CN101106977A - 直接压片的配方及方法 - Google Patents
直接压片的配方及方法 Download PDFInfo
- Publication number
- CN101106977A CN101106977A CNA2006800025892A CN200680002589A CN101106977A CN 101106977 A CN101106977 A CN 101106977A CN A2006800025892 A CNA2006800025892 A CN A2006800025892A CN 200680002589 A CN200680002589 A CN 200680002589A CN 101106977 A CN101106977 A CN 101106977A
- Authority
- CN
- China
- Prior art keywords
- vildagliptin
- tablet
- weight
- compacting
- dpp
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images




Abstract
本发明涉及片剂,特别是通过直接压制二肽基肽酶IV(DPP-IV)抑制剂化合物而形成的片剂、其制备方法、新药物配方以及能够直接压制成片剂的包含DPP-IV抑制剂的新制片粉末。本发明进一步涉及通过将活性成分与特定赋形剂混合于新配方中然后直接将所述配方压制成为直接压制的片剂的片剂制备法。本发明还涉及维格列汀粒度分布和一种维格列汀的新晶型,其特别适用于制备改良的片剂和其它药物组合物。
Description
本发明涉及片剂,特别是通过直接压制二肽基肽酶IV(DPP-IV)抑制剂化合物而形成的片剂、其制备方法、新药物配方以及能够直接压制成片剂的包含DPP-IV抑制剂的新制片粉末。本发明进一步涉及通过将活性成分与特定赋形剂混合于新配方中然后直接将所述配方压制成为直接压制的片剂的片剂制备法。本发明还涉及维格列汀粒度分布和一种维格列汀的新晶型,其特别适用于制备改良的片剂和其它药物组合物。
以下描述本发明主要涉及的优选DPP-IV抑制剂化合物:
在本文中,“DPP-IV抑制剂”还包括它的活性代谢物和前药,如DPP-IV抑制剂的活性代谢物和前药。“代谢物”是指当DPP-IV抑制剂被代谢时生成的DPP-IV抑制剂的活性衍生物。“前药”是指被代谢为DPP-IV抑制剂或者被代谢为与DPP-IV抑制剂的代谢物相同的代谢物的化合物。
DPP-IV抑制剂是本领域已知的。例如,在WO 98/19998、DE 19616486 A1、WO 00/34241、WO 95/15309、WO 01/72290、WO 01/52825、WO 9310127、WO 9925719、WO 9938501、WO 9946272、WO 9967278和WO 9967279中一般性或具体性地公开了DPP-IV抑制剂。
优选的DPP-IV抑制剂描述于以下专利申请中:WO 02053548,特别是化合物1001至1293和实施例1至24,WO 02067918,特别是化合物1000至1278和2001至2159,WO 02066627,特别是所述的实施例,WO02/068420,特别是实施例I至LXIII中具体列出的全部化合物以及所述的相应类似物,更优选的化合物是报告IC50的表中的2(28)、2(88)、2(119)、2(136),WO 02083128,特别是实施例1至13,US 2003096846,特别是具体描述的化合物,WO 2004/037181,特别是实施例1至33和权利要求3至5的化合物,WO 0168603,特别是实施例1至109的化合物,EP 1258480,特别是实施例1至60的化合物,WO 0181337,特别是实施例1至118的化合物,WO 02083109,特别是实施例1A至1D的化合物,WO 030003250,特别是实施例1至166、最优选1至8的化合物,WO 03035067,特别是实施例中所述的化合物,WO 03/035057,特别是实施例中所述的化合物,US2003216450,特别是实施例1至450,WO 99/46272,特别是权利要求12、14、15和17的化合物,WO 0197808,特别是权利要求2的化合物,WO 03002553,特别是实施例1至33的化合物,WO 01/34594,特别是实施例1至4所述的化合物,WO 02051836,特别是实施例1至712,EP1245568,特别是实施例1至7,EP1258476,特别是实施例1至32,US 2003087950,特别是所述的实施例,WO 02/076450,特别是实施例1至128,WO 03000180,特别是实施例1至162,WO 03000181,特别是实施例1至66,WO 03004498,特别是实施例1至33,WO 0302942,特别是实施例1至68,US 6482844,特别是所述的实施例,WO 0155105,特别是实施例1和2中列出的化合物,WO 0202560,特别是实施例1至166,WO 03004496,特别是实施例1至103,WO 03/024965,特别是实施例1至54,WO 0303727,特别是实施例1至209,WO 0368757,特别是实施例1至88,WO 03074500,特别是实施例1至72,实施例4.1至4.23,实施例5.1至5.10,实施例6.1至6.30,实施例7.1至7.23,实施例8.1至8.10,实施例9.1至9.30,WO 02038541,特别是实施例1至53,WO 02062764,特别是实施例1至293,优选实施例95的化合物(2-{{3-(氨基甲基)-4-丁氧基-2-新戊基-1-氧代-1,2-二氢-6-异喹啉基}氧基}乙酰胺盐酸盐),WO02308090,特别是实施例1-1至1-109,实施例2-1至2-9,实施例3,实施例4-1至4-19,实施例5-1至5-39,实施例6-1至6-4,实施例7-1至7-10,实施例8-1至8-8,第90页的实施例7-1至7-7,第91至95页的实施例8-1至8-59,实施例9-1至9-33,实施例10-1至10-20,US 2003225102,特别是化合物1至115,实施例1至121的化合物,优选化合物a)至z)、aa)至az)、ba)至bz)、ca)至cz)和da)至dk),WO 0214271,特别是实施例1至320和US 2003096857,WO 2004/052850,特别是具体描述的化合物,如实施例1至42和权利要求1的化合物,DE 10256264 A1,特别是所述的化合物如实施例1至181以及权利要求5的化合物,WO 04/076433,特别是具体描述的化合物,如表A中列出的化合物,优选表B中列出的化合物,优选化合物I至XXXXVII或权利要求6至49的化合物,WO 04/071454,特别是具体描述的化合物如化合物1至53或表Ia至If的化合物,或权利要求2至55的化合物,WO 02/068420,特别是具体描述的化合物如化合物I至LXIII或实施例I和类似物1至140或实施例2和类似物1至174或实施例3和类似物1或实施例4至5,或实施例6和类似物1至5,或实施例7和类似物1-3,或实施例8和类似物1,或实施例9,或实施例10和类似物1至531,更优选的是权利要求13的化合物,WO 03/000250,特别是具体描述的化合物如化合物1至166,优选实施例1至9的化合物,WO 03/024942,特别是具体描述的化合物如化合物1至59,表1的化合物(1至68),权利要求6、7、8、9的化合物,WO 03024965,特别是具体描述的化合物如化合物1至54,WO 03002593,特别是具体描述的化合物如表1或权利要求2至15的化合物,WO 03037327,特别是具体描述的化合物如实施例1至209的化合物,WO 03/000250,特别是具体描述的化合物如化合物1至166,优选实施例1至9的化合物,WO 03/024942,特别是具体描述的化合物如化合物1至59,表1的化合物(1至68),权利要求6、7、8、9的化合物,WO 03024965,特别是具体描述的化合物如化合物1至54,WO 03002593特别是具体描述的化合物如表1或权利要求2至15的化合物,WO03037327,特别是具体描述的化合物如实施例1至209的化合物,WO 0238541,WO 0230890,2001年2月16日递交的美国申请09/788,173(律师档案LA50)特别是所述的实施例,WO99/38501,特别是所述的实施例,W099/46272,特别是所述的实施例和DE19616 486 A1,特别是val-pyr、val-噻唑烷、异亮氨酰基噻唑烷、异亮氨酰基吡咯烷、和异亮氨酰基噻唑烷和异亮氨酰基吡咯烷的反式异构盐,WO 0238541,特别是具体描述的化合物如化合物实施例1至53,WO 03/002531,特别是具体描述的化合物优选第9至13页列出的化合物,最优选实施例1至46的化合物,更优选实施例9的化合物,美国专利6,395,767,优选实施例1至109的化合物,最优选实施例60的化合物。
进一步优选的DPP-IV抑制剂包括美国专利6124305和6107317、国际专利申请公开号为WO 9819998、WO 95153 09和WO 9818763的申请中公开的具体实施例,例如1-[2-[(5-氰基吡啶-2-基)氨基乙基氨基]乙酰基-2-氰基-(S)-吡咯烷和(2S)-1-[(2S)-2-氨基-3,3-二甲基丁酰基]-2-吡咯烷甲腈。
WO 9819998公开了N-(N′-取代的甘氨酰)-2-氰基吡咯烷,特别是1-[2-[5-氰基吡啶-2-基]氨基]-乙基氨基]乙酰基-2-氰基-(S)-吡咯烷。WO03/002553中的优选化合物列在第9至11页上,本申请引入这些内容以供参考。公开的专利申请WO 0034241和公开的美国专利6110949中分别公开了N-取代的金刚烷基-氨基-乙酰基-2-氰基吡咯烷和N-(取代的甘氨酰)-4-氰基吡咯烷。令人感兴趣的DPP-IV抑制剂特别是权利要求1至4中引用的化合物。具体而言,这些申请描述了化合物1-[[(3-羟基-1-金刚烷基)氨基]乙酰基]-2-氰基-(S)-吡咯烷(也称为LAF237或维格列汀)。
WO 9515309公开了作为DPP-IV抑制剂的氨基酸2-氰基吡咯烷酰胺,WO 9529691公开了α-氨基烷基磷酸二酯的肽基衍生物,特别是带有脯氨酸或相关结构的衍生物。令人感兴趣的DPP-IV抑制剂特别是表1至8中引用的化合物。WO 01/72290中,令人感兴趣的DPP-IV抑制剂特别是实施例1和权利要求1、4和6中引用的化合物。WO 9310127公开了可用作DDP-IV抑制剂的脯氨酸硼酸酯。令人感兴趣的DPP-IV抑制剂特别是实施例1至19中引用的化合物。公开的专利申请WO 9925719公开了sulphostin,sulphostin是一种通过培养链霉菌属微生物制备的DPP-IV抑制剂。WO 9938501公开了N-取代的4-至8-元杂环。令人感兴趣的DPP-IV抑制剂特别是权利要求15至20中引用的化合物。
WO 9946272公开了作为DPP-IV抑制剂的含磷化合物。令人感兴趣的DPP-IV抑制剂特别是权利要求1至23中引用的化合物。
其它优选的DPP-IV抑制剂是专利申请WO 03/057200中第14至27页公开的式I、II或III的化合物。最优选的DPP-IV抑制剂是第28和29页上具体公开的化合物。
公开的专利申请WO 9967278和WO 9967279公开了A-B-C形式的DPP-IV前药和抑制剂,其中C是稳定或不稳定的DPP-IV抑制剂。
优选N-肽基-O-芳酰基羟基胺是式VII的化合物或其可药用盐

其中
j是0、1或2;
Rε1是天然氨基酸的侧链;且
Rε2是低级烷氧基、低级烷基、卤素或硝基。
在本发明非常优选的实施方案中,N-肽基-O-芳酰基羟基胺是式VIIa的化合物或其可药用盐

H.U.Demuth等在J.Enzyme Inhibition 1988,Vol.2,第129-142页,特别是第130-132页上描述了N-肽基-O-芳酰基羟基胺例如式VII或VIIa的化合物以及它们的制备。
最优选的抑制剂是游离形式或酸加成盐形式的式(I)的N-(取代的甘氨酰)-2-氰基吡咯烷

其中
R是取代的金刚烷基;且
n是0至3。
术语“取代的金刚烷基”是指被一个或多个如两个选自于烷基、-OR1或-NR2R3的取代基取代的金刚烷基,即1-或2-金刚烷基,其中R1、R2和R3独立地是卤素、烷基、(C1-C8烷酰基)、氨基甲酰基或-CO-NR4R5,其中R4和R5独立地是烷基、未取代的或取代的芳基且其中R4和R5之一还是卤素或R4和R5一起是C2-C7亚烷基。
术语“芳基”优选是苯基。取代的苯基优选是被一个或多个例如两个选自于例如烷基、烷氧基、卤素和三氟甲基的取代基取代的苯基。
术语“烷氧基”是指烷基-O-。
术语“卤素”或“卤代”是指氟、氯、溴和碘。
术语“亚烷基”是指2至7个碳原子、优选3至6个碳原子、最优选5个碳原子的直链桥。
优选的一组本发明的化合物是式(I)化合物,其中金刚烷基上的取代基在桥头或与桥头相邻的亚甲基上键合。其中甘氨酰-2-氰基吡咯烷部分与桥头键合的式(I)化合物中,金刚烷基上的R′取代基优选是3-羟基。其中甘氨酰-2-氰基吡咯烷基团在与桥头相邻的亚甲基上键合的式(I)化合物,金刚烷基上的R′取代基优选是5-羟基。
本发明特别涉及游离形式或可药用酸加成盐形式的式(IA)或(IB)的化合物
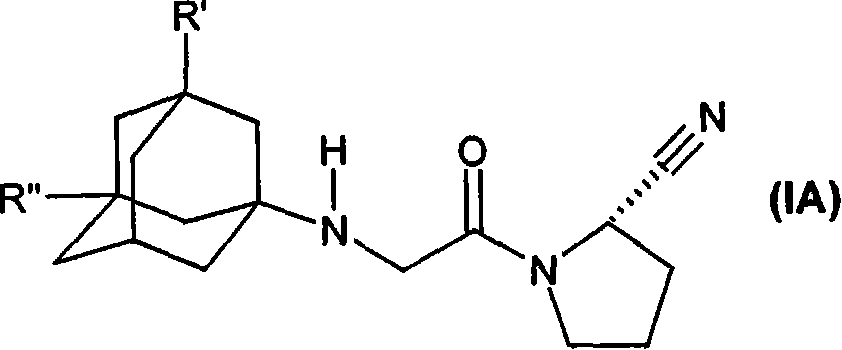

其中
R′是羟基、C1-C7烷氧基、C1-C8烷酰基氧基或R5R4N-CO-O-,其中R4和R5独立地是C1-C7烷基或苯基,其是未取代的或被选自于C1-C7烷基、C1-C7烷氧基、卤素和三氟甲基的取代基取代,其中R4还可以是氢;或
R4和R5一起是C3-C6亚烷基;且
R″是氢;或
R′和R″独立地是C1-C7烷基。
这些式(I)、(IA)或(IB)的DDP-IV抑制剂在2000年12月26日授权的美国专利6,166,063和WO 01/52825中有所描述。特别公开的是(S)-1-{2-[5-氰基吡啶-2-基)氨基]乙基-氨基乙酰基)-2-氰基-吡咯烷或(S)-1-[(3-羟基-1-金刚烷基)氨基]乙酰基-2-氰基-吡咯烷(LAF237或维格列汀)。它们可以以游离形式或酸加成盐形式存在。优选可药用盐,即无毒且在药理学上可以接受的盐,但是其它盐也可用于例如分离或纯化本发明的化合物。虽然优选的酸加成盐是盐酸盐,但也可以使用甲磺酸盐、硫酸盐、磷酸盐、柠檬酸盐、乳酸盐和乙酸盐。
优选的DDP-IV抑制剂是Mona Patel和col.(Expert Opinion InvestigDrugs.2003 Apr;12(4):623-33)在第5段所述的化合物,特别是P32/98、K-364、FE-999011、BDPX、NVP-DDP-728等,本文引入该公开物特别是所述的DDP-IV抑制剂以供参考。
专利申请WO 95/15309第14页描述了FE-999011,其为第18号化合物。
另一个优选的抑制剂是美国专利6,395,767公开的化合物BMS-477118(实施例60的化合物),也称为(1S,3S,5S)-2-[(2S)-2-氨基-2-(3-羟基三环[3.3.1.13,7]癸-1-基)-1-氧代乙基]-2-氮杂二环[3.1.0]己烷-3-腈,苯甲酸盐(1∶1),如专利申请WO 2004/052850第2页上的式M的化合物以及相应的游离碱,(1S,3S,5S)-2-[(2S)-2-氨基-2-(3-羟基-三环[3.3.1.13,7]癸-1-基)-1-氧代乙基]-2-氮杂二环-[3.1.0]己烷-3-腈(M′)以及专利申请WO 2004/052850第3页上描述的它的一水合物(M″)。
另一个优选的抑制剂是WO 03/002531中公开的化合物GSK23A(实施例9),也称为(2S,4S)-1-((2R)-2-氨基-3-[(4-甲氧基苄基)磺酰基]-3-甲基丁酰基)-4-氟代吡咯烷-2-甲腈盐酸盐。
其它非常优选的本发明的DDP-IV抑制剂描述于国际专利申请WO02/076450(特别是实施例1至128)和Wallace T.Ashton(Bioorganic&Medicinal Chemistry Letters 14(2004)859-863)中,特别是化合物1和表1和2中列出的化合物。优选的化合物是式21e的化合物(表1)。
P32/98或P3298(CAS号:251572-86-8)也称为3-[(2S,3S)-2-氨基-3-甲基-1-氧代戊基]噻唑烷,其可以3-[(2S,3S)-2-氨基-3-甲基-1-氧代戊基]噻唑烷和(2E)-2-丁烯二酸盐(2∶1)的混合物的形式使用,如下所示

其在WO 99/61431中以生物前药的名称描述,还有化合物P 93/01。
其它优选的DDP-IV抑制剂是专利申请WO 02/083128中公开的化合物,如权利要求1至5中描述的化合物。最优选的DDP-IV抑制剂是实施例1至13和权利要求6至10中特别描述的化合物。
其它优选的DDP-IV抑制剂描述于专利申请WO 2004/037169中,特别是实施例1至48中所述的化合物、描述于WO 02/062764中,特别是实施例1至293中所述的化合物,更优选的是第7页描述的化合物3-(氨基甲基)-2-异丁基-1-氧代-4-苯基-1,2-二氢-6-异喹啉甲酰胺和2-{[3-(氨基甲基)-2-异丁基-4-苯基-1-氧代-1,2-二氢-6-异喹啉基]氧基}乙酰胺,以及专利申请WO2004/024184中,特别是实施例1至4中所描述的化合物。
其它优选的DDP-IV抑制剂描述于专利申请WO 03/004498中,特别是实施例1至33,最优选的实施例7所述的下式化合物

也称为MK-0431。
优选的DDP-IV抑制剂也描述于专利申请WO 2004/037181,特别是实施例1至33中,最优选的化合物是权利要求3至5中所述的化合物。
优选的DDP-IV抑制剂是N-取代的金刚烷基-氨基-乙酰基-2-氰基吡咯烷、N(取代的甘氨酰)-4-氰基吡咯烷、N-(N’-取代的甘氨酰)-2-氰基吡咯烷、N-氨基酰基噻唑烷、N-氨基酰基吡咯烷、L-赤-异亮氨酰基噻唑烷、L-苏-异亮氨酰基吡咯烷和L-赤-异亮氨酰基吡咯烷、1-[2-[(5-氰基吡啶-2-基)氨基]乙基氨基]乙酰基-2-氰基-(S)-吡咯烷及其可药用盐。
特别优选的是下式的1-{2-[(5-氰基吡啶-2-基)氨基]乙基氨基}乙酰基-2(S)-氰基-吡咯烷二盐酸盐(DPP728)

特别是它的二盐酸盐,
和下式的(S)-1-[(3-羟基-1-金刚烷基)氨基]乙酰基-2-氰基-吡咯烷(LAF237或维格列汀(非专利名称-INN))
以及L-苏-异亮氨酰基噻唑烷(根据生物前药的化合物编码:如上所述的P32/98)、MK-0431、GSK23A、BMS-477118、3-(氨基甲基)-2-异丁基-1-氧代-4-苯基-1,2-二氢-6-异喹啉甲酰胺和2-{[3-(氨基甲基)-2-异丁基-4-苯基-1-氧代-1,2-二氢-6-异喹啉基]氧基}乙酰胺,在任何情况下其任选是其药学盐。
DPP728和LAF237是最优选的化合物,在WO 98/19998的实施例3和WO 00/34241的实施例1中分别具体公开了它们。DDP-IV抑制剂P32/98(见上)具体公开于Diabetes 1998,47,1253-1258中。DPP728和LAF237可以如WO 98/19998第20页或WO 00/34241中所述进行配制。优选的LAF237的施用配方描述于美国临时申请60/604274中。在本申请中,术语“维格列汀”是指维格列汀的任何形式,如无定形维格列汀、维格列汀的结晶形式、维格列汀的A晶型、维格列汀的部分结晶形式、维格列汀的多晶型形式、维格列汀的溶剂合物形式或维格列汀的水合物形式或它的任何盐。
