CN103781490A - 用于配制肽蛋白酶体抑制剂的环糊精络合法 - Google Patents
用于配制肽蛋白酶体抑制剂的环糊精络合法 Download PDFInfo
- Publication number
- CN103781490A CN103781490A CN201280001354.7A CN201280001354A CN103781490A CN 103781490 A CN103781490 A CN 103781490A CN 201280001354 A CN201280001354 A CN 201280001354A CN 103781490 A CN103781490 A CN 103781490A
- Authority
- CN
- China
- Prior art keywords
- combination
- compound
- cyclodextrin
- acid
- approximately
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 CC(C[C@@]1C=C*CC1)=I Chemical compound CC(C[C@@]1C=C*CC1)=I 0.000 description 2
- WXUHRLNTLMRATM-UHFFFAOYSA-N CC(C(N=C)=C)=O Chemical compound CC(C(N=C)=C)=O WXUHRLNTLMRATM-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y5/00—Nanobiotechnology or nanomedicine, e.g. protein engineering or drug delivery
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6949—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit inclusion complexes, e.g. clathrates, cavitates or fullerenes
- A61K47/6951—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit inclusion complexes, e.g. clathrates, cavitates or fullerenes using cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Abstract
本公开内容提供用于配制包含一种或多种肽蛋白酶体抑制剂和环糊精特别是经取代环糊精的组合物的方法。所述方法大大提高了这些蛋白酶体抑制剂的溶解度和稳定性,并促进其制备和给药两者。
Description
相关申请的交叉引用
本申请主张于2012年5月8日提交的美国临时申请号61/644,122的权益,其全部通过引用并入本文。
技术领域
本公开内容提供用于配制包含一种或多种肽蛋白酶体抑制剂和环糊精(特别是经取代环糊精)或环糊精混合物的组合物的环糊精络合法。所述方法大大提高了这些蛋白酶体抑制剂的溶解度和稳定性,并促进其制备和给药两者。
背景
业已确认蛋白酶体可作为治疗靶标用于治疗包括多发性骨髓瘤在内的多种癌症适应症,其如经FDA批准的硼酸蛋白酶体抑制剂硼替佐米(bortezomib)所证明。然而,最近阐述了可能具有更小的毒副作用的对蛋白酶体更加高度特异的其它抑制剂。这些化合物包括肽环氧酮,例如美国专利号6,831,099中所阐述的环氧甲酮四肽蛋白酶体抑制剂(epoxomicin),其内容通过引用并入本文;和美国专利号7,232,818中所阐述的那些,其内容通过引用并入本文。然而,这些化合物中一些的低水溶性使得难以将其配制为足够高浓度的组合物,从而使其难以以所期需的抗肿瘤或其它的药理学效应来实际给药。因此,需要配制肽环氧酮的另外方法。
简述
本文提供用环糊精配制肽蛋白酶体抑制剂(例如式(1)-(5)的化合物或其药学上可接受的盐)的环糊精络合法。已证明很多肽蛋白酶体抑制剂在水中具有低溶解度。可通过用本文所提供的方法让所述化合物与环糊精络合来克服这种低溶解度。例如,可在药学上有用的pH (例如约3.5)下和在比无环糊精及无本文所提供的化合物与环糊精之间的络合作用过程可获得的浓度更高的浓度下(例如约5 mg/mL),获得式(5) (卡非佐米(carfilzomib))化合物的均质溶液。通过本文所提供的方法制备的制剂除增加肽蛋白酶体抑制剂在溶液中的溶解度之外,还产生具有令人惊奇的稳定性的药用溶液。络合的抑制剂的稳定性反映在:经过长期和在热应力下,均质的络合抑制剂溶液中没有沉淀。例如,在超过那些无菌制备的可注射药用产品实际使用时的典型时间周期和热应力下,络合抑制剂仍可保持可溶。虽然通过本文所提供的处理方法获得的高浓度可能并未预期其热力学稳定,但业已显示该溶液的物理稳定性不受储存温度(例如,溶液从-20℃至25℃是稳定的)、冻融循环和冻干及重构的影响。络合的肽蛋白酶体抑制剂和环糊精的过饱和溶液的稳定性足以耐受络合作用后的pH调节而无沉淀。例如,在pH 2.5-3范围内进行络合作用,然后用氢氧化钠溶液滴定pH至pH 3.5。这种溶液物理稳定性使得可在对于注射和其它药用目的可接受的pH范围内使用络合的物质,还使得可在获得适宜的化学稳定性和保质期时的pH范围内表现出稳定性。因此,通过本文所提供的方法制备的药物组合物,可以是在许多医药应用的使用期间不沉淀或不降低浓度到显著程度的过饱和溶液(例如,在无菌产品制备期间,原液(bulk solution)在无菌过滤后尽管保持在瓶灌装无菌贮存罐中若干天但可不沉淀。同样,最终重构的药物组合物可稳定数小时至数天,这便于将其用作药用试剂)。
除产生稳定的、高度浓缩的肽蛋白酶体抑制剂溶液外,还可获得通过本文所提供的络合法制备的制剂,且无其它配制方法的化学降解和稳定性限制。例如,本文所提供的方法在络合作用期间避免使用强酸(例如HCl)来降低pH。虽然将制剂的pH降低到小于2的值可在络合作用之前促进肽蛋白酶体抑制剂的溶解并产生均质溶液,但该溶液的酸度可导致肽蛋白酶体抑制剂的降解。例如,在肽蛋白酶体抑制剂卡非佐米的情况下,使用强酸例如HCl可导致药理学环氧化物的水解,并通过氯离子的亲核攻击,导致形成降解产物(degradant)氯醇加合物(CDP):
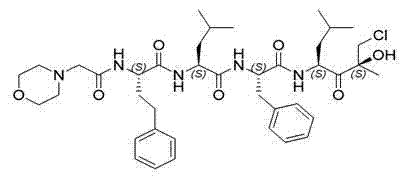
基于其结构,将这种降解产物归类为烷化物(alkylator),其被FDA认为是有潜在遗传毒性的杂质的一类化合物。重要的是,从控制产品安全性这点来看,使用本文所提供的方法避免了这类强酸,因此可显著减少肽蛋白酶体抑制剂降解为此类化合物的降解反应,并且在一些情况下,甚至可消除这种降解反应。
在一个方面,用于制备药物组合物的方法的特征包括:
(i)提供包括以下的第一组合(first combination):
(a)一种(或多种)肽蛋白酶体抑制剂(例如式(1)-(5)的化合物或其药学上可接受的盐);
(b)一种或多种环糊精(“CD”);和
(c)水;
其中第一组合是异质的,并且化合物或盐在第一组合中具有低溶解度;和
(ii)让第一组合与酸接触以形成第二组合(second combination),其中化合物在第二组合中比在第一组合中更加可溶。
在另一个方面,用于制备药物组合物的方法的特征包括:
(i)提供包括以下的第一组合:
(a)如下化合物或其药学上可接受的盐:
;
(b)一种或多种环糊精(“CD”);和
(c)水;
其中第一组合是异质的,并且化合物或盐在第一组合中具有低溶解度;和
(ii)让第一组合与酸接触以形成第二组合,其中化合物在第二组合中比在第一组合中更加可溶。
在又一方面,用于制备药物组合物的方法的特征包括:
(i)提供包括以下的第一组合:
(a)如下化合物或其药学上可接受的盐:
;
(b) SBECD;和
(c)注射用水;
其中第一组合是异质的,并且化合物或盐在第一组合中具有低溶解度;和
(ii)让第一组合与柠檬酸水溶液接触以形成第二组合,其中化合物在第二组合中比在第一组合中更加可溶。
在一个方面,药物组合物特征为通过本文所述方法中的任何一种来制备。
在一个方面,用于治疗患者癌症(例如多发性骨髓瘤,例如复发性和/或顽固性的多发性骨髓瘤)的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
在另一方面,用于治疗患者自身免疫病的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
在另一方面,用于治疗患者移植或移植-相关病况的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
在另一方面,用于治疗患者神经退行性疾病的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
在另一方面,用于治疗患者纤维化-相关病况的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
在另一方面,用于治疗患者纤维化-相关病况的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
在另一方面,用于治疗患者缺血相关病况的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
在另一方面,用于治疗患者感染的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
在另一方面,用于治疗患者感染的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
在另一方面,用于治疗患者骨丢失相关疾病的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
在另一方面,用于治疗患者感染的方法的特征包括,将治疗有效量的通过本文所述方法中的任何一种制备的药物组合物给予患者。
实施方案可包括下列一个或多个特征。
第一组合不包含可察觉量的任何有机溶剂。在一些实施方案中,第一组合不包含美国专利7,232,818和/或7,417,042和/或7,737,112和/或US-2009-0105156和/或US-2011-0236428中所阐述的任何量或种类的有机溶剂,其每一个通过引用并入本文。在一些实施方案中,第一组合不含任何有机溶剂(例如,含有小于5%、小于4%、小于3%、小于2%、小于1% (w/w或 w/v)的任何有机溶剂)。在一些实施方案中,第一组合基本上不含任何有机溶剂(例如,含有小于0.5%、小于0.2、小于0.1、小于0.05% (w/w或w/v)的任何有机溶剂)。在某些实施方案中,第一组合不包含可检测的量的任何有机溶剂。
第一组合不包含可察觉量的任何缓冲剂。在一些实施方案中,第一组合不包含美国专利7,232,818和/或7,417,042和/或7,737,112和/或US-2009-0105156和/或US-2011-0236428中所阐述的任何量或种类的任何缓冲剂,这些专利的每一个通过引用并入本文。在一些实施方案中,第一组合不含任何缓冲剂(例如,含有小于5%、小于4%、小于3%、小于2%、小于1% (w/w或w/v)的任何缓冲剂)。在一些实施方案中,第一组合基本上不含任何缓冲剂(例如,含有小于0.5%、小于0.2、小于0.1、小于0.05% (w/w或w/v)的任何缓冲剂)。在一些实施方案中,第一组合不包含可检测量的任何缓冲剂。
第二组合包含化合物与一种或多种环糊精的络合物。
以水溶液形式加入酸。
一种或多种环糊精的至少一种为HPBCD或SBECD (例如SBECD)。
本发明人已经发现,使本文所述方法和药物组合物中的氯离子(或其它亲核阴离子)的量最小化可为有利的。
在一些实施方案中,一种或多种环糊精的至少一种(加入到第一组合中的)为低氯化物环糊精。本文所用“低氯化物环糊精”指具有小于或等于0.05% w/w氯化钠的环糊精,或如果氯化钠以外的氯化物源存在(或除氯化钠外还存在其它氯化物源),则“低氯化物环糊精”指这样的环糊精,其所具有的氯离子含量小于或等于应存在于含有0.05% w/w氯化钠的环糊精中的氯化物量。在一些实施方案中,低氯化物环糊精为低氯化物SBECD。可通过本领域已知的多种方法来测定氯化物浓度(例如对于市购获得的环糊精,根据厂家产品说明书的方法,例如通过比重测定技术,例如通过电势测定技术)。
在一些实施方案中,存在的氯离子的量(例如氯离子与化合物的摩尔比)足够低以便当储存在2-8摄氏度时提供2年的保质期。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于2.0。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于1.5。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于1.2。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于1.0。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.9。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.8。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.7。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.6。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.5。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.4。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.3。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.2。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.1。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比为0.2-1.2 (例如0.3-1.2、例如0.2-0.4、例如0.3-0.4、例如0.32)。
在实施方案中,本文所述的氯离子与化合物的摩尔比亦可存在于第二和/或第三组合(third combination)中。
举例而言,可如下所示,用小瓶卡非佐米(“CFZ”)干粉作为计算的基础来计算第一组合中氯离子与化合物的摩尔比:
小瓶内含物质量= 3.212 g
CFZ质量= 61.8 mg
氯化物最大质量(0.03 % w/w氯离子时) = 0.0009636 g
氯化物最大摩尔质量= 2.714 x 10^-5
(原子质量Cl = 35.5)
CFZ摩尔质量= 8.584 x 10^-5
(MW CFZ = 719.9)
小瓶中固态Cl/CFZ的摩尔比= 0.32
亦可用例如环糊精的氯化物含量(和任何其它的氯离子源)和被加入以制造第一组合的化合物的质量来确定第一组合的这种计算。
如技术人员可理解的一样,期望该比率在用于依次存入(file)小瓶中的前体原液(冻干前)以及当所述干粉小瓶中的内含物在无菌水中重构供患者给药时是一样的。
提供第一组合(步骤(i))包括将化合物加入到一种或多种环糊精和水的溶液中。
化合物为结晶固体。在实施方案中,化合物的晶形具有的X-射线粉末衍射图样包含在6.10、9.32、10.10、12.14、13.94、18.44、20.38和23.30的2θ角处表示的2至8个特征峰。
所述方法进一步包括在让第一组合与酸接触之前混合第一组合。
步骤(i)和(ii)两步都在单个容器中进行。
方法进一步包括将第二组合混合足够时间以获得均质的第三组合。
第三组合中溶解和络合的化合物浓度为1 mg/mL-20 mg/mL。
第三组合中溶解和络合的化合物浓度为4-8 mg/mL。
第三组合的pH为2-4。
所述方法进一步包括将第三组合过滤。
所述方法进一步包括将第三组合冻干以提供冻干物(lyophilizate)。
所述方法进一步包括将冻干物与药学上可接受的载体混合。
药学上可接受的载体包含无菌注射用水。在实施方案中,药学上可接受的载体进一步包括柠檬酸。
除非另外定义,否则本文所使用的所有技术和科学术语具有与本公开内容所属领域的普通技术人员通常理解一样的意思。本文阐述了用于本公开内容的方法和材料;亦可使用本领域所知的其它适宜的方法和材料。材料、方法和实例仅为阐明性的而非意欲限制。本文所提到的所有出版物、专利申请、专利、序列、数据库条目和其它参考文献,全部以整体内容通过引用并入本文。在有冲突的情况下,以包括定义在内的本说明书为准。
根据以下详述和附图及根据权利要求书,本公开内容的其它特征和优势将清楚明了。
附图简述
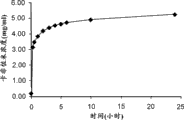
图1为显示随时间变化SBECD络合CFZ-API的线图。
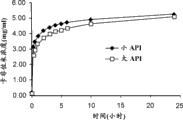
图2图示说明了本文所制备的药物组合物不依赖于蛋白酶体抑制剂的生理化学性质(例如粒度)。
图3为显示随SBECD浓度增加CFZ-API溶解作用增加的线图。
图4图示说明了CFZ-API/SBECD络合物的溶解度不依赖处理或储存温度。
图5图示说明了在pH 3.5下氯醇降解产物(CDP)的水平与水及氯化物含量的二因子交互效应之间的相关性。
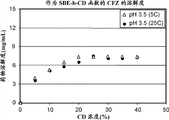
图6图示说明了在pH 1.5和pH 3.5、25℃和5℃时卡非佐米在SBECD中的溶解度(5.9 mg/mL柠檬酸)。
详述
本文提供用环糊精配制肽蛋白酶体抑制剂(例如式(1)-(5)的化合物或其药学上可接受的盐)的环糊精络合法。本文亦提供包含肽蛋白酶体抑制剂和环糊精的药物组合物,其中该组合物具有如本文各处所述的氯离子(例如,用低氯化物环糊精制备组合物;例如,氯离子与化合物的摩尔比为0.32)。在一些实施方案中,具有如本文所述的低氯离子含量的制剂可导致不希望的降解产物的形成减少。
定义
术语“Cx-y烷基”指取代的或未被取代的饱和烃基,包括在链中含有x-y个碳的直链烷基和支链烷基,包括卤代烷基,例如三氟甲基和2,2,2-三氟乙基等。术语“C2-y烯基”和“C2-y炔基”指取代的或未被取代的不饱和脂肪族基团,其在长度和可能的取代上与上述烷基类似,但其各自含有至少一个二键或三键。
术语“烷氧基”指其上连接有氧的烷基。代表性的烷氧基包括甲氧基、乙氧基、丙氧基、叔-丁氧基等等。“醚”是通过氧共价连接的两个烃。因此,致使烷基成为醚的烷基取代基是烷氧基或类似烷氧基。
术语“C1-6烷氧基烷基”指用烷氧基取代的C1-6烷基,藉此形成醚。
本文所用术语“C1-6芳烷基”指用芳基取代的C1-6烷基。
术语“胺”和“氨基”为本领域公认,指未被取代的胺和取代的胺及其盐,例如可用以下通式表示的部分:
或
其中R9、R10和R10′各自独立代表氢、烷基、烯基、-(CH2)m-R8,或R9和R10与其所连接的N原子一起完成环结构中具有4-8个原子的杂环;R8代表芳基、环烷基、环烯基、杂环基或多环基;m为零或1-8的整数。在一些实施方案中,R9或R10中仅有一个为羰基,例如R9、R10和氮一起不形成酰亚胺。在一些实施方案中,R9和R10 (和任选R10′)各自独立代表氢、烷基、烯基或-(CH2)m-R8。在某些实施方案中,氨基为碱性,意即其质子化形式的pKa高于7.00。
术语“酰胺”和“酰氨基”本领域公认为取代氨基的羰基,包括可用以下通式表示的部分:
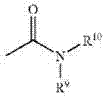
其中R9、R10为如上所定义。在一些实施方案中,酰胺将不包括可能不稳定的酰亚胺。
本文所用术语“芳基”包括取代或未被取代的5-、6-和7-元单环芳族基,其中环中每一个原子都是碳。术语“芳基”亦包括具有两个或多个环状环的多环环系统,其中两个或多个碳原子为两个相邻的环共有,其中至少一个环为芳香环,例如其它的环状环可为环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。芳基包括苯、萘、菲、苯酚、苯胺等等。
术语“缓冲剂”为这样的物质,其在溶液中的存在增加必须加入以引起pH单位变化的酸或碱的量。因此,缓冲剂是辅助调节组合物的pH的物质。通常基于所需的pH和与组合物中其它组分的相容性来选择缓冲剂。总的来说,缓冲剂具有的pKa比组合物所需的(或组合物将在溶解时产生的) pH低或高不到1个单位。
本文所用术语“水”指具有pH为约7.0的H2O的液体溶液。
本文所用术语“碳环”和“碳环基”,指取代的或未被取代的非芳香环,其中环中每一个原子都是碳。术语“碳环”和“碳环基”亦包括具有两个或多个环状环的多环环系统,其中两个或多个碳原子为两个相邻的环共有,其中至少一个环为碳环,例如其它的环状环可为环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。
术语“羰基”为本领域公认,包括诸如可用以下通式表示的部分:
或
其中X为键或代表氧或硫,R11代表氢、烷基、烯基、-(CH2)m-R8或药学上可接受的盐,R11′代表氢、烷基、烯基或-(CH2)m-R8,其中m和R8如上所定义。当X为氧且R11或R11′不为氧时,所述式表示“酯”。当X为氧且R11为氢时,所述式表示“羧酸”。
本文所用术语“C1-6杂芳烷基”,指用杂芳基取代的C1-6烷基。
术语“杂芳基”包括取代的或未被取代的芳香族5-至7-元环结构,例如5-至6-元环,其环结构包括一至四个杂原子。术语“杂芳基”亦包括具有两个或多个环状环的多环环系统,其中两个或多个碳原子为两个相邻的环共有,其中至少一个环为杂芳香环,例如其它的环状环可为环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。杂芳基包括例如吡咯、呋喃、噻吩、咪唑、噁唑、噻唑、三唑、吡唑、吡啶、吡嗪、哒嗪和嘧啶等等。
