1中所概述的相同方式培养,并且设计不同变化以增强抗原加工,以利于T细胞与亚优势表位的产生发生反应。
图7:以20:1的效应物:靶标(E/T)比(CTL系:HLA匹配成纤维细胞)的(51Cr释放)Cr释放,HLA匹配的成纤维细胞用代表优势抗原(EBNA-3A)、亚优势抗原(LMP2)和来自LMP2的特定HLA A2限制性亚优势表位的肽预先脉冲,并且CTLs使用两种不同方法从相同患者中制成:一种在抗原呈递过程中加入100nM-300nM的硼替佐米,另一种则不加入。
结论:在CTL抗原呈递过程中加入蛋白酶体拮抗物硼替佐米修饰了抗原加工以产生针对更广泛数目的亚优势表位的CTL,并且在针对优势表位的应答中适度降低。
图8:以20:1的效应物:靶标(E/T)比(CTL系:HLA匹配的成纤维细胞)的(51Cr释放)Cr释放,HLA匹配的成纤维细胞用代表优势抗原(EBNA-3A)、亚优势抗原(LMP2)和来自LMP2的特异性HLA A2限制性亚优势表位的肽预先脉冲,并且CTLs使用两种不同方法从相同患者中制成:一种在抗原呈递之前12小时和呈递过程中加入10 ng/ml的干扰素γ (IFNγ),另一种则不加入。
结论:在CTL抗原呈递过程中加入IFNγ修饰了抗原加工以产生针对更广泛数目的亚优势表位的CTL,并且在针对优势表位的应答中适度降低。该效应在巨噬细胞中比在树突细胞中更显著。
c.实施例3:肝炎和肝细胞癌
近来已开发了慢性肝炎的小鼠模型,其允许研究乙肝中的原发性和继发性免疫应答(Publicover J等2011)。该HBVtgRAG小鼠是在C57BL/6背景下HBV复制15代的转基因小鼠(HBVRpl)(其组成性地允许病毒复制和病毒体的释放)与RAG-1缺失小鼠(使其无法产生T细胞和B细胞)的杂交小鼠。当从C57BL/6小鼠转移108 脾细胞时,免疫系统重建并且针对肝炎感染的原发性免疫应答建模。如果施用脾细胞至年幼(3-4周龄)小鼠,则动物(与感染有乙肝的幼童类似)会发展慢性肝炎。和人一样,它们清除血清中的HBc,但血清中的HBs保留高水平(图1)。将该慢性肝炎的模型用于以下实验中。
使用ALT-L3K试剂盒(Diagnostic Chemicals Ltd)在Cobas Miras Plus分析仪(Roche diagnostics)上测量丙氨酸转氨酶。使用ETI-MAX 2 Plus (Diasorin)测量HBsAg;使用ETI-AB-AUK-PLUS和ABAU标准组(Diasorin)测量HBs抗体。使用ETI-AB-COREK-PLUS(Diasorin)测量HBcAB。测定在ELx800 (Biotek Instruments)上波长450nm和630nm处读取。
图9:慢性乙肝的HBVtgRAG小鼠模型。
使用我们的方案先体外后体内将T细胞培养为HBs,并在原始脾细胞转移后3-4周通过尾静脉注射。从1×105-1×108细胞进行滴定,在1×106细胞时达到平台。如下文可见,我们的T细胞重新平衡治疗清除了HBVtgRAG模型中的慢性肝炎。
图10:用对HBs Ag反应性的T细胞处理HBVtgRAG模型导致急性炎症和之前的慢性感染的清除。
当在抗原呈递过程中将IL-4或IL-21用作向培养基中的添加物时,达到平台所需的T细胞的量分别为5×105和7×104。该细胞数低于在标准IL-15方案中扩增的T细胞多于一个对数,并且表明在培养过程中向滤泡T辅助细胞和TH2细胞的极化是有利的。另一方面,IL-12的极化增加达到平台所需的细胞数至1×107,并且用IL-2和雷帕霉素的对Treg的极化消除了对治疗的应答。
在清除HBVtgRAG小鼠中的肝炎中不同的培养条件导致对不同T细胞亚群的极化和不同数目的T细胞达到平台。
| 培养条件 | 达到平台的T细胞数目 |
| IL-15 | 1×10<sup>6</sup> |
| IL-15+IL-4 | 5×10<sup>5</sup> |
| IL-15+IL-12 | 1×10<sup>7</sup> |
| IL-15+IL-21 | 7×10<sup>4</sup> |
| IL-2+雷帕霉素 | 无应答 |
因此,T细胞对不同T细胞亚群的极化具有重要的治疗作用。
为了确定T细胞治疗对针对不同表位的T细胞频率的作用,通过ELISPOT测定针对不同表位的IFNγ产生表位的%。在移植后第6周,从HBVtgRAG小鼠中收集脾细胞,所述HBVtgRAG小鼠未用或者用平台水平的针对HBs Ag生成的T细胞处理。用肝炎核心抗原(HBc)、肝炎表面抗原 (HBs)或HBs的两种Kb限制肽对脾细胞脉冲:ILS或WWL。
HBs Ag 190-197 VWLSVIWM Kb
HBs Ag 208-215 ILSPFLPL Kb
观察到以下结果:
图11:用HBs T细胞处理将免疫系统重新平衡至新的优势等级。
如可见的,使用我们的方案制备的T细胞系产生明显更高的针对HBs的T细胞应答和略微降低的针对HBc的应答。HBs应答主要通过针对之前的亚优势表位的从新应答所驱动(WWL)。因此,HBs内的优势等级由T细胞重新平衡治疗所转换,这是由于存在于未处理的动物中的应答(尽管微弱)是针对优势表位(ILS),其相对于之前优势的HBc抗原转换HBV应答的总体平衡以利于之前的亚优势HBs抗原。
还使用HBVtgRAG小鼠模型以研究体内方法是否还能够将免疫优势等级重新平衡。在一种方法中,本发明人通过尾静脉注射(IV)、肌内(IM)或腹腔内(IP)注射弗氏佐剂中的HBs抗原。或者,每只小鼠施用100μg质粒DNA(数据未显示)。
图12:在免疫接种过程中用蛋白酶抑制剂的剂量处理时,IM途径施用导致针对亚优势抗原的更广泛的应答。
如可见的,相比于另外两种,通过IM施用产生了4倍更强的应答,并且这与针对亚优势表位的应答相关。此外,当用蛋白酶体拮抗物硼替佐米处理该小鼠并且抗原经IM施用时,针对该亚优势表位的应答进一步增强。
在第二组实验中,单独以及与不同同种型(IgG1对IgG2a)的两种HBs反应性小鼠抗体复合进行应答。
针对HBs Ag的单克隆抗体
10-H05小鼠IgG1 HBsAg (Fitzgerald, Concord, MA)
