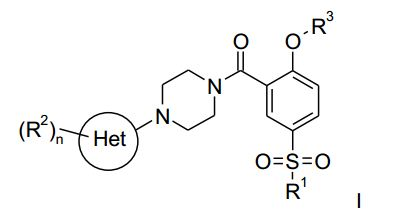
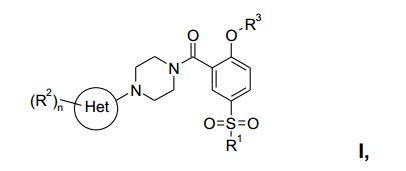
[0001] A presente invenção refere-se a uma nova síntese escalá- vel de compostos da fórmula geral:
na qual: Het é um grupo heteroarila de 6 membros, contendo um, dois ou três átomos de nitrogênio; R1 é (Ci-Ce)-alquila, (C3-C6)-cicloalquila, NR4R5 ou (Ci- Ce)-alquila substituída por halogênio; R2 é hidróxi, halogênio, NO2, CN, (Ci-Ce)-alquila, (Cs-Cβ)- cicloalquila, (Ci-C6)-alquila substituída por halogênio, (Ci-C6)-alquila substituída por hidróxi, (CH2)o-(Ci-C6)-alcóxi, (Ci-Ce)-alcóxi substituída por halogênio, NR4R5, C(O)R6ou SO2R7; R3 é (Ci-C6)-alquila, (C3-C6)-cicloalquila ou (Ci-C6)-alquila substituída por halogênio; R4 e R5, independentemente um do outro, são hidrogênio ou (Ci-Ce)-alquila; R6 é hidrogênio, (Ci-Ce)-alquila, (Ci-C6)-alcóxi ou NR4R5; R7 é (Ci-C6)-alquila, (Ci-C6)-alquila opcionalmente substituída por halogênio, (CH2)o-(C3-C6)-cicloalquila, (CH2)o-(C3-C6)-alcóxi ou NR4R5; n é 1,2 ou 3; 0 é 0, 1 ou 2; e a sais de adição de ácido farmaceuticamente aceitáveis dos mesmos.
[0002] O composto mais preferido, preparado pela nova síntese escalável, é o composto de fórmula:
[0003] Conforme definido na fórmula I, o termo "halogênio" denota cloro, iodo, flúor e bromo.
[0004] O termo "alquila" denota uma cadeia de carbono linear ou ramificada, contendo 1 a 6 átomos de carbono.
[0005] O termo "alcóxi"denota um grupo, no qual o resíduo de alquila é conforme definido acima, e que está ligado via um átomo de oxigênio.
[0006] O termo "heteroarila de 6 membros contendo um, dois ou três átomos de nitrogênio"denota um radical aromático monovalente, por exemplo, piridila, pirazinila, pirimidinila, piridazinila ou 1,3,5- triazinila.
[0007] O termo "alcóxi substituído com halogênio" denota um resíduo de alcóxi, conforme definido acima, em que pelo menos um átomo de hidrogênio é substituído por halogênio.
[0008] O termo "alquila substituída por halogênio" denota um resíduo de alquila, conforme definido acima, em que pelo menos um átomo de hidrogênio é substituído por halogênio, por exemplo, os seguintes grupos: CF3, CHF2, CH2F, CH2CF3, CH2CHF2, CH2CH2F, CH2CH2CF3, CH2CH2CH2CF3, CH2CF2CF3, CH2CF2CHF2, CF2CHFCF3, C(CH3)2CF3, CH(CH3)CF3OU CH(CH2F)CH2F.
[0009] O termo "alquila substituída por hidróxi" denota um resíduo de alquila, conforme definido acima, em que pelo menos um átomo de hidrogênio é substituído por um grupo hidróxi, por exemplo, CH(OH)CH3, CH2CH(OH)CH3, CH2CH(CH3)CH2OH, (CH2)2OH, (CH2)3OH OU CH2C[(CH3)]2-CH2OH.
[00010] O termo "sais de adição de ácido farmaceuticamente aceitáveis" engloba sais com ácidos inorgânicos e orgânicos, tais como ácido clorídrico, ácido nítrico, ácido sulfúrico, ácido fosfórico, ácido cítrico, ácido fórmico, ácido fumárico, ácido maleico, ácido acético, ácido succínico, ácido tartárico, ácido metanossulfônico, ácido p- toluenossulfônico e similares.
[00011] A presente invenção refere-se a uma nova e eficiente síntese em 5 etapas escaláveis, para compostos de fórmula geral I, que são bons inibidores do transportador 1 de glicina (GlyT-1) e que são seletivos para inibidores de transportador 2 de glicina (GlyT-2).
[00012] Inibidores de transportadores de glicina são adequados para o tratamento de distúrbios neurológicos e neuropsiquiátricos. A maioria dos estados patológicos implicados são psicoses, esquizofrenia (Armer RE e Miller DJ, Exp. Opin. Ther. Patents, 11 (4): 563-572, 2001), distúrbios de humor psicóticos, tais como distúrbio depressivo principal severo, distúrbios de humor associados a distúrbios psicóticos, tais como mania ou depressão agudas, associadas a distúrbios bipolares e distúrbios de humor, associados à esquizofrenia (Pralong ET et al., Prog. Neurobiol., 67: 173-202, 2002), distúrbios autísticos (Carlsson ML, J. Neural Trans,. 105: 525-535, 1998), distúrbios cognitivos, tais como demências, incluindo demência relacionada à idade e demência senil do tipo Alzheimer, distúrbios de memória em um mamífero, incluindo um ser humano, distúrbios de déficit de atenção e dor (Armer RE e Miller DJ, Exp. Opin. Ther. Patents, 11 (4): 563-572, 2001).
[00013] A indicação mais preferida para compostos de fórmula I é esquizofrenia.