本文所用术语“杂原子”意为非碳或氢的任何元素的原子。例如,杂原子包括氮、氧、磷和硫。
术语“杂环基”或“杂环基团”指取代的或未被取代的非芳香族的3-至10-元环结构,例如,3-至7-元环,其环结构包括一至四个杂原子。术语“杂环基”或“杂环基团”亦包括具有两个或多个环状环的多环环系统,其中两个或多个碳原子为两个相邻的环共有,其中至少一个环为杂环,例如其它的环状环可为环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。杂环基包括例如哌啶、哌嗪、吡咯烷、吗啉、内酯、内酰胺等等。
术语“C1-6羟烷基”指用羟基取代的C1-6烷基。
术语“硫醚”指其上连接有硫部分的如上所定义的烷基。在一些实施方案中,“硫醚”用-S-烷基来表示。代表性的硫醚基包括甲硫基、乙硫基等等。
术语“取代的”指在分子的一个或多个非氢原子上具有取代氢的取代基的部分。应理解“取代的”或“用……取代”包括暗示的前提条件,即所述取代依照被取代原子和取代基所允许的化合价,并且取代导致形成稳定的化合物,例如,其不会自发例如通过重排、环化、消除等进行转化。本文所用术语“被取代的”考虑包括有机化合物的所有可容许的取代基。在广泛方面,可容许的取代基包括有机化合物的非环状的和环状的、分支的和非分支的、碳环的和杂环的、芳香族的和非芳香族的取代基。可容许的取代基可为一种或多种,并且对于适当的有机化合物可相同或不同。对于本公开内容的目的而言,杂原子例如氮可具有氢取代基和/或本文所述有机化合物的满足杂原子的化合价的任何可容许的取代基。取代基可包括例如卤素、羟基、羰基(例如羧基、烷氧羰基、甲酰基或酰基)、硫代羰基(例如硫酯、硫代乙酸酯或硫代甲酸酯)、烷氧基、磷酰基、磷酸酯、膦酸酯、亚膦酸酯、氨基、酰氨基、脒、亚胺、氰基、硝基、叠氮基、巯基、烷硫基、硫酸基、磺酸基、氨磺酰基、磺酰氨基、磺酰基、杂环基、芳烷基或芳香族或杂芳香族部分。本领域技术人员应了解,若合适,烃链上取代的部分本身可被取代。
在一些实施方案中,本文所提供的化合物或其盐为基本上分离的或纯化的。“基本上分离的”意即化合物至少部分或大体上自其所形成或检出的环境分离。部分分离可包括例如富含本文所提供的化合物的组合物。基本上分离可包括含有至少约50%、至少约60%、至少约70%、至少约80%、至少约90%、至少约95%、至少约97%或至少约99%重量的化合物或其盐的组合物。用于分离化合物及其盐的方法是本领域的常规技术。
本文所用术语“肽”指长度为约二至约十个氨基酸的氨基酸链。
本文所用术语“天然的”或“天然存在的”氨基酸,指二十种最普遍存在的氨基酸之一。用其标准的一个或三个字母的缩写来指天然氨基酸。
术语“非天然氨基酸”或“非天然的”指天然氨基酸的任何衍生物或结构类似物,包括D型和β及γ氨基酸衍生物。应注意本文中将其归类为非天然氨基酸的某些氨基酸,例如羟脯氨酸,可天然存在于某些生物体或特定蛋白质中。非天然氨基酸的非限制性实例包括:β-丙氨酸(β-Ala)、γ-氨基丁酸(GABA)、2-氨基丁酸(2-Abu)、α,β-脱氢-2-氨基丁酸(Δ-Abu)、1-氨基环丙烷-1-羧酸(ACPC)、氨基异丁酸(Aib)、2-氨基-噻唑啉-4-羧酸、5-氨基戊酸(5-Ava)、6-氨基己酸(6-Ahx)、8-氨基辛酸(8-Aoc)、11-氨基十一酸(11-Aun)、12-氨基十二酸(12-Ado)、2-氨基苯甲酸(2-Abz)、3-氨基苯甲酸(3-Abz)、4-氨基苯甲酸(4-Abz)、4-氨基-3-羟基-6-甲基庚酸(Statine,Sta)、氨基氧乙酸(Aoa)、2-氨基四氢萘-2-羧酸(Atc)、4-氨基-5-环己基-3-羟基戊酸(ACHPA)、对-氨基苯丙氨酸(4-NH2-Phe)、联苯丙氨酸(Bip)、对-溴苯丙氨酸(4-Br-Phe)、邻-氯苯丙氨酸(2-Cl-Phe)、间-氯苯丙氨酸(3-Cl-Phe)、对-氯苯丙氨酸(4-Cl-Phe)、间-氯酪氨酸(3-Cl-Tyr)、对-苯甲酰基苯丙氨酸(Bpa)、叔-丁基甘氨酸(Tle)、环己基丙氨酸(Cha)、环己基甘氨酸(Chg)、2,3-二氨基丙酸(Dpr)、2,4-二氨基丁酸(Dbu)、3,4-二氯苯丙氨酸(3,4-Cl2-Phe)、3,4-二氟苯丙氨酸(3,4-F2-Phe)、3,5-二碘酪氨酸(3,5-12-Tyr)、邻-氟苯丙氨酸(2-F-Phe)、间-氟苯丙氨酸(3-F-Phe)、对-氟苯丙氨酸(4-F-Phe)、间-氟酪氨酸(3-F-Tyr)、高丝氨酸(Hse)、高苯丙氨酸(Hfe)、高酪氨酸(Htyr)、5-羟色氨酸(5-OH-Trp)、羟脯氨酸(Hyp)、对-碘苯丙氨酸(4-1-Phe)、3-碘酪氨酸(3-I-Tyr)、二氢吲哚-2-羧酸(Idc)、异哌啶酸(Inp)、间-甲基酪氨酸(3-Me-Tyr)、1-萘基丙氨酸(1-Nal)、2-萘基丙氨酸(2-Nal)、对-硝基苯丙氨酸(4-NO2-Phe)、3-硝基酪氨酸(3-NO2-Tyr)、正亮氨酸(Nle)、正缬氨酸(Nva)、鸟氨酸(Orn)、邻-磷酸酪氨酸(H2PO3-Tyr)、八氢吲哚-2-羧酸(Oic)、青霉胺(Pen)、五氟苯丙氨酸(F5-Phe)、苯基甘氨酸(Phg)、哌可酸(Pip)、炔丙基甘氨酸(Pra)、焦谷氨酸(pGlu)、肌氨酸(Sar)、四氢异喹啉-3-羧酸(Tic)和噻唑烷-4-羧酸(硫代脯氨酸,Th)。视情况可通过在名称或缩写之前标识“D”或“d”或“L”或“l”来标明氨基酸的立体化学形式。或者,可用传统的(S)-或(R)-标识来表示手性中心。另外,亦可使用αN-烷基化的氨基酸,以及具有含胺侧链的氨基酸(例如Lys和Orn),其中胺被酰化或烷基化。参见例如Robert A. Meyers编辑的Molecular Biology and Biotechnology: A Comprehensive Desk Reference (分子生物学和生物技术:综合性案头参考)一书中Hruby和Boteju所写的“Peptides and Mimics, Design of Conformationally Constrained” (肽和模拟物,构象上受约束的设计)一文,VCH出版社(1995),第658-664页,其通过引用并入本文。
本文所用术语“络合作用”,指在溶液中和在一种或多种肽蛋白酶体抑制剂和一种或多种环糊精分子之间,形成分子间包合络合物或分子间缔合物。包含物和/或缔合物作为以下机理提供效用:与无络合剂(即一种或多种环糊精分子)时在相似pH范围下的水相溶解相比,其大大增加了水溶液中可获得的抑制剂的浓度。
术语“预防性的或治疗性的”治疗为本领域公认,包括将一种或多种受试组合物给予宿主。如果是在临床上表现出不想要的病况(例如宿主动物的疾病或其它不想要的状态)之前给予,则治疗是预防性的(即其保护宿主避免出现不想要的病况);而如果是在表现出不想要的病况之后给予,则治疗是治疗性的(即意欲消除、改善或稳定现有不想要的病况或其副作用)。
本文所用术语“蛋白酶体”意即包括免疫型和组成型蛋白酶体。
本文所用术语“抑制剂”意即描述阻断或降低酶或酶系统、受体或其它药理学靶标的活性的化合物(例如,抑制标准产荧光的肽底物例如suc-LLVY-AMC、Box-LLR-AMC和Z-LLE-AMC的蛋白酶剪切,抑制20S蛋白酶体的各种催化活性)。抑制剂可表现为竞争性、无竞争性或非竞争性抑制。抑制剂可以可逆或不可逆地结合,因此该术语包括为酶的自杀底物的化合物。抑制剂可修饰位于或接近酶的活性位点的一个或多个位点,或其可导致酶在别处的构象变化。本文对术语抑制剂的使用比科学文献中的使用更加广泛以便还包含其它类别的在药理学上或治疗上有用的试剂,例如激动剂、拮抗剂、刺激剂、辅因子等等。
本文所用“低溶解度”指在例如水或另一溶液中(例如第一组合)略溶、微溶、极微溶、几乎不溶或不溶;术语“略溶、微溶、极微溶、几乎不溶或不溶”在含意上对应于美国药典(USP)中对近似溶解度表述的通用术语。参见例如Avis, K.E., Lackman, L和Lieberman, H.A.编辑的DeLuca和Boylan的Pharmaceutical Dosage Forms: Parenteral Medications,第1卷,Marcel Dekkar:1084,第141-142页:
| USP术语 | 溶解1份溶质的溶剂的相对量 |
| 略溶 | 30-100 |
| 微溶 | 100-1,000 |
| 极微溶 | 1,000-10,000 |
| 几乎不溶或不溶 | >10,000 |
本文所用“异质的”指具有非均一(多相)组成的溶液。例如,异质溶液可包括液体中固体颗粒的悬浮液(例如浆液)。
本文所用“均质的”指其整个体积中是一致的或均一的溶液(单相,观察为透明溶液)。
对于主题治疗方法而言,化合物的“治疗有效量”指制剂中化合物的这样的量,当将其作为所需的给药方案的部分给予(患者例如人)时,根据待治疗的病症或病况的临床上可接受的标准或美容目的,例如以适用于任何医学治疗的合理的效益/风险比值,减轻症状、改善病况或延缓疾病病况的发作。
本文所用术语“治疗(teating或treatment)”包括以改善或稳定患者病况的方式来逆转、减少或阻止病况的症状、临床征兆和潜在病理。
化合物
本文提供用于制备在水中具有低溶解度特征的肽蛋白酶体抑制剂制剂的方法。肽蛋白酶体抑制剂包含含有环氧化物或氮杂环丙烷的部分,所述部分含有邻近含杂原子的三元环的基团,从而促进含杂原子的三元环的开环反应。这类基团包括例如吸电子基团,例如羰基。在一些实施方案中,肽蛋白酶体抑制剂为肽环氧蛋白酶体抑制剂。本文所用“肽环氧蛋白酶体抑制剂”包含在酮的一侧具有环氧基团且在另一侧具有肽的酮部分。
肽蛋白酶体抑制剂的肽包括2-10个氨基酸。例如,该肽可具有2-8个氨基酸、2-6个氨基酸、2-5个氨基酸、2-4个氨基酸、3-10个氨基酸、4-10个氨基酸、6-10个氨基酸、8-10个氨基酸、3-4个氨基酸、3-5个氨基酸和4-6个氨基酸。在一些实施方案中,所述肽具有3或4个氨基酸。
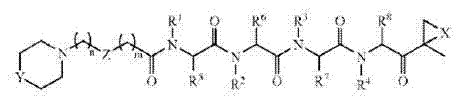
在一些实施方案中,肽蛋白酶体抑制剂为式(1)化合物或其药学上可接受的盐:

其中:
X为氧、NH或N(C1-6烷基);
W为包含二至十个氨基酸的肽,其中氨基酸可为天然的、非天然的或其组合;和
R为氢原子或C1-4烷基,其可用以下官能团来取代:羟基、卤素、氨基、羧基、羰基、硫、硫化物、酯、酰胺或醚官能团中的一种或多种。
在一些实施方案中,配置X以促进与Ntn水解酶的N-末端亲核基团相互作用。例如,导致抑制的酶抑制剂与20S蛋白酶体的β5/Pre2亚基的不可逆相互作用,似乎受到以上阐明的构型的促进。在其它Ntn水解酶的情况下,肽环氧化物或肽氮杂环丙烷类的α-碳的相反的空间化学可能有用。在一些实施方案中,X为氧。
α′-碳(即形成环氧化物或氮杂环丙烷环部分的碳)的立体化学可为(R)或(S)。注意化合物可具有多个立体中心,所述立体中心具有标明的上-下(或β-α,其中本文所绘的β在页面平面之上)或(R)-(S)关系(即不需要化合物中的每一立体中心符合陈述的偏好)。在一些实施方案中,α′碳的立体化学为(R),即当如式(1)中所绘时,X原子为β,或在分子平面之上。
在式(1)化合物的情况下,β′碳被两个氢原子取代。关于立体化学,用星号标明手性α′碳,并遵照用于确定绝对立体化学的Cahn-Ingold-Prelog规则。这些规则例如在以下文献中阐述:Fox和Whitesell的Organic Chemistry (有机化学),Jones and Bartlett出版社,Boston, Mass. (1994)的第5-6部分,第177-178页,通过引用将该部分并入本文。当氧或氮具有最高优先性、肽-酮基团具有第二高优先性而-CH2-X-基团第三高优先性时,α′碳的立体化学为(R)。对于一些实施方案,如果肽-酮、-CH2-X-和R基团的相对优先性改变,则标称的立体化学可改变,但基团的基本构型仍可保留一样。也就是说,提及上面刚刚所述的一般结构,肽-酮从左边与手性α′碳结合,R从右边与手性α′碳结合,而X原子凸出于页面平面。原则上,氮杂环丙烷环的氮原子亦可为手性的,其在以下文献中讨论:March的Advanced Organic Chemistry (高等有机化学),第四版(1992),Wiley-Interscience出版社,New York,第98-100页,这几页通过引用并入本文。
W为包含二至十个氨基酸的肽,其中氨基酸可为天然的、非天然的或其组合。例如,肽可具有2-8个氨基酸、2-6个氨基酸、2-5个氨基酸、2-4个氨基酸、3-10个氨基酸、4-10个氨基酸、6-10个氨基酸、8-10个氨基酸、3-4个氨基酸、3-5个氨基酸和4-6个氨基酸。在一些实施方案中,肽具有3或4个氨基酸。在可用于抑制蛋白酶体的糜蛋白酶-类(CT-L)活性的一些实施方案中,存在四到八个氨基酸,而在用于CT-L抑制的一些实施方案中,存在四到六个氨基酸。在可用于抑制蛋白酶体的PGPH活性的其它实施方案中,存在二到八个氨基酸,而在用于PGPH抑制的一些实施方案中,存在三到六个氨基酸。可在肽的任一末端之间形成式(1)中W和酮部分之间的键。例如,在一些实施方案中,让酮与肽的羧基末端键合。或者,酮可与肽的氨基末端键合。在一些实施方案中,酮可与肽的侧链键合。
可在美国专利号7,737,112中找到式(1)化合物的实例,其全部通过引用并入本文。在一些实施方案中,式(1)化合物在水中具有低溶解度。
抑制Ntn的糜蛋白酶-类(CT-L)活性的肽蛋白酶体抑制剂可包括具有至少四个氨基酸的肽。在一些CT-L抑制剂的实施方案中,抑制剂具有有至少四个氨基酸的肽和α′,β′-环氧酮或α′,β′-氮杂环丙烷酮部分(四肽环氧酮或四肽氮杂环丙烷酮)。
在一些实施方案中,具有低水溶解性的肽蛋白酶体抑制剂可为式(II)化合物或其药学上可接受的盐:
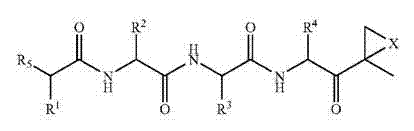
其中:
每一个A独立选自C═O、C═S和SO2;或
当毗邻出现的Z时,A为任选共价键;
L不存在或选自C═O、C═S和SO2;
M不存在或为C1-12烷基;
Q不存在或选自O、NH和N(C1-6烷基);
X选自O、NH和N(C1-6烷基);
Y不存在或选自O、NH、N(C1-6烷基)、S、SO、SO2、CHOR10和CHCO2R10;
每一个Z独立选自O、S、NH和N(C1-6烷基);或
当毗邻出现的A时,Z为任选共价键;
R1、R2、R3和R4每一个独立选自C1-6烷基、C1-6羟烷基、C1-6烷氧基烷基、芳基和C1-6芳烷基,其任何一个任选用酰胺、胺、羧酸(或其盐)、酯、硫醇基或硫醚取代基中的一种或多种取代;
R5为N(R6)LQR7;
R6选自氢、OH和C1-6烷基;
R7选自氢、C1-6烷基、C1-6烯基、C1-6炔基、芳香基、C1-6芳烷基、杂芳基、C1-6杂芳烷基、R8ZAZ-C1-8烷基-、R11Z-C1-8烷基-、(R8O)(R9O)P(═O)O-C1-8烷基-ZAZ-C1-6烷基-、R8ZAZ-C1-8烷基-ZAZ-C1-8烷基-、杂环基MZAZ-C1-8烷基-、(R8O)(R9O)P(═O)O-C1-8烷基-、(R10)2N-C1-12烷基-、(R10)3N+-C1-12烷基-、杂环基M-、碳环基M-、R11SO2C1-8烷基-和R11SO2NH;或
R6和R7一起为C1-6烷基-Y-C1-6烷基、C1-6烷基-ZAZ-C1-6烷基、ZAZ-C1-6烷基-ZAZ-C1-6烷基、ZAZ-C1-6烷基-ZAZ或C1-6烷基-A,藉此形成环;
R8和R9独立选自氢、金属阳离子、C1-6烷基、C1-6烯基、C1-6炔基、芳基、杂芳基、C1-6芳烷基和C1-6杂芳烷基,或R8和R9一起为C1-6烷基,藉此形成环;
每一个R10独立选自氢和C1-6烷基;和
R11独立选自氢、C1-6烷基、C1-6烯基、C1-6炔基、碳环基、杂环基、芳基、杂芳基、C1-6芳烷基和C1-6杂芳烷基;
前提为当R6为H或CH3且Q不存在时,LR7不为氢、未被取代的C1-6烷基C═O、氨基酸的另外链、叔-丁氧羰基(Boc)、苯酰基(Bz)、芴-9-基甲氧羰基(Fmoc)、三苯甲基(trityl)、苄氧羰基(Cbz)、三氯乙氧羰基(Troc)、或取代的或未被取代的芳基或杂芳基;和
在序列ZAZ的任何出现时,序列中的至少一个成员必须不是共价键。
在某些实施方案中,当R6为H、L为C═O且Q不存在时,R7不为氢、C1-6烷基、或取代的或未被取代的芳香基或杂芳基。在某些实施方案中,当R6为H且Q不存在时,R7不为保护基团,例如以下文献中所述的保护基团:Greene, T. W.和Wuts, P. G. M. “Protective Groups in Organic Synthesis” (有机合成中的保护基团),John Wiley & Sons,1999;或Kocienfski, P. J. “Protecting Groups” (保护基团),Georg Thieme Verlag,1994。
在一些实施方案中,R1、R2、R3和R4选自C1-6烷基或C1-6芳烷基。例如,R2和R4为C1-6烷基,R1和R3为C1-6芳烷基。在一些实施方案中,R2和R4为异丁基,R1为2-苯乙基,R3为苯甲基。
在一些实施方案中,L和Q不存在,R7选自C1-6烷基、C1-6烯基、C1-6炔基、C1-6芳烷基和C1-6杂芳烷基。例如,R6为C1-6烷基,R7选自丁基、烯丙基、炔丙基、苯甲基、2-吡啶基、3-吡啶基和4-吡啶基。
在一些实施方案中,L为SO2,Q不存在,R7选自C1-6烷基和芳基。例如,R7可选自甲基和苯基。
在一些实施方案中,L为C═O,R7选自C1-6烷基、C1-6烯基、C1-6炔基、芳基、C1-6芳烷基、杂芳基、C1-6杂芳烷基、R8ZA-C1-8烷基-R11Z-C1-8烷基-、(R8O)(R9O)P(═O)O-C1-8烷基-、(R8O)(R9O)P(═O)O-C1-8烷基-ZAZ-C1-8烷基-、(R8O)(R9O)P(═O)O-C1-8烷基-Z-C1-8烷基-、R8ZA-C1-8烷基-ZAZ-C1-8烷基-、杂环基MZAZ-C1-8烷基-、(R10)2N-C1-8烷基-、(R10)3N+-C1-8烷基-、杂环基-M碳环基M-、R11SO2C1-8烷基-和R11SO2NH-,其中Z和A的每一个的出现独立地不是共价键。在一些实施方案中,L为C═O,Q不存在,R7为H。
在一些实施方案中,R6为C1-6烷基,R7为C1-6烷基,Q不存在,L为C═O。在某些所述实施方案中,R7为乙基、异丙基、2,2,2-三氟乙基或2-(甲磺酰)乙基。
在一些实施方案中,L为C═O,Q不存在,R7为C1-6芳烷基。例如,R7可选自2-苯乙基、苯甲基、(4-甲氧苯基)甲基、(4-氯苯基)甲基和(4-氟苯基)甲基。
在一些实施方案中,L为C═O,Q不存在,R6为C1-6烷基,R7为芳基。例如,R7可为取代的或未被取代的苯基。
在一些实施方案中,L为C═O,Q不存在或为O,n为0或1,R7为-(CH2)n碳环基。例如,R7可为环丙基或环己基。
在一些实施方案中,L和A为C═O,Q不存在,Z为O,n为1-8的整数(例如1),R7选自R8ZA-C1-8烷基-、R11Z-C1-8烷基-、R8ZA-C1-8烷基-ZAZ-C1-8烷基-、(R8O)(R9O)P(═O)O-C1-8烷基-ZAZ-C1-8烷基-、(R8O)(R9O)P(═O)O-C1-8烷基-Z-C1-8烷基-和杂环基MZAZ-C1-8烷基-,其中每一个A的出现独立地不是共价键。例如,R7可为杂环基MZAZ-C1-8烷基-,其中杂环基为取代的或未被取代的氧代间二氧杂环戊烯基(oxodioxolenyl)或N(R12)(R13),其中R12和R13一起为C1-6烷基-Y-C1-6烷基,例如C1-3烷基-Y-C1-3烷基,藉此形成环。
在一些实施方案中,L为C═O,Q不存在,n为1-8的整数,R7选自(R8O)(R9O)P(═O)O-C1-8烷基-、(R10)2NC1-8烷基、(R10)3N+(CH2)n-和杂环基-M-。在某些所述实施方案中,R7为-C1-8烷基N(R10)2或-C1-8烷基N+(R10)3,其中R10为C1-6烷基。例如,R7为杂环基M-,其中杂环基选自吗啉代、哌啶子基、哌嗪基(piperazino)和吡咯基(pyrrolidino)。
在一些实施方案中,L为C═O,R6为C1-6烷基,Q选自O和NH,R7选自C1-6烷基、环烷基-M、C1-6芳烷基和C1-6杂芳烷基。在一些实施方案中,L为C═O,R6为C1-6烷基,Q选自O和NH,R7为C1-6烷基,其中C1-6烷基选自甲基、乙基和异丙基。在一些实施方案中,L为C═O,R6为C1-6烷基,Q选自O和NH,R7为C1-6芳烷基,其中芳烷基为苯甲基。在一些实施方案中,L为C═O,R6为C1-6烷基,Q选自O和NH,R7为C1-6杂芳烷基,其中杂芳烷基为(4-吡啶基)甲基。
在一些实施方案中,L不存在或为C═O,R6和R7一起为C1-6烷基-Y-C1-6烷基、C1-6烷基-ZA-C1-6烷基或C1-6烷基-A,其中每一个Z和A的出现独立地不是共价键,藉此形成环。在一些实施方案中,L为C═O,Q和Y不存在,R6和R7一起为C1-3烷基-Y-C1-3烷基。在一些实施方案中,L和Q不存在,R6和R7一起为C1-3烷基-Y-C1-3烷基。在一些实施方案中,L为C═O,Q不存在,Y选自NH和N-C1-6烷基,R6和R7一起为C1-3烷基-Y-C1-3烷基。在一些实施方案中,L为C═O,Y不存在,R6和R7一起为C1-3烷基-Y-C1-3烷基。在一些实施方案中,L和A为C═O,R6和R7一起为C1-2烷基-ZA-C1-2烷基。在一些实施方案中,L和A为C═O,R6和R7一起为C2-3烷基-A。
式(2)化合物可具有以下立体化学:
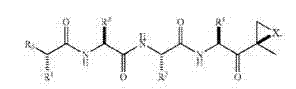
可例如在美国专利号7,232,818中找到式(2)化合物的更多非限制性实例,其全部通过引用并入本文。在一些实施方案中,式(2)化合物在水中具有低溶解度。
在一些实施方案中,肽蛋白酶体抑制剂可为式(3)化合物或其药用上可接受盐:
其中:
X为氧、NH或N(C1-6烷基);
Y为NH、N(C1-6烷基)、O或C(R9)2;
Z为O或C(R9)2;
R1、R2、R3和R4都为氢;
