CN111032043A - 2-(4-氯苯基)-n-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的组合物和使用方法 - Google Patents
2-(4-氯苯基)-n-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的组合物和使用方法 Download PDFInfo
- Publication number
- CN111032043A CN111032043A CN201880056213.2A CN201880056213A CN111032043A CN 111032043 A CN111032043 A CN 111032043A CN 201880056213 A CN201880056213 A CN 201880056213A CN 111032043 A CN111032043 A CN 111032043A
- Authority
- CN
- China
- Prior art keywords
- inhibitor
- formulation
- compound
- amount
- leukemia
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images































































































































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/20—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing sulfur, e.g. dimethyl sulfoxide [DMSO], docusate, sodium lauryl sulfate or aminosulfonic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/40—Cyclodextrins; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6949—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit inclusion complexes, e.g. clathrates, cavitates or fullerenes
- A61K47/6951—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit inclusion complexes, e.g. clathrates, cavitates or fullerenes using cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
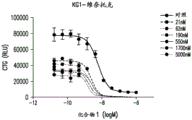
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/0006—Homoglycans, i.e. polysaccharides having a main chain consisting of one single sugar, e.g. colominic acid
- C08B37/0009—Homoglycans, i.e. polysaccharides having a main chain consisting of one single sugar, e.g. colominic acid alpha-D-Glucans, e.g. polydextrose, alternan, glycogen; (alpha-1,4)(alpha-1,6)-D-Glucans; (alpha-1,3)(alpha-1,4)-D-Glucans, e.g. isolichenan or nigeran; (alpha-1,4)-D-Glucans; (alpha-1,3)-D-Glucans, e.g. pseudonigeran; Derivatives thereof
- C08B37/0012—Cyclodextrin [CD], e.g. cycle with 6 units (alpha), with 7 units (beta) and with 8 units (gamma), large-ring cyclodextrin or cycloamylose with 9 units or more; Derivatives thereof
- C08B37/0015—Inclusion compounds, i.e. host-guest compounds, e.g. polyrotaxanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L5/00—Compositions of polysaccharides or of their derivatives not provided for in groups C08L1/00 or C08L3/00
- C08L5/16—Cyclodextrin; Derivatives thereof
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Dermatology (AREA)
- Inorganic Chemistry (AREA)
- Polymers & Plastics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Materials Engineering (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本文提供了2‑(4‑氯苯基)‑N‑((2‑(2,6‑二氧代哌啶‑3‑基)‑1‑氧代异吲哚啉‑5‑基)甲基)‑2,2‑二氟乙酰胺或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物的制剂和使用方法。
Description
相关申请
本申请要求以下美国临时申请号的权益:2017年6月30日提交的62/527,744、2018年4月5日提交的62/653,436,以及2018年5月17日提交的62/673,064,它们中每一者的公开内容都全文以引用方式并入。
技术领域
提供了2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物的制剂和剂型。本文还提供了使用所述制剂和剂型来治疗、管理和/或预防癌症的方法。因此,本文提供了在治疗、管理和/或预防癌症的方法中使用的所述制剂和剂型。
本文还提供了用2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺或其立体异构体或立体异构体混合物、同位素体、药学上可接受的盐、互变异构体、溶剂合物、水合物、共晶体、笼形包合物或多晶型物和第二剂的组合来治疗、预防、管理和/或改善癌症的方法。因此,本文提供了在此类方法中使用的2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺或其立体异构体或立体异构体混合物、同位素体、药学上可接受的盐、互变异构体、溶剂合物、水合物、共晶体、笼形包合物或多晶型物和第二剂的组合。
背景技术
2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物已显示具有抗癌活性。该化合物的示例性制剂公开于2017年1月6日提交的美国公布号2017-0196847中。
需要2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物的另外的方法和制剂来治疗癌症。
发明内容
本文的制剂和方法中所使用的化合物1在美国专利号9,499,514和国际公布号WO2016/007848中进行了描述,这两份专利各自的公开内容全文以引用方式并入本文。在一个实施方案中,化合物1是2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的多晶型物A型、B型、C型、D型、E型或无定形形式。在一个实施方案中,化合物1是2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的多晶型物C型。2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的多晶型物在本文和2017年1月6日提交的美国公布号2017-0197934中进行了描述,该美国公布号的公开内容全文以引用方式并入本文。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.2%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂、量为约92%至98%的羟丙基β-环糊精,和不超过约1%的二甲基亚砜。在一个实施方案中,柠檬酸盐缓冲剂包括无水柠檬酸和无水柠檬酸钠。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.25%的化合物1、量为约99.1%至99.9%的羟丙基β-环糊精,和不超过约0.5%的甲酸。
在一个实施方案中,本文提供以下制剂:其包含量为约0.01%至0.15%的化合物1、量为约99.1%至99.99%的羟丙基β-环糊精。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.01%至0.15%的化合物1、量为约99.1%至99.99%的羟丙基β-环糊精,和不超过约0.5%的甲酸。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.2%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂、量为约92%至98%的磺丁基醚-β-环糊精,和不超过约1%的二甲基亚砜。在一个实施方案中,柠檬酸盐缓冲剂包括无水柠檬酸和无水柠檬酸钠。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.25%的化合物1、量为约99.1%至99.9%的磺丁基醚-β-环糊精,和不超过约0.5%的甲酸。
在一个实施方案中,本文提供了治疗、预防、管理和/或改善癌症(包括实体肿瘤和血液学癌症)或者癌症的一种或多种症状或起因的方法,方式为将化合物1与选自下列的一种或多种第二剂组合施用:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。因此,本文提供了用于此类方法中的化合物1,其中该方法包括将化合物1与选自下列的一种或多种第二剂组合施用:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供的方法包括将化合物1的制剂与选自下列的一种或多种第二剂组合施用:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在某些实施方案中,本文提供的制剂包含2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的固体形式。在某些实施方案中,本文提供的制剂包含2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的无定形形式。
在某些实施方案中,本文提供了包含制剂的单位剂型,其中该制剂包含化合物1、缓冲剂和增量剂。
在一个方面,将包含治疗有效浓度的化合物1的制剂施用于表现出待治疗的疾病或疾患的症状的个体。这些量能有效地改善或消除该疾病或疾患的一种或多种症状。
还提供了药物包装或药盒,其包括填充有药物组合物的一种或多种成分的一个或多个容器。任选地,与一个或多个此类容器相关联的可以是由管理药物产品或生物产品的制造、使用或销售的政府机构规定的形式的通告,该通告反映获得了该机构对制造、使用或销售用于向人施用的批准。可以用有关施用方式、药物施用顺序(例如,分别、相继或同时)等的信息来标记该包装或药盒。
参考以下具体实施方式,本文所述主题的这些方面和其它方面将变得显而易见。
附图说明
图1描绘了化合物1的A型、B型、C型、D型和E型的X-射线粉末衍射图叠加图。
图2描绘了化合物1的A型的X-射线粉末衍射(XRPD)图。
图3描绘了化合物1的A型的SEM图像。
图4描绘了化合物1的A型的热重分析(TGA)图。
图5描绘了化合物1的A型的差示扫描量热(DSC)热谱图。
图6提供了化合物1的A型的动态蒸气吸附(DVS)等温线图。
图7提供了化合物1的A型的1H NMR谱。
图8描绘了化合物1的A型在压缩之前(a)和之后(b)的X-射线粉末衍射图的比较。
图9描绘了化合物1的B型的XRPD图。
图10描绘了化合物1的B型的SEM图像。
图11描绘了化合物1的B型的TGA热谱图。
图12描绘了化合物1的B型的DSC热谱图。
图13提供了化合物1的B型的DVS等温线图。
图14提供了化合物1的B型的1H NMR谱。
图15描绘了化合物1的B型在压缩之前(a)和之后(b)的X-射线粉末衍射图的比较。
图16描绘了化合物1的C型的XRPD图。
图17描绘了化合物1的C型的SEM图像。
图18描绘了化合物1的C型的TGA热谱图。
图19描绘了化合物1的C型的DSC热谱图。
图20提供了化合物1的C型的DVS等温线图。
图21提供了化合物1的C型的1H NMR谱。
图22描绘了化合物1的C型在压缩之前(a)和之后(b)的X-射线粉末衍射图的比较。
图23描绘了化合物1的D型的XRPD图。
图24描绘了化合物1的D型的TGA热谱图。
图25描绘了化合物1的E型的XRPD图。
图26描绘了化合物1的E型的TGA热谱图。
图27描绘了无定形化合物1的经调制DSC热谱图。
图28描绘了无定形化合物1的XRPD图。
图29描绘了无定形化合物1的1H NMR谱。
图30提供了化合物1在各种类型、品牌和百分比的环糊精中,连同在具有不同的溶剂和溶剂与环糊精比率的情况下的溶解度。
图31提供了本体溶液的最终pH相对于柠檬酸盐缓冲剂的pH和强度的关系。
图32提供了制剂Ib的第一按比例放大批次的冻干曲线。
图33提供了制剂Ib的残留溶剂随冻干工艺时间的变化。
图34提供了制剂Ib的第二按比例放大批次的冻干曲线。
图35提供了制剂Ia的过程图。
图36提供了制剂Ib的过程图。
图37示出了在25℃下化合物1的溶解度随Kleptose浓度的增加(自下而上)。
图38证明了Kleptose浓度增加对化合物1沉淀情况的作用。
图39示出了在冷藏条件下,Kleptose溶液中的化合物1沉淀情况。
图40展示了化合物1和Kleptose的制剂的设计空间的轮廓。
图41证明了借助同一个冻干周期,随着Kleptose量增加,甲酸的去除量减少。
图42证明了粉饼厚度对残留甲酸水平的影响。
图43示出了制剂Ic的原型中每mg剂量的残留甲酸相对于Kleptose浓度的关系。
图44证明了复原溶液的克分子渗透压浓度与Kleptose浓度线性相关。
图45提供了制剂Ic的实验室规模批次1的冻干工艺的产品温度曲线。
图46提供了制剂Ic的实验室规模批次2的冻干工艺的产品温度曲线。
图47提供了制剂Ic的开发批次Ic-1的冻干工艺的产品温度曲线。
图48提供了制剂Ic的开发批次Ic-2的冻干工艺的产品温度曲线。
图49提供了制剂Ic的开发批次Ic-3的冻干工艺的产品温度曲线。
图50提供了制剂Ic的开发批次Ic-1-F1、Ic-1-F2、Ic-2-F1和Ic-2-F2的残留水分随冻干周期时间而变化的图。
图51提供了制剂Ic的开发批次Ic-1-F1、Ic-1-F2、Ic-2-F1、Ic-2-F2、Ic-3-F1和Ic-3-F2的残留甲酸随二次干燥时间而变化的图。
图52提供了制剂Ic的批次C1的冻干工艺的产品温度曲线。
图53提供了制剂Ic的批次C2的冻干工艺的产品温度曲线。
图54展示了制剂Ic的批次C1和C2的冻干粉饼外观。
图55提供了用于制备制剂Ic的过程图。
图56提供了在各种浓度的第二剂存在下,化合物1的细胞增殖剂量响应(EC50)。数据证明,在第二剂存在下EC50向更低的值移动,这表明了化合物1与第二剂的协同活性。该协同作用通过Bliss分析得到证实。
图57提供了化合物1与米哚妥林(midostaurin)和鲁索替尼(ruxolitinib)的组合在MOLM-13细胞系中的细胞增殖剂量响应曲线。
图58提供了化合物1在与依维莫司(everolimus)或替西罗莫司(temsirolimus)组合使用时观察到的协同作用的汇总,如通过在实体肿瘤细胞系中的EC50移动和Bliss分析所测量的。
图59提供了化合物1与依维莫司的组合在各种实体肿瘤细胞系中的细胞增殖剂量响应曲线。
图60提供了单独的化合物1、以及化合物1与依维莫司(RAD,2nM、20nM和200nM)的组合在处理后24h对BON细胞增殖的作用。
图61提供了单独的化合物1、以及化合物1与依维莫司(RAD,2nM、20nM和200nM)的组合在2D板上处理后120h对BON细胞增殖的作用。
图62提供了单独的化合物1、以及化合物1与依维莫司(RAD,2nM、20nM和200nM)的组合在处理后120h对BON细胞增殖的作用。
图63提供了单独的化合物1、以及化合物1与依维莫司(RAD,2nM、20nM和200nM)的组合在3D板上处理后96h对BON细胞增殖的作用。
图64A和图64B提供了单独的化合物1、以及化合物1与依维莫司(RAD,2nM、20nM和200nM)的组合在3D板上处理后120h对BON细胞增殖的作用。
图65A和图65B提供了3D离体增殖测定中化合物1和依维莫司的剂量响应,其示出了所述化合物在GA0087模型中在重复实验中的IC50和最大抑制。
图66提供了3D离体GA0087增殖测定中顺铂(参考化合物)的剂量依赖性响应,其示出了顺铂模型的IC50和最大抑制(重复实验)。
图67A和图67B提供了在基质组合测定中,化合物1与依维莫司在3D GA0087细胞增殖模型(重复实验)中的作用。
图68A和图68B提供了在3D GA0087细胞增殖模型(重复实验)中,如通过Chou和Talalay方法计算的化合物1和依维莫司的组合指数。
图69提供了在GA0087模型中,化合物1和依维莫司单独地和组合地对肿瘤体积的作用。
图70提供了在集落形成测定中,化合物1对来自骨髓纤维化患者的样品中的集落数的作用。
图71提供了化合物1对表达hCRBN、hCRNB和野生型JAK2、或hCRBN和JAK2-V617F的BaF3细胞的细胞活力的作用。
图72提供了鲁索替尼对表达hCRBN、hCRNB和野生型JAK2、或hCRBN和JAK2-V617F的BaF3细胞的细胞活力的作用。
图73提供了NS-18对表达hCRBN、hCRNB和野生型JAK2、或hCRBN和JAK2-V617F的BaF3细胞的细胞活力的作用。
图74提供了莫美罗替尼(momelotinib)对表达hCRBN、hCRNB和野生型JAK2、或hCRBN和JAK2-V617F的BaF3细胞的细胞活力的作用。
图75提供了帕克替尼(pacritinib)对表达hCRBN、hCRNB和野生型JAK2、或hCRBN和JAK2-V617F的BaF3细胞的细胞活力的作用。
图76提供了菲卓替尼(fedratinib)对表达hCRBN、hCRNB和野生型JAK2、或hCRBN和JAK2-V617F的BaF3细胞的细胞活力的作用。
图77提供了依维莫司对表达hCRBN、hCRNB和野生型JAK2、或hCRBN和JAK2-V617F的BaF3细胞的细胞活力的作用。
图78提供了化合物1和NS-018的组合对hCRBN、JAK2、JAK2-V617F新转导的BaF细胞系、IL3依赖性和JAK2-V617F BaF细胞系以及IL3非依赖性BaF细胞系的细胞活力的作用。
图79提供了化合物1和低剂量NS-018的组合对hCRBN、JAK2、JAK2-V617F新转导的BaF细胞系、IL3依赖性和JAK2-V617F BaF细胞系以及IL3非依赖性BaF细胞系的细胞活力的作用。
图80提供了化合物1和鲁索替尼的组合对hCRBN、JAK2、JAK2-V617F新转导的BaF细胞系、IL3依赖性和JAK2-V617F BaF细胞系以及IL3非依赖性BaF细胞系的细胞活力的作用。
图81提供了化合物1和低剂量鲁索替尼的组合对hCRBN、JAK2、JAK2-V617F新转导的BaF细胞系、IL3依赖性和JAK2-V617F BaF细胞系以及IL3非依赖性BaF细胞系的细胞活力的作用。
图82提供了化合物1和莫美罗替尼的组合对hCRBN、JAK2、JAK2-V617F新转导的BaF细胞系、IL3依赖性和JAK2-V617F BaF细胞系以及IL3非依赖性BaF细胞系的细胞活力的作用。
图83提供了化合物1和帕克替尼的组合对hCRBN、JAK2、JAK2-V617F新转导的BaF细胞系、IL3依赖性和JAK2-V617F BaF细胞系以及IL3非依赖性BaF细胞系的细胞活力的作用。
图84提供了化合物1和菲卓替尼的组合对hCRBN、JAK2、JAK2-V617F新转导的BaF细胞系、IL3依赖性和JAK2-V617F BaF细胞系以及IL3非依赖性BaF细胞系的细胞活力的作用。
图85提供了化合物1和依维莫司的组合对hCRBN、JAK2、JAK2-V617F新转导的BaF细胞系、IL3依赖性和JAK2-V617F BaF细胞系以及IL3非依赖性BaF细胞系的细胞活力的作用。
图86提供了作为单一剂的NS-018和鲁索替尼对JAK2V617F AML细胞系的细胞活力的作用。
图87提供了化合物1和NS-018的组合对HEL、SET-2和MUTZ-8AML细胞系中的JAK2V617F的细胞活力的作用。
图88提供了化合物1和鲁索替尼的组合对HEL、SET-2和MUTZ-8AML细胞系中的JAK2V617F的细胞活力的作用。
图89提供了化合物1和依维莫司的组合对HEL、SET-2和MUTZ-8AML细胞的细胞活力的作用。
图90提供了JAK2抑制剂与化合物1组合在JAK2 V617F细胞中的作用的概览,其中协同作用通过EC50移动和Bliss方法进行评分。
图91提供了化合物1和IDH2抑制剂恩西地平(enasidenib)(AG-221)的组合的给药计划。
图92提供了在流式细胞术测定中对表型的定性分析的结果。
图93提供了化合物1和恩西地平(AG-221)的组合对TF-1:IDH2R140Q干细胞和祖细胞(CD34+)以及CD34-/CD235+成红血细胞的分化作用,如来自计划A的散点图中所示。
图94提供了在使用计划A、B或C的情况下,化合物1和恩西地平对TF-1:IDH2R140Q干细胞和祖细胞(CD34+/CD38+)、HSC(CD34+/CD38-)以及CD34-/CD38-非干细胞/祖细胞的分化作用。
图95提供了化合物1和恩西地平对CD235a+(血型糖蛋白A)成红血细胞的分化作用。
图96提供了在TF1测定中化合物1和恩西地平对几个细胞亚群中的GSPT1降解的作用。
图97提供了在TF1测定中化合物1和恩西地平对几个细胞亚群中的GSPT1降解的作用。
图98提供了化合物1和恩西地平对细胞增殖的作用,如总细胞计数以及未分化的HSC(CD34+/CD38-)和祖细胞(CD34+/CD38+)细胞计数所示。
图99示出了化合物1与RAD组合导致GSPT1蛋白显著减少,以及调节翻译和代谢的磷酸化蛋白的变化。对用指示浓度的媒介物、RAD和/或化合物1处理120h的BON细胞裂解物执行蛋白质印迹分析,然后用指示抗体进行探测。肌动蛋白用作上样对照。
图100A至图100E示出了在U937 AML细胞中用化合物1和靶向mTOR、FLT3、JAK2或JAK3的抑制剂处理对细胞增殖的组合作用。图100A示出了化合物1与mTOR抑制剂依维莫司的组合;图100B示出了化合物1与FLT3抑制剂奎扎替尼(quizartinib)的组合;图100C示出了化合物1与JAK2抑制剂鲁索替尼的组合;图100D示出了化合物1与JAK2抑制剂AZD1480的组合;并且100E示出了化合物1与JAK3抑制剂托法替尼(tofacitinib)的组合。
图101示出了在U937 AML细胞中用化合物1和mTOR、FLT3、JAK2或JAK3抑制剂处理对GSPT1表达、mTOR活化、ATF4诱导和半胱天冬酶-3裂解的组合作用。
图102A至图102G示出了化合物1在与和不与指示浓度的维奈托克(venetoclax)一起使用的情况下,在温育48小时时对AML细胞系增殖的作用。
图103示出了相对ATP水平,其作为响应于化合物1和维奈托克的几种剂量组合的活力的量度。
图104示出了测量在用一组剂量的化合物1、维奈托克以及化合物1和维奈托克的组合处理后16小时,KG-1细胞中的GSPT1、Mcl-1、Bcl-2、经裂解的半胱天冬酶3和GAPDH蛋白水平的蛋白质印迹分析。
图105A和图105B示出了在用化合物1、维奈托克以及化合物1和维奈托克的组合处理后,对KG-1汇合度的活细胞分析(图105A)和对细胞凋亡事件计数的活细胞分析(图105B)。
图106示出了用化合物1和依维莫司处理对GSPT1表达、mTOR活化、Mcl-1表达和半胱天冬酶-3裂解的组合作用。
图107示出了通过液体培养测定的化合物1对来自2名不同的骨髓增生异常综合征患者的骨髓单核细胞或分离的CD34+母细胞的作用。
图108示出了通过液体培养(A)或集落形成测定(B)测定的化合物1对来自骨髓增生异常综合征患者的骨髓单核细胞的作用。
图109示出了在暴露于一种或多种化合物24小时之后,作为单一剂测试或与111nM依维莫司组合测试时,化合物1对来自骨髓增生异常综合征患者的骨髓单核细胞中的半胱天冬酶-3活化和GSPT1降解的作用。
具体实施方式
定义
一般来讲,本文所用的命名法和本文所述的有机化学、药物化学以及药理学中的实验程序是那些熟知的实验程序并且在本领域中是常用的。除非另外定义,否则本文所用的所有技术术语和科学术语通常都具有与本公开所属领域的普通技术人员通常所理解相同的含义。一般来讲,一个实施方案的技术教导内容可以与本文提供的其它实施方案中所公开的技术教导内容组合。
在权利要求书和/或说明书中,词语“一个”或“一种”当连同术语“包括”使用时,可以意味着“一个/种”,不过也与“一个/种或多个/种”、“至少一个/种”以及“一个/种或多于一个/种”的含义一致。
如本文所用,术语“包括”和“包含”可以互换使用。术语“包括”和“包含”应被解释为指定存在所提及的规定特征或组成部分,但并不排除存在和添加一个或多个特征、或组成部分、或它们的组合。此外,术语“包括”和“包含”旨在包括术语“由……组成”所涵盖的实例。因此,可以使用术语“由……组成”来代替术语“包括”和“包含”,以提供本发明的更具体的实施方案。
术语“由……组成”意味着由规定特征或组成部分组成的某个主题具有至少90%、95%、97%、98%或99%的所述规定特征或组成部分。在另一个实施方案中,术语“由……组成”从任何随后的列举内容的范围中排除了任何其它的特征或组成部分,对于要实现的技术效果不是必需的那些特征或组成部分除外。
如本文所用,术语“或”应被解释为包含性的“或”,其意味着任何一个或任何组合。因此,“A、B或C”意味着以下各项中的任何一种:“A;B;C;A和B;A和C;B和C;A、B和C”。只有当要素、功能、步骤或动作的组合在某种程度上固有地相互排斥时,才会出现该定义的例外情况。例如,“治疗、预防或管理”或类似的清单意味着:“治疗;预防;管理;治疗和预防;治疗和管理;预防和管理;治疗、预防和管理”。
术语“化合物1”是指具有以下结构的“2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺”:

及其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物。在某些实施方案中,化合物1是指2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺及其互变异构体。在某些实施方案中,化合物1是指2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的多晶型物,诸如A型、B型、C型、D型或E型,或它们的混合物。在某些实施方案中,化合物1是指2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的多晶型物C型。在某些实施方案中,化合物1是指2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的无定形形式。在一个实施方案中,立体异构体是对映异构体。
除非另外明确规定,否则在化合物可以采取替代性的互变异构、区域异构和/或立体异构形式的情况下,所有的替代性异构体均旨在被涵盖于要求权利的主题的范围之内。例如,在化合物可以具有两种互变异构形式之一的情况下,两种互变异构体都旨在涵盖于本文中。
因此,本文的化合物可以是对映异构纯的,或者是立体异构或非对映异构混合物。如本文所用并且除非另外指明,否则术语“立体异构纯的”意味着包含化合物的一种立体异构体并且基本上不含该化合物的其它立体异构体的组合物。例如,具有一个手性中心的化合物的立体异构纯组合物将基本上不含该化合物的相反对映异构体。具有两个手性中心的化合物的立体异构纯组合物将基本上不含该化合物的其它非对映异构体。典型的立体异构纯化合物包含大于约80重量%的该化合物的一种立体异构体和小于约20重量%的该化合物的其它立体异构体、更优选地大于约90重量%的该化合物的一种立体异构体和小于约10重量%的该化合物的其它立体异构体、甚至更优选地大于约95重量%的该化合物的一种立体异构体和小于约5重量%的该化合物的其它立体异构体,以及最优选地大于约97重量%的该化合物的一种立体异构体和小于约3重量%的该化合物的其它立体异构体。如本文所用的立体异构纯化合物包含大于约80重量%的该化合物的一种立体异构体、更优选地大于约90重量%的该化合物的一种立体异构体、甚至更优选地大于约95重量%的该化合物的一种立体异构体,以及最优选地大于约97重量%的该化合物的一种立体异构体。如本文所用并且除非另外指明,否则术语“立体异构富集的”意味着包含大于约60重量%的化合物的一种立体异构体,优选地大于约70重量%、更优选地大于约80重量%的化合物的一种立体异构体的组合物。如本文所用并且除非另外指明,否则术语“对映异构纯的”意味着具有一个手性中心的化合物的立体异构体纯的组合物。类似地,术语“立体异构富集的”意味着具有一个手性中心的化合物的立体异构富集的组合物。如本文所用,立体异构或非对映异构混合物意味着包含化合物的多于一种立体异构体的组合物。化合物的典型立体异构混合物包含约50重量%的该化合物的一种立体异构体和约50重量%的该化合物的其它立体异构体、或包含大于约50重量%的该化合物的一种立体异构体和小于约50重量%的该化合物的其它立体异构体、或包含大于约45重量%的该化合物的一种立体异构体和小于约55重量%的该化合物的其它立体异构体、或包含大于约40重量%的该化合物的一种立体异构体和小于约60重量%的该化合物的其它立体异构体,或包含大于约35重量%的该化合物的一种立体异构体和小于约65重量%的该化合物的其它立体异构体。
如本文所用,API是指化合物1。在某些实施方案中,API是指2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺。
如本文所用,除非另外指明,否则任何保护基团、氨基酸和其它化合物的缩写都符合它们的通用用途、公认缩写或IUPAC-IUB生物化学命名委员会(IUPAC-IUB Commissionon Biochemical Nomenclature)(参见Biochem.1972,11:942-944)。
如本文所用,并且除非另外指明,否则术语“冻干”是指从溶液中分离固体物质和/或除去溶剂的过程。在一些实施方案中,这可以通过本领域技术人员已知的各种技术来实现,包括例如蒸发(例如,在真空下,例如通过冷冻干燥,和/或将溶液冷冻并在真空或减压条件下使冷冻的溶剂汽化,等等。)
如本文所用,术语“共溶剂”是指在制造本文所提供的制剂期间有助于活性剂在水中增溶的溶剂。共溶剂可以是还为制造期间的中间制剂提供足够大的稳定性的溶剂。在制造期间,共溶剂也可以从制剂中除去,或减少到可接受的水平。共溶剂的实例包括乙腈、氯仿、叔丁醇、甲醇、四氢呋喃、甲酸、醋酸、丙酮、苯甲醚、丁醇、乙酸丁酯、叔丁基甲基醚、乙醇、乙酸乙酯、乙醚、甲酸乙酯、庚烷、乙酸异丁酯、乙酸异丙酯、乙酸甲酯、3-甲基-丁醇、甲基乙基酮、甲基异丁基酮、2-甲基-1-丙醇、戊烷、1-戊醇、1-丙醇、2-丙醇和乙酸丙酯。
如本文所用,并且除非另外指明,否则术语“基本上不含”意味着含有不超过微不足道的量。在一些实施方案中,如果组合物或制备物含有少于5重量%、4重量%、3重量%、2重量%或1重量%的所列举要素,则其“基本上不含”该要素。在一些实施方案中,该组合物或制备物含有少于0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或更少的所列举要素。在一些实施方案中,该组合物或制备物含有不可检测量的所列举要素。
如本文所用,“复原水性溶液”或“复原水性组合物”或“复原水性制剂”是指通过将本文提供的冻干制剂溶解在水性溶剂中而获得的水性溶液。
本文所用的术语“水性稀释剂”是指能够被包含在肠胃外制剂中的水性液体。如果需要,此类水性稀释剂可以包括例如水、盐水、1/2生理盐水或右旋糖,以及通常作为肠胃外制剂的一部分发现的任何已知的辅助防腐剂或赋形剂。示例性的水性稀释剂包括水、5%右旋糖溶液,等等。
如本文所用,并且除非另外指明,否则术语“肠胃外”包括皮下、静脉内、肌内、关节内、滑膜内、胸骨内、鞘内、肝内、病灶内和颅内注射或输注技术。
如本文所用,并且除非另外指明,否则表达“单位剂量”是指适合于待治疗的对象的制剂的物理上离散的单位(例如,对于单剂量);每个单位含有预定量的被选择用于产生期望的治疗作用的活性剂(可以理解,可能需要多剂量来实现期望的或最佳的作用),任选地连同含有可以以预定量提供的药学上可接受的载剂。单位剂量可以是例如含有预定量的一种或多种治疗剂的一定体积的液体(例如可接受的载剂)、预定量的一种或多种固体形式治疗剂、含有预定量的一种或多种治疗剂的缓释制剂或药物递送装置,等等。应当理解,单位剂量除一种或多种治疗剂之外还可以含有多种组分。例如,可以如下文所述包含可接受的载剂(例如,药学上可接受的载剂)、稀释剂、稳定剂、缓冲剂、防腐剂等。然而,应当理解,本公开制剂的总日用量将由主治医师在合理的医学判断范围内决定。任何特定对象或生物体的具体有效剂量水平可以取决于多种因素,包括正在治疗的疾患和该疾患的严重程度;采用的具体活性化合物的活性;采用的具体组合物;对象的年龄、体重、总体健康状况、性别和饮食;采用的具体活性化合物的施用时间和排泄速率;治疗持续时间;与采用的一种或多种具体化合物组合或同时使用的药物和/或附加疗法,以及医学领域众所周知的类似因素。
如本文所用,术语“固体形式”是指2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的晶体形式或无定形形式或它们的混合物,或该化合物的立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物。
如本文所用,除非另外指明,否则如本文所用的术语“一种或多种药学上可接受的盐”包括但不限于化合物1的酸性部分或碱性部分的盐。碱性部分能够与各种无机酸和有机酸形成多种多样的盐。可以用于制备此类碱性化合物的药学上可接受的酸加成盐的酸是形成无毒酸加成盐(例如,含有药理学上可接受的阴离子的盐)的那些酸。合适的有机酸包括但不限于马来酸、富马酸、苯甲酸、抗坏血酸、琥珀酸、醋酸、甲酸、草酸、丙酸、酒石酸、水杨酸、柠檬酸、葡糖酸、乳酸、扁桃酸、肉桂酸、油酸、单宁酸、天冬氨酸、硬脂酸、棕榈酸、乙醇酸、谷氨酸、葡糖酸、葡糖醛酸、糖质酸、异烟酸、甲磺酸、乙磺酸、对甲苯磺酸、苯磺酸或双羟萘酸(例如,1,1’-亚甲基-双-(2-羟基-3-萘甲酸酯)。合适的无机酸包括但不限于盐酸、氢溴酸、氢碘酸、硫酸、磷酸或硝酸。除上文提到的酸之外,包含胺部分的化合物可以与各种氨基酸形成药学上可接受的盐。本质上是酸性的化学部分能够与各种药理学上可接受的阳离子形成碱盐。此类盐的实例是碱金属盐或碱土金属盐,特别地是钙盐、镁盐、钠盐、锂盐、锌盐、钾盐或铁盐。
如本文所用,并且除非另外指明,否则术语“溶剂合物”意味着还包含由分子内非共价力结合的化学计量或非化学计量量的溶剂的本文所提供的化合物或其盐。在溶剂是水的情况下,溶剂合物是水合物。
如本文所用,并且除非另外指明,否则术语“前药”意味着化合物的可以在生物条件(体外或体内)下水解、氧化或以其它方式反应以提供该化合物的衍生物。前药的实例包括但不限于本文所述化合物(例如化合物1)的衍生物,其包括可生物水解部分诸如可生物水解酰胺、可生物水解酯、可生物水解氨基甲酸酯、可生物水解碳酸酯、可生物水解酰脲和生物可水解磷酸酯类似物。
“药学上可接受的赋形剂”是指通过例如改变活性剂的稳定性或改变施用后被对象的吸收,来有助于向对象施用活性剂的物质。药学上可接受的赋形剂典型地对患者没有显著的不良毒理学作用。药学上可接受的赋形剂的实例包括例如水、NaCl(包括盐溶液)、生理盐水溶液、1/2生理盐水、蔗糖、葡萄糖、增量剂、缓冲剂、粘结剂、填充剂、崩解剂、润滑剂、包衣、甜味剂、风味剂、醇、油、明胶、碳水化合物诸如直链淀粉或淀粉、脂肪酸酯、羟甲基纤维素、聚乙烯吡咯烷和色素,等等。本领域技术人员应当认识到,本领域已知的其它药用赋形剂可用于本发明中并且包括例如Handbook of Pharmaceutical Excipients,RoweR.C.,Shesky P.J.,and Quinn M.E.,第6版,The Pharmaceutical Press,RPS Publishing(2009)中列出的那些。术语“增量剂”和“缓冲剂”根据本领域内的平常和普通的含义使用。
如本文所用,并且除非另外指明,否则术语“约”在结合组合物或剂型的成分的剂量、量或重量百分比使用时,意味着涵盖本领域普通技术人员所认识到的用于提供与从指定的剂量、量或重量百分比获得的药理作用等效的药理作用的剂量、量或重量百分比。具体地讲,术语“约”设想,涵盖了与指定的剂量、量或重量百分比相差不到30%、25%、20%、15%、10%或5%的剂量、量或重量百分比。
如本文所用,并且除非另外指明,否则术语“稳定的”在结合液体制剂或剂型使用时,意味着该制剂或剂型的活性成分在指定量的时间内保持增溶,并且不显著劣化或聚集或以其它方式改变(例如,如例如通过HPLC测定)。在一些实施方案中,在指定时段之后,约70%或更多、约80%或更多、或约90%或更多的化合物保持增溶。稳定性也可以指本文所述的药学上可接受的赋形剂的相容性。因此,当本文所述的组合的药学上可接受的赋形剂和一种或多种活性剂不劣化或不以其它方式(例如,与其发生反应)改变本文所述活性剂的有效性或治疗价值时,剂型可以被认为是稳定的。
如本文所用,并且除非另外指明,否则术语“稳定的”在结合固体制剂或剂型使用时,意味着该制剂或剂型的活性成分不显著劣化、分解或以其它方式改变(例如,如例如通过HPLC测定)。在一些实施方案中,在指定时段之后,约85%或更多、约90%或更多、约95%或更多、或约98%或更多的活性成分保持不变。稳定性也可以指本文所述的药学上可接受的赋形剂的相容性。因此,当本文所述的组合的药学上可接受的赋形剂和一种或多种活性剂不劣化或不以其它方式(例如,与其发生反应)改变本文所述活性剂的有效性或治疗价值时,剂型可以被认为是稳定的。
如本文所用,“施用”是指将存在于身体外部的物质物理地递送到对象中的行为。施用包括本领域已知的用于递送治疗剂的所有形式,包括但不限于:局部、粘膜、注射、真皮内、静脉内、肌内递送,或者本文所述或本领域已知的其它物理递送方法(例如,将缓释装置诸如微渗透泵植入对象;脂质体制剂;口腔;舌下;腭;牙龈;鼻;阴道;直肠;小动脉内;腹膜内;心室内;颅内;或透皮)。
“抗癌剂”是指抗代谢物(例如5-氟-尿嘧啶、甲氨蝶呤、氟达拉滨)、抗微管剂(例如长春花生物碱,诸如长春新碱、长春花碱;紫杉烷类,诸如紫杉醇、多西他赛)、烷化剂(例如,环磷酰胺,美法仑、卡莫司汀、亚硝基脲诸如双氯乙基亚硝基脲和羟基脲)、铂剂(例如顺铂、卡铂、奥沙利铂、JM-216或沙铂、CI-973)、蒽环类药物(例如多柔比星、柔红霉素)、抗肿瘤抗生素(例如丝裂霉素、伊达比星、阿霉素、道诺霉素)、拓扑异构酶抑制剂(例如依托泊苷、喜树碱)、抗血管生成剂(例如 苹果酸舒尼替尼和贝伐单抗)或任何其它细胞毒性剂(磷酸雌莫司汀、泼尼莫司汀)、激素或激素激动剂、拮抗剂、部分激动剂或部分拮抗剂、激酶抑制剂、检查点抑制剂,以及辐射治疗。
苹果酸舒尼替尼和贝伐单抗)或任何其它细胞毒性剂(磷酸雌莫司汀、泼尼莫司汀)、激素或激素激动剂、拮抗剂、部分激动剂或部分拮抗剂、激酶抑制剂、检查点抑制剂,以及辐射治疗。
所谓“共同施用”,意味着本文所述的化合物、组合物或剂在施用一种或多种附加的化合物、组合物或剂(包括例如抗癌剂)的同时、紧临所述施用之前或紧临所述施用之后施用。共同施用意在包括个别或组合地同时或相继施用化合物、组合物或剂(多于一种化合物或剂)。共同施用包括同时、大致同时(例如,彼此相差约1、5、10、15、20或30分钟)或以任何顺序相继施用两种化合物、组合物或药剂。因此,共同施用可以包括与第二活性剂相差0.5、1、2、4、6、8、10、12、16、20或24小时施用一种活性剂(例如,本文所述的化合物)。共同施用还可以通过共同配制(例如,制备包含两种活性剂的单一剂型)来实现。这些活性剂可以单独配制。在此类情况下,这些活性剂一起混合和包含在最终形式的剂量单位中。替代性地,如本文所述的共同施用可以包括施用至少两种单独的活性剂(例如,化合物1和本文所述的第二活性剂)的两种单独的单位剂型。
如本文所用,术语“每日”旨在意味着治疗化合物(诸如化合物1)每天施用一次或多于一次持续一定时间段。术语“连续”旨在意味着治疗化合物(诸如化合物1)每天施用持续至少10天至52周的不间断时间段。如本文所用,术语“间歇”或“间歇地”旨在意味着以规则或不规则的间隔停止和开始。例如,化合物1的间歇施用是每周施用1至6天、周期性地施用(例如,每日施用,持续28天周期的连续1至10天,然后是停用期,其中该28天周期的其余时间不施用;或每日施用,持续连续的2至8周,然后是停用期,其中最长一周不施用),或隔天施用。如本文所用,术语“周期性”旨在意味着治疗化合物(诸如化合物1)每日施用或连续施用,但具有停用期。
“周期性疗法”是指包括如本文所述的施用期和如本文所述的停用期的方案或疗法。
如本文所用,术语“施用期”是指将本文所述的化合物或组合物连续或主动施用于对象的一段时间。
如本文所用,术语“停用期”是指通常在施用期后,本文所述的化合物或组合物不施用于对象(例如停止治疗)的一段时间。在某些实施方案中,“停用期”是指单一药剂不施用于对象或停止使用特定化合物治疗的一段时间。在此类实施方案中,可以将第二治疗剂(例如,不同于此前施用期所施用的化合物或组合物的剂)施用于对象。
“有效量”是足以实现施用效果(例如,治疗疾病或减轻疾病或病症的一种或多种症状)的量。因此,向对象施用一定“量”的本文所述化合物是指施用“能有效地”实现期望的治疗结果的“量”。因此,出于本文目的,本文所述化合物的“治疗有效量”是通过本领域已知的此类考虑来确定的。本文所述组合物的术语“治疗有效量”是指该组合物在施用时足以治疗本文所述疾病(例如癌症,例如AML、MDS、MPN或实体肿瘤)的一种或多种症状的量。本文所述化合物的施用可以根据诸如个体的疾病状态、年龄、性别和体重的因素来确定。治疗有效量还指化合物1的任何毒性或有害作用均被治疗有益作用超过。
如本文所用,并且除非另外指明,否则术语“治疗”是指根除或改善疾病或疾患,或者与疾病或疾患相关联的一种或多种症状。在某些实施方案中,该术语是指由于将一种或多种预防剂或治疗剂施用于患有这种疾病或疾患的患者,而最大程度减轻该疾病或疾患的传播或恶化。在一些实施方案中,该术语是指在特定疾病的症状发作之后,在存在或不存在其它附加的活性剂的情况下,施用本文提供的化合物。在一个实施方案中,疾病是白血病,包括但不限于慢性淋巴细胞性白血病(CLL)、慢性髓细胞性白血病(CML)、急性淋巴母细胞性白血病(ALL)、急性骨髓性白血病或急性骨髓母细胞性白血病(AML)。在一个实施方案中,白血病可以是复发性的,至少一种抗癌疗法难治性的或对至少一种抗癌疗法具有耐药性的。在一个实施方案中,疾病是AML,包括本文讨论的AML的亚型。在一个实施方案中,疾病是骨髓增生异常综合征MDS,包括本文讨论的MDS的亚型。
如本文所用,并且除非另外指明,否则术语“预防”是指预防疾病或疾患,或者其一种或多种症状的发作、复发或传播。在某些实施方案中,该术语是指在症状发作之前,在存在或不存在其它附加的活性化合物的情况下,用本文提供的化合物治疗或施用本文提供的化合物,特别是施用于有本文提供的疾病或疾患的风险的患者。该术语涵盖抑制或减轻特定疾病的症状。在某些实施方案中,具有疾病家族史的患者尤其是预防性方案的候选者。此外,具有复发性症状史的患者也是预防的潜在候选者。就此而言,术语“预防”可以与术语“预防性治疗”互换使用。在一个实施方案中,疾病是白血病,包括但不限于慢性淋巴细胞性白血病、慢性髓细胞性白血病、急性淋巴母细胞性白血病、急性骨髓性白血病和急性骨髓母细胞性白血病。在一个实施方案中,白血病可以是复发性的,至少一种抗癌疗法难治性的或对至少一种抗癌疗法具有耐药性的。在一个实施方案中,疾病是AML,包括本文讨论的AML的亚型。在一个实施方案中,疾病是MDS,包括本文讨论的MDS的亚型。
如本文所用,并且除非另外指明,否则术语“管理”是指预防或减缓疾病或疾患,或者其一种或多种症状的发展、传播或恶化。通常,患者从预防剂和/或治疗剂得到的有益作用不会产生疾病或疾患被治愈的结果。就此而言,术语“管理”涵盖治疗患有特定疾病的患者,以试图预防或最大程度减少该疾病的复发,或延长维持缓解持续的时间。在一个实施方案中,疾病是白血病,包括但不限于慢性淋巴细胞性白血病、慢性髓细胞性白血病、急性淋巴母细胞性白血病、急性骨髓性白血病和急性骨髓母细胞性白血病。在一个实施方案中,白血病可以是复发性的,至少一种抗癌疗法难治性的或对至少一种抗癌疗法具有耐药性的。在一个实施方案中,疾病是AML,包括本文讨论的AML的亚型。在一个实施方案中,疾病是MDS,包括本文讨论的MDS的亚型。
如本文所用,“诱导疗法”是指针对疾病给予的第一次治疗,或旨在诱导疾病(诸如癌症)的完全缓解而给予的第一次治疗。在单独使用时,诱导疗法是被接纳为最佳可用治疗的疗法。例如,针对AML的诱导疗法包括用阿糖胞苷治疗7天,加上用蒽环类药物(诸如柔红霉素或伊达比星)治疗3天。如果检测到残留的白血病,则使用另一种化学疗法疗程(称为再诱导)对患者进行治疗。如果患者在诱导疗法后完全缓解,则给予附加的巩固疗法和/或维持疗法,以延长缓解时间或潜在地治愈患者。
如本文所用,“巩固疗法”是指首次实现缓解之后针对疾病给予的治疗。例如,针对癌症的巩固疗法是在初始疗法后癌症已消失之后给予的治疗。巩固疗法可以包括放射疗法、干细胞移植,或用癌症药物疗法进行治疗。巩固疗法也被称为强化疗法和缓解后疗法。
如本文所用,“维持疗法”是指在实现缓解或最佳响应之后针对疾病给予的治疗,以便预防或延迟复发。维持疗法可以包括化学疗法、激素疗法或靶向疗法。
如本文所用,“缓解”是指癌症(例如多发性骨髓瘤)的征象和症状减少或消失。在部分缓解中,癌症的一些但不是所有的征象和症状已经消失。在完全缓解中,尽管癌症仍可能存在于体内,但癌症的所有病征和症状都已经消失。
术语“对象”、“患者”、“有需要的对象”和“有需要的患者”在本文中可互换使用,并且是指患有可以通过施用本文所述的组合物来治疗的本文所述的一种或多种疾病(例如,AML)的活生物体。生物体的非限制性实例包括人、其它哺乳动物、牛、大鼠、小鼠、狗、猴、山羊、绵羊、母牛、鹿和其它非哺乳动物。在一些实施方案中,对象是人。人对象的年龄可以介于约1岁至约100岁之间。在一些实施方案中,本文的对象可以由正在治疗的疾病来表征(例如,“AML对象”、“癌症对象”或“白血病对象”)。
如本文所用,术语“肿瘤”是指所有赘生性细胞生长和增殖(无论是恶性还是良性),以及所有癌前和癌性细胞和组织。如本文所用,“赘生性”是指导致异常的组织生长的任何形式的失调或未经调节的细胞生长(无论是恶性还是良性)。因此,“赘生性细胞”包括细胞生长失调或未经调节的恶性细胞和良性细胞。
如本文所用,“血液系统恶性肿瘤”是指身体的造血和免疫系统即骨髓和淋巴组织的癌症。这些癌症包括白血病、淋巴瘤(非霍奇金氏淋巴瘤(Non-Hodgkin’s Lymphoma))、霍奇金氏病(也称为霍奇金氏淋巴瘤(Hodgkin’s Lymphoma))和骨髓瘤。在一个实施方案中,骨髓瘤是多发性骨髓瘤。在一些实施方案中,白血病是例如急性髓源性白血病(AML)、急性淋巴细胞性白血病(ALL)、成人T细胞白血病、慢性淋巴细胞性白血病(CLL)、毛细胞白血病、骨髓增生异常、骨髓增殖性障碍或骨髓增殖性赘生物(MPN)、慢性髓源性白血病(CML)、骨髓增生异常综合征(MDS)、人嗜淋巴细胞病毒-1型(HTLV 1)白血病、肥大细胞增生症,或B细胞急性淋巴母细胞性白血病。在一些实施方案中,淋巴瘤是例如弥散性大B细胞淋巴瘤(DLBCL)、B细胞免疫母细胞性淋巴瘤、小无核裂细胞淋巴瘤、人嗜淋巴细胞病毒-1型(HTLV-1)白血病/淋巴瘤、成人T细胞淋巴瘤、外周T细胞淋巴瘤(PTCL)、皮肤T细胞淋巴瘤(CTCL)、套细胞淋巴瘤(MCL)、霍奇金淋巴瘤(HL)、非霍奇金淋巴瘤(NHL)、AIDS相关淋巴瘤、滤泡性淋巴瘤、小淋巴细胞性淋巴瘤、富含T细胞/组织细胞的大B细胞淋巴瘤、转化型淋巴瘤、原发性纵隔(胸腺)大B细胞淋巴瘤、脾边缘区淋巴瘤、里希特氏转化(Richter’stransformation)、结节边缘区淋巴瘤,或ALK阳性大B细胞淋巴瘤。在一个实施方案中,血液学癌症是惰性淋巴瘤,包括例如DLBCL、滤泡性淋巴瘤或边缘区淋巴瘤。在一个实施方案中,血液学恶性肿瘤是AML。在另一个实施方案中,血液学恶性肿瘤是MDS。
术语“白血病”是指造血组织的恶性赘生物。白血病包括但不限于慢性淋巴细胞性白血病、慢性髓细胞性白血病、急性淋巴母细胞性白血病、急性骨髓性白血病和急性骨髓母细胞性白血病。白血病可以是复发性的,至少一种抗癌疗法难治性的或对至少一种抗癌疗法具有耐药性的。
在一个实施方案中,对象患有AML,包括例如以下AML的亚型。术语“急性髓源性或骨髓性白血病”是指特征在于主要未分化型或最小分化型骨髓细胞在骨髓中增殖和累积的血液学病症,并且包括由FAB(French,American,British)或WHO分类系统分类的亚型。如本文所述,基于FAB分类,AML包括以下亚型:M0(AML最小分化型);M1(最小成熟型AML);M2(成熟型AML);M3(急性早幼粒细胞性白血病);M4(急性髓单核细胞性白血病);M4(eos急性髓单核细胞性白血病伴嗜酸性粒细胞增多);M5(急性单核细胞性白血病);M6(急性红细胞系白血病);和M7(急性巨核母细胞性白血病)。如本文所述,基于WHO分类,AML包括以下亚型:具有复现性遗传异常的AML(具有染色体8和染色体21之间的易位的AML);具有染色体16中的易位或倒位的AML;具有染色体9和染色体11之间的易位的AML;具有染色体15和染色体17之间的易位的APL(M3);具有染色体6和染色体9之间的易位的AML;具有染色体3中的易位或倒位的AML);具有染色体1和染色体22之间的易位的AML(巨核母细胞性);具有骨髓增生异常相关变化的AML;涉及前期化学疗法或辐射的AML(烷化剂相关AML;拓扑异构酶II抑制剂相关AML);不可另外分类的AML(不属于上述类别的AML,即最小分化型AML(M0);最小成熟型AML(M1);成熟型AML(M2);急性髓单核细胞性白血病(M4);急性单核细胞性白血病(M5);急性红细胞系白血病(M6);急性巨核母细胞性白血病(M7);急性嗜碱粒细胞性白血病;急性全骨髓增生症伴纤维化);骨髓性肉瘤(也称为粒细胞性肉瘤、绿色瘤或髓外成骨髓细胞瘤);以及未分化型和双表型急性白血病(也称为混合表型急性白血病)。(请参见https://www.cancer.org/cancer/acute-myeloid-leukemia/detection-diagnosis-staging/how-classified.html,最近访问时间:2017年5月25日)。
在某些实施方案中,基于细胞遗传学的AML风险组如下所述:


a该表中包含的分子异常反映了经验证的测定在标准化商业实验室中可以针对其使用的那些分子异常。
b新出现的数据表明,在具有t(8;21)的患者中存在KIT突变,并且在具有inv(16)的患者中以较小的程度存在,带来较高的复发风险。这些患者被认为是中等风险,如果可以的话,应当考虑进行造血干细胞移植(HSCT)或临床试验。除这些发现之外的其它细胞遗传学异常没有改变风险状态。
cPaschka P等人,Blood 2013;121:170-177。
d除这些发现之外的其它细胞遗传学异常没有改变较好的风险状态
e对于费城(Philadelphia)+急性骨髓性白血病(AML)t(9;22),通过添加酪氨酸激酶抑制剂,作为慢性骨髓性白血病(CML)中的骨髓性急变期加以管理。
在一个实施方案中,对象患有MDS,包括例如以下MDS亚型。术语“骨髓增生异常综合征”是指特征在于血液的一种或多种细胞组分(红细胞、白细胞(除淋巴细胞之外)和血小板(或其祖细胞、巨核细胞))的产生出现异常的血液学病症。在MDS中,骨髓(BM)的无效造血和外周血细胞减少在临床上表现为贫血、中性粒细胞减少和/或血小板减少,其发生频率和严重程度各不相同。贫血是实验室最常发现的现象,它经常进展为对红血细胞(RBC)输注的依赖性。与血细胞减少相关的其它较不常见的临床特征是感染和/或出血的风险增加,以及有进展为急性骨髓性白血病(AML)的倾向(Catenacci等人,Blood Rev 2005;19:301-319)。
MDS包括以下疾患:难治性贫血(RA);RA伴环形铁粒幼细胞(RARS);RA伴母细胞过量(RAEB);难治性血细胞减少伴多系增生异常(RCMD)、难治性血细胞减少伴单系增生异常(RCUD);不可分类型骨髓增生异常综合征(MDS-U)、孤立del(5q)染色体异常相关联的骨髓增生异常综合征、疗法相关的骨髓赘生物和慢性髓单核细胞性白血病(CMML)。如本文所用的MDS还包括极低风险、低风险、中等风险、高风险和极高风险MDS。在一些实施方案中,MDS是原发性或新发MDS。在其它实施方案中,MDS是继发性的。
在某些实施方案中,MDS基于如下所述的世界卫生组织(WHO)MDS分类进行分类:
WHO关于MDS的分类


a血细胞减少被定义为:血红蛋白<10g/dL,血小板计数<100x109/L;并且中性粒细胞绝对计数<1.8x109/L。极少情况下,MDS可见这些水平以上的轻度贫血或血小板减少。外周血单核细胞必须<1x109/L。
b根据定义,环形铁粒幼细胞≥15%的病例具有明显的红细胞系增生异常,被归类为MDS-RS-SLD。
c1%的PB母细胞必须有至少两次不同场合检查的记录。
d异常必须由常规核型分析证明,而不是由FISH或测序证明。在没有MDS的诊断形态学特征的情况下,存在del(20q)的+8、-Y不被认为是定义MDS的。Arber等人,Blood 2016;127(20):2391-2405,以及Vardiman等人,Blood.2009;114(5):937-51。
如本文所用,“早幼粒细胞性白血病”或“急性早幼粒细胞性白血病”是指骨髓的恶性肿瘤,其中细胞的髓系中缺失成熟的血细胞,并且称为早幼粒细胞的未成熟细胞过量。它通常由染色体15和17的区域交换进行标记。
如本文所用,“急性淋巴细胞性白血病(ALL)”,也称为“急性淋巴母细胞性白血病”,是指由早期非粒状白血细胞或淋巴细胞的异常生长和发育引起的恶性疾病。
如本文所用,“T细胞白血病”是指其中淋巴系统的称为T淋巴细胞或T细胞的某些细胞为恶性的疾病。T细胞是通常可以攻击被病毒感染的细胞、外来细胞和癌细胞并且产生调节免疫响应的物质的白血细胞。
术语“复发性”是指在治疗后白血病已经得到缓解的患者,其骨髓中的白血病细胞恢复并且正常血细胞减少的情况。
术语“难治性或耐药性”是指即使在强化治疗之后,患者的骨髓中也有残留的白血病细胞的情况。
术语“抗药性”是指疾病对一种或多种某些药物的治疗无响应的病症。抗药性可以是固有的,这意味着疾病从未对一种或多种特定药物产生响应,或者抗药性可以是获得的,这意味着疾病停止对该疾病此前产生响应的一种或多种特定药物产生响应。在某些实施方案中,抗药性是固有的。在某些实施方案中,抗药性是获得的。
如本文所用,并且除非另外指明,否则化合物的“治疗有效量”是足以在疾病或疾患的治疗或管理中提供治疗有益效果,或者延缓或最小化与该疾病或疾患相关联的一种或多种症状的量。化合物的治疗有效量意味着单独或与其它疗法组合的治疗剂在疾病或疾患的治疗或管理中提供了治疗有益效果的量。术语“治疗有效量”可以涵盖改善总体疗法、减少或避免疾病或疾患的症状或起因,或者增强另一种治疗剂的治疗功效的量。
如本文所用,并且除非另外指明,否则化合物的“预防有效量”是足以预防疾病或疾患、或者预防其复发的量。化合物的预防有效量意味着单独或与其它剂组合的治疗剂在疾病的预防中提供了预防有益效果的量。术语“预防有效量”可以涵盖改善总体预防或增强另一种预防剂的预防功效的量。
如本文所用,ECOG状态是指美国东部肿瘤协作组(Eastern CooperativeOncology Group,ECOG)行为状态(Oken M等人,Toxicity and response criteria of theEastern Cooperative Oncology Group.Am J Clin Oncol 1982;5(6):649-655),如下所示:

在癌症的背景下,治疗或抑制可以通过下列各项来评定:抑制疾病进展、抑制肿瘤生长、减少原发性肿瘤、减轻肿瘤相关症状、抑制肿瘤分泌因子、延迟原发性或继发性肿瘤的出现、减缓原发性或继发性肿瘤的发展、减少原发性或继发性肿瘤的发生、疾病的继发性效应减缓或严重程度降低、肿瘤生长停滞和肿瘤消退、进展时间(TTP)增加、无进展存活期(PFS)增加、总体存活期(OS)增加,等等。本文所用的OS意味着从治疗开始直到由于任何原因而死亡的时间。如本文所用的TTP意味着从治疗开始直到肿瘤进展的时间;TTP不包括死亡。如本文所用的缓解时间(TTR)意味着从治疗开始直到缓解(例如完全或部分缓解)的时间。如本文所用,PFS意味着从治疗开始直到肿瘤进展或死亡的时间。在一个实施方案中,将使用卡普兰-梅尔(Kaplan-Meier)估计值来计算PFS率。无事件存活期(EFS)意味着从研究进入直到任何治疗失败(包括疾病进展、由于任何原因而停止治疗、或死亡)的时间。无复发存活期(RFS)意味着治疗结束后患者存活而没有癌症的任何征象或症状的时间长度。总体响应率(ORR)意味着实现完全响应和部分响应的患者的百分比的总和。完全缓解率(CRR)是指实现完全缓解(CR)的患者的百分比。响应持续时间(DoR)是从实现响应直到复发或疾病进展的时间。缓解持续时间是从实现缓解(例如完全或部分缓解)直到复发的时间。在极端情况下,完全抑制在本文中被称为预防或化学预防。在该背景中,术语“预防”包括完全预防临床上明显的癌症的发作,或预防癌症的临床前明显的阶段的发作。该定义还旨在涵盖预防转化成恶性细胞,或者阻止或逆转恶化前细胞进展为恶性细胞。这包括对有患上癌症的风险的个体的预防性治疗。
对于白血病,特别是AML,可以基于国际工作组AML响应标准(InternationalWorking Group Response Criteria in AML)来评定对治疗的响应(Cheson等人,J ClinOncol 2003;21(24):4642-9)。
根据IWG针对AML的标准的血液学响应:

关键词:CR=完全缓解;EMD=髓外病;IWG=国际工作组;NA=不适用。
淋巴瘤的治疗可以通过针对NHL的国际研讨会标准(International WorkshopCriteria(IWC))(参见Cheson BD等人,J.Clin.Oncol:2007:(25)579-586),使用下文示出的响应和终点定义来评定:


缩写:CR,完全缓解;FDG,[18F]氟脱氧葡萄糖;PET,正电子发射断层摄影术;CT,计算机断层摄影术;PR,部分缓解;SPD,直径乘积总和;SD,疾病稳定;PD,疾病进展。


缩写:CR:完全缓解;PR:部分缓解。
在一个实施方案中,淋巴瘤的终点是临床益处的证据。临床益处可以反映生活质量的改善,或患者症状、输注需求、频繁感染或其它参数减少。到淋巴瘤相关症状的再现或进展的时间也可以用于该终点中。
CLL的治疗可以通过针对CLL的国际研讨会指南(参见Hallek M等人,Blood,2008;(111)12:5446-5456),使用其中示出的响应和终点定义来评定,具体为:

A组标准限定肿瘤负荷;B组标准限定造血系统(或骨髓)的功能。CR(完全缓解):必须满足所有标准,并且患者必须缺少疾病相关的体质性症状;PR(部分缓解):必须满足A组的至少两个标准加上B组的一个标准;SD是没有疾病进展(PD)且未实现至少PR;PD:必须满足以上A组或B组的标准中的至少一个。多个淋巴结的乘积之和(如通过临床试验中的CT扫描、或通过全科诊疗中的体格检查来评估的)。这些参数对于一些响应类别是不相关的。
MM的治疗可以通过针对多发性骨髓瘤的国际统一响应标准(IURC)(参见Durie等人,Leukemia,2006;(10)10:1-7),使用下文示出的响应和终点定义来评定:

缩写:CR,完全响应;FLC,游离轻链;PR,部分响应;SD,疾病稳定;sCR,严格完全响应;VGPR,极好的部分响应;a所有反应类别均需要在建立任何新疗法之前的任何时间进行的两个连续评定;如果进行了射线照相研究,则所有类别还需要无已知的进行性或新的骨病变的迹象。不需要射线照相研究来满足这些响应要求;b不需要用重复骨髓活检来证实;c克隆细胞的存在/不存在基于κ/λ比。通过免疫组织化学和/或免疫荧光得到的异常κ/λ比需要最少100个浆细胞用于分析。反映异常克隆的存在的异常比率是κ/λ>4:1或<1:2。d可测量的疾病由以下测量值中的至少一个来定义:骨髓浆细胞≥30%;血清M-蛋白≥1g/dl(≥10gm/l)[10g/l];尿液M-蛋白≥200mg/24h;血清FLC测定:涉及的FLC水平≥10mg/dl(≥100mg/l);条件是血清FLC比异常。
癌症的治疗也可以通过实体肿瘤响应评估标准(RECIST 1.1)来评定(参见Thereasse P.等人,J.of the National Cancer Institute;2000;(92)205-216,以及Eisenhauer等人,European J.Cancer;2009;(45)228–247)。靶标病变和非靶标病变中的肿瘤响应与出现或不出现新病变的所有可能组合的总体响应如下:
| 靶标病变 | 非靶标病变 | 新病变 | 总体响应 |
| CR | CR | 无 | CR |
| CR | 不完全响应/SD | 无 | PR |
| PR | 非-PD | 无 | PR |
| SD | 非-PD | 无 | SD |
| PD | 任何 | 有或无 | PD |
| 任何 | PD | 有或无 | PD |
| 任何 | 任何 | 有 | PD |
CR=完全响应;PR=部分响应;SD=疾病稳定;PD=疾病进展。
关于靶标病变的评估,完全响应(CR)是所有靶标病变消失;部分响应(PR)是以基线总最长直径作为参照,靶标病变的最长直径总和至少降低30%;疾病进展(PD)是以从开始治疗或出现一个或多个新病变以来所记录的最小总最长直径作为参照,靶标病变的最长直径的总和至少增大20%;并且疾病稳定(SD)是以从开始治疗以来所记录的最小总最长直径作为参照,既不足以降低至对部分响应合格,也不足以增大至对疾病进展合格。
关于非靶标病变的评估,完全响应是所有非靶标病变消失且肿瘤标志物水平正常化;不完全响应/疾病稳定是一个或多个非靶标病变持续存在并且/或者肿瘤标志物水平维持在正常限度以上;并且疾病进展(PD)是出现一个或多个新病变并且/或者现有的非靶标病变明确进展。
MDS的治疗可以通过针对骨髓增生异常的国际工作组(IWG)响应标准来评定。
针对MDS的经修改IWG响应标准