特别优选的是口服活性的DDP-IV抑制剂。
在每种情况下,特别是化合物权利要求和工作实施例的终产物中,本申请中参考本文提及的出版物或专利申请引入终产物的主题物质、药物制剂和权利要求。
DPP-IV抑制剂化合物例如式(I)的化合物及其相应的可药用酸加成盐可以与一种或多种可药用载体和任选的一种或多种其它药学辅剂组合,可经肠如通过口服途径以片剂、胶囊剂、软胶囊剂等形式施用,或经胃肠外途径如静脉内以无菌注射溶液剂或混悬剂的形式施用。经肠组合物和胃肠外组合物可通过常规方法制备。
DPP-IV抑制剂化合物例如式(I)的化合物及其相应的可药用酸加成盐可以配制成为其中活性成分含量可有效地治疗由DPP-IV抑制剂介导的病症的经肠和胃肠外组合物、单元剂量形式的这种组合物和包含可药用载体的这种组合物。
DPP-IV抑制剂化合物例如式(I)的化合物,包括每个亚范围和每个实施例的化合物,可以以对映体纯如>98%、优选>99%的形式施用,或与R对映体一起施用如以外消旋形式施用。上述剂量范围基于式I化合物,不包括R对映体的量。
由于抑制DPP-IV的能力,DPP-IV抑制剂化合物例如式(I)的化合物及其相应的可药用酸加成盐可用于治疗由DPP-IV抑制介导的病症,基于以上所述和文献中的结果,预期本文公开的化合物可用于治疗诸如非胰岛素依赖性糖尿病、关节炎、肥胖症、同种异体移植和降钙素型骨质疏松。此外,基于胰高血糖素样肽如GLP-1和GLP-2的作用及其与DPP-IV的关系,预期本文公开的化合物可用于例如产生镇静作用或抗焦虑作用,或减少术后分解代谢变化和应激引起的激素反应,或降低心肌梗塞后的死亡率和发病率,或治疗与上述作用有关的可能由GLP-1和/或GLP-2水平介导的病症。
更具体而言,DPP-IV抑制剂化合物例如式(I)的化合物及其相应的可药用酸加成盐可改善对口服葡萄糖应激的早期胰岛素反应,因此可用于治疗非胰岛素依赖性糖尿病。
可用于本发明的DPP-IV抑制剂化合物、特别是式I、IA或IB的化合物具有吸湿性,存在稳定性的问题,本身不可压片,因此,需要提供一种自由流动的粘性组合物,其能够直接压制成为体外溶出特征可接受的坚实片剂。片剂可以定义为包含药物物质、含有或不含适当填充剂的固体药物剂型。它们通过压制或压实(compaction)包含活性成分或某些用于辅助加工并改进产品性质的赋形剂的配方来制备。片剂可以包衣或不包衣,并且从粉末状晶体物质制备。它们可能包含各种稀释剂、粘合剂、崩解剂、润滑剂、助流剂,很多情况下还包含着色剂。使用的赋形剂根据其功能分类,例如助流剂可用于改进进料斗中粉末混合物的流动和向压片冲模中的流动。
片剂应用广泛,大部分药物剂型都以片剂销售。片剂受欢迎的主要原因是剂型简单、生产成本低、速度快。其它原因包括药物产品的稳定性、包装、运输和分配方便。对于患者或使用者而言,片剂施用方便、容易控制准确剂量、轻便、便于携带、味道平衡、易于施用、外观优雅、易于识别。
片剂可以是普通片剂、包膜或糖衣对切、印字、分层或延时释放片剂。它们可以制备为不同大小、形状和颜色。片剂可以吞服、咀嚼或溶解于口腔中或舌下。它们可溶于水中用于局部施用。无菌片剂通常用于胃肠外溶液和用于植入皮下。
除了活性成分或治疗性成分之外,片剂还可以含有多种被称为赋形剂的惰性物质。它们可以根据其在最终片剂中发挥的作用来进行分类。主要的组分包括填充剂、粘合剂、润滑剂和助流剂。
可以赋予最终片剂物理特征的其它赋形剂对于咀嚼片剂而言还有着色剂和矫味剂。不加入赋形剂时多数药物和药物成分都不能直接压制为片剂。这主要是因为多数药物的流动性和粘性较差。通常将赋形剂加至配方中以赋予被压制物质良好的流动性和压制特征。通过预处理步骤如湿法制粒、预压片、喷雾干燥滚圆或结晶赋予这些赋形剂这种性质。
通常加入润滑剂以防止制片物质粘在冲头上,在片剂压制过程中将摩擦力降至最低并使压制后的片剂能够从冲模中移出。这种润滑剂通常在最终片剂混合物中的含量低于1重量%。
此外,片剂通常含有稀释剂,它的加入是为了增加混合物的堆重量,其形成可以压制的粒度。通常当药物剂量相对较小时需要这种物质。
片剂中通常使用的另一类赋形剂是粘合剂。粘合剂是赋予粉末物质粘附性质的物质。通常使用的粘合剂包括淀粉和糖如蔗糖、葡萄糖、右旋糖和乳糖。
崩解剂通常用于保证片剂具有可接受的崩解速率。典型的崩解剂包括淀粉衍生物和羧甲基纤维素盐。
其它期望的赋形剂特征包括以下各项;
●高度可压性,使得在较低压力下即可形成坚实的片剂;
●流动性好,能够改进配方中其它赋形剂的流动性;和
●粘性(防止片剂在加工、运输和装卸过程中粉碎)。
制备压制的片剂有三个商业上很重要的过程:湿法制粒、直接压片和干法制粒(预压片或滚筒碾压)。制备方法和赋形剂类型的选择是为了给予片剂期望的物理特征,使之能够快速压片。压片后,片剂必须具有多种其它的属性如外观、硬度、崩解力和可接受的溶出特征。填充剂和其它赋形剂的选择取决于药物的理化性质、混合物在加工过程中的表现以及最终片剂的性质。进行处方设计前研究以确定活性组分与建议赋形剂的理化兼容性。
药物性质、其剂型和运行经济学将决定压片的最佳过程。一般而言,湿法制粒和直接压片都用于研制片剂。
干法制粒法可用于其中一种组分即药物或稀释剂的粘性足以压片的情况。该方法由混合、各成分预压片、干燥过筛、润滑和压片等步骤组成。
湿法制粒用于将粉末混合物转化为流动性和粘性适合于进行压片的颗粒。该方法包括将粉末在适当的混合器中混合,然后在搅拌下将制粒溶液加至混合粉末中得到制粒混合物。然后将湿块通过适当的筛,并通过托盘干燥法或流化床干燥进行干燥。或者,湿块可以干燥并通过研磨机。总体过程包括称重、干粉混合、湿法制粒、干燥、研磨、混合、润滑和压片。
一般而言,粉末的粘附或粘合性质不足以形成坚硬、坚实的颗粒。由于多数粉末的粘合性质较差,通常需要粘合剂将粉末颗粒粘合在一起。对热和湿度敏感的药物通常不能用湿法制粒制备。由于增加制备成本,加工步骤多,加工时间长是该法的问题。也已知湿法制粒能够降低一些药物赋形剂如微晶纤维素的可压性。
直接压片被认为是相对较快的过程,其中不改变药物的理化性质而对粉末物质直接压片。活性成分、直接压片赋形剂和其它辅剂如助流剂和润滑剂在双壳拌合器或相似的低切应力设备中混合,然后压制为片剂。据信这类混合是制备“可药用”剂型所必需的。一些药学科学家认为必需小心控制向配方中加入润滑剂的方式。相应地,通常通过轻轻地混合将润滑剂加至制粒混合物中。另外还认为润滑剂与制粒混合物混合时间过长会影响所得片剂的的硬度和崩解时间。由于这些原因,直接压片剂型的制备中不使用高切变力的混合条件。
直接压片的优点包括混合均匀、涉及的制备步骤少,即总体过程包括粉末称重、混合和压片,因此成本较低;消除热量和湿度、主要的粒子分离和物理稳定性。
由于加工时间短和成本优势,药物生产商更愿意使用直接压片技术而不愿使用湿法制粒或干法制粒法。但是,直接压片通常限于药物或活性成分具有形成可药用片剂所需物理特征的情况。但是,通常必须将一种或多种赋形剂与活性成分混合,然后才能使用直接压片法,因为很多成分不具有所需的性质。由于向配方中加入的每种赋形剂都会增加终产品的片剂大小,所以生产商通常仅用直接压片法来制备每片压制的片剂中含有低剂量活性成分的制剂。
含有高剂量药物的固体剂型,即药物本身占压制片剂总重量的较大部分时,只有当药物本身具有足够使该成分可以直接压片的物理特征如粘性时才能直接压片。
例如,DPP-IV抑制剂如式(I)化合物被认为是高剂量药物。多数片剂配方中每片包含70-85重量%的DPP-IV抑制剂。这种高剂量药物,加上其不利于直接压片的物理特征,还没有被允许使用直接压片法作为制备最终片剂的方法。此外,活性成分在水存在时稳定性较差,该因素是不利于使用湿法制粒的另一因素。
直接压片法作为制备片剂的方法的另一限制是压制片剂的潜在大小。若活性成分的量较高,药学制剂人员可以选择与其它赋形剂一起将活性成分进行湿法制粒,以获得含有期望活性成分量的可接受大小的片剂。湿法制粒中需要的填充剂、粘合剂或其它赋形剂的量低于直接压片法中需要的量,因为湿法制粒的过程有助于使片剂具有期望的物理性质。
羟丙基甲基纤维素在药学工业中已用作固体剂型直接压片的赋形剂。羟丙基甲基纤维素是一种加工过的纤维素,能够控制药物从固体剂型中的释放。
尽管直接压片法有诸多优点,如加工时间和成本低,但湿法制粒还是广泛地在工业中用于制备固体剂型。湿法制粒通常优选于直接压片法的原因是湿法制粒克服与配方中各成分的物理特征有关的任何问题的机会更大。这样可以提供具有想获得可接受的固体剂型所必需的流动性和粘性的材料。
湿法制粒相对于直接压片法的受欢迎程度至少基于三个优点。首先,湿法制粒提供的待压片材料具有更好的湿润性质,特别是对于疏水性药物物质更是如此。加入亲水性赋形剂使疏水性药物表面的亲水性更高,减少崩解和溶出中的问题。第二,湿法制粒中固体剂型中含量均一性通常得到改进,因为通常所有的颗粒都含有相同量的药物。最后,能够避免药物与赋形剂的离析。
直接压片法中,离析可能是潜在的问题。含有待压制颗粒的粒子的大小和形状在湿法制粒中的过程中得到最优化。这是因为,当干固体进行湿制粒时,粘合剂将各粒子“胶接”在一起,使它们聚集形成球形颗粒。
虽然一般而言,湿法制粒具有诸多优点,但是由于化合物在水的存在下不稳定的原因,期望直接压制含有高剂量DPP-IV抑制剂、即式(I)中定义的抑制剂的片剂。在制药工业中需要允许生产商通过直接压片法制备高剂量DPP-IV抑制剂片剂的技术和药物赋形剂。
本发明的一个目的是提供自由流动、粘性制片粉末形式的能够被直接压制成为片剂的DPP-IV抑制剂配方。
本发明的另一个目的是提供直接压制的单元剂型的DPP-IV抑制剂片剂,其具有可接受的溶出特征、可接受的硬度、不易碎裂、崩解时间短。
本发明的另一个目的是提供通过直接压片法制备单元剂型的压制DPP-IV抑制剂片剂的方法。
本发明提供一种制片粉末形式的直接制片、自由流动的DPP-IV抑制剂配方,其能够直接被压制为具有充分硬度、较短的崩解时间和可接受的溶出特征的片剂。
除活性成分外,制片粉末还含有多种被称为赋形剂的惰性材料。可以按照它们在最终片剂中所起的作用进行分类。主要包括填充剂、粘合剂或稀释剂、润滑剂、崩解剂和助流剂。其它赋予最终片剂物理特性的赋形剂是着色剂以及咀嚼片中的矫味剂。通常将赋形剂加至配方中以赋予被压制物质良好的流动性和压制特征。
本发明的优选配方包含以下物质:活性成分,即DPP-IV抑制剂化合物,粘合剂或稀释剂微晶纤维素和乳糖,崩解剂淀粉乙醇酸钠,以及润滑剂硬脂酸镁。
可以选择一种、两种、三种或多种稀释剂。可药用填充剂和可药用稀释剂的实例包括但不限于糖果用糖、可压性糖、葡萄糖结合剂、糊精、右旋糖、乳糖、甘露醇、微晶纤维素、纤维素粉末、山梨糖醇、蔗糖和滑石。填充剂和/或稀释剂例如可以以组合物的约15%至约40重量%的量存在。优选的稀释剂包括微晶纤维素,其通过控制用稀无机酸水溶液进行的α-纤维素(从纤维性植物材料中作为浆体获得)的水解来来制备。水解后,水解纤维素经过滤纯化,将水性浆体喷雾干燥至形成干燥、粒度分布广泛的多孔颗粒。合适的微晶纤维素平均粒度为约20nm至约200nm。微晶纤维素可以从多个供应商获得。合适的微晶纤维素包括FMC公司生产的AvicelPH 101、Avicel PH 102、Avicel PH 103、Avicel PH 105和Avicel PH 200。本发明实践中特别优选的是Avicel PH 102,其具有最小的表面积和多孔结构。优选微晶纤维素在片剂中的存在量为约25%至约70重量%。该材料的另一优选范围为约30%至约35重量%;另一优选的范围为约30%至约32重量%。
另一种稀释剂是乳糖。优选配制前将乳糖研磨至平均粒度为约50μm至约500μm。乳糖在片剂配方中存在的量为约5%至约40重量%,可以是约18%至约35重量%,最优选可以是约20%至约25重量%。
可以选择一种、两种、三种或多种崩解剂。可药用崩解剂的实例包括但不限于淀粉、粘土、纤维素、褐藻酸盐、树胶、交联聚合物如交联聚乙烯吡咯烷酮、交联羧甲基纤维素钙和交联羧甲基纤维素钠;大豆多糖;和瓜尔胶。崩解剂例如可在组合中存在的量为组合物的约2重量%至约20重量%,例如约5重量%至约10重量%,例如约7重量%。崩解剂也是片剂配方中任选但有用的组分。崩解剂用于确保片剂具有可接受的崩解速率。典型的崩解剂包括淀粉衍生物和羧甲基纤维素盐。淀粉乙醇酸钠是该配方优选的崩解剂。优选崩解剂在片剂配方中的存在量为约0%至约10重量%,可以是约1%至约4重量%,最优选可以是约1.5%至约2.5重量%。
可以选择一种、两种、三种或多种润滑剂。可药用润滑剂和可药用助流剂的实例包括但不限于硅胶、三硅酸镁、淀粉、滑石、三碱价磷酸钙、硬脂酸镁、硬脂酸铝、硬脂酸钙、碳酸镁、氧化镁、聚乙二醇、纤维素粉末和微晶纤维素。润滑剂,例如在组合物中的存在量可以为约0.1%至约5重量%,而助流剂的存在量可以为约0.1%至约10重量%。通常加入润滑剂防止制片材料粘在冲头上,在压片过程中将摩擦力降至最低,使压制后的片剂能够从冲模中取出。这种润滑剂通常在最终片剂混合物中的含量低于1重量%。润滑剂组分可以是疏水性或亲水性组分。这种润滑剂的实例包括硬脂酸、滑石和硬脂酸镁。硬脂酸镁在压制片剂和弹出片剂的过程中减小冲模壁与片剂混合物之间的摩擦力。它有助于防止片剂粘在冲头和冲模上。硬脂酸镁还有助于粉末在进料斗中的流动和向冲模中的流动。其粒度为450-550微米,密度为1.00-1.80g/mL。其稳定,在制片混合物中不会聚合。优选的润滑剂硬脂酸镁还用于配方中。优选润滑剂在片剂配方中存在的量为约0.25%至约6%;还优选约0.5%至约4重量%;最优选约0.1%至约2重量%。其它可能的润滑剂包括滑石、聚乙二醇、硅胶和硬化植物油。在本发明的任选实施方案中,润滑剂不存在于配方中,即,不是直接加至配方中,而是喷雾至冲模或冲头上。
可以任选使用其它常规的固体填充剂或载体如玉米淀粉、磷酸钙、硫酸钙、硬脂酸钙、硬脂酸镁、硬脂酸、硬脂酸单甘油酯和硬脂酸甘油二酯、山梨糖醇、甘露醇、明胶、天然树胶或合成树胶如羧甲基纤维素、甲基纤维素、褐藻酸盐、葡聚糖、阿拉伯胶、梧桐胶、豆角胶、西黄耆胶等、稀释剂、粘合剂、润滑剂、崩解剂、着色剂和矫味剂。
可药用粘合剂的实例包括但不限于淀粉;纤维素及其衍生物,如微晶纤维素、羟丙基纤维素、羟乙基纤维素和羟丙基甲基纤维素;蔗糖;右旋糖;玉米糖浆;多糖和明胶。粘合剂例如可以在组合物中存在的量为约10%至约40重量%。
有用的赋形剂的实例描述于Handbook of pharmaceutical excipients,第三版,A.H.Kibbe编,American Pharmaceutical Association出版,Washington DC,ISBN:0-917330-96-X中,或Handbook of PharmaceuticalExcipients(第四版),Raymond C Rowe编-出版商:Science and Practice,本文引入这些文献以供参考。
因此,在第一个实施方案中,本发明涉及一种药物组合物,其包含:
(a)游离形式或酸加成盐形式的DPP-IV抑制剂,优选LAF237;
(b)可药用稀释剂,其中在单元剂型中,DPP-IV抑制剂、优选LAF237的干重与稀释剂总重量的比例为0.5至0.25,优选0.4至0.28。
换言之,本发明涉及一种药物组合物,其包含:
(a)游离形式或酸加成盐形式的DPP-IV抑制剂,优选LAF237;
(b)可药用稀释剂,其中在单元剂型中,DPP-IV抑制剂、优选LAF237的重量与稀释剂重量的比例为0.5至0.25,优选0.4至0.28。
上述组合物,其中至少一种稀释剂是微晶纤维素且在单元剂型中,DPP-IV抑制剂、优选LAF237的干重与片剂中微晶纤维素的总重量比例为2至0.333,优选为1至0.333,最优选0.7至0.333。换言之,上述组合物,其中至少一种稀释剂是微晶纤维素且在单元剂型中,DPP-IV抑制剂、优选LAF237的重量与微晶纤维素的重量比例为2至0.333,优选为1至0.333,最优选0.7至0.333。
上述组合物,包含20至120mg、优选25至100mg的LAF237或其可药用酸加成盐。
上述组合物,其中稀释剂选自于微晶纤维素和乳糖,优选微晶纤维素和乳糖存在于组合物中。
上述组合物,其还含有:
(c)以干重计为0-20重量%的可药用崩解剂;
(d)以干重计为0.1-10重量%的可药用润滑剂。
优选上述组合物,其还含有:
(c)以干重计为1-6重量%的可药用崩解剂;
(d)以干重计为0.25-6重量%的可药用润滑剂。
上述比例基于DPP-IV抑制剂和稀释剂的干重。
单元剂型是任何类型的药物剂型如胶囊剂、片剂、颗粒剂、咀嚼片剂等。
在另一实施方案中,本发明涉及一种药物组合物,其包含:
(a)以干重计为5-60重量%的DPP-IV抑制剂,其为游离形式或酸加成盐形式,优选LAF237;
(b)以干重计为40-95重量%的可药用稀释剂;
(c)以干重计为0-20重量%的可药用崩解剂;和任选的
(d)以干重计为0.1-10重量%的可药用润滑剂。
优选地,本发明涉及一种药物组合物,其包含:
(a)以干重计为20-40重量%的DPP-IV抑制剂,其为游离形式或酸加成盐形式,优选LAF237;
(b)以干重计为40-95重量%的可药用稀释剂;
(c)以干重计为0-10重量%的可药用崩解剂;和任选地
(d)以干重计为0.25-6重量%的可药用润滑剂。
优选地,本发明涉及一种药物组合物,其包含:
(a)以干重计为20-40重量%的DPP-IV抑制剂,其为游离形式或酸加成盐形式,优选LAF237;
(b)以干重计为40-80重量%的可药用稀释剂;
(c)以干重计为0-10重量%的可药用崩解剂;和任选地
(d)以干重计为0.25-6重量%的可药用润滑剂。
最优选本发明涉及一种药物组合物,其包含:
(a)以干重计为20-35重量%的DPP-IV抑制剂,其为游离形式或酸加成盐形式,优选LAF237;
(b)以干重计为40-95重量%的可药用稀释剂;
(c)以干重计为0-10重量%的可药用崩解剂;和任选地
(d)以干重计为0.25-6重量%的可药用润滑剂。
最优选本发明涉及一种药物组合物,其包含:
(a)以干重计为20-35重量%的DPP-IV抑制剂,其为游离形式或酸加成盐形式,优选LAF237;
(b)以干重计为62-78重量%的可药用稀释剂;
(c)以干重计为0-10重量%的可药用崩解剂;和任选地
(d)以干重计为0.1-10重量%的可药用润滑剂。
最优选本发明涉及一种药物组合物,其包含:
(a)以干重计为20-35重量%的DPP-IV抑制剂,其为游离形式或酸加成盐形式,优选LAF237;
(b)以干重计为62-78重量%的可药用稀释剂;
(c)以干重计为1-6重量%的可药用崩解剂;和任选地
(d)以干重计为0.25-6重量%的可药用润滑剂。
最优选本发明涉及一种药物组合物,其包含:
(a)以干重计为22-28重量%的DPP-IV抑制剂,其为游离形式或酸加成盐形式,优选LAF237;
(b)以干重计为66-76重量%的可药用稀释剂;
(c)以干重计为0-6重量%的可药用崩解剂;和任选地
(d)以干重计为0.25-6重量%的可药用润滑剂。
最优选本发明涉及一种药物组合物,其包含:
(a)以干重计为22-28重量%的DPP-IV抑制剂,其为游离形式或酸加成盐形式,优选LAF237;
(b)以干重计为66-76重量%的可药用稀释剂;
(c)以干重计为1-6重量%的可药用崩解剂;和任选地
(d)以干重计为0.25-6重量%的可药用润滑剂。
上述组合物,其还包含:
(c)以干重计为0-6重量%的可药用崩解剂;和任选地