10-H05A小鼠IgG2a HBsAg (Fitzgerald, Concord, MA)
图13:对HBs Ag特异性的IgG2a单克隆抗体而非IgG1抗体导致不同的优势等级和通过IV途径的明显更好的抗原呈递。
当将抗原抗体复合物静脉内和IM施用时,IgG2a复合物产生更多的针对亚优势表位的应答。此外,该方案产生了通过IV施用的与通过IM施用获得的几乎相等的免疫应答水平,表明IgG2a免疫复合物的IV施用可以能够重新平衡体内免疫优势等级。
本发明人假设可能该观察的机制是由于IgG2a结合了优先表达于树突细胞中的高亲和力FcR,并因此将HBs抗原靶向树突细胞。此外,由于HBs Ag a 决定簇第一环(aa 124-147)是由中和抗体识别的主要表位,并且ILS表位离得较远,因此可能是对于ILS的抗原加工得到增强。
T细胞重新平衡治疗在HBV相关的肝细胞癌中的临床试验
对5位具有HBV和HCC的为HLA-A *0201的患者进行它们的体液和细胞免疫应答的免疫概况分析。治疗前的临床实验室测试证明针对乙肝核心抗原(HBc Ag)而非针对乙肝表面抗原(HBs Ag)的高效价抗体(图10)。
将PBMC诱导成为树突细胞,用乙肝核心抗原(HBc Ag)和乙肝表面抗原(HBs Ag)脉冲,并用于培养来自PBMC的CTL,其通过ELISPOT测试IFNγ。
图14:来自患者3的105 PBMCs中斑点形成细胞的数目
通过在ELISPOT中产生IFNγ的T细胞的频率,观察到等级:无抗原(10 sfc)<HBsAg(15sfc)<HBcAg(45 sfc)。这表明乙肝表面抗原相对于乙肝核心抗原是亚优势的。此外,研究了3种HBs肽 (FLL、GLS和ILS):
HBs Ag 20-28 FLLTRILTI HLA-A*201
HBs Ag 185-194 GLSPTVWLSV HLA-A*201
HBs Ag 208-216 ILSPFLPLL HLA-A*201
就针对HBs的微弱应答是可检测到的而言,FLL肽似乎稍微是优势的,但由于应答微弱,因此通过ICS测定来确定优势等级:
胞内细胞因子染色(ICS)
将5×105 CTL重悬并在100 μl的含有肽(10-5-10-9 M终浓度)的1XPBS 1% FCS中孵育1小时。随后加入Golgi Plug (BD Biosciences, San Diego, CA),将细胞在37摄氏度 5%CO2下孵育5小时,沉淀,用200μl PBS 1%FCS洗涤,并在4摄氏度下对表面抗原(CD4异硫氰酸荧光素和CD8别藻蓝蛋白(Pharmingen, Becton Dickinson)染色30分钟。在固定和透性溶液中重悬后,将细胞在冰上用1/20稀释的抗人IFNγ藻红蛋白(Pharmingen, BD)染色30分钟,洗涤一次,并在PBS 1% FCS中重悬并在FACS Canto (Becton Dickinson)上分析。
图15:针对HBs表位的应答的ICS分析
尽管通过ICS可检测到的应答并不强,但FLL表位的确表现出相对于其他两种表位为优势的。
将5×106 PBMC从每一患者中分离,并将T细胞按照我们的方案使用HBs 15聚体肽混合物作为刺激抗原进行培养,其中所述肽排除了肽15-30(以消除FLL)。在扩增过程中使用IL-4 + IL-15。将1×1010 T细胞获取并冷冻,并取一等分试样用于ELISPOT测定。
图16:HBs细胞是大部分向患者施用的T细胞,并且这些细胞对之前亚优势的抗原应答。
如可见的,产生的CTL系几乎仅对之前亚优势的HBs具有反应性,其是主要由之前的亚优势表位驱动的应答。将CTL经IV输注以5×107-1×108细胞/m2给药,并监测患者。
图17:患者具有急性发作并随后清除了肝炎。
在发展成急性肝炎后,患者缓解并完全清除了肝炎。在第14天,再次收集PBMC并且再次确定细胞免疫应答的概况。
图18:患者的免疫优势等级已经重新平衡为之前亚优势抗原(HBs)和该抗原上之前亚优势的表位。
通过由我们的方法培养的T细胞已成功地将免疫优势等级重新平衡。除了清除慢性肝炎之外,患者还具有针对他的肝细胞癌的完全应答。
图19:T细胞完全缓解了患者的肝细胞癌。
d.施例4:治疗癌症的系统方法
癌症的临床诊断和治疗涉及肿瘤的活组织检查和成像,以确定它的组织的来源、分化及局部和全身转移的程度。尽管有时进行对癌基因或肿瘤抑制基因中的缺陷的诊断以及确定对化学治疗剂或生物制剂的敏感性的测定,但通常根据患者的具体癌症和阶段,使他们用手术、化学治疗和放射治疗的组合进行治疗。类似地,尽管已经使用了不同技术研究了多种患者中的免疫系统,但这一信息尚无法用于患者的临床处理中。随着他们的细胞治疗以重新平衡免疫系统的来临,本发明人可以建立免疫概况分析在癌症患者的处理中的用途。本发明人已经开发了标准化的免疫概况分析方法学,其用于选择在该患者中是亚优势的抗原并且可以用于体外培养T细胞,所述T细胞能够重新输注入患者中以重新平衡针对肿瘤的免疫应答。治疗后,对患者进行免疫概况分析以确定是否该治疗成功地将免疫应答重新平衡。这样的治疗方法是新颖的,并且能导致临床应答和增进存活。同样的方法可用于传染病和自身免疫和器官移植。
第一步是鉴定哪一种与肿瘤相关的抗原存在于患者的肿瘤中。这通常通过对来自患者肿瘤的活体样本上进行免疫组织化学来完成。检测抗原组将依赖于肿瘤类型。例如,在黑素瘤中,抗原包括NY-ESO-1、SSX-2、Melan A、gp100、MAGE A4、MAGE A1、酪氨酸酶,并且当描述了新的肿瘤相关抗原时将被补充。并非所有肿瘤都将表达组内的所有抗原。对于患者肿瘤中具有的抗原,使用如上所述对体液和细胞免疫应答的系统性概况分析来确定针对不同抗原的基线免疫概况。使用Elisa来确定血清中针对每一抗原的抗体滴度,并且通过IFNγ ICS或ELISPOT测定对抗原相应的T细胞的%。以这种方式,确定基线概况。
图20:患者1中对肿瘤抗原组的IFNγ ICS