BM=骨髓;CR=完全缓解;FAB=法-美-英;Hgb=血红蛋白;HI=血液学改善;IWG=国际工作组;MDS=骨髓增生异常综合征;PB=外周血;PD=疾病进展;PR=部分缓解;RBC=红血细胞。
a增生异常改变应当考虑增生异常改变的正常范围(修改)。
b对IWG响应标准的修改。
c在一些情况下,方案疗法可能需要在4周时期之前开始进一步治疗(例如,巩固、维持)。此类对象可以被包括在该疗法开始时他们适合的响应类别中。重复化学疗法疗程期间出现的短暂性血细胞减少,只要恢复到前期疗程的改善计数,就不应视为中断响应的持久性。
d申办方对IWG标准的修改。
资料来源:Cheson,2006和Vardiman,2008。
RBC和血小板输注独立性

RBC=红血细胞;Hgb=血红蛋白。
aRBC输注独立性和RBC输注依赖性根据经修改的IWG标准进行定义。
b血小板输注独立性和血小板输注依赖性由申办方定义。
资料来源:Cheson等人,Blood.2006;108(2):419-25。
经修订的国际预后评分系统用于MDS的预后,如下所示:
IPSS-R细胞遗传学风险组


资料来源:Greenburg等人,Blood.2012;120(12):2454-65。
IPSS-R预后评分值

资料来源:Greenburg等人,Blood.2012;120(12):2454-65。
总IPSS-R评分被计算为细胞遗传学、骨髓母细胞百分比、血红蛋白、血小板和ANC个体评分的总和。
IPSS-R预后风险类别/评分
| 风险类别 | 风险评分 |
| 极低 | ≤1.5 |
| 低 | >1.5–3 |
| 中等 | >3–4.5 |
| 高 | >4.5–6 |
| 极高 | >6 |
资料来源:Greenburg等人,Blood.2012;120(12):2454-65。
IPSS-R:预后风险类别临床结果

资料来源:Greenberg等人,Blood.2012;120(12):2454-65
化合物
适合在本文提供的方法和制剂中使用的化合物是具有以下结构的化合物1:2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺:

或其立体异构体或立体异构体混合物、同位素体、药学上可接受的盐、互变异构体、溶剂合物、水合物、共晶体、笼形包合物或多晶型物。在某些实施方案中,化合物1是指2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺。
化合物1可以根据本文提供的实施例中所述或如美国专利号9,499,514中所述的方法制备,该专利的公开内容全文以引用方式并入本文。基于本文的教导,该化合物也可以根据对于本领域技术人员来说显而易见的其它方法合成。
在某些实施方案中,化合物1是固体。在某些实施方案中,化合物1是水合物。在某些实施方案中,化合物1是溶剂合物。在某些实施方案中,化合物1是无水的。
在某些实施方案中,化合物1是无定形的。在某些实施方案中,化合物1是结晶的。在某些实施方案中,化合物1是2017年1月6日提交的美国公布号2017-0197934中所述的结晶形式,该美国公布全文以引用方式并入本文。示例性的固体形式在第86至101页上有所描述。
化合物1的固体形式可以根据2017年1月6日提交的美国公布号2017-0197934的公开内容中所述的方法制备。参见第86至101页。这些固体形式也可以根据对于本领域技术人员来说显而易见的其它方法制备。
在一个实施方案中,化合物1是2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的多晶型物A型、B型、C型、D型、E型或无定形形式。2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的多晶型物在本文中简要描述。
2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的A型
在某些实施方案中,本文提供的制剂由化合物1的A型制备。
在一个实施方案中,A型是化合物1的无水形式。在另一个实施方案中,化合物1的A型是结晶的。
在某些实施方案中,A型从某些溶剂体系通过结晶来获得,所述溶剂体系例如包含以下溶剂中的一种或多种:丙酮以及室温下异丙醇和水的溶剂混合物。在某些实施方案中,A型在升高的温度下从浆液,例如约50℃,乙醇/水(1:1)、丙酮或乙腈中,以中间固体形式获得。
在某些实施方案中,A型是基本上结晶的,如通过例如X-射线粉末衍射测量所指示。在一个实施方案中,化合物1的A型具有基本上如图2中所示的X-射线粉末衍射图。
在一个实施方案中,如图2中所描绘,化合物1的A型在大约11.5、15.6、16.6、17.2、18.1、19.0、19.6、21.1、23.2或24.8度2θ的2θ角下具有一个或多个特征X-射线粉末衍射峰。在另一个实施方案中,化合物1的A型在大约15.6、16.6、17.2或24.8度2θ的2θ角下具有一个、两个、三个或四个特征X-射线粉末衍射峰。在另一个实施方案中,如表A中所示,化合物1的A型具有一个、两个、三个、四个、五个、六个或七个特征X-射线粉末衍射峰。在另一个实施方案中,如表A中所示,化合物1的A型具有一个、两个或三个特征X-射线粉末衍射峰。
表A

在一个实施方案中,化合物1的A型具有如图3中所示的SEM图。
在一个实施方案中,化合物1的结晶形式具有基本上对应于如图4中所描绘的代表性热重(TGA)热谱图的TGA温度过程线。在某些实施方案中,未观察到A型的TGA重量损失。
在一个实施方案中,化合物1的结晶A型具有基本上与如图5中所描绘对应的DSC热谱图。在某些实施方案中,A型的特征在于包括起始温度为229℃并且熔化热为118J/g的熔融事件的DSC图。
在某些实施方案中,A型的特征在于动态蒸气吸附分析。代表性动态蒸气吸附(DVS)等温线图在图6中示出。在某些实施方案中,当相对湿度(“RH”)从约0%增加至约90%RH时,A型表现出小于1.5重量%、小于1.2重量%或约1.2重量%的水摄取。在某些实施方案中,如配备有设定为225℃的炉样品处理器的库伦卡尔·费歇尔(Karl Fischer,KF)滴定器所确定,A型包含小于0.1%的水。
在某些实施方案中,通过1H NMR观察到A型无显著降解或残留溶剂(图7)。
在某些实施方案中,化合物1的A型的特征在于其在压缩时的稳定性曲线。在某些实施方案中,A型是稳定的,例如,在施加2000-psi压力约1分钟时,其XRPD图保持基本上不变,但具有更宽的衍射峰(图8)。
在又一个实施方案中,化合物1的A型是基本上纯的。在某些实施方案中,化合物1的基本上纯的A型基本上不含其它固体形式,例如无定形形式。在某些实施方案中,化合物1的基本上纯的A型的纯度为不小于约95%纯的、不小于约96%纯的、不小于约97%纯的、不小于约98%纯的、不小于约98.5%纯的、不小于约99%纯的、不小于约99.5%纯的,或不小于约99.8%纯的。
在某些实施方案中,化合物1的A型是基本上纯的。在本文的某些实施方案中,化合物1的A型基本上不含包含化合物1的其它固体形式,包括例如,包含化合物1的B型、C型、D型、E型和/或无定形固体形式。在某些实施方案中,A型是包含化合物1的固体形式的混合物,包括例如,包含下列中的一种或多种的混合物:包含化合物1的B型、C型、D型、E型和无定形固体形式。
2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的B型
在某些实施方案中,本文提供的制剂由化合物1的无水B型制备。
在某些实施方案中,B型从某些溶剂体系通过反溶剂再结晶来获得,所述溶剂体系例如包含以下溶剂中的一种或多种:甲醇/水、DMSO/异丙醇、DMSO/甲苯,和DMSO/水。在某些实施方案中,B型从THF/水(1:1)通过冷却再结晶来获得。
在某些实施方案中,B型是结晶的,如通过例如X-射线粉末衍射测量所指示。在一个实施方案中,化合物1的B型具有基本上如图9中所示的X-射线粉末衍射图。
在一个实施方案中,如图9中所描绘,化合物1的B型在大约15.4、16.3、16.7、17.7、20.4、25.6或27.5度2θ的2θ角下具有一个或多个特征X-射线粉末衍射峰。在另一个实施方案中,化合物1的B型在大约16.7、25.6、15.4或16.3度2θ的2θ角下具有一个、两个、三个或四个特征X-射线粉末衍射峰。在另一个实施方案中,如表B中所示,化合物1的B型具有一个、两个、三个、四个、五个、六个或七个特征X-射线粉末衍射峰。在另一个实施方案中,如表B中所示,化合物1的B型具有一个、两个或三个特征X-射线粉末衍射峰。
表B

在一个实施方案中,化合物1的B型具有如图10中所示的SEM图。在一个实施方案中,化合物1的结晶形式具有基本上对应于如图11中所描绘的代表性热重(TGA)热谱图的TGA温度过程线。在某些实施方案中,在低于170℃下,B型未示出TGA重量损失。在某些实施方案中,在170至230℃之间,B型示出0.4%的TGA重量损失。
在一个实施方案中,化合物1的结晶B型具有基本上与如图12中所描绘对应的DSC热谱图。在某些实施方案中,B型的特征在于包括在219至224℃下的熔融/再结晶事件和峰值温度为231℃的主要熔融事件的DSC图。
在某些实施方案中,B型的特征在于动态蒸气吸附分析。代表性动态蒸气吸附(DVS)等温线图在图13中示出。在某些实施方案中,当相对湿度(“RH”)从约0%增加至约90%RH时,B型表现出约1.4重量%的水摄取。在某些实施方案中,如配备有设定为225℃的炉样品加热处理器的库伦卡尔·费歇尔(KF)滴定器所确定,B型包含小于0.1%的水。
在某些实施方案中,通过1H NMR检测到,B型未示出显著降解或残留溶剂(图14)。
在某些实施方案中,化合物1的B型的特征在于其在压缩时的稳定性曲线。在某些实施方案中,B型是稳定的,例如,在施加2000-psi压力约1分钟时,其XRPD图保持基本上不变,但具有更宽的衍射峰(图15)。
在又一个实施方案中,化合物1的B型是基本上纯的。在某些实施方案中,化合物1的基本上纯的B型基本上不含其它固体形式,例如无定形形式。在某些实施方案中,化合物1的基本上纯的B型的纯度为不小于约95%纯的、不小于约96%纯的、不小于约97%纯的、不小于约98%纯的、不小于约98.5%纯的、不小于约99%纯的、不小于约99.5%纯的,或不小于约99.8%纯的。
在某些实施方案中,化合物1的B型是基本上纯的。在某些实施方案中,化合物1的B型基本上不含包含化合物1的其它固体形式,包括例如,包含化合物1的A型、C型、D型、E型和/或无定形固体形式。在某些实施方案中,B型是包含化合物1的固体形式的混合物,包括例如,包含下列中的一种或多种的混合物:包含化合物1的A型、C型、D型、E型和无定形固体形式。
2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的C型
在某些实施方案中,本文提供的制剂由化合物1的无水C型制备。在某些实施方案中,C型是化合物1的晶体形式中热力学最稳定的无水物。
在某些实施方案中,C型通过使化合物1在某些溶剂体系中长时间浆化来获得,所述溶剂体系例如包含以下溶剂中的一种或多种:乙腈/水、丙酮或乙醇/水。
在某些方面,C型通过在升高的温度下(例如在60至80℃或70至75℃下)将B型(1X重量)在丙酮(30X体积)中浆化至少24小时,然后将混合物冷却至室温来获得。在一个方面,浆化在70至75℃的温度和50至55-psi的氮气压力下进行。在一个方面,将该混合物在至少6小时内冷却至室温。
在某些实施方案中,C型是结晶的,如通过例如X-射线粉末衍射测量所指示。在一个实施方案中,化合物1的C型具有基本上如图16中所示的X-射线粉末衍射图。
在一个实施方案中,如图16中所描绘,化合物1的C型在大约7.4、11.5、15.8、16.7、16.9、17.7、18.4、19.2、19.5、21.1、23.4、24.7或29.9度2θ的2θ角下具有一个或多个特征X-射线粉末衍射峰。在另一个实施方案中,化合物1的C型在大约16.7、16.9、17.7或24.7度2θ的2θ角下具有一个、两个、三个或四个特征X-射线粉末衍射峰。在另一个实施方案中,如表C中所示,化合物1的C型具有一个、两个、三个、四个、五个、六个或七个特征X-射线粉末衍射峰。在另一个实施方案中,如表C中所示,化合物1的C型具有一个、两个或三个特征X-射线粉末衍射峰。
表C

在一个实施方案中,化合物1的C型具有如图17中所示的SEM图。在一个实施方案中,化合物1的结晶形式具有基本上对应于如图18中所描绘的代表性热重(TGA)热谱图的TGA温度过程线。在某些实施方案中,C型未示出TGA重量损失。
在一个实施方案中,化合物1的结晶C型具有基本上与如图19中所描绘对应的DSC热谱图。在某些实施方案中,C型的特征在于包括起始温度为232℃并且熔化热为126J/g的熔融事件的DSC图。
在某些实施方案中,C型的特征在于动态蒸气吸附分析。代表性动态蒸气吸附(DVS)等温线图在图20中示出。在某些实施方案中,当相对湿度(“RH”)从约0%增加至约90%RH时,C型表现出约0.6重量%的水摄取。在某些实施方案中,如配备有设定为225℃的炉样品加热处理器的库伦卡尔·费歇尔(KF)滴定器所确定,C型包含小于0.1%的水。
在某些实施方案中,通过1H NMR检测到,C型未示出显著降解或残留溶剂(图21)。
在某些实施方案中,化合物1的C型的特征在于其在压缩时的稳定性曲线。在某些实施方案中,C型是稳定的,例如,在施加2000-psi压力约1分钟时,其XRPD图保持基本上不变,但具有更宽的衍射峰(图22)。
在又一个实施方案中,化合物1的C型是基本上纯的。在某些实施方案中,化合物1的基本上纯的C型基本上不含其它固体形式,例如无定形形式。在某些实施方案中,化合物1的基本上纯的C型的纯度为不小于约95%纯的、不小于约96%纯的、不小于约97%纯的、不小于约98%纯的、不小于约98.5%纯的、不小于约99%纯的、不小于约99.5%纯的,或不小于约99.8%纯的。
在某些实施方案中,化合物1的C型是基本上纯的。在某些实施方案中,化合物1的C型基本上不含包含化合物1的其它固体形式,包括例如,包含化合物1的A型、B型、D型、E型和/或无定形固体形式。在某些实施方案中,C型是包含化合物1的固体形式的混合物,包括例如,包含下列中的一种或多种的混合物:包含化合物1的A型、B型、D型、E型和无定形固体形式。
2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的D型
在某些实施方案中,本文提供的制剂由化合物1的D型制备。在某些实施方案中,化合物1的D型是DMSO溶剂合物。
在某些实施方案中,D型通过加热溶于DMSO/甲基异丁基酮的B型并冷却溶液来获得。
在某些实施方案中,D型是结晶的,如通过例如X-射线粉末衍射测量所指示。在一个实施方案中,化合物1的D型具有基本上如图23中所示的X-射线粉末衍射图。
在一个实施方案中,如图23中所描绘,化合物1的D型在大约14.1、14.3、18.8、19.1、23.6或24.0度2θ的2θ角下具有一个或多个特征X-射线粉末衍射峰。在另一个实施方案中,化合物1的D型在大约14.1、14.3、18.8或19.1度2θ的2θ角下具有一个、两个、三个或四个特征X-射线粉末衍射峰。在另一个实施方案中,如表D中所示,化合物1的D型具有一个、两个、三个、四个、五个、六个或七个特征X-射线粉末衍射峰。在另一个实施方案中,如表D中所示,化合物1的D型具有一个、两个或三个特征X-射线粉末衍射峰。
表D

在一个实施方案中,本文提供了化合物1的具有基本上对应于如图24中所描绘的代表性热重(TGA)热谱图的TGA温度过程线的结晶形式。在某些实施方案中,D型在最高140℃下示出约14.1%的TGA重量损失。
在某些实施方案中,如通过气相色谱法所测量的,D型包含约14.3重量%的DMSO。
在又一个实施方案中,化合物1的D型是基本上纯的。在某些实施方案中,化合物1的基本上纯的D型基本上不含其它固体形式,例如无定形形式。在某些实施方案中,化合物1的基本上纯的D型的纯度为不小于约95%纯的、不小于约96%纯的、不小于约97%纯的、不小于约98%纯的、不小于约98.5%纯的、不小于约99%纯的、不小于约99.5%纯的,或不小于约99.8%纯的。
在某些实施方案中,化合物1的D型是基本上纯的。在某些实施方案中,化合物1的D型基本上不含包含化合物1的其它固体形式,包括例如,如本文提供的包含化合物1的A型、B型、C型、E型和/或无定形固体形式。在某些实施方案中,D型是包含化合物1的固体形式的混合物,包括例如,包含下列中的一种或多种的混合物:包含化合物1的A型、B型、C型、E型和无定形固体形式。
2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的E型
在某些实施方案中,本文提供的制剂由化合物1的E型制备。在某些实施方案中,化合物1的E型是DMSO溶剂合物。
在某些实施方案中,E型在室温下从溶于DMSO/MIBK或DMSO/IPA或DMSO/苯甲醚的C型获得。
在某些实施方案中,E型是结晶的,如通过例如X-射线粉末衍射测量所指示。在一个实施方案中,化合物1的E型具有基本上如图25中所示的X-射线粉末衍射图。
在一个实施方案中,如图25中所描绘,化合物1的E型在大约10.5、12.5、16.1、17.0、18.5、21.2、21.7、22.6、22.9、23.4、23.8、24.1、25.1或26.7度2θ的2θ角下具有一个或多个特征X-射线粉末衍射峰。在另一个实施方案中,化合物1的E型在大约16.1、17.0、21.2或22.9度2θ的2θ角下具有一个、两个、三个或四个特征X-射线粉末衍射峰。在另一个实施方案中,如表E中所示,化合物1的E型具有一个、两个、三个、四个、五个、六个或七个特征X-射线粉末衍射峰。在另一个实施方案中,如表E中所示,化合物1的E型具有一个、两个或三个特征X-射线粉末衍射峰。
表E