(d)以干重计为0.1-10重量%的可药用润滑剂。
本申请中,术语“可药用稀释剂”是指至少一种稀释剂,也包括例如两种或三种稀释剂的混合物。
优选上述组合物包含:
i)一种或两种选自于微晶纤维素和乳糖的稀释剂
ii)两种稀释剂微晶纤维素和乳糖
iii)以干重计为25-70重量%、优选35-55重量%的可药用微晶纤维素,或
iv)以干重计为25-70重量%、优选35-55重量%的可药用微晶纤维素和5-40重量%、优选18-35重量%的乳糖。
最优选上述组合物包含一种或两种选自于微晶纤维素的稀释剂如Avicel PH 102和乳糖。
最优选所述药物组合物包含可药用润滑剂(d)。
本申请中,术语“可药用崩解剂”是指至少一种崩解剂,也包括例如两种或三种崩解剂的混合物。
本申请中,术语“可药用润滑剂”是指至少一种润滑剂,也包括例如两种或三种润滑剂的混合物。
优选的DPP-IV抑制剂是LAF237,优选的稀释剂是微晶纤维素或乳糖或优选微晶纤维素和乳糖的组合,优选的崩解剂是淀粉乙醇酸钠,优选的润滑剂是硬脂酸镁。
优选组合物中的具体组分如下:
(a)以干重计为20-35重量%的DPP-IV抑制剂如LAF237;
(b)以干重计为25-70重量%的可药用微晶纤维素;
(c)以干重计为5-40重量%的可药用乳糖;
(d)以干重计为0-10重量%的可药用淀粉乙醇酸钠;
(e)以干重计为0.25-6重量%的硬脂酸镁。
优选组合物中的具体组分如下:
(a)以干重计为20-35重量%的DPP-IV抑制剂如LAF237;
(b)以干重计为25-70重量%的可药用微晶纤维素;
(c)以干重计为5-40重量%的可药用乳糖;
(d)以干重计为0-10重量%的可药用淀粉乙醇酸钠;
(e)以干重计为0.25-6重量%的硬脂酸镁。
另一优选的组合物如下:
(a)以干重计为约30-约32重量%的DPP-IV抑制剂或式(I)的DPP-IV抑制剂;
(b)以干重计为约40-约45重量%的可药用微晶纤维素;
(c)以干重计为约20-约25重量%的可药用乳糖;
(d)以干重计为约1.5-至约2.5重量%的可药用淀粉乙醇酸钠;
(e)以干重计为约0.1-约2重量%的硬脂酸镁。
另一优选组合物如下:
(a)以干重计为约20-35重量%、优选22-28重量%的DPP-IV抑制剂例如LAF237;
(b)以干重计为35-55重量%的可药用微晶纤维素;
(c)以干重计为18-35重量%的可药用乳糖;
(d)以干重计为1-4重量%的可药用淀粉乙醇酸钠;
(e)以干重计为0.5-4重量%的硬脂酸镁。
另一优选组合物如下:
(a)以干重计为约22-28重量%、优选24-26重量%的DPP-IV抑制剂或式(I)的DPP-IV抑制剂;
(b)以干重计为约45-约50重量%的可药用微晶纤维素;
(c)以干重计为约20-约25重量%的可药用乳糖;
(d)以干重计为约1.5-至约2.5重量%的可药用淀粉乙醇酸钠;
(e)以干重计为约0.1-约2重量%的硬脂酸镁。
另一优选组合物如下:
(a)以干重计为24-26重量%的DPP-IV抑制剂或式(I)的DPP-IV抑制剂;
(b)以干重计为约46-约48重量%的可药用微晶纤维素;
(c)以干重计为约23-约24.5重量%的可药用乳糖;
(d)以干重计为约1.5-至约2.5重量%的可药用淀粉乙醇酸钠;
(e)以干重计为约0.1-约2重量%的硬脂酸镁。
另一组优选组合物如下:
(a)以干重计为30-35重量%的DPP-IV抑制剂如LAF237;
(b)以干重计为35-50重量%的可药用微晶纤维素;
(c)以干重计为18-35重量%的可药用乳糖;
(d)以干重计为1-4重量%的可药用淀粉乙醇酸钠;
(e)以干重计为0.5-4重量%的硬脂酸镁。
在另一实施方案中,本发明涉及上述任一组合物,其中可药用润滑剂(d)在配方中只是任选存在。但优选可药用润滑剂(d)包含于组合物中。
优选对于压制的片剂、特别是直接压制的片剂而言,上述组合物包含以干重计为20至35重量%、最优选22至28重量%的DPP-IV抑制剂、特别是LAF237,其是游离形式或酸加成盐形式。
本申请中,术语组合物和配方具有相同的含义。
其它的常规赋形剂可任选加至本文描述的配方中,例如本文以上描述的常规填充剂或载体。
上述组合物特别适用于生产药物片剂例如压制的片剂或优选直接压制的片剂、囊形片剂或胶囊剂,并提供本领域技术人员要求的必需的物理特征、溶出特征和药物释放特征。因此,在另一实施方案中,本发明涉及上述任一配方用于制备药物片剂、囊形片剂或胶囊剂、特别是用于制粒、直接压片和干法制粒(预压片或滚筒碾压)的用途。
上述组合物还特别可用于生产片剂特别是压制的片剂,非常优选直接压制的片剂。
具体而言,用上述配方获得的片剂特别是以直接压制片剂的形式加工时,或以下所述的直接压制的片剂,很少发生脆碎性问题,具有很强的断裂强度,生产坚实性得到改善,片剂厚度与片剂重量的比例(直接压制的片剂)最优化,配方中、特别是直接压制的片剂中水分含量较低,根据英国药典1988年版中的良好分散质量确定分散崩解时间良好。
本发明中的直接压制DPP-IV抑制剂涉及混合和压制。赋形剂级别的选择考虑粒度应保持在一定范围内以使DPP-IV抑制剂粉末混合均匀、DPP-IV抑制剂含量均一。其在直接压片过程中防止进料斗中粉末的离析。使用这些赋形剂的优点在于它们赋予粉末混合物可压性、粘性和流动性。此外,使用直接压片法在单元生产成本、货架寿命、除去热量和水分、允许主要的粒子分离、物理稳定性和确保粒度均一的方面可提供竞争力。
要求保护的组合物的所述优点对于例如滚筒碾压法或湿法制粒或填充胶囊都非常有用。
在研制本文描述的药物组合物时,申请人发现如果满足以下条件,则压制的片剂、特别是直接压制的片剂具有特别优势:
i)含有DPP-IV抑制剂的粒子粒度分布低于250μm,优选为10至250μm,和/或
ii)在25℃、60%室内湿度(RH)下一周后片剂中水分含量低于10%,和/或
iii)片剂厚度与片剂重量比为0.002至0.06mm/mg。
本发明涉及一种压制的药物片剂、优选直接压制的片剂,其包含游离形式或酸加成盐形式的DPP-IV抑制剂,所述的DPP-IV抑制剂的物理性质使其不可能或很难压制、优选直接压制为药物片剂。优选的DPP-IV抑制剂是LAF237。物理性质可以是例如松散性(bulkiness)和蓬松性(fluffiness)等。在进一步研制所述的药物组合物的过程中,申请人发现如果包含DPP-IV抑制剂的粒子粒度分布低于250μm、或为10至250μm、或为50至150μm,则配方的加工性质或物理性质如吸湿性、流动性、松散性、蓬松性等会惊人地得以改善。申请人还惊人地发现如果遵循上述标准i)、ii)和/或iii)中的至少一个,则片剂的物理特征如溶解度、脆碎性、吸湿性、硬度等都会得到改善。
因此在第一个实施方案(a)中,本发明涉及压制的片剂、优选直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子,其中片剂中至少40%、优选60%、最优选80%、更优选90%的粒度分布低于250μm或优选为10至250μm。
本发明涉及压制的片剂、优选直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子,其中片剂中至少40%、优选60%、最优选80%、更优选90%的粒度分布高于10μm。
术语“其中至少40%、优选60%、最优选80%、更优选90%”是指至少40%、优选至少60%、最优选至少80%、更优选至少90%。
术语“其中至少25%、优选35%、最优选45%”是指至少25%、优选至少35%、最优选至少45%。
具体而言,本发明涉及压制的片剂、优选直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子,其中至少片剂中25%、优选35%、最优选45%的粒度分布为50至150μm。
在第二个实施方案(b)中,本发明涉及压制的片剂、优选直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子,其中片剂厚度与片剂重量的比例为0.002至0.06mm/mg、优选为0.01至0.03mm/mg。
上述第一和第二个实施方案(a)和(b)的组合,提供了具有良好压实特征的压制的片剂、优选直接压制的片剂。
因此本发明还涉及压制的片剂、优选直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子,其中:
i)片剂中至少40%、优选60%、最优选80%、更优选90%的粒度分布为10至250μm,且
ii)片剂厚度与片剂重量的比例为0.002至0.06mm/mg或0.01至0.03mm/mg
优选其中:
i)片剂中至少25%、优选35%、最优选45%的粒度分布为50至150μm,且
ii)片剂厚度与片剂重量的比例为0.002至0.06mm/mg或0.01至0.03mm/mg。
在第三个实施方案中,本发明涉及压制的片剂、优选直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子,其中:
i)片剂中至少40%、优选60%、最优选80%、更优选90%的粒度分布为10至250μm,
ii)在25℃、60%RH下一周后片剂中水分含量低于10%,且
iii)片剂厚度与片剂重量的比例为0.002至0.06mm/mg。
优选地,本发明涉及压制的片剂、最优选直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子,其中:
i)片剂中至少25%、优选35%、最优选45%的粒度分布为50至150μm,
ii)在25℃、60%RH下一周后片剂中水分含量低于10%,且
iii)片剂厚度与片剂重量的比例为0.002至0.06mm/mg。
优选地,本发明涉及压制的片剂、最优选直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子,其中:
i)片剂中至少25%、优选35%、最优选45%的粒度分布为50至150μm,
ii)在25℃、60%RH下一周后片剂中水分含量低于5%,且
iii)片剂厚度与片剂重量的比例为0.002至0.06mm/mg。
优选本发明涉及压制的片剂、最优选直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子,其中:
i)片剂中至少25%、优选35%、最优选45%的粒度分布为50至150μm,
ii)在25℃、60%RH下一周后片剂中水分含量低于5%,且
iii)片剂厚度与片剂重量的比例为0.01至0.03mm/mg。
在一个非常优选的方面,上述的三个实施方案,即压制的片剂和直接压制的片剂包含本文描述的组合物如药物组合物,其包含:
(a)以干重计为5-60重量%的游离形式或酸加成盐形式的DPP-IV抑制剂,优选LAF237;
(b)以干重计为40-95%或40-80重量%的可药用稀释剂;
(c)以干重计为0-20重量%的可药用崩解剂;和任选地
(d)以干重计为0.1-10重量%的可药用润滑剂,或如下药物组合物,其包含:
(a)以干重计为20-35重量%的游离形式或酸加成盐形式的DPP-IV抑制剂,优选LAF237;
(b)以干重计为40-95%或40-80重量%、优选62-78重量%的可药用稀释剂;
(c)以干重计为0-10重量%的可药用崩解剂;和任选地
(d)以干重计为0.25-6重量%的可药用润滑剂。
优选DPPIV粒子、特别是LAF237粒子包含高于60%、最优选高于90%或95%、更优选高于98%的DPPIV抑制剂。或者,DPPIV粒子、特别是LAF237粒子可以通过本领域熟知的微制粒法形成,并且含有高至40%的可药用赋形剂。
优选LAF237粒子含有高于60%、最优选高于90%或95%、更优选高于98%的LAF237。
已发现所选择的DPPIV抑制剂、特别是LAF237的粒度分布对于提供最佳片剂压实条件特别重要。
在另一优选的实施方案中,所选择的赋形剂(b)、(c)和/或(d)的粒度分布与DPPIV抑制剂粒子、特别是LAF237粒子的粒度分布相似。
术语“相似”是指片剂中赋形剂的粒度分布为约5至400μm、或10至300μm、优选10至250μm。
优选的具有合适粒度分布的赋形剂可从例如Handbook ofPharmaceutical Excipients(第四版),Raymond C Rowe编-出版商:Science and Practice中选择。
药物的粒度、如LAF237的粒度由结晶、干燥和/或碾磨/过筛(非限制性实例如以下所述)过程控制。粒度还可使用滚筒碾压和碾磨/过筛方法进行研细。制备合适的粒度在本领域是熟知的,描述于例如“Pharmaceuticaldosage forms:第2卷,第2版,Ed.:H.A.Lieberman,L.Lachman,J.B.Schwartz(第3章:SIZE REDUCTION)”中。期望的粒度分布可以从任何形式的DPPIV抑制剂、特别是任何形式的LAF237中获得,例如从无定形LAF237、结晶形式的LAF237、“A”晶型的LAF237、部分结晶形式的LAF237、多晶型态的LAF237、溶剂合物形式的LAF237或水合物形式的LAF237或从它的任何盐获得。优选的粒子从“A”晶型的LAF237获得。
已研究了多种粒度,发现本文描述的具体粒度范围对于直接压片可获得出乎意料的好结果。
通过筛分析法估计粒度分布:粒度分布用筛分析法、光子相关光谱法或激光衍射法(国际标准ISO 13320-1)、电子传感带法、光阻法、沉淀法或显微镜法测量,这些方法都是本领域技术人员熟知的方法。筛分析法是通过粒度分布将粉末分类的最老的方法之一。这种方法是熟知的,在现有技术中有所描述,如描述于分析化学课本中或美国药典(USP)的出版物USP-NF(2004-第786章-(The United States Pharmacopeial Convention,Inc.,Rockville,MD))中,其中描述了美国食品药品管理局(FDA)实施的标准。使用的技术描述于例如Pharmaceutical dosage forms:第2卷,第2版,Ed.:H.A.Lieberman,L.Lachman,J.B.Schwartz中,该书是很好的实例。其中还提及(第187页)了其它方法:电子传感带法、光阻法、空气渗透法、在气体或液体中的沉淀法。
在用空气喷射筛测量粒度时,向上抽吸空气,使之从旋转的狭缝通过筛使筛上的材料流化。同时,在筛的底部施加负压,将微细粒子转移至收集装置中。通过连续使用单个筛从粒度分布的较细端取出粒子进行粒度分析和平均粒度测定。更多详情另请参见″Particle Size Measurement″,5thEd.,p 178,vol.1;T.Allen,Chapman&Hall,London,UK,1997。对于本领域技术人员而言,这种粒度测量是常规方法。
片剂中水分含量可以使用本领域技术人员熟知的干燥失重法或Karl-Fischer法测量(例如可以通过热重量分析法测量干燥失重测量水分含量)。这种方法是熟知的,在现有技术中有所描述,如描述于分析化学课本中或美国药典(USP)的出版物USP-NF(2004)中,其中描述了美国食品药品管理局(FDA)实施的标准(2004-USP-Chapter 921)。
片剂厚度可以使用刻度尺、游标卡尺、螺纹量规或任何测量尺寸的电子方法测量。我们取片剂厚度的mm值除以片剂重量的mg数得到比例。这种方法是熟知的,在现有技术中有所描述,如描述于分析化学课本中或美国药典(USP)的出版物USP-NF(2004)中,其中描述了美国食品药品管理局(FDA)实施的标准。
具体而言,本发明提供压制的片剂或直接压制的片剂,根据本文定义的英国药典对可分散片剂的测试,其能够在5至15分钟的时间内分散于水中从而提供能够通过孔径为710μm的筛网的分散系。
本发明的片剂不仅可以迅速分散于水中,其附加优点是其满足英国药典(B.P.)对可分散片剂关于分散时间和分散质量(即通过710μm筛)的测试。
优选本发明的片剂分散时间少于15分钟、更优选少于12分钟、最优选少于10分钟。
本发明片剂的另一优点是因为形成的分散系相对较细,因此片剂溶出时间短,药物可以更快地吸收入血流中。此外,利用本发明的片剂获得的较短的分散时间和相对较细的分散系对于可吞服片剂而言也是优点。因此本发明的片剂既可分散于水中也可直接吞服。本发明的旨在膨胀的这些片剂优选进行包膜以辅助吞服。
本发明的另一实施方案涉及溶出速率(药物溶出速率)得以改进的压制的片剂,其中分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子,即DPPIV粒子特别是LAF237粒子,其中片剂中至少40%、优选60%、最优选80%、更优选90%的粒度分布为10至250μm,
且其中
i)在0至10分钟释放85至99.5%的活性成分,且
ii)在10至15分钟释放90至99.5%的活性成分,
优选其中
i)在0至10分钟释放88至99.5%的活性成分,且
ii)在10至15分钟释放95至99.5%的活性成分,
或优选地
i)在0至10分钟释放89至94%的活性成分,且
ii)在10至15分钟释放96至99%的活性成分。
使用桨法用1000ml 0.01N HCl测定药物溶出速率(释放%)。这种方式是熟知的,在现有技术中有所描述,如描述于任何分析化学课本中或美国药典(USP)的出版物USP-NF(2004-Chapter 711),其中描述了美国食品药品管理局(FDA)实施的标准。
本发明还涉及DPPIV抑制剂、特别是维格列汀用于制备压制片剂或直接压制片剂的用途,其中片剂中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm。
在另一实施方案中,本发明涉及上述任一药物组合物,其中DPP-IV抑制剂、特别是维格列汀或“A型”维格列汀晶体的粒度分布如以上描述压制片剂时所述。
因此,在另一实施方案中,本发明还涉及一种本文描述的药物组合物,其中所述分散系含有包含DPP-IV抑制剂、特别是维格列汀或维格列汀的结晶形式或“A型”维格列汀晶型或其药学盐的粒子,其中:
i)配方中至少40%、优选60%的粒度分布低于250μm,和/或
ii)配方中至少40%、优选60%的粒度分布为10至250μm,和/或
iii)配方中至少60%、优选80%的粒度分布为10至250μm,和/或
iv)配方中至少25%、优选35%的粒度分布为50至150μm。
在另一个实施方案中,药物赋形剂在上述配方中的粒度分布为5至400μm。
本发明还提供了一种制备包含DPP-IV抑制剂、特别是维格列汀的压制片剂或直接压制片剂的方法,其中该方法中使用的至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm。
本发明还涉及一种用于制备压制的、单元剂型的DPP-IV抑制剂片剂、优选直接压制的片剂的方法,
其包含:
(a)将以干重计为以下重量%的以下物质混合:
(i)以干重计为5-60重量%的DPP-IV抑制剂、特别是维格列汀,其中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm;和
(ii)至少一种选自于稀释剂、崩解剂和润滑剂的赋形剂,以形成制片粉末形式的DPP-IV抑制剂配方,其能够压制成、优选直接压制成片剂;和
(b)将步骤(a)中制备的配方压制成为压制的单元剂型形式的DPP-IV抑制剂片剂。
本发明还提供了制备压制的、单元剂型的DPP-IV抑制剂片剂、优选直接压制的片剂的方法
其中
(i)片剂厚度与片剂重量的比例为0.002至0.06mm
其包含:
(a)将以干重计为以下重量%的以下物质混合:
(i)以干重计为5-60重量%的DPP-IV抑制剂例如维格列汀,其中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm;和
(ii)至少一种选自于稀释剂、崩解剂和润滑剂的赋形剂,以形成制片粉末形式的DPP-IV抑制剂配方,其能够压制成、优选直接压制成片剂;和
(b)将步骤(a)中制备的配方压制成为压制的单元剂型形式的DPP-IV抑制剂片剂。
本发明还提供一种用于制备压制的单元剂型形式的DPP-IV抑制剂片剂、优选直接压制的片剂的方法,其中
(i)片剂中至少40%、优选60%、最优选80%、更优选90%的游离形式或酸加成盐形式的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm;
(ii)在25℃、60%RH下一周后片剂中水分含量低于10%,和
(iii)片剂厚度与片剂重量的比例为0.002至0.06mm
该方法包括:
(a)将以干重计为以下重量%的以下物质混合:
(i)以干重计为5-60重量%或6-60重量%的DPP-IV抑制剂例如维格列汀,其中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm;和
(ii)至少一种选自于稀释剂、崩解剂和润滑剂的赋形剂,
以形成制片粉末形式的DPP-IV抑制剂配方,其能够压制成、优选直接压制成片剂;和
(b)将步骤(a)中制备的配方压制成为压制的单元剂型形式的DPP-IV抑制剂片剂。
在另一优选的实施方案中,本文所述的方法中使用的DPP-IV抑制剂、特别是维格列汀的至少25%、优选35%、最优选45%的粒度分布为50至150μm。
优选上述方法包括:
(a)将以干重计为以下重量%的以下物质混合:
(i)以干重计为5-60重量%的DPP-IV抑制剂例如LAF237,其中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm;
(ii)以干重计为40-95%的可药用稀释剂;
(iii)以干重计为0-20重量%的可药用崩解剂;和
(iv)以干重计为0.1-10重量%的可药用润滑剂,
以形成制片粉末形式的DPP-IV抑制剂配方,其能够压制成、优选直接压制成片剂;和
(b)将步骤(a)中制备的配方压制成为压制的单元剂型形式的DPP-IV抑制剂片剂。
最优选该方法包括:
(a)将以干重计为以下重量%的以下物质混合:
(i)以于重计为20-35重量%或25-35重量%的DPP-IV抑制剂例如LAF237,其中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm;
(ii)以干重计为40-80%或40-95重量%的可药用稀释剂;
(iii)以干重计为0-10重量%的可药用崩解剂;和
(iv)以干重计为0.25-6重量%的可药用润滑剂,
以形成制片粉末形式的DPP-IV抑制剂配方,其能够压制成、优选直接压制成片剂;和
(b)将步骤(a)中制备的配方压制成为压制的单元剂型形式的DPP-IV抑制剂片剂。
优选步骤(a)中使用的混合组合物选自于本文描述的优选配方。
优选的DPP-IV抑制剂是LAF237,优选的稀释剂是微晶纤维素或乳糖或优选微晶纤维素和乳糖的组合,优选的崩解剂是淀粉乙醇酸钠,优选的润滑剂是硬脂酸镁。
在最优选实施方案中,该方法包括:
(a)将以干重计为以下重量%的以下物质混合:
(i)以干重计为20-35重量%或25-35重量%的游离形式或酸加成盐形式的DPP-IV抑制剂、优选维格列汀,其中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm;
(ii)以干重计为25-70%或优选35-50重量%的可药用微晶纤维素如Avicel PH 102;
(iii)以干重计为5-40重量%或优选18-35重量%的可药用乳糖;
(iv)以干重计为0-10重量%或优选1-4重量%的可药用淀粉乙醇酸钠;和
(v)以干重计为0.25-6重量%或优选0.5-4重量%的可药用硬脂酸镁,
以形成制片粉末形式的DPP-IV抑制剂配方,其能够压制成、优选直接压制成片剂;和
(b)将步骤(a)中制备的配方压制成为压制的单元剂型形式的DPP-IV抑制剂片剂。
本发明还提供一种用于制备压制的单元剂型形式的DPP-IV抑制剂片剂的方法,其包括
(a)将以干重计为以下重量%的以下物质混合:
(i)以干重计为30-32重量%的游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237,其中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm;
(ii)以干重计为40-45%的可药用微晶纤维素(Avicel PH 102);
(iii)以干重计为20-25重量%的可药用乳糖;
(iv)以干重计为1.5-2重量%的可药用淀粉乙醇酸钠;和
(v)以干重计为0.1-2重量%的硬脂酸镁,
以形成制片粉末形式的DPP-IV抑制剂配方,其能够压制成、优选直接压制成片剂;和
(b)将步骤(a)中制备的配方压制成为压制的单元剂型形式的DPP-IV抑制剂片剂。
本发明还提供一种用于制备压制的单元剂型形式的DPP-IV抑制剂片剂的方法,其包括
(a)将以干重计为以下重量%的以下物质混合:
(i)以干重计为23-28重量%的游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237,其中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂、特别是维格列汀的粒度分布低于250μm或优选为10至250μm;
(ii)以干重计为40-45%的可药用微晶纤维素(Avicel PH 102);
(iii)以干重计为20-25重量%的可药用乳糖;
(iv)以干重计为1.5-2重量%的可药用淀粉乙醇酸钠;和
(v)以干重计为0.1-2重量%的硬脂酸镁,
以形成制片粉末形式的DPP-IV抑制剂配方,其能够压制成、优选直接压制成片剂;和
(b)将步骤(a)中制备的配方压制成为压制的单元剂型形式的DPP-IV抑制剂片剂。
压制步骤(b)之前优选对配方进行过筛步骤以基本除去团块,即除去任何团聚物/结块。
在另一实施方案中,本发明包括包含药物组合物如上述的药物组合物的胶囊剂,优选其中:
i)胶囊中至少60%、优选80%、最优选90%的包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子的粒度分布为10至500μm,
ii)在25℃、60%RH下一周后片剂中水分含量低于10%。
更优选包含药物组合物如上述的药物组合物的胶囊剂,优选其中:
i)胶囊中至少40%、优选60%、最优选80%、更优选90%的包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选LAF237的粒子的粒度分布低于250μm,优选为10至250μm,
ii)在25℃、60%RH下一周后片剂中水分含量低于5%。
通过使用常规制片机或相似机器将终产品制备为片剂、胶囊剂等形式。
最优选用于本文描述的配方、胶囊剂、压制的片剂、用途或方法的DPP-IV抑制剂选自于1-{2-[(5-氰基吡啶-2-基)氨基]乙基氨基}乙酰基-2(S)-氰基-吡咯烷二盐酸盐、(S)-1-[(3-羟基-1-金刚烷基)氨基]乙酰基-2-氰基-吡咯烷、L-苏-异亮氨酰基噻唑烷、MK-0431、GSK23A、BMS-477118、3-(氨基甲基)-2-异丁基-1-氧代-4-苯基-1,2-二氢-6-异喹啉甲酰胺和2-{[3-(氨基甲基)-2-异丁基-4-苯基-1-氧代-1,2-二氢-6-异喹啉基]氧基}乙酰胺,在任何情况下任选它们的药学盐。
最优选的DPP-IV抑制剂是1-[3-羟基-金刚烷-1-基氨基)-乙酰基]-吡咯烷-2(S)-甲腈(LAF237或维格列汀)例如维格列汀的无定形状态或结晶形式。
优选包含维格列汀的单元剂型例如片剂或胶囊剂含有约10至150mg、优选25至100mg、最优选50至100mg的维格列汀或其A型晶体。优选50mg或100mg的维格列汀或其A型晶体。
最优选本文描述的组合物、胶囊剂、压制的片剂或直接压制的片剂含有LAF237的形式是结晶形式、优选“A型”晶型,如本文以下的定义,且优选至少1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或98%的LAF237化合物可以是结晶形式,优选A型晶型。
本发明还涉及一种如本文所述的药物组合物(药物配方)、胶囊剂、压制的片剂或直接压制的片剂,其含有LAF237的形式是结晶形式、优选“A型”晶型,如本文以下的定义。配方中优选至少1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或98%的LAF237化合物可以是结晶形式,优选A型晶型。
本发明还涉及一种如本文所述的组合物(药物配方),其中低于1%、或低于0.4%的LAF237是A型晶型,高于99%或99.6%的LAF237是无定形形式。
优选至少20或50%、最优选至少80%的活性成分LAF237是“A型”晶型。
因此在另一方面中,本发明涉及LAF237(维格列汀)的固态物理性质。可通过控制获得固体形式LAF237的条件影响这些性质。固态物理性质包括例如碾磨固体的流动性。流动性影响将该材料加工为药物产品过程中操作的难易程度。当化合物粉末的粒子不能容易地流过彼此时,配方专家在研制片剂或胶囊剂配方时必须考虑该事实,可能有必要使用助流剂如胶态二氧化硅、滑石、淀粉或三碱价磷酸钙。
药物化合物的另一重要固态性质是在水性液体中的溶出速率或药物的生物利用度。活性成分在患者胃液中的溶出速率可能产生治疗上的后果,因为溶出速率限制了口服施用的活性成分到达患者血流中的速率的上限。
例如,同一药物的不同晶型或无定形形式可能在这种药学上重要的性质如溶出速率和生物利用度上具有显著差异。同样,不同晶型或无定形形式可能具有不同的加工性质如吸湿性、流动性等,这些可能影响它们是否适合用于活性药物的商业生产。
溶出速率在配制糖浆剂、酏剂和其它液体药物时也是一个考虑因素。化合物的固态形式可能也会影响它的压实行为和储存稳定性。
这些实际的物理特征受单元晶胞中分子的构象和方向的影响,所述单元晶胞定义了一种物质的具体多晶型形式。多晶型形式可能导致热学行为不同于无定形材料或另一种多晶型形式。热学行为在实验室内用诸如毛细管熔点、热重量分析(TGA)和差示扫描热计(DSC)等技术测量,热学行为可用于将一些多晶型形式与另一些形式区分开来。一种具体的多晶型形式还可能导致不同的光谱学性质,这些性质可以通过粉末X射线衍射晶体分析法、固态13C NMR光谱测定法和红外光谱测定法检测。用于描述晶型特征的方法:IR、X射线粉末衍射、熔点测定。
在研制本文所述的配方和粒度分布时,申请人发现一种新的维格列汀晶型具有出乎意料的优秀理化特征,其特别适合于:改进包含维格列汀的药物配方的质量和制备过程(容易加工、装卸和给药),改进制备其粒度分布特别适合于压制片剂的粒子的过程,通过例如改进维格列汀的吸湿特征来改进维格列汀在配方中的稳定性,改进其它性质如生物利用度、溶解度。这些惊人的优秀理化性质使这种晶型特别适合于生产各种药物剂型。
因此在第一方面,本发明提供一种制备维格列汀或其盐的结晶形式的方法,该方法包括以下步骤:
i)加热维格列汀或其盐在有机溶剂中的溶液,
ii)诱导维格列汀结晶,和
iii)回收维格列汀晶体。
在优选的实施方案中,本发明提供一种制备维格列汀“A型”晶型的方法,该晶型的X射线衍射图具有在16.6°、17.1°、17.2°+/-0.3度2-θ,或优选在12.0°、13.5°、16.6°、17.1°、17.2°、20.1°、22.5°、27.4°、28.1°+/-0.3度2-θ的峰,该方法包括以下步骤:
i)加热维格列汀在有机溶剂中的溶液,
ii)诱导维格列汀结晶,和
iii)回收维格列汀晶体。
优选溶剂选自于2-丁酮、2-丙醇/乙酸乙酯、2-丙醇、丙酮。
优选结晶包括以下步骤:
i)加热LAF237在有机溶剂中的溶液,有机溶剂优选选自于2-丁酮、2-丙醇/乙酸乙酯、2-丙醇、丙酮,
ii)将溶液冷却至约-20℃至约20℃,优选至约-10℃至约10℃以诱导结晶,和
iii)回收维格列汀晶体,优选不加热。
优选如上所述,在加热步骤后i),在冷却步骤中将溶液的温度降低至(-)20℃至约(+)20℃的范围内,优选至约(-)10℃至约(+)10℃的范围内。
在另一个实施方案中,可通过向溶液中加入反溶剂(冷却或不冷却)诱导结晶ii)。
本文使用的反溶剂是一种当加至化合物X(即维格列汀)在溶剂中的溶液中时可诱导X沉淀的液体。当加入反溶剂后引起X从溶液中沉淀的速度快于或程度大于在相同条件下放置相同时间但不加入反溶剂时X从等浓度X在相同溶剂中的溶液中沉淀的速度或程度时,认为反溶剂诱导X的沉淀。视觉上可以观察到沉淀,溶液变得浑浊或形成明晰的X粒子悬浮于溶液中或在装有该溶液的容器底部收集沉淀。
优选地,将溶液逐渐冷却至约-20℃至约20℃的温度范围、优选至约-10℃至约10℃的温度范围诱导结晶。
优选地,将溶液逐渐冷却至约-20℃至约20℃的温度范围、优选至约-10℃至约10℃的温度范围诱导结晶,例如在一段确定的时间内冷却至50℃、然后在一段确定的时间内冷却至30℃、在一段确定的时间内冷却至0℃。
优选地,将溶液在100至500分钟内、优选250至450分钟内逐渐冷却至约-10℃至约10℃。
优选溶液在1至3小时、优选2小时内冷却至50℃、然后在40至120分钟、优选80分钟内冷却至30℃、然后在30至120分钟、优选72分钟内冷却至0℃
然后可以通过本领域熟知的技术如过滤、离心、倾析等回收所得的晶体。
然后可将晶体干燥。干燥可在常压下进行或减压进行。优选干燥在约20℃至约60℃的温度下进行,更优选结合低于约30mm Hg的压力。
取决于条件,大约几个小时例如约2至约5小时的干燥可能足够了。
本文使用的术语干燥是指通过加热除去溶剂,优选在常压或减压下进行。
本文使用的术语减压是指低于一个大气压的压力,更优选低于约100mmHg。
本文使用的术语沉淀是指在混合物中固体小粒子混悬液的形成。
本文使用的术语结晶是指从液体或气体中形成晶体的过程。
上述的过程中至少40%、优选60%、更优选80%的所得维格列汀的“A型”晶型的粒度分布低于250μm,优选为10至250μm。
在另一方面中,本发明涉及维格列汀(LAF237)“A型”晶型,其可通过以下方法获得,其中:
i)向装有机械搅拌器的1500ml反应器中加入120克(g)LAF237(1-[(3-羟基-金刚烷-1-基氨基)-乙酰基]-吡咯烷-2(S)-甲腈)、3.6g活性炭、2.4gcellflock 40、3.6g 1,8-二氮杂二环[5.4.0]十一-7-烯和483g 2-丁酮,
ii)将混合物加热至回流(夹套温度(JT):95℃)并搅拌30分钟,
iii)将混合物滤至温热的反应器(JT:75℃)中,用48g 2-丁酮洗涤滤饼,
iv)然后将IT调至70℃,将0.102g所得的重结晶的LAF237在1.1ml2-丁酮中的混悬液加至溶液中,
v)将所得混悬液搅拌30分钟,在2小时内冷却至内部温度(IT)50℃,然后在80分钟内冷却至30℃,
vi)最后将混悬液在72分钟内冷却至0℃,再搅拌1小时,
vii)此后将混悬液过滤,粗产物用冷的(0℃)37g 2-丁酮和34g叔-丁基甲基醚的混合物洗涤,
viii)粗产物(维格列汀晶型A)最后在约JT 55℃下减压干燥。
在另一方面中,本发明涉及维格列汀的结晶形式。
根据本发明,术语“DPP-IV抑制剂的结晶形式”、特别是术语“维格列汀的结晶形式”还包括无水结晶形式、部分结晶形式、多种晶型的混合物、水合物结晶形式或溶剂合物结晶形式。
-无定形态:非晶体,(固态中原子或分子为随机无序排列的三维结构)。通过水溶液低压冻干法获得LAF237无定形形式。
-晶体态:晶体材料是原子或分子精确几何排列的三维周期阵列。
-无水结晶形式:其三维周期排列中不含溶剂或水分子的晶型。
-水合物:其三维周期排列中含有一个或多个水分子的晶型。
-溶剂合物:其三维周期排列中含有一个或多个不同于水的溶剂分子的晶型。
-半晶型:固态中仅有一部分原子或分子为有序三维排列。
本发明中,术语“有峰”是指“包含峰”,无限制性。
本发明中,术语“多晶型”或“多晶型的”是指不同于“A型”晶型的晶型。
优选本发明涉及热力学最稳定的维格列汀晶型(高度理化稳定性)。
优选本发明涉及一种维格列汀的晶型,其中40%、优选60%、最优选80%的维格列汀晶体的粒度分布低于250μm,优选为10至250μm。
优选本文描述的维格列汀粒子是“A型”晶型的维格列汀。“A型”粒子优选包含60%以上、最优选90%或95%以上、更优选98%以上的“A型”晶型的维格列汀。
术语“热力学最稳定的”是指通过例如溶解度测试、溶解热、DSC等方法研究不同的维格列汀形式以检测当前晶型不同形式之间发生过渡转变时的热力学关系(单向转变、互变现象)。基于这一分析可以确定例如在室温下或在整个温度范围内哪种是最稳定的晶型。
在优选的方面,本发明涉及“A型”晶型的维格列汀,其特征为X射线衍射图具有在16.6°、17.1°、17.2°+/-0.3度2-θ,或优选在12.0°、13.5°、16.6°、17.1°、17.2°、20.1°、22.5°、27.4°、28.1°+/-0.3度2-θ的峰。
在另一方面,本发明涉及“A型”晶型的维格列汀,其特征为它的X射线衍射图基本如图1所示。
可以通过如以下实施例7.2.i中所述的方法获得X射线晶型衍射数据。
在另一方面,本发明涉及“A型”晶型的维格列汀,其特征为在液体石蜡中的IR光谱具有以下以倒数波数(cm-1)表示的显著的吸收带:约3293cm-1、2925-2853cm-1、2238cm-1、1658cm-1、1455/1354cm-1、1254cm-1、1121cm-1、1054-1035cm-1+/-2cm-1。FT-IR偏差:+/-2cm-1。
在另一方面,本发明涉及“A型”晶型的维格列汀,其特征为在液体石蜡中的IR光谱具有基本如图2所示的以倒数波数(cm-1)表示的吸收带。
可以通过如以下实施例7.2.ii中所述的方法获得IR(红外)数据。
在另一方面,本发明涉及“A型”晶型的维格列汀,其特征为它的熔点是147℃+/-4℃(例如通过差示扫描量热法(DSC)获得,10K/min)。优选约149℃+/-2℃。
在另一方面,本发明涉及新的LAF237(维格列汀)晶型“A型”晶型,其特征为DSC差示热分析图在25C至140C之间无过渡转变,而无定形形式的玻璃化转变温度为27℃(样品从干燥变为糊状),然后从50℃开始至110℃结束是重结晶放热,随后在约127℃熔化转变。特别是其中在约140C至约150C的区域内没有熔点。
在优选的实施方案中,维格列汀晶型、特别是“A型”晶型的粒度分布低于250μm,优选为10至250μm。
本发明还涉及维格列汀晶型、特别是“A型”晶型用于制备相应的维格列汀无定形形式的用途,或者维格列汀晶型、特别是“A型”晶型用于制备另一种多晶型形式的用途。
本发明还涉及维格列汀“A型”晶型用于制备相应的维格列汀元定形形式的用途,或者维格列汀“A型”晶型用于制备另一种多晶型形式的用途。
在另一方面,本发明还涉及制备维格列汀无定形形式的方法,其中使用维格列汀“A型”晶型作为原料或结晶过程的中间体。
可以通过X射线衍射和/或红外光谱或本领域已知的任何其它方法识别或区分新的“A型”晶型。
“A型”晶型的维格列汀可以通过X射线粉末衍射进行特征描述。X射线衍射图对于具体的晶型是特有的。每种晶型的衍射图都具有独特的一套衍射峰,其可表达为2θ角度、晶面间距d值和相对峰强度。2-θ衍射角和相应的晶面间距d值可说明各峰在X射线粉末衍射图中的位置。用观察的2-θ衍射角和铜K(al)波长根据本领域技术人员熟知的Brag方程计算晶面间距d值。
图1表示维格列汀“A型”晶型的X射线粉末衍射图的实例。X射线数据通过以下实施例1中所述方法获得。
仪器测量衍射的X射线强度(每秒计数,cps),测量针对的是X射线源的角度。只有晶体样品具有明确的衍射角,因此取决于晶型的性质可以观察到尖锐的峰。每种形式都有独特的衍射图。峰强度取决于粒度和形状,因此它是该批的性质而不是该晶型的性质。衍射峰(图)定义每个原子在分子中的位置,定义晶体对称性和给定晶体系统的间距基团(space group)。
应该理解,基于具体使用的衍射仪、分析人员和样品制备技术,观察的2-θ衍射角和晶面间距d值会有轻度的变化。预期相对峰强度变化较大。
对化合物确切晶型的识别应该主要基于观察到的2-θ衍射角,不应强调相对峰强度。
由于在指定2-θ衍射角和晶面间距d值时可能有些误差,所以比较X射线粉末衍射图以识别具体晶型时优选的方法是将未知形式的X射线粉末衍射图覆盖于已知形式的X射线粉末衍射图上。
例如,本领域技术人员可将使用本文所述方法获得的未识别的LAF237 A型的X射线粉末衍射图覆盖于图1上,容易地确定未识别形式的X射线粉末衍射图是否与A型的X射线粉末衍射图基本相同。若X射线粉末衍射图基本与图1相同,先前未知的LAF237晶型可以容易并准确地识别为A型。虽然2-θ衍射角或晶面间距d值是识别晶型的主要方法,但是可能也期望比较相对峰强度。如上所述,相对峰强度可随具体使用的衍射仪和分析人员的样品制备技术而变化。峰强度报告为相对于最强峰峰强度的相对强度。峰强度可用于质量控制,但不应用于晶型的识别。
X射线衍射为定量确定固体混合物中晶型和/或无定形形式的相对量提供了一种方便、实用的方法。X射线衍射可适用于定量应用,因为混合物中给定衍射峰的强度与混合物中相应粉末的份数成正比。可在未知组合物中确定LAF237晶体组分的百分比。
优选对LAF237固体粉末进行测量。未知组分的X射线粉末衍射图可以与已知的含有纯晶型的A型LAF237的定量标准品进行比较以识别A型LAF237的百分比。这可通过将未知固体粉末组合物的衍射图中相对峰强度与从已知纯样品的X射线粉末衍射图获得的校正曲线相比较来进行。可基于LAF237纯晶体样品的最强峰的X射线粉末衍射图对曲线进行校正。校正曲线的建立可采用本领域技术人员已知的方式。例如可以制备不同量的LAF237晶型的五种或更多种人工混合物。在一个非限制性实例中,这种混合物可以含有2%、5%、7%、8%和10%的LAF237的每种晶型。然后使用标准X射线衍射技术获得每个人工混合物的X射线粉末衍射图。如果峰位存在轻度变化,则可通过调整待测量峰的位置进行补偿。然后用每个人工混合物中所选择的特征峰的强度对已知晶型的重量百分比作图。所得图是校正曲线,可确定未知样品中晶体LAF237的量。对于晶体和无定形形式LAF237的未知的混合物,所选择的混合物中的特征峰强度与校正混合物中该峰强度的相对值可用于确定组合物中给定晶型的百分比,剩余的部分确定为无定形材料。
维格列汀“A型”晶型还可以通过红外光谱进行特征描述。本发明的发明人获得的维格列汀“A型”晶型的红外光谱见图2。可以通过如以下实施例7.2.ii中所述的方法获得IR(红外)数据。
在另一方面中,本发明涉及包含维格列汀“A型”晶型的药物组合物。
优选所述配方含有10至150mg、优选25至100mg、最优选50至100mg维格列汀,优选结晶形式的维格列汀、最优选维格列汀“A型”晶型或其药学盐。
优选本发明涉及本文以上所述的药物组合物或压制的片剂,其包含维格列汀的结晶形式、优选“A型”晶型,或在任何情况下它的药学盐。
优选维格列汀的结晶形式或维格列汀“A型”晶型是本文所述的粒子形式。
本发明的包含维格列汀“A型”晶型的药物组合物适合于向哺乳动物包括人类经肠如口服或直肠;透皮和胃肠外施用以治疗由DPP-4抑制剂介导的病症。这种病症包括下文提及的可使用本发明化合物治疗的病症。所述的药物组合物包含有效量的药理学活性的本发明的维格列汀“A型”晶型,其单独使用或与一种或多种可药用载体组合使用。
本发明的药理学维格列汀“A型”晶型可用于制备药物组合物,其包含有效量的维格列汀“A型”晶型与适合于经肠或胃肠外施用的赋形剂或载体的混合物。所述的组合物可以灭菌和/或含有辅剂如防腐剂、稳定剂、湿润剂或乳化剂、助溶剂、渗透压调节剂和/或缓冲剂。此外,它们还可以含有其它治疗上有价值的物质。所述的组合物分别通过常规的混合、制粒或包衣方法制备,含有约0.1-75%、优选约1-50%的维格列汀“A型”晶型。
适合于透皮施用的配方包括治疗有效量的本发明的化合物和载体。优选载体包括可吸收的药理学可接受的溶剂以帮助通过宿主皮肤。典型的透皮装置是绷带形式,其包含支持性部件、含有化合物并任选含有载体的贮器、任选的控速屏障用于在一段持续时间内以控制的、预定的速率向宿主皮肤递送化合物、以及将该装置固定于皮肤上的手段。
维格列汀“A型”晶型或以上定义的包含维格列汀“A型”晶型的药物组合物可以单独施用或与另一种(例如一种或两种)治疗活性剂(在相同或不同的剂量单元中)以例如现有技术中报告的有效治疗剂量组合施用。本文描述的压制片剂或直接压制的片剂或配方也可以包含其它治疗活性剂。这种治疗活性剂包括胰岛素、胰岛素衍生物和类似物、胰岛素促分泌剂如磺酰基脲如格列吡嗪和亚莫利;促胰岛素磺酰脲受体配体如氯茴苯酸类那格列奈和瑞格列奈;胰岛素敏化剂如蛋白质酪氨酸磷酸酶-1B(PTP-1B)抑制剂、GSK3(糖原合成酶激酶-3)抑制剂或RXR配体;双胍类如二甲双胍;格列酮类如匹格列酮或罗格列酮、α-糖苷酶抑制剂如阿卡波糖;GLP-1(胰高血糖素-1)、GLP-1类似物如肠促胰岛素类似物(Exendin-4)和GLP-1类似物;DPPIV(二肽基肽酶IV)抑制剂如异亮氨酸-噻唑烷;DPP728和LAF237,降血脂活性剂如3-羟基-3-甲基-戊二酰辅酶A(HMG-CoA)还原酶抑制剂如洛伐他汀、匹伐他汀、辛伐他汀、帕伐他汀、西立伐他汀、米法斯丁、velostatin、氟伐他汀、达尔伐他汀、阿伐他汀、罗苏伐他汀、fluindostatin和雷伐他汀、角鲨烯合酶抑制剂或FXR(肝X受体)和LXR(法尼酯X受体)配体、消胆胺、氯贝特、烟酸、缬沙坦和阿司匹林。本发明的LAF237“A型”晶型可以与其它活性成分同时、在其它成分之前或之后施用,通过相同或不同施用途径分别施用或在同一药物配方(同一剂量单元)中一起给药。
维格列汀“A型”晶型优选与一种或两种选自于二甲双胍、格列酮类(如匹格列酮或罗格列酮)、胰岛素、磺酰脲类、那格列奈或缬沙坦的化合物组合施用。
在另一方面,本发明涉及本文描述的配方、胶囊剂、片剂、压制的片剂、直接压制的片剂用于治疗病症如非胰岛素依赖性糖尿病、关节炎、肥胖症、同种异体移植、降钙素性骨质疏松、心力衰竭、葡萄糖代谢紊乱、IGT(葡萄糖耐量降低)、神经变性疾病如阿尔茨海默病和帕金森病、调节高脂血症、调节与高脂血症有关的病症或用于降低VLDL、LDL和Lp(a)水平、心血管疾病或肾病如糖尿病性心肌病、左心室或右心室肥大、动脉和/或大血管中层肥大性增厚、肠系膜血管肥大、肾小球膜肥大、用于治疗神经变性病症和认知障碍、产生镇静或抗焦虑作用、用于减小术后分解代谢变化和对应激的激素反应、降低心肌梗塞后死亡率和发病率、治疗与上述作用有关的可能由GLP-1和/或GLP-2水平介导的病症的用途。
在另一方面,本发明涉及立即释放剂型,其中在每日施用一次50mg维格列汀或其盐后10.5小时平均DPP-4抑制至少为79%、优选至少为83%或在83%至94.5%之间或为89.34+/-3.02%。
立即释放剂型在每日施用一次50mg维格列汀或其盐后0.25至10.5小时平均DPP-4抑制为84%至98%。
立即释放剂型在每日施用一次50mg维格列汀或其盐后24小时平均DPP-4抑制为64.2%+/-12.7%。
立即释放剂型在每日施用一次50mg维格列汀或其盐后24小时的DPP-4抑制基本如图7所示。
如上所述的立即释放剂型,其中的剂型是本文描述的任何剂型和要求保护的药物组合物、片剂、压制的片剂。
本发明还涉及一种立即释放剂型,其中每日施用一次100mg维格列汀或其盐后10.5小时平均DPP-4抑制为至少83%、优选至少90%或为90%至95.2%。
立即释放剂型,其中在每日施用一次100mg维格列汀或其盐后0.25至10.5小时平均DPP-4抑制为84%至98.8%。
立即释放剂型,其中在每日施用一次100mg维格列汀或其盐后24小时平均DPP-4抑制为76.3%+/-13.7%。
立即释放剂型,其中在每日施用一次100mg维格列汀或其盐后24小时的DPP-4抑制基本如图7所示。
如上所述的立即释放剂型,其中的剂型是本文描述的任何配方、片剂或胶囊剂。
如上所述的立即释放剂型,其中剂型施用于患有2型糖尿病的患者。
立即释放剂型,其中在每日施用两次50mg维格列汀或其盐后10小时的平均DPP-4抑制为至少75%、优选80%。
立即释放剂型,其中在每日施用两次50mg维格列汀或其盐后24小时平均DPP-4抑制为至少50%、优选60%或64.2%。
立即释放剂型,其中在每日施用两次50mg维格列汀或其盐后10小时平均DPP-4抑制为至少70%、优选80%。
立即释放剂型,其中在每日施用两次50mg维格列汀或其盐后24小时平均DPP-4抑制为至少60%、优选70%或76.3%。
如上所述的立即释放剂型,其中的剂型是本文描述的任何剂型和要求保护的药物组合物、片剂、压制的片剂。
术语“每日施用两次50mg维格列汀或其盐”是指分两次施用维格列汀,其中第二次施用在第一次施用50mg后8至12小时、优选9至11小时进行。
术语“立即释放剂型”是指口服施用单剂量25至200mg维格列汀后其中维格列汀tmax的算术平均值为2.0hr+/-1.9hr或+/-1.4hr的剂型。
在另一实施方案中,本发明提供了:
i)包含约50mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在单剂量口服施用50mg维格列汀后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约77.3ng/mL+/-20.8ng/mL至约195ng/mL+/-89.1ng/mL。
ii)包含约50mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在单剂量口服施用50mg维格列汀后提供的维格列汀AUC(0-∞)算术平均值为约839至约1221ng·h/mL即1030ng·h/mL+/-191ng·h/mL。
iii)包含约50mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在单剂量口服施用50mg维格列汀后提供的维格列汀tmax的算术平均值为2.1hr+/-1.3hr。
iv)包含约50mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,其中所述剂型
- 在单剂量口服施用50mg维格列汀后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约77.3ng/mL+/-20.8ng/mL至约195ng/mL+/-89.1ng/mL,和/或
- 在单剂量口服施用50mg维格列汀后提供的维格列汀AUC(0-∞)算术平均值为约839至约1221ng·h/mL即1030ng·h/mL+/-191ng·h/mL,和/或
- 在单剂量口服施用50mg维格列汀后提供的维格列汀tmax的算术平均值为2.1hr+/-1.3hr。
v)包含约50mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在单剂量口服施用50mg维格列汀后提供的药代动力学特征基本如图3或4所示。
优选施用口服剂量在健康人类个体中进行。
在另一实施方案中,本发明提供:
i)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在单剂量口服施用50mg维格列汀后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约186ng/mL+/-64.9ng/mL至约428ng/mL+/-165ng/mL。
ii)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在单剂量口服施用100mg维格列汀后提供的维格列汀AUC(0-∞)算术平均值为约2071至约2629ng·h/mL即2350ng·h/mL+/-279ng·h/mL。
iii)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在单剂量口服施用100mg维格列汀后提供的维格列汀tmax的算术平均值为2.0hr+/-1.4hr。
iv)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,其中所述剂型
- 在单剂量口服施用50mg维格列汀后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约186ng/mL+/-64.9ng/mL至约428ng/mL+/-165ng/mL,和/或
- 在单剂量口服施用100mg维格列汀后提供的维格列汀AUC(0-∞)算术平均值为约2071至约2629ng·h/mL即2350ng·h/mL+/-279ng·h/mL,和/或
- 在单剂量口服施用100mg维格列汀后提供的维格列汀tmax的算术平均值为2.0hr+/-1.4hr。
v)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在单剂量口服施用100mg维格列汀后提供的药代动力学特征基本如图3或4所示。
优选施用口服剂量在健康人类个体中进行。
在另一实施方案中,本发明提供:
i)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约188ng/mL+/-132ng/mL至约327ng/mL+/-87.6ng/mL。
ii)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后提供的维格列汀AUC(0-24)算术平均值为1840ng·h/mL+/-360ng·h/mL。
iii)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后提供的维格列汀tmax的算术平均值为2.5hr+/-1.3hr。
iv)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,其中所述剂型
- 在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约约188ng/mL+/-132ng/mL至约327ng/mL+/-87.6ng/mL,和/或
- 在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后提供的维格列汀AUC(0-24)算术平均值为1840ng·h/mL+/-360ng·h/mL,和/或
- 在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后提供的维格列汀tmax的算术平均值为2.5hr+/-1.3hr。
v)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后提供的药代动力学特征基本如图5所示。
优选施用口服剂量在患有2型糖尿病的人类个体中进行。
在另一实施方案中,本发明提供:
i)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约123ng/mL+/-51.5ng/mL至约455ng/mL+/-217ng/mL。
ii)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后提供的维格列汀平均AUC(0-∞)值为2090ng·h/mL+/-446ng·h/mL。
iii)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后提供的维格列汀tmax的算术平均值为1hr+/-1.3hr。
iv)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,其中所述剂型
- 在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约123ng/mL+/-51.5ng/mL至约455ng/mL+/-217ng/mL,
和/或
- 在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后提供的维格列汀平均AUC(0-∞)值为2090ng·h/mL+/-446ng·h/mL,
和/或
- 在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后提供的维格列汀tmax的算术平均值为1hr+/-1.3hr。
v)包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后提供的药代动力学特征基本如图6所示。
优选施用口服剂量在患有2型糖尿病的人类个体中进行。
上文i)至v)中所述的包含约100mg或50mg维格列汀的固体剂型,其中的剂型是本文描述的剂型之一和要求保护的药物组合物、片剂、压制的片剂。
在每种情况下,特别是在化合物权利要求中,本申请中参考本文提及的发表物或专利申请引入工作实施例的终产品、终产品终的主体物质、分析和测量方法(如USP文件)、获得正确粒度的方法、药物制剂、赋形剂和权利要求。
本发明通过实施例具体进行举例说明,本发明还涉及实施例中提及的新化合物、它们的用途以及它们的制备方法。
以下实施例用于举例说明本发明而不以任何方式限制本发明。
实施例1
为制备25mg大小的片剂(直接压制的片剂),用对应于以下量的每单元量制备7kg的批量:每单元25mg的1-[3-羟基-金刚烷-1-基氨基)-乙酰基]-吡咯烷-2(S)-甲腈与35.1mg微晶纤维素、17.5mg无水乳糖和1.6mg淀粉乙醇酸钠混合。将各成分在商业料箱混合器中预先混合在一起,然后通过500μm或850μm的筛网。将混合物在料箱混合器中再次混合,然后混加入必需量的硬脂酸镁使每25mg大小的片剂含0.8mg硬脂酸镁。每个步骤中的混合步骤在约150-450转下进行以保证混合物的均匀性。在料箱混合器中再次混合后,可将混合物在常规压片机中压片。25mg片剂的每个片剂重量为80mg。含有50mg活性成分的片剂重160mg,含有100mg活性成分的片剂重320mg。混合物是具有优秀可压性的粉末,可以压制成为期望大小的片剂。
实施例2
以上实施例1中所述的方法可用于制备以下所述的优选50mg片剂(直接压片)。
组分 每单元组成 每批的量
(mg) (kg)
LAF 237药物物质 50.00 65.0
微晶纤维素,PH102(Ph.Eur.,NF) 95.68 124.38
无水乳糖DT(USP,Ph.Eur.) 47.82 62.17
淀粉乙醇酸钠(USP,Ph.Eur.) 4.00 5.2
硬脂酸镁(Ph.Eur,NF) 2.50 3.25
每片或每批总重 200.0 260.0
制备相当于100mg LAF237的片剂,即100mg LAF237、191.36mg微晶纤维素、95.64mg无水乳糖、8mg淀粉乙醇酸钠、5mg硬脂酸镁。
实施例3:如以上实施例所述制备的片剂可进行如下测试。
片剂评价方法
1.平均片重。在分析天平上称重20片片剂,计算平均片重。
2.片剂断裂强度(kilo bond-kp)。施用Schleuniger抗压强度测试仪分别对5个片剂进行测试,计算平均断裂强度。
3.脆碎性(失重%)。使用Roche脆碎性测定仪对准确称重的10片片剂进行10分钟脆碎性测试。除去片剂上面的粉尘,重新称重,计算由于脆碎性导致的失重占起始重量的百分比。
4.分散崩解时间DT(英国药典,1988,Volume II,895页-BP 1988中定义的对可分散片剂的测试)。根据上述定义的可分散片剂的BP测试法(无碟)测试6片片剂。使用的水温为19°-21℃。
5.分散质量。根据BP对可分散片剂的均匀分散测试法(BP 1988 VolumeII,895页),将两片片剂置于19°-21℃ 的100mL水中,使之分散。
颗粒剂评价方法
1.干燥失重(LOD)。颗粒中残余水分含量(LOD)可以用3-4g样品测定,使用Computrac水分分析仪,设定为90℃,根据生产商的程序进行。
2.重量中数直径(WMD)。根据生产商的说明,在Allen Bradley超声筛粉机中将10g颗粒样品在合适的脉冲和筛幅下筛动2分钟。使用300μm、250μm、200μm、150μm、100μm、53μm和40μm的筛。使用计算机程序从筛下颗粒的粒度分布累积百分比计算WMD。
实施例4
改进的生产坚实性
用Carver压片机使用不同配方以及LAF 237与不同赋形剂如微晶纤维素(Avicel PH102)的二元混合物进行初步的压实性能评价。
数据证明我们要求保护的组合物随着压制压力增加(压片力)其片剂强度显著增强。具体而言,例如LAF237和Avicel的混合物的片剂强度显著增强。这些结果表明从压实性能的角度而言,微晶纤维素例如Avicel将是与LAF237组合的优选赋形剂。随着压力(压片力)增加,我们要求保护的配方和所选择的范围在片剂强度方面显著增强。
用仪器化的Korsch单工位压片机进行压实性能研究,该压片机的上冲头和下冲头都带有压力传感器和位移传感器。
这些数据清楚地表明除非用足量的具有优秀压实性能的填充剂稀释,否则LAF237片剂很可能具有较差的片剂硬度/脆碎强度。我们要求保护的配方和选择的范围特别适合于提供所要求的压实性能。微晶纤维素例如Avicel在这方面是很好的填充剂选择。
实施例5:脆碎性
使用Manesty Betapress在6种不同设置下进行评价:牵拉速率设置为66-90rpm(63,000-86,000TPH),牵拉力为7.5-15kN。这些试验使用平底斜面(FFBE)模具,直径9mm的模具用于250mg片剂,直径10mm的模具用于310mg的片剂(根据受试片剂使用其它直径)。片剂总重的选择使得9和10mm FFBE片剂都含有100mg LAF237和具有相同的片剂厚度。脆碎性、压制特征、牵拉速率特征和重量差异是测量的结果。实验设计和实验所得脆碎性结果用于确定影响硬度结果的各变量(配方中的粒度分布、片剂重量、片剂厚度和重量、片剂中的水分含量等)。
实施例7:粒度分布和晶型A(非限制性实施例)
特别适合于制备本文描述的配方、特别是压制的片剂的维格列汀的粒度分布为10至250μm,可如下所述获得该粒度分布:
1.通过应用于直接压制片剂的维格列汀晶型制备粒度分布
本发明的申请人发现了特别适合于直接压制片剂的维格列汀的粒度分布(10至250μm)。
由激光衍射法或等同的方法确定的粒度分布具体如下:x10大于或等于5μm,x50大于或等于35μm,x90小于或等于380μm。
通过夫琅禾费光衍射法测量粒度。
使用的试剂:
助分散剂:例如Antistatic Additive AA3,Shell,约1%的己烷溶液。
分散液:例如异己烷,Merck目录号1.04333,含约1ml助分散剂
设备:
测量装置:例如Sympatec HELOS,Sympatec GmbH,Germany
分散装置:混悬池,例如QUIXEL,Sympatec GmbH,Germany
条件:
焦距:1000mm;光学浓度:≥5%;测量时长:60s;通过测量杯距离:6mm;泵速:15-30%;超声时间:30s。
方法:
原料的分散:向约0.5g的受试物质中加入几滴助分散剂。剧烈混合,例如在涡旋混合器上混合,使受试物质彻底湿润形成光滑、均匀的糊。用分散液稀释该糊至终体积为3-6ml,再次将分散系混合。
测量:制备受试分散系,使用激光衍射仪根据说明手册确定累积体积分布。
可以调整参数使受试分散系具有代表性、均匀、分散充分。
评价/评估:从累积体积分布确定10%、50%和90%筛下物值的粒度(X10,X50,X90)和所述的其它值。
可通过以下所述的方法获得这一粒度分布。这一粒度分布可使用维格列汀的任何形式如无定形维格列汀、结晶形式的维格列汀、优选A型晶型的维格列汀获得。
以下的非限制性实施例将维格列汀A型晶型的制备和随后的机械应力结合起来。
A.维格列汀A型晶型的制备
向装有机械搅拌器的1500ml反应器中加入120克(g)维格列汀(1-[(3-羟基-金刚烷-1-基氨基)-乙酰基]-吡咯烷-2(S)-甲腈)、3.6g活性炭、2.4gcellflock 40、3.6g 1,8-二氮杂二环[5.4.0]十一-7-烯和483g 2-丁酮。
将混合物加热至回流(夹套温度(JT):95℃)并搅拌30分钟。将混合物滤至温热的反应器(JT:75℃)中,用48g 2-丁酮洗涤滤饼。
然后将IT调至70℃,将0.102g所得的重结晶的维格列汀在1.1ml 2-丁酮中的混悬液加至溶液中。将所得混悬液搅拌30分钟,在2小时内冷却至内部温度(IT)为50℃,然后在80分钟内冷却至30℃。最后将混悬液在72分钟内冷却至0℃,再搅拌1小时。此后将混悬液过滤,粗产物用冷的(0℃)37g 2-丁酮和34g叔-丁基甲基醚的混合物洗涤。
粗产物(晶型A)最后在约JT 55℃下减压干燥。
所得的“A型”晶型的维格列汀的粒度分布的物理特征特别适合于通过随后的碾磨步骤获得期望的粒度分布。所得的物质是白色至灰白色晶体粉末。
B.机械应力
在期望粒度范围内的材料可以通过机械应力从无定形维格列汀、结晶形式的维格列汀、“A型”晶型的维格列汀、部分结晶形式的维格列汀、多晶型形式的维格列汀、溶剂合物形式的维格列汀或水合物形式的维格列汀制备。这种应力可以由冲击力、切应力或压制力产生。优选使用“A型”晶型的维格列汀。
在多数可商购获得的碾磨设备中,发生这些原理的组合。对于通过上述结晶方法获得的LAF237晶体而言,优选使用机械冲击磨或气流磨。最优选的机械冲击磨可装备有不同种类的锤头(beater)、叶片(screen)、衬板(liner)或针板(pin plate)。对于我们的方法,优选冲击磨使用板式锤头(platebeater)和5*2.5cm的狭缝叶片(slit screen)。冲击速度应该在20至100m/s之间(外周速度)变化以适应于任何批次与批次之间的差异。在我们的情况中,使用的锤头外周速度为约40-50m/s。
通过将结晶形式、优选“A型”晶型的维格列汀的制备与随后的机械应力如滚筒碾压、碾磨和/或过筛组合,获得了最佳结果(粒度分布)。
2.A型晶体的特征描述
i)X射线粉末衍射(XRPD)
使用的粉末衍射仪是XDS 2000或X1型,Scintag,Santa Clara,USA。
方法:将受试物质置于样品容器中。记录2°至35°(2θ)之间的X射线衍射图,辐射为CuKα辐射(45kV,40mA)。
测量在约45kV和40mA时在以下条件下进行:
扫描速率 0.5°(2θ)/min
斩波器增幅 0.02°
狭缝(从左至右): 2,3,0.3,0.2mm
将受试物质的X射线衍射图中的所有线与参照物质的X射线衍射图中的所有线进行比较。
如果与参照物质比较,强带和中强带的位置和相对强度一致,且没有其它峰,没有无定形背景出现,则受试物质的X射线衍射图与参照物质相符。
重要带列表:约12.0°,13.5°,16.6°,17.1°,17.2°,20.1°,22.5°,27.4°,28.1°
对另一批次的LAF237 A型晶型进行了进一步X射线粉末衍射分析(XRPD)。
| XRPD方法 | |
| 仪器 | X1或XDS2000;Scintag INC |
| 辐射 | CuKα(45kV,40mA) |
| CuKα1 | λ=1.540598 |
| 发散片 | 3mm and 2mm |
| 测量片 | 0.3mm and 0.2mm |
| 斩波器 | 0.02 grd |
| 扫描类型 | 连续扫描 |
| 扫描速率 | 0.5°/min(2θ值) |
| 仪器 | Stoe粉末衍射系统 |
| 辐射 | CuKα(50kV,30mA) |
| CuKα1 | λ=1.540598 |
| 检测器 | 线性PSD |
| 扫描模式 | 传播(Transmission) |
| 扫描范围 | 2°-40°(2θ值) |
A型晶体的最显著衍射峰和从单晶结构计算的结果的列表
| A型晶体,批次0344012 | 从A型晶体的单晶结构计算的值 | ||
| 2θ | d-间距 | 相对 | 2θ |
| (度) | () | 强度 | (度) |
| 10.1710.4211.8313.2916.4716.9617.1417.5518.1719.0419.5819.9720.5121.7622.2822.6423.9124.3224.6925.7326.1526.4627.1627.9329.1231.13 | 8.698.487.486.665.385.225.175.054.884.664.534.444.334.083.993.923.723.663.603.463.413.373.283.193.062.87 | 461911306510031443134612233443410944 | 10.2602510.5365611.9477213.4083516.5815117.0678917.2668017.6701218.3159319.1720619.6859620.0739720.6039321.1635522.3665022.7564624.0278524.4278424.7612025.7315326.2601326.4702027.1805527.8646929.2158531.12078 |
ii)在液体石蜡(Nujol)中的IR光谱
使用的试剂:光谱学液体石蜡(Nujol),例如Uvasol Merck No.107161。KBr或NaCl片。
设备:IR分光光度计,例如Perkin-Elmer 1725-X或Bruker IFS-55。
方法:将受试物质(若需要,还有参照物质)与液体石蜡混合,在最小4000-600cm-1的范围内记录光谱。
若由于过度光散射导致主要吸收带过强或基线过于倾斜,则重复较低浓度的该制剂。
评价/评估:将受试物质光谱中各带的位置和相对强度与参照物质光谱进行比较。
如果各带的位置和相对强度一致,则受试物质的光谱与参照物质的光谱一致。
重要带的列表:
| 波数(cm-1) | 归属 |
| 32932925-2853223816581455/1354125411211054-1035 | v O-H和v N-Hv CH(Nujol的脂族CH)v CN(腈)v C=O叔酰胺δ CH(Nujol的脂族CH)vC-NvC-O(H)v C-O(H)环烷烃3-羟基金刚烷 |
v=伸缩振动
δ=形变振动
对另一批LAF237 A晶型的分析得到以下重要带的列表:
| 波数(cm-1) | 归属 |
| ~3380(宽)/32942993/2915/2849223816571405/135412541120/11021054/1034 | v O-H和v N-Hv CH脂族v CN(腈)v C=O叔酰胺δ CH脂族v C-Nv C-O(H)v C-O(H)环烷烃3-羟基金刚烷 |
v=伸缩振动
δ=形变振动
iii)晶体学分析
已经通过标准晶体学分析成功地澄清了LAF237-NXA、A型晶体的单晶结构。
使用带有CuKα辐射和石墨单色器的Nonius CAD4自动衍射仪收集数据。通过直接法解析结构(SHELXS)。用全部非氢原子的各向异性位移参数经全矩阵最小二乘法精修计算(SHELXL)各参数。随后的傅立叶图谱差值显示所有25个氢原子的位置。从差值图谱上得到氢原子参数,保持固定。所有其它氢原子的参数都理想化,未加以精修。绝对构型由合成给出。
LAF237A型晶体的晶体数据和精修细节
样品参照 LAF237
化学式 C17H25N3O2
fw 303.40
晶体大小,mm 0.59×0.45×0.32
晶体系统 正交晶
空间群 P212121
a, 10.263(1)
b, 10.684(1)
c, 14.564(1)
V,3 1596.9(2)
Z 4
D(calc],g/cm3 1.262
辐射, 1.54178(CuKα)
强度衰减,% ±1
∝,mm-1 0.669
数据采集范围,° 3-74.0
变量数目 199
测量的反射数目 3547
最小二乘反射的数目 3222
R 0.065
最大差值,峰/孔 0.381/-0.245
分子中可区分出三种不同类型的C-N键:长度为1.462至1.475的C-N单键、为1.352的酰胺C-N键和1.129的C-N三键。氮原子N4是sp3杂化的。它的孤电子对作为质子接受体参与分子内氢键。
金刚烷的六元环部分采取几乎完美的椅式构象。吡咯烷环则通过其它四个环原子在平面外与C8585形成轻度变形的信封构象。
表LAF237碱的晶体学数据
| 辐射, | 1.5406 |
| 结晶系统空间群a,b,c,Vcell,3ZDcalc,g cm-3N-H…Ointra aO-H…NaC-H…OaC-H…Na | 正交晶P21212110.263(1)10.684(1)14.564(1)1596.9(2)41.2622.691,109°3.134,175°3.361,137°3.525,167° |
a:对于每个X-H…Y氢键,报告X…Y间距和X-H…Y角度。
LAF237碱的晶格特征是正交晶胞,两个约10的a轴与b轴几乎相等,空间群为P212121。键长和角度都在标准值内。氨基NH基团参与与相邻羰基氧形成的分子内短氢键,N…O和N-H…O值请参见上表。由于该氮原子是sp3杂化的,它的孤电子对在O-H…N分子间接触中是氢键接受体(O…N=3.134,O-H…N=175°),该分子间接触的方向是沿着[001]晶体学方向。固态中有两种其它的较弱相互作用,沿c轴方向的C-H…N接触,和在a方向上与分子结合的C-H…O氢键。这种分子间接触的各向同性分布表明LAF237碱在固态非常稳定。
该化合物在晶格中形成氢键的三维网络,表明该化合物作为晶体相非常稳定。模拟粉末和实验粉末衍射图表明当前的批次是纯相。SEM的形态学预测和实验特征描述得出一些分歧,这已用溶剂作用进行解释。若LAF237碱从2-丙醇中生成,由于(002)面对(011)面的稳定性好,则最终形态学是棱形而不是六角形(如在2-丁酮中)。
3.水吸附/解吸等温线
使用表面测量系统(Surface Measurement Systems)动态蒸汽吸附装置(DVS-1)测量水吸附/解吸等温线。测量在25℃进行。该技术测量样品重量作为相对湿度(RH)的函数。维格列汀A型晶体只有轻微的吸湿性,因为在85%RH下它仅仅吸收0.9%的水分,而无定形样品具有吸湿性,它在85%RH下吸收4.2%的水分。具有吸湿性的形式需要防止接触空气,这样它们才不会吸收水分。水分会引起配方、稳定性和分析方面的问题。因此我们的新维格列汀A型晶体比已知的维格列汀无定形形式又有额外的优势。由于维格列汀易溶于水,使用维格列汀A型晶体在盖仑配方中可改进活性成分的稳定性。
AUC 浓度时间曲线下面积
AUC0-t 血浆浓度时间曲线从0时间至最后一个可定量时间点
t的曲线下面积[ng*hr/mL]
AUC0-inf或 血浆浓度时间曲线从0时间至无穷大时间的曲线下面
AUC(0-∞) 积[ng*hr/mL]
BAPK 生物分析和药代动力学部分
Cmax 最高血浆浓度
CRF 案例报告/记录表格
CRO 临床研究组织
CV 变异系数
ECG 心电图
DPP-4 二肽基肽酶4;二肽基肽酶IV
FMI 最终市场形象
GLP-1 胰高血糖素样肽1
ICH 国际协调委员会
IRB 机构审查委员会
LAF237 维格列汀
LC-MS/MS 液相色谱-质谱/质谱
LOQ 定量限
o.d. 每日一次
PD 药效动力学
PK 药代动力学
p.o 口服
QC 质量控制
SOP 标准操作规程
SD 标准偏差
tmax 达峰Cmax时间
t1/2 消除半衰期
Vd/f 分布容积,经绝对生物利用度校正
实施例8:临床研究获得的DPP-4抑制剂活性如下所述。
研究题目:向接受75g口服葡萄糖耐量测试的2型糖尿病患者单剂量口服给药10、25、50、100、200和400mg维格列汀制剂后评价剂量反应关系的随机、开放、安慰剂对照、七周期的交叉实验。维格列汀以本文所述的剂型即配方、片剂和胶囊剂施用。
目的:
旨在评价维格列汀在接受75g口服葡萄糖耐量测试的2型糖尿病患者中对DPP-IV抑制的剂量依赖性作用。
设计:这是一项随机、开放、安慰剂对照、七周期的交叉实验。14名2型糖尿病患者完成了实验。有29 的筛选期,包括21天的先前使用的降血糖活性剂的洗脱期。先前进行二甲双胍治疗的主体要求进行28天的洗脱期。主体平均禁食血浆葡萄糖水平为7.0-10mmol/L(126-180mg/dl),这是给药前两周内的三天分别进行的三次测量的平均值。筛选HbA1c为7.5-10%。
将合格的研究对象随机分配至14个序列。第一剂量之前有36小时的基线期,7个治疗周期结束前至少3个星期的隔离停留期,在最后的药效动力学评价之后进行实验结束评价。研究对象进食标准BDA餐,在第-1天进行基线评价。
剂量间间隔72小时。在给药当日,在禁食过夜后向研究对象施用指定的剂量。研究对象在给药后30分钟口服摄入75g的葡萄糖。在特定时间进行药代动力学和药效动力学取样。
在给药当日,研究对象不吃早餐。给药后5.5hr和10hr分别进标准午餐和晚餐。在剩余的停留天数内,研究对象保持进标准BDA餐。实验结束评价在第7个治疗周期最后的药效动力学评价结束后进行。
研究对象数目:
十四名研究对象完成了实验。在该实验中共向十六名研究对象给药。其中有2名中断实验,14名完成实验。
入组标准
患有2型糖尿病且疾病持续至少3个月的男性和非生育女性(即绝经后、子宫切除后或经输卵管结扎绝育的女性),年龄为30至70岁,愿意进行3周的降血糖药物洗脱期。在洗脱期最后两周进行的三次评价的平均禁食血浆葡萄糖水平应该为7.0至10mmol/L(126-180mg/dL),筛选HbA1c为7.5-10%,C-肽>0.3nmol/L,体重指数≤40kg/m2。
研究药物:维格列汀
治疗持续时间:每个研究对象在每个治疗周期内随机接受单剂量的10mg、25mg、50mg、100mg、200mg、400mg维格列汀和安慰剂。连续治疗的给药间隔为72小时。
评价标准
安全性和耐受性:安全线和耐受性评价包括生命征象、ECG、生化、血液学、尿检,如下所述。
●血液学;血液化学;尿检:筛选、基线、第3和第5周期的预给药以及
实验结束评价
●潜血检测:筛选、基线、第3和第5周期以及实验结束
●不良反应;共同用药/重要的非药物治疗:从第一次施用实验药物至实验
结束
药代动力学
OGTT看作是0小时。
●采集血样进行LAF237的测定[每个样品1mL血液,肝素试管(血浆)]:-0.5hr(给药维格列汀之前)、OGTT后0.5、1.5、5和8hr
●分析物、介质和方法:血浆中的维格列汀,用LC-MS/MS;LOQ约为2ng/mL
●LAF237的PK参数:AUC、AUC0-t、t1/2、Cmax、tmax,、CL/F药效动力学
AM给药时间~0800hr
AM OGTT时间在给药后~0830hr
注:以下列出的所有PD时间都是w.r.t.OGTT
血浆DPP-IV肽酶活性(1mL血液样品)
在每个给药日:
OGTT前1hr和0.75hr
OGTT后:-0.25、0(OGTT前)、0.25、0.5、1、1.5、2、4、6、8、10、12、16和24hr。
统计学方法
葡萄糖、胰岛素、胰高血糖素和GIP的药效动力学参数AUE和Emax的统计学比较基于方差分析。对于两个参数,使用线性混合作用模型分析对数转化数据,包括治疗、周期和序列作为固定因素,序列内患者作为随机因素。对于每次比较,为原始规模的给药方法比例提供点估计和90%的置信区间。给药组和安慰剂组之间的比较是主要的分析。也进行其它分析以在各活性给药组之间进行比较。
数据分析
在施用维格列汀之前和之后的各时间点测量DPP-4活性,直至24小时。图7表示DPP-4抑制的百分比。DPP-4抑制的百分比从测量的DPP-4活性根据以下方程计算:

其中DPP-4活性(t)是在t时间测量的DPP-4活性,DPP-4活性(0)是在施用维格列汀之前测量的基线DPP-4活性。
DPP-4抑制的平均持续时间(MRT)从每个给药方案DPP-4抑制的百分比与时间之间的关系上进行估计,使用WinNonlin(4.1版,Pharsight,CA)基于非房室模型进行分析。DPP-4抑制的平均持续时间使用以下方程进行估计:

通过用DPP-4百分比对时间曲线下的面积除以时间间隔来估算24小时间隔内的平均DPP-4抑制。使用以下方程计算24小时内的平均DPP-4抑制:

实施例9:
进行一项开放、单剂量、四周期、四组给药、随机的交叉实验以比较25、50、100和200mg维格列汀(使用本文描述的配方、片剂和胶囊剂)在健康志愿者中的血浆浓度,每个周期之间有2天的洗脱期。共有20名健康志愿者入选,20名都完成了全部实验步骤和给药。对研究对象在21天的期间内进行筛选,若合格,则在每组给药前继续进行基线访问供进行四次基线评价)。在离开实验中心之前进行实验结束评价。研究对象随机分配至4个给药序列组,每个序列有5名研究对象。每个周期首次给药之前至少12个小时研究对象就入住实验中心进行基线评价,每个周期给药后至少24小时都限制于研究中心内。给药间隔至少2天后,每个研究对象回到实验中心接受根据他们随机时间表进行的其它的给药。所有的研究对象在实验期间都根据随机时间表接受一次25、50、100和200mg的给药。
在每个给药组给药后24小时内采集用于测定维格列汀的血浆样品。对于所有的给药周期,研究对象在给药前10小时至给药后4小时禁食。完成全部安全性和药代动力学评价后认为研究对象完成了实验。
单剂量口服给药25、50、100和200mg维格列汀后收集血样测定药代动力学。使用维格列汀的血浆浓度根据非房室模型确定药代动力学参数,数据总结于图3和图4和表1中。
表1.单剂量口服给药25、50、100和200mg FMI片剂后维格列汀药代动力学参数的算术平均值
| 剂量(mg) | tmax(h)中数(min,max) | Cmax(ng/mL)平均值±SD(CV%) | AUC0-t(h.ng/ml)平均值±SD(CV%) | AUC0-∞(h·ng/mL)平均值±SD(CV%) | t1/2(h)平均值±SD(CV%) |
| 25 | 1.5(1.0,6.0) | 117±41(35) | 453±91(20) | 461±91(20) | 1.7±0.35(21) |
| 50 | 1.5(0.5,6.0) | 245±87(36) | 1020±193(19%) | 1030±191(19) | 2.3±1.42(62) |
| 100 | 1.75(0.75,6.0) | 505±120(24) | 2330±278(12%) | 2350±279(54) | 2.5±1.34(54) |
| 200 | 1.25(0.75,4.0) | 1100±280(26) | 5060±722(14) | 5080±721(14) | 3.1±1.06(34) |
实施例10
在2型糖尿病患者中进行一项开放的3周期实验以比较单独给药或组合给药100mg维格列汀qd与1000mg二甲双胍qd 5天后它们之间的药代动力学药物-药物相互作用。共有17名健康志愿者入选,全部完成了全部实验步骤和给药。首次给药之前至少12个小时研究对象就入住实验中心进行基线评价,整个实验期间都限制于研究中心内。在第二周期的最后一天(第20天)对研究对象进行实验结束评价。对于所有的给药周期,研究对象在给药前10小时至给药后4小时禁食。完成全部安全性和药代动力学评价后认为研究对象完成了实验。
在24小时内采集用于测定维格列汀和二甲双胍药代动力学的血液样品用于药代动力学评价。药代动力学特征见图5。使用非房室模型确定药代动力学参数,数据总结于表2中。
表2.在2型糖尿病患者中给药多剂量的维格列汀100mg qd FMI片剂后稳态药代动力学参数
| 给药 | Tmax(h)中数(min,max) | Cmax(ng/mL)平均值±SD(CV%) | AUC0-24(h·ng/mL)平均值±SD(CV%) | CL/F(L/h)平均值±SD(CV%) | t1/2(h)平均值±SD(CV%) |
| LAF237单独(N=17)LAF237+二甲双胍(N=17) | 1.00(0.50,4.00)2.50(0.50,4.00) | 467±134(29)381±103(27) | 1960±413(21)1840±360(20) | 53.3±11.3(21)56.6±12.3(22) | 1.68±0.259(15)1.86±0.689(37) |
实施例11
该实验是一项开放、三周期、多剂量的设计,旨在评价向2型糖尿病患者多次单独给药或组合给药100mg维格列汀qd与45mg匹格列酮qd 28天和7天后它们之间的药代动力学药物-药物相互作用。筛选后,共有15名患者入选,全部完成了实验。首次给药之前至少12个小时研究对象就入住实验中心进行基线评价。若研究对象在基线水平满足全部合格标准,则将他们随机分配至实验中。所有的实验药物都在早餐前30分钟服用。在给药周期最后一天进行实验结束评价,完成实验。
对于单独给药或与匹格列酮组合给药,分别在第7天和第28天的24小时内采集用于测定维格列汀的药代动力学血液样品。单独给药或与匹格列酮组合给药的维格列汀药代动力学见图6。使用非房室模型确定药代动力学参数,数据总结于表3中。
表3.单独给药或与匹格列酮组合给药后稳态维格列汀的药代动力学参数
| 给药 | Tmax(hr)中数(Min,Max) | Cmax(ng/mL)平均值±SD(CV%) | AUC0-24(ng/mLxhr)平均值±SD(CV%) | AUC0-inf(ng/mLxhr)平均值±SD(CV%) | t1/2(hr)平均值±SD(CV%) | CL/F(L/hr)平均值±SD(CV%) |
| LAF237单独LAF237+匹格列酮 | 1.75(0.5-4.0)1.00(0.5-4.0) | 531±115(22)505±117(35) | 2190±425(19)2080±448(22) | 2210±425(19)2090±446(21) | 3.71±2.14(58)3.82±1.64(43) | 47.5±10.3(22)50.6±12.7(25) |
Claims (102)
1.一种压制的药物片剂或直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂的粒子,其中片剂中至少40%、优选60%或80%的粒度分布低于250μm。
2.一种压制的药物片剂或直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂的粒子,其中片剂厚度与片剂重量的比例为0.002至0.06mm/mg、优选为0.01至0.03mm/mg。
3.一种压制的药物片剂或直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂的粒子,其中:
i)片剂中至少40%、优选60%的粒度分布为10至250μm,且
ii)片剂厚度与片剂重量的比例为0.002至0.06mm/mg或0.01至0.03mm/mg。
4.一种压制的药物片剂或直接压制的药物片剂,其中所述分散系含有包含游离形式或酸加成盐形式的DPP-IV抑制剂、优选维格列汀的粒子,其中:
i)片剂中至少40%、优选60%的粒度分布为10至250μm,
ii)在25℃、60%RH下一周后片剂中水分含量低于10%,且
iii)片剂厚度与片剂重量的比例为0.002至0.06mm/mg。
5.权利要求1-4中任一项的压制的药物片剂或直接压制的药物片剂,其中片剂中的粒度分别为50至150μm。
6.权利要求1-5中任一项的压制的药物片剂或直接压制的药物片剂,其中在25℃、60%RH下一周后片剂中水分含量低于5%。
7.权利要求1-6中任一项的压制的药物片剂或直接压制的药物片剂,其中片剂厚度与片剂重量的比例为0.01至0.03mm/mg。
8.权利要求1-7中任一项的压制的药物片剂或直接压制的药物片剂,其中片剂中至少60%、优选至少80%或90%的粒度分布为10至250μm。
9.权利要求1-7中任一项的压制的药物片剂或直接压制的药物片剂,其中片剂中至少25%或至少35%的粒度分布为50至150μm。
10.权利要求1-9中任一项的压制的药物片剂或直接压制的药物片剂,其中所述片剂还含有另一种治疗活性剂,优选二甲双胍、格列酮类或缬沙坦。
11.权利要求1-10中任一项的压制的药物片剂或直接压制的药物片剂,其中
i)在0至10分钟释放85至99.5%的活性成分,且
ii)在10至15分钟释放90至99.5%的活性成分。
12.权利要求1-10中任一项的压制的药物片剂或直接压制的药物片剂,其中片剂中药物赋形剂的粒度分布为5至400μm。
13.一种药物组合物,其中所述分散系含有包含DPP-IV抑制剂或其药学盐的粒子,其中:
i)组合物中至少40%、优选60%的粒度分布低于250μm,和/或
ii)组合物中至少40%、优选60%的粒度分布为10至250μm,和/或
iii)组合物中至少60%、优选至少80%的粒度分布为10至250μm,和/或
iv)组合物中至少25%或至少35%的粒度分布为50至150μm。
14.权利要求13的组合物,其中组合物中药物赋形剂的粒度分布为5至400μm。
15.权利要求1-12中任一项的压制的药物片剂或直接压制的药物片剂,或权利要求13或14中任一项的药物组合物,其中DPP-IV抑制剂选自于1-{2-[(5-氰基吡啶-2-基)氨基]乙基氨基}乙酰基-2(S)-氰基-吡咯烷二盐酸盐、维格列汀、L-苏-异亮氨酰基噻唑烷、MK-0431、GSK23A、BMS-477118、3-(氨基甲基)-2-异丁基-1-氧代-4-苯基-1,2-二氢-6-异喹啉甲酰胺和2-{[3-(氨基甲基)-2-异丁基-4-苯基-1-氧代-1,2-二氢-6-异喹啉基]氧基}乙酰胺以及在任何情况下任选的其药学盐。
16.权利要求1-12中任一项的压制的药物片剂或直接压制的药物片剂,或权利要求13或14中任一项的药物组合物,其中DPP-IV抑制剂选自于维格列汀、结晶形式的维格列汀、或“A型”晶型的维格列汀或其药学盐。
17.权利要求1-12或权利要求15至16中任一项的压制的药物片剂,其是直接压制的药物片剂。
18.一种压制的片剂、优选直接压制的片剂,其包含DPP-IV抑制剂或在任何情况下其药学盐。
19.一种压制的片剂、优选直接压制的片剂,其包含维格列汀、结晶形式的维格列汀、或“A型”晶型的维格列汀或在任何情况下其药学盐。
20.制备权利要求1至12或15至19中任一项的单元剂型形式的、压制的片剂、优选直接压制的片剂的方法,其包含:
(a)将以干重计为以下重量%的以下物质混合:
(i)以干重计为5-60重量%或以干重计为6-60重量%的DPP-IV抑制剂,其中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂的粒度分布低于250μm或优选为10至250μm;或至少25%或至少35%的粒度分布为50至150μm,和
(ii)至少一种选自于稀释剂、崩解剂和润滑剂的赋形剂,以形成制片粉末形式的DPP-IV抑制剂组合物,其能够压制成、优选直接压制成片剂;和
(b)将步骤(a)中制备的组合物压制成为单元剂型形式的压制的DPP-IV抑制剂片剂。
21.制备权利要求1至12或15至19中任一项的单元剂型形式的、压制的片剂、优选直接压制的片剂的方法,其包含:
(a)将以干重计为以下重量%的以下物质混合:
(i)以干重计为25-35重量%、优选20-35重量%的DPP-IV抑制剂,其中至少40%、优选60%、最优选80%、更优选90%的DPP-IV抑制剂的粒度分布低于250μm或优选为10至250μm;或至少25%或至少35%的粒度分布为50至150μm。
(ii)以干重计为40-95%、优选40-80重量%的可药用稀释剂;
(iii)以干重计为0-10重量%的可药用崩解剂;和
(iv)以干重计为0.25-6重量%的可药用润滑剂,以形成制片粉末形式的DPP-IV抑制剂组合物,其能够压制成、优选直接压制成片剂;和
(b)将步骤(a)中制备的组合物压制成为单元剂型形式的压制的DPP-IV抑制剂片剂。
22.权利要求21的方法,其中混合的组合物包含:
(i)以干重计为20-35重量%或25-30重量%的游离形式或酸加成盐形式的DPP-IV抑制剂,
(ii)以干重计为25-70%或优选35-50重量%的可药用微晶纤维素如AvicelPH102;
(iii)以干重计为5-40重量%或优选18-35重量%的可药用乳糖;
(iv)以干重计为0-10重量%或优选1-4重量%的可药用淀粉乙醇酸钠;和
(v)以干重计为0.25-6重量%或优选0.5-4重量%的可药用硬脂酸镁。
23.权利要求20的方法,其中步骤(a)中使用的混合的组合物选自于包含以下成分的组合物:
(i)以干重计为5-60重量%或优选25-35重量%的游离形式或酸加成盐形式的DPP-IV抑制剂,
(ii)以干重计为40-95%或40-80重量%或优选62-78重量%的可药用稀释剂;
(iii)以干重计为0-20重量%的可药用崩解剂;和任选地
(iv)以干重计为0.1-10重量%的可药用润滑剂。
24.权利要求23的方法,其中所述组合物包含:
i)一种或两种选自于微晶纤维素和乳糖的稀释剂
ii)两种稀释剂微晶纤维素和乳糖
iii)以干重计为25-70重量%、优选35-55重量%的可药用微晶纤维素,或
iv)以干重计为25-70重量%、优选35-55重量%的可药用微晶纤维素和以干重计为5-40重量%、优选18-35重量%的乳糖。
25.权利要求20至24中任一项的方法,其中DPP-IV抑制剂选自于1-{2-[(5-氰基吡啶-2-基)氨基]乙基氨基}乙酰基-2(S)-氰基-吡咯烷二盐酸盐、维格列汀、L-苏-异亮氨酰基噻唑烷、MK-0431、GSK23A、BMS-477118、3-(氨基甲基)-2-异丁基-1-氧代-4-苯基-1,2-二氢-6-异喹啉甲酰胺和2-{[3-(氨基甲基)-2-异丁基-4-苯基-1-氧代-1,2-二氢-6-异喹啉基]氧基}乙酰胺,以及在任何情况下任选的它们的药学盐。
26.权利要求20至24中任一项的方法,其中DPP-IV抑制剂是维格列汀、优选结晶形式的维格列汀、最优选“A型”晶型的维格列汀或在任何情况下的其药学盐。
27.前述权利要求中任一项的药物组合物、压制的药物片剂、直接压制的药物片剂或方法,其中维格列汀或其药学盐是无定形态或结晶形式。
28.前述权利要求中任一项的药物组合物、压制的药物片剂、直接压制的药物片剂或方法,其中DPP-IV抑制剂是维格列汀“A型”晶型或其药学盐。
29.维格列汀或其药学盐的结晶形式。
30.权利要求29的结晶形式,其是热力学上最稳定的维格列汀晶型。
31.维格列汀的结晶形式(“A型”晶型),其特征为X射线衍射图具有在约16.6°、17.1°、17.2°+/-0.3度2-θ的峰。
32.维格列汀的结晶形式(“A型”晶型),其特征为X射线衍射图具有在约12.0°、13.5°、16.6°、17.1°、17.2°、20.1°、22.5°、27.4°、28.1°+/-0.3度2-θ的峰。
33.权利要求32或31的结晶形式,其特征为它的X射线粉末衍射图基本如图1所示。
34.维格列汀的结晶形式(“A型”晶型),其特征为在液体石蜡中的IR光谱具有以下以倒数波数(cm-1)表示的显著的吸收带:约3293cm-1、2925-2853cm-1、2238cm-1、1658cm-1、1455/1354cm-1、1254cm-1、1121cm-1、1054-1035cm-1+/-2cm-1。
35.权利要求34的结晶形式,其特征为在液体石蜡中的IR光谱具有基本如图2所示的以倒数波数(cm-1)表示的吸收带。
36.维格列汀的结晶形式(“A型”晶型),其特征为它的熔点是147℃+/-4℃、优选约149℃+/-2℃。
37.权利要求29至36中任一项的维格列汀的结晶形式用于制备相应的维格列汀无定形形式的用途。
38.权利要求30至36中任一项的维格列汀“A型”晶型用于制备维格列汀的另一种晶体(多晶型)形式或制备相应的维格列汀无定形形式或在任何情况下其盐的用途。
39.一种制备维格列汀多晶型形式的方法,其中使用维格列汀“A型”晶型作为原料或结晶过程的中间体。
40.一种制备维格列汀或其盐的结晶形式的方法,该方法包括以下步骤:
i)加热维格列汀或其盐在有机溶剂中的溶液,
ii)诱导维格列汀结晶,和
iii)回收维格列汀晶体。
41.一种制备维格列汀“A型”晶型的方法,该晶型的X射线衍射图具有在16.6°、17.1°、17.2°+/-0.3度2-θ,优选在12.0°、13.5°、16.6°、17.1°、17.2°、20.1°、22.5°、27.4°、28.1°+/-0.3度2-θ的峰,该方法包括以下步骤:
i)加热维格列汀在有机溶剂中的溶液,
ii)诱导维格列汀结晶,和
iii)回收维格列汀晶体。
42.权利要求40或41所述的方法,其中的溶剂选自于2-丁酮、2-丙醇/乙酸乙酯、2-丙醇、丙酮。
43.权利要求40或41的方法,其中的结晶包括以下步骤:
i)加热维格列汀在有机溶剂中的溶液,所述有机溶剂优选选自于2-丁酮、2-丙醇/乙酸乙酯、2-丙醇、丙酮,
ii)将溶液冷却至约-20℃至约20℃,优选至约-10℃至约10℃以诱导结晶,和
iii)回收维格列汀晶体,优选不加热。
44.权利要求40至43中任一项的方法,可通过向溶液中加入反溶剂诱导结晶ii)。
45.权利要求41至44中任一项的方法,其中至少40%、优选60%、更优选80%的所得维格列汀“A型”晶型的粒度分布低于250μm,优选为10至250μm。
46.权利要求29至30中任一项的结晶形式,其中至少40%、优选60%、最优选80%的维格列汀晶型的粒度分布低于250μm,优选为10至250μm。
47.权利要求31至36中任一项的结晶形式,其中至少40%、优选60%、最优选80%的维格列汀“A型”晶型的粒度分布低于250μm,优选为10至250μm。
48.一种药物组合物,其包含:
(a)游离形式或酸加成盐形式的DPP-IV抑制剂;
(b)可药用稀释剂,
其中在单元剂型中,DPP-IV抑制剂的重量与稀释剂重量的比例为0.5至0.25,优选0.4至0.28;
