CN102724996A - 血管内皮生长因子(vegf)受体的抑制剂及其使用方法 - Google Patents
血管内皮生长因子(vegf)受体的抑制剂及其使用方法 Download PDFInfo
- Publication number
- CN102724996A CN102724996A CN2010800599677A CN201080059967A CN102724996A CN 102724996 A CN102724996 A CN 102724996A CN 2010800599677 A CN2010800599677 A CN 2010800599677A CN 201080059967 A CN201080059967 A CN 201080059967A CN 102724996 A CN102724996 A CN 102724996A
- Authority
- CN
- China
- Prior art keywords
- composition
- antibody
- vegf receptor
- amino acid
- receptor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images


























































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2/00—Peptides of undefined number of amino acids; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/01—Preparation of mutants without inserting foreign genetic material therein; Screening processes therefor
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/573—Immunoassay; Biospecific binding assay; Materials therefor for enzymes or isoenzymes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/34—Identification of a linear epitope shorter than 20 amino acid residues or of a conformational epitope defined by amino acid residues
Abstract
本发明提供了与血管内皮生长因子(VEGF)受体胞外域(D7)的最近膜Ig-样结构域结合的成分,其中该成分拮抗该VEGF受体的活性。
Description
对相关申请的交叉引用
本申请涉及并要求2009年12月29日提交的美国临时申请序列号61/290,789的优先权,其全部内容通过引用特此引入本文。
关于政府赞助研究或开发的声明
本发明是利用由国家卫生研究院以合同号R01-AR 051448、R01-AR 051886和P50 AR054086资助的政府支持而完成的。政府可以拥有本发明的某些权利。
发明背景
血管内皮生长因子(VEGF)通过结合并激活受体酪氨酸激酶(RTK)的VEGF受体(VEGFR)家族的三名成员来调节血管和淋巴管的发育和内环境稳定性(Olsson等人,Nat.Rev.Mol.Cell.Biol.,7(5):359-371(2006))。VEGFR1(Flt1)、VEGFR2(KDR/Flk1)和VEGFR3(Flt4)是V型RTK的成员;V型RTK是包含由7个Ig-样结构域(D1-D7)组成的大的细胞外区域、单一的跨膜(TM)螺旋和具有酪氨酸激酶活性的胞质区域以及另外的调控序列的家族。VEGFR的胞外域的第二和第三Ig-样结构域,例如D2和D3,起到细胞因子的VEGF家族的五个成员(即,VEGF-A、B、C、D和胎盘生长因子(PLGF))的结合位点的作用(Barleon等人,J.Biol.Chem.,272(16):10382-10388(1997);和Shinkai等人,J.Biol.Chem.,273(47):31283-31288(1998))。这些生长因子是共价连接的同型二聚体。各原体是由在名为半胱氨酸结(cysteine-knot)生长因子的结构中以反向平行的方式排列的四链β折叠片构成的(Weismann等人,Cell,91(5):695-704(1997))。细胞因子的半胱氨酸结家族的其他成员包括神经生长因子(NGF)和血小板源生长因子(PDGF)。但是,RTK的PDGFR家族(III型)的胞外域由5个Ig-样重复序列构成,其中D1、D2和D3起到PDGFR和家族的其他成员(即KIT、CSF1R和Flt3)的配体结合区域的作用。结构和生化实验已经表明,SCF结合细胞外区域诱导KIT二聚化,该步骤随后是邻近的KIT分子的两个近膜Ig-样结构域D4和D5之间的同型接触(Yuzawa等人,Cell,130(2):323-334(2007))。野生型和致癌KIT突变体的生化研究已经表明,同型D4和D5接触对于KIT二聚体在胞质区中以有利于反式自磷酸化、激酶活化和细胞信号传导的距离和方向来定位起到关键作用。然而,有必要更好地表征VEGF受体的结构。这种表征将会导致可以作为药物、药品或其他生物药剂靶向的区域的有意鉴定。
发明概述
本发明提供了结合血管内皮生长因子受体(VEGF受体)例如,VEGFR1(Flt1)、VEGFR2(KDR/Flk1)和VEGFR3(Flt4)的胞外域的成分(moiety),例如,抗体或其抗原结合部分、小分子、肽类分子、适体和adnectin。本发明的成分可以锁定VEGF受体的胞外域处于非活性状态,从而抑制VEGF受体的活性。在本发明的一个实施方案中,该成分锁定VEGF受体的胞外域为单体状态。在本发明的另一个实施方案中,该成分允许VEGF受体的胞外域二聚化,但影响两个单体的Ig-样结构域(例如,VEGF受体的D7-D7结构域)之间的定位、定向和/或距离,从而抑制VEGF受体的活性。换句话说,该成分可以允许配体诱导的VEGF受体胞外域的二聚化,但影响两个胞外域在细胞表面界面处的定位或改变或阻止VEGF受体中的构象变化,从而抑制VEGF受体的活性(例如,抑制受体内化和/或抑制受体的酪氨酸自磷酸化和/或抑制受体激活下游信号传导途径的能力)。本发明至少部分地基于VEGF2受体胞外域部分的晶体结构的解译。这种晶体结构的解译使得能够鉴别本发明的成分可以靶向的表位,例如,构象表位。
本发明还至少部分地基于发现邻近受体之间的同型D7相互作用不是在受体二聚化中起作用,而是两个受体的近膜区以能够实现导致酪氨酸激酶激活的胞质域之间的相互作用的距离和定向精确定位所必需的。
因此,一方面,本发明提供了一种结合人血管内皮生长因子受体(VEGF受体)的胞外域的成分,其中该成分锁定VEGF受体的胞外域于非活性状态,从而拮抗VEGF受体的活性。在一个实施方案中,该成分结合人VEGF受体的Ig-样结构域。在一个实施方案中,该Ig-样结构域不负责配体与VEGF受体的结合。在另一个实施方案中,该Ig-样结构域负责配体与VEGF受体的结合。在一个实施方案中,该成分不阻断VEGF受体和VEGF受体配体之间的相互作用。在另一个实施方案中,该成分阻断VEGF受体和VEGF受体配体之间的相互作用。在一个实施方案中,该成分不阻止VEGF受体的二聚化。在另一个实施方案中,该成分阻止VEGF受体的二聚化。
在一个实施方案中,该成分阻止VEGF受体各原体的胞外域的近膜区之间的相互作用。在另一个实施方案中,该相互作用是同型的。而在另一个实施方案中,该相互作用是异型的。
在一个实施方案中,胞外域的近膜区是VEGF受体的第七Ig-样结构域(D7)。在另一个实施方案中,该成分结合到VEGF受体的D7结构域的下列共有序列:L/IX1 R Φ X2 X3 X4 D/E X5 G(SEQ ID NO:158),其中L是亮氨酸,I是异亮氨酸,R是精氨酸,Φ是疏水性氨基酸,D是天冬氨酸,E是谷氨酸,G是甘氨酸以及X1、X2、X3、X4和X5是任何氨基酸。在具体的实施方案中,Φ是缬氨酸;X1选自于精氨酸、谷氨酰胺、谷氨酸和天冬氨酸;X2选自于精氨酸、赖氨酸和苏氨酸;X3选自于赖氨酸、谷氨酸、谷氨酰胺和缬氨酸;X4选自于谷氨酸和缬氨酸;以及X5选自于谷氨酸、甘氨酸、丝氨酸和谷氨酰胺。
在另一个实施方案中,该成分引起VEGF受体各原体的胞外域的近膜区以约 或者
或者 的距离间隔。在一个实施方案中,该成分锁定VEGF受体的胞外域于非活性状态。
的距离间隔。在一个实施方案中,该成分锁定VEGF受体的胞外域于非活性状态。
在一个实施方案中,VEGF受体是VEGFR1(Flt1)。在另一个实施方案中,VEGF受体是VEGFR2(KDR/Flk1)。在另一个实施方案中,VEGF受体是VEGFR3(Flt4)。
在另一个实施方案中,该成分结合VEGFR2的氨基酸残基Arg726。在另一个实施方案中,该成分结合VEGFR2的氨基酸残基Asp731。在另一个实施方案中,该成分结合VEGFR2的氨基酸残基Arg726和Asp731。在再另一个实施方案中,该成分结合选自于包括VEGFR2的氨基酸残基724、725、726、727、728、729、730、731、732和733的组中的一个或多个氨基酸残基。该成分可以结合在任何上述氨基酸残基的 或
或 内。
内。
在一个实施方案中,该成分结合VEGFR1的氨基酸残基Arg720。在另一个实施方案中,该成分结合VEGFR1的氨基酸残基Asp725。在另一个实施方案中,该成分结合VEGFR1的氨基酸残基Arg720和Asp725。在另一个实施方案中,该成分结合选自于包括VEGFR1的氨基酸残基718、719、720、721、722、723、724、725、726和727的组中的一个或多个氨基酸残基。该成分可以在结合上述任何氨基酸残基的 或
或 内。
内。
在一个实施方案中,该成分结合VEGFR3的氨基酸残基Arg737。在另一个实施方案中,该成分结合VEGFR3的氨基酸残基Asp742。在另一个实施方案中,该成分结合VEGFR3的氨基酸残基Arg737和Asp742。在再另一个实施方案中,该成分结合选自于包括VEGFR3的氨基酸残基735、736、737、738、739、740、741、742、743和744的组中的一个或多个氨基酸残基。该成分可以结合在上述任何氨基酸残基的 或内。
或内。
在一个实施方案中,该成分结合VEGF受体胞外域上的构象表位。在一个实施方案中,该构象表位由VEGF受体的D7结构域中的两个或更多个残基构成。在再另一个实施方案中,构象表位包含或由氨基酸残基Arg726和Asp731;Arg 720和Asp 725;或者Arg737和Asp742组成。在某些实施方案中,该成分结合在上述构象表位的
 或内。
或内。
在另一个实施方案中,该成分结合VEGF受体上的连续表位。在一个实施方案中,连续表位由VEGF受体D7结构域中的两个或更多个残基组成。在另一个实施方案中,连续表位是选自于VEGFR1的672VAISSS677、VEGFR1的678TTLDCHA684、VEGFR1的685NGVPEPQ691、VEGFR1的700KIQQEPG706、VEGFR1的707IILG710、VEGFR1的711PGS713、VEGFR1的714STLFI718、VEGFR1的719ERVTEEDEGV728、VEGFR3的689VNVSDS694、VEGFR3的695LEMQCLV701、VEGFR3的702AGAHAPS708、VEGFR3的717LLEEKSG723、VEGFR3的724VDLA727、VEGFR3的728DSN730、VEGFR3的731QKLSI735和VEGFR3的736QRVREEDAGR745、VEGFR2的678TSIGES683、VEGFR2的684IEVSCTA690、VEGFR2的691SGNPPPQ697、VEGFR2的706TLVEDSG712、VEGFR2的713IVLK716、VEGFR2的717DGN719、VEGFR2的720RNLTI724和VEGFR2的725RRVRKEDEGL734的表位。在一些实施方案中,该成分可以结合在任何上述表位的 或
或 内。
内。
在一个实施方案中,该成分阻断配体诱导的VEGF受体酪氨酸自磷酸化作用。在另一个实施方案中,该成分阻断配体诱导的VEGF受体的内化。
在一个实施方案中,结合VEGF受体胞外域的成分是分离的抗体或其抗原结合部分。在另一个实施方案中,该抗体或其抗原结合选自于人抗体、人源化抗体、双特异性抗体和嵌合抗体。在另一个实施方案中,该抗体或其抗原结合部分包含选自于IgG1、IgG2、IgG3、IgG4、IgM、IgA和IgE恒定区的重链恒定区。在一个实施方案中,该抗体重链恒定区是IgG1。在另一个实施方案中,抗体或其抗原结合部分选自于包括Fab片段、F(AB′)2片段、单链Fv片段、SMIP、亲和体(affibody)、亲和性多聚体(avimer)、纳米抗体(nanobody)以及单域抗体的组。在再另一个实施方案中,抗体或其抗原结合部分以选自于包括1x 10-7M或更小、更优选地5x 10-8M或更小、更优选地1x 10-8M或更小、更优选地5x 10-9M或更小的KD结合受体酪氨酸激酶的Ig-样结构域。
一方面,本发明提供了产生结合本文所描述的VEGF受体胞外域的抗体或其抗原结合部分的杂交瘤。
在一个实施方案中,结合VEGF受体胞外域的部分是小分子。在另一个实施方案中,该小分子结合VEGFR2的氨基酸残基Arg726或Asp731中的至少一个。在另一个实施方案中,该小分子结合VEGFR1的氨基酸残基Arg720或Asp725中的至少一个。在另一个实施方案中,该小分子结合VEGFR3的氨基酸残基Arg737或Asp742中的至少一个。
在另一个实施方案中,结合VEGF受体胞外域的成分是一种肽类分子。在一个实施方案中,该肽类分子是基于VEGF受体的Ig-样结构域设计的。在另一个实施方案中,该肽类分子是基于人VEGF受体的D7结构域设计的。在一个实施方案中,肽类分子包含结构:L/I X1R Φ X2 X3 X4 D/E X5 G(SEQ ID NO:158),其中L是亮氨酸,I是异亮氨酸,R为精氨酸,Φ是疏水性氨基酸,D是天冬氨酸,E是谷氨酸,G是甘氨酸以及X1、X2、X3、X4和X5是任何氨基酸。在具体的实施方案中,Φ是缬氨酸;X1选自于精氨酸、谷氨酰胺、谷氨酸和天冬氨酸;X2选自于精氨酸、赖氨酸和苏氨酸;X3选自于赖氨酸、谷氨酸、谷氨酰胺和缬氨酸;X4选自于谷氨酸和缬氨酸;且X5选自于谷氨酸、甘氨酸、丝氨酸和谷氨酰胺。
在另一个实施方案中,肽类分子包含与人VEGFR2的氨基酸残基724-733、678-683、684-690、691-697、706-712、713-716、717-719、720-724或725-734至少80%、85%、90%或95%相同的结构。在另一个实施方案中,肽类分子包含与人VEGFR1的氨基酸残基718-727、672-677、678-684、685-691、700-706、707-710、711-713、714-718或719-728至少80%、85%、90%或95%相同的结构。在另一个实施方案中,肽类分子包含与人VEGFR3的氨基酸残基735-744、689-694、695-701、702-708、717-723、724-727、728-730、731-735或736-745至少80%、85%、90%或95%相同的结构。在另一个实施方案中,肽类分子包含至少一个D-氨基酸残基。
在一个实施方案中,结合VEGF受体胞外域的成分是adnectin。
在另一个方面,本发明提供了结合人类VEGF受体的D7结构域上的构象表位并拮抗人VEGF受体的活性的成分,其中所述的构象表位包含VEGFR2的残基Arg726和Asp731;VEGFR1的残基Arg720和Asp725;或VEGFR3的残基Arg737和Asp742。
在另一个方面,本发明提供结合VEGFR2的氨基酸残基Arg726和Asp731;VEGFR1的氨基酸残基Arg720和Asp725;或VEGFR3的氨基酸残基Arg737和Asp742,从而拮抗人VEGF受体的活性的成分。
在另一个方面,发明提供了包含如本文所述的结合VEGF受体的胞外域的成分和药学上可接受的载体的药物组合物。
在另一个方面,本发明提供了一种治疗或预防受试者中VEGF受体相关疾病的方法,该方法包括向受试者施用有效量的本发明的成分,从而治疗或预防该疾病。在一个实施方案中,VEGF受体酪氨酸激酶相关疾病选自于癌症、年龄相关性黄斑变性(AMD)、动脉粥样硬化、类风湿性关节炎、糖尿病性视网膜病、淋巴系统的疾病和疼痛相关疾病。在一个实施方案中,癌症选自于GIST、AML、SCLC、肾癌、结肠癌、乳腺癌、淋巴癌和其生长由基质支持的癌症。
在一个方面,本发明提供了用于鉴定结合VEGF受体的胞外域,如Ig-样结构域的成分的方法,该方法包括:将VEGF受体与候选成分接触;同时地或相继地将VEGF受体与VEGF受体的配体接触;和测定该成分是否影响配体诱导的二聚VEGF受体的Ig-样结构域之间的定位、定方和/或距离,从而鉴定结合VEGF受体的胞外域,如Ig-样结构域的成分。在一个实施方案中,该成分锁定VEGF受体的胞外域于非活性状态。在另一个实施方案中,该成分结合VEGF受体的第七Ig-样结构域(D7)。
在另一个方面,本发明提供了一种结合人VEGF受体的D7结构域上的构象表位的分离的抗体或其抗原结合部分,其中该抗体或其抗原结合部分拮抗人VEGF受体的活性,并且其中所述构象表位包含VEGFR2的残基Arg726和Asp731残基。在另一个方面,本发明提供了一种结合人VEGF受体的D7结构域上的构象表位的分离的抗体或其抗原结合部分,其中该抗体或其抗原结合部分拮抗人VEGF受体的活性,并且其中所述构象表位包含VEGFR1的残基Arg720和Asp725。在另一个方面,本发明提供了一种结合人VEGF受体的D7结构域上的构象表位的分离的抗体或其抗原结合部分,其中该抗体或其抗原结合部分拮抗人VEGF受体的活性,并且其中所述构象表位包含VEGFR3的残基Arg737和Asp742。
在另一个方面,本发明提供了一种结合VEGFR2的氨基酸残基724-733,从而拮抗VEGFR2的活性的分离的抗体或其抗原结合部分。在一个方面,本发明提供了一种结合VEGFR1的氨基酸残基718-727,从而拮抗VEGFR1的活性的分离的抗体或其抗原结合部分。在另一个方面,本发明提供了一种结合VEGFR3的氨基酸残基735-744,从而拮抗VEGFR3的活性的分离的抗体或其抗原结合部分。
在一个方面,本发明提供了一种结合选自于人VEGFR2的Arg726和Asp731的至少一个氨基酸残基,从而拮抗人VEGFR2的活性的分离的抗体或其抗原结合部分。在另一个方面,本发明提供了一种结合选自于人VEGFR1的Arg720和Asp725的至少一个氨基酸残基,从而拮抗人类VEGFR1的活性的分离的抗体或其抗原结合部分。在另一个方面,本发明提供了一种结合选自于人类VEGFR3的Arg737和Asp742的至少一个氨基酸残基,从而拮抗人VEGFR3的活性的分离的抗体或其抗原结合部分。
在另一个方面,本发明提供了结合人受体酪氨酸激酶的胞外域,例如,Ig-样结构域或铰链区的成分,其中该成分锁定受体酪氨酸激酶的胞外域于非活性状态,从而拮抗受体酪氨酸激酶的活性。在一个实施方案中,该Ig-样结构域可以负责或不负责配体与受体酪氨酸激酶的结合。在另一个实施方案中,该成分可以阻断或不阻断受体酪氨酸激酶和受体酪氨酸激酶的配体之间的相互作用。在再另一个实施方案中,本发明的成分可以阻止或不阻止受体酪氨酸激酶的二聚化。在进一步的实施方案中,本发明的成分可以不阻止配体诱导的受体二聚化,但会阻止受体酪氨酸激酶活化所需的近膜区之间的同型或异型相互作用。
在一些实施方案中,本发明的成分阻止受体酪氨酸激酶各原体的胞外域的近膜区之间的同型或异型相互作用。例如,本发明的成分可以导致受体酪氨酸激酶各原体的胞外域末端(胞外域最接近于细胞膜的端部)以大于约 约
约 约约约
约约约 或约
或约 的距离间隔。
的距离间隔。
在优选的实施方案中,受体酪氨酸激酶是III型受体酪氨酸激酶,如Kit、PDGFRα、PDGFRβ、CSF1R、Fms、Flt3或Flk2。
在其他的实施方案中,被本发明的成分结合的Ig-样结构域是III型受体酪氨酸激酶的D4结构域。在一个具体的实施方案中,该成分结合D4相互作用位点的以下共有序列:LX1RX2X3X4X5X6X7G,其中L为亮氨酸,R为精氨酸,G是甘氨酸,且X1、X2、X3、X4、X5、X6和X7是任何氨基酸。在具体的实施方案中,X1选自于苏氨酸、异亮氨酸、缬氨酸、脯氨酸、天冬酰胺或赖氨酸;X2选自于亮氨酸、缬氨酸、丙氨酸和甲硫氨酸;X3选自于赖氨酸、组氨酸、天冬酰胺和精氨酸;X4选自于甘氨酸、缬氨酸、丙氨酸、谷氨酸、脯氨酸和甲硫氨酸;X5选自于苏氨酸、丝氨酸、谷氨酸、丙氨酸、谷氨酰胺和天冬氨酸;X6选自于谷氨酸、天冬氨酸和谷氨酰胺;以及X7选自于甘氨酸、丝氨酸、丙氨酸、赖氨酸、精氨酸、谷氨酰胺和苏氨酸。
在另一个实施方案中,被本发明的成分结合的Ig-样结构域是III型受体酪氨酸激酶的D5结构域,如人Kit的氨基酸残基309-413或410-519。在具体的实施方案中,本发明的成分可以结合到D5相互作用位点的保守氨基酸的共有序列。
在另一个实施方案中,本发明的成分结合III型受体酪氨酸激酶D4或D5结构域的突变体或结合V型受体酪氨酸激酶D7结构域的突变体。在具体的实施方案中,该成分结合在人Kit的突变D5结构域中的点突变,其中该突变选自于Thr417、Tyr418、Asp419、Leu421、Arg420、Tyr503和Ala502。
在一些实施方案中,III型受体酪氨酸激酶是人Kit和本发明的成分结合从下面表4所示的那些氨基酸残基组成的组中选择的1个或多个氨基酸残基,如2个或更多、3个或更多、4个或更多、5个或更多、6个或更多、7个或更多、8个或更多、9个或更多、10个或更多、11个或更多、12个或更多、13个或更多、14个或更多、15个或更多、16个或更多、17个或更多或18个或更多个氨基酸残基。例如,本发明的成分可以结合以下的一个或多个残基:Y125、G126、H180、R181、K203、V204、R205、P206、P206、F208、K127、A207、V238、S239、S240、S241、H263、G265、D266、F267、N268、Y269、T295、L222、L222、L223、E306、V308、R224、V308、K310、K218、A219、S220、K218、A220、Y221、A339、D327、D398、E338、E368、E386、F312、F324、F340、F355、G311、G384、G387、G388、I371、K342、K358、L382、L379、N326、N367、N370、N410、P341、S369、T385、V325、V407、V409、Y373、Y350、Y408、T380、T390、R381、R353、T411、K412、E414、K471、F433、G470、L472、V497、F469、A431或G432。在具体的实施方案中,本发明的成分结合选自于K218、S220、Y221、L222、F340、P341、K342、N367、E368、S369、N370、I371和Y373中的Kit受体的至少一个氨基酸残基,或选自于Y350、R353、F355、K358、L379、T380、R381、L382、E386和T390中的Kit受体的至少一个氨基酸残基。本发明的成分可以结合形成表4中确定的口袋或空腔的所有残基或者他们可以结合形成口袋或空腔的残基的亚集。本领域的技术人员可以理解,在一些实施方案中,本发明的成分可以很容易地靶向其他III型RTK中与以上列出的那些残基对应的残基,例如,那些形成类似的口袋或空腔的残基或者通过结构比对或序列比对处于相同位置的残基。
在另一个实施方案中,本发明的成分结合人Kit的氨基酸残基381Arg和386Glu。在再另一个实施方案中,本发明的成分结合人Kit的氨基酸残基418Tyr和/或505Asn。
在进一步的实施方案中,本发明的成分结合PDGFRα或PDGFRβ受体。在类似的实施方案中,本发明的成分结合人PDGFRβ的氨基酸残基385Arg和/或390Glu,或PDGFRα中的相应残基。
在再另一个实施方案中,本发明的成分结合III型RTK的构象表位。在具体的实施方案中,该构象表位由III型RTK,例如人Kit受体或PDGF受体,的D3、D4或D5结构域或铰链区中的两个或更多个残基构成。在进一步的具体实施方案中,本发明的成分可以结合由选自于表4中列出的那些氨基酸残基中的两个或更多个残基,如2个或更多、3个或更多、4个或更多、5个或更多、6个或更多、7个或更多、8个或更多、9个或更多、10个或更多、11个或更多、12个或更多、13个或更多、14个或更多、15个或更多、16个或更多、17个或更多或18个或更多个氨基酸残基构成的人Kit受体中的构象表位。在特定的实施方案中,本发明的成分结合由选自于Y125、H180、R181、K203、V204、R205、P206、V238、S239、S240、H263、G265、D266、F267、N268和Y269的2个或更多个氨基酸组成的构象表位。在类似的实施方案中,本发明的成分结合由选自于下组的氨基酸的2个或更多个氨基酸组成的构象表位:P206、F208、V238和S239;K127、A207、F208和T295;L222、A339、F340、K342、E368、S369、N370、I371和Y373;L222、L223、E306、V308、F312、E338、F340和I371;R224、V308、K310、G311、F340、P341和D398;K218、A219、S220、N367、E368和S369;K218、A220、E368和S369;G384、T385、T411、K412、E414和K471;Y408、F433、G470、K471和L472;F324、V325、N326和N410;D327、N410、T411、K412和V497;G384、G387、V409和K471;L382、G387、V407和V409;Y125、G126、H180、R181、K203、V204、R205、P206、F208、V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269;P206、F208、V238和S239;K218、S220、Y221、L222、F340、P341、K342、N367、E368、S369、N370、I371和Y373;G384、G387、G388、Y408、V409、T411、F433、F469、G470和K471;D327、T411、K412、E414、A431、G432和K471;Y350、F355、K358、L379、T380、R381、L382、E386和T390;Y350、R353和F355。如上所述,本发明的成分可以结合形成表4中确定的口袋或空腔的所有残基或者他们可以结合形成口袋或空腔的残基的亚集。
在进一步的实施方案中,本发明的成分结合构象表位,其中该构象表位是由选自于表5中列出的肽中的两个或更多个氨基酸残基组成的。在具体的实施方案中,该构象表位是由选自于第一肽的一个或多个氨基酸残基和选自于第二肽的一个或多个氨基酸残基组成的,其中该第一和第二肽选自于表5中所列的肽。因此,本发明的成分可以结合其中第一和第二肽组如下的构象表位:Ala219-Leu222和Thr304-Val308;Asp309-Gly311和Arg224-Gly226;Thr303-Glu306和Ala219-Leu222;Asn367-Asn370和Ser217-Tyr221;Ala339-Pro343和Asn396-Val399;Ala339-Pro343和Glu368-Arg372;Lys358-Tyr362和Val374-His378;Asp357-Glu360和Leu377-Thr380;Met351-Glu360和His378-Thr389;His378-Thr389和Val323-Asp332;Val409-Ile415和Ala493-Thr500;Val409-Ile415和Ala431-Thr437;Val409-Ile415和Phe469-Val473;Val409-Ile415和Val325-Asn330;Val409-Ile415和Arg381-Gly387;Gly466-Leu472和Gly384-gly388;Val325-Glu329和Tyr494-Lys499;Thr411-Leu416和Val497-Ala502;Ile415-Leu421和Ala502-Ala507;Ala502-Ala507和Lys484-Thr488;以及Ala502-Ala507和Gly445-Cys450。本发明的成分可以结合形成上述的第一和第二肽组的所有氨基酸残基,或他们可以结合形成上述的第一和第二肽组的残基的亚集。
在其他实施方案中,本发明的成分结合作为VEGF受体家族(V型受体酪氨酸激酶)成员的受体酪氨酸激酶,例如,VEGFR-1(Flt1)、VEGFR-2(Flk1)和VEGFR-3(Flt4)。在一些实施方案中,本发明的成分所结合的Ig-样结构域可以是VEGF受体家族成员的D7结构域。在具体的实施方案中,该成分结合VEGF受体家族成员的D7结构域的以下共有序列:IX1RVX2X3EDX4G,其中I是异亮氨酸,R是精氨酸,E是谷氨酸,D是天冬氨酸,G是甘氨酸;且X1、X2、X3和X4是任何氨基酸。在具体的实施方案中,X1选自于谷氨酸、精氨酸和谷氨酰胺;X2选自于精氨酸和苏氨酸;X3选自于谷氨酸和赖氨酸;且X4选自于谷氨酸和丙氨酸(SEQ ID NO:1)。
在一些实施方案中,本发明的成分是分离的抗体或其抗原结合部分。该抗体或其抗原结合部分可以是人抗体、人源化抗体、双特异性抗体或嵌合抗体。在一些实施方案中,该抗体或其抗原结合部分包含选自于IgG1、IgG2、IgG3、IgG4、IgM、IgA和IgE恒定区的重链恒定区。在优选的实施方案中,该抗体重链恒定区是IgG1。此外,本发明的成分可以是抗体或其抗原结合部分,其中该抗体或其抗原结合部分选自于Fab片段、F(ab′)2片段、单链Fv片段、SMIP、亲和体、亲和性多聚体、纳米抗体以及单域抗体。在特定的实施方案中,本发明的抗体或其抗原结合部分以1x 10-7M或更小、更优选地5x 10-8M或更小、更优选地1x 10-8M或更小、更优选地5x 10-9M或更小的KD结合受体酪氨酸激酶的Ig-样结构域。
在一些实施方案中,本发明的分离的抗体或其抗原结合部分结合人Kit的氨基酸残基309-413或410-519,从而使人Kit的胞外域锁定于非活性状态并拮抗人Kit的活性。
在进一步的实施方案中,本发明包括产生本发明的抗体或其抗原结合部分的杂交瘤。
在另一优选的实施方案中,本发明的成分是小分子。
在一些优选的实施方案中,本发明的小分子结合选自于表4所示的那些氨基酸残基中的一个或多个氨基酸残基。例如,本发明的小分子可以与下面的一个或多个残基结合:Y125、G126、H180、R181、K203、V204、R205、P206、P206、F208、K127、A207、V238、S239、S240、S241、H263、G265、D266、F267、N268、Y269、T295、L222、L222、L223、E306、V308、R224、V308、K310、K218、A219、S220、K218、A220、Y221、A339、D327、D398、E338、E368、E386、F312、F324、F340、F355、G311、G384、G387、G388、I371、K342、K358、L382、L379、N326、N367、N370、N410、P341、S369、T385、V325、V407、V409、Y373、Y350、Y408、T380、T390、R381、R353、T411、K412、E414、K471、F433、G470、L472、V497、F469、A431或G432。在具体的实施方案中,本发明的小分子结合选自于K218、S220、Y221、L222、F340、P341、K342、N367、E368、S369、N370、I371和Y373的Kit受体中的至少一个氨基酸残基。在相关的实施方案中,本发明的小分子结合选自于Y350、R353、F355、K358、L379、T380、R381、L382、E386和T390的Kit受体中的至少一个氨基酸残基。本领域的技术人员可以理解,在一些实施方案中,本发明的小分子可以很容易地靶向其他的III型RTK中与上述所列出的那些残基对应的残基,例如,那些形成类似的口袋或空腔的残基或那些通过结构比对或序列比对处于相同位置的残基。
在进一步的实施方案中,本发明的成分是肽类分子。在一些实施方案中,该肽类分子是基于受体酪氨酸激酶的Ig-样结构域设计的。在具体的实施方案中,本发明的肽类分子是基于Kit的D4结构域设计的。本发明的肽类分子可以包含保守的D4相互作用位点,例如,如上所述的D4共有序列(LX1RX2X3X4X5X6X7G),或通过比对或比较III型受体酪氨酸激酶的D4结构域所产生的其他序列。在另外的实施方案中,本发明的肽类分子包含与人Kit的氨基酸残基309-413至少80%相同的结构,或与人Kit的氨基酸残基410-519至少80%相同的结构。该肽类成分也可以基于Kit的D5结构域而设计,且在进一步的优选实施方案中,可以包含通过比对或比较III型受体酪氨酸激酶的D5结构域产生的共有序列。在可选的实施方案中,肽类分子可以基于突变的D5结构域的序列或共有序列而设计。
本发明的肽类成分可以是包含或由本文所确认的任何氨基酸序列组成的肽(例如,SEQ ID NO:1-89、92、93和105-157)。
在一些实施方案中,本发明的肽类分子包含至少一个D-氨基酸残基。
在另一优选的实施方案中,本发明的成分是adnectin。
此外,在一些实施方案中,本发明的小分子和肽类分子结合靶RTK中的构象表位。在其他实施方案中,本发明的小分子和肽类分子结合到靶RTK中作为非构象构象表位的表位。
在另一个方面,本发明提供了包含本发明的任何成分以及药学上可接受的载体的药物组合物。
在其他方面,本发明提供了治疗或预防受试者中受体酪氨酸激酶相关疾病的方法。该方法包括向受试者施用有效量的本发明的成分(例如,结合III型受体酪氨酸激酶的D4或D5结构域或V型受体酪氨酸激酶的D7结构域的成分),从而治疗或预防该疾病。在优选的实施方案中,受体酪氨酸激酶相关疾病是淋巴疾病或癌症,如GIST、AML、SCLC、黑色素瘤、肾癌、结肠癌、乳腺癌、淋巴癌及其他癌症。
在另一个方面,本发明提供了治疗或预防受试者中受体酪氨酸激酶相关疾病的方法,该方法通过向受试者施用有效量的结合人III型受体酪氨酸激酶的D3-D4和/或D4-D5铰链区的成分从而治疗或预防该疾病。在具体的实施方案中,该受体酪氨酸激酶相关疾病是癌症,如GIST、AML、SCLC、黑色素瘤、肾癌、结肠癌、乳腺癌、淋巴癌及其他癌症。
在另一个方面,本发明提供了用于鉴定结合受体酪氨酸激酶的Ig-样结构域和使受体酪氨酸激酶的胞外域锁定于非活性状态的成分的方法。该方法包括将受体酪氨酸激酶与候选成分接触;同时地或相继地将受体酪氨酸激酶与受体酪氨酸激酶的配体接触;和测定该成分是否影响配体诱导的二聚受体酪氨酸激酶的Ig-样结构域之间的定位、定向和/或距离,从而鉴定结合受体酪氨酸激酶的Ig-样结构域和使受体酪氨酸激酶的胞外域锁定于非活性状态的成分。
在进一步的方面,本发明提供了用于鉴定将III型受体酪氨酸激酶的胞外域锁定于非活性状态的成分的方法。该方法包括将III型受体酪氨酸激酶与候选成分接触;同时地或相继地将受体酪氨酸激酶与受体酪氨酸激酶的配体接触;和测定该成分是否影响配体诱导的二聚受体酪氨酸激酶的D4-D4或D5-D5结构域之间的定位、定向和/或距离,从而鉴定将III型受体酪氨酸激酶的胞外域锁定于非活性状态的成分。
从下面的详细描述和权利要求能够清楚本发明的其他特征和优势。
附图简要说明
该专利或申请案包含至少一个以彩色完成的附图。带有彩色附图的本专利或专利申请公开文本将根据要求和缴纳必要费用后由专利局提供。
图1A-E描绘了Kit胞外域的晶体结构。图1A显示了Kit胞外域单体的带状栅图(ribbon diagram)(左)和表面示意图(surface representation)(右)。右图显示的是左图中显示的视图沿垂直轴旋转了90°后的视图。D1标为蓝色、D2标为绿色、D3标为黄色、D4标为橙色和D5标为粉色,标记了N和C末端。D1和D5中的二硫键用球-棍表示,使硫原子标为橙色。天冬酰胺连接的糖以棍模型显示。图1B-E提供了D1-D2(B)、D2-D3(C)、D3-D4(D)和D4-D5(E)界面的详细视图。颜色编码与图1A相同。参与结构域-结构域相互作用的氨基酸被标记,并且氢键用黄色虚线画出。根据IgSF命名法命名二级结构元件。
图2A-B描绘了SCF-Kit胞外域2:2复合体的晶体结构。图2A显示了SCF-Kit 2:2复合体的带状栅图。D1到D5的颜色编码与图1相同,并且SCF标为红紫色。标记了Kit以及SCF的N和C末端。D1和D5中的二硫键用球-棍表示,使硫原子标为橙色。天冬酰胺连接的糖显示为棍模型。箭头标记SCF-Kit 2:2复合体中大的空腔。图2B显示了SCF-Kit胞外域2:2复合体的表面示意图。该图显示了俯视图(顶图)、正视图(中左图)、侧视图(中右图)和仰视图(下图)。颜色编码与图A相同。视图显示SCF二聚体与两个相应的Kit胞外域的D1、D2和D3对称地相互作用。此外,Kit胞外域通过两个相邻受体的D4(橙色)之间和D5(粉红色)之间的侧向接触形成同型相互作用。
图3A-E描绘了Kit的SCF识别。图3A显示了SCF-Kit界面的视图。通过拉开两个分子使SCF和Kit胞外域的包埋的表面中的氨基酸可视化。该图显示了Kit D1-D2-D3(左图)和SCF(右图)的分子表面。酸性氨基酸用红色表示,碱性氨基酸用蓝色表示,极性氨基酸用橙色表示和疏水性氨基酸用黄色表示。Kit上的SCF结合位点-I、位点-II和位点-III被圈起来。图3B描绘了在配体-受体界面中静电势的互补性。右图显示了沿左图呈现的静电表面的垂直轴旋转180°后的视图。D1-D2-D3的静电表面电位叠加在具有以标为绿色的结合SCF的草图印记的分子表面上。右图描绘了SCF-结合Kit的静电表面电位,标为蓝色(正)和红色(负)。Kit以蓝绿色的带状栅图形式表示。图3C-E显示SCF-Kit界面的位点-I(C)、位点-II(D)和位点-III(E)的近视图。SCF标为绿色和Kit标为蓝绿色。标记了相互作用的氨基酸,用黄色虚线画出氢键和用色和条标记二级结构元件。
图4A-C描绘了SCF结合Kit时的构象变化。图4A显示当Kit结合时两个SCF原体之间的角度发生了改变。该视图显示了游离的SCF(绿色)和结合Kit的SCF(红紫色)的草图。一个SCF原体(左)的叠加揭示了第二个原体(右)大约5°的角移动,正如对αC螺旋所测量的。标记了螺旋并用圆柱体表示。图4B描绘了当Kit结合时SCF的N-末端的构象变化。Kit的位点-III显示为分子表面(灰色)、游离SCF的N-末端显示为绿色和结合Kit的SCF的N-末端为红紫色。Cys4’和Cys89’之间的二硫键显示为黄色球。标记了关键氨基酸并显示为棍模型。图4C描绘了当结合Kit的位点-I时SCF的αC-β2环的构象变化。颜色编码与图B相同。
图5A-B描绘了SCF结合时Kit D4和D5的重构。图5A显示了SCF-Kit复合体中D4和D5的重构。Kit单体的D3与结合SCF的Kit的D3(都标为蓝色)的叠加显示,结合形式的D4(红色)相对于游离形式的D4(绿色)的位置移动了22°。两种形式的D4(都为蓝色)的叠加(右图)表明SCF结合形式的D5(红色)相对于游离形式的D5(绿色)的位置移动了27°。两幅底图显示了单体(绿色)和同型二聚体(红色)形式的D3-D4和D4-D5界面的铰链区的近视图。图5B显示了SCF占据的Kit的D4和D5的表面示意图(顶图),以与图2相同的方向观察。黑色轮廓显示通过结合到配体结合区域的SCF桥接的Kit胞外域单体的D4和D5的位置。D4和D5的重构导致了两个相邻的胞外域的C-末端彼此之间的 到
到 的移动。下图显示SCF-Kit复合体的细胞膜的视图(仰视图)。注意沿x轴的90°的旋转。D1到D5的颜色编码同图1。
的移动。下图显示SCF-Kit复合体的细胞膜的视图(仰视图)。注意沿x轴的90°的旋转。D1到D5的颜色编码同图1。
图6A-D描绘了D4-D4和D5-D5界面的视图。图6A(顶图)显示了呈现D4-D4界面的1.1σ水平下勾画的2Fo-Fc电子密度图。Kit原体的骨架分别呈现为粉红色和黄色管。两个相邻胞外域的D4-D4界面的近视图(下图)。在两个相邻D4的Arg381和Glu386之间形成的链间氢键标为黄色。标记了关键氨基酸并用棍模型显示。根据IgSF命名法标记二级结构元件。图6B描绘了III型和V型RTK家族的成员间的D4-D4二聚化基序的保守性。人Kit残基370-398(AAC50969.1)(SEQ ID NO:94)与小鼠(AAH75716.1)(SEQ ID NO:95)、鸡(NP_989692.1)(SEQ IDNO:96)、爪蟾(AAH61947)(SEQ ID NO:97)、真螈(AAS91161.1)(SEQID NO:98)和斑马鱼(A型(SEQ ID NO:99)和B型(SEQ ID NO:100)(NP_571128,XP_691901)同源物的序列比对。也显示了人CSF1R(P07333)(SEQ ID NO:101)、小鼠(P09581)(SEQ ID NO:102)及河豚(torafugu)A型(SEQ ID NO:103)和B型(SEQ ID NO:104)(P79750、Q8UVR8)的氨基酸序列以及人PDGFRα和PDGFRβ(分别为SEQ IDNO:105和107)(P16234、P09619)及小鼠(分别为SEQ ID NO:106和108)(NP_035188,P05622)的序列。也显示了人VEGFR 1-3型的V型RTK的氨基酸序列(以出现先后为序分别为SEQ ID NO:109-111)(第七Ig样结构域)(P17948、P35968和P35916)。在序列比对的顶部标记Kit的二级结构元件。保守残基Arg381和Lys383、Leu382和Leu379、Glu386和Gly388分别标为蓝色、黄色、红色和绿色。图6C描绘了D5-D5界面的带状栅图。两个邻近Kit原体的A链和G链参与了D5-D5界面的形成。D5-D5界面可能是通过离子或水分子由两个相邻受体的Tyr418和Asn505之间的侧向相互作用维持的。图6D描绘了Kit的D4和D5的静电势表面。图中显示了D4-D4相互作用表面的正视图(右图)和沿垂直轴旋转90°后的视图(左图)。圈出了酸性补丁和D4-D4界面的位置并标记了相互作用残基Arg381和Glu386。
图7A-C描绘了涉及到癌症和其他疾病以及Kit和其它RTK激活的机理的Kit胞外域突变。图7A描绘了在左图中显示的导致花斑性状的功能缺失突变。D1(蓝色)、D2(绿色)和D3(黄色)的带状栅图和SCF(灰色)的表面示意图。突变的氨基酸标为红色。右图显示导致GIST、SCLC和AML的功能获得突变。同型二聚体形式的D4和D5的表面示意图以灰色显示。在GIST中加倍的Ala502和Tyr503标为蓝色且接近Asp419的缺失和插入突变(AML和NCLL)标为绿色。注意,激活的Kit突变局限于D5-D5界面。图7B显示,Kit活化被D4-D4界面中的点突变减弱。正如所标明的(左上图),瞬时表达野生型Kit(WT)、D4中的R381A或E386A点突变的HEK293细胞用10ng/ml的SCF在37℃下刺激6分钟。未刺激的或SCF刺激的细胞的裂解液用抗-Kit抗体进行免疫沉淀(IP),然后进行SDS-PAGE和用抗-Kit抗体或抗磷酸酪氨酸(p-Tyr)抗体进行免疫印迹(IB)。抗-p-Tyr-免疫印迹的Kit酪氨酸自磷酸化的密度定量(右上图)。用不同浓度的SCF处理稳定表达野生型Kit(WT)或R381A突变体的3T3细胞。来自未刺激的或SCF刺激的细胞的裂解液用抗-Kit抗体进行免疫沉淀,然后进行SDS-PAGE并用抗-Kit抗体或抗-p-Tyr抗体进行免疫印迹(左下图)。用天然SCF进行的细胞结合125I-SCF的置换分析。存在渐增浓度的天然的SCF情况下用125I-SCF处理表达WT(■)、R381A(▼)、R381A/E386A(◆)或激酶阴性Kit(▲)的3T3细胞。SCF对于WT Kit的EC50(置换50%的结合c-Kit的SCF的配体浓度)(1.1nM)与SCF对于R381A(1.0nM)、R381A/E386A(0.8nM)或激酶阴性Kit突变体(1.4nM)的EC50相当。图7C显示了由可溶性的(左图)或在相邻细胞的细胞表面上表达的膜锚定的(右图)SCF分子驱动的Kit和其它RTK的活化的模型。SCF结合D1-D2-D3配体结合模块使得两个结合的Kit胞外域单体的C-末端彼此相距 以外。D3-D4和D4-D5铰链的灵活性使得能够实现使两个相邻胞外域的C-末端彼此在
以外。D3-D4和D4-D5铰链的灵活性使得能够实现使两个相邻胞外域的C-末端彼此在 以内的横向D4-D4和D5-D5相互作用。因此,增加的接近性和Kit胞质域的局部浓度提高导致激酶结构域中调控性酪氨酸残基的自磷酸化,从而导致PTK活化。(注意PTK活化未在模型中画出)。胞质域中关键酪氨酸的磷酸化后,将进行细胞信号传导分子的补体的募集和活化。该模型是基于游离SCF的结构、无配体Kit、SCF-Kit复合体和Kit PTK结构(PDB项目1QZJ、1R01和1T45)。对结构还未确定的区域用二级结构预测进行建模(绿色螺旋和黑色环)。SCF标为紫红色、Kit胞外域标为蓝色和kit PTK标为淡蓝色。
以内的横向D4-D4和D5-D5相互作用。因此,增加的接近性和Kit胞质域的局部浓度提高导致激酶结构域中调控性酪氨酸残基的自磷酸化,从而导致PTK活化。(注意PTK活化未在模型中画出)。胞质域中关键酪氨酸的磷酸化后,将进行细胞信号传导分子的补体的募集和活化。该模型是基于游离SCF的结构、无配体Kit、SCF-Kit复合体和Kit PTK结构(PDB项目1QZJ、1R01和1T45)。对结构还未确定的区域用二级结构预测进行建模(绿色螺旋和黑色环)。SCF标为紫红色、Kit胞外域标为蓝色和kit PTK标为淡蓝色。
图8描绘了III型RTK的基于结构的序列比对,基于Kit胞外域的结构,和基于结构的III型家族RTK的配体的比对。各排显示单个Ig样结构域的比对。基于IgSF折叠特征手动比对氨基酸序列,如由(Harpaz等人.(1994)J Mol Biol 238:528-539)确定的,并且与Jpred(Cuff等人.(1998)Bioinformatics 14:892-893)计算的家族成员的二级结构预测相一致。红色标记的氨基酸代表IgSF折叠决定氨基酸。在序列上方用箭头标记β链和用弹簧标记α-螺旋,以及人Kit和人SCF的编号。用星号标记配体结合时表现出溶剂可达性降低的配体结合位点的残基。位点-I标为黑色、位点-II标为红色和位点-III标为绿色。相同的颜色编码被用于标记SCF中的相互作用的氨基酸残基。用蓝绿色的方框圈出负责D4-D4相互作用的D4EF环。用于比对的序列是:人Kit(AAC50969)、Kit小鼠(AAH75716)、CSFR1人(P07333)、PDGFRα人(P16234)、PDGFRβ人(P09619)和Flt3人(P36888)。对于配体结构比对,用Lsqman(Kleywegt和Jones,1995)对SCF(1EXZ)、CSF(1HMC)、Flt3L(1ETE)的PDB项进行叠加,并用ClustalW比对小鼠SCF的序列(NP_038626)和人SCF。图分别按出现的先后顺序显示了SEQ ID NO:112-147。
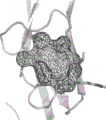
图9提供了2:2SCF-Kit复合体的结构的立体视图。2:2SCF-Kit复合体的带模型以立体示意图显示。视图和颜色编码与图2A相同。
图10A-B描绘了SCF-Kit复合体表面的氨基酸保守性。图10A显示彩色编码的SCF-Kit晶体结构复合体的保守性模式。蓝绿色至栗色用来标记从可变氨基酸到保守氨基酸。图10B显示了通过将两个分子彼此拉开而得到的SCF和Kit可视化。圈出了位点I、位点II和位点III以及D4-D4相互作用区域(D4-D4界面)。
图11A-B描绘了SCF-Kit界面的电子密度。图11A显示了具有以2σ水平在Kit周围绘出的电子密度图的2:2 SCF-Kit复合体的位点II的局部视图。除标记的侧链外,用黄色管绘出Kit主链。图11B描绘了SCF-Kit界面的电子密度,显示了具有以1.5σ水平在Kit周围绘出的实验图的游离Kit的局部视图。方向和颜色编码与图12A相同。
图12A-D描绘了游离的和SCF结合的Kit的Ig样结构域的叠加对的视图。游离的和SCF结合的Kit的单独D1、D2、D3和D4叠加在一起。显示的是Ig样结构域(A)D1和D2、(B)D2和D3、(C)D3和D4以及(D)D4和D5对的结构,其中各对中叠加的Ig样结构域标为蓝色且第二(未叠加)Ig样结构域对于游离的胞外域标为绿色和对于SCF结合的胞外域标为红色。这些图显示,当SCF结合时五个单独Kit Ig样结构域的各结构域的结构实际上没发生变化,且D1-D2-D3作为准备用于SCF结合的配体结合单元发挥作用。相反,在SCF结合的Kit中D3-D4和D4-D5界面发生大的重排。
图13A-B描绘了SCF-Kit复合体结构的静电表面电位。图13A具体地显示了SCF-Kit 2:2复合体。图13B描绘了SCF-Kit复合体结构的静电表面电位,具体地说在SCF被从SCF-Kit 2:2复合体中拉开后,Kit的静电表面电位的可视化。带正和负电荷的表面分别标为蓝色和红色。圈出了SCF结合区域和D4-D4界面。
图14描绘了抗Kit-D5抗体抑制SCF诱导的Kit激活。表达Kit的3T3细胞用渐增浓度的抗Kit-D5(针对Kit的第五Ig样结构域)孵育或作为对照用抗-SCF(针对SCF配体)或抗-Kit胞外域(针对整个Kit胞外域)孵育。
图15描绘了用重组Kit D4抑制SCF诱导的Kit激活。表达Kit的3T3细胞用渐增浓度的重组Kit-D4室温下孵育10分钟,然后SCF刺激10分钟。
图16A显示D4中的点突变阻止PDGF诱导的PDGFR激活。对表达WT PDGFR或D4突变体(R385A和E390A)的PDGFR-/-MEF血清饥饿过夜并用注明浓度的PDGF BB刺激5分钟。细胞裂解液用抗-PDGFR抗体进行免疫沉淀,然后进行SDS-PAGE和用抗-磷酸酪氨酸抗体4G10进行免疫印迹。膜剥离,并用抗-flag标签抗体再印迹分析以确定总PDGFR水平。
图16B表明D4中的点突变阻止经由PDGFR的信号传导。对表达WT PDGFR和D4突变体(R385A和E390A)的PDGFR-/-MEF血清饥饿过夜,并用注明浓度的PDGF BB在23℃下刺激5分钟。等量的总细胞裂解液(TCL)进行SDS-PAGE和分别用抗-磷酸-MAPK、MAPK、磷酸-Akt和Akt进行免疫印迹。这个实验表明,D4中的点突变阻止了MAPK反应和Akt活化。
图16C表明阻止PDGFR激活的D4点突变不干扰PDGF诱导的PDGFR二聚化。对表达WT或E390A突变体的PDGFR-/-MEF血清饥饿过夜,随后用含有注明量的PDGF的DMEM/50mM HEPES缓冲液(pH7.4)在4℃孵育90分钟。去除未结合的配体后,细胞用PBS中的0.5mM的辛二酸二琥珀酰亚胺(DSS)孵育30分钟。用抗-PDGFR抗体对未刺激的或刺激的细胞的裂解液进行免疫沉淀,然后进行SDS-PAGE分析和用抗-flag抗体(左图)或抗-pTyr抗体(右图)进行免疫印迹。
图17显示D3-D4铰链区中的空腔。几个空腔散布在胞外域单体结构的D3-D4界面上。参与限定空腔的氨基酸总结于表4(如下)中。当两个Kit受体之间形成同型相互作用时,D3-D4铰链区发生改变导致形成由下列残基形成的浅空腔:D3的K218、S220、Y221、L222和D4的F340、P341、K342、N367、E368、S369、N370、I371、Y373。图17显示了未占据的单体(A)和SCF-结合的二聚体(B)的D3-D4铰链区的带状栅图和D3-D4口袋的网格示意图。
图18显示了D4-D5铰链区中的空腔。小空腔由Kit单体的D4的AB环和EF环、连接D4-D5的接头和D5的部分DE环和FG环形成。限定空腔的残基总结于表4(如下)中。在Kit胞外域二聚体结构中空腔的形状和大小是改变的。由D4的EF环和G链、D4-D5接头和D5的B链和DE环形成的主要空腔位于D4的EF环之下,D4同型界面形成的关键区域。注意,如未结合的和占据的Kit结构的电子密度的较低质量所揭示的,靠近空腔定位的D5的DE环可具有更高的灵活性。图18显示了未占据的单体(A)和SCF-二聚体(B)的带状栅图和D4-D5铰链区周围的浅空腔的网格示意图。
图19显示了介导D4同型相互作用的区域处的空腔。Kit D4的CD环和EF环形成的凹面位于D4同型界面正上方。D4的残基Y350、R353、F355、K358、L379、T380、R381、L382、E386和T390为胞外域二聚结构中的凹面提供了大约130A2的表面积。在D4同型界面中起着重要作用的Glu386的侧链向表面的中心突出。凹面的特征性结构是由带电的残基(Glu386和Lys358)围绕的小的疏水补丁。当同型D4:D4相互作用时该表面的大小和可接近性发生改变,CD环的构象中发生朝向结构域顶部折叠的变化。参与形成凹面的残基总结于表4(如下)中。下图中图A显示了覆盖在配体-占据的Kit D4(未示出)上的未占据的KitD4结构域(金色)的带状栅图,配体-占据(绿色)和未占据的胞外域结构(红色)之间的CD环和EF环的构象不同。D4:D4相互作用的关键残基以棍模型形式表示。图B和C显示了未占据的Kit(图19B)和SCF-占据的Kit结构(图19C)的带状栅图和D4同型界面之上的浅空腔的网格示意图。
图20显示了配体-结合的D2和D3区域处的凹面。浅凹面位于D2和D3的部分配体结合表面。包括于小口袋中的残基是D2的Y125、G126、H180、R181、K203、V204、R205、P206和F208以及D3的V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269。口袋是通过由亲水性残基包围的小疏水补丁形成的。未占据的和SCF-占据的Kit结构之间没有大的变化,整体包埋的表面积约为500A2。图A和图B显示了未占据的Kit(A)和SCF-结合的Kit(B)的带状栅图和D2-D3口袋的网格示意图。
图21描绘了PDGF受体近膜区的基于结构的序列分析和同源性建模。图21A描绘了PDGFRα、PDGFRβ和Kit的D4的氨基酸序列(按出现顺序分别为SEQ ID NO:148-157)的比对。人Kit结构D4的IgSF折叠的关键残基和Ig折叠的核心残基的氨基酸相应地标为红色和绿色。负责D4同型相互作用的两个关键碱性和酸性残基分别用蓝色和红色框出。对应于在Ig样结构域(B5和F5)上保守的二硫键形成半胱氨酸残基的位置用星号标记。在Kit序列下面用箭头标记β-链。根据IgSF命名法标记二级结构元件。图21B描绘了PDGFR胞外结构域的近膜域模型。PDGFRβ胞外域的近膜区域标为白色并且显示为具有透明的分子表面的带(D4标为橙色和D5标为粉色;左图)。两个相邻的PDGFRβ分子的D4-D4界面的放大图(右图)表明,D4之间的相互作用是由从两个邻近的EF环伸出的残基Arg385和Glu390介导的。标记了关键氨基酸并以棍模型表示。
图22描绘了证明PDGF诱导的PDGFR激活被D4中的突变影响的实验结果。图22A显示了表明PDGF诱导的PDGFRβ的酪氨酸自磷酸化在表达PDGFRβ的E390A、R385A、RE/AA和RKE/AAA突变体的细胞中受到强烈影响的实验结果。图22B是显示了野生型和突变体PDGFRβ的置换曲线的曲线图。用Prism4通过曲线拟合确定IC50值。图22C描绘了免疫印迹的结果,其证明R385A、E390A或RE/AA突变体不影响PDGFR的固有酪氨酸激酶活性。
图23描绘了免疫沉淀实验的结果,其表明PDGF-刺激的D4结构域突变的PDGFRβ在细胞表面上以非活性的二聚体形式表达。细胞裂解液用抗-PDGFR抗体进行免疫沉淀,并通过SDS-PAGE分析免疫沉淀物和分别用抗-flag抗体(左图)和抗磷酸酪氨酸抗体(右图)进行免疫印迹。
图24描绘了免疫沉淀实验的结果,其表明PDGF诱导的细胞反应受PDGFRβ D4突变体中突变的影响。
图25描绘表明在表达PDGFR D4突变体的MEF中,肌动蛋白环形成的PDGF刺激受到影响的实验结果。大约83%的表达WT PDGFR的MEF表现出环形肌动蛋白环形成,但只有5%的PDGFR D4突变体细胞在用50ng/ml的PDGF刺激2分钟后表现出相似的环形肌动蛋白环形成。此外,在PDGF刺激后2-5分钟在表达WT PDGFR的MEF中达到峰值的瞬时环形肌动蛋白环形成在表达R385A、E390A或RE/AA的PDGFR突变体的细胞中微弱地检测到。
图26描绘了表明PDGFR D4中的突变影响PDGFR内化和泛素介导的PDGFR的降解的实验结果。图26A是表明与表达WT PDGFR的MEF结合的125I标记PDGF的内化动力学要比与表达E390A、R385A或RE/AAPDGFR的细胞结合的125I标记PDGF的内化动力学快得多的曲线图。图26B表明,R385A、E390A或RE/AA PDGFR突变体的降解动力学大大减弱;虽然一半的WT PDGFR在PDGF刺激的1.5小时之内降解,但是PDGFR D4突变体的半衰期延长到大约4至6小时。26C图描绘了显示在相同条件下与WT PDGFR相比,PDGF诱导的E390A PDGFR的泛素化也大大降低的实验。
图27描绘了表明D4界面的破坏阻断了KIT中的致癌突变的实验结果。野生型Kit的SCF刺激导致了由KIT的酪氨酸自磷酸化的增强显示的KIT活化的增强。实验进一步表明,KIT的致癌性D5重复突变体是组成型酪氨酸自磷酸化的。相反,D5-重复/E386A突变体阻断了由致癌的D5-重复突变介导的KIT的组成型酪氨酸自磷酸化。
图28描绘了免疫印迹实验的结果,其表明抗对应于KIT-D4的同型相互作用基序的肽的抗体识别全长KIT受体。
图29A描绘了来自不同物种的VEGFR1和VEGFR2的D7的预测EF环区域的基于结构的多序列比对。I-群Ig框中的关键氨基酸用绿色突出显示,以及EF环中的保守Arg/Asp对用红色突出显示。图29B描绘了VEGFR的D4及KIT、CSF1R和PDGFR(III型RTK)的D4的预测EF环区域的比较。I-群Ig框中的关键氨基酸以绿色突出显示,并且EF环中的保守Arg/Asp或Glu对以红色突出显示。EF环中带相反电荷的非保守氨基酸以蓝色突出显示。保守的Y-conner基序用*标记。
图30表明配体诱导的VEGFR2的活化被D7的EF环区域中突变弱化但不受D4的EF环区域中突变的影响。图30A表明用注明量的VEGF在37℃刺激瞬时表达野生型VEGFR2、R726A或E731A VEGFR2突变体的HEK293细胞5分钟。未刺激的或VEGF刺激的细胞的裂解液用抗-VEGFR2抗体进行免疫沉淀,然后与抗-pTyr或与抗-VEGFR2抗体进行免疫印迹(IB)。用SDS-PAGE分析由同一实验得到的总细胞裂解液,然后用抗-磷酸MAPK(pMAPK)或抗-MAPK抗体进行免疫印迹。图30B表明,稳定表达WT VEGFR2-PDGFR嵌合受体或携带D7区域中的突变(R726A、D731A或R726/D731双突变体RD/2A)的嵌合受体的血清饥饿的3T3细胞在37℃下用VEGF刺激5分钟。未刺激的或VEGF刺激的细胞的裂解液用抗嵌合受体的胞质区的抗体进行免疫沉淀,随后分别用抗-pTyr或抗-标签(FLAG)抗体进行免疫印迹。图30C表明,稳定表达WT VEGFR1-PDGFR嵌合受体或携带D7区域中的突变(R721A、D725A或R721D725/2A双突变)的嵌合受体的血清饥饿的3T3细胞在37℃下用VEGF刺激5分钟。未刺激的或VEGF刺激的细胞的裂解液用抗嵌合受体的胞质区的抗体进行免疫沉淀,随后分别用抗-pTyr或抗-标签(FLAG)抗体进行免疫印迹。图30D表明,如图30A中所述的分析了表达WT VEGFR2-PDGFR嵌合受体或携带在D4区中的突变(D392A或D387/R391A双突变)的嵌合受体的3T3细胞。。
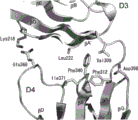
图31描绘了VEGFR2胞外域D7二聚体的结构。图31A描绘了D7同型二聚体结构的带状栅图和透明分子表面(侧视图)。Asp731和Arg726以棍模型显示。图31B描绘了两个相邻分子(粉色和绿色)的同型D7界面的近视图。由Asp731和Arg726形成的盐桥显示为虚线。图31C描绘了作为表面电位模型的D7二聚体(侧视图)的电荷分布(左图)。介导同型接触的D7表面的视图(右图)。图31D描绘了以1.1σ水平描绘的2Fo-Fc电子密度图,呈现出D7-D7界面的视图。VEGFR D7原体的骨架分别表示为粉色和黄色管。
图32描绘了VEGFR2的D7结构与二聚的KIT-SCF复合体的D4结构的叠加。VEGFR D7结构(PDB ID代码:3KVQ)和与SCF的复合体的KIT二聚体(PDB ID代码:2E9W)的覆盖图(左图)。叠加的D7和D4区域的放大图(右图)显示了结构域排列和同型接触的高度相似性。VEGFR D7显示为绿色和EF环为黄色。KIT的D4显示为灰色以及它的EF环为橙色。
图33描绘了VEGFR1和VEGFR2的系统发生分析。图33A描绘了来自各种不同物种的Ⅲ型和V型RTK的保守EF环的位置。含有保守的EF环基序的Ig样结构域标记为蓝色。图33B描绘了VEGFR2 D7区域的颜色编码的保守模式。人VEGFR的氨基酸序列作为查询序列使用PSI-BLAST来对非冗余数据库(nr)进行同源序列检索(Altschul等人,J.Mol.Boiol.,215(3):403-410(1990))。用ClustalW2(Thompson等人,Nucleic Acids Res.,22(22):4673-4680(1994))进行D7的序列比对,基于对20个关键残基的IgSF折叠限制进行手动调节。氨基酸序列的比对提交到Consurf 3.0服务器(Landau等人,Nucleic Acids Res.,33(Web Server号):W299-302(2005))以产生各个位置的最大似然归一进化速率。蓝绿色至栗色用来标记从可变氨基酸到保守氨基酸。图33C描绘了使用Clustal W2基于邻接法生成的VEGFR1和VEGFR2的系统发生树。在分析中使用的氨基酸序列包括:VEGFR2_HUMAN(gi:11321597)、VEGFR2_DOG(gi:114158632)、VEGFR2_HORSE(gi:194209154)、VEGFR2_CATTLE(gi:158508551)、VEGFR2_RAT(gi:56269800)、VEGFR2_MOUSE(gi:27777648)、VEGFR2_CHICK(gi:52138639)、VEGFR2_QUAIL(gi:1718188)、VEGFR2_ZEBRARISH(gi:46401444)、VEGFR1_HUMAN(gi:143811474)、VEGFR1_MOUSE(gi:148673892)、VEFGR1_RAT(gi:149034835)、VEFGR1_HORSE(gi:149730119)、VEGFR1_CHICK(gi:82105132)、VEGFR1_ZEBRAFISH(gi:72535148)、VEGFR_SEAURCHIN(gi:144226988)、VER1_C_ELEGANS(gi:6003694)、VER3_C_ELEGANS(gi:3877967)、VER4_C_ELEGANS(gi:3877968)、PVR_DROSOPHILA(gi:45552252)、VEGFR_SEASQUIRT(gi:198434052)。
发明详述
本发明提供了结合受体酪氨酸激酶,例如VEGF受体如VEGFR1(Flt1)、VEGFR2(KDR/Flk1)和VEGFR3(Flt4),的胞外域,例如Ig样结构域或Ig样结构域之间的铰链,的成分,例如抗体或其抗原结合部分、小分子、肽类分子、适体和Adnectin。本发明的成分可以使VEGF受体的胞外域锁定在非活性状态,从而抑制VEGF受体的活性。在本发明的一个实施方案中,该成分使VEGF受体的胞外域锁定在单体状态。在本发明的另一个实施方案中,该成分允许VEGF受体的胞外域二聚化,但影响两个单体的Ig-样结构域之间的定位、定向和/或距离(例如,VEGF受体的D7-D7结构域),从而抑制VEGF受体的活性。换句话说,该成分可以允许VEGF受体胞外域的配体诱导的二聚化,但影响两个胞外域在细胞表面界面处的定位或者改变或阻止VEGF受体中的构象变化,从而抑制VEGF受体的活性(例如,抑制受体内化和/或抑制受体的酪氨酸自磷酸化和/或抑制受体激活下游信号传导途径的能力)。本发明至少部分地基于VEGF受体VEGFR2整个胞外域的晶体结构的破译。这种晶体结构的破译使得能够鉴别本发明的成分可以靶向于的表位,例如,构象表位。
如本文中所使用,术语“成分”意图包括任何结合受体酪氨酸激酶的胞外域(例如Ig样结构域)的成分,其中所述成分将受体酪氨酸激酶的胞外域锁定在非活性状态,例如单体状态,从而拮抗受体酪氨酸激酶的活性。所述成分可以是分离的抗体或其抗原结合部分;小分子;肽类分子(例如基于受体酪氨酸激酶的Ig样结构域的结构设计的肽类分子);适体或adnectin。在一些方面,所述成分结合连接受体酪氨酸激酶的Ig样结构域的铰链区(例如Ⅲ型RTK的D3-D4或D4-D5铰链区)。
在一些实施方式中,所述成分将结合人VEGF受体的特定序列,例如,VEGFR1的718-727残基、VEGFR1的Arg720和Asp725残基、VEGFR2的724-733残基、VEGFR2的Arg726和Asp731残基、VEGFR3的735-744残基或VEGFR3的Arg737和Asp742残基。所述成分可选地结合人Kit受体的特定序列,例如人Kit的残基309-413、残基410-519、381Arg和386Glu或418Tyr和505Asn。残基309-413包含D4结构域,残基410-519包含人Kit的D5结构域,并在本文中表明对于Kit受体二聚化至关重要。残基381Arg和386Glu是Kit的D4结构域中的残基,在本文中表明对于D4结构域的非共价连接是重要的,因此,对于受体的二聚化是重要的。类似地,残基418Tyr和505Asn是Kit的D5结构域中的残基,在本文中表明对于受体的二聚化是重要的。本领域技术人员将理解,特异性结合上述残基的成分可以通过例如阻止两个单体Kit或VEGF受体分子的二聚化而拮抗受体的活性。
在另外的实施方案中,所述成分结合人VEGF受体的突变的氨基酸残基,其中该氨基酸残基是至少VEGFR1的Arg720或ASP725、VEGFR2的Arg726或Asp731或VEGFR3的Arg737或Asp742之一。在另外的实施方式中,所述成分结合人Kit中突变的氨基酸残基,其中所述氨基酸残基是417Thr、418Tyr、419Asp、421Leu、420Arg、503Tyr或502Ala中的至少之一。
在优选实施方式中,本发明的成分结合Kit受体中的一或多个残基,这些残基构成在表4(下文)中所述的小腔或口袋。例如,本发明的成分可以结合Kit受体的D3-D4铰链区中的一个或多个以下残基:来自D3结构域的K218、S220、Y221、L222和来自D4结构域的F340、P341、K342、N367、E368、S369、N370、I371、Y373。本发明的成分也可以结合一个或多个以下残基,这些残基构成Kit受体的D4结构域中的凹面:Y350、R353、F355、K358、L379、T380、R381、L382、E386和T390。在另一实施方式中,本发明的成分结合一个或多个以下残基,这些残基形成Kit受体的D2-D3铰链区中的口袋:来自D2结构域的Y125、G126、H180、R181、K203、V204、R205、P206和F208,以及来自D3结构域的V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269。
因此,在一些实施方式中,本发明的成分可以结合连续的或不连续的氨基酸残基,并起到阻止RTK的近膜区以使酪氨酸激酶能够活化的距离和定向进行定位所需的运动分子楔(molecular wedge)的作用。本发明的成分也可以起到阻止同型或异型的D4或D5受体相互作用或使配体-受体相互作用位点不稳定的作用。在一些优选实施方式,本发明的成分结合Kit受体上的一个或多个以下残基:Y125、G126、H180、R181、K203、V204、R205、P206、P206、F208、K127、A207、V238、S239、S240、S241、H263、G265、D266、F267、N268、Y269、T295、L222、L222、L223、E306、V308、R224、V308、K310、K218、A219、S220、K218、A220、Y221、A339、D327、D398、E338、E368、E386、F312、F324、F340、F355、G311、G384、G387、G388、I371、K342、K358、L382、L379、N326、N367、N370、N410、P341、S369、T385、V325、V407、V409、Y373、Y350、Y408、T380、T390、R381、R353、T411、K412、E414、K471、F433、G470、L472、V497、F469、A431或G432。本领域技术人员将理解,在一些实施方式中,本发明的成分可以容易地靶向于其它Ⅲ型RTK中的相应残基,例如形成相似的口袋或空腔的那些残基或通过结构比对或序列比对处于相同位置的那些残基。
在一种特定实施方式中,本发明的成分结合Ⅲ型RTK上的构象表位或非连续表位。构象表位或非连续表位可以由来自Ⅲ型RTK(例如人Kit受体或PDGF受体)的D3、D4和/或D5结构域或者D4-D5或D3-D4铰链区的两个或更多个残基组成。例如,所述构象表位或非连续表位可以由表4中所列的两个或更多个残基组成。
在一种具体实施方式中,本发明的成分结合由2个或多个选自Y125、H180、R181、K203、V204、R205、P206、V238、S239、S240、H263、G265、D266、F267、N268和Y269的氨基酸组成的构象表位。在相似的实施方式中,本发明的成分可以结合由选自以下的氨基酸组之一的2个或多个氨基酸组成的构象表位:P206、F208、V238和S239;K127、A207、F208和T295;L222、A339、F340、K342、E368、S369、N370、I371和Y373;L222、L223、E306、V308、F312、E338、F340和I371;R224、V308、K310、G311、F340、P341和D398;K218、A219、S220、N367、E368和S369;K218、A220、E368和S369;G384、T385、T411、K412、E414和K471;Y408、F433、G470、K471和L472;F324、V325、N326和N410;D327、N410、T411、K412和V497;G384、G387、V409和K471;L382、G387、V407和V409;Y125、G126、H180、R181、K203、V204、R205、P206、F208、V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269;P206、F208、V238和S239;K218、S220、Y221、L222、F340、P341、K342、N367、E368、S369、N370、I371和Y373;G384、G387、G388、Y408、V409、T411、F433、F469、G470和K471;D327、T411、K412、E414、A431、G432和K471;Y350、F355、K358、L379、T380、R381、L382、E386和T390;Y350、R353和F355。如以上所指出的,本发明的成分可以结合形成如表4中确定的口袋或空腔的所有氨基酸残基,或者它们可以结合形成口袋或空腔的残基的一部分。应当理解,在特定实施方式中,当提到结合表位例如构象表位的本发明的成分时,意图是所述成分仅结合那些构成该表位(例如表4中确定的口袋或空腔)的那些特定残基,而不是在该受体的线性氨基酸序列中的其它残基。
在进一步的实施方式中,本发明的成分结合构象表位,其中所述表位由选自表5中所列的肽的两个或更多个氨基酸残基组成。在一种具体实施方式中,构象表位由选自第一肽的一个或多个氨基酸残基和选自第二肽的一个或多个氨基酸残基组成,其中第一和第二肽选自表5中所列的肽的组。因而,本发明的成分可以结合构象表位,其中来自表5的所述第一和第二肽组如下:Ala219-Leu222和Thr304-Val308;Asp309-Gly311和Arg224-Gly226;Thr303-Glu306和Ala219-Leu222;Asn367-Asn370和Ser217-Tyr221;Ala339-Pro343和Asn396-Val399;Ala339-Pro343和Glu368-Arg372;Lys358-Tyr362和Val374-His378;Asp357-Glu360和Leu377-Thr380;Met351-Glu360和His378-Thr389;His378-Thr389和Val323-Asp332;Val409-Ile415和Ala493-Thr500;Val409-Ile415和Ala431-Thr437;Val409-Ile415和Phe469-Val473;Val409-Ile415和Val325-Asn330;Val409-Ile415和Arg381-Gly387;Gly466-Leu472和Gly384-Gly388;Val325-Glu329和Tyr494-Lys499;Thr411-Leu416和Val497-Ala502;Ile415-Leu421和Ala502-Ala507;Ala502-Ala507和Lys484-Thr488;及Ala502-Ala507和Gly445-Cys450。本发明的成分可以结合形成前述第一和第二肽组的所有氨基酸残基,或者它们可以结合形成第一和第二肽组的残基的一部分。应当理解,在特定实施方式中,当提到结合表位例如构象表位的本发明的成分时,意图是所述成分仅结合那些构成该表位(例如表5中确定的特定肽)的那些特定残基,而不是在该受体的线性氨基酸序列中的其它残基。
在另一实施方式中,本发明的成分结合由2个或多个选自E33、P34、D72、E76、N77、K78、Q79、K158、D159、N250、S251、Q252、T253、K254、L255、N260、W262、H264、G265、E344、N352、R353、F355、T356、D357、Y362、S365、E366、N367、N370和G466的氨基酸组成的构象表位或非连续表位。
在另一个实施方案中,本发明的成分结合VEGF受体的连续表位。在一个实施方案中,连续表位是由VEGF受体D7结构域中的两个或更多个残基组成的。在另一个实施方案中,连续表位是选自于VEGFR1的672VAISSS677、VEGFR1的678TTLDCHA684、VEGFR1的685NGVPEPQ691、VEGFR1的700KIQQEPG706、VEGFR1的707IILG710、VEGFR1的711PGS713、VEGFR1的714STLFI718、VEGFR1的719ERVTEEDEGV728、VEGFR3的689VNVSDS694、VEGFR3的695LEMQCLV701、VEGFR3的702AGAHAPS708、VEGFR3的717LLEEKSG723、VEGFR3的724VDLA727、VEGFR3的728DSN730、VEGFR3的731QKLSI735和VEGFR3的736QRVREEDAGR745、VEGFR2的678TSIGES683、VEGFR2的684IEVSCTA690、VEGFR2的691SGNPPPQ697、VEGFR2的706TLVEDSG712、VEGFR2的713IVLK716、VEGFR2的717DGN719、VEGFR2的720RNLTI724和VEGFR2的725RRVRKEDEGL734的表位。
在另一实施方式中,本发明的成分结合人PDGFRβ的氨基酸残基385Arg和390Glu,或者PDGFRα中的相应残基。人PDGFRβ的残基385Arg和390Glu与Kit受体的残基381Arg和386Glu类似,并介导PDGFRβ的同型D4-D4相互作用。本发明的成分可以通过阻止Ⅲ型RTK的近膜区之间的关键同型相互作用(例如人PDGFRβ的385Arg和390Glu之间形成的盐桥)而发挥其对受体活化的抑制作用,所述相互作用对于胞质域以对于酪氨酸激酶活化至关重要的距离和定向进行定位是不可或缺的。本文中讨论的实验证明:同型D4-D4相互作用对于PDGFRβ二聚化不是必要的,而PDGFRβ二聚化对于受体活化是必要的然而并非足够的。因此,本发明的成分可以允许PDGFRβ二聚化而阻止活化。基于结构的序列比对已经表明:在Kit、PDGFRα、PDGFRβ和CSF1R中,EF环的大小以及包含重要氨基酸的D4-D4界面是保守的。因此,在一些实施方式中,本发明的成分可以靶向于Ⅲ型RTK的D4或D5结构域的保守区。本领域技术人员也会理解,本发明的成分可以结合糖残基,所述糖残基可以出现在RTK的糖基化形式上。本发明的成分也可以结合由氨基酸残基和糖残基两者组成的表位。
术语“受体酪氨酸激酶”和“RTK”在本文中可互换地使用以表示公知的使酪氨酸残基磷酸化的膜受体的家族。很多受体酪氨酸激酶在发育或者细胞分裂中起到重要作用。受体酪氨酸激酶具有细胞外配体结合结构域、跨膜结构域和胞内催化结构域。胞外结构域结合细胞因子、生长因子或其它配体,并通常由一或多个可识别的结构基序组成,包括富半胱氨酸区、纤连蛋白Ⅲ样结构域、免疫球蛋白样结构域、EGF样结构域、钙粘着蛋白样结构域、kringle样结构域、因子VIII样结构域、富甘氨酸区、富亮氨酸区、酸性区和盘状(discoidin)结构域。胞内激酶结构域的活化通过配体结合细胞外结构域实现,这引起受体二聚化。以这种方式活化的受体能够自磷酸化催化结构域外面的酪氨酸残基,从而促进活性受体构象的稳定。磷酸化残基也作为随后在细胞内传导信号的蛋白质的结合位点。RTK的实例包括但不限于Kit受体(亦称干细胞因子受体或SCF受体)、成纤维细胞生长因子(FGF)受体、肝细胞生长因子(HGF)受体、胰岛素受体、胰岛素样生长因子-1(IGF-1)受体、神经生长因子(NGF)受体、血管内皮生长因子(VEGF)受体、PDGF受体-α、PDGF受体-β、CSF-1受体(亦称M-CSF受体或Fms)和Flt3-受体(亦称Flk2)。
在本发明的优选实施方式中,RTK是Ⅲ型RTK。在本发明另一实施方式中,RTK是V型,即VEGF受体家族的成员。
如本文中所使用,术语“受体酪氨酸激酶的Ⅲ型家族”或“Ⅲ型RTK”意于包括一般地在其胞外域中包含五个免疫球蛋白像结构域或Ig样结构域的受体酪氨酸激酶。Ⅲ型RTK的实例包括但不限于PDGF受体、M-CSF受体、FGF受体、Flt3受体(亦称Flk2)和Kit受体。在本发明的优选实施方式中,Ⅲ型RTK是Kit(在本领域也称为SCF受体)。Kit,与其它Ⅲ型RTK相似,由通过单一跨膜(TM)结构域连接于胞质区糖基化的细胞外配体结合结构域(胞外域)构成(综述于Schlessinger(2000)Cell 103:211-225)。Ⅲ型RTK,例如Kit或PDGFR,的另一标志是带有大的激酶插入区的胞质蛋白酪氨酸激酶(PTK)结构域。已知存在至少两种Kit受体的剪切同工型,较短的利用框内的剪接位点。本发明包括Kit以及其它上述RTK的所有同工型。
本文中所用的受体酪氨酸激酶(RTK)的“Ig样结构域”意于包括本领域公知的存在于RTK胞外域中的结构域。在Ⅲ型受体酪氨酸激酶(Ⅲ型RTK)家族例如Kit的胞外域中,存在5个这样的结构域,已知为D1、D2、D3、D4和D5。Ⅲ型RTK的D1、D2和D3结构域负责结合RTK的配体(综述于Ullrich和Schlessinger(1990)Cell 61:203-212)。因此,在本发明的一种实施方式中,术语“Ig样结构域”不意于包括负责配体结合的RTK的结构域。在本发明的优选实施方式中,Ig样结构域是Ⅲ型RTK的D4和/或D5结构域。在VEGF受体家族的胞外域中,存在7个Ig样结构域,已知为D1、D2、D3、D4、D5、D6和D7。在本发明的一种优选实施方式中,Ig样结构域是VEGF受体家族的D7结构域。
本文使用的术语“血管内皮生长因子受体”、“VEGF受体”或“VEGF受体家族”,亦称V型RTK,包括血管内皮生长因子的RTK受体。如上所述,这些RTK在其胞外域中具有7个Ig样结构域。VEGF家族受体的实例为VEGFR-1(亦称Flt-1)、VEGFR-2(亦称KDR或Flk-1)和VEGFR-3(亦称Flt-4)。
术语受体酪氨酸激酶(RTK)的“胞外域”是本领域公知的,并且指RTK的细胞外部分,即在质膜外的RTK的部分。
术语受体酪氨酸激酶的胞外域的“近膜区”指的是接近质膜的RTK的细胞外部分,并优选地不直接负责配体与RTK的结合。近膜区的实例包括但不限于Ⅲ型受体酪氨酸激酶的D4结构域、Ⅲ型受体酪氨酸激酶的D5结构域、Ⅲ型受体酪氨酸激酶的D3-D4铰链区、Ⅲ型受体酪氨酸激酶的D4-D5铰链区和V型受体酪氨酸激酶的D7结构域。
本文中所用的术语“同型相互作用”指的是来自两个单体受体的两个相同的近膜区之间的相互作用。
本文中所用的术语“异型相互作用”指的是来自两个单体受体的两个不同的近膜区之间的相互作用。异型相互作用可以是两个不同类型的单体受体二聚化的结果,或者同种单体受体的野生型和突变体形式二聚化的结果。例如,本领域公知癌症患者可以携带某一受体的野生型等位基因和突变体等位基因。
本文中所用的术语“单体状态”指的是其中RTK分子由单一多肽链组成的RTK的状态,该单一多肽链不与相同或不同类型的第二RTK多肽结合。RTK二聚化导致自磷酸化和受体活化。因此,单体状态的RTK是非活性状态。单体状态也是其中单一RTK的D4、D5或D7结构域不与第二RTK的D4、D5或D7结构域分别结合的状态。
本文所使用的,“原体”是寡聚蛋白如RTK的结构单元。原体是可以以确定的化学计量组装以形成低聚物的蛋白质亚基。受体酪氨酸激酶的VEGFR家族是共价连接的二聚体,并且各个VEGFR原体是以名为“半胱氨酸结生长因子”的结构由按反向平行方式排列的四链β-折叠片组成的。
短语“锁定受体酪氨酸激酶的胞外域在非活性状态”指的是本发明的成分抑制受体酪氨酸激酶的活性的能力。换言之,该短语包括本发明的成分将平衡转向形成非活性或抑制性的受体构型的能力。例如,与不存在本发明的成分时受体活性相比,本发明的成分可以抑制受体酪氨酸激酶的活性至少5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%或95%。
本文中所用的术语“非活性状态”指的是其中RTK分子不能激活下游的信号传导的RTK的状态。非活性状态可以是这样的状态,其中受体酪氨酸激酶的胞外域被允许二聚化,但是两个单体的Ig样结构域(例如III型受体酪氨酸激酶的D4-D4或D5-D5结构域或者V型受体酪氨酸激酶的D7-D7结构域)之间的定位、定向、构象和/或距离发生改变,从而抑制了受体酪氨酸激酶的活性(例如受体内化受抑制和/或受体的酪氨酸自磷酸化受抑制和/或受体激活下游信号传导途径的能力受抑制)。非活性状态也包括如上所述的单体状态。非活性状态也可以是这样的状态,其中受体酪氨酸激酶的胞外域结合于受体配体并二聚化,但是还没有发生允许受体活化的构象变化。实施例22-25进一步讨论了表明存在对于RTK活化至关重要(例如通过介导D4或D5同型相互作用)但是对于受体二聚化不是必要的特定保守氨基酸残基的实验。术语“非活性状态”包括这样的状态,其中与没有本发明的成分时的受体活性相比,本发明的成分可以降低或抑制受体酪氨酸激酶的活性至少5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%或95%。本文中描述的任何功能试验可用于确定本发明的成分抑制受体酪氨酸激酶活性的能力。在一些实施方式中,本发明的成分可以表现出宽的效应,例如当大部分或全部目标RTK灭活时。在其他的实施方式中,本发明的成分可以表现出较窄的效应,例如当部分靶RTK灭活时。在这样的实施方式中,与没有所述成分时的受体相比,至少5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%或95%的受体被锁定在非活性状态。
本文中所使用的术语“构象表位”或“非线性表位”或“不连续表位”可互换地使用以指由至少两个不是单蛋白质链中的连续氨基酸的氨基酸组成的表位。例如,构象表位可以是由一段(strech)插入氨基酸分开,但是足够靠近以被本发明的成分作为单一表位识别的两个或更多个氨基酸组成。作为进一步的实例,在单蛋白质链上由插入氨基酸分开的氨基酸或者存在于分离的蛋白质链上的氨基酸可以由于蛋白质结构或复合体的构象形状而拉近,以成为可以被本发明的成分结合的构象表位。特定的不连续表位和构象表位在本文中进行描述(参见例如表4和5)。
本领域技术人员将理解,通常,本发明的成分结合的线性表位可以取决于或不取决于RTK的二级、三级或四级结构。例如,在一些实施方式中,本发明的成分可以结合一组氨基酸而无论它们是否以天然的三维蛋白质结构进行折叠。在其他的实施方式中,本发明的成分可能不识别组成该表位的单个氨基酸残基,而是可能需要特别的构象(弯曲、盘旋、转角或折叠)以识别和结合表位。
如本文使用的,“邻近表位”或“连续表位”可互换使用,指的是由单肽链中的连续氨基酸的至少两个氨基酸组成的表位。本文中描述了特定的连续表位(见,例如,表8)。在另一个实施方案中,本发明的成分结合VEGF受体上的连续表位。在一个实施方案中,连续表位由VEGF受体D7结构域中的两个或更多个残基组成。在另一个实施方案中,连续表位是选自于VEGFR1的672VAISSS677、VEGFR1的678TTLDCHA684、VEGFR1的685NGVPEPQ691、VEGFR1的700KIQQEPG706、VEGFR1的707IILG710、VEGFR1的711PGS713、VEGFR1的714STLFI718、VEGFR1的719ERVTEEDEGV728、VEGFR3的689VNVSDS694、VEGFR3的695LEMQCLV701、VEGFR3的702AGAHAPS708、VEGFR3的717LLEEKSG723、VEGFR3的724VDLA727、VEGFR3的728DSN730、VEGFR3的731QKLSI735和VEGFR3的736QRVREEDAGR745、VEGFR2的678TSIGES683、VEGFR2的684IEVSCTA690、VEGFR2的691SGNPPPQ697、VEGFR2的706TLVEDSG712、VEGFR2的713IVLK716、VEGFR2的717DGN719、VEGFR2的720RNLTI724和VEGFR2的725RRVRKEDEGL734的表位。
本文所用的短语“疏水氨基酸”是指包含疏水性能的氨基酸,如丙氨酸、半胱氨酸、苯丙氨酸、甘氨酸、组氨酸、异亮氨酸、赖氨酸、亮氨酸、甲硫氨酸、精氨酸、苏氨酸、缬氨酸、色氨酸、酪氨酸、丝氨酸、脯氨酸和本文列出的其他氨基酸。
本发明的各个方面将在以下小节中进一步详细描述:
I.结合人受体酪氨酸激酶胞外域的抗体
在本发明的一个方面,结合人受体酪氨酸激酶的胞外域例如Ig样结构域或铰链区的成分是抗体或其抗原结合片段。
在本文中所指的“抗体”包括整个的抗体和任何抗原结合片段(即抗原结合部分”)或其单链。抗体”指包含通过二硫键相互连接的至少两个重(H)链和两个轻(L)链的糖蛋白,或其抗原结合部分。各个重链由重链可变区(在这里缩写为VH)和重链恒定区组成。重链恒定区由三个结构域组成,CH1、CH2和CH3。各个轻链由轻链可变区(在这里缩写为VL)和轻链恒定区组成。轻链恒定区由一个结构域,CL,组成。VH和VL区可以进一步细分成称为互补决定区(CDR)的超变区,其间交替散布着更加保守的称为构架区(FR)的区域。各个VH和VL由三个CDR和四个FR组成,按以下顺序从氨基端至羧基端排列:FR1,CDR1,FR2,CDR2,FR3,CDR3,FR4。重链和轻链的可变区含有与抗原相互作用的结合结构域。抗体的恒定区可以介导免疫球蛋白与宿主组织或因子(包括免疫系统的各种细胞(例如效应细胞)和经典补体系统的第一成分(C1q))的结合。
本文中所用的术语抗体的“抗原结合部分”(或简称“抗体部分”)指的是保留特异性结合抗原(例如Kit的D4或D5结构域或VEGF受体的D7结构域)能力的抗体的一个或多个片段。已经表明,全长抗体的片段可以执行抗体的抗原结合功能。包含在术语抗体的“抗原结合部分”中的结合片段的实例包括(i)Fab片段,由VL、VH、CL和CH1结构域构成的单价片段;(ii)F(ab′)2片段,包含在铰链区通过二硫桥连接的两个Fab片段的二价片段;(iii)Fab’片段,主要是带有部分铰链区的Fab(参见,FUNDAMENTAL IMMUNOLOGY(Paul编著,第三版1993);(iv)由VH和CH1结构域构成的Fd片段;(v)由抗体单臂的VL和VH结构域构成的Fv片段,(vi)dAb片段(Ward等,(1989)Nature341:544-546),其由VH结构域构成;(vii)分离的互补决定区(CDR);以及(viii)纳米体,含有单一的可变结构域和两个恒定结构域的重链可变区。而且,尽管Fv片段的两个结构域VL和VH由独立的基因编码,但是它们可以使用重组方法通过使得它们能够形成单蛋白质链的合成接头连接的,其中VL和VH区配对以形成单价分子(已知为单链Fv(scFv);参见例如Bird等(1988)Science 242:423-426;和Huston等(1988)Proc.Natl.Acad.Sci.USA 85:5879-5883)。这样的单链抗体也意于包括在术语抗体的“抗原结合部分”中。这些抗体片段使用本领域技术人员已知的常规方法获得,并且片段以与完整抗体同样的方法进行用途筛选。
本文中所用的“分离的抗体”意于指基本上没有具有不同抗原特异性的其它抗体的抗体(例如特异性结合RTK的Ig样结构域的分离的抗体基本上没有特异性结合除了RTK的Ig样结构域之外的抗原的抗体)。而且,分离的抗体可以是基本上没有其它的细胞物质和/或化学物质。但是,“分离的抗体”可以包括全都特异性结合例如RTK的Ig样结构域的多克隆抗体。
本文中所用的术语“单克隆抗体”或“单克隆抗体组成”指的是单分子组成的抗体分子的制剂。单克隆抗体组成显示对特定表位的单一结合特异性和亲合性。
本文中所用的术语“人抗体”意于包括具有其中框架区和CDR区均源自人种系免疫球蛋白序列的可变区的抗体。而且,如果抗体含有恒定区,该恒定区也源自于人种系免疫球蛋白序列。本发明的人抗体可以包括不是由人种系免疫球蛋白序列编码的氨基酸残基(例如体外随机或定点诱变或者体内体细胞突变引入的突变)。然而,本文中所用的术语“人抗体”不意于包括其中来自另一哺乳动物种(例如小鼠)的种系的CDR序列嫁接于人框架序列的抗体。
术语“人单克隆抗体”是指具有其中框架区和CDR区均源自人种系免疫球蛋白序列的可变区的显示单一结合特异性的单克隆抗体。在一种实施方式中,人单克隆抗体由包括由非人类转基因动物例如转基因小鼠获得的B细胞的杂交瘤产生,所述非人类转基因动物具有融入无限增殖细胞的包含人重链转基因和轻链转基因的基因组。
本文中所用的术语“重组人抗体”包括通过重组方法制备、表达、创造或分离的所有人抗体,例如(a)从对于人免疫球蛋白基因进行转基因或转染色体的动物(例如小鼠)或由其制备的杂交瘤分离的抗体(以下进一步描述),(b)从经转化以表达人抗体的宿主细胞分离的抗体,例如从转染瘤的抗体,(c)从重组、组合的人抗体文库分离的抗体,和(d)通过包括将人免疫球蛋白基因序列与其它DNA序列剪接的任何其它方法制备、表达、创造或分离的抗体。这样的重组人抗体具有其中框架区和CDR区域源自于人种系免疫球蛋白序列的可变区。然而,在某些实施方式中,这样的重组人抗体可以进行体外诱变(或在使用人Ig序列转基因的动物时,进行体内体细胞诱变),从而尽管重组抗体的VH和VL区的氨基酸序列源自人种系VH和VL序列和与之相关,但它们可以并不是天然存在于体内人抗体种系库内。
本文中所用的“同种型”指的是由重链恒定区基因编码的抗体类(例如IgM或IgG1)。
短语“识别抗原的抗体”和“抗原特异性的抗体”在本文中与术语“特异性结合抗原的抗体”互换地使用。
术语“人抗体衍生物”指的是人抗体的任何修饰形式,例如抗体和另一种物质或抗体的缀合物。
术语“人源化抗体”意于表示其中源自另一哺乳动物种例如小鼠的CDR序列嫁接于人框架序列上的抗体。可以在人框架序列内进行另外的构架区修饰。本领域技术人员将理解,当序列“源自于”特定的种时,所述序列可以是蛋白质序列,例如当可变区氨基酸取自鼠抗体时,或者所述序列可以是DNA序列,例如当编码可变区的核酸取自鼠DNA时。也可以基于人和非人(例如鼠或兔)抗体来设计人源化抗体。所述设计的抗体(可能结合了人的和非人的残基)可以是化学合成的。所述序列也可以在DNA水平合成,并在体外或体内表达以产生人源化抗体。
术语“嵌合抗体”意于指其中可变区序列源自于一个种而恒定区序列源自于另一个种的抗体,例如其中可变区序列源自于小鼠抗体,而恒定区序列源自于人抗体的抗体。
术语“抗体模拟物”或“抗体类似物”意于指的是能够模拟抗体结合抗原的能力的分子,但它不局限于天然的抗体结构。此类抗体模拟物的实例包括但不限于Adnectin(即基于纤连蛋白的结合分子)、亲和体、DARPin、抗运载蛋白(Anticalin)、高亲合性多聚体和Versabody,尽管它们它们模拟传统的抗体结合,它们全部采用通过完全不同的机制产生的结构并通过完全不同的机制发挥作用。当本发明的实施方式涉及抗体或其抗原结合部分时,它们也可以应用于如上所述的抗体模拟物。
本文中所用的“特异性结合”RTK的Ig样结构域的抗体意指以1x10-7M或更低、更优选5x 10-8M或更低、更优选1x 10-8M或更低、更优选5x 10-9M或更低的的KD结合RTK的Ig样结构域的抗体。
本文中所用的术语“基本上不结合”蛋白质或细胞表示不结合或不以高亲和性结合蛋白质或细胞,即以1x 10-6M或更高、更优选1x 10-5M或更高、更优选1x 10-4M或更高、更优选1x 10-3M或更高、甚至更优选1x 10-2M或更高的KD结合蛋白质或细胞。
本文中所用的术语″K结合″或“Ka”意指特定抗体-抗原相互作用的结合速率,而本文中所用的术语″K解离″或“Kd”意指特定抗体-抗原相互作用的解离速率。本文中所用的术语“KD”意指解离常数,其从Kd与Ka的比率(即Kd/Ka)获得,并表示为摩尔浓度(M)。可以使用本领域公认的方法确定抗体的KD值。测定抗体的KD的优选方法是使用表面等离子体共振,优选使用生物传感器系统,例如Biacore 系统。
系统。
本文中所用的术语“高亲和性”在涉及IgG型抗体时,指的是对于RTK的Ig样结构域,抗体具有10-8M或更低、更优选10-9M或更低、甚至更优选10-10M或更低的KD。然而,对于其它抗体同种型“高亲和性”结合可能有所改变。例如,IgM同种型的“高亲和性”结合指的是抗体具有10-7M或更低、更优选10-8M或更低、更优选10-9M或更低的KD。
抗体
本发明的抗体特异性结合RTK的Ig样结构域,例如人受体酪氨酸激酶Ⅲ型家族的成员。在优选实施方式中,本发明的抗体或其抗原结合部分与RTK单体的Ig样结构域的结合将胞外域锁定在非活性状态,例如单体状态,并因此拮抗RTK二聚化和激活下游信号传导途径的能力。例如,所述抗体可以阻断配体诱导的受体酪氨酸激酶的酪氨酸自磷酸化和/或受体内化。
选择或设计本发明的抗体以结合RTK的特定Ig样结构域,例如人Kit的D4结构域或D5结构域或者VEGF受体的D7结构域。在其它实施方式中,选择或设计抗体或其抗原结合部分以结合与RTK(例如Kit受体或VEGF受体)的结构域具有同源性的蛋白质。例如,可以选择或设计抗体以结合与Kit受体的结构域(例如D4或D5结构域)或者VEGF受体的D7结构域具有至少50%的同一性、至少60%的同一性、至少70%的同一性、至少80%的同一性、至少90%的同一性、或至少95%、96%、97%、98%或99%的同一性的结构域。这样的抗体或其抗原结合部分将能够结合与Kit的D4或D5结构域或者VEGF受体的D7结构域在功能上类似的蛋白质结构域(可能在Kit、VEGF受体和其它RTK中)。
也可以选择或设计本发明的抗体或其抗原结合部分以结合由RTK(例如人Ⅲ型RTK)的Ig样结构域衍生的特殊基序或共有序列,从而使抗体或其抗原结合部分特异性结合人RTK Ⅲ型家族的成员之间以及Ⅲ型RTK和其它RTK(例如V型RTK)之间共有的表位或结构域。例如,可以通过使用序列比对算法来比对各种RTK的结构域(例如在RTK类型或各物种之间D4结构域的结构域)找到这类线性共有序列(参见图6B)。本领域技术人员可以比对例如来自各种物种(例如人、小鼠、大鼠)的Kit D4结构域的蛋白质序列以确定在被比对的序列的至少40%、至少50%、至少60%、至少70%、至少80%、至少90%或至少100%中哪些蛋白质残基是保守的。然后,这样的共有序列可以用于产生特异性结合该共有序列的抗体或其它成分,并因此结合Kit RTK的最保守的残基。类似地,人们也可以比对V型RTK的D7结构域的蛋白质序列(参见图6)以获得可以用于产生本发明的成分的共有序列。本领域技术人员应理解,最高度保守的残基是哪些通过进化保留并最可能对于蛋白质功能具有重要性的残基。或者,如果比对在各种不同类的RTK之间进行,对于这样的共有序列产生的抗体将允许抗体结合多种RTK类型中的相似Ig样结构域。
在具体实施方式中,本发明的成分(例如抗体或其抗原结合部分)结合D4相互作用位点的以下共有序列:LX1RX2X3X4X5X6X7G,其中L是亮氨酸,R是精氨酸,G是甘氨酸,和X1、X2、X3、X4、X5、X6和X7是任何氨基酸。在具体的实施方案中,X1选自苏氨酸、异亮氨酸、缬氨酸、脯氨酸、天门冬酰胺或赖氨酸;X2选自亮氨酸、缬氨酸、丙氨酸和甲硫氨酸;X3选自赖氨酸、组氨酸、天门冬酰胺和精氨酸;X4选自甘氨酸、缬氨酸、丙氨酸、谷氨酸、脯氨酸和甲硫氨酸;X5选自苏氨酸、丝氨酸、谷氨酸、丙氨酸、谷氨酰胺和天冬氨酸;X6选自谷氨酸、天冬氨酸和谷氨酰胺;以及X7选自甘氨酸、丝氨酸、丙氨酸、赖氨酸、精氨酸、谷氨酰胺和苏氨酸。
在另一实施方式中,本发明的成分(例如抗体或其抗原结合部分)结合VEGF受体家族成员的D7结构域的以下共有序列:IX1RVX2X3EDX4G,其中I是异亮氨酸,R是精氨酸,E是谷氨酸,D是天冬氨酸,G是甘氨酸;和X1、X2、X3和X4是任何氨基酸。在具体的实施方案中,X1选自谷氨酸、精氨酸和谷氨酰胺;X2选自精氨酸和苏氨酸;X3选自谷氨酸和赖氨酸;以及X4选自谷氨酸和丙氨酸(SEQ ID NO:1)。
在另一个实施方案中,本发明的成分(例如,抗体或其抗原结合部分)结合VEGF受体的D7结构域的以下共有序列:L/I X1 R Φ X2 X3X4 D/E X5 G(SEQ ID NO:158),其中L是亮氨酸,I是异亮氨酸,R是精氨酸,Φ是疏水性氨基酸,D是天冬氨酸,E是谷氨酸,G是甘氨酸;以及X1、X2、X3、X4和X5是任何氨基酸。在具体的实施方案中,Φ是缬氨酸;X1选自于精氨酸、谷氨酰胺、谷氨酸和天冬氨酸;X2选自于精氨酸、赖氨酸和苏氨酸;X3选自于赖氨酸、谷氨酸、谷氨酰胺和缬氨酸;X4选自于谷氨酸和缬氨酸;以及X5选自于谷氨酸、甘氨酸、丝氨酸和谷氨酰胺。
本发明的抗体不结合RTK的配体结合位点,例如Kit受体的SCF结合位点。因此,本文中描述的抗体不拮抗受体结合其目标配体的能力。
在一些实施方式中,抗体或其抗原结合部分结合人Kit受体的特定序列,例如人Kit受体的残基309-413、残基410-519、381Arg和386Glu或者418Tyr和505Asn。
在其它实施方式中,抗体或其抗原结合部分结合代表蛋白质中三维结构的蛋白质基序或共有序列。这样的基序或共有序列不代表氨基酸的连续链,而是导致RTK的三维折叠的非连续的氨基酸排列(即“结构基序”或“非线性表位”)。这样的基序的实例是Kit受体的D4-D4或D5-D5结合界面或VEGF受体D7-D7结合界面。在一种实施方式中,本发明的抗体结合例如D4-D4、D5-D5或D7-D7界面中的非线性表位,从而阻止RTK的活化。
在优选实施方式中,本发明的抗体或其抗原结合部分可以结合构成表4(下文中)中描述的小腔或口袋的Kit受体中的一个或多个残基。例如,本发明的抗体或其抗原结合部分可以结合Kit受体的D3-D4铰链区中的一个或多个以下残基:来自D3结构域的K218、S220、Y221、L222和来自D4结构域的F340、P341、K342、N367、E368、S369、N370、I371、Y373。本发明的抗体或其抗原结合部分也可以结合构成Kit受体的D4结构域中的凹面的一个或多个以下残基:Y350、R353、F355、K358、L379、T380、R381、L382、E386和T390。在另一实施方式中,本发明的抗体或其抗原结合部分可以结合形成Kit受体的D2-D3铰链区中的口袋的一个或多个以下残基:来自D2结构域的Y125、G126、H180、R181、K203、V204、R205、P206和F208以及来自D3结构域的V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269。
因此,在一些实施方式中,本发明的抗体或其抗原结合部分可以结合连续的或不连续的氨基酸残基,并起到阻止RTK的近膜区以使酪氨酸激酶能够活化的距离和定向进行定位所需的运动的分子楔的作用。本发明的抗体或其抗原结合部分也可以起到阻止同型D4或D5受体的相互作用或使配体-受体相互作用位点不稳定的作用。在一些优选实施方式,本发明的抗体或其抗原结合部分可以结合Kit受体上的一个或多个以下残基:Y125、G126、H180、R181、K203、V204、R205、P206、P206、F208、K127、A207、V238、S239、S240、S241、H263、G265、D266、F267、N268、Y269、T295、L222、L222、L223、E306、V308、R224、V308、K310、K218、A219、S220、K218、A220、Y221、A339、D327、D398、E338、E368、E386、F312、F324、F340、F355、G311、G384、G387、G388、I371、K342、K358、L382、L379、N326、N367、N370、N410、P341、S369、T385、V325、V407、V409、Y373、Y350、Y408、T380、T390、R381、R353、T411、K412、E414、K471、F433、G470、L472、V497、F469、A431或G432。
本领域技术人员将理解,在一些实施方式中,本发明的抗体或其抗原结合部分可以容易地靶向于其它Ⅲ型RTK中的相应残基,例如形成相似的口袋或空腔的那些残基或通过结构比对或序列比对处于相同的位置的那些残基。
在一种具体实施方式中,本发明的抗体或其抗原结合部分结合Ⅲ型RTK上的构象表位或非连续表位。构象表位或非连续表位可以由来自Ⅲ型RTK(例如人Kit受体或PDGF受体)的D3、D4或D5结构域或者D4-D5或D3-D4铰链区的两个或更多个残基组成。例如,所述构象表位或非连续表位可以由下面表4中所列的两个或更多个残基组成。
在一种特定实施方式中,本发明的抗体或其抗原结合部分结合由2个或多个选自Y125、H180、R181、K203、V204、R205、P206、V238、S239、S240、H263、G265、D266、F267、N268和Y269的氨基酸组成的构象表位。在相似的实施方式中,本发明的抗体或其抗原结合部分可以结合由选自以下的氨基酸组之一的2个或多个氨基酸组成的构象表位:P206、F208、V238和S239;K127、A207、F208和T295;L222、A339、F340、K342、E368、S369、N370、I371和Y373;L222、L223、E306、V308、F312、E338、F340和I371;R224、V308、K310、G311、F340、P341和D398;K218、A219、S220、N367、E368和S369;K218、A220、E368和S369;G384、T385、T411、K412、E414和K471;Y408、F433、G470、K471和L472;F324、V325、N326和N410;D327、N410、T411、K412和V497;G384、G387、V409和K471;L382、G387、V407和V409;Y125、G126、H180、R181、K203、V204、R205、P206、F208、V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269;P206、F208、V238和S239;K218、S220、Y221、L222、F340、P341、K342、N367、E368、S369、N370、I371和Y373;G384、G387、G388、Y408、V409、T411、F433、F469、G470和K471;D327、T411、K412、E414、A431、G432和K471;Y350、F355、K358、L379、T380、R381、L382、E386和T390;Y350、R353和F355。如以上所指出的,本发明的抗体可以结合形成在表4中确定的口袋或空腔的所有氨基酸残基,或者它们可以结合形成口袋或空腔的残基的一部分。应当理解,在某些实施方式中,当说明本发明的抗体结合表位例如构象表位时,意图是抗体仅结合那些构成该表位(例如表4中确定的口袋或空腔)的那些特定残基,而不是在该受体的线性氨基酸序列中的其它残基。
在进一步的实施方式中,本发明的抗体或其抗原结合部分结合构象表位,其中所述构象表位由选自表5中所列的肽的两个或更多个氨基酸残基组成。在一种具体实施方式中,构象表位由选自第一肽的一个或多个氨基酸残基和选自第二肽的一个或多个氨基酸残基组成,其中第一和第二肽选自表5中所列的肽的组。因而,本发明的抗体或其抗原结合部分结合构象表位,其中所述第一和第二肽组如下:Ala219-Leu222和Thr304-Val308;Asp309-Gly311和Arg224-Gly226;Thr303-Glu306和Ala219-Leu222;Asn367-Asn370和Ser217-Tyr221;Ala339-Pro343和Asn396-Val399;Ala339-Pro343和Glu368-Arg372;Lys358-Tyr362和Val374-His378;Asp357-Glu360和Leu377-Thr380;Met351-Glu360和His378-Thr389;His378-Thr389和Val323-Asp332;Val409-Ile415和Ala493-Thr500;Val409-Ile415和Ala431-Thr437;Val409-Ile415和Phe469-Val473;Val409-Ile415和Val325-Asn330;Val409-Ile415和Arg381-Gly387;Gly466-Leu472和Gly384-Gly388;Val325-Glu329和Tyr494-Lys499;Thr411-Leu416和Val497-Ala502;Ile415-Leu421和Ala502-Ala507;Ala502-Ala507和Lys484-Thr488;以及Ala502-Ala507和Gly445-Cys450。
本发明的抗体可以结合形成前述第一和第二肽组的所有氨基酸残基,或者它们可以结合形成第一和第二肽组的残基的一部分。应当理解,在某些实施方式中,当说到本发明的抗体结合表位例如构象表位时,意图是抗体仅结合构成该表位(例如表5中确定的特定肽)的那些特异性残基,而不是在该受体的线性氨基酸序列中的其它残基。
在另一实施方式中,本发明的抗体或其抗原结合部分结合由选自E33、P34、D72、E76、N77、K78、Q79、K158、D159、N250、S251、Q252、T253、K254、L255、N260、W262、H264、G265、E344、N352、R353、F355、T356、D357、Y362、S365、E366、N367、N370和G466的2个或多个氨基酸组成的构象表位或非连续表位。
在另一实施方式中,本发明的抗体或其抗原结合部分结合人PDGFRβ的氨基酸残基385Arg和390Glu,或者PDGFRα中相应的残基。人PDGFRβ的残基385Arg和390Glu类似于Kit受体的残基381Arg和386Glu,并介导PDGFRβ的同型D4-D4相互作用。本发明的抗体或其抗原结合部分可以通过阻止对于胞质结构域以酪氨酸激酶活化必要的距离和定向进行定位所必需的III型RTK的近膜区之间的关键同型相互作用(例如人PDGFRβ的385Arg和390Glu之间形成的盐桥)而发挥其对受体活化的抑制作用。本文中讨论的实验证明:同型D4-D4相互作用对于PDGFRβ的二聚化不是必要的,而PDGFRβ二聚化对于受体活化是必要的然而并非足够的。因此,本发明的抗体或其抗原结合部分可以允许PDGFRβ二聚化而阻止活化。基于结构的序列比对已经表明:在Kit、PDGFRα、PDGFRβ和CSF1R中,EF环的大小以及包含重要氨基酸的D4-D4界面是保守的。因此,在一些实施方式中,本发明的抗体或其抗原结合片段可以靶向于Ⅲ型RTK的D4或D5结构域的保守区。
在一些实施方案中,本发明的抗体或其抗原结合部分结合人VEGF受体的特定序列,例如,VEGFR1的残基718-727、VEGFR1的Arg720和Asp725、VEGFR2的残基724-733、VEGFR2的Arg726和Asp731、VEGFR3的残基735-744或VEGFR3的残基Arg737和Asp742。
在另一个实施方案中,本发明的抗体或其抗原结合部分结合VEGF受体的连续表位。在一个实施方案中,连续表位由VEGF受体D7结构域中的两个或更多个残基组成。在另一个实施方案中,连续表位是选自VEGFR1的672VAISSS677、VEGFR1的678TTLDCHA684、VEGFR1的685NGVPEPQ691、VEGFR1的700KIQQEPG706、VEGFR1的707IILG710、VEGFR1的711PGS713、VEGFR1的714STLFI718、VEGFR1的719ERVTEEDEGV728、VEGFR3的689VNVSDS694、VEGFR3的695LEMQCLV701、VEGFR3的702AGAHAPS708、VEGFR3的717LLEEKSG723、VEGFR3的724VDLA727、VEGFR3的728DSN730、VEGFR3的731QKLSI735和VEGFR3的736QRVREEDAGR745、VEGFR2的678TSIGES683、VEGFR2的684IEVSCTA690、VEGFR2的691SGNPPPQ697、VEGFR2的706TLVEDSG712、VEGFR2的713IVLK716、VEGFR2的717DGN719、VEGFR2的720RNLTI724和VEGFR2的725RRVRKEDEGL734的表位。
在另外的实施方式中,选择或设计本发明的抗体或其抗原结合部分以特异性结合突变体RTK。在优选实施方式中,突变体RTK是致瘤的或致癌的突变体。在一种具体实施方式中,选择或设计抗体或其抗原结合部分以结合致癌的Kit受体突变体。可以被本发明的抗体靶向的几种Kit受体突变体是具有一个或多个以下氨基酸中的突变的Kit受体:Thr417、Tyr418、Asp419、Leu421、Arg420、Tyr503或Ala502。本领域技术人员应理解,本发明的方法将可以应用于Kit中的其它突变或其它RTK中的突变。靶向突变体RTK的一个优点是治疗抗体可以仅结合包含突变的细胞上的RTK,从而保持健康细胞大部分或全部不受影响。因此,在突变是致瘤性突变的情况下,仅肿瘤细胞被作为治疗靶标,从而潜在地减少副作用和剂量要求。
优选地,抗体以5x 10-8M或更低的KD、1x 10-8M或更低的KD、5x 10-9M或更低的KD,或者1x 10-8M至1x 10-10M之间或更低的KD结合人RTK的Ig样结构域。本领域已知用于评价抗体对于RTK(例如Kit或VEGF受体)的Ig样结构域的结合能力的标准试验,包括例如ELISA、蛋白质印迹和RIA。抗体的结合动力学(例如结合亲合力)也可以通过本领域已知的标准试验来评定,例如通过ELISA、Scatchard和Biacore分析。
工程化和修饰的抗体
根据本发明的方法制备的抗体的VH和/或VL序列可以用作工程化修饰的抗体的起始材料,所述修饰的抗体可以具有从起始抗体发生改变的性质。抗体可以通过修饰一个或两个原始可变区(即VH和/或VL)内(例如一或多个CDR区和/或一或多个构架区内)的一或多个残基进行工程化。另外地或者可选地,可以通过修饰恒定区内的残基来进行工程化,例如改变抗体的效应子功能。
一种可以进行的可变区工程化是CDR接枝。抗体主要地通过位于6个重链和轻链互补决定区(CDR)中的氨基酸残基与靶抗原相互作用。为此,在各抗体之间CDR中的氨基酸序列比CDR外的序列更加多样化。因为CDR序列负责大多数抗体-抗原相互作用,可以通过构建表达载体来表达模拟特定的天然存在抗体的性质的重组抗体,所述表达载体包括来自特定的天然存在抗体的CDR序列,这些CDR序列被接枝到来自具有不同性质的不同抗体的框架序列上(参见,例如Riechmann,L.等(1998)Nature 332:323-327;Jones,P.等(1986)Nature 321:522-525;Queen,C.等(1989)Proc.Natl.Acad.Sei.U.S.A.86:10029-10033;授予Winter的美国专利No.5,225,539,以及授予Queen等的美国专利No.5,530,101;5,585,089;5,693,762和6,180,370)。
可以从公共的DNA数据库或包括种系抗体基因序列的发表的参考文献来获得抗体的框架序列。例如,人重链和轻链可变区基因的种系DNA序列可以从“VBase”人种系序列数据库中找到(可在互联网上在mrc-cpe.cam.ac.uk/vbase网站获得),以及在Kabat,E.A.等(1991)Sequences of Proteins of Immunological Interest,Fifth Edition,U.S.Department of Health and Human Services,NIH Publication No.91-3242;Tomlinson,I.M.等(1992)″The Repertoire of Human Germline VHSequences Reveals about Fifty Groups of VH Segments with DifferentHypervariable Loops″J.Mol.Biol.227:776-798;和Cox,J.P.L.等(1994)″A Directory of Human Germ-line VH Segments Reveals a StrongBias in their Usage″Eur.J.Immunol.24:827-836中找到;每一篇文献的内容均通过引用的方式明确并入本文中。作为另一实例,人重链和轻链可变区基因的种系DNA序列可以从Genbank数据库中找到。
使用一种称为Gapped BLAST的序列相似性检索方法相对于编译蛋白质序列数据库比较抗体蛋白质序列(Altschul等(1997)NucleicAcids Research 25:3389-3402),这是本领域技术人员公知的。BLAST是一种探试算法(heuristic algorithm),其中抗体序列和数据库序列之间的统计上显著的比对很可能含有比对字段(aligned word)的高分片段对(high-scoring segment pair)(HSP)。不能通过延伸或者修剪提高分值的片段对称为命中(hit)。简言之,VBASE来源的核苷酸序列(vbase.mrc-cpe.cam.ac.uk/vbase1/list2.php)被翻译,且FR1-FR3框架区之间和包括上述框架区之间的区域得到保持。数据库序列平均长度为98个残基。去除在蛋白质全长范围内精确匹配的重复序列。使用具有缺省的标准参数(除了被关闭的低复杂性过滤器)的程序blastp和BLOSUM62的替代矩阵进行蛋白质的BLAST检索过滤出产生序列匹配的前5个命中。核苷酸序列在所有6个框架中被翻译,而在数据库序列的匹配片段中没有终止密码子的框架被认为是潜在的命中。这随后使用BLAST程序tblastx进行证实,其在所有6个框架中翻译抗体序列,并将那些翻译与在所有6个框架中动态地翻译的VBASE核苷酸序列进行比较。可以如上所述类似地对VBASE进行其它人种系序列数据库,例如可从IMGT(http://imgt.cines.fr)获得的那些的检索。
同一性是在全长序列上抗体序列和蛋白质数据库之间的精确的氨基酸匹配。阳性结果(同一性+置换匹配)不是相同的,但是氨基酸置换由BLOSUM62替代矩阵指导。如果抗体序列以相同的同一性与两个数据库序列相匹配,那么具有最多阳性结果的命中将被确定为匹配序列命中。
确认的VH CDR1、CDR2和CDR3序列以及VK CDR1、CDR2和CDR3序列可以接枝到构架区上,所述框架区具有与框架序列所来源的种系免疫球蛋白基因中发现的那些序列相同的序列,或者CDR序列可以接枝到与种系序列相比含有一或多个突变的构架区上。例如,已经发现在某些情况下,在构架区内的突变残基对于维持或提高抗体的抗原结合能力是有益的(参见例如授予Queen等的美国专利No.5,530,101;5,585,089;5,693,762和6,180,370)。
另一类型的可变区修饰是突变VH和/或VK CDR1、CDR2和/或CDR3区内的氨基酸残基,从而改进目的抗体的一种或多种结合性质(例如亲合性)。可以进行定点诱变或PCR介导的诱变来引入突变,并且可以在本领域已知的体外或体内试验中评价对抗体结合的影响或其它感兴趣的功能性质。例如,可以突变本发明的抗体来产生文库,然后可以就与RTK的Ig样结构域,例如人Kit RTK的D4或D5结构域或VEGF受体的D7结构域,的结合对文库进行筛选。优选地,引入保守修饰(如以上讨论的)。突变可以是氨基酸置换、添加或缺失,但优选是置换。而且,一般CDR区内不超过一个、两个、三个、四个或五个残基被改变。
另一种类型的框架修饰包括突变构架区内、或者甚至一个或多个CDR区内的一个或多个残基以去除T细胞表位,从而降低抗体潜在的免疫原性。该方法也称为“脱免疫(deimmunization)”,并在Carr等人的美国专利公开No.20030153043中更加详细地进行了描述。
在框架或CDR区内进行的修饰之外或者替代以上修饰,本发明的抗体可以工程化以包含Fc区内的修饰,从而通常改变抗体的一种或多种功能性质,例如血清半衰期、补体结合、Fc受体结合和/或抗原依赖性细胞毒性。而且,本发明的抗体可以进行化学修饰(例如一个或多个化学部分可以附着于抗体)或者进行修饰以改变它的糖基化作用,而再改变抗体的一种或多种功能性质。在下文中更加详细地描述这些实施方式中的每一个。Fc区中残基的编号是Kabat的EU索引的编号。
在一种实施方式中,修饰CH1的铰链区从而改变(例如增加或减少)铰链区中半胱氨酸残基的数量。该方法在Bodmer等人的美国专利No.5,677,425中进一步描述。改变CH1铰链区中半胱氨酸残基的数目例如以帮助轻链和重链的组装或者提高或降低抗体的稳定性。
在另一实施方式中,使抗体的Fc铰链区突变以降低抗体的生物半衰期。更具体地说,将一个或多个氨基酸突变引入Fc铰链片段的CH2-CH3结构域界面区,使得抗体相对于天然的Fc铰链结构域的葡萄球菌A蛋白(SpA)结合具有受损的SpA结合。该方法在Ward等人的美国专利No.6,165,745中进一步描述。
在另一实施方式中,抗体经修饰以增加其生物半衰期。多种方法均可行。例如,如授予Ward的美国专利No.6,277,375中描述的,可以引入一个或多个以下突变:T252L、T254S、T256F。或者,为了提高生物半衰期,如在Presta等人的美国专利No.5,869,046和6,121,022中所述,可以在CH1或CL区内改变抗体以包含从IgG的Fc区CH2结构域的两个环获得的补救受体(salvage receptor)结合表位。只要抗体与RTK的Ig样结构域的结合不受损,这些策略将是有效的。
在再其它实施方式中,通过以不同的氨基酸残基来置换至少一个氨基酸残基而改变Fc区,以改变抗体的效应子功能。例如,选自氨基酸残基234、235、236、237、297、318、320和322的一个或多个氨基酸可以被不同的氨基酸残基取代,从而抗体具有对效应子配体的改变的亲和性,但保持亲本抗体的抗原结合能力。其亲和性发生改变的效应子配体可以是例如Fc受体或补体的C1成分。该方法在Winter的美国专利No.5,624,821和5,648,260中有进一步的详细描述。
在另一实例中,选自氨基酸残基329、331和322的一个或多个氨基酸可以被不同的氨基酸残基置换,从而抗体具有改变的C1q结合和/或降低的或消除的补体依赖性细胞毒性(CDC)。该方法在Idusogie等人的美国专利No.6,194,551中进一步详细描述。
在另一实例中,改变氨基酸位置231和239内的一个或多个氨基酸残基,从而改变抗体固定补体的能力。该方法在Bodmer等人的PCT公开WO 94/29351中进一步描述。
在又一个实例中,通过修饰以下位置的一个或多个氨基酸来修饰Fc区以提高抗体介导抗体依赖性细胞毒性(ADCC)的能力和/或提高抗体对Fcγ受体的亲和力:238、239、248、249、252、254、255、256、258、265、267、268、269、270、272、276、278、280、283、285、286、289、290、292、293、294、295、296、298、301、303、305、307、309、312、315、320、322、324、326、327、329、330、331、333、334、335、337、338、340、360、373、376、378、382、388、389、398、414、416、419、430、434、435、437、438或439。该方法在Presta的PCT公开WO 00/42072中进一步描述。人IgG1上与FcγR1、FcγRII、FcγRIII和FcRn的结合位点已经作图,并且具有改进的结合的变异体已经描述(参见Shields,R.L.等(2001)J.Biol.Chem.276:6591-6604)。位置256、290、298、333、334和339的特定突变显示出提高了对FcγRIII的结合。另外,以下结合突变体显示出提高了FcγRIII的结合:T256A/S298A,S298A/E333A、S298A/K224A和S298A/E333A/K334A。
在再另一实施方式中,如美国临时申请No.60/957,271(通过引用的方式将该专利以其全部内容并入本文)中描述的那样,通过引入半胱氨酸残基来修饰本发明抗体的C-末端。这样的修饰包括但是不局限于在全长重链序列的C-末端或其附近置换现有的氨基酸残基,以及将含有半胱氨酸的延伸段引入全长重链序列的C-末端。在优选实施方式中,含有半胱氨酸的延伸段包括序列丙氨酸-丙氨酸-半胱氨酸(从N-末端至C-末端)。
在优选实施方式中,这样的C-末端半胱氨酸修饰的存在提供了结合伴体分子例如治疗剂或标记分子的位置。特别是,由于C-末端半胱氨酸修饰周到反应性硫醇基的存在可如下文详述的用于缀合使用二硫接头的伴体分子。用这样的方式结合抗体与伴体分子能够提高对连接的特定位点的控制。此外,通过在C-末端处或其附近引入连接位点,可以优化结合,从而降低或消除对抗体功能性质的干扰,并能够简化对缀合物制剂的分析和质量控制。
在再另一实施方式中,抗体的糖基化作用被修饰。例如,可以产生无糖基化抗体(即抗体缺少糖基化)。可以改变糖基化作用以例如提高抗体对抗原的亲和性。这样的糖修饰可以通过例如改变抗体序列内的一个或多个糖基化位点而实现。例如,可以进行导致消除一个或多个可变区框架糖基化位点的一个或多个氨基酸置换,从而消除该位点的糖基化。这样的无糖基化可以增加抗体对抗原的亲和性。在授予Co等的美国专利No.5,714,350和6,350,861中进一步详细描述了这样的方法。在授予Hanai等人的美国专利7,214,775、授予Presta的美国专利No.6,737,056、授予Presta的美国公开No.20070020260、授予Dickey等的PCT公开No.WO/2007/084926、授予Zhu等人的PCT公开No.WO/2006/089294和授予Ravetch的PCT公开No.WO/2007/055916中进一步详细描述了用于改变糖基化的其它方法,上述每一篇文献的全部内容均通过引用的方式并入本文。
另外地或可选地,可以产生具有改变的糖基化类型的抗体,例如具有减少量的岩藻糖残基的低岩藻糖化(hypofucosylated)抗体或具有增加的对开(bisecting)GlcNac结构的抗体。已经证明这种改变的糖基化模式提高了抗体的ADCC能力。通过例如在具有改变的糖基化机构的宿主细胞中表达抗体可以实现这种糖修饰。具有改变的糖基化机构的细胞在本领域中有描述,并且可用作表达本发明的重组抗体的宿主细胞,从而产生具有改变的糖基化的抗体。例如,细胞系Ms704、Ms705和Ms709缺少岩藻糖基转移酶基因,FUT8(α(1,6)岩藻糖基转移酶),从而在Ms704、Ms705和Ms709细胞系中表达的抗体在其糖类化合物上缺少岩藻糖。使用两个置换载体通过靶向破坏CHO/DG44细胞中的FUT8基因来产生Ms704、Ms705和Ms709FUT8-/-细胞系。(参见Yamane等人的美国专利公开No.20040110704和Yamane-Ohnuki等(2004)Biotechnol Bioeng 87:614-22)。作为另一实例,Hanai等的EP 1,176,195描述了具有功能受损的FUT8基因(其编码岩藻糖转移酶)的细胞系,从而在这样的细胞系中表达的抗体通过减少或消除α-1,6键-相关的酶而表现出低岩藻糖化。Hanai等也描述了具有将岩藻糖添加至N-乙酰葡糖胺(其结合抗体的Fc区)的低的酶活性或不具有所述酶活性的细胞系,例如大鼠骨髓瘤细胞系YB2/0(ATCC CRL 1662)。Presta的PCT公开WO 03/035835描述了变异CHO细胞系、Lec13细胞,其具有降低的将岩藻糖连接到Asn(297)-连接的糖上的能力,也导致在该宿主细胞中表达的抗体的低岩藻糖化(也参见Shields,R.L.等(2002)J.Biol.Chem.277:26733-26740)。Umana等的PCT公开WO 99/54342描述了工程化以表达糖蛋白-修饰的糖基转移酶(例如β(1,4)-N-乙酰葡糖氨基转移酶III(GnTIII))的细胞系,使得在该工程化细胞系中表达的抗体表现出对开GlcNac结构增加,这引起所述抗体的ADCC活性增加(也参见Umana等(1999)Nat.Biotech.17:176-180)。可选地,所述抗体的岩藻糖残基可以使用岩藻糖苷酶切割。例如,岩藻糖苷酶α-L-岩藻糖苷酶从抗体除去岩藻糖残基。(Tarentino,A.L.等(1975)Biochem.14:5516-23)。
另外地或可选地,可以制备具有改变类型的糖基化的抗体,其中该改变涉及所述抗体的唾液酸化水平。这样的改变被描述于Dickey等的PCT公开WO/2007/084926号和Ravetch等的PCT公开WO/2007/055916号,两者都通过引用以其全部内容并入本文。例如,可使用利用唾液酸酶(诸如产脲节杆菌(Arthrobacter ureafacens)唾液酸酶)的酶促反应。这种反应的条件被一般地描述于美国专利5,831,077中,其通过引用以其全部内容由此并入。适当的酶的其它非限制性实例是神经氨糖酸苷酶和N-糖苷酶F,如分别在Schloemer等J.Virology,15(4),882-893(1975)中和在Leibiger等,Biochem J.,338,529-538(1999)中描述的。去唾液酸化抗体可以通过使用亲和层析进一步纯化。可选地,可使用增加唾液酸化水平的方法,例如通过使用唾液酸转移酶进行。这种反应的条件被一般地描述于Basset等,Scandinavian Journal of Immunology,51(3),307-311(2000)中。
在本文中本发明考虑的另一种抗体修饰是聚乙二醇化。例如,抗体可以被聚乙二醇化,以例如增加所述抗体的生物(例如血清)半衰期。为使抗体聚乙二醇化,所述抗体或其片段通常在其中一个或多个PEG基团形成与所述抗体或抗体片段的连接的条件下与聚乙二醇(PEG)(诸如PEG的活性酯或醛衍生物)反应。优选地,聚乙二醇化经由与反应性PEG分子(或类似的反应性水溶性聚合物)的酰化反应或烷基化反应进行。如本文所使用,术语“聚乙二醇”意图包括已经用于衍生其它蛋白质的任何形式的PEG,诸如单(C1-C10)烷氧基-或芳氧基-聚乙二醇或聚乙二醇-马来酰亚胺。在某些实施方式,待乙二醇化的抗体是无糖基化抗体。用于蛋白质聚乙二醇化的方法是本领域已知的并且可以被应用于本发明的抗体。例如,参见Nishimura等的EP 0154 316和Ishikawa等的EP 0 401 384。同样,这里描述的聚乙二醇化方法也适用如下所述的本发明的肽类分子。
抗体片段和抗体模拟物
本发明不限于传统的抗体,而是可以通过利用抗体片段和抗体模拟物进行实施。如下文所详述的,各种抗体片段和抗体模拟物技术现在已经被开发出来并且在本领域中是广为人知的。尽管许多的这些技术,诸如结构域抗体、纳米体和UniBody,使用传统抗体结构的片段或对传统抗体结构的其它修饰,但仍存在采用结合结构的替代的技术,诸如Adnectin、亲和体、DARPin、抗运载蛋白、高亲合性多聚体和Versabody,尽管它们模拟传统的抗体结合,但是通过完全不同的机理产生并经由不同的机理起作用。这些替代结构的一些在Gill和Damle(2006)17:653-658中进行了综述。
结构域抗体(dAb)是抗体的最小功能结合单元,相应于人抗体的重(VH)链或轻(VL)链的可变区。结构域抗体具有大约13kDa的分子量。Domantis已经开发出完全人VH和VL dAb的一系列大的和高度功能性的文库(在各文库中超过一百亿不同的序列),并运用这些文库来选择特异于治疗靶标的dAb。与许多常规抗体相反,结构域抗体在细菌、酵母和哺乳动物细胞系统中良好地表达。通过参考美国专利6,291,158、6,582,915、6,593,081、6,172,197、6,696,245;美国专利申请No.2004/0110941;欧洲专利申请No.1433846和欧洲专利0368684和0616640;WO05/035572、WO04/101790、WO04/081026、WO04/058821、WO04/003019和WO03/002609可获得结构域抗体和其制备方法的更多细节,上述文献每一篇通过引用以其全部内容并入本文.
纳米体是包含天然存在的重链抗体的独特结构性和功能性特性的源自抗体的治疗性蛋白质。这些重链抗体包含单一可变结构域(VHH)和两个恒定结构域(CH2和CH3)。重要的是,克隆的和分离的VHH结构域是具有原始重链抗体的完全的抗原结合能力的完全稳定的多肽。纳米体与人抗体的VH结构域具有高同源性,并可以被进一步人源化而没有任何活性损失。重要的是,纳米体具有低的致免疫能力,这已经在采用纳米体前导化合物的灵长类动物研究中得到了证实。
纳米体将常规抗体的优点和小分子药物的重要特征相结合。像常规抗体一样,纳米体显示出高的靶特异性、对它们的靶的高亲合力和低的固有毒性。然而,如小分子药物一样,它们可以抑制酶并容易地到达受体裂缝(receptor cleft)。而且,纳米体极其稳定,可以通过除了注射之外的方式进行施用(例如,参见WO 04/041867,其通过引用以其全部内容并入本文)并易于制造。纳米体的其它优点包括由于它们的小尺寸而识别不常见的或隐藏的表位,从而以高亲合力、由于独特的三维结构而导致的选择性及药物形式灵活性结合到蛋白质靶的空腔或活性部位,因而调整半衰期和药物开发的容易度和速度。
纳米体通过单一基因进行编码并在几乎所有的原核和真核宿主中高效产生,例如大肠杆菌(例如见U.S.6,765,087,其通过引用以其全部并入本文)、霉菌(例如曲霉属(Aspergillus)或木霉属(Trichoderma))和酵母(例如酵母属(Saccharomyces)、克鲁维酵母属(Kluyveromyces)、汉逊酵母属(Hansenula)或毕赤氏酵母属(Pichia))(例如见美国6,838,254,其通过引用以其全部并入本文)。该产生方法可扩大规模并已经产生出数公斤量的纳米体。因为纳米体相比于常规抗体表现出优异的稳定性,所以它们可以被配制成长贮存期的即用溶液。
Nanoclone法(例如见WO 06/079372,其通过引用以其全部并入本文)是基于B细胞的自动化高通量选择的,用于产生针对期望靶的纳米体的专用方法,并且可被用于本发明。
UniBody是另一种抗体片段技术,然而这个技术是基于去除IgG4抗体的铰链区。该铰链区的缺失产生大小基本上为常规IgG4抗体的一半并具有单价结合区而不是IgG4抗体的二价结合区的分子。也众所周知的是,IgG4抗体是惰性的,因此不与免疫系统相互作用,这对于其中不期望免疫应答的疾病的治疗可能是有利的,并且该优点传递到UniBody。例如,UniBody可起到抑制或沉默而不是杀死它们所结合的细胞的作用。另外,结合癌细胞的UniBody不刺激它们增殖。而且,因为UniBody的大小为常规IgG4抗体大小的一半左右,所以它们可以潜在有利的效力在较大的实体瘤上显示出更好的分布。UniBody以与整个IgG4抗体相似的速率从机体清除,并且能够以与全抗体相似的对抗原的亲合性结合。通过参考专利申请WO2007/059782——其通过引用以其全部并入本文,可获得UniBody的更多细节。
Adnectin分子是来源于纤连蛋白的一个或多个结构域的工程化结合蛋白。纤连蛋白在人体内天然存在。它作为不溶性糖蛋白二聚物存在于细胞外基质中并还充当连接蛋白。它还以可溶的形式作为二硫连接的二聚体存在于血浆中。通过肝脏细胞(肝细胞)合成纤连蛋白的血浆形式,而通过软骨细胞、巨噬细胞、内皮细胞、成纤维细胞和上皮的一些细胞产生ECM形式(见Ward M.和Marcey,D.,callutheran.edu/Academic_Programs/Departments/BioDev/omm/fibro/fibro.htm)。如早先提及,纤连蛋白可天然地作为细胞粘附分子发挥其作用,或者它可通过在细胞外基质中进行接触而介导细胞的相互作用。通常,纤连蛋白由三个不同的蛋白质模块——I型、II型和III型模块——组成。对于纤连蛋白的结构或功能的综述,见Pankov和Yamada(2002)J Cell Sci.;115(Pt 20):3861-3,Hohenester和Engel(2002)21:115-128,和Lucena等(2007)Invest Clin.48:249-262。
在优选的实施方式中,adnectin分子通过改变由分布在两个β折叠之间的多个β链组成的天然蛋白质而衍生于纤连蛋白III型结构域。取决于起源的组织,纤连蛋白可包含多个III型结构域,其可以被表示为例如1Fn3、2Fn3、3Fn3等。10Fn3结构域包含整联蛋白结合基序,并进一步包含连接所述β链的三个环。这些环可以被认为相当于IgG重链的抗原结合环,并且它们可以通过下文讨论的方法加以改变以特异性结合目标靶,例如RTK的Ig样结构域,诸如人Kit RTK的D4或D5结构域或VEGF受体的D7结构域。优选地,可用于本发明的目的的纤连蛋白III型结构域是表现出与编码纤连蛋白III型分子的结构的序列(其可从Protein Data Bank(PDB,rcsb.org/pdb/home/home.do)以登录代码:1ttg获取)具有至少30%、至少40%、至少50%、至少60%、至少70%、至少80%、至少90%或至少95%的序列同一性的序列。Adnectin分子还可来源于10Fn3相关分子的聚合物,而不是简单的单体10Fn3结构。
尽管天然10Fn3结构域通常结合整联蛋白,但经改装变成adnectin分子的10Fn3蛋白被改变以结合目标抗原,例如RTK的Ig样结构域,诸如人Kit的D4或D5结构域。在一个实施方式中,对10Fn3分子的改变包括对β链的至少一个突变。在优选的实施方式中,连接10Fn3分子的β链的环区被改变以结合人受体酪氨酸激酶(例如VEGF受体或III型受体酪氨酸激酶,诸如人Kit)的Ig样结构域。
10Fn3中的改变可通过本领域已知的任何方法进行,包括但不限于易错PCR、定点诱变、DNA改组或在本文提及的其它类型的重组诱变。在一个实例中,编码10Fn3序列的DNA的变异体可以直接体外合成,并且之后在体外或体内转录和翻译。可选地,天然10Fn3序列可使用标准方法从基因组分离或克隆(如在例如美国专利申请No.20070082365中进行的),然后使用本领域已知的诱变方法进行突变。
在一个实施方式中,靶蛋白(例如,RTK的Ig样结构域如Kit RTK的D4或D5结构域或VEGF受体的D7结构域)可以固定在固相载体上,例如柱树脂或微孔滴定板中的孔。然后,将靶与潜在结合蛋白的文库接触。该文库可包含通过10Fn3序列的诱变/随机化或通过10Fn3环区(不是β链)的诱变/随机化而来源于野生型10Fn3的10Fn3克隆或adnectin分子。在优选的实施方式中,所述文库可以是通过Szostak等的美国专利申请No.09/007,005和09/247,190、Szostak等的WO989/31700以及Roberts & Szostak(1997)94:12297-12302描述的技术产生的RNA-蛋白质融合文库。所述文库也可以是DNA-蛋白质文库(例如,如描述于Lohse,美国专利申请No.60/110,549、美国专利申请No.09/459,190和WO 00/32823)。然后,所述融合文库与固定的靶(例如人Kit RTK的D4或D5结构域或人VEGF受体的D7结构域)一起温育,并洗涤固相载体以除去非特异性结合的成分。然后,在严格条件下洗脱紧密结合物,并且使用PCR来扩增遗传信息或产生结合分子的新的文库,以重复所述过程(有或者没有另外的诱变)。可以重复选择/诱变过程,直到获得对靶具有足够的亲合性的结合物。用于本发明的Adnectin分子可以使用Adnexus(Briston-Myers Squibb公司)采用的PROfusionTM技术进行工程化。PROfusion技术是基于上文所述的技术建立的(例如Roberts & Szostak(1997)94:12297-12302)。产生改变的10Fn3结构域的文库和选择可用于本发明的适当结合物的方法被充分描述于下列美国专利和专利申请文件中并通过引用并入本文:美国专利7,115,396、6,818,418、6,537,749、6,660,473、7,195,880、6,416,950、6,214,553、6623926、6,312,927、6,602,685、6,518,018、6,207,446、6,258,558、6,436,665、6,281,344、7,270,950、6,951,725、6,846,655、7,078,197、6,429,300、7,125,669、6,537,749、6,660,473号和美国专利申请20070082365、20050255548、20050038229、20030143616、20020182597、20020177158、20040086980、20040253612、20030022236、20030013160、20030027194、20030013110、20040259155、20020182687、20060270604、20060246059、20030100004、20030143616和20020182597号。纤连蛋白III型结构域(诸如10Fn3)的多样性的产生及随后的选择步骤可以使用本领域已知的其它方法进行,诸如噬菌体展示、核糖体展示或酵母表面展示,例如 等(2007)Journal of Molecular Biology368:1024-1041;Sergeeva等(2006)Adv Drug Deliv Rev.58:1622-1654;Petty等(2007)Trends Biotechnol.25:7-15;Rothe等(2006)ExpertOpin Biol Ther.6:177-187;和Hoogenboom(2005)Nat Biotechnol.23:1105-1116。
等(2007)Journal of Molecular Biology368:1024-1041;Sergeeva等(2006)Adv Drug Deliv Rev.58:1622-1654;Petty等(2007)Trends Biotechnol.25:7-15;Rothe等(2006)ExpertOpin Biol Ther.6:177-187;和Hoogenboom(2005)Nat Biotechnol.23:1105-1116。
本领域普通技术人员应该理解的是,上文引用的参考文献中的方法可用来从优选的10Fn3结构域之外的蛋白质衍生抗体模拟物。可用于经由上述引用的方法产生抗体模拟物的其他分子非限制性地包括人纤连蛋白模块1Fn3-9Fn3和11Fn3-17Fn3以及来自非人类动物和原核生物的相关Fn3模块。此外,也可使用来自与10Fn3具有序列同源性的其它蛋白质——诸如腱生蛋白(tenascin)和粗纤维调节素(undulin)——的Fn3模块。具有免疫球蛋白样折叠(但是具有与VH结构域无关的序列)的其它示例性蛋白质包括N-钙粘蛋白、ICAM-2、肌联蛋白(titin)、GCSF受体、细胞因子受体、糖苷酶抑制剂、E-钙粘蛋白和抗生素色蛋白。具有相关结构的其它结构域可以来源于髓磷脂膜粘附分子P0、CD8、CD4、CD2、I类MHC、T细胞抗原受体、CD1、VCAM-1的C2和I-组结构域、肌球蛋白-结合蛋白C的I-组免疫球蛋白折叠、肌球蛋白-结合蛋白H的I-组免疫球蛋白折叠、端蛋白(telokin)的I-组免疫球蛋白折叠、telikin、NCAM、twitchin、神经胶质蛋白、生长激素受体、红细胞生成素受体、催乳素受体、GC-SF受体、干扰素-γ受体、β-半乳糖苷酶/葡糖苷酸酶、β-葡糖苷酸酶和转谷氨酰胺酶。可选地,包括一个或多个免疫球蛋白样折叠的任何其他的蛋白质可以用于产生adnectin样结合成分。这样的蛋白质可以例如使用程序SCOP加以确定(Murzin等,J.Mol.Biol.247:536(1995);Lo Conte等,NucleicAcids Res.25:257(2000)。
适体是本发明包括的另一种类型的抗体模拟物。适体通常是小的核苷酸聚合物,其结合特定的分子靶。适体可以是单链或双链核酸分子(DNA或RNA),尽管DNA基适体最通常是双链的。对于适体核酸没有限定的长度;然而,适体分子最常见具有在15和40个核苷酸之间的长度。
适体经常形成复合三维结构,其决定它们对靶分子的亲合性。适体相比于简单的抗体可以提供许多优势,主要是因为它们可以几乎完全在体外被工程化和被扩增。而且,适体通常很少或不诱导免疫应答。
适体可以使用各种技术产生,但最初使用体外选择(Ellington和Szostak.(1990)Nature.346(6287):818-22)和SELEX法(通过指数富集导致系统性配体进化)(Schneider等1992.J Mol Biol.228(3):862-9)——其内容通过引用并入本文——进行开发。制备和使用适体的其它方法已经公开,包括Klussmann.The Aptamer Handbook:Functional Oligonucleotides and Their Applications.ISBN:978-3-527-31059-3;Ulrich等2006.Comb Chem High ThroughputScreen 9(8):619-32;Cerchia和de Franciscis.2007.Methods Mol Biol.361:187-200;Ireson和Kelland.2006.Mol Cancer Ther.20065(12):2957-62;美国专利5582981;5840867;5756291;6261783;6458559;5792613;6111095号;以及美国专利申请11/482,671;11/102,428;11/291,610;和10/627,543号,其都通过引用并入本文。
SELEX法明显是最流行的并且以三个基本步骤进行。首先,针对与特定分子靶的结合对候选核酸分子的文库进行挑选。其次,从非结合物分离出对于所述靶具有足够亲合性的核酸。第三,扩增结合的核酸,形成第二文库,并且重复该过程。在各次重复时,选择对于靶分子具有越来越高的亲合性的适体。SELEX法被更充分描述于通过引用并入本文的下列出版物中:Bugaut等2006.4(22):4082-8;Stoltenburg等2007 Biomol Eng.200724(4):381-403;和Gopinath.2007.Anal Bioanal Chem.2007.387(1):171-82。
本发明的“适体”也包括由肽而不是核苷酸制成的适体。肽适体与核苷酸适体具有一些共同的性质(例如,小的尺寸和以高亲合性结合靶分子的能力)并且它们可以通过与用于产生核苷酸适体的选择法具有相似原理的选择法产生,例如Baines和Colas.2006.Drug DiscovToday.11(7-8):334-41;和Bickle等2006.Nat Protoc.1(3):1066-91,其通过引用并入本文。
亲和体分子表示来源于葡萄球菌蛋白A的IgG结合结构域之一的基于58个氨基酸残基蛋白质结构域的一类新的亲合性蛋白质。这一种三螺旋束结构域已经用作构建组合噬菌粒文库的支架,可使用噬菌体展示技术从该文库选择靶向于期望分子的亲和体变体(Nord K,Gunneriusson E,Ringdahl J,Stahl S,Uhlen M,Nygren PA,Bindingproteins selected from combinatorial libraries of an α-helical bacterialreceptor domain,Nat Biotechnol 1997;15:772-7.Ronmark J,Gronlund H,Uhlen M,Nygren PA,Human immunoglobulin A(IgA)-specific ligandsfrom combinatorial engineering of protein A,Eur J Biochem2002;269:2647-55)。与它们的低分子量(6kDa)结合,亲和体分子的简单、稳定的结构使得它们适于各式各样的应用,例如作为检测试剂(Ronmark J,Hansson M,Nguyen T等,Construction and characterizationof affibody-Fc chimeras produced in Escherichia coli,J ImmunolMethods 2002;261:199-211),以及用于抑制受体相互作用(SandstormK,Xu Z,Forsberg G,Nygren PA,Inhibition of the CD28-CD80co-stimulation signal by a CD28-binding Affibody ligand developed bycombinatorial protein engineering,Protein Eng 2003;16:691-7)。通过参考美国专利No.5,831,012(其通过引用以其全部并入本文)可获得亲和体和其制备方法的更多细节。
DARPin(设计的锚蛋白重复序列蛋白质)是抗体模拟DRP(设计的重复序列蛋白质)技术的一个实例,该技术已经被开发为利用非抗体多肽的结合能力。重复序列蛋白质如锚蛋白或富亮氨酸重复序列蛋白质是遍在的结合分子,其在细胞内和细胞外存在,这与抗体不同。它们的独特的模块结构的特征在于重复结构单元(重复序列),其堆叠在一起以形成显示出可变的和模块的靶-结合表面的细长重复序列结构域。基于该模块性,具有高度多样化结合特异性的多肽的组合文库可以被产生。该策略包括展示可变表面残基的自相容性重复序列的共有设计以及它们随机装配成重复序列结构域。
DARPin可以在细菌表达系统中以很高的产量产生并且它们属于已知的最稳定的蛋白质。对宽范围的靶蛋白质——包括人受体、细胞因子、激酶、人蛋白酶、病毒和膜蛋白质——具有高度特异性、高亲合性的DARPin已经被选择。可以获得具有单数位纳摩尔至皮摩尔范围内的亲合性的DARPin。
DARPin已经用于各种各样的应用,包括ELISA、夹心ELISA、流式细胞术分析(FACS)、免疫组织化学(IHC)、芯片应用、亲和纯化或蛋白质印迹。DARPin也已证明在细胞内空间具有高度的活性,例如作为融合于绿色荧光蛋白(GFP)的胞内标记蛋白。DARPin被进一步用于以pM范围内IC50抑制病毒进入。DARPin不仅对于阻断蛋白质相互作用是理想的,而且对于抑制酶也是理想的。蛋白酶、激酶和转运蛋白已经被成功抑制,最经常变构性抑制模式。在肿瘤上的极快和特异性富集以及非常有利的肿瘤-血液比率使得DARPin很适于体内诊断或治疗方法。
关于DARPin及其他DRP技术的其它信息可发现于美国专利申请公开No.2004/0132028和国际专利申请公开No.WO 02/20565,两者都通过引用以其全部内容并入本文。
抗运载蛋白是另外的抗体模拟技术,然而,在这种情况下,结合特异性源自于脂钙蛋白(lipocalin)——在人组织和体液中天然存在且大量表达的低分子量蛋白质家族。脂钙蛋白已进化为执行与化学上敏感的或不溶性的化合物的生理运输和储存有关的许多体内功能。脂钙蛋白具有稳固的内在结构,其包含高度保存的β桶,该β桶在所述蛋白质一个末端支持四个环。这些环形成结合袋的入口,并且在所述分子的这个部分的构象差异造成在各个脂钙蛋白之间结合特异性的变化。
尽管由保守β折叠框架支持的超变环的整体结构类似于免疫球蛋白,但是脂钙蛋白与抗体在大小、由160-180个氨基酸(其比单一免疫球蛋白结构域稍大)的单一多肽链组成方面显著不同。
脂钙蛋白被克隆并且它们的环进行工程化以产生抗运载蛋白。结构上多样的抗运载蛋白的文库已经被产生,并且抗运载蛋白展示允许对结合功能进行选择和筛选,之后在原核或真核系统中表达和产生用于进一步分析的可溶性蛋白质。研究已经成功地证明,可以开发对于几乎任何可以被分离的人靶蛋白特异性的抗运载蛋白,并且可以获得在纳摩尔或以上的范围内的结合亲合性。
抗运载蛋白还可以被设计成双靶向蛋白质,所谓的Duocalin。Duocalin结合在使用标准制造方法容易产生的一个单体蛋白质中的两个独立的治疗靶标上,同时保持靶特异性和亲合性,而无论它的两个结合结构域的结构取向如何。
通过单一分子对多个靶的调节在已知涉及一种以上病因的疾病中是特别有利的。而且,二价或多价结合形式如Duocalin在靶向疾病中的细胞表面分子、介导对信号转导途径的激动效应或诱导经由细胞表面受体的结合和成簇而增强的内化效应方面具有显著的能力。而且,Duocalin的高内在稳定性与单体抗运载蛋白相当,从而为Duocalin提供灵活的制刘和输送可能。
关于抗运载蛋白的其它信息可发现于美国专利7,250,297和国际专利申请公布No.WO 99/16873中,两者都通过引用以其全部内容并入本文。
可用于本发明的另一个抗体模拟技术是高亲合性多聚体。高亲合性多聚体是通过体外外显子改组和噬菌体展示从人的细胞外受体结构域的大家族发展而来,从而产生具有结合和抑制性质的多结构域蛋白。连接多个独立的结合结构域已经表明产生亲合力(avidity),并且相比于常规的单一表位结合蛋白质,引起亲合性和特异性的改善。其它潜在优点包括在大肠杆菌中多靶特异性分子的简单和有效的产生、热稳定性改善和蛋白酶抗性。已经获得针对各种靶的、具有亚纳摩尔亲合性的高亲合性多聚体。
关于高亲合性多聚体的其它信息可发现于美国专利申请公开2006/0286603、2006/0234299、2006/0223114、2006/0177831、2006/0008844、2005/0221384、2005/0164301、2005/0089932、2005/0053973、2005/0048512、2004/0175756号,其全部都通过引用以其全部内容并入于此。
Versabody是可用于本发明的另一个抗体模拟技术。Versabody是具有>15%的半胱氨酸的3-5kDa的小蛋白质,其形成高二硫化密度的骨架,取代典型蛋白质具有的疏水核心。用少量二硫化物置换大量疏水性氨基酸(包括疏水核心)产生较小、更亲水(较少聚集和非特异性结合)、更耐蛋白酶和更耐热的蛋白质,并且所述蛋白质具有较低密度的T细胞表位,因为对MHC呈递贡献最大的残基是疏水性的。这四个性质已知全部影响免疫原性,并且预期它们一起引起大的免疫原性降低。
Versabody的灵感来自通过水蛭、蛇、蜘蛛、蝎子、蜗牛和海葵产生的天然可注射生物药物,其已知出乎意料地表现出低免疫原性。从选择的天然蛋白质家族开始,通过设计和通过筛选尺寸、疏水性、蛋白水解抗原加工和表位密度得以最小化至远低于天然可注射蛋白质的平均水平。
给定Versabody的结构,这些抗体模拟物提供多种多样的形式,包括多价、多特异性、半衰期机理的多样性、组织靶向模块和抗体Fc区的缺失。而且,Versabody在大肠杆菌中以高产率制造,并且因为它们的亲水性和小尺寸,Versabody高度可溶并可以配制成高浓度。Versabody是异常耐热的(它们可以被煮沸),因而提供延长的贮存期。
关于Versabody的其它信息可发现于美国专利申请公开No.2007/0191272,其通过引用以其全部内容由此并入。
SMIPTM(Small Modular ImmunoPharmaceuticals-TrubionPharmaceuticals)被工程化以保持并优化靶结合、效应子功能、体内半衰期和表达水平。SMIPS由三个不同的模块结构域组成。首先,它们包含可以由赋予特异性的任何蛋白质(例如细胞表面受体、单链抗体、可溶蛋白质等等)组成的结合结构域。其次,它们包含在结合结构域和效应子结构域之间充当柔性接头,以及帮助控制SMIP药物的多聚化的铰链结构域。最后,SMIP包含可以来源于各种分子的效应子结构域,包括Fc结构域或其它特别地设计的蛋白质。所述设计的模块化(其允许采用各种不同的结合、铰链和效应子结构域简单地构建SMIP)提供了快速和个性化的药物设计。
关于SMIP的更多信息,包括如何设计它们的实例,可以发现于Zhao等(2007)Blood 110:2569-77和下列美国专利申请20050238646、20050202534、20050202028、20050202023、20050202012、20050186216、20050180970和20050175614号。
上文提供的抗体片段和抗体模拟物技术的详细描述不意图是可被用于本说明书的全部技术的完全列表。例如,并且非限制性地,各种另外的技术,包括可选的基于多肽的技术,诸如在Qui等,NatureBiotechnology,25(8)921-929(2007)(通过引用以其全部内容由此并入)中概述的互补决定区的融合,以及基于核酸的技术,如描述于美国专利5,789,157、5,864,026、5,712,375、5,763,566、6,013,443、6,376,474、6,613,526、6,114,120、6,261,774和6,387,620(全部通过引用由此并入)中的RNA适体技术,可用于本发明中。
抗体的物理性质
本发明的抗体,其结合RTK的Ig样结构域,可以进一步通过各种物理特性加以表征。基于这些物理特性,各种测定方法可用来检测和/或区别不同类的抗体。
在一些实施方式中,本发明的抗体可以在轻链或重链可变区中包含一个或多个糖基化位点。一个或多个糖基化位点在可变区中的存在可以导致抗体的免疫原性增加或由于抗原结合改变而引起抗体的pK改变(Marshall等(1972)Annu Rev Biochem 41:673-702;Gala FA和Morrison SL(2004)J Immunol 172:5489-94;Wallick等(1988)J ExpMed168:1099-109;Spiro RG(2002)Glycobiology 12:43R-56R;Parekh等(1985)Nature 316:452-7;Mimura等(2000)Mol Immunol37:697-706)。已知糖基化在包含N-X-S/T序列的基序上发生。使用Glycoblot测定可检测可变区糖基化,该测定切割抗体以产生Fab,然后使用测量高碘酸盐氧化和希夫碱形成的测定方法检测糖基化。可选地,可以使用Dionex薄层色谱(Dionex-LC)检测可变区糖基化,其从Fab切割糖以成为单糖并分析各种糖含量。在一些情况下,可优选具有不包含可变区糖基化的抗体。这可以通过选择在可变区不包含糖基化基序的抗体或者通过使用本领域熟知的标准技术在糖基化基序内使残基突变加以实现。
各抗体具有独特的等电点(pI),但是通常抗体将落在6和9.5之间的pH范围内。IgG1抗体的pI通常落入7-9.5的pH范围内,而IgG4抗体的pI通常落入6-8的pH范围内。抗体可具有在该范围之外的pI。尽管效应通常是未知的,但推测具有在正常范围之外的pI的抗体可能在体内条件下产生一些展开和不稳定性。可以使用毛细管等电聚焦分析检测等电点,所述测定建立pH梯度并可利用激光聚焦以提高准确度(Janini等(2002)Electrophoresis 23:1605-11;Ma等(2001)Chromatographia 53:S75-89;Hunt等(1998)J Chromatogr A800:355-67)。在一些情况下,优选具有包含落于正常范围内的pI值的抗体。这可以通过选择具有在正常范围内的pI的抗体或者通过使用本领域熟知的标准技术突变带电表面残基加以实现。
各抗体具有作为热稳定性指标的解链温度(Krishnamurthy R和Manning MC(2002)Curr Pharm Biotechnol 3:361-71)。较高的热稳定性表示体内总体抗体稳定性更高。使用诸如差示扫描量热法的技术可以测量抗体的熔点(Chen等(2003)Pharm Res 20:1952-60;Ghirlando等(1999)Immunol Lett 68:47-52)。TM1表示抗体最初展开的温度。TM2表示抗体完全展开的温度。通常,优选的是,本发明的抗体的TM1大于60℃、优选大于65℃、甚至更优选大于70℃。可选地,可以使用圆二色性测量抗体的热稳定性(Murray等(2002)J.Chromatogr Sci40:343-9)。
在优选的实施方式,可以期望不迅速地降解的抗体。可以使用毛细管电泳(CE)和MALDI-MS测量抗体的断裂,如本领域充分了解的(Alexander AJ和Hughes DE(1995)Anal Chem 67:3626-32)。
在另一个优选的实施方式中,选择具有最小聚集效应的抗体。聚集可能导致引发不需要的免疫应答和/或导致改变的或不利的药物代谢动力学性质。通常,具有25%或以下的聚集的抗体是可接受的,优选20%或以下,甚至更优选15%或以下,甚至更优选10%或以下并且甚至更优选5%或以下。可以通过本领域熟知的几种技术测量聚集,包括尺寸排阻柱(SEC)高效液相色谱法(HPLC)和光散射以鉴定单体、二聚体、三聚物或多聚体。
本发明多克隆抗体的制备
本发明的多克隆抗体可以通过本领域熟知的多种技术产生。多克隆抗体源自于不同的B细胞系,因此可识别同一抗原上的多个表位。通常通过用目标抗原(例如RTK的Ig样结构域,诸如人Kit的D4或D5结构域或人VEGF的D7结构域)免疫适当的哺乳动物产生多克隆抗体。经常被用来产生多克隆抗体的动物是鸡、山羊、豚鼠、仓鼠、马、小鼠、大鼠、绵羊和最常用的兔子。在下文的实施例14中,通过用Kit的第四(D4)或第五(D5)Ig样结构域或Kit的完整胞外域免疫兔子产生多克隆抗Kit抗体。产生多克隆抗体的标准方法是本领域广泛熟知的,并可以与本发明的方法结合(例如research.cm.utexas.edu/bkitto/Kittolabpage/Protocols/Immunology/PAb.html;美国专利4,719,290、6,335,163、5,789,208、2,520,076、2,543,215和3,597,409号,其完整内容通过引用并入本文)。
本发明的单克隆抗体的制备
本发明的单克隆抗体(mAb)可以通过多种技术产生,包括常规单克隆抗体方法,例如Kohler和Milstein(1975)Nature 256:495的标准体细胞杂交技术。尽管体细胞杂交法是优选的,但原则上,其它产生单克隆抗体的技术可以被使用,例如B淋巴细胞的病毒性或致癌性转化。应该注意到,抗体(单克隆或多克隆)或其抗原结合部分可以针对RTK的Ig样结构域、更优选人Kit RTK的D4或D5结构域或VEGF受体的D7结构域上的任何表位而产生,或者针对在本文论述的共有序列而产生,或者针对在本文描述的任何构象、非连续或线性表位而产生。
本领域已知的几种方法可用于特异性选择特异性结合目标非连续表位的抗体或其抗原结合片段。例如,在通过引用并入本文的美国公开No.2005/0169925中公开的技术允许选择结合蛋白质序列内两个不同的肽的抗体。这样的方法可根据本发明用于特异性靶向于在本文公开的构象和非连续表位。如果构象表位是蛋白质二级结构,这样的结构通常很容易在较小的肽(例如<50个氨基酸)中形成。因此,用较小的肽免疫动物可以捕获一些构象表位。可选地,包含构象表位的两个小肽(例如表5中确定的肽)可以经由柔性接头(例如,聚乙二醇或一段极性的、不带电的氨基酸)连接。所述接头允许肽试探各种相互作用取向。采用这种构建体进行免疫,继之以适当的筛选,可允许鉴定针对构象表位的抗体。在优选的实施方式中,通过用RTK的特定结构域(例如,Kit胞外域的结构域4或结构域5或VEGF受体的D7结构域)免疫动物,接着筛选结合目标表位的抗体,可以产生针对特定构象或线性表位的肽。在一个实施方式中,冷冻电镜术(Jiang等(2008)Nature 451,1130-1134;Joachim(2006)Oxford University PressISBN:0195182189)可用来鉴定由本发明的抗体或抗原结合片段结合的表位。在另一个实施方案中,RTK或其结构域可以与结合的抗体或其抗原结合片段一起结晶并通过X-射线晶体衍射法分析,以确定被结合的精确表位。此外,可以通过用来自小鼠或另一个物种的相应序列替换RTK序列的部分对表位进行作图。针对目标表位的抗体选择性地结合人序列区,因此,有可能对靶表位进行顺序作图。该基于嵌合体的表位作图技术已经成功地用于鉴定各种情况下的表位(参见Henriksson和Pettersson(1997)Journal of Autoimmunity.10(6):559-568;Netzer等(1999)J Biol Chem.1999 Apr 16;274(16):11267-74;Hsia等(1996)Mol.Microbiol.19,53-63,其完整内容通过引用并入本文)。
据认为,靶RTK(例如Kit RTK或VEGF受体)中的目标表位未被糖基化。然而,如果目标RTK被糖基化,抗体或其抗原结合部分(及本发明的其他成分)可被产生以使得它们结合有关的氨基酸和/或糖残基。例如,本领域已知Kit蛋白质具有最少10个潜在的N-连接糖基化位点。(Morstyn,Foote,Lieschke(2004)Hematopoietic Growth Factorsin Oncology:Basic Science and Clinical Therapeutics.Humana Press.ISBN:1588293025)。进一步认为,Kit可表现出O-连接糖基化以及与唾液酸残基的连接(Wypych J等(1995)Blood.85(1):66-73)。因此,本发明考虑,抗体或其抗原结合部分(及本发明的其他成分)可被产生以使得它们也结合可以连接于在本文鉴定的任何表位的糖残基。出于该目的,可以在动物细胞中产生抗原性目标肽以使得它被适当糖基化,并且然后糖基化的抗原肽可用于免疫动物。产生糖基化肽的适当的细胞和技术是本领域已知的并在下文进一步描述(参见例如,可从GlycoFi,Inc.,Lebanon,NH and BioWa;Princeton,NJ获得的技术)。可使用任何标准方法诸如等电点聚焦(IEF)、酸水解(以确定单糖组成)、化学或酶促裂解和质谱法(MS)鉴定聚糖而检测肽的适当糖基化。也可使用由Procognia(procognia.com)提供的技术——其使用基于植物凝血素的阵列来加速聚糖分析。特别地,可以使用如还原性碱性裂解或“β-消除”、肽作图、液相色谱和质谱或这些技术的任何组合的技术检测O-糖基化。
用于制备杂交瘤的优选的动物系统是鼠系统。在小鼠中产生杂交瘤是非常完善的方法。免疫方案和分离用于融合的免疫脾细胞的技术是本领域已知的。融合伴体(例如鼠骨髓瘤细胞)和融合方法也是已知的。
本发明的嵌合或人源化抗体可以基于如上所述制备的鼠单克隆抗体的序列进行制备。编码重链和轻链免疫球蛋白的DNA可以使用标准分子生物学技术从感兴趣的的鼠杂交瘤获得并工程化以包含非鼠(例如人)的免疫球蛋白序列。例如,为产生嵌合抗体,可以使用本领域已知的方法(例如见Cabilly等的美国专利No.4,816,567)将鼠可变区连接到人恒定区。为产生人源化抗体,可以使用本领域已知的方法将鼠CDR区插入人框架中(例如,见Winter的美国专利5,225,539和Queen等的美国专利5,530,101、5,585,089、5,693,762和6,180,370)。可选地,在已知人和非人序列的情况下,可以在DNA或蛋白水平设计人源化抗体。这样的抗体可以直接进行化学合成,或者DNA可以被合成并进行体外或体内表达以产生人源化抗体。
在优选的实施方式中,本发明的抗体是人单克隆抗体。可以使用携带部分人免疫系统而不是小鼠系统的转基因或转染色体小鼠产生针对RTK的Ig样结构域(例如Kit的D4或D5结构域或VEGF受体的D7结构域)的这类人单克隆抗体。这些转基因和转染色体小鼠包括在本文被分别称为HuMAb小鼠和KM miceTM的小鼠,并且在本文被统称为“人Ig小鼠”。
HuMAb mouse(Medarex,Inc.)包含编码未重排的人重链(μ和γ)和κ轻链免疫球蛋白序列的人免疫球蛋白基因小基因座(miniloci),连同灭活内源性μ和κ链基因座的靶向突变(见例如Lonberg等(1994)Nature 368(6474):856-859)。因此,所述小鼠表现出小鼠IgM或κ的表达减少,并且响应于免疫时引入的人重链和轻链转基因经历类别转换和体细胞突变以产生高亲合性人IgGκ单克隆(Lonberg,N.等(1994),同上;在Lonberg,N.(1994)Handbook of Experimental Pharmacology113:49-101中综述;Lonberg,N.和Huszar,D.(1995)Intern.Rev.Immunol.13:65-93,以及Harding,F.和Lonberg,N.(1995)Ann.N.Y.Acad.Sci.764:536-546)。HuMab小鼠的制备和使用以及这样的小鼠携带的基因组修饰被进一步描述在Taylor,L.等(1992)Nucleic AcidsResearch 20:6287-6295;Chen,J.等(1993)International Immunology 5:647-656;Tuaillon等(1993)Proc.Natl.Acad.Sci.USA 90:3720-3724;Choi等(1993)Nature Genetics 4:117-123;Chen,J.等(1993)EMBO J.12:821-830;Tuaillon等(1994)J.Immunol.152:2912-2920;Taylor,L.等(1994)International Immunology 6:579-591;和Fishwild,D.等(1996)Nature Biotechnology 14:845-851中,其全部内容都通过引用明确地由此并入。进一步参见美国专利5,545,806;5,569,825;5,625,126;5,633,425;5,789,650;5,877,397;5,661,016;5,814,318;5,874,299;和5,770,429,其全部授予Lonberg和Kay;授予Surani等的美国专利5,545,807;PCT公开WO 92/03918、WO 93/12227、WO 94/25585、WO 97/13852、WO 98/24884和WO 99/45962,其全部授予Lonberg和Kay;和授予Korman等的PCT公开WO 01/14424。
在另一个实施方式中,可以使用在转基因和转染色体上携带人免疫球蛋白序列的小鼠(如携带人重链转基因和人轻链转染色体的小鼠)产生本发明的人抗体。这类小鼠,在本文称为“KM miceTM”,被详细描述于授予Ishida等的PCT公开WO 02/43478中。
更进一步,可选择的表达人免疫球蛋白基因的转基因动物系统在本领域中是可获得的,并可用于产生本发明的抗体。例如,称为Xenomouse(Abgenix,Inc.)的可选转基因系统可被使用;这样的小鼠被描述于例如授予Kucherlapati的美国专利5,939,598、6,075,181、6,114,598、6,150,584和6,162,963中。
而且,可选择的表达人免疫球蛋白基因的转染色体动物系统在本领域中是可获得的,并可用于产生本发明的抗体。例如,携带人重链转染色体和人轻链转染色体的小鼠,称为“TC小鼠”,可以被使用;这类小鼠被描述于Tomizuka等(2000)Proc.Natl.Acad.Sci.USA97:722-727中。而且,携带人重链和轻链转染色体的母牛已经在本领域中进行了描述(Kuroiwa等(2002)Nature Biotechnology 20:889-894)并且可用于产生本发明的抗体。
本发明的人单克隆抗体还可以使用用于筛选人免疫球蛋白基因文库的噬菌体展示方法加以制备。用于分离人抗体的这类噬菌体展示方法在本领域中被建立。例如,参见:授予Ladner等的美国专利5,223,409、5,403,484和5,571,698号;授予Dower等的美国专利5,427,908和5,580,717号;授予McCafferty等的美国专利5,969,108和6,172,197号;以及授予Griffiths等的美国专利5,885,793、6,521,404、6,544,731、6,555,313、6,582,915和6,593,081号。
本发明的人单克隆抗体还可以使用SCID小鼠进行制备,其中人免疫细胞已经被重构以使得在免疫时可以产生人抗体应答。这样的小鼠被描述在例如授予Wilson等的美国专利5,476,996和5,698,767中。
在另一个实施方式中,本发明的抗体可以使用熟知的噬菌体展示技术产生,如描述于Marks,J.D.等((1991).J.Mol.Biol.222,581),Nissim,A.等((1994).EMBO J.13,692)以及美国专利6,794,132、6562341、6057098、5821047和6512097中。
在进一步的实施方式中,本发明的抗体可使用酵母细胞表面展示技术进行寻找,如在例如美国专利6,423,538、6,300,065、6,696,251、6,699,658号中所描述的。
制备本发明人类单克隆抗体的杂交瘤的产生
为产生制备本发明的人单克隆抗体的杂交瘤,来自免疫小鼠的脾细胞和/或淋巴结细胞可以被分离并且融合到适当的无限增殖化细胞系,如小鼠骨髓瘤细胞系。可以对得到的杂交瘤进行抗原特异性抗体产生的筛选。例如,可以采用50%PEG将来自免疫小鼠的脾淋巴细胞的单细胞悬液融合于六分之一数目的P3X63-Ag8.653非分泌性小鼠骨髓瘤细胞(ATCC,CRL 1580)。可选地,可以使用基于电场的电融合法,使用CytoPulse大室细胞融合电感受器(CytoPulse Sciences,Inc.,Glen Burnie Maryland)对来自免疫小鼠的脾淋巴细胞的单细胞悬液进行融合。在平底微孔滴定板上以大约2x 105接种细胞,接下来在含有20%胎儿克隆血清、18%“653”条件培养基、5%origen(IGEN)、4mML-谷氨酰胺、1mM丙酮酸钠、5mM HEPES、0.055mM 2-巯基乙醇、50单位/ml青霉素、50mg/ml链霉素、50mg/ml庆大霉素和1X HAT(Sigma;HAT在融合之后24小时添加)的选择培养基中培养两周。在大约两周之后,可以在HAT被替换为HT的培养基中培养细胞。然后,可以通过ELISA对各细胞进行人单克隆IgM和IgG抗体筛选。一旦大规模的杂交瘤生长发生,则通常可以在10-14天之后观察培养基。分泌抗体的杂交瘤可以被重新接种,再次筛选,并且如果仍然是人IgG阳性的,则单克隆抗体可以通过有限稀释进行至少两次分种。然后,体外培养稳定的亚克隆,以在组织培养基中产生少量的用于鉴定的抗体。
为纯化人单克隆抗体,可以将选择的杂交瘤在两升旋转烧瓶中培养以用于单克隆抗体纯化。在用蛋白A-琼脂糖(Pharmacia,Piscataway,N.J.)进行亲和层析之前,可以过滤并浓缩上清液。洗脱的IgG可以通过凝胶电泳和高效液相色谱进行检验以确保纯度。缓冲溶液可以被替换成PBS,并且可以使用1.43的消光系数通过OD280确定浓度。单克隆抗体可以被等分并且储存在-80℃。
制备本发明单克隆抗体的转染瘤的产生
也可以使用例如本领域所熟知的重组DNA技术和基因转染方法的组合(例如Morrison,S.(1985)Science 229:1202)在宿主细胞转染瘤(杂交瘤的一种类型)中产生本发明的抗体。
例如,为表达所述抗体或其抗体片段,编码部分或全长轻链和重链的DNA可以通过标准分子生物学技术(例如,PCR扩增或使用表达目标抗体的杂交瘤进行cDNA克隆)获得并且所述DNA可以被插入表达载体中以使得基因被可操作地连接于转录和翻译调控序列。在本文中,术语“可操作地连接”意图指抗体基因被接合入载体以使得在所述载体内的转录和翻译调控序列发挥它们调节所述抗体基因的转录和翻译的预期功能。选择表达载体和表达调控序列以与所使用的表达宿主细胞相容。抗体轻链基因和抗体重链基因可以被插入独立的载体中,或者更通常地,两种基因被插入同一表达载体中。所述抗体基因通过标准方法(例如在抗体基因片段和载体上的互补限制性位点的接合,或者如果不存在限制性位点的话通过平端连接)被插入所述表达载体中。所描述的抗体的轻链和重链可变区可用于通过将它们插入已经编码需要的同种型的重链恒定区和轻链恒定区的表达载体中以使得VH片段被可操作地连接于所述载体内的CH片段,而VK片段被可操作地连接于所述载体内的CL片段而产生任何抗体同种型的全长抗体基因。另外地或替代地,重组表达载体可以编码促进抗体链从宿主细胞分泌的信号肽。抗体链基因可以被克隆入所述载体以使得信号肽在框架内(in-frame)被连接到抗体链基因的氨基端。信号肽可以是免疫球蛋白信号肽或异源信号肽(即,来自非免疫球蛋白的信号肽)。
除了所述抗体链基因之外,本发明的重组表达载体携带调控序列,其调控所述抗体链基因在宿主细胞中的表达。术语“调控序列”意图包括启动子、增强子及其他控制抗体链基因的转录或翻译的表达调控元件(例如多腺苷酸化信号)。这样的调控序列被描述在例如Goeddel(Gene Expression Technology.Methods in Enzymology 185,AcademicPress,San Diego,CA(1990))中。本领域普通技术人员将理解,表达载体的设计,包括调控序列的选择,可取决于如待转化宿主细胞的选择、期望的蛋白质表达水平等因素。用于哺乳动物宿主细胞表达的优选调控序列包括指导哺乳动物细胞中高水平的蛋白质表达的病毒元件,如来源于巨细胞病毒(CMV)、猴病毒40(SV40)、腺病毒(例如腺病毒主要晚期启动子(AdMLP))和多瘤病毒的启动子和/或增强子。可选地,可以使用非病毒调控序列,如遍在启动子或β-球蛋白启动子。更进一步,由来自不同来源的序列组成的调控元件,如SRa启动子系统,其包含来自SV40早期启动子和人T细胞白血病毒1型的长末端重复序列的序列(Takebe,Y.等(1988)Mol.Cell.Biol.8:466-472)。
除了抗体链基因和调控序列之外,本发明的重组表达载体可携带另外的序列,诸如调节载体在宿主细胞中的复制的序列(例如复制起点)和选择性标记基因。选择性标记基因帮助选择所述载体已经被引入的宿主细胞(参见例如Axel等的美国专利4,399,216、4,634,665和5,179,017)。例如,通常,选择性标记基因赋予所述载体已经被引入的宿主细胞对药物诸如G418、潮霉素或氨甲喋呤的抗性。优选的选择性标记基因包括二氢叶酸还原酶(DHFR)基因(用于以氨甲喋呤进行选择/扩增的dhfr-宿主细胞)和neo基因(用于G418选择)。
为了表达轻链和重链,编码重链和轻链的表达载体通过标准技术转染到宿主细胞中。术语“转染”的各种形式意图包括常用来将外源性DNA引入真核或原核宿主细胞的各种技术,例如电穿孔、磷酸钙沉淀、DEAE-葡聚糖转染等等。尽管理论上在原核或真核宿主细胞中表达本发明的抗体是可能的,但是在真核细胞中表达抗体是最优选的,并且最优选哺乳动物宿主细胞,因为这类真核细胞、特别是哺乳动物细胞比原核细胞更可能装配和分泌适当折叠的免疫活性抗体。已经报告,抗体基因的原核生物表达对于以高产量产生活性抗体是无效的。(Boss,M.A.和Wood,C.R.(1985)Immunology Today 6:12-13)。
表达本发明的重组抗体的优选哺乳动物宿主细胞包括中国仓鼠卵巢细胞(CHO细胞)(包括dhfr-CHO细胞,其描述于Urlaub和Chasin,(1980)Proc.Natl.Acad.Sci.USA 77:4216-4220中,与DHFR选择性标记一起使用,如描述于R.J.Kaufman和P.A.Sharp(1982)Mol.Biol.159:601-621)中)、NSO骨髓瘤细胞、COS细胞和SP2细胞。特别地,对于采用NSO骨髓瘤细胞,另一个优选的表达系统是GS基因表达系统,其公开于WO 87/04462、WO 89/01036和EP 338,841中。当编码抗体基因的重组表达载体被引入哺乳动物宿主细胞时,通过培养宿主细胞一段足以允许所述抗体在宿主细胞中表达或更优选所述抗体分泌入宿主细胞在其中生长的培养基中的一段时间产生所述抗体。可以使用标准蛋白质纯化方法从培养基回收抗体。
结合RTK的Ig-样结构域的抗体的表征
通过例如标准ELISA,可以检测本发明的抗体与胞外域例如RTK的Ig样结构域(或任何选择的区域,如在本文论述的共有序列)的结合。简言之,用在PBS中的0.25μg/ml纯化的Ig样结构域(或优选的受体结构域)包被微孔滴定板,然后用在PBS中的5%牛血清白蛋白封闭。抗体的稀释物(例如,来自免疫小鼠——例如用人Kit的D4或D5结构域免疫的小鼠——的血浆的稀释物)被添加至每一孔并在37℃温育1-2小时。用PBS/Tween洗涤所述板,然后与偶联于碱性磷酸酶的第二试剂(例如对于人抗体,为山羊抗人IgG Fc特异性多克隆试剂)在37℃温育1小时。在洗涤之后,用pNPP底物(1mg/ml)对所述板进行显色,并在405-650的OD下分析。优选地,显示出最高滴度的小鼠将用于融合。
如上所述的ELISA测定还可以用来筛选显示出与免疫原的阳性反应性的杂交瘤。以高亲合力结合例如RTK的Ig样结构域的杂交瘤被分种并进一步进行鉴定。来自各杂交瘤的一个克隆(其保持亲本细胞的反应性(通过ELISA))可以被选择来用于制备储存于-140℃的5-10小瓶细胞库和用于抗体纯化。
为纯化抗RTK抗体,可以使选择的杂交瘤在两升旋转烧瓶中培养用于单克隆抗体纯化。在用蛋白A-琼脂糖(Pharmacia,Piscataway,NJ)进行亲和层析之前,可以过滤并浓缩上清液。洗脱的IgG可以通过凝胶电泳和高效液相色谱进行检验以确保纯度。缓冲溶液可以被替换成PBS,并且可以使用1.43消光系数通过OD280测定浓度。单克隆抗体可以被等分并且储存在-80℃。
为确定所选择的单克隆抗体是否结合独特表位,可以使用市售试剂(Pierce,Rockford,IL)生物素化各抗体。可以使用如上所述以RTK的Ig样结构域(例如Kit-D4结构域、Kit-D5结构域或VEGF受体D7结构域)包被的RTK包被ELISA板进行使用未标记的单克隆抗体和生物素化单克隆抗体的竞争研究。生物素化mAb结合可以采用链霉-抗生物素蛋白-碱性磷酸酶探针进行检测。
为确定纯化抗体的同种型,可以使用对于具有特定同种型的抗体具有特异性的试剂进行同种型ELISA。例如,为确定人单克隆抗体的同种型,可以用1μg/ml的抗人免疫球蛋白在4℃包被微孔滴定板的孔过夜。在用1%BSA封闭之后,所述板与1μg/ml或更低的测试单克隆抗体或纯化的同种型对照在环境温度反应一至两个小时。然后,所述孔与人IgG1或人IgM-特异性碱性磷酸酶偶联的探针反应。如上所述对板进行显色和分析。
可以通过蛋白质印迹法进一步测试抗RTK人IgG与RTK的Ig样结构域或在本文给出的共有序列的反应性。简言之,RTK的Ig样结构域,如Kit RTK的D4或D5结构域或者VEGF受体的D7结构域,可以被制备并进行十二烷基硫酸钠聚丙烯酰胺凝胶电泳。在电泳之后,将分离的抗原转移到硝酸纤维素膜、用10%胎牛血清封闭、并用待测试的单克隆抗体探测。可以使用抗人IgG碱性磷酸酶检测人IgG结合,并用BCIP/NBT底物片(Sigma Chem.Co.,St.Louis,Mo.)进行显色。
表位作图可用来确定本发明的抗体或其抗原结合片段的结合位点。可获得进一步允许构象表位作图的几种方法。例如,可以使用公开于Timmerman等(Mol Divers.2004;8(2):61-77)的方法。Timmerman等能够使用两种新的技术Domain Scan和Matrix Scan成功地对非连续/构象表位作图。也可以使用公开于Ansong等(J Thromb Haemost.2006.4(4):842-7)中的技术。Ansong等使用亲合性定向质谱(affinitydirected mass spectrometry)来对抗体R8B12识别的非连续表位进行作图。此外,成像技术如Protein Tomography可用于使靶RTK的抗体或肽结合成像。Protein Tomography先前已经用于观察分子相互作用,并用来显示抑制性抗体通过结合EGFR的结构域III起作用,从而将EGFR锁定为非柔性和非活性的构象(Lammerts等Proc Natl Acad SciU S A.2008.105(16):6109-14)。更多的传统方法诸如定点诱变也可被用于对非连续表位作图。据认为参与非连续表位的氨基酸区可以被选择性突变并分析其与本发明的抗体或其抗原结合片段的结合。当任一个区被突变时,所述抗体丧失结合能力可表明所述结合取决于两个氨基酸片段。如上所述,一些线性表位的特征在于为了结合本发明的成分必须存在的特定三维结构。当RTK处于其天然或折叠状态时以及再次当RTK变性时,这样的表位可以通过分析所述抗体(或另一成分)的结合发现。结合仅仅在折叠状态发生这一观察结果表明所述表位或者是特征在于特定折叠结构的线性表位或者是仅仅存在于折叠蛋白质中的非连续表位。
除了在本文描述的活性分析之外,Protein Tomography还可用来确定本发明的抗体或其抗原结合片段是否能结合和灭活受体酪氨酸激酶。结合相互作用的可视化可表明,所述抗体的结合可影响两个胞外域在细胞表面界面的定位或者改变或阻止在所述受体酪氨酸激酶中的构象变化。
II.结合人受体酪氨酸激酶的Ig样结构域或铰链区的小分子
在本发明的另一个方面,结合人受体酪氨酸激酶的胞外域例如Ig样结构域或铰链区的成分是小分子。
本发明的小分子的特征在于特定功能特征或性质。例如,所述小分子结合RTK的Ig样结构域,例如Kit RTK的D4或D5结构域或者VEGF受体的D7结构域,或者RTK的铰链区,例如Kit RTK的D3-D4或D4-D5铰链区。在优选的实施方式,小分子抑制剂与D3-D4或D4-D5铰链区的结合将阻止使得近膜D4和D5结构域能够处于允许酪氨酸激酶结构域的反式自磷酸化和活化,随后是下游信号传导通路的募集和活化的距离和取向(位置)的运动。在一些实施方式中,小分子结合可允许受体酪氨酸激酶的胞外域二聚化,但影响在两个单体的Ig样结构域(例如,III型受体酪氨酸激酶的D4-D4或D5-D5结构域或者V型受体酪氨酸激酶的D7-D7结构域)之间的定位、取向和/或距离,从而抑制受体酪氨酸激酶的活性。换句话说,所述成分或小分子可允许配体诱导的受体酪氨酸激酶胞外域的二聚化,但影响两个胞外域在细胞表面界面的定位,从而抑制受体酪氨酸激酶的活性(例如,抑制受体内化作用和/或抑制所述受体的酪氨酸自磷酸化和/或抑制所述受体激活下游信号传导通路的能力)。
术语“小分子化合物”、“小分子药物”、“小分子”或“小分子抑制剂”在本文可互换使用以用来指对其对RTK的影响和它们抑制RTK(例如Kit RTK或VEGF受体)的二聚化或活性的能力进行筛选的本发明的化合物。这些化合物可包含在PubChem数据库(pubchem.ncbi.nlm.nih.gov/)、Molecular Libraries Screening CenterNetwork(MLSCN)数据库中的化合物、相关数据库中的化合物、或者它们的衍生物和/或功能类似物。
如本文所使用,“类似物”或“功能类似物”是指化合物或小分子抑制剂,其在结构上类似于母体化合物,但在组成上稍微不同(例如,一个或多个原子或功能团被添加、除去或改变)。相比于初始化合物,类似物可具有或不具有不同的化学或物理特性,并可具有或不具有改善的生物学和/或化学活性。例如,与母体化合物相比,类似物可以是更疏水性的或者它可具有改变的活性(增加、降低或与母体化合物相同)。所述类似物可以是初始化合物的天然或非天然发生(例如重组)的变体。其它类型的类似物包括化合物的异构体(对映异构体、非对映异构体等等)和其它类型的化合物手性变体,以及结构异构体。所述类似物可以是线性化合物的分支或环状变体。例如,线性化合物可具有支化的或另外取代以赋予某些期望的性质(例如改善的亲水性或生物利用度)的类似物。
如本文所使用,“衍生物”是指化合物或小分子抑制剂的化学上或生物学上的改良形式,其结构上类似于母体化合物并且(实际上或理论上)可衍生于该母体化合物。“衍生物”与“类似物”或“功能类似物”的区别在于母体化合物可以是产生“衍生物”的起始物质,而母体化合物不是必须被用作产生“类似物”或“功能类似物”的起始物质。衍生物可以具有或不具有与所述母体化合物不同的化学或物理特性。例如,与母体化合物相比,衍生物可以是更亲水性的或者它可具有改变的反应性。衍生化(即,通过化学或其它手段修饰)可包括在分子内一个或多个部分的取代(例如,官能团的改变)。例如,氢可以用卤素如氟或氯取代,或者羟基(--OH)可以用羧酸部分(--COOH)取代。术语“衍生物”也包括偶联物和母体化合物的前药(即化学修饰衍生物,其在生理条件下可转化为原始化合物)。例如,前药可以是活性剂的无活性形式。在生理条件下,前药可以转化为所述化合物的活性形式。例如,可以通过用酰基基团(酰基前药)或氨基甲酸酯基团(氨基甲酸酯前药)置换氮原子上的一个或两个氢原子形成前药。关于前药的更多详细信息可例如发现于Fleisher等,Advanced Drug Delivery Reviews 19(1996)115;Design of Prodrugs,H.Bundgaard(编辑),Elsevier,1985;和H.Bundgaard,Drugs of the Future 16(1991)443。术语“衍生物”也用于描述全部溶剂合物,例如水合物或加合物(例如与醇的加合物)、活性代谢物和母体化合物的盐。可以制备的盐的类型取决于在化合物内的部分的性质。例如,酸性基团如羧酸基团可以形成碱金属盐或碱土金属盐(例如钠盐、钾盐、镁盐、钙盐)和与生理上可容许的季铵离子的盐以及与氨水和生理上可容许的有机胺如三乙胺、乙醇胺或三(2-羟乙基)胺)的酸加成盐。碱性基团可以例如与无机酸如盐酸(“HCl”)、硫酸或磷酸形成酸加成盐,或者与有机羧酸和磺酸如乙酸、柠檬酸、苯甲酸、马来酸、富马酸、酒石酸、甲磺酸或对甲苯磺酸形成酸加成盐。除了碱性氮原子之外同时包含碱性基团和酸性基团(如羧基)的化合物可以作为两性离子存在。可以通过本领域普通技术人员已知的惯用方法获得盐,例如通过在溶剂或稀释剂中结合化合物和无机或有机酸或碱,或者通过阳离子交换或阴离子交换从其它盐获得所述盐。
小分子已知具有1200或以下、1000或以下、900或以下、800或以下、700或以下、600或以下、500或以下、400或以下、300或以下、200或以下、100或以下、50或以下、25或以下或者10或以下的分子量。
本发明的小分子抑制剂被选择或设计来结合RTK的胞外域,例如Ig样结构域或铰链区。在一些实施方式中,小分子抑制剂被选择或设计来结合人Kit、人VEGF受体或PDGFR的Ig样结构域或铰链区,例如人Kit受体的D4或D5结构域、或D3-D4和/或D4-D5铰链区,从而拮抗受体二聚化和转变成活性形式的能力,例如自磷酸化和活化细胞内信号传导途径。在其它实施方式中,小分子抑制剂被选择来结合与Kit受体或VEGF受体的结构域具有同源性的结构域。例如,本发明的小分子可以针对与RTK的Ig样结构域(例如Kit的D4或D5结构域或VEGF受体的D7结构域)、RTK的铰链区(例如Kit或PDGFR受体的D3-D4或D4-D5铰链区)具有至少50%同一性、至少60%同一性、至少70%同一性、至少80%同一性、至少90%同一性、或至少95%或99%同一性的结构域。这样的小分子将能够结合在功能上类似于例如Kit或PDGF受体的D4、D5或D7结构域或D3-D4和/或D4-D5铰链区的蛋白结构域——可能在Kit、VEGF受体和其它RTK中
本发明的小分子抑制剂也可结合来源于RTK(例如人VEGF受体或人III型RTK)的Ig样结构域或铰链区的特定基序或共有序列,这允许所述小分子抑制剂特异性结合RTK家族的成员(例如人RTK的III型家族的成员)中共有的结构域。
在一种具体实施方式中,本发明的小分子结合D4相互作用位点的以下共有序列:LX1RX2X3X4X5X6X7G,其中L是亮氨酸,R是精氨酸,G是甘氨酸,和X1、X2、X3、X4、X5、X6和X7是任何氨基酸。在一种具体实施方式中,X1选自苏氨酸、异亮氨酸、缬氨酸、脯氨酸、天门冬酰胺或赖氨酸;X2选自亮氨酸、缬氨酸、丙氨酸和甲硫氨酸;X3选自赖氨酸、组氨酸、天门冬酰胺和精氨酸;X4选自甘氨酸、缬氨酸、丙氨酸、谷氨酸、脯氨酸和甲硫氨酸;X5选自苏氨酸、丝氨酸、谷氨酸、丙氨酸、谷氨酰胺和天冬氨酸;X6选自谷氨酸、天冬氨酸和谷氨酰胺;以及X7选自甘氨酸、丝氨酸、丙氨酸、赖氨酸、精氨酸、谷氨酰胺和苏氨酸。
在另一实施方式中,本发明的小分子结合VEGF受体家族成员的D7结构域的以下共有序列:IX1RVX2X3EDX4G,其中I是异亮氨酸,R是精氨酸,E是谷氨酸,D是天冬氨酸,G是甘氨酸;和X1、X2、X3和X4是任何氨基酸。在一个具体的实施方案中,X1选自谷氨酸、精氨酸和谷氨酰胺;X2选自精氨酸和苏氨酸;X3选自谷氨酸和赖氨酸;以及X4选自谷氨酸和丙氨酸(SEQ ID NO:1)。
在另一个实施方案中,本发明的成分(例如,小分子)结合VEGF受体的D7结构域的以下共有序列:L/I X1 R Φ X2 X3 X4 D/E X5 G(SEQ ID NO:158),其中L是亮氨酸,I是异亮氨酸,R是精氨酸,Φ是疏水性氨基酸,D是天冬氨酸,E是谷氨酸,G是甘氨酸;以及X1、X2、X3、X4和X5是任何氨基酸。在一个具体的实施方案中,Φ是缬氨酸;X1选自于精氨酸、谷氨酰胺、谷氨酸和天冬氨酸;X2选自于精氨酸、赖氨酸和苏氨酸;X3选自于赖氨酸、谷氨酸、谷氨酰胺和缬氨酸;X4选自于谷氨酸和缬氨酸;以及X5选自于谷氨酸、甘氨酸、丝氨酸和谷氨酰胺。
在其它实施方式中,小分子抑制剂结合代表蛋白质的三维结构的蛋白质基序或共有序列。这样的基序或共有序列不表示氨基酸的连续链,而是产生于RTK的三维折叠的非线性的氨基酸排列(即“结构基序”)。这样的基序的实例是基于Kit受体的D3-D4和/或D4-D5铰链区设计的基序。这样的基序和共有序列可以根据关于抗体的章节I中论述的方法设计。
重要的是,本发明的小分子抑制剂不结合RTK的配体结合位点,例如Kit受体的SCF结合位点。换言之,小分子抑制剂不结合负责配体结合的RTK的Ig样结构域。
在另一个实施方案中,本发明的小分子抑制剂结合VEGF受体的连续表位。在一个实施方案中,连续表位由VEGF受体D7结构域中的两个或更多个残基组成。在另一个实施方案中,连续表位是选自VEGFR1的672VAISSS677、VEGFR1的678TTLDCHA684、VEGFR1的685NGVPEPQ691、VEGFR1的700KIQQEPG706、VEGFR1的707IILG710、VEGFR1的711PGS713、VEGFR1的714STLFI718、VEGFR1的719ERVTEEDEGV728、VEGFR3的689VNVSDS694、VEGFR3的695LEMQCLV701、VEGFR3的702AGAHAPS708、VEGFR3的717LLEEKSG723、VEGFR3的724VDLA727、VEGFR3的728DSN730、VEGFR3的731QKLSI735和VEGFR3的736QRVREEDAGR745、VEGFR2的678TSIGES683、VEGFR2的684IEVSCTA690、VEGFR2的691SGNPPPQ697、VEGFR2的706TLVEDSG712、VEGFR2的713IVLK716、VEGFR2的717DGN719、VEGFR2的720RNLTI724和VEGFR2的725RRVRKEDEGL734的表位。
在另外的实施方式中,选择或设计本发明的小分子抑制剂来特异性结合RTK的突变胞外域,例如突变Ig样结构域或突变铰链区。在优选实施方式中,突变体RTK是致瘤的或致癌的突变体。在一种具体实施方式中,选择或设计小分子抑制剂来结合致癌的Kit受体突变体。可以被本发明的小分子靶向的Kit受体突变体具有一个或多个以下氨基酸中的突变的Kit受体:Thr417、Tyr418、Asp419、Leu421、Arg420、Tyr503或Ala502。本领域技术人员应理解,本发明的方法可以应用于Kit中的其它突变或其它RTK中的突变。靶向于突变体RTK的一个优点是治疗性小分子可以仅结合包含突变的细胞上的RTK,而使得健康细胞大部分或全部不受影响。因此,在突变是致瘤性突变的情况下,仅肿瘤细胞将被用作治疗靶标,潜在地减少副作用和剂量要求。
在一些实施方式中,所述小分子结合人Kit受体的特定序列,例如人Kit受体的残基309-413、残基410-519、381Arg和386Glu或418Tyr和505Asn。在一些实施方案中,本发明的小分子可以结合人VEGF受体的特定序列,例如,VEGFR1的残基718-727、VEGFR1的Arg720和Asp725、VEGFR2的残基724-733、VEGFR2的Arg726和Asp731、VEGFR3的残基735-744或VEGFR3的残基Arg737和Asp742。
在优选实施方式中,本发明的小分子可以结合Kit受体中构成在表4(下文中)中所描述的小腔或口袋的一个或多个残基。例如,本发明的小分子可以结合Kit受体的D3-D4铰链区中的一个或多个以下残基:来自D3结构域的K218、S220、Y221、L222和来自D4结构域的F340、P341、K342、N367、E368、S369、N370、I371、Y373。本发明的小分子也可以结合构成Kit受体的D4结构域中的凹面的一个或多个以下残基:Y350、R353、F355、K358、L379、T380、R381、L382、E386和T390。在另一实施方式中,本发明的小分子可以结合形成Kit受体的D2-D3铰链区中的口袋的一个或多个以下残基:来自D2结构域的Y125、G126、H180、R181、K203、V204、R205、P206和F208以及来自D3结构域的V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269。
因此,在一些实施方式中,本发明的小分子可以结合连续的或不连续的氨基酸残基,并起到阻止RTK的近膜区以使酪氨酸激酶能够活化的距离和定向来定位所需的运动的分子楔的作用。本发明的小分子也可以起到阻止同型D4或D5受体相互作用或使配体-受体相互作用位点不稳定的作用。在一些优选实施方式中,本发明的小分子可以结合Kit受体上的的一个或多个以下残基:Y125、G126、H180、R181、K203、V204、R205、P206、P206、F208、K127、A207、V238、S239、S240、S241、H263、G265、D266、F267、N268、Y269、T295、L222、L222、L223、E306、V308、R224、V308、K310、K218、A219、S220、K218、A220、Y221、A339、D327、D398、E338、E368、E386、F312、F324、F340、F355、G311、G384、G387、G388、I371、K342、K358、L382、L379、N326、N367、N370、N410、P341、S369、T385、V325、V407、V409、Y373、Y350、Y408、T380、T390、R381、R353、T411、K412、E414、K471、F433、G470、L472、V497、F469、A431或G432。本领域技术人员将理解,在一些实施方式中,本发明的小分子可以容易地被靶向于其它Ⅲ型RTK中的相应残基,例如形成相似的口袋或空腔的那些残基或通过结构比对或序列比对处于相同的位置的那些残基。
在一种具体实施方式中,本发明的小分子结合Ⅲ型RTK或V型RTK上的构象表位或非连续表位。构象表位或非连续表位可以由来自Ⅲ型RTK(例如人Kit受体或PDGF受体)的D3、D4或D5结构域或者D4-D5或D3-D4铰链区的两个或更多个残基组成,或者由来自VEGF受体的D7结构域的两个或更多个残基组成。例如,所述构象表位或非连续表位可以由下表4中所列的两个或更多个残基组成。在一种具体实施方式中,本发明的小分子结合由选自Y125、H180、R181、K203、V204、R205、P206、V238、S239、S240、H263、G265、D266、F267、N268和Y269的2个或多个氨基酸组成的构象表位。在相似的具体实施方式中,本发明的小分子可以结合由选自以下氨基酸组之一的2个或多个氨基酸组成的构象表位:P206、F208、V238和S239;K127、A207、F208和T295;L222、A339、F340、K342、E368、S369、N370、I371和Y373;L222、L223、E306、V308、F312、E338、F340和I371;R224、V308、K310、G311、F340、P341和D398;K218、A219、S220、N367、E368和S369;K218、A220、E368和S369;G384、T385、T411、K412、E414和K471;Y408、F433、G470、K471和L472;F324、V325、N326和N410;D327、N410、T411、K412和V497;G384、G387、V409和K471;L382、G387、V407和V409;Y125、G126、H180、R181、K203、V204、R205、P206、F208、V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269;P206、F208、V238和S239;K218、S220、Y221、L222、F340、P341、K342、N367、E368、S369、N370、I371和Y373;G384、G387、G388、Y408、V409、T411、F433、F469、G470和K471;D327、T411、K412、E414、A431、G432和K471;Y350、F355、K358、L379、T380、R381、L382、E386和T390;Y350、R353和F355。如以上所指出的,本发明的小分子可以结合形成在表4中确定的口袋或空腔的所有氨基酸残基,或者它们可以结合形成口袋或空腔的残基的一部分。应理解,在某些实施方式中,当涉及结合表位(例如构象表位)的本发明的小分子时,意图是所述小分子仅结合那些构成该表位(例如表4中确定的口袋或空腔)的那些特定残基,而不是在该受体的线性氨基酸序列中的其它残基。
在进一步的实施方式中,本发明的小分子结合构象表位,其中所述构象表位由选自表5中所列的肽的两个或更多个氨基酸残基组成。在一种具体实施方式中,构象表位由选自第一肽的一个或多个氨基酸残基和选自第二肽的一个或多个氨基酸残基组成,其中第一和第二肽选自表5中所列的肽的组。因而,本发明的小分子可以结合其中第一和第二肽组如下的构象表位:Ala219-Leu222和Thr304-Val308;Asp309-Gly311和Arg224-Gly226;Thr303-Glu306和Ala219-Leu222;Asn367-Asn370和Ser217-Tyr221;Ala339-Pro343和Asn396-Val399;Ala339-Pro343和Glu368-Arg372;Lys358-Tyr362和Val374-His378;Asp357-Glu360和Leu377-Thr380;Met351-Glu360和His378-Thr389;His378-Thr389和Val323-Asp332;Val409-Ile415和Ala493-Thr500;Val409-Ile415和Ala431-Thr437;Val409-Ile415和Phe469-Val473;Val409-Ile415和Val325-Asn330;Val409-Ile415和Arg381-Gly387;Gly466-Leu472和Gly384-Gly388;Val325-Glu329和Tyr494-Lys499;Thr411-Leu416和Val497-Ala502;Ile415-Leu421和Ala502-Ala507;Ala502-Ala507和Lys484-Thr488;及Ala502-Ala507和Gly445-Cys450。本发明的小分子可以结合形成上述第一和第二肽组的所有氨基酸残基,或者它们可以结合形成第一和第二肽组的残基的一部分。应理解,在某些实施方式中,当涉及结合表位(例如构象表位)的本发明的小分子时,意图是所述小分子仅结合那些构成该表位(例如表5中确定的特定肽)的那些特定残基,而不是在该受体的线性氨基酸序列中的其它残基。
在另一实施方式中,本发明的小分子可以结合由2个或多个选自E33、P34、D72、E76、N77、K78、Q79、K158、D159、N250、S251、Q252、T253、K254、L255、N260、W262、H264、G265、E344、N352、R353、F355、T356、D357、Y362、S365、E366、N367、N370和G466的氨基酸组成的构象表位或非连续表位。
在另一实施方式中,本发明的小分子结合人PDGFRβ的氨基酸残基385Arg和390Glu或PDGFRα中的相应残基。人PDGFRβ的残基385Arg和390Glu类似于Kit受体残基381Arg和386Glu并介导PDGFRβ的同型D4-D4相互作用。本发明的小分子可以通过阻止Ⅲ型RTK的近膜区之间的关键同型相互作用(例如人PDGFRβ的385Arg和390Glu之间形成的盐桥)发挥其对受体活化的抑制作用,所述近膜区之间的关键同型相互作用对于将胞质结构域定位在对于酪氨酸激酶活化至关重要的距离和定向是是必要的。本文中论述的实验表明,同型D4-D4相互作用对于PDGFRβ二聚化不是必要的,而PDGFRβ二聚化对于受体活化是必要的然而并非足够的。因此,本发明的小分子可以允许PDGFRβ的二聚化而同时阻止活化。基于结构的序列比对已经表明EF环的大小以及包含关键氨基酸的D4-D4界面在Kit、PDGFRα、PDGFRβ和CSF1R中是保守的。因此,在一些实施方式中,本发明的小分子可以靶向于Ⅲ型RTK的D4或D5结构域的保守区。
在优选实施方式中,本发明的小分子以高亲和性结合Kit的Ig样结构域或铰链区(例如Kit受体的D3-D4和/或D4-D5铰链区或D4-D4和/或D5-D5界面结合位点),例如KD为1x 10-7M或更低、KD为5x10-8M或更低、KD为1x 10-8M更低、KD为5x 10-9M更低、或者KD为1x 10-8M和1x 10-10M之间或更低的亲和性。
本发明的小分子抑制剂可以通过本领域已知的几种方法进行制备或选择。筛选方法可用于从文库鉴定结合希望的RTK的Ig样结构域或铰链区(例如人Kit RTK的D4或D5结构域)的小分子。一种方法(Chemetics (Nuevolutions))利用文库中各分子的DNA标签来辅助选择。Chemetics
(Nuevolutions))利用文库中各分子的DNA标签来辅助选择。Chemetics 系统允许筛选用于靶向结合的数百万化合物。涉及基于小分子文库和标签的筛选的专利是美国专利申请20070026397;20060292603;20060269920;20060246450;20060234231;20060099592;20040049008;20030143561,将这些专利以其全文通过引用的方式并入本文。
系统允许筛选用于靶向结合的数百万化合物。涉及基于小分子文库和标签的筛选的专利是美国专利申请20070026397;20060292603;20060269920;20060246450;20060234231;20060099592;20040049008;20030143561,将这些专利以其全文通过引用的方式并入本文。
其它可以用于从文库鉴定结合希望的RTK的Ig样结构域或铰链区(例如人Kit RTK D4或D5结构域或VEGF受体的D7结构域)的小分子的公知方法包括利用其中文库成员以鉴定标记进行标记(即,文库中存在的各个标记与文库中存在的不同化合物结构相关,从而鉴别标记得知标记分子的结构)的文库的方法。标记的文库的一种方法利用寡核苷酸标签,如在例如以下文献中描述:PCT公开WO2005/058479A2(Direct SelectTM技术)和美国专利5,573,905;5,708,153;5,723,598,6,060,596号;公开的PCT申请WO 93/06121;WO 93/20242;WO 94/13623;WO 00/23458;WO 02/074929和WO 02/103008,以及Brenner和Lerner(Proc.Natl.Acad.Sci.USA 89,5381-5383(1992);Nielsen和Janda(Methods:A Companion to Methods in Enzymology 6,361-371(1994);以及Nielsen、Brenner和Janda(J.Am.Chem.Soc.115,9812-9813(1993)),每一篇文献的全部内容均通过引用的方式并入本文中。这样的标签可以使用例如聚合酶链式反应进行扩增,以产生很多标签拷贝并通过测序鉴定所述标签。然后标签的序列确认结合分子的结构,所述结合分子可以纯化形式进行合成并测试活性。
组合化学文库的预备和筛选是本领域技术人员公知的。可用于鉴定本发明的成分的这类组合化学文库包括但不局限于肽文库(参见,例如美国专利No.5,010,175,Furka,Int.J.Pept.Prot.Res.37:487493(1991)和Houghton等,Nature 354:8488(1991))。用于产生化学多样性文库的其它化学物质是本领域公知的并且可以使用。这样的化学物包括但不局限于:类肽(例如PCT公开No.WO 91/19735),编码肽(例如PCT公开WO 93/20242),随机生物寡聚物(例如PCT公开No.WO92/00091),苯并二氮 (例如美国专利No.5,288,514),diversomer如乙内酰脲、苯并二氮
(例如美国专利No.5,288,514),diversomer如乙内酰脲、苯并二氮 和二肽(Hobbs等,Proc.Nat.Acad.Sci.USA90:6909 6913(1993)),插烯多肽(vinylogous polypeptide)(Hagihara等,J.Amer.Chem.Soc.114:6568(1992))、具有葡萄糖骨架的非肽类肽模拟物(Hirschmann等,J.Amer.Chem.Soc.114:9217 9218(1992)),小化合物文库的类似有机合成(Chen等,J.Amer.Chem.Soc.116:2661(1994))、寡氨酯(oligocarbamate)(Cho等,Science 261:1303(1993))和/或肽基膦酸酯(Campbell等,J.Org.Chem.59:658(1994))、核酸文库(参见Ausubel,Berger和Russell & Sambrook,所有同上)、肽核酸文库(参见例如美国专利No.5,539,083)、碳水化合物文库(参见例如Liang等,Science,274:1520 1522(1996)和美国专利No.5,593,853)、小有机分子文库(参见例如苯并二氮
和二肽(Hobbs等,Proc.Nat.Acad.Sci.USA90:6909 6913(1993)),插烯多肽(vinylogous polypeptide)(Hagihara等,J.Amer.Chem.Soc.114:6568(1992))、具有葡萄糖骨架的非肽类肽模拟物(Hirschmann等,J.Amer.Chem.Soc.114:9217 9218(1992)),小化合物文库的类似有机合成(Chen等,J.Amer.Chem.Soc.116:2661(1994))、寡氨酯(oligocarbamate)(Cho等,Science 261:1303(1993))和/或肽基膦酸酯(Campbell等,J.Org.Chem.59:658(1994))、核酸文库(参见Ausubel,Berger和Russell & Sambrook,所有同上)、肽核酸文库(参见例如美国专利No.5,539,083)、碳水化合物文库(参见例如Liang等,Science,274:1520 1522(1996)和美国专利No.5,593,853)、小有机分子文库(参见例如苯并二氮 Baum C&EN,Jan 18,33页(1993);类异戊二烯,美国专利5,569,588号;噻唑烷酮和间噻嗪酮,美国专利5,549,974号;吡咯烷,美国专利5,525,735和5,519,134号;吗啉代化合物,美国专利5,506,337号;苯并二氮
Baum C&EN,Jan 18,33页(1993);类异戊二烯,美国专利5,569,588号;噻唑烷酮和间噻嗪酮,美国专利5,549,974号;吡咯烷,美国专利5,525,735和5,519,134号;吗啉代化合物,美国专利5,506,337号;苯并二氮 5,288,514等)。各前述出版物通过引用的方式并入本文。公众数据库也是可利用的,并常用于小分子筛选,例如PubChem(pubchem.ncbi.nlm.nih.gov),Zinc(Irwin和Shoichet(2005)J.Chem.Inf.Model.45(1):177-82)和ChemBank(Seiler等(2008)Nucleic Acids Res.36(数据库发行号):D351-D359)。
5,288,514等)。各前述出版物通过引用的方式并入本文。公众数据库也是可利用的,并常用于小分子筛选,例如PubChem(pubchem.ncbi.nlm.nih.gov),Zinc(Irwin和Shoichet(2005)J.Chem.Inf.Model.45(1):177-82)和ChemBank(Seiler等(2008)Nucleic Acids Res.36(数据库发行号):D351-D359)。
用于制备组合文库的装置是市场上可买到的(见,例如357 MPS,390 MPS,Advanced Chem Tech,Louisville Ky.,Symphony,Rainin,Woburn,Mass.,433A Applied Biosystems,Foster City,Calif.,9050 Plus,Millipore,Bedford,Mass.)。此外,许多组合文库本身是市场上可买到的(见例如,ComGenex,Princeton,N.J.,Tripos,Inc.,St.Louis,Mo.,3DPharmaceuticals,Exton,Pa.,Martek Biosciences,Columbia,Md.,等)。而且,因为筛选方法被非常好地限定,通常与专业公司签订合同以鉴定用于目标靶的特定化合物(例如BioFocus DPI(biofocus.com)和Quantum Lead(q-lead.com))。
本领域众所周知的并且可应用于本发明的方法的其它选择小分子的方法是Huang和Stuart L.Schreiber(1997)Proc Natl Acad Sci U SA.94(25):13396-13401;Hung等(2005)Science 310:670-674;Zhang等(2007)Proc Natl Acad Sci 104:4606-4611;或者在Gordon(2007)ACS Chem.Biol.2:9-16中综述的任何一种方法,其全部都通过引用以其全部内容并入本文。
除了实验筛选法之外,本发明的小分子还可以使用虚拟筛选法加以选择。虚拟筛选技术使用统计分析和蛋白质对接(docking)模拟来预测来自文库的哪些小分子将结合蛋白质或其中的特定表位。最常见地,虚拟筛选方法比较蛋白质的三维结构和文库中小分子的三维结构。使用对蛋白质-分子相互作用进行建模的不同策略,尽管常用的是模拟在原子之间的结合能——包括氢键、静电力和范德瓦耳斯相互作用——的算法。通常,虚拟筛选方法可以扫描超过一百万个化合物的文库并返回可能是强结合物的小分子的短的名单。对虚拟筛选的若干综述可以获得,其详述了可用来鉴定本发明的小分子的技术(Engel等(2008)J.Am.Chem.Soc.,130(15),5115-5123;McInnes.(2007).Curr Opin Chem Biol.Oct;11(5):494-502;Reddy等(2007)Curr ProteinPept Sci.Aug;8(4):329-51;Muegge和Oloff.(2006)Drug DiscoveryToday.3(4):405-411;Kitchen等(2004)Nature Reviews DrugDiscovery 3,935-949)。小分子筛选的进一步的实例可以在美国专利申请2005/0124678中找到,其通过引用并入本文。
本发明的小分子可以包含在下表中描绘的骨架结构之一。在该表中引用的参考文献均通过引用的方式以其全文并入本文中。基团R1、R2、R3和R4的仅限制于它们不应干扰或显著抑制所指出的反应,并且可以包括氢、烷基、取代烷基、杂烷基、取代杂烷基、环烷基、杂环烷基、取代环烷基、取代杂环烷基、芳基、取代芳基、芳烷基、杂芳烷基、取代芳烷基、取代杂芳烷基、杂方基、取代杂芳基、卤素、烷氧基、芳氧基、氨基、取代氨基和本领域已知的其它基团。适宜的取代基包括但不局限于烷基、烷氧基、硫代烷氧基、硝基、羟基、巯基、芳氧基、芳基-S-、卤素、羧基、氨基、烷基氨基、二烷基氨基、芳基氨基、氰基、氰酸酯、腈、异氰酸酯、硫氰酸酯、氨基甲酰基和取代氨基甲酰基。


III.结合人受体酪氨酸激酶的Ig样结构域的肽类分子
在本发明的另一方面,结合人受体酪氨酸激酶的胞外域(例如Ig样结构域或铰链区)的成分是肽类分子。
所述肽类分子可以基于RTK的Ig样结构域或源自这样的结构域的共有序列进行设计。
在具体实施方式中,本发明的肽类分子结合D4相互作用位点的以下共有序列:LX1RX2X3X4X5X6X7G,其中L是亮氨酸,R是精氨酸,G是甘氨酸,和X1、X2、X3、X4、X5、X6和X7是任何氨基酸。在具体的实施方案中,X1选自苏氨酸、异亮氨酸、缬氨酸、脯氨酸、天门冬酰胺或赖氨酸;X2选自亮氨酸、缬氨酸、丙氨酸和甲硫氨酸;X3选自赖氨酸、组氨酸、天门冬酰胺和精氨酸;X4选自甘氨酸、缬氨酸、丙氨酸、谷氨酸、脯氨酸和甲硫氨酸;X5选自苏氨酸、丝氨酸、谷氨酸、丙氨酸、谷氨酰胺和天冬氨酸;X6选自谷氨酸、天冬氨酸和谷氨酰胺;以及X7选自甘氨酸、丝氨酸、丙氨酸、赖氨酸、精氨酸、谷氨酰胺和苏氨酸。
因而,在一种实施方式中,本发明的肽类分子包含或由匹配上述共有序列(LX1RX2X3X4X5X6X7G)的序列组成,其中L是亮氨酸,R是精氨酸,G是甘氨酸,和X1、X2、X3、X4、X5、X6和X7是任何氨基酸。在具体的实施方案中,X1选自苏氨酸、异亮氨酸、缬氨酸、脯氨酸、天门冬酰胺或赖氨酸;X2选自亮氨酸、缬氨酸、丙氨酸和甲硫氨酸;X3选自赖氨酸、组氨酸、天门冬酰胺和精氨酸;X4选自甘氨酸、缬氨酸、丙氨酸、谷氨酸、脯氨酸和甲硫氨酸;X5选自苏氨酸、丝氨酸、谷氨酸、丙氨酸、谷氨酰胺和天冬氨酸;X6选自谷氨酸、天冬氨酸和谷氨酰胺;以及X7选自甘氨酸、丝氨酸、丙氨酸、赖氨酸、精氨酸、谷氨酰胺和苏氨酸。
在另一个实施方案中,本发明的肽类分子包含或由VEGF受体的D7结构域的以下共有序列组成:L/I X1 R Φ X2 X3 X4 D/E X5 G(SEQ ID NO:158),其中L是亮氨酸,I是异亮氨酸,R是精氨酸,Φ是疏水性氨基酸,D是天冬氨酸,E是谷氨酸,G是甘氨酸;以及X1、X2、X3、X4和X5是任何氨基酸。在具体的实施方案中,Φ是缬氨酸;X1选自于精氨酸、谷氨酰胺、谷氨酸和天冬氨酸;X2选自于精氨酸、赖氨酸和苏氨酸;X3选自于赖氨酸、谷氨酸、谷氨酰胺和缬氨酸;X4选自于谷氨酸和缬氨酸;以及X5选自于谷氨酸、甘氨酸、丝氨酸和谷氨酰胺。
在另一实施方式中,所述肽类分子结合VEGF受体家族成员的D7结构域的以下共有序列:IX1RVX2X3EDX4G,其中I是异亮氨酸,R是精氨酸,E是谷氨酸,D是天冬氨酸,G是甘氨酸,且X1、X2、X3和X4是任何氨基酸。在具体的实施方案中,X1选自谷氨酸、精氨酸和谷氨酰胺;X2选自精氨酸和苏氨酸;X3选自谷氨酸和赖氨酸;和X4选自谷氨酸和丙氨酸(SEQ ID NO:1)。
因而,在一种实施方式中,本发明的肽类成分包含或由匹配该共有序列IX1RVX2X3EDX4G的序列组成,其中I是异亮氨酸,R是精氨酸,E是谷氨酸,D是天冬氨酸,G是甘氨酸,且X1、X2、X3和X4是任何氨基酸。在具体的实施方案中,X1选自谷氨酸、精氨酸和谷氨酰胺;X2选自精氨酸和苏氨酸;X3选自谷氨酸和赖氨酸;以及X4选自谷氨酸和丙氨酸(SEQ ID NO:1)。
在一种实施方式中,本发明的肽类成分可以包含整个蛋白质结构域,例如人Kit的D4或D5结构域,如D4结构域(残基309-413)或D5结构域(残基410-519)。作为进一步的实例,本发明的肽类成分可以包含V型RTK的D7结构域(或其片段),例如VEGFR的D7结构域(VEGFR1的残基718-727、VEGFR2的残基724-733或VEGFR3的残基735-744)。这样的肽类分子结合RTK并通过阻止RTK活化而起到拮抗剂的作用(参见下面实施例16)。在一些实施方式中,本发明的肽类成分可以与RTK(例如Ⅲ型RTK)的结构域具有低至50%的同一性,例如,本发明的肽类成分可以与RTK的D4、D5或D7结构域具有至少50%的同一性、至少60%的同一性、至少70%的同一性、至少80%的同一性、至少90%的同一性、或至少95%、96%、97%或98%的同一性。在具体实施方式中,本发明的肽类成分与人Kit RTK的氨基酸残基309-413、VEGFR1的氨基酸残基718-727、VEGFR2的氨基酸残基724-733或VEGFR3的氨基酸残基735-744具有至少80%的同一性、至少90%的同一性或至少95%、96%、97%或98%的同一性。在类似的实施方式中,本发明的肽类成分与人Kit RTK的氨基酸残基410-519、VEGFR1的氨基酸残基718-727、VEGFR2的氨基酸残基724-733或VEGFR3的氨基酸残基735-744具有至少80%的同一性、至少90%的同一性或至少95%、96%、97%或98%的同一性。
在一些实施方式中,本发明的肽类成分结合或包含人Kit受体的特定序列,例如人Kit受体的残基309-413、残基410-519、381Arg和386Glu或418Tyr和505Asn。在其它实施方式中,本发明的肽类成分结合或包含VEGF受体的特定序列,例如VEGFR1的残基718-727、VEGFR1的Arg720和Asp725、VEGFR2的残基724-733、VEGFR2的Arg726和Asp731、VEGFR3的残基735-744或VEGFR3的残基Arg737和Asp742。
在优选实施方式中,本发明的肽类成分可以结合(或包含或由其组成)Kit受体中的一或多个残基,这些残基构成在表4(下文)中所描述的小腔或口袋。例如,本发明的肽类分子可以结合(或包含或由其组成)Kit受体的D3-D4铰链区中的一个或多个以下残基:来自D3结构域的K218、S220、Y221、L222和来自D4结构域的F340、P341、K342、N367、E368、S369、N370、I371、Y373。本发明的肽类分子也可以结合(或包含或由其组成)构成Kit受体的D4结构域中的凹面的一个或多个以下残基:Y350、R353、F355、K358、L379、T380、R381、L382、E386和T390。在另一实施方式中,本发明的肽类分子可以结合(或包含或由其组成)形成Kit受体的D2-D3铰链区的一个或多个以下残基:来自D2结构域的Y125、G126、H180、R181、K203、V204、R205、P206和F208以及来自D3结构域的V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269。
本发明的肽类成分可以结合连续的或不连续的氨基酸残基,并起到阻止RTK的近膜区以使酪氨酸激酶能够活化的距离和定向进行定位所需的运动的分子楔的作用。本发明的肽类分子也可以起到阻止同型D4、D5或D7受体相互作用或使配体-受体相互作用位点不稳定的作用。在一些优选实施方式中,本发明的肽类分子可结合(或包含或由其组成)Kit受体上的一个或多个以下残基:Y125、G126、H180、R181、K203、V204、R205、P206、P206、F208、K127、A207、V238、S239、S240、S241、H263、G265、D266、F267、N268、Y269、T295、L222、L222、L223、E306、V308、R224、V308、K310、K218、A219、S220、K218、A220、Y221、A339、D327、D398、E338、E368、E386、F312、F324、F340、F355、G311、G384、G387、G388、I371、K342、K358、L382、L379、N326、N367、N370、N410、P341、S369、T385、V325、V407、V409、Y373、Y350、Y408、T380、T390、R381、R353、T411、K412、E414、K471、F433、G470、L472、V497、F469、A431或G432。本发明的肽类成分可以结合(或包含或由其组成)形成在表4中确定的口袋或空腔的所有氨基酸残基,或者它们可以结合(或包含或由其组成)形成口袋或空腔的残基的一部分。本领域技术人员将理解,在一些实施方式中,本发明的肽类分子可以容易地被靶向于其它Ⅲ型RTK中的相应残基,例如形成相似的口袋或空腔的那些残基或通过结构比对或序列比对处于相同的位置的那些残基。
在具体实施方式中,本发明的肽类分子结合Ⅲ型RTK或V型RTK上的构象表位或非连续表位。构象表位或非连续表位可以由来自Ⅲ型RTK(例如人Kit受体或PDGF受体)或V型RTK(例如人VEGF受体)的D3、D4、D5或D7结构域或者D4-D5或D3-D4铰链区的两个或更多个残基组成。例如,所述构象表位或非连续表位可以由下表4中所列的两个或更多个残基组成。在一种特定实施方式中,本发明的肽类分子结合由选自Y125、H180、R181、K203、V204、R205、P206、V238、S239、S240、H263、G265、D266、F267、N268和Y269的2个或多个氨基酸组成的构象表位。在相似的实施方式中,本发明的肽类分子可以结合由选自以下氨基酸组中的一种的2个或多个氨基酸组成的构象表位:P206、F208、V238和S239;K127、A207、F208和T295;L222、A339、F340、K342、E368、S369、N370、I371和Y373;L222、L223、E306、V308、F312、E338、F340和I371;R224、V308、K310、G311、F340、P341和D398;K218、A219、S220、N367、E368和S369;K218、A220、E368和S369;G384、T385、T411、K412、E414和K471;Y408、F433、G470、K471和L472;F324、V325、N326和N410;D327、N410、T411、K412和V497;G384、G387、V409和K471;L382、G387、V407和V409;Y125、G126、H180、R181、K203、V204、R205、P206、F208、V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269;P206、F208、V238和S239;K218、S220、Y221、L222、F340、P341、K342、N367、E368、S369、N370、I371和Y373;G384、G387、G388、Y408、V409、T411、F433、F469、G470和K471;D327、T411、K412、E414、A431、G432和K471;Y350、F355、K358、L379、T380、R381、L382、E386和T390;Y350、R353和F355。
在进一步的实施方式中,本发明的肽类分子结合由选自表5中所列的肽的两个或更多个氨基酸残基组成的构象表位。在一种具体实施方式中,构象表位由选自第一肽的一个或多个氨基酸和选自第二肽的一个或多个氨基酸组成,其中第一和第二肽选自表5中所列的肽的组。因而,本发明的肽类分子可以结合其中第一和第二肽组如下的构象表位:Ala219-Leu222和Thr304-Val308;Asp309-Gly311和Arg224-Gly226;Thr303-Glu306和Ala219-Leu222;Asn367-Asn370和Ser217-Tyr221;Ala339-Pro343和Asn396-Val399;Ala339-Pro343和Glu368-Arg372;Lys358-Tyr362和Val374-His378;Asp357-Glu360和Leu377-Thr380;Met351-Glu360和His378-Thr389;His378-Thr389和Val323-Asp332;Val409-Ile415和Ala493-Thr500;Val409-Ile415和Ala431-Thr437;Val409-Ile415和Phe469-Val473;Val409-Ile415和Val325-Asn330;Val409-Ile415和Arg381-Gly387;Gly466-Leu472和Gly384-Gly388;Val325-Glu329和Tyr494-Lys499;Thr411-Leu416和Val497-Ala502;Ile415-Leu421和Ala502-Ala507;Ala502-Ala507和Lys484-Thr488;及Ala502-Ala507和Gly445-Cys450。本发明的肽类成分可以结合形成上述第一和第二肽组的所有氨基酸残基,或者它们可以结合形成第一和第二肽组的残基的一部分。
在另一实施方式中,本发明的肽类成分可以结合(或包含或由其组成)2个或多个选自E33、P34、D72、E76、N77、K78、Q79、K158、D159、N250、S251、Q252、T253、K254、L255、N260、W262、H264、G265、E344、N352、R353、F355、T356、D357、Y362、S365、E366、N367、N370和G466的氨基酸。
在另一个实施方案中,本发明的肽类成分结合VEGF受体的连续表位。在一个实施方案中,连续表位由VEGF受体D7结构域中的两个或更多个残基组成。在另一个实施方案中,连续表位是选自VEGFR1的672VAISSS677、VEGFR1的678TTLDCHA684、VEGFR1的685NGVPEPQ691、VEGFR1的700KIQQEPG706、VEGFR1的707IILG710、VEGFR1的711PGS713、VEGFR1的714STLFI718、VEGFR1的719ERVTEEDEGV728、VEGFR3的689VNVSDS694、VEGFR3的695LEMQCLV701、VEGFR3的702AGAHAPS708、VEGFR3的717LLEEKSG723、VEGFR3的724VDLA727、VEGFR3的728DSN730、VEGFR3的731QKLSI735和VEGFR3的736QRVREEDAGR745、VEGFR2的678TSIGES683、VEGFR2的684IEVSCTA690、VEGFR2的691SGNPPPQ697、VEGFR2的706TLVEDSG712、VEGFR2的713IVLK716、VEGFR2的717DGN719、VEGFR2的720RNLTI724和VEGFR2的725RRVRKEDEGL734的表位。
在另一实施方式中,本发明的肽类分子结合或包含人PDGFRβ的氨基酸残基385Arg和390Glu或PDGFRα中的相应残基。人PDGFRβ的残基385Arg和390Glu类似于Kit受体的残基381Arg和386Glu并介导PDGFRβ的同型D4-D4相互作用。本发明的肽类分子可以通过阻止Ⅲ型RTK的近膜区之间的关键同型相互作用(例如人PDGFRβ的385Arg和390Glu之间形成的盐桥)发挥其对受体活化的抑制作用,所述近膜区之间的关键同型相互作用对于将胞质结构域定位在对于酪氨酸激酶活化至关重要的距离和定向上是至关重要的。本文中论述的实验表明同型D4-D4相互作用对于PDGFRβ二聚化不是必要的,而PDGFRβ二聚化对于受体活化是必要的然而并非足够的。因此,本发明的肽类分子可以允许PDGFRβ的二聚化而同时阻止活化。基于结构的序列比对已经表明EF环的大小以及包含关键氨基酸的D4-D4界面在Kit、PDGFRα、PDGFRβ和CSF 1R中是保守的。因此,在一些实施方式中,本发明的肽类分子可以靶向于Ⅲ型RTK的D4或D5结构域的保守区。
本发明的肽类成分可以是包含或由本文中确定的任一氨基酸序列(例如SEQ ID NO:1-89、92、93和105-157)组成。例如,本发明的肽类成分可以是包含或由以下任一氨基酸序列组成的肽:EVVDKGFIN(SEQ ID NO:2)、ASYL(SEQ ID NO:3)、TLEVV(SEQID NO:4)、ASYLTLEVV(SEQ ID NO:5)、DKG,REG,DKGREG(SEQID NO:6)、VVSVSKASYLL(SEQ ID NO:7)、VTTTLEVVD(SEQ IDNO:8)、REGEEFTVTCTI(SEQ ID NO:9)、TTLE(SEQ ID NO:10)、TTLEASYL(SEQ ID NO:11)、KSENESNIR(SEQ ID NO:12)、NESN(SEQ ID NO:13)、SKASY(SEQ ID NO:14)、NESNSKASY(SEQ IDNO:15)、AFPKP(SEQ ID NO:16)、NSDV(SEQ ID NO:17)、AFPKPNSDV(SEQ ID NO:18)、ESNIR(SEQ ID NO:19)、AFPKPESNIR(SEQ ID NO:20)、DKWEDYPKSE(SEQ ID NO:21)、IRYVSELHL(SEQ ID NO:22)、LTRLKGTEGGT(SEQ ID NO:23)、GENVDLIVEYE(SEQ ID NO:24)、MNRTFTDKWE(SEQ ID NO:25)、KWEDY(SEQ ID NO:26)、VSELH(SEQ ID NO:27)、KWEDYVSELH(SEQ ID NO:28)、DKWE(SEQ ID NO:29)、LHLT(SEQ ID NO:30)、DKWELHLT(SEQ ID NO:31)、HLTRLKGTEGGT(SEQ ID NO:32)、MNRTFTDKWE(SEQ ID NO:25)、HLTRLKGTEGGT(SEQ ID NO:32)、MNRTFTDKWEHLTRLKGTEGGT(SEQ ID NO:33)、VFVNDGENVD(SEQ ID NO:34)、VNTKPEI(SEQ ID NO:35)、AYNDVGKT(SEQ ID NO:36)、VNTKPEIAYNDVGKT(SEQ ID NO:37)、AGFPEPT(SEQ ID NO:38)、VNTKPEIAGFPEPT(SEQ ID NO:39)、FGKLV(SEQ ID NO:40)、VNTKPEI FGKLV(SEQ ID NO:41)、VNDGEN(SEQ ID NO:42)、VNTKPEIVNDGEN(SEQ ID NO:43)、RLKGTEG(SEQ ID NO:44)、VNTKPEIRLKGTEG(SEQ ID NO:45)、GPPFGKL(SEQ ID NO:46)、GTEGG(SEQ ID NO:47)、GPPFGKLGTEGG(SEQ ID NO:48)、VNDGE(SEQ ID NO:49)、YNDVGK(SEQ ID NO:50)、VNDGEYNDVGK(SEQ ID NO:51)、TKPEILTYDRL(SEQ ID NO:52)、DRLVNGMLQC(SEQ ID NO:53)、GKTSAYFNFAFK(SEQ ID NO:54)、CPGTEQRCSAS(SEQ ID NO:55)、CSASVLPVDVQ(SEQ ID NO:56)、DSSAFKHNGT(SEQ ID NO:57)、GTVECKAYND(SEQ ID NO:58)、LNSSGPPFGKL(SEQ ID NO:59)、FAFKGNNKEQI(SEQ ID NO:60)、TKPEIL(SEQ ID NO:61)、VGKTSA(SEQ ID NO:62)、TKPEILVGKTSA(SEQ ID NO:63)、ILTYDRL(SEQ ID NO:64)、AYFNFA(SEQ ID NO:65)、ILTYDRLAYFNFA(SEQ ID NO:66)、KHNGT(SEQ ID NO:67)、AYFNFAKHNGT(SEQ ID NO:68)、GTEQRC(SEQ ID NO:69)、AYFNFAGTEQRC(SEQ ID NO:70)、YHRKVRPVS SHGDFNY(SEQID NO:71)、PFVS(SEQ ID NO:72)、KAFT(SEQ ID NO:73)、LAFKESNIY(SEQ ID NO:74)、LLEVFEFI(SEQ ID NO:75)、RVKGFPD(SEQ ID NO:76)、KASNES(SEQ ID NO:77)、KAES(SEQID NO:78)、GTTKEK(SEQ ID NO:79)、YFGKL(SEQ ID NO:80)、FVNN(SEQ ID NO:81)、DNTKV(SEQ ID NO:82)、GGVK(SEQ IDNO:83)、LGVV(SEQ ID NO:84)、YGHRKVRPFVSSSHGDFNY(SEQID NO:85)、PFVS(SEQ ID NO:72)、KSYLFPKNESNIY(SEQ ID NO:86)、GGGYVTFFGK(SEQ ID NO:87)、DTKEAGK(SEQ ID NO:88)、YFKLTRLET(SEQ ID NO:89)和YRF。
本发明的肽类分子可以被进一步修饰以增加其稳定性、生物利用度或溶解度。例如,在所述肽类分子内的一个或多个L-氨基酸残基可以用D-氨基酸残基替换。如应用于本发明的肽类分子的术语“模拟物”意图包括模拟D-肽结构的化学结构并保持D-肽结构的功能特性的模拟物。术语“模拟物”进一步意图包括如下所述的肽的“类似物”和/或“衍生物”。设计肽类似物、衍生物和模拟物的方法是本领域已知的。例如,见Farmer,P.S.in Drug Design(E.J.Ariens编辑)Academic Press,New York,1980,vol.10,pp.119-143;Ball.J.B.和Alewood,P.F.(1990)J.Mol.Recognition 3:55;Morgan,B.A.和Gainor,J.A.(1989)Ann.Rep.Med.Chem.24:243;以及Freidinger,R.M.(1989)Trends Pharmacol.Sci.10:270。也参见Sawyer,T.K.(1995)″Peptidomimetic Design andChemical Approaches to Peptide Metabolism″in Taylor,M.D.和Amidon,G.L.(编辑)Peptide-Based Drug Design:Controlling Transport andMetabolism,Chapter 17;Smith,A.B.3rd等(1995)J.Am.Chem.Soc.117:11113-11123;Smith,A.B.3rd等(1994)J.Am.Chem.Soc.116:9947-9962;和Hirschman,R.等(1993)J.Am.Chem.Soc.115:12550-12568。
如本文所使用,本发明的肽类分子的“衍生物”是指其中在所述分子上的一个或多个反应基团已经用取代基团衍生化的肽类分子的一种形式。肽衍生物的实例包括其中氨基酸侧链、肽主链或氨基或羧基末端进行衍生化的肽(例如具有甲基化酰胺键的肽化合物)。如本文所使用,本发明的肽类分子的“类似物”是指保持所述分子的功能活性所需的所述分子的化学结构,但还包含不同于所述分子的某些化学结构的肽类分子。天然存在的肽的类似物的实例是包括一个或多个非天然存在的氨基酸的肽。如本文所使用,本发明的肽类分子的“模拟物”是指其中对于所述分子的功能活性所需的所述分子的化学结构已经被其它模拟所述分子的构象的化学结构替换的肽类分子。肽模拟物的实例包括其中肽主链被一个或多个苯并二氮 分子取代的肽化合物。(例如,见James,G.L.等(1993)Science 260:1937-1942)。
分子取代的肽化合物。(例如,见James,G.L.等(1993)Science 260:1937-1942)。
本发明的肽类分子的类似物意图包括其中肽结构的一个或多个L-或D-氨基酸被类似氨基酸置换而使得所述分子的性质得以保持的分子。优选地,在一个或多个氨基酸残基处进行保守的氨基酸置换。“保守的氨基酸置换”是其中氨基酸残基被具有相似侧链的氨基酸残基替换的置换。具有相似侧链的氨基酸残基族已经在本领域定义,包括碱性侧链(例如赖氨酸、精氨酸、组氨酸)、酸性侧链(例如天冬氨酸、谷氨酸)、不带电极性侧链(例如甘氨酸、天门冬酰胺、谷氨酰胺、丝氨酸、苏氨酸、酪氨酸、半胱氨酸)、非极性侧链(例如丙氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、甲硫氨酸、色氨酸)、β-分支侧链(例如苏氨酸、缬氨酸、异亮氨酸)和芳香族侧链(例如酪氨酸、苯丙氨酸、色氨酸、组氨酸)。可以在本发明的肽类分子的结构中进行的同系置换的非限制实例包括用D-酪氨酸、D-吡啶基丙氨酸或D-高苯丙氨酸置换D-苯丙氨酸,用D-缬氨酸或其它具有脂族侧链的天然或非天然氨基酸置换D-亮氨酸和/或用D-亮氨酸或其它具有脂族侧链的天然或非天然氨基酸置换D-缬氨酸。
术语模拟物,特别是肽模拟物,意图包括等排体。如本文所使用的,术语“等排体”意图包括,因为第一化学结构的立体构象适合对于第二结构特异的结合位点,可用来替换第二化学结构的化学结构。所述术语明确地包括本领域普通技术人员熟知的肽主链修饰(即酰胺键模拟物)。这样的修饰包括酰胺氮、α-碳、酰胺羰基、酰胺键的完全置换,延伸、缺失或主链交联的修饰。几种肽主链修饰是已知的,包括ψ[CH2S]、ψ[CH2NH]、ψ[CSNH2]、ψ[NHCO]、ψ[COCH2]和ψ[(E)或(Z)CH=CH]。在上文使用的命名中,ψ表示酰胺键不存在。替代酰胺基的结构在括号内具体说明。
其它可能的修饰包括N-烷基(或芳基)取代(ψ[CONR])、或主链交联以构建内酰胺及其它环状结构。本发明的调节化合物的其它衍生物包括C-末端羟甲基衍生物、O-修饰衍生物(例如C-末端羟甲基二甲苯醚)、N-末端修饰的衍生物,包括取代的酰胺如烷基酰胺和酰肼。
本发明的肽类分子可以通过本领域已知的标准方法进行制备。所述肽类分子,例如人Kit RTK的D4结构域或人VEGF受体的D7结构域,可以使用标准技术从人细胞克隆,插入重组载体,并在体外细胞系统中表达(例如,通过将所述载体转染入酵母细胞中)。可选地,可以设计并经由已知的合成法从头合成所述肽类分子,例如Atherton等(1989)Oxford,England:IRL Press.ISBN 0199630674;Stewart等(1984).第二版,Rockford:Pierce Chemical Company,91.ISBN0935940030;Merrifield(1963)J.Am.Chem.Soc.85:2149-2154。
然后,使用在本文描述的任何分析方法,例如在下文实施例部分描述的那些,测试所述肽类分子的功能活性。
IV.用于鉴定本发明的成分的筛选分析
可以使用在本文中描述的任何一种测定和本领域熟知的那些测定方法筛选本发明的成分的RTK抑制活性。例如,可测定受体内化作用、受体自磷酸化和/或激酶信号传导的分析方法可用来鉴定阻止目标RTK(例如Kit受体或人VEGF受体)的活化的成分。可以通过使用本领域已知的标准方法,例如通过使用phosphoELISATM方法(可在Invitrogen获得)来确定RTK或下游分子的磷酸化状态,而实现新的抑制剂成分的筛选。所述受体(例如Kit受体或VEGF受体)的磷酸化状态可以使用市场上可买到的试剂盒,例如C-Kit[pY823]ELISA KIT,HU(BioSourceTM;Catalog Number-KHO0401);c-KIT[TOTAL]ELISAKIT,HU(BioSourceTM;Catalog Number-KHO0391),加以测定。本发明的抗体、小分子和其它成分可以使用这样的试剂盒来测定它们的RTK抑制活性而进行筛选。例如,在用适当的配体和本发明的成分处理之后,可以进行phosphoELISATM以确定磷酸化状态,从而确定目标RTK的活化状态。本发明的成分可以被鉴定为阻止RTK活化的那些成分。下文的实施例15和16描述了包括使用抗磷酸酪氨酸抗体检测RTK活化的分析方法。下文的实施例20描述了使用phosphoELISATM系统检测受体活化的一种可能的分析方法。实施例22-25(包括与其相关的方法和介绍)进一步描述了本文使用的确定RTK的活化状态的方法。
因为受体活化可导致胞吞作用和受体内化作用,所以在一些实施方式中,通过测量本发明的成分阻止受体内化作用的能力而测定本发明的成分抑制目标RTK的能力是有用的。下文的实施例25(和与其相关的方法)描述了对PDGF受体突变体的内化和降解的测量。受体内化测定是本领域熟知的,并描述于如Fukunaga等(2006)LifeSciences.80(1).p.17-23;Bernhagen等(2007)Nature Medicine 13,587-596;natureprotocols.com/2007/04/18/receptor_internalization_assay.php),其中各文献的全部内容以引用的方式并入本文。测定受体内化的一个众所周知的方法是用荧光蛋白(例如绿色荧光蛋白(GFP))或其它适当的标记剂标记配体。在配体结合受体后,荧光显微术可用来使受体内化作用可视化。类似地,本发明的成分可以用标记剂进行标记,并且荧光显微术可用来使受体内化可视化。如果所述成分能够抑制所述受体的活性,则相比于适当的对照,在配体存在的情况下降低荧光的内化(例如仅可在其中所述成分与受体结合的细胞边缘而不是在内涵体或囊泡中观察到荧光)。
除了上述的这些之外,各种其它受体活化测定是本领域已知的,其任何一个可用于评价本发明的成分的功能。根据本发明可使得的进一步的受体活化测定被描述于美国专利6,287,784;6,025,145;5,599,681;5,766,863;5,891,650;5,914,237;7,056,685和许多科学出版物中,包括但不限于:Amir-Zaltsman等(2000)Luminescence15(6):377-80;Nakayama和Parandoosh(1999)Journal of ImmunologicalMethods.225(1-2),27,67-74;Pike等(1987)Methods of Enzymology146:353-362;Atienza等(2005)Journal of Biomolecular Screening.11(6):634-643;Hunter等(1982).Journal of Biological Chemistry257(9):4843-4848;White和Backer(1991)Methods in Enzymology 201:65-67;Madden等(1991)Anal Biochem 199:210-215;Cleaveland等(1990)Analytical Biochemistry 190:249-253;Lazaro等(1991)Analytical Biochemistry 192:257-261;Hunter和Cooper(1985)Ann RevBiochem 54:897-930;Ullrich和Schlessinger(1990)Cell 61:203-212;Knutson和Buck(1991)Archives of Biochemistry and Biophysics 285(2):197-204);King等(1993)Life Sciences 53:1465-1472;Wang.(1985)Molecular and Cellular Biology 5(12):3640-3643;Glenney等(1988)Journal of Immunological Methods 109:277-285;Kamps(1991)Methodsin Enzymology 201:101-110;Kozma等(1991)Methods in Enzymology201:28-43;Holmes等(1992)Science 256:1205-10;及Corfas等(1993)PNAS,USA 90:1624-1628。
通过配体结合的受体活化通常启动后续的细胞内事件,例如第二信使如IP3增加,第二信使再释放钙离子的细胞内储存。因此,受体活性可以通过测量第二信使,如IP3、环核苷酸、细胞内钙或磷酸化信号传导分子,如STAT、PI3K、Grb2或其它本领域已知的可能目标,的量加以测定。美国专利7,056,685描述并引用了几种根据本发明可用来检测受体活性的方法,其通过引用的方式并入本文。
上面描述的许多分析方法,如受体内化作用测定或受体活化测定,可包括使用免疫结合测定检测或定量目标RTK(例如当在受体内化作用测定期间使用放射性标记的抗体检测在细胞表面上的RTK的量时)。免疫结合测定在本领域中被广泛地描述(参见,例如美国专利4,366,241;4,376,110;4,517,288和4,837,168)。对于一般性免疫测定的综述,也参见Methods in Cell Biology:Antibodies in Cell Biology,volume 37(Asai,ed.1993);Basic and Clinical Immunology(Stites &Terr编辑,第七版1991)。
免疫分析,如可以用于受体内化作用研究、受体活化研究或受体检测分析的那些,经常采用标记剂来特异性结合和标记由检测抗体和RTK形成的复合物(见美国专利7,056,685,其通过引用并入本文)。标记剂本身可以是用于检测受体的抗体(这里所述的抗体可以是或不是本发明的成分)。可选地,标记剂可以是第三试剂,诸如第二或第三抗体(例如结合对于目标RTK特异性的小鼠单克隆抗体的抗小鼠抗体)。能够特异性结合免疫球蛋白恒定区的蛋白质,如A蛋白或G蛋白,在免疫结合分析中也可用作标记剂。这些蛋白质表现出强的与来自多个物种的免疫球蛋白恒定区的非免疫原性反应性(例如见Kronval等(1973),J.Immunol.111:1401-1406;Akerstrom等(1985),J.Immunol.135:25892542)。所述标记剂还可以用一个可检测试剂(如生物素)加以修饰,另一个分子(如链霉抗生物蛋白)可以特异性结合该可检测试剂。多种可检测成分为本领域普通技术人员所熟知。
通常使用的分析方法包括非竞争性分析(例如夹心式分析)以及竞争性分析。通常使用的分析形式包括蛋白质印迹(免疫印迹),其用来检测和定量蛋白质在样品中的存在。用于所述测定中的特定标记或可检测基团不是本发明的关键方面,只要它不显著地干扰用于检测RTK的免疫球蛋白或设计用来结合和灭活RTK的本发明的成分的特异性结合。可检测基团可以是具有可检测的物理或化学性质的任何材料。这样的可检测标记在免疫分析领域已经得到充分的发展,并且一般而言,大多数可用于这类方法的任何标记可以被用于本发明。因此,标记是可通过光谱学、光化学、生物化学、免疫化学、电学、光学或化学方法检测的任何组成。在本发明中有用的标记包括荧光染料(例如异硫氰酸荧光素、德克萨斯红、若丹明等等)、放射性标记(例如3H、125I、35S、14C或32P)、酶(例如辣根过氧化物酶、碱性磷酸酶及其他ELISA中常用的酶)和比色标记如胶体金或有色玻璃或塑料珠(例如聚苯乙烯、聚丙烯或胶乳)。
可以根据本领域熟知的方法将所述标记直接或间接地偶联到所述测定的期望成分上。所述标记还可以被直接结合到信号产生化合物上,例如通过与酶或荧光团结合。作为标记的目标酶主要是水解酶,特别是磷酸酶、酯酶和糖苷酶,或氧化酶(oxidotase),特别是过氧化物酶。荧光化合物包括荧光素及其衍生物、若丹明及其衍生物、丹磺酰、伞形酮等等。化学发光化合物包括萤光素和2,3-二氢邻苯二甲酰肼,例如鲁米诺。对于可使用的各种标记或信号产生系统的综述见美国专利4,391,904。
检测标记的方法是本领域普通技术人员所熟知的。因此,例如,在所述标记是放射性标记的情况下,检测手段包括闪烁计数器或如在放射自显影术中的摄影胶片。在标记是荧光标记时,可以通过用适当波长的光激发荧光团并检测所得到的荧光进行检测。荧光可以目测、利用摄影胶片、利用电子检测器(如电荷耦合器件(CCD)或光电倍增器等等)进行检测。类似地,可以通过提供酶的适当底物并检测所得到的反应产物检测酶标记。最后,简单的比色标记可以简单地通过观察与所述标记相关的颜色进行检测。因此,在各种浸量尺(dipstick)测定中,结合的金通常显粉色,而各种结合珠显现所述珠的颜色。
在本发明的进一步的方面,本发明的成分可结合在目标RTK上的表位上并仍允许受体酪氨酸激酶的胞外域二聚化。在该实施方式中,所述成分的结合可影响两个单体的Ig样结构域(例如,III型受体酪氨酸激酶的D4-D4或D5-D5结构域或者V型受体酪氨酸激酶的D7-D7结构域)之间的定位、取向和/或距离,从而抑制受体酪氨酸激酶的活性。换句话说,所述成分可允许配体诱导的受体酪氨酸激酶胞外域的二聚化,但影响两个胞外域在细胞表面界面的定位或者改变或阻止受体酪氨酸激酶的构象变化,从而抑制受体酪氨酸激酶的活性(例如,抑制受体内化作用和/或抑制所述受体的酪氨酸自磷酸化和/或抑制所述受体激活下游信号传导通路的能力)。
因此,在一些实施方式中,使用能够鉴定允许受体二聚化但仍使受体无活性的成分的测定方法是有用的。这样的测定方法在下文中描述。例如,实施例18描述了采用PDGF受体进行的实验,由此使用交联检测受体二聚化,并使用磷酸酪氨酸特异性抗体测定受体活化。而且,实施例23表明PDGFR的突变体对配体诱导的酪氨酸自磷酸化有影响,这不是由配体诱导的受体二聚化中的缺陷引起的(也参见实施例22-25的方法和介绍)。
通过荧光能量共振转移(FRET)分析也可以确定RTK的构象状态。关于确定蛋白质构象和相互作用的荧光方法的综述可以发现于Johnson(2005)Traffic.2005Dec;6(12):1078-92,其通过引用并入本文。在FRET测定中,用合适的FRET荧光团标记目标RTK。在标记RTK之后,表达标记的RTK的细胞与本发明的测试成分以及RTK的配体(例如,对于Kit RTK是SCF)一起温育。FRET分析将允许观察在RTK中与配体结合、RTK二聚化和/或受体活化相关的构象变化。通过该方法,本领域普通技术人员可直接评价表明RTK二聚化而无下游活化的蛋白质构象变化。存在许多可用来进行FRET分析的方法,并且大部分的不同源于使用不同的荧光团或将荧光团掺入目标蛋白质的不同技术。FRET荧光团和分析方法在本领域中是熟知的,并且FRET技术的简述可获自Heyduk.(2002)Current Opinion inBiotechnology.13(4).292-296及其中的参考文献。下列出版物详述FRET方法并通过引用的方式并入本文:Kajihara等(2006)NatMethods.3(11):923-9;Biener-Ramanujan等(2006)Growth Horm IGFRes.16(4):247-57;Taniguchi等(2007)Biochemistry.46(18):5349-57;美国专利No.6,689,574;5,891,646和WIPO公开WO/2002/033102。FRET荧光团可以被掺入RTK的任何结构域或铰链区以检测构象变化(例如,III型RTK的D4或D5结构域或者V型RTK的D7结构域),条件是所述荧光团不干扰RTK的功能或本发明的成分结合RTK的能力。
可用于FRET的荧光团通常与可用于如下文讨论的生物发光能量共振转移(BRET)的那些荧光团相同。最常用的FRET方法是将反应性半胱氨酸残基工程化到目标蛋白质中。然后,荧光团可以容易地与选择的半胱氨酸残基反应。通常,构建融合蛋白质,由此将目标蛋白质与绿色荧光蛋白融合(见Neininger等(2001)EMBO Reports.2(8):703-708)。另外的方法和可用于FRET的荧光团被描述于Huebsch和Mooney(2007)Biomaterials.28(15):2424-37;Schmid和Birbach(2007)Thromb Haemost.97(3):378-84;Jares-Erijman和Jovin(2006)Curr Opin Chem Biol.10(5):409-16;Johansson(2006)Methods Mol Biol.335:17-29;Wallrabe和Periasamy(2005)Curr Opin Biotechnol.16(1):19-27;以及Clegg RM(1995)Curr Opin Biotechnol.6(1):103-10,其通过引用并入本文。
在其它实施方式中,可能不知道或难以确定(取决于受体)是哪种RTK构象特别地表示在无活化的情况下的二聚化。在这类情况下,本领域普通技术人员可将确定受体二聚化的测定方法和确定受体活化的测定方法结合。例如,可结合上述讨论到的任何受体活化测定方法使用传统的交联研究(由Rodriguez等(1990)MolecularEndocrinology,4(12),1782-1790示例)检测RTK二聚化。FRET和相似系统也可用于直接测量受体活化或二聚化。例如,通过将适当的FRET荧光团掺入RTK的胞质域和掺入磷酸化目标蛋白(即下游信号传导分子),FRET将能够确定下游信号传导分子是否正被RTK募集。因此,在一个实施方式中,成功的本发明的成分是允许受体二聚化(如通过交联或FRET所测量的),但阻止受体活化(通过FRET或BRET分析或通过其它受体活化测定(例如使用抗磷酸酪氨酸抗体的自磷酸化测定和蛋白质印迹)检测为不存在荧光)的成分。因此,使用在本文描述的技术,本领域普通技术人员可以容易地测试成分(例如小分子、肽或抗体),以确定它们是否抑制RTK活性和它们是否允许受体二聚化。
特别地,生物发光能量共振转移(BRET)分析可用来鉴定抑制RTK活性的成分。美国专利公开20060199226、WIPO公开WO/2006/094073和Tan等(2007.Molecular Pharmacology.72:1440-1446)具体描述鉴定活化RTK的配体的方法,并因此通过引用的方式并入本文。这些技术已经被用于确定体外和体内蛋白质相互作用(Pfleger等(2006)Nature Protocols 1 337-345;Kroeger等(2001),J.Biol.Chem.,276(16):12736-43;和Harikumar等(2004)Mol Pharmacol 65:28-35;其通过引用的方式全部并入本文)。
通过筛选阻止RTK活化的那些成分,BRET可用于从测试化合物中鉴定本发明的成分。
如在通过引用并入本文的美国专利公开2006/0199226中讨论的,基于BRET的测定可用于监测具有生物发光供体分子(DM)的蛋白质与具有荧光受体部分(AM)的蛋白质的相互作用。简言之,表达RTK-DM融合体的细胞将底物的化学能转化为光。如果存在非常接近RTK-DM融合体的AM(例如,信号传导蛋白质-AM融合体),则细胞将在发现特定波长的光。例如,基于BRET的测定可用于评价在RTK-荧光素酶融合体和GFP-信号传导蛋白融合体之间的相互作用。这稍微不同于FRET分析(其中供体分子可通过特定波长的光激发而不是通过化学能转化)。可用于BRET分析的具有荧光素酶活性的生物发光蛋白质的实例可以在美国专利5,229,285、5,219,737、5,843,746、5,196,524、5,670,356中找到。可选的DM包括酶,其可以作用于适当的底物以产生荧光信号。这样的酶的具体实例是β-半乳糖苷酶、碱性磷酸酶、β-葡糖苷酸酶和β-葡糖苷酶。这些酶的合成发光底物是本领域所熟知的,并且可从公司如Tropix Inc.(Bedford,Mass.,USA)商购获得。DM也可以从昆虫分离或进行工程化(美国专利5,670,356)。
取决于底物,DM发射不同的波长的光。DM底物的非限制性实例包括肠腔素(coelenterazine)、苯并噻唑、萤光素、烯醇甲酸酯(enolformate)、萜和醛等等。DM部分可以被直接与RTK蛋白质的氨基末端或羧基末端部分融合。优选地,BDM结构域在RTK-DM融合体内的位置不改变天然蛋白质的活性或本发明的成分的结合。RTK-DM融合蛋白质可以被检测,以确保它保持生化特性,如配体结合和与所述天然蛋白质的下游信号传导分子相互作用的能力。
在BRET分析中AM可以再发射荧光形式的转移能量。AM的实例包括绿色荧光蛋白(GFP),或其同工型和衍生物,诸如YFP、EGFP、EYFP等等(R.Y.Tsien,(1998)Ann.Rev.Biochem.63:509-544)。优选地,AM结构域在AM-蛋白质融合体内的位置不改变所述天然蛋白质的活性。AM-第二蛋白质融合蛋白可以被检测以确保它保持同源天然蛋白质的生化特性,诸如与RTK的相互作用。例如,可以使用GFP蛋白质与通过目标RTK磷酸化或可以结合目标RTK的任何底物的氨基末端融合体。
V.包含本发明的成分的药物组合物
在另一个方面,本发明提供包含与药学上可接受的载体一起配制的本发明的成分(例如单克隆抗体或其抗原结合部分、抗体模拟物、小分子或本发明的肽类分子)之一或组合的组合物例如药物组合物。这样的组合物可包括一种或组合的(例如,两种或多种)本发明的抗体、或者免疫缀合物、小分子或肽类分子。例如,本发明的药物组合物可以包含结合在靶RTK上不同的表位或者具有互补的活性的抗体和小分子的组合,例如结合III型RTK的D3-D4铰链区的小分子与结合III型RTK的D4结构域的单克隆抗体组合。
本发明的药物组合物还可以联合治疗施用,即与其它药剂结合。例如,联合治疗可以包括与至少一种其它抗癌剂联合的本发明的抗RTK抗体(或者小分子或肽类分子)。可被用于联合治疗的治疗剂的实例在下文更详细地描述。
如本文所使用的,“药学上可接受的载体”包括生理上相容的任何和所有溶剂、分散介质、包衣、抗菌剂和抗真菌剂、等渗剂和吸收延迟剂等等。优选地,所述载体适于静脉内、肌内、皮下、非肠道、脊柱或表皮施用(例如通过注射或输注)。取决于给药途径,活性化合物,即本发明的成分,可以包被在材料中以防止所述化合物免受酸和其它可能灭活所述化合物的自然条件的作用。
本发明的药物化合物可包括一种或多种药学上可接受的盐。“药学上可接受的盐”是指保持母体化合物的期望的生物活性而不施加任何不想要的毒理效应的盐(例如见Berge,S.M.等(1977)J.Pharm.Sci.66:1-19)。这样的盐的实例包括酸加成盐和碱加成盐。酸加成盐包括源于非毒性无机酸如盐酸、硝酸、磷酸、硫酸、氢溴酸、氢碘酸、亚磷酸等等的那些盐,以及源自非毒性有机酸如脂族单羧酸和二羧酸、苯基取代的链烷酸、羟基链烷酸、芳香酸、脂族和芳族磺酸等等的那些盐。碱加成盐包括源于碱土金属如钠、钾、镁、钙等等的那些盐以及源自非毒性有机胺如N,N′-二苄基乙二胺、N-甲基葡糖胺、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、普鲁卡因等等的那些盐。
本发明的药物组合物还可包括药学上可接受的抗氧化剂。药学上可接受的抗氧化剂的实例包括:(1)水溶性抗氧化剂,例如抗坏血酸、盐酸半胱氨酸、硫酸氢钠、偏亚硫酸氢钠、亚硫酸钠等等;(2)油溶性抗氧化剂,例如抗坏血酸棕榈酸酯、丁羟茴醚(BHA)、丁羟甲苯(BHT)、卵磷脂、没食子酸丙酯、α-生育酚等等;和(3)金属螯合剂,例如柠檬酸、乙二胺四乙酸(EDTA)、山梨醇、酒石酸、磷酸等等。
可用于本发明的药物组合物中的适当的水性载体和非水性载体的实例包括水、乙醇、多元醇(例如甘油、丙二醇、聚乙二醇等等)和其适当的混合物、植物油如橄榄油及可注射的有机酯如油酸乙酯。例如,可以通过利用包衣材料(如卵磷脂)、在分散剂的情况下通过维持所需要的颗粒大小和通过利用表面活性剂维持适当的流动性。
这些组合物还可包含辅助剂如防腐剂、润湿剂、乳化剂和分散剂。通过灭菌方法和通过包含各种抗菌剂和抗真菌剂(例如对羟苯甲酸酯、氯代丁醇、苯酚、山梨酸等等)可以确保防止微生物的存在。将等渗剂如糖、氯化钠等等包括入所述组合物也可能是期望的。此外,通过包含延迟吸收的物质如单硬脂酸铝和明胶,可以引起注射药物形式的吸收延长。
药学上可接受的载体包括无菌水溶液或分散液和用于临时制备无菌注射溶液或分散液的无菌粉末。用于药学上活性物质的这类介质和试剂在本领域中是已知的。除非任何常规介质或试剂与活性化合物不相容,可以考虑任何常规介质或试剂在本发明的药物组合物中的使用。补充活性化合物也可以被加入所述组合物中。
药物组合物通常必须是无菌的并且在制造和储存的条件下是稳定的。所述组合物可以被配制为溶液、微乳剂、脂质体或其它适于高药物浓度的有序结构。所述载体可以是溶剂或分散介质,其包含例如水、乙醇、多元醇(例如甘油、丙二醇和液体聚乙二醇等等)及其适当的混合物。例如,通过利用包衣(如卵磷脂)、在分散剂的情况下通过维持所需要的颗粒大小和通过利用表面活性剂可以维持适当的流动性。在很多情况下,优选在所述组合物中包括等渗剂例如糖、多元醇(如甘露糖醇、山梨醇)或氯化钠。通过将延迟吸收的物质(例如单硬脂酸盐和明胶)包括在所述组合物内可以引起可注射组合物的吸收延长。
可以通过将活性化合物以需要的量与按照需要的上文列举的组分之一或其组合一起加入适当的溶剂中,然后无菌微过滤制备无菌注射溶液。通常,可以通过将所述活性化合物加入包含基本分散介质和来自上文列举的物质的其它需要的组分的无菌载体中制备分散液。在用于制备无菌注射液的无菌粉末的情况中,优选的制备方法是真空干燥和冷冻干燥(冻干),其从先前无菌过滤的溶液产生活性成分加任何附加的需要成分的粉末。
可以与载体物质结合以产生单一剂型的活性成分的量取决于所治疗的对象和特定施用模式。可以与载体结合以产生单一剂型的活性成分的量一般是产生治疗效应的组合物的量。通常,以100%算,该量的范围为约0.01%至约99%的活性成分,优选从约0.1%至约70%,最优选约1%至约30%的活性成分与药学上可接受的载体组合。
调节给药方案以提供最佳的期望反应(例如治疗反应)。例如,可以施用单一大丸剂,可以随着时间施用几个分开的剂量,或者可以根据治疗情况的紧迫性而按比例减少或增加所述剂量。由于便于施用和剂量的均匀性,将非肠道组合物配制成单位剂型是特别有利的。如本文所使用,单位剂型是指适合作为用于待治疗对象的单一剂量的物理上分散的单位;各单元包含与需要的药物载体结合的适于产生期望治疗效应的预定量的活性化合物。本发明的单位剂型的规格由以下因素规定并直接取决于以下因素:(a)活性化合物的独特性质和待实现的特定治疗效应,和(b)在配制这类活性化合物用于治疗个体敏感性的领域中固有的限制。
对于所述抗体、小分子或肽类分子的施用,剂量范围为宿主体重的约0.0001至100mg/kg,并更通常是0.01至5mg/kg。例如,剂量可以是0.3mg/kg体重、1mg/kg体重、3mg/kg体重、5mg/kg体重或10mg/kg体重或在1-10mg/kg的范围内。示例性的治疗方案要求每周施用一次、每两周一次、每三周一次、每四周一次、一月一次、每3个月一次或每三至6个月一次。对于本发明的成分,优选的给药方案包括经由静脉内施用,1mg/kg体重或3mg/kg体重,其中使用下列给药时间表之一给予所述抗体:(i)每四周给予六个剂量,然后每三个月;(ii)每三周;(iii)3mg/kg体重一次,然后每三周1mg/kg体重。
可选地,所述抗体、小分子或肽类分子可以作为缓释制剂进行施用,在这种情况下需要较低频度的施用。剂量和频率随所施用的物质在患者中的半衰期而变化。通常,人抗体显示出最长的半衰期,接下来是人源化抗体、嵌合抗体和非人类抗体。施用的剂量和频率可以取决于治疗是预防性或者是治疗性的。在预防性应用中,以相对不频繁的间隔在长时间内施用以相对低的剂量施用。一些患者在他们的余生中持续接受治疗。在治疗性应用中,有时要求相对短的时间间隔和相对高的剂量,直到疾病的进展被减缓或终止,并优选直到患者显示出疾病症状的部分或完全改善。其后,可以给予患者预防性方案。
活性成分和小分子在本发明的药物组合物中的实际剂量水平可以有所变化,以便获得对特定患者、组成和施用模式有效实现期望治疗响应而对所述患者无毒性的活性成分的量。所选择的剂量水平取决于各种药代动力学因素,包括所使用的本发明的特定组合、或者其酯、盐或酰胺的活性、给药途径、施用时间、使用的特定化合物的排泄速率、治疗持续时间、与所使用的特定组合联合使用的其它药物、化合物和/或材料、待治疗患者的年龄、性别、体重、病症、总体健康状况和在先病史、以及医学领域内熟知的类似因素。
本发明的抗RTK成分的“治疗有效剂量”优选引起病症严重程度的降低、无疾病症状期间的频率和持续时间增加或者防止由于疾病折磨而导致损伤或残疾。例如,对于肿瘤的治疗,相对于未治疗的对象,“治疗有效剂量”优选抑制细胞生长或肿瘤生长至少约20%、更优选至少约40%、甚至更优选至少约60%和更优选至少约80%。化合物抑制肿瘤生长的能力可以在预测在人肿瘤中的功效的动物模型中进行评价。可选地,可以通过用普通技术人员已知的测定方法检查化合物的抑制能力,如这类体外抑制,评价组合物的这一性质。治疗有效量的治疗化合物可以降低肿瘤大小,或者以其它方式改善对象的症状。基于如对象身材、对象症状的严重程度和所选择的特定组合或给药途径等因素,本领域普通技术人员能够确定这样的量。
本发明的抗RTK成分可以被测试以确定其是否有效拮抗RTK。测试抗RTK成分的一个方法是证实在抗RTK成分和RTK之间存在相互作用。例如,本领域普通技术人员可测试本发明的抗体、小分子或肽类分子是否结合人Kit RTK的D4或D5结构域或VEGF受体的D7结构域。这样的对结合的测试是本领域熟知的并且可包括标记(例如放射性标记)抗RTK成分,在结合可发生的条件下温育抗RTK成分和RTK,然后在凝胶或荧光屏上分离/显像复合物。类似地,ELISA技术可用来测定结合。
确定本发明的成分是否拮抗RTK的另一种方法是检测RTK的胞质域的磷酸化状态。在具体实施方式中,有效拮抗剂将阻止RTK的活化和自磷酸化。使用本领域已知的标准方法例如通过使用特异性结合RTK的磷酸化残基的抗体,可以检测RTK的磷酸化。检测磷酸化事件的其它方法包括描述于美国专利6548266;或Goshe等(2006)Brief Funct Genomic Proteomic.4:363-76;de Graauw等(2006)Electrophoresis.27:2676-86;Schmidt等(2007)J Chromatogr B AnalytTechnol Biomed Life Sci.849:154-62中的方法;或利用用于PerkinElmer的采用[γ-33P]ATP的激酶磷酸化分析的FlashPlates(SMP200)方案。本领域普通技术人员将理解,这些方法以及在实施例中示范的那些方法,也可用来测定通过RTK磷酸化并且作为在所述细胞内的信号传导物的蛋白质的磷酸化状态。检测这样的蛋白质的磷酸化状态还表明RTK是否已经被本发明的成分有效地拮抗。
可以使用本领域已知的各种方法的一种或多种,经由一种或多种给药途径施用本发明的组合物。如普通技术人员理解的,施用路径和/或模式将取决于期望的结果。本发明的结合成分的优选施用途径包括静脉内、肌内、真皮内、腹膜内、皮下、脊柱或其它非肠道施用途径,例如通过注射或输注。如本文所使用的,短语“非肠道施用”是指除了肠内和局部施用之外的施用模式,通常通过注射进行,并且非限制性地包括静脉内、肌内、动脉内、鞘内、囊内、眶内、心内、真皮内、腹膜内、经气管、皮下、表皮下、关节内、囊下、蛛网膜下、脊柱内、硬膜外和胸骨内注射和输注。
可选地,本发明的抗RTK结合成分可以经由诸如局部、表皮或黏膜给药途径的非肠道途径施用,例如鼻内、口腔、阴道、直肠、舌下或局部给药。
可以用防止活性化合物快速释放的载体制备所述活性化合物,诸如控释制剂,包括植入物、经皮贴剂和微胶囊输送系统。可使用可生物降解的生物相容性聚合物,如乙烯乙酸乙烯酯、聚酐、聚乙醇酸、胶原、聚原酸酯和聚乳酸。制备这样的制剂的许多方法被授予专利或通常是本领域普通技术人员已知的。参见例如Sustained andControlled Release Drug Delivery Systems,J.R.Robinson,ed.,MarcelDekker,Inc.,New York,1978。
可以用本领域已知的医疗器械施用药物组合物。例如,在优选的实施方式中,本发明的药物组合物可以用无针皮下注射装置诸如在美国专利5,399,163、5,383,851、5,312,335、5,064,413、4,941,880、4,790,824或4,596,556中公开的装置进行施用。可用于本发明的公知的植入物和模块的实例包括:美国专利4,487,603,其披露了用于以控制的速率分配药物的可植入微输液泵;美国专利4,486,194,其披露了用于经皮肤施用药物的治疗装置;美国专利4,447,233,其披露了用于以精确的输注速率输送药物的药物输液泵;美国专利4,447,224,其披露了用于连续输送药物的可变流植入式输注装置;美国专利4,439,196,其披露了具有多腔隔室的蠕动药物输送系统;和美国专利4,475,196,其披露蠕动药物输送系统。这些专利通过引用的方式并入本文。许多其它这样的植入物、输送系统和模块是本领域普通技术人员已知的。
V.使用本发明的成分的方法
在另一个方面,本发明提供治疗对象的RTK相关疾病的方法,包括向所述对象施用治疗有效量的本发明的成分。本发明的抗RTK成分例如抗体、小分子或肽类分子具有许多涉及受体酪氨酸激酶相关疾病的诊断和治疗的体外和体内诊断和治疗用途。本发明的结合成分可以被施用于在培养中、体外或活体外的细胞,或者施用于人类对象(例如体内施用)以治疗、预防和诊断受体酪氨酸激酶相关疾病。
如本文所使用的,“受体酪氨酸激酶相关疾病”是由RTK活性介导或与异常RTK表达或活化有关的疾病或病症。受体酪氨酸激酶相关疾病的实例包括与例如FGF受体、HGF受体、胰岛素受体、IGF-1受体、NGF受体、VEGF受体、PDGF受体-α、PDGF受体-β、CSF-1受体和Flt3受体有关的疾病或病症,如年龄相关性黄斑变性(AMD)、动脉粥样硬化、类风湿性关节炎、糖尿病性视网膜病或疼痛相关疾病。受体酪氨酸激酶相关疾病的具体实例包括但不限于胃肠间质瘤(GIST)、急性骨髓性白血病(AML)、小细胞性肺癌(SCLC)、乳腺癌、骨转移乳腺癌、淋巴病和腱鞘巨细胞瘤。受体酪氨酸激酶相关疾病的其它实例包括结肠癌(包括小肠癌)、肺癌、乳腺癌、胰腺癌、黑素瘤(例如转移性恶性黑色素瘤)、急性骨髓性白血病、肾癌、膀胱癌、卵巢癌和前列腺癌。可以使用本发明方法治疗的其它癌的实例包括肾癌(例如肾细胞癌)、胶质母细胞瘤、淋巴病、脑肿瘤、慢性或急性白血病包括急性淋巴细胞性白血病(ALL)、成人T细胞性白血病(T-ALL)、慢性粒细胞性白血病、急性淋巴母细胞性白血病、慢性淋巴细胞性白血病、淋巴瘤(例如霍奇金和非霍奇金淋巴瘤、淋巴细胞性淋巴瘤、原发性CNS淋巴瘤、T细胞淋巴瘤、伯基特氏肿瘤、间变大细胞性淋巴瘤(ALCL)、皮肤T细胞淋巴瘤、结节性小裂细胞性淋巴瘤(nodularsmall cleaved-cell lymphomas)、外周T细胞淋巴瘤、伦纳特淋巴瘤、免疫母细胞性淋巴瘤、T细胞白血病/淋巴瘤(ATLL)、中心母细胞(entroblastic)/中心细胞性(cb/cc)滤泡性淋巴瘤癌、弥散性B系大细胞淋巴瘤、血管免疫母细胞性淋巴结病(AILD)样T细胞淋巴瘤和HIV相关体腔基淋巴瘤)、胚胎性癌、鼻咽的未分化性癌(例如Schmincke氏肿瘤)、Castleman氏病、卡波济氏肉瘤、多发性骨髓瘤、Waldenstrom氏巨球蛋白血症及其他B细胞淋巴瘤、鼻咽癌、骨癌、皮肤癌、头颈癌、皮肤或眼内恶性黑色素瘤、子宫癌、直肠癌、肛区癌、胃癌、睾丸癌、子宫癌、输卵管癌、子宫内膜癌、宫颈癌、阴道癌、外阴癌、食管癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、尿道癌、阴茎癌、儿童期实体瘤、膀胱癌、肾或输尿管癌、肾盂癌、中枢神经系统(CNS)的肿瘤、肿瘤血管发生、脊柱轴(spinalaxis)肿瘤、脑干神经胶质瘤、垂体腺瘤、表皮样癌、鳞状上皮细胞癌、环境诱导型癌包括由石棉诱导的那些癌(例如间皮瘤)和所述癌的组合。可以使用本发明的方法治疗的淋巴病或“淋巴系统的疾病”包括纤维蛋白原缺乏血症、贫血症、再生障碍性贫血、溶血性贫血、先天性非球形红细胞性贫血、巨红细胞性贫血、恶性贫血、镰状细胞贫血、肾性贫血、伴有嗜酸粒细胞增多的血管淋巴样增生、抗凝血酶III缺乏、Bernard-Soulier综合征、凝血障碍、蓝色橡皮疱样痣综合征、Chediak-Higashi综合征、冷球蛋白血症、弥散性血管内凝血、嗜酸粒细胞增多、Erdheim-Chester病、新生儿幼红细胞增多病、伊文思综合征、因子V缺乏、因子VII缺乏、因子X缺乏、因子XI缺乏、因子XII缺乏、范科尼贫血、大淋巴结增生、血液病、血红蛋白病、阵发性血红蛋白尿、血友病A、血友病B、新生儿出血性疾病、组织细胞增多病、朗格汉斯细胞组织细胞增多病、非朗格汉斯细胞组织细胞增多病、乔布综合征、白细胞减少症、淋巴腺炎、淋巴管平滑肌增多症、淋巴水肿、高铁血红蛋白症、骨髓增生异常综合征、骨髓纤维变性、骨髓组织异生、骨髓增生性疾病、嗜中性白细胞减少症、病变蛋白血症、血小板贮存池缺陷(platelet storage pool deficiency)、真性红细胞增多、蛋白C缺乏、蛋白S缺乏、血小板减少性紫癜、血栓性血小板减少性紫癜、RH-同种免疫、肉样瘤病、肉样瘤病、球形红细胞增多症、脾破裂、地中海贫血、血小板机能不全、血小板减少症、Waldenstrom氏巨球蛋白血症或Von Willebrand病。
而且,假设III型或V型RTK在各种肿瘤细胞上表达,本发明的结合成分、组合物和方法可用于治疗患有致瘤障碍(例如以存在表达Kit的肿瘤细胞为特征的障碍,例如胃肠间质瘤、肥大细胞病和急性骨髓性白血病)的对象。其他的患有致瘤障碍的对象的实例包括患有肾癌(例如肾细胞癌)、胶质母细胞瘤、脑肿瘤、慢性或急性白血病(包括急性淋巴细胞性白血病(ALL)、成人T细胞性白血病(T-ALL)、慢性粒细胞性白血病、急性淋巴母细胞性白血病、慢性淋巴细胞性白血病)、淋巴瘤(例如霍奇金和非霍奇金淋巴瘤、淋巴细胞性淋巴瘤、原发性CNS淋巴瘤、T细胞淋巴瘤、伯基特氏肿瘤、间变大细胞性淋巴瘤(ALCL)、皮肤T细胞淋巴瘤、结节性小裂细胞性淋巴瘤、外周T细胞淋巴瘤、伦纳特淋巴瘤、免疫母细胞性淋巴瘤、T细胞白血病/淋巴瘤(ATLL)、中心母细胞/中心细胞性(cb/cc)滤泡性淋巴瘤癌、弥散性B系大细胞淋巴瘤、血管免疫母细胞性淋巴结病(AILD)样T细胞淋巴瘤和HIV相关体腔基淋巴瘤)、胚胎性癌、鼻咽的未分化性癌(例如Schmincke氏肿瘤)、Castleman氏病、卡波济氏肉瘤、多发性骨髓瘤、Waldenstrom氏巨球蛋白血症及其他B细胞淋巴瘤、鼻咽癌、骨癌、皮肤癌、头颈癌、皮肤或眼内恶性黑色素瘤、子宫癌、直肠癌、肛区癌、胃癌、睾丸癌、子宫癌、输卵管癌、子宫内膜癌、宫颈癌、阴道癌、外阴癌、食管癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、尿道癌、阴茎癌、儿童期实体瘤、膀胱癌、肾或输尿管癌、肾盂癌、中枢神经系统(CNS)的肿瘤、肿瘤血管发生、脊柱轴肿瘤、脑干神经胶质瘤、垂体腺瘤、表皮样癌、鳞状上皮细胞癌、环境诱导型癌包括由石棉诱导的那些癌(例如间皮瘤)和所述癌的组合的对象。
如本文所使用的,术语“对象”意图包括人和非人动物。非人动物包括所有脊椎动物例如哺乳动物和非哺乳动物,诸如非人灵长类、绵羊、狗、猫、牛、马、鸡、两栖动物和爬行动物。优选的对象包括患有受体酪氨酸激酶相关疾病的人类对象。
本发明的成分(例如抗体、其抗原结合部分、小分子、肽类分子、抗体模拟物和组合物)在RTK相关疾病的治疗和诊断中具有用途。例如,人单克隆抗体、多特异性或双特异性分子、小分子或肽类分子可用于在体内或体外引发一种或多种下列生物学活性:抑制表达RTK(例如Kit、VEGF受体或PDGFR)的细胞的生长和/或杀死所述细胞;在人效应细胞存在的情况下介导表达RTK(例如Kit、VEGF受体或PDGFR)的细胞的吞噬ADCC或;或将RTK(例如RTK III型或V型家族成员)的胞外域锁定至无活性状态和/或单体状态,从而拮抗所述受体的活性。
体内和体外施用本发明的抗RTK成分的适当路径是本领域所熟知的,并且可以由普通技术人员加以选择。例如,可以通过注射(例如静脉内或皮下注射)施用所述抗RTK成分。所使用分子的适当剂量将取决于对象的年龄和体重以及结合成分组合物的浓度和/或配制。
如先前描述的,本发明的抗RTK成分可以与一种或其它更多种治疗剂(例如细胞毒性剂、放射毒性剂或免疫抑制剂)共同施用。所述成分可以与所述药剂连接或可以与所述药剂分开施用。在后一种情况(分开施用)下,所述结合成分可以在所述剂之前、之后或同时施用,或者可以与其它已知的治疗例如抗癌治疗(例如放射)共同施用。这样的治疗剂特别包括抗肿瘤剂如多柔比星(阿霉素)、顺铂、博莱霉素硫酸盐、卡莫司汀、苯丁酸氮芥和环磷酰胺羟基脲等,它们本身仅仅在对患者具有毒性或亚毒性的水平下才有效。顺铂以100mg/剂,每四周一次进行静脉施用,而阿霉素以60-75mg/ml剂量,每21天一次进行静脉施用。本发明的抗RTK结合成分与化疗剂的共同施用提供经由对人肿瘤细胞产生细胞毒性效应的不同机理起作用的两种抗癌药。这样的共同施用可以解决由于抗药性的产生或肿瘤细胞的抗原性变化(其将使它们不与所述结合成分起反应)带来的问题。
当施用本发明的抗RTK成分-伴体分子结合物预防和/或治疗与异常细胞增殖有关的疾病时,可以使用约0.001μM至20μM或约0.01μM至5μM的施用化合物循环浓度。
对于患者经口施用在本文描述的化合物,通常剂量范围为约1mg/日至约10,000mg/日,更通常从约10mg/日至约1,000mg/日,以及最通常从约50mg/日至约500mg/日。从患者体重方面来说,典型的剂量范围为约0.01至约150mg/kg/日,更通常从约0.1至约15mg/kg/日,以及最通常从约1至约10mg/kg/日,例如5mg/kg/日或3mg/kg/日。
在至少一些实施方式中,延迟或抑制肿瘤生长的患者剂量可以是1μmol/kg/日或更低。例如,患者剂量可以是0.9、0.6、0.5、0.45、0.3、0.2、0.15或0.1μmol/kg/日或更低(指药物的摩尔数)。优选地,当以每日剂量在至少五天的期间内施用时,抗RTK成分-药物结合物延迟肿瘤的生长。
在一个实施方式中,本发明的结合物可通过将这类化合物连接至抗RTK结合成分而用于将化合物(例如治疗剂、标记、细胞毒素、放射性毒素、免疫抑制剂等等)靶向于具有RTK细胞表面受体的细胞。例如,抗RTK成分可以被结合至在美国专利6,281,354和6,548,530、美国专利公开20030050331、20030064984、20030073852和20040087497中描述的或者公布于WO 03/022806中的毒素化合物的任何一种,这些文献通过引用的方式以全部内容由此并入。因此,本发明还提供活体外或体内定位表达RTK的细胞的方法(例如利用可检测标记,如放射性同位素、荧光化合物、酶或酶辅因子)。
靶特异性效应细胞,例如连接至本发明的组分(例如抗体、其抗原结合部分、小分子或肽类分子)的效应细胞,也可以用作治疗剂。用于靶向的效应细胞可以是人白细胞,如巨噬细胞、嗜中性白细胞或单核细胞。其它细胞包括嗜酸性粒细胞、天然杀伤细胞及其他IgG或IgA受体携带细胞。如果需要,效应细胞可以从待治疗的对象获得。靶特异性效应细胞可以作为在生理上可接受的溶液中的细胞悬液进行施用。施用的细胞数目可以在108-109的数量级,但将随治疗目的而变化。通常,所述量将足以获得在靶细胞(例如表达RTK的肿瘤细胞)的定位并且通过例如吞噬作用实现细胞杀伤。施用途径也可以变化。
用靶特异性效应细胞治疗可以连同其它去除靶细胞的技术一起进行。例如,使用本发明的成分和/或装备有这些组分的效应细胞进行的抗肿瘤治疗可与化疗一起使用。
本发明进一步提供检测样品中人RTK抗原的存在或者测量人RTK抗原(例如人Kit RTK、人VEGF受体或PDGFR的Ig样结构域)的量的方法,包括使所述样品和对照样品与RTK结合成分(例如人单克隆抗体)或特异性结合人RTK的其它结合成分在允许所述抗体或其它成分与人RTK(如Kit或人VEGF受体)之间形成复合物的条件下接触。然后,检测复合物的形成,其中在样品之间相比于对照样品的复合物形成差异指示RTK(例如人Kit RTK、人VEGF受体或PDGFRRTK)在所述样品中的存在。
包含抗RTK结合成分(例如,抗体、其抗原结合部分、小分子或肽类分子)和使用说明的试剂盒也在本发明范围内。所述试剂盒可进一步包含一种或多个另外的试剂(如免疫抑制剂、细胞毒性剂或放射性毒剂)或者一种或多种另外的本发明的抗RTK成分(例如具有结合与第一抗RTK成分不同的RTK抗原中的表位的互补活性的抗RTK结合成分)。通常,试剂盒包括指明所述试剂盒的内含物的指定用途的标签。术语标签包括任何文字或者在所述试剂盒上,或者与所述试剂盒一起提供的记录材料,或者以另外的方式与所述试剂盒结合的记录材料。
本发明通过下列实施例进一步阐述,其不应该被解释为进一步限制。全部附图的内容和本申请通篇引用的全部参考文献、专利和公开的专利申请以及附图通过引用的方式以它们的全部内容明确并入本文。
实施例
实施例1-19的引言
干细胞因子(SCF)是一种通过结合并激活受体酪氨酸激酶Kit(又被称为SCF-受体)而介导其各种各样的细胞应答的细胞因子。Kit最初是作为捕获该表面受体的活化的和截短的形式的猫逆转录病毒中的致癌基因被发现的(Besmer等人.(1986)J Virol 60:194-203.)。SCF是由鼠steel(SI)基因座编码的而Kit在小鼠中是由显性白斑(W)基因座编码的(Copeland等人.(1990)Cell 63:175-183;Huang等人.(1990)Cell 63:225-233;Flanagan and Leder(1990)Cell 63:185-194.;Tan等人.(1990)Science 247:209-212;Bemstein等人.(1990)Ciba Found Symp148:158-166;讨论166-172)。SCF作为非共价同型二聚体发挥功能,并且已经描述了由可变RNA剪接和蛋白水解加工产生的两种形式的膜锚定的和可溶性的SCF(在Ashman(1999)Int J Biochem Cell Biol31:1037-1051中综述)。Kit是受体酪氨酸激酶(RTK)III型家族的成员,该家族还包括PDGF-受体-α、和β、CSF-1-受体(也称为M-CSF-受体或Fms),以及Flt3-受体(也称为Flk2)(在Ullrich和Schlessinger(1990)Cell 61:203-212;Blume-Jensen等人.(2001)Nature 411:355-365中综述)。Kit由糖基化的细胞外配体结合域(胞外域)组成,其通过单跨膜(TM)结构域连接到胞质区(在Schlessinger,(2000)Cell 103:211-225中综述)。Kit的胞外域和其他III型RTK成员都包含5个Ig-样结构域,其中第二和第三膜远端结构域表现出在配体识别方面发挥作用(在Ullrich和Schlessinger(1990)Cell 61:203-212中综述)。其他的其胞外配体结合结构域完全是由多Ig-样重复序列组成的RTK包括VEGF-受体家族(7个Ig-样重复序列)、CCK4受体(7个Ig-样重复序列)和FGF受体(3个Ig-样重复序列)的成员。Kit胞质区包含具有大的激酶插入区域(III型RTK的另一个标志)的蛋白酪氨酸激酶(PTK)结构域。SCF与Kit的结合导致了受体的二聚化、分子间自磷酸化和PTK激活。已经提出Kit的第四个Ig-样结构域负责Kit响应于单价或二价SCF的结合的二聚化(Lev等人.(1992b)J Biol Chem 267:15970-15977;Blechman等人.(1995)Cell 80:103-113)。然而,其他研究已经证实Kit的配体诱导的二聚化是被SCF的双价结合驱动的(Philo等人.(1996)J Biol Chem271:6895-6902;Lemmon等人.(1997)J Biol Chem 272:6311-6317)。
SCF或Kit基因座处突变的小鼠的表征已表明SCF和Kit是造血干细胞、黑色素细胞、生殖细胞和肠起搏细胞发育所必需的(在Ashman(1999)Int J Biochem Cell Biol 31:1037-1051中综述)。在人类中,Kit的功能缺失突变引起的花斑性状,其表现出胸部和腹部色素减退、毛发白化、耳聋及便秘的特征(Fleischman等人.(1991)Proc Natl Acad SciU S A 88:10885-10889)。在不同类型的人类癌症中发现多种Kit的功能获得突变。在胃肠道间质瘤(GIST)、急性髓细胞性白血病(AML)和肥大细胞白血病以及其他癌症中发现激活的Kit突变。在近膜Ig-样结构域(D5)(8和9外显子)中、在近膜(JM)结构域(外显子11)和在酪氨酸激酶(PTK)结构域(外显子17)中确认了突变(见Forbes等人.(2006)COSMIC 2005.BR J.CANCER,94:318-22.体细胞突变数据库:癌症中体细胞突变的目录(Catalogue of Somatic Mutations in Cancer)http://www.sanger.ac.uk/genetics/CGP/cosmic/)。虽然有很好的证据证明,在JM和PTK结构域中的功能获得的突变导致Kit的组成型活化,这是通过解除自动抑制性的限制(Mol等人.(2004)J Biol Chem.279:31655-31663),但作为胞外域的D5中的功能突变获得的分子机制未知。有必要更好地表征RTK如Kit和PDGFR以及SCF、PDGFα/β和结合的Kit/SCF复合物的结构。这样的表征会引起可能作为药品、药物剂或其他生物制剂所靶向的区域的知情鉴定。
干细胞因子(SCF)通过结合Kit胞外域而导致酪氨酸激酶激活从而启动了它的多种细胞应答。在下面的一些实施例中描述了SCF刺激前后Kit整个胞外域的晶体结构。结构表明Kit的二聚化被SCF结合(其仅有的作用是使两个Kit分子会合到一起)。受体二聚化后发生使两个Kit分子的近膜的Ig-样结构域D4和D5之间能够发生横向相互作用的构象变化。培养细胞的实验表明Kit激活受到对于D4-D4相互作用关键的氨基酸中的点突变影响。此外,多个致癌基因突变映射到D5-D5界面上。由于Kit结构的关键标志、配体诱导的受体二聚化和D4-D4界面的关键氨基酸在其它受体中是保守的,所以在本报告中揭示的Kit刺激的机制可能应用于其他受体的激活。这表明,靶向于这些界面的药物或生物制剂可以用作治疗剂。
本文所述的SCF刺激前后的整个Kit胞外域的X-射线晶体结构的阐明已经提供了关于SCF诱导的Kit二聚化和激活的机制有价值的认识。该结构显示,命名为D1、D2和D3的Kit的前三个Ig-样结构域负责SCF结合。SCF结合的主要作用是交联两个Kit分子以提高细胞膜上Kit的局部浓度。这促进Kit近膜区中大的构象变化从而引起相邻Kit分子的D4或D5之间的同型相互作用。经由通过来自各个D4原体的EF环的两对盐桥的直接接触发生两个相邻的Kit分子的D4之间的横向相互作用。近膜区D5结构域提供了相邻Kit分子之间的另外的间接相互作用以进一步以使胞质酪氨酸激酶能够激活的距离和方向稳定和定位胞外域的近膜部分。
在下面的一些实施例中,描述了单体的和SCF诱导的同型二聚体(SCF-Kit 2:2复合体)形式的Kit完整胞外域的晶体结构。未占据的单体形式分辨率为的详细视图和SCF-诱导的同型二聚体形式分辨率为 的详细视图提供了关于Kit和其他RTK的激活机制的新的见解。本领域的技术人员应该了解,下面描述的实验可能用其他RTK完成。可被本发明的方法使用的示例RTK序列包括,但不限于,Kit mRNA的Genbank参考序列NM_000222.2(编码蛋白质NP_000213.1;
的详细视图提供了关于Kit和其他RTK的激活机制的新的见解。本领域的技术人员应该了解,下面描述的实验可能用其他RTK完成。可被本发明的方法使用的示例RTK序列包括,但不限于,Kit mRNA的Genbank参考序列NM_000222.2(编码蛋白质NP_000213.1;
MRGARGAWDFLCVLLLLLRVQTGSSQPSVSPGEPSPPSIHPGKSDLIVRVGDEIRLLCTDPGFVKWTFEILDETNENKQNEWITEKAEATNTGKYTCTNKHGLSNSIYVFVRDPAKLFLVDRSLYGKEDNDTLVRCPLTDPEVTNYSLKGCQGKPLPKDLRLIPDPKAGIMIKSVKRAYHRLCLHC SVDQEGKSVLSEKFILKVRPAFKAVPVVSVSKASYLLREGEEFTVTCTIKDVSSSVYSTWKRENSQTKLQEKYNSWHHGDFNYERQATLTISSARVNDSGVFMCYANNTFGSANVTTTLEVVDKGFINIFPMINTTVFVNDGENVDLIVEYEAFPKPEHQQWIYMNRTFTDKWEDYPKSENESNIRYVSELHLTRLKGTEGGTYTFLVSNSDVNAAIAFNVYVNTKPEILTYDRLVNGMLQCVAAGFPEPTIDWYFCPGTEQRCSASVLPVDVQTLNSSGPPFGKLVVQSSIDSSAFKHNGTVECKAYNDVGKTSAYFNFAFKGNNKEQIHPHTLFTPLLIGFVIVAGMMCIIVMILTYKYLQKPMYEVQWKVVEEINGNNYVYIDPTQLPYDHKWEFPRNRLSFGKTLGAGAFGKVVEATAYGLIKSDAAMTVAVKMLKPSAHLTEREALMSELKVLSYLGNHMNIVNLLGACTIGGPTLVITEYCCYGDLLNFLRRKRDSFICSKQEDHAEAALYKNLLHSKESSCSDSTNEYMDMKPGVSYVVPTKADKRRSVRIGSYIERDVTPAIMEDDELALDLEDLLSFSYQVAKGMAFLASKNCIHRDLAARNILLTHGRLTKICDFGLARDIKNDSNYVVKGNARLPVKWMAPESIFNCVYTFESDVWSYGIFLWELFSLGSSPYPGMPVDSKFYKMIKEGFRMLSPEHAPAEMYDIMKTCWDADPLKRPTFKQIVQLIEKQISESTNHIYSNLANCSPNRQKPVVDHSVRINSVGSTASSSQPLLVHDDV(SEQ ID NO:92))或Kit mRNA突变体2的Genbank参考序列NM_001093772.1(编码蛋白质NP_001087241.1;
MRGARGAWDFLCVLLLLLRVQTGSSQPSVSPGEPSPPSIHPGKSDLIVRVGDEIRLLCTDPGFVKWTFEILDETNENKQNEWITEKAEATNTGKYTCTNKHGLSNSIYVFVRDPAKLFLVDRSLYGKEDNDTLVRCPLTDPEVTNYSLKGCQGKPLPKDLRFIPDPKAGIMIKSVKRAYHRLCLHC SVDQEGKSVLSEKFILKVRPAFKAVPVVSVSKASYLLREGEEFTVTCTIKDVSSSVYSTWKRENSQTKLQEKYNSWHHGDFNYERQATLTISSARVNDSGVFMCYANNTFGSANVTTTLEVVDKGFINIFPMINTTVFVNDGENVDLIVEYEAFPKPEHQQWIYMNRTFTDKWEDYPKSENESNIRYVSELHLTRLKGTEGGTYTFLVSNSDVNAAIAFNVYVNTKPEILTYDRLVNGMLQCVAAGFPEPTIDWYFCPGTEQRC SASVLPVDVQTLNSSGPPFGKLVVQSSIDSSAFKHNGTVECKAYNDVGKTSAYFNFAFKEQIHPHTLFTPLLIGFVIVAGMMCIIVMILTYKYLQKPMYEVQWKVVEEINGNNYVYIDPTQLPYDHKWEFPRNRLSFGKTLGAGAFGKVVEATAYGLIKSDAAMTVAVKMLKPSAHLTEREALMSELKVLSYLGNHMNIVNLLGACTIGGPTLVITEYCCYGDLLNFLRRKRDSFICSKQEDHAEAALYKNLLHSKES SCSDSTNEYMDMKPGVSYVVPTKADKRRSVRLGSYIERDVTPAIMEDDELALDLEDLLSFSYQVAKGMAFLASKNCIHRDLAARNILLTHGRITKICDFGLARDIKNDSNYVVKGNARLPVKWMAPESIFNCVYTFESDVWSYGIFLWELFSLGSSPYPGMPVDSKFYKMIKEGFRMLSPEHAPAEMYDIMKTCWDADPLKRPTFKQIVQLIEKQISESTNHIYSNLANCSPNRQKPVVDHSVRINSVGSTASSSQPLLVHDDV(SEQ ID NO:93)),其中蛋白质使用标准的单字母氨基酸代码来标明。
实施例1:SCF和Kit的表达、纯化和结晶
利用杆状病毒表达系统,由命名为D1、D2、D3、D4和D5的5个Ig-样结构域组成的Kit完整胞外域在昆虫细胞中表达。纯化的Kit胞外域单体或SCF-诱导的Kit胞外域二聚体(SCF-Kit 2:2复合体)进行晶体生长和优化的大规模筛选,然后测定其晶体结构。
蛋白质表达与纯化
用杆状病毒表达系统,将在C-末端含有多组氨酸标签的可溶性Kit胞外域(氨基酸1-519)在昆虫细胞(Sf9)中表达。Kit胞外域通过镍螯合剂纯化,之后进行大小排阻层析(Superdex 200,GE Healthcare)。使用糖苷内切酶F1部分去糖基化后,用阴离子交换色谱(MonoQ,GEHealthcare)进一步纯化胞外域。SCF(1-141)进行表达、重折叠及纯化,如先前所描述的(Langley等人.(1994)Arch Biochem Biophys 311:55-61;Zhang等人.(2000)Proc Natl Acad Sci U S A 97:7732-7737)。
细胞系和表达载体
HEK和NIH3T3细胞分别在补充10%FCS and 10%CS的DMEM中培养。SCF刺激之前,细胞在无血清培养基中饥饿过夜,如前所述(Kouhara等人(1997)Cell 30:693-702)。根据制造商的说明书用Lipofectamin(Invitrogen)转染。全长Kit的cDNA被亚克隆到RK5表达载体中用于瞬时转染和亚克隆到pBABE/puro载体中用于稳定表达(Kouhara等人(1997)Cell 30:693-702)。通过用重组Kit胞外域免疫兔子产生抗-Kit抗体。单克隆抗-Kit抗体(Santa Cruz)用于免疫印迹。抗-磷酸酪氨酸抗体(抗-pTyr)购自Upstate Biotechnology。
结晶和数据收集
,对单独的Kit胞外域或与SCF复合的样本进行晶体生长和优化的大规模筛选。在4°下以聚乙二醇(PEG)为沉淀剂(0.1M Na-Pi缓冲液pH 6.0、0.2M KCl、12%PEG 400)的磷酸盐缓冲液中,获得尺寸大约为0.12x0.1x0.05mm的去糖基化胞外域的晶体。所有的晶体浸没在补充有5-18%甘油的储存溶液中几秒钟;快速冷却并在数据收集期间保持在的氮气流中。晶体属于菱形空间群R3,其晶胞尺寸为和 为六角晶格设置,每个不对称单元一个分子。通过在277K下将晶体浸泡在的含有浓度范围为0.1mM到50mM的重原子试剂的保存溶液中几秒到10天制备Kit的铂、溴和碘衍生物。
为六角晶格设置,每个不对称单元一个分子。通过在277K下将晶体浸泡在的含有浓度范围为0.1mM到50mM的重原子试剂的保存溶液中几秒到10天制备Kit的铂、溴和碘衍生物。
SCF-Kit复合体晶体在4℃下以聚乙二醇(PEG)为沉淀剂(0.2M硫酸铵,8-12%PEG 8000,5-8%乙二醇,pH 7.0-8.5)生长,并用ADSDquantum-210 CCD检测器在NSLS,Brookhaven National Laboratory的X25辐射束线(beamline)下收集分辨率达的衍射数据。该晶体属于单斜空间群C2,其晶胞尺寸为 
 β=108.2°,其是由两组SCF和Kit分子以不对称单元构成的。用DENZO和SCALEPACK以及HKL2000程序包对所有的数据集进行了处理和换算(Otwinowski等人.(1997)Methods Enzymol.276:307-326)。数据收集统计总结于表1A中。
β=108.2°,其是由两组SCF和Kit分子以不对称单元构成的。用DENZO和SCALEPACK以及HKL2000程序包对所有的数据集进行了处理和换算(Otwinowski等人.(1997)Methods Enzymol.276:307-326)。数据收集统计总结于表1A中。
实施例2:结构测定
利用反常散射的多重同晶置换(MIRAS)和利用分辨率达到 的多波长异常衍射(MAD)来计算实验相(表1A)。由此产生的电子密度图显示了β三明治结构的连续电子密度和清晰的溶剂-蛋白质边界。将单体Kit胞外域的分子模型人工建立成实验性的电子密度图。用25.4%的晶体学R-因子和29.6%的自由R-因子的原始数据集将结构优化到
的多波长异常衍射(MAD)来计算实验相(表1A)。由此产生的电子密度图显示了β三明治结构的连续电子密度和清晰的溶剂-蛋白质边界。将单体Kit胞外域的分子模型人工建立成实验性的电子密度图。用25.4%的晶体学R-因子和29.6%的自由R-因子的原始数据集将结构优化到 分辨率(表1B)。使用本报告中描述的单体形式的结构和SCF结构(Zhang等人.(2000)Proc Natl Acad Sci USA 97:7732-7737;可从Protein Data Bank以代码:1EXZ获得)作为检索模型通过分子置换解析SCF-Kit 2:2复合体的结构。用24.9%晶体学R-因子和29.5%自由R-因子的原始数据集将结构优化到
分辨率(表1B)。使用本报告中描述的单体形式的结构和SCF结构(Zhang等人.(2000)Proc Natl Acad Sci USA 97:7732-7737;可从Protein Data Bank以代码:1EXZ获得)作为检索模型通过分子置换解析SCF-Kit 2:2复合体的结构。用24.9%晶体学R-因子和29.5%自由R-因子的原始数据集将结构优化到 分辨率(表1A和1B)。用Pymol(pymol.sourceforge.net)和CCP4MG软件(Potterton等人.(2004)ActaCrystallogr D Biol Crystallogr 60:2288-2294)制作分子图像。Kit单体和SCF-Kit复合体的原子坐标和结构因子被保存在Protein Data Bank(rcsb.org/pdb)中,登录码分别是2EC8和2E9W。
分辨率(表1A和1B)。用Pymol(pymol.sourceforge.net)和CCP4MG软件(Potterton等人.(2004)ActaCrystallogr D Biol Crystallogr 60:2288-2294)制作分子图像。Kit单体和SCF-Kit复合体的原子坐标和结构因子被保存在Protein Data Bank(rcsb.org/pdb)中,登录码分别是2EC8和2E9W。
表1A.数据收集与定相统计(phasing statistics)

括号中的值表示最高分辨率壳(highest resolution shell)的统计值。(a)完全性=(独立偏转数)/(总理论偏转)。(b)Rmerge=∑|li<i>|/l∑li,其中li是观察的强度和<li>是从对称相关偏转的多个观察获得的平均强度。(c)Riso=||Fph|-|Fp||/∑|Fp|。(d)Rculls=∑|Fh-(Fph-Fp)/∑|Fph-Fp|。(e)定相能力=r.m.s.(|Fh|/E),分别地其中Fph和Fp是导数和原始结构因子幅度,Fh是重原子结构振幅。E是缺乏闭合差的余数。(f)<FOM>是平均优值(figure of merit)。
表1B.优化统计

(a)Rcryst=∑|Fobs-Fcalc|/∑Fobs,其中Fobs和Fcalc分别是观察的和计算的结构因子。(b)Rfree由优化前移除的5%偏转计算得到。(c)r.m.s.d.,根均方差。
实施例3:Kit胞外域的结构分析
胞外域结构的一般分析
Kit胞外域显示细长蛇形,尺寸约为(图1A)。Kit的D1、D2、D3、D4和D5结构域表现出典型的免疫球蛋白超家族(IgSF)折叠,其由包含组装成2个反向平行β折叠组成的β夹心结构(图1A)的命名为ABCC′DEFG的8个β链构成。D1、D2、D3和D5各含有连接B5和F5的半胱氨酸残基(分别为B和F链的第五个氨基酸)的保守的二硫键;桥接该两个β折叠以形成Ig-样折叠的疏水核心的中心的位置(Harpaz和Chothia(1994)J Mol Biol 238:528-539)。D2和D5包含两个二硫键和D4不含任何半胱氨酸残基,不过,D4的Ig-样折叠的完整性得以保持,即使B5和F5处的保守半胱氨酸残基分别被缬氨酸和苯丙氨酸残基替换。
D1和D2之间沿两个结构域的轴的角度是76°(图1A、B),与白介素-1β受体的第一和第二Ig-样结构域之间的取向相似(Vigers等人.(1997)Nature,386:190-194)。相比之下,D2和D3之间的角度为150°、D3和D4之间是119°以及D4和D5之间是162°。不同的Ig-样结构域的ABED和A’GFC β-折叠之间的定向D1-D2是~180°、D2-D3是~180°、D3-D4是~90°和D4-D5是~180°(图1)。
Kit胞外域的所有5个Ig-样结构域与用作为Ig折叠的标准的端蛋白叠合(Holden等人.(1992)J.Mol.Biol.227:840-851)揭示了 的等效Cα原子的均方根(r.m.s.)差。正如其与端蛋白叠合时D2的较高r.m.s.d.值所揭示的,5个Kit Ig-样结构域中D2是最趋异的(图8)。根据Ig-样结构域中关键氨基酸的结构保守性和其二级结构的拓扑结构(Harpaz等人.(1994)J Mol Biol 238:528-539;Halaby等人.(1999)Protein Eng 12:563-571),D1、D2、D3和D4属于I-亚群而D5与IgSF的C2和IgCAM亚群相关。此外,在IgSF的结构保守的20个指纹残基中(Harpaz等人.(1994)J Mol Biol 238:528-539),Kit的5个Ig-样结构域中10-14个残基是保守的(表2)。
的等效Cα原子的均方根(r.m.s.)差。正如其与端蛋白叠合时D2的较高r.m.s.d.值所揭示的,5个Kit Ig-样结构域中D2是最趋异的(图8)。根据Ig-样结构域中关键氨基酸的结构保守性和其二级结构的拓扑结构(Harpaz等人.(1994)J Mol Biol 238:528-539;Halaby等人.(1999)Protein Eng 12:563-571),D1、D2、D3和D4属于I-亚群而D5与IgSF的C2和IgCAM亚群相关。此外,在IgSF的结构保守的20个指纹残基中(Harpaz等人.(1994)J Mol Biol 238:528-539),Kit的5个Ig-样结构域中10-14个残基是保守的(表2)。
表2.Kit结构域和作为典型I-群IgSF的端蛋白(PDB代码:1TLK)的IgSF的20个关键指纹残基(1)


Kit Ig-样结构域结构的详细分析
Kit D1.D1折叠是由两个β折叠片组成的β夹心结构。一个折叠片由三条链A、B和E形成,而第二个折叠片由五条链A’、G、F、C和C’构成(ABE/A’GFCC’)。第一条链(被Pro41处的顺式构象中断)分成分别与B和G链配对的两个较短链A和A′。连接B5的Cys58和F5的Cys97的二硫键桥接两个β折叠片。相当长的C’链(与C链相互作用)引导多肽链的C-末端朝向直接连接E链的D1的上方,根据Ig-样结构域命名法,D1属于IgSF的I2-亚群(Casasnovas等人.(1998)Proc Natl Acad Sci USA 95:4134-4139)。
Kit D2.D2包含由B、E和D链形成的小的β-折叠片和由A’、G、F和C链形成的第二β-折叠片(BED/A’GFC),以及在E和F链交叉处(残基177-179)的附加螺旋。虽然在D2中确认了IgSF I-群中的20个标志残基中的11个,但是该Ig-样结构域在很多方面不同于标准的IgSF I1-群。D2在C4位置有Leu残基,而IgSF的其他I1-群有保守的Trp。由于两个短β链(称为B和B′链)的形成,B链中的氢键模式发生改变。另外的B’链与A链对齐,形成具有AB拓扑结构的短β折叠片。由于在氨基酸197-199位置处的插入(其导致形成与A’链的β折叠片),使G链分裂成两个短链,G(底侧)和G′(顶侧)。在残基197-199处由G链中的“纽结”引起的氢键模式破坏由Ser197的侧链和Cys186的主链酰胺之间的氢键补偿。值得注意地是Ser197在来自不同物种的Kit中和其他III型RTK中作为Ser或Thr残基是保守的。D2包含桥接CD环与F链末端的在Cys151和Cys183之间的额外二硫键以提供额外的稳定性给C链和CD环。额外的二硫桥可以弥补C和F链之间减少的氢键网络。从斑马鱼到人类的Kit中,这两个半胱氨酸是高度保守的。
Kit D3.D3由两组属于IgSF的I1-亚群的β折叠片(ABED/A’GFC)组成。两个β折叠片是由B链的Cys233和F链的Cys290之间的二硫键桥接的。端蛋白(PDB代码:1TLK)和D3结构的比较显示,D3的98个比对的Cα残基的Z评分为10.4和r.m.s.差为
Kit D4.虽然D4缺乏B5和F5的半胱氨酸之间特征二硫键,但是D4维持了IgSF拓扑结构。此外,D4中I-群IgSF的20个指纹残基中的13个是保守的。通过分别在B5和F5处存在的包埋的脂肪族(Val335)和芳香族的(Phe392)残基(其构成结构域疏水核心的一部分)之间的相互作用保持了D4结构的完整性。采用DALI进行的结构比较显示在Kit Ig-样结构域中,D4是与端蛋白最为相似的(Protein Data Bank代码:1TLK),89个比对的Cα残基的Z-评分为11.9和r.m.s.d.为 Val335和Phe392的Cα-Cα之间的距离
Val335和Phe392的Cα-Cα之间的距离 在缺乏连接B5和F5的二硫键的IgSF结构域中相似位置之间观察到的距离范围内。例如,也缺乏二硫键的肌联蛋白Ig-样结构域M5(Protein Data Bank代码:1TNM)以
在缺乏连接B5和F5的二硫键的IgSF结构域中相似位置之间观察到的距离范围内。例如,也缺乏二硫键的肌联蛋白Ig-样结构域M5(Protein Data Bank代码:1TNM)以 的r.m.s.d与D4叠合并且B5和F5位置之间有的距离。D4由两个β折叠片构成,各折叠片包含具有ABED/A’GFC排列的四条链。Thr321,A’链的第一个残基,与高度保守的Phe405的芳香环形成范德瓦耳斯接触。值得注意的是,向上折叠到结构域的顶侧的CD环由三个主要的相互作用的来稳定。Thr354的侧链与Gln347的侧链和Trp348的主链的羰基形成氢键。位于疏水核心边缘的疏水残基(Trp348、Tyr350、Trp359Val377、Leu379和Tyr390)为Phe355提供了疏水环境。虽然CD环没有显现明显的序列保守性,但该环在所有III-型家族RTK中包含8个氨基酸。
的r.m.s.d与D4叠合并且B5和F5位置之间有的距离。D4由两个β折叠片构成,各折叠片包含具有ABED/A’GFC排列的四条链。Thr321,A’链的第一个残基,与高度保守的Phe405的芳香环形成范德瓦耳斯接触。值得注意的是,向上折叠到结构域的顶侧的CD环由三个主要的相互作用的来稳定。Thr354的侧链与Gln347的侧链和Trp348的主链的羰基形成氢键。位于疏水核心边缘的疏水残基(Trp348、Tyr350、Trp359Val377、Leu379和Tyr390)为Phe355提供了疏水环境。虽然CD环没有显现明显的序列保守性,但该环在所有III-型家族RTK中包含8个氨基酸。
Kit D5.D5属于IgSF的C2和IgCAM亚群并且在该模块中20个指纹残基中的10个是保守的。D5显示了ABED/CFG拓扑结构,B5的Cys428和F5的Cys491之间的二硫键(其桥接两个β折叠片)和桥接C链和CD环的第二二硫键。在所有Kit和III型RTK中,该两个二硫键是保守的。值得注意的是,D5的上半部类似于神经细胞粘附分子Axonin-1/TAG-1的第三Ig(Protein Data Bank代码:1CS6)。几个标志可被识别,尽管在端蛋白(Protein Data Bank代码1FHG)、FGFR(Protein Data Bank代码1CVS)和RTK Musk(Protein Data Bank代码2IEP)中程度较低。这些包括接近于连接F5和B5的二硫键的两个Ala残基(Ala430和Ala493),该区域中小侧链的存在使得能够在结构域顶部封闭包装。第二个标志是分别在A、B、C和G链中的Pro和Gly残基Pro413、Gly432、Pro436和Gly498的环状排列。第三个标志是F9中的Asn残基(Asn495)的存在,其分别与FG和BC环的Val497和Pro434的主链形成氢键。总而言之,位于D5顶部的这三个标志导致与细胞粘附蛋白的Ig-样结构域构型类似的紧密包装构型。
实施例4:Kit单体形式的Ig-样结构域间相互作用
Kit的5个Ig-样结构域之间的结构域间相互作用负责维持Kit胞外域单体的整体拓扑结构(图1)。D1相对于D2的定向是通过由两个Ig-样结构域间的数目众多的相互作用引起的大范围包埋表面决定的(图1B)。D1-D2界面中 的包埋表面积远大于杆状多结构域IgSF结构的大多数Ig-样结构域间界面的包埋表面积(Su等人.(1998)Science281:991-995),包括介于500和
的包埋表面积远大于杆状多结构域IgSF结构的大多数Ig-样结构域间界面的包埋表面积(Su等人.(1998)Science281:991-995),包括介于500和 之间的Kit胞外域中的三个其它Ig-样间界面。该界面主要由D1的A’链和G链、EF环和CC’环与D2的A链的N末端区域、B链的C末端、BC环和DE环之间的疏水性和静电相互作用形成的(图1B)。此外,在来自不同物种的Kit中,D1-D2界面的许多残基(包括D1的G链、连接D1和D2的接头以及D2的BC环的氨基酸)是保守的(图1B)。
之间的Kit胞外域中的三个其它Ig-样间界面。该界面主要由D1的A’链和G链、EF环和CC’环与D2的A链的N末端区域、B链的C末端、BC环和DE环之间的疏水性和静电相互作用形成的(图1B)。此外,在来自不同物种的Kit中,D1-D2界面的许多残基(包括D1的G链、连接D1和D2的接头以及D2的BC环的氨基酸)是保守的(图1B)。
D2-D3界面的包埋表面积大约是D2-D3界面由两个静电相互作用包围的小的疏水补丁组成。该界面是由D2的EF环和D3的DE环之间的相互作用和D2-D3的接头与D3的FG和BC环之间的相互作用形成的(图1C)。D3-D4界面的包埋表面积大约是 D3和D4主要通过D3的A’链和G链与D4的BC环和DE环相互作用(图1D)。由于D4相对于D3具有沿两个Ig-样结构域的长轴的119°夹角的角排列,D3-D4界面的长度约为
D3和D4主要通过D3的A’链和G链与D4的BC环和DE环相互作用(图1D)。由于D4相对于D3具有沿两个Ig-样结构域的长轴的119°夹角的角排列,D3-D4界面的长度约为 D4-D5界面形成约为
D4-D5界面形成约为 的包埋表面积,主要由疏水相互作用介导(图1E)。界面是由D4的A链、G链和F链与D5的BC环和DE环以及与D4-D5的接头之间的相互作用形成(图1E)。
的包埋表面积,主要由疏水相互作用介导(图1E)。界面是由D4的A链、G链和F链与D5的BC环和DE环以及与D4-D5的接头之间的相互作用形成(图1E)。
关于Kit单体的Ig-样结构域间相互作用的详细的结构域-结构域信息
D1-D2界面.D1的残基Ile47、Ile70、Leu71、Ala89、Tyr108和Phe110与D2的Leu119、Pro137、Leu138、Pro141、Pro166以及Lys167的侧链之间的疏水性相互作用稳定了结构域间的相互作用(图1B)。在疏水补丁的周边区域中存在两个主要的静电相互作用,包括D1的Arg112与D2的Asp140之间的相互作用及D1的Asp72与D2的Arg135之间的相互作用(图1B)。
D2-D3界面.疏水补丁由Arg177的脂族部分及Pro206、Phe208、Val238和Phe267的侧链组成。静电相互作用涉及D2的Glu128和Asp129的侧链与D3的Lys209之间的氢键(图1C)。Arg177的侧链和Glu128的侧链之间的盐桥稳定了D2中Arg177侧链和Pro206侧链以及D3中Phe267的位置,以为D2的Arg177侧链的脂族部分创建疏水环境(图1C)。第二静电相互作用是由D2中Arg181的侧链与D3的Asp266的侧链介导的。
D3-D4界面.D3-D4界面的疏水相互作用包括D3的Val308和Leu222和D4的Phe312、Phe340和Ile371之间的疏水相互作用。D3-D4界面覆盖比其他Ig-样结构域间界面小的包埋面积(图1D)。
D4-D5界面.D4-D5界面上的疏水补丁分别包括D4的A链和G链的Phe324和Tyr408和D5的BC环的Phe433。此外,范德瓦耳斯接触有利于疏水补丁周围的界面的稳定;D4的Phe324、Gly384、Thr389、Tyr408、Asn410、Thr411和Met351与D5的Val497、Phe433、Gly470、Phe649和Lys471相互作用(图1E)。
实施例5:结合的SCF-Kit复合体的整体结构的分析
SCF-Kit复合体的结构显示了2:2的化学计量,其中不对称单元中两组1:1复合体按照非晶体学双面对称相关(图2)。在晶格中观测到的2:2复合体与表明Kit二聚化受二聚的SCF配体驱动的实验一致(Philo等人.(1996)J Biol Chem 271:6895-6902;Lemmon等人.(1997)J BiolChem 272:6311-6317)。这两组Kit胞外域和SCF分子像尺寸约为 的倒置字母“A”(图2A和图9)。
的倒置字母“A”(图2A和图9)。
结合于Kit的SCF的整体结构与先前所描述的游离SCF的结构相似(Zhang等人.(2000)Proc Natl Acad Sci U S A 97:7732-7737;Jiang等人.(2000)Embo J 19:3192-3203)。SCF-Kit 2:2复合体的结构显示,单个SCF原体直接结合单个Kit原体的D1、D2和D3(图2B)。因此,单一受体原体形成具有SCF原体上类似的双折叠相关表面的对称复合体。Kit二聚化是由Kit的两个近膜Ig-样结构域之间的同型相互作用介导的,即D4-D4和D5-D5相互作用(图2B)。这导致相对于将C-末端引到他们与跨膜结构域连接处相互接近的 内的分子其余部分的D4和D5的显著改变的构型(图2B和图9)。该结构特征也在于尺寸约为
内的分子其余部分的D4和D5的显著改变的构型(图2B和图9)。该结构特征也在于尺寸约为 的在复合物中心处较大的空腔的存在(图2B)。晶体结构表明,SCF的各原体专门结合单一Kit分子和受体二聚化是由促进额外的受体-受体相互作用的SCF二聚体驱动的。
的在复合物中心处较大的空腔的存在(图2B)。晶体结构表明,SCF的各原体专门结合单一Kit分子和受体二聚化是由促进额外的受体-受体相互作用的SCF二聚体驱动的。
实施例6:Kit的SCF结合区的分析
SCF以其中SCF的四螺旋束垂直于D1、D2和D3的长轴定向和SCF和Kit的C-末端的面向相反方向的构型上结合到由Kit的D1、D2和D3形成的凹面上(图2、图3和图9)。在Kit和各SCF原体之间的界面处包埋的溶剂可接触表面积约为 在已知配体受体界面范围内的包埋表面积。有可能将SCF-Kit界面分为三个结合位点(图3A、B、表2和表3)。位点-I位于D1上、位点-II位于D2和D2-D3接头中以及位点-III位于D3中。位点I、II和III的包埋表面积分别为大约280、770和
在已知配体受体界面范围内的包埋表面积。有可能将SCF-Kit界面分为三个结合位点(图3A、B、表2和表3)。位点-I位于D1上、位点-II位于D2和D2-D3接头中以及位点-III位于D3中。位点I、II和III的包埋表面积分别为大约280、770和
位点-I
SCF的αC-β2环垂直于D1的C’链排列,如图3C所表示的。D1的Asp72、Glu73和Thr74与SCF的Lys99’、Ser101’和Phe102’以的Cα距离紧密地定位在,表明这些残基可能参与D1和SCF之间的相互作用。由于αC-β2环的较低侧链电子密度,不能确定特定的相互作用。
位点II
SCF结合大部分是由Kit的带电表面的互补静电相互作用介导的(图3A、B、D)。Kit的碱性氨基酸Arg122、Arg181、Lys203和Arg205与SCF的酸性氨基酸Asp54’、Asp77’、Asp84’和Glu88’之间形成盐桥。Kit的Glu198与SCF的Asp54’之间的盐桥使Arg122的构象稳定。图3D显示,D2上的三个主要相互作用的残基Tyr125、Arg181和Lys203排列在同一平面上并与SCF的αB和αC的Asp77’、Asn81’、Asp84’、Ser53’和Thr57’形成氢键。D2的Ser123和Ile201与SCF的Val50’和Thr57’之间的范德瓦耳斯接触也有助于配体-受体复合体的形成。然而,来自其它物种的Kit和SCF的位点II中的残基有明显不同(图3、图8和图10)。尽管在哺乳动物中Arg181和Lys203作为碱性氨基酸是不变的,在小鼠和大鼠中Tyr125被苯丙氨酸替代,这很可能导致氢键的损失。在小鼠和大鼠中Kit的Arg205是高度保守的氨基酸,而Glu88’分别被亮氨酸和丙氨酸残基替代。此外,人类中Kit的Arg122和SCF的Asp54’在小鼠和大鼠中分别被亮氨酸和缬氨酸替代。这些置换可能说明啮齿动物的SCF与人Kit的亲和性降低的原因(Lev等人.(1992b)J Biol Chem 267:15970-15977)。
位点-III
SCF的N末端片段与D3的D链相互作用(图3A、E)。氢键在SCF的Asn10’的侧链与Ser261的主链酰胺和羰基基团之间以及与D3上的Asp260和Trp262的侧链形成。此外,SCF的Thr9’和Asn11’分别结合Kit的Ser261和His263的侧链和主链酰胺。SCF的突变分析表明,用丙氨酸或谷氨酸残基替换Asn10’降低了SCF对Kit的结合亲和力大约10倍和Asn10’(或其他物种的Asp)对于生物活性是必需的(Hsu等人.(1998)Biochemistry 37:2251-2262)。不同物种的SCF中受体结合界面的比较表明,Asn10’(或Asp)是高度保守的残基(图8)。额外的重要相互作用是由SCF的Asn6’和Arg7’经由与Kit的D3的Tyr259、Thr269、Ser240、Val242、Ser241、Ser244的范德瓦耳斯接触介导的。
表3.两个Kit原体之间的SCF-Kit相互作用和亲同种抗原相互作用


实施例7:与结合相关的Kit/SCF结构和构象变化的分析
Kit的配体结合结构域准备用于SCF结合
Kit单体形式的单个D1、D2和D3的结构与相应的SCF诱导的同型二聚体形式的结构叠合揭示了分别对于在D1、D2和D3中82、92和100个比对的Cα残基的0.5、0.8和 的r.m.s.d.值。类似地,Kit单体的整个D1-D2-D3区域的结构与SCF-Kit 2:2复合体中的相应结构叠合揭示了对于在D1-D2-D3区域的274个比对Cα残基的
的r.m.s.d.值。类似地,Kit单体的整个D1-D2-D3区域的结构与SCF-Kit 2:2复合体中的相应结构叠合揭示了对于在D1-D2-D3区域的274个比对Cα残基的 的r.m.s.d.值(图3和图11)。引人注目的是,Kit的SCF结合口袋的结构中没有明显的骨架变化。然而,SCF结合时在SCF结合裂缝中检测到几个小结构变化。SCF结合后在D2的G、F和C链的上半部(氨基酸167-187和143-166)中观察到结构变化(图1A、2A)。这些链位于与SCF结合界面的相对侧,并且不参与介导任何与SCF的直接接触。总的来说,Kit单体的结构与SCF-占据的Kit二聚体的结构的比较表明,Kit的D1-D2-D3区域可以被视为准备用于SCF结合的功能单元,之后是由二聚的SCF分子驱动的后续Kit二聚化。
的r.m.s.d.值(图3和图11)。引人注目的是,Kit的SCF结合口袋的结构中没有明显的骨架变化。然而,SCF结合时在SCF结合裂缝中检测到几个小结构变化。SCF结合后在D2的G、F和C链的上半部(氨基酸167-187和143-166)中观察到结构变化(图1A、2A)。这些链位于与SCF结合界面的相对侧,并且不参与介导任何与SCF的直接接触。总的来说,Kit单体的结构与SCF-占据的Kit二聚体的结构的比较表明,Kit的D1-D2-D3区域可以被视为准备用于SCF结合的功能单元,之后是由二聚的SCF分子驱动的后续Kit二聚化。
结合Kit的SCF分子的构象变化
虽然与Kit结合的SCF的总体结构与游离SCF的结构相似,但两个原体之间的角度、连接环的构象和分子的柔性N末端的结构中有明显差异(图4)。公开的SCF二聚体的结构的比较(Protein Data Bank中的登录号1EXZ和1SCF)显示,游离的SCF同型二聚体的两个原体之间的角度(αC螺旋之间的角)可能在不同结构中有2°到6°的变化,从而显示了SCF二聚体中存在一定程度的灵活性。在Kit结合的SCF原体与游离SCF之间的角度差异增加了3-9°。图4显示了Kit结合的SCF结构,其中SCF原体之间的夹角增加了5°。
图4B表明,从Cys4’到Asn11’的游离SCF的N末端有无规卷曲构型(Zhang等人.(2000)Proc Natl Acad Sci U S A 97:7732-7737)。它还表明,前四个氨基酸的缺失导致SCF对Kit的结合亲和力降低大约25%,表明Cys4’和Cys89’之间的二硫桥在保持SCF功能完整性方面发挥作用(Langley等人.(1994)Arch Biochem Biophys 311:55-61)。图4B也表明结合Kit的SCF的N-末端区域的Thr9’和Asn10’经历构象变化,其中当受体结合时他们的Cα位置移位3至 (图4B)。在介导SCF的N-末端发生的构象变化方面,在N末端的Cys4’和Cα螺旋的Cys89’之间的二硫键发挥重要作用。如Cα位
(图4B)。在介导SCF的N-末端发生的构象变化方面,在N末端的Cys4’和Cα螺旋的Cys89’之间的二硫键发挥重要作用。如Cα位 的均方根偏差(r.m.s.d.)所显示的,当受体占据时游离SCF的Cys24’位置没有改变。最后,游离SCF的αC-β2是无序的或者有与结合Kit的SCF中的αC-β2环结构不同的结构。图4C显示,受体结合时SCF的αC-β2环发生了大的构象变化,一个对于SCF-Kit界面位点-I的建立关键的构象变化。
的均方根偏差(r.m.s.d.)所显示的,当受体占据时游离SCF的Cys24’位置没有改变。最后,游离SCF的αC-β2是无序的或者有与结合Kit的SCF中的αC-β2环结构不同的结构。图4C显示,受体结合时SCF的αC-β2环发生了大的构象变化,一个对于SCF-Kit界面位点-I的建立关键的构象变化。
在SCF结合的Kit中D4和D5定向的大的重排
Kit单体形式的单个D1、D2、D3、D4和D5的结构与相应的SCF诱导的同型二聚体形式的单个Ig-样结构域的叠合表现出SCF结合后Kit
Ig-样结构域结构的小变化。相比之下,Kit单体形式的D3-D4-D5区域与同型二聚体形式的相应区域的叠合表现出了D4和D5相对于彼此和相对于Kit的配体结合区域的定向的大的结构变化(图5A和图12)。单体Kit的单个结构域D3、D4和D5中各结构域可以与SCF-占据的Kit中其相应部分叠合,其中D3、D4和D5的98、101和85Ca原子的r.m.s.d值分别为0.9、0.9和 然而,Kit单体的D3结构与配体-占据同型二聚体形式的D3结构的叠合揭示在SCF结合的Kit中D4和D5方向的显著移动(图5A)。分别通过沿连接D3至D4的接头的轴旋转和沿穿过D3-D4和D4-D5界面的连接D4至D5的接头的轴旋转(图5A),发生相对于配体结合区的D4和D5的方向的重新定向。游离和配体结合Kit比较表明,配体占据的Kit的D4相对于D3旋转22°,和配体占据的Kit的D5相对于D4旋转27°(图5A)。SCF占据的Kit的D4和D5的重排导致了由两个相邻Kit分子的D4-D4和D5-D5相互作用介导的受体-受体相互作用(图5B)。D5的DE环的构象在SCF占据的胞外域中发生改变。受体二聚化驱动的D4和D5的重新定向赋予D5的DE环新的构象(图5A)。
然而,Kit单体的D3结构与配体-占据同型二聚体形式的D3结构的叠合揭示在SCF结合的Kit中D4和D5方向的显著移动(图5A)。分别通过沿连接D3至D4的接头的轴旋转和沿穿过D3-D4和D4-D5界面的连接D4至D5的接头的轴旋转(图5A),发生相对于配体结合区的D4和D5的方向的重新定向。游离和配体结合Kit比较表明,配体占据的Kit的D4相对于D3旋转22°,和配体占据的Kit的D5相对于D4旋转27°(图5A)。SCF占据的Kit的D4和D5的重排导致了由两个相邻Kit分子的D4-D4和D5-D5相互作用介导的受体-受体相互作用(图5B)。D5的DE环的构象在SCF占据的胞外域中发生改变。受体二聚化驱动的D4和D5的重新定向赋予D5的DE环新的构象(图5A)。
Kit同型二聚体的D4:D4相互作用
两个相邻的Kit分子的D4之间的同型相互作用是由SCF-Kit 2:2复合体的D4-D4界面介导的。D4-D4界面是由各Kit原体的D4的ABED链形成的两个β折叠片介导以形成近似平面的排列,其中各原体的Arg381彼此相向,从而形成360A2的包埋表面积。图6表明,Arg381和Glu386形成横跨Kit二聚体的双折叠轴的盐桥和范德瓦耳斯接触。此外,各原体Arg381的侧链与相邻的Kit分子的相应残基的主链羰基形成氢键。
基于结构的序列分析表明D4-D4界面在大多数III-型RTK(包括CSF1R、PDGFRα和PDGFRβ)中是保守的(图6B和图8)。在PDGFRα中,Glu386被天冬氨酸替换,一个也可以作为盐桥配体发挥功能的残基。在不同物种的III-型RTK中一对碱性(Arg381)和酸性(Glu386)残基是严格保守的。D4-D4界面中发现的序列基序在V-型RTK(VEGFR家族)的所有成员(包括VEGFR-1(Flt1)、VEGFR-2(Flk1)和VEGFR-3(Flt4))的近膜第七Ig-样结构域(D7)中也是保守的。在VEGFR中,碱性(Arg)和酸性(Asp)残基位于EF环中。尽管负责III型RTK的D4-D4界面的核心序列基序位于VEGFR的不同Ig-样结构域中(即,III型的D7对D4),与那些在Kit的D4-D4界面中观察到的相似的受体-受体相互作用也有可能在RTK的VEGFR家族所有成员中通过相似的D7-D7界面(图6A)发生(Ruch等人.(2007)Nature.Struct.Mol.Biol.14:249-250)。
Kit同型二聚体中的D5-D5相互作用
图2B和图5B、6C表明,在SCF-Kit 2:2复合体中相邻的D5原体是平行的且彼此接近的,并且处在很可能垂直于细胞膜的方向中。D5的β-折叠片拓扑结构遵循与大多数I-群IgSF不同的非典型排列,其中A链被分成A链和A’链。D5的A链与B链配对,从而形成ABED/CFG的β折叠片拓扑结构。因此,位于两个β折叠片(ABED/CFG)边缘的A链和G链以彼此之间Cα的 的距离是几乎平行的。此外,一个原体的A链和G链以双面对称形式面对相邻D5的A链和G链。两个相邻Kit原体的Asn505的侧链彼此之间大约是
的距离是几乎平行的。此外,一个原体的A链和G链以双面对称形式面对相邻D5的A链和G链。两个相邻Kit原体的Asn505的侧链彼此之间大约是 但可以介导两个天冬酰胺之间的间接相互作用的水或金属离子不能在该弱电子密度区域中被检测到。额外的D5-D5相互作用是由两个相邻Kit分子的Tyr418介导的(图6C)。相邻的Tyr418侧链的羟基之间的相互作用能够由水分子介导。也表明,相邻D5结构域的相对位置是由相邻原体的Tyr418和Asn505形成的间接相互作用介导的。D5的G链经由短连接体连接到Kit的跨膜结构域。
但可以介导两个天冬酰胺之间的间接相互作用的水或金属离子不能在该弱电子密度区域中被检测到。额外的D5-D5相互作用是由两个相邻Kit分子的Tyr418介导的(图6C)。相邻的Tyr418侧链的羟基之间的相互作用能够由水分子介导。也表明,相邻D5结构域的相对位置是由相邻原体的Tyr418和Asn505形成的间接相互作用介导的。D5的G链经由短连接体连接到Kit的跨膜结构域。
实施例8:受体激活的机制
Kit的胞外域单体和SCF诱导的二聚体的结构提供了关于Kit和其他在其胞外域中包含5或7个Ig-样结构域的RTK的配体诱导激活机制的新的见解。Kit胞外域单体的D1、D2和D3结构与SCF诱导的胞外域二聚体中相应的区域的比较显示SCF结合后在SCF结合口袋和D1、D2和D3的其他部分中很少的结构变化。根据他们显著不同的生化功能,我们将Kit的胞外域分为三个独立的功能单元。第一单元由三个膜远端Ig-样结构域D1、D2和D3组成。D1-D2-D3区域作为独立的模块发挥功能以起到特异性SCF结合单元的作用。SCF-结合单元是由柔性接头(D3-D4界面)连接到D4;D4是由另外的柔性接头(D4-D5界面)连接到D5上的第二独立单元,D5定义为第三独立单元。D4和D5的功能是分别介导横向的D4-D4和D5-D5相互作用,其两个相邻的Kit胞外域的近膜区拉近并稳定二者之间的相互作用。
根据这种观点,Kit的二聚化是受二价SCF结合的驱动,它唯一的功能是结合SCF并将两个Kit分子集合到一起。SCF诱导的Kit二聚化之后是D4和D5定向相对于D1-D2-D3SCF-结合单元的位置的大的变化。本文提供的数据表明,D3-D4和D4-D5界面的柔性接头使横向相互作用成为可能,这导致在受体二聚化时发生大的构象变化。不是诱导Kit的构象变化,而是二聚化可以在由柔性连接的单体向刚性二聚体的过渡中选择特定的构象。这最终导致Kit的两个相邻的D4和两个相邻的D5之间的复合体形成,将D5的C末端引到其中两个相邻Kit分子的跨膜结构域彼此之间在 之内的细胞膜的一点。事实上,SCF诱导的Kit酪氨酸自磷酸化(图7B)和下游信号传导途径的刺激受到Kit D4内的Arg381或Glu386的点突变的强烈损害。PDGF受体激活和下游信号传导途径的刺激也受PDGFR的D4中的点突变损害。本文提供的数据表明,Kit近膜区之间的同型相互作用主要受D4-D4界面的介导,和D5-D5界面通过促进细胞表面界面处两个Kit胞外域的精确定位发挥协同的次要作用。
之内的细胞膜的一点。事实上,SCF诱导的Kit酪氨酸自磷酸化(图7B)和下游信号传导途径的刺激受到Kit D4内的Arg381或Glu386的点突变的强烈损害。PDGF受体激活和下游信号传导途径的刺激也受PDGFR的D4中的点突变损害。本文提供的数据表明,Kit近膜区之间的同型相互作用主要受D4-D4界面的介导,和D5-D5界面通过促进细胞表面界面处两个Kit胞外域的精确定位发挥协同的次要作用。
SCF-Kit复合体显示具有以下特征的静电场强极化:(i)整体带负电荷的表面;(ii)SCF(负)和配体结合的D1-D2-D3单元(正)之间的互补性;和(iii)D4-D4界面正上方和围绕D4-D4界面的强负极化的表面(图6D、3B和图13)。该数据表明,SCF与Kit的结合以至少2步发生:首先,SCF和D1-D2-D3之间的静电吸引会将SCF沿Kit上的相对配体结合区域对准。由于Steering效应,静电吸引力也可以导致更快的SCF缔合速率(Muellera等人.(2002)Biochina and Biophysica Acta.1592:237-250)。随后,SCF-Kit复合体的形成将由另外的相互作用(包括那些由结合的SCF分子的构象变化介导的相互作用)来稳定。D4的强极化静电表面也可以在通过诱导相邻Kit受体的D4结构域之间的排斥而将Kit维持在单体的非活化构型中发挥作用(图6D)。在通过SCF-驱动的受体二聚化和维度效应(dimensionality effect)提高细胞表面的局部受体浓度之前,相邻受体的D4对D4和D5对D5的结合亲和力可能太低以至于不利于Kit胞外域的二聚化。一旦达到这种局部浓度的阈值,相邻的D4之间的吸引力将克服静电斥力达到两个相邻D4单元能够互相结合的程度。有趣的是,维持D4-D4界面的主要相互作用,即相邻Kit分子的Arg381和Glu386之间的双重盐桥也是由静电相互作用介导的。
Kit和C-钙粘蛋白的胞外域(Boggon等人.(2002)Science 296:1308-1313)各由五个串联的Ig-样结构域组成并且都表现相似的细长拓扑结构,Kit为 和C-钙粘蛋白为
和C-钙粘蛋白为 此外,细菌粘附分子侵入呈现出非常相似的细长结构和Ig-样结构域间的拓扑结构(Hamburger等人.(1999)Science 286:291-295)。Kit胞外域可能从编码介导细胞-细胞相互作用的蛋白的共同祖先基因进化而来。尽管经典-钙粘蛋白利用他们的最膜远端Ig-样结构域进行介导细胞-细胞相互作用的同型结合,Kit的胞外域已演为起到细胞信号传导受体的功能,其结合膜锚定或可溶的SCF同型体来诱导受体二聚化和激活(图7C)。
此外,细菌粘附分子侵入呈现出非常相似的细长结构和Ig-样结构域间的拓扑结构(Hamburger等人.(1999)Science 286:291-295)。Kit胞外域可能从编码介导细胞-细胞相互作用的蛋白的共同祖先基因进化而来。尽管经典-钙粘蛋白利用他们的最膜远端Ig-样结构域进行介导细胞-细胞相互作用的同型结合,Kit的胞外域已演为起到细胞信号传导受体的功能,其结合膜锚定或可溶的SCF同型体来诱导受体二聚化和激活(图7C)。
由于Kit结构的标志、配体结合和受体二聚化在其他受体中是保守的,本文描述的Kit激活机制可能是许多受体激活的普遍机制(图7C)。此外,本文描述的结构信息可以应用于设计用于治疗癌症和受激活受体驱动的其他疾病新的治疗介入。
实施例9:人类疾病的Kit突变分析
多种人类疾病是由Kit基因突变引起的。在人类中,Kit的胞外域的功能缺失突变引起花斑性状(Fleischman等人.(1996)J Invest Dermatol107:703-706;Murakami等人.(2005)J Invest Dermatol.124:670-672)。Kt基因座中这些外显子-2和外显子-3的点突变导致Cys136被精氨酸残基替换和Ala178被苏氨酸残基替换。两个突变都发生在D2(Kit上SCF结合位点的关键组件)中(图7A)。花斑Cys136Arg突变会造成在维持D2的结构和功能完整性中发挥关键作用的重要二硫键的缺失,由此造成其识别SCF能力的缺失。Ala178位于邻近D2-D3界面的D2的EF环中(图7A)。花斑Ala178Thr突变可以破坏对于维持D2-D3界面完整性的必要的相互作用和D2和/或D3结合SCF必需的相互作用(图7A)。
在不同的癌症中发现多种Kit基因座中的功能获得突变,包括GIST、AML和SCLC(参见Forbes等人.(2006)COSMIC 2005.BR J.CANCER,94:318-22.体细胞突变数据库:癌症中体细胞突变目录http://www.sanger.ac.uk/genetics/CGP/cosmic/)。在JM中和Kit的PTK结构域中鉴定了很多致癌突变。在Kit胞外域中也发现了许多种致癌突变(图7A),包括共同导致形成Kit的激活形式的框内缺失、点突变、框内重复和插入。在外显子-8处的框内缺失和插入突变(涉及Asp419的缺失或替代)在患有AML的患者中记述,同时在GIST中确认了Ala502-Tyr503和Ala502-Phe506序列的重复(图7A)。Asp419位于连接D5的A链和AB的区域中和Ala502-Tyr503位于Kit的D5的G链上。有趣的是,实际上所有在Kit胞外域中发现的活化致癌基因突变被映射到D5-D5界面上(图7A)。对致癌的D5突变的作用模式的最合理的解释是,这些突变通过提高on-rate或降低off-rate或以利于增强D5-D5相互作用的方式改变两个过程的速度来提高相邻D5结构域之间的结合亲和力和同型相互作用。
以上分析表明,D4和D5区域是治疗剂靶向的良好候选区域。可以使用药品、药物或生物制剂结合Kit以促进Kit二聚化/激活或更理想地阻止二聚化/激活。
实施例10:Kit胞外域的表达、纯化和部分去糖基化
将编码人Kit的氨基酸1-519的DNA构建体(Lemmon等人.(1997)J Biol Chem 272:6311-6317)(在C末端包含额外的五个组氨酸残基)连接到pFastBac1(Invitrogen,Inc.)中。根据Bac-to-Bac指导手册(Invitrogen)中描述的程序制备表达胞外域Kit蛋白质的杆状病毒。昆虫Sf9细胞用Wave Bioreactor(Wave Biotech,LLC,System 20/50)在15L补充有10%热灭活胎牛血清的Grace’s昆虫培养基中生长至2~3×106细胞/毫升,然后用携带Kit胞外域基因的重组杆状病毒感染。虽然胞外域Kit包含来自人Kit的信号序列,蛋白质积累在昆虫细胞中而不是分泌出来。72小时后收获细胞并在冰上1.4L的包含200mM氯化钠、10%的甘油、1%的NDP-40和2mM PMSF的50mM磷酸钾缓冲液(pH 8)中裂解20分钟。离心和过滤后,用Ni-NTA珠的亲和层析纯化Kit胞外域,之后使用Superdex 200进行凝胶过滤。在含有25mM氯化钠和1%甘油的25mMTris缓冲液pH 8.5中的纯化胞外域用加入到Kit溶液中达到10∶1 w/w最终比例的重组内切糖苷酶F1在4℃下处理12小时。然后将核酸内切酶F1处理的Kit胞外域加载到预平衡的16/10 Mono Q柱上并用包含400mM NaCl和1%甘油的Tris缓冲液pH 8.5的浅(shallow)梯度洗脱。收集去糖基化的Kit胞外域的级分并用离心浓缩器(spin concentrator)浓缩到35mg/ml。将纯化的、部分去糖基化的Kit胞外域制备物分成等分并在液氮中速冻。使用这种方法,从15升培养细胞中纯化~10毫克部分去糖基化的Kit胞外域。如前所述(Zhang等人.(2000)Proc Natl Acad SciU S A 97:7732-7737),SCF(1-141)被表达、重折叠和纯化。如前所述(Lemmon等人.(1997)J Biol Chem 272:6311-6317),Kit胞外域(氨基酸1-514)也利用杆状病毒表达系统以分泌形式在Sf9昆虫细胞中表达和纯化。
实施例11:结构的确定和优化
实验相由Kit胞外域单体晶体的多波长异常衍射(MAD)和反常散射的多重同晶置换(MIRAS)的组合来测定。使用CNS(Brunger等人.(1998)Acta Crystallogr D Biol Crystallogr 54:905-921)和SHARP(Bricogne等人.(2003)Acta Crystallogr D Biol Crystallogr 59:2023-2030)程序套装完成重原子检索和定相。对于铂衍生物(K2Pt(NO2)4)检测到一个主要的和两个次要的位点且对于碘浸泡晶体检测到一个主要的和五个次要的位点。使用CNS在三个波长下计算铂衍生物的MAD相最高达 分辨率。使用CNS和SHARP计算铂和碘衍生物的MIRAS相最高达
分辨率。使用CNS和SHARP计算铂和碘衍生物的MIRAS相最高达 分辨率。溶剂翻转(solvent flipping)密度改变导致带有连续电子密度和非常明确的溶剂蛋白质边界的可判断质量的电子密度图。通过比较由MIRAS和MAD定相计算的电子密度图确认了低电子密度质量区域,包括F、G和C链的上半部以及D2中的CD环和D5中的CD环、D链和EF环和F链的下半部。数据收集和定相统计总结于表1A和1B中。用COOT将Kit分子模型人工建立成实验电子密度图(Emsley和Cowtan(2004)Acta Crystallogr D Biol Crystallogr 60:2126-2132)。为计算自由R-因子,在优化过程中略去5%的数据。使用CNS对原始数据进行优化以达到
分辨率。溶剂翻转(solvent flipping)密度改变导致带有连续电子密度和非常明确的溶剂蛋白质边界的可判断质量的电子密度图。通过比较由MIRAS和MAD定相计算的电子密度图确认了低电子密度质量区域,包括F、G和C链的上半部以及D2中的CD环和D5中的CD环、D链和EF环和F链的下半部。数据收集和定相统计总结于表1A和1B中。用COOT将Kit分子模型人工建立成实验电子密度图(Emsley和Cowtan(2004)Acta Crystallogr D Biol Crystallogr 60:2126-2132)。为计算自由R-因子,在优化过程中略去5%的数据。使用CNS对原始数据进行优化以达到 的分辨率。在优化的最终阶段,在具有使用TLSMD网络服务器生成的三个TLS组的CCP4程序套装中的Refmac5(Murshudov等人.(1997)Acta Crystallogr D Biol Crystallogr53:240-255)进行翻译/释放/螺旋(translation/liberation/screw)(TLS)优化。
的分辨率。在优化的最终阶段,在具有使用TLSMD网络服务器生成的三个TLS组的CCP4程序套装中的Refmac5(Murshudov等人.(1997)Acta Crystallogr D Biol Crystallogr53:240-255)进行翻译/释放/螺旋(translation/liberation/screw)(TLS)优化。
使用PHASER通过分子置换解析了SCF-Kit复合体的结构(McCoy等人.(2005)Acta Crystallogr D Biol Crystallogr 61:458-464)。使用PHASER,分别利用Kit和SCF的D1D2D3和D4作为用于原始数据集的检索模型发现了对于Kit胞外域和SCF的D1D2D3D4的明确分子置换方案。该Kit(D1D2D3D4)-SCF复合体结构经过从20到 的刚性体优化,从而得到43.8%的Rcryst。使用CNS进行模型重建和优化以使Rcryst和Rfree值分别达到31.6%和34.0%。在2σ2Fo-Fc和3σFo-Fc图中发现D5区域中的连续电子密度。使用COOT将D5链人工描绘在图中,每步结束后都进行优化。通过初步优化,非晶体学对称(NCS)的限制施加于残基上。对原始的X射线衍射数据进行进一步优化以达到
的刚性体优化,从而得到43.8%的Rcryst。使用CNS进行模型重建和优化以使Rcryst和Rfree值分别达到31.6%和34.0%。在2σ2Fo-Fc和3σFo-Fc图中发现D5区域中的连续电子密度。使用COOT将D5链人工描绘在图中,每步结束后都进行优化。通过初步优化,非晶体学对称(NCS)的限制施加于残基上。对原始的X射线衍射数据进行进一步优化以达到 分辨率。在构建几乎整个SCF-Kit复合体分子后,解除NCS限制从而导致R和Rfree值的降低和电子密度的改进。在优化的最后阶段,NCS限制被完全解除。用PROCHECK对模型的立体化学进行了分析(Laskowski等人.(1993)J Appl Cryst 26:283-291)。优化统计学概括于表1B中。
分辨率。在构建几乎整个SCF-Kit复合体分子后,解除NCS限制从而导致R和Rfree值的降低和电子密度的改进。在优化的最后阶段,NCS限制被完全解除。用PROCHECK对模型的立体化学进行了分析(Laskowski等人.(1993)J Appl Cryst 26:283-291)。优化统计学概括于表1B中。
实施例12:SCF的放射性标记和配体置换试验
根据制造商的说明书,利用Iodo-Gen Iodination Tubes(Pierce)用1mCi的125I(PerkinElmer)对人SCF(10μg)进行标记。对于置换结合试验,表达WT Kit或Kit突变体的3T3细胞在含10%FCS的DMEM中生长。用含10mM HEPES pH 7.4和0.1%BSA的DMEM(DMEM-BSA)洗涤细胞三次,然后在存在渐增浓度的天然SCF的情况下用2ng的125I-标记的SCF室温下孵育1小时。然后细胞用冰冷的DMEM-BSA洗涤三次,室温下在0.5ml的0.5M NaOH中裂解1小时,并将100μl细胞裂解液加入到10ml Opti-Fluor闪烁液(Perkin Elmer)中以使用LS6500闪烁计数器(Beckman Coulter)来测量细胞相关放射性。
实施例13:保守性分析
使用PSI-BLAST(Altschul等人.(1990)J Mol Biol 215:403-410),用人SCF和Kit的氨基酸序列作为查询序列以检索非冗余数据库(nr)的同源序列。采用ClustalW在SCF序列或Kit序列上进行序列比对(Higgins(1994)Methods Mol Biol 25:307-318),然后根据Kit Ig-样结构域中的20个关键残基的IgSF折叠限制人工调整。由SCF-Kit复合体晶体结构揭示的氨基酸序列的比对提交到Consurf 3.0服务器(Landau等人.(2005)Nucleic Acids Res 33:W299-302)以产生比对的各位置的最大似然性归一化进化率,其中低趋异率对应于高序列保守性。对于Consurf输出,将连续保守性评分分配到9项的离散标度中以实现可视化,从而第9项包含了最保守(栗色)的位置和第1项包含最可变(蓝绿色)的位置。
实施例14:蛋白表达、纯化及抗体制备
使用PCR反应从全长人Kit的cDNA扩增编码人Kit第四Ig-样结构域(残基309-413;Kit D4)的DNA。在16℃孵育过夜后,用指导Kit D4合成的细菌表达载体(pET-NusA带组氨酸标签的)转化BL 21(DE 3)大肠杆菌密码子+细胞。使用金属螯合亲和层析柱(Ni-NTA;QIAGEN)从BL 21裂解液纯化Kit D4-NusA融合蛋白后,用阴离子交换层析进一步纯化(Source Q柱;GE Healthcare)。然后为了将NusA和组氨酸标签与D4切除,Kit D4-NusA在4℃下与TEV蛋白酶孵育过夜。使用凝胶过滤色谱(Ni-NTA;QIAGEN)对Kit D4进行附加的纯化步骤。
在大肠杆菌BL 21株(DE 3)细胞中表达人Kit的第五Ig-样结构域(残基410-519;Kit D5)并使用含有6.0M盐酸胍的10mM Tris缓冲液pH 8.0通过重折叠步骤将其从细菌包涵体纯化。用阴离子交换层析(Q琼脂糖柱;GE Healthcare)进一步纯化重折叠的Kit D5后,进行凝胶过滤色谱(Superdex 200柱;GE Healthcare)的纯化和阴离子交换层析(Q琼脂糖柱;GE Healthcare)的附加纯化步骤。
用本研究领域熟知的技术如实施例1中所描述的方法生成抗分离的D4、D5或抗整个Kit胞外域(氨基酸1-519;Kit EC)或抗包含人Kit的C-末端的片段(残基876-976)的GST-融合蛋白的兔多克隆抗体。例如,可以通过用纯化Kit胞外域免疫兔并用标准方法收集所产生的抗体来制备Kit胞外域的多克隆抗体。其中测试抗体对Kit的激活的效应的实验(如实施例15和图14中)用经过A蛋白亲和层析纯化的抗体制剂进行。
实施例15:使用针对Kit的D5结构域的抗体抑制SCF-诱导的Kit活化
表达人Kit的3T3细胞与包含增加浓度的针对分离的重组Kit的D5产生的多克隆兔抗体一起温育(图14)。作为对照,细胞用针对SCF的兔多克隆抗体或针对完整Kit胞外域的兔多克隆抗体处理,所述完整Kit胞外域在使用杆状病毒表达系统的昆虫细胞中产生。采用抗Kit抗体使细胞溶解产物进行免疫沉淀,接下来进行SDS-PAGE并用抗Kit抗体或抗p-Tyr抗体进行免疫印迹(图14)。
该实验表明抗D5抗体阻断Kit的SCF诱导的酪氨酸自磷酸化。
实施例16:通过分离的重组Kit D4结构域抑制SCF-诱导的Kit活化
表达Kit的3T3细胞与增加浓度的、在大肠杆菌中表达的纯化重组D4在23℃温育10分钟,接着进行SCF温育。采用抗Kit抗体对未刺激或刺激的细胞的溶解产物进行免疫沉淀反应,接下来进行SDS-PAGE并用抗Kit抗体或抗p-Tyr抗体进行免疫印迹(图15)。
该实验表明分离的D4的存在干扰Kit的SCF诱导的酪氨酸自磷酸化。
实施例17:SCF诱导的Kit刺激实验
在包含10%小牛血清的DMEM中培养表达人Kit的3T3细胞。在SCF刺激前,如Yuzawa et al(2007)Cell,130:323所述在无血清培养基中使细胞饥饿过夜。用包含10mM的HEPES(pH 7.4)和0.1%BSA的冷DMEM洗涤饥饿细胞三次,接下来与增加浓度的抗体或与Kit-D4在23℃温育10分钟,如在图14或图15中所示。细胞用100ng/ml SCF在23℃刺激10分钟,并用冷PBS洗涤三次。采用抗Kit抗体,使未刺激或SCF刺激的细胞的溶解产物发生免疫沉淀,接下来进行SDS-PAGE并用抗Kit抗体或抗p-Tyr抗体进行免疫印迹。
实施例18:在PDGFRβ的D4中关键氨基酸的点突变阻止PDGF诱导的PDGF受体β的活化和经由PDGFRβ的信号传导
来源于表达WT PDGFRβ或在D4中关键氨基酸的点突变体(基于在Kit胞外域X射线晶体结构中与D4-D4界面的序列相似性)的PDGFR-/-小鼠的小鼠胚胎成纤维细胞(MEF)被用来证明,R385或E390的突变阻止PDGF诱导的受体活化(图16A)、或者PDGF-诱导的MAP激酶应答和Akt刺激(图16B)。而且,使用采用共价交联剂的交联实验,我们证明,E390A点突变不干扰PDGF诱导的受体二聚化。然而,与以活化状态存在于细胞表面的WT PDGFRβ共价交联的二聚体不同,E390A突变体的共价交联的二聚体是无活性的(图16C)。该实验表明在D4中的关键E390残基的突变阻止对于PDGFR活化必需的D4-D4相互作用。然而,PDGF诱导的PDGFR二聚化不受阻止受体活化的D4中的点突变影响,表明D4-D4在介导胞外域的近膜区的定位以能够活化PDGFR的酪氨酸激酶结构域中起重要作用。
因此,本发明的一个实施方式包括结合或靶向PDGFR中的残基R385或E390的成分。所述成分可用于灭活所述受体,而保持受体二聚化。本实施例也表明,基于一个RTK的晶体结构(在该情况下是Kit胞外域晶体结构)的信息可以被容易地转移到其它RTK。这里,在鉴定对于PDGF受体的活化而言重要的氨基酸方面,关于Kit D4的认识是正确的。一组涉及PDGFR的更详细的实验被描述于实施例22-25中。
实施例19:Kit胞外域的分子表面分析
在本文描述的SCF结合之前和之后测定Kit的完整胞外域的晶体结构已经表明,SCF诱导的受体二聚化之后是在两个相邻Kit分子的近膜Ig样结构域D4和D5之间的同型侧向相互作用。同型D4和D5相互作用使得两个相邻受体的胞质酪氨酸激酶结构域以能够进行酪氨酸自磷酸化和激酶活化的距离和取向进行定位。在本文也已证明,对于D4同型相互作用关键的单一氨基酸残基的突变影响SCF诱导的Kit活化和PDGF诱导的PDGF受体活化(参见实施例22-25)。
在本文描述的结构分析提供了关于如何设计抑制性成分(诸如结合由RTK胞外域(例如D3、D4或D5区域)形成的空腔的浅区域中的构象表位或非连续表位的单克隆抗体或结合Kit和其它类型RTK的胞外域的D3-D4和D4-D5铰链区的小分子抑制剂)的新的见解。在所述胞外域中四个区域最初被定靶:(A)可以产生结合D3-D4铰链区并作为分子楔起作用的本发明的成分,所述分子楔阻止近膜区以能够活化酪氨酸激酶的距离和取向进行定位所需的运动(参见图17);(B)可以产生结合D4-D5铰链区并作为分子楔起作用的成分,所述分子楔阻止近膜区以能够活化酪氨酸激酶的距离和取向进行定位所需的运动(参见图18);(C)可以产生结合D4:D4界面而阻止同型D4受体相互作用的成分(参见图19);(D)可以产生结合D2-D3铰链区处的凹面而导致配体-受体相互作用不稳定的成分(参见图20);和(E)可以产生结合在Kit表面上的形成各种连续和非连续表位的肽区域(表5)的成分。
使用Computed Atlas of Surface Topography of proteins(CASTp)服务器分析Kit胞外域和SCF-Kit复合物(PDB码:2EC8和2E9W)的分子表面,以提供与在蛋白质三维结构上的凹面区域的位置有关的信息,并使得能够描绘和测量蛋白质三维结构上的凹面区域(Dundas等,(2006)Nucl Acids Res,34:W116-W118)。使用Pymol可视化并检查所鉴定的空腔(DeLano.(2002)DeLano Scientific,San Carlos,CA,USA)。
(A)在D3-D4铰链区中的空腔(图17)
若干空腔分散在胞外域单体结构中的D3-D4界面上。参与界定所述空腔的氨基酸在表4中概述。在两个Kit受体之间形成同型相互作用时,D3-D4铰链区被改变,导致形成由下列残基产生的浅的空腔:来自D3的K218、S220、Y221、L222和来自D4的F340、P341、K342、N367、E368、S369、N370、I371、Y373。图17显示了未被占据的单体(图17A)和SCF结合的二聚体(图17B)D3-D4铰链区的带状栅图以及D3-D4口袋的网格表示。
(B)在D4-D5铰链区中的空腔(图18)
通过Kit单体中D4的AB环和EF环、D4-D5连接接头和D5的DE环和FG环的部分形成小空腔。界定上述空腔的残基在表4中概述。所述空腔的形状和大小在Kit胞外域二聚结构中被改变。由D4的EF环和G链、D4-D5接头和D5的B链和DE环形成的主要空腔位于D4的EF环之下,一个对于D4同型界面的形成很关键的区域。注意,位置靠近所述空腔的D5的DE环可具有较高的灵活性,如通过来自非结合的和占据的Kit结构两者的较低电子密度特性所揭示的。图18显示了未被占据的单体(图18A)和SCF二聚体(图18B)的带状栅图以及在D4-D5铰链区周围的浅空腔的网格表示。
(C)在介导D4同型相互作用的区域中的空腔
由Kit D4的CD环和EF环形成的凹面正好位于D4同型界面的上方。来自D4的残基Y350、R353、F355、K358、L379、T380、R381、L382、E386和T390为胞外域二聚结构中的凹面提供了大约130A2的表面积。在D4同型界面中起重要作用的Glu386的侧链伸向所述表面的中心。所述凹面的特征是由带电残基(Glu386和Lys358)围绕的小的疏水性补丁。在同型D4:D4相互作用时所述表面的尺寸和可及性被改变,使得变化发生在CD环的构象中,其变成向着所述结构域顶部向上折叠。参与凹面形成的残基在表4中概述。图19A描绘了覆盖在配体占据的Kit D4(未示出)之上的Kit的未被占据的D4结构域(金色)的带状栅图,其中在配体占据的(绿色)和未被占据的胞外域结构(红色)之间CD和EF环的构象不同。用于D4:D4相互作用的关键残基以棍型形式示出。图19B和19C示出了未被占据的Kit(图19B)和SCF占据的Kit结构(图19C)的带状栅图以及D4同型界面之上的浅空腔的网格表示。
(D)配体结合D2和D3区域的凹面
浅的凹面位于D2和D3结构域的一部分配体结合表面上。包括在小口袋中的残基是来自Kit的D2结构域的Y125、G126、H180、R181、K203、V204、R205、P206和F208,以及来自D3结构域的V238、S239、S240、S241、H263、G265、D266、F267、N268和Y269。通过由亲水性残基围绕的小的疏水性补丁产生所述口袋。在未被占据的和SCF占据的Kit结构之间没有大的改变,其中总包埋表面积为大约500A2。图20A和20B显示了未被占据的Kit(A)和SCF结合的Kit(B)的带状栅图以及D2-D3口袋的网格表示。
表4


SCF-Kit复合体molA,C(表4续)

(E)如上所述进行KIT酪氨酸激酶的结构分析。所述分析显示出可以是本发明的成分的靶标的连续和非连续表位。在表5中,表位1、4、5、6、8、12-16、19、22-23和31-39是连续表位。这些表位由KIT蛋白质中的连续氨基酸组成。表5中的表位2、3、7、9-11、17、18、20、21、24-30和40-43是非连续构象表位,其由通过KIT蛋白质的折叠而使其接近的KIT蛋白质的至少2个肽组成。
表5




实施例20:RTK活性测定
使包含目标RTK的细胞暴露于所述受体的活化配体和本发明的成分。通过标准分子生物学方法(例如抗体纯化)可以分离目标RTK。在纯化之后,结合RTK的抗体(不是本发明的成分,而仅仅是结构结合物,如用于纯化)被预包被在96孔微量滴定板上。然后,将RTK和校准标准物添加至RTK蛋白质被捕获的单独的孔中。接下来添加检测抗体,其可以是磷酸位点特异性的(例如c-Kit pY823或在活化时被磷酸化的Kit的其它残基;phosphoELISATM系统使用兔抗体)。使用被结合到标记或酶(例如辣根过氧化酶被用于phosphoELISATM系统)的二抗(例如抗兔Ab,以检测兔来源的一抗),之后使用比色底物检测抗体-Kit复合物。然后,添加终止溶液,并对所述板进行读数(例如使用450nm光源和检测器)。phosphoELISATM的详细方案可从Invitrogen获得(invitrogen.com/content.cfm?pageid=11655;invitrogen.com/downloads/F1027_BN_pELISA1006.pdf;invitrogen.com/downloads/F1028_BN_pELISA1006.pdf C-KIT[pY823]ELISA KIT,HU(BioSourceTM)Catalog Number-KHO0401;c-KIT[TOTAL]ELISA KIT,HU(BioSourceTM)Catalog Number-KHO0391)。
实施例21:受体内化作用测定
表达目标RTK的细胞首先与适当的配体一起温育(例如表达Kit的细胞与SCF一起温育),从而诱导受体内化作用。通过在冷PBS中洗涤细胞终止受体内化过程。然后,通过在具有足以解离所述配体盐浓度和/或pH水平的溶液中洗涤所述细胞除去残留的表面结合的配体。然后,将细胞重悬于适当的缓冲液中。此时,所述细胞将包含内化的受体,因此较少量的受体仍保持在细胞表面上。
进行另一组相似的实验,其中所述细胞暴露于适当的配体和本发明的测试成分。如果所述测试成分阻止靶RTK的活化,则受体内化作用将被抑制。当相比于上述实验中描述的细胞(其中受体活化发生)时,这些细胞显示出降低的内化作用和在细胞表面上更大量的受体。也设立对照组,其中细胞仅仅用缓冲液或乙醇溶液——一种药物溶解的常见载体——处理。
在上述实验中在细胞表面上的受体的量的测定可以通过使所述细胞与对所述受体具有特异性的小鼠抗体一起温育,接下来与结合于荧光团诸如绿色荧光蛋白(GFP)的抗小鼠抗体一起温育而实现。然后,荧光显微术可用来显像和对细胞表面上的受体进行定量。
细胞表面受体的定量或显像的可选技术在本领域中是熟知的,并包括各种荧光和放射性技术。例如,一种方法包括使细胞与放射性标记的抗受体抗体一起温育。可选地,所述受体的天然配体可以被结合于荧光分子或放射性标记并与所述细胞一起温育。另外的受体内化作用测定方法是本领域熟知的并描述于例如:Jiménez等(1999)Biochemical Pharmacology.57(10):1125-1131;Bernhagen等(2007)Nature Medicine.13(5):587-596;和Conway等(2001)J.Cell Physiol.189(3):341-55,其中每一个文献的全部内容通过引用的方式并入本文。
实施例22-25的介绍
受体酪氨酸激酶(RTK)活化的一般公认机理是,配体诱导的受体二聚化促进在催化核心的活化环中关键调节性酪氨酸残基的反式自磷酸化;这是一个对于酪氨酸激酶活化必需的步骤。这之后进行所述胞质域中的多个酪氨酸残基的自磷酸化,所述多个酪氨酸残基充当各种信号传导蛋白质的SH2(Src同源物2)或PTB(磷酸酪氨酸结合)结构域的结合位点,所述信号传导蛋白质在募集和/或酪氨酸磷酸化时以受控方式发送信号至各种胞内区室(Schlessinger,J.(2000)Cell 103,211-225;Pawson,T.& Nash,P.(2003)Science 300,445-452;和Hunter,T.(2000)Cell 100,113-127)。
尽管全部RTK都通过二聚化而活化,但不同的RTK家族已经进化成利用不同的分子策略来进行配体诱导的受体二聚化和活化(Burgess,A.W.等(2003)Mol Cell 12,541-552;Schlessinger,J.等(2000)Molecular Cell 6,743-750)。III型RTK(包括PDGF、SCF、CSF和Flt3L)的全部配体是能够通过二价结合于两个相邻受体分子的等同位置而交联它们的同族受体的二聚分子。PDGF原体由中央的四链β-折叠组成,其在分子的一端具有特征性的半胱氨酸结。两个PDGF原体以反平行的方式排列并且通过两个链间二硫桥彼此连接(Oefner,C.等(1992)EMBO J.11,3921-3926.)。相反,各SCF、CSF或Flt3L原体由短螺线折叠组成并且通过非共价相互作用彼此连接(Jiang,X.等(2000)Embo J 19,3192-3203;Zhang,Z.等(2000)Proc Natl Acad Sci US A 97,7732-7737;Pandit,J.等(1992)Science 258,1358-62;和Savvides,S.N.等(2000)Nat Struct Mol Biol 7,486-491)。尽管它们的多样性折叠,这两种生长因子亚型结合并以基本上相同的方式激活它们的同族RTK,导致活化配体/RTK 2:2复合物的形成(Savvides,S.N.等(2000)Nat Struct Mol Biol 7,486-491)。所有III型RTK由细胞外配体结合区和胞质酪氨酸激酶结构域组成,所述细胞外配体结合区包含五个串联Ig样结构域,继之以单个跨膜螺旋,而所述胞质酪氨酸激酶结构域具有与发生自磷酸化和通过异源蛋白质激酶的磷酸化的调节区侧邻的大的激酶插入区域(Hubbard,S.R.(1999)Prog BiophysMol Biol 71,343-358)。
通过分析基于相关受体酪氨酸激酶Kit的细胞外区域的晶体结构设计的突变体受体的性质探索PDGF受体β(PDGFRβ)活化的机理。基于这些实验,证明了在D4(细胞外区的第四Ig样结构域)中Arg385或Glu390突变的PDGFRβ的PDGF诱导型活化受到影响,导致各种PDGF诱导的细胞应答受损。这些实验也表明,同型D4相互作用(其可能通过在Arg385和Glu390之间的盐桥介导)在PDGFRβ和全部III型RTK的活化中起重要作用。化学交联剂也被用于共价交联PDGF-刺激的细胞,以证明PDGFRβ的Glu390Ala突变体经历典型的PDGF诱导的受体二聚化。然而,与在配体刺激的细胞的表面上表达的以活化态的WT PDGFR不同,PDGF诱导的Glu390Ala二聚体是无活性的。尽管介导D4同型相互作用所需的保守氨基酸对于PDGFRβ活化(以及III型RTK中的相似相互作用)是关键的,但是这些相互作用对于PDGFRβ二聚化不是必要的。而且,PDGFRβ二聚化对于酪氨酸激酶活化是必需的,但不足以活化酪氨酸激酶。
类似于Kit的D4结构域,PDGFRα和PDGFRβ的D4结构域缺乏桥接位于Ig样结构域的B5和F5中的半胱氨酸残基的特征性二硫键。在图21中给出的氨基酸序列比对表明,I组IgSF折叠的20个指纹残基中的13个在PDGFR的D4结构域中是保守的,并且相应于指纹残基的链的编号和长度在Kit、PDGFRα、PDGFRβ和CSF1R的D4结构域中是高度保守的。这表明,本发明的抑制剂可以被设计来抑制包括全部III型RTK的各种受体分子。
Kit的D4结构域由两个β折叠组成,各包含具有ABED/A’GFC排列的四个链,并且同型D4接触通过从两个相邻Kit分子伸出的D4的EF环介导。在本文披露的Kit结构表明,EF环中的Arg381和Glu386跨过Kit二聚体的二重轴形成盐桥和范德瓦耳斯接触。此外,各原体的Arg381的侧链与相邻Kit分子的相应残基的主链羰基形成氢键。基于结构的序列对比已经表明,EF环的尺寸和D4-D4界面包含的关键氨基酸在Kit、PDGFRα、PDGFRβ和CSF1R中是保守的。在PDGFRα中,Glu386被天冬氨酸替代,这是也可起盐桥伴体作用的残基。此外,一对碱性和酸性(Glu/Asp)残基在从红鳍东方鲀(Takifugu rubripes)到人的不同物种的PDGFRα和PDGFRβ中是严格保守的(图21),从而为该区域的功能重要性提供了进一步的支持。如此,靶向于具有不同氨基酸序列的RTK(例如III型RTK)或靶向于在本文描述的那些具有相似功能的变体结构域的本发明的成分也属于本发明的范围。
与实施例22-25相关的方法
序列对比和同源性建模
使用CONSEQ服务器(Berezin,C.等(2004)Bioinformatics 20,1322-1324)以及根据IgSF折叠特征(Harpaz,Y.& Chothia,C.(1994)Journal of Molecular Biology 238,528-539)和根据人Kit结构的D4的Ig折叠的核心残基(Yuzawa,S.等(2007)Cell 130,323-334)进行氨基酸序列比对。各序列的获取码是:PDGFRα人(P 16234)、小鼠(P26618)、鸡(Q9PUF6)、蛙(P26619)和河豚(Q8AXC7);PDGFRβ人(P09619)、狗(Q6QNF3)、小鼠(P05622)、河豚(P79749)和Kit人(Q96RW7)。使用WHAT IF服务器(Rodriguez,R.等(1998)Bioinformatics 14,523-528)基于D4Kit结构(PDB码:2E9W)产生PDGFRβ的D4的同源性模型。使用PyMOL(Delano,W.L.;pymol.org)制图。
试剂和抗体
从Sigma购买L-组氨醇和抗标记抗体。抗MAPK、磷酸-MAPK、Akt、磷酸-Akt和磷脂酶Cγ的抗体购自Cell Signaling Technology。抗磷酸酪氨酸(4G 10)抗体来自于Upstate Technology。抗泛素抗体(P4D1)来自Santa Cruze。抗PDGFRβ的抗体通过用来自PDGFRβ的胞质域的合成肽免疫兔子而产生。PDGF BB cDNA获自Stuart Aaronson。PDGF BB购自Invitrogen,以及如原先所描述的在细菌中产生(Hoppe,J.等(1990)European Journal of Biochemistry 187,207-214)。125I放射性核素购自Perkin Elmer。Bolton-Hunter试剂和IODO-GEN预包被碘化管来自Piece。FITC-鬼笔环肽购自Invitrogen。
细胞系和逆转录病毒感染
来源于PDGFRα和PDGFRβ缺陷(PDGFRα/β)的小鼠胚的成纤维细胞由Philip Sariano和Andrius Kazlauskas提供。PDGFRβcDNA由Daniel DeMaio提供。PDGFRβcDNA被亚克隆到pLXSHD逆转录病毒载体中,且标记-标签被添加至所述受体的C-末端。根据生产商的说明(Stratagen),通过定点诱变产生在D4中的所有突变体。编码WT和突变体PDGFRβ的逆转录病毒在293GPG细胞中产生(Ory,D.S.等(1996)Proc.Nat.Acad.Sci.93,11400-11406)。感染之后,用L-组氨醇选择细胞,并且所选择细胞的集合被用于实验。
免疫沉淀和免疫印迹
在包含50mM Hepes、150mM NaCl、1mM EDTA、1%TritonX-100、25mM氟化钠、1mM原钒酸盐、1mM苯基-甲磺酰氟、5μg的抑肽酶和亮抑酶肽的缓冲液(pH 7.5)中裂解未刺激的或PDGF刺激的细胞。用所示的抗体免疫沉淀等量的细胞溶解产物,通过SDS-PAGE解析免疫沉淀物并转移到硝酸纤维素膜。膜用不同的抗体进行免疫印迹。使用光密度计(Amersham)扫描膜并用Imagequant软件(Molecular dynamics)进行定量。
PDGFR的体外磷酸化测定
使细胞进行血清饥饿16小时,并在包含150mM NaCl、50mMHepes(pH 7.4)、1mM EDTA、25mM NaF、0.1mM原钒酸钠、5μg/ml亮抑酶肽和抑肽酶、1mM PMSF和1%NP40的裂解缓冲液中溶解。溶解产物用抗PDGFRβ抗体进行免疫沉淀,而免疫沉淀物在包含50mM Hepes(pH7.4)、1mM ATP和10mM MgCl2的反应缓冲液中在室温下温育5分钟。在温育之后,通过SDS-PAGE分析沉淀物,继之以采用抗磷酸酪氨酸抗体的免疫印迹分析。剥去膜,用抗标记标签抗体进行再印迹分析以测定总PDGFRβ水平。
受体二聚体的化学交联
在150mm板中生长细胞直到达到80%汇合,并且在与所示浓度的PDGF在包含50mM Hepes(pH 7)的DMEM中在4℃温育之前,使细胞血清饥饿16小时。在90分钟之后,用PBS(pH 7.4)彻底洗涤细胞。板被转移到室温下并且辛二酸二琥珀酰亚胺酯(DSS)被添加至0.5mM的终浓度。在30分钟之后通过与10mM Tris缓冲液温育15分钟淬灭交联反应,继之以深度的PBS洗涤火。在50mM Hepes、150mM NaCl、1mM EDTA、1%Triton X-100、25mM氟化钠、1mM原钒酸钠、1mM苯基甲磺酰氟、5μg/ml抑肽酶和5μg/ml亮抑酶肽(pH7.4)中的细胞溶解产物用抗PDGFR抗体进行免疫沉淀,并通过SDS-PAGE解析。硝酸纤维素膜用针对标记-标签的抗体或抗磷酸酪氨酸(4G10)抗体进行免疫印迹,以分别检测总受体和磷酸化受体的水平。
PDGF诱导的肌动蛋白细胞骨架重组
将MEF在盖玻片上接种24小时这到亚汇合,继之以血清饥饿过夜。细胞或者用50ng/ml PDGF处理2、5、10或30分钟,或者不进行处理。细胞在4%低聚甲醛PBS溶液中固定、用0.1%Triton的PBS溶液渗透,并用在包含1%BSA的PBS中用FITC-鬼笔环肽(Sigma)染色30分钟。用Prolong Antifade封固剂(Invitrogen)固定盖片,并用Nikon荧光显微镜获得图像。分析各盖片上的约400个细胞,计算显示出肌动蛋白环形成的细胞的百分比,并线性地呈现。
PDGF结合和内化作用实验
根据生产商的说明,在使用Iodo-gen碘化管(Pierce)碘化之前使用Bolton-Hunter试剂(Pierce)标记PDGF。将细胞接种入24孔板,并使其在补充有胎牛血清的DMEM中生长至80%汇合。在包含20mMHepes(pH7.4)和0.1%BSA的冷DMEM中洗涤细胞两次。在增加量的天然PDGF存在的情况下,用5ng/ml的125I-PDGF在孔中进行温育,一式三份。在25℃使结合进行1小时。然后,在冷PBS中洗涤细胞,并使其溶解在0.5M NaOH中。使用LS6500闪烁计数器(BeckmanCoulter)测定样品的放射性含量,并使用PRISM软件(GraphPad)分析数据。
对于内化作用实验,细胞被接种入24孔板,使其生长至80%汇合,并饥饿过夜。在4℃,用在DMEM/0.1%BSA/50mM Hepes pH7.4中的5ng/ml 125I-PDGF温育细胞90分钟。通过用冰冷的PBS(pH 7.4)洗涤除去未结合的配体。预加热的DMEM/0.1%BSA/50mM Hepes被添加至所述细胞并在37℃温育所示的时间。用包含PBS(pH 3)和0.1%BSA的冰冷酸性缓冲液收集细胞表面结合配体10分钟。通过用0.5MNaOH溶解收集内化的配体。通过用10%三氯乙酸(TCA)沉淀培养基并对上清液进行TCA可溶组分计数测定降解的125I-PDGF的量。通过液体闪烁计数器测定细胞表面结合的、内化的和释放的放射性。表面结合的、细胞内的和降解的PDGF的量被表示为在冰上温育90分钟之后总细胞结合放射性的百分比(t=0分钟)。每一个时间点进行三次,结果表示为平均值±SE。
实施例22:PDGF诱导的PDGF受体活化受到D4结构域中突变的影响。
图21A中给出的氨基酸序列比对表明,在PDGFR的EF环中的Arg385和Glu390可介导同型D4相互作用,其类似于在Kit的Arg381和Glu386之间负责介导在相邻Kit受体之间的同型D4相互作用的盐桥。为研究是否PDGFRβ采用相似的机理,Arg385和Glu390各自单独地(R385A、E390A)或组合地(R385E390/AA)被丙氨酸残基置换。在环区中另外的保守Lys387残基也被丙氨酸残基置换(R385K387E390/AAA),以检验其在调控PDGF诱导的PDGFRβ活化中的潜在作用。野生型和突变体PDGFRβ在来源于PDGFRα和PDGFRβ缺陷的小鼠胚的成纤维细胞(MEF)中稳定地表达(Soriano,P.(1994)Genes Dev.8,1888-1896;Soriano,P.(1997)Development 124,2691-2700;和Andrews,A.等(1999)Invest.Ophthalmol.Vis.Sci.40,2683-2689)。表达野生型或突变体PDGFRβ的MEF——其在表达水平方面相当——用于如下所述的实验。来自未刺激的或PDGF刺激的细胞的溶解产物用抗PDGFR抗体进行免疫沉淀,接下来用抗磷酸酪氨酸抗体进行免疫印迹。
接着剥去膜,并用抗PDGFR抗体再印迹以用于定量PDGFR表达。在图22A中给出的实验表明,PDGFRβ的PDGF诱导的酪氨酸自磷酸化在表达PDGFRβ的E390A、R385A、(R385E390/AA)和(R385K387E390/AAA)突变体的细胞中受到强烈损害;酪氨酸自磷酸化的程度和动力学分别被降低和渐弱。这些实验表明,D4的EF环中的Arg385和Glu390在PDGFRβ的PDGF诱导的刺激中起重要作用,这表明与在Kit结构中鉴定的那些类似的盐桥对也存在于活化的PDGFR和其它III型RTK中。在配体-受体复合物内相邻受体的D4结构域之间的直接相互作用可代表用于配体诱导的III型RTK活化的共同机理。已经一致地和可再现地观察到,与表达R385A、(R385E390/AA)或(R385K387E390/AAA)突变体的细胞相比,PDGF诱导的受体自磷酸化在表达E390A的细胞中受到更强烈的影响。尽管造成这些突变体之间的差异的精确机理尚不清楚,但可能的是,在D4界面处的局部正表面电荷可引起静电推斥,以在配体刺激之前保持相邻受体的D4分开。而Arg385被丙氨酸残基置换将阻止盐桥形成,该变化也可降低D4-D4界面中的净正电荷,导致对PDGFR活化的抑制较弱。
为了检验PDGFR的D4结构域中的突变是否可能影响突变体PDGFRβ的细胞膜表达和配体结合亲和性,接下来对表达野生型或突变体PDGFRβ的细胞进行定量PDGF结合实验。在存在浓度增加的天然PDGF的情况下,在4℃下,表达野生型、R385A、E390A或(R385E390/AA)PDGFRβ突变体的细胞与包含125I-PDGF的缓冲液一起温育90分钟。使用闪烁计数器测量细胞结合放射性。通过用Prism4进行曲线拟合分析野生型和突变体PDGFRβ的置换曲线的EC50值(图22B)。在转染的MEF中表达的野生型和突变体PDGFRβ的量也通过用抗PDGFR的抗体或抗标签抗体进行总细胞溶解产物的免疫印迹而进行比较(图22A和C)。合起来,这些实验表明,相似量的野生型或突变体PDGFRβ被表达于转染细胞的细胞表面上。而且,对于表达野生型(3.7nM)、R385A(6.0nM)、E390A(2.8nM)或RE/AA(3.0nM)突变体的细胞获得相似的IC50值(置换50%的125I-PDGF结合的PDGF浓度)。通过比较野生型和突变体受体的体外酪氨酸激酶活性,突变体PDGFRβ的固有酪氨酸激酶活性是否受到不利地影响也被检验。在该实验中,用抗PDGFR抗体对来自血清饥饿细胞的细胞溶解产物进行免疫沉淀,并且使固定的PDGFR在存在1mM ATP和10mM氯化镁的情况下进行体外激酶分析。在温育之后,通过采用抗磷酸酪氨酸抗体的免疫印迹分析样品。在图22C中给出的实验表明,R385A、E390A或RE/AA突变不影响PDGFR的固有酪氨酸激酶活性。总而言之,这些实验表明在D4中影响PDGF诱导的PDGFRβ刺激的突变不改变PDFGRβ在细胞表面上的表达、不影响PDFGRβ的配体结合亲和性且不改变突变体PDGFRβ的固有酪氨酸激酶活性。
实施例23:PDGF受体D4点突变体在PDGF刺激的细胞的表面上以无活性二聚体的形式表达
因为受体二聚化已经被认为是受体酪氨酸激酶活化的关键机理,所以我们研究了突变体PDGFRβ响应PDGF刺激的酪氨酸自磷酸化减少是否由受体二聚化缺陷所引起。化学交联剂早先已经用来监测和追踪几种细胞膜受体(包括野生型和各种EGF受体突变体)在活细胞的细胞表面上的配体诱导二聚化(Cochet,C.等(1988)J Biol Chem263,3290-3295)。在该实验中,表达野生型PDGFRβ或E390A突变体的细胞被血清饥饿过夜,继之以在4℃进行PDGF温育90分钟。若干次洗涤被用来除去非结合的PDGF,并且用在PBS中的0.5mM辛二酸二琥珀酰亚胺酯(DSS)在25℃温育细胞30分钟。使来自未刺激的或PDGF刺激的细胞的细胞溶解产物用抗PDGFR抗体进行免疫沉淀,继之以SDS-PAGE,并采用抗标记抗体进行免疫印迹以监控PDGFR二聚化的状态,或者采用抗磷酸酪氨酸抗体的免疫印迹以监控PDGFR活化的状态(图23)。
在图23中描绘的实验表明,在未刺激的细胞的溶解产物中,在SDS凝胶上迁移的具有相当于PDGFR单体的180kDa的表观分子量的带在来自表达野生型PDGFRβ或E390A突变体的细胞的溶解产物中检测到。在PDGF刺激后,在SDS凝胶上迁移的具有相当于PDGFR二聚体的360kDa的表观分子量的另外的带在表达野生型PDGFRβ和E390A突变体的细胞中都检测到。然而,采用抗磷酸酪氨酸抗体的样品免疫印迹表明,尽管相当于野生型PDGFR的二聚体的带是强酪氨酸磷酸化的,但是检测到相应于E390A突变体二聚体的带的酪氨酸磷酸化非常弱(图23)。
该实验表明,E390A突变体的配体诱导酪氨酸自磷酸化的削弱不是由配体诱导的受体二聚化缺陷所引起的。该实验也表明,共价交联的野生型PDGFRβ以活性二聚体的形式展示在PDGF刺激的细胞的细胞表面上,而E390A突变体以无活性二聚体的形式展示在PDGF刺激的细胞的表面上。上述数据表明,PDGFR中的D4同型相互作用不是受体二聚化所必需的,并且PDGF诱导的受体二聚是活化酪氨酸激酶必需的,但不足以活化酪氨酸激酶。
实施例24:在表达D4PDGF-受体突变体的细胞中细胞信号传导的刺激削弱
PDGFR D4突变对响应PDGF刺激的细胞信号传导的影响被检验。采用抗磷脂酶Cγ(抗PLCγ)抗体对来自未刺激的或PDGF刺激的表达WT或PDGFR D4突变体的细胞的溶解产物进行免疫沉淀,接下来进行SDS-PAGE,并用抗PLCγ或抗Tyr抗体进行免疫印迹。在图24A中给出的实验表明,在表达R385A、E390A、RE/AA或RKE/AAA的PDGFR突变体的细胞中,PLCγ的酪氨酸磷酸化受到严重损害。在表达PDGFR D4突变体的细胞中观察到另外的PDGF诱导的细胞应答的刺激削弱。在图24B中给出的实验表明,与在表达WT PDGFR的MEF中通过PDGF诱导的相似应答相比,在表达R385A、E390A、R385E390/AA或R385K387E390/AAA PDGFR突变体的细胞中MAP-激酶应答和Akt刺激受到强烈损害。总的说来,为在表达E390A、R385E390/AA(即RE/AA)或R385K387E390/AAA(即RKE/AAA)PDGFR突变体的细胞中达到相似的MAP-激酶应答和Akt刺激水平,需要大约10倍高的PDGF浓度。
培养的成纤维细胞的PDGF刺激的标志之一是在PDGF-刺激的细胞的背面上通常形成膜皱褶和圆形肌动蛋白环结构。在图25中给出的实验表明,肌动蛋白环形成的PDGF刺激在表达PDGFR D4突变体的MEF中受到损害。尽管大约83%的表达WT PDGFR的MEF表现出圆形肌动蛋白环形成,但是仅仅5%的PDGFR D4突变体细胞在用50ng/ml的PDGF刺激2分钟之后显示出相似的圆形肌动蛋白环形成。而且,在2-5分钟的PDGF刺激之后在表达WT PDGFR的MEF中达到峰值的瞬时圆形肌动蛋白环形成在表达R385A、E390A或RE/AAPDGFR突变体的细胞中被微弱地检测到。
实施例25:D4 PDGF受体突变体的内化作用和降解减少
PDGFR D4突变对PDGF内化作用、PDGFR降解和PDGFR泛素化的影响也被检验。在4℃用5ng/ml的125I标记的PDGF处理表达WT PDGFR或PDGFR D4突变体的MEF 90分钟,继之以用PBS(pH7.4)短暂清洗以除去培养基中的过量配体。将预标记的细胞温热至37℃,以最长4小时的各种时间长度开始配体-受体复合物的胞吞作用。细胞表面结合的、细胞内的和在培养基中的降解的125I-PDGF被收集、使用闪烁计数器定量、并作为在4℃温育90分钟(t=0)之后总细胞结合125I PDGF放射性的百分比(平均值±SD)给出。在图26A中给出的实验表明,结合于表达WT PDGFR的MEF的125I标记的PDGF的内化作用动力学比结合于表达E390A、R385A或R385E390/AAPDGFR突变体的细胞的125I标记的PDGF的内化作用动力学快得多。在30分钟之后,约75-80%的125I-PDGF被从细胞表面除去并在表达WT受体的细胞内部累积,这与在表达突变体的细胞中的50%以下的比率形成对比。
125I-PDGF的低分子量降解产物在30分钟之后变成可检测的。降解的125I-PDGF的释放在E390A突变体细胞中比在WT中慢得多(图26A)。PDGF内化作用和降解减少被反映在PDGFR D4突变体的降解减少。表达WT或R385A、E390A或R385E390/AA PDGFR突变体的细胞首先与放线菌酮温育30分钟,以防止在降解实验期间新的PDGFR分子的生物合成。采用抗PDGFR抗体使未刺激的或PDGF刺激的细胞的溶解产物进行免疫沉淀,继之以SDS-PAGE,并采用针对连接于WT或PDGFR D4突变体的C-末端的标签的抗体进行免疫印迹。图26B中给出的实验表明,R385A、E390A或R385E390/AA PDGFR突变体的降解动力学被大大削弱;尽管一半的WT PDGFR在PDGF刺激的1.5小时内被降解,PDGFR D4突变体的半衰期延长至大约4至6小时。在图26C中给出的实验表明,在相似条件下,相比于WTPDGFR,E390A PDGFR的PDGF诱导的泛素化刺激也被大大降低。总的说来,这些实验表明PDGFR内化和泛素介导的PDGFR降解受到PDGFR的D4中突变的损害。
实施例22-25的讨论
III型RTK的全部成员——包括PDGFRα、PDGFRβ、CSF1R、Flt3和Kit——的细胞外结构域由五个Ig样结构域组成,其中前三个用作二聚配体分子的结合位点,二聚配体分子在结合后刺激受体二聚经和活化。由于III型RTKs的分子结构、配体结合特征和受体二聚化的机理是高度保守的,由刺激之前和之后Kit的完整胞外域的晶体结构所揭示的SCF诱导的Kit活化的机理代表全部III型RTK的一般性活化机理。而且,在它们的细胞外结构域中包含Ig样结构域的RTK的系统发育分析显示出III型和IV型RTK的共同进化起源:一个包括VEGFR1(Flt1)、VEGFR2(KDR)和VEGFR3(Flt4)的家族。而且,VEGF和PDGF都属于同一半胱氨酸节家族:同型二聚化生长因子,共享相似的拓扑结构、大小和受体结合策略。因此,由它的细胞外结构域X-射线结构分析(在本文首次披露的)所揭示的Kit活化的显著特征也可适用于V型RTK的配体诱导活化。
Kit的结构分析已经表明,在两个相邻D4结构域的Glu386和Arg381之间形成的一对盐桥负责介导对于SCF诱导的Kit活化必需的同型D4相互作用。III型RTK的氨基酸序列的比较表明,相同序列基序存在于PDGFRα、PDGFRβ和CSF1R的D4的EF环中(图21),这提供了证据证明相似的盐桥也在III型RTK的D4之间形成。实际上,在PDGFRβ的D4结构域中Arg385或Asp390被丙氨酸置换已经影响PDGFRβ活化的PDGF刺激,导致在表达WT PDGFRβ的细胞中由PDGF刺激的多种细胞应答受损。由Kit结构的分析揭示的配体诱导的Kit活化的机理适用于全部III型RTK的活化。与负责D4同型相互作用的序列基序相同的序列基序也在RTK的VEGFR家族(IV型)的全部三个成员的近膜第七Ig样结构域(D7)的EF环中被鉴定。尽管负责在Kit和其它III型RTK中介导同型D4相互作用的保守序列基序位于IV型RTK的D7结构域中,VEGFR的D7可能在介导IV型RTK的近膜区之间的同型相互作用中起相似的作用。实际上,VEGFR2的细胞外结构域结构的电子显微镜分析已经在VEGF结合的VEGFR2二聚体中显示出D7之间的直接接触(Ruch,C.等(2007)NatStruct Mol Biol 14,249-250)。在近膜Ig样结构域之间的直接接触代表III型和IV型RTK都使用的一般机理。
研究各种受体突变体或使用特异性结合Kit(Blechman等(1995)Cell 80,103-113),PDGF受体(Miyazawa,K.等(1998)J.Biol.Chem.273,25495-25502)和其它III型RTK的单个Ig样结构域的单克隆抗体的研究已经揭示,即使当Kit由单价SCF配体刺激时,D4也在介导受体二聚化中起作用(Lev,S.等(1992)J Biol Chem 267,15970-15977)。然而,使用SCF结合的微量量热学和SCF化学计量法对由前三个Ig样结构域(D1-D3)或全部五个Ig样结构域(D1-D5)组成的Kit的纯化细胞外结构域的定量分析已经表明,D4和D5对于Kit二聚化的SCF刺激不是必需的。换句话说,这些报道已经表明,Kit二聚化主要由结合Kit的SCF的二聚性质驱动(Lemmon,M.A.等(1997)J.Biol.Chem.272,6311-6317.)。
然而,在本文中描述的工作表明,对于两个受体的近膜区以使它们的胞质域之间的相互作用能导致酪氨酸激酶活化的距离和取向进行精确定位而言,在相邻受体之间的同型D4(以及同型D5)相互作用是必需的,而不是在受体二聚化中起作用。因此,本发明的成分(例如单克隆抗体)通过阻止在III型RTK的近膜区之间的关键同型相互作用而对受体活化发挥它们的抑制作用,而不是干扰受体二聚化,其中所述同型相互作用对于胞质域以酪氨酸激酶活化必需的距离和取向定位而言是不可缺少的。
在本文给出的实验表明,PDGFRβ、Kit及其他III型RTK的二聚化完全由配体结合驱动,并且配体结合的唯一作用是交联两个受体分子,以增加它们在细胞膜中的局部浓度。负责介导同型D4相互作用的两个盐桥(具有包埋表面积为 的界面)太弱以致不能在没有配体介导的受体二聚化支持的情况下支持受体相互作用,在Kit的情况下,配体介导的受体二聚化由各种强相互作用介导的,对于各SCF原体具有
的界面)太弱以致不能在没有配体介导的受体二聚化支持的情况下支持受体相互作用,在Kit的情况下,配体介导的受体二聚化由各种强相互作用介导的,对于各SCF原体具有 的总包埋表面积。在每细胞表达20,000个受体的未刺激细胞的细胞膜中受体的表观浓度估计为大约1-3μM(Klein,P.等(2004)Proc Natl Acad Sci U S A 101,929-934;Chandrasekhar,S.(1943)Reviews of Modern Physics 15,1)。在结合二聚配体(如SCF)时,两个占据的受体以
的总包埋表面积。在每细胞表达20,000个受体的未刺激细胞的细胞膜中受体的表观浓度估计为大约1-3μM(Klein,P.等(2004)Proc Natl Acad Sci U S A 101,929-934;Chandrasekhar,S.(1943)Reviews of Modern Physics 15,1)。在结合二聚配体(如SCF)时,两个占据的受体以 的距离保持在一起。在这些条件下,使用平均距离,以至最近邻域的平均距离法计算的细胞膜中的表观受体浓度提高超过两个数量级,达到4-6x 10-4M。该计算表明,即使解离常数在10-410-5M范围内的弱相互作用(如由两个盐桥介导的那些)也可介导在两个相邻受体的近膜区之间的结合和直接接触。细胞膜内的高局部浓度连同连接D4和D5到其余受体分子的接头的灵活性使得将受体的近膜区以能够进行相邻胞质域之间的相互作用的精确定向和距离(在Kit情况下为
的距离保持在一起。在这些条件下,使用平均距离,以至最近邻域的平均距离法计算的细胞膜中的表观受体浓度提高超过两个数量级,达到4-6x 10-4M。该计算表明,即使解离常数在10-410-5M范围内的弱相互作用(如由两个盐桥介导的那些)也可介导在两个相邻受体的近膜区之间的结合和直接接触。细胞膜内的高局部浓度连同连接D4和D5到其余受体分子的接头的灵活性使得将受体的近膜区以能够进行相邻胞质域之间的相互作用的精确定向和距离(在Kit情况下为 )定位的同型D4以及同型D5的移动和形成成为可能。
)定位的同型D4以及同型D5的移动和形成成为可能。
最后,通过应用化学交联剂以在未刺激的或PDGF刺激的细胞上共价交联WT或突变体受体,本发明已经证明,E390A PDGFRβ突变体发生与PDGF诱导的WT受体二聚化类似的PDGF诱导的二聚化。然而,与在PDGF刺激的细胞的细胞表面上以活化的形式表达WTPDGFRβ不同,E390A突变体在PDGF刺激的细胞上以无活性二聚体的形式表达。该实验表明,同型D4-D4相互作用对于PDGFRβ二聚化不是必需的,并且PDGFRβ二聚是活化受体必需的,但对于活化受体不是足够的。
实施例26:D4-D4界面的破坏克服致癌KIT活化
稳定表达野生型(WT)KIT、其中D5的Ala502和Tyr503被重复的致癌KIT突变体(D5-重复序列突变体)或其中D5的Ala502和Tyr503(D5重复序列)被重复的KIT突变体以及其中D4的Glu386由Ala残基置换的另外的点突变(D5-重复序列/E386A突变体)的鼠3T3细胞用1、5或10ng/ml的SCF在37℃刺激5分钟。
使未刺激的或SCF刺激的细胞的溶解产物与抗KIT抗体进行免疫沉淀,继之以SDS-PAGE,并且采用抗KIT或抗磷酸酪氨酸(抗pY)抗体进行免疫印迹。
在图27中给出的实验证明,野生型KIT的SCF刺激导致由响应SCF刺激的KIT酪氨酸自磷酸化增强揭示的KIT活化增强。实验也表明,KIT的致癌D5-重复序列突变体被组成型地酪氨酸自磷酸化(即,它在SCF刺激不存在的情况下被活化)。相比之下,在D4中携带另外的点突变(其在正常受体蛋白的背景下表现出削弱KIT的SCF活化)的D5-重复序列/E386A突变体阻断由该致癌D5-重复序列突变介导的组成型酪氨酸自磷酸化。
该实验为D4-D4同型相互作用在介导通过D5中的致癌突变和据推测通过该KIT分子的不同部分中的其它致癌突变产生的KIT活化方面的重要性提供了遗传确认。而且,该实验对这样的观点提供了进一步的确认:通过由本发明的成分(例如抗体、或其抗原结合部分、小分子或肽类分子)的药理学干预破坏D4-D4界面将阻断D5中的致癌突变、KIT分子的其它部分中和在致癌III型和IV型RTK中的致癌突变的活性。
实施例27:针对相应于参与介导D4同型相互作用的KIT特征基序的合成肽的抗体识别完整的KIT蛋白质
在这个实施例中,针对三个不同的KIT抗原产生兔多克隆抗体:
1.人KIT的全长细胞外结构域(氨基酸1-510)。
2.由氨基酸308-411组成的KIT Ig样结构域4(D4)(KIT-D4)
3.相应于氨基酸375-391的17-聚体肽,其包括结合于KLH的KITD4的特征基序(SELHLTRLKGTEGGTYT)。
以两周间隔,用三个抗原的每一种免疫兔子,并分析测试血。给出的结果来自在第三次免疫之后收集的血清样品。
表达野生型人KIT的3T3细胞的溶解产物与包含下列抗体之一的30μl血清一起温育:1.抗KIT抗体,其针对全长KIT细胞外结构域。2.抗D4抗体,其针对KIT-D4,和3.抗肽抗体,其针对对应于KITD4的氨基酸375-391的肽。表达野生型KIT的3T3细胞的溶解产物与各种抗体一起,连同A蛋白琼脂糖在4℃温育2小时,然后用包含20mM Hepes、150mM NaCl、0.1%TritonX-100和5%丙三醇的洗涤缓冲液洗涤三次。在SDS-PAGE上分离免疫沉淀物,将其转移到硝酸纤维素上并与如图28所描述的各抗体进行免疫印迹分析。在图28中给出的数据表明,各抗体,包括针对对于将KIT二聚物定位在活化构型中必需的D4的同型相互作用区的抗肽抗体,在所述实验的免疫沉淀和免疫印迹步骤中识别完整的天然KIT。
值得注意的是,该实验表明,抗肽抗体识别野生型KIT与针对KIT的完整细胞外区或D4区的抗体一样有效。
实施例28:细胞外近膜结构域之间的直接接触是VEGF-受体激活和细胞信号传导所需的
与SCF复合的KIT的细胞外区域的结构分析显示第四Ig-样结构域(D4)的EF-环中的序列基序,其负责形成同型受体接触和负责配体诱导的KIT激活和细胞信号传导。在大多数VEGFR1、2和3的近膜第七Ig-样结构域(D7)中鉴定了相同的基序。该实施例表明,配体诱导的酪氨酸自磷酸化和经由携带D7的关键碱基(Arg726或Asp731)的突变的VEGFR1或VEGFR2的细胞信号传导受到严重损害。VEGFR2的D7的晶体结构也描绘到的分辨率。结构显示同型D7接触是由一个原体的Arg726和另一个原体的Asp731之间形成的盐桥和范德瓦耳斯接触介导的。D7二聚体的结构非常类似于与SCF复合的KIT细胞外区域的晶体结构中所见的D4二聚体的结构。两个结构中的EF环和盐桥的位置几乎相同,且在两个结构中它们的C-末端之间的距离大约为 在两者结构和功能中VEGFR D7和KIT D4之间的高度相似性为III型和V型RTK的共同的祖先起源提供了进一步的证据。也揭示了RTK激活的保守机制和病理性激活RTK的药物介入的新靶标。
在两者结构和功能中VEGFR D7和KIT D4之间的高度相似性为III型和V型RTK的共同的祖先起源提供了进一步的证据。也揭示了RTK激活的保守机制和病理性激活RTK的药物介入的新靶标。
血管内皮生长因子(VEGF)通过结合和激活受体酪氨酸激酶(RTK)的VEGF-受体(VEGFR)家族的三个成员来调控血管和淋巴管的发育和内环境的稳定(Olsson等人.,Nat.Rev.Mol.Cell.Biol.,7(5):359-371(2006))。VEGFR1(Flt1)、VEGFR2(KDR/Flk1)和VEGFR3(Flt4)是V型RTK的成员,V型RTK是包含由7个Ig-样结构域(D1-D7)组成的大细胞外区域、单一跨膜(TM)螺旋及具有酪氨酸激酶活性和另外的调控序列的胞质区的家族。VEGFR胞外域的第二和第三Ig-样结构域,D2和D3,对于细胞因子VEGF家族五个成员(即VEGF-A、B、C、D和胎盘生长因子(PlGF))作为结合位点发挥功能(Barleon等人.,J.Biol.Chem.,272(16):10382-10388(1997);和Shinkai等人.,J.Biol.Chem.,273(47):31283-31288(1998))。这些生长因子是共价键连接的二聚体。各个原体是由在名为半胱氨酸结(cysteine-knot)生长因子的结构中以反向平行的方式排列的四链β折叠片构成的(Weismann等人,Cell,91(5):695-704(1997))。细胞因子的半胱氨酸结家族的其他成员包括神经生长因子(NGF)和血小板源生长因子(PDGF)。但是,RTK的PDGFR家族(III型)的胞外域由5个Ig-样重复序列构成,其中D1、D2和D3起到PDGFR和家族的其他成员(即KIT、CSF1R和Flt3)的配体结合区域的作用。结构和生化实验已经表明,SCF结合细胞外区域诱导KIT二聚化,该步骤随后是邻近的KIT分子的两个近膜Ig-样结构域D4和D5之间的同型接触(Yuzawa等人,Cell,130(2):323-334(2007))。野生型和致癌KIT突变体的生化研究已经表明,同型D4和D5接触对于KIT二聚体在胞质区中以有利于反式自磷酸化、激酶活化和细胞信号传导的距离和方向来定位起到关键作用。
在该实施例中,结构和生化证据表明,VEGF-受体的胞外域最近膜Ig-样结构域(D7)之间的同型接触在VEGF诱导的激活和经由VEGF-受体的细胞信号传导方面发挥了至关重要的作用。
VEGFR2 D4和D7的序列分析
根据基于KIT的D4的序列比对通过结构鉴定了负责介导Ig-样结构域之间的同型接触的进化保守序列基序(L/IxRΦxxxD/ExG)(Yuzawa等人,Cell,130(2):323-334(2007))。在PDGFRα、PDGFR β和CSF1R的D4中以及在VEGFR1、VEGFR2和VEGFR3的最近膜Ig-样结构域(D7)中发现相似的基序(图29)。L/IxRΦxxxD/ExG基序位于连接D7的βE和βF链的环区域处,其为显示出负责介导同型D4KIT接触所需的盐桥的区域。在VEGFR1和VEGFR2中从海胆到人类该Arg和Asp是进化保守的(图29),表明这些残基在VEGFR活性中的重要性。然而与KIT和PDGFR的D4相反,VEGFR1和VEGFR2的D7在B5和F5位置包含两个保守的半胱氨酸残基,其形成β链之间的二硫键,这是造成I-群Ig-样结构域的疏水核心的相互作用(Harpaz和Chothia,J.Mol.Biol.,238(4):528-539(1994))。
与PDGFR和KIT的D4相似,VEGFR2的D4缺乏B5和F5位的负责β链之间的二硫键形成的保守半胱氨酸。在VEGFR2的D4中,与其它典型的I-群Ig结构域相比,连接βC与βE的区域更短,或许是因为该区域缺乏β链中的一条。氨基酸序列分析表明,VEGFR2D4与肌间蛋白(myomesin)结构域D13(2R15)和端蛋白(1TLK)同源,序列同一性分别为30%和33%。手动序列比对显示VEGFR2的D4和PDGFRα的D4之间20%的序列同一性。KIT的D4和PDGFR的D4都包含由D/E-x-G/A/D-x-Y-x-C基序组成的βF中的“Y-角基序”周围的保守“D/E-x-G”氨基酸(Hemmingsen等人,Protein Sci.,3(11):1927-1937(1994))(在D4中Cys残基被疏水氨基酸替换)。在D4中,一个分子的Glu残基与第二KIT分子-5位的Arg残基之间形成盐桥(图29B)。在VEGFR2的D4中发现Asp残基,但不是-5位置,这个Ig-样结构域在相对于Asp残基的-2和-6位包含带相反电荷的一对氨基酸(图29B)。虽然在VEGF-A诱导的VEGFR2的胞外域二聚体的电镜(EM)图像中观察到D4之间的直接接触(Ruch等人,Nat.Struct.Mol.Biol.,14(3):249-250(2007)),但VEGFR2 D4的功能仍未知。因此在EF环区域中,D7与KIT和PDGFR的D4中的相应序列比VEGFR2的D4更相似。
同型D7接触对于配体诱导的VEGFR2激活是必需的
这一序列分析和与KIT的比较显示,VEGFR2的R726和D731残基可以介导受体间盐桥的形成和可以改变对配体的反应。为研究在配体诱导的VEGFR2的激活和信号传导中D7区域的保守残基的作用,生成了VEGFR2突变体,其中Arg726、Asp731或两个氨基酸被Ala残基(R726A、D731A和RD2A)替换。用指导WT VEGFR2或携带D7突变的VEGFR2的表达的pCDNA3表达载体瞬时转染HEK 293细胞。温育24小时后,在VEGF-A刺激之前饥饿转染细胞过夜。分别用抗-磷酸酪氨酸抗体(抗-pTyr)或抗-磷酸-MAPK抗体分析未刺激或者VEGF-A刺激或未刺激的细胞的VEGFR2酪氨酸自磷酸化和和MAPK反应。图30A表明,被预测参与介导受体间盐桥形成的Arg或Glu残基的突变明显降低了VEGF-A诱导的VEGFR2的自磷酸化和MAPK刺激。
为克服VEGFR2相对弱的激酶活性,采用了嵌合受体方法(Fambrough等人,Cell,97(6):727-741(1999)和Meyer等人,J.Biol.Chem.,281(2):867-875(2006))。制备由连接到PDGFR的跨膜和胞质区(氨基酸528-1106)的VEGFR2的胞外域(氨基酸1-761)组成的嵌合受体并用来进一步研究D7在VEGF-A诱导的VEGFR2激活中所发挥的作用。用抗-PDGFR的抗体对VEGF-A刺激的或未刺激的稳定表达嵌合VEGFR2/PDGFR或携带D7突变的嵌合VEGFR2/PDGFR的NIH-3T3细胞裂解液进行免疫沉淀,之后用抗-pTyr抗体进行免疫印迹。图30B显示响应于VEGF-A刺激的嵌合VEGFR2/PDGFR的稳定酪氨酸自磷酸化(图30B,WT)。相反,VEGF-A诱导的携带D7突变(R726A、D731A、RD2A)的嵌合受体的酪氨酸自磷酸化被强烈减弱(图30B)。还生成由融合到TM的VEGFR1细胞外区域和PDGFR的细胞内区域组成的嵌合受体。稳定表达野生型嵌合受体的3T3细胞显示出响应于配体刺激的自磷酸化反应(图30C,WT)。相反,在表达分别携带Arg720、Asp725或两个氨基酸的突变,R720A、D725A和RD2A,的嵌合受体的3T3细胞中配体诱导的激酶活性的刺激被强烈地减弱(图30C)。上述数据表明,III型和V型RTK的近膜Ig-样结构域之间的同型接触对于配体诱导的受体激活和细胞信号传导是必需的。
通过交联配体刺激的细胞然后用SDS-PAGE分析配体刺激或未刺激的细胞裂解液使用共价交联剂研究D7突变对配体诱导的受体二聚化的影响(见,例如,Cochet等人,J.Biol.Chem.,263(7):3290-3295(1988))。该实验表明,VEGF-A诱导的嵌合受体二聚化不被D7突变影响(数据未显示)。与以前关于PDGFR和Kit的报告相似(Yuzawa等人,Cell,130(2):323-334(2007)和Yang等人,Proc.Nat’l.Acad.Sci.U.S.A.,105(220:7681-7686(2008)),介导同型接触的D7对于受体激活是必需的,但对于受体二聚化不是必不可少的。此外,配体诱导的受体二聚化对于酪氨酸自磷酸化和受体激活是必要的但不是充分的。相反,VEGF-A诱导的带有VEGFR2D4中的突变(包括D392A突变或其中Ala残基替换Asp387和Arg391的突变(DR2A))的嵌合受体的酪氨酸自磷酸化保持不变(图30D),从而表明不同的界面可能参与介导在VEGF-A诱导的VEGFR2胞外域二聚体的EM图像中看到的D4相互作用(Ruch等人,Nat.Struct.Mol.Biol.,14(3):249-250(2007))。
分析离心应用于测定分离的D7区域二聚化的解离常数。使用4x10-5、8x10-5和1.6x10-4M蛋白质浓度进行的分析离心实验显示分离的D7在高达10-4M浓度的溶液中保持单体形式,表明D7二聚化的解离常数超过10-4M。在分离的KIT或PDGFR的D4或D5二聚化中发现了相似的高解离常数。先前已证明,随着SCF或PDGF诱导的二聚化,细胞膜中两个相邻KIT或PDGFR原体的局部浓度范围是4-6x10-4M。这连同降低的维度使得能够实现有效的横向相互作用和在细胞膜中以低亲和力互相结合的Ig-样结构域对之间稳定同型接触的形成。此外,III型和V型RTK的近膜Ig-样结构域之间的同型接触受到由以协同方式发生在相邻受体的TM和胞质区之间的另外的横向相互作用支持。
VEGFR细胞外结构域D7的结构
为了确定D7在配体诱导的VEGFR2激活中发挥作用的分子基础,确定了该Ig-样结构域的晶体结构。在空间群I212121中获得了晶体,每个不对称单元具有单一D7分子连同28个水分子。D7结构由VEGFR2的氨基酸667至756组成,衍射X-射线的分辨率达到 用基于端蛋白(PDB代码:1TLK)的结构的模型通过分子置换来确定该结构(Holden等人,J.Mol.Biol.,227(3):840-851(1992))。在复合体中D7的两个拷贝彼此非常相似,r.m.s.差为
用基于端蛋白(PDB代码:1TLK)的结构的模型通过分子置换来确定该结构(Holden等人,J.Mol.Biol.,227(3):840-851(1992))。在复合体中D7的两个拷贝彼此非常相似,r.m.s.差为 D7假定典型的IgSF折叠,其由2个四链折叠片,一个由A、B、D和E链组成的折叠片和由A’、G、F和C链组成的第二折叠片形成的β-夹心结构构成。A链的第一半与B链形成氢键,A’链与G链形成氢键,与受体酪氨酸激酶MuSK的细胞外区域的Ig-样结构域Ig1和Ig2结构类似(Stiegler等人,J.Mol.Biol.,364(3):424-433(2006))。βE和βF链之间的跨接连接在729-731残基处包括单螺旋转角。VEGFR2的D7展示了IgSF折叠的几个特征,包括βB的Cys688和βF的Cys737之间的保守二硫键,和包裹二硫键以形成疏水核心的标志色氨酸残基。用DALI(Holm等人,Curr.Protoc.Bioinformatics,Chapter 5,Unit 5 5(2006))进行的结构比较显示在VEGFR2的Ig-样结构域中,D7与端蛋白(PDB代码:1TLK)最为相似,89个比对的Cβ残基的Z-评分为13.4和r.m.s.d.为D7包含限定I-群的V-框分布(V-frame profile)的20个关键位置中的16个(Harpaz和Chothia,J.Mol.Biol.,238(4):528-539(1994))。定位在βF(Cys740)和βG(Cys745)中的一对半胱氨酸形成另外的暴露跨链二硫键。这一特征在VEGFR2和VEGFR3中是高度保守的,但VEGFR1中不是高度保守的。
D7假定典型的IgSF折叠,其由2个四链折叠片,一个由A、B、D和E链组成的折叠片和由A’、G、F和C链组成的第二折叠片形成的β-夹心结构构成。A链的第一半与B链形成氢键,A’链与G链形成氢键,与受体酪氨酸激酶MuSK的细胞外区域的Ig-样结构域Ig1和Ig2结构类似(Stiegler等人,J.Mol.Biol.,364(3):424-433(2006))。βE和βF链之间的跨接连接在729-731残基处包括单螺旋转角。VEGFR2的D7展示了IgSF折叠的几个特征,包括βB的Cys688和βF的Cys737之间的保守二硫键,和包裹二硫键以形成疏水核心的标志色氨酸残基。用DALI(Holm等人,Curr.Protoc.Bioinformatics,Chapter 5,Unit 5 5(2006))进行的结构比较显示在VEGFR2的Ig-样结构域中,D7与端蛋白(PDB代码:1TLK)最为相似,89个比对的Cβ残基的Z-评分为13.4和r.m.s.d.为D7包含限定I-群的V-框分布(V-frame profile)的20个关键位置中的16个(Harpaz和Chothia,J.Mol.Biol.,238(4):528-539(1994))。定位在βF(Cys740)和βG(Cys745)中的一对半胱氨酸形成另外的暴露跨链二硫键。这一特征在VEGFR2和VEGFR3中是高度保守的,但VEGFR1中不是高度保守的。
晶体结构表明,同型D7接触是由各个原体的D7的ABED链形成的两个β折叠片介导的,其中一个原体的Arg726指向另一个原体的Asp731,导致形成大约360A2的包埋表面积。图31B显示了Arg726和Asp731形成盐桥和范德瓦耳斯接触。D7二聚体结构与KIT细胞外二聚体(PDB代码:2E9W)结构中见到的同型D4接触非常相似(Yuzawa等人,Cell,130(2):323-334(2007))。此外,VEGFR2的D7展现了除了恰好沿D7-D7界面的带正电荷中心具有整体带负电表面的静电场的强极化(图31C)。强带电界面可以防止在配体刺激前单体受体分子的异常缔合。
用DALI(Holm等人,Curr.Protoc.Bioinformatics,Chapter 5,Unit 5 5(2006))比较KIT的D4的结构与VEGFR2的D7结构,显示出83个比对的Cα残基的显著的相似性,其中Z-评分为10.4和r.m.s.d.为 两个结构中的EF环的位置也几乎相同,D4和D7二聚体结构的C-末端之间的距离大约为
两个结构中的EF环的位置也几乎相同,D4和D7二聚体结构的C-末端之间的距离大约为 (图33)。VEGFR D7和KIT D4之间结构和功能的高相似性表明RTK激活的非常保守的机制,并为Ⅲ型和V型RTK共同的祖先起源提供了进一步的证据。有趣的是,果蝇(Cho等人,Cell,108(6):865-876(2002))、秀丽线虫(Plowman等人,Proc.Nat’l.Acad.Sci.U.S.A.,96(24):13603-13610(1999))、海鞘(Satou等人,Dev.Genes Evol.,213(5-6):254-263(2003))和海胆(Duloquin等人,Development,134(12):2293-2302(2007))的基因组包含VEGFR/PDGFR样RTK的单一家族,其在细胞外区域中包含7个Ig-样结构域。脊椎动物中III型和V型RTK基因功能上分离但在染色体上的位置是彼此邻近的(Shibuya,Biol.Chem.,383(10):1573-1579(2002)和Grassot等人,Mol.Biol.Evol.,23(6):1232-1241(2006))。在人类中,染色体4q12(KIT、PDGFRα和VEGFR2),5q33(FMS、PDGFRβ和VEGFR3)和13q 12(FLT3和VEGFR1)上发现III类和V类RTK的基因在3个簇中。III类和V类RTK的系统发育表明这8个RTK是由2轮顺式复制和2轮反式复制生成的(Grassot等人,Mol.Biol.Evol.,23(6):1232-1241(2006))。VER3和VER4的D7中也鉴定到了EF环区域中的高度保守基序,秀丽线虫的两个VEGFR/PDGFR样受体基因。海胆的VEGFR/PDGFR样受体的D7中发现类似的基序,但在果蝇的VEGFR/PDGFR样受体中没有发现。有趣的是,海鞘的VEGFR/PDGFR样受体的10个Ig-样结构域中的3个含有典型EF环基序。VEGFR/PDGFR样海鞘受体的D3、D6和D9之间的同型接触可能对于在其细胞外区域中包含10个Ig-样结构域的RTK的配体诱导激活是必需的。
(图33)。VEGFR D7和KIT D4之间结构和功能的高相似性表明RTK激活的非常保守的机制,并为Ⅲ型和V型RTK共同的祖先起源提供了进一步的证据。有趣的是,果蝇(Cho等人,Cell,108(6):865-876(2002))、秀丽线虫(Plowman等人,Proc.Nat’l.Acad.Sci.U.S.A.,96(24):13603-13610(1999))、海鞘(Satou等人,Dev.Genes Evol.,213(5-6):254-263(2003))和海胆(Duloquin等人,Development,134(12):2293-2302(2007))的基因组包含VEGFR/PDGFR样RTK的单一家族,其在细胞外区域中包含7个Ig-样结构域。脊椎动物中III型和V型RTK基因功能上分离但在染色体上的位置是彼此邻近的(Shibuya,Biol.Chem.,383(10):1573-1579(2002)和Grassot等人,Mol.Biol.Evol.,23(6):1232-1241(2006))。在人类中,染色体4q12(KIT、PDGFRα和VEGFR2),5q33(FMS、PDGFRβ和VEGFR3)和13q 12(FLT3和VEGFR1)上发现III类和V类RTK的基因在3个簇中。III类和V类RTK的系统发育表明这8个RTK是由2轮顺式复制和2轮反式复制生成的(Grassot等人,Mol.Biol.Evol.,23(6):1232-1241(2006))。VER3和VER4的D7中也鉴定到了EF环区域中的高度保守基序,秀丽线虫的两个VEGFR/PDGFR样受体基因。海胆的VEGFR/PDGFR样受体的D7中发现类似的基序,但在果蝇的VEGFR/PDGFR样受体中没有发现。有趣的是,海鞘的VEGFR/PDGFR样受体的10个Ig-样结构域中的3个含有典型EF环基序。VEGFR/PDGFR样海鞘受体的D3、D6和D9之间的同型接触可能对于在其细胞外区域中包含10个Ig-样结构域的RTK的配体诱导激活是必需的。
该实施例中给出的实验表明,III型和V型RTK通过共同机制激活,其中近膜Ig-样结构域介导的同型接触保证了两个受体单体的TM和胞质区达到紧密接近和正确的定向,以能够实现有效的反式-自磷酸化、激酶激活和细胞信号传导。配体诱导的受体二聚化与近膜Ig-样结构域之间的多重低亲和力同型缔合的组合提供了简单但有效的配体诱导跨膜信号传导的机制。此外,单个Ig-样结构域对于彼此的低亲和力防止在配体接合前受体单体的意外受体激活。同型接触区为病理性RTK激活和细胞信号传导的药物干预提供了理想的靶标。
VEGFR1、VEGFR2和VEGFR3的D7区域的连续表位
进行了基于结构的序列比对。该比对揭示了可被本发明的成分识别的VEGFR1、VEGFR2和VEGFR3D7区域上的潜在连续表位(表8)。该表位定位在B链、D链、E链、A’B环、CD环、DE环和EF环中。这些表位位于介导同型D7接触的界面中。
表8:VEGFR1、VEGFR2和VEGFR3 D7区域上的连续表位

实施例28的材料与方法
1.蛋白质的表达、纯化和结晶
用PET28a载体在大肠杆菌中表达含有N末端6xHis-标签的VEGFR2的D7(氨基酸657-765)。收集包涵体并在6M的盐酸胍(pH 8.0)中溶解。通过蛋白质在含有10mM Tris(pH8.0)、2mM还原型谷胱甘肽和0.2mM氧化型谷胱甘肽的重折叠缓冲液中逐滴稀释对D7进行重折叠,最终蛋白质浓度为80-100μg/ml。在4℃通过搅动进行重折叠48小时。通过用0.45μm过滤器装置过滤使重折叠溶液澄清,并用FastQ琼脂糖柱纯化和之后进行尺寸排阻(S200,GE Healthcare)和阴离子交换色谱(MonoQ,GE Healthcare)。用凝血酶消化去除N-末端6xHis标签。D7蛋白质在含有25mM Tris(pH 8.0)和200mM NaCl的缓冲液中被浓缩到15mg/ml;并进行结晶和优化的大规模筛选(Hampton研究,晶体筛选)。在4℃下,大约尺寸为300×75×20μM的胞外域D7晶体在0.2M丁二酸和16%PEG3350中生长。所有晶体浸没在添加有5-18%甘油的储存溶液中几秒钟、快速冷却并且在收集数据过程中保持在100K的氮气流中。晶体属于I212121空间群,其晶胞尺寸为
 和
和 每个不对称单元一个分子。用在NSLS,Brookhaven National Laboratory的X29A线束上的ADSD quantum-210 CCD检查器收集分辨率达到
每个不对称单元一个分子。用在NSLS,Brookhaven National Laboratory的X29A线束上的ADSD quantum-210 CCD检查器收集分辨率达到 的衍射数据。用HKL2000程序包对所有数据集进行处理和换算(Otwinowski和Minor,Methods in Enzymology,276(part A):307-326(1997))。数据收集统计总结于表7中。使用基于来自MUSK(2IEP)(Stiegler等人,J.Mol.Biol.,364(3):424-433(2006))、端蛋白(1TLK)(Holden等人,J.Mol.Biol.,227(3):840-851(1992))和KIT的D4(2E9W)(Yuzawa等人,Cell,130(2):323-344(2007))的Ig结构域结构的模型为检索模型通过Phaser的分子置换解析结构。用22.7%的晶体学R-因子和27.7%的自由R-因子优化结构达到
的衍射数据。用HKL2000程序包对所有数据集进行处理和换算(Otwinowski和Minor,Methods in Enzymology,276(part A):307-326(1997))。数据收集统计总结于表7中。使用基于来自MUSK(2IEP)(Stiegler等人,J.Mol.Biol.,364(3):424-433(2006))、端蛋白(1TLK)(Holden等人,J.Mol.Biol.,227(3):840-851(1992))和KIT的D4(2E9W)(Yuzawa等人,Cell,130(2):323-344(2007))的Ig结构域结构的模型为检索模型通过Phaser的分子置换解析结构。用22.7%的晶体学R-因子和27.7%的自由R-因子优化结构达到 分辨率。VEGFR2 D7的原子坐标保存于Protein Data Bank,其登录代码为XXX。用Pymol和CCP4MG软件生成分子图像(Potterton等人,Acta Crystallogr.D.Biol.Crystallogr.,60(Pt.12Pt.1):2288-2294(2004))。
分辨率。VEGFR2 D7的原子坐标保存于Protein Data Bank,其登录代码为XXX。用Pymol和CCP4MG软件生成分子图像(Potterton等人,Acta Crystallogr.D.Biol.Crystallogr.,60(Pt.12Pt.1):2288-2294(2004))。
表7:VEGFR-D7的数据收集和优化统计

a括号中的值是SEB的最高分辨率壳的统计
b Rmerge=∑Ij-<I>/∑Ij,Ij为单个偏转的强度且是偏转强度的平均值。
cRwork=∑||Fo|-<|Fc||>/∑|Fo|,其中Fc是计算的结构因子。
dRfree如Rwork,但对于不包括在优化内的随机选择的10%偏转进行计算。
2.氨基酸序列比对
用ClustalW对进行D7的氨基酸序列比对(Thompson等人,NucleicAcids Res.,22(220:4673-4680(1994))并随后根据I-群IgSF折叠对20个关键残基的限制进行人工调整。人VEGFR的氨基酸序列用作查询以使用PSI-BLAST检索非冗余数据库(nr)的同源序列(Altschul等人,J.Mol.Biol.,215(3):403-410(1990))。氨基酸序列的比对以及D7 PDB文件提交至Consurf 3.0服务器(Landau等人.,Nucleic Acids Res.,33(WebServer Issue),W299-302(2005))来生成比对的各个位置的最大似然性归一化进化率,其中低趋异率对应高的序列保守性。对于Consurf输出,将连续保守性评分分配到9项的离散标度中以用于可视化,使得第9项包含了最保守(栗色)位置和第1项包含最可变(蓝绿色)的位置
3.VEGFR表达载体和嵌合受体的产生
人VEGFR1和VEGFR2的cDNA由Dr.Masabumi Shibuya惠赠(Sawano等人.,Blood,97(3):785-791(2001))。VEGFR2的cDNA通过PCR被亚克隆到pcDAN3表达载体中并插入到XhoI/XbaI位点。由VEGFR1或VEGFR2的细胞外区域组成的嵌合受体与PDGFR-β的跨膜区和胞质区融合。在C-末端添加flag-标签并将嵌合受体克隆到pLXSHD逆转录病毒表达载体的EcoRI/XhoI位点中。
4.细胞系和表达载体
如前所述,通过逆转录病毒感染生成稳定表达VEGFR1/2-PDGFR嵌合受体的3T3细胞系(Yuzawa等人,2007和Cochet等人,1988)。用L-组氨醇选择细胞且合并类似表达水平匹配的细胞用于实验中。
在VEGF刺激之前,用1μg DNA瞬时转染HEK 293细胞并对细胞血清饥饿过夜。用200ng/ml VEGF处理细胞然后用抗VEGFR1或VEGFR2的抗体对细胞裂解液进行免疫沉淀,之后用抗-pTyr抗体进行免疫印迹(PY20,Santa Cruz)。用SDS-PAGE分析全细胞裂解物并分别用抗-磷酸MAPK和抗-MAPK的抗体(Cell Signaling)进行免疫印迹。
用杆状病毒表达载体pFastBac1在sf9细胞中产生VEGF,如前所述(Cohen等人,Growth Factors,7(2):131-138(1992))。用肝素琼脂糖珠纯化VEGF,通过考马斯亮蓝染色的SDS-PAGE实验达到>80%的纯度。
5.分析超离心
用大分子装配分析超离心中心(Center for AnalyticalUltracentrifugation of Macromolecular Assemblies)(Department ofBiochemistry,University of Texas Health Science Center,San Antonio,TX)的Beckman Optima XL-I进行沉降速度实验。D7蛋白质以于含有25mMTris pH 8和100mM NaCl的缓冲液中的4x10-5M、8x10-5M和1.6x10-4M浓度,在20℃下进行50,000rpm的离心。用二维光谱分析结合MonteCarlo分析对速率数据进行分析。
等同
本领域普通技术人员将认识到或能够仅使用常规试验确定在本文描述的本发明的具体实施方式的许多等同方案。这样的等同方案意图被以下权利要求包括在内。





































Claims (74)
1.一种结合人血管内皮生长因子受体(VEGF受体)的胞外域的成分,其中所述成分拮抗VEGF受体的活性。
2.权利要求1的成分,其中所述的成分结合人VEGF受体的Ig-样结构域。
3.权利要求2的成分,其中所述Ig-样结构域不负责配体与所述VEGF受体的结合。
4.权利要求2的成分,其中所述Ig-样结构域负责配体与所述VEGF受体的结合。
5.权利要求1的成分,其中所述成分不阻断所述VEGF受体和VEGF受体配体之间的相互作用。
6.权利要求1的成分,其中所述成分阻断所述VEGF受体和VEGF受体配体之间的相互作用。
7.权利要求1的成分,其中所述成分不阻止所述VEGF受体的二聚化。
8.权利要求1的成分,其中所述成分阻止所述VEGF受体的二聚化。
9.权利要求1的成分,其中所述成分阻止所述胞外域的近膜区与所述VEGF受体的各原体之间的相互作用。
10.权利要求9的成分,其中所述相互作用是同型的。
11.权利要求9的成分,其中所述相互作用是异型的。
12.权利要求9的成分,其中所述胞外域的近膜区是VEGF受体的第七Ig-样结构域(D7)。
13.权利要求12的成分,其中所述成分结合VEGF受体的D7结构域的以下共有序列:
L/I X1 R Φ X2 X3 X4 D/E X5 G(SEQ ID NO:158),其中L是亮氨酸,I是异亮氨酸,R是精氨酸,Φ是疏水性氨基酸,D是天冬氨酸,E是谷氨酸,G是甘氨酸;和X1、X2、X3、X4和X5是任何氨基酸。
14.权利要求13的成分,其中Φ是缬氨酸;X1选自于精氨酸、谷氨酰胺、谷氨酸和天冬氨酸;X2选自于精氨酸、赖氨酸和苏氨酸;X3选自于赖氨酸、谷氨酸、谷氨酰胺和缬氨酸;X4选自于谷氨酸和缬氨酸;和X5选自于谷氨酸、甘氨酸、丝氨酸和谷氨酰胺(SEQ IDNO:159)。
15.权利要求9的成分,其中所述成分使得所述胞外域的近膜区与VEGF受体的各原体分隔大于 的距离。
的距离。
16.权利要求1的成分,其中所述VEGF受体是VEGFR1。
17.权利要求1的成分,其中所述VEGF受体是VEGFR2。
18.权利要求1的成分,其中所述VEGF受体是VEGFR3。
19.权利要求1的成分,其中所述成分将VEGF受体的胞外域锁定在非活性状态。
20.权利要求1的成分,其中所述成分结合VEGFR2的氨基酸残基Arg726。
21.权利要求1的成分,其中所述成分结合VEGFR2的氨基酸残基Asp731。
22.权利要求1的成分,其中所述成分结合VEGFR2的氨基酸残基Arg726和Asp731。
23.权利要求1的成分,其中所述成分结合选自于VEGFR2的氨基酸残基724、725、726、727、728、729、730、731、732和733的一个或多个氨基酸残基。
24.权利要求1的成分,其中所述成分结合VEGFR1的氨基酸残基Arg720。
25.权利要求1的成分,其中所述成分结合VEGFR1的氨基酸残基Asp725。
26.权利要求1的成分,其中所述成分结合VEGFR1的氨基酸残基Arg720和Asp725。
27.权利要求1的成分,其中所述成分结合选自于VEGFR1的氨基酸残基718、719、720、721、722、723、724、725、726和727的一个或多个氨基酸残基。
28.权利要求1的成分,其中所述成分结合VEGFR3的氨基酸残基Arg737。
29.权利要求1的成分,其中所述成分结合VEGFR3的氨基酸残基Asp742。
30.权利要求1的成分,其中所述成分结合VEGFR3的氨基酸残基Arg737和Asp742。
31.权利要求1的成分,其中所述成分结合选自于VEGFR3的氨基酸残基735、736、737、738、739、740、741、742、743和744的一个或多个氨基酸残基。
32.权利要求1的成分,其中所述成分结合VEGF受体上的构象表位。
33.权利要求32的成分,其中所述构象表位由VEGF受体的D7结构域中的两个或更多个残基构成。
34.权利要求32的成分,其中所述构象表位包含氨基酸残基Arg726和Asp731;Arg 720和Asp 725;或者Arg737和Asp742。
35.权利要求1的成分,其中所述成分阻断配体诱导的VEGF受体酪氨酸自磷酸化。
36.权利要求1的成分,其中所述成分阻碍配体诱导的VEGF受体内化。
37.权利要求1的成分,其中所述成分结合VEGF受体上的连续表位。
38.权利要求37的成分,其中所述连续表位由VEGF受体D7结构域中的两个或更多个残基构成。
39.权利要求38的成分,其中所述连续表位是选自于VEGFR1的672VAISSS677、VEGFR1的678TTLDCHA684、VEGFR1的685NGVPEPQ691、VEGFR1的700KIQQEPG706、VEGFR 1的707IILG710、VEGFR1的711PGS713、VEGFR1的714STLFI718、VEGFR1的719ERVTEEDEGV728、VEGFR3的689VNVSDS694、VEGFR3的695LEMQCLV701、VEGFR3的702AGAHAPS708、VEGFR3的717LLEEKSG723、VEGFR3的724VDLA727、VEGFR3的728DSN730、VEGFR3的731QKLSI735和VEGFR3的736QRVREEDAGR745、VEGFR2的678TSIGES683、VEGFR2的684IEVSCTA690、VEGFR2的691SGNPPPQ697、VEGFR2的706TLVEDSG712、VEGFR2的713IVLK716、VEGFR2的717DGN719、VEGFR2的720RNLTI724和VEGFR2的725RRVRKEDEGL734的表位。
40.权利要求1的成分,其中所述成分是分离的抗体或其抗原结合成分。
41.权利要求40的成分,其中所述抗体或其抗原结合成分选自于人抗体、人源化抗体、双特异性抗体和嵌合抗体。
42.权利要求41的成分,其中所述抗体或其抗原结合成分包含选自于IgG1、IgG2、IgG3、IgG4、IgM、IgA和IgE恒定区的重链恒定区。
43.权利要求42的成分,其中所述抗体重链恒定区是IgG1。
44.权利要求40的成分,其中所述抗体或其抗原结合成分选自于Fab片段、F(AB′)2片段、单链Fv片段、SMIP、亲和体、亲和性多聚体、纳米抗体以及单域抗体。
45.权利要求40的成分,其中所述抗体或其抗原结合成分以选自于1x 10-7M或更低、更优选地5x 10-8M或更低、更优选地1x 10-8M或更低、更优选地5x 10-9M或更低的KD结合受体酪氨酸激酶的Ig-样结构域。
46.一种产生权利要求40至45中任一项的抗体或其抗原结合成分的杂交瘤。
47.权利要求1的成分,其中所述成分是小分子。
48.权利要求47的成分,其中所述成分结合选自于氨基酸残基VEGFR2的Arg 726、VEGFR2的Asp731、VEGFR1的Arg720、VEGFR1的Asp725、VEGFR3的Arg737和VEGFR3的Asp742中的至少一个氨基酸残基。
49.权利要求1的成分,其中所述成分是肽类分子。
50.权利要求49的成分,其中所述肽类分子是基于VEGF受体的Ig-样结构域设计的。
51.权利要求50的成分,其中所述肽类分子是基于人VEGF受体的D7结构域设计的。
52.权利要求51的成分,其中所述肽类分子包含结构:L/I X1 R ΦX2 X3 X4 D/E X5 G(SEQ ID NO:158),其中L是亮氨酸,I是异亮氨酸,R为精氨酸,Φ是疏水性氨基酸,D是天冬氨酸,E是谷氨酸,G是甘氨酸;以及X1、X2、X3、X4和X5是任何氨基酸。
53.权利要求52的成分,其中Φ是缬氨酸;X1选自于精氨酸、谷氨酰胺、谷氨酸和天冬氨酸;X2选自于精氨酸、赖氨酸和苏氨酸;X3选自于赖氨酸、谷氨酸、谷氨酰胺和缬氨酸;X4选自于谷氨酸和缬氨酸;X5选自于谷氨酸、甘氨酸、丝氨酸和谷氨酰胺(SEQ ID NO:159)。
54.权利要求50的成分,其中所述肽类分子包含与人VEGFR2的氨基酸残基724-733至少80%相同的结构。
55.权利要求50的成分,其中所述肽类分子包含与人VEGFR1的氨基酸残基718-727至少80%相同的结构。
56.权利要求50的成分,其中所述肽类分子包含与人VEGFR3的氨基酸残基735-744至少80%相同的结构。
57.权利要求50的成分,其中所述肽类分子包含至少一个D-氨基酸残基。
58.权利要求1的成分,其中所述成分是adnectin。
59.一种结合人VEGF受体的第7Ig-样结构域上的构象表位的成分,其拮抗人VEGF受体的活性,并且其中所述构象表位包含VEGFR2的残基Arg726和Asp731;VEGFR1的残基Arg720和Asp725;或VEGFR3的残基Arg737和Asp742。
60.一种结合VEGFR2的氨基酸残基Arg726和Asp731;VEGFR1的氨基酸残基Arg720和Asp725;或VEGFR3的氨基酸残基Arg737和Asp742,从而拮抗人VEGF受体的活性的成分。
61.一种包含权利要求1至权利要求60中任一项的成分和药学上可接受的载体的药物组合物。
62.有效量的权利要求1至60中任一项的成分在制备用于治疗或预防受试者中VEGF受体酪氨酸激酶相关疾病的药物中的用途。
63.权利要求62的用途,其中所述VEGF受体酪氨酸激酶相关疾病选自于癌症、年龄相关性黄斑变性(AMD)、动脉粥样硬化、类风湿性关节炎、糖尿病性视网膜病、淋巴疾病和疼痛相关疾病。
64.权利要求63的用途,其中所述癌症选自于GIST、AML、SCLC、肾癌、结肠癌、淋巴癌和乳腺癌。
65.一种用于鉴定结合VEGF受体的Ig-样结构域的成分的方法,该方法包括:
将VEGF受体与候选成分接触;
同时地或相继地将所述VEGF受体与VEGF受体的配体接触;和
测定所述成分是否影响配体诱导的二聚VEGF受体的Ig-样结构域之间的定位、定向和/或距离,从而鉴定结合VEGF受体的Ig-样结构域的成分。
66.权利要求65的方法,其中所述成分锁定VEGF受体的胞外域于非活性状态。
67.权利要求65的方法,其中所述成分结合VEGF受体的第七Ig-样结构域(D7)。
68.一种结合人VEGF受体的第7Ig-样结构域上的构象表位的分离的抗体或其抗原结合部分,其中所述抗体或其抗原结合部分拮抗人VEGF受体的活性,并且其中所述构象表位包含VEGFR2的残基Arg726和Asp731残基;VEGFR1的残基Arg720和Asp725;或VEGFR3的残基Arg737和Asp742。
69.一种结合VEGFR2的氨基酸残基724-733从而拮抗VEGFR2的活性的分离的抗体或其抗原结合部分。
70.一种结合VEGFR1的氨基酸残基Arg720和Asp725从而拮抗VEGFR1的活性的分离的抗体或其抗原结合部分。
71.一种结合VEGFR3的氨基酸残基Arg737和Asp742从而拮抗VEGFR3的活性的分离的抗体或其抗原结合部分。
72.一种结合选自于人VEGF受体的Arg726和Asp731的至少一个氨基酸残基从而拮抗人VEGF受体的活性的分离的抗体或其抗原结合部分。
73.一种结合选自于人VEGF受体的Arg720和Asp725的至少一个氨基酸残基从而拮抗人VEGF受体的活性的分离的抗体或其抗原结合部分。
74.一种结合选自于人VEGF受体的Arg737和Asp742的至少一个氨基酸残基从而拮抗人VEGF受体的活性的分离的抗体或其抗原结合部分。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US29078909P | 2009-12-29 | 2009-12-29 | |
| US61/290,789 | 2009-12-29 | ||
| PCT/US2010/061296 WO2011090648A2 (en) | 2009-12-29 | 2010-12-20 | Inhibitors of vascular endothelial growth factor (vegf) receptors and methods of use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN102724996A true CN102724996A (zh) | 2012-10-10 |
Family
ID=44307462
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2010800599677A Pending CN102724996A (zh) | 2009-12-29 | 2010-12-20 | 血管内皮生长因子(vegf)受体的抑制剂及其使用方法 |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US20130071397A1 (zh) |
| EP (1) | EP2519253A4 (zh) |
| JP (1) | JP2013515776A (zh) |
| KR (1) | KR20120115348A (zh) |
| CN (1) | CN102724996A (zh) |
| AU (1) | AU2010343193A1 (zh) |
| BR (1) | BR112012016309A2 (zh) |
| CA (1) | CA2785723A1 (zh) |
| IL (1) | IL220147A0 (zh) |
| IN (1) | IN2012DN05014A (zh) |
| MX (1) | MX2012007745A (zh) |
| RU (1) | RU2012132470A (zh) |
| SG (1) | SG181495A1 (zh) |
| WO (1) | WO2011090648A2 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104356226A (zh) * | 2014-11-04 | 2015-02-18 | 深圳市海兰深生物医学技术有限公司 | 一种检测血浆免疫标志物-vegfr1自身抗体的抗原多肽及应用 |
| CN105820244A (zh) * | 2015-01-06 | 2016-08-03 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN108148134A (zh) * | 2015-01-06 | 2018-06-12 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN109476746A (zh) * | 2016-07-19 | 2019-03-15 | F星贝塔有限公司 | Egfr结合分子 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BRPI0812400A2 (pt) | 2007-06-05 | 2014-10-29 | Univ Yale | Unidade, hibridoma, composição farmacêutica, método para identificar uma unidade, anticorpo isolado a uma unidade de ligação de antígeno do mesmo, molécula peptídica, e, uso da unidade. |
| WO2012103165A2 (en) | 2011-01-26 | 2012-08-02 | Kolltan Pharmaceuticals, Inc. | Anti-kit antibodies and uses thereof |
| CN104650229A (zh) * | 2011-11-02 | 2015-05-27 | 埃派斯进有限公司 | 抗-kdr抗体和使用方法 |
| EP4063391A1 (en) | 2012-07-25 | 2022-09-28 | Celldex Therapeutics, Inc. | Anti-kit antibodies and uses thereof |
| US10188495B2 (en) * | 2013-11-01 | 2019-01-29 | Atrium Medical Corporation | Positioning agent and method of using the same |
| CN113908269A (zh) | 2014-05-23 | 2022-01-11 | 塞尔德克斯医疗公司 | 嗜酸性粒细胞或肥大细胞相关病症的治疗 |
| WO2018213848A1 (en) * | 2017-05-19 | 2018-11-22 | The Regents Of The University Of California | Antibody chemically induced dimerizer (abcid) as molecular switches for regulating cellular therapies |
| CA3068121A1 (en) * | 2017-06-23 | 2018-12-27 | Baxalta Incorporated | Purification of factor viii subspecies |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050004066A1 (en) * | 1994-02-10 | 2005-01-06 | Patricia Rockwell | Monoclonal antibodies specific to VEGF receptors and uses thereof |
| WO2008153926A2 (en) * | 2007-06-05 | 2008-12-18 | Yale University | Inhibitors of receptor tyrosine kinases and methods of use thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2478169C (en) * | 2002-03-04 | 2013-04-16 | Imclone Systems Incorporated | Human antibodies specific to kdr and uses thereof |
| KR100883430B1 (ko) * | 2007-06-13 | 2009-02-12 | 한국생명공학연구원 | 혈관내피성장인자 수용체를 중화하는 인간 단클론항체 및그 용도 |
-
2010
- 2010-12-20 AU AU2010343193A patent/AU2010343193A1/en not_active Abandoned
- 2010-12-20 WO PCT/US2010/061296 patent/WO2011090648A2/en active Application Filing
- 2010-12-20 CA CA2785723A patent/CA2785723A1/en not_active Abandoned
- 2010-12-20 US US13/519,839 patent/US20130071397A1/en not_active Abandoned
- 2010-12-20 JP JP2012547128A patent/JP2013515776A/ja not_active Withdrawn
- 2010-12-20 BR BR112012016309A patent/BR112012016309A2/pt not_active IP Right Cessation
- 2010-12-20 CN CN2010800599677A patent/CN102724996A/zh active Pending
- 2010-12-20 MX MX2012007745A patent/MX2012007745A/es not_active Application Discontinuation
- 2010-12-20 RU RU2012132470/10A patent/RU2012132470A/ru not_active Application Discontinuation
- 2010-12-20 EP EP10844218.7A patent/EP2519253A4/en not_active Withdrawn
- 2010-12-20 SG SG2012040861A patent/SG181495A1/en unknown
- 2010-12-20 KR KR1020127019855A patent/KR20120115348A/ko not_active Application Discontinuation
- 2010-12-20 IN IN5014DEN2012 patent/IN2012DN05014A/en unknown
-
2012
- 2012-06-04 IL IL220147A patent/IL220147A0/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050004066A1 (en) * | 1994-02-10 | 2005-01-06 | Patricia Rockwell | Monoclonal antibodies specific to VEGF receptors and uses thereof |
| WO2008153926A2 (en) * | 2007-06-05 | 2008-12-18 | Yale University | Inhibitors of receptor tyrosine kinases and methods of use thereof |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104356226A (zh) * | 2014-11-04 | 2015-02-18 | 深圳市海兰深生物医学技术有限公司 | 一种检测血浆免疫标志物-vegfr1自身抗体的抗原多肽及应用 |
| WO2016070662A1 (zh) * | 2014-11-04 | 2016-05-12 | 青岛海兰深生物科技有限公司 | 一种检测血浆免疫标志物-vegfr1自身抗体的抗原多肽及应用 |
| KR101976297B1 (ko) | 2014-11-04 | 2019-05-07 | 칭다오 하일란쉔 바이오테크놀로지 컴퍼니 리미티드 | 혈장 면역 표지물-vegfr1 자가 항체를 검출하기 위한 항원 폴리펩티드 및 용도 |
| KR20170082152A (ko) * | 2014-11-04 | 2017-07-13 | 칭다오 하일란쉔 바이오테크놀로지 컴퍼니 리미티드 | 혈장 면역 표지물-vegfr1 자가 항체를 검출하기 위한 항원 폴리펩티드 및 용도 |
| CN108148134A (zh) * | 2015-01-06 | 2018-06-12 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN108148136A (zh) * | 2015-01-06 | 2018-06-12 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN105820244B (zh) * | 2015-01-06 | 2018-03-13 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN108148135A (zh) * | 2015-01-06 | 2018-06-12 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN108285484A (zh) * | 2015-01-06 | 2018-07-17 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN105820244A (zh) * | 2015-01-06 | 2016-08-03 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN108148135B (zh) * | 2015-01-06 | 2020-06-12 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN108285484B (zh) * | 2015-01-06 | 2020-06-12 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN108148136B (zh) * | 2015-01-06 | 2020-06-12 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN108148134B (zh) * | 2015-01-06 | 2020-06-12 | 珠海亿胜生物制药有限公司 | 抗vegf抗体 |
| CN109476746A (zh) * | 2016-07-19 | 2019-03-15 | F星贝塔有限公司 | Egfr结合分子 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013515776A (ja) | 2013-05-09 |
| WO2011090648A2 (en) | 2011-07-28 |
| IL220147A0 (en) | 2012-07-31 |
| IN2012DN05014A (zh) | 2015-10-02 |
| EP2519253A4 (en) | 2013-08-07 |
| WO2011090648A3 (en) | 2012-01-05 |
| RU2012132470A (ru) | 2014-02-10 |
| EP2519253A2 (en) | 2012-11-07 |
| AU2010343193A1 (en) | 2012-06-21 |
| CA2785723A1 (en) | 2011-07-28 |
| US20130071397A1 (en) | 2013-03-21 |
| KR20120115348A (ko) | 2012-10-17 |
| BR112012016309A2 (pt) | 2017-04-18 |
| MX2012007745A (es) | 2012-11-23 |
| SG181495A1 (en) | 2012-07-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101801407B (zh) | 受体酪氨酸激酶抑制剂及其使用方法 | |
| CN102724996A (zh) | 血管内皮生长因子(vegf)受体的抑制剂及其使用方法 | |
| CN102762221A (zh) | 受体酪氨酸激酶(rtk)的抑制剂及其使用方法 | |
| JP6979971B2 (ja) | 抗pd−l1抗体 | |
| JP2020515239A (ja) | 抗icosアゴニスト抗体およびそれらの使用 | |
| CN108699153A (zh) | Pd-l1的结合成员 | |
| CN109476740A (zh) | 利用抗cd73抗体的联合治疗 | |
| CN108350076A (zh) | 抗-bcma抗体,结合bcma和cd3的双特异性抗原结合分子及其用途 | |
| SA516370515B1 (ar) | أجسام مضادة ضد cxcr4 ومتقارنات جسم مضاد- عقار | |
| CN108473569A (zh) | 针对人白介素-2的免疫刺激性人源化单克隆抗体及其融合蛋白 | |
| JP2019506844A (ja) | CD32bを標的とする抗体およびその使用方法 | |
| CN102482353A (zh) | 特异于钙粘素-17的抗体 | |
| CN109069639A (zh) | Gitr抗体、方法及用途 | |
| CN114206929A (zh) | 一种抗tigit免疫抑制剂及应用 | |
| AU2014203645A1 (en) | Inhibitors of receptor tyrosine kinases and methods of use thereof | |
| Jalil | The Development of Multivalent Nano-Self Peptides as Antagonists for Antibody-Dependent Macrophage Phagocytosis | |
| WO2020176605A1 (en) | Antibodies to neoantigens and uses thereof | |
| WO2011140295A2 (en) | Modulators of notch receptor signaling and methods of use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20121010 |