每一R5、R6、R7、R8和R9独立选自氢、C1-6烷基、C1-6羟烷基、C1-6烷氧基烷基、芳基和C1-6芳烷基,其每一个任选用烷基、酰胺、胺、羧酸或其药学上可接受的盐、羧基酯、硫醇基和硫醚中的一种或多种取代;
m为0-2的整数;和
n为0-2的整数。
在一些实施方案中,X为O。在一些实施方案中,Y为N(C1-6烷基)、O或C(R9)2。在一些实施方案中,Z为C(R9)2。在一些实施方案中,R5、R6、R7和R8独立选自C1-6烷基、C1-6羟烷基和C1-6芳烷基,并且每一R9为氢。例如,R6和R8独立为C1-6烷基,R5和R7独立为C1-6芳烷基,每一R9为H。在一些实施方案中,n为0或1。
在一些实施方案中,X为O,并且R5、R6、R7和R8独立选自C1-6烷基、C1-6羟烷基和C1-6芳烷基。例如,R6和R8独立为C1-6烷基,R5和R7独立为C1-6芳烷基。
在一些实施方案中,X为O,并且R6和R8二者都为异丁基,R5为苯乙基,R7为苯甲基。
在一些实施方案中,R5、R6、R7和R8独立选自氢、C1-6烷基、C1-6羟烷基、C1-6烷氧基烷基、芳香基和C1-6芳烷基,其每一个任选用选自烷基、酰胺、胺、羧酸或其药学上可接受的盐、羧基酯、硫醇基和硫醚的基团取代。在一些实施方案中,R5和R7的至少一个为用烷基(例如全卤烷基)取代的C1-6芳烷基。例如,R7为用三氟甲基取代的C1-6芳烷基。
在一些实施方案中,Y选自N-烷基、O和CH2。在某些所述实施方案中,Z为CH2,m和n二者都为0。在一些实施方案中,Z为CH2,m为0,n为2或3。在一些实施方案中,Z为O,m为1,n为2。
在一些实施方案中,式(3)化合物为式(4)化合物或其药用上可接受盐:

其中:
X为O、NH或N-烷基,优选O;
R1、R2、R3和R4都为氢;和
R5、R6、R7和R8独立选自氢、C1-6烷基、C1-6羟烷基、C1-6烷氧基烷基、芳香基和C1-6芳烷基,其每一个任选用选自酰胺、胺、羧酸或其药学上可接受的盐、羧基酯、硫醇基和硫醚的基团取代。
在一些实施方案中,R5、R6、R7和R8独立选自C1-6烷基、C1-6羟烷基和C1-6芳烷基。例如,R6和R8独立为C1-6烷基,R5和R7独立为C1-6芳烷基。
在一些实施方案中,X为O,并且R5、R6、R7和R8独立选自C1-6烷基、C1-6羟烷基和C1-6芳烷基。例如,R6和R8独立为C1-6烷基,R5和R7独立为C1-6芳烷基。
在一些实施方案中,X为O,并且R6和R8二者都为异丁基,R5为苯乙基,R7为苯甲基。
在一些实施方案中,式III化合物具有以下立体化学:
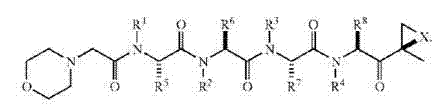
可例如在美国专利号7,417,042中找到式(3)和(4)的化合物的非限制性实例,其全部通过引用并入本文。在一些实施方案中,式(3)或(4)的化合物在水中具有低溶解度。
在一些实施方案中,肽蛋白酶体抑制剂为式(5)化合物或其药学上可接受的盐:
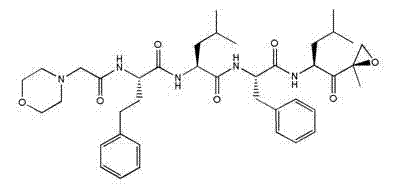
式(5)化合物亦称为卡非佐米。
本文所述任何化合物可分离为无定形或结晶形式。可如本领域所知来进行本文所提供的晶体化合物的制备和纯化,其例如在美国专利公开号2009/0105156中所述,其全部通过引用并入本文。
在一些实施方案中,式(5)晶体化合物基本上纯。在一些实施方案中,式(5)晶体化合物的熔点为约200-约220℃、约205-约215℃、约211-约213℃范围或甚至约212℃。在一些实施方案中,式(5)晶体化合物可具有约205-约215℃的熔点。例如,化合物可具有约211-约213℃的熔点。在一些实施方案中,式(5)晶体化合物的DSC例如因所述化合物的晶形的熔化和分解而在约212℃下具有急剧的最高吸热温度。
式(5)晶体化合物的X-射线粉末衍射图样在角度2θ具有特征衍射峰。例如,式(5)晶体化合物可具有在6.10的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在9.32的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在10.10的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在12.14的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在13.94的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在18.44的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在20.38的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在23.30的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有这样的X射线粉末衍射图样,其包含在6.10、9.32、10.10、12.14、13.94、18.44、20.38和23.30的2θ角处表示的2-8个特征峰。例如,式(5)晶体化合物具有这样的X射线粉末衍射图样,其包含在6.10、9.32、10.10、12.14、13.94、18.44、20.38和23.30的2θ角处表示的特征峰。
在一些实施方案中,式(5)晶体化合物具有在约6.1的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在约9.3的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在约10.1的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在约12.1的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在约13.9的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在约18.4的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在约20.4的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有在约23.3的2θ角处表示的特征峰。在一些实施方案中,式(5)晶体化合物具有这样的X射线粉末衍射图样,其包含在约6.1、9.3、10.1、12.1、13.9、18.4、20.4和23.3的2θ角处表示的2-8个特征峰。在一些实施方案中,式(5)晶体化合物具有这样的X射线粉末衍射图样,其包含在约6.1、9.3、10.1、12.1、13.9、18.4、20.4和23.3的2θ角处表示的特征峰。
在一些实施方案中,式(5)晶体化合物具有这样的X射线粉末衍射图样,其具有在6.10、8.10、9.32、10.10、11.00、12.14、12.50、13.64、13.94、17.14、17.52、18.44、20.38、21.00、22.26、23.30、24.66、25.98、26.02、27.84、28.00、28.16、29.98、30.46、32.98、33.22、34.52和39.46的2θ角处表示的特征峰。
在一些实施方案中,式(5)晶体化合物具有这样的X射线粉末衍射图样,其具有在6.1、8.1、9.3、10.1、11.0、12.1、12.5、13.6、13.9、17.1、17.5、18.4、20.4、21.0、22.3、23.3、24.7、25.9、26.0、27.8、28.0、28.2、30.0、30.5、33.0、33.2、34.5和39.5的2θ角处表示的特征峰。
用Shimadzu XRD-6000 X射线粉末衍射仪使用Cu Kα辐射来实施X射线粉末衍射(XRPD)分析。仪器装备有长细聚焦X射线管。将管电压和电流强度分别设为40 kV和40 mA。将发散和散射狭缝设为1?,将接收狭缝设为0.15 mm。通过碘化钠闪烁探测器来检测衍射辐射。使用从2.5-40? 2θ以3?/分钟(0.4秒/0.02?)的θ-2θ连续扫描。分析硅标准品以检查仪器校准。使用XRD-6100/7000 v.5.0来收集和分析数据。通过将样品置入具有硅嵌入物的铝容器中来制备用于分析的样品。
在一些实施方案中,式(5)晶体化合物为式(5)化合物的晶体盐。例如,式(5)化合物的晶体盐可选自:柠檬酸盐、酒石酸盐、三氟乙酸盐、甲磺酸盐、甲苯磺酸盐、盐酸盐和氢溴酸盐。在一些实施方案中,式(5)化合物的晶体盐为柠檬酸盐。在一些实施方案中,结晶固体可以以共晶体存在。
在一些实施方案中,式(5)化合物的晶体柠檬酸盐基本上纯。在一些实施方案中,式(5)化合物的晶体柠檬酸盐的熔点为约180-约190℃范围,例如约184-约188℃。在一些实施方案中,式(5)化合物的晶体柠檬酸盐的DSC例如因其结晶形式的熔化和分解而在187℃下具有急剧的最高吸热温度。
在一些实施方案中,式(5)晶体化合物具有这样的X射线粉末衍射图样,其包含在4.40、7.22、9.12、12.36、13.35、14.34、15.54、16.14、16.54、17.00、18.24、18.58、19.70、19.90、20.30、20.42、21.84、22.02、23.34、23.84、24.04、24.08、24.48、24.76、25.48、26.18、28.14、28.20、28.64、29.64、31.04、31.84、33.00、33.20、34.06、34.30、34.50、35.18、37.48、37.90和39.48的2θ角处表示的两个或多个特征峰。例如,式(5)化合物的晶体柠檬酸盐可具有这样的X射线粉末衍射图样,其具有在4.40、7.22、9.12、12.36、13.35、14.34、15.54、16.14、16.54、17.00、18.24、18.58、19.70、19.90、20.30、20.42、21.84、22.02、23.34、23.84、24.04、24.08、24.48、24.76、25.48、26.18、28.14、28.20、28.64、29.64、31.04、31.84、33.00、33.20、34.06、34.30、34.50、35.18、37.48、37.90和39.48的2θ角处表示的特征峰。
药物组合物
本文提供的方法包括药物组合物的制备和使用,所述药物组合物包括本文所提供的任何化合物。亦包括所述药物组合物本身。
在一些实施方案中,可如美国专利号7,737,112所述来配制本文所提供的化合物。
本文还提供用于制备肽蛋白酶体抑制剂的药物组合物(例如式(1)-(5)的化合物或其药学上可接受的盐、溶剂化物、水合物、共晶体或多形体)的环糊精络合法。所述方法包括提供含有肽蛋白酶体抑制剂、环糊精和水的第一组合,其中第一组合为异质的,并且肽蛋白酶体抑制剂或盐在第一组合中具有低溶解度。所述方法进一步包括改变第一组合的pH以形成第二组合,其中肽蛋白酶体抑制剂在第二组合中的溶解度大于肽蛋白酶体抑制剂在第一组合中的溶解度。例如,所述方法可包括让第一组合与酸接触以形成第二组合。第二组合仍可为异质的,但其仍可促进溶解度的充分增加以便络合过程可以启动和进行。这可使大部分的抑制剂被络合,然而通过部分络合而为异质混合物,或完全络合形成均质溶液。在异质络合混合物的情况下,一旦达到所需要的溶解和络合程度,可过滤掉多余的固体以产生均质溶液。
本文所用术语“络合”指在溶液中和在一种或多种肽蛋白酶体抑制剂和一种或多种环糊精分子之间,形成分子间包合络合物或分子间缔合物。包含物和/或缔合物作为以下机理提供效用:与无络合剂(即一种或多种环糊精分子)时在相似pH范围下的水相溶解相比,其大大增加了水溶液中可获得的抑制剂的浓度。
当可经由合适的常规分析方法例如HPLC测量溶解的抑制剂浓度并且所述浓度大大超过了无环糊精存在时将抑制剂溶解在水中可获得的浓度时,络合或缔合状态是明显的。可制备抑制剂和环糊精的络合或缔合溶液,以便超过不存在环糊精时的水溶液中的浓度,这可用于配制方便的注射体积和递送剂量的药用化合物。而且,抑制剂的络合或缔合溶液表现出物理稳定性(或另外阐述为亚稳定性),其中抑制剂在均质溶液(没有沉淀或固体颗粒的结晶)中所保持的时间期限比在无环糊精存在时抑制剂的溶液中通常所保持的时间期限更长。由于在此延长期间保持为透明溶液,在用作医药制剂的所有实际条件下,不会发生晶体成核作用和随后的过饱和损耗。
很多小分子有机化合物药物具有pH依赖性的溶解度。通常适于给予药物的pH范围(例如通过注射,通常认为静脉内给予可容许的pH范围为3–10.5)与在水溶液中可发现药物的足够溶解性的pH (例如为或低于pH 2)不一样。为使得药物在给药(例如通过注射)可接受的和可容许的pH范围下在溶液中达到药学上有用的浓度水平,用本文要求保护的环糊精络合或缔合药物是实用的方法。其可增加溶液浓度,这可在给药容许的pH范围内实现。所述浓度增加可从例如最初无环糊精时的每毫升1–100微克,增加到有环糊精时的多达每毫升500–10,000微克。从而络合或缔合是这样的技术,其使得原本水溶性差的化合物变得充分溶解并被开发成药学上有用的化合物。本领域技术人员了解,获得所需浓度和物理稳定性状态所需的环糊精的量可改变。因此,可在个体组合的基础上用熟知的方法确定环糊精的量。
对于碱性药物分子,通常在较低pH下其溶解度增加。如果在不含络合或缔合剂例如环糊精时使用,在一些情况下这亦呈现稳定性和保质期挑战。例如,经由用酸降低溶液的pH可获得充分的溶解度,但所述pH降低可导致酸性条件下的降解反应。卡非佐米固有的水溶解度数据见表1,其显示随pH的降低,溶解度有一些适度的增加。
表1:无环糊精时作为pH函数的卡非佐米的水溶解度
| 溶剂 | 溶解度(mg/mL) |
| 水 | 0.002 |
| 水/pH 5 | 0.002 |
| 水/pH 3 | 0.02 |
| 水/pH 1 | 1.8 |
对于小分子药物和生物学分子,存在很多酸介导的降解反应途径,例如酰胺在较小的无活性肽片段中的水解,或功能性环氧化物部分的水解开环。酸介导的降解的产物可能缺乏药理学活性,甚至在痕量级水平也可能是毒性或遗传毒性化合物。在避免显著降解的pH条件下对化合物的络合和缔合更加拓宽了环糊精的效用,促进具有pH依赖性稳定特征的化合物的临床和商业开发。
为了平衡以下竞争性需要:避免在低pH下发生的酸介导的降解副反应与经由降低pH增加络合效率,找到了独特的pH条件。令人惊讶的是,发现经由加入一定浓度的酸,例如柠檬酸(pH约2.5-3.0),获得的水溶液的pH足以使pH降低到引发络合作用而不引发显著水平的降解副反应。在这种状态下,通过pH条件部分而非全部溶解抑制剂。因此,存在以部分溶解于环糊精和柠檬酸水溶液中的抑制剂的异质混合物(例如浆液),并且其部分以抑制剂的固体颗粒(晶体)存在。随时间推移(通常为数小时至一天),抑制剂的溶解部分将与环糊精络合或缔合。该过程可使更多的抑制剂固体颗粒溶解,然后变成络合状态。随时间推移,可发生质量转移:从最初的固相抑制剂,至溶解相抑制剂,至环糊精-抑制剂的溶解络合状态。更常用的是,经由形成待络合的化合物的均质溶液来实现环糊精的络合作用。对于卡非佐米,形成均质溶液将需要极低的pH,此时会发生降解反应,例如用强酸盐酸形成潜在的遗传毒性杂质的降解反应。在这种情况下,在使用弱羧酸柠檬酸的2.5–3.0的更温和pH条件下在异质状态实施络合过程是实用的和有用的。一旦获得络合抑制剂的靶浓度,则通过过滤掉任何未溶解的固体颗粒的抑制剂来终止浆液的络合过程。然后可按照需要将所得的均质溶液的pH调节到适于静脉内给予的pH范围(例如,使用氢氧化钠水溶液调节到pH 3.5)。而且,可用水将均质的pH经调节的络合溶液稀释到产品制备的下一步所需的确切浓度,以确保药物产品的标签强度精确。
环糊精浓度和pH对络合作用的联合效应比任何一项技术单独使用时具有更强的增溶能力。增溶作用的程度相对独立于温度并使任何温度加速的降解反应最小化,这便于使制备维持在冷条件下,更优选用于无菌产品制备。
第二组合包括肽蛋白酶体抑制剂和环糊精的络合物。所述络合物比单独的肽蛋白酶体抑制剂具有改进的水溶解性。例如,可在药学上有用的pH (例如约3.5)和比无环糊精及无本文所提供的化合物和环糊精之间的络合作用过程时可获得的浓度更高的浓度下(例如约5 mg/mL),获得式(5)化合物(卡非佐米)的均质溶液。
除增加肽蛋白酶体抑制剂在溶液中的溶解度外,通过本文所提供的方法制备的制剂产生具有惊人稳定性的药用溶液。虽然可能未预料到通过本文所提供的处理方法而获得的高浓度蛋白酶体抑制剂是热力学稳定的,但溶液已显示出不受储存温度(例如,溶液可在-20℃-25℃是稳定的)、冻融循环和冻干及重构的影响。络合的肽蛋白酶体抑制剂和环糊精的稳定性足以耐受络合作用后pH的调节而无沉淀。该溶液的稳定性使得可在对于注射、产品的稳定性和其它药用目可接受的pH范围内使用络合的物质。因此,对于药物用途,通过本文所提供的方法制备的药物组合物可被认为是过饱和溶液,其在任何次数医药应用的使用期间不沉淀或浓度不显著程度地降低(例如,最终的药物组合物在至少1-5天或可能更长范围内是稳定的)。
可通过将固体形式的肽蛋白酶体抑制剂加入到一种或多种环糊精的水溶液中来制备第一组合。在一些实施方案中,当肽蛋白酶体抑制剂为式(5)化合物或其药学上可接受的盐时,一种或多种环糊精在溶液中的浓度为从小于约1%至多达尽可能和环糊精的溶解度极限一样高,例如约40%。在一些实施方案中,为制备目的,溶液中的一种或多种环糊精浓度为约15%-约30%。在一些实施方案中,为了将成品药物重构为溶液,用于治疗性给药或准备在给药之前进一步稀释这一目的,溶液中的一种或多种环糊精浓度为约5%-约15%,例如,大约10%。进一步稀释时,这一浓度可进一步降低至认为适于注射或其它药物递送途径的浓度。溶液中一种或多种环糊精与式(5)化合物的摩尔比为约0.5-约100。在一些实施方案中,这一比值以过量摩尔数的环糊精存在,以使络合稳定性平衡移动,使之倾向络合状态而非未络合状态。例如,摩尔比(环糊精摩尔数除以蛋白酶体抑制剂摩尔数)为约10-约20。在一些实施方案中,环糊精与蛋白酶体抑制剂的重量/重量比为约30-约60。环糊精溶液的过度起泡可为稳健(robust)生产过程中的难题。另人惊讶的是,加入肽蛋白酶体抑制剂到环糊精的水溶液中可控制第一组合溶液中的起泡。
在一些实施方案中,第一组合基本上由肽蛋白酶体抑制剂、环糊精和水组成。
加入到环糊精和水的溶液中的固体形式肽蛋白酶体抑制剂可为本文所阐述化合物的晶形(例如,化合物可为如本文所述的多形体或特定的多形体)。在一些实施方案中,肽蛋白酶体抑制剂的固体形式为无定形的。
第一组合是异质的(例如,悬液或浆液)。所述溶液可通过溶液中总固体重量百分比和粒度分布来表征。例如,当肽蛋白酶体抑制剂为式(5)化合物或其药学上可接受的盐时,第一组合的总固体重量百分比可为约1%-约45% (例如,约1%-约40%、约1%-约35%、约1%-约30%、约1%-约25%、约1%-约20%、约1%-约15%、约1%-约10%、约5%-约45%、约10%-约45%、约12%-约45%、约15%-约45%、约20%-约45%、约25%-约45%、约30%-约45%、约35%-约45%、约5%-约35%、约10%-约40%、约15%-约37%和约18%-约36%)。在一些实施方案中,第一组合的固体重量百分比可为约20%-约33%。在一些实施方案中,第一组合的固体重量百分比可为约30%-约33%。随着制备时程的推移,溶解的固体部分与未溶解部分之比可视溶解度和络合程度而变化。最初,一种或多种环糊精极溶于水,而抑制剂为略溶,藉此保持大部分为异质的混合物或浆液。
在一些实施方案中,第一组合具有这样的粒度分布,其初级粒子的直径范围为小于约1微米至约300微米或更多(例如,约1 μm-约200 μm、约1 μm-约150 μm、约1 μm-约125 μm、约1 μm-约100 μm、约1 μm-约50 μm、约1 μm-约10 μm、约5 μm-约300 μm、约25 μm-约300 μm、约50 μm-约300 μm、约60 μm-约300 μm、约75 μm-约300 μm、约100 μm-约300 μm、约125 μm-约300 μm、约150 μm-约300 μm、约200 μm-约300 μm、约225 μm-约300 μm、约250 μm-约300 μm、约5 μm-约150 μm、约25 μm-约200 μm、约50 μm-约125 μm、约10 μm-约100 μm、约75 μm-约225 μm和约100 μm-约200 μm)。初级粒子可以作为离散颗粒或作为包含一个或多个初级粒子的附聚物存在。初级粒子的附聚物可具有显著大于初级粒子的尺寸。因此,除普通悬浮叶轮混合器外,还引入高能量混合装置例如高剪切混合器(常常配置为转子定子混合器)是有用的。高能量混合器经约5分钟-约90分钟(例如,约5分钟-约80分钟、约5分钟-约75分钟、约5分钟-约60分钟、约5分钟-约45分钟、约5分钟-约30分钟、约10分钟-约90分钟、约15分钟-约90分钟、约30分钟-约90分钟、约45分钟-约90分钟、约50分钟-约90分钟、约75分钟-约90分钟、约15分钟-约75分钟、约20分钟-约70分钟、约30分钟-约70分钟、约45分钟-约75分钟和约10分钟-约45分钟)的时程,例如经约60分钟的时程,将环糊精溶液中的大附聚物破碎为分散的初级粒子。可通过将初级粒子破碎为更小的初级粒子片段协助进一步的混合。这一过程设计促进了稳健的方法,其中混合系统获得基本上分散的初级粒子,其尺寸分布范围为小于约1微米-多达约30微米,例如,多达约10微米,该尺寸分布不依赖于蛋白体抑制剂固体的尺寸分布和附聚程度。因此,由于混合系统将附聚物和初级粒子通常降低为更优选的粒度分布范围,所以对于处理效果来说,蛋白体抑制剂的粒度分布的批与批之间的变异性不显著。例如,第一组合可具有粒度分布为最初的小于约1微米-多达约10,000微米,到应用高能混合步骤之后的尺寸分布为小于约1微米-多达约30微米。
在一些实施方案中,第一组合基本上无有机溶剂。例如,第一组合中的水可为注射用水(WFI)。在一些实施方案中,第一组合基本上无缓冲剂(例如,第一组合缺乏缓冲酸或缓冲碱)。
所述方法可进一步包括在改变第一组合的pH之前让第一组合混合,例如通过使用高剪切混合器和常规叶轮。可以例如以足以维持颗粒悬浮而不沉于混合槽底部的任何转速来操作一般的混合器。混合速度在其它因素中是槽和叶轮几何形状的函数,且本领域技术人员经由混合浆液或溶液的目测外观足以确定该混合速度。同样,高剪切混合器的速度取决于例如混合元件的直径、定子几何形状、间隙宽度和其它因素。可经由理论计算或经由实验测量来确定输入到浆液中的能量。或者,本领域技术人员可在各种混合速度和时间组合后经由浆液样品的显微观测来确定必需的高剪切混合速度和高速操作的持续时间。一旦解聚并且初级粒子已减少,则可施用对过程无损害的过高的剪切混合速度和过长的时间。例如,在一些实施方案中,混合可包括以约500rpm-约10,000 rpm的速率搅拌第一组合。例如,可以以速度为约2,000 rpm-约3,500 rpm来实施高剪切混合。对于更小和更大的混合器和槽直径,相关的速度可显著变化。
可在温度为约0℃-约30℃下(例如,约5℃-约25℃、约10℃-约30℃、约15℃-约25℃、约5℃-约20℃、约2℃-约22℃和约20℃-约30℃)时实施第一组合的混合。在一些实施方案中,在足以获得小于约1微米-约30微米的第一组合中的粒度分布范围的时间内,实施第一组合的混合。对第一组合进行混合的时间为约30分钟-约90分钟,例如60分钟。
改变第一溶液的pH可包括通过加入酸或碱来增加或减小第一溶液的pH。在一些实施方案中,当肽蛋白酶体抑制剂为式(5)化合物或其药学上可接受的盐时,第一组合的pH为约4-约7。在一些实施方案中,加入酸以改变pH,例如无机或有机酸。酸的非限制性实例包括乳酸、乙酸、甲酸、柠檬酸、草酸、尿酸、琥珀酸、马来酸、富马酸、苯甲酸、酒石酸、盐酸甘氨酸、硫酸氢盐(例如以钠盐、钾盐或铵盐存在)和磷酸或磷酸盐。在一些实施方案中,酸为有机酸。在一些实施方案中,酸为柠檬酸。适宜的酸可具有一个或多个pKa值,其中第一pKa为约-6至约+5。例如,酸的第一pKa在约+1至约+4.5的范围内。在一些实施方案中,酸的第一pKa在约+1.5至约+3.5的范围内。参见例如P. Heinrich Stahl和Camille G. Wermuth编辑的Handbook of Pharmaceutical Salts: Properties, Selection, and Use (药用盐类手册:特性、选择和使用),Verlag Helvetica Chimica Acta (Switzerland) 2002, 336-341,其内容以其整体通过引用并入本文。
在一些实施方案中,对于其中经由增加pH而事实上增加溶解度和络合作用的化合物,通过加入碱例如无机或有机碱来改变pH。无机碱的非限制性实例包括氢氧化钠、氢氧化钾、氢氧化铵、氢氧化钙、氢氧化镁和钠、钾或铵的碳酸盐或碳酸氢盐。有机碱的非限制性实例包括吡啶、甲胺、三乙胺、咪唑、苯并咪唑、组氨酸和磷腈碱(phosphazene base)。有机碱可具有约-6至约+10的pKb或第一pKb。酸或碱各自相关pKa或pKb需要在足以使抑制剂的溶解度获得某些增加的范围内。在一些实施方案中,以水溶液(例如酸的水溶液)的形式加入酸或碱。
改变第一溶液的pH导致形成第二组合,其中肽蛋白酶体抑制剂比在第一组合中更加可溶。例如,与第一组合中抑制剂的溶解度相比,第二组合中的肽蛋白酶体抑制剂可多溶解至少约10% (例如多溶解至少约100%、至少约150%、至少约200%、至少约250%、至少约400%、至少约500%、至少约1000%、至少约1250%、至少约1500%、至少约2000%、至少约2500%、至少约3000%、至少约4000%、至少约5000%、至少约5500%、至少约6000%、至少约7500%、至少约8000%、至少约9000%和至少约10,000%)。
不受理论的约束,改变第一组合的pH引发一种或多种环糊精和肽蛋白酶体抑制剂的络合作用。渐增的络合作用改变溶液的平衡,触发另外的络合作用,最终导致肽蛋白酶体抑制剂的溶解。加入添加剂后,可让第二组合混合足够时间,以获得具有充分溶解和络合的抑制剂的异质混合物,或获得其中所有抑制剂都已络合且无任何保留为不溶解固体的均质的第三组合。例如,第三组合中的蛋白酶体抑制剂的浓度可为约1-约18 mg/mL,例如约2-约8 mg/mL、约4-约6 mg/mL或约5-约6 mg/mL。在一些实施方案中,第三组合的pH为约1.5-约4,例如约2-约3.5或约2.5-约3.5。考虑到其中无需溶解和络合作为浆液存在的抑制剂整个质量就可获得充分络合的情况,一旦达到靶浓度便终止络合过程可能是有用的。在这些情况下,可经由过滤掉过多固体抑制剂含量来获得期需抑制剂浓度的均质溶液。这使络合的抑制剂和环糊精处于功能稳定的溶液中,尽管络合作用和增溶作用的动态平衡可能意味着非热力学稳定的状态。
第三组合中肽蛋白酶体抑制剂的络合作用为至少约50% (例如,至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%、至少约92%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%、至少约99%)。在一些实施方案中,第三组合中肽蛋白酶体抑制剂的络合作用为至少约99%。可以想象到,对于一些环糊精浓度、抑制剂浓度、pH和络合时间的组合,可制备100%的抑制剂络合溶液,此时混合物变得均质。
在一些实施方案中,在单个容器中实施上述方法。例如,可在温控夹套混合槽中使用探针式高剪切混合器(例如匀浆器)来实施所述方法中络合浆液的混合。
本文提供用于制备式(5)化合物或其药学上可接受的盐形式的药物组合物的方法,所述方法包括提供式(5)化合物、环糊精和水的第一组合,其中第一组合为异质的,并且该化合物或盐在第一组合中具有低溶解度。在一些实施方案中,环糊精为SBECD,水为WFI。方法进一步包括让第一组合与酸接触以形成第二组合,其中化合物在第二组合中比在第一组合中更加可溶。在一些实施方案中,酸为柠檬酸(例如柠檬酸的水溶液)。
所述方法的非限制性实例包括在容器中提供包括水(例如WFI)、SBECD和式(5)化合物或其药学上可接受的盐的第一组合。在一些实施方案中,在加入化合物之前让水和SBECD混合。可混合第一组合直至获得异质的溶液(例如约30-约90分钟、约40-约80分钟和约50-约70分钟)。在一些实施方案中,混合第一组合约60分钟。若化合物在第一组合中凝聚,则可减小任何凝聚的化合物的粒度。一旦获得异质的混合物(例如浆液),就将酸(例如有机酸,例如柠檬酸)加入到第一组合中以制备第二组合。在一些实施方案中,酸作为水溶液加入。然后可继续混合直至制备均质的第三组合,或混合更短的时间仍保持为异质混合物,但获得了期需程度的络合作用和增溶作用。在一些实施方案中,进行第二组合混合的时间范围为约1-约48小时,例如多达18小时。在一些实施方案中,进行第二组合混合的时间为约12小时。例如,可进行约六小时混合。在一些实施方案中,第三组合中化合物的浓度范围为约1-约15 mg/mL (例如约3-约12 mg/mL、约4-约8 mg/mL、约5 mg/mL)。在一些实施方案中,使用所述方法来制备注射用的化合物溶液。在其它实施方案中,使用所述方法来制备溶液以供冻干成为无菌的药物成品,其可储存、运输,当准备给患者注射时用水或其它溶媒重构。
除非制备涉及到灭菌步骤,否则使用本文所述的程序获得的无菌产品形式的药物组合物,通常在装入初级包装单元(例如玻璃小瓶)之前应用无菌技术和过滤除菌来制造,并在使用之前不发生任何污染。
例如,在过滤除菌后溶解在水性缓冲剂或水溶液中的肽蛋白酶体抑制剂组合物,可任选被冻干(在无污染和不能透过污染物的容器中),并在临用前在合适的水性稀释剂中重构。在一些实施方案中,稀释剂为无菌注射用水(WFI)。在一些实施方案中,稀释剂为无菌缓冲剂(例如柠檬酸盐缓冲剂)。在一些实施方案中,稀释剂包含柠檬酸。
在本文提供的组合物中,一种pH控制源为缓冲剂。通常缓冲剂以酸或碱及其各自的共轭碱或酸存在。在一个实施方案中,缓冲盐的范围为1-100 mM。例如,缓冲盐的范围可为5-50 mM (例如约10 mM (在固体制剂中,选择缓冲剂的量以在重构/稀释后产生这一浓度))。可选择缓冲剂的浓度和溶液的pH以赋予溶解度和稳定性的最佳平衡。
适宜的缓冲剂的实例包括弱酸和弱酸的共轭碱的碱金属盐(例如钠盐、钾盐)混合物,例如酒石酸钠和柠檬酸钠。在一些实施方案中,缓冲剂为柠檬酸钠/柠檬酸。
已大量研究了环糊精络合作用对水溶性差的药物的增溶作用。环糊精为由6、7或8个葡萄糖单元(α-CD、β-CD和γ-CD)以α-1,4键连接组成的环状寡糖。α-CD、β-CD和γ-CD的内径各自为约5?、6?和8?。由于CH2和醚基团,内腔为相对疏水;而由伯羟基或仲羟基组成的外层,具有更大极性。腔内的水倾向于被更加非极性的分子替代。环糊精与部分适合其非极性腔内的分子形成非共价包合络合物的能力导致药物的增溶作用。
两种有药用价值的水溶性的β-CD衍生物为磺丁基醚β-环糊精(SBECD)和羟丙基-β环糊精(HPCD),其两者都被证明为安全的和良好耐受。SBECD (商标名Captisol?)和HPCD (商标名Kleptose?)二者都为可市购的静脉内产品。
本文所提供的环糊精包括α-、β-和γ-环糊精。在一个实施方案中,一种或多种环糊精为例如以5-35% (w/v)存在的取代的或未被取代的β-环糊精。在一些实施方案中,环糊精的量为约25% (w/v)。在某些实施方案中,适于注射的制剂中环糊精的量为约10% (w/v)。在另一个实施方案中,一种或多种环糊精为取代的β-环糊精。取代的环糊精增加环糊精的溶解度,减轻与未被取代的环糊精有关的毒性作用。取代的β-环糊精的实例包括用一个或多个亲水基团取代的那些,例如单糖(例如葡萄糖基、麦芽糖基)、羧基烷基(例如羧甲基、羧乙基)、羟烷基-取代的(例如羟乙基、2-羟丙基)和磺烷基醚-取代的β-环糊精。特别适宜的β-环糊精包括羟丙基β-环糊精(HPBCD)和磺烷基醚β-环糊精(SBECD)。在一些实施方案中,环糊精为SBECD。然而,应该理解,通常环糊精的任何取代,包括用疏水基团例如烷基的取代,将通过破坏固体环糊精的晶格内的氢键网络来增加其水性溶解度,从而降低固体的晶格能。认为取代的程度并非关键;然而,在一些实施方案中,取代的程度为至少1%,通常为2%-10%,例如3%-6%。
在一些实施方案中,可使用一种或多种环糊精。例如,可使用两种或多种环糊精来络合本文所提供的肽蛋白酶体抑制剂。在一些实施方案中,可使用captisol和kleptose来络合肽蛋白酶体抑制剂例如卡非佐米。
本发明人已经发现使本文所述的方法和药物组合物中氯离子(或其它亲核阴离子)的量最小化可为有利的。
在一些实施方案中,一种或多种环糊精的至少一种(加入到第一组合中的)为低氯化物环糊精。本文所用“低氯化物环糊精”指具有小于或等于0.05% w/w氯化钠的环糊精,或如果氯化钠以外的氯化物源存在(或除氯化钠外还存在其它氯化物源),则“低氯化物环糊精”指这样的环糊精,其所具有的氯离子含量小于或等于应存在于含有0.05% w/w氯化钠的环糊精中的氯化物量。在一些实施方案中,低氯化物环糊精为低氯化物SBECD。可通过本领域已知的多种方法来测定氯化物浓度(例如对于市购获得的环糊精,根据厂家产品说明书的方法,例如通过比重测定技术,例如通过电势测定技术)。
在一些实施方案中,存在的氯离子的量足够低以便当储存在2-8摄氏度时提供2年的保质期。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于2.0。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于1.5。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于1.2。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于1.0。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.9。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.8。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.7。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.6。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.5。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.4。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.3。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.2。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比不大于0.1。
在一些实施方案中,第一组合中氯离子与化合物的摩尔比为0.2-1.2 (例如0.3-1.2、例如0.2-0.4、例如0.3-0.4、例如0.32)。
在实施方案中,本文所述的氯离子与化合物的摩尔比亦可存在于第二和/或第三组合中。
在本文所述的方法中,本文所提供的组合物(例如环糊精、第一组合、第二组合、第三组合和药物组合物的溶液)具有低浓度的任何强亲核离子(例如氯离子、溴离子、氟离子和碘离子)。例如,溶液可具有多达并包括8.5×10-3 M的亲核离子浓度。在一些实施方案中,具有低亲核离子的溶液可市购,或可使用本领域所知的技术来制备,所述技术包括例如纳米过滤、超滤、透析过滤、离子交换色谱、反渗透和电解。
在一些实施方案中,本文所提供的药物组合物包含多达并包括8.5×10-3 M的亲核离子。在一些实施方案中,亲核离子作为盐存在,例如钠盐,但是亲核的盐可与其它非钠阳离子(例如氢、钾、镁和钙阳离子)存在于溶液中。在一些实施方案中,本文所提供的药物组合物包含多达8.5×10-3 M的亲核离子。例如,药物组合物包含小于8.5×10-3 M的亲核离子。
在本文所述的方法中,本文所提供的组合物(例如环糊精、第一组合、第二组合、第三组合和药物组合物的溶液)具有低浓度的氯离子。例如,溶液可具有多达并包括0.03% (w/v)的氯离子浓度(例如0-0.03%、0.01-0.03%、0.015-0.03%、0.02-0.03%、0.025-0.03%、0-0.025%、0-0.2%、0-0.01%、0.005%-0.025%和0.015%-0.025%)。在一些实施方案中,具有低氯离子的溶液可市购,或可使用本领域所知的技术来制备,所述技术包括例如纳米过滤、超滤、透析过滤、离子交换色谱、反渗透和电解。
在一些实施方案中,本文所提供的药物组合物包含多达并包括0.03% (w/v)的氯离子。在一些实施方案中,氯离子作为盐存在,例如氯化钠,但是氯化物盐可与其它非钠阳离子(例如氢、钾、镁和钙阳离子)存在于溶液中。在一些实施方案中,本文所提供的药物组合物包含多达0.03% (w/v)的氯离子。例如,药物组合物包含小于0.03% (w/v)的氯离子。
在本文所述的方法中,本文所提供的组合物(例如环糊精、第一组合、第二组合、第三组合和药物组合物的溶液)具有低浓度的氯化钠。例如,溶液可具有多达并包括0.05% (w/v)的氯化钠浓度(例如,0-0.05%、0.01-0.05%、0.015-0.05%、0.02-0.05%、0.025-0.05%、0.03-0.05%、0.04-0.05%、0-0.045%、0-0.04%、0-0.035%、0-0.03%、0-0.025%、0-0.2%、0-0.01%、0.01%-0.04%、0.025%-0.045%和0.02%-0.03%)。在一些实施方案中,具有低氯化钠的溶液可市购,或可使用本领域所知的技术来制备,所述技术包括例如纳米过滤、超滤、透析过滤、离子交换色谱、反渗透和电解。
在一些实施方案中,本文所提供的药物组合物包含多达并包括0.05% (w/v)的氯化钠。在一些实施方案中,本文所提供的药物组合物包含多达 0.05% (w/v)的氯化钠。例如,药物组合物包含小于0.05% (w/v)的氯化钠。
在一些实施方案中,使用具有低浓度的任何强亲核离子(例如氯离子、溴离子、氟离子和碘离子)的环糊精溶液来配制本文所提供的肽蛋白酶体抑制剂(例如式(1)-(5)的化合物或其药学上可接受的盐)。例如,用于配制肽蛋白酶体抑制剂的环糊精溶液可具有多达并包括8.5×10-3 M的亲核离子浓度。所述溶液可市购或可使用本领域所知的技术来制备,所述技术例如纳米过滤、超滤、透析过滤、离子交换色谱、反渗透和电解。
在一些实施方案中,用于配制肽蛋白酶体抑制剂的一种或多种环糊精的溶液包含多达并包括8.5×10-3 M的亲核离子。在一些实施方案中,亲核离子作为盐存在,例如钠盐,但是亲核的盐可与其它非钠阳离子(例如氢、钾、镁和钙阳离子)存在于溶液中。在一些实施方案中,本文所提供的药物组合物包含多达8.5×10-3 M的亲核离子。例如,药物组合物包含小于8.5×10-3 M的亲核离子。
在一些实施方案中,使用具有低浓度的氯离子的环糊精溶液来配制本文所提供的肽蛋白酶体抑制剂(例如式(1)-(5)的化合物或其药学上可接受的盐)。例如,用于配制肽蛋白酶体抑制剂的环糊精溶液可具有多达并包括0.03% (w/v)的氯离子浓度(例如0-0.03%、0.01-0.03%、0.015-0.03%、0.02-0.03%、0.025-0.03%、0-0.025%、0-0.2%、0-0.01%、0.005%-0.025%和0.015%-0.025%)。所述溶液可市购,或可使用本领域所知的技术来制备,所述技术例如纳米过滤、超滤、透析过滤、离子交换色谱、反渗透和电解。
在一些实施方案中,用于配制肽蛋白酶体抑制剂的一种或多种环糊精的溶液包含多达并包括0.03% (w/v)的氯离子。在一些实施方案中,氯离子作为盐存在,例如氯化钠,但是氯化物盐可与其它非钠阳离子(例如氢、钾、镁和钙阳离子)存在于溶液中。在一些实施方案中,本文所提供的药物组合物包含多达0.03% (w/v)的氯离子。例如,药物组合物包含小于0.03% (w/v)的氯离子。
在一些实施方案中,使用具有低浓度的氯化钠的环糊精溶液来配制本文所提供的肽蛋白酶体抑制剂(例如式(1)-(5)的化合物或其药学上可接受的盐)。例如,用于配制肽蛋白酶体抑制剂的环糊精溶液可具有多达并包括0.05% (w/v)的氯化钠浓度(例如,0-0.05%、0.01-0.05%、0.015-0.05%、0.02-0.05%、0.025-0.05%、0.03-0.05%、0.04-0.05%、0-0.045%、0-0.04%、0-0.035%、0-0.03%、0-0.025%、0-0.2%、0-0.01%、0.01%-0.04%、0.025%-0.045%和0.02%-0.03%)。所述溶液可市购或可使用本领域已知的脱盐技术来制备,所述技术例如纳米过滤、超滤、透析过滤、离子交换色谱、反渗透和电解。
在一些实施方案中,用于配制肽蛋白酶体抑制剂的一种或多种环糊精的溶液包含多达并包括0.05% (w/v)的氯化钠。在一些实施方案中,本文所提供的药物组合物包含多达0.03% (w/v)的氯化钠。例如,药物组合物包含小于0.03% (w/v)的氯化钠。
除产生稳定的、高度浓缩的肽蛋白酶体抑制剂溶液外,还可在没有其它络合和配制方法的化学降解和稳定性限制的情况下获得通过本文所提供的方法制备的制剂。例如,本文所提供的方法在络合作用期间避免使用强酸(例如HCl)来降低pH。虽然将制剂的pH降低到小于2的值可在络合作用之前促进肽蛋白酶体抑制剂的溶解和产生均质溶液,但溶液的酸度可导致肽蛋白酶体抑制剂的降解。此外,肽蛋白酶体抑制剂含有环氧酮官能团,并且抑制剂易受强亲核离子例如氯离子的水解。环氧化物环的水解和酸-催化的环氧化物部分的亲核开环是化合物降解的途径。例如,式(5)化合物的降解导致形成氯醇降解产物(CDP)杂质。基于其结构,将这种降解产物归类为烷化物,因此全球监管机构认为其为潜在的遗传毒性杂质。另外,在一些实施方案中,氯离子亦可降解环氧化物,这导致形成氯醇加合物。如实例2所示,式(5)化合物的制剂中氯离子水平的减小可使所述水解途径最小化或消除所述水解途径,这导致产品的稳定性和质量增加。然而,使用本文所提供的方法,避免了所述强酸和亲核离子,因此可显著降低肽蛋白酶体抑制剂降解为所述降解产物,在一些情况下,甚至可消除所述降解。
适于注射的药物组合物可包括无菌水溶液(在可水溶的情况下)或分散剂和可临时制备为无菌可注射溶液或分散体的无菌粉末。对于静脉内给药,适宜的载体包括无菌注射用水、无菌缓冲剂例如柠檬酸盐缓冲剂、抑菌的水和Cremophor EL? (BASF, Parsippany, NJ)。在所有情况下,组合物必须是无菌,并且应为存在轻易可注射性程度的流体。组合物在制备和储存条件下应稳定,必须保护其免受微生物例如细菌和真菌的污染作用。载体可为含有例如水、乙醇、多元醇(例如丙三醇、丙二醇、液态聚乙二醇等等)及其适宜混合物的溶剂或分散介质。例如,可通过使用包衣例如卵磷脂、在分散体情况下通过保持所需的粒度和通过使用表面活性剂,来维持合适的流动性。可通过各种抗细菌和抗真菌剂例如对羟苯甲酸酯类、氯丁醇、苯酚、抗坏血酸、硫柳汞等等,来获得对微生物的作用的预防。在很多情况下,优选在组合物中包括等渗剂,例如糖类,多元醇例如甘露醇、山梨醇和氯化钠。可通过将延迟吸收的剂例如单硬脂酸铝和明胶包括在组合物中来使可注射组合物的吸收延长。
可通过将活性化合物与上面列举的一种成分或成分组合以所需量用适当的溶剂掺合,若需要的话接着灭菌,来制备无菌的可注射溶液。通常通过将活性化合物掺入到无菌溶媒中来制备分散体,所述无菌溶媒含有基本分散介质和上面所列举的其它所需成分。在用于制备无菌可注射溶液的无菌粉末的情况下,优选的制备方法为冷冻干燥(冻干),其产生活性成分加上来自其先前的无菌滤过溶液的任何另外所需成分的粉末。
口服组合物通常包括惰性稀释剂或可食用载体。为了口服治疗给药目的,可让活性化合物与赋形剂掺合,并呈片剂、锭剂或胶囊剂例如明胶胶囊形式来使用。亦可使用流体载体来制备作为漱口剂使用的口服组合物。可作为组合物的部分包括药学上相容的粘合剂和/或辅助物质。片剂、丸剂、锭剂等等可含有任何以下成分或类似性质的化合物:粘合剂,例如微晶纤维素、黄蓍胶或明胶;赋形剂,例如淀粉或乳糖;崩解剂,例如藻酸、Primogel或玉米淀粉;润滑剂,例如硬脂酸镁或Sterote;助流剂,例如胶体二氧化硅;甜味剂,例如蔗糖或糖精;或矫味剂,例如薄荷、水杨酸甲酯或橙子矫味剂。
对于吸入给药,可呈喷雾剂形式从压力容器或含有适宜抛射剂例如气体例如二氧化碳的分配器或喷雾器递送化合物。所述方法包括美国专利号6,468,798中所阐述的那些。
本文所述的治疗化合物的全身给药亦可为通过经粘膜或透皮的方式。对于经粘膜或透皮给药,在制剂中使用适于要渗透的屏障的渗透剂。所述渗透剂为本领域通常所知,包括例如,对于经粘膜给药为去垢剂、胆盐和夫西地酸衍生物。可通过使用鼻喷雾剂或栓剂来完成经粘膜给药。对于透皮给药,将活性化合物配制为本领域通常所知的软膏、药膏、凝胶或乳膏。
亦可将药物组合物制备为栓剂(例如,用传统的栓剂基质例如可可脂或其它甘油酯)形式或用于直肠递送的保留灌肠。
另外,鼻内递送是可能的,如尤其是Hamajima等在Clin. Immunol. Immunopathol, 88(2), 205-10 (1998)中所述。亦可使用脂质体(例如如美国专利号6,472,375中所述)和微胶囊化包封。亦可使用生物可降解的可靶向的微粒递送系统(例如,如美国专利号6,471,996中所述)。
在一个实施方案中,用可保护治疗性化合物免受从身体中快速消除的载体来制备治疗性化合物,例如控释制剂,包括植入物和微胶囊化递送系统。可使用可生物降解的、生物相容的聚合物,例如乙烯乙酸乙烯酯、聚酐、聚乙醇酸、胶原、聚原酸酯和聚乳酸。可使用标准技术来制备所述制剂,或可例如从Alza Corporation和Nova Pharmaceuticals, Inc市购获得所述制剂。亦可用脂质体悬液(包括用针对细胞抗原的单克隆抗体靶向所选择细胞的脂质体)作为药学上可接受的载体。可按照本领域技术人员所知的方法来制备这些,其例如美国专利号4,522,811所述。
可一次性给予药物组合物,或可将其分为许多小剂量以在间隔时间给予。应该理解,精确的剂量和治疗持续时间是待治疗的疾病的函数,可使用已知的测试方案或通过从体内或体外测试数据的外推来实验性确定。应注意到浓度和剂量值亦可随待减轻的病况的严重性而变化。更要了解对于任何特定患者,应根据个体需要和给予组合物或监督组合物给予的人的专业判断来随时间推移调整具体的给药方案,本文所示的浓度范围仅为例示性的,并非意欲限制要求权利保护的组合物的范围或实施。
可制备剂型或组合物,其含有在0.005%-100%范围内的本文所述化合物,且用无毒载体补足来平衡。用于制备这些组合物的方法为本领域技术人员所知。所考虑的组合物可含有0.001%-100%的活性成分,在一个实施方案中为0.1-95%,在另一个实施方案中为75-85%。
可将药物组合物连同给药说明一起包括在容器、包装或分配器之中。
使用方法
蛋白酶体抑制的生物学后果是很多的。已建议将蛋白酶体抑制作为众多疾病的预防和/或治疗,所述疾病例如但不限于增生性疾病、神经毒性/退行性疾病、阿尔茨海默病(Alzheimer's)、缺血性病况、炎症、自身免疫疾病、HIV、癌症、器官移植排斥、脓毒性休克、抗原提呈抑制、病毒基因表达的减少、寄生虫感染、与酸中毒有关的病况、黄斑变性、肺的病况、肌萎缩疾病(muscle wasting disease)、纤维化疾病、骨和毛发生长疾病。因此,非常有效的蛋白酶体-特异性化合物(例如环氧酮类的分子)的药物制剂,提供将药物给予患者和治疗这些病况的方式。
已报道当用多种蛋白酶体抑制剂处理细胞时,在细胞水平多泛素化蛋白质积累、细胞形态学变化和细胞凋亡。亦已建议将蛋白酶体抑制作为可能的抗肿瘤治疗策略。在抗肿瘤化合物的筛选中最先鉴定出环氧甲酮四肽蛋白酶体抑制剂(epoxomicin)这一事实,证实了蛋白酶体作为抗肿瘤化学疗法的靶标。因此,这些组合物可用于治疗癌症。
总之,体外和体内模型二者都显示恶性细胞对蛋白酶体抑制敏感。事实上,已证实将蛋白酶体抑制作为治疗多发性骨髓瘤的治疗策略。这可能部分是由于高度增殖性恶性细胞对蛋白酶体系统快速去除蛋白质的依赖性(Rolfe等,J. Mol. Med. (1997) 75: 5-17;Adams,Nature (2004) 4: 349-360)。因此,本文提供治疗癌症的方法,其包括将治疗有效量的本文所提供的肽蛋白酶体抑制剂给予需要所述治疗的患者。
本文所用术语“癌症”包括但不限于血源性和实体瘤。癌症指血液、骨、器官、皮肤组织和血管系统的疾病,其包括但不限于:膀胱癌、血癌、骨癌、脑癌、乳癌、宫颈癌、胸癌、结肠癌、子宫内膜癌、食道癌、眼癌、头癌、肾癌、肝癌、肺癌、淋巴结癌、口腔癌、颈癌、卵巢癌、胰腺癌、前列腺癌、直肠癌、肾癌、皮肤癌、胃癌、睾丸癌、咽喉癌和子宫癌。特定的癌症包括但不限于白血病(急性淋巴细胞性白血病(ALL)、急性髓性白血病(acute myelogenous leukemia, AML)、慢性淋巴细胞性白血病(CLL)、慢性髓性白血病(CML)、毛细胞白血病)、成熟B细胞肿瘤(小淋巴细胞淋巴瘤、B细胞幼淋巴细胞白血病、淋巴质浆细胞淋巴瘤(例如Waldenstr?m巨球蛋白血症)、脾边缘区淋巴瘤、浆细胞骨髓瘤、浆细胞瘤、单克隆免疫球蛋白沉积病、重链病、结节外边缘区B细胞淋巴瘤(MALT淋巴瘤)、结节边缘区B细胞淋巴瘤(NMZL)、滤泡淋巴瘤、套细胞淋巴瘤、弥散B细胞淋巴瘤、纵膈(胸腺的)大B细胞淋巴瘤、血管内大B细胞淋巴瘤、原发性渗出性淋巴瘤和Burkitt淋巴瘤/白血病)、成熟T细胞和自然杀伤(NK)细胞肿瘤(T细胞幼淋巴细胞白血病、T细胞大粒淋巴细胞白血病、攻击性NK细胞白血病、成体T细胞白血病/淋巴瘤、结节外NK/T细胞淋巴瘤、肠病型T细胞淋巴瘤、肝脾T细胞淋巴瘤、母细胞性NK细胞淋巴瘤、蕈样真菌病(Sezary综合征)、原发性皮肤间变性大细胞淋巴瘤、淋巴瘤样丘疹病、血管免疫母细胞的T细胞淋巴瘤、非特指外周T细胞淋巴瘤和间变性大细胞淋巴瘤)、霍奇金淋巴瘤(结节性硬化型、混合细胞性、淋巴细胞丰富型、淋巴细胞衰竭或未衰竭的、结节性淋巴细胞为主型)、骨髓瘤(多发性骨髓瘤、无痛性骨髓瘤、冒烟性骨髓瘤)、慢性骨髓增生性疾病、脊髓发育不良性/骨髓增生性疾病、骨髓增生异常综合征、免疫缺陷相关的淋巴增殖性病症、组织细胞和树突细胞肿瘤、肥大细胞增多症、软骨肉瘤、Ewing肉瘤、纤维肉瘤、恶性巨细胞瘤、骨髓瘤骨病、骨肉瘤、乳腺癌(激素依赖型、激素非依赖型)、妇科癌症(宫颈癌、子宫内膜癌、输卵管癌、妊娠期滋养层疾病、卵巢癌、腹膜癌、子宫癌、阴道癌和外阴癌)、基底细胞癌(BCC)、鳞状细胞癌(SCC)、恶性黑素瘤、隆凸性皮肤纤维肉瘤、Merkel细胞癌、卡波济氏肉瘤、星形细胞瘤、纤维状星形细胞瘤、胚胎发育不良性(dysembryoplastic)神经上皮瘤、少突神经胶质瘤、室管膜瘤、多形性胶质母细胞瘤、混合性神经胶质瘤、少突星状细胞瘤(oligoastrocytoma)、髓母细胞瘤、视网膜母细胞瘤、成神经细胞瘤、生殖细胞瘤、畸胎瘤、恶性间皮瘤(腹膜间皮瘤、心包间皮瘤、胸膜间皮瘤)、胃-肠-胰或胃肠胰神经内分泌肿瘤(GEP-NET)、类癌、胰腺内分泌肿瘤(PET)、结肠直肠腺癌、结直肠癌、攻击性神经内分泌肿瘤、平滑肌肉瘤粘液腺癌、印戒细胞腺癌、肝细胞癌、胆管癌、肝母细胞瘤、血管瘤、肝腺瘤、灶性结节状增生(结节状再生性增生、错构瘤)、非小细胞肺癌(NSCLC)(鳞状上皮细胞肺癌、腺癌、大细胞肺癌)、小细胞肺癌、甲状腺癌、前列腺癌(激素顽固性、雄激素非依赖性、雄激素依赖性、激素-非敏感性)和软组织肉瘤(纤维肉瘤、恶性纤维性组织细胞瘤、皮肤纤维肉瘤、脂肪肉瘤、横纹肌肉瘤、平滑肌肉瘤、血管肉瘤、滑膜肉瘤、恶性外周神经鞘肿瘤/神经纤维肉瘤、骨外骨肉瘤)。
在一些实施方案中,可给予本文所提供的肽蛋白酶体抑制剂或包含所述抑制剂的药物组合物来治疗患者的多发性骨髓瘤。例如,多发性骨髓瘤可包括顽固性和/或顽固性多发性骨髓瘤。
很多造血或淋巴组织的肿瘤以细胞增殖或特定类型细胞的增加为特征。慢性骨髓增生性疾病(CMPD)是以一种或多种骨髓谱系的骨髓增殖为特征的无性系造血干细胞病症,所述增殖导致外周血中粒细胞、红细胞和/或血小板的数量增加。如此,使用蛋白酶体抑制剂来治疗这些疾病是有吸引力的,并正在检测中(Cilloni等,Haematologica (2007) 92: 1124-1229)。CMPD可包括慢性髓性白血病、慢性嗜中性白血病、慢性嗜酸性白血病、真性红细胞增多症、慢性特发性骨髓纤维化、特发性血小板增多症和未分类的慢性骨髓增生性疾病。本文提供治疗CMPD的方法,其包括将有效量的本文所公开的蛋白酶体抑制剂化合物给予需要所述治疗的患者。
骨髓发育不良/骨髓增生性疾病,例如慢性髓单核细胞性白血病、非典型性慢性髓样白血病、幼髓单核细胞性白血病和未分类的骨髓发育不良/骨髓增生性疾病,表征为因一种或多种骨髓谱系的增殖而致的骨髓细胞过多。用本文所述组合物抑制蛋白酶体,可用于治疗这些骨髓发育不良/骨髓增生性疾病,所述治疗通过将有效量的组合物提供给需要所述治疗的患者来进行。
骨髓增生异常综合征(MDS)指一组以一种或多种成年髓样细胞系的发育不良和不能造血为特征的造血干细胞病症。在这些血液恶性肿瘤中用蛋白酶体抑制剂靶向NF-kB诱导凋亡,从而杀死恶性细胞(Braun等,Cell Death and Differentiation (2006) 13: 748-758)。本文还提供了治疗MDS的方法,其包括将有效量的本文所提供的化合物给予需要所述治疗的患者。MDS包括顽固性贫血、伴环形成高铁红细胞的顽固性贫血、伴多谱系增生异常的顽固性细胞减少、原始细胞过多性顽固性贫血、未分类的骨髓增生异常综合征和与分离的del (5q)染色体异常有关的骨髓增生异常综合征。
肥大细胞增多症是肥大细胞的增殖及其随后在一种或多种器官系统中的积累。肥大细胞增多症包括但不限于:皮肤性肥大细胞增多症、静止性系统性肥大细胞增多症(ISM)、与无性系的血液学上非肥大细胞谱系疾病有关的系统性肥大细胞增多症(SM-AHNMD)、攻击性系统性肥大细胞增多症(ASM)、肥大细胞白血病(MCL)、肥大细胞肉瘤(MCS)和皮肤外肥大细胞瘤。本文进一步提供治疗肥大细胞增多症的方法,其包括将有效量的本文所公开的化合物给予诊断为有肥大细胞增多症的患者。
蛋白酶体调控NF-κB,进而调控涉及免疫和炎症应答的基因。例如,NF-κB为表达以下基因所必需:免疫球蛋白轻链κ基因、IL-2受体α-链基因、I型主要组织相容性复合体基因和编码诸如IL-2、IL-6、粒细胞集落刺激因子和IFN-β等多种细胞因子的基因(Palombella等,Cell (1994) 78: 773-785)。因此,本文提供影响IL-2、MHC-I、IL-6、TNFα和IFN-β或前面提及的任何其它蛋白质的表达水平的方法,每一方法包括将有效量的本文所公开的蛋白酶体抑制剂组合物给予患者。
本文亦提供治疗患者自身免疫疾病的方法,其包括给予治疗有效量的本文所述化合物。本文“自身免疫疾病”是由个体自身组织引起并针对个体自身组织的疾病或病症。自身免疫疾病或病症的实例包括但不限于:炎症应答,例如炎性皮肤疾病,包括银屑病和皮炎(例如特应性皮炎);系统性硬皮病和硬化;与炎性肠病(例如克罗恩病和溃疡性结肠炎)相关的应答;呼吸窘迫综合征(包括成人呼吸窘迫综合征,ARDS);皮炎;脑膜炎;脑炎;葡萄膜炎;结肠炎;肾小球肾炎;过敏性病况,例如湿疹和哮喘和其它涉及T细胞浸润和慢性炎症应答的病况;动脉粥样硬化;白细胞黏附缺陷;类风湿性关节炎;系统性红斑狼疮(SLE);糖尿病(例如I型糖尿病或胰岛素依赖性糖尿病);多发性硬化症;Reynaud综合征;自身免疫性甲状腺炎;变应性脑脊髓炎;Sjorgen综合征;青少年型糖尿病;和与由细胞因子或T-淋巴细胞介导的急性和迟发性超敏反应有关的免疫应答,其通常在肺结核、结节病、多肌炎、肉芽肿病和血管炎中发现;恶性贫血(阿狄森氏病);涉及白细胞渗出的疾病;中枢神经系统(CNS)炎性病症;多器官损害综合征;溶血性贫血(包括但不限于冷球蛋白血症(cryoglobinemia)或Coombs阳性贫血);重症肌无力;抗原-抗体复合物介导的疾病;抗肾小球基膜病;抗磷脂综合征;变应性神经炎;格雷夫斯病;Lambert-Eaton肌无力综合征;大疱性类天疱疮;天疱疮;自身免疫性多内分泌腺疾病;Reiter疾病;僵人综合征;Beheet疾病;巨大细胞动脉炎;免疫复合物肾炎;IgA肾病;IgM多发性神经病;免疫性血小板减少性紫癜(ITP)或自身免疫性血小板减少症。
免疫系统筛查以下自体细胞:受病毒感染的、已经历致癌转化的或在其表面提呈不熟悉肽。细胞内蛋白水解产生提呈至T-淋巴细胞的小肽以诱导I型MHC介导的免疫应答。因此,本文提供使用本文所提供的蛋白酶体抑制剂作为免疫调节剂来抑制或改变细胞中的抗原提呈的方法,所述方法包括将细胞与本文所述的化合物接触(或给予患者本文所述的化合物)。具体的实施方案包括治疗移植或移植相关的疾病例如患者中的移植物抗宿主病或宿主抗移植物病的方法,其包括给予治疗有效量的本文所述化合物。本文所用术语“移植物”指衍生自供体以移植进入受体的生物学材料。移植物包括诸如以下的多种多样的材料:例如分离的细胞,例如岛细胞;组织,例如新生儿的羊膜、骨髓、造血前体细胞和眼睛组织例如角膜组织;和器官,例如皮肤、心脏、肝、脾、胰、甲状腺叶、肺、肾脏、管状器官(例如肠、血管或食道)。可用管状器官替代食道、血管或胆管的受损部分。皮肤移植物不但可用于烧伤,亦可作为受损肠道或愈合某些缺损例如膈疝的敷料。移植物来源于任何哺乳动物源,包括人,不论其来自尸体还是活体供体。在一些情况下,供体和受体是同一个患者。在一些实施方案中,移植物为骨髓或器官例如心脏,移植物的供体和宿主的HLA II型抗原是匹配的。
组织细胞和树突细胞肿瘤衍生自吞噬细胞和辅助细胞,所述吞噬细胞和辅助细胞在处理和提呈抗原至淋巴细胞中具有重要作用。已表明在树突细胞中消耗蛋白酶体含量可改变其抗原诱导的应答(Chapatte等,Cancer Res. (2006) 66: 5461-5468)。在一些实施方案中,可将本文所提供的组合物给予有组织细胞和树突细胞肿瘤的患者。组织细胞和树突细胞肿瘤包括组织细胞肉瘤、朗格汉斯细胞组织细胞增生症、朗格汉斯细胞肉瘤、指突状树突细胞肉瘤/肿瘤、小结树突细胞肉瘤/肿瘤和非特指树突细胞肉瘤。
已表明抑制蛋白酶体有益于治疗其中某一细胞类型增殖的疾病和免疫病症;因此,在一些实施方案中,提供对与原发性免疫病症(PID)有关的淋巴细胞增生性疾病(LPD)的治疗,其包含将有效量的所公开的化合物给予有需要治疗的患者。与包括B-细胞和T-细胞肿瘤和淋巴瘤在内的淋巴细胞增生性病症的发病率增加有关的最常见的免疫缺陷临床情况,为原发性免疫缺陷综合征和其它原发性免疫病症、人免疫缺陷病毒(HIV)感染、接受过固体器官或骨髓异体移植物的患者中的医源性免疫抑制和与甲氨蝶呤治疗有关的医源性免疫抑制。其它通常与LPD有关的PID为(但不限于)共济失调性毛细血管扩张症(AT)、Wiskott-Aldrich综合征(WAS)、普通可变型免疫缺陷病(CVID)、重症联合免疫缺陷(SCID)、X连锁淋巴细胞增生性疾病(XLP)、Nijmegen染色体断裂综合征(NBS)、高-IgM综合征和自身免疫淋巴细胞增生性综合征(ALPS)。
蛋白酶体抑制亦与NF-κB激活的抑制和p53水平的稳定有关。因此,本文所提供的组合物亦可用于在细胞培养物中抑制NF-κB的激活和稳定p53水平。因为NF-κB是炎症的关键调控子,所以它是抗炎治疗介入的有吸引力的靶标。因此,本文所提供的组合物可能对治疗与炎症有关的病况有用,所述病况包括但不限于COPD、银屑病、哮喘、支气管炎、肺气肿和囊性纤维化。
所公开的组合物可用于治疗由蛋白酶体的蛋白水解功能直接介导的病况,例如肌萎缩;或经由通过蛋白酶体处理的蛋白质例如NF-κB间接介导的病况。蛋白酶体参与涉及细胞调控(例如细胞周期、基因转录和代谢途径)、细胞间通讯和免疫应答(例如抗原提呈)的蛋白质(例如酶)的快速清除和翻译后加工。下面所讨论的具体实例包括β-淀粉样蛋白和调控蛋白,例如细胞周期蛋白和转录因子NF-κB。
在一些实施方案中,本文所提供的组合物可用于治疗包括但不限于以下的神经变性疾病或病况:中风、神经系统的缺血性损害、神经创伤(例如冲击性脑损害、脊髓损伤和神经系统的外伤损害)、多发性硬化和其它免疫介导的神经病(例如Guillain-Barre综合征及其变异型、急性运动轴突神经病、急性炎症性脱髓鞘性多发性神经病和Fisher综合征)、HIV/AIDS痴呆综合征(complex)、axonomy、糖尿病神经病变、帕金森病、亨廷顿舞蹈病、多发性硬化症、细菌性、寄生虫性、真菌性、和病毒性脑膜炎、脑炎、血管性痴呆、多发梗塞性痴呆、路易体痴呆、额叶痴呆例如Pick疾病、皮质下痴呆(例如Huntington或进行性核上麻痹)、灶性皮质萎缩综合征(例如原发性失语症)、代谢毒性痴呆(例如慢性甲状腺功能减退或B12缺陷症)和由感染(例如梅毒或慢性脑膜炎)导致的痴呆。
阿尔茨海默病以β-淀粉样蛋白(β-AP)在衰老斑和脑血管中的细胞外沉积为特征。β-AP是衍生自淀粉样蛋白前体(APP)的39至42个氨基酸的肽片段。已知至少三种APP的同工型(695、751和770个氨基酸)。mRNA的选择性剪接产生同工型;正常加工影响β-AP序列的部分,从而防止产生β-AP。认为通过蛋白酶体的异常蛋白加工有助于阿尔茨海默病脑中富含β-AP。大鼠中APP-加工酶含有约十个不同的亚基(22 kDa-32 kDa)。25 kDa亚基具有N-末端序列X-Gln-Asn-Pro-Met-X-Thr-Gly-Thr-Ser,其与人巨蛋白因子(macropain)的β-亚基完全一致(Kojima, S等,Fed. Eur. Biochem. Soc., (1992) 304: 57-60)。APP-加工酶切开Gln15--Lys16键;在钙离子存在时,该酶亦可切开Met-1--Asp1键和Asp1--Ala2键以释放β-AP的胞外域。
因此,一个实施方案为治疗阿尔茨海默病的方法,其包括将有效量的本文所提供的组合物给予患者。所述治疗包括降低β-AP的加工速率、降低β-AP的蚀斑形成速率、降低β-AP产生的速率和降低阿尔茨海默病的临床病征。
本文亦提供治疗恶病质和肌萎缩疾病的方法。蛋白酶体在成熟网状细胞和成长中的成纤维细胞中降解很多蛋白质。在剥夺了胰岛素或血清的细胞中,蛋白水解的速率几乎翻倍。抑制蛋白酶体降低蛋白水解,从而降低肌肉蛋白损失和肾脏或肝脏的氮荷载。本文提供的肽蛋白酶体抑制剂可用于治疗例如以下病况:癌症、慢性传染病、发烧、肌肉废用(萎缩)和去神经支配、神经损伤、禁食、与酸中毒有关的肾衰竭、和肝衰竭。参见例如Goldberg的美国专利号5,340,736。治疗的方法包括:降低细胞中肌肉蛋白的降解速率;降低胞内蛋白的降解速率;降低细胞中p53蛋白的降解速率;和抑制p53-相关的癌症的生长。这些方法的每一种包括让细胞(体内或体外,例如患者的肌肉)与有效量的本文所公开的药物组合物接触。
纤维化是因成纤维细胞的过多增生性生长而起的瘢痕组织的过量和持久形成,其与TGF-β信号转导途径的激活有关。纤维化涉及到胞外基质的广泛沉积,可在实际上任何组织内或几个不同组织间发生。通常在TGF-β刺激时激活靶基因转录的胞内信号转导蛋白(Smad)的水平受蛋白酶体活性的调控。然而,已在癌症和其它过多增生性病况中观察到TGF-β信号转导组分的加速降解。因此,在某些实施方案中,提供了用于治疗过多增生性病况的方法,所述病况例如糖尿病视网膜病变、黄斑变性、糖尿病肾病、肾小球硬化症、IgA肾病、肝硬化、胆道闭锁、充血性心力衰竭、硬皮病、辐射诱发的纤维化和肺纤维化(特发性肺纤维化、胶原血管病、结节病、间质性肺疾病和非固有肺病症)。烧伤受害者的治疗通常受到纤维化的妨碍,因此,在一些实施方案中,可通过局部或全身性给药来给予本文所提供的抑制剂以治疗烧伤。手术后的创伤愈合通常与毁容性疤痕有关,该疤痕可通过抑制纤维化来防止。因此,在某些实施方案中,本文提供用于防止或减少疤痕的方法。
由蛋白酶体加工的另一蛋白质为NF-κB,其为Rel蛋白家族的成员。Rel家族的转录激活因子蛋白可分为两类。第一类需要蛋白水解加工,包括p50 (NF-κB1、105 kDa)和p52 (NF-κ2、100 kDa)。第二类不需要蛋白水解加工,包括p65 (RelA、Rel (c-Rel)和RelB)。同型二聚体和异二聚体二者都可由Rel家族成员形成;例如,NF-κB为p50-p65异二聚体。在IκB和p105的磷酸化和泛素化之后,将这两种蛋白质各自降解和加工,以产生从细胞质转移至细胞核的活性NF-κB。亦可通过纯化的蛋白酶体来加工泛素化的p105 (Palombella等,Cell (1994) 78: 773-785)。活性NF-κB与其它转录激活因子和例如HMG I(Y)形成立体特异性的增强子复合物,这诱导特定基因的选择性表达。
NF-κB调控涉及免疫和炎症应答及有丝分裂事件的基因。例如,NF-κB为表达以下基因所必需:免疫球蛋白轻链κ基因、IL-2受体α-链基因、I型主要组织相容性复合体基因和编码例如IL-2、IL-6、粒细胞集落刺激因子和IFN-β等多种细胞因子的基因(Palombella等,Cell (1994) 78: 773-785)。一些实施方案包括影响IL-2、MHC-I、IL-6、TNFα、IFN-β或前面提及的任何其它蛋白质的表达水平的方法,每一方法包括将有效量的本文所公开的组合物给予患者。包括p50的复合物是急性炎症和免疫应答的快速介导物(mediator)(Thanos, D.和Maniatis, T.,Cell (1995) 80: 529-532)。
NF-κB亦参与表达编码E-选择素、P-选择素、ICAM、和VCAM-1的细胞粘附基因(Collins, T.,Lab. Invest. (1993) 68: 499-508)。在一些实施方案中,提供了用于抑制细胞粘附(例如由E-选择素、P-选择素、ICAM或VCAM-1介导的细胞粘附)的方法,其包括让细胞与有效量的本文所公开的药物组合物接触(或给予患者有效量的本文所公开的药物组合物)。
缺血和再灌注损伤导致缺氧,其为到达身体组织的氧不足的病况。这种病况导致Iκ-Bα的降解增加,从而导致NF-κB的激活。已证明可用给予蛋白酶体抑制剂来降低导致缺氧的损伤的严重性。因此,本文提供治疗缺血病况或再灌注损伤的方法,其包括将有效量的本文所公开的化合物给予需要所述治疗的患者。所述病况或损伤的实例包括但不限于:急性冠状动脉综合征(脆性斑块)、动脉阻塞性疾病(心脏、脑、外周动脉和血管闭塞)、动脉粥样硬化(冠状动脉硬化、冠状动脉病)、梗塞、心力衰竭、胰腺炎、心肌肥大、狭窄和再狭窄。