在患者1中,患者的肿瘤具有NYESO-1、SSX-2、Melan A和MAGE A4。ICS证明对MAGE A4的强应答,但对NYESO-1、SSX-2和Melan A的中度/无应答。因此选择NYESO-、SSX-2和Melan A抗原使用以下方案来在体外培养中生长并扩增CTL:
从血液中分离5×106 PBMC。通过将PBMCs铺于组织培养瓶中2小时以允许单核细胞的粘附,从患者的外周血单核细胞(PBMCs)体外产生单核细胞来源的树突细胞。此时,将未粘附的细胞去除并冷冻于-80中后期用作T细胞来源。用白细胞介素4(IL-4)和粒细胞-巨噬细胞菌落刺激因子(GM-CSF)处理粘附的单核细胞导致在约1周内分化为未成熟的树突细胞(iDCs)。用肿瘤坏死因子(TNF)的后续处理进一步将iDCs分化为成熟的树突细胞。随后将这些细胞分至3个单独的瓶(或者无论多少希望用于培养T细胞的抗原数量)中,其含有添加有45% Click培养基(Irvine Scientific, Santa Ana, CA)、2mM Glutamax 1和5%人血清的RPMI 1640培养基。用包含目标抗原的编码序列(在该患者1中含有NY-ESO-1,1位患者含有SSX-2,并且第三位含有Melan A)的3种质粒(或pepmix)之一将每一瓶脉冲。将细胞储存在37摄氏度下2小时。同时,将PBMC融化并以PBMC对树突细胞比例1:20-1:100加入经脉冲的树突细胞,并在37摄氏度下孵育18小时。随后合并三瓶细胞并在含有IL-15 (5ng/ml)相同的培养基中重悬,以产生已识别抗原的T细胞的体外扩增。使用Grex气体渗透生物反应器(Wilson Wolf Manufacturing Minneapolis, MN),从而不需更换培养基并实现指数生长动力学。通常在3-6周内获得3×108-1.5×1010细胞的足够细胞以向患者施用。
T细胞在气体渗透生物反应器中生长。
T细胞在气体渗透膜上生长为单层以优良的气体交换,并具有足以生长至所需密度的培养基体积。
在体外培养结束时,用相同抗原在ICS测定中测定T细胞:NY-ESO-1、SSX-2、MelanA、gp100、MAGE A4、MAGE A1、酪氨酸酶。
图21:患者1的ICS测定
基于测定,在该情况下确定针对每一亚优势抗原/表位(NY-ESO-1、SSX-2和Melan A)应答的细胞的%。对每一亚优势抗原或表位应答细胞总数可以使用该%和培养物中CTLs的数目来计算。在患者1的情况下,61.4%的细胞对NY-ESO-1应答(图21)。
基于这一点,可以将对亚优势抗原应答的CTLs的已知剂量向患者施用。将CTL以5×106-2×108细胞/m2给药。
向患者输注后2-3周,再次收集PBMC,并通过ICS再次确定细胞免疫应答的概况:
图22:患者1-治疗后通过ICS的细胞免疫概况分析
图23:患者1-通过ELISA的体液免疫概况分析(体液免疫应答的倒数效价在下文作图)。
如同从免疫等级测定的细胞和体液概况中可见,免疫优势等级被重新平衡以利于之前的亚优势抗原。
临床发现:
临床上,患者为52岁男性,其被诊断患有IV期转移性黑素瘤。他对组合有IL-2的达卡巴嗪(DTIC)和Temodar(替莫唑胺(Temolzolomide))化学治疗剂的组合方案无应答。
T细胞治疗两年后,患者存活并且已进行完全应答。如胸部CT扫描中可见,在免疫重新平衡治疗的6个月内肺中的转移完全缓解并且已保持稳定。
图24:T细胞治疗前和后的CT扫描
e. 实施例5:淋巴瘤的T细胞治疗
将150位患有复发性侵袭性非霍奇金淋巴瘤的成年患者随机分为3组:A:Rituxan +CHOP; B:测试肿瘤中的EBV LMP1和LMP2抗原,随后为EBV LMP1和LMP2 T细胞重新平衡和C:泛淋巴瘤:不测定抗原;用对EBV LMP1、LMP2、存活、MAGE A3应答生长的T细胞治疗。40%的淋巴瘤活组织检查对EBV LMP2测试为阳性,50%的测试对存活为阳性,并且15%的测试对MAGE A3为阳性。使用Care R-CHOP方案的标准,并以5×107-2×108 T细胞/m2给药。
图25:无疾病进展的存活
如可见的,对肿瘤应答的T细胞提供了优于目前的护理标准的应答率。尽管在第1年中发生失败,但第1年后CTL使患者维持减轻。这是免疫系统重新平衡后正常行使功能的证据。最后,尽管最初EBV LMP T细胞产生了较好的应答,在到第3年,无疾病进展的存活已达到泛淋巴瘤产品的应答。此外,如今的淋巴瘤是复发性-减轻性疾病,患者通常在18个月至2年内复发。CTL重新平衡治疗改变了这一过程:一旦患者的免疫系统重新平衡,患者会发展出维持长期减轻的记忆应答。因此,与淋巴瘤的其他治疗不同,T细胞治疗提供了持久的减轻。
图26:T细胞治疗改变了疾病的自然史
f.实施例6:自身免疫疾病的T细胞治疗
胶原诱导的关节炎模型(CIA)是类风湿关节炎(RA)模型,其能够由用异源胶原II(CII)在DBA/1小鼠中免疫来诱导。
DBA/1雄性6-8周龄的小鼠获自Jackson Laboratories (Bar Harbor, ME)。将100μg含有4 mg/ml结核分枝杆菌(Chondrex)在CFA中乳化的牛CII (Chondrex, Redmond, WA)经皮下注射入尾中。到注射后第5周,80-100%的未处理的小鼠显示出完全发生的疾病。
使用对T细胞的体外生长所述的方案将T细胞以下肽培养。
II型胶原
用于刺激Treg生长的263-270免疫优势肽(IL-2 + 雷帕霉素)
用于刺激T细胞生长的286-300亚优势肽(IL-15)
在诱导后第20天将5×106细胞单独或与相等部分组合向每一动物施用。见图27。CD25是Treg细胞的标志物。对照动物接受PBS。见图28。
每周三次对小鼠进行临床疾病评分,使用每肢0-3分,最大可能总分为12分:0-1正常;1 单趾轻微发红或肿胀;2 踝或腕明显肿胀并有红斑;3 多个关节的严重肿胀及红斑。组中具有关节炎损伤的动物的百分比代表关节炎的发生率。组中平均临床分数反映了疾病的严重性。参见图28和图29。
关节的组织病理学
图30A显示了正常大鼠,图30B显示了用人蛋白聚糖免疫的大鼠,并且图30C显示了用T细胞处理的大鼠。
