CN103118710A - 基于羊毛硫抗生素肽的凋亡成像剂 - Google Patents
基于羊毛硫抗生素肽的凋亡成像剂 Download PDFInfo
- Publication number
- CN103118710A CN103118710A CN2011800462575A CN201180046257A CN103118710A CN 103118710 A CN103118710 A CN 103118710A CN 2011800462575 A CN2011800462575 A CN 2011800462575A CN 201180046257 A CN201180046257 A CN 201180046257A CN 103118710 A CN103118710 A CN 103118710A
- Authority
- CN
- China
- Prior art keywords
- preparation
- chelating agen
- group
- conjugate
- lbp
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/088—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins conjugates with carriers being peptides, polyamino acids or proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/04—X-ray contrast preparations
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Optics & Photonics (AREA)
- Pharmacology & Pharmacy (AREA)
- Physics & Mathematics (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Peptides Or Proteins (AREA)
Abstract
本发明涉及凋亡的放射性药物体内成像。本发明提供经由与在凋亡细胞的表面上暴露的氨基磷脂磷脂酰乙醇胺(PE)选择性结合,靶向凋亡细胞的成像剂。所述放射性药物包含PE-结合肽的螯合剂缀合物的放射性金属络合物。还提供了药物组合物、试剂盒和体内成像的方法。
Description
发明领域
本发明涉及凋亡和其它形式的细胞死亡的放射性药物体内成像。本发明提供经由与在凋亡细胞的表面上暴露的氨基磷脂磷脂酰乙醇胺(PE)选择性结合,靶向凋亡细胞的成像剂。还提供了药物组合物、试剂盒和体内成像的方法。
发明背景
凋亡或程序性细胞死亡(PCD)为最普遍的细胞死亡路径,并且经由高度调节的、能量守恒的机制进行。在健康状态下,凋亡在控制细胞生长、调节细胞数量、促进形态发生和除去有害或异常细胞中起关键作用。PCD过程的失调牵涉多种疾病状态,包括与抑制凋亡相关的那些,例如癌症和自身免疫病症,以及与活动过强凋亡相关的那些,包括神经变性疾病、血液疾病、AIDS,局部缺血和同种异体移植排斥。因此,凋亡的显影和定量可用于诊断这样的与凋亡相关的病理生理学。
对于这些疾病治疗性处理目的是通过适当地刺激或抑制PCD过程来恢复平衡的凋亡。因此,在细胞和组织中,凋亡的体内非侵入性成像对于治疗性介入响应的早期评价具有巨大价值,并且对于破坏性病理过程可提供新的深刻了解。特别感兴趣的是早期监测癌症治疗的功效,以确保在病况变为晚期之前控制恶性生长。
因此特别感兴趣开发用于凋亡的成像剂[参见例如Zeng等人,Anti-cancer Agent Med.Chem.,9(9),986-995 (2009);Zhao,ibid,9(9),1018-1023 (2009)和M. De Saint-Hubert等人,Methods,48,178-187 (2009)]。在可用于对细胞死亡成像的探针中,放射性标记的膜联蛋白V最受关注。膜联蛋白V仅与带负电荷的磷脂结合,这使其不能区分凋亡与坏死。
含有羊毛硫氨酸的抗生素肽(“羊毛硫抗生素”)耐久霉素和肉桂霉素为两种具有紧凑四环结构的密切相关的19-聚体肽[Zhao,Amino Acids,DOI 10.1007/s00726-009-0386-9,Springer-Verlag (2009),以及其中引用的参考文献]。它们经由四个共价、分子内桥交联,并且区别仅在于在2位的单一氨基酸残基。耐久霉素和肉桂霉素的结构在以下示意性显示,其中编号是指在19-聚体序列中连接的氨基酸残基的位置:

程序性细胞死亡或凋亡为细胞的胞内、能量-依赖性自身毁灭。磷脂跨过细胞质膜的双层的再分布是凋亡的重要标记。因此,在活细胞中,氨基磷脂磷脂酰乙醇胺(PE或PtdE)和磷脂酰丝氨酸(PS)为细胞质膜的内部小叶的主要组分。在凋亡的细胞中,存在PE和PS的同步表面化。
通过形成在PE头部基团周围相适应的疏水性袋,耐久霉素和肉桂霉素二者均以类似的特异性和高亲和力与中性氨基磷脂PE结合。通过β-羟基天冬氨酸残基(HO-Asp15)与乙醇胺基之间的离子相互作用,使结合稳定。已知对该残基的修饰使耐久霉素失活[Zhao等人,J.Nucl.Med,49,1345-1352 (2008)]。Zhao [Amino Acids,DOI 10.1007/s00726-009-0386-9,Springer-Verlag (2009)]引用Wakamatsu等人[Biochemistry,29,113-188 (1990)]的早期研究,其中NMR研究显示,在与PE结合时,肉桂霉素的5个末端氨基酸的1H NMR共振无一偏移,这表明它们不涉及与PE的相互作用。
US 2004/0147440 A1 (University of Texas System)描述了标记的抗-氨基磷脂抗体,其可用于检测预先-凋亡的或凋亡的细胞,或者用于癌症成像。还提供了耐久霉素与用于癌症治疗的生物素、蛋白质或抗病毒药物的缀合物。
WO 2006/055855公开了使用放射性标记的化合物对凋亡成像的方法,所述放射性标记的化合物包含蛋白质的磷脂酰丝氨酸-结合C2结构域。
WO 2009/114549公开了通过以下方法制备的放射性药物,所述方法包括:
(i) 提供与CKQSCSFGPFTFVCDGNTK具有至少70%序列相似性的多肽,
其中多肽在氨基酸残基1-18、4-14和5-11之间包含硫醚键以及在氨基酸残基6-19之间包含酰胺键,并且其中一个或多个具有以下结构的末端部分

在多肽的1位、2位或者1位和2位处与氨基酸共价结合,并且其中R1和R2各自独立地为直链或支链、饱和或不饱和的C1-4烷基;和
(II) 将一个或多个末端部分与99mTcx、(99mTc=O)3+、(99mTc≡N)2+、(O=99mTc=O)+或[99mTc(CO)3]+或其盐、溶剂合物或水合物螯合,其中x为选自+7、+6、+5、+4、+3、+2、+1、0和-1的氧化还原或氧化态。
WO 2009/114549的‘末端部分’为放射性同位素99mTc的络合剂,其基于肼基烟酰胺(通常简称为“HYNIC”)。HYNIC在文献[参见例如Banerjee等人,Nucl.Med.Biol,32,1-20 (2005)]中众所周知,并且为使用99mTc标记肽和蛋白质的优选方法[R.Alberto,第2章,第19-40页,IAEA Radioisotopes and Radiopharmaceuticals Series 1: “Technetium-99m Radiopharmaceuticals Status and Trends”(IAEA放射性同位素和放射性药物系列1:“锝-99m放射性药物状况和趋势”)(2009)]。
WO 2009/114549特别公开了99mTc-HYNIC-耐久霉素,并且提出其中教导的放射性药物可用于对凋亡和/或坏死、动脉粥样硬化斑或急性心肌梗死成像。
Zhao等人[J.Nucl.Med,49,1345-1352 (2008)]公开了99mTc-HYNIC-耐久霉素的制备。Zhao等人注意到耐久霉素具有2个可用于与HYNIC缀合的胺基:在N-末端(Cys1残基)和Lys2残基的ε-胺侧链。它们通过HPLC纯化HYNIC-耐久霉素缀合物,以除去双-HYNIC-官能化的耐久霉素,然后用99mTc进行放射性标记。Zhao等人承认99mTc-标记的单-HYNIC-耐久霉素缀合物研究可能是异构体的混合物形式。
虽然HYNIC形成稳定的99mTc络合物,但是其需要另外的共同-配体以完成锝金属络合物的配位层。HYNIC可用作单齿配体或二齿螯合剂,这取决于在附近的氨基酸侧链官能团的性质[King等人,Dalton Trans.,4998-5007 (2007);Meszaros等人[Inorg.Chim.Acta,363,1059-1069 (2010)]。因此,取决于环境,HYNIC形成具有1个或2个金属供体原子的金属络合物。Meszaros等人注意到与HYNIC一起使用的共同-配体的性质可对系统的行为具有显著作用,并且陈述没有一个共同-配体是理想的。
发明内容
本发明提供放射性药物成像剂,特别是用于对其中涉及异常凋亡的哺乳动物身体的疾病状态成像。所述成像剂包含羊毛硫抗生素肽的放射性金属螯合剂缀合物。
本发明提供放射性金属络合物,其以高放射性化学纯度(RCP)可再现地形成,而无需共同-配体。本发明人还已证实在本文式II的羊毛硫抗生素肽的N-末端(Cysa残基)处连接放射性金属络合物是强烈优选的,这是由于在甚至与N-末端(式II的Xaa)相邻的氨基酸处连接未络合的螯合剂对于与磷脂酰乙醇胺结合具有有害作用。该作用先前在现有技术中未认识到,因此,认为对结合亲和力的影响程度是新的。
发明详述
在第一方面,本发明提供了一种包含式I化合物的成像剂:
Z1-(L)n-[LBP]-Z2
(I)
其中:
LBP为式II的羊毛硫抗生素肽:
Xaa为Arg或Lys;
Cysa-Thra、Serb-Cysb和Cysc-Thrc经由硫醚键共价连接;
Serd-Lysd经由赖氨酸丙氨酸键共价连接;
HO-Asp为β-羟基天冬氨酸;
Z1-(L)n-与Cysa连接并且还任选与LBP的Xaa连接,其中Z1包含具有至少4个金属供体原子的螯合剂的放射性金属络合物;
Z2与LBP的C-末端连接,并且为OH、OBc或MIG,
其中Bc为生物相容的阳离子;和
MIG为代谢抑制基团,其为抑制或遏抑LBP肽的体内代谢的生物相容的基团;
L为式-(A)m-的合成的接头基团,其中每个A独立地为-CR2-、-CR=CR-、-C≡C-、-CR2CO2-、-CO2CR2-、-NRCO-、-CONR-、-NR(C=O)NR-、-NR(C=S)NR-、-SO2NR-、-NRSO2-、-CR2OCR2-、-CR2SCR2-、-CR2NRCR2-、C4-8环杂亚烷基、C4-8环亚烷基、C5-12亚芳基或C3-12杂亚芳基、氨基酸、糖或单分散聚乙二醇(PEG)结构单元;
每个R独立地选自H、C1-4烷基、C2-4烯基、C2-4炔基、C1-4烷氧基烷基或C1-4羟基烷基;
m为数值1-20的整数;
n为数值0或1的整数。
术语“成像剂”是指适用于对哺乳动物身体成像的化合物。优选,哺乳动物为体内完整的哺乳动物身体,并且更优选为人受试者。优选,可将成像剂以最小侵入性方式给予哺乳动物身体,即,当在专业医疗专家下进行时,对哺乳动物受试者不会有显著健康风险。这样的最小侵入性给药优选向所述受试者的外周静脉静脉内给药,无需局部或全身麻醉。第一方面的成像剂特别适用于对凋亡和其它形式的细胞死亡成像,如在(以下)第六方面中所描述。
本文使用的术语“体内成像”是指非侵入性产生哺乳动物受试者的全部或部分内部方面的图像的那些技术。
术语“氨基酸”是指L-或D-氨基酸、氨基酸类似物(例如,萘基丙氨酸)或氨基酸模拟物,其可为天然存在的或纯粹合成起源,并且可为光学纯的,即,单一的对映异构体,因此为手性的,或者为对映异构体的混合物。本文中使用氨基酸的常规3-字母或单一字母缩写。优选本发明的氨基酸为光学纯的。
术语“肽”是指包含两个或更多个通过肽键(即,连接一个氨基酸的胺与另一个氨基酸的羧基的酰胺键)连接的如上定义的氨基酸的化合物。
术语“羊毛硫抗生素肽”是指含有至少一个羊毛硫氨酸键的肽。“羊毛硫氨酸”具有其常规的含义,并且是指具有所示化学结构的胱氨酸的硫化物类似物:
术语“经由硫醚键共价连接”是指相关的Cys残基的硫醇官能团作为硫醚键经由Ser或Thr残基的羟基官能团脱水与所示的Ser或Thr残基连接,以得到羊毛硫氨酸或甲基羊毛硫氨酸键。这样的键由Willey等人[Ann.Rev.Microbiol.,61,477-501 (2007)]描述。
术语“赖氨酸丙氨酸键”是指Lys残基的ε胺基作为胺键经由Ser的羟基官能团脱水与所示的Ser残基连接,得到连接氨基酸残基的两个α-碳原子的-(CH2)-NH-(CH2)4-键。
术语“放射性金属络合物”是指放射性金属与螯合剂的配位金属络合物,其中所述螯合剂经由式I的接头基团(L)与LBP肽共价键合。配位络合物不包含与放射性金属结合的肼基烟酰胺(HYNIC)配体。因此,螯合剂为与放射性金属结合的主要物类,其不仅仅是HYNIC的共同-配体。
术语“螯合剂”具有其常规的含义,并且是指两个或更多个金属供体原子,其排列使得当金属配位时,得到螯合环,优选5-元至7-元螯合环,更优选5-元或6-元螯合环。金属供体原子通过碳原子或非配位杂原子的非配位骨架而共价连接。螯合剂可为大环或开链。本发明的螯合剂包含至少4个金属供体原子,适宜地4-8个金属供体原子,其中至少4个这样的金属供体原子与放射性金属络合物中的放射性金属结合。
本发明的合适放射性金属包括:99mTc、94mTc、186Re、188Re、64Cu、67Cu、67Ga、68Ga、105Rh、101mRh、111In、89Zr或45Ti。
当Z1与Cysa连接时,其与LBP肽的N-末端连接。当Z1也与Xaa连接时,这意味着Xaa为Lys,并且Z1与Lys残基的ε氨基连接。
Z2基团取代LBP的最后的氨基酸残基的羰基,即,羧基末端。因此,当Z2为OH时,LBP的羧基末端终止于最后的氨基酸残基的游离CO2H基团,并且当Z2为OBc时,该末端羧基电离作为CO2Bc基团。
术语“生物相容的阳离子”(Bc)是指与电离的、带负电荷的基团形成盐的带正电荷的反荷离子,其中所述带正电荷的反荷离子也是无毒的,因此适于给予哺乳动物身体,尤其是人体。合适的生物相容的阳离子的实例包括:碱金属钠或钾;碱土金属钙和镁;和铵离子。优选的生物相容的阳离子为钠和钾,最优选钠。
术语“代谢抑制基团”(MIG)是指在羧基末端(Z2)抑制或遏抑LBP肽的体内代谢的生物相容的基团。这样的基团为本领域技术人员众所周知的,并且适宜地选自:甲酰胺、叔丁酯、苄酯、环己酯、氨基醇或聚乙二醇(PEG)结构单元。已知本发明的LBP肽呈现高体内代谢稳定性(在60分钟时,95%),因此,Z2优选为OH或OBc。
优选的实施方案。
螯合剂优选经设计使得在与放射性金属络合时形成的螯合环包含至少一个5-元或6-元环,更优选2-4个这样的环,最优选3或4个这样的环。
优选螯合剂选自:具有至少6个供体原子的氨基羧化物(aminocarboxylate)配体;或具有N3S、N2S2或N4供体组(donor set)的四齿螯合剂。更优选螯合剂为具有至少6个供体原子的氨基羧化物配体或具有N4供体组的四齿螯合剂,最优选具有N4供体组的四齿螯合剂。
术语“氨基羧化物配体”具有其常规的含义,并且是指EDTA、DTPA类型的螯合剂。这样的螯合剂的供体原子为胺(N)供体和羧酸(O)供体的混合物。这样的螯合剂可为开链(例如,EDTA、DTPA或HBED)或大环(例如,DOTA或NOTA)。合适的这样的螯合剂包括DOTA、HBED和NOTA,其为本领域众所周知的,并且对于放射性金属,例如67Ga或68Ga、111In、铜的放射性同位素、89Zr和45Ti是优选的。
术语“四齿螯合剂”具有其常规的含义,并且是指其中放射性金属被四齿螯合剂的四个金属供体原子配位的螯合剂。
术语“N3S供体组”是指四齿螯合剂的四个金属供体原子由三个氮供体原子和一个硫供体原子组成。合适的这样的N供体原子类型的实例为:胺(尤其是伯或仲胺);酰胺或肟,或它们的组合。合适的这样的S供体原子类型的实例为:硫醇和硫醚。优选的这样的N3S螯合剂具有硫醇三酰胺供体组,并且优选为开链螯合剂,例如MAG3 (巯基乙酰基三甘氨酸)。
术语“N2S2供体组”是指四齿螯合剂的四个金属供体原子由2个氮供体原子和2个硫供体原子组成。合适的N和S供体原子如对于(以上)N3S所描述。优选的这样的N2S2螯合剂具有二胺二硫醇或酰胺胺二硫醇供体组,并且优选为开链螯合剂,例如BAT或N,N'-亚乙基二-L-半胱氨酸[Inorg Chem.,35(2):404-414 (1996)]。
术语“N4供体组”是指四齿螯合剂的四个金属供体原子均基于氮。合适的这样的N供体原子类型的实例为:胺(尤其是伯或仲胺);酰胺或肟,或它们的组合。
优选N4供体组选自:二胺二肟;四胺;酰胺三胺或二酰胺二胺。N4螯合剂可为开链或大环(例如,1,4,8,11-四氮杂环十四烷(cyclam)、1,4,7,10-四氮杂环十二烷(cyclen)、monoxocyclam或dioxocyclam)。优选的本发明的N4四齿螯合剂具有二胺二肟或四胺供体组,更优选开链二胺二肟或开链四胺。
优选的二胺二肟螯合剂具有下式:

其中E1-E6各自独立地为R′基团;
每个R′独立地为H或C1-10烷基、C3-10烷基芳基、C2-10烷氧基烷基、C1-10羟基烷基、C1-10氟烷基、C2-10羧基烷基或C1-10氨基烷基,或者两个或更多个R′基团与它们连接的原子共同形成碳环、杂环、饱和或不饱和环;
并且Q为式-(J)f-的桥接基团;
其中f为3、4或5,并且每个J独立地为-O-、-NR′-或-C(R′)2-,条件是-(J)f-可最多含有一个J基团,其为-O-或-NR′-。
优选的Q基团如下:
Q=-(CH2)(CHR′)(CH2)-,即,丙胺肟(propyleneamine oxime)或PnAO衍生物;
Q=-(CH2)2(CHR′)(CH2)2-,即,戊胺肟(pentyleneamine oxime)或PentAO衍生物;
Q=-(CH2)2NR′(CH2)2-。
E1-E6优选选自:C1-3烷基、C2-4烷氧基烷基、C1-3羟基烷基、C1-3氟烷基、C2-6羧基烷基或C1-3氨基烷基。最优选,每个E1-E6基团为CH3。
Q优选为-(CH2)(CHR′)(CH2)-、-(CH2)2(CHR′)(CH2)2-或-(CH2)2NR′(CH2)2-,最优选-(CH2)2(CHR′)(CH2)2-。尤其优选的二胺二肟螯合剂具有下式:

其中桥头伯胺基与(L)n (即,接头基团)和/或LBP肽缀合。
优选的四胺螯合剂具有下式:

其中桥头羧基与接头基团和/或LBP肽缀合。
在螯合剂2中,[接头]优选为式(A′)m1的基团,其中m1为数值0-6的整数,并且每个A′独立地为CH2或对亚苯基,其中不多于一个A′基团为对亚苯基。优选,每个A′基团为CH2,并且m1为1-6。优选的这样的螯合剂为螯合剂2A,其中[接头]为-(CH2)-。
成像剂的放射性金属优选为94mTc或99mTc,更优选99mTc。对于这些锝放射性同位素,螯合剂优选为具有上文定义的N4供体组的四齿。
Z2优选为OH或OBc。
在第一方面的成像剂中,Z1优选仅与LBP的Cysa连接。当Xaa为Arg时,这意味着在Cysa残基的游离氨基处,Z1与LBP的N-末端连接。当Xaa为Lys时,这意味着采取步骤以:
(i) 优先于Xaa残基的ε胺基,在Cysa残基处选择性官能化LBP肽;或者
(ii) 制备包含在Cysa和Xaa两处被Z1官能化的LBP的组合物,随后将Xaa-官能化的物类除去。
在第一方面的成像剂中,Xaa优选为Arg。
第一方面的成像剂优选包含接头基团(L),即,在式(I)中n优选为1。优选L包含式-(OCH2CH2)x-的PEG基团,其中x为数值6-18的整数,优选8-14,更优选10-12。这样的接头基团在降低肝背景保留和提高成像剂的体内尿排泄方面是有利的。优选,L包含式IA或IB的生物修饰剂基团:
式IA的17-氨基-5-氧代-6-氮杂-3,9,12,15-四氧杂十七烷酸
其中p为1-10的整数。在式IA中,p优选为1、2或3。或者,可使用基于式IB的丙酸衍生物的PEG-样结构:

其中p如对于式IA所定义,并且q为3-15的整数。
在式IB中,p优选为1或2,更优选为1,并且q优选为5-12,更优选为12。
术语“生物修饰剂”是指对于试剂的体内生物分布具有影响的基团。
第一方面的成像剂可如在第三方面中所描述得到。
在第二方面,本发明提供了一种式III的螯合剂缀合物:

其中:
Z3为具有至少4个金属供体原子的螯合剂;和
L、n、LBP和Z2如在第一方面中所定义。
在第二方面中,L、n、LBP和Z2和螯合剂(Z3)的优选方面如在(以上)第一方面中所定义。
某些LBP肽市售可得。因此,肉桂霉素和耐久霉素可得自Sigma-Aldrich。耐久霉素由菌株产生:D3168耐久霉素得自肉桂链轮丝菌(Streptoverticillium cinnamoneus)。肉桂霉素可由若干菌株通过生物化学方法产生,例如,得自肉桂链霉菌(Strptomyces cinnamoneus)或得自灰轮丝链轮丝菌(Streptoverticillium griseoverticillatum)。参见C. Chatterjee等人的综述[Chem. Rev.,105,633-683 (2005)]。其它肽可通过固相肽合成得到,如在P. Lloyd-Williams,F. Albericio和E. Girald;Chemical Approaches to the Synthesis of Peptides and Proteins (合成肽和蛋白质的化学方法),CRC Press,1997中描述的。
第二方面的螯合剂缀合物可如下得到。当螯合剂为二胺二肟时,通过适当的二胺与以下之一反应:
(i) 适当的氯亚硝基衍生物Cl-C(R1)2-CH(NO)R1;
(ii) 式Cl-C(R1)2-C(=NOH)R1的α-氯肟;
(iii) 式Br-C(R1)2-C(=O)R1的α-溴酮,接着使用羟基胺将二胺二酮产物转化为二胺二肟。
路线(i)由S. Jurisson等人[Inorg. Chem.,26,3576-82 (1987)]描述。通过用亚硝酰氯(NOCl)处理适当的烯烃,可得到氯亚硝基化合物,如本领域已知的。氯亚硝基化合物的其它合成细节由以下给出:Ramalingam [Synth. Commun.,25(5),743-752 (1995)];Glaser [J. Org. Chem.,61(3),1047-48 (1996)];Clapp [J. Org Chem.,36(8) 1169-70 (1971)];Saito [Shizen Kagaku,47,41-49 (1995)]和Schulz [Z. Chem.,21(11),404-405 (1981)]。路线(iii)由Nowotnik等人[Tetrahedron,50(29),第8617-8632页(1994)]概括性地描述。α-氯-肟可通过市售可得的相应的α-氯-酮或醛的肟化得到。α-溴酮市售可得。
更优选的四胺螯合剂具有下式:

其中:
L、LBP、n和Z2如在第一方面中所定义;
Q1-Q6独立地为Q基团,其中Q为H或胺保护基。
术语“保护基”是指这样的基团,其抑制或遏抑不期望的化学反应,但是经设计具有足够的反应性,使其可在不改变分子的其余部分的足够温和的条件下从目标官能团裂解。在脱保护后,得到期望的产物。胺保护基为本领域技术人员众所周知的,并且适宜地选自:Boc (其中Boc为叔丁氧基羰基)、Fmoc (其中Fmoc为芴基甲氧基羰基)、三氟乙酰基、烯丙氧基羰基、Dde [即,1-(4,4-二甲基-2,6-二氧代环亚己基)乙基]或Npys (即,3-硝基-2-吡啶氧硫基(3-nitro-2-pyridine sulfenyl))。在一些情况下,保护基的性质可使得Q1/Q2或Q5/Q6基团二者,即,在关联的胺氮原子上不存在NH键。其他保护基的使用描述于‘Protective Groups in Organic Synthesis(有机合成中的保护基)’,第4版,Theorodora W. Greene和Peter G. M. Wuts,[Wiley Blackwell,(2006)]。优选的胺保护基为Boc和Fmoc,最优选Boc。当使用Boc时,Q1和Q6均为H,并且Q2、Q3、Q4和Q5各自为叔丁氧基羰基。
在螯合剂3中,L、LBP、n和Z2的优选方面如在(以上)第一方面中所定义。优选的螯合剂3螯合剂具有(L)n=(A')m1,其中A'和m1及其优选方面如对于(以上)螯合剂2所描述。
四胺螯合剂可如(以下)流程1中描述的而得到。关于氨基-和羧基-官能化的四胺螯合剂的其它合成信息由Abiraj等人[Chem.Eur.J.,16,2115-2124 (2010)]提供。用-(CH2)5OH桥头取代基合成Boc-保护的四胺类似物由Turpin等人[J.Lab.Comp.Radiopharm.,45,379-393 (2002)]描述。四胺螯合剂与生物学靶向肽的缀合由以下描述:Nock等人[Eur.J.Nucl.Med.,30(2),247-258 (2003)]以及Maina等人[Eur.J.Nucl.Med.,30(9),1211-1219 (2003)]。具有侧基活性酯基的双官能HBED衍生物由Eder等人[Eur.J.Nucl.Med.Mol.Imaging,35,1878-1886 (2008)]教导。
流程1:Boc-保护的螯合剂2A的合成。
N3S双官能螯合剂可通过Sudhaker等人[Bioconj. Chem.,第9卷,108-117(1998)]的方法制备。N2S2二酰胺二硫醇化合物可通过Kung等人[Tetr. Lett.,第30卷,4069-4072 (1989]的方法制备。
单酰胺单胺双硫醇化合物可通过Hansen等人[Inorg. Chem.,第38卷,5351-5358 (1999)]的方法制备。
在第三方面,本发明提供了制备第一方面的成像剂的方法,所述方法包括使第二方面的螯合剂缀合物与期望的放射性金属的供应在合适的溶剂中反应。
在第三方面中,螯合剂缀合物和放射性金属的优选方面如在(以上)本发明的第一和第二方面中所描述。
合适的溶剂通常为水性的,并且优选为在(以下)第四方面中定义的生物相容的载体溶剂。
在第四方面,本发明提供了放射性药物组合物,所述组合物包含第一方面的成像剂以及生物相容的载体,所述组合物呈适用于哺乳动物给药的形式。
在第四方面中,成像剂的优选方面为在(以上)本发明的第一方面中所描述。
“生物相容的载体”为流体,尤其是液体,其中成像剂可悬浮或优选溶解,使得组合物在生理学上可耐受,即,可给予哺乳动物身体,而没有毒性或过度不适。生物相容的载体适宜地为注射用载体液体,例如用于注射的无菌无热原的水;水溶液,例如盐水(其可有利地平衡,使得用于注射的最终产品为等渗的);包含生物相容缓冲剂(例如,磷酸盐缓冲剂)的水性缓冲溶液;一种或多种张度-调节物质(例如等离子体阳离子与生物相容的反荷离子的盐)的水溶液、糖(例如葡萄糖或蔗糖)、糖醇(例如山梨糖醇或甘露糖醇)、二醇(例如甘油)或其它非离子多元醇物质(例如聚乙二醇、丙二醇等)。优选生物相容的载体为用于注射的无热原的水、等渗盐水或磷酸盐缓冲剂。
短语“适用于哺乳动物给药的形式”是指无菌、无热原的组合物,其缺乏产生毒性或有害作用的化合物,并且在生物相容的pH (约pH 4.0-10.5)下配制。这样的组合物缺乏可能具有体内引起栓塞的风险的颗粒,并且经配制使得在与生物学流体(例如,血液)接触时不发生沉淀。这样的组合物还含有仅生物学上相容的赋形剂,并且优选为等渗的。
成像剂和生物相容的载体各自提供在包括密封容器加上任选惰性顶空气体(例如氮气或氩气)的合适的小瓶或容器中,所述密封容器使得可保持无菌完整性和/或放射性安全,所述惰性顶空气体同时使得可通过注射器或插管加入及取出溶液。优选这样的容器为隔膜密封小瓶,其中用顶封(通常为铝)压紧气密封盖。所述封盖适于用皮下注射器针头一次或多次穿刺同时保持无菌完整性(例如压紧的隔膜封封盖)。这样的容器具有以下另外的优点:需要时封盖可承受真空(例如以改换顶空气体或除去溶液中的气体)以及承受压力变化,例如降低压力而不使外部大气气体例如氧气或水蒸气进入。
优选的多剂量容器包含单一的大包装(bulk)瓶(例如,10-30 cm3体积),其含有多个患者剂量,由此在制剂的可用期限期间,在不同的时间间隔,可将单一的患者剂量取出至临床级别注射器中,以适于临床状况。设计预先填充的注射器以含有单人剂量或“单位剂量”,因此优选为一次性注射器或其它适用于临床使用的注射器。本发明的药物组合物优选具有适用于单个患者的剂量,并且在合适的注射器或容器中提供,如以上描述的。
药物组合物可含有另外的任选的赋形剂,例如:抗微生物防腐剂、pH-调节剂、填充剂、辐射防护剂、增溶剂或重量克分子渗透压浓度调节剂。术语“辐射防护剂”是指抑制降解反应的化合物,例如氧化还原过程,通过捕获高度-反应性自由基,例如由水的辐射分解引起的含氧自由基。本发明的辐射防护剂适宜地选自:抗坏血酸、对氨基苯甲酸(即,4-氨基苯甲酸)、龙胆酸(即,2,5-二羟基苯甲酸)及其与上述生物相容的阳离子的盐。术语“增溶剂”是指提高成像剂在溶剂中的溶解性的存在于组合物中的添加剂。优选的这样的溶剂为水性介质,因此,增溶剂优选改进在水中的溶解性。合适的这样的增溶剂包括:C1-4醇;甘油;聚乙二醇(PEG);丙二醇;聚氧乙烯脱水山梨醇单油酸酯;脱水山梨醇单油酸酯(monooloeate);聚山梨酸酯;聚(氧乙烯)聚(氧丙烯)聚(氧乙烯)嵌段共聚物(PluronicsTM);环糊精(例如,α、β或γ环糊精、羟丙基-β-环糊精或羟丙基-γ-环糊精)和卵磷脂。
术语“抗微生物防腐剂”是指抑制可能有害的微生物(例如细菌、酵母或霉菌)的生长的试剂。抗微生物防腐剂还可呈现一些杀菌性质,取决于采用的剂量。本发明的抗微生物防腐剂的主要作用是抑制任何这样的微生物在药物组合物中的生长。然而,在给药之前,抗微生物防腐剂还可任选用于抑制用于制备所述组合物的试剂盒的一种或多种组分中的可能有害的微生物的生长。合适的抗微生物防腐剂包括:对羟基苯甲酸酯,即,对羟基苯甲酸甲酯、对羟基苯甲酸乙酯、对羟基苯甲酸丙酯或对羟基苯甲酸丁酯或它们的混合物;苄醇;苯酚;甲酚;溴化十六烷基三甲铵和硫柳汞。优选的抗微生物防腐剂为对羟基苯甲酸酯。
术语“pH-调节剂”是指可用于确保组合物的pH在用于人或哺乳动物给药的可接受的限度(约pH 4.0-10.5)内的化合物或多种化合物的混合物。合适的这样的pH-调节剂包括药学上可接受的缓冲剂,例如曲辛(tricine)、磷酸盐或TRIS [即,三(羟甲基)氨基甲烷],以及药学上可接受的碱例如碳酸钠、碳酸氢钠或它们的混合物。当组合物以试剂盒形式使用时,pH调节剂可任选在单独的小瓶或容器中提供,因此试剂盒的使用者可调节pH作为多步程序的一部分。
术语“填充剂”是指在生产和冻干期间可促进材料处理的药学上可接受的膨胀剂。合适的填充剂包括无机盐例如氯化钠,和水溶性糖或糖醇例如蔗糖、麦芽糖、甘露醇或海藻糖。
第二方面的药物组合物可在无菌制造(即,净化室)条件下制备,以得到期望的无菌、无热原产物。优选关键组分,尤其是相关试剂加上与成像剂接触的设备的那些部件(例如,小瓶)无菌。组分和试剂可通过本领域已知的方法灭菌,包括:无菌过滤、使用例如γ-照射的最终灭菌、高压灭菌、干热或化学处理(例如使用环氧乙烷)。优选事先灭菌一些组分,使得需要进行最少数量的操作。然而,作为预防,优选包括至少无菌过滤步骤作为制备药物组合物的最终步骤。
如上所述,本发明的药物组合物优选包含增溶剂,使得可使用无菌过滤步骤而不会过度损失过滤材料上吸附的放射性。类似的考虑适用于在临床级别注射器中处理药物组合物,或者使用塑料管,其中在不使用增溶剂时,吸附可引起放射性损失。
本发明的放射性药物组合物可通过各种方法制备:
(i) 无菌制造技术,其中在净化室环境中进行放射性金属络合物形成;
(ii) 最终灭菌,其中不使用无菌制造进行放射性金属络合物形成,随后在最后的步骤灭菌[例如,通过γ照射、高压灭菌、干热或化学处理(例如使用环氧乙烷)];
(iii) 试剂盒方法,其中包含式III的螯合剂缀合物和任选的赋形剂的无菌、非放射性试剂盒制剂与期望的放射性金属供应反应。
优选方法(iii),并且用于该方法的试剂盒描述于(以下)第五实施方案。
在第五方面,本发明提供了用于制备第四方面的放射性药物组合物的试剂盒,所述放射性药物组合物包含无菌、固体形式的第二方面的螯合剂缀合物,使得当与放射性金属的无菌供应在生物相容的载体中重构时,发生溶解,以得到期望的放射性药物组合物。
在第五方面中,螯合剂缀合物的优选方面如在(以上)本发明的第二方面中所描述。
术语“试剂盒”是指一个或多个非放射性药物级别容器,包含所需的化学品以制备期望的放射性药物组合物,以及操作用法说明。将试剂盒设计为与期望的放射性金属重构,以用最少的操作,得到适用于人给药的溶液。
无菌、固体形式优选为冻干的固体。
对于99mTc,试剂盒优选为冻干的,并且设计为与得自99mTc放射性同位素发生器的无菌99mTc-高锝酸盐(TcO4 -)重构,以得到适用于人给药的溶液,而无需其它操作。合适的试剂盒包含含有游离碱式或酸式盐形式的螯合剂缀合物的容器(例如,隔膜-密封的小瓶),以及生物相容的还原剂例如连二亚硫酸钠、亚硫酸氢钠、抗坏血酸、甲脒亚磺酸、亚锡离子、Fe(II)或Cu(I)。生物相容的还原剂优选为亚锡盐,例如氯化亚锡或酒石酸亚锡。或者,试剂盒可任选含有非放射性金属络合物,当加入锝时,进行金属转移(即,金属交换),得到期望的产物。非放射性试剂盒可任选还包含另外的组分,例如转移螯合剂(transchelator)、辐射防护剂、抗微生物防腐剂、pH-调节剂或填充剂,如上定义的。
在第六方面,本发明提供了对人或动物身体成像的方法,所述方法包括使用PET或SPECT产生已分布第一方面的成像剂或第四方面的组合物的所述身体的至少部分的图像,其中所述成像剂或组合物已预先给予所述身体。
在第六方面中,成像剂或组合物的优选方面分别如在(以上)本发明的第一和第四方面中所描述。当一部分身体为其中涉及异常凋亡的疾病状态时,优选进行第六方面的方法。术语“异常凋亡”是指程序性细胞死亡(PCD)过程的失调。这样的失调牵涉多种疾病状态,包括与抑制凋亡相关的那些,例如癌症和自身免疫病症,以及与活动过强凋亡相关的那些,包括:神经变性疾病;血液疾病;AIDS;局部缺血;同种异体移植排斥和心脏病学(心肌梗死、动脉粥样硬化和/或药物疗法后的心脏毒性(cardiotoxicity follow drug therapy))。因此,凋亡的显影和定量可用于诊断这样的与凋亡-相关的病理生理学。
第六方面的成像方法可任选重复进行,以监测用药物治疗人或动物身体的作用,所述成像在用所述药物治疗之前和之后,以及任选还在用所述药物治疗期间实现。用于这些疾病的治疗性处理目的是恢复平衡的凋亡,适当时通过刺激或抑制PCD过程。特别感兴趣的是早期监测癌症治疗的功效以在病况变为晚期之前确保控制恶性生长。
在第七方面,本发明提供第一方面的成像剂、第四方面的组合物或第五方面的试剂盒在诊断人或动物身体的方法中的用途。
在第八方面,本发明提供了诊断人或动物身体的方法,所述方法包括第六方面的成像方法。
在第七和第八方面中,成像剂或组合物的优选方面分别如在(以上)本发明的第一和第四方面中所描述。两方面的人或动物身体的诊断优选为其中涉及异常凋亡的疾病状态的诊断。这样的“异常凋亡”如在(以上)第六方面所描述。
通过以下详述的非限制性实施例来说明本发明。实施例1-3提供本发明的螯合剂1 (二胺二肟)的合成,实施例4提供螯合剂1A (用戊二酸官能化的二胺二肟)的合成和相应的活性酯螯合剂1A-TFTP酯的合成。实施例5提供螯合剂1B (用戊二酰基-氨基-PEG12丙酸官能化的二胺二肟)的合成。实施例6提供本发明的Boc-保护的四胺螯合剂(螯合剂2A)的合成。实施例7提供HYNIC-耐久霉素缀合物(现有技术)的合成,用于比较目的。实施例8提供用螯合剂1A官能化的耐久霉素(缀合物3A和缀合物3B)的合成。实施例9提供具有螯合剂1A的肉桂霉素(缀合物5)的合成。实施例10提供具有螯合剂1B的耐久霉素(缀合物6)的合成。实施例11提供具有螯合剂1B的肉桂霉素(缀合物6)的合成。实施例12提供用螯合剂2A官能化的耐久霉素(缀合物2A和缀合物2B)的合成,实施例13提供被螯合剂2A官能化的肉桂霉素(缀合物4)的合成。实施例14提供使用放射性金属99mTc放射性标记本发明的螯合剂缀合物。99mTc络合物作为具有高RCP的单一物类形成。这比起HYNIC具有优势,其中当使用HYNIC/膦/曲辛标记时,形成多种物类。程序简单,且在室温下有效标记。RCP非常良好,即使在高放射性浓度(在> 500MBq/mL下,>90% RCP)下。
实施例15提供测定螯合剂的缀合部位,实施例16证明螯合剂的缀合部位对于对磷脂酰乙醇胺的结合亲和力具有显著作用,具有18倍差异(Kd 5 nM相对于90 nM)。这提供了比起在式II的Xaa处连接,优选在N-末端(式II的Cysa)处连接放射性金属络合物的证据。实施例17的EL4淋巴瘤小鼠异种移植肿瘤模型用作模仿在化疗后凋亡响应的模型。与溶媒对照处理的动物相比,经治疗-处理的小鼠(依托泊苷/环磷酰胺)显示肿瘤凋亡的4倍提高。实施例17的生物分布结果显示在经化疗-处理的肿瘤中每种试剂较高的吸收,而关联分析表明在具有较高水平凋亡的肿瘤中较高的结合剂吸收趋势。相对于99mTc-[缀合物3A],99mTc-[缀合物5]具有类似的肿瘤和改进的肝性能。相对于99mTc-[缀合物5],99mTc-[缀合物2A]显示类似的肿瘤但是低等的肺性能。使用99mTc-[缀合物5]重复成像研究显示,在治疗之后肿瘤:肌肉比一致提高。实施例18显示PEG接头基团在降低肝背景和提高体内尿排泄中是有利的。
缩写
使用常规的单一字母或3-字母氨基酸缩写。
% id:注射剂量的百分比。
Ac:乙酰基。
Acm:乙酰氨基甲基。
ACN:乙腈。
Boc:叔丁氧基羰基。
Bz:苄基。
DCM:二氯甲烷。
DIPEA:N-二异丙基乙胺。
DMF:N,N-二甲基甲酰胺。
DMSO:二甲基亚砜。
Fmoc:9-芴基甲氧基羰基。
Glut:戊二酸。
HATU:O-(7-氮杂苯并三唑-1-基)-N,N,N',N'-四甲基脲 六氟磷酸盐。
六氟磷酸盐。
HOAt:7-氮杂-1-羟基苯并三唑。
HPLC:高效液相色谱法。
IBX:1-羟基-1,2-碘杂氧杂苯并环戊烯(benziodoxole)-3(1H)-酮-1-氧化物。
MDP:亚甲基二膦酸。
NaPABA:对氨基苯甲酸钠。
NHS:N-羟基-琥珀酰亚胺。
NMM:N-甲基吗啉。
NMP:1-甲基-2-吡咯烷酮。
PBS:磷酸盐缓冲盐水。
PEG12:-(OCH2CH2)12-。
PyAOP:(7-氮杂苯并三唑-1-基-氧基-三-吡咯烷子基(pyrrolidino)-磷 六氟磷酸盐。
六氟磷酸盐。
PyBOP:苯并三唑-1-基-氧基三吡咯烷子基六氟磷酸盐。
RAC:放射性浓度。
RCP:放射性化学纯度。
tBu:叔丁基。
TFA:三氟乙酸。
TFTP:四氟苯硫酚。
THF:四氢呋喃。
TIS:三异丙基硅烷。
Trt:三苯甲基。
表1:本发明的化合物。
式II (具有在第一方面中指定的桥):

实施例1:1,1,1-三(2-氨基乙基)甲烷的合成。
步骤1(a):3(甲氧基羰基亚甲基)戊二酸二甲酯。
甲苯(600ml)中的甲酯基亚甲基三苯基磷烷(167g,0.5mol)用3-氧代戊二酸二甲酯(87g,0.5mol)处理,在氮气气氛下,在120℃的油浴上,将反应物加热至100℃保持36小时。随后将反应物真空浓缩,油性残余物用40/60石油醚/乙醚1:1(600ml)研磨。将三苯基氧化膦沉淀出,将上清液液体倾析/过滤。将真空蒸发的残余物在高真空Bpt下库格尔若蒸馏(烘箱温度180-200℃,0.2托),以得到3-(甲氧基羰基亚甲基)戊二酸二甲酯(89.08g,53%)。
NMR 1H(CDCl3):δ 3.31 (2H,s,CH2),3.7(9H,s,3×OCH3),3.87 (2H,s,CH2),5.79 (1H,s,=CH) ppm。
NMR 13C(CDCl3),δ 36.56,CH3,48.7,2×CH3,52.09和52.5 (2×CH2);122.3和146.16 C=CH;165.9,170.0和170.5 3×COO ppm。
步骤1(b):3-(甲氧基羰基亚甲基)戊二酸二甲酯的氢化。
甲醇(200ml)中的3-(甲氧基羰基亚甲基)戊二酸二甲酯(89g,267mmol)与(10%披钯木炭:50%水) (9 g)在氢气气氛(3.5巴)下振动30小时。将溶液通过硅藻土过滤,真空浓缩,以得到油状的3-(甲氧基羰基甲基)戊二酸二甲酯,产量(84.9g,94 %)。
NMR 1H(CDCl3),δ 2.48 (6H,d,J=8Hz,3×CH2),2.78 (1H,六重峰,J=8Hz CH,) 3.7 (9H,s,3×CH3)。
NMR 13C(CDCl3),δ 28.6,CH;37.50,3×CH3;51.6,3×CH2;172.28,3×COO。
步骤1(c):将三甲酯还原和酯化为三乙酸酯。
在氮气气氛下,在1小时内,在3颈2L圆底烧瓶中,THF (400ml)中的氢化铝锂(20g,588mmol)用THF (200ml)中的三(甲氧基羰基甲基)甲烷(40g,212mmol)小心处理。发生强放热反应,引起溶剂强烈回流。将反应物在90℃的油浴上加热回流3天。通过小心逐滴加入乙酸(100ml)淬灭反应,直至停止放出氢气。用乙酸酐溶液(500ml)小心处理搅动的反应混合物,其速率引起温和回流。配备烧瓶用于蒸馏和搅拌,随后在90℃(油浴温度)下加热,以蒸馏出THF。加入另一部分乙酸酐(300ml),反应物返回至回流配置,搅拌,并在140℃的油浴中加热5小时。让反应物冷却,过滤。氧化铝沉淀用乙酸乙酯洗涤,将合并的滤液在旋转蒸发仪上在50℃水浴温度下真空(5 mmHg)浓缩,以得到油。油被乙酸乙酯(500ml)吸收,用饱和碳酸钾水溶液洗涤。将乙酸乙酯溶液分离,经硫酸钠干燥,真空浓缩,以得到油。将油在高真空下库格尔若蒸馏,以得到油状的三(2-乙酰氧基乙基)甲烷(45.3g,95.9%)。在0.1 mmHg的沸点220℃。
步骤1(d):从三乙酸酯除去乙酸酯基。
甲醇(200ml)中的三(2-乙酰氧基乙基)甲烷(45.3g,165mM)与880氨(100ml)在80℃的油浴上加热2天。反应物用另一部分的880氨(50ml)处理,并且在80℃的油浴上加热24小时。加入另一部分的880氨(50ml),将反应物于80℃下加热24小时。随后将反应物真空浓缩,以除去所有溶剂,以得到油。其被880氨(150ml)吸收,并于80℃下加热24小时。随后将反应物真空浓缩,以除去所有溶剂,以得到油。库格尔若蒸馏得到乙酰胺,沸点170-180℃/0.2mm。将含有乙酰胺的球管洗涤干净,并且继续蒸馏。在沸点220℃/0.2mm下蒸馏三(2-羟基乙基)甲烷(22.53g,92%)。
NMR 1H(CDCl3),δ 1.45(6H,q,3×CH2),2.2(1H,五重峰,CH);3.7(6H,t,3×CH2OH);5.5(3H,brs,3×OH)。
NMR 13C(CDCl3),δ 22.13,CH;33.95,3×CH2;57.8,3×CH2OH。
步骤1(e):将三醇转化为三(甲磺酸酯)。
在氮气下,向三(2-羟基乙基)甲烷(10g,0.0676mol)在二氯甲烷(50ml)中的搅动的冰冷溶液中缓慢滴加甲磺酰氯(40g,0.349mol)在二氯甲烷(50ml)中的溶液,其速率使得温度不上升超过15℃。随后逐滴加入溶解于二氯甲烷(50ml)中的吡啶(21.4g,0.27mol,4当量),其速率使得温度不上升超过15℃,为放热反应。让反应物在室温下搅拌24小时,随后用5N盐酸溶液(80ml)处理,将各层分离。水层用另外的二氯甲烷(50ml)萃取,将有机萃取物合并,经硫酸钠干燥,过滤,真空浓缩,以得到被过量的甲磺酰氯污染的三[2-(甲基磺酰基氧基)乙基]甲烷。理论产量为25.8g。
NMR 1H(CDCl3),δ 4.3(6H,t,2×CH2),3.0(9H,s,3×CH3),2(1H,六重峰,CH),1.85(6H,q,3×CH2)。
步骤1(f):制备1,1,1-三(2-叠氮基乙基)甲烷。
在氮气下,在15分钟内,三[2-(甲基磺酰氧基)乙基]甲烷[得自步骤1(e),被过量的甲基磺酰氯污染](25.8g,67mmol,理论)在无水DMF(250ml)中的搅拌溶液用叠氮化钠(30.7g,0.47mol)分批处理。观察到放热,将反应物在冰浴上冷却。30分钟后,将反应混合物在50℃的油浴上加热24小时。反应物变为褐色。让反应物冷却,用稀碳酸钾溶液(200ml)处理,用40/60石油醚/乙醚10:1(3×150ml)萃取三次。有机萃取物用水(2×150ml)洗涤,经硫酸钠干燥,过滤。向石油/醚溶液中加入乙醇(200ml),以保持三叠氮化物在溶液中,将体积真空降至不小于200ml。加入乙醇(200ml),再次真空浓缩,以除去最后痕量的石油,留下不小于200ml的乙醇溶液。三叠氮化物的乙醇溶液直接用于步骤1(g)。
注意:不要除去所有的溶剂,由于叠氮化物可能爆炸,应始终保持在稀溶液中。
将小于0.2ml的溶液真空蒸发,以除去乙醇,并且对该小的样品进行NMR操作:
NMR 1H(CDCl3),δ 3.35(6H,t,3×CH2),1.8(1H,七重峰,CH,),1.6(6H,q,3×CH2)。
步骤1(g):制备1,1,1-三(2-氨基乙基)甲烷。
在乙醇(200ml)中的三(2-叠氮基乙基)甲烷(15.06g,0.0676 mol)(假定由先前的反应100%收率)用10%披钯木炭(2g,50%水)处理,并且氢化12小时。每2小时抽空反应容器,以除去氮由反应放出的氮气,并且用氢气再次填充。取样品用于NMR分析,以证实三叠氮化物完全转化为三胺。
警告:未还原的叠氮化物在蒸馏时可能爆炸。将反应物过滤通过硅藻土垫,以除去催化剂,真空浓缩,以得到油状的三(2-氨基乙基)甲烷。通过库格尔若蒸馏将其进一步纯化,沸点180-200℃/0.4mmHg,以得到无色油(8.1g,82.7%,由三醇的总收率)。
NMR 1H(CDCl3),δ 2.72(6H,t,3×CH2N),1.41(H,七重峰,CH),1.39(6H,q,3×CH2)。
NMR 13C(CDCl3),δ 39.8(CH2NH2),38.2(CH2),31.0(CH)。
实施例2:制备3-氯-3-甲基-2-亚硝基丁烷。
将2-甲基丁-2-烯(147ml,1.4mol)和异戊基腈(156ml,1.16mol)的混合物在cardice和甲醇的浴中冷却至-30℃,使用顶空空气搅拌器剧烈搅拌,用浓盐酸(140ml,1.68mol)逐滴处理,其速率使得温度保持低于-20℃。由于存在显著放热,这需要约1小时,并且必须注意防止过热。加入乙醇(100ml),以降低在加入结束时形成的浆液的粘度,并且在-20至-10℃下将反应物再搅拌2小时,以完成反应。通过在真空下过滤收集沉淀物,用4×30ml冷(-20℃)乙醇和100ml冰冷的水洗涤,真空干燥,以得到白色固体状的3-氯-3-甲基-2-亚硝基丁烷。将乙醇滤液和洗涤液合并,用水(200ml)稀释,冷却,当另一个产物3-氯-3-甲基-2-亚硝基丁烷结晶出时,在-10℃下静置1小时。通过过滤收集沉淀物,用最少的水洗涤,真空干燥,以得到总产量3-氯-3-甲基-2-亚硝基丁烷(115g 0.85mol,73%),通过NMR测定纯度>98%。
NMR 1H(CDCl3),作为异构体的混合物(异构体1,90%) 1.5 d,(2H,CH3),1.65 d,(4H,2×CH3),5.85,q和5.95,q,共1H。(异构体2,10%),1.76 s,(6H,2×CH3),2.07(3H,CH3)。
实施例3:双[N-(1,1-二甲基-2-N-羟基亚胺丙基)2-氨基乙基]-(2-氨基乙基)甲烷(螯合剂1)的合成。
在氮气气氛下,剧烈搅拌下,在室温下,向三(2-氨基乙基)甲烷(4.047g,27.9mmol)在无水乙醇(30ml)中的溶液中加入无水碳酸钾(7.7g,55.8mmol,2当量)。将3-氯-3-甲基-2-亚硝基丁烷(7.56g,55.8mol,2当量)的溶液溶解于无水乙醇(100ml)中,将75ml该溶液缓慢滴加至反应混合物中。反应物用TLC在二氧化硅上追踪[板在二氯甲烷/甲醇/浓(0.88sg)氨(100/30/5)中运行,通过喷水合茚三酮展开TLC板,并加热]。随着RF的提高,按顺序看到单-、二-和三-烷基化的产物。使用RPR反相柱运行分析型HPLC,梯度为7.5-75%乙腈/3%氨水。将反应物真空浓缩,以除去乙醇,并且再次悬浮于水(110ml)中。水性浆液用乙醚(100ml)萃取,以除去一些三烷基化的化合物和亲脂性杂质,将单烷基化的产物和期望的二烷基化的产物留在水层。水溶液用乙酸铵(2当量,4.3g,55.8mmol)缓冲,以确保良好的色谱法。将水溶液在4℃下储存过夜,随后通过自动化制备型HPLC纯化。
产量(2.2g,6.4mmol,23%)。
质谱;正离子10V锥形电压。实测值:344;计算值M+H=344。
HPLC条件:流速8ml/分钟,使用25mm PRP柱[A=3%氨溶液(sp.gr=0.88)/水;B=乙腈]。

每次运行载荷3ml水溶液,在12.5-13.5分钟的时间窗收集。
实施例4:螯合剂1-戊二酸的四氟硫苯基酯(螯合剂1A-TFTP酯)的合成。
a) [螯合剂1]-戊二酸(螯合剂1A)的合成。

搅拌下,将螯合剂1(100 mg,0.29 mmol)溶解于DMF(10ml)中,分批加入戊二酸酐(33 mg,0.29 mmol)。将反应物搅拌23小时,以提供完全转化为期望的产物。RP-HPLC后,以良好的收率得到纯酸。
b) 螯合剂1A-TFTP酯的合成。

向螯合剂1A(得自步骤a;300 mg,0.66 mmol)/DMF(2 ml)中加入HATU(249 mg,0.66 mmol)和NMM(132 μL,1.32 mmol)。将混合物搅拌5分钟,随后加入四氟苯硫酚(0.66 mmol,119 mg)。将溶液搅拌10分钟,随后反应混合物用20%乙腈/水(8 ml)稀释,通过RP-HPLC纯化产物,得到110 mg期望的产物(冷冻干燥之后)。
实施例5:螯合剂1-戊二酰基-氨基-PEG12丙酸(螯合剂1B)的合成。

Boc-氨基-PEG12丙酸(Polypure;45 mg,0.060mmol)用TFA/水(19:1)(1 mL)处理30分钟。随后将TFA真空蒸发,将残余物真空干燥过夜,得到52 mg粗制氨基-PEG12丙酸。将螯合剂1A(46 mg,0.10mmol)和PyAOP(31 mg,0.060mmol)溶解于NMP(1 mL)中。加入DIPEA(42 μL,0.24 mmol),将溶液振动5分钟,并加入到氨基-PEG12丙酸(0.06 mmol)中。将反应混合物振动过夜。将另外的螯合剂1A(0.03 mmol)加入到反应混合物中,1小时后,混合物用水/0.1% TFA(7 mL)稀释,通过制备型RP-HPLC纯化产物。
纯化和表征
通过RP-HPLC(梯度:10-30% B,经40分钟,tR:34.5分钟)纯化得到44 mg(67%收率)的螯合剂1B (冻干后)。螯合剂1B通过LC-MS表征(梯度:10-40% B,经5分钟,tR:2.2分钟;计算值m/z 1057.7 [MH]+,实测值m/z 1058.0)。
实施例6:螯合剂2A的合成。
步骤(a):[2-(苄氧基)乙基]丙二酸二乙酯。
通过Ramalingam等人[Tetrahedron,51,2875-2894(1995)]的方法的修改,制备化合物。因此,在氩气下,将钠(1.20g)溶解于无水乙醇(25 ml)中。加入丙二酸二乙酯(14.00g),将混合物回流30分钟。加入苄基溴乙基乙醚(10g),将混合物搅拌回流16小时。通过旋转蒸发除去乙醇,将残余物在乙醚(100ml)和水(50ml)之间分配。乙醚层用水(3×50ml)洗涤,经硫酸钠干燥。通过旋转蒸发除去乙醚,将残余物真空蒸馏。将在40-55℃下蒸馏的馏分(未反应的丙二酸二乙酯)丢弃。产物在140-150℃(1mm)下蒸馏[lit. bp 138-140℃(1mm)]。产量为12.60g无色油。

步骤(b):N,N'-双(2-氨基乙基)-2-(2-苄氧基-乙基)丙二酰胺。
将[2-(苄氧基)乙基]丙二酸二乙酯(4.00g)加入到乙二胺(30ml)中,将溶液在室温下搅拌2天。通过旋转蒸发除去过量的乙二胺,将残余物在高真空下干燥2天,以得到黄色油(4.28g),在静置时结晶。产物仍含有痕量的乙二胺,如在NMR光谱中检测到。

步骤(c):N,N'-双(2-氨基-乙基)-2-(2-苄氧基乙基)-1,3-二氨基丙烷。
将N,N'-双-(2-氨基乙基)-2-(2-苄氧基-乙基)丙二酰胺(3.80g)溶解于THF(20ml)中,将烧瓶浸没在冰浴中。烧瓶用氩气冲洗,通过注射器加入THF硼烷络合物(80ml,1M,在THF中)。让反应混合物升温至室温,随后在40℃下搅拌2天,并回流1小时。逐滴加入甲醇(50ml),将溶液在40℃下搅拌过夜。通过旋转蒸发仪除去溶剂,将残余物溶解于甲醇(20ml)中。加入氢氧化钠(10g在15ml水)中,将甲醇煮干。将分离的无色油在CH2Cl2(3×50ml)中萃取。溶液经Na2SO4干燥。除去溶剂,得到3.40g无色油。
步骤(d):N,N'-双(2-叔丁氧基羰基氨基-乙基)-2-(2-苄氧基乙基)-1,3-二(叔丁氧基羰基氨基)丙烷。
将N,N'-双(2-氨基乙基)-2-(2-苄氧基-乙基)-1,3-二氨基丙烷(3.30g)溶解于CH2Cl2(100ml)中,加入三乙胺(5.40g)和二碳酸叔丁酯(10.30g)。将反应混合物在室温下搅拌2天。混合物用水(100ml)、柠檬酸溶液(100ml,10%在水中)和水(2×100ml)洗涤。有机层经Na2SO4干燥,通过旋转蒸发除去溶剂,得到黄色油,在高真空下将其干燥至恒定质量。粗产物(7.70g)在硅胶柱上纯化(250g,230-400目,CH2Cl2,CH2Cl2-Et2O 1:1),以得到6.10g(78.3%)澄清的油。
步骤(e):N,N'-双(2-叔丁氧基羰基氨基-乙基)-2-(2-羟基乙基)-1,3-二(叔丁氧基羰基氨基)丙烷。
将N,N'-双(2-叔丁氧基羰基氨基-乙基)-2-(2-苄氧基-乙基)-1,3-二(叔丁氧基羰基氨基)丙烷(3.16g)溶解于无水乙醇(100ml)中,加入披Pd活性碳(1.00g,干燥,10%)。将混合物在Parr氢化设备中在35 psi下氢化两天。将催化剂过滤掉,用乙醇(3×20ml)洗涤。通过旋转蒸发除去乙醇,以得到无色油,在高真空下将其干燥至恒定质量(2.67g,97.1%)。
步骤(f):N,N'-双(2-叔丁氧基羰基氨基-乙基)-2-(2-羧基甲基)-1,3-二(叔丁氧基羰基氨基)丙烷(Boc-保护的螯合剂2A)。
使用Mazitschek等人的方法[Ang. Chem. Int. Ed.,41,4059-4061(2002)]。因此,将N,N'-双(2-叔丁氧基羰基氨基-乙基)-2-(2-羟基乙基)-1,3-二(叔丁氧基羰基氨基)丙烷(2.60g)溶解于DMSO(15 ml)中,加入1-羟基-1,2-碘杂氧杂苯并环戊烯-3(1H)-酮-1-氧化物(IBX,3.50g)。将混合物在室温下搅拌1小时,随后加入N-羟基琥珀酰亚胺(2.50g)。将反应混合物在室温下搅拌2天。加入氢氧化钠溶液(2M,40ml),将混合物在室温下搅拌4小时。将溶液浸没在冰浴中,使用2M盐酸酸化至pH 2。水层用乙醚(4×100ml)萃取,合并的乙醚萃取物用水(3×50ml)洗涤。乙醚层经Na2SO4干燥,通过旋转蒸发除去溶剂,以得到含有产物和2-亚碘酰基苯甲酸的黄色固体残余物。通过从氯仿-己烷(1:3)(80ml)结晶,除去大多数亚碘酰基苯甲酸(2.1g)。蒸发氯仿-己烷母液,得到黄色油(3g),将其装载于二氧化硅柱(300g,CH2Cl2-Et2O,1:1)上。剩余的亚碘酰基苯甲酸用乙醚洗脱。产物用乙醚-甲醇(9:1)洗脱。将含有产物的级分合并,除去溶剂,得到1.5g浅黄色油。将其在二氧化硅柱(50g,Et2O)上再次进行色谱法。产物用乙醚-乙酸(95:5)洗脱。将含有产物的级分合并,通过旋转蒸发除去溶剂,以得到油,将其在高真空下干燥。产量为1.10g(41.3%)。

实施例7:HYNIC-耐久霉素(缀合物1A和缀合物1B)的合成。
将耐久霉素(Sigma-Aldrich;5.0 mg,2.5 μmol)和N-Boc-HYNIC琥珀酰亚胺基酯(ABX Advanced Biochemical Compounds;1.0 mg,2.8 μmol)溶解于DMF(1 ml)中,将DIPEA(2.0 μL,13 μmol)加入到混合物中。通过LC-MS分析监测反应进程。3小时后加入HOAt(1.1当量),以驱动缓慢反应(sluggish reaction),得到约60%产物形成过夜。在随后的一天于室温下和3天于4℃下之后,需要另外的N-Boc-HYNIC琥珀酰亚胺基酯(1当量)和HOAt(2当量)以得到约80% HYNIC-缀合物形成。除了双-缀合物(约35%)以外,通过LC-MS分析还观察到相应于单-缀合物的两个基线分离的峰。
纯化和表征。
将水/0.1% TFA(4 ml)加入到反应混合物中,两种单-缀合的产物通过制备型RP-HPLC纯化(梯度:0% B,经15分钟;0-45% B,经10分钟;45-60% B,经40分钟,tR:47.9和49.3分钟),随后在TFA中Boc-脱保护,得到两种单-缀合的耐久霉素异构体,产量分别为1.7 mg和1.1 mg。
两种异构体通过LC-MS表征(梯度:20-40% B,经5分钟,tR:2.8分钟(缀合物1A),实测值m/z:1074.8,预期为MH2 2+:1074.4,tR:2.9分钟(缀合物1B),实测值m/z:1074.8,预期为MH2 2+:1074.4。
实施例8:[螯合剂1A]-耐久霉素单-缀合物(缀合物3A)和[螯合剂1A]-耐久霉素双-缀合物(缀合物3B)的合成。
将螯合剂1A(实施例4;3.0 mg,6.6 μmol)、PyBOP(2.6 mg,5.0 μmol)和DIPEA(1.7 μL,9.7 μmol)溶解于NMP(0.7 ml)中。将混合物振动5分钟,并且加入到耐久霉素(Sigma-Aldrich;5.0 mg,2.5 μmol)在NMP(0.5 ml)的溶液中。将反应混合物振动40分钟,随后用水/0.1% TFA(6 ml)稀释,使用制备型HPLC纯化产物。
通过制备型HPLC纯化(梯度:5-35% B,经40分钟,其中A=H2O/0.1% HCOOH,B=ACN/0.1% HCOOH)得到2.5 mg纯的缀合物3A(产率41%)和1.7 mg纯的缀合物3B(产率28%)。
通过分析型LC-MS分析纯化的缀合物3A(梯度:25-35% B,经5分钟,tR:1.93分钟,实测值m/z:1227.0,预期为MH2 2+:1226.6)。
通过分析型LC-MS分析纯化的缀合物3B(梯度:25-35% B,经5分钟,tR:2.35分钟,实测值m/z:1446.7,预期为MH2 2+:1446.3)。
使用分析型或制备型HPLC中的任一个不能实现两种单-缀合物(缀合物3A)的分离。在每一种情况下,两种区域异构体作为一个单一的峰洗脱。
实施例9:[螯合剂1A]-肉桂霉素缀合物(缀合物5)的合成。
将肉桂霉素(Sigma-Aldrich;2.0 mg,1.0 μmol)、螯合剂1A(实施例4;0.9 mg,1.5 μmol)和DIPEA(0.5 μL,2.9 μmol)溶解于NMP(0.2 ml)、DMF(0.2 ml)和DMSO(0.6 ml)的溶液中。将反应混合物振动过夜。混合物随后用10% ACN/水/0.1% TFA(7 ml)稀释,使用制备型HPLC纯化产物。
纯化和表征
通过制备型HPLC纯化(梯度:20-40% B,经40分钟)得到1.9 mg纯的缀合物5(产率78%)。通过分析型LC-MS分析纯化的物质(梯度:20-40% B,经5分钟,tR:2.86分钟,实测值m/z:1241.0,预期为MH2 2+:1240.6)。
实施例10:[螯合剂1B]-耐久霉素缀合物(缀合物6)的合成。
将螯合剂1B(实施例5;1.6 mg,1.5 μmol)、PyBOP(0.4 mg,0.8 μmol)和DIPEA(1 μL,6 μmol)溶解于NMP(0.5 mL)中。将混合物振动5分钟,并加入到耐久霉素(3.0 mg,1.5 μmol)在NMP(0.5 mL)的溶液中。将反应混合物振动30分钟。以30分钟间隔加入两份另外的等分的活化螯合剂1B(2×1.6 mg)。混合物用水/0.1% TFA(6 mL)稀释,使用制备型RP-HPLC纯化产物。
纯化和表征
通过制备型HPLC纯化(梯度:20-50% B,经40分钟,其中A=水/0.1%乙酸铵,B=ACN)得到3.9 mg纯的缀合物6(产率87%)。通过LC-MS分析纯化的物质(梯度:20-40% B,经5分钟,tR:2.89分钟,实测值m/z:1526.5,预期为MH2 2+:1526.2)。
实施例11:[螯合剂1B]-肉桂霉素缀合物(缀合物7)的合成。
将螯合剂1B(实施例5;4.8 mg,4.4 μmol)、PyBOP(2.1 mg,4.0 μmol)和DIPEA(2.3 μL,13.2 μmol)溶解于DMF(0.5 mL)中。将混合物振动5分钟,并加入到固体肉桂霉素(4.5 mg,2.2 μmol)中。2小时后和3.5小时后,加入另外的预先-活化的螯合剂1B,以驱动反应在4小时内接近完成。混合物用20% ACN/水/0.1% TFA(8 mL)稀释,使用制备型RP-HPLC纯化产物。
纯化和表征
通过制备型RP-HPLC纯化(梯度:25-35% B,经40分钟;tR 38.6分钟)得到3.9 mg 纯化的缀合物7(产率58%)。
通过LC-MS分析纯化的物质(梯度:20-40% B,经5分钟:tR 2.9分钟,实测值m/z:1028.0,预期为MH2 2+:1027.5(纯度约93.5%,约3%未反应的原料)。
实施例12:[螯合剂2A]-耐久霉素缀合物(缀合物2A和缀合物2B)的合成。
将耐久霉素(Sigma-Aldrich;7.5 mg,3.8 μmol)、Boc-保护的螯合剂2A(实施例6;5.0 mg,6.9 μmol)、HOAt(1.9 mg,8.8 μmol)和DIPEA(4.1 μL,20.0 μmol)溶解于NMP(1.5 ml)中。将反应混合物振动过夜。混合物随后用20% ACN/水/0.1% TFA(6 ml)稀释,使用制备型HPLC纯化产物。
纯化和表征。
通过制备型HPLC纯化(梯度:0% B,经10分钟;0-30% B,经5分钟;30-70% B,经40分钟,tR:42.4和45.0分钟),接着在TFA中Boc-脱保护,得到两种单-缀合的耐久霉素异构体,产量分别为2.0 mg和0.4 mg。
两种异构体通过LC-MS表征(梯度:20-60% B,经5分钟,tR:1.7分钟(缀合物2A),实测值m/z:1107.5,预期为MH2 2+:1107.0,tR:1.6分钟(缀合物2B),实测值m/z:1107.5,预期为MH2 2+:1107.0)。
实施例13:[螯合剂2A]-肉桂霉素缀合物(缀合物4)的合成。
将肉桂霉素(Sigma-Aldrich;2.0 mg,1.0 μmol)、Boc-保护的螯合剂2A(实施例6;1.1 mg,1.5 μmol)和DIPEA(0.5 μL,2.9 μmol)溶解于DMF(1.0ml)中。将反应混合物振动过夜。混合物随后用20% ACN/水/0.1% TFA(6 ml)稀释,使用制备型HPLC纯化产物。
纯化和表征。
通过制备型HPLC纯化(梯度:30-70% B,经40分钟)得到1.8 mg纯的Boc-保护的缀合物4。将纯化的物质在TFA/4%水(2 ml)中Boc-脱保护45分钟,由50% ACN/水冻干,得到1.6 mg缀合物4(产率73%)。该物质通过分析型LC-MS分析(梯度:10-40% B,经5分钟,tR:3.7分钟,实测值m/z:1120.9,预期为MH2 2+:1121.0)。
实施例14:制备99mTc-标记的螯合剂-缀合物。
放射性标记的制备物如下使用:(i) 未经纯化(高RAC时的高RCP);或(ii) 经过纯化,以除去未标记的LBP肽。
将缀合物3A(0.1 mg,40 nmol)溶解于乙醇(100 μL)和水(100 μL)的混合物中,并放置在声波浴中约20分钟,以帮助溶解。将溶液加入到冻干的试剂盒中[配方:SnCl2.2H2O(0.016 mg,0.07 μmol),MDP(H4)(0.025 mg,0.14 μmol),NaHCO3(4.5 mg,53.6 μmol ),Na2CO3(0.6 mg,5.66 μmol)和NaPABA(0.2 mg,1.26 μmol)]。
随后加入得自99Mo/99mTc发生器的[99mTcO4]-洗出液(约1 ml),将混合物于室温下静置约10分钟。将一部分粗产物(约400 μL)注入HPLC柱(参见以下HPLC条件)。将保留时间为约18分钟的放射性峰“切割”到含有PBS(各种体积,取决于期望的RAC)的小瓶中,随后真空干燥,以除去过量的流动相。
粗RCP=93 ± 6%(n=13)。配制的RCP(t=0)=99 ± 1%(n=13)。配制的RCP(t=120分钟)=97 ± 3%(n=13)。
比活=4.2 ± 0.5 GBq/nmol(n=13)。
RT(99mTc-缀合物3A)=18分钟。
按照与缀合物3A相同的程序制备99mTc-缀合物5:
粗RCP=> 85%(n=12)。配制的RCP(t=0)=93 ± 7%(n=12)。配制的RCP(t=120分钟)=91 ± 7%(n=6)。
比活=3.5 ± 0.5 GBq/nmol(n=13)。
RT(99mTc-缀合物5)=18分钟。
HPLC条件

现有技术HYNIC对应物99mTc-[缀合物1]的RCP为78-89%(粗品)。
类似地制备四胺螯合剂(缀合物2A和4)的缀合物,不同之处在于使用0.1% TFA作为流动相A代替50mM乙酸铵。99mTc-[缀合物2A]的保留时间为12.2分钟,而99mTc-[缀合物4]的保留时间为12.4分钟。
实施例15:缀合物2A和2B的埃德曼降解。
发现使用单独的MS-分析技术不可用于测定螯合剂的缀合部位,应用手动埃德曼降解化学联合LC-MS分析。
使用修改的文献方法[Xu等人;PNAS,106,第19310-19315页(2009);Onisko等人,J. Am. Soc. Mass Spectrom.,18,第1070-1079页(2007)和Hayashi等人,J Antibiotics,43,1421-1430(1990)]。
得到的数据证明缀合物2A相应于Nα氨基缀合的异构体,而缀合物2B相应于Lys2 Nε-氨基缀合物。数据不符合对于仲氨基缀合物(式II的Lysd)所预期的降解产物,证明该部位在用于螯合缀合的条件下不具有反应性。然而,注意到,在埃德曼降解循环期间,在所用的更加强迫的偶联条件下,仲氨基不与苯基异硫氰酸酯反应。
实施例16:对磷脂酰乙醇胺的亲和力。
Biacore 3000(GE Healthcare,Uppsala)配备有L1芯片。使用制造商推荐的捕获技术,施用由POPE/POPC(20% PE)制成的脂质体,用于亲和力研究。每次运行由活化芯片表面、固定脂质体、结合肽和洗去脂质体和肽(再生)二者组成。类似应用可参见Frostell-Karlsson等人[Pharm. Sciences,V.94(1),(2005)]。在每个循环之后,用运行缓冲液对针、管和液体处理系统进行充分洗涤。
BIACORE软件:应用包括所有方法指令的BIACORE控制软件。带有命令的方法亦以BIACORE方法定义语言(Method Definition Language,MDL)编写,以对整个预先程序化指令具有完全控制。应用BIACORE评价软件用于分析传感图(sensorgrams)。发现所有物质对于磷脂酰乙醇胺是良好的结合剂。所有物质的KD小于100 nM。结果在表2中给出:
表2
| 耐久霉素 | 缀合物2A | 缀合物2B | 缀合物4 | |
| kd(1/s) | 约8·10-5 | 约7·10-5 | 约8·10-4 | 约1.5·10-4 |
| ka(1/Ms) | 约2·104 | 约1·104 | 约4·103 | 约8·103 |
| KD(nM) | 约5 | 约6 | 约73 | 约16 |
实施例17:肿瘤吸收研究。
在EL4小鼠淋巴瘤异种移植模型中,通过生物分布评价99mTc-[缀合物2A]、99mTc-[缀合物3A]和99mTc-[缀合物5]。简要地,在C57/Bl6小鼠中建立肿瘤生长之后,将动物用如下之一处理:
(i) 盐水/DMSO溶液;或
(ii) 化疗(67 mg/kg依托泊苷和100 mg/kg环磷酰胺在50%盐水50% DMSO中)。
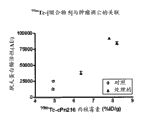
在治疗或溶媒处理24小时后,评价动物的经适当放射性标记的化合物的生物分布。此外,提取肿瘤,并且通过测量胱天蛋白酶活性(胱天蛋白酶-Glo测定)评价凋亡水平。随后绘制在各个时间点结合剂吸收与胱天蛋白酶活性的关系。在注射后120分钟时的结果示于(以下)表3,并且99mTc-[缀合物5]示于图1和2:
表3:

实施例18:99mTc-标记的螯合剂-缀合物。
评价99mTc-缀合物在幼稚大鼠中的生物分布,以确定不同化合物的药代动力学概况。随后绘制在各个时间点在不同的器官/组织中结合剂保留的关联。产生的数据证明,在LBP1和LBP2缀合物中包括PEG通过降低肝保留改进了药代动力学(参见下表4):
表4:






Claims (18)
1. 一种成像剂,所述成像剂包含式I的化合物:

其中:
LBP为式II的羊毛硫抗生素肽:

Xaa为Arg或Lys;
Cysa-Thra、Serb-Cysb和Cysc-Thrc经由硫醚键共价连接;
Serd-Lysd经由赖氨酸丙氨酸键共价连接;
HO-Asp为β-羟基天冬氨酸;
Z1-(L)n-与Cysa连接并且任选还与LBP的Xaa连接,其中Z1包含具有至少4个金属供体原子的螯合剂的放射性金属络合物;
Z2与LBP的C-末端连接,并且为OH、OBc或MIG,
其中Bc为生物相容的阳离子;和
MIG为代谢抑制基团,其为抑制或遏抑LBP肽的体内代谢的生物相容的基团;
L为式-(A)m-的合成的接头基团,其中每个A独立地为-CR2-、-CR=CR-、-C≡C-、-CR2CO2-、-CO2CR2-、-NRCO-、-CONR-、-NR(C=O)NR-、-NR(C=S)NR-、-SO2NR-、-NRSO2-、-CR2OCR2-、-CR2SCR2-、-CR2NRCR2-、C4-8环杂亚烷基、C4-8环亚烷基、C5-12亚芳基或C3-12杂亚芳基、氨基酸、糖或单分散聚乙二醇(PEG)结构单元;
每个R独立地选自H、C1-4烷基、C2-4烯基、C2-4炔基、C1-4烷氧基烷基或C1-4羟基烷基;
m为数值1-20的整数;
n为数值0或1的整数。
2. 权利要求1的成像剂,其中所述螯合剂为具有至少6个供体原子的氨基羧化物配体或具有N4供体组的四齿螯合剂。
3. 权利要求2的成像剂,其中所述N4供体组为二胺二肟螯合剂或四胺螯合剂。
4. 权利要求1-3中任一项的成像剂,其中Z1仅与LBP的Cysa连接。
5. 权利要求1-4中任一项的成像剂,其中Xaa为Arg。
6. 权利要求1-5中任一项的成像剂,其中Z2为OH或OBc。
7. 权利要求1-6中任一项的成像剂,其中n为1并且L包含式-(OCH2CH2)x-的PEG基团,其中x为数值6-18的整数。
8. 权利要求1-7中任一项的成像剂,其中所述放射性金属为99mTc。
9. 式III的螯合剂缀合物:

其中:
Z3为具有至少4个金属供体原子的螯合剂;和
L、n、LBP和Z2如权利要求1-7中任一项所定义。
10. 一种制备权利要求1-8中任一项的成像剂的方法,所述方法包括使权利要求9的螯合剂缀合物与期望的放射性金属供应在合适的溶剂中反应。
11. 一种放射性药物组合物,所述组合物包含权利要求1-8中任一项的成像剂以及生物相容的载体,所述组合物呈适用于哺乳动物给药的形式。
12. 一种用于制备权利要求11的放射性药物组合物的试剂盒,所述试剂盒包含无菌、固体形式的权利要求9的螯合剂缀合物,使得当与放射性金属的无菌供应在生物相容的载体中重构时,发生溶解,以得到期望的放射性药物组合物。
13. 权利要求12的试剂盒,其中所述无菌、固体形式为冻干的固体。
14. 一种对人或动物身体成像的方法,所述方法包括使用PET或SPECT产生已分布权利要求1-8中任一项的成像剂或权利要求11的组合物的所述身体的至少部分的图像,其中所述成像剂或组合物已预先给予所述身体。
15. 权利要求14的方法,其中所述部分身体为其中涉及异常凋亡的疾病状态。
16. 权利要求14或权利要求15的方法,将所述方法重复进行,以监测用药物治疗人或动物身体的作用,在用所述药物治疗之前和之后并且还任选在用所述药物治疗期间,实现所述成像。
17. 权利要求1-8中任一项的成像剂、权利要求11的组合物或权利要求12的试剂盒在诊断人或动物身体的方法中的用途。
18. 一种诊断人或动物身体的方法,所述方法包括权利要求14或权利要求15的成像方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1016206.3A GB201016206D0 (en) | 2010-09-27 | 2010-09-27 | Apoptosis imaging agents |
| GB1016206.3 | 2010-09-27 | ||
| PCT/EP2011/066789 WO2012041862A1 (en) | 2010-09-27 | 2011-09-27 | Apoptosis imaging agents based on lantibiotic peptides |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN103118710A true CN103118710A (zh) | 2013-05-22 |
Family
ID=43128013
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2011800462575A Pending CN103118710A (zh) | 2010-09-27 | 2011-09-27 | 基于羊毛硫抗生素肽的凋亡成像剂 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20130189186A1 (zh) |
| EP (1) | EP2621545A1 (zh) |
| JP (1) | JP2013538819A (zh) |
| KR (1) | KR20130097780A (zh) |
| CN (1) | CN103118710A (zh) |
| AU (1) | AU2011310663A1 (zh) |
| BR (1) | BR112013006536A2 (zh) |
| CA (1) | CA2810573A1 (zh) |
| GB (1) | GB201016206D0 (zh) |
| MX (1) | MX2013003398A (zh) |
| RU (1) | RU2013113677A (zh) |
| SG (1) | SG188464A1 (zh) |
| WO (1) | WO2012041862A1 (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108144073A (zh) * | 2017-12-28 | 2018-06-12 | 中山大学附属第医院 | 靶向磷脂酰乙醇胺的放射性标记三聚乙二醇修饰耐久霉素多肽药物 |
| CN112203698A (zh) * | 2018-03-26 | 2021-01-08 | 通用电气医疗集团股份有限公司 | 制剂及制备方法 |
| CN113368264A (zh) * | 2020-03-09 | 2021-09-10 | 南方医科大学南方医院 | 放射性标记肉桂霉素,其制备方法及其用途 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101128219A (zh) * | 2004-07-19 | 2008-02-20 | 通用电气健康护理有限公司 | 改进的n4螯合剂缀合物 |
| WO2009114549A2 (en) * | 2008-03-10 | 2009-09-17 | Mcw Research Foundation, Inc. | 99mtc-labeled 19 amino acid containing peptide for use as phosphatidylethanolamine binding molecular probe and radiopharmaceutical |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0105224D0 (en) * | 2001-03-02 | 2001-04-18 | Nycomed Amersham Plc | Improved peptide-chelate conjugates |
| US7034113B2 (en) * | 2002-02-22 | 2006-04-25 | Paradigm Diagnostics, Llc | Bacteriocin-metal complexes in the detection of pathogens and other biological analytes |
| US7879801B2 (en) | 2002-07-15 | 2011-02-01 | Board Of Regents, The University Of Texas System | Compositions comprising cell-impermeant duramycin derivatives |
| EP1827509A2 (en) | 2004-11-19 | 2007-09-05 | MCW Research Foundation, Inc. | Method of imaging cell death in vivo |
-
2010
- 2010-09-27 GB GBGB1016206.3A patent/GB201016206D0/en not_active Ceased
-
2011
- 2011-09-27 SG SG2013017306A patent/SG188464A1/en unknown
- 2011-09-27 KR KR1020137010727A patent/KR20130097780A/ko not_active Application Discontinuation
- 2011-09-27 US US13/876,187 patent/US20130189186A1/en not_active Abandoned
- 2011-09-27 CN CN2011800462575A patent/CN103118710A/zh active Pending
- 2011-09-27 WO PCT/EP2011/066789 patent/WO2012041862A1/en active Application Filing
- 2011-09-27 AU AU2011310663A patent/AU2011310663A1/en not_active Abandoned
- 2011-09-27 JP JP2013529674A patent/JP2013538819A/ja not_active Withdrawn
- 2011-09-27 CA CA2810573A patent/CA2810573A1/en not_active Abandoned
- 2011-09-27 MX MX2013003398A patent/MX2013003398A/es unknown
- 2011-09-27 BR BR112013006536A patent/BR112013006536A2/pt not_active Application Discontinuation
- 2011-09-27 EP EP11761385.1A patent/EP2621545A1/en not_active Withdrawn
- 2011-09-27 RU RU2013113677/04A patent/RU2013113677A/ru not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101128219A (zh) * | 2004-07-19 | 2008-02-20 | 通用电气健康护理有限公司 | 改进的n4螯合剂缀合物 |
| WO2009114549A2 (en) * | 2008-03-10 | 2009-09-17 | Mcw Research Foundation, Inc. | 99mtc-labeled 19 amino acid containing peptide for use as phosphatidylethanolamine binding molecular probe and radiopharmaceutical |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108144073A (zh) * | 2017-12-28 | 2018-06-12 | 中山大学附属第医院 | 靶向磷脂酰乙醇胺的放射性标记三聚乙二醇修饰耐久霉素多肽药物 |
| CN112203698A (zh) * | 2018-03-26 | 2021-01-08 | 通用电气医疗集团股份有限公司 | 制剂及制备方法 |
| CN113368264A (zh) * | 2020-03-09 | 2021-09-10 | 南方医科大学南方医院 | 放射性标记肉桂霉素,其制备方法及其用途 |
| CN113368264B (zh) * | 2020-03-09 | 2022-09-30 | 南方医科大学南方医院 | 放射性标记肉桂霉素,其制备方法及其用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| MX2013003398A (es) | 2013-05-30 |
| RU2013113677A (ru) | 2014-11-10 |
| EP2621545A1 (en) | 2013-08-07 |
| WO2012041862A1 (en) | 2012-04-05 |
| AU2011310663A1 (en) | 2013-03-28 |
| GB201016206D0 (en) | 2010-11-10 |
| US20130189186A1 (en) | 2013-07-25 |
| CA2810573A1 (en) | 2012-04-05 |
| JP2013538819A (ja) | 2013-10-17 |
| KR20130097780A (ko) | 2013-09-03 |
| BR112013006536A2 (pt) | 2016-05-31 |
| SG188464A1 (en) | 2013-04-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN100509061C (zh) | 改进的螯合剂缀合物 | |
| Smith et al. | Radiochemical investigations of gastrin-releasing peptide receptor-specific [99mTc (X)(CO) 3-Dpr-Ser-Ser-Ser-Gln-Trp-Ala-Val-Gly-His-Leu-Met-(NH2)] in PC-3, tumor-bearing, rodent models: syntheses, radiolabeling, and in vitro/in vivo studies where Dpr= 2, 3-diaminopropionic acid and X= H2O or P (CH2OH) 3 | |
| Maresca et al. | Novel polar single amino acid chelates for technetium-99m tricarbonyl-based radiopharmaceuticals with enhanced renal clearance: application to octreotide | |
| US6200546B1 (en) | Gastrin receptor-avid peptide conjugates | |
| WO1998047524A9 (en) | Gastrin receptor-avid peptide conjugates | |
| AU2002317317A1 (en) | Improved chelator conjugates | |
| CN103228298A (zh) | 凋亡pet成像剂 | |
| CN101128219B (zh) | 改进的n4螯合剂缀合物 | |
| US9259496B2 (en) | Technetium labelled peptides | |
| JPH07149799A (ja) | 新規化合物及びその製造方法並びに診断用薬剤 | |
| WO2002087631A1 (en) | Gastrin-receptor-avid peptide conjugates | |
| CN103118710A (zh) | 基于羊毛硫抗生素肽的凋亡成像剂 | |
| Pathuri et al. | Synthesis and in vivo evaluation of Tc-99m-labeled cyclic CisoDGRC peptide conjugates for targeting αvβ3 integrin expression | |
| Lee et al. | Synthesis and application of a novel cysteine-based DTPA-NCS for targeted radioimmunotherapy | |
| EP1385556A1 (en) | Gastrin-receptor-avid peptide conjugates | |
| CN104066454A (zh) | 螯合剂 | |
| Kasten | Technetium-99m Carbonyl Complexes and Conjugation Strategies for Targeted Molecular Imaging |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20130522 |