在一个实施方案中,本文提供了化合物1的具有基本上对应于如图26中所描绘的代表性热重(TGA)热谱图的TGA温度过程线的结晶形式。在某些实施方案中,E型在最高120℃下示出约19.4%的TGA重量损失。在某些实施方案中,E型在120℃与220℃之间示出24.9%的附加重量损失。
在一个实施方案中,化合物1的E型是基本上纯的。在某些实施方案中,化合物1的基本上纯的E型基本上不含其它固体形式,例如无定形形式。在某些实施方案中,化合物1的基本上纯的E型的纯度为不小于约95%纯的、不小于约96%纯的、不小于约97%纯的、不小于约98%纯的、不小于约98.5%纯的、不小于约99%纯的、不小于约99.5%纯的,或不小于约99.8%纯的。
在某些实施方案中,化合物1的E型是基本上纯的。在本文的某些实施方案中,化合物1的E型基本上不含包含化合物1的其它固体形式,包括例如,包含化合物1的A型、B型、C型、D型和/或无定形固体形式。在某些实施方案中,E型是包含化合物1的固体形式的混合物,包括例如,包含下列中的一种或多种的混合物:包含化合物1的A型、B型、C型、D型和无定形固体形式。
2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的无定形形式
在某些实施方案中,本文提供的制剂包含无定形化合物1。
在某些实施方案中,本文提供了通过在THF和水中加热化合物1并冷却溶液来用于制备无定形形式的方法。
在一个实施方案中,本文提供了化合物1的具有如图27中所描绘的经调制DSC热谱图的无定形固体形式。
在一个实施方案中,无定形化合物1具有基本上如图28中所示的X-射线粉末衍射图。
在一个实施方案中,无定形化合物1具有基本上如图29中所示的1H NMR谱。
在又一个实施方案中,无定形化合物1是基本上纯的。在某些实施方案中,基本上纯的无定形化合物1基本上不含其它固体形式,例如,A型、B型、C型、D型或E型。在某些实施方案中,基本上纯的无定形化合物1的纯度为不小于约95%纯的、不小于约96%纯的、不小于约97%纯的、不小于约98%纯的、不小于约98.5%纯的、不小于约99%纯的、不小于约99.5%纯的,或不小于约99.8%纯的。
化合物1的制剂
在一个方面,本文提供了稳定的化合物1的制剂。在一个实施方案中,这些化合物1的制剂包含2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的固体形式。在一个实施方案中,这些化合物1的制剂包含2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的无定形形式。
在某些实施方案中,这些制剂用二甲基亚砜作为共溶剂或加工助剂来制备。在某些实施方案中,这些制剂用甲酸作为共溶剂或加工助剂来制备。在某些实施方案中,这些制剂在没有任何共溶剂或加工助剂的情况下制备。
在某些实施方案中,这些制剂包含二甲基亚砜作为共溶剂或加工助剂。在某些实施方案中,这些制剂包含甲酸作为共溶剂或加工助剂。在某些实施方案中,这些制剂不包含任何共溶剂或加工助剂。
在某些实施方案中,本文提供的这些制剂是冻干制剂。在某些实施方案中,本文提供的制剂是在药学上可接受的溶剂中获得以产生药学上可接受的溶液的复原制剂。
制剂Ia
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.2%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂,和量为约92%至98%的羟丙基β-环糊精(HPBCD)。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.2%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂,和量为约92%至98%的磺丁基醚-β-环糊精。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.2%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂、量为约92%至98%的HPBCD,和不超过约1%的二甲基亚砜。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.2%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂、量为约92%至98%的磺丁基醚-β-环糊精,和不超过约1%的二甲基亚砜。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.08%至0.15%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂,和量为约94%至96%的HPBCD。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.08%至0.15%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂,和量为约94%至96%的磺丁基醚-β-环糊精。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.08%至0.15%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂、量为约94%至96%的HPBCD,和不超过约1%的二甲基亚砜。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.08%至0.15%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂、量为约94%至96%的磺丁基醚-β-环糊精,和不超过约1%的二甲基亚砜。
在一个方面,基于该制剂的总重量,本文提供的该制剂包含量为约0.08%至约0.15%的化合物1。在某些实施方案中,基于该制剂的总重量,化合物1的量为约0.09%至约0.15%、约0.1%至约0.13%,或约0.11%至约0.12%。在某些实施方案中,基于该制剂的总重量,化合物1的量为约0.05%、0.07%、0.09%、0.11%、0.12%、0.13%或0.15%。在一个实施方案中,基于该制剂的总重量,该制剂中化合物1的量为约0.12%。
在另一个方面,本文提供了在20cc小瓶中包含量为约0.5mg至约2mg的化合物1的制剂。在又一个方面,是在20cc小瓶中包含量为约0.5mg至约1.5mg、约0.75mg至约1.25mg、或约0.8mg至约1.1mg的化合物1的制剂。在一个方面,化合物1以约0.7、0.75、0.76、0.8、0.9、1.0、1.05或1.2mg的量存在于20cc小瓶中。在一个方面,化合物1以约1.05mg的量存在于20cc小瓶中。
在一个方面,本文提供的这些制剂含有柠檬酸盐缓冲剂。在一个方面,基于该制剂的总重量,本文提供的这些制剂中的柠檬酸盐缓冲剂的量为约3%至约6%。在一个方面,基于该制剂的总重量,本文提供的这些制剂中的柠檬酸盐缓冲剂的量为约3%、3.5%、4%、4.2%、4.5%或5%。在一个方面,基于该制剂的总重量,本文提供的这些制剂中的柠檬酸盐缓冲剂的量为约4.2%。在一个方面,在20cc小瓶中,本文提供的这些制剂中的柠檬酸盐缓冲剂的量为约37mg。
在一个实施方案中,柠檬酸盐缓冲剂包括无水柠檬酸和无水柠檬酸钠。在某些实施方案中,基于该制剂的总重量,无水柠檬酸的量为约1.5%至约3%、约1.75%至约2.75%,或约2%至约2.5%。在某些实施方案中,基于该制剂的总重量,该制剂中无水柠檬酸的量为约1.5%、1.75%、2%、2.1%或2.5%。在一个实施方案中,基于该制剂的总重量,该制剂中无水柠檬酸的量为约2%、2.1%、2.22%或2.3%。在一个实施方案中,基于该制剂的总重量,该制剂中无水柠檬酸的量为约2.10%。
在又一个方面,是在20cc小瓶中包含量为约16mg至约20mg的无水柠檬酸的制剂。在一个实施方案中,在20cc小瓶中,无水柠檬酸的量为约16、17、18、18.2、18.4、18.6、18.8、19或20mg。在一个实施方案中,在20cc小瓶中,无水柠檬酸的量为约18.6mg。
在某些实施方案中,基于该制剂的总重量,无水柠檬酸钠的量为约1.5%至约3%、约1.75%至约2.75%,或约2%至约2.5%。在某些实施方案中,基于该制剂的总重量,该制剂中无水柠檬酸钠的量为约1.5%、1.75%、2%、2.1%或2.5%。在一个实施方案中,基于该制剂的总重量,该制剂中无水柠檬酸钠的量为约2%、2.05%、2.08%或2.1%。在一个实施方案中,基于该制剂的总重量,该制剂中无水柠檬酸钠的量为约2.08%。
在又一个方面,是在20cc小瓶中包含量为约16mg至约20mg的无水柠檬酸钠的制剂。在一个实施方案中,在20cc小瓶中,无水柠檬酸钠的量为约16、17、18、18.2、18.4、18.6、18.8、19或20mg。在一个实施方案中,在20cc小瓶中,无水柠檬酸钠的量为约18.4mg。
在某些实施方案中,基于该制剂的总重量,本文提供的这些制剂中的HPBCD的量为约94%至约97%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的HPBCD的量为约94.5%、95%、95.5%或96%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的HPBCD的量为约95%。
在某些实施方案中,基于该制剂的总重量,本文提供的这些制剂中的磺丁基醚-β-环糊精的量为约94%至约97%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的磺丁基醚-β-环糊精的量为约94.5%、95%、95.5%或96%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的磺丁基醚-β-环糊精的量为约95%。
在另一个方面,是在20cc小瓶中包含量为约800至900mg的HPBCD的制剂。在另一个方面,是在20cc小瓶中包含量为约810至880mg、820至860mg或830至850mg的HPBCD的制剂。在另一个方面,是在20cc小瓶中包含量为约840mg的HPBCD的制剂。
在另一个方面,是在20cc小瓶中包含量为约800至900mg的磺丁基醚-β-环糊精的制剂。在另一个方面,是在20cc小瓶中包含量为约810至880mg、820至860mg或830至850mg的磺丁基醚-β-环糊精的制剂。在另一个方面,是在20cc小瓶中包含量为约840mg的磺丁基醚-β-环糊精的制剂。
在另一个方面,是在20cc小瓶中包含量为约840mg的 的制剂。
的制剂。
在一个实施方案中,基于该制剂的总重量,这些制剂包含量不超过约1.5%的二甲基亚砜。在一个实施方案中,基于该制剂的总重量,这些制剂包含量至多0.1%、0.2%、0.3%、0.4%、0.6%、0.7%、0.8%、0.9%或1%的二甲基亚砜。在一个实施方案中,基于该制剂的总重量,这些制剂包含不超过约0.1%、0.2%、0.3%、0.4%、0.6%、0.7%、0.8%、0.9%或1%的二甲基亚砜。在一个实施方案中,基于该制剂的总重量,这些制剂包含量至多约0.1%至约1.5%的二甲基亚砜。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的二甲基亚砜的量为约0.1%至约1.3%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的二甲基亚砜的量为约0.1%、0.2%、0.3%、0.4%、0.6%、0.7%、0.8%、0.9%或1%。在一个实施方案中,本文提供的这些制剂不包含任何二甲基亚砜。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的二甲基亚砜的量为约0.4%至约0.8%。
在另一个方面,是在20cc小瓶中包含量为约4至7mg的二甲基亚砜的制剂。在另一个方面,是在20cc小瓶中包含量为约4.5至6.5mg、或5至6mg的二甲基亚砜的制剂。
在某些实施方案中,本文提供的制剂是冻干的,并且该冻干制剂在复原后具有约4至5的pH。在某些实施方案中,该制剂在复原后具有约4.2至4.4的pH。在一个实施方案中,该冻干制剂在复原后具有约4、4.1、4.2、4.3、4.4、4.5、4.6、4.7、4.8、4.9或5的pH。
在某些实施方案中,该冻干制剂在复原后具有约250mOsm/kg至290mOsm/kg的克分子渗透压浓度。在某些实施方案中,该冻干制剂在复原后具有约260mOsm/kg至280mOsm/kg的克分子渗透压浓度。
在某些实施方案中,本文提供了包含本文提供的制剂的容器。在一个方面,该容器是玻璃小瓶。在一个方面,该容器是20cc玻璃小瓶。
在一个方面,本文提供了在20cc小瓶中的制剂,其包含:量为提供1.05mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1,和药学上可接受的载剂或赋形剂(包括如本文所述的增量剂)。在一个实施方案中,该制剂还包含不超过约7mg作为残留溶剂的二甲基亚砜。在一个实施方案中,该制剂包含不超过约6mg作为残留溶剂的二甲基亚砜。在一个实施方案中,该制剂包含不超过约5mg作为残留溶剂的二甲基亚砜。在一个实施方案中,该制剂包含不超过约4mg作为残留溶剂的二甲基亚砜。在一个实施方案中,该制剂包含约3mg至约7mg、约4mg至约6mg、约4mg至约5mg或约5mg至约6mg作为残留溶剂的二甲基亚砜。在一个实施方案中,该制剂包含约4、4.5、5、5.3、5.5、5.7、6或6.5mg作为残留溶剂的二甲基亚砜。
在一个实施方案中,本文提供的制剂基本上由下列组分组成:基于该制剂的总重量,量为约0.05%至0.2%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂,和量为约92%至98%的HPBCD。
在一个实施方案中,本文提供的制剂基本上由下列组分组成:基于该制剂的总重量,量为约0.05%至0.2%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂,和量为约92%至98%的磺丁基醚-β-环糊精。
在一个实施方案中,本文提供的制剂基本上由下列组分组成:基于该制剂的总重量,量为约0.05%至0.2%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂、量为约92%至98%的HPBCD,和不超过约1%的二甲基亚砜。
在一个实施方案中,本文提供的制剂基本上由下列组分组成:基于该制剂的总重量,量为约0.05%至0.2%的化合物1、量为约3%至6%的柠檬酸盐缓冲剂、量为约92%至98%的磺丁基醚-β-环糊精,和不超过约1%的二甲基亚砜。
在一个方面,本文提供了在20cc小瓶中的制剂,其包含:量为提供1.05mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、药学上可接受的载剂或赋形剂(包括如本文所述的缓冲剂和增量剂),和约5mg至约6mg作为残留溶剂的二甲基亚砜。缓冲剂和增量剂可以以如本文所述的量存在。
在一个方面,本文提供了在20cc小瓶中的制剂,其包含:量为提供1.05mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、18.6mg无水柠檬酸、18.4mg无水柠檬酸钠、840mg HPBCD,和约5mg至约6mg作为残留溶剂的二甲基亚砜,如本文所述。在一个实施方案中,用3.8mL无菌注射用水复原20cc小瓶中的制剂。
在一个方面,本文提供了在20cc小瓶中的制剂,其基本上由下列组分组成:量为提供1.05mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、18.6mg无水柠檬酸、18.4mg无水柠檬酸钠、840mg HPBCD,和约5mg至约6mg作为残留溶剂的二甲基亚砜,如本文所述。在一个实施方案中,用3.8mL无菌注射用水复原20cc小瓶中的制剂。
在一个方面,本文提供了在20cc小瓶中的制剂,其由下列组分组成:量为提供1.05mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、18.6mg无水柠檬酸、18.4mg无水柠檬酸钠、840mg HPBCD,和约5mg至约6mg作为残留溶剂的二甲基亚砜,如本文所述。在一个实施方案中,用3.8mL无菌注射用水复原20cc小瓶中的制剂。
在一个实施方案中,本文提供了水性制剂,其包含:基于固体的总重量,量为约0.05%至0.2%的化合物1;基于固体的总重量,量为约3%至6%的柠檬酸盐缓冲剂;基于固体的总重量,量为约92%至98%的HPBCD;和稀释剂。
在一个实施方案中,本文提供了水性制剂,其基本上由下列组分组成:基于固体的总重量,量为约0.05%至0.2%的化合物1;基于固体的总重量,量为约3%至6%的柠檬酸盐缓冲剂;基于固体的总重量,量为约92%至98%的HPBCD;和稀释剂。
在一个方面,本文提供了水性制剂,其包含:量为提供1.05mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、18.6mg无水柠檬酸、18.4mg无水柠檬酸钠、840mg HPBCD、约5mg至约6mg作为残留溶剂的二甲基亚砜和约3.8mL稀释剂。
在一个方面,本文提供了水性制剂,其基本上由下列组分组成:量为提供1.05mg2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、18.6mg无水柠檬酸、18.4mg无水柠檬酸钠、840mg HPBCD、约5mg至约6mg作为残留溶剂的二甲基亚砜和约3.8mL稀释剂。
在一个方面,本文提供了水性制剂,其由下列组分组成:量为提供1.05mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、18.6mg无水柠檬酸、18.4mg无水柠檬酸钠、840mg HPBCD、约5mg至约6mg作为残留溶剂的二甲基亚砜和约3.8mL稀释剂。
在某些实施方案中,该制剂具有如表43中所述的组成。
制剂Ib
在一个实施方案中,本文提供以下制剂:其包含量为约0.01%至0.15%的化合物1、量为约99.1%至99.99%的羟丙基β-环糊精。在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.01%至0.15%的化合物1、量为约99.1%至99.99%的羟丙基β-环糊精,和不超过约0.5%的甲酸。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.25%的化合物1和量为约99.1%至99.9%的HPBCD。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.25%的化合物1、量为约99.1%至99.9%的HPBCD,和不超过约0.5%的甲酸。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.25%的化合物1和量为约99.75%至99.9%的HPBCD。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.25%的化合物1、量为约99.75%至99.9%的HPBCD,和不超过约0.5%的甲酸。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.25%的化合物1、量为约99.75%至99.9%的HPBCD,和不超过约0.2%的甲酸。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.08%至0.15%的化合物1和量为约99.8%至99.9%的HPBCD。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.08%至0.15%的化合物1、量为约99.8%至99.9%的HPBCD,和不超过约0.5%的甲酸。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.08%至0.15%的化合物1、量为约99.8%至99.9%的HPBCD,和不超过约0.12%的甲酸。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.12%的化合物1和量为约99.88%的HPBCD。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.25%的化合物1和量为约99.1%至99.9%的磺丁基醚-β-环糊精。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.25%的化合物1、量为约99.1%至99.9%的磺丁基醚-β-环糊精,和不超过约0.5%的甲酸。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.05%至0.25%的化合物1和量为约99.75%至99.9%的磺丁基醚-β-环糊精。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.08%至0.15%的化合物1和量为约99.8%至99.9%的磺丁基醚-β-环糊精。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.08%至0.15%的化合物1、量为约99.8%至99.9%的磺丁基醚-β-环糊精,和不超过约0.5%的甲酸。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.12%的化合物1和量为约99.88%的磺丁基醚-β-环糊精。
在一个方面,基于该制剂的总重量,本文提供的该制剂包含量为约0.08%至约0.15%的化合物1。在某些实施方案中,基于该制剂的总重量,化合物1的量为约0.09%至约0.15%、约0.1%至约0.13%,或约0.11%至约0.12%。在某些实施方案中,基于该制剂的总重量,化合物1的量为约0.05%、0.07%、0.09%、0.11%、0.12%、0.13%或0.15%。在一个实施方案中,基于该制剂的总重量,该制剂中化合物1的量为约0.12%。
在另一个方面,本文提供了在20cc小瓶中包含量为约0.5mg至约2mg的化合物1的制剂。在又一个方面,是在20cc小瓶中包含量为约0.5mg至约1.5mg、约0.75mg至约1.25mg、或约0.8mg至约1.1mg的化合物1的制剂。在一个方面,化合物1以约0.7、0.75、0.76、0.8、0.9、1.0、1.05或1.2mg的量存在于20cc小瓶中。在一个方面,化合物1以约1mg的量存在于20cc小瓶中。
在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的HPBCD的量为约97%至约99.9%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的HPBCD的量为约98%至约99.9%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的HPBCD的量为约99.1%、99.3%、99.5%、99.7%或99.9%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的HPBCD的量为约99.5%。在另一个方面,是在20cc小瓶中包含量为约750至850mg的HPBCD的制剂。在另一个方面,是在20cc小瓶中包含量为约790至840mg、780至830mg或790至810mg的HPBCD的制剂。在另一个方面,是在20cc小瓶中包含量为约800mg的HPBCD的制剂。
在另一个方面,是在20cc小瓶中包含量为约800mg的 的制剂。
的制剂。
在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的磺丁基醚-β-环糊精的量为约97%至约99.9%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的磺丁基醚-β-环糊精的量为约98%至约99.9%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的磺丁基醚-β-环糊精的量为约99.1%、99.3%、99.5%、99.7%或99.9%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的磺丁基醚-β-环糊精的量为约99.5%。
在另一个方面,是在20cc小瓶中包含量为约750至850mg的磺丁基醚-β-环糊精的制剂。在另一个方面,是在20cc小瓶中包含量为约790至840mg、780至830mg或790至810mg的磺丁基醚-β-环糊精的制剂。在另一个方面,是在20cc小瓶中包含量为约800mg的磺丁基醚-β-环糊精的制剂。
在另一个方面,是在20cc小瓶中包含量为约800mg的 的制剂。
的制剂。
在一个实施方案中,基于该制剂的总重量,这些制剂包含不超过约0.5%的甲酸。在一个实施方案中,基于该制剂的总重量,这些制剂包含量至多约0.05%、0.07%、0.09%、0.1%、0.2%、0.3%、0.4%或0.5%的甲酸。在一个实施方案中,基于该制剂的总重量,这些制剂包含不超过约0.05%、0.07%、0.09%、0.1%、0.2%、0.3%、0.4%或0.5%的甲酸。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的甲酸的量为约0.05%至约0.5%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的甲酸的量为约0.05%至约0.1%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的甲酸的量为约0.05%、0.07%、0.09%、0.1%、0.2%、0.3%、0.4%或0.5%。在一个实施方案中,本文提供的这些制剂不包含任何甲酸。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的甲酸的量为约0.05%至0.09%。
在另一个方面,是在20cc小瓶中包含量不超过约1mg的甲酸的制剂。在另一个方面,是在20cc小瓶中包含量至多约0.2、0 5、0.7、0.9mg或1mg的甲酸的制剂。在另一个方面,是在20cc小瓶中包含量为约0.3至0.9mg或0.4至0.8mg的甲酸的制剂。
在另一个方面,本文提供了在20cc小瓶中包含量为约1mg的化合物1和量为约800mg的HPBCD的制剂。
在另一个方面,本文提供了在20cc小瓶中包含量为约1mg的化合物1、量为约800mg的HPBCD和量为约0.9mg的甲酸的制剂。
在某些实施方案中,该制剂具有如表43中所述的组成。
制剂Ic
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.01%至0.08%的化合物1和量为约99.40%至99.99%的HPBCD。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.01%至0.08%的化合物1、量为约99.40%至99.99%的HPBCD,和不超过约0.5%的甲酸。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.03%至0.06%的化合物1和量为约99.60%至99.99%的HPBCD。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含约0.01%至约0.08%的化合物1、约99.40%至约99.99%的羟丙基β-环糊精,和约0.1%至约0.3%的甲酸。
在一个方面,基于该制剂的总重量,本文提供的该制剂包含量为约0.02%至约0.06%的化合物1。在某些实施方案中,基于该制剂的总重量,化合物1的量为约0.03%至约0.06%、或约0.04%至约0.06%。在某些实施方案中,基于该制剂的总重量,化合物1的量为约0.03%、0.04%、0.05%或0.06%。在一个实施方案中,基于该制剂的总重量,该制剂中化合物1的量为约0.05%。
在另一个方面,本文提供了在20cc小瓶中包含量为约0.75mg至约1.5mg的化合物1的制剂。在又一个方面,是在20cc小瓶中包含量为约0.75mg至约1.25mg的化合物1的制剂。在一个方面,化合物1以约0.75、0.8、0.9、1.0、1.05或1.2mg的量存在于20cc小瓶中。在一个方面,化合物1以约1mg的量存在于20cc小瓶中。
在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的HPBCD的量为约99.40%至约99.99%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的HPBCD的量为约99.5%、99.6%、99.7%、99.8%、99.9%、99.95%或99.99%。在另一个方面,是在20cc小瓶中包含量为约1800至1900mg的HPBCD的制剂。在另一个方面,是在20cc小瓶中包含量为约1850至1900mg的HPBCD的制剂。在另一个方面,是在20cc小瓶中包含量为约1875mg的HPBCD的制剂。
在一个实施方案中,基于该制剂的总重量,这些制剂包含不超过约0.5%的甲酸。在一个实施方案中,基于该制剂的总重量,这些制剂包含量至多约0.05%、0.07%、0.09%、0.1%、0.2%、0.3%、0.4%或0.5%的甲酸。在一个实施方案中,基于该制剂的总重量,这些制剂包含不超过约0.05%、0.07%、0.09%、0.1%、0.2%、0.3%、0.4%或0.5%的甲酸。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的甲酸的量为约0.05%至约0.3%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的甲酸的量为约0.05%至约0.25%。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的甲酸的量为约0.05%、0.07%、0.09%、0.1%、0.2%或0.3%。在一个实施方案中,本文提供的这些制剂不包含任何甲酸。在一个实施方案中,基于该制剂的总重量,本文提供的这些制剂中的甲酸的量为约0.11%至0.3%。
在另一个方面,是在20cc小瓶中包含量不超过约4mg的甲酸的制剂。在另一个方面,是在20cc小瓶中包含量至多约1、1.8、2、2.1、2.5、3、3.5、3.8、3.9、4、4.5、4.9mg或5mg的甲酸的制剂。在另一个方面,是在20cc小瓶中包含量为约1至1.8mg、2.1至3.8mg或3.9至4.9mg的甲酸的制剂。
在另一个方面,本文提供了在20cc小瓶中包含量为约1mg的化合物1和量为约1875mg的HPBCD的制剂。
在另一个方面,本文提供了在20cc小瓶中包含量为约1mg的化合物1、量为约1875mg的HPBCD和量为约2.1至3.8mg的甲酸的制剂。
在某些实施方案中,该制剂具有如表64中所述的组成。
不使用共溶剂的制剂
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.15%至0.5%的化合物1、量为约15%至约35%的柠檬酸盐缓冲剂,和量为约92%至约98%的HPBCD。在一个实施方案中,柠檬酸盐缓冲剂包括无水柠檬酸和无水柠檬酸钠。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.25%至0.30%的化合物1、量为约30%至32%的柠檬酸盐缓冲剂,和量为约67%至69%的HPBCD。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其包含量为约0.30%至0.33%的化合物1、量为约17%至18%的柠檬酸盐缓冲剂,和量为约80%至85%的HPBCD。
示例性制剂
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其基本上由下列组分组成:量为约0.05%至0.25%的化合物1和量为约99.75%至99.95%的HPBCD。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其基本上由下列组分组成:量为约0.05%至0.25%的化合物1和量为约99.75%至99.99%的HPBCD。
在一个实施方案中,本文提供以下制剂:基于该制剂的总重量,其基本上由下列组分组成:量为约0.05%至0.25%的化合物1和量为约99.75%至99.95%的磺丁基醚-β-环糊精。
在一个方面,本文提供了在20cc小瓶中的制剂,其包含:量为提供1mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、800mg HPBCD和约0.6mg甲酸,如本文所述。在一个实施方案中,用4.5mL无菌注射用水复原20cc小瓶中的制剂。
在一个方面,本文提供了在20cc小瓶中的制剂,其基本上由下列组分组成:量为提供1mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、800mg HPBCD和约0.6mg甲酸,如本文所述。在一个实施方案中,用4.5mL无菌注射用水复原20cc小瓶中的制剂。
在一个方面,本文提供了在20cc小瓶中的制剂,其由下列组分组成:量为提供1mg2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、800mg HPBCD和约0.6mg甲酸,如本文所述。在一个实施方案中,用4.5mL无菌注射用水复原20cc小瓶中的制剂。
在一个方面,本文提供了在20cc小瓶中的制剂,其包含:量为提供1mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、800mg磺丁基醚-β-环糊精和约0.6mg甲酸,如本文所述。在一个实施方案中,用4.5mL无菌注射用水复原20cc小瓶中的制剂。
在一个方面,本文提供了在20cc小瓶中的制剂,其基本上由下列组分组成:量为提供1mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、800mg磺丁基醚-β-环糊精和约0.6mg甲酸,如本文所述。在一个实施方案中,用4.5mL无菌注射用水复原20cc小瓶中的制剂。
在一个方面,本文提供了在20cc小瓶中的制剂,其由下列组分组成:量为提供1mg2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、800mg磺丁基醚-β-环糊精和约0.6mg甲酸,如本文所述。在一个实施方案中,用4.5mL无菌注射用水复原20cc小瓶中的制剂。
在一个方面,本文提供了在20cc小瓶中的制剂,其包含:量为提供1mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、1875mg HPBCD和约2.1至3.8mg甲酸,如本文所述。在一个实施方案中,用12.5ml注射用生理盐水复原20cc小瓶中的制剂。
在一个方面,本文提供了在20cc小瓶中的制剂,其基本上由下列组分组成:量为提供1mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、1875mg HPBCD和约2.1至3.8mg甲酸,如本文所述。在一个实施方案中,用12.5ml注射用生理盐水复原20cc小瓶中的制剂。
在一个方面,本文提供了在20cc小瓶中的制剂,其由下列组分组成:量为提供1mg2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、1875mg HPBCD和约2.1至3.8mg甲酸,如本文所述。在一个实施方案中,用12.5ml注射用生理盐水复原20cc小瓶中的制剂。
在一个实施方案中,本文提供了水性制剂,其包含:基于固体的总重量,量为约0.05%至0.25%的化合物1;基于固体的总重量,量为约99.1%至99.9%的HPBCD;和稀释剂。
在一个实施方案中,本文提供了水性制剂,其包含:基于固体的总重量,量为约0.05%至0.25%的化合物1;基于固体的总重量,量为约99.75%至99.95%的HPBCD;和稀释剂。
在一个实施方案中,本文提供了水性制剂,其基本上由下列组分组成:基于固体的总重量,量为约0.05%至0.25%的化合物1;基于固体的总重量,量为约99.75%至99.95%的HPBCD;和稀释剂。
在一个方面,本文提供了水性制剂,其包含:量为提供1mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、800mgHPBCD、约0.6mg甲酸和约4.5mL稀释剂。
在一个方面,本文提供了水性制剂,其由下列组分组成:量为提供1mg2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、800mg HPBCD、约0.6mg甲酸和约4.5mL稀释剂。
在一个实施方案中,本文提供了水性制剂,其包含:基于固体的总重量,量为约0.01%至0.08%的化合物1;基于固体的总重量,量为约99.50%至99.99%的HPBCD;和稀释剂。
在一个实施方案中,本文提供了水性制剂,其包含:基于固体的总重量,量为约0.01%至0.08%的化合物1;基于固体的总重量,量为约99.50%至99.99%的HPBCD;和稀释剂。
在一个实施方案中,本文提供了水性制剂,其基本上由下列组分组成:基于固体的总重量,量为约0.01%至0.08%的化合物1;基于固体的总重量,量为约99.50%至99.99%的HPBCD;和稀释剂。
在一个方面,本文提供了水性制剂,其包含:量为提供1mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、800mgHPBCD、约0.6mg甲酸和约4.5mL稀释剂。
在一个方面,本文提供了水性制剂,其由下列组分组成:量为提供1mg2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1、800mg HPBCD、约0.6mg甲酸和约4.5mL稀释剂。
在某些实施方案中,该制剂具有如表43中所述的组成。在某些实施方案中,该制剂具有如表64中所述的组成。
在某些实施方案中,本文提供的制剂是冻干的,并且该冻干制剂在复原后具有约2.5至4的pH。在某些实施方案中,该冻干制剂在复原后具有约2.5至3.5的pH。在某些实施方案中,该冻干制剂在复原后具有约3.0至3.6的pH。在一个实施方案中,该冻干制剂在复原后具有约2.5、3、3.2、3.4、3.6、3.8或4的pH。在一个实施方案中,该冻干制剂在复原后具有约2.5、2.8、3、3.2、3.4、3.6、3.8或4的pH。
在某些实施方案中,该冻干制剂在复原后具有约260mOsm/kg至290mOsm/kg的克分子渗透压浓度。在某些实施方案中,该冻干制剂在复原后具有约280mOsm/kg的克分子渗透压浓度。在某些实施方案中,该冻干制剂在复原后具有约260mOsm/kg至370mOsm/kg的克分子渗透压浓度。在某些实施方案中,该冻干制剂在复原后具有约360mOsm/kg的克分子渗透压浓度。在某些实施方案中,该冻干制剂在复原后具有约350mOsm/kg至450mOsm/kg的克分子渗透压浓度。在某些实施方案中,该冻干制剂在复原后具有约416mOsm的克分子渗透压浓度。
在某些实施方案中,该冻干制剂用1/2生理盐水(0.45%氯化钠无菌注射用溶液)复原,并且在复原后具有约280mOsm/kg至320mOsm/kg的克分子渗透压浓度。在某些实施方案中,该冻干制剂用1/2生理盐水(0.45%氯化钠无菌注射用溶液)复原,并且在复原后具有3.0至3.2的pH和约280mOsm/kg至320mOsm/kg的克分子渗透压浓度。在某些实施方案中,该冻干制剂用4.5mL1/2生理盐水(0.45%氯化钠无菌注射用溶液)复原,并且在复原后具有3.0至3.2的pH和约280mOsm/kg至320mOsm/kg的克分子渗透压浓度。在一个实施方案中,在输液袋中将所需剂量的复原溶液用生理盐水(0.9%氯化钠无菌注射用溶液)稀释至50mL体积,用于30分钟静脉内施用。
在某些实施方案中,该冻干制剂用生理盐水复原,并且在复原后具有约440mOsm/kg的克分子渗透压浓度。在一个实施方案中,将所需剂量的复原溶液用生理盐水稀释至50mL体积,以获得具有约310mOsm/kg至380mOsm/kg的克分子渗透压浓度的给药溶液。在一个实施方案中,将所需剂量的复原溶液用生理盐水稀释至50mL体积,以获得具有约310mOsm/kg至355mOsm/kg的克分子渗透压浓度的给药溶液。在一个实施方案中,将所需剂量的复原溶液用生理盐水稀释至50mL体积,以获得具有约317mOsm/kg至371mOsm/kg的克分子渗透压浓度的给药溶液。在一个实施方案中,将所需剂量的复原溶液用生理盐水稀释至50mL体积,以获得具有约317mOsm/kg的克分子渗透压浓度的给药溶液。在一个实施方案中,将所需剂量的复原溶液用生理盐水稀释至50mL体积,以获得具有约371mOsm/kg的克分子渗透压浓度的给药溶液。在一个实施方案中,给药溶液的克分子渗透压浓度不超过352mOsm/kg。在一个实施方案中,具有4.8mg剂量的化合物1的给药溶液的克分子渗透压浓度为352mOsm/kg。
在某些实施方案中,本文提供了包含本文提供的制剂的容器。在一个方面,该容器是玻璃小瓶。在一个方面,该容器是20cc玻璃小瓶。
在一个方面,本文提供了在20cc小瓶中的制剂,其包含:量为提供1mg 2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的化合物1,和如本文所述的增量剂。在一个实施方案中,该制剂还包含不超过约5mg作为残留溶剂的甲酸。在一个实施方案中,该制剂还包含不超过约4mg作为残留溶剂的甲酸。在一个实施方案中,该制剂还包含不超过约3mg作为残留溶剂的甲酸。在一个实施方案中,该制剂还包含不超过约2mg作为残留溶剂的甲酸。在一个实施方案中,该制剂还包含不超过约1.5mg作为残留溶剂的甲酸。在一个实施方案中,该制剂还包含不超过约1mg作为残留溶剂的甲酸。在一个实施方案中,该制剂还包含不超过约0.8mg作为残留溶剂的甲酸。在一个实施方案中,该制剂包含约0.4mg至约1.5mg、约0.5mg至约1mg、或约0.5mg至约0.9mg作为残留溶剂的甲酸。在一个实施方案中,该制剂包含约0.4mg、约0.6mg、约0.8mg、约1mg或约1.5mg作为残留溶剂的甲酸。在一个实施方案中,该制剂包含作为残留溶剂的甲酸,其量为约1.0mg/mg化合物1至约1.8mg/mg化合物1、约2.1mg/mg化合物1至约3.8mg/mg化合物1、或约3.9mg/mg化合物1至约4.9mg/mg化合物1。
可以使用用于递送化合物1的标准治疗方法将本文提供的化合物1的制剂施用于有需要的患者,所述方法包括但不限于本文所述的方法。在一个实施方案中,将本文提供的制剂在药学上可接受的溶剂中复原以产生药学上可接受的溶液,其中将该溶液施用(诸如通过静脉内注射)于患者。
在一个方面,本文提供的制剂是冻干的,并且这些冻干制剂适合于在施用前用合适的稀释剂复原为适当的浓度。在一个实施方案中,该冻干制剂在室温下稳定。在一个实施方案中,该冻干制剂在室温下稳定至多约24个月。在一个实施方案中,该冻干制剂在室温下稳定至多约24个月、至多约18个月、至多约12个月、至多约6个月、至多约3个月或至多约1个月。在一个实施方案中,该冻干制剂在40℃/75%RH的加速条件下储存时稳定至多约12个月、至多约6个月或至多约3个月。
本文提供的冻干制剂可以使用任何药学上可接受的稀释剂复原,用于肠胃外施用于患者。此类稀释剂包括但不限于无菌注射用水(SWFI)、5%右旋糖水溶液(D5W),或共溶剂体系。可以使用任何量的稀释剂来复原该冻干制剂,以制备适用于注射的溶液。因此,稀释剂的量必须足以溶解该冻干制剂。在一个实施方案中,使用1至5mL或1至4mL稀释剂复原该冻干制剂,以得到约0.05至0.3mg/mL或约0.15至0.25mg/mL的化合物1的最终浓度。在某些实施方案中,该复原溶液中化合物1的最终浓度为约0.25mg/mL。在某些实施方案中,该复原溶液中化合物1的最终浓度为约0.20mg/mL。在某些实施方案中,该复原稀释剂的体积在3ml与5ml之间变化,以得到0.15至0.3mg/mL的最终浓度。在某些实施方案中,根据所需的剂量,可以使用多个小瓶进行复原。
可以储存冻干制剂的复原溶液并在至多约24小时、约12小时或约8小时内使用。在一个实施方案中,该复原水性溶液在复原后在室温下稳定约1至24小时、2至20小时、2至15小时、2至10小时。在一个实施方案中,该复原水性溶液在复原后在室温下稳定至多约20、15、12、10、8、6、4或2小时。在一些实施方案中,该溶液在制备后的8小时内使用。在一些实施方案中,该溶液在制备后的5小时内使用。在一些实施方案中,该溶液在制备后的1小时内使用。
用于制备制剂的工艺
本文提供的制剂可以通过本领域已知的和如本文所述的任何方法制备,但是所有方法都包括使活性成分与药学上可接受的赋形剂缔合的步骤,所述赋形剂构成一种或多种必要成分(诸如增量剂和/或缓冲剂)。
在一个方面,本文提供的制剂通过以下方式制备:将化合物1、增量剂和柠檬酸盐缓冲剂溶解在水和二甲基亚砜(DMSO)中以获得溶液,然后任选地冻干该溶液。图37和图38提供了流程图,其展示了制备本文提供的制剂的示例性工艺。
在一个实施方案中,用于制备该制剂的工艺包括:将HPBCD溶解在柠檬酸盐缓冲剂中以获得缓冲溶液,将化合物1溶解在DMSO中以获得预混物,将该预混物添加至缓冲溶液以获得溶液;然后任选地冻干该溶液以产生冻干制剂。
在一个实施方案中,该工艺包括:将 HPB溶解在20mM、pH 4–4.5的柠檬酸盐缓冲剂中以获得缓冲溶液,将化合物1溶解在DMSO中以获得活性预混物,将该预混物添加至缓冲溶液以获得混合物,将水添加至该混合物以获得本体溶液,通过一个或多个0.45μm过滤器和0.22μm过滤器过滤该本体溶液以获得经过滤的溶液,将该经过滤的溶液填充到小瓶中,然后冻干该溶液。在一个实施方案中,通过一个0.45μm过滤器和两个0.22μm过滤器过滤所述溶液。在一个实施方案中,该工艺包括:将
HPB溶解在20mM、pH 4–4.5的柠檬酸盐缓冲剂中以获得缓冲溶液,将化合物1溶解在DMSO中以获得活性预混物,将该预混物添加至缓冲溶液以获得混合物,将水添加至该混合物以获得本体溶液,通过一个或多个0.45μm过滤器和0.22μm过滤器过滤该本体溶液以获得经过滤的溶液,将该经过滤的溶液填充到小瓶中,然后冻干该溶液。在一个实施方案中,通过一个0.45μm过滤器和两个0.22μm过滤器过滤所述溶液。在一个实施方案中,该工艺包括:将 HPB溶解在20mM、pH 4.3的柠檬酸盐缓冲剂中以获得缓冲溶液,将化合物1溶解在DMSO中以获得活性预混物,将该预混物添加至缓冲溶液以获得混合物,将水添加至该混合物以获得本体溶液,通过一个0.45μm过滤器和两个0.22μm过滤器过滤该本体溶液以获得经过滤的溶液,将该经过滤的溶液填充到20cc玻璃小瓶中,然后任选地冻干该溶液。在一个实施方案中,冻干之后在氮气下将小瓶密封。
HPB溶解在20mM、pH 4.3的柠檬酸盐缓冲剂中以获得缓冲溶液,将化合物1溶解在DMSO中以获得活性预混物,将该预混物添加至缓冲溶液以获得混合物,将水添加至该混合物以获得本体溶液,通过一个0.45μm过滤器和两个0.22μm过滤器过滤该本体溶液以获得经过滤的溶液,将该经过滤的溶液填充到20cc玻璃小瓶中,然后任选地冻干该溶液。在一个实施方案中,冻干之后在氮气下将小瓶密封。
在一个方面,本文提供的制剂通过以下方式制备:将化合物1溶解在甲酸中以获得预混物,将HPBCD溶解在水中以获得溶液,将该预混物添加至溶液以获得药物溶液;然后任选地冻干该药物溶液以产生冻干制剂。
在一个方面,本文提供的制剂通过以下方式制备:将化合物1溶解在甲酸中以获得活性预混物,将 HPB溶解在水中以获得Kleptose溶液,将该预混物添加至Kleptose溶液以获得混合物,将水添加至该混合物以获得本体溶液,通过一个或多个0.45μm过滤器和0.22μm过滤器过滤该本体溶液以获得经过滤的溶液,将该经过滤的溶液填充到小瓶中,然后冻干该溶液。在一个实施方案中,通过一个0.45μm过滤器和两个0.22μm过滤器过滤所述溶液。在一个实施方案中,该工艺包括将化合物1溶解在甲酸中以获得活性预混物,将
HPB溶解在水中以获得Kleptose溶液,将该预混物添加至Kleptose溶液以获得混合物,将水添加至该混合物以获得本体溶液,通过一个或多个0.45μm过滤器和0.22μm过滤器过滤该本体溶液以获得经过滤的溶液,将该经过滤的溶液填充到小瓶中,然后冻干该溶液。在一个实施方案中,通过一个0.45μm过滤器和两个0.22μm过滤器过滤所述溶液。在一个实施方案中,该工艺包括将化合物1溶解在甲酸中以获得活性预混物,将 HPB溶解在水中以获得Kleptose溶液,将该预混物添加至Kleptose溶液以获得混合物,将水添加至该混合物以获得本体溶液,通过一个0.45μm过滤器和两个0.22μm过滤器过滤该本体溶液以获得经过滤的溶液,将该经过滤的溶液填充到20cc小瓶中,然后冻干该溶液。在一个实施方案中,冻干之后在氮气下将小瓶密封。
HPB溶解在水中以获得Kleptose溶液,将该预混物添加至Kleptose溶液以获得混合物,将水添加至该混合物以获得本体溶液,通过一个0.45μm过滤器和两个0.22μm过滤器过滤该本体溶液以获得经过滤的溶液,将该经过滤的溶液填充到20cc小瓶中,然后冻干该溶液。在一个实施方案中,冻干之后在氮气下将小瓶密封。
在一个方面,冻干工艺包含三个阶段:冷冻、初次干燥和二次干燥。通过冷冻阶段中的完全固化、初次干燥中冰和溶剂的升华,以及二次干燥中残留水分和溶剂的解吸,将液体制剂转变为冻干粉末形式。控制初次干燥和二次干燥中的搁板温度和腔室压力,以获得所需质量的成品药物产品。在该工艺的一个方面,通过目视检查来表征粉饼的外观和结构。
药盒
还提供了包含本文提供的药物组合物或剂型的药物包装或药盒。示例性药盒包括由管理药物产品的制造、使用或销售的政府机构规定的形式的通告,该通告反映获得了该机构对制造、使用或销售用于向人施用的批准。
使用方法和在此类方法中使用的化合物1
如本文提供的化合物1可以用于本文提供的所有方法。在一个实施方案中,本文提供了治疗、预防、管理和/或改善癌症(包括实体肿瘤和血液学癌症)或者癌症的一种或多种症状或起因的方法,方式为将化合物1与选自下列的一种或多种第二剂组合施用:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在治疗、预防、管理和/或改善癌症(包括实体肿瘤和血液学癌症)或者癌症的一种或多种症状或起因的此类方法中使用的化合物1,其中化合物1与选自下列的一种或多种第二剂组合施用:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了治疗和预防癌症的方法,其包括向患者施用本文提供的化合物1的制剂。本文提供了在治疗和预防癌症的这种方法中使用的化合物1的制剂。
在另一个实施方案中,本文提供了管理癌症的方法,其包括向患者施用本文提供的化合物1的制剂。本文提供了在管理癌症的这种方法中使用的化合物1。
在一个实施方案中,本文提供的方法包括将化合物1的制剂与选自下列的一种或多种第二剂组合施用:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
本文还提供了治疗先前已针对癌症进行过治疗但对癌症疗法无响应的患者以及那些先前未治疗过的患者的方法。尽管某些疾病或疾患在某些年龄组中更常见,但本文还涵盖治疗患者而不考虑患者年龄的方法。还涵盖治疗已经进行手术以试图治疗所讨论的疾病或疾患的患者以及那些尚未进行手术的患者的方法。因为患有癌症的患者具有各种各样的临床表现和变化的临床结果,所以给予患者的治疗可能有所不同,具体取决于他的/她的预后。熟练的临床医生将能够在没有过度实验的情况下容易地确定具体的第二剂、手术类型以及可以有效地用于治疗患有癌症的个体患者的基于非药物的标准疗法的类型。
在一个实施方案中,本文提供了用于改善癌症患者的美国东部肿瘤协作组行为状态(ECOG)的方法,包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于改善癌症患者的美国东部肿瘤协作组行为状态(ECOG)的方法中使用的化合物1,所述方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于改善癌症患者的美国东部肿瘤协作组行为状态(ECOG)的方法,包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于改善癌症患者的美国东部肿瘤协作组行为状态(ECOG)的方法中使用的化合物1的制剂,所述方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于改善癌症患者的美国东部肿瘤协作组行为状态(ECOG)的方法,包括将有效量的化合物1的制剂施用于所述患者。本文提供了用于改善癌症患者的美国东部肿瘤协作组行为状态(ECOG)的化合物1的制剂。
在一个实施方案中,本文提供了用于在癌症患者中抑制疾病进展、抑制肿瘤生长、减少原发性肿瘤、减轻肿瘤相关症状、抑制肿瘤分泌因子、延迟原发性或继发性肿瘤的出现、减缓原发性或继发性肿瘤的发展、减少原发性或继发性肿瘤的发生、减缓疾病的继发性效应或降低其严重程度、使肿瘤生长停滞和肿瘤消退、增加至进展的时间、增加无进展存活期、增加总体存活期或者它们中的一项或多项的方法,包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于癌症患者的所有此类方法中使用的化合物1,所述方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在癌症患者中抑制疾病进展、抑制肿瘤生长、减少原发性肿瘤、减轻肿瘤相关症状、抑制肿瘤分泌因子、延迟原发性或继发性肿瘤的出现、减缓原发性或继发性肿瘤的发展、减少原发性或继发性肿瘤的发生、减缓疾病的继发性效应或降低其严重程度、使肿瘤生长停滞和肿瘤消退、增加至进展的时间、增加无进展存活期、增加总体存活期的方法,或者它们中的一种或多种,包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了用于癌症患者中的所有此类方法、或者它们中的一种或多种中的化合物1,所述方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在癌症患者中抑制疾病进展、抑制肿瘤生长、减少原发性肿瘤、减轻肿瘤相关症状、抑制肿瘤分泌因子、延迟原发性或继发性肿瘤的出现、减缓原发性或继发性肿瘤的发展、减少原发性或继发性肿瘤的发生、减缓疾病的继发性效应或降低其严重程度、使肿瘤生长停滞和肿瘤消退、增加至进展的时间、增加无进展存活期、增加总体存活期,或者它们中的一种或多种的方法,包括将有效量的化合物1的制剂施用于所述患者。本文提供了用于癌症患者的所有此类方法、或者它们中的一种或多种中的化合物1,所述方法包括将有效量的化合物1的制剂施用于所述患者。
在某些实施方案中,癌症是实体肿瘤或血液学癌症。在某些实施方案中,癌症是白介素-3(IL-3)非依赖性的。在某些实施方案中,癌症是实体肿瘤。在某些实施方案中,实体肿瘤是转移性的。在某些实施方案中,实体肿瘤具有抗药性。
在某些实施方案中,癌症是指皮肤组织、器官、血液和脉管的疾病。在某些实施方案中,癌症是实体肿瘤,包括但不限于膀胱癌、骨癌、血癌、脑癌、乳腺癌、子宫颈癌、胸癌、结肠癌、子宫内膜癌、食管癌、眼癌、头癌、肾癌、肝癌、淋巴结癌、肺癌、口腔癌、颈癌、卵巢癌、胰腺癌、前列腺癌、直肠癌、胃癌、睾丸癌、咽喉癌和子宫癌。具体癌症包括但不限于晚期恶性肿瘤、淀粉样变性病、神经母细胞瘤、脑膜瘤、血管外皮细胞瘤、多发性脑转移瘤、多形性成胶质细胞瘤、成胶质细胞瘤、脑干胶质瘤、不良预后恶性脑肿瘤、恶性胶质瘤、复发性恶性胶质瘤、间变性星形细胞瘤、间变性少突神经胶质瘤、神经内分泌肿瘤、直肠腺癌、结肠直肠癌(包括3期和4期)、不可切除的结肠直肠癌、转移性肝细胞癌、卡波西氏肉瘤(Kaposi’ssarcoma)、核型急性骨髓母细胞性白血病、霍奇金氏淋巴瘤、非霍奇金氏淋巴瘤、皮肤T细胞淋巴瘤、皮肤B细胞淋巴瘤、弥漫性大B细胞淋巴瘤、低级滤泡性淋巴瘤、恶性黑素瘤、恶性间皮瘤、恶性胸腔积液间皮瘤综合征、腹膜癌、乳头状浆液性癌、妇科肉瘤、软组织肉瘤、硬皮病、皮肤血管炎、朗格汉斯细胞组织细胞增多症(Langerhans cell histiocytosis)、平滑肌肉瘤、进行性骨化性纤维发育不良、激素难治性前列腺癌、切除性高风险软组织肉瘤、不可切除的肝细胞癌、瓦尔登斯特伦氏巨球蛋白血症(Waldenstrom’s macroglobulinemia)、冒烟型骨髓瘤、无痛性骨髓瘤、输卵管癌、雄激素非依赖性前列腺癌、雄激素依赖性IV期非转移性前列腺癌、激素不敏感前列腺癌、化学疗法不敏感前列腺癌、上皮癌,包括甲状腺乳头状癌、甲状腺滤泡癌、甲状腺髓样癌和平滑肌瘤。
在某些实施方案中,癌症是实体肿瘤,包括但不限于皮肤癌、中枢神经系统癌、软组织癌、唾液腺癌、卵巢癌、肾癌、肺癌、骨癌、胃癌、子宫内膜癌、胰腺癌、泌尿道癌、甲状腺癌、上呼吸消化道癌、乳腺癌、大肠癌、食道癌、前列腺癌、肝癌、自主神经节癌和恶性胸膜间皮瘤。
在某些实施方案中,实体肿瘤是肝细胞癌、前列腺癌、卵巢癌或成胶质细胞瘤。
在某些实施方案中,实体肿瘤是乳腺癌、肾癌、胰腺癌、胃肠道癌、肺癌、神经内分泌肿瘤(NET)或肾细胞癌(RCC)。
在某些实施方案中,癌症是血液学癌症。在某些实施方案中,血液学癌症是转移性的。在某些实施方案中,血液学癌症对至少一种抗癌疗法具有抗药性。在某些实施方案中,血液学癌症是复发性的或至少一种抗癌疗法难治性的。
在一个实施方案中,血液学癌症是多发性骨髓瘤(MM)。在一个实施方案中,血液学癌症是复发性/难治性的(R/R)MM。在一个实施方案中,患有R/R MM的患者肾功能受损。
在一个实施方案中,本文提供了用于在MM患者中实现严格完全缓解(sCR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MM患者中实现严格完全缓解(sCR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MM患者中实现严格完全缓解(sCR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MM患者中实现严格完全缓解(sCR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MM患者中实现严格完全缓解(sCR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在MM患者中实现严格完全缓解(sCR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂施用于所述患者。
在一个实施方案中,本文提供了用于在MM患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MM患者中实现完全缓解(CR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MM患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MM患者中实现完全缓解(CR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MM患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在MM患者中实现完全缓解(CR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂施用于所述患者。
在一个实施方案中,本文提供了用于在MM患者中实现极好的部分响应(VGPR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MM患者中实现极好的部分响应(VGPR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MM患者中实现极好的部分响应(VGPR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MM患者中实现极好的部分响应(VGPR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MM患者中实现极好的部分响应(VGPR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在MM患者中实现极好的部分响应(VGPR)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在MM患者中实现部分响应(PR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MM患者中实现部分响应(PR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MM患者中实现部分响应(PR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MM患者中实现部分响应(PR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MM患者中实现部分响应(PR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在MM患者中实现部分响应的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在MM患者中实现疾病稳定(SD)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MM患者中实现疾病稳定(SD)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MM患者中实现疾病稳定(SD)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MM患者中实现疾病稳定(SD)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MM患者中实现疾病稳定(SD)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在MM患者中实现疾病稳定的方法中使用的化合物1的制剂。
在一个实施方案中,血液学癌症是急性髓源性白血病(AML)。在一个实施方案中,血液学癌症是急性淋巴细胞性白血病(ALL)。在一个实施方案中,血液学癌症是成人T细胞白血病。在一个实施方案中,血液学癌症是慢性淋巴细胞性白血病(CLL)。在一个实施方案中,血液学癌症是毛细胞白血病。在一个实施方案中,血液学癌症是骨髓增生异常。在一个实施方案中,血液学癌症是骨髓增殖性障碍或骨髓增殖性赘生物(MPN)。在一个实施方案中,血液学癌症是慢性髓源性白血病(CML)。在一个实施方案中,血液学癌症是骨髓增生异常综合征(MDS)。在一个实施方案中,血液学癌症是人嗜淋巴细胞病毒-1型(HTLV-1)白血病。在一个实施方案中,血液学癌症是肥大细胞增生症。在一个实施方案中,血液学癌症是B细胞急性淋巴母细胞性白血病。在一个实施方案中,血液学癌症是CLL。
在一个实施方案中,本文提供了治疗、预防、管理和/或改善对象中的选自下列的癌症的方法:弥散性大B细胞淋巴瘤(DLBCL)、B细胞免疫母细胞性淋巴瘤、小无核裂细胞淋巴瘤、人嗜淋巴细胞病毒-1型(HTLV-1)白血病/淋巴瘤、成人T细胞淋巴瘤、套细胞淋巴瘤(MCL)、霍奇金淋巴瘤(HL)、非霍奇金淋巴瘤(NHL)、AIDS相关淋巴瘤、滤泡性淋巴瘤、小淋巴细胞性淋巴瘤、富含T细胞/组织细胞的大B细胞淋巴瘤、转化型淋巴瘤、原发性纵隔(胸腺)大B细胞淋巴瘤、脾边缘区淋巴瘤、里希特氏转化、结节边缘区淋巴瘤和ALK阳性大B细胞淋巴瘤,所述方法包括将能有效地治疗、预防和/或管理癌症的一定量的本文提供的化合物1的制剂施用于所述对象的步骤。因此,本文提供了在治疗、预防、管理和/或改善对象中的癌症的所有所述方法中使用的化合物1的制剂,其中该癌症选自弥散性大B细胞淋巴瘤(DLBCL)、B细胞免疫母细胞性淋巴瘤、小无核裂细胞淋巴瘤、人嗜淋巴细胞病毒-1型(HTLV-1)白血病/淋巴瘤、成人T细胞淋巴瘤、套细胞淋巴瘤(MCL)、霍奇金淋巴瘤(HL)、非霍奇金淋巴瘤(NHL)、AIDS相关淋巴瘤、滤泡性淋巴瘤、小淋巴细胞性淋巴瘤、富含T细胞/组织细胞的大B细胞淋巴瘤、转化型淋巴瘤、原发性纵隔(胸腺)大B细胞淋巴瘤、脾边缘区淋巴瘤、里希特氏转化、结节边缘区淋巴瘤和ALK阳性大B细胞淋巴瘤,在一些实施方案中,所述方法包括将本文提供的化合物1的制剂与第二活性剂以能有效地治疗、预防和/或管理癌症的量组合施用于所述对象的步骤。在一个实施方案中,血液学癌症是HL。在一个实施方案中,血液学癌症是NHL。在一个实施方案中,血液学癌症是惰性淋巴瘤,包括例如DLBCL、滤泡性淋巴瘤和边缘区淋巴瘤。
在一个实施方案中,本文提供了用于在NHL患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在NHL患者中实现完全缓解(CR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在NHL患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在NHL患者中实现完全缓解(CR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在NHL患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在NHL患者中实现完全缓解(CR)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在NHL患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在NHL患者中实现部分缓解(PR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在NHL患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在NHL患者中实现部分缓解(PR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在NHL患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在NHL患者中实现部分缓解(PR)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在NHL患者中实现疾病稳定(SD)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在NHL患者中实现疾病稳定(SD)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在NHL患者中实现疾病稳定(SD)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在NHL患者中实现疾病稳定(SD)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在NHL患者中实现疾病稳定(SD)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在NHL患者中实现疾病稳定(SD)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了通过将治疗活性量的化合物1施用于对象来治疗、预防、管理和/或改善白血病的方法。因此,本文提供了在治疗、预防、管理和/或改善白血病的此类方法中使用的化合物1。在一个实施方案中,白血病是急性骨髓性白血病(AML)。在一个实施方案中,AML是复发性或难治性AML。在一个实施方案中,AML是新诊断的AML。在另一个实施方案中,AML具有FAB分类M0/1。在另一个实施方案中,AML具有FAB分类M2。在另一个实施方案中,AML具有FAB分类M3。在另一个实施方案中,AML具有FAB分类M4。在另一个实施方案中,AML具有FAB分类M5。在一个实施方案中,AML是具有至少一种复现性遗传异常的AML(例如,具有染色体8和染色体21之间的易位的AML;具有染色体16中的易位或倒位的AML;具有染色体9和染色体11之间的易位的AML;具有染色体15和染色体17之间的易位的APL(M3);具有染色体6和染色体9之间的易位的AML;具有染色体3中的易位或倒位的AML);具有染色体1和染色体22之间的易位的AML(巨核母细胞性);具有骨髓增生异常相关变化的AML;涉及前期化学疗法或辐射的AML(例如,烷化剂相关AML;或拓扑异构酶II抑制剂相关AML);不可另外分类的AML(例如,不属于上述类别的AML,即最小分化型AML(M0);最小成熟型AML(M1);成熟型AML(M2);急性髓单核细胞性白血病(M4);急性单核细胞性白血病(M5);急性红细胞系白血病(M6);急性巨核母细胞性白血病(M7);急性嗜碱粒细胞性白血病;或急性全骨髓增生症伴纤维化);骨髓性肉瘤(也称为粒细胞性肉瘤、绿色瘤或髓外成骨髓细胞瘤);或未分化型和双表型急性白血病(也称为混合表型急性白血病)。在一个实施方案中,AML的特征在于IDH2的突变等位基因。在该实施方案的一个方面,IDH2的突变等位基因具有R140X突变。在该实施方案的另一个方面,R140X突变是R140Q突变。在该实施方案的另一个方面,R140X突变是R140W突变。在该实施方案的另一个方面,R140X突变是R140L突变。在该实施方案的另一个方面,IDH2的突变等位基因具有R172X突变。在该实施方案的另一个方面,R172X突变是R172K突变。在该实施方案的另一个方面,R172X突变是R172G突变。
在一个实施方案中,AML是同种异体HSCT之后的复发性AML。在一个实施方案中,AML是第二次或后来复发的AML。在一个实施方案中,AML是初始诱导或再诱导治疗难治性的。在某些实施方案中,AML是至少一种诱导/再诱导或巩固疗法难治性的。在一个实施方案中,AML是低甲基化剂(HMA)难治性的或在施用低甲基化剂(HMA)之后复发的。如本文所用,HMA失败被定义为在最少6个周期后出现原发性进展或缺乏临床益处,或由于毒性而不能耐受HMA。在一个实施方案中,AML在初始治疗后的1年内复发(排除具有有利风险状态的AML)。
在某些实施方案中,治疗、预防和/或管理对象中的急性骨髓性白血病的方法包括将能有效地治疗、预防和/或管理急性骨髓性白血病的一定量的本文提供的化合物1的制剂施用于所述对象的步骤。在一些实施方案中,所述方法包括将本文提供的化合物1的制剂与第二活性剂以能有效地治疗、预防和/或管理急性骨髓性白血病的量组合施用于所述对象的步骤。
在一个实施方案中,本文提供了用于在AML患者中实现形态学无白血病状态的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现形态学无白血病状态的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现形态学无白血病状态的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现形态学无白血病状态的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现形态学无白血病状态的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在AML患者中实现形态学无白血病状态的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在AML患者中实现形态学完全缓解的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现形态学完全缓解的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现形态学完全缓解的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现形态学完全缓解的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现形态学完全缓解的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在AML患者中实现形态学完全缓解的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在AML患者中实现细胞遗传学完全缓解(CRc)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现细胞遗传学完全缓解(CRc)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现细胞遗传学完全缓解(CRc)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现细胞遗传学完全缓解(CRc)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在AML患者中实现细胞遗传学完全缓解(CRc)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在AML患者中实现分子完全缓解(CRm)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现分子完全缓解(CRm)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现分子完全缓解(CRm)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现分子完全缓解(CRm)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现分子完全缓解(CRm)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在AML患者中实现分子完全缓解(CRm)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在AML患者中实现形态学完全缓解伴不完全血液恢复(CRi)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现形态学完全缓解伴不完全血液恢复(CRi)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现形态学完全缓解伴不完全血液恢复(CRi)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现形态学完全缓解伴不完全血液恢复(CRi)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现形态学完全缓解伴不完全血液恢复(CRi)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在AML患者中实现形态学完全缓解伴不完全血液恢复(CRi)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在AML患者中实现部分缓解的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现部分缓解的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现部分缓解(PR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在AML患者中实现部分缓解(PR)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在AML患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现完全缓解(CR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在AML患者中实现完全缓解(CR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在AML患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在AML患者中实现完全缓解(CR)的方法中使用的化合物1的制剂。
在一些实施方案中,本文提供的方法涵盖治疗、预防和/或管理对象中的急性淋巴细胞性白血病(ALL)。在一些实施方案中,ALL包括起源于骨髓的母细胞(B细胞)、胸腺(T细胞)和淋巴结的白血病。可以根据法-美-英(FAB)形态学分类方案将ALL分类为:L1–外观成熟的淋巴母细胞(T细胞或前B细胞)、L2–未成熟和多形性(各种形状的)淋巴母细胞(T细胞或前B细胞),和L3–淋巴母细胞(B细胞;伯基特氏细胞(Burkitt’s cell))。在一个实施方案中,ALL起源于骨髓的母细胞(B细胞)。在一个实施方案中,ALL起源于胸腺(T细胞)。在一个实施方案中,ALL起源于淋巴结。在一个实施方案中,ALL是L1型,其特征在于外观成熟的淋巴母细胞(T细胞或前B细胞)。在一个实施方案中,ALL是L2型,其特征在于未成熟和多形性(各种形状的)淋巴母细胞(T细胞或前B细胞)。在一个实施方案中,ALL是L3型,其特征在于淋巴母细胞(B细胞;伯基特氏细胞)。在某些实施方案中,ALL是T细胞白血病。在一个实施方案中,T细胞白血病是外周T细胞白血病。在另一个实施方案中,T细胞白血病是T细胞淋巴母细胞性白血病。在另一个实施方案中,T细胞白血病是皮肤T细胞白血病。在另一个实施方案中,T细胞白血病是成人T细胞白血病。在某些实施方案中,治疗、预防和/或管理对象中的ALL的方法包括将能有效地治疗、预防和/或管理ALL的一定量的本文提供的化合物1的制剂施用于所述对象的步骤。在一些实施方案中,所述方法包括将本文提供的化合物1的制剂与第二活性剂以能有效地治疗、预防和/或管理ALL的量组合施用于所述对象的步骤。
在一些实施方案中,本文提供的方法涵盖治疗、预防和/或管理对象中的慢性髓源性白血病(CML)。所述方法包括将能有效地治疗、预防和/或管理CML的一定量的本文提供的化合物1的制剂施用于所述对象的步骤。在一些实施方案中,所述方法包括将本文提供的化合物1的制剂与第二活性剂以能有效地治疗、预防和/或管理CML的量组合施用于所述对象的步骤。
在一些实施方案中,本文提供的方法涵盖治疗、预防和/或管理对象中的慢性淋巴细胞性白血病(CLL)。所述方法包括将能有效地治疗、预防和/或管理慢性淋巴细胞性白血病的一定量的本文提供的化合物1的制剂施用于所述对象的步骤。在一些实施方案中,所述方法包括将本文提供的化合物1的制剂与第二活性剂以能有效地治疗、预防和/或管理CLL的量组合施用于所述对象的步骤。
在一个实施方案中,本文提供了用于在CLL患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在CLL患者中实现完全缓解(CR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在CLL患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在CLL患者中实现完全缓解(CR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在CLL患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在CLL患者中实现完全缓解(CR)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在CLL患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在CLL患者中实现部分缓解(PR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在CLL患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在CLL患者中实现部分缓解(PR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在CLL患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在CLL患者中实现部分缓解(PR)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在CLL患者中实现疾病稳定(SD)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在CLL患者中实现疾病稳定(SD)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在CLL患者中实现疾病稳定(SD)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在CLL患者中实现疾病稳定(SD)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在CLL患者中实现疾病稳定(SD)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在CLL患者中实现疾病稳定(SD)的方法中使用的化合物1。
在一个实施方案中,本文提供了通过将治疗活性量的化合物1施用于对象来治疗、预防、管理和/或改善骨髓增生异常综合征(MDS)的方法。在一个实施方案中,本文提供了治疗MDS的方法。因此,本文提供了在治疗、预防、管理和/或改善MDS的此类方法中使用的化合物1。在一个实施方案中,MDS是复发性、耐药性或难治性MDS。在一个实施方案中,MDS是难治性贫血(RA);RA伴环形铁粒幼细胞(RARS);RA伴母细胞过量(RAEB);难治性血细胞减少伴多系增生异常(RCMD)、难治性血细胞减少伴单系增生异常(RCUD);不可分类型骨髓增生异常综合征(MDS-U)、孤立del(5q)染色体异常相关联的骨髓增生异常综合征、疗法相关的骨髓赘生物或慢性髓单核细胞性白血病(CMML)。在一些实施方案中,MDS是极低风险、低风险、中等风险、高风险或极高风险MDS。在一个实施方案中,MDS是极低风险的。在另一个实施方案中,MDS是低风险的。在另一个实施方案中,MDS是中等风险的。在另一个实施方案中,MDS是高风险的。在另一个实施方案中,MDS是极高风险MDS。在一个实施方案中,MDS是复发性或难治性高风险MDS。在一个实施方案中,MDS在经修订的国际预后评分系统(IPSS-R)中的评分>3.5分(例如,IPSS-R中等风险(与超过10%的骨髓母细胞或者差或极差的IPSS-R细胞遗传学风险组合)、IPSS-R高风险和IPSS-R极高风险]。在一个实施方案中,MDS不适合其它已建立疗法(例如,移植或低甲基化剂)。在一些实施方案中,MDS是原发性或新发MDS。在其它实施方案中,MDS是继发性MDS。在一个实施方案中,MDS是初始诱导或再诱导治疗难治性的。在某些实施方案中,MDS是至少一种诱导/再诱导或巩固疗法难治性的。在某些实施方案中,治疗、预防和/或管理对象中的MDS的方法包括将能有效地治疗、预防和/或管理MDS的一定量的本文提供的化合物1的制剂施用于所述对象的步骤。在一些实施方案中,所述方法包括将本文提供的化合物1的制剂与第二活性剂以能有效地治疗、预防和/或管理MDS的量组合施用于所述对象的步骤。
在一个实施方案中,本文提供了用于在MDS患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MDS患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MDS患者中实现完全缓解(CR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MDS患者中实现完全缓解(CR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在MDS患者中实现完全缓解(CR)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在MDS患者中实现骨髓完全缓解(mCR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MDS患者中实现骨髓完全缓解(mCR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MDS患者中实现骨髓完全缓解(mCR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MDS患者中实现骨髓完全缓解(mCR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MDS患者中实现骨髓完全缓解(mCR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在MDS患者中实现骨髓完全缓解(mCR)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在MDS患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MDS患者中实现部分缓解(PR)的方法中使用的化合物1,其中该方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MDS患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MDS患者中实现部分缓解(PR)的方法中使用的化合物1的制剂,其中该方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MDS患者中实现部分缓解(PR)的方法,其中该方法包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在MDS患者中实现部分缓解(PR)的方法中使用的化合物1的制剂。
在一个实施方案中,本文提供了用于在MDS患者中增加总体存活期、增加无复发存活期、增加无进展存活期、增加无事件存活期、增加缓解持续时间、增加响应持续时间或增加至转化为AML的时间的方法,包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MDS患者中增加总体存活期、增加无复发存活期、增加无进展存活期、增加无事件存活期、增加缓解持续时间、增加响应持续时间或增加至转化为AML的时间的方法中使用的化合物1,所述方法包括将有效量的化合物1与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MDS患者中增加总体存活期、增加无复发存活期、增加无进展存活期、增加无事件存活期、增加缓解持续时间、增加响应持续时间或增加至转化为AML的时间的方法,包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。本文提供了在用于在MDS患者中增加总体存活期、增加无复发存活期、增加无进展存活期、增加无事件存活期、增加缓解持续时间、增加响应持续时间或增加至转化为AML的时间的方法中使用的化合物1的制剂,所述方法包括将有效量的化合物1的制剂与选自下列的一种或多种第二剂组合施用于所述患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个实施方案中,本文提供了用于在MDS患者中增加总体存活期、增加无复发存活期、增加无进展存活期、增加无事件存活期、增加缓解持续时间、增加响应持续时间或增加至转化为AML的时间的方法,包括将有效量的化合物1的制剂施用于所述患者。本文提供了在用于在MDS患者中增加总体存活期、增加无复发存活期、增加无进展存活期、增加无事件存活期、增加缓解持续时间、增加响应持续时间或增加至转化为AML的时间的方法中使用的化合物1的制剂。
在一些实施方案中,本文提供的方法涵盖治疗、预防和/或管理骨髓增殖性赘生物。在一个实施方案中,骨髓增殖性赘生物是真性红细胞增多、原发性或自发性血小板增多、骨髓纤维化、慢性髓源性白血病、慢性中性粒细胞性白血病、幼年型髓单核细胞性白血病、慢性嗜酸性粒细胞性白血病或高嗜酸性粒细胞综合征。在一个实施方案中,骨髓增殖性赘生物是真性红细胞增多、原发性或自发性血小板增多、原发性或特发性骨髓纤维化、继发性骨髓纤维化、真性红细胞增多后骨髓纤维化、自发性血小板增多后骨髓纤维化、慢性髓源性白血病、慢性中性粒细胞性白血病、幼年型髓单核细胞性白血病、慢性嗜酸性粒细胞性白血病或高嗜酸性粒细胞综合征。在一个实施方案中,骨髓增殖性赘生物是真性红细胞增多。在一个实施方案中,骨髓增殖性赘生物是原发性或自发性血小板增多。在一个实施方案中,骨髓增殖性赘生物是骨髓纤维化。在一个实施方案中,骨髓增殖性赘生物是原发性或特发性骨髓纤维化。在一个实施方案中,骨髓增殖性赘生物是继发性骨髓纤维化。在一个实施方案中,骨髓增殖性赘生物是真性红细胞增多后骨髓纤维化。在一个实施方案中,骨髓增殖性赘生物是自发性血小板增多后骨髓纤维化。在一个实施方案中,骨髓增殖性赘生物是慢性髓源性白血病。在一个实施方案中,骨髓增殖性赘生物是慢性中性粒细胞性白血病。在一个实施方案中,骨髓增殖性赘生物是幼年型髓单核细胞性白血病。在一个实施方案中,骨髓增殖性赘生物是慢性嗜酸性粒细胞性白血病。在一个实施方案中,骨髓增殖性赘生物是高嗜酸性粒细胞综合征。在某些实施方案中,骨髓增殖性赘生物是白介素-3(IL-3)非依赖性的。在一些实施方案中,骨髓增殖性赘生物的特征在于JAK突变,例如V617突变,诸如V617F。
在某些实施方案中,治疗、预防和/或管理对象中的骨髓增殖性赘生物的方法包括将能有效地治疗、预防和/或管理骨髓增殖性赘生物的一定量的本文提供的化合物1的制剂施用于所述对象的步骤。在一些实施方案中,所述方法包括将本文提供的化合物1的制剂与第二活性剂以能有效地治疗、预防和/或管理骨髓增殖性赘生物的量组合施用于所述对象的步骤。
在一个实施方案中,本文提供的治疗、预防和/或管理癌症的方法包括静脉内施用化合物1的制剂。在一个实施方案中,将化合物1的制剂溶解在水中,以形成在本文提供的治疗、预防和/或管理癌症的方法中用于静脉内施用的水性溶液。
在一些实施方案中,所述方法包括将本文提供的化合物1的制剂与第二活性剂以能有效地治疗、预防和/或管理癌症的量组合施用于所述对象的步骤。
在某些实施方案中,本文提供了治疗、预防和/或管理肾功能受损的患者中的癌症的方法。在某些实施方案中,本文提供了为由于但不限于疾病、衰老或其它患者因素而导致肾功能受损的患者提供适当的剂量调整的方法。
在一个实施方案中,本文提供了降低对象中的GSPT1水平的方法,所述方法包括将化合物1与如本文所述的第二剂组合施用于所述对象。因此,本文提供了在降低对象中的GSPT1水平的方法中使用的化合物1,所述方法包括将化合物1与如本文所述的第二剂组合施用于所述对象。在一些实施方案中,本文提供了监测用化合物1与第二剂组合治疗对象中的癌症的功效的方法,包括:(a)将化合物1和第二剂施用于对象;(b)从对象获得样品;(c)确定样品中的GSPT1水平;(d)将样品中的GSPT1水平与参考样品中的GSPT1水平进行比较,其中样品中的GSPT1水平相比于参考样品降低指示用化合物1和第二剂治疗对象中的癌症的功效。因此,本文提供了在监测用化合物1与第二剂组合治疗对象中的癌症的功效的这种方法中使用的化合物1。在又一个方面,本文提供了预测患有或怀疑患有癌症的对象对用化合物1和第二剂治疗的响应性的方法,该方法包括(a)将化合物1和第二剂施用于对象;(b)从对象获得样品;(c)确定样品中的GSPT1水平;(d)如果样品中的GSPT1水平与参考样品中的GSPT1水平相比降低,则诊断对象可能对用化合物1和第二剂治疗癌症有响应。因此,本文提供了在预测患有或怀疑患有癌症的对象对用化合物1和第二剂治疗的响应性的方法中使用的化合物1。
在一个实施方案中,本文提供了降低对象中的Mcl-1水平的方法,所述方法包括将化合物1与如本文所述的第二剂组合施用于所述对象。因此,本文提供了在降低对象中的Mcl-1水平的此类方法中使用的化合物1,所述方法包括将化合物1与如本文所述的第二剂组合施用于所述对象。在一些实施方案中,本文提供了监测用化合物1与第二剂组合治疗对象中的癌症的功效的方法,包括:(a)将化合物1和第二剂施用于对象;(b)从对象获得样品;(c)确定样品中的Mcl-1水平;(d)将样品中的Mcl-1水平与参考样品中的Mcl-1水平进行比较,其中样品中的Mcl-1水平相比于参考样品降低指示用化合物1和第二剂治疗对象中的癌症的功效。因此,本文提供了在监测用化合物1与第二剂组合治疗对象中的癌症的功效的方法中使用的化合物1。在又一个方面,本文提供了预测患有或怀疑患有癌症的对象对用化合物1和第二剂治疗的响应性的方法,该方法包括(a)将化合物1和第二剂施用于对象;(b)从对象获得样品;(c)确定样品中的Mcl-1水平;(d)如果样品中的Mcl-1水平与参考样品中的Mcl-1水平相比降低,则诊断对象可能对用化合物1和第二剂治疗癌症有响应。因此,本文提供了在预测患有或怀疑患有癌症的对象对用化合物1和第二剂治疗的响应性的方法中使用的化合物1。
在本文提供的方法的一些实施方案中,先从对象获得参考样品,之后将化合物1和第二剂施用于对象,并且参考样品来自与样品相同的来源。在本文提供的方法的其它实施方案中,参考样品从患有癌症的第二对象获得,并且参考样品来自与样品相同的来源。在本文提供的方法的还有其它实施方案中,参考样品从患有癌症的一组对象获得,并且参考样品来自与样品相同的来源。
在一个实施方案中,本文提供了鉴定适合于用化合物1和第二剂治疗的癌症对象的方法,包括:(a)从患有癌症的对象获得样品;(b)确定样品中的GSPT1水平;(c)使样品与化合物1和第二剂接触;(d)在接触步骤之后确定样品中的GSPT1水平;(e)如果步骤(d)中的GSPT1水平与步骤(b)中的GSPT1水平相比降低,则鉴定该对象可能对用化合物1和第二剂治疗癌症有响应。因此,本文提供了在鉴定适合于用化合物1和第二剂治疗的癌症对象的方法中使用的化合物1。
在一个实施方案中,本文提供了鉴定适合于用化合物1和第二剂治疗的癌症对象的方法,包括:(a)从患有癌症的对象获得样品;(b)确定样品中的Mcl-1水平;(c)使样品与化合物1和第二剂接触;(d)在接触步骤之后确定样品中的Mcl-1水平;(e)如果步骤(d)中的Mcl-1水平与步骤(b)中的Mcl-1水平相比降低,则鉴定该对象可能对用化合物1和第二剂治疗癌症有响应。因此,本文提供了在鉴定适合于用化合物1和第二剂治疗的癌症对象的方法中使用的化合物1。
如本文所用,术语“样品”是指从对象获得的材料或材料混合物,包括在体内或在原位获得、收到或收集的组织样品或流体来源样品。样品还包括来自对象的包含癌前细胞或癌细胞或者癌前组织或癌组织的区域的样品。此类样品可以是但不限于从哺乳动物分离的器官、组织和细胞。示例性样品包括但不限于细胞裂解物、细胞培养物、细胞系、组织、口腔组织、胃肠道组织、器官、细胞器、生物流体、血液样品、尿液样品、皮肤样品,等等。在一个实施方案中,样品包括但不限于全血、部分纯化血液、PBMC、组织活检(包括骨髓核活检)、骨髓穿刺液、分离的骨髓单核细胞、循环肿瘤细胞,等等。
在一些这样的实施方案中,如本文所述,第二剂选自JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在某些实施方案中,化合物1的治疗或预防有效量是每天约0.005至约20mg、每天约0.05至20mg、每天约0.01至约10mg、每天约0.01至约7mg、每天约0.01至约5mg、每天约0.01至约3mg、每天约0.05至约10mg、每天约0.05至约7mg、每天约0.05至约5mg、每天约0.05至约3mg、每天约0.1至约15mg、每天约0.1至约10mg、每天约0.1至约7mg、每天约0.1至约5mg、每天约0.1至约3mg、每天约0.5至约10mg、每天约0.05至约5mg、每天约0.5至约3mg、每天约0.5至约2mg、每天约0.3至约10mg、每天约0.3至约8.5mg、每天约0.3至约8.1mg、每天约0.6至约10mg,或每天约0.6至约5mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.005至约20mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.05至20mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.01至约10mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.01至约7mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.01至约5mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.01至约3mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.05至约10mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.05至约7mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.05至约5mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.05至约3mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.1至约15mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.1至约10mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.1至约7mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.1至约5mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.1至约3mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.5至约10mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.5至约5mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.5至约3mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.5至约2mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.3至约10mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.3至约8.5mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.3至约8.1mg。在一个实施方案中,化合物1的治疗或预防有效量是每天约0.6至约10mg或每天约0.6至约5mg。
在某些实施方案中,所述治疗或预防有效量是每天约0.1、约0.2、约0.5、约1、约2、约3、约4、约5、约6、约7、约8、约9或约10mg。在一些这样的实施方案中,所述治疗或预防有效量是每天约0.5、约0.6、约0.75、约1、约2、约3、约4、约5、约6或约7mg。在一些这样的实施方案中,所述治疗或预防有效量是每天约0.6、约1.2、约1.8、约2.4或约3.6mg。在某些实施方案中,所述治疗或预防有效量是每天约0.1mg。在某些实施方案中,所述治疗或预防有效量是每天约0.2mg。在某些实施方案中,所述治疗或预防有效量是每天约0.5mg。在某些实施方案中,所述治疗或预防有效量是每天约1mg。在某些实施方案中,所述治疗或预防有效量是每天约2mg。在某些实施方案中,所述治疗或预防有效量是每天约3mg。在某些实施方案中,所述治疗或预防有效量是每天约4mg。在某些实施方案中,所述治疗或预防有效量是每天约5mg。在某些实施方案中,所述治疗或预防有效量是每天约6mg。在某些实施方案中,所述治疗或预防有效量是每天约7mg。在某些实施方案中,所述治疗或预防有效量是每天约8mg。在某些实施方案中,所述治疗或预防有效量是每天约9mg。在某些实施方案中,所述治疗或预防有效量是每天约10mg。
在一个实施方案中,对于本文所述的病症,化合物1的推荐日剂量范围在每天约0.01mg至约20mg的范围内,优选地作为每天一次的单剂量给予,或者在整个一天中以分剂量给予。在一个实施方案中,对于本文所述的病症,化合物1的推荐日剂量范围在每天约0.01mg至约15mg的范围内,优选地作为每天一次的单剂量给予,或者在整个一天中以分剂量给予。在一个实施方案中,对于本文所述的病症,化合物1的推荐日剂量范围在每天约0.01mg至约12mg的范围内,优选地作为每天一次的单剂量给予,或者在整个一天中以分剂量给予。在一些实施方案中,该剂量在每天约0.1mg至约10mg的范围内。在其它实施方案中,该剂量在每天约0.5mg至约5mg的范围内。每天的具体剂量包括每天0.1、0.2、0.5、0.6、1、1.2、1.5、1.8、2、2.4、2.5、3、3.5、3.6、4、4.5、5、5.5、6、6.5、7、7.2、7.5、8、8.5、9、9.5、10、10.5、11、11.5、12、12.5、13、13.5、14、14.4、14.5或15mg。在其它实施方案中,该剂量在每天约0.5mg至约5mg的范围内。每天的具体剂量包括每天0.1、0.2、0.5、0.6、1、1.2、1.5、1.8、2、2.4、2.5、3、3.5、3.6、4、4.5、5、5.5、6、6.5、7、7.5、8、8.5、9、9.5或10mg。在一个实施方案中,每天的剂量是每天0.1mg。在一个实施方案中,每天的剂量是每天0.2mg。在一个实施方案中,每天的剂量是每天0.5mg。在一个实施方案中,每天的剂量是每天0.6mg。在一个实施方案中,每天的剂量是每天1mg。在一个实施方案中,每天的剂量是每天1.2mg。在一个实施方案中,每天的剂量是每天1.5mg。在一个实施方案中,每天的剂量是每天1.8mg。在一个实施方案中,每天的剂量是每天2mg。在一个实施方案中,每天的剂量是每天2.4mg。在一个实施方案中,每天的剂量是每天2.5mg。在一个实施方案中,每天的剂量是每天3mg。在一个实施方案中,每天的剂量是每天3.5mg。在一个实施方案中,每天的剂量是每天3.6mg。在一个实施方案中,每天的剂量是每天4mg。在一个实施方案中,每天的剂量是每天4.5mg。在一个实施方案中,每天的剂量是每天5mg。在一个实施方案中,每天的剂量是每天5.5mg。在一个实施方案中,每天的剂量是每天6mg。在一个实施方案中,每天的剂量是每天6.5mg。在一个实施方案中,每天的剂量是每天7mg。在一个实施方案中,每天的剂量是每天7.2mg。在一个实施方案中,每天的剂量是每天7.5mg。在一个实施方案中,每天的剂量是每天8mg。在一个实施方案中,每天的剂量是每天8.5mg。在一个实施方案中,每天的剂量是每天9mg。在一个实施方案中,每天的剂量是每天9.5mg。在一个实施方案中,每天的剂量是每天10mg。在一个实施方案中,每天的剂量是每天12mg。在一个实施方案中,每天的剂量是每天10mg。在一个实施方案中,每天的剂量是每天12mg。在一个实施方案中,每天的剂量是每天14.4mg。在一个实施方案中,每天的剂量是每天15mg。
在一个具体的实施方案中,推荐的起始剂量可以是每天0.1、0.5、0.6、0.7、1、1.2、1.5、1.8、2、2.4、2.5、3、3.5、3.6、4、4.5、5、5.5、6、6.5或7mg。在另一个实施方案中,推荐的起始剂量可以是每天0.1、0.5、0.6、1、1.2、1.8、2、2.4、3、3.6、4或5mg。在一个实施方案中,该剂量可以递增至7、8、9、10、12或15mg/天。在一个实施方案中,该剂量可以递增至7、8、9或10mg/天。
在一个具体的实施方案中,化合物1可以以约0.1mg/天的量施用于患有白血病(包括AML)的患者。在一个特定的实施方案中,化合物1可以以约1mg/天的量施用于患有白血病(包括AML)的患者。在一个特定的实施方案中,化合物1可以以约3mg/天的量施用于患有白血病(包括AML)的患者。在一个特定的实施方案中,化合物1可以以约4mg/天的量施用于患有白血病(包括AML)的患者。在一个特定的实施方案中,本文提供的化合物1可以以约5mg/天的量施用于患有白血病(包括AML)的患者。在一个特定的实施方案中,本文提供的化合物1可以以约6mg/天的量施用于患有白血病(包括AML)的患者。在一个特定的实施方案中,本文提供的化合物1可以以约7mg/天的量施用于患有白血病(包括AML)的患者。在一个特定的实施方案中,本文提供的化合物1可以以约10mg/天的量施用于患有白血病(包括AML)的患者。在一个特定的实施方案中,本文提供的化合物1可以以约12mg/天的量施用于患有白血病(包括AML)的患者。在一个特定的实施方案中,本文提供的化合物1可以以约15mg/天的量施用于患有白血病(包括AML)的患者。
在一个具体的实施方案中,化合物1可以以约0.1mg/天的量施用于患有MDS的患者。在一个特定的实施方案中,化合物1可以以约1mg/天的量施用于患有MDS的患者。在一个特定的实施方案中,化合物1可以以约3mg/天的量施用于患有MDS的患者。在一个特定的实施方案中,化合物1可以以约4mg/天的量施用于患有MDS的患者。在一个特定的实施方案中,本文提供的化合物1可以以约5mg/天的量施用于患有MDS的患者。在一个特定的实施方案中,本文提供的化合物1可以以约6mg/天的量施用于患有MDS的患者。在一个特定的实施方案中,本文提供的化合物1可以以约7mg/天的量施用于患有MDS的患者。在一个特定的实施方案中,本文提供的化合物1可以以约10mg/天的量施用于患有MDS的患者。在一个特定的实施方案中,本文提供的化合物1可以以约12mg/天的量施用于患有MDS的患者。在一个特定的实施方案中,本文提供的化合物1可以以约15mg/天的量施用于患有MDS的患者。
在某些实施方案中,所述治疗或预防有效量是约0.001至约20mg/kg/天、约0.01至约15mg/kg/天、约0.01至约10mg/kg/天、约0.01至约9mg/kg/天、0.01至约8mg/kg/天、约0.01至约7mg/kg/天、约0.01至约6mg/kg/天、约0.01至约5mg/kg/天、约0.01至约4mg/kg/天、约0.01至约3mg/kg/天、约0.01至约2mg/kg/天、约0.01至约1mg/kg/天、或约0.01至约0.05mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.001至约20mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约15mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约10mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约9mg/kg/天。在某些实施方案中,所述治疗或预防有效量是0.01至约8mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约7mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约6mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约5mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约4mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约3mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约2mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约1mg/kg/天。在某些实施方案中,所述治疗或预防有效量是约0.01至约0.05mg/kg/天。
施用的剂量还可以以除mg/kg/天之外的单位表示。例如,肠胃外施用的剂量可以表示为mg/m2/天。本领域的普通技术人员将容易地知道如何针对对象的给定身高或体重或这两者将剂量从mg/kg/天转换为mg/m2/天(参见www.fda.gov/cder/cancer/animalframe.htm)。例如,对于65kg的人,1mg/kg/天的剂量大约等于38mg/m2/天。
在某些实施方案中,所施用的化合物1的量足以提供在约0.001至约500μM、约0.002至约200μM、约0.005至约100μM、约0.01至约50μM、约1至约50μM、约0.02至约25μM、约0.05至约20μM、约0.1至约20μM、约0.5至约20μM、或约1至约20μM范围内的该化合物的稳态血浆浓度。在某些实施方案中,所施用的化合物1的量足以提供在约0.001至约500μM、约0.002至约200μM、约0.005至约100μM、约0.01至约50μM、约1至约50μM、约0.02至约25μM、约0.05至约20μM、约0.1至约20μM、约0.5至约20μM、或约1至约20μM范围内的该化合物的稳态血浆浓度。
在其它实施方案中,所施用的化合物1的制剂的量足以提供在约5至约100nM、约5至约50nM、约10至约100nM、约10至约50nM或约50至约100nM范围内的该化合物的稳态血浆浓度。在其它实施方案中,所施用的化合物1的制剂的量足以提供在约5至约100nM范围内的该化合物的稳态血浆浓度。在其它实施方案中,所施用的化合物1的制剂的量足以提供在约5至约50nM范围内的该化合物的稳态血浆浓度。在其它实施方案中,所施用的化合物1的制剂的量足以提供在约10至约100nM范围内的该化合物的稳态血浆浓度。在其它实施方案中,所施用的化合物1的制剂的量足以提供在约10至约50nM范围内的该化合物的稳态血浆浓度。在其它实施方案中,所施用的化合物1的制剂的量足以提供在约50至约100nM范围内的该化合物的稳态血浆浓度。
如本文所用,术语“稳态血浆浓度”是在施用本文提供的制剂一段时间后达到的浓度。一旦达到稳态,在固体形式的血浆浓度的时间依赖性曲线上将只有微小的峰和谷。
在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.001至约500μM、约0.002至约200μM、约0.005至约100μM、约0.01至约50μM、约1至约50μM、约0.02至约25μM、约0.05至约20μM、约0.1至约20μM、约0.5至约20μM、或约1至约20μM范围内的该化合物的最大血浆浓度(峰浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.001至约500μM范围内的该化合物的最大血浆浓度(峰浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.002至约200μM范围内的该化合物的最大血浆浓度(峰浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.005至约100μM范围内的该化合物的最大血浆浓度(峰浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.01至约50μM范围内的该化合物的最大血浆浓度(峰浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约1至约50μM范围内的该化合物的最大血浆浓度(峰浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.02至约25μM范围内的该化合物的最大血浆浓度(峰浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.05至约20μM范围内的该化合物的最大血浆浓度(峰浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.1至约20μM范围内的该化合物的最大血浆浓度(峰浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.5至约20μM范围内的该化合物的最大血浆浓度(峰浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约1至约20μM范围内的该化合物的最大血浆浓度(峰浓度)。
在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.001至约500μM、约0.002至约200μM、约0.005至约100μM、约0.01至约50μM、约1至约50μM、约0.01至约25μM、约0.01至约20μM、约0.02至约20μM、约0.02至约20μM、或约0.01至约20μM范围内的该化合物的最小血浆浓度(谷浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.001至约500μM范围内的该化合物的最小血浆浓度(谷浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.002至约200μM范围内的该化合物的最小血浆浓度(谷浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.005至约100μM范围内的该化合物的最小血浆浓度(谷浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.01至约50μM范围内的该化合物的最小血浆浓度(谷浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约1至约50μM、约0.01至约25μM范围内的该化合物的最小血浆浓度(谷浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.01至约20μM范围内的该化合物的最小血浆浓度(谷浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.02至约20μM范围内的该化合物的最小血浆浓度(谷浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.02至约20μM范围内的该化合物的最小血浆浓度(谷浓度)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约0.01至约20μM范围内的该化合物的最小血浆浓度(谷浓度)。
在某些实施方案中,所施用的化合物1的制剂的量足以提供在约100至约100,000ng*h/mL、约1,000至约50,000ng*h/mL、约5,000至约25,000ng*h/mL、或约5,000至约10,000ng*h/mL范围内的该化合物的曲线下面积(AUC)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约100至约100,000ng*h/mL范围内的该化合物的曲线下面积(AUC)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约1,000至约50,000ng*h/mL范围内的该化合物的曲线下面积(AUC)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约5,000至约25,000ng*h/mL范围内的该化合物的曲线下面积(AUC)。在某些实施方案中,所施用的化合物1的制剂的量足以提供在约5,000至约10,000ng*h/mL范围内的该化合物的曲线下面积(AUC)。
在某些实施方案中,待用本文提供的方法之一治疗的患者在施用本文提供的化合物1的制剂之前未用抗癌疗法治疗过。在某些实施方案中,待用本文提供的方法之一治疗的患者在施用本文提供的化合物1的制剂之前用抗癌疗法治疗过。在某些实施方案中,待用本文提供的方法之一治疗的患者已产生对该抗癌疗法的抗药性。
尽管某些疾病或疾患在某些年龄组中更常见,但本文提供的方法涵盖治疗患者而不考虑患者的年龄。
本文提供的化合物1的制剂可以作为单剂量递送,诸如,单次推注;或者随时间推移,诸如,随时间推移连续输注或随时间推移分次推注剂量。如有必要,可以重复施用化合物1的制剂,例如,直到患者经历疾病稳定或消退,或直到患者经历疾病进展或不可接受的毒性。例如,对于实体肿瘤而言的疾病稳定,通常意味着可测量病变的垂直直径从最后一次测量开始未增加25%或更多。Response Evaluation Criteria in Solid Tumors(RECIST)Guidelines,Journal of the National Cancer Institute 92(3):205-216(2000)。通过本领域已知的方法,诸如患者症状评估、身体检查,已使用X射线、CAT、PET或MRI扫描成像的肿瘤的可视化和其它被普遍接受的评估模式来确定疾病稳定或欠缺疾病稳定。
本文提供的化合物1的制剂可以每日一次(QD)施用,或者分成多个日剂量,诸如每日两次(BID)、每日三次(TID)和每日四次(QID)。此外,施用可以是连续的(即,在几个连续日的每日,或每一天)、间歇的,例如周期性地施用(即,包括几天、几周或几个月的无药物停用期)。如本文所用,术语“每日”旨在意味着治疗化合物在例如一段时间内每天施用一次或多于一次。术语“连续”旨在意味着治疗化合物在至少10天至52周的不间断时段内每日施用。如本文所用,术语“间歇”或“间歇地”旨在意味着以规则或不规则的间隔停止和开始。例如,化合物1的制剂的间歇施用是每周施用1至6天、周期性地施用(例如,每日施用,持续28天周期的连续1至10天,然后是停用期,其中该28天周期的其余时间不施用;或每日施用,持续连续的2至8周,然后是停用期,其中最长一周不施用),或隔天施用。使用化合物1的周期性疗法在本文的其它地方进行讨论。
