CN101341578B - 加工多光子可固化光活性组合物的方法及装置 - Google Patents
加工多光子可固化光活性组合物的方法及装置 Download PDFInfo
- Publication number
- CN101341578B CN101341578B CN2006800482953A CN200680048295A CN101341578B CN 101341578 B CN101341578 B CN 101341578B CN 2006800482953 A CN2006800482953 A CN 2006800482953A CN 200680048295 A CN200680048295 A CN 200680048295A CN 101341578 B CN101341578 B CN 101341578B
- Authority
- CN
- China
- Prior art keywords
- layer
- substrate
- optical system
- light beam
- interface
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images










Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/20—Exposure; Apparatus therefor
- G03F7/2051—Exposure without an original mask, e.g. using a programmed deflection of a point source, by scanning, by drawing with a light beam, using an addressed light or corpuscular source
- G03F7/2053—Exposure without an original mask, e.g. using a programmed deflection of a point source, by scanning, by drawing with a light beam, using an addressed light or corpuscular source using a laser
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/027—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds
- G03F7/028—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds with photosensitivity-increasing substances, e.g. photoinitiators
- G03F7/031—Organic compounds not covered by group G03F7/029
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/038—Macromolecular compounds which are rendered insoluble or differentially wettable
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/70—Microphotolithographic exposure; Apparatus therefor
- G03F7/70375—Multiphoton lithography or multiphoton photopolymerization; Imaging systems comprising means for converting one type of radiation into another type of radiation
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/70—Microphotolithographic exposure; Apparatus therefor
- G03F7/70483—Information management; Active and passive control; Testing; Wafer monitoring, e.g. pattern monitoring
- G03F7/70605—Workpiece metrology
- G03F7/70608—Monitoring the unpatterned workpiece, e.g. measuring thickness, reflectivity or effects of immersion liquid on resist
Landscapes
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Optics & Photonics (AREA)
- Laser Beam Processing (AREA)
- Investigating, Analyzing Materials By Fluorescence Or Luminescence (AREA)
- Application Of Or Painting With Fluid Materials (AREA)
- Heating, Cooling, Or Curing Plastics Or The Like In General (AREA)
- Manufacturing Optical Record Carriers (AREA)
Abstract
一种方法,所述方法包括提供其上具有包含多光子可固化光活性组合物的层的基底,用光束照射所述层的至少一个区域,其中所述光束可固化或引发固化所述多光子可固化光反应组合物;以及处理所述基底反射的光束的一部分,以获得每个区域所述层与所述基底之间界面的位置信号。
Description
优先权声明
本专利申请要求对2005年12月21日提交的美国临时申请No.60/752,529的优先权,该临时申请的内容在此以引用的方式并入本文。
技术领域
本发明涉及在过程中检测多光子可固化光活性组合物与基底之间界面的方法及装置。
背景技术
在多光子引发固化工艺(例如,在美国专利No.6,855,478中描述的工艺,该专利以引用的方式并入本文)中,在基底上(例如硅晶片)施加包含多光子可固化光活性组合物的层,并使用聚焦光源(例如激光束)进行固化。所施加层中的多光子可固化光活性组合物包括至少一种能够经受酸或基引发的化学反应的活性物质和一个多光子引发剂系统。利用具有适当波长和足够强度的光对该层进行成像曝光,会在多光子引发剂系统中导致双光子吸收,从而在层的曝光区域中诱导活性物质中酸或基引发的化学反应。该化学反应会导致曝光区域中形成交联、聚合或溶解度特性发生变化(在本文中将其称为固化),从而形成固化物。在固化步骤之后,可以通过以下方式可选地形成层:移除该层的非固化部分以获得固化物,或从该层上移除固化物本身。
包含多光子可固化光活性组合物的层通常具有约10-500μm的相对均匀的厚度,并且该层的任何位置都可发生固化以形成固化物。然而,要确保固化物附连到基底上,应在固化组合物与基底之间的界面上开始固化工艺。该界面应处于位置的准确度变化很大,取决于要形 成的具体固化结构,但通常界面应位于约100nm至1μm的范围内。
常规多光子固化工艺利用表面映射技术,该技术在固化步骤之前映射整个基底表面,使界面位于基底与包含多光子可固化光活性组合物的层之间。在可供选择的技术中,使用第二光束来跟踪基底表面的变化并定位该界面,所述第二光束不同于用于在包含多光子可固化光活性组合物的层中进行固化或引发固化的光束。然而,这些技术没有考虑表面测量的用时与固化工艺引发阶段之间或固化工艺进行过程中的环境变化和工艺变化。因此,这些方法提供的界面位置信息不够准确,尤其是在制造尺寸大于约1cm2的大型固化物的工艺中。
发明内容
参见图1A,图中示出了结构体10,其包括基底12,所述基底上具有多光子可固化光活性组合物的层14。如果引发固化反应的光束瞄准深度高于层14与基底12之间界面17的层14的区域,所得的固化物16的位置将过高,并且固化物16将在形成过程中被冲洗掉。如图1B所示,如果引发固化反应的光束瞄准深度低于层24与基底22之间界面27的层24的区域,那么光束会聚焦在不可光聚合的材料上,并且将不会形成所得固化物26的部分28。参见图1C,如果引发固化反应的光束瞄准具有合适深度的层34的区域,那么将在邻近基底32与组合物层34之间界面37的地方形成所得的结构体36。假定多光子聚合结构体的尺寸可小至几微米,并且结构体应该只与基底相交,沿其总高度的一小部分有效附连在基底上,图1C中定位界面37的工艺窗口非常小。
此外,施加了组合物层的基底表面的不规则性进一步加大了定位多光子可固化光活性组合物与基底之间界面的难度。例如,基底的表面粗糙度在不同区域可能不同,基底可能是倾斜或弯曲的,基底可能在固化过程中轻微移动,或者固化过程中的温度变化可能会引起热漂移。在制造尺寸大于约1cm2的固化物的工艺中,这些基底不规则性变得更加麻烦,因为基底表面的特性在这样大的区域内可能大不相同。
在一个方面,本发明涉及就地定位和/或跟踪多光子可固化光活性组合物与基底之间界面的方法,所述方法可以使固化工艺更加准确。在一个实施例中,该方法包括:提供其上具有包含多光子可固化光活性组合物的层的基底,用光束照射该层的至少一个区域,其中光束可固化或引发固化多光子可固化光活性组合物;以及处理基底反射的光束的一部分,以获得在每个区域所述层与所述基底之间界面的位置信号。
在另一方面,本发明涉及一种用于加工多光子可固化光活性组合物的方法,该方法包括:提供其上具有包含多光子可固化光活性组合物的层的基底;通过第一光学系统将光束照射在该层的至少第一区域上;在每个第一区域从基底反射光束的一部分,以形成反射光束;在第二光学系统中处理每个第一区域的反射光束,第二光学系统包括光学检测器,其中光学检测器的输出包括在每个第一区域所述层与所述基底之间界面的位置信号;响应位置信号调整第一光学系统;以及通过第一光学系统用光束固化固化区域中的组合物。
在另一方面,本发明涉及一种用于加工多光子可固化光活性组合物的方法,该方法包括:在基底上提供层,所述层包含多光子可固化光活性组合物,并且其中基底在可调平台上;通过第一光学系统将光束照射在所述层的至少第一区域上;在第二光学系统中处理每个第一区域的反射光束,第二光学系统包括光学检测器,其中光学检测器的输出包括在每个第一区域所述层与所述基底之间界面的位置信号;响应信号调整第一光学系统和平台中的至少一个;以及用光束固化固化区域中的组合物。
在该方法中,优选地,所述光束在不同的第一、第二和第三区域照射所述层,获得在每个区域所述基底与所述层之间界面的位置信号,基于在所述第一、第二和第三区域中每个区域生成的位置信号生成复合位置信号,并基于所述复合位置信号调整所述第一光学系统和所述平台中的至少一个。
在又一方面,本发明涉及一种用于通过使用界面的位置信号调整光学系统或平台的方法,该方法包括:接收基底与层的至少一个区域之间界面的位置信号,所述层包含多光子可固化光活性组合物,其中位置信号是由通过第一光学系统用聚焦光束照射所述层,并在光学检测器中处理基底反射的光束的一部分而生成的;基于每个区域所生成的位置信号生成复合位置信号;以及基于复合位置信号调整第一光学系统和支撑基底的平台中的至少一个。
在另一方面,本发明涉及一种用于固化包含多光子可固化光活性组合物的层的区域的装置,其中装置包括第一光学系统和第二光学系统,第一光学系统将聚焦激光束的第一部分射入所述层,第二光学系统处理基底反射的光束的第二部分以生成所述基底与所述层之间界面的输出信号。
当多光子可固化光活性物质层的区域被固化以形成第一固化物时,可以使用界面位置信号来调整光束,当制造了多个固化物时,可以使用该信号来连续调整固化光束;或者可以使用该信号来制造紧邻第一固化物的第二固化物。由于是通过该装置是在刚刚引发固化之前或之时获得并使用该位置信号的,因此与界面位置信息生成与固化步骤之间存在明显时间滞后的常规测量技术相比,环境和基底表面变化基本上很少影响固化工艺。
附图和以下具体实施方式给出了本发明的一个或多个实施例的详细说明。从具体实施方式、附图以及从权利要求书中,本发明的其它特征、对象和优点将显而易见。
附图说明
图1A-1C是通过固化紧邻基底的包含多光子可固化光活性组合物的层而形成的固化物的示意性剖视图。
图2是用于固化多光子可固化光活性物质的装置的示意图。
