CN102675346A - 一种左旋咪唑有机酸盐,其合成方法和它的药物组合物 - Google Patents
一种左旋咪唑有机酸盐,其合成方法和它的药物组合物 Download PDFInfo
- Publication number
- CN102675346A CN102675346A CN201210168103XA CN201210168103A CN102675346A CN 102675346 A CN102675346 A CN 102675346A CN 201210168103X A CN201210168103X A CN 201210168103XA CN 201210168103 A CN201210168103 A CN 201210168103A CN 102675346 A CN102675346 A CN 102675346A
- Authority
- CN
- China
- Prior art keywords
- levamisole
- levamisole hcl
- organic acid
- acid salt
- free alkali
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明涉及一种左旋咪唑有机酸盐其合成方法和它的药物组合物。该左旋咪唑有机酸盐用通式(Ⅰ)表示。本发明用盐酸左旋咪唑作为起始原料,滴加氢氧化钠水溶液反应一段时间后用有机溶剂提取,干燥浓缩,得到白色固体状的左旋咪唑游离碱。然后将左旋咪唑游离碱溶于反应溶媒中,于一定温度下,投入有机酸,搅拌反应、过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加析出溶剂,析出结晶后再过滤、洗涤、干燥,即得左旋咪唑有机酸盐。本发明方法制备工艺简捷、生产周期短、成本低、反应条件缓和、收率高。本发明的左旋咪唑有机酸盐具有良好的水溶性、稳定性及生物利用度。本发明的药物组合物包含有治疗有效量的式(Ⅰ)左旋咪唑有机酸盐和药学上可接受的载体。
Description
技术领域
本发明涉及一种医药领域,具体涉及一种左旋咪唑有机酸盐,其合成方法和以该左旋咪唑有机酸盐为活性成分的药物组合物。
背景技术
机体免疫系统主要承担防御和消除外来异物和体内衰老突变细胞的功能。机体免疫功能降低可导致感染、肿瘤或免疫缺陷疾病的发生。对免疫功能低下所导致的感染、肿瘤以及免疫缺陷病,可使用特异性或者非特异性免疫增强剂进行治疗,这种治疗有利于加强机体的免疫系统功能,缓解由环境应激造成的免疫功能紊乱,有利于预防和治疗传染性及条件性疾病。
左旋咪唑(Levamisole,LMS),又名左旋四咪唑或左旋噻咪唑,其化学名为(S)-(-)-6-苯基-2,3,5,6-四氢咪唑并[2,1-b]噻唑,分子式:C11H12N2S,分子量:204,结构式如下:

左旋咪唑是目前国内外研究最广,也是使用最广的免疫增强剂,具有免疫调节和免疫兴奋功能,可使免疫功能低下者的免疫功能恢复正常,左旋咪唑的分子结构中含有两个药理活性区:一个是咪唑环,另一个是含硫区,左旋咪唑通过活性区发挥免疫作用。左旋咪唑的免疫增强功能主要表现为对机体体液免疫有一定的作用,口服左旋咪唑后,能增加患者血清免疫球蛋白,同时使血清中总补体水平明显增高。LMS对细胞免疫和抗病能力也有影响,用于免疫系统缺陷或免疫系统抑制的患者改善其免疫功能,由于具有胆碱能作用,通过刺激淋巴组织的T细胞系统,调节B细胞产生抗体,促使淋巴细胞分化增殖,加强淋巴细胞因子的产生,提高巨噬细胞、T细胞等细胞内的环鸟苷酸水平和E玫瑰花结形成率,加强单核巨噬细胞和多核白细胞的吞噬功能和杀菌活力,改善细胞免疫力,使原来免疫力较低者得到恢复。同时具有拟胸腺素的性质,增强嗜中性粒细胞的活性及数量和诱导T细胞分化成熟,从而增加干扰素和淋巴因子的产生,对宿主具有明显的免疫兴奋作用、补体受体活性都有恢复作用,使受抑制的细胞免疫功能恢复正常,从而提高机体的免疫力,恢复免疫防御功能。左旋咪唑还能增强机体对温度应激的耐受性。左旋咪唑作为一种免疫增强剂在医学领域的用途日趋广泛,已被用于治疗乙脑和免疫性神经病。此外,左旋咪唑在肿瘤化疗中与抗癌药合用,通过增强机体免疫活性细胞功能,达到抗瘤作用,其次对某些慢性复发性感染性疾病有很好的辅助治疗作用,常见的是与预拉米夫定合并治疗慢性乙型肝炎,被认为是一种可提高疗效的方法,还用于治疗流行性出血热和布氏病以及变态反应性和自身免疫性疾病如类风湿关节炎、红斑狼疮。由于左旋咪唑在水中的溶解度极小,照常规工艺制成注射剂和可溶性颗粒有相当的难度,所以目前左旋咪唑的大多数制剂为口服给药,但口服制剂经过消化道吸收一定时间,无法像注射剂一样迅速起效,在现有技术中主要靠增加其分散性来提高起效速度,比如分散片。
目前,在临床上治疗免疫力低下的药物有以下几种:四咪唑、左旋咪唑、盐酸左旋咪唑、磷酸左旋咪唑,它们都以其稳定的疗效在临床上得到广泛的应用。虽然这些药物有良好的免疫增强作用,但它们具有不足之处,其中四咪唑的右旋体副反应比较大,因此该药的疗效不如同类;左旋咪唑水溶解性不好,给制剂生产带来许多不便,且容易发生易构化,从而增加了本品的有关物质,使含量不准确,从而可能达不到治疗效果;盐酸左旋咪唑和磷酸左旋咪唑均为无机盐,存在氯离子、磷酸根离子,增加了药品的不稳定性从而降低了成品的稳定性,且无机强酸盐对人体有一定的刺激作用,体内吸收差,生物利用度低。
发明内容
针对目前药物的不足,本发明所要解决的第一个技术问题就是提供一种稳定性好、生物利用度高,溶解性好,便于人体吸收的增强机体免疫功能的左旋咪唑有机酸盐。
本发明所要解决的第二个技术问题提供一种左旋咪唑有机酸盐的合成方法。
本发明所要解决的第三个技术问题提供一种增强机体免疫功能的左旋咪唑有机酸盐的药物组合物。
本发明的技术方案如下:
本发明的左旋咪唑有机酸盐用下述通式(Ⅰ)表示:
式(Ⅰ)中:RCOOH代表有机酸,分别选自如下医药上允许的有机酸:酒石酸、乙酸、熊果酸、水杨酸、苹果酸、琥珀酸、抗坏血酸、乳酸、柠檬酸、咖啡酸、绿原酸、阿魏酸、甘油酸、赖氨酸、葡萄糖酸、山梨酸、烟酸、乳糖酸、精氨酸。
优选为:左旋咪唑的酒石酸盐,其中酒石酸优选为DL-酒石酸或D-酒石酸,结构式如下:
或
或:左旋咪唑的乙酸盐,结构式如下:

或:左旋咪唑的熊果酸盐,结构式如下:

或:左旋咪唑的水杨酸盐,结构式如下:
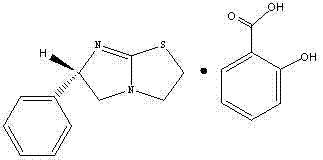
或:左旋咪唑的苹果酸盐,其中苹果酸优选为L-苹果酸,结构式如下:

或:左旋咪唑的琥珀酸盐,结构式如下:

或:左旋咪唑的抗坏血酸盐,其中抗坏血酸优选为L-抗坏血酸,结构式如下:

或:左旋咪唑的乳酸盐,其中乳酸优选为DL-乳酸或L-乳酸,结构式如下:
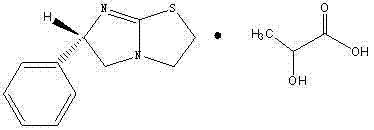

或:左旋咪唑的柠檬酸盐,结构式如下:
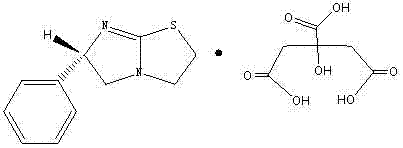
或:左旋咪唑的咖啡酸盐,结构式如下:

或:左旋咪唑的绿原酸盐,结构式如下:

或:左旋咪唑的阿魏酸盐,其中的阿魏酸优选为反式体,结构式如下:

或:左旋咪唑的甘油酸盐,其中甘油酸优选为DL-甘油酸,结构式如下:

或:左旋咪唑的赖氨酸盐,其中赖氨酸优选为L-赖氨酸,结构式如下:

或:左旋咪唑的葡萄糖酸盐,其中葡萄糖酸优选为D-葡萄糖酸,结构式如下:

或:左旋咪唑的山梨酸盐,结构式如下:

或:左旋咪唑的烟酸盐,结构式如下:

或:左旋咪唑的乳糖酸盐,结构式如下:

或:左旋咪唑的精氨酸盐,其中精氨酸优选为DL-精氨酸或L-精氨酸,结构式如下:


由于左旋咪唑具有机体免疫增强功能、治疗免疫力低下的药学功能,左旋咪唑与有机酸反应获得质量可控的左旋咪唑有机酸盐,大大提高了药用物质的熔点,使它的热稳定性明显提高;而且某些有机酸本身也是药物有效成分,左旋咪唑有机酸盐的药效作用比单一的左旋咪唑的药效作用还要高。本发明的左旋咪唑有机酸盐具有生物利用度高、易溶于水的特点,可根据临床需要制成各种制剂,水溶液呈弱酸性,人体易于接受,能减轻对胃肠道的刺激性。
上述左旋咪唑有机酸盐(Ⅰ)的合成路线为:

本发明提供的左旋咪唑有机酸盐的合成方法,包括以下步骤:
(1)反应容器中加入盐酸左旋咪唑(Ⅱ)和蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液搅拌直至溶液pH=7~12,盐酸左旋咪唑(Ⅱ)与NaOH的摩尔比为1:1~9,搅拌反应结束后,过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ);
(2)按照每毫升反应溶媒中加入0.01~0.5克左旋咪唑游离碱(Ⅲ),待完全溶解后,搅拌下加入有机酸,有机酸的用量为左旋咪唑游离碱(Ⅲ)1-3倍摩尔量,于20~70℃条件下搅拌反应0.5~6h,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加反应溶媒的2~8倍体积比的析出溶剂,冷冻析晶5~24h,过滤,滤饼用少量析出溶剂洗涤,干燥,即得左旋咪唑有机酸盐(Ⅰ)。
优选地,步骤(1)中,过滤后的滤液用有机溶剂萃取三次,每次使用的有机溶剂与盐酸左旋咪唑的重量比为1~50:1,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥有机溶剂层1~12h,过滤,减压蒸除溶剂,又得白色固体状的左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
所述有机溶剂为乙醚(DEE)、二氯甲烷(DCM)、乙酸乙酯(EAC)或上述三溶剂的混合。
进一步,步骤(1)的反应温度为5~50℃。
步骤(1)的搅拌反应时间为0.5~6h。
步骤(2)中所述的反应溶媒是蒸馏水或醇类,优选甲醇、乙醇或者丙醇。
步骤(2)中所述的析出溶剂是低沸点易挥发的酮类、酯类或醚类有机溶剂,优选丙酮、乙酸乙酯、丁酮、乙醚、石油醚或者异丙醚。
本发明的左旋咪唑有机酸盐合成方法通过游离碱直接中和或通过复分解反应(双分解)制备,尤以中和方法为佳。左旋咪唑的咪唑并[2,1-b]噻唑环上的碱基与有机酸的-H通过离子方式相结合,得到固态左旋咪唑有机酸盐,具有如下优点:
1、制备简单,目前大部分左旋咪唑衍生物的制备都需要比较严格的反应条件,可能与其引入特定的共价结构有关。而本文左旋咪唑有机酸盐以离子方式相结合,所以制备条件比较温和,简单易操作。
2、制备的左旋咪唑有机酸盐具有较好的油水分配比,解决了水溶性和生物吸收的问题,弥补了左旋咪唑水溶性差的缺点,可根据临床需要制成各种剂型,能制成无菌水溶液用于静脉注射或肌肉注射,使之起效更加迅速。
3、制备的左旋咪唑有机酸盐不含金属离子,因此,体内电解质平衡不受影响。
4、酸性有机酸与碱性左旋咪唑成盐后其水溶液接近弱酸性,有利于人体吸收。制备所得的左旋咪唑有机酸盐还能提高药效,作为一种品质优良的左旋咪唑材料,它是一类具有潜在应用价值的免疫增强药。
本发明的药物组合物含有治疗有效量的上述(Ⅰ)式左旋咪唑有机酸盐为活性成分,以及含有一种或多种药学上可接受的载体。
所述药学上可接受的载体是指药学领域常规的载体,例如:稀释剂、赋形剂如水等,填充剂如淀粉、蔗糖等;黏合剂如纤维素衍生物、藻盐酸、明胶等;润湿剂如甘油;还有崩解剂、吸收促进剂、表面活性剂、润滑剂等。另外还可以在组合物中加入其他辅剂如香味剂、甜味剂等。
本发明的左旋咪唑有机酸盐具有良好的机体免疫增强功能,用于治疗免疫力低下的生物体。其药用的制剂形式可选片剂、胶囊剂、口服液、合剂、口含剂、颗粒剂、冲剂、丸剂、散剂、膏剂、丹剂、混悬剂、溶液剂、注射剂、粉针剂、冻干粉针剂、栓剂、软膏剂、霜剂、喷雾剂、气雾剂、滴剂或贴剂。
具体实施方式
实施例1:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液200ml,搅拌直至溶液pH=7~12,用水浴加温至40℃,继续搅拌30分钟。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用二氯甲烷萃取三次,第一次160ml,第二、三次各120ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥6h,过滤。减压蒸除二氯甲烷,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入D-酒石酸4.5g,于40℃搅拌反应3h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加90ml异丙醚,冷冻析晶12h,过滤,滤饼用少量异丙醚洗涤,干燥至恒重,得白色粉末状左旋咪唑酒石酸盐(Ⅰ),产率约82.99%。元素分析:理论值C50.84%,H5.12%,N7.91%;实测值C50.73%,H5.28%,N7.85%。用ESI-MS测得分子量为354.08(计算值354.37)。
实施例2:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液360ml,搅拌直至溶液pH=7~12,用水浴加温至40℃,继续搅拌5.5h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙醚萃取三次,第一次240ml,第二、三次各90ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥6h,过滤。减压蒸除乙醚,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水甲醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入乙酸4g,于67℃搅拌反应4.5h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加190ml石油醚,冷冻析晶9h,过滤,滤饼用少量石油醚洗涤,干燥至恒重,得白色粉末状左旋咪唑乙酸盐(Ⅰ),产率约79.53%。元素分析:理论值C59.06%,H6.10%,N10.59%;实测值C59.24%,H6.31%,N10.46%。用ESI-MS测得分子量为264.12(计算值264.34)。
实施例3:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液400ml,搅拌直至溶液pH=7~12,用水浴加温至25℃,继续搅拌3h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用二氯甲烷与乙醚(1:1)混合溶剂萃取三次,第一次320ml,第二、三次各160ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥12h,过滤。减压蒸除溶剂,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水甲醇55ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入熊果酸19.5g,于50℃搅拌反应4h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加145ml丁酮,冷冻析晶18h,过滤,滤饼用少量丁酮洗涤,干燥至恒重,得白色粉末状左旋咪唑熊果酸盐(Ⅰ),产率约75.13%。元素分析:理论值C74.50%,H9.15%,N4.24%;实测值C74.41%,H9.07%,N4.36%。用ESI-MS测得分子量为660.21(计算值660.99)。
实施例4:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液270ml,搅拌直至溶液pH=7~12,用水浴温度至10℃,继续搅拌3.5h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙醚、二氯甲烷与乙酸乙酯混合溶剂(1:1:1)萃取三次,第一次430ml,第二、三次各210ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥5.4h,过滤。减压蒸除溶剂,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入水杨酸11.5g,于34℃搅拌反应1.5h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加175ml丁酮,冷冻析晶16h,过滤,滤饼用少量丁酮洗涤,干燥至恒重,得白色粉末状左旋咪唑水杨酸盐(Ⅰ),产率约87.35%。元素分析:理论值C63.13%,H5.29%,N8.18%;实测值C63.09%,H5.51%,N8.63%。用ESI-MS测得分子量为342.28(计算值342.41)。
实施例5:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液370ml,搅拌直至溶液pH=7~12,用水浴加温至34℃,继续搅拌45分钟。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙酸乙酯萃取三次,第一次220ml,第二、三次各90ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥6h,过滤。减压蒸除乙酸乙酯,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入丙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入L-苹果酸9.3g,于48℃搅拌反应1h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加90ml异丙醚,冷冻析晶12h,过滤,滤饼用少量异丙醚洗涤,干燥至恒重,得白色粉末状左旋咪唑苹果酸盐(Ⅰ),产率约73.52%。元素分析:理论值C53.24%,H5.36%,N8.27%;实测值C53.32%,H5.60%,N8.39%。用ESI-MS测得分子量为338.43(计算值338.37)。
实施例6:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液310ml,搅拌直至溶液pH=7~12,用水浴加温至40℃,继续搅拌4.5h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用二氯甲烷与乙酸乙酯(1:1)混合溶剂萃取三次,第一次200ml,第二、三次各90ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥7.5h,过滤。减压蒸除溶剂,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入琥珀酸5.5g,于40℃搅拌反应3h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加120ml异丙醚,冷冻析晶5h,过滤,滤饼用少量异丙醚洗涤,干燥至恒重,得白色粉末状左旋咪唑琥珀酸盐(Ⅰ),产率约90.13%。元素分析:理论值C56.23%,H5.03%,N8.74%;实测值C56.85%,H5.14%,N8.56%。用ESI-MS测得分子量为320.59(计算值320.36)。
实施例7:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液300ml,搅拌直至溶液pH=7~12,用水浴加温至45℃,继续搅拌1h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙酸乙酯萃取三次,第一次200ml,第二、三次各100ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥8h,过滤。减压蒸除乙酸乙酯,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入蒸馏水50ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入L-抗坏血酸7g,于35℃搅拌反应1.5h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加100ml丙酮,冷冻析晶16h,过滤,滤饼用少量丙酮洗涤,干燥至恒重,得淡黄色粉末状左旋咪唑抗坏血酸盐(Ⅰ),产率约78.34%。元素分析:理论值C53.67%,H5.30%,N7.36%;实测值C53.59%,H5.36%,N7.42%。用ESI-MS测得分子量为380.19(计算值380.41)。
实施例8:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液250ml,搅拌直至溶液pH=7~12,用水浴加温至30℃,继续搅拌2h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙醚萃取三次,第一次150ml,第二、三次各100ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥5h,过滤。减压蒸除乙醚,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水丙醇60ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入L-乳酸5.4g,于30℃搅拌反应2h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加150ml无水乙醚,冷冻析晶24h,过滤,滤饼用少量无水乙醚洗涤,干燥至恒重,得白色粉末状左旋咪唑乳酸盐(Ⅰ),产率约86.15%。元素分析:理论值C57.12%,H6.16%,N9.52%;实测值C57.26%,H6.22%,N9.41%。用ESI-MS测得分子量为294.05(计算值294.36)。
实施例9:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液320ml,搅拌直至溶液pH=7~12,用水浴加温至35℃,继续搅拌1.5h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用二氯甲烷与乙酸乙酯(1:1)混合溶剂萃取三次,第一次250ml,第二、三次各140ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥6h,过滤。减压蒸除溶剂,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇45ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入柠檬酸7.5g,于45℃搅拌反应2.5h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加180ml石油醚,冷冻析晶20h,过滤,滤饼用少量石油醚洗涤,干燥至恒重,得白色粉末状左旋咪唑柠檬酸盐(Ⅰ),产率约80.26%。元素分析:理论值C51.51%,H5.09%,N7.07%;实测值C51.67%,H5.14%,N6.93%。用ESI-MS测得分子量为396.40(计算值396.42)。
实施例10:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液460ml,搅拌直至溶液pH=7~12,用水浴加温至47℃,继续搅拌2.5h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙酸乙酯与乙醚(1:1)混合溶剂萃取三次,第一次420ml,第二、三次各150ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥8h,过滤。减压蒸除溶剂,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入咖啡酸14.8g,于62℃搅拌反应1.5h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加230ml异丙醚,冷冻析晶12h,过滤,滤饼用少量异丙醚洗涤,干燥至恒重,得白色粉末状左旋咪唑咖啡酸盐(Ⅰ),产率约86.24%。元素分析:理论值C62.48%,H5.24%,N7.28%;实测值C62.26%,H5.06%,N7.33%。用ESI-MS测得分子量为384.19(计算值384.23)。
实施例11:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液500ml,搅拌直至溶液pH=7~12,用水浴加温至32℃,继续搅拌4h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙酸乙酯萃取三次,第一次410ml,第二、三次各150ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥5.5h,过滤。减压蒸除乙酸乙酯,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入绿原酸4.5g,于25℃搅拌反应3h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加180ml异丙醚,冷冻析晶15h,过滤,滤饼用少量异丙醚洗涤,干燥至恒重,得白色粉末状左旋咪唑绿原酸盐(Ⅰ),产率约86.41%。元素分析:理论值C58.05%,H5.41%,N5.01%;实测值C58.31%,H5.67%,N7.17%。用ESI-MS测得分子量为558.75(计算值558.60)。
实施例12:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液460ml,搅拌直至溶液pH=7~12,用水浴加温至35℃,继续搅拌3.5h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用二氯甲烷萃取三次,第一次130ml,第二、三次各100ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥8h,过滤。减压蒸除二氯甲烷,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入反式阿魏酸6.12g,于30℃搅拌反应4h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加60ml无水乙醚,冷冻析晶24h,过滤,滤饼用少量无水乙醚洗涤,干燥至恒重,得淡绿色粉末状左旋咪唑阿魏酸盐(Ⅰ),产率约78.14%。元素分析:理论值C63.30%,H5.56%,N7.03%;实测值C63.21%,H5.48%,N7.15%。用ESI-MS测得分子量为398.32(计算值398.47)。
实施例13:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液370ml,搅拌直至溶液pH=7~12,用水浴加温至35℃,继续搅拌5.5h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙醚与二氯甲烷(1:1)混合溶剂萃取三次,第一次140ml,第二、三次各110ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥3h,过滤。减压蒸除溶剂,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入DL-甘油酸6.36g,于45℃搅拌反应4h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加185ml乙酸乙酯,冷冻析晶12h,过滤,滤饼用少量乙酸乙酯洗涤,干燥至恒重,得白色粉末状左旋咪唑甘油酸盐(Ⅰ),产率约75.12%。元素分析:理论值C54.17%,H5.84%,N9.02%;实测值C54.25%,H5.47%,N9.34%。用ESI-MS测得分子量为310.71(计算值310.36)。
实施例14:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液520ml,搅拌直至溶液pH=7~12,用水浴温度调至5℃,继续搅拌6h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙醚萃取三次,第一次180ml,第二、三次各90ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥11h,过滤。减压蒸除乙醚,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入L-赖氨酸12g,于65℃搅拌反应1h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加120ml异丙醚,冷冻析晶12h,过滤,滤饼用少量异丙醚洗涤,干燥至恒重,得白色粉末状左旋咪唑赖氨酸盐(Ⅰ),产率约83.42%。元素分析:理论值C58.25%,H7.47%,N15.98%;实测值C58.64%,H7.06%,N15.79%。用ESI-MS测得分子量为350.15(计算值350.47)。
实施例15:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液250ml,搅拌直至溶液pH=7~12,用水浴加温至40℃,继续搅拌30分钟。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙醚、二氯甲烷与乙酸乙酯(1:1:1)混合溶剂萃取三次,第一次180ml,第二、三次各95ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥9h,过滤。减压蒸除溶剂,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入D-葡萄糖酸13g,于40℃搅拌反应3h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加230ml石油醚,冷冻析晶12h,过滤,滤饼用少量石油醚洗涤,干燥至恒重,得白色粉末状左旋咪唑葡萄糖酸盐(Ⅰ),产率约88.73%。元素分析:理论值C50.98%,H6.04%,N6.99%;实测值C50.43%,H6.38%,N6.65%。用ESI-MS测得分子量为400.53(计算值400.44)。
实施例16:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液420ml,搅拌直至溶液pH=7~12,用水浴加温至27℃,继续搅拌2.5h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用二氯甲烷萃取三次,第一次280ml,第二、三次各110ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥5h,过滤。减压蒸除二氯甲烷,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入蒸馏水30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入山梨酸6.7g,于55℃搅拌反应4.5h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加90ml丙酮,冷冻析晶12h,过滤,滤饼用少量丙酮洗涤,干燥至恒重,得白色粉末状左旋咪唑山梨酸盐(Ⅰ),产率约79.53%。元素分析:理论值C64.52%,H6.37%,N8.85%;实测值C64.36%,H6.24%,N8.61%。用ESI-MS测得分子量为316.22(计算值316.41)。
实施例17:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液250ml,搅拌直至溶液pH=7~12,用水浴加温至40℃,继续搅拌1h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用二氯甲烷与乙酸乙酯(1:1)混合溶剂萃取三次,第一次140ml,第二、三次各100ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥12h,过滤。减压蒸除溶剂,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入烟酸3.7g,于66℃搅拌反应5.5h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加110ml丁酮,冷冻析晶10h,过滤,滤饼用少量丁酮洗涤,干燥至恒重,得白色粉末状左旋咪唑烟酸盐(Ⅰ),产率约85.42%。元素分析:理论值C62.36%,H5.23%,N12.83%;实测值C62.44%,H5.27%,N12.59%。用ESI-MS测得分子量为327.76(计算值327.40)。
实施例18:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液360ml,搅拌直至溶液pH=7~12,用水浴加温至50℃,继续搅拌30分钟。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙醚萃取三次,第一次320ml,第二、三次各150ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥9h,过滤。减压蒸除乙醚,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水丙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入乳糖酸12g,于70℃搅拌反应1h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加90ml石油醚,冷冻析晶24h,过滤,滤饼用少量石油醚洗涤,干燥至恒重,得白色粉末状左旋咪唑乳糖酸盐(Ⅰ),产率约74.35%。元素分析:理论值C49.10%,H6.09%,N4.97%;实测值C49.05%,H6.47%,N4.56%。用ESI-MS测得分子量为562.41(计算值562.58)。
实施例19:反应瓶中加入盐酸左旋咪唑(Ⅱ)48g和200ml蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液400ml,搅拌直至溶液pH=7~12,用水浴加温至25℃,继续搅拌2.5h。过滤,蒸馏水洗涤滤饼直至中性,减压干燥,得左旋咪唑游离碱(Ⅲ)。滤液用乙醚萃取三次,第一次200ml,第二、三次各100ml,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥8h,过滤。减压蒸除乙醚,又得白色固体状左旋咪唑游离碱(Ⅲ),合并左旋咪唑游离碱。
在干燥反应瓶中,加入无水乙醇30ml,于室温下搅拌溶解左旋咪唑游离碱(Ⅲ)6.12g,然后投入L-精氨酸6g,于45℃搅拌反应3h后,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加90ml丁酮,冷冻析晶24h,过滤,滤饼用少量丁酮洗涤,干燥至恒重,得白色粉末状左旋咪唑精氨酸盐(Ⅰ),产率约80.51%。元素分析:理论值C53.94%,H6.92%,N22.20%;实测值C53.23%,H6.52%,N22.14%。用ESI-MS测得分子量为378.68(计算值378.49)。
实施例20:注射用左旋咪唑抗坏血酸盐冻干粉针剂的制备
称取抗氧剂硫代硫酸钠0.25g,加入注射用水200ml(处方量80%的注射用水),搅拌使之溶解,备用;另称取处方量的左旋咪唑抗坏血酸盐12.5g,加入上述溶液中,搅拌使其充分溶解,再加入甘露醇20g,搅拌溶解,用0.1mol/L的氢氧化钠溶液或0.1mol/L的稀盐酸调节pH值约为6.5,加入配液量0.01%针用活性炭,搅拌30min,用普通滤纸过滤脱碳,补加注射用水至全量,定容至250ml,搅拌均匀,于超净工作台上先用0.45μm微孔滤膜粗滤,滤液再通过0.22μm和0.22μm微孔滤膜两次无菌精滤,检查澄明度合格后,滤液灌装,2ml/瓶,加塞,冻干,压盖,压铝盖,即得规格100mg/瓶。
实验:左旋咪唑有机酸盐对小鼠免疫功能的影响
清洁级昆明种小白鼠随机分组,每组10只,体质量19~23g,雌雄各半。用正常小白鼠建立免疫功能比照模型;通过腹腔注射环磷酰胺(CTX)建立的小鼠免疫功能低下模型,在免疫功能低下模型中,又分别采用腹腔注射盐酸左旋咪唑和各左旋咪唑有机酸盐(10mg/(kg·d))建立免疫功能恢复模型,连续给药21天;于末次给药后,禁食不禁水12h,眼眶取血,颈椎脱臼处死,采用酶联免疫法测定了各模型组的小鼠血清免疫球蛋白IgA、IgG、IgM含量和免疫器官指数等实验指标,验证和评价了其免疫增强效果,实验数据用平均数表示。结果如下:
各组小鼠的血清IgA、IgG和IgM含量
| 组别 | IgA(mg/ml) | IgG(mg/ml) | IgM(mg/ml) |
| 空白对照组 | 1.04 | 9.47 | 0.54 |
| CTX模型组 | 0.94 | 5.83 | 0.35 |
| 盐酸左旋咪唑组 | 0.97 | 9.13 | 0.47 |
| 左旋咪唑酒石酸盐组 | 1.03 | 10.17 | 0.56 |
| 左旋咪唑乙酸盐组 | 1.07 | 10.38 | 0.59 |
| 左旋咪唑熊果酸盐组 | 0.99 | 10.79 | 0.58 |
| 左旋咪唑水杨酸组 | 1.05 | 9.27 | 0.49 |
| 左旋咪唑苹果酸组 | 0.97 | 9.76 | 0.51 |
| 左旋咪唑琥珀酸组 | 1.08 | 10.56 | 0.57 |
| 左旋咪唑抗坏血酸盐组 | 0.99 | 9.92 | 0.51 |
| 左旋咪唑乳酸盐组 | 1.05 | 9.59 | 0.49 |
| 左旋咪唑柠檬酸盐组 | 0.97 | 9.46 | 0.53 |
| 左旋咪唑咖啡酸组 | 0.98 | 9.93 | 0.50 |
| 左旋咪唑绿原酸组 | 1.04 | 10.16 | 0.58 |
| 左旋咪唑阿魏酸组 | 1.01 | 10.64 | 0.53 |
| 左旋咪唑甘油酸组 | 0.98 | 9.29 | 0.47 |
| 左旋咪唑赖氨酸组 | 1.06 | 10.01 | 0.55 |
| 左旋咪唑葡萄糖酸组 | 1.08 | 9.95 | 0.48 |
| 左旋咪唑山梨酸组 | 0.99 | 10.78 | 0.50 |
| 左旋咪唑烟酸组 | 0.97 | 9.98 | 0.61 |
| 左旋咪唑乳糖酸组 | 1.03 | 9.81 | 0.52 |
| 左旋咪唑精氨酸组 | 1.05 | 9.15 | 0.59 |
各组小鼠体质量、胸腺、脾脏和肝脏指数的比较
| 组别 | 体质量变化(g) | 胸腺指数(mg/g) | 脾脏指数(mg/g) | 肝脏指数(mg/g) |
| 空白对照组 | 6.29 | 4.62 | 6.44 | 58.60 |
| CTX模型组 | 3.45 | 2.88 | 4.95 | 57.85 |
| 盐酸左旋咪唑组 | 3.91 | 3.03 | 5.47 | 59.34 |
| 左旋咪唑酒石酸盐组 | 5.72 | 4.31 | 6.06 | 60.19 |
| 左旋咪唑乙酸盐组 | 4.13 | 4.70 | 6.29 | 59.62 |
| 左旋咪唑熊果酸盐组 | 5.53 | 4.56 | 6.45 | 60.72 |
| 左旋咪唑水杨酸组 | 6.21 | 4.27 | 6.07 | 59.98 |
| 左旋咪唑苹果酸组 | 6.27 | 3.59 | 6.29 | 59.86 |
| 左旋咪唑琥珀酸组 | 5.63 | 3.21 | 5.75 | 59.58 |
| 左旋咪唑抗坏血酸盐组 | 3.93 | 3.66 | 5.86 | 60.04 |
| 左旋咪唑乳酸盐组 | 4.77 | 3.92 | 5.63 | 59.73 |
| 左旋咪唑柠檬酸盐组 | 4.05 | 3.54 | 6.25 | 61.07 |
| 左旋咪唑咖啡酸组 | 4.20 | 4.56 | 6.17 | 60.11 |
| 左旋咪唑绿原酸组 | 4.42 | 4.55 | 6.08 | 59.61 |
| 左旋咪唑阿魏酸组 | 5.86 | 4.04 | 5.96 | 59.83 |
| 左旋咪唑甘油酸组 | 5.01 | 3.60 | 5.70 | 60.09 |
| 左旋咪唑赖氨酸组 | 6.22 | 4.24 | 6.13 | 59.89 |
| 左旋咪唑葡萄糖酸组 | 5.08 | 3.93 | 5.73 | 60.21 |
| 左旋咪唑山梨酸组 | 5.32 | 4.15 | 6.24 | 60.38 |
| 左旋咪唑烟酸组 | 6.38 | 4.39 | 6.08 | 60.25 |
| 左旋咪唑乳糖酸组 | 4.42 | 4.24 | 6.14 | 59.64 |
| 左旋咪唑精氨酸组 | 4.36 | 4.60 | 5.61 | 59.71 |
从以上数据可以看出,本发明的各种左旋咪唑有机酸盐相对于参与对比的盐酸左旋咪唑对小鼠具有更好的免疫增强效果,对CTX所致的免疫损伤小鼠的免疫功能均具有一定的恢复或增强作用,使胸腺指数、脾脏指数、IgG和IgM产生能力均有所回升,能对抗CTX引起的脾脏和胸腺萎缩。
Claims (10)
1.一种左旋咪唑有机酸盐,用下述通式(Ⅰ)表示:

式(Ⅰ)中:RCOOH代表有机酸,分别选自如下所示医药上允许的有机酸:酒石酸、乙酸、熊果酸、水杨酸、苹果酸、琥珀酸、抗坏血酸、乳酸、柠檬酸、咖啡酸、绿原酸、阿魏酸、甘油酸、赖氨酸、葡萄糖酸、山梨酸、烟酸、乳糖酸、精氨酸。
2.一种权利要求1所述的左旋咪唑有机酸盐的合成方法,其特征在于包括以下步骤:
(1)反应容器中加入盐酸左旋咪唑(Ⅱ)和蒸馏水,搅拌溶解,在冰浴的条件下,滴加1mol/LNaOH水溶液搅拌直至溶液pH=7~12,盐酸左旋咪唑(Ⅱ)与NaOH的摩尔比为1: 1~9;搅拌反应结束后,过滤,蒸馏水洗涤滤饼直至中性;减压干燥,得左旋咪唑游离碱(Ⅲ);


(2)按照每毫升反应溶媒中加入0.01~0.5克左旋咪唑游离碱(Ⅲ),待完全溶解后,搅拌下加入有机酸,有机酸的用量为左旋咪唑游离碱(Ⅲ)1-3倍摩尔量,于20~70℃条件下搅拌反应0.5~6h,过滤,滤液冷冻至2~10℃后,搅拌下缓慢滴加反应溶媒的2~8倍体积比的析出溶剂,冷冻析晶5~24h,过滤,滤饼用少量析出溶剂洗涤,干燥,即得左旋咪唑有机酸盐(Ⅰ)。
3.如权利要求2所述的方法,其特征在于:步骤(1)中,过滤后的滤液用有机溶剂萃取三次,每次使用的有机溶剂与盐酸左旋咪唑的重量比为1~50:1,合并萃取液,再向该萃取液中加入蒸馏水,搅拌清洗、静置分离,重复清洗直至洗液呈弱碱性,用无水氯化钙干燥有机溶剂层1~12h,过滤,减压蒸除有机溶剂,又得白色固体状的左旋咪唑游离碱(Ⅲ)。
4.如权利要求3所述的方法,其特征在于:所述有机溶剂为乙醚、二氯甲烷、乙酸乙酯或上述三溶剂的混合。
5.如权利要求2或3所述的方法,其特征在于步骤(1)的反应温度为5~50℃。
6.如权利要求2或3所述的方法,其特征在于步骤(1)的搅拌反应时间为0.5~6h。
7.如权利要求2或3所述的方法,其特征在于步骤(2)中所述的反应溶媒是蒸馏水或醇类。
8.如权利要求2或3所述的方法,其特征在于步骤(2)中所述的析出溶剂是低沸点易挥发的酮类、酯类或醚类有机溶剂。
9.用于增强机体免疫功能的药物组合物,其特征在于:该药物组合物包含有治疗有效量的权利要求1的左旋咪唑有机酸盐和药学上可接受的载体。
10.根据权利要求9所述的药物组合物,其特征在于:该药物组合物的制剂形式有片剂、胶囊剂、口服液、合剂、口含剂、颗粒剂、冲剂、丸剂、散剂、膏剂、丹剂、混悬剂、溶液剂、注射剂、粉针剂、冻干粉针剂、栓剂、软膏剂、霜剂、喷雾剂、气雾剂、滴剂或贴剂。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210168103XA CN102675346A (zh) | 2012-05-28 | 2012-05-28 | 一种左旋咪唑有机酸盐,其合成方法和它的药物组合物 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210168103XA CN102675346A (zh) | 2012-05-28 | 2012-05-28 | 一种左旋咪唑有机酸盐,其合成方法和它的药物组合物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN102675346A true CN102675346A (zh) | 2012-09-19 |
Family
ID=46807955
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201210168103XA Pending CN102675346A (zh) | 2012-05-28 | 2012-05-28 | 一种左旋咪唑有机酸盐,其合成方法和它的药物组合物 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102675346A (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104906036A (zh) * | 2015-06-12 | 2015-09-16 | 重庆市畜牧科学院 | 阿魏酸左旋咪唑注射液的制备方法 |
| CN113677649A (zh) * | 2019-03-27 | 2021-11-19 | 费尔蒂纳格罗生物技术有限公司 | 包含植物可同化磷及钙增效剂的施肥组合物及其用途 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0142191A2 (en) * | 1983-11-09 | 1985-05-22 | Janssen Pharmaceutica N.V. | A process for isolating levamisole from tetramisole |
| US5516647A (en) * | 1993-11-05 | 1996-05-14 | Abbott Laboratories | Compounds useful as alkaline phosphatase inhibitors and therapeutic agents |
| CN101107255A (zh) * | 2005-01-28 | 2008-01-16 | 詹森药业有限公司 | 抗蠕虫咪唑-噻唑衍生物 |
-
2012
- 2012-05-28 CN CN201210168103XA patent/CN102675346A/zh active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0142191A2 (en) * | 1983-11-09 | 1985-05-22 | Janssen Pharmaceutica N.V. | A process for isolating levamisole from tetramisole |
| EP0142191B1 (en) * | 1983-11-09 | 1990-03-14 | Janssen Pharmaceutica N.V. | A process for isolating levamisole from tetramisole |
| US5516647A (en) * | 1993-11-05 | 1996-05-14 | Abbott Laboratories | Compounds useful as alkaline phosphatase inhibitors and therapeutic agents |
| CN101107255A (zh) * | 2005-01-28 | 2008-01-16 | 詹森药业有限公司 | 抗蠕虫咪唑-噻唑衍生物 |
Non-Patent Citations (4)
| Title |
|---|
| A.K. SIWICKI,等: "Comparisons of nonspecific and specific immunomodulation by oxolinic acid, oxytetracycline and levamisole in salmonids", 《VETERINARY IMMUNOLOGY AND IMMUNOPATHOLOGY》, vol. 23, no. 12, 30 November 1989 (1989-11-30), pages 195 - 200 * |
| 张寄銮,等: "左旋咪唑临床应用现况", 《中国医院药学杂志》, vol. 3, no. 2, 2 March 1983 (1983-03-02) * |
| 李宁: "盐酸右旋咪唑消旋化及拆分工艺研究", 《中国优秀硕士学位论文全文数据库工程科技I辑》, 13 September 2006 (2006-09-13) * |
| 陈钟瑛,等: "四咪唑环合工艺的改进", 《医药工业》, no. 08, 15 August 1981 (1981-08-15) * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104906036A (zh) * | 2015-06-12 | 2015-09-16 | 重庆市畜牧科学院 | 阿魏酸左旋咪唑注射液的制备方法 |
| CN113677649A (zh) * | 2019-03-27 | 2021-11-19 | 费尔蒂纳格罗生物技术有限公司 | 包含植物可同化磷及钙增效剂的施肥组合物及其用途 |
| CN113677649B (zh) * | 2019-03-27 | 2023-04-14 | 费尔蒂纳格罗生物技术有限公司 | 包含植物可同化磷及钙增效剂的施肥组合物及其用途 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6014695B2 (ja) | 癌および他の疾患または障害の治療のための医薬組成物 | |
| CZ291231B6 (cs) | Farmaceutický prostředek obsahující mofetil mykofenolát nebo kyselinu mykofenolovou, a způsob jeho přípravy | |
| CN110054624A (zh) | 盐酸小檗碱与咖啡酸共晶物及制备方法和其组合物与用途 | |
| JP2008501038A (ja) | 10−プロパルギル−10−デアザアミノプテリンを用いるt細胞リンパ腫の治療 | |
| US20170050975A1 (en) | Tetrandrine family pharmaceutical formulations and method | |
| CN102166219A (zh) | 一种鼻腔给药制剂及其应用 | |
| JPH10298093A (ja) | 内用液剤 | |
| CN101066980A (zh) | 富马酸泰诺福韦酯新晶型及其药用制剂 | |
| WO2017092230A1 (zh) | 双黄酮化合物及其治疗癌症和制备药物的用途 | |
| CN102675346A (zh) | 一种左旋咪唑有机酸盐,其合成方法和它的药物组合物 | |
| CN101597272A (zh) | 艾拉莫德的钾盐化合物,其制备方法和药物应用 | |
| CN107613984A (zh) | 药物组合物及其用途 | |
| CN109988104A (zh) | 山奈酚与异烟酰胺共晶物及制备方法和其药物组合物与用途 | |
| RU2296581C2 (ru) | Капсула nlkj для лечения заболеваний предстательной железы | |
| CN109232293A (zh) | 芬乐胺晶g型、制备方法和其组合物与用途 | |
| RU2237475C1 (ru) | Комбинированный препарат для устранения симптомов простудных заболеваний и гриппа (варианты) | |
| WO2014187185A1 (zh) | 一种药物组合物作为制备治疗皮炎湿疹中的应用 | |
| CN103330933A (zh) | 含有米卡芬净或其盐的药物组合物 | |
| CN112457291B (zh) | 苯并硫代吡喃酮类化合物的盐及其制备方法和用途 | |
| CN109232297A (zh) | 芬乐胺晶b型、制备方法和其组合物与用途 | |
| US20090275654A1 (en) | Pharmaceutical Gallium Compositions and Methods | |
| KR20210064247A (ko) | 독성이 감소된 혼성 암포테리신 b 유도체 | |
| WO2015073958A1 (en) | Ephedra alata extracts and methods of use thereof | |
| JP2021503460A (ja) | B型肝炎ウイルス感染症の治療におけるシアノバクテリアバイオマスの使用 | |
| RU2805751C2 (ru) | Со-кристаллические формы теобромина |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C12 | Rejection of a patent application after its publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20120919 |