在一些实施方案中,施用频率在约日剂量至约月剂量的范围内。在某些实施方案中,施用是每天一次、每天两次、每天三次、每天四次、每隔一天一次、每周两次、每周一次、每两周一次、每三周一次,或每四周一次。在一个实施方案中,化合物1每天施用一次。在另一个实施方案中,化合物1每天施用两次。在又一个实施方案中,本文提供的化合物1每天施用三次。在还有一个实施方案中,本文提供的化合物1每天施用四次。在还有一个实施方案中,本文提供的化合物1每隔一天施用一次。在还有一个实施方案中,本文提供的化合物1每周施用两次。在还有一个实施方案中,本文提供的化合物1每周施用一次。在还有一个实施方案中,本文提供的化合物1每两周施用一次。在还有一个实施方案中,本文提供的化合物1每三周施用一次。在还有一个实施方案中,本文提供的化合物1每四周施用一次。
在某些实施方案中,本文提供的化合物1的制剂每天施用一次,持续一天至六个月、一周至三个月、一周至四周、一周至三周,或一周至两周。在某些实施方案中,本文提供的化合物1的制剂每天施用一次,持续一周、两周、三周或四周。在一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续1天。在一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续2天。在一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续3天。在一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续4天。在一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续5天。在一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续6天。在一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续一周。在一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续至多10天。在另一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续两周。在又一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续三周。在还有一个实施方案中,本文提供的化合物1的制剂每天施用一次,持续四周。
组合疗法
在一个实施方案中,本文提供了治疗、预防和/或管理癌症的方法,包括将化合物1与选自JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂的一种或多种第二剂组合,并且任选地与放射疗法、输血或手术组合施用于患者。本文公开了第二活性剂的实例。
在一个实施方案中,本文提供了治疗、预防和/或管理癌症的方法,包括将本文提供的化合物1的制剂与一种或多种第二活性剂组合,并且任选地与放射疗法、输血或手术组合施用于患者。本文公开了第二活性剂的实例。
如本文所用,术语“组合”包括使用多于一种疗法(例如,一种或多种预防剂和/或治疗剂)。然而,术语“组合”的使用并不限制其中疗法(例如,预防剂和/或治疗剂)施用于患有疾病或疾患的患者的顺序。例如,“组合”可以包括作为混合物施用、使用分开的制剂同时施用,以及以任何顺序连续施用。“连续的”意味着在两次施用活性剂之间经过了具体的时间。例如,“连续的”可以是在施用分开的活性剂之间经过了超过10分钟。此外,该时间段可以是超过10分钟、超过30分钟、超过1小时、超过3小时、超过6小时或超过12小时。例如,第一疗法(例如,预防剂或治疗剂,诸如本文提供的化合物1的制剂)可以在第二疗法(例如,预防剂或治疗剂)施用于对象之前(例如,之前5分钟、15分钟、30分钟、45分钟、1小时、2小时、4小时、6小时、12小时、24小时、48小时、72小时、96小时、1周、2周、3周、4周、5周、6周、8周或12周)、同时或之后(例如,之后5分钟、15分钟、30分钟、45分钟、1小时、2小时、4小时、6小时、12小时、24小时、48小时、72小时、96小时、1周、2周、3周、4周、5周、6周、8周或12周)施用。本文还设想了三联疗法。
在一个实施方案中,将化合物1(包括本文提供的化合物1的制剂)和一种或多种第二活性剂施用于患者可以通过相同或不同的施用途径同时或相继地进行。在一个实施方案中,将化合物1(包括本文提供的化合物1的制剂)和一种或多种第二活性剂施用于患者可以通过相同或不同的施用途径同时或相继地进行。用于特定活性剂的特定施用途径的适用性将取决于活性剂本身(例如,它是否可以口服施用且在进入血流之前不分解)和正在治疗的癌症。
化合物1(包括本文提供的化合物1的制剂)的施用途径独立于第二疗法的施用途径。因此,在一个实施方案中,化合物1(包括本文提供的化合物1的制剂)通过静脉内施用,并且第二疗法可以通过口服、肠胃外、腹膜内、静脉内、动脉内、经皮、舌下、肌肉内、直肠、经口含化、鼻内、脂质体、经由吸入、阴道内、眼内、通过导管或支架经由局部递送、皮下、脂肪内、关节内、鞘内或以缓释剂型施用。在一个实施方案中,化合物1(包括本文提供的化合物1的制剂)和第二疗法通过相同的施用方式(通过IV)施用。在另一个实施方案中,化合物1(包括本文提供的化合物1的制剂)通过一种施用方式(例如通过IV)施用,而第二剂(抗癌剂)通过另一种施用方式(例如口服)施用。
在一个实施方案中,第二活性剂通过静脉内或皮下施用并且每日施用一次或两次,施用量为约1至约1000mg、约5至约500mg、约10至约350mg或约50至约200mg。第二活性剂的具体量将取决于所使用的具体药剂、正在治疗和/或管理的疾病的类型、疾病的严重程度和阶段,以及化合物1和同时施用于患者的任何可选的附加活性剂的量。
一种或多种第二活性成分或活性剂可以与化合物1一起用于本文提供的方法和组合物中。第二活性剂可以是大分子(例如,蛋白质)或小分子(例如,合成无机分子、有机金属分子或有机分子)。
大分子活性剂的实例包括但不限于造血生长因子、细胞因子以及单克隆抗体和多克隆抗体(特别是癌症抗原的治疗抗体)。典型的大分子活性剂是生物分子,诸如天然存在的蛋白质或合成蛋白质或重组蛋白质。特别可用于本文提供的方法和组合物中的蛋白质包括在体外或在体内刺激造血前体细胞和免疫活性生成细胞的存活和/或增殖的蛋白质。其它可用的蛋白质在体外或体内刺激细胞中定向红细胞系祖细胞的分裂和分化。特定的蛋白质包括但不限于:白介素,诸如IL-2(包括重组IL-II(“rIL2”)和金丝雀痘IL-2)、IL-10、IL-12和IL-18;干扰素,诸如干扰素α-2a、干扰素α-2b、干扰素α-n1、干扰素α-n3、干扰素β-I a和干扰素γ-I b;GM-CF和GM-CSF;以及EPO。
在某些实施方案中,GM-CSF、G-CSF、SCF或EPO在四周或六周周期的约五天期间以约1至约750mg/m2/天、约25至约500mg/m2/天、约50至约250mg/m2/天、或约50至约200mg/m2/天范围内的量皮下施用。在某些实施方案中,GM-CSF可以以约60至约500mcg/m2的量静脉内施用达2小时或以约5至约12mcg/m2/天的量皮下施用。在某些实施方案中,G-CSF最初可以以约1mcg/kg/天的量皮下施用,并且可以根据总粒细胞计数的增长进行调整。G-CSF的维持剂量可以以约300(在较小的患者中)或480mcg的量皮下施用。在某些实施方案中,EPO可以以10,000个单位的量皮下施用,每周3次。
可以用于所述方法和组合物中的特定蛋白质包括但不限于:非格司亭(filgrastim),其在美国以商品名 (Amgen,Thousand Oaks,CA)出售;沙格司亭(sargramostim),其在美国以商品名
(Amgen,Thousand Oaks,CA)出售;沙格司亭(sargramostim),其在美国以商品名 (Immunex,Seattle,WA)出售;和重组EPO,其在美国以商品名
(Immunex,Seattle,WA)出售;和重组EPO,其在美国以商品名 (Amgen,Thousand Oaks,CA)出售。
(Amgen,Thousand Oaks,CA)出售。
GM-CSF的重组形式和突变形式可以如美国专利号5,391,485、5,393,870和5,229,496中所述进行制备;所有这些专利都以引用方式并入本文。G-CSF的重组形式和突变形式可以如美国专利号4,810,643、4,999,291、5,528,823和5,580,755中所述进行制备;这些专利的全文以引用方式并入本文。
还提供了与化合物1(包括化合物1的制剂)组合使用的自然蛋白质、天然存在的蛋白质和重组蛋白质。还涵盖天然存在的蛋白质的突变体和衍生物(例如,经修饰形式),这些突变体和衍生物在体内表现出它们所基于的蛋白质的至少一些药理活性。突变体的实例包括但不限于具有与蛋白质的天然存在形式中的对应残基不同的一个或多个氨基酸残基的蛋白质。术语“突变体”还涵盖缺乏天然存在的形式(例如,非糖基化形式)中通常存在的碳水化合物部分的蛋白质。衍生物的实例包括但不限于聚乙二醇化衍生物和融合蛋白,诸如通过将IgG1或IgG3融合至蛋白质或感兴趣的蛋白质的活性部分来形成的蛋白质。参见例如,Penichet,M.L.和Morrison,S.L.,J.Immunol.Methods 248:91-101(2001)。
可以与化合物1(包括本文提供的化合物1的制剂)组合使用的抗体包括单克隆抗体和多克隆抗体。抗体的实例包括但不限于曲妥珠单抗(trastuzumab, )、利妥昔单抗(rituximab,
)、利妥昔单抗(rituximab, )、贝伐珠单抗(bevacizumab,AvastinTM)、帕妥珠单抗(pertuzumab,OmnitargTM)、托西莫单抗(tositumomab,
)、贝伐珠单抗(bevacizumab,AvastinTM)、帕妥珠单抗(pertuzumab,OmnitargTM)、托西莫单抗(tositumomab, )、依决洛单抗(edrecolomab,
)、依决洛单抗(edrecolomab, )和G250。化合物1的制剂也可以与抗TNF-α抗体和/或抗EGFR抗体,诸如
)和G250。化合物1的制剂也可以与抗TNF-α抗体和/或抗EGFR抗体,诸如 或帕尼单抗(panitumumab)组合或与它们组合使用。
或帕尼单抗(panitumumab)组合或与它们组合使用。
大分子活性剂可以以抗癌症疫苗的形式施用。例如,分泌细胞因子(诸如IL-2、G-CSF和GM-CSF)或引起细胞因子分泌的疫苗可以用于所提供的方法和药物组合物中。参见例如,Emens,L.A.,et al.,Curr.Opinion Mol.Ther.3(1):77-84(2001)。
为小分子的第二活性剂也可以用于减轻与施用本文提供的化合物1的制剂相关联的副作用。然而,与一些大分子类似,据信很多小分子在与本文提供的化合物1(包括化合物1的制剂)一起(例如,在其之前、之后或同时)施用时,能够提供协同作用。小分子第二活性剂的实例包括但不限于抗癌剂、抗生素、免疫抑制剂和类固醇。
在某些实施方案中,第二剂是HSP抑制剂、蛋白酶体抑制剂、FLT3抑制剂或mTOR抑制剂。在一些实施方案中,mTOR抑制剂是mTOR激酶抑制剂。
待在本文所述的方法或组合物中使用的抗癌剂的实例包括但不限于:阿西维辛;阿柔比星;盐酸阿考达唑;阿克罗宁(acronine);阿多来新;阿地白介素(aldesleukin);六甲蜜胺;安波霉素(ambomycin);醋酸阿美蒽醌;安吖啶;阿那曲唑;氨茴霉素;天冬酰胺酶;曲林菌素(asperlin);阿扎胞苷;阿扎替派(azetepa);阿佐霉素(azotomycin);巴马司他;苯佐替派(benzodepa);比卡鲁胺;盐酸比生群;二甲磺酸双奈法德(bisnafidedimesylate);比折来新(bizelesin);硫酸博莱霉素;布喹那钠;溴匹立明;白消安;放线菌素C(cactinomycin);卡普睾酮;卡醋胺(caracemide);卡贝替姆(carbetimer);卡铂;卡莫司汀;盐酸卡柔比星;卡折来新(carzelesin);西地芬戈(cedefingol);塞来昔布(COX-2抑制剂);苯丁酸氮芥;西罗霉素(cirolemycin);顺铂;克拉屈滨;克罗拉滨;甲磺酸克立那托(crisnatol mesylate);环磷酰胺;Ara-C;达卡巴嗪;放线菌素D;盐酸柔红霉素;地西他滨;右奥马铂(dexormaplatin);地扎胍宁(dezaguanine);甲磺酸地扎胍宁、地吖醌(diaziquone);多西他赛;多柔比星;盐酸多柔比星;屈洛昔芬;枸橼酸屈洛昔芬;丙酸屈他雄酮;达佐霉素(duazomycin);依达曲沙;盐酸依氟鸟氨酸;依沙卢星;恩洛铂(enloplatin);恩普氨酯(enpromate);依匹哌啶(epipropidine);盐酸表柔比星;厄布洛唑(erbulozole);盐酸依索比星;雌莫司汀;雌莫司汀磷酸钠;依他硝唑;依托泊苷;磷酸依托泊苷;艾托卜宁(etoprine);盐酸法倔唑;法扎拉滨;芬维A胺;氟尿苷;磷酸氟达拉滨;氟尿嘧啶;氟西他滨(flurocitabine);磷喹酮(fosquidone);福斯曲星钠;吉西他滨;盐酸吉西他滨;羟基脲;盐酸伊达比星;异环磷酰胺;伊莫福新;异丙铂;伊立替康;盐酸伊立替康;醋酸兰瑞肽;来曲唑;醋酸亮丙瑞林;盐酸利阿唑;洛美曲索钠(Lometrexol sodium);洛莫司汀;盐酸洛索蒽醌;马索罗酚(masoprocol);美登素;盐酸氮芥;醋酸甲地孕酮;醋酸美仑孕酮;美法仑;美诺立尔(menogaril);巯基嘌呤;甲氨蝶呤;甲氨蝶呤钠;氯苯氨啶(metoprine);美妥替哌(meturedepa);米丁度胺(mitindomide);米托卡星(mitocarcin);米托罗明(mitocromin);米托洁林(mitogillin);米托马星(mitomalcin);丝裂霉素;米托司培(mitosper);米托坦;盐酸米托蒽醌;霉酚酸;诺考达唑;诺加霉素(nogalamycin);omacetaxine;奥马铂;奥昔舒仑(oxisuran);紫杉醇;培门冬酰胺酶(pegaspargase);培利霉素(peliomycin);奈莫司汀(pentamustine);硫酸培洛霉素;培磷酰胺(perfosfamide);哌泊溴烷;哌泊舒凡;盐酸吡罗蒽醌;普卡霉素;普洛美坦(plomestane);卟菲尔钠;泊非霉素;泼尼莫司汀;盐酸丙卡巴肼;嘌呤霉素;盐酸嘌呤霉素;吡唑呋喃;利波腺苷(riboprine);沙芬戈(safingol);盐酸沙芬戈(safingol hydrochloride);司莫司汀;辛曲秦(simtrazene);索拉非尼;磷乙酰天冬氨酸钠(sparfosate sodium);稀疏霉素;盐酸锗螺胺(spirogermanium hydrochloride);螺莫司汀(spiromustine);螺铂;链黑菌素;链脲佐菌素;磺氯苯脲(sulofenur);他利霉素(talisomycin);替可加兰钠(tecogalan sodium);泰索帝(taxotere);替加氟(tegafur);盐酸替洛蒽醌(teloxantrone hydrochloride);替莫泊芬;替尼泊苷;替罗昔隆(teroxirone);睾内酯;硫唑嘌呤胺(thiamiprine);硫鸟嘌呤;噻替派;噻唑呋林;替拉扎明;柠檬酸托瑞米芬;醋酸曲托龙(trestoloneacetate);磷酸曲西立滨;三甲曲沙;葡萄糖醛酸三甲曲沙;曲普瑞林;盐酸妥布氯唑(tubulozolehydrochloride);尿嘧啶氮芥;乌瑞替派(uredepa);伐普肽;维替泊芬;硫酸长春花碱;硫酸长春甘酯(vinglycinate sulfate);硫酸环氧长春碱(vinleurosine sulfate);酒石酸长春瑞滨;硫酸长春罗定;硫酸长春利定;伏氯唑;折尼铂(zeniplatin);净司他丁;和盐酸佐柔比星。
待被包含在本文的方法内的其它抗癌药物包括但不限于:20-表-1,25二羟基维生素D3;5-乙炔基尿嘧啶;阿比特龙;阿柔比星;阿弗韦恩(acylfulvene);腺环戊醇(adecypenol);阿多来新;阿地白介素(aldesleukin);ALL-TK拮抗剂;六甲蜜胺;氨莫司汀(ambamustine);美沙酮(amidox);氨磷汀;氨基乙酰丙酸;氨柔比星;安吖啶;阿那格雷;阿那曲唑;穿心莲内酯;血管生成抑制剂;拮抗剂D;拮抗剂G;安雷利克斯(antarelix);抗背部化形态发生蛋白-1;抗雄激素、前列腺癌药;抗雌激素;抗瘤酮;反义寡核苷酸;甘氨酸阿非迪霉素(aphidicolin glycinate);凋亡基因调控剂;凋亡调节剂;脱嘌呤核酸;ara-CDP-DL-PTBA;精氨酸脱氨酶;阿苏拉林(asulacrine);阿他美坦;阿莫司汀(atrimustine);阿西他汀1(axinastatin 1);阿西他汀2(axinastatin 2);阿西他汀3(axinastatin 3);阿扎司琼;阿扎霉素(azatoxin);重氮酪氨酸(azatyrosine);浆果赤霉素III衍生物;巴拉诺(balanol);巴马司他;BCR/ABL拮抗剂;苯并卟吩(benzochlorin);苯甲酰十字孢碱(benzoylstaurosporine);β内酰胺衍生物;β-艾森(alethine);β-克拉霉素B(betaclamycin B);桦木酸;b-FGF抑制剂;比卡鲁胺;比生群;二吖丙啶精胺(bisaziridinylspermine);双奈法德(bisnafide);双枸缘酸环己噻卓酯A(bistrateneA);比折来新(bizelesin);贝弗雷(breflate);溴匹立明;布度钛;丁硫氨酸亚砜亚胺;卡泊三醇;钙磷酸蛋白C(calphostin C);喜树碱衍生物;卡培他滨;甲酰胺-氨基-三唑;羧胺三唑;CaRest M3;CARN 700;软骨衍生的抑制剂;卡折来新(carzelesin);酪蛋白激酶抑制剂(ICOS);栗树精胺(castanospermine);杀菌肽B;西曲瑞克;二氢卟酚(chlorln);氯喹喔啉磺酰胺;西卡前列素;顺卟啉;克拉屈滨;克罗米芬类似物;克霉唑;碰撞霉素A(collismycinA);碰撞霉素B(collismycin B);考布他汀A4;考布他汀类似物;康纳京尼(conagenin);卡那贝西汀(crambescidin)816;克雷斯托(crisnatol);缩酚酸肽类药(cryptophycin)8;自念珠藻环肽A(cryptophycin A)衍生物;库拉欣A(curacin A);环戊蒽醌(cyclopentanthraquinone);环钼(cycloplatam);西配霉素(cypemycin);Ara-C十八烷基磷酸盐(Ara-C ocfosfate);细胞溶解因子;磷酸己烷雌酚(cytostatin);达昔单抗(dacliximab);地西他滨;脱氢膜海鞘素B(dehydrodidemnin B);德舍瑞林;地塞米松;多巴右异环磷酰胺(dexifosfamide);右丙亚胺;右维拉帕米(dexverapamil);地吖醌(diaziquone);膜海鞘素B(didemnin B);地多西(didox);二乙基去甲精胺(diethylnorspermine);二氢-5-氮杂胞苷;二氢紫杉醇(dihydrotaxol),9-;二氧霉素(dioxamycin);二苯基螺莫司汀;多西他赛;二十二烷醇;多拉司琼;脱氧氟尿苷;多柔比星;屈洛昔芬;屈大麻酚;倍癌霉素SA(duocarmycin SA);依布硒啉;依考莫司汀(ecomustine);依地福新;依决洛单抗;依氟鸟氨酸;榄香烯;乙嘧替氟;表柔比星;爱普列特;雌莫司汀类似物;雌激素激动剂;雌激素拮抗剂;依他硝唑;磷酸依托泊苷;依西美坦;法倔唑;法扎拉滨;芬维A胺;非格司亭;非那雄胺;夫拉平度;氟卓斯汀(flezelastine);16A-氟-脱氢表雄酮(fluasterone);氟达拉滨;盐酸氟代柔红霉素;福酚美克(forfenimex);福美坦;福司曲星(fostriecin);福莫司汀;莫特沙芬钆(gadolinium texaphyrin);硝酸镓;加洛他滨(galocitabine);加尼瑞克;明胶酶抑制剂;吉西他滨;谷胱甘肽抑制剂;赫舒方(hepsulfam);调节蛋白(heregulin);六亚甲基二乙酰胺;金丝桃素;伊班膦酸;伊达比星;艾多昔芬;伊决孟酮(idramantone);伊莫福新;伊洛马司他(ilomastat);伊马替尼(例如, ),咪喹莫特;免疫刺激剂肽;胰岛素样生长因子-1受体抑制剂;干扰素激动剂;干扰素;白细胞介素;碘苄胍(iobenguane);碘阿霉素(iododoxorubicin);苯并三嗪类(ipomeanol),4-;伊罗普拉(iroplact);伊索拉定;伊索本唑(isobengazole);伊索高软海绵素(isohomohalicondrin)B;伊他司琼(itasetron);盐酸药根碱(jasplakinolide);十九元环酯肽(kahalalide)F;三乙酸层状素-N;兰瑞肽;雷拉霉素(leinamycin);来格司亭;硫酸香菇多糖;莱托斯汀(leptolstatin);来曲唑;白血病抑制因子;白细胞α干扰素;醋酸亮丙瑞林+雌激素+孕酮;亮丙瑞林;左旋咪唑;利阿唑;线性多胺类似物;亲脂性二糖肽;亲脂性铂化合物;利索迈德(lissoclinamide)7;洛铂;蚯蚓磷脂(lombricine);洛美曲索;氯尼达明;洛索蒽醌;洛索立宾;勒托替康;镥特沙弗林(lutetium texaphyrin);利夫林(lysofylline);裂解肽;美坦新(maitansine);马诺他汀(mannostatin)A;马马司他;马索罗酚(masoprocol);乳腺丝抑蛋白(maspin);基质溶解素抑制剂;基质金属蛋白酶抑制剂;美诺立尔(menogaril);美巴龙(merbarone);美替瑞林(meterelin);蛋氨酸酶(methioninase);胃复安;MIF抑制剂;米非司酮;米替福新;米立司亭(mirimostim);米托胍腙(mitoguazone);二溴卫矛醇;丝裂霉素类似物;米托萘胺(mitonafide);米托蒽醌毒素成纤维细胞生长因子-皂草素;米托蒽醌;莫法罗汀(mofarotene);莫拉司亭(molgramostim);爱必妥,人绒毛膜促性腺激素;单磷酰脂质A+肌杆菌(myobacterium)细胞壁sk;莫哌达醇(mopidamol);芥末抗癌剂;迈佩昔德(mycaperoxide)B;分枝杆菌细胞壁提取物;迈泼龙(myriaporone);N-乙酰基地那林(N-acetyldinaline);N-取代的苯甲酰胺;那法瑞林;那雷蒂普(nagrestip);纳洛酮+喷他佐辛;那帕文(napavin);那非特平(naphterpin);那托司亭(nartograstim);奈达铂;奈莫柔比星;奈立膦酸;尼鲁米特;尼萨霉素(nisamycin);一氧化氮调节剂;硝基氧化物抗氧化剂;尼图林(nitrullyn);奥利默森(oblimersen)
),咪喹莫特;免疫刺激剂肽;胰岛素样生长因子-1受体抑制剂;干扰素激动剂;干扰素;白细胞介素;碘苄胍(iobenguane);碘阿霉素(iododoxorubicin);苯并三嗪类(ipomeanol),4-;伊罗普拉(iroplact);伊索拉定;伊索本唑(isobengazole);伊索高软海绵素(isohomohalicondrin)B;伊他司琼(itasetron);盐酸药根碱(jasplakinolide);十九元环酯肽(kahalalide)F;三乙酸层状素-N;兰瑞肽;雷拉霉素(leinamycin);来格司亭;硫酸香菇多糖;莱托斯汀(leptolstatin);来曲唑;白血病抑制因子;白细胞α干扰素;醋酸亮丙瑞林+雌激素+孕酮;亮丙瑞林;左旋咪唑;利阿唑;线性多胺类似物;亲脂性二糖肽;亲脂性铂化合物;利索迈德(lissoclinamide)7;洛铂;蚯蚓磷脂(lombricine);洛美曲索;氯尼达明;洛索蒽醌;洛索立宾;勒托替康;镥特沙弗林(lutetium texaphyrin);利夫林(lysofylline);裂解肽;美坦新(maitansine);马诺他汀(mannostatin)A;马马司他;马索罗酚(masoprocol);乳腺丝抑蛋白(maspin);基质溶解素抑制剂;基质金属蛋白酶抑制剂;美诺立尔(menogaril);美巴龙(merbarone);美替瑞林(meterelin);蛋氨酸酶(methioninase);胃复安;MIF抑制剂;米非司酮;米替福新;米立司亭(mirimostim);米托胍腙(mitoguazone);二溴卫矛醇;丝裂霉素类似物;米托萘胺(mitonafide);米托蒽醌毒素成纤维细胞生长因子-皂草素;米托蒽醌;莫法罗汀(mofarotene);莫拉司亭(molgramostim);爱必妥,人绒毛膜促性腺激素;单磷酰脂质A+肌杆菌(myobacterium)细胞壁sk;莫哌达醇(mopidamol);芥末抗癌剂;迈佩昔德(mycaperoxide)B;分枝杆菌细胞壁提取物;迈泼龙(myriaporone);N-乙酰基地那林(N-acetyldinaline);N-取代的苯甲酰胺;那法瑞林;那雷蒂普(nagrestip);纳洛酮+喷他佐辛;那帕文(napavin);那非特平(naphterpin);那托司亭(nartograstim);奈达铂;奈莫柔比星;奈立膦酸;尼鲁米特;尼萨霉素(nisamycin);一氧化氮调节剂;硝基氧化物抗氧化剂;尼图林(nitrullyn);奥利默森(oblimersen) O6-苄基鸟嘌呤(benzylguanine);奥曲肽;奥克恩(okicenone);寡核苷酸;奥那司酮;昂丹司琼;昂丹司琼;奥拉新(oracin);口服细胞因子诱导剂;奥马铂;奥沙特隆;奥沙利铂;奥诺霉素(oxaunomycin);紫杉醇;紫杉醇类似物;紫杉醇衍生物;帕劳民(palauamine);帕米佐星(palmitoylrhizoxin);帕米膦酸;人参炔三醇;帕诺米芬(panomifene);帕拉贝新(parabactin);帕折普汀(pazelliptine);天门冬酰胺酶;佩德星(peldesine);戊聚糖多硫酸钠;喷司他丁;喷唑(pentrozole);全氟溴烷;培磷酰胺(perfosfamide);紫苏醇;非那霉素(phenazinomycin);苯乙酸;磷酸酶抑制剂;溶血链球菌制剂;盐酸毛果芸香碱;吡柔比星;吡曲克辛;帕瑟亭(placetin)A;帕瑟亭B;纤溶酶原激活物抑制剂;铂络合物;铂化合物;铂-三胺复合物;卟菲尔钠;泊非霉素;泼尼松;丙基双吖啶酮;前列腺素J2;蛋白酶体抑制剂;基于蛋白质A的免疫调节剂;蛋白激酶C抑制剂;蛋白激酶C抑制剂,微藻;蛋白酪氨酸磷酸酶抑制剂;嘌呤核苷磷酸化酶抑制剂;红紫素;吡唑啉吖啶(pyrazoloacridine);吡哆酰基化血红蛋白聚氧乙烯缀合物;raf拮抗剂;雷替曲塞;雷莫司琼;ras法尼基蛋白转移酶抑制剂;ras抑制剂;ras-GAP抑制剂;去甲基化瑞替普汀(retelliptine demethylated);依替膦酸铼Re 186;根霉素;核酶;RII维胺(RIIretinamide);罗希吐碱(rohitukine);罗莫肽(romurtide);罗喹美克;卢比格酮B1(rubiginone B1);鲁波西(ruboxyl);沙芬戈(safingol);森托平(saintopin);SarCNU;肌肉叶绿醇(sarcophytol)A;沙格司亭;Sdi 1模拟物;司莫司汀;衰老衍生抑制剂1;正义寡核苷酸;信号转导抑制剂;西佐喃;索布佐生;硼卡钠(sodium borocaptate);苯乙酸钠;索维洛(solverol);生长调节素结合蛋白;索纳明(sonermin);膦门冬酸(sparfosic acid);穗霉素D(Spicamycin D);螺莫司汀(spiromustine);斯兰罗皮汀(splenopentin);海绵斯他丁1;角鲨胺;斯皮迈德(stipiamide);溶基质素抑制剂;苏诺星(sulfinosine);超活性血管活性肠肽拮抗剂;苏迪塔(suradista);苏拉明;苦马豆素;他莫司汀(tallimustine);他莫昔芬甲碘化物;牛磺莫司汀(tauromustine);他扎罗汀;替可加兰钠(tecogalan sodium);替加氟;特鲁皮留(tellurapyrylium);端粒酶抑制剂;替莫泊芬;替尼泊苷;四氯十氧化物(tetrachlorodecaoxide);替挫明(tetrazomine);沙利拉亭(thaliblastine);硫考拉林(thiocoraline);血小板生成素;血小板生成素模拟物;胸腺法新;胸腺生成素受体激动剂;胸腺曲南(thymotrinan);促甲状腺激素;乙基锡初紫红素(tin ethyl etiopurpurin);替拉扎明;二氯环戊二烯钛(titanocene bichloride);托森亭(topsentin);托瑞米芬;翻译抑制剂;维甲酸;三乙酰尿苷;曲西立滨;三甲曲沙;曲普瑞林;托烷司琼;妥罗雄脲(turosteride);酪氨酸激酶抑制剂;酪氨酸磷酸化抑制剂;UBC抑制剂;乌苯美司;泌尿生殖窦来源的生长抑制因子;尿激酶受体拮抗剂;伐普肽;凡瑞林(variolin)B;维拉雷琐;藜芦胺(veramine);维尔丁(verdins);维替泊芬;长春瑞滨;维卡亭(vinxaltine);整合素拮抗剂(vitaxin);伏氯唑;扎诺特隆(zanoterone);折尼铂(zeniplatin);亚苄维(zilascorb);和净司他丁斯酯(zinostatin stimalamer)。
O6-苄基鸟嘌呤(benzylguanine);奥曲肽;奥克恩(okicenone);寡核苷酸;奥那司酮;昂丹司琼;昂丹司琼;奥拉新(oracin);口服细胞因子诱导剂;奥马铂;奥沙特隆;奥沙利铂;奥诺霉素(oxaunomycin);紫杉醇;紫杉醇类似物;紫杉醇衍生物;帕劳民(palauamine);帕米佐星(palmitoylrhizoxin);帕米膦酸;人参炔三醇;帕诺米芬(panomifene);帕拉贝新(parabactin);帕折普汀(pazelliptine);天门冬酰胺酶;佩德星(peldesine);戊聚糖多硫酸钠;喷司他丁;喷唑(pentrozole);全氟溴烷;培磷酰胺(perfosfamide);紫苏醇;非那霉素(phenazinomycin);苯乙酸;磷酸酶抑制剂;溶血链球菌制剂;盐酸毛果芸香碱;吡柔比星;吡曲克辛;帕瑟亭(placetin)A;帕瑟亭B;纤溶酶原激活物抑制剂;铂络合物;铂化合物;铂-三胺复合物;卟菲尔钠;泊非霉素;泼尼松;丙基双吖啶酮;前列腺素J2;蛋白酶体抑制剂;基于蛋白质A的免疫调节剂;蛋白激酶C抑制剂;蛋白激酶C抑制剂,微藻;蛋白酪氨酸磷酸酶抑制剂;嘌呤核苷磷酸化酶抑制剂;红紫素;吡唑啉吖啶(pyrazoloacridine);吡哆酰基化血红蛋白聚氧乙烯缀合物;raf拮抗剂;雷替曲塞;雷莫司琼;ras法尼基蛋白转移酶抑制剂;ras抑制剂;ras-GAP抑制剂;去甲基化瑞替普汀(retelliptine demethylated);依替膦酸铼Re 186;根霉素;核酶;RII维胺(RIIretinamide);罗希吐碱(rohitukine);罗莫肽(romurtide);罗喹美克;卢比格酮B1(rubiginone B1);鲁波西(ruboxyl);沙芬戈(safingol);森托平(saintopin);SarCNU;肌肉叶绿醇(sarcophytol)A;沙格司亭;Sdi 1模拟物;司莫司汀;衰老衍生抑制剂1;正义寡核苷酸;信号转导抑制剂;西佐喃;索布佐生;硼卡钠(sodium borocaptate);苯乙酸钠;索维洛(solverol);生长调节素结合蛋白;索纳明(sonermin);膦门冬酸(sparfosic acid);穗霉素D(Spicamycin D);螺莫司汀(spiromustine);斯兰罗皮汀(splenopentin);海绵斯他丁1;角鲨胺;斯皮迈德(stipiamide);溶基质素抑制剂;苏诺星(sulfinosine);超活性血管活性肠肽拮抗剂;苏迪塔(suradista);苏拉明;苦马豆素;他莫司汀(tallimustine);他莫昔芬甲碘化物;牛磺莫司汀(tauromustine);他扎罗汀;替可加兰钠(tecogalan sodium);替加氟;特鲁皮留(tellurapyrylium);端粒酶抑制剂;替莫泊芬;替尼泊苷;四氯十氧化物(tetrachlorodecaoxide);替挫明(tetrazomine);沙利拉亭(thaliblastine);硫考拉林(thiocoraline);血小板生成素;血小板生成素模拟物;胸腺法新;胸腺生成素受体激动剂;胸腺曲南(thymotrinan);促甲状腺激素;乙基锡初紫红素(tin ethyl etiopurpurin);替拉扎明;二氯环戊二烯钛(titanocene bichloride);托森亭(topsentin);托瑞米芬;翻译抑制剂;维甲酸;三乙酰尿苷;曲西立滨;三甲曲沙;曲普瑞林;托烷司琼;妥罗雄脲(turosteride);酪氨酸激酶抑制剂;酪氨酸磷酸化抑制剂;UBC抑制剂;乌苯美司;泌尿生殖窦来源的生长抑制因子;尿激酶受体拮抗剂;伐普肽;凡瑞林(variolin)B;维拉雷琐;藜芦胺(veramine);维尔丁(verdins);维替泊芬;长春瑞滨;维卡亭(vinxaltine);整合素拮抗剂(vitaxin);伏氯唑;扎诺特隆(zanoterone);折尼铂(zeniplatin);亚苄维(zilascorb);和净司他丁斯酯(zinostatin stimalamer)。
在某些实施方案中,第二剂选自一种或多种检查点抑制剂。在一个实施方案中,在本文提供的方法中,将一种检查点抑制剂与化合物1或化合物1的制剂组合使用。在另一个实施方案中,结合本文提供的方法,将两种检查点抑制剂与化合物1或化合物1的制剂组合使用。在又一个实施方案中,结合本文提供的方法,将三种或更多种检查点抑制剂与化合物1或化合物1的制剂组合使用。
如本文所用,术语“免疫检查点抑制剂”或“检查点抑制剂”是指完全或部分地减少、抑制、干扰或调控一种或多种检查点蛋白的分子。不受特定理论的限制,检查点蛋白调节T细胞活化或功能。已知许多检查点蛋白,诸如CTLA-4及其配体CD80和CD86;PD-1及其配体PD-Ll和PD-L2(Pardoll,Nature Reviews Cancer,2012,12,252-264)。这些蛋白质显示出负责T细胞响应的共刺激或抑制相互作用。免疫检查点蛋白显示出调节和维持自身耐受性以及生理免疫响应的持续时间和幅度。免疫检查点抑制剂包括抗体或来源于抗体。
在一个实施方案中,检查点抑制剂是CTLA-4抑制剂。在一个实施方案中,CTLA-4抑制剂是抗CTLA-4抗体。抗CTLA-4抗体的实例包括但不限于美国专利号5,811,097、5,811,097、5,855,887、6,051,227、6,207,157、6,682,736、6,984,720和7,605,238中所述的那些,所有这些专利均全文并入本文。在一个实施方案中,抗CTLA-4抗体是曲美目单抗(tremelimumab)(也称为替西木单抗(ticilimumab)或CP-675,206)。在另一个实施方案中,抗CTLA-4抗体是伊匹单抗(ipilimumab)(也称为MDX-010或MDX-101)。伊匹单抗是与CTLA-4结合的完全人单克隆IgG抗体。伊匹单抗以商品名YervoyTM销售。
在一个实施方案中,检查点抑制剂是PD-1/PD-L1抑制剂。PD-l/PD-L1抑制剂的实例包括但不限于美国专利号7,488,802、7,943,743、8,008,449、8,168,757、8,217,149以及PCT专利申请公布号WO2003042402、WO2008156712、WO2010089411、WO2010036959、WO2011066342、WO2011159877、WO2011082400和WO2011161699中所述的那些,所有这些专利均全文并入本文。
在一个实施方案中,检查点抑制剂是PD-1抑制剂。在一个实施方案中,PD-1抑制剂是抗PD-1抗体。在一个实施方案中,抗PD-1抗体是BGB-A317、纳武单抗(nivolumab)(也称为ONO-4538、BMS-936558或MDX1106)或帕博利珠单抗(pembrolizumab)(也称为MK-3475、SCH900475或拉姆布罗力珠单抗(lambrolizumab))。在一个实施方案中,抗PD-1抗体是纳武单抗。纳武单抗是人IgG4抗PD-1单克隆抗体,并且以商品名OpdivoTM出售。在另一个实施方案中,抗PD-1抗体是帕博利珠单抗。帕博利珠单抗是人源化单克隆IgG4抗体,并且以商品名KeytrudaTM出售。在又一个实施方案中,抗PD-1抗体是CT-011,一种人源化抗体。单独施用的CT-011在复发时治疗急性骨髓性白血病(AML)未能示出响应。在又一个实施方案中,抗PD-1抗体是融合蛋白AMP-224。在另一个实施方案中,PD-1抗体是BGB-A317。BGB-A317是单克隆抗体,其中与Fcγ受体I结合的能力被特异性地工程化,并且具有以高亲和力和优异的靶标特异性结合PD-1的独特结合特征。
在一个实施方案中,检查点抑制剂是PD-L1抑制剂。在一个实施方案中,PD-L1抑制剂是抗PD-L1抗体。在一个实施方案中,抗PD-L1抗体是MEDI4736(德瓦鲁单抗(durvalumab))。在另一个实施方案中,抗PD-L1抗体是BMS-936559(也称为MDX-1105-01)。在又一个实施方案中,PD-L1抑制剂是阿特珠单抗(atezolizumab)(也称为MPDL3280A和 )。
)。
在一个实施方案中,检查点抑制剂是PD-L2抑制剂。在一个实施方案中,PD-L2抑制剂是抗PD-L2抗体。在一个实施方案中,抗PD-L2抗体是rHIgM12B7A。
在一个实施方案中,检查点抑制剂是淋巴细胞活化基因-3(LAG-3)抑制剂。在一个实施方案中,LAG-3抑制剂是可溶性Ig融合蛋白IMP321(Brignone等人,J.Immunol.,2007,179,4202-4211)。在另一个实施方案中,LAG-3抑制剂是BMS-986016。
在一个实施方案中,检查点抑制剂是B7抑制剂。在一个实施方案中,B7抑制剂是B7-H3抑制剂或B7-H4抑制剂。在一个实施方案中,B7-H3抑制剂是抗B7-H3抗体MGA271(Loo等人,Clin.Cancer Res.,2012,3834)。
在一个实施方案中,检查点抑制剂是TIM3(T细胞免疫球蛋白结构域和粘蛋白结构域3)抑制剂(Fourcade等人,J.Exp.Med.,2010,207,2175-86;Sakuishi等人,J.Exp.Med.,2010,207,2187-94)。
在一个实施方案中,检查点抑制剂是OX40(CD134)激动剂。在一个实施方案中,检查点抑制剂是抗OX40抗体。在一个实施方案中,抗OX40抗体是抗OX-40。在另一个实施方案中,抗OX40抗体是MEDI6469。
在一个实施方案中,检查点抑制剂是GITR激动剂。在一个实施方案中,检查点抑制剂是抗GITR抗体。在一个实施方案中,抗GITR抗体是TRX518。
在一个实施方案中,检查点抑制剂是CD137激动剂。在一个实施方案中,检查点抑制剂是抗CD137抗体。在一个实施方案中,抗CD137抗体是乌瑞鲁单抗(urelumab)。在另一个实施方案中,抗CD137抗体是PF-05082566。
在一个实施方案中,检查点抑制剂是CD40激动剂。在一个实施方案中,检查点抑制剂是抗CD40抗体。在一个实施方案中,抗CD40抗体是CF-870,893。
在一个实施方案中,检查点抑制剂是重组人白介素-15(rhIL-15)。
在一个实施方案中,检查点抑制剂是IDO抑制剂。在一个实施方案中,IDO抑制剂是INCB024360。在另一个实施方案中,IDO抑制剂是吲哚莫德(indoximod)。
在某些实施方案中,本文提供的组合疗法包括两种或更多种本文所述的检查点抑制剂(包括相同或不同种类的检查点抑制剂)。此外,本文所述的组合疗法可以与如本文所述的第二活性剂组合使用,适用于治疗本文所述和本领域所理解的疾病。
在某些实施方案中,化合物1可以与在表面上表达一种或多种嵌合抗原受体(CAR)的一种或多种免疫细胞(例如,经修饰的免疫细胞)组合使用。通常,CAR包含来自第一蛋白例如,抗原结合蛋白)的细胞外结构域、跨膜结构域和细胞内信号传导结构域。在某些实施方案中,一旦细胞外结构域与靶蛋白(诸如肿瘤相关抗原(TAA)或肿瘤特异性抗原(TSA))结合,便经由细胞内信号传导结构域产生信号,该信号活化免疫细胞,例如以靶向和杀死表达靶蛋白的细胞。
细胞外结构域:CAR的细胞外结构域与感兴趣的抗原结合。在某些实施方案中,CAR的细胞外结构域包含与所述抗原结合的受体或受体的一部分。在某些实施方案中,细胞外结构域包含或为抗体或其抗原结合部分。在具体实施方案中,细胞外结构域包含或为单链Fv(scFv)结构域。单链Fv结构域可以包含例如通过柔性接头连接至VH的VL,其中所述VL和VH来自与所述抗原结合的抗体。
在某些实施方案中,由本文所述多肽的细胞外结构域识别的抗原是肿瘤相关抗原(TAA)或肿瘤特异性抗原(TSA)。在各种具体实施方案中,肿瘤相关抗原或肿瘤特异性抗原是但不限于Her2、前列腺干细胞抗原(PSCA)、甲胎蛋白(AFP)、癌胚抗原(CEA)、癌症抗原-125(CA-125)、CA19-9、钙网膜蛋白、MUC-1、B细胞成熟抗原(BCMA)、上皮膜蛋白(EMA)、上皮肿瘤抗原(ETA)、酪氨酸酶、黑素瘤-24相关抗原(MAGE)、CD19、CD22、CD27、CD30、CD34、CD45、CD70、CD99、CD117、EGFRvIII(表皮生长因子变体III)、间皮素、PAP(前列腺酸性磷酸酶)、prostein、TARP(T细胞受体γ交替阅读框蛋白)、Trp-p8、STEAPI(前列腺的六次跨膜上皮抗原1)、嗜铬粒蛋白、细胞角蛋白、结蛋白、胶质纤维酸性蛋白(GFAP)、巨囊性病液体蛋白(GCDFP-15)、HMB-45抗原、蛋白质melan-A(T淋巴细胞识别的黑素瘤抗原;MART-I)、myo-D1、肌肉特异性肌动蛋白(MSA)、神经丝、神经元特异性烯醇化酶(NSE)、胎盘碱性磷酸酶、突触小泡蛋白、甲状腺球蛋白、甲状腺转录因子-1、丙酮酸激酶同工酶M2型(肿瘤M2-PK)的二聚体形式、异常ras蛋白或异常p53蛋白。在某些其它实施方案中,由CAR的细胞外结构域识别的TAA或TSA是整合素αvβ3(CD61)、泌乳素或Ral-B。
在某些实施方案中,由CAR的细胞外结构域识别的TAA或TSA是癌症/睾丸(CT)抗原,例如,BAGE、CAGE、CTAGE、FATE、GAGE、HCA661、HOM-TES-85、MAGEA、MAGEB、MAGEC、NA88、NY-ES0-1、NY-SAR-35、OY-TES-1、SPANXBI、SPA17、SSX、SYCPI或TPTE。
在某些其它实施方案中,由CAR的细胞外结构域识别的TAA或TSA是碳水化合物或神经节苷脂,例如,fuc-GMI、GM2(癌胚抗原免疫原性-1;OFA-I-1);GD2(OFA-I-2)、GM3、GD3等。
在某些其它实施方案中,由CAR的细胞外结构域识别的TAA或TSA是α-辅肌动蛋白-4、Bage-l、BCR-ABL、Bcr-Abl融合蛋白、β-连环蛋白、CA 125、CA 15-3(CA 27.29\BCAA)、CA195、CA 242、CA-50、CAM43、Casp-8、cdc27、cdk4、cdkn2a、CEA、coa-l、dek-can融合蛋白、EBNA、EF2、爱泼斯坦-巴尔(Epstein Barr)病毒抗原、ETV6-AML1融合蛋白、HLA-A2、HLA-All、hsp70-2、KIAA0205、Mart2,Mum-1、2和3,neo-PAP、肌球蛋白I类、OS-9、pml-RARα融合蛋白、PTPRK、K-ras、N-ras、磷酸丙糖异构酶,Gage 3、4、5、6、7,GnTV、Herv-K-mel、Lage-1、NA-88、NY-Eso-1/Lage-2、SP17、SSX-2、TRP2-Int2、gp100(Pmel17)、酪氨酸酶、TRP-1、TRP-2、MAGE-l、MAGE-3、RAGE、GAGE-l、GAGE-2、p15(58)、RAGE、SCP-1、Hom/Mel-40、PRAME、p53、HRas、HER-2/neu、E2A-PRL、H4-RET、IGH-IGK、MYL-RAR、人乳头瘤病毒(HPV)抗原E6和E7、TSP-180、MAGE-4、MAGE-5、MAGE-6、p185erbB2、p180erbB-3、c-met、nm-23H1、PSA、TAG-72-4、CA 19-9、CA 72-4、CAM 17.1、NuMa、K-ras、13-连环蛋白、Mum-1、p16、TAGE、PSMA、CT7、端粒酶、43-9F、5T4、791Tgp72、13HCG、BCA225、BTAA、CD68\KP1、C0-029、FGF-5、G250、Ga733(EpCAM)、HTgp-175、M344、MA-50、MG7-Ag、MOV18、NB\70K、NY-C0-1、RCAS1、SDCCAG16、TA-90、TAAL6、TAG72、TLP或TPS。
在各种具体实施方案中,肿瘤相关抗原或肿瘤特异性抗原是AML相关肿瘤抗原,如S.Anguille等人,Leukemia(2012),26,2186-2196中所述。
其它肿瘤相关抗原和肿瘤特异性抗原是本领域技术人员已知的。
可用于构建嵌合抗原受体的结合TSA和TAA的受体、抗体和scFv是本领域已知的,编码它们的核苷酸序列也是本领域已知的。
在某些具体实施方案中,由嵌合抗原受体的细胞外结构域识别的抗原是通常不被视为TSA或TAA的抗原,但是它与肿瘤细胞或肿瘤导致的损伤相关联。例如,在某些实施方案中,抗原是例如生长因子、细胞因子或白介素,例如与血管新生或血管发生相关联的生长因子、细胞因子或白介素。此类生长因子、细胞因子或白介素可以包括例如血管内皮生长因子(VEGF)、碱性成纤维细胞生长因子(bFGF)、血小板源性生长因子(PDGF)、肝细胞生长因子(HGF)、胰岛素样生长因子(IGF)或白介素-8(IL-8)。肿瘤也可以在肿瘤局部产生缺氧环境。同样地,在其它具体实施方案中,抗原是缺氧相关因子,例如HIF-1α、HIF-1β、HIF-2α、HIF-2β、HIF-3α或HIF-3β。肿瘤也可能导致对正常组织的局部损伤,引起被称为损伤相关分子模式分子(DAMP;也称为警报素)的分子释放。因此,在某些其它具体实施方案中,抗原是DAMP,例如热休克蛋白、染色质相关蛋白高迁移率族框1(HMGB 1)、S100A8(MRP8,钙粒蛋白A)、S100A9(MRP14,钙粒蛋白B)、血清淀粉样蛋白A(SAA),或者可以是脱氧核糖核酸、三磷酸腺苷、尿酸或硫酸肝素。
跨膜结构域:在某些实施方案中,CAR的细胞外结构域通过接头、间隔区或铰链多肽序列(例如来自CD28的序列或来自CTLA4的序列)连接至所述多肽的跨膜结构域。跨膜结构域可以从任何跨膜蛋白的跨膜结构域获得或来源,并且可以包括这种跨膜结构域的全部或一部分。在具体实施方案中,跨膜结构域可以从例如CD8、CD16、细胞因子受体和白介素受体或生长因子受体等获得或来源。
细胞内信号传导结构域:在某些实施方案中,CAR的细胞内结构域是或包括在T细胞的表面上表达并且触发所述T细胞的活化和/或增殖的蛋白质的细胞内结构域或基序。此类结构域或基序能够传递T淋巴细胞响应于抗原结合到CAR的细胞外部分而活化所必需的初级抗原结合信号。典型地,该结构域或基序包括或为ITAM(基于免疫受体酪氨酸的活化基序)。适用于CAR的含ITAM多肽包括例如ζCD3链(CD3ζ)或其含ITAM部分。在一个具体实施方案中,细胞内结构域是CD3ζ细胞内信号传导结构域。在其它具体实施方案中,细胞内结构域来自淋巴细胞受体链、TCR/CD3复合蛋白、Fe受体亚基或IL-2受体亚基。在某些实施方案中,CAR另外包含一个或多个共刺激结构域或基序,例如,作为所述多肽的细胞内结构域的一部分。所述一个或多个共刺激结构域或基序可以是或可以包含共刺激CD27多肽序列、共刺激CD28多肽序列、共刺激OX40(CD134)多肽序列、共刺激4-1BB(CD137)多肽序列或共刺激诱导型T细胞共刺激(ICOS)多肽序列或其它共刺激结构域或基序中的一者或多者,或它们的任何组合。
CAR也可以包含T细胞存活基序。T细胞存活基序可以是在受到抗原刺激后促进T淋巴细胞存活的任何多肽序列或基序。在某些实施方案中,T细胞存活基序是或来源于CD3、CD28、IL-7受体(IL-7R)的细胞内信号传导结构域、IL-12受体的细胞内信号传导结构域、IL-15受体的细胞内信号传导结构域、IL-21受体的细胞内信号传导结构域,或转化生长因子β(TGFβ)受体的细胞内信号传导结构域。
表达CAR的经修饰的免疫细胞可以是例如T淋巴细胞(T细胞,例如CD4+T细胞或CD8+T细胞)、细胞毒性淋巴细胞(CTL)或自然杀伤(NK)细胞。本文提供的组合物和方法中所使用的T淋巴细胞可以是天然T淋巴细胞或MHC限制性T淋巴细胞。在某些实施方案中,T淋巴细胞是肿瘤浸润性淋巴细胞(TIL)。在某些实施方案中,T淋巴细胞已从肿瘤活检分离,或已从由肿瘤活检分离的T淋巴细胞扩增。在某些其它实施方案中,T细胞已从外周血、脐带血或淋巴分离,或从由外周血、脐带血或淋巴分离的T淋巴细胞扩增。用于产生表达CAR的经修饰的免疫细胞的免疫细胞可以使用本领域公认的常规方法分离,例如血液收集,然后进行血液成分单采术以及任选地抗体介导的细胞分离或分选。
经修饰的免疫细胞优选地对于经修饰的免疫细胞待施用的个体是自体的。在某些其它实施方案中,经修饰的免疫细胞对于经修饰的免疫细胞待施用的个体是同种异体的。当同种异体的T淋巴细胞或NK细胞用于制备经修饰的T淋巴细胞时,优选地选择将减小在个体中产生移植物抗宿主病(GVHD)的可能性的T淋巴细胞或NK细胞。例如,在某些实施方案中,选择病毒特异性T淋巴细胞来制备经修饰的T淋巴细胞;预期此类淋巴细胞结合任何受体抗原,从而被任何受体抗原活化的天然能力将大大降低。在某些实施方案中,受体介导的同种异体T淋巴细胞排斥可以通过将一种或多种免疫抑制剂(例如环孢素、他克莫司(tacrolimus)、西罗莫司(sirolimus)、环磷酰胺等)共施用于宿主来减少。
可以使用CD3和CD28的抗体,例如附接到珠粒的抗体来扩增T淋巴细胞,例如未经修饰的T淋巴细胞,或者表达CD3和CD28、或包含含有CD3ζ信号传导结构域和CD28共刺激结构域的多肽的T淋巴细胞;参见例如美国专利号5,948,893、6,534,055、6,352,694、6,692,964、6,887,466和6,905,681。
经修饰的免疫细胞(例如经修饰的T淋巴细胞)可以任选地包含在需要时能够杀死基本上全部经修饰的免疫细胞的“自杀基因”或“安全开关”。例如,在某些实施方案中,经修饰的T淋巴细胞可以包含HSV胸苷激酶基因(HSV-TK),其在接触更昔洛韦(gancyclovir)时导致经修饰的T淋巴细胞死亡。在另一个实施方案中,经修饰的T淋巴细胞包含诱导型半胱天冬酶,例如诱导型半胱天冬酶9(icaspase9),例如允许使用特定小分子药物进行二聚化的位于半胱天冬酶9与人FK506结合蛋白之间的融合蛋白。参见Straathof等人,Blood 105(11):4247-4254(2005)。
可用于所述方法或组合物的具体的第二活性剂包括但不限于利妥昔单抗、奥利默森 英利昔单抗(remicade)、多西他赛、塞来昔布、美法仑、地塞米松
英利昔单抗(remicade)、多西他赛、塞来昔布、美法仑、地塞米松 类固醇、吉西他滨、顺铂、替莫唑胺(temozolomide)、依托泊苷、环磷酰胺、替莫唑胺(temodar)、卡铂、甲基芐肼、格立得(gliadel)、他莫昔芬、拓扑替康、甲氨蝶呤、
类固醇、吉西他滨、顺铂、替莫唑胺(temozolomide)、依托泊苷、环磷酰胺、替莫唑胺(temodar)、卡铂、甲基芐肼、格立得(gliadel)、他莫昔芬、拓扑替康、甲氨蝶呤、 紫杉酚、克癌易、氟尿嘧啶、甲酰四氢叶酸、伊立替康、希罗达(xeloda)、干扰素α、聚乙二醇化干扰素α(例如,PEG INTRON-A)、卡培他滨、顺铂、噻替哌、氟达拉滨、卡铂、脂质体道诺霉素、Ara-C、多西紫杉醇(doxetaxol)、紫杉醇、长春花碱、IL-2、GM-CSF、达卡巴嗪、长春瑞滨、唑来膦酸、帕米膦酸盐(palmitronate)、甲红霉素(biaxin)、白消安、泼尼松、双磷酸盐、三氧化二砷、长春新碱、阿霉素
紫杉酚、克癌易、氟尿嘧啶、甲酰四氢叶酸、伊立替康、希罗达(xeloda)、干扰素α、聚乙二醇化干扰素α(例如,PEG INTRON-A)、卡培他滨、顺铂、噻替哌、氟达拉滨、卡铂、脂质体道诺霉素、Ara-C、多西紫杉醇(doxetaxol)、紫杉醇、长春花碱、IL-2、GM-CSF、达卡巴嗪、长春瑞滨、唑来膦酸、帕米膦酸盐(palmitronate)、甲红霉素(biaxin)、白消安、泼尼松、双磷酸盐、三氧化二砷、长春新碱、阿霉素 紫杉醇、更昔洛韦、亚德里亚霉素(adriamycin)、雌莫司汀磷酸钠
紫杉醇、更昔洛韦、亚德里亚霉素(adriamycin)、雌莫司汀磷酸钠 舒林酸(sulindac)和依托泊苷。
舒林酸(sulindac)和依托泊苷。
在本文提供的方法的某些实施方案中,如本领域技术人员认为适当的那样,在施用化合物1(包括本文提供的化合物1的制剂)期间或之后不久,可以修改或延缓第二活性剂与化合物1(包括本文提供的化合物1的制剂)的组合使用。在某些实施方案中,正被施用单独的化合物1(包括本文提供的化合物1的制剂)或所述化合物1与其它疗法的组合的对象可以在适当时接受支持性护理,包括止吐药、骨髓生长因子和血小板输注。在一些实施方案中,根据本领域技术人员的判断,可以向正被施用化合物1(包括本文提供的化合物1的制剂)的对象施用作为第二活性剂的生长因子。在一些实施方案中,提供了化合物1(包括本文提供的化合物1的制剂)与红细胞生成素或达贝泊汀(darbepoetin,Aranesp)的组合施用。
在一个方面,本文提供了治疗、预防、管理和/或改善局部晚期或转移性移行细胞膀胱癌的方法,包括将化合物1的制剂与吉西他滨、顺铂、5-氟尿嘧啶、丝裂霉素、甲氨蝶呤、长春花碱、多柔比星、卡铂、噻替派、紫杉醇、多西他赛、阿特珠单抗、阿利库单抗(avelumab)、德瓦鲁单抗、keytruda(派姆单抗(pembrolizumab))和/或纳武单抗一起施用。
在一个方面,本文提供的治疗、预防、管理和/或改善癌症的方法包括将化合物1的制剂与如下的第二活性成分组合施用:替莫唑胺,施用于患有复发性或进行性脑肿瘤或者复发性神经母细胞瘤的小儿患者;塞来昔布、依托泊苷和环磷酰胺,施用于复发性或进行性CNS癌症;替莫唑胺,施用于患有复发性或进行性脑膜瘤、恶性脑膜瘤、血管外皮细胞瘤、多发性脑转移瘤、复发性脑肿瘤或新诊断的多形性成胶质细胞瘤的患者;伊立替康,施用于患有复发性成胶质细胞瘤的患者;卡铂,施用于患有脑干胶质细胞瘤的小儿患者;丙卡巴肼,施用于患有进行性恶性神经胶质瘤的小儿患者;环磷酰胺,施用于患有预后不良的恶性脑肿瘤,新诊断的或复发性多形性成胶质细胞瘤的患者; 施用于高级别复发性恶性神经胶质瘤;替莫唑胺和他莫西芬,施用于间变性星形细胞瘤;或拓扑替康,施用于神经胶质瘤、成胶质细胞瘤、间变性星形细胞瘤或间变性少突神经胶质瘤。
施用于高级别复发性恶性神经胶质瘤;替莫唑胺和他莫西芬,施用于间变性星形细胞瘤;或拓扑替康,施用于神经胶质瘤、成胶质细胞瘤、间变性星形细胞瘤或间变性少突神经胶质瘤。
在一个方面,本文提供的治疗、预防、管理和/或改善转移性乳腺癌的方法包括将化合物1的制剂与下列药剂一起施用于患有转移性乳腺癌的患者:甲氨蝶呤、环磷酰胺、卡培他滨、5-氟尿嘧啶、紫杉烷、替西罗莫司、 (用于注射用混悬液的紫杉醇蛋白质结合颗粒)(与白蛋白结合)、拉帕替尼、赫塞汀、帕米膦酸二钠、甲磺酸艾瑞布林、依维莫司、吉西他滨、帕博西尼、伊沙匹隆、卡德西拉(kadcyla)、帕妥珠单抗、特奥替帕(theotepa)、阿那曲唑、多西他赛、盐酸多柔比星、盐酸表柔比星、托瑞米芬(toremifene)、氟维司群(fulvestrant)、醋酸戈舍瑞林、瑞博西尼(ribociclib)、醋酸甲地孕酮、长春花碱、芳香酶抑制剂(诸如来曲唑、依西美坦)、选择性雌激素调控剂、雌激素受体拮抗剂、蒽环类药物、恩坦辛(emtansine)和/或培西达替尼。
(用于注射用混悬液的紫杉醇蛋白质结合颗粒)(与白蛋白结合)、拉帕替尼、赫塞汀、帕米膦酸二钠、甲磺酸艾瑞布林、依维莫司、吉西他滨、帕博西尼、伊沙匹隆、卡德西拉(kadcyla)、帕妥珠单抗、特奥替帕(theotepa)、阿那曲唑、多西他赛、盐酸多柔比星、盐酸表柔比星、托瑞米芬(toremifene)、氟维司群(fulvestrant)、醋酸戈舍瑞林、瑞博西尼(ribociclib)、醋酸甲地孕酮、长春花碱、芳香酶抑制剂(诸如来曲唑、依西美坦)、选择性雌激素调控剂、雌激素受体拮抗剂、蒽环类药物、恩坦辛(emtansine)和/或培西达替尼。
在一个方面,本文提供的治疗、预防、管理和/或改善神经内分泌肿瘤的方法包括将化合物1的制剂与下列中的至少一者一起施用于患有神经内分泌肿瘤的患者:依维莫司、阿利库单抗、舒尼替尼、多吉美(nexavar)、亚叶酸、奥沙利铂、替莫唑胺、卡培他滨、贝伐珠单抗、多柔比星(阿霉素)、氟尿嘧啶(Adrucil,5-氟尿嘧啶)、链脲霉素(Zanosar)、达卡巴嗪、善得定(sandostatin)、兰瑞肽和/或帕瑞肽。
在一个方面,本文提供的治疗、预防、管理和/或改善转移性乳腺癌的方法包括将化合物1的制剂与下列药剂一起施用于患有复发性或转移性头部或颈部癌症的患者:甲氨蝶呤、吉西他滨、顺铂、西妥昔单抗、5-氟尿嘧啶、博来霉素、多西他赛、卡铂、羟基脲、派姆单抗和/或纳武单抗。
在一个方面,本文提供的治疗、预防、管理和/或改善胰腺癌的方法包括将化合物1的制剂与下列药剂一起施用于患有胰腺癌的患者:吉西他滨、 5-氟尿嘧啶、伊维莫司(afinitor)、伊立替康、丝裂霉素C、舒尼替尼、苹果酸舒尼替尼和/或特罗凯(tarceva)。
5-氟尿嘧啶、伊维莫司(afinitor)、伊立替康、丝裂霉素C、舒尼替尼、苹果酸舒尼替尼和/或特罗凯(tarceva)。
在一个方面,本文提供的治疗、预防、管理和/或改善结肠癌或直肠癌的方法包括将化合物1的制剂与下列药剂一起施用: 阿伐他汀(avastatin)、奥沙利铂、5-氟尿嘧啶、伊立替康、卡培他滨、西妥昔单抗、雷莫芦单抗(ramucirumab)、帕尼单抗、贝伐珠单抗、亚叶酸钙、lonsurf、瑞格拉非尼(regorafenib)、阿柏西普(ziv-aflibercept)、紫杉醇和/或泰索帝。
阿伐他汀(avastatin)、奥沙利铂、5-氟尿嘧啶、伊立替康、卡培他滨、西妥昔单抗、雷莫芦单抗(ramucirumab)、帕尼单抗、贝伐珠单抗、亚叶酸钙、lonsurf、瑞格拉非尼(regorafenib)、阿柏西普(ziv-aflibercept)、紫杉醇和/或泰索帝。
在一个方面,本文提供的治疗、预防、管理和/或改善难治性结肠直肠癌的方法包括将化合物1的制剂与卡培他滨和/或威罗菲尼(vemurafenib)一起施用于患有难治性结肠直肠癌的患者,或者一线疗法失败或在结肠或直肠腺癌中表现较差的患者。
在一个方面,本文提供的治疗、预防、管理和/或改善结肠直肠癌的方法包括将化合物1的制剂与氟尿嘧啶、亚叶酸和/或伊立替康施用于患有结肠直肠癌(包括3期和4期)的患者,或先前已针对转移性结肠直肠癌进行治疗的患者。
在某些实施方案中,将本文提供的化合物1的制剂与卡培他滨、希罗达和/或伊立替康组合施用于患有难治性结肠直肠癌的患者。
在某些实施方案中,将本文提供的化合物1的制剂与卡培他滨和伊立替康一起施用于患有难治性结肠直肠癌的患者,或者患有不可切除的或转移性结肠直肠癌的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与干扰素α或卡培他滨一起施用于患有不可切除的或转移性肝细胞癌的患者;或与顺铂和噻替派一起、或与甲苯磺酸索拉非尼一起施用于患有原发性或转移性肝癌的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与多柔比星、紫杉醇、长春花碱、聚乙二醇化干扰素α和/或重组干扰素α-2b一起施用于患有卡波西氏肉瘤的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与下列中的至少一者一起施用于患有急性骨髓性白血病(包括难治性或复发性或高风险急性骨髓性白血病)的患者:恩西地平、三氧化二砷、氟达拉滨、卡铂、柔红霉素、环磷酰胺、阿糖胞苷、多柔比星、伊达比星、盐酸米托蒽醌、硫鸟嘌呤、长春新碱、米哚妥林和/或拓扑替康。
在一个方面,本文提供的方法包括将化合物1的制剂与下列中的至少一者一起施用于患有不利的核型急性骨髓母细胞性白血病的患者:恩西地平、脂质体柔红霉素、拓扑替康和/或阿糖胞苷。
在一个方面,本文提供的方法包括将化合物1与IDH2抑制剂一起施用于患有白血病的患者,其中所述白血病的特征在于存在IDH2的突变等位基因。示例性的IDH2抑制剂公开于美国专利号9,732,062、9,724,350、9,738,625和9,579,324,和美国公布号2016-0159771和US 2016-0158230A1中。在一个方面,本文提供的方法包括将化合物1与恩西地平一起施用于患有白血病的患者,其中所述白血病的特征在于存在IDH2的突变等位基因。在某些实施方案中,化合物1和IDH2抑制剂的组合增加了患有急性骨髓性白血病的患者中的分化细胞(CD34-/CD38)和成红血细胞,其中急性骨髓性白血病的特征在于存在IDH2R140Q。在某些实施方案中,化合物1和IDH2抑制剂的组合减少了患有急性骨髓性白血病的患者中的祖细胞(CD34+/CD38+)和HSC,其中急性骨髓性白血病的特征在于存在IDH2R140Q。
在一个方面,本文提供的方法包括将化合物1与恩西地平一起施用于患有急性骨髓性白血病的患者,其中急性骨髓性白血病的特征在于存在IDH2的突变等位基因。在一个实施方案中,IDH2的突变等位基因是IDH2R140Q或R172K。
在一个方面,本文提供的方法包括将化合物1的制剂与恩西地平一起施用于患有白血病的患者,其中所述白血病的特征在于存在IDH2的突变等位基因。在一个方面,本文提供的方法包括将化合物1的制剂与恩西地平一起施用于患有急性骨髓性白血病的患者,其中急性骨髓性白血病的特征在于存在IDH2的突变等位基因。在一个实施方案中,IDH2的突变等位基因是IDH2 R140Q或R172K。
在一个方面,本文提供的方法包括将化合物1与6-(6-(三氟甲基)吡啶-2-基)-N2-(2-(三氟甲基)吡啶-4-基)-1,3,5-三嗪-2,4-二胺(化合物2)一起施用于患有白血病的患者,其中所述白血病的特征在于存在IDH2的突变等位基因。在一个方面,本文提供的方法包括将化合物1与化合物2一起施用于患有急性骨髓性白血病的患者,其中急性骨髓性白血病的特征在于存在IDH2的突变等位基因。在一个实施方案中,IDH2的突变等位基因是IDH2R140Q或R172K。
在一个方面,本文提供的方法包括将化合物1的制剂与化合物2一起施用于患有白血病的患者,其中所述白血病的特征在于存在IDH2的突变等位基因。在一个方面,本文提供的方法包括将化合物1的制剂与化合物2一起施用于患有急性骨髓性白血病的患者,其中急性骨髓性白血病的特征在于存在IDH2的突变等位基因。在一个实施方案中,IDH2的突变等位基因是IDH2 R140Q或R172K。
在一个方面,本文提供的方法包括将化合物1的制剂与甲氨蝶呤、盐酸氮芥、阿法替尼双马来酸盐(afatinib dimaleate)、培美曲塞、贝伐珠单抗、卡铂、顺铂、赛立替尼(ceritinib)、克唑替尼(crizotinib)、雷莫芦单抗、派姆单抗、多西他赛、酒石酸长春瑞滨、吉西他滨、 厄洛替尼(erlotinib)、吉非替尼(geftinib)、伊立替康、依维莫司、阿来替尼(alectinib)、布加替尼(brigatinib)、纳武单抗、奥西替尼(osimertinib)、阿特珠单抗、耐昔妥珠单抗(necitumumab)和/或一起施用于患有非小细胞肺癌的患者。
厄洛替尼(erlotinib)、吉非替尼(geftinib)、伊立替康、依维莫司、阿来替尼(alectinib)、布加替尼(brigatinib)、纳武单抗、奥西替尼(osimertinib)、阿特珠单抗、耐昔妥珠单抗(necitumumab)和/或一起施用于患有非小细胞肺癌的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与卡铂和伊立替康一起施用于患有非小细胞肺癌的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与多西紫杉醇一起施用于先前已用卡铂/依托泊苷和放射疗法治疗过的患有非小细胞肺癌的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与卡铂和/或泰索帝一起,或与卡铂、紫杉醇和/或胸部放射疗法组合施用于患有非小细胞肺癌的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与泰索帝一起施用于患有IIIB或IV期非小细胞肺癌的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与奥利默森 甲氨蝶呤、盐酸氮芥、依托泊苷、拓扑替康和/或多柔比星一起施用于患有小细胞肺癌的患者。
甲氨蝶呤、盐酸氮芥、依托泊苷、拓扑替康和/或多柔比星一起施用于患有小细胞肺癌的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与维奈托克、ABT-737(AbbottLaboratories)和/或obatoclax(GX15-070)一起施用于患有淋巴瘤和其它血液癌症的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与第二活性成分一起施用于患有各种类型的淋巴瘤的患者,其中第二活性成分诸如长春花碱或氟达拉滨adcetris、瘤可宁(ambochlorin)、becenum、博来霉素、本妥昔单抗(brentuximab vedotin)、卡莫司汀苯丁酸氮芥(carmustinem chlorambucil)、环磷酰胺、达卡巴嗪、多柔比星、洛莫司汀、甲苯肼、盐酸氮芥、泼尼松、盐酸丙卡巴肼、长春新碱、甲氨蝶呤、奈拉滨(nelarabin)、贝利司他(belinostat)、盐酸苯达莫司汀(bendamustine HCl)、托西莫单抗和碘131托西莫单抗、地尼白介素(denileukin diftitox)、地塞米松、普拉曲沙(pralatrexate)、普乐沙福(prelixafor)、阿妥珠单抗(obinutuzumab)、替伊莫单抗(ibritumomab)、tiuxefan、依鲁替尼(ibritinib)、艾代拉里斯(idelasib)、intron A、罗米地辛(romidepsin)、来那度胺、利妥昔单抗和/或伏立诺他(vorinostat),所述淋巴瘤包括但不限于霍奇金氏淋巴瘤、非霍奇金氏淋巴瘤、皮肤T细胞淋巴瘤、皮肤B细胞淋巴瘤、弥漫性大B细胞淋巴瘤,或者复发性或难治性低级滤泡性淋巴瘤。
在一个方面,本文提供的方法包括将化合物1的制剂与泰索帝、达拉非尼(dabrafenib)、imlygic、伊匹单抗、派姆单抗、纳武单抗、曲美替尼(trametinib)、威罗菲尼、他莫替尼拉赫帕普韦克(talimogene laherparepvec)、IL-2、IFN、GM-CSF和/或达卡巴嗪、阿地白介素、考比替尼(cobimetinib)、Intron 聚乙二醇干扰素α-2b和/或曲美替尼一起施用于患有各种类型或阶段的黑素瘤的患者。
聚乙二醇干扰素α-2b和/或曲美替尼一起施用于患有各种类型或阶段的黑素瘤的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与长春瑞滨或培美曲塞二钠一起施用于患有恶性间皮瘤、或IIIB期非小细胞肺癌伴胸腔植入物或恶性胸腔积液间皮瘤综合征的患者。
在一个方面,本文提供的治疗患有各种类型或阶段的多发性骨髓瘤的患者的方法包括将化合物1的制剂与地塞米松、唑来膦酸、帕米磷酸(palmitronate)、GM-CSF、克拉霉素(biaxin)、长春花碱、美法仑、白消安(busulphan)、环磷酰胺、IFN、泼尼松、双膦酸盐、塞来昔布、三氧化二砷、PEG INTRON-A、长春新碱、becenum、硼替佐米、卡非佐米、多柔比星、帕比司他、来那度胺、泊马度胺、沙利度胺、莫佐比尔(mozobil)、卡莫司汀、达雷木单抗(daratumumab)、埃洛妥珠单抗(elotuzumab)、艾沙佐米柠檬酸盐(ixazomib citrate)、普乐沙福(plerixafor)或它们的组合一起施用。
在某些实施方案中,将本文提供的化合物1的制剂与嵌合抗原受体(CAR)T细胞组合施用于患有各种类型或阶段的多发性骨髓瘤的患者。在某些实施方案中,该组合中的CART细胞靶向B细胞成熟抗原(BCMA),并且在更具体的实施方案中,CAR T细胞是bb2121或bb21217。在一些实施方案中,CAR T细胞是JCARH125。
在某些实施方案中,将本文提供的化合物1的制剂与多柔比星 长春新碱和/或地塞米松
长春新碱和/或地塞米松 组合施用于患有复发性或难治性多发性骨髓瘤的患者。
组合施用于患有复发性或难治性多发性骨髓瘤的患者。
在某些实施方案中,本文提供的方法包括将化合物1的制剂与紫杉酚、卡铂、多柔比星、吉西他滨、顺铂、希罗达、紫杉醇、地塞米松、阿瓦斯汀(avastin)、环磷酰胺、拓扑替康、奥拉帕尼(olaparib)、噻替派、美法仑、甲苯磺酸尼拉帕尼一水合物、卢布拉卡(rubraca)或它们的组合组合施用于患有各种类型或阶段的卵巢癌的患者,所述卵巢癌诸如腹膜癌、乳头状浆液性癌、难治性卵巢癌或复发性卵巢癌。
在某些实施方案中,本文提供的方法包括将化合物1的制剂与希罗达、5FU/LV、吉西他滨、伊立替康加吉西他滨、环磷酰胺、长春新碱、地塞米松、GM-CSF、塞来昔布、泰索帝、更昔洛韦、紫杉醇、阿霉素、多西他赛、雌莫司汀、Emcyt、denderon、zytiga、比卡鲁胺、卡巴他赛(cabazitaxel)、地加瑞克(degarelix)、恩扎鲁胺(enzalutamide)、诺雷德(zoladex)、醋酸亮丙瑞林、盐酸米托蒽醌、泼尼松、西普鲁塞-T(sipuleucel-T)、镭223二氯化物或它们的组合组合施用于患有各种类型或阶段的前列腺癌的患者。
在某些实施方案中,本文提供的方法包括将化合物1的制剂与卡培他滨、IFN、他莫昔芬、IL-2、GM-CSF、 氟他胺、醋酸戈舍瑞林、尼鲁米特或它们的组合组合施用于患有各种类型或阶段的肾细胞癌的患者。
氟他胺、醋酸戈舍瑞林、尼鲁米特或它们的组合组合施用于患有各种类型或阶段的肾细胞癌的患者。
在某些实施方案中,本文提供的方法包括将化合物1的制剂与IFN、放线菌素D、多柔比星、甲磺酸伊马替尼、盐酸帕唑帕尼、曲贝替定(trabectedin)、甲磺酸艾瑞布林、奥拉单抗(olaratumab)、COX-2抑制剂(诸如塞来昔布)和/或舒林酸组合施用于患有各种类型或阶段的妇科癌症、子宫癌症或软组织肉瘤癌症的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与塞来昔布、依托泊苷、环磷酰胺、多西他赛、阿培他滨(apecitabine)、IFN、他莫西芬、IL-2、GM-CSF或它们的组合组合施用于患有各种类型或阶段的固体肿瘤的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与西乐葆(celebrex)、依托泊苷、环磷酰胺、多西他赛、阿培他滨、IFN、他莫昔芬、IL-2、GM-CSF或它们的组合组合施用于患有硬皮病或皮肤血管炎的患者。
在一个方面,本文提供的方法包括将化合物1的制剂与阿扎胞苷、阿糖胞苷、柔红霉素、地西他滨、伊达比星、来那度胺、恩西地平或它们的组合组合施用于患有MDS的患者。
在一个方面,本文提供的方法包括将化合物1与选自下列的一种或多种第二剂组合施用于患有血液学癌症的患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。在一个方面,本文提供的方法包括将化合物1的制剂与选自下列的一种或多种第二剂组合施用于患有血液学癌症的患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个方面,本文提供的方法包括将化合物1与选自下列的一种或多种第二剂组合施用于患有白血病的患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。在某些实施方案中,将本文提供的化合物1的制剂与选自下列的一种或多种第二剂组合施用于患有白血病的患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个方面,本文提供的方法包括将化合物1与选自下列的一种或多种第二剂组合施用于患有AML的患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。在某些实施方案中,将本文提供的化合物1的制剂与选自下列的一种或多种第二剂组合施用于患有AML的患者:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
在一个方面,本文提供的方法包括将化合物1与mTOR抑制剂组合施用于患有白血病的患者。在某些实施方案中,将本文提供的化合物1的制剂与mTOR抑制剂组合施用于患有白血病的患者。在某些实施方案中,mTOR抑制剂选自依维莫司、MLN-0128和AZD8055。在一些实施方案中,mTOR抑制剂是mTOR激酶抑制剂。在某些实施方案中,mTOR激酶抑制剂选自7-(6-(2-羟基丙烷-2-基)吡啶-3-基)-1-((反式)-4-甲氧基环己基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-223)和1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-115)。在某些实施方案中,将化合物1与7-(6-(2-羟基丙烷-2-基)吡啶-3-基)-1-((反式)-4-甲氧基环己基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-223)组合施用于患有白血病的患者。在某些实施方案中,将化合物1与1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-115)组合施用于患有白血病的患者。在某些实施方案中,将化合物1与依维莫司组合施用于患有白血病的患者。在某些实施方案中,将化合物1与MLN-0128组合施用于患有白血病的患者。在某些实施方案中,将化合物1与AZD8055组合施用于患有白血病的患者。
在一个方面,本文提供的方法包括将化合物1与mTOR抑制剂组合施用于患有AML的患者。在某些实施方案中,将本文提供的化合物1的制剂与mTOR抑制剂组合施用于患有AML的患者。在某些实施方案中,mTOR抑制剂选自依维莫司、MLN-0128和AZD8055。在一些实施方案中,mTOR抑制剂是mTOR激酶抑制剂。在某些实施方案中,mTOR激酶抑制剂选自7-(6-(2-羟基丙烷-2-基)吡啶-3-基)-1-((反式)-4-甲氧基环己基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-223)和1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-115)。在某些实施方案中,将化合物1与1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮组合施用于患有AML的患者。在某些实施方案中,将化合物1与依维莫司组合施用于患有AML的患者。在某些实施方案中,先将依维莫司施用于患有AML的患者,之后再施用化合物1。在某些实施方案中,将化合物1与MLN-0128组合施用于患有AML的患者。在某些实施方案中,将化合物1与AZD8055组合施用于患有AML的患者。
在一个方面,本文提供的方法包括将化合物1与JAK抑制剂组合施用于患有MPN的患者。在某些实施方案中,将本文提供的化合物1的制剂与JAK抑制剂组合施用于患有MPN的患者。在一个方面,JAK抑制剂选自JAK1抑制剂、JAK2抑制剂和JAK3抑制剂。在某些实施方案中,JAK抑制剂选自托法替尼、莫美罗替尼、非戈替尼(filgotinib)、地西罗替尼(decernotinib)、巴瑞替尼(barcitinib)、鲁索替尼、菲卓替尼、NS-018和帕克替尼。在某些实施方案中,JAK抑制剂选自托法替尼、莫美罗替尼、鲁索替尼、菲卓替尼、NS-018和帕克替尼。在某些实施方案中,将化合物1与托法替尼组合施用于患有MPN的患者。在某些实施方案中,将化合物1与莫美罗替尼组合施用于患有MPN的患者。在某些实施方案中,将化合物1与非戈替尼组合施用于患有MPN的患者。在某些实施方案中,将化合物1与地西罗替尼组合施用于患有MPN的患者。在某些实施方案中,将化合物1与巴瑞替尼组合施用于患有MPN的患者。在某些实施方案中,将化合物1与鲁索替尼组合施用于患有MPN的患者。在某些实施方案中,将化合物1与菲卓替尼组合施用于患有MPN的患者。在某些实施方案中,将化合物1与NS-018组合施用于患有MPN的患者。在某些实施方案中,将化合物1与帕克替尼组合施用于患有MPN的患者。在某些实施方案中,MPN是IL-3非依赖性的。在某些实施方案中,MPN的特征在于JAK 2突变,例如,JAK2V617F突变。
在一个方面,本文提供的方法包括将化合物1与JAK抑制剂组合施用于患有骨髓纤维化的患者。在某些实施方案中,将本文提供的化合物1的制剂与JAK抑制剂组合施用于患有骨髓纤维化的患者。在一个方面,JAK抑制剂选自JAK1抑制剂、JAK2抑制剂和JAK3抑制剂。在某些实施方案中,JAK抑制剂选自托法替尼、莫美罗替尼、鲁索替尼、菲卓替尼、NS-018和帕克替尼。在某些实施方案中,将化合物1与托法替尼组合施用于患有骨髓纤维化的患者。在某些实施方案中,将化合物1与莫美罗替尼组合施用于患有骨髓纤维化的患者。在某些实施方案中,将化合物1与鲁索替尼组合施用于患有骨髓纤维化的患者。在某些实施方案中,将化合物1与菲卓替尼组合施用于患有骨髓纤维化的患者。在某些实施方案中,将化合物1与NS-018组合施用于患有骨髓纤维化的患者。在某些实施方案中,将化合物1与帕克替尼组合施用于患有骨髓纤维化的患者。在某些实施方案中,骨髓纤维化的特征在于JAK 2突变,例如JAK2V617F突变。在一些实施方案中,骨髓纤维化是原发性骨髓纤维化。在其它实施方案中,骨髓纤维化是继发性骨髓纤维化。在一些这样的实施方案中,继发性骨髓纤维化是真性红细胞增多后骨髓纤维化。在其它实施方案中,继发性骨髓纤维化是自发性血小板增多后骨髓纤维化。
在一个方面,本文提供的方法包括将化合物1与JAK抑制剂组合施用于患有白血病的患者。在某些实施方案中,将本文提供的化合物1的制剂与JAK抑制剂组合施用于患有白血病的患者。在一个方面,JAK抑制剂选自JAK1抑制剂、JAK2抑制剂和JAK3抑制剂。在某些实施方案中,JAK抑制剂选自托法替尼、莫美罗替尼、非戈替尼、地西罗替尼、巴瑞替尼、鲁索替尼、菲卓替尼、NS-018和帕克替尼。在某些实施方案中,JAK抑制剂选自莫美罗替尼、鲁索替尼、菲卓替尼、NS-018和帕克替尼。在某些实施方案中,将化合物1与托法替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与莫美罗替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与非戈替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与地西罗替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与巴瑞替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与鲁索替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与菲卓替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与NS-018组合施用于患有白血病的患者。在某些实施方案中,将化合物1与帕克替尼组合施用于患有白血病的患者。在某些实施方案中,MPN的特征在于JAK 2突变,例如JAK2V617F突变。
在一个方面,本文提供的方法包括将化合物1与JAK抑制剂组合施用于患有AML的患者。在某些实施方案中,将本文提供的化合物1的制剂与JAK抑制剂组合施用于患有AML的患者。在一个方面,JAK抑制剂选自JAK1抑制剂、JAK2抑制剂和JAK3抑制剂。在某些实施方案中,JAK抑制剂选自托法替尼、莫美罗替尼、非戈替尼(filgotinib)、地西罗替尼(decernotinib)、巴瑞替尼(barcitinib)、鲁索替尼、菲卓替尼、NS-018和帕克替尼。在某些实施方案中,JAK抑制剂选自莫美罗替尼、鲁索替尼、菲卓替尼、NS-018和帕克替尼。在某些实施方案中,将化合物1与托法替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与莫美罗替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与非戈替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与地西罗替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与巴瑞替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与鲁索替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与菲卓替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与NS-018组合施用于患有AML的患者。在某些实施方案中,将化合物1与帕克替尼组合施用于患有AML的患者。在某些实施方案中,MPN的特征在于JAK2突变,例如JAK2V617F突变。
在一个方面,本文提供的方法包括将化合物1与FLT3激酶抑制剂组合施用于患有白血病的患者。在某些实施方案中,将本文提供的化合物1的制剂与FLT3激酶抑制剂组合施用于患有白血病的患者。在某些实施方案中,FLT3激酶抑制剂选自奎扎替尼、舒尼替尼、苹果酸舒尼替尼、米哚妥林、培西达替尼、来他替尼(lestaurtinib)、坦度替尼(tandutinib)和克诺拉尼(crenolanib)。在某些实施方案中,将化合物1与奎扎替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与舒尼替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与米哚妥林组合施用于患有白血病的患者。在某些实施方案中,将化合物1与培西达替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与来他替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与坦度替尼组合施用于患有白血病的患者。在某些实施方案中,将化合物1与克诺拉尼组合施用于患有白血病的患者。在某些实施方案中,患者携带FLT3-ITD突变。
在一个方面,本文提供的方法包括将化合物1与FLT3激酶抑制剂组合施用于患有AML的患者。在某些实施方案中,将本文提供的化合物1的制剂与FLT3激酶抑制剂组合施用于患有AML的患者。在某些实施方案中,FLT3激酶抑制剂选自奎扎替尼、舒尼替尼、苹果酸舒尼替尼、米哚妥林、培西达替尼、来他替尼、坦度替尼、奎扎替尼和克诺拉尼。在某些实施方案中,将化合物1与奎扎替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与舒尼替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与米哚妥林组合施用于患有AML的患者。在某些实施方案中,将化合物1与培西达替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与来他替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与坦度替尼组合施用于患有AML的患者。在某些实施方案中,将化合物1与克诺拉尼组合施用于患有AML的患者。在某些实施方案中,患者携带FLT3-ITD突变。
在某些实施方案中,将化合物1与剪接体抑制剂组合施用于患有白血病的患者。在某些实施方案中,将化合物1与剪接体抑制剂组合施用于患有AML的患者。在某些实施方案中,剪接体抑制剂是普拉二烯内酯B、6-脱氧普拉二烯内酯D或H3B-8800。
在一个方面,本文提供的方法包括将化合物1与SMG1激酶抑制剂组合施用于患有白血病的患者。在某些实施方案中,将本文提供的化合物1的制剂与SMG1激酶抑制剂组合施用于患有白血病的患者。在一个方面,本文提供的方法包括将化合物1与SMG1激酶抑制剂组合施用于患有AML的患者。在某些实施方案中,将本文提供的化合物1的制剂与SMG1激酶抑制剂组合施用于患有AML的患者。在某些实施方案中,SMG1抑制剂是1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮、氯-N,N-二乙基-5-((4-(2-(4-(3-甲基脲基)苯基)吡啶-4-基)嘧啶-2-基)氨基)苯磺酰胺(化合物Ii),或A.Gopalsamy等人,Bioorg.Med Chem Lett.2012,22:6636-66412中公开的化合物(例如,氯-N,N-二乙基-5-((4-(2-(4-(3-甲基脲基)苯基)吡啶-4-基)嘧啶-2-基)氨基)苯磺酰胺。
在一个方面,本文提供的方法包括将化合物1与BCL2抑制剂组合施用于患有白血病的患者。在某些实施方案中,将本文提供的化合物1的制剂与BCL2抑制剂组合施用于患有白血病的患者。在某些实施方案中,将化合物1与BCL2抑制剂组合施用于患有AML的患者。在某些实施方案中,将本文提供的化合物1的制剂与BCL2抑制剂(例如,维奈托克或纳维托克斯(navitoclax))组合施用于患有AML的患者。在某些实施方案中,BCL2抑制剂是维奈托克。
在一个实施方案中,本文提供了用于治疗对用BCL2抑制剂治疗具有耐药性的AML的方法,包括施用化合物1。在一个实施方案中,本文提供了用于治疗已经获得了对维奈托克治疗的耐药性的AML的方法,包括施用化合物1。在一个实施方案中,本文提供了用于治疗已经获得了对维奈托克治疗的耐药性的AML的方法,包括施用化合物1和BCL2抑制剂的组合。在一个实施方案中,本文提供了用于治疗已经获得了对维奈托克治疗的耐药性的AML的方法,包括施用化合物1和维奈托克的组合。
在一个方面,本文提供的方法包括将化合物1与拓扑异构酶抑制剂组合施用于患有白血病的患者。在某些实施方案中,将本文提供的化合物1的制剂与拓扑异构酶抑制剂组合施用于患有白血病的患者。在某些实施方案中,将化合物1与拓扑异构酶抑制剂组合施用于患有AML的患者。在某些实施方案中,将本文提供的化合物1的制剂与拓扑异构酶抑制剂(例如,伊立替康、拓扑替康、喜树碱、片螺素D、依托泊苷、替尼泊苷、多柔比星、柔红霉素、米托蒽醌、安吖啶、玫瑰树碱、金精三羧酸或HU-331)组合施用于患有AML的患者。在某些实施方案中,拓扑异构酶抑制剂是拓扑替康。
在某些实施方案中,将化合物1与BET抑制剂组合施用于患有白血病的患者。在某些实施方案中,将化合物1与BET抑制剂组合施用于患有AML的患者。在某些实施方案中,该BET抑制剂选自GSK525762A、OTX015、BMS-986158、TEN-010、CPI-0610、INCB54329、BAY1238097、FT-1101、C90010、ABBV-075、BI 894999、GS-5829、GSK1210151A(I-BET-151)、CPI-203、RVX 208、XD46、MS436、PFI-1、RVX2135、ZEN3365、XD14、ARV-771、MZ-1、PLX5117、4-[2-(环丙基甲氧基)-5-(甲磺酰基)苯基]-2-甲基异喹啉-1(2H)-酮(化合物A)、EP11313和EP11336。
在某些实施方案中,将化合物1与LSD1抑制剂组合施用于患有白血病的患者。在某些实施方案中,将化合物1与LSD1抑制剂组合施用于患有AML的患者。在某些实施方案中,LSD1抑制剂选自ORY-1001、ORY-2001、INCB-59872、IMG-7289、TAK 418、GSK-2879552和4-[2-(4-氨基-哌啶-1-基)-5-(3-氟-4-甲氧基-苯基)-1-甲基-6-氧代-1,6-二氢嘧啶-4-基]-2-氟-苄腈或其盐(例如苯磺酸盐,化合物B)。
在一个方面,本文提供的方法包括将化合物1与雷公藤甲素、瑞他霉素、阿螺旋霉素、7-(6-(2-羟基丙烷-2-基)吡啶-3-基)-1-((反式)-4-甲氧基环己基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-223)、1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-115)、雷帕霉素、MLN-0128、依维莫司、AZD8055、普拉二烯内酯B、拓扑替康、硫鸟嘌呤、米托蒽醌、依托泊苷、地西他滨、柔红霉素、克罗拉滨、克拉屈滨、6-巯基嘌呤、氯-N,N-二乙基-5-((4-(2-(4-(3-甲基脲基)苯基)吡啶-4-基)嘧啶-2-基)氨基)苯磺酰胺(化合物Ii)、菲卓替尼、舒尼替尼、培西达替尼、米哚妥林、来他替尼、莫美罗替尼、奎扎替尼和克诺拉尼组合施用于患有白血病的患者。
在一个方面,本文提供的方法包括将化合物1与雷公藤甲素、瑞他霉素、阿螺旋霉素、7-(6-(2-羟基丙烷-2-基)吡啶-3-基)-1-((反式)-4-甲氧基环己基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-223)、1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-115)、雷帕霉素、MLN-0128、依维莫司、AZD8055、普拉二烯内酯B、拓扑替康、硫鸟嘌呤、米托蒽醌、依托泊苷、地西他滨、柔红霉素、克罗拉滨、克拉屈滨、6-巯基嘌呤、氯-N,N-二乙基-5-((4-(2-(4-(3-甲基脲基)苯基)吡啶-4-基)嘧啶-2-基)氨基)苯磺酰胺(化合物Ii)、菲卓替尼、舒尼替尼、培西达替尼、米哚妥林、来他替尼、莫美罗替尼、奎扎替尼和克诺拉尼组合施用于患有AML的患者。
在一个方面,本文提供的方法包括将化合物1与mTOR抑制剂组合施用于患有癌症的患者,其中所述癌症选自乳腺癌、肾癌、胰腺癌、胃肠道癌、肺癌、神经内分泌肿瘤(NET)和肾细胞癌(RCC)。在某些实施方案中,将本文提供的化合物1的制剂与拓扑异构酶抑制剂组合施用于患有癌症的患者。在某些实施方案中,将本文提供的化合物1的制剂与mTOR抑制剂组合施用于癌症患者,其中所述癌症选自乳腺癌、肾癌、胰腺癌、胃肠道癌、肺癌、神经内分泌肿瘤(NET)和肾细胞癌。在某些实施方案中,mTOR抑制剂选自依维莫司、MLN-0128和AZD8055。在一些实施方案中,mTOR抑制剂是mTOR激酶抑制剂。在某些实施方案中,mTOR激酶抑制剂选自7-(6-(2-羟基丙烷-2-基)吡啶-3-基)-1-((反式)-4-甲氧基环己基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-223)和1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-115)。在一个实施方案中,mTOR激酶抑制剂是7-(6-(2-羟基丙烷-2-基)吡啶-3-基)-1-((反式)-4-甲氧基环己基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-223)。在一个实施方案中,mTOR激酶抑制剂是1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-115)。在一个实施方案中,mTOR抑制剂是依维莫司。在一个实施方案中,mTOR抑制剂是替西罗莫司。在一个实施方案中,mTOR抑制剂是MLN-0128。在一个实施方案中,mTOR抑制剂是AZD8055。
在某些实施方案中,将化合物1与依维莫司组合施用于乳腺癌患者。在某些实施方案中,将本文提供的化合物1的制剂与依维莫司组合施用于乳腺癌患者。
在某些实施方案中,将化合物1与依维莫司组合施用于肾癌患者。在某些实施方案中,将本文提供的化合物1的制剂与依维莫司组合施用于肾癌患者。
在某些实施方案中,将化合物1与依维莫司组合施用于胰腺癌患者。在某些实施方案中,将本文提供的化合物1的制剂与依维莫司组合施用于胰腺癌患者。
在某些实施方案中,将化合物1与依维莫司组合施用于胃肠道癌患者。在某些实施方案中,将本文提供的化合物1的制剂与依维莫司组合施用于胃肠道癌患者。
在某些实施方案中,将化合物1与依维莫司组合施用于肺癌患者。在某些实施方案中,将本文提供的化合物1的制剂与依维莫司组合施用于肺癌患者。
在某些实施方案中,将化合物1与依维莫司组合施用于神经内分泌肿瘤患者。在某些实施方案中,将本文提供的化合物1的制剂与依维莫司组合施用于神经内分泌肿瘤患者。
在某些实施方案中,将化合物1与依维莫司组合施用于肾细胞癌患者。在某些实施方案中,将本文提供的化合物1的制剂与依维莫司组合施用于肾细胞癌患者。
本文还涵盖增加可以安全且有效地施用于患者的抗癌药物或剂的剂量的方法,其包括将化合物1(例如本文提供的化合物1的制剂)与第二抗癌药物组合施用于患者(例如,人)可以从该方法受益的患者是那些可能遭受与用于治疗皮肤、皮下组织、淋巴结、脑、肺、肝、骨、肠、结肠、心脏、胰腺、肾上腺、肾、前列腺、乳腺、结肠直肠或它们的组合的具体癌症的抗癌药物相关联的副作用的患者。施用化合物1(例如本文提供的化合物1的制剂)减轻或减少了其严重程度原本会限制抗癌药物的量的副作用。
本文还涵盖减少可以安全且有效地施用于患者的抗癌药物或剂的剂量的方法,其包括将化合物1(例如本文提供的化合物1的制剂)与第二抗癌药物组合施用于患者(例如,人)可以从该方法受益的患者是那些可能遭受与用于治疗皮肤、皮下组织、淋巴结、脑、肺、肝、骨、肠、结肠、心脏、胰腺、肾上腺、肾、前列腺、乳腺、结肠直肠或它们的组合的具体癌症的抗癌药物相关联的副作用的患者。施用化合物1(例如本文提供的化合物1的制剂)增强了抗癌药物的活性,这在维持功效的同时允许减少抗癌药物的剂量,这进而可以减轻或减少其严重程度限制抗癌药物的量的副作用。
在一个实施方案中,化合物1在与向患者施用抗癌药物相关联的副作用发生之前、期间或之后,以在约0.1至约20mg、约1至约15mg、约1至约10mg或约1至约15mg范围内的量每日施用。在某些实施方案中,将化合物1与具体剂诸如肝素、阿司匹林、香豆素或G-CSF组合施用,以避免与抗癌药物相关联的副作用,诸如但不限于中性粒细胞减少或血小板减少。
在一个实施方案中,将化合物1(例如本文提供的化合物1的制剂)与附加的活性成分(包括但不限于抗癌药物、抗炎药、抗组胺药、抗生素和类固醇)组合施用于患有与不期望的血管生成相关联或以不期望的血管生成为特征的疾病和疾患的患者。
在另一个实施方案中,本文涵盖治疗、预防和/或管理癌症的方法,其包括将化合物1(例如,本文提供的化合物1的制剂)与至少一种抗癌疗法(包括但不限于手术、免疫疗法、生物疗法、放射疗法,或目前用于治疗、预防和/或管理癌症的其它基于非药物的疗法)结合(例如,在其之前、期间或之后)施用。本文提供的化合物和其它抗癌疗法的组合使用可以提供在某些患者中出乎意料地有效的独特治疗方案。不受理论限制,据信当与至少一种抗癌疗法同时给予时,化合物1可以提供累加或协同的作用。
如本文其它地方所讨论的,本文涵盖减少、治疗和/或预防与其它抗癌疗法(包括但不限于手术、化学疗法、放射疗法、激素疗法、生物疗法和免疫疗法)相关联的副作用或不期望的作用的方法。化合物1(例如本文提供的化合物1的制剂)和其它活性成分可以在与其它抗癌疗法相关联的副作用发生之前、期间或之后施用于患者。
在某些实施方案中,本文提供的方法包括将钙、骨化三醇或维生素D补充剂中的一者或多者与化合物1一起施用。在某些实施方案中,本文提供的方法包括在用化合物1治疗之前施用钙、骨化三醇和维生素D补充剂。在某些实施方案中,本文提供的方法包括在每个周期中施用第一剂量的化合物1之前施用钙、骨化三醇和维生素D补充剂。在某些实施方案中,本文提供的方法包括在用化合物1治疗之前施用钙、骨化三醇和维生素D补充剂至少达到3天。在某些实施方案中,本文提供的方法包括在每个周期中施用第一剂量的化合物1之前施用钙、骨化三醇和维生素D补充剂。在某些实施方案中,本文提供的方法包括在每个周期中施用第一剂量的化合物1之前施用钙、骨化三醇和维生素D补充剂至少达到3天。在某些实施方案中,本文提供的方法包括在每个周期中施用第一剂量的化合物1之前施用钙、骨化三醇和维生素D补充剂,并且在每个周期中在施用最后剂量的化合物1之后继续施用。在某些实施方案中,本文提供的方法包括在每个周期中施用第一剂量的化合物1之前施用钙、骨化三醇和维生素D补充剂至少达到3天,并且在每个周期中施用最后剂量的化合物1之后继续施用至少达到3天(例如,当在第1至5天施用化合物1时,至少达到第8天)。在一个实施方案中,本文提供的方法包括在每个周期的第1天施用之前施用钙、骨化三醇和维生素D补充剂至少达到3天,并且在每个周期中施用最后剂量的化合物1之后继续施用直到≥3天(例如,当在第1至5天施用化合物1时,≥第8天;当在第1至3天和第8至10天施用化合物1时,≥第13天)。
在某些实施方案中,施用钙补充剂,以便每天递送至少1200mg元素钙,这些元素钙以分剂量给予。在某些实施方案中,钙补充剂以500mg剂量的碳酸钙施用,每天口服(PO)施用三次。
在某些实施方案中,施用骨化三醇补充剂,以便每日一次递送0.25μg骨化三醇(PO)。
在某些实施方案中,施用维生素D补充剂,以便每日一次递送约500IU至约50,000IU维生素D。在某些实施方案中,施用维生素D补充剂,以便每日一次递送约1000IU维生素D。在某些实施方案中,施用维生素D补充剂,以便每周递送约50,000IU维生素D。在某些实施方案中,施用维生素D补充剂,以便每日一次递送约1000IU维生素D2或D3。在某些实施方案中,施用维生素D补充剂,以便每日一次递送约500IU维生素D。在某些实施方案中,施用维生素D补充剂,以便每周递送约50,000IU维生素D。在某些实施方案中,施用维生素D补充剂,以便每周递送约20,000IU维生素D。在某些实施方案中,施用维生素D补充剂,以便每日一次递送约1000IU维生素D2或D3。在某些实施方案中,施用维生素D补充剂,以便每周递送约50,000IU维生素D2或D3。在某些实施方案中,施用维生素D补充剂,以便每周递送约20,000IU维生素D2或D3。
在某些实施方案中,将本文提供的化合物1的制剂和多西紫杉醇施用于先前用卡铂/VP 16和放射疗法治疗过的患有非小细胞肺癌的患者。
与移植疗法一起使用
化合物1(例如本文提供的化合物1的制剂)可以用于降低移植物抗宿主病(GVHD)的风险。因此,本文涵盖治疗、预防和/或管理癌症的方法,其包括将化合物1(例如本文提供的化合物1的制剂)与移植疗法结合施用。
如本领域的普通技术人员所知道的,癌症的治疗通常基于该疾病的阶段和机制。例如,随着在癌症的某些阶段中不可避免地产生白血病转化,则可能需要移植外周血干细胞、造血干细胞制剂或骨髓。化合物1(例如本文提供的化合物1的制剂)与移植疗法的组合使用提供了独特且出乎意料的协同作用。特别地,本文提供的化合物1的制剂表现出免疫调节活性,当与移植疗法同时给予患有癌症的患者时,其可以提供累加或协同的作用。
化合物1(例如本文提供的化合物1的制剂)可以与移植疗法组合起效,从而减少与移植的侵犯性程序相关联的并发症并降低GVHD风险。本文涵盖治疗、预防和/或管理癌症的方法,其包括在移植脐带血、胎盘血、外周血干细胞、造血干细胞制剂或骨髓之前、期间或之后将本文提供的化合物1的制剂施用于患者(例如,人)。适合在本文提供的方法中使用的干细胞的一些实例公开于美国专利号7,498,171中,该专利的公开内容全文以引用方式并入本文。
在一个实施方案中,在移植之前、期间或之后将化合物1(例如本文提供的化合物1的制剂)施用于患有急性骨髓性白血病的患者。
在一个实施方案中,在移植自体外周血祖细胞之前、期间或之后将化合物1(例如本文提供的化合物1的制剂)施用于患有多发性骨髓瘤的患者。
在一个实施方案中,在移植自体外周血祖细胞之前、期间或之后将化合物1(例如本文提供的化合物1的制剂)施用于患有NHL(例如,DLBCL)的患者。
周期性疗法
在某些实施方案中,将化合物1(例如本文提供的化合物1的制剂)周期性地施用于与经治疗的癌症无关的患者。周期性疗法涉及施用活性剂一段时间,然后停用一段时间,并重复该相继施用。周期性疗法可以减少对一种或多种疗法产生耐药性、避免或减少其中一种疗法的副作用,并且/或者改善治疗的功效。
在某些实施方案中,化合物1(例如本文提供的化合物1的制剂)在四至六周的周期(其中停药期为约一周或两周)内以单剂量或分剂量每日施用。在某些实施方案中,化合物1(例如本文提供的化合物1的制剂)以单剂量或分剂量每日施用,持续28天周期的连续1至10天,然后是停用期,其中该28天周期的其余时间不施用。周期性方法还允许增加给药周期的频率、数目和长度。因此,在某些实施方案中,本文涵盖的是化合物1(例如本文提供的化合物1的制剂)的施用周期数比单独施用时典型的周期数更多。在某些实施方案中,化合物1(例如本文提供的化合物1的制剂)以更大的周期数施用,这典型地将在还未施用第二活性成分的患者中导致剂量限制毒性。
在一个实施方案中,化合物1(例如本文提供的化合物1的制剂)每日并且连续施用三或四周,以便施用约0.1至约20mg/d剂量的化合物1,然后中断一或两周。
在另一个实施方案中,静脉内施用化合物1(例如本文提供的化合物1的制剂)并且口服施用第二活性成分,其中在四至六周的周期期间,施用化合物1(例如本文提供的化合物1的制剂)在第二活性成分之前30至60分钟发生。在某些实施方案中,化合物1(例如本文提供的化合物1的制剂)与第二活性成分的组合通过每个周期约90分钟的静脉内输注来施用。在某些实施方案中,一个周期包括每日施用约0.1至约150mg/天的化合物1(例如本文提供的化合物1的制剂),以及约50至约200mg/m2/天的第二活性成分,持续三至四周,然后停用一或两周。在某些实施方案中,在其期间向患者施用组合治疗的周期的数目在约1至约24个周期、约2至约16个周期、或约4至约3个周期的范围内。
在一个实施方案中,本文提供的周期性疗法包括在包括最长5天的施用期和随后的停用期的治疗周期中施用化合物1(例如本文提供的化合物1的制剂)。在一个实施方案中,治疗周期包括5天的施用期,然后是停用期。在一个实施方案中,治疗周期包括至多10天的施用期,然后是停用期。在一个实施方案中,停用期为约10天至至多约40天。在一个实施方案中,治疗周期包括至多10天的施用期,然后是约10天至至多约40天的停用期。在一个实施方案中,治疗周期包括至多10天的施用期,然后是约23天至至多约37天的停用期。在一个实施方案中,停用期为约23天至至多约37天。在一个实施方案中,停用期为23天。在一个实施方案中,治疗周期包括至多10天的施用期,然后是23天的停用期。在一个实施方案中,停用期为37天。在一个实施方案中,治疗周期包括至多10天的施用期,然后是37天的停用期。
在一个实施方案中,治疗周期包括在28天周期的第1至5天施用化合物1,例如,本文提供的化合物1的制剂。在另一个实施方案中,治疗周期包括在28天周期的第1至10天施用化合物1,例如,本文提供的化合物1的制剂。在一个实施方案中,治疗周期包括在42天周期的第1至5天施用。在另一个实施方案中,治疗周期包括在42天周期的第1至10天施用。在另一个实施方案中,治疗周期包括在28天周期的第1至5天和第15至19天施用。在另一个实施方案中,治疗周期包括在28天周期的第1至3天和第8至10天施用。
在一个实施方案中,治疗周期包括在28天周期的第1至21天施用化合物1,例如,本文提供的化合物1的制剂。在另一个实施方案中,治疗周期包括在7天周期的第1至5天施用。在另一个实施方案中,治疗周期包括在7天周期的第1至7天施用。
本文所述的任何治疗周期可以重复至少2个、3个、4个、5个、6个、7个、8个或更多个周期。在某些情况下,如本文所述的治疗周期包括1个至约24个周期、约2个至约16个周期,或约2个至约4个周期。在某些情况下,如本文所述的治疗周期包括1个至约4个周期。在某些实施方案中,周期1至周期4均为28天周期。在某些实施方案中,周期1为42天周期,周期2至周期4为28天周期。在一些实施方案中,将化合物1(例如,本文提供的化合物1的制剂)施用1至13个28天周期(例如约1年)。在某些情况下,周期性疗法的周期数无限制,并且疗法持续直到疾病进展。在某些情况下,周期可以包括本文所述的施用期和/或停用期的不同持续时间。
在一个实施方案中,治疗周期包括以约0.3mg/天、0.6mg/天、1.2mg/天、1.8mg/天、2.4mg/天、3.6mg/天、5.4mg/天、7.2mg/天、8.1mg/天、9.0mg/天、10.0mg/天、10.8mg/天或12.2mg/天的剂量施用化合物1,每天施用一次。在一个实施方案中,治疗周期包括以约0.3mg/天、0.6mg/天、1.2mg/天、1.8mg/天、2.4mg/天、3.6mg/天、5.4mg/天、7.2mg/天、8.1mg/天、9.0mg/天、10.0mg/天、10.8mg/天、12.2mg/天或20mg/天的剂量施用化合物1,每天施用一次。在一个实施方案中,治疗周期包括以约0.6mg/天、1.2mg/天、1.8mg/天、2.4mg/天或3.6mg/天的剂量施用化合物1,每天施用一次。在一些这样的实施方案中,治疗周期包括在28天周期的第1至3天以约0.6mg、1.2mg、1.8mg、2.4mg或3.6mg的剂量施用化合物1。在另一个实施方案中,治疗周期包括在28天周期的第1至5天和第15至19天以约0.6mg、1.2mg、1.8mg、2.4mg或3.6mg的剂量施用化合物1。在其它实施方案中,治疗周期包括在28天周期的第1至5天和第15至19天以约0.6mg、1.2mg、1.8mg、2.4mg、3.6mg、5.4mg/天、7.2mg/天、8.1mg/天、9.0mg/天或10.0mg/天的剂量施用化合物1。
对于一个治疗周期的所有施用期,化合物1(例如本文提供的化合物1的制剂)可以以相同的量施用。替代性地,在一个实施方案中,该化合物在施用期中以不同的剂量施用。
在一个实施方案中,在一个周期中将本文提供的化合物1的制剂施用于对象,其中该周期包括在28天周期中施用该制剂至少5天。在一个实施方案中,在一个周期中将本文提供的化合物1的制剂施用于对象,其中该周期包括在28天周期的第1至5天施用该制剂。在一个实施方案中,在28天周期的第1至5天施用该制剂,以便以约0.1mg至约20mg的剂量递送化合物1。在一个实施方案中,在28天周期的第1至5天施用该制剂,以便以约0.5mg至约5mg的剂量递送化合物1。在一个实施方案中,在28天周期的第1至5天施用该制剂,以便以约0.5mg至约10mg的剂量递送化合物1。在一个实施方案中,在一个周期中将本文提供的化合物1的制剂施用于对象,其中该周期包括在28天周期的第1至5天和第15至19天施用该制剂。在一个实施方案中,在28天周期的第1至5天和第15至19天施用该制剂,以便以约0.1mg至约20mg的剂量递送化合物1。在一个实施方案中,在28天周期的第1至5天和第15至19天施用该制剂,以便以约0.5mg至约5mg的剂量递送化合物1。在一个实施方案中,在28天周期的第1至5天和第15至19天施用该制剂,以便以约0.5mg至约10mg的剂量递送化合物1。
在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗AML的方法,其中该周期包括在28天周期中施用该制剂至少5天,以便以约0.1mg至约20mg的剂量递送化合物1。在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗AML的方法,其中该周期包括在28天周期的第1至5天施用该制剂,以便以约0.1mg至约20mg的剂量递送化合物1。在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗AML的方法,其中该周期包括在28天周期的第1至5天施用该制剂,以便以约0.1mg至约5mg的剂量递送化合物1。在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗AML的方法,其中该周期包括在28天周期的第1至5天施用该制剂,以便以约0.5mg至约5mg的剂量递送化合物1。在另一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗AML的方法,其中该周期包括在28天周期的第1至5天和第15至19天施用该制剂,以便以约0.1mg至约20mg的剂量递送化合物1。在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗AML的方法,其中该周期包括在28天周期的第1至5天和第15至19天施用该制剂,以便以约0.1mg至约5mg的剂量递送化合物1。在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗AML的方法,其中该周期包括在28天周期的第1至5天和第15至19天施用该制剂,以便以约0.5mg至约5mg的剂量递送化合物1。
在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗MDS的方法,其中该周期包括在28天周期中施用该制剂至少5天,以便以约0.1mg至约20mg的剂量递送化合物1。在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗MDS的方法,其中该周期包括在28天周期的第1至5天施用该制剂,以便以约0.1mg至约20mg的剂量递送化合物1。在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗MDS的方法,其中该周期包括在28天周期的第1至5天施用该制剂,以便以约0.1mg至约5mg的剂量递送化合物1。在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗MDS的方法,其中该周期包括在28天周期的第1至5天施用该制剂,以便以约0.5mg至约5mg的剂量递送化合物1。在另一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗MDS的方法,其中该周期包括在28天周期的第1至5天和第15至19天施用该制剂,以便以约0.1mg至约20mg的剂量递送化合物1。在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗MDS的方法,其中该周期包括在28天周期的第1至5天和第15至19天施用该制剂,以便以约0.1mg至约5mg的剂量递送化合物1。在一个实施方案中,本文提供了通过在一个周期中向对象施用本文提供的化合物1的制剂来治疗MDS的方法,其中该周期包括在28天周期的第1至5天和第15至19天施用该制剂,以便以约0.5mg至约5mg的剂量递送化合物1。
患者群体
在本文提供的方法的某些实施方案中,对象是动物,优选地是哺乳动物,更优选地是非人灵长类动物。在特定实施方案中,对象是人。对象可以是男性或女性对象。
对于本文提供的方法特别有用的对象包括人癌症患者,例如,已经被诊断为患有白血病的患者,白血病包括急性骨髓性白血病、急性淋巴细胞性白血病、慢性髓源性白血病和慢性髓源性白血病。在某些实施方案中,对象未被诊断为患有急性早幼粒细胞性白血病。
在一些实施方案中,对象具有高于正常的母细胞群体。在一些实施方案中,对象具有至少为10%的母细胞群体。在一些实施方案中,对象具有介于10%与15%之间的母细胞群体。在一些实施方案中,对象具有至少为15%的母细胞群体。在一些实施方案中,对象具有介于15%与20%之间的母细胞群体。在一些实施方案中,对象具有至少为20%的母细胞群体。在一些实施方案中,对象具有为约10%至15%、约15%至20%或约20%至25%的母细胞群体。在其它实施方案中,对象具有小于10%的母细胞群体。在本文所述方法的上下文中,具有小于10%的母细胞群体的有用对象包括根据本领域技术人员的判断,出于任何原因需要用本文提供的化合物单独治疗或与第二活性剂组合治疗的那些对象。
在一些实施方案中,基于白血病对象的美国东部肿瘤协作组(ECOG)行为状态评分对对象进行治疗。ECOG行为状态可以按0至5的等级评分,其中0表示无症状;1表示有症状但完全能行走;2表示有症状且白天在床上的时间<50%;3表示有症状且在床上的时间>50%,但不局限于床上;4表示局限于床上;5表示死亡。在一些实施方案中,对象的ECOG行为状态评分为0或1。在一些实施方案中,对象的ECOG行为状态评分为0。在一些实施方案中,对象的ECOG行为状态评分为1。在其它实施方案中,对象的ECOG行为状态评分为2。
在某些实施方案中,本文提供的方法涵盖治疗先前未针对白血病进行过治疗的对象。在一些实施方案中,对象未经历过同种异体骨髓移植。在一些实施方案中,对象未经历过干细胞移植。在一些实施方案中,对象未接受过羟基脲治疗。在一些实施方案中,对象未用任何用于白血病的研究产品治疗过。在一些实施方案中,对象未用全身性糖皮质激素治疗过。
在其它实施方案中,所述方法涵盖治疗先前已针对白血病进行治疗或目前正在针对白血病进行治疗的对象。例如,该对象先前可能已使用针对白血病的标准治疗方案治疗过或目前正在使用针对白血病的标准治疗方案治疗。该对象可能已使用本领域技术人员已知的任何标准白血病标准方案治疗过。在某些实施方案中,该对象先前已用至少一种诱导/再诱导或巩固AML方案治疗过。在一些实施方案中,对象已经历了作为巩固方案的一部分的自体骨髓移植或干细胞移植。在一些实施方案中,骨髓或干细胞移植发生在根据本文提供的方法治疗之前至少3个月。在一些实施方案中,对象已经历了羟基脲治疗。在一些实施方案中,羟基脲治疗不迟于根据本文提供的方法治疗之前24小时发生。在一些实施方案中,对象已经历了用阿糖胞苷(Ara-C)进行的先前的诱导或巩固疗法。在一些实施方案中,对象已经历了用全身性糖皮质激素治疗。在一些实施方案中,糖皮质激素治疗不迟于根据本文所述的方法治疗之前24小时发生。在其它实施方案中,所述方法涵盖治疗先前已针对癌症进行治疗但对标准疗法无响应的对象。
还涵盖治疗患有复发性或难治性白血病的对象的方法。在一些实施方案中,该对象已经被诊断为患有复发性或难治性AML亚型,如世界卫生组织(WHO)所定义。复发性或难治性疾病可能是新发AML或继发性AML,例如,疗法相关AML(t-AML)。
在一些实施方案中,本文提供的方法用于治疗特征在于存在IDH2的突变等位基因的白血病。在一个实施方案中,IDH2的突变等位基因是IDH2R140Q或R172K。
在一些实施方案中,本文提供的方法用于治疗特征在于存在IDH2的突变等位基因的AML。在一个实施方案中,IDH2的突变等位基因是IDH2R140Q或R172K。
因此,用本文提供的化合物治疗可以为对其它治疗方法无响应的患者提供替代方案。在一些实施方案中,此类其它治疗方法涵盖用 (甲磺酸伊马替尼)治疗。在一些实施方案中,本文提供了治疗费城染色体阳性慢性髓源性白血病(Ph+CML)的方法。在一些实施方案中,本文提供了治疗
(甲磺酸伊马替尼)治疗。在一些实施方案中,本文提供了治疗费城染色体阳性慢性髓源性白血病(Ph+CML)的方法。在一些实施方案中,本文提供了治疗 (甲磺酸伊马替尼)耐药性费城染色体阳性慢性髓源性白血病(Ph+CML)的方法。
(甲磺酸伊马替尼)耐药性费城染色体阳性慢性髓源性白血病(Ph+CML)的方法。
在一些实施方案中,本文提供的方法用于治疗抗药性白血病,诸如CML。因此,用本文提供的化合物治疗可以为对其它治疗方法无响应的患者提供替代方案。在一些实施方案中,此类其它治疗方法涵盖用 (甲磺酸伊马替尼)治疗。在一些实施方案中,本文提供了治疗Ph+CML的方法。在一些实施方案中,本文提供了治疗
(甲磺酸伊马替尼)治疗。在一些实施方案中,本文提供了治疗Ph+CML的方法。在一些实施方案中,本文提供了治疗 (甲磺酸伊马替尼)耐药性Ph+CML的方法。
(甲磺酸伊马替尼)耐药性Ph+CML的方法。
尽管某些疾病或疾患在某些年龄组中更常见,但本文还涵盖治疗对象而不考虑对象年龄的方法。在一些实施方案中,对象为至少18岁。在一些实施方案中,对象为18岁、25岁、35岁、40岁、45岁、50岁、55岁、60岁、65岁或70岁以上。在其它实施方案中,对象为65岁以下。在一些实施方案中,对象为18岁以下。在一些实施方案中,对象为18岁、15岁、12岁、10岁、9岁、8岁或7岁以下。
在一些实施方案中,尽管较年轻的对象也可以从所述方法中受益,但是所述方法可以在至少50岁的对象中得到应用。在其它实施方案中,对象为至少55岁、至少60岁、至少65岁和至少70岁。在另一个实施方案中,对象患有具有不利细胞遗传学的癌症。“不利细胞遗传学”被定义为任何非二倍体核型,或者大于或等于3个染色体异常。在另一个实施方案中,对象为至少60岁并且患有具有不利细胞遗传学的癌症。在另一个实施方案中,对象为60至65岁并且患有具有不利细胞遗传学的癌症。在另一个实施方案中,对象为65至70岁并且患有具有不利细胞遗传学的癌症。
在某些实施方案中,所治疗的对象在根据本文提供的方法治疗的三个月内没有心肌梗塞病史。在某些实施方案中,该对象在根据本文提供的方法治疗的三个月内没有脑血管意外或短暂性脑缺血发作的病史。在一些实施方案中,该对象在根据本文提供的方法治疗的28天内没有遭受过血栓栓塞事件,包括深层静脉血栓形成或肺栓塞。在其它实施方案中,该对象没有经历过或没有经历不受控制的弥漫性血管内凝血。
因为患有癌症的对象具有多样的临床表现和不同的临床结局,所以给予患者的治疗可以根据他/她的预后而变化。熟练的临床医生将能够在不进行过度实验的情况下容易地确定可以有效地用于治疗患有癌症的个体对象的具体的第二剂、手术类型和基于非药物的标准疗法的类型。
应当理解,本文设想了本文提供的化合物与一种或多种前述化合物和任选地一种或多种另外的药理活性物质的每种合适组合。
活性的评估
标准的生理、药理和生化程序可用于测试化合物,以鉴定具有所需活性的那些化合物。
此类测定包括例如基于细胞的测定,包括实施例部分中所述的测定。
参考以下实施例可以更充分地理解本文提供的实施方案。这些实施例意在说明本文提供的药物组合物和剂型,但并非以任何方式进行限制。
实施例
以下实施例以说明而非限制的方式提出。在描述和实施例中使用以下缩写:
SWFI–无菌注射用水
WFI–注射用水
D5W–5%右旋糖水溶液
HPβCD或HPBCD–羟丙基-β-环糊精
SBEβCD–磺丁基醚-β-环糊精钠盐
CD–环糊精
DMSO–二甲基亚砜
FDM–冷冻干燥显微镜
SEM–扫描电子显微镜
LT-DSC–低温差示扫描量热法
DSC–差示扫描量热法
DVS动态蒸气吸附
TGA–热重分析
GC–气相色谱法
KF–卡尔·费歇尔
本文的实施例中的“化合物1,C型”或“C型”或“API”是指2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的多晶型物C型。本文的实施例中的“化合物1,A型”或“A型”是指2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的多晶型物A型。2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的理化性质汇总在表1中。
表1:2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的理化性质汇总