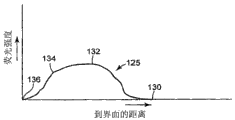
图3是图2中荧光强度与到基底106与层104之间界面的距离之间的图线。
图4是可以在图2的装置中使用的共焦界面定位器系统的示意图。
图5是使用图4中系统和检测器的图2中装置沿Z方向位移与PMT输出之间的图线。
图6是用于固化多光子可固化光活性物质的装置的示意图,该装置包括干涉测量检测器(interferometric detector)。
图7是用于固化多光子可固化光活性物质的装置的示意图,该装置包括分光检测器焦点测量系统(split detector focus measurementsystem)。
图8是用于固化多光子可固化光活性物质的装置的示意图,该装置包括静态或动态可调正透镜和工作台。
图9是图8中装置的计算机处理器的工作流程图。
在这些附图中,类似的符号表示类似的元件。
具体实施方式
参见图2,图中示出了可以用于精确固化施加在基底106上的层104的所选区域102的装置100。层104包括多光子可固化光活性组合物,下文将详细讨论该组合物。在本发明工艺的执行中,在可进行多光子吸收的条件下,光活性组合物可暴露在光中,从而导致该层区域的化学或物理特性发生变化。此类变化的实例包括聚合、交联和/或与多光子组合物曝光之前相比溶解度特性的变化(例如,在特定溶剂中溶解性减小或增大)。可使用任何已知能够获得足够强度光的方法实现此类曝光,但通常使用激光发出的聚焦光源。
装置100包括带有光源110(通常为激光)、分束器112和聚焦正透镜114的第一光学系统。发自光源110的光束116进入分束器112,通过透镜114聚焦,然后进入包含多光子可固化光活性物质的层104。当正透镜114沿Z方向相对于层104和基底106向下移动时,光束116的焦点穿过层104的厚度同样向下移动。如果需要,可以调整光束116以获得合适的波长和足够的强度,以使得光束 的焦点固化层104中区域102的任何范围内的多光子可固化光活性物质。无论是否在区域102中固化层104,进入层104的光束116的一部分都会在层104与基底106之间的界面118处被反射。光束116从界面118反射之后进入第二光学系统,该光学系统包括透镜114、分束器112和检测器120。
检测器120可以根据预期应用而各不相同,在一个实施例中包括荧光检测器,在最简单的情况下可以是人的裸眼。当光束116的焦点向着界面118向下穿过层104时,可观察到固化过程中发出的荧光强度有所变化,并且这种强度变化可以用来确定光束116的焦点何时位于界面118上。参见图2-3,当正透镜116沿Z方向向下移动时,光束116的焦点进入层104(参见图3中曲线125上的130)。荧光强度起初会增强,然后当光束116的焦点完全进入层104后(参见图3中的132)保持相对恒定。当光束116的焦点接触到层104与基底106之间的界面118时,荧光强度迅速下降(参见图3中的134),直至荧光信号在光束116在基底106(参见图3中的136)内完全聚焦的点处实际消失。
当光束116的焦点位于界面118上时,荧光强度在曲线125上的点134与136之间的某个地方。因此,当光束116的焦点位于界面118上时,曲线125上检测不到可以准确确定的清晰和明确的点,例如最小或最大荧光强度。确定荧光相符程度的另一个复杂因素是在固化层104中多光子可固化光活性物质时可能使用的曝光程度不同。根据层104中的多光子可固化光活性材料以及使用固化工艺形成的固化物类型,总激光功率及因此产生的总荧光量可能有巨大的变化。因此,对应于图3中的曲线125上点134与136之间的所需强度下降,正透镜114沿Z方向的位置范围也大不相同。
此外,利用荧光强度确定界面118的位置还需要沿Z方向移动正透镜114及多次穿过层104。如果是在层104的相同区域102进 行这种多次穿过,会消耗该区域内多光子可固化光活性物质中的光引发剂,从而导致荧光减弱。要克服此问题,可以将基底106的区域102转换为另一区域以找到新的可固化材料,但这种转换工序会在工艺中引起晶片平坦度变化,从而对工序的精确度产生不利影响。
因此,尽管荧光强度检测是有用的,但在需要高水平的精确度时,相对欠缺精确度和可预测性使该工序成为次优选工序。然而,对于需要相对粗略地估计界面118的位置的应用而言,荧光检测的低成本和简易性使上述工序成为可行的选择。
参见图4,该图示出了图2中装置的检测器120的另一个实施例。图4中示出的检测器120(在本文中是指共焦界面定位器系统)包括正透镜150、小孔152和检测器154,其中正透镜用于聚焦进入光束116,检测器通常为光电倍增管(PMT)。共焦界面定位器系统120是空间滤波器,如果光束从透镜114的焦点发出(参见图2),则只允许光束116穿过小孔152。因此,仅正透镜114焦点处发生的反射能被捕获并传输至PMT 154。仅当焦点穿过层104与基底106之间的界面118时(参见图2),PMT 154上才会有信号。
当正透镜114的焦点穿过界面118时,图4所示共焦界面定位器系统120的空间滤波效应会产生尖峰信号。可清楚而明确地检测到该尖峰,并且激光110的功率、在第一或第二光学系统中的损耗或共焦界面定位器系统120本身的特性都不会影响峰值出现与界面118位置之间的相关性。此外,共焦界面定位器系统120并不依赖于荧光,并且为提供更明确的界面118的位置,可以可选地包括滤波器122(图2),以在光束116进入检测器120之前从光束116中去除荧光波长。即使基底106上没有层104,仍可以准确定位界面118,从而在层104中找到不必耗尽光引发剂的界面。
由于图2和图4所示装置使用了相同的正透镜114,该正透镜 用于聚焦层104中的光束116,并且检测器120在继续进行层104中区域102的固化工艺之时或之后立即接收界面位置信号,因此检测器120获得的界面位置信息非常准确。使用图4中的装置和上述方法,可以在约100nm至1μm的范围内确定界面118的位置。当层104的第一区域中的多光子可固化光活性物质被固化以形成第一固化物时,界面位置信息可以用来保持正透镜114的位置,或者可以用来定位正透镜114以在不同于第一区域的层104的另一区域形成另一个固化物。
图5示出了图4中共焦显微镜检测器的代表性反应曲线160。PMT输出信号中的尖峰162对应于层104与基底106之间界面118的位置。
参见图6,该图示出了装置200的另一个实施例,该装置可以用于固化包含多光子可固化光活性物质的层204的区域202。短相干光源210(例如准直弧光灯)发出的光束216 A首先穿过包含分束器212和聚焦正透镜214的第一光学系统。光束216可选地固化或引发固化层204的区域202中的多光子可固化光活性物质。然后光束216的一部分从层204与基底206之间的界面218反射回来,进入包括正透镜214、分束器212和反射镜215的第二光学系统。从反射镜215反射回来的光再次穿过分束器212,并进入检测器220(例如PMT)。当正透镜214的焦点位于界面218上时,穿过第一和第二光学系统的光波之间的相长干涉会在检测器220上产生强烈的信号。如果正透镜214的焦点不在界面218上,穿过第一和第二光学系统的光波之间的相长干涉会在检测器220上产生相对较弱的信号。
在图7中所示的又一个实施例中,示出了分光检测器焦点测量系统和装置300,该装置可以用于固化包含多光子可固化光活性物质的层304的区域302。准直光源310发出的光束316穿过包括分束器312和聚焦目标314的第一光学系统。光束316可选地固化或引发 固化层304的区域302中的多光子可固化光活性物质。然后光束316的一部分从层304与基底306之间的界面318反射回来,进入包括目标314、分束器312、可阻挡一半光束的不透明屏幕321、聚焦透镜319和分光检测器320的第二光学系统。当光束聚焦在层304与基底306之间的界面318上时,那么所得光束位于分光检测器320的中心。分光检测器两侧之间的信号差值将为零。当光束聚焦在层304与基底306之间界面318的上方或下方时,那么所得光束将聚焦在分光检测器的一侧或另一侧。分光检测器两侧之间的信号差值将为非零,其大小表示散焦量,差异信号的特征表示散焦方向。
参见图8,该图示出了装置400,其可用于固化包含多光子可固化光活性物质的层404的区域402。层404施加在基底406上,基底406放置在可调平台或工作台405上。可手动或通过控制线472由数字计算机470来调整工作台405沿Z方向的高度以及层404在X-Y平面之上或之下的倾斜度。聚焦光源410发出的光束416首先穿过包括分束器412和聚焦正透镜414的第一光学系统。也可手动或通过控制线474由数字计算机470来调整聚焦正透镜414沿Z方向的高度。光束416离开正透镜414之后进入层404,可选地固化或引发固化层404的区域402中的多光子可固化光活性物质,并且其中一部分光束在基底406的界面418处被反射。然后光束416进入包含正透镜414和检测器420的第二光学系统。检测器420可以是图2和图4所示的共焦界面定位器系统120、图6所示的干涉检测系统或图7所示的分光检测器。
检测器420的输出然后沿线471提供给数字计算机470。再参见图9,在步骤500进行初始化后,计算机470在步骤502接收来自正透镜414和可调工作台405的位置数据以及来自检测器420的数据。在步骤504,计算机470使来自检测器的信号与正透镜414沿Z方向相对于界面418的高度以及工作台405的倾斜度信号关联起来。例如,如图9所示,如果检测器470是共焦界面定位器系 统并带有PMT,PMT的最大输出可与正透镜414沿Z方向的位置相关,而该位置对应于正透镜414的焦点位于层404与基底406之间的界面418上。在步骤506,将位置数据和检测器数据输入计算机470,并且在步骤508,计算机评估检测器数据以确定PMT信号是否为最大值。如果是,正透镜414沿Z方向的位置不变。如果不是,在步骤510计算机会调整透镜414沿Z方向的高度,直到PMT信号再次达到最大值。
使用该连续反馈系统,上述方法和装置可用于以用多种方式准确找到界面418。例如,采用静态方法时,检测器470可用作光学探针,在基底406的表面上的几个不同点(通常至少三个点)对层404/基底406的界面进行采样。对于基底406上的所有采样位置来说,如果正透镜414沿Z方向相对于界面418的位置不在约±0.5μm内,计算机470会执行必要的计算以调整正透镜414和/或工作台405。当基底406对于正透镜414下方沿X轴的运动平面足够水平时,可使用光束416固化层404的区域402。该方法仅可用于足够平坦的基底,因为仅可校正基底相对于X-Y平面的倾斜度。
当正透镜414移动并且层404中的多光子可固化光活性物质被固化形成固化物时,也可以使用动态方法提供有关界面418位置的连续反馈。该方法可对基底406的表面的任何不平坦情况进行校正,因为在从层404的第一区域到第二区域等的固化过程中,可连续校正正透镜414沿Z方向的位置。这种连续的动态反馈还可以在通常非平坦表面(例如球体和非球面)的顶部构建结构。
可用于本文所述工艺的多光子可固化光活性组合物在3M代理人档案号为60893US002的共同未决和共同提交的专利申请中有详细讨论,该专利申请全文以引用的方式并入本文。
所施加层中的多光子可固化光活性组合物包括至少一种能够经受 酸或基引发的化学反应的活性物质和多光子引发剂系统。利用具有适当波长和足够强度的光对该层进行成像曝光,会在多光子引发剂系统中导致双光子吸收,从而在层暴露于光的区域中诱发活性物质中酸或基引发的化学反应。该化学反应会导致在暴露于光的层的区域中发生可检测到的化学或物理特性变化。可检测到的变化的实例包括(例如)暴露区域内的交联、聚合或溶解度特性变化。发生任何这些可检测到的变化在本文中被称为固化,并且固化会继续直至形成固化物。固化步骤可以发生在包含多光子可固化光活性组合物的层中的任何区域,包括与施加在该层的基底之间的界面附近。在固化步骤之后,可以通过以下方式可选地形成层:移除该层的非固化部分以获得固化物,或从该层上移除固化物本身。
活性物质
适用于光活性组合物的活性物质包括可固化和非可固化物质。通常优选可固化物质,其包括(例如)可加成聚合单体和低聚物及可加成交联聚合物(诸如自由基可聚合或可交联烯键不饱和物质,其包括(例如)丙烯酸酯、异丁烯酸酯和某些乙烯基化合物(诸如苯乙烯)),以及阳离子可聚合单体和低聚物及阳离子可交联聚合物(这些物质是最普遍的酸引发物质,并且其包括(例如)环氧树脂、乙烯基醚、氰酸酯等)等等,以及它们的混合物。
