WO2024204287A1 - 修飾ヌクレオチドを有するcrRNA - Google Patents
修飾ヌクレオチドを有するcrRNA Download PDFInfo
- Publication number
- WO2024204287A1 WO2024204287A1 PCT/JP2024/012128 JP2024012128W WO2024204287A1 WO 2024204287 A1 WO2024204287 A1 WO 2024204287A1 JP 2024012128 W JP2024012128 W JP 2024012128W WO 2024204287 A1 WO2024204287 A1 WO 2024204287A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- sequence
- crrna
- crispr
- cells
- repeat
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images





























































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/686—Polymerase chain reaction [PCR]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6897—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids involving reporter genes operably linked to promoters
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPR]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/50—Vector systems having a special element relevant for transcription regulating RNA stability, not being an intron, e.g. poly A signal
Definitions
- the present invention relates to pre-crRNAs having modified nucleotides. More specifically, the present invention relates to pre-crRNAs having specific modifications, allele-specific pre-crRNAs, and methods for producing double-stranded DNA modified cells using these pre-crRNAs.
- CRISPR-Cas system which is mainly composed of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) proteins, an adaptive immune mechanism found in eubacteria and archaea, has been developed as a genome editing technology, including gene destruction (knockout), by freely designing the target sequence of guide RNA, and is widely used.
- CRISPR-Cas systems are broadly divided into class 1 CRISPR-Cas systems in which the effector that works in the process of cutting DNA in a guide RNA-dependent manner functions as a complex of multiple proteins, and class 2 CRISPR-Cas systems that function as a single protein.
- the CRISPR-Cas9 system which belongs to class 2 type II, is currently the most commonly used due to its simplicity and wide range of applications.
- the CRISPR-Cas12a system which belongs to type V, is also known as another CRISPR-Cas system that belongs to class 2 and is used for genome editing.
- Patent Document 1 the type I-E CRISPR-Cas3 system developed at Osaka University (Patent Document 1) and the type I-D CRISPR-Cas10 (TiD) system developed at Tokushima University (Patent Document 2) are known.
- RNA encoding Cas proteins and Cascade complexes rather than plasmid DNA, which may be unexpectedly integrated into the cell genome, or RNP (ribonucleoprotein), which requires protein purification, and have succeeded in introducing deletions of more than 100 nucleotides into the target region of genomic DNA using the CRISPR-Cas3 system (Patent Document 3).
- CRISPR-Cas3 system While the class 1 CRISPR-Cas system requires multiple proteins, it has a DNA cutting mechanism different from the class 2 CRISPR-Cas system, and is therefore highly useful in a variety of situations, such as being able to induce unique DNA deletion patterns.
- the CRISPR-Cas3 system is known to induce long-stranded DNA deletions of several hundred bases to more than 100 kilobases in one direction, starting from a point several tens to several hundred bases away from the guide RNA recognition sequence.
- the guide RNA of CRISPR-Cas3 has a longer recognition sequence than the guide RNA of the CRISPR-Cas9 system, suggesting high specificity.
- PCR does not accurately reflect the length of the deletion mutation and its efficiency because the amplification efficiency varies depending on the length of the amplified product, and deletion mutations (several tens of kb or more) that exceed the amplification range cannot be amplified in the first place and cannot be detected as a result. It is possible to analyze the break pattern to a certain extent by using whole genome sequence analysis using a next-generation sequencer or by using sequence analysis by pulling down the target vicinity sequence (Non-Patent Document 1), but the analysis cost is high and it requires technology and time to analyze a large amount of data.
- an objective of the present invention is to improve the editing efficiency in genome editing and to provide a method for analyzing genome editing patterns by the CRISPR-Cas system, as well as tools that can be used for said method.
- DM1 myotonic dystrophy type I
- DM1 myotonic dystrophy type I
- DM1 myotonic dystrophy type I
- genome editing basically occurs in both alleles, and even if an attempt is made to induce genome editing only in the target allele by targeting a region containing a SNP sequence specific to the target allele, there is a problem that there is no suitable heterozygous SNP in the immediate vicinity of the target site for genome editing. Therefore, it is also an objective of the present invention to provide a method for performing genome editing specific to one allele by targeting a heterozygous SNP away from the target site for genome editing.
- RNA modification can change the physical properties and secondary structure of RNA, as well as interactions with proteins, and may cause the original function to be lost, and even experts cannot predict which modification should be introduced at which position.
- RNA modifications have been tried in Cas9 guide RNA (sgRNA or gRNA-crRNA), but it is difficult to predict which modification and position are optimal.
- the pre-crRNA of the E. coli CRISPR-Cas3 system is a 90-base RNA that contains two repeat sequences, each consisting of 29 bases, and a 32-base spacer sequence ( Figure 3-1), but there are no precedents for examining the chemical modification mode when using it in mammalian cells.
- the pre-crRNA of the CRISPR-Cas3 system and the guide RNA of the CRISPR-Cas9 system do not match in sequence, structure, or functional characteristics, so it is not possible to use this as a reference for which position is optimal for modification.
- biochemical experiments have reported that processing of the pre-crRNA by Cas6 is required in the CRISPR-Cas3 system, and the complexity of the mechanism makes it difficult to predict the appropriate modification position.
- the inventors therefore introduced chemical modifications into the pre-crRNA of the CRISPR-Cas3 system to search for modification positions that would improve activity. As a result, they found that modifying specific regions of the pre-crRNA significantly improved genome editing efficiency without losing the function of the crRNA. Surprisingly, although the pre-crRNA has a nearly symmetrical structure, it was suggested that the effect of modification by the pre-crRNA is asymmetrical. They also found that by introducing modifications into the pre-crRNA similar to those applied to mRNA in vivo, the activity of the pre-crRNA could be improved.
- SSA single-strand annealing
- the inventors also attempted to develop a technique for measuring the length distribution and proportion of deletion mutations caused by the CRISPR-Cas system by applying droplet digital PCR (ddPCR) technology.
- ddPCR droplet digital PCR
- a pre-crRNA of a Type I CRISPR-Cas system having modified nucleotides A pre-crRNA, wherein at least one nucleotide in the region consisting of the 5' arm region of the first repeat sequence, the 5' stem-forming region of the first repeat sequence, and the loop-forming region of the first repeat sequence is a modified nucleotide.
- a pre-crRNA according to [1-1] wherein at least one nucleotide in the 5' arm region of the first repeat sequence is a modified nucleotide.
- [3] The pre-crRNA according to any one of [1-1] to [2-3], wherein the first to third nucleotides from the 5'-end and the second to fourth nucleotides from the 3'-end are modified nucleotides.
- [4-1] The pre-crRNA according to any one of [1-1] to [3], wherein at least one, and optionally all, nucleotides in the loop-forming region of the first repeat sequence are modified nucleotides.
- [4-2] The pre-crRNA according to any one of [1-1] to [4-1], wherein at least one, and optionally all, nucleotides in the loop-forming region of the second repeat sequence are modified nucleotides.
- [4-5] The pre-crRNA according to any one of [1-1] to [4-4], wherein the internucleoside bond on the 3' side of at least one or all of the nucleosides having a modification in the sugar portion is a modified internucleoside bond.
- [4-9] The pre-crRNA according to any one of [1-1] to [4-8], wherein the 3' stem-forming region of the first repeat sequence, the 3' arm region of the first repeat sequence, the 5' arm region of the second repeat sequence, and the stem-forming region of the second repeat sequence are all ribonucleotides.
- [4-10] The pre-crRNA according to any one of [1-1] to [4-9], wherein the spacer sequence is entirely ribonucleotides.
- [4-11] The pre-crRNA according to any one of [1-1] to [4-10], wherein the site cleaved by Cas6 or Cas5 is unmodified.
- [5-1] The pre-crRNA according to any one of [1-1] to [4-11], wherein the first to third nucleotides from the 5'-end and the second to fourth nucleotides from the 3'-end are modified nucleotides, all nucleotides constituting the 3' arm region of the first repeat sequence, the 5' arm region of the second repeat sequence, and the spacer region are ribonucleotides, and the site cleaved by Cas6 or Cas5 is unmodified.
- [5-2] The pre-crRNA according to [5-1], wherein the internucleoside bond on the 3' side of at least one or all of the nucleosides having a modification in the sugar portion is a modified internucleoside bond.
- [5-3] The pre-crRNA according to [5-1] or [5-2], wherein the bond on the 3' side of the ribonucleoside is a phosphodiester bond.
- [6] The method according to any one of [1-1] to [5-3], which has two or more spacer regions.
- a method for designing a pre-crRNA for an allele-specific Type I CRISPR-Cas system comprising: A method comprising the steps of: designing a pre-crRNA that has a deletion target site upstream of a PAM sequence adjacent to a sequence targeted by the pre-crRNA; and that targets a position within the sequence targeted by the pre-crRNA excluding the 6nth position (n is a positive integer) counting from the downstream terminal base of the PAM sequence, or a sequence on the PAM sequence where there is at least one base that differs between alleles.
- [9-2] The method according to [9-1], wherein the type I CRISPR-Cas system is a type I-E CRISPR-Cas system, optionally a CRISPR-Cas system derived from Escherichia coli.
- [11-1] The method according to any one of [9-1] to [10], wherein the deletion target site comprises a repeat.
- [12-1] A type I CRISPR-Cas system comprising the following (1) and (2). (1) A pre-crRNA according to any one of [1-1] to [8-3] or a pre-crRNA designed by the method according to any one of [9-1] to [11-2]; (2) Cascade-constituting proteins or nucleic acids encoding said proteins [12-2]
- the CRISPR-Cas system according to [12-1] further comprises (3) Cas3 and/or Cas10d or a nucleic acid encoding Cas3 and/or Cas10d.
- a double-stranded nucleic acid for evaluating the activity of a type I CRISPR-Cas system wherein one nucleic acid strand is (1) a sequence encoding a portion of a reporter protein, optionally a fluorescent protein; (2) an insert sequence comprising a sequence targeted by the crRNA or a sequence complementary to said sequence; and (3) a sequence different from (1) that encodes a portion of a reporter protein, in this order; (1) and (3) have an overlapping sequence, and a sequence other than the overlapping sequence of (1), the overlapping sequence, and a sequence other than the overlapping sequence of (3) constitute a sequence encoding a full-length reporter protein; (2) The base length of the insertion sequence is 100 to 3,000; A double-stranded nucleic acid.
- a double-stranded nucleic acid for evaluating the activity of a type I CRISPR-Cas system wherein one nucleic acid strand is (1) a sequence encoding a portion of a reporter protein, optionally a fluorescent protein; (2) an insert sequence including a sequence targeted by the crRNA and a sequence complementary to said sequence; and (3) a sequence different from (1) that encodes a portion of a reporter protein, in this order; (1) and (3) have an overlapping sequence, and a sequence other than the overlapping sequence of (1), the overlapping sequence, and a sequence other than the overlapping sequence of (3) constitute a sequence encoding a full-length reporter protein.
- a double-stranded nucleic acid is (1) a sequence encoding a portion of a reporter protein, optionally a fluorescent protein; (2) an insert sequence including a sequence targeted by the crRNA and a sequence complementary to said sequence; and (3) a sequence different from (1) that encodes a portion of a reporter protein, in this order; (1) and (3) have an overlapping sequence, and
- [15-1] A method for producing a cell in which double-stranded DNA has been modified, the method comprising a step of introducing the CRISPR-Cas system according to [12-1] or [12-2] into the cell.
- [15-2] A method for modifying double-stranded DNA contained in a cell, the method comprising a step of introducing the CRISPR-Cas system according to [12-1] or [12-2] into the cell.
- [16] The method according to [15-1] or [15-2], wherein the step of introducing the CRISPR-Cas system into a cell is repeated multiple times.
- [17] The method according to any one of [15-1] to [16], comprising a step of introducing into a cell the double-stranded nucleic acid according to [13] or [14-1], or the expression vector according to [14-2].
- [18] The method according to [17], wherein the double-stranded nucleic acid to be introduced is of two or more types.
- [20-1] The method according to any one of [15-1] to [19], wherein the cell is a pluripotent stem cell, optionally an induced pluripotent stem cell.
- the type I CRISPR-Cas system is a type I-E CRISPR-Cas system, optionally a CRISPR-Cas system derived from Escherichia coli.
- [22-1] A method for extending a deletion site, comprising the step of introducing a type I CRISPR-Cas system into a cell multiple times.
- the type I CRISPR-Cas system is a type I-E CRISPR-Cas system, optionally a CRISPR-Cas system derived from Escherichia coli.
- the cell is a pluripotent stem cell, optionally an induced pluripotent stem cell.
- [24-1] A modifier for modifying a target sequence of double-stranded DNA, comprising the CRISPR-Cas system of [12-1] or [12-2].
- [24-2] A target gene expression regulator comprising the CRISPR-Cas system of [12-1] or [12-2].
- [25] The agent according to [24-1] or [24-2] for treating a disease.
- the present invention makes it possible to improve the efficiency of genome editing using the type I CRISPR-Cas system and to analyze genome editing patterns using this system. It also makes it possible to provide tools that can concentrate genome-edited cells and perform genome editing specifically for one allele.
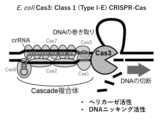
- FIG. 1 A schematic diagram of DNA cleavage by the class 1 CRISPR-Cas system.
- Cas3 has helicase activity and DNA nicking activity, and introduces a nick into the DNA strand not bound by the crRNA or Cascade complex.
- the figure shows the kinetics of expression of each protein constituting the Cascade complex (hereinafter sometimes referred to as the "Cascade protein group") by mRNA.
- the Cascade protein group was introduced into iPS cell lines (I01s04 or I14s04) in the form of mRNA ( Figure 29 of Patent Document 3) or plasmid ( Figure 9d of Patent Document 3) using Lipofectamine Stem. NC indicates the negative control.
- Nucleotides with an MS modification have the second hydroxyl group of the sugar moiety replaced with an O-methyl group, and the bond between the sugar moiety of the nucleotide and the sugar moiety of the next nucleotide is a phosphorothioate bond (where the oxygen atom at the phosphodiester bond site is replaced with a sulfur atom (S)), as below.
- the base sequence of pre-crRNA is shown as sequence number 168.
- the repeat structures of pre-crRNAs other than E. coli Cas3 pre-crRNA are shown (Source: Zheng Y. et al., Front Bioeng Biotechnol. 2020 Mar 4;8:62).
- each repeat structure is shown in sequence numbers 169 to 174 from top to bottom.
- the circled numbers in the graph correspond to the circled numbers at the modification positions in Figure 2-1.
- the human B2M gene was targeted and genome editing (B2M gene knockout) efficiency was measured by HLA-A/B/C staining of the cell surface, and high activity was observed with modifications at sites (1) and (4). This shows the change in activity depending on the RNA modification position in E.
- coli Cas3 pre-crRNA electroporation using 4D-Nucleofector.
- the circled numbers in the graph correspond to the circled numbers at the modification positions in Figure 2-1.
- the human B2M gene was targeted and genome editing efficiency was measured by HLA staining, and high activity was observed with modifications at sites (1) and (4).
- a schematic diagram of additional RNA modification locations in E. coli Cas3 pre-crRNA is shown. Nucleotides marked with * have 2'-O-methyl 3'phosphorothioate (MS) modifications. Arrows indicate the cleavage site by Cas6.
- a schematic diagram of additional RNA modification locations in E. coli Cas3 pre-crRNA is shown.
- Nucleotides marked with * have 2'-O-methyl 3'phosphorothioate (MS) modifications. Arrows indicate the cleavage site by Cas6.
- a schematic diagram of additional RNA modification locations in E. coli Cas3 pre-crRNA is shown. Nucleotides marked with * have 2'-O-methyl 3'phosphorothioate (MS) modifications. Arrows indicate the cleavage site by Cas6.
- a schematic diagram of additional RNA modification locations in E. coli Cas3 pre-crRNA is shown. Nucleotides marked with # have 2'-O-methyl (M) modifications and nucleotides marked with * have 2'-O-methyl 3'phosphorothioate (MS) modifications.
- FIG. 5-1 to 5-4 The figure shows the change in activity of E. coli Cas3 pre-crRNA depending on the RNA modification positions shown in Figures 5-1 to 5-4.
- I14s04 iPS cells were transfected with mRNA encoding Cas3 and Cascade proteins and pre-crRNA targeting the human B2M gene using Lipofectamine Stem, and genome editing efficiency was measured by HLA-A/B/C staining.
- ⁇ indicates an activity-increasing effect
- X indicates an activity-decreasing effect
- N indicates little or no effect on activity.
- the results of genome editing using crRNA (B2M#1) in a different iPS cell line (1383D2) are shown.
- the target sequence is the sequence shown in SEQ ID NO:4.
- the figure shows the change in activity of E. coli Cas3 pre-crRNA depending on the RNA modification positions shown in Figures 5-1 to 5-4. 1383D2 iPS cells were transfected with mRNA encoding Cas3 and Cascade proteins and pre-crRNA targeting exon 45 (Ex45) of the human dystrophin (DMD) gene using Lipofectamine Stem, and genome editing efficiency was measured by ddPCR.
- the target sequence is the sequence shown in SEQ ID NO:55.
- a schematic diagram of the addition of a 5'-Cap analog and polyA tail to E. coli Cas3 pre-crRNA is shown.
- Pre-crRNA with an ARCA (anti-reverse cap analog) cap analog was synthesized by in vitro transcription (IVT) reaction from a plasmid carrying a T7 promoter encoding the pre-crRNA, and polyA was added to the 3' end of the RNA.
- IVT in vitro transcription
- PolyA Rxn+ is a sample to which PolyA was added using E. coli Poly(A) Polymerase (E-PAP) and ATP
- PolyA Rxn- is a sample before PolyA was added.
- E-PAP E. coli Poly(A) Polymerase
- PolyA Rxn- is a sample before PolyA was added.
- the effect of adding a 5'-Cap analog and a polyA tail to E. coli Cas3 pre-crRNA in HEK293T cells is shown.
- HEK293T cells were transfected with mRNA expressing Cas3 and the Cascade protein group and pre-crRNA (B2M#4) using Lipofectamine 2000.
- Pre-crRNA with a 5'-Cap analog and a polyA tail showed higher genome editing efficiency than pre-crRNA without these.
- the effect of adding a 5'-Cap analog and a polyA tail to E. coli Cas3 pre-crRNA in the I14s04 strain is shown.
- mRNA expressing Cas3 and Cascade proteins and pre-crRNA (B2M#1 or B2M#4) were introduced into the I14s04 strain using Lipofectamine Stem.
- Pre-crRNA with a 5'-Cap analog and a polyA tail showed a higher genome editing effect than pre-crRNA without these. This shows the accumulation of effects from multiple Cas3 genome editing.
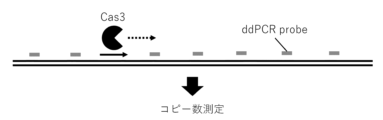
- FIG. 1 shows a schematic diagram of the technique for analyzing the deletion induction pattern of Cas3 by ddPCR. The results of analyzing Cas3 genome editing patterns in HEK293T cells using ddPCR are shown.
- HEK293T cells stable strain
- the pPV-C1-crRNA(B2M#1)-EF1a-G2ABA piggyBac vector expressing pre-crRNA targeting the B2M gene and the pPV-TRE3G-Bi(263+751)-EF1a-Tet3G-iPA piggyBac vector which allows DOX-regulated expression of Cas3 and Cascade proteins.
- DOX concentrations were varied (0.1 ⁇ M, 0.3 ⁇ M, or 1 ⁇ M) and DOX-induced expression of Cas3 and Cascade proteins was performed for multiple days (0, 1, 2, 3, or 4 days).
- the results of an analysis of Cas3 genome editing patterns in iPS cells using ddPCR are shown below.
- CiRA00213 iPS cells stable strain
- B2M#1 ddPCR
- the pattern of deletion induction after RNA-mediated Cas3 genome editing is shown.
- the top graph shows the results after introducing the CRISPR-Cas3 system in the form of RNA (1, 2 or 3 times) into I14s04 iPS cells (the copy numbers at approximately 1kb and 10kb from the crRNA target sequence were measured by ddPCR), and the bottom graph shows the results after DOX-induced expression of Cas3 and Cascade proteins in HEK293T cells and I14s04 iPS cells for multiple days (0, 1, 2, 3 or 4 days) or with different DOX concentrations (0, 0.5, 2 ⁇ M) (partially quoted from Figures 13 and 14 for comparison).
- LF Stem is an abbreviation for Lipofectamine Stem. "ul" in the figure means “ ⁇ l". The same applies below.
- Cas3 vector All-in-one-Puro
- crRNA vector or Cas9 vector (Cas9-Puro) and sgRNA vector
- a schematic diagram of the genome editing method for HLA-A*24:02 and B*52:01 in iPSCs (I01s04 and I14s04) derived from Japanese HLA homozygous donors is shown.
- the AAG+32nt sequence was extracted and converted to AAG+XXXXNXXXXXXNXXXXXNXXXXXXNXXXXXXNXXXXXXNXXXXXNXXXXXNXXXXXNXXXXXNXXXXXNXXXXXNXXXXXNXXXXXNXXXXXNXXXXXXNXXXXXXNXXXXXXNXXXXXXNXXXXX, after which off-target prediction (i.e., examination of other perfect match sites, especially matches in other HLA genes) was performed using GGGenome. The distance and direction were then narrowed down to remove the cluster of exons 1-3.
- the crRNA sequences (A24:02 F1/R2/R4, B52:01 F1/R5/R7) are examples. ddPCR probes and primers for measuring genome editing efficiency were designed on exon 2 of each
- mRNA expressing Cas3 and Cascade proteins and crRNA expression vectors (pBSIIKS-U6-crRNA or pPV-C1-crRNA) were expressed in I14s04 iPS cells using Lipofectamine Stem, and the resulting genome samples were analyzed by PCR (LA Taq HS). Bands smaller than 7.7 kb represent deletions caused by Cas3 genome editing. The results of measuring the efficiency of HLA-A*24:02 and B*52:01 exon 2 deletion by ddPCR are shown (single crRNA).
- I14s04 iPS cells were transfected multiple times using Lipofectamine Stem with mRNA expressing Cas3 and Cascade proteins, and Pre-both3 pre-crRNA targeting the B2M gene, HLA-A*24:02, and B*52:01 ( Figure 5-1), and the collected genome samples were analyzed by ddPCR.
- the experiment using crRNA (B2M#1) was the control experiment (left panel).
- the results of measuring the efficiency of exon 2 deletion by ddPCR (double crRNA) are shown.
- two types of pre-both3 pre-crRNA targeting HLA-A*24:02 and pre-both3 pre-crRNA targeting HLA-B*52:01 were added.
- SSA single-strand annealing
- HEK293T cells in which expression of Cas3 and Cascade proteins can be induced by the addition of DOX, were added with DOX and an mRFP-SSA vector (mRFP is expressed by SSA) containing a non-targeted crRNA (AAVS1#1) or a target crRNA (DMPK DownSNP1T) and the target DMPK gene sequence (DMPK DownSNP1T) was introduced, and the percentage of mRFP-positive cells was measured by flow cytometry three days later.
- SSA insertion sequences containing the target sequence were compared in length, approximately 0.5, 1, and 2 kb.
- the Puro-SSA reporter vector shows approximately six-fold enrichment of gene-deficient cells.
- HEK293T cells were used, with pPV-Dual_Promoter-EF1a-2xNLS-Cascade+Cascade(RD)-iCA (All-in-one-mCherry) as the Cas3 and Cascade protein expression vector, pPV-C1-crRNA(B2M#4)-EF1a-BA as the crRNA, and pHL-EF1a-EGFP-IRES-Puro-SSA(B2M-1kb)-A as the SSA reporter vector. 2 ⁇ g/ml puromycin was added 3 to 5 days after transfection (+Puro sample), and the efficiency of B2M deletion by genome editing was measured by HLA-A2 staining.
- HEK293T cells were transfected with All-in-one-mCherry as the Cas3 and Cascade protein expression vector, pPV-C1-crRNA(AAVS1#1)-EF1a-BA (control crRNA) or pPV-C1-crRNA(B2M#1)-EF1a-BA as the crRNA expression vector, and pHL-EF1a-Puro-SSA(B2M-1kb)-A as the Puro-SSA reporter vector, and 2 ⁇ g/ml puromycin was added 2 to 4 days after transfection.
- a Cas3 expression vector (All-in-one-Puro) and a crRNA expression vector (DMDex45 and 55 were used as target crRNAs, and B2M#1 was used as non-target crRNA) were transfected by lipofection using Lipofectamine 2000.
- Genome-edited cells were enriched using both HEK293T cells (left) and iPS cells derived from DMD patients (FF12020: right).
- Cas3 vector All-ine-one-Puro
- crRNA vector DMDex45&55
- Cas9 vector Cas9-Puro
- crRNA vector SSA reporter vector
- Cas3 vector All-ine-one-Puro
- crRNA vector DMDex45&55
- SSA reporter vector SSA-EGFP and SSA-mRFP
- DMD patient-derived iPS cells (FF12020: exon 46,47 deletion, CiRA00458: exon 51-53 deletion, CiRA00646: exon 48-52 deletion) were transfected with Lipofectamine Stem 24 hours prior to drug selection with Puromycin for two days, after which EGFP/mRFP double-positive cells were sorted using a cell sorter to obtain subclones. Genotyping was performed by PCR of the dystrophin gene exon 45-55 region, and the presence or absence of deletion was confirmed by Sanger sequencing. As a result, a subclone of iPS cells in which the exon 45-55 region had been deleted was obtained.
- the base sequences in the left diagram are shown in sequence numbers 175 to 177 from top to bottom, and the base sequences in the right diagram are shown in sequence numbers 178 to 180 from top to bottom.
- the signal-to-noise ratio is compared with that of a conventional SSA reporter vector with a short insertion sequence.
- HEK293T cells were transfected with a Cas3 vector (All-in-one-Puro), a crRNA vector (DMDex45&55 as target crRNA and B2M#1 as non-target crRNA), and SSA reporter vectors, SSA-EGFP and SSA-mRFP, by lipofection using Lipofectamine 2000, and the percentage of EGFP- and mRFP-positive cells was measured by flow cytometry.
- the length of the SSA insertion sequence was 0 kb (target sequence only), 0.5 kb, 1.0 kb, or 1.7 kb.
- the underlined sequences are the sequences of the targeted disease alleles.
- ND Not characterized.
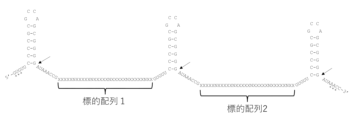
- a schematic diagram of the method for designing allele-specific crRNA is shown.
- the sequences marked with X are those that can be recognized by crRNA.
- the design results of allele-specific crRNA (DMPK CTG repeat upstream: DM1_DownSNP1C) are shown.
- the base sequences are shown from top to bottom as SEQ ID NOs: 181 to 184.
- the crRNA with an SNP at the 12th base (DM1_DownSNP1C_12nt) is a control crRNA that does not show allele specificity.
- the results of activity evaluation of allele-specific crRNA (DM1_DownSNP1C/T and DM1_DownSNP1C_12nt) are shown.
- pPV-Dual_Promoter-EF1a-2xNLS-Cascade+Cascade3(RD)-iPA vector expressing Cas3 and Cascade protein group and pPV-C1-crRNA-EF11-G2ABA vector expressing each pre-crRNA were integrated into the genome of DM1 patient-derived iPS cells HPS1051 carrying heterozygous SNP (C/T) of rs934739524 (stable strain). The genomes of these cell samples were amplified by SNP-specific (allele-specific) PCR and analyzed by TapeStation. An example histogram of the case where the disease allele was targeted using crRNA (DownSNP1C) is shown (right).
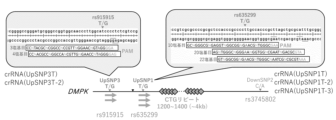
- CiRA00213 iPS cells (stable line) were generated from DM1 patient-derived iPS cells carrying heterozygous SNPs rs915915 (UpSNP3T/G) and rs635299 (UpSNP1T/G) by incorporating the pPV-C1-crRNA-EF1a-BA vector expressing pre-crRNA targeting the DMPK CTG repeat shown in Figure 37 and the pPV-TRE3G-Bi(263+751)-EF1a-Tet3G-iPA vector capable of inducing the expression of Cas3 and Cascade proteins with DOX into the genomic DNA.
- Plasmid-based genome editing was performed, and bands obtained by allele-specific nested PCR were analyzed by Sanger sequencing. Detailed results using crRNA (UpSNP3T). Plasmid-based genome editing was performed, and bands obtained by allele-specific nested PCR were analyzed by Sanger sequencing. Detailed results using crRNA(UpSNP1T). RNA-based Cas3 genome editing was performed and bands obtained by allele-specific nested PCR were analyzed by Sanger sequencing. Detailed results using crRNA (UpSNP3T). RNA-based Cas3 genome editing was performed, and cells were separated at approximately 10 cells/well. Bands obtained by allele-specific PCR from these samples were analyzed by Sanger sequencing (UpSNP3T well #62).
- RNA-based Cas3 genome editing was performed, and bands obtained by allele-specific nested PCR were analyzed by Sanger sequencing.
- the results of allele-specific dual-Cas3 CTG repeat removal are shown (Example 2).
- RNA-based Cas3 genome editing was performed, and bands obtained by allele-specific nested PCR were analyzed by Sanger sequencing.
- the results of allele-specific dual-Cas3 CTG repeat removal are shown (Example 2).
- Plasmid-based Cas3 genome editing was performed, and bands obtained by allele-specific nested PCR were analyzed by Sanger sequencing.
- RNA-based Cas3 genome editing was performed, cells were separated at approximately 10 cells/well, and bands obtained by allele-specific PCR from the samples were analyzed by Sanger sequencing (UpSNP3T x AGG DownSNP2C well #57).
- Sanger sequencing UpSNP3T x AGG DownSNP2C well #57.
- a schematic diagram (left) and analytical results (right) of the method for evaluating allele specificity using a two-color SSA reporter vector are shown.
- DOX 1 ⁇ M DOX was added to HEK293T stable cells, which can express Cas3 and Cascade proteins using DOX, and the crRNA expression vector pPV-C1-crRNA-EF1a-BA (non-target: DMD#20, target: DownSNP1-C/T), mRFP SSA vector (pPV-EF1a-mRxxFP-iPA), and EGFP SSA vector (pPV-EF1a-EGxxFP-iPA) were simultaneously transfected using Lipofectamine 2000. Three days later, the percentage of mRFP- and EGFP-positive cells was measured by flow cytometry.
- a sequence containing DownSNP1-T/C (rs934739524 heterozygous SNP) was used as the insertion sequence for the SSA vector.
- a schematic diagram of the method for improving the specificity of allele-specific crRNA is shown. We show that the specificity can be regulated by the length of the crRNA. 1 ⁇ M DOX was added to HEK293T stable cells, which can express Cas3 and Cascade proteins using DOX.
- the crRNA expression vector pPV-C1-crRNA-EF1a-BA (non-target: AAVS1#1, target: DownSNP1-C), mRFP SSA vector (pPV-EF1a-mRxxFP-iPA) and EGFP SSA vector (pPV-EF1a-EGxxFP-iPA) were simultaneously transfected using Lipofectamine 2000, and the percentage of mRFP- and EGFP-positive cells was measured by flow cytometry three days later.
- the mRFP SSA vector contained an insertion sequence containing DownSNP1-T (rs934739524 heterozygous SNP-T), and the EGFP SSA vector contained DownSNP1-C.
- the crRNA(DownSNP1-C 32nt) has the same sequence as the normal crRNA(DownSNP1-C). This shows that specificity can be adjusted by intentionally introducing mismatches.
- the experimental conditions were the same as in Figure 50.
- crRNA(DownSNP1-C 0MM) has the same sequence as normal crRNA(DownSNP1-C). Since measurements were taken in the same batch as Figure 50, the data for crRNA(AAVS1#1) and crRNA(DownSNP1-C 0MM) are quoted from Figure 50.
- Nucleotides marked with # have 2'-O-methyl (M) modifications, nucleotides marked with + have 3'phosphorothioate (PS) modifications and nucleotides marked with * have 2'-O-methyl 3'phosphorothioate (MS) modifications.
- Arrows indicate the cleavage site by Cas6. This shows the change in activity in E. coli Cas3 pre-crRNA depending on the RNA modification positions shown in Figure 52. I14s04 iPS cells were transfected with mRNA expressing Cas3 and Cascade proteins, and B2M#1 crRNA (each having the chemical modifications corresponding to Figure 52), and the KO efficiency of the B2M gene was measured by staining for HLA-A, B, and C.
- Nucleotides marked with # have 2'-O-methyl (M) modifications, nucleotides marked with + have 3'phosphorothioate (PS) modifications and nucleotides marked with * have 2'-O-methyl 3'phosphorothioate (MS) modifications.
- Arrows indicate the cleavage site by Cas6. The figure shows the change in activity of E. coli Cas3 pre-crRNA depending on the RNA modification positions shown in Figure 52. I14s04 iPS cells were transfected with mRNA expressing Cas3 and Cascade proteins, and B2M#1 crRNA (each having the chemical modifications corresponding to Figure 52), and the KO efficiency of the B2M gene was measured by staining for HLA-A, B, and C.
- I14s04 iPS cells were transfected multiple times with mRNA expressing Cas3 and Cascade proteins and Pre-both3 pre-crRNA targeting HLA-A*24:02 and B*52:01 ( Figure 5-1) using Lipofectamine Stem, and the collected genome samples were analyzed by ddPCR.
- "Dual A24 F1 & B52 R5" represents tandem pre-crRNA with MS modification at both ends.
- a schematic diagram of an example of chemical modification of tandem pre-crRNA is shown. Since Cas3 pre-crRNA is processed by Cas6, two or more target recognition sequences can be linked in tandem as shown in the figure. In this case, it is also possible to apply modifications such as three bases at both ends.
- the present invention provides a pre-crRNA (pre-crRNA) of a type I CRISPR-Cas system having modified nucleotides.
- the pre-crRNA (hereinafter, sometimes referred to as the "modified crRNA of the present invention") has at least one (e.g., 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) nucleotide that is a modified nucleotide in the region consisting of the 5' arm region of the first repeat sequence, the 5' side region of the stem-forming region of the first repeat sequence (hereinafter, sometimes referred to as the "5' stem-forming region”), and the loop-forming region of the first repeat sequence.
- a pre-crRNA of a type I CRISPR-Cas system having a 5' cap and polyA (hereinafter sometimes referred to as the "5'-capped crRNA of the present invention") is provided.
- the term "crRNA of the present invention” may be used to collectively refer to the "modified crRNA of the present invention” and the "5'-capped crRNA of the present invention”.
- type I CRISPR-Cas systems There are six known types of type I CRISPR-Cas systems, A to F, and any type of system can be used as the crRNA of the present invention as long as it has an arm region and a stem-loop forming region. Among them, type I-E (especially type I-E derived from Escherichia coli) or type I-D (especially type I-D derived from Microcystis aeruginosa) pre-crRNA is preferred. In the vicinity of CRISPR, there is a cas operon that encodes Cas (CRISPR-associated) proteins such as Cas1, Cas2, and proteins that constitute the Cascade complex.
- Cas Cas (CRISPR-associated) proteins
- the Cascade complex is composed of a group of proteins other than Cas3, which is a nuclease/helicase (however, type I-D Cas3 does not have these activities, and Cas10d functions as a nuclease/helicase), and Cas1, Cas2, and Cas4, which are involved in the cleavage of foreign genes during immunity acquisition.
- the Cascade complex includes Cas3 (or a complex of Cas3 and Cas10d) and exerts the same DNA recognition and cleavage function as Cas9 or Cas12a in the type II CRISPR-Cas system.
- Types I-A, I-B and I-D are relatively common in archaea, while types I-C, I-E and I-F are relatively common in eubacteria.
- Type I-A has been analyzed in S. solfataricus, T. tenax, etc., type I-B in Haloferax volcanii, etc., type I-C in B. halodurans, etc., type I-D in M. aeruginosa, etc., type I-E in E. coli, etc., and type I-F in P. aeruginosa, E. coli, P. atospeticum, etc.
- Cse1 also known as CasA, Cas8e
- Cse2 also known as CasB, Cas11
- Cas7 also known as CasC, Cas4
- Cas5 also known as CasD
- Cas6 also known as CasE
- Cascade complex in the ratio of 1:2:6:1:1 for one molecule of crRNA.
- Cas7, Cas5 (Csc1) and Cas6 form a Cascade complex in a ratio of 6:1:1 for one crRNA molecule.
- PAM sequences such as GTA, GTC and GTT can be used, while in type I-E CRISPR-Cas systems, ATG, AAG, AGG, GAG, TAG and AAA can be used.
- TCN N is A, T, G or C
- TTC ACT, TAA, TAT, TAG and CAC
- NTTC N is A, T, G or C
- CC can be used
- the base sequence is represented in the 5' to 3' direction.
- the Cascade complex is made up of Cas8a1, Csa5 (Cas11), Cas5, Cas6, and Cas7; in type I-B CRISPR-Cas systems, the Cascade complex is made up of Cas8b1, Cas5, Cas6, and Cas7; in type I-C CRISPR-Cas systems, the Cascade complex is made up of Cas8c, Cas5, and Cas7; in type I-F CRISPR-Cas systems, the Cascade complex is made up of Csy1 (Cas8f), Csy2 (Cas5), Cas6, and Csy3 (Cas7); and in type I-G CRISPR-Cas systems, the Cascade complex is made up of Cst1 (Cas8a1), Cas5, Cas6, and Cst2 (Cas7).
- Cas3, Cas10d and the Cascade protein group may be
- pre-crRNA when there are multiple spacer sequences, it is called repeat, spacer 1, repeat, spacer 2, repeat, (hereinafter referred to as repeat)) which has a structure consisting of repeat sequences and target recognition regions (spacer regions) as shown in Figure 3-1, is cleaved by Cas6 (in the case of types I-A, B, D-E) or Cas5 (in the case of type I-C) to become mature crRNA.
- pre-crRNA means an RNA which has at least repeat, spacer 1 and repeat, and which is cleaved by Cas6 or Cas5 in the cell to become functional crRNA.
- Figure 3-1 shows the pre-crRNA of type I-E of E. coli, which consists of repeat, spacer 1 and repeat.
- Other types of pre-crRNA repeat structures are illustrated in Figure 3-2 (Source: Zheng Y. et al., Front Bioeng Biotechnol. 2020 Mar 4;8:62).
- the sequence of the target recognition region (hereinafter sometimes referred to as the "spacer sequence") is originally a sequence derived from foreign DNA incorporated during the natural adaptation process, but the sequence can be designed based on the sequence of the target DNA.
- a pre-crRNA with one spacer region typically comprises a 5' arm region of the first repeat sequence (corresponding to the region consisting of nucleotides 1 to 5, GUGUU, in FIG.
- a stem-loop forming region of the first repeat sequence (corresponding to the region consisting of nucleotides 6 to 21, CCCCGCGCCAGCGGGG (SEQ ID NO: 1), in FIG. 3-1), a 3' arm region of the first repeat sequence (corresponding to the region consisting of nucleotides 22 to 29, AUAAACCG, in FIG. 3-1), a spacer region (corresponding to the region consisting of nucleotides 30 to 61, in FIG. 3-1), and a 5' arm region of the first repeat sequence (corresponding to the region consisting of nucleotides 31 to 35, AUAAACCG, in FIG. 3-1).
- the pre-crRNA includes a 5' arm region of the second repeat sequence (corresponding to the region consisting of nucleotides XXXXXNXXXXXXNXXXXXXNXXXXXXNXXXXXNXXXXXNXXXXXNXXXXXX), a 5' arm region of the second repeat sequence (corresponding to the region consisting of nucleotides 62-66 GUGUU in FIG. 3-1), a stem-loop forming region of the second repeat sequence (corresponding to the region consisting of nucleotides 67-82 CCCCGCGCCAGCGGGG in FIG. 3-1), and a 3' arm region of the second repeat sequence (corresponding to the region consisting of nucleotides 83-90 AUAAACCG in FIG. 3-1).
- FIG. 58 An example of a pre-crRNA with two spacer regions is shown in FIG. 58.
- a pre-crRNA is shown that includes the 5' arm region of the first repeat sequence, the stem loop forming region of the first repeat sequence, the 3' arm region of the first repeat sequence, the first spacer region (target sequence 1), the 5' arm region of the second repeat sequence, the stem loop forming region of the second repeat sequence, the 3' arm region of the second repeat sequence, as well as the second spacer region (target sequence 2), the 5' arm region of the third repeat sequence, the stem loop forming region of the third repeat sequence, and the 3' arm region of the third repeat sequence.
- the Cas3 protein (Cas10d in the case of Type I-D) has DNA nickase activity and DNA helicase activity, and introduces a nick into the non-target strand (the strand to which the target recognition region of the crRNA is not complementary (hybridized)).
- the PAM is present on the non-target strand, and typically, a nick is introduced at the upstream (5' side) site of the PAM sequence of the non-target strand, and then a defect occurs upstream of the PAM sequence due to a winding reaction by the helicase.
- target sequence refers to a sequence targeted by pre-crRNA, crRNA, or guide RNA, also called a protospacer sequence, and means a sequence adjacent to the 3' side of the PAM on the non-target strand where the PAM is present.
- the target sequence is a sequence that is homologous to the sequence of the target sequence recognition region present in the crRNA (sometimes referred to as a "spacer sequence") (however, U in the RNA sequence should be read as T in the DNA sequence).
- crRNAs having a spacer sequence in which at least one (e.g., two, three, four, five or more) bases have been substituted, deleted, added, and/or inserted in the base sequence portion represented by N above can also be used in the present invention.
- "homologous sequences” include not only sequences that are completely identical to the sequence of interest (e.g., a target sequence), but also sequences in which at least one (e.g., 2, 3, 4, 5 or more) bases have been substituted, deleted, added, and/or inserted.
- the target sequence may be appropriately selected depending on the purpose.
- Specific target sequences include, for example, the sequence of the ⁇ 2-microglobulin (B2M) gene and its control region (e.g., the sequence shown in SEQ ID NO: 4 or 5), the sequence of the Human Leukocyte Antigen (HLA) gene and its control region (e.g., the sequence shown in any of SEQ ID NOs: 6 to 38), the sequence of the dystrophin (DMD) gene and its control region (e.g., the sequence shown in any of SEQ ID NOs: 55 to 68), the sequence of the DMPK gene and its control region (e.g., the sequence shown in any of SEQ ID NOs: 39 to 53), the sequence of the AAVS1 (Adeno-associated virus integration site 1) region (e.g., the sequence shown in SEQ ID NO: 54), a repeat sequence and a sequence adjacent thereto, etc.
- B2M ⁇ 2-microglobulin
- HLA Human Leukocyte Antigen
- DMD dys
- HLA genes include HLA-A genes, HLA-B genes, HLA-C genes, HLA-E genes, HLA-F genes, HLA-G genes, HLA-DRA genes, HLA-DRB genes, HLA-DPA genes, HLA-DPB genes, HLA-DQA genes, and HLA-DQB genes.
- HLA-A genes HLA-A genes, HLA-B genes, HLA-C genes, HLA-E genes, HLA-F genes, HLA-G genes, HLA-DRA genes, HLA-DRB genes, HLA-DPA genes, HLA-DPB genes, HLA-DQA genes, and HLA-DQB genes.
- HLA-A genes HLA-A genes, HLA-B genes, HLA-C genes, HLA-E genes, HLA-F genes, HLA-G genes, HLA-DRA genes, HLA-DRB genes, HLA-DPA genes, HLA-DPB genes, HLA-DQA genes, and HLA-D
- repeat sequences include a sequence in which three bases of "CTG” are repeated, a sequence in which three bases of "CGG” are repeated, a sequence in which three bases of "CAG” are repeated, a sequence in which three bases of "GAA” are repeated, a sequence in which four bases of "CCTG” are repeated, and a sequence in which five bases of "TTTCA” are repeated.
- a pre-crRNA targeting any of the sequences shown in SEQ ID NOs: 4 to 68 is also provided.
- These pre-crRNAs can be used in combination with multiple pre-crRNAs having the same target gene or target exon.
- These pre-crRNAs may be wild-type pre-crRNAs or non-wild-type pre-crRNAs.
- These pre-crRNAs may be provided in the form of a kit or agent (e.g., a therapeutic agent, a reagent, etc.).
- the pre-crRNA before the introduction of modified nucleotides may be a wild-type pre-crRNA, or may have at least one (e.g., 2, 3, 4, 5 or more) base substituted, deleted, added and/or inserted compared to the wild-type pre-crRNA in at least any of the following regions (e.g., the 5' arm region of the first repeat sequence and/or the 3' arm region of the second repeat sequence): the 5' arm region of the first repeat sequence, the stem-loop forming region of the first repeat sequence, the 3' arm region of the first repeat sequence (the region corresponding to the 5' handle of the mature crRNA), the spacer region, the 5' arm region of the second repeat sequence, the stem-loop forming region of the second repeat sequence and the 3' arm region of the second repeat sequence (e.g., the 5' arm region of the first repeat sequence and/or the 3' arm region of the second repeat sequence), as long as the ability to recognize the target sequence and the ability to recruit Cascade are maintained; such
- At least one (e.g., 2, 3, 4, 5 or more) base may be substituted, deleted, added and/or inserted in at least one of the other regions (e.g., the second spacer region (target sequence 2), the 5' arm region of the third repeat sequence, the stem-loop forming region of the third repeat sequence, and the 3' arm region of the third repeat sequence) compared to the wild-type pre-crRNA.
- the second spacer region target sequence 2
- the 5' arm region of the third repeat sequence the stem-loop forming region of the third repeat sequence
- the 3' arm region of the third repeat sequence are also included in the "mutant pre-crRNA".
- the length of the spacer sequence and target sequence of the type 1 CRISPR-Cas system is not particularly limited, but may be, for example, 30 to 45 nucleotides, preferably 32 to 33 nucleotides for types I-C, I-E, and I-F, and preferably 34 to 44 nucleotides for types I-A, I-B, and I-D.
- modified nucleotide refers to a nucleotide other than a ribonucleotide in which at least one of the components of the ribonucleotide is modified.
- the components of the nucleotide include a sugar portion (e.g., ribose), a base, and a phosphate group, and the modified nucleotide has a modification at at least one of the sugar portion, the base, and the phosphate group (particularly the sugar portion).
- a nucleoside having a modification at at least one of the sugar portion and the base is also referred to as a "modified nucleotide", and in this specification, “modified nucleotide” can be read as “modified nucleoside” as appropriate.
- “modification” includes, for example, substitution, addition, and/or deletion in the components and/or internucleoside bonds, and substitution, addition, and/or deletion of atoms and/or functional groups in the components and/or internucleoside bonds.
- the modified nucleotide also includes deoxyribonucleotides, which are natural nucleotides.
- the modified crRNA of the present invention may contain only one type of modified nucleotide, or may contain multiple types of modified nucleotides.
- Natural bases include adenine, cytosine, guanine, thymine, and uracil.
- Modified bases obtained by modifying the bases include, but are not limited to, 5-methylcytosine, 5-fluorocytosine, 5-bromocytosine, 5-iodocytosine, or N4-methylcytosine; N6-methyladenine or 8-bromoadenine; and N2-methylguanine or 8-bromoguanine.
- modifications of the sugar moiety include 2'-O-methoxyethyl modification of the sugar moiety, 2'-O-methyl modification of the sugar moiety, 2' fluoro modification of the sugar moiety, and cross-linking between the 2' and 4' positions of the sugar moiety (the nucleotide having the cross-linked structure is BNA).
- the nucleotides contained in the modified crRNA of the present invention may be partially (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) nucleotides having 2'-O-methyl modification (i.e., 2'-O-methyl ribonucleotides), or may be entirely 2'-O-methyl ribonucleotides.
- BNA include locked artificial nucleic acid (LNA: locked nucleic acid) and 2'-O, 4'-C-ethylene bridged nucleic acid (ENA: 2'-O, 4'-C-ethylene bridged nucleic acid). More specifically, BNAs include those having the following nucleoside structure:
- R represents a hydrogen atom, an alkyl group having 1 to 7 carbon atoms which may be branched or cyclic, an alkenyl group having 2 to 7 carbon atoms which may be branched or cyclic, an aryl group having 3 to 12 carbon atoms which may contain a heteroatom, an aralkyl group having an aryl portion having 3 to 12 carbon atoms which may contain a heteroatom, or a protecting group for an amino group in nucleic acid synthesis.
- R is a hydrogen atom, a methyl group, an ethyl group, an n-propyl group, an isopropyl group, a phenyl group, or a benzyl group, and more preferably, R is a hydrogen atom or a methyl group.
- Base is a natural base or a modified base.
- some (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) or all of the internucleoside bonds may be bonds other than phosphodiester bonds.
- bonds other than phosphodiester bonds include, but are not limited to, phosphorothioate bonds, phosphorodithioate bonds, phosphotriester bonds, methylphosphonate bonds, methylthiophosphonate bonds, boranophosphate bonds, phosphoramidate bonds, etc.
- nucleosides having a modification e.g., 2'-O-methyl modification
- a modified internucleoside bond preferably, the 3' internucleoside bond
- a pre-crRNA is provided in which the phosphodiester bond at the 3' side of all modified nucleosides is a phosphorothioate bond.
- the modified crRNA of the present invention may contain only one type of modified internucleoside bond or multiple types of modified internucleoside bonds.
- the modified crRNA of the present invention at least one (e.g., 2, 3, 4, 5 or more) modified nucleotide is preferably present in the 5' arm region of the first repeat sequence, and all nucleotides of the 5' arm region may be modified nucleotides. In this case, at least one (e.g., 2, 3, 4, 5, 6, 7, 8 or more) nucleotides in the 3' arm region of the second repeat sequence may be modified nucleotides.
- the modified crRNA of the present invention has a modified nucleotide at the first nucleotide from the 5' end and a modified nucleotide at the second nucleotide from the 3' end.
- a pre-crRNA in which all nucleotides from the 5' end (corresponding to GUG in Figure 3-1) are modified nucleotides and all nucleotides from the 3' end (corresponding to ACC in Figure 3-1) are modified nucleotides is more preferred.
- at least one (e.g., two, three, four or more) or all nucleotides may be modified nucleotides.
- the Mth nucleotide (M is a positive integer) from the 5' end means the Mth nucleotide toward the 3' end, with the nucleotide located at the 5' end in Figure 3-1 as the first nucleotide.
- a leader sequence may be added to the 5' side of the pre-crRNA, and in that case, the position is defined based on the sequence excluding the leader sequence.
- the Mth nucleotide (M is a positive integer) from the 3' end means the Mth nucleotide toward the 5' end, with the nucleotide located at the 3' end as the first nucleotide.
- modified nucleotides in the 3' region of the stem-forming portion of the first repeat sequence also referred to as the "3' stem-forming region" and the stem-forming portion of the second repeat sequence.
- these regions may or may not contain modified nucleotides, but from a cost perspective, it is preferable that they do not contain modified nucleotides (i.e., all ribonucleotides).
- modified nucleotides reduces genome editing efficiency in the 3' arm region of the first repeat sequence, as well as the 5' arm region and 5' stem-forming region of the second repeat sequence.
- the 3' arm region of the first repeat sequence, the 5' arm region of the second repeat sequence, and the 5' stem-forming region of the second repeat sequence of the modified crRNA of the present invention are composed only of ribonucleotides, and further, the 3' stem-forming region of the first repeat sequence and the stem-forming region of the second repeat sequence may also be composed only of ribonucleotides.
- the spacer sequence at least one nucleotide (e.g., 2, 3, 4, 5, 6, 7, 8 or more) may be a modified nucleotide, but in one embodiment, all of the nucleotides are ribonucleotides.
- the modified crRNA of the present invention typically has a phosphodiester bond at the 3' end of the ribonucleoside.
- cleavage of pre-crRNA into mature crRNA in cells may be important for activation of genome editing. Therefore, it is preferable that the sites cleaved by Cas6 or Cas5 (specifically, the phosphodiester bond between the 3'-terminal nucleoside of the stem-forming region of the first repeat sequence and the 5'-terminal nucleoside of the 3'-arm region of the first repeat sequence, and the phosphodiester bond between the 3'-terminal nucleoside of the stem-forming region of the second repeat sequence and the 5'-terminal nucleoside of the 3'-arm region of the second repeat sequence) and/or the nucleosides adjacent to these sites (particularly the nucleosides on the 3' side) are not modified.
- the 5' cap includes not only 7-methyl guanosine (m 7 G) found in natural mRNA, but also its analogs (e.g., ARCA (anti-reverse cap analog) in which the 3'-OH group of the sugar of m 7 G is methylated).
- the length of the polyA conferred to the crRNA of the present invention is not particularly limited, but since a polyA consisting of 200 to 250 adenines is usually added in mammals, the polyA may typically be 10 or more (e.g., 20, 30, 40, 50 or more) and 300 or less (e.g., 250, 240, 230, 220, 210, 200 or less).
- the crRNA of the present invention may be bound to one or more (e.g., 1, 2, 3, 4 or more) functional molecules.
- the functional molecules may typically be bound to the 5'-terminus and/or 3'-terminus.
- the bond between the crRNA of the present invention and the functional molecules may be a direct bond or an indirect bond mediated by another substance.
- the functional molecules are directly bound to the oligonucleotide via a covalent bond, an ionic bond, a hydrogen bond, etc., and a covalent bond is more preferred in terms of obtaining a more stable bond.
- the functional molecules may also be bound to the crRNA via a cleavable linking group.
- the functional molecules may be linked via a disulfide bond. Only one type of functional molecule may be used, or multiple types may be used in combination.
- Desired functions include, for example, labeling, purification, and delivery functions.
- molecules that confer a labeling function include compounds such as fluorescent dyes (Cy3, Alexa, etc.), fluorescent proteins, and luciferase.
- moieties that confer a purification function include compounds such as biotin, avidin, His tag peptide, GST tag peptide, and FLAG tag peptide.
- Molecules that confer delivery function include, for example, arginine-rich peptides P007 and B peptide (HaiFang Yin et al., Human Molecular Genetics, Vol.
- m3G-CAP Pedro M. D. Moreno et al., Nucleic Acids Res., Vol. 37, 1925-1935 (2009)
- TAT peptide lipids such as N-acetylgalactosamine (GalNAc), cholesterol and fatty acids (e.g., vitamin E (tocopherol, tocotrienol), vitamin A, and vitamin D); fat-soluble vitamins such as vitamin K (e.g., acylcarnitines); intermediate metabolites such as acyl-CoA; glycolipids, glycerides, and their derivatives or analogs.
- vitamin K e.g., acylcarnitines
- intermediate metabolites such as acyl-CoA
- the crRNA of the present invention may be produced by chemical synthesis or by in vitro transcription (IVT) method. Alternatively, it may be expressed in an organism (Escherichia coli or cultured mammalian cells) and then purified. Examples of chemical synthesis methods include a method using nucleoside phosphoramidites and solid-phase supports. In addition, modified nucleotides can be introduced into any base site of the crRNA sequence by using nucleoside phosphoramidites that have been chemically modified (e.g., 2'-O-methylated phosphoramidites of the sugar moiety, GalNAc phosphoramidite, etc.).
- nucleoside phosphoramidites that have been chemically modified (e.g., 2'-O-methylated phosphoramidites of the sugar moiety, GalNAc phosphoramidite, etc.).
- single-stranded RNA can be synthesized by T7 RNA polymerase from DNA encoding pre-crRNA linked under the control of a T7 promoter, but by including a 5' cap in the reaction solution, pre-crRNA with a 5' cap structure can be synthesized. Furthermore, by including a modified nucleoside triphosphate in the reaction solution, pre-crRNA containing a modified nucleotide can be synthesized.
- PolyA can be added by reacting the crRNA with polyA polymerase.
- pre-crRNA with polyA added can be synthesized by linking polyT to the DNA encoding the pre-crRNA by the IVT reaction.
- a 5' cap can also be added to the pre-crRNA by chemical synthesis.
- a 5' cap can be added to the pre-crRNA by reacting a chemically synthesized pre-cRNA with an imidazole-activated form of m7G diphosphate in an organic solvent (e.g., dimethyl sulfoxide).
- an organic solvent e.g., dimethyl sulfoxide.
- a pre-crRNA in which a 5' cap and polyA are further added to the modified crRNA of the present invention can also be suitably used.
- a desired pre-crRNA can be obtained by specifying the base sequence and the modification site or type and ordering it from a manufacturer.
- a pre-crRNA other than the wild type which includes a mutant nucleotide, a 5' cap structure, a 3' polyA, etc., as in the crRNA of the present invention, is also referred to as an "artificial pre-crRNA".
- the artificial pre-crRNA may have a region added thereto that is not found in the wild-type pre-crRNA.
- DNA encoding the pre-crRNA can be chemically synthesized, for example, by designing a sequence encoding the pre-crRNA that contains a base sequence homologous to the target sequence (i.e., a spacer sequence) and using a DNA/RNA synthesizer.
- a pre-crRNA can be designed to target a sequence in which a heterozygous SNP exists on the target sequence other than the base.
- the position of the heterozygous SNP is preferably close to the PAM sequence, and more preferably exists in the seed region or its vicinity.
- the seed region is a region that is highly homologous (typically completely homologous) to the spacer sequence of the crRNA.
- Non-Patent Document 1 it is the first to eighth region counting from the terminal base on the downstream (3') side of the PAM sequence (Non-Patent Document 1), and the vicinity of the seed region is typically the ninth to eleventh region counting from the terminal base on the downstream (3') side of the PAM sequence.
- a pre-crRNA may be designed to target a sequence in which a heterozygous SNP exists on the PAM sequence.
- a crRNA is designed to target a sequence in which a valid PAM sequence (e.g., ATG in the case of a type IE CRISPR-Cas system) exists on the allele in which it is desired to introduce a deletion (target allele), and the corresponding sequence on the other allele is an invalid PAM sequence (e.g., CTG).
- the cRNA designed in this way is also called an "allele-specific crRNA", and a deletion site can be introduced upstream (5' side) of the PAM sequence in the target allele by using a CRISPR-Cas system containing an allele-specific crRNA.
- the deletion site targeted by such a system is also called a deletion target site.
- a method for designing an allele-specific pre-crRNA is provided.
- a method for designing an allele-specific pre-crRNA for a type I CRISPR-Cas system (hereinafter sometimes referred to as the "design method of the present invention") is provided, which comprises the steps of designing a pre-crRNA that targets a sequence in which a deletion target site exists upstream (5') of a PAM sequence adjacent to a target sequence, and which is within the target sequence and has at least one base (e.g., 1, 2, 3, 4, 5 or more bases) that differs between alleles at a position other than the 6nth position (n is a positive integer) counting from the terminal base downstream (3') of the PAM sequence, or on the PAM sequence.
- a base e.g., 1, 2, 3, 4, 5 or more bases
- the pre-crRNA designed by the design method of the present invention is not particularly limited as long as it is a crRNA that constitutes a type I CRISPR-Cas system, but is preferably a type I-E (particularly, type I-E derived from Escherichia coli) or type I-D (particularly, type I-D derived from Microcystis aeruginosa) pre-crRNA, with type I-E pre-crRNA being more preferred.
- allele-specific means targeting only one allele (usually the allele that causes a disease).
- targeting only one allele includes not only aspects in which the other allele is not targeted at all (or is predicted not to be targeted at all), but also aspects in which the genome editing efficiency of the other allele is lower (or is predicted to be lower) than the genome editing efficiency of the gene of the targeted allele.
- Single nucleotide polymorphism generally refers to a single nucleotide mutation that is frequently (e.g., at a frequency of 1% or more) found in the genomic sequence of a certain biological population, but in this specification, it refers to at least one nucleotide that differs between alleles (also referred to as a “heterogeneous SNP").
- heterogeneous SNPs When there are two or more heterogeneous SNPs, all of the nucleotides may be present at positions other than the 6nth (n is a positive integer) nucleotide counting from the downstream terminal nucleotide of the PAM sequence or on the PAM sequence, or only some of the nucleotides may be present at that position or on the PAM sequence.
- Heterogeneous SNPs can be identified, for example, by searching for frequently occurring SNPs in an SNP database and, for the SNPs found, checking whether the SNPs are also present in cells derived from disease patients. Examples of the SNP databases mentioned above include dbSNP, Kaviar, SNPedia, OMIM database, dbSAP, The Human Gene Mutation Database, The International HapMap Project, and GWAS Central.
- the alleles targeted by the design method of the present invention are not particularly limited, but include, for example, alleles that cause repeat diseases caused by abnormal expansion of a repeat sequence of a specific base sequence. It is believed that abnormality in the number of repeats causes disease through mechanisms such as loss of function or gain of function of the gene product (protein or RNA), abnormal liquid-liquid phase separation formation, or non-classical translation.
- repeat diseases diseases in which a triplet repeat sequence is abnormally expanded are known as triplet diseases. It is known that triplet diseases typically occur when there are 20 or more triplet repeats (typically 35 or more repeats).
- the CRISPR-Cas9 system in which base recognition and DNA cleavage occur at approximately the same site, it was difficult to shorten such abnormally expanded repeats by deleting the repeat sequence allele-specifically using hetero SNPs at distant positions.
- the CRISPR-Cas3 system can treat triplet disease by inducing deletions to shorten the abnormally expanded repeats.
- the deletion target site is a region that contains a triplet repeat.
- the above repeat diseases include, for example, fragile X syndrome (repeat: CGC, causative gene: FMR1), fragile XE syndrome (repeat: CCG, causative gene: AFF2), Friedreich's ataxia (repeat: GAA, causative gene: FXN), Huntington's disease (repeat: CAG, causative gene: HTT), spinal-bulbar muscular atrophy (repeat: CAG, causative gene: AR), spinocerebellar ataxia type 1 (repeat: CAG, causative gene: ATXN1), spinocerebellar ataxia type 2 (repeat: CAG, causative gene: ATXN2), spinocerebellar ataxia type 3 (repeat: CAG, causative gene: ATXN3), and spinocerebellar ataxia type 6 (repeat: CAG, causative gene: CACN A1A), spinocerebellar ataxia type 7 (repeat: CA
- the repeat disease targeted by the design method of the present invention is myotonic dystrophy type 1 (DM1), known as one of the triplet diseases.
- DM1 is a dominant genetic disease caused by abnormal expansion of the CTG repeat in only one allele in the 3'-UTR region of the DMPK gene.
- heterozygous SNPs for DM1 in the dbSNP database examples include SNP ID: rs16939 (distance from repeat: 2.5 kb), SNP ID: rs915915 (distance from repeat: 1.5 kb), SNP ID: rs558794490 (distance from repeat: 0.8 kb), SNP ID: rs635299 (distance from repeat: 0.6 kb), SNP ID: rs934739524 (distance from repeat: 1.6 kb), and SNP ID: rs3745802 (distance from repeat: 2.3 kb).
- sequences targeted by the pre-crRNA prepared by the design method of the present invention include sequences shown in any of SEQ ID NOs: 39, 40, and 42 to 47, which contain SNP ID: rs934739524 in the target sequence, sequences shown in any of SEQ ID NOs: 48 to 50, which contain SNP ID: rs635299 in the target sequence, sequences shown in SEQ ID NOs: 51 or 52, which contain SNP ID: rs915915 in the target sequence, and sequences shown in SEQ ID NO: 53, which contains SNP ID: rs3745802 in the target sequence.
- the activity and specificity of the crRNA designed by the design method of the present invention may differ depending on the spacer sequence. Therefore, the target allele specificity may be increased by adjusting the crRNA and decreasing the activity of non-target alleles. Examples of such adjustment methods include shortening the length of the crRNA (e.g., deleting 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more nucleotides) and intentionally introducing a base mismatch at a position distant from the PAM.
- the design method of the present invention may include a step of adjusting the length of the pre-crRNA (in one embodiment, the length of the spacer sequence) and/or a step of introducing mismatches into the pre-crRNA (in one embodiment, the spacer sequence).
- the number of mismatches introduced may be, for example, one to several (e.g., 2, 3, 4, 5 or more).
- the position at which the mismatch is introduced may be adjusted to further adjust the allele specificity.
- the design method of the present invention it is not necessary to actually synthesize the pre-crRNA; it is sufficient to imagine it in one's mind.
- the imagined nucleic acid is embodied in a program that operates on a computer (e.g., nucleic acid design software, graphic design tools, office software, etc.) or on paper.
- the designed pre-crRNA may be actually synthesized, and the allele specificity and genome editing activity of the synthesized pre-crRNA may be evaluated.
- the crRNA designed by the design method of the present invention may be a wild-type pre-crRNA or an artificial pre-crRNA including a mutant pre-crRNA.
- the definitions and types of mutant nucleotides, 5' cap structure, and 3' polyA, the definitions and preparation methods of each pre-crRNA, etc. are all as described in "1. Pre-crRNA having modified nucleotides" above.
- a type I CRISPR-Cas system which includes the crRNA of the present invention.
- a type I CRISPR-Cas system (hereinafter, sometimes referred to as the "CRISPR-Cas system of the present invention") is provided, which includes (1) the crRNA of the present invention, and (2) a Cascade protein group (Cse1, Cse2, Cas7, Cas5 and Cas6 in the case of type IE) or a nucleic acid encoding the protein group.
- the CRISPR-Cas system of the present invention may include (3) a Cas3 protein or a nucleic acid encoding the protein.
- the CIRSPR-Cas system of the present invention may include a Cas3 protein or a nucleic acid encoding the protein, as well as a Cas10d protein or a nucleic acid encoding the protein.
- the nucleic acid encoding the Cas protein may be RNA or DNA.
- the CRISPR-Cas system of the present invention is a type I CRISPR-Cas system, but is preferably a type IE (particularly, type IE derived from Escherichia coli) or type ID (particularly, type ID derived from Microcystis aeruginosa) CRISPR-Cas system, and more preferably a type IE CRISPR-Cas system.
- type I CRISPR-Cas are as described in "1. Pre-crRNA having modified nucleotides”.
- the CIRSPR-Cas system of the present invention can be used to modify double-stranded DNA contained in a cell.
- a method for modifying double-stranded DNA contained in a cell or a method for producing a cell with modified double-stranded DNA (hereinafter sometimes referred to as the "method of the present invention") is provided, which includes a step of introducing the CRISPR-Cas system of the present invention into the cell.
- double-stranded DNA contained in a cell examples include chromosomal DNA, mitochondrial DNA, chloroplast DNA (hereinafter, these are also collectively referred to as “genomic DNA”), and exogenous DNA (e.g., plasmid DNA, viral DNA), but genomic DNA, particularly chromosomal DNA, is preferred.
- genomic DNA particularly chromosomal DNA
- “modification” means that a certain nucleotide or nucleotide sequence on a DNA strand is deleted, replaced with another nucleotide and/or nucleotide sequence, and/or a nucleotide or nucleotide sequence is inserted into a certain region on a DNA strand. Modification on genomic DNA is also sometimes referred to as "genome editing".
- nuclease activity and helicase activity of Cas3 and Cas10d are not necessarily required, so Cas and Cas10d do not have to be used, or mutants that have lost some or all of these activities (e.g., D domain H74A mutant (dnCas3), SF2 domain motif 1 K320N mutant (dhCas3), and SF2 domain motif 3 S483A/T485A double mutant (dh2Cas3)) can also be used.
- D domain H74A mutant dnCas3
- dhCas3 SF2 domain motif 1 K320N mutant
- dh2Cas3 SF2 domain motif 3 S483A/T485A double mutant
- a fusion protein of a mutant in which Cas3 nuclease activity has been partially or completely eliminated and a deaminase as a component of the CRISPR-Cas3 system of the present invention precise genome editing is possible by replacing bases without causing large deletions at the target site.
- pinpoint base modification is also possible by fusing a Cascade protein (e.g., Cas6) with a deaminase (e.g., hAID, PmCDA1, etc.) without using Cas3.
- DNA modification without changing the base sequence is also possible by using a protein with methyltransferase activity (e.g., DNA methyltransferase, histone methyltransferase, etc.), a protein with demethylase activity (e.g., DNA demethylase, histone demethylase, etc.), histone acetyltransferase, or histone deacetylase.
- a protein with methyltransferase activity e.g., DNA methyltransferase, histone methyltransferase, etc.
- a protein with demethylase activity e.g., DNA demethylase, histone demethylase, etc.
- histone acetyltransferase e.g., histone deacetylase
- the DNA encoding the Cas protein is typically provided in the form of an expression vector containing the DNA.
- expression vectors include viral vectors such as retroviruses, lentiviruses, adenoviruses, adeno-associated viruses, herpes viruses, and Sendai viruses, as well as plasmid vectors, episomal vectors, artificial chromosome vectors, and transposon vectors (piggyBac, piggyBat, TolII), etc.
- Promoters used in expression vectors include, for example, the EF1 ⁇ promoter, ACTB promoter, UbqC promoter, PGK promoter, CAG promoter, SR ⁇ promoter, SV40 promoter, LTR promoter, CMV (cytomegalovirus) promoter, RSV (Rous sarcoma virus) promoter, MoMuLV (Moloney murine leukemia virus) LTR, HIV LTR, and HSV-TK (herpes simplex virus thymidine kinase) promoter.
- the EF1 ⁇ promoter, ACTB promoter, UbqC promoter, PGK promoter, CAG promoter, and SR ⁇ promoter are preferred.
- the expression vector may contain an enhancer, a terminator, an IRES, a 2A coding sequence enhancer, a polyA addition signal, an SV40 replication origin, a selection marker gene, etc., as desired.
- selection marker genes include drug resistance genes and fluorescent protein genes.
- the expression vector may be a combination of individual expression vectors capable of expressing each component of the CRISPR-Cas system (e.g., in the case of type I-E, the Cse1 protein, Cse2 protein, Cas7 protein, Cas5 protein, Cas6 protein, Cas3 protein, and crRNA factors), or one expression vector may be prepared to be capable of expressing multiple of these factors, or one expression vector may be prepared to be capable of expressing all of these factors.
- each component of the CRISPR-Cas system may be linked by a 2A sequence that induces self-cleavage, an IRES (Internal Ribosome Entry Site) sequence having a ribosome binding site, etc.
- IRES Internal Ribosome Entry Site
- 2A sequences include the P2A sequence derived from Porcine teschovirus, the T2A sequence derived from Thosea asigne, the F2A sequence derived from foot-and-mouth disease virus, and the E2A sequence derived from equine rhinitis A virus.
- the IRES sequence may be a sequence derived from a virus such as Encephalomyocarditis virus or Foot-and-mouth disease virus, or may be a sequence derived from an mRNA in a cell. This allows two or more proteins to be expressed individually from a single mRNA.
- RNA DNA editing can be performed with high efficiency in pluripotent stem cells by introducing three types of RNA: (1) two types of mRNA that individually express each of the three constituent proteins of the Cse1 protein, Cse2 protein, Cas7 protein, Cas5 protein, Cas6 protein, and Cas3 protein, and (2) wild-type pre-crRNA. Therefore, in an embodiment in which the Cascade protein group is introduced into stem cells in the form of mRNA, for example, an mRNA that polycistronically codes for 2 to 4 proteins that constitute Cascade may be used, or an mRNA that polycistronically codes for the three constituent proteins may be used.
- the present inventors further revealed that in a mode of genome editing by introducing two types of expression vectors, (1) a single expression vector having two promoters and in which three genes each of the Cse1 gene, Cse2 gene, Cas7 gene, Cas5 gene, Cas6 gene, and Cas3 gene are driven by one promoter, and (2) an expression vector of pre-crRNA, the efficiency of genome editing in pluripotent stem cells is significantly different between when the transcription directions of the two promoters are in the same direction (uni-directional promoter) and when they are in opposite directions (bi-directional promoter). They also revealed that the genome editing efficiency is highest when the two promoters are in opposite directions.
- an expression cassette mode in which three genes are expressed by one promoter each (two promoters in total) is particularly preferred, and the two expression cassettes may be on the same expression vector or on different expression vectors.
- the most preferred embodiment is one in which two expression cassettes carrying the three genes are arranged in opposite directions on the same expression vector (bi-directional promoter).
- the promoter for expressing the Cas protein may be an expression-inducible promoter.
- a promoter whose expression can be induced by the addition or removal of an expression control substance to the medium, by light irradiation, by a temperature change, or the like can be used as the expression-inducible promoter.
- the expression-inducible promoter may be one in which the expression of a protein is induced by the addition of an expression control substance to the medium, or one in which the expression of a protein is induced by the removal of an expression control substance from the medium.
- expression-inducible promoters include, but are not limited to, Tet-ON/Tet-OFF promoters (induced by the addition or removal of tetracycline or its derivatives (e.g., doxycycline)), metallothionein promoters (induced by heavy metal ions), heat shock protein promoters (induced by heat shock), and steroid-responsive promoters (induced by steroid hormones or their derivatives).
- Nucleic acids e.g., the crRNA of the present invention
- expression vectors or proteins e.g., Cas3, Cas10d, proteins constituting the Cascade complex (hereinafter also referred to as "Cascade proteins"
- Cascade proteins proteins constituting the Cascade complex
- Such methods include, for example, calcium phosphate-mediated transfection, electroporation, liposome transfection, lipofection, gene gun, microinjection, viral vector method, virus-like particle method, Agrobacterium method, agroinfiltration method, PEG-calcium method, sonoporation method, lipid nanoparticle method, etc.
- the CRISPR-Cas system of the present invention may be introduced into cells multiple times.
- a single introduction is the norm because deletions and mutations occur in the target sequence and the risk of off-target is high.
- the type I CRISPR-Cas system a large deletion is introduced into the PAM sequence side of the target sequence, but the sequence targeted by the crRNA (i.e., the target sequence) is often preserved, so by introducing the same crRNA multiple times into the same cell, it is possible to improve the deletion efficiency and extend (accumulate) the deletion site.
- the number of times it is introduced is not particularly limited, but is typically 2 to 10 times (e.g., 2, 3, or 4 times).
- the interval at which the CRISPR-Cas system is introduced is not particularly limited, but typically, the CRISPR-Cas system is introduced at intervals of 1 to 15 days, preferably every 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 days.
- the cells into which the CRISPR-Cas system has been introduced may be cryopreserved, and after several days to several years, the cells may be thawed and the CRISPR-Cas system may be introduced again into the cells.
- a method for extending a deletion site which includes a step of introducing a type I CRISPR-Cas system into a cell multiple times.
- Such a CRISPR-Cas system typically includes (I) a wild-type pre-crRNA, (II) a Cas3 protein or a nucleic acid encoding the protein, and (III) a Cascade protein group or a nucleic acid encoding the protein group.
- the CRISPR-Cas system is preferably a type I-E (particularly, type I-E derived from Escherichia coli) or type I-D (particularly, type I-D derived from Microcystis aeruginosa) CRISPR-Cas system, and a type I-E CRISPR-Cas system is more preferable.
- the CRISPR-Cas system may further include a Cas10d protein or a nucleic acid encoding the protein.
- the types and definitions of Type I CRISPR-Cas are as described in "1. Pre-crRNA with modified nucleotides.”
- DNA encoding Cas proteins can be obtained, for example, by isolating regions containing the ORF of the desired Cas protein from the cas operon by genomic PCR using genomic DNA from the above-mentioned bacterial species as a template.
- DNA encoding Cas proteins can also be cloned by synthesizing oligo DNA primers based on the cDNA sequence information or amino acid sequence information of the protein to be used (for example, information from databases such as NCBI GenBank), and amplifying the protein by RT-PCR using total RNA or mRNA fractions prepared from cells that produce the protein as a template.
- NCBI accession numbers for Cse1 protein belonging to type I-E derived from E. coli include, for example, NP_417240.1.
- NCBI accession numbers for Cse2 protein include, for example, NP_417239.1.
- NCBI accession numbers for Cas7 protein include, for example, NP_417238.1.
- NCBI accession numbers for Cas5 protein include, for example, NP_417237.2.
- NCBI accession numbers for Cas6 protein include NP_417236.1, and for Cas3 protein include NP_417241.1.
- Other Cas proteins can be obtained from NCBI GenBank and other databases. Sequences from new microbial species can also be obtained using the BLAST program from microbial genome data obtained by metagenomic analysis.
- the Cas protein may have one or several (e.g., 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) amino acids substituted, deleted, added and/or inserted with respect to the amino acid sequence described in the above-mentioned accession number, so long as the function of the protein is maintained.
- the Cas protein may also have an amino acid sequence that has an identity of 70% or more, preferably 80% or more, more preferably 90% or more, and even more preferably 95% or more (e.g., 96%, 97%, 98%, 99% or more) with the amino acid sequence described in the above-mentioned accession number.
- the identity of the amino acid sequence can be calculated using the homology calculation algorithm NCBI BLAST (National Center for Biotechnology Information Basic Local Alignment Search Tool).
- the cloned DNA can be used as is, or after digestion with restriction enzymes as desired, or after adding a sequence encoding an appropriate linker and/or nuclear localization signal (or an organelle localization signal if the target double-stranded DNA is mitochondrial or chloroplast DNA), it can be ligated with Cas3, Cas10d, or Cascade protein to prepare DNA encoding a fusion protein. Only one or more organelle localization signals including a nuclear localization signal may be added (e.g., added to both the N-terminus and C-terminus of a protein).
- the method for preparing DNA encoding pre-crRNA is as described above in "1. Pre-crRNA with modified nucleotides.”
- RNA encoding Cas3, Cas10d, or Cascade protein can be synthesized, for example, by an IVT reaction using DNA encoding Cas3, Cas10d, or Cascade protein as a template.
- Cas3, Cas10d or Cascade protein can be produced by an in vitro translation system. Alternatively, it can be obtained by expressing the protein in a cell using an expression vector or the like, and isolating and purifying the protein from the cell.
- Cells used in the method of the present invention include, for example, Escherichia genus, Bacillus genus, yeast, insect cells, insects, animal cells, plant cells, and the like.
- Escherichia bacteria that can be used include Escherichia coli K12-DH1 [Proc. Natl. Acad. Sci.
- yeast examples include Saccharomyces cerevisiae AH22, AH22R - , NA87-11A, DKD-5D, and 20B-12, Schizosaccharomyces pombe NCYC1913 and NCYC2036, and Pichia pastoris KM71.
- insect cells include Spodoptera frugiperda cells (Sf cells) derived from the larvae of the night thief moth, MG1 cells derived from the midgut of Trichoplusia ni, High Five TM cells derived from the eggs of Trichoplusia ni, cells derived from Mamestra brassicae, cells derived from Estigmena acrea, and cell lines derived from silkworms (Bombyx mori N cells (BmN cells)).
- Sf cells include Sf9 cells (ATCC CRL1711) and Sf21 cells (see In Vivo, 13, 213-217 (1977)).
- Plant cells that can be used include suspension culture cells, calli, protoplasts, leaf segments, and root segments prepared from various plants (e.g., grains such as rice, wheat, and corn; commercial crops such as tomato, cucumber, and eggplant; horticultural plants such as carnation and lisianthus; and experimental plants such as tobacco and Arabidopsis).
- plants e.g., grains such as rice, wheat, and corn; commercial crops such as tomato, cucumber, and eggplant; horticultural plants such as carnation and lisianthus; and experimental plants such as tobacco and Arabidopsis).
- Animal cells that can be used include, for example, cells isolated from a living body, cells in a living body, cell lines such as monkey COS-7 cells, monkey Vero cells, Chinese hamster ovary (CHO) cells, mouse L cells, mouse AtT-20 cells, mouse myeloma cells, rat GH3 cells, HeLa cells, and human FL cells, pluripotent stem cells from humans and other mammals (mouse, rat, dog, monkey, etc.), and primary cultured cells prepared from various tissues. Furthermore, zebrafish embryos and Xenopus oocytes can also be used.
- cell lines such as monkey COS-7 cells, monkey Vero cells, Chinese hamster ovary (CHO) cells, mouse L cells, mouse AtT-20 cells, mouse myeloma cells, rat GH3 cells, HeLa cells, and human FL cells, pluripotent stem cells from humans and other mammals (mouse, rat, dog, monkey, etc.), and primary cultured cells prepared from various tissues.
- Mammals are preferred as animal cells for use in the method of the present invention, and examples of such mammals include rodents such as mice, rats, hamsters, and guinea pigs, primates such as humans, rhesus monkeys, cynomolgus monkeys, Japanese monkeys, and chimpanzees, cows, horses, dogs, and cats.
- rodents such as mice, rats, hamsters, and guinea pigs
- primates such as humans, rhesus monkeys, cynomolgus monkeys, Japanese monkeys, and chimpanzees, cows, horses, dogs, and cats.
- the method of the present invention is expected to produce a high genome editing effect even in pluripotent stem cells, which have traditionally exhibited low genome editing efficiency. Therefore, pluripotent stem cells are preferred as the cells to be used in the method of the present invention.
- pluripotent stem cells refer to stem cells that can differentiate into tissues and cells with various different morphologies and functions in the body, and have the ability to differentiate into cells of any lineage of the three germ layers (endoderm, mesoderm, and ectoderm).
- pluripotent stem cells used in the present invention include induced pluripotent stem cells (iPS cells), embryonic stem cells (ES cells), embryonic stem cells derived from cloned embryos obtained by nuclear transfer (ntES cells), multipotent germline stem cells (mGS cells), and embryonic germline stem cells (EG cells), with iPS cells (more preferably human iPS cells) and ES cells (more preferably human ES cells) being preferred.
- the pluripotent stem cells are ES cells or any cells derived from a human embryo, the cells may be cells produced by destroying an embryo or cells produced without destroying an embryo, but preferably cells produced without destroying an embryo.
- ES cells are stem cells that are pluripotent and have the ability to proliferate through self-renewal and are established from the inner cell mass of (early) embryos (e.g., blastocysts) of mammals such as humans and mice.
- ES cells were discovered in mice in 1981 (M.J. Evans and M.H. Kaufman (1981), Nature 292:154-156), and subsequently, ES cell lines were established in humans, monkeys, and other primates (J.A. Thomson et al. (1998), Science 282:1145-1147; J.A. Thomson et al. (1999), Science 282:1145-1147). (1995), Proc. Natl. Acad. Sci. USA, 92:7844-7848; J.A.
- ES cells can be established by extracting the inner cell mass from the blastocyst of a fertilized egg of a target animal and culturing the inner cell mass on fibroblast feeders.
- ES cells can be established using only a single blastomere from an embryo at the cleavage stage prior to the blastocyst stage (Chung Y. et al. (2008), Cell Stem Cell 2: 113-117) or from developmentally arrested embryos (Zhang X. et al. (2006), Stem Cells 24: 2669-2676.).
- ES cell lines used in the present invention for example, various mouse ES cell lines established by inGenious targeting laboratory, RIKEN (Riken) and the like can be used for mouse ES cells, and for human ES cell lines, for example, various human ES cell lines established by University of Wisconsin, NIH, RIKEN, Kyoto University, National Center for Child Health and Development, Cellartis and the like can be used.
- human ES cell lines include CHB-1 to CHB-12 strains, RUES1 strain, RUES2 strain, HUES1 to HUES28 strains, etc. distributed by ESI Bio, H1 strain, H9 strain, etc. distributed by WiCell Research, and KhES-1 strain, KhES-2 strain, KhES-3 strain, KhES-4 strain, KhES-5 strain, SSES1 strain, SSES2 strain, SSES3 strain, etc. distributed by RIKEN.
- iPS cells are cells that can be obtained by reprogramming mammalian somatic cells or undifferentiated stem cells through the introduction of specific factors (nuclear reprogramming factors).
- iPSCs established by Yamanaka et al. by introducing the four factors Oct3/4, Sox2, Klf4, and c-Myc into mouse fibroblasts (Takahashi K, Yamanaka S., Cell, (2006) 126: 663-676), human cell-derived iPSCs established by introducing the same four factors into human fibroblasts (Takahashi K, Yamanaka S., et al.
- Nanog-iPSCs established by selecting using the expression of Nanog as an indicator after the introduction of the above four factors
- Other examples that can be used include iPSCs produced by a method that does not include c-Myc (Ichisaka, T., and Yamanaka, S. (2007). Nature 448, 313-317.), iPSCs produced by a method that does not include c-Myc (Nakagawa M, Yamanaka S., et al. Nature Biotechnology, (2008) 26, 101-106), and iPSCs established by introducing six factors using a virus-free method (Okita K et al. Nat.
- induced pluripotent stem cells established by introducing the four factors OCT3/4, SOX2, NANOG, and LIN28 created by Thomson et al. (Yu J., Thomson JA. et al., Science (2007) 318: 1917-1920.), induced pluripotent stem cells created by Daley et al. (Park IH, Daley GQ. et al., Nature (2007) 451: 141-146), and induced pluripotent stem cells created by Sakurada et al. (JP Patent Publication No. 2008-307007) can also be used.
- human iPSC strains include RIKEN's HiPS-RIKEN-1A strain, HiPS-RIKEN-2A strain, HiPS-RIKEN-12A strain, Nips-B2 strain, etc., and Kyoto University's 253G1 strain, 253G4 strain, 1201C1 strain, 1205D1 strain, 1210B2 strain, 1383D2 strain, 1383D4 strain, 1383D6 strain, 201B7 strain, 409B2 strain, 454E2 strain, 606A1 strain, 610B1 strain, 648A1 strain, 1231A3 strain, Ff-I01s04 strain, Ff-I14s04 strain, etc.
- Cells into which the CIRSPR-Cas system has been introduced can be cultured according to known methods depending on the type of cells.
- the medium used for the culture is preferably a liquid medium.
- the medium preferably contains a carbon source, a nitrogen source, inorganic substances, etc. necessary for the growth of the transformant.
- examples of carbon sources include glucose, dextrin, soluble starch, sucrose, etc.
- nitrogen sources include inorganic or organic substances such as ammonium salts, nitrates, corn steep liquor, peptone, casein, meat extract, soybean meal, potato extract, etc.
- examples of inorganic substances include calcium chloride, sodium dihydrogen phosphate, magnesium chloride, etc.
- yeast extract, vitamins, growth promoting factors, etc. may be added to the medium.
- the pH of the medium is preferably about 5 to about 8.
- E. coli for example, M9 medium containing glucose and casamino acids [Journal of Experiments in Molecular Genetics, 431-433, Cold Spring Harbor Laboratory, New York 1972] or LB medium is preferable. If necessary, a drug such as 3 ⁇ -indolylacrylic acid may be added to the medium to make the promoter work efficiently.
- E. coli is usually cultured at about 15 to about 43°C. If necessary, aeration or stirring may be performed. Bacillus bacteria are usually cultured at about 30 to about 40° C. If necessary, aeration or stirring may be performed. Examples of media for culturing yeast include Burkholder's minimal medium [Proc. Natl. Acad. Sci.
- the pH of the medium is preferably about 5 to about 8. Cultivation is usually performed at about 20°C to about 35°C. Aeration and stirring may be performed as necessary.
- a medium for culturing insect cells or insects for example, Grace's Insect Medium [Nature, 195, 788 (1962)] with additives such as inactivated 10% bovine serum, etc., is used.
- the pH of the medium is preferably about 6.2 to about 6.4.
- the culture is usually carried out at about 27°C. Aeration and stirring may be performed as necessary.
- Examples of media for culturing animal cells include Minimum Essential Medium (MEM) containing about 5 to about 20% fetal bovine serum [Science, 122, 501 (1952)], Dulbecco's Modified Eagle Medium (DMEM) [Virology, 8, 396 (1959)], RPMI 1640 medium [The Journal of the American Medical Association, 199, 519 (1967)], and 199 medium [Proceeding of the Society for the Biological Medicine, 73, 1 (1950)].
- Examples of media for culturing pluripotent stem cells such as human iPS cells include mTeSR medium, Essential-8 medium, and StemFit AK03N medium.
- the pH of the medium is preferably about 6 to about 8.
- the culture is usually performed at about 30°C to about 40°C.
- Media for culturing plant cells include MS medium, LS medium, B5 medium, etc.
- the pH of the medium is preferably about 5 to about 8.
- Cultivation is usually carried out at about 20°C to about 30°C.
- Aeration and stirring may be performed as necessary.
- a double-stranded DNA target sequence modifying agent and a target gene expression control agent are also provided, which include the CRISPR-Cas system of the present invention.
- these agents can also be used to treat or prevent mammalian diseases (e.g., the above-mentioned repeat diseases, etc.).
- mammals include rodents such as mice, rats, hamsters, and guinea pigs, primates such as humans, rhesus monkeys, cynomolgus monkeys, Japanese macaques, and chimpanzees, cows, horses, dogs, and cats.
- the agent of the present invention can be administered orally or parenterally (e.g., subcutaneous injection, intramuscular injection, intravenous injection, local injection (topical application, local coating), intraventricular administration, intrathecal administration, intraperitoneal administration, etc.) to mammals.
- Agents for oral administration include solid or liquid dosage forms, specifically tablets (including sugar-coated tablets and film-coated tablets), pills, granules, powders, capsules (including soft capsules), syrups, emulsions, suspensions, etc.
- agents for parenteral administration include, for example, topical agents (e.g., ointments, creams, topical liquid agents, etc.), injections, suppositories, etc., and injections may include dosage forms such as intravenous injections, subcutaneous injections, intradermal injections, intramuscular injections, and drip injections.
- topical agents e.g., ointments, creams, topical liquid agents, etc.
- injections may include dosage forms such as intravenous injections, subcutaneous injections, intradermal injections, intramuscular injections, and drip injections.
- the CRISPR-Cas system of the present invention may be formulated with any carrier, for example, a pharmacologically acceptable carrier.
- pharmacologically acceptable carriers include, but are not limited to, excipients such as sucrose and starch, binders such as cellulose and methylcellulose, disintegrants such as starch and carboxymethylcellulose, lubricants such as magnesium stearate and aerosil, fragrances such as citric acid and menthol, preservatives such as sodium benzoate and sodium bisulfite, stabilizers such as citric acid and sodium citrate, suspending agents such as methylcellulose and polyvinylpyrrolide, dispersing agents such as surfactants, diluents such as water and saline, base waxes, etc.
- the agent of the present invention may further contain a nucleic acid introduction reagent to promote the introduction of the nucleic acid into target cells.
- a nucleic acid introduction reagent examples include calcium chloride, calcium enrichment reagent, atelocollagen; liposomes; nanoparticles; lipofectin, lipofectamine, DOGS (transfectam), DOPE, DOTAP, DDAB, DHDEAB, HDEAB, polybrene, and cationic lipids such as poly(ethyleneimine) (PEI).
- a SSA reporter nucleic acid for a type I CRISPR-Cas system is provided.
- the SSA reporter nucleic acid is a double-stranded nucleic acid for evaluating the activity of a type I CRISPR-Cas system, wherein one nucleic acid strand is: (1) A sequence encoding a portion of a reporter protein (i.e., a portion of the sequence of a reporter gene (also called a "selection marker gene”)), (2) an insert sequence containing a sequence targeted by the crRNA or a sequence complementary to said sequence; and (3) a sequence different from (1) that encodes a portion of a reporter protein, in this order; (1) and (3) have an overlapping sequence, and a sequence other than the overlapping sequence of (1), the overlapping sequence, and a sequence other than the overlapping sequence of (3) constitute a sequence encoding a full-length reporter protein; (2) The length
- a double-stranded nucleic acid for evaluating the activity of a type I CRISPR-Cas system, wherein one nucleic acid strand is (I) a sequence encoding a portion of a reporter protein; (II) an insert sequence including a sequence targeted by the crRNA and a sequence complementary to said sequence; and (III) a sequence different from (I) that encodes a portion of a reporter protein, in this order;
- a double-stranded nucleic acid (hereinafter sometimes referred to as "the double-nicking nucleic acid of the present invention") is provided in which (I) and (III) have an overlapping sequence, and the sequence other than the overlapping sequence of (I), the overlapping sequence, and the sequence other than the overlapping sequence of (III) constitute a sequence encoding a full-length reporter protein.
- the term "the long sequence insertion nucleic acid of the present invention” and the “double-nicking nucleic acid of the present invention” may be collectively referred to
- DNA cleavage occurs near the target sequence by the type I CRISPR-Cas system, and then a single-stranded DNA region is formed.
- (1) and (3), or (I) and (III) have overlapping sequences, and an annealing reaction occurs between the overlapping sequences, repairing the DNA damage that occurred in the SSA reporter nucleic acid.
- a sequence encoding the full-length reporter protein is generated in the SSA reporter nucleic acid, and the protein is expressed.
- the presence or absence of expression of the reporter protein and its expression level can be used as an indicator to evaluate the presence or absence and the degree of activity of the type I CRISPR-Cas system in cells.
- the SSA reporter nucleic acid of the present invention is applicable to type I CRISPR-Cas systems, but is preferably type I-E (particularly type I-E derived from Escherichia coli) or type I-D (particularly type I-D derived from Microcystis aeruginosa) CRISPR-Cas systems, and type I-E CRISPR-Cas systems are more preferable.
- type I-E CRISPR-Cas are as described in "1. Pre-crRNA with modified nucleotides.”
- Reporter proteins used in the SSA reporter nucleic acid of the present invention include, but are not limited to, fluorescent proteins, luminescent enzymes, chromogenic enzymes, drug resistance proteins, etc.
- Fluorescent proteins include, for example, blue fluorescent proteins such as Sirius, TagBFP, EBFP, etc., cyan fluorescent proteins such as mTurquoise, TagCFP, AmCyan, mTFP1, MidoriishiCyan, CFP, etc., green fluorescent proteins such as TurboGFP, AcGFP, TagGFP, Azami-Green (e.g., hmAG1, etc.), GFP, ZsGreen, EmGFP, EGFP, GFP2, HyPer, etc., TagYFP, EYFP, Venus, YFP, PhiYFP, PhiYFP-m, TurboYFP, ZsYellow, mBa
- fluorescent enzymes include, but are not limited to, yellow fluorescent proteins such as nana, orange fluorescent proteins such as KusabiraOrange (e
- luminescent enzymes include firefly luciferase, bacterial luciferase, synthetic Renilla luciferase, Oplophorus gracilirostris luciferase (e.g., NanoLuc (registered trademark), etc.), and secreted luciferase.
- chromogenic enzymes include ⁇ -galactosidase, ⁇ -glucuronidase, and alkaline phosphatase.
- drug resistance proteins include kanamycin resistance protein, ampicillin resistance protein, neomycin resistance protein, puromycin resistance protein, blasticidin resistance protein, and zeocin resistance protein.
- the length of the inserted sequence is typically 100 bases or more (e.g., 200, 300, 400, 500 or more bases) and 3000 bases or less (e.g., 2500, 2000, 1900, 1800, 1700 or less bases), preferably 500 to 2000 bases, and more preferably 1000 to 1700 bases. In one embodiment, the length of the inserted sequence is 1000 bases.
- a schematic diagram of an example of a long-sequence-inserted nucleic acid is shown on the left of Figure 27.
- the insertion sequence includes a sequence targeted by the crRNA (i.e., the target sequence) and a sequence complementary to the sequence.
- the target sequence is included in both strands of the double-stranded nucleic acid, so that the two target sequences are arranged in a batch sequence.
- the CRISPR-Cas system acts on each strand to cause DNA cleavage (such as a DNA nick), which causes a DNA repair reaction via SSA.
- a spacer sequence (hereinafter sometimes referred to as a "spacer sequence for SSA”) may or may not be present between the 3'-terminal base of the target sequence of the crRNA and the 5'-terminal base of the sequence complementary to the target sequence, but it is preferable that it does not exist.
- a spacer sequence for SSA its length is not particularly limited, but is typically 1 to 100 bases long, preferably 1 to 50 bases long, and more preferably 15 to 35 bases long.
- the double-nicked nucleic acid has a spacer sequence for SSA (typically 1-100 bases long) between the 3'-terminal base of the target sequence of the crRNA (II) and the 3'-terminal base of the sequence complementary to said sequence.
- a schematic diagram of an example of a double-nicked nucleic acid is shown on the right side of Figure 27.
- the length of the inserted sequence is not particularly limited, but is typically 64 bases or more (e.g., 100 bases, 150 bases, 200 bases or more) and 1000 bases or less (e.g., 800 bases, 600 bases, 400 bases or less).
- complementary refers to a relationship in which nucleic acid bases can form so-called Watson-Crick base pairs (natural base pairs) or non-Watson-Crick base pairs (Hoogsteen base pairs, wobble base pairs, etc.) through hydrogen bonds. Therefore, “complementary sequence” is used to mean not only a sequence that is completely complementary (i.e., hybridizes without mismatches) to the target sequence (e.g., the target sequence of crRNA, etc.), but also a sequence that contains one or several (e.g., 2, 3, 4, 5 or more) mismatches as long as it can hybridize with the target sequence under stringent conditions or under the physiological conditions of mammalian cells.
- a sequence that is completely complementary to the target sequence has an identity of 80% or more (e.g., 85% or more, 90% or more, 95% or more, 96% or more, 97% or more, 98% or more, 99% or more), and most preferably 100%, to the target sequence.
- the identity of the base sequences in this specification can be calculated using the homology calculation algorithm NCBI BLAST.
- the degree of activity of the type I CRISPR-Cas system can be evaluated using the SSA reporter nucleic acid of the present invention.
- a method for evaluating the degree of activity of the type I CRISPR-Cas system comprising: (A) a step of introducing the SSA reporter nucleic acid of the present invention into a cell to be evaluated for activity of the type I CRISPR-Cas system; and (B) a step of evaluating the degree of activity of the type I CRISPR-Cas system by detecting or measuring the expression of a reporter protein in the cell into which the SSA reporter nucleic acid has been introduced in step (A).
- the cells into which the type I CRISPR-Cas system in step (A) has been introduced may be cells into which the type I CRISPR-Cas system (i.e., pre-crRNA or a nucleic acid encoding said RNA, each Cas protein or a nucleic acid encoding said protein) has been introduced in advance, or may be cells into which the type I CRISPR-Cas system has been introduced simultaneously with step (A), or may be cells into which the type I CRISPR-Cas system has been introduced after step (A).
- the nucleic acid encoding the protein may be RNA or DNA.
- the cells into which the type I CRISPR-Cas system in step (A) has been introduced may be cells having a nucleic acid to which a Cas protein is linked under the control of an expression-inducing promoter.
- the type I CRISPR-Cas system can be expressed at the desired timing by adding or removing an expression-inducing factor corresponding to each promoter to the medium.
- step (B) can be carried out by detecting or measuring the fluorescence intensity, luminescence intensity or color intensity of the cells using a known method. If the reporter protein is a drug-resistant protein, step (B) can be carried out by determining whether the protein survives in a medium containing the corresponding drug, or by adjusting the concentration of the drug in the medium. Alternatively, the protein can be detected or measured by Western blot, immunostaining, enzyme immunoassay (e.g., EIA, ELISA) or the like using an antibody that specifically recognizes the protein.
- EIA enzyme immunoassay
- a cell selection method comprising: (a) a step of introducing the SSA reporter nucleic acid of the present invention into a target cell; and (b) a step of selecting cells expressing a reporter protein from the cells into which the SSA reporter nucleic acid has been introduced in step (a).
- Step (b) is not particularly limited as long as it allows the selection of cells expressing the reporter protein, but if the reporter protein is a fluorescent protein, the cells can be selected using a cell sorter such as a FACS (Fluorescence-Activated Cell Sorter) with the fluorescence as an indicator.
- a cell sorter such as a FACS (Fluorescence-Activated Cell Sorter) with the fluorescence as an indicator.
- a substance such as an antibody
- the reporter protein is a drug-resistant protein, it can be performed by adding a drug to the culture medium and selecting surviving cells.
- the SSA reporter nucleic acid of the present invention is typically provided in the form of an expression vector containing the nucleic acid.
- the types of expression vectors, the types and descriptions of promoters (including expression-inducible promoters) and other components used in the vectors, the method for introducing the SSA reporter nucleic acid of the present invention, the expression vector containing the nucleic acid, or the Cas protein into cells, and the types of cells and the method for culturing the cells used in the present invention are all incorporated by reference in the contents described above in "1.
- Pre-crRNA having modified nucleotides and "3. Type I CRISPR-Cas system and uses thereof.”
- only one type of SSA reporter nucleic acid of the present invention may be used, or two or more types may be used.
- two or more types are used, for example, by using SSA reporter nucleic acids with different crRNA target sequences and reporter protein-encoding sequences, it is possible to evaluate the target recognition abilities of multiple crRNAs in a single operation, or to select only cells in which genome editing has occurred simultaneously at multiple target sites.
- a combination of different fluorescent colors e.g., a combination of a green fluorescent protein such as EGFP and a red fluorescent protein such as mRFP
- a combination of different types of reporter proteins e.g., a combination of a fluorescent protein and a drug resistance protein
- the specificity of the crRNA can be evaluated by using two or more types of SSA reporter nucleic acids of the present invention to evaluate the crRNA designed by the design method of the present invention.
- SSA reporter nucleic acids of the present invention for example, in two types of SSA vectors expressing EGFP or mRFP as reporter proteins, an insertion sequence with a difference of one base in the target sequence of the crRNA can be used to analyze whether EGFP or mRFP emits a dominant signal.
- the SSA reporter nucleic acid of the present invention may be provided in the form of a kit.
- the kit may contain only one type of SSA reporter nucleic acid of the present invention, or may contain multiple types.
- the kit may also contain agents for cell selection, cell culture media, cells, reagents for introducing nucleic acids or proteins, and instructions describing the procedures of the evaluation method and cell selection method of the present invention.
- Type I-E CRISPR-Cas systems typically introduce long deletions on the 5' side of the target sequence
- type I-D CRISPR-Cas systems typically introduce indels (insertion/deletions) at or near the target site, and long deletions on the 5' and 3' sides of the target sequence.
- Class 1 CRISPR-Cas systems are preferably type I-E (particularly type I-E derived from Escherichia coli) or type I-D (particularly type I-D derived from Microcystis aeruginosa) CRISPR-Cas systems, with type I-E CRISPR-Cas systems being more preferred.
- target deletion site refers to a site that is expected to be deleted by the introduced CRISPR-Cas system.
- CRISPR-Cas system it is possible to delete regions consisting of 100 or more (e.g., 1,000, 10,000 or more) nucleotides upstream and/or downstream of the PAM sequence, and probe and primer pairs can be designed for these regions.
- probe and primer pairs can be designed for these regions.
- the region surrounded by the target regions of these RNAs is also the deletion target site.
- probe spacing can be adjusted as appropriate depending on the purpose of the analysis, but typically, probes are set at intervals of 100 to 2,000,000 bases, for example, at intervals of 1,000 to 100,000 bases, 3,000 to 70,000 bases, etc. Furthermore, each probe is typically 5 to 40 bases long, preferably 10 to 35 bases long, and more preferably 18 to 30 bases long.
- a fluorescent dye can be bound to each probe.
- Various such fluorescent dyes are commercially available, including 6-FAM (fluorescein), HEX, TE, Quasar 670, Quasar 570, Quasar 705, Pulsar 650, TET, HEX, VIC, JOE, CAL Fluor Orange, CAL Fluor Gold, CAL Fluor Red, Texas Red, Cy, and Cy5. It is preferable that each probe is further bound to a quencher capable of quenching the fluorescence from the fluorescent dye.
- quencher there are no particular limitations on the quencher as long as it is capable of quenching the fluorescence from the fluorescent dye, and it may be a fluorescent dye or a non-fluorescent dye, although a non-fluorescent dye is preferable from the viewpoint of detection accuracy.
- quenchers include Black Hole Quencher 1 (BHQ1), 6-carboxytetramethylrhodamine (TAMRA), 6-carboxy-X-rhodamine (ROX), Eclipse Dark Quencher, Iowa black FQ (IBFQ), minor groove binder (MGB), and non-fluorescent quencher (NFQ).
- combinations of fluorescent dyes and quenchers include, but are not limited to, combinations of 6-FAM and BHQ1, and combinations of HEX and BHQ1.
- a fluorescent dye is attached to the 5' or 3' end of each probe, and a quencher is attached to the opposite end.
- Digital PCR methods used in the present invention include, for example, droplet digital PCR (ddPCR) and chip-based digital PCR (cdPCR).
- Digital PCR is performed, for example, by the following procedure.
- a reaction solution containing a probe set, a DNA sample, a PCR primer set, and DNA polymerase is placed in a digital PCR device.
- the mixing ratio of each reaction solution component can be appropriately selected and optimized within a known range, and can be appropriately changed depending on the primer set, probe set, etc. used.
- the reaction solution is made into minute droplets (e.g., nanoliter size) using a water-oil emulsion, and in the case of cdPCR, it is distributed by the digital PCR device to a large number of minute reaction wells (e.g., with an opening size of several tens of ⁇ m) provided on the chip.
- the amount of DNA sample is adjusted so that the reaction mix thus fractionated into minute fractions contains the region on the DNA to be measured (preferably about one copy) and does not contain it, and PCR is performed.
- a PCR amplification reaction occurs in the reaction droplet or well where the measured DNA region exists, and during this, the nuclease activity of the DNA polymerase cleaves the probe bound to the target site, and the fluorescent dye dissociates from the quencher, emitting fluorescence.
- the number of droplets or wells in which fluorescence is detected is measured using the reader of the digital PCR device, and the DNA copy number can be quantified by statistical calculation from the total number of droplets or wells.
- copy numbers of two or more DNA regions can be compared using the same sample (e.g., comparing regions where DNA deletions are expected to occur with regions where they are not expected to occur).
- the DNA loss rate at the distance from the crRNA or guide RNA can be calculated.
- digital PCR probes and primers are designed at approximately 1 kb, 10 kb, and 20 kb in the direction of progression from the crRNA target sequence, and 1 kb (-1 kb) and 3.5 kb (-3.5 kb) in the opposite direction from the direction of progression, and digital PCR probes and primers are also designed for a region that is not considered to be in the range of genome editing by Cas3 (for example, on another gene Y) as a comparison.
- gene X is an autosomal gene, the cell retains two copies, so if genome editing is not performed, the measurement result will be approximately two copies regardless of the position.
- the copy number of the comparison target (gene Y) remains approximately 2 copies, but the copy number of gene X decreases depending on its position (for example, if the copy number at a position 1 kb away from the crRNA is 1.5 copies, this indicates a 25% copy number decrease, i.e., a deletion of 1 kb or more has occurred by 25%).
- the extent to which the copy number at which position on gene X is decreased depends on the cleavage pattern that Cas3 shows from the crRNA target sequence, so the deletion mutation induction pattern of Cas3 can be measured.
- probes that can be used for digital PCR include probes containing any of the sequences shown in SEQ ID NOs: 102, 105, 108, 111, 114, 117, 120, and 123 for the human B2M gene, probes containing any of the sequences shown in SEQ ID NOs: 126 or 129 for the human HLA gene, and probes containing any of the sequences shown in SEQ ID NOs: 134, 137, 140, 143, 146, 149, 152, 155, 158, 161, 164, and 167 for the human DMD gene. Two or more of these probes can also be used in combination.
- primers comprising any of the sequences shown in SEQ ID NOs: 59, 160, 162, 163, 165, and 166, and/or probes comprising any of the sequences shown in SEQ ID NOs: 102, 105, 108, 111, 114, 117, 120, 123, 126, 129, 134, 137, 140, 143, 146, 149, 152, 155, 158, 161, 164, and 167.
- These primers and probes may be
- RNA synthesis was requested from Intergrated DNA Technologies (IDT).
- Pre-crRNA sequences containing 5' Cap analog (ARCA) and PolyA tail were synthesized by in vitro transcription (IVT) using Invitrogen mMESSAGE mMACHINE T7 Ultra Transcription Kit.
- the template for IVT was prepared by PCR using a PCR forward primer (GAAATTAATACGACTCACTATAGGATGTGTTGTTTGTGTGATACTA; SEQ ID NO: 2) containing a T7 promoter sequence and a reverse primer (CCCGGGCtgcaggaattc; SEQ ID NO: 3) and a plasmid (pPV-C1-crRNA-EF1a-G2ABA) containing the pre-crRNA sequence (B2M#1 or B2M#4).
- the final pre-crRNA sequence contained a leader sequence at the 5′ end.
- target sequence is as follows:
- cells were transfected with a total of 1000-2000 ng of plasmid DNA or RNA using 2-4 ⁇ L of Lipofectamine Stem (Thermo Fisher Scientific).
- Lipofectamine Stem (Thermo Fisher Scientific).
- 2 x 105 cells were seeded into wells of 12-well plates or 1 x 106 cells were seeded into wells of 6-well plates and transfected the next day with a total of 1000-2000 ng of plasmid DNA or RNA using 4-8 ⁇ L of Lipofectamine 2000 (Thermo Fisher Scientific).
- the CRISPR-Cas3 and Cascade protein group expression plasmids used were pPV-Dual_promoter-EF1 ⁇ -2xNLS-Cascade+Cas3(RD)-iPA (expressing Puromycin resistance gene), pPV-Dual_promoter-EF1 ⁇ -2xNLS-Cascade+Cas3(RD)-iZA (expressing Zeocin resistance gene), pPV-Dual_promoter-EF1 ⁇ -2xNLS-Cascade+Cas3(RD)-iCA (expressing mCherry), and pPV-TRE3G-Bi(263+751)-EF1a-Tet3G-iPA (DOX-inducible, expressing Puromycin resistance gene).
- the crRNA expression plasmids used were pBSIIKS-U6-crRNA or pPV-C1-crRNA vectors (including those that simultaneously express EGFP and drug resistance genes by EF1 ⁇ ) in which pre-crRNA is expressed by the U6 promoter.
- pPV-EF1a-EGxxFP-iPA For SSA reporter selection, pPV-EF1a-EGxxFP-iPA, pPV-EF1a-EGxxFP-iBA, pPV-EF1a-mRxxFP-iPA, pHL-EF1a-EGxxFP-iPA, pHL-EF1a-mRxxFP-iPA, pHL-EF1a-Puro-SSA-A, or pHL-EF1a-EGFP-IRES-Puro-SSA-A containing the appropriate target sequences were used.
- drug selection Puromycin, Blasticidin, Zeocin
- cells were cultured in medium containing the desired drug concentration 1 to 3 days after transfection.
- RNA was introduced by electroporation 3x105 cells were introduced using P4 Primary Cell 4D-Nucleofector X Kit S from 4D-Nucleofector (LONZA) and electroporation program CA-189.
- transfection When transfection was performed multiple times, after transfection using the above method, the cells were cultured for several days depending on the cytotoxicity at that time, and then transfection was repeated from seeding the cells. The interval between transfections was adjusted to between approximately 3 and 15 days, taking into consideration the cell growth rate, transfection conditions, and sensitivity to transfection. In some cases, cells were frozen and stored during the multiple transfection process, and transfection was resumed after thawing.
- HLA-A/B/C staining of the cell surface.
- the cells were cultured for a while (preferably for at least 3-4 days until HLA molecules disappear from the cell surface), and then isolated and collected using trypsin (including trypsin alternatives such as TrypLE Select).
- trypsin including trypsin alternatives such as TrypLE Select.
- the collected cells were washed with PBS and stained with anti-HLA-A/B/C antibodies (iPS cells: BioLegend Pacific Blue anti-human HLA-A,B,C Antibody, HEK293T cells: BD BV421 Mouse anti-human HLA-A2) diluted 50-100 times in 2% FBS/PBS (30 minutes on ice).
- HLA-stained cells were analyzed by flow cytometry, and the percentage of HLA-negative cells was calculated.
- SSA reporter vectors Construction of SSA reporter vectors
- the reporter genes EGFP, mRFP, and Puro R were split into two, and sequences were added to ensure that they had approximately 150-350 bp of homology with each other.
- Two restriction enzyme sites (AfeI or Esp3I sites) were also added for insertion of target sequences.
- These split reporter genes were inserted into vectors with pHL-EF1a or pPV-EF1a as backbones.
- a crRNA target sequence of any length was inserted between the split reporter genes by digestion with restriction enzymes and In-Fusion reaction.
- SSA reporter vector When performing cell sorting using the SSA reporter vector, cells were transfected with the SSA reporter vector in addition to the vector expressing Cas3 and Cascade proteins and the vector expressing crRNA.
- Puro-SSA vector When the Puro-SSA vector was used as the reporter gene of the SSA vector, cells were selected by culturing in a medium containing Puromycin.
- the fluorescent protein EGFP or mRFP was used as the reporter gene, cells were harvested after transfection using trypsin (or a trypsin substitute reagent), and EGFP- and mRFP-positive cells were separated using a BD FACSAria II (BD) cell sorter.
- Single-cell cloning To isolate subclones of iPS cells in which genome editing had occurred, limiting dilution or colony picking methods were used ( Figure 36). When using SSA vectors carrying EGFP or mRFP fluorescent proteins, single-cell sorting was performed into wells of a 96-well plate precoated with iMatrix-511 silk for cell sorting of EGFP- and mRFP-positive cells. The isolated cells were expanded and collected for cryopreservation and genomic DNA extraction. The target regions were PCR amplified for genotyping using the primers in Table 2 or Table 3 below. Sanger sequencing was performed to analyze the deletion pattern.
- ddPCR probes and primer pairs were designed using Primer3Plus (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) software.
- DNA probes labeled with 5'-6-FAM fluorophore, 3'-BHQ1 were used to detect the target genomic regions (e.g., B2M gene, dystrophin exon 44-55 locus, etc.).
- the reference probe was a primer labeled with 5'-HEX fluorophore, 3'-BHQ1. All primer pairs and probes are listed in Table 4 below.
- the isolated genomic DNA was first digested with EcoRI restriction enzyme (TaKaRa) overnight at 37°C.
- the ddPCR reaction solution was prepared by mixing 11 ⁇ L of ddPCR SUpermix for Probes (No dUTP) (Bio-Rad Laboratories), 1 ⁇ L of 20 ⁇ M target forward and reverse primer pair, 0.6 ⁇ L of 10 ⁇ M target probe, 1 ⁇ L of 20 ⁇ M reference forward and reverse primer pair, 0.6 ⁇ L of 10 ⁇ M reference probe, and EcoRI-digested genomic DNA, and adjusted to a total volume of 22 ⁇ L with ultrapure water.
- Droplets were generated using a QX200 Droplet Generator or QX200 Automated Droplet Generator (Bio-Rad Laboratories) and PCR reactions were performed in a C1000 Thermal Cycler (Bio-Rad Laboratories). ddPCR reactions were performed under the following three thermal steps: step 1, 95°C for 10 min; step 2, 94°C for 30 s; step 3, 55-60°C for 1-2 min; steps 2 and 3 were repeated 40 times; step 4, 98°C for 10 min; and step 5, storage at 4°C. After the PCR reaction, the fluorescence of the droplets was measured using a QX200 Droplet reader (Bio-Rad Laboratories) and analyzed using Bio-Rad QuantaSoft software.
- Example 1 Chemical modification of crRNA in RNA-based Cas3 genome editing
- Many experimental cancer cell lines such as HEK293T cells, have high gene transfer efficiency and are often used as model lines for genome editing.
- iPS cells and primary culture cell lines intended for practical application in regenerative medicine do not necessarily have high gene transfer efficiency, and the number of cell lines selected for clinical use is small, so it is not always possible to select the optimal line for gene transfer every time. For these reasons, it is important to develop a method that allows stable and efficient genome editing even in cell lines with low gene transfer efficiency. Therefore, the present inventors chemically modified crRNA and verified the genome editing efficiency by the modification.
- the mRNA expressing a total of six proteins (Cas3, Cse1, Cse2, Cas5, Cas6, Cas7) was assembled into two mRNAs, Cse2-P2A-Cas6-T2A-Cas3 and Cas7-P2A-Cas5-T2A-Cse1, using the 2A peptide, which can express multiple proteins from one mRNA.
- this mRNA was modified with 5-methyl-cytosine and pseudo-uracil to suppress cytotoxicity due to natural immune reactions and to increase translation efficiency (also used in the examples of Patent Document 3: TriLink).
- RNA modification can change the physical properties and secondary structure of RNA, and interactions with proteins, and may cause the original function to be lost, and even experts cannot predict which modification should be introduced at which position.
- the present inventors introduced 2'-O-methyl 3'phosphorothioate modification (MS modification) into the pre-crRNA of the E. coli CRISPR-Cas3 system and searched for a modification position that would improve activity.
- MS modification 2'-O-methyl 3'phosphorothioate modification
- pre-crRNA activity could be improved by methods other than chemical modification of the RNA bases and backbone.
- the results in HEK293T cells (Fig. 9) and I14s04 iPS cells (Fig. 10) showed that pre-crRNA activity could also be improved by adding 5'-Cap and a polyA tail.
- the desired genome editing efficiency is often not achieved by a single genome editing. In this case, it was possible to accumulate the effect of genome editing by repeating gene introduction (Fig. 11).
- Example 2 Analysis of deletion mutation patterns using ddPCR
- Cas3 is known to induce various long-chain deletions of different lengths, but a method for easily analyzing which length of deletion mutation occurs at what rate has not yet been developed. Therefore, the present inventors attempted to develop a technology for measuring the length distribution and rate of deletion mutations caused by Cas3 by applying the technology of droplet digital PCR (ddPCR).
- ddPCR droplet digital PCR
- this measurement technique is useful for a variety of evaluations, such as analyzing deletion patterns by changing the position of the ddPCR probe according to the purpose, evaluating the activity of the designed crRNA, and measuring the rate at which mutations are induced in the opposite direction to the desired direction.
- Example 3 Enrichment of genome-edited cell groups using SSA reporter vector for CRISPR-Cas3 system
- SSA reporter vector for CRISPR-Cas3 system
- the present inventors have developed a technology to specifically enrich cell groups in which Cas3 is activated as a result of sufficient expression of these necessary factors.
- Cas9 genome-edited cells are enriched by sorting cells in which Cas9 is activated using a reporter vector that utilizes the DNA repair mechanism by Single Strand Annealing (SSA).
- SSA Single Strand Annealing
- the present inventors designed an SSA reporter vector based on a fluorescent protein (such as EGFP or mRNP) and a drug resistance gene (such as Puro R ) ( Figure 22), and searched for an optimal insertion sequence for the CRISPR-Cas3 system.
- a fluorescent protein such as EGFP or mRNP
- a drug resistance gene such as Puro R
- Figure 22 the reporter gene might also be deleted by Cas3. Therefore, it was decided to evaluate the activity of SSA vectors for the CRISPR-Cas3 system incorporating relatively long insertion sequences of 0.5 kb, 1 kb, or 2 kb. In this case, the reporter gene was mRFP.
- HEK293T stable expression line which can express Cas3 and Cascade proteins by DOX
- crRNA expression vector non-target: AAVS1#1, target: DownSNP1T
- mRFP SSA vector DownSNP1T is the target insertion sequence, the length of the insertion sequence is 0.5-2kb: pPV-EF1a-mRxxFP-iPA) using Lipofectamine 2000.
- the percentage of mRFP-positive cells was analyzed by flow cytometry. Compared to the case of non-target crRNA, the case of target crRNA gave a higher reporter signal (Figure 23).
- the background signal tended to be lower with longer insertion sequences, but a decrease in on-target signal was also observed with 2 kb insertion sequences.
- an insertion sequence of about 1 kb is suitable for Cas3.
- SSA vector developed for the CRISPR-Cas3 system could be used to enrich for cells in which genome editing had actually occurred.
- HEK293T cells were transfected with Cas3 and Cascade protein group expression vectors (same as above), crRNA expression vectors (non-target: AAVS1#1, target: pPV-C1-crRNA-EF1a-BA vector of B2M#1), and Puro-SSA vector (pHL-EF1a-Puro-SSA(B2M 1kb)-A) containing the target sequence of B2M#1 using Lipofectamine 2000, and treated with 2 ⁇ g/ml Puromycin for 2 days from day 2 to day 4 after transfection.
- This SSA vector can also be used in combination with multiple different reporter genes (e.g., EGFP and mRFP), and can also be applied to genome editing using multiple crRNAs simultaneously (Figure 27).
- reporter genes e.g., EGFP and mRFP
- Figure 27 For example, in the case of inducing multi-exon skipping to delete exons 45-55 (approximately 340 kb) of the human dystrophin (DMD) gene as a model for the treatment of Dischenne muscular dystrophy, even if two crRNAs are designed facing each other (crRNAs targeting Ex45 and Ex55), the deletion efficiency predicted from the approximate midpoint copy number is low due to the breadth of the deletion range (Figure 29: SSA unsorted).
- DMD human dystrophin
- genome-edited cells could be enriched from various iPS cell lines (FF12020, CiRA00458, CiRA00646), and multiple subclones containing approximately 340 kb deletion mutations were successfully obtained (Figure 30).
- This multi-exon skipping method which deletes the exon 45-55 region, is known to be able to restore the reading frame of dystrophin protein, and is also useful as a treatment for Duchenne muscular dystrophy.
- Example 4 Allele-specific genome editing technology using the CRISPR-Cas3 system
- Genes on autosomes are usually composed of two copies that are paired, but in the treatment of genetic diseases, allele-specific genome editing technology that targets only one allele of the gene is also important.
- myotonic dystrophy type I (DM1) is a genetic disease caused by abnormal expansion of the CTG repeat in the 3'-UTR region of the DMPK gene in only one allele.
- DM1 myotonic dystrophy type I
- the present inventors focused on heterozygous single nucleotide polymorphisms (SNPs) and developed a CRISPR-Cas3 system technology that can delete the CTG repeat expanded only by the disease allele.
- SNPs with high allele frequency were searched for in the SNP database, and at the same time, it was confirmed whether those SNPs actually existed in iPS cells derived from multiple DM1 patients ( Figure 32).
- the Protospacer Adjacent Motif (PAM) sequences used were AAG, ATG, TAG, AGG, GAG, AAC, AAT, AAA, GTG, TTG, and GGG, and the heterozygous SNP sequence was located in the recognizable sequence of the crRNA (a position excluding 6n (n is a positive integer) bases counting from the 3'-terminal base of the PAM sequence) and in the direction in which Cas3 proceeds toward the CTG repeat ( Figure 33).
- PAM Protospacer Adjacent Motif
- a crRNA was designed for the rs934739524 SNP (C: disease allele, T: healthy allele) downstream of the CTG repeat found in the DM1 patient-derived cell line HPS1051 iPS cell line ( Figure 34).
- the crRNA with the heterozygous SNP at the 12th base is a control crRNA that does not (does not) easily distinguish between alleles.
- HPS1051 a stable expression line expressing Cas3, Cascade proteins, and a crRNA expression vector
- RNA-based genome editing mRNA and Pre-both3 pre-crRNA
- plasmid-based genome editing Figures 38-41; RNA-based: 42, 43.
- this allele-specific genome editing can also be performed using two heterozygous SNPs and a crRNA pair that faces each other across the target sequence (allele-specific dual-Cas3).
- RNA-based editing Figure 44
- RNA-based editing Figures 45 and 47
- plasmid-based editing Figure 46
- allele-specific crRNA can be designed by placing a heterozygous SNP at the crRNA recognition sequence site, and its activity and specificity can be confirmed using an SSA reporter vector ( Figure 48, left).
- an insertion sequence with a one-base difference in the target sequence for crRNA can be used to analyze whether EGFP or mRFP emits a dominant signal.
- crRNAs targeting "C” of rs934739524 crRNA-C
- targeting "T” crRNA-T
- NT crRNA non-targeting
- the activity and specificity of the designed allele-specific crRNA are expected to differ depending on the sequence, but they can be adjusted to some extent. Methods for this include shortening the length of the crRNA or intentionally introducing a mismatch at a position distant from the PAM (Figure 49). For example, in an experiment in which the crRNA targets "C” of the rs934739524 SNP mentioned above, and the EGFP SSA targets "C” and the mRFP SSA targets "T", activity gradually decreases as the length of the crRNA is shortened ( Figure 50) or the intentional mismatch is moved closer to the PAM from a distant position ( Figure 51), but nonspecific activity (mRFP signal) also decreases at the same time.
- Example 5 Chemical modification of crRNA based on protein structure Based on the crystal structure of E. coli Cascade/crRNA (Zhao et al. Nature, vol. 515, 147-150 (2014), Jackson et al. Science, 345(6203), 1473-1479 (2014)), sites that could be modified with 2'-O-Me (M) or Phosphorothioate (PS) were modified as shown in Figure 52 (Structure-guided: SG).
- M 2'-O-Me
- PS Phosphorothioate
- the genome editing activity was measured in I14s04 iPS cells using the above pre-crRNA.
- 1.5x105 cells were seeded in a 12-well plate and transfected with mRNA expressing Cas3 and Cascade proteins, and B2M#1 crRNA (each with chemical modifications corresponding to FIG. 52), and the KO efficiency of the B2M gene was measured by HLA-A, B, C staining (FIG. 53) or ddPCR (FIG. 54).
- ddPCR used the B2M_1kb/PATL2 probe.
- SG+Edges3 crRNA In SG+Edges3 crRNA, whose activity was not confirmed in Figures 53 and 54, further modification patterns were examined, focusing on which modifications at the sites adversely affect activity.
- pre-crRNA the following were produced: one in which the modification near the Cas6 processing site was removed (SG+Edges3-A), one in which the target sequence recognition site was further removed (SG+Edges3-A-Target), one in which the modification of the inner arm portion of SG+Edges3-A was removed (SG+Edges3-A-Inner arm), one in which the modification of the inner arm portion and the target sequence recognition site were removed from SG+Edges3-A (SG+Edges3-A-Center), one in which the three bases at both ends, both loops, and the left stem sequence of the left repeat, which have the effect of enhancing activity, were simultaneously modified (Edge3+Loops+Left Stem), and one in which the three base modifications at both ends were added
- Example 6 Measurement of HLA-A*24:02 and B*52:01 exon 2 deletion efficiency by ddPCR I14s04 iPS cells were transfected multiple times with mRNA expressing Cas3 and Cascade proteins and Pre-both3 pre-crRNA targeting HLA-A*24:02 and B*52:01 (Figure 5-1) using Lipofectamine Stem, and the collected genome samples were analyzed by ddPCR. "Dual A24 F1 & B52 R5" indicates tandem pre-crRNA with MS modification at both ends. The target sequence information of the crRNA used in this example is shown in Table 5.
- Example 7 Evaluation of the effect of Puro-SSA vector in 1383D4 iPS cells 1.5x105 cells were seeded in a 12-well plate and transfected with mRNA expressing Cas3 and Cascade proteins, B2M#1 crRNA (both ends modified with three bases MS), and pHL-EF1a-Puro-SSA(B2M#1)-A or pHL-EF1a-Puro-SSA(B2M#1)-2A-EGFP-A as a Puro-SSA reporter vector.
- the present invention makes it possible to improve the efficiency of genome editing using the Type I CRISPR-Cas system and to analyze genome editing patterns using this system. It also makes it possible to develop tools that can concentrate genome-edited cells and perform genome editing specifically for one allele. Therefore, it is particularly useful for genome editing of pluripotent stem cells, which was difficult to achieve using conventional methods.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- General Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Physics & Mathematics (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Analytical Chemistry (AREA)
- Immunology (AREA)
- Plant Pathology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2025510989A JPWO2024204287A1 (https=) | 2023-03-27 | 2024-03-26 | |
| CN202480035109.0A CN121195066A (zh) | 2023-03-27 | 2024-03-26 | 具有修饰核苷酸的crRNA |
| KR1020257035001A KR20250166996A (ko) | 2023-03-27 | 2024-03-26 | 수식 뉴클레오티드를 갖는 crRNA |
| EP24780394.3A EP4692346A1 (en) | 2023-03-27 | 2024-03-26 | Crrna having modified nucleotide |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023-050681 | 2023-03-27 | ||
| JP2023050681 | 2023-03-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024204287A1 true WO2024204287A1 (ja) | 2024-10-03 |
Family
ID=92905577
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2024/012128 Ceased WO2024204287A1 (ja) | 2023-03-27 | 2024-03-26 | 修飾ヌクレオチドを有するcrRNA |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP4692346A1 (https=) |
| JP (1) | JPWO2024204287A1 (https=) |
| KR (1) | KR20250166996A (https=) |
| CN (1) | CN121195066A (https=) |
| WO (1) | WO2024204287A1 (https=) |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008307007A (ja) | 2007-06-15 | 2008-12-25 | Bayer Schering Pharma Ag | 出生後のヒト組織由来未分化幹細胞から誘導したヒト多能性幹細胞 |
| WO2017043573A1 (ja) | 2015-09-09 | 2017-03-16 | 国立大学法人神戸大学 | 標的化したdna配列の核酸塩基を特異的に変換するゲノム配列の改変方法及びそれに用いる分子複合体 |
| WO2018225858A1 (ja) | 2017-06-08 | 2018-12-13 | 国立大学法人大阪大学 | Dnaが編集された真核細胞を製造する方法、および当該方法に用いられるキット |
| WO2019039417A1 (ja) | 2017-08-21 | 2019-02-28 | 国立大学法人徳島大学 | ヌクレオチド標的認識を利用した標的配列特異的改変技術 |
| WO2020111229A1 (ja) * | 2018-11-30 | 2020-06-04 | 国立大学法人京都大学 | 溶液を用いた薬物送達系 |
| WO2020122104A1 (ja) | 2018-12-11 | 2020-06-18 | 国立大学法人京都大学 | ゲノムdnaに欠失を誘導する方法 |
| JP2023050681A (ja) | 2021-09-30 | 2023-04-11 | Tpr株式会社 | ピストンリングの組合せ、及び内燃機関 |
-
2024
- 2024-03-26 JP JP2025510989A patent/JPWO2024204287A1/ja active Pending
- 2024-03-26 EP EP24780394.3A patent/EP4692346A1/en active Pending
- 2024-03-26 KR KR1020257035001A patent/KR20250166996A/ko active Pending
- 2024-03-26 CN CN202480035109.0A patent/CN121195066A/zh active Pending
- 2024-03-26 WO PCT/JP2024/012128 patent/WO2024204287A1/ja not_active Ceased
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008307007A (ja) | 2007-06-15 | 2008-12-25 | Bayer Schering Pharma Ag | 出生後のヒト組織由来未分化幹細胞から誘導したヒト多能性幹細胞 |
| WO2017043573A1 (ja) | 2015-09-09 | 2017-03-16 | 国立大学法人神戸大学 | 標的化したdna配列の核酸塩基を特異的に変換するゲノム配列の改変方法及びそれに用いる分子複合体 |
| WO2018225858A1 (ja) | 2017-06-08 | 2018-12-13 | 国立大学法人大阪大学 | Dnaが編集された真核細胞を製造する方法、および当該方法に用いられるキット |
| WO2019039417A1 (ja) | 2017-08-21 | 2019-02-28 | 国立大学法人徳島大学 | ヌクレオチド標的認識を利用した標的配列特異的改変技術 |
| WO2020111229A1 (ja) * | 2018-11-30 | 2020-06-04 | 国立大学法人京都大学 | 溶液を用いた薬物送達系 |
| WO2020122104A1 (ja) | 2018-12-11 | 2020-06-18 | 国立大学法人京都大学 | ゲノムdnaに欠失を誘導する方法 |
| JP2023050681A (ja) | 2021-09-30 | 2023-04-11 | Tpr株式会社 | ピストンリングの組合せ、及び内燃機関 |
Non-Patent Citations (39)
| Title |
|---|
| "Journal of Experiments in Molecular Genetics", vol. 431-433, 1972, COLD SPRING HARBOR LABORATORY |
| "NCBI", Database accession no. NP_417241.1 |
| ABE N. ET AL., ACS CHEM BIOL., vol. 17, no. 6, 2022, pages 1308 - 1314 |
| CHEN YUXI, LIU JIAQI, ZHI SHENGYAO, ZHENG QI, MA WENBIN, HUANG JUNJIU, LIU YIZHI, LIU DAN, LIANG PUPING, SONGYANG ZHOU: "Repurposing type I–F CRISPR–Cas system as a transcriptional activation tool in human cells", NATURE COMMUNICATIONS, NATURE PUBLISHING GROUP, UK, vol. 11, no. 1, UK, XP093213869, ISSN: 2041-1723, DOI: 10.1038/s41467-020-16880-8 * |
| CHUNG Y. ET AL., CELL STEM CELL, vol. 2, 2008, pages 113 - 117 |
| GENE, vol. 24, 1983, pages 255 |
| HAIFANG YIN ET AL., HUMAN MOLECULAR GENETICS, vol. 17, no. 24, 2008, pages 3909 - 3918 |
| J.A. THOMSON ET AL., BIOL. REPROD., vol. 55, 1996, pages 254 - 259 |
| J.A. THOMSON ET AL., PROC. NATL. ACAD. SCI., vol. 92, 1995, pages 7844 - 7848 |
| J.A. THOMSON ET AL., SCIENCE, vol. 282, 1998, pages 1145 - 1147 |
| J.A. THOMSONVS. MARSHALL, CURR. TOP. DEV. BIOL., vol. 38, 1998, pages 133 - 165 |
| JACKSON ET AL., SCIENCE, vol. 345, no. 6203, 2014, pages 1473 - 1479 |
| JOURNAL OF BIOCHEMISTRY, vol. 95, 1984, pages 87 |
| JOURNAL OF MOLECULAR BIOLOGY, vol. 120, 1978, pages 517 |
| JOURNAL OF MOLECULAR BIOLOGY, vol. 41, 1969, pages 459 |
| LIU TINA Y., DOUDNA JENNIFER A.: "Chemistry of Class 1 CRISPR-Cas effectors: Binding, editing, and regulation", JOURNAL OF BIOLOGICAL CHEMISTRY, AMERICAN SOCIETY FOR BIOCHEMISTRY AND MOLECULAR BIOLOGY, US, vol. 295, no. 42, 1 October 2020 (2020-10-01), US , pages 14473 - 14487, XP093145607, ISSN: 0021-9258, DOI: 10.1074/jbc.REV120.007034 * |
| M.J. EVANSM.H. KAUFMAN, NATURE, vol. 292, 1981, pages 154 - 156 |
| MORISAKA H. ET AL., NAT COMMUN, vol. 10, no. 1, 2019, pages 5302 |
| NAKAGAWA MYAMANAKA S. ET AL., NATURE BIOTECHNOLOGY, vol. 26, 2008, pages 101 - 106 |
| NATURE, vol. 195, 1962, pages 788 |
| NUCLEIC ACIDS RESEARCH, vol. 9, 1981, pages 309 |
| OKITA K ET AL., NAT. METHODS, vol. 8, no. 5, May 2011 (2011-05-01), pages 409 - 12 |
| OKITA K ET AL., STEM CELLS, vol. 31, no. 3, pages 458 - 66 |
| OKITA, K.ICHISAKA, T.YAMANAKA, S., NATURE, vol. 451, 2007, pages 141 - 146 |
| PEDRO M. D. MORENO ET AL., NUCLEIC ACIDS RES., vol. 37, 2009, pages 1925 - 1935 |
| PROC. NATL. ACAD. SCI., vol. 60, 1968, pages 160 |
| PROC. NATL. ACAD. SCI., vol. 77, 1980, pages 4505 |
| PROC. NATL. ACAD. SCI., vol. 81, 1984, pages 5330 |
| PROCEEDING OF THE SOCIETY FOR THE BIOLOGICAL MEDICINE, vol. 73, 1950, pages 1 |
| SCIENCE, vol. 122, 1952, pages 501 |
| TAKAHASHI KYAMANAKA S. ET AL., CELL, vol. 131, 2007, pages 861 - 872 |
| TAKAHASHI KYAMANAKA S., CELL, vol. 126, 2006, pages 663 - 676 |
| THE JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION, vol. 199, 1967, pages 519 |
| VIROLOGY, vol. 8, 1959, pages 396 |
| VIVO, vol. 13, 1977, pages 213 - 217 |
| YU J.THOMSON JA. ET AL., SCIENCE, vol. 318, 2007, pages 1917 - 1920 |
| ZHANG X. ET AL., STEM CELLS, vol. 24, 2006, pages 2669 - 2676 |
| ZHAO ET AL., NATURE, vol. 515, 2014, pages 147 - 150 |
| ZHENG Y. ET AL., FRONT BIOENG BIOTECHNOL, vol. 8, 4 March 2020 (2020-03-04), pages 62 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2024204287A1 (https=) | 2024-10-03 |
| CN121195066A (zh) | 2025-12-23 |
| EP4692346A1 (en) | 2026-02-11 |
| KR20250166996A (ko) | 2025-11-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2022201266B2 (en) | Targeted rna editing | |
| AU2021204304B2 (en) | CRISPR-based compositions and methods of use | |
| CN115651927B (zh) | 编辑rna的方法和组合物 | |
| Frey et al. | RNA-mediated interaction of Cajal bodies and U2 snRNA genes | |
| Weiss et al. | Erythroid Cell-Specific mRNA Stability Elements in the α2-Globin 39 Nontranslated Region | |
| EP3250689B1 (en) | Methods and compositions for labeling a single-stranded target nucleic acid | |
| US8513401B2 (en) | Double stranded nucleic acid targeting low copy promoter-specific RNA | |
| KR20220004674A (ko) | Rna를 편집하기 위한 방법 및 조성물 | |
| JP6952315B2 (ja) | ゲノム編集方法 | |
| JP7641952B2 (ja) | 環状一本鎖ポリヌクレオチドを含む核酸送達ベクター | |
| JPH08502408A (ja) | 特定のrna鎖の切断方法 | |
| AU2024216321B2 (en) | Crispr/cas screening platform to identify genetic modifiers of tau seeding or aggregation | |
| US12018297B2 (en) | Nuclease-mediated nucleic acid modification | |
| WO2019041344A1 (en) | METHODS AND COMPOSITIONS FOR THE TRANSFECTION OF SINGLE STRANDED DNA | |
| JP2024506016A (ja) | 免疫療法のためのt細胞免疫グロブリン及びムチンドメイン3(tim3)組成物及び方法 | |
| EP3896155A1 (en) | Production method for genome-edited cells | |
| CN119630788A (zh) | 可编程腺嘌呤碱基编辑器及其用途 | |
| WO2024204287A1 (ja) | 修飾ヌクレオチドを有するcrRNA | |
| AU2021329295B2 (en) | Nuclease-mediated nucleic acid modification | |
| Gong et al. | Reversal of aberrant splicing of β‐thalassaemia allele (IVS‐2‐654 C→ T) by antisense RNA expression vector in cultured human erythroid cells | |
| Reza et al. | Triplex-mediated genome targeting and editing | |
| CA2289907C (en) | Eukaryotic and retroviral antisense initiator elements | |
| WO2026057649A1 (en) | Small artificial rna (smartrna) oligonucleotide for modulating protein expression | |
| WO2025032401A1 (en) | Method for identifying 3'p rna fragments as modulators of ribosomes and protein synthesis | |
| CN120826469A (zh) | 一种向导rna及其使用方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24780394 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2025510989 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2025510989 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 1020257035001 Country of ref document: KR Free format text: ST27 STATUS EVENT CODE: A-0-1-A10-A15-NAP-PA0105 (AS PROVIDED BY THE NATIONAL OFFICE) |
|
| WWE | Wipo information: entry into national phase |
Ref document number: KR1020257035001 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024780394 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2024780394 Country of ref document: EP Effective date: 20251027 |
|
| ENP | Entry into the national phase |
Ref document number: 2024780394 Country of ref document: EP Effective date: 20251027 |
|
| ENP | Entry into the national phase |
Ref document number: 2024780394 Country of ref document: EP Effective date: 20251027 |
|
| ENP | Entry into the national phase |
Ref document number: 2024780394 Country of ref document: EP Effective date: 20251027 |
|
| ENP | Entry into the national phase |
Ref document number: 2024780394 Country of ref document: EP Effective date: 20251027 |
|
| ENP | Entry into the national phase |
Ref document number: 2024780394 Country of ref document: EP Effective date: 20251027 |
|
| ENP | Entry into the national phase |
Ref document number: 2024780394 Country of ref document: EP Effective date: 20251027 |
|
| ENP | Entry into the national phase |
Ref document number: 2024780394 Country of ref document: EP Effective date: 20251027 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2024780394 Country of ref document: EP |