[00014] A esquizofrenia é uma doença neurológica progressiva e devastadora caracterizada por sintomas positivos episódicos, tais como delírios, alucinações, distúrbios de pensamento e psicose, e por sintomas negativos persistentes, tais como afeto atenuado, atenção prejudicada e retiro social, e prejuízos cognitivos (Lewis DA e Lieberman JA, Neuron, , 28:325-33, 2000). Durante décadas, a pesquisa se focalizou sobre a hipótese de "hiperatividade dopaminérgica", que conduziu a intervenções terapêuticas envolvendo o bloqueio do sistema dopaminérgico (Vandenberg RJ e Aubrey KR., Exp. Opin. Ther. Targets, 5(4): 507-518, 200T, Nakazato A e Okuyama S, et al., Exp. Opin. Ther. Patents, 10(1): 75-98, 2000). Essa abordagem farmacológica enfrenta pobremente sintomas negativos e cognitivos, que são os melhores indicadores do resultado funcional (Sharma T., Br. J. Psychiatry, 174 (supl. 28): 44-51, 1999).
[00015] Os compostos de fórmula I são compostos conhecidos, descritos no documento de número WO 2005/014563.
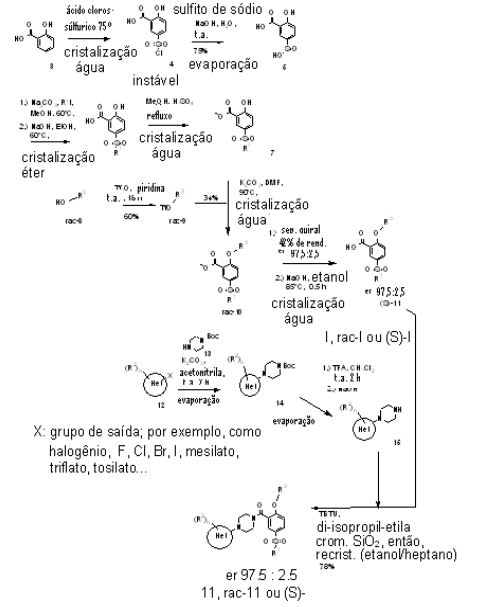
[00016] Os compostos, aqui descritos, têm sido preparados, por exemplo, de acordo com o seguinte esquema geral 1:
Esquema I
[00017] Os compostos de fórmula geral I têm sido preparados por reação de derivados de piperazina de fórmula 15 com um ácido correspondente de fórmula 11, na presença de um agente de ativação como TBTU (tetrafluoroborato de (2-(1 H-benzotriazol-1-il)-1,1,3,3- tetrametil-urônio). Derivados de piperazina de fórmula 15 têm sido preparados por aquecimento da piperazina 13 N-protegida correspon-dente com HetX 12, na presença de uma base, seguido por clivagem do grupo protetor. O grupo protetor era, tipicamente, t-butóxi-carbonila (Boc).
[00018] Mais especificamente, um composto de fórmula I poderia ser preparado, conforme descrito no Esquema I, em uma síntese de 12 etapas.
[00019] A síntese do bloco de construção de piperazina 15 partiu de um composto de fórmula 12, por exemplo, a partir de 2,3-dicloro-5- triflúor-metil-piridina ou a partir do oneroso composto 2-CI, 3-F, 5- triflúor-metil-piridina via troca de halogênio para formar 14. A substituição nucleofílica com a onerosa Boc-piperazina 13 e subsequente desproteção de Boc forneceu o derivado de piperazina 15, em cerca de 23 a 30%.
[00020] As principais desvantagens da síntese mostrada acima, com respeito à escalabilidade, eram: a) o manejo de ácido clorossulfúrico para a preparação de 4, b) a instabilidade de 4, c) o baixo rendimento global para formar 7, d) a separação por HPLC quiral de 10, e) a síntese onerosa e de baixo rendimento de 14, f) a purificação muito difícil de 14, g) a onerosa Boc-piperazina 13 e h) a purificação cromatográfica do bloco de construção 15.
[00021] Objeto da presente invenção é uma nova, curta e eficiente síntese escalável de inibidores de Glyt-1 de fórmula I, especialmente para o composto específico [4-(3-flúor-5-triflúor-metil-piridin-2-il)- piperazin-1-il]-[5-metanos-sulfonil-2-(S)-(2,2,2-triflúor-1-metil-etóxi)-fenil]- metanona com uma fonte barata e uma síntese barata e praticável dos materiais de partida.
[00022] Esse problema foi solucionado por uso da síntese conforme descrita nos Esquemas 2 e 3:
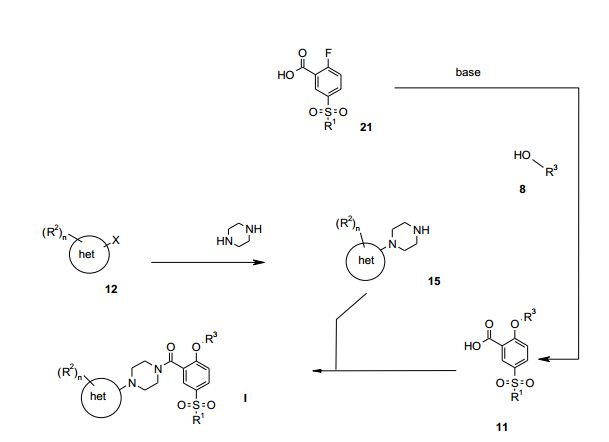
[00023] Compostos racêmicos de fórmula I pode ser preparada de acordo com o Esquema 2: Esquema 2
em que X é um grupo de saída, tal como halogênio (F, Cl, Br, I, mesilato, triflato ou tosilato), R1, R2, het e n são conforme descritos acima e R3é (C1-C6)-alquila ou (C1-C6)-alquila substituída por halo- gênio.
[00024] Os S-enanciômeros correspondentes podem ser preparados de acordo com o Esquema 3.