例如,在Palazzotto等人的美国专利No.5,545,676第1列第65行至第2列第26行中对适用的烯键不饱和物质有所描述,其包括:单、二及聚丙烯酸酯和异丁烯酸酯(例如甲基丙烯酸酯、甲基丙烯酸甲酯、丙烯酸乙酯、异丙基甲基丙烯酸酯、正-己基丙烯酸酯、丙烯酸十八酯、丙烯酸烯丙酯、丙三醇二丙烯酸酯、丙三醇三丙烯酸酯、乙二醇二丙烯酸酯、二乙二醇二丙烯酸酯、三乙二醇二甲基丙烯酸酯、1,3-丙二醇二丙烯酸酯、1,3-丙二醇二甲基丙烯酸酯、三羟甲基丙烷三丙烯酸酯、1,2,4-丁三醇三甲基丙烯酸酯、1,4-环己二醇二丙烯酸酯、季戊四醇三丙烯酸酯、季戊四醇四丙烯酸酯、季戊四醇四甲基丙烯酸 酯、山梨醇六丙烯酸酯、双[1-(2-丙烯酰氧基)]-对乙氧苯基二甲基甲烷、双[1-(3-丙烯酰氧基-2-羟基)]-对丙氧苯基二甲基甲烷、三羧乙基-异氰脲酸酯三甲基丙烯酸酯、分子量为约200-500的聚乙二醇类的二丙烯酸酯和二甲基丙烯酸酯、诸如美国专利No.4,652,274中描述的丙烯酸化单体的共聚混合物以及诸如美国专利No.4,642,126中描述的丙烯酸化低聚物)、不饱和酰胺(例如亚甲基双丙烯酰胺、亚甲基双甲基丙烯酰胺、1,6-六亚甲基双丙烯酰胺、二亚乙基三胺三丙烯酰胺和β-甲基丙烯酰胺基乙基甲基丙烯酸酯)、乙烯基化合物(例如苯乙烯、邻苯二甲酸二烯丙酯、丁二酸二乙烯酯、己二酸二乙烯酯和邻苯二甲酸二乙烯酯)等等,以及它们的混合物。适用的活性聚合物包括含有侧(甲基)丙烯酸基的聚合物,例如每条聚合物链含有1到约50个(甲基)丙烯酸基。这类聚合物的实例包括芳族酸(甲基)丙烯酸半酯树脂,例如可得自Sartomer的SarboxTM树脂(例如SarboxTM 400、401、402、404和405)。其它可用的可由自由基化学作用固化的活性聚合物包括那些含有烃基主链和侧肽基(其上连接有自由基可聚合官能团)的聚合物,例如美国专利No.5,235,015(Ali等人)中描述的那些聚合物。如果需要,可使用两个或更多个单体、低聚物和/或活性聚合物的混合物。优选的烯键不饱和物质包括丙烯酸酯、芳族酸(甲基)丙烯酸半酯树脂和含有烃基主链和侧肽基(其上连接有自由基可聚合官能团)的聚合物。
适用的阳离子活性物质在例如Oxman等人的美国专利No.5,998,495和6,025,406中有所描述,并且包括环氧树脂。此类物质广义地被称为环氧化物,其包括单体环氧化合物和聚合物型的环氧化物,并且可以是脂肪族、脂环烃、芳族或杂环。此类物质通常平均每个分子含有至少1个可聚合环氧基(优选的是至少为约1.5个,并且更优选的是至少为约2个)。聚合环氧化物包括具有末端环氧基的线型聚合物(例如聚氧化烯乙二醇的二缩水甘油醚)、含有环氧乙烷骨架单位的聚合物(例如聚丁二烯聚环氧化合物)以及含有侧环氧基的聚合物(例如缩水甘油基甲基丙烯酸酯聚合物或共聚物)。环氧化物可以 是纯化合物,或者可以是每个分子包含一个、两个或更多个环氧基的化合物的混合物。这些包含环氧基的物质的主链和取代基性质可以大不相同。例如,主链可为任何类型,并且其上的取代基可为基本上不干扰室温下阳离子固化的任何基团。可用的取代基示例包括卤素、酯基、醚、磺酸基、硅氧烷基、硝基、磷酸基等。包含环氧基的物质的分子量可在约58至约100,000或更大之间变化。
其它可用的包含环氧基的物质包括具有以下化学式的缩水甘油醚单体:

其中,R′为烷基或芳基,n为整数1至8。实例有通过多元酚与过量氯醇(诸如环氧氯丙烷,例如2,2-双(2,3-环氧基丙氧基苯酚)丙烷的二缩水甘油醚)反应得到的多元酚的缩水甘油醚。此类环氧化物的其它实例在美国专利No.3,018,262和Handbook of Epoxy Resins (环氧树脂手册)(Lee和Neville(McGraw-Hill Book Co.,New York(1967))中有所描述。
可使用多种市售的环氧单体或树脂。易得的环氧化物包括,但不限于:十八烯氧化物、环氧氯丙烷、苯乙烯氧化物、乙烯基环己烯氧化物、缩水甘油、缩水甘油甲基丙烯酸酯、双酚A的二缩水甘油醚(例如,可以商品名“EPON 815C”、“EPON 813”、“EPON 828”、“EPON1004F”和“EPON 1001F”得自Hexion Specialty Chemicals,Inc.(Columbus,OH)的产品)以及双酚F的二缩水甘油醚(例如,可以商品名“ARALDITE GY281”得自Ciba Specialty Chemicals HoldingCompany(Basel,Switzerland)的产品和可得自Hexion SpecialtyChemicals,Inc.的“EPON 862”)。其它芳族环氧树脂包括可得自 MicroChem Corp.(Newton,MA)的SU-8树脂。
其它示例性环氧单体包括:二氧化乙烯基环己烯(可得自SPISupplies(West Chester,PA))、4-乙烯基-1-环己烯二环氧化物(可得自Aldrich Chemical Co.(Milwaukee,WI))、3,4-环氧基环己基甲基-3,4-环氧基环己烯羧酸酯(例如,可以商品名“CYRACURE UVR-6110”得自Dow Chemical Co.(Midland,MI)的产品)、3,4-环氧基-6-甲基环己基甲基-3,4-环氧基-6-甲基-环己烯羧酸酯、2-(3,4-环氧基环己基-5,5-螺-3,4-环氧基)环己烷-间二氧己环、双(3,4-环氧基环己基甲基)己二酸酯(例如,可以商品名“CYRACURE UVR-6128”得自DowChemical Co.的产品)、双(3,4-环氧基-6-甲基环己基甲基)己二酸酯、3,4-环氧基-6-甲基环己烷羧酸酯及二氧化二戊烯。
其它示例性的环氧树脂还包括:环氧化聚丁二烯(例如,可以商品名“POLY BD 605E”得自Sartomer Co.,Inc.(Exton,PA)的产品)、环氧硅烷(例如,可从Aldrich Chemical Co.(Milwaukee,WI)商购获得的3,4-环氧环己基乙基三甲氧基硅烷和3-缩水甘油氧基丙基三甲氧基硅烷)、阻燃环氧单体(例如,可以商品名“DER-542”得自DowChemical Co.(Midland,MI)的溴化处理双酚型环氧单体)、1,4-丁二醇二缩水甘油醚(例如,可以商品名“ARALDITE RD-2”得自CibaSpecialty Chemicals的产品)、基于氢化双酚A-环氧氯丙烷的环氧单体(例如,可以商品名“EPONEX 1510”得自Hexion SpecialtyChemicals,Inc.的产品)、苯酚甲醛酚醛树脂的聚缩水甘油醚(例如,可以商品名“DEN-431”和“DEN-438”得自Dow Chemical Co.的产品)以及环氧化植物油(例如,可以商品名“VIKOLOX”和“VIKOFLEX”得自Atofina Chemicals(Philadelphia,PA)的环氧化亚麻籽和大豆油)。
其它适用的环氧树脂包括可以商品名“HELOXY”从HexionSpecialty Chemicals,Inc.(Columbus,OH)商购获得的烷基缩水甘油醚。示例性单体包括“HELOXY MODFIER 7”(C8-C10烷基缩水甘油醚)、 “HELOXY MODIFIER 8”(C12-C14烷基缩水甘油醚)、“HELOXYMODIFIER 61”(丁基缩水甘油醚)、“HELOXY MODIFER 62”(甲苯基缩水甘油醚)、“HELOXY MODIFER 65”(对叔丁基苯基缩水甘油醚)、“HELOXY MODIFER 67”(1,4-丁二醇的二缩水甘油醚)、“HELOXY 68”(新戊二醇的二缩水甘油醚)、“HELOXY MODIFER107”(环己烷二甲醇的二缩水甘油醚)、“HELOXY MODIFER 44”(三羟甲基乙烷三缩水甘油醚)、“HELOXY MODIFIER 48”(三羟甲基丙烷三缩水甘油醚)、“HELOXY MODIFER 84”(脂肪族多元醇的聚缩水甘油醚)以及“HELOXY MODIFER 32”(聚乙二醇二环氧化物)。
其它可用的环氧树脂包括含有一个或多个共聚乙烯基化合物的缩水甘油丙烯酸酯的共聚物(例如缩水甘油丙烯酸酯和缩水甘油甲基丙烯酸酯)。此类共聚物的实例有1∶1的苯乙烯-缩水甘油甲基丙烯酸酯和1∶1的甲基丙烯酸甲酯-缩水甘油丙烯酸酯。其它可用的环氧树脂都是熟知的,并且包含环氧化物,诸如环氧氯丙烷、烯化氧(例如环氧丙烷)、苯乙烯氧化物、烯基氧化物(例如丁二烯氧化物)和缩水甘油酯(例如缩水甘油乙酯)。
可用的环氧官能团聚合物包括环氧官能团硅氧烷,例如美国专利No.4,279,717(Eckberg等人)中所述的那些环氧官能团硅氧烷,它们可从General Electric Company商购获得。这些聚合物是聚二甲基硅氧烷,其中1-20摩尔%的硅原子被环氧烷基(优选的是美国专利No.5,753,346(Leir等人)中所述的环氧环己基乙基)取代。
还可使用各种含环氧基物质的共混物。此类共混物可包括两个或更多个含环氧基化合物的重均分子量分布(例如低分子量(小于200)、中等分子量(约200至1000)和高分子量(大于约1000))。作为另外一种选择或除此之外,环氧树脂可包含具有不同化学性质(例如脂肪族和芳族)或功能(例如极性和非极性)的含环氧基物质的共聚 物。如果需要,还可另外掺入其它阳离子活性聚合物(例如乙烯基醚等)。
优选的环氧树脂包括芳族缩水甘油环氧树脂(例如,可得自Hexion Specialty Chemicals,Inc.的EPON树脂和可得自MicroChemCorp.(Newton,MA)的SU-8树脂)等,以及它们的混合物。更优选的是SU-8树脂及其混合物。
适用的阳离子活性物质还包括:乙烯基醚单体、低聚物及活性聚合物(例如甲基乙烯基醚、乙基乙烯基醚、叔丁基乙烯基醚、异丁基乙烯基醚、三乙二醇二乙烯基醚(RAPI-CURE DVE-3,可得自International Specialty Products(Wayne,NJ))、三羟甲基丙烷三乙烯醚及可得自Morflex,Inc.(Greensboro,NC)的VECTOMER二乙烯醚树脂(例如VECTOMER 1312、VECTOMER 4010、VECTOMER 4051和VECTOMER 4060以及可得自其它制造商的等同产品))以及它们的混合物。也可使用一种或多种乙烯基醚树脂和/或一种或多种环氧树脂的共混物(以任意比例)。也可以结合环氧和/或乙烯基醚功能性材料使用多羟基功能性材料(例如在美国专利No.5,856,373(Kaisaki等人)中所述的材料)。
非可固化物质包括,例如通过酸或基引发的反应增大溶解度的活性聚合物。此类活性聚合物包括,例如含有酯基的非水溶性聚合物,酯基团可通过光生酸转换为水溶性的酸基团(例如聚(4-叔丁氧基羰氧基苯乙烯))。非可固化物质还包括在由R.D.Allen、G.M.Wallraff、W.D.Hinsberg及L.L.Simpson所著的“High Performance AcrylicPolymers for Chemically Amplified Photoresist Applications”(化学放大光致抗蚀剂应用中的高性能丙烯酸类聚合物)(J.Vac.Sci.Technol.B,9,3357(1991))中所述的化学放大光致抗蚀剂。化学放大光致抗蚀剂的概念现已广泛用于微芯片制造,尤其是具有0.5微米以下(或甚至0.2微米以下)部件的微芯片。在此类光致抗蚀剂系统中,通过照射可 以产生催化物质(典型地为氢离子),引发级联化学反应。当氢离子引发产生更多氢离子或其它酸性物质的反应时,会发生这一级联反应,从而增大反应速率。代表性的酸催化化学放大光致抗蚀剂系统的实例包括脱保护(例如美国专利No.4,491,628所述的叔丁氧基羰氧基苯乙烯抗蚀剂、基于四氢吡喃(THP)甲基丙烯酸酯的材料、THP-酚材料(例如美国专利No.3,779,778中所述的材料)、基于异丁烯酸叔丁酯的材料(例如R.D Allen等人的Proc.SPIE 2438,474(1995)中所述的材料)等)、解聚(例如基于聚苯二醛的材料)及与重排(例如根据频哪醇重排而得到的材料)。
如果需要,可在光活性组合物中使用不同类型活性物质的混合物。例如,也可使用自由基活性物质和阳离子活性物质的混合物。
光引发剂系统
光引发剂系统是多光子光引发剂系统,使用此类系统能将聚合反应禁闭或限制在聚焦光束的聚焦区域。此类系统优选的是二组分或三组分系统,其中包含至少一种多光子光敏剂、至少一种光引发剂(或电子受体)以及可选的至少一种电子供体。此类多组分系统可提供增大的灵敏度,使光致反应在更短的时间内发生,从而降低因样本和/或曝光系统的一个或多个组分的移动而产生问题的可能性。
优选的是,多光子光引发剂系统包含光学有效量的(a)至少一种多光子光敏剂,该光敏剂能够同时吸收至少两个光子,并且可选但优选地含有一个大于荧光素吸收横截面的双光子吸收横截面;(b)可选地,至少一种电子供体化合物,该化合物不同于多光子光敏剂并且能够为光敏剂的电子激发态提供电子;(c)至少一种光引发剂,该引发剂能够通过从光敏剂的电子激发态接受电子而感光,从而形成至少一种自由基和/或酸。