实施例1:溶剂选择
进行了溶剂筛选,以鉴定ICH(国际医药法规协和会)Q3C指南中列出的合适的第3类溶剂,因为所述第3类溶剂具有较高的允许每日暴露(PDE)水平,为50mg/天。良好的候选溶剂被认为具有:1)与水的良好混溶性和2)足够低的沸点,因此可以轻易地在冻干工艺期间将其除去。在具有20mM pH 4.2柠檬酸盐缓冲剂的一系列溶剂混合物中测试化合物1的溶解度;结果在表2中示出。
表2:化合物1在溶剂和缓冲剂的混合物中的溶解度

据发现,在大多数测试的溶剂体系中,药物溶解度均低于10mg/mL,在100%甲酸或100%DMSO中除外,这两者提供高于100mg/mL的药物溶解度。目的是实现在溶剂中的高溶解度,使得在加工期间使用少量溶剂(因而只需要有限的溶剂去除)。因此,甲酸和DMSO被鉴定为进一步的制剂开发的两种主导溶剂。
随后对不同比例的甲酸/DMSO混合物进行了溶解度测试,以评估通过使用两种溶剂是否存在任何协同作用。
表3:化合物1在甲酸和DMSO的混合物中的溶解度


如表3中所示,所有甲酸/DMSO混合物的溶解度均远低于任一种单独溶剂的溶解度。因此,决定应在单独的甲酸或DMSO中制备API预混物。
实施例2:增溶剂选择
环糊精(CD)是最常用的络合剂,用于增加水溶性差的药物化合物的水溶性。在所有环糊精衍生物中,只有羟丙基-β-环糊精(HPβCD)和磺丁基醚β-环糊精(SBEβCD)已被用于经批准的肠胃外产品中。因此,该研究仅评估了这两种环糊精类型。
第一步,在环境温度下,在1、2和6天的时间点,测量水性溶液中的药物溶解度随HPβCD(Roquette生产的 )或SBEβCD(Cyclolab生产的
)或SBEβCD(Cyclolab生产的 )的浓度的变化。结果在表4中示出。
)的浓度的变化。结果在表4中示出。
表4:化合物1在含有Dexolve或Kleptose的水性溶液中的溶解度

一般来讲,化合物1在这两种类型的CD中的溶解度相当。化合物1的溶解度随CD浓度增加而增加。使用40%或更高的CD浓度时,溶液变得非常粘稠,这可能阻碍药物在溶液中的动力学溶解度。含有>25%CD的溶液通常很难冻干。因此,随后的溶解度研究在四种CD浓度水平下进行:3重量%、9重量%、15重量%和25重量%。除了 和
和 之外,还评估了另外两个可商购获得的CD品牌,即Ligand生产的
之外,还评估了另外两个可商购获得的CD品牌,即Ligand生产的 (SBEβCD)和AcrosOrganics提供的
(SBEβCD)和AcrosOrganics提供的 CD仿制版本。在该研究中还评估了溶剂类型和水平对药物溶解度的影响。将甲酸或DMSO以各种溶剂与CD的摩尔比(0.4:1、1:1、2.5:1、5:1和10:1)添加到溶液中。收集24小时和48小时的溶解度数据;由于存在药物化学稳定性风险,所以未考虑更长的时段。因此,这里仅呈现了48小时的数据。图30示出了在使用不同的环糊精品牌和浓度、不同的溶剂类型以及变化的溶剂与环糊精比率的情况下化合物1的溶解度。据发现,化合物1的溶解度主要受溶液中CD浓度的影响,而CD的类型和品牌对溶解度的影响最小。关于溶剂类型,在将甲酸而不是DMSO添加到环糊精溶液中时,化合物1示出稍高的溶解度。对于甲酸和DMSO这两者,药物溶解度随溶剂与环糊精的比率增加而略有增加。同CD浓度的作用相反,溶剂对药物溶解度的作用最小。
CD仿制版本。在该研究中还评估了溶剂类型和水平对药物溶解度的影响。将甲酸或DMSO以各种溶剂与CD的摩尔比(0.4:1、1:1、2.5:1、5:1和10:1)添加到溶液中。收集24小时和48小时的溶解度数据;由于存在药物化学稳定性风险,所以未考虑更长的时段。因此,这里仅呈现了48小时的数据。图30示出了在使用不同的环糊精品牌和浓度、不同的溶剂类型以及变化的溶剂与环糊精比率的情况下化合物1的溶解度。据发现,化合物1的溶解度主要受溶液中CD浓度的影响,而CD的类型和品牌对溶解度的影响最小。关于溶剂类型,在将甲酸而不是DMSO添加到环糊精溶液中时,化合物1示出稍高的溶解度。对于甲酸和DMSO这两者,药物溶解度随溶剂与环糊精的比率增加而略有增加。同CD浓度的作用相反,溶剂对药物溶解度的作用最小。
实施例3:缓冲剂选择
在IV给药时,药物溶液的pH通常优选地在4至7范围内,和或在5至7范围内。进一步考虑了包含醋酸盐、苯甲酸盐、柠檬酸盐、乳酸盐和酒石酸盐的多种药学上可接受的缓冲体系的缓冲能力以及它们对药物溶解度的影响。由于醋酸盐缓冲剂可能在冻干工艺期间升华并导致pH改变,所以该缓冲剂随后从评估中移除。
为了评估替代缓冲剂,制备了一系列药物溶液,其中Kleptose的含量为3%或20%。将甲酸或DMSO以约0.1重量%(对应于其API预混物浓度)添加到溶液中。由于大于50mM的缓冲剂强度可能对患者产生刺激,所以在2至20mM范围内的各种缓冲剂强度下评估了苯甲酸盐、柠檬酸盐、乳酸盐和酒石酸盐。也将初始pH为4.2或4.7的20mM柠檬酸盐、酒石酸盐、苯甲酸盐或乳酸盐缓冲剂添加到溶液中。随后测量每种溶液的最终pH和溶解度。结果在表5和表6中示出。
表5:含有Kleptose、溶剂和不同缓冲剂的溶液的pH值

表6:化合物1在含有Kleptose、溶剂和不同缓冲剂的溶液中的溶解度

据观察,在相同的Kleptose水平下,无论溶剂是何种类型,所有缓冲溶液的溶解度均相当,但苯甲酸盐溶液除外,其表现出的溶解度要低得多。此外,在存在甲酸的情况下,缓冲溶液中的药物溶解度略低于未缓冲溶液中的药物溶解度。相比之下,在存在DMSO的情况下,缓冲溶液的溶解度略高于未缓冲溶液的溶解度。这意味着不同类型的缓冲盐可能与溶剂分子具有不同的分子相互作用,这因而可能影响药物络合到CD腔中。含有DMSO的溶液在除苯甲酸盐以外的所有测试缓冲剂的情况下均表现出保持良好的pH,为4.2或4.7。然而,无论缓冲剂类型和初始pH是怎样的,含有甲酸的所有测试溶液均示出低于4的pH。为了在存在甲酸的情况下将本体溶液的最终pH保持在4以上,可能需要缓冲剂的pH高于4.7,以承受pH的变化。由于当pH接近或等于其pKa时,缓冲剂提供最佳的缓冲能力,并且由于苯甲酸盐、乳酸盐和酒石酸盐的pKa分别为4.2、3.86和4.4,所以认为它们几乎不可能支持大于4的pH。因此,不再考虑苯甲酸盐、乳酸盐和酒石酸盐缓冲剂。柠檬酸盐具有三个pKa,分别为3.13、4.76和6.39,在高pH值下产生良好的缓冲能力。因此,在进一步的制剂开发中选择了柠檬酸盐缓冲剂。
在后续的研究中,将10%Dexolve溶解在具有各种pH和强度的柠檬酸盐缓冲剂中,并添加了相当于150mg/mL的API预混物浓度的一定量甲酸。图31示出了最终溶液pH随柠檬酸盐缓冲剂强度(在2至20mM范围内)和初始缓冲剂pH(在4.2至5.3范围内)而变化。正如预期的那样,溶液的pH随缓冲溶液的pH和强度增加而增加。当使用pH为5.3的20mM柠檬酸盐缓冲剂时,添加甲酸之后溶液的pH能够保持为4.3的最终pH。因此,当使用甲酸作为溶剂时,建议将pH为5.3的20mM柠檬酸盐缓冲剂添加到该制剂中,以保持复原溶液的pH高于4。
实施例4:冻干制剂的热分析
在该研究中,使用冷冻干燥显微镜(FDM)和低温差示扫描量热法(LT-DSC)进行了一系列热分析,以确定含有不同的CD类型、CD浓度、溶剂类型、溶剂浓度和缓冲剂强度的各种本体溶液制剂的塌陷温度(collapse temperature)和Tg’。一般来讲,随着制剂体系的Tg’和塌陷温度升高,冻干周期时间缩短,并且在冻干期间获得良好的粉饼稳定性。
表7汇总了作为该研究的一部分获得的热表征结果,连同美国公布号2017-0196847中所公开的示例性制剂。
表7:基于环糊精的制剂的塌陷温度


表7中所公开的最后四种制剂在美国公布号US 2017-0196847中有所描述。
如表7中所示,通过LT-DSC测量的所有测试制剂的Tg’都非常接近通过FDM确定的它们的塌陷温度。一般来讲,Tg’比它们对应的塌陷温度低1至2℃。随着Kleptose浓度从3%增加到15%,塌陷温度从-14.7℃升高到-8.6℃,然后在Kleptose浓度从15%增加到25%时保持不变。在10%的相同Kleptose水平下,随着缓冲剂强度从2mM增加到20mM,塌陷温度从-7.7℃降低到-10.7℃。溶剂类型和溶剂水平示出对塌陷温度的影响最小,没有观察到明显的趋势。结果证明,环糊精的类型是对塌陷温度具有最显著影响的变量,且SBEβCD 或
或 )制剂表现出比它们的HPβCD
)制剂表现出比它们的HPβCD 对应物低的塌陷温度。与美国公布号2017-0196847中所公开的塌陷温度为-18℃的制剂相比,本文所公开的包含10%Kleptose的制剂呈现-8.2℃至-11.1℃的较高塌陷温度。这表明对于本文所公开的制剂,具有-16℃的初次干燥搁板温度的冻干周期非常保守。然而,相同的冻干周期被用作最小化加工相关风险的起始点,并且可以根据需要选择对其进行进一步修改。
对应物低的塌陷温度。与美国公布号2017-0196847中所公开的塌陷温度为-18℃的制剂相比,本文所公开的包含10%Kleptose的制剂呈现-8.2℃至-11.1℃的较高塌陷温度。这表明对于本文所公开的制剂,具有-16℃的初次干燥搁板温度的冻干周期非常保守。然而,相同的冻干周期被用作最小化加工相关风险的起始点,并且可以根据需要选择对其进行进一步修改。
实施例5:制剂Ia的制剂和工艺评估
实验室规模批次以3-kg的批次大小制备,其制剂组成在表8中示出。
表8:3-kg试验批次的制剂组成

a:在干燥时除去;b:5%过量填充(填充体积为8.4mL/小瓶)
首先在DMSO预混物中以220mg/mL的浓度制备化合物1,然后将其滴加到Kleptose缓冲溶液中。配混完成后,将溶液通过0.22μm的PVDF过滤器过滤,并填充到20cc I型玻璃小瓶中,填充重量为8.4g/小瓶,然后在表9中示出的冻干周期参数下装载到冻干机中。
在初次干燥后和二次干燥期间取出样品,以了解溶剂水平与干燥时间的关系。成品药物产品的分析结果在表10中示出。在67℃下二次干燥24小时之后,残留的DMSO水平为约6mg/小瓶,相当于去除率为约29%(初始水平:约8.6mg/mL)。注意到大部分的溶剂是在初次干燥期间去除的,而在二次干燥期间溶剂水平的降低最小。留意了冻干粉饼和复原溶液的外观,并且发现是可接受的。
表9:冻干周期参数