NF-κB亦特异性地结合HIV-增强子/启动子。当与mac239的Nef相比较时,HIV调控蛋白pbj14的Nef在控制蛋白激酶结合的区域中有两个氨基酸的差异。认为蛋白激酶发出IκB磷酸化的信号,这通过泛素-蛋白酶体途径触发IκB的降解。降解后,NF-κB释放到细胞核中,从而增加HIV的转录(Cohen, J.,Science, (1995) 267: 960)。本文提供用于抑制或降低患者中HIV感染的方法,和用于减少病毒基因表达水平的方法,每一方法包括将有效量的本文所公开的组合物给予患者。
病毒感染促成很多疾病的病理学。心脏病况例如进行性心肌炎和扩张型心肌病与柯萨奇病毒B3有关。在受感染小鼠心脏的比较性全基因组微阵列分析中,特异的蛋白酶体亚基在发生慢性心肌炎的小鼠心脏中一致上调(Szalay等,Am J Pathol 168: 1542-52, 2006)。一些病毒在病毒从核内体释放到胞质溶胶的病毒侵入步骤中利用了泛素-蛋白酶体系统。小鼠肝炎病毒(MHV)属于冠状病毒科,其亦包括严重急性呼吸器官综合征(SARS)冠状病毒。Yu和Lai (J Virol 79: 644-648, 2005)证明用蛋白酶体抑制剂治疗受MHV感染的细胞导致病毒复制的减少,这关联到与未治疗的细胞相比病毒效价的降低。人乙型肝炎病毒(HBV)是肝DNA病毒科(Hepadnaviridae virus family)的成员,同样需要病毒编码的包膜蛋白来繁殖。抑制蛋白酶体的降解途径导致分泌型包膜蛋白在数量上的显著降低(Simsek等,J Virol 79: 12914-12920, 2005)。除HBV外,其它肝炎病毒(A、C、D和E)亦可利用泛素-蛋白酶体降解途径来分泌、导致形态发生和致病。因此,在某些实施方案中,提供了用于治疗病毒感染例如SARS或甲、乙、丙、丁和戊型肝炎的方法,其包括让细胞与有效量的本文所公开的化合物接触(或给予患者有效量的本文所公开的化合物)。
认为脂多糖(LPS)-诱导的细胞因子例如TNFα的过度产生对与脓毒性休克有关的进程是极为重要的。此外,公认为LPS激活细胞的第一个步骤是将LPS结合到特异的膜受体上。已鉴定出20S蛋白酶体复合物的α-和β-亚基为LPS-结合蛋白,这表明LPS-诱导的信号转导可能是脓毒症治疗或预防中的重要治疗靶标(Qureshi, N.等,J. Immun. (2003) 171: 1515-1525)。因此,在某些实施方案中,可用本文所提供的组合物抑制TNFα来预防和/或治疗脓毒性休克。
细胞内蛋白水解产生小肽并提呈至T-淋巴细胞以诱导I型MHC-介导的免疫应答。免疫系统筛查受病毒感染或已经历致癌性转化的自体细胞。一个实施方案为用于抑制细胞中抗原提呈的方法,其包括让细胞与本文所述组合物接触。另一实施方案是用于抑制患者免疫系统(例如抑制移植排斥、变态反应、哮喘)的方法,其包括将有效量的本文所述组合物给予患者。本文所提供的组合物亦可用于治疗自身免疫疾病,例如狼疮、类风湿性关节炎、多发性硬化症和炎性肠病例如溃疡性结肠炎和克罗恩病。
另一个实施方案是用于改变由蛋白酶体或其它有多催化活性的Ntn产生的抗原肽的全部(repertoire)的方法。例如,如果20S蛋白酶体的PGPH活性受到选择性抑制,则与没有任何酶抑制或有例如选择性抑制蛋白酶体的糜蛋白酶-样活性时可产生和存在的相比,通过蛋白酶体会产生不同的抗原肽组并存在于细胞表面的MHC分子上。
某些蛋白酶体抑制剂阻断泛素化NF-κB在体内和体外的降解和加工二者。蛋白酶体抑制剂亦阻断IκB-α的降解和NF-κB的激活(Palombella等,Cell (1994) 78: 773-785;和Traenckner等,EMBO J. (1994) 13: 5433-5441)。在一些实施方案中,提供了用于抑制IκB-α降解的方法,其包括让细胞与本文所述组合物接触。另一实施方案为用于降低细胞、肌肉、器官或患者中NF-κB的胞内含量的方法,其包括让细胞、肌肉、器官或患者与本文所述组合物接触。
需要蛋白水解加工的其它真核转录因子包括通用转录因子TFIIA、单纯疱疹病毒VP16辅助蛋白(宿主细胞因子)、病毒-可诱导的IFN调控因子2蛋白和膜结合的固醇调控元件-结合蛋白1。
本文另外提供用于影响细胞周期蛋白-依赖性真核细胞周期的方法,其包括将细胞(体外或体内)与本文所公开的组合物接触。细胞周期蛋白是涉及细胞周期控制的蛋白质。蛋白酶体参与细胞周期蛋白的降解。细胞周期蛋白的实例包括有丝分裂细胞周期蛋白、G1细胞周期蛋白和细胞周期蛋白B。细胞周期蛋白的降解使细胞退出一个细胞周期阶段(例如有丝分裂),进入另一个阶段(例如分裂)。认为所有的细胞周期蛋白与p34cdc2蛋白激酶或相关的激酶相关。蛋白水解靶向信号位于氨基酸42-RAALGNISEN-50 (破坏框)。这是细胞周期蛋白转换为易受泛素连接酶影响的形式的证据,或有丝分裂期间激活了细胞周期蛋白-特异性连接酶的证据(Ciechanover, A., Cell, (1994) 79: 13-21)。抑制蛋白酶体抑制了细胞周期蛋白的降解,因此抑制例如细胞周期蛋白-相关的癌症中的细胞增殖(Kumatori等, Proc. Natl. Acad. Sci. USA (1990) 87: 7071-7075)。本文提供用于治疗患者中增殖性疾病(例如癌症、银屑病或再狭窄)的方法,其包括将有效量的本文所公开的组合物给予患者。本文亦提供用于治疗患者的细胞周期蛋白-相关炎症的方法,其包括将治疗有效量的本文所述组合物给予患者。
另外的实施方案包括用于影响癌蛋白的蛋白酶体-依赖性调控的方法和治疗或抑制癌症生长的方法,每一方法包括将细胞(例如在患者体内,或体外)与本文所公开的组合物接触。HPV-16和HPV-18-衍生的E6蛋白刺激未成熟网织红细胞裂解物中p53的ATP-和泛素-依赖性缀合和降解。已表明在有突变的不耐热E1的细胞系中,隐性癌基因p53在非允许的温度下积累。p53水平的提高可导致细胞凋亡。通过泛素系统降解的原癌蛋白的实例包括c-Mos、c-Fos和c-Jun。一个实施方案是用于治疗p53-相关的细胞凋亡的方法,其包括将有效量的本文所公开的组合物给予患者。
在另一个实施方案中,所公开的组合物可用于治疗寄生虫感染例如由原生动物寄生虫引起的感染。认为这些寄生虫的蛋白酶体主要涉及到细胞分化和复制活性(Paugam等,Trends Parasitol. 2003, 19(2): 55-59)。此外,已表明当与蛋白酶体抑制剂接触时,内阿米巴属物种丧失成囊能力(Gonzales等,Arch. Med. Res. 1997, 28, Spec No: 139-140)。在某些所述实施方案中,所公开的组合物可用于治疗由选自以下的原生动物寄生虫引起的人寄生虫感染:疟原虫属(Plasmodium sps.) (包括导致疟疾的恶性疟原虫(P. falciparum)、间日疟原虫(P. vivax)、三日疟原虫(P. malariae)和卵形疟原虫(P. ovale))、锥虫属(Trypanosoma sps.) (包括导致Chaga病的美洲锥虫(T. cruzi)和导致非洲昏睡病的布鲁氏锥虫(T. brucei))、利什曼原虫属(Leishmania sps.) (包括亚马逊利什曼原虫(L. amazonesis)、杜氏利什曼原虫(L. donovani)、婴儿利什曼原虫(L. infantum)、墨西哥利什曼原虫(L. mexicana)等)、卡氏肺囊虫(Pneumocystis carinii) (一种已知引起AIDS和其它免疫抑制患者中肺炎的原生动物)、鼠弓形体(Toxoplasma gondii)、溶组织内阿米巴(Entamoeba histolytica)、侵袭性内阿米巴(Entamoeba invadens)和兰伯贾第虫(Giardia lamblia)。在某些实施方案中,所公开的组合物可用于治疗由选自以下的原生动物寄生虫引起的动物和家畜寄生虫感染:赫尔曼疟原虫(Plasmodium hermani)、隐孢子虫属(Cryptosporidium sps.)、细粒棘球绦虫(Echinococcus granulosus)、禽艾美球虫(Eimeria tenella)、神经肉孢子虫(Sarcocystis neurona)和粗糙链孢霉(Neurospora crassa)。在WO 98/10779中阐述了在治疗寄生虫病中可用作蛋白酶体抑制剂的其它化合物,其内容以其整体并入本文。
在某些实施方案中,所公开的组合物在寄生虫中不可逆地抑制蛋白酶体活性。已表明所述不可逆抑制诱导红细胞和白细胞中酶活性不可恢复地丧失。在某些所述实施方案中,关于针对重新与寄生虫接触的治疗,血细胞的长半衰期可提供延长的保护作用。在某些实施方案中,关于化学预防未来的感染,血细胞的长半衰期可提供延长的保护作用。
原核生物具有真核生物20S蛋白酶体颗粒的等效物。虽然原核生物的20S颗粒的亚基组合物比真核生物简单,但其具有以相似的方式水解肽键的能力。例如,在β-亚基的N-末端通过苏氨酸残基发生对肽键的亲核攻击。在一些实施方案中,提供了治疗原核生物感染的方法,其包括将有效量的本文所公开的蛋白酶体抑制剂组合物给予患者。原核生物感染可包括由分枝杆菌(例如结核、麻风病或布路里溃疡(Buruli Ulcer))或古细菌导致的疾病。
已证明结合到20S蛋白酶体的抑制剂刺激骨器官培养物中骨的形成。此外,当将所述抑制剂全身性给予小鼠时,某些蛋白酶体抑制剂对骨体积和骨形成速率的增加超过70% (Garrett, I. R. 等,J. Clin. Invest. (2003) 111: 1771-1782),因此表明泛素-蛋白酶体机构调控成骨细胞分化和骨形成。因此,所公开的组合物可能在治疗和/或预防与骨丢失有关的疾病例如骨质疏松症中有用。
本文提供治疗选自以下的疾病或病况的方法:癌症、自身免疫病、移植物或移植-相关病况、神经变性疾病、纤维化相关的病况、缺血相关的病况、感染(病毒的、寄生虫的或原核生物的)和与骨丢失有关的疾病,所述方法包括给予本文所提供的蛋白酶体抑制剂。例如,式(5)化合物。
骨组织是具有刺激骨细胞能力的因子的良好来源。因此,牛骨组织提取物不仅含有负责维持骨结构完整性的结构蛋白,而且还含有可刺激骨细胞增殖的生物学上活性的骨生长因子。这后一类因子为最近阐述的蛋白质家族,称作骨形态生成蛋白(BMP)。所有这些生长因子对其它类型的细胞以及骨细胞具有影响,包括Hardy, M. H等,Trans Genet (1992) 8: 55-61阐述的证据,即骨形态生成蛋白(BMP)在发育期间的毛囊中差异表达。Harris, S. E.等,J Bone Miner Res (1994) 9: 855-863阐述TGF-β在骨细胞中对BMP-2和其它物质的表达的影响。在成熟过程中和细胞增殖期之后,在成熟滤泡中亦发生BMP-2的表达(Hardy, 等(1992, 见上)。因此,本文所提供的化合物亦可能对毛囊生长刺激有用。
最后,所公开的组合物亦可用作诊断剂(例如在诊断试剂盒中或在临床实验室中使用),以筛查由包括蛋白酶体在内的Ntn水解酶加工的蛋白质(例如酶、转录因子)。所公开的组合物亦可用作研究试剂,以特异性结合X/MB1亚基或α-链和抑制与其有关的蛋白水解活性。例如,可测定蛋白酶体其它亚基(和特异的抑制剂)的活性。
在成熟或激活期间大多数的细胞蛋白质经受蛋白水解加工。可用本文所公开的酶抑制剂来测定细胞、发育或生理学过程或输出是否受特定Ntn水解酶的蛋白水解活性的调控。一种所述方法包括:获得生物体、完整的细胞制备物或细胞提取物;让生物体、细胞制备物或细胞提取物与本文所公开的组合物接触;让已接触过化合物的生物体、细胞制备物或细胞提取物与信号接触,并监测过程或输出。本文所公开的化合物的高度选择性允许在给定的细胞、发育或生理学过程中快速和精确地消除或牵涉Ntn (例如20S蛋白酶体)。
给药
如本领域所熟知,视待治疗的病症和患者的年龄、病况和体重,可给予呈各种形式的如本文所述制备的组合物。例如,当要口服给予组合物时,可将其制成片剂、胶囊剂、颗粒剂、粉剂或糖浆剂;或对于胃肠外给药,可将其制成注射剂(静脉内的、肌内的或皮下的)、滴注制剂或栓剂。对于经眼睛粘膜途径施用,可将其制成滴眼剂或眼膏。可通过常规方式与本文所述方法联合来制备这些制剂,若需要可让活性成分与任何传统添加剂或赋形剂混合,其例如除环糊精和缓冲剂外,还有粘合剂、崩解剂、润滑剂、矫味剂、增溶剂、助悬剂、乳化剂或涂层剂。虽然剂量将视患者的症状、年龄和体重、待治疗或预防的病变的性质和严重性、给药途径和药物的形式而变化,但总的来说,对于成年人患者,推荐化合物的日用剂量为0.01-2000 mg,该剂量可以以单次剂量或分开成多次剂量给予。可与载体材料联合产生单一剂型的活性成分的量将通常为可产生治疗作用的化合物的量。总的来说,意欲胃肠外使用(例如静脉、皮下注射)的组合物包括取代的环糊精。经由其它途径特别是口服途径给予的组合物,包括取代的或未被取代的环糊精。
在给定的患者中从治疗功效上可产生最有效结果的给药精确时间和/或组合物的量将视以下因素而定:具体化合物的活性、药代动力学和生物利用度;患者的生理条件(包括年龄、性别、疾病类型和阶段、总的身体健康情况、对给定剂量的应答性和药物的类型);给药途径等。然而,上面的准则可用作细调治疗的基础,例如确定给药的最佳时间和/或数量,只是这将需要进行例行实验,所述例行实验由监测患者和调整剂量和/或时间组成。
本文采用短语“药学上可接受的”指那些在合理的医学判断范围内适于与人类和动物的组织接触使用而无过度毒性、刺激、过敏反应或其它问题或并发症且有同等合理的利益/风险比的配体、材料、组合物和/或剂型。
本文所用短语“药学上可接受的载体”意为药学上可接受的材料、组合物或溶媒,例如液体或固体填充剂、稀释剂、赋形剂、溶剂或包封材料。每一种载体在与制剂中其它成分相容并对患者无害意义上必须是“可接受的”。可用作药学上可接受的载体的材料的一些实例包括:(1)糖类,例如乳糖、葡萄糖和蔗糖;(2)淀粉,例如玉米淀粉、土豆淀粉和取代的或未被取代的β-环糊精;(3)纤维素及其衍生物,例如羧甲基纤维素钠、乙基纤维素和乙酸纤维素;(4)粉末化的黄蓍胶;(5)麦芽;(6)明胶;(7)滑石;(8)赋形剂,例如可可脂和栓剂蜡类;(9)油类,例如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆油;(10)二醇类,例如丙二醇;(11)多元醇,例如甘油、山梨醇、甘露醇和聚乙二醇;(12)酯类,例如油酸乙酯和十二烷酸乙酯;(13)琼脂;(14)缓冲剂,例如氢氧化镁和氢氧化铝;(15)藻酸;(16)无热原的水;(17)等渗盐;(18) 林格溶液;(19)乙醇;(20)磷酸盐缓冲溶液;和(21)在药物制剂中采用的其它无毒相容物质。在某些实施方案中,本文提供的药物组合物为无热原的,即当给予患者时不会诱导显著的温度升高。
术语“药学上可接受的盐”指抑制剂的相对无毒的无机或有机酸加成盐。可在抑制剂最终的分离和纯化期间原位制备这些盐,或通过让呈游离碱形式的纯化的肽蛋白酶体抑制剂与适宜的有机或无机酸单独反应并分离因此形成的盐来制备这些盐。代表性的盐包括氢溴酸盐、盐酸盐、硫酸盐、硫酸氢盐、磷酸盐、硝酸盐、乙酸盐、戊酸盐、油酸盐、棕榈酸盐、硬脂酸盐、月桂酸盐、苯甲酸盐、乳酸盐、磷酸盐、甲苯磺酸盐、柠檬酸盐、马来酸盐、富马酸盐、琥珀酸盐、酒石酸盐、萘酸盐(naphthylate)、甲磺酸盐、葡庚糖酸盐、乳糖酸盐、月桂基磺酸盐和氨基酸盐等等。(参见例如Berge等,(1977) “Pharmaceutical Salts” (药用盐类),J. Pharm. Sci. 66: 1-19)。
在一些实施方案中,本文所提供的肽蛋白酶体抑制剂可含有一种或多种酸官能团,因此能够与药学上可接受的碱形成药学上可接受的盐。在这些情况下术语“药学上可接受的盐”指抑制剂的相对无毒的无机和有机碱加成盐。可同样在抑制剂最终的分离和纯化期间原位制备这些盐,或通过让呈游离酸形式的纯化的抑制剂与以下物质单独反应来制备这些烟:适宜的碱,例如氢氧化物、药学上可接受的金属阳离子的碳酸盐或碳酸氢盐;铵;或药学上可接受的有机伯胺、仲胺或叔胺。代表性的碱金属或碱土金属盐包括锂、钠、钾、钙、镁和铝盐等等。可用于形成碱加成盐的代表性有机胺包括乙胺、二乙胺、乙二胺、乙醇胺、二乙醇胺、哌嗪等等(参见例如Berge等,见上)。
组合物中亦可存在湿润剂、乳化剂和润滑剂例如十二烷基硫酸钠和硬脂酸镁以及着色剂、释放剂、涂层剂、甜味剂、矫味剂和芳香剂、防腐剂和抗氧化剂。
药学上可接受的抗氧化剂的实例包括:(1)水溶性的抗氧化剂,例如抗坏血酸、盐酸半胱氨酸、硫酸氢钠、偏亚硫酸氢钠、亚硫酸钠等等;(2)油溶性的抗氧化剂,例如抗坏血酸棕榈酸酯、丁羟茴醚(BHA)、丁羟甲苯(BHT)、卵磷脂、没食子酸丙酯、α-生育酚等等;和(3)金属螯合剂,例如柠檬酸、乙二胺四乙酸(EDTA)、山梨醇、酒石酸、磷酸等等。
适于口服给予的制剂可为胶囊剂、扁胶囊、丸剂、片剂、锭剂(使用调味基质,通常为蔗糖和阿拉伯胶或黄蓍胶)、粉剂、颗粒剂形式;或为水或非水液体的溶液剂或混悬剂;或为水包油或油包水的液态乳剂;或为酏剂或糖浆剂;或为软锭剂(使用惰性基质,例如明胶和甘油,或蔗糖和阿拉伯胶)和/或为漱口剂等等,其每一种含有预设的量的抑制剂作为活性成分。亦可以大丸剂、药糖剂或糊剂来给予组合物。
在口服给予的固体剂型中(胶囊剂、片剂、丸剂、锭剂、粉剂、颗粒剂等等),活性成分与一种或多种药学上可接受的载体混合,所述载体例如柠檬酸钠或磷酸二钙;和/或任何下述物质:(1)填充剂或增量剂,例如淀粉、环糊精、乳糖、蔗糖、葡萄糖、甘露糖和/或硅酸;(2)粘合剂,例如羧甲基纤维素、藻酸盐、明胶、聚乙烯吡咯烷酮、蔗糖和/或阿拉伯胶;(3)保湿剂,例如甘油;(4)崩解剂,例如琼脂-琼脂、碳酸钙、马铃薯淀粉或木薯淀粉、藻酸、某些硅酸盐类和碳酸钠;(5)溶液阻滞剂,例如石蜡;(6)吸收加速剂,例如季铵化合物;(7)湿润剂,例如乙酰基醇和单硬脂酸甘油酯;(8)吸收剂,例如高岭土和膨润土;(9)润滑剂,例如滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、十二烷基硫酸钠及其混合物;和(10)着色剂。在胶囊剂、片剂和丸剂的情况下,药物组合物亦可包含缓冲剂。使用诸如乳糖或奶糖等赋形剂以及高分子量聚乙二醇等等,亦可将相似类型的固体组合物用作软和硬明胶胶囊中的填充剂。
可通过压缩或模制来制造片剂,任选用一种或多种辅助成分。可使用粘合剂(例如明胶或羟丙甲基纤维素)、润滑剂、惰性稀释剂、防腐剂、崩解剂(例如淀粉羟乙酸钠或交联的羧甲基纤维素钠)、表面活性剂或分散剂来制备压制片。可通过在适宜的机器中让用惰性液体稀释剂弄湿的粉状抑制剂混合物成模来制造模制的片剂。
片剂和其它固体剂型,例如锭剂、胶囊剂、丸剂和颗粒剂可任选经刻痕或用包被物和外壳例如肠衣和药物配制领域熟知的其它包被物来制备。亦可配制它们以便于提供缓释或控释的活性成分,其中使用例如不同比例以提供所需的释放特征的羟丙甲基纤维素、其它聚合物基质、脂质体和/或微球体。可将其灭菌,例如通过细菌-阻留性过滤器过滤或通过引入呈无菌固体组合物形式的灭菌剂来实现灭菌,所述组合物可在临用前溶于无菌水或一些其它可注射介质中。这些组合物亦可任选含有乳浊剂,并且可为仅在胃肠道的某些部分或优选在胃肠道的某些部分任选以延迟的方式释放活性成分的组合物。可使用的包埋组合物的实例包括聚合物质和蜡类。活性成分亦可为微胶囊化形式,如果合适的话,可带有一种或多种如上所述的赋形剂。
供口服给予的液体剂型包括药学上可接受的乳剂、微乳剂、溶液剂、混悬剂、糖浆剂和酏剂。除活性成分外,液体剂型还可含有本领域通常使用的惰性稀释剂,例如水或其它溶剂;增溶剂;和乳化剂,例如乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苯甲醇、苯甲酸苄酯、丙二醇、1,3-丁二醇、油类(具体来说,棉籽油、落花生油、玉米油、胚芽油、橄榄油、蓖麻油和芝麻油)、甘油、四氢糠醇、聚乙二醇和脱水山梨醇的脂肪酸酯及其混合物。
除了惰性稀释剂,口服组合物亦可包括辅助剂,例如湿润剂、乳化剂和悬浮剂、甜味剂、矫味剂、着色剂、芳香剂和防腐剂。
混悬剂除活性抑制剂外还可含有悬浮剂,例如乙氧基化的异硬脂醇、聚氧乙烯山梨醇和脱水山梨醇酯、微晶纤维素、偏氢氧化铝、膨润土、琼脂-琼脂和黄蓍胶及其混合物。
供直肠或阴道给予的制剂可呈现为栓剂,其可通过将一种或多种抑制剂与一种或多种适宜的非刺激性赋形剂或载体混合来制备,所述赋形剂或载体包含例如可可脂、聚乙二醇、栓剂蜡或水杨酸酯,这些物质在室温下是固体,但在体温下是液体,因此,将在直肠或阴道腔内融化和释放活性剂。
适于阴道给予的制剂亦包括阴道栓、卫生栓、乳膏剂、凝胶剂、糊剂、泡沫剂或喷雾制剂,其含有本领域所知的合适的所述载体。
供局部或透皮给予抑制剂的剂型包括粉剂、喷雾剂、软膏剂、糊剂、乳膏剂、洗剂、凝胶剂、溶液剂、贴剂和吸入剂。可在无菌条件下让活性组分与药学上可接受的载体并与可能需要的任何防腐剂、缓冲剂或喷射剂混合。
软膏剂、糊剂、乳膏剂和凝胶剂除抑制剂外还可含有赋形剂,例如动物和植物脂肪、油类、蜡类、石蜡、淀粉、黄蓍胶、纤维素衍生物、聚乙二醇、硅酮类、膨润土、硅酸、滑石和氧化锌或其混合物。
粉剂和喷雾剂除抑制剂外亦可含有赋形剂,例如乳糖、滑石、硅酸、氢氧化铝、硅酸钙和聚酰胺粉末或这些物质的混合物。喷雾剂可另外含有惯用的喷射剂,例如氯氟烃和挥发性未被取代的烃类,例如丁烷和丙烷。
可通过气雾剂来给予肽蛋白酶体抑制剂。这通过制备含有组合物的水性气雾剂、脂质体制剂或固体颗粒来实现。可使用非水性(例如,碳氟化合物喷射剂)混悬剂。在一些实施方案中,优选声波雾化器,因为其可使试剂对剪切的暴露最小化,剪切可导致化合物的降解。
通常可通过配制药剂与传统的药学上可接受的载体和稳定剂在一起的水溶液或混悬液来制备水性气雾剂。载体和稳定剂随具体组合物的需要而变化,但通常包括非离子型表面活性剂(吐温类、Pluronics,脱水山梨醇酯类、卵磷脂、Cremophor)、药学上可接受的共溶剂例如聚乙二醇、无害的蛋白质如血清白蛋白、脱水山梨醇酯类、油酸、卵磷脂、氨基酸例如甘氨酸、缓冲剂、盐类、糖类或糖醇类。通常自等渗溶液制备气雾剂。
透皮贴剂具有向身体提供抑制剂的可控递送的另外优势。可通过将药剂溶解或分散于适当的介质中来制备所述剂型。亦可使用吸收促进剂来增加抑制剂穿过皮肤的流量。可通过提供速率可控的膜或将抑制剂分散于聚合物基质或凝胶中来控制所述流量的速率。
适于胃肠外给予的药物组合物包含与以下物质组合的一种或多种肽蛋白酶体抑制剂:一种或多种药学上可接受的无菌水或非水溶液、分散体、混悬液或乳液;或在临用前可重构为无菌可注射溶液或分散体的无菌粉末;其可含有抗氧化剂、缓冲剂、抑菌剂、可使制剂与预期受体的血液等渗的溶质或悬浮剂或增稠剂。
可用于本文所提供的药物组合物的适宜的水性和非水性载体的实例包括注射用水(例如无菌注射用水)、乙醇、多元醇(例如甘油、丙二醇、聚乙二醇等等)、缓冲剂(例如柠檬酸盐缓冲剂)及其适宜的混合物、植物油例如橄榄油和可注射的有机酯类例如油酸乙酯。可例如通过使用涂层材料例如卵磷脂、通过在分散体的情况下维持所需的粒度和通过使用表面活性剂,来保持适当的流动性。
药物组合物通常包括药学上可接受的载体。本文所用术语“药学上可接受的载体”包括与药物给予相容的缓冲剂、无菌注射用水、溶剂、分散介质、涂层剂、抗细菌和抗真菌剂、等渗剂和吸收延迟剂等等。在一些实施方案中,药学上可接受的载体为缓冲剂(例如柠檬酸盐缓冲剂)。在一些实施方案中,药学上可接受的载体为无菌注射用水。在一些实施方案中,药学上可接受的载体包含柠檬酸。
这些组合物亦可含有辅助剂,例如防腐剂、湿润剂、乳化剂和分散剂。可通过包含各种抗细菌和抗真菌剂例如对羟苯甲酸酯、氯丁醇、苯酚山梨酸等等确保防止微生物的作用。亦希望在组合物中包括张度调节剂例如糖类等等。另外,可通过包含延迟吸收的剂例如单硬脂酸铝和明胶来使可注射药物形式的吸收延长。
在一些情况下,为了延长药物的作用,希望减缓经皮下或肌内注射的药物吸收。例如,通过将药物溶解或悬浮在油性溶媒中来实现胃肠外给予的药物形式的延迟吸收。
通过在生物可降解的聚合物例如聚交酯-聚乙交酯中形成抑制剂的微胶囊基质来制造可注射的贮库形式。可视药物与聚合物的比值、所使用的具体聚合物的性质,来控制药物的释放速率。其它生物可降解的聚合物的实例包括聚(原酸酯)和聚(酐)。亦可通过将药物陷入可与身体组织相容的脂质体或微胶囊中来制备可注射的贮库制剂。
可经口、胃肠外、局部或直肠来给予药剂的制剂。当然,通过适于每一给药途径的形式来给予它们。例如,它们可以如下给予:以片剂或胶囊剂形式;通过注射、吸入、洗眼、软膏、栓剂、输注;通过洗剂或软膏局部给予;通过栓剂直肠给予。在一些实施方案中,给予为口服。
本文所用的短语“胃肠外给药”和“胃肠外给予”,意即除肠道和局部给予外的给药方式,其通常通过注射,包括但不限于:静脉内、肌内、动脉内、鞘内、囊内、眶内、心内、皮内、腹膜内、经气管、皮下、表皮下、关节内、囊下、蛛网膜下、脊柱内和胸骨内注射和输注。
本文所用短语“全身给药”、“全身给予”、“外周给药”和“外周给予”,意为非直接进入中枢神经系统给予配体、药物或其它物质,以便其进入患者的循环系统,并因此经受代谢和其它类似的过程,例如皮下给予。
可通过任何适宜的给药途径将本文所述的肽蛋白酶体抑制剂给予人和其它动物用于治疗,所述给药途径包括经口给药、经鼻给药(如通过例如喷雾剂)给药、直肠给药、阴道内给药、胃肠外给药、脑池内给药和局部(如通过粉剂、软膏剂和滴剂)给药,包括含服和舌下给药。
不管所选择的给药途径如何,可通过本领域技术人员所知的传统方法,将可呈适宜的含水形式使用的肽蛋白酶体抑制剂和/或本文所提供的药物组合物配制为药学上可接受的剂型。
本文所提供的药物组合物中的活性成分的实际剂量水平可变化,以便对于特定患者、组合物和给药方式,获得可有效达到所需治疗应答而对患者没有毒性的活性成分的量。
药学上可接受的混合物中的所公开化合物的浓度将视若干因子而变化,包括待给予的化合物的剂量、所使用的化合物的药代动力学特征和给药途径。总的来说,对于胃肠外给药,可以在含有约0.1-10% w/v的本文所公开化合物(除其它物质之外)的水溶液中提供本文所提供的组合物。通常的剂量范围为约0.01-约50 mg/kg体重每天,以1-4次分开的剂量给予。每一分开的剂量可含有相同或不同的化合物。剂量将为视若干因素而定的有效量,所述因素包括患者的总体健康和所选择的化合物的制剂和给药途径。
在另一实施方案中,药物组合物为口服溶液或胃肠外溶液。另一实施方案为可在给药前重构的冻干制剂。作为固体,该制剂亦可包括片剂、胶囊剂或粉剂。
本文亦提供联合治疗,其中将一种或多种其它的治疗剂与肽蛋白酶体抑制剂或包含肽蛋白酶体抑制剂的药物组合物一起给予。可通过同时、序贯或分开给予治疗的单独组分的方式来实现所述联合治疗。
在某些实施方案中,本文提供的组合物与一种或多种其它的蛋白酶体抑制剂联合给予。
在某些实施方案中,本文所提供的组合物与化疗剂一起给予。适宜的化疗剂可包括:天然产物,例如长春花生物碱类(即长春碱(vinblastine)、长春新碱(vincristine)和长春瑞滨(vinorelbine))、紫杉醇、表鬼臼毒素(epidipodophyllotoxins)(即依托泊苷(etoposide)、替尼泊甙(teniposide))、抗生素(更生霉素(dactinomycin) (放线菌素D (actinomycin D))、柔红霉素(daunorubicin)、多柔比星(doxorubicin)和伊达比星(idarubicin))、蒽环类(anthracyclines)、米托蒽醌(mitoxantrone)、博莱霉素(bleomycin)、普卡霉素(plicamycin) (光辉霉素(mithramycin))和丝裂霉素(mitomycin)、酶(可系统代谢L-天冬酰胺和剥夺没有能力合成其自身天冬酰胺的细胞的L-天冬酰胺酶);抗血小板剂;抗增殖的/抗有丝分裂的烷化剂例如氮芥(氮芥(mechlorethamine)、环磷酰胺和类似物、美法仑(melphalan)、苯丁酸氮芥(chlorambucil))、乙烯亚胺和甲基三聚氰胺(methylmelamine )(六甲三聚氰胺和塞替派(thiotepa))、烷基磺酸盐类(白消安(busulfan))、亚硝基脲类(卡莫司汀(carmustine) (BCNU)和类似物、链脲菌素(streptozocin))、三氮烯-达卡巴嗪(trazenes-dacarbazinine)(DTIC);抗增殖的/抗有丝分裂的抗代谢物例如叶酸类似物(氨甲喋呤(methotrexate))、嘧啶类似物(氟尿嘧啶、氟尿苷和阿糖胞苷)、嘌呤类似物和相关的抑制剂(巯嘌呤、硫鸟嘌呤、喷司他丁(pentostatin)和2-氯脱氧腺苷);芳香酶抑制剂(阿那曲唑(anastrozole)、依西美坦(exemestane)和来曲唑(letrozole));和铂配位络合物(顺铂、卡铂(carboplatin))、丙卡巴肼(procarbazine)、羟基脲、米托坦(mitotane)、氨鲁米特(aminoglutethimide);组蛋白脱乙酰基酶(HDAC)抑制剂(曲古抑菌素(trichostatin)、丁酸钠、apicidan、辛二酰基苯胺异羟肟酸(suberoyl anilide hydroamic acid));激素类(即雌激素)和激素激动剂例如促黄体激素释放激素(LHRH)激动剂(戈舍瑞林(goserelin)、亮丙瑞林(leuprolide)和曲普瑞林(triptorelin))。其它的化学治疗剂可包括氮芥、喜树碱(camptothecin)、异环磷酰胺(ifosfamid)、他莫昔芬(tamoxifen)、雷洛昔芬(raloxifene)、吉西他滨(gemcitabine)、诺维本(navelbine)或前述物质的任何类似物或衍生变体。
在某些实施方案中,本文所提供的药物组合物与细胞因子一起给予。细胞因子包括但不限于干扰素-γ、-α和-β,白细胞介素1-8、10和12,粒细胞单核细胞集落刺激因子(GM-CSF),TNF-α和-β,和TGF-β。
在某些实施方案中,本文所提供的药物组合物与类固醇一起给予。适宜的类固醇包括但不限于21-乙酰氧基孕烯醇酮、阿氯米松(alclometasone)、阿尔孕酮(algeston)、安西奈德(amcinonid)、倍氯米松(beclomethasone)、倍他米松(betamethason)、布地奈德(budesonide)、氯泼尼松(chloroprednisone)、氯倍他索(clobetasol)、氯可托龙(clocortolone)、氯泼尼醇(cloprednol)、皮质酮、可的松、可的伐唑(cortivazol)、地夫可特(deflazacort)、地奈德(desonide)、去羟米松(desoximetasone)、地塞米松(dexamethasone)、二氟拉松(diflorasone)、二氟可龙(diflucortolone)、二氟泼尼酯(difuprednate)、甘草次酸(enoxolone)、氟扎可特(fluazacort)、氟氯奈德(flucloronide)、氟米松(flumethasone)、氟尼缩松(flunisolide)、氟轻松(fluocinolone acetonide)、乙酸氟轻松(fluocinonide)、氟可丁酯(fluocortin butyl)、氟可龙(fluocortolone)、氟米龙(fluorometholone)、乙酸甲氟龙(fluperolone acetate)、乙酸氟泼尼定(fluprednidene acetate)、氟泼尼龙(fluprednisolone)、氟氢缩松(flurandrenolide)、丙酸氟替卡松(fluticasone propionate)、福莫可他(formocortal)、氯氟舒松(halcinonide)、丙酸卤倍他索(halobetasol propionate)、卤米松(halometasone)、氢化可的松(hydrocortisone)、氯替泼诺碳酸乙酯(loteprednol etabonate)、马泼尼酮(mazipredone)、甲羟松(medrysone)、甲泼尼松(meprednisone)、甲基强的松龙(methylprednisolone)、糠酸莫米松(mometasone furoate)、帕拉米松(paramethasone)、泼尼卡酯(prednicarbate)、泼尼松龙(prednisolone)、25-二乙氨基醋酸泼尼松龙(prednisolone 25-diethylaminoacetate)、泼尼松龙磷酸钠(prednisolone sodium phosphate)、泼尼松(prednisone)、强的松龙戊酸酯(prednival)、泼尼立定(prednylidene)、利美索龙(rimexolone)、替可的松(tixocortol)、曲安西龙(triamcinolone)、曲安奈德(triamcinolone acetonide)、苯曲安奈德(triamcinolone benetonide)、己曲安奈德(triamcinolone hexacetonide)及其盐和/或衍生物。
在一些实施方案中,本文所提供的药物组合物与免疫治疗剂一起给予。适宜的免疫治疗剂可包括但不限于MDR调节剂(维拉帕米(verapamil)、伐司朴达(valspordar)、比立考达(biricodar)、塔立奎达(tariquidar)、拉立奎达(laniquidar))、环孢菌素(cyclosporine)、沙立度胺(thalidomide)和单克隆抗体。单克隆抗体可为裸露的或缀合的,例如利妥昔单抗(rituximab)、托西莫单抗(tositumomab)、阿仑珠单抗(alemtuzumab)、依帕珠单抗(epratuzumab)、替伊莫单抗(ibritumomab tiuxetan)、吉妥珠单抗奥佐米星(gemtuzumab ozogamicin)、贝伐单抗(bevacizumab)、西妥昔单抗(cetuximab)、厄洛替尼(erlotinib)和曲妥珠单抗(trastuzumab)。
实施例
实施例1. 磺丁基醚β-环糊精(SBECD)中卡非佐米-活性药用成分(CFZ-API)混悬液的制备
本实施例阐述在400 L批量大小下SBECD中CFZ-API混悬液的制备。以同等比例的组分实施更小批量大小,例如290L、90 L和1-3 L批量大小。
在控制为2℃–8℃的525 L不锈钢夹套冷却槽中,制备含2.0 kg卡非佐米-API (CFZ-API)、246 kg注射用水(WFI)和100 kg磺丁基醚β-环糊精(SBECD)的混悬液。具体地,在控制为2℃–8℃的525 L不锈钢夹套冷却槽中,将100 kg SBECD溶解于246 kg WFI中。然后用2.0 kg的CFZ-API来制备卡非佐米悬液。使用叶轮混合器实施混合以维持CFZ-API固体的悬浮并溶解SBECD。在同一容器中,使用探针式转子-定子高剪切混合器(均质器)以及低剪切叶轮。开动高剪切混合器大约1个小时,产生均匀的混悬液并减小任何较大的初级粒子或聚结的API的粒度。获得混悬液后,将1.96 kg的柠檬酸作为16%的水溶液加入。然后降低溶液的pH以诱导CFZ-API的部分溶解,接着因SBECD的存在而致络合。用叶轮和高剪切混合器二者继续再混合24小时,获得溶解浓度大于5.1 mg/mL的CFZ-API。用0.45微米澄清过滤器过滤含有大于5.1 mg/mL的溶解的络合CFZ-API的混悬液,然后精确稀释至溶解浓度为5.0 mg/mL,用1 N氢氧化钠调节pH以获得pH 3.5。用两个连续的0.22微米无菌过滤器让溶液无菌过滤,然后装入每一小瓶12.36 mL,使每小瓶含有61.8 mg的CFZ-API。将小瓶部分塞上,置于冻干机中,用-45℃冷冻温度冷冻干燥103小时以上,初次干燥温度-15℃,第二次干燥为+30℃。将冻干小瓶完全塞紧,加盖,然后储存在产品稳定性温度2℃-8℃下多达两年直至使用。使用时,用无菌注射用水将小瓶重构以产生2 mg/mL的重构溶液以供注射,其具有pH 3.5和用于直接注射给患者的可接受张度。或者,为了进一步稀释和输注,在静脉输液袋中将重构溶液进一步稀释而不诱导沉淀。
如图1所示,基于浆液的络合作用过程导致CFZ-API的溶解作用随时间推移增加(大于5毫克每毫升,其大大高于CFZ-API的固有的小于10微克每毫升的水溶解度)。另外,所述过程更少依赖CFZ-API的物理化学性质(例如粒度、表面积、凝聚程度、多态形式等)。不像大多数药用产品或测试,该过程的溶解率(或增溶率)有效独立于API的粒度(参见例如图2),因为无论API起初具有大还是小的平均API粒度(分别为21.1微米和7.5微米),在络合作用发生的24小时时间周期内,该过程产生等同程度的增溶作用。在上述过程中进一步确定,较高浓度的SBECD增加CFZ-API的溶解度(参见图3)。最后,观测到CFZ/SBECD络合溶解性有效独立于加工或储存温度(参见例如图4,其中显示在pH 3.5下作为SBECD浓度的函数的溶解程度,在两个温度5℃和25℃下显示无明显差别)。因此,优选较低的加工温度(2℃-8℃)以使可能发生的任何热诱导的降解反应最小化。在其它的过程中,通常更高温度是增加溶解度所必须的,然而在该过程中,经由增加环糊精的浓度和/或pH而不是通过增加温度来获得较高的溶解度,这使得在该过程中热降解产物最小化。
实施例2.氯离子对卡非佐米的稳定性的影响
实施多变量统计设计来评估控制氯醇降解产物水平的因素,所述氯醇降解产物水平为加工参数和超过六个月的储存时间的函数。以实施例1所给的比例和参数来实施络合,且有下列改变:(i)在2 L批量大小下实施络合过程;(ii)装入小瓶之前溶液的最终pH因实验目的而在3.0-4.0之间变化;(iii)在一些实验中,将氯化钠掺入SBECD中以创造高氯化钠条件;(iv)在高和低氯化钠条件下经由早期终止冻干和给小瓶加塞子以创造较高的残留水含量条件,来产生加塞小瓶中的冻干终产品中的水含量。
材料。
表2. 材料
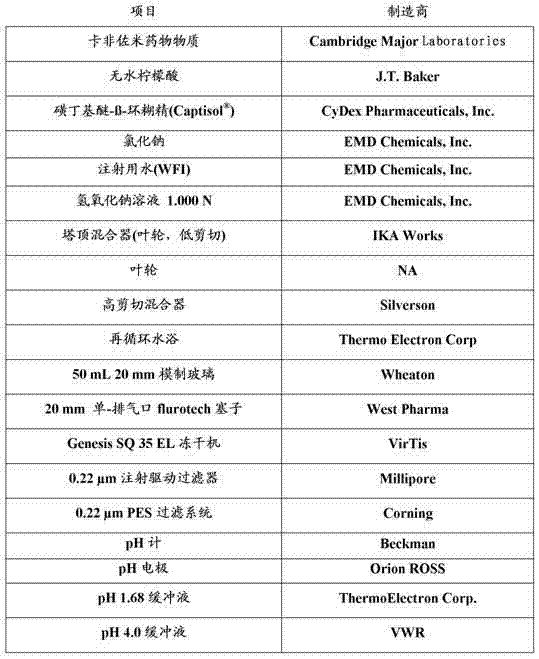
方法
络合作用过程:
用于注射原液预先冻干的络合卡非佐米溶液包括5 mg/mL水性卡非佐米、250 mg/mL Captisol? (SBECD)和4.86 mg/mL柠檬酸,用氢氧化钠水溶液调节pH。让实施例1中所详述程序后的用于冻干的原液与下列操作联合以产生具不同特定属性的溶液:
1. 将pH调节至3.0和4.0
2. 将氯化钠掺入到Captisol?中以创造“高氯化物”条件
由Cydex(Ligand的子公司)制造的Captisol?,其中氯化钠的标准产品分析范围为0.05%-0.2% (w/v)。一批Captisol?可用于实验,其具有作为氯化钠的仅0.05% (w/v)的低氯化物含量。为实施2L规模批次的络合加工(与每一实施例1相同的比例和总体参数)过程,每一批次需要400 g的这种Captisol?。为创造“高氯化物”条件,将0.6 g的NaCl加入到399.4 g的Captisol?中,从而使其模仿由含有0.2%氯化物的Captisol?组成的批次。
冻干:
为在最终的冻干小瓶中产生二(2)含水量条件,冻干二(2)组的61.8 mg/小瓶(CFZ-API)样品。第一轮每一实施例1冻干参数产生含有大约0.6%残留水的小瓶“干”样品组。对于第二样品组,在第二次冻干阶段的早期终止冻干并塞紧小瓶以产生“湿润”条件小瓶,且每一小瓶最初残留水分为大约2.4%的水。
制备一(1)批次的安慰剂用作对照,其含有250 mg/mL Captisol?和4.86 mg/mL柠檬酸,用NaOH调节至pH 3.5。
分析试验:
在制备过程中通过高效液相色谱(HPLC)分析络合的卡非佐米的原液来精确定量溶解的和络合的卡非佐米药物物质的浓度。随后将另外的水加入来精确稀释原液的络合溶液。在该稀释步骤后,再次使用HPLC来确保获得5.0 mg/mL的靶浓度。通过HPLC来分析三(3)最终原液样品的功效和纯度确认试验。通过HPLC来分析于5℃和25℃下储存六个月后的稳定性样品的功效和纯度。使用Karl Fischer比色法来测定冻干的药物产品中的水含量。
数据处理:
使用Stat-Ease DX7来分析结果。
结果
将于5℃和25℃下六个月后形成氯醇降解产物(CDP)的结果总结于下表3。
表3. 于5℃和25℃下六个月后形成的CDP的结果
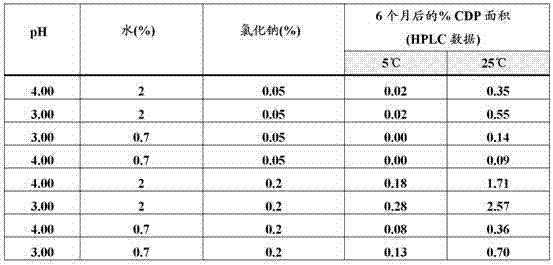
下面对CDP的ANOVA分析(表4和5)显示氯化物含量是CDP形成的主要因素。较高的氯化物含量导致更高水平的CDP。即使在低水平的氯化物含量(0.05% (w/v))下,仍可观测到氯醇的形成,但与0.2%氯化物相比,为可接受的低浓度。另外,含有低水平氯离子的药物产品显示在25℃下储存6个月后形成不可接受的氯醇产物。图5说明CDP和水及氯化物含量的二因素交互效应之间的关系。顶部的线为高氯化物含量,底部的线为低氯化物含量。x-轴代表水含量,左端为0.7%和右端为2%。在较高的氯化物水平下,CDP产物的水平增加。在较高的水含量条件下,这种增加甚至更明显,这可从顶部曲线的斜率看出。在低氯化物水平下,低和高水含量条件之间几乎没有差异。
表4. ANOVA分析 – 5℃下6个月的CDP (RRT 0.86)

表5. ANOVA分析–25℃下6个月的CDP (RRT 0.86)

实施例3. 盐酸和柠檬酸对氯醇类降解产物的影响
通过比较无HCl时所产生并在相同时间期间储存的批次的降解产物CDP随储存时间推移的杂质水平,来实施研究以测定在络合过程中使用盐酸的影响。在生产过程中,在过程结束时用氢氧化钠将所有批次的pH调节至3.5。
如表6所示,加入HCl所产生的批次(2、3和4)显示,随储存时间推移,明显形成氯醇降解产物(CDP),而当处于5℃的推荐储存温度时,在批次1和5 (其中没有使用HCl)中,CDP大部分处于HPLC的报告极限(0.1%)之下或不能检测到(ND)。很明显,来自HCl(作为用于引发络合作用的酸)的更多氯化物含量导致更多(和不可接受水平的) CDP形成。因此,单独使用较弱的酸柠檬酸来引发SBECD中的络合作用使CDP的形成最小化。
表6. 于5℃和25℃下,CDP形成的结果(%面积)

实施例4
在pH 1.5和pH 3.5和包括5℃和25℃的温度下,在含有柠檬酸(30 mM)的水溶液中测定作为SBECD环糊精浓度的函数的卡非佐米的溶解度。溶解度概况见图6。观测到在低和高受试温度之间溶解度无显著差异。实验在目标pH值之下的酸条件下,并用氢氧化钠水溶液滴定至pH 1.5或3.5。对平衡24小时时间之后的样品进行分析,测量其溶解浓度。
其它实施方案
要了解尽管本公开内容与其详述联合在一起阅读,但前述阐述意欲阐明而非限制本公开内容的范围,该范围由所附权利要求书的范围限定。其它方面、优势和改进在所附权利要求书的范围内。
Claims (55)
1.一种用于制备药物组合物的方法,所述方法包括:
(i) 提供第一组合,其包含:
(a)以下化合物或其药学上可接受的盐:
(b)一种或多种环糊精(“CD”);和
(c)水;
其中所述第一组合为异质的,并且所述化合物或盐在所述第一组合中具有低溶解度;和
(ii)让所述第一组合与酸接触以形成第二组合,其中所述化合物在所述第二组合中比在所述第一组合中更加可溶。
2.权利要求1的方法,其中所述第一组合基本上不含有机溶剂。
3.权利要求1的方法,其中所述第一组合基本上不含缓冲剂。
4.权利要求1的方法,其中所述第二组合包含所述化合物和所述一种或多种环糊精的络合物。
5.权利要求1的方法,其中将所述酸以水溶液的形式加入。
6.权利要求1的方法,其中所述一种或多种环糊精的至少一种为HPBCD或SBECD。
7.权利要求1的方法,其中所述一种或多种环糊精的至少一种为低氯化物环糊精。
8.权利要求7的方法,其中所述低氯化物环糊精为低氯化物SBECD。
9.权利要求1的方法,其中所述第一组合中氯离子与化合物的摩尔比不大于0.32。
10.权利要求1的方法,其中提供第一组合(步骤(i))包含将所述化合物加入到所述一种或多种环糊精和水的溶液中。
11.权利要求10的方法,其中所述化合物为结晶固体。
12.权利要求11的方法,其中所述化合物的晶形具有这样的X射线粉末衍射图样,其包含在6.10、9.32、10.10、12.14、13.94、18.44、20.38和23.30的2θ角处表示的2-8个特征峰。
13.权利要求1的方法,其中所述方法进一步包括混合所述第一组合,然后让所述第一组合与酸接触。
14.权利要求1的方法,其中(i)和(ii)二者均在单个容器中进行。
15.权利要求1的方法,其中方法进一步包括将所述第二组合混合足够时间以获得均质的第三组合。
16.权利要求15的方法,其中所述第三组合中化合物的溶解和络合的浓度为1 mg/mL-20 mg/mL。
17.权利要求16的方法,其中所述第三组合中化合物的溶解和络合的浓度为4-8 mg/mL。
18.权利要求15的方法,其中所述第三组合的pH为2-4。
19.权利要求15的方法,其中所述方法进一步包括将所述第三组合过滤。
20.权利要求15的方法,其中所述方法进一步包括冻干所述第三组合以提供冻干物。
21.权利要求20的方法,其中所述方法进一步包括将所述冻干物与药学上可接受的载体混合。
22.权利要求21的方法,其中所述药学上可接受的载体包含无菌注射用水。
23.权利要求22的方法,其中所述药学上可接受的载体进一步包含柠檬酸。
24.一种通过权利要求1中要求保护的方法制备的药物组合物。
25.一种通过权利要求7中要求保护的方法制备的药物组合物。
26.一种通过权利要求9中要求保护的方法制备的药物组合物。
27.一种通过权利要求15中要求保护的方法制备的药物组合物。
28.一种通过权利要求20中要求保护的方法制备的药物组合物。
29.一种通过权利要求21中要求保护的方法制备的药物组合物。
30.一种用于制备药物组合物的方法,所述方法包括:
(i)提供第一组合,其包含:
(a)以下化合物或其药学上可接受的盐:
(b) SBECD;和
(c)注射用水;
其中所述第一组合为异质的,并且所述化合物或盐在所述第一组合中具有低溶解度;和
(ii)让所述第一组合与柠檬酸的水溶液接触以形成第二组合,其中所述化合物在所述第二组合中比在所述第一组合中更加可溶。
31.权利要求30的方法,其中所述第一组合基本上不含有机溶剂。
32.权利要求30的方法,其中所述第一组合基本上不含缓冲剂。
33.权利要求30的方法,其中所述第二组合包含所述化合物和SBECD的络合物。
34.权利要求30的方法,其中所述SBECD为低氯化物SBECD。
35.权利要求30的方法,其中所述第一组合中氯离子与化合物的摩尔比不大于0.32。
36.权利要求30的方法,其中提供第一组合(步骤(i))包含将所述化合物加入到所述一种或多种环糊精和水的溶液中。
37.权利要求36的方法,其中所述化合物为结晶固体。
38.权利要求37的方法,其中所述化合物的晶形具有这样的X射线粉末衍射图样,其包含在6.10、9.32、10.10、12.14、13.94、18.44、20.38和23.30的2θ角处表示的2-8个特征峰。
39.权利要求30的方法,所述方法进一步包括混合所述第一组合,然后让所述第一组合与酸接触。
40.权利要求30的方法,其中(i)和(ii)二者均在单个容器中进行。
41.权利要求30的方法,其中方法进一步包括将第二组合混合足够时间以获得均质的第三组合。
42.权利要求41的方法,其中所述第三组合中化合物的溶解和络合的浓度为1 mg/mL-20 mg/mL。
43.权利要求42的方法,其中所述第三组合中化合物的溶解和络合的浓度为4-8 mg/mL。
44.权利要求41的方法,其中所述第三组合的pH为2-4。
45.权利要求41的方法,其中所述方法进一步包括将所述第三组合过滤。
46.权利要求41的方法,其中所述方法进一步包括冻干所述第三组合以提供冻干物。
47.权利要求46的方法,其中所述方法进一步包括让所述冻干物与药学上可接受的载体混合。
48.权利要求47的方法,其中所述药学上可接受的载体包含无菌注射用水。
49.权利要求48的方法,其中所述药学上可接受的载体进一步包含柠檬酸。
50.一种通过权利要求30中要求保护的方法制备的药物组合物。
51.一种通过权利要求34中要求保护的方法制备的药物组合物。
52.一种通过权利要求35中要求保护的方法制备的药物组合物。
53.一种通过权利要求41中要求保护的方法制备的药物组合物。
54.一种通过权利要求46中要求保护的方法制备的药物组合物。
55.一种通过权利要求47中要求保护的方法制备的药物组合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261644122P | 2012-05-08 | 2012-05-08 | |
| US61/644,122 | 2012-05-08 | ||
| PCT/US2012/055127 WO2013169282A1 (en) | 2012-05-08 | 2012-09-13 | Cylodextrin complexation methods for formulating peptide proteasome inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN103781490A true CN103781490A (zh) | 2014-05-07 |
Family
ID=49549072
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280001354.7A Pending CN103781490A (zh) | 2012-05-08 | 2012-09-13 | 用于配制肽蛋白酶体抑制剂的环糊精络合法 |
Country Status (21)
| Country | Link |
|---|---|
| US (2) | US20130303465A1 (zh) |
| JP (1) | JP2015516416A (zh) |
| KR (1) | KR20150007361A (zh) |
| CN (1) | CN103781490A (zh) |
| AR (2) | AR087863A1 (zh) |
| AU (1) | AU2012238318B2 (zh) |
| BR (1) | BR112012028726B1 (zh) |
| CA (1) | CA2793894A1 (zh) |
| CO (1) | CO6571868A2 (zh) |
| CR (1) | CR20120485A (zh) |
| CU (1) | CU20120159A7 (zh) |
| DO (1) | DOP2012000252A (zh) |
| EA (1) | EA201201519A1 (zh) |
| EC (1) | ECSP12012167A (zh) |
| MA (1) | MA35238B1 (zh) |
| MX (1) | MX2012010891A (zh) |
| MY (2) | MY196510A (zh) |
| SG (1) | SG194417A1 (zh) |
| TW (1) | TWI603737B (zh) |
| WO (1) | WO2013169282A1 (zh) |
| ZA (1) | ZA201207384B (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105919972A (zh) * | 2015-12-18 | 2016-09-07 | 重庆两江药物研发中心有限公司 | 一种包载卡非佐米的纳米粒制剂及其制备方法 |
| CN109415353A (zh) * | 2016-05-24 | 2019-03-01 | 美国安进公司 | 聚乙二醇化卡非佐米化合物 |
| CN113406183A (zh) * | 2021-06-29 | 2021-09-17 | 宁波大学 | 基于离子淌度质谱仪高效识别青霉胺手性对映体的方法 |
| CN113406183B (zh) * | 2021-06-29 | 2024-04-23 | 常州磐诺仪器有限公司 | 基于离子淌度质谱仪高效识别青霉胺手性对映体的方法 |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7635773B2 (en) | 2008-04-28 | 2009-12-22 | Cydex Pharmaceuticals, Inc. | Sulfoalkyl ether cyclodextrin compositions |
| DK2814849T3 (da) | 2012-02-15 | 2020-03-09 | Cydex Pharmaceuticals Inc | Fremgangsmåde til fremstilling af cyclodextrin-derivater |
| AR095426A1 (es) | 2013-03-14 | 2015-10-14 | Onyx Therapeutics Inc | Inhibidores tripeptídicos de la epoxicetona proteasa |
| DK2970225T3 (en) | 2013-03-14 | 2018-09-24 | Onyx Therapeutics Inc | DIPEPTIDE AND TRIPEPTIDE AS EPOXY-KETONE PROTEASE INHIBITORS |
| GB201312737D0 (en) | 2013-07-17 | 2013-08-28 | Univ Greenwich | Cyclodextrin |
| WO2015009888A2 (en) | 2013-07-19 | 2015-01-22 | Onyx Therapeutics, Inc. | Peptide epoxyketone proteasome inhibitors in combination with pim kinase inhibitors for treatment of cancers |
| CN104945470B (zh) * | 2014-03-30 | 2020-08-11 | 浙江大学 | 杂环构建的三肽环氧酮类化合物及制备和应用 |
| CN103936828A (zh) * | 2014-05-12 | 2014-07-23 | 苏州科耐尔医药科技有限公司 | 卡非佐米中间体及卡非佐米的制备方法 |
| UA125648C2 (uk) | 2016-08-05 | 2022-05-11 | Емджен Інк. | Синтез (s)-2-аміно-4-метил-1-((r)-2-метилоксиран-2-іл)-пентан-1-ону та його фармацевтично прийнятних солей |
| WO2018038687A1 (en) | 2016-08-22 | 2018-03-01 | Mustafa Nevzat Ilaç Sanayii A.Ş. | Pharmaceutical formulations comprising a bortezomib-cyclodextrin complex |
| US20180282260A1 (en) * | 2017-03-31 | 2018-10-04 | Valent Biosciences Llc | 1-aminocyclopropane-1-carboxylic acid polymorphs |
| US10975121B2 (en) | 2017-06-24 | 2021-04-13 | Cytogel Pharma, Llc | Analgesic mu-opioid receptor binding peptide pharmaceutical formulations and uses thereof |
| CN111344018A (zh) | 2017-11-16 | 2020-06-26 | 美国安进公司 | 聚乙二醇化卡非佐米化合物的稳定的组合物 |
| EP3716948B1 (en) * | 2017-11-30 | 2023-11-22 | Cytogel Pharma, LLC | Novel analgesic pharmaceutical formulations and uses thereof |
| AU2021292417A1 (en) * | 2020-06-19 | 2023-01-19 | Amgen Inc. | Methods of measuring carfilzomib |
| US11246874B1 (en) | 2021-04-20 | 2022-02-15 | Oxygen Biotech LLC | Treatment of COVID-19 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE499109T1 (de) * | 2004-12-07 | 2011-03-15 | Proteolix Inc | Zusammensetzung zur proteasomhemmung |
| KR101274417B1 (ko) * | 2005-11-09 | 2013-06-17 | 프로테올릭스, 인코퍼레이티드 | 효소 저해를 위한 화합물 |
| MY173938A (en) * | 2007-10-04 | 2020-02-27 | Onyx Therapeutics Inc | Crystalline peptide epoxy ketone protease inhibitors and the synthesis of amino acid keto-epoxides |
-
2012
- 2012-09-13 BR BR112012028726-5A patent/BR112012028726B1/pt active IP Right Grant
- 2012-09-13 TW TW101133449A patent/TWI603737B/zh active
- 2012-09-13 JP JP2015511431A patent/JP2015516416A/ja active Pending
- 2012-09-13 EA EA201201519A patent/EA201201519A1/ru unknown
- 2012-09-13 MX MX2012010891A patent/MX2012010891A/es not_active Application Discontinuation
- 2012-09-13 MY MYPI2017001061A patent/MY196510A/en unknown
- 2012-09-13 AR ARP120103377A patent/AR087863A1/es not_active Application Discontinuation
- 2012-09-13 WO PCT/US2012/055127 patent/WO2013169282A1/en active Application Filing
- 2012-09-13 KR KR1020127028681A patent/KR20150007361A/ko not_active Application Discontinuation
- 2012-09-13 MY MYPI2012005407A patent/MY165002A/en unknown
- 2012-09-13 CA CA2793894A patent/CA2793894A1/en not_active Abandoned
- 2012-09-13 SG SG2012084075A patent/SG194417A1/en unknown
- 2012-09-13 US US13/614,599 patent/US20130303465A1/en not_active Abandoned
- 2012-09-13 AU AU2012238318A patent/AU2012238318B2/en active Active
- 2012-09-13 CN CN201280001354.7A patent/CN103781490A/zh active Pending
- 2012-09-13 US US13/614,829 patent/US20130303482A1/en not_active Abandoned
- 2012-09-26 CR CR20120485A patent/CR20120485A/es unknown
- 2012-09-26 EC ECSP12012167 patent/ECSP12012167A/es unknown
- 2012-09-27 DO DO2012000252A patent/DOP2012000252A/es unknown
- 2012-09-28 CO CO12170407A patent/CO6571868A2/es not_active Application Discontinuation
- 2012-10-02 ZA ZA2012/07384A patent/ZA201207384B/en unknown
- 2012-11-12 CU CU2012000159A patent/CU20120159A7/es unknown
-
2013
- 2013-12-04 MA MA36520A patent/MA35238B1/fr unknown
-
2022
- 2022-12-02 AR ARP220103323A patent/AR127861A2/es unknown
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105919972A (zh) * | 2015-12-18 | 2016-09-07 | 重庆两江药物研发中心有限公司 | 一种包载卡非佐米的纳米粒制剂及其制备方法 |
| CN109415353A (zh) * | 2016-05-24 | 2019-03-01 | 美国安进公司 | 聚乙二醇化卡非佐米化合物 |
| CN109415353B (zh) * | 2016-05-24 | 2023-01-10 | 美国安进公司 | 聚乙二醇化卡非佐米化合物 |
| CN113406183A (zh) * | 2021-06-29 | 2021-09-17 | 宁波大学 | 基于离子淌度质谱仪高效识别青霉胺手性对映体的方法 |
| CN113406183B (zh) * | 2021-06-29 | 2024-04-23 | 常州磐诺仪器有限公司 | 基于离子淌度质谱仪高效识别青霉胺手性对映体的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| SG194417A1 (en) | 2013-12-30 |
| MA35238B1 (fr) | 2014-07-03 |
| EA201201519A1 (ru) | 2013-11-29 |
| AR087863A1 (es) | 2014-04-23 |
| AU2012238318A1 (en) | 2013-11-28 |
| AR127861A2 (es) | 2024-03-06 |
| TW201345543A (zh) | 2013-11-16 |
| ECSP12012167A (es) | 2013-02-28 |
| CO6571868A2 (es) | 2012-11-30 |
| US20130303465A1 (en) | 2013-11-14 |
| MY196510A (en) | 2023-04-18 |
| BR112012028726A2 (pt) | 2016-07-19 |
| MY165002A (en) | 2018-02-28 |
| BR112012028726B1 (pt) | 2021-07-13 |
| ZA201207384B (en) | 2018-12-19 |
| WO2013169282A1 (en) | 2013-11-14 |
| US20130303482A1 (en) | 2013-11-14 |
| CR20120485A (es) | 2013-12-18 |
| NZ602490A (en) | 2016-03-31 |
| KR20150007361A (ko) | 2015-01-21 |
| CU20120159A7 (es) | 2014-03-26 |
| MX2012010891A (es) | 2014-03-05 |
| DOP2012000252A (es) | 2013-12-31 |
| JP2015516416A (ja) | 2015-06-11 |
| AU2012238318B2 (en) | 2014-02-13 |
| TWI603737B (zh) | 2017-11-01 |
| CA2793894A1 (en) | 2013-11-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103781490A (zh) | 用于配制肽蛋白酶体抑制剂的环糊精络合法 | |
| CN104411334A (zh) | 用于配制肽蛋白酶体抑制剂的环糊精络合法 | |
| AU2018201445B2 (en) | Liposomal compositions of epoxyketone-based proteasome inhibitors | |
| EP4087536A1 (en) | Stable cyclodextrin free carfilzomib formulation | |
| JP2023510258A (ja) | 安定なシクロデキストリン不含カルフィルゾミブ処方物 | |
| AU2013204448A1 (en) | Cylodextrin complexation methods for formulating peptide proteasome inhibitors | |
| NZ602490B2 (en) | Cylodextrin complexation methods for formulating peptide proteasome inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1197579 Country of ref document: HK |
|
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20140507 |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: WD Ref document number: 1197579 Country of ref document: HK |