如可见的,T细胞治疗明显降低了动物关节中炎症的发生率、严重性和量。在重新平衡针对亚优势表位生长的T细胞和针对优势表位生长的Treg之间的免疫应答中似乎存在协同效应。
序列表
<110> 金纽斯生物科技投资有限责任公司(GENEIUS BIOTECHNOLOGY INVESTMENTS,LLC)
<120> 调节的免疫优势疗法
<130> 44270-501001WO
<140> PCT/US2012/039605
<141> 2012-05-25
<150> US 61/490,505
<151> 2011-05-26
<160> 19
<170> PatentIn version 3.5
<210> 1
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 1
Leu Leu Trp Thr Leu Val Val Leu Leu
1 5
<210> 2
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 2
Cys Leu Gly Gly Leu Leu Thr Met Val
1 5
<210> 3
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 3
Ile Glu Asp Pro Pro Phe Asn Ser Leu
1 5
<210> 4
<211> 11
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 4
Ser Ser Cys Ser Ser Cys Pro Leu Ser Lys Ile
1 5 10
<210> 5
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 5
Thr Tyr Gly Pro Val Phe Met Cys Leu
1 5
<210> 6
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 6
Leu Leu Ser Ala Trp Ile Leu Thr Ala
1 5
<210> 7
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 7
Leu Thr Ala Gly Phe Leu Ile Phe Leu
1 5
<210> 8
<211> 8
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 8
Phe Leu Tyr Ala Leu Ala Leu Leu
1 5
<210> 9
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 9
Tyr Leu Leu Glu Met Leu Trp Arg Leu
1 5
<210> 10
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 10
Tyr Leu Gln Gln Asn Trp Trp Thr Leu
1 5
<210> 11
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 11
Thr Leu Leu Val Asp Leu Leu Trp Leu Leu
1 5 10
<210> 12
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 12
Leu Leu Val Asp Leu Leu Trp Leu Leu
1 5
<210> 13
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 13
Leu Leu Leu Ile Ala Leu Trp Asn Leu
1 5
<210> 14
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 14
Arg Leu Gly Ala Thr Ile Trp Gln Leu
1 5
<210> 15
<211> 8
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 15
Val Trp Leu Ser Val Ile Trp Met
1 5
<210> 16
<211> 8
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 16
Ile Leu Ser Pro Phe Leu Pro Leu
1 5
<210> 17
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 17
Phe Leu Leu Thr Arg Ile Leu Thr Ile
1 5
<210> 18
<211> 10
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 18
Gly Leu Ser Pro Thr Val Trp Leu Ser Val
1 5 10
<210> 19
<211> 9
<212> PRT
<213> 人工序列
<220>
<223> 合成的肽
<400> 19
Ile Leu Ser Pro Phe Leu Pro Leu Leu
1 5
Claims (16)
1.制备用于改变患者的免疫优势等级的T细胞群体的方法,所述方法包括以下步骤:
a. 鉴定获自所述患者的样品中至少一种亚优势抗原或亚优势表位,
b. 获得各自包含抗原呈递细胞(APC)和/或T细胞的一份或多份患者样品;
c. 使来自所述患者样品的APC暴露于一种或多种亚优势抗原或表位;
c. 通过以下从所述患者样品培养包含能够识别所述亚优势抗原或表位的T细胞的T细胞群体:
(i) 使T细胞在包含IL-7、IL-12、IL-15和IL-21中的一种或多种的培养基中暴露于来自步骤(c)的APC,
(ii) 在所述培养基中扩增T细胞,和
(iii) 任选地通过暴露于来自步骤(c)的APC,再次刺激T细胞。
2.权利要求1的方法,其中所述患者样品包含外周血单核细胞(PBMC)。
3.权利要求1的方法,其还包括分离APC的步骤。
4.权利要求1的方法,其中所述抗原呈递细胞是树突细胞。
5.权利要求4的方法,其中步骤(c)包括使多个等分试样的树突细胞各自暴露于不同的亚优势抗原。
6.权利要求1的方法,其中使T细胞暴露于来自步骤(c)的APC的步骤包括使T细胞在进一步包含选自IL-2、IL-4和IL-6中的细胞因子的培养基中暴露于来自步骤(c)的APC。
7.权利要求1的方法,其中使T细胞暴露于来自步骤(c)的APC的步骤包括使T细胞在包含选自IL-15的培养基中暴露于来自步骤(c)的APC。
8.权利要求1的方法,其中使T细胞暴露于来自步骤(c)的APC的步骤包括使T细胞在包含选自IL-15和IL-21的培养基中暴露于来自步骤(c)的APC。
9.权利要求1的方法,其中使T细胞暴露于来自步骤(c)的APC的步骤包括使T细胞在包含选自IL-14和IL-15的培养基中暴露于来自步骤(c)的APC。
10.权利要求1的方法,其中在培养基中扩增T细胞的步骤包括使用包含IL-2、IL-4、IL-6、IL-7、IL-12、IL-15和IL-21中的一种或多种的培养基。
11.权利要求1的方法,其中在培养基中扩增T细胞的步骤包括使用包含IL-7和IL-15中的一种或多种的培养基。
12.权利要求1的方法,其中所述亚优势抗原或表位包括病毒抗原、真菌抗原、细菌抗原、寄生虫抗原、朊病毒抗原或肿瘤抗原。
13.权利要求12的方法,其中所述亚优势抗原或表位作为一种或多种肽或作为表达亚优势抗原或表位的质粒或载体呈递给APC。
14.权利要求1的方法,其中在培养基中扩增T细胞的步骤包括在气体可渗透的生物反应器中培养T细胞。
15.通过权利要求1的方法获得的T细胞群体。
16.权利要求15的T细胞群体,其中至少一种亚优势抗原或表位是针对其的细胞或体液免疫应答在患者样品中是不可检测的或仅低水平可测的抗原或表位。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161490505P | 2011-05-26 | 2011-05-26 | |
| US61/490505 | 2011-05-26 | ||
| CN201280037470.4A CN103906531A (zh) | 2011-05-26 | 2012-05-25 | 调节的免疫优势疗法 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280037470.4A Division CN103906531A (zh) | 2011-05-26 | 2012-05-25 | 调节的免疫优势疗法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN111529697A true CN111529697A (zh) | 2020-08-14 |
Family
ID=46208182
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280037470.4A Pending CN103906531A (zh) | 2011-05-26 | 2012-05-25 | 调节的免疫优势疗法 |
| CN201911266293.7A Pending CN111529697A (zh) | 2011-05-26 | 2012-05-25 | 调节的免疫优势疗法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280037470.4A Pending CN103906531A (zh) | 2011-05-26 | 2012-05-25 | 调节的免疫优势疗法 |
Country Status (12)
| Country | Link |
|---|---|
| US (7) | US20140099341A1 (zh) |
| EP (2) | EP2714072A1 (zh) |
| JP (1) | JP2014516538A (zh) |
| KR (2) | KR20160104753A (zh) |
| CN (2) | CN103906531A (zh) |
| AU (4) | AU2012258603A1 (zh) |
| CA (1) | CA2874431A1 (zh) |
| HK (1) | HK1199404A1 (zh) |
| RU (1) | RU2013157923A (zh) |
| SG (2) | SG10202012534VA (zh) |
| TW (3) | TW201313902A (zh) |
| WO (1) | WO2012162620A1 (zh) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2018013959A (es) * | 2016-05-25 | 2019-08-22 | Council Queensland Inst Medical Res | Metodos para tratar enfermedad autoinmune usando celulas t alogenicas. |
| NZ749136A (en) | 2016-05-25 | 2023-11-24 | Council Queensland Inst Medical Res | Methods of immunotherapy |
| AU2017283480A1 (en) | 2016-06-13 | 2019-01-24 | Torque Therapeutics, Inc. | Methods and compositions for promoting immune cell function |
| US20200237819A1 (en) * | 2016-06-28 | 2020-07-30 | Geneius Biotechnology, Inc. | T cell compositions for immunotherapy |
| KR20210032924A (ko) | 2017-09-05 | 2021-03-25 | 토크 테라퓨틱스, 인코포레이티드 | 치료용 단백질 조성물 및 그의 제조 및 사용 방법 |
| EP3700553A1 (en) * | 2017-10-23 | 2020-09-02 | Atara Biotherapeutics, Inc. | Methods of managing tumor flare in adoptive immunotherapy |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998033888A1 (en) * | 1997-01-31 | 1998-08-06 | Epimmune, Inc. | Peptides and peptide-loaded antigen presenting cells for the activation of ctl |
| CN1323217A (zh) * | 1998-10-05 | 2001-11-21 | M&E生物技术公司 | 治疗性接种的新方法 |
| CN1440462A (zh) * | 2000-04-28 | 2003-09-03 | 麦康公司 | 鉴定和生产抗原肽的方法并且将其用作疫苗 |
| CN101031641A (zh) * | 2004-08-19 | 2007-09-05 | 伯哈德·莫瑟 | 呈递抗原的人γδT细胞的制备和在免疫治疗中的用途 |
| CN101146550A (zh) * | 2004-12-29 | 2008-03-19 | 曼康公司 | 用于预防或治疗目的的激发、增强和维持对i类mhc-限制性表位的免疫应答的方法 |
| CN101198620A (zh) * | 2005-06-17 | 2008-06-11 | 曼康公司 | 激发针对在癌细胞和肿瘤基质上表达的结构域和亚结构域表位的多价免疫应答的方法和组合物 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0914142B9 (en) * | 1996-03-11 | 2012-01-04 | Epimmune Inc. | Peptides with increased binding affinity for at least three hla-a3-like molecules |
| ATE437366T1 (de) | 1998-05-11 | 2009-08-15 | Miltenyi Biotec Gmbh | Verfahren zur direkten auswahl von antigen- spezifischen t-zellen |
| FR2812087B1 (fr) * | 2000-07-21 | 2007-05-11 | Inst Nat Sante Rech Med | Procede de criblage de peptides utilisables en immunotherapie |
| CA2505379A1 (en) | 2002-11-07 | 2004-05-21 | Johnson & Johnson Research Pty Limited | A means of producing and utilising a population of disease specific cytotoxic t-lymphocytes |
| AU2010289785A1 (en) | 2009-08-24 | 2012-03-15 | Baylor College Of Medicine | Generation of CTL lines with specificity against multiple tumor antigens or multiple viruses |
-
2012
- 2012-05-25 RU RU2013157923/10A patent/RU2013157923A/ru not_active Application Discontinuation
- 2012-05-25 SG SG10202012534VA patent/SG10202012534VA/en unknown
- 2012-05-25 EP EP12725589.1A patent/EP2714072A1/en not_active Withdrawn
- 2012-05-25 CN CN201280037470.4A patent/CN103906531A/zh active Pending
- 2012-05-25 WO PCT/US2012/039605 patent/WO2012162620A1/en active Application Filing
- 2012-05-25 CN CN201911266293.7A patent/CN111529697A/zh active Pending
- 2012-05-25 KR KR1020167023641A patent/KR20160104753A/ko active Search and Examination
- 2012-05-25 EP EP23158828.6A patent/EP4285922A1/en active Pending
- 2012-05-25 JP JP2014512143A patent/JP2014516538A/ja active Pending
- 2012-05-25 US US14/122,036 patent/US20140099341A1/en not_active Abandoned
- 2012-05-25 AU AU2012258603A patent/AU2012258603A1/en not_active Abandoned
- 2012-05-25 SG SG10201604236RA patent/SG10201604236RA/en unknown
- 2012-05-25 KR KR1020137034616A patent/KR20140068810A/ko active Application Filing
- 2012-05-25 CA CA2874431A patent/CA2874431A1/en active Pending
- 2012-05-28 TW TW101118979A patent/TW201313902A/zh unknown
- 2012-05-28 TW TW108117367A patent/TWI774957B/zh active
- 2012-05-28 TW TW111127515A patent/TW202321442A/zh unknown
-
2014
- 2014-12-29 HK HK14113011.2A patent/HK1199404A1/zh unknown
-
2016
- 2016-10-28 US US15/337,415 patent/US20170042998A1/en not_active Abandoned
- 2016-10-28 US US15/337,506 patent/US20170042999A1/en not_active Abandoned
- 2016-10-28 US US15/337,556 patent/US20170101625A1/en not_active Abandoned
- 2016-10-28 US US15/337,661 patent/US20170043001A1/en not_active Abandoned
- 2016-10-28 US US15/337,620 patent/US20170043000A1/en not_active Abandoned
-
2017
- 2017-04-13 US US15/486,864 patent/US11723921B2/en active Active
- 2017-05-31 AU AU2017203671A patent/AU2017203671A1/en not_active Abandoned
-
2019
- 2019-03-01 AU AU2019201456A patent/AU2019201456A1/en not_active Abandoned
-
2021
- 2021-02-10 AU AU2021200831A patent/AU2021200831A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998033888A1 (en) * | 1997-01-31 | 1998-08-06 | Epimmune, Inc. | Peptides and peptide-loaded antigen presenting cells for the activation of ctl |
| CN1323217A (zh) * | 1998-10-05 | 2001-11-21 | M&E生物技术公司 | 治疗性接种的新方法 |
| CN1440462A (zh) * | 2000-04-28 | 2003-09-03 | 麦康公司 | 鉴定和生产抗原肽的方法并且将其用作疫苗 |
| CN101031641A (zh) * | 2004-08-19 | 2007-09-05 | 伯哈德·莫瑟 | 呈递抗原的人γδT细胞的制备和在免疫治疗中的用途 |
| CN101146550A (zh) * | 2004-12-29 | 2008-03-19 | 曼康公司 | 用于预防或治疗目的的激发、增强和维持对i类mhc-限制性表位的免疫应答的方法 |
| CN101198620A (zh) * | 2005-06-17 | 2008-06-11 | 曼康公司 | 激发针对在癌细胞和肿瘤基质上表达的结构域和亚结构域表位的多价免疫应答的方法和组合物 |
Non-Patent Citations (3)
| Title |
|---|
| FRAIA MELCHIONDA等: "Adjuvant IL-7 or IL-15 overcomes immunodominance and improves survival of the CD8+ memory cell pool" * |
| HANH K. LE等: "Incubation of antigen-sensitized T lymphocytes activated with bryostatin 1 + ionomycin in IL-7 + IL-15 increases yield of cells capable of inducing regression of melanoma metastases compared to culture in IL-2" * |
| REDCHENKO等: "Accessing Epstein-Barr Virus-Specifific T-Cell Memory with Peptide-Loaded Dendritic Cells" * |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20160104753A (ko) | 2016-09-05 |
| US20170042999A1 (en) | 2017-02-16 |
| SG10201604236RA (en) | 2016-07-28 |
| US20170043000A1 (en) | 2017-02-16 |
| AU2012258603A1 (en) | 2014-01-16 |
| US20170216357A1 (en) | 2017-08-03 |
| US11723921B2 (en) | 2023-08-15 |
| RU2013157923A (ru) | 2015-07-10 |
| WO2012162620A1 (en) | 2012-11-29 |
| US20140099341A1 (en) | 2014-04-10 |
| TWI774957B (zh) | 2022-08-21 |
| US20170042998A1 (en) | 2017-02-16 |
| TW201313902A (zh) | 2013-04-01 |
| TW201934749A (zh) | 2019-09-01 |
| CN103906531A (zh) | 2014-07-02 |
| AU2017203671A1 (en) | 2017-06-15 |
| AU2019201456A1 (en) | 2019-03-21 |
| KR20140068810A (ko) | 2014-06-09 |
| AU2021200831A1 (en) | 2021-03-04 |
| JP2014516538A (ja) | 2014-07-17 |
| TW202321442A (zh) | 2023-06-01 |
| CA2874431A1 (en) | 2012-11-29 |
| HK1199404A1 (zh) | 2015-07-03 |
| EP2714072A1 (en) | 2014-04-09 |
| US20170043001A1 (en) | 2017-02-16 |
| EP4285922A1 (en) | 2023-12-06 |
| US20170101625A1 (en) | 2017-04-13 |
| SG10202012534VA (en) | 2021-01-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11723921B2 (en) | Modulated immunodominance therapy | |
| ES2292619T3 (es) | Composiciones y metodos para inducir respuestas citologicas especificas de la celula t. | |
| RU2734784C2 (ru) | Способы отбора t-клеточной линии и ее донора для адоптивной клеточной терапии | |
| WO2016179573A1 (en) | Variant survivin vaccine for treatment of cancer | |
| US6821778B1 (en) | Methods for using dendritic cells to activate gamma/delta-T cell receptor-positive T cells | |
| KR20080048455A (ko) | 기억 티 림프구들의 가시화, 분리 및 유전자 변형에사용되는 통상의 감마 사슬 사이토카인들의 용도 | |
| JP2019530449A (ja) | 幹細胞様メモリーt細胞の生成および養子免疫療法における使用 | |
| Gary et al. | Clinical-grade generation of peptide-stimulated CMV/EBV-specific T cells from G-CSF mobilized stem cell grafts | |
| US20140234353A1 (en) | Methods of obtaining antigen-specific t cell populations | |
| WO2006026746A2 (en) | Methods to separate and expand antigen-specific t cells | |
| AU2011240752A1 (en) | Methods and compositions for inhibition of Treg cells | |
| van Essen et al. | Role of B cells in maintaining helper T–cell memory | |
| JP2005538167A (ja) | インターロイキン12(il−12)を発現する樹状細胞(dc)の使用 | |
| Cornforth et al. | Autologous peripheral blood mononuclear cell recognition of autologous proliferating tumor cells in the context of a patient-specific vaccine trial | |
| KR101777252B1 (ko) | T 세포의 prrsv 증식 억제능 평가 방법 | |
| Bollard et al. | Manufacture of GMP-grade cytotoxic T lymphocytes specific for LMP1 and LMP2 for patients with EBV-associated lymphoma | |
| WO2021119293A2 (en) | T-cell epitopes of human parainfluenza virus 3 for adoptive t-cell immunotherapy | |
| WO2003008558A2 (en) | Cd4+cd25+ inhibitory hybridoma clones |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 40035507 Country of ref document: HK |