表10:试验批次药物产品的表征

注意:PD=初次干燥;SD=二次干燥
注意到冻干粉饼的测定处于低端,考虑到5%过量填充,其实际测定值为91.9%。在残留溶剂为约6mg/mL的情况下,该制剂可能潜在地支持高达8mg/天的剂量(如前所述,DMSO的PDE水平为50mg/天)。
基于以上的研究结果,以1-kg实验室规模制备了几个相同制剂的附加试验批次用于配混,以仅用于评估药物沉淀的潜在风险。制剂信息和测定结果在表11中示出。前三个试验批次在开始时的低含量测定(low assay)和在环境条件下储存8小时内随时间推移的递减测定(decreasing assay)的情况下并不成功。储存4小时之后,在容器底部目视观察到沉淀的药物颗粒。在后续的三个试验批次中,通过减少初始水装料量、减少API预混物的分配体积以及在添加API预混物期间进行适度混合,来修改配混过程。结果,最后三个批次的初始测定得到极大改善。然而,对于所有三个批次,在环境条件下储存19小时之后,均观察到显著的测定下降。总而言之,数据表明该制剂在物理上不稳定,并且药物沉淀的风险相当高。因此,不考虑使用3%CD水平的“溶剂交换”制剂进行进一步评估。
表11:“溶剂交换”制剂的本体溶液稳定性评估(DMSO中的3%CD和135mg/mL API)

*:在T=19h的时间点接收到数据
基于先前的溶解度研究,以下三个1-kg的试验批次(1-B、1-C和1-D)将Kleptose水平提高到10%至20%。每种本体溶液的制剂组成在表12中描述。在药物浓度和缓冲剂组成恒定的情况下,这三种制剂的唯一变量是Kleptose水平,和与API预混物浓度相对应的初始DMSO装料。冻干周期参数在表13中示出;使用较低的二次干燥温度,为50℃。
表12:1-kg试验批次的制剂组成


a:5%过量填充;b:在干燥时除去。表中的数字代表初始溶剂含量。
表13:1-kg试验批次的冻干周期参数

*在LTI批次中使用50℃;在BSP批次中使用60℃
将这三个批次的过滤后本体溶液在环境条件下储存最长8小时。在t=0、4和8小时测量每个批次的测定。结果在表14中示出。通过目视检查,这三个批次的本体溶液看起来是澄清的,并且该测定在8小时的持续时间内保持稳定。分析了这三个批次的冻干粉饼,并且结果在表15中示出。这三个批次的测定均为约100%,并且每种制剂中的单独降解物/杂质均低于0.1%。这三个批次的残留DMSO水平下降至约4mg/小瓶,示出溶剂去除率为约25%至40%。
表14:试验批次的本体溶液稳定性评估

表15:基于DMSO的制剂的表征


除上述批次之外,还以与批次1-B相同的制剂(10%Kleptose、170mg/mL DMSO预混物),以1-kg批次大小制备了两个重复试验批次(3-B1和3-B2)用于配混,仅用于评估本体溶液的保持时间。将过滤后的本体溶液在环境条件下储存19小时的持续时间。溶液保持澄清,没有可见的未溶解颗粒,并且测定保持稳定,如表16中所示。类似地,以与批次1-C相同的制剂(10%Kleptose、220mg/mL DMSO预混物),以1-kg批次大小制备了另外两个重复试验批次(4-C1和4-C2)用于配混,仅用于评估本体溶液的保持时间。将过滤后的本体溶液在环境条件下储存8小时的持续时间。溶液保持澄清,没有可见的未溶解颗粒,并且测定保持稳定,如表17中所示。配混研究证实,制剂中含有10%Kleptose的本体溶液可以在制造时间框架期间提供足够大的稳定性。因此,选择1-C作为最终制剂,CD水平为10%。
表16:试验批次的本体溶液稳定性评估

表17:试验批次的本体溶液稳定性评估

根据以上关于溶液稳定性和残留溶剂水平的结果,人们做出了更多努力,通过减少制剂中的初始DMSO量并使用不同类型的环糊精来进一步降低残留DMSO水平。制备了两个10-L试验批次,其中API-DMSO预混物的浓度从220mg/mL增加到270mg/mL(使用DMSO冲洗1mL)。这两个批次具有相同的制剂组成(10%CD和20mM pH 5.3柠檬酸盐缓冲剂),但使用了不同类型的环糊精,如表18中所示。冻干周期参数和冻干药物产品特性分别在表19和表20中展示。无论CD是何种类型,这两个批次均获得了4.6mg/小瓶的相同残留DMSO水平,与5.2mg/小瓶的初始DMSO装料的11%去除率相对应。由于与先前的试验相比,这两个试验运行均未在残留溶剂减少方面取得重大进步,因而不再采用该方法,并且在随后的过程开发工作中将API-DMSO预混物浓度固定为220mg/mL。
表18:基于DMSO的试验批次的制剂组成

a:5%过量填充(填充体积为8.4mL/小瓶);b:在干燥时部分除去。表中的数字代表初始溶剂含量。
表19:试验批次的冻干周期参数

表20:基于DMSO的冻干试验批次的表征
| 测试 | 5-F1 | 6-F1 |
| 外观 | 粉饼 | 粉饼 |
| 颜色 | 白色 | 白色 |
| 异物 | 相符 | 相符 |
| 测定(%LC) | 100.5 | 未测试 |
| 相关杂质 | <0.05% | 未测试 |
| 残留DMSO(mg/小瓶) | 4.6 | 4.6 |
| 复原溶液pH | 5.2 | 5.2 |
使用与批次1-C相同的制剂(CD除外)和过程,以500-mL实验室规模,用10%Dexolve和220mg/mL DMSO预混物制备一个附加的试验批次7-Dex1。将该本体溶液在环境条件下储存最长24小时,通过目视检查没有观察到药物沉淀。成品药物产品的测试结果包含在表21中,以与批次1-C的测试结果进行直接比较。这两个批次之间的相当的药物产品特征表明,当前的冻干周期也可以适用于基于SBEβCD的制剂,但基于SBEβCD的冻干剂的塌陷温度比其基于Kleptose的对应物低得多。即使对于基于Dexolve的制剂和基于Kleptose的制剂这两者获得了相当的结果,但是选择了Kleptose作为对环糊精的选择,原因是使用这类制剂的经验更丰富,并且Kleptose具有比Dexolve更好的塌陷温度曲线。
表21:基于Dexolve和DMSO的制剂的表征
| 测试 | 7-Dex1 |
| 外观 | 粉饼 |
| 颜色 | 白色 |
| 异物 | 相符 |
| 复原时间 | 82秒 |
| 复原外观 | 澄清溶液 |
| 测定(%LC) | 112.4 |
| 相关杂质(%) | 总杂质<0.07 |
| 残留DMSO(mg/小瓶) | 4.7 |
| pH | 4.0 |
实施例6:制剂Ia的复原方案
当基于DMSO的制剂Ia的组成最终确定时,对复原方案进行了评估,以确保复原溶液呈现对于IV施用的可接受的pH和克分子渗透压浓度。优选地,pH应当保持在4至7范围内,并且克分子渗透压浓度应当保持尽可能接近285mOsm/kg至295mOsm/kg的人血浆克分子渗透压浓度。评估了三种最常用的可商购获得的IV流体,包括注射用水(WFI)、生理盐水(NS)和5%右旋糖(D5W)。使用变化的复原方案的复原溶液的克分子渗透压浓度和pH值在表22中示出。据发现,单独的体积为5至15mL的NS或D5W提供高于370mOsm/kg的克分子渗透压浓度值。在所有测试的媒介物中,只有4mL WFI、或4mL WFI与等体积NS或D5W的混合物能够提供克分子渗透压浓度值为260mOsm/kg至280mOsm/kg的复原溶液。所有复原溶液的pH都在4.2至4.5范围内。为了使复原方案尽可能简单,认为4mL WFI是最有利的选择。最后,将复原媒介物的体积调整为3.8mL,使得复原溶液的浓度为0.25mg/mL的约整数,实际载药量为1.05mg/小瓶(5%过量填充)且最终复原溶液体积为4.2mL/小瓶。
表22:制剂Ia的复原溶液的克分子渗透压浓度和pH测量

实施例7:制剂Ia的工艺按比例放大
一旦在实验室试验中证明了制剂Ia的可行性,便以8.5L规模制造出工程化批次(批次A)。使用的冻干周期与表13中展现的相同。为了确保成功地按比例放大制剂Ia,对美国公布号2017-0196847中所公开的工艺的关键操作程序进行了全面的风险评定。已鉴定的风险和已实现的过程改进汇总在下表23中。
表23:在制剂Ia按比例放大批次中实现的工艺改进


该批次的发布测试结果在表24中示出。
表24:来自按比例放大批次的制剂Ia的批次分析结果

下表25中汇总的以下工艺改进在另外的按比例放大批次中实现:
表25:在制剂Ia按比例放大批次中实现的工艺改进


示例性批次的批次分析结果在表26中示出。
表26:制剂Ia的批次分析结果


NT=未测试
实施例8:制剂Ib的制剂和工艺评估
以含有甲酸作为共溶剂和10%Dexolve作为环糊精的制剂,按500-mL实验室规模制备了两个试验批次1-Dex2和1-Dex3。两个批次中均使用Dexolve,因为它在先前的试验中已证明具有与Kleptose相当的制剂性能。由于甲酸中的药物溶解度为约170mg/mL,因此以150mg/mL(相当于8.54mg/小瓶)和100mg/mL(相当于12.81mg/小瓶)的浓度分别在这两个批次中制备了甲酸中的API预混物。柠檬酸盐缓冲剂pH从4.2增加到5.3,以使复原溶液的pH高于4.0。冻干周期保持与美国公布号2017-0196847中所公开的制剂中使用的相同。这两个批次的制剂组成在表27中示出。成品药物产品的分析结果在表28中示出。
表27:基于甲酸的试验批次的实验室规模制剂组成


a:5%过量填充
b:在干燥时部分除去。表中的数字代表初始溶剂含量,包括1mL冲洗液
表28:基于甲酸的试验批次的冻干药物产品的表征

由于甲酸的沸点为100.8℃,远低于DMA的沸点(165℃)或DMSO的沸点(189℃),因此认为在冷冻干燥期间更容易除去。发现甲酸的减少率仅为约35%。进一步的分析表明,这可能是由于形成了甲酸钠。由于甲酸的pKa为3.75,接近本体溶液的pH,所以甲酸可以解离成其离子形式。现在,在存在缓冲剂的情况下,这些离子可以与游离金属离子反应形成更稳定的产物。在这种情况下,柠檬酸钠被用作缓冲体系的一部分,因此可能导致形成甲酸钠。一旦形成了甲酸钠,由于其具有极高的熔点(253℃),所以可能很难通过升华除去。为了有效地除去甲酸,关键是避免形成甲酸钠,并使甲酸处于其天然形式。这可以通过使本体溶液的pH保持较低(至少比其pKa低1至2个单位)、或更确切地说避免该体系中存在任何抗衡离子(诸如钠)(例如,通过从该体系中除去缓冲剂)来实现。在考虑了这两种选择之后,决定首先评估从该体系中除去缓冲剂。
后续进行了可行性研究,以调查研究溶液pH对去除溶剂的作用。在第一试验批次1-F2中,使用10%Captisol,并从制剂中除去缓冲剂。第二试验批次2-F2具有与批次1-F2相同的制剂组成,不同之处在于将10%Captisol替换为10%Kleptose。这些制剂组成在表29中示出。每10L批次添加1-mL溶剂冲洗液,计算出溶剂组成。冻干周期在表19中示出。成品药物产品的分析结果在表30中示出。据观察,批次1-F2中不存在缓冲剂,导致本体溶液的pH低于3,从而使残留甲酸水平从先前的5.6mg/小瓶明显降低至2.3mg/小瓶。在2-F2制剂中看到了甚至更令人鼓舞的结果,该结果示出残留甲酸水平低至0.4mg/小瓶,或溶剂减少了95%。
不希望受到任何理论的束缚,这两个试验运行之间的残留甲酸水平的差异归因于Captisol与Kleptose之间的化学结构差异。Captisol是由丁基醚基团拴系至亲脂性腔的磺酸钠盐的β环糊精衍生物的混合物。即使在低pH条件下,Captisol分子中存在钠离子也可能在某种程度上导致甲酸钠的形成。相比之下,Kleptose是不与任何钠离子缔合的β环糊精的部分取代的聚羟丙基醚。基于第二试验批次的残留溶剂结果,选择制剂2-F2作为最终制剂Ib。
表29:10-kg基于甲酸的试验运行的制剂组成

a:无5%过量填充(每个小瓶的填充体积为8.0mL)
b:在干燥时部分除去。表中的数字代表初始溶剂含量。
表30:基于甲酸的试验批次的冻干药物产品的表征
| 测试 | 1-F2 | 2-F2 |
| 外观 | 粉饼 | 粉饼 |
| 颜色 | 白色 | 白色 |
| 异物 | 相符 | 相符 |
| 测定(%LC) | 101.6 | 99.4 |
| 相关杂质 | <0.02% | <0.05% |
| 残留甲酸 | 2.3mg/小瓶 | 0.4mg/小瓶 |
| 复原溶液pH | 3.0 | 3.4 |
将两个试验批次的本体溶液在环境条件下储存7天的持续时间,然后评估它们的物理稳定性和化学稳定性。表31示出了每种制剂在初始时间点处和几天之后的测定结果和杂质结果。这两种制剂在环境条件下保持物理稳定和化学稳定持续至少7天。
表31:试验批次的本体溶液稳定性评估
| 1-F2 | 2-F2 | |
| t=0过滤前测定(%) | 100.8 | 99.2 |
| t=0过滤后测定(%) | 100.8 | 99.2 |
| t=7天过滤后测定(%) | 100.0 | 100.0 |
| t=7天水解1(%) | 0.13% | 0.13% |
| t=7天水解2(%) | 0.04% | 0.04% |
除了基于甲酸的制剂之外,在开发期间还评估了“双重溶剂”制剂的可行性。在这里假设允许每种溶剂的PDE水平为50mg/天。以10-L规模制备了四种“双重溶剂”制剂,其中一半制剂的API溶解在270mg/mL DMSO溶液中,另一半制剂的API溶解在150mg/mL甲酸溶液中。然后将两种API预混物溶液依次添加到本体溶液中。表32示出了四种制剂的组成。批次1-F3和1-F4均使用10%的Captisol。它们之间的唯一区别是F3在pH 5.3处缓冲,而F4没有缓冲。批次2-F3和2-F4使用10%的Kleptose,且F3在pH5.3处缓冲,而F4没有缓冲。
表32:“双重溶剂”制剂的组成

a:无5%过量填充(每个小瓶的填充体积为8.0mL)
b:在干燥时部分除去。表中的数字代表初始溶剂含量。
这四种制剂的冻干药物产品特性在表33中示出。尽管与使用单独的单一溶剂的制剂相比,DMSO或甲酸的初始量都减少了,但所有这四种制剂中的残留DMSO水平仅为约2.5mg/小瓶,而残留甲酸水平非常接近制剂Ib的该水平。基于该结果,没有进一步考虑“双重溶剂”制剂,因为它们与制剂Ib相比没有提供任何优势。
表33:冻干“双重溶剂”制剂的表征


实施例9:制剂Ib的复原方案
使用可商购获得的IV流体(包括WFI、NS、D5W和乳酸林格氏液(Lactate Ringers,LR))进行复原研究。测量了在进行和未进行NS稀释的情况下,复原溶液的克分子渗透压浓度。结果在表34中示出。3mL或更大体积的WFI提供低于220mOsm/kg的克分子渗透压浓度,而4至20mL的D5W或LR提供高于400mOsm/kg的克分子渗透压浓度。体积小于13mL的单独的NS也产生高于300mOsm/kg的克分子渗透压浓度。当使用13至19mL NS进行复原时,克分子渗透压浓度接近290mOsm/kg。然而,对于最高达20mg的剂量,这将导致总给药体积接近400mL,这将需要较长的输注时间。由于需要较低的复原体积,因此还评估了另一种可商购获得的IV流体,即1/2生理盐水(0.45%氯化钠)。据发现,4至5mL的1/2生理盐水能够实现290mOsm/kg的克分子渗透压浓度。制剂Ib的最终复原方案被确定为4.5mL1/2生理盐水,以便在复原后小瓶中的最终溶液体积为5.0mL并且载药量为1.0mg/小瓶(无过量填充)的前提下,递送精确浓度为0.2mg/mL的复原溶液。
表34:制剂Ib的复原溶液的克分子渗透压浓度和pH测量


注意到制剂Ib的复原溶液的pH处于下侧(<3.6),但这被认为是可接受的。
实施例10:制剂Ib的工艺按比例放大
制造了三个开发冻干运行,且每个冻干批次中有多个批次,以将批次大小从10L按比例放大至临床批次大小30L。
第一按比例放大批次:
在第一按比例放大批次中,制备了一个30-L和两个10-L的配混批次。这些制剂组成在表35中示出。用150mg/mL API预混物在甲酸中以30-L的批次大小制备批次3-F2-1。通过蠕动泵和0.6mm硅胶管,以每10秒每次添加50μL的速率将预混物添加到配混器皿中。批次3-F2-2和3-F2-3均用100mg/mL API预混物在甲酸中以10-L批次大小制备。F2-2与F2-3之间的唯一差别是API预混物添加方法。在F2-2中,API预混物以非常快的速度添加(预混物被一次性立即直接倒入配混器皿中以进行测试,作为最坏的情况),而F2-3则遵循与批次F2-1相同的API预混物添加程序。使用如表19中示出的相同冻干周期,在实验室冻干机中将所有这三个批次冻干。对于按比例放大批次F2-1,从配混器皿以及从接收器皿的开始、中部和结束处取出本体溶液样品,以进行工艺测定测试。还测试了批次F2-2和F2-3的过滤前和过滤后本体溶液测定。将F2-1和F2-2的经过滤本体溶液在环境条件下在接收器皿中储存7天。检查了这两个批次的物理稳定性和化学稳定性。
表35:第一按比例放大运行的制剂组成

a:无5%过量填充(每个小瓶的填充体积为8.0mL)
b:在干燥时部分除去。表中的数字对应于初始溶剂含量。
表36和表37分别呈现了本体稳定性和成品药物产品的结果。一般来讲,所有这三个批次均展现了可接受的质量属性。批次F2-2,尽管其API预混物的添加速率极高,但仍实现了良好的过程中测定,并且本体溶液稳定性与另外两个批次相当。这表明配混过程是可靠的,并且对API预混物的添加速率不敏感。不管甲酸的初始装料量有多大,所有这三个批次的残留甲酸水平均以<2.5mg/小瓶为目标。据推测,残留甲酸去除动力学可能已经达到由冷冻干燥过程条件而非初始溶剂水平确定的平台期。此外,成功完成了按比例放大至30-L配混批次大小(3-F2-1)。
注意到这些批次的残留甲酸水平高于制剂开发批次2-F2的观察值。这些按比例放大批次与制剂开发批次存在细微差异,然而,3-F2-3和2-F2具有相同的制剂和相同的工艺,但残留溶剂水平却有很大差异(1.4mg/小瓶对0.4mg/小瓶)。作为找到该现象的潜在原因的一部分,评估了冻干数据曲线,如图32中所展示。该曲线示出,在初次干燥结束时,皮拉尼(pirani)压力未与腔室压力(通过电容压力计测量)完全趋同,这表明升华未完成。因此,在下一个按比例放大批次中,做出了更多努力来了解残留溶剂的可变性。
表36:第一按比例放大批次的本体溶液稳定性

表37:第一按比例放大批次的药物产品表征


第二按比例放大批次:
用两个配混批次4-F1和4-F2评估了100mg/mL和50mg/mL的化合物1浓度。这两个批次的制剂组成在表38中示出。
表38:第二按比例放大运行的制剂组成

a:无5%过量填充(每个小瓶的填充体积为8.0mL)
b:在干燥时部分除去。表中的数字对应于初始溶剂含量。
评估了更保守的冻干周期(以最大程度减小加工风险,从而使得能够在GMP生产线中直接制造临床批次)。如表40中所示,在冻干工艺中作出了三个主要改变:1)将冷冻保持时间从4小时延长至6小时;2)将初次干燥保持时间从70小时延长至90小时;3)将二次干燥时间从12小时延长至18小时。
表39:第二试验批次的冻干循环参数

从冻干机腔室的各个位置和各个时间点取出样品。更具体地讲,在初次干燥保持的65小时、75小时和90小时,以及在二次干燥保持的6小时、12小时和18小时的每个时间点,由thief从中部搁板的前边缘取出三个小瓶。
在受控的生物负荷条件下制备批次,以实现生物负荷方法的开发。
对该批次进行成功加工。成品药物产品的结果在表40中示出。发现所有的质量属性都是可接受的。注意到尽管初始溶剂水平不同,但这两个配混批次具有相同的残留溶剂水平,为0.6mg/小瓶。还发现残留溶剂水平低于第一按比例放大批次,这可能是由于额外还有6小时的二次干燥时间。如上所述,该批次还调查研究了残留溶剂水平随时间的变化;结果在图33中示出。
表40:第二按比例放大批次的药物产品表征
| 测试 | 4-F1 | 4-F2 |
| 外观 | 粉饼 | 粉饼 |
| 颜色 | 白色 | 白色 |
| 异物 | 相符 | 相符 |
| 测定 | 100.5 | 101.6 |
| 相关杂质(%) | 总计<0.05% | 总计<0.05% |
| 水含量 | 0.07% | 0.07% |
| 残留溶剂 | 0.6mg/小瓶 | 0.6mg/小瓶 |
| 复原时间(s) | 36 | 39 |
| 复原溶液pH | 3.2 | 3.2 |
在初始溶剂水平为13.2mg/小瓶的情况下,在初次干燥保持65小时之后,冻干小瓶中的残留溶剂降低至4.5mg/小瓶,且初次干燥期间此后的变化很小。后续进行二次干燥,残留溶剂连续下降,在干燥18小时之后达到约0.6mg/小瓶。该发现支持将二次干燥保持时间增加至18小时可以帮助减少残留溶剂水平。冻干数据曲线(如图34中所展示)示出,在初次干燥结束时,皮拉尼压力完全与腔室压力趋同,如通过电容压力计测量的,这表明已完全升华。因此,建议临床批次的初次干燥保持时间为90小时,以便处于保守侧(如之前提到的,用于临床批次的冻干机为约开发批次的3倍)。
在该批次中,还调查研究了来自不同托盘位置(中心对边缘)和不同搁板位置(仅中部和底部搁板,顶部三个搁板由于搁板定位(thief positioning)而塌陷)的残留溶剂水平。中心小瓶包含的残留溶剂水平比边缘小瓶略高,这是因为边缘小瓶通常从冻干机的门和壁接收更多的热辐射。总体而言,残留溶剂水平随冻干腔室中的小瓶位置的变化不大。这示出取样位置不是早期批次中观察到的残留溶剂水平变化的可能原因。
第三按比例放大批次:
第三也是最后一个按比例放大运行是以30-L批次规模进行的确认批次,其制剂组成和工艺参数与批次4-F1相同。该批次在受控的生物负荷条件下制备,并且用于支持稍后的ICH稳定性研究。批次发布测试结果在表41中示出。
表41:按比例放大批次的药物产品表征


在几次冻干试验运行中,通过改变载药量、小瓶大小、药物浓度和Kleptose浓度来筛选附加的制剂。如表42中所示,所有测试的制剂均示出可接受的产品质量属性。
表42

实施例11:评估制剂稳定性
所选择的制剂(Ia和Ib)在下表43中提供:
表43:制剂Ia和Ib的组成


*5%过量填充
进一步评估了制剂Ia和制剂Ib的以下方面:1)成品药物产品的物理稳定性和化学稳定性,2)复原后药物产品的使用中稳定性,以及3)施用期间的材料相容性研究。在这里,目标是使药物产品的货架期至少为12个月。对于复原后的药物溶液,需要最少8小时的稳定性,以允许临床医生在向患者给药之前有足够的准备时间。此外,在施用时间框架期间,预期稀释后(如果需要)的这种复原溶液与接触材料(诸如注射器、IV管、输液袋、肝素、在线过滤器等)具有可接受的化学稳定性和物理稳定性。
成品药物产品的稳定性
评估制剂Ia的药物产品稳定性,并将1个月和3个月的稳定性结果示于表44中。在40℃/75%RH的加速条件下储存3个月之后,药物产品保持稳定,外观、测定和杂质以及复原都没有重大变化。该结果被认为可以在环境条件下为制剂Ia提供最长达12个月的货架期。
表44:制剂Ia的药物产品稳定性

类似地,评估了制剂Ib的药物产品稳定性。如表45中所示,在40℃/75%RH的加速条件下储存一个月之后,没有观察到外观、测定和杂质以及复原的重大变化。
表45:制剂Ib的药物产品稳定性

由于制剂Ia的药物产品稳定性早于制剂Ib确立,因此建立了加速稳定性评定程序(ASAP),以将制剂Ib与制剂Ia进行比较。在几种高温/低湿度条件下,在敞口小瓶中有意地进行短时段的研究,以便预测每种制剂的降解可能性和温度/湿度敏感性。测定结果和杂质结果在表46中示出。杂质的百分比随温度、湿度和储存时间增加而增加,如所预期的。总体而言,制剂Ia和制剂Ib在所有测试条件下均示出非常相似的降解曲线。仅在80℃/21%RH的最强压力条件下,制剂Ib在7天的时间点才示出比制剂Ia(3%)稍高的降解(5%)。后来通过质谱仪证实,所有降解物都是由水解降解产生的,并且不涉及未知的降解机理。基于ASAP结果,得出结论,制剂Ib具有与制剂Ia相当的药物产品稳定性。因此,预期制剂Ib具有与制剂Ia相同的货架期和相同的储存条件。
表46:制剂Ia和Ib的ASAP稳定性结果


ND=未检测到
复原溶液的使用中稳定性
大量评估了产品小瓶以及注射器、IV管线和IV袋中的复原溶液的使用中稳定性。对于制剂Ia和制剂Ib这两者,发现在环境条件下和5℃下,复原溶液在产品小瓶中在物理和化学上稳定最长达8小时。此外,在模拟剂量施用时间框架期间,该复原溶液在注射器、IV管和输液袋中证实了可接受的化学稳定性和物理稳定性,且与肝素、稀释剂或在线过滤器没有相容性问题。
对用于制剂Ia和制剂Ib这两者的产品小瓶中的复原溶液进行了一项附加的稳定性研究,以了解该溶液的物理稳定性风险。这两种制剂的小瓶均在5℃下储存,并在不同的时间点测试了测定/相关杂质。3个月的数据示出,所有测试样品的测定均无重大变化(表47)。在冷藏溶液储存55天之后,制剂Ia和制剂Ib这两者的总降解保持低于1%。由于两种水解降解物随时间推移增多,制剂Ia在超出62天示出总杂质>1%,而制剂Ib由于其复原溶液的pH低,所以在最长达96天仍保持其总杂质低于1%。这些结果示出,基于该数据,在制剂Ia和制剂Ib的复原溶液中药物沉淀的风险较低。
表47:5℃下小瓶中的复原溶液稳定性(制剂Ia和制剂Ib)

实施例12:制剂Ia和Ib的工艺程序
制剂Ia的工艺程序的详细描述如下:
配混:将柠檬酸、柠檬酸钠和羟丙基-β-环糊精 溶解在适当大小的器皿(器皿1)中的注射用水(WFI)中。将化合物1溶解在单独的器皿(器皿2)中的二甲基亚砜(DMSO)中。然后将这种化合物1-DMSO预混物溶液通过电子移液器或蠕动泵以恒定速率(每10秒每次添加约50μL)添加到器皿1中,同时使用良好的涡旋对器皿1中的溶液进行混合。目视检查溶液,以确保器皿1中不存在未溶解的颗粒。混合后,用WFI将批次重量调整为目标重量。
溶解在适当大小的器皿(器皿1)中的注射用水(WFI)中。将化合物1溶解在单独的器皿(器皿2)中的二甲基亚砜(DMSO)中。然后将这种化合物1-DMSO预混物溶液通过电子移液器或蠕动泵以恒定速率(每10秒每次添加约50μL)添加到器皿1中,同时使用良好的涡旋对器皿1中的溶液进行混合。目视检查溶液,以确保器皿1中不存在未溶解的颗粒。混合后,用WFI将批次重量调整为目标重量。
过滤:然后使用两个串联的0.2μm除菌过滤器过滤本体溶液。在该步骤之前,可以任选地使用一个0.45μm或0.2μm除菌过滤器对本体溶液进行预过滤。
无菌填充、冻干和小瓶封盖:在20-ml的小瓶中进行无菌填充,其中目标填充体积为8.40mL(5%过量填充)。然后将冻干塞部分放置在每个已填充的小瓶上(至第一个凹口)。然后装入冻干机并执行冷冻干燥周期(表13)。冻干完成之后,将小瓶在氮气气氛中减压加塞并密封。
制剂Ia的工艺流程图在图35中展示,并且制剂Ia的冻干工艺参数在表48中示出。
表48:制剂Ia的冻干周期参数

*:在LTI批次中使用50℃;在BSP批次中使用60℃
制剂Ib的工艺程序的详细描述如下:
配混:将羟丙基-β-环糊精 溶解在适当大小的器皿(器皿1)中的注射用水(WFI)中。将化合物1溶解在单独的器皿(器皿2)中的甲酸(FA)中。然后将这种化合物1-FA预混物溶液通过电子移液器或蠕动泵以恒定速率(每10秒每次添加约50μL)添加到器皿1中,同时使用良好的涡旋对器皿1中的溶液进行混合。目视检查溶液,以确保器皿1中不存在未溶解的颗粒。混合后,用WFI将批次重量调整为目标重量。
溶解在适当大小的器皿(器皿1)中的注射用水(WFI)中。将化合物1溶解在单独的器皿(器皿2)中的甲酸(FA)中。然后将这种化合物1-FA预混物溶液通过电子移液器或蠕动泵以恒定速率(每10秒每次添加约50μL)添加到器皿1中,同时使用良好的涡旋对器皿1中的溶液进行混合。目视检查溶液,以确保器皿1中不存在未溶解的颗粒。混合后,用WFI将批次重量调整为目标重量。
过滤:然后使用两个串联的0.2μm除菌过滤器过滤本体溶液。在该步骤之前,可以任选地使用一个0.45μm或0.2μm除菌过滤器对本体溶液进行预过滤。
无菌填充、冻干和小瓶封盖:在20ml的小瓶中进行无菌填充,其中目标填充重量为8.0mL(无过量填充)。然后将冻干塞部分放置在每个已填充的小瓶上(至第一个凹口)。然后装入冻干机并执行冷冻干燥周期。冻干完成之后,将小瓶在氮气气氛中减压加塞并密封。
制剂Ib的工艺流程图在图36中展示,并且制剂Ib的冻干工艺参数在表49中示出。
表49:制剂Ib的冻干周期参数

实施例13:制剂Ib的过饱和风险评定
在实施例11中描述的制剂Ib中,本体溶液和复原溶液在其平衡溶解度之上约2倍过饱和。进行了两项研究以评估基于Kleptose的制剂的过饱和风险。第一项研究的目的是使用“自上而下”和“自下而上”这两种方法确定化合物1在不同浓度的Kleptose溶液中的平衡溶解度。第二项研究旨在评估制剂Ib中的药物沉淀的动力学。
测定化合物1在Kleptose溶液中的平衡溶解度
采用“自下而上”方法来测量化合物1在Kleptose溶液中的平衡溶解度。在该实验中,通过将Kleptose和甲酸溶解在WFI中制备了十种溶液。所有这十种溶液中的Kleptose浓度从30mg/mL到250mg/mL不等,而甲酸浓度则如制剂Ib的本体溶液中所用,保持恒定为1.65mg/mL。将约25mg的过量化合物1添加到每种制备的100-mL溶液中,并在25℃下混合48或72小时。然后取出每种溶液的样品并离心以进行测定测试。化合物1在每种媒介物中的溶解度在表50中列出。
表50:化合物1在Kleptose溶液中的溶解度

a:在25℃下保持72小时之后测得的溶解度
如图37中所示,据发现化合物1的溶解度随Kleptose浓度线性增加。
使用“自上而下”方法进行了另一个溶解度实验,即,将结晶化合物1添加到过饱和溶液中以诱导化合物1沉淀,直到该溶液达到平衡溶解度为止。在该实验中,通过将甲酸中的预混物中的100mg/mL化合物1添加到Kleptose溶液中,以使最终药物浓度为约125μg/mL,而制备了六种溶液制剂。Kleptose浓度从30到100mg/mL不等。然后将包含6.25mg化合物1(约占总化合物1溶液的约10%)的1mL化合物1的水中浆液添加到500mL化合物1/Kleptose溶液中,并在25℃下连续混合9天。在9天结束时测试该溶液的测定,结果在表51中示出。相比之下,还在25℃下监测了不添加化合物1浆液的相同溶液的测定最长达4周。4周结束时的测定结果在表51中示出。
表51化合物1溶解度的“自上而下”方法

结果示出,除了其中一种具有30mg/mL Kleptose的溶液外,所有这六种溶液在25℃下都保持相对稳定,并且在未向这些溶液中添加化合物1晶体的情况下,4周之后的测定没有变化。在将结晶化合物1材料添加到溶液中时,观察到具有30-80mg/mL Kleptose的溶液具有明显的药物沉淀。溶液中的Kleptose浓度越低,混合9天之后出现的测定减少就越多。即使在存在化合物1晶体的情况下,具有90-100mg/mL Kleptose的溶液中也没有测定变化。通过XRPD证实沉淀的化合物1是起始材料的B型而不是C型(数据未示出)。这表明由“自上而下”方法测得的溶解度为B型的溶解度。因此,这些值高于由“自下而上”方法获得的C型溶解度。“自上而下”方法可能需要更长的时间才能达到热力学最稳定的化合物1形式(C型)的平衡溶解度。
过饱和Kleptose溶液中的药物沉淀动力学
在第一项沉淀研究中,以100mg/mL的Kleptose浓度制备了四种溶液制剂。在每种制剂中,化合物1的药物浓度从50μg/mL到480μg/mL不等。配混完成后,将相同量的结晶化合物1材料添加到每种制剂中(1.25mg化合物1至500g溶液)。然后将溶液储存在三种条件下:(1)在环境条件下,伴随轻柔混合;(2)在环境条件下,不混合;(3)在2至8℃下,不混合。测试了每种溶液在t=48小时的测定。结果在表52中示出。
表52:化合物1在100mg/mL Kleptose溶液中的药物沉淀


制剂F1在所有这三种储存条件下均保持稳定,且在48小时内没有测定变化。在所有这三种储存条件下储存48小时之后,制剂F2的最终浓度为约148μg/mL,从t=0开始测定减少了2%至3%。尽管化合物1接种的比率较低,制剂F3和制剂F4仍示出从最初开始的显著测定降低。混合加快了这两种制剂的药物沉淀动力学。得出结论,100mg/mL Kleptose溶液中的药物沉淀风险在较高的过饱和比下以及在向溶液施加混合时增加。
在第二项沉淀研究中,制备了三种溶液制剂,每种制剂中的Kleptose浓度分别为10%、15%和20%。每种制剂的对应药物浓度为300、400和500μg/mL,因此所有这三种制剂的过饱和比为约5倍。在添加2%的化合物1晶种之后,每种溶液制剂均在两种不同的条件下储存:(1)在环境条件下,伴随轻柔混合;(2)在2至8℃下,不混合。监测所有溶液的测定最长达34天。在该研究中,将相同的实验重复两次,以评估结果的重现性。测定结果在图38和图39中示出。
如图38中所示,随着Kleptose浓度增加,药物沉淀变慢。对于具有10%Kleptose的制剂F1,在室温下放置7天之后,药物浓度达到约98μg/mL的平台期。具有15%Kleptose的制剂F2在34天之后看起来似乎达到了平台浓度184μg/mL,而具有20%Kleptose的制剂F3的浓度下降至290μg/mL,并且仍处于下降趋势。如图39中所示,当在将过饱和溶液储存在2至8℃下且不混合时,药物沉淀速率甚至更慢。这三种制剂在34天之后的测定值分别变为97μg/mL、133μg/mL和385μg/mL。
基于这两项沉淀研究加上在“自上而下”溶解度研究中进行的一项研究,化合物1在室温下在10%Kleptose溶液中的表观溶解度在2天之后为135μg/mL、在9天之后为115μg/mL,并且在34天之后为98μg/mL。这表明由10%Kleptose和125μg/mL的药物浓度组成的制剂Ib虽然处于过饱和状态,但由于Kleptose作为络合剂的稳定化作用,该制剂可能在数天至数周内经历非常缓慢的沉淀动力学以达到其平衡溶解度。
制剂Ib本体溶液和复原溶液的物理稳定性
在该研究中,制备了制剂Ib的本体溶液(10%Kleptose、125μg/mL化合物1)和复原溶液(16%Kleptose、200μg/mL化合物1),然后用0.5%化合物1晶种攻击。两种溶液均在室温下伴随连续混合储存48小时。在不同的时间点监测这些溶液的测定。随后用2.5%化合物1晶种重复该实验。这两次实验的结果在表53中示出。
表53:具有化合物1晶种的制剂Ib的本体溶液和复原溶液的测定

结果表明,即使在存在化合物1晶种的情况下,无论添加到该制剂中的化合物1晶种的量有多少(0.5%或2.5%),本体溶液和复原溶液在室温下在48小时内均保持相对稳定,没有明显的随时间推移测定减少的趋势。从T0开始的测定变化为3%或更小,完全在90%至110%的测定规格之内。这意味着在制造和患者使用中施用期间,制剂Ib的药物沉淀风险相当低,尤其是当溶液基本上没有异物参与控制良好的无菌条件时。
实施例14:制剂Ic的制剂开发
制剂设计空间的定义
ICP制剂的设计有两个主要限制条件:(1)制剂的过饱和比,其影响药物产品的稳定性;(2)最终剂量中的Kleptose摄入总量,其影响药物产品的安全曲线。过饱和比和Kleptose摄入总量可以表示为药物浓度、Kleptose浓度和总药物剂量的函数,方程如下:


基于图40中示出的溶解度数据,平衡溶解度与Kleptose浓度线性相关,并且可以通过以下公式计算:

前提是100cc小瓶中的最大填充体积为50mL,并且最多可以向患者给药2个小瓶,则过饱和比、总Kleptose摄入量和要递送的最大剂量之间的相互依存关系在图40中绘出。
优选地,该制剂的过饱和比应当低于1(即非过饱和)。Kleptose摄入量应当低于其允许的每日暴露(PDE)阈值300mg/kg/天。基于商业IV药物产品的毒理学评定表明,Kleptose日剂量低于8g/天。在制剂设计空间内可以递送的最大剂量小于6mg。由于预期的商业产品(ICP)制剂的最高剂量被设定为3.6mg/天,因此可以在该设计空间内鉴定基于Kleptose的制剂,以在过饱和水平与总Kleptose摄入量之间实现平衡。
原型制剂的评估
以实验室规模(3至15L)制造了一系列可行性批次,它们具有不同的药物浓度、Kleptose浓度、填充体积和小瓶大小。对于所有可行性批次,均采用与如制剂Ib中所使用相同的冻干周期。该研究有两个目标:(1)证明在基于Kleptose的制剂中递送1mg至最多6mg剂量的可行性;(2)评估制剂表现(填充体积和小瓶大小)对药物产品的关键质量属性(诸如粉饼外观、复原时间和残留甲酸水平)的影响。这些制剂组成在表54中示出。
表54:在可行性批次中评估的原型制剂


测试了每种制剂的冻干样品的粉饼外观、测定和杂质、残留溶剂和复原时间。测试结果汇总在表55中。由于那些原型制剂的制剂组分和制造工艺与制剂Ib的非常相似,所以预期冻干药物产品的稳定性曲线与制剂Ib的相当。因此,在这些可行性批次中未进行稳定性评估。
表55可行性批次中的原型制剂的测试结果

二十五种原型制剂涵盖了宽泛的载药量范围,为1至6.25mg/小瓶。在不同的小瓶大小中,Kleptose浓度从10%到20%不等,药物浓度从0.075mg/mL到0.125mg/mL不等,填充体积从8mL到50mL不等。所有的冻干小瓶均示出精美的粉饼外观和可接受的测定/杂质值。所有原型制剂的复原时间均少于5分钟。具有20%Kleptose的制剂的复原时间往往比具有10%或15%Kleptose的那些制剂更长,这可能归因于在较高的Kleptose浓度下复原溶液的粘度增加。总体而言,尽管Kleptose浓度、药物浓度、填充体积和小瓶大小存在差异,但基于Kleptose的制剂仍证明了其稳健性。
残留甲酸水平在各种不同的原型制剂中在宽泛的范围内变化。据发现,残留甲酸去除效率与小瓶中Kleptose的总量负相关。如图41中所示,通过相同的冻干周期除去的甲酸的百分比随着Kleptose量增加而降低。这可以通过Kleptose将溶剂捕集在其复合腔中的倾向来解释。
还调查研究了粉饼厚度对残留甲酸水平的影响。如图42中所示,在小瓶中的Kleptose量相同的前提下,残留甲酸水平与填充高度无关。这表明在较厚的冻干粉饼中增大的质量传递阻力可能对残留甲酸水平具有最小的影响。相反,残留甲酸水平随Kleptose量增加而增加,这与图41中观察到的趋势相符。
已知针对20cc小瓶表现中的1mg剂量,制剂Ib中的残留甲酸水平为约0.9mg/小瓶,并且残留甲酸的释放规格为NMT 2.5mg/mg API,因此针对20mg的最高剂量,甲酸的总日摄入量可以被保持低于50mg/天的ICH限值。为了与制剂Ib进行比较,在图43中将所有这25种原型制剂中的每mg化合物1的残留甲酸作为Kleptose浓度的函数绘图。考虑到各种载药量和小瓶大小表现,残留甲酸水平在100mg/ml的Kleptose浓度下为1.0至1.8mg/mg化合物1(与制剂Ib相同)、在150mg/ml的Kleptose浓度下为2.1至3.8mg/mg化合物1,并且在200mg/ml的Kleptose浓度下为3.9至4.9mg/mg化合物1。因此,无论Kleptose浓度有多大,所有这些原型制剂中的残留甲酸都应当远低于3.6mg最高剂量下的ICH阈值。
最终制剂选择
由于所有这些原型制剂在可接受的测定和杂质、残留溶剂水平以及复原时间的条件下均被认为是可行的,DPD研究小组选择了这样的制剂:其中借助甲酸将0.08mg/mL化合物1溶解在150mg/mL Kleptose中。由于药物浓度降低并且Kleptose浓度增加,所以这种新制剂提供了未饱和的本体溶液和复原溶液。因此,它消除了制剂Ib中潜在的药物沉淀风险。该制剂称为制剂Ic。替代性地,也可以考虑其中借助甲酸将0.08mg/mL API溶解在100mg/mLKleptose中的制剂。该制剂称为制剂Ibm,将制剂Ib的过饱和比从2x降至1x,从而减轻了潜在的药物沉淀风险。表56中列出了制剂Ic的制剂组成与制剂Ibm的比较。
表56:制剂Ic的组成与制剂Ibm的比较


实施例15:评估制剂稳定性
制剂稳定性评估包含三个部分:(1)本体溶液的物理稳定性和化学稳定性,其决定制造过程期间的最大保持时间;(2)冻干粉饼的ICH稳定性测试,其确定成品药物产品的货架期;(3)小瓶和IV袋中的复原药物溶液的物理稳定性和化学稳定性,其决定向患者施用药物的使用中时间。
本体溶液的稳定性
制剂Ic的本体溶液由150mg/ml Kleptose、0.08mg/ml化合物1和0.98mg/ml甲酸组成。该本体溶液的pH为2.8,并且该本体溶液的药物浓度低于其平衡溶解度,使得嵌入制剂Ib中的过饱和风险在制剂Ic中得到消除。因此,预期本体溶液Ic的物理稳定性和化学稳定性优于制剂Ib的物理稳定性和化学稳定性。本体溶液的“保持时间”研究随后在工程批次中进行,并证实了在环境条件下最长达24小时的溶液稳定性。
成品药物产品的稳定性
使用来自开发批次的小瓶评估了制剂Ic的药物产品稳定性。稳定性结果在表57中示出。在25℃/60%RH和40℃/75%RH这两种条件下储存6个月之后,药物产品保持稳定,外观、测定和杂质以及复原时间都没有重大变化。该结果用于在环境条件下为制剂Ic提供最长达18个月的货架期。
表57:制剂Ic的药物产品稳定性

实施例16:制剂Ic的冻干工艺优化
I.制剂Ic的热表征
用冷冻干燥显微镜(FDM)和低温差示扫描量热法(LT-DSC)对制剂Ib和制剂Ic进行热分析。制剂Ic具有为0.125mg/ml的药物浓度。结果的汇总在表58中示出。制剂Ic表现出与制剂Ib类似的塌陷温度和Tg’。基于该结果,在冻干工艺的初次干燥期间,推荐的产品温度为-11℃至-12℃,比塌陷温度低2至3度。
表58:制剂Ib和Ic的塌陷温度和Tg’:
| 制剂Ib | 制剂Ic | |
| Tg’(DSC) | -11.4℃ | -11.0° |
| 塌陷起始温度(FDM) | -9.3℃ | -9.0° |
II.冻干建模
代替运行试误法(trial and error)冻干实验,首先利用了由Michael Pikal教授开发的冻干模型来预测不同干燥条件下的产品温度曲线。将从三个制剂Ib开发批次收集的现有热电偶数据用作模型输入,以反算出传质阻力系数(Rp),作为干层厚度(Ldry)以及传热系数(kv)的函数。然后,基于以下方程由曲线拟合推导出系数R0、A1和A2。计算出的参数在表59中示出。

表59:计算出的传热系数和产品阻力

由于制剂Ib和制剂Ic之间的Tg’和塌陷温度相似,所以将来自表59的计算出的Rp值和Kv值连同表60中列出的其它已知参数一起应用于Gen2c制剂的模型预测中。
由于中心小瓶的Kv低于边缘小瓶(4.04x10-4对9.49x10-4),所以中心小瓶需要比边缘小瓶更长的干燥时间,而边缘小瓶的产品温度往往高于中心小瓶,这导致较高的粉饼塌陷风险。因此,在建模中最坏的情况是使用中心小瓶的Kv来估计干燥时间,并且使用边缘小瓶的Kv来估计一种或多种不同搁板温度(Ts)条件下的产品温度。如表61中所示,在10℃的搁板温度下,预测的产品温度为约-12℃,接近来自上述FDM研究的推荐产品温度。在相同的搁板条件下,对于20cc小瓶中的1.0mg剂量,预测的初次干燥时间为约20小时,这与制剂Ib冻干周期中的70小时形成对照。
表60:冻干建模中使用的关键输入参数

表61:预测的初次干燥时间和产品温度

实施例17:3-L批次的冻干周期开发
基于建模结果,使用实验室规模的冻干机(型号SP Virtis Genesis 25EL)制造了一个3-L冻干批次的制剂Ic。药物产品的载药量为2mg/小瓶,或50cc小瓶中填充25mL。冻干循环参数在表62中示出。初次干燥阶段中的搁板温度和腔室压力分别设定为10℃和250微米。
表62:实验室规模批次1的冻干周期参数


将四个热电偶分别放入两个中心小瓶和两个边缘小瓶中。实验室规模批次1的产品温度曲线在图45中示出。在初次干燥期间,两个边缘小瓶显示约-11℃的产品温度,这与两个中心小瓶的约-14℃产品温度形成对照。边缘小瓶的“中断点”或产品温度开始接近搁板温度的时间点是24小时,中心小瓶是35小时。在冻干周期结束时未观察到粉饼塌陷。该实验结果与表61中示出的预测性建模结果高度吻合。
随后,为了更好地了解产品温度随搁板温度的变化,使相同的已填充小瓶经受另一个冻干周期,其中初次干燥的搁板温度从5℃逐步升高到最高14℃。每个步骤的保持时间为2至6小时,并且每个步骤中的温度升高3℃。然后将搁板温度降低至10℃并保持30小时。冻干循环参数在表63中示出。
表63:实验室规模批次2的冻干周期参数

将三个热电偶分别放置于一个边缘小瓶、一个中心小瓶以及边缘与中心之间的一个小瓶中。实验室规模批次2的产品温度曲线在图46中示出。在每种搁板温度条件下由每个热电偶测得的产品温度汇总在表64中。据发现,搁板温度每次递增3℃,产品温度就升高1℃。当搁板温度达到11℃时,中部和中心的小瓶的产品温度仍低于-11℃,而边缘小瓶则示出-9.6℃的产品温度,超过了塌陷温度。尽管如此,在任何冻干小瓶(包括边缘小瓶)中均未观察到粉饼塌陷的迹象。当搁板温度升至14℃时,产品温度已超过“断点”,这意味着初次干燥接近完成。因此,升高的温度没有引起任何粉饼塌陷。
表64:实验室规模批次2的冻干周期参数


组合来自这两次冻干运行的结果,初次干燥的推荐搁板温度和腔室压力分别被确定为10℃和250微米。
实施例18:按比例放大批次的冻干周期开发
在这两次试验运行之后,使用实验室冻干机(型号:Millrock )制造了三个15L批次(Ic-1、Ic-2和Ic-3)的制剂Ic。每个批次包括两个子批次,一个子批次用于1mg/小瓶剂量,另一个子批次用于4mg/小瓶剂量。批次Ic-1和Ic-2针对1.0mg剂量使用20cc小瓶,并且针对4mg剂量使用100cc小瓶。由于1.0mg剂量子批次中的小百分比小瓶破损在这两个批次中均被观察到,并且归因于20cc小瓶中相对高的填充体积,所以在批次Ic-3中作为替代将较大的50cc小瓶大小用于1.0mg剂量。每个子批次的制剂组成和表现在表65中示出。
)制造了三个15L批次(Ic-1、Ic-2和Ic-3)的制剂Ic。每个批次包括两个子批次,一个子批次用于1mg/小瓶剂量,另一个子批次用于4mg/小瓶剂量。批次Ic-1和Ic-2针对1.0mg剂量使用20cc小瓶,并且针对4mg剂量使用100cc小瓶。由于1.0mg剂量子批次中的小百分比小瓶破损在这两个批次中均被观察到,并且归因于20cc小瓶中相对高的填充体积,所以在批次Ic-3中作为替代将较大的50cc小瓶大小用于1.0mg剂量。每个子批次的制剂组成和表现在表65中示出。
表65:开发批次的制剂表现

正在这三个开发批次中评估的主要变量是:初次干燥的搁板温度和腔室压力,以及二次干燥的搁板温度和保持时间。每个批次的冻干周期条件汇总在表66中。
表66:开发批次的冻干周期参数

I.初次干燥对产品温度和周期时间的影响
这三个开发批次的产品温度曲线在图47至图49中示出。关键特征(包括初次干燥期间的产品温度和达到初次干燥终点的时间)汇总在表67中。产品温度分别为-10℃、-11℃和-13℃,对应于14℃、10℃和8℃的搁板温度,这与实施例37中所述的先前批次的发现相符。在这三个批次中的任一者中均未观察到粉饼塌陷,包括具有14℃的搁板温度的一个批次,其具有-10℃的对应产品温度。这是示出制剂Ic的过程可靠性的证据。
表67冻干开发批次的表征

有不同的方法可以确定初次干燥的终点。最保守的方法是查看皮拉尼规压力曲线与电容压力计压力曲线完全趋同的时间点,这表明不再有水蒸气从干燥的粉饼升华。替代性的方法是查看产品温度与搁板温度交叉的时间点,这也表明升华完成。后一种方法的仅有注意事项是,必须将热电偶放置于小瓶的底部,并且带有热电偶的小瓶的产品温度往往比没有热电偶的小瓶略高。因此,在达到交叉温度之后始终额外增加10%至30%的干燥时间,来作为初次干燥结束时的缓冲。在该研究中,由于每个批次包含两个具有不同的填充体积和小瓶大小的子批次,第一种方法无法反映每个子批次的初次干燥终点,因此采用第二种方法来确定每个单独的子批次的初次干燥时间。如表67中所示,在前两个批次中,由于粉饼厚度增加,所以子批次Ic-1-F2和Ic-2-F2(4mg剂量)需要为子批次Ic-1-F1和Ic-2-F1(1.0mg剂量)的干燥时间的约1.5倍的干燥时间。由于搁板温度较低,所以批次Ic-2显示的干燥时间比批次Ic-1长20%至40%。在批次Ic-3中,尽管子批次Ic-3-F1的搁板温度较低,为10℃,但其干燥时间却缩短至17小时,这与批次Ic-1-F1中的27小时形成对照,这主要是因为在较大的50cc小瓶大小中,粉饼厚度减小为10mm,这与20cc小瓶中的18mm粉饼厚度形成对照。基于该结果,推荐的初次干燥条件是:针对50cc小瓶中填充的1.0mg剂量,搁板温度为10℃、腔室压力为250微米,并且保持时间为20小时。
II.周期条件对残留水的影响
最终冻干药物产品的两个关键质量属性是水含量和残留溶剂水平。为了评估冻干期间水和甲酸的去除率,在批次Ic-1和Ic-2中在干燥步骤的不同时间点取出样品小瓶。通过卡尔·费歇尔方法测量每个样品的残留水含量,并且结果在图50中示出。在初次干燥步骤中,在达到交叉温度时的时间点与初次干燥结束时的时间点之间取出前四个样品。在二次干燥步骤中,在二次干燥持续四至六个小时的时间点与二次干燥结束时的时间点之间取出最后两个或三个样品。数据示出,到达到交叉温度的时候为止,冻干粉饼中的残留水分已经低于0.2%。附加的初次干燥时间使残留水分能够减少至0.06%。这表明除去大部分的水是在初次干燥步骤期间发生的。在二次干燥步骤中,残留水分水平在最初的四至六小时内进一步下降至0.04%,然后达到平台期。不管粉饼大小或干燥温度差是怎样的,这两个批次的成品药物产品中的残留水分最终都保持在0.04%的相同水平。同样,在批次Ic-3中,尽管二次干燥时间延长为36小时,但这两个子批次的成品药物产品的残留水分水平均为0.03%。总而言之,18小时的二次干燥时间和40℃至60℃的干燥温度足以除去制剂Ic的残留水分。
III.周期条件对残留甲酸的影响
还通过在从初次干燥结束至二次干燥结束的不同时间点取出样品小瓶来评估残留甲酸随干燥时间的变化。结果在图51中示出。批次Ic-1和Ic-2的二次干燥时间均为18小时。然而,批次Ic-1的二次干燥温度为60℃,比批次Ic-2的40℃高。因此,批次Ic-1表现出比批次Ic-2更快的残留甲酸去除率。对于两个子批次,Ic-1中的最终残留甲酸仅为批次Ic-2中的最终残留甲酸的60%。因此,二次干燥温度是关键的工艺参数,对成品药物产品的残留溶剂水平有重大影响。在同一个冻干批次中,两个子批次之间的残留甲酸水平也有所不同。在两个批次中,子批次F1(1.0mg剂量)仅示出子批次F2(4mg剂量)中残留甲酸水平的80%。这并不奇怪,因为据发现,残留甲酸水平随冻干小瓶中的Kleptose量增加而增加。
从前两个批次中还观察到,在18小时的二次干燥结束时,残留甲酸水平未达到平台期。因此,在批次Ic-3中,二次干燥在60℃下延长至36小时。如图51中所示,当干燥时间加倍至36小时时,子批料F1中的残留甲酸水平从0.15%降低至0.08%。子批次F2遵循相同的趋势。冻干周期完成时,从冻干机上卸下小瓶之后,将几个F1小瓶和F2小瓶放置于60℃真空烘箱中再保持24小时。残留甲酸在真空烘箱中在16小时时间点开始显示平台期,并且在24小时的烘箱干燥结束时分别达到子批次F1和F2的最低水平,为0.01%和0.03%。
根据ICH Q3指南,甲酸的PDE作为3类溶剂为50mg/天。在最高剂量为4mg/天的前提下,最大允许残留甲酸水平为12.5mg/mg API,或粉饼重量的0.6重量%。基于图51中示出的数据,在二次干燥18小时之后,残留甲酸水平低于0.2%,远低于ICH阈值。将干燥时间加倍至36小时仅使残留甲酸从0.2%有限地下降至约0.1%。因此,研究小组建议在60℃下对不同小瓶表现的制剂Ic的二次干燥时间为18至24小时,以使残留甲酸水平保持在低于0.4重量%。制剂Ic的该残留甲酸水平与制剂Ib中的0.3重量%NMT释放规格相当。
IV.冷冻速率对循环时间和残留甲酸的影响
在一些文献中报道过,由于冰晶形态的变化,冷冻速率可能对初次干燥和/或二次干燥的效率有影响。因此,进行了一项研究,以评估冷冻速率对成品药物产品的干燥时间和残留溶剂水平的作用。制造了制剂Ic的两个10-L冻干批次。每个冻干批次包含三个子批次,即50cc小瓶中的1.0mg、50cc小瓶中的2mg,以及100cc小瓶中的4mg。这两个冻干批次中使用的冷冻速率分别为0.1℃/min和1.0℃/min,与先前的冻干批次中使用的0.5℃/min的冷冻速率形成对照。这两个批次的冻干循环参数在表68中描述。
表68:冻干循环参数

*:达到的实际冷冻速率为0.85℃/min。
如图52和图53中所示,尽管冷冻速率不同,但这两个批次的产品温度曲线彼此相当。所有子批次在“中断”点处的产品温度和初次干燥时间汇总在表69中。不同子批次的“中断”产品温度相似。正如预期的那样,“中断”时间随着剂量或粉饼大小增加而增加,并且在两个批次中,中心小瓶都显示比边缘小瓶更长的升华时间。具有较高冷冻速率的批次C1示出的初次干燥时间比批次C2略短。
表69:“中断点”产品温度和干燥时间

来自这两个批次的最终冻干小瓶示出相似的粉饼外观,如图54中所示。还测试了来自这两个批次的最终冻干小瓶的残留甲酸、残留水分和复原时间。测试结果在表70中示出。这两个批次之间的复原时间相当。批次C2中的每个子批次的残留甲酸水平比批次C1中的它们的对应物略低。有趣的是,残留水分水平证明了相反的趋势,示出批次C2中的水平更高。已知的是,由于成核时间较短,所以较快的冷冻速率导致冰晶较小。因此,由于干燥粉饼的孔径大小较小,所以初次干燥期间的传递阻力可以增加。在另一个方面,由于干燥粉饼的表面积增加,所以二次干燥的解吸效率可以增加。据推测,水的去除主要依赖于初次干燥,而甲酸的去除则更多地依赖于二次干燥。这可以解释两个批次之间残留甲酸水平和残留水分水平的差异。尽管如此,认为这两个批次之间的差异不大,从而表明冷冻速率对成品药物产品的关键质量属性没有重大影响。因此,研究小组决定将以后的批次的冷冻速率保持在0.5℃/min。
表70:批次C1和C2的成品药物产品测试

实施例19:制剂Ic的最终工艺
制剂Ic的工艺程序的详细描述如下:
配混:将 溶解在适当大小的器皿(器皿1)中的注射用水(WFI)中。将化合物1溶解在单独的器皿(器皿2)中的甲酸(FA)中。然后将这种化合物1-FA预混物溶液通过电子移液器或蠕动泵以恒定速率(每10秒每次添加约50μL)添加到器皿1中,同时使用良好的涡旋对器皿1中的溶液进行混合。目视检查溶液,以确保器皿1中不存在未溶解的颗粒。混合后,用WFI将批次重量调整为目标重量。
溶解在适当大小的器皿(器皿1)中的注射用水(WFI)中。将化合物1溶解在单独的器皿(器皿2)中的甲酸(FA)中。然后将这种化合物1-FA预混物溶液通过电子移液器或蠕动泵以恒定速率(每10秒每次添加约50μL)添加到器皿1中,同时使用良好的涡旋对器皿1中的溶液进行混合。目视检查溶液,以确保器皿1中不存在未溶解的颗粒。混合后,用WFI将批次重量调整为目标重量。
过滤:然后使用两个串联的0.2μm除菌过滤器过滤本体溶液。在该步骤之前,将使用一个0.45μm或0.2μm除菌过滤器对本体溶液进行预过滤。
无菌填充、冻干和小瓶封盖:在50cc的小瓶中进行无菌填充,针对1.0mg的剂量强度,目标填充重量为12.5mL。然后将冻干塞部分放置在每个已填充的小瓶上(至第一个凹口)。然后装入冻干机并执行冷冻干燥周期。冻干完成之后,将小瓶在氮气气氛中减压加塞并密封。
制剂Ic(1.0mg剂量强度)的冻干工艺参数在表71中示出。制剂Ic(1.0mg剂量强度)的总冻干周期时间为约2.6天,仅为制剂Ib的冻干周期时间的一半。
表71:制剂Ic的冻干周期参数

制剂Ic的过程流程图在图55中展示。
实施例20:具有生理盐水的复原制剂的克分子渗透压浓度
在研究中使用制剂1b(1mg/小瓶)。每个小瓶都用4.5mL生理盐水(NS)复原,制成浓度为0.2mg/mL的药物溶液(复原后的最终溶液体积为5mL)。用渗透压计( 3250型,Advanced Instruments Inc.)测量小瓶中的复原溶液的克分子渗透压浓度,并且获得了440mOsm/kg的克分子渗透压浓度值。然后从三个小瓶中取出12mL的复原溶液,用38mL生理盐水稀释至50mL的最终体积,以获得代表2.4mg临床剂量的给药溶液。经稀释的给药溶液的克分子渗透压浓度被测得为317mOsm/kg。随后,从六个小瓶中取出30mL的复原溶液,用20mL生理盐水稀释至50mL的最终体积,以获得代表6.0mg临床剂量的给药溶液。经稀释的给药溶液的克分子渗透压浓度被测得为371mOsm/kg。克分子渗透压浓度的测量结果汇总在下表72中。
3250型,Advanced Instruments Inc.)测量小瓶中的复原溶液的克分子渗透压浓度,并且获得了440mOsm/kg的克分子渗透压浓度值。然后从三个小瓶中取出12mL的复原溶液,用38mL生理盐水稀释至50mL的最终体积,以获得代表2.4mg临床剂量的给药溶液。经稀释的给药溶液的克分子渗透压浓度被测得为317mOsm/kg。随后,从六个小瓶中取出30mL的复原溶液,用20mL生理盐水稀释至50mL的最终体积,以获得代表6.0mg临床剂量的给药溶液。经稀释的给药溶液的克分子渗透压浓度被测得为371mOsm/kg。克分子渗透压浓度的测量结果汇总在下表72中。
此外,用10mL生理盐水将药物产品小瓶复原。该复原溶液的克分子渗透压浓度被测得为352mOsm/kg(表72中的样品4)。得到了约50mL的复原溶液,剂量为4.8mg。
表72:制剂Ib复原溶液的克分子渗透压浓度

纯生理盐水的克分子渗透压浓度被测得为285mOsm/kg。据发现,复原溶液的克分子渗透压浓度与溶液中的Kleptose浓度线性相关,如图44中所示。
因此,当用4.5mL生理盐水复原时,制剂Ib产生440mOsm/kg的复原溶液。对于2.4至6mg的剂量范围,在50mL生理盐水袋中的最终给药溶液的克分子渗透压浓度范围为318mOsm/kg至371mOsm/kg。当复原NS或经稀释NS的体积针对具体剂量改变时,可以从图44基于计算出的HPBCD浓度通过插值获得所得溶液的克分子渗透压浓度。
基于该数据,只要药物浓度对克分子渗透压浓度几乎没有贡献,就可以计算出制剂Ic在生理盐水中的复原溶液的克分子渗透压浓度。对于制剂Ic,每个小瓶包含1mg药物和1875mg Kleptose。当一个小瓶用12.5ml生理盐水复原时,复原溶液的克分子渗透压浓度为约416mOsm/kg,且产品小瓶中的Kleptose为150mg/ml。用生理盐水将3.6mg药物的45-mL复原溶液稀释至50ml时,IV袋中的给药溶液的克分子渗透压浓度为约383mOsm/kg,且Kleptose为112.5mg/ml。尽管该克分子渗透压浓度范围高于血浆克分子渗透压浓度,但仍被认为是IV输注可接受的。
制剂Ic的复原溶液由0.08mg/ml化合物1和150mg/ml Kleptose组成。与制剂Ib的复原溶液相比,其具有相似的溶液pH和更高的Kleptose:药物比率。在某些实施方案中,与制剂Ib的复原溶液相比,制剂Ic的复原溶液具有相当的化学稳定性曲线和改善的物理稳定性。
实施例21:无溶剂组合
如表73和表74中所示,制备了无溶剂化合物1的制剂的两个1-kg试验批次。批次B-1用10重量%的Kleptose制备,目标药物浓度为40μg/mL。批次B-2用20重量%的Kleptose制备,目标药物浓度为80μg/mL。这两个批次的目标药物浓度均为室温下其饱和溶解度的约60%。因为已知化合物1在高于pH 5的溶液中化学不稳定,所以在制剂中使用柠檬酸盐缓冲剂将溶液pH调节至4.2。
表73:无溶剂可行性批次的制剂组成
| 批号 | B-1 | B-2 |
| 化合物1(mg/g) | 0.040 | 0.080 |
| Kleptose(mg/g) | 10 | 20 |
| 无水柠檬酸(mg/g) | 2.21 | 2.19 |
| 无水柠檬酸钠(mg/g) | 2.21 | 2.19 |
| WFI | 定量至1000mg/g | 定量至1000mg/g |
在无溶剂配混过程中,首先将Kleptose溶解在水中。然后将化合物1粉末直接添加到Kleptose溶液中,并用台式顶置式匀浆器(POLYTRON PT3100,Kinematica AG,Switzerland)以6800rpm混合。在混合过程期间,使用带夹套配混器皿将溶液温度保持在20至25℃。对于批次B-1,继续混合24小时。然后停止混合,再将溶液保持在室温下24小时。对于批次B-2,继续混合48小时。在t=4、24和48小时取出溶液样品用于过程中测定测试。测定结果在表74中示出。
表74:无溶剂可行性批次的过程中测定结果

a:目标浓度=0.040mg/g或0.041mg/mL,且溶液密度为1.033g/mL
b:目标浓度=0.080mg/g或0.086mg/mL,且溶液密度为1.069g/mL
c:在t=24h时停止混合。
实施例22:体外组合研究
在以下AML细胞系和实体肿瘤细胞系中进行了体外组合研究,以评定化合物1的活性:
AML细胞系:MOLM-13、MV-4-11、OCI-AML-2、F-36-P、OCT-AML-3、NOMO-1、ML-2、KG-1、HNT-34和HL-60。
乳腺癌细胞系:AU565、ZR-75-30、SK-BR-3、MCF-7(E545K)、BT-474(K111N)和CAL-51(E542K)。
神经内分泌肿瘤(NET)细胞系:COLO320DM、NCI-H727和QGP-1;以及
肾细胞癌(RCC)细胞系:786-O、A-498、ACHN和CAKI-1。
将细胞以最佳接种密度(每孔50微升)接种到384孔组织培养板中。在37℃下用5%CO2使细胞生长1天,然后用化合物进行处理,方式为:向每个孔中添加5微升浓缩的化合物溶液,并在37℃下用5%二氧化碳温育3天。然后测量细胞活力,方式为:将 发光细胞活力试剂添加到经化合物处理的细胞中(每孔20微升),在室温下温育至少20分钟,然后用光度计对发光信号进行定量。
发光细胞活力试剂添加到经化合物处理的细胞中(每孔20微升),在室温下温育至少20分钟,然后用光度计对发光信号进行定量。
对于初步的协同作用分析,使用布利斯独立性组合指数(Bliss IndependenceCombination Index),使用以下方程(Foucquier,Pharma Res Per,3(3),2015)计算每种化合物1组合治疗的协同作用:

其中EA和EB代表每种单一剂的作用,并且EAB代表在给定剂量下该组合的作用。
后续的协同作用分析是使用洛伊相加性(Loewe additivity)、布利斯独立性、最高单一剂(HSA)和协同效应协同作用(CES)这些模型进行的,如先前所述(Veroli G.,Bioinformatics,32(18),2016;Geary N.,Am J Physiol Endocrinol Metab,2012)。
使用CES方程分析所有数据:
CES=EAB-max(EA,EB)
在单剂剂量响应符合希尔斜率(Hill slope)的情况下,将CES分析与洛伊模型、布利斯模型和HSA模型进行比较。
图56A提供了化合物1与1)依维莫司、2)菲卓替尼、3)米哚妥林和4)普拉二烯内酯B的组合的作用。如图56A中所见,剂量响应曲线示出了使用测试化合物时的EC50移动。例如,在所有的测试剂量水平下,与依维莫司的组合相比单一剂化合物1具有较低的EC50。
图56B提供了化合物1与普拉二烯内酯B的组合在AML细胞系中的作用。
图56C提供了化合物1与化合物A的组合在AML细胞系中的作用。
图56D提供了化合物1与化合物B的组合在AML细胞系中的作用。
表75提供了化合物1与示例性第二剂(包括mTOR抑制剂、JAK2抑制剂、FLT3抑制剂、剪接体抑制剂、BET抑制剂和LSD-1抑制剂)的组合在AML细胞系中的协同作用分析的汇总。
表75:与化合物1组合的协同作用分析


表76提供了在MOLM-13细胞系和NOMO-1细胞系中与化合物1组合测试的FLT3抑制剂和JAK抑制剂的清单。
表76
| 化合物 | 协同作用 |
| 索拉非尼 | 无 |
| 舒尼替尼 | 有 |
| 米哚妥林 | 有 |
| 培西达替尼 | 有 |
| 来他替尼 | 有 |
| 坦度替尼 | 有 |
| 奎扎替尼 | 有 |
| 克诺拉尼 | 有 |
| 非戈替尼 | 有 |
| 地西罗替尼 | 有 |
| 巴瑞替尼(Baricitinib) | 有 |
| 鲁索替尼 | 有 |
| 菲卓替尼 | 有 |
| NS-018 | 有 |
| 帕克替尼 | 有 |
| 莫美罗替尼 | 有 |
如上所见,大多数测试组合在AML细胞系MOLM-13和NOMO-1中证实了协同作用。图57展示了在FLT3活化的细胞系(诸如携带FLT3内部串联重复(ITD)突变的细胞系)中,化合物1与米哚妥林(FLT3抑制剂)和鲁索替尼(JAK抑制剂)的组合的协同作用。
化合物1和TOR抑制剂依维莫司、替西罗莫司、1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-115)和7-(6-(2-羟基丙烷-2-基)吡啶-3-基)-1-((反式)-4-甲氧基环己基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮(CC-223)的组合治疗在BrCa(5/6)、RCC(4/4)和NET(2/3)细胞系中示出了协同作用,如图58和图59中所展示。
实施例23:BON细胞中的体外研究
使用 测定进行了体外研究,以评定作为单一剂施用以及与依维莫司(在图60至74中称为RAD)组合施用的化合物1对BON细胞系的信号传导和增殖的活性。
测定进行了体外研究,以评定作为单一剂施用以及与依维莫司(在图60至74中称为RAD)组合施用的化合物1对BON细胞系的信号传导和增殖的活性。
在2D板上处理后24小时、在2D板上处理后120小时、在3D板上处理后120小时以及在2D板上处理后96小时研究了单独的化合物1以及化合物1与依维莫司的组合对信号传导和增殖的作用。
图60提供了单独的化合物1以及化合物1与依维莫司的组合在2nM、20nM和200nM下的相对发光单位(RLU)值。
图61提供了单独的化合物1以及化合物1与依维莫司的组合在2nM、20nM和200nM下的相对发光单位(RLU)值。
图62提供了单独的化合物1以及化合物1与依维莫司的组合在2nM、20nM和200nM下的相对发光单位(RLU)值。
图63提供了单独的化合物1以及化合物1与依维莫司的组合在2nM、20nM和200nM下的相对发光单位(RLU)值。
图64A和图64B提供了两次运行的图线,描绘了单独的化合物1以及化合物1与依维莫司的组合在2nM、20nM和200nM下的相对发光单位(RLU)值。
数据证明,化合物1对BON1没有独立的活性。化合物1与依维莫司组合治疗导致生长抑制的协同作用,如通过使生长停滞为药物前水平,且没有细胞凋亡所见。
实施例24:化合物1和依维莫司在GA0087 PDX模型中的作用–离体测定
使用离体3D测定,调查研究了化合物1和依维莫司对GA0087 PDX模型的细胞活力的组合作用。使用离体3D甲基纤维素测定确定了这两种化合物的50%抑制浓度(IC50),然后使用组合指数确定了在基质组合中的协同作用。
研究设计:用单独的化合物1、化合物1和依维莫司的基质组合,以及一种参考对照化合物以所需的剂量对每种细胞系进行接种和处理。
材料和方法:该研究中使用了胃癌细胞系GA0087。使用含有DMEM/F12+10%FBS+Pen/Strep+补充生长因子的生长培养基,在37℃温度、5%CO2和95%湿度下对细胞进行培养。培养基购自美国的GIBCO或Sigma。
使用了以下材料和试剂:
· 发光细胞活力测定(目录号:G7572,Promega。储存在-20℃)
发光细胞活力测定(目录号:G7572,Promega。储存在-20℃)
·96孔聚苯乙烯微孔板(目录号655096,Greiner bio-one)
·微孔板封盖(目录号656171,Greiner bio-one)
·甲基纤维素(目录号M0512,Sigma)
·后密封黑色粘合剂底部密封件(目录号6005189,Perkin Elmer)
·胶原酶(目录号:17100-017,Invitrogen)
·Falcon细胞滤网(目录号352340,BD Falcon)
将化合物1在DMSO中复原,以制备5mM溶液,以便每次使用15μl等分试样。依维莫司从Selleck获得。顺铂用作参考药物,并且从Hospira Australia pty Ltd获得。
方法:
1%甲基纤维素制备物:将1g甲基纤维素量入带封盖的玻璃容器中。在标准灭菌条件下对该容器进行高压灭菌。将容器冷却,然后添加100mL适当的细胞培养基。用无菌细胞刮刀从底部除去任何残留的甲基纤维素,并剧烈混合内容物。将该容器储存在4℃的摇床平台上,以完成溶解,持续最长48小时,从而实现完全溶解。将甲基纤维素溶液储存在4℃下。
1.单细胞分离
1.在Crownbio HuPrime动物设施中维护PDX肿瘤模型。每周监测肿瘤生长。使用卡尺以二维方式测量肿瘤体积,并使用以下公式,以mm3表示体积:V=0.5axb2,其中a和b分别是肿瘤的长径和短径。
2.用无菌手术工具容纳肿瘤体积为约800mm3的小鼠异种移植物,并用剪刀将肿瘤组织剪碎成小块置于新的组织培养板上的少量PBS中。
3.通过Falcon细胞滤网过滤细胞悬液。用PBS将尼龙网洗涤3至5次。
4.将细胞悬液以1000rpm离心5分钟,然后用PBS洗涤沉淀并再次离心。
5.使用红血细胞裂解缓冲液除去红血细胞。
6.将细胞悬液以1000rpm离心5分钟,然后用PBS洗涤沉淀并再次离心。
7.用适合于不同细胞的细胞培养基重悬细胞沉淀。
8.通过台盼蓝排除法(trypan blue exclusion),用Countstar对细胞进行计数。
2. 细胞活力测定(3D甲基纤维素形式)
细胞活力测定(3D甲基纤维素形式)
第-1天:细胞铺板
1.在对数生长期期间收获细胞。将收获的细胞用适当的细胞培养基培养,并以1000rpm离心3分钟。将细胞重悬,并使用CountStar计数(通过台盼蓝排除法测定,细胞活力应当满足>90%的标准)。
2.用相应的培养基将细胞浓度调整为2×105个细胞/ml。(根据数据库或密度优化测定调整细胞浓度)。
3.将3.5mL细胞悬液与6.5mL 1%甲基纤维素混合。该步骤在0.65%甲基纤维素溶液中产生10ml细胞悬液。
4.根据具有最终细胞密度的板图,将90μL细胞悬液添加到96孔板中。设置两个重复板:一个用于第0天读数(T0),另一个在温育箱中培养以便在终点读数。
将这两个板置于加湿温育箱中,在37℃和5%CO2下温育过夜。
第0天:T0板读数和化合物处理
5.将10μL培养基添加到T0板的每个孔中,以读取T0。
6.将100μl 试剂添加到每个孔中。
试剂添加到每个孔中。
7.将内容物置于轨道式震荡器上混合2分钟,以促进细胞裂解。
8.让这些板在室温下温育10分钟,以使发光信号稳定。
9.将后密封黑色贴纸放置于每个板的底部。
10.使用EnVision多标签读数器读取发光。
11.将化合物1、依维莫司和参考药物溶液按以下指示的浓度稀释。
将化合物1和依维莫司溶解在DMSO中,以制成10mM储备溶液和等分试样。将这些溶液储存在-20℃。将九种浓度的化合物1(3倍稀释)与6种浓度的依维莫司组合。通过相对于DMSO对照归一化来计算响应评分。使用Chou和Talalay方法计算组合指数(CI),其中CI小于1指示协同作用。顺铂在单一化合物剂量响应中用作对照,但未包含在组合测试中。
第7天:7天化合物处理的板读数。
12.在药物温育之后,使用相差显微镜为代表性的孔拍照。
13.将100μL 试剂添加到每个孔中。
试剂添加到每个孔中。
14.将内容物置于轨道式震荡器上混合2分钟,以促进细胞裂解。
15.让这些板在室温下温育10分钟,以使发光信号稳定。
16.将后密封黑色贴纸放置于每个板的底部。
17.使用EnVision多标签读数器记录发光。
数据分析
使用GraphPad Prism 5.0以图形方式显示数据。为了计算IC50,使用具有S形剂量响应的非线性回归模型拟合了剂量响应曲线。存活率的公式如下所示,IC50则由GraphPadPrism 5.0自动生成。
存活率(%)=(Lum测试物品-Lum培养基对照)/(Lum未处理-Lum培养基对照)×100%。
协同作用测定
化合物的相互作用通过多重药物作用分析计算,并根据Chou和Talalay描述的方法通过中位数方程原理进行。Fa是受剂量影响的分数。组合指数(CI)由Chou等人的方程计算,该方程同时考虑了效力(Dm或IC50)和剂量作用曲线的形状(m值)。
这两种化合物的CI的通用方程通过下列方程给出:
其中:分母中的(Dx)1和(Dx)2是单独的化合物1和化合物2的剂量(或浓度),它们展示x%的抑制作用。而分子中的(D)1和(D)2是两种化合物(1和2)的组合剂量,其也有x%的抑制作用(同效)。CI<1、=1和>1分别指示协同作用、相加作用和拮抗作用。
(Dx)1和(Dx)2可以由Chou等人的中位数作用方程计算:

其中:Dm是从中位数作用图的x截距的反对数获得的中位数作用剂量,x=log(D)对y=log{fa/(1-fa)},或Dm=10-(y-截距)/m;并且m是中位数作用图的斜率,fa是受处理影响的细胞的分数。
使用CalcuSyn软件,从每种药物比率浓度下的平均受影响分数计算每个CI。对于2种化合物在7种浓度下的固定比率组合,获得了7个CI值。
表77
| CI范围 | 描述 |
| 0.1 | 极强的协同作用 |
| 0.1-0.3 | 强烈的协同作用 |
| 0.3-0.7 | 协同作用 |
| 0.7-0.85 | 中度的协同作用 |
| 0.85-0.90 | 轻微的协同作用 |
| 0.90-1.10 | 几乎为相加 |
| 1.10-1.20 | 轻微的拮抗作用 |
| 1.20-1.45 | 中度的拮抗作用 |
| 1.45-3.3 | 拮抗作用 |
| 3.3-10 | 强烈的拮抗作用 |
| >10 | 极强的拮抗作用 |
DRI是在给定的作用水平下,与单独使用每种药物的剂量相比,可以将协同作用组合中的每种药物的剂量降低至几分之一的量度。对于两种药物的组合




结果:下表78提供了GA0087模型的IC50和最大抑制率的汇总。
表78

表79

图65A和图65B提供了剂量响应曲线(3D离体克隆形成测定),描绘了在2个实验的GA0087模型中,化合物1和依维莫司的最大抑制。图66以重复实验提供了在GA0087模型中,参考化合物的IC50和最大抑制。
图67A和图67B提供了这两个实验的基质组合抑制作用测定,并且图68A和图68B提供了这两个实验的组合指数。
从数据可以看出,化合物1在GA0087模型中示出活性,并且在大多数测试的浓度下示出与依维莫司的协同作用。特别地,在化合物1的中等剂量(0.5至111.1nM)的多个浓度下,化合物1示出与依维莫司的协同作用。
实施例25:化合物1和依维莫司在GA0087 PDX模型中的作用–体内测定
在雌性BALB/c裸鼠中的 胃癌异种移植物GA-0087(神经内分泌肿瘤)模型中测试了化合物1和依维莫司的组合治疗。收获来自stock小鼠的肿瘤片段并用于接种到小鼠中。用原发性人肿瘤异种移植物模型GA0087肿瘤片段(P7,直径2至3mm)接种于每只小鼠的右侧腹部皮下,以发展为肿瘤。为了进行功效研究,当平均肿瘤体积达到约196mm3时,基于肿瘤体积和体重,将小鼠随机分为9组(每组有10只小鼠,1个媒介物对照组、2个依维莫司单一剂治疗组和3个化合物1单一剂治疗组,以及3个组合治疗组),并在同一天(研究的第0天)开始给药。给药和计划在图69中示出。在接种肿瘤之后,每日检查动物的发病率和死亡率。在常规监测时,检查动物的肿瘤生长和治疗对正常行为诸如活动能力、食物和水消耗、体重增加/减少、眼睛/毛发消光和任何其它异常作用的任何作用。记录被安乐死小鼠的死亡。所有组均在第40天终止。每个组收集三个肿瘤,肿瘤的体积接近每个组的中位数。收集相同小鼠的血浆。为了在三个或更多个组之间进行比较,执行了单因素ANOVA,然后进行多重比较程序。使用SPSS 18.0分析所有数据。P值<0.05被认为是统计学显著的。
胃癌异种移植物GA-0087(神经内分泌肿瘤)模型中测试了化合物1和依维莫司的组合治疗。收获来自stock小鼠的肿瘤片段并用于接种到小鼠中。用原发性人肿瘤异种移植物模型GA0087肿瘤片段(P7,直径2至3mm)接种于每只小鼠的右侧腹部皮下,以发展为肿瘤。为了进行功效研究,当平均肿瘤体积达到约196mm3时,基于肿瘤体积和体重,将小鼠随机分为9组(每组有10只小鼠,1个媒介物对照组、2个依维莫司单一剂治疗组和3个化合物1单一剂治疗组,以及3个组合治疗组),并在同一天(研究的第0天)开始给药。给药和计划在图69中示出。在接种肿瘤之后,每日检查动物的发病率和死亡率。在常规监测时,检查动物的肿瘤生长和治疗对正常行为诸如活动能力、食物和水消耗、体重增加/减少、眼睛/毛发消光和任何其它异常作用的任何作用。记录被安乐死小鼠的死亡。所有组均在第40天终止。每个组收集三个肿瘤,肿瘤的体积接近每个组的中位数。收集相同小鼠的血浆。为了在三个或更多个组之间进行比较,执行了单因素ANOVA,然后进行多重比较程序。使用SPSS 18.0分析所有数据。P值<0.05被认为是统计学显著的。
图69提供了单独地和组合地使用化合物1和依维莫司时的平均肿瘤体积。从数据可以看出,化合物1示出肿瘤生长抑制。与单独使用任一种剂相比,化合物1与1.25mg/kg依维莫司的组合显著增加了肿瘤生长抑制。用5mg/kg化合物1和1.25mg/kg依维莫司的组合实现了短暂的肿瘤消退。
总而言之,使用依维莫司(1.25mg/kg和5mg/kg)和化合物1(1.25mg/kg和2.5mg/kg)的单一剂治疗产生中度的抗肿瘤活性,而使用化合物1(5mg/kg)的单一剂治疗和使用组合治疗(依维莫司,1.25mg/kg,和化合物1,1.25mg/kg、2.5mg/kg和5mg/kg)针对 胃癌异种移植物模型GA0087产生显著的抗肿瘤活性。
胃癌异种移植物模型GA0087产生显著的抗肿瘤活性。
实施例26:化合物1对骨髓纤维化祖细胞的作用
在使用骨髓纤维化患者样品的集落形成测定中研究了化合物1的作用。将来自骨髓纤维化患者的外周血单核细胞(PBMC)接种在有和没有化合物1的甲基纤维素(methocult)培养基中14天,之后测定集落形成细胞的数量。
使用了以下样品:
MF13:新发MF,其特征在于野生型JAK2、野生型FLT3、野生型NPM1、野生型CEBPA
MF14:新发MF,其特征在于野生型JAK2、突变型CALR外显子9、野生型ASXL1、野生型MPL
图70中提供的数据证明,在来自骨髓纤维化患者的样品中,化合物1以剂量依赖性方式减少了集落形成细胞的数量。在这些样品中实现的IC50低于来自健康志愿者的样品中观察到的IC50,从而表明化合物1有潜力用于治疗骨髓纤维化。
实施例27:骨髓增殖性赘生物的组合研究
在被工程化为稳定表达外源蛋白质的BaF3细胞中进行了体外组合研究,以评定化合物1与以下JAK2抑制剂组合的活性。使用了以下细胞系:hCRBN是表达人CRBN的BaF3细胞系;EF1a-GFP-P2A-Nluc-P2A-JAK2是hCRBN,和表达野生型JAK2的BaF3细胞系;新转导的IL3依赖性EF1a-GFP-P2A-Nluc-P2A-JAK2-V617F是hCRBN和仍依赖于表达IL3的BaF3细胞系的突变型JAK2V617F;并且EF1a-GFP-P2A-Nluc-P2A-JAK2-V617F;IL3非依赖性克隆是hCRBN和已经适合于变得独立于IL3 BaF3细胞系的突变型JAK2V617F。该测定中使用的JAK2抑制剂是NS-018、INCB018424(鲁索替尼;Jakafi)、CYT387(莫美罗替尼)、TG101348(菲卓替尼)和帕克替尼。
通过添加化合物1的系列稀释液(起始浓度为50nM)和JAK2抑制剂的系列稀释液(所有JAK2抑制剂的起始浓度均为10μM)的组合,来用化合物对细胞进行处理。依维莫司用作对照。3天后,通过 测量细胞活力。
测量细胞活力。
如通过图71至图77中的数据所证明,化合物1或JAK2抑制剂当在IL3依赖性BaF3细胞系中用作单一剂时,未观察到差异。IL3非依赖性JAK2V617F细胞对大多数JAK2抑制剂、化合物1和依维莫司更为敏感。
如图78中所证明,化合物1和NS-018的组合示出了亲本JAK2野生型细胞和JAK2V617F细胞中的EC50移动。观察到hCRBN的野生型细胞系与新转导的JAK2 V617F细胞系之间的协同作用谱没有明显差异。与所有三个IL3依赖性细胞系相比,IL3非依赖性JAK2V617F细胞对NS-018单一剂更为敏感。在NS-018的临床Cmax(2.57μM,如在患有骨髓纤维化的患者中用JAK2抑制剂NS-018进行的I期、开放标签、剂量递增、多中心研究中所述,Leukemia(2017)31,393–402)下观察到协同作用。
如图79中所示,在小于100nM的NS-018下,观察到所有4个细胞系均没有明显的EC50移动。
如图80中所证明,化合物1和鲁索替尼的组合示出了亲本JAK2野生型细胞和JAK2V617F细胞中的强烈EC50移动。观察到hCRBN的野生型细胞系与新转导的JAK2 V617F细胞系之间的协同作用谱没有明显差异。与所有三个IL3依赖性细胞系相比,IL3非依赖性JAK2 V617F细胞对鲁索替尼单一剂更为敏感。在鲁索替尼的临床Cmax(约0.5至1μM,如Blood(2011)118:5162中所述)下观察到协同作用。
如图81中所示,在小于100nM的鲁索替尼下,观察到所有4个细胞系均没有明显的EC50移动。
如图82中所证明,化合物1和莫美罗替尼的组合示出了亲本JAK2野生型细胞和JAK2V617F细胞中的强烈EC50移动。观察到hCRBN的野生型细胞系与新转导的JAK2 V617F细胞系之间的协同作用谱没有明显差异。与所有三个IL3依赖性细胞系相比,IL3非依赖性JAK2 V617F细胞对莫美罗替尼单一药剂更为敏感。
如图83中所证明,化合物1和帕克替尼的组合示出了亲本JAK2野生型细胞和JAK2V617F细胞中的强烈EC50移动。观察到hCRBN的野生型细胞系与新转导的JAK2 V617F细胞系之间的协同作用谱没有明显差异。
如图84中所证明,化合物1和菲卓替尼的组合示出了亲本JAK2野生型细胞和JAK2V617F细胞中的强烈EC50移动。观察到hCRBN的野生型细胞系与新转导的JAK2 V617F细胞系之间的协同作用谱没有明显差异。
如图85中所证明,化合物1和依维莫司的组合示出了亲本JAK2野生型细胞和JAK2V617F细胞中的轻微EC50移动。观察到在IL3非依赖性JAK2V617F细胞中存在更强烈的EC50移动。在所有的BaF3细胞系中,使用依维莫司时EC50移动的程度比使用与JAK2抑制剂的组合时弱,这表明mTOR活性不是负责JAK2抑制剂与化合物1之间的协同作用的主要下游机制。
结论:这些数据证明了在被工程化为表达具有野生型JAK2或JAK2V617F的hCRBN的BaF3细胞中化合物1与JAK2抑制剂之间的协同作用。由于与mTOR抑制剂依维莫司组合示出了相比与JAK2抑制剂组合降低的协同作用,所以这些结果表明,在使用化合物1的情况下JAK2介导的协同作用很可能是通过除了mTOR活性之外的其它机制发生的。
实施例28:在AML细胞系中使用化合物1和JAK2抑制剂的组合研究
在12种AML细胞系中进行了体外组合研究,以评定化合物1与以下JAK2抑制剂组合的活性:NS-018、INCB018424(鲁索替尼;Jakafi)、CYT387(莫美罗替尼)、TG101348(菲卓替尼)和帕克替尼。
使用了下列AML细胞系:
JAK2V617F:HEL、SET-2、MUTZ-8
JAK野生型(WT):HL-60、HNT-34、KG-1、ML-2、NOMO-1、MOLM-13、MV4-11、F36P、OCI-AML2
3天后,通过 测量细胞活力,如本文其它地方所述。
测量细胞活力,如本文其它地方所述。
如通过图86中的数据所证明,NS-018和鲁索替尼作为单一剂抑制JAK2V617F AML细胞系的细胞活力。
如图87中所证明,化合物1和NS-018的组合示出了抑制JAK2 V617F在HEL、SET-2和MUTZ-8细胞系中的活力的协同作用。
如图88中所证明,化合物1和鲁索替尼的组合示出了抑制JAK2 V617F在HEL、SET-2和MUTZ-8细胞系中的活力的协同作用。协同作用在对化合物1单一剂较不敏感的细胞系(HEL)上较明显。
如图89中所证明,化合物1和依维莫司的组合在抑制HEL细胞的活力方面示出协同作用,但是在SET-2或MUTZ-8细胞上未观察到明显的IC50移动。
如图90中所证明,所有五种JAK2抑制剂在抑制JAK2 V617F细胞活力方面均示出与化合物1的协同作用。
结论:使用所有5种临床阶段的JAK2抑制剂均证明了与化合物1的强烈协同作用。
实施例29:使用化合物1和IDH2抑制剂的组合研究
在IDH2突变细胞系TF1-R140Q中进行了体外组合研究,以评定化合物1与IDH2抑制剂恩西地平(AG-221)组合的活性。
将细胞以0.2e6个细胞/ml 2ml培养物的密度铺板在6孔板中。恩西地平以0、200nM和1000nM的浓度测试,而化合物1则以0、10nM、30nM、100nM的浓度测试。
图91提供了在该研究中使用的给药计划。
在以下培养基中培养TF1细胞:含有HEPES和L-谷氨酰胺、10%FBS、Pen/Strep、G418(最终浓度500μg/ml)和GM-CSF(最终浓度5ng/ml)的RPMI。在使用或未使用如图91中所述的化合物处理的情况下将细胞培养7天。
对于血红蛋白生成测定,用PBS将TF1细胞洗涤三次,以去除残留的GM-CSF,然后铺板(以100,000个细胞/ml)。然后使用EPO(2单位/ml)诱导细胞分化。诱导持续7天,第4天更换培养基,并添加具有新鲜EPO的培养基。收集细胞沉淀并对其成像以分析血红蛋白生成含量(作为分化成血统的替代品)。对细胞进行沉淀、洗涤、用一组抗体染色并通过流式细胞术进行分析,使用了以下抗体:
| 抗体 | 克隆 | 荧光团 |
| CD34 | 8G12 | PE-Cy7 |
| CD38 | HIT2 | BV421 |
| CD235a | GA-R2(HIR2) | BV711 |
| 波形蛋白 | RV202 | FITC |
| c半胱天冬酶3 | C92-605 | PE |
| GSPT1 | AF647(APC) |
在血红蛋白生成测定中,恩西地平和化合物2产生了增加血红蛋白表达的预期作用,如图92中所见。此外,用化合物1和恩西地平进行组合治疗示出血红蛋白生成和细胞分化增强,如图93至图97中所示的HSC(CD34+/CD38-)和祖细胞CD34+/CD38+减少以及CD34-/CD38-和CD34-/CD235a+成红血细胞增加所示。
图93至图94中提供的来自EPO诱导的分化测定的结果证明了化合物1和恩西地平在减少TF-1:IDH2R140Q祖细胞(CD34+/CD38+)和造血干细胞(HSC)(CD34+/CD38-)、以及增加分化的CD34-/CD38-细胞和成红血细胞方面的增强的作用。图95示出了CD235a+(血型糖蛋白A)的增加,其中CD235a+是红细胞群体的标志物。这种增强作用主要体现在计划A和计划C中。
图96和图97示出了在TF1测定中,GSPT1降解在CD34+细胞中优先于在分化细胞中发生。计划A(在恩西地平之前24小时添加化合物1)由于分化增加而导致CD34+的绝对计数最低(在计划C中也可见),并且具有优先杀死CD34+细胞的持续作用。
图98证明了化合物1对总细胞计数和HSC计数(CD34+/CD38-)和祖细胞计数(CD34+/CD38+)的增殖的抑制作用。如图所示,计划A对细胞增殖具有强烈且持久的抑制作用,并且计划A导致未分化的CD34+细胞的细胞数最少。图98示出恩西地平具有温和的促进生长作用,但是与化合物1的组合却导致总细胞计数(左边)以及干细胞组和祖细胞组的细胞计数(右边的两个图)明显减少。
结论:数据证明,恩西地平和化合物1的组合导致干细胞和祖细胞中的GSPT1水平与CD34-/CD38-和成红血细胞的分化程度更高的亚群相比较低。此外,示出恩西地平具有温和的促进生长作用,但是与化合物1的组合却导致总细胞计数以及干细胞组和祖细胞组的细胞计数减少。干细胞和祖细胞的细胞计数净损失可能是由于这些细胞死亡或CD34+细胞分化为CD34-细胞所致。
实施例30:化合物1在实体肿瘤细胞系中的作用
通过Luminex测定测试了化合物1在563个实体肿瘤细胞系中的活性。下表80中提供的AUC值是基于中位数荧光强度值(PRISM中Luminex测定的主要读出值)计算得出的。该测定中使用了以下参数:
-一式三份地使用了8点剂量
-利用连续4倍稀释,使用了10μM最大浓度(最低约1nM)
-23个池中的563个贴壁细胞系。
-处理持续时间为5天。
表80:







结论:这些数据证明了化合物1对实体肿瘤细胞系的活性,对肺、肠、胃、肝、胰和肾的细胞系的活性最高。
实施例31:化合物1和依维莫司在QGP1模型中的作用–体内测定
在雌性NOD/SCID小鼠中的人QGP1胰腺生长抑素瘤异种移植物中测试了化合物1和依维莫司的组合治疗。从第1天开始,对已形成皮下QGP1肿瘤的9组小鼠(n=10)进行治疗。依维莫司每日以3或10mg/kg腹膜内(i.p.)给药,持续21天(qdx21)。每天两次(bidx21)腹膜内(i.p.)给药媒介物(5%NMP/45%PEG400/50%盐水)和化合物1(2.5、5或10mg/kg),间隔3小时。第1组接受媒介物;第2组和第3组分别接受3和10mg/kg依维莫司;第4、5和6组分别接受2.5、5和10mg/kg化合物1;第7、8和9组分别接受3mg/kg依维莫司加上2.5、5和10mg/kg化合物1。到第52天研究结束时,每周使用卡尺测量肿瘤两次。从每个组中取样处于或接近组中位数肿瘤体积2000mm3终点的三个肿瘤。
治疗功效基于肿瘤生长抑制(TGI),被定义为受治疗小鼠和对照小鼠的最终(第52天)中位数肿瘤体积(MTV)之间的差异。利用曼-惠特尼U检验(Mann-Whitney U-test)对结果进行了分析,认为结果在P≤0.05时具有统计学显著性。通过体重测量和对受治疗动物的频繁观察来评定药物耐受性。
用媒介物治疗的第1组对照小鼠的第52天中位数肿瘤体积(MTV)为2181mm3,其中单个肿瘤体积在650至5000mm3范围内。接受依维莫司单一疗法的第2组和第3组分别导致肿瘤生长抑制41%和39%。另外,第2组比接受化合物1单一疗法的第4、5和6组更有效(P≤0.01,相对于第4组和第5组的P≤0.001),即使第2组和第3组与对照相比没有达到统计学意义。达到有统计学显著功效的唯一治疗方案是第9组(3mg/kg依维莫司和10mg/kg化合物1),与对照相比,其实现了两次局部肿瘤消退(PR)和70%肿瘤生长抑制(TGI),其中第52天的MTV为650mm3,并且单个肿瘤体积在446至1688mm3范围内。在接受化合物1的所有组中,所有方案在第14天与第17天之间均具有良好的耐受性,且体重减少适中(1.6%至6.8%)。
结论:在该研究的条件下,用依维莫司给药的组证明,某些肿瘤生长抑制没有统计学显著性。另外,依维莫司(3mg/kg)和化合物1(10mg/kg)的组合相比对应的单一疗法显著延迟了雌性NOD/SCID中的人QGP1胰腺生长抑素瘤异种移植物的生长。
实施例32:用化合物1和依维莫司的组合处理后,BON细胞中GSPT1水平的降低
方法:在含有10%胎牛血清和青霉素/链霉素抗生素的DMEM:F12K培养基中培养BON细胞。将细胞以3e6个细胞铺板在10cm培养皿中,并在120小时内每日用媒介物、RAD(依维莫司)和/或单独的浓度增加的化合物1处理。收获细胞并用抗体探测以进行蛋白质印迹分析,然后使用Li-Cor Odyssey成像系统成像。所有总抗体和磷酸化抗体均获自CellSignaling Technology。
结果:化合物1引起剂量依赖性GSPT1降解,并且对mTORC1效应子(诸如p4EBP1或S6K1介导的核糖体蛋白S6的磷酸化)没有直接作用。相比之下,RAD通过抑制mTORC1显著降低了S6的磷酸化和4EBP1的磷酸化,并通过负反馈增加了pAKT,图99。与单独的单一化合物相比,RAD和化合物1的组合显著增加了GSPT1降解、增加了对细胞生长(pS6)和cap依赖性翻译的抑制(p4EBP1、p-elF2、pAKT、elFG1与周期蛋白D1减少)并且活化了AMPK途径(pAMPK增加)。
结论:与使用单独的RAD或化合物1处理相比,使用RAD和化合物1的组合处理胰腺神经内分泌肿瘤细胞导致GSPT1降解增加,这可能导致RAD在细胞生长、帽依赖性翻译和代谢活动中的作用增强。
实施例33:用依维莫司或激酶抑制剂与化合物1组合处理对AML细胞的作用
为了评定对mTOR、FLT3、JAK2或JAK3的抑制是否可以增强AML细胞对化合物1的响应,用单独的化合物1或化合物1与一组激酶抑制剂的组合对U937 AML细胞进行处理。在存在或不存在指定浓度的各种抑制剂的情况下,用DMSO媒介物对照或化合物1对细胞处理(参见图101A至图101E)5天。通过Cell-Titer Glo测量细胞增殖,并且将细胞增殖的百分比相对于用DMSO对照处理的亲代细胞归一化。
此外,用100nM化合物1单独地或与指定浓度(参见图102)的依维莫司、奎扎替尼、鲁索替尼、AZD1480和托法替尼组合处理U937 AML细胞16小时。收集全细胞提取物并进行免疫印迹分析。
结果:这5种激酶抑制剂和依维莫司增强了化合物1的抗增殖作用,如通过增殖IC50值向左移动所示(图100A至图100E)。化合物1的生长抑制作用通过与依维莫司、鲁索替尼、AZD1480和托法替尼组合处理而得到增强,并且与GSPT1耗竭增加和半胱天冬酶-3裂解有关联(图101)。
结论:与用单独的化合物1或抑制剂处理相比,用化合物1和mTOR、FLT3、JAK2或JAK3抑制剂的组合处理AML细胞导致GSPT1降解增加和细胞凋亡增加。
实施例34:用化合物1作为单一剂或与维奈托克组合来处理AML细胞系。
方法:Cell TitreGlo:将5000个细胞以每孔50μl铺板在容纳有指定浓度的化合物组合的384孔板中。处理48小时之后,测量相对ATP水平,方式为:添加Cell TitreGlo试剂、在黑暗中温育30分钟,然后在Envision读板仪上测量发光。
结果:示出了使用化合物1和维奈托克组合处理多种AML细胞系的协同作用。
结论:用维奈托克处理使AML细胞对化合物1处理敏感。在存在亚致死剂量的维奈托克时,化合物1剂量响应曲线向左移动示出了这种协同作用。图102A至图102G示出了在有和没有指定浓度的维奈托克的情况下,当与浓度增加的化合物1一起温育48小时之后对AML细胞系增殖的作用。
实施例35:用化合物1和维奈托克对AML细胞进行组合处理。
方法:Cell TitreGlo:将KG-1细胞接种(每ml 200,000个细胞,每孔0.05mL)在384孔组织培养板(Corning,3764)中,然后使其在37℃和5%CO2下生长1天。然后在37℃和5%CO2下,用指定浓度的维奈托克和/或化合物1处理细胞3天。处理之后,测量相对ATP水平,方式为:添加Cell TitreGlo试剂(每孔0.025mL)、在黑暗中温育20分钟,然后在Envision读板仪上测量发光。
蛋白质印迹:将KG-1细胞以每mL 500,000个细胞、每孔4mL接种在6孔组织培养板中。在37℃和5%CO2下,用指定剂量的维奈托克和/或化合物1处理细胞16小时。然后用冷PBS将细胞洗涤两次,并在冰上用补充有150mM NaCl和Halt蛋白酶及磷酸酶抑制剂混合物(Thermo)的M-PER裂解缓冲液(Thermo)裂解30分钟。通过离心(14000rpm,10min,4℃)使粗制裂解物澄清,并通过BCA测定对总蛋白进行定量。通过蛋白质印迹法探测了GSPT1(Abcam,ab49878)、Mcl-1(CST,4572)、Bcl-2(CST,4223)、经切割的半胱天冬酶3(CST,9664)和GAPDH(Santa Cruz,sc-47724)的水平,并且在LiCor红外成像仪上成像。
半胱天冬酶3/7和汇合度活细胞分析:将KG-1细胞接种(每ml 200,000个细胞,每孔0.05mL)在预先涂有纤连蛋白(Sigma,F1141)的384孔组织培养板(Corning,3764)中,然后使其在37℃和5%CO2下生长1天。然后用指定浓度的维奈托克和/或化合物1和半胱天冬酶-3/7绿色细胞凋亡测定试剂(Incucyte,4440)处理细胞。在37℃和5%CO2下温育的同时,在3天的时间里每4小时用IncuCyte活细胞分析成像系统对细胞成像。图103示出了相对ATP水平,其作为响应于化合物1和维奈托克的几种剂量组合的活力的量度。图104示出了测量在用一组剂量的化合物1、维奈托克或化合物1和维奈托克的组合处理后16小时,KG-1细胞中的GSPT1、Mcl-1、Bcl-2、经切割的半胱天冬酶3和GAPDH蛋白水平的蛋白质印迹分析。图105A和图105B示出了KG-1汇合度的活细胞分析(图106A)和细胞凋亡事件计数的活细胞分析(图106B),显示化合物1和维奈托克组合处理与用单一剂处理相比增强了细胞凋亡。
结果:示出了使用化合物1和维奈托克组合处理FLT3-ITD AML细胞系(KG-1)的协同作用。示出了单独的化合物1以及与维奈托克的组合降低KG-1细胞中Mcl-1的水平。此外,与使用单一药剂进行处理相比,化合物1和维奈托克对KG-1细胞的组合处理增强了细胞凋亡。
结论:用化合物1处理AML细胞降低了MCL-1的水平。与用单独的维奈托克或化合物1处理相比,用维奈托克和化合物1组合处理具有协同作用,并导致细胞凋亡增加。
实施例36:用依维莫司与化合物1组合处理对AML细胞系的作用
为了确定对mTOR的抑制是否可以促进化合物1诱导的GSPT1耗竭并诱导细胞凋亡,将AML细胞用单独的化合物1、单独的依维莫司或用依维莫司和化合物1的组合处理。收集全细胞提取物并进行免疫印迹分析。
结果:尽管依维莫司处理显著阻断了mTOR途径的活化,这通过S6K1和4E-BP1的磷酸化水平降低示出,但是对GPST1表达和半胱天冬酶-3裂解的作用微乎其微(参见图106)。然而,与使用单独的化合物1处理相比,依维莫司显著增强了化合物1对GSPT1降解、Mcl-1损耗和凋亡细胞诱导的作用。
在多种AML细胞系(MOLM-13、MV-4-11、NB-4、U937、UT-7、OCI-AML2和F-36P)中观察到依维莫司赋予的对化合物1的致敏作用。在AML细胞系HNT-34和KG1中,单独进行化合物1处理足以诱导强烈的GSPT1降解、半胱天冬酶-3裂解和Mcl-1损耗。
结论:与使用单独的化合物1处理相比,用化合物1和依维莫司的组合处理AML细胞导致GSPT1降解增加、细胞凋亡增加和Mcl-1损耗。
实施例37:化合物1对来自MDS患者的细胞的作用
方法。对来自MDS患者的骨髓单核细胞(BMMC)或从来自相同患者样品的BMMC分离的匹配CD34+细胞进行离体培养,以评定其对化合物1的敏感性。将BMMC以106个细胞/mL接种在完全培养基(补充有LDL、FLT3L、IL-6、IL-3和TPO的CellGenix培养基)中,并且将CD34+细胞以5x105个细胞/mL接种在完全干细胞培养基(补充有CC100细胞因子混合物的无血清StemSpan培养基)中。在最长达10天期间,让所有培养物暴露于0、37和111nM的化合物1,并且每周收集细胞两次,用于细胞培养基更换和细胞计数评定。
结果。如图107中所示,暴露于化合物1减少了来自2名不同MDS患者的样品中的总BMMC中的细胞数量,但同时也减少了CD34+母细胞中的细胞数量。所需的动力学和浓度因患者和细胞类型不同而异。
结论。化合物1使MDS样品中的细胞数量减少,这示出大量BMMC与分离的CD34+母细胞的敏感性谱之间存在差异
实施例38:用化合物1和依维莫司处理对来自MDS患者的细胞的作用
方法。对来自MDS患者的BMMC进行离体培养,以评定其对用化合物1和依维莫司组合处理的敏感性。为了评估对MDS骨髓细胞的总体作用,将总BMMC以106个细胞/mL接种在完全培养基(补充有LDL、FLT3L、IL-6、IL-3和TPO的CellGenix培养基)中,然后暴露于单独的化合物1(0、37、111和333nM)或其与固定浓度111nM的依维莫司的组合。维持培养1周,并在培养的第1、3和7天通过活细胞计数来测量细胞数。在暴露之后24小时,通过流式细胞术测量对细胞凋亡和GSPT1降解的作用。
为了更好地定义化合物1对MDS祖细胞/干细胞的作用,将这些BMMC接种在Methocult培养基中,然后暴露于单独的化合物1(0、37、111、333和1000nM)或其与111nM依维莫司的组合,以评定它们的克隆形成潜力。在培养14天之后,使用StemVision对集落数进行评分。
结果。图108A示出了用化合物1处理对来自MDS患者样品的BMMC的作用。用37nM化合物1处理在培养3天之后使细胞数减少了约70%,并且在较高浓度下,这种作用被增强至出现最多95%的细胞数减少。在37nM下未观察到单一剂的作用。有趣的是,当与依维莫司组合时,该作用显著增强,并且该低剂量的37nM化合物1与依维莫司组合处理实现了用111nM化合物1单一剂处理时观察到的相似抑制作用。图108B示出了MDS祖细胞/干细胞中的相同趋势。在暴露于化合物1 24小时之后,GSPT1降解,并且通过与依维莫司组合处理,该作用在较低剂量下略有增加(图109)。在暴露于化合物1后,半胱天冬酶3被活化,并且该作用通过与依维莫司组合而得到增强。
结论。总而言之,与依维莫司组合处理提高了MDS患者样品中对低剂量化合物1的敏感性,并且可以改善治疗指数。这种作用通过作为GSPT1降解的结果诱导细胞凋亡来加以介导。
实施例39:化合物1—新型Cereblon E3连接酶调控药物在患有复发性或难治性急性骨髓性白血病的对象中的1期、开放标签、剂量发现研究
主要目标:确定化合物1的安全性和耐受性。定义化合物1的非耐受剂量(NTD)、最大耐受剂量(MTD)和/或推荐2期剂量(RP2D)。
次要目标:提供关于化合物1的初步功效的信息。表征化合物1的药代动力学(PK)。
研究设计:该研究是化合物1在患有复发性或难治性AML的对象中的开放标签、1期、剂量递增和扩大、首次人体试验(first-in-human)临床研究。该研究的剂量递增部分(A部分)将评估静脉内施用的化合物1的递增剂量的安全性和耐受性,并确定化合物1的MTD。在A部分中,将测试两种制剂。扩大部分(B部分)将进一步评估所选择的扩大群组中以等于或小于MTD施用的化合物1的安全性和功效,以确定RP2D,其中每个群组包括最多大约20名可评估对象。可以选择一个或多个给药方案用于群组扩大。A部分和B部分将由3期构成:筛选期、治疗期和随访期。白血病响应将由研究者确定。疾病评定将基于国际工作组AML响应标准(Cheson等人,J Clin Oncol 2003;21(24):4642-9)。
筛选期:筛选期始于第一剂量的化合物1之前28天。在开始任何其它研究程序之前,对象和管理人员必须签署知情同意书并注明日期。所有筛选测试和程序必须在第一剂量的化合物1之前28天内完成。
治疗期:在治疗期中,在不存在疾病进展、复发、不可接受的毒性或对象/内科医生决定退出的情况下,将在每个28天周期的第1至5天静脉内施用化合物1最多4个周期。(疾病进展被定义为:与基线(筛选)骨髓母细胞计数相比,骨髓母细胞计数百分比增加了>50%,持续存在间隔至少1个月的至少2次骨髓评定,除非基线骨髓母细胞计数>70%,在这种情况下,发现母细胞>70%持续存在间隔至少1个月的2次基线后骨髓评定将被视为进展,或者基线绝对外周血母细胞计数翻倍持续存在≥7天并且最终绝对外周血母细胞计数>10x109/L。)在A部分期间,如果对象显示出临床有益效果(SD或PR)并且耐受研究药物,且无不可接受的毒性,则可以允许在第4周期之外再进行2个治疗周期。如有必要,可以在附加的群组中基于毒性、PK曲线和PD发现评估经修改的给药计划(例如,给药从5天增加至最多10天,或增加输注长度)。所有对象都将被要求在每个周期的第1天之前开始钙、骨化三醇和维生素D补充剂至少3天,并且继续直到每个周期中化合物1的最后剂量之后≥3天(例如,当在第1至5天施用化合物1时,≥第8天)。
在周期1中,骨髓评估将在第28天(±3天)进行。根据第28天的骨髓评估,将跟踪骨髓发育不全、无持续性白血病迹象、中性粒细胞减少≥3级的对象另外2周,以进行周期1的安全性监测(总持续时间42天)。在血液恢复时或第42天(±3天)将进行附加的骨髓评定。因此,在A部分中,周期1期间的剂量限制性毒性(DLT)评估窗口将为最多42天(28或42天)。≥2个周期的长度将为28天。后续的周期应当在前一个周期的最后一天之后≤7天开始。
随访期:在随访期中,在化合物1的最后剂量之后,将跟踪所有对象28天(±3天),以评估安全性。未记录疾病进展(或复发)的对象将在第1年的后续每8周(±1周)和第2年的每12周(±2周)进行全血计数和外周血涂片的功效评估,或者直到疾病进展(或复发)、开始新抗癌疗法、同意退出研究、死亡或试验结束,以先出现者为准。将在第1年结束时完成骨髓评估,并且在随访期期间根据临床状况进行。
将跟踪所有对象以进行生存随访,该生存随访根据功效长期随访计划进行最多2年或直到死亡、失访或试验结束,以先出现者为准。生存随访可以通过记录查阅(包括公开记录)和/或与对象、家庭或对象的主治医生电话联系进行。
A部分-剂量递增。在递增期(A部分)期间,将使用经修改的加速滴定设计(Simon等人,J Natl Cancer Inst 1997;89(15):1138-47)来建立初始毒性。将给每个一名或多名对象的群组施用化合物1,每个群组的剂量将以100%递增的方式增加,直到DLT窗口中≥2名对象经历化合物1相关的≥2级不良事件(可以是不同的群组),或者DLT窗口中≥1名对象经历DLT。此时,当前群组和所有后续群组将扩大招募3至6名对象。同时将开始剂量递增不超过50%的剂量递增计划,以便建立NTD和MTD。初始剂量将是0.3mg。在研究开始时,将利用化合物1的初始制剂(制剂A),并且在剂量递增期间,将引入化合物1的第二制剂(表43中所述的制剂Ib)来代替制剂A。制剂A具有以下组成(参见美国公布号2017/0196847-A1)。


制剂A中的DMA残留溶剂必须不超过ICH Q3C Impurities:Residual Solvents(ICH Q3C杂质:残留溶剂)中设定的允许每日暴露(PDE)限值,以便使用剂量递增群组进行大于2.4mg的每日化合物1剂量。制剂Ib中的残留溶剂(甲酸)水平允许化合物1的日剂量最高达20mg,而不会超过ICH Q3C指南中设定的其PDE。
在审查了在制剂A的初始剂量水平中观察到的毒性之后,将以新的剂量群组(根据研究剂量递增指南)引入制剂Ib。
可以决定评估较高剂量群组、剂量群组内的另外对象、中等剂量群组、更小的剂量递增、替代给药计划(例如,化合物1施用从5天增加至最多10天,或更长的输注时间),并且/或者基于可用的临床和实验室安全性数据、PK曲线和PD发现的综述发布MTD。在评估替代给药计划的事件中,起始剂量和计划将不超过先前已满足剂量递增标准的剂量群组的剂量强度。
在剂量递增期间在任何群组中施用第一剂量之后,观察每个群组中的对象至少28天和最多42天(周期1,DLT窗口),然后可以开始下一个更高剂量群组。在给定剂量递增群组中每天将招募不超过一名对象。可评估DLT的对象被定义为:在周期1期间已接受化合物1的总计划周期1剂量的至少80%(例如,5天剂量计划的4个完整化合物1剂量;在剂量缺失的情况下,在第7天或之前完成≥4个剂量),不经历DLT,或者在接受化合物1的至少一个剂量(或其部分)之后经历DLT。
在A部分中评估替代剂量计划(例如,给药从5天增加至最多10天)的事件中,将适用用于确定可评估DLT的对象的相同标准。不可评估DLT的对象将被替换。
如果剂量群组中>33%的可评估对象在周期1期间经历DLT,则剂量将被认为是不可容忍的。MTD将被定义为小于NTD的最后剂量,其中在周期1期间≤33%的可评价对象经历DLT。如果在第一剂量群组中6名可评估对象中的2名或更多名经历DLT,则可以探究更低剂量的群组(即,0.1mg化合物1)。可以评估中等剂量的化合物1(在NTD与NTD之前的最后剂量水平之间的剂量),以准确确定MTD。
在DLT评定期期间将不允许对象内剂量递增;然而,在≥2个周期中,无疾病进展迹象、耐受化合物1的分配剂量的对象可以递增至该研究中的至少一个群组的对象示出充分耐受的最高剂量水平(即,≤33%的可评估对象在该剂量水平经历了DLT)。如果最高耐受剂量水平是制剂Ib,则允许当前关于制剂A招募的对象转换到较新的制剂。
B部分-群组扩大:在完成剂量递增(A部分)之后,可以招募附加的对象进入扩大期(B部分),其中每个群组最多约20名可评估对象。扩大可以基于A部分的安全性、PK和PD数据的综述,以MTD和剂量递增期建立的计划,和/或以替代性的可耐受剂量和计划进行。可以选择一个或多个给药方案(剂量、计划)用于群组扩大。
研究群体:不适合其它已建立疗法的患有由世界卫生组织(WHO)标准定义的复发性或难治性AML的18岁或以上的男性和女性将被招募于该研究中。
研究的长度。预期招募需要大约18至24个月来完成(12至15个月剂量递增,6至9个月扩展)。预期完成积极治疗和治疗后随访还需要6至24个月。预期整个研究持续最多大约3至4年。根据方案所预先确定,试验结束被定义为最后一名对象完成治疗后随访的最后一次访问的日期,或者主要、次要和/或探索性分析所需的最后一名对象的最后一个数据点的接收日期,以较晚的日期为准。
研究性治疗。将提供根据相关国家卫生主管部门的规定被适当标记用于调查研究用途的调查研究产品,IV注射用化合物1。研究药物将如上文的治疗期部分中所概述施用。
如果存在具有临床意义的疾病进展(或复发)的迹象、不可接受的毒性或对象/内科医生决定退出,则研究治疗可以停止。根据研究者在与Celgene医学监测员磋商后的判断,除疾病进展之外,对象可以继续接受研究药物。
关键功效评定的概述。主要功效变量是白血病响应率。所有受治疗的对象都将包括在功效分析中。白血病响应将由研究者确定。评定将基于国际工作组AML响应标准(Cheson等人,J Clin Oncol 2003;21(24):4642-9)。
抗白血病活性证据的描述性分析将由研究者基于临床评定、实验室评定、分子评定和细胞遗传学评定来提供,这些评定包括对骨髓母细胞百分比、骨髓细胞遗传学、用于评估分子响应的分子遗传学研究、骨髓流式细胞术、血小板计数和绝对中性粒细胞计数的评定。响应标准将通过最佳总体响应类别来汇总:完全缓解率(CRR)和客观响应率(ORR)。ORR包括完全缓解(CR)(即,形态学无白血病状态、形态学CR、细胞遗传学CR、分子CR和具有不完全血液恢复的形态学CR)和部分缓解的所有响应。
所关注的功效变量将为ORR和CRR。将汇总其它临床活性度量,包括总体存活期(OS)、无复发存活期(RFS)、无进展存活期(PFS)、无事件存活期、缓解持续时间、响应持续时间,以及至缓解/响应的时间。
关键安全性评定的概述。该研究的安全性变量包括不良事件、安全性临床实验室变量、12导程心电图、美国东部肿瘤协作组行为状态、左心室射血分数评定、体格检查、生命体征、研究治疗暴露、合并药物治疗评定,以及针对有生育潜能的女性的怀孕测试。
关键药代动力学评定的概述。所确定的化合物1的血浆PK参数将为最大观察血浆浓度(Cmax)、给药后0至24小时时间的血浆浓度-时间曲线下面积(AUC24)、末期消除半衰期(t1/2)、总血浆清除率(CL)、达到峰值(最大)血浆浓度的时间(tmax)、稳态时的分布体积(Vss)。将酌情估算化合物1的R-对映异构体和S-对映异构体的所选择PK参数(例如,Cmax、AUC24、t1/2)。
统计学方法。根据需要或适用时,将通过剂量水平(A部分)和群组(B部分)进行统计学分析。所有分析在本质上都将是描述性的。将使用接受任何化合物1的对象(受治疗群体)进行所有安全性数据的汇总。
主要关注的功效变量是ORR和CRR。将汇总其它初步功效变量,包括OS、RFS、PFS、无事件存活期、缓解持续时间、响应持续时间,以及至缓解/响应的时间。将对受治疗群体和功效可评估群体(接受基线白血病评定评估,至少一个研究治疗周期或周期1中至少80%的计划剂量,以及一次研究中白血病评定评估)重复进行功效分析,其中使用受治疗群体的结果被认为是主要的。
除非另外指明,否则所有生物标志物相关数据呈现都将基于采用至少一种生物标志物评定的受治疗对象。将通过给药方案和/或疾病亚组以及总体呈现连续生物标志物终点的基线和从基线的变化的描述性统计。
该研究的进行将遵守国际医药法规协和会(ICH)/优良临床试验(InternationalCouncil for Harmonisation of Technical Requirements for Pharmaceuticals forHuman Use(ICH)/Good Clinical Practice)以及适用的法规要求。
纳入标准。对象必须满足该研究的以下剂量递增(A部分)或剂量扩大(B部分)招募标准。
1.在签署知情同意书(ICD)时,男性和女性≥18岁。
2.对象在进行任何研究相关评定/程序之前必须理解并自愿签署ICD。
3.对象愿意并且能够遵守研究访问计划和其它方案要求。
4.由世界卫生组织(WHO)标准定义的复发性或难治性AML,不适合其它已建立疗法。
5.美国东部肿瘤协作组行为状态(ECOG PS)为0至2。
6.供体淋巴细胞输注(DLI)已过去至少4周(从第一剂量开始),但无改善。
7.对象必须具有以下筛选实验室值:
·校正血清Ca或游离(离子化)血清Ca在正常限度内(WNL)。
ο校正Ca(mg/dL)=总Ca(mg/dL)–0.8(白蛋白[g/dL]–4)
·在第一次输注之前总白血细胞计数(WBC)<25x109/L。允许使用羟基脲进行前期或同时治疗来实现该水平。
·钾和镁在正常限度内或可用补充剂校正。
·天冬氨酸氨基转移酶/血清谷氨酸草酰乙酸转氨酶(AST/SGOT)或丙氨酸氨基转移酶/血清谷氨酸丙酮酸转氨酶(ALT/SGPT)≤2.5x正常值上限(ULN)。
·尿酸≤7.5mg/dL(446μmol/L)。允许使用低尿剂(例如,别嘌呤醇、拉布立酶)进行前期和/或同时治疗。
·血清胆红素≤1.5xULN。
·使用Cockcroft-Gault公式估算血清肌酸酐清除率≥60mL/min。
·INR<1.5xULN,并且PTT<1.5xULN。
8.根据化合物1的怀孕预防计划(PPP):
·有生育潜能的女性(FCBP)必须基于PPP中概述的频率经过怀孕测试,并且怀孕结果必须为阴性。
·除非实施完全杜绝异性性交,否则性活跃的FCBP必须同意使用如PPP中所指定的适当避孕方法。
οFCBP必须同意在开始化合物1之前28天、在化合物1治疗的整个持续时间中、在剂量中断期间和在化合物1的最后剂量之后至少28天,同时不间断使用两种可靠的避孕形式(或实施完全禁欲)。
ο在异性性交是对象的优选和通常生活方式的情况下,完全禁欲是唯一可接受的方式。
ο周期性禁欲(排卵日历法、症状体温法、排卵后方法)和体外射精是不可接受的方式。
·除非实施完全杜绝异性性交,否则性活跃的男性(包括已进行输精管切除术的那些男性)在与FCBP从事性活动时必须使用屏障避孕(避孕套),如PPP中所指定。
ο在异性性交是对象的优选和通常生活方式的情况下,完全禁欲是唯一可接受的方式。
·女性必须同意在PPP指定的持续时间内放弃母乳喂养和放弃提供母乳。
·男性必须同意在PPP指定的持续时间内不捐献精液和精子。
·所有对象必须:
ο理解化合物1可能具有潜在致畸风险。
ο同意在PPP指定的持续时间内放弃献血。
ο被告知怀孕预防措施和胎儿暴露风险(请参阅PPP)。
排除标准存在以下任一种情况将从招募中排除对象:
1.患有急性早幼粒细胞性白血病(APL)的对象
2.具有提示活动性中枢神经系统(CNS)白血病或已知的CNS白血病的临床症状的对象。只有在筛查期间临床怀疑白血病累及CNS的情况下才需要进行脑脊液的评估。
3.具有白血病的直接危及生命的严重并发症(诸如弥散性/难以控制的感染、难以控制的出血,和/或难以控制的弥散性血管内凝血)的对象。
4.具有破坏正常钙稳态或阻止钙补充的疾患或病症,包括:
·破坏钙吸收的任何已知病症。
·甲状旁腺功能减退或甲状旁腺功能亢进的临床证据。
·在开始化合物1之前最后4周内实施双磷酸盐或地诺单抗(denosumab)疗法。
·活动性肾结石或近期肾结石(在开始化合物1之前≤1年)。
·血清25-羟基维生素D水平<12ng/mL(30nmol/L)。
5.心脏功能受损或患有具有临床意义的心脏疾病,包括以下任一种:
·如多门控采集(MUGA)扫描或超声心动图(ECHO)所确定的左心室射血分数(LVEF)<45%。
·左束支或双束支完全阻滞。
·先天性长QT综合征。
·持续性或具有临床意义的室性心律失常。
·心电图(ECG)筛查时QTcF≥470毫秒(在第1天前≥72小时进行,取三次记录的平均值)。
·在开始化合物1之前≤3个月具有不稳定性心绞痛或心肌梗塞。
6.进行前期自体造血干细胞移植的患者,根据研究者的判断,尚未完全从最后一次移植的影响(例如,移植相关副作用)恢复。
7.在开始化合物1之前≤6个月,进行前期同种异体造血干细胞移植(HSCT),使用标准或降低的强度调节。
8.在筛查时进行HSCT后全身免疫抑制疗法或患有具有临床意义的移植物抗宿主病(GVHD)的对象。允许使用局部类固醇治疗持续的皮肤或眼部GVHD。
9.在开始化合物1之前≤5个半衰期或4周(以较短者为准)进行前期全身癌症定向治疗或研究方式。允许使用羟基脲来控制外周白血病母细胞。
10.在开始化合物1之前≤2周进行白细胞去除术。
11.在开始化合物1之前≤2周进行大手术。对象必须已从最近手术的任何具有临床意义的影响恢复。
12.怀孕或哺乳女性。
13.已知的人免疫缺陷病毒(HIV)感染。
14.已知的慢性、活动性乙肝或丙肝病毒(HBV/HCV)感染。
15.进行持续的抗凝血剂(例如,华法林、低分子量肝素、因子Xa抑制剂)的长期、治疗性给药治疗。
16.具有需要积极、持续的全身治疗的第二并发癌症史。
17.对象对于钙、骨化三醇和/或维生素D补充剂或它们的任何成分具有已知的过敏性/超敏性。
18.对象具有阻止对象参与研究的任何显著医学病症、实验室异常或精神疾病。
19.对象具有如果该对象参与研究,则会使他/她处于不可接受的风险中的任何病症,包括存在实验室异常。
20.对象具有会破坏解释研究数据的能力的任何病症。
实施例40:化合物1—新型Cereblon E3连接酶调控药物在患有复发性或难治性急性骨髓性白血病、复发性或难治性较高风险骨髓增生异常综合征的对象中的1期、开放标签、剂量发现研究
主要目标:确定化合物1的安全性和耐受性。定义化合物1的非耐受剂量(NTD)、最大耐受剂量(MTD)和/或推荐2期剂量(RP2D)。
次要目标:提供关于化合物1在R/R AML和R/R HR-MDS中的初步功效的信息。表征化合物1在血浆和尿液中的药代动力学(PK)。
研究设计:该研究是化合物1在患有复发性或难治性AML的对象中或者在患有复发性或难治性较高风险MDS的对象中的开放标签、1期、剂量递增和扩大、首次人体试验临床研究。该研究的剂量递增部分(A部分)将评估静脉内施用的化合物1的递增剂量的安全性和耐受性,并确定化合物1的MTD。在A部分中,将测试两种制剂。扩大部分(B部分)将进一步评估所选择的扩大群组中以等于或小于MTD施用的化合物1的安全性和功效,以确定RP2D,其中每个群组包括最多大约20名可评估对象。可以选择一种或多种给药方案用于群组扩大(在R/R AML中最少一种,并且在R/R HR-MDS中最少一种)。MDS对象将仅在B部分期间招募。A部分和B部分将由3期构成:筛选期、治疗期和随访期。白血病响应将由研究者确定。疾病评定将基于国际工作组(IWG)AML响应标准(Cheson等人,J Clin Oncol2003;21(24):4642-9)。MDS响应将基于IWG骨髓增生异常响应标准(Cheson BD等人,Blood.2006;108(2):419-25)。
筛选期:筛选期始于第一剂量的化合物1之前28天。在开始任何其它研究程序之前,对象和管理人员必须签署知情同意书并注明日期。所有筛选测试和程序必须在第一剂量的化合物1之前28天内完成。对于该研究的B部分,将基于中心实验室诊断确认将对象分配到R/R AML或R/R HR-MDS群组中。
治疗期:在治疗期中,在不存在疾病进展、复发、不可接受的毒性或对象/内科医生决定退出的情况下,将在每个28天周期的第1至5天静脉内施用化合物1最多4个周期。(疾病进展被定义为:与基线(筛选)骨髓母细胞计数相比,骨髓母细胞计数百分比增加了>50%,持续存在间隔至少1个月的至少2次骨髓评定,除非基线骨髓母细胞计数>70%,在这种情况下,发现母细胞>70%持续存在间隔至少1个月的2次基线后骨髓评定将被视为进展,或者基线绝对外周血母细胞计数翻倍持续存在≥7天并且最终绝对外周血母细胞计数>10x109/L。)如有必要,可以在附加的群组中基于毒性、PK曲线和PD发现评估经修改的给药计划(例如,给药从5天增加至最多10天,或增加输注长度)。可以探究在每个28天周期的第1至3天和第8至10天每日施用一次化合物1的附加计划。那些显示出从治疗受益而又没有不可接受的毒性(与医学监测员讨论过,为完全缓解[CR]、部分缓解[PR]或疾病稳定)的患者可以继续治疗至第4周期之后,直到丧失该益处、出现不可接受的毒性或对象/内科医生决定退出。所有对象都将必须在每个周期的第1天之前开始钙、骨化三醇和维生素D补充剂至少3天,并且继续直到每个周期中化合物1的最后剂量之后≥3天(例如,当在第1至5天施用化合物1时,≥第8天;当在第1至3天/第8至10天施用化合物1时,≥第13天)。
在A部分的周期1中,骨髓评估将在第28天(±3天)进行。根据第28天的骨髓评估,将跟踪骨髓发育不全、无持续性白血病迹象、中性粒细胞减少≥3级的对象另外2周,以进行周期1的安全性监测(总持续时间42天)。在血液恢复时或第42天(±3天)将进行附加的骨髓评定。因此,在A部分中,周期1期间的剂量限制性毒性(DLT)评估窗口将为最多42天(28或42天)。在B部分中,周期1的长度将为28天。≥2个周期的长度将为28天。后续的周期应当在前一个周期的最后一天之后≤7天开始。
随访期:在随访期中,在化合物1的最后剂量之后,将跟踪所有对象28天(±3天),以评估安全性。记录疾病进展(或复发)的对象将在第1年的后续每8周(±1周)和第2年的每12周(±2周)进行全血计数和外周血涂片的功效评估,或者直到疾病进展(或复发)、开始新抗癌疗法、同意退出研究、死亡或试验结束,以先出现者为准。将在第1年结束时完成骨髓评估,并且在随访期期间根据临床状况进行。
将跟踪所有对象以进行生存随访,该生存随访根据功效长期随访计划进行最多2年或直到死亡、失访或试验结束,以先出现者为准。生存随访可以通过记录查阅(包括公开记录)和/或与对象、家庭或对象的主治医生电话联系进行。
A部分-剂量递增。在递增期(A部分)期间,将使用经修改的加速滴定设计(Simon等人,J Natl Cancer Inst 1997;89(15):1138-47)来建立初始毒性。将给每个一名或多名对象的群组施用化合物1,每个群组的剂量将以100%递增的方式增加,直到DLT窗口中≥2名对象经历化合物1相关的≥2级不良事件(可以是不同的群组),或者DLT窗口中≥1名对象经历DLT。此时,当前群组和所有后续群组将扩大招募3至6名对象。同时将开始剂量递增不超过50%的剂量递增计划,以便建立NTD和MTD。初始剂量将是0.3mg。在研究开始时,将利用初始制剂(制剂A),并且在剂量递增期间,将引入第二制剂(表43中所述的制剂Ib)来代替制剂A。制剂A具有以下组成(参见美国公布号2017/0196847-A1)。

制剂A中的DMA残留溶剂必须不超过ICH Q3C Impurities:Residual Solvents(ICH Q3C杂质:残留溶剂)中设定的允许每日暴露(PDE)限值,以便使用剂量递增群组进行大于2.4mg的每日化合物1剂量。制剂Ib中的残留溶剂(甲酸)水平允许化合物1的日剂量最高达20mg,而不会超过ICH Q3C指南中设定的其PDE。
在审查了在制剂A的初始剂量水平中观察到的毒性之后,将以新的剂量群组(根据研究剂量递增指南)引入制剂Ib。
可以决定评估较高剂量群组、剂量群组内的另外对象、中等剂量群组、更小的剂量递增、替代给药计划(例如,化合物1施用从5天增加至最多10天,或更长的输注时间),并且/或者基于可用的临床和实验室安全性数据、PK曲线和PD发现的综述发布MTD。在评估替代给药计划的事件中,起始剂量和计划将不超过先前已满足剂量递增标准的剂量群组的剂量强度。可以探究在每个28天计划的第1至3天和第8至10天每日施用一次化合物1的附加计划。
在剂量递增期间在任何群组中施用第一剂量之后,观察每个群组中的对象至少28天和最多42天(周期1,DLT窗口),然后可以开始下一个更高剂量群组。在给定剂量递增群组中每天将招募不超过一名对象。可评估DLT的对象被定义为:在周期1期间已接受化合物1的总计划周期1剂量的至少80%(例如,5天剂量计划的≥4个化合物1剂量;在剂量缺失的情况下,在第10天或之前完成≥4个剂量,或到D1-3/D8-10计划的第14天为止完成≥5个剂量),不经历DLT,或者在接受化合物1的至少一个剂量(或其部分)之后经历DLT。
在A部分中评估替代剂量计划(例如,给药从5天增加至最多10天)的事件中,将适用用于确定可评估DLT的对象的相同标准。不可评估DLT的对象将被替换。
如果剂量群组中>33%的可评估对象在周期1期间经历DLT,则剂量水平(剂量/计划)将被认为是不可容忍的。MTD将被定义为小于NTD的最后剂量,其中在周期1期间≤33%的可评价对象经历DLT。如果在第一剂量群组中6名可评估对象中的2名或更多名经历DLT,则可以探究更低剂量的群组(即,0.1mg化合物1)。可以评估中等剂量的化合物1(在NTD与NTD之前的最后剂量水平之间的剂量),以准确确定MTD。
在DLT评定期期间将不允许对象内剂量递增;然而,在≥2个周期中,无疾病进展迹象、耐受化合物1的分配剂量的对象可以递增至该研究中的至少一个群组的对象示出充分耐受的最高剂量水平(即,≤33%的可评估对象在该剂量水平经历了DLT)。如果最高耐受剂量水平是制剂Ib,则允许当前关于制剂A招募的对象转换到较新的制剂。
B部分-群组扩大:在完成剂量递增(A部分)之后,可以招募附加的对象进入扩大期(B部分),其中每个群组最多约20名可评估对象。扩大可以基于A部分的安全性、PK和PD数据的综述,以MTD和剂量递增期建立的计划,和/或以替代性的可耐受剂量和计划进行。可以选择一个或多个给药方案(剂量、计划)用于群组扩大。
研究群体:不适合其它已建立疗法的患有由世界卫生组织(WHO)标准定义的复发性或难治性AML或者复发性或难治性较高风险MDS的18岁或以上的男性和女性将被招募于该研究中。
在A部分中,将只招募R/R AML对象。在B部分中,至少一个群组将包括R/R AML对象,包括以下对象:在同种异体HSCT之后复发、第二次或后来复发、初始诱导或再诱导治疗难治性的、低甲基化剂难治性的或在施用低甲基化剂之后复发(HMA失败被定义为在最少6个周期后出现原发性进展或缺乏临床益处,或由于毒性而不能耐受HMA),或在初始治疗后的1年内复发(排除具有有利风险状态的那些)。
在B部分中,将治疗至少一个群组的R/R HR-MDS对象,包括以下对象:在经修订的国际预后评分系统(IPSS-R)中的评分>3.5分[例如,IPSS-R中等风险(与超过10%的骨髓母细胞或者差或极差的IPSS-R细胞遗传学风险组合)、IPSS-R高风险和IPSS-R极高风险],并且不适合其它已建立疗法(例如,移植或低甲基化剂)。
研究的长度。预期招募需要大约27至36个月来完成(18至24个月剂量递增,9至12个月扩大)。预期完成积极治疗和治疗后随访还需要6至24个月。预期整个研究持续最多大约3至5年。根据方案所预先确定,试验结束被定义为最后一名对象完成治疗后随访的最后一次访问的日期,或者主要、次要和/或探索性分析所需的最后一名对象的最后一个数据点的接收日期,以较晚的日期为准。
研究性治疗。将提供根据相关国家卫生主管部门的规定被适当标记用于调查研究用途的调查研究产品,IV注射用化合物1。研究药物将如上文的治疗期部分中所概述施用。
如果存在具有临床意义的疾病进展(或复发)的迹象、不可接受的毒性或对象/内科医生决定退出,则研究治疗可以停止。根据研究者在与Celgene医学监测员磋商后的判断,除疾病进展之外,对象可以继续接受研究药物。
关键功效评定的概述。主要功效变量是响应率。所有受治疗的对象都将包括在功效分析中。白血病响应将由研究者确定。疾病评定将基于国际工作组(IWG)AML响应标准(Cheson等人,J Clin Oncol 2003;21(24):4642-9)。总体响应将使用针对HR-MDS群组的IWG骨髓增生异常响应标准来确定(Cheson BD等人,Blood.2006;108(2):419-25)。
抗白血病活性证据的描述性分析将由研究者基于临床评定、实验室评定、分子评定和细胞遗传学评定来提供,这些评定包括对骨髓母细胞百分比、骨髓细胞遗传学、用于评估分子响应的分子遗传学研究、骨髓流式细胞术、血小板计数和绝对中性粒细胞计数的评定。AML响应标准将通过最佳总体响应类别来汇总:完全缓解率(CRR)和客观响应率(ORR)。ORR包括完全缓解(CR)(即,形态学无白血病状态、形态学CR、细胞遗传学CR、分子CR和具有不完全血液恢复的形态学CR)和部分缓解的所有响应。对于MDS,ORR包括所有响应(CR、骨髓完全缓解mCR和PR)。
所关注的功效变量将为ORR和CRR。将汇总其它临床活性度量,包括总体存活期(OS)、无复发存活期(RFS)、无进展存活期(PFS)、无事件存活期、缓解持续时间、响应持续时间、转化为AML的时间(仅HR-MDS对象),以及至缓解/响应的时间。
关键安全性评定的概述。该研究的安全性变量包括不良事件、安全性临床实验室变量、12导程心电图、美国东部肿瘤协作组行为状态、左心室射血分数评定、体格检查、生命体征、研究治疗暴露、合并药物治疗评定,以及针对有生育潜能的女性的怀孕测试。
关键药代动力学评定的概述。所确定的化合物1的关键血浆PK参数将包括最大观察浓度(Cmax)、给药后0至24小时时间的血浆浓度-时间曲线下面积(AUC24)、末期消除半衰期(t1/2)、总血浆清除率(CL)、达到峰值(最大)血浆浓度的时间(tmax)、稳态时的分布体积(Vss)、在尿液中以不变的形式排泄的剂量的百分比(Fe)和肾清除率(CLR)。将酌情估算化合物1的R-对映异构体和S-对映异构体的所选择PK参数(例如,Cmax、AUC24、t1/2)。对于HPBCD,还将估计如上所述的关键血浆和尿液PK参数。
统计学方法。根据需要或适用时,将通过剂量水平(A部分)和群组(B部分)进行统计学分析。所有分析在本质上都将是描述性的。将使用接受任何化合物1的对象(受治疗群体)进行所有安全性数据的汇总。
主要关注的功效变量是ORR和CRR。将汇总其它初步功效变量,包括OS、RFS、PFS、无事件存活期、缓解持续时间、响应持续时间,以及至缓解/响应的时间。将对受治疗群体和功效可评估群体(接受基线白血病评定评估,至少一个研究治疗周期或周期1中至少80%的计划剂量,以及一次研究中白血病评定评估)重复进行功效分析,其中使用受治疗群体的结果被认为是主要的。
除非另外指明,否则所有生物标志物相关数据呈现都将基于采用至少一种生物标志物评定的受治疗对象。将通过给药方案和/或疾病亚组以及总体呈现连续生物标志物终点的基线和从基线的变化的描述性统计。
该研究的进行将遵守国际医药法规协和会(ICH)/优良临床试验(InternationalCouncil for Harmonisation of Technical Requirements for Pharmaceuticals forHuman Use(ICH)/Good Clinical Practice)以及适用的法规要求。
纳入标准。对象必须满足该研究的以下剂量递增(A部分)或剂量扩大(B部分)招募标准。
1.在签署ICD时,男性和女性≥18岁。
2.对象在进行任何研究相关评定/程序之前必须理解并自愿签署ICD。
3.对象愿意并且能够遵守研究访问计划和其它方案要求。
4.由世界卫生组织(WHO)标准定义的复发性或难治性AML(A部分和B部分)或R/RHR-MDS(仅B部分),不适合其它已建立疗法。
a.在A部分中,R/R AML
b.在B部分中,R/R AML包括
-在同种异体HSCT之后复发或
-第二次或后来复发或
-初始诱导或再诱导治疗难治性的或
-HMA治疗难治性的或在HMA治疗之后复发(HMA失败被定义为在最少6个周期后出现原发性进展或缺乏临床益处,或由于毒性而不能耐受HMA)或
-在初始治疗后的1年内复发(排除具有基于细胞遗传学的有利风险的那些)
c.在B部分中,R/R HR-MDS(IPSS-R>3.5点):
-IPSS-R中等风险(与超过10%的骨髓母细胞或者差或极差的IPSS-R细胞遗传学风险组合)或
-IPSS-R高风险或
-IPSS-R极高风险。
5.美国东部肿瘤协作组行为状态(ECOG PS)为0至2。
6.供体淋巴细胞输注(DLI)已过去至少4周(从第一剂量开始),但无改善。
7.对象必须具有以下筛选实验室值:
·校正血清Ca或游离(离子化)血清Ca在正常限度内(WNL)。
ο校正Ca(mg/dL)=总Ca(mg/dL)–0.8(白蛋白[g/dL]–4)
·在第一次输注之前总白血细胞计数(WBC)<25x109/L。允许使用羟基脲进行前期或同时治疗来实现该水平。
·钾和镁在正常限度内或可用补充剂校正。
·天冬氨酸氨基转移酶/血清谷氨酸草酰乙酸转氨酶(AST/SGOT)或丙氨酸氨基转移酶/血清谷氨酸丙酮酸转氨酶(ALT/SGPT)≤2.5x正常值上限(ULN)。
·尿酸≤7.5mg/dL(446μmol/L)。允许使用低尿剂(例如,别嘌呤醇、拉布立酶)进行前期和/或同时治疗。
·血清胆红素≤1.5xULN。
·使用Cockcroft-Gault公式估算血清肌酸酐清除率≥60mL/min。如果临床上有指征,则从收集的24小时尿液测得的肌酸酐清除率是可接受的。
·INR<1.5xULN,并且PTT<1.5xULN。
8.根据化合物1的怀孕预防计划(PPP):
·有生育潜能的女性(FCBP)必须基于PPP中概述的频率经过怀孕测试,并且怀孕结果必须为阴性。
·除非实施完全杜绝异性性交,否则性活跃的FCBP必须同意使用如PPP中所指定的适当避孕方法。
οFCBP必须同意在开始化合物1之前28天、在化合物1治疗的整个持续时间中、在剂量中断期间和在化合物1的最后剂量之后至少28天,同时不间断使用两种可靠的避孕形式(或实施完全禁欲)。
ο在异性性交是对象的优选和通常生活方式的情况下,完全禁欲是唯一可接受的方式。
ο周期性禁欲(排卵日历法、症状体温法、排卵后方法)和体外射精是不可接受的方式。
·除非实施完全杜绝异性性交,否则性活跃的男性(包括已进行输精管切除术的那些男性)在与FCBP从事性活动时必须使用屏障避孕(避孕套),如PPP中所指定。
ο在异性性交是对象的优选和通常生活方式的情况下,完全禁欲是唯一可接受的方式。
ο男性患者必须告知有生育潜能的女性伴侣在治疗的整个持续过程中、在剂量中断期间以及在最后剂量的化合物1之后至少90天使用两种可靠的避孕方法,如PPP中所指定。
·女性必须同意在PPP指定的持续时间内放弃母乳喂养和放弃提供母乳。
·男性必须同意在接受化合物1的同时、在剂量中断期间或在最后剂量的化合物1之后至少90天内不捐献精液和精子,如PPP所指定。
·所有对象必须:
ο理解化合物1可能具有潜在致畸风险。
ο同意在PPP指定的持续时间内放弃献血。
ο被告知怀孕预防措施和胎儿暴露风险(请参阅PPP)。
排除标准。
存在以下任一种情况将从招募中排除对象:
1.患有急性早幼粒细胞性白血病(APL)的对象
2.具有提示活动性中枢神经系统(CNS)白血病或已知的CNS白血病的临床症状的对象。只有在筛查期间临床怀疑白血病累及CNS的情况下才需要进行脑脊液的评估。
3.具有白血病的直接危及生命的严重并发症(诸如弥散性/难以控制的感染、难以控制的出血,和/或难以控制的弥散性血管内凝血)的对象。
4.具有破坏正常钙稳态或阻止钙补充的疾患或病症,包括:
·破坏钙吸收的任何已知病症。
·甲状旁腺功能减退或甲状旁腺功能亢进的临床证据。
·在开始化合物1之前最后4周内实施双磷酸盐或地诺单抗(denosumab)疗法。
·活动性肾结石或近期肾结石(在开始化合物1之前≤1年)。
·血清25-羟基维生素D水平<12ng/mL(30nmol/L)。
5.心脏功能受损或患有具有临床意义的心脏疾病,包括以下任一种:
·如多门控采集(MUGA)扫描或超声心动图(ECHO)所确定的左心室射血分数(LVEF)<45%。
·左束支或双束支完全阻滞。
·先天性长QT综合征。
·持续性或具有临床意义的室性心律失常。
·心电图(ECG)筛查时QTcF≥470毫秒(在第1天前≥72小时进行,取三次记录的平均值)。
·在开始化合物1之前≤3个月具有不稳定性心绞痛或心肌梗塞。
6.进行前期自体造血干细胞移植的患者,根据研究者的判断,尚未完全从最后一次移植的影响(例如,移植相关副作用)恢复。
7.在开始化合物1之前≤6个月,进行前期同种异体造血干细胞移植(HSCT),使用标准或降低的强度调节。
8.在筛查时进行HSCT后全身免疫抑制疗法或患有具有临床意义的移植物抗宿主病(GVHD)的对象。允许使用局部类固醇治疗持续的皮肤或眼部GVHD。
9.在开始化合物1之前≤5个半衰期或4周(以较短者为准)进行前期全身癌症定向治疗或研究方式。允许使用羟基脲来控制外周白血病母细胞。
10.在开始化合物1之前≤2周进行白细胞去除术。
11.在开始化合物1之前≤2周进行大手术。对象必须已从最近手术的任何具有临床意义的影响恢复。
12.怀孕或哺乳女性。
13.已知的人免疫缺陷病毒(HIV)感染。
14.已知的慢性、活动性乙肝或丙肝病毒(HBV/HCV)感染。
15.进行持续的抗凝血剂(例如,华法林、低分子量肝素、因子Xa抑制剂)的长期、治疗性给药治疗。
16.具有需要积极、持续的全身治疗的第二并发癌症史。
17.对象对于钙、骨化三醇和/或维生素D补充剂或它们的任何成分具有已知的过敏性/超敏性。
18.对象具有阻止对象参与研究的任何显著医学病症、实验室异常或精神疾病。
19.对象具有如果该对象参与研究,则会使他/她处于不可接受的风险中的任何病症,包括存在实验室异常。
20.对象具有会破坏解释研究数据的能力的任何病症。
上文所述的实施方案旨在仅仅是示例性的,并且本领域的技术人员将认识到、或将能够在仅使用常规实验的情况下探知具体化合物、材料和程序的许多等同物。所有此类等同物都被认为是在本发明的范围之内,并且由所附权利要求书涵盖。
Claims (89)
1.一种制剂,其包含:基于所述制剂的总重量,量为约0.01%至约0.15%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物,和量为约99.1%至约99.99%的羟丙基β-环糊精或磺丁基醚-β-环糊精。
2.如权利要求1所述的制剂,其包含:基于所述制剂的总重量,量为约0.08%至约0.15%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物,和量为约99.1%至约99.9%的羟丙基β-环糊精或磺丁基醚-β-环糊精。
3.如权利要求1或2所述的制剂,其包含:基于所述制剂的总重量,量为约0.1%至约0.13%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物。
4.如权利要求1至3中任一项所述的制剂,其包含:基于所述制剂的总重量,量为约0.12%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物。
5.如权利要求1所述的制剂,其包含:基于所述制剂的总重量,量为约0.01%至约0.08%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物,和量为约99.40%至约99.99%的羟丙基β-环糊精。
6.如权利要求1或5所述的制剂,其包含:基于所述制剂的总重量,量为约0.03%至约0.06%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物。
7.如权利要求1和5至6中任一项所述的制剂,其包含:基于所述制剂的总重量,约0.1%至约0.13%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物;约99.1%至约99.9%的羟丙基β-环糊精;和约0.05%至约0.1%的甲酸。
8.如权利要求1和5至7中任一项所述的制剂,其包含:基于所述制剂的总重量,约0.01%至约0.08%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物;约99.40%至约99.99%的羟丙基β-环糊精;和约0.1%至约0.3%的甲酸。
9.如权利要求1至8中任一项所述的制剂,其还包含量不超过约0.5%的甲酸。
10.如权利要求1至9中任一项所述的制剂,其包含(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)的固体形式。
11.如权利要求1至9中任一项所述的制剂,其包含(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)的无定形形式。
12.一种制剂,其包含:基于所述制剂的总重量,量为约0.08%至约0.15%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物,量为约3%至约6%的柠檬酸盐缓冲剂,和量为约94%至约96%的羟丙基β-环糊精或磺丁基醚-β-环糊精。
13.如权利要求12所述的制剂,其包含(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)的固体形式。
14.如权利要求12所述的制剂,其包含(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)的无定形形式。
15.如权利要求12至14中任一项所述的制剂,其包含:基于所述制剂的总重量,量为约0.1%至约0.13%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物。
16.如权利要求12至15中任一项所述的制剂,其包含:基于所述制剂的总重量,量为约0.12%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物。
17.如权利要求12至16中任一项所述的制剂,其包含:基于所述制剂的总重量,量为约3%至约6%的柠檬酸盐缓冲剂。
18.如权利要求12至17中任一项所述的制剂,其中柠檬酸盐缓冲剂包括无水柠檬酸和无水柠檬酸钠。
19.如权利要求18所述的制剂,其包含:基于所述制剂的总重量,量为约2%至约2.5%的无水柠檬酸。
20.如权利要求18所述的制剂,其包含:基于所述制剂的总重量,量为约2.1%的无水柠檬酸。
21.如权利要求18所述的制剂,其包含:基于所述制剂的总重量,量为约2%至约2.5%的无水柠檬酸钠。
22.如权利要求21所述的制剂,其包含:基于所述制剂的总重量,量为约2.08%的无水柠檬酸钠。
23.如权利要求12至22中任一项所述的制剂,其包含:基于所述制剂的总重量,量为约94%至约97%的羟丙基β-环糊精。
24.如权利要求12至23中任一项所述的制剂,其包含:基于所述制剂的总重量,量为约95%的羟丙基β-环糊精。
25.如权利要求12至24中任一项所述的制剂,其还包含量不超过约1.5%的二甲基亚砜。
26.如权利要求12至25中任一项所述的制剂,其包含:基于所述制剂的总重量,约0.1%至约0.13%的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物;约2%至约2.5%的无水柠檬酸;约2%至约2.5%的无水柠檬酸钠;约94%至约96%的羟丙基β-环糊精;和约0.4%至约1.5%的二甲基亚砜。
27.一种水性制剂,其包含如权利要求1至26中任一项所述的制剂和稀释剂。
28.如权利要求27所述的水性制剂,其中所述稀释剂是水或1/2生理盐水。
29.如权利要求27所述的水性制剂,其中所述稀释剂是生理盐水。
30.如权利要求27至29中任一项所述的水性制剂,其包含量为约0.1至0.3mg/mL的(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺),或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物。
31.如权利要求27至30中任一项所述的水性制剂,其中所述水性溶液具有在约3.0至约3.6范围内的pH。
32.如权利要求27至30中任一项所述的水性制剂,其中所述水性溶液具有在约4.2至约4.4范围内的pH。
33.如权利要求27至32中任一项所述的水性制剂,其中所述水性溶液具有约260mOsm/kg至280mOsm/kg的克分子渗透压浓度。
34.如权利要求27至32中任一项所述的水性制剂,其中所述水性溶液具有约310mOsm/kg至380mOsm/kg的克分子渗透压浓度。
35.一种治疗哺乳动物的癌症的方法,其中所述方法包括将如权利要求1至26中任一项所述的制剂或如权利要求27至34中任一项所述的水性制剂施用于所述哺乳动物。
36.如权利要求35所述的方法,其中所述方法包括静脉内施用如权利要求27至34中任一项所述的水性制剂。
37.如权利要求35或36所述的方法,其中所述癌症是白血病。
38.如权利要求37所述的方法,其中所述白血病是慢性淋巴细胞性白血病、慢性髓细胞性白血病、急性淋巴母细胞性白血病或急性骨髓性白血病。
39.如权利要求35至38中任一项所述的方法,其还包括施用治疗有效量的另外的第二活性剂或支持性护理疗法。
40.如权利要求39所述的方法,其中所述另外的第二活性剂是特异性地结合至癌症抗原、造血生长因子、细胞因子、抗癌剂、抗生素、cox-2抑制剂、免疫调节剂、免疫抑制剂、皮质类固醇或它们的药理活性突变体或衍生物的治疗性抗体。
41.一种治疗哺乳动物的白血病的方法,其中所述方法包括将(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物与选自下列的第二剂组合施用于所述哺乳动物:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、ERK抑制剂、LSD1抑制剂、SMG1抑制剂、BH3模拟物和拓扑异构酶抑制剂。
42.如权利要求41所述的方法,其中所述第二剂选自普拉二烯内酯B、氯-N,N-二乙基-5-((4-(2-(4-(3-甲基脲基)苯基)吡啶-4-基)嘧啶-2-基)氨基)苯磺酰胺(化合物Ii)、维奈托克、拓扑替康和依维莫司。
43.如权利要求41所述的方法,其中所述第二剂是JAK抑制剂。
44.如权利要求43所述的方法,其中所述JAK抑制剂选自托法替尼、莫美罗替尼、非戈替尼、地西罗替尼、巴瑞替尼、鲁索替尼、菲卓替尼、NS-018和帕克替尼。
45.如权利要求41所述的方法,其中所述第二剂是FLT3抑制剂。
46.如权利要求45所述的方法,其中所述FLT3抑制剂选自奎扎替尼、舒尼替尼、米哚妥林、培西达替尼、来他替尼、坦度替尼和克诺拉尼。
47.如权利要求41所述的方法,其中所述第二剂是依维莫司。
48.如权利要求41至47中任一项所述的方法,其中所述白血病是急性骨髓性白血病。
49.如权利要求41至48中任一项所述的方法,其中所述白血病是复发性、难治性或耐药性的。
50.一种治疗哺乳动物的骨髓增殖性赘生物的方法,其中所述方法包括将(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物与JAK抑制剂组合施用于所述哺乳动物。
51.如权利要求50所述的方法,其中所述JAK抑制剂选自托法替尼、莫美罗替尼、非戈替尼、地西罗替尼、巴瑞替尼、鲁索替尼、菲卓替尼、NS-018和帕克替尼。
52.一种治疗哺乳动物的选自乳腺癌、神经内分泌肿瘤和肾细胞癌的癌症的方法,其中所述方法包括将(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物与选自下列的第二剂组合施用于所述哺乳动物:依维莫司、替西罗莫司、1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮和7-(6-(2-羟基丙烷-2-基)吡啶-3-基)-1-((反式)-4-甲氧基环己基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮。
53.如权利要求52所述的方法,其中所述第二剂是依维莫司。
54.如权利要求41至53中任一项所述的方法,其包括将如权利要求1至26中任一项所述的制剂或如权利要求27至34中任一项所述的水性制剂施用于所述哺乳动物。
55.一种治疗哺乳动物的白血病的方法,其中所述方法包括将(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物与IDH2抑制剂组合施用于所述哺乳动物,其中所述白血病的特征在于存在IDH2的突变等位基因。
56.如权利要求54所述的方法,其中所述IDH2抑制剂是恩西地平或6-(6-(三氟甲基)吡啶-2-基)-N2-(2-(三氟甲基)吡啶-4-基)-1,3,5-三嗪-2,4-二胺。
57.如权利要求55或56所述的方法,其中所述白血病是特征在于存在IDH2的突变等位基因的急性骨髓性白血病。
58.如权利要求55至57中任一项所述的方法,其中所述白血病是复发性、难治性或耐药性的。
59.一种降低对象中的GSPT1水平的方法,其包括将(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物与第二剂的组合施用于所述对象。
60.一种降低对象中的Mcl-1水平的方法,其包括将(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物与第二剂的组合施用于所述对象。
61.如权利要求59或60所述的方法,其中所述第二剂选自JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、BET抑制剂、SMG1抑制剂、ERK抑制剂、LSD1抑制剂、BH3模拟物、拓扑异构酶抑制剂和RTK抑制剂。
62.如权利要求1至26中任一项所述的制剂或如权利要求27至34中任一项所述的水性制剂,其用作药物。
63.如权利要求1至26中任一项所述的制剂或如权利要求27至34中任一项所述的水性制剂,其在治疗哺乳动物的癌症的方法中使用。
64.如权利要求63所述的使用的水性制剂,其中所述方法包括静脉内施用所述水性制剂。
65.如权利要求63所述的使用的制剂或如权利要求63或64所述的使用的水性制剂,其中所述癌症是白血病。
66.如权利要求65所述的使用的制剂或如权利要求65所述的使用的水性制剂,其中所述白血病是慢性淋巴细胞性白血病、慢性髓细胞性白血病、急性淋巴母细胞性白血病或急性骨髓性白血病。
67.如权利要求62、63或65所述的使用的制剂或如权利要求62至66中任一项所述的使用的水性制剂,其还包括施用治疗有效量的另外的第二活性剂或支持性护理疗法。
68.如权利要求67所述的使用的制剂或如权利要求67所述的使用的水性制剂,其中所述另外的第二活性剂是特异性地结合至癌症抗原、造血生长因子、细胞因子、抗癌剂、抗生素、cox-2抑制剂、免疫调节剂、免疫抑制剂、皮质类固醇或它们的药理活性突变体或衍生物的治疗性抗体。
69.在治疗哺乳动物的白血病的方法中使用的化合物1,其中化合物1是(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物,并且其中所述方法包括将化合物1与选自下列的第二剂组合施用于所述哺乳动物:JAK抑制剂、FLT3抑制剂、mTOR抑制剂、剪接体抑制剂、ERK抑制剂、LSD1抑制剂、SMG1抑制剂、BH3模拟物和拓扑异构酶抑制剂。
70.如权利要求69所述的使用的化合物1,其中所述第二剂选自普拉二烯内酯B、氯-N,N-二乙基-5-((4-(2-(4-(3-甲基脲基)苯基)吡啶-4-基)嘧啶-2-基)氨基)苯磺酰胺(化合物Ii)、维奈托克、拓扑替康和依维莫司。
71.如权利要求69所述的使用的化合物1,其中所述第二剂是JAK抑制剂。
72.如权利要求71所述的使用的化合物1,其中所述JAK抑制剂选自托法替尼、莫美罗替尼、非戈替尼、地西罗替尼、巴瑞替尼、鲁索替尼、菲卓替尼、NS-018和帕克替尼。
73.如权利要求69所述的使用的化合物1,其中所述第二剂是FLT3抑制剂。
74.如权利要求73所述的使用的化合物1,其中所述FLT3抑制剂选自奎扎替尼、舒尼替尼、米哚妥林、培西达替尼、来他替尼、坦度替尼和克诺拉尼。
75.如权利要求69所述的使用的化合物1,其中所述第二剂是依维莫司。
76.如权利要求69至75中任一项所述的使用的化合物1,其中所述白血病是急性骨髓性白血病。
77.如权利要求69至76中任一项所述的使用的化合物1,其中所述白血病是复发性、难治性或耐药性的。
78.在治疗哺乳动物的骨髓增殖性赘生物的方法中使用的化合物1,其中化合物1是(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物,并且其中所述方法包括将化合物1与JAK抑制剂组合施用于所述哺乳动物。
79.如权利要求78所述的使用的化合物1,其中所述JAK抑制剂选自托法替尼、莫美罗替尼、非戈替尼、地西罗替尼、巴瑞替尼、鲁索替尼、菲卓替尼、NS-018和帕克替尼。
80.在治疗哺乳动物的选自乳腺癌、神经内分泌肿瘤和肾细胞癌的癌症的方法中使用的化合物1,其中化合物1是(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物,并且其中所述方法包括将化合物1与选自下列的第二剂组合施用于所述哺乳动物:依维莫司、替西罗莫司、1-乙基-7-(2-甲基-6-(1H-1,2,4-三唑-3-基)吡啶-3-基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮和7-(6-(2-羟基丙烷-2-基)吡啶-3-基)-1-((反式)-4-甲氧基环己基)-3,4-二氢吡嗪并[2,3-b]吡嗪-2(1H)-酮。
81.如权利要求80所述的使用的化合物1,其中所述第二剂是依维莫司。
82.如权利要求69至81中任一项所述的使用的化合物1,包括将如权利要求1至26中任一项所述的制剂或如权利要求27至34中任一项所述的水性制剂施用于所述哺乳动物。
83.在治疗哺乳动物的白血病的方法中使用的化合物1,其中化合物1是(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)或其立体异构体或立体异构体混合物、药学上可接受的盐、互变异构体、前药、溶剂合物、水合物、共晶体、笼形包合物或多晶型物,其中所述方法包括将化合物1与IDH2抑制剂组合施用于所述哺乳动物,并且其中所述白血病的特征在于存在IDH2的突变等位基因。
84.如权利要求83所述的使用的化合物1,其中所述IDH2抑制剂是恩西地平或6-(6-(三氟甲基)吡啶-2-基)-N2-(2-(三氟甲基)吡啶-4-基)-1,3,5-三嗪-2,4-二胺。
85.如权利要求83或84所述的使用的化合物1,其中所述白血病是特征在于存在IDH2的突变等位基因的急性骨髓性白血病。
86.如权利要求83至85中任一项所述的使用的化合物1,其中所述白血病是复发性、难治性或耐药性的。
87.一种用于制备如权利要求1至11中任一项所述的制剂的工艺,其包括:将(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)溶解在甲酸中以获得预混物,将羟丙基β-环糊精溶解在水中以获得溶液,将所述预混物添加至所述溶液中以获得药物溶液。
88.如权利要求89所述的工艺,其还包括将所述溶液冻干以产生冻干制剂。一种用于制备如权利要求12至26中任一项所述的制剂的工艺,其包括:将羟丙基β-环糊精溶解在柠檬酸盐缓冲剂中以获得缓冲溶液,将(2-(4-氯苯基)-N-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺)溶解在DMSO中以获得预混物,将所述预混物添加至所述缓冲溶液中以获得溶液。
89.如权利要求87所述的工艺,其还包括将所述溶液冻干以产生冻干制剂。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762527744P | 2017-06-30 | 2017-06-30 | |
| US62/527,744 | 2017-06-30 | ||
| US201862653436P | 2018-04-05 | 2018-04-05 | |
| US62/653,436 | 2018-04-05 | ||
| US201862673064P | 2018-05-17 | 2018-05-17 | |
| US62/673,064 | 2018-05-17 | ||
| PCT/US2018/040286 WO2019006299A1 (en) | 2017-06-30 | 2018-06-29 | COMPOSITIONS AND METHODS FOR THE USE OF 2- (4-CHLOROPHENYL) -N - ((2- (2,6-DIOXOPIPERIDIN-3-YL) -1-OXOISOINDOLIN-5-YL) METHYL) -2,2-DIFLUOROACETAMIDE |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN111032043A true CN111032043A (zh) | 2020-04-17 |
Family
ID=64741897
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201880056213.2A Pending CN111032043A (zh) | 2017-06-30 | 2018-06-29 | 2-(4-氯苯基)-n-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的组合物和使用方法 |
Country Status (24)
| Country | Link |
|---|---|
| US (1) | US20190030018A1 (zh) |
| EP (2) | EP4201399A3 (zh) |
| JP (2) | JP2020526501A (zh) |
| KR (1) | KR20200020899A (zh) |
| CN (1) | CN111032043A (zh) |
| AU (1) | AU2018294327A1 (zh) |
| BR (1) | BR112019028101A2 (zh) |
| CA (1) | CA3068591A1 (zh) |
| CL (1) | CL2019003857A1 (zh) |
| DK (1) | DK3644999T3 (zh) |
| ES (1) | ES2939739T3 (zh) |
| FI (1) | FI3644999T3 (zh) |
| HR (1) | HRP20230210T1 (zh) |
| HU (1) | HUE061356T2 (zh) |
| IL (1) | IL271726A (zh) |
| LT (1) | LT3644999T (zh) |
| MX (2) | MX2019015886A (zh) |
| PL (1) | PL3644999T3 (zh) |
| PT (1) | PT3644999T (zh) |
| RS (1) | RS64029B1 (zh) |
| SG (1) | SG11201913008TA (zh) |
| SI (1) | SI3644999T1 (zh) |
| WO (1) | WO2019006299A1 (zh) |
| ZA (1) | ZA201908605B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024074699A1 (en) * | 2022-10-07 | 2024-04-11 | Oculis Operations Sarl | Eye drop microsuspensions of mtor inhibitors |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3125753A1 (en) | 2019-01-09 | 2020-07-16 | Celgene Corporation | Antiproliferative compounds and second active agents for use in treating multiple myeloma |
| JP2022553427A (ja) * | 2019-10-28 | 2022-12-22 | セルジーン コーポレーション | 白血病を処置するための方法および治療への臨床的感度を予測するための白血病幹細胞シグネチャーの使用 |
| MX2022005159A (es) * | 2019-10-28 | 2022-06-08 | Celgene Corp | Uso de biomarcadores para predecir la sensibilidad clinica a 2-(4-clorofenil)-n-((2-(2,6-dioxopiperidin-3-il)-1-oxoisoindolin- 5-il)metil)-2,2-difluoroacetamida. |
| WO2021095835A1 (en) * | 2019-11-13 | 2021-05-20 | Taiho Pharmaceutical Co., Ltd. | Novel salt of terphenyl compound |
| BR112022018515A2 (pt) * | 2020-03-16 | 2022-11-16 | Celgene Corp | Terapia de combinação para leucemia mieloide aguda |
| CN116583509A (zh) * | 2020-10-07 | 2023-08-11 | 上海睿跃生物科技有限公司 | 治疗癌症的化合物和方法 |
| EP4277901A1 (en) | 2021-01-13 | 2023-11-22 | Monte Rosa Therapeutics, Inc. | Isoindolinone compounds |
| CN113125602A (zh) * | 2021-04-16 | 2021-07-16 | 山东铂源药业有限公司 | 一种哌柏西利中残留溶剂的检测方法 |
| WO2024015855A1 (en) * | 2022-07-13 | 2024-01-18 | Monte Rosa Therapeutics, Inc. | COMBINATION THERAPY COMPRISING GSPT1-DIRECTED MOLECULAR GLUE DEGRADERS AND PI3K/AKT/mTOR PATHWAY INHIBITORS |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140045843A1 (en) * | 2012-08-09 | 2014-02-13 | Celgene Corporation | Methods of treating cancer using 3-(4-((4-(morpholinomethyl)benzyl)oxy)-1-oxoisoindolin-2-yl)piperidine-2,6-dione |
| US20150196562A1 (en) * | 2014-01-15 | 2015-07-16 | Celgene Corporation | Formulations of 3-(5-amino-2-methyl-4-oxo-4h-quinazolin-3-yl)-piperidine-2,6-dione |
| CN106660991A (zh) * | 2014-07-11 | 2017-05-10 | 细胞基因公司 | 抗增殖化合物及其使用方法 |
Family Cites Families (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0011312A1 (en) | 1977-07-09 | 1980-05-28 | LUCAS INDUSTRIES public limited company | Road vehicle electrical systems |
| DE2848746A1 (de) | 1978-11-10 | 1980-05-22 | Wolfgang Arheiliger | Dichtung fuer eine schwellenlose tuere mit drehfluegel |
| DE3346123A1 (de) * | 1983-12-21 | 1985-06-27 | Janssen Pharmaceutica, N.V., Beerse | Pharmazeutische praeparate von in wasser schwerloeslichen oder instabilen arzneistoffen und verfahren zu ihrer herstellung |
| US5391485A (en) | 1985-08-06 | 1995-02-21 | Immunex Corporation | DNAs encoding analog GM-CSF molecules displaying resistance to proteases which cleave at adjacent dibasic residues |
| US4810643A (en) | 1985-08-23 | 1989-03-07 | Kirin- Amgen Inc. | Production of pluripotent granulocyte colony-stimulating factor |
| JPS63500636A (ja) | 1985-08-23 | 1988-03-10 | 麒麟麦酒株式会社 | 多分化能性顆粒球コロニー刺激因子をコードするdna |
| US6352694B1 (en) | 1994-06-03 | 2002-03-05 | Genetics Institute, Inc. | Methods for inducing a population of T cells to proliferate using agents which recognize TCR/CD3 and ligands which stimulate an accessory molecule on the surface of the T cells |
| US6534055B1 (en) | 1988-11-23 | 2003-03-18 | Genetics Institute, Inc. | Methods for selectively stimulating proliferation of T cells |
| KR0166088B1 (ko) * | 1990-01-23 | 1999-01-15 | . | 수용해도가 증가된 시클로덱스트린 유도체 및 이의 용도 |
| GB9126209D0 (en) * | 1991-12-10 | 1992-02-12 | Orion Yhtymae Oy | Drug formulations for parenteral use |
| US5360352A (en) | 1992-12-24 | 1994-11-01 | The Whitaker Corporation | Wire retainer for current mode coupler |
| US6692964B1 (en) | 1995-05-04 | 2004-02-17 | The United States Of America As Represented By The Secretary Of The Navy | Methods for transfecting T cells |
| US6051227A (en) | 1995-07-25 | 2000-04-18 | The Regents Of The University Of California, Office Of Technology Transfer | Blockade of T lymphocyte down-regulation associated with CTLA-4 signaling |
| US5855887A (en) | 1995-07-25 | 1999-01-05 | The Regents Of The University Of California | Blockade of lymphocyte down-regulation associated with CTLA-4 signaling |
| US5811097A (en) | 1995-07-25 | 1998-09-22 | The Regents Of The University Of California | Blockade of T lymphocyte down-regulation associated with CTLA-4 signaling |
| US5948893A (en) | 1996-01-17 | 1999-09-07 | The United States Of America As Represented By The Secretary Of The Navy | Murine hybridoma and antibody binding to CD28 receptor secreted by the hybridoma and method of using the antibody |
| US6207157B1 (en) | 1996-04-23 | 2001-03-27 | The United States Of America As Represented By The Department Of Health And Human Services | Conjugate vaccine for nontypeable Haemophilus influenzae |
| US6682736B1 (en) | 1998-12-23 | 2004-01-27 | Abgenix, Inc. | Human monoclonal antibodies to CTLA-4 |
| NZ517202A (en) | 1999-08-24 | 2004-05-28 | Medarex Inc | Human CTLA-4 antibodies and their uses |
| US7605238B2 (en) | 1999-08-24 | 2009-10-20 | Medarex, Inc. | Human CTLA-4 antibodies and their uses |
| CA2466279A1 (en) | 2001-11-13 | 2003-05-22 | Dana-Farber Cancer Institute, Inc. | Agents that modulate immune cell activation and methods of use thereof |
| US7498171B2 (en) | 2002-04-12 | 2009-03-03 | Anthrogenesis Corporation | Modulation of stem and progenitor cell differentiation, assays, and uses thereof |
| ES2367430T3 (es) | 2002-12-23 | 2011-11-03 | Wyeth Llc | Anticuerpos contra pd-1 y sus usos. |
| RU2406760C3 (ru) | 2005-05-09 | 2017-11-28 | Оно Фармасьютикал Ко., Лтд. | Моноклональные антитела человека к белку программируемой смерти 1 (pd-1) и способы лечения рака с использованием анти-pd-1-антител самостоятельно или в комбинации с другими иммунотерапевтическими средствами |
| CN101248089A (zh) | 2005-07-01 | 2008-08-20 | 米德列斯公司 | 抗程序性死亡配体1(pd-l1)的人单克隆抗体 |
| SI2170959T1 (sl) | 2007-06-18 | 2014-04-30 | Merck Sharp & Dohme B.V. | Protitelesa proti receptorjem pd-1 za humano programirano smrt |
| EP2262837A4 (en) | 2008-03-12 | 2011-04-06 | Merck Sharp & Dohme | PD-1 BINDING PROTEINS |
| EP3133086B1 (en) | 2008-09-26 | 2018-08-01 | Dana-Farber Cancer Institute, Inc. | Human anti-pd-1, pd-l1, and pd-l2 antibodies and uses thereof |
| CN104479018B (zh) | 2008-12-09 | 2018-09-21 | 霍夫曼-拉罗奇有限公司 | 抗-pd-l1抗体及它们用于增强t细胞功能的用途 |
| JP5844159B2 (ja) | 2009-02-09 | 2016-01-13 | ユニヴェルシテ デクス−マルセイユUniversite D’Aix−Marseille | Pd−1抗体およびpd−l1抗体ならびにその使用 |
| JP2013512251A (ja) | 2009-11-24 | 2013-04-11 | アンプリミューン、インコーポレーテッド | Pd−l1/pd−l2の同時阻害 |
| US20130022629A1 (en) | 2010-01-04 | 2013-01-24 | Sharpe Arlene H | Modulators of Immunoinhibitory Receptor PD-1, and Methods of Use Thereof |
| JP2013532153A (ja) | 2010-06-18 | 2013-08-15 | ザ ブリガム アンド ウィメンズ ホスピタル インコーポレイテッド | 慢性免疫病に対する免疫治療のためのtim−3およびpd−1に対する二重特異性抗体 |
| US8907053B2 (en) | 2010-06-25 | 2014-12-09 | Aurigene Discovery Technologies Limited | Immunosuppression modulating compounds |
| CN107417667B (zh) | 2012-01-06 | 2020-04-10 | 安吉奥斯医药品有限公司 | 治疗活性化合物及其使用方法 |
| US9579324B2 (en) | 2013-07-11 | 2017-02-28 | Agios Pharmaceuticals, Inc | Therapeutically active compounds and their methods of use |
| BR112016000489A8 (pt) | 2013-07-11 | 2020-01-07 | Agios Pharmaceuticals Inc | composto, composição farmacêutica que o compreende, uso da composição e métodos para fabricar compostos |
| JP6529492B2 (ja) | 2013-07-11 | 2019-06-12 | アジオス ファーマシューティカルズ, インコーポレイテッド | 癌の処置のためのidh2突然変異体阻害剤としての2,4−または4,6−ジアミノピリミジン化合物 |
| WO2015003355A2 (en) | 2013-07-11 | 2015-01-15 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| EA030428B1 (ru) | 2013-08-02 | 2018-08-31 | Аджиос Фармасьютикалз, Инк. | Терапевтически активные соединения и способы их применения |
| AR107320A1 (es) * | 2016-01-08 | 2018-04-18 | Celgene Corp | Formas sólidas de 2-(4-clorofenil)-n-((2-(2,6-dioxopiperidin-3-il)-1-oxoindolin-5-il)metil)-2,2-difluoroacetamida y sus composiciones farmacéuticas y usos |
| US10052315B2 (en) * | 2016-01-08 | 2018-08-21 | Celgene Corporation | Formulations of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide |
-
2018
- 2018-06-29 HU HUE18823719A patent/HUE061356T2/hu unknown
- 2018-06-29 CA CA3068591A patent/CA3068591A1/en active Pending
- 2018-06-29 KR KR1020207002231A patent/KR20200020899A/ko not_active Application Discontinuation
- 2018-06-29 EP EP22212562.7A patent/EP4201399A3/en active Pending
- 2018-06-29 FI FIEP18823719.2T patent/FI3644999T3/fi active
- 2018-06-29 HR HRP20230210TT patent/HRP20230210T1/hr unknown
- 2018-06-29 WO PCT/US2018/040286 patent/WO2019006299A1/en active Application Filing
- 2018-06-29 PL PL18823719.2T patent/PL3644999T3/pl unknown
- 2018-06-29 ES ES18823719T patent/ES2939739T3/es active Active
- 2018-06-29 SG SG11201913008TA patent/SG11201913008TA/en unknown
- 2018-06-29 PT PT188237192T patent/PT3644999T/pt unknown
- 2018-06-29 AU AU2018294327A patent/AU2018294327A1/en active Pending
- 2018-06-29 EP EP18823719.2A patent/EP3644999B1/en active Active
- 2018-06-29 US US16/024,581 patent/US20190030018A1/en not_active Abandoned
- 2018-06-29 JP JP2019572475A patent/JP2020526501A/ja active Pending
- 2018-06-29 DK DK18823719.2T patent/DK3644999T3/da active
- 2018-06-29 MX MX2019015886A patent/MX2019015886A/es unknown
- 2018-06-29 RS RS20230153A patent/RS64029B1/sr unknown
- 2018-06-29 CN CN201880056213.2A patent/CN111032043A/zh active Pending
- 2018-06-29 BR BR112019028101-0A patent/BR112019028101A2/pt not_active Application Discontinuation
- 2018-06-29 LT LTEPPCT/US2018/040286T patent/LT3644999T/lt unknown
- 2018-06-29 SI SI201830859T patent/SI3644999T1/sl unknown
-
2019
- 2019-12-20 MX MX2022001410A patent/MX2022001410A/es unknown
- 2019-12-23 ZA ZA2019/08605A patent/ZA201908605B/en unknown
- 2019-12-26 CL CL2019003857A patent/CL2019003857A1/es unknown
- 2019-12-26 IL IL271726A patent/IL271726A/en unknown
-
2023
- 2023-05-16 JP JP2023080568A patent/JP2023113681A/ja active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140045843A1 (en) * | 2012-08-09 | 2014-02-13 | Celgene Corporation | Methods of treating cancer using 3-(4-((4-(morpholinomethyl)benzyl)oxy)-1-oxoisoindolin-2-yl)piperidine-2,6-dione |
| US20150196562A1 (en) * | 2014-01-15 | 2015-07-16 | Celgene Corporation | Formulations of 3-(5-amino-2-methyl-4-oxo-4h-quinazolin-3-yl)-piperidine-2,6-dione |
| TW201540323A (zh) * | 2014-01-15 | 2015-11-01 | Celgene Corp | 3-(5-胺基-2-甲基-4-側氧基-4h-喹唑啉-3-基)-哌啶-2,6-二酮之調配物 |
| CN106660991A (zh) * | 2014-07-11 | 2017-05-10 | 细胞基因公司 | 抗增殖化合物及其使用方法 |
Non-Patent Citations (2)
| Title |
|---|
| MASAKO YOKOO等: "2-Hydroxypropyl-β-Cyclodextrin Acts as a Novel Anticancer Agent", 《PLOS ONE》 * |
| THORSTEINN LOFTSSON等: "Cyclodextrins in drug delivery", 《EXPERT OPIN. DRUG DELIV.》 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024074699A1 (en) * | 2022-10-07 | 2024-04-11 | Oculis Operations Sarl | Eye drop microsuspensions of mtor inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| MX2019015886A (es) | 2020-09-10 |
| PL3644999T3 (pl) | 2023-05-08 |
| DK3644999T3 (da) | 2023-03-06 |
| BR112019028101A2 (pt) | 2020-07-28 |
| CL2019003857A1 (es) | 2020-07-03 |
| PT3644999T (pt) | 2023-03-10 |
| HUE061356T2 (hu) | 2023-06-28 |
| AU2018294327A1 (en) | 2020-01-23 |
| JP2023113681A (ja) | 2023-08-16 |
| MX2022001410A (es) | 2022-06-09 |
| WO2019006299A1 (en) | 2019-01-03 |
| JP2020526501A (ja) | 2020-08-31 |
| US20190030018A1 (en) | 2019-01-31 |
| RS64029B1 (sr) | 2023-03-31 |
| SI3644999T1 (sl) | 2023-04-28 |
| ZA201908605B (en) | 2023-04-26 |
| LT3644999T (lt) | 2023-03-10 |
| HRP20230210T1 (hr) | 2023-04-14 |
| SG11201913008TA (en) | 2020-01-30 |
| ES2939739T3 (es) | 2023-04-26 |
| IL271726A (en) | 2020-02-27 |
| FI3644999T3 (fi) | 2023-03-19 |
| EP3644999A1 (en) | 2020-05-06 |
| EP3644999B1 (en) | 2022-12-14 |
| EP4201399A3 (en) | 2023-08-09 |
| EP4201399A2 (en) | 2023-06-28 |
| CA3068591A1 (en) | 2019-01-03 |
| EP3644999A4 (en) | 2021-08-04 |
| KR20200020899A (ko) | 2020-02-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3644999B1 (en) | Compositions and methods of use of 2-(4-chlorophenyl)-n-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl) methyl) -2,2-difluoroacetamide | |
| CN108601777B (zh) | 2-(4-氯苯基)-n-((2-(2,6-二氧代哌啶-3-基)-1-氧代异吲哚啉-5-基)甲基)-2,2-二氟乙酰胺的制剂 | |
| CN108430473B (zh) | 抗增殖化合物以及其药物组合物和用途 | |
| CN115282149A (zh) | 用抗癌剂治疗血液恶性肿瘤 | |
| JP2022515670A (ja) | 2-(4-クロロフェニル)-n-((2-(2,6-ジオキソピペリジン-3-イル)-1-オキソイソインドリン-5-イル)メチル)-2,2-ジフルオロアセトアミドの組成物及び使用方法 | |
| US20210128545A1 (en) | Combination therapy with 2-(4-chlorophenyl)-n-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide | |
| US20230149379A1 (en) | Combination therapy for acute myeloid leukemia |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |