WO2024201260A1 - 二次電池 - Google Patents
二次電池 Download PDFInfo
- Publication number
- WO2024201260A1 WO2024201260A1 PCT/IB2024/052815 IB2024052815W WO2024201260A1 WO 2024201260 A1 WO2024201260 A1 WO 2024201260A1 IB 2024052815 W IB2024052815 W IB 2024052815W WO 2024201260 A1 WO2024201260 A1 WO 2024201260A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- positive electrode
- active material
- electrode active
- lithium
- secondary battery
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images

































































Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
- H01M10/0564—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes the electrolyte being constituted of organic materials only
- H01M10/0566—Liquid materials
- H01M10/0569—Liquid materials characterised by the solvents
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/131—Electrodes based on mixed oxides or hydroxides, or on mixtures of oxides or hydroxides, e.g. LiCoOx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/133—Electrodes based on carbonaceous material, e.g. graphite-intercalation compounds or CFx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/50—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese
- H01M4/505—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese of mixed oxides or hydroxides containing manganese for inserting or intercalating light metals, e.g. LiMn2O4 or LiMn2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/583—Carbonaceous material, e.g. graphite-intercalation compounds or CFx
- H01M4/587—Carbonaceous material, e.g. graphite-intercalation compounds or CFx for inserting or intercalating light metals
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M50/00—Constructional details or processes of manufacture of the non-active parts of electrochemical cells other than fuel cells, e.g. hybrid cells
- H01M50/40—Separators; Membranes; Diaphragms; Spacing elements inside cells
- H01M50/409—Separators, membranes or diaphragms characterised by the material
- H01M50/411—Organic material
- H01M50/414—Synthetic resins, e.g. thermoplastics or thermosetting resins
- H01M50/417—Polyolefins
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M50/00—Constructional details or processes of manufacture of the non-active parts of electrochemical cells other than fuel cells, e.g. hybrid cells
- H01M50/40—Separators; Membranes; Diaphragms; Spacing elements inside cells
- H01M50/46—Separators, membranes or diaphragms characterised by their combination with electrodes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the present invention relates to a secondary battery.
- the present invention is not limited to the above fields, and relates to a semiconductor device, a display device, a light-emitting device, a power storage device, a lighting device, an electronic device, a vehicle, and a manufacturing method thereof.
- the above-mentioned semiconductor device, display device, light-emitting device, power storage device, lighting device, electronic device, and vehicle can use the secondary battery of the present invention as a necessary power source.
- the above-mentioned electronic device includes an information terminal device equipped with a secondary battery.
- the above-mentioned power storage device includes a stationary power storage device.
- the positive electrode active material with a layered rock salt crystal structure used in lithium ion secondary batteries is expected to have a high capacity because the diffusion paths of lithium ions exist two-dimensionally within the crystal structure.
- the positive electrode active material with a layered rock salt crystal structure is prone to thermal runaway if too many lithium ions are desorbed during charging because the crystal structure is broken, and this has been a problem from the perspective of safety.
- Safety tests include nail penetration tests, and in order to suppress the rise in battery temperature during abnormal conditions such as nail penetration, for example, Patent Document 1 proposes a configuration in which a protective layer is provided between the positive electrode composite layer and the positive electrode current collector.
- Lithium cobalt oxide (LiCoO 2 ) and the like are known as positive electrode active materials with a layered rock salt crystal structure.
- Lithium cobalt oxide has a layered rock salt crystal structure, and lithium ions can move two-dimensionally between layers made of CoO 6 octahedra, so the cycle characteristics are also good.
- lithium cobalt oxide has a problem of phase change accompanying charge and discharge. For example, when lithium ions are desorbed to a certain extent during charging, lithium cobalt oxide undergoes a phase change from hexagonal to monoclinic. Therefore, in order to use lithium cobalt oxide with good cycle characteristics, the amount of lithium ions desorbed is limited.
- Patent Documents 2 to 4 propose a configuration in which an additive element is added to lithium cobalt oxide.
- Non-Patent Documents 1 to 4 research is being conducted on the crystal structure of positive electrode active materials (Non-Patent Documents 1 to 4).
- One of the methods used to analyze the crystal structure of positive electrode active materials is XRD (X-ray diffraction).
- XRD can be analyzed using the Inorganic Crystal Structure Database (ICSD) introduced in Non-Patent Document 5.
- ICSD Inorganic Crystal Structure Database
- the lattice constant of lithium cobalt oxide described in Non-Patent Document 6 can be referenced from the ICSD.
- the analysis program RIETAN-FP Non-Patent Document 7 can be used.
- ImageJ (Non-Patent Documents 8 to 10) is known as an example of image processing software. By using this software, for example, the shape of the positive electrode active material can be analyzed.
- Microelectron diffraction is also effective for identifying the crystal structure of the positive electrode active material, especially the crystal structure of the surface layer.
- the analysis program ReciPro can be used to analyze the electron diffraction pattern.
- fluorides such as fluorite (calcium fluoride) have long been used as fluxes in steelmaking and other processes, and their physical properties have been studied (Non-Patent Document 12).
- Non-Patent Document 13 It is known that when the temperature of a lithium-ion secondary battery rises during charging, it goes through several states and then reaches thermal runaway.
- Non-Patent Document 14 describes the thermal stability of positive electrode active materials and electrolytes.
- Lithium cobalt oxide (LiCoO 2 , sometimes written as LCO) shown in Patent Documents 2 to 4 is said to have low thermal stability.
- Lithium cobalt oxide (LiCoO 2 , sometimes written as LCO) shown in Patent Documents 2 to 4 is said to have low thermal stability.
- a nail penetration test which is one of the safety tests for lithium ion secondary batteries, Joule heat is generated and may lead to thermal runaway. Therefore, when using lithium cobalt oxide as described above, safety measures for the secondary battery are essential.
- Patent Document 1 discloses a configuration having a protective layer between the positive electrode current collector and the positive electrode mixture layer in order to suppress the rise in battery temperature during nail penetration.
- one aspect of the present invention aims to provide a highly safe battery.
- Another aspect of the present invention aims to provide a high-capacity and highly safe battery.
- One aspect of the present invention is a secondary battery having a positive electrode and a negative electrode, the positive electrode having a positive electrode active material, the positive electrode active material having lithium cobalt oxide containing magnesium, the magnesium concentration in the surface layer of the positive electrode active material being higher than the magnesium concentration inside the positive electrode active material, the negative electrode having a negative electrode active material, the negative electrode active material having a carbon material, and when AC impedance measurement is performed on the secondary battery in a state where the secondary battery is charged to a voltage of 4.5 V, the AC impedance value at a frequency of 1 kHz satisfies the condition of less than 90 m ⁇ .
- Another aspect of the present invention is a secondary battery having a positive electrode, a negative electrode, a separator, and an electrolyte, in which the positive electrode has a positive electrode active material, the positive electrode active material has lithium cobalt oxide containing magnesium, the magnesium concentration in the surface layer of the positive electrode active material is higher than the magnesium concentration inside the positive electrode active material, the negative electrode has a negative electrode active material, the negative electrode active material has a carbon material, the electrolyte solvent has ethylene carbonate and diethyl carbonate, the separator has polypropylene, and when the secondary battery is charged to a voltage of 4.5 V and an AC impedance measurement is performed, the AC impedance value at a frequency of 1 kHz satisfies less than 90 m ⁇ .
- Another aspect of the present invention is a secondary battery having a positive electrode and a negative electrode, the positive electrode having a positive electrode active material, the positive electrode active material having lithium cobalt oxide containing magnesium, the magnesium concentration in the surface layer of the positive electrode active material being higher than the magnesium concentration inside the positive electrode active material, the negative electrode having a negative electrode active material, the negative electrode active material having a carbon material, and when the secondary battery is charged to a voltage of 4.5 V and AC impedance measurement is performed in a state where the capacity is 2000 mAh or more, the AC impedance value at a frequency of 1 kHz satisfies less than 90 m ⁇ .
- Another aspect of the present invention is a secondary battery having a positive electrode, a negative electrode, a separator, and an electrolyte, in which the positive electrode has a positive electrode active material, the positive electrode active material has lithium cobalt oxide containing magnesium, the magnesium concentration in the surface layer of the positive electrode active material is higher than the magnesium concentration inside the positive electrode active material, the negative electrode has a negative electrode active material, the negative electrode active material has a carbon material, the electrolyte solvent has ethylene carbonate and diethyl carbonate, the separator has polypropylene, and when the secondary battery is charged to a voltage of 4.5 V and AC impedance measurement is performed in a state where the capacity is 2000 mAh or more, the AC impedance value at a frequency of 1 kHz satisfies less than 90 m ⁇ .
- the temperature rise ⁇ T is 50°C or less.
- the lithium cobalt oxide further contains aluminum.
- the lithium cobalt oxide further contains nickel.
- the lithium cobalt oxide further contains fluorine.
- One aspect of the present invention makes it possible to provide a highly safe secondary battery. Furthermore, one aspect of the present invention makes it possible to provide a high-capacity and highly safe secondary battery.
- FIGS. 1A and 1B are diagrams illustrating a nail penetration test. 2A and 2B are diagrams illustrating the nail penetration operation.
- FIG. 3 is a graph showing changes in the internal temperature when the internal temperature of a secondary battery in which an internal short circuit occurs increases.
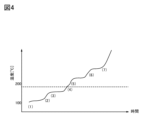
- FIG. 4 is a graph showing changes when the internal temperature of the secondary battery increases.
- 5A to 5C show an example of a cross-sectional structure of a positive electrode active material.
- FIG. 6 is an example of a TEM image in which the crystal orientations are roughly consistent.
- Fig. 7A is an example of an STEM image in which the crystal orientations are roughly consistent
- Fig. 7B is an FFT pattern of a region of rock-salt crystal RS, and Fig.
- FIG. 7C is an FFT pattern of a region of layered rock-salt crystal LRS.
- FIG. 8 is a diagram illustrating the crystal structure of the positive electrode active material.
- FIG. 9 is a diagram illustrating the crystal structure of a conventional positive electrode active material.
- FIG. 10 is a diagram illustrating the charge depth and the c-axis length of the positive electrode active material.
- FIG. 11 is a diagram showing an XRD pattern calculated from the crystal structure.
- FIG. 12 shows an XRD pattern calculated from the crystal structure.
- 13A and 13B are diagrams showing XRD patterns calculated from the crystal structure.
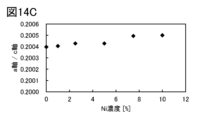
- 14A to 14C show the lattice constants calculated from XRD.
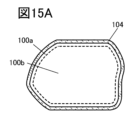
- 15A and 15B are cross-sectional views of the positive electrode active material.
- FIG. 16A and 16B are diagrams illustrating a laminated secondary battery.
- FIG. 17 is a phase diagram showing the relationship between the composition of lithium fluoride and magnesium fluoride and the temperature.
- FIG. 18 is a diagram illustrating the results of the DSC measurement.
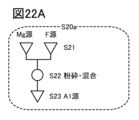
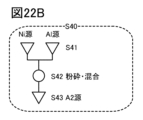
- 19A to 19C are diagrams illustrating a method for manufacturing a positive electrode active material.
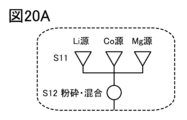
- 20A to 20C are diagrams illustrating a method for manufacturing a positive electrode active material.
- FIG. 21 is a diagram illustrating a method for manufacturing a positive electrode active material.
- 22A to 22C are diagrams illustrating a method for manufacturing a positive electrode active material.
- FIG. 23 is a diagram for explaining the heating furnace and heating conditions.
- 24A and 24B are diagrams illustrating the positive electrode.
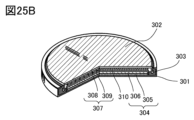
- 25A to 25C are diagrams illustrating a coin-type secondary battery.
- 26A to 26D are diagrams illustrating a cylindrical secondary battery.
- 27A and 27B are diagrams illustrating a wound type secondary battery.
- FIG. 28 is a diagram illustrating a wound type secondary battery.
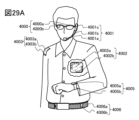
- 29A to 29D are diagrams illustrating electronic devices.
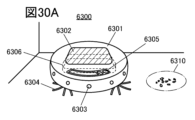
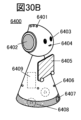
- 30A to 30C are diagrams illustrating an electronic device.
- 31A to 31C are diagrams illustrating a vehicle.
- 32A to 32C are graphs showing charge/discharge curves.
- FIG. 33A is a graph showing the results of the impedance test
- FIG. 33B is a diagram showing the equivalent circuit used in the analysis.
- 34A-34C are photographs illustrating the results of the nail penetration test.
- 35A-35C are graphs illustrating the results of the nail penetration test.
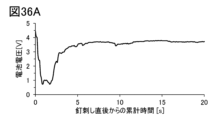
- 36A-36C are graphs illustrating the results of the nail penetration test.
- 37A-37C are graphs illustrating the results of the nail penetration test.
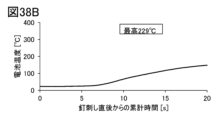
- 38A-38C are graphs illustrating the results of the nail penetration test.
- the space group is expressed using short notation of the international notation (or Hermann-Mauguin notation).
- the crystal plane and crystal direction are expressed using Miller indices.
- the space group, crystal plane, and crystal direction are expressed by adding a superscript bar to the number, but in this specification, due to format restrictions, instead of adding a bar above the number, a - (minus sign) may be added before the number.
- Individual directions indicating directions within a crystal are expressed with [ ]
- collective directions indicating all equivalent directions are expressed with ⁇ >
- individual faces indicating crystal faces are expressed with ( )
- collective faces with equivalent symmetry are expressed with ⁇ ⁇ .
- trigonal crystals represented by the space group R-3m are generally expressed as a composite hexagonal lattice of hexagonal crystals for ease of understanding of the structure, and in this specification, unless otherwise specified, the space group R-3m will be expressed as a composite hexagonal lattice.
- the space group R-3m will be expressed as a composite hexagonal lattice.
- not only (hkl) but also (hkil) may be used as Miller indices.
- i is -(h+k).
- the space group of the positive electrode active material, etc. is identified by X-ray diffraction (XRD), electron beam diffraction, neutron beam diffraction, etc. Therefore, in this specification, etc., belonging to a certain space group, belonging to a certain space group, or being a certain space group can be rephrased as being identified with a certain space group.
- XRD X-ray diffraction
- electron beam diffraction electron beam diffraction
- neutron beam diffraction etc. Therefore, in this specification, etc., belonging to a certain space group, belonging to a certain space group, or being a certain space group can be rephrased as being identified with a certain space group.
- the anions do not have to be strictly cubic lattices.
- the analysis results do not necessarily have to be the same as the theory.
- FFT fast Fourier transform
- TEM Transmission Electron Microscope
- particles are not limited to those having a spherical shape (a circular cross-sectional shape), but may have an elliptical, rectangular, trapezoidal, triangular, rectangular with rounded corners, asymmetrical shape, or the like in cross-sectional shape of each particle, and each particle may also have an irregular shape.
- the theoretical capacity of a positive electrode active material refers to the amount of electricity when all of the lithium that can be inserted and removed from the positive electrode active material is removed.
- the theoretical capacity of LiCoO2 is 274 mAh/g per weight of the positive electrode active material
- the theoretical capacity of LiNiO2 is 275 mAh/g per weight of the positive electrode active material
- the theoretical capacity of LiMn2O4 is 148 mAh/g per weight of the positive electrode active material.
- the amount of lithium that can be inserted and removed from the positive electrode active material is indicated by x in the composition formula, for example, x in Li x MO 2.
- M represents a transition metal, and unless otherwise specified in this specification, M is cobalt and/or nickel.
- x in Li x MO 2 is small means 0.1 ⁇ x ⁇ 0.24.
- the lithium cobalt oxide in a lithium ion secondary battery that has completed discharge is LiCoO2 , and x may be said to be 1.
- the completion of discharge here refers to a state in which the current per weight of the positive electrode active material is 100 mA/g or less and the voltage is 3.0 V or 2.5 V or less.
- the charge capacity and/or discharge capacity used to calculate x in Li x MO 2 is preferably calculated under conditions where there is no effect of short circuit and/or no effect of decomposition of the electrolyte in the lithium ion secondary battery, or where such effects are minimal. For example, data from a lithium ion secondary battery that has experienced a sudden change in capacity that is considered to be due to a short circuit should not be used to calculate x.
- segregation of an element refers to a state in which an element (e.g., A) is distributed unevenly in a solid composed of multiple elements (e.g., A, B, C).
- the distribution of a certain element refers to the continuous presence of that element when the element is detected in a range that is not noise using any analytical method.
- the continuous change in concentration of the element is sometimes called a concentration gradient.
- the maximum value in the distribution is sometimes called a peak.
- identifying a peak a distribution limited to a certain region can be targeted.
- the above-mentioned distribution is not limited to a normal distribution. When it corresponds to a normal distribution, the half-width of the distribution can also be determined.
- uneven distribution means that the concentration of an element in one area is different from that in other areas. It is synonymous with bias, or the presence of a mixture of areas of high concentration and areas of low concentration. Uneven distribution through solid solution is called segregation.
- the surface layer of the positive electrode active material refers to a region within 20 nm or within 50 nm from the surface toward the inside in a direction perpendicular or nearly perpendicular to the surface.
- the surface layer is synonymous with the surface vicinity and the surface vicinity region. Note that perpendicular or nearly perpendicular specifically refers to an angle with the surface of 80° or more and 100° or less.
- the region deeper than the surface layer of the positive electrode active material is called the interior.
- the interior is synonymous with the bulk or core.
- the positive electrode active material may be expressed as a composite oxide, a positive electrode material, a positive electrode material, a positive electrode material for secondary batteries, a positive electrode material for lithium ion secondary batteries, etc.
- the positive electrode active material of one embodiment of the present invention preferably has a compound.
- the positive electrode active material of one embodiment of the present invention preferably has a composition.
- the positive electrode active material of one embodiment of the present invention preferably has a composite.
- the characteristics of the positive electrode active material may be described in this specification, it is not necessary that all of the positive electrode active material in the positive electrode have that characteristic. For example, if three or more of five or more randomly selected particles of positive electrode active material have that characteristic, it can be said that there is a sufficient effect of improving the characteristics of the positive electrode active material and the secondary battery containing it.
- an internal short circuit in a secondary battery refers to contact between the positive and negative electrodes inside the battery.
- An external short circuit in a secondary battery which is intended to occur in the event of misuse, refers to contact between the positive and negative electrodes outside the battery.
- An internal short circuit in a secondary battery can be intentionally induced by a nail penetration test.
- thermal runaway is said to have occurred when thermal decomposition products of the positive electrode and/or negative electrode are observed in an area 2 cm or more away from the nail insertion point after the nail penetration test is completed.
- Thermal decomposition products of the positive electrode and/or negative electrode include, for example, aluminum oxide formed by the oxidation of aluminum when aluminum foil is used as the positive electrode current collector, and copper oxide formed by the oxidation of copper when copper foil is used as the negative electrode current collector.
- the materials (positive electrode active material, negative electrode active material, electrolyte, separator, etc.) of the secondary battery are described in the state before degradation. Note that the decrease in charge capacity and/or discharge capacity due to aging and burn-in treatments during the secondary battery manufacturing stage is not included in degradation.
- a secondary battery consisting of a single cell or a battery pack has a discharge capacity of 97% or more of the rated capacity, it can be said to be in the state before degradation.
- the rated capacity complies with JIS C 8711:2019.
- other secondary batteries it is not limited to the above JIS standards, but also complies with various JIS and IEC standards for electric vehicle propulsion, industrial use, etc.
- a lithium ion secondary battery refers to a battery that uses lithium ions as carrier ions, but in the present invention, the carrier ions are not limited to lithium ions.
- the carrier ions are not limited to lithium ions.
- alkali metal ions or alkaline earth metal ions can be used as carrier ions, and specifically, sodium ions and the like can be applied.
- the present invention can be understood by reading lithium ions as sodium ions and the like.
- the carrier ions it may be referred to as a secondary battery.
- the (001) plane and the (003) plane may be collectively referred to as the (00l) plane.
- the (00l) plane may also be referred to as the C plane, the basal plane, etc.
- lithium ions have a two-dimensional diffusion path. In other words, it can be said that the diffusion path of lithium ions exists along the plane.
- a plane on which the diffusion path of lithium ions is exposed that is, a plane other than the plane on which lithium is inserted and removed (specifically the (001) plane), may be referred to as an edge plane.
- the amount of support is the weight of active material per unit surface area of the current collector.
- the amount of negative electrode active material supported can be adjusted according to the capacity of the positive electrode. In the case of double-sided coating in which a slurry containing active material is applied to both sides of the current collector, the above amount of support is considered per side.
- secondary particles refer to particles formed by aggregation of primary particles.
- single particles refer to particles that do not have grain boundaries on the outside.
- single crystal particles refer to crystal particles in which there are no grain boundaries inside the particle, and polycrystalline particles refer to crystal particles in which there are grain boundaries inside the particle.
- Polycrystalline particles may be said to be an aggregate of multiple crystallites, and grain boundaries may be said to be the interface that exists between two or more crystallites.
- a and/or B may be used, but this is an example of a description that includes only A, only B, and both A and B.
- the nail penetration test is a test in which a secondary battery is fully charged and a nail having a predetermined diameter selected from 2 mm to 20 mm is inserted into the secondary battery at a predetermined speed.
- Full charge refers to a state in which the charging rate, expressed as State Of Charge (hereinafter referred to as SOC), is 100%.
- SOC State Of Charge
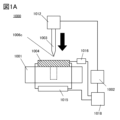
- FIG. 1A shows a cross-sectional view of a nail penetration test device 1000.
- the nail penetration test device 1000 includes a stage 1001, a driving unit 1002, a nail 1003, a voltage measuring device 1015, a temperature measuring device 1016, and a control unit 1018.
- the driving unit 1002 includes a driving mechanism 1012 that moves the nail 1003 in the direction of the arrow in the figure, and the driving mechanism 1012 operates so that the nail 1003 penetrates a secondary battery 1004 installed on the stage 1001. At this time, the secondary battery 1004 is in a fully charged state, and this operation is called a nail penetration operation.
- the dashed line shown in FIG. 1A indicates a recess in the stage 1001 that is provided to accommodate the nail 1003 that has penetrated the secondary battery 1004 during the nail penetration operation.
- the voltage measuring device 1015 transmits information about the voltage of the secondary battery during the nail penetration operation to the control unit 1018. Specifically, the amount of voltage change and the like are transmitted to the control unit 1018. In addition, the temperature measuring device 1016 transmits information about the temperature during the nail penetration operation to the control unit 1018. When controlling the operating conditions of the nail 1003, the control unit 1018 can transmit a control signal to the drive unit 1002.
- Figure 1B is a perspective view illustrating the vicinity of the upper part of the stage 1001 of the nail penetration test device 1000.
- the secondary battery 1004 placed on the stage 1001 is electrically connected to the wiring 1005a and wiring 1005b.
- the wiring 1005a and wiring 1005b belong to the voltage measuring device 1015, and the wiring 1005a and wiring 1005b are electrically connected to the positive electrode tab and the negative electrode tab of the secondary battery 1004, respectively, and the voltage of the secondary battery 1004 can be measured.
- the voltage of the secondary battery 1004 is simply called the voltage, the voltage value between the positive and negative electrodes, the battery voltage, the cell voltage, or the open voltage.
- the temperature sensor is provided so as to be in contact with the surface of the exterior body of the secondary battery 1004.
- a first temperature sensor 1006a and a second temperature sensor 1006b are arranged on the secondary battery 1004, and a third temperature sensor 1006c is further provided on the nail 1003 shown in FIG. 1A.
- one or more temperature sensors may be arranged on the secondary battery 1004.
- the first temperature sensor 1006a is arranged on the side where the wiring 1005a and wiring 1005b are not arranged
- the second temperature sensor 1006b is arranged on the side where the wiring 1005a and wiring 1005b are arranged. In this way, when two or more temperature sensors are arranged, it is preferable because the other temperature sensors can be used even if one temperature sensor becomes unusable due to expansion of the exterior body or the like.
- the exterior body is folded back on the side where the wiring 1005a and wiring 1005b are not arranged, so there is no adhesive area. Therefore, even if the exterior body expands, the expansion is suppressed on the side where the wiring 1005a and wiring 1005b are not arranged, and the first temperature sensor 1006a is less likely to peel off than the second temperature sensor 1006b, which is preferable.
- the dashed ellipse in FIG. 1B is the area where the nail 1003 penetrates the secondary battery 1004 during the nail penetration operation.
- the first temperature sensor 1006a and the second temperature sensor 1006b provided on the secondary battery 1004 should be provided at equal distances from the area where the nail 1003 penetrates.
- the first temperature sensor 1006a and the second temperature sensor 1006b should be provided within 5 cm, preferably within 2 cm, from the area where the nail 1003 penetrates.
- the vicinity here refers to an area within 1 cm from the penetration area.
- the nail penetration test is a test in which the secondary battery 1004 is fully charged and a nail 1003 is inserted into the secondary battery 1004 at a predetermined speed.
- the diameter of the nail 1003 is 2 mm or more and 10 mm or less.
- FIG. 2A shows a cross-sectional view of the secondary battery 1004 in a state in which the nail 1003 is inserted.
- the secondary battery 1004 has a structure in which a positive electrode 503, a separator 508, a negative electrode 506, and an electrolyte 530 are contained in an exterior body 531.
- the positive electrode 503 has a positive electrode current collector 501 and a positive electrode active material layer 502 formed on both sides thereof, and the negative electrode 506 has a negative electrode current collector 511 and a negative electrode active material layer 512 formed on both sides thereof.
- the positive electrode active material layer is a layer having at least a positive electrode active material, and has, for example, a conductive material and/or a binder.
- the negative electrode active material layer is a layer having at least a negative electrode active material, and has, for example, a conductive material and/or a binder.
- Fig. 2B shows an enlarged view of the nail 1003 and the positive electrode current collector 501 and their vicinity, and also clearly shows the positive electrode active material 100 and the conductive material 553 contained in the positive electrode active material layer 502.
- a nail 1003 is driven into a secondary battery 1004, and the nail 1003 penetrates the positive electrode 503 and the negative electrode 506, causing an internal short circuit. Then, the potential of the nail 1003 becomes equal to the potential of the negative electrode 506, and electrons (e ⁇ ) flow to the positive electrode 503 through the nail 1003 and the like as shown by the black arrow, and Joule heat is generated at the internal short circuit location and its vicinity.
- carrier ions typically lithium ions (Li + )
- Li + lithium ions
- the electrolyte begins to be reduced and decomposed on the surface of the negative electrode before the lithium ions from the negative electrode are completely released. This is one of the electrochemical reactions, and is called a reduction reaction of the electrolyte by the negative electrode.
- the lithium cobalt oxide when the temperature of the secondary battery 1004 rises due to Joule heat, if lithium cobalt oxide is used as the positive electrode active material, the lithium cobalt oxide may undergo a phase change (i.e., a structural change) to an H1-3 type crystal structure or an O1 type crystal structure, and heat may be generated due to the phase change.
- a phase change i.e., a structural change
- the H1-3 type crystal structure and the O1 type crystal structure will be described later. If an internal short circuit occurs, heat will be generated one after another.
- the electrons ( e- ) flowing to the positive electrode 503 reduce the tetravalent Co in the lithium cobalt oxide in the charged state to a trivalent or divalent Co, and oxygen is released from the lithium cobalt oxide by this reduction reaction, and the electrolyte 530 is decomposed by an oxidation reaction caused by the oxygen.
- This is one of the electrochemical reactions, and is called an oxidation reaction of the electrolyte by the positive electrode. It is thought that the speed at which the current flows into the positive electrode active material 100 etc. affects the electrochemical reaction, and it is possible to delay the speed at which the current flows by the insulating property of the positive electrode active material.
- FIG. 3 is a graph of the temperature (specifically, the internal temperature) of a secondary battery versus time, which is a partially modified version of the graph shown on page 70 [FIG. 2-12] of Non-Patent Document 13.
- P0 the temperature of the secondary battery rises with time.
- P1 heat generation due to Joule heat continues until the temperature of the secondary battery reaches 100° C. or close thereto, exceeding the reference temperature (Ts) of the secondary battery.
- the positive electrode active material which is one aspect of the present invention, can have both the above-mentioned stable structure and a structure that slows down the rate of current flow.
- the positive electrode active material 100 may be a compound having a transition metal and oxygen, capable of inserting and removing carrier ions, typically lithium ions (Li + ).
- the transition metal may be one or more selected from cobalt (Co), nickel (Ni), manganese (Mn), iron (Fe), and the like.
- FIG. 5A shows an example of a cross-sectional structure of the positive electrode active material 100.
- the positive electrode active material 100 has a surface layer 100a and an interior 100b, and the (001) surface, which is the diffusion surface of lithium ions, is indicated by a dashed line.
- the positive electrode active material 100 of one embodiment of the present invention preferably uses cobalt as a main component of the transition metal M responsible for the redox reaction.
- the main component of the transition metal M refers to the transition metal M having the highest atomic ratio.
- lithium cobalt oxide can be applied to the positive electrode active material 100 as a compound using Co as the transition metal.
- the positive electrode active material 100 preferably has lithium cobalt oxide (LiCoO 2 ) to which an additive element described below is added.
- the positive electrode active material 100 of one embodiment of the present invention preferably has an insulating region or a region with high resistance.
- the above region is preferably present in a narrow width of 1 nm to 20 nm, preferably 2 nm to 10 nm, and more preferably 2 nm to 5 nm in a cross-sectional view of the positive electrode active material 100.
- the narrow region may be referred to as a "shell” in this specification and the like.
- a cross-sectional STEM (Scanning Transmission Electron Microscope) image can be used for the above cross-sectional view.
- the positive electrode active material undergoes a reaction in which cobalt is reduced from tetravalent to divalent due to the electrons that flow into the positive electrode active material rapidly, causing oxygen to be released from the positive electrode active material.
- This reaction is an exothermic reaction, which accelerates thermal runaway.
- the positive electrode active material 100 of the present invention has a structure that makes it difficult to release oxygen by using a shell 100s described below. If oxygen is not released from the positive electrode active material, the above reduction reaction (for example, the reaction from Co 4+ to Co 2+ ) is also suppressed.
- Figure 5B shows a positive electrode active material 100 having a shell 100s.
- the positive electrode active material 100 having a shell 100s has increased resistance, so that even when a nail penetration test is performed, the speed of the current flowing into the positive electrode active material 100 can be slowed down, which is preferable because it can suppress ignition, etc.
- the shell 100s with high resistance is located in the surface layer portion 100a.
- the positive electrode active material 100 preferably contains an additive element.
- the additive element include magnesium, fluorine, nickel, aluminum, titanium, zirconium, vanadium, iron, manganese, chromium, niobium, arsenic, zinc, silicon, sulfur, phosphorus, boron, bromine, and beryllium, and one or more additive elements selected from these may be used.
- the additive elements do not necessarily have to include magnesium, fluorine, nickel, aluminum, titanium, zirconium, vanadium, iron, manganese, chromium, niobium, arsenic, zinc, silicon, sulfur, phosphorus, boron, bromine, and beryllium.
- the weight of manganese contained in the positive electrode active material 100 is preferably, for example, 600 ppm or less, and more preferably 100 ppm or less.
- Magnesium is one of the elements suitable for forming the shell 100s. Specifically, magnesium is suitable as an additive element that is less likely to release oxygen because the closer the oxygen is to the magnesium, the greater the energy required to desorb it. Magnesium also has the function of stabilizing the crystal structure of the positive electrode active material, making it a suitable additive element. Magnesium can keep the crystal structure stable while slowing down the speed of the current flowing into the positive electrode active material 100.
- magnesium When viewed in cross section of the positive electrode active material 100, magnesium is preferably present in a narrow area of 1 nm to 20 nm, preferably 2 nm to 10 nm, and more preferably 2 nm to 5 nm, from the surface. Magnesium should be present in greater amounts, i.e., at a higher concentration, in the shell 100s on the edge surface than in the shell 100s on the basal surface.
- the shell 100s contains at least magnesium, oxygen release from this region is suppressed and thermal stability can be improved, making it a structure that is less likely to lead to thermal runaway. Therefore, the shell 100s is preferably provided so as to fully cover the entire positive electrode active material 100 as shown in FIG. 5A, but it is preferable that it is formed on 60% or more of the region in cross-sectional view, and preferably 80% or more. Furthermore, it is preferable that the shell 100s is formed uniformly, but it may be formed so that specific regions of the positive electrode active material 100, for example, faces other than the (00l) face, are thicker.
- the shell 100s may be arranged in any manner relative to the positive electrode active material 100, and as long as it enables the insertion and removal of lithium ions (Li + ) while slowing down the rate at which current flows due to an internal short circuit, magnesium may be present outside the shell 100s, for example, in the interior 100b.
- the positive electrode active material 100 may have an area in which the shell 100s is not provided.
- magnesium fluoride When magnesium is added to the positive electrode active material 100, it is preferable to use magnesium fluoride as the magnesium source. When magnesium fluoride is used, fluorine can also be added to the positive electrode active material 100. In other words, it may be preferable for the positive electrode active material 100 to have fluorine in addition to magnesium as an added element. Lithium and fluorine may react during a nail penetration test, etc., but this produces less heat than when lithium reacts with oxygen, so the temperature rise of the secondary battery can be suppressed.
- nickel in addition to magnesium as an additive element in the positive electrode active material 100, which is one embodiment of the present invention.
- nickel is also present in the shell 100s. It is preferable that nickel is present in greater amounts, that is, in a higher concentration, in the shell 100s on the edge surface than in the shell 100s on the basal surface. This configuration can suppress the release of oxygen from the positive electrode active material, or suppress structural changes in the positive electrode active material.
- the above-mentioned additive elements are present at least in the shell 100s. In other words, it is preferable that the above-mentioned additive elements are present at least in the surface layer 100a.
- the additive elements that contribute to the stability of the crystal structure of the positive electrode active material 100 are preferably present in the surface layer 100a where degradation is likely to begin. It is preferable that the additive elements are present in greater amounts, i.e., at a higher concentration, in the shell 100s of the edge surface than in the shell 100s of the basal surface.
- a method for checking whether or not a shell 100s is formed on the positive electrode active material 100 there is a method for measuring the resistance (called powder resistance) of the powder that becomes the positive electrode active material.
- powder resistance the resistance of the powder that becomes the positive electrode active material.
- the powder resistance value will be higher than when the shell 100s is not formed.
- the powder resistance of a positive electrode active material having an additive element shows a higher value compared to the powder resistance of a positive electrode active material not having an additive element, there is a possibility that a shell 100s has been formed.
- the shell 100s contains cobalt in addition to the additive element.
- the shell 100s enables the insertion and desorption of lithium ions (Li + ).
- the shell 100s can slow down the rate of current flow caused by an internal short circuit.
- the surface layer 100a also contains the additive element and cobalt.
- the magnesium concentration in the lithium cobalt oxide shell 100s is preferably greater than 0 and less than 10 atomic%, preferably greater than 0 and less than 5 atomic%, more preferably greater than 0 and less than 2 atomic%.
- the magnesium concentration can be determined by line analysis of energy dispersive X-ray spectroscopy (EDX). If magnesium is present throughout the entire surface layer 100a and at a high concentration, the insulating properties will be high, making it difficult to obtain favorable battery characteristics in charge-discharge cycle tests and the like.
- the presence of magnesium in the surface layer 100a can stabilize the lithium cobalt oxide and suppress ignition and the like in the above-mentioned nail penetration test and the like, which is preferable. Furthermore, the presence of magnesium at an appropriate concentration in the shell 100s is expected to improve the hardness of the lithium cobalt oxide.
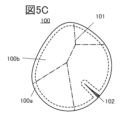
- Figure 5C shows a positive electrode active material 100 having a crystal grain boundary 101.
- the crystal grain boundary 101 refers to, for example, a portion where the particles of the positive electrode active material 100 are adhered to each other, a portion where the crystal orientation changes inside the positive electrode active material 100, that is, a portion where the repetition of bright and dark lines in an STEM image or the like becomes discontinuous, a portion containing many crystal defects, a portion where the crystal structure is disordered, etc.
- the crystal defect refers to a defect that can be observed in a cross-sectional TEM or cross-sectional STEM image, that is, a structure in which other atoms have entered between the lattices, a cavity, etc.
- the crystal grain boundary 101 can be said to be one type of planar defect.
- the vicinity of the crystal grain boundary 101 refers to a region within 10 nm of the crystal grain boundary 101.
- Figure 5C also shows a positive electrode active material 100 having a crack 102.
- a crack includes an area where the crystal plane is misaligned or an area where the crystal plane is broken, and the crack often occurs along the (00l) plane. It is preferable that such cracks 102 are not observed, but cracks 102 may be present as long as they do not ignite during a nail penetration test. In order to prevent ignition during a nail penetration test, it is preferable that a shell is also formed on the newly formed surface layer when crack 102 occurs. Furthermore, the vicinity of crack 102 refers to a region within 10 nm of crack 102.
- the positive electrode active material 100 is preferably highly crystalline, and more preferably single crystalline or polycrystalline. In particular, when the positive electrode active material 100 has a single crystal, even if the volume of the positive electrode active material 100 changes due to charging and discharging, cracks are less likely to occur, which is preferable. In other words, when the positive electrode active material 100 is a single crystal, a secondary battery using the positive electrode active material 100 is considered to be less likely to ignite, and safety can be improved. After the initial heating described below, the crystallinity of the positive electrode active material 100 becomes high, which is preferable. In addition, the positive electrode active material 100 is preferably a single particle (also called a primary particle) rather than a secondary particle.
- ⁇ Median diameter (D50) of positive electrode active material The median diameter (D50) of the positive electrode active material of a highly safe secondary battery will be described. If the positive electrode active material is too small, it may be difficult to apply the material when preparing the positive electrode. If the positive electrode active material is too small, the surface area may become too large, and the reaction between the positive electrode active material surface and the electrolyte may become excessive. If the positive electrode active material is too small, it may be necessary to mix a large amount of conductive material, which may lead to a decrease in capacity. In these respects, the median diameter (D50) of the positive electrode active material is preferably 1 ⁇ m or more, preferably 5 ⁇ m or more, and more preferably 9 ⁇ m or more.
- a positive electrode active material with a small median diameter (D50) is preferable because it is less likely to cause a slippage region. Also, a positive electrode active material with a small median diameter (D50) is preferable because it is less likely to cause cracks even after a pressing process.
- the median diameter (D50) of the positive electrode active material should be 20 ⁇ m or less, preferably 18 ⁇ m or less, and more preferably 15 ⁇ m or less.
- the median diameter (D50) of the positive electrode active material can be any combination of the upper and lower limits described above.
- the median diameter is 1 ⁇ m or more and 20 ⁇ m or less, preferably 1 ⁇ m or more and 18 ⁇ m or less, and more preferably 1 ⁇ m or more and 15 ⁇ m or less.
- the above-mentioned median diameter (D50) can be measured, for example, by observation using an SEM or TEM, or by a particle size distribution analyzer using a laser diffraction/scattering method.
- the median diameter (D50) is the particle diameter when the cumulative amount in the cumulative curve of the particle size distribution measurement results accounts for 50%.
- a method for measuring the median diameter (D50) from analysis using an SEM or TEM, for example, is to measure 20 or more particles, create a cumulative curve, and determine the particle diameter when the cumulative amount accounts for 50%.
- the surface of the positive electrode active material 100 refers to the surface of the composite oxide including the surface layer 100a and the interior 100b. Such a surface can be confirmed in a cross-sectional view. Therefore, the surface of the positive electrode active material 100 does not include metal oxides such as aluminum oxide (Al 2 O 3 ) that do not have lithium sites that can contribute to charging and discharging, carbonates that are chemically adsorbed after the preparation of the positive electrode active material, and hydroxyl groups.
- the attached metal oxide refers to, for example, a metal oxide whose crystal structure does not match that of the interior 100b.
- the positive electrode active material 100 is a composite oxide containing oxygen and a transition metal capable of inserting and removing lithium
- the interface between the region where the transition metal M (e.g., Co, Ni, Mn, Fe, etc.) that is oxidized and reduced with the insertion and removal of lithium and oxygen is present and the region where it is not present is the surface of the positive electrode active material.
- a protective film may be attached to the surface, but the protective film is not included in the positive electrode active material.
- the protective film a single layer or multilayer film of carbon, metal, oxide, resin, etc. may be used.
- the reference point in STEM-EDX-ray analysis etc. is a point that is 50% of the average value M AVE of the amount of characteristic X-rays of transition metal M inside the positive electrode active material.
- the reference point may be called the position of the surface of the positive electrode active material.
- the amount of detection of characteristic X-rays of transition metal M when the amount of detection of characteristic X-rays of transition metal M does not drop sufficiently to the left of the reference point, the amount of detection of characteristic X-rays of transition metal M to the left of the reference point is called background, and the point that is 50% of the sum of the average value M BG of the amount of detection of characteristic X-rays of transition metal M in the background and the average value M AVE of the amount of detection of transition metal M inside may be used as the reference point.
- the amount of detection of characteristic X-rays of oxygen inside the positive electrode active material may be used, and the reference point can be obtained by replacing the transition metal M with oxygen.
- the reference point can be obtained using the M AVE of the element with the largest amount of detection of the characteristic X-ray inside, or the M AVE and M BG .
- the average value M AVE of the internal detected amount of characteristic X-rays can be obtained by averaging a range of 2 nm or more, preferably 3 nm or more, in a region where the detected amount of characteristic X-rays of the transition metal M or oxygen is stable, in other words, a saturated region, specifically, a region 30 nm or more, preferably 50 nm or more deep from the region where the detected amount of characteristic X-rays of the transition metal M starts to increase or the region where the detected amount of characteristic X-rays of oxygen starts to increase.
- the average value M BG of the background can be obtained by averaging a range of 2 nm or more, preferably 3 nm or more, in a stable region avoiding, for example, the vicinity where the detected amount of characteristic X-rays of the transition metal M starts to increase.
- the surface of the positive electrode active material 100 in a cross-sectional STEM image or the like is the boundary between an area where an image originating from the crystal structure of the positive electrode active material is observed and an area where it is not observed, and is the outermost area where atomic columns originating from the atomic nuclei of metal elements having atomic numbers larger than lithium among the metal elements that make up the positive electrode active material can be confirmed.
- the spatial resolution of STEM-EDX is at least about 1 nm. Therefore, the peak position (also called the maximum value) of the detected amount of characteristic X-rays corresponding to the added element may be shifted by about 1 nm. For example, even if the peak position of the detected amount of characteristic X-rays corresponding to an added element such as magnesium is to the left of the surface determined above, it can be considered an error as long as the difference between the peak and the surface is less than 1 nm.
- a peak in STEM-ED X-ray analysis refers to the maximum or local maximum value of the characteristic X-rays corresponding to each element.
- noise in STEM-ED X-ray analysis can be a measured value with a half-width less than the spatial resolution (R), for example, less than R/2.
- the effects of noise can be reduced by scanning the same location multiple times under the same conditions.
- the integrated values measured over six scans can be used to graph the characteristic X-rays of each element.
- the number of scans is not limited to six, and more scans can be performed and the average can be used to graph the characteristic X-rays of each element.
- STEM-EDX analysis can be performed, for example, as follows. First, a protective film is deposited on the surface of the positive electrode active material in the atmosphere. For example, carbon can be deposited using a carbon coating unit of an ion sputtering device (Hitachi High-Tech MC1000).
- the positive electrode active material is then sliced to prepare a STEM cross-sectional sample.
- the slice processing can be performed using a FIB-SEM device (Hitachi High-Tech XVision 200TBS).
- the pickup is performed using an MPS (micro-probing system), and the finishing processing conditions can be, for example, an acceleration voltage of 10 kV.
- EDAX's Octane T Ultra W can be used as the EDX detector mounted on the STEM device.
- the acceleration voltage of the STEM device is set to 200 kV, and the emission current is set to 6 ⁇ A or more and 10 ⁇ A or less, and a portion of the sliced sample with minimal depth and unevenness is measured.
- the magnification is, for example, about 150,000 times.
- the conditions for EDX-ray analysis can be drift correction, line width 42 nm, pitch 0.2 nm, and frame number 6 or more.
- Crystal structure changes continuously from the inside 100b toward the surface due to the concentration gradient of the added element as described above.
- crystal orientation of the surface layer 100a and the inside 100b are approximately the same.
- the crystal structure changes continuously from the inside 100b of the layered rock salt type toward the surface and surface layer 100a that has characteristics of the rock salt type or both the rock salt type and the layered rock salt type.
- the crystal orientation of the surface layer 100a that has characteristics of the rock salt type or both the rock salt type and the layered rock salt type and the inside 100b of the layered rock salt type are roughly the same.
- a rock-salt crystal structure is a cubic crystal structure, including those belonging to the space group Fm-3m, in which cations and anions are arranged alternately. Note that there may be a deficiency of cations or anions.
- the distance between the bright spots on the (003) plane of LiCoO 2 is observed to be about half the distance between the bright spots on the (111) plane of MgO. Therefore, when the analysis region has two phases, for example, rock salt type MgO and layered rock salt type LiCoO2 , the electron beam diffraction pattern shows a plane orientation in which bright spots with strong brightness and bright spots with weak brightness are arranged alternately. Bright spots common to the rock salt type and layered rock salt type have strong brightness, while bright spots occurring only in the layered rock salt type have weak brightness.
- the layered rock salt type crystal and the anions in the rock salt type crystal have a cubic close-packed structure (face-centered cubic lattice structure). It is presumed that the anions in the O3' type and monoclinic O1(15) crystals described below also have a cubic close-packed structure. Therefore, when a layered rock salt type crystal comes into contact with a rock salt type crystal, there is a crystal face where the cubic close-packed structure formed by the anions is oriented in the same direction.
- the anions on the ⁇ 111 ⁇ plane of the cubic crystal structure have a triangular lattice.
- the layered rock salt type is in space group R-3m and has a rhombohedral structure, but is generally represented as a compound hexagonal lattice to make the structure easier to understand, and the (0001) plane of the layered rock salt type has a hexagonal lattice.
- the triangular lattice of the cubic ⁇ 111 ⁇ plane has the same atomic arrangement as the hexagonal lattice of the (0001) plane of the layered rock salt type. When the two lattices are compatible, it can be said that the orientation of the cubic close-packed structure is aligned.
- the space group of layered rock salt crystals and O3' type crystals is R-3m, which is different from the space group Fm-3m (the space group of general rock salt crystals) of rock salt crystals, so the Miller indices of the crystal planes that satisfy the above conditions are different between layered rock salt crystals and O3' type crystals and rock salt crystals.
- the orientation of the cubic close-packed structure formed by anions in layered rock salt crystals, O3' type crystals and rock salt crystals is aligned, it may be said that the crystal orientations are roughly the same.
- the three-dimensional structural similarity in which the crystal orientations are roughly the same, or the same crystallographic orientation is called topotaxis.
- the fact that the crystal orientation of the two regions roughly coincides can be determined from TEM images, STEM images, HAADF-STEM (High-angle Annular Dark Field STEM, high-angle scattering annular dark-field scanning transmission electron microscope) images, ABF-STEM (Annular Bright-Field STEM, annular bright-field scanning transmission electron microscope) images, electron diffraction patterns, FFT patterns of TEM images and STEM images, etc.
- XRD electron diffraction, neutron diffraction, etc. can also be used to make the determination.
- Figure 6 shows an example of a TEM image in which the orientation of the layered rock salt crystal LRS and the rock salt crystal RS roughly coincides. Images reflecting the crystal structure are obtained in TEM images, STEM images, HAADF-STEM images, ABF-STEM images, etc.
- a contrast originating from a crystal plane is obtained.
- the contrast originating from the (0003) plane is obtained as a repetition of a bright band (bright strip) and a dark band (dark strip). Therefore, when a repetition of bright lines and dark lines is observed in a TEM image and the angle between the bright lines (for example, L RS and L LRS shown in FIG.
- lithium cobalt oxide having a layered rock-salt crystal structure is observed perpendicular to the c-axis
- the arrangement of the cobalt atoms is observed perpendicular to the c-axis as a bright line or an arrangement of dots with high brightness
- the arrangement of the lithium atoms and oxygen atoms is observed as a dark line or a region with low brightness.
- fluorine (atomic number 9) and magnesium (atomic number 12) are added to the lithium cobalt oxide.
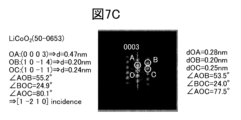
- Figure 7A shows an example of an STEM image in which the orientations of the layered rock-salt crystal LRS and the rock-salt crystal RS are roughly the same.
- Figure 7B shows the FFT pattern of the region of the rock-salt crystal RS
- Figure 7C shows the FFT pattern of the region of the layered rock-salt crystal LRS.
- the left side of Figures 7B and 7C show the composition, JCPDS card number, and the d value and angle calculated from the JCPDS card data.
- the right side shows the actual measured values.
- the spot marked with an O is the zeroth order diffraction.
- the spot marked A in Figure 7B is due to the 11-1 reflection of the cubic crystal.
- the spot marked A in Figure 7C is due to the 0003 reflection of the layered rock salt type. From Figures 7B and 7C, it can be seen that the orientation of the 11-1 reflection of the cubic crystal and the orientation of the 0003 reflection of the layered rock salt type roughly coincide. In other words, it can be seen that the line passing through AO in Figure 7B and the line passing through AO in Figure 7C are roughly parallel. Here, roughly coincident and roughly parallel mean that the angle between the above lines is 5 degrees or less, or 2.5 degrees or less.
- the ⁇ 0003> orientation of the layered rock salt type may roughly match the ⁇ 11-1> orientation of the rock salt type.
- these reciprocal lattice points are spot-like, that is, not continuous with other reciprocal lattice points. The fact that the reciprocal lattice points are spot-like and not continuous with other reciprocal lattice points indicates high crystallinity.
- a spot not originating from the 0003 reflection of the layered rock salt type may be observed in a reciprocal lattice space different from the orientation of the 0003 reflection of the layered rock salt type.
- the spot marked B in FIG. 7C is originating from the 1014 reflection of the layered rock salt type. This may be observed at an angle of 52° to 56° (i.e., ⁇ AOB is 52° to 56°) from the orientation of the reciprocal lattice point (A in FIG.
- a spot not originating from the 11-1 reflection of the cubic crystal may be observed.
- the spot marked B in FIG. 7B originates from the 200 reflection of the cubic crystal. This is a location at an angle of 54° to 56° (i.e., ⁇ AOB is 54° to 56°) from the orientation of the reciprocal lattice point (A in FIG. 7B) originating from the 11-1 reflection of the cubic crystal, and a diffraction spot may be observed at this location.
- this index is only an example, and does not necessarily have to match this.
- a reciprocal lattice point equivalent to 11-1 and 200 may be used.
- layered rock-salt type positive electrode active materials such as lithium cobalt oxide
- the (0003) plane and its equivalent as well as the (10-14) plane and its equivalent, as crystal planes. Therefore, when observing the (0003) plane with a TEM or the like, first select a particle of the positive electrode active material in which a crystal plane expected to be the (0003) plane is observed with a SEM or the like, and then slice the particle of the positive electrode active material with a FIB (Focused Ion Beam) or the like so that the (0003) plane can be observed with an electron beam incident in the [12-10] direction in the TEM or the like. When it is desired to determine the coincidence of the crystal orientations, it is preferable to slice the layered rock-salt type (0003) plane so that it is easy to observe.
- FIB Flucused Ion Beam
- the internal resistance has an AC impedance value of 100 m ⁇ or less, preferably less than 90 m ⁇ , at a frequency of 1 kHz.
- a secondary battery having such an internal resistance can be said to be highly safe.
- the charge capacity of the secondary battery is preferably 2000 mAh or more, preferably 2400 mAh or more. Since the secondary battery in the fully charged state may discharge over time, it is preferable to perform AC impedance measurement within 24 hours, preferably within 12 hours, and more preferably within 6 hours after the secondary battery is fully charged.
- the temperature rise of the secondary battery i.e., the difference between the temperature before the nail penetration test and the maximum temperature reached after the nail penetration (also referred to as the temperature rise ⁇ T)
- the temperature is the temperature within 5 cm, preferably within 2 cm, of the nail hole, and specifically, the value output by a temperature sensor placed within 5 cm, preferably within 2 cm, of the nail hole.
- the temperature sensor is provided so as to be in contact with the exterior body of the secondary battery, this temperature is equal to the temperature of the exterior body.
- the temperature of the nail 1003 is preferably 100°C or less, more preferably 80°C or less, and even more preferably 60°C or less. This is the value output by a temperature sensor placed on the nail 1003.
- the maximum temperature of the secondary battery during the nail penetration test is preferably 150°C or less, more preferably 100°C or less, and even more preferably 80°C or less. More preferably, it is lower than the temperature at which oxidation of the electrolyte occurs due to the positive electrode. Even more preferably, the maximum temperature is lower than the flash point of the mixed organic solvent used in the electrolyte. If the flash point of the mixed organic solvent is unknown, the flash point of each organic solvent can be used as a reference.
- the amount of the positive electrode active material carried in the positive electrode of the secondary battery is preferably 15 mg/cm 2 or more and 25 mg/cm 2 or less, more preferably 18 mg/cm 2 or more and 23 mg/cm 2 or less, and more preferably 20 mg/cm 2 or more and 22 mg/cm 2 or less. With such a carrying amount, a highly safe secondary battery can be provided.
- the positive and negative electrode capacity ratio In a secondary battery, it is preferable to set the positive and negative electrode capacity ratio to 75% or more and 100% or less, and preferably 80% or more and 90% or less. With such a positive and negative electrode capacity ratio, a highly safe secondary battery can be provided.
- the positive and negative electrode capacity ratio will be described in detail in the examples.
- the positive electrode active material particles in the secondary battery have very few cracks.
- the number of observable cracks per positive electrode active material particle is preferably 0 to 5.
- the pressure of the press is, for example, a linear pressure of 500 kN/m or less, preferably a linear pressure of 300 kN/m or less, and more preferably a linear pressure of 250 kN/m or less.
- the thickness of the positive electrode active material layer should be 70 ⁇ m or more and 80 ⁇ m or less, preferably 75 ⁇ m or more and 78 ⁇ m or less.
- ⁇ Crystal structure of positive electrode active material The crystal structure of the positive electrode active material 100 according to one embodiment of the present invention will be described in comparison with that of a conventional positive electrode active material.
- FIG. 8 shows the crystal structure of the positive electrode active material 100 of one embodiment of the present invention.
- the positive electrode active material 100 of one embodiment of the present invention preferably has a layered rock salt type crystal structure belonging to the space group R-3m.
- a layered rock salt type composite oxide has a high discharge capacity, has a two-dimensional lithium ion diffusion path, is suitable for the insertion and desorption of lithium ions, and is excellent as a positive electrode active material for a secondary battery.
- the inner portion 100b which occupies most of the volume of the positive electrode active material 100, has a layered rock salt type crystal structure.
- FIG. 8 shows the layered rock salt type crystal structure with R-3m O3.
- O3 is added under the space group, but in this crystal structure, lithium occupies an octahedral site, and three MO2 layers exist in the unit cell, so this crystal structure is sometimes called an O3 type crystal structure.
- the MO2 layer refers to a structure in which an octahedral structure in which oxygen is coordinated to a transition metal M six times is continuous on a plane in an edge-sharing state. This may also be called a layer consisting of an octahedron of transition metal M and oxygen.
- lithium ions are shown to exist at all lithium sites, but as described above, ions of an added element, such as magnesium ions, may be located at the lithium sites.
- the surface layer 100a of the positive electrode active material 100 preferably has a function of reinforcing the layered structure of the transition metal M and the oxygen octahedron in the inner portion 100b so that it is not broken even if lithium is removed from the positive electrode active material 100 by charging.
- the surface layer 100a functions as a barrier film for the positive electrode active material 100.
- the surface layer 100a which is the outer periphery of the positive electrode active material 100, reinforces the positive electrode active material 100.
- the reinforcement here means suppressing structural changes in the surface layer 100a and the inner portion 100b of the positive electrode active material 100, such as oxygen elimination and/or shifting of the layered structure of the transition metal M and the oxygen octahedron, and/or suppressing decomposition of the organic electrolyte solution or the like on the surface of the positive electrode active material 100.
- the surface layer portion 100a has a different crystal structure from the inner portion 100b. It is also preferable that the surface layer portion 100a has a composition and crystal structure that are more stable at room temperature (25°C) than the inner portion 100b. For example, it is preferable that at least a part of the surface layer portion 100a of the positive electrode active material 100 of one embodiment of the present invention has a rock salt type crystal structure. Alternatively, it is preferable that the surface layer portion 100a has both a layered rock salt type crystal structure and a rock salt type crystal structure. Alternatively, it is preferable that the surface layer portion 100a has the characteristics of both a layered rock salt type crystal structure and a rock salt type crystal structure.
- the surface layer 100a is the region where lithium ions are first desorbed during charging, and is the region where the lithium concentration is likely to be lower than that of the inside 100b.
- the atoms on the surface of the particles of the positive electrode active material 100 that the surface layer 100a has can be said to be in a state where some bonds are broken. Therefore, the surface layer 100a is likely to become unstable, and can be said to be a region where the deterioration of the crystal structure is likely to begin.
- the crystal structure of the layered structure consisting of the transition metal M and oxygen octahedrons in the surface layer 100a is shifted, the influence is linked to the inside 100b, and the crystal structure of the layered structure is also shifted in the inside 100b, which is thought to lead to the deterioration of the crystal structure of the entire positive electrode active material 100.
- the surface layer 100a can be sufficiently stabilized, even when x in Li x CoO 2 is small, for example, even if x is 0.24 or less, the layered structure consisting of the transition metal M and oxygen octahedrons in the inside 100b can be made less likely to be broken. Furthermore, it is possible to suppress the misalignment of the layer consisting of the transition metal M and oxygen octahedrons in the inner portion 100b.
- the interior 100b of the positive electrode active material 100 preferably has a low density of defects such as dislocations.
- the positive electrode active material 100 preferably has a large crystallite size measured by XRD.
- the interior 100b preferably has high crystallinity.
- the surface of the positive electrode active material 100 preferably is smooth.
- Dislocations in the interior 100b can be observed, for example, with a TEM. If the density of defects such as dislocations is sufficiently low, defects such as dislocations may not be observed in a specific 1 ⁇ m square area of the observation sample. Note that dislocations are a type of crystal defect and are different from point defects.
- the crystallite size measured by XRD is preferably, for example, 300 nm or more. As the crystallite size is larger, it is easier to maintain the O3' type crystal structure when x in Li x CoO 2 is small, as described below, and shortening of the c-axis length is more easily suppressed.
- the XRD diffraction pattern for calculating the crystallite size is preferably obtained from only the positive electrode active material, but may also be obtained from the positive electrode containing the current collector, binder, conductive material, etc. in addition to the positive electrode active material.
- the particles of the positive electrode active material may be oriented so that the crystal planes of the particles of the positive electrode active material are aligned in one direction due to the influence of pressure and the like in the manufacturing process. If the orientation is too strong, the crystallite size may not be calculated accurately, so it is more preferable to obtain the XRD diffraction pattern by removing the positive electrode active material layer from the positive electrode, removing the binder, etc.
- Another method is to apply grease to a silicon non-reflective plate and attach a powder sample of the positive electrode active material, etc. to the silicon non-reflective plate.
- Crystallite size can be calculated, for example, using the Scherrer formula below.
- the crystallite size can be calculated using, for example, Bruker D8 ADVANCE, Cuka ray as X-ray, 2 ⁇ is 15° to 90°, increment 0.005, LYNXEYE XE-T as detector, and the diffraction pattern obtained using ICSD coll. code. 172909 as the literature value of lithium cobalt oxide. Analysis can be performed using DIFFRAC. TOPAS ver. 6 as crystal structure analysis software, and can be set, for example, as follows. Emission Profile: CuKa5.
- LVol-IB which is the crystallite size calculated by the above method, as the crystallite size. Note that if the calculated Preferred Orientation is less than 0.8, the orientation of the sample may be too strong and the sample may not be suitable for determining the crystallite size.
- the surface layer portion 100a has an additive element, and more preferably has a plurality of additive elements. It is also preferable that the surface layer portion 100a has a higher concentration of one or more selected from the additive elements than the inside 100b. It is also preferable that one or more selected from the additive elements possessed by the positive electrode active material 100 have a concentration gradient. It is also more preferable that the distribution of the positive electrode active material 100 differs depending on the additive element.
- the depth from the surface of the peak of the detection amount differs depending on the additive element.
- the peak of the detection amount here is the maximum value of the detection amount
- the peak of the detection amount in the surface layer portion 100a is the maximum value of the detection amount in the surface layer portion 100a.
- the detection amount may be called, for example, the intensity when the vertical axis in EDX-ray analysis is count, or the concentration when the vertical axis is atomic%.
- the amount of magnesium detected in the surface layer 100a is greater than the amount detected in the interior 100b. Furthermore, it is preferable that the peak of the amount of magnesium detected in the surface layer 100a is in a region closer to the surface.
- the amount of fluorine detected in the surface layer 100a is greater than the amount detected in the interior 100b. It is also preferable that the peak of the amount of fluorine detected in the surface layer 100a is in a region closer to the surface.
- the amount of nickel detected in the surface layer 100a is greater than the amount detected in the interior 100b. Furthermore, it is preferable that the surface layer 100a has a peak of the amount of nickel detected in a region closer to the surface. For example, in the surface layer 100a, it is preferable that the amount of nickel detected in the shell is greater than the amount of nickel detected in the region inside the shell, and that the shell has a peak of the amount of nickel detected.
- the ratio (Ni/Co) of the number of nickel Ni atoms in the shell of the surface layer 100a to the number of cobalt Co atoms is less than 1.
- the number of nickel Ni atoms in the shell of the surface layer 100a is less than the number of cobalt Co atoms. Furthermore, the ratio (Ni/Co) of the number of nickel Ni atoms to the number of cobalt Co atoms at the peak of the amount of nickel detected is less than 1. Furthermore, the ratio (Ni/Co) of the number of nickel (Ni) atoms to the number of cobalt (Co) atoms in the region inside the shell is smaller than the ratio (Ni/Co) of the number of nickel (Ni) atoms to the number of cobalt (Co) atoms in the shell. Note that the amount of nickel detected in the interior 100b may be very small compared to the surface layer 100a.
- the number of nickel (Ni) atoms is smaller than the number of cobalt (Co) atoms in both the surface layer 100a and the interior 100b. Furthermore, the number of nickel (Ni) atoms in the positive electrode active material 100 is smaller than the number of cobalt (Co) atoms.
- overlapping distributions includes the peaks of the detected amounts of magnesium and nickel being the same or the difference between the peaks being 3 nm or less, and the peaks do not have to overlap as a whole.
- the peak of the detected amount of magnesium may be closer to the surface, or the peak of the detected amount of nickel may be closer to the surface.
- the amount of titanium detected in the surface layer 100a is greater than the amount detected in the interior 100b. It is also preferable that the peak of the amount of titanium detected in the surface layer 100a is in a region closer to the surface.
- the detected amount of silicon, phosphorus, boron and/or calcium in the surface layer 100a is greater than the detected amount in the interior 100b. It is also preferable that the detected amount of silicon, phosphorus, boron and/or calcium in the surface layer 100a has a peak in a region closer to the surface.
- aluminum preferably has a peak of detection amount further in than magnesium.
- the distributions of magnesium and aluminum may overlap, or there may be little overlap between the distributions of magnesium and aluminum.
- overlapping distributions includes the detection amount peaks of magnesium and aluminum being the same or the difference between the peaks being 3 nm or less, and the peaks may not overlap as a whole.
- the detection amount peak of aluminum may be present in the surface layer 100a, or may be deeper than the surface layer 100a. If the detection amount peak of aluminum is deeper than the surface layer 100a, there may be little overlap between the distributions of magnesium and aluminum.
- aluminum preferably has a peak in a region of 5 nm to 30 nm from the surface toward the inside. The reason that aluminum is distributed deeper into the inside than magnesium is thought to be because aluminum is more easily diffused than magnesium.
- the distribution of aluminum may not be a normal distribution.
- the length of the tail may be different between the surface side and the inner side.
- the peak width at 1/5 the height (1/5 Max Al ) of the maximum height of the detected amount of aluminum (Max Al ) is divided in half by a perpendicular line drawn from the maximum value to the horizontal axis, the peak width (W c ) on the inner side may be larger than the peak width (W s ) on the surface side.
- the amount of aluminum detected in the area closest to the surface is small, which is presumably because aluminum is more stable in areas where magnesium and the like are dissolved in solid solution at high concentrations than in areas where they are not. More specifically, in areas of the layered rock salt type of space group R-3m or the rock salt type of cubic crystal system, in areas where magnesium is dissolved in solid solution at high concentrations, the distance between the cation and oxygen is longer than in the layered rock salt type LiAlO 2 , making it difficult for aluminum to exist stably. In addition, the valence change caused by Li + being replaced by Mg 2+ around cobalt can be compensated for by changing from Co 3+ to Co 2+ , thereby achieving cation balance. However, since Al can only take a trivalent value, it is thought that it is difficult for it to coexist with magnesium in the rock salt type or layered rock salt type structure.
- manganese like aluminum, has a peak of detectable amount inside magnesium.
- the additive elements do not necessarily have to be distributed in the same manner throughout the entire surface layer 100a of the positive electrode active material 100.
- the region of the positive electrode active material 100 having a (001) oriented surface may have a different distribution of the additive element compared to the region having a surface other than the (001) orientation.
- the (001) oriented surface and its surface layer 100a may have a lower detection amount of one or more selected from the additive elements compared to the surface other than the (001) orientation.
- the detection amount of nickel may be low.
- the region having a (001) oriented surface may have one or more peaks selected from the additive elements located shallower from the surface compared to the region having a surface other than the (001) orientation. Specifically, the peaks of magnesium and aluminum may be located shallower from the surface. The region having the above-mentioned surface is included in the surface layer 100a.
- the main diffusion path of lithium ions during charging and discharging is not exposed on the (001) plane.
- the diffusion path of lithium ions is exposed on surfaces other than the (001) orientation. Therefore, the surface other than the (001) orientation and its surface layer 100a are important regions for maintaining the diffusion path of lithium ions, and at the same time, they are prone to become unstable because they are the regions from which lithium ions are first desorbed. Therefore, in order to maintain the crystal structure of the entire positive electrode active material 100, it is preferable to reinforce at least the surface other than the (001) orientation and its surface layer 100a.
- the concentration distribution of the added element on the surface other than the (001) orientation and on the surface layer 100a attention may be paid to the concentration distribution of the added element on the surface other than the (001) orientation and on the surface layer 100a.
- the added elements it is preferable that magnesium is detected on the surface other than the (001) orientation and on the surface layer 100a.
- the concentration of the added element may be low or absent as described above. Nickel may not be present on the (001) oriented surface and on the surface layer 100a.
- the half-width of the magnesium distribution in the (001) oriented surface and its surface layer 100a is preferably 10 nm to 200 nm, more preferably 50 nm to 150 nm, and even more preferably 80 nm to 120 nm.
- the half-width of the magnesium distribution in the non-(001) oriented surface and its surface layer 100a is preferably 200 nm to 500 nm, more preferably 200 nm to 300 nm, and even more preferably 230 nm to 270 nm.
- the half-width of the magnesium distribution may be wider in the non-(001) oriented surface and its surface layer 100a than in the (001) oriented surface and its surface layer 100a.
- the half-width of the nickel distribution in the surface that is not (001) oriented and in the surface layer 100a thereof is preferably 30 nm or more and 150 nm or less, more preferably 50 nm or more and 130 nm or less, and even more preferably 70 nm or more and 110 nm or less.
- the additive elements in which the additive elements are mixed and then heated, the additive elements may spread through the diffusion path of lithium ions. Therefore, in order to make the distribution of the additive elements in the surface other than the (001) orientation and in the surface layer 100a thereof within a preferred range, it is better to mix the additive elements and heat them after manufacturing lithium cobalt oxide with high purity or lithium cobalt oxide with a smooth surface.
- Magnesium is divalent, and since the additive element aluminum or nickel is stably present at the cobalt site in the layered rock salt type crystal structure, magnesium ions are likely to be present at the lithium site rather than the cobalt site, that is, to enter the lithium site.
- the presence of magnesium at an appropriate concentration at the lithium site of the surface layer portion 100a makes it easier to maintain the layered rock salt type crystal structure. This is presumed to be because the magnesium present at the lithium site functions as a pillar supporting the CoO 2 layers.
- the presence of magnesium can suppress the detachment of oxygen around magnesium when x in Li x CoO 2 is, for example, 0.24 or less, and can suppress the thermal decomposition reaction.
- the presence of magnesium can be expected to increase the density of the positive electrode active material 100. Furthermore, if the magnesium concentration of the surface layer portion 100a is high, it can be expected that the corrosion resistance to hydrofluoric acid generated by the decomposition of the organic electrolyte solution or the like will be improved.
- magnesium does not adversely affect the insertion and desorption of lithium during charging and discharging, and the above benefits can be enjoyed. However, if there is an excess of magnesium, it may have an adverse effect on the insertion and desorption of lithium. Furthermore, the effect of stabilizing the crystal structure may be reduced. This is thought to be because magnesium enters the cobalt site in addition to the lithium site. In addition, unnecessary magnesium compounds (oxides, fluorides, etc.) that do not substitute for either the lithium site or the cobalt site may segregate on the surface of the positive electrode active material, and may become resistance components of the secondary battery. In addition, as the magnesium concentration of the positive electrode active material increases, the discharge capacity of the positive electrode active material may decrease. This is thought to be because too much magnesium enters the lithium site, reducing the amount of lithium that contributes to charging and discharging.
- the amount of magnesium contained in the entire positive electrode active material 100 is appropriate.
- the number of magnesium atoms is preferably 0.002 to 0.06 times the number of cobalt atoms, more preferably 0.005 to 0.03 times, and even more preferably about 0.01 times.
- the amount of magnesium contained in the entire positive electrode active material 100 may be a value obtained by performing an elemental analysis of the entire positive electrode active material 100 using, for example, GD-MS (glow discharge mass spectrometry) or ICP-MS (inductively coupled plasma mass spectrometry), or may be based on the value of the composition of raw materials in the process of producing the positive electrode active material 100.
- Fluorine is a monovalent anion, and when fluorine is adsorbed on the surface of the positive electrode active material 100, the energy of lithium desorption from the positive electrode active material 100 is reduced. As long as the energy is reduced, fluorine may be substituted for a part of the oxygen in the surface layer portion 100a. This is because the redox potential of the cobalt ion accompanying lithium desorption differs depending on the presence or absence of fluorine. In other words, when there is no fluorine, the cobalt ion changes from trivalent to tetravalent with lithium desorption. On the other hand, when there is fluorine, the cobalt ion changes from divalent to trivalent with lithium desorption.
- the redox potential of the cobalt ion is different between the two. Therefore, the desorption and insertion of lithium ions near the fluorine is likely to occur smoothly, and it is preferable to have fluorine on the surface or surface layer portion of the positive electrode active material 100.
- the positive electrode active material 100 having fluorine is used in a secondary battery, the charge/discharge characteristics, large current characteristics, etc. can be improved.
- the presence of fluorine on the surface or surface layer that is in contact with the electrolyte, or the adsorption or attachment of fluoride to the surface can suppress excessive reaction between the positive electrode active material 100 and the electrolyte, and can effectively improve corrosion resistance to hydrofluoric acid.
- Nickel can exist on either the cobalt site or the lithium site. When nickel exists on the cobalt site, it has a lower redox potential than cobalt, so it can be said that it is easier to release lithium during charging, for example. This is expected to result in faster charging and discharging speeds.
- the shift of the layered structure consisting of octahedra of cobalt and oxygen can be suppressed.
- the change in volume accompanying charging and discharging is suppressed.
- the elastic modulus increases, that is, the material becomes hard. This is presumably because nickel present at the lithium site also functions as a pillar supporting the CoO 2 layers. Therefore, it is expected that the crystal structure will be more stable, particularly in a charged state at high temperatures, for example, 45°C or higher, which is preferable.
- Nickel can also exist on either the cobalt site or the lithium site. When nickel exists on the cobalt site, it has a lower redox potential than cobalt, so it can be said that it is easier to release lithium during discharging, for example. This is expected to result in faster charging and discharging speeds, i.e. improved charge and discharge rate characteristics.
- NiO nickel oxide
- the order of ionization tendency is lowest for magnesium, aluminum, cobalt, and nickel (Mg>Al>Co>Ni). Therefore, nickel is thought to be less likely to dissolve into the electrolyte during charging than the other elements listed above. Therefore, it is thought to be highly effective in stabilizing the crystal structure of the surface layer when in a charged state.
- Ni2 + is the most stable, and nickel has a higher trivalent ionization energy than cobalt. Therefore, it is known that nickel and oxygen alone do not form a spinel crystal structure. Therefore, nickel is thought to have the effect of suppressing the phase change from the layered rock salt type to the spinel type crystal structure.
- an excess of nickel is undesirable because it increases the influence of distortion due to the Jahn-Teller effect. Also, an excess of nickel may adversely affect the insertion and extraction of lithium.
- the amount of nickel contained in the entire positive electrode active material 100 is appropriate.
- the number of nickel atoms contained in the positive electrode active material 100 is less than the number of cobalt atoms, and is preferably more than 0% and less than 7.5% of the number of cobalt atoms, preferably 0.05% to 4%, preferably 0.1% to 2%, and more preferably 0.2% to 1%.
- more than 0% and less than 4% is preferable.
- more than 0% and less than 2% is preferable.
- more than 0.05% to 7.5% is preferable.
- more than 0.05% to 2% is preferable.
- Or more than 0.1% to 7.5% is preferable.
- Or more than 0.1% to 4% is preferable.
- the amount of nickel shown here may be, for example, a value obtained by performing elemental analysis of the entire positive electrode active material using GD-MS, ICP-MS, or the like, or may be based on the value of the raw material composition in the process of producing the positive electrode active material.
- Aluminum may also be present at the cobalt site in the layered rock salt crystal structure. Since aluminum is a typical trivalent element and its valence does not change, lithium around the aluminum is unlikely to move during charging and discharging. Therefore, aluminum and its surrounding lithium function as columns, and can suppress changes in the crystal structure. Therefore, as described below, even if the positive electrode active material 100 is subjected to a force that causes it to expand and contract in the c-axis direction due to the insertion and desorption of lithium ions, that is, even if a force that causes it to expand and contract in the c-axis direction is applied by changing the charging depth or charging rate, deterioration of the positive electrode active material 100 can be suppressed.
- Aluminum also has the effect of suppressing the dissolution of surrounding cobalt and improving continuous charging resistance. Furthermore, since the Al-O bond is stronger than the Co-O bond, it can suppress the desorption of oxygen from around the aluminum. These effects improve thermal stability. Therefore, by having aluminum as an added element, it is possible to improve safety when using the positive electrode active material 100 in a secondary battery. Furthermore, it is possible to obtain a positive electrode active material 100 whose crystal structure is less likely to collapse even when repeatedly charged and discharged.
- the amount of aluminum contained in the entire positive electrode active material 100 is appropriate.
- the number of aluminum atoms contained in the entire positive electrode active material 100 is preferably 0.05% to 4% of the number of cobalt atoms, preferably 0.1% to 2% and more preferably 0.3% to 1.5%.
- 0.05% to 2% is preferable.
- 0.1% to 4% is preferable.
- the amount contained in the entire positive electrode active material 100 here may be, for example, a value obtained by performing elemental analysis of the entire positive electrode active material 100 using GD-MS, ICP-MS, etc., or may be based on the value of the composition of raw materials in the process of producing the positive electrode active material 100.
- Titanium oxide is also known to have superhydrophilic properties. Therefore, by making the surface layer 100a of the positive electrode active material 100 have titanium oxide, it is possible that the positive electrode active material 100 has good wettability with highly polar solvents. When used as a secondary battery, the positive electrode active material 100 has good contact at the interface with the highly polar electrolyte, which may prevent an increase in internal resistance.
- phosphorus is present in the surface layer 100a, it is preferable because short circuits can be suppressed when x in Li x CoO 2 is kept small.
- phosphorus it is preferable for phosphorus to be present in the surface layer 100a as a compound containing phosphorus and oxygen.
- the hydrogen fluoride generated by the decomposition of the electrolyte or lithium salt reacts with the phosphorus, which may reduce the hydrogen fluoride concentration in the electrolyte, which is preferable.
- hydrogen fluoride When the lithium salt contains LiPF 6 , hydrogen fluoride may be generated by hydrolysis. In addition, hydrogen fluoride may be generated by the reaction of polyvinylidene fluoride (PVDF), which is used as a component of the positive electrode, with an alkali.
- PVDF polyvinylidene fluoride
- a decrease in adhesion due to gelation and/or insolubilization of PVDF may be suppressed.
- the positive electrode active material 100 When the positive electrode active material 100 has phosphorus together with magnesium, the stability of the crystal structure in the state where x in Li x CoO 2 is small becomes extremely high, which is preferable.
- the number of phosphorus atoms is preferably 1% to 20% of the number of cobalt atoms, more preferably 2% to 10%, and even more preferably 3% to 8%. Or 1% to 10% is preferable. Or 1% to 8% is preferable. Or 2% to 20% is preferable. Or 2% to 8% is preferable. Or 3% to 20% is preferable. Or 3% to 10% is preferable.
- the number of magnesium atoms is preferably 0.1% to 10% of the number of cobalt atoms, more preferably 0.5% to 5%, and more preferably 0.7% to 4%. Or 0.1% to 5% is preferable. Or 0.1% to 4% is preferable. Or 0.5% to 10% is preferable. Alternatively, 0.5% or more and 4% or less is preferable. Alternatively, 0.7% or more and 10% or less is preferable. Alternatively, 0.7% or more and 5% or less is preferable.
- the phosphorus and magnesium concentrations shown here may be values obtained by performing elemental analysis of the entire positive electrode active material 100 using, for example, GD-MS, ICP-MS, or the like, or may be based on values of the composition of raw materials in the process of producing the positive electrode active material 100.
- the progression of the crack can be suppressed by the presence of phosphorus, or more specifically, a compound containing phosphorus and oxygen, in the embedded portion that contacts the portion of the surface.
- the divalent nickel may be more stable near the divalent magnesium. Therefore, even when x in Li x CoO 2 is small, the elution of magnesium may be suppressed. This may contribute to the stabilization of the surface layer 100a.
- magnesium is added in a step before nickel.
- magnesium and nickel are added in the same step.
- Magnesium has a large ionic radius and tends to remain in the surface layer of lithium cobalt oxide regardless of the step in which it is added, whereas nickel can diffuse widely inside the lithium cobalt oxide if magnesium is not present. Therefore, if nickel is added before magnesium, there is a concern that nickel will diffuse into the lithium cobalt oxide and not remain in the surface layer in the desired amount.
- the positive electrode active material 100 has additive elements with different distributions, it is preferable because it can stabilize the crystal structure in a wider region.
- the positive electrode active material 100 has magnesium, nickel, and aluminum, it can stabilize the crystal structure in a wider region than if it had only one of these elements.
- aluminum is not essential on the surface because the stabilization of the surface can be sufficiently achieved by magnesium, nickel, etc. Rather, it is preferable for aluminum to be distributed widely in a deeper region.
- it is preferable that aluminum is continuously detected in a region from the surface to a depth of 1 nm to 25 nm. It is preferable to distribute aluminum widely in a region from 0 nm to 100 nm from the surface, preferably from 0.5 nm to 50 nm from the surface, because it can stabilize the crystal structure in a wider region.
- each additive element When multiple additive elements are included as described above, the effects of each additive element are synergistic, which can contribute to further stabilization of the surface layer 100a.
- the effect of achieving a stable composition and crystal structure is high and is therefore preferable.
- the surface layer 100a is occupied only by a compound of the added element and oxygen, it is not preferable because it makes it difficult to insert and remove lithium.
- the surface layer 100a it is not preferable for the surface layer 100a to be occupied only by MgO, a structure in which MgO and NiO(II) are solid-solved, and/or a structure in which MgO and CoO(II) are solid-solved. Therefore, the surface layer 100a must contain at least cobalt, and in the discharged state, it must also contain lithium, and must have a path for the insertion and removal of lithium.
- the surface layer 100a has a higher concentration of cobalt than magnesium.
- the ratio Mg/Co of the number of magnesium atoms Mg to the number of cobalt atoms Co is preferably 0.62 or less. It is also preferable that the surface layer 100a has a higher concentration of cobalt than nickel. It is also preferable that the surface layer 100a has a higher concentration of cobalt than aluminum. It is also preferable that the surface layer 100a has a higher concentration of cobalt than fluorine.
- the surface layer 100a has a higher concentration of magnesium than nickel.
- the number of nickel atoms is preferably 1/6 or less of the number of magnesium atoms.
- some of the added elements particularly magnesium, nickel, and aluminum, have a higher concentration in the surface layer 100a than in the interior 100b, but it is also preferable that they are present randomly and dilutely in the interior 100b.
- magnesium and aluminum are present at appropriate concentrations in the lithium sites of the interior 100b, it has the effect of making it easier to maintain the layered rock-salt crystal structure, as described above.
- nickel is present at an appropriate concentration in the interior 100b, it is possible to suppress the shifting of the layered structure consisting of transition metal M and oxygen octahedra, as described above.
- magnesium and nickel are present together, a synergistic effect of suppressing the elution of magnesium can be expected, as described above.
- the positive electrode active material 100 of one embodiment of the present invention has a crystal structure in a state where x in Li x MO 2 is small, that is, in a charged state at a high voltage, which is different from that of conventional positive electrode active materials due to the distribution and/or crystal structure of the added elements as described above.
- small x means 0.1 ⁇ x ⁇ 0.24.
- high voltage in a charged state means 4.5V or more, preferably 4.6V or more, and more preferably 4.8V or more.
- a change in crystal structure with a change in x in Li x MO 2 will be described with reference to FIGS. 8 and 9 while comparing a conventional positive electrode active material with the positive electrode active material 100 of one embodiment of the present invention.
- the change in the crystal structure of a conventional positive electrode active material is shown in Fig. 9.
- the conventional positive electrode active material shown in Fig. 9 is lithium cobalt oxide ( LiCoO2 ) that does not have any added elements.
- the change in the crystal structure of lithium cobalt oxide that does not have any added elements is described in Non-Patent Documents 1 to 3, etc.
- conventional lithium cobalt oxide has a crystal structure that is highly symmetrical to lithium when x is about 0.5, and belongs to the monoclinic space group P2/m.
- This structure has one CoO2 layer in the unit cell. Therefore, it is sometimes called O1 type or monoclinic (denoted as monoclinic in the figure) O1 type.
- the positive electrode active material has a crystal structure of the trigonal space group P-3m1, and one CoO2 layer is present in the unit cell. Therefore, this crystal structure may be called O1 type or trigonal (denoted as trigonal in the figure) O1 type.
- the trigonal crystal may be converted to a composite hexagonal lattice and called a hexagonal O1 type.
- conventional lithium cobalt oxide has a crystal structure of space group R-3m.
- This structure can be said to be a structure in which a CoO 2 structure such as trigonal O1 type and a LiCoO 2 structure such as R-3m O3 are alternately stacked. Therefore, this crystal structure may be called an H1-3 type crystal structure (H1-3 in the figure).
- H1-3 type crystal structure H1-3 in the figure.
- the number of cobalt atoms per unit cell in the H1-3 type crystal structure is twice that of other structures.
- the coordinates of cobalt and oxygen in the unit cell can be expressed as Co (0,0,0.42150 ⁇ 0.00016), O1 (0,0,0.27671 ⁇ 0.00045), and O2 (0,0,0.11535 ⁇ 0.00045).
- O1 and O2 are oxygen atoms.
- Which unit cell should be used to express the crystal structure of the positive electrode active material can be determined, for example, by Rietveld analysis of the XRD pattern. For example, it is preferable to adopt a unit cell with a GOF (goodness of fit) value close to 1.
- the H1-3 type crystal structure has a structure in which two CoO layers are continuous, such as the trigonal O1 type, and is therefore highly likely to be unstable.
- the crystal structure of conventional lithium cobalt oxide breaks down.
- the breakdown of the crystal structure causes a deterioration in cycle characteristics. This is because the breakdown of the crystal structure reduces the number of sites where lithium can exist stably and makes it difficult to insert and remove lithium.
- the change in the crystal structure in the discharge state where x in Li x MO 2 is 1 and the state where x is 0.24 or less is smaller than that of the conventional positive electrode active material. More specifically, the deviation of the MO 2 layer in the state where x is 1 and the state where x is 0.24 or less can be reduced.
- the volume change of the positive electrode active material 100 is smaller than the volume change of the conventional positive electrode active material. Therefore, the positive electrode active material 100 of one embodiment of the present invention is less likely to collapse in crystal structure even when charging and discharging such that x is 0.24 or less are repeated, the site where lithium can exist stably is maintained, and excellent cycle characteristics can be realized.
- the positive electrode active material 100 of one embodiment of the present invention can have a more stable crystal structure than conventional positive electrode active materials when x in Li x MO 2 is 0.24 or less. Therefore, even when the positive electrode active material 100 of one embodiment of the present invention maintains a state in which x in Li x MO 2 is 0.24 or less, oxygen is less likely to be released and a thermal decomposition reaction can be suppressed. In other words, a secondary battery using the positive electrode active material 100 of one embodiment of the present invention is preferable because it has improved safety.
- FIG. 8 shows the crystal structure of the interior 100b of the positive electrode active material 100 when x in Li x MO2 is 1, approximately 0.2, and approximately 0.15.
- the interior 100b occupies most of the volume of the positive electrode active material 100 and is the part that contributes greatly to charge and discharge, so it can be said that the shift of the MO2 layer and the change in volume are the most problematic parts.
- the positive electrode active material 100 has the same R-3m O3 crystal structure as conventional lithium cobalt oxide.
- x is 0.24 or less, for example, about 0.2 or about 0.15, at which point conventional lithium cobalt oxide has an H1-3 type crystal structure, the positive electrode active material 100 has a crystal with a different structure.
- the symmetry of the MO2 layer is the same as that of O3. Therefore, this crystal structure is called an O3' type crystal structure.
- a pattern similar to a spinel structure may appear in the XRD pattern, and this crystal structure may be called a pseudo-spinel structure. This crystal structure is shown in FIG. 8 with R-3m O3'.
- the positive electrode active material 100 that has passed through the O3' type crystal structure can have the effect of suppressing oxygen release even if it becomes, for example, a H1-3 type crystal structure, so it is estimated that ignition is suppressed even if a nail penetration test is performed on a lithium ion secondary battery using the positive electrode active material 100.
- the coordinates of cobalt and oxygen in the unit cell can be expressed in the range of Co(0,0,0.5), O(0,0,x), 0.20 ⁇ x ⁇ 0.25.
- this crystal structure is called a monoclinic O1(15) type crystal structure. This crystal structure is shown in FIG. 9 with P2/m monoclinic O1(15).
- the coordinates of cobalt and oxygen in the unit cell are: Co1(0.5,0,0.5), Co2 (0, 0.5, 0.5), O1 (X O1 , 0, Z O1 ), 0.23 ⁇ X O1 ⁇ 0.24, 0.61 ⁇ Z O1 ⁇ 0.65, O2( XO2,0.5 , ZO2 ),
- this crystal structure can show the lattice constant even in the space group R-3m if a certain degree of error is allowed in the Rietveld analysis, etc.
- the coordinates of cobalt and oxygen in the unit cell are as follows: Co(0,0,0.5), O(0,0,Z O ), The range of Z O can be expressed as 0.21 ⁇ Z O ⁇ 0.23.
- the difference in volume per the same number of cobalt atoms between R-3m O3 in a discharged state and the O3' type crystal structure is 2.5% or less, more specifically 2.2% or less, typically 1.8%.
- the difference in volume per the same number of cobalt atoms between R-3m O3 in a discharged state and the monoclinic O1(15) crystal structure is 3.3% or less, more specifically 3.0% or less, typically 2.5%.
- the table below shows the difference in volume per cobalt atom between R-3m O3 in a discharged state, O3', monoclinic O1(15), H1-3 type, and trigonal O1.
- ICSD coll. code. 172909 and 88721 For the lattice constants of the crystal structures of R-3m O3 in a discharged state and trigonal O1 used in the calculation of the table below, refer to ICSD coll. code. 172909 and 88721.
- H1-3 refer to Non-Patent Document 3.
- O3' and monoclinic O1(15) calculations can be made from experimental values of XRD.
- the positive electrode active material 100 of one embodiment of the present invention when x in Li x CoO 2 is small, that is, when a large amount of lithium is released, the change in the crystal structure is suppressed more than in the conventional positive electrode active material.
- the volume change of the positive electrode active material 100 is smaller than the volume change of the conventional positive electrode active material. Therefore, the crystal structure of the positive electrode active material 100 is not easily broken even when charging and discharging are repeated so that x is 0.24 or less. Therefore, the positive electrode active material 100 suppresses a decrease in the charge and discharge capacity in the charge and discharge cycle.
- the positive electrode active material 100 since more lithium can be stably used than in the conventional positive electrode active material, the positive electrode active material 100 has a large discharge capacity per weight and per volume. Therefore, by using the positive electrode active material 100, a secondary battery with a high discharge capacity per weight and per volume can be manufactured.
- the positive electrode active material 100 may have an O3' type crystal structure when x in Li x MO 2 is 0.15 or more and 0.24 or less, and it is estimated that the positive electrode active material 100 may have an O3' type crystal structure even when x is more than 0.24 and 0.27 or less. It has also been confirmed that the positive electrode active material 100 may have a monoclinic O1 (15) type crystal structure when x in Li x MO 2 is more than 0.1 and 0.2 or less, typically when x is 0.15 or more and 0.17 or less.
- the crystal structure is not necessarily limited to the above range of x because it is affected not only by x in Li x MO 2 but also by the number of charge/discharge cycles, charge/discharge current, temperature, electrolyte, etc.
- the positive electrode active material 100 may have only O3' type, may have only monoclinic O1(15) type, or may have both crystal structures. Furthermore, all of the particles in the interior 100b of the positive electrode active material 100 do not have to have O3' type and/or monoclinic O1(15) type crystal structures. They may contain other crystal structures, or may be partially amorphous.
- the state in which x in Li x MO 2 is small can be said to be a state in which it is charged at a high charging voltage.
- a charging voltage of 4.6 V or more based on the potential of lithium metal can be said to be a high charging voltage.
- the charging voltage is expressed based on the potential of lithium metal.
- the positive electrode active material 100 of one embodiment of the present invention is preferable because it can maintain a crystal structure with the symmetry of R-3m O3 even when charged at a high charging voltage, for example, a voltage of 4.6 V or more at 25° C.
- a high charging voltage for example, a voltage of 4.6 V or more at 25° C.
- the positive electrode active material 100 of one embodiment of the present invention may be able to adopt the O3' type crystal structure even when the charging voltage is lower, for example, at a charging voltage of 4.5 V or more and less than 4.6 V at 25°C. Similarly, when charged at a voltage of 4.65 V or more and 4.7 V or less at 25°C, the positive electrode active material 100 of one embodiment of the present invention may be able to adopt the monoclinic O1(15) type crystal structure.
- the voltage of the secondary battery is lower than the above by the amount of the graphite potential.
- the potential of graphite is about 0.05 V to 0.2 V based on the potential of lithium metal. Therefore, in the case of a secondary battery using graphite as the negative electrode active material, when the voltage is the above voltage minus the graphite potential, the battery has the same crystal structure as above.
- lithium is shown to exist at all lithium sites with equal probability, but this is not limited thereto.
- Lithium may be present biasedly at some lithium sites, or may have lithium symmetry, such as monoclinic O1( Li0.5CoO2 ) shown in Fig. 9.
- the distribution of lithium can be analyzed, for example, by neutron diffraction.
- the O3' and monoclinic O1(15) type crystal structures have random lithium between the layers, but can be said to be similar to the CdCl2 type crystal structure.
- This CdCl2 type -like crystal structure is close to the crystal structure of lithium nickel oxide when it is charged to Li0.06NiO2 , but it is known that pure lithium cobalt oxide or layered rock salt type positive electrode active materials containing a large amount of cobalt do not usually have the CdCl2 type crystal structure.
- the crystal structure of conventional lithium cobalt oxide and the positive electrode active material 100 changes with the change in the charge depth, i.e., with the change in x in Li x CoO 2.
- the change in the c-axis length of conventional lithium cobalt oxide described in Non-Patent Document 12 is shown in Figure 10.
- the round markers indicate the hexagonal phase
- the diamond markers indicate the monoclinic phase.
- the change in the c-axis length of lithium cobalt oxide corresponds to the change in the angle at which the peak of, for example, the (003) plane of lithium cobalt oxide appears in the XRD pattern. It is known that in XRD using CuK ⁇ 1 radiation (CuK ⁇ radiation excluding CuK ⁇ 2 radiation), the peak of the (003) plane of lithium cobalt oxide appears at 2 ⁇ of approximately 19° to 20°.
- the additive element in the positive electrode active material 100 of one embodiment of the present invention is unevenly distributed in and around the crystal grain boundary 101.
- the vicinity of the crystal grain boundary 101 refers to a region within 10 nm from the crystal grain boundary 101.
- the magnesium concentration at and near the grain boundaries 101 of the positive electrode active material 100 is higher than that in other regions of the interior 100b. It is also preferable that the fluorine concentration at and near the grain boundaries 101 is higher than that in other regions of the interior 100b. It is also preferable that the nickel concentration at and near the grain boundaries 101 is higher than that in other regions of the interior 100b. It is also preferable that the aluminum concentration at and near the grain boundaries 101 is higher than that in other regions of the interior 100b.
- the grain boundary 101 is a type of planar defect. Therefore, like the particle surface, it is prone to become unstable and changes in the crystal structure are likely to begin. Therefore, if the concentration of the added element at and near the grain boundary 101 is high, changes in the crystal structure can be more effectively suppressed.
- the magnesium concentration and fluorine concentration are high at and near the grain boundaries 101, even if cracks occur along the grain boundaries 101 of the positive electrode active material 100 of one embodiment of the present invention, the magnesium concentration and fluorine concentration are high on the surface created by the cracks and in its vicinity. Therefore, even in the positive electrode active material after cracks have occurred, the corrosion resistance to hydrofluoric acid can be improved.
- a certain positive electrode active material is the positive electrode active material 100 of one embodiment of the present invention having an O3′ type and/or monoclinic O1(15) type crystal structure when x in Li x CoO 2 is small can be determined by analyzing a positive electrode having a positive electrode active material with a small x in Li x CoO 2 using XRD, electron beam diffraction, neutron beam diffraction, electron spin resonance (ESR), nuclear magnetic resonance (NMR), or the like.
- XRD is particularly preferred because it can analyze the symmetry of transition metals such as cobalt contained in the positive electrode active material with high resolution, can compare the degree of crystallinity and the orientation of the crystals, can analyze the periodic distortion of the lattice and the crystallite size, and can provide sufficient accuracy even when measuring the positive electrode obtained by disassembling the secondary battery.
- powder XRD can provide diffraction peaks that reflect the crystal structure of the interior 100b of the positive electrode active material 100, which occupies most of the volume of the positive electrode active material 100.
- the measurement When analyzing the crystallite size by powder XRD, it is preferable to perform the measurement without the influence of the orientation of the positive electrode active material particles due to pressure, etc. For example, it is preferable to perform the measurement after removing the positive electrode active material layer from the positive electrode obtained by dismantling the secondary battery. It is also possible to perform the measurement after removing the positive electrode active material from the positive electrode active material layer.
- the positive electrode active material 100 of one embodiment of the present invention is characterized in that there is little change in the crystal structure when x in Li x CoO 2 is 1 and when it is 0.24 or less.
- a material in which 50% or more of the crystal structure exhibits a large change in the crystal structure when charged at a high voltage is not preferable because it cannot withstand repeated charging and discharging at a high voltage.
- the O3' type or monoclinic O1(15) type crystal structure is not obtained by simply adding an additive element.
- lithium cobalt oxide having magnesium and fluorine, or lithium cobalt oxide having magnesium and aluminum is common, depending on the concentration and distribution of the additive element, there are cases where x in Li x CoO 2 is 0.24 or less and the O3' type and/or monoclinic O1(15) type crystal structure is 60% or more, and cases where the H1-3 type crystal structure is 50% or more.
- the positive electrode active material 100 of one embodiment of the present invention Even in the case of the positive electrode active material 100 of one embodiment of the present invention, if x is too small, such as 0.1 or less, or under conditions where the charging voltage exceeds 4.9 V, an H1-3 type or trigonal O1 type crystal structure may be produced. Therefore, to determine whether or not a positive electrode active material 100 of one embodiment of the present invention is present, analysis of the crystal structure, such as XRD, and information such as the charging capacity or charging voltage are required.
- the crystal structure may change when exposed to air.
- the crystal structure may change from O3' type and monoclinic O1(15) type to H1-3 type. Therefore, it is preferable to handle all samples used for crystal structure analysis in an inert atmosphere such as an argon atmosphere.
- Whether the distribution of added elements in a certain positive electrode active material is as described above can be determined by analyzing it using, for example, XPS, EDX, EPMA (electron probe microanalysis), etc.
- the crystal structure of the surface layer 100a, the grain boundaries 101, etc. can be analyzed by electron beam diffraction of a cross section of the positive electrode active material 100.
- Whether or not a certain composite oxide is the positive electrode active material 100 of one embodiment of the present invention can be determined by, for example, preparing a coin cell (CR2032 type, diameter 20 mm, height 3.2 mm) using the composite oxide for the positive electrode and lithium metal for the counter electrode, and charging the coin cell.
- the coin cell has an electrolyte, a separator, a positive electrode can, and a negative electrode can.
- the positive electrode can be prepared by coating a positive electrode current collector made of aluminum foil with a slurry containing the composite oxide as the positive electrode active material, a conductive material, and a binder.
- lithium metal can be used for the counter electrode, but materials other than lithium metal may also be used.
- materials other than lithium metal are used, the potential of the secondary battery and the potential of the positive electrode are different. Unless otherwise specified, the voltage and potential in this specification are the potential of the positive electrode.
- EC ethylene carbonate
- DEC diethyl carbonate
- a 25 ⁇ m thick polypropylene porous film can be used as the separator.
- the positive and negative electrode cans can be made of stainless steel (SUS).
- the coin cell prepared under the above conditions is charged to an arbitrary voltage (for example, 4.5V, 4.55V, 4.6V, 4.65V, 4.7V, 4.75V, or 4.8V).
- the charging method is not particularly limited as long as it can be charged to the arbitrary voltage over a sufficient time.
- the current in CC charging can be 20mA/g or more and 100mA/g or less.
- CV charging can be completed at 2mA/g or more and 10mA/g or less.
- the temperature of 25°C is an example.
- the coin cell is disassembled in a glove box in an argon atmosphere and the positive electrode is taken out, and a positive electrode active material with an arbitrary charging capacity can be obtained.
- XRD measurement can be performed with the positive electrode active material sealed in a sealed container in an argon atmosphere.
- After charging is completed it is preferable to quickly remove the positive electrode and start the XRD analysis described below. Specifically, it is preferable to perform the XRD analysis within 1 hour after charging is completed, and more preferably within 30 minutes.
- the conditions for the multiple charge/discharge cycles may be different from the above-mentioned charging conditions.
- charging can be performed by constant current charging at a current value of 20 mA/g to 100 mA/g up to an arbitrary voltage (e.g., 4.6 V, 4.65 V, 4.7 V, 4.75 V, or 4.8 V), followed by constant voltage charging until the current value is 2 mA/g to 10 mA/g, and discharging at a constant current of 20 mA/g to 100 mA/g up to 2.5 V.
- the discharge conditions for the multiple charge/discharge cycles can be, for example, constant current discharge at 2.5 V and a current value of 20 mA/g or more and 100 mA/g or less.
- XRD> As long as appropriate adjustment and calibration are performed, the apparatus and conditions for XRD measurement are not particularly limited. For example, the measurement can be performed using the following apparatus and conditions.
- XRD device Bruker AXS, D8 ADVANCE X-ray: Cuk ⁇ ray, Cuk ⁇ 1 ray Output: 40 kV, 40 mA Divergence angle: Div.
- the measurement sample is a powder, it can be set up by placing it on a glass sample holder or sprinkling the sample on a greased silicone anti-reflective plate. If the measurement sample is a positive electrode, the positive electrode can be attached to the substrate with double-sided tape, and the positive electrode active material layer can be set to match the measurement surface required by the device.
- the characteristic X-rays may be monochromated using a filter or may be monochromated using XRD data analysis software after obtaining an XRD pattern.
- XRD data analysis software manufactured by Bruker
- DIFFRAC.EVA XRD data analysis software manufactured by Bruker
- the same software can also be used to remove background.
- Figure 11 shows XRD patterns corresponding to the O3 type crystal structure, the O3' type crystal structure, and the monoclinic O1(15) type crystal structure when CuK ⁇ 1 radiation is used.
- Figures 13A and 13B show all the above-mentioned XRD patterns. However, the range of 2 ⁇ is 18° to 21°, and the range of 2 ⁇ is 42° to 46°.
- the pattern of the H1-3 type crystal structure was similarly created from the crystal structure information described in Non-Patent Document 3.
- the crystal structure patterns of the O3′ type and the monoclinic O1(15) type were estimated from the XRD pattern of the positive electrode active material 100 of one embodiment of the present invention and fitted using TOPAS ver. 3 (crystal structure analysis software manufactured by Bruker).
- the positive electrode active material 100 of one embodiment of the present invention has an O3'-type and/or monoclinic O1 (15)-type crystal structure, but not all of the particles may have an O3'-type and/or monoclinic O1 (15)-type crystal structure. It may contain other crystal structures, or may be partially amorphous.
- the O3'-type and/or monoclinic O1 (15)-type crystal structure is 50% or more, more preferably 60% or more, and even more preferably 66% or more. If the O3'-type and/or monoclinic O1 (15)-type crystal structure is 50% or more, more preferably 60% or more, and even more preferably 66% or more, it can be a positive electrode active material with sufficiently excellent cycle characteristics.
- the O3' type and/or monoclinic O1(15) type crystal structure is 35% or more, more preferably 40% or more, and even more preferably 43% or more.
- the H1-3 type and O1 type crystal structures are 50% or less. Or, more preferably, they are 34% or less. Or, more preferably, they are substantially not observed.
- each diffraction peak after charging is sharp, that is, the half-width, for example, the full width at half maximum is narrow.
- the half-width varies depending on the XRD measurement conditions and the value of 2 ⁇ , even for peaks arising from the same crystal phase.
- the full width at half maximum is preferably, for example, 0.2° or less, more preferably 0.15° or less, and even more preferably 0.12° or less. Note that not all peaks necessarily meet this requirement. If some peaks meet this requirement, it can be said that the crystallinity of the crystal phase is high. Such high crystallinity contributes to the stabilization of the crystal structure after sufficient charging.
- the crystallite size of the O3' type and monoclinic O1 (15) crystal structures of the positive electrode active material 100 is only reduced to about 1/20 of LiCoO 2 (O3) in the discharged state. Therefore, even under the same XRD measurement conditions as the positive electrode before charging and discharging, a clear peak of the O3' type and/or monoclinic O1 (15) crystal structure can be confirmed when x in Li x CoO 2 is small.
- conventional LiCoO 2 even if a part of it can have a structure similar to the O3' type and/or monoclinic O1 (15) crystal structure, the crystallite size becomes small and the peak becomes broad and small. The crystallite size can be determined from the half-width of the XRD peak.
- the influence of the Jahn-Teller effect is small.
- transition metals such as nickel and manganese may be added as additive elements as long as the influence of the Jahn-Teller effect is small.
- Figures 14A to 14C show the results of calculating the lattice constants of the a-axis and c-axis by XRD when the positive electrode active material 100 of one embodiment of the present invention has a layered rock salt crystal structure and contains cobalt and nickel.
- Figure 14A shows the result for the a-axis
- Figure 14B shows the result for the c-axis. Note that the XRD patterns used in these calculations are for the powder after the synthesis of the positive electrode active material, and before it is incorporated into the positive electrode.
- the nickel concentration on the horizontal axis indicates the nickel concentration when the sum of the number of atoms of cobalt and nickel is taken as 100%.
- Figure 14C shows the value obtained by dividing the a-axis lattice constant by the c-axis lattice constant (a-axis/c-axis) for the positive electrode active materials whose lattice constant results are shown in Figures 14A and 14B.
- the above nickel concentration range does not necessarily apply to the surface layer 100a.
- the concentration in the surface layer 100a may be higher than the above concentration.
- the preferable range of the lattice constant was considered, and it was found that, in the positive electrode active material of one embodiment of the present invention, in the layered rock salt crystal structure of the positive electrode active material 100 in a state where no charging or discharging is performed or in a discharged state, which can be estimated from the XRD pattern, the a-axis lattice constant is preferably greater than 2.814 ⁇ 10 ⁇ 10 m and smaller than 2.817 ⁇ 10 ⁇ 10 m, and the c-axis lattice constant is preferably greater than 14.05 ⁇ 10 ⁇ 10 m and smaller than 14.07 ⁇ 10 ⁇ 10 m.
- the state where no charging or discharging is performed may be, for example, a powder state before the positive electrode of a secondary battery is prepared.
- the value obtained by dividing the lattice constant of the a-axis by the lattice constant of the c-axis is greater than 0.20000 and less than 0.20049.
- a first peak may be observed at 2 ⁇ of 18.50° or more and 19.30° or less, and a second peak may be observed at 2 ⁇ of 38.00° or more and 38.80° or less.
- XPS In the case of inorganic oxides, when monochromatic aluminum K ⁇ rays are used as X-rays, XPS can analyze a region from the surface to a depth of about 2 to 8 nm (usually 5 nm or less), so that the concentration of each element can be quantitatively analyzed in a region about half the depth of the surface layer 100a. In addition, narrow scan analysis can be used to analyze the bonding state of elements.
- the concentration of one or more selected from the additive elements is preferably higher in the surface layer 100a than in the interior 100b.
- concentration of one or more selected from the additive elements in the surface layer 100a is preferably higher than the average of the entire positive electrode active material 100. Therefore, for example, it can be said that the concentration of one or more additive elements selected from the surface layer 100a measured by XPS or the like is preferably higher than the average concentration of the additive elements in the entire positive electrode active material 100 measured by ICP-MS or GD-MS or the like.
- the magnesium concentration of at least a part of the surface layer 100a measured by XPS or the like is higher than the average magnesium concentration of the entire positive electrode active material 100.
- the nickel concentration of at least a part of the surface layer 100a is higher than the average nickel concentration of the entire positive electrode active material 100.
- the aluminum concentration of at least a part of the surface layer 100a is higher than the average aluminum concentration of the entire positive electrode active material 100.
- the fluorine concentration in at least a portion of the surface layer 100a is higher than the average fluorine concentration in the entire positive electrode active material 100.
- the surface and surface layer 100a of the positive electrode active material 100 do not contain carbonates, hydroxyl groups, etc. that are chemically adsorbed after the preparation of the positive electrode active material 100. Also, they do not contain electrolyte, binder, conductive material, or compounds derived from these that are attached to the surface of the positive electrode active material 100. Therefore, when quantifying the elements contained in the positive electrode active material, corrections may be made to exclude carbon, hydrogen, excess oxygen, excess fluorine, etc. that can be detected by surface analysis such as XPS. For example, XPS allows the type of bond to be separated by analysis, and corrections may be made to exclude C-F bonds derived from the binder.
- the samples such as the positive electrode active material and the positive electrode active material layer may be washed to remove the electrolyte, binder, conductive material, or compounds derived from these that are attached to the surface of the positive electrode active material.
- lithium may dissolve in the solvent used for washing, but even in this case, the added element is unlikely to dissolve, so the atomic ratio of the added element is not affected.
- the concentration of the added element may also be compared in terms of the ratio to cobalt.
- Using the ratio to cobalt is preferable because it allows comparisons to be made while reducing the influence of carbonates and the like that are chemically adsorbed after the positive electrode active material is produced.
- the ratio Mg/Co of magnesium to the number of cobalt atoms as determined by XPS analysis is preferably 0.4 or more and 1.5 or less.
- the ratio Mg/Co as determined by ICP-MS analysis is preferably 0.001 or more and 0.06 or less.
- the concentrations of lithium and cobalt in the surface layer portion 100a are higher than the concentrations of one or more additive elements selected from the additive elements contained in the surface layer portion 100a measured by XPS or the like.
- the concentration of at least a part of the cobalt in the surface layer portion 100a measured by XPS or the like is higher than the concentration of at least a part of the magnesium in the surface layer portion 100a measured by XPS or the like.
- the concentration of lithium is higher than the concentration of magnesium.
- the concentration of cobalt is higher than the concentration of nickel.
- the concentration of lithium is higher than the concentration of nickel. It is also preferable that the concentration of cobalt is higher than the concentration of aluminum. It is also preferable that the concentration of lithium is higher than the concentration of aluminum. It is also preferable that the concentration of cobalt is higher than the concentration of fluorine. It is also preferable that the concentration of lithium is higher than the concentration of fluorine.
- the additive elements such as aluminum
- a deep region for example, a region having a depth of 5 nm to 50 nm from the surface. Therefore, when the entire positive electrode active material 100 is analyzed using ICP-MS, GD-MS, or the like, additive elements, such as aluminum, are detected, but the concentration may differ from the analysis results obtained by XPS, etc., which targets a region about 5 nm from the surface.
- the number of magnesium atoms is preferably 0.4 times or more and 1.2 times or less, more preferably 0.65 times or more and 1.0 times or less, relative to the number of cobalt atoms.
- the number of nickel atoms is preferably 0.15 times or less, more preferably 0.03 times or more and 0.13 times or less, relative to the number of cobalt atoms.
- the number of aluminum atoms is preferably 0.12 times or less, more preferably 0.09 times or less, relative to the number of cobalt atoms.
- the number of fluorine atoms is preferably 0.1 times or more and 1.1 times or less, more preferably 0.3 times or more and 0.9 times or less, relative to the number of cobalt atoms.
- the above ranges indicate that these additive elements are not attached to a narrow range on the surface of the positive electrode active material 100, but are widely distributed in the surface layer 100a of the positive electrode active material 100 at a preferred concentration.
- monochromated aluminum K ⁇ rays can be used as X-rays.
- the take-off angle is, for example, 45°, and the measurement can be performed, for example, with the following apparatus and conditions.
- the peak showing the bond energy between fluorine and other elements is preferably 682 eV or more and less than 685 eV, and more preferably about 684.3 eV. This value is different from both the bond energy of lithium fluoride, 685 eV, and the bond energy of magnesium fluoride, 686 eV.
- the peak showing the bond energy between magnesium and other elements is preferably 1302 eV or more and less than 1304 eV, and more preferably about 1303 eV. This is a value different from the bond energy of magnesium fluoride, which is 1305 eV, and is close to the bond energy of magnesium oxide.
- ⁇ EDX> It is preferable that one or more selected from the additive elements contained in the positive electrode active material 100 have a concentration gradient. It is more preferable that the depth from the surface of the concentration peak differs depending on the additive element in the positive electrode active material 100.
- the concentration gradient of the additive element can be evaluated, for example, by exposing a cross section of the positive electrode active material 100 by FIB or the like and analyzing the cross section by EDX, EPMA, or the like.
- EDX area analysis In EDX measurements, performing measurements while scanning an area and evaluating the area in two dimensions is called EDX area analysis. Performing measurements while scanning linearly and evaluating the distribution of atomic concentrations within the positive electrode active material is called line analysis. Furthermore, data extracted from a linear area from EDX area analysis is sometimes called line analysis. Performing measurements without scanning an area is called point analysis.
- EDX surface analysis can quantitatively analyze the concentration of the added element in the surface layer 100a, the interior 100b, the grain boundary 101, and the vicinity thereof of the positive electrode active material 100.
- EDX ray analysis can analyze the concentration distribution and maximum value of the added element.
- analysis using a thinned sample such as STEM-EDX is more suitable because it can analyze the concentration distribution in the depth direction from the surface to the center of the positive electrode active material in a specific region without being affected by the distribution in the depth direction.
- the concentration of the added element, such as magnesium, in the surface layer portion 100a is higher than that in the interior portion 100b.
- the magnesium concentration in the surface layer 100a is higher than the magnesium concentration in the interior 100b.
- the peak of the magnesium concentration in the surface layer 100a is preferably present at a depth of 3 nm from the surface of the positive electrode active material 100 toward the center, more preferably at a depth of 1 nm, and even more preferably at a depth of 0.5 nm. It is preferable that the magnesium concentration decays to 60% or less of the peak at a point 1 nm deep from the peak top. It is also preferable that the magnesium concentration decays to 30% or less of the peak at a point 2 nm deep from the peak top. Note that the peak concentration here refers to the maximum value of the concentration.
- the distribution of fluorine overlaps with the distribution of magnesium.
- the difference in the depth direction between the peak of the fluorine concentration and the peak of the magnesium concentration is preferably within 10 nm, more preferably within 3 nm, and even more preferably within 1 nm.
- the fluorine concentration peak of the surface layer 100a is preferably present at a depth of up to 3 nm from the surface toward the center of the positive electrode active material 100, more preferably at a depth of up to 1 nm, and even more preferably at a depth of up to 0.5 nm. Furthermore, it is more preferable for the fluorine concentration peak to be slightly closer to the surface than the magnesium concentration peak, as this increases resistance to hydrofluoric acid. For example, it is more preferable for the fluorine concentration peak to be 0.5 nm or more closer to the surface than the magnesium concentration peak, and even more preferable for it to be 1.5 nm or more closer to the surface.
- the nickel concentration peak in the surface layer 100a is preferably present at a depth of up to 3 nm from the surface toward the center of the positive electrode active material 100, more preferably at a depth of up to 1 nm, and even more preferably at a depth of up to 0.5 nm.
- the distribution of nickel preferably overlaps with the distribution of magnesium.
- the difference in the depth direction between the nickel concentration peak and the magnesium concentration peak is preferably within 10 nm, more preferably within 3 nm, and even more preferably within 1 nm.
- the magnesium, nickel, or fluorine concentration peak is closer to the surface than the aluminum concentration peak of the surface layer 100a when EDX-ray analysis is performed.
- the aluminum concentration peak is preferably present at a depth of 0.5 nm to 50 nm from the surface toward the center of the positive electrode active material 100, and more preferably at a depth of 5 nm to 50 nm.
- the ratio of magnesium Mg to the number of atoms of cobalt Co (Mg/Co) at the peak of the magnesium concentration is less than 1, preferably 0.05 to 0.6, and more preferably 0.1 to 0.4.
- the ratio of the number of atoms of aluminum Al to cobalt Co (Al/Co) at the peak of the aluminum concentration is less than 1, preferably 0.05 to 0.6, and more preferably 0.1 to 0.45.
- the ratio of the number of atoms of nickel Ni to cobalt Co (Ni/Co) at the peak of the nickel concentration is less than 1, preferably 0 to 0.2, and more preferably 0.01 to 0.1.
- Ni/Co is preferably 0.1 to 0.5.
- the atomic ratio (F/Co) of fluorine (F) to cobalt (Co) at the peak of the fluorine concentration is less than 1, preferably 0 to 1.6, more preferably 0.1 to 1.4.
- the surface of the positive electrode active material 100 in the EDX analysis results can be estimated, for example, as follows: For an element that is uniformly present in the interior 100b of the positive electrode active material 100, such as oxygen or cobalt, the point where the amount detected in the interior 100b is half that of the element is defined as the surface.
- the surface can be estimated using the amount of detected oxygen. Specifically, first, the average oxygen concentration O ave is obtained from the region in the interior 100b where the amount of detected oxygen is stable. At this time, if oxygen O bg that is thought to be due to chemical adsorption or background is detected in a region that can be clearly determined to be outside the surface, the average oxygen concentration O ave can be obtained by subtracting O bg from the measured value. The measurement point showing a measured value that is 1/2 of this average value O ave , that is, closest to O ave /2, can be estimated to be the surface of the positive electrode active material.
- the surface can also be estimated in the same manner as above using the amount of cobalt detected.
- the sum of the amounts of multiple transition metals detected can be used to make a similar estimation.
- the amount of transition metals detected, including cobalt, is suitable for estimating the surface because it is less susceptible to the effects of chemical adsorption.
- the ratio of the number of atoms of the added element A to the number of atoms of cobalt Co at or near the grain boundary 101 is preferably 0.020 or more and 0.50 or less. More preferably, it is 0.025 or more and 0.30 or less. Even more preferably, it is 0.030 or more and 0.20 or less. Or it is preferably 0.020 or more and 0.30 or less. Or it is preferably 0.020 or more and 0.20 or less. Or it is preferably 0.025 or more and 0.50 or less. Or it is preferably 0.025 or more and 0.20 or less. Or it is preferably 0.030 or more and 0.50 or less. Or it is preferably 0.030 or more and 0.30 or less.
- the ratio of the number of atoms of magnesium to cobalt (Mg/Co) at or near the grain boundary 101 is preferably 0.020 or more and 0.50 or less. Further, it is preferably 0.025 or more and 0.30 or less. Further, it is preferably 0.030 or more and 0.20 or less. Or it is preferably 0.020 or more and 0.30 or less. Or it is preferably 0.020 or more and 0.20 or less. Or it is preferably 0.025 or more and 0.50 or less. Or it is preferably 0.025 or more and 0.20 or less. Or it is preferably 0.030 or more and 0.50 or less.
- the positive electrode active material 100 is preferably 0.030 or more and 0.30 or less. Furthermore, if the above range is found in multiple locations, for example, three or more locations, of the positive electrode active material 100, it can be said that this indicates that the added element is not attached to a narrow area on the surface of the positive electrode active material 100, but is widely distributed in a preferred concentration in the surface layer 100a of the positive electrode active material 100.
- a coating portion may be attached to at least a portion of the surface of the positive electrode active material 100.
- Figures 15A and 15B show structures in which a coating portion 104 is attached to the positive electrode active material 100 shown in Figures 5A and 5C, respectively.
- the coating portion 104 is preferably formed by, for example, deposition of decomposition products such as lithium salt and organic electrolyte solution accompanying charge and discharge.
- decomposition products such as lithium salt and organic electrolyte solution accompanying charge and discharge.
- x in Li x CoO 2 is 0.24 or less
- the coating portion 104 preferably has, for example, carbon, oxygen, and fluorine.
- the coating portion 104 having one or more selected from boron, nitrogen, sulfur, and fluorine may be a good quality coating portion and is preferable.
- the coating portion 104 does not have to cover all of the positive electrode active material 100. For example, it is sufficient that 50% or more of the surface of the positive electrode active material 100 is covered, more preferably 70% or more, and even more preferably 90% or more. In the area where the covering portion 104 is not present, fluorine may be adsorbed on the surface of the positive electrode active material 100.
- the secondary battery 1004 has multiple positive electrodes 503, multiple negative electrodes 506, and multiple separators 508.
- the separators 508 are provided between the positive electrodes 503 and the negative electrodes 506, and are shown by dotted lines in FIG. 16A for ease of viewing.
- the separators 508 may contain an electrolyte, specifically a liquid electrolyte (also called an electrolyte solution). Note that if a solid electrolyte or a semi-solid electrolyte is used as the electrolyte, the secondary battery 1004 does not need to have the separators 508.
- the positive electrode 503 and the negative electrode 506 each have a protruding tab portion and a portion other than the tab portion.
- the tab portion can electrically connect the wiring 1005a and the wiring 1005b, etc., in a nail penetration test device.
- the positive electrode 503 has a positive electrode current collector and a positive electrode active material layer formed on the positive electrode current collector, and the positive electrode active material layer is preferably formed on both sides of the positive electrode current collector.
- the negative electrode 506 has a negative electrode current collector and a negative electrode active material layer formed on the negative electrode current collector, and the negative electrode active material layer is preferably formed on both sides of the negative electrode current collector.
- a plurality of positive electrodes 503, a plurality of negative electrodes 506, and a plurality of separators 508 are stacked, and this may be referred to as a laminate in the present specification.
- the tab portions of the plurality of negative electrodes 506 are joined together with the lead 512b at the joint 515b, and are electrically connected to each other.
- the tab portions of the plurality of positive electrodes 503 are joined together with the lead 512a at the joint 515a, and are electrically connected to each other.
- the positive electrode active material layer and the negative electrode active material layer (these are simply called active material layers) have high insulation, so it is better not to form an active material layer on the tab portion.
- the leads 512a and 512b can be made of a material selected from aluminum, nickel, copper, titanium, or alloys thereof. Ultrasonic bonding can be used for bonding at the joints. In addition, in the nail penetration test, it is not necessary to provide leads 512a and 512b, but if they are provided, wiring 1005a and wiring 1005b are electrically connected to leads 512a and 512b, respectively.
- the secondary battery 1004 has an exterior body (not shown), and the laminate shown in FIG. 16A is housed in the exterior body. After that, an electrolyte solution in which lithium salt is dissolved is injected into the exterior body.
- the electrolyte solution has carrier ions, typically lithium ions.
- a secondary battery that has such lithium ions is called a lithium ion secondary battery.
- the exterior body is preferably in the form of a film from the viewpoint of weight reduction, and a secondary battery having a film-shaped exterior body is called a laminate-type secondary battery.
- the exterior body may have a laminated structure of a polymer and a metal with excellent thermal conductivity.
- polypropylene is preferably used as the polymer and aluminum is preferably used as the metal, and nylon or the like may be further arranged on the outside of the exterior body.
- a metal can case may be used as the exterior body, and when a circular can case is used, it is called a coin-type secondary battery.
- This embodiment can be used in combination with other embodiments.
- the method of adding the added elements is important. At the same time, it is also important that the crystallinity of the interior 100b is good.
- the process of producing the positive electrode active material 100 there is a method in which lithium cobalt oxide is synthesized, and then an additive element source is mixed and heat treatment is performed.
- a method in which the additive element source is mixed with the cobalt source in the same step as the lithium source to synthesize lithium cobalt oxide containing the additive element may be used.
- the heat treatment after mixing the additive element source is important.
- the heat treatment after mixing the additive element source is sometimes called firing or annealing.
- a material that functions as a flux it is preferable to mix a material that functions as a flux together with or as the additive element source.
- a material that functions as a flux a substance with a melting point lower than that of lithium cobalt oxide can be used.
- fluorine compounds such as lithium fluoride are suitable as fluxes. Adding a flux lowers the melting point of the additive element source and lithium cobalt oxide. Lowering the melting point makes it easier to distribute the additive element well at a temperature where cation mixing is unlikely to occur.
- the initial heating causes lithium to be released from part of the surface layer 100a of the lithium cobalt oxide, resulting in a better distribution of the added elements.
- the distribution of the additive elements is easily differentiated by initial heating through the following mechanism.
- lithium is released from a part of the surface layer 100a by initial heating.
- the lithium cobalt oxide having the lithium-deficient surface layer 100a is mixed with additive element sources such as nickel source, aluminum source, and magnesium source and heated.
- additive element sources such as nickel source, aluminum source, and magnesium source and heated.
- magnesium is a typical divalent element
- nickel is a transition metal but is prone to becoming a divalent ion. Therefore, a rock salt phase containing Mg 2+ , Ni 2+ , and Co 2+ reduced by the deficiency of lithium is formed in a part of the surface layer 100a.
- this phase is formed in a part of the surface layer 100a, it may not be clearly confirmed in an electron microscope image such as STEM and an electron beam diffraction pattern.
- nickel is likely to dissolve in the surface layer 100a of layered rock-salt lithium cobalt oxide and diffuse to the interior 100b, but is likely to remain in the surface layer 100a if part of the surface layer 100a is of the rock-salt type. Therefore, by performing initial heating, it is possible to make it easier for divalent additive elements such as nickel to remain in the surface layer 100a.
- the effect of this initial heating is particularly large on the surface other than the (001) orientation of the positive electrode active material 100 and on its surface layer 100a.
- the Me-O distance in rock salt Ni0.5Mg0.5O is 2.09x10-10 m
- the Me-O distance in rock salt MgO is 2.11x10-10 m.
- the Me-O distance in spinel NiAl2O4 is 2.0125x10-10 m
- the Me-O distance in spinel MgAl2O4 is 2.02x10-10 m . In both cases, the Me-O distance exceeds 2x10-10 m.
- the bond distance between metals other than lithium and oxygen is shorter than the above.
- the Al-O distance in layered rock salt type LiAlO2 is 1.905 ⁇ 10-10 m (Li-O distance is 2.11 ⁇ 10-10 m).
- the Co-O distance in layered rock salt type LiCoO2 is 1.9224 ⁇ 10-10 m (Li-O distance is 2.0916 ⁇ 10-10 m).
- the ionic radius of hexacoordinated aluminum is 0.535 ⁇ 10 ⁇ 10 m
- the ionic radius of hexacoordinated oxygen is 1.4 ⁇ 10 ⁇ 10 m
- the sum of these is 1.935 ⁇ 10 ⁇ 10 m.
- Initial heating is also expected to have the effect of increasing the crystallinity of the layered rock salt type crystal structure in the interior 100b.
- x in Li x CoO 2 is, for example, 0.15 or more and 0.17 or less, it is preferable to perform this initial heating in order to produce a positive electrode active material 100 having a monoclinic O1(15) type crystal structure.
- initial heating is not necessarily required. In other heating steps, by controlling the atmosphere, temperature, time, etc., it may be possible to produce a positive electrode active material 100 having O3′ type and/or monoclinic O1(15) type when x in Li x CoO 2 is small.
- ⁇ Eutectic point> When the melting point of a fluorine compound (sometimes called a fluoride) such as lithium fluoride is lower than that of another additive element source, the fluorine compound can function as a flux (also called a fluxing agent) that lowers the melting point of the other additive element source.
- the fluorine compound has LiF and MgF2 , as shown in Figure 17 (quoted and added from Figure 6 of Non-Patent Document 12), the eutectic point P of LiF and MgF2 is around 742°C (T1), so it is preferable to set the heating temperature to 742°C or higher in the heating step after mixing the additive element.
- the heating temperature after mixing the additive element is preferably 742°C or higher, more preferably 830°C or higher. Also, 800°C (T2 in Fig. 17) or higher, which is between these, is preferable.
- Method 1 for producing positive electrode active material 100 that undergoes initial heating will be described with reference to FIGS. 19A to 19C.
- Step S11 In step S11 shown in FIG. 19A, a lithium source (Li source) and a cobalt source (Co source) are prepared as starting materials, that is, lithium and transition metal materials, respectively.
- Li source Li source
- Co source cobalt source
- the lithium source it is preferable to use a compound containing lithium, such as lithium carbonate, lithium hydroxide, lithium nitrate, or lithium fluoride. It is preferable that the lithium source has high purity, for example, a material with a purity of 99.99% or more.
- cobalt source it is preferable to use a compound containing cobalt, for example, cobalt oxide such as tricobalt tetroxide, cobalt hydroxide, etc.
- cobalt oxide such as tricobalt tetroxide, cobalt hydroxide, etc.
- the cobalt source is preferably of high purity, for example, a material with a purity of 3N (99.9%) or more, preferably 4N (99.99%) or more, more preferably 4N5 (99.995%) or more, and even more preferably 5N (99.999%) or more may be used.
- a high purity material impurities in the positive electrode active material can be controlled. As a result, the capacity of the secondary battery is increased and/or the reliability of the secondary battery is improved.
- the cobalt source has high crystallinity, for example, it is preferable that the cobalt source has single crystal grains.
- the crystallinity of the cobalt source can be evaluated using TEM images, STEM images, HAADF-STEM images, ABF-STEM images, etc., or evaluation using XRD, electron beam diffraction, neutron beam diffraction, etc. Note that the above-mentioned methods for evaluating crystallinity can be applied not only to the evaluation of cobalt sources, but also to the evaluation of other crystallinity.
- step S12 the lithium source and the cobalt source are pulverized and mixed to prepare a mixed material.
- the pulverization and mixing can be performed in a dry or wet manner.
- the wet method is preferable because it can be crushed into smaller pieces.
- a solvent is prepared.
- ketones such as acetone, alcohols such as ethanol and isopropanol, ether, dioxane, acetonitrile, N-methyl-2-pyrrolidone (NMP), etc. can be used. It is more preferable to use an aprotic solvent that is less likely to react with lithium.
- dehydrated acetone with a purity of 99.5% or more is used. It is preferable to mix the lithium source and the cobalt source with dehydrated acetone with a purity of 99.5% or more, in which the moisture content is suppressed to 10 ppm or less, and then pulverize and mix them.
- dehydrated acetone with the above-mentioned purity it is possible to reduce impurities that may be mixed in.
- a ball mill, a bead mill, or the like can be used as a means for grinding and mixing.
- a ball mill it is advisable to use aluminum oxide balls or zirconium oxide balls as the media. Zirconium oxide balls are preferable because they emit less impurities.
- the peripheral speed is preferably 100 mm/s or more and 2000 mm/s or less in order to suppress contamination from the media. In this embodiment, the peripheral speed is set to 838 mm/s (rotation speed 400 rpm, ball mill diameter 40 mm).
- step S13 shown in FIG. 19A the mixed material is heated.
- the heating is preferably performed at 800° C. or more and 1100° C. or less, more preferably at 900° C. or more and 1000° C. or less, and even more preferably at about 950° C. If the temperature is too low, the decomposition and melting of the lithium source and the cobalt source may be insufficient. On the other hand, if the temperature is too high, defects may occur due to lithium transpiration from the lithium source and/or cobalt being excessively reduced. For example, cobalt may change from trivalent to divalent, inducing oxygen defects, etc.
- the heating time is preferably 1 hour or more and 100 hours or less, and more preferably 2 hours or more and 20 hours or less.
- the heating rate depends on the heating temperature reached, but is preferably 80°C/h or more and 250°C/h or less. For example, when heating at 1000°C for 10 hours, a heating rate of 200°C/h is preferable.
- the heating is preferably performed in an atmosphere with little water, such as dry air, for example, an atmosphere with a dew point of ⁇ 50° C. or less, more preferably an atmosphere with a dew point of ⁇ 80° C. or less. In this embodiment, the heating is performed in an atmosphere with a dew point of ⁇ 93° C.
- the impurity concentrations of CH 4 , CO, CO 2 , H 2 , and the like in the heating atmosphere are preferably each 5 ppb (parts per billion) or less.
- the heating atmosphere is preferably an atmosphere containing oxygen.
- the heating atmosphere is preferably an atmosphere containing oxygen.
- the flow rate of the dry air is preferably 10 L/min.
- the method of continuously introducing oxygen into the reaction chamber and having oxygen flow through the reaction chamber is called flow.
- the heating atmosphere is an atmosphere containing oxygen
- One method of preventing flow is, for example, reducing the pressure in the reaction chamber and then filling it with oxygen (which can also be called purging) to prevent the oxygen from escaping the reaction chamber.
- oxygen which can also be called purging
- the material After heating, the material can be allowed to cool naturally, but it is preferable that the time it takes to cool from the specified temperature to room temperature is within a range of 10 to 50 hours.
- the cooling rate (hereinafter also referred to as the cooling rate) should be 80°C/h to 250°C/h, and more preferably 180°C/h to 210°C/h.
- cooling to room temperature is not necessarily required, as long as the material is cooled to a temperature that is acceptable for the next step.
- the heating in this process may be performed using a rotary kiln or a roller hearth kiln. Heating using a rotary kiln can be performed while stirring, whether it is a continuous or batch type.
- the crucible used for heating is preferably an aluminum oxide crucible.
- An aluminum oxide crucible is a material that does not easily release impurities. In this embodiment, an aluminum oxide crucible with a purity of 99.9% is used. It is preferable to place a lid on the crucible when heating. This can prevent the material from volatilizing or sublimating. Placing a lid on the crucible means that it is possible to prevent the material from volatilizing or sublimating from the time the temperature is increased to the time the temperature is decreased in this step, and it is not necessary to seal the crucible with a lid. For example, as described above, by filling the reaction chamber with oxygen, it is possible to carry out this step without sealing the crucible.
- a used crucible refers to one that has undergone the process of putting in and heating materials containing lithium, transition metal M, and/or additive elements two or less times.
- a used crucible refers to one that has undergone the process of putting in and heating materials containing lithium, transition metal M, and/or additive elements three or more times. This is because when a new crucible is used, there is a risk that some of the materials, including lithium fluoride, may be absorbed, diffused, moved, and/or attached to the sheath during heating.
- the material After heating, the material may be crushed and sieved as necessary. When recovering the heated material, it may be transferred from the crucible to a mortar and then recovered.
- the mortar is preferably made of aluminum oxide or zirconium oxide.
- Aluminum oxide mortars are made of a material that does not easily release impurities. Specifically, an aluminum oxide mortar with a purity of 90% or more, preferably 99% or more, is used. Note that the same heating conditions as those in step S13 can be applied to the heating steps described below other than step S13.
- lithium cobalt oxide (LiCoO 2 ) can be synthesized as shown in step S14 of Fig. 19A.
- the median diameter (D50) is used as the particle diameter of lithium cobalt oxide, it is preferable to pulverize the lithium cobalt oxide in order to obtain a positive electrode active material 100 having a relatively small median diameter (D50).
- the composite oxide is produced by a solid phase method, but the composite oxide may also be produced by a coprecipitation method.
- the composite oxide may also be produced by a hydrothermal method.
- step S15 shown in FIG. 19A the lithium cobalt oxide is heated. Since this is the first heating of the lithium cobalt oxide, the heating in step S15 may be called initial heating. Or, since it is heating before step S20 described below, it may be called preheating or pretreatment.
- the crucible and/or lid used in this step are the same as those used in step S13. Although the following effects are expected from the initial heating, the initial heating is not essential to obtain the positive electrode active material which is one aspect of the present invention.
- initial heating causes lithium to be released from part of the surface layer 100a of the lithium cobalt oxide. It is also expected to have the effect of increasing the crystallinity of the interior 100b. Impurities may be mixed into the lithium source and/or cobalt source prepared in step S11, etc. Initial heating can reduce the amount of impurities in the lithium cobalt oxide completed in step S14.
- the initial heating has the effect of smoothing the surface of the lithium cobalt oxide.
- a smooth surface of a composite oxide means that there are few irregularities, the composite oxide is rounded overall, and the corners are also rounded. Furthermore, a surface is called smooth when there is little foreign matter adhering to it. Foreign matter is thought to be a cause of unevenness, so it is preferable that it does not adhere to the surface.
- the heating conditions can be selected from those described in step S13.
- the heating temperature of this step is preferably lower than the temperature of step S13 in order to maintain the crystal structure of the complex oxide.
- the heating time of this step is preferably shorter than the time of step S13 in order to maintain the crystal structure of the complex oxide. For example, it is preferable to heat at a temperature of 700°C or higher and 1000°C or lower for 2 hours or higher and 20 hours or lower.
- the effect of increasing the crystallinity of the inner portion 100b is, for example, the effect of mitigating distortion, misalignment, etc. resulting from differences in shrinkage of the lithium cobalt oxide produced in step S13.
- the heating in step S13 may cause a temperature difference between the surface and the inside of the lithium cobalt oxide.
- the temperature difference may induce a shrinkage difference. It is also believed that the temperature difference causes the difference in fluidity between the surface and the inside, which leads to the shrinkage difference.
- the energy related to the shrinkage difference gives the lithium cobalt oxide a difference in internal stress.
- the internal stress difference is also called strain, and this energy is sometimes called strain energy.
- the internal stress is removed by the initial heating in step S15, or in other words, the strain energy is thought to be homogenized by the initial heating in step S15. When the strain energy is homogenized, the strain of the lithium cobalt oxide is alleviated. As a result, the surface of the lithium cobalt oxide may become smooth. This is also called the surface being improved. In other words, it is believed that after step S15, the shrinkage difference caused in the lithium cobalt oxide is alleviated, and the surface of the composite oxide becomes smooth.
- the shrinkage difference may cause microscopic misalignment, such as crystal misalignment, in the lithium cobalt oxide. It is preferable to carry out this step in order to reduce this misalignment. By going through this step, it is possible to equalize the misalignment of the composite oxide. When the misalignment is equalized, the surface of the composite oxide may become smooth. This is also referred to as the alignment of crystal grains. In other words, it is believed that by going through step S15, the misalignment of the crystals and the like that has occurred in the composite oxide is alleviated, and the surface of the composite oxide becomes smooth.
- lithium cobalt oxide which has a smooth surface, it reduces deterioration during charging and discharging as a secondary battery and prevents cracking of the positive electrode active material.
- step S14 lithium cobalt oxide that has been synthesized in advance may be used. In this case, steps S11 to S13 can be omitted. By carrying out step S15 on lithium cobalt oxide that has been synthesized in advance, lithium cobalt oxide with a smooth surface can be obtained.
- step S20 it is preferable to add the additive element A to the lithium cobalt oxide that has been initially heated.
- the additive element A can be added evenly. Therefore, it is preferable to add the additive element A after the initial heating.
- the step of adding the additive element A will be described with reference to FIG. 19B and FIG. 19C.
- Steps S21 to S23> 19B and 19C the steps of preparing a source of the additional element A (A source) will be described.
- a lithium source may be prepared together with the source of the additional element A.
- the additive element A can be one or more of the additive elements described in the previous embodiment, such as magnesium, fluorine, nickel, aluminum, titanium, zirconium, vanadium, iron, manganese, chromium, niobium, arsenic, zinc, silicon, sulfur, phosphorus, and boron. It is also possible to use one or two of the additive elements selected from bromine and beryllium.
- the additive element source can be called a magnesium source (Mg source).
- Mg source magnesium fluoride, magnesium oxide, magnesium hydroxide, magnesium carbonate, or the like can be used.
- a plurality of the above-mentioned magnesium sources may be used.
- the source of the additive element can be called a fluorine source (F source).
- fluorine source examples include lithium fluoride (LiF), magnesium fluoride ( MgF2 ), aluminum fluoride ( AlF3 ), titanium fluoride ( TiF4 ), cobalt fluoride ( CoF2 , CoF3 ), nickel fluoride ( NiF2 ), zirconium fluoride ( ZrF4 ), vanadium fluoride ( VF5 ), manganese fluoride, iron fluoride, chromium fluoride, niobium fluoride, zinc fluoride ( ZnF2 ), calcium fluoride ( CaF2 ), sodium fluoride (NaF), potassium fluoride (KF), barium fluoride ( BaF2 ), cerium fluoride ( CeF3 , CeF4 ), lanthanum fluoride ( LaF3 ), and sodium aluminum hexafluoride ( Na3
- Magnesium fluoride can be used as both a fluorine source and a magnesium source.
- Lithium fluoride can be used as both a fluorine source and a lithium source.
- Another lithium source that can be used in step S21 is lithium carbonate.
- the fluorine source may also be a gas, such as fluorine ( F2 ), carbon fluoride, sulfur fluoride, or oxygen fluoride ( OF2 , O2F2 , O3F2 , O4F2 , O5F2 , O6F2 , O2F ) , which may be mixed into the atmosphere in the heating step described below.
- F2 fluorine
- OF2 , O2F2 , O3F2 , O4F2 , O5F2 , O6F2 , O2F oxygen fluoride
- a plurality of the above-mentioned fluorine sources may also be used .
- lithium fluoride (LiF) is prepared as the fluorine source
- magnesium fluoride (MgF 2 ) is prepared as the fluorine source and magnesium source.
- the amount of lithium fluoride increases, there is a concern that the lithium becomes excessive and the cycle characteristics deteriorate.
- the vicinity is a value that is greater than 0.9 times and less than 1.1 times the value.
- Step S22> 19B the magnesium source and the fluorine source are pulverized and mixed. This step can be performed under the pulverization and mixing conditions selected from those described in step S12.
- Step S23> 19B the material crushed and mixed as described above is collected to obtain a source of additive element A (source A).
- source A a source of additive element A
- the source of additive element A shown in step S23 has a plurality of starting materials and can be called a mixture.
- the particle size of the mixture is preferably a median diameter (D50) of 600 nm or more and 10 ⁇ m or less, more preferably 1 ⁇ m or more and 5 ⁇ m or less. Even when a single material is used as the additive element source, the median diameter (D50) is preferably 600 nm or more and 10 ⁇ m or less, more preferably 1 ⁇ m or more and 5 ⁇ m or less.
- Such a finely powdered mixture makes it easier to uniformly attach the mixture to the surface of the lithium cobalt oxide particles when mixed with the lithium cobalt oxide in a later process. If the mixture is uniformly attached to the surface of the lithium cobalt oxide particles, it is preferable because it makes it easier to uniformly distribute or diffuse the additive element in the surface layer portion 100a of the composite oxide after heating.
- Step S21 A process different from that shown in FIG. 19B will be described with reference to FIG. 19C.
- step S21 shown in FIG. 19C four types of additive element sources to be added to lithium cobalt oxide are prepared. That is, the types of additive element sources shown in FIG. 19C are different from those shown in FIG. 19B.
- a lithium source may be prepared together with the additive element sources.
- a magnesium source Mg source
- a fluorine source F source
- a nickel source Ni source
- an aluminum source Al source
- the magnesium source and the fluorine source can be selected from the compounds described in FIG. 19B.
- Nickel oxide, nickel hydroxide, etc. can be used as the nickel source.
- Aluminum oxide, aluminum hydroxide, etc. can be used as the aluminum source.
- Steps S22 and S23 shown in FIG. 19C are similar to the steps described in FIG. 19B.
- lithium cobalt oxide is mixed with a source of additive element A.
- the mixing conditions in step S31 are preferably milder than those in step S12 in order not to destroy the shape of the lithium cobalt oxide particles.
- the mixing conditions are preferably lower in rotation speed or shorter in time than those in step S12.
- the dry method provides milder conditions than the wet method.
- a ball mill, a bead mill, etc. can be used for mixing.
- zirconium oxide balls it is preferable to use zirconium oxide balls as the media.
- the mixture is mixed dry in a ball mill using zirconium oxide balls with a diameter of 1 mm at 150 rpm for 1 hour.
- the mixture is performed in a dry room with a dew point of -100°C or higher and -10°C or lower.
- Step S32> 19A the mixed material is collected to obtain a mixture 903.
- the material may be crushed and then sieved, if necessary.
- Figures 19A to 19C illustrate a fabrication method in which an additive element is added after initial heating
- the additive element may be added at a different timing, or may be added multiple times. The timing may be changed depending on the additive element.
- the additive elements may be added to the lithium source and the cobalt source in step S11, i.e., at the stage of the starting material for the composite oxide.
- Figure 20A shows a flow of adding a magnesium source to a lithium source and a cobalt source.
- Figure 20B shows a flow of adding a magnesium source and an aluminum source to a lithium source and a cobalt source.
- Figure 20C shows a flow of adding a magnesium source and a nickel source to a lithium source and a cobalt source.
- the additive element sources shown in Figures 20A to 20C are examples.
- step S12 the process proceeds to step S12, and then to step S13, and in step S14, lithium cobalt oxide having the additive element can be obtained. It is also possible to control the distribution of the additive element according to the timing of adding the additive element.
- the additive element added as shown in Figures 20A to 20C is expected to be located inside the positive electrode active material 100.
- the above-mentioned steps S11 to S14 do not need to be separated from the steps S21 to S23, so it can be said to be a simple and highly productive method.
- a new additive element may be added in step S20.
- lithium cobalt oxide that already contains some of the added elements may be used.
- steps S11 to S14 and some of the steps in step S20 can be omitted. This is a simple and highly productive method.
- a magnesium source and a fluorine source or a magnesium source, a fluorine source, a nickel source, and an aluminum source may be added as in step S20.
- step S33 shown in Fig. 19A the mixture 903 is heated.
- the heating can be performed under a heating condition selected from those described in step S13.
- the heating time is preferably 2 hours or more.
- the pressure inside the furnace may be higher than atmospheric pressure in order to increase the oxygen partial pressure of the heating atmosphere. This is because if the oxygen partial pressure of the heating atmosphere is insufficient, cobalt, etc. will be reduced, and lithium cobalt oxide, etc. may not be able to maintain the layered rock salt type crystal structure.
- the lower limit of the heating temperature in step S33 must be equal to or higher than the temperature at which the reaction between the lithium cobalt oxide and the additive element source proceeds.
- the temperature at which the reaction proceeds is preferably a temperature at which mutual diffusion between the lithium cobalt oxide and the elements contained in the additive element source occurs, and may be lower than the melting temperature of these materials.
- An oxide is used as an example for explanation, and it is known that solid-phase diffusion occurs at a temperature 0.757 times the melting temperature Tm (Tammann temperature Td ). Therefore, the heating temperature in step S33 is preferably 650°C or higher.
- the reaction proceeds more easily if the temperature is equal to or higher than the melting temperature of one or more of the materials contained in the mixture 903.
- the eutectic point of LiF and MgF2 is around 742°C (see eutectic point P in FIG. 17), so that the lower limit of the heating temperature in step S33 is preferably 742°C or higher.
- the lower limit of the heating temperature is more preferably 830° C. or higher.
- the upper limit of the heating temperature is below the decomposition temperature of lithium cobalt oxide (melting point 1130°C). At temperatures close to the decomposition temperature, there is concern that lithium cobalt oxide may decompose, albeit only in small amounts. Therefore, the upper limit of the heating temperature is preferably 1000°C or lower, more preferably 950°C or lower, and even more preferably 900°C or lower.
- the heating temperature in step S33 is preferably 650°C to 1130°C, more preferably 650°C to 1000°C, even more preferably 650°C to 950°C, and even more preferably 650°C to 900°C.
- 742°C to 1130°C is preferred, more preferably 742°C to 1000°C, even more preferably 742°C to 950°C, and even more preferably 742°C to 900°C.
- 830°C to 1130°C is preferred, more preferably 830°C to 1000°C, even more preferably 830°C to 950°C, and even more preferably 830°C to 900°C.
- the heating temperature in step S33 is preferably lower than the heating temperature in step S13.
- step S33 an example of a heating furnace used in step S33 will be described with reference to FIG. 23.
- the heating furnace 220 shown in FIG. 23 has a heating furnace space 202, a hot plate 204, a pressure gauge 221, a heater unit 206, and a heat insulating material 208. It is preferable to heat the container 216, which corresponds to a crucible or a scabbard, with a lid 218 placed thereon. With this configuration, the space 219 formed by the container 216 and the lid 218 can be made into an atmosphere containing fluoride. During heating, if the state is maintained by closing the lid so that the concentration of gasified fluoride in the space 219 remains constant or does not decrease, fluorine and magnesium can be contained near the particle surface of the mixture 903.
- the space 219 has a smaller volume than the heating furnace space 202, a small amount of fluoride volatilizes, making it possible to make the atmosphere containing fluoride.
- the atmosphere of the reaction system can be made into an atmosphere containing fluoride without significantly reducing the amount of fluoride contained in the mixture 903.
- the lid 218, the mixture 903 can be heated in an atmosphere containing fluoride easily and inexpensively.
- a process of making the heating furnace space 202 an atmosphere containing oxygen and a process of placing the container 216 containing the mixture 903 in the heating furnace space 202 are performed before heating in the heating furnace space 202.
- the mixture 903 can be heated in an atmosphere containing oxygen and fluoride.
- the heating is performed while gas is flowing (flow).
- the gas can be introduced from the bottom of the heating furnace space 202 and exhausted to the top.
- the heating furnace space 202 can be sealed during heating to make it a closed space so that the gas is not transported to the outside (purging).
- the method for making the heating furnace space 202 into an oxygen-containing atmosphere includes a method of evacuating the heating furnace space 202 and then introducing an oxygen-containing gas such as oxygen gas or dry air, or a method of injecting an oxygen-containing gas such as oxygen gas or dry air for a certain period of time.
- an oxygen-containing gas such as oxygen gas or dry air
- the atmosphere in the heating furnace space 202 may be considered to be an oxygen-containing atmosphere.
- fluorides and the like that are attached to the inner walls of the container 216 and the lid 218 can be re-emitted by heating and attached to the mixture 903.
- the mixture 903 should be arranged when placed in the container 216, but it is preferable to arrange the mixture 903 so that the top surface of the mixture 903 is flat with respect to the bottom surface of the container 216, in other words, so that the height of the top surface of the mixture 903 is uniform, as shown in FIG. 23.
- the heating in step S33 is preferably performed while controlling the pressure inside the furnace with the pressure gauge 221.
- the inside of the furnace is preferably in an atmospheric pressure state or a pressurized state.
- the surface of lithium cobalt oxide melts when exposed to a pressurized state. Therefore, the surface of lithium cobalt oxide heated together with LiF and MgF2 can melt by applying pressure.
- the cooling after heating in step S33 may be allowed to cool naturally, but it is preferable that the time required for the temperature to drop from the specified temperature to room temperature is within a range of 10 to 50 hours.
- the cooling rate is preferably 80°C/h to 250°C/h, and more preferably 180°C/h to 210°C/h.
- the cooling rate in step S33 is preferably faster than that in step S13.
- a fast cooling rate is called rapid cooling.
- the partial pressure of fluorine or fluorine compounds due to the fluorine source, etc. within an appropriate range. It is also possible to control the partial pressure by placing a lid on the crucible used in this step and heating it. As described above, the lid can prevent the material from volatilizing or sublimating. In other words, it is not necessary to seal the crucible with a lid as long as it is possible to prevent the material from volatilizing or sublimating during the temperature increase and decrease periods in this step. For example, by filling the reaction chamber in which the crucible is placed with oxygen, it is also possible to carry out this step without sealing the crucible. A positive electrode active material having an appropriate amount of fluorine or fluorine compounds is preferable because it can suppress heat generation and smoke generation even in the event of an internal short circuit.
- some materials for example LiF, which is a fluorine source, may function as a flux.
- This function allows the heating temperature to be lowered below the decomposition temperature of lithium cobalt oxide, for example to 742°C or higher and 950°C or lower, and additive elements such as magnesium can be distributed in the surface layer to produce a positive electrode active material with good characteristics.
- LiF has a lower specific gravity in a gaseous state than oxygen
- LiF may volatilize or sublime when heated, and if it volatilizes, the amount of LiF in the mixture 903 will decrease. This weakens its function as a flux. Therefore, it is necessary to heat while suppressing the volatilization of LiF.
- LiF is not used as the fluorine source, etc.
- Li on the LiCoO2 surface may react with F of the fluorine source to produce LiF, which may volatilize. Therefore, even if a fluorine compound with a melting point higher than LiF is used, it is necessary to suppress the volatilization in the same way.
- the mixture 903 it is preferable to heat the mixture 903 in an atmosphere containing LiF, that is, to heat the mixture 903 in a state where the partial pressure of LiF in the heating furnace is high. By heating in this manner, it is possible to suppress the volatilization of LiF in the mixture 903. It is also preferable to place a lid on the crucible in order to suppress the volatilization of LiF.
- the heating in this process is preferably performed so that the particles of mixture 903 do not stick to each other. If the particles of mixture 903 stick to each other during heating, the contact area with oxygen in the atmosphere decreases, and the route for the added element (e.g., fluorine) to diffuse is blocked, which may result in poor distribution of the added element (e.g., magnesium and fluorine) in the surface layer. In order to promote the reaction with oxygen in the atmosphere, the crucible does not need to be sealed with a lid.
- the added element e.g., fluorine
- the additive element e.g., fluorine
- the additive element e.g., fluorine
- the particles of mixture 903 do not stick to each other.
- the flow rate of the oxygen-containing atmosphere in the kiln When heating in a rotary kiln, it is preferable to control the flow rate of the oxygen-containing atmosphere in the kiln. For example, it is preferable to reduce the flow rate of the oxygen-containing atmosphere, or to first purge the atmosphere and not flow the atmosphere after introducing the oxygen atmosphere into the kiln. Flowing oxygen can cause the fluorine source to evaporate, which is not preferable in terms of maintaining the smoothness of the surface.
- the mixture 903 can be heated in an atmosphere containing LiF by, for example, placing a lid on the container containing the mixture 903. This is similar to the lid placed on a crucible.
- the heating time varies depending on conditions such as the heating temperature, the size of the lithium cobalt oxide obtained in step S14, and the composition. When the lithium cobalt oxide is small, a lower temperature or a shorter heating time may be more preferable than when the lithium cobalt oxide is large.
- the heating temperature is preferably, for example, 650° C. or more and 950° C. or less.
- the heating time is preferably, for example, 3 hours or more and 60 hours or less, more preferably 10 hours or more and 30 hours or less, and even more preferably about 20 hours.
- the cooling time after heating is preferably, for example, 10 hours or more and 50 hours or less.
- the heating temperature is preferably, for example, 650° C. or more and 950° C. or less.
- the heating time is preferably, for example, 1 hour or more and 10 hours or less, and more preferably about 5 hours.
- the cooling time after heating is preferably, for example, 10 hours or more and 50 hours or less.
- Step S34> 19A the heated material is collected to obtain the positive electrode active material 100.
- the collected particles may be sieved.
- the positive electrode active material 100 of one embodiment of the present invention can be produced.
- the positive electrode active material of one embodiment of the present invention has a smooth surface.
- a method for producing a positive electrode active material which is one embodiment of the present invention and is different from the method for producing a positive electrode active material, is described below.
- the method for producing a positive electrode active material which is different from the method for producing a positive electrode active material, is mainly different from the method for producing a positive electrode active material, in the number of times that additive elements are added and the mixing method.
- the description of the method for producing a positive electrode active material can be referred to.
- steps S11 to S15 are carried out in the same manner as in FIG. 19A to prepare lithium cobalt oxide that has undergone initial heating.
- step S20a an additive element A1 source is prepared for adding the additive element A1 to the lithium cobalt oxide that has been subjected to the initial heating.
- the process of preparing the additive element A1 source will be described with reference to FIG.
- Step S21 shown in Fig. 22A will be described.
- the additive element A1 can be selected from the elements exemplified as the additive element A described in step S21 shown in Fig. 19B and used.
- one or more selected from magnesium, fluorine, and calcium can be suitably used as the additive element A1.
- Fig. 22A shows an example in which a magnesium source (Mg source) and a fluorine source (F source) are prepared in step S21 in the case where magnesium and fluorine are selected as the additive element A1.
- Mg source magnesium source
- F source fluorine source
- Steps S21 to S23 shown in FIG. 22A can be performed under the same conditions as steps S21 to S23 shown in FIG. 19B.
- the additive element A1 source (A1 source) can be obtained in step S23.
- steps S31 to S33 shown in FIG. 21 can be performed in the same manner as steps S31 to S33 shown in FIG. 19A.
- step S33 the heated material is collected to produce lithium cobalt oxide containing the additive element A1. To distinguish this from the composite oxide in step S14, this is also called a second composite oxide.
- step S40 shown in Fig. 21 an additive element A2 source used for adding the additive element A2 to the second composite oxide is prepared.
- the steps of preparing the additive element A2 source will be described with reference to Figs. 22B and 22C.
- the additive element A2 can be selected from the elements exemplified as the additive element A described in step S21 shown in Fig. 19C.
- the additive element A2 can be one or more selected from nickel, titanium, boron, zirconium, and aluminum.
- Fig. 22B shows an example in which a nickel source (Ni source) and an aluminum source (Al source) are prepared in the case where nickel and aluminum are selected as the additive element A2.
- Steps S41 to S43 shown in FIG. 22B can be performed under the same conditions as steps S21 to S23 shown in FIG. 19B.
- the additive element A2 source (A2 source) can be obtained in step S43.
- FIG. 22C shows a modified example of the process for preparing the additive element A2 source described with reference to FIG. 22B.
- step S41 shown in FIG. 22C a nickel source (Ni source) and an aluminum source (Al source) are prepared, and in step S42a, they are each crushed independently.
- step S43 multiple additive element A2 sources (A2 sources) are prepared.
- the steps in FIG. 22C differ from FIG. 22B in that the additive elements are crushed independently in step S42a.
- steps S51 to S54 shown in Fig. 21 can be performed under the same conditions as steps S31 to S34 shown in Fig. 19A, and a mixture 904 is obtained in step S52.
- the conditions for step S53 relating to the heating step can be a lower temperature and a shorter time than those for step S33.
- the positive electrode active material 100 of one embodiment of the present invention can be produced in step S54.
- the positive electrode active material of one embodiment of the present invention has a smooth surface.
- the additive element is introduced into lithium cobalt oxide in two parts, additive element A1 and additive element A2.
- the location of each additive element in the depth direction can be changed. For example, it is possible to position additive element A1 so that it has a higher concentration in the surface layer than in the interior, and to position additive element A2 so that it has a higher concentration in the interior than in the surface layer.
- the initial heating shown in this embodiment is performed on lithium cobalt oxide. Therefore, the initial heating is preferably performed under conditions that are lower than the heating temperature for obtaining lithium cobalt oxide and shorter than the heating time for obtaining lithium cobalt oxide.
- the step of adding an additive element to lithium cobalt oxide is preferably performed after the initial heating. This addition step can be divided into two or more steps. Following this order of steps is preferable because it maintains the smoothness of the surface obtained by the initial heating.
- Positive electrode active material 100 with a smooth surface may be more resistant to physical destruction caused by pressure, etc., than positive electrode active material that does not have a smooth surface.
- the positive electrode active material 100 is less likely to be destroyed in a test involving pressure, such as a nail penetration test, which may result in increased safety.
- This embodiment can be used in combination with other embodiments.
- [Positive electrode] 24A shows an example of a cross-sectional view of a positive electrode 503 used in a secondary battery 1004 or the like.
- the positive electrode 503 has a positive electrode active material layer 502 on a positive electrode current collector 501.
- the positive electrode active material layer 502 contains a positive electrode active material 100, a positive electrode active material 562, a conductive material 553, a conductive material 554, and an electrolyte solution 530.
- the positive electrode active material layer 502 also has a binder (not shown).
- the secondary battery preferably has a structure including either the conductive material 553 or the conductive material 554.
- the median diameter (D50) of the positive electrode active material 100 is 1 ⁇ m or more and 50 ⁇ m or less, preferably 5 ⁇ m or more and 30 ⁇ m or less. In order to increase the packing density, it is advisable to add a positive electrode active material 562 having a different median diameter (D50).
- the median diameter (D50) of the positive electrode active material 562 is preferably 1/10 to 1/6 of the median diameter (D50) of the positive electrode active material 100.
- both the positive electrode active material 100 and the positive electrode active material 562 have a shell.
- a positive electrode active material with a shell can increase insulation properties and is less likely to cause thermal runaway.
- FIG. 24A a dotted line is drawn at the boundary between the surface layer and the interior, but the boundary is not necessarily as clear as in FIG. 24A.
- the shell can have the configuration shown in FIG. 5B, etc.
- the positive electrode active material is not limited to FIG. 24A, and for example, either the positive electrode active material 100 or the positive electrode active material 562 may have a shell.
- the positive electrode active material 100 may have the same composition as the positive electrode active material 562, or may have a different composition.
- the same composition includes positive electrode active materials having the same main composition, with the difference being the presence or absence of added elements, etc.
- the different composition includes positive electrode active materials having different main compositions.
- the positive electrode active material 100 and the positive electrode active material 562 have an additive element, and it is particularly preferable that the shell has an additive element.
- the additive element may be unevenly distributed in the shell, or may be sparsely distributed inside. "Unevenly distributed” refers to the additive element being present unevenly or unevenly. Therefore, a state in which the concentration of the additive element is higher toward the shell than the inside is sometimes referred to as the additive element being unevenly distributed in the shell.
- the surface layer may contain the additive element.
- the concentration of the additive element in the surface layer may differ from the concentration of the additive element in the interior, and it is preferable that the concentration of the additive element in the surface layer is higher than the concentration in the interior. This is sometimes called the additive element being unevenly distributed in the surface layer.
- the positive electrode active material 100 and the positive electrode active material 562 are sometimes called positive electrode active material particles, but the positive electrode active material can take a variety of shapes other than particulate.
- FIG. 24B shows a positive electrode 503 having a positive electrode active material in a shape other than particulate.
- the description is omitted because it is the same as FIG. 24A.
- the positive electrode active material 100 and the positive electrode active material 562 shown in FIG. 24A and FIG. 24B are shown as primary particles, but may be secondary particles.
- the positive electrode has a positive electrode active material layer and a positive electrode current collector.
- the positive electrode active material layer has a positive electrode active material, and may have a conductive material (synonymous with a conductive assistant) and a binder.
- a positive electrode active material prepared using the preparation method described in the previous embodiment may be used.
- a positive electrode active material having a relatively small median diameter (D50) and a positive electrode active material having a relatively large median diameter (D50) may be mixed and used.
- the positive electrode active material described in the previous embodiment may also be mixed with other positive electrode active materials.
- positive electrode active material examples include composite oxides having an olivine type crystal structure, a layered rock salt type crystal structure, or a spinel type crystal structure, such as LiFePO4 , LiFeO2 , LiNiO2 , LiMn2O4 , V2O5 , Cr2O5 , and MnO2 .
- LiMn2O4 lithium nickel oxide
- a lithium manganese composite oxide that can be expressed by the composition formula Li a Mn b M c O d can be used.
- the element M is preferably a metal element selected from lithium and manganese, or silicon or phosphorus, and more preferably nickel.
- the composition of metal, silicon, phosphorus, etc. of the entire particle of the lithium manganese composite oxide can be measured using, for example, ICP-MS.
- the composition of oxygen of the entire particle of the lithium manganese composite oxide can be measured using, for example, EDX. In addition, it can be obtained by using valence evaluation of melt gas analysis and XAFS (X-ray absorption fine structure) analysis in combination with ICP-MS analysis.
- the lithium manganese composite oxide refers to an oxide containing at least lithium and manganese, and may contain one or more elements selected from the group consisting of chromium, cobalt, aluminum, nickel, iron, magnesium, molybdenum, zinc, indium, gallium, copper, titanium, niobium, silicon, phosphorus, and the like.
- the conductive material serves to assist the current path between the active material and the current collector, or the current path between a plurality of active materials.
- the conductive material is preferably made of a material having a lower resistance than the active material. Due to its role, the conductive material is also called a conductive assistant or a conductive agent.
- the conductive material is typically a carbon material or a metal material.
- the conductive material is particulate, and examples of particulate conductive materials include carbon black (furnace black, acetylene black, graphite, etc.). Most carbon blacks have a smaller particle size than the positive electrode active material.
- the conductive material may be fibrous, and examples of such fibrous conductive assistants include carbon nanotubes (CNT) and VGCF (registered trademark).
- the conductive material may be sheet-shaped, and an example of a sheet-shaped conductive assistant is multilayer graphene. Sheet-shaped conductive assistants may appear thread-like on the cross section of the positive electrode.
- Particulate conductive materials can penetrate into gaps in the positive electrode active material and are also prone to agglomeration. Therefore, particulate conductive materials can assist the conductive paths between nearby positive electrode active materials. Fibrous conductive materials also have bent regions, but are larger than the positive electrode active materials. Therefore, fibrous conductive materials can assist the conductive paths between distant positive electrode active materials in addition to adjacent positive electrode active materials. In this way, it is preferable to mix two or more shapes of conductive additives.
- the weight of the carbon black in the mixed slurry state should be 1.5 to 20 times, preferably 2 to 9.5 times, that of the multi-layer graphene.
- the carbon black does not aggregate and is easily dispersed. Furthermore, when the mixing ratio of multi-layer graphene and carbon black is within the above range, the electrode density can be made higher than when only carbon black is used as the conductive additive. By increasing the electrode density, the capacity per unit weight can be increased.
- graphene includes multi-layer graphene and multi-graphene.
- graphene has carbon, has a shape such as a plate or sheet, and has a two-dimensional structure formed of six-membered carbon rings.
- the two-dimensional structure formed of six-membered carbon rings may be called a carbon sheet.
- Graphene compounds include graphene oxide, multi-layer graphene oxide, multi-graphene oxide, reduced graphene oxide, reduced multi-layer graphene oxide, reduced multi-graphene oxide, graphene quantum dots, and the like.
- graphene compounds may have functional groups.
- Graphene or graphene compounds preferably have a curved shape.
- Graphene or graphene compounds may be rolled up, and rolled up graphene may be called carbon nanofibers.
- graphene oxide refers to a material that contains carbon and oxygen, has a sheet-like shape, and has functional groups, particularly epoxy groups, carboxy groups, or hydroxy groups.
- reduced graphene oxide refers to a material that has carbon and oxygen, has a sheet-like shape, and has a two-dimensional structure formed of six-membered carbon rings. Although reduced graphene oxide can function as a single sheet, multiple sheets may be stacked. Reduced graphene oxide preferably has a portion where the carbon concentration is greater than 80 atomic% and the oxygen concentration is 2 atomic% or more and 15 atomic% or less. By setting such carbon and oxygen concentrations, it can function as a highly conductive conductive material even in small amounts. In addition, reduced graphene oxide preferably has an intensity ratio G/D of the G band and the D band in the Raman spectrum of 1 or more. Reduced graphene oxide with such an intensity ratio can function as a highly conductive conductive material even in small amounts.
- Fluorine-containing graphene may be used as the graphene compound.
- the fluorine in the graphene compound may be adsorbed on the surface.
- the fluorine-containing graphene may be produced by contacting graphene with a fluorine compound (called fluorination treatment).
- fluorination treatment fluorine (F 2 ) or a fluorine compound may be used.
- fluorine compound hydrogen fluoride, halogen fluoride (ClF 3 , IF 5 , etc.), gaseous fluoride (BF 3 , NF 3 , PF 5 , SiF 4 , SF 6 , etc.), metal fluoride (LiF, NiF 2 , AlF 3 , MgF 2 , etc.), etc. are preferable.
- gaseous fluoride is preferably used, and the gaseous fluoride may be diluted with an inert gas.
- the temperature of the fluorination treatment is preferably room temperature, but is preferably 0° C. or more and 250° C. or less, which includes the room temperature. When the fluorination treatment is performed at 0° C. or more, fluorine can be adsorbed on the surface of the graphene.
- the graphene compound may have excellent electrical properties such as high electrical conductivity, and excellent physical properties such as high flexibility and high mechanical strength.
- the graphene compound may have a sheet-like shape.
- the graphene compound may have a curved surface, which allows for surface contact with low contact resistance.
- even if the graphene compound is thin it may have very high electrical conductivity, and a conductive path can be efficiently formed in the active material layer with a small amount. Therefore, by using the graphene compound as a conductive material, the contact area between the active material and the conductive material can be increased.
- the graphene compound may cover 80% or more of the area of the active material. It is preferable that the graphene compound is wrapped around at least a part of the active material.
- the graphene compound is overlapped on at least a part of the active material. It is also preferable that the shape of the graphene compound matches at least a part of the shape of the active material.
- the shape of the active material refers to, for example, the unevenness of a single active material or the unevenness formed by multiple active materials. It is also preferable that the graphene compound surrounds at least a part of the active material.
- the graphene compound may have holes.
- an active material with a small particle size for example an active material with a particle size of 1 ⁇ m or less, more conductive paths are required to connect the active materials together. In such cases, it is preferable to use a graphene compound, which can efficiently form conductive paths even in small amounts.
- Rapid charging and discharging refers to, for example, charging and discharging at 200 mA/g, 400 mA/g, or 1000 mA/g or more.
- sheet-like graphene or graphene compounds are dispersed approximately uniformly inside the positive electrode active material layer 502.
- the graphene or graphene compounds are shown in bold lines, but in reality, they are thin films having the thickness of a single layer or multiple layers of carbon molecules.
- the multiple graphene or graphene compounds are formed so as to partially cover the multiple granular positive electrode active materials 100 or to be attached to the surfaces of the multiple granular positive electrode active materials 100, and are therefore in surface contact with each other.
- a mesh-like graphene compound sheet (hereinafter referred to as a graphene compound net or graphene net) can be formed by bonding multiple graphenes or graphene compounds together.
- the graphene net can also function as a binder that bonds the active materials together. Therefore, the amount of binder can be reduced or no binder can be used, and the ratio of active material to the electrode volume and electrode weight can be improved. In other words, the discharge capacity of the secondary battery can be increased.
- graphene oxide As the graphene or graphene compound, mix it with the active material to form a layer that becomes the positive electrode active material layer 502, and then reduce it. In other words, it is preferable that the completed active material layer has reduced graphene oxide.
- graphene oxide which has extremely high dispersibility in a polar solvent, to form the graphene or graphene compound, the graphene or graphene compound can be dispersed approximately uniformly inside the positive electrode active material layer 502.
- the graphene or graphene compound remaining in the positive electrode active material layer 502 is dispersed to such an extent that it partially overlaps and is in surface contact with each other, thereby forming a three-dimensional conductive path.
- the reduction of the graphene oxide may be performed, for example, by heat treatment or by using a reducing agent.
- graphene or graphene compounds enable surface contact with low contact resistance, so that a smaller amount than that of a normal conductive material can improve the electrical conductivity between the granular positive electrode active material 100 and graphene or graphene compounds. Therefore, the ratio of the positive electrode active material 100 in the positive electrode active material layer 502 can be increased. This can increase the discharge capacity of the secondary battery.
- the active material can be entirely covered with a conductive graphene compound as a coating, and further conductive paths can be formed between the active material particles with the graphene compound.
- a material used in forming the graphene compound may be mixed into the positive electrode active material layer 502 together with the graphene compound.
- particles used as a catalyst in forming the graphene compound may be mixed with the graphene compound.
- catalysts used in forming the graphene compound include particles having silicon oxide (SiO 2 , SiO x (x ⁇ 2)), aluminum oxide, iron, nickel, ruthenium, iridium, platinum, copper, germanium, or the like.
- the particles preferably have a median diameter (D50) of 1 ⁇ m or less, and more preferably 100 nm or less.
- acetylene black (AB) can be used as a conductive material. Fluorine-containing acetylene black may also be used. The fluorine in the fluorine-containing acetylene black is preferably adsorbed on the surface. Fluorine-containing acetylene black can be produced by contacting acetylene black with a fluorine compound (called a fluorination treatment). The fluorination treatment described for graphene can be applied to acetylene black.
- carbon fiber materials also referred to as carbon nanotubes, or CNTs
- CNTs carbon nanotubes
- Fluorine-containing carbon nanotubes may also be used.
- the fluorine in the fluorine-containing carbon nanotubes is preferably adsorbed on the surface.
- Fluorine-containing carbon nanotubes can be produced by contacting carbon nanotubes with a fluorine compound (called a fluorination process).
- the fluorination process described for graphene can also be applied to carbon nanotubes.
- the binder is necessary to strengthen the adhesion of the powdered active material without covering the surface of the active material. Furthermore, the binder must be adhesive to the current collector. In other words, it is preferable to use a material having an adhesive component as the binder. Furthermore, in consideration of the expansion of the active material, the binder should be sufficiently flexible and should be able to respond to changes in the state of the active material. The binder must also be compatible with the electrolyte. Furthermore, since extremely strong oxidation and reduction reactions occur in secondary batteries, a binder that does not deteriorate or has low reactivity to these reactions is desired.
- the binder it is preferable to use a rubber material such as styrene-butadiene rubber (SBR), styrene-isoprene-styrene rubber, acrylonitrile-butadiene rubber, butadiene rubber, or ethylene-propylene-diene copolymer.
- SBR styrene-butadiene rubber
- fluororubber can be used as the binder.
- a water-soluble polymer as the binder.
- polysaccharides can be used as the water-soluble polymer.
- the polysaccharide one or more of cellulose derivatives such as carboxymethyl cellulose (CMC), methyl cellulose, ethyl cellulose, hydroxypropyl cellulose, diacetyl cellulose, regenerated cellulose, and starch can be used.
- CMC carboxymethyl cellulose
- methyl cellulose methyl cellulose
- ethyl cellulose methyl cellulose
- hydroxypropyl cellulose diacetyl cellulose
- regenerated cellulose regenerated cellulose
- polystyrene polymethyl acrylate, polymethyl methacrylate (polymethyl methacrylate, PMMA), sodium polyacrylate, polyvinyl alcohol (PVA), polyethylene oxide (PEO), polypropylene oxide, polyimide, polyvinyl chloride, polytetrafluoroethylene, polyethylene, polypropylene, polyisobutylene, polyethylene terephthalate, nylon, polyvinylidene fluoride (PVDF), polyacrylonitrile (PAN), ethylene propylene diene polymer, polyvinyl acetate, and nitrocellulose as the binder.
- PVDF polyvinylidene fluoride
- PAN polyacrylonitrile
- the binders may be a combination of more than one of the above.
- a material with particularly excellent viscosity adjustment effect may be used in combination with other materials.
- rubber materials have excellent adhesive strength and/or elasticity, it may be difficult to adjust the viscosity when mixed with a solvent. In such cases, it is preferable to mix with a material with particularly excellent viscosity adjustment effect.
- a water-soluble polymer may be used as a material with particularly excellent viscosity adjustment effect.
- the above-mentioned polysaccharides for example, carboxymethylcellulose (CMC), methylcellulose, ethylcellulose, hydroxypropylcellulose, and diacetylcellulose, cellulose derivatives such as regenerated cellulose, starch, etc. may be used.
- CMC carboxymethylcellulose
- methylcellulose methylcellulose
- ethylcellulose methylcellulose
- hydroxypropylcellulose hydroxypropylcellulose
- diacetylcellulose cellulose derivatives such as regenerated cellulose, starch, etc.
- the solubility of cellulose derivatives such as carboxymethylcellulose can be increased by converting them into salts, such as sodium salt and ammonium salt of carboxymethylcellulose, making them more effective as viscosity adjusters. Increasing the solubility can also increase the dispersibility of the active material and other components when preparing the electrode slurry.
- the cellulose and cellulose derivatives used as electrode binders include their salts.
- Water-soluble polymers stabilize the viscosity by dissolving in water, and can stably disperse active materials and other materials combined as binders, such as styrene-butadiene rubber, in an aqueous solution.
- binders such as styrene-butadiene rubber
- binders such as styrene-butadiene rubber
- Many cellulose derivatives, such as carboxymethyl cellulose have functional groups such as hydroxyl and carboxyl groups, and because they have functional groups, the polymers are expected to interact with each other and widely cover the surface of the active material.
- a passive film is a film with no electrical conductivity or a film with extremely low electrical conductivity.
- a passive film when a passive film is formed on the surface of the active material, it can suppress decomposition of the electrolyte at the battery reaction potential. Furthermore, it is even more desirable for the passive film to suppress electrical conductivity while still being able to conduct lithium ions.
- the current collector As the current collector, a material having high electrical conductivity, such as metals such as stainless steel, gold, platinum, aluminum, and titanium, and alloys thereof, can be used. In addition, it is preferable that the material used for the positive electrode current collector does not dissolve at the potential of the positive electrode. In addition, an aluminum alloy to which an element that improves heat resistance, such as silicon, titanium, neodymium, scandium, or molybdenum, is added can be used. In addition, it may be formed of a metal element that reacts with silicon to form a silicide.
- Examples of metal elements that react with silicon to form a silicide include zirconium, titanium, hafnium, vanadium, niobium, tantalum, chromium, molybdenum, tungsten, cobalt, and nickel.
- the current collector can be appropriately used in a shape such as a foil, plate, sheet, mesh, punched metal, or expanded metal. It is preferable to use a current collector having a thickness of 5 ⁇ m or more and 30 ⁇ m or less.
- the negative electrode includes a negative electrode active material layer and a negative electrode current collector.
- the negative electrode active material layer may include a conductive material and a binder.
- Negative electrode active material for example, an alloy-based material and/or a carbon-based material can be used.
- an element capable of carrying out a charge/discharge reaction by alloying/dealloying reaction with lithium can be used.
- a material containing one or more selected from silicon, tin, gallium, aluminum, germanium, lead, antimony, bismuth, silver, zinc, cadmium, indium, etc. can be used.
- Such elements have a larger charge/discharge capacity than carbon, and silicon in particular has a high theoretical capacity of 4200 mAh/g. For this reason, it is preferable to use silicon as the negative electrode active material. Compounds containing these elements may also be used.
- Examples include SiO, Mg2Si , Mg2Ge , SnO, SnO2 , Mg2Sn , SnS2 , V2Sn3 , FeSn2, CoSn2 , Ni3Sn2 , Cu6Sn5, Ag3Sn , Ag3Sb, Ni2MnSb, CeSb3, LaSn3, La3Co2Sn7 , CoSb3 , InSb , SbSn , etc.
- elements capable of carrying out charge/discharge reactions by alloying/dealloying reactions with lithium, and compounds containing such elements, etc. are sometimes called alloy - based materials.
- SiO refers to, for example, silicon monoxide.
- SiO can be expressed as SiO x .
- x is preferably 0.2 or more and 1.5 or less, more preferably 0.3 or more and 1.2 or less, or preferably 0.2 or more and 1.2 or less, or preferably 0.3 or more and 1.5 or less.
- carbon-based materials examples include graphite, easily graphitizable carbon (soft carbon), non-graphitizable carbon (hard carbon), carbon nanotubes, graphene, carbon black, etc.
- Examples of graphite include artificial graphite and natural graphite.
- Examples of artificial graphite include mesocarbon microbeads (MCMB), coke-based artificial graphite, and pitch-based artificial graphite.
- MCMB mesocarbon microbeads
- spherical graphite having a spherical shape can be used as the artificial graphite.
- MCMB may have a spherical shape, which is preferable.
- it is relatively easy to reduce the surface area of MCMB which may be preferable.
- Examples of natural graphite include flake graphite and spheroidized natural graphite.
- graphite When lithium ions are inserted into graphite (when a lithium-graphite intercalation compound is formed), graphite exhibits a low potential (0.05 V to 0.3 V vs. Li/Li + ) similar to that of lithium metal. This allows lithium ion secondary batteries to exhibit a high operating voltage. Furthermore, graphite is preferable because it has the advantages of a relatively high charge/discharge capacity per unit volume, a relatively small volume expansion, low cost, and high safety compared to lithium metal.
- oxides such as titanium dioxide ( TiO2 ), lithium titanium oxide ( Li4Ti5O12 ), lithium graphite intercalation compound ( LixC6 ), niobium pentoxide ( Nb2O5 ), tungsten dioxide ( WO2 ), and molybdenum dioxide ( MoO2 ) can be used as the negative electrode active material.
- TiO2 titanium dioxide
- Li4Ti5O12 lithium titanium oxide
- LixC6 lithium graphite intercalation compound
- Nb2O5 niobium pentoxide
- WO2 tungsten dioxide
- MoO2 molybdenum dioxide
- Li2.6Co0.4N is preferable because it shows a large charge/discharge capacity (900mAh/g, 1890mAh/ cm3 ).
- the nitride of lithium and a transition metal When a nitride of lithium and a transition metal is used, lithium ions are contained in the negative electrode active material, and therefore it is preferable that the nitride of lithium and a transition metal is combined with a material that does not contain lithium ions as a positive electrode active material, such as V 2 O 5 or Cr 3 O 8. Even when a material that contains lithium ions is used as the positive electrode active material, the nitride of lithium and a transition metal can be used as the negative electrode active material by desorbing the lithium ions contained in the positive electrode active material in advance.
- a material that undergoes a conversion reaction can be used as the negative electrode active material.
- a transition metal oxide that does not form an alloy with lithium such as cobalt oxide (CoO), nickel oxide (NiO), or iron oxide (FeO) may be used as the negative electrode active material.
- materials that undergo a conversion reaction include oxides such as Fe2O3 , CuO, Cu2O , RuO2 , and Cr2O3 , sulfides such as CoS0.89 , NiS, and CuS, nitrides such as Zn3N2 , Cu3N , and Ge3N4 , phosphides such as NiP2 , FeP2 , and CoP3 , and fluorine compounds such as FeF3 and BiF3 .
- oxides such as Fe2O3 , CuO, Cu2O , RuO2 , and Cr2O3
- sulfides such as CoS0.89 , NiS, and CuS
- nitrides such as Zn3N2 , Cu3N , and Ge3N4
- phosphides such as NiP2 , FeP2 , and CoP3
- fluorine compounds such as FeF3 and BiF3 .
- the conductive material and binder that can be used in the negative electrode active material layer can be the same materials as the conductive material and binder that can be used in the positive electrode active material layer.
- the negative electrode current collector may be made of the same material as the positive electrode current collector, but it is preferable that the negative electrode current collector is made of a material that does not form an alloy with carrier ions such as lithium.
- An electrolyte solution can be used as one form of electrolyte.
- the electrolyte solution has a solvent and a lithium salt.
- the above-mentioned solvent also called the electrolyte solvent
- the organic solvent contained in the electrolyte can be a mixed organic solvent containing a fluorinated cyclic carbonate or a fluorinated chain carbonate. Furthermore, it is preferable that the mixed organic solvent contains both a fluorinated cyclic carbonate and a fluorinated chain carbonate. Both the fluorinated cyclic carbonate and the fluorinated chain carbonate have a substituent that exhibits electron-withdrawing properties, and are preferable because they lower the solvation energy of lithium ions. Therefore, both the fluorinated cyclic carbonate and the fluorinated chain carbonate are suitable for the electrolyte, and these mixed organic solvents are suitable.
- fluorinated cyclic carbonate for example, fluoroethylene carbonate (fluoroethylene carbonate, FEC, F1EC), difluoroethylene carbonate (DFEC, F2EC), trifluoroethylene carbonate (F3EC), or tetrafluoroethylene carbonate (F4EC) can be used.
- DFEC has isomers such as cis-4,5 and trans-4,5. Since all of the fluorinated cyclic carbonates have substituents that exhibit electron-withdrawing properties, it is believed that the solvation energy of lithium ions is low.
- the following structural formula (H10) is the structural formula of FEC.
- the electron-withdrawing substituent is an F group.
- Methyl 3,3,3-trifluoropropionate is an example of a fluorinated chain carbonate.
- the following structural formula (H22) is the structural formula of methyl 3,3,3-trifluoropropionate.
- the abbreviation for methyl 3,3,3-trifluoropropionate is "MTFP".
- MTFP the electron-withdrawing substituent is a CF3 group.
- FEC is one of the cyclic carbonates and has a high dielectric constant, so when used in an organic solvent, it has the effect of promoting the dissociation of lithium salts.
- FEC has a substituent that shows electron-withdrawing properties, so it is easier to desolvate with lithium ions than ethylene carbonate (EC).
- the solvation energy of lithium ions in FEC is smaller than that of ethylene carbonate (EC), which does not have a substituent that shows electron-withdrawing properties. Therefore, it is easier to separate lithium ions on the surface of the positive electrode active material and the surface of the negative electrode active material, and the internal resistance of the secondary battery can be reduced.
- FEC is thought to have a deep highest occupied molecular orbital (HOMO), and if the HOMO level is deep, it is less likely to be oxidized and the oxidation resistance is improved.
- HOMO deep highest occupied molecular orbital
- FEC has a high viscosity. Therefore, it is recommended to use a mixed organic solvent that further contains MTFP in the electrolyte, rather than just FEC.
- MTFP is a type of chain carbonate, and can reduce the viscosity of the electrolyte, or maintain the viscosity at room temperature (typically 25°C) even at low temperatures (typically 0°C).
- MTFP has a smaller solvation energy than methyl propionate (abbreviated as "MP"), which does not have a substituent that exhibits electron-withdrawing properties, so it may form a solvate with lithium ions when used in an electrolyte.
- MP methyl propionate
- the mixed organic solvent containing FEC and MTFP having such physical properties is taken as 100 vol%, it is preferable to mix them so that the volume ratio is x:100-x (where 5 ⁇ x ⁇ 30, preferably 10 ⁇ x ⁇ 20). In other words, it is preferable to mix them so that there is more MTFP than FEC in the mixed organic solvent.
- ionic liquids room-temperature molten salts
- the ionic liquid is composed of a cation and an anion, and includes an organic cation and an anion.
- Examples of the organic cation used in the electrolyte include aliphatic onium cations such as quaternary ammonium cations, tertiary sulfonium cations, and quaternary phosphonium cations, and aromatic cations such as imidazolium cations and pyridinium cations.
- Examples of the anion used in the electrolyte include monovalent amide anions, monovalent methide anions, fluorosulfonate anions, perfluoroalkylsulfonate anions, tetrafluoroborate anions, perfluoroalkylborate anions, hexafluorophosphate anions, and perfluoroalkylphosphate anions.
- the above-mentioned electrolyte solvent preferably has a low content of granular dust or molecules other than the constituent molecules of the organic solvent (hereinafter, also referred to simply as "impurities", including oxygen (O 2 ), water (H 2 O) or moisture) and is highly purified.
- impurities including oxygen (O 2 ), water (H 2 O) or moisture
- the amount of the above impurities contained in the electrolyte solvent is 100 ppm or less, preferably 50 ppm or less, and more preferably less than 10 ppm.
- moisture can be detected by Karl Fischer titration.
- the above-mentioned organic solvent has almost no peaks due to impurities confirmed by NMR measurement or the like. Almost no peaks can be confirmed includes that the ratio of the integrated area of the peak due to the impurity to the integrated area of the peak due to the main component (simply called integral ratio) is 0.005 or less, preferably 0.002 or less.
- integral ratio the ratio of the integrated area of the peak due to the impurity to the integrated area of the peak due to the main component
- integral ratio 0.005 or less, preferably 0.002 or less.
- the central peak can be 1.94 ppm.
- lithium salts also called electrolytes
- examples of lithium salts (also called electrolytes) dissolved in the solvent include LiPF6 , LiClO4 , LiAsF6 , LiBF4 , LiAlCl4 , LiSCN, LiBr, LiI , Li2SO4, Li2B10Cl10 , Li2B12Cl12, LiCF3SO3 , LiC4F9SO3 , LiC ( CF3SO2 ) 3 , LiC ( C2F5SO2 ) 3 , LiN ( CF3SO2 )2 , LiN ( C4F9SO2 ) ( CF3SO2 ) , LiN ( C2F5SO2 ) .
- Lithium salts such as LiPF6 , LiBF4, etc. can be used alone or in any combination and ratio of two or more of these.
- the lithium salt is preferably 0.5 mol/L or more and 3.0 mol/L or less relative to the solvent.
- the safety of the lithium ion secondary battery is improved by using fluorides such as LiPF6 , LiBF4 , etc.
- VC vinylene carbonate
- PS propane sultone
- TB tert-butylbenzene
- FEC fluoroethylene carbonate
- LiBOB lithium bis(oxalate)borate
- dinitrile compounds such as succinonitrile and adiponitrile to the electrolyte.
- concentration of the added material is preferably 0.1 wt % or more and 5 wt % or less with respect to the entire solvent.
- VC or LiBOB is particularly preferable because it is easy to form a good coating portion.
- Gel electrolyte As the gel electrolyte, a polymer gel in which a polymer is swollen with an electrolytic solution may be used. By using a polymer gel electrolyte, a semi-solid electrolyte layer can be provided, and safety against leakage and the like can be improved. In addition, it is possible to make the secondary battery thinner and lighter.
- Polymers that can be gelled include silicone gel, acrylic gel, acrylonitrile gel, polyethylene oxide gel, polypropylene oxide gel, fluorine-based polymer gel, etc.
- polymer for example, a polymer having a polyalkylene oxide structure such as polyethylene oxide (PEO), PVDF, polyacrylonitrile, etc., and copolymers containing these can be used.
- PEO polyethylene oxide
- PVDF-HFP which is a copolymer of PVDF and hexafluoropropylene (HFP)
- the polymer formed may have a porous shape.
- Solid electrolyte instead of the electrolyte, a solid electrolyte having an inorganic material such as a sulfide or oxide, or a solid electrolyte having a polymer material such as a PEO (polyethylene oxide) can be used.
- a solid electrolyte When a solid electrolyte is used, the installation of a separator and/or a spacer becomes unnecessary.
- the entire battery can be solidified, there is no risk of leakage, and safety is dramatically improved.
- the secondary battery preferably has a separator.
- the separator may be made of, for example, paper, nonwoven fabric, glass fiber, ceramics, or synthetic fibers using nylon (polyamide), vinylon (polyvinyl alcohol fiber), polyester, acrylic, polyolefin, or polyurethane.
- the separator is preferably processed into an envelope shape and disposed so as to encase either the positive electrode or the negative electrode.
- the separator may have a multi-layer structure.
- an organic material film such as polypropylene or polyethylene may be coated with a ceramic material, a fluorine material, a polyamide material, or a mixture of these.
- the ceramic material for example, aluminum oxide particles or silicon oxide particles may be used.
- the fluorine material for example, PVDF or polytetrafluoroethylene may be used.
- the polyamide material for example, nylon or aramid (meta-aramid or para-aramid) may be used.
- Coating with ceramic-based materials improves oxidation resistance, suppressing the deterioration of the separator during high-voltage charging and improving the reliability of the secondary battery. Coating with fluorine-based materials also makes it easier for the separator and electrodes to adhere to each other, improving output characteristics. Coating with polyamide-based materials, especially aramid, improves heat resistance, improving the safety of the secondary battery.
- both sides of a polypropylene film may be coated with a mixture of aluminum oxide and aramid.
- the surface of the polypropylene film that comes into contact with the positive electrode may be coated with a mixture of aluminum oxide and aramid, and the surface that comes into contact with the negative electrode may be coated with a fluorine-based material.
- the safety of the secondary battery can be maintained even if the overall thickness of the separator is thin, so the discharge capacity per volume of the secondary battery can be increased.
- the exterior body of the secondary battery can be made of, for example, a metal material such as aluminum and/or a resin material.
- a film-shaped exterior body can also be used.
- As the exterior body a three-layer structure film can be used in which a thin metal film having excellent flexibility such as aluminum, stainless steel, copper, nickel, etc. is provided on a film made of a material such as polyethylene, polypropylene, polycarbonate, ionomer, polyamide, etc., and a synthetic resin such as polyamide resin or polyester resin in a fibrous form is provided on the thin metal film as the outer surface of the exterior body. Fibers made of polyamide resin are called nylon.
- Such a three-layer structure film containing aluminum is sometimes referred to as an aluminum laminate film.
- Fig. 25A is an external view of a coin-type (single-layer flat) secondary battery
- Fig. 25B is a cross-sectional view thereof.
- Coin-type secondary batteries are mainly used in small electronic devices. In this specification and the like, the coin-type battery includes a button-type battery.
- the coin-type secondary battery 300 has a positive electrode can 301, which also serves as a positive electrode terminal, and a negative electrode can 302, which also serves as a negative electrode terminal, which are insulated and sealed with a gasket 303 made of polypropylene or the like.
- the positive electrode 304 is formed of a positive electrode current collector 305 and a positive electrode active material layer 306 provided in contact with the positive electrode current collector.
- the negative electrode 307 is formed of a negative electrode current collector 308 and a negative electrode active material layer 309 provided in contact with the negative electrode current collector.
- a separator 310 is provided between the positive electrode 304 and the negative electrode 307.
- each of the positive electrode 304 and negative electrode 307 used in the coin-type secondary battery 300 is formed only on one side of the current collector.
- the positive electrode can 301 and the negative electrode can 302 can be made of metals such as nickel, aluminum, titanium, etc., which are corrosion-resistant to the electrolyte, or alloys of these metals and/or alloys of these metals with other metals (e.g., stainless steel, etc.). In order to prevent corrosion by the electrolyte, it is preferable to coat them with nickel and/or aluminum, etc.
- the positive electrode can 301 is electrically connected to the positive electrode 304
- the negative electrode can 302 is electrically connected to the negative electrode 307.
- the negative electrode 307, positive electrode 304, and separator 310 are impregnated with an electrolyte, and as shown in FIG. 25B, the positive electrode can 301 is placed on the bottom, and the positive electrode 304, separator 310, negative electrode 307, and negative electrode can 302 are stacked in this order, and the positive electrode can 301 and the negative electrode can 302 are crimped together via a gasket 303 to produce a coin-shaped secondary battery 300.
- FIG. 25C When a secondary battery using lithium is regarded as a closed circuit, the movement of lithium ions and the flow of current are in the same direction.
- the anode (positive electrode) and the cathode (negative electrode) are interchanged during charging and discharging, and the oxidation reaction and the reduction reaction are interchanged, so the electrode with a high reaction potential is called the positive electrode, and the electrode with a low reaction potential is called the negative electrode.
- the positive electrode will be called the “positive electrode” or “+ electrode (plus electrode),” and the negative electrode will be called the “negative electrode” or “- electrode (minus electrode).”
- the terms anode (positive electrode) and cathode (negative electrode) related to the oxidation reaction and reduction reaction are used, the terms will be reversed during charging and discharging, which may cause confusion. Therefore, the terms anode (positive electrode) and cathode (negative electrode) will not be used in this specification. If the terms anode and cathode are used, it should be specified whether they are used during charging or discharging, and also whether they correspond to a positive electrode (plus pole) or a negative electrode (minus pole).
- a charger is connected to the two terminals shown in FIG. 25C, and the secondary battery 300 is charged. As the secondary battery 300 is charged, the potential difference between the electrodes increases.
- Fig. 26A shows an external view of a cylindrical secondary battery 600.
- Fig. 26B is a schematic diagram showing a cross section of the cylindrical secondary battery 600.
- the cylindrical secondary battery 600 has a positive electrode cap (battery lid) 601 on the top surface, and a battery can (external can) 602 on the side and bottom surfaces.
- the positive electrode cap 601 and the battery can 602 are insulated by a gasket (insulating packing) 610.
- a battery element Inside the hollow cylindrical battery can 602, a battery element is provided in which a strip-shaped positive electrode 604 and a negative electrode 606 are wound with a separator 605 sandwiched therebetween. Although not shown, the battery element is wound around a center pin. One end of the battery can 602 is closed and the other end is open.
- metals such as nickel, aluminum, and titanium that are resistant to corrosion by the electrolyte, or alloys of these and/or alloys of these with other metals (e.g., stainless steel, etc.) can be used.
- the battery element in which the positive electrode, negative electrode, and separator are wound is sandwiched between a pair of opposing insulating plates 608 and 609.
- a nonaqueous electrolyte (not shown) is injected into the inside of the battery can 602 in which the battery element is provided.
- the non-aqueous electrolyte can be the same as that used in coin-type secondary batteries.
- a positive electrode terminal (positive electrode current collector lead) 603 is connected to the positive electrode 604, and a negative electrode terminal (negative electrode current collector lead) 607 is connected to the negative electrode 606.
- the positive electrode terminal 603 can be made of a metal material such as aluminum.
- the negative electrode terminal 607 can be made of a metal material such as copper.
- the positive electrode terminal 603 is resistance-welded to a safety valve mechanism 612, and the negative electrode terminal 607 is resistance-welded to the bottom of the battery can 602.
- the safety valve mechanism 612 is electrically connected to the positive electrode cap 601 via a PTC (Positive Temperature Coefficient) element 611.
- the safety valve mechanism 612 cuts off the electrical connection between the positive electrode cap 601 and the positive electrode 604 when the rise in the internal pressure of the battery exceeds a predetermined threshold value.
- the PTC element 611 is a thermosensitive resistor whose resistance increases when the temperature rises, and the increase in resistance limits the amount of current to prevent abnormal heat generation. Barium titanate (BaTiO 3 )-based semiconductor ceramics or the like can be used for the PTC element.
- multiple secondary batteries 600 may be sandwiched between conductive plates 613 and 614 to form a module 615.
- the multiple secondary batteries 600 may be connected in parallel, in series, or in parallel and then further in series.
- FIG 26D is a top view of the module 615.
- the conductive plate 613 is shown with a dotted line.
- the module 615 may have a conductor 616 that electrically connects the multiple secondary batteries 600.
- the conductive plate 613 can be superimposed on the conductor 616.
- a temperature control device 617 may be provided between the multiple secondary batteries 600. When the secondary battery 600 is overheated, it can be cooled by the temperature control device 617, and when the secondary battery 600 is too cold, it can be heated by the temperature control device 617. Therefore, the performance of the module 615 is less susceptible to the influence of the outside air temperature. It is preferable that the heat medium of the temperature control device 617 is insulating and non-flammable.
- the secondary battery 913 shown in FIG. 27A has a wound body 950 with terminals 951 and 952 provided inside the housing 930.
- the wound body 950 is impregnated with an electrolyte inside the housing 930.
- the terminal 952 is in contact with the housing 930, and the terminal 951 is not in contact with the housing 930 by using an insulating material or the like.
- the housing 930 is shown separated for convenience, but in reality, the wound body 950 is covered by the housing 930, and the terminals 951 and 952 extend outside the housing 930.
- the housing 930 can be made of a metal material (e.g., aluminum) or a laminate of the metal material and a resin material.
- the housing 930 shown in FIG. 27A may be formed from a plurality of materials.
- the secondary battery 913 shown in FIG. 27B has housings 930a and 930b bonded together, and a wound body 950 is provided in the area surrounded by the housings 930a and 930b.
- the housing 930a can be made of a metal material (e.g., aluminum, etc.) or a laminate of the metal material and a resin material.
- the housing 930b can be made of a metal material (e.g., aluminum, etc.) or a laminate of the metal material and a resin material.
- the wound body 950 has a negative electrode 931, a positive electrode 932, and a separator 933.
- the wound body 950 is a wound body in which the negative electrode 931 and the positive electrode 932 are stacked on top of each other with the separator 933 in between, and the laminated sheet is wound. Note that the stack of the negative electrode 931, the positive electrode 932, and the separator 933 may be stacked multiple times.
- Figure 29A shows an example of a wearable device.
- Wearable devices use secondary batteries as a power source. Furthermore, when used by a user at home or outdoors, there is a demand for wearable devices that can be charged wirelessly as well as via wired charging, with an exposed connector, in order to improve splash-proof, water-resistant, or dust-proof performance.
- a secondary battery according to one embodiment of the present invention can be mounted on a glasses-type device 4000 as shown in FIG. 29A.
- the glasses-type device 4000 has a frame 4000a and a display unit 4000b.
- the glasses-type device 4000 can be made lightweight, well-balanced in weight, and capable of long continuous use.
- a configuration can be realized that can accommodate space-saving features associated with a smaller housing.
- the headset type device 4001 can be equipped with a secondary battery which is one embodiment of the present invention.
- the headset type device 4001 has at least a microphone unit 4001a, a flexible pipe 4001b, and an earphone unit 4001c.
- a secondary battery can be provided in the flexible pipe 4001b and/or the earphone unit 4001c.
- the secondary battery according to one embodiment of the present invention can be mounted on the device 4002 that can be directly attached to the body.
- the secondary battery 4002b can be provided inside the thin housing 4002a of the device 4002.
- the secondary battery according to one embodiment of the present invention can be mounted on the device 4003 that can be attached to clothing.
- the secondary battery 4003b can be provided inside the thin housing 4003a of the device 4003.
- the belt-type device 4006 can be equipped with a secondary battery according to one embodiment of the present invention.
- the belt-type device 4006 has a belt portion 4006a and a wireless power receiving portion 4006b, and a secondary battery can be mounted inside the belt portion 4006a.
- a configuration that can accommodate space saving associated with a miniaturized housing can be realized.
- the wristwatch device 4005 can be equipped with a secondary battery according to one embodiment of the present invention.
- the wristwatch device 4005 has a display portion 4005a and a belt portion 4005b, and a secondary battery can be provided in the display portion 4005a or the belt portion 4005b.
- a configuration that can accommodate space saving associated with a miniaturized housing can be realized.
- the display unit 4005a can display not only the time but also various other information such as incoming emails and phone calls.
- the wristwatch type device 4005 is a wearable device that is worn directly on the arm, it may be equipped with sensors that measure the user's pulse, blood pressure, etc. Data on the user's amount of exercise and health can be accumulated to manage the user's health.
- Figure 29B shows an oblique view of the wristwatch device 4005 removed from the wrist.
- FIG. 29C shows a state in which a secondary battery 913 is built in.
- the secondary battery 913 is the secondary battery described in embodiment 4.
- the secondary battery 913 is provided in a position overlapping with the display portion 4005a, and is small and lightweight.
- Figure 29D shows an example of a wireless earphone.
- the wireless earphone is shown having a pair of main bodies 4100a and 4100b, but this does not necessarily have to be a pair.
- the main bodies 4100a and 4100b each have a driver unit 4101, an antenna 4102, and a secondary battery 4103. They may also have a display unit 4104. They also preferably have a substrate on which a circuit such as a wireless IC is mounted, a charging terminal, and the like. They may also have a microphone.
- the case 4110 has a secondary battery 4111. It also preferably has a board on which circuits such as a wireless IC and a charging control IC are mounted, and a charging terminal. It may also have a display unit, buttons, etc.
- the main units 4100a and 4100b can wirelessly communicate with other electronic devices such as smartphones. This allows sound data and the like sent from other electronic devices to be played on the main units 4100a and 4100b. Furthermore, if the main units 4100a and 4100b have a microphone, the sound picked up by the microphone can be sent to the other electronic device, and the sound data after processing by the electronic device can be sent back to the main units 4100a and 4100b for playback. This allows the device to be used as a translator, for example.
- the secondary battery 4111 in the case 4110 can charge the secondary battery 4103 in the main body 4100a and the main body 4100b.
- the coin-type secondary battery, the cylindrical secondary battery, or the like described in the previous embodiment can be used as the secondary battery 4111 and the secondary battery 4103.
- the secondary battery using the positive electrode active material 100 obtained in embodiment 1 as the positive electrode has a high energy density, and by using the secondary battery 4103 and the secondary battery 4111 as the positive electrode, a configuration that can accommodate space saving associated with miniaturization of wireless earphones can be realized.
- FIG 30A shows an example of a cleaning robot.
- the cleaning robot 6300 has a display unit 6302 arranged on the top surface of a housing 6301, multiple cameras 6303 arranged on the side, a brush 6304, an operation button 6305, a secondary battery 6306, various sensors, and the like.
- the cleaning robot 6300 is equipped with tires, a suction port, and the like.
- the cleaning robot 6300 can move by itself, detect dirt 6310, and suck up the dirt from a suction port provided on the bottom surface.
- the cleaning robot 6300 can analyze an image captured by the camera 6303 and determine whether or not there is an obstacle such as a wall, furniture, or a step. When an object that may become entangled in the brush 6304, such as a wire, is detected by image analysis, the rotation of the brush 6304 can be stopped.
- the cleaning robot 6300 includes a secondary battery 6306 according to one embodiment of the present invention and a semiconductor device or electronic component therein. By using the secondary battery 6306 according to one embodiment of the present invention in the cleaning robot 6300, the cleaning robot 6300 can be an electronic device with long operating time and high reliability.
- Figure 30B shows an example of a robot.
- the robot 6400 shown in Figure 30B includes a secondary battery 6409, an illuminance sensor 6401, a microphone 6402, an upper camera 6403, a speaker 6404, a display unit 6405, a lower camera 6406, an obstacle sensor 6407, a movement mechanism 6408, a computing device, etc.
- the microphone 6402 has a function of detecting the user's voice and environmental sounds.
- the speaker 6404 has a function of emitting sound.
- the robot 6400 can communicate with the user using the microphone 6402 and the speaker 6404.
- the display unit 6405 has a function of displaying various information.
- the robot 6400 can display information desired by the user on the display unit 6405.
- the display unit 6405 may be equipped with a touch panel.
- the display unit 6405 may also be a removable information terminal, and by installing it in a fixed position on the robot 6400, charging and data transfer are possible.
- the upper camera 6403 and the lower camera 6406 have the function of capturing images of the surroundings of the robot 6400.
- the obstacle sensor 6407 can detect the presence or absence of obstacles in the direction of travel when the robot 6400 moves forward using the moving mechanism 6408.
- the robot 6400 can recognize the surrounding environment and move safely using the upper camera 6403, the lower camera 6406, and the obstacle sensor 6407.
- the robot 6400 includes a secondary battery 6409 according to one embodiment of the present invention and a semiconductor device or electronic component.
- the robot 6400 can be an electronic device with a long operating time and high reliability.
- Figure 30C shows an example of an aircraft.
- the aircraft 6500 shown in Figure 30C has a propeller 6501, a camera 6502, a secondary battery 6503, etc., and has the ability to fly autonomously.
- the flying object 6500 includes therein a secondary battery 6503 according to one embodiment of the present invention.
- the flying object 6500 can be an electronic device with a long operating time and high reliability.
- next-generation clean energy vehicles such as hybrid vehicles (HVs), electric vehicles (EVs), or plug-in hybrid vehicles (PHVs) can be realized.
- HVs hybrid vehicles
- EVs electric vehicles
- PVs plug-in hybrid vehicles
- FIG. 31A to 31C illustrate an example of a vehicle using a secondary battery according to one embodiment of the present invention.
- the automobile 8400 illustrated in FIG. 31A is an electric automobile using an electric motor as a power source for traveling.
- the automobile 8400 is a hybrid automobile that can appropriately select and use an electric motor and an engine as a power source for traveling.
- the automobile 8400 also includes a secondary battery. It is preferable to use the secondary battery by arranging the secondary battery modules illustrated in FIG. 26C and FIG. 26D on the floor of the vehicle.
- a battery pack in which a plurality of secondary batteries illustrated in FIG. 28 are combined may be installed on the floor of the vehicle.
- the secondary battery can not only drive the electric motor 8406, but also supply power to light-emitting devices such as the headlight 8401 and a room light (not shown).
- the secondary battery can also supply power to display devices such as a speedometer and a tachometer that the automobile 8400 has.
- the secondary battery can also supply power to semiconductor devices such as a navigation system that the automobile 8400 has.
- the automobile 8500 shown in FIG. 31B can charge the secondary battery of the automobile 8500 by receiving power supply from an external charging facility by a plug-in method and/or a non-contact power supply method.
- FIG. 31B shows a state in which a secondary battery 8024 mounted on the automobile 8500 is being charged from a ground-mounted charging device 8021 via a cable 8022.
- the charging method and connector standards may be appropriately performed using a predetermined method such as CHAdeMO (registered trademark) or combo.
- the charging device 8021 is preferably a charging station installed in a commercial facility, and is also preferably a home power source.
- the secondary battery 8024 mounted on the automobile 8500 can be charged by an external power supply using plug-in technology. Charging can be performed by converting AC power to DC power via a conversion device such as an AC-DC converter.
- a power receiving device can be mounted on the vehicle and charging can be performed by supplying power contactlessly from a power transmitting device on the ground.
- charging can be performed not only while the vehicle is stopped but also while it is moving.
- This contactless power supply method can also be used to transmit and receive power between vehicles.
- solar cells can be provided on the exterior of the vehicle to charge the secondary battery when the vehicle is stopped and/or moving. Electromagnetic induction and/or magnetic resonance methods can be used for such contactless power supply.
- FIG. 31C shows an example of a two-wheeled vehicle using the secondary battery of one embodiment of the present invention.
- a scooter 8600 shown in FIG. 31C includes a secondary battery 8602, a side mirror 8601, and a turn signal light 8603.
- the secondary battery 8602 can supply electricity to the turn signal light 8603.
- the scooter 8600 shown in FIG. 31C can store a secondary battery 8602 in the under-seat storage 8604.
- the secondary battery 8602 can be stored in the under-seat storage 8604 even if the under-seat storage 8604 is small.
- the secondary battery 8602 is removable, and when charging, the secondary battery 8602 can be carried indoors, charged, and stored before riding.
- the cycle characteristics of the secondary battery are improved, and the discharge capacity of the secondary battery can be increased. Therefore, the secondary battery itself can be made smaller and lighter. If the secondary battery itself can be made smaller and lighter, it will contribute to reducing the weight of the vehicle, and the cruising distance can be improved.
- the secondary battery mounted on the vehicle can be used as a power supply source for something other than the vehicle. In this case, for example, it is possible to avoid using a commercial power source during peak power demand. If it is possible to avoid using a commercial power source during peak power demand, it can contribute to energy conservation and reduction of carbon dioxide emissions. Furthermore, if the cycle characteristics are good, the secondary battery can be used for a long period of time, and the amount of rare metals used, including cobalt, can be reduced.
- AC impedance test and nail penetration test A secondary battery using the positive electrode active material of one embodiment of the present invention was subjected to an AC impedance test in a fully charged state, and then to a nail penetration test as a safety test. A method for manufacturing the secondary battery used in the nail penetration test is described below.
- lithium cobalt oxide (Cellseed C-10N, manufactured by Nippon Chemical Industry Co., Ltd.) having cobalt as the transition metal M and no particular additive element was prepared and sieved with an automatic sieving machine.
- this lithium cobalt oxide was placed in a sheath, covered, and heated at 850°C for 2 hours using a roller hearth kiln simulator furnace (manufactured by Noritake Company, Ltd.). Air (compressed air, sufficiently dried) was flowed in the furnace at 10 L/min. Specifically, the opening width of the exhaust port was adjusted so that the differential pressure gauge of the furnace indicated 5 Pa. When cooling the inside of the furnace, it was cooled at a rate of 200°C/hour, and the air flow was not stopped until it reached 200°C.
- LiF was prepared as the F source and MgF2 was prepared as the Mg source according to step S21 in Fig. 22A.
- LiF: MgF2 was weighed out to be 1:3 (molar ratio) and sieved with an automatic sieving machine.
- LiF and MgF2 were mixed in dehydrated acetone at a rotation speed of 500 rpm for 20 hours to prepare an additive element source (A1 source).
- step S31 the A1 source and the lithium cobalt oxide after the initial heating were mixed in a dry state.
- a Picobond manufactured by Hosokawa Micron
- the mixture was stirred at a rotation speed of 3000 rpm for 10 minutes and sieved with an automatic sieve to obtain mixture 903 (step S32).
- step S33 the mixture 903 was heated.
- the heating conditions were 900°C and 20 hours.
- a lid was placed on the scabbard containing the mixture 903.
- the inside of the scabbard was made into an oxygen-containing atmosphere, and the leakage of the oxygen could be blocked ( O2 purge).
- a roller hearth kiln simulator furnace manufactured by Noritake Company, Ltd.
- Oxygen was flowed into the furnace at 10 L/min ( O2 flow). Specifically, the opening width of the exhaust port was adjusted so that the differential pressure gauge of the furnace was 5 Pa (positive pressure inside the furnace).
- the inside of the furnace was cooled, it was cooled at a rate of 200°C/hour, and the flow of oxygen was not stopped until it reached 200°C. In this way, a composite oxide containing Mg and F was obtained (step S34a).
- step S51 nickel hydroxide that had undergone a crushing process was prepared as the nickel source, and aluminum hydroxide that had undergone a crushing process was prepared as the aluminum source, each of which was used as an additive element source (A2 source).
- A2 source additive element source
- step S51 the nickel hydroxide was weighed so that the nickel was 0.5 mol% of the cobalt, and the aluminum hydroxide was weighed so that the aluminum was 0.5 mol% of the cobalt, and the A2 source and the composite oxide were mixed in a dry state.
- a Picobond manufactured by Hosokawa Micron
- was used as the mixing means was used as the mixing means, and the mixture was stirred at a rotation speed of 3000 rpm for 10 minutes to obtain mixture 904 (step S52).
- step S53 the mixture 904 was heated.
- the heating conditions were 850° C. and 10 hours.
- a lid was placed on the scabbard containing the mixture 904.
- a roller hearth kiln simulator furnace manufactured by Noritake Company, Ltd.
- Oxygen was flowed into the furnace at 10 L/min ( O2 flow).
- the opening width of the exhaust port was adjusted so that the differential pressure gauge of the furnace was 5 Pa.
- the furnace was cooled it was cooled at a rate of 200° C./hour, and the flow of oxygen was not stopped until it reached 200° C. In this way, lithium cobalt oxide having Mg, F, Ni, and Al was obtained (step S54). In this way, the positive electrode active material 100 was made into sample 1.
- the above sample 1 was prepared as the positive electrode active material, acetylene black (AB) was prepared as the conductive material, and polyvinylidene fluoride (PVDF) was prepared as the binder. PVDF was prepared by dissolving it in N-methyl-2-pyrrolidone (NMP) at a weight ratio of 5% in advance.
- NMP N-methyl-2-pyrrolidone
- the positive electrode active material: AB: PVDF was mixed to be 95: 3: 2 (weight ratio) to prepare a slurry.
- NMP was used as the solvent for the slurry.
- the slurry was applied to an aluminum positive electrode current collector, and the solvent was then volatilized. It was confirmed that the thickness of the aluminum positive electrode current collector was 20 ⁇ m, and both sides were mirror surfaces.
- a pressing process was performed using a roll press machine after the solvent had evaporated.
- the pressing process was performed under conditions of a linear pressure of 210 kN/m.
- the upper and lower rolls of the roll press machine were both set at 120°C. 120°C is the temperature at which PVDF melts.
- the thickness of the positive electrode active material layer was 76 ⁇ m or more and 77 ⁇ m or less.
- a carbon material specifically, spherical natural graphite, was prepared as the negative electrode active material.
- SBR was prepared as a binder
- CMC was prepared as a thickener.
- Carbon fiber (VGCF (registered trademark), manufactured by Showa Denko K.K.) was prepared as a conductive material.
- VGCF registered trademark
- the spherical natural graphite: VGCF: CMC: SBR were mixed at a ratio of 97: 1: 1: 1 (weight ratio) to prepare a slurry. Water was used as the solvent for the slurry. The slurry was applied to a copper negative electrode current collector, and then the solvent was volatilized. The thickness of the copper negative electrode current collector was 18 ⁇ m.
- a pressing process was performed using a roll press machine after the solvent had evaporated.
- the pressing process was performed under a linear pressure of 28 kN/m, which was lower than that of the positive electrode.
- the thickness of the negative electrode active material layer was 122 ⁇ m or more and 124 ⁇ m or less.
- the negative electrode was obtained through the above process.
- the following table shows the structural formulas, flash points, boiling points, and melting points of EC and DEC.
- electrolyte A injected amount 100%
- electrolyte B was 75% of the injected amount of electrolyte A (25% reduction from electrolyte A)
- electrolyte C 50% of the injected amount of electrolyte A (50% reduction from electrolyte A) to satisfy the appropriate amount of electrolyte injected for the secondary battery subjected to the nail penetration test. That is, the injected amounts of electrolyte B and electrolyte C were appropriately smaller than that of electrolyte A.
- the electrolyte A was 89% of the volume
- the electrolyte B was 75% of the volume
- the electrolyte C was 45% of the volume.
- ⁇ Separator> As the separator, a polypropylene film having a thickness of 25 ⁇ m was used.
- An aluminum laminate film was used as the exterior body, which has, from the inside of the exterior body, a first polypropylene (thickness 22.5 ⁇ m), a second polypropylene (thickness 22.5 ⁇ m), an aluminum foil (thickness 40 ⁇ m), an adhesive layer (thickness 3 ⁇ m), and nylon (thickness 25 ⁇ m).
- a lithium ion secondary battery (cell A) was produced using the positive electrode having the sample 1 produced above, the negative electrode produced above, the electrolyte A, a separator, and an exterior body.
- a lithium ion secondary battery (cell B) was produced by replacing the electrolyte A with electrolyte B.
- a lithium ion secondary battery (cell C) was produced by replacing the electrolyte A with electrolyte C. After a predetermined amount of electrolyte was injected into cells A to C, a degassing sealer capable of reducing pressure was used to reduce the pressure to -60 kPa and seal the cells.
- the outer dimensions of cell A, cell B, and cell C were 76 mm x 89 mm.
- the area of the positive electrode coated on one side of the current collector was 20.5 cm 2
- the area of the negative electrode was 23.8 cm 2.
- the area of the negative electrode is preferably larger than the area of the positive electrode, and may be 1.1 times or more and 1.3 times or less.
- 15 positive electrodes coated with a positive electrode active material layer on both sides were prepared.
- 14 negative electrodes coated with a negative electrode active material layer on both sides and 2 negative electrodes coated with a negative electrode active material layer on one side were prepared.
- the positive electrode and the negative electrode were stacked so as to face each other.
- a separator was also placed between the positive electrode and the negative electrode.
- the negative electrode coated with a negative electrode active material layer on one side was located on the outermost side, so there was an area where the current collector of the negative electrode was in contact with the exterior body.
- the secondary batteries were assembled so that the amounts of positive and negative active materials carried were the same in Cell A, Cell B, and Cell C. Specifically, the amount of positive active material carried was adjusted to be 20 mg/cm2 or more and 22 mg/cm2 or less, and the amount of negative active material carried to be 13 mg/cm2 or more and 15 mg/cm2 or less. When active material layers are formed on both sides of the current collector, the amounts of positive and negative active materials carried are indicated as values per active material layer on one side of the current collector.
- the positive/negative electrode capacity ratio (%) in Cell A, Cell B, and Cell C is set to 80% or more and 90% or less, and preferably 83% or more and 85% or less.
- the positive/negative electrode capacity ratio (%) is calculated by considering the capacity as the product of the amount of support, the charge capacity of the active material (when charged at a charge voltage of 4.5 V (counter electrode graphite), a capacity of 200 mAh/g per weight of positive electrode active material and 300 mAh/g per weight of negative electrode active material is used), and the area.
- Fig. 32A shows the charge/discharge curves of cell A during the aging treatment (steps A7-A8) and the charge curve immediately before nail penetration, superimposed on each other.
- Fig. 32B shows the charge/discharge curves of cell B during the aging treatment (steps A7-A8) and the charge curve immediately before nail penetration, superimposed on each other.
- Fig. 32C shows the charge/discharge curves of cell A during the aging treatment (steps A7-A8) and the charge curve immediately before nail penetration, superimposed on each other.
- the horizontal axis shows capacity (mAh/g, capacity per weight of positive electrode active material), and the vertical axis shows voltage (V). From Figs.
- AC impedance measurement Cells A to C were subjected to alternating current impedance measurement (AC impedance measurement) in the above-mentioned fully charged state. Specifically, AC impedance measurement was started within 2 hours for cells A to C that were fully charged.
- the device used for AC impedance measurement was Biologic's VMP3 (multi-channel potentiostat/galvanostat).
- the measurement conditions were an amplitude voltage of 10 mV and a frequency sweep in the range from 200 kHz to 10 mHz, and then an impedance value corresponding to 1 kHz was extracted. 1001.6 Hz was used as the frequency closest to 1 kHz. When sweeping, 10 points per digit were measured.
- the measurement environment temperature was 25°C.
- Figure 33A shows a Nyquist diagram as the measurement result of the AC impedance measurement.
- Figure 33B shows an equivalent circuit used to analyze the measurement data of the AC impedance measurement.
- a fitting calculation was performed using the equivalent circuit of Figure 33B based on the Nyquist diagram of Figure 33A.
- R1 corresponds to the resistance due to the battery leads and wiring
- L corresponds to the inductance due to the battery leads and wiring
- R2 corresponds to the resistance of the electrolyte and the electrical resistance of the electrodes
- R3 corresponds to the film resistance on the positive electrode surface, the film resistance on the negative electrode surface, and the charge transfer resistance on the negative electrode surface (resistance associated with solvation and desolvation of Li ions and de-insertion of Li ions into the active material)
- R4 corresponds to the charge transfer resistance on the positive electrode surface (resistance associated with solvation and desolvation of Li ions and de-insertion of Li ions into the active material)
- CPE constant phase element
- the table below shows the impedance values at a frequency of 1 kHz for cells A to C as the analysis results of the AC impedance measurement.
- the variation in impedance at 1 kHz was small for cell A, but large for cells B and C. This suggests that the variation in impedance may be due to the amount of liquid injected.
- ⁇ Nail penetration test> After the AC impedance measurement, a nail penetration test was performed on Cell A to Cell C in a fully charged state.
- an Advanced Safety Tester manufactured by Espec Corporation
- Espec Corporation was used as a tester as shown in Figures 1A and 1B, and the tester was placed in an environment of 25°C.
- the temperature of the exterior bodies of Cell A, Cell B, and Cell C had reached 22°C.
- a temperature sensor was placed 2 cm away from the nail hole, and the value obtained from the temperature sensor was taken as the temperature of the exterior body. Even when the temperature gradient in the exterior body is taken into account by aligning the placement conditions of the temperature sensor, it is believed that there is no or only an extremely small difference in the temperature of the exterior body between Cell A, Cell B, and Cell C.
- a nail with a diameter of 3 mm was used as the nail 1003 in FIG. 1A.
- the nail penetration speed was 5 mm/sec.
- the nail penetration depth was the thickness of the secondary battery + 5 mm.
- the nail penetration test was performed in accordance with the description in SAE J2464 "Safety and Abuse Testing for Energy Storage Systems in Electric and Hybrid Vehicles.”
- the impedance of a safe secondary battery that will not ignite in the nail penetration test of this embodiment is less than 90 m ⁇ at 1 kHz, preferably 80 m ⁇ or less, and more preferably 60 m ⁇ or less.
- the condition for a secondary battery to ignite in a nail penetration test is surprisingly related to the impedance value, not the amount of electrolyte injected.
- Figures 36A to 36C are graphs in which the maximum value on the horizontal axis of Figures 35A to 35C has been aligned to 20 seconds, respectively.
- Figures 35A to 35C show that the battery voltage of cells A and C dropped gradually immediately after the nail was inserted.
- Figures 36A to 36C show that the battery voltage of cells A to C dropped sharply to approximately 1.0 V or less immediately after the nail was inserted, specifically within 2 seconds, but cells A and C showed a unique behavior in that the battery voltage rose to approximately 3.0 V immediately thereafter.
- the battery voltage of cell B also rose to approximately 3.0 V, but then dropped sharply to 0 V.
- Figures 38A to 38C are graphs in which the maximum value on the horizontal axis of Figures 37A to 37C is aligned to 20 seconds. No difference was observed in the temperature change up to 20 seconds for cells A to C, and the temperature rose gradually after nail penetration, with no sudden temperature change observed.
- the temperature that is considered to have been generated during the nail penetration test is the difference between the highest value obtained from the temperature sensor and 22°C (called the temperature rise ⁇ T).
- the temperature rise ⁇ T of cell A was 43°C
- the temperature rise ⁇ T of cell B was 207°C
- the temperature rise ⁇ T of cell C was 35°C.
- the cell temperature rise ⁇ T should be 50°C or less, preferably 45°C or less, and more preferably 40°C or less.
- 100 Positive electrode active material
- 100a Surface layer
- 100b Interior
- 100s Shell
- 101 Grain boundary
- 102 Crack
- 104 Coating
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Inorganic Chemistry (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- General Physics & Mathematics (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- Physics & Mathematics (AREA)
- Battery Electrode And Active Subsutance (AREA)
- Secondary Cells (AREA)
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2025509040A JPWO2024201260A1 (https=) | 2023-03-30 | 2024-03-25 | |
| CN202480017728.7A CN120858477A (zh) | 2023-03-30 | 2024-03-25 | 二次电池 |
| KR1020257029272A KR20250167577A (ko) | 2023-03-30 | 2024-03-25 | 이차 전지 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023055823 | 2023-03-30 | ||
| JP2023-055823 | 2023-03-30 | ||
| JP2023112480 | 2023-07-07 | ||
| JP2023-112480 | 2023-07-07 | ||
| JP2023-178349 | 2023-10-16 | ||
| JP2023178349 | 2023-10-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024201260A1 true WO2024201260A1 (ja) | 2024-10-03 |
Family
ID=92903893
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2024/052815 Ceased WO2024201260A1 (ja) | 2023-03-30 | 2024-03-25 | 二次電池 |
Country Status (4)
| Country | Link |
|---|---|
| JP (1) | JPWO2024201260A1 (https=) |
| KR (1) | KR20250167577A (https=) |
| CN (1) | CN120858477A (https=) |
| WO (1) | WO2024201260A1 (https=) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022229776A1 (ja) * | 2021-04-29 | 2022-11-03 | 株式会社半導体エネルギー研究所 | 二次電池、および電子機器 |
| JP2023033572A (ja) * | 2018-08-03 | 2023-03-10 | 株式会社半導体エネルギー研究所 | リチウムイオン二次電池の作製方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112201844A (zh) | 2017-06-26 | 2021-01-08 | 株式会社半导体能源研究所 | 正极活性物质的制造方法及二次电池 |
| JP6933149B2 (ja) | 2018-01-22 | 2021-09-08 | トヨタ自動車株式会社 | 非水電解質二次電池 |
| US20220029159A1 (en) | 2018-12-13 | 2022-01-27 | Semiconductor Energy Laboratory Co., Ltd. | Positive electrode active material, method for manufacturing the same, and secondary battery |
-
2024
- 2024-03-25 CN CN202480017728.7A patent/CN120858477A/zh active Pending
- 2024-03-25 KR KR1020257029272A patent/KR20250167577A/ko active Pending
- 2024-03-25 WO PCT/IB2024/052815 patent/WO2024201260A1/ja not_active Ceased
- 2024-03-25 JP JP2025509040A patent/JPWO2024201260A1/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2023033572A (ja) * | 2018-08-03 | 2023-03-10 | 株式会社半導体エネルギー研究所 | リチウムイオン二次電池の作製方法 |
| WO2022229776A1 (ja) * | 2021-04-29 | 2022-11-03 | 株式会社半導体エネルギー研究所 | 二次電池、および電子機器 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20250167577A (ko) | 2025-12-01 |
| JPWO2024201260A1 (https=) | 2024-10-03 |
| CN120858477A (zh) | 2025-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7466718B2 (ja) | リチウムイオン二次電池 | |
| JP2025163250A (ja) | 二次電池 | |
| JP2024028210A (ja) | 正極活物質および二次電池 | |
| JP7756147B2 (ja) | 正極、リチウムイオン二次電池、電子機器、蓄電システム、及び移動体 | |
| KR20250034949A (ko) | 양극 활물질의 제조 방법 및 양극 활물질 | |
| WO2023209474A1 (ja) | 正極活物質、リチウムイオン電池、電子機器、および車両 | |
| WO2022219456A1 (ja) | 二次電池、電子機器および車両 | |
| WO2024201260A1 (ja) | 二次電池 | |
| US20240038985A1 (en) | Battery | |
| WO2025083508A1 (ja) | 正極活物質の製造方法 | |
| CN117199500A (zh) | 电池 | |
| WO2024170994A1 (ja) | 正極活物質、リチウムイオン二次電池、電子機器、車両および複合酸化物の作製方法 | |
| WO2026022539A1 (ja) | リチウムイオン二次電池 | |
| WO2025177116A1 (ja) | 正極活物質 | |
| WO2024150082A1 (ja) | 電池 | |
| WO2024105514A1 (ja) | 二次電池 | |
| WO2025224596A1 (ja) | 正極活物質の作製方法 | |
| KR20250025616A (ko) | 양극 활물질, 이차 전지, 전자 기기, 및 차량 | |
| JP2025098971A (ja) | 正極活物質の作製方法、及び電池 | |
| WO2025083509A1 (ja) | 二次電池の作製方法 | |
| JP2025025701A (ja) | 二次電池、電子機器および車両 | |
| WO2025017418A1 (ja) | 二次電池の作製方法又は正極活物質の作製方法 | |
| WO2025083507A1 (ja) | 正極、二次電池および電子機器 | |
| WO2024161292A1 (ja) | 正極活物質、正極、二次電池、電子機器および車両 | |
| WO2024228087A1 (ja) | 二次電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24778392 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2025509040 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 1020257029272 Country of ref document: KR Free format text: ST27 STATUS EVENT CODE: A-0-1-A10-A15-NAP-PA0105 (AS PROVIDED BY THE NATIONAL OFFICE) |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202480017728.7 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 202480017728.7 Country of ref document: CN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 24778392 Country of ref document: EP Kind code of ref document: A1 |