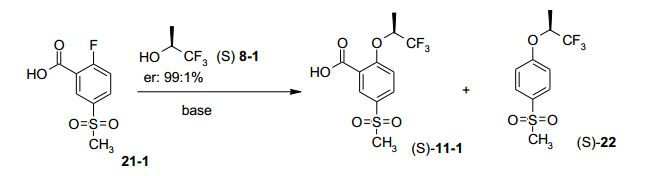
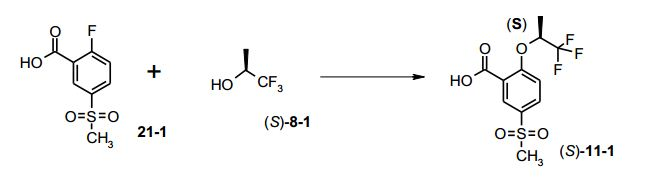
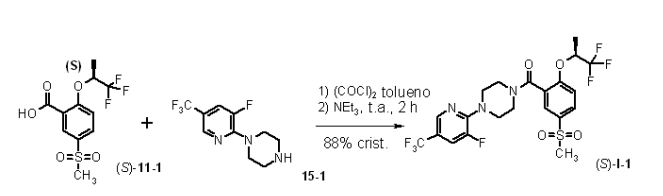
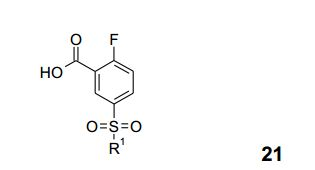
[00025] Uma nova, curta e eficiente síntese em 5 (2+2+1) etapas escaláveis, para o inibidor de Glyt-1 de fórmula I, foi estabelecida por substituição da síntese conhecida não-escalável. A síntese se inicia com a transformação do ácido flúor-metanossulfonil-benzóico 21 ou 21- 1 para formar o derivado de ácido benzóico 11, rac 11 ou (SJ-11-1, aplicando HOR3 8 ou triflúor-isopropanol (S)-8-1. (S)-8-1 é produzido via a redução assimétrica de trifluoroacetona (16) com levedura de Baker em rendimento de 83% depois de destilação, ou via redução assimétrica com catalisadores de Ru. O bloco de construção de piperazina 15 ou 15-1 é sintetizado em duas etapas a partir de um composto de fórmula 12 ou 12-1, tal como dicloro-triflúor-metil-piridina (12-1). A reação de 12- 1 com CsF e K2CO3 em NMP dá a diflúor-triflúor-metil-piridina correspondente (12-2), a qual, depois de reação com piperazina, conduziu a 15 ou 15-1. O acoplamento de 15 ou 15-1 com 0 cloreto de ácido correspondente de 11, rac-11 ou (SJ-11-1 fornece, depois de cristalização, 0 composto final I, rac-l ou (S)-l-1, em rendimento global de cerca de 74%.
[00026] Esse novo processo é descrito abaixo em mais detalhes: 1. Redução Assimétrica de Trifluoroacetona (16):
a) com levedura de Baker:
[00027] Embora (S)-8-1 fosse preparado de maneira bem sucedida por resolução de racemato enzimática, 0 desenvolvimento da redução assimétrica de 16 com levedura da Baker foi continuada para diminuir 0 custo de processo. O objetivo foi também aumentar a pureza enan- ciomérica de (S)-8-1 por otimização da biotransformação catalisada por levedura. A levedura de Baker comprada de Klipfel AG foi escolhida (dentre aproximadamente 60 leveduras testadas) como biocatalisa- dor, por razões de custo e seletividade. Um pré-tratamento com calor da levedura, a cerca de 50°C durante duas horas, aumentou 0 ee desde 96 até > 99%. A otimização de parâmetros resultou em um proces- so em escala de 10 L com uma concentração de substrato de 3% (p/v) e com rendimentos em biotransformação de cerca de 83 a 96%, depois de 5 a 6 dias. O principal subproduto formado durante o tratamento com calor da levedura, seguido pela biotransformação descrita acima, foi o etanol. Um processo de isolamento de produto foi desenvolvido com base unicamente em destilação e retificação. (S)-Triflúor- isopropanol (S)-8-1 (< 0,1% em etanol) altamente purificado foi obtido como um azeótropo com 5% de água. Depois de aumento de escala do processo para a escala de 800 L, 21,8 Kg (rendimento isolado de 83%) de (S)-triflúor-isopropanol (S)-8-1, (er = 99,7 : 0,3) foram produzidos.
[00028] Em adição à redução microbiana, o potencial químico de álcool desidrogenases (ADH) isoladas foi também investigado. Foi factível produzir quantidades em g tanto do enanciômero S quanto do enanciômero R, em elevado excesso enanciomérico. O (S)-8-1 necessário foi obtido de maneira reprodutível com um er > 99,5 : 0,5. No entanto, como a melhor ADH a partir de Sacharomyces cerivisiae é vendida unicamente como uma enzima diagnóstica (Roche Penzberg), a enzima foi considerada mais do que onerosa demais.