且其中DPP-IV抑制剂是维格列汀结晶形式、优选维格列汀“A型”晶型或在任何情况下的其药学盐。
49.一种药物组合物,其包含:
(a)游离形式或酸加成盐形式的DPP-IV抑制剂;
(b)可药用稀释剂,
其中在单元剂型中,DPP-IV抑制剂的重量与稀释剂重量的比例为0.5至0.25,优选0.4至0.28;
且其中所述组合物分散系含有包含DPP-IV抑制剂或其药学盐的粒子,其中:
i)组合物中至少40%、优选60%的粒度分布低于250μm,和/或
ii)组合物中至少40%、优选60%的粒度分布为10至250μm,和/或
iii)组合物中至少60%、优选80%的粒度分布为10至250μm,和/
或
iv)组合物中至少25%或至少35%的粒度分布为50至150μm。
50.权利要求48或49的组合物,其中的稀释剂选自于微晶纤维素和乳糖。
51.权利要求48至50中任一项的组合物,其中至少一种稀释剂是微晶纤维素,且其中在单元剂型中,DPP-IV抑制剂的重量与微晶纤维素的重量的比例为2至0.333,优选为1至0.333,最优选0.7至0.333。
52.权利要求48至51中任一项的组合物,其除微晶纤维素稀释剂外还包含乳糖作为稀释剂。
53.权利要求49至52中任一项的组合物,其中DPP-IV抑制剂选自于1-{2-[(5-氰基吡啶-2-基)氨基]乙基氨基}乙酰基-2(S)-氰基-吡咯烷二盐酸盐、(S)-1-[(3-羟基-1-金刚烷基)氨基]乙酰基-2-氰基-吡咯烷、L-苏-异亮氨酰基噻唑烷、MK-0431、GSK23A、BMS-477118、3-(氨基甲基)-2-异丁基-1-氧代-4-苯基-1,2-二氢-6-异喹啉甲酰胺和2-{[3-(氨基甲基)-2-异丁基-4-苯基-1-氧代-1,2-二氢-6-异喹啉基]氧基}乙酰胺,以及在任何情况下的任选的它们的药学盐。
54.权利要求49至52中任一项的组合物,其中DPP-IV抑制剂是维格列汀或其药学盐。
55.前述权利要求中任一项的药物组合物、压制的药物片剂或直接压制的药物片剂,其包含20至120mg、优选25至100mg或50至100mg的维格列汀或其可药用酸加成盐。
56.权利要求48至55中任一项的组合物,其进一步包含:
(c)以干重计为0-20重量%的可药用崩解剂;
(d)以干重计为0.1-10重量%的可药用润滑剂。
57.权利要求48至56中任一项的组合物,其进一步包含:
(c)以干重计为1-6重量%的可药用崩解剂;
(d)以干重计为0.25-6重量%的可药用润滑剂。
58.权利要求48至57中任一项的组合物,其进一步包含:
(c)以干重计为1-4重量%的可药用淀粉乙醇酸钠;和
(d)以干重计为0.5-4重量%的可药用硬脂酸镁。
59.权利要求48至58中任一项的组合物,其中DPP-IV抑制剂是结晶形式的维格列汀、优选“A型”晶型的维格列汀或在任何情况下的其药学盐。
60.权利要求1至12中任一项的压制的药物片剂或直接压制的药物片剂,或包含权利要求48至59中任一项的组合物。
61.一种压制的药物片剂、优选直接压制的药物片剂,其包含游离形式或酸加成盐形式的DPP-IV抑制剂。
62.权利要求61的压制的药物片剂或直接压制的药物片剂,其中DPP-IV抑制剂选自于1-{2-[(5-氰基吡啶-2-基)氨基]乙基氨基}乙酰基-2(S)-氰基-吡咯烷二盐酸盐、(S)-1-[(3-羟基-1-金刚烷基)氨基]乙酰基-2-氰基-吡咯烷、L-苏-异亮氨酰基噻唑烷、MK-0431、GSK23A、BMS-477118、3-(氨基甲基)-2-异丁基-1-氧代-4-苯基-1,2-二氢-6-异喹啉甲酰胺和2-{[3-(氨基甲基)-2-异丁基-4-苯基-1-氧代-1,2-二氢-6-异喹啉基]氧基}乙酰胺,以及在任何情况下的任选的它们的药学盐。
63.权利要求61的压制的药物片剂或直接压制的药物片剂,其中DPP-IV抑制剂是维格列汀、结晶形式的维格列汀、优选“A型”晶型的维格列汀或其药学盐。
64.前述权利要求中任一项的方法、药物组合物、压制的药物片剂或直接压制的药物片剂,其中DPPIV抑制剂粒子、尤其是维格列汀粒子包含60%以上、最优选90%或95%以上、更优选98%以上的DPPIV抑制剂,尤其是维格列汀。
65.DPP-IV抑制剂用于制备压制的或直接压制的片剂的用途,其中至少40%的DPP-IV抑制剂的粒度分布低于250μm,优选为10至250μm。
66.权利要求65的用途,其中DPP-IV抑制剂选自于1-{2-[(5-氰基吡啶-2-基)氨基]乙基氨基}乙酰基-2(S)-氰基-吡咯烷二盐酸盐、维格列汀、L-苏-异亮氨酰基噻唑烷、MK-0431、GSK23A、BMS-477118、3-(氨基甲基)-2-异丁基-1-氧代-4-苯基-1,2-二氢-6-异喹啉甲酰胺和2-{[3-(氨基甲基)-2-异丁基-4-苯基-1-氧代-1,2-二氢-6-异喹啉基]氧基}乙酰胺,以及在任何情况下的任选的它们的药学盐。
67.权利要求65的用途,其中的DPP-IV抑制剂是维格列汀、结晶形式的维格列汀、优选“A型”晶型的维格列汀或在任何情况下的其药学盐。
68.权利要求65至67中任一项的用途,其中60%、优选80%的DPP-IV抑制剂的粒度分布为10至250μm。
69.一种药物组合物,其包含维格列汀的结晶形式、优选“A型”晶型。
70.前述权利要求中任一项的药物组合物、压制的药物片剂或直接压制的药物片剂,其中至少1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或98%的维格列汀化合物是结晶形式,优选A型晶型。
71.一种药物组合物,其中低于1%、或低于0.4%的维格列汀是A型晶型,高于99%或99.6%的维格列汀是无定形形式。
72.前述权利要求中任一项的药物组合物、压制的药物片剂或直接压制的药物片剂,其中低于1%、或低于0.4%的维格列汀是A型晶型,高于99%或99.6%的维格列汀是无定形形式。
73.包含维格列汀“A型”晶型和一种或两种治疗活性剂或在任何情况下的其药学盐的组合。
74.包含维格列汀“A型”晶型和一种或两种选自于二甲双胍、格列酮类、胰岛素、磺酰脲类、那格列奈或缬沙坦的治疗活性剂的组合。
75.权利要求74的组合,其中格列酮类是匹格列酮或罗格列酮。
76.权利要求73至75中任一项的组合,其中活性成分在同一药物组合物中给药或在不同的剂量单元中给药。
77.前述权利要求中任一项的维格列汀粒子,其中所述粒子包含60%以上、最优选90%或95%以上、更优选98%以上的维格列汀。
78.前述权利要求中任一项的药物组合物、压制的药物片剂或直接压制的药物片剂,其包含20至120mg、优选25至100mg或50至100mg的维格列汀或其可药用酸加成盐。
79.前述权利要求中任一项的药物组合物、压制的药物片剂或直接压制的药物片剂,其包含50至100mg的维格列汀或其可药用酸加成盐。
80.前述权利要求中任一项的药物组合物、压制的药物片剂或直接压制的药物片剂,其中,在0.01N HCl溶液中,
i)在0至10分钟释放85至99.5%的活性成分,且
ii)在10至15分钟释放90至99.5%的活性成分,
或
i)在0至10分钟释放88至99.5%的活性成分,且
ii)在10至15分钟释放95至99.5%的活性成分,
或
i)在0至10分钟释放89至94%的活性成分,且
ii)在10至15分钟释放96至99%的活性成分。
81.前述权利要求中任一项的药物组合物或压制的药物片剂,其中组合物还包含另一种治疗活性剂,优选二甲双胍、格列酮类或缬沙坦。
82.一种立即释放剂型,其在向2型糖尿病患者每日施用一次50mg维格列汀或其盐10.5小时后,平均DPP-4抑制为至少79%、优选至少83%或为83%至94.5%或89.34+/-3.02%。
83.一种立即释放剂型,其在向2型糖尿病患者每日施用一次50mg维格列汀或其盐后0.25至10.5小时之间,平均DPP-4抑制为84%至98%。
84.一种立即释放剂型,其在向2型糖尿病患者每日施用一次50mg维格列汀或其盐后24小时,平均DPP-4抑制为64.2%+/-12.7%。
85.一种立即释放剂型,其在向2型糖尿病患者每日施用一次50mg维格列汀或其盐后24小时,DPP-4抑制基本如图7所示。
86.权利要求82至85中任一项的立即释放剂型,其中所述剂型是权利要求1至19、27、28、48至64、69至72或78至81中任一项的药物组合物、片剂或胶囊剂。
87.一种立即释放剂型,其在向2型糖尿病患者每日施用一次100mg维格列汀或其盐10.5小时后,平均DPP-4抑制为至少83%、优选至少90%或为90%至95.2%。
88.一种立即释放剂型,其在向2型糖尿病患者每日施用一次100mg维格列汀或其盐后0.25至10.5小时之间,平均DPP-4抑制为84%至98.8%。
89.一种立即释放剂型,其在向2型糖尿病患者每日施用一次100mg维格列汀或其盐后24小时,平均DPP-4抑制为76.3%+/-13.7%。
90.一种立即释放剂型,其在向2型糖尿病患者每日施用一次100mg维格列汀或其盐后24小时,DPP-4抑制基本如图7所示。
91.权利要求97至90中任一项的立即释放剂型,其中所述剂型是权利要求1至19、27、28、48至64、69至72或78至81中任一项的药物组合物、片剂或胶囊剂。
92.包含约50mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,其中所述剂型:
-在单剂量口服施用50mg维格列汀后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约77.3ng/mL+/-20.8ng/mL至约195ng/mL+/-89.1ng/mL,和/或
-在单剂量口服施用50mg维格列汀后提供的维格列汀AUC(0-∞)算术平均值为约839至约1221ng·h/mL,即1030ng·h/mL+/-191ng·h/mL,和/或
-在单剂量口服施用50mg维格列汀后提供的维格列汀tmax的算术平均值为2.1hr+/-1.3hr。
93.包含约50mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在单剂量口服施用50mg维格列汀后提供的药代动力学特征基本如图3或4所示。
94.包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,其中所述剂型
-在单剂量口服施用50mg维格列汀后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约186ng/mL+/-64.9ng/mL至约428ng/mL+/-165ng/mL,和/或
-在单剂量口服施用100mg维格列汀后提供的维格列汀AUC(0-∞)算术平均值为约2071至约2629ng·h/mL,即2350ng·h/mL+/-279ng·h/mL,和/或
-在单剂量口服施用100mg维格列汀后提供的维格列汀tmax的算术平均值为2.0hr+/-1.4hr。
95.包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在单剂量口服施用100mg维格列汀后提供的药代动力学特征基本如图3或4所示。
96.权利要求92至95中任一项的固体口服剂型,其中施用口服剂量在健康人类个体中进行。
97.包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,其中所述剂型
-在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约188ng/mL+/-132ng/mL至约327ng/mL+/-87.6ng/mL,和/或
-在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后提供的维格列汀AUC(0-24h)算术平均值为1840ng·h/mL+/-360ng·h/mL,和/或
-在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后提供的维格列汀tmax的算术平均值为2.5hr+/-1.3hr。
98.包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在同时单剂量口服施用100mg维格列汀和1000mg二甲双胍后提供的药代动力学特征基本如图5所示。
99.包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,其中所述剂型
-在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后约0.5至约6小时之间提供的维格列汀最高血浆浓度的算术平均值为约123ng/mL+/-51.5ng/mL至约455ng/mL+/-217ng/mL,和/或
-在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后提供的维格列汀平均AUC(0-∞)值为2090ng·h/mL+/-446ng·h/mL,和/或
-在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后提供的维格列汀tmax的算术平均值为1hr+/-1.3hr。
100.包含约100mg维格列汀游离碱或相应量的它的可药用盐和载体介质的固体剂型,所述剂型在同时单剂量口服施用100mg维格列汀和45mg匹格列酮后提供的药代动力学特征基本如图6所示。
101.权利要求97至100中任一项的固体口服剂型,其中施用口服剂量在患有2型糖尿病的人类个体中进行。
102.权利要求92至101中任一项的固体口服剂型,其中所述口服剂型是权利要求1至19、27、28、48至64、69至72或78至81中任一项的药物组合物、片剂或胶囊剂的形式。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US64464505P | 2005-01-18 | 2005-01-18 | |
| US60/644,645 | 2005-01-18 | ||
| US60/690,484 | 2005-06-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN101106977A true CN101106977A (zh) | 2008-01-16 |
Family
ID=39000509
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2006800025892A Pending CN101106977A (zh) | 2005-01-18 | 2006-01-17 | 直接压片的配方及方法 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN101106977A (zh) |
| ZA (1) | ZA200704693B (zh) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103610661A (zh) * | 2013-11-22 | 2014-03-05 | 迪沙药业集团有限公司 | 一种组合物 |
| TWI483943B (zh) * | 2009-02-13 | 2015-05-11 | Boehringer Ingelheim Int | 醫藥組合物、醫藥劑型、其製備方法及其用途 |
| CN104856970A (zh) * | 2015-06-23 | 2015-08-26 | 孙丽华 | 一种治疗ⅱ型糖尿病的维格列汀片剂 |
| CN105193752A (zh) * | 2015-10-27 | 2015-12-30 | 石家庄康贺威药业有限公司 | 一种维格列汀片剂及其制备方法 |
| CN109350603A (zh) * | 2018-12-28 | 2019-02-19 | 珠海和凡医药股份有限公司 | 一种治疗痛风的压制药物片剂 |
| CN112957355A (zh) * | 2021-04-07 | 2021-06-15 | 烟台万润药业有限公司 | 一种维格列汀片剂及其制备方法 |
-
2006
- 2006-01-17 CN CNA2006800025892A patent/CN101106977A/zh active Pending
-
2007
- 2007-06-06 ZA ZA200704693A patent/ZA200704693B/en unknown
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI483943B (zh) * | 2009-02-13 | 2015-05-11 | Boehringer Ingelheim Int | 醫藥組合物、醫藥劑型、其製備方法及其用途 |
| CN103610661A (zh) * | 2013-11-22 | 2014-03-05 | 迪沙药业集团有限公司 | 一种组合物 |
| CN103610661B (zh) * | 2013-11-22 | 2017-09-12 | 威海迪素制药有限公司 | 一种组合物 |
| CN104856970A (zh) * | 2015-06-23 | 2015-08-26 | 孙丽华 | 一种治疗ⅱ型糖尿病的维格列汀片剂 |
| CN105193752A (zh) * | 2015-10-27 | 2015-12-30 | 石家庄康贺威药业有限公司 | 一种维格列汀片剂及其制备方法 |
| CN105193752B (zh) * | 2015-10-27 | 2018-03-30 | 石家庄康贺威药业有限公司 | 一种维格列汀片剂及其制备方法 |
| CN109350603A (zh) * | 2018-12-28 | 2019-02-19 | 珠海和凡医药股份有限公司 | 一种治疗痛风的压制药物片剂 |
| CN112957355A (zh) * | 2021-04-07 | 2021-06-15 | 烟台万润药业有限公司 | 一种维格列汀片剂及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| ZA200704693B (en) | 2008-05-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7194153B2 (ja) | 新規製剤 | |
| AU2006206670B2 (en) | Direct compression formulation and process | |
| KR101259645B1 (ko) | 직접 압축 제제 및 방법 | |
| EP1893236A2 (en) | Direct compression formulation of dpp-iv inhibitors and glitazones, and process | |
| CN101106977A (zh) | 直接压片的配方及方法 | |
| CN101618216B (zh) | 直接压片配方和方法 | |
| MX2008003974A (en) | Formulation comprising metformin and vildagli ptin |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20080116 |