作为另外一种选择,多光子光引发剂系统可以是包含至少一种光 引发剂的单组分系统。可用作单组分多光子光引发剂系统的光引发剂包括酰基氧化膦(例如,Ciba销售的商品名为IrgacureTM 819的产品,以及BASF Corporation销售的商品名为LucirinTM TPO-L的2,4,6-三甲基苯甲酰乙氧苯基氧化膦)和带有共价键合锍盐部分的二苯乙烯衍生物(例如W.Zhou等人在Science 296,1106(2002)中所述的物质)。还可使用其它常规紫外线(UV)光引发剂(例如联苯酰缩酮),但它们的多光子光反应敏感性一般相对较低。
下文描述了可用于双组分和三组分多光子光引发剂系统的多光子光敏剂、电子供体及光引发剂(或电子受体)。
(1)多光子光敏剂
适合在光活性组合物的多光子光引发剂系统中使用的多光子光敏剂是那些暴露于充足光线下时能够同时吸收至少两个光子的光敏剂。优选的是,光敏剂具有大于荧光素吸收横截面的双光子吸收横截面(即,大于3′,6′-二羟基螺旋(异苯并呋喃-1(3H),9′-[9H]占吨)3-酮的吸收横截面)。一般来讲,优选的横截面可以大于约50×10-50cm4秒/光子,测量方法是C.Xu和W.W.Webb在J.Opt.Soc.Am.B,13,481(1996)(Marder和Perry等人在国际专利公开No.WO 98/21521第85页第18-22行引用)描述的方法。
更优选的是,光敏剂的双光子吸收横截面大于比荧光素吸收横截面的约1.5倍(或作为另外一种选择,采用上述方法测量时,大于约75×10-50cm4sec/光子);甚至更优选的是,大于约荧光素吸收横截面的约两倍(或作为另外一种选择,大于约100×10-50cm4sec/光子);最优选的是,大于荧光素吸收横截面的约三倍(或作为另外一种选择,大于约150×10-50cm4sec/光子);以及最理想的是,大于荧光素吸收横截面的约四倍(或作为另外一种选择,大于约200×10-50cm4sec/光子)。
优选的是,光敏剂可溶解于活性物质(如果活性物质是液体),或者与活性物质及包含在组合物中的任何粘结剂(如下所述)相容。最优选地是,采用美国专利No.3,729,313所描述的测试步骤,在波长范围重叠光敏剂的单光子吸收光谱(单光子吸收状态)的光的连续照射下,光敏剂还能够敏化2-甲基-4,6-双(三氯甲基)-S-三嗪。
优选的是,还可部分基于架藏稳定性选择光敏剂。因此,具体光敏剂的选择在一定程度上取决于所用的具体活性物质(以及取决于所选择的电子供体化合物和/或光引发剂)。
尤其优选的多光子光敏剂包括那些显示具有较大多光子吸收横截面的光敏剂,例如若丹明B(即,N-[9-(2-羧基苯基)-3,6-(二乙氨基)占吨翁氯化物或六氟锑酸盐)以及Marder和Perry等人在国际专利公布Nos.WO 98/21521和WO 99/53242中描述的四类光敏剂。这四类光敏剂可以描述如下:(a)其中两个供体连接到共轭π(pi)电子桥的分子;(b)其中两个供体连接到可由一个或多个电子接收基团取代的共轭π(pi)电子桥的分子;(c)其中两个受体连接到共轭π(pi)电子桥的分子;以及(d)其中两个受体连接到可被一个或多个供电子基所取代的共轭π(pi)电子桥的分子(其中“桥”是指连接两个或更多个化学基团的分子片段,“供体”是指可连接到共轭π(pi)电子桥、具有低电离势的原子或原子基团,“受体”是指可连接到共轭π(pi)电子桥、具有高电子亲和势的原子或原子基团)。
如国际专利公布No.WO 98/21521中详细描述的,利用麦克默立反应使醛与叶立德在标准维蒂希条件下进行反应制备上述四类光敏剂。
Reinhardt等人(例如在美国专利No.6,100,405、5,859,251和5,770,737中)还描述了具有大的多光子吸收横截面的其它化合物,但要通过除上述方法之外的其它方法来测定这些横截面。
优选的光敏剂包括以下化合物(以及它们的混合物):

(2)电子供体化合物
可在光活性组合物的多光子光引发剂系统中使用的电子供体化合 物是那些能够为光敏剂的电子激发状态提供电子的化合物(光敏剂本身除外)。此类化合物可以可选地用来提高光引发剂系统的多光子光敏性,从而减少使光活性组合物进行光致反应所需的曝光。电子供体化合物优选地具有的氧化电势大于零并且小于或等于对苯二酚二甲醚的氧化电势。优选的是,相对于标准饱和甘汞电极(“S.C.E.”)的氧化电势在约0.3至1伏特之间。
电子供体化合物还优选地可溶解于活性物质,并在一定程度上根据架藏稳定性进行选择(如上文所述)。当暴露于所需波长的光时,适用的供体通常能够提高光活性组合物的固化速度或成像密度。
当采用阳离子活性物质时,本领域内的技术人员将认识到如果电子供体化合物具有显著的碱度,会对阳离子反应产生不利影响。(参见,例如,美国专利No.6,025,406(Oxman等人)中第7列第62行至第8列第49行的讨论。)
一般来讲,可以通过比较三种组分的氧化和还原电势选择适合与具体光敏剂和光引发剂一起使用的电子供体化合物(例如,在美国专利No.4,859,572(Farid等人)中有所描述)。可通过实验测量这些电势(例如,通过R.J.Cox,Photographic Sensitivity,Chapter 15,AcademicPress(1973)中所述方法),或者可从诸如N.L.Weinburg,Ed.的 Technique of Electroorganic Synthesis Part II Techniques of Chemistry,Vol.V(1975)和C.K.Mann和K.K.Barnes的ElectrochemicalReactions in Nonaqueous Systems(1970)等参考文献中获得这些电势。这些电势可反映相对能量关系,并且可用于指导对电子供体化合物的选择。
适用的电子供体化合物包括(例如)那些在以下文献中描述的化合物:D.F.Eaton在B.Voman等人编辑的Advances inPhotochemistry,Volume 13,pp.427-488,John Wiley and Sons,New York (1986);Oxman等人的美国专利No.6,025,406第7列42-61行;以及Palazzotto等人的美国专利No.5,545,676第4列第14行至第5列第18行。此类电子供体化合物包括胺(包括三乙醇胺、肼、1,4-二氮杂二环[2.2.2]辛烷、三苯胺(及其三苯基膦和三苯胂类似物)、氨基醛及氨基硅烷)、酰胺(包括磷酰胺)、醚(包括硫醚)、脲(硫脲)、亚磺酸及其盐、氰亚铁酸盐、抗坏血酸及其盐、氨基二硫代甲酸及其盐、黄酸盐、1,2-乙二胺四乙酸盐、(烷基)n(芳基)m硼酸盐(n+m=4)(优选四烷基铵盐)、各种有机金属化合物(例如SnR4化合物(其中每个R是从烷基、芳烷基(尤其是苯甲基)、芳基和烷芳基基团中独立选择的)(例如,n-C3H7Sn(CH3)3、(烯丙基)Sn(CH3)3和(苯甲基)Sn(n-C3H7)3等化合物))、二茂络铁等等,以及它们的混合物。电子供体化合物可以是未取代的,或可以被一个或多个无干扰取代基取代。尤其优选的电子供体化合物包含电子配位原子(例如氮、氧、磷或硫原子)和键合到位于电子配位原子α位的碳或硅原子上的可夺取的氢原子。
优选的胺电子供体化合物包括烷基、芳基、烷芳基和芳烷基胺(例如,甲胺、乙胺、丙胺、丁胺、三乙醇胺、戊胺、己胺、2,4-二甲基苯胺、2,3-二甲基苯胺、o-、m-与p-甲苯胺、苄胺、氨基吡啶、N,N′-二甲基乙二胺、N,N′-二乙基乙二胺、N,N′-二苄基乙二胺、N,N′-二乙基-1,3-丙二胺、N,N′-二乙基-2-丁烯-1,4-二胺、N,N′-二甲基-1,6-己二胺、哌嗪、4,4′-三亚甲基双哌啶、4,4′-乙烯双哌啶、p-N,N-二甲基-氨基苯乙醇和p-N-二甲基氨基苯甲腈)、氨基醛(例如,p-N,N-二甲氨基苯甲醛、p-N,N-二乙氨基苯甲醛、9-久洛尼定甲醛和4-吗啉苯甲醛)、氨基硅烷(例如,三甲基硅基吗啉、三甲基硅基哌啶、双(二甲胺基)二苯基硅烷、三(二甲胺基)甲基硅烷、N,N-二乙基胺三甲基硅烷、三(二甲基氨基)苯硅烷、三(甲基甲硅烷基)胺、三(二甲基甲硅烷基)胺、二(二甲基甲硅烷基)胺、N,N-二(二甲基甲硅烷基)苯胺、N-苯基-N-二甲基甲硅烷基苯胺和N,N-二甲基-N-二甲基硅烷胺)、以及它们的混合物。已发现叔芳族烷基胺,尤其是那些在芳族环上含有至少一个吸电子基团 的叔芳族烷基胺可提供非常好的架藏稳定性。使用室温下为固体的胺也能获得良好的架藏稳定性。使用含有一个或多个久洛尼定基部分的胺可获得良好的光敏性。
优选的酰胺电子供体化合物包括N,N-二甲基乙酰胺、N,N-二乙基乙酰胺、N-甲基-N-苯乙酰胺、六甲基磷酰胺、六乙基磷酰胺、六丙基磷酰胺、三(4-吗啉代)膦、磷酸三酰哌啶以及它们的混合物。
优选的烷基芳基硼酸盐包括
Ar3B-(n-C4H9)N+(C2H5)4
Ar3B-(n-C4H9)N+(CH3)4
Ar3B-(n-C4H9)N+(n-C4H9)4
Ar3B-(n-C4H9)Li+
Ar3B-(n-C4H9)N+(C6H13)4
Ar3B--(C4H9)N+(CH3)3(CH2)2CO2(CH2)2CH3
Ar3B--(C4H9)N+(CH3)3(CH2)2OCO(CH2)2CH3
Ar3B--(sec-C4H9)N+(CH3)3(CH2)2CO2(CH2)2CH3
Ar3B--(sec-C4H9)N+(C6H13)4
Ar3B--(C4H9)N+(C8H17)4
Ar3B--(C4H9)N+(CH3)4
(p-CH3O-C6H4)3B-(n-C4H9)N+(n-C4H9)4
Ar3B--(C4H9)N+(CH3)3(CH2)2OH
ArB-(n-C4H9)3N+(CH3)4
ArB-(C2H5)3N+(CH3)4
Ar2B-(n-C4H9)2N+(CH3)4
Ar3B-(C4H9)N+(C4H9)4
Ar4B-N+(C4H9)4
ArB-(CH3)3N+(CH3)4
(n-C4H9)4B-N+(CH3)4
Ar3B-(C4H9)P+(C4H9)4
(其中Ar为苯基、萘基、取代(优选的是氟取代)苯基、取代萘基及含有更多稠合芳族环的类似基团),以及四甲铵正丁基三苯基硼酸盐和四丁基铵正己基-三(3-氟代苯基)硼酸盐,以及它们的混合物。
适合的醚电子供体化合物包括4,4′-二甲氧基联苯基、1,2,4-三甲氧基苯、1,2,4,5-四甲氧基苯等等,以及它们的混合物。适合的脲电子供体化合物包括N,N′-二甲基脲、N,N-二甲基脲、N,N′-二苯基脲、四甲基硫脲、四乙基硫脲、四-正-丁基硫脲、N,N-二-正-丁基硫脲、N,N′-二-正-丁基硫脲、N,N-二苯基硫脲、N,N′-二苯基-N,N′-二乙基硫脲等等,以及它们的混合物。
用于自由基引发反应的优选电子供体化合物包括含有一个或多个久洛尼定基部分的胺、烷基芳基硼酸盐及芳族亚磺酸盐。然而,对于此类反应,如果需要(例如,为了提高光活性组合物的架藏稳定性或为了修正分辨率、对比度和可逆性),也可不使用电子供体化合物。用于酸引发反应的优选电子供体化合物包括4-二甲基氨基苯甲酸、乙基4-二甲基氨基苯甲酸盐、3-二甲基氨基苯甲酸、4-二甲基氨基安息香、4-二甲基氨基苯甲醛、4-二甲基氨基苯腈、4-二甲基氨基苯乙醇及1,2,4-三甲氧基苯。
(3)光引发剂
适用于光活性组合物活性物质的光引发剂(即电子受体化合物)是那些能够通过接受来自多光子光敏剂电子激发状态的电子以形成至少一种自由基和/或酸而被感光的化合物。此类光引发剂包括碘鎓盐(例如二芳基碘鎓盐)、锍盐(例如三芳基硫盐,其可选地被烷基或烷氧基取代,并且可选地含有桥接相邻芳基部分的2,2′氧基团)等等,以及它们的混合物。
光引发剂优选地可溶解于活性物质,并且优选地具有架藏稳定性(即,当溶解于含有光敏剂和电子供体化合物的活性物质中时,不会 自发促进活性物质反应)。因此,如上所述,具体光引发剂的选择在一定程度上取决于所选择的具体活性物质、光敏剂和电子供体化合物。如果活性物质能够经受酸引发的化学反应,那么该光引发剂为鎓盐(例如碘鎓盐或锍盐)。
适用的碘鎓盐包括Palazzotto等人在美国专利No.5,545,676第2列第28行至46行所描述的碘鎓盐。适用的碘鎓盐在美国专利No.3,729,313、3,741,769、3,808,006、4,250,053和4,394,403中也有所描述。碘鎓盐可以是简单盐(例如,包含阴离子,如Cl-、Br-、I-或C4H5 SO3 -)或金属复合盐(例如,包含SbF6、PF6 -、BF4 -、四(全氟苯基)硼酸盐、SbF5 OH-或AsF6 -)。如果需要,可以使用碘鎓盐的混合物。
可用的芳族碘鎓复合盐光引发剂的实例包括:二苯基碘鎓四氟硼酸盐、二(4-甲基苯基)碘鎓四氟硼酸盐、苯基-4-甲基苯基碘鎓四氟硼酸盐、二(4-庚基苯基)碘鎓四氟硼酸盐、二(3-硝基苯基)碘鎓六氟磷酸盐、二(4-氯苯基)碘鎓六氟磷酸盐、二(萘基)碘鎓四氟硼酸盐、二(4-三氟代甲基苯基)碘鎓四氟硼酸盐、二苯基碘鎓六氟磷酸盐、二(4-甲基苯基)碘鎓六氟磷酸盐、二苯基碘鎓六氟砷酸盐、二(4-苯氧基苯基)碘鎓四氟硼酸盐、苯基-2-噻吩基碘鎓六氟磷酸盐、3,5-二甲基吡唑基-4-苯基碘鎓六氟磷酸盐、二苯基碘鎓六氟锑酸盐、2,2′-二苯基碘鎓四氟硼酸盐、二(2,4-二氯苯基)碘鎓六氟磷酸盐、二(4-溴苯)碘鎓六氟磷酸盐、二(4-甲氧基苯基)碘鎓六氟磷酸盐、二(3-羧基苯基)碘鎓六氟磷酸盐、二(3-甲氧基羧基苯)碘鎓六氟磷酸盐、二(3-甲氧基磺酰基苯基)碘鎓六氟磷酸盐、二(4-乙酰胺基苯基)碘鎓六氟磷酸盐、二(2-苯并噻吩基)碘鎓六氟磷酸盐及二苯基碘鎓六氟锑酸盐等等,以及它们的混合物。可根据Beringer等人在J.Am.Chem.Soc. 81,342(1959)的教导内容,通过对应芳族碘鎓简单盐(例如二苯基碘鎓硫酸氢盐)的复分解制备芳族碘鎓复合盐。
优选的碘鎓盐包括二苯基碘鎓盐(例如二苯基碘鎓氯化物、二苯 基碘鎓六氟磷酸盐和二苯基碘鎓四氟硼酸盐)、二芳基碘鎓六氟锑酸盐(例如可得自Sartomer Company的SarCatTM SR 1012),以及它们的混合物。
可用的锍盐包括美国专利No.4,250,053(Smith)中第1列第66行至第4列第2行所描述的锍盐,可用以下化学式表示:


其中R1、R2和R3各自独立地选自含有约4个至约20个碳原子的芳族基团(例如,取代或未取代的苯基、萘基、噻吩基和呋喃,其中可用烷氧基、烷硫基、芳硫基、卤素等这类基团取代)和含有1个至约20个碳原子的烷基。此处使用的术语“烷基”包括取代的烷基(例如,用卤素、羟基、烷氧基或芳基这类基团取代)。R1、R2和R3 中的至少一个为芳族,并且优选的是,每一个都是独立的芳族。Z选自由共价键、氧、硫、-S(=O)-、-C(=O)-、-(O=)S(=O)-和-N(R)-组成的群组,其中R为芳基(含有约6个至约20个碳,例如苯基)、酰基(含有约2个至约20个碳,例如乙酰基、苯甲酰等等),碳-碳键或-(R4-)C(-R5)-,其中R4和R5独立地选自由氢、含有1个至约4个碳原子的烷基以及含有约2个至约4个碳原子的烯基基团组成的群组。X-是阴离子,如下所述。
适用于锍盐(以及适用于任何其它类型光引发剂)的阴离子X-包括多种阴离子类型,例如酰亚胺、甲基化物、以硼为中心、以磷为中心、以锑为中心、以砷为中心和以铝为中心的阴离子。
适用的酰亚胺和甲基化阴离子的示例性的、但并非限制性的实例包括(C2F5SO2)2N-、(C4F9SO2)2N-、(C8F17SO2)3C-、(CF3SO2)3C-、(CF3SO2)2N-、(C4F9SO2)3C-、(CF3SO2)2(C4F9SO2)C-、(CF3SO2)(C4F9SO2)N-、((CF3)2NC2F4SO2)2N-、(CF3)2NC2F4SO2C-(SO2 CF3)2、(3,5-bis(CF3)C6H3)SO2N-SO2CF3、C6H5SO2C-(SO2CF3)2、 C6H5SO2N-SO2CF3等等。优选的此类阴离子包括那些由化学式(RfSO2)3C-表示的阴离子,其中Rf为含有1个至约4个碳原子的全氟烷基。
适用的以硼为中心的阴离子的示例性的、但并非限制性的实例包括F4B-、(3,5-bis(CF3)C6H3)4B-、(C6F5)4B-、(p-CF3C6H4)4B-、(m-CF3C6H4)4B-、(p-FC6H4)4B-、(C6F5)3(CH3)B-、(C6F5)3(n-C4H9)B-、(p-CH3C6H4)3(C6F5)B-、(C6F5)3FB-、(C6H5)3(C6F5)B-、(CH3)2(p-CF3C6H4)2B-、(C6F5)3(n-C18H37O)B-等等。优选的以硼为中心的阴离子通常含有3个或更多个卤素取代的芳族烃基,该基连接到硼,其中氟是最优选的卤素。优选阴离子的示例性的、但并非限制性的实例包括(3,5-bis(CF3)C6H3)4B-、(C6F5)4B-、(C6F5)3(n-C4H9)B-、(C6F5)3FB-和(C6F5)3(CH3)B-。
适用的含有其它金属或准金属中心的阴离子包括,例如(3,5-bis(CF3)C6H3)4Al-、(C6F5)4Al-、(C6F5)2F4P-、(C6F5)F5P-、F6P-、(C6F5)F5Sb-、F6Sb-、(HO)F5Sb-和F6As-。上述列表并非旨在详尽列出,因为(上述通式)对本领域内的技术人员而言,其它可用的以硼为中心的非亲核盐以及其它包含其它金属或准金属的可用阴离子将显而易见。
优选的是,阴离子X-应选自四氟硼酸盐、六氟磷酸盐、六氟砷酸盐、六氟锑酸盐和羟基五氟锑酸盐(例如,与阳离子活性物(如环氧树脂)一起使用)。
适用的锍盐光引发剂实例包括:
三苯基锍四氟硼酸盐
甲基二苯基锍四氟硼酸盐
二甲基苯基锍六氟磷酸盐
三苯基锍六氟磷酸盐
三苯基锍六氟锑酸盐
二苯基萘基锍六氟砷酸盐
三甲苯基锍六氟磷酸盐
甲氧基二苯基锍六氟锑酸盐
4-丁氧基苯基二苯基锍四氟硼酸盐
4-氯苯基二苯基锍六氟磷酸盐
三(4-苯氧基苯基)锍六氟磷酸盐
二(4-乙氧基苯基)甲基锍六氟砷酸盐
4-丙酮基苯基二苯基锍四氟硼酸盐
4-硫甲氧基苯基二苯基锍六氟磷酸盐
二(甲氧基磺酰苯基)甲基锍六氟锑酸盐
二(硝基苯)苯基锍六氟锑酸盐
二(甲酯基苯基)甲基锍六氟磷酸盐
4-乙酰胺基苯基二苯基锍四氟硼酸盐
二甲基萘基锍六氟磷酸盐
三氟甲基二苯基锍四氟硼酸盐
间(苯基苯硫基)二苯基锍六氟锑酸盐
10-甲基苯基氧硫杂环己二烯基六氟磷酸盐
5-甲基硫蒽基六氟磷酸盐
10-苯基-9,9-二甲基硫氧蒽基六氟磷酸盐
10-苯基-9-氧代硫氧蒽基四氟硼酸盐
5-甲基-10-氧代硫蒽基四氟硼酸盐
5-甲基-10,10-二氧代硫蒽基六氟磷酸盐
优选的锍盐包括三芳基取代盐,例如三芳基锍六氟锑酸盐(例如可得自Sartomer Company的SarCatTM SR1010)、三芳基锍六氟磷酸盐(例如可得自Sartomer Company的SarCatTM SR 1011)及三芳基锍六氟磷酸盐(例如可得自Sartomer Company的SarCatTM KI85)。
优选的光引发剂包括碘鎓盐(更优选的是芳基碘鎓盐)、锍盐以及它们的混合物。更优选的是芳基碘鎓盐以及它们的混合物。
光活性组合物的制备
可通过上述方法或本领域中已知的其它方法制备活性物质、多光子光敏剂、电子供体化合物和光引发剂,其中许多是市售的产品。可以任何混合顺序和方式(可选的是搅拌或搅动)在“安全光”条件下混合这四种组分,但有时更可取的是(从储存寿命和热稳定性方面考虑)最后(及在所有用于加速其它组分溶解的可选加热步骤之后)添加光引发剂。如果需要,可使用溶剂,前提条件是所选溶剂不会与组合物中的组分发生任何反应。适用的溶剂包括(例如)丙酮、二氯甲烷和乙腈。活性物质本身有时也可作为其它组分的溶剂。
光引发剂系统的三种组分都以光化学有效量(如上文所定义的)存在。一般来讲,基于固体总重量(即,除溶剂之外的组分总重量),组合物可包含:至少约5重量%(优选地至少约10重量%;更优选地至少约20重量%)直至最多约99.79重量%(优选地最多约95重量%;更优选地最多约80重量%)的一种或多种活性物质;至少约0.01重量%(优选地至少约0.1重量%;更优选地至少约0.2重量%)直至最多约10重量%(优选地最多约5重量%;更优选地最多约2重量%)的一种或多种光敏剂;可选地,最多约10重量%(优选地最多约5重量%)的一种或多种电子供体化合物(优选地至少约0.1重量%;更优选地约0.1重量%至约5重量%);以及约0.1重量%至约10重量%的一种或多种电子受体化合物(优选地,约0.1重量%至约5重量%)。
根据所需最终的用途,光活性组合物中可以包含多种辅剂。适用的辅剂包括溶剂、稀释剂、树脂、粘结剂、增塑剂、颜料、染料、无机或有机加强或延伸填充剂(优选量为占组合物总重量的约10重量%至90重量%)、触变剂、指示剂,抑制剂、稳定剂、紫外线吸收剂等等。这些辅剂的量和类型及其添加到组合物中的方式是本领域内的技术人员所熟知的。
例如,在本发明的范围内,可在组合物中包括非反应聚合物粘合剂,以控制粘度并提供成膜特性。通常可选择与活性物质相容的此类聚合物粘合剂。例如,使用可溶解于用于活性物质的相同溶剂中的聚合物粘合剂,并且该粘合剂没有对活性物质反应过程产生不利影响的官能团。粘合剂可具有适于达到所需成膜特性和溶液流变特性的分子量(例如,分子量在约5,000至1,000,000道尔顿之间;优选地在约10,000至500,000道尔顿之间;更优选地在约15,000至250,000道尔顿之间)。适用的聚合物粘合剂包括(例如)聚苯乙烯、聚(甲基丙烯酸甲酯)、苯乙烯-丙烯腈共聚物、乙酸纤维素丁酸酯等等。
在曝光之前,根据需要,使用本领域内技术人员已知的多种涂覆方法中的任一种方法(包括,例如刮涂和旋转涂覆)将所得的光活性组合物涂覆在基底上。可根据具体应用和所用曝光方法,从各种膜、薄片及其它平面(包括硅晶片和玻璃板)中选择基底。优选的基底一般要足够平坦,以便能够制备具有均匀厚度的光活性组合物层。对于涂层不太适用的应用,作为另外一种选择,光活性组合物可以块状曝光。
实例
以下实例对本发明的目的和优点进行了进一步的说明,但这些实例中列举的具体物质和用量以及其它条件和细节不应当理解为对本发明不当限制。除非另有说明,否则所有工序都是在干燥氮气氛下使用干燥并且脱氧的溶剂和试剂进行。除非另有说明,否则所有溶剂和试剂均可得自Sigma-Aldrich Corp.(St.Louis,MO)。
若丹明B六氟锑酸盐是通过若丹明B氯化物与六氟锑酸纳的复分解制备的。
如本文所用,
“SR368”是指三(2-羟基乙基)异氰脲酸酯三丙烯酸酯,可得自Sartomer Co.Inc.(Exton PA);
“SR9008”是指三官能丙烯酸酯,可得自Sartomer Co.,Inc.(ExtonPA);
“SR1012”是指二芳基錪鎓六氟锑酸盐,可得自Sartomer Co.,Inc.(Exton PA);
“SU-8 R2150”是指环氧负光致抗蚀剂,可得自MicroChemCorp.,(Newton,MA);
“PGMEA”是指聚(乙二醇)甲醚乙酸酯;
“THF”是指四氢呋喃;
“PHOTOMER 6210”是指脂肪族聚氨酯二丙烯酸酯,由CognisCorp.(USA Cincinnati OH)制造;
“SARTOMER 238”是指己二醇二丙烯酸酯,可得自Sartomer Co.Inc.(Exton PA);
“IRGACURE 819”是指氧化酰基磷化氢光引发剂,可得自CibaSpecialty Chemicals(Tarrytown NY)。
“CGI 7460”是指四丁基氨正-己基-三(3-氟代苯基)硼酸盐,可得自Ciba Specialty Chemicals(Tarrytown NY)。
实例1
使用共焦界面定位器系统制备微透镜列阵
将得自Wafer World,Inc.(West Palm Beach,Florida)的圆形硅晶片(直径为10.2cm(4英寸))在体积/体积(v/v)比为3∶1的浓硫酸和30重量%过氧化氢水溶液的混合物中浸泡约10分钟,进行清洗和准备。然后依次用去离子水和异丙醇冲洗晶片,之后用气流干燥。随后将晶片浸入重量百分比为2%的3-(三甲氧基硅烷)丙基甲基丙烯酸酯的190-proof乙醇溶液(已用乙酸配制为酸性(pH 4-5))中。晶片然后用无水乙醇冲洗,再在130℃的烘箱中加热10分钟。
将数均分子量为约120,000的聚(甲基丙烯酸甲酯)、SR9008和SR368以30∶35∶35的重量比混合,形成单体混合物,然后将该单体混合物溶解于足量的1,2-二氯乙烷中,得到重量百分比为54%的单体混合物溶液。然后在该溶液中加入光敏剂染料N,N,N-三(7-(2-苯并噻唑基)-9,9-二乙基-2-芴基)胺(在美国专利No.6,300,502(Kannan等人)实例20中描述了该物质及其合成)的THF浓缩液、SR1012的THF浓缩液以及CGI 7460的浓缩液的等分试样,以获得按固体总重量计,光敏剂染料占0.5%、SR1012占1.0%和CGI 7460占0.5%的涂层溶液。用1微米注射器过滤器过滤该涂层溶液,然后旋转喷涂到硅晶片上。将晶片在60℃的鼓风烘箱下放置18小时,得到基本上无溶剂(在下文中用“干燥”表示)的带涂层的硅晶片,涂层厚度约20微米。用异丙醇冲洗晶片的未涂覆面,然后将晶片安装在多孔碳真空吸头上,吸头安装在三点水平基座上,晶片的未涂覆面与吸头接触。通过调节设置在基座周边附近三个调节点的相应水平螺丝调平水平基座。
使用二极管泵浦钛:兰宝石激光器(Spectra-Physics(MountainView,CA)),在波长为800nm、标称脉冲宽度为80fs、脉冲重复率为80MHz、平均功率为约1W的操作条件下,用以下方式进行干燥涂层的双光子聚合。将带涂层的晶片放置在计算机控制的三轴工作台(可得自Aerotech,Inc.(Pittsburgh,PA))上。激光束被中性密度滤光器减弱,然后使用电流扫描仪将光束聚焦在干燥涂层中,该扫描仪带有控制x、y和z轴的望远镜(可得自Nutfield Technology,Inc.(Windham,NH))以及浸在可直接应用于干燥涂层表面的A型浸渍油(可得自Cargille Laboratories(Cedar Grove,NJ))中的望远镜物镜镜头(具有0.9的数值孔径)。用波长已校准的光电二极管(可得自OphirOptronics,Ltd.(Wilmington,MA))在物镜镜头的输出处测量平均功率,并确定平均过滤为约8mW。
上述双光子光聚反应系统首先与共焦界面定位系统(如下所述) 一起使用以调平晶片表面(根据下述工序),以使得曝光系统的焦平面在约500纳米以内。因此可相对于硅晶片表面与可聚合涂层之间的界面,保持激光束的聚焦,以使得阵列中微透镜与硅晶片表面之间基本上未留下未聚合的涂层。晶片的片厚度为600nm,填充系数为71.1%,平均表面粗糙度为38nm,垂度为8.3μm。
共焦界面定位器系统是带有分束器(型号10RQ00UB.2,可得自Newport Irvine,CA)、反射镜、10微米小孔、安装在用型号为PS 300的直流电源(可得自Stanford Research Systems Inc.(Sunnyvale,CA))供电的型号为PR1405.C6的壳体中(可得自Products for Research,Inc.(Danvers,MA))的光电倍增管(型号为R3898,可得自Hamamatsu Corp.(Bridgewater,NJ))、放大器(型号为3312,可得自Pomona Electronics(Everett,WA))和万用表(Fluke,型号为73III)的光学系统。激活双光子光聚反应系统,并且激光信号在用于制造微透镜阵列之前将被用于检测硅晶片的表面与可聚合涂层之间的界面,如下文所述。
移动三点水平基座,以使得聚焦激光束指向预期微透镜阵列区域之外的点。从硅晶片表面(即涂层界面)反射回来的聚焦激光信号向回穿过电流扫描仪,然后经过分束器,被反射镜反射,穿过带通滤波器。带通滤波器只能通过聚焦激光信号,其它波长的光(例如从晶片涂层中光敏剂发出的荧光)不能通过带通滤波器。穿过10微米小孔后,聚焦激光信号被光电倍增管放大,并用万用表监控光电倍增管发出的电信号的电位(电压)。最大强度的反射激光信号为聚焦激光信号,万用表上显示为最大电压。低于最大电压的电压表示激光的焦点未被硅晶片反射,即,激光的焦点基本上高于或基本上低于涂层与晶片之间的界面。
用这种方式检测晶片的表面之后,将支撑晶片的水平基座移到激光下,然后调整三点水平基座,直至聚焦激光束再次从晶片表面在预期微透镜阵列区域之外的点反射,这时用万用表测得的电压为最大电 压。重复该过程两次,以在曝光系统光学平面的500纳米以内形成硅晶片表面。
然后基本上用上述系统和工序进行干燥涂层的双光子聚合反应,以便生成1760×2490的非球面、径向对称的微透镜的六方填充阵列,微透镜尺寸约为4.4cm×4.4cm。
然后将1760×2490的微镜片六方填充阵列用作复制阵列工艺中的靠模。使用双组分硅氧烷树脂(RTV 615A/B,可得自GE Silicones(Waterford,NY))来制作微透镜阵列的硅氧烷模具。将十重量份的RTV615A与一重量份的RTV 615B搅拌约30分钟。将高度为约14.3毫米(0.56英寸)、直径大于微透镜阵列最大尺寸的铝环放置在阵列上方。然后将硅氧烷混合物倒在阵列上方,在铝环限定的范围内。随后将所得样品放置在真空室中,在约4000Pa(30mmHg)的压力下保持约一小时。之后将样品放置在鼓风烘箱中,在55℃的温度下保持约100分钟。然后将样品冷却至室温,将所得的固化硅氧烷树脂从微透镜阵列母模上移除,得到微透镜阵列的硅氧烷模具。
用0.2微米的针头式过滤器过滤IRGACURE 819(0.3g)和SARTOMER 238(5.0g)的混合物,然后与加热至约55℃并保持约30分钟的PHOTOMER 6210(15.0g)混合。将所得的固化混合物倒入硅氧烷模具中,然后将填充的硅氧烷模具放入真空室,在约4000Pa(30mmHg)的压力下保持大约45分钟。随后将填充的硅氧烷模具放置在玻璃板上,以使得固化混合物与玻璃板接触。在硅氧烷模具顶部用手轻压片刻。在型号为DRS-120的紫外线处理器(可得自FusionUV Systems Inc.(Gaithersburg,MD))中用H型灯泡照射固化混合物。填充的硅氧烷模具通过处理器(硅氧烷模具面向紫外线灯泡)七次,处理器(皮带)的速度为每分钟4.5m(15英尺)。然后将填充的硅氧烷模具冷却至室温,然后从所得的硬化混合物上移除硅氧烷模具,得到微透镜阵列的复制品。
实例2
使用共焦定位器系统制备倾斜的微透镜阵列
基本上采用实例1中所述方法,清洗和准备圆形硅晶片(直径10.2cm(4英寸),可得自Wafer World,Inc.(West Palm Beach,Florida))
将数均分子量为约120,000的聚(甲基丙烯酸甲酯)、SR9008和SR368以30∶35∶35的重量比混合,形成单体混合物,然后将该单体混合物溶解于足量的1,2-二氯乙烷中,得到重量百分比为54%的单体混合物溶液。然后在该溶液中加入足量的光敏剂若丹明B六氟锑酸盐的TNF浓缩液和SR1012的TNF浓缩液的等分试样,以便获得按固体总重量计,若丹明B六氟锑酸盐占0.5%,SR1012占1.0%的涂层溶液。使用1微米针头式过滤器过滤该涂层溶液,然后旋转涂布到硅晶片上。将晶片在60℃的鼓风烘箱下放置18小时,得到基本上无溶剂(在下文中简称“干燥”)的带涂层的硅晶片,涂层厚度为约300微米。双光子光聚反应系统与实例1所描述的基本相同,不同的是使用了数值孔径为1.4的显微镜物镜镜头,然后用实例1中所述的共焦界面定位器系统制备倾斜透镜(片厚度为560nm)的3×3阵列。
实例3
使用共焦定位器系统制备六方填充微透镜阵列
基本上用实例2所述方法制备带涂层硅晶片,不同的是单体混合物的涂层厚度为约10微米。然后使用实例2所述的双光子光聚反应系统与实例1所述的共焦界面定位器系统共同制备2273×3214非球面、径向对称的微透镜阵列,微透镜尺寸为约5.0cm×5.0cm(填充系数为91.8%,垂度为8.3微米)。
已描述了本发明的多个实施例。
这些及其它实施例均在以下权利要求书的范围内。
Claims (13)
1.一种用于检测多光子可固化光活性组合物与基底之间界面的方法,包括:
提供基底,所述基底上具有包含多光子可固化光活性组合物的层;
用光束照射所述层的至少一个区域,其中所述光束固化或引发固化所述多光子可固化光活性组合物;以及
处理所述基底反射的所述光束的一部分,以获得每个区域的所述层与所述基底之间界面的位置信号。
2.根据权利要求1所述的方法,其中所述光束在所述处理步骤之后固化所述区域的所述多光子可固化活性组合物,以形成固化物。
3.根据权利要求1所述的方法,其中所述位置信号是从光学装置中获取的,所述光学装置包括共焦界面定位器系统、干涉测量检测器和分光检测器焦点测量系统中的至少一个。
4.根据权利要求2所述的方法,还包括从所述基底上移除已固化材料的至少一部分。
5.一种用于加工多光子可固化光活性组合物的方法,包括:
提供基底,所述基底上具有包含多光子可固化光活性组合物的层;
通过第一光学系统将光束照射在所述层的至少第一区域上;
在每个第一区域从所述基底反射所述光束的一部分,以提供反射光束;
在第二光学系统中处理每个第一区域的所述反射光束,所述第二光学系统包括光学检测器,其中所述光学检测器的输出包括每个第一区域的所述基底与所述层之间界面的位置信号;
响应所述位置信号调整所述第一光学系统;以及
通过所述第一光学系统用所述光束固化固化区域内的所述组合 物。
6.根据权利要求5所述的方法,其中所述固化区域不同于所述第一区域。
7.根据权利要求5所述的方法,其中所述信号在所述光束照射所述固化区域时连续施加在所述第一光学系统上。
8.一种用于加工多光子可固化光活性组合物的方法,包括
在基底上提供层,其中所述层包含多光子可固化光活性组合物,并且其中所述基底在可调平台上;
通过第一光学系统将光束照射到至少第一区域中的所述层上;
在第二光学系统中处理所述光束,所述第二光学系统包括光学检测器,其中所述光学检测器的输出包括每个区域的所述基底与所述层之间界面的位置信号;
响应所述信号调整所述第一光学系统和所述平台中的至少一个;
用所述光束固化固化区域内的所述组合物。
9.根据权利要求8所述的方法,其中所述调整步骤包括更改所述平台相对于所述第一光学系统的高度。
10.一种用于通过使用界面的位置信号调整光学系统或平台的方法,所述方法包括:
接收基底与层的至少一个区域之间界面的位置信号,所述层包含多光子可固化光活性组合物,其中所述位置信号是由通过第一光学系统将聚焦光束照射在所述层上,并在光学检测器中处理所述基底上反射的所述聚焦光束的一部分而生成的;
基于在每个区域生成的所述位置信号生成复合位置信号;以及
基于所述复合位置信号调整所述第一光学系统和支撑所述基底的平台中的至少一个。
11.根据权利要求10所述的方法,其中所述复合位置信号是基于至少三个位置信号的。
12.一种用于固化包含多光子可固化光活性组合物的层的区域的装置,其中所述装置包括第一光学系统和第二光学系统,所述第一光学系统将聚焦激光束的第一部分导入所述层,所述第二光学系统处理所述光束被基底反射的第二部分以生成所述基底与所述层之间界面的输出信号。
13.根据权利要求12所述的装置,其中所述第二光学系统包括光学检测器,所述光学检测器选自共焦界面定位器系统、干涉测量检测器和分光检测器焦点测量系统中的一种。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US75252905P | 2005-12-21 | 2005-12-21 | |
| US60/752,529 | 2005-12-21 | ||
| PCT/US2006/048498 WO2007073482A2 (en) | 2005-12-21 | 2006-12-20 | Method and apparatus for processing multiphoton curable photoreactive compositions |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101341578A CN101341578A (zh) | 2009-01-07 |
| CN101341578B true CN101341578B (zh) | 2010-12-08 |
Family
ID=38189117
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2006800482953A Expired - Fee Related CN101341578B (zh) | 2005-12-21 | 2006-12-20 | 加工多光子可固化光活性组合物的方法及装置 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US7893410B2 (zh) |
| JP (1) | JP4880701B2 (zh) |
| CN (1) | CN101341578B (zh) |
| DE (1) | DE112006003494T5 (zh) |
| WO (1) | WO2007073482A2 (zh) |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7583444B1 (en) | 2005-12-21 | 2009-09-01 | 3M Innovative Properties Company | Process for making microlens arrays and masterforms |
| EP2018263B1 (en) | 2006-05-18 | 2017-03-01 | 3M Innovative Properties Company | Process for making light guides with extraction structures |
| JP5951928B2 (ja) * | 2007-09-06 | 2016-07-13 | スリーエム イノベイティブ プロパティズ カンパニー | 光出力の領域制御を提供する光抽出構造体を有する光ガイド |
| CN101795838B (zh) | 2007-09-06 | 2014-02-12 | 3M创新有限公司 | 形成模具的方法以及使用所述模具形成制品的方法 |
| CN101795961B (zh) * | 2007-09-06 | 2013-05-01 | 3M创新有限公司 | 用于制备微结构化制品的工具 |
| WO2009048808A1 (en) * | 2007-10-11 | 2009-04-16 | 3M Innovative Properties Company | Chromatic confocal sensor |
| JP5524856B2 (ja) | 2007-12-12 | 2014-06-18 | スリーエム イノベイティブ プロパティズ カンパニー | エッジ明瞭性が向上した構造の製造方法 |
| US8605256B2 (en) * | 2008-02-26 | 2013-12-10 | 3M Innovative Properties Company | Multi-photon exposure system |
| GB2489722B (en) | 2011-04-06 | 2017-01-18 | Precitec Optronik Gmbh | Apparatus and method for determining a depth of a region having a high aspect ratio that protrudes into a surface of a semiconductor wafer |
| DE102011051146B3 (de) | 2011-06-17 | 2012-10-04 | Precitec Optronik Gmbh | Prüfverfahren zum Prüfen einer Verbindungsschicht zwischen waferförmigen Proben |
| CN104303108A (zh) * | 2012-02-28 | 2015-01-21 | 3M创新有限公司 | 利用负对比组合物的多光子固化方法 |
| US9040921B2 (en) * | 2012-07-28 | 2015-05-26 | Harvard Apparatus Regenerative Technology, Inc. | Analytical methods |
| DE102012111008B4 (de) | 2012-11-15 | 2014-05-22 | Precitec Optronik Gmbh | Optisches Messverfahren und optische Messvorrichtung zum Erfassen einer Oberflächentopographie |
| US9772552B2 (en) * | 2013-03-19 | 2017-09-26 | Eastman Kodak Company | Thiosulfate polymer compositions and articles |
| US9005878B2 (en) | 2013-03-19 | 2015-04-14 | Eastman Kodak Company | Forming patterns using thiosulfate polymer compositions |
| US9499650B2 (en) | 2013-03-19 | 2016-11-22 | Eastman Kodak Company | Thiosulfate polymers |
| US8986924B2 (en) | 2013-03-19 | 2015-03-24 | Eastman Kodak Company | Method of sequestering metals using thiosulfate polymers |
| US8916336B2 (en) | 2013-03-19 | 2014-12-23 | Eastman Kodak Company | Patterning method using thiosulfate polymer and metal nanoparticles |
| DE102014008584B4 (de) | 2013-06-17 | 2021-05-27 | Precitec Optronik Gmbh | Optische Messvorrichtung zum Erfassen von Abstandsdifferenzen und optisches Messverfahren |
| EP3077421B1 (en) | 2013-12-06 | 2018-01-31 | 3M Innovative Properties Company | Liquid photoreactive composition and method of fabricating structures |
| TWI583916B (zh) * | 2015-10-08 | 2017-05-21 | 財團法人工業技術研究院 | 雷射共焦量測方法及應用此方法之雷射共焦量測裝置 |
| US10234265B2 (en) | 2016-12-12 | 2019-03-19 | Precitec Optronik Gmbh | Distance measuring device and method for measuring distances |
| DE102017126310A1 (de) | 2017-11-09 | 2019-05-09 | Precitec Optronik Gmbh | Abstandsmessvorrichtung |
| US11150484B2 (en) * | 2018-05-18 | 2021-10-19 | Lawrence Livermore National Security, Llc | System and method for curved light sheet projection during two-photon polymerization |
| DE102018130901A1 (de) | 2018-12-04 | 2020-06-04 | Precitec Optronik Gmbh | Optische Messeinrichtung |
| DE102021113189A1 (de) | 2021-05-20 | 2022-11-24 | Nanoscribe Holding Gmbh | Verfahren zum Erzeugen einer dreidimensionalen Zielstruktur in einem Lithographiematerial mittels einer Laserlithographie-Vorrichtung |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1692313A (zh) * | 2002-10-02 | 2005-11-02 | 3M创新有限公司 | 多光子光敏化方法 |
Family Cites Families (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3018262A (en) | 1957-05-01 | 1962-01-23 | Shell Oil Co | Curing polyepoxides with certain metal salts of inorganic acids |
| US3729313A (en) | 1971-12-06 | 1973-04-24 | Minnesota Mining & Mfg | Novel photosensitive systems comprising diaryliodonium compounds and their use |
| US3808006A (en) | 1971-12-06 | 1974-04-30 | Minnesota Mining & Mfg | Photosensitive material containing a diaryliodium compound, a sensitizer and a color former |
| US3779778A (en) | 1972-02-09 | 1973-12-18 | Minnesota Mining & Mfg | Photosolubilizable compositions and elements |
| US3741769A (en) | 1972-10-24 | 1973-06-26 | Minnesota Mining & Mfg | Novel photosensitive polymerizable systems and their use |
| AU497960B2 (en) | 1974-04-11 | 1979-01-25 | Minnesota Mining And Manufacturing Company | Photopolymerizable compositions |
| US4250053A (en) | 1979-05-21 | 1981-02-10 | Minnesota Mining And Manufacturing Company | Sensitized aromatic iodonium or aromatic sulfonium salt photoinitiator systems |
| US4279717A (en) | 1979-08-03 | 1981-07-21 | General Electric Company | Ultraviolet curable epoxy silicone coating compositions |
| US4491628A (en) | 1982-08-23 | 1985-01-01 | International Business Machines Corporation | Positive- and negative-working resist compositions with acid generating photoinitiator and polymer with acid labile groups pendant from polymer backbone |
| US4642126A (en) | 1985-02-11 | 1987-02-10 | Norton Company | Coated abrasives with rapidly curable adhesives and controllable curvature |
| US4652274A (en) | 1985-08-07 | 1987-03-24 | Minnesota Mining And Manufacturing Company | Coated abrasive product having radiation curable binder |
| CA1323949C (en) | 1987-04-02 | 1993-11-02 | Michael C. Palazzotto | Ternary photoinitiator system for addition polymerization |
| US4859572A (en) | 1988-05-02 | 1989-08-22 | Eastman Kodak Company | Dye sensitized photographic imaging system |
| JP2912721B2 (ja) * | 1991-02-19 | 1999-06-28 | 日本電信電話株式会社 | 三次元物体の形成方法 |
| US5235015A (en) | 1991-02-21 | 1993-08-10 | Minnesota Mining And Manufacturing Company | High speed aqueous solvent developable photopolymer compositions |
| TW268969B (zh) | 1992-10-02 | 1996-01-21 | Minnesota Mining & Mfg | |
| US5856373A (en) | 1994-10-31 | 1999-01-05 | Minnesota Mining And Manufacturing Company | Dental visible light curable epoxy system with enhanced depth of cure |