b) com catalisadores:
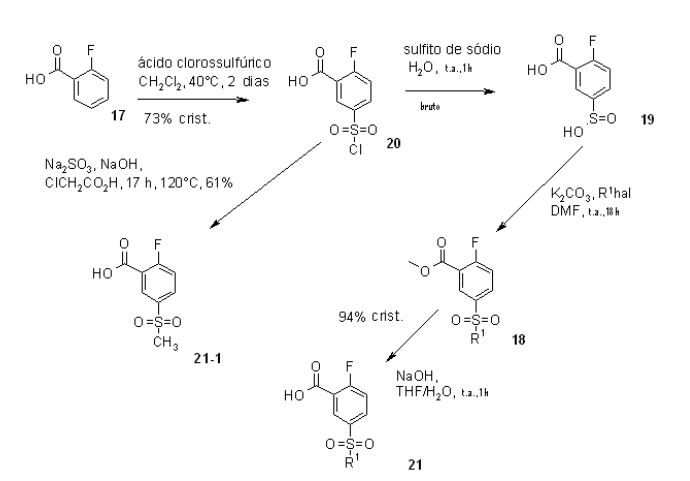
[00029] (S)-1,1,1-Triflúor-2-propanol (S)-8-1, química e enanciome- ricamente puro, também pode ser preparado por um hidrogênio assimétrico de 1,1,1-triflúor-acetona com complexos de rutênio-fosfina na ausência de uma base e de um aditivo. 2. Síntese de Material de Saída 21 ou 21-1:
[00030] A correção de problemas intensiva da sequência de 4 etapas a partir de ácido 2-flúor-benzóico (17) a 21 deu-se em um rendimento global a partir de 17 a 50%. O principal aperfeiçoamento foi conseguido por otimização das condições de reação para formar 19 com sulfato de sódio, seguido pela reação de alquilação com R1hal (hal = I, Cl, Br) produzindo 21 depois de saponificação e cristalização. Em uma reação não-otimizada, um procedimento de pote único a partir de 20 a 21-1 foi demonstrado por aplicação de sulfito de sódio em NaOH a 32% seguida pelo tratamento de CICH2CO2H produzindo 21-1 em 61%, análogo ao documento de número W002/07238. 3. Síntese Aperfeiçoada de (S)-11-1:
[00031] Várias condições foram testadas para aperfeiçoar a condi- ção não-técnica original durante a conversão de 21-1 para formar (S)- 11-1 por aplicação K2CO3 (3 eq.) e (S)-triflúor-isopropanol (5 eq.) em DMA, irradiação com micro-ondas a 150°C durante duas horas, produzindo cerca de 35% até 71%. Devido à volatilidade do (S)-triflúor- isopropanol, a reação foi realizada em um vaso fechado. Substituindo K2CO3 (3 eq., restando 40% de material de partida) por CS2CO3 (3 eq.), a reação foi completada dentro de 3 horas a 150°C, sem irradiação. Temperatura de reação mais baixa e menos CS2CO3 conduziu a tempo de reação mais longo (até 20 horas). O foco dos inventores era reduzir a quantidade do (S)-triflúor-isopropanol oneroso, resultando na redução de (S)-8-1 a partir de 5 eq. para 1,25 eq. As condições aplicadas CS2CO3 (1,9 eq.) e (S)-triflúor-isopropanol (1,4 eq.) em DMA a 120°C 0,15 MPa (1,5 bar), durante 72 horas, forneceram, depois de processamento, cristais brancos de (S)-11-1 em 84-90%. Tempo de reação estendido (90 horas) a 150°C e 0,5 MPa (5 bar), conduziram ao produto descarboxilado (S)-22 (até 30%), 0 qual foi separado do intermediário desejado (S)-11 -1 via extração básica.
[00032] Em detalhes, a reação é realizada com 1 a 5 eq. de bases como Na2COs, K2CO3, LÍ2CO3 ou CS2CO3, de preferência, 2-3 eq. de CS2CO3, em solventes de elevado ponto de ebulição, como NMP ou DMA, de preferência, DMA em temperatura, por exemplo, na faixa entre 60°C e 0 ponto de ebulição do solvente, de preferência, entre 100°C e 150°C, durante 1 a 90 horas, de preferência, 24 - 48 horas, ou com 1 a 5 eq. de bases, como NatOBu, LitOBu ou KtOBu, de preferência, 1 a 1,5 eq. de KtOBu em solventes, por exemplo, DMF ou THF, de preferência, THF, em temperatura, por exemplo, na faixa entre 0°C e 0 ponto de ebulição do solvente, de preferência, entre 20°C e 50°C, durante 1 a 30 horas, de preferência, 3 a 8 horas. 4. Procedimento Otimizado para Diflúor-triflúor-metil-piridina (12-2):
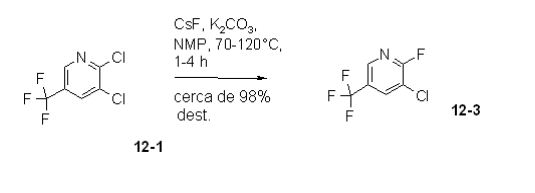
[00033] A síntese da 12-2 foi elaborada partindo-se do composto de dicloro 12-1 correspondente ou do composto de cloro-flúor 12-3 muito oneroso. A reatividade do átomo de cloro na posição 2 de 12-1 é significativamente elevada, comparada com o átomo de cloro na posição 3. Com base na conhecida questão de segurança de DMSO, em combinação com bases como K2CO3, em temperaturas elevadas como 120°C, DMSO foi substituído por N-metil-pirrolidinona (NMP). A reação heterogênica é muito sensível à água. Traços de água conduziram a tempo de reação mais longo e/ou conversão incompleta. Tempo de reação mais longo (maior do que 17 horas, a 120°C) ou temperatura mais elevada conduziram, devido à instabilidade do produto 12-2, a vários subprodutos desconhecidos, findando como um alcatrão negro no vaso de reação. Além disso, foi necessário trabalhar com solvente livre de água. Uma quantidade substancial de CsF foi necessária para essa reação. CsF é muito hidroscópico e contaminou a mistura de rea-ção com água. Portanto, para eliminar água completamente da mistura de reação, uma quantidade definida de NMP foi evaporada antes da adição do composto de dicloro 12-1 à suspensão de K2CO3 e CsF em NMP.
[00034] Durante o aumento de escala, foi difícil controlar a reação e para conseguir 12-2 livre de solvente, puro, fora da mistura de reação devido à pequena diferença entre os pontos de ebulição de 12-3 e 12- 2. Sob condições de destilação otimizadas, foi possível obter material em uma razão de 12-3 a 12-2 de cerca de 0,3 a 99,7 contendo DMSO. 5. Síntese Curta do Bloco de Construção de Piperazina 15:
em que X é um grupo de saída, tais como F, Cl, Br, I, mesilate, triflato ou tosilato.
[00035] De maneira usual, o bloco de construção de piperazina 15 pode ser sintetizado por aplicação de 12, bases como 1 a 3 eq. de K2CO3, de preferência, 1,5 eq., e 1 a 3 eq. da onerosa Boc-piperazina, de preferência, 1,1 eq., em solventes como, por exemplo, THF, toluene, acetonitrila, de preferência, acetonitrila em temperatura, por exemplo, na faixa entre 0°C e 0 ponto de ebulição do solvente, de preferência, entre 40°C e 70°C, durante 1 a 16 horas, de preferência, 3 horas. A subsequente desproteção com Boc sob as condições de ácido triflú- or-acético em CH2CI2, à temperatura ambiente, durante 3 horas e processamento básico, forneceu 15 em cerca de 88%, sobre duas etapas.
[00036] Além disso, uma modificação do procedimento de desproteção com Boc usando HCl em MeOH, à temperatura ambiente, durante 3 horas, forneceu o 15 HCl cristalino, em rendimento global de 93%, o qual foi diretamente usado como sal de HCl, na etapa de acoplamento final. Devido a seu preço elevado, Boc-piperazina foi substituída pela barata acetil-piperazina, fornecendo 24, em 91%, depois de cristalização. A desproteção com N-acetila, usando NaOH aquoso em MeOH, sob refluxo, durante 18 horas, conduziu a 15, em 99% de rendimento.
[00037] Um procedimento de uma etapa utilizando piperazina barata, em solventes como, por exemplo, THF, tolueno, acetonitrila, de preferência, THF, em temperatura, por exemplo, na faixa entre 0°C e o ponto de ebulição do solvente, de preferência, à temperatura ambiente, durante 1 a 16 horas, de preferência, 1 hora, foi finalmente desenvolvido, fornecendo o 15 bruto, depois de processamento aquoso em rendimento quantitativo.
[00038] O 15 bruto foi diretamente acoplado com 11, rac-11 ou (S)- 11-1 na etapa final, para dar API I, rac-l ou (S)-l. Vários reagentes de acoplamento, como TBTU, HBTU, CDI e EDCI (em DMF, THF ou CH2CI2) foram testados para esse tipo de acoplamento, por meio do quê, em todos os casos, uma purificação cromatográfica foi necessária para se conseguir composto final puro de fórmula I, rac-l ou (S)-l em 35 a 78% de rendimento. O acoplamento via anidrido misto, utilizando cloroformiato de etila em CH2CI2, forneceu, depois de cristalização, 0 I, rac-l ou (S)-l, em 75 a 80%.
[00039] De acordo com 0 novo processo acima descrito, as seguintes vantagens sobre 0 procedimento conhecido podem ser fornecidas: - A síntese foi encurtada de 12 para 5 etapas. - O rendimento global aumentou desde cerca de 7% para 74%. - Uma fonte barata e uma síntese barata e praticável dos mate-riais de partida 21,15 e (S)-8-1 foram identificadas. - O uso da onerosa piperazina 13 protegida foi evitada. - Um procedimento eficiente foi desenvolvido para sintetizar o composto 15. - Todas a purificações cromatográficas foram eliminadas.
[00040] As seguintes abreviações foram usadas no relatório descritivo e nas reivindicações: TBTU (2-(1 H-benzotriazol-1 -il)-1,1,3,3-tetrametil-urônio- tetraflúor-borato) NMP N-metil-pirrolidinona DMF N,N-dimetil-formamida TFA ácido triflúor-acético DMA dimetilamina THF tetra-hidrofurano DMSO sulfóxido de metila CDI 1,T-carbonil-di-imidazol EDCI 1 -(3-dimetil-amino-propil)-3-etil-carbodi-imida Exemplo 1 Ácido 5-metanossulfonil-2-(2,2,2-triflúor-1- metil-etóxi)-benzóico ((S)- 11-1)
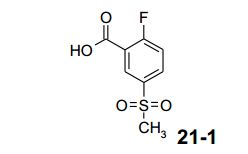
[00041] Uma solução incolor de 700,0 g de ácido 2-flúor-5- metanossul-fonil-benzóico (21-1, 3,2 mols) em 7,7 L de N,N-dimetil- acetamida foi tratada com 1.965,0 g de carbonato de césio (6,0 mols) e 522,8 g de (S)-triflúor-isopropanol (S)-8-1 (4,5 mols). A suspensão de reação branca foi aquecida para 120°C e agitada sob argônio durante 72 horas 0,15 MPa (1,5 bar).
[00042] Depois de resfriamento para 20°C, a suspensão branca foi filtrada, a torta de filtração foi lavada com 500 ml_ de N,N-dimetil- acetamida e o filtrado foi evaporado. Ao resíduo, foram adicionados 9 L de água e a solução foi extraída 3 vezes com 7 L, no total com 21 L, de acetato de etila. A fase aquosa foi aquecida no evaporador rotativo para remover completamente acetato de etila residual da fase aquosa. O pH da fase aquosa foi ajustado para 1,5 por adição de 600 ml_ de HCI a 37%, por meio do qual o produto se precipitou. A suspensão foi agitada à temperatura ambiente durante uma hora, filtrada, os cristais foram lavados com 5 L de água e secados sob vácuo elevado durante 24 horas, a 50°C, para dar 840,0 g (84,0%) de (S)-11 -1 como cristais brancos. Análise por HPLC 99,6 área-% de (S)-11 -1. er = 99,2 : 0,8% (HPLC) ou:
[00043] Equipamento: 500 mL de vaso de camisa dupla, equipado com uma sonda de temperatura, um agitador mecânico, um resfriador e um suprimento de gás inerte.
[00044] Uma solução incolor de 65,5 g de ácido 2-flúor-5-metanos- sulfonil-benzóico (21-1, 300 mmols) em 300 mL de THF foi tratada à temperatura ambiente com 38,0 g de (S)-triflúor-isopropanol (S)-8-1 (330 mmols). A mistura de reação foi tratada dentro de uma hora com uma solução de 71,4 g de KOtBu (630 mmols) em 300 mL de THF (reação exotérmica). A suspensão amarelo-clara foi aquecida para 50°C dentro de uma hora e agitada sob argônio durante duas horas.
[00045] À mistura de reação, foram adicionados, a 50°C, dentro de 15 minutos, 48 g de ácido fórmico. O solvente da mistura foi evapora- do (50°C, 30-15 KPa (300 - 150 mbar). Ao resíduo, foram adicionados 40 ml_ de EtOH, agitou-se durante 5 minutos a 40°C e tratou-se dentro de 5 minutos, a 46 a 48°C, com 150 ml_ de água, agitou-se durante 5 minutos e foram adicionados, dentro de 20 minutos, a 46 a 48°C, outros 350 ml_ de água. A solução foi resfriada, dentro de uma hora, para 20°C, e agitou-se durante duas horas. A suspensão formada foi filtrada, os cristais foram lavados duas vezes com 50 ml_ de água e secados sob vácuo elevado durante 18 horas, a 45°C, para fornecer 91,6 g (91,5%) de (S)-11-1, como cristais brancos. Exemplo 1b
[00046] Equipamento: frasco de fundo redondo de quatro gargalos de 100 ml_, equipado com uma sonda de temperatura, um agitador mecânico e um suprimento de gás inerte.
[00047] A uma suspensão branca de 1,0 g de ácido 2-flúor-5- metanossulfonil-benzóico (21-1, 4,6 mmols) em 30 ml_ de 2-propanol 8-2, foram adicionados 4,5 g de carbonato de césio (13,8 mols). A suspensão de reação branca foi aquecida para 80°C e agitada sob argon io durante 67 horas. O solvente da mistura de reação foi evaporado e o resíduo foi tratado com 20 ml_ de CH2CI2 e 10 mL de água. O pH da fase aquosa foi ajustado para 1,5 por adição de cerca de 14 mL de HCI a 2 N. Depois de extração, as fases foram separadas e a fase aquosa foi extraída duas vezes com 10 mL de CH2CI2. As fases orgânicas combinadas foram evaporadas, para se conseguir 0 produto bruto em rendimento quantitativo. A cristalização a partir de EtO- Ac/hexano forneceu 1,02 g de 11-2, como cristais brancos, em 87%. (Análise por HPLC > 98 área-%)- Exemplo 1c
[00048] Equipamento: frasco de fundo redondo de quatro gargalos de 500 mL, equipado com uma sonda de temperatura, um agitador mecânico, um resfriador e um suprimento de gás inerte.
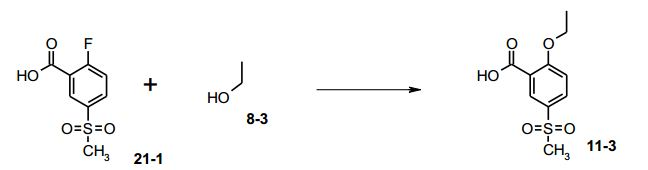
[00049] A uma suspensão branca de 15,1 g de ácido 2-flúor-5- metanossulfonil-benzóico (21-1, 69,2 mmols) em 302 mL de etanol 8- 3, foram adicionados 68,3 g de carbonato de césio (207,6 mols). A suspensão de reação branca foi aquecida para 80°C e agitada sob ar- gônio durante 18 horas. O solvente da mistura de reação foi evaporado e o resíduo foi tratado com 150 mL de EtOAc e 150 mL de água. O pH da fase aquosa foi ajustado para 1,5 por adição de cerca de 75 mL de HCl a 25%. Depois de extração, as fases foram separadas e a fase aquosa foi extraída com 150 mL de EtOAc. As fases orgânicas combinadas foram evaporadas para um volume de 150 mL e tratadas com 150 mL de heptano. A suspensão formada foi filtrada, os cristais foram lavados com 150 mL de EtOAc/heptano 1:1 e secados para fornecer 14,4 g de 11-3, como cristais brancos em 85% (Análise por HPLC 99,7% área-%). Exemplo 2 (5) - Triflúor- isopropanol ((S)-8-1):

Equipamento: Vaso de 800 L equipado com uma sonda de temperatura no fundo do vaso, mergulhando uma sonda de temperatura na mistura de reação, um agitador mecânico e um suprimento de gás inerte.
[00050] Uma suspensão marrom de 240 L de tampão de fosfato de pH 7,5 e 240 Kg de levedura de Baker (Klipfel AG (Rheinfelden), Sackhefe 104020, armazenado em 4°C) foi agitada à temperatura ambiente durante uma hora, aquecida para até 50°C dentro de 85 minutos e mantida a 50,3°C (± 0,5°C) durante 1,5 hora. O pH da suspensão foi mantido a 7,5 por adição de KOH (a 50%), com o auxílio de um estabilizador de pH. A suspensão foi resfriada para 10°C dentro de 120 minutos, diluída com 320 L de tampão de fosfato de pH 7,5 e agitada durante 24 horas a 10°C. À mistura, foram adicionados, dentro de 100 minutos, 24,7 Kg de triflúor-acetona (16, 220,4 mmols, pré-resfriados para < 10°C). A mistura de reação foi aquecida para 20°C e agitada durante 159 horas nesta temperatura (o pH da suspensão foi mantido em 7,5, por adição de KOH (a 50%), com o auxílio de um estabilizador de pH).
[00051] À mistura, foram adicionados 0,5 Kg de antiespumante BC 86/013 (Basildon Chemical Company (Inglaterra), antiespumante BC 86/013, composto antiespumante à base de silicone/não-silicone), aquecidos para 60°C, e o produto foi removido por destilação a 14 KPa (140 mbar), para se obter 101 Kg de mistura de (S)-triflúor-isopropanol (S)-8, água e etanol. 101 Kg de mistura de (S)-triflúor-isopropanol (S)- 8, água e etanol foram destilados em um evaporador rotativo de 50 L em 3 porções a 90°C, partindo de 101,3 KPa para 50 KPa (1,013 mbar para 500 mbar). As frações combinadas forneceram 28,5 Kg de mistura de (S)-triflúor-isopropanol (S)-8-1, água e etanol. 28,5 Kg de (S)- triflúor-isopropanol (S)-8-1 foram destilados em uma coluna Sulzer(5 x 150 cm BX recheio Sulzer) em 2 porções, a 115°C e 101,3 KPa (1,013 mbar), para fornecer (incluindo as frações laterais redestiladas) 21,8 Kg (82,9%) de (S)-triflúor-isopropanol ((S)-8-1). Análise por CG: 95,1 m/m-% de (S)-8-1 er = 99,7 : 0,3 Exemplo 3 2,3-Diflúor-5-triflúor-metil-piridina (12-2):

[00052] Equipamento: Frasco de fundo redondo com quatro gargalos de 2,5 L, equipado com um termômetro, um agitador mecânico, um funil de gotejamento e um suprimento de gás inerte.
[00053] 150 mL de N-metil-2-pirrolidinona foram evaporados a 110°C e 2,5-3,0 KPa (25-30 mbar) a partir de uma suspensão de 2 L de N-metil- 2-pirrolidinona, 28 g de carbonato de potássio (202,6 mmols) e 615,0 g de fluoreto de césio (4,0 mols). A mistura de reação foi tratada com 170,0 g de 2,3-dicloro-5-triflúor-metil-piridina (12-1, 779,2 mmols) e agitada a 120°C durante 24 h. O produto 12-2 foi diretamente removido por destilação da suspensão de reação a 95 a 110°C e 4,0-5,0 KPa (40-50 mbar), fornecendo 190 g de 12-2, como uma mistura. 190 g dessa mistura foram extraídos com 200 mL de pentano e 400 mL de água. Depois de separação das fases, a fase aquosa foi extraída com 2 L de pentano. A fase de pentano combinada foi destilada em uma coluna Sulzer a 40 até 100°C fornecendo 60,0 g (40,4%) de 12-2. Análise por CG: 99,9 área-% de 12-2 Exemplo 3b
[00054] Equipamento: Frasco de fundo redondo com quatro gargalos de 250 mL, equipado com um termômetro, um agitador mecânico, um funil de gotejamento e um suprimento de gás inerte.
[00055] 25 mL de DMSO foram evaporados, a 120°C e 2,5-3,0 KPa (25-30 mbar), a partir de uma suspensão de 150 mL de DMSO, 2,5 g de carbonato de potássio (17,9 mmols) e 25,0 g de fluoreto de césio (162,9 mmols). A mistura de reação foi tratada com 25,0 g de 2,3- dicloro-5-triflúor-metil-piridina (12-1, 112,3 mmols) e agitada a 120°C durante 4 horas. A suspensão foi filtrada e o produto 12-3 foi diretamente removido por destilação do destilado, a 95 até 115°C e 4,0-6,0 KPa (40-60 mbar), para dar 12-3 em rendimento quantitativo. Análise por CG: 96,9 área-% de 12-3 Exemplo 4 Síntese de 1-(3-flúor-5-triflúor-metil-piridin-2-il)-piperazina (15-1):
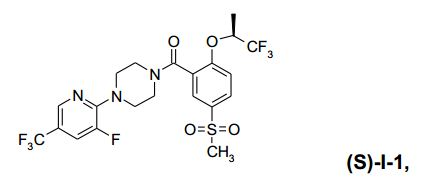
[00056] Uma suspensão de 1,0 Kg de piperazina (12,1 mols) em 15,0 L de THF foi tratada, a 0°C dentro de 30 minutos, com uma solução de 732,0 g de 2,3-diflúor-5-triflúor-metil-piridina 12-2, (4,0 mols) em 2,0 L de THF. A mistura de reação foi agitada durante 30 minutos, a 0°C, e aquecida para a temperatura ambiente, dentro de 30 minutos. A mistura de reação branca foi extraída com 15 L de água e 15 L de tolueno. Depois de separação das fases, a fase aquosa foi extraída com 10 L de tolueno. As fases orgânicas combinadas foram lavadas duas vezes com 10 L, no total, com 20 L de água. O solvente da fase orgânica foi evaporado a 45°C e 5 KPa (50 mbar), para fornecer 984,0 g (99,3%) de 15-1, como sólido branco. Análise por CG: 98,9 área-% de 15-1 Exemplo 5 Síntese de [4-(3-flúor-5-triflúor-metil-piridin-2-il)-piperazin-1 -il]-[5- metanos-sulfonil-2-(2,2,2-triflúor-1-metil-etóxi)-fenil]-metanona ((S)-l-1)
[00057] Uma suspensão de 1,2 Kg de ácido 5-metanossulfonil-2- (2,2,2-triflúor-1-metil-etóxi)-benzóico ((S)-11-1, 4,0 mols) e 50 mL de DMF em 15 L de tolueno foi tratada, à temperatura ambiente, dentro de 1 hora, com uma solução de 485,2 g de cloreto de oxalila (3,7 mols) em 650 mL de tolueno. A suspensão foi agitada durante uma hora, à temperatura ambiente, e gotejada, à temperatura ambiente, dentro de 30 a 45 minutos, a uma solução de 1,0 Kg de 1-(3-flúor-5-triflúor-metil- piridin-2-il)-piperazina (15-1, 4,0 mols) em 12 L de tolueno e 1,1 L de trietilamina (7,9 mols). A mistura de reação foi agitada à temperatura ambiente, durante 30 minutos.
[00058] A suspensão foi filtrada e o resíduo foi lavado, em porções, com 5 L de tolueno. O filtrado foi extraído com 15 L de água. Depois de separação das fases, a fase orgânica foi lavada com 15 L de bicarbonato de sódio a 5% e 7 L de solução a 5% de NaCI. O solvente da fase orgânica foi evaporado (50°C, 40 KPa (400 mbar)) e tratado com 20 L de EtOH. A solução foi filtrada a quente e o solvente foi evaporado a 50°C para um volume de cerca de 10 L. A solução foi aquecida para 60°C, tratada dentro de 30 minutos com 25 L de heptano e resfriada, dentro de 4 horas, para 20°C. A suspensão branca foi agitada nesta temperatura durante a noite, resfriada a 0°C e agitada durante uma hora a 0°C. Depois de filtração, os cristais foram lavados, em porções, com uma mistura resfriada de 3 L de EtOH e 7 L de heptano, para fornecer 1.795 g (88,2%) de (S)-l-1, como cristais brancos. Análise por HPLC 99,8 área-% de (S)-l-1 er = 99,4: 0,6% Exemplo 5b
[00059] Equipamento: Frasco de fundo redondo com três gargalos de 100 mL, equipado com um termômetro, um agitador mecânico e um suprimento de gás inerte.
[00060] Uma solução de 200 mg de ácido 5-metanossulfonil-2- (2,2,2-triflúor-1-metil-etóxi)-benzóico ((S)-11-1, 0,63 mmol) em 20 mL de CH2CI2 foi tratada, à temperatura ambiente, com 166 mg de di- isopropil-etilamina. À mistura, foram adicionados, a 0°C, 70 mg de cloroformiato de etila (0,63 mmol) e agitou-se durante 60 minutos. A mistura de reação foi tratada com 166,8 mg de 1-(3-cloro-5-triflúor-metil-piridin-2-il)- piperazina (15-2, 0,63 mmol) e agitada durante cerca de duas horas. A mistura foi aquecida para temperatura ambiente e tratada com 15 mL de CH2CI2 e 5 mL de água. Depois de extração, as fases foram separadas e a fase aquosa foi extraída com 5 mL de CH2CI2. As fases orgânicas combinadas foram evaporadas sob pressão reduzida, para fornecer 0 produto bruto, como um óleo. Depois de purificação croma- tográfica, 120 mg de (S)-l-2 foram obtidos (cristalização a partir de hexano também funciona). Análise por HPLC 96,6 área-% de (S)-l-2 Exemplo 5c
[00061] Equipamento: Frasco de fundo redondo com quatro gargalos de 100 mL, equipado com um termômetro, um agitador mecânico e um suprimento de gás inerte.
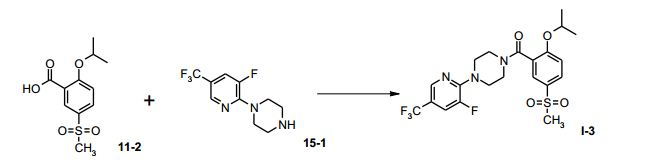
[00062] Uma solução de 200 mg de ácido 2-isopropóxi-5- metanossul-fonil-benzóico (11-2, 0,77 mmol) em 20 mL de CH2CI2 foi tratada, à temperatura ambiente, com 214,4 mg de di-isopropil- etilamina (1,63 mmol). A mistura de reação foi resfriada para -5°C, tratada com 85,7 mg de cloroformiato de etila (0,77 mmol) e agitada durante 60 minutos nesta temperatura. Uma solução de 221,1 mg de 1- (3-flúor-5-triflúor-metil-piridin-2-il)-piperazina (15-1, 0,77 mmol) e 102,1 mg de di-isopropil-etilamina (0,77 mmol) em 10 mL de CH2CI2 foram adicionados a -5°C. A mistura de reação foi agitada durante 4 horas, aquecida para a temperatura ambiente e tratada com 15 mL de água. Depois de extração, as fases foram separadas e a fase aquosa foi extraída duas vezes com 5 mL de CH2CI2. As fases orgânicas combinadas foram evaporadas sob pressão reduzida, para fornecer 0 produto bruto, como um óleo. Depois de purificação cromatográfica, 40 mg de I-3 foram obtidos. Análise por HPLC 96,6 área-% de (S)-l-3 Exemplo 5d
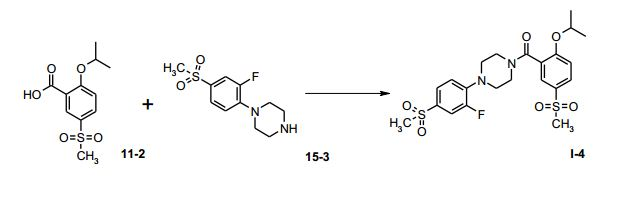
[00063] Equipamento: Frasco de fundo redondo com quatro gargalos de 250 mL, equipado com um termômetro, um agitador mecânico e um suprimento de gás inerte.
[00064] Uma solução de 5,0 g de ácido 2-isopropóxi-5- metanossulfonil-benzóico (11-2, 19,4 mmols) em 150 mL de CH2CI2 foi tratada, à temperatura ambiente, com 2,8 g de di-isopropil-etilamina (21,3 mmols). A mistura de reação foi resfriada para 0°C, tratada com uma solução de 2,1 g de cloroformiato de etila (19,4 mmols) em 50 mL de CH2CI2 e agitada durante duas horas nesta temperatura. Uma solução de 5,1 g de 1-(4-metanossulfonil-2-flúor-fenil)-piperazina (15-3, 19,36 mmols) em 50 mL de CH2CI2 foi adicionada a 0°C, dentro de 15 minutos. A mistura de reação foi agitada durante duas horas, aquecida para a temperatura ambiente e tratada com 15 mL de água. Depois de extração, as fases foram separadas e a fase aquosa foi extraída duas vezes com 10 mL de CH2CI2. As fases orgânicas combinadas foram evaporadas sob pressão reduzida, para fornecer 0 produto bruto, como um óleo. A cristalização a partir de EtOAc forneceu 6,25 g de I-4, como pó branco. Análise por HPLC 96,6 área-% de I-4 Exemplo 5e
[00065] Equipamento: Frasco de fundo redondo com quatro gargalos de 350 mL, equipado com um termômetro, um agitador mecânico e um suprimento de gás inerte.
[00066] Uma suspensão de 11,0 g de ácido 2-etóxi-5- metanossulfonil-benzóico (11-3, 19,4 mmols), em 110 mL de tolueno, foi tratada à temperatura ambiente com 0,5 mL de DMF. A mistura de reação foi tratada com uma solução de 3,7 mL de cloreto de oxalila (42,7 mmols) em 10 mL de tolueno. A suspensão foi agitada durante uma hora, à temperatura ambiente, e foi gotejada para uma solução de 11,3 g de 1-(3-flúor-5-triflúor-metil-piridin-2-il)-piperazina (15-1, 44,8 mmols) e 12,0 mL de trietilamina (85,8 mmols) em 140 mL de tolueno. A suspensão foi agitada durante 30 minutos, à temperatura ambiente, filtrada e o resíduo foi enxaguado com 100 mL de tolueno. O filtrado foi lavado três vezes com 400 mL de água. O solvente da fase orgânica foi evaporado e o resíduo foi tratado com 250 mL de EtOH. Cerca de 150 mL de EtOH foram evaporados a 60°C e 300 mL de heptano foram adicionados dentro de 30 minutos. A mistura foi resfriada para temperatura ambiente dentro de 4 horas, a suspensão formada foi resfriada para 0°C e agitada durante uma hora. Os cristais foram filtrados, lavados com 120 mL de EtOH/heptano 1:2 e secados durante 24 horas a 50°C, para fornecer 16,5 g (81,3%) de produto I-5, como cristais brancos. Análise por HPLC 99,8 área-% de I-5