| US6608228B1 (en) | 1997-11-07 | 2003-08-19 | California Institute Of Technology | Two-photon or higher-order absorbing optical materials for generation of reactive species |
| WO1998021521A1 (en) | 1996-11-12 | 1998-05-22 | California Institute Of Technology | Two-photon or higher-order absorbing optical materials and methods of use |
| US6025406A (en) | 1997-04-11 | 2000-02-15 | 3M Innovative Properties Company | Ternary photoinitiator system for curing of epoxy resins |
| US5998495A (en) | 1997-04-11 | 1999-12-07 | 3M Innovative Properties Company | Ternary photoinitiator system for curing of epoxy/polyol resin compositions |
| US5770737A (en) | 1997-09-18 | 1998-06-23 | The United States Of America As Represented By The Secretary Of The Air Force | Asymmetrical dyes with large two-photon absorption cross-sections |
| US5859251A (en) | 1997-09-18 | 1999-01-12 | The United States Of America As Represented By The Secretary Of The Air Force | Symmetrical dyes with large two-photon absorption cross-sections |
| WO1999054784A1 (en) * | 1998-04-21 | 1999-10-28 | University Of Connecticut | Free-form nanofabrication using multi-photon excitation |
| US6100405A (en) | 1999-06-15 | 2000-08-08 | The United States Of America As Represented By The Secretary Of The Air Force | Benzothiazole-containing two-photon chromophores exhibiting strong frequency upconversion |
| KR100810546B1 (ko) | 2000-06-15 | 2008-03-18 | 쓰리엠 이노베이티브 프로퍼티즈 캄파니 | 삼차원 광학 소자의 가공 방법 |
| KR100754813B1 (ko) | 2000-06-15 | 2007-09-04 | 쓰리엠 이노베이티브 프로퍼티즈 캄파니 | 다중통과 다광자 흡수 방법 및 장치 |
| JP4689936B2 (ja) * | 2000-06-15 | 2011-06-01 | スリーエム イノベイティブ プロパティズ カンパニー | 構造を製作するか又は物品に構造を付加するための方法 |
| US6300502B1 (en) | 2000-12-08 | 2001-10-09 | The United States Of America As Represented By The Secretary Of The Air Force | Multi-armed chromophores with very large two-photon absorption cross-sections |
| JP2001287273A (ja) * | 2001-03-12 | 2001-10-16 | Three D Syst Inc | 立体造形方法及び装置 |
| US20050254035A1 (en) * | 2004-05-11 | 2005-11-17 | Chromaplex, Inc. | Multi-photon lithography |
| US8304313B2 (en) * | 2004-08-23 | 2012-11-06 | Semiconductor Energy Laboratory Co., Ltd. | Semiconductor device and its manufacturing method |
| DE102005009188A1 (de) * | 2005-03-01 | 2006-09-07 | Carl Zeiss Jena Gmbh | Punktscannendes Laser-Scanning-Mikroskop sowie Verfahren zur Einstellung eines Mikroskopes |
| US20090245066A1 (en) * | 2005-12-12 | 2009-10-01 | Mempile Inc. | Optical data carrier, and method for reading/recording data therein |
| US7583444B1 (en) | 2005-12-21 | 2009-09-01 | 3M Innovative Properties Company | Process for making microlens arrays and masterforms |
-
2006
- 2006-12-20 DE DE112006003494T patent/DE112006003494T5/de not_active Withdrawn
- 2006-12-20 WO PCT/US2006/048498 patent/WO2007073482A2/en active Application Filing
- 2006-12-20 JP JP2008547458A patent/JP4880701B2/ja not_active Expired - Fee Related
- 2006-12-20 US US12/158,143 patent/US7893410B2/en not_active Expired - Fee Related
- 2006-12-20 CN CN2006800482953A patent/CN101341578B/zh not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1692313A (zh) * | 2002-10-02 | 2005-11-02 | 3M创新有限公司 | 多光子光敏化方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009521315A (ja) | 2009-06-04 |
| WO2007073482A2 (en) | 2007-06-28 |
| US20090250635A1 (en) | 2009-10-08 |
| US7893410B2 (en) | 2011-02-22 |
| CN101341578A (zh) | 2009-01-07 |
| JP4880701B2 (ja) | 2012-02-22 |
| WO2007073482A3 (en) | 2007-09-27 |
| DE112006003494T5 (de) | 2008-10-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101341578B (zh) | 加工多光子可固化光活性组合物的方法及装置 | |
| CN101448632B (zh) | 用于制备具有提取结构的光导的方法以及由此方法生产的光导 | |
| US10400057B2 (en) | Liquid, hybrid UV/vis radiation curable resin compositions for additive fabrication | |
| CN101821302A (zh) | 高功能性多光子可固化反应性物质 | |
| AU751062B2 (en) | Viscosity stabilization of radiation-curable filled compositions | |
| JP2022070865A (ja) | 付加造形用液状ハイブリッドUV/vis線硬化性樹脂組成物 | |
| CN101346644B (zh) | 用于制造微透镜阵列和仿型模的方法 | |
| US7592376B2 (en) | Photopolymerizable epoxide and oxetane compositions | |
| US6177232B1 (en) | Sedimentation stabilized radiation-curable filled compositions | |
| KR100808954B1 (ko) | 특히 입체리소그래피에 적합한 액체 방사선―경화성 조성물 | |
| AU704046B2 (en) | Stabilization of liquid radiation-curable compositions against undesired premature polymerization | |
| US20170355857A1 (en) | Liquid, hybrid uv/vis radiation curable resin compositions for additive fabrication | |
| JP4689936B2 (ja) | 構造を製作するか又は物品に構造を付加するための方法 | |
| ES2564145T3 (es) | Composición de resina sensible a rayos de energía activa, película de resina sensible a rayos de energía activa y método para formar un motivo usando dicha película | |
| US20220282003A1 (en) | Liquid, hybrid uv/vis radiation curable resin compositions for additive fabrication | |
| GB2099825A (en) | Photopolymerisable mixtures, and processes for the photopolymerisation of cationically polymerisable compounds | |
| KR20220069877A (ko) | 전자 디바이스용 광 경화성 수지 조성물 | |
| KR100776363B1 (ko) | 광 도파로 형성용 광경화성 수지 조성물, 광 도파로 형성용 광경화성 드라이 필름 및 광 도파로 | |
| US20240092946A1 (en) | Liquid, hybrid uv/vis radiation curable resin compositions for additive fabrication | |
| MXPA00000422A (en) | Sedimentation stabilized radiation-curable filled compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20101208 Termination date: 20161220 |
|
| CF01 | Termination of patent right due to non